Embed Size (px)
Citation preview

Thermal AnalysisInformation for Users
User Com
Um homogene feuchte Polymerproben zu erhalten, ist eine Konditionierung von mehre-ren Tagen in definierter relativer Luftfeuchtigkeit notwendig. Die Abhängigkeit der Glas-übergangstemperatur und des mechanischen Modulus vom Wassergehalt der Probe wird mittels Dynamisch Mechanischer Analyse (DMA) im Schermodus gemessen. Es konnte gezeigt werden, dass während einer Messung keine signifikante Wasserdesorption statt-findet, so dass die Messergebnisse nicht beeinflusst werden.
Einfluss von absorbiertem Wasser auf die mechanischen Eigenschaften von Polyamid 6Marco Zappa
24
Sehr geehrter Kunde,2005 haben wir unser DSC823e zusammen mit dem MultiSTARe HSS7 DSC-Sensor eingeführt. Dieses Jahr wurde das Gerät in den USA mit dem „R&D 100 Award“ ausgezeichnet. Der „R&D 100 Award“ wird jedes Jahr an Personen überreicht, die ein neues Produkt mit der grössten technischen Verbesserung entwickelt haben. Seit 44 Jahren werden Produkte aus 16 Kategorien ausgezeichnet.Wir freuen uns sehr über den „R&D 100 Award“, der laut R&D Magazin beweist, dass unser DSC823e eines der innovativsten Produkte ist.
Inhalt 2/2006
TA-Tipp
- Einfluss von absorbiertem Wasser auf die mechanischen Eigenschaf- ten von Polyamid 6 1
Neu im Verkaufsprogramm - Duroplast-Handbuch 6- Poster 7- GA-FTIR-MS Interface 8- IntraCooler 8- Mikroskop-Heiztisch-Kühlung 9- Zertifizierte Referenzmaterialien für
die Thermische Analyse von LGC 9- Präzisionsgewichte
(0.2 g, 1 g und 5 g) 10
Applikationen - Auswertung und Interpretation von
Peaktemperaturen bei DSC-Kurven: Beispiele 11
- Curie-Temperaturmessungen an nanokristallinen mechanisch legierten Eisen-Basis-Materialien 16
- Thermische Charakterisierung von Nahrungs-mittelprodukten am Beispiel von Gummibärchen 18
- Bestimmung des Gehalts an orga- nischen Stoffen in Keramikton 21
Daten - Exhibitions 23- Courses and Seminars 23

2 METTLER TOLEDO UserCom 2/2006
TA-T
ipp
EinleitungWasser wirkt für viele amorphe und se-mikristalline Kunststoffe als Weichma-cher und ändert die technisch relevanten mechanischen Materialparameter wie Elastizitätsmodul und Glasübergangs-temperatur signifikant in Abhängigkeit der absorbierten Menge. Vertreter von Kunststoffen, welche spontan aus ihrer Umgebung Wasser aufnehmen, sind die hygroskopischen Epoxydharze, Polyure-thane, ABS-Polymere, Polykarbonate und alle Polyamid-Derivate.Anhand von Polyamid 6 (Nylon 6) wird hier gezeigt,
wie mit Dynamischer Mechanischer Analyse (DMA) die Abhängigkeit des Elastizitätsmoduls und der Glasüber-gangstemperatur von der absorbierten Wassermenge bestimmt wird,wie die maximale Sättigung und der zeitliche Verlauf der Wasseraufnah-me bei unterschiedlichen relativen Luftfeuchtigkeiten und Temperaturen bestimmt wirdund welche Folgerungen sich daraus für eine korrekte Probenkonditionie-rung in feuchter Umgebung ableiten lassen.
Probenmaterial: Polyamid 6Polyamid 6-Proben (GoodFellow) von 1 mm Dicke, 3 mm Länge und 3 mm Breite wurden für alle DMA-, TGA- und
•
•
•
Sorptions-Experimente verwendet. Die Probengeometrie entspricht einer ty-pischen Probengrösse für diese Tech-niken. Zur Herstellung definierter Aus- gangsproben wurde das Probenmate-rial unter Vakuum (0.1 mbar) bei 60 °C während 24 Stunden getrocknet. Der Masseverlust der Proben durch Trock-nung wurde mit TGA bestimmt und be-trug bis 240 °C (Schmelzbereich Endset: 230 °C) weniger als 0.1 Gewichtsprozent. Das trockene Probenmaterial wurde luft-dicht verschlossen und bei 25 °C aufbe-wahrt.
Probenkonditionierung im Ex-sikkatorZum Einstellen definierter Wassergehalte wurde die Probenkonditionierung in Ex-sikkatoren durchgeführt, deren unterer Teil übersättigte Salzlösungen enthielt, um eine definierte relative Luftfeuchtig-keit (r.F.) einzustellen. Es wurde Natri-umchlorid (75% r.F.), Natriumbromid (58% r.F.), Kaliumkarbonat (43% r.F.), Kaliumacetat (22% r.F.) und reines Was-ser (100% r.F.) verwendet.
Die relative Luftfeuchtigkeit im Exsik-kator wurde mit dem Feuchtemessgerät Hygropalm 2 von Rotronic AG überwacht. Die Genauigkeit war über den gesamten Verlauf des Experiments (≈ 200 Stunden) besser als ± 2% r.F. Die Exsikkatoren mit 100% r.F. wurden bei Raumtemperatur (25 °C) bzw. im Wärmeschrank bei 40 °C und 65 °C gehalten. Damit die Proben mit ihrer ganzen Oberfläche der Umge-bung ausgesetzt sind, wurden sie im Ex-sikkator auf einem Metallnetz gelagert.
Der Wassergehalt ww als Funktion der Konditionierungszeit ist für die unter-schiedlichen Bedingungen in Abbildung 1 dargestellt. ww ist definiert als:
wobei mw die Masse absorbierten Was-sers und mp die Trockenmasse des Poly-mers ist. mw wurde durch Messung der durch die Konditionierung bedingten Massenänderung bestimmt. Um Oberflä-chenwasser auf der Probe zu entfernen, wurden diese vor dem Wägen mit einem Fliespapier getrocknet. Entnahme aus dem Exsikkator, Abkühlen, Trocknen,
Wägen und Transport zurück in den Ex-sikkator dauerten pro Probe weniger als zwei Minuten.Bei Raumtemperatur beträgt die Zeit bis zur Sättigung unabhängig von der re-lativen Luftfeuchtigkeit für Polyamid 6 mehr als 100 Stunden. Bei 100% r.F. lässt sich die Zeit bis zum Gleichgewicht durch erhöhte Konditionierungstemperaturen von 40 °C bzw. 65 °C auf 20 bzw. fünf Stunden reduzieren.
Konsequenzen des Sorptions-verhaltens
Um nahezu gesättigte Proben zu erhalten, müssen diese unabhängig von der relativen Luftfeuchtigkeit ungefähr 200 Stunden der definierten Umgebung ausgesetzt werden. Selbst bei mehrfach dünneren Proben oder stark erhöhten Konditionierungstem-peraturen liegt diese Zeit über fünf Stunden.Wenn Proben während eines Experi-ments im Messinstrument konditio-niert werden, um die Änderung der Materialeigenschaften zu verfolgen, muss sichergestellt werden, dass innerhalb der Messzeit Aussicht auf einen Gleichgewichtszustand zwischen Probe und Umgebung besteht.Wenn die komplexe Kinetik der Was-seraufnahme noch nicht abgeschlos-sen ist, zeigen Proben Feuchtigkeits-gradienten in ihrem Innern, sodass inhomogene Probeneigenschaften und Randeffekte gemessen werden.
Befeuchtung bis zur SättigungFür die folgenden Messungen wurden nur Proben verwendet, die 200 Stunden im Exsikkator der relativen Luftfeuchtigkeit ausgesetzt waren. Bis zum Experiment wurden die Proben in hermetisch ver-schlossenen Aluminium-Tiegeln aufbe-wahrt.Im gesättigten Zustand kann die Bezie-hung zwischen maximaler Sättigung ww, max und relativer Luftfeuchtigkeit r.F. dargestellt werden (Abbildung 2).Daraus geht hervor, dass zwischen 0% und 55% r.F. vom Probenmaterial nur we-nig Wasser absorbiert wird (ww ≤ 1%). Ab einer relativen Luftfeuchtigkeit r.F. von 55% ändert die Abhängigkeit zwischen ww, max und r.F. deutlich. Bei 55% r.F. ist ww, max = 1%. Die Glasübergangstem-
•
•
•
Abbildung 1: Sorptionsverhalten von Polyamid 6: Der Massenanteil des Wassers ww bei unterschiedlichen relativen Luftfeuch-tigkeiten und Tempe-raturen aufgetragen über die Zeit.
Abbildung 2: Sorptionsisotherme von Polyamid 6: Die maximale Sorptions-kapazität ww, max in Funktion der rela-tiven Luftfeuchtigkeit r.F. (hier für Raum-temperatur).
3METTLER TOLEDO UserCom 2/2006
peratur Tg ist dann 27 °C (siehe Abbil-dung 4). Tg entspricht dann praktisch der Konditionierungstemperatur (25 °C). Da-raus kann geschlossen werden, dass die maximale Sorptionskapazität markant ansteigt, wenn die Umgebungstemperatur der Glasübergangstemperatur entspricht.ww, max zeigt eine geringe Temperaturab-hängigkeit. Bei 100 r.F. % nimmt ww, max zwischen 25 °C und 65 °C um 1% ab (sie-he Abbildung 1).
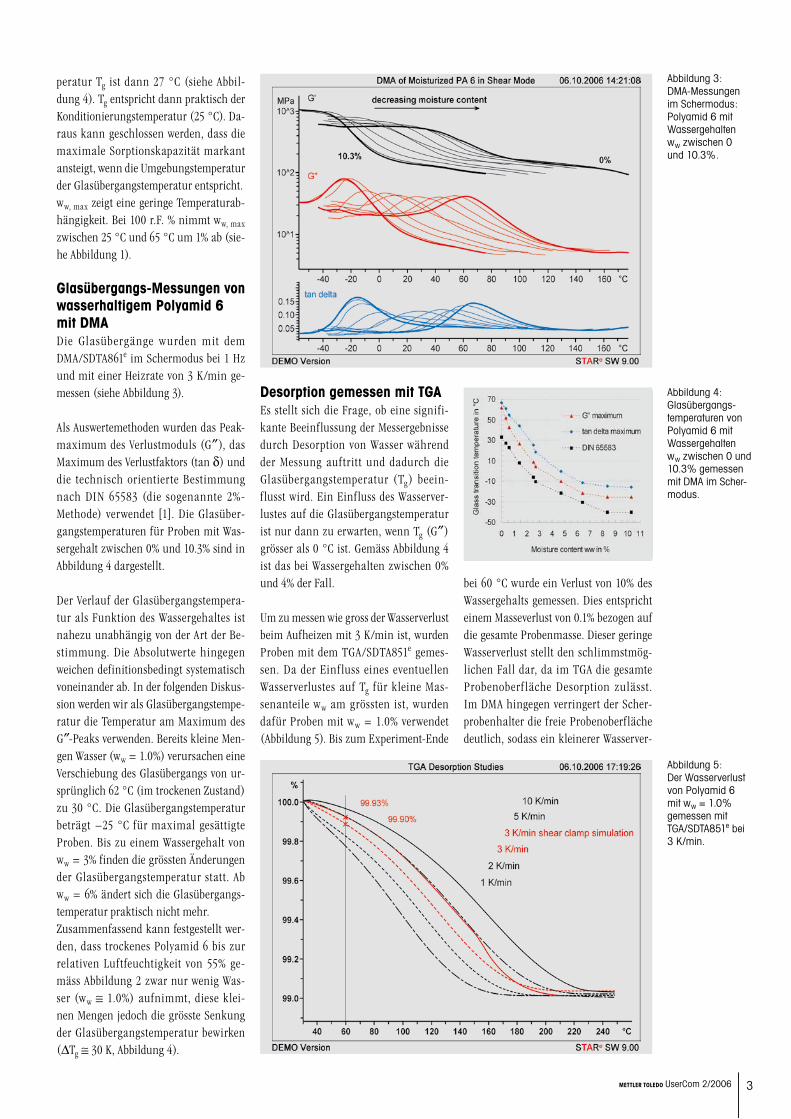
Glasübergangs-Messungen von wasserhaltigem Polyamid 6 mit DMADie Glasübergänge wurden mit dem DMA/SDTA861e im Schermodus bei 1 Hz und mit einer Heizrate von 3 K/min ge-messen (siehe Abbildung 3).
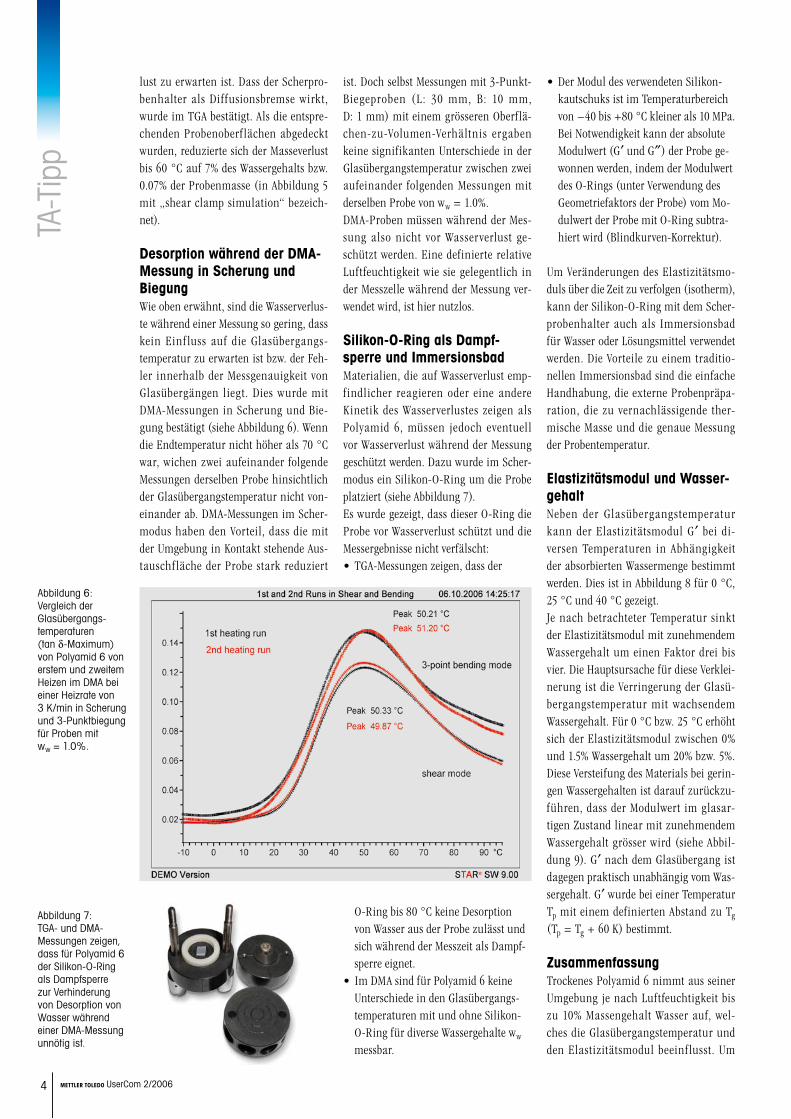
Als Auswertemethoden wurden das Peak-maximum des Verlustmoduls (G″), das Maximum des Verlustfaktors (tan d) und die technisch orientierte Bestimmung nach DIN 65583 (die sogenannte 2%- Methode) verwendet [1]. Die Glasüber-gangstemperaturen für Proben mit Was-sergehalt zwischen 0% und 10.3% sind in Abbildung 4 dargestellt.
Der Verlauf der Glasübergangstempera-tur als Funktion des Wassergehaltes ist nahezu unabhängig von der Art der Be-stimmung. Die Absolutwerte hingegen weichen definitionsbedingt systematisch voneinander ab. In der folgenden Diskus-sion werden wir als Glasübergangstempe-ratur die Temperatur am Maximum des G″-Peaks verwenden. Bereits kleine Men-gen Wasser (ww = 1.0%) verursachen eine Verschiebung des Glasübergangs von ur-sprünglich 62 °C (im trockenen Zustand) zu 30 °C. Die Glasübergangstemperatur beträgt −25 °C für maximal gesättigte Proben. Bis zu einem Wassergehalt von ww = 3% finden die grössten Änderungen der Glasübergangstemperatur statt. Ab ww = 6% ändert sich die Glasübergangs-temperatur praktisch nicht mehr.Zusammenfassend kann festgestellt wer-den, dass trockenes Polyamid 6 bis zur relativen Luftfeuchtigkeit von 55% ge-mäss Abbildung 2 zwar nur wenig Was-ser (ww ≅ 1.0%) aufnimmt, diese klei-nen Mengen jedoch die grösste Senkung der Glasübergangstemperatur bewirken (DTg ≅ 30 K, Abbildung 4).
Desorption gemessen mit TGAEs stellt sich die Frage, ob eine signifi-kante Beeinflussung der Messergebnisse durch Desorption von Wasser während der Messung auftritt und dadurch die Glasübergangstemperatur (Tg) beein-flusst wird. Ein Einfluss des Wasserver-lustes auf die Glasübergangstemperatur ist nur dann zu erwarten, wenn Tg (G″) grösser als 0 °C ist. Gemäss Abbildung 4 ist das bei Wassergehalten zwischen 0% und 4% der Fall.
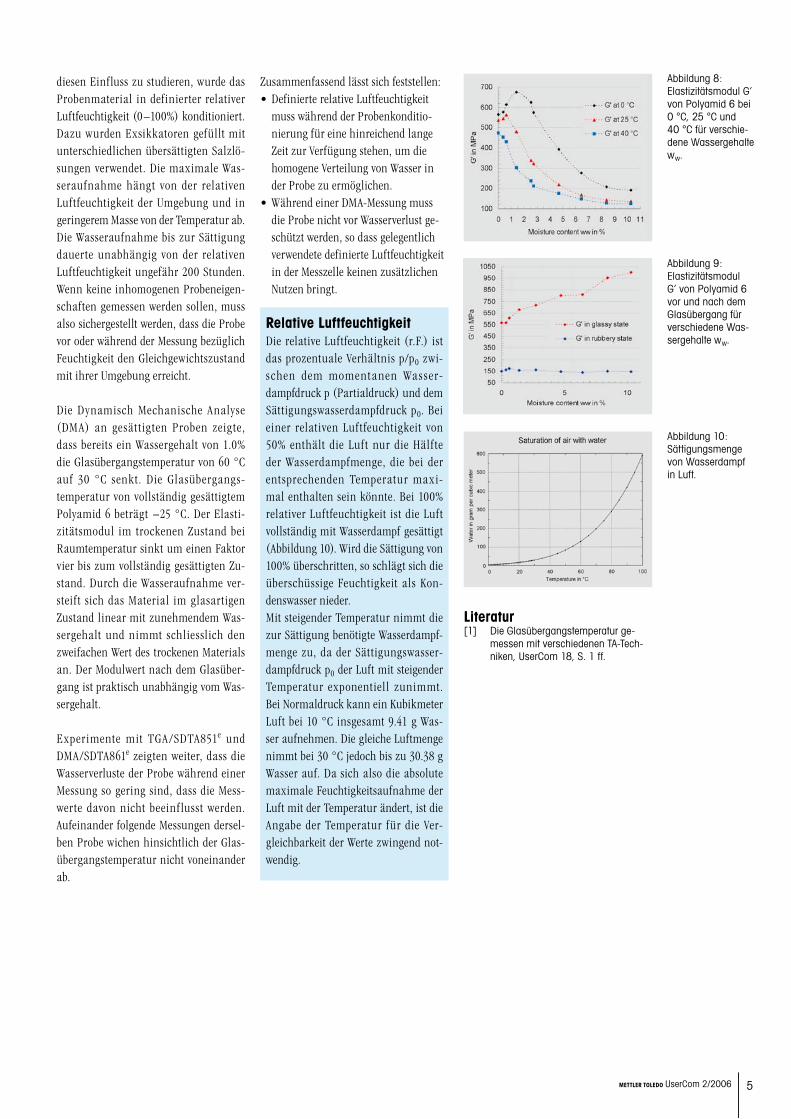
Um zu messen wie gross der Wasserverlust beim Aufheizen mit 3 K/min ist, wurden Proben mit dem TGA/SDTA851e gemes-sen. Da der Einfluss eines eventuellen Wasserverlustes auf Tg für kleine Mas-senanteile ww am grössten ist, wurden dafür Proben mit ww = 1.0% verwendet (Abbildung 5). Bis zum Experiment-Ende
bei 60 °C wurde ein Verlust von 10% des Wassergehalts gemessen. Dies entspricht einem Masseverlust von 0.1% bezogen auf die gesamte Probenmasse. Dieser geringe Wasserverlust stellt den schlimmstmög-lichen Fall dar, da im TGA die gesamte Probenoberfläche Desorption zulässt. Im DMA hingegen verringert der Scher-probenhalter die freie Probenoberfläche deutlich, sodass ein kleinerer Wasserver-
Abbildung 5: Der Wasserverlust von Polyamid 6 mit ww = 1.0% gemessen mit TGA/SDTA851e bei 3 K/min.
