Embed Size (px)
Citation preview

Fresenius Zeitschrift fiir Fresenius Z. Anal. Chem. 289, 353-357 (1978) . . . .
.~) by Springer-Verlag 1978
Riintgentluorescenzspektrometrische Untersuchung der Miffiillung von Kobalt und Nickel bei der Eisenhydroxidfiillung mit Urotropin
A. Jangen* und D. Tenbrink
Fachbereieh Chemie-Ingerlieurwesen der FH Mtinster, Abt. Steinfurt, Stegerwaldstr. 13 D-4430 Steinfurt- 1
X-Ray Fluorimetric Study of the Coprecipitation of Cobalt and Nickel with Ferric Hydroxide in the Precipitation with Urotropine
Summary. The adsorption (coprecipitation) of cobalt and nickel on ferric hydroxide, precipitated by urotropine, was studied. The quantities of cobalt and nickel in the ferric hydroxide were determined by X-ray fluorimetric measurements of borate fusions of the hydroxides. It has been found, that fivefold more nickel than cobalt is adsorbed with equal ratios of concentration. Therefore the separation of ferric hydroxide with urotropine is not suitable for a following determination of nickel and only conditionally suitable for a following determination of cobalt, if the ferric hydroxide is not purified by reprecipi- tation with ammonia.
Zusammenfassung. Es wurde die Adsorption (Mitf/il- lung) yon Kobalt und Nickel an mit der Urotropinme- thode gefhlltem Eisenhydroxid untersucht. Die Mengen an Kobalt und Nickel im Eisenhydroxid wurden dutch R6ntgenfluorescenzmessungen an Boratschmelzen der Hydroxide bestimmt. Dabei wurde festgestellt, dab bei gleichen Konzentrationsverhhltnissen ffinfmal soviel Nickel wie Kobalt adsorbiert wird und dab die Fiillung yon Eisenhydroxid mit der Urotropinmethode fiir eine Abtrennung des Eisens vor einer Nickelbestimmung nicht und vor einer Kobaltbestimmung nur bedingt ge- eignet ist, wenn nicht das Eisenhydroxid durch eine Um- fhllung, z.B. mit Ammoniak, gereinigt wird.
Key words: Mitfhllung von Kobalt, Nickel mit Eisenhy- droxid; Urotropin
* Korrespondenz-Anschrift
Trotz der hohen Leistungsfghigkeit und des hohen Auto- matisierungsgrades der instrumentellen Analysenver- fahren, vor allem der Emissionsspektrometrie und der R6ntgenfluorescenzanalyse, haben naBchemische Ana- lysenmethoden in der Stahlanalyse noch nicht entschei- dend an Bedeutung verloren, da diese Methoden vor allem zur Analyse der Eichstandards herangezogen werden. Dadurch ist jedoch die Forderung nach geringem Zeit- aufwand ersetzt worden durch die Forderung nach hoher Analysengenauigkeit. Da die Selektivitht der nagchemi- schen Analysenverfahren meist nicht sehr hoch ist, muB sehr hiiufig vor der eigentlichen Bestimmung eine Ab- trennung st6render Begleitionen erfolgen. Handett es sich bei den St6rionen um solche Metallionen, die drei- und h6herwertig sind, so werden diese hhufig als Hydro- xide abgetrennt, wenn Metalle, die als zweiwertige Ionen in der Analysenl6sung vorliegen, bestimmt werden sollen. Ftir derartige Abscheidungen von Hydroxiden der drei- und h6herwertigen Metallionen haben das sog. Acetatverfahren und die Fhllung mit Urotropin weite Verbreitung gefunden. Von diesen beiden Methoden ist das Acetatverfahren [6, 11] insofern problematisch, als die Abscheidung durch Hydrolyse der Metall-Acetato~ komplexe in der Siedehitze erfolgt, diese Hydrolysenre- aktion aber beim Abkiihlen zumindest teilweise wieder riicklfiufig ist. Bei der Urotropinmethode [14, 15] erfolgt die Hydroxidfhllung dagegen durch kontinuierliche Stei- gerung der OH - -Ionenkonzentration auf Grund der Hy- drolyse von Urotropin, wobei reproduzierbare Fhllungs- bedingungen erzielt werden k6nnen.
