Embed Size (px)
Citation preview

Uber die Darstellung von wasserfreiem Hydrazin
Von F. FEHER, ,J. CREMER~) und W. TROMM~)
&fit 3 Abbildungen
Inhalt,subersicht In der vorliegenden Arbei t werden die bisher bekannten Methoden zur Darstellung
hochkonzentrierter Hydrazinlosungen gepriift und zwei neue Verfahren zur Gewinnung groBerer Mengen der reinen wasserfreien Verbindung entwickelt.
Nach deni ersten Verfahren wird Monohydrazinsulfat N2H, . H,S04 in StahlgefaBen unter Druck und Kiihlung (auf 0' bis 10' C) mit flussigem Aminoniak umgesetzt, das als Nebenprodukt entstandene, in der Fliissigkei t unlosliche Ammoniumsulfat-Triammo- niakat abfiltriert und das Hydrazin durch Verdanipfung des uberschiissigen Ammoniaks und anschlieaende Vakuumdestillation isoliert. Man erh< dabei ein Produkt mit 99,5 bis 99,6proz. Hydrazingehalt in einer Ausbeute von 85%.
Bei dem zweiten Verfahren wird Hydraziniumdichlorid N,H4. 2 HCl im Vakuum (12-20 Torr) auf 180-190" C erhitzt und rnit gasformigem Ammoniak das Hydrazin aus dem hierbei gebildeten, flussig vorliegenderi Monochlorid unter Normaldruck bei 160" C in Freiheit gesetzt. Nach einer fraktionierten Kondensatiou und anschlieRenden Vakuumdestillation, bei der das Produkt, von restlichen Mengen NH3 und Spuren HCl befreit wird, gewinnt man auf diesem Wege reiues Hydrazin in einer Ausbeute von 90%.
Die Bestimmung des Hydrazingehaltes wird durch Titration mit KJO, in 3- bis 5-normaler HC1-Losung mit eiiier Genauigkeit von IIZ 0,l yo durchgefiihrt,.
Bei unseren Arbeiten iiber Siliciuni- und Germaniurnwa~serstoffe~) wurde gefunden, dal3 sich diese Verbindungen in Analogie zu dem Ver- faliren in fliissigem Ammoniak 4, vorteilhaft unter Verwendung von I-Iydra,zin als Reaktionsmedium darstellen lassen. Es war dabei er- strebenswert, ein moglichst wssserfreies Hydrazin zu den Umsetzungen heranziehen zu konnen. Itn folgenden werden die Versuche zur Ge- winnung dieses Losungsmittels in der erforderlichen Menge und Reinheit behandelt.
l) J. CREMER, Diplomarbeit Koln 1952. e, W. TROMM, Diplomarbeit Koln 1952. 3) F. FEHER u. W. TROMM, Z. anorg. allg. Chem. 282, 29 (1955); F. FEH$R u. J.
CREMER, erscheint in Kiirze; J. CREMER, Diss. Koln, 1954; W. TROMM, Diss. Koln, 1954. 4, W. C. JOHNSON u. Mitarb., .J. Amer. chem. SOC. 56, 1252 (1934); 57, 1349 (1935).

176 Zeitschrift fur anorganische und allgemeine Chemie. Band 287. 1956
Von den in der Literatur 5, beschriebenen Verfahren wurde zunachst die Anreicherung der im Handel erhaltlichen verdunnten wafirigen N,H,-Losungen durch Destillation gepriift.
Wir verwendeten hierzu eine 50 cm lange, elektrisch beheizte Fiillkorper-Kolonne- Die Beheizung der Kolonne ermoglichte eine Senkung der olbad-Temperatur des Siede- kolbens und beugte damit Zersetzungserscheinungen des Hydrazins vor. Um die Mog- lichkeit des Auftretens weiterer Nebenreaktionen abzuschwachen, wurde in Stickstoff- oder Wasserstoffatmosphare gearbeitet. Die einzelnen Versuche egaben bei einem Ruck- laufverhaltnis von 1 : 7 bis 1 : 10 eine Erhohung des Hydrazingehaltes der eingesetzten 15proz. Losungen auf rund 64% und eine Ausbeute von 85-90y0. Eine Anwendung der von HURD und BENNET 6, beschriebenen azeotropen Destillation mit Xylol brachte keine besseren Ergebnisse ; dagegen fiihrte das bekannte Verfahren von RASCHIG, bei welchem Hydrazinhydrat nach Zusatz von Natriumhydroxyd einer normalen Destillation unter- worfen wird, bei Einsatz von gleichen Mengen Hydrat und NaOH und Verwendung einer Glasschliffapparatur unter gberleiten von Wassarstoff zu Kondensaten mit einem N,H,-Gehalt von 90-98% bei einer Ausbeute von 85-90%.
Eine weitere Entwasserung der RAscHIG-Losungen wurde durch die Einwirkung von geschmolzenem NaOH, CaO, BaO, Ca bzw. Na er- reicht.
