Embed Size (px)
Citation preview

AUS DEM LEHRSTUHL FÜR PHYSIOLOGIE
PROFESSOR DR. MED. ARMIN KURTZ DER NATURWISSENSCHAFTLICHEN FAKULTÄT III
DER UNIVERSITÄT REGENSBURG
REGULATION DER RENINFREISETZUNG DURCH DEN BLUTDRUCK:
ROLLE VON ADENOSIN
Inaugural - Dissertation zur Erlangung des Doktorgrades
der Medizin
der medizinischen Fakultät
der Universität Regensburg
vorgelegt von Florian Segerer
unter Anleitung von Professor Dr. med. Frank Schweda
2010

2

3
AUS DEM LEHRSTUHL FÜR PHYSIOLOGIE
PROFESSOR DR. MED. ARMIN KURTZ DER NATURWISSENSCHAFTLICHEN FAKULTÄT III
DER UNIVERSITÄT REGENSBURG
REGULATION DER RENINFREISETZUNG DURCH DEN BLUTDRUCK:
ROLLE VON ADENOSIN
Inaugural - Dissertation zur Erlangung des Doktorgrades
der Medizin
der medizinischen Fakultät
der Universität Regensburg
vorgelegt von Florian Segerer
unter Anleitung von Professor Dr. med. Frank Schweda
2010

4
Dekan:................................................................................Prof. Dr. Bernhard Weber 1. Berichterstatter.............................................................. Prof. Dr. Frank Schweda 2. Berichterstatter...........................................................Prof. Dr. Andreas Luchner Tag der mündlichen Prüfung.........................................................19. Oktober 2010

5
A. Einleitung ........................................................................................................................... 7
B. Physiologische Grundlagen .......................................................................................... 8 B.1 Das Renin-Angiotensin-Aldosteron System.................................................................... 8
B.1.1 Funktionsweise ......................................................................................................... 9
B.1.2 Physiologische Bedeutung.......................................................................................... 10
B.1.3 Regulation der Reninfreisetzung ............................................................................ 12
B.2 Adenosin ........................................................................................................................ 15
B.2.1 Metabolismus und Rezeptoren ............................................................................... 15
B.2.2 Adenosin in der Niere............................................................................................. 17
C. Fragestellung................................................................................................................... 17
D. Vorbefunde an der isoliert perfundierten Mausniere ........................................... 19
E. Methoden .......................................................................................................................... 22 E.1 Versuchstiere ................................................................................................................. 22
E.1.1 Art Herkunft und Haltung....................................................................................... 22
E.1.2 Genotypisierung...................................................................................................... 22
E.2 Blutdruckmessung ......................................................................................................... 23
E.2.1 Nicht invasiv ........................................................................................................... 23
E.2.2 Invasiv..................................................................................................................... 24
E.3 Bestimmung der Plasmareninaktivität ........................................................................... 24
E.3.1 Blutabnahme ........................................................................................................... 24
E.3.2 Messung der Plasmareninaktivität .......................................................................... 24
E.4 Quantitative RNA-Messung .......................................................................................... 25
E.4.1 RNA-Isolierung ...................................................................................................... 25
E.4.2 Reverse Transkription............................................................................................. 25
E.4.3 Quantitative Vermessung........................................................................................ 26
E.5 Organentnahme.............................................................................................................. 26
E.6 Experimentelle Blutdrucksteigerung ............................................................................. 26
E.7 Statistische Methoden .................................................................................................... 28
F. Versuchsdurchführung ................................................................................................. 28 F.1 Experiment zum Kurzzeiteffekt ..................................................................................... 28
F.1.1 Blutdruckanstieg ..................................................................................................... 28
F.1.2 Vorbereitung und Durchführung des Versuchs ...................................................... 30
F.2 Experimente zum Langzeiteffekt ................................................................................... 31
F.2.1 Medikamentöse Blutdrucksteigerung ..................................................................... 31
F.2.2 Experimentelle Nierenarterienstenose .................................................................... 31
G. Ergebnisse ....................................................................................................................... 32 G.1 Blutdruckanstieg:........................................................................................................... 32
G.2 Kurzfristige Blutdruckstimulierung: ............................................................................. 34
G.3 Längerfristige Blutdruckstimulierung: .......................................................................... 36
G.3.1 Medikamentöse Blutdrucksteigerung..................................................................... 36
G.3.2 Experimentelle Nierenarterienstenose.................................................................... 38
H. Diskussion ....................................................................................................................... 40
Anhang:.................................................................................................................................. 45 Verwendete Medikamente und Hilfsmittel .......................................................................... 45
Standardmethoden ................................................................................................................ 47
Isolation von DNA aus Gewebe....................................................................................... 47
DNA-Amplifikation mittels Polymerase-Ketten-Reaktion.............................................. 48
Bestimmung der Plasmareninkonzentration mittels RIA................................................. 48
RNA-Isolation aus Nieren................................................................................................ 49
Reverse Transkiption........................................................................................................ 50
Quantitative Vermessung von cDNA mittels Light-Cycler ............................................. 51

6
Literaturnachweis: ................................................................................................................. 52
Lebenslauf ............................................................................................................................... 59
Danksagung: ........................................................................................................................... 60

7
A. Einleitung
Der Blutdruck ist für Aufrechterhaltung der Organfunktion von entscheidender
Bedeutung. Entsprechend reguliert der Organismus den Blutdruck in engen Grenzen.
Kommt es zu einer Störung dieser Regulation, sei es nach oben oder nach unten, so
kann das zu schweren gesundheitlichen Beeinträchtigungen führen. Ein zu starkes
Abfallen des Blutdrucks, beispielsweise durch massiven Flüssigkeitsverlust, führt zu
Unwohlsein, Schwindel bis hin zum hypovolämischen Schock mit Gewebsischämien,
Organversagen und schließlich zum Tod. Ein zu starkes Ansteigen des Blutdrucks
kann zu Überlastungserscheinungen des Herz- Kreislaufsystems mit vielfältigen
Folgeschäden führen. Die American Heart Association fasst die Risiken erhöhten
Blutdrucks folgendermaßen zusammen: „Unkontrollierter hoher Blutdruck kann zu
Schlaganfall, Herzinfarkt, Herz- und Niereninsuffizienz führen. Deshalb wird hoher
Blutdruck oft „silent killer“ genannt.“
Im Organismus erfolgt die Regulation des Blutdrucks kurz- und langfristig auf
mehreren Ebenen. Zentrale Bedeutung kommen dabei dem intravasalen
Flüssigkeitsvolumen, gesteuert durch Flüssigkeitsaufnahme und –ausscheidung, der
Herzleistung, sowie dem durch den Gefäßtonus regulierten Verteilungsvolumen des
Blutes zu.
Im Februar 1933 veröffentlichte John Loesch im „Zentralblatt für innere Medizin“ den
Artikel „Ein Beitrag zur experimentellen Nephritis und zum arteriellen Hochdruck“, in
dem er die These vertrat, dass eine renale Ischämie zu einer persistierenden
Hypertension führe39.
Ebendies veröffentlichte ein Jahr später Harry Goldblatt in seinem wesentlich
bekannteren Artikel „The Production of Persistent Elevation of Systolic Blood
Pressure“ im „Journal of Experimental Medicine“ 33. Er hatte an Hunden gezeigt, dass
durch eine Stenose der Nierenarterien ein dauerhafter Bluthochdruck induziert wird.
Er war der Meinung, dass sich dieser Mechanismus auf den Menschen übertragen
ließe, nämlich, dass entweder die Stenose einer Hauptnierenarterie, oder aber
organische Veränderungen in den kleineren renalen Arterien bis hin zu glomerulären
afferenten Arteriolen zu einem Hypertonus führe. Goldmann war sich bewusst, dass
zwischen dem Trigger der renalen Ischämie und dem Effekt der Hypertonie ein
Bindeglied fehlte, und forderte dafür einen humoralen Mechanismus. Tigersted und
Bergmann hatten bereits 1898 publiziert, dass sie ein renales Hormon mit

8
blutdrucksteigernden Eigenschaften gefunden hatten, welches sie Renin nannten34.
Inzwischen ist die Annahme gut belegt, dass renale Minderdurchblutung über das
Bindeglied Renin zu einem systemischen Bluthochdruck führt, nämlich der renalen
Hypertonie,.
Mittlerweile ist bekannt, dass nicht das Renin selbst eine Hypertonie induziert,
sondern dass es eine enzymatische Kaskade in Gang setzt, an deren Ende das
Peptid Angiotensin II steht. Angiotensin II hat eine ausgeprägte blutdrucksteigernde
Wirkung. Die große Bedeutung dieser sogenannten Renin-Angiotensin-Kaskade für
die Entwicklung anderer Hypertonieformen wird daran deutlich, dass Medikamente,
die die Bildung von Angiotensin II hemmen (ACE-Hemmer) oder seine Rezeptoren
blockieren (AT1-Antagonisten), zu den wirksamsten antihypertensiven
Medikamenten gehören. Mittlerweile hat auch ein Reninantagonist, der die
enzymatische Aktivität von Renin hemmt, Einzug in die Therapie der Hypertonie
gefunden.
Obwohl das Renin-Angiotensin-System, entsprechend seiner großen
physiologischen und pathophysiologischen Bedeutung, ausgiebig untersucht wurde
und insbesondere auch die Regulation der Reninfreisetzung durch den renalen
Blutdruck Gegenstand zahlreicher Studien war, sind die Signalwege zwischen der
Freisetzung von Renin und der Steuerung des Blutdrucks bis heute nicht hinreichend
verstanden.
In der vorliegenden Arbeit soll, basierend auf zahlreichen Vorbefunden, die Frage
geklärt werden, ob Adenosin und der A1 Adenosin Rezeptor an der
blutdruckabhängigen Regulation des Reninsystems beteiligt sind.
B. Physiologische Grundlagen
B.1 Das Renin-Angiotensin-Aldosteron System
Das Renin-Angiotensin-Aldosteron-System (RAAS) spielt eine zentrale Rolle in der
Kontrolle des arteriellen Blutdrucks. Eine Stimulation des RAAS führt zu einer
Steigerung, eine Hemmung zu einer Senkung des Blutdrucks.
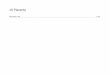
Renin wird in den reninbildendenen juxtaglomerulären Zellen (JG-Zellen) der Niere
gebildet. Diese sind in der Wand des blutzuführenden Vas afferents des Glomerulus
9
lokalisiert. Die JG-Zellen treten in engen Kontakt zu Zellen des distalen Tubulus, den
Macula densa-Zellen.
Graphik B.1 juxtaglomerulärer Apparat
B.1.1 Funktionsweise
Das RAAS umfasst eine Reihe von Proenzymen, Enzymen und Hormonen, nämlich
Renin, das Angiotensin-converting-enzyme (ACE), Angiotensinogen, Angiotensin I,
Angiotensin II und Aldosteron.
Renin ist eine Aspartylprotease mit einer Halbwertszeit von wenigen Minuten. Es wird
in der Niere im juxtaglomerulären Apparat gebildet und von den Zellen des Vas
afferents in die Blutbahn sezerniert. Renin spaltet von dem in der Leber gebildeten
Precursor-Peptid Angiotensinogen das Dekapeptid Angiotensin I ab; dieses wird
wiederum von der Dipeptidyl-Carboxypeptidase „angiotensin-converting enzyme“
(ACE) in das Oktapeptid Angiotensin II umgewandelt. Der größte Teil des ACE
befindet sich in Endothelzellen der Lunge, so dass besonders während der Passage
durch die Lunge, jedoch auch durch viele andere Gewebe, die Umwandlung von im
Blut zirkulierenden Angiotensin I in Angiotensin II stattfindet. Angiotensin II besitzt
eine Halbwertszeit von 1-2 Minuten.

10
Graphik B.1.1 Schema des Renin-Angiotensin-Aldosteronsystems, (Funktionen von Angiotensin II und Aldosteron s. Tabelle B.1.2)
B.1.2 Physiologische Bedeutung
Wie oben gezeigt steht am Ende der Renin-Angiotensin-Kasakade das Angiotensin
II, der eigentliche biologische Effektor des Systems. Angiotensin II besitzt eine
Vielzahl physiologisch und pathophysiologisch bedeutsamer Funktionen. Von
zentraler Bedeutung ist seine blutdrucksteigernden Wirkung. Diese hypertensive
Wirkung vermittelt Angiotensin II sowohl über direkte als auch indirekte Effekte. Zum
einen ist Angiotensin II ist ein potenter Vasokonstriktor, erhöht daher direkt den
vaskulären Gefäßwiderstand und damit den Blutdruck1. Darüber hinaus stimuliert
Angiotensin II die Kochsalzrückresorption in der Niere. Hierbei spielen wiederum
direkte und indirekte Mechanismen eine Rolle. Direkt erhöht Angiotensin II
rezeptorvermittelt unter anderem die Aktivität des Natrium-Protonen-Austauschers
NHE3 des proximalen Tubulus und damit die Natrium-Rückresorption1. Außerdem
steigert Angiotensin II die Aldosteronsekretion aus dem Nebennierenkortex1. Dieses

11
Mineralokortikoid stimuliert im Nierentubulus die Reabsorption von Natrium aus dem
Primärharn (sowie aus Speichel, Schweiß und Magensaft), wobei im Austausch
Kalium und Protonen in erhöhtem Maße ausgeschieden werden. In Folge des
veränderten osmotischen Verhältnisses zwischen Harn und Interstitium folgt der
gesteigerten Salzresorption eine erhöhte Wasserretention im Sammelrohr der
Nierentubuli. Dies resultiert in einer Zunahme des intravasalen Volumens und damit
in einer Blutdrucksteigerung.
Schließlich bewirkt Angiotensin II eine erleichterte Noradrenalinsekretion durch
direkte Beeinflussung der postganglionären sympathischen Neuronen1 und eine
Kontraktion mesangialer Zellen mit der Folge einer verminderten glomerulären
Filtrationsrate (GFR)1. Im Gehirn wirkt es auf die circumventrikulären Organe und löst
eine Erhöhung des Blutdrucks1, ein gesteigertes Durstgefühl1, sowie eine vermehrte
ADH/Vasopressin- und ACTH-Ausschüttung aus1.
Zusammenfassend lässt sich also feststellen, dass eine Aktivierung des Renin-
Angiotensin-Systems, mit der Reninaktivität als geschwindigkeitsbestimmendem
Schritt, den Blutdruck erhöht.
Funktionen von Angiotensin II Funktionen von Aldosteron
Peripher Niere
Vasokonstriktion erhöhte Natrium-Retention
Gehirn Kalium- und Protonendiurese
Blutdruckstimulation Schweiß, Speichel, Magensäure
Durstgefühl verminderte Natriumausscheidung
ACTH-Freisetzung Muskeln, Gehirn
ADH-Freisetzung Kalium-Einstrom
Sympathikus
erleichterte Noradrenalinfreisetzung
Niere
verminderte glomeruläre Filtration
Nebenniere
Aldosteronfreisetzung
Tabelle B.1.2 Funktionen von Angiotensin II und Aldosteron, nach William F.Ganong1
Neben Angiotensin II, welches sicher den bedeutsamsten biologischen Effektor des
RAAS darstellt, gibt es weitere aktive Peptide, die als Degradationsprodukte von
Angiotensin I oder Angiotensin II entstehen. Ein Degradationsprodukt von
Angiotensin II ist Angiotensin III, das noch die volle Aldosteron-stimulierende