Obwohl die Ffillung aus homogener L6sung in der Siedehitze optimal fiir das Erhalten eines grobteiligen Niederschlags von gleichmggiger Morphologie ist, wird doch wegen der geringen Kristallisationsfhhigkeit yon Schwermetallhydroxiden auch unter optimalen Bedin- gungen ein Niederschlag erhalten, der an der Oberflfiche stets noch von Ionen aus der L6sung, aus der er gefhllt wurde, verunreinigt ist. Wie welt diese Verunreinigungen
0016-1152/78/0289/0353/$ 01.00

354 Fresenius Z. Anal. Chem., Band 289 (1978)
die Anwendbarkei t derartiger Hydroxidffillungen zu einer Vor t rennung im Rahmen der Stahlanalyse beein- tr~chtigen, sollte durch Best immung des Kobalt- und Nickelgehaltes im Eisenoxid, das durch Vergliihen von Eisenhydroxid erhalten wurde, festgestellt werden, wenn dieses Eisenhydroxid nach der Urot ropinmethode aus
L6sungen ausgef~illt wird, die gleichzeitig steigende Mengen Nickel und/oder Kobalt enthalten.
Daf3 n~mlich Eisenhydroxidniederschlfige, wie die meisten Hydroxidniederschlfige, auch Ionen solcher Elemente enthalten, deren Hydroxidl6slichkeitsprodukt noch nicht fiberschritten ist, zeigen die Untersuchungen yon Kurbatov u.a. [9, 10] fiber Mitffil- lungen mit Eisenhydroxid in Abh~ngigkeit vom pH-Wert; insbe- sondere die dabei ffir Kobalt gefundenen Mitf~llungskurven zeigen auch ffir kleine pH-Werte noch Kobaltgehalte im Niederschlag. Diese Erscheinungen wurden auch von Kolarik u. a. [7] und yon Schulze u.a. [18,19] bei ihren Untersuchungen fiber Sorptions- effekte an Eisenhydroxidffillungen beobachtet, wobei Schulze [ 18] ferner feststeIlen konnte, dag die Sorptionserscheinungen zumin- dest teilweise nicht auf reversiblen Gleichgewichten beruhen, was f fir die Praxis bedeutet, dag solche Niederschlfige nicht vollst~indig durch Auswaschen gerinigt werden k6nnen. Diese Untersuchungen befaf3ten sich jedoch nicht mit den Auswirkungen dieser Erschei- nungen auf die Durchfiihrung yon Trennungen, sondern dienten vor allem der Aufklfirung der Sorptionsmechanismen.
Die Best immung von Nickel und/oder Kobalt im Eisenoxid wurde durchgetiihrt mittels der R6ntgenfluo- rescenzanalyse (RFA) nach Aufl6sen des Oxids in einer
Schmelze aus Lithiumtetraborat und Bortrioxid und di- rekter Messung des nach dem Abktihlen erhal tenen Bo- ratglases. Die R F A ist fiir die Untersuchung derartiger pulverf6rmiger feinteiliger Substanzen die optimale Analysenmethode, da sie ausreichende Empfindlichkeit bei geringem Zeitbedarf aufweist. Und mit der Borat- schmelztechnik ist eine Probenvorbere i tung gegeben, die mit geringem Aufwand homogene Proben liefert, in denen kaum noch Matrixeffekte auftreten und die wegen ihrer glasartigen Struktur und damit reproduzierbar her- stellbaren Oberfl~iche fiir die R F A geradezu ideal sind [3,4,13,17, 21]. Die Schmelztechnik als Probenvorbe- reitung hat zudem den Vorteil, dal3 dadurch der ur- spriingliche Zustand der Probe aufgehoben wird und dadurch auch die Eichung unproblematisch wird, weil man - wie bei der Analyse von fRissigen L6sungen - die Eichproben aus Reins tverbindungen der Kationen, z.B. entsprechenden Oxiden, herstellen kann [3,4,13].
