Embed Size (px)
Citation preview

Structural and Spectroscopic Characterization of Tin−Tin DoubleBonds in Cyclic DistannenesJens Henning,† Klaus Eichele,† Reinhold F. Fink,‡ and Lars Wesemann*,†
†Institut fur Anorganische Chemie and ‡Institut fur Physikalische und Theoretische Chemie, Universitat Tubingen, Auf derMorgenstelle 18, 72076 Tubingen, Germany
*S Supporting Information
ABSTRACT: Three cyclic distannenes, 1, 3, and 4, and one spacer-bridgedbis(stannylene), 2, were prepared and thoroughly investigated by single-crystal X-raydiffraction in the solid state, by variable-temperature (VT) 119Sn NMR, VT 1HNMR, 13C NMR, and UV−vis spectroscopy in solution, and by quantum chemicalcalculations. The tin(II) compounds feature rigid 9,9-dimethylxanthene ornaphthalene backbones and very bulky m-terphenyl substituents ArR [=C6H3-2,6-{C6H2-2,4,6-R3}2; R = Me (1, 3), i-Pr (2,4)]. For distannenes 3 and 4, the strain of the naphthalene backbone results in rather short tin−tin distances of 2.7299(3) and2.7688(2) Å, respectively, whereas the xanthene backbone produces long tin−tin distances of 3.0009(7) Å for distannene 1 and4.2779(7) Å for the spacer-bridged bis(stannylene) 2. In comparison to the AriPr substituents, the less bulky ArMe substituentsgive rise to stronger trans-bending of the distannenes; moreover, DFT calculations indicate that, in contrast to AriPr, the ArMe
substituents allow for asymmetric distortion of the trans-bending in dynamic processes. The oxidation products of distannene 1and bis(stannylene) 2 reveal rare structural motifs: dihydroxydistannoxane 5 and bis(dihydroxystannane) 6, respectively, whichfeature terminal Sn−OH functionalities. The reaction of distannene 1 with 1 equiv of potassium chloride in the presence of thecryptating agent 222 results in the formation of the unusual stannyl stannide 7. A modified synthesis protocol for the preparationof distannene 1 yields in one step the stannyl stannylene 8 with a center of chirality at the stannyl tin atom. The series 1, 7, and 8represents a variation of electronic tin−tin interactions.
■ INTRODUCTION
In 1976, Lappert and co-workers reported the first structuralcharacterization of an alkene homologue of heavier group 14elements. The distannene R2SnSnR2 with bulky R =CH(SiMe3)2 substituents revealed pyramidalization of the tinatoms, in contrast to the planar structure of alkenes, and theformal tin−tin double bond readily dissociated in solution toform monomeric stannylenes.1,2 Some examples of compoundsfeaturing formal double bonds between tin and also silicon,germanium, and lead followed and have been reviewed.3−7 Ingeneral, the structural deviation from planarity becomes morepronounced with increasing element number and the formalelement−element double bond becomes more labile. For tin,dissociation of SnSn is observed often; hence, the availableamount of physical data characterizing the formal tin−tindouble bond is very limited. Furthermore, reactivity studiesreflect the lability of the SnSn bond because distannenesoften behave as expected for monomeric stannylenes.Previously, we reported the cyclic distannene 1 featuring a
9,9-dimethylxanthene backbone and the bulky terphenylsubstituents ArMe (=C6H3-2,6-Mes2; Mes = C6H2-2,4,6-Me3).Furthermore, we found an unusual nickel(0) coordinationcompound in which distannene 1 coordinates side-on towardthe nickel atom.8
In order to further investigate the tin−tin interaction of suchintramolecular cyclic distannenes, we prepared compoundswith variation of the terphenyl substituent as well as thebackbone. Herein, we report the synthesis of the spacer-bridged
bis(stannylene) 2, which is a derivative of 1 comprising thebulkier AriPr (=C6H3-2,6-Trip2; Trip = C6H2-2,4,6-i-Pr3)substituents instead of ArMe, and cyclic distannenes 3 and 4.The two distannenes feature a naphthalene backbone instead of9,9-dimethylxanthene and either ArMe (3) or AriPr (4)substituents on the tin atoms (Chart 1). Compounds 1−4were thoroughly investigated by single-crystal X-ray diffractionin the solid state and by 119Sn NMR, 1H NMR, 13C{1H} NMR,and UV−vis spectroscopy in solution. In addition, quantumchemical calculations were performed.
■ RESULTS
Compounds 1−4 were prepared straightforwardly fromdilithium 9,9-dimethylxanthene-4,5-diide or dilithium naphtha-lene-1,8-diide and [ArMeSnCl]2
9 or AriPrSnCl10 (Scheme 1).The lithium reagents were prepared in situ from 9,9-dimethylxanthene with n-butyllithium in the presence ofTMEDA or from 1,8-diiodonaphthalene with n-butyllithium.The products were isolated in moderate yield (39−64%) asintensely colored (2, red-brown; 3, green; 4, purple) crystallineor microcrystalline materials by recrystallization from n-hexaneor n-pentane. All compounds are readily soluble in hydrocarbonsolvents and give intensely colored solutions. The lithiumnaphthalene reagent requires careful removal of all volatilecomponents prior to the reaction with the tin(II) reactants.
Received: June 18, 2014Published: July 10, 2014
Article
pubs.acs.org/Organometallics
© 2014 American Chemical Society 3904 dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−3918
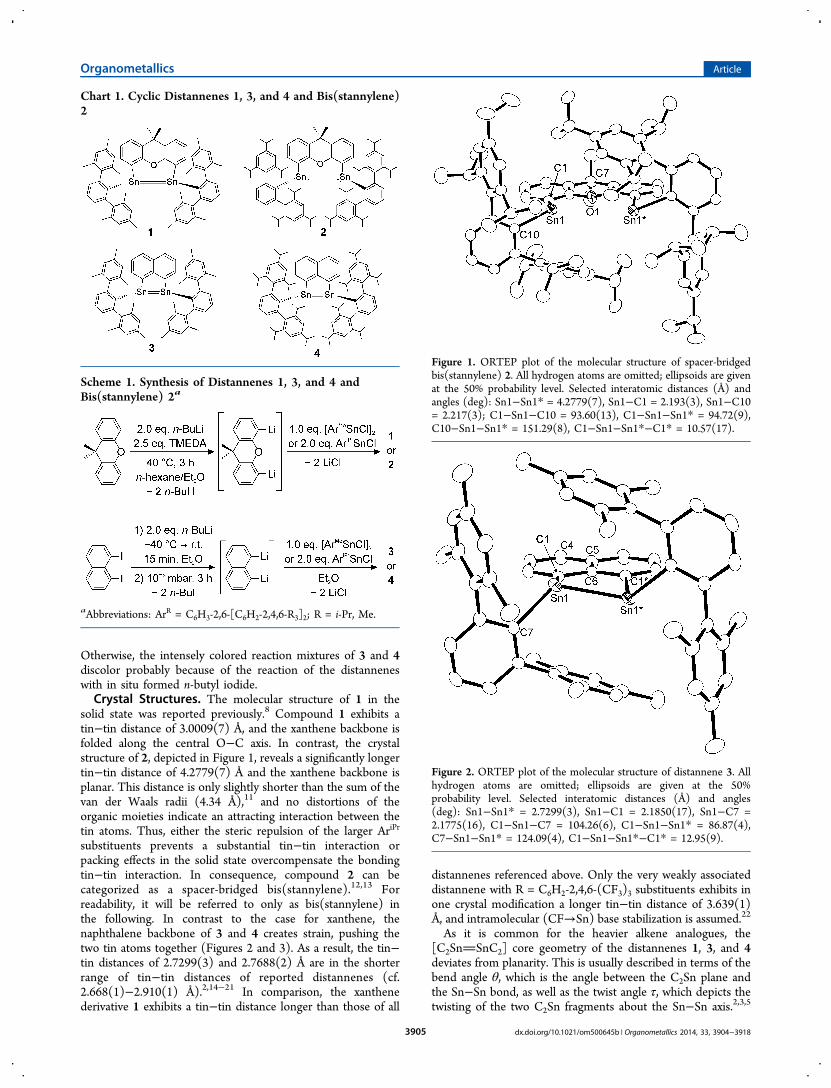
Otherwise, the intensely colored reaction mixtures of 3 and 4discolor probably because of the reaction of the distanneneswith in situ formed n-butyl iodide.Crystal Structures. The molecular structure of 1 in the
solid state was reported previously.8 Compound 1 exhibits atin−tin distance of 3.0009(7) Å, and the xanthene backbone isfolded along the central O−C axis. In contrast, the crystalstructure of 2, depicted in Figure 1, reveals a significantly longertin−tin distance of 4.2779(7) Å and the xanthene backbone isplanar. This distance is only slightly shorter than the sum of thevan der Waals radii (4.34 Å),11 and no distortions of theorganic moieties indicate an attracting interaction between thetin atoms. Thus, either the steric repulsion of the larger AriPr
substituents prevents a substantial tin−tin interaction orpacking effects in the solid state overcompensate the bondingtin−tin interaction. In consequence, compound 2 can becategorized as a spacer-bridged bis(stannylene).12,13 Forreadability, it will be referred to only as bis(stannylene) inthe following. In contrast to the case for xanthene, thenaphthalene backbone of 3 and 4 creates strain, pushing thetwo tin atoms together (Figures 2 and 3). As a result, the tin−tin distances of 2.7299(3) and 2.7688(2) Å are in the shorterrange of tin−tin distances of reported distannenes (cf.2.668(1)−2.910(1) Å).2,14−21 In comparison, the xanthenederivative 1 exhibits a tin−tin distance longer than those of all
distannenes referenced above. Only the very weakly associateddistannene with R = C6H2-2,4,6-(CF3)3 substituents exhibits inone crystal modification a longer tin−tin distance of 3.639(1)Å, and intramolecular (CF→Sn) base stabilization is assumed.22
As it is common for the heavier alkene analogues, the[C2SnSnC2] core geometry of the distannenes 1, 3, and 4deviates from planarity. This is usually described in terms of thebend angle θ, which is the angle between the C2Sn plane andthe Sn−Sn bond, as well as the twist angle τ, which depicts thetwisting of the two C2Sn fragments about the Sn−Sn axis.2,3,5
Chart 1. Cyclic Distannenes 1, 3, and 4 and Bis(stannylene)2
Scheme 1. Synthesis of Distannenes 1, 3, and 4 andBis(stannylene) 2a
aAbbreviations: ArR = C6H3-2,6-[C6H2-2,4,6-R3]2; R = i-Pr, Me.
Figure 1. ORTEP plot of the molecular structure of spacer-bridgedbis(stannylene) 2. All hydrogen atoms are omitted; ellipsoids are givenat the 50% probability level. Selected interatomic distances (Å) andangles (deg): Sn1−Sn1* = 4.2779(7), Sn1−C1 = 2.193(3), Sn1−C10= 2.217(3); C1−Sn1−C10 = 93.60(13), C1−Sn1−Sn1* = 94.72(9),C10−Sn1−Sn1* = 151.29(8), C1−Sn1−Sn1*−C1* = 10.57(17).
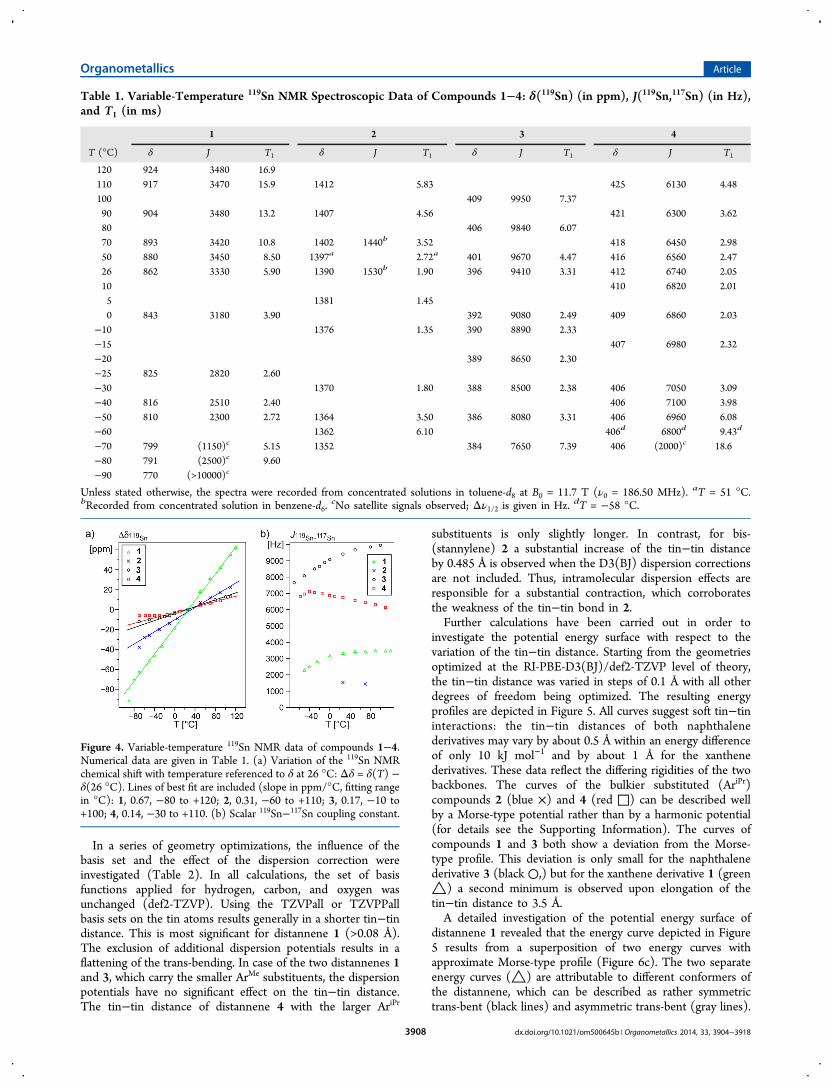
Figure 2. ORTEP plot of the molecular structure of distannene 3. Allhydrogen atoms are omitted; ellipsoids are given at the 50%probability level. Selected interatomic distances (Å) and angles(deg): Sn1−Sn1* = 2.7299(3), Sn1−C1 = 2.1850(17), Sn1−C7 =2.1775(16), C1−Sn1−C7 = 104.26(6), C1−Sn1−Sn1* = 86.87(4),C7−Sn1−Sn1* = 124.09(4), C1−Sn1−Sn1*−C1* = 12.95(9).
