Embed Size (px)
Citation preview

Psoriasiforme entz€undliche Dermatosen
H. Beltraminelli* und R. BlumUniversitätsklinik f€ur Dermatologie, Inselspital, Bern University Hospital, University of Bern, Bern, Schweiz
1 Psoriasis
Definition und klinisches Bild Die Psoriasis ist eine chronische, teils immunologisch bedingte papulos-quamöse Krankheit mit polygener Prädisposition, kombiniert mit Umweltfaktoren (Traumata, Infekte,Stress, Arzneimittel). Pathophysiologisch spielen Antigen-präsentierende Zellen, T-Lymphozyten, dendri-tische Zellen und Zellen der angeborenen Immunität zusammen mit Keratinozyten und Mechanismen derAngiogenese eine wesentliche Rolle (Nestlé et al. 2009). Die Psoriasis zählt zu den häufigsten Hauter-krankungen, die Prävalenz liegt zwischen 2–4 %. Eine positive Familienanamnese wurde bei 35–90 % derPatienten gefunden. Die meisten involvierten Gene haben immunologische Kodierungsfunktionen(z. B. f€ur TNF, IFN, IL-23). In bis zu 30 % manifestiert sich die Krankheit an Stellen geschädigter Haut(mechanisch, physikalisch, chemisch, immunologisch), auch isomorpher Reizeffekt oder Koebner Phäno-men genannt. Infekte (vor allem Streptokokken) sind insbesondere bei Kindern ein bekannter Auslöser vonErkrankungssch€uben. Bei pädiatrischen Patienten beobachtet man häufiger die exanthematischen Formender Krankheit, bei Erwachsenen vor allem die Plaqueform. Paradoxerweise wird manchmal das Auftreteneiner Psoriasis (de novo oder Exazerbation) im Rahmen einer Therapie mit verschiedenenTNF-a-Inhibitoren beobachtet. Auch das Auftreten der palmo-plantaren Pustulose wurde beschrieben.Die Krankheit ist charakterisiert durch eine hyperproliferative Epidermis mit fr€uhzeitiger Reifung derKeratinozyten durch verk€urzten Zellzyklus (Mitoserate etwa 8-fach erhöht) mit daraus resultierenderFehlverhornung die sich durch Präsenz von Kernresten im Stratum corneum (Parakeratose) auszeichnet.
Die psoriatische Arthritis ist die häufigste systemische Manifestation der Krankheit (5–30 %) und kannden Hauterscheinungen vorausgehen, diese begleiten, oder selten auch isoliert auftreten. Sie manifestiertsich am häufigsten mit einer asymmetrischen Oligoarthritis der kleinen Hand- und Fußgelenke. Patientenmit mittelschwerer bis schwerer Psoriasis haben ein erhöhtes relatives Risiko an metabolischem Syndromund kardiovaskulären Krankheiten zu erkranken. Auch Vitiligo, Morbus Crohn, Colitis ulcerosa, rheu-matoide Arthritis, Depression, Tumoren und Lymphome werden gehäuft bei Psoriasis gesehen(Christophers 2001). Die Psoriasis hat einen erheblichen Effekt auf die Lebensqualität der Patienten.Folgende Veränderungen zeigen klinisch und/oder histologisch gemeinsame Charakteristiken mit derPsoriasis und werden in der Literatur als deren Varianten diskutiert:
– ILVEN– Reaktive Arthritis (fr€uher Reiter-Syndrom)– Subkorneale pustulöse Dermatose (Sneddon-Wilkinson)– Klarzellakanthom
Die Assoziation von Psoriasis und reaktiver Arthritis wird später in diesem Kapitel diskutiert.Klinisch beobachtet man meist (85–90 %) scharf begrenzte erythematöse Plaques mit groblamellärer
silbriger Schuppung. Die Läsionen sind symmetrisch verteilt, Prädilektionsstellen sind die Streckseitenvon Ellbogen und Knien, präsakral, Nabel, retroaurikulär, okzipitale Kopfhaut. Die Patienten haben
*E-Mail: [email protected]
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 1 von 23