Abbildung 4: Glasübergangs-temperaturen von Polyamid 6 mit Wassergehalten ww zwischen 0 und 10.3% gemessen mit DMA im Scher-modus.
Abbildung 3: DMA-Messungen im Schermodus: Polyamid 6 mit Wassergehalten ww zwischen 0 und 10.3%.
4 METTLER TOLEDO UserCom 2/2006
TA-T
ipp
lust zu erwarten ist. Dass der Scherpro-benhalter als Diffusionsbremse wirkt, wurde im TGA bestätigt. Als die entspre-chenden Probenoberflächen abgedeckt wurden, reduzierte sich der Masseverlust bis 60 °C auf 7% des Wassergehalts bzw. 0.07% der Probenmasse (in Abbildung 5 mit „shear clamp simulation“ bezeich-net).
Desorption während der DMA-Messung in Scherung und BiegungWie oben erwähnt, sind die Wasserverlus-te während einer Messung so gering, dass kein Einfluss auf die Glasübergangs- temperatur zu erwarten ist bzw. der Feh-ler innerhalb der Messgenauigkeit von Glasübergängen liegt. Dies wurde mit DMA-Messungen in Scherung und Bie-gung bestätigt (siehe Abbildung 6). Wenn die Endtemperatur nicht höher als 70 °C war, wichen zwei aufeinander folgende Messungen derselben Probe hinsichtlich der Glasübergangstemperatur nicht von-einander ab. DMA-Messungen im Scher-modus haben den Vorteil, dass die mit der Umgebung in Kontakt stehende Aus-tauschfläche der Probe stark reduziert
ist. Doch selbst Messungen mit 3-Punkt-Biegeproben (L: 30 mm, B: 10 mm, D: 1 mm) mit einem grösseren Oberflä-chen-zu-Volumen-Verhältnis ergaben keine signifikanten Unterschiede in der Glasübergangstemperatur zwischen zwei aufeinander folgenden Messungen mit derselben Probe von ww = 1.0%.DMA-Proben müssen während der Mes-sung also nicht vor Wasserverlust ge-schützt werden. Eine definierte relative Luftfeuchtigkeit wie sie gelegentlich in der Messzelle während der Messung ver-wendet wird, ist hier nutzlos.
Silikon-O-Ring als Dampf- sperre und ImmersionsbadMaterialien, die auf Wasserverlust emp-findlicher reagieren oder eine andere Kinetik des Wasserverlustes zeigen als Polyamid 6, müssen jedoch eventuell vor Wasserverlust während der Messung geschützt werden. Dazu wurde im Scher-modus ein Silikon-O-Ring um die Probe platziert (siehe Abbildung 7). Es wurde gezeigt, dass dieser O-Ring die Probe vor Wasserverlust schützt und die Messergebnisse nicht verfälscht:
TGA-Messungen zeigen, dass der
O-Ring bis 80 °C keine Desorption von Wasser aus der Probe zulässt und sich während der Messzeit als Dampf-sperre eignet.Im DMA sind für Polyamid 6 keine Unterschiede in den Glasübergangs-temperaturen mit und ohne Silikon-O-Ring für diverse Wassergehalte ww messbar.
•
•
Der Modul des verwendeten Silikon-kautschuks ist im Temperaturbereich von −40 bis +80 °C kleiner als 10 MPa. Bei Notwendigkeit kann der absolute Modulwert (G′ und G″) der Probe ge-wonnen werden, indem der Modulwert des O-Rings (unter Verwendung des Geometriefaktors der Probe) vom Mo-dulwert der Probe mit O-Ring subtra-hiert wird (Blindkurven-Korrektur).
Um Veränderungen des Elastizitätsmo-duls über die Zeit zu verfolgen (isotherm), kann der Silikon-O-Ring mit dem Scher-probenhalter auch als Immersionsbad für Wasser oder Lösungsmittel verwendet werden. Die Vorteile zu einem traditio-nellen Immersionsbad sind die einfache Handhabung, die externe Probenpräpa-ration, die zu vernachlässigende ther-mische Masse und die genaue Messung der Probentemperatur.
Elastizitätsmodul und Wasser-gehaltNeben der Glasübergangstemperatur kann der Elastizitätsmodul G′ bei di-versen Temperaturen in Abhängigkeit der absorbierten Wassermenge bestimmt werden. Dies ist in Abbildung 8 für 0 °C, 25 °C und 40 °C gezeigt.Je nach betrachteter Temperatur sinkt der Elastizitätsmodul mit zunehmendem Wassergehalt um einen Faktor drei bis vier. Die Hauptsursache für diese Verklei-nerung ist die Verringerung der Glasü-bergangstemperatur mit wachsendem Wassergehalt. Für 0 °C bzw. 25 °C erhöht sich der Elastizitätsmodul zwischen 0% und 1.5% Wassergehalt um 20% bzw. 5%. Diese Versteifung des Materials bei gerin-gen Wassergehalten ist darauf zurückzu-führen, dass der Modulwert im glasar-tigen Zustand linear mit zunehmendem Wassergehalt grösser wird (siehe Abbil-dung 9). G′ nach dem Glasübergang ist dagegen praktisch unabhängig vom Was-sergehalt. G′ wurde bei einer Temperatur Tp mit einem definierten Abstand zu Tg (Tp = Tg + 60 K) bestimmt.
ZusammenfassungTrockenes Polyamid 6 nimmt aus seiner Umgebung je nach Luftfeuchtigkeit bis zu 10% Massengehalt Wasser auf, wel-ches die Glasübergangstemperatur und den Elastizitätsmodul beeinflusst. Um
•
Abbildung 7: TGA- und DMA-Messungen zeigen, dass für Polyamid 6 der Silikon-O-Ring als Dampfsperre zur Verhinderung von Desorption von Wasser während einer DMA-Messung unnötig ist.
Abbildung 6: Vergleich der Glasübergangs- temperaturen (tan d-Maximum) von Polyamid 6 von erstem und zweitem Heizen im DMA bei einer Heizrate von 3 K/min in Scherung und 3-Punktbiegung für Proben mit ww = 1.0%.
5METTLER TOLEDO UserCom 2/2006
diesen Einfluss zu studieren, wurde das Probenmaterial in definierter relativer Luftfeuchtigkeit (0−100%) konditioniert. Dazu wurden Exsikkatoren gefüllt mit unterschiedlichen übersättigten Salzlö-sungen verwendet. Die maximale Was-seraufnahme hängt von der relativen Luftfeuchtigkeit der Umgebung und in geringerem Masse von der Temperatur ab. Die Wasseraufnahme bis zur Sättigung dauerte unabhängig von der relativen Luftfeuchtigkeit ungefähr 200 Stunden. Wenn keine inhomogenen Probeneigen-schaften gemessen werden sollen, muss also sichergestellt werden, dass die Probe vor oder während der Messung bezüglich Feuchtigkeit den Gleichgewichtszustand mit ihrer Umgebung erreicht.
Die Dynamisch Mechanische Analyse (DMA) an gesättigten Proben zeigte, dass bereits ein Wassergehalt von 1.0% die Glasübergangstemperatur von 60 °C auf 30 °C senkt. Die Glasübergangs-temperatur von vollständig gesättigtem Polyamid 6 beträgt −25 °C. Der Elasti-zitätsmodul im trockenen Zustand bei Raumtemperatur sinkt um einen Faktor vier bis zum vollständig gesättigten Zu-stand. Durch die Wasseraufnahme ver-steift sich das Material im glasartigen Zustand linear mit zunehmendem Was-sergehalt und nimmt schliesslich den zweifachen Wert des trockenen Materials an. Der Modulwert nach dem Glasüber-gang ist praktisch unabhängig vom Was-sergehalt.
Experimente mit TGA/SDTA851e und DMA/SDTA861e zeigten weiter, dass die Wasserverluste der Probe während einer Messung so gering sind, dass die Mess-werte davon nicht beeinflusst werden. Aufeinander folgende Messungen dersel-ben Probe wichen hinsichtlich der Glas-übergangstemperatur nicht voneinander ab.
Abbildung 8: Elastizitätsmodul G’ von Polyamid 6 bei 0 °C, 25 °C und 40 °C für verschie-dene Wassergehalte ww.
Abbildung 10: Sättigungsmenge von Wasserdampf in Luft.
Zusammenfassend lässt sich feststellen:Definierte relative Luftfeuchtigkeit muss während der Probenkonditio-nierung für eine hinreichend lange Zeit zur Verfügung stehen, um die homogene Verteilung von Wasser in der Probe zu ermöglichen.Während einer DMA-Messung muss die Probe nicht vor Wasserverlust ge-schützt werden, so dass gelegentlich verwendete definierte Luftfeuchtigkeit in der Messzelle keinen zusätzlichen Nutzen bringt.
Relative LuftfeuchtigkeitDie relative Luftfeuchtigkeit (r.F.) ist das prozentuale Verhältnis p/p0 zwi-schen dem momentanen Wasser-dampfdruck p (Partialdruck) und dem Sättigungswasserdampfdruck p0. Bei einer relativen Luftfeuchtigkeit von 50% enthält die Luft nur die Hälfte der Wasserdampfmenge, die bei der entsprechenden Temperatur maxi-mal enthalten sein könnte. Bei 100% relativer Luftfeuchtigkeit ist die Luft vollständig mit Wasserdampf gesättigt (Abbildung 10). Wird die Sättigung von 100% überschritten, so schlägt sich die überschüssige Feuchtigkeit als Kon-denswasser nieder.Mit steigender Temperatur nimmt die zur Sättigung benötigte Wasserdampf-menge zu, da der Sättigungswasser-dampfdruck p0 der Luft mit steigender Temperatur exponentiell zunimmt. Bei Normaldruck kann ein Kubikmeter Luft bei 10 °C insgesamt 9.41 g Was-ser aufnehmen. Die gleiche Luftmenge nimmt bei 30 °C jedoch bis zu 30.38 g Wasser auf. Da sich also die absolute maximale Feuchtigkeitsaufnahme der Luft mit der Temperatur ändert, ist die Angabe der Temperatur für die Ver-gleichbarkeit der Werte zwingend not-wendig.
•
•
Literatur[1] Die Glasübergangstemperatur ge-
messen mit verschiedenen TA-Tech-niken, UserCom 18, S. 1 ff.
Abbildung 9: Elastizitätsmodul G’ von Polyamid 6 vor und nach dem Glasübergang für verschiedene Was-sergehalte ww.
6 METTLER TOLEDO UserCom 2/2006
Neu
im V
erka
ufsp
rogr
amm
Wir freuen uns, dass wir Ihnen ein neues Applikations-Handbuch anbieten können. Mit dem Duroplast-Handbuch vervollständigen wir unsere sehr umfangreiche Applikations-Sammlung. Praktisch zu jedem Gebiet gibt es nun eine umfassende Applikations-Sammlung mit zusätzlichen Informationen zu den einzelnen Spezialgebieten.
Duroplast-Handbuch
Titel Sprache / Bestell-Nummer Inhalt
Neu: Duroplast- Handbuch (300 Seiten)
Englisch: 51 725 069 (Band 1 + 2)
Teil 1: 51 725 067 Teil 2: 51 725 068
Das Applikations-Handbuch gibt einen Einblick in die thermische Analyse (TA) von Duroplasten und stellt über 90 praktische Beispiele in zwei Bänden vor. Band 1 gibt zuerst einen Überblick über die TA-Techniken und eine Ein-führung in die Chemie der Harze und deren Verwendung. Danach werden hauptsächlich die Eigenschaften und Effekte vorgestellt, die mit den TA-Techniken DSC, TGA, thermomechanische Analyse (TMA) und dyna-misch-mechanische Analyse (DMA) untersucht werden können. Band 2 konzentriert sich auf praktische Beispiele, in denen mehr als 10 verschiede Harztypen wie Epoxide, Polyester, Formaldehyde und Polyurethane diskutiert werden. Die Anwendungen beschreiben die ver-schiedenen Eigenschaften, die während dem Lebenszyklus eines Duro-plasten untersucht, geprüft oder gemessen werden können.
Thermoplast- Handbuch (150 Seiten)
Englisch: 51 725 002 Das thermische Verhalten von über 20 Thermoplasten wird in 59 Anwen-dungsbeispielen diskutiert, wobei neben vielen DSC-, TGA- und TMA- neu auch 8 DMA-Beispiele enthalten sind. Die untersuchten Effekte sind: Schmelzen, Kristallisieren, Glasübergang, Erweichen, Trocknen, ther-mische Zersetzung, Oxidationsstabilität, Ausdehnungs- und Schrumpf-verhalten. Mit Dynamisch Mechanischer Analyse (DMA) wird das viskoelastische Verhalten von Festkörpern untersucht. Praxisnahe Frage-stellungen wie die thermogravimetrische Gehaltsbestimmung oder das Vermeiden von Verwechslungen von Materialien bilden einen wichtigen Bestandteil des Buches.
Elastomer- Handbuch (275 Seiten)
Englisch: 51 725 061 (Band 1 + 2)
Band 1: 51 725 057 Band 2: 51 725 058
Nach einer Einführung in die Thermische Analyse und die Struktur und Eigenschaften der Elastomere werden über 50 Beispiele der Elastomer- Analytik vorgestellt. Dabei werden neben DSC, TGA und TMA auch ge-koppelte Methoden der Gasanalyse und die Dynamisch-Mechanische Analyse (DMA) berücksichtigt. Das Handbuch ist in 2 Bände aufgeteilt. Im ersten Band werden die Grundlagen der thermischen Effekte von Elastomeren und deren Auswertung behandelt. Dabei werden neben der Analyse der Zusammensetzung mittels TGA auch Messungen zur Vulkani-sation, Kristallisation und Glasübergang besprochen sowie Hinweise zur Optimierung von Messung und Auswertung gegeben. Band 2 enthält viele praktische Beispiele der Elastomeranalyse, ange-fangen bei relativ einfachen Beispielen bis zu komplexen Analysen an komplizierten Systemen.
Pharmazeutik-Handbuch (100 Seiten)
Englisch: 51 725 006 Anhand von 47 ausgewählten Beispielen werden die Anwendungsmög-lichkeiten der Thermischen Analyse in der pharmazeutischen Industrie aufgezeigt. Mittels DSC, TGA, EGA und TOA wird unter anderem das Schmelzverhalten, die Polymorphie, die Reinheit, die Feuchtigkeit sowie die Stabilität von Wirk- und Hilfsstoffen analysiert. Daneben werden auch Einflüsse von Messbedingungen und die Gerätekalibrierung diskutiert.
Lebensmittel-Handbuch (50 Seiten)
Englisch: 51 725 004 Die Anwendungen der Thermischen Analyse von Proteinen, Kohlenhy-draten, Fetten und Ölen werden anhand von 53 DSC-, 2 TGA und einer TMA-Kurve aufgezeigt. Die wichtigsten untersuchten Effekte sind: Denaturieren von Proteinen, Quellen von Stärke in Wasser, Schmelzen von Zucker, thermische Zerset-zung von Zucker und Stärke, Schmelzen und Kristallisieren von Fetten, Ölen und Schokolade
7METTLER TOLEDO UserCom 2/2006
TGA-EGA- Handbuch (65 Seiten)
Englisch: 51 725 056 Das Handbuch behandelt die Kopplung von Thermogravimetrie (TGA) und Gasanalyse. Im ersten Teil werden die Grundlagen der Massenspek-trometrie (MS), FTIR-Spektroskopie und deren Interpretation behandelt. Im praktischen Teil folgen 17 Beispiele der Anwendung dieser Kopplungs-techniken. Es werden dabei organische und anorganische Proben sowie Polymer-Systeme untersucht. Neben den konventionellen TGA-MS und TGA-FTIR Kopplungen gibt es auch ein Beispiel einer TMA-MS Kopplung.
Tutorial- Handbuch (25 Seiten)
Handbuch Deutsch: 51 709 919 Englisch: 51 709 920 Französisch: 51 709 921
Handbuch mit Test- substanzen: Deutsch: 51 140 877 Englisch: 51 140 878 Französisch: 51 140 879
Das Applikations-Handbuch, passend zu den Testsubstanzen, eignet sich sehr gut für das Selbst-Studium in der Thermischen Analyse. Anhand von 22 Beispielen wird gezeigt, was mit der Thermischen Analy-se alles gemacht werden kann.
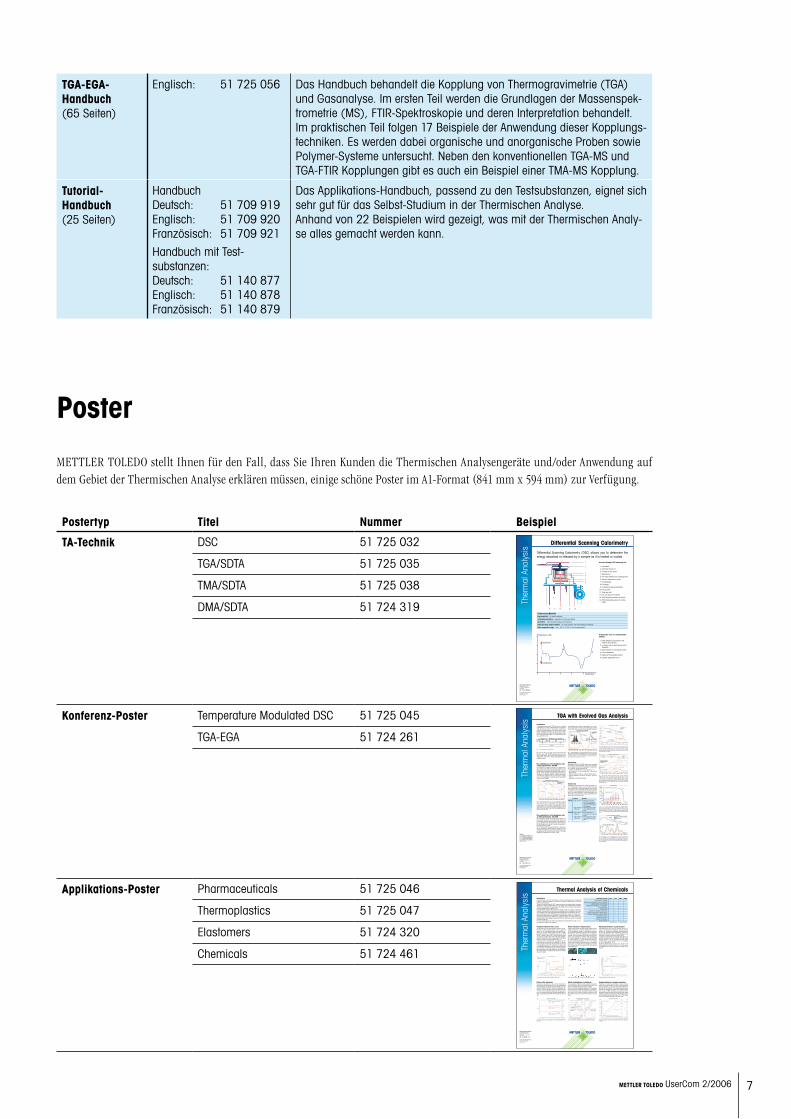
METTLER TOLEDO stellt Ihnen für den Fall, dass Sie Ihren Kunden die Thermischen Analysengeräte und/oder Anwendung auf dem Gebiet der Thermischen Analyse erklären müssen, einige schöne Poster im A1-Format (841 mm x 594 mm) zur Verfügung.