Experimenteller Teil
Die r6ntgenfluorescenzspektrometrischen Messungen wurden durchgeffihrt mit einem R6ntgenfluorescenz-Spektrometer, Modell PW 1410 (Philips Elektronic Industrie GmbH, Hamburg) unter Verwendung einer Goldr6hre, Modell PW 2181/00 (Philips), als Anregungsquelle und Litbiumfluorid (2 d = 0,4028 nm) als Analysator-Kristall. Da die statistischen Untersuchungen von Birks [1] gezeigt haben, dab die Standardabweiehung bei der Arbeits- weise mit Zeitvorwahl das 1,1-fache der Impulsvorwahl niemals fibersteigt, wurden ffir alle Proben die Impulse pro 4 s (Zeitvor-
Tabelle 1. Daten der r6ntgenfluorimetrischen Messungen
Fe Co Ni
Anregungsspannung 20 kV 25 kV 30 kV R6hrenstrom 15 mA 30 mA 30 mA Fluorescenzlinie:
Art K a K a K~ Wellenlfinge (nm) 0,1936 0,1790 0,1659 Goniometerwinkel 20 57,45 ~ 52,74 ~ 48,61 ~
wahl) gez~ihlt, also nicht die zu z~ihlende Impulsrate jeweils opti- miert. Die Z~ihlung erfolgte mit einem Szintillationsz~ihler, dessen Zfihlrohrspannung 320 V betrug, w/ihrend sich die Probe im Vakuum (0,11 Torr) befand und mit 60 Upm gedreht wurde. Die Anregung der Fluorescenzstrahlung erfolgte nicht bei maximal zu- liissiger R6hrenleistung, sondern bei den in Tabelle 1 angegebenen Werten, da bei h6herer R6hrenleistung ein Schwarzwerden der Probenoberfl~iche beobachtet wurde. Die Tabelle 1 enth/ilt ferner die Angabe der R6ntgenlinie, deren Intensit/it zur quantitativen Analyse der Proben ausgewertet wurde sowie die daraus resultie- rende Goniometereinstellung.
Die Herstellung der Eichstandards und Proben erfolgte durch ein Schmelzgiegverfahren unter Verwendung eines Gemisches aus 5 g Di-Lithiumtetraborat (wasserfrei, zur Analyse ffir die R6ntgen- fluorescenzanalyse) und 1,5 g Di-Bortrioxid, (wasserfrei, zur Ana- lyse ffir die Silicatanalyse) als Schmelzaufschlugmittel [4,13]. Dabei erwies es sich als am g/instigsten, wenn das eingewogene Oxid oder Oxidgemisch zun~ichst mit dem Bortrioxid vermischt wurde und diese Mischung dann erst mit dem Lithiumtetraborat gemischt wurde. Die Mischung wurde direkt in einen 20 ml-Platintiegel, hohe Form, eingewogen und bei einer Temperatur zwischen 1200 und 1300~ im elektrischen Muffelofen zur Homogenitfit geschmolzen, wobei dies durch gelegentliches Umschwenken des Tiegelinbaltes beschleunigt werden konnte. Die homogene Schmelze wurde dann in Tiegeldeckel von 36 mm Durchmesser aus Platin-Gold-Legie- rung (95/5) gegossen und darin zu klaren Scheiben abkiihlen ge- lassen. Zumindest das Erkalten der Schmelzen mug in Platin- Gold-Legierungen geschehen, da diese von dem Aufschluf3mittel nicht benetzt werden und somit die erstarrten Boratgl~iser nach dem Erkalten leicht herausgenommen werden k6nnen. Stehen auch Platin-Gold-Tiegel zur Verffigung, kann die Schmelze direkt darin abkiihlen gelassen werden. Die so erhaltenen Proben waren im Exsiccator fiber ,,Blaugel" unbegrenzt haltbar; an der Laborluft verwitterte die glatte Oberfl~iche jedoch schnell, wodurch die Proben fiir RFA-Messungen unbrauchbar wurden. - Zur Herstel- lung der Eichstandards wurden entsprechende Mengen von Eisen- oxid zur Analyse, Kobaltoxid (Kobaltschwarz, Co304) und/oder Nickeloxid, reinst (NiO) eingewogen.
0,10 M Eisen(IIl)-nitratlOsung: 40,400 g Fe(NO3) 3 . 9 H~O wurden in ca. 600 ml Wasser gel6st, mit 2 N Salpeters~ure auf pH 2,0 gebracht und mit Wasser auf 1000 ml L6sung aufgeffillt.
0,10 M Kobalt(II)-sulfatl6sung; 28,110 g CoSO 4 . 7 H20 wurden in Wasser zu 1000 ml L6sung gel6st.
0,10 M Nickel(lI)-nitratlOsung; 29,081 g Ni(NO3)2 " 6 H20 wurden in Wasser zu 1000 ml L6sung gelfst.
lO%ige Urotropinl6sung; 100 g Hexamethylentetramin wurden in Wasser zu 1000 ml L6sung gel6st.