manchmal Juckreiz. Nach Entfernung der Schuppenkruste sieht man eine diagnostisch wichtige punkt-förmige Blutung (Auspitz-Phänomen). Die Psoriasis guttata tritt typischerweise nach einerStreptokokken-Infektion auf und ist meist bei pädiatrischen Patienten zu sehen. Hier sieht man mehrerekleine gerötete fein-schuppende Papeln am Stamm und an den proximalen Extremitäten. Die Psoriasisinversa, mit Befall der großen Falten stellt eine Sonderform dar, sie wird häufig durch eine lokaleSuperinfektion (Candida) ausgelöst (Koebner-Phänomen). Den scharf begrenzten roten Plaques fehlt indieser besonderen Lokalisation häufig die Schuppung. Seltener (2 %) sieht man die erythrodermatischeForm. Die Lingua geographica ist eine enorale Manifestation der Krankheit. Die Nägel werden bis zu80 % der Fälle befallen, man sieht eine Leukonychie, Ölflecken, Splitterhämorrhagien, kleine Gr€ubchen(auch T€upfelnägel genannt), und eine subunguale distale Hyperkeratose; diese Patienten leiden häufigzusätzlich an einer Psoriasis-Arthritis. Die palmoplantare Psoriasis tritt in etwa 20 % der Fälle auf.
Klinische Differenzialdiagnose Bei einer Lokalisation ausschließlich amRumpf sollteman die Diagnosen
– reaktive Arthritis (andere Vorgeschichte und Begleiterscheinungen),– Pityriasis rubra pilaris (follikuläre keratotische Papeln),– nummuläres Ekzem (starke Exsudation) und– Mycosis fungoides in die Differenzialdiagnose einbeziehen.
Ihnen allen fehlt das Auftreten im Bereich der Prädilektionsstellen.Bei der Psoriasis guttata sind eine Pityriasis lichenoides chronica, Pityriasis rosea (Primärplaque,
stammbetont) und Syphilis (positive Serologie) abzugrenzen.Am Kopf sollte – sofern möglich – das seborrhoische Ekzem abgegrenzt werden; es wurden jedoch
Mischformen beschrieben und als „Sebopsoriasis“ benannt.An den Unterschenkeln kann ein Lichen planus verrucosus ähnlich aussehen, die Ganzkörperunter-
suchung inklusive der bukkalen Mukosa ermöglicht in der Regel eine Klärung.An Händen und F€ußen kann ein chronisch-hyperkeratotisches Ekzem häufig schwierig abzugrenzen
sein, sowohl klinisch als auch histologisch.Auch eine Dyshidrosis kann in diesem Zusammenhang bei beiden Erkrankungen auftreten.Im intertriginösen Bereich muss eineCandida-Infektion ausgeschlossen werden, vor allem auch, weil
beide Entitäten gleichzeitig auftreten können.Bei Erythrodermie ist die Unterscheidung von einem T-Zell-Lymphom oder einem Arzneimittele-
xanthem manchmal sehr schwierig. Seltener geht die erythroderme Psoriasis sogar in ein T-Zell-Lymphom €uber.
Histologie Das histologische Bild ist sehr charakteristisch, kann jedoch je nach Lokalisation, Stadiumund Manifestationsform variieren (Ragaz und Ackerman 1979; Gordon und Johnson 1967).
Histopathologische Kriterien der Psoriasis vulgaris
– Hyperkeratose mit diffuser und konfluierender Parakeratose– Unregelmäßige Ausd€unnung, bzw. fehlendes Stratum granulosum– Gleichmäßige Akanthose durch Verlängerung der Reteleisten– Verschmälerte suprapapilläre Epidermis– Munro-Mikroabszesse– Erweiterte, gewundene Kapillaren in der papillären Dermis
(Fortsetzung)
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 2 von 23
– Superfizielle perivaskuläre Entz€undung mit Lymphozyten und neutrophilen Granulozyten– Vermehrt Nachweis von Mitosen im Stratum basale
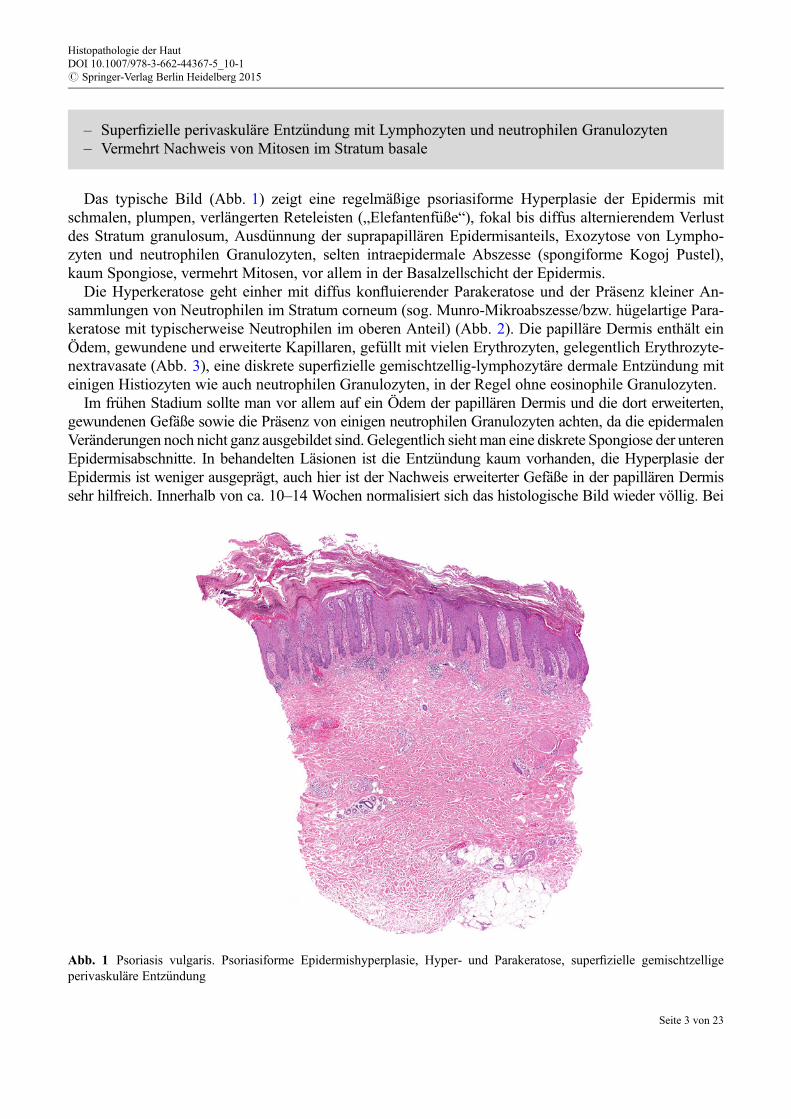
Das typische Bild (Abb. 1) zeigt eine regelmäßige psoriasiforme Hyperplasie der Epidermis mitschmalen, plumpen, verlängerten Reteleisten („Elefantenf€uße“), fokal bis diffus alternierendem Verlustdes Stratum granulosum, Ausd€unnung der suprapapillären Epidermisanteils, Exozytose von Lympho-zyten und neutrophilen Granulozyten, selten intraepidermale Abszesse (spongiforme Kogoj Pustel),kaum Spongiose, vermehrt Mitosen, vor allem in der Basalzellschicht der Epidermis.
Die Hyperkeratose geht einher mit diffus konfluierender Parakeratose und der Präsenz kleiner An-sammlungen von Neutrophilen im Stratum corneum (sog. Munro-Mikroabszesse/bzw. h€ugelartige Para-keratose mit typischerweise Neutrophilen im oberen Anteil) (Abb. 2). Die papilläre Dermis enthält einÖdem, gewundene und erweiterte Kapillaren, gef€ullt mit vielen Erythrozyten, gelegentlich Erythrozyte-nextravasate (Abb. 3), eine diskrete superfizielle gemischtzellig-lymphozytäre dermale Entz€undung miteinigen Histiozyten wie auch neutrophilen Granulozyten, in der Regel ohne eosinophile Granulozyten.
Im fr€uhen Stadium sollte man vor allem auf ein Ödem der papillären Dermis und die dort erweiterten,gewundenen Gefäße sowie die Präsenz von einigen neutrophilen Granulozyten achten, da die epidermalenVeränderungen noch nicht ganz ausgebildet sind. Gelegentlich sieht man eine diskrete Spongiose der unterenEpidermisabschnitte. In behandelten Läsionen ist die Entz€undung kaum vorhanden, die Hyperplasie derEpidermis ist weniger ausgeprägt, auch hier ist der Nachweis erweiterter Gefäße in der papillären Dermissehr hilfreich. Innerhalb von ca. 10–14 Wochen normalisiert sich das histologische Bild wieder völlig. Bei
Abb. 1 Psoriasis vulgaris. Psoriasiforme Epidermishyperplasie, Hyper- und Parakeratose, superfizielle gemischtzelligeperivaskuläre Entz€undung
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 3 von 23
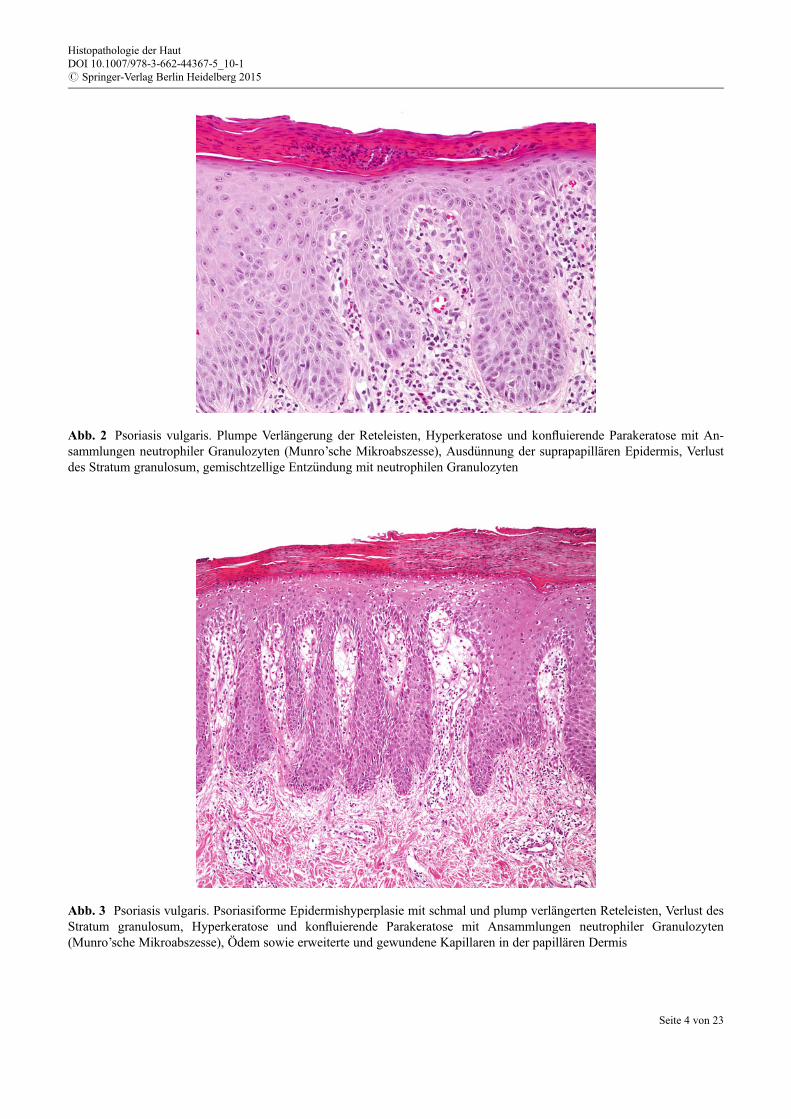
Abb. 2 Psoriasis vulgaris. Plumpe Verlängerung der Reteleisten, Hyperkeratose und konfluierende Parakeratose mit An-sammlungen neutrophiler Granulozyten (Munro’sche Mikroabszesse), Ausd€unnung der suprapapillären Epidermis, Verlustdes Stratum granulosum, gemischtzellige Entz€undung mit neutrophilen Granulozyten
Abb. 3 Psoriasis vulgaris. Psoriasiforme Epidermishyperplasie mit schmal und plump verlängerten Reteleisten, Verlust desStratum granulosum, Hyperkeratose und konfluierende Parakeratose mit Ansammlungen neutrophiler Granulozyten(Munro’sche Mikroabszesse), Ödem sowie erweiterte und gewundene Kapillaren in der papillären Dermis
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 4 von 23
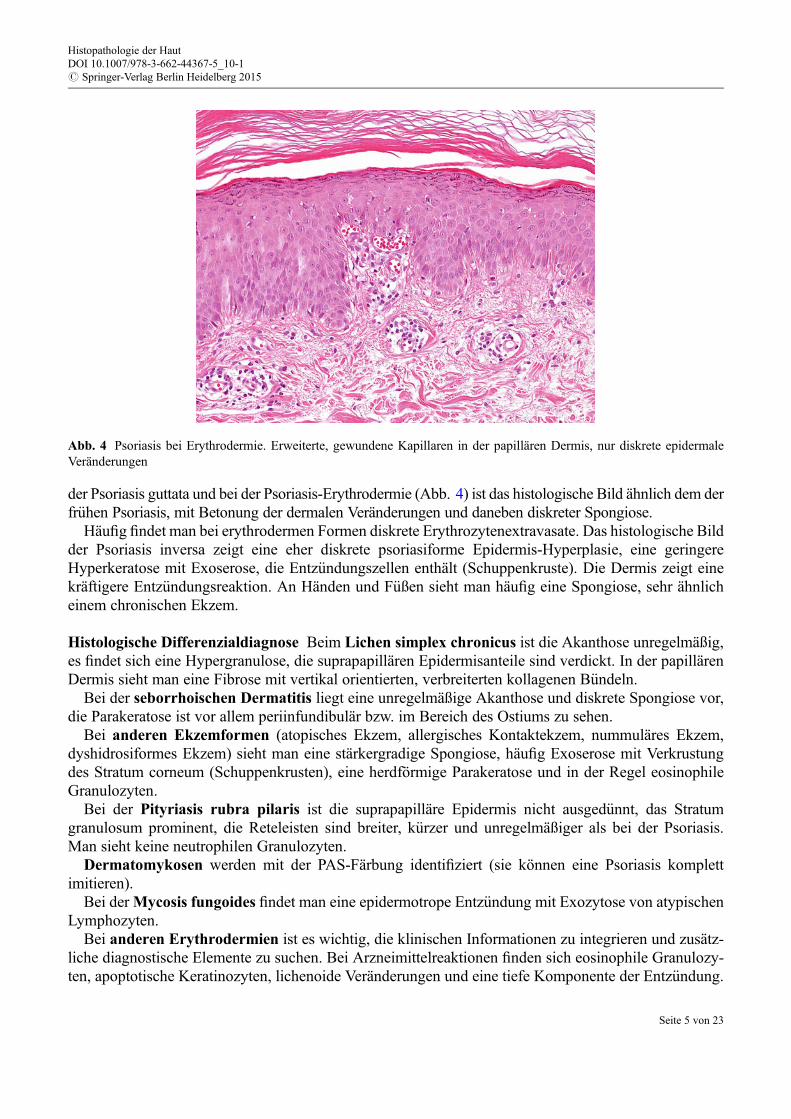
der Psoriasis guttata und bei der Psoriasis-Erythrodermie (Abb. 4) ist das histologische Bild ähnlich dem derfr€uhen Psoriasis, mit Betonung der dermalen Veränderungen und daneben diskreter Spongiose.
Häufig findet man bei erythrodermen Formen diskrete Erythrozytenextravasate. Das histologische Bildder Psoriasis inversa zeigt eine eher diskrete psoriasiforme Epidermis-Hyperplasie, eine geringereHyperkeratose mit Exoserose, die Entz€undungszellen enthält (Schuppenkruste). Die Dermis zeigt einekräftigere Entz€undungsreaktion. An Händen und F€ußen sieht man häufig eine Spongiose, sehr ähnlicheinem chronischen Ekzem.
Histologische Differenzialdiagnose Beim Lichen simplex chronicus ist die Akanthose unregelmäßig,es findet sich eine Hypergranulose, die suprapapillären Epidermisanteile sind verdickt. In der papillärenDermis sieht man eine Fibrose mit vertikal orientierten, verbreiterten kollagenen B€undeln.
Bei der seborrhoischen Dermatitis liegt eine unregelmäßige Akanthose und diskrete Spongiose vor,die Parakeratose ist vor allem periinfundibulär bzw. im Bereich des Ostiums zu sehen.
Bei anderen Ekzemformen (atopisches Ekzem, allergisches Kontaktekzem, nummuläres Ekzem,dyshidrosiformes Ekzem) sieht man eine stärkergradige Spongiose, häufig Exoserose mit Verkrustungdes Stratum corneum (Schuppenkrusten), eine herdförmige Parakeratose und in der Regel eosinophileGranulozyten.
Bei der Pityriasis rubra pilaris ist die suprapapilläre Epidermis nicht ausged€unnt, das Stratumgranulosum prominent, die Reteleisten sind breiter, k€urzer und unregelmäßiger als bei der Psoriasis.Man sieht keine neutrophilen Granulozyten.
Dermatomykosen werden mit der PAS-Färbung identifiziert (sie können eine Psoriasis komplettimitieren).
Bei derMycosis fungoides findet man eine epidermotrope Entz€undung mit Exozytose von atypischenLymphozyten.
Bei anderen Erythrodermien ist es wichtig, die klinischen Informationen zu integrieren und zusätz-liche diagnostische Elemente zu suchen. Bei Arzneimittelreaktionen finden sich eosinophile Granulozy-ten, apoptotische Keratinozyten, lichenoide Veränderungen und eine tiefe Komponente der Entz€undung.
Abb. 4 Psoriasis bei Erythrodermie. Erweiterte, gewundene Kapillaren in der papillären Dermis, nur diskrete epidermaleVeränderungen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 5 von 23