Poster
Postertyp Titel Nummer Beispiel
TA-Technik DSC 51 725 032
TGA/SDTA 51 725 035
TMA/SDTA 51 725 038
DMA/SDTA 51 724 319
Konferenz-Poster Temperature Modulated DSC 51 725 045
TGA-EGA 51 724 261
Applikations-Poster Pharmaceuticals 51 725 046
Thermoplastics 51 725 047
Elastomers 51 724 320
Chemicals 51 724 461
Ther
mal
Ana
lysi
s
Mettler-Toledo AG AnalyticalSonnenbergstrasse 74CH-8603 SchwerzenbachSwitzerlandTel. ++41 44 806 77 11Fax ++41 44 806 72 40
© 09/2006 Mettler-Toledo AGPrinted in Switzerland ME-51725032A
Differential Scanning Calorimetry
A section through a DSC measuring cell:
1 Heat shield
2 Automatic furnace lid
3 Crucible on DSC sensor
4 Silver furnace
5 Flat heater between two insulating disks
6 Thermal resistance for cooler
7 Cooling flange
8 Cold finger
9 Compression spring construction
10 Dry gas inlet
11 Purge gas inlet
12 DSC raw signal for amplifier
13 Pt100 temperature sensor of furnace
14 Pt100 temperature sensor of cooling flange
1
2
3
4
5
6
10 11 12 13 14
7 8
9
A typical DSC curve of a semicrystalline polymer:
1 initial deflection proportional to the sample’s heat capacity
2 cp change with no other thermal effect (baseline)
3 glass transition of amorphous fraction
4 cold crystallization
5 melting of the crystalline fraction
6 oxidative degradation in air
Features and BenefitsHigh sensitivity – for weak transitions
Outstanding resolution – separation of close-lying effects
Automation – high sample throughput and efficiency
Small and large sample volumes – for small samples and inhomogeneous materials
Wide temperature range – from –150 °C to 700 °C in one measurement
Differential Scanning Calorimetry (DSC) allows you to determine the energy absorbed or released by a sample as it is heated or cooled.
Heat flow in mW
Temperature
exothermic
endothermic
12
3
4
5
6
Ther
mal
Ana
lysi
s
Mettler-Toledo AG, AnalyticalSonnenbergstrasse 74CH-8603 SchwerzenbachSwitzerlandTel. ++41 44 806 77 11Fax ++41 44 806 72 40
© 09/2006 Mettler-Toledo AGPrinted in Switzerland ME-51724261A
The resulting curve, known as a chemigram, is a very use-ful way to compare the results of the spectroscopic analysis with the TGA mass loss curve. This is illustrated in Figure 3.
Fig. 3. Thermal degradation of PVC measured by TGA-FTIR. The TGA curve and the chemigram show two clearly defined steps. A FTIR spectrum mea-sured at the first maximum corresponds to HCl. The spectrum measured at the second maximum is, however, very different and is identified as benzene formed through the cyclization of (-CH=CH-)n.
ApplicationsHyphenated TGA-MS or TGA-FTIR analysis is an invaluable aid in research and development, and is also a very useful tool for quality control and the investigation of material fail-ure or damage. Typical applications are:• detection and identification of compounds (Fig. 4)• characterization of raw materials and final products
(Fig. 5 and 6)• chemical reactions (catalysis, synthesis, polymerization)• thermal degradation processes (oxidation, pyrolysis)
(Fig. 7)• degassing and adsorption behavior
ConclusionsCombining a thermobalance with a mass spectrometer or an FTIR spectrometer opens up many important new appli-cation possibilities for thermogravimetric analysis. Qualita-tive information on the substances evolved can be obtained in addition to the quantitative results from the mass loss steps. The online combination of thermogravimetric and spectrometric measurements provides comprehensive de-tails on the processes that occur.
Features Benefits
TGA-MS • High sensitivity
• High resolution (timescale)
• Extremely low concen-trations of evolved gases can still be identified (e.g. impurities in pharmaceuti-cal substances
• Overlapping mass losses can be qualitatively inter-preted
TGA-FTIR • High chemical specificity
• High resolution (timescale)
• Direct identification of compounds and functional groups
• Overlapping mass losses can be qualitatively inter-preted
Table 1. Features and benefits of TGA-MS and TGA-FTIR
TGA with Evolved Gas AnalysisIntroduction In thermogravimetric analysis (TGA) the mass of a sample is recorded as a function of temperature or time under de-fined atmospheric conditions. Quantitative compositional analysis can be performed and the reaction kinetics inves-tigated. Qualitative information on the gaseous products evolved is obtained by coupling the thermobalance online with a mass spectrometer (MS) or a Fourier transform infra-red spectrometer (FTIR).
Fig. 1. The coupling of TGA and gas analysis
The data from the gas analyzer is then compared with the TGA mass loss curve. The DTG (derivative mass loss) and SDTA (single DTA) curves are also often displayed to aid interpretation. SDTA monitors temperature differences due to enthalpy changes.
The combination of a thermobalance with a mass spectrometer, TGA-MSThe TGA/SDTA is coupled to the MS via a heated quartz glass capillary tube. One end of the glass capillary is posi-tioned close to the sample in the thermobalance. Part of the evolved gases is sucked into the capillary by the vacuum in the MS. The MS repeatedly measures either the entire mass spectrum or, as shown in Figure 2, monitors the intensity of characteristic fragment ions (m/z, the mass to charge ratio). The decomposition of calcium oxalate monohydrate is shown as an example.
Fig. 2. The decomposition of CaC2O4 · H2O was investigated by monitoring the m/z values 18, 28 and 44. The first step in the TGA curve corresponds to the elimination of water of crystallization, the second step to the release of carbon monoxide from the anhydrous calcium oxalate, and the third step to the liberation of CO2 from the calcium carbonate formed in the second reaction step. The m/z 44 curve also shows that a small amount of CO2 is formed in the second step. This effect is due to the disproportionation reac-tion of CO to CO2 and carbon.
The combination of a thermobalance with an FTIR spectrometer, TGA-FTIRThe TGA/SDTA is coupled to the FTIR spectrometer via a glass-coated transfer line. This transports the volatile prod-ucts evolved during the decomposition of the sample to a gas cell installed in the FTIR spectrometer. Both the transfer line and the gas cell are heated to prevent condensation of the decomposition products.The FTIR spectrometer measures the spectra of the gases in the gas cell rapidly at frequent intervals. Afterward, a spec-tral range characteristic for a particular functional group can be selected and the infrared absorption bands in this range integrated and displayed as a function of time.
Fig. 4. Methanol and acetone were used to recrystallize a pharmaceutical substance. Residues of both solvents can be clearly detected by TGA-MS. The high temperature observed for the elimination of the relatively large amount of acetone indicates that acetone is more firmly bound in the sub-stance, possibly as a solvate.
Fig. 5. The TGA curve shows three mass loss steps. The first is due to the evaporation of moisture. The combustion of the coal takes place in the two steps that follow. The MS data shows that appreciably more water is evolved in the first of these two steps. Besides carbon, a greater proportion of hydro-gen and hydrogen-containing compounds (e.g. CH4) is burned. In addition, the formation of SO2 (m/z 64) proves that sulfur-containing substances are present in the coal.
Fig. 6. ETFE is used as cable insulation material. An important point is whether hydrogen fluoride is formed when the material undergoes thermal degradation. The TGA-FTIR data of a sample of ETFE shows that volatile ad-ditives are evolved from about 200 °C onward. Degradation begins at about 440 °C. The FTIR spectra prove that hydrogen fluoride is formed above about 450 °C.
Fig. 7. (NH4)6Mo7 O24 · 4H2O decomposes in three steps with the elimina-tion of 6 molecules of NH3 and 7 molecules of H2O. Except in the last mass loss step, it can be seen that water and ammonia are formed simultaneously but not in a fixed ratio to each other. This indicates that the decomposition is non-stoichiometric.
Literature
A more detailed description of the use of evolved gas analysis is giv-en in the Collected Applicationsbooklet Evolved Gas Analysisavailable from METTLER TOLEDO (ME-51725056).
Computer
FTIR or MS
Gas AnalyzerTransfer Line
Heater
TG Analyzer
Balance Furnace
Purgegas
Purgegas +
products
Application Overview DSC TGA TMA DMAGlass transition, softening • • •
Temperature and enthalpy of fusion, crystallinity •Melting behavior, fraction melted • • •
Temperature and enthalpy of crystallization •Cold crystallization • • •
Polymorphism • • •Evaporation, desorption, vaporization • •
Thermal decomposition, stability, kinetics • • • •Oxidative degradation, oxidation stability • • •
Compositional analysis • •Specific heat capacity •
Coefficient of expansion •Young’s modulus • •
Mettler-Toledo AG, AnalyticalSonnenbergstrasse 74CH-8603 SchwerzenbachSwitzerlandTel. ++41 44 806 77 11Fax ++41 44 806 72 40
© 09/2006 Mettler-Toledo AGPrinted in Switzerland ME-51724461
Thermal Analysis of Chemicals
Oxidation Induction Time, of oilsAn important test in the petrochemical industry is the de-termination of the oxidation stability of oils. It allows the behavior of oils to be predicted under actual operating con-ditions, e.g. in motor vehicle engines. The test is usually performed according to an appropriate standard, e.g. ASTM D6186. A sample is held at 180 °C under increased oxygen pressure until oxidation begins. The onset of the exothermic oxidation (intersection of the baseline with the inflectional tangent) is called the oxidation induction time, OIT.The curves show OIT measurements on samples of two dif-ferent motor oils: a mineral oil and a synthetic oil. The min-eral oil oxidizes after about 35 minutes (shown by the onset of the exothermic peak). The synthetic oil is stable at the same temperature during the 120-minute period prescribed by the standard. The inserted diagram on the right displays the measurement curve of the synthetic oil. The synthetic oil takes much longer to oxidize than the mineral oil, namely about 237 minutes.
Mechanical behavior of polysiloxanesPolysiloxanes have been used for decades mainly in in-dustry for the production of refrigerants, lubricants, silicone sealants, etc. The mechanical properties of these substanc-es are vitally important for their manufacture, storage, pro-cessing and application. Many of these properties can be measured by dynamic mechanical analysis (DMA).A sample of a polysiloxane was prepared in the shear sam-ple holder for liquids. It was then installed in the DMA, which had been cooled to –150 °C. On heating, the shock-cooled material exhibits a glass transition at –115 °C, crystallizes at –100 °C and melts at –40 °C. Above this temperature it is liquid (G”>G’). At 120 °C, the phase angle almost reaches the limit of /2 radians typical for a Newtonian fluid. The storage modulus changes by 7.5 decades.
Sorption behavior of organic moleculesThe behavior of substances with regard to drying, moisture uptake, and moisture content is a topic of major importance because moisture can very often have adverse effects on the properties of materials. A TGA sorption analyzer system was used to investigate the influence of the relative humidity (RH) on pure (> 98%) N-amidino-3,5-diamino-6-chloro-pyrazine-carboxamide hydrochloride dihydrate. After drying the RH was stepwise increased and then decreased again while holding the sample isothermally at 25 °C. The original mass of the sample is reached at about 50% RH.
IntroductionThermal Analysis (TA) is the name given to a group of techniques used to measure the physical or chemical properties of a sample as it is heated, cooled or held at a constant (isothermal) temperature.Differential Scanning Calorimetry (DSC) measures the amount of energy (heat) absorbed or released by a sample. Thermal effects such as melting, solid-solid transitions or chemical reactions can be studied. The appearance of the sample can be observed under the micro-scope using thermal-optical analysis (TOA).Thermogravimetric Analysis (TGA) measures the change in mass of a sample in a defined at-mosphere. Processes such as evaporation or decomposition can be investigated. Evolved gas-es are analyzed on line (EGA) using hyphenated techniques such as TGA-MS and TGA-FTIR.Thermomechanical Analysis (TMA) measures the dimensional change of a sample under a defined load. Depending on the applied load, softening, creep or expansion is observed.Dynamic Mechanical Analysis (DMA) allows viscoelastic behavior to be studied over a wide frequency range. The technique provides information on mechanical moduli, compliances and damping behavior.The various TA techniques are widely used in research and development as well as in rou-tine analysis for quality control purposes.
Safety investigations of explosivesTo ensure safety in chemical plants, a detailed understand-ing of the thermal behavior of potentially hazardous and explosive materials like 2-nitrophenol is essential.As part of a safety investigation samples of this substance were measured at four different heating rates. The advanced model free kinetics software calculates the activation energy as a function of the reaction conversion. Advanced model free kinetics allows the course of the reaction to be pre-dicted.
Purity of fine chemicalsThe purity of substances is a topic of major importance in the production of fine chemicals. Purity can be determined by evaluating the peak shape of the DSC melting curve. The method is based on the van’t Hoff law of melting point de-pression of eutectic systems. Purity levels between 90 and 100 mol% can be determined rapidly and with good accu-racy. The example below shows the purity determination of ethyl-4-hydroxybenzoate (EHB) contaminated with benzoic acid.
Phase transition of liquid crystalsLiquid crystals undergo reversible phase changes when a voltage is applied at a particular temperature, or as a re-sult of a temperature change. It is therefore very important to characterize the thermal properties of such compounds. The DSC curve shows the phase behavior of cholesteryl myristate, which on heating exhibits three liquid-liquid tran-sitions. If the substance is observed under polarized light, the solvent-crystallized form appears as translucent flakes (a). These change at 71 °C to the smectic form, which dif-fracts the polarized light diffusely (b). At 78 °C the crystals change to the cholesteric form and present a structureless gray picture (c). This phase finally undergoes a transition to an isotropic liquid that no longer transmits polarized light and appears black.
(a) (b) (c)Ther
mal
Ana
lysi
s
• preferred technique • alternative technique
OIT measurements of mineral and synthetic oils by HP DSC
(a) (b) (c)
Phase transitions of cholesteryl myristate measured by DSC and TOA Shear measurement of silicone oil by DMA
DSC measurements and purity evaluation of EHB containing different levels of impurity
DSC curves and kinetic evaluations are used to predict thermal stability Release and uptake of water under controlled relative humidity conditions using TGA

8 METTLER TOLEDO UserCom 2/2006
Neu
im V
erka
ufsp
rogr
amm
Abbildung 1: TGA-FTIR-MS Interface.
Abbildung 1: Kühlgeschwindig-keiten der 3 neuen IntraCooler.
Minimal-Temperatur Maximal-Temperatur
TC45 –35 °C 700 °C
TC100 –85 °C 700 °C
TC125 –100 °C 550 °C
Heute kommt es oft vor, dass man bei einer TGA-Messung die Zersetzungspro-dukte weiter analysieren will um den Zersetzungsvorgang besser verstehen zu können. Meist wird dafür entweder ein Massenspektrometer (MS) oder Fou-
rier-Transformations-IR-Spektroskopie (FTIR) eingesetzt.
Neu gibt es die Möglichkeit, die Zerset-zungsprodukte mittels MS und FTIR gleichzeitig zu messen und zu analysie-
ren. Dazu wird das FTIR wie bisher an das TGA-Gerät gekoppelt. Am Ausgang des FTIR kann man mit einem neuen Interface auf einfache Weise ein MS an-koppeln. Da die FTIR-Gaszelle mit der Transferline nur über ein sehr kleines Volumen verfügt, ist die zeitliche Verzö-gerung zwischen FTIR und MS-Messung vernachlässigbar.
Die Vorteile der Serieschaltung sind:Praktisch keine Verschmutzung der MS-Kapillare Kein Zuschmelzen der MS-Kapillare (ist nicht mehr den hohen Tempera-turen im Ofen der TGA ausgesetzt)
•
•
TGA-FTIR-MS Interface
METTLER TOLEDO kann neue IntraCoo-ler mit einem grösseren Temperaturbe-reich anbieten. Diese neuen IntraCooler sind zu allen DSC82x kompatibel.
IntraCooler sind geschlossene Kühlsys-teme und sind demzufolge praktisch wartungsfrei. Dank den tieferen Mini-maltemperaturen gibt es auch eine Ver-besserung bei den Kühlraten.
IntraCooler
9
Bestellnummern:Mikroskop-Heiztisch Isolation: ME 51 141 438Kaltgasgenerator: ME 51 191 722
METTLER TOLEDO UserCom 2/2006
Für die Mikroskop-Heiztische FP82HT und FP84HT gibt es neu einen Kaltgasge-nerator, mit dem Messungen bis −100 °C möglich sind. Der für die Mikroskop-Heiztische benötigte kalte Dampf wird
mit einem 50-Liter-Flüssigstickstoffbe-hälter durch Verdampfen von flüssigem Stickstoff an dessen Aussenwand erzeugt.Um Vereisungen zu vermeiden, muss der Mikroskop-Heiztisch isoliert werden.
Mikroskop-Heiztisch-Kühlung
In der dynamische Differenzkalorimetrie (DSC) muss zusätzlich auch der Wär-mestrom kalibriert und justiert werden. Dabei gelangen Referenzmaterialien mit einer zertifizierten Schmelzenthalpie zum Einsatz.
Die DSC wird insbesondere in der Phar-maindustrie häufig zur Reinheitsbestim-mung von leicht verunreinigten Substan-zen verwendet. Für die Validierung einer
DSC-Methode zur Reinheitsbestimmung bietet LGC Referenzmaterialen mit zerti-fizierten Verunreinigungen an.