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183905

Compound 1 exhibits a strongly trans-bent core geometry withθ = 45 and 59°. The mean value (52°) is quite similar to thetrans-bending of 3: θ = 55°. Distannene 4 shows a slightlyflatter trans-bending of θ = 49 and 46°. This is probably a resultof the increased steric demand of the terphenyl substituent.Along with the flattening of the trans-bending, the dispositionof the tin atoms to opposite sides of the naphthalene planeincreases slightly from 0.269(3) Å (3, Sn1) to 0.304(3) Å (4,Sn1) and 0.318(3) Å (4, Sn2). In addition, the naphthalenebackbone shows slightly stronger distortions from planarity,which is indicated by the central torsion angles C1*−C6−C5−C4 = 179.66(12)° (3) and C1−C10−C5−C6 = 177.7(2)° (4).These structural differences might be understood as acounteraction in order to maintain the trans-bending of thedistannene moiety.Bis(stannylene) 2 still shows a trans-bending of θ = 27°,
which may be seen as a result of the packing of the terphenylsubstituents in the solid state rather than as a result of the tin−tin interaction. For comparison, reported nontwisted dis-tannenes exhibit trans-bending in the range of θ = 41−50°.2,14−16 Still, pronounced distortions are known as well:asymmetric trans-bending for R2SnSnR2 with R = C6H-2-tBu-4,5,6-Me3 (θ = 64 and 21°, τ = 5°),20 nonbent andtwisted with R = SiMetBu2 (θ = 1°, τ = 45°),18 and trans-bentand highly twisted with R = Si(SiMe3)3 (θ = 29°, τ = 63°).19
The connection of the two tin atoms in each of compounds 1−4 by rigid organic linkers results in strong twisting of the
[C2SnSnC2] core geometries: τ = 72°(1), 112° (2), 76° (3),and 86° (4). Moreover, because of the rigid linkers, the twoSnR2 units are bent within the plane of the backbone, which isindicated by the C1−Sn1−Sn1* (94.72(9)°, 2; 86.87(4)°, 3)and Sn2−Sn1−C1 (85.74(7)°, 4) angles, respectively.
119Sn NMR Spectroscopy. Compounds 1−4 all give rise toonly one singlet resonance with weak satellite signals due toJ(119Sn,117Sn) coupling in 119Sn NMR spectra in solution. Thisis expected for compounds 2 and 3, which exhibit molecular C2symmetry in the solid state, as well as for compound 4, whichexhibits only slight conformational deviations from C2symmetry in the solid state. In contrast, a comparable 2-foldmolecular symmetry of compound 1 is disrupted because of thefolding of the xanthene backbone in the solid state. Hence,dynamic processes inverting the folding or the planarization ofthe xanthene backbone in solution have to be responsible forthe observed higher symmetry.Significant differences are observed at room temperature
among the 119Sn chemical shift values of bis(stannylene) 2(1390 ppm), distannene 1 (862 ppm), and the distannenes 3(396 ppm) and 4 (412 ppm). In general, the 119Sn NMRchemical shift is known to correlate with the coordinationnumber of the observed tin species: a higher coordinationnumber results in a chemical shift at lower frequencies.24,25
Moreover, chemical shifts at lower frequencies have beenshown to reflect weak interactions between tin atoms anddonor sites.24,25 This is explained by paramagnetic deshieldingcontributions to the magnetic shielding tensor arising frommagnetically induced charge circulations. For low-coordinatedtin, this contribution is dominated by electronic promotions ofSn−R σ-bond electrons into the vacant p orbital.25 Regardingdistannenes as two stannylene fragments [R2Sn:] which interactin terms of a double donor−acceptor bond,2,26,27 one mayassume similar Sn−R σ-bond energetic levels for compounds1−4. Hence, the chemical shift difference allows for conclusionson the different participations of the vacant p orbital of the tinatoms in accepting electrons from the respective second tinatom. The participation is greater when the chemical shift issmaller; thus, the extent of the tin−tin electronic interactioncan be given in the order 3 ≈ 4 > 1 > 2. These solution NMRspectroscopic data are consistent with expectations derivedfrom structural data: for geometric and steric reasons, the sameorder would have been postulated mainly because of theobserved tin−tin distances in the solid state.Comparison of the 119Sn NMR chemical shifts with literature
data corroborates the categorization of the four compounds asbis(stannylene) (2) and distannenes (1, 3, and 4): the chemicalshifts of Masamune’s distannenes with R = Trip (δ 427 ppm)28
and the b i cyc l i c d i s t annene w i th two [S i(S i -Me3)2(SiMe2)2Si(SiMe3)2] bridges (δ 545 ppm)21 comparewith data of the naphthalene derivatives 3 and 4. In addition,the significantly higher chemical shift of xanthene derivative 1 isexemplified in data reported for distannenes: Lappert’sdistannene with R = CH(SiMe3)2 (δA 735 ppm, δB 750 ppm;two species were observed at −91 °C, attributed to con-formers)29,30 and the distannene with R = C6H-2-
tBu-4,5,6-Me3(δ 798 ppm at −63 °C).20,31 The latter example dissociates intomonomers at room temperature. At 25 °C, a chemical shift of1331 ppm was observed.20,31 This value compares well with thechemical shift of bis(stannylene) 2. However, other monomericstannylenes with bulky organyl substituents reveal 119Snresonances at frequencies considerably higher (δ 1904−2323ppm)29,30,32−36 than those for bis(stannylene) 2. This might
Figure 3. ORTEP plot of the molecular structure of distannene 4. Allhydrogen atoms and cocrystallized hexamethyldisiloxane (crystals wereobtained from hexamethyldisiloxane solution at −40 °C) are omitted;ellipsoids are given at the 50% probability level. Selected interatomicdistances (Å) and angles (deg): Sn1−Sn2 = 2.7688(2), Sn1−C1 =2.185(3), Sn2−C9 = 2.186(3), Sn1−C11 = 2.187(2), Sn2−C47 =2.190(2); C1−Sn1−C11 = 97.80(9), C9−Sn2−C47 = 98.86(9), C1−Sn1−Sn2 = 85.74(7), C9−Sn2−Sn1 = 85.80(7), C11−Sn1−Sn2 =133.49(6), C47−Sn2−Sn1 = 131.26(6), C1−Sn1−Sn2−C9 =15.36(9).
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183906

indicate that 2 comprises yet a weak mutual tin−tin interactionin solution.In addition to a main singlet resonance, all four compounds
give rise to satellite signals which are consistent withJ(119Sn,117Sn) coupling. This is expected for the threedistannenes 1, 3, and 4 yet surprising for bis(stannylene) 2.In principle, the sequence of the coupling constants reproducesthe sequence of the 119Sn NMR chemical shift data: 3 (9410Hz) > 4 (6740 Hz) > 1 (3330 Hz) > 2 (1530 Hz). However,the difference in the coupling constants between the twonaphthalene derivatives is more significant in comparison to thechemical shift values (3 (396 ppm) ≈ 4 (412 ppm)). Mostremarkably, the observed coupling for bis(stannylene) 2 clearlyindicate an electronic interaction of the two tin atoms insolution. Literature data on scalar tin−tin coupling ofdistannenes are fairly scarce; J(119Sn,117Sn) values are reportedin the range of 1240−3317 Hz, including Lappert’s distannenewith R = CH(SiMe3)2,
29,30 Masamune’s distannene28 with R =Trip, and the doubly [Si(SiMe3)2(SiMe2)2Si(SiMe3)2] bridgeddistannene.37 The corresponding values of the xanthenederivatives 1 and 2 compare well with these, and the couplingconstants of the naphthalene derivatives are remarkably largerthan those in the xanthenes.Variable-Temperature NMR Spectroscopy. Within this
section, we confine to summarize our conclusions. Details onthe 1H and 13C{1H} NMR spectroscopic investigation are givenin the Supporting Information. For the distannenes 1, 3, and 4,inversion of the trans-bending is observed in 1H NMR spectraas a result of dynamic processes in solution (Chart 2).
However, the underlying processes are frozen out on the NMRtime scale at temperatures below −15 °C. The investigation ofbis(stannylene) 2 similarly reveals inversion of the trans-bending which, in contrast, is also observed at temperaturesdown to −70 °C. This indicates a lower activation barrier forthe inversion of the trans-bending of bis(stannylene) 2 incomparison to the distannenes and thus supports thecategorization into bis(stannylene) and distannenes.In addition, 119Sn NMR spectra were recorded at variable
temperatures, and spin−lattice relaxation times (T1) weredetermined by inversion recovery experiments (Table 1). Therelative variation of the 119Sn NMR chemical shift is plotted inFigure 4a. It is found that the chemical shift variesapproximately linearly with the temperature. The naphthalenederivatives 3 and 4 each reveal two linear regions, and the slopeis greater at high temperatures. In comparison, the slope of thechemical shift variation, Δδ119
Sn(T), corresponds to the expectedmolecular flexibility: 1 > 2 > 3 ≥ 4.The scalar J(119Sn,117Sn) coupling constant also varies with
temperature (Figure 4b). The distannenes 1 and 3, which carrythe smaller ArMe substituents, show essentially a similar
behavior: the coupling constant increases with increasingtemperature. The slope is steeper at low temperatures andflattens at high temperatures. For 1, no significant change isobserved above 50 °C. In contrast, distannene 4, carrying thelarger AriPr moieties, reveals a decrease of the 119Sn−117Sncoupling constant with increasing temperature in the range −40to +110 °C. A maximum is observed at −40 °C, and cooling tolower temperatures results again in a decrease of the couplingconstant. Because of the broad main resonance of bis-(stannylene) 2, the coupling constants were not completelyresolved in spectra recorded from toluene-d8 solution.However, the line width of the resonances was smaller inbenzene-d6 solution, and two reliable J(119Sn,117Sn) values weredetermined at 26 and 70 °C. Still, no pronounced temperaturedependence is evident. Quantum chemical calculations suggesta different dynamic behavior of the ArMe substituted derivativeson the one side and the AriPr substituted derivatives on theother side, which might account for the observed temperaturedependence (see below).In order to get more detailed information on the interplay of
structural parameters and 119Sn NMR spectroscopic data, wetried to obtain 119Sn CP/MAS NMR spectra. However, noresonance could be observed probably because of disadvanta-geous cross-polarization behavior and a large anisotropy of thechemical shift tensor. Large chemical shift anisotropy (CSA,defined as Δδ = [δ11 + δ22]/2 − δ33) is known for low-coordinated tin(II) compounds.25,36 An evaluation of theminimum of the spin−lattice relaxation time (T1) (according toWasylishen) allows for estimation of the CSA (for details seethe Supporting Information).38 The distannenes 1, 3, and 4reveal equal values for the CSA within the accuracy of theestimation: 1, Δδ = 2200 ± 200 ppm; 3, Δδ = 2200 ± 200ppm; 4, Δδ = 2300 ± 200 ppm. In contrast, the CSA ofbis(stannylene) 2 is estimated to be significantly higher: Δδ =2900 ± 300 ppm. This difference also reflects the strongermutual tin−tin interaction of the distannenes in contrast to thebis(stannylene). A rationale is given in the SupportingInformation.
Quantum Chemical Calculations. In order to gain deeperinsight into the tin−tin interactions of compounds 1−4, DFTcalculations were performed using the program TURBO-MOLE6.4. All calculations were run on the completemolecules. The molecular geometries were optimized startingfrom X-ray diffraction data by applying the PBE functional,39
additional D3(BJ) dispersion corrections,40,41 the RI42,43 andMARIJ44 options, the def2-TZVP basis set,45,46 and theStuttgart small core pseudopotential.47 Crucial parameters forthe tin−tin interactions are the tin−tin distances and the trans-bending θ values of the two R2Sn fragments toward the tin−tinaxis. The optimized geometries of the distannenes 1, 3, and 4are in good agreement with the experimental structures. Thecalculated tin−tin distances exceed the X-ray structure data by2−4%. However, the C2-symmetric geometry of distannene 3 isslightly distorted into a conformation which can be described asasymmetric trans-bent in the optimized structure. This isindicated by the difference between the trans-bent angles Δθ =17.9° (Table 2). In contrast to the distannenes, the calculatedtin−tin distance of bis(stannylene) 2 is 0.361 Å (8.4%)significantly shorter in comparison to the X-ray structure data.As discussed before, this SnSn bond is substantially weakerthan those in 1, 3, and 4. Thus, packing effects of the large ArR
moieties in the solid state might compensate for a rather weaktin−tin interaction which is yet observable in solution.