Die Ansatze wurden bei Verwendung yon NaOH (geschm.), CaO, BaO und Ca in einer sauerstofffreien und trockenen Wasserstoffatmosphare 3-5 Stunden lang unter RiickfluB gekocht und anschlieaend bei 50-150 mm Hg - bzw. bei dem Einsatz von ge- schmolzenem NaOH unter Normaldruck - destilliert. Bei der Behandlung mit metalli- sohem Natrium folgten wir wegen der Bildung des hochexplosiven Na-hydrazids streng der Arbeitsvorschrift yon ~ C H L E N K und WEICHSELFELDER').
Die erhaltenen Produkte zeigten folgenden Hydrazingehalt 8) :
Natriumhydroxyd (geschmolzen) . . . .98,3-99,3y0 Calciumoxyd . . . . . . . . . . . . 98,3-99,2y0 Bariumoxyd . . . . . . . . . . . .99,2-99,7y0 Calcium. . . . . . . . . . . . . . . 99,3-99,6y0 Natriuni . . . . . . . . . . . . . . 100,Oyo
Der Hydrazingehalt der durch Eintragen von Ca gewonnenen Pro- dukte konnte bei der Durchfuhrung der Reaktion in fliissigem Ammoniak auf nahezu 100 % erhoht werden. Das hierbei sich bildende Ca-hydrazid verursachte aber gelegentlich (besonders beim Arbeiten in der Warme) heftige Explosionen, so da13 dieses Verfahren ebenso wie die Methode
6 ) Einen zusammenfassenden Bericht sowie eine umfangreiche Literaturiibersicht iiber die bisher entwickelten Methoden findet man bei L. F. AUDRIETH und B. ACKERSON Ogg ,,The Chemistry of Hydrazine", John Wile) and Sons, NewYork 1951.
6) C. D. HURD u. C. W. BENNET, J. Amer. chem. Soc. 51, 265 (1928). 7) W. SCHLENK u. TH. WEICHSELFELDER, Ber. dtsch. chem. Ges. 48, 669 (1915). 8 ) Die Hydrazingehalte wurden nach der von R. A. PENNEMANN u. L. F. AUDRIETH,
Analytic. Chem. 20, 1058 (1948) beschriebenen direkten Jodat-Methode bestimmt. (Siehe axperimenteller Teil.)

FEHER, CBEMER u. TROMM, uber die Darstellung von wasserfreiem Hydrazin 177
von SCHLEKK und WEICHSELFELDER fur eine Darstellung grol3erer Mengen Hydrazins als zu gefahrvoll angesehen wurde. Da aul3erdem die oben aufgefuhrten Gehaltsangaben - welche durch N,H,-Analysen mit einer Genauigkeit von f O , l % besti,mmt wurden - keineswegs die. vollstandige Wasserfreiheit der Produkte bewiesen, wendeten wir uns daraufhin Methoden zu, die als Ausgangsstoffe wasserfreie Hydrazin- salze verwenden und es gestatten, die Base in wasserfreien Medien in Freiheit, zu setzen.
Den ersten Weg in dieser Richtung zeigte LOBRY DE B R U Y N ~ ) , indem er Natriuminethylat in absolutem Methylalkohol auf Hydrazinium- monochlorid einwirken lie& Die Trennung des Hydrazin-Alkohol- Gemisches gelang ihm jedoch nur bis zu einer Hydrazinkonzentration von 92%.
Ein weit~eres Verfahren ist die Umsetzung von Bariumoxyd rnit hydrazincarbonsaurem Hydrazin (N2H,COON,H,) oder rnit Hydrazin- carbonsaure (N,H,COOH), die von STOLLO und HOFMANN lo) unter- sucht wurde.
Die Darstellung des Salzes sowie der freien Saure erfolgte durch Einleiten von Kohlen- dioxyd in gekuhlte Hydrazinlosung, wobei sich unter Erwarmung zunachst das Salz und erst nach langerem Einleiten yon Kohlendioxyd aus diesem die Saure bildet"). Da die Saure jedoch leichter wasserfrei zu erhalten ist, wurden von uns nur mit dieser Versuche durchgefiihrt. Die Zeit zu ihrer Darstellung konnte dadurch abgekiirzt werden, daO wir in 50proz. Bydrazinlosung unter Umschwenken nach und nach kleine Stuckchen festes Kohlendioxyd eintrugen, bis die Losung erstrlrrt war. Die Losung erwarmte sich dabei nur unwesentlich so daO eine Kiihlung von auBen nicht erforderlich war. Nach mehr- stiindigem Stehen wurde die auegefallene Hydrazincarbonsaure bei Zimmertemperatur unter AusschluO von Luft vorsichtig abgenutscht, mit etwas kohlendioxydhaltigem Wasser gewaschen und in C0,-Atmosphare uber konz. Schwefelsaure getrocknet. Aus dem Filtrat lieB sich in der beschriebenen Weise weitere Hydrazincarbonsaure ausfallen. Die wasserfreie Saure wurde durch Erhitzen auf etwa 140" C in das kohlendioxydarmere Salz iiberfuhrt und dieses nach Zugabe von Bariumoxyd in IOproz. UberschuB 3-5 Stunden lang unter RiickfluO in einer Wasserstoffatmosphare gekocht. Die nachfolgende Vakuum- destillation ergab ein kohlendioxydhaltiges 98proz. Hydrazin mit einer Ausbeute von 70%. Die niedrige Ausbeute erklarte sich durch Einschlusse in der verhaltnismaOig groOen Bariumoxyd- bzw. Bariumcarbonatmenge. Znr Entfernung des restlichen Kohlen- dioxyds hatte es einer erneuten Behandlung rnit Bariumoxyd bedurft, YOU der wir jedoch abgesehen haben.