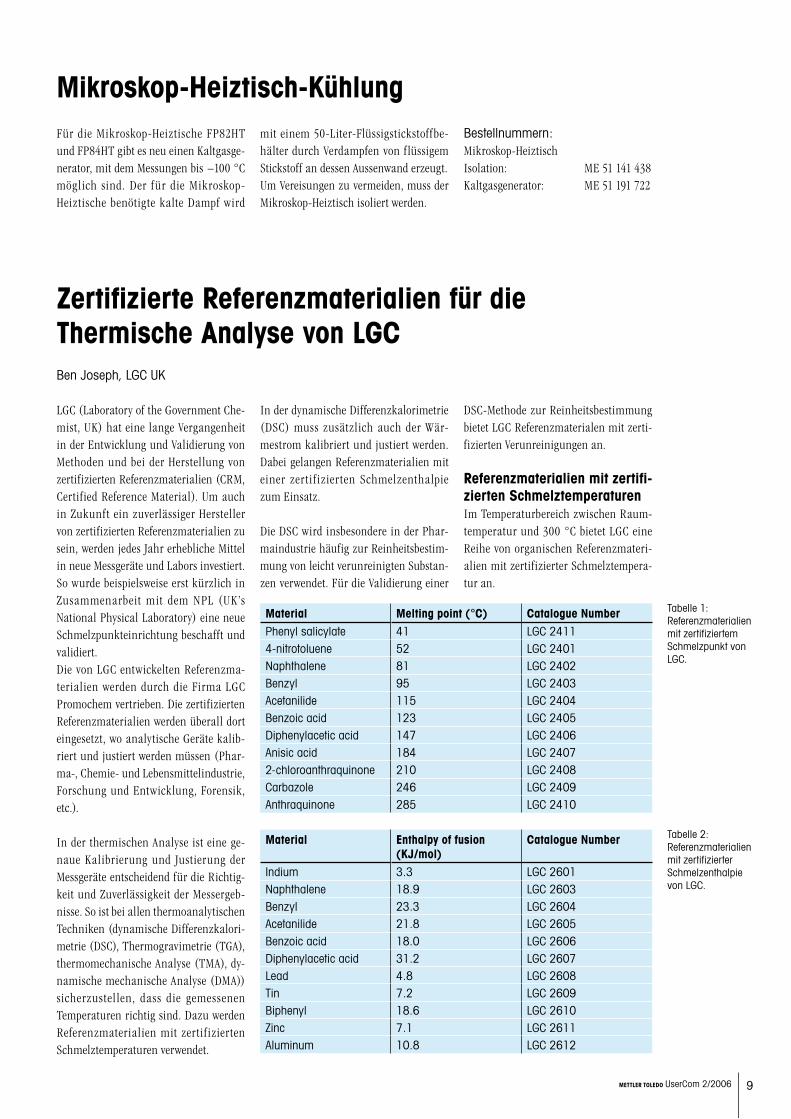
Referenzmaterialien mit zertifi-zierten SchmelztemperaturenIm Temperaturbereich zwischen Raum-temperatur und 300 °C bietet LGC eine Reihe von organischen Referenzmateri-alien mit zertifizierter Schmelztempera-tur an.
LGC (Laboratory of the Government Che-mist, UK) hat eine lange Vergangenheit in der Entwicklung und Validierung von Methoden und bei der Herstellung von zertifizierten Referenzmaterialien (CRM, Certified Reference Material). Um auch in Zukunft ein zuverlässiger Hersteller von zertifizierten Referenzmaterialien zu sein, werden jedes Jahr erhebliche Mittel in neue Messgeräte und Labors investiert. So wurde beispielsweise erst kürzlich in Zusammenarbeit mit dem NPL (UK’s National Physical Laboratory) eine neue Schmelzpunkteinrichtung beschafft und validiert.Die von LGC entwickelten Referenzma-terialien werden durch die Firma LGC Promochem vertrieben. Die zertifizierten Referenzmaterialien werden überall dort eingesetzt, wo analytische Geräte kalib-riert und justiert werden müssen (Phar-ma-, Chemie- und Lebensmittelindustrie, Forschung und Entwicklung, Forensik, etc.).
In der thermischen Analyse ist eine ge-naue Kalibrierung und Justierung der Messgeräte entscheidend für die Richtig-keit und Zuverlässigkeit der Messergeb-nisse. So ist bei allen thermoanalytischen Techniken (dynamische Differenzkalori-metrie (DSC), Thermogravimetrie (TGA), thermomechanische Analyse (TMA), dy-namische mechanische Analyse (DMA)) sicherzustellen, dass die gemessenen Temperaturen richtig sind. Dazu werden Referenzmaterialien mit zertifizierten Schmelztemperaturen verwendet.
Zertifizierte Referenzmaterialien für die Thermische Analyse von LGCBen Joseph, LGC UK
Material Melting point (°C) Catalogue NumberPhenyl salicylate 41 LGC 24114-nitrotoluene 52 LGC 2401Naphthalene 81 LGC 2402Benzyl 95 LGC 2403Acetanilide 115 LGC 2404Benzoic acid 123 LGC 2405Diphenylacetic acid 147 LGC 2406Anisic acid 184 LGC 24072-chloroanthraquinone 210 LGC 2408Carbazole 246 LGC 2409Anthraquinone 285 LGC 2410
Material Enthalpy of fusion (KJ/mol)
Catalogue Number
Indium 3.3 LGC 2601Naphthalene 18.9 LGC 2603Benzyl 23.3 LGC 2604Acetanilide 21.8 LGC 2605Benzoic acid 18.0 LGC 2606Diphenylacetic acid 31.2 LGC 2607Lead 4.8 LGC 2608Tin 7.2 LGC 2609Biphenyl 18.6 LGC 2610Zinc 7.1 LGC 2611Aluminum 10.8 LGC 2612
Tabelle 1: Referenzmaterialien mit zertifiziertem Schmelzpunkt von LGC.
Tabelle 2: Referenzmaterialien mit zertifizierter Schmelzenthalpie von LGC.

10 METTLER TOLEDO UserCom 2/2006
Neu
im V
erka
ufsp
rogr
amm
Material Impurity (mole%) Catalogue NumberBiphenyl 0.1
1.1 1.6 2.1 2.6 3.1
LGC 2013
Tabelle 3: Reinheit von zertifizierten Referenzmaterial.
Referenzmaterialien mit zertifi-zierten Schmelzenthalpien Referenzmaterialien mit zertifizierter Schmelzenthalpie werden von LGC mit einem adiabatischen Kalorimeter ver-messen. Dadurch ergibt sich eine direkte Rückführbarkeit der Schmelzenthalpie auf Kelvin (SI-Einheit der absoluten Temperatur).
Reinheit von zertifizierten ReferenzmaterialFür die Validierung der DSC-Reinheits-methode stellt LGC einen Satz mit 6 Refe-renzmaterialien mit zertifizierten Verun-reinigungen zur Verfügung.
Alle in diesem Artikel erwähnten Refe-renzmaterialien wurden mit der Unter-
stützung des Valid Analytical Measure-ment Program (www.vam.co.uk), der Abteilung für Handel und Industrie in UK vorbereitet und geprüft.Für weitere Informationen wenden Sie sich bitte an LGC Promochem:
LGC PromochemTel: ++44 (0) 20 8943 8480E-mail: [email protected]: www.lgcpromochem.comLGC Promochem - Supporting Laborato-ry Standards
Abbildung 3: Präzisionsgewichte 1 mg bis 500 mg.
Abbildung 1: TA-Präzisions- gewichte.
Präzisionsgewichte für die Thermische AnalyseSpeziell für die Thermische Analyse stellt METTLER TOLEDO ein Set von Präzi- sionsgewichten zur Verfügung, mit der die TGA-Waage mit dem Probenwechsler automatisiert mit kalibrierten Präzisi-onsgewichten kalibriert werden kann. Das neue 200-mg-Gewicht ist so gebaut, dass es mit dem automatischen Proben-wechsler verwendet werden kann.
Die zwei internen Ringgewichte der TGA-Mikrowaage ermöglichen das automa-tische Justieren der Waagenkennlinie. Mit den externen Gewichten lässt sich die Kalibrierung auf das Urgewicht in Paris zurückführen.
Bestellangaben:Gewichtsklasse E2: 11116761 mit Kalib-rierzertifikat in einem Holzetui (0.2 g, 1 g, 5 g).
Präzisionsgewichtssatz 1 mg bis 500 mg Für die Kalibrierung und den Test von Micro-Waagen in dem interessierenden Wägebereich für die thermische Analyse stellt METTLR TOLEDO auch einen Satz mit Präzisionsgewichten von 1 bis 500 mg zur Verfügung.
Bestellangaben:Gewichtsklasse E2: ME- 00158801 mit Kalibrierzertifikat in einem Holzetui (1 bis 500 mg).
Beide Gewichts-Sets bestehen aus einem Holzetui, einer Pinzette und den Präzi-sionsgewichten mit den entsprechenden Zertifikaten.
Weitere Gewichte und Gewichts-Sets finden Sie im Gewichtsprospekt von METTLER TOLEDO (D: 117 95 460, E: 117 95 461, F: 117 95 462).
Präzisionsgewichte (0.2 g, 1 g und 5 g)
Abbildung 2: Präzisions-Ring- gewichte der METTLER TOLEDO Mikro-Waage.
EinleitungIm ersten Teil dieser Artikelserie [1] wer-den die Grundlagen der Entstehung von Peaks in der DSC diskutiert. Es wird ge-zeigt, dass die Peaktemperatur von der Heizrate, der Probenmasse und dem Wär-mekontakt zwischen Probe und Sensor (und somit auch vom verwendeten Tiegel und Gas) abhängt. Ausserdem erhält man unterschiedliche Peaktemperaturen, je nachdem, ob über die Probentemperatur oder die Programmtemperatur (in der STARe Nomenklatur die Referenztem-peratur) ausgewertet wird. Schliesslich wird gezeigt, dass beim Schmelzprozess die Peaktemperatur der Gleichung
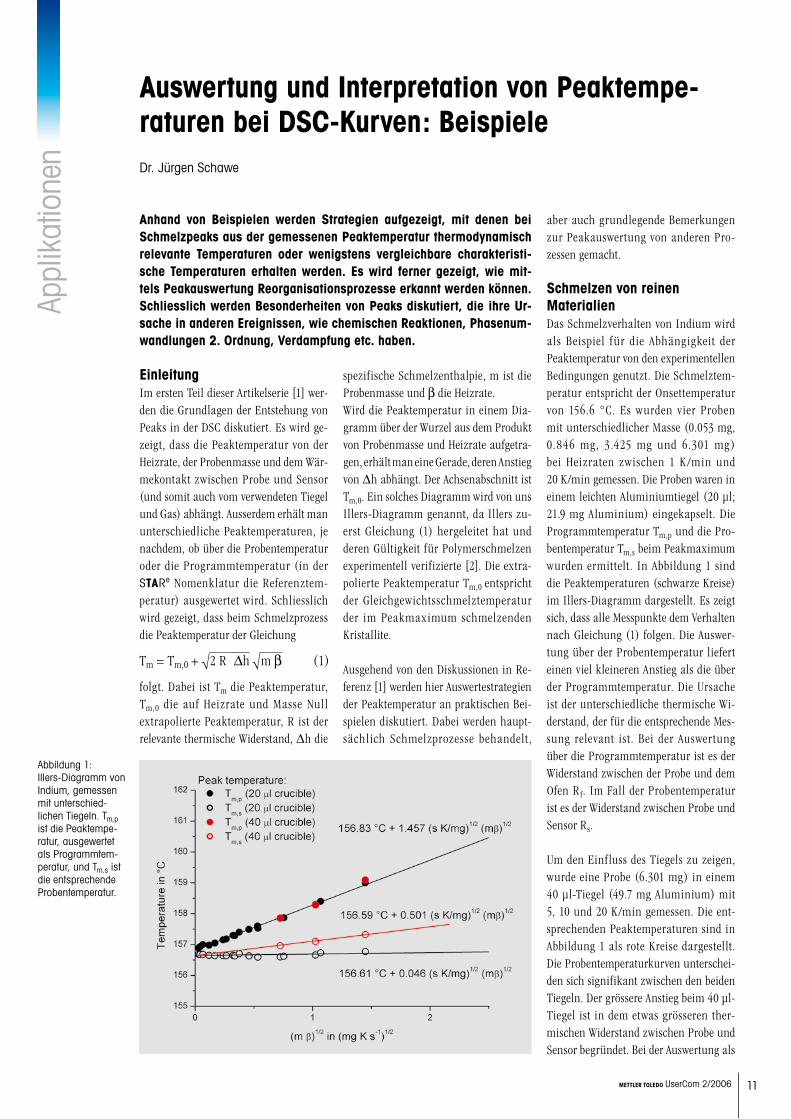
folgt. Dabei ist Tm die Peaktemperatur, Tm,0 die auf Heizrate und Masse Null extrapolierte Peaktemperatur, R ist der relevante thermische Widerstand, Dh die
spezifische Schmelzenthalpie, m ist die Probenmasse und b die Heizrate. Wird die Peaktemperatur in einem Dia-gramm über der Wurzel aus dem Produkt von Probenmasse und Heizrate aufgetra-gen, erhält man eine Gerade, deren Anstieg von Dh abhängt. Der Achsenabschnitt ist Tm,0. Ein solches Diagramm wird von uns Illers-Diagramm genannt, da Illers zu-erst Gleichung (1) hergeleitet hat und deren Gültigkeit für Polymerschmelzen experimentell verifizierte [2]. Die extra-polierte Peaktemperatur Tm,0 entspricht der Gleichgewichtsschmelztemperatur der im Peakmaximum schmelzenden Kristallite.
Ausgehend von den Diskussionen in Re-ferenz [1] werden hier Auswertestrategien der Peaktemperatur an praktischen Bei-spielen diskutiert. Dabei werden haupt-sächlich Schmelzprozesse behandelt,
aber auch grundlegende Bemerkungen zur Peakauswertung von anderen Pro-zessen gemacht.
Schmelzen von reinen MaterialienDas Schmelzverhalten von Indium wird als Beispiel für die Abhängigkeit der Peaktemperatur von den experimentellen Bedingungen genutzt. Die Schmelztem-peratur entspricht der Onsettemperatur von 156.6 °C. Es wurden vier Proben mit unterschiedlicher Masse (0.053 mg, 0.846 mg, 3.425 mg und 6.301 mg) bei Heizraten zwischen 1 K/min und 20 K/min gemessen. Die Proben waren in einem leichten Aluminiumtiegel (20 µl; 21.9 mg Aluminium) eingekapselt. Die Programmtemperatur Tm,p und die Pro-bentemperatur Tm,s beim Peakmaximum wurden ermittelt. In Abbildung 1 sind die Peaktemperaturen (schwarze Kreise) im Illers-Diagramm dargestellt. Es zeigt sich, dass alle Messpunkte dem Verhalten nach Gleichung (1) folgen. Die Auswer-tung über der Probentemperatur liefert einen viel kleineren Anstieg als die über der Programmtemperatur. Die Ursache ist der unterschiedliche thermische Wi-derstand, der für die entsprechende Mes-sung relevant ist. Bei der Auswertung über die Programmtemperatur ist es der Widerstand zwischen der Probe und dem Ofen Rf. Im Fall der Probentemperatur ist es der Widerstand zwischen Probe und Sensor Rs.
Um den Einfluss des Tiegels zu zeigen, wurde eine Probe (6.301 mg) in einem 40 µl-Tiegel (49.7 mg Aluminium) mit 5, 10 und 20 K/min gemessen. Die ent-sprechenden Peaktemperaturen sind in Abbildung 1 als rote Kreise dargestellt. Die Probentemperaturkurven unterschei-den sich signifikant zwischen den beiden Tiegeln. Der grössere Anstieg beim 40 µl-Tiegel ist in dem etwas grösseren ther-mischen Widerstand zwischen Probe und Sensor begründet. Bei der Auswertung als
11
Abbildung 1: Illers-Diagramm von Indium, gemessen mit unterschied-lichen Tiegeln. Tm,p ist die Peaktempe-ratur, ausgewertet als Programmtem-peratur, und Tm,s ist die entsprechende Probentemperatur.
Appl
ikat
ione
n
METTLER TOLEDO UserCom 2/2006
Anhand von Beispielen werden Strategien aufgezeigt, mit denen bei Schmelzpeaks aus der gemessenen Peaktemperatur thermodynamisch relevante Temperaturen oder wenigstens vergleichbare charakteristi-sche Temperaturen erhalten werden. Es wird ferner gezeigt, wie mit-tels Peakauswertung Reorganisationsprozesse erkannt werden können. Schliesslich werden Besonderheiten von Peaks diskutiert, die ihre Ur-sache in anderen Ereignissen, wie chemischen Reaktionen, Phasenum-wandlungen 2. Ordnung, Verdampfung etc. haben.
Auswertung und Interpretation von Peaktempe- raturen bei DSC-Kurven: BeispieleDr. Jürgen Schawe
12 METTLER TOLEDO UserCom 2/2006
Funktion der Programmtemperatur ist der Unterschied zwischen diesen Tiegeln zu vernachlässigen, da der thermische Widerstand im Ofen um ein Vielfaches grösser ist.
Die Ausgleichsgeraden in Abbildung 1 liefern als Achsenabschnitt die Peaktem-peratur extrapoliert für Heizrate Null. Das sollte die tatsächliche Schmelztem-peratur sein. Wie erwartet liegt sie bei beiden Tiegeln bei 156.6 °C. Bei der Aus-wertung über der Programmtemperatur ist dieser Wert etwas höher (156.8 °C), weil die Probentemperatur und nicht die Programmtemperatur kalibriert wurde. Diese Temperaturdifferenz DT von 0.2 K entspricht dem Unterschied zwischen der Probentemperatur Ts und der Programm-temperatur Tp [1].Aus diesen Messungen kann man erken-nen, dass aus der Peaktemperatur die entsprechende Schmelztemperatur bei Extrapolation auf Heizrate Null im Illers- Diagramm bestimmt werden kann. Die Auswertung sollte über die Probentempe-ratur erfolgen. Der Schmelzpeak bei meta- stabilen Kristalliten (Polymeren)Im Gegensatz zum Schmelzverhalten von reinen Metallen, ist der Schmelzpeak von semikristallinen Polymeren häufig sehr breit. Daher liefert eine Auswertung der Onsettemperatur keine repräsenta-tiven und vergleichbaren Werte. Die Ur-sache für den breiten Schmelzbereich ist die Kristallitgrössenverteilung. Kleine
Kristallite schmelzen früher als grosse perfektere Kristallite. Zur Charakterisie-rung des Schmelzverhaltens wird im All-gemeinen die Peaktemperatur genutzt. Analog zum Schmelzen von reinen Me-tallen ist die Peaktemperatur bei diesen breiten Peaks von der Probenmasse und der Heizrate abhängig. Sollen also Mate-rialien lediglich miteinander verglichen werden, sind Messungen an Proben mit ähnlicher Masse und gleicher Heizrate zu vergleichen.