Die „psoriasiforme Keratose“ ist klinisch charakterisiert durch eine solitäre Läsion.
2 Psoriasis pustulosa
Definition und klinisches Bild Die pustulöse Psoriasis ist eine seltene Manifestation der Krankheit.Prädisponierende Faktoren sind eine Schwangerschaft (bekannt als Impetigo herpetiformis), abrupterAbbruch von systemischen Steroiden, Hypokalzämie, Infektionen und lokal irritierende Substanzen. Beider pustulösen Psoriasis steht ein Erythem mit sterilen Pusteln im Vordergrund. Die Läsionen könnenlokalisiert oder generalisiert auftreten. Es werden mehrere Typen unterschieden:
– Von-Zumbusch-Typ: mit einer schnell-auftretenden pustulösen Erythrodermie, Fieber, Arthralgien,reduziertem Allgemeinzustand und Abheilung unter Ausbildung einer charakteristischen lamellärenSchuppung bei konfluierten, eigetrockneten Pusteln
– Anulärer Typ: mit erythematös-anulären Läsionen, die sich zentrifugal ausbreiten und im Zentrumabheilen
– Exanthematischer Typ: mit einem akut-auftretenden Exanthem mit kleinen Pusteln und schnellerAbheilung, häufig nach einer Infektion oder Einnahme von besonderen Arzneimitteln; Überlappungenmit der akuten generalisierten exanthematösen Pustulose (AGEP) werden in der Literatur diskutiert
– Lokalisierter Typ: mit Auftreten von Pusteln in „klassischen“ Psoriasis-Plaques; die palmoplantar-pustulöse Psoriasis zeichnet sich durch chronische Präsenz steriler Pusteln an Handflächen und Fuß-sohlen aus. Häufig ist sie Teil des SAPHOSyndroms (Synovitis, Akne, Pustulose, Hyperostose, Osteitis)
– Akrodermatitis Hallopeau ist ein zusätzlicher Typ, charakterisiert durch Pusteln an den distalenFingern (und Zehen) und am Nagelbett
– Subkorneale Pustulose (Sneddon-Wilkinson) ist ein chronisch verlaufender Psoriasis-Typ mit großengruppierten Pusteln, die häufig zur Ausbildung eines Hypopyons f€uhren, im Verlauf bildet sich einecharakteristische krustöse Schuppung; assoziiert sind häufig eine IgA Gammopathie, ein Myelom oderandere neutrophile Dermatosen
Klinische Differenzialdiagnose Dermatomykosen, Impetigo, Arzneimittelexantheme vom TypAGEP und eine dyshidrotische Dermatitis (palmoplantar-pustulöse Variante) sollten abgegrenzt werden.
Histologie Gemeinsam charakteristisch f€ur die pustulöse Psoriasis ist die Akkumulation von neutrophi-len Granulozyten in der Epidermis. In den meisten Fällen sieht man eine intraepidermale spongiformePustel (Kogoj), es werden aber auch einzelne Neutrophile, subkorneale und intrakorneale AnsammlungenNeutrophiler (Munro´sche Mikroabszesse) beobachtet (Abb. 5 und 6).
Histopathologische Kriterien der Psoriasis pustulosa
– Kogoj´sche Makropustel– Gelegentlich spongiforme Pusteln– Munro-Mikroabszesse– Verlängerte Reteleisten– Hyperkeratose mit Parakeratose– Superfizielle perivaskuläre gemischtzellige dermale Entz€undung mit Neutrophilen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 6 von 23
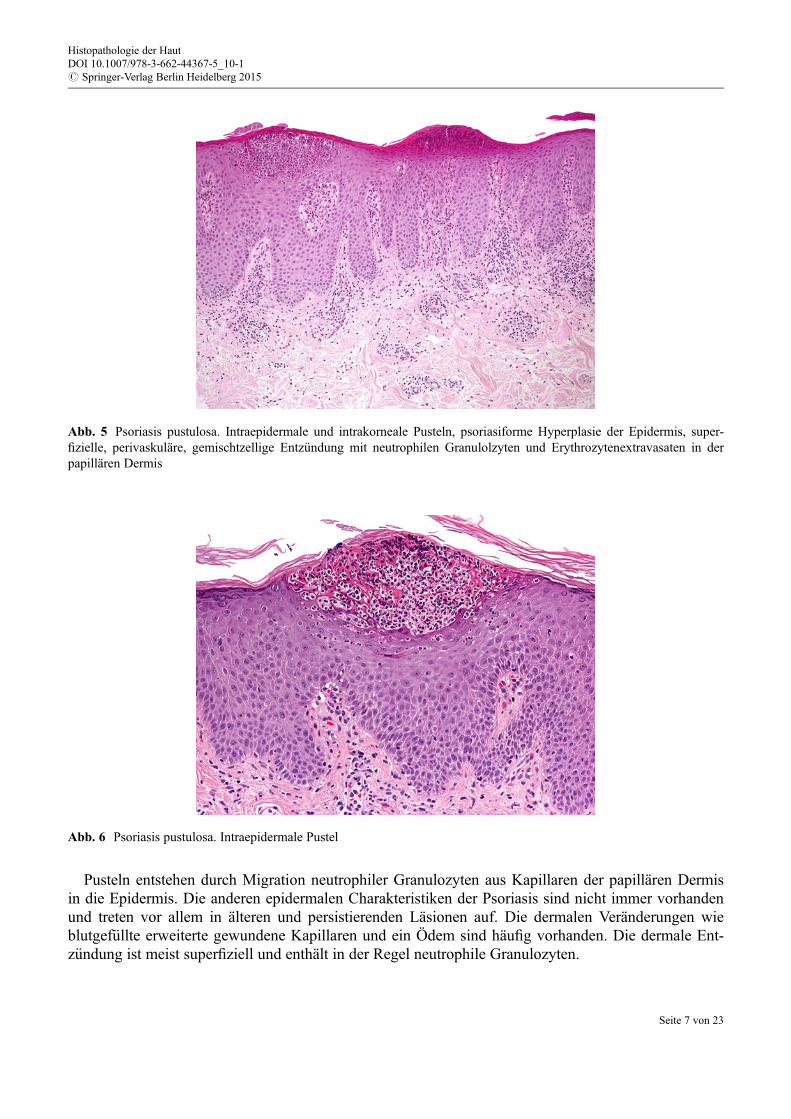
Pusteln entstehen durch Migration neutrophiler Granulozyten aus Kapillaren der papillären Dermisin die Epidermis. Die anderen epidermalen Charakteristiken der Psoriasis sind nicht immer vorhandenund treten vor allem in älteren und persistierenden Läsionen auf. Die dermalen Veränderungen wieblutgef€ullte erweiterte gewundene Kapillaren und ein Ödem sind häufig vorhanden. Die dermale Ent-z€undung ist meist superfiziell und enthält in der Regel neutrophile Granulozyten.
Abb. 6 Psoriasis pustulosa. Intraepidermale Pustel
Abb. 5 Psoriasis pustulosa. Intraepidermale und intrakorneale Pusteln, psoriasiforme Hyperplasie der Epidermis, super-fizielle, perivaskuläre, gemischtzellige Entz€undung mit neutrophilen Granulolzyten und Erythrozytenextravasaten in derpapillären Dermis
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 7 von 23

Histologische Differenzialdiagnose Intraepidermale Pusteln können bei folgenden Erkrankungen beo-bachtet werden: Dermatomykose, Impetigo, IgA Pemphigus und pustulösen Arzneimittelexanthe-men. Immer sollte in diesen Fällen eine Gram- und PAS-Färbung durchgef€uhrt werden. Bei Arznei-exanthemen ist das Auftreten einer lichenoiden Entz€undungskomponente mit apoptotischenKeratinozyten und eosinophilen Granulozyten in der dermalen Entz€undung oft richtungsweisend. BeimIgA Pemphigus findet man mit der direkten Immunofluoreszenz diagnostische intraepidermaleIgA-Ablagerungen, histologisch sieht man meist eine Akantholyse.
Beim dyshidrotischen Ekzem können ebenfalls intraepidermale Pusteln auftreten, in der Regel ist dieSpongiose jedoch ausgeprägter außerdem sind in der Dermis eosinophile Granulozyten nachzuweisen(Yoon et al. 2013).
3 Pityriasis rubra pilaris
Definition und klinisches Bild Die Pityriasis rubra pilaris (PRP) ist eine seltene erythrosquamöseErkrankung unklarer Genese. Manchmal wird die PRP durch UV-Strahlen ausgelöst. Man unterscheidetadulte, juvenile und HIV-assoziierte Formen (Sehgal und Srivastava 2006). Die meisten Patienten sindKaukasier, Männer und Frauen werden in gleichemMaße befallen. Typisch sind rötlich-orange schuppen-de Papeln und Plaques mit einem hyperkeratotischen follikulär-gebundenen Pfropf und einer besonderenpityriasiformen Schuppung an Gesicht und Kopf. Die Läsionen können konfluieren bis zum Auftreteneiner Erythrodermie mit typischen ausgesparten Zonen, sog. nappes claires.
Die PRP wird in 5 Typen klassifiziert (mit einem 6. HIV-assoziierten Typ; Griffiths 1980):
– Typ I: Die klassische adulte Form (>50 % der Fälle) beginnt typischerweise in der oberen Hälfte desKörpers, häufig im Gesicht oder an der Kopfhaut, und dehnt sich €uber den restlichen Körper aus (sog.kraniokaudale Ausbreitung). Anfangs sieht man erythematöse Papeln und Plaques mit follikulärenHyperkeratosen. Durch die Konfluenz der Herde breitet sich die Erkrankung symmetrisch aus, mehrerePatienten entwickeln im Verlauf eine Erythrodermie. Das Erythem hat eine besondere orange-gelbeFarbe, vor allem an den Handflächen und Fußsohlen. Typischerweise bleiben kleine Inseln nichtbefallener Haut inmitten der erythematokeratotischen Areale (nappes claires) €ubrig. Die Hautver-änderungen können jucken oder brennen. Follikulär gebundene Papeln auf der Dorsalfläche der Hände,Armen und Beinen sind charakteristisch. Die Läsionen zeigen im Verlauf eine diffuse Schuppung amKörper und eine betont groblamelläre Schuppung der ausgeprägten Hyperkeratosen an Händen undF€ußen. Die Kopfhaut zeigt häufig ein diskretes Erythem, ähnlich wie ein seborrhoisches Ekzem, mitfeinlamellärer Schuppung im Verlauf. Ein Ektropium ist häufig vorhanden. Die Nägel sind gelblich-bräunlich verfärbt, es bilden sich subunguale Hyperkeratosen, Splitterblutungen, longitudinale Fur-chen und Nagelverdickungen. Orale Manifestationen mit Rötungen und feinen, weißlichen Streifen,ähnlich wie beim Lichen planus, sind selten. Die Prognose ist in dieser Gruppe sehr gut mit Genesungder meisten Fälle innerhalb von 3–5 Jahren.
– Typ II. Dieser Typ ist eine seltene adulte Form (5 %) mit atypischem klinischen Bild und protrahier-tem, eher chronischen Verlauf. Die Schuppung ist mehr ichthyosiform, es bilden sich auch ekzematöseHerde und selten eine Alopezie. Die Prognose ist schlecht, nur 20% der Fälle zeigen eine Heilung nach3 Jahren.
– Typ III. Die klassische juvenile Form (10 %) ähnelt in ihrem klinischen Bild der klassischen adultenForm. Sie tritt gewöhnlich vor dem zweiten Lebensjahr auf und besteht oft nur im Kleinkindesalter,selten beobachtet man R€uckfälle im Verlauf.
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 8 von 23

