Embed Size (px)
Citation preview

22
daft die M. v. S m o l u c h o w s k i ' s c h e Formel der elektroendosmotischen Geschwindigkeiten der FlfissigkeitsstrSme in parallelepipedischen Kam- mern auch air diese 3 bis 4 mm dicke Kfivette gfiltig ist. Es wird eine offene Kammer beschrie- ben, die ohne Aenderung ihrer Beobachtungs- stelle mit Fltissigkeit wahrend der Messungen neu geffillt werden kann. Durch wiederholtes Neuffillen kann man je nach Belieben die stBrende Wirkung der Abbauprodukte der Elektrolyse aus- schalten. Es kfnnen in dieser Kfivette schon an einer einzigen Beobachtungsstelle so viele Messungen ausgeffihrt werden, dab sie zur ge-
nauen Ermittlung der wahren kataphoretischen Geschwindigkeit der Kolloidteilchen geniJgen.
Diese Arbeff wurde im agrikulturchemischen Laboratorium der EidgenBssischen Technischen Hochschule in Zfirich ausgeffihrt. Es sei mir an dieser Stelle gestattet, dem Vorstand des erwahn- ten Laboratoriums, Herrn Prof. Dr. G. W'i egn e r, meinen herzlichen Dank auszusprechen f/ir sein unermfidliches Interesse, seine wertvollen Rat- schlage, sein weitgehendes Entgegenkommen, sowie far die Erleichterungen bei der Durch- f~hrung der praktischen Arbeiten in seinem La- boratorium.
/Jeber die Methodik der kataphoretischen Messungen bei Suspensoiden. (Eingegangen am 10. Oktobor 1927.)
Von H. R. K r u y t und P. C. v a n d e r W i l l i g e n (Utrecht).
Der eine 1) yon uns hat vor einigen Jahren in dieser Zeitschrift eine Uebersicht gegeben yon den Methoden, die bis dahin angewandt worden sind zur Messung der elektrischen La- dung kolloider Teilchen.
In dieser Abhandlung werden wir einige neuere Methoden besprechen, welche angewandt worden sind bei der Untersuchung der katapho- retischen Geschwindigkeit (K.G.) yon Suspen- soiden. Far die Bestimmung der K.G. yon Suspensoiden eignen sich prinzipiell sowohl die makroskopische, als auch die mikroskopische Methode. Dennoch 1/iBt sich in vielen Fallen nut eine der beiden Methoden erfolgreich an- wenden. Amikroskopische Sole, wie das AszSs-Sol und das F%O3-Sol kann man ja nur makroskopisch messen, ebenso wie solche Sole, welche zu konzentriert sind, daft sich die Einzel- teilchen gen~gend genau beobachten lassen. Anderseits wird man vorzugsweise die mikro- skopische Methode anwenden bei Solen, welche ungenfigend farbig sind oder welche in so ge- ringer Konzentration vorliegen, dab sich eine Trennungsflache nicht scharf beobachten 1/iBt.
Wir haben ein Goldsol und ein Selensol nach beiden Methoden untersucht, damit wir imstande waren, die Resultate beider Methoden an ein und demselben Sole zu vergleichen (vgl. S. 31).
I. Makroskopische Methode. Der Schwierigkeiten, welche vorliegen bei
der Anwendung des B u r t o n'schen Apparates, sind so viele und so verschiedener Art, daft wir
1) H. R. Kruy t , Koll.-Zeitschr. 37, 358 (1925).
die Methodik getrennt besprechen werden f/Jr reine elektrolytarme Sole und f/Jr solche, zu denen man Elektrolyte hinzugesetzt hat.
K a t a p h o r e s e e l e k t r o l y t a r m e r S o l e . Die Bildung von Elektrolyseprodukten an
den Elektroden und die Warmeentwicklung durch den elektrischen Strom sind bei diesen Solen so geringffigig, dab man mit diesen beiden Fak- toren in erster Linie keine Rechnung zu halten braucht. Vorlaufig machen wir darum die Voraus- setzung, dab man den ursprfinglichen B u r t o n - schen Apparat 2) in diesem Falle anwenden kann. Auf S. 24 wird abet diskutiert werden, dab in vielen Fallen der Gebrauch dieses Apparates Komplikationen herbeiffihrt; daraus zeigt sich, dab auch bei elektrolytarmen Solen der B u r t o n - sche Apparat sich nicht verwenden lafit. Derselbe wird in einem quadratischen glasernen Behalter aufgestellt, welcher Wasser bekannter Temperatur enthalt. Die K/icken der Hahne sind mit gla- semen St~iben versehen, um Oeffnung der H/ihne unter dem Wasser zu ermSglichen. Die beiden Rohre des Kataphoreserohres tragen eine Skala in Millimetern. Die Beobachtung der Grenz- flache wird erleichtert, indem man, wie bei einer Bfirette, einen Spiegel hinter den Beh~ilter stellt, welcher um eine vertikale Achse drehbar ist. Aufierdem stellt man hinter den Behalter ein St/ick Pappkarton in weiB oder schwarz zur Er- leichterung der Beobachtung bei dunkelgefarbten bzw. hellgefarbten Solen.
2) E. F. B u r t o n , Phil. Mag. [6] 11, 425 (1906); siehe E. F. B u r t o n , The Physical Properties of Colloidal Solutions (1916), 130.

23
AuBerordentlich wichtig ist die Frage, welche Flfissigkeit fiber das Sol geschichtet werden soll. Nach B u r t o n S ) soll man dazu eine LSsung verwenden, die gleiche Leitf[ihigkeit hat wie das Sol. Z u r Berechnung der PotentialgeEille pro Zentimeter wird die Spannungsdifferenz zwischen den Elektroden dividiert durch deren Distanz. Diese Berechnungsweise ist nut dann richtig, wenn die LeitEihigkeit im ganzen Rohr konstant ist. Daraus geht die Forderung hervor, eine Flfissigkeit mit gleicher Leitf~ihigkeit zur Ueber- schichtung zu verweriden. B u r t o n l~iI~t dahin- gestellt sein, welche Elektro]yte man verwenden soll zur Erhaltung der gleichen Leitfahigkeit.
Mu k h e r j e e 4) empfiehlt dazu Salzs~iure. In dieserWeise erzielt man, dat~ die Leitfiihigkeit im ganzen Rohr gleich ist, aber einer anderen Forderung wird kein Genfige geleistet, denn die hinzugesetzten Elektrolyte werden nur aus- nahmsweise dieselben sein und in derselben Konzentration hinzugeffigt werden kSnnen, als wie sie im Sol vorliegen. Deshalb wird die Grenzfl~iche der Sitz eines Diffusionspotentials sein. Die Potentialgefalle, welche man wie oben angegeben berechnet hat, werden dann an den Trennungsfl[ichen einen abnormen Wert haben, und gerade die Potentialgef[ille dort sind bestimmend ffir die K.G. Aui~erdem ist zu be- ftirchten, dab das hinzugesetzte Fremdelektrolyt die K.G. an der Grenzfl~iche beeinflut~t.
Es ist vielmehr angemessen, daffir Sorge zu tragen, dab d i e Kolloidteilchen sich im selben Medium bewegen, als sie im Sol zuvor hatten. Das ist" eben der Vorteil bei der mikroskopischen Methode, und man erreicht gleiches, wenn man bei der makroskopischen Methode die intermi- zellare Flfissigkeit fiber das Sol schichtet.
Zwei Methoden erm6glichen solches, Ultra- filtration und Zentrifugation. Die letztere Methode ist aber unbrauchbar, insoweit es sich um Ultra- mikronen handelt, wenigstens wenn man nicht eine Superzentrifuge anwendet. Nur grobe Suspensionen, wie Glas und Kohle oder Emul- sionen, werden sich ffir diese Methode eignen. Die Ultrafiltration aber eignet sich daftir viel besser, vor allem, seit B e c h h o l d und K 6 n i g 5) eine ganz besonders geeignete Methode vorge- schlagen haben.