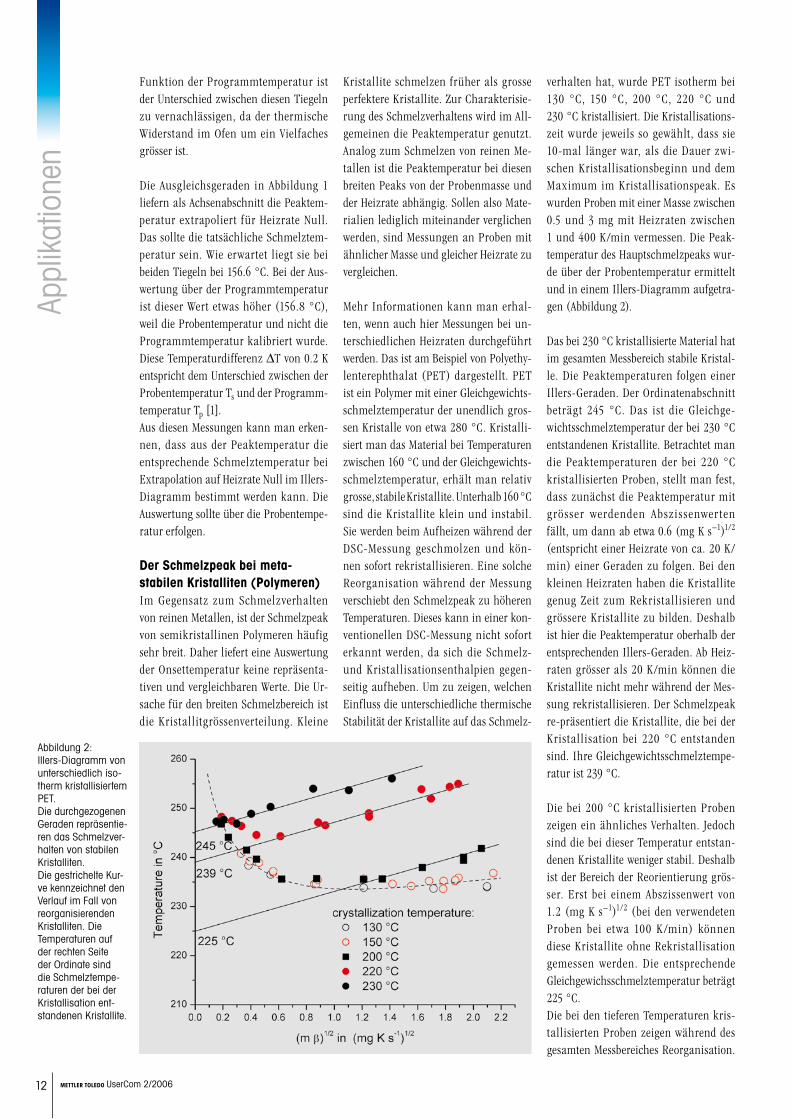
Mehr Informationen kann man erhal-ten, wenn auch hier Messungen bei un-terschiedlichen Heizraten durchgeführt werden. Das ist am Beispiel von Polyethy-lenterephthalat (PET) dargestellt. PET ist ein Polymer mit einer Gleichgewichts-schmelztemperatur der unendlich gros-sen Kristalle von etwa 280 °C. Kristalli-siert man das Material bei Temperaturen zwischen 160 °C und der Gleichgewichts-schmelztemperatur, erhält man relativ grosse, stabile Kristallite. Unterhalb 160 °C sind die Kristallite klein und instabil. Sie werden beim Aufheizen während der DSC-Messung geschmolzen und kön-nen sofort rekristallisieren. Eine solche Reorganisation während der Messung verschiebt den Schmelzpeak zu höheren Temperaturen. Dieses kann in einer kon-ventionellen DSC-Messung nicht sofort erkannt werden, da sich die Schmelz- und Kristallisationsenthalpien gegen-seitig aufheben. Um zu zeigen, welchen Einfluss die unterschiedliche thermische Stabilität der Kristallite auf das Schmelz-
verhalten hat, wurde PET isotherm bei 130 °C, 150 °C, 200 °C, 220 °C und 230 °C kristallisiert. Die Kristallisations-zeit wurde jeweils so gewählt, dass sie 10-mal länger war, als die Dauer zwi-schen Kristallisationsbeginn und dem Maximum im Kristallisationspeak. Es wurden Proben mit einer Masse zwischen 0.5 und 3 mg mit Heizraten zwischen 1 und 400 K/min vermessen. Die Peak-temperatur des Hauptschmelzpeaks wur-de über der Probentemperatur ermittelt und in einem Illers-Diagramm aufgetra-gen (Abbildung 2).
Das bei 230 °C kristallisierte Material hat im gesamten Messbereich stabile Kristal-le. Die Peaktemperaturen folgen einer Illers-Geraden. Der Ordinatenabschnitt beträgt 245 °C. Das ist die Gleichge-wichtsschmelztemperatur der bei 230 °C entstandenen Kristallite. Betrachtet man die Peaktemperaturen der bei 220 °C kristallisierten Proben, stellt man fest, dass zunächst die Peaktemperatur mit grösser werdenden Abszissenwerten fällt, um dann ab etwa 0.6 (mg K s−1)1/2 (entspricht einer Heizrate von ca. 20 K/min) einer Geraden zu folgen. Bei den kleinen Heizraten haben die Kristallite genug Zeit zum Rekristallisieren und grössere Kristallite zu bilden. Deshalb ist hier die Peaktemperatur oberhalb der entsprechenden Illers-Geraden. Ab Heiz-raten grösser als 20 K/min können die Kristallite nicht mehr während der Mes-sung rekristallisieren. Der Schmelzpeak re-präsentiert die Kristallite, die bei der Kristallisation bei 220 °C entstanden sind. Ihre Gleichgewichtsschmelztempe-ratur ist 239 °C.
Die bei 200 °C kristallisierten Proben zeigen ein ähnliches Verhalten. Jedoch sind die bei dieser Temperatur entstan-denen Kristallite weniger stabil. Deshalb ist der Bereich der Reorientierung grös-ser. Erst bei einem Abszissenwert von 1.2 (mg K s−1)1/2 (bei den verwendeten Proben bei etwa 100 K/min) können diese Kristallite ohne Rekristallisation gemessen werden. Die entsprechende Gleichgewichsschmelztemperatur beträgt 225 °C.Die bei den tieferen Temperaturen kris-tallisierten Proben zeigen während des gesamten Messbereiches Reorganisation.
Appl
ikat
ione
n
Abbildung 2: Illers-Diagramm von unterschiedlich iso-therm kristallisiertem PET. Die durchgezogenen Geraden repräsentie-ren das Schmelzver-halten von stabilen Kristalliten. Die gestrichelte Kur-ve kennzeichnet den Verlauf im Fall von reorganisierenden Kristalliten. Die Temperaturen auf der rechten Seite der Ordinate sind die Schmelztempe-raturen der bei der Kristallisation ent-standenen Kristallite.
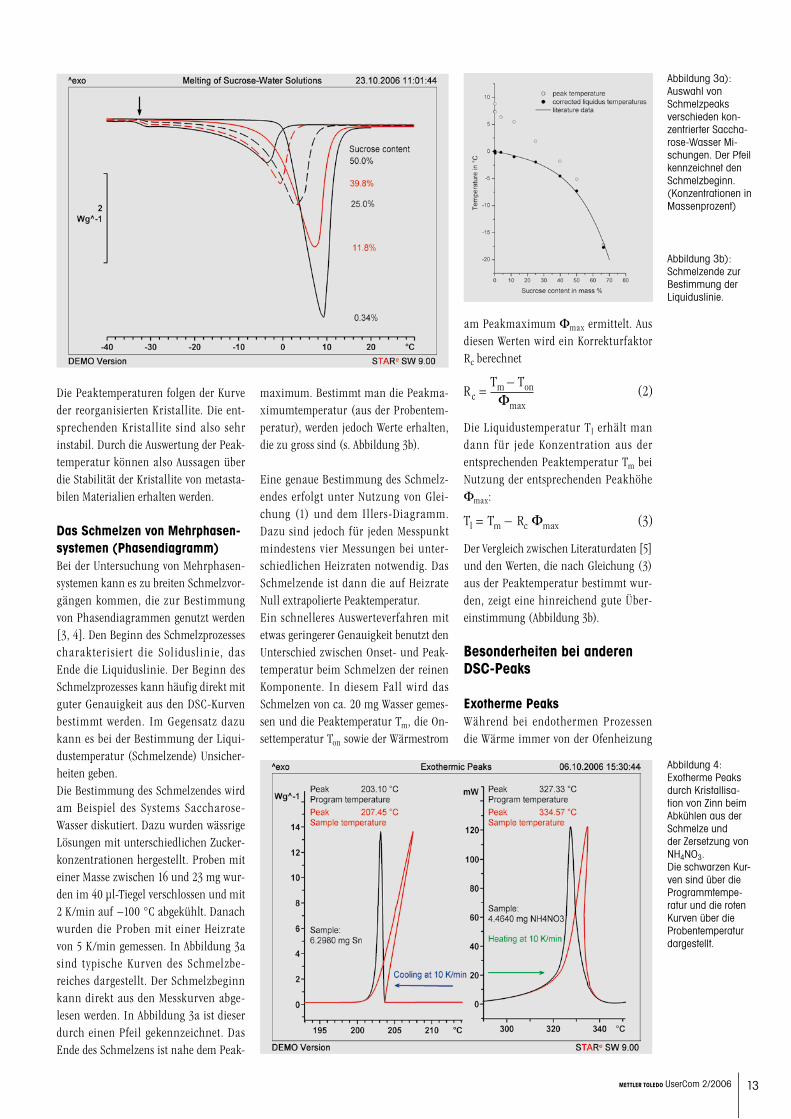
13METTLER TOLEDO UserCom 2/2006
Die Peaktemperaturen folgen der Kurve der reorganisierten Kristallite. Die ent-sprechenden Kristallite sind also sehr instabil. Durch die Auswertung der Peak-temperatur können also Aussagen über die Stabilität der Kristallite von metasta-bilen Materialien erhalten werden.
Das Schmelzen von Mehrphasen-systemen (Phasendiagramm)Bei der Untersuchung von Mehrphasen-systemen kann es zu breiten Schmelzvor-gängen kommen, die zur Bestimmung von Phasendiagrammen genutzt werden [3, 4]. Den Beginn des Schmelzprozesses charakterisiert die Soliduslinie, das Ende die Liquiduslinie. Der Beginn des Schmelzprozesses kann häufig direkt mit guter Genauigkeit aus den DSC-Kurven bestimmt werden. Im Gegensatz dazu kann es bei der Bestimmung der Liqui-dustemperatur (Schmelzende) Unsicher-heiten geben. Die Bestimmung des Schmelzendes wird am Beispiel des Systems Saccharose-Wasser diskutiert. Dazu wurden wässrige Lösungen mit unterschiedlichen Zucker-konzentrationen hergestellt. Proben mit einer Masse zwischen 16 und 23 mg wur-den im 40 µl-Tiegel verschlossen und mit 2 K/min auf −100 °C abgekühlt. Danach wurden die Proben mit einer Heizrate von 5 K/min gemessen. In Abbildung 3a sind typische Kurven des Schmelzbe-reiches dargestellt. Der Schmelzbeginn kann direkt aus den Messkurven abge-lesen werden. In Abbildung 3a ist dieser durch einen Pfeil gekennzeichnet. Das Ende des Schmelzens ist nahe dem Peak-
maximum. Bestimmt man die Peakma-ximumtemperatur (aus der Probentem-peratur), werden jedoch Werte erhalten, die zu gross sind (s. Abbildung 3b).
Eine genaue Bestimmung des Schmelz-endes erfolgt unter Nutzung von Glei-chung (1) und dem Illers-Diagramm. Dazu sind jedoch für jeden Messpunkt mindestens vier Messungen bei unter-schiedlichen Heizraten notwendig. Das Schmelzende ist dann die auf Heizrate Null extrapolierte Peaktemperatur. Ein schnelleres Auswerteverfahren mit etwas geringerer Genauigkeit benutzt den Unterschied zwischen Onset- und Peak-temperatur beim Schmelzen der reinen Komponente. In diesem Fall wird das Schmelzen von ca. 20 mg Wasser gemes-sen und die Peaktemperatur Tm, die On-settemperatur Ton sowie der Wärmestrom
am Peakmaximum Fmax ermittelt. Aus diesen Werten wird ein Korrekturfaktor Rc berechnet
Die Liquidustemperatur Tl erhält man dann für jede Konzentration aus der entsprechenden Peaktemperatur Tm bei Nutzung der entsprechenden Peakhöhe Fmax:
Der Vergleich zwischen Literaturdaten [5] und den Werten, die nach Gleichung (3) aus der Peaktemperatur bestimmt wur-den, zeigt eine hinreichend gute Über-einstimmung (Abbildung 3b).
Besonderheiten bei anderen DSC-Peaks
Exotherme PeaksWährend bei endothermen Prozessen die Wärme immer von der Ofenheizung
Abbildung 3a): Auswahl von Schmelzpeaks verschieden kon-zentrierter Saccha-rose-Wasser Mi-schungen. Der Pfeil kennzeichnet den Schmelzbeginn. (Konzentrationen in Massenprozent)
Abbildung 3b): Schmelzende zur Bestimmung der Liquiduslinie.
Abbildung 4: Exotherme Peaks durch Kristallisa-tion von Zinn beim Abkühlen aus der Schmelze und der Zersetzung von NH4NO3. Die schwarzen Kur-ven sind über die Programmtempe-ratur und die roten Kurven über die Probentemperatur dargestellt.
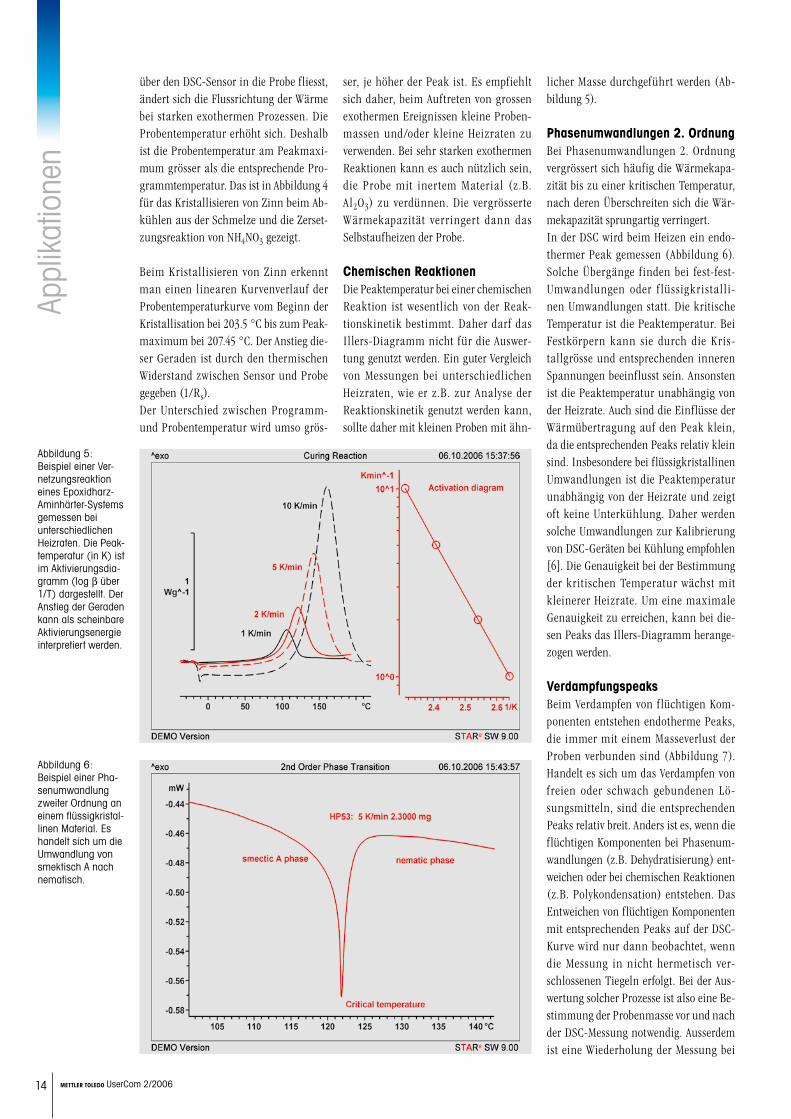
14 METTLER TOLEDO UserCom 2/2006
über den DSC-Sensor in die Probe fliesst, ändert sich die Flussrichtung der Wärme bei starken exothermen Prozessen. Die Probentemperatur erhöht sich. Deshalb ist die Probentemperatur am Peakmaxi-mum grösser als die entsprechende Pro-grammtemperatur. Das ist in Abbildung 4 für das Kristallisieren von Zinn beim Ab-kühlen aus der Schmelze und die Zerset-zungsreaktion von NH4NO3 gezeigt.
Beim Kristallisieren von Zinn erkennt man einen linearen Kurvenverlauf der Probentemperaturkurve vom Beginn der Kristallisation bei 203.5 °C bis zum Peak-maximum bei 207.45 °C. Der Anstieg die-ser Geraden ist durch den thermischen Widerstand zwischen Sensor und Probe gegeben (1/Rs). Der Unterschied zwischen Programm- und Probentemperatur wird umso grös-
ser, je höher der Peak ist. Es empfiehlt sich daher, beim Auftreten von grossen exothermen Ereignissen kleine Proben-massen und/oder kleine Heizraten zu verwenden. Bei sehr starken exothermen Reaktionen kann es auch nützlich sein, die Probe mit inertem Material (z.B. Al2O3) zu verdünnen. Die vergrösserte Wärmekapazität verringert dann das Selbstaufheizen der Probe.
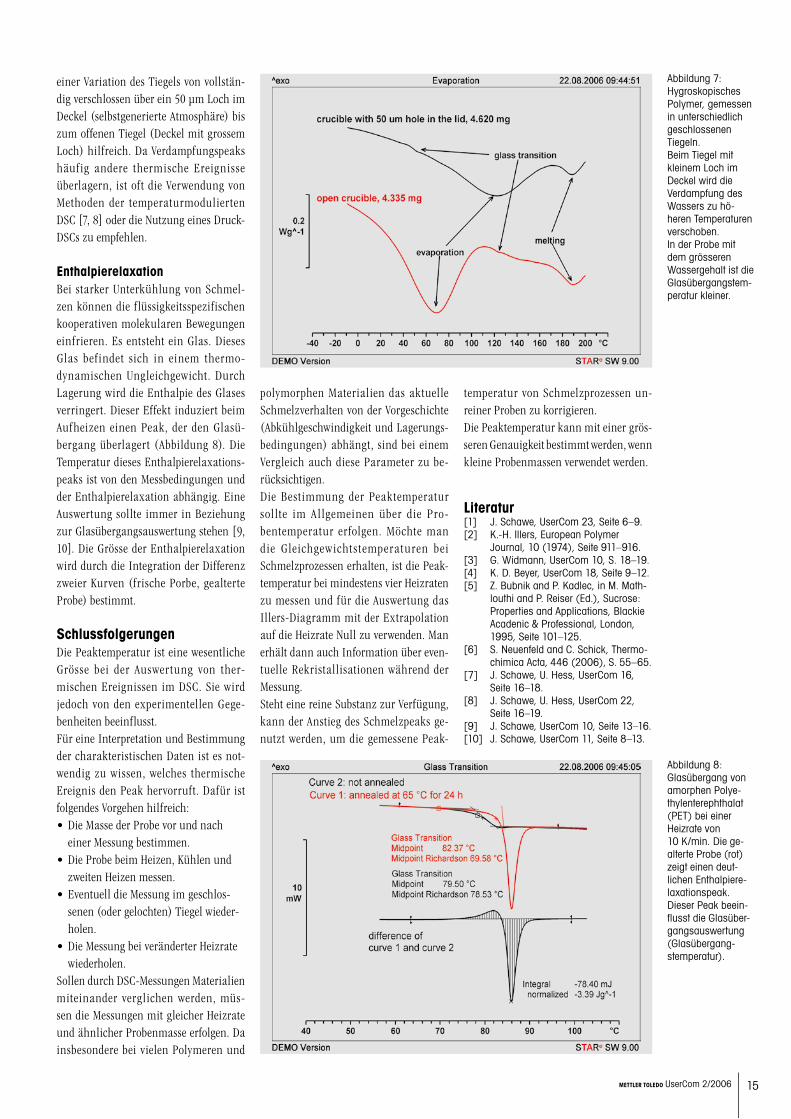
Chemischen ReaktionenDie Peaktemperatur bei einer chemischen Reaktion ist wesentlich von der Reak- tionskinetik bestimmt. Daher darf das Illers-Diagramm nicht für die Auswer-tung genutzt werden. Ein guter Vergleich von Messungen bei unterschiedlichen Heizraten, wie er z.B. zur Analyse der Reaktionskinetik genutzt werden kann, sollte daher mit kleinen Proben mit ähn-
licher Masse durchgeführt werden (Ab-bildung 5).
Phasenumwandlungen 2. OrdnungBei Phasenumwandlungen 2. Ordnung vergrössert sich häufig die Wärmekapa-zität bis zu einer kritischen Temperatur, nach deren Überschreiten sich die Wär-mekapazität sprungartig verringert. In der DSC wird beim Heizen ein endo-thermer Peak gemessen (Abbildung 6). Solche Übergänge finden bei fest-fest-Umwandlungen oder flüssigkristalli-nen Umwandlungen statt. Die kritische Temperatur ist die Peaktemperatur. Bei Festkörpern kann sie durch die Kris-tallgrösse und entsprechenden inneren Spannungen beeinflusst sein. Ansonsten ist die Peaktemperatur unabhängig von der Heizrate. Auch sind die Einflüsse der Wärmübertragung auf den Peak klein, da die entsprechenden Peaks relativ klein sind. Insbesondere bei flüssigkristallinen Umwandlungen ist die Peaktemperatur unabhängig von der Heizrate und zeigt oft keine Unterkühlung. Daher werden solche Umwandlungen zur Kalibrierung von DSC-Geräten bei Kühlung empfohlen [6]. Die Genauigkeit bei der Bestimmung der kritischen Temperatur wächst mit kleinerer Heizrate. Um eine maximale Genauigkeit zu erreichen, kann bei die-sen Peaks das Illers-Diagramm herange-zogen werden.