Eine weitere Moglichkeit zur Dars tellung des wasserfreien Hydra- zins in einfacher Weise bietet das Hydrazinsulfat. Nach Feststellungen
9 , LOBRY DE BRUTN, Rec. Trav. chim. Pays-Bas 15, 174 (1896). lo) R. S T O L L ~ u. K. HOFMANN, Ber. dtsch. chem. Ges. 87, 4523 (1904). 11) Vgl. hierzu F. EPHRAIM u. E. LASOCKI, Ber. dtsch. chem. Ges. 44, 395 (1911).

178 Zeitschrift fur anorganische und allgemeine Chemie. Band 287. 1956
von BROWNE und WELSH^^) sowie LOBI~T DE B R U I - S ~ ~ ) ruft die Ein- wirkung von gasformigem Ammoniak auf Monohydraziiisulfat eine Gleichgewichtsreaktion hervor, bei der sich freies Hydrazin bildet :
N,H, . H,SO, + 2 N H 3 s N,H, + (NH,),SO,.
Fuhrt man diese Reaktion mit fliissigem Ammoniali durch, so kann ein quantitativer Ablauf des Vorganges nach der rechten Seite der Gleichung erreicht werden, da Ammoniumsulfat in der Form des sich bildenden Triammoniakats nach den Untersuchungeii von BROW N E
und WELSH 12) in fliissigem NH, unloslich ist und deshalb fortlaufend dem Reaktionssystem entzogen wird. Zur praparativen Darstellung des freien Hydrazins nach dieser Reaktion beschreibt FRIEDRICHS 14) eineri Extraktionsapparat, dessen Wirkungsweise auf dem Soxhlet-Prinzip beruht und dessen Aufbau das Arbeiten bei tiefen Teinperaturen erlaubt. Der Apparat gestattet einen Eiiisatz von 8-10 g Hydrazinsulfat ent- sprechend 2,O- 2,5 g Hydrazin- Aus beut e .
Urn grofiere Mengen absoluten Hydrazins in dieser Weise dar- stellen zu konnen, war es notwendig, Apparat,uren mit griifierem Fassungs- vermogen zu entwickeln. Die vorgenonimene VergroBerung des FRIED- zucasschen Extraktors fuhrte zwar zu einer Steigerung des Umsatzes, jedoch lieBen sich auch hiermit noch niclit die fiir unsere Arbeiten er- forderlichen Mengen darstellen. Der hohe Verbrauch an Troekeneis wahrend der mehrstundigen Extraktionen lie13 es aber nicht zweck- maBig erscheinen, die Apparatur in dieser Form noch weiter zu ver- grofiern. Wir versuchten daher, die Unisetzung zwischen Monohy- drazinsulfat und flussigern dmmoniak in griifieren Stahlgefsfien unter Druck durchzufuhren, wobei die Kondensation des Ammoniaks durch Kuhlung mit Wasser oder Eis erreicht werden konnte. Auch unter diesen Bedingungen fuhrte die Reaktion zu unliislichem 15) Ammonium- sulfat-Triarnmoniakat und dein in der flussigen Phase verteilten Hy- drazin, welches nach Filtration aus der klaren 1,osung durch Verdampfung des Arnmoiiiaks isoliert und durch Vakuumdestillation gereinigt wurde. Einzelheit,eii uber das nach vielen Vorversuchen entwickelte Verfahren und die Apparatur sind aus dem experimentellen Teil zu ersehen. Die erhalteiien Produkte weisen Hydrazirlgehalte von 99,5-99,6 % auf : die Ausbeute betrug etwa 85 %. - ~ ~
12) A. W. BROWNE u. T. W. B. WELSH, J . Amer. chem. S O ~ . 33, 1728.(1911). 13) LOBRY DE BRUYN, Rec. Trav. chim. Pays-Bas 15, 179 (1896). l4) F. FRIEDRICHS, Z. angew. Chem. 26, 201 (1913). 15) Vgl. hierzu F. FRIEDRICHS, FuBnote 14 .