Der Ultrafiltrationstiegel ist aus Porzellan herge- stellt, das an der Augenseite zum Teile glasiert ist.
a) E. F. B u r t o n , loc. cit. 4) j .N. M u k h e r j e e , Proc. Roy. Soc. London
[A] 103, 102 (1923). 5) H. B e c h h o l d u. L. G u t l o h n , Zeitschr. f.
angew. Chem. 37, 494 (1924).
Der Tiegel spielt die Rolle einer Untersttitzung fiir die Kollodiummembran, die im Inneren des Tiegels angebracht wird.
Als Impr~gnierungsfltissigkeit wird Kollodium (de Hahn) verwendet, welches in Alkohol und Aether gel6st ist. 0,5prozentige LOsungen geniigen in den meisten F~illen zur Erhaltung eines klaren Filtrates:
Wit benu~zten folgende Arbeitsmethode: DerTiegel wird auf einen Absauge-Erlenmeyer gestellt und Wasser wird durchgesaugt. Naehdem dieses abgesaugt worden war, wurde tier Tiegel mit der Kollodiumltisung geftillt und sofort wieder entleert. Die geringe Menge, welche zuriickbleibt, wurde w/ihrend 5 Minuten an der Luft getrocknet, dann wurde Wasser durchgesaugt zur Ent- fernung des Alkohols und des Aethers. Wit haben immer den Gebrauch der KollodiumlOsung in Alkohol und Aether dem in Eisessig vorgezogen, weil im ersten Falle kein Elektrolyt ausgewaschen zu werden braucht. Bei der Ultrafiltration wurden immer die ersten durchgehenden 200 ccm weggeworfen. Bei dem Ultrafiltrationstiegel in Form eines Btichner-Trichters war es mOglich, bei einem Druck yon 2--4 cm Queck- silber ca. 0,5 Liter Sol pro Stunde zu ultrafiltrieren. Zur Entfernung einer gebildeten Kollodiumschicht mit Teilchen veffahrt man so, dab man den Tiegel trocknet. Es bilden sich dann Risse in der Schicht, welche sich dann leicht entfemen l~it~t. Das Durchsaugen yon Wasser, bevor man den Tiegel benutzt, bezweckt, die Poren des Porzellans mit Wasser zu ftillen. Unterl/iBt man solches, so wird die Kollodiummembran nicht nur an der Oberfl~iche, sondern auch innerhalb des Porzellans gebildet. Nach der Trocknung ist es dann nicht mehr moglich, die Kollodiummembran mit den Kolloidteilchen zu entfernen. Zur endgiiltigen Reini- gung des Tiegels kann man denselben mit heifier Salpetersaure oder KOnigswasser extrahieren. Es zeigt sich nicht moglich, die letzten Spuren dieser S~iuren durch Auswaschen mit Wasser zu entfemen. Dazu ist es unbedingt notwendig, den Tiegel w~ihrend einiger Zeit zu erhitzen.
Die Leitfiihigkeit von verschiedenen Solen bzw. intermizellaren Fltissigkeiten wurde bei 25 0 gemessen in einem Gef~it~ mit platinierlen Platin- elektroden.
Wir geben einige Beispiele in Tabelle I.
T a b e l l e I:
~Sol X 10 5. ~int. F1. X I 0 5 in Ohm--1 in Ohm--1
Au-Sol (0,13 g pro L.) HgS-Sol (7,5 g pro L.) HgS-Sol (7,5 g pro L.) As2S~-Sol (6 g pro L.) As2S3-Sol (6 g pro L. As2Ss-Sol (22 g pro L As~Ss-Sol (22 g pro LI
7,60 5,95 5,35
21,0 20,8 10,76 16,15
7,57 5,43 4,97
19,3 17,9 8,55 6,21
Wie die Tabelle zeigt, sind die Unterschiede dann und wann recht grot~; zwar ist der Unter- schied bei dem erw~ihnten Goldsol sehr gering- ffigig, bei dem As 2 $3-Sol aber recht groB, Wenn man K.G.-Messungen an diesem Sole macht, wird also der oben angegebenen Forderung

24
kein Gen/ige geleistet: die Leitf/ihigkeiten des Sols und der fiberschichtenden Fl/issigkeit sind ungleich. Wir werden aber bald sehen, dai~ es m6glich ist, eine Korrektur anzubringen f/Jr die genaue Berechnung der K.G.
In einem B u r t o n'schen Apparat wurde die K.O. des letzten in Tabelle I angegebenen Sols gemessen. Die Entfernung der Elektroden war 236 ram. Die Trennungsfl/khe zwischen Sol und intermizellarer Fltissigkeit war in beiden R6hren 60,5 mm unter den Elektroden.
Tabelle II gibt die Resultate:
T a b e l l e II. F
Zeit Volt Linkes Rohr ~Rechtes Rohr Temp.
4,434'28 --[-75+75 66,3 . ~ . 52,8}1o, a 64,9/~, 13,9 70,0) ~'i 13,9
Man entnimmt dieser Tabelle sofort, dab die Verschiebung der Trennungsfltiche w I im linken Rohr viel gr6Ber ist als diese im rechten w2. Die emporsteigende Orenzflttche schreitet in einem Medium, das eine Leitftihigkeit hat, welche kleiner ist als die des Sols. Das Potentialgef/ille pro Zentimeter wird deshalb dort gr6Ber sein und deshalb auch die Oeschwindigkeit der Tren- nungsfltiche. Es ist bemerkenswert, dab das Sol, dort wo es gestiegen ist, oberhalb der ursprting- lichen Trennungsfl/iche immer durchsichtiger und verdtinnter ist als das tibrige Sol. Aus dem eben Oesagten ist das verstttndlich: das Sol ist dort mehr oder weniger ,ausgezogen".
Die Oesamtltinge der intermizellaren Flfissig- keitskolumne ist hi = 2 x 6 0 , 5 ----- 121 mm, die L~nge des Solkolumnes h2 = 236- - 121 = 115 ram.
Die angebrachte Potentialdifferenz E k6nnen wir uns denken getrennt in Teilen yon Elektro- den bis zu Grenzfltichen, je 1/2 el, und yon einer Trennungsfltiche zur anderen Trennungsfl/khe e2 ; deshalb ist
e l + e2 = E. (1) Das Potentialgefiille pro Zentimeter ist der
Leitfiihigkeit umgekehrt proportional, deshalb:
ei e2 - - : - - ~ XSol :~int . Fl. ( 2 ) hi h2
Die Werte den Tabellen entnehmend, erh~ilt man f/ir (1) und (2)
el + e~ = 75
ei e~ - -16 ,15 :6 ,21 . 12,1 : 11,5
Hieraus e2
e--L = 4,55 und -h~=l ,75Vol tp roZen t ime te r . hi
Die Oeschwindigkeit vi im linken Rohr ist dann (wenn Z die Zeit angibt)
wi 1,35 - - - - 3 , 3 ~ .
Z ei 900 X 4,55
hi Ebenso findet man ffir die Oeschwindigkeit im rechten Rohr
0,51 - - 3 , 2 ~ . v2 -- 9 0 0 x l , 7 5
DiesevorztiglicheUebereinstimmungzwischen den beiden Oeschwindigkeiten ist eine Besttiti- gung unserer Annahme, dab die Oeschwindig- keiten den Leitftihigkeiten umgekehrt proportional sind.