2%ige Ammoniumnitratl6sung; 20 g NH4NO 3 wurden in Wasser zu 1000 ml L6sung gel6st.
In einem 250 ml-Becherglas wurden zu 5 ml 0,1 M Eisenni- tratl6sung Volumina der Kobalt- und/oder Nickelsalzl6sung ent-
A. Jangen und D. Tenbrink: Mitf~illung yon Kobalt und Nickel bei der Eisenhydroxidf~illung 355
O.L
o~ 0 0 . 3 g '
14..
--~0.2 x
-6 2~ EO.I
| x=Ni
x= Co
/ . ~ / •
1 2 3 Z, MoL x / Mot Fe
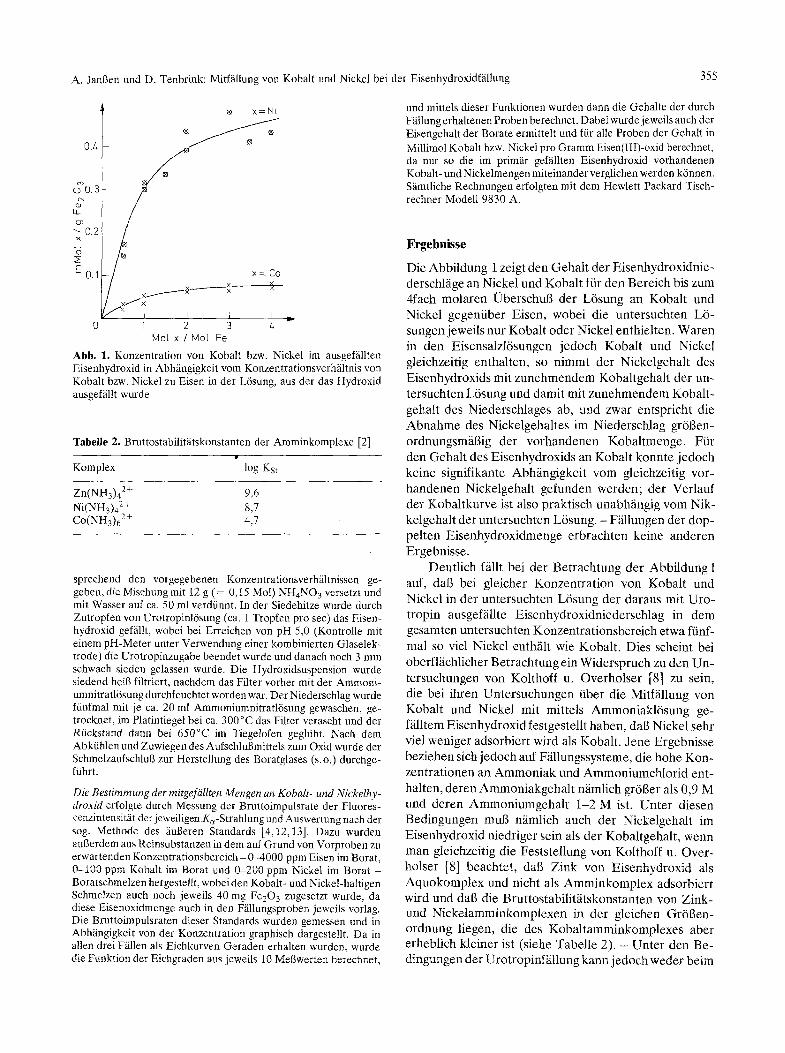
Abb. 1. Konzentration von Kobalt bzw. Nickel im ausgefiillten Eisenhydroxid in Abh~ingigkeit vom Konzentrationsverhfiltnis yon Kobalt bzw. Nickel zu Eisen in der L6sung, aus der das Hydroxid ausgef~illt wurde
Tabelle 2. Bruttostabilit/itskonstanten der Amminkomplexe [2]
Komplex log Kst
Zn(NH3)4 ~+ 9,6 Ni(NH3)42+ 8,7 Co(NH3)62+ 4,7
sprechend den vorgegebenen Konzentrationsverh~iltnissen ge- geben, die Mischung mit 12 g (= 0,15 Mol) NH4NO3 versetzt und mit Wasser auf ca. 50 ml verdtinnt. In der Siedehitze wurde durch Zutropfen yon Urotropinl6sung (ca. 1 Tropfen pro sec) das Eisen- hydroxid gef~illt, wobei bei Erreichen yon pH 5,0 (Kontrolle mit einem pH-Meter unter Verwendung einer kombinierten Glaselek- trode) die Urotropinzugabe beendet wurde und danach noch 3 min schwach sieden gelassen wurde. Die Hydroxidsuspension wurde siedend heig filtriert, nachdem das Filter vorher mit der Ammoni- umnitratl6sung durchfeuchtet worden war. Der Niederschlag wurde fiinfmal mit je ca. 20 ml Ammoniumnitratl6sung gewaschen, ge- trocknet, im Platintiegel bei ca. 300~ das Filter verascht und der R/ickstand dann bei 650~ im Tiegelofen gegl/iht. Nach dem Abkiihlen und Zuwiegen des Aufschlugmittels zum Oxid wurde der Schmelzaufschlul3 zur Herstellung des Boratglases (s.o.) durchge- fiihrt.