Chart 2. Schematic Illustration of the Inversion of the Trans-Bending of Distannenes 1, 3, and 4 and Bis(stannylene) 2
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183907
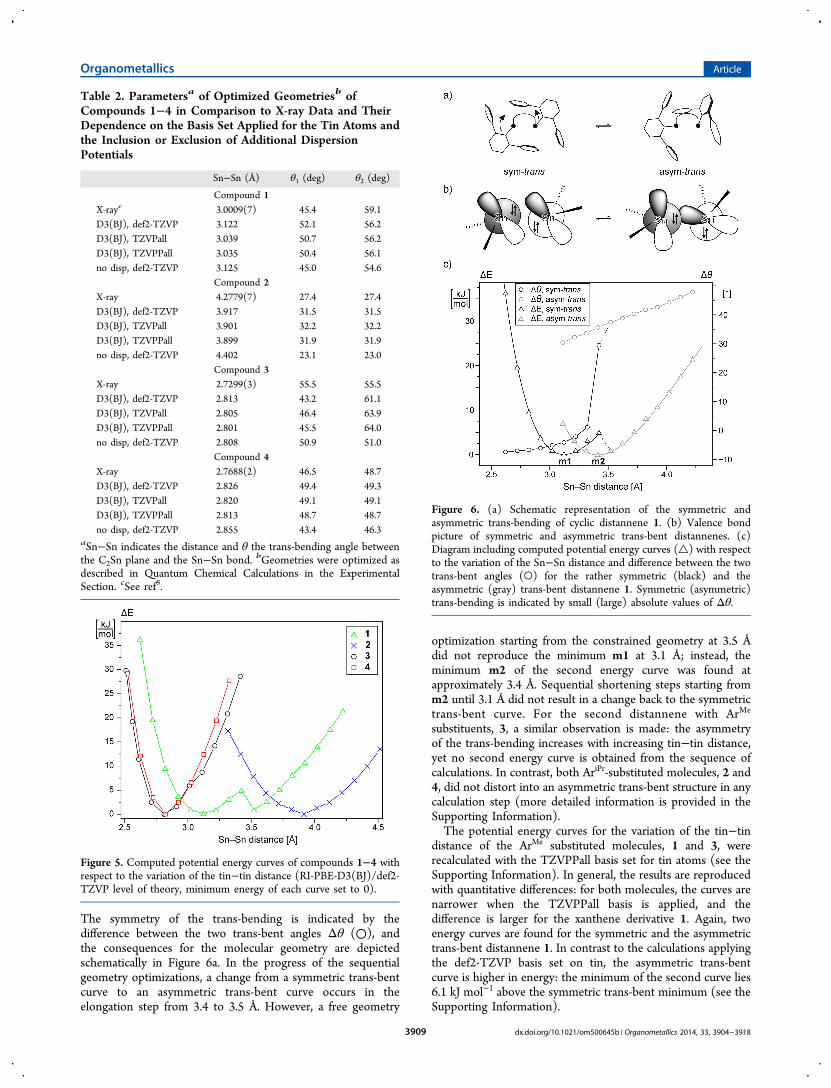
In a series of geometry optimizations, the influence of thebasis set and the effect of the dispersion correction wereinvestigated (Table 2). In all calculations, the set of basisfunctions applied for hydrogen, carbon, and oxygen wasunchanged (def2-TZVP). Using the TZVPall or TZVPPallbasis sets on the tin atoms results generally in a shorter tin−tindistance. This is most significant for distannene 1 (>0.08 Å).The exclusion of additional dispersion potentials results in aflattening of the trans-bending. In case of the two distannenes 1and 3, which carry the smaller ArMe substituents, the dispersionpotentials have no significant effect on the tin−tin distance.The tin−tin distance of distannene 4 with the larger AriPr
substituents is only slightly longer. In contrast, for bis-(stannylene) 2 a substantial increase of the tin−tin distanceby 0.485 Å is observed when the D3(BJ) dispersion correctionsare not included. Thus, intramolecular dispersion effects areresponsible for a substantial contraction, which corroboratesthe weakness of the tin−tin bond in 2.Further calculations have been carried out in order to
investigate the potential energy surface with respect to thevariation of the tin−tin distance. Starting from the geometriesoptimized at the RI-PBE-D3(BJ)/def2-TZVP level of theory,the tin−tin distance was varied in steps of 0.1 Å with all otherdegrees of freedom being optimized. The resulting energyprofiles are depicted in Figure 5. All curves suggest soft tin−tininteractions: the tin−tin distances of both naphthalenederivatives may vary by about 0.5 Å within an energy differenceof only 10 kJ mol−1 and by about 1 Å for the xanthenederivatives. These data reflect the differing rigidities of the twobackbones. The curves of the bulkier substituted (AriPr)compounds 2 (blue ×) and 4 (red □) can be described wellby a Morse-type potential rather than by a harmonic potential(for details see the Supporting Information). The curves ofcompounds 1 and 3 both show a deviation from the Morse-type profile. This deviation is only small for the naphthalenederivative 3 (black ○,) but for the xanthene derivative 1 (green△) a second minimum is observed upon elongation of thetin−tin distance to 3.5 Å.A detailed investigation of the potential energy surface of
distannene 1 revealed that the energy curve depicted in Figure5 results from a superposition of two energy curves withapproximate Morse-type profile (Figure 6c). The two separateenergy curves (△) are attributable to different conformers ofthe distannene, which can be described as rather symmetrictrans-bent (black lines) and asymmetric trans-bent (gray lines).
Table 1. Variable-Temperature 119Sn NMR Spectroscopic Data of Compounds 1−4: δ(119Sn) (in ppm), J(119Sn,117Sn) (in Hz),and T1 (in ms)
1 2 3 4
T (°C) δ J T1 δ J T1 δ J T1 δ J T1
120 924 3480 16.9110 917 3470 15.9 1412 5.83 425 6130 4.48100 409 9950 7.3790 904 3480 13.2 1407 4.56 421 6300 3.6280 406 9840 6.0770 893 3420 10.8 1402 1440b 3.52 418 6450 2.9850 880 3450 8.50 1397a 2.72a 401 9670 4.47 416 6560 2.4726 862 3330 5.90 1390 1530b 1.90 396 9410 3.31 412 6740 2.0510 410 6820 2.015 1381 1.450 843 3180 3.90 392 9080 2.49 409 6860 2.03
−10 1376 1.35 390 8890 2.33−15 407 6980 2.32−20 389 8650 2.30−25 825 2820 2.60−30 1370 1.80 388 8500 2.38 406 7050 3.09−40 816 2510 2.40 406 7100 3.98−50 810 2300 2.72 1364 3.50 386 8080 3.31 406 6960 6.08−60 1362 6.10 406d 6800d 9.43d
−70 799 (1150)c 5.15 1352 384 7650 7.39 406 (2000)c 18.6−80 791 (2500)c 9.60−90 770 (>10000)c
Unless stated otherwise, the spectra were recorded from concentrated solutions in toluene-d8 at B0 = 11.7 T (ν0 = 186.50 MHz). aT = 51 °C.bRecorded from concentrated solution in benzene-d6.
cNo satellite signals observed; Δν1/2 is given in Hz. dT = −58 °C.
Figure 4. Variable-temperature 119Sn NMR data of compounds 1−4.Numerical data are given in Table 1. (a) Variation of the 119Sn NMRchemical shift with temperature referenced to δ at 26 °C: Δδ = δ(T) −δ(26 °C). Lines of best fit are included (slope in ppm/°C, fitting rangein °C): 1, 0.67, −80 to +120; 2, 0.31, −60 to +110; 3, 0.17, −10 to+100; 4, 0.14, −30 to +110. (b) Scalar 119Sn−117Sn coupling constant.
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183908
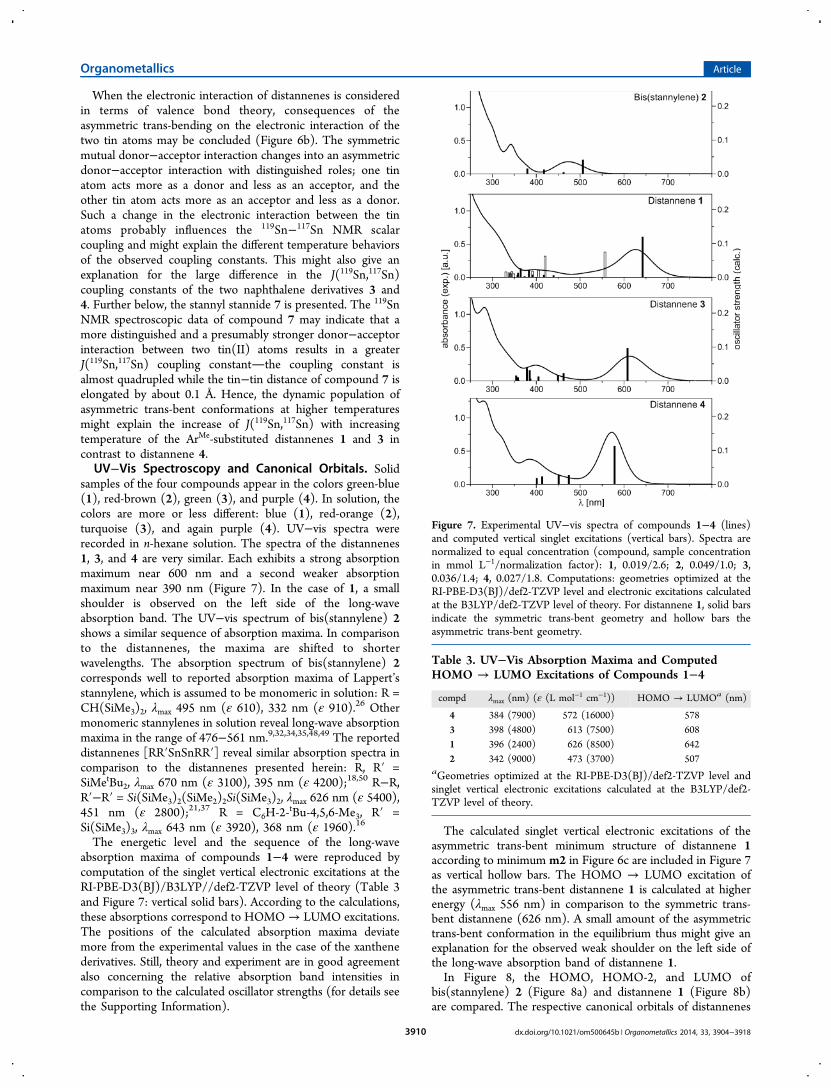
The symmetry of the trans-bending is indicated by thedifference between the two trans-bent angles Δθ (○), andthe consequences for the molecular geometry are depictedschematically in Figure 6a. In the progress of the sequentialgeometry optimizations, a change from a symmetric trans-bentcurve to an asymmetric trans-bent curve occurs in theelongation step from 3.4 to 3.5 Å. However, a free geometry
optimization starting from the constrained geometry at 3.5 Ådid not reproduce the minimum m1 at 3.1 Å; instead, theminimum m2 of the second energy curve was found atapproximately 3.4 Å. Sequential shortening steps starting fromm2 until 3.1 Å did not result in a change back to the symmetrictrans-bent curve. For the second distannene with ArMe
substituents, 3, a similar observation is made: the asymmetryof the trans-bending increases with increasing tin−tin distance,yet no second energy curve is obtained from the sequence ofcalculations. In contrast, both AriPr-substituted molecules, 2 and4, did not distort into an asymmetric trans-bent structure in anycalculation step (more detailed information is provided in theSupporting Information).The potential energy curves for the variation of the tin−tin
distance of the ArMe substituted molecules, 1 and 3, wererecalculated with the TZVPPall basis set for tin atoms (see theSupporting Information). In general, the results are reproducedwith quantitative differences: for both molecules, the curves arenarrower when the TZVPPall basis is applied, and thedifference is larger for the xanthene derivative 1. Again, twoenergy curves are found for the symmetric and the asymmetrictrans-bent distannene 1. In contrast to the calculations applyingthe def2-TZVP basis set on tin, the asymmetric trans-bentcurve is higher in energy: the minimum of the second curve lies6.1 kJ mol−1 above the symmetric trans-bent minimum (see theSupporting Information).
Table 2. Parametersa of Optimized Geometriesb ofCompounds 1−4 in Comparison to X-ray Data and TheirDependence on the Basis Set Applied for the Tin Atoms andthe Inclusion or Exclusion of Additional DispersionPotentials
Sn−Sn (Å) θ1 (deg) θ2 (deg)
Compound 1X-rayc 3.0009(7) 45.4 59.1D3(BJ), def2-TZVP 3.122 52.1 56.2D3(BJ), TZVPall 3.039 50.7 56.2D3(BJ), TZVPPall 3.035 50.4 56.1no disp, def2-TZVP 3.125 45.0 54.6
Compound 2X-ray 4.2779(7) 27.4 27.4D3(BJ), def2-TZVP 3.917 31.5 31.5D3(BJ), TZVPall 3.901 32.2 32.2D3(BJ), TZVPPall 3.899 31.9 31.9no disp, def2-TZVP 4.402 23.1 23.0
Compound 3X-ray 2.7299(3) 55.5 55.5D3(BJ), def2-TZVP 2.813 43.2 61.1D3(BJ), TZVPall 2.805 46.4 63.9D3(BJ), TZVPPall 2.801 45.5 64.0no disp, def2-TZVP 2.808 50.9 51.0
Compound 4X-ray 2.7688(2) 46.5 48.7D3(BJ), def2-TZVP 2.826 49.4 49.3D3(BJ), TZVPall 2.820 49.1 49.1D3(BJ), TZVPPall 2.813 48.7 48.7no disp, def2-TZVP 2.855 43.4 46.3
aSn−Sn indicates the distance and θ the trans-bending angle betweenthe C2Sn plane and the Sn−Sn bond. bGeometries were optimized asdescribed in Quantum Chemical Calculations in the ExperimentalSection. cSee ref8.
Figure 5. Computed potential energy curves of compounds 1−4 withrespect to the variation of the tin−tin distance (RI-PBE-D3(BJ)/def2-TZVP level of theory, minimum energy of each curve set to 0).
Figure 6. (a) Schematic representation of the symmetric andasymmetric trans-bending of cyclic distannene 1. (b) Valence bondpicture of symmetric and asymmetric trans-bent distannenes. (c)Diagram including computed potential energy curves (△) with respectto the variation of the Sn−Sn distance and difference between the twotrans-bent angles (○) for the rather symmetric (black) and theasymmetric (gray) trans-bent distannene 1. Symmetric (asymmetric)trans-bending is indicated by small (large) absolute values of Δθ.
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183909
When the electronic interaction of distannenes is consideredin terms of valence bond theory, consequences of theasymmetric trans-bending on the electronic interaction of thetwo tin atoms may be concluded (Figure 6b). The symmetricmutual donor−acceptor interaction changes into an asymmetricdonor−acceptor interaction with distinguished roles; one tinatom acts more as a donor and less as an acceptor, and theother tin atom acts more as an acceptor and less as a donor.Such a change in the electronic interaction between the tinatoms probably influences the 119Sn−117Sn NMR scalarcoupling and might explain the different temperature behaviorsof the observed coupling constants. This might also give anexplanation for the large difference in the J(119Sn,117Sn)coupling constants of the two naphthalene derivatives 3 and4. Further below, the stannyl stannide 7 is presented. The 119SnNMR spectroscopic data of compound 7 may indicate that amore distinguished and a presumably stronger donor−acceptorinteraction between two tin(II) atoms results in a greaterJ(119Sn,117Sn) coupling constantthe coupling constant isalmost quadrupled while the tin−tin distance of compound 7 iselongated by about 0.1 Å. Hence, the dynamic population ofasymmetric trans-bent conformations at higher temperaturesmight explain the increase of J(119Sn,117Sn) with increasingtemperature of the ArMe-substituted distannenes 1 and 3 incontrast to distannene 4.UV−Vis Spectroscopy and Canonical Orbitals. Solid
samples of the four compounds appear in the colors green-blue(1), red-brown (2), green (3), and purple (4). In solution, thecolors are more or less different: blue (1), red-orange (2),turquoise (3), and again purple (4). UV−vis spectra wererecorded in n-hexane solution. The spectra of the distannenes1, 3, and 4 are very similar. Each exhibits a strong absorptionmaximum near 600 nm and a second weaker absorptionmaximum near 390 nm (Figure 7). In the case of 1, a smallshoulder is observed on the left side of the long-waveabsorption band. The UV−vis spectrum of bis(stannylene) 2shows a similar sequence of absorption maxima. In comparisonto the distannenes, the maxima are shifted to shorterwavelengths. The absorption spectrum of bis(stannylene) 2corresponds well to reported absorption maxima of Lappert’sstannylene, which is assumed to be monomeric in solution: R =CH(SiMe3)2, λmax 495 nm (ε 610), 332 nm (ε 910).26 Othermonomeric stannylenes in solution reveal long-wave absorptionmaxima in the range of 476−561 nm.9,32,34,35,48,49 The reporteddistannenes [RR′SnSnRR′] reveal similar absorption spectra incomparison to the distannenes presented herein: R, R′ =SiMetBu2, λmax 670 nm (ε 3100), 395 nm (ε 4200);18,50 R−R,R′−R′ = Si(SiMe3)2(SiMe2)2Si(SiMe3)2, λmax 626 nm (ε 5400),451 nm (ε 2800);21,37 R = C6H-2-
tBu-4,5,6-Me3, R′ =Si(SiMe3)3, λmax 643 nm (ε 3920), 368 nm (ε 1960).16
The energetic level and the sequence of the long-waveabsorption maxima of compounds 1−4 were reproduced bycomputation of the singlet vertical electronic excitations at theRI-PBE-D3(BJ)/B3LYP//def2-TZVP level of theory (Table 3and Figure 7: vertical solid bars). According to the calculations,these absorptions correspond to HOMO→ LUMO excitations.The positions of the calculated absorption maxima deviatemore from the experimental values in the case of the xanthenederivatives. Still, theory and experiment are in good agreementalso concerning the relative absorption band intensities incomparison to the calculated oscillator strengths (for details seethe Supporting Information).