Nun da wir diese Hypothese als bewiesen erachten kOnnen, ItiBt sich eine Beziehung ab- leiten, welche die Oeschwindigkeit v angibt aus den Oeschwindigkeiten in den beiden ROhren, ohne dab man die Leifftihigkeiten der intermi- zellaren Fltissigkeit und des Sols zu wissen braucht.
wi hi wi , also e 1 - -
z e l Z v hi
w2 w2 h2 v - - - - , also e 2 - -
Z e~ Z v h2
zusammen :
el'+" e2 ~ E -- w l h l + w2 h2 oder v = wlhi-+- w~ h~ Z v ZE
(3) Wendet man dieses Resultat 6) an auf oben-
stehendes Beispiel, so findet man v = 3,29 ~t.
Ffir hi = h2 = 1/2 H ergibt sich ~/~ (wl + w~) H
V EZ
Dieser letzte Wert ist derjenige, welchen man bisher im allgemeinen in Rechnung brachte : die mittlere Verschiebung ~/2 (wi + w2) dividiert
E durch das Potentialgefiille pro Zentimeter -H-
und durch die Zeit Z. Wir wir gesehen haben, ist solches nur erlaubt im Spezialfalle hi = h2, d. h. wenn die Summe der L~ingen der beiden Saulen der intermizellaren Fliissigkeit der Liinge der Solsiiule gleich ist.
s) Neuerdings hat J. B i k e r m a n, Koll.- Zeitschr. 42, 293 (1927), eine ahnliche Formel gegeben, ohne die Herkunft anzugeben.
Es ist ja
V - - - -

25
Nach B u r t o n 7) sollte man die Verschiebung der Grenzfl~ichen nur messen, nachdem die Stromrichtung umgekehrt sei. Augenscheinlich bevorzugt er solches, weft man in dieser Weise mehr fihnliche Resultate erhalt. Nun da wir aber wissen, wie die Verhfiltnisse quantitativ liegen, liegt kein Grund mehr vor, diese Kom- plikation beizubehalten.
Zur Prfifung obenstehender Formel (3) haben wir Messungen gemacht an einem As~S3-Sol mit 17,5 g As~S3 pro Liter. Die benutzte Appa- ratur wird auf S. 27 beschrieben werden. Die Messungen sind in einem Thermostaten bei 200 ausgeffihrt. W~ihrend der Messungen soll man den Rfihrer im Thermostaten abstellen, damit die Trennungsfl/ichen nicht zerstSrt werden; die Temperaturschwankungen fibersteigen dennoch 0,10 nicht. Die Skala auf den R6hren war in Millimetern mit 0 oben und 70 unten. Der Ab- stand H zwischen den Elektroden, welche genau auf die Skala 0 gestellt waren, wurde zuerst genau gemessen. Das Rohr war geffillt mit einer KCI-LSsung von 34 m Mol pro Liter. Dann wurde der Widerstand gemessen; dieser betrug R 1 = 2940 -(2-. Dann wurden die Elek- troden auf Skala 35 gestellt und der Widerstand wieder gemessen, Re = 1957,(2-; eine dritte Messung wurde gemacht, indem die Elektroden auf Skala 70 gestellt wurden: das Resultat war R3 = 950 ~. Wir nennen das Lumen des Rohres r und g die spezifische Leitfiihigkeit der KCI-L6sung. Man hat dann:
H 1 R 1 - - - - 2940
r 2
H - - 7 , 0 1 R ~ - - - 1957
r 2 z
H - - 1 4 , 0 1 _ 950. Ra - - u r 2 z
T a b e l l e II1.
Zeit
11,35 12,16
E Linkes Rohr
~2 28,3`(77 36,oj""
2,42 65,3J ~176
Rechtes Rohr
29,6 10,1} 19,5
v in ~ bei 200
4,21
61,0 , ~ 44,4} l~176
3,29 22 56,7)~. 62,5 4,10 62,2j~,o 47,1}15,4 4,28
12,01 22 39517^ 47,8 12,46 46:Sj,,v 28,4}19,4 4,26
4,27
7) E. F. B u r t o n , loc. cit.
Aus diesen Gleichungen findet man drei Werte f/ir H mit einem Mittelwert von 20,7cm.
In Tabelle III geben wir nun die Resultate der K.O.-Messungen, welche alle innerhalb zweier Tage gemacht worden sind. Bei jeder Messung wurden die Trennungsfl~ichen an einem anderen Punkt der Skala angebracht; h 1 und h 2 wurden also variiert. Die letzte Kolumne gibt nun K.G., berechnet aus obenstehender Formel.
Die Werte ffir v stimmen gut fiberein. Aus der Tabelle findet ~man, dab die Versuchsdauer 41 bzw. 45 Minuten war. Im allgemeinen ist es nfitzlich, die Dauer des Versuches nicht zu lang zu machen, denn die Erniedrigung der Leitf~.hig- keit in den Seitenr6hren (vgl. S. 27) soll nicht bis zu den Hilfselektroden heranragen. AuBer- dem ~indern sich selbstverstandlich hi und h2 allm~ihlich, und auch diese Aenderung soll einen gewissen Wert nicht fibersteigen. DasVerhfiltnis zwischen den "Leitffihigkeiten des Soles und der intermizellaren Flfissigkeit war bei diesem As2 S3- Sol 2,7. Man kann nun der Tabelle. wieder ent- nehmen, dab die Verschiebungen tatsfichlich der Leitfahigkeit umgekehrt proportional sind. Ffir das Verh~iltnis der Verschiebungen berechnet man n~imlich aus der Tabelle ffir die vier Versuche bzw. 2,5, 2,8, 2,8 und 2,6.
Der gr6Beren erreichten Genauigkeit zufolge war es nun auch m6glich zu prfifen, ob wirklich die gefundene K.G. unabh~ingig vom Potential- gefiille sei. Neben der in Tabelle III erw~ihnten Messung bei 22 Volt wurden nun Messungen bei 44 bzw. 11 Volt zwischen den Hilfselektroden gemacht.
Die Resultate sind in Tabelle IV zusammen- gestellt.
T a b e l l e IV.
Zeit E Linkes Rohr Rechtes Rohr vin~bei20 o
11,31 44 50,3]~. 55,4 . ~ . 11,51 44 55,7J ~# 39,7}Io,I 4,18
11,304444 55,6"(. ~ 50,8`(lo 11,54 61,3[ ~ 31,Oj . . . . 4,23
3,30 11 5,06 11
1,50 11 3,21 11
52,0/5 8 57,8J,
5o,9v. 57,4J ~176
35012}19,8
54,0 , o 36,0} l o,v
4,15
4,25
:~;~ Die Tabelle zeigt, dab die K.G., gemessen bei Potentialgeffillen yon ungeffihr 2 ~ 0 , 5 Volt pro Zentimeter, innerhalb der Versuchsfehler gleich sind.