Die Bestimmung der mitgefiillten Mengen an Kobalt- und Nickelhy- droxid effolgte durch Messung der Bruttoimpulsrate der Fluores- cenzintensit/it der jeweiligen K~-Strahlung und Auswertung nach der sog. Methode des ~iugeren Standards [4,12,13]. Dazu wurden aul3erdem aus Reinsubstanzen in dem auf Grund von Vorproben zu erwartenden Konzentrationsbereich - 0-4000 ppm Eisen im Borat, 0-100 ppm Kobalt im Borat und 0-200 ppm Nickel im Borat - Boratschrnelzen hergestellt, wobei den Kobalt- und Nickel-haltigen Schmelzen auch noch jeweils 40 rng Fe203 zugesetzt wurde, da diese Eisenoxidmenge auch in den Ffillungsproben jeweils vorlag. Die Bruttoimpulsraten dieser Standards wurden gemessen und in Abh~ingigkeit yon der Konzentration graphisch dargestellt. Da in allen drei Ffillen als Eichkurven Geraden erhalten wurden, wurde die Funktion der Eichgraden aus jeweils 10 MeBwerten berechnet,
und mittels dieser Funktionen wurden dann die Gehalte der durch F~illung erhaltenen Proben berechnet. Dabei wurde jeweils auch der Eisengehalt der Borate ermittelt und fiir alle Proben der Gehalt in Millimol Kobalt bzw. Nickel pro Gramm Eisen(llI)-oxid berechnet, da nur so die im primiir gef~illten Eisenhydroxid vorhandenen Kobalt- und Nickelmengen miteinander verglichen werden k6nnen. S~imtliche Rechnungen erfolgten mit dem Hewlett Packard Tisch- rechner Modell 9830 A.
Ergebnisse
Die Abbildung i zeigt den Gehalt der Eisenhydroxidnie- derschlfige an Nickel und Kobalt fiir den Bereich bis zum 4fach molaren l~berschul3 der L6sung an Kobalt und Nickel gegenfiber Eisen, wobei die untersuchten L6- sungen jeweils nur Kobalt oder Nickel enthielten. Waren in den Eisensalzl6sungen jedoch Kobalt und Nickel gleichzeitig enthalten, so nimmt der Nickelgehalt des Eisenhydroxids mit zunehmendem Kobaltgehalt der un- tersuchten L6sung und damit mit zunehmendem Kobalt- gehalt des Niederschlages ab, und zwar entspricht die Abnahme des Nickelgehaltes im Niederschlag gr6gen- ordnungsm~igig der vorhandenen Kobaltmenge. Ffir den Gehalt des Eisenhydroxids an Kobalt konnte jedoch keine signifikante Abhfingigkeit vom gleichzeitig vor- handenen Nickelgehalt gefunden werden; der Verlauf der Kobaltkurve ist also praktisch unabhgngig vom Nik- kelgehalt der untersuchten L6sung. - F~illungen der dop- pelten Eisenhydroxidmenge erbrachten keine anderen Ergebnisse.