– Typ IV. Bei der zirkumskripten Form (25 %) werden vor allem präpubertäre Kinder befallen. Klinischsieht man lokalisierte follikuläre Hyperkeratosen auf erythematösem Grund an Ellbogen und Knien,seltener an anderen Körperzonen. Gelegentlich zeigen sich auch palmoplantare Hyperkeratosen.
– Typ V. Die atypische juvenile Form (5–10 %) ist hereditär autosomal rezessiv. Eine fr€uhe Manifesta-tion und ein chronischer Verlauf sowie charakteristische follikuläre Hyperkeratosen und ein diskretesErythem sind kennzeichnend. Selten wird eine ichthyosiforme Schuppung beobachtet, sowie sklero-dermieartige Verdickungen der Haut an Händen und F€ußen.
– Typ VI. Dieser Typ ist HIV-assoziiert. Teilweise spricht der HIV-assoziierte Typ auf antiretroviraleTherapie an, ist aber generell sehr therapieresistent unter konventionellen PRP-Therapieschemata.
Klinische Differenzialdiagnose Die Psoriasis vulgaris stellt die wichtigste Differenzialdiagnose dar.Die follikulären Hyperkeratosen zusammen mit der orange-gelben Färbung palmo-plantar und die aus-gesparten Zonen helfen bei der Unterscheidung. Der Lichen planopilaris unterscheidet sich durch seinePrädilektionsstellen. Eine Erythrodermie mit feinlamellärer Schuppung der Kopfhaut und follikulärenHyperkeratosen kann auch bei kutanen T-Zell-Lymphomen vorkommen. Der Lichen spinulosus (alsVariante der Keratosis pilaris) stellt eine andere Differenzialdiagnose dar. Gewisse Formen von Ichthyoseund einKeratoderm können lokal klinisch ähnlich aussehen, der Verlauf und das gesamte Bild sind in derDifferenzierung von wesentlicher Bedeutung.
Histologie Die Diagnose wird vor allem klinisch gestellt, weil das histologische Bild weniger spezifischist. Um die follikulären Hyperkeratosen nachzuweisen, wird eine Biopsie im Bereich eines Haarfollikelsempfohlen, das charakteristische histologische Bild der PRP ist jedoch in einer geröteten Plaque zuerwarten (Soeprono 1986).
Histopathologische Kriterien der Pityriasis rubra pilaris
– Ortho- und Parakeratose alternierend in vertikaler und horizontaler Richtung (Schachbrettmuster)– Alternierende Hypergranulose– Mäßige Akanthose mit verbreiterten Reteleisten– Verdickte suprapapilläre Epidermis– Erweiterte Infundibula mit orthokeratotischen Hornpfropfen, Parakeratose der infundibulären
Öffnung– Superfizielle und perivaskuläre gemischtzellige Entz€undung in der Dermis
Zu sehen sind psoriasiforme und unregelmäßige epidermale Akanthose mit breiter, aber irregulärerHypergranulose, selten Spongiose mit Exozytose von vereinzelten Lymphozyten in der Epidermis(Abb. 7). Außerdem zeigt sich ein Stratum corneum mit einer vertikal und horizontal alternierendenHyper- und Parakeratose (sog. Schachbrettmuster). Letzteres Kriterium wird unterschiedlich interpretiertund ist gelegentlich auch bei klinisch eindeutiger PRP nicht nachzuweisen. Typisch ist auch eineCollerette-artige, perifollikuläre Hyper- und Parakeratose. Die Parakeratose ist nur diskret in fr€uhenLäsionen vorhanden. Die Reteleisten sind breiter und weniger verlängert als bei der Psoriasis, man siehtkeine suprapapilläre Ausd€unnung der Epidermis und keineMikroabszesse (Braun-Falco et al. 1983). Einesuprabasale Akantholyse und Dyskeratose werden beschrieben (Ko et al. 2011). Die erythrodermischenAreale zeigen gelegentlich ein verd€unntes oder fehlendes Stratum corneum, ein vermindertes Stratumgranulosum sowie ein Ödem in der oberen Dermis. In der Dermis kann eine diskrete gemischte super-
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 9 von 23

fizielle perivaskuläre und perifollikuläre Entz€undung nachgewiesen werden, selten auchmit Beimischungvon eosinophilen Granulozyten und Plasmazellen.
Histologische Differenzialdiagnose Psoriasis vulgaris: die Hypergranulose, die k€urzeren und verbrei-terten Reteleisten und das Fehlen von neutrophilen Granulozyten in Epidermis und Stratum corneumhelfen bei der Unterscheidung der PRP von der Psoriasis (Magro und Crowson 1997). Sofern vorhanden,sind die Akantholyse und Dyskeratose bei der PRP diagnostisch sehr hilfreich.
Die fr€uhe PRP am Kopf kann ein seborrhoisches Ekzem imitieren.Bei Vitamin A-Mangel sieht man keine fokale Parakeratose und keine dermale Entz€undung.
4 Nummuläres Ekzem
Synonym: nummulär-mikrobielles Ekzem, nummuläre Dermatitis
Definition und klinisches Bild Es handelt sich um eine idiopathische, klinisch-definierte und besondereErscheinungsform eines Ekzems. Die genaue Trennung von anderen Ekzemen wie atopisches Ekzem,Stauungsekzem oder asteatotisches Ekzem ist manchmal schwierig, weil Überlappungsformen immerwieder beobachtet werden. Obwohl pathogenetisch vermutet, ist eine Infektion der Ekzemherde kaumnachweisbar. Es werden mehrere Ursachen bzw. prädisponierenden Faktoren diskutiert (Jiamtonet al. 2013; Schubert 2002; Buonamonte et al. 2012): atopische Diathese, atopisches Ekzem, Kontaktal-lergie (Nickel, Chrom, Kobalt), Xerodermie und Alkoholabusus. Vor allem erwachsene Männer sindbetroffen. Zu den typischen Lokalisationen gehören die Extremitäten-Streckseiten, vor allem Unterschen-kel, Unterarme, Stamm und Handr€ucken. Das nummuläre Ekzem ist durch mehrere, 1–3 cm messende,rundlich-ovale (nummuläre, m€unzförmige) Ekzemherde mit feinlamellärer, festhaftender Schuppunggekennzeichnet. In der Entwicklung häufig vesikulös-nässend-impetiginisiert oder krustös, manchmal
Abb. 7 Pityriasis rubra pilaris. Alternierende Hyper- und Parakeratose (Schachbrettmuster), mäßige Akanthose der Epidermismit verbreiterten Reteleisten
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 10 von 23

hyperkeratotisch und lichenifiziert, typischerweise streuend. Der Verlauf ist chronisch-rezidivierend undes zeigt sich eine hartnäckige Therapieresistenz. Die meisten Patienten haben einen starken Juckreiz, esbilden sich Kratzexkoriationen und Superinfektionen. Das Sulzberger-Garbe-Syndrom (Oid-oiddisease – diskoide und lichenoide chronische Dermatitis) stellt eine besondere, therapieresistente Variantemit größeren Läsionen und Gesichtsbefall dar (Sulzberger und Garbe 1985).
Klinische Differenzialdiagnose Schwierig ist eine eindeutige Differenzierung nummulärer Läsionenvon anderen Ekzemformen. Einzelne Psoriasisherdeweisen in der Regel keine Exsudation auf. Bei derImpetigo contagiosa entstehen sehr schnell rupturierende, zarte Vesikel. Die Dermatomykose wird mitHilfe eines einfachen Direktpräparates der Schuppen schnell ausgeschlossen, beim nummulären Ekzemsind außerdem die Ränder kaum erhaben. Bei differenzialdiagnostischem Verdacht auf einen MorbusBowen sollte die Diagnose histologisch verifiziert werden.
Histologie Das histologische Bild kann im Verlauf variieren. Fr€uhe Läsionen zeigen eine epidermaleSpongiose, spongiotische Bläschen, eine diskrete Akanthose mit eher breiten Reteleisten und Exozytosevon Lymphozyten und neutrophilen Granulozyten. In der Folge zeigt sich eine psoriasiforme Hyperplasieder Epidermis, verglichen mit der Psoriasis jedoch viel unregelmäßiger.
Histopathologische Kriterien des nummulären Ekzems
– Variable Akanthose– Fokale Spongiose, spongiotische Bläschen– Orthokeratose mit fokaler Parakeratose und Exoserose, teils mit Neutrophilen (Schuppenkrusten)– Ödem der papillären Dermis– Superfizielles perivaskuläres und interstitielles Infiltrat
Häufig beobachtet man eine Hyperkeratose mit fokaler Parakeratose, Exoserose und Krustenbildung.Die Dermis enthält eine superfizielle, perivaskuläre, gemischtzellige Entz€undung mit Beteiligung voneosinophilen Granulozyten, vereinzelten Neutrophilen und Plasmazellen. Gelegentlich findet sich imAnfangsstadium ein Ödem der papillären Dermis. Durch chronisches Reiben und Kratzen wird dieEpidermis stärker akanthotisch und imitiert das histologische Bild des Lichen simplex chronicus.
Die Histologie der Sulzberger-Garbe-Krankheit ist häufig mit dem nummulären Ekzem identisch.Zusätzlich zeigen sich manchmal apoptotische Keratinozyten entlang der Basalzellschicht. Ein perivas-kuläres Ödem und erweiterte Kapillaren mit geschwollenen Endothelien in der papillären Dermis stellenzusätzliche Charakteristiken dar.
Histologische Differenzialdiagnose Das allergische Kontaktekzem, die atopische Dermatitis wieauch das dyshidrosiforme Ekzem sind histologisch nicht immer eindeutig vom nummulären Ekzemabgrenzbar. Bei toxischen Dermatitiden finden sich Einzelzellnekrosen von Keratinozyten, gelegentlichauch Defekte des Stratum corneums. Bei allergischen Kontaktekzemen sieht man in der Regel vieleeosinophile Granulozyten beim oberflächlichen Infiltrat. Bei der Dermatomykose sind in der Regelneutrophile Granulozyten im Stratum corneum nachzuweisen, Eosinophile Granulozyten werden dage-gen kaum gesehen. Die PAS-Färbung stellt Hyphen und Sporen gut dar.
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 11 von 23