12
Wirkung, jedoch verminderte zentrale Effekte zeigt1. Renin kann über die Bindung an
den Renin/Proreninrezeptor auch unabhängig von seiner enzymatischen Aktivität
biologische Effekte ausüben40, 41. Neben dem oben skizzierten Renin-Angiotensin-
System existieren auch lokale Gewebs-Renin-Angiotensin-Systeme, die Angiotensin
II produzieren und in der Niere, in Blutgefäßen, Uterus, Plazenta, den Augen, dem
Herz und anderen Organen vorkommen und bei verschiedenen physiologischen und
vor allem pathophysiologischen Prozessen wirksam werden können42, 43. Der Beitrag
dieser lokalen Systeme zur Plasmareninaktivität ist jedoch sehr gering, so dass sie
nach bilateraler Nephrektomie auf nicht messbare Werte abfällt1.
B.1.3 Regulation der Reninfreisetzung
Die Regulation der Reninkonzentration im Blut findet auf der Ebene der
Reninausschüttung in der Niere statt; die Elimination erfolgt durch unspezifische
Proteasen. Die Einflussgrößen auf die Sekretion von Renin sind in der Tabelle B.1.3
aufgelistet.
a) Faktoren mit Einfluss auf die Reninsekretion b) Konditionen mit erhöhter Reninsekretion
stimulierend Hyponatriämie
erhöhter Sympathikotonus über renale Nerven Diuretika
erhöhter Katecholaminspiegel Hypotension
Prostaglandine Hämorrhagie
hemmend Orthostase
erhöhte Na- und Cl-Reabsorption über die Macula densa Dehydration
erhöhter Druck in der afferenten Arteriole Herzinsuffizienz
Angiotensin II Cirrhose
Vasopressin/ADH Stenose der Nierenarterie oder Aorta abdominalis
verschiedene psychologische Stimuli
Tabelle B.1.3 Einflussfaktoren auf die Reninsekretion, nach William F.Ganong 1
Die Rate der Reninsekretion wird also ständig durch die Summe der verschiedenen
Einflussfaktoren bestimmt.
Entsprechend seiner biologischen Potenz wird die Reninfreisetzung durch mehrere
negative Feedbackschleifen kontrolliert. So hemmt Angiotensin II, als biologisches
Endprodukt der Renin-Angiotensin-Kaskade, die Reninfreisetzung. Auch ein Anstieg
des Kochsalzbestandes des Körpers, beispielsweise durch eine vermehrte

13
Kochsalzaufnahme, hemmt die Bildung und Freisetzung von Renin. Schließlich
hemmt, wie schon erwähnt, ein Anstieg des Blutdrucks die Reninfreisetzung. Hierbei
wird ein intrarenaler Barorezeptormechanismus postuliert, der bei erhöhtem Druck in
der afferenten Arteriole des juxtaglomerulären Apparates (JGA) eine hemmende
Wirkung, bei vermindertem Druck eine Stimulation der Reninausschüttung bewirkt 61.
Für die Kontrolle des Reninsystems durch den Kochsalzbestand des Körpers spielt
nach aktuellem Wissensstand die Macula densa im Bereich des distalen Tubulus
eine entscheidende Rolle. Die Reninsekretion ist umgekehrt proportional zum NaCl-
Gehalt des Harnes in diesem Abschnitt des Tubulus. Eine niedrige NaCl-
Konzentration im distalen Tubulus stimuliert also die Reninsekretion, während eine
hohe Kochsalzkonzentration die Reninfreisetzung hemmt 1,60.
Schon 1960 wurde die Hypothese aufgestellt, dass neben dem salzabhängigen
Macula densa-Signal ein zweiter, vasculärer Mechanismus existiere, der direkt auf
Druckänderungen in der A. renalis reagiere und das Reninsystem blutdruckabhängig
reguliere 19, 28. Inzwischen ist das Phänomen der druckabhängigen Reninsekretion in
der Niere funktionell phänomenologisch gut charakterisiert; ein Anstieg des Drucks in
den afferenten Arteriolen führt zu einer Verminderung der Reninfreisetzung und
umgekehrt. Die Wirkung dieses Mechanismus wird in der Pathophysiologie des
Bluthochrucks bei einseitiger Nierenarterienstenose deutlich (vgl. Graphik 1.2). Durch
die Stenose einer Nierenarterie sinkt der Blutdruck in der betroffenen Niere. Dies
registiert der Barosensor und stimuliert die Reninfreisetzung, was über die
Aktivierung der Renin-Angiotensin-Kaskade zu einer Steigerung des systemischen
Blutdrucks führt. Hierdurch steigt auch der Druck in der stenosierten Niere etwas,
wodurch die Reninsekretion wieder leicht sinkt. Die kontralaterale Niere dagegen ist
einem zu hohen Blutdruck ausgesetzt und reguliert ihre Reninsekretion herunter. So
stellt sich ein neues Gleichgewicht ein, bei dem der systemische Blutdruck erhöht
bleibt.

14
Graphik B.1.3 renale oder Goldblatt-Hypertension
a) frische Stenose der linken Nierenarterie, normaler Perfusionsdruck in der Aorta und der rechten Niere, normale Reninsekretion rechts. Der verminderte Perfusionsdruck in der linken Niere führt zu erhöhter Reninsekretion links.
b) neue Gleichgewichtssituation: Erhöhter Druck in der Aorta, nur leicht verminderter Druck distal der Stenose in der linken Niere, leicht erhöhte Reninsekretion links, erhöhter Perfusionsdruck in der rechten Niere, verminderte Reninsekretion rechts
Obwohl dieser renale Barorezeptor-Mechanismus in vielen Untersuchungen und
verschiedenen Spezies nachgewiesen wurde, ist bis heute unklar, über welche
Signalkaskaden der Blutdruck dabei mit dem Reninsystem verbunden ist.
Zunächst wurde vermutet, dass die Hemmung des Renins bei erhöhtem Blutdruck
über eine erhöhte Salzbeladung des Tubulus zustande käme, da in Folge des
erhöhten Drucks mehr Primärharn entsteht, und dadurch eine tubuläre
Salzüberladung erfolgen könnte. Somit würde die druckabhängige Reninregulation in
der Niere auf dem gleichen morphologischen Mechanismus beruhen wie die
salzabhängige. Da jedoch die druckabhängige Reninsekretion auch in nominell nicht
mehr filtrierenden Hundenieren erhalten blieb, wurde vermutet, dass es tatsächlich
einen unabhängigen vaskulären Barorezeptormechanismus zur Kontrolle der

15
Reninfreisetzung gibt31, 32. In einer anderen Studie konnte gezeigt werden, dass auch
bei Blockade des salzabhängigen Macula densa-gesteuerten Mechanismus eine
Steuerung der Reninfreisetzung durch den renalen Perfusionsdruck möglich war.
Insbesondere bei hohem Perfusionsdruck von 140 mmHg war diese druckabhängige
Inhibition einer Stimulation über den Macula densa-Mechanismus überlegen18. Noch
immer jedoch bleiben sowohl die zelluläre Lokalisation dieses geforderten
Barorezeptors, als auch die genauen Signalübertragungswege für die
druckabhängige Regulation der Reninfreisetzung unvollständig geklärt.
B.2 Adenosin
N
N N
NC
C
C
CC
NH2
CHHC
HC CH
O
H2COH
HO OH
N
N N
NC
C
C
CC
NH2
CHHC
HC CH
O
H2COH
HO OH Graphik B.2: Strukturformel von Adenosin
B.2.1 Metabolismus und Rezeptoren
Unter normalen Bedingungen wird Adenosin kontinuierlich sowohl intra-, als auch
extrazellulär gebildet.
Die intrazelluläre Bildung geschieht entweder mit Hilfe einer intrazellulären 5’-
Nukleotidase, die Adenosin-Monophosphat (AMP) dephosphoryliert 51,50, oder durch
Hydrolyse von S-Adenosyl-Homocystein 52.
Extrazellulär kann Adenosin durch Dephosphorylierung von Adenosin-Triphosphat
(ATP) durch Ecto-ATPasen zu AMP und dessen Spaltung durch das Enzym Ecto-5’-
Nucleotidase entstehen 49.
Die dazu notwendigen Enzyme werden auch im juxtaglomerulären Apparat exprimiert 43. Dieser Weg der Adenosinbildung wird besonders bei einem Energiedefizit

16
aktiviert, bei dem ein vermehrter Abbau des energiereichen ATPs stattfindet. In den
Macula densa-Zellen wird die ATP-Freisetzung vermutlich durch erhöhte NaCl-
Konzentration 45 und Schwellung der Zellen 46-48 stimuliert.
Außerdem kann Adenosin de novo aus der Nukleinbase Adenin und dem Zucker β-
D-Ribose synthetisiert werden, wie es in der Zelle zur Bildung von RNA und DNA
geschieht.
Die Halbwertszeit von Adenosin im Blut oder Interstitium beträgt nur wenige
Sekunden. Der Metabolismus ist in Graphik B.2.1 dargestellt.
Graphik B.2.1 Beziehung zwischen ATP und Adenosin und Metabolismus von Adenosin zu Harnsäure, nach William F.Ganong 1
Je nach Wirkort und Rezeptorausstattung wirkt Adenosin als Neurotransmitter
(Gehirn), Vasodilatator (z.B. Herz) oder hormoneller Faktor, wie beispielsweise in der
Niere, wo Adenosin eine parakrine Funktion im tubuloglomerulären Feedback
einnimmt 44. Adenosin wirkt dabei über die vier Rezeptoren A1, A2A, A2B und A3.
Alle vier Adenosinrezeptoren sind transmembranäre, G-Protein-gekoppelte
Rezeptoren, die die Adenylatcyclaseaktivität und somit die intrazelluläre Bildung von
cAMP steuern. Dabei wirken die Rezeptoren A1 und A3 inhibitorisch, die Rezeptoren
A2a und A2b stimulatorisch auf die cAMP-Bildung53. Das „second messenger“ cAMP
ist ein intrazelluläres Signalmolekül, das z.B. den zentralen Stimulator der
Reninfreisetzung aus den JG-Zellen darstellt.

17
B.2.2 Adenosin in der Niere
In der Niere hat Adenosin vielfältige Effekte.
Es ist ein potenter Inhibitor des Reninsystems, ein Effekt, der durch direkte
Stimulation der A1-Adenosin-Rezeptor (A1AR) auf den reninproduzierenden Zellen
des JGA vermittelt wird 16, 17,56, 57, 58,59. In Experimenten mit Ratten und Hunden
konnte ein inhibitorischer Effekt von Adenosin auf die Plasmareninkonzentration
festgestellt werden, der nahe legt, dass Adenosin eine Rolle in der
blutdruckabhängigen Reninregulation spielt 16, 22, 23, 36, 37,54, 55.
C. Fragestellung
Obwohl die inverse Beziehung zwischen renalem Perfusionsdruck und Reninfreisetzung in vielen Untersuchungen zweifelsfrei nachgewiesen wurde, konnte bis jetzt nicht hinreichend geklärt werden, über welche Steuerungsmechanismen beide Parameter miteinander verbunden sind.
Vorangegangene Studien lassen vermuten, dass entweder die reninproduzierenden
Zellen selbst, oder die Macula densa in Abhängigkeit vom Perfusionsdruck ein Signal
auszusenden, das die Renin-Produktion und -Ausschüttung steuert 2-5. Darüber
hinaus legen einige Studien nahe, dass die Cyclooxygenaseprodukte Prostaglandin
E2 sowie Prostacyclin eine Rolle bei der Stimulation der Reninsynthese und -
freisetzung durch eine Verminderung des renalen Perfusionsdrucks spielen 6-10. Da
jedoch die Blockade der Prostaglandinbildung in anderen Studien keine
Auswirkungen auf die blutdruckabhängige Reninregulation zeigte11-14, bleibt die Rolle
dieser Arachidonsäurederivate im renalen Barorezeptormechanismus unklar.
Neben der Kontrolle der Reninfreisetzung setzen Blutdruckveränderungen weitere
Regulationsmechanismen in der Niere in Gang. So wird über einen weiten
Druckbereich die Durchblutung und die Filtrationsrate der Niere (glomeruläre
Filtrationsrate, GFR) konstant gehalten. Dieser sogenannten renalen Autoregulation
liegen mindestens zwei Mechanismen zu Grunde. Zum einen löst ein Druckanstieg
direkt an den afferenten Arteriolen eine Vasokonstriktion aus (myogene
Komponente), zum anderen führt die initial vermehrte tubuläre Salzbeladung indirekt
zur Induktion einer afferenten Vasokonstriktion tubuläre Komponente)60,61. Grundlage
dieser indirekten Komponente ist der sogenannte tubuloglomeruläre Feedback
(TGF). Der TGF ist ein spezifischer intrarenaler Kontrollmechanismus für die NaCl-
Ausscheidung, der sich im juxtaglomerulären Apparat jedes Nephrons abspielt.

18
Dabei werden Veränderungen in der Salzkonzentration des tubulären Harnes an der
Macula densa registriert und davon abhängig der Gefäßtonus in der afferenten
Arteriole verändert. Ein Erhöhung der tubulären Salzkonzentration führt dabei zu
einer Vasokonstriktion der afferenten Arteriole und damit einer verminderten
glomerulären Filtrationsrate (GFR) und tubulären Salzbeladung. Eine Abnahme der
NaCl-Konzentation über der Macula densa bewirkt einen umgekehrten Effekt61. Über
diesen TGF kann also sehr schnell auf Veränderungen in der tubulären
Salzkonzentration, wie sie z.B. bei Veränderungen des Blutdrucks auftreten, reagiert
werden60. TGF vermittelt also eine sehr effektive kurzfristige salzabhängige
Regulation der GFR. Dieser TGF-Mechanismus wird durch genetische Deletion des
A1-Adenosinrezeptor (A1AR) oder dessen pharmakologische Blockade komplett
aufgehoben. Er ist Adenosin-abhängig 20, 24.
Wie bereits erwähnt, konnte in früheren Studien gezeigt werden, dass Adenosin über
den A1AR die Reninsekretion hemmt 16, 22, 23, 36, 37,54,55. Da die reninbildenden Zellen
zudem in unmittelbarer Nachbarschaft zu den glatten Gefäßmuskelzellen der
afferenten Arteriole liegen, die den Angriffspunkt von Adenosin im TGF-
Mechanismus darstellen, kann man vermuten, dass Adenosin auch bei der
blutdruckabhängigen Reninregulation, also einer verminderten Reninsekretion in
Folge erhöhten Blutdrucks und umgekehrt, eine wichtige Rolle spielt.
Bei in vitro-Voruntersuchungen unserer Arbeitsgruppe, die im folgenden Absatz
detaillierter vorgestellt werden, stellte sich heraus, dass durch pharmakologische
Blockade oder genetische Deletion des A1AR der inhibitorische Effekt eines
Blutdruckanstiegs auf die Reninsekretion völlig aufgehoben wird.
Nun war Ziel dieser Arbeit herauszufinden, welche Rolle Adenosin und der A1-
Adenosinrezeptor bei der blutdruckabhängigen Reninregulation in vivo spielt.