Deutlich ffillt bei der Betrachtung der Abbildung 1 auf, dab bei gleicher Konzentration yon Kobalt und Nickel in der untersuchten L6sung der daraus mit Uro- tropin ausgef~illte Eisenhydroxidniederschlag in dem gesamten untersuchten Konzentrationsbereich etwa ffinf- mal so viel Nickel enth/ilt wie Kobalt. Dies scheint bei oberflfichlicher Betrachtung ein Widerspruch zu den Un- tersuchungen von Kolthoff u. Overholser [81 zu sein, die bei ihren Untersuchungen fiber die Mifffillung von Kobalt und Nickel mit mittels Ammoniakl6sung ge- f~illtem Eisenhydroxid festgestellt haben, dab Nickel sehr viel weniger adsorbiert wird als Kobalt. Jene Ergebnisse beziehen sich jedoch auf Ffillungssysteme, die hohe Kon- zentrationen an Ammoniak und Ammoniumchlor id ent- halten, deren Ammoniakgehal t n~imlich gr6ger als 0,9 M und deren Ammoniumgehal t 1-2 Mis t . Unter diesen Bedingungen mug niimlich auch der Nickelgehalt im Eisenhydroxid niedriger sein als der Kobaltgehalt, wenn man gleichzeitig die Feststellung yon Kolthoff u. Over- holser [8] beachtet, dag Zink yon Eisenhydroxid als Aquokomplex und nicht als Amminkomplex adsorbiert wird und dab die Bruttostabilitfitskonstanten von Zink- und Nickelamminkomplexen in der gleichen Gr6gen- ordnung liegen, die des Kobaltamminkomplexes aber erheblich kleiner ist (siehe Tabelle 2). - Unter den Be- dingungen der Urotropinf~llung kann jedoch weder beim
356 Fresenius Z. Anal. Chem., Band 289 (1978)
4 Z
~ 2 H_ c3~
x
t J I 0.B 1.0 1.5 2.0
Mo[ F e / M o t Ni
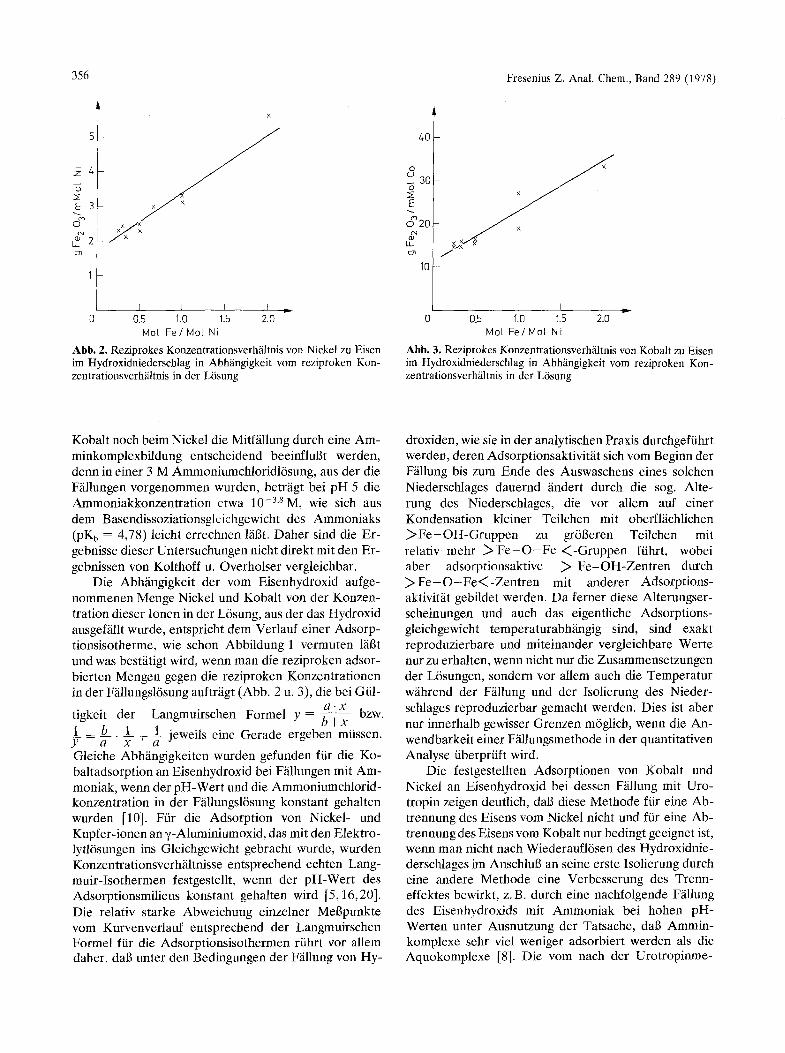
A b b . 2. Reziprokes Konzentrationsverh/iltnis von Nickel zu Eisen im Hydroxidniederschlag in Abh/ingigkeit yore reziproken Kon- zentrationsverh/iltnis in der L6sung
z,0
O C) _ 30 o
g kL Cin
10 J
_1 [ I I 0.5 1.0 1.5 2.0
Mol Fe / Mol Ni
A b b . 3. Reziprokes Konzentrationsverhiiltnis von Kobalt zu Eisen im Hydroxidniederschlag in Abhfingigkeit vom reziproken Kon- zentrationsverh/iltnis in der L6sung
Kobalt noch beim Nickel die Mitf~illung durch eine Am- minkomplexbildung entscheidend beeinfiu6t werden, denn in einer 3 M Ammoniumchloridl6sung, aus der die Ffillungen vorgenommen wurden, betrfigt bei pH 5 die Ammoniakkonzentration etwa 10 -3,8 M, wie sich aus dem Basendissoziationsglcichgewicht des Ammoniaks (pKb = 4,78) leicht errechnen lfif3t. Daher sind die Er- gebnisse dieser Untersuchungen nicht direkt mit den Er- gebnissen yon Kolthoff u. Overholser vergleichbar.