FEHER, CREMER u. TROMM, Uber die Darstellung von wasserfreiem Hydrazin 179
Obwohl im ProzeB nur vollig wasserfreie Ausgangstoffe eingesetzt wurden, konnte kein 100proz. Hydrazin gewonnen werden. Die Er- klarung hierfur ist durch die Annahme von Nebenreaktionen gegeben, beispielsweise einer Reduktion der Schwefelsaure zu Schwefelwasser- stoff und Schwefel unter gleichzeitiger Bildung von Wasser. Schon CURTIUS la) hatte beini Erhitzen von Monohydrazinsulfat festgestellt, daB das Salz unter explosionsartiger Gasentwicklung schmilzt und bei weiterer Warmezufuhr in Animoniumsulfit, Schwefeldioxyd, Schwefel- wasserstoff und Schwefel zerfallt. Im vorliegenden Fall sind die Neben- reaktionen im wesentlichen auf eine Erwarmung wahrend der Um- setzung zuruckzufuhren uiid in ihrem AusmaB denizufolge weitgehend von der Geschwindigkeit der Ammoniakzufuhr und der dabei wirk- sainen AuBenkuhlung abhangig. Leitet man das NH, zu schnell eiii, so kann es zu betrachtlicher Selbsterwarmung und schnellem Druck- anstieg kommen, indem sich die Zersetzung auf da.s gesainte Hydraziii- sulfat ausdehnt. Versuche. hei denen wir das Ammoniak sehr langsain und bei -30" C auf das Hydrazinsulfat kondensierten, liefcrteii in uber- einstimmung mit o bigen Uberlegungen 99,8proz. Hydrazin. Offenbar war aber auch jetzt noch eine geringfugige Reduktion des Sulfates er-
Fiir die Darstellung von Hydrazin in griif3erem MaBstah ist das Verfahren dann geeignet, wenn fur schnelle Ableitung der Reaktions- wii,rme Sorge getragen wird. Es besitzt gegeniiber anderen Methoden den Vorteil, daf3 aus dem leicht durch Umkristallisatiori zu reinigenden und zu trocknenden Monohydrazinsulfat in eineni ArbeitsprozeB hochst- konzentriertes Hydrazin gewonnen wird.
Da. fur das Dihydrazinsulfat (N,H,), . H,80, sowie fur das Mono- hydrazinoxalat N,H, . H,C,O, voii BEOWNE und Mitarbeit8ern lT) eben- € a h Amnionolyse beobachtet wurde, haben wir auch mit diesen Salzen Versuche in flussigem Ammoniak durchgefuhrt. Zusatzlich wurde noch das Dihydrazinosalat (N,H,), . H,C,O, untersucht. Es zeigte sich, da13 jedes dieser Salze analog den1 Monohydrazinsulfat fur die Extraktion geeignet ist, jedoch ergaben sicli im Vergleich zu letzterem bei ihrer Verwendung Produkte mit etwas hoherem Wassergehalt. Der hohere Hydrazingehalt der eingesetzten Salze sowie die Tatsache, daB Ammo- niumoxalat mit flussigem Ainmoriiak keine Additionsverbindungen eingeht, gestatten jedoch bei gleichem Volumen des Ext,raktors die
folgt.
l6) TH. CURTIUS, Ber. dtsch. chem. Ges. SO, 1632 (1887); TH. CURTIUS u. R. JAY,
17) A. W. BROWNE 11. T. W. B. WELSH, J. Amer. chem. SOC. 33, 1728 (1911); A. W. J. prakt. Chem. [2] 39, 27 (1889).
BROWNE u. A. E. HOULEHAN, J. Amer. chem. SOC. 33, 1734 (1911).

180 Zeitachrift fur anorganische und allgemeine Chenlie. Band 287. 1956
Gewinnung grol3erer Mengen der Base. So konnten z. B. bei Verwendung von Dihydrazinoxalat 1,5 Mol eingesetzt werden, die mit 80proz. Aus- kleute rund 75 g 98,4proz. Hydrazin lieferten, wahrend mit Monohydrazin- sulfat in der gleichen Apparatur hei einem Ansatz 28 g Hydrazin er- halten wurden.
Nachdem die Versuche mit Hydrazinsalzen sauerstoffhaltiger Sauren gezeigt hatten, da13 selbst bei tiefen Temperaturen eine Wasser- bildung nicht vollig auszuschlieflen ist, gingen wir dazu uber, sauer- stofffreie Hydrazinverbindungen als Ausgangsstoffe zu verwenden. Von diesen war zu fordern, da13 sie leicht zuganglich und wasserfrei darzustellen sind. Diesen Anspruchen werderi das Hydraziniumdi- bzw. -monochlorid gerecht. Da das hei der Reaktion rnit Ammoniak nach der Gleichung
N2H* - HCl + N H 3 Z N,H, + NH,Cl
gebildete Aninioniumchlorid jedoch in flussigem Animoniak betrachtlich loslich ist und dadurch nicht aus den1 Reaktionsgleichgewicht ver- schwindet, fuhrte die Extralition init flussigem Ammoniak hier nicht zum Erfolg. Wir versuchten daher, die Einwirkung von gasformigem Ammoniak auf die Salze bei Temperaturen oberhalb von 100" C vorzu- nehmen, uni auf diese Weise zu erreichen, da13 das in Freiheit gesetzte Hydrazin im Ammoniakstrom fortlaufend abdestillierte und in Konden- sationsfallen bei -20 bis -30" C vom Ammoriiak getrennt wurde.