5 Lichen simplex chronicus
Synonyme Lichen Vidal, Neurodermitis circumscripta
Definition und klinisches Bild Der Lichen simplex chronicus (LSC) ist ein besonderes, jedoch un-spezifisches Reaktionsmuster der Haut infolge chronisch-mechanischer Irritation, das lokalisiert oder anmehreren Hautstellen im Rahmen zahlreicher verschiedener, vor allem juckender Hauterkrankungenauftreten kann. Je nach Ausmaß, Art und Dauer der Irritation (in der Regel Reiben und Kratzen) entstehteine wechselnd ausgeprägte Verdickung oder gar knotige und exkoriierte Veränderung der Haut. DieHautläsionen des LSC jucken meist extrem und spontan und f€uhren zu einem „Teufelskreis“ (Jucken-Kratzen-Verletzen-Jucken). Pathophysiologisch wurde eine erhöhte Proliferation der Keratinozyten in derEpidermis diskutiert. Mikrotraumen durch Kratzen erklären teilweise den lokalisierten und starkenJuckreiz. Die Prurigo (nodularis) kann als Maximalvariante des LSC gesehen werden.
Der LSC ist durch umschriebene, flache, infiltrierte, lichenifizierte und leicht hyperpigmentierte Haut-läsionen gekennzeichnet. Je nach Situation können noch folgende zusätzliche Elemente gesehen werden:eine variable Rötung, Kratzexkoriationen, feine Krusten. Die Läsionen können solitär oder multipel sein.Prädilektionsstellen sind die Streckseiten der Extremitäten, die Dorsalseite der Hände und F€uße, derNacken, die okzipitale Kopfhaut, die Vulva, die Analregion, das Skrotum. Es gibt viele prädisponierendeFaktoren, die wichtigsten sind trockene Haut, Atopie, Stauungsdermatitis, Pruritus als Folge von ver-schiedenen internistischen Erkrankungen und psychologische Faktoren wie Stress, Angstzustände,obsessiv-kompulsive Störungen. Wenn der LSC im Rahmen einer juckenden Dermatose auftritt, zeigtdie klinische Untersuchung zusätzlich die Morphe der zugrundeliegenden Hauterkrankung.
Klinische Differenzialdiagnose Der hypertrophe Lichen planus (der gelegentlich auch als Mischbilddes Lichen planus mit juckreizbedingten Sekundärveränderungen eines LSC interpretiert wird) kann imBereich der Unterschenkel sehr ähnlich aussehen. Eine sorgfältige Untersuchung des gesamten Integu-ments inklusive der Nägel und Mundschleimhaut kann in diesem Fall sehr hilfreich sein.
Histologie Das histologische Bild ist gelegentlich wenig spezifisch und verlangt zur korrekten Interpre-tation eine klinisch-pathologische Korrelation. Die Epidermis zeigt eine unregelmäßige Akanthose mitsägezahnartiger Verlängerung der Reteleisten, eine Hypergranulose und kompakte Hyperkeratose(Abb. 8; Marks undWells 1973). Dieses Bild wird auch mit der Bezeichnung „hairy palm“ umschrieben:im Bereich einer scheinbar akral differenzierten Epidermis offenbart der Nachweis von Haarfollikeln (diepalmoplantar nicht vorhanden sind) eine zwingend vorausgegangene, chronische mechanische Irritation.
Histopathologische Kriterien des Lichen simplex chronicus
– Kompakte Hyperkeratose mit fokaler Parakeratose– Diffuse und breite Hypergranulose– Unregelmäßige Akanthose mit breiten verlängerten Reteleisten– „Hairy palm sign“– Dermale Fibrose mit vermehrt Fibroblasten
Seltener sieht man Exkoriationen und eine fokale Parakeratose, gelegentlich auch eine diskreteSpongiose, eine Papillomatose und manchmal eine Zunahme der Anzahl der basalen Melanozyten
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 12 von 23
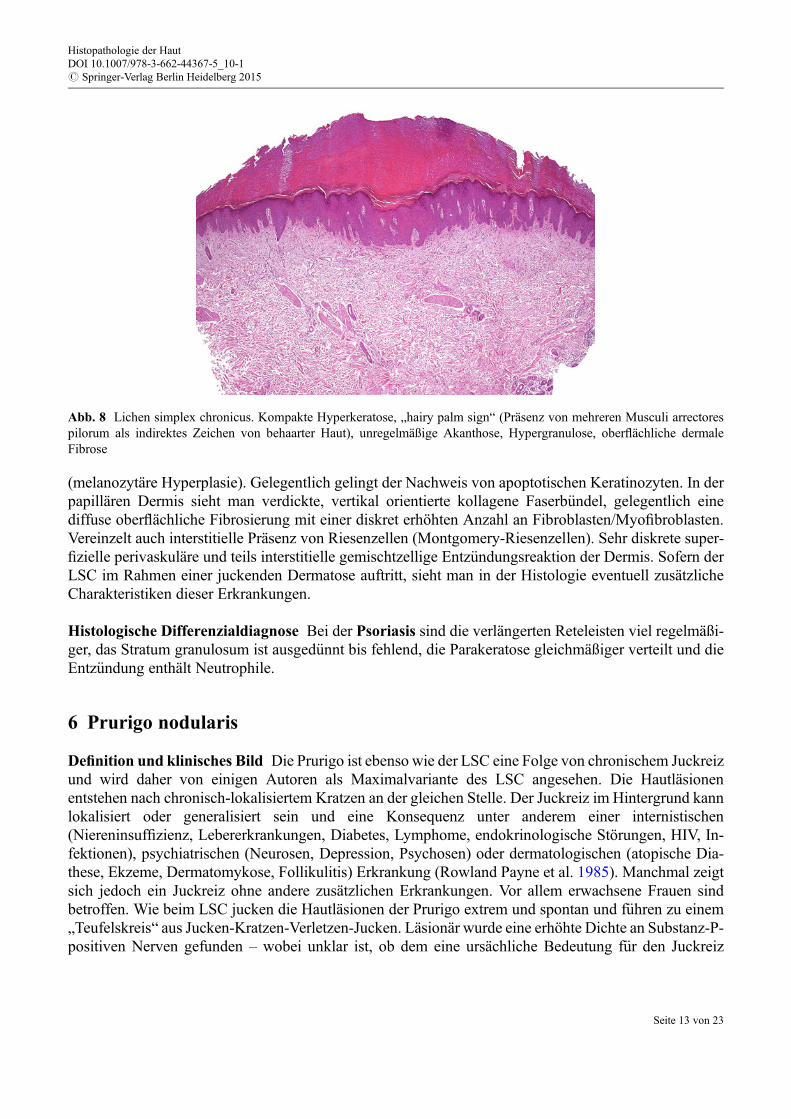
(melanozytäre Hyperplasie). Gelegentlich gelingt der Nachweis von apoptotischen Keratinozyten. In derpapillären Dermis sieht man verdickte, vertikal orientierte kollagene Faserb€undel, gelegentlich einediffuse oberflächliche Fibrosierung mit einer diskret erhöhten Anzahl an Fibroblasten/Myofibroblasten.Vereinzelt auch interstitielle Präsenz von Riesenzellen (Montgomery-Riesenzellen). Sehr diskrete super-fizielle perivaskuläre und teils interstitielle gemischtzellige Entz€undungsreaktion der Dermis. Sofern derLSC im Rahmen einer juckenden Dermatose auftritt, sieht man in der Histologie eventuell zusätzlicheCharakteristiken dieser Erkrankungen.
Histologische Differenzialdiagnose Bei der Psoriasis sind die verlängerten Reteleisten viel regelmäßi-ger, das Stratum granulosum ist ausged€unnt bis fehlend, die Parakeratose gleichmäßiger verteilt und dieEntz€undung enthält Neutrophile.
6 Prurigo nodularis
Definition und klinisches Bild Die Prurigo ist ebenso wie der LSC eine Folge von chronischem Juckreizund wird daher von einigen Autoren als Maximalvariante des LSC angesehen. Die Hautläsionenentstehen nach chronisch-lokalisiertem Kratzen an der gleichen Stelle. Der Juckreiz im Hintergrund kannlokalisiert oder generalisiert sein und eine Konsequenz unter anderem einer internistischen(Niereninsuffizienz, Lebererkrankungen, Diabetes, Lymphome, endokrinologische Störungen, HIV, In-fektionen), psychiatrischen (Neurosen, Depression, Psychosen) oder dermatologischen (atopische Dia-these, Ekzeme, Dermatomykose, Follikulitis) Erkrankung (Rowland Payne et al. 1985). Manchmal zeigtsich jedoch ein Juckreiz ohne andere zusätzlichen Erkrankungen. Vor allem erwachsene Frauen sindbetroffen. Wie beim LSC jucken die Hautläsionen der Prurigo extrem und spontan und f€uhren zu einem„Teufelskreis“ aus Jucken-Kratzen-Verletzen-Jucken. Läsionär wurde eine erhöhte Dichte an Substanz-P-positiven Nerven gefunden – wobei unklar ist, ob dem eine ursächliche Bedeutung f€ur den Juckreiz
Abb. 8 Lichen simplex chronicus. Kompakte Hyperkeratose, „hairy palm sign“ (Präsenz von mehreren Musculi arrectorespilorum als indirektes Zeichen von behaarter Haut), unregelmäßige Akanthose, Hypergranulose, oberflächliche dermaleFibrose
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 13 von 23

zukommt oder es sich nur um ein reaktives Phänomen handelt (Lee und Shumack 2005). Das vermehrt inNervenfasern der Prurigo exprimierte Calzitonin-Gene-Related-Peptide (CGRP) spielt eine gewisse Rollebei der Rekrutierung und Regulation von eosinophilen Granulozyten und Mastzellen.
Die Läsionen sind oft symmetrisch, manchmal gruppiert an den Streckseiten der Extremitäten, R€uckenSchulterregion, lumbosakral, Gesäß. Typischerweise ist eine mit den Händen schlecht erreichbare Zoneam zentralen oberen R€ucken ausgespart (Schmetterling-Zeichen), ebenso auch die volaren Zonen derExtremitäten und das Gesicht. Klinisch sieht man eine indurierte, erhabene papulo-noduläre Läsion mitmanchmal verrukösem Aspekt und zentraler Exkoriation/Erosion/Ulzeration oder Kruste. Die Farbevariiert zwischen Hautfarbe, rot und braun. Die Läsionen heilen oft mit einer hypopigmentierten Narbeab. Die umgebende Haut kann lichenifiziert sein.
Klinische Differenzialdiagnose Perforierende Dermatosen, hypertropher Lichen planus, Skabies, per-sistierende Arthropodenstich-Reaktionen, multiple Keratoakanthome m€ussen ausgeschlossen werden.
Histologie Die Histologie zeigt eine ausgeprägte und irreguläre Hyperplasie der Epidermis, gelegentlichsieht man eine pseudokarzinomatöse Hyperplasie, das Stratum granulosum ist verdickt (Abb. 9; Weigeltet al. 2010). Manchmal werden vereinzelt atypische Keratinozyten und Mitosen nachgewiesen, die indiesem Kontext als reaktiv interpretiert werden. Häufig sieht man eine Exkoriation/Erosion oder Ulze-ration mit aufgeworfenen epidermalen Rändern. Gelegentlich findet sich eine diskrete Spongiose, einStratum corneum mit kompakter Hyperkeratose und fokaler Parakeratose, Exoserose und Krustenbil-dung. Manchmal werden apoptotische Keratinozyten nachgewiesen.
In der papillären Dermis sieht man verdickte, vertikal orientierte Kollagenfaserb€undel, gelegentlichfindet sich eine diffuse oberflächliche Fibrosierung mit einer diskreten Zunahme der Anzahl der Fibrob-lasten/Myofibroblasten. Vereinzelt wird auch die interstitielle Präsenz von Riesenzellen (Montgomery-Riesenzellen) beobachtet. Die Kapillaren sind vermehrt und erweitert (Doyle et al. 1979). Manchmal
Abb. 9 Prurigo. Unregelmäßige Akanthose mit Verlängerung der Reteleisten und pseudokarzinomatöser Hyperplasie derEpidermis im Zentrum der Läsion, fokale Exkoriation mit aufgeworfenen Rändern, diffuse dermale Fibrose und gemischt-zellige Entz€undung
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 14 von 23