19
D. Vorbefunde an der isoliert perfundierten Mausniere
Der erste Teil der Studie wurde am Modell der isoliert perfundierten Mausniere
durchgeführt.
Die Organe wurden homozygoten A1AR-knockout-Mäusen (A1AR-/-) und deren
Wildtyp-Geschwistern (A1AR+/+) beiden Geschlechts, oder männlichen C57B1/6
Mäusen (Charles River, Deutschland) mit einem Körpergewicht zwischen 23 und 30
g entnommen.
Die Tiere wurden mit einer intraperitonealen Gabe von 12 mg/kg Xylazin und 80
mg/kg Ketamin-HCl anästhesiert. Die Aorta abdominalis wurde katheterisiert und
und proximal abgebunden, ebenso die linke Nierenarterie. Die rechte Niere war somit
über den Katheter in der Bauchaorta perfundiert, wurde entnommen und in eine
Kammer mit 37°C und 100% Luftfeuchtigkeit überführt. Schließlich wurde die
Nierenvene kanüliert, um das venöse Blut aufzufangen, aus dem die Reninaktivität
und der renale Blutfluss bestimmt wurden.
Über eine Pumpe mit elektronischer Feedback-Kontrolle konnte der Perfusionsdruck
stufenweise zwischen 40 und 140 mmHg variiert werden.
Die Reninaktivität im gesammelten venösen Blut wurde mit einem
Radioimmunoassay gemessen (siehe unten). Die Reninsekretionsrate wurde aus
dem Produkt der Reninkonzentration und des renalen Blutflusses berechnet
[ml/min*g Nierenmasse].
Die in vitro-Experimente lieferten, kurz zusammengefasst, folgende Ergebnisse:
In den Nieren von C57BL/6 Mäusen oder A1AR Wildtypmäusen führte eine
Verminderung des renalen Perfusionsdrucks von 90 auf 40 mmHg zu einer sofortigen
deutlichen Steigerung der Reninsekretionsrate (RSR). Die schrittweise Erhöhung des
renalen Perfusionsdrucks bis auf den Ausgangswert führte auch die RSR
schrittweise auf den Ursprungswert zurück. Eine weitere Steigerung des renalen
Perfusionsdrucks auf bis zu 140 mmHg verminderte die RSR auf bis zu 40% ihres
Ausgangswerts.
Um die Rolle des A1AR in dieser Regulation zu untersuchen, wurde der Rezeptor
entweder pharmakologisch blockiert (8- Cyclopentyl-1,3-dipropylxanthine (DPCPX),
1µmol/L), oder es wurden Nieren von A1AR-Knockout-Mäusen untersucht. Wie aus
Graphik D.1. ersichtlich ist, veränderte die Deletion des A1AR nicht die

20
Reninstimulation bei niedrigem renalen Perfusionsdruck. Ein niedriger
Perfusionsdruck von 40 oder 65 mmHg stimulierte also die RSR in A1AR -/- Mäusen
in gleichem Maße wie in der Wildtyp-Gruppe. Im Gegensatz dazu war die
Verminderung der Reninsekretionsrate bei hohem renalen Perfusionsdruck bei den
A1AR-/- Tieren völlig aufgehoben.
A1AR +/+
A1AR -/-
mmHg
Ren
inse
kret
ions
rate
(ng
Ag
I / (
h*m
in*g
))
Graphik D.1: Reninsekretion in isoliert perfundierten Nieren von A1AR Knockout-Mäusen (A1AR -/-, n=7) und ihren Wildtypgeschwistern (A1AR +/+, n=7). Schrittweise Änderung des Perfusionsdrucks zwischen 40 mmHg und 140 mmHg.
Um die Notwendigkeit des A1AR bei der druckabhängigen Renininhibition zu
untermauern, wurden zusätzliche Experimente durchgeführt, bei denen ohne
vorherige Senkung des renalen Perfusionsdrucks vom Basisdruck 90 mmHg direkt
auf 115 mmHg oder 140 mmHg gesteigert wurde.
Die direkte Steigerung des renalen Perfusionsdrucks von 90 auf 115 oder auf 140
mmHg führte zu einer deutlichen Verminderung die Reninsekretionsraten in
Wildtypnieren (Graphik D.2). Diese Inhibition wurde sowohl durch die
pharmakologische Blockade als auch durch die genetische Deletion (Graphik D.2)
des A1AR völlig aufgehoben.

21
A1AR +/+
A1AR -/-
DPCPX
Perfusionsdruck (mmHg)
Ren
inse
kret
ions
rate
(ng
Ag
I/(h*
min
*g))
Graphik D.2: Reninsekretion in isoliert perfundierten Nieren von A1AR Knockoutmäusen (A1AR -/-, n=4) und ihren Wildtypgeschwistern (A1AR +/+, n=4), sowie Wildtypnieren unter pharmakologsicher Blockade der A1AR mit DPCPX. Der Perfusionsdruck wurde in den isolierten Nieren direkt vom Basisdruck (90 mmHg) auf 115 mmHg (oberes Diagramm), bzw. 140 mmHg (unteres Diagramm) angehoben.
* p<0,05 gegenüber 90 mmHg

22
E. Methoden
E.1 Versuchstiere
E.1.1 Art Herkunft und Haltung
Es kamen A1AR -/- und A1AR +/+ Mäuse beiden Geschlechts (Körpergewicht 25 –
30g) und männliche Ratten (230 - 260 g) zum Einsatz.
Der in dieser Studie verwendete A1AR-knockout-Mäusestamm wurde an den
National Institutes of Health (Prof .J. Schnermann) Bethesda, USA generiert20 und
uns als heterozygote Zuchtpaare zur Verfügung gestellt. Für die Versuche wurden
die homozygoten A1AR-knockout- (A1AR -/-) oder Wildtyp-Nachkommen (A1AR +/+)
der heterozygoten Zuchtpaare verwendet.
Bei den Rattenexperimenten wurden Sprague-Dawley-Ratten (Charles River,
Deutschland) verwendet.
Alle Tierexperimente wurden gemäß den Richtlinien für Haltung und Gebrauch von
Labortieren durchgeführt, die von „US National Institutes of Health“ veröffentlicht
wurden.
E.1.2 Genotypisierung
Zur Genotypisierung der Nachkommen der heterozygoten A1AR Zuchtpaare wurden
2 Wochen alten Mäusen die Schwanzspitze abgeschnitten und aus diesem Gewebe
die DNA gewonnen.
Von jeder Probe wurde ein Ansatz mit Primern des A1AR-Gens und ein Ansatz mit
Primern für das Neomycinresistenz-Gen mittels Polymerase-Chain-Reaktion
amplifiziert. Das Neomycinresistenz-Gen wurde bei der genetischen Veränderung in
das A1AR-Gen eingebaut, so dass dieses nicht mehr funktionsfähig ist, und damit
zum A1AR-ko-Gen wurde. Deshalb dient der Nachweis des Neomycinresistenz-Gen
der Identifikation eines A1AR-ko-Gensatzes. Da das diploide Genom der Maus
jedoch auch heterozygot sein, also einen intakten A1AR-Gensatz und einen A1AR-
ko-Gensatz enthalten kann, testeten wir das Genom auf das Vorhandensein beider
Gene (s. Graphik E.1.3).
Die PCR-Produkte wurden auf ein 2% Agaroselaufgel aufgetragen und
elektrophoretisch getrennt. Die DNA wurde mittels Ethidiumbromid gefärbt und durch
UV-Licht sichtbar gemacht.

23
Die genaue Beschreibung der DNA-Isolation und der Amplifikation ist im Anhang zu
finden.
Graphik E.1.2: Bild eines Laufgels: In jeder Probe wurde in einem Ansatz das A1AR-Gen und in einem weiteren Ansatz das Neomycingen, durch das das A1AR-Gen zerstört wurde, amplifiziert. Beide Ansätze (oben A1AR, unten Neomycin) wurden nach der Amplifikation übereinander in einem 2%-Agarosegel aufgetragen und bei 120V über 30 Minuten getrennt. War das jeweilige Gen in der Probe vorhanden, wurde es amplifiziert und erschien als helle Bande.
Die Ergebnisse von links nach rechts:
A1AR +/- (Kontrolle), 5x A1AR -/-; A1AR +/-; A1AR -/-; A1AR +/-; 3x A1AR +/+
E.2 Blutdruckmessung
E.2.1 Nicht invasiv
Zur nicht-invasiven Blutdruckmessung an den Versuchsmäusen wurde den Tieren
am Schwanz proximal eine hydraulische Druckmanschette angelegt und distal der
Manschette durch einen Druckfühler die Pulsamplitude registriert.
Die Manschette war mit einem Manometer und einer Aufpumpvorrichtung verbunden.
Der Druckfühler übertrug das Signal auf einen Computerbildschirm. Das
sinusähnliche Pulssignal am Bildschirm konnte eingefroren werden, so dass sich

24
über der Zeitachse die Herzfrequenz ablesen ließ. Wurde bei laufender Übertragung
des Signals die Druckmanschette aufgepumpt, so verschwand das wellenförmig
oszillierende Signal beim Erreichen des systolischen Blutdrucks.
E.2.2 Invasiv
Die Versuchsmäuse wurden durch intraperitoneale Injektion narkotisiert (0,18
ml/100g, gemischt aus 10 ml Ketamin (100mg/ml), 1,6 ml Xylazine (20mg/ml) und 1,6
ml H2O; Wiederholungsgabe 1/3 nach 40 Minuten) und auf eine Wärmeplatte (37°C)
gelegt.
Nun wurde in die linke A. carotis communis ein PE-Katheter gelegt, über den der
mittlere arterielle Blutdruck gemessen wurde. Um eine Thrombosierung des
Katheters zu vermeiden, war dieser mit einer NaCl-Heparinlösung gefüllt.
An dem Katheter war ein Drucktransducer angeschlossen, der den mittleren
arteriellen Druck anzeigte und an einen Schreiber weiterleitete, so dass der Blutdruck
konstant aufgezeichnet wurde.
E.3 Bestimmung der Plasmareninaktivität
E.3.1 Blutabnahme
Die Blutentnahme erfolgte aus der Vene des Mäuseschwanzes direkt in 75µl-
Hämatokritröhrchen, die innen mit 1µl 125 mM EDTA benetzt waren.
Bis zur Zentrifugation wurden die blutgefüllten Röhrchen auf Eis gelagert; der
Plasma-Überstand wurde anschließend bis zur Weiterverarbeitung bei -20°
aufbewahrt.
E.3.2 Messung der Plasmareninaktivität
Zur Messung der Reninkonzentration in den Plasmaproben wurden diese zusammen
mit Reninsubstrat (= Angiotensinogen im Plasma von beidseitig nephrektomierten
Ratten) 90 Minuten inkubiert. Das dabei vom Renin gebildete Angiotensin I (aus dem
in hoher Konzentration vorhandenem Angiotensinogen) wurde mit Hilfe eines
Radioimmunoassays quantifiziert. Durch Vergleich mit der Aktivität von
Standardproben konnten wir daraus die Reninkonzentration bestimmen.

25
Für diese Messung kam ein Standard-Kit (Fa. Byk & DiaSorin Diagnostics) zum
Einsatz. Der genaue Ablauf ist im Anhang beschrieben.
E.4 Quantitative RNA-Messung
Durch Messung der Renin-mRNA-Konzentration in der Niere (relativ zur Aktin-
mRNA-Konzentration) verglichen wir die Transkription des Reningens in
Rattennieren verschiedener Versuchsgruppen.
Bei der Verarbeitung von RNA wurden RNAse-freie Utensilien (Pipettenspitzen,
Eppendorfcups) verwendet.
E.4.1 RNA-Isolierung
Die Isolierung der Renin-RNA erfolgte aus in flüssigem Stickstoff gefrorenen Nieren.
Das Prinzip der RNA-Isolierung beruht darauf, dass das homogenisierte Gewebe in
eine Lösung gebracht wird, die aus zwei Phasen besteht. In der einen Phase lösen
sich Proteine und DNA, in der anderen die RNA. In den nachfolgenden Schritten
werden die Phasen getrennt und die RNA isoliert. TRIZOL ist so eine Lösung, die
chaotrope Salze enthält, welche Proteine denaturieren, so dass DNA und RNA frei
werden. Außerdem enthält TRIZOL eine wässrige Phase, in der sich die RNA löst
und eine Phenolphase, in der Proteine und DNA verbleiben. Zur Trennung der
Phasen wird Chloroform verwendet, das sich kaum in Wasser, jedoch sehr gut in
Phenol löst, so dass die beiden Phasen aufgrund ihrer unterschiedlichen Dichte
durch Zentrifugation voneinander geschieden werden können.
Der genaue Ablauf ist im Anhang beschrieben.
E.4.2 Reverse Transkription
Bei der reversen Transkription wird RNA durch das Enzym reverse Transkriptase in
c-DNA umgeschrieben. Dieser Prozess wird sowohl für die Renin-m-RNA, als auch
für die Aktin-m-RNA durchgeführt, da diese im Ergebnis in Relation zueinander
gesetzt werden.
Zur reversen Transkription wurde ein Standard-Kit verwendet. Der genaue Ablauf ist
im Anhang beschrieben.