The calculated singlet vertical electronic excitations of theasymmetric trans-bent minimum structure of distannene 1according to minimum m2 in Figure 6c are included in Figure 7as vertical hollow bars. The HOMO → LUMO excitation ofthe asymmetric trans-bent distannene 1 is calculated at higherenergy (λmax 556 nm) in comparison to the symmetric trans-bent distannene (626 nm). A small amount of the asymmetrictrans-bent conformation in the equilibrium thus might give anexplanation for the observed weak shoulder on the left side ofthe long-wave absorption band of distannene 1.In Figure 8, the HOMO, HOMO-2, and LUMO of
bis(stannylene) 2 (Figure 8a) and distannene 1 (Figure 8b)are compared. The respective canonical orbitals of distannenes
Figure 7. Experimental UV−vis spectra of compounds 1−4 (lines)and computed vertical singlet excitations (vertical bars). Spectra arenormalized to equal concentration (compound, sample concentrationin mmol L−1/normalization factor): 1, 0.019/2.6; 2, 0.049/1.0; 3,0.036/1.4; 4, 0.027/1.8. Computations: geometries optimized at theRI-PBE-D3(BJ)/def2-TZVP level and electronic excitations calculatedat the B3LYP/def2-TZVP level of theory. For distannene 1, solid barsindicate the symmetric trans-bent geometry and hollow bars theasymmetric trans-bent geometry.
Table 3. UV−Vis Absorption Maxima and ComputedHOMO → LUMO Excitations of Compounds 1−4
compd λmax (nm) (ε (L mol−1 cm−1)) HOMO → LUMOa (nm)
4 384 (7900) 572 (16000) 5783 398 (4800) 613 (7500) 6081 396 (2400) 626 (8500) 6422 342 (9000) 473 (3700) 507
aGeometries optimized at the RI-PBE-D3(BJ)/def2-TZVP level andsinglet vertical electronic excitations calculated at the B3LYP/def2-TZVP level of theory.
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183910

3 and 4 largely correspond to those of the distannene 2 and areincluded in the Supporting Information. We have pointed outthat the canonical orbitals of distannene 2 can be derived fromthe electronic interaction of a twisted trans-bent distannene.8
HOMO-2 comprises to some extent a Sn−Sn σ-bondinginteraction, and the HOMO may be interpreted as a rathernonbonding orbital with lone-pair character being distributedequally to both tin atoms. In contrast, the correspondingorbitals of bis(stannylene) 2 rather resemble the combinationsof isolated stannylene fragment orbitals. The HOMO-2 may beseen as an in-phase combination and the HOMO as out-of-phase combination of the stannylene lone pair orbitals.Oxidation with Air. Intensely colored solid samples or
solutions of compounds 1−4 discolor immediately uponexposure to air. Oxidation products 5 and 6 of distannene 1and bis(stannylene) 2, respectively, were isolated andcharacterized by NMR spectroscopy, X-ray diffraction, andelemental analysis (Scheme 2).For both compounds, single-crystalline material suitable for
X-ray diffraction was obtained by very slow diffusion of air intoinert vessels equipped with solutions of the respective reactantin n-hexane. Both oxidized compounds reveal rare structuralmotifs comprising terminal hydroxyl substituents (Figure 9).Compound 5 can be categorized as a dihydroxytetraorganyldi-stannoxane, merely two examples of which have been reportedto date: the first carries bulky R = CH(SiMe3)2 substituents
51
and the second R = C6H3-2,6-(CH2O-t-Bu)2.52 In contrast,
compound 6, carrying the bulkier AriPr substituents, comprisestwo dihydroxydiorganotin(IV) moieties which are doublyhydrogen bridged. Examples of dihydroxydiorganotin(IV)compounds carry R = ArMe substituents or R = C6H2-2,6-CH(SiMe3)2-4-C(SiMe3)3 and R′ = C6H3-2,6-(C6H32,4-i-Pr2)2.
53,54 Terminal hydroxyl groups are rather uncommon in
stannoxane chemistry. Usually, condensation to oligomers orpolymers is observed.55,56 For all cited examples except for R =CH(SiMe3)2, intramolecular stabilization of the terminalhydroxyl functions is considered via classical hydrogen bridgebonds to oxygen or nonclassical hydrogen−π-aryl bridges. Thelatter is also observed for the compounds 5 and 6 presentedherein.Results of the crystallographic investigation on compounds 5
and 6 are depicted in Figure 9. Probably because of theoxidation, the Sn−C bonds of compound 5 are contracted by2−4% in comparison to those of distannene 1. The Sn−C andSn−O bond lengths correspond well to reported values of thedihydroxydistannoxanes cited above. Because of the insertion ofthe μ-oxo bridge, the direct Sn−Sn distance is elongated andthus allows for planarization of the xanthene backbone. Still, thebackbone causes strain, as indicated by the Sn1−O3−Sn2 angleof 141.88(5)°, which is significantly wider than those forcomparable dihydroxydistannoxanes: 125.0(5)° (R = CH-(SiMe3)2) and 134.4(1)° (R = C6H3-2,6-(CH2O-t-Bu)2).However, the geometrical parameters of compound 6 are notdiscussed in detail because the quality of the data is too low(details are given in the Supporting Information). Nevertheless,the configuration of the non-hydrogen atoms was unambigu-ously determined and the position of the hydrogen atoms mostprobably corresponds to the depicted conformation.By means of 119Sn{1H} NMR, one singlet resonance at δ
−155 ppm with satellite signals corresponding to2J(119Sn,117Sn) = 709 Hz is observed for distannoxane 5, andone singlet resonance at δ −115 ppm (benzene-d6) or −112ppm (CD2Cl2) is observed for compound 6, but without anysatellite signals. These data are consistent with two symmetri-cally equivalent tin atoms for each compound. The chemicalshift values are in the usual range for four-coordinated tin. Inagreement with the 119Sn{1H} NMR data, 1H NMR ofdistannoxane 5 reveals one singlet for the SnOH protons at δ0.32 ppm with satellite signals consistent with 2J(119Sn,1H) =37.8 Hz and 2J(117Sn,1H) = 36.2 Hz. For the organic moieties,additional symmetric equivalencies are indicated on the 1HNMR time scale: three instead of six singlet resonances areobserved for the mesityl methyl protons and two instead of fourfor the methyl aryl protons. Broadening of these resonances ofup to Δν1/2 = 16 Hz indicates dynamic processes to beresponsible. Hence, the OH···π-aryl interactions can be
Figure 8. Selected canonical orbitals including orbital energies of (a)bis(stannylene) 2 and (b) distannene 1. Geometries optimized at theRI-PBE-D3(BJ)/def2-TZVP level and electronic structures calculatedat the B3LYP/def2-TZVP level of theory. Isovalue = 0.05.
Scheme 2. Reaction of Distannene 1 and Bis(stannylene) 2with Ambient Atmospherea
aAbbreviations: Mes = C6H2-2,4,6-Me3, Trip = C6H2-2,4,6-i-Pr3.
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183911

concluded to be too weak to preserve the solid-stateconformation in solution. In addition, the 1H NMR ofcompound 6 indicates higher symmetry equivalence of theorganic moieties in solution. Furthermore, only one singletresonance is observed for all four SnOH protons at higherchemical shift in comparison to the distannoxane 5 (δ 2.77 ppmin C6D6, δ 2.17 ppm in CD2Cl2) but with a rather similarcoupling constant (2J(119Sn,1H) = 34 Hz).Stannyl Stannide 7. Distannene 1 forms a chloride adduct,
that is, a stannyl stannide, when it is stirred with 1 equiv ofpotassium chloride in the presence of cryptating agent 222
(crypt-222) in tetrahydrofuran solution (Scheme 3). Thecryptating agent acts as a chelating agent for the potassiumcation. Within a few hours, the deep blue green solutionchanges color to deep red.
Large deep red single crystals can be obtained by slow gas-phase diffusion of n-pentane into solutions of compound 7 inbenzene. The crystal structure (7·0.5C6H6) contains half amolecule of benzene per formula unit. Single crystals of asecond polymorph (7), which contains no cocrystallizedsolvent, were obtained from a crude reaction mixture ofdistannene 1 with potassium metal in the presence of crypt-222in tetrahydrofuran by slow gas-phase diffusion. The origin ofchloride in the latter is unclear, however. The molecularstructure of the anionic part of compound 7 is depicted inFigure 10. Both polymorphs crystallize in space group P21/c
Figure 9. (a) ORTEP plot of the molecular structure of distannoxane5. The solvent molecule, aromatic methyl groups, and hydrogen atomsexcept for OH are omitted; ellipsoids are given at the 50% probabilitylevel. Interatomic distances (Å) and angles (deg): Sn1−O1 =1.9751(9), Sn2−O2 = 1.9692(10), Sn1−O3 = 1.9573(9), Sn2−O3= 1.9596(9), Sn1−C1 = 2.1411(13), Sn1−C17 = 2.1486(13), Sn2−C11 = 2.1436(13), Sn2−C41 = 2.1530(13), Sn1−Sn2 = 3.7022(5);Sn1−O3−Sn2 = 141.88(5), O1−Sn1−O3 = 104.57(4), O2−Sn2−O3= 102.01(4), C1−Sn1−C17 = 111.61(5), C11−Sn2−C41 =111.73(5).(b) ORTEP plot of the molecular structure of bis-(dihydroxystannane) 6. Isopropyl groups and hydrogen atoms exceptfor OH are omitted; ellipsoids are given at the 50% probability level.The quality of the crystal structure analysis was insufficient for adetailed discussion; hence, the structural parameters are given with lowaccuracy and without standard deviations. Interatomic distances (Å)and angles (deg): Sn1−O1 = 1.93, Sn1−O2 = 1.98, Sn1−C1 = 2.13,Sn1−C10 = 2.15, O1−O2* = 2.71, Sn1−Sn1* = 4.63; O1−Sn1−O2 =102.3, C1−Sn1−C10 = 110.5.
Scheme 3. Reaction of Distannene 1 with PotassiumChloride in the Presence of crypt-222a
aAbbreviation: ArMe = C6H3-2,6-[C6H2-2,4,6-Me3]2.
Figure 10. ORTEP plot of the molecular anion of compound 7 of thesolvent-free polymorph. The complex cation [crypt-222]K+ and allhydrogens are omitted; ellipsoids are given at the 50% probabilitylevel. Selected interatomic distances (Å) and angles (deg),corresponding values of the polymorph 7·0.5C6H6 given in braces:Sn1−Sn2 = 3.1191(4) {3.0916(3)}, Sn1−Cl1 = 2.4802(6){2.4699(5)}, Sn1−C1 = 2.203(2) {2.1906(18)}, Sn1−C17 =2.247(2) {2.232(3)}, Sn2−C11 = 2.255(2) {2.2428(18)}, Sn2−C41= 2.259(2) {2.2628(18)}; Sn2−Sn1−Cl1 = 138.892(16){138.343(13)}, C1−Sn1−C17 = 109.31(8) {108.24(8)}, C11−Sn2−C41 = 87.18(8) {91.04(6)}, C17−Sn1−Sn2 = 112.52(5){109.55(11)}, C41−Sn2−Sn1 = 117.75(6) {117.51(5)}, C1−Sn1−Sn2−C11 = 5.15(8) {6.66(7)}.