Es sei noch darauf hingewiesen, dab immer die emporsteigende Grenzfl~iche sich scharf
26
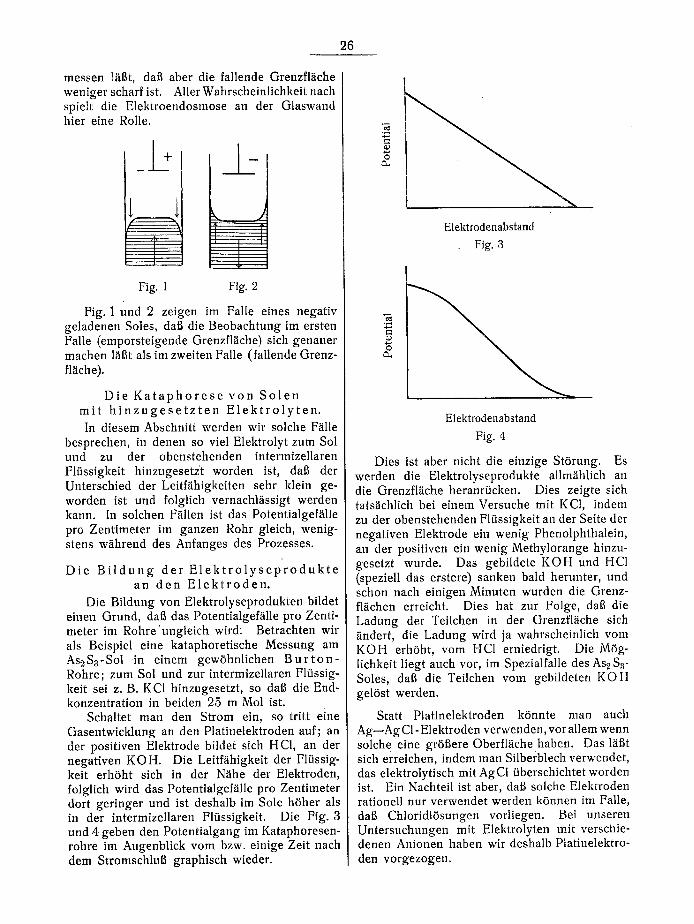
messen 1/iBt, dab aber die fallende Grenzfl~iche weniger scharf ist. AllerWahrscheinlichkeit nach spielt die Elektroendosmose an der Glaswand hier eine Rolle.
I I
[ I I I
H- il
I , , l i Fig. 1 Fig. 2
Fig. 1 und 2 zeigen im Falle eines negativ geladenen Soles, dab die Beobachtung im ersten Falle (emporsteigende Grenzfl~iche) sich genauer machen l~iBt als im zweiten Falle (fallende Grenz- fl/iche).
D i e K a t a p h o r e s e v o n S o l e n m i t h i n z u g e s e t z t e n E l e k t r o l y t e n . In diesem Abschnitt werden wit solche F~ille
besprechen, in denen so viel Elektrolyt zum Sol und zu der obenstehenden intermizellaren Fl/issigkeit hinzugesetz't worden ist, dat~ der Unterschied der Leitf/ahigkeiten sehr klein ge- worden ist und folglich vernachl~issigt werden kann. In solchen F/illen ist das Potentialgef/ille pro Zentimeter im ganzen Rohr gleich, wenig- stens w~ihrend des Anfanges des Prozesses.
D i e B i l d u n g d e r E l e k t r o l y s e p ' r o d u k t e an d e n E l e k t r o d e n .
Die Bildung von Elektrolyseprodukten bildet einen Grund, dab das Potentialgef/ille pro Zenti- meter im Rohre'ungleich wird: Betrachten wir als Beispiel eine kataphoretische Messung am A%S3-Sol in einem gew6hnlichen B u r t o n - Rohre; zum Sol und zur intermizellaren Fl/issig- keit sei z. B. KCI hinzugesetzt, so dab die End- konzentration in beiden 25 m Mol ist.
Schaltet man den Strom ein, so tritt eine Gasentwicklung an den Platinelektroden auf; an der positiven Elektrode bildet sich HC1, an der negativen KOH. Die Leitf~ihigkeit der Fltissig- keit erh6ht sich in der N/ihe der Elektroden, folglich wird das Potentialgef/ille pro Zentimeter dort geringer und ist deshalb im Sole h6her als in der intermizellaren FKissigkeit. Die Fig. 3 und 4 geben den Potentialgang im Kataphoresen- rohre im Augenblick vom bzw. einige Zeit nach dem StromschluB graphisch wieder.
O a ~
Elektrodenabstand
Fig. 3
rq
Elektrodenabstand
Fig. 4
Dies ist abet nicht die einzige St6rung. Es werden die Elektrolyseprodukte allm/ihlich an die Orenzfl~iche heranrticken. Dies zeigte sich tats~ichlich bei einem Versuche mit KC1, indem zu der obenstehenden F1/issigkeit an der Seite der negativen Elektrode ein wenig Phenolphthalein, an der positiven ein wenig Methylorange hinzu- gesetzt wurde. Das gebildete KOH und HC1 (speziell das erstere) sanken bald herunter, und schon nach einigen Minuten wurden die Grenz- flgtchen erreicht. Dies hat zur Folge, dat~ die Ladung der Teilchen in der Orenzfl~iche sich ~indert, die Ladung wird ja wahrscheinlich vom K O H erh6ht, vom HC1 erniedrigt. Die M6g- lichkeit liegt auch vor, im Spezialfalle des As 2 S 3- Soles, dab die Teilchen vom gebildeten K O H gel6st werden.
Statt Platinelektroden k6nnte man auch Ag--AgCI- Elektroden verwenden, vor allem wenn solche eine gr6t~ere Oberfl~che haben. Das i/iBt sich erreichen, indem man Silberblech verwendet, das elektrolytisch mit AgCI/iberschichtet worden ist. Ein Nachteil ist aber, dab solche Elektroden rationell nut verwendet werden k6nnen im Falle, dab Chloridl6sungen vorliegen. Bei unseren Untersuchungen mit Elektrolyten mit verschie- denen Anionen haben wir deshalb Platinelektro- den vorgezogen.
7
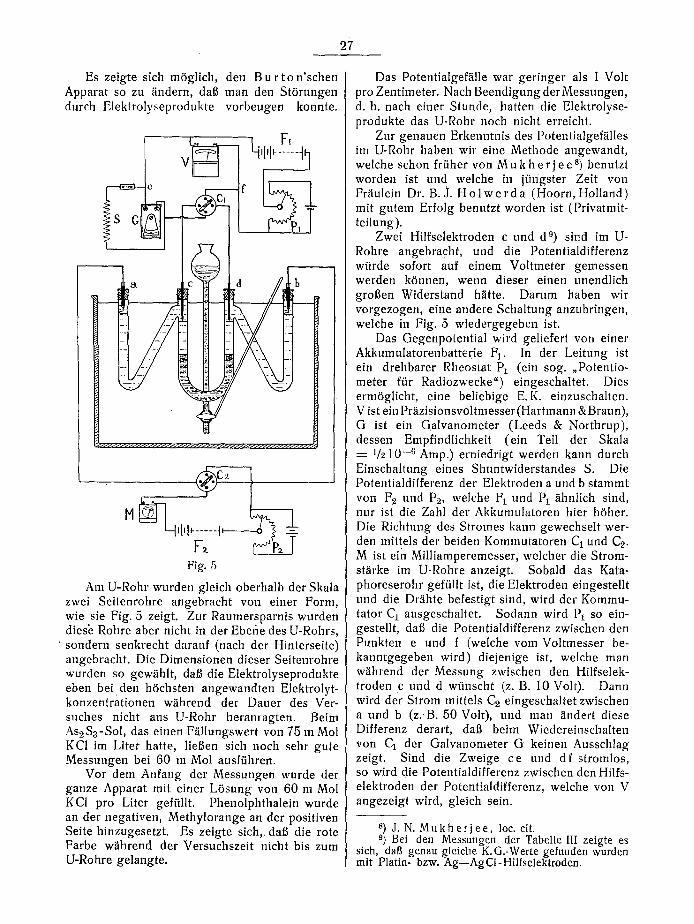
Es zeigte sich m6glich, den B u r t o n ' s c h e n Apparat so zu ~indern, daf man den St6rungen dutch Elektrolyseprodukte vorbeugen konnte.
,I,I~ ..... ~ I
Fig. 5
Am U-Rohr wurden gleich oberhalb der Skala zwei Seitenrohre angebracht yon einer Form, wie sie Fig. 5 zeigt. Zur Raumersparnis wurden diesh Rohre aber nicht in der Ebefie des U-Rohrs,
' sondern senkrecht darauf (nach der Hinterseite ) angebracht. Die Dimensionen dieser Seitenrohre wurden so gew~ihlt, daft die Elektrolyseprodukte eben bei den h6chsten angewandten Elektrolyt- konzentrationen w/ihrend der Dauer des Ver- suches nicht ans U-Rohr heranragten. Beim As~S3-Sol, das einen F/~llungswert yon 75 m Mol KCI im Liter hatte, lieBen sich noch sehr gute Messungen bei 60 m Mol ausffihren.