26
E.4.3 Quantitative Vermessung
Die Quantitative Vermessung der cDNA-Menge erfolgte im Light-Cycler durch
mehrfache abwechselnde Reduplikation der DNA durch PCR und fluorometrische
Messung der cDNA-Menge. Pro PCR-Zyklus wird die DNA-Konzentration annähernd
verdoppelt. Um keinen Fehler durch Schwankungen der gesamten m-RNA-Menge
der Proben in Kauf nehmen zu müssen, werden bei jedem Zyklus die
Konzentrationen der Renin- und der Aktin-cDNA gemessen und am Ende die Renin-
cDNA-Konzentration in Relation zur Aktin-cDNA-Konzentration angegeben. Da Aktin
in allen Nierenzellen in sehr gleichmäßiger Konzentration vorhanden ist, werden die
auf diese Weise gewonnenen Ergebnisse gut miteinander vergleichbar. Bei jeder
Messung wurde eine Leerprobe mitgeführt, die den gleichen Ansatz wie die zu
vermessenden Proben enthielt, jedoch keine DNA. Um eine zuverlässige Messung
zu erhalten, darf die Leerprobe über mindestens 35 Zyklen keine messbare DNA-
Konzentration enthalten, da sonst von einer Verunreinigung des Ansatzes
ausgegangen werden muss.
Die quantitative Vermessung der DNA erfolgte mit Hilfe eines Standard-Kits sowie
eines Light-Cyclers (Fa. Roche). Der genaue Ablauf ist im Anhang beschrieben.
E.5 Organentnahme
Zur Organentnahme wurden die Tiere mit Sevofluran betäubt und decapitiert. Nieren
und Herz wurden entnommen, gewogen, in flüssigem Stickstoff eingefroren und bei -
80°C bis zur Verarbeitung gelagert.
E.6 Experimentelle Blutdrucksteigerung
Zur gezielten Steigerung des Blutdruckes der Versuchstiere wurden mehrere
Methoden in Erwägung gezogen.
Durch eine Injektion einer NaCl-Lösung in den Peritonealraum ließe sich durch die
daraus folgende Steigerung des Intravasalvolumens der Blutdruck steigern. Jedoch
nimmt dieses Manöver großen Einfluss auf den Salzgehalt des Körpers, der ja selbst
einen wesentlichen Kontrollfaktor der Reninfreisetzung darstellt. Bei Auswirkungen
der Kochsalzinjektion auf die Reninfreisetzung kann daher schlecht zwischen salz-
und druckabhängigen Mechanismen unterschieden werden.

27
Eine weitere Möglichkeit, den Blutdruck gezielt zu steigern, ist die Applikation von
Vasokonstriktoren. Idealerweise erreicht man damit einen isolierten Anstieg des
Blutdrucks.
Verschiedene Vasokonstriktoren standen hierfür prinzipiell zur Verfügung, die jeweils
verschiedene Vor- und Nachteile aufweisen:
N-omega-Nitro-L-arginine-methylester-hydrochloride (L-NAME) ist ein Inhibitor der
endothelialen NO-Synthetase. Da unter normalen Bedingungen ständig NO in
Endothelien gebildet wird, dessen Funktion eine Vasodilatation ist, führt eine
Hemmung dieser Bildung zu einer Vasokonstriktion.
Der Vorteil von L-NAME ist die orale Bioverfügbarkeit. Es reicht, den Tieren die
Substanz in das Trinkwasser zu mischen, um den Blutdruck zu steigern. So werden
die Tiere weniger unter Stress gesetzt, wodurch Störfaktoren, wie erhöhte
Katecholaminausschüttung, die direkt die Reninausschüttung beeinflussen,
vermieden werden.
Die Nachteile sind erstens, dass der Blutdruckanstieg relativ schwach ist. Außerdem
aktiviert NO direkt das Reninsystem, so dass L-NAME mit der Hemmung der NO-
Synthese das Reninsystem auch blutdruckunabhängig inhibiert.
Angiotensin II ist ein Peptidhormon, das als starker endogener Vasokonstriktor wirkt.
Es lässt sich nicht oral verabreichen; es hemmt direkt die Reninausschüttung.
Phenylephrin ist ein selektiver alpha-Agonist. Es hat starke vasokonstriktorische
Wirkung. Es kann nicht oral verabreicht werden. Da Phenylephrin keinen direkten
Einfluss auf das Reninsystem nimmt, ist es für die Untersuchung der
druckabhängigen Regulation des Reninsystems geeignet.
Um herauszufinden, wie stark der Blutdruckanstieg nach der einer subcutanen
Injektion von Phenylephrin ist, wurden verschiedene Versuche durchgeführt (siehe
G.2, Kurzfristige Blutdruckstimulierung)
Durch eine künstliche Nierenarterienstenose lässt sich der Blutdruck in
Versuchstieren ebenfalls steigern. (zum Mechanismus s. Kapitel „Reninregulation“)
Der Versuch, den Blutdruckanstieg durch nicht-invasive Messung am Schwanz der
wachen Maus zu messen, scheiterte nach Injektion von Phenylephrin an der
induzierten peripheren Vasokonstriktion. Ca. 15 Minuten nach der Injektion wurde

28
das Drucksignal am Schwanz zu schwach, um noch eine zuverlässige Messung
zuzulassen. Deshalb wichen wir auf die intravasale Blutdruckmessung aus.
E.7 Statistische Methoden
Die Aufteilung der Tiere auf die Gruppen erfolgte durch Randomisierung, wobei nach
Geschlecht (m, w) und genetischem Status für den A1A-Rezeptor (ko, wt) eingeteilt
wurde.
Verglichen wurden Gruppenmittelwerte und Standardfehler des Mittelwerts.
Bei den Experimenten mit isoliert perfundierten Nieren wurden die letzten zwei in
einer Experimentengruppe erhobenen Werte gemittelt und für die statistische
Analyse verwertet.
Als Signifikanzgrenze wurde eine Irrtumswahrscheinlichkeit p < 5 % im Student’s
paired t-test festgesetzt.
F. Versuchsdurchführung
F.1 Experiment zum Kurzzeiteffekt
F.1.1 Blutdruckanstieg
Um Blutdruckänderungen möglichst genau registrieren und damit möglichst präzise
ihren Einfluss auf die Plasmareninaktivität beschreiben zu können, führten wir
zunächst an anästhesierten Mäusen Vorversuche zur intravasalen Blutdruckmessung
durch.
Nach Präparation der Versuchstiere und Stabilisierung des Blutdrucks wurden 1,6
mg/kg KG Phenylephrin (1mg/ml in NaCl) subcutan injiziert. Graphik F.1 zeigt ein
Beispiel einer raschen und ausgeprägten Steigerung des arteriellen Mitteldrucks
nach Phenylephrininjektion.

29
Graphik F.1: Blutdruckverlauf an der anästhesierten Maus nach Injektion von 1,6 mg/kg KG Phenylephrin s.c.;
Zeitpunkt A: Ende der Präparationsphase, Injektion der Erhaltungsnarkose; B: Blutentnahme und Injektion von Phenylephrin; C: Blutabnahme
Obwohl sich mit diesem experimentellen Protokoll gut reproduzierbar und stabil der
Blutdruck erhöhen ließ, wiesen die Plasmareninkonzentrationen sehr starke
Schwankungen auf, sowohl basal, als auch nach Phenylephrinapplikaiton (230 bis
2100 ng Ag I/(h*ml)), so dass letztlich keine verwertbaren Ergebnisse erhalten
wurden. Eine Ursache für die starke Streuung der Plasmareninaktivitäten könnte in
der unterschiedlichen Narkosetiefe und einer möglichen Atemdepression der Tiere
liegen. Nachdem auch eine Tracheotomie mit kontrollierter Beatmung keine
wesentliche Verbesserung der Reproduzierbarkeit erbrachte, wechselten wir zu
einem Protokoll zur Blutdrucksteigerung an wachen Mäusen.
Der wachen Versuchsmaus wurden im Bereich des Nackens 1,6 mg/kg Phenylephrin
1 mg/ml, bzw. in der Kontrollgruppe das gleiche Volumen 0,9% NaCl subcutan
gespritzt. 30 Minuten später wurde dem Tier eine Blutprobe entnommen. Aus den
Versuchen an betäubten Mäusen wussten wir, wie schnell und stark der Blutdruck
nach diesem Manöver anstieg.
0
20
40
60
80
100
120
140
160
180
1 3 5 7 9 11 13 15 17 19 21 23 25 27 29 31
Zeit in Minuten
Blu
tdru
ck in m
mH
g
A
B
C

30
F.1.2 Vorbereitung und Durchführung des Versuchs
Für die Blutabnahme wurden die Tiere in einer Haltevorrichtung fixiert, die einen
ungehinderten Zugang zum Schwanz der Maus ermöglichte.
Zehn Minuten vor der Blutabnahme wurde das Versuchstier in die vorgewärmte
Haltevorrichtung gesetzt und in eine Kammer mit einer Innentemperatur von 39°C
gestellt. Durch den vasokonstriktorischen Effekt von Phenylephrin war diese
Maßnahme zur peripheren Vasodilatation, die die Blutabnahme aus dem Schwanz
ermöglichte, notwendig.
Zur Verminderung des Stressfaktors für die Tiere im Experiment wurden die Mäuse
auf die Prozedur des Experiments trainiert. Dazu wurden sie an den vier dem
Versuch vorausgehenden Tagen je einmal aus dem Käfig gehoben, wie zum
Spritzen notwendig, und anschließend zehn Minuten in der Haltevorrichtung in die
Wärmekammer gesetzt.
Am Versuchstag wurden die Mäuse am Nackenfell angehoben und in die
entstehende Hautfalte subcutan entweder 1,6 mg/kg Phenylephrinlösung 1mg/ml in
0,9% NaCl oder nur 40µl NaCl gespritzt. 25 Minuten später wurden sie in die
Haltevorrichtung und in die Wärmekammer gesetzt und nach 30 Minuten Blut
abgenommen.
Am dritten, vierten und fünften Tag nach dem Versuch wurden die Mäuse wieder
trainiert und am sechsten Tag derselbe Versuch durchgeführt, diesmal jedoch mit
getauschten Gruppen, d.h. der Phenylephringruppe wurde nun NaCl injiziert und
umgekehrt.
Beide Plasmaproben wurden im selben Ansatz vermessen.
Tag 1 2 3 4 5 6 7 8 9 10 11
Manöver Tr Tr Tr Tr Versuch Tr Tr Tr Kontrollversuch
Tr = Training

31
F.2 Experimente zum Langzeiteffekt
F.2.1 Medikamentöse Blutdrucksteigerung
Um die Abhängigkeit der langfristigen Reninbildung und -sekretion nach einem
Blutdruckanstieg vom A1AR zu untersuchen, wurde den Versuchstieren im Bereich
des Nackens subcutan eine osmotische Minipumpe implantiert, die über einen
Zeitraum von 72 Stunden Phenylephrin (40 µg/kg Körpergewicht pro Stunde)
freisetzte. Der Blutdruck wurde vor und nach Einsetzen der Pumpen nicht-invasiv
gemessen.
Die Mäuse wurden unter Inhalationsanästhesie (Sevofluran) operiert. Nach
Entfernung der Haare im Bereich des Nackens und Hautdesinfektion wurde ein ca.
5mm langer Hautschnitt gesetzt. Anschließend wurde die Haut stumpf von der
Muskulatur gelöst, so dass ein subcutane Tasche entstand. In diese wurde die
Pumpe eingesetzt, die Wunde einschichtig genäht und zusätzlich verklammert.
Bei keinem der Versuchstiere traten im Versuchszeitraum Zeichen einer Entzündung
auf.
Nach 72 Stunden wurden erst Schwanzblutproben und dann die Organe
entnommen.
F.2.2 Experimentelle Nierenarterienstenose
Um die physiologische Relevanz des A1-Adenosinrezeptors in der
blutdruckabhängigen Regulation des Renin-Angiotensin-Aldosteron-Systems weiter
zu untersuchen, führten wir schließlich noch Experimente mit künstlicher einseitiger
Nierenarterienstenose bei Ratten durch. Der Mechanismus der entstehenden
Hypertonie ist unter B.1.3 erläutert.
In diesem Versuch wurden vier Gruppen miteinander verglichen; zwei
Kontrollgruppen und zwei Versuchsgruppen von Ratten, deren A1-Adenosinrezeptor
mit 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX) 1 mg/kg KG intraperitoneal blockiert
wurde. Jede Gruppe wurde wiederum in je eine Untergruppe mit bzw. ohne
einseitiger Nierenarterienstenose unterteilt. Im ersten Schritt des Versuchs wurden
diese vier Gruppen durch Randomisierung aus männlichen Ratten gebildet.

32
Nun wurden die Tiere zunächst vier Mal an den Vorgang der unter E.2.1
beschriebenen nicht-invasiven Blutdruckmessung gewöhnt. Nach dieser Periode
wurde den dafür vorgesehenen Tieren eine einseitige Nierenarterienstenose gesetzt.
Dazu wurden die Versuchstiere mit Ketamin + Xylazin intraperitoneal betäubt (0,18
ml/100g, gemischt aus 10 ml Ketamine (100mg/ml), 1,6 ml Xylazine (20mg/ml) und
1,6 ml H2O). Der Zugang zur linken Nierenarterie erfolgte unter sterilen Bedingungen
über einen Schnitt unterhalb des Rippenbogens an der dorsolateralen Flanke links.
Die Niere wurde freigelegt, deren Arterie von der Nierenvene und anliegendem
Bindegewebe getrennt und mit einem Clip versehen. Dieser Clip bestand aus einem
U-förmig gebogenen Silberblechstreifen (Länge 4 mm, Breite 1 mm) mit einem freien
Lumen vom 0,2 mm. Dieser Clip wurde so weit wie möglich über das Gefäß
geschoben und in dieser Position, vom Perfusionsdruck der Arterie festgehalten,
ohne weitere Fixierung belassen. Nach Reposition des Organs wurde der Schnitt
zweischichtig mit je zwei Einzelknopfnähten und Metallklammern verschlossen. Bis
zum Ende des Versuches zeigte keine Ratte Entzündungszeichen oder andere
Beeinträchtigungen.
Nach zehn Tagen wurde über einen Zeitraum von 4 Tagen täglich Blutdruck
gemessen. Am 14. Tag nach dem Einsetzen der Nierenarterienstenose wurden die
Ratten durch Dekapitation getötet, das austretende Blut aufgefangen, beide Nieren
entnommen und diese sofort in flüssigem Stickstoff eingefroren.
Zur Auswertung wurde aus der Blutprobe die Plasmareninkonzentration und aus den
Nieren die Renin-mRNA-Konzentratoin gemessen.
G. Ergebnisse
G.1 Blutdruckanstieg:
Wie bereits erwähnt ist die Untersuchung des Effektes einer pharmakologischen
Blutdrucksteigerung auf die Reninsekretion dadurch erschwert, dass die meisten
Vasokonstriktoren einen direkt hemmenden Einfluss auf die Reninfreisetzung
ausüben. Damit wird es unmöglich zu unterscheiden, ob der Effekt eines solchen
Vasokonstrikotors auf die Plasmareninkonzentration in vivo auf seinen
blutdrucksteigerenden Effekt oder auf einen direkten Effekt an den reninbildenden
Zellen zurückzuführen ist. Daher war es zunächst erforderlich, einen Vasokonstriktor
zu identifizieren, der keinen blutdruckunabhängigen Effekt auf die Reninfreisetzung