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183912
with one formula unit in the asymmetric unit. The molecularstructures of the stannyl stannide are almost equal, and thegeometric parameters deviate only slightly (see the caption ofFigure 10). In comparison to distannene 1, the tin−tin distanceelongates by about 0.1 Å upon addition of the chloride. Thestannyl stannide may be formally described as a chlorodiorganylstannide at Sn1 acting as a Lewis base toward a diorganylstannylene at Sn2. This may correspond to the smaller C11−Sn2−C41 angle (1, 93.7°; 7, 87.2°; 7·0.5C6H6, 91.0°). Theformal localization of the negative charge on Sn2 might beconsistent with the elongation of the Sn2−carbon bond lengthsby about 0.03−0.06 Å. However, the orientation of thestannylene moiety at Sn2 with respect to the Sn1−Sn2 axis isalmost unchanged: the bend angle θ(Sn2) is only marginallysteeper (1, 59°; 7, 61°; 7·0.5C6H6, 60°). This may be explainedby the steric requirement of the large terphenyl substituents.The coordination of the chloride to Sn1 causes the C1−Sn1−C17 angle to widen from 100.5° (1) to an approximatelytetrahedral angle. In addition, the terphenyl moiety at Sn1twists about the Sn1−C17 axis, which is indicated by the C1−Sn1−C17−C22 torsion angle: 1, 95.6°; 7, 16.1°; 7·0.5C6H6,25.3°. For comparison, potassium chloride cocrystallized withthe intramolecularly coordinated distannyne [{2,6-(Me2NCH2)2C6H3}Sn]2 and formed infinite ···K···Cl···Sn−Sn···K···Cl··· chains with significantly larger Sn···Cl separa-tions.57
In 1H and 13C{1H} NMR spectra, the resonances of thestannyl stannide are broad, which is probably due to dynamicprocesses at room temperature. The mesityl methyl protonsgive rise to only two broad singlet resonances, and for themesityl aryl protons only one broad singlet is observed. Thus,no lowering of symmetry equivalency is observed on the NMRtime scale because of the coordination of the chloride. Incontrast, the nonequivalence of two tin atoms is detected by119Sn NMR spectroscopy: two singlets are observed at δ 325and −138 ppm carrying satellite signals consistent withJ(119Sn,119Sn) = 13100 Hz (AB spin system) andJ(119Sn,117Sn) = 12400 Hz (AX spin system). The strongresonance shift of both tin species to lower frequencies (cf. 1: δ862 ppm) is consistent with stronger Lewis base interactions onthe Sn 5p orbital. This is evident for Sn1 because of thecoordination of the chloride ligand, which is tentativelyassigned to the resonance at −138 ppm. Moreover, the formalchlorodiorganyl stannide fragment may be considered astronger Lewis base toward Sn2 in comparison to the mutualdonor−acceptor interaction of distannene 1. The scalarcoupling constant between 119Sn and 117Sn approximatelyquadruples as result of the coordination of the chloride (cf. 1:J(119Sn,117Sn) = 3330 Hz).UV−vis spectra of compound 7 were recorded at three
different concentrations in benzene solution using single-crystalline material of the polymorph 7·0.5C6H6 (Figure 11).Each spectrum shows two absorption maxima in the wave-length range of visible light at about λ 620 and 460 nm. Theabsorbances of both maxima are about equal at the lowestconcentration, and at higher concentrations the absorbance atλmax 460 nm is more than twice the absorbance at λmax 620 nm.This observation is in disagreement with the Beer−Lambert lawand might hence indicate too high a concentration of thesample and thus a loss of linearity due to molecularinteractions. Still, the disagreement might also be attributedto concentration-dependent dynamic processes: The long-waveabsorption band corresponds well to the absorption maximum
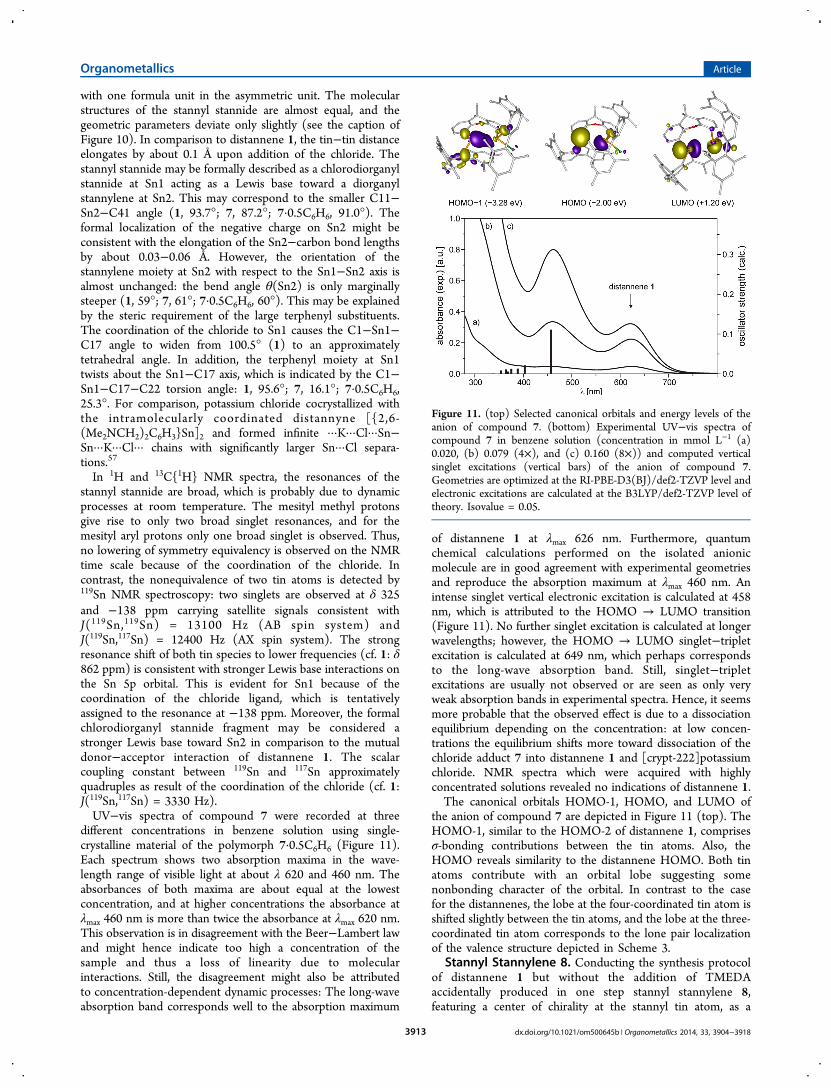
of distannene 1 at λmax 626 nm. Furthermore, quantumchemical calculations performed on the isolated anionicmolecule are in good agreement with experimental geometriesand reproduce the absorption maximum at λmax 460 nm. Anintense singlet vertical electronic excitation is calculated at 458nm, which is attributed to the HOMO → LUMO transition(Figure 11). No further singlet excitation is calculated at longerwavelengths; however, the HOMO → LUMO singlet−tripletexcitation is calculated at 649 nm, which perhaps correspondsto the long-wave absorption band. Still, singlet−tripletexcitations are usually not observed or are seen as only veryweak absorption bands in experimental spectra. Hence, it seemsmore probable that the observed effect is due to a dissociationequilibrium depending on the concentration: at low concen-trations the equilibrium shifts more toward dissociation of thechloride adduct 7 into distannene 1 and [crypt-222]potassiumchloride. NMR spectra which were acquired with highlyconcentrated solutions revealed no indications of distannene 1.The canonical orbitals HOMO-1, HOMO, and LUMO of
the anion of compound 7 are depicted in Figure 11 (top). TheHOMO-1, similar to the HOMO-2 of distannene 1, comprisesσ-bonding contributions between the tin atoms. Also, theHOMO reveals similarity to the distannene HOMO. Both tinatoms contribute with an orbital lobe suggesting somenonbonding character of the orbital. In contrast to the casefor the distannenes, the lobe at the four-coordinated tin atom isshifted slightly between the tin atoms, and the lobe at the three-coordinated tin atom corresponds to the lone pair localizationof the valence structure depicted in Scheme 3.
Stannyl Stannylene 8. Conducting the synthesis protocolof distannene 1 but without the addition of TMEDAaccidentally produced in one step stannyl stannylene 8,featuring a center of chirality at the stannyl tin atom, as a
Figure 11. (top) Selected canonical orbitals and energy levels of theanion of compound 7. (bottom) Experimental UV−vis spectra ofcompound 7 in benzene solution (concentration in mmol L−1 (a)0.020, (b) 0.079 (4×), and (c) 0.160 (8×)) and computed verticalsinglet excitations (vertical bars) of the anion of compound 7.Geometries are optimized at the RI-PBE-D3(BJ)/def2-TZVP level andelectronic excitations are calculated at the B3LYP/def2-TZVP level oftheory. Isovalue = 0.05.
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183913
racemic mixture. Probably because of initially mere mono-lithiation of 9,9-dimethylxanthene, purple stannyl stannylene 8is gained instead of blue-green distannene 1 and can be isolatedin 32% yield (Scheme 4).
Single-crystal-quality material of compound 8 was obtainedby slow evaporation of the solvent from concentrated solutionsin n-hexane. Molecules of 8 comprise a chiral center at the four-coordinated tin atom and crystallized in the noncentrosym-metric space group Cc. However, the investigated crystal wasdetermined to be a racemic twin. The molecular structure isdepicted in Figure 12. Consistent with the formal oxidationstate +I, the substituents at the two-coordinated tin atom Sn2enclose an angle of 97.37(7)°. The coordination geometryaround Sn1 corresponds to a tetrahedron which is slightlystretched in the direction of the Sn1−Sn2 axis. The tin−tindistance of 2.9020(8) Å is between the values of the distannene
1 on the one side and the distannenes 3 and 4 on the otherside. It is in the range of comparable stannyl stannylenes(2.865(1)−2.9688(5) Å).35,58−63 A contact between Sn2 andone mesityl ring is observed and is indicated as a dashed line inFigure 12. The Sn2−C distances, 3.533(3)−3.707(3) Å, areshorter than the sum of the van der Waals radii (3.87 Å).11
By means of 119Sn NMR spectroscopy, two resonances at δ109 and 1973 ppm are observed. The resonance at lowerchemical shift may be attributed to the four-coordinated tinatom, and satellite signals consistent with J(119Sn,117/119Sn) =9400 Hz are observed. The second signal is attributable to thetwo-coordinated tin atom and exhibits a considerable line widthof approximately Δν1/2 = 4000 Hz; hence, the expected satellitesignals are not observed. The scalar coupling constant iscomparable to those of other stannyl stannylenes (6240−9011Hz)59−62 and is very similar to the value for distannene 3. Incontrast to the distannenes 1, 3, and 4, stannyl stannylene 8exhibits no significant temperature-dependent variation of thecoupling constant within the investigated range of −50 to +70°C (see the Supporting Information). The chemical shift valueof the 2-fold-coordinated tin is between values of other stannylstannylenes, for example, δ(119Sn) 2857 ppm ((AriPr)-Me2Sn
I I ISnIAr iP r)59 and 1302 ppm ({( iPrO)2-2,6-C6H3}3Sn
IIISn0SnIII{C6H3-2,6-(iPrO)2}3).
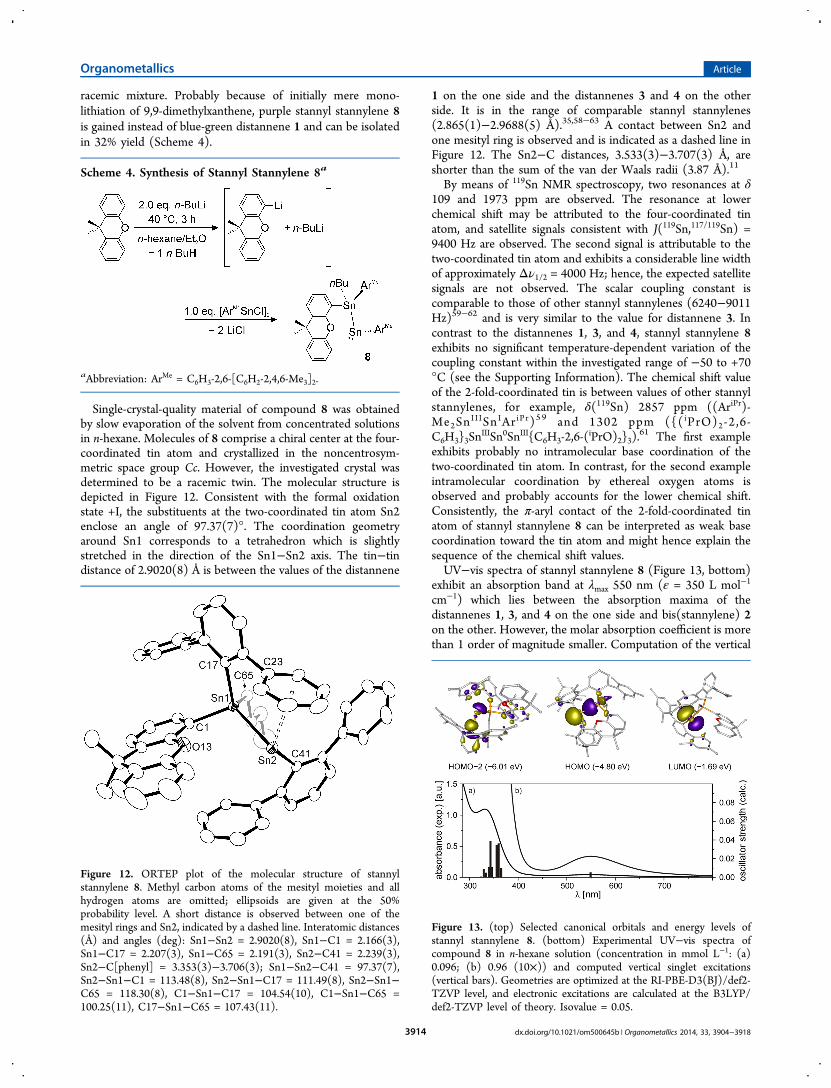
61 The first exampleexhibits probably no intramolecular base coordination of thetwo-coordinated tin atom. In contrast, for the second exampleintramolecular coordination by ethereal oxygen atoms isobserved and probably accounts for the lower chemical shift.Consistently, the π-aryl contact of the 2-fold-coordinated tinatom of stannyl stannylene 8 can be interpreted as weak basecoordination toward the tin atom and might hence explain thesequence of the chemical shift values.UV−vis spectra of stannyl stannylene 8 (Figure 13, bottom)
exhibit an absorption band at λmax 550 nm (ε = 350 L mol−1
cm−1) which lies between the absorption maxima of thedistannenes 1, 3, and 4 on the one side and bis(stannylene) 2on the other. However, the molar absorption coefficient is morethan 1 order of magnitude smaller. Computation of the vertical
Scheme 4. Synthesis of Stannyl Stannylene 8a
aAbbreviation: ArMe = C6H3-2,6-[C6H2-2,4,6-Me3]2.
Figure 12. ORTEP plot of the molecular structure of stannylstannylene 8. Methyl carbon atoms of the mesityl moieties and allhydrogen atoms are omitted; ellipsoids are given at the 50%probability level. A short distance is observed between one of themesityl rings and Sn2, indicated by a dashed line. Interatomic distances(Å) and angles (deg): Sn1−Sn2 = 2.9020(8), Sn1−C1 = 2.166(3),Sn1−C17 = 2.207(3), Sn1−C65 = 2.191(3), Sn2−C41 = 2.239(3),Sn2−C[phenyl] = 3.353(3)−3.706(3); Sn1−Sn2−C41 = 97.37(7),Sn2−Sn1−C1 = 113.48(8), Sn2−Sn1−C17 = 111.49(8), Sn2−Sn1−C65 = 118.30(8), C1−Sn1−C17 = 104.54(10), C1−Sn1−C65 =100.25(11), C17−Sn1−C65 = 107.43(11).
Figure 13. (top) Selected canonical orbitals and energy levels ofstannyl stannylene 8. (bottom) Experimental UV−vis spectra ofcompound 8 in n-hexane solution (concentration in mmol L−1: (a)0.096; (b) 0.96 (10×)) and computed vertical singlet excitations(vertical bars). Geometries are optimized at the RI-PBE-D3(BJ)/def2-TZVP level, and electronic excitations are calculated at the B3LYP/def2-TZVP level of theory. Isovalue = 0.05.
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183914

singlet electronic excitations reproduces the weak absorption atλ 549 nm and attributes it to the HOMO → LUMO transition.Selected canonical orbitals are depicted in Figure 13 (top).Additionally, the Supporting Information contains a compara-tive depiction of canonical orbitals of distannene 1, stannylstannide 7, and stannyl stannylene 8. The HOMO of stannylstannylene 8 reveals contributions from both the lone pair onthe two-coordinated tin atom and the tin−tin σ-bondinginteraction. In contrast to the case for compounds 1 and 7, nofurther orbital is found, which is predominantly attributable tothese contributions. In the sequence 1, 7, and 8, the LUMO isdelocalized over the two tin atoms in 1 and localized on thetwo-coordinated tin atom in 8. The LUMO of anion 7 liessomehow between with reduced contributions from the four-coordinated tin atom.