können prominente Musculi arrectores pilorum nachgewiesen werden. Eine superfizielle, gemischtzelligeEntz€undungsreaktion ist häufig vorhanden, manchmal sogar unter Ausbildung von vereinzelten Lymph-follikeln. Bei epithelialen Defekten sind zusätzlich neutrophile Granulozyten und einige Plasmazellennachzuweisen. Eine Hypertrophie und Vermehrung von Nervenfasern wurde in der Literatur diskutiert(Harris et al. 1992). Wenn die Prurigo im Rahmen einer juckenden Dermatose auftritt, sind zusätzlich inder Histologie Charakteristiken der jeweiligen Erkrankung zu sehen.
Histopathologische Merkmale der Prurigo nodularis
– ausgeprägte Hyperkeratose mit Parakeratose– unregelmäßige Akanthose mit starker Verlängerung der Reteleisten bis hin zu pseudokarzinoma-
töser Hyperplasie– häufig Erosion/Ulzeration– dermale Fibrose mit vermehrt Fibroblasten– verdickte, vertikal ausgerichtete Kollagenfaserb€undel in der papillären Dermis– gemischtzellige dermale Entz€undung
Histologische Differenzialdiagnose Beim hypertrophen Lichen planus ist der Nachweis einerInterface-Dermatitis wegweisend. Beim chronisch-aufgekratzten Ekzem sieht man häufig zusätzlicheosinophile Granulozyten. Manchmal ist die diffenzialdiagnostische Abgrenzung von einem gut diffe-renzierten Plattenepithelkarzinom schwierig, die klinischen Informationen ermöglichen hier die wei-tere Zuordnung.
7 Nekrolytisches migratorisches Erythem (Glukagonom Syndrom)
Definition und klinisches Bild Die Bezeichnung nekrolytisches migratorisches Erythem leitet sich vonder morphologischen Ähnlichkeit zum TEN-Syndrom (toxische epidermale Nekrolyse) ab und stellt eineklinisch definierte Entität dar (Binnick et al. 1977). In den allermeisten Fällen ist ursächlich ein Glukagon-produzierender Pankreas-Tumor der a-Zellen assoziiert. Das nekrolytische migratorische Erythem wirddaher auch synonym als Glukagonom-Syndrom gef€uhrt und als obligate Präkanzerose angesehen(Hautveränderungen in rund 70 % der Glukagonome). Beide Sichtweisen sind jedoch insofern fraglich,als die Hauterscheinungen selten auch bei Leberzirrhose, chronischer Hepatitis, Pankreasinsuffizienzoder auch anderen intestinalen oder sonstigen Tumoren (Adenokarzinom des Jejunum, Kolonkarzinom,neuroendokriner Tumor) vorkommen können (Tierny und Badger 2004). Ist der Glukagonspiegel normal,so spricht man von einem Pseudoglukagonom-Syndrom. Außerdem ist das klinische und histologischeBild speziell auch von demjenigen bei Zinkmangel-Syndromen oder Biotin-Mangel (gelegentlich auchMalabsorption, Diarrhoen, Verlust an essentiellen Fettsäuren und Aminosäuren) nicht zu unterscheiden,sodass diese Erkrankungen mit unterschiedlicher Ätiologie klinisch und histologisch immer differenzial-diagnostisch zu betrachten sind.
Das Glukagonom-Syndrom ist charakterisiert durch ein migratorisches Erythem: typisch sind inguinal-perineal schuppende, erythematös-erosive Hautveränderungen, teils anulär bzw. randbetont und zirzinär,gelegentlich mit Pusteln, Krusten und Pigmentverschiebungen. Selten sind im weiteren Verlauf Stamm,Beine und Perioralregion (anguläre Cheilitis) betroffen. Es treten ein Diabetes mellitus, Gewichtsverlustund Anämie auf. Mit einer Operation des zugrunde liegenden Pankreastumors verschwinden die Haut-
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 15 von 23
veränderungen ebenso wie durch Substitutionsernährung mit Aminosäuren beim Pseudoglukagonom-Syndrom. Pathophysiologisch ursächlich wird daher eine Störung des Proteinstoffwechsels angenommen(Tierny und Badger 2004; van Beck et al. 2004).
Klinische Differenzialdiagnose Das nekrolytische migratorische Erythem wird häufig als Intertrigobzw. auch speziell Candida-Intertrigo oder seborrhoisches Ekzem verkannt, wobei die Histologie in derRegel eine Abgrenzung ermöglicht. Aufgrund der These der multifaktoriellen Mangelernährung mitpraktisch identischer klinischer Präsentation an der Haut bleiben Glukagonom-Syndrom,Acrodermatitis enteropathica, Pellagra wie auch angeborene und nutritive Mangelzustände unter-schiedlichster Art letztlich immer Differenzialdiagnosen.
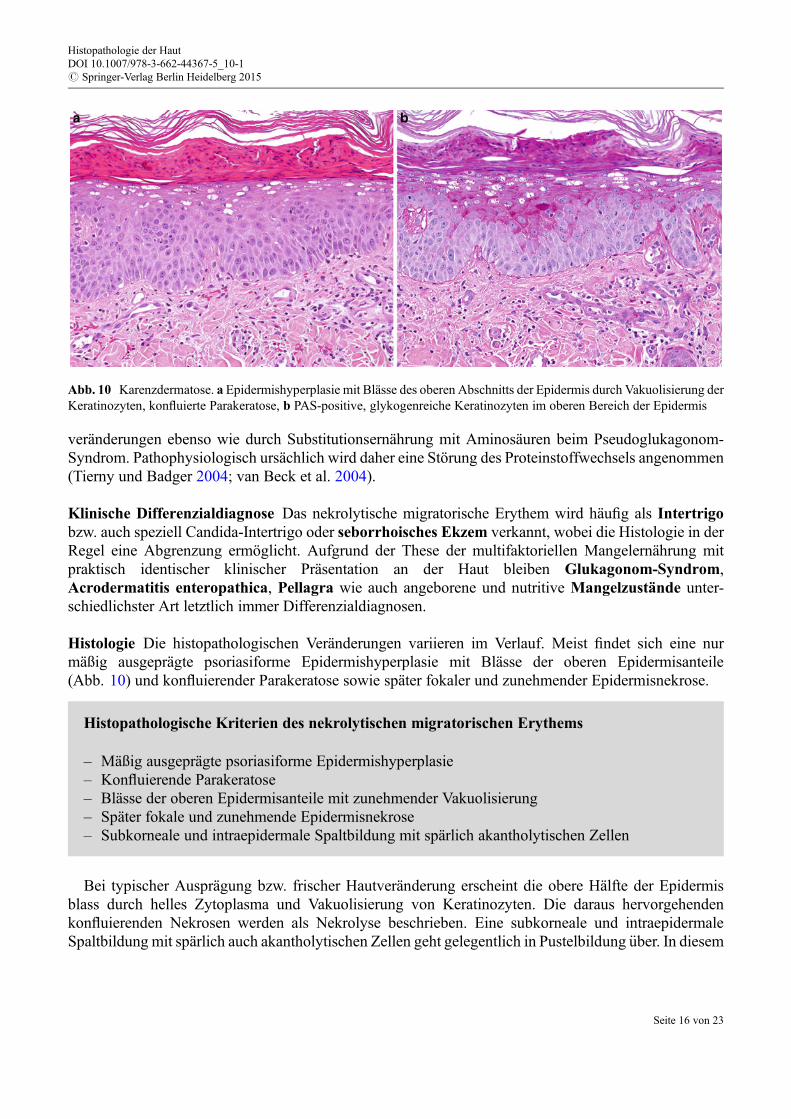
Histologie Die histopathologischen Veränderungen variieren im Verlauf. Meist findet sich eine nurmäßig ausgeprägte psoriasiforme Epidermishyperplasie mit Blässe der oberen Epidermisanteile(Abb. 10) und konfluierender Parakeratose sowie später fokaler und zunehmender Epidermisnekrose.
Histopathologische Kriterien des nekrolytischen migratorischen Erythems
– Mäßig ausgeprägte psoriasiforme Epidermishyperplasie– Konfluierende Parakeratose– Blässe der oberen Epidermisanteile mit zunehmender Vakuolisierung– Später fokale und zunehmende Epidermisnekrose– Subkorneale und intraepidermale Spaltbildung mit spärlich akantholytischen Zellen
Bei typischer Ausprägung bzw. frischer Hautveränderung erscheint die obere Hälfte der Epidermisblass durch helles Zytoplasma und Vakuolisierung von Keratinozyten. Die daraus hervorgehendenkonfluierenden Nekrosen werden als Nekrolyse beschrieben. Eine subkorneale und intraepidermaleSpaltbildung mit spärlich auch akantholytischen Zellen geht gelegentlich in Pustelbildung €uber. In diesem
Abb. 10 Karenzdermatose. a Epidermishyperplasie mit Blässe des oberen Abschnitts der Epidermis durch Vakuolisierung derKeratinozyten, konfluierte Parakeratose, b PAS-positive, glykogenreiche Keratinozyten im oberen Bereich der Epidermis
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 16 von 23

Fall enthält das sonst eher geringgradige, perivaskulär lymphozytäre Begleitinfiltrat auch neutrophileGranulozyten.
Histologische Differenzialdiagnose Die Histologie der Acrodermatitis enteropathica und Pellagrawie auch des nekrolytischen akralen Erythems ist mit der des nekrolytischen migratorischen Erythemspraktisch identisch. Liegen eine Vesikel- und Pustelbildung vor, spricht dies allerdings gegen einePellagra. Hingegen sind bei Nachweis von Pusteln differenzialdiagnostisch eine Impetigo, Dermato-mykose, Psoriasis pustulosa, und ein Pemphigus foliaceus auszuschließen.
8 Acrodermatitis enteropathica
Definition und klinisches Bild Die Acrodermatitis enteropathica ist eine seltene autosomal-rezessiveErkrankung mit Störung der intestinalen Zink-Resorption (Genmutation des Zinktransporters, lokalisiertauf Chromosom 8q24). Ihre klinisch charakteristischen Hautveränderungen unterscheiden sich nicht vondenen bei Zinkmangel-Syndromen anderer Ursache. Mit €uber 200 Zink-abhängigen Metalloproteasenund einem altersabhängig stark variierenden Grundbedarf ist der Zinkstoffwechsel äußerst komplex. DasMonitoring wird außerdem aufgrund des geringen Verteilungsvolumens im Plasma (nur 0,1 % derZinkvorräte) erschwert. Neben der seltenen genetischen Form stellt der erworbene Zinkmangel vor allemunter Kindern in Entwicklungsländern ein weit verbreitetes Problem dar (bis zu rund 1=3 der Bevölkerungin ärmeren Länder), das mit einer deutlich erhöhten Morbidität und Mortalität einhergeht. In Industrie-ländern sind die Risikogruppen f€ur Zinkmangel besonders Vegetarier, Alkoholiker, Mangelernährte undFr€uhgeburten (Maverakis et al. 2007).
Charakteristischerweise beginnt die (genetische) Acrodermatitis enteropathica nach dem Abstillen desSäuglings in periorifizieller und akraler Lokalisation: neben relativ scharf begrenzten, gelegentlich näs-senden Erythemen mit Schuppung und sekundärer Krustenbildung treten teilweise auch Vesikel, Blasenund Nekrosen auf, was Verbrennungswunden ähneln kann. Häufig findet sich eine Superinfektion mitCandida und Staphylokokken. Klassischerweise zeigen sich Lichtempfindlichkeit, Diarrhoen und eineAlopezie, seltener auch Nagelveränderungen (pustulöse Paronychie mit Nageldystrophie), Blepharitis/Konjunktivitis, Cheilitis und Stomatitis. Auch Wachstumsverzögerungen, Störungen der Immunabwehrund eingeschränkte Wundheilung stellen ein Problem dar (Maverakis et al. 2007).
Klinische Differenzialdiagnose Eine primäre Candidose kann gelegentlich schwer abgrenzbar sein, dadie Acrodermatitis enteropathica häufig Superinfektionen mit Candida zeigt, ansonsten wird auf die unterAbschn. 7 aufgef€uhrten Differenzialdiagnosen verwiesen. Sehr seltene Differenzialdiagnosen sind auchAminoacidopathien.
Histologie Zunächst findet sich wechselnd mit Orthokeratose eine fokale Parakeratose, später prominen-ter und konfluierend bei zunehmend schwindendem Stratum granulosum. Es liegt eine geringe Spongioseund Akanthose oder gelegentlich auch ausgeprägtere psoriasiforme Epidermishyperplasie vor. Die Blässeder Keratinozyten zeigt einen Übergang in zytoplasmatische Vakuolisierung und Nekrosen, gelegentlichunter Ausbildung intraepidermaler Vesikel oder gar Blasenbildung (Borroni et al. 1992). In fortge-schrittenen Läsionen verschwindet die epidermale Blässe. Nicht regelhaft finden sich neutrophile Gra-nulozyten sowie apoptotische oder akantholytische Keratinozyten. Das Begleitinfiltrat ist gering, zumTeil sind die Gefäße der papillären Dermis etwas dilatiert (Gonzalez et al. 1982; Maverakis et al. 2007).
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 17 von 23