33
ausübt. Hierzu wurden Versuche an der isoliert perfundierten Niere durchgeführt.
Dieses Modell bietet durch eine elektronische Pumpensteuerung die Möglichkeit,
Nieren mit einem konstanten Perfusionsdruck zu perfundieren. Ein Vasokonstriktor
vermindert hier also den Blutfluss durch die Niere, ohne den Perfusionsdruck zu
verändern.
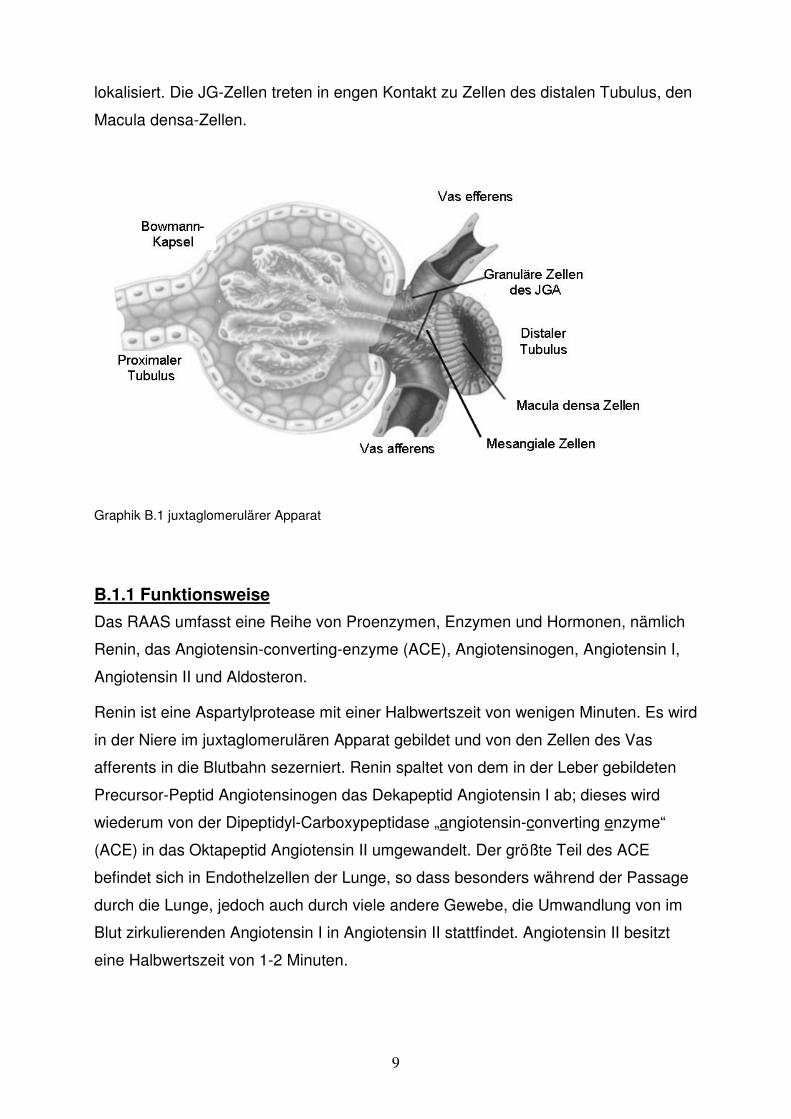
Wie die Graphik G.1 zeigt, induzieren die Blockade der NO-Synthethasen mittels L-
NAME, der Alpha-Adrenorezeptoragonist Phenylephrin und Angiotensin II deutliche
Vasokonstriktionen, erkennbar an der Abnahme des Perfusatflusses. Während L-
NAME und Angiotensin II zudem die Reninsekretionsrate vermindern, weist
Phenylephrin keinen hemmenden Effekt auf die Reninsekretion auf. Phenylephrin
scheint also keinen blutdruckunabhängigen Effekt auf die Reninfreisetzung zu
besitzen. Deshalb kam bei dieser Studie Phenylephrin zur pharmakologischen
Blutdrucksteigerung zum Einsatz.
Flu
ssra
te (
(ml /
min
) /
g N
iere
ngew
icht
)R
enin
sekr
etio
nsra
te(n
g A
g I/h
*min
*g)
Perfusionszeit (min)
Phenylephrin L-NAME Angiotensin II
Kontrolle
Kontrolle
Vasokonstriktor
Vasokonstriktor
34
Graphik G.1 oben: Blutflussrate durch die isoliert perfundierte Niere. Der Abfall der Flussrate nach Applikation des Vasokonstriktors spiegelt umgekehrt den Anstieg der Vasokonstriktion wieder. Alle drei Substanzen führen zu einer deutlichen Vasokonstriktion.
unten: Reninsekretionsrate über der Zeitachse ohne und unter Einfluss der Vasokonstriktoren bei konstantem Perfusionsdruck von 100 mmHg. Sowohl L-NAME, als auch Ag II führen zu einer direkten Inhibition der Reninsekretionsrate, Phenylephrin nicht.
G.2 Kurzfristige Blutdruckstimulierung:
Unter Kontrollbedingungen fanden sich keine Unterschiede im Blutdruck von A1AR
+/+ und A1AR -/- Mäusen (A1AR +/+ 118 +/- 13 mmHg , A1AR -/- 121 +/- 14 mmHg,
p~0,14, n=33 jeder Genotyp).
Die subcutane Injektion von Phenylephrin (1,6 mg/kg KG) erhöhte den Blutdruck von
A1AR +/+ Mäusen signifikant von 106.9+/-4.6 auf 138.4+/-5.6 mmHg (p<0,05, n=7)
und bei A1AR -/- Mäusen von 101.0+/-2.6 auf 135.6+/-5.8 mmHg (p<0.01, n=5; vgl.
Graphik G.2.1). Signifikante Unterschiede zwischen den Gruppen fanden sich nicht.
A1AR +/+ A1AR -/-
MA
D (
mm
Hg
)
NaCl Phenyl-ephrin
NaCl Phenyl-ephrin
Graphik G.2.1: Auswirkungen einer einzelnen subcutanen Phenylephrininjektion auf den Blutdruck.
A1AR -/- und A1AR +/+ Mäusen wurden 1,6 mg/kg Körpergewicht subcutan injiziert, wobei zur Kontrolle NaCl verwendet wurde.
Die Injektion von Phenylephrin führte bei Wildtypmäusen zu einem deutlichen Abfall
der Plasmareninkonzentration (Weibchen 121 +/- 22, n=12 gegenüber 297 +/- 30 ng
angI/h*ml in der NaCl- Kontrollgruppe, p<0.05, Männchen 119 +/- 41, n=5 gegenüber

35
590 +/- 100 ng angI/h*ml, p<0.01). Im Gegensatz fiel bei A1AR-knockout-Mäusen die
Plasmareninkonzentration nicht signifikant ab (Weibchen 359 +/- 48, n=12
gegenüber 464 +/- 57 ng angI/h*ml; nicht signifikant; Männchen 878 +/- 96, n=5
gegenüber 950 +/- 60 ng angI/h*ml; nicht signifikant). Vgl. Graphik G.2.2.
Dieses Ergebnis ist daher mit der Schlussfolgerung der in vitro-Vorbefunde
vereinbar, dass Adensoin und der A1-Adensoinrezeptor für die Hemmung der
Reninfreisetzung durch einen Blutdruckanstieg erforderlich sind.
Pla
sm
are
nin
ko
nze
ntr
ati
on
(ng
Ag
I /
(h
*ml)
)
NaCl Phenyl-ephrin
NaCl Phenyl-ephrin
Weibchen
A1AR +/+ A1AR -/-

36
NaCl Phenyl-ephrin
NaCl Phenyl-ephrin
Pla
sm
are
nin
ko
nz
en
tra
tio
n(n
g A
g I
/ (
h*m
l))
Männchen
A1AR +/+ A1AR -/-
Graphik G.2.2: Auswirkungen einer einzelnen subcutanen Phenylephrininjektion auf die Plasmareninkonzentration in A1AR-knockout-Mäusen
A1AR -/- und A1AR +/+ Mäusen wurden 1,6 mg/kg Körpergewicht subcutan injiziert, wobei zur Kontrolle NaCl verwendet wurde.
Die obere Graphik zeigt die Plasmareninkonzentration wacher weiblicher Versuchstiere 30 Minuten nach der Injektion von Phenylephrin (A1AR+/+ n=12; A1AR-/-, n = 12) oder NaCl (A1AR+/+, n = 14; A1AR-/-, n = 12).
Dasselbe ist in der unteren Graphik für männliche Versuchstiere gezeigt (A1AR+/+ n = 6 je Gruppe, A1AR-/- n = 5 je Gruppe).
G.3 Längerfristige Blutdruckstimulierung:
G.3.1 Medikamentöse Blutdrucksteigerung
Durch die konstante Phenylephrininfusion (1,6 mg/kg/h) über osmotischen
Minipumpen wurde der Blutdruck weiblicher Mäuse über einen Zeitraum von 72
Stunden gesteigert. Den Kontrolltieren wurden mit 0,9% NaCl gefüllte Pumpen
implantiert.
Der signifikante Blutdruckanstieg fiel in der A1AR -/- Gruppe (von 130,5 +/- 3,2
mmHg auf 139,5 +/- 4,3 mmHg, p<0,05, n = 5) gleich aus, wie in der A1AR +/+
Gruppe (von 127,7 +/- 1.7 mmHg auf 135,9 +/- 1,8mmHg, p<0,05, n = 5). In der
Wildtypgruppe fiel daraufhin die Plasmareninkonzentration auf 40% des Wertes der

37
Wildtypkontrollgruppe. In der A1AR-knockout-Gruppe hingegen fiel die
Plasmareninkonzentration wiederum nicht signifikant ab (vgl. Graphik G.3.1).
Dieses Ergebnis unterstreicht weiter die Notwendigkeit des A1AR für die Inhibition
der Reninfreisetzung durch einen arteriellen Hypertonus.
SB
D (
mm
Hg
)
NaCl Phenyl-ephrin
NaCl Phenyl-ephrin
A1AR +/+ A1AR -/-
NaCl Phenyl-ephrin
NaCl Phenyl-ephrin
Pla
sm
are
nin
ko
nz
en
tra
tio
n(n
g A
g I
/ (
h*m
l))
A1AR +/+ A1AR -/-

38
Graphik G.3.1: Auswirkungen einer langfristigen Phenylephrinperfusion auf Blutdruck und Plasmareninaktivität bei A1AR -/- Mäusen
Über osmotische Minipumpen wurden entweder 1,6 mg/kg/h oder 0,9% NaCl über einen Zeitraum von 72 h in weibliche A1AR -/- (n = 5 je Gruppe) oder A1AR +/+(n = 5 je Gruppe) Mäuse infundiert.
Am letzten Tag der Infusion wurden systolischer Blutdruck (SBD, oben) und Plasmareninkonzentration (unten) bestimmt.
G.3.2 Experimentelle Nierenarterienstenose
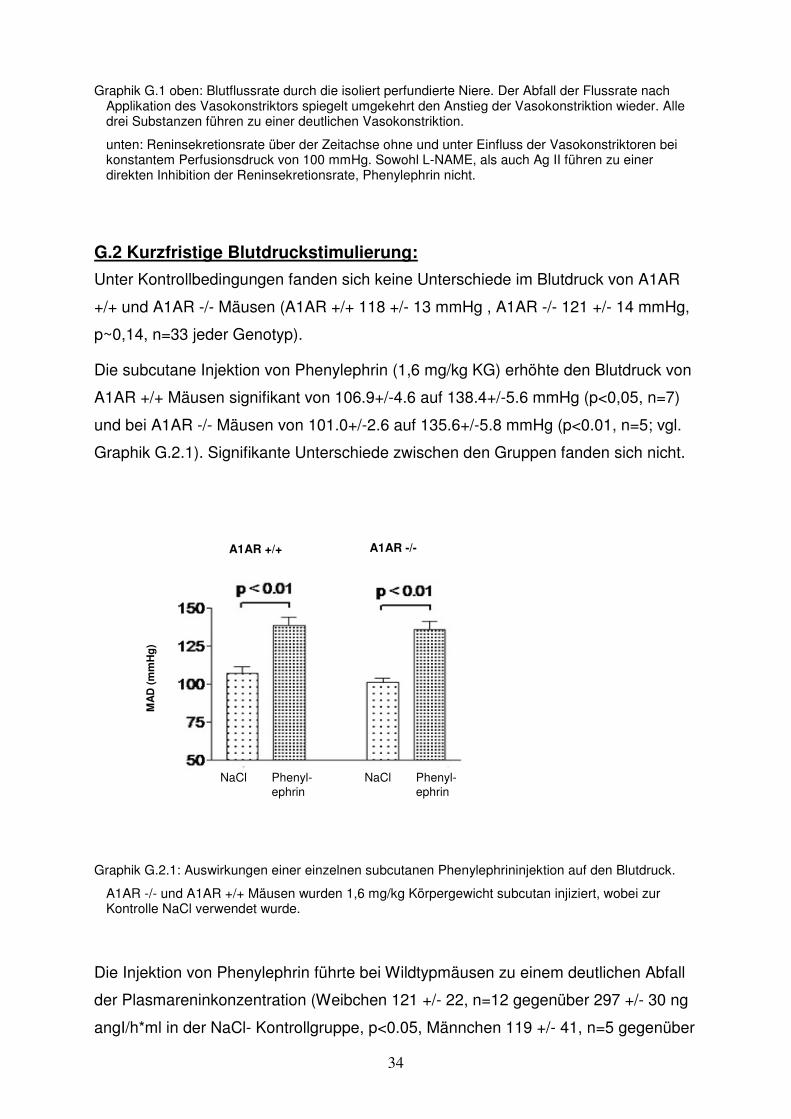
Die pharmakologische Blockade der A1-Adenosinrezeptoren hatte bei Kontrolltieren
ohne Nierenarterienstenose keinen Einfluss auf den Blutdruck (DPCPX: 133,4 +/- 6
mmHg, Kontrolle mit NaCl: 133,3 +/- 6,1 mmHg, n = 4 vs. 4), die Plasmareninaktivität
(DPCPX: 9,1+/- 1,4, Kontrolle: 6,6 +/- 2,3, p = 0,4, n.s.) und die Renin-mRNA-
Expression.
Die Anlage einer Nierenarterienstenose führte zu einem deutlichen Anstieg des
Blutdrucks bei unbehandelten Ratten von 133,3 +/- 6,1 mmHg auf 163,0 +/- 3,3
mmHg (n= 5, p<0,05). Dieser Blutdruckanstieg wurde durch die Blockade der A1-
Adenosinrezeptoren noch verstärkt. So betrug der Blutdruck bei DPCPX-behandelten
Ratten mit Nierenarterienstenose 175,0 +/- 2,5 mmHg (n = 6) (Kontrolle ohne
Stenose 133,4 +/- 6,0 mmHg, p< 0,05 ) und war damit signifikant höher als in der
Gruppe ohne A1AR-Blockade (p<0,05) (vgl. Graphik G.3.2 a).
Auch die Plasmareninaktivität wurde durch die Nierenarterienstenose erhöht, wobei
der Anstieg in der mit DPCPX behandelten Gruppe signifikant größer war als in der
Gruppe ohne A1AR-Blockade ([ng Ag I/(h*ml))] DPCPX: Stenose 48,4 +/- 7,3, n=6
vs. ohne Stenose 9,1 +/- 1,4, n=4, p<0,05; Kontrolle: Stenose 19,9 +/- 4,4, n=5 vs.
ohne Stenose 6,6 +/- 2,3, n=4, p<0,05) (vgl. Graphik G.3.2 b).
Wie erwartet führte die Anlage einer Nierenarterienstenose zu einer Stimulation der
Renin-Genexpression in der betroffenen linken Niere und einer Hemmung der Renin-
Genexpression in der kontralateralen rechten Niere. Signifikante Unterschiede
zwischen DPCPX-behandelten und -unbehandelten Tieren fanden sich dabei nicht
([% Aktin-m-RNA] DPCPX: Stenose li. 1,53 +/- 0,29 / re. 0,06 +/- 0,01, n=6, - vs.
ohne Stenose li 0,57 +/- 0,08 / re. vs. 0,49 +/- 0,04, n=4, pli<0,05, pre<0,05; Kontrolle:
Stenose li. 1,25 +/- 0,19 / re 0,08 +/- 0,01n=5, - vs. ohne Stenose li. 0,51 +/- 0,05, re.
0,45 +/- 0,04, n=4, pli<0,05, pre<0,05).
39
0,0
10,0
20,0
30,0
40,0
50,0
60,0
Basiswert Stenose Basiswert Stenose
NaCl DPCPX
Pla
sm
are
nin
akti
vit
ät
(ng
Ag
I/(
h*m
l))
P<0,05 P<0,05
P<0,05
n.s.
Basiswert BasiswertStenose Stenose
10
60
50
40
30
20