Die AbNingigkeit der vom Eisenhydroxid aufge- nommenen Menge Nickel und Kobalt yon der Konzen- tration dieser Ionen in der L6sung, aus der das Hydroxid ausgef~illt wurde, entspricht dem Verlauf einer Adsorp- tionsisotherme, wie schon Abbildung 1 vermuten lfigt und was bestfitigt wird, wenn man die reziproken adsor- bierten Mengen gegen die reziproken Konzentrationen in der F~llungsl6sung auftrfigt (Abb. 2 u. 3), die bei Gill-
a . X tigkeit der Langmuirschen Formel y = b+-~- bzw.
I _ b 1 + I jeweils eine Gerade ergeben miissen. Y - h - ' Y- h- Gleiche Abhfingigkeiten wurden gefunden fiir die Ko- baltadsorption an Eisenhydroxid bei F~illungen mit Am- moniak, wenn der pH-Wert und die Ammoniumchlorid- konzentration in der Ffillungsl6sung konstant gehalten wurden [10]. Ffir die Adsorption von Nickel- und Kupfer-ionen an •-Aluminiumoxid, das mit den Elektro- lytl6sungen ins Gleichgewicht gebracht wurde, wurden Konzentrationsverhfiltnisse entsprechend echten Lang- muir-Isothermen festgestellt, wenn der pH-Wert des Adsorptionsmilieus konstant gehalten wird [5,16,20]. Die relativ starke Abweichung einzelner Mel3punkte vom Kurvenverlauf entsprechend der Langmuirschen Formel fiir die Adsorptionsisothermen riihrt vor allem daher, dab unter den Bedingungen der Ffillung yon Hy-
droxiden, wie sie in der analytischen Praxis durchgefiihrt werden, deren Adsorptionsaktivitfit sich vom Beginn der F~illung bis zum Ende des Auswaschens eines solchen Niederschlages dauernd ~indert durch die sog. Alte- rung des Niederschlages, die vor allem auf einer Kondensation kleiner Teilchen mit oberfl~ichlichen
Fe -OH-Gruppen zu gr6geren Teilchen mit relativ mehr ~ F e - O - F e ( - G r u p p e n ftihrt, wobei aber adsorptionsaktive ) Fe-OH-Zent ren dutch ~ F e - O - F e < - Z e n t r e n mit anderer Adsorptions- aktivitfit gebildet werden. Da ferner diese Alterungser- scheinungen und auch das eigentliche Adsorptions- gleichgewicht temperaturabhfingig sind, sind exakt reproduzierbare und miteinander vergleichbare Werte nur zu erhalten, wenn nicht nur die Zusammensetzungen der L6sungen, sondern vor allem auch die Temperatur wfihrend der F~illung und der Isolierung des Nieder- schlages reproduzierbar gemacht werden. Dies ist aber nur innerhalb gewisser Grenzen m6glich, wenn die An- wendbarkeit einer Ffillungsmethode in der quantitativen Analyse iiberprtift wird.