Histopathologische Kriterien der Acrodermatitis enteropathica
– Wechselnd fokale Ortho- und Parakeratose, im Verlauf prominenter und konfluierend bei schwin-dendem Stratum granulosum
– Geringe Spongiose und Akanthose, gelegentlich auch ausgeprägtere psoriasiforme Epidermishy-perplasie
– Blässe der Keratinozyten mit Übergang in zytoplasmatische Vakuolisierung und Nekrosen;gelegentlich auch Vesikel und Blasen
– Gelegentlich neutrophile Granulozyten, apoptotische Keratinozyten, akantholytische Keratinozy-ten
Histologische Differenzialdiagnose Wie erwähnt sind das nekrolytische migratorische Erythem unddie Pellagra histologisch-differenzialdiagnostisch nicht abzugrenzen. Die chronische oder abklingendeAcrodermatitis enteropathica zeigt gelegentlich eine Ähnlichkeit zur Psoriasis (bei der auch ein Zink-mangel vorliegen kann).
Ebenso wurden f€ur Kwashiorkor, Hartnup Syndrom, Ahornsirup-Krankheit, nichtketonischeHyperglykämie und Marasmus ähnliche histologische Veränderungen beschrieben (ein gemeinsamerNenner könnte hier ebenfalls eine Störung des Zinkstoffwechsels bei teils kataboler Stoffwechsellage sein).
Das Phänomen blasser Keratinozyten ist beschrieben f€ur: Psoriasis (v. a. fr€uhe Läsionen), Hepatitis-C-assoziiertes nekrolytisches akrales Erythem, Pityriasis rubra pilaris, Pityriasis lichenoides, Lues II,lichenoide Keratose, irritative Kontaktdermatitis, phototoxische Dermatitis, Mazeration.
9 Pellagra
Definition und klinisches Bild Die Pellagra ist historisch gesehen die älteste dokumentierte Photoder-matose (Willan 1798: „ Eczema solare“ ) und als Multisystemerkrankung Folge eines Mangels an Niacin(Nikotinsäureamid bzw. Vitamin B3) oder dessen Vorstufe Tryptophan (Wan et al. 2011). Niacin wird vorallem durch Verzehr von Leber, Lachs, Gefl€ugel und rotem Fleisch zugef€uhrt. Mangelzustände treten inEntwicklungsländern unter getreidereicher Kost und Fehlernährung, in Industrieländern dagegen beiAlkoholismus, gastrointestinalen Erkrankungen/Malabsorption (z. B. Morbus Crohn), Anorexie, HIV,unter Medikamenten (Isoniazid, Azathioprin, Chloramphenicol, 6-Mercaptopurin, 5-Flurouracil) odereinem Karzinoid-Syndrom auf (erhöhter Tryptophan-Verbrauch bei Serotonin-Synthese). Klassischer-weise werden die Kardinalsymptome der Pellagra in der angloamerikanischen Literatur mit 4 Ds benannt:dermatitis, diarrhoea, dementia, death. Die Hautveränderungen beginnen symmetrisch in den licht-exponierten Arealen. Es finden sich scharf umschriebene Erytheme mit Desquamation und Krusten.Dunkle, schuppige Hyperpigmentierungen treten besonders auch bei verdickter Haut und €uber Knochen-vorspr€ungen auf. Hinzukommen können Blasenbildung, palmoplantare Fissuren, perianale Entz€undun-gen, eine Cheilitis und atrophe Glossittis, wobei diese Veränderungen außerhalb der lichtexponiertenAreale als Ausdruck anderer, assoziierter Mangelerscheinungen diskutiert werden. Systemische Mani-festationen umfassen Übelkeit, Erbrechen, Abdominalschmerzen, Gastritis und Diarrhoen. Zeichen einerneurologischen Beteiligung sind Kopfschmerzen, Depression und Ataxie. Erst bei länger anhaltendemVitamin-B3-Mangel treten auch dementielle Veränderungen bis hin zu Koma und Tod auf. Die Besserungunter Substitution von Niacin ist f€ur die Pellagra diagnostisch (Hendricks 1991; Piqué-Duran et al. 2012;Wan et al. 2011).
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 18 von 23

Klinische Differenzialdiagnose Ein klinisch identisches Bild zeigt die seltene Hartnup-Erkrankung(autosomal rezessive Störung des Aminosäurenstoffwechsel). Ähnlichkeiten kann es auch mit einerPorphyria cutanea tarda oder einer chronisch photoallergischen Dermatitis geben. Bei unklarenHautveränderungen in lichtexponierten Arealen sollte daher die Diagnose Pellagra ausgeschlossenwerden.
Histologie Das histopathologische Bild der Pellagra ist in der Regel nicht diagnostisch. Bei eher atropherEpidermis mit Blässe der Keratinozyten im oberen Drittel der Epidermis und gelegentlich basalerHyperpigmentierung finden sich Parakeratose und Hyperkeratose. In fr€uhen Läsionen können sichvakuolisierte Keratinozyten zeigen, eine psoriasiforme Epidermishyperplasie tritt eher bei älteren Läsio-nen auf. Auch Nekrosen und intra- oder subepidermale Blasenbildung als Folge ballonierter Keratinozy-ten können vorkommen. Falls vorhanden ist ein oberflächliches Begleitinfiltrat nur gering ausgeprägt,häufig sind jedoch dilatierte Gefäße, auch mit Erythrozytenextravasaten nachweisbar (Hendricks 1991;Piqué-Duran et al. 2012).
Histopathologische Kriterien der Pellagra
– Eher atrophe Epidermis, später psoriasiforme Epidermishyperplasie– Blässe der Keratinozyten im oberen Drittel der Epidermis– In fr€uhen Läsionen gelegentlich vakuolisierte Keratinozyten– Basale Hyperpigmentierung– Parakeratose und Hyperkeratose– Nekrosen und intra- oder subepidermale Blasenbildung als Folge ballonierter Keratinozyten
möglich– Häufig dilatierte Gefäße mit Erythrozytenextravasaten
Histologische Differenzialdiagnose Die kaum von der Pellagra zu unterscheidende Histologie desnekrolytischen migratorischen Erythems und der Acrodermatitis enteropathica muss durchklinisch-pathologische Korrelation zugeordnet werden, der Nachweis von Pusteln spricht in diesemZusammenhang gegen eine Pellagra.
10 Reaktive Arthritis
Synonym Morbus Reiter)
Definition und klinisches Bild Die reaktive Arthritis ist auch als Morbus Reiter bekannt. Der NameMorbus Reiter wird in der Literatur immer weniger benutzt bzw. aktiv gemieden, unter anderem auch,weil Hans Reiter während des Zweiten Weltkrieges wissenschaftliche Experimente an Gefangenen inKonzentrationslagern durchgef€uhrt hatte (Lu und Katz 2005). Die Krankheit wird durch die Trias alsnicht-gonorrhoische Urethritis, okkuläre Entz€undung und Arthritis definiert (Wu und Schwarz 2008).Allerdings erf€ullen nur 30 % aller Patienten diese Definition. Es handelt sich um eine parainfektiöseErkrankung, meist mit einer nicht-gonorrhoischen Urethritis assoziiert (Chlamydien, Ureaplasma, Myco-plasmen). Gelegentlich tritt sie nach einer gastrointestinalen Infektion (Shigellen, Salmonellen, Yersinien,Campylobacter) oder einer HIV-Infektion auf. Somit ist fraglich, ob es sich bei dieser Entität nichtletztlich um die Sonderform einer infektgetriggerten Psoriasis arthropatica handelt. Ackermann stellt
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 19 von 23