40
NaCl DPCPX
m-R
NA
-Ko
nzen
trati
on
(Rela
tio
n z
ur
Akti
n-m
RN
A)
10
60
50
40
30
20
100
100
100
100
P<0,05
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
1,80
2,00
Sham + vehicle Clip+vehicle Sham+DPCPX Clip+DPCPXBasiswert BasiswertStenose li. Stenose li.
li. Niere re. Niere
P<0,05( jeweils li. und re.)
P<0,05( jeweils li. und re.)
n.s.
( jeweils li. und re.)
Graphik G.3.2: Auswirkungen einer einseitigen Nierenarterienstenose auf Blutdruck, Plasmareninaktivität und relativer Renin-m-RNA-Konzentration bei Ratten.
Der Versuchsgruppe wurde täglich DPCPX (1 mg/kg KG i.p.) gespritzt, der Kontrollgruppe wurde NaCl gespritzt. In der Versuchsgruppe erhielten sechs Tiere eine einseitige Nierenarterienstenose (n=6), vier Tiere dienten der Ermittlung der Basiswerte (n=4). In der Kontrollgruppe gab es fünf Tiere mit Nierenarterienstenose (n=5) und vier Tiere ohne (n=4).
Die Stenose blieb 14 Tage bestehen. In den letzten vier Tagen dieser Zeit wurde den Ratten je einmal am Tag Blutdruck gemessen (pro Messung und Tier wurden dazu 10 Einzelwerte erhoben) und daraus der Mittelwert errechnet (obere Graphik). Am Tag 14 wurde den Tieren Blut und die beiden Nieren entnommen, um die Plasmareninaktivität (mittlere Graphik), sowie die Renin-m-RNA-Konzentration relativ zur Aktin-m-RNA-Konzentration (untere Graphik) zu bestimmen.
H. Diskussion
Die Blutdruckabhängigkeit der Reninsekretion ist phänomenologisch gut
charakterisiert. Bis heute bleibt jedoch der zu Grunde liegende intrarenale
Mechanismus, der Änderungen des renalen Perfusionsdrucks mit der Freisetzung
von Renin verbindet, unklar.
Die Ergebnisse der vorliegenden Studie zeigen erstmalig, dass die
blutdruckabhängige Kontrolle der Reninsekretion in vitro und in vivo auf die
Anwesenheit von Adenosin und den A1-Adenosinrezeptor angewiesen ist.
In vitro konnte am Modell der isoliert perfundierten Niere die völlige Aufhebung des
inhibitorischen Effektes einer Blutdrucksteigerung auf die Reninsekretion gezeigt
werden.

41
Auch in vivo senkte die Blutdrucksteigerung in Folge einer kurzfristigen oder
langfristigen Phenylephringabe die Plasmareninkonzentration bei Wildtypmäusen
signifikant. Bei A1AR-knockout-Mäusen blieb die Plasmareninkonzentration sowohl
nach kurz- als auch nach langfristiger Stimulation des Blutdruckes auf gleichem
Niveau. Damit konnten wir zeigen, dass auch in vivo Adenosin eine wichtige Rolle in
der blutdruckabhängigen Reninsekretion inne hat.
Ein signifikanter Unterschied in der Plasmareninaktivität lässt sich auch bei den
Ratten mit einseitiger Nierenarterienstenose zwischen der Kontrollgruppe und den
Tieren mit pharmakologischer Blockade des A1AR beobachten. Obwohl der arterielle
Blutdruck, der normalerweise die Reninausschüttung hemmt, in der DPCPX-Gruppe
durch einseitige Nierenarterienstenose stärker ansteigt als in der Kontrollgruppe, fiel
die Plasmareninkonzentration deutlich höher aus. Hier lässt sich allerdings nicht
sagen, ob dieser stärkere Plasmareninkonzentrations-Anstieg auf eine fehlende
Hemmung in der nicht-stenosierten Niere, oder auf eine Disinhibition der
Reninausschüttung in der stenosierten Niere zurückzuführen ist. Die Annahme der
Disinhibition beruht auf der Vorstellung, dass die Reninsekretion immer gleichzeitig
einerseits stimuliert, andererseits durch andere Faktoren inhibiert wird, so dass sich
ein Gleichgewicht einstellt, das durch Veränderung der stimulierenden oder
inhibierenden Faktoren verschoben werden kann. Eine verstärkte Disinhibition
bedeutet also, dass inhibierende Faktoren weniger stark wirken.
Dass es sich bei den DPCPX-behandelten Ratten mit Nierenarterienstenose jedoch
ausschließlich um eine Hemmung der Reninausschüttung in der nicht-stenosierten
Niere handelt, erscheint aus folgender Überlegung heraus unwahrscheinlich: Unter
Basalbedingungen produzieren beide Nieren zusammen in der DPCPX-Gruppe eine
Reninmenge, die eine Plasmareninaktivität von 9,1 mg Ag I I/(h*ml) besitzt, also pro
Niere eine Aktivität von 4,6 mg Ag I I/(h*ml). Mit Stenose steigert sich diese Aktivität
auf insgesamt 48,4 mg Ag I I/(h*ml). In der Kontrollgruppe liegt die
Plasmareninaktivität mit Stenose bei 18,9 mg Ag I I/(h*ml). Unter der Annahme, dass
diese Aktivität ausschließlich von Renin aus der stenosierten Niere produziert wird
und die kontralaterale Niere ihre Reninsekretion auf Null verringert hat, dürfte in der
DPCPX-Gruppe bei gleicher Stimulation der stenosierten Niere und fehlender
Inhibition der Reninsekretion der kontralateralen Niere die gesamte
Plasmareninaktivität nicht wesentlich über (18,9 + 4,6 =) 23,5 mg Ag I/(h*ml)
hinausgehen. Da die tatsächlich gemessene Plasmareninaktivität jedoch über das

42
Doppelte dieses Wertes beträgt, ließe sich vermuten, dass in diesem Versuch nach
Blockade des A1AR die Disinhibition der Reninsekretion in der stenosierten Niere
deutlich stärker ausfällt, als in der Kontrollgruppe.
Dieser Schluss steht in Widerspruch zu den aus isoliert perfundierten Nieren
gewonnenen Ergebnissen. Dort fiel unter genetischer oder pharmakologischer
Blockade des A1AR der Plasmareninaktivität-Anstieg in Folge eines niedrigen
renalen Perfusionsdrucks nicht anders aus, als in den Kontrollen mit intaktem A1AR
(vgl. Kap D). Bei einer einseitigen Nierenarterienstenose wird die Steigerung der
Reninsekretion in der stenosierten Niere durch einen verminderten Perfusionsdruck
distal der Stenose ausgelöst, so dass man gemäß den Ergebnissen aus der isoliert
perfundierten Niere erwarten sollte, dass die Steigerung dieser Reninsekretion
unabhängig vom A1AR in beiden Gruppen gleich ausfallen sollte. Allerdings ist hier
noch zu berücksichtigen, dass die Hemmung der Reninsekretion aus den Nieren
nicht nur durch einen erhöhten Blutdruck ausgelöst wird, sondern auch durch erhöhte
Angiotensin II-Spiegel im Blut in Folge der erhöhten Plasmareninaktivität (vgl. Tab.
B.1.3). Diese Hemmung wirkt der Stimulation durch verminderten Perfusionsdruck in
der stenosierten Niere entgegen. Es wäre also denkbar, dass diese Hemmung bei
Blockade des A1AR schwächer ausfällt als sonst. Aber auch andere unspezifische
Effekte, die in vitro nicht auftreten, können für diese vermutete stärkere Disinhibition
der Reninsekretion verantwortlich sein. Einer dieser Effekte könnte der
Sympathikotonus sein, der nur in vivo besteht und eine entscheidende Rolle bei der
Regulation des Reninsystems spielt, da eine Aktivierung des Sympathikus eine
direkte Stimulation der Reninfreisetzung bewirkt.
Im Gegenzug bieten die höheren Plasmareninaktivität-Werte in der DPCPX-Gruppe
bei einseitiger Nierenarterienstenose eine mögliche Erklärung für die signifikant
höheren Blutdruckwerte im Vergleich zur Kontrollgruppe.
Die Werte der relativen Renin-m-RNA-Konzentration in den Nieren der Versuchstiere
ändern sich in beiden Gruppen durch eine einseitige Nierenarterienstenose
signifikant. In der stenosierten Niere steigt die Renin-m-RNA-Konzentration stark an,
während sie in der kontralateralen Niere abfällt. Jedoch wurde dies durch eine
pharmakologische Blockade des A1AR weder unter Basalbedingungen, noch bei
einseitiger Nierenarterienstenose beeinflusst. In keiner Situation gab es, weder in der
stenosierten noch in der kontralateralen Niere, zwischen der DPCPX- und der
Kontrollgruppe signifikante Unterschiede. Dieses Ergebnis deutet darauf hin, dass

43
Adenosin über den A1AR zwar die Sekretion von Renin aus der Niere, jedoch nicht
deren Renin-m-RNA-Expression beeinflusst. Hier könnten verschiedene Faktoren
eine Rolle spielen, beispielsweise der direkt hemmende Einfluss von AT II, welches
ja in Tieren mit einseitiger Nierenarterienstenose durch die hohe Reninsekretion aus
der stenosierten Niere hoch ist, auf die Reninsynthese.
Ein Haupteffekt von Adenosin in der druckabhängigen Regulation ist also die
kurzfristige Inhibition der Reninsekretion durch Aktivierung der A 1-Adenosin-
Rezeptoren bei plötzlich erhöhtem Blutdruck, während die Rolle in der langfristigen
Reninregulation sich durch diese Studie nicht eindeutig zuordnen lässt.
In anderen in vivo- und in vitro-Experimenten führte die direkte Stimulation der A1AR
durch selektive A1AR-Agonisten zu einer verminderten Reninsekretion16, 21, 22,
während deren Blockade die Reninfreisetzung erhöhte17, 23, was mit unseren
Ergebnissen übereinstimmt.
Während der A1-Adenosin-Rezeptor für die kurzfristige Hemmung des Reninsystems
durch einen hohen Blutdruck essentiell zu sein scheint, wird die Reninstimulation
infolge erniedrigten Blutdrucks hingegen offenbar durch einen anderen Mechanismus
gesteuert. Der Anstieg der Reninsekretion durch niedrigen renalen Perfusionsdruck
war in vitro durch eine Blockade (medikamentös oder pharmakologisch) des A1AR
nicht beeinflusst, was einen A1AR-unabhängigen Mechanismus vermuten lässt.
Frühere Studien legen nahe, dass Prostaglandine bei niedrigem renalen
Perfusionsdruck eine Renin-stimulierende Rolle einnehmen6-10. Die Hemmung der
Prostaglandinbildung durch Indomethazin bremste die Reninfreisetzung infolge
niedrigen renalen Perfusionsdrucks, ohne diese Reaktion jedoch vollständig zu
unterdrücken. Dies könnte entweder an einer unvollständigen Blockade der COX-
Aktivität liegen, oder darauf hindeuten, dass noch andere Transmitter, wie NO6, 26, 27
in diesen Mechanismus eingebunden sind.
Aus diesem Ergebnis ergeben sich nun andere Fragen:
Ist Adenosin tatsächlich das einzige Bindeglied zwischen einer messbaren
Blutdrucksteigerung und einer verminderten Reninsekretion, oder ist es nur Teil einer
längeren Signalkaskade?
Wo wird das Adenosin, das die Reninsekretion steuert, gebildet? Findet in den
reninbildenden Zellen der Tunica media der afferenten Arteriole eine autokrine

44
Regulation statt, oder werden diese Zellen durch andere spezialisierte Sensorzellen
gesteuert?
Wir fanden außerdem einen erhöhten Plasmareninspiegel unter Basalbedingungen
bei A1AR-knockout-Mäusen gegenüber ihren Wildtyp-Geschwistern, was die
Ergebnisse vorangegangener Studien, die an A1AR -/- Mäusen anderen genetischen
Hintergrunds durchgeführt wurden, bestätigt 24. Für diesen Befund erscheinen
mehrere Erklärungsversuche plausibel:
Einerseits ließe er sich gut mit der Annahme vereinen, dass bereits unter
Basalbedingungen über eine tonische Aktivierung des A1AR die Reninausschüttung
gebremst wird, was bei den knockout-Mäusen nicht mehr der Fall ist. Der erhöhte
Reninspiegel wäre dann Folge einer Disinhibition der Reninsekretion.
Zudem wurde beschrieben, dass A1AR -/- Mäuse bei Fütterung mit Normal- und
Niedrigsalzkost eine höhere Salzausscheidung über den Urin haben als
Wildtypmäuse38. Dies könnte auch eine Erklärung für die höheren basalen
Plasmareninspiegel sein, da ja eine Veränderung des Salzbestandes des Körpers
die Reninfreisetzung steuert, also ein erhöhter Salzverlust über den Urin zu einem
erhöhten Plasmareninspiegel führt. Bei diesem Erklärungsansatz wäre der erhöhte
Reninspiegel also eine Antwort auf den erhöhten Salzverlust.
Dem gegenüber muss betont werden, dass sich im Modell der isoliert perfundierten
Niere bei einem renalen Perfusionsdruck von 90 mmHg die Reninsekretionsrate
zwischen A1AR +/+ und A1AR -/- Nieren nicht signifikant unterschied. Dieser Befund
lässt es unwahrscheinlich erscheinen, dass eine fehlende basale Inhibition der
Reninsekretion Grund für die unterschiedlich hohen Plasmareninspiegel ist.
Es gab jedoch auch eine andere Studie, die keinen Unterschied in der systemischen
Plasmareninkonzentration zwischen A1AR +/+ und A1AR -/- Mäusen finden konnte25.
Insgesamt scheint es daher wahrscheinlich, dass unter basalen Bedingungen die
Reninsekretion durch noch nicht bestimmte Faktoren reguliert wird und die Adenosin-
vermittelte Hemmung der Reninfreisetzung erst bei deutlichen Blutdruckerhöhungen
über den Normaldruck hinaus zum Tragen kommt.
Interessant ist auch die Feststellung, dass sich der basale Blutdruck der knockout-
Tiere nicht von dem der Wildtypen unterschied. Eigentlich könnte man erwarten,
dass infolge erhöhter Reninausschüttung in den A1AR -/- Mäusen auch der basale
Blutdruck höher liegt. Dies ist nicht der Fall, was darauf schließen lässt, dass bei den