Vorversuche zeigten, daR sich Hydraziniumdichlorid nicht als Aus- gangssubstanz eignet. Die Einwirkung von Ammoniak fuhrte unter den genannten Bedingungen entweder zur Zersetzung des Salzes oder sogar ZUI Explosion. Demgegenuber lie13 sich mit Hydraziniummono- chlorid der stete Austausch von A4nimoniak gegen Hydrazin erreichen.
Die Darstellung des Monochlorids erfolgte uber das Dichlorid, welches durch uberschussige Salzsaure aus verdunnten Hydrazinlosungen aus- gefallt wurde. Durch Erhitzeri in1 Vakuum lie13 sich das Dichlorid unter Abspaltung von Chlorwasserstoff iii eine Schmelze von Monochlorid um- wandeln. Die gewahlten Arbeitsbedingungen (12-20 Torr, 180-190" C) gewahrleisteten selbstverstandlich auch die restlose Entfernung der letzten derri Salz eveiituell noch anhaftenden Spuren Wasser, so da13 eine besondere Trocknung des eingesetzten Dichlorids nicht erforderlich war. Die Einwirkung des Ammoniaks auf das schmelzflussige Mono- chlorid erfolgte durch Uherleiten des Gases bei etwa 160" C in derselben Apparatur, in der auch die Tiniwandlung des Salzes vorgenomnien wurde. (Nahere Einzelheiten sind aus dem experimentellen Teil zu erseheri.) Das gewonnene Hydrazin war wasserfrei, enthielt jedoch noch geringe

FEHBR, CREMER u. TROMM, Uber die Darstellung von wasserfreiem Hydrazin 181
Mengen Animoniak und Spuren von gebundenem Chlorwasserstoff. Die Entfernung dieser Verunreinigungen erfolgte ohne Schwierigkeiten durch Vakuum-Destillation. Zur Darstelluiig grol3erer Hydrazin- mengen im Laboratorium la Bt sich das Verfahren durch Parallelschalten mehrerer Reaktionsrohre beliebig erweitern. So lieferte eine Anlage von 4 Rohren bei Einsatz von 400-500 g feuchtem Hydraziniumdichlorid je Rohr in 50 Stunderi insgesanit etwa 500 em3 100proz. Hydrazin. Die Ausbeute an Hydrazin betrug etwa 90% bezogen auf die im Salz ge- bundene Base.
Um die Geschwindigkeit der Reaktion zu erhohen, versuchten wir Snimoniak bei Teniperaturen zwischen 130" und 190" C direkt in das geschniolzene Hydraziniummonochlorid (Schmelzpunkt etwa 90 O C) einzuleiten l a ) . Durch das gebildete Ammoniumchlorid wurde die Schmelze jedoch allmahlich fest, so dal3 Verstopfung des Einleitungs- rohres eine quantitative Umsetzung verhinderte. Versuche, das ge- schmolzene Monochlorid im Ammoniakstrom zu zerstauben, zeigten, daB die Reaktionsgeschwindigkeit hierdurch erheblich gesteigert werden kann. Dabei stellten sich aber hinsichtlich der Konstruktion des Zer- staubers im LabormaBstab zahlreiche Schwierigkeiten ein.
E xperimentelles 1. Extraktion des Monohydrazinsulfats mit
fliissigem Ammoniak unter Druck2) Der Extraktor besteht aus einer eisernen
Druckflasche von 1200 om3 Inhalt (Abb. 1) mit den1 aufschraubbarem Deckel D, der zwei mit Gewinde versehene Durchbohrungen besitzt. Die eine dient als Einfulloffnung fur das Hydrazinsulfat und zur Aufnahme eines in die Abbildung nicht eingezeich- neten Thermoelements, wahrend in der anderen ein Kreuzstuck mit den beiden Ventilen V, und V, und dem Manometer &I befestigt ist. An das Ventil V, ist die Steigleitung L angeschlossen, welche unten in dem kleinen Teller T endet, uber den die mit Glaswolle dicht gestopfte und mit dem Spreng- ring R gehaltene Hulse geschoben ist.
Zur Ausfuhrung einer Extraktion wurde fol- gende Arbeitsweise angewendet:
130 g (1 Mol) mehrfach aus heiBem Wasser umkristallisiertes, wasserfreies Monohydrazinsulfat
'*) Fur die rasche Darstellung wasserfreien Hydrazins besitzt das Verfahren den Vorzug, da5 kein besonderer apparativer Aufwarid erforderlich ist.