■ SUMMARY
Bis(stannylene) 2 and distannenes 3 and 4 were structurallycharacterized in the solid state. We established the catego-rization of compounds 1−4 into a spacer-bridged bis-(stannylene) and distannenes also in solution by means of119Sn NMR chemical shifts, 119Sn spin−lattice relaxation times,UV−vis spectroscopy, and quantum chemical calculations.However, a weaker electronic tin−tin interaction was alsoproven for bis(stannylene) 2. The formal tin−tin double bondcan be characterized as a rather soft and fluxional interactioneven for distannenes 3 and 4, which comprise the very rigidnaphthalene backbone. NMR spectroscopy at variable temper-atures indicated dynamic inversion of the trans-bending, whichhas higher activation barriers for the distannenes than for thebis(stannylene). Furthermore, the 119Sn−117Sn scalar couplingat variable temperatures, UV−vis spectroscopy of distannene 1,and quantum chemical calculations are conclusive in that thesmaller ArMe substituents allow for an asymmetric trans-bentgeometry in dynamic processes in solution.Oxidation products of distannene 1 and bis(stannylene) 2
reveal rare structural motifs, dihydroxydistannoxane andbis[dihydroxtin(IV)], respectively, with terminal Sn−OHfunctions. The two structural motifs reflect the steric demandof the terphenyl substituents.Stannyl stannide 7 and stannyl stannylene 8 represent
additional variations of the tin−tin interaction. Stannyl stannide7 exhibits the longest tin−tin distance within this work;however, the scalar 119Sn−117Sn coupling constant is signifi-cantly larger than those in the other compounds. The formallydoubly bonded distannenes 1, 3, and 4 exhibit strongtemperature dependence of the scalar 119Sn−117Sn couplingconstant. In contrast, the singly bonded stannyl stannylene 8reveals no significant variation of the coupling constant withtemperature.The presented compounds 1−4 represent a readily accessible
model system for alkene homologues of tin, which allows forsystematic investigation of the tin−tin interaction. Thevariation of the organic moieties allows for influencing thegeometric parameters, the dynamic behavior of the molecules,and thus the electronic structure and properties. In contrast tomost known distannenes, which usually react in terms of themonomeric stannylenes after dissociation of the tin−tin bond,these compounds have high potential for evaluating thechemical properties of the formal tin−tin double bond.
■ EXPERIMENTAL SECTIONGeneral Considerations. Unless otherwise stated, inert con-
ditions (vacuum or argon atmosphere) were maintained throughout alloperations. All solvents were dried and degassed by standardtechniques. Distannene 1,8 [AriPrSnCl],10 and [ArMeSnCl]2
9 wereprepared according to literature procedures. Further chemicals werepurchased and used as received. Elemental analyses were performed atthe Institute of Inorganic Chemistry, University of Tubingen, using aVario MICRO EL analyzer. UV−vis measurements were performed ona PerkinElmer Lambda 35 instrument using cuvettes from Hellma.
NMR Spectroscopy. NMR spectra were recorded on a BrukerDRX-250 spectrometer (1H, 250.13 MHz; 119Sn, 93.28 MHz)equipped with a 5 mm ATM probe head, a Bruker AvanceII+400spectrometer (1H, 400.13 MHz; 13C, 100.61 MHz) equipped with a 5mm QNP (quad nucleus probe) head, and a Bruker AvanceII+500spectrometer (1H, 500.13 MHz; 13C, 125.76 MHz; 119Sn, 186.50MHz) equipped with a 5 mm ATM probe head. The chemical shiftsare reported in δ values in ppm relative to external SiMe4 (
1H, 13C) orSnMe4 (
119Sn) using the chemical shift of the solvent 2H resonancefrequency and Ξ = 25.145020% for 13C and 37.290632% for 119Sn.64
Coupling constants J are given in Hz as absolute values. Themultiplicities of the signals are indicated as s = singlet, d = doublet, t =triplet, q = quartet, sept = septet, and m = multiplet or unresolved.The proton and carbon signals were assigned via 1H, 13C{1H}, 1H−1HCOSY, 1H−13C HSQC, and 1H−13C HMBC spectra. For variable-temperature measurements the sample temperature was stabilized witha Bruker BVT 3200 temperature controller and equilibrated for 10 minprior to acquisition. The temperatures given are uncorrected.
Crystallography. X-ray diffraction data were collected with aBruker APEX-II CCD diffractometer equipped with a sealed-tubesource with molybdenum anode and graphite monochromator (5, 6, 7·0.5C6H6), a Bruker APEX-II CCD Duo diffractometer equipped withan Mo IμS microfocus tube and TRIUMPH monochromator (3, 4, 7),or a Stoe IPDS 2T diffractometer equipped with a sealed-tube sourcewith molybdenum anode and graphite monochromator (2, 8). Fordata reduction and absorption correction Bruker’s APEX2, includingthe programs SAINT, SADABS, and XPREP, or Stoe’s X-Area, withthe programs X-Red and X-Shape, was used. For structure solution andrefinement SHELXS-97 and Shelxle GUI for SHELXL-97 have beenused.65−73 Details on the production of the crystals and the structurerefinements are given in the Supporting Information.
Quantum Chemical Calculations. The quantum chemicalcalculations were performed using the computer cluster of theTheoretical Chemistry group of the University of Tubingen, theinfrastructure of the bwGRiD project,74 and the TURBOMOLE V6.4program package.75−77 The input files were generated, and the resultswere evaluated with the program TmoleXClient Version 3.3 or3.4.78,79 All calculations were performed with C1 molecular symmetryon the complete molecules. All molecules were treated as closed-shellmolecules. Geometries were optimized using the jobex script at the RI-PBE-D3(BJ)39−43 level of theory using the basis set def2-TZVP45,46 onH, C, O, and Cl in all calculations and the corresponding auxiliarybasis sets.42,43 For Sn the basis set def2-TZVP and the Stuttgart smallcore pseudopotential,47 TZVPall, or TZVPPall80 were applied.Electronic vertical excitations were computed using the ecsfmodule,81,82 the B3LYP functional,83−86 the def2-TZVP basis set,and the Stuttgart small core pseudopotential.47
Synthesis of Bis(stannylene) 2. A 0.15 g amount (0.71 mmol) of9,9-dimethylxanthene and 0.27 mL (0.21 g, 1.8 mmol) of TMEDA in amixture of 3.0 mL of diethyl ether and 4.7 mL of n-hexane weretreated with 0.89 mL (1.4 mmol) of a 1.6 M n-butyllithium solution inn-hexane. Subsequent heating of the mixture to 40 °C for 3 h yieldedan orange solution. After it was cooled to −40 °C, the solution wasadded to a solution of 0.91 g (1.4 mmol) of [AriPrSnCl] in a mixture of20 mL of diethyl ether and 28 mL of n-hexane. Stirring overnightyields an intense red-brown solution. The mixture was filtered, and thesolvent was removed in vacuo. The residue was treated with 40 mL ofn-hexane, and filtration yielded product 2 as a light brown powder.The filtrate was reduced in volume to 20 mL and stored for several
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183915

days at ambient temperature for crystallization. Additional product 2was obtained by filtration as dark-brow crystals. Combined yield: 394mg (39%). 1H NMR (400.11 MHz, C6D6, 26 °C, δ): 0.79 [d, 12H,3JHH = 6.7 Hz, −CH(CH3)2], 0.97 [d, 12H, 3JHH = 6.9 Hz,−CH(CH3)2], 1.05 [d, 12H, 3JHH = 6.9 Hz, −CH(CH3)2], 1.13 [d,12H, 3JHH = 6.8 Hz, −CH(CH3)2], 1.19 [d, 12H, 3JHH = 6.7 Hz,−CH(CH3)2], 1.45 (s, 6H, Xant-CH3), 1.74 [d, 12H, 3JHH = 6.9 Hz,−CH(CH3)2], 2.61 (q of q, 4H, 3JHH = 6.9 Hz, −CHMe2), 3.12 (q ofq, 4H, 3JHH = 6.7 Hz, −CHMe2), 3.62 (q of q, 4H, 3JHH = 6.8 Hz,−CHMe2), 6.98 (d, 4H,
4JHH = 1.7 Hz, Trip-H), 7.00 (d of d, 2H, 3JHH= 7.6 Hz, 3JHH = 6.9 Hz, Xant-H), 7.13 (d of d, 2H, 3JHH = 7.6 Hz, 4JHH= 1.5 Hz, Xant-H), 7.17 (d, 4H, 4JHH = 1.6 Hz, Trip-H), 7.34−7.40 (m,6H, aryl-H), 7.66 (d of d, 2H, 3JHH = 6.8 Hz, 4JHH = 1.6 Hz, Xant-H).13C{1H} NMR (100.61 MHz, C6D6, 26 °C, δ): 22.6 [−CH(CH3)2],24.0 [−CH(CH3)2], 24.0 [−CH(CH3)2], 24.5 [−CH(CH3)2], 26.6[−CH(CH3)2], 27.7 [−CH(CH3)2], 31.4 (−CHMe2), 31.6(−CHMe2), 32.4 (Xant-Cprim), 34.7 (2 s superimposed, -CHMe2 andXant-Cquat), 121.8 (AriPr-Ctert), 124.4 (Xant-Ctert), 126.6 (AriPr-Ctert),127.4 (Xant-Ctert), 129.1 (Xant-Cquat), 130.6 (AriPr-Ctert), 134.3 (Xant-Ctert), 134.5 (AriPr-Cquat), 145.6 (AriPr-Cquat), 146.4 (AriPr-Cquat), 148.2(AriPr-Cquat), 149.1 (AriPr-Cquat), 155.9 (Xant-Cquat), 167.6 (Xant-Cquat),180.4 (AriPr-Cquat). 119Sn NMR (93.28 MHz, C6D6, 26 °C, δ): 1390 (s+ d, J119Sn−117
Sn = 1517 Hz). UV−vis (n-hexane, c = 0.049 mmol L−1;λmax, nm (ε)): 473 (3700), 342 (9000). Anal. Calcd for C87H110OSn2:C, 74.15; H, 7.87. Found: C, 73.94; H, 7.65.Synthesis of Distannene 3. After being precooled to −40 °C,
200 mg (0.53 mmol) of 1,8-diiodonaphthalene in 6 mL of diethylether was treated with 0.7 mL (1.1 mmol) of a 1.6 M n-butyllithiumsolution in n-hexane at ambient temperature and the solution wasstirred for 20 min. The mixture was evacuated for 3 h at a pressure of10−3 mbar to remove the n-butyl iodide. The residue was dissolved in6 mL of diethyl ether. After it was cooled to −40 °C, this solution wasadded dropwise to a solution of 492 mg (1.05 mmol) of [ArMeSnCl]2in 40 mL of diethyl ether. Stirring overnight yielded an intense greenmixture. The solvent was removed in vacuo, and the residue wastreated with 60 mL of n-hexane. The suspension was filtered throughCelite, and the residue was washed three times with 5 mL of n-hexane.The volume of the filtrate was reduced in vacuo to incipientcrystallization, and the mixture was allowed to crystallize at −40 °C forseveral days. Cold filtration yielded the intense green product (250 mg,48%). The filtrate was further concentrated, and additional productwas obtained by repeated crystallization. 1H NMR (500.13 MHz,toluene-d8, 26 °C, δ): 1.95 (two singlets superimposed, 24H, Mes-CH3), 2.25 (s, Δν1/2 = 50 Hz, 12H, Mes-CH3), 6.28 (s, Δν1/2 = 60 Hz,4H, Mes-H), 6.45 (s, Δν1/2 = 50 Hz, 4H, Mes-H), 7.01−7.04 (m, 4H,aryl-H), 7.27−7.31 (m, 4H, Nap-H and aryl-H), 7.58 (d of d, 2H, JHH= 8.1 Hz, JHH = 1.1 Hz, Nap-H), 7.83 (d of d, 2H, JHH = 6.5 Hz, JHH =1.3 Hz, Nap-H). 13C{1H} NMR (125.76 MHz, toluene-d8, 26 °C, δ):20.6 (Mes-CH3), 21.2 (Mes-CH3), 124.1 (Nap-Ctert), 127.8 (ArMe-Ctert), 128.6−128.8 (superimposed, Xant-Ctert, ArMe-Ctert and Mes-Ctert),132.9 (Nap-Cquat), 133.1 (Nap-Ctert), 135.8−136.4 (Mes-Cquat), 137.1(Mes-Cquat), 140.5 (ArMe-Cquat), 141.5 (Nap-Cquat), 149.0 (ArMe-Cquat),161.5 (ArMe-Cquat), 179.8 (Nap-Cquat). 119Sn NMR (186.50 MHz,toluene-d8, 26 °C, δ): 396 (s + d, J119Sn−117
Sn = 9470 ± 30 Hz). UV−vis(n-hexane, c = 0.036 mmol L−1; λmax, nm (ε)): 613 (7500), 398(4800), 282 (22000). Anal. Calcd for C58H56Sn2: C, 70.33; H, 5.70.Found: C, 70.41; H, 6.18.Synthesis of Distannene 4. The same protocol was applied as for
3 using 669 mg (1.1 mmol) of [AriPrSnCl]. The first cold filtrationyielded 450 mg (64%) of the intense purple product. 1H NMR(500.13 MHz, toluene-d8, 26 °C, δ): −0.01 [s, Δν1/2 = 300 Hz, 3H,−CH(CH3)2], 1.00−1.08 [m, Δν1/2 ≤ 25 Hz, approximately 24H,−CH(CH3)2], 1.18−1.28 [m, Δν1/2 ≤ 25 Hz, approximately 48H,−CH(CH3)2], 2.78 (s, Δν1/2 = 120 Hz, 8H, −CHMe2), 3.31 (s, Δν1/2= 140 Hz, 4H, −CHMe2), 6.92 (s, Δν1/2 = 140 Hz, 8H, Trip-H),7.12−7.16 (m, 6H, aryl-H), 7.25 (d of d, 2H, JHH = 8.0 Hz, JHH = 6.6Hz, Nap-H), 7.52 (d of d, 2H, JHH = 8.0 Hz, JHH = 1.1 Hz, Nap-H),7.82 (d of d, 2H, JHH = 6.6 Hz, JHH = 1.1 Hz, Nap-H). 13C{1H} NMR(125.76 MHz, toluene-d8, 26 °C, δ): 23.5 [Δν1/2 = 45 Hz,−CH(CH3)2], 24.8 [Δν1/2 = 8 Hz, −CH(CH3)2], 25.2 [Δν1/2 = 30
Hz, −CH(CH3)2], 26.0 [Δν1/2 = 30 Hz, −CH(CH3)2], 26.2 [Δν1/2 =120 Hz, −CH(CH3)2], 31.0 [Δν1/2 = 35 Hz, -CHMe2], 31.1 [Δν1/2 =4 Hz, -CHMe2], 34.1 [Δν1/2 = 27 Hz, -CHMe2], 120.6 (Δν1/2 = 150Hz, AriPr-Ctert), 122.2 (Δν1/2 = 20 Hz, AriPr-Ctert), 125.8 (Nap-Ctert),127.2 (AriPr-Ctert), 129.7 (Nap-Ctert), 130.8 (Δν1/2 = 12 Hz, AriPr-Ctert),133.1 (Nap-Cquat), 134.4 (Nap-Ctert), 138.9 (AriPr-Cquat), 143.3 (Nap-Cquat), 146.7 (Δν1/2 = 65 Hz, AriPr-Cquat), 147.8 (Δν1/2 = 25 Hz, AriPr-Cquat), 148.5 (AriPr-Cquat), 164.9 (AriPr-Cquat), 179.8 (Nap-Cquat). 119SnNMR (186.50 MHz, toluene-d8, 26 °C, δ): 412 (s + d, J119Sn−117
Sn =6740 ± 50 Hz). UV−vis (n-hexane, c = 0.027 mmol L−1; λmax, nm(ε)): 572 (16000), 384 (7900), 278 (26000). Anal. Calcd forC82H104Sn2·C6H14: C, 74.79; H, 8.42. Found: C, 74.42; H, 8.27. Thesample for elemental analysis was obtained by slow crystallization fromn-hexane solution at −40 °C; ∼1 equiv of n-hexane was detected by 1HNMR spectroscopy).