45
knockout-Mäusen eine Gegenregulation durch andere Mechanismen stattfindet, die
ausreichend potent sind, die Einstellung des Blutdrucks entgegen den Abweichungen
in der Reninsekretion beim Sollwert zu erhalten.
In einer vorangegangenen Studie wurde bei knockout -Tieren eine gegenüber
Wildtypen erhöhte Salzausscheidung gefunden38. Würde dieser Salzausscheidung
nicht entgegengesteuert werden, fände nach und nach ein intravasaler
Volumenverlust statt, der auf Dauer zu einem Absinken des Blutdrucks führen
müsste. Dies könnte erklären, dass bei den knockout -Tieren eine erhöhte
Reninsekretion nicht im Widerspruch zu einem unveränderten Blutdruck stünde,
sondern für diesen gerade notwendig sei. Eine endgültige Antwort auf diese Frage
wird durch die vorliegende Studie nicht gegeben. Konsequenz dieser Feststellung
muss jedoch sein, dass Absolutwerte der Plasmareninkonzentration zwischen den
verschiedenen Genotypen nur mit größter Vorsicht direkt miteinander verglichen
werden dürfen.
In der vorliegenden Studie wurden zur Untersuchung der Hypothese zwischen
verschiedenen Genotypen relative Veränderungen der gemessenen Werte
miteinander verglichen. Außerdem konnten die an A1AR -/- Mausnieren gewonnenen
Ergebnisse durch Versuche an Nieren von Wildtypmäusen nach pharmakologischer
Blockade des A1AR bestätigt werden. Die unterschiedlichen Absolutwerte der
Plasmareninkonzentration zwischen Wildtyp- und knockout -Gruppen stellt also die
Schlussfolgerung unserer Studie nicht in Frage, nämlich dass der A1-
Adenosinrezeptor eine unverzichtbare Rolle in der druckabhängigen Verminderung
der Reninfreisetzung spielt.
Anhang:
Verwendete Medikamente und Hilfsmittel
Nicht-invasive Blutdruckmessung: Blood-pressure Monitor, Fa. TSE Gemany
Invasive Blutdruckmessung: Harvard Apparatus, Fa. Hugo-Sachs-Elektronik
Photometer: GeneQuant II, Fa. Pharmacia Biotec
DNA-Isolierung und Amplifikation:
Proteinase K: QIAGEN, Cat. No. 19133

46
Polymerase + MgCl2: GoTaq, Fa. PROMEGA
5xPuffer, dNTP, Primer: Fa. Roche
TNES-Puffer:
5ml Tris (1M, pH 7,5), Fa. Merck KGaA
66,7ml NaCl, 3M, Fa. Merck KGaA
100ml EDTA, 0,5M, Fa. Merck KGaA
15ml SDS, 20%, Fa. Merck KGaA
mit ddH2O auf 500ml auffüllen
TBE-Puffer:
Tris 216g, Fa. Merck KGaA
H3BO3 110g, Fa. Merck KGaA
EDTA 18,9g, Fa. Merck KGaA
mit ddH2O auf 2 Liter auffüllen
Maleatpuffer:
4,06g Tris, Fa. Merck KGaA
5,8g Maleinsäure, Fa. Merck KGaA
2,96g EDTA, Fa. Merck KGaA
mit NaOH 1M auf pH 6 einstellen
RNA-Messung:
Mixer: Ultra-Tarrex T 25, Fa. Janke & Kunkel, IKA-Labortechnik
RNA-Isolierung: Quiagen-Kit
Reverse Transkription: Roche-Kit
Amplifikation: Roche-Kit
Lightcycler: RealTime PCR, Fa. Roche
Bestimmung der Plasmareninaktivität:
Reninsubstrat: Plasma aus bilateral nephrectomierten Ratten.
Radioimmunoassey: Kit RIAP2721, Byk & DiaSorin Diagnostics, Deutschland

47
Alktivitätsmessung (Counter): 1282 Compugamma CS, Fa. LKB-WALLAC
L-NAME: N-omega-Nitro-L-arginine methyl ester hydrochloride, minimum 98% TLC,
Fa. Sigma, N5751-10G, Aufbewahrung bei -20°C, M 269,7
Phenylephrin: L-Phenylephrin Hydrochloride, Fa. SIGMA, P-6126, M 203,7
DPCPX: 8- Cyclopentyl-1,3-dipropylxanthine, Fa. SIGMA, C-101, M 304,4
Heparin: Liquemin, Fa. Roche
Xylazine: RompunR, Bayer, Deutschland
Ketamin-HCl: Curamed, Deutschland)
EDTA disodium 125mM, Fa. SERVA
Sevoflouran: Sevorane, Abbott
Osmotische Pumpen: Fa. DURECT Corporation, alzet reg., MICRO-OSMOTIC
PUMP MODEL 1003D, Bezug via Charles River
Standardmethoden
Isolation von DNA aus Gewebe
Die Gewebeprobe wurde zur Verdauung und Freisetzung der DNA bei 55°C unter
Schütteln mindestens 4h in 600µl TNES-Puffer + 10µl Proteinase K inkubiert.
Zur Ausfällung der Proteine wurden der Probe 167µl NaCl 6M hinzugefügt,
geschüttelt, und der Überstand abzentrifugiert (6000 rpm, 15 min, RT). Vom
Überstand wurden 500µl in ein neues Gefäß überführt.
Durch Hinzufügen von 500µl Ethanol 100% (-20°C) und vorsichtiges Schütteln wurde
die DNA ausgefällt, 3min bei 10000 rpm abzentrifugiert, und der Überstand
verworfen.
Zur Reinigung wurde das Pellet mit 500µl Ethanol, 80% (-20°C) gewaschen, 15min
bei 14000 rpm abzentrifugiert und der Überstand verworfen.
Nach dem Trocknen des Pellets (Raumtemperatur 10 min) wurde es ca. 1h bei 55°C
unter Schütteln in 150µl H2O gelöst, davon 5µl abgenommen, mit 95µl H2O verdünnt
und photometrisch die DNA-Konzentration vermessen.

48
DNA-Amplifikation mittels Polymerase-Ketten-Reaktion
Zur DNA-Amplifikation wurde folgender Ansatz hergestellt:
100ng DNA
Je 3 µl Sense und Antisense-Primer 10µM
3µl dNTP 2,5µM
5µl 5xPuffer
3µl MgCl2
0,25µl Taq
Mit H2O auf 50 µl verdünnen.
Im Cycler wurde die DNA bei folgendem Programm vervielfältigt:
5 min 94°C
Dann 34 Cyclen mit
1 min 94°C
1 min 58°C
1 min 72°C
Bestimmung der Plasmareninkonzentration mittels RIA
Das zu vermessende Plasma wurde 1:20, das Reninsubstrat (= Rattenplasma) 1:5
mit Maleatpuffer verdünnt.
Nun wurden 50µl verdünnte Probe, 50µl Mix aus 4 Teilen verdünntem Reninsubstrat
und 5 Teilen RIA-Reaktionspuffer (Kit), sowie 2µl Enzyminhibitor (PMSF, Kit)
vermengt.
Von diesen 102µl Mix wurden jeweils 51µl bei 37°C und bei 4°C 90 Minuten lang
inkubiert.
Anschließend wurden davon je 45µl mit je 500µl Tracer (Kit) in RIA-Röhrchen (Kit)
überführt. Diese Röhrchen wurden mit 5 Standardproben (Kit) 3-24 Stunden bei RT
inkubiert.

49
Während dieser Inkubationsphase binden sich proportional zur Angiotensin I–
Konzentration Angiotensin I-Moleküle an die anti-Angiotensin I-Antikörper, mit der die
RIA-Röhrchen innen beschichtet sind.
An die übrigen Bindestellen im Röhrchen, die nicht von Angiotensin I-Molekülen
besetzt sind, binden sich die radioaktiven Tracermoleküle.
Nach der Inkubationsphase wurde die Flüssigkeit aus Tracer und Probe abgesaugt,
in einem Counter die Radioaktivität der Röhrchen gemessen, und anhand einer aus
den Standardwerten berechneten Standardkurve die Angiotensin I-Konzentratoin
berechnet.
Durch Subtraktion des Warm- und Kaltwertes einer Probe kamen wir schließlich auf
die tatsächliche Angiotensin I-Konzentration der Plasmaproben.
RNA-Isolation aus Nieren
In Sarstedt-Tubes mit rundem Boden werden 2,5 ml TRIZOL vorgelegt und auf Eis
gestellt.
Je eine halbe Rattenniere wird direkt aus flüssigem Stickstoff in das Röhrchen
überführt und 30 s mit einem feinen Mixer homogenisiert.
Der Mix wird in Eppendorfcups überführt und 5 Minuten bei Raumtemperatur
stehengelassen. Während dieser Zeit denaturieren die Proteine und lösen sich mit
der DNA im Phenol.
Es wird 1/5 des TRIZOL-Volumens Chloroform hinzugefügt und von Hand
geschüttelt. Hier sollte nicht mit einem automatischen Schüttler („Vortexer“)
gearbeitet werden, da sonst die DNA zerstört wird und sich dann in der wässrigen
Phase löst.
Nun wird über 20 Minuten bei 12000 rpm und 4°C zentrifugiert, wobei sich die
Phenol/Chloroform- von der Wasserphase trennt. Es entstehen drei Schichten: oben
der wässrige Überstand, unten die rote Phenolphase und dazwischen eine weiße
Interphase aus relativ hydrophilen Proteinen. Der wässrige Überstand wird
abgenommen und in ein Eppendorfcup überführt.
Zur Ausfällung der RNA wird dem Überstand 100 Volumenprozent Isopropanol 100%
mit Raumtemperatur hinzugefügt, kurz mit einem Vortex-Gerät geschüttelt, die

50
Lösung 10 Minuten bei Raumtemperatur stehengelassen und schließlich 10 Minuten
bei 12000 rpm und 4°C zentrifugiert.
Der Überstand wird verworfen, das Eppendorfcup mit dem glasigen Pellet 5 Minuten
bei Raumtemperatur umgedreht auf saugfähiges Papier gestellt und verbleibende
Tropfen mit einer Pipette abgesaugt.
Zur weiteren Reinigung wird das Pellet mit 2,5ml Äthanol 75% bei Raumtemperatur
gewaschen. Der Alkohol verdrängt die Hydrathülle der RNA, so dass diese nicht in
Lösung geht, während das Wasser die verbleibenden Salze löst. Nach 5 Minuten
wird über 15 Minuten bei 7500 rpm und 4°C zentrifugiert. Der Überstand wird
verworfen, das Eppendorfcup mit dem nun weißen Pellet 5 Minuten bei
Raumtemperatur umgedreht auf saugfähiges Papier gestellt und verbleibende
Tropfen mit einer Pipette abgesaugt. Das Pellet wird 10 Minuten bei Raumtemperatur
trocknen gelassen.
Das Pellet wird abschließend durch Vortexen, anschließend maximal 2 Minuten
Inkubation im Wärmeschüttler bei 65°C und nochmaligem Vortexen in 750µl Wasser
gelöst und auf Eis gestellt.
Zur Konzentrationsbestimmung werden 2µl Probe mit 98µl Wasser verdünnt und bei
260 und 280nm Wellenlänge photometrisch vermessen. Die Ratio 260/280 sollte 1,8
bis 2,0 betragen. Da aromatische Aminosäuren ein Absorptionsmaximum bei 280nm
haben, spricht eine Ratio unter 1,8 für einen hohen Proteinanteil, der die
enzymatische Aktivität bei der folgenden reversen Transkription mindern kann.
Reverse Transkiption
Es wurden 2µg RNA mit 1µl polT (0,5µg/µl, Kit) und H20 auf ein Endvolumen von
10µl vermischt, 5 Minuten bei 65°C inkubiert und auf Eis gestellt. Dann wurde ein Mix
aus 1µl reverser Transkriptase (Kit), 4µl dNTP (2,5mMol/l, Kit), 4µl Puffer (Kit) und
3µl H20 hergestellt. Nach dem Vermischen beider Ansätze wurden die Proben eine
Stunde lang in einen auf 37°C vorgeheizten Block gestellt. Schließlich wurde der
Prozess durch zweiminütiges Erhitzen auf 94°C beendet.

51
Quantitative Vermessung von cDNA mittels Light-Cycler
Zunächst wurde der doppelte Reaktionsansatz für alle Proben, die Kontrolle und die
Eicheinheiten hergestellt. Dieser bestand aus je 10 µl Mastermix (Kit), der die
Polymerase, dNTP, sowie die für das richtige Reaktionsmilieu wichtigen Puffer und
Elektrolyte enthält, und schließlich 6µl H2O. Der Hälfte dieses Reaktionsansatzes
wurden je 1µl Primer und Antiprimer (je 10µM) des Reningens und der anderen
Hälfte des Aktingens hinzugefügt.
Nun wurde die erforderliche Anzahl an Kapillaren mit je 18µl Reaktionsansatz
beschickt, und je einer Kapillare mit dem Aktin- und einer mit dem Renin-
Reaktionsansatz 2µl der zu quantifizierenden c-DNA-Lösung hinzugefügt.
Der Programmablauf des Lightcyclers sah folgendermaßen aus:
Im ersten Schritt wurde das System aktiviert. Dazu wurden die Proben 900 Sekunden
bei 95°C inkubiert, wodurch eventuell vorhandene unspezifische Bindungen
zwischen DNA-Strängen oder zwischen DNA und Polymerasemolekülen
aufgeschmolzen wurden. Nach dieser Phase wurde das System mit 20°C/s
abgekühlt. Diese Abkühlgeschwindigkeit ist notwendig, um die erneute Bildung
unspezifischer Bindungen zu vermeiden.
Es folgte das zyklische Reduplikationsprogramm, bei dem nach jedem Zyklus eine
photometrische Vermessung der DNA-Konzentration stattfand und welches 50 mal
wiederholt wurde. Jeder Zyklus bestand aus einer Trennungsphase, bei der die
Proben 15 s bei 95°C gehalten wurden und die die Bindung von Polymerase an
DNA, sowie Bindungen zwischen DNA-Strängen aufschmilzt. Nun erfolgte die
Bindungsphase mit 20 s bei 58°C, welche die gezielte Bindung von Primer und
Antiprimer an den Anfang der DNA-Stränge und der Polymerasemoleküle an den
Reduplikationsstart erlaubt. In der folgenden Phase mit 20 s bei 72°C ist die
Polymerase aktiv und verdoppelt diejenigen DNA-Stränge, an die Primer und
Polymerase gebunden haben. Die Temperaturänderungen zwischen den Phasen
verliefen mit einer Geschwindigkeit von 20°C/s.
Für die Erstellung der Schmelzkurve wurden am Ende der Potenzierungsphase die
Proben nochmals für 1 s auf 95°C erhitzt, schnell auf 60°C abgekühlt und nach 15 s
mit 0,1°C/s bis auf 95°C erwärmt, wobei die Proben ständig photometrisch
vermessen wurden. An dem Temperaturpunkt, an welchem doppelsträngige DNA in
einzelsträngige aufgeschmolzen wird, findet ein deutlicher Abfall der Absorption statt.