VerdampfungspeaksBeim Verdampfen von flüchtigen Kom-ponenten entstehen endotherme Peaks, die immer mit einem Masseverlust der Proben verbunden sind (Abbildung 7). Handelt es sich um das Verdampfen von freien oder schwach gebundenen Lö-sungsmitteln, sind die entsprechenden Peaks relativ breit. Anders ist es, wenn die flüchtigen Komponenten bei Phasenum-wandlungen (z.B. Dehydratisierung) ent-weichen oder bei chemischen Reaktionen (z.B. Polykondensation) entstehen. Das Entweichen von flüchtigen Komponenten mit entsprechenden Peaks auf der DSC-Kurve wird nur dann beobachtet, wenn die Messung in nicht hermetisch ver-schlossenen Tiegeln erfolgt. Bei der Aus-wertung solcher Prozesse ist also eine Be-stimmung der Probenmasse vor und nach der DSC-Messung notwendig. Ausserdem ist eine Wiederholung der Messung bei
Appl
ikat
ione
n
Abbildung 6: Beispiel einer Pha-senumwandlung zweiter Ordnung an einem flüssigkristal-linen Material. Es handelt sich um die Umwandlung von smektisch A nach nematisch.
Abbildung 5: Beispiel einer Ver-netzungsreaktion eines Epoxidharz-Aminhärter-Systems gemessen bei unterschiedlichen Heizraten. Die Peak-temperatur (in K) ist im Aktivierungsdia-gramm (log b über 1/T) dargestellt. Der Anstieg der Geraden kann als scheinbare Aktivierungsenergie interpretiert werden.
15METTLER TOLEDO UserCom 2/2006
einer Variation des Tiegels von vollstän-dig verschlossen über ein 50 µm Loch im Deckel (selbstgenerierte Atmosphäre) bis zum offenen Tiegel (Deckel mit grossem Loch) hilfreich. Da Verdampfungspeaks häufig andere thermische Ereignisse überlagern, ist oft die Verwendung von Methoden der temperaturmodulierten DSC [7, 8] oder die Nutzung eines Druck-DSCs zu empfehlen.
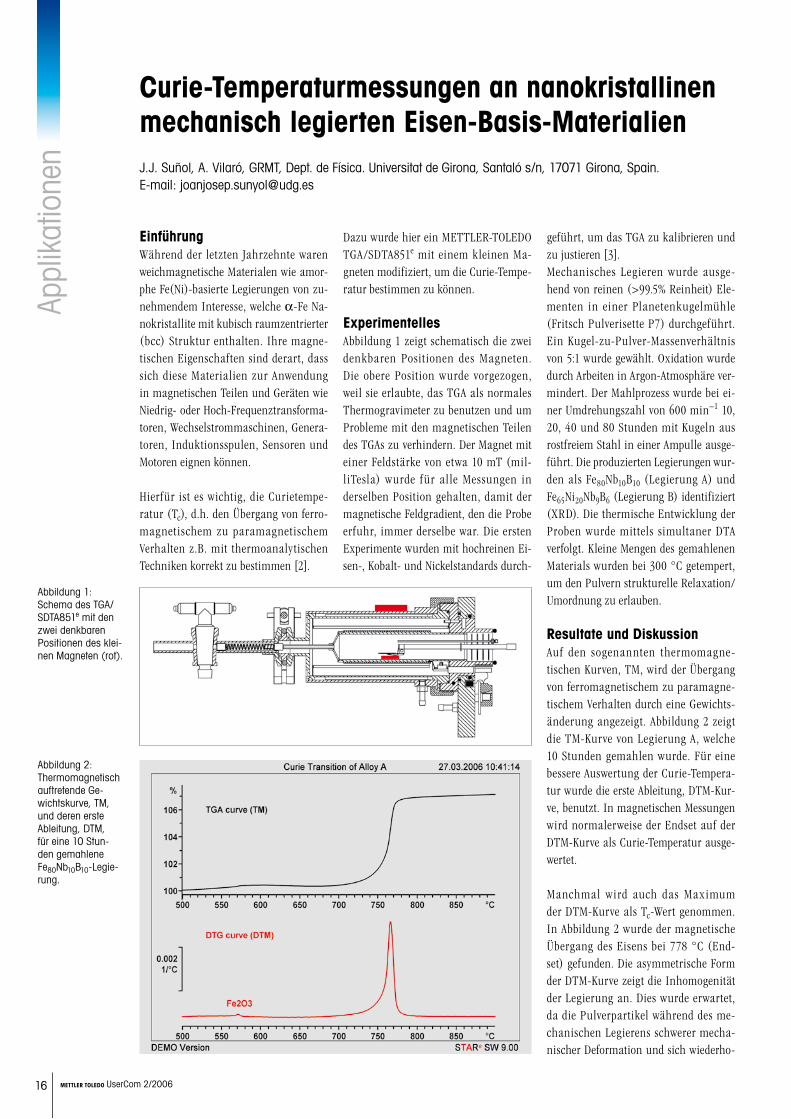
EnthalpierelaxationBei starker Unterkühlung von Schmel-zen können die flüssigkeitsspezifischen kooperativen molekularen Bewegungen einfrieren. Es entsteht ein Glas. Dieses Glas befindet sich in einem thermo-dynamischen Ungleichgewicht. Durch Lagerung wird die Enthalpie des Glases verringert. Dieser Effekt induziert beim Aufheizen einen Peak, der den Glasü-bergang überlagert (Abbildung 8). Die Temperatur dieses Enthalpierelaxations-peaks ist von den Messbedingungen und der Enthalpierelaxation abhängig. Eine Auswertung sollte immer in Beziehung zur Glasübergangsauswertung stehen [9, 10]. Die Grösse der Enthalpierelaxation wird durch die Integration der Differenz zweier Kurven (frische Porbe, gealterte Probe) bestimmt.
SchlussfolgerungenDie Peaktemperatur ist eine wesentliche Grösse bei der Auswertung von ther-mischen Ereignissen im DSC. Sie wird jedoch von den experimentellen Gege-benheiten beeinflusst. Für eine Interpretation und Bestimmung der charakteristischen Daten ist es not-wendig zu wissen, welches thermische Ereignis den Peak hervorruft. Dafür ist folgendes Vorgehen hilfreich:
Die Masse der Probe vor und nach einer Messung bestimmen.Die Probe beim Heizen, Kühlen und zweiten Heizen messen.Eventuell die Messung im geschlos-senen (oder gelochten) Tiegel wieder-holen.Die Messung bei veränderter Heizrate wiederholen.
Sollen durch DSC-Messungen Materialien miteinander verglichen werden, müs-sen die Messungen mit gleicher Heizrate und ähnlicher Probenmasse erfolgen. Da insbesondere bei vielen Polymeren und
•
•
•
•
polymorphen Materialien das aktuelle Schmelzverhalten von der Vorgeschichte (Abkühlgeschwindigkeit und Lagerungs-bedingungen) abhängt, sind bei einem Vergleich auch diese Parameter zu be-rücksichtigen.Die Bestimmung der Peaktemperatur sollte im Allgemeinen über die Pro-bentemperatur erfolgen. Möchte man die Gleichgewichtstemperaturen bei Schmelzprozessen erhalten, ist die Peak-temperatur bei mindestens vier Heizraten zu messen und für die Auswertung das Illers-Diagramm mit der Extrapolation auf die Heizrate Null zu verwenden. Man erhält dann auch Information über even-tuelle Rekristallisationen während der Messung.Steht eine reine Substanz zur Verfügung, kann der Anstieg des Schmelzpeaks ge-nutzt werden, um die gemessene Peak-
temperatur von Schmelzprozessen un-reiner Proben zu korrigieren.Die Peaktemperatur kann mit einer grös-seren Genauigkeit bestimmt werden, wenn kleine Probenmassen verwendet werden.
Literatur[1] J. Schawe, UserCom 23, Seite 6–9.[2] K.-H. Illers, European Polymer
Journal, 10 (1974), Seite 911–916.[3] G. Widmann, UserCom 10, S. 18–19.[4] K. D. Beyer, UserCom 18, Seite 9–12.[5] Z. Bubnik and P. Kadlec, in M. Math-
louthi and P. Reiser (Ed.), Sucrose: Properties and Applications, Blackie Acadenic & Professional, London, 1995, Seite 101–125.
[6] S. Neuenfeld and C. Schick, Thermo-chimica Acta, 446 (2006), S. 55–65.
[7] J. Schawe, U. Hess, UserCom 16, Seite 16–18.
[8] J. Schawe, U. Hess, UserCom 22, Seite 16–19.
[9] J. Schawe, UserCom 10, Seite 13–16.[10] J. Schawe, UserCom 11, Seite 8–13.
Abbildung 7: Hygroskopisches Polymer, gemessen in unterschiedlich geschlossenen Tiegeln. Beim Tiegel mit kleinem Loch im Deckel wird die Verdampfung des Wassers zu hö-heren Temperaturen verschoben. In der Probe mit dem grösseren Wassergehalt ist die Glasübergangstem-peratur kleiner.
Abbildung 8: Glasübergang von amorphen Polye-thylenterephthalat (PET) bei einer Heizrate von 10 K/min. Die ge- alterte Probe (rot) zeigt einen deut-lichen Enthalpiere-laxationspeak. Dieser Peak beein-flusst die Glasüber-gangsauswertung (Glasübergang-stemperatur).
16 METTLER TOLEDO UserCom 2/2006
EinführungWährend der letzten Jahrzehnte waren weichmagnetische Materialen wie amor-phe Fe(Ni)-basierte Legierungen von zu-nehmendem Interesse, welche a-Fe Na-nokristallite mit kubisch raumzentrierter (bcc) Struktur enthalten. Ihre magne-tischen Eigenschaften sind derart, dass sich diese Materialien zur Anwendung in magnetischen Teilen und Geräten wie Niedrig- oder Hoch-Frequenztransforma-toren, Wechselstrommaschinen, Genera-toren, Induktionsspulen, Sensoren und Motoren eignen können.
Hierfür ist es wichtig, die Curietempe-ratur (Tc), d.h. den Übergang von ferro-magnetischem zu paramagnetischem Verhalten z.B. mit thermoanalytischen Techniken korrekt zu bestimmen [2].
Dazu wurde hier ein METTLER-TOLEDO TGA/SDTA851e mit einem kleinen Ma-gneten modifiziert, um die Curie-Tempe-ratur bestimmen zu können.
ExperimentellesAbbildung 1 zeigt schematisch die zwei denkbaren Positionen des Magneten. Die obere Position wurde vorgezogen, weil sie erlaubte, das TGA als normales Thermogravimeter zu benutzen und um Probleme mit den magnetischen Teilen des TGAs zu verhindern. Der Magnet mit einer Feldstärke von etwa 10 mT (mil-liTesla) wurde für alle Messungen in derselben Position gehalten, damit der magnetische Feldgradient, den die Probe erfuhr, immer derselbe war. Die ersten Experimente wurden mit hochreinen Ei-sen-, Kobalt- und Nickelstandards durch-
geführt, um das TGA zu kalibrieren und zu justieren [3].Mechanisches Legieren wurde ausge-hend von reinen (>99.5% Reinheit) Ele-menten in einer Planetenkugelmühle (Fritsch Pulverisette P7) durchgeführt. Ein Kugel-zu-Pulver-Massenverhältnis von 5:1 wurde gewählt. Oxidation wurde durch Arbeiten in Argon-Atmosphäre ver-mindert. Der Mahlprozess wurde bei ei-ner Umdrehungszahl von 600 min−1 10, 20, 40 und 80 Stunden mit Kugeln aus rostfreiem Stahl in einer Ampulle ausge-führt. Die produzierten Legierungen wur-den als Fe80Nb10B10 (Legierung A) und Fe65Ni20Nb9B6 (Legierung B) identifiziert (XRD). Die thermische Entwicklung der Proben wurde mittels simultaner DTA verfolgt. Kleine Mengen des gemahlenen Materials wurden bei 300 °C getempert, um den Pulvern strukturelle Relaxation/Umordnung zu erlauben.
Resultate und DiskussionAuf den sogenannten thermomagne-tischen Kurven, TM, wird der Übergang von ferromagnetischem zu paramagne-tischem Verhalten durch eine Gewichts-änderung angezeigt. Abbildung 2 zeigt die TM-Kurve von Legierung A, welche 10 Stunden gemahlen wurde. Für eine bessere Auswertung der Curie-Tempera-tur wurde die erste Ableitung, DTM-Kur-ve, benutzt. In magnetischen Messungen wird normalerweise der Endset auf der DTM-Kurve als Curie-Temperatur ausge-wertet.
Manchmal wird auch das Maximum der DTM-Kurve als Tc-Wert genommen. In Abbildung 2 wurde der magnetische Übergang des Eisens bei 778 °C (End-set) gefunden. Die asymmetrische Form der DTM-Kurve zeigt die Inhomogenität der Legierung an. Dies wurde erwartet, da die Pulverpartikel während des me-chanischen Legierens schwerer mecha-nischer Deformation und sich wiederho-
Appl
ikat
ione
n
Abbildung 2: Thermomagnetisch auftretende Ge-wichtskurve, TM, und deren erste Ableitung, DTM, für eine 10 Stun-den gemahlene Fe80Nb10B10-Legie-rung.
Abbildung 1: Schema des TGA/SDTA851e mit den zwei denkbaren Positionen des klei-nen Magneten (rot).
Curie-Temperaturmessungen an nanokristallinen mechanisch legierten Eisen-Basis-MaterialienJ.J. Suñol, A. Vilaró, GRMT, Dept. de Física. Universitat de Girona, Santaló s/n, 17071 Girona, Spain. E-mail: [email protected]
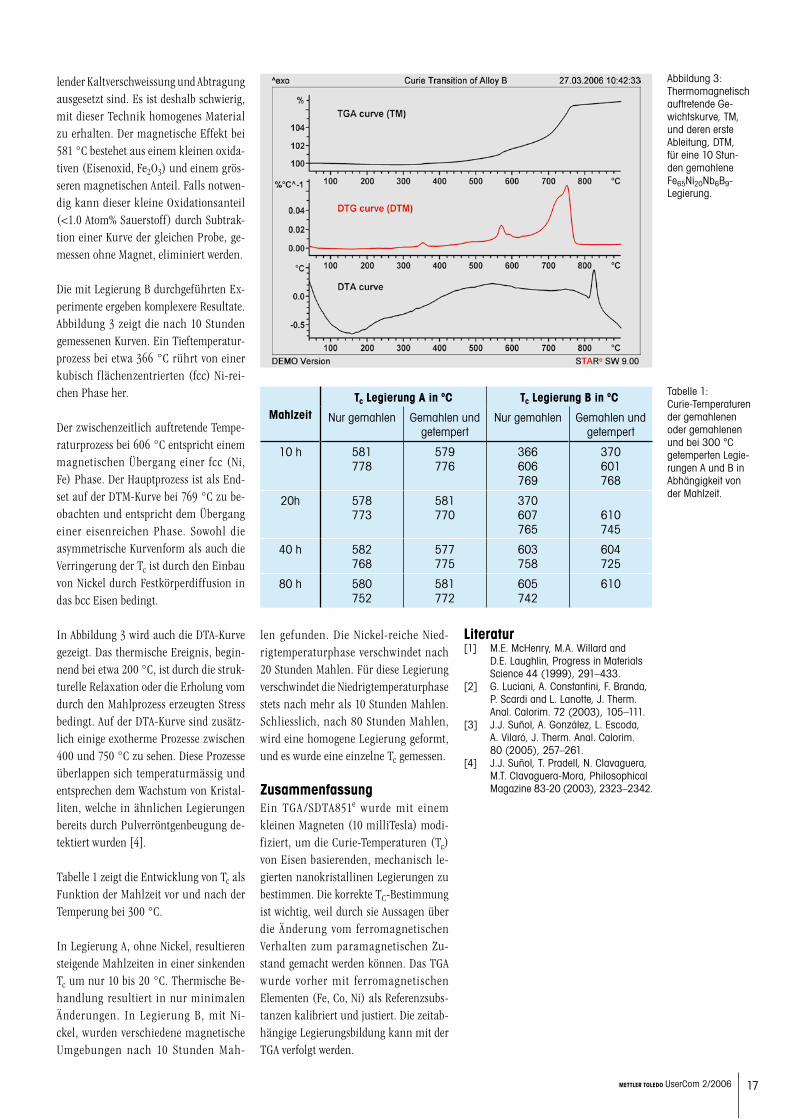
17METTLER TOLEDO UserCom 2/2006
Abbildung 3: Thermomagnetisch auftretende Ge-wichtskurve, TM, und deren erste Ableitung, DTM, für eine 10 Stun-den gemahlene Fe65Ni20Nb6B9- Legierung.
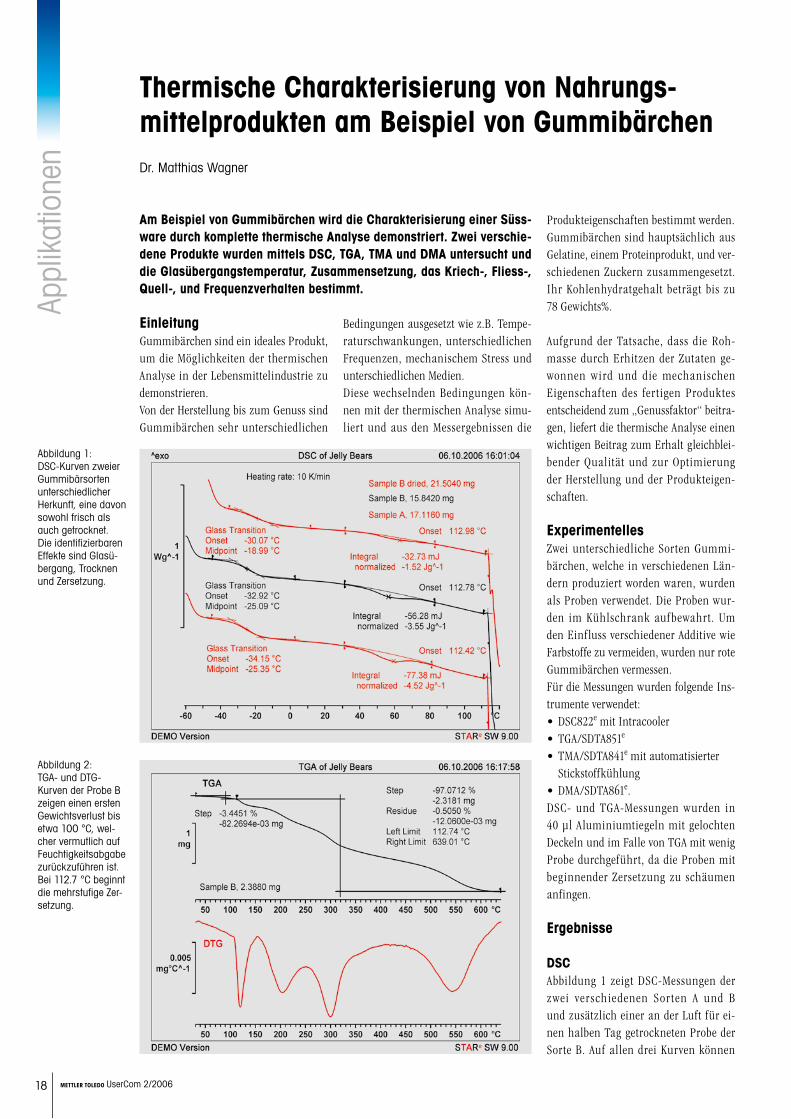
Tabelle 1: Curie-Temperaturen der gemahlenen oder gemahlenen und bei 300 °C getemperten Legie-rungen A und B in Abhängigkeit von der Mahlzeit.
lender Kaltverschweissung und Abtragung ausgesetzt sind. Es ist deshalb schwierig, mit dieser Technik homogenes Material zu erhalten. Der magnetische Effekt bei 581 °C bestehet aus einem kleinen oxida-tiven (Eisenoxid, Fe2O3) und einem grös-seren magnetischen Anteil. Falls notwen-dig kann dieser kleine Oxidationsanteil (<1.0 Atom% Sauerstoff) durch Subtrak-tion einer Kurve der gleichen Probe, ge-messen ohne Magnet, eliminiert werden.