Vor dem Anfang der Messungen wurde der ganze Apparat mit einer L6sung yon 60 m Mol KCI pro Liter geffillt. Phenolphthalein wurde an der negativen, Methylorange an der positiven Seite hinzugesetzt. Es zeigte sich,.daB die rote Farbe w~ihrend der Versuchszeit nicht bis zum U-Rohre gelangte.
Das Potentialgef/ille war geringer als 1 Volt pro Zentimeter. Nach Beendigung derMessungen, d. h. nach einer Stunde, hatten die Elektrolyse- produkte das U-Rohr noch nicht erreicht.
Zur genauen Erkenntnis des Potentialgef~illes im U-Rohr haben wit eine Methode angewandt, welche schon fr/iher yon M u k h e r j e e s) benutzt worden ist und welche in j/Jngster Zeit yon Fr~iulein Dr. B.J. H o l w e r d a (Hoorn, Holland) mit gutem Erfolg benutzt worden ist (Privatmit- teilung).
Zwei Hilfselektroden c und d 9) sind im U- Rohre angebracht, und die Potentialdifferenz wtirde sofort auf einem Voltmeter gemessen werden k6nnen, wenn dieser einen unendlich grofen Widerstand h/itte. Datum haben wir vorgezogen, eine andere Schaltung anzubringen, welche in Fig. 5 wiedergegeben ist.
Das Gegenpotential wird geliefert von einer Akkumulatorenbatterie F 1. In der Leitung ist ein drehbarer Rheostat Pl (ein sog. ,Potentio- meter ffir Radiozwecke") eingeschaltet. Dies erm6glicht, eine beliebige E.K. einzuschalten. V ist ein Priizisionsvoltmesser (Hartmann &Braun), G ist ein Galvanometer (Leeds & Northrup), dessert Empfindlichkeit (ein Teil der Skala
1/210 -6 Amp.) erniedrigt werden kann durch Einschaltung eines Shuntwiderstandes S. Die Potentialdifferenz der Elektroden a und b stammt yon F~ und P2, welche F 1 und t)1/ihnlich sind, nut ist die Zahl der Akkumulatoren hier h6her. Die Richtung des Stromes kann gewechselt wer- den mittels der beiden Kommutatoren C1 und C~. Mi s t ein Milliamperemesser, welcher die Strom- st/irke im U-Rohre anzeigt. Sobald das Kata- phoreserohr geffillt ist, die Elektroden eingestellt und die Dr~ihte befestigt sind, wil:d der Kommu- tator C 1 ausgeschaltet. Sodann wird P1 so ein- gestellt, daft die Potentialdifferenz zwischen den Punkten e und f (welche vorn Voltmesser be- kanntgegeben wird) diejenige ist, welche man w~ihrend der Messung zwischen den Hilfselek- troden c und d wtinscht (z. B. 10 Volt). Dann wird der Strom mittels C~ eingeschaltet zwischen a urld b (z.,B. 50 Volt), und man /indert diese Differenz derart, daft beim Wiedereinschalten von C1 der Galvanometer G keinen Ausschlag zeigt. Sind die Zweige ce und df stromlos, so wird die Potentialdifferenz zwischen den Hilfs- elektroden der Potentialdifferenz, welche von V angezeigt wird, gleich sein.
e) j. N. M u k h e r j e e , loc. cit. 9) Bei den Messungen der Tabelle III zeigte es
sich, dab genau gleiche K.G.-Werte gefunden wurden mit Platin- bzw. Ag--AgCI-Hilfselektroden.
28
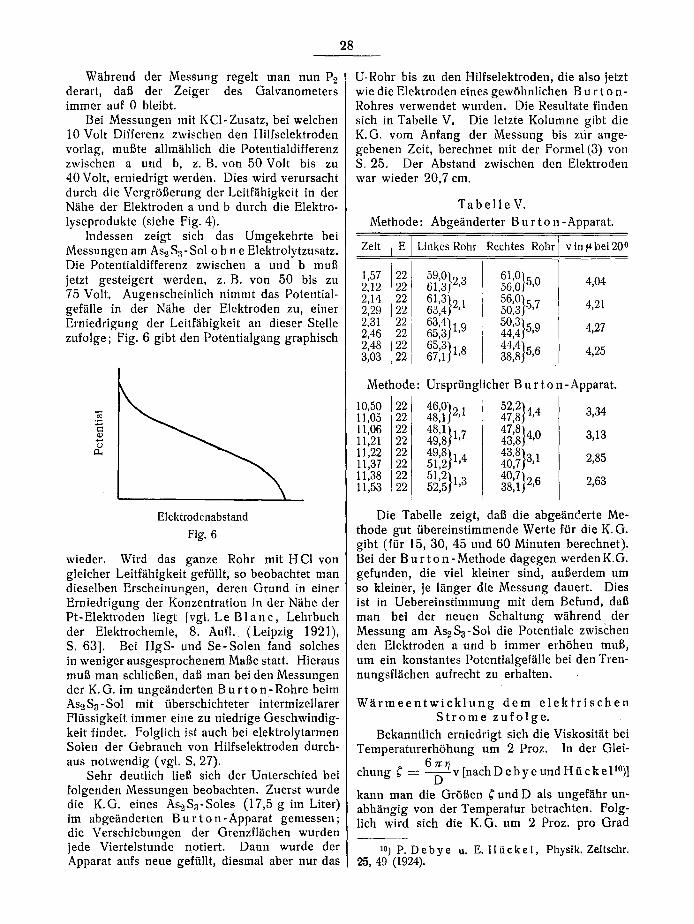
W~ihrend der Messung regelt man nun P~ derart, dat~ der Zeiger des Galvanometers immer auf 0 bleibt.
Bei Messungen mit KCI-Zusatz, bei welchen 10 Volt Differenz zwischen den Hilfselektroden vorlag, mui~te allm~ihlich die Potentialdifferenz zwischen a und b, z. B. von 50 Volt bis zu 40 Volt, erniedrigt werden. Dies wird verursacht durch die Vergr51~erung der Leitf~ihigkeit in tier N~ihe der Elektroden a und b durch die Elektro- lyseprodukte (siehe Fig. 4).
Indessen zeigt sich das Umgekehrte bei Messungen am As2 $3-Sol o h n e Elektrolytzusatz. Die Potentiatdifferenz zwischen a und b mut~ jetzt gesteigert werden, z .B. von 50 bis zu 75 Volt. Augenscheinlich nimmt das Potential- gef~ille in der N~ihe der Elektroden zu, einer Erniedrigung der Leitf~ihigkeit an dieser Stelle zufolge; Fig. 6 gibt den Potentialgang graphisch
Elektrodenabstand
Fig. 6
wieder. Wird das ganze Rohr mit H CI von gleicher Leitf~ihigkeit gef/illt, so beobachtet man dieselben Erscheinungen, deren Grund in einer Erniedrigung der Konzentration in der Niihe der Pt-Elektroden liegt [vgl. Le B l a n c , Lehrbuch der Elektrochemie, 8. Aufl. (Leipzig 1921), S. 63]. Bei HgS- und Se-Solen fand solches in weniger ausgesprochenem Mat~e statt. Hieraus muff man schliet~en, daft man bei den Messungen der K.G. im unge~inderten B u r t o n - Rohre beim As2Ss-Sol mit tiberschichteter intermizellarer Flfissigkeit immer eine zu niedrige Geschwindig- keit findet. Folglich ist auch bei elektrolytarmen Solen der Gebrauch von Hilfselektroden durch- aus notwendig (vgl. S. 27).