1 "2 M
? D
Abb. 1. Extraktor zur Darstellung yon wasserfreiem Hydrazin aus Monohydrazinsulfat und flussigem
Ammoniak

182 Zeitschrift fur anorganische und allgemeine Chemie. Band 287. 1956
N,H,. H,SO, werden mit etwa 100 Stahlkugeln (10 mm Durchmesser) zusammen in den Extraktor eingefiillt, der daraufhin verschlossen und mittels einer Eis-Kochsalz- IVIischung auf -10" C abgekiihlt wird. Nun 1aBt man trockenes Ammoniakgas langsam durch das Ventil Vl in die Steigleitung einstrBmen. Die Trocknung desselben erfolgt zweckmaoig durch Vorkondensation uber Natriurn 19). Das Ventil V, bleibt zu- nachst geoffnet und dieiit dazu, die in der Apparatur vorhandene Luft entweichen zu lassen. Auf diese Weise sattigt sich das Salz noch unter Normaldruck mit Ammoniak. Nachdem alle Luft vertrieben ist, wird das Vent,il V, geschlossen und weiter langsam Ammoniak zugefuhrt. Urn die Ammonolyse des Hydrazinsulfats nur ganz allmahlich verlaufen zu lassen, wird zunachst noch kein Ammoniak-Uberdruck erzeugt. Die Tem- peratur im Innern des Extraktors sol1 nicht uber + l o " C ansteigen, was durch Regelung der Ammoniakzufuhr leicht erreicht werdeu kann. Zur Ableitung der Reaktionswarme an die GefaRwandung leisten die Stahlkugeln gute Dienste.
Nach etwa 1 Stunde ist die Animonolyse weitgehend beendet, und es kann das zur Losung des Hydrazins noch benotigte Ammoniclk schneller und bei etwa 5 atii aufkon- densiert werden (insgesami etwa 600 g). Der Druckbehalter wird daraufhin 15 Minuten lang kraftig gschuttelt, um eine vollstandige Umsetzuug des Hydrazinsulfats mit Am- moniak und Auflosung des in Freiheit gesetzten Hydrazins zu bewirken. Die Stahlkugeln zerschlagen dabei Salzzusammenballungen und ersetzen so den Einbau eines Ruhrwerks. AuBenkiihlung ist hierzu nicht mehr erforderlich.
Vor dem Umfiillen der Hydrazin-Ammoniak-Losung kiihlt man den Behalter in einem groRen Dewar-Gefa5 auf etwa -35" C ab, so daR in seinem Innern kein Oberdruck mehr herrscht. Am Ventil Vl wird nun ein Metallschliffkern angeschraubt, der die Appara- tur uber eine Gasleitung an eine Destillationsapparatur anschliellt. Ventil V, wird ge- offnet uud durch Ventil V, mit getrocknetem Stickstoff ein geringer Uberdruck erzeugt. Der Extrakt laRt sich auf diese Weise in wenigen Minuten in den vorgekuhlten Destilla- tionskolben uberfiihren. War geniigend Ammoniak vorhanden, und die Losung gut, durchmischt, so erubrigt sich ein Nachspulen durch nochnialiges Aufkondensieren yon Ammoniak.
Das Ammoniak laBt man bei kraftiger Kiihlung der Destillationsbriicke langsam ab- dampfen, wobei der Gasaustritt gegen Eintritt von Luft und Feuchtigkeit entsprechend zu sichern ist. Das Hydrazin kann durch eine anschliefiende Vakuumdestillation weiter gcreinigt werden.
Die Ausbente hetragt etwa 28 g Hydrazin.