Die mit Legierung B durchgeführten Ex-perimente ergeben komplexere Resultate. Abbildung 3 zeigt die nach 10 Stunden gemessenen Kurven. Ein Tieftemperatur-prozess bei etwa 366 °C rührt von einer kubisch flächenzentrierten (fcc) Ni-rei-chen Phase her.
Der zwischenzeitlich auftretende Tempe-raturprozess bei 606 °C entspricht einem magnetischen Übergang einer fcc (Ni, Fe) Phase. Der Hauptprozess ist als End-set auf der DTM-Kurve bei 769 °C zu be-obachten und entspricht dem Übergang einer eisenreichen Phase. Sowohl die asymmetrische Kurvenform als auch die Verringerung der Tc ist durch den Einbau von Nickel durch Festkörperdiffusion in das bcc Eisen bedingt.
In Abbildung 3 wird auch die DTA-Kurve gezeigt. Das thermische Ereignis, begin-nend bei etwa 200 °C, ist durch die struk-turelle Relaxation oder die Erholung vom durch den Mahlprozess erzeugten Stress bedingt. Auf der DTA-Kurve sind zusätz-lich einige exotherme Prozesse zwischen 400 und 750 °C zu sehen. Diese Prozesse überlappen sich temperaturmässig und entsprechen dem Wachstum von Kristal-liten, welche in ähnlichen Legierungen bereits durch Pulverröntgenbeugung de-tektiert wurden [4].
Tabelle 1 zeigt die Entwicklung von Tc als Funktion der Mahlzeit vor und nach der Temperung bei 300 °C.
In Legierung A, ohne Nickel, resultieren steigende Mahlzeiten in einer sinkenden Tc um nur 10 bis 20 °C. Thermische Be-handlung resultiert in nur minimalen Änderungen. In Legierung B, mit Ni-ckel, wurden verschiedene magnetische Umgebungen nach 10 Stunden Mah-
len gefunden. Die Nickel-reiche Nied- rigtemperaturphase verschwindet nach 20 Stunden Mahlen. Für diese Legierung verschwindet die Niedrigtemperaturphase stets nach mehr als 10 Stunden Mahlen. Schliesslich, nach 80 Stunden Mahlen, wird eine homogene Legierung geformt, und es wurde eine einzelne Tc gemessen.
ZusammenfassungEin TGA/SDTA851e wurde mit einem kleinen Magneten (10 milliTesla) modi-fiziert, um die Curie-Temperaturen (Tc) von Eisen basierenden, mechanisch le-gierten nanokristallinen Legierungen zu bestimmen. Die korrekte TC-Bestimmung ist wichtig, weil durch sie Aussagen über die Änderung vom ferromagnetischen Verhalten zum paramagnetischen Zu-stand gemacht werden können. Das TGA wurde vorher mit ferromagnetischen Elementen (Fe, Co, Ni) als Referenzsubs-tanzen kalibriert und justiert. Die zeitab-hängige Legierungsbildung kann mit der TGA verfolgt werden.
Literatur[1] M.E. McHenry, M.A. Willard and
D.E. Laughlin, Progress in Materials Science 44 (1999), 291–433.
[2] G. Luciani, A. Constantini, F. Branda, P. Scardi and L. Lanotte, J. Therm. Anal. Calorim. 72 (2003), 105–111.
[3] J.J. Suñol, A. González, L. Escoda, A. Vilaró, J. Therm. Anal. Calorim. 80 (2005), 257–261.
[4] J.J. Suñol, T. Pradell, N. Clavaguera, M.T. Clavaguera-Mora, Philosophical Magazine 83-20 (2003), 2323–2342.
MahlzeitTc Legierung A in ºC Tc Legierung B in ºC
Nur gemahlen Gemahlen und getempert
Nur gemahlen Gemahlen und getempert
10 h 581 778
579 776
366 606 769
370 601 768
20h 578 773
581 770
370 607 765
610 745
40 h 582 768
577 775
603 758
604 725
80 h 580 752
581 772
605 742
610
18 METTLER TOLEDO UserCom 2/2006
EinleitungGummibärchen sind ein ideales Produkt, um die Möglichkeiten der thermischen Analyse in der Lebensmittelindustrie zu demonstrieren.Von der Herstellung bis zum Genuss sind Gummibärchen sehr unterschiedlichen
Bedingungen ausgesetzt wie z.B. Tempe-raturschwankungen, unterschiedlichen Frequenzen, mechanischem Stress und unterschiedlichen Medien.Diese wechselnden Bedingungen kön- nen mit der thermischen Analyse simu-liert und aus den Messergebnissen die
Produkteigenschaften bestimmt werden.Gummibärchen sind hauptsächlich aus Gelatine, einem Proteinprodukt, und ver-schiedenen Zuckern zusammengesetzt. Ihr Kohlenhydratgehalt beträgt bis zu 78 Gewichts%.
Aufgrund der Tatsache, dass die Roh-masse durch Erhitzen der Zutaten ge-wonnen wird und die mechanischen Eigenschaften des fertigen Produktes entscheidend zum „Genussfaktor“ beitra-gen, liefert die thermische Analyse einen wichtigen Beitrag zum Erhalt gleichblei-bender Qualität und zur Optimierung der Herstellung und der Produkteigen-schaften.
ExperimentellesZwei unterschiedliche Sorten Gummi-bärchen, welche in verschiedenen Län-dern produziert worden waren, wurden als Proben verwendet. Die Proben wur-den im Kühlschrank aufbewahrt. Um den Einfluss verschiedener Additive wie Farbstoffe zu vermeiden, wurden nur rote Gummibärchen vermessen.Für die Messungen wurden folgende Ins-trumente verwendet:
DSC822e mit IntracoolerTGA/SDTA851e
TMA/SDTA841e mit automatisierter StickstoffkühlungDMA/SDTA861e.
DSC- und TGA-Messungen wurden in 40 µl Aluminiumtiegeln mit gelochten Deckeln und im Falle von TGA mit wenig Probe durchgeführt, da die Proben mit beginnender Zersetzung zu schäumen anfingen.
Ergebnisse
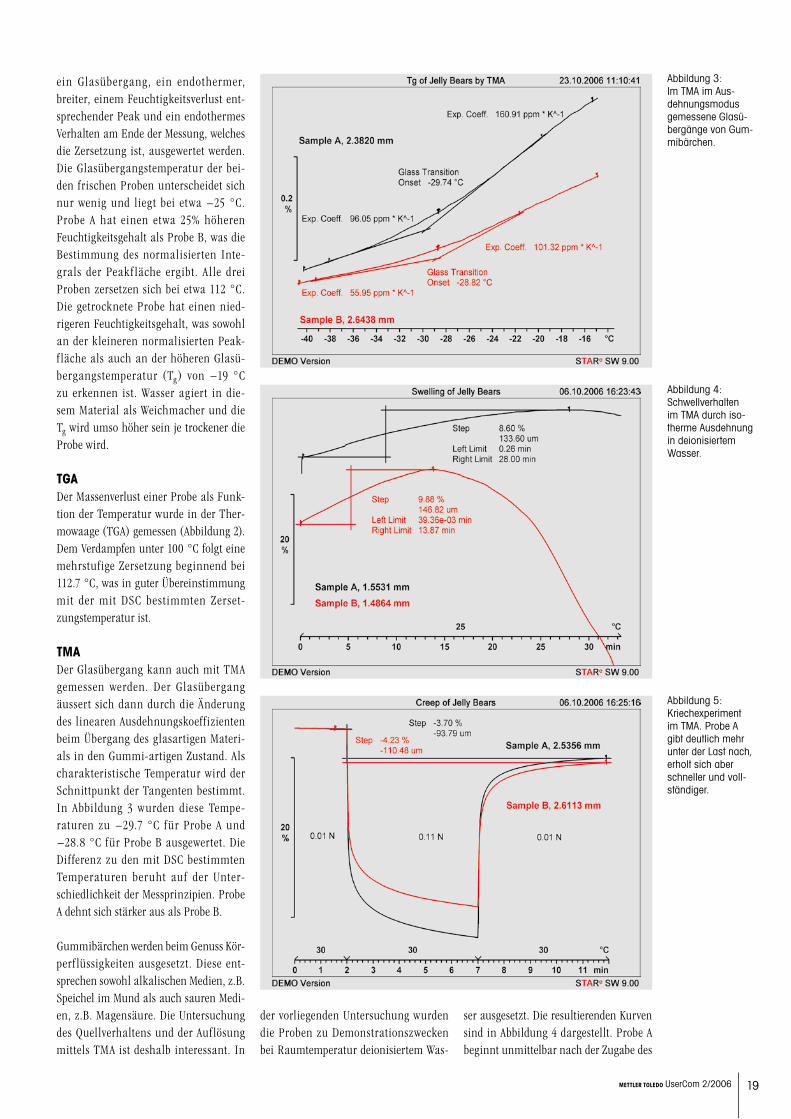
DSCAbbildung 1 zeigt DSC-Messungen der zwei verschiedenen Sorten A und B und zusätzlich einer an der Luft für ei-nen halben Tag getrockneten Probe der Sorte B. Auf allen drei Kurven können
•••
•
Appl
ikat
ione
n
Abbildung 1: DSC-Kurven zweier Gummibärsorten unterschiedlicher Herkunft, eine davon sowohl frisch als auch getrocknet. Die identifizierbaren Effekte sind Glasü-bergang, Trocknen und Zersetzung.
Abbildung 2: TGA- und DTG-Kurven der Probe B zeigen einen ersten Gewichtsverlust bis etwa 100 °C, wel-cher vermutlich auf Feuchtigkeitsabgabe zurückzuführen ist. Bei 112.7 °C beginnt die mehrstufige Zer-setzung.
Am Beispiel von Gummibärchen wird die Charakterisierung einer Süss-ware durch komplette thermische Analyse demonstriert. Zwei verschie-dene Produkte wurden mittels DSC, TGA, TMA und DMA untersucht und die Glasübergangstemperatur, Zusammensetzung, das Kriech-, Fliess-, Quell-, und Frequenzverhalten bestimmt.
Thermische Charakterisierung von Nahrungs- mittelprodukten am Beispiel von GummibärchenDr. Matthias Wagner
19METTLER TOLEDO UserCom 2/2006
der vorliegenden Untersuchung wurden die Proben zu Demonstrationszwecken bei Raumtemperatur deionisiertem Was-
ser ausgesetzt. Die resultierenden Kurven sind in Abbildung 4 dargestellt. Probe A beginnt unmittelbar nach der Zugabe des
Abbildung 3: Im TMA im Aus-dehnungsmodus gemessene Glasü-bergänge von Gum-mibärchen.
Abbildung 4: Schwellverhalten im TMA durch iso-therme Ausdehnung in deionisiertem Wasser.
Abbildung 5: Kriechexperiment im TMA. Probe A gibt deutlich mehr unter der Last nach, erholt sich aber schneller und voll-ständiger.
ein Glasübergang, ein endothermer, breiter, einem Feuchtigkeitsverlust ent-sprechender Peak und ein endothermes Verhalten am Ende der Messung, welches die Zersetzung ist, ausgewertet werden. Die Glasübergangstemperatur der bei-den frischen Proben unterscheidet sich nur wenig und liegt bei etwa −25 °C. Probe A hat einen etwa 25% höheren Feuchtigkeitsgehalt als Probe B, was die Bestimmung des normalisierten Inte-grals der Peakfläche ergibt. Alle drei Proben zersetzen sich bei etwa 112 °C. Die getrocknete Probe hat einen nied-rigeren Feuchtigkeitsgehalt, was sowohl an der kleineren normalisierten Peak-fläche als auch an der höheren Glasü-bergangstemperatur (Tg) von −19 °C zu erkennen ist. Wasser agiert in die-sem Material als Weichmacher und die Tg wird umso höher sein je trockener die Probe wird.
TGADer Massenverlust einer Probe als Funk-tion der Temperatur wurde in der Ther-mowaage (TGA) gemessen (Abbildung 2). Dem Verdampfen unter 100 °C folgt eine mehrstufige Zersetzung beginnend bei 112.7 °C, was in guter Übereinstimmung mit der mit DSC bestimmten Zerset-zungstemperatur ist.
TMADer Glasübergang kann auch mit TMA gemessen werden. Der Glasübergang äussert sich dann durch die Änderung des linearen Ausdehnungskoeffizienten beim Übergang des glasartigen Materi-als in den Gummi-artigen Zustand. Als charakteristische Temperatur wird der Schnittpunkt der Tangenten bestimmt. In Abbildung 3 wurden diese Tempe-raturen zu −29.7 °C für Probe A und −28.8 °C für Probe B ausgewertet. Die Differenz zu den mit DSC bestimmten Temperaturen beruht auf der Unter-schiedlichkeit der Messprinzipien. Probe A dehnt sich stärker aus als Probe B.
Gummibärchen werden beim Genuss Kör-perflüssigkeiten ausgesetzt. Diese ent-sprechen sowohl alkalischen Medien, z.B. Speichel im Mund als auch sauren Medi-en, z.B. Magensäure. Die Untersuchung des Quellverhaltens und der Auflösung mittels TMA ist deshalb interessant. In
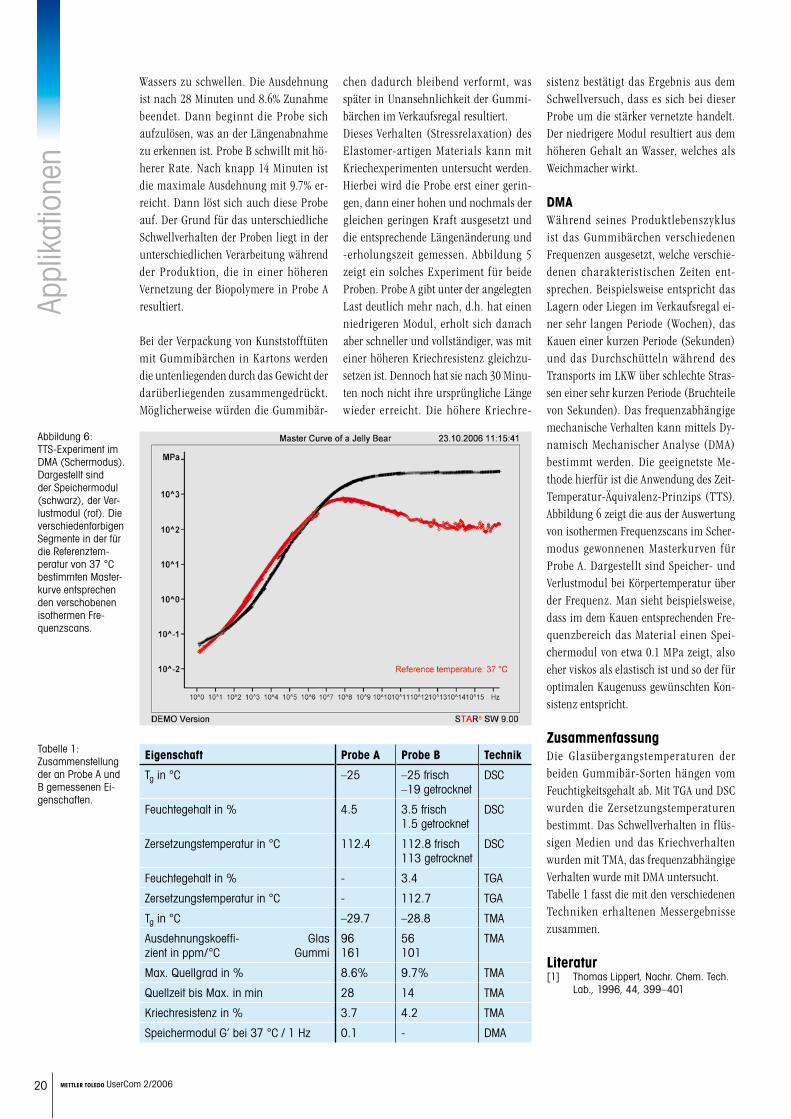
20 METTLER TOLEDO UserCom 2/2006
Appl
ikat
ione
nWassers zu schwellen. Die Ausdehnung ist nach 28 Minuten und 8.6% Zunahme beendet. Dann beginnt die Probe sich aufzulösen, was an der Längenabnahme zu erkennen ist. Probe B schwillt mit hö-herer Rate. Nach knapp 14 Minuten ist die maximale Ausdehnung mit 9.7% er-reicht. Dann löst sich auch diese Probe auf. Der Grund für das unterschiedliche Schwellverhalten der Proben liegt in der unterschiedlichen Verarbeitung während der Produktion, die in einer höheren Vernetzung der Biopolymere in Probe A resultiert.
Bei der Verpackung von Kunststofftüten mit Gummibärchen in Kartons werden die untenliegenden durch das Gewicht der darüberliegenden zusammengedrückt. Möglicherweise würden die Gummibär-
chen dadurch bleibend verformt, was später in Unansehnlichkeit der Gummi-bärchen im Verkaufsregal resultiert.Dieses Verhalten (Stressrelaxation) des Elastomer-artigen Materials kann mit Kriechexperimenten untersucht werden. Hierbei wird die Probe erst einer gerin-gen, dann einer hohen und nochmals der gleichen geringen Kraft ausgesetzt und die entsprechende Längenänderung und -erholungszeit gemessen. Abbildung 5 zeigt ein solches Experiment für beide Proben. Probe A gibt unter der angelegten Last deutlich mehr nach, d.h. hat einen niedrigeren Modul, erholt sich danach aber schneller und vollständiger, was mit einer höheren Kriechresistenz gleichzu- setzen ist. Dennoch hat sie nach 30 Minu-ten noch nicht ihre ursprüngliche Länge wieder erreicht. Die höhere Kriechre-
sistenz bestätigt das Ergebnis aus dem Schwellversuch, dass es sich bei dieser Probe um die stärker vernetzte handelt. Der niedrigere Modul resultiert aus dem höheren Gehalt an Wasser, welches als Weichmacher wirkt.
DMAWährend seines Produktlebenszyklus ist das Gummibärchen verschiedenen Frequenzen ausgesetzt, welche verschie-denen charakteristischen Zeiten ent-sprechen. Beispielsweise entspricht das Lagern oder Liegen im Verkaufsregal ei-ner sehr langen Periode (Wochen), das Kauen einer kurzen Periode (Sekunden) und das Durchschütteln während des Transports im LKW über schlechte Stras-sen einer sehr kurzen Periode (Bruchteile von Sekunden). Das frequenzabhängige mechanische Verhalten kann mittels Dy-namisch Mechanischer Analyse (DMA) bestimmt werden. Die geeignetste Me-thode hierfür ist die Anwendung des Zeit-Temperatur-Äquivalenz-Prinzips (TTS). Abbildung 6 zeigt die aus der Auswertung von isothermen Frequenzscans im Scher-modus gewonnenen Masterkurven für Probe A. Dargestellt sind Speicher- und Verlustmodul bei Körpertemperatur über der Frequenz. Man sieht beispielsweise, dass im dem Kauen entsprechenden Fre-quenzbereich das Material einen Spei-chermodul von etwa 0.1 MPa zeigt, also eher viskos als elastisch ist und so der für optimalen Kaugenuss gewünschten Kon-sistenz entspricht.