Sehr deutlich lie[~ sich der Unterschied bei folgenden Messungen beobachten. Zuerst wurde die K.G. eines As~S3-Soles (17,5 g im Liter) im abgeiinderten B u r t o n - A p p a r a t gemessen; die Verschiebungen der Grenzfl~ichen wurden jede Viertelstunde notiert. Dann wurde der Apparat aufs neue geftillt, diesmal aber nut das
U-Rohr bis zu den Hilfselektroden, die also jetzt wie die Elektroden eines gewShnlichen B u r t o n- Rohres verwendet wurden. Die Resultate linden sich in Tabelle V. Die letzte Kolumne gibt die K.G. vom Anfang der Messung bis ztir ange- gebenen Zeit, berechnet mit der Formel (3) yon S. 25. Der Abstand zwischen den Elektroden war wieder 20,7 cm.
T a b e l l e V . Methode: Abge~inderter B u r t o n - Apparat.
Zeit E l Linkes Rohr Rechtes Rohr vin~bei20 o
1,57 2,12 2,14 2,29 2.31 2,46 2,48 3,03
ii 59,0/. o
22 61,3J z'o 22 61,3~9 1 22 63,4}-'" 22 63,4]~ 22 65,3J 1'~ 22 65,3] 1
67,1J ",-
61,0/. 56.0/~176 56,0]~. 50,3~ v,/ 5o,3/. 44,4~ ~ 44,4/. 38,8j'a, ~
4,04
4,21
4,27
4,25
Methode: Ursprtinglicher B u r t o n-Apparat.
10,50 11,05 11,06 11,21 11,22 11,37 11,38 11,53
22 22 22 22 22 22 22 22
46,0]9 1 48,1U'" 48,1]1 7 49,8J"" 49'8/1 4 51,2/ ' 51,2] 1 52,5~ ,v
5 2 2 ] . . 47,8[ ~'a 47,8 / . ^ 43,8J %U 43,8]~ 1 40,7j v," 40,7~ 38,1J z,~
3,34
3,13
2,85
2,63
Die Tabelle zeigt, dat~ die abge~inderte Me- thode gut tibereinstimmende Werte ffir die K.G. gibt (ftir 15, 30, 45 und 60 Minuten berechnet). Bei der B u r t o n - Methode dagegen werden K.O. gefunden, die viel kleiner sind, aut~erdem um so kleiner, ]e lfinger die Messung dauert. Dies ist in Uebereinstimmung mit dem Befund, daft man bei der neuen Schaltung w~ihrend der Messung am As2Ss-Sol die Potentiale zwischen den Elektroden a und b immer erh5hen mut~, um ein konstantes Potentialgef~ille bei den Tren- nungsfl~ichen aufrecht zu erhalten.
W ~ i r m e e n t w i c k l u n g d e m e l e k t r i s c h e n S t r o m e z u f o l g e .
Bekanntlich erniedrigt sich die Viskosit~it bei Temperaturerh6hung um 2 Proz. In der Glei-
67~,] [nach D e b y e und H fi ck ell0)] chung ~" = D v
kann man die GrSt~en ~" und D als ungef~ihr un- abh~ingig v o n d e r Temperatur betrachten. Folg- lich wird sich die K.G. um 2 Proz. pro Grad
10) p. D e b y e u. E. H t i cke l , Physik. Zeitschr. 25, 49 (1924).

29
erh6hen. Die W/irme, welche von dem elektri- schen Strom ent.wickelt wird, kann sich nur aus- gleichen durch die Wand, und demzufolge ist die Temperatur in der Achse des Rohres maximal. Die Oeschwindigkeit der Teilchen an dieser Stelle wird deshalb etwas gr6Ber sein, abet ein zweiter Effekt soll mit betrachtet werden. Da die Flfissigkeit in der Achse des Rohres w~irmer ist als die an der Olaswand, wird sie empor- steigen. Dieser EinfluB ist viel gr6Ber als der erstgenannte, denn bei zu groBen Stromstiirken kann man immer beobachten, dab die beiden Trennungsfl/ichen eine Form annehmen, so wie sie Fig. 7 zeigt. Da die beiden genannten Effekte einander an der fallenden Grenzfl~iche entgegen- wirken und die Form dieser Grenzfl/ichen dennoch den Charakter der Fig. 7 annimmt, muB der erstgenannte Einflut~ ganz zurficktreten im Ver- gleich zu dem zweiten.
I ....
Fig. ?
Aut~erdem sei bemerkt, dab das Emporsteigen in beiden R6hren stattfindet; wenn man deshalb die Verschiebung in beiden R6hren mit~t, so wie wir das oben beftirwortet haben, so wird der beztigliche Fehler kompensiert. Gleiches gilt ftir ein eventuelles Heruntersinken der Teil- chen, so wie man das bei sehr grobdispersen Solen dann und warm antrifft.
Zur Vermeidung aller Komplikationen soll man sich im voraus tiberzeugen, bei welcher Elektrodenspannung die Grenzfl~ichen noch flach bleiben wiihrend der Messung.
Bei Messungen am As2S3-Sol nach Hinzu- ftigung yon 60 m Mol ~(C1 blieben die Fliichen z.B. nur dann flach, wenn die Stromstiirke 10 Milliampere nicht tiberstieg. Dazu war eine Potentialdifferenz zwischen den Hilfselektroden erforderlich yon 14 Volt, d. h. das Potentialgef/ille pro Zentimeter war ungefiihr 0,7 Volt. Bei den Messungen am Se-Sol zeigte es sich notwendig, die Potentialdifferenz noch niedriger zu nehmen, niimlich 7 Volt. Es ist dann der Nachteil mit in Kauf zu nehmen, dab die Messungen zwei
bis drei Stunden dauern zur Erhaltung einer gentigenden Verschiebung der Orenzfl~iche. Schon d e J o n g : 1) hat auf die Unumg~inglichkeit kleiner Potentialdifferenzen hingewiesen.
AuBerdem kann man einen guten W/irme- austausch erhalten, indem man U-Rohre aus sehr d/inhere Glas benutzt und solche mit kleinem Durchmesser. Wir benutzten Rohre mit einem inneren Durchmesser yon 1,4 cm. Dieselben waren hergestellt yon Reagenzr6hrenglas.
H. Adikroskopische Adethoden. Zur Eliminierung des Einflusses der B r o w n -
schen Bewegung soll man hier immer den Mittelwert aus einer groBen Anzahl Messungen nehmen. Der Strom wird mittels eines Kommu- tators nach jeder Messung gedreht, um Ein- flfissen der Elektrolyse an den Elektroden vor- zubeugen und zur Kompensierung einer eventuellen Bewegung der Gesamtfliissigkeit in einer Richtung.
Bei der Untersuchung fiber den EinfluB yon Elektrolyten auf die K.G. des alkalischen Selen- soles wurde die Ktivette nach K r u y t und v a n Ar k el 12) benutzt. Zur Vorbeugung einer sehr st6renden Gasentwicklung an den Elekfroden wurden bei diesem elektrolythaltenden Sole Elektroden benutzt aus Kupfer-Kupferoxyd. Nut bei den Messungen mit K4Fe(CN)6 wurden diese Elektr0den unbrauchbar wegen der Bildung un- 16slicher Cu2 Fe (C N)6, well solches das Potential- gef~ille beeinflul~t. Bei diesen letzten Messungen wurden datum Pt-Elektroden benutzt.
Im allgemeinen sind Suspensoidsole sehr empfindlich gegenVerunreinigungen. Dies ist der Grund, warum man immer versucht, das Glas so weit wie nut m6glich zu reinigen. Dies f/illt abet schwer bei allen bis jetzt gebr/iuchlichen ultramikroskopischen Kiivetten. Die benutzten Kittsubstanzen, in unserem Falle Pizein, erlauben nicht, dab man Chromsiiure, alkoholische Lauge usw. anwendet. Nur eine Reinigung mittels Wasser ist m6glich, h6chstens kann man sehr verdtinnte Alkoholl6sung verwenden.