denMorbus Reiter lediglich als eine Sonderform der Psoriasis dar. Die reaktive Arthritis wurde auch unterInterferon und Adalimumab Behandlungen gesehen. Die genauen pathophysiologischen Mechanismensind noch nicht ganz bekannt, die Krankheit zeigt eine genetische Veranlagung mit Assoziation zumHistokompatibilitätsantigen HLA-B27 (Leirisalo et al. 1982; Keat 1983; Calin 1977). Meist manifestiertsich die Erkrankung bei jungen Männern in der dritten Lebensdekade. Das klinische Bild, Schweregradund Prognose können breit variieren. 15–20 % der Patienten zeigen einen chronischen, schubförmigenVerlauf mit Komplikationen.
Einige Wochen nach der genitalen oder auslösenden Infektion kommt es zu einem akuten Krankheits-geschehen mit Fieber, Arthritiden und Allgemeinzustandsverminderung, das Wochen bis Monate dauernkann. Es werden gelegentlich die Haut (5–30 %) und die Schleimhäute befallen. Die Haut zeigtpsoriasiforme schuppende erythematöse Papeln und Plaques, die meist an Genitalien, Perineum, Gesäß,Kopf und Streckseiten der Extremitäten zu sehen sind. Typisch sind auch dicke hyperkeratotische Plaquesan Handflächen und Fußsohlen, sie können konfluieren und häufig auch sterile Pusteln aufweisen(Keratoderma blenorrhagicum). Die Nagelregion ist zu 20–30 % mit subungualen Hyperkeratosen undVerlust der Durchsichtigkeit des Nagels, Nageldystrophie und eventuell kompletter Onycholyse betrof-fen. Der Genitalbefall zeichnet sich durch kleine keratotische Papeln und Pusteln aus, die sich zu scharfbegrenzten polyzyklischen Plaques mit randständiger Schuppung gruppieren (Balanitis circinata). An derMundschleimhaut zeigen sich erythematöse Makulae, selten Erosionen oder superfizielle Ulzerationen.Der Augenbefall mainifestiert sich in erster Linie mit einer Konjunktivitis, seltener mit einer Iritis,Uveitis, Keratitis und Glaukom. Eine Polyarthritis und eine Sakroilitis sind die häufigsten Gelenk-manifestationen.
Klinische Differenzialdiagnose Die Hautveränderungen ähneln denen der Psoriasis vulgaris oder derPsoriasis pustulosa, die Abgrenzung erfolgt im Kontext der unterschiedlichen Anamnese (Perry undMayne 1965). Rheumatoide und septische Arthritis sollen auch abgegrenzt werden.
Histologie Die reaktive Arthritis zeigt häufig ein sehr ähnliches Bild wie die pustulöse Psoriasis. Einepsoriasiforme Hyperplasie der Epidermis mit akanthotisch verlängerten Reteleisten und ausged€unntersuprapapillären Epidermis wird beobachtet.
Histopathologische Kriterien der reaktiven Arthritis (Morbus Reiter)
– Psoriasiforme Akanthose der Epidermis mit verlängerten Reteleisten– Hyper- und Parakeratose mit vielen neutrophilen Granulozyten– Spongiforme Pusteln im Bereich der oberen Epidermis– D€unne suprapapilläre Epidermis– Superfizielle dermale Entz€undung mit Neutrophilen
Es zeigt sich häufig eine Spongiose und eine Exozytose von neutrophilen Granulozyten, teilweise dieBildung von Mikroabszessen und Pusteln (spongiforme Pusteln; Abb. 11). Das Stratum corneum isthyperkeratotisch mit einer variablen Parakeratose, Beimischung von vielen neutrophilen Granulozyten(Abb. 12). Superfizielle perivaskuläre gemischtzellige Entz€undung mit Neutrophilen findet sich in derDermis.
Histologische Differenzialdiagnose Pustulöse Formen der Psoriasis sind zu unterscheiden. Die band-förmige Präsenz von Neutrophilen im Stratum corneum wird eher dem Morbus Reiter zugeschrieben.
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 20 von 23
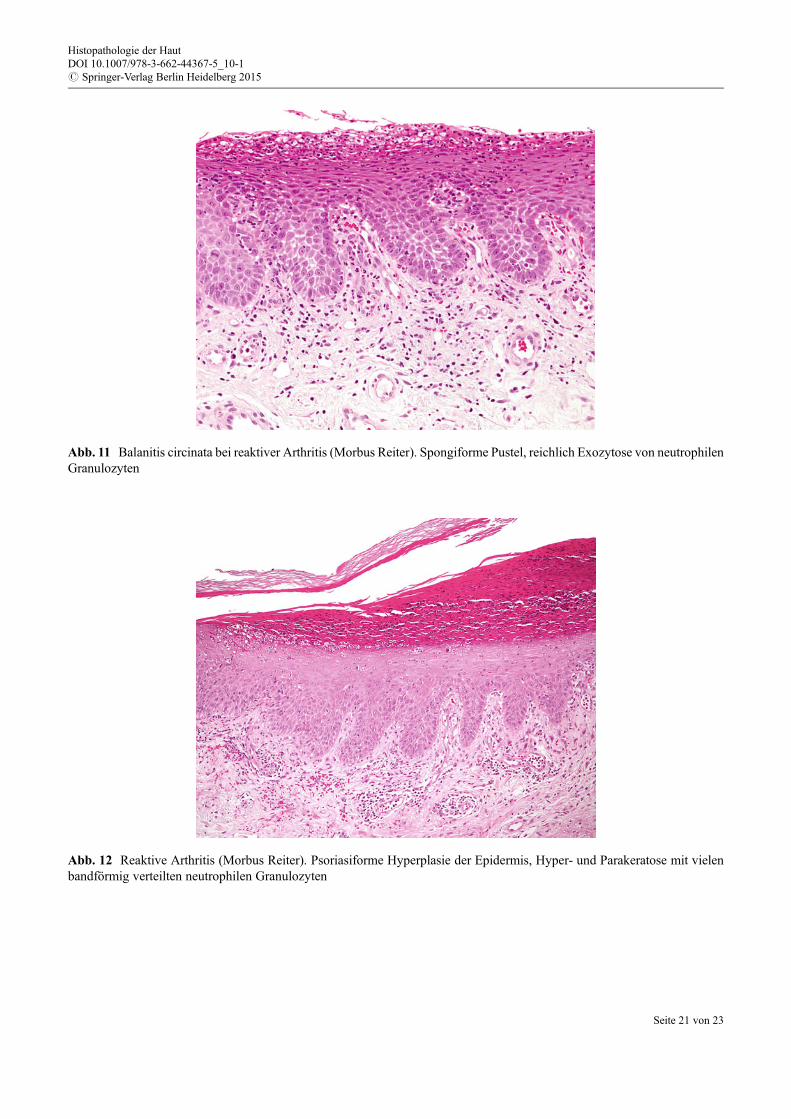
Abb. 12 Reaktive Arthritis (Morbus Reiter). Psoriasiforme Hyperplasie der Epidermis, Hyper- und Parakeratose mit vielenbandförmig verteilten neutrophilen Granulozyten
Abb. 11 Balanitis circinata bei reaktiver Arthritis (Morbus Reiter). Spongiforme Pustel, reichlich Exozytose von neutrophilenGranulozyten
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 21 von 23

Literatur
Binnick AN, Spencer SK, Dennison WL Jr et al (1977) Glucagonoma syndrome. Arch Dermatol113:749–754
Borroni G, Brazzelli V, Vignati G et al (1992) Bullous lesions in acrodermatitis enteropathica. Histopa-thologic findings regarding two patients. Am J Dermatopathol 14:304–309
Braun-Falco O, Ryckmanns F, Schmoeckel C et al (1983) Pityriasis rubra pilaris: a clinico-pathologicaland therapeutic study with special reference to histochemistry, autoradiography, and electron micros-copy. Arch Dermatol Res 275:287–295
Buonamonte D, Foti C, Vestita M et al (2012) Nummular eczema and contact allergy: a retrospectivestudy. Dermatitis 23:153–157
Calin A (1977) Reiter's syndrome. Med Clin North Am 61:365–376Christophers E (2001) Psoriasis-epidemiology and clinical spectrum. Clin Exp Dermatol 26:314–320Doyle JA, Connolly SM, Hunziker N et al (1979) Prurigo nodularis: a reappraisal of the clinical and
histologic features. J Cutan Pathol 16:392–403Gonzalez JR, Botet MV, Sanchez JL (1982) The histopathology of acrodermatitis enteropathica. Am
J Dermatopathol 4:303–311Gordon M, Johnson WC (1967) Histopathology and histochemistry of psoriasis I. Arch Dermatol
95:402–407Griffiths WAD (1980) Pityriasis rubra pilaris. Clin Exp Dermatol 5:105–112Harris B, Harri K, Penneys NS (1992) Demonstration by S-100 protein staining of increased number of
nerves in the papillary dermis of patients with prurigo nodularis. J Am Acad Dermatol 26:56Hendricks WM (1991) Pellagra and pellagralike dermatoses: etiology, differential diagnosis, dermatopa-
thology, and treatment. Semin Dermatol 10:282–292Jiamton S, Tangjaturonrusamee C, Kulthanan K (2013) Clinical features and aggravating factors in
nummular eczema in Thais. Asian Pac J Allergy Immunol 31:36–42Keat A (1983) Reiter's syndrome and reactive arthritis in perspective. N Engl J Med 309:1606–1615Ko CJ, Milstone LM, Choi J et al (2011) Pityriasis rubra pilaris: the clinical context of acantholysis and
other histologic features. Int J Dermatol 50:1480–1485Lee MR, Shumack S (2005) Prurigo nodularis: a review. Australas J Dermatol 46:211–218, quiz 219–20Leirisalo M, Skylv G, Kousa M et al (1982) Followup study on patients with Reiter's disease and reactive
arthritis, with special reference to HLA-B27. Arthritis Rheum 25:249–259Lu DW, Katz KA (2005) Declining use of the eponym ‘Reiter's syndrome’ in the medical literature,
1998–2003. J Am Acad Dermatol 53:720–723Magro CM, Crowson AN (1997) The clinical and histomorphological features of pityriasis rubra pilaris.
A comparative analysis with psoriasis. J Cutan Pathol 24:416–424Marks R, Wells GC (1973) Lichen simplex: morphodynamic correlates. Br J Dermatol 88:249–256Maverakis E, Fung MA, Lynch PJ et al (2007) Acrodermatitis enteropathica and an overview of zinc
metabolism. J Am Acad Dermatol 56:116–124Nestlé FO, Kaplan DH, Barker J (2009) Psoriasis. N Engl J Med 361:496–509Perry HO, Mayne JG (1965) Psoriasis and Reiter’s syndrome. Arch Dermatol 92:129–136Piqué-Duran E, Pérez-Cejudo JA, Cameselle D et al (2012) Pellagra: a clinical, histopathological and
epidemiological study. Actas Dermosifiliogr 103:51–58Ragaz A, Ackerman AB (1979) Evolution, maturation, and regression of lesions of psoriasis. Am
J Dermatopathol 1:199–214Rowland Payne CME, Wilkinson JD, McKee PH et al (1985) Nodular prurigo: a clinicopathological
study of 46 patients. Br J Dermatol 113:431–439
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 22 von 23

Schubert C (2002) Entz€undliche Dermatosen. Pathologe 23:9–19Sehgal VN, Srivastava G (2006) (Juvenile) Pityriasis rubra pilaris. Int J Dermatol 45:438–446Soeprono FF (1986) Histologic criteria for the diagnosis of pityriasis rubra pilaris. Am J Dermatopathol
8:277–283Sulzberger MB, Garbe W (1985) Nine cases of distinctive exudative discoid and lichenoid dermatosis.
Arch Dermatol Venereol 65:164–167Tierny EP, Badger J (2004) Etiology and pathogenesis of necrolytic migratory erythema: review of the
literature. MedGenMed 6:4Van Beck AP, de Haas ERM, van Vloten WA et al (2004) The glucagonoma syndrome and necrolytic
migratory erythema: a clinical review. Eur J Endocrinol 151:531–537Wan P, Moat S, Anstey A (2011) Pellagra: a review with emphasis on photosensitivity. Br J Dermatol
164:1188–1200Weigelt N, Metze D, Ständer S (2010) Prurigo nodularis: systematic analysis of 58 histological criteria in
136 patients. J Cutan Pathol 37:578–586Willan R (1798) Description and treatment of cutaneous diseases. Order I.J. Johnson, LondonWu IB, Schwarz RA (2008) Reiter’s syndrome: the classic triad and more. J Am Acad Dermatol
59:113–121Yoon SY, Park HS, Lee JH et al (2013) Histological differentiation between palmoplantar pustulosis and
pompholyx. J Eur Acad Dermatol Venereol 27:889–893
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_10-1# Springer-Verlag Berlin Heidelberg 2015
Seite 23 von 23