ZusammenfassungDie Glasübergangstemperaturen der beiden Gummibär-Sorten hängen vom Feuchtigkeitsgehalt ab. Mit TGA und DSC wurden die Zersetzungstemperaturen bestimmt. Das Schwellverhalten in flüs-sigen Medien und das Kriechverhalten wurden mit TMA, das frequenzabhängige Verhalten wurde mit DMA untersucht.Tabelle 1 fasst die mit den verschiedenen Techniken erhaltenen Messergebnisse zusammen.
Literatur[1] Thomas Lippert, Nachr. Chem. Tech.
Lab., 1996, 44, 399–401
Abbildung 6: TTS-Experiment im DMA (Schermodus). Dargestellt sind der Speichermodul (schwarz), der Ver-lustmodul (rot). Die verschiedenfarbigen Segmente in der für die Referenztem-peratur von 37 °C bestimmten Master-kurve entsprechen den verschobenen isothermen Fre-quenzscans.
Tabelle 1: Zusammenstellung der an Probe A und B gemessenen Ei-genschaften.
Eigenschaft Probe A Probe B Technik
Tg in °C –25 –25 frisch –19 getrocknet
DSC
Feuchtegehalt in % 4.5 3.5 frisch 1.5 getrocknet
DSC
Zersetzungstemperatur in °C 112.4 112.8 frisch 113 getrocknet
DSC
Feuchtegehalt in % - 3.4 TGA
Zersetzungstemperatur in °C - 112.7 TGA
Tg in °C –29.7 –28.8 TMA
Ausdehnungskoeffi- Glas zient in ppm/°C Gummi
96 161
56 101
TMA
Max. Quellgrad in % 8.6% 9.7% TMA
Quellzeit bis Max. in min 28 14 TMA
Kriechresistenz in % 3.7 4.2 TMA
Speichermodul G’ bei 37 °C / 1 Hz 0.1 - DMA
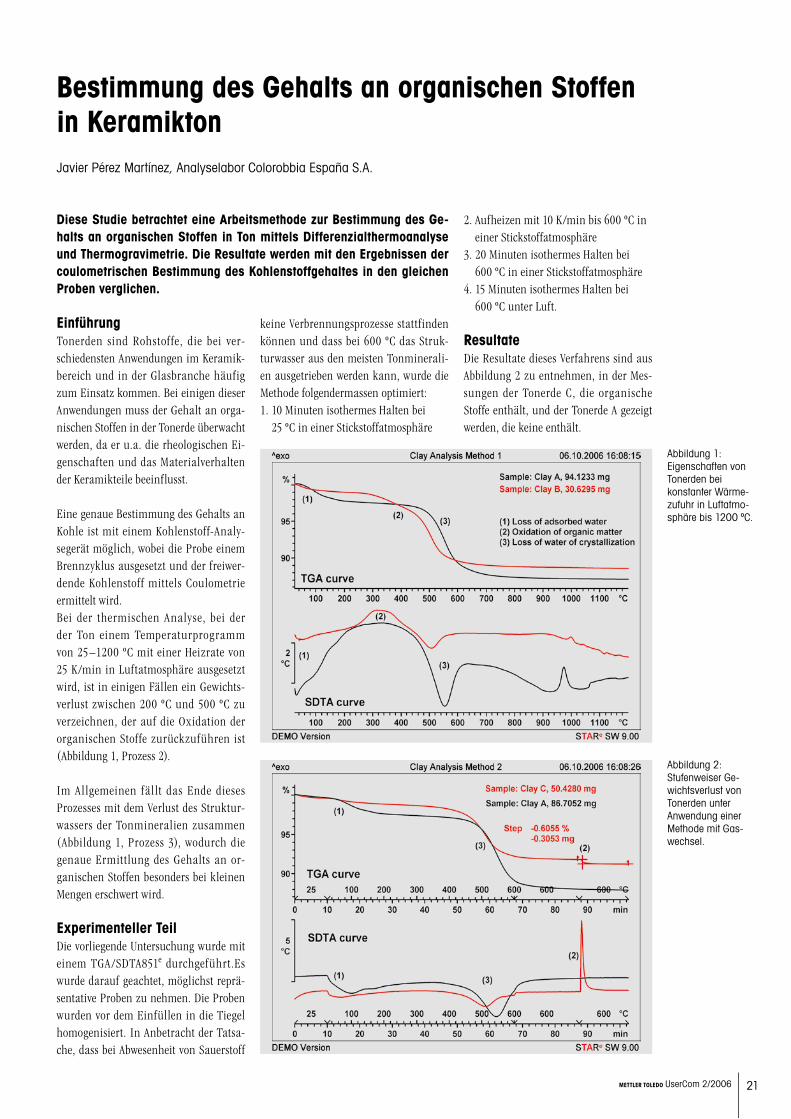
21METTLER TOLEDO UserCom 2/2006
keine Verbrennungsprozesse stattfinden können und dass bei 600 ºC das Struk-turwasser aus den meisten Tonminerali-en ausgetrieben werden kann, wurde die Methode folgendermassen optimiert:
10 Minuten isothermes Halten bei 25 ºC in einer Stickstoffatmosphäre
1.
Aufheizen mit 10 K/min bis 600 ºC in einer Stickstoffatmosphäre20 Minuten isothermes Halten bei 600 ºC in einer Stickstoffatmosphäre15 Minuten isothermes Halten bei 600 ºC unter Luft.
ResultateDie Resultate dieses Verfahrens sind aus Abbildung 2 zu entnehmen, in der Mes-sungen der Tonerde C, die organische Stoffe enthält, und der Tonerde A gezeigt werden, die keine enthält.
2.
3.
4.
EinführungTonerden sind Rohstoffe, die bei ver-schiedensten Anwendungen im Keramik-bereich und in der Glasbranche häufig zum Einsatz kommen. Bei einigen dieser Anwendungen muss der Gehalt an orga-nischen Stoffen in der Tonerde überwacht werden, da er u.a. die rheologischen Ei-genschaften und das Materialverhalten der Keramikteile beeinflusst.
Eine genaue Bestimmung des Gehalts an Kohle ist mit einem Kohlenstoff-Analy-segerät möglich, wobei die Probe einem Brennzyklus ausgesetzt und der freiwer-dende Kohlenstoff mittels Coulometrie ermittelt wird.Bei der thermischen Analyse, bei der der Ton einem Temperaturprogramm von 25−1200 ºC mit einer Heizrate von 25 K/min in Luftatmosphäre ausgesetzt wird, ist in einigen Fällen ein Gewichts-verlust zwischen 200 ºC und 500 ºC zu verzeichnen, der auf die Oxidation der organischen Stoffe zurückzuführen ist (Abbildung 1, Prozess 2).
Im Allgemeinen fällt das Ende dieses Prozesses mit dem Verlust des Struktur-wassers der Tonmineralien zusammen (Abbildung 1, Prozess 3), wodurch die genaue Ermittlung des Gehalts an or-ganischen Stoffen besonders bei kleinen Mengen erschwert wird.
Experimenteller TeilDie vorliegende Untersuchung wurde mit einem TGA/SDTA851e durchgeführt.Es wurde darauf geachtet, möglichst reprä-sentative Proben zu nehmen. Die Proben wurden vor dem Einfüllen in die Tiegel homogenisiert. In Anbetracht der Tatsa-che, dass bei Abwesenheit von Sauerstoff
Abbildung 1: Eigenschaften von Tonerden bei konstanter Wärme-zufuhr in Luftatmo-sphäre bis 1200 ºC.
Diese Studie betrachtet eine Arbeitsmethode zur Bestimmung des Ge-halts an organischen Stoffen in Ton mittels Differenzialthermoanalyse und Thermogravimetrie. Die Resultate werden mit den Ergebnissen der coulometrischen Bestimmung des Kohlenstoffgehaltes in den gleichen Proben verglichen.
Bestimmung des Gehalts an organischen Stoffen in KeramiktonJavier Pérez Martínez, Analyselabor Colorobbia España S.A.
Abbildung 2: Stufenweiser Ge-wichtsverlust von Tonerden unter Anwendung einer Methode mit Gas-wechsel.

22 METTLER TOLEDO UserCom 2/2006
ringe Abweichungen im Gehalt an orga-nischen Stoffen innerhalb desselben Pro-duktloses und innerhalb verschiedener, im Laufe der Zeit hergestellter Lose er-mittelt. Versuche zur Wiederholbarkeit, welche an Tonerde B ausgeführt wurden, ergaben einen Gehalt an organischen Stoffen von 0.90 ± 0.02 %.
Beispielhafte Ergebnisse, welche in Ab-bildung 3 gezeigt werden, zeigen die im Allgemeinen gute Wiederholbarkeit.
SchlussfolgerungenSowohl diese Studie als auch die Erfah-rungswerte aus Messungen zahlreicher Proben im industriellen Bereich zeigen, dass die mit dem TGA/SDTA851e erzielten Resultate unter Anwendung des oben dargelegten Verfahrens mit Atmosphä-renwechsel geeignet sind, um geringe Abweichungen im Gehalt an organischen Stoffen in Tonerden zu ermitteln.
Literatur[1] „Materias primas para la fabricación
de soportes de baldosas cerámicas”, Instituto de Tecnología Cerámica (ITC) - AICE, 1997, Castellón, Spanien.
[2] R. Riesen, UserCom 14, Seite 18–20
Appl
ikat
ione
nDie Tonerde C wurde ausgewählt, da sie einen geringen Gehalt an organischen Stoffen aufweist. Es wurde eine Kohlen-stoffbestimmung mittels Coulometrie durchgeführt und das Ergebnis mit dem mit Hilfe der Differenzialthermoanalyse und der Thermogravimetrie erhaltenen Resultat verglichen (Tabelle 1).
Die Ergebnisse, welche aus den Mes-sungen mit thermischer Analyse erhalten wurden, weichen wenig (6.5%) von denen der Coulometrie ab. Dies zeigt, dass die thermische Analyse durchaus zur ver-lässlichen Bestimmung des Kohlenstoff-gehaltes eingesetzt werden kann.
Die optimierte Methode hat folgende Vor-teile gegenüber der in Abbildung 1 aufge-zeigten Methode:
Der Prozess der Verbrennung orga-nischer Stoffe (2) wird von den üb-rigen (1 und 3) getrennt.Eindeutige TGA- und SDTA-Signale werden durch Beschleunigung des Ver-brennungsprozesses erhalten.
Thermische Analyse wird bei Colorobbia España bei der Routineüberwachung von Tonerden eingesetzt. Damit werden ge-
1.
2.
Abbildung 3: Erfassung der Ab-weichungen im Gehalt an orga-nischen Stoffen in Ton zeitlich verschie-dener Produktlose.
Tabelle 1: Vergleich der Bestim- mung organischer Stoffe mittels Ther-moanalyse mit der mittels Coulometrie bei 500 ºC.
Probe % Organische Stoffe
TGA/SDTA Coulometrisch
Ton A 0 0
Ton C 0.61 0.65
METTLER TOLEDO UserCom 2/2006 23
Dat
en
International and Swiss TA Customer Courses:
TA-Kundenkurse
und Seminarein Deutschland
Cours et séminaires
d’Analyse Thermique en
France
Exhibitions, Conferences and Seminars – Veranstaltungen, Konferenzen und SeminarePittsburgh Conference February 26–March 1, 2007 Chicago, IL (USA)Ulm-Freiberger Kalorimetrietage March 21–23, 2007 Freiberg (Germany)Analytical Sciences: Industrial Problem Solving May 16–18, 2007 Newark, NJ (USA)12th International Congress on the Chemistry of Cement July 8–13, 2007 Montreal, CanadaFederated Society for Coatings, Coating woods and Wood Composites for Durability Symposium July 23–25, 2007 Seattle, WA (USA)NATAS 2007 August 26–29, 2007 State University, Lansing, MI (USA)ILMAC 2007 September 25–28, 2007 Basel (Switzerland)K 2007 October 24–31, 2007 Düsseldorf (Germany)
Local TA Customer Courses:
Für nähere Informationen wenden Sie sich bitte an: Frau Petra Fehl, Mettler-Toledo GmbH, Giessen, Tel: ++49 641 507 404, e-mail: [email protected]
Anwenderworkshop DSC 06./07.03.2007 Giessen 04./05.09.2007 GiessenAnwenderworkshop TGA 08./09.03.2007 Giessen
Thermische Analyse und Rheologie in Forschung und Qualitäts- 13.02.2007 Hannover 28.03.2007 Wien sicherung – Eine gemeinsame Veranstaltung der Unternehmen 20.03.2007 Potsdam 08.05.2007 Düsseldorf Thermo Electron und METTLER TOLEDO auf Anfrage: Zürich, Basel
Renseignements et inscriptions par: Christine Fauvarque, Mettler-Toledo S.A., 18-20 Av. de la pépinière, 78222 Viroflay Cedex, Tél: ++33 1 3097 1439, Fax: ++33 1 3097 1660
Principe de la TMA/DMA 1 octobre 2007 Viroflay (France) Principe de la TGA 4 octobre 2007 Viroflay (France)Principe de la DSC : les bases 2 octobre 2007 Viroflay (France) Logiciel STARe : perfectionnem. 5 octobre 2007 Viroflay (France)Principe de la DSC : perfectionnem. 3 octobre 2007 Viroflay (France)
Per ulteriori informazioni Vi preghiamo di contattare: Simona Ferrari, Mettler-Toledo S.p.A., Novate Milanese, Tel: ++39 02 333 321, Fax: ++39 02 356 2973, e-mail: [email protected]
DSC base 6 Febbraio 2007 12 Giugno 2007 25 Settembre 2007 Novate MilaneseDSC avanzato 7 Febbraio 2007 13 Giugno 2007 26 Settembre 2007 Novate MilaneseTGA 8 Febbraio 2007 14 Giugno 2007 27 Settembre 2007 Novate MilaneseTMA 9 Febbraio 2007 15 Giugno 2007 28 Settembre 2007 Novate Milanese
La corretta interpretazione dei tracciati termoanalitici 13 Marzo 2007 Milano 20 Marzo 2007 NapoliL’Analisi Termica nel Controllo Qualità e nella Ricerca e Sviluppo 22 Marzo 2007 Roma 29 Marzo 2007 Padova
TA Customer Courses and Seminars in Switzerland – Information and Course Registration:TA-Kundenkurse und Seminare in der Schweiz – Auskunft und Anmeldung bei:Frau Esther Andreato, Mettler-Toledo AG, Analytical, Schwerzenbach,Tel: ++41 44 806 73 57, Fax: ++41 44 806 72 60, e-mail: [email protected]
Courses / KurseSW Basic (Deutsch) 12. Feb. 2007 17. Sep. 2007 SW Basic (English) Feb 19, 2007 Sep 24, 2007TMA (Deutsch) 12. Feb. 2007 17. Sep. 2007 TMA (English) Feb 19, 2007 Sep 24, 2007DMA Basic (Deutsch) 12. Feb. 2007 17. Sep. 2007 DMA Basic (English) Feb 19, 2007 Sep 24, 2007DMA Advanced (Deutsch) 13. Feb. 2007 18. Sep. 2007 DMA Advanced (English) Feb 20, 2007 Sep 25, 2007TGA (Deutsch) 13. Feb. 2007 18. Sep. 2007 TGA (English) Feb 20, 2007 Sep 25, 2007TGA-MS (Deutsch) 14. Feb. 2007 19. Sep. 2007 TGA-MS (English) Feb 21, 2007 Sep 26, 2007DSC Basic (Deutsch) 14. Feb. 2007 19. Sep. 2007 DSC Basic (English) Feb 21, 2007 Sep 26, 2007DSC Advanced (Deutsch) 15. Feb. 2007 20. Sep. 2007 DSC Advanced (English) Feb 22, 2007 Sep 27, 2007TGA-FTIR (Deutsch) 15. Feb. 2007 20. Sep. 2007 TGA-FTIR (English) Feb 22, 2007 Sep 27, 2007SW Advanced (Deutsch) 16. Feb. 2007 21. Sep. 2007 SW Advanced (English) Feb 23, 2007 Sep 28, 2007
Corsi e Seminari di Analisi
Termica in Italia

Editorial team
Dr. J. Schawe Dr. R. Riesen J. Widmann Dr. M. Schubnell Dr. M. Wagner Dr. D. P. May Ni Jing Marco Zappa Urs JörimannPhysicist Chem. Engineer Chem. Engineer Physicist Chemist Chemist Chemist Material Scientist Electr. Engineer
www.mt.com/ta
Mettler-Toledo AG, AnalyticalPostfach, CH-8603 SchwerzenbachTel. ++41 44 806 73 87Fax ++41 44 806 72 60Kontact: [email protected]
©11/2006 Mettler-Toledo AGME-51724501, Gedruckt in der SchweizMarCom Analytical
Cursos y Seminarios
de TA en España
TA Customer Courses
and Seminars in the UK
TA Customer Courses
and Seminars in Singapore
TA School and Seminars
in Korea
Para detalles acerca de los cursos y seminarios, por favor, contacte con: Francesc Catala, Mettler-Toledo S.A.E., Tel: ++34 93 223 76 00, e-mail: [email protected] Aplicaciones del Análisis Térmico Febrero, 13 de 2007 Zaragoza Septiembre, 25 de 2007 Barcelona Octubre, 2 de 2007 MadridUso del sistema STARe Septiembre, 26 de 2007 Barcelona Octubre, 3 de 2007 Madrid
Für nähere Informationen wenden Sie sich bitte an: Frau Geraldine Braun, Mettler-Toledo GmbH, Wien, Tel: ++43 1 604 19 80 - 33DW, e-mail: [email protected]
Thermische Analyse und Rheologie in Forschung und Qualitätssicherung 28. März 2007 Wien
For details of training courses and seminars, please contact: Rod Bottom, Mettler-Toledo Ltd, Leicester, Tel: ++44 116 234 5025, Fax: ++44 116 236 5500, e-mail: [email protected]
DSC BASIC February 27, 2007 October 25, 2007 Leicester
For details of training courses and seminars, please contact: Bryan Yiew, Mettler-Toledo (S) Pte Ltd, 28 Ayer Rajah Crescent, #05 - 01, Singapore 139959 Tel: +65-68900011, Fax: +65-68900012, e-mail: [email protected]
STARe Infoday Seminar April 2007 SingaporeDSC BASIC August 2007 November 2007 Singapore
For details of training courses and seminars, please contact: KiHun Lee at Mettler-Toledo Korea, Tel: ++82 2 3498 3500, e-mail: [email protected], Homepage: www.kr.mt.com
TA School for MT Users April 2007 Seoul and DaeJeonTA Public Seminar in 2007 October 2007 Seoul and Ulsan
For details of training courses and seminars, please contact: Lu LiMing at Mettler-Toledo Instruments (Shanghai) Co., Ltd., Tel: ++86 21 6485 0435, Fax: ++86 21 6485 3351, or by e-mail: [email protected] STARe Customer Seminar March 2007 Hangzhou April 2007 Hefei and Tianjin May 2007 Guangzhou and Kunming
For details of seminars, please contact: Tomoko Wakabayashi, Mettler-Toledo KK, 3-8 Sanbancho, Chiyoda-ku, Tokyo 102-0075, Japan, Tel : +81-3-3222-7164, Fax : +82-3-3222-7124, e-mail: [email protected]
STARe Infoday Seminar April 2007 Tokyo and Osaka
TA-Seminare in Österreich
TA Customer Courses
and Seminars in China
TA Seminars in Japan
Für mehr Informationen





