K. v a n d e r G r i n t e n 18) hat nun vor kurzem eine Ktivette beschrieben, welche zusammenge- stellt ist ohne Verwendung eines jeden Kittmittels. Dieselbe ist ganz einfach : auf einem Objekttr/iger liegen zwei Glasstreifen gleicher L~inge. Der offene Raum zwischen diesen beiden Streifen
11) H. (i. de J o n g , Diss. (Utrecht 1921), 62. 12) H. R. Kruy t u. A.E. van Arke I , Koll.-
Zeitschr. 32, 91 (1923). 18) K. van der O r i n t e n , Diss. (Ziirich 1925);
Compt. rend. 178, 2083 (1924).

30
wird an der Oberseite mittels eines anderen Objekttr~igers abgeschlossen. Der letztere hat eine L~inge yon 4 cm. In den Oeffnungen, welche in dieser Weise entstehen, sind zwei Platinbleche geschoben, welche mit einer Akku-
mulatorenbatterie von 8 Volt verbunden sind. Da in der ursprfinglichen Abhandlung dieses Glas nicht angegeben ist, wird in Fig. 8 die Ktivette (yon oben gesehen) wiedergegeben, das obere Glas ist asziert.
Fig.
Man ffillt die Kfivette mittels einer Pipette, welche man auf eines der Bleche h/ilt. Ist die Kfivette geffillt, so wird die Plfissigkeit von Kapillarkr~iften an der Stelle gehalten. Die Reinigung der Gl~iser geschieht, indem man die Gl~iser in einem Becherglas mit heiger Chrom- s/iure /ibergieBt. Nachdem dies so fiber Nacht gestanden hat, gief~t man doppelt destilliertes Wasser an Stelle der Chroms~iure. Nach einigen Stunden wird mit warmem, doppelt destilliertem Wasser abgespritzt und die Gl/iser auf Piltrier- papier bei 100 ~ getrocknet. Man kann deshalb mit einer K/ivette nur eine Messung pro Tag machen. Es ist datum notwendig, eine Anzahl dieser Kiivetten gleichzeitig in Gebrauch zu haben. Nur die Elektroden, welche sich schnell reinigen lassen, braucht man nur in einem Paar zu haben.
Eine Unannehmlichkeit der ultramikroskopi- schen Methoden bleibt, dag es so schwer h/ilt, die Temperatur konstant zu halten und genau zu messen. Wie oben gesagt, betr/igt der dies- bezfigliche Fehler ungefiihr 2 Proz. pro Grad. Wir haben ein Thermometer sehr nahe an die K/ivette gestellt, sind aber fiberzeugt, dab man in dieser Weise Fehler von ca. 1 ~ nicht aus- schliegen kann. Bedenkt man, dag das kleine Volumen der Flfissigkeit leicht.erw/irmt werden kann yon Wiirmestrahlung (vom K6rper des Be- obachters; Strahlung seitens der Bogenlampe wird soviel wie mSglich vermieden mittels einer Kfivette mit Wasser in der Lichtbahn), so ver- steht man das Unzureichende dieses Megver- fahrens.
Bei dieser Kfivette migt man ausschlieglich auf halber H6he zwischen beiden Q1/isern. Van d e r G r i n t e n meint n~imlich, da~ die Geschwindigkeit an hatber H6he der wahren Geschwindigkeit gleich sei, wenn die H6he der
[
8
Kfivette grSBer i s t als 0,5 ram. Wir arbeiteten mit Kfivetten zwischen 0,52 und 0,64 mm; beim Gebrauch einer Oelimmersion zwischen Para- boloidkondensor und Kfivette und bei Benutzung des Objektives 4 C von R e i e h e r t und Okular 18 konnten wir noch gerade auf der Unterseite des oberen Glases fokusieren.
Die MSglichkeit, auf halber H6he die wahre K.G. messen zu kSnnen, basiert auf folgen- den Grtinden. Man soil einen Unterschied machen zwischen den Verh~iltnissen in einer ge- schlossenen und in einer offenen.Kfivette. Bei ersterer wird der elektroendosmotische Transport an der Wand kompensiert von einem Gegenstrom auf halber H6he. Die Geschwindigkeit dieses Gegenstromes ist eine Funktion derKfivetten- hShe. M.v. S m o l u c h o w s k i 14) hat berechnet, wie sich in solch einer Kfivette die wahre K.G. berechnen l~it~t aus der K.G. auf verschiedene Niveaus,
Bei einer offenen Ktivette wird die ganze Flfissigkeit zum negativen Pole strSmen mit einer Oeschwindigkeit, welche tiberall gleich ist. Dies ist aber ein idealer Fall, welcher bei den ge- br~iuchlichen Kfivetten nie vorkommt; die kapil- laren Kr~ifte werden immer an den Enden der Flfissigkeitss~iule der Kfivette einen mehr oder weniger geschlossenen Charakter geben. Die Kfivette S v e d b e r g ' s 15) ist ein Beispiel einer ab- solut geschlossenen Kfivette. Die meisten Kfi- vetten werden Zwischenf~ille darstellen, bei denen der elektroendosmotische Gegenstrom gr6ger, gleich bzw. kleiner sein kann als die Geschwin- digkeit der Flfissigkeit an der Wand. Zuf~illiger- weise zeigt sich, dag bei einer Kfivette nach
1~) M.v. S m o l u c h o w s k i , Grae tz 'Handb . d. Elektr_ u. Magn. II (1914), 379.
1~) T h e S v e d b e r g, Nova Acta Upsaliensis [41 2, Nr. 1, 149 (1907).

van d e r G r i n t e n die Oeschwindigkeiten gleich sind und daf~ deshalb die Fltissigkeit in halber H6he gerade unbeweglich ist. Die be- obachtete Geschwindigkeit der Teilchen ist darum in dieser H6he der wahren Geschwindigkeit gleich.
Aus den Resultaten von S. 29 wird folgen, dab die beobachteten Geschwindigkeiten in der Mitte der I~tivette bei denselben Solen immer gr613er sind als diejenigen, welche im neuen Apparat gemessen sind. Dies macht es doch zweifelhaft, ob wirklich die gemessenen Geschwindigkeiten die wahren K.G. darstellen. Aller Wahrscheinlichkeit nach wird die Beein-
L
, J
31
%
flussung yon kapillaren Kraften am Ende der F1/issigkeitss~iule abh~ingig sein vonder Ffillung und der genauen Stelle, an der die Platinbleche angebracht sind. Es scheint uns darum, dag man mit dem Vorteile der Abwesenheit eines Kittmittels eine methodische Unsicherheit mit in Kauf nimmt.
Wir haben auch eine Kfivette benutzt, welche in Fig. 9 angegeben ist. Schon v a n A r k e l und K r u y t haben bei ihren Untersuchungen an dem Selensol eine solche Ktivette benutzt (nicht publiziert). Auch M a t t s o n 16) benutzte einen derartigen Apparat.
Fig.
An einer Bi l tz ' schen Ktivette ffir das Spalt- Ultramikroskop sind zwei Seitenrohre angebracht, in denen Platindr~ihte stecken, welche an der unteren Seite spiralf6rmig gewunden sind. Die- selben sind an der Oberseite festgekittet. Ein Seitenrohr an diesem Rohr erm6glicht Druck- austausch mit der Atmosph~ire. Zuf~illigerweise f6rdern diese Seitenrohre die Spfilung des Appa- rats in hohem Mat~e, weil sich die Flfissigkeit in den R6hren erniedrigt, sobald man den Quetsch- hahn 6ffnet. Die K~vette erm6glicht Beobach- tungen an vielen Solen in kurzer Zeit. Wie immer bei ultramikroskopischen Wahrnehmungen kann man nur stark verdtinnte Sole gentigend genau beobachten.