2. Reaktion zwischen geschmolzenem Hydraziniummonochlorid und gas- formigem Ammoniak l)
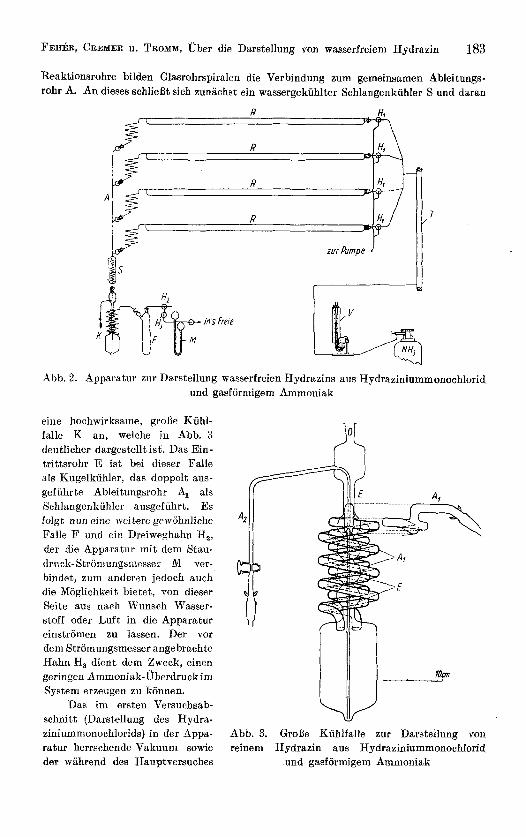
Die fur die Versuche mit 4 parallel geschalteten Reaktionsrohren verwendet,e Apparatur ist in Abb. 2 schematisch dargestellt. Die aus Aluminium angefertigten Rohre R besitzen einen au5eren Durchmesser von 45.mm und eine Lange von 1200 mm. An ihreni Eingang befindet sich je ein Dreiweghahn HI, der den wahlweisen AnschluB an die Ammoniakapparatur oder eine Wasserstrahlpumpe ermoglicht. Zur scharfen Trock- nung dea gasformigeii Ammoniaks dient ein mit Natriumdraht gefdlltes Rohr T. Wird jedoch - wie oben beschrieben - Natriuni in einer geeigneten Stahlbombelg) vorgelegt und Ammoniak aufkondensiert, so erubrigt sich das Trockenrohr. Am Ausgang der
l9) In unserem Falle diente hierzu eine eigens hergestellte Druckflasche aus Stahl (1,5 1 Inhalt), die zur Einfiillung des Natriums iind Entfernung des bei der Reaktion mit Wasser gebildeten Wasserstoffs mit einem leicht abschraubbaren Ventil versehen war.
FEHER, CAEMER 11. TROMM, uber die Darstellung yon wasserfreiem Hydrazin 183
Reaktionsrohre bilden Glasrohrspiralen die Verbindung zum gemeinsamen Ableitungs- rohr A. An dieses schlieBt sich zunachst ein wassergekiihlter Schlangenkuhler S und daran
Abb. 2. Apparatur zur Darstellung wasserfreien Hydrazins aus Hydraziniummonochlorid und gasformigem Ammouiak
eine hochwirksame, grolje Kiihl- falle K an, welche in Abb. 3 deutlieher dargestellt ist. Das Ein- trittsrohr E ist hei dieser Falle als Kngelkuhler, das doppelt aus- gefiihrte Ableitungsrohr 4 als Schlangenkiihler ausgefiihrt. Es folgt nun eine weitere gewohnliche Falle F iind ein Dreiweghahn H,, der die Apparatur mit dem Stau- druck-Stroniungsiiiesser M ver- bindet, zum anderm jedoch such die Moglichkeit bietet, von dieser Aeite aus nach Wunsch Wasser- stoff oder Luft in die Apparatur einstromen zu lassen. Der vor dem Stromungsmesser angebrachte Hahn H, dient dem Zweck, eiuen geringen Ammoniak-Uberdruckim System erzeugen zu kounen.
Das im ersten Versuchsab- schnitt (Darstellung des Hydra- ziniummonochlorids) in der Appa- ratur herrschende Vakuum sowie der wahrend des Hauptversuches
Abb. 3. GroRe Kiihlfalle zur Darstellung yon reinem Hydrazin ans Hydraziniummonochlorid
und gasformigem Ammoniak

184 Zeitschrift fur anorganische und allgemeine Chemie. Band 287. 1956
angewandte Ammoniak-uberdruck wird durch ein kombiniertes Druck-Vakuum-Meter V angezeigt, welches den Vorteil aufweist, durch sein groBes Ausgleichvolumen die bei anderen Vakuummetern notwendige Skalenverschiebung weitgehend ersparen zu konnen.
Samtliche Schliffverbindungen der Apparatur sind durch Stahlfedern gegen starken Uberdruck gesichert.
Zur Erwarmung der Reaktionsrohre werden elektrisch beheizte Aluminiumrohre (1260 x 50 mm) benutzt. Wahrend ein derartiger Rohrenofen bei AnschluB seiner beiden auReren Klemmen an 220 V Wechselstrom eine Hochsttemperatur von 125" C erreicht, kann diese durch geeignete Schaltung unter , Verwendung einer Mittelanzapfung der Heizwicklung verdoppelt und auBerdem jede RohrhaIfte getrennt eiugeschaltet werden. Ein Schiebewiderstand erlaubt die kontinuierliche Temperatureinstellung in jedem Bereich.