Synthesis of Distannoxane 5. A blue-green solution of 150 mg(0.1 mmol) of 1 in 30 mL n-hexane was exposed to air and colorlessproduct precipitated. The precipitate was washed three times with 2mL of n-pentane and then dried in vacuo. Yield: 60 mg (36%). 1HNMR (400.11 MHz, C6D6, 26 °C, δ): 0.32 (s + 2d, 2H, 2J119Sn−1
H =37.8 Hz, 2J117Sn−1
H = 36.2 Hz, SnOH), 1.50 (s, 6H, Xant-CH3), 1.84 (s,Δν1/2 = 9 Hz, 12H, Mes-CH3), 2.07 (s, Δν1/2 = 13 Hz, 12H, Mes-CH3), 2.25 (s, 12H, Mes-CH3), 6.56 (s, Δν1/2 = 16 Hz, 4H, Mes-H),6.80 (d of d + d of d of d, 2H, 3JHH = 7.6 Hz, 3JHH = 7.2 Hz, 4J119/117Sn−1
H= 24.5 Hz, Xant-H), 6.89 (s, Δν1/2 = 4 Hz, 4H, Mes-H), 6.93 (d + d ofd, 4H, 3JHH = 7.5 Hz, 4J119/117Sn−1
H = 27.2 Hz, aryl-H), 7.00 (d of d + d ofd of d, 2H, 3JHH = 7.7 Hz, 4JHH = 1.3 Hz, 5J119/117Sn−1
H = 2.5 Hz, Xant-H), 7.20 (t + d of t, 2H, 3JHH = 7.6 Hz, 5J119/117Sn−1
H = 4.8 Hz, aryl-H),7.24 (d of d + d of d of d, 2H, 3JHH = 7.2 Hz, 4JHH = 1.4 Hz, 3J119/117Sn−1
H= 72.0 Hz, Xant-H). 13C{1H} NMR (125.76 MHz, C6D6, 26 °C, δ):20.9 (s, 2C, Mes-CH3), 21.8 (s, 2C, Mes-CH3), 21.8 (s, 2C, Mes-CH3),31.9 (s, 2C, Xant-Cprim), 35.5 (s + d, 1C, 4J119/117Sn−13
C = 8 Hz), Xant-Cquat), 123.8 (s + d, 2C, 3J119/117Sn−13
C = 81 Hz, Xant-Ctert), 126.8 (s + d,2C, 4J119/117Sn−13
C = 11 Hz, Xant-Ctert), 128.1 (ArMe-Ctert), 128.5 (4C,ArMe-Ctert), 129.0 (s +d, 2C, 3J119/117Sn−13
C = 34 Hz, Xant-Cquat), 129.2 (s,4C, ArMe-Ctert), 130.1 (s + 2d, 2C, 1J119Sn−13
C = 832 Hz, 1J117Sn−13C = 795
Hz, Xant-Cquat), 130.6 (s + d, 2C, 4J119/117Sn−13C = 13 Hz, ArMe-Ctert),
134.1 (s + d, 2C, 2J119/117Sn−13C = 45 Hz, Xant-Ctert), 136.3 (s, Δν1/2 = 7
Hz, 4C, ArMe-Cquat), 137.4 (s, 4C, ArMe-Cquat), 137.6 (s, Δν1/2 = 7 Hz,4C, ArMe-Cquat), 139.9 (s + d, 4C, 3J119/117Sn−13
C = 28 Hz, ArMe-Cquat),145.5 (s + 2d, 2C, 1J119Sn−13
C = 852 Hz, 1J117Sn−13C = 815 Hz, ArMe-Cquat),
149.5 (s + d, 4C, 2J119/117Sn−13C = 51 Hz, ArMe-Cquat), 153.4 (s, 2C, Xant-
Cquat). 119Sn{1H} NMR (186.50 MHz, C6D6, 26 °C, δ): −155 (s + d,2J119Sn−117
Sn = 709 ± 3 Hz). Anal. Calcd for C63H64O4Sn2·0.75(C6H14):C, 68.29; H, 6.32. Found: C, 67.99; H, 6.36; N, 0.24. The crystalstructure contains one molecule n-hexane per formula unit of 5.Drying in vacuo fractionally removed the cocrystallized solvent. Theintegration ratio of the 1H NMR spectrum of the CHN sample wasused to determine approximately 0.75 equiv of n-hexane. The smallamount of nitrogen is probably caused by residual TMEDA from thesynthesis of the distannene 1.
Synthesis of Bis(dihydroxystannane) 6. A brown solution ofbis(stannylene) 2 in n-hexane was exposed to air. The solution becamepale yellow, and a pale yellow solid precipitated. The solvent wasremoved in vacuo. Recrystallization from acetonitrile with a temper-ature gradient of ambient temperature to −29 °C yielded micro-crystalline white product. 1H NMR (400.13 MHz, CD2Cl2, 26 °C, δ):0.99−1.03 [2 d superimposed, 48H, −CH(CH3)2], 1.26 [d, 24H,
3JHH= 6.9 Hz, −CH(CH3)2], 1.49 (s, 6H, Xant−CH3), 2.28 (s + d, 4H,2J119/117Sn−1
H = 34 Hz, SnOH), 2.77 (q of q, 8H, 3JHH = 6.6 Hz,−CHMe2), 2.86 (q of q, 4H,
3JHH = 6.9 Hz, −CHMe2), 6.84−6.90 (m,4H, Xant-H), 6.96 (s, 8H, Trip-H), 7.19 (d + d of d, 4H, 3JHH = 7.6Hz, 3J119/117Sn−1
H = 26 Hz, aryl-H), 7.30 (d of d + d of d of d, 2H, JHH =6.8 Hz, JHH = 2.7 Hz, J119/117Sn−1
H = 16 Hz, Xant-H), 7.47 (t + d of t, 2H,3JHH = 7.6 Hz, J119/117Sn−1
H = 9 Hz, aryl-H). 13C{1H} NMR (100.61MHz, CD2Cl2, 26 °C, δ): 23.2 [−CH(CH3)2], 24.1 [−CH(CH3)2],28.9 [−CH(CH3)2], 31.2 (−CHMe2), 34.4 (−CHMe2), 34.4 (Xant-Cprim), 34.4 (Xant-Cquat), 121.2 (AriPr-Ctert), 123.8 (s + d, J119/117Sn−13
C =81 Hz, Xant-Ctert), 129.0 (s + d, 4J119/117Sn−13
C = 14 Hz, AriPr-Ctert), 129.5(s + d, J119/117Sn−13
C = 11 Hz, Xant-Ctert), 129.8 (s + d, 3J119/117Sn−13C = 35
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183916

Hz, Xant-Cquat), 130.6 (s + d, 3J119/117Sn−13C = 69 Hz, AriPr-Ctert), 135.6 (s
+ d, J119/117Sn−13C = 54 Hz, Xant-Ctert), 137.3 (s + d, J119/117Sn−13
C = 24 Hz,AriPr-Cquat), 140.4 (s + 2d, 1J119Sn−13
C = 834 Hz, 1J117Sn−13C = 798 Hz,
AriPr-Cquat), 147.7 (AriPr-Cquat), 148.8 (s + d, 2J119/117Sn−13C = 46 Hz, AriPr-
Cquat), 149.1 (AriPr-Cquat), 153.6 (Xant-Cquat). 119Sn{1H} NMR (93.28MHz, CD2Cl2, 26 °C, δ): −112. Anal. Calcd for C87H114O5Sn2: C,70.73; H, 7.78. Found: C, 70.67; H, 7.65; N, 0.02. The minimalamount of nitrogen is probably caused by residual acetonitrile.Synthesis of Stannyl Stannide 7. A 305 mg portion (0.28
mmol) of 1, 22 mg (0.29 mmol) of KCl, and 105 mg (0.29 mmol) of[2.2.2]cryptand were treated with 18 mL of THF, and the mixture wasstirred for 20 h. The intense blue-green solution changed color to deepred. The solvent was removed in vacuo, the residue was dissolved in 50mL of benzene, and the solution was filtered. Large deep red singlecrystals were obtained in good yield by slow diffusion of n-pentane viagas phase into the benzene solution. 1H NMR (500.13 MHz, THF-d8,26 °C, δ): 1.47 (s, Δν1/2 = 100 Hz, 6H, Xant-CH3), 1.96 (2 ssuperimposed, Δν1/2 > 50 Hz, 36H, Mes-CH3), 2.50−2.52 (m, 12H,cryptand), 3.48−3.50 (m, 12H, cryptand), 3.53 (s, 12H, cryptand), 6.49(s, Δν1/2 = 80 Hz, 8H, Mes-H), 6.53 (d of d, 3JHH = 7.4 Hz, 3JHH = 7.1Hz, 2H, Xant-H), 6.62 (d, Δν1/2 = 4 Hz, JHH = 7.5 Hz, 4H, aryl-H),6.79 (d of d, 3JHH = 7.5 Hz, 4JHH = 1.4 Hz, 2H, Xant-H), 6.8−7.1(elevated underground, Xant-H), 7.03 (t, JHH = 7.5 Hz, 2H, aryl-H).13C{1H} NMR (125.76 MHz, THF-d8, 26 °C, δ): 21.5 (Mes-CH3),22.9 (Δν1/2 = 50 Hz, Mes-CH3), 30−33 (elevated underground, Xant-Cprim), 35.3 (Xant-Cquat), 54.6 (cryptand), 68.4 (cryptand), 71.3(cryptand), 120.8 (Δν1/2 = 16 Hz, Xant-Ctert), 122.6 (Δν1/2 = 50 Hz,ArMe-Ctert), 126.3 (Δν1/2 = 50 Hz, ArMe-Ctert), 127.8 (Δν1/2 = 15 Hz,ArMe-Ctert), 127.5−129.5 (ArMe-Ctert), 134.5 (Δν1/2 = 17 Hz), 135.2(Δν1/2 = 28 Hz, ArMe-Ctert), 136.6 (Δν1/2 = 80 Hz), 143.5 (Δν1/2 = 10Hz), 150.6 (Δν1/2 = 19 Hz), 157.5−161.1 (elevated underground).119Sn NMR (186.50 MHz, toluene-d8, 26 °C, δ): −138 (s + 2 d,J119Sn−119
Sn = 13150 ± 200 Hz, J119Sn−117Sn = 12500 ± 200 Hz), 325 (s + 2
d, J119Sn−119Sn = 13050 ± 200 Hz, J119Sn−117
Sn = 12300 ± 200 Hz). UV−vis(benzene, λmax, nm (A in au)): c = 0.160 mmol L−1, 622 (0.323), 462(0.800); c = 0.079 mmol L−1, 623 (0.224), 461 (0.335); c = 0.020mmol L−1, 624 (0.047), 457 (0.048). Anal. Calcd forC81H98ClKN2O7Sn2·0.5C6H6: C, 64.56; H, 6.51; N, 1.79. Found:C,64.68; H, 6.76; N, 1.84. Crystalline material of 7·0.5C6H6 was usedfor elemental analysis containing half a molecule of benzene performula unit. The 1H NMR spectrum of the sample revealed acorresponding benzene resonance.Synthesis of Stannyl Stannylene 8. Compound 8 was prepared
by applying a modified protocol of the preparation of 2 using 225 mg(1.1 mmol) of 9,9-dimethylxanthene in a mixture of 4.5 mL of diethylether and 6.3 mL of n-hexane, 1.3 mL (2.2 mmol) of a 1.6 M n-butyllithium solution in n-hexane, and 1.00 g (4.2 mmol) of[ArMeSnCl]2 in a mixture of 20 mL of diethyl ether and 30 mL ofn-hexane, but no (!) TMEDA. The reaction mixture changed color tointense purple overnight. Subsequently, it was filtered through Celite,and the solvent of the filtrate was removed in vacuo. The residue wasdissolved in 60 mL of n-hexane, and the solution was placed for severaldays in a refrigerator at −40 °C for crystallization. The product wasgained by cold filtration, yielding 400 mg (32%). The filtrate wasfurther concentrated, and additional product was obtained byrepeating the crystallization. 1H NMR (400.11 MHz, C6D6, 26 °C,δ): 0.94 (t, 3H, 3JHH = 6.9 Hz, butyl-CH3), 1.20−1.52 (m, 6H, butyl-CH2), 1.37 (s, 3H, Xant-CH3), 1.56 (s, 6H, Mes-CH3), 1.72 (s, 3H,Xant-CH3), 1.87 (s, Δν1/2 = 37 Hz, 6H, Mes-CH3), 2.20 (s, 6H, Mes-CH3), 2.23 (s, 6H, Mes-CH3), 2.24 (s, Δν1/2 = 20 Hz, 6H, Mes-CH3),2.38 (s, 6H, Mes-CH3), 6.35 (s, Δν1/2 = 4.5 Hz, 2H, Mes-H), 6.56 (s,Δν1/2 = 40 Hz, 2H, Mes-H), 6.78−6.84 (m, 3H, aryl-H), 6.90 (s, Δν1/2= 4.5 Hz, 2H, Mes-H), 6.91−6.97 (m, 3H, 2 × Xant-H and 1 × aryl-H), 7.03 (d of d, 1H, 3JHH = 8.2 Hz, 4JHH = 1.2 Hz, Xant-H), 7.09−7.17 (m, 5H, 1 × Xant-H and 4 × aryl-H), 7.21 (d of d, 1H, 3JHH = 7.8Hz, 4JHH = 1.5 Hz, Xant-H). 13C{1H} NMR (125.76 MHz, C6D6, 26°C, δ): 14.0 (butyl-CH3), 20.3 (Mes-CH3), 21.0 (Mes-CH3), 21.0(Mes-CH3), 21.2 (Mes-CH3), 21.4 (butyl-CH2), 21.6 (Mes-CH3), 22.0(Mes-CH3), 22.1 (Mes-CH3), 28.9 (Δν1/2 = 10 Hz, Xant-CH3), 29.1 (s+ d, J119/117Sn−13
C = 78 Hz, butyl-CH2), 31.9 (butyl-CH2), 34.6 (Xant-
CMe2), 35.7 (Xant-CH3), 118.3 (Xant-Ctert), 122.8 (Xant-Ctert), 123.5(s + d, J119/117Sn−13
C = 28 Hz, aryl-Ctert), 125.2 (aryl-Ctert), 125.8 (Xant-Ctert), 126.8 (Xant-Ctert), 127.1 (aryl-Ctert), 128.0 (Xant-Cquat), 128.3(aryl-Ctert), 128.4 (aryl- Cquat), 128.5 (aryl-Ctert), 129.4 (aryl-Ctert),129.5 (aryl-Ctert), 129.5 (aryl-Ctert), 130.0 (Δν1/2 = 10 Hz, aryl-Ctert),130.9 (Xant-Cquat), 135.9 (Δν1/2 = 14 Hz, aryl-Cquat), 136.0 (aryl-Cquat), 136.6 (Δν1/2 = 6 Hz, aryl-Cquat), 137.1 (Δν1/2 = 5 Hz, aryl-Cquat), 137.6 (Δν1/2 = 10 Hz, aryl-Cquat), 137.7 (aryl-Ctert), 143.2(Δν1/2 = 10 Hz, aryl-Cquat), 146.1 (Δν1/2 = 140 Hz, aryl-Cquat), 150.0(aryl-Cquat), 150.7 (s + d, Δν1/2 = 4 Hz, J119/117Sn−13
C = 28 Hz, aryl-Cquat),151.5 (Xant-Cquat), 155.0 (s + d, J119/117Sn−13
C = 10 Hz, aryl-Cquat), 179.9(s + d, J119/117Sn−13
C = 74 Hz, aryl-Cquat). 119Sn NMR (186.50 MHz,C6D6, 26 °C, δ): 109 (s + d, Δν1/2 = 220 Hz, J119Sn−119/117
Sn = 9400 Hz,SnIII), 1973 (s, Δν1/2 = 4000 Hz, SnI). UV−vis (n-hexane; λmax, nm(ε)): c = 0.960 mmol L−1, 550 (350); c = 0.096 mmol L−1, 545 (440),332 (11000). Anal. Calcd for C67H72OSn2: C, 71.17; H, 6.42. Found:C, 71.16; H, 6.49.