Eine Unannehmlichkeit dieser Kfivette ist, dab der Durchmesser der eigentlichen Ktivette nicht /iberall gleich ist, dab gerade an der Be- obachtungsstelle eine Ausbauchung vorliegt. Es ist also unm6glich, das Potentialgef~ille an der Beobachtungsstelle genau zu messen. Die Kti- vette l~it~t sich aut~erdem nur anwenden bei kleinenElektrolytkonzentrationen ; bis zu 5 m/Mol pro Liter erh~ilt man aber gut reproduzierbare Werte. Die Anforderung kleiner Elektrolytkon- zentrationen macht, dab die K/ivette sich vor
9
allem eignet ffir Messungen mit polivalenten Ionen. Es zeigt sich aber, dat~ nach Messungen mit AI-bzw. Th-Salzen einfaches Sp/ilen mit Wasser nicht ausreicht, um den ursprfinglichen Wert des Sols wieder hervorzurufen. Dieses Verhalten ist auch yon E l i s s a f o f f 17) und anderen bei der Messung elektrokinetischer Er- scheinungen beobachtet worden. Wir fanden aber, dat~ Sptilung mit 1/10 oder 1/100 n HCI eine genfigende Reinigung erm6glicht. Gew6hnlich verwendet man beim Spalt-Ultramikroskop recht starke Objektive. Da wir aber w/inschten, die Mitre der Ktivette zu beobachten, zogen wir vor, das Objektiv 4C yon R e i c h e r t zu benutzen. Zusammen mit Okular 18 erh~ilt man scharfe Bilder. Grof3er Wert soll gelegt werden auf eine feste Montierung. Das Ultramikroskop ist auf einen eingemauerten Tisch gestellt. In Ta- belle VI geben wir nun die Werte f/ir die K.O. , gemessen bei einem Goldsol und einem Selenso! mittels makroskopischen und mikroskopischen Methoden.
16) S. E. Ma t t so n , Kolloidchem. Beih. 14, 309
(192~~ O. v. E l i s s a f o f f , Zeitschr. f. physik. Chem. 79, 385 (1912).

32
T a b e l l e VI.
I i K.G. in ~ IK.Q. in/~ Methode i bei 20 0 des b. 20 0 des
i Goldsols Selensols
Abge~nderter B u r t o n - Apparat 3.6 ~-~4~4,5 - Kiivettenachvan der G r i n t e n 6,9 ] 8,9
Z u s a m m e n f a s s u n g .
Die makroskopische Methode zur Bestimmung der K.G. wird diskutiert und zur gleichen Zeit die verschiedenen Fehler der Messungen an reinen bzw. elektrolythaltigen Solen. Es ist not- wendig, die Sole mit intermizellarer Flfissigkeit zu fiberschichten. Haben intermizellare Flfissig-
keit und Sol ungleiche Leitf~ihigkeit, so kann man in einfacher Weise aus der Bewegung der Trennungsfl~ichen in beiden RShren die wahre K.G. berechnen. Eine verbesserte Form des B u r t o n ' s c h e n Apparats wird beschrieben. In dieser sind die St6rungen wegen den Elektrolyse- produkten ausgeschlossen, mit beliebigen Elek- trolyten in Konzentrationen bis zu 100 m/Mol. AuBerdem ist das Potentialgefiille an den Tren- nungsfRichen genau bekannt.
Einige mikroskopische Methoden werden diskutiert und eine Kfivette fiir relativeMessungen beschrieben.
Utrecht, September 1927. van't Hoff Laboratorium.
Ueber die Diffusion yon Methylenblau in Eielatinegallerten. Von S. G, M o k r u s c h i n 1) (Swerdlowsk). (Eingeg~ngen am 24. Juni1927.)
Bei seinen Untersuchungen fiber die Diffusion von Farbstoffen bemerkte R. Au e r b ach2), dab Melhylenblau durch Gelatinegallerte adsorbiert wird und dab infolgedessen die Methylenblau- konzentration in der Grenzzone zwischen der w~isserigen LSsung und der Gallerte stark an- steigtS). Da R. A u e r b a c h ' s Diffusionsversuche hie Ringer als 48 Stunden dauerten, so war das Bild der Diffusion nach diesen Versuchen viel- leicht nicht vollst~indig. Als der Verfasser die Dauer der Versuche fiber 48 Stunden ausdehnte, stieg er auf folgende interessante Erscheinung: Nach 7 0 - 8 0 Stunden fiillt die Methylenblau- konzentration an der Grenze zwischen Gallerte und LSsung bedeutend und wird nach ungefiihr 100 Stunden gleich Null. Zwischen der stark gefiirbten Gelatineschicht und der darfiber stehenden w~isserigen L6sung von Methylenblau befindet sich eine farblose Schicht. Die Breite dieser farblosen Zone wird allm~ihlich grSBer, auch die F~irbung der Gelatine und der fiber- stehenden LSsung wird schw~icher. Sie ver- schwindet schliefilich ganz oder es bleibt nur
1) Deutsch bearbeitet von R. K 6 h l e r (Leipzig). ~) R. A u e r b a c h , Koll.-Zeitschr. 35, 202 (1924). a) Die Adsorption yon Methylenblau durch Gela-
tinesol ist ktirzlich yon A. Fodor , Joum. chem. Soc. 102 (1926), untersucht worden. Er kommt auf Grund seiner spektrometrischen Untersuchungen zu dem Er- gebnis, daft diese Adsorption recht unbedeutend sei. Nach Ansicht des Verfassers braucht aber eine Farben- ~inderung w~ihrend der Adsorption nicht stattzufinden, wenn dabei keine Deformation der adsorbierten Mole- kfile des Methylenblaus und keine Ver~inderung des Dispersionsgrades eintritt.
eine geringe F~irbung oben im Reagenzglase zurfick. Nach Umschfitteln des Glases sammelt sich die gefiirbte Flfissigkeit wieder oben.
Der Verfasser f/ihrte nun eingehendere Ver- suche fiber diese merkwfirdige Erscheinung aus. Er verfuhr dabei so, wie es R. A u e r b a c h 4) angegeben hat. Die Diffusion wurde in Reagenz- gl~isern dutch kolorimetrische Bestimmung in verschiedenen HShen fiber dem Boden des Reagenzglases gemessen. Um einen EinfluB der Konzentration zu finden, wurden Gelatine- gallerten yon 2,5 Proz., 5 Proz. und 10 Proz. und Methylenblaul6sungen von 1/4 Promille, 1/8 Promille, 1/16 Promille und 0 Promille (d. h. die Gelatine wurde mit reinem Wasser fiberge- gossen) untersucht.
Dabei wurde folgendes beobachtet: W~ihrend der ersten 24 Stunden verlief die Diffusion normal, in den n~ichsten 24 Stunden ergab sich eine merkliche VergrSBerung der Konzentration in der Gelatinezone [wie dies R. A u e r b a c h beobachtete 5)]. Besonders stark war dieser Effekt an konzentrierteren L6sungen yon Methylenblau (114 Promille bis 1/8 Promille) und an konzen- trierteren Gelatinegallerten (5 Proz. bis 10 Proz.). Nach ungeffihr ftinf Tagen erschien an der Grenze zwischen der Gelatine und der LSsung in der LSsung eine farblose Schicht, die sich mit der Zeit vergr6Berte und nach ungef~ihr 14 Tagen die Breite von 50 mm erreichte. Die Konzentration der fiberstehenden Schicht ver-
4) R. A u e r b a c h , loc. cit. 5) R. A u e r b a c h , loc. cit.