52
Ist nur die gewollte DNA in größeren Mengen vorhanden, findet sich in der
Schmelzkurve an einem einzigen Punkt eine deutliche Änderung der Absorption.
Sind verschiedene DNA-Stücke amplifiziert worden, so haben diese unterschiedliche
Längen und Basenzusammensetzung und damit unterschiedliche
Schmelztemperaturen; die Schmelzkurve weist dann mehrere Änderungen in der
Absorption auf.
Literaturnachweis:
1. William F.Ganong. Review of Medical Physiology, eighteenth edition, LANGE
1997
A: Page 425, Chap. 24
B: Page 428, Chap. 24
2. Fray JC. Regulation of renin secretion by calcium and chemiosmotic forces:
(patho) physiological considerations. Biochim Biophys Acta. 1991; 1097:243-
262.
3. Carey RM, McGrath HE, Pentz ES, Gomez RA, Barrett PQ. Biomechanical
coupling in renin-releasing cells. J Clin Invest. 1997; 100:1566-1574.
4. Vander AJ, Miller R. Control Of Renin Secretion In The Anesthetized Dog. Am
J Physiol. 1964; 207:537-546.
5. Schricker K, Hamann M, Kaissling B, Kurtz A. Role of the macula densa in the
control of renal renin gene expression in two-kidney/one-clip rats. Pflugers
Arch. 1994; 427:42-46.
6. Schricker K, Hamann M, Kaissling B, Kurtz A. Renal autacoids are involved in
the stimulation of renin gene expression by low perfusion pressure. Kidney Int.
1994; 46:1330-1336.
7. Schricker K, Hamann M, Kurtz A. Prostaglandins are involved in the
stimulation of renin gene expression in 2 kidney-1 clip rats. Pflugers Arch.
1995; 430:188-194.

53
8. Seymour AA, Zehr JE. Influence of renal prostaglandin synthesis on renin
control mechanisms in the dog. Circ Res. 1979; 45:13-25.
9. Wang JL, Cheng HF, Harris RC. Cyclooxygenase-2 inhibition decreases renin
content and lowers blood pressure in a model of renovascular hypertension.
Hypertension. 1999; 34:96-101.
10. Fujino T, Nakagawa N, Yuhki K, Hara A, Yamada T, Takayama K, Kuriyama
S, Hosoki Y, Takahata O, Taniguchi T, Fukuzawa J, Hasebe N, Kikuchi K,
Narumiya S, Ushikubi F. Decreased susceptibility to renovascular
hypertension in mice lacking the prostaglandin I2 receptor IP. J Clin Invest.
2004; 114:805-812.
11. Freeman RH, Davis JO, Dietz JR, Villarreal D, Seymour AA, Echtenkamp SF.
Renal prostaglandins and the control of renin release. Hypertension. 1982;
4:106-112.
12. Villarreal D, Davis JO, Freeman RH, Sweet WD, Dietz JR. Effects of
meclofenamate on the renin response to aortic constriction in the rat. Am J
Physiol. 1984; 247:R546-551.
13. Mann B, Hartner A, Jensen BL, Hilgers KF, Hocherl K, Kramer BK, Kurtz A.
Acute upregulation of COX-2 by renal artery stenosis. Am J Physiol Renal
Physiol. 2001; 280:F119-125.
14. Hartner A, Goppelt-Struebe M, Hilgers KF. Coordinate expression of
cyclooxygenase-2 and renin in the rat kidney in renovascular hypertension.
Hypertension. 1998; 31:201-205.
15. Navar LG. Integrating multiple paracrine regulators of renal microvascular
dynamics. Am J Physiol. 1998;274:F433-444.
16. Albinus M, Finkbeiner E, Sosath B, Osswald H. Isolated superfused
juxtaglomerular cells from rat kidney: a model for study of renin secretion. Am
J Physiol. 1998; 275:F991-997.
17. Weihprecht H, Lorenz JN, Schnermann J, Skott O, Briggs JP. Effect of
adenosine1-receptor blockade on renin release from rabbit isolated perfused
juxtaglomerular apparatus. J Clin Invest. 1990; 85:1622-1628.

54
18. Scholz,Vogel,Kurtz.Interrelation between baroreceptor and macula densa
mechanisms in the control of rennin secretion. Journal oh Physiology; 1993,
511-524
19. Tobian, L. 1960. Interrelationship of electrolytes, juxtaglomerular cells and
hypertension; Physiological reviews, 40, 280-312
20. Sun D, Samuelson LC, Yang T, Huang Y, Paliege A, Saunders T, Briggs J,
Schnermann J. Mediation of tubuloglomerular feedback by adenosine:
evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci U S A.
2001; 98:9983-9988.
21. Schweda F, Wagner C, Kramer BK, Schnermann J, Kurtz A. Preserved
macula densa-dependent renin secretion in A1 adenosine receptor knockout
mice. Am J Physiol Renal Physiol. 2003; 284:F770-777.
22. Churchill PC, Bidani A. Renal effects of selective adenosine receptor agonists
in anesthetized rats. Am J Physiol. 1987; 252:F299-303.
23. Churchill PC, Rossi NF, Churchill MC. Renin secretory effects of N-6-
cyclohexyladenosine: effects of dietary sodium. Am J Physiol Renal Fluid
Electrolyte Physiol 252: F872-F876, 1987.
24. Pfeifer CA, Suzuki F, Jackson EK. Selective A1 adenosine receptor
antagonism augments beta-adrenergic-induced renin release in vivo. Am J
Physiol.1995; 269:F469-479.
25. Brown R, Ollerstam A, Johansson B, Skott O, Gebre-Medhin S, Fredholm B,
Persson AE. Abolished tubuloglomerular feedback and increased plasma
renin in adenosine A1 receptor-deficient mice. Am J Physiol Regul Integr
Comp Physiol. 2001; 281:R1362-1367.
26. Hansen PB, Yang T, Huang Y, Mizel D, Briggs J, Schnermann J. Plasma renin
in mice with one or two renin genes. Acta Physiol Scand.2004; 181:431-437.
27. Scholz H, Kurtz A. Involvement of endothelium-derived relaxing factor in the
pressure control of renin secretion from isolated perfused kidney. J Clin Invest.
1993; 91:1088-1094.

55
28. Knoblich PR, Freeman RH, Villarreal D. Pressure-dependent renin release
during chronic blockade of nitric oxide synthase. Hypertension. 1996; 28:738-
742.
29. Skinner, S.L., McCubbin, J.W., Page, I.H. 1964. Control of rennin secretion.
Circulation Research 15, 1618 – 1620.
30. Hackenthal, E., Paul, M., Ganten & Taugner, 1990. Morphology physiology,
and molecular biology of renin secretion. Physiological Reviews 70, 1067-
1116
31. Blaine, Davis &Witty, 1970. Renin release after hemorrhage and after
suprarenal aortic constriction in dogs without sodium delivery to the macula
densa. Circulation Research 27, 1081-1089
32. Blaine, Davis & Prewitt, 1971. Evidence for a renal vascular receptor in control
of rennin secretion. American Journal oh Physiology 220, 1593-1597
33. Glodny B., Glodny De., John Loesch, discoverer of renovascular hypertension,
and Harry Goldblatt: two great pioneers in circulation research. Ann Intern
Med. 2006 Feb 21;144(4):286-95.
34. Timio M., Goldblatt, kidney and hypertension: three steps in the history of
nephrology. Historical Archives of Italian Nephrology. G Ital Nefrol. 2003 Sep-
Oct;20(5):512-5.
35. Keeton, T.K., Campbell, W.B. 1981. The pharmacologic alteration of rennin
release. Pharmacological Reviews 31, 81-a227
36. Itoh S, Carretero OA, Murray RD. Possible role of adenosine in the macula
densa mechanism of rennin release in rabbits. J Clin invest 76: 1412-1417,
1985.
37. Lorenz JN, Weihprecht H, Schnermann J, Skott O, Briggs JP. Renin release
from isolated juxtaglomerular apparatus depends on macula densa chloride
transport. Am J Physiol Renal Fluid Electrolyte Physiol 260: F486-F493, 1991.
38. Brown D, Thorén P, Steege A, Mrowka R, Sällström, Skott O, Fredholm B,
Persson A. Influence of the adenosine A1 receptor on bood pressure

56
regulation and rennin release. Am J Physiol Renal Fluid Electrolyte Physiol
290:1324-1329, 2006
39. Loesch J, Ein Beitrag zur experimentellen Nephritis und zum arteiellen
Hochdruck I. Zentralblatt für Innere Medizin 1933; 7: 144-69
40. Nguyen G. Renin/prorenin receptors. Kidney Int 69: 1503-1506, 2006;
41. Jan Danser AH, Batenburg WW, van Esch JH. Prorenin and the (pro)renin
receptor--an update. Nephrol Dial Transplant 22: 1288-1292, 2007
42. Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin
systems. Physiol Rev 86: 747-803, 2006;
43. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-
angiotensin system: from physiology to the pathobiology of hypertension and
kidney disease. Pharmacol Rev 59: 251-287, 2007.
44. Castrop H, Huang Y, Hashimoto S, Mizel D, Hansen P, Theilig F, Bachmann
S, Deng C, Briggs J, Schnermann J. Impairment of tubuloglomerular feedback
regulation of GFR in ecto-5'-nucleotidase/CD73-deficient mice. J Clin Invest
114: 634-642, 2004
45. Bell, PD, et al. Macula densa cell signaling involves ATP release through a
maxi anion channel. Proc. Natl. Acad. Sci. U. S. A. 2003. 100:4322-4327
46. Sabirov, RZ, Dutta, AK, Okada, Y. Volume-dependent ATP-conductive large-
conductance anion channel as a pathway for swelling-induced ATP release. J.
Gen. Physiol. 2001. 118:251-266.
47. Peti-Peterdi, J, Morishima, S, Bell, PD, Okada, Y. Two-photon excitation
fluorescence imaging of the living juxtaglomerular apparatus. Am. J. Physiol.
Renal Physiol. 2002. 283:F197-F201.
48. Liu, R, Pittner, J, Persson, AE. Changes of cell volume and nitric oxide
concentration in macula densa cells caused by changes in luminal NaCl
concentration. J. Am. Soc. Nephrol. 2002. 13:2688-2696.

57
49. Zimmermann H. Extracellular metabolism of ATP and other nucleotides.
Naunyn Schmiedebergs Arch Pharmacol 2000; 362: 299-309
50. Zimmermann H, Braun N, Kegel B and Heine P (1998) New insights into
molecular structure and function of ectonucleotidases in the nervous system.
Neurochem Int 32: 421-425[Medline]
51. Schubert P, Komp W and Kreutzberg GW (1979) Correlation of 5'-
nucleotidase activity and selective transneuronal transfer of adenosine in the
hippocampus. Brain Res 168: 419-424[Medline].
52. Broch OJ and Ueland PM (1980) Regional and subcellular distribution of S-
adenosylhomocysteine hydrolase in the adult rat brain. J Neurochem 35: 484-
488[Medline].
53. Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J.
International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001 Dec;53(4):527-52. Review.
54. Osswald, H., H. J. Schmitz, & R. Kemper. Renal action of adenosine: effect on
renin secretion in the rat. Naunyn Schmiedebergs Arch. Pharmacol. 303: 95-
99, 1978
55. Tagawa, H., & A. J. Vander. Effects of adenosine compounds on renal function
and renin secretion in dogs. Circ. Res. 26: 327-338, 1970
56. Kurtz, A., R. Della Bruna, J. Pfeilschifter, & C. Bauer. Role of cGMP as second
messenger of adenosine in the inhibition of renin release. Kidney Int. 33: 798-
803, 1988
57. Spielman, W. S. Antagonistic effect of theophylline on the adenosine-induced
decrease in renin release. Am. J. Physiol. 247 (Renal Fluid Electrolyte Physiol.
16): F246-F251, 1984.
58. Spielman, W. S., L. J. Arend. Adenosine receptors and signaling in the kidney.
Hypertension 17: 117-130, 1991
59. Churchill, P. C., and M. C. Churchill. A1 and A2 adenosine receptor activation
inhibits and stimulates renin secretion of rat renal cortical slices. J. Pharmacol.
Exp. Ther. 232: 589-594, 1985

58
60. Pavlov AN, Sosnovtseva OV, Pavlova ON, Mosekilde E, Holstein-Rathlou NH.
Characterizing multimode interaction in renal autoregulation.
Physiol Meas. 2008 Aug;29(8):945-58. Epub 2008 Jul 4.
61. Iliescu R, Cazan R, McLemore GR Jr, Venegas-Pont M, Ryan MJ. Renal blood
flow and dynamic autoregulation in conscious mice. Am J Physiol Renal
Physiol. 2008 Sep;295(3):F734-40. Epub 2008 Jun 25.

59
Lebenslauf
Name: Florian Johannes Hugo Segerer Geburtsdatum: 3. September 1981 Geburtsort: Fürth Staatsangehörigkeit: deutsch Schulausbildung: August 1988 bis Juni 2001 Grundschule und Gymnasium in Berlin und Regensburg Studium: S 2002 – SS 2009 Studium der Medizin in Regensburg
und Limoges (Frankreich) Facharztausbildung: Seit Januar 2010 an der Kinderklinik der Universität
Würzburg, Direktor: Prof. Dr. C. Speer Stipendien: 2005 - 2009 Stipendiat des Cusanuswerks
(Studienförderung der deutschen Bischofskonferenz)

60
Danksagung:
Ich danke allen meinen wissenschaftlichen und medizinischen Lehrern und
Vorbildern, insbesondere Professor Frank Schweda, Professor Peter Gruß,
Profesor Johannes Wolff, Professor Hugo Segerer und vielen anderen für ihre
Lehre, Anleitung und die Begeisterung für Medizin und medizinische Forschung,
die sie mir vermitteln konnten.
Ich danke meinen Eltern Angelika und Hugo Segerer, meinen Freunden und
Geschwistern für ihre Unterstützung.