■ ASSOCIATED CONTENT
*S Supporting InformationText, figures, tables, and CIF and xyz files giving more detailedinformation on experimental preparations, crystal structuredeterminations of 2−7, 7·0.5C6H6, and 8, variable-temperatureNMR data, UV−vis data, and theoretical calculations. Thismaterial is available free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATION
Corresponding Author*E-mail for L.W.: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
This work was financially supported by the Fonds derChemischen Industrie.
■ REFERENCES(1) Davidson, P. J.; Lappert, M. F. J. Chem. Soc., Chem. Commun.1973, 317.(2) Goldberg, D. E.; Harris, D. H.; Lappert, M. F.; Thomas, K. M. J.Chem. Soc., Chem. Commun. 1976, 261.(3) Driess, M.; Grutzmacher, H. Angew. Chem., Int. Ed. 1996, 35, 828.(4) Wang, Y.; Robinson, G. H. Chem. Commun. 2009, 7345, 5201.(5) Fischer, R. C.; Power, P. P. Chem. Rev. 2010, 110, 3877.(6) Kira, M.; Iwamoto, T. Adv. Organomet. Chem. 2006, 54, 73.(7) Power, P. P. Chem. Rev. 1999, 99, 3463.(8) Henning, J.; Wesemann, L. Angew. Chem., Int. Ed. 2012, 51,12869.(9) Simons, R. S.; Pu, L.; Olmstead, M. M.; Power, P. P.Organometallics 1997, 16, 1920.(10) Eichler, B. E.; Pu, L.; Stender, M.; Power, P. P. Polyhedron 2001,20, 551.(11) Bondi, A. J. Phys. Chem. 1964, 68, 441.(12) Zabula, A. V.; Hahn, F. E. Eur. J. Inorg. Chem. 2008, 2008, 5165.(13) Henn, M.; Schurmann, M.; Mahieu, B.; Zanello, P.; Cinquantini,A.; Jurkschat, K. J. Organomet. Chem. 2006, 691, 1560.(14) Stanciu, C.; Richards, A. F.; Power, P. P. J. Am. Chem. Soc. 2004,126, 4106.(15) Klinkhammer, K. W.; Fassler, T. F.; Grutzmacher, H. Angew.Chem., Int. Ed. 1998, 37, 124.(16) Sturmann, M.; Saak, W.; Klinkhammer, K. W.; Weidenbruch, M.Z. Anorg. Allg. Chem. 1999, 625, 1955.(17) Kurzbach, D.; Yao, S.; Hinderberger, D.; Klinkhammer, K. W.Dalton Trans. 2010, 39, 6449.
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183917

(18) Fukawa, T.; Lee, V. Y.; Nakamoto, M.; Sekiguchi, A. J. Am.Chem. Soc. 2004, 126, 11758.(19) Klinkhammer, K. W.; Schwarz, W. Angew. Chem., Int. Ed. 1995,34, 1334.(20) Weidenbruch, M.; Kilian, H.; Peters, K.; von Schnering, H. G.;Marsmann, H. Chem. Ber. 1995, 128, 983.(21) Arp, H.; Baumgartner, J.; Marschner, C.; Muller, T. J. Am. Chem.Soc. 2011, 133, 5632.(22) Lay, U.; Pritzkow, H.; Grutzmacher, H. J. Chem. Soc., Chem.Commun. 1992, 260.(23) Power, P. P. J. Chem. Soc., Dalton Trans. 1998, 2939.(24) Wrackmeyer, B. Annu. Rep. NMR Spectrosc. 1985, 16, 73.(25) Wrackmeyer, B. Annu. Rep. NMR Spectrosc. 1999, 38, 203.(26) Davidson, P. J.; Harris, D. H.; Lappert, M. F. J. Chem. Soc.,Dalton Trans. 1976, 2268.(27) Goldberg, D. E.; Hitchcock, P. B.; Lappert, M. F.; Thomas, K.M.; Thorne, A. J.; Fjeldberg, T.; Haaland, A.; Schilling, B. E. R. J.Chem. Soc., Dalton Trans. 1986, 2387.(28) Masamune, S.; Sita, L. R. J. Am. Chem. Soc. 1985, 107, 6390.(29) Zilm, K. W.; Lawless, G. A.; Merrill, R. M.; Millar, J. M.; Webb,G. G. J. Am. Chem. Soc. 1987, 109, 7236.(30) Masamune, S.; Eriyama, Y.; Kawase, T. Angew. Chem., Int. Ed.1987, 26, 584.(31) Della Bona, M. A.; Cassani, M. C.; Keates, J. M.; Lawless, G. A.;Lappert, M. F.; Stuermann, M.; Weidenbruch, M. J. Chem. Soc., Chem.Commun. 1998, 1187.(32) Kira, M.; Yauchibara, R.; Hirano, R.; Kabuto, C.; Sakurai, H. J.Am. Chem. Soc. 1991, 113, 7785.(33) Kira, M.; Ishida, S.; Iwamoto, T.; Yauchibara, R.; Sakurai, H. J.Organomet. Chem. 2001, 636, 144.(34) Eaborn, C.; Hill, M. S.; Hitchcock, P. B.; Patel, D.; Smith, J. D.;Zhang, S. Organometallics 2000, 19, 49.(35) Eichler, B. E.; Power, P. P. Inorg. Chem. 2000, 39, 5444.(36) Eichler, B. E.; Phillips, B. L.; Power, P. P.; Augustine, M. P.Inorg. Chem. 2000, 39, 5450.(37) Arp, H.; Baumgartner, J.; Marschner, C.; Zark, P.; Muller, T. J.Am. Chem. Soc. 2012, 134, 10864.(38) Grindley, T. B.; Curtis, R. D.; Thangarasa, R.; Wasylishen, R. E.Can. J. Chem. 1990, 68, 2102.(39) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77,3865.(40) Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys.2010, 132, 154104.(41) Grimme, S.; Ehrlich, S.; Goerigk, L. J. Comput. Chem. 2011, 32,1456.(42) Weigend, F. Phys. Chem. Chem. Phys. 2006, 8, 1057.(43) Eichkorn, K.; Treutler, O.; Ohm, H.; Haser, M.; Ahlrichs, R.Chem. Phys. Lett. 1995, 242, 652.(44) Sierka, M.; Hogekamp, A.; Ahlrichs, R. J. Chem. Phys. 2003, 118,9136.(45) Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297.(46) Hellweg, A.; Hattig, C.; Hofener, S.; Klopper, W. Theor. Chem.Acc. 2007, 117, 587.(47) Metz, B.; Stoll, H.; Dolg, M. J. Chem. Phys. 2000, 113, 2563.(48) Tokitoh, N.; Saito, M.; Okazaki, R. J. Am. Chem. Soc. 1993, 115,2065.(49) Weidenbruch, M.; Schlaefke, J.; Schafer, A.; Peters, K.; vonSchnering, H. G.; Marsmann, H. Angew. Chem., Int. Ed. 1994, 33, 1846.(50) Lee, V. Y.; Fukawa, T.; Nakamoto, M.; Sekiguchi, A.; Tumanskii,B. L.; Karni, M.; Apeloig, Y. J. Am. Chem. Soc. 2006, 128, 11643.(51) Edelman, M. A.; Hitchcock, P. B.; Lappert, M. F. J. Chem. Soc.,Chem. Commun. 1990, 1116.(52) Kasna, B.; Jambor, R.; Schurmann, M.; Jurkschat, K. J.Organomet. Chem. 2007, 692, 3555.(53) Pu, L.; Hardman, N. J.; Power, P. P. Organometallics 2001, 20,5105.(54) Tajima, T.; Takeda, N.; Sasamori, T.; Tokitoh, N. Organo-metallics 2006, 25, 3552.
(55) Chandrasekhar, V.; Nagendran, S.; Baskar, V. Coord. Chem. Rev.2002, 235, 1.(56) Davies, A. G. J. Chem. Res. 2004, 2004, 309.(57) Jambor, R.; Kasna, B.; Kirschner, K. N.; Schurmann, M.;Jurkschat, K. Angew. Chem., Int. Ed. 2008, 47, 1650.(58) Eichler, B. E.; Phillips, A. D.; Power, P. P. Organometallics 2003,22, 5423.(59) Phillips, A. D.; Hino, S.; Power, P. P. J. Am. Chem. Soc. 2003,125, 7520.(60) Cardin, C. J.; Cardin, D. J.; Constantine, S. P.; Todd, A. K.;Teat, S. J.; Coles, S. Organometallics 1998, 17, 2144.(61) Drost, C.; Hildebrand, M.; Lonnecke, P.Main Group Met. Chem.2002, 25, 93.(62) Setaka, W.; Hirai, K.; Tomioka, H.; Sakamoto, K.; Kira, M.Chem. Commun. 2008, 0, 6558.(63) Lei, H.; Fettinger, J. C.; Power, P. P. Organometallics 2010, 29,5585.(64) Harris, R. K.; Becker, E. D.; Cabral de Menezes, S. M.;Goodfellow, R.; Granger, P. Pure Appl. Chem. 2001, 73, 1795.(65) APEX2 v2011.8-0; Bruker AXS, Madison, WI, 2011.(66) SAINT V7.68A; Bruker AXS, Madison, WI, 2009.(67) SADABS V2008/1; Bruker AXS, Madison, WI, 2008.(68) Sheldrick, G. M. SHELXS 97, Program for the Solution of CrystalStructures; University of Gottingen, Gottingen, Germany, 1997.(69) Sheldrick, G. M. SHELXL 97, Program for Crystal StructureRefinement; University of Gottingen, Gottingen, Germany, 1997.(70) Hubschle, C. B.; Sheldrick, G. M.; Dittrich, B. J. Appl.Crystallogr. 2011, 44, 1281.(71) X-AREA 1.26; Stoe & Cie, Darmstadt, Germany, 2004.(72) X-RED 1.26; Stoe & Cie, Darmstadt, Germany, 2004.(73) X-SHAPE 2.05; Stoe & Cie, Darmstadt, Germany, 2004.(74) bwGRiD (http://www.bw-grid.de), member of the German D-Grid initiative, founded by the Ministy for Education and Research andthe Ministry for Science, Research, and Arts Baden-Wurtemberg.(75) TURBOMOLE V6.4; a development of University of Karlsruheand Forschungszentrum Karlsruhe GmbH, 1989−2007, TURBO-MOLE GmbH, since 2007: available from http://www.turbomole.com, 2012.(76) Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Chem.Phys. Lett. 1989, 162, 165.(77) Haser, M.; Ahlrichs, R. J. Comput. Chem. 1989, 10, 104.(78) TmoleXClient 3.3; COSMOlogic, Leverkusen, Germany, 2012.(79) TmoleXClient 3.4; COSMOlogic, Leverkusen, Germany, 2013.(80) Ahlrichs, R.; May, K. Phys. Chem. Chem. Phys. 2000, 2, 943.(81) Bauernschmitt, R.; Ahlrichs, R. Chem. Phys. Lett. 1996, 256, 454.(82) Bauernschmitt, R.; Haser, M.; Treutler, O.; Ahlrichs, R. Chem.Phys. Lett. 1997, 264, 573.(83) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.(84) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.(85) Vosko, S. H.; Wilk, L.; Nusair, M. Can. J. Phys. 1980, 58, 1200.(86) Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J.Phys. Chem. 1994, 98, 11623.
Organometallics Article
dx.doi.org/10.1021/om500645b | Organometallics 2014, 33, 3904−39183918





