Die Temperaturmessung erfolgt in jedem Rohr durch zwei eingebaute Thermo- elemente mit einer Genauigkeit von &5" C. Die Versuche zur Darstellung von Hydrazin wurden wie folgt ausgefuhrt:
Nachdem die 4 Reaktionsrohre mit je etwa 500 g nutschenfeuchtem Hydrazinium- dichlorid so gefullt sind, daR das obere Drittel des Rohrquerschnittes frei von Salz bleibt, werden die Rohre auf 120 bis 150" C erhitzt und zur schnelleren Trocknung des Salzes mit Hilfe von Wasserstrahlpumpen ein lebEafter Luftstrom durch die Apparatur gesaugt. 1st das Salz trocken und in den Verbindungsleitungen keine Feuchtigkeit mehr zu erkennen, wird der Dreiweghahn H, geschlossen und die Temperatur der Rohre auf etwa 190" C erhoht. Bei dem sich nun eiustellenden Vakuum wandelt sioh das Hydraziniumdichlorid unter Abspaltung von Chlorwasserstoff im Verlaufe von 4-6 Stunden in das Mono- chlorid um. Da dieses einen Schmelzpunkt von ungefahr 90" C besitzt, liegt es nun in flussiger Form in den Reaktionsrohren vor. Die Temperatur wird jetzt auf 160" C herab- gesetzt und das Vakuum nach Umschaltung der vier Dreiweghahne langsam mit Ammo- niak aufgehoben. Nachdem daraufhin die Apparatur uber den Dreiweghahn H, mit den Stromungsmessern verbunden worden ist, kann durch wechselseitiges Bedienen des Nadelventils der Ammoniak-Bombe und des Hahnes H, ein geringer Uberdruck (0,2 bis .0,5 atii) sowie die geforderte Stromungsgeschwindigkeit (2-4 1/Min) eingestellt werden. Die beiden Kondensationsfallen werden auf -30" C abgekuhlt. Der ordnungsgemaBe Ablauf der Reaktion wird durch Kontrolle dcs Druckes, der Stromungsgeschwindigkeit, der Temperatur und der Tropfgeschwindigkeit des Hydrazins verfolgt. Die Umsetzung ist uach Ablauf von etwa 50 Stunden beendet.
Wahrend oder nach der Reaktion kann Hydrazin unter LuftabschluB uber das Ab- fiillrohr A, aus der groBen Kuhlfalle entnommen werden. Der zweite Stutzen eines hierzu verwendeten Zweihalskolbens dient zur Ableitung des freiwerdenden Ammoniaks. Die restlose Entfernung des gelosten Ammoniaks und auBerst geringer Mengen Hydrazinium- nionochlorides geschieht durch Vakuumdestillation. Wird nur die Beseitigung des Am- moniaks gofordert, so erwarmt man das Hydrazin unter RiickfluB im Wasserstoffstrom auf 100" C und laBt anschlieaend langsam abkuhlen.
Der Versuch liefert so in einem ArbeitsprozeB bei Einsatz von feuchtem Hydra- ziniumdichlorid und trockenem Ammoniak etwa 0,5 Liter 100proz. Hydrazin in etwa 90proz. Ausbeute. Eine Explosion ist bei Einhaltung der beschriebenen Reaktions- bedingungen nicht zu befiirchten.
3. Die quantitative Analyse des Hydrazins und seiner konzentrierten LSsungen Zur quantitativen Analyse samtlicher im Rahmen unserer Untersuchungen dar-
gestellten Hydrazinprodukte wurde die von PENNEMANN und AUDRIETH~~) beschriebene
20) R. A. P E N N E M A N u. L. F. AUDRIETH, Analytic. Chem. 20, 10.58 (1948).

FEHBR, CREMER u. TROMM, uber die Darstellung von wasserfreiem Hydrazin 185
direkte Jodat-Methode angewendet, bei der das Hydrazin in saurer Losung mit 0 , l mo- larer Kaliumjodat-Losung titriert wird. Der Vorteil dieser Methode beruht darauf, daIj durch die Titration in saurem Medium der EinfluW des Kohlendioxyds der Luft auf das Hydrazin ausgeschlossen und die Einwirkung des Luftsauerstoffs gemindert wird, die beide in akalisehem Medium Anla6 zu nicht unerheblichen Fehiern sein konnen. Die Reaktionsgleichung lautet :
N,H, + KJO, + 2 HCI + N, + JC1 + KCI + 3 HzO.
Fur den glatten Ablauf der Reaktion ist es erforderlicb, da13 die Losung eine Salzslure- konzentration von 3- bis 5-n aufweistZ1). Um eine hohe Genauigkeit zu erreichen, ist es ferner unerlafllich, sowohl die Einwaagen als auch das Verdunnen der Proben unter Luft- abschlufl durchzufuhren. Die Einwaagen wurden in besonders konstruierten Mikropipetten vorgenommen, und zum Verdiinnen wurde luftfreies, mit HCl angesauertes Wasser vorgelegt. Nachdem wir einige Erfahrung in der Handhabung des Hydrazins und der Durchfiihrung der Analysen gewonnen hatten, gelang es uns schlielllich, den Hydrazin- gehalt vorgelegter Proben mit einem maximalen Fehler vou iO,lyo zu bestimmen.
”) G. S . JAMIESOX, Z. analyt. Chem. 62, 782 (1913).
Koln, Anorganische und Analytische Abteilung des Chemischen Instituts der Universitat.
Bei der Redaktion eingegangen am 13. Juni 1956.
Z. anorg. allg. Chemie. Bd. 287. 13





