Die festgestellten Adsorptionen von Kobalt und Nickel an Eisenhydroxid bei dessen Ffillung mit Uro- tropin zeigen deutlich, dab diese Methode ftir eine Ab- trennung des Eisens vom Nickel nicht und f/ir eine Ab- trennung des Eisens vom Kobalt nur bedingt geeignet ist, wenn man nicht nach Wiederaufl6sen des Hydroxidnie- derschlages im Anschlu[3 an seine erste Isolierung durch eine andere Methode eine Verbesserung des Trenn- effektes bewirkt, z.B. durch eine nachfolgende F/illung des Eisenhydroxids mit Ammoniak bei hohen pH- Werten unter Ausnutzung der Tatsache, dal3 Ammin- komplexe sehr viel weniger adsorbiert werden als die Aquokomplexe [8]. Die yore nach der Urotropinme-

A. Jangen und D. Tenbrink: Mitf~illung von Kobalt und Nickel bei der Eisenhydroxidf~illung 357
thode gef~illten Eisenhydroxid adsorbierte Menge an Kobalt und Nickel ist n~imlich so grog, dag z. B. bei einer entsprechenden Analyse einer Stahlprobe mit 5 % Nickel und 5 % Kobalt bei ca. 90 % Eisen nach dem Aufl6sen des Stahls beim Ausf~illen des Eisenhydroxids zu dessen Ab- trennung fiir eine nachfolgende Best immung yon Kobalt und Nickel im Filtrat 1,6% des Kobalts und sogar 8,2% des vorhandenen Nickels vom Eisenhydroxid mitgerissen werden, wobei diese Mengen zwar yon den F/illungsbe- dingungen abhiingen, aber durch deren Variation nicht entscheidend verringert werden k6nnen. Diese systema- tischen Fehler nehmen mit steigendem Gehalt der Proben an Kobalt und Nickel ab, entsprechend mit ab- nehmendem Gehalt zu, jedoch ist in keinem Gehaltsbe- reich der Fehler ffir Nickelbestimmungen zu tolerieren, w~ihrend fiir Kobaltbest immungen bei der Untersuchung von Proben mit weniger als 1% ein systematischer Fehler von dann knapp 2 % relativ eventuell in Kauf genommen werden kann.
Literatur
1. Birks, L. S., Brown, D. M.: Anal. Chem. 34, 240 (1962) 2. Christen, H. R.: Grundlagen der allgemeinen und anorgani-
schen Chemic. Aarau: Sauerl~inder, Frankfurt a.M.: Salle 1968 3. Dtirr, W.: Merck-Kontakte 2/74, 23 (1974)
4. Emmermann, R., Obi, D. V. C.: Fresenius Z. Anal. Chem. 254, 1 (1971)
5. Fischer, W., Kulling, A., Umland, F.: Angew. Chem. 61, 386 (1950)
6. H61tje, R.: Z. Anorg. Chem. 243, 252 (1940) 7. Kolarik, Z. u.a.: Coll. Czechoslov. Chem Commun. 25, 1000
(1960); 26, 1082 (1961); 30, 724 (1965); 28, 2818 (1963) 8. Kolthoff, I. M., Overholser, L. G.: J. Phys. Chem. 43, 767
(1938) 9. Kurbatov, J. D., Kulp, J. L., Mack, E.: J. Am. Chem. Soc. 67,
1923 (1945) 10. Kurbatov, M. H,, Wood, G. B., Kurbatov, J. D.: J. Phys. Chem.
55, 1170 (1951) 11. Mittasch, A.: Fresenius Z. Anal. Chem 42, 492 (1903) 12. Plesch, R.: Fresenius Z. Anal. Chem. 261, 97 (1972); 262, 84
(1972); 265, 114 (1973); 266, 9 (1973) 13. Plesch, R., Ebersp~icher, O.: Siemens Analysentechn. Mitt. Nr.
116 14. Ray, P.: Fresenius Z. Anal. Chem. 86, 13 (1931) 15. Ray, P., Chattopadhya, A. K.: Z. Anorg. Chem. 169, 99 (1928) 16. Sch~ifer, H., Neugebauer, W.: Naturwissenschaften 38, 561
(1951) 17. Schneider, H.: Fresenius Z. Anal. Chem. 249, 225 (1970) 18. Schulze, W.: Fresenius Z. Anal. Chem. 241, 207 (1968) 19. Schulze, W., Scheffler, M.: Fresenius Z. Anal. Chem. 226, 395
(1967); 229, 161 (1967) 20. Umland, F.: Z. Elektrochem. 60, 711(1956) 21. Willigen, J. H. H. G. van, Kruidhof, H., Dahmen, E. A. M. F.:
Talanta 18, 450 (1971)
Eingegangen am 9. August 1977





























