Embed Size (px)
Citation preview

315
Journal of Organometallic Chemistry, 258 (1983) 315-329
Elsevier Sequoia S.A., Lausanne - Printed in The Netherlands
VI *. SYNTHESE DER FUNF- UND SECHSZAHNIGEN THIOETHERTHIOL-LIGANDEN dpttd-H, = 2,3,11,12-DIBENZO-1,4,7,10,13-
PENTATI-IIATRIDECAN BZW. dhtid-H, = 2,3,14,15-DIBENZO-1,4,7,1O,l3,16- HEXATHIAHEXADECAN SOWIE IHRER Fe”-, Moo- UND MO”-CARBONYL-KOMPLEXE **
DIETER SELLMANN* und ULRICH KLEINE-KLEFFMANN
Institut fiir Anorganische Chemie der Universitiir, Egerlandstrasse I, D - 8520 Erlangen (BR Deutschland)
(Eingegangen den 5. Juli 1983)
Hydrolysis of [Fe(dpttd)CO] by HCl in THF yields the pentadentate thioether- thiol ligand dpttd-H, = 2,3,11,12-dibenzo-1,4,7,10,13-pentathiatridecane in the free state. The same principle of synthesis leads also to the hexadentate ligand dhthd-H, = 2,3,14,15-dibenzo-1,4,7,10,13,16-hexathiahexadecane; [Fe(CO),(C,H,$)J2- is alkylated by BrC,H,SC,H,SC,H,Br to[Fe(dhthd’)CO], which is subsequently hy- drolyzed. The reaction of dpttd2- salts with Mo(CO), yields [Mo(dpttd)(CO),]‘-, where the dpttd ligand functions as tridentate ligand; [Mo(CO),Cl,] or [Mo(CO),(PPh,),Cl,] yield [Mo(dpttd)(CO),] with pentadentate dpttd. This com- plex with PMe, yields [Mo(dpttd)(PMe,)CO]. The reaction of the potentially hexa- dentate dhthd-H, with Mot’-carbonyl complexes yields a mixture of [Mo(dhthd)CO] and [Mo(dhthd)(CO),]. Furthermore, [Fe(Cl,-dpttd)CO], which contains totally chlorinated benzene rings as well as the methylhydrazine complex [Fe(dpttd)- (CH3N2H3)], were obtained as new derivatives of [Fe(dpttd)CO].
Zusammenfassung
Hydrolyse von [Fe(dpttd)CO] mit HCl in THF ergibt den ftinfzahnigen Thioether-thiol-Liganden dpttd-H, = 2,3,11,12-Dibenzo-1,4,7,10,13-pentathiatri- decan in freier Form. Das gleiche Syntheseprinzip ftihrt such zu dem sechszalmigen Liganden dhthd-H, = 2,3,14,15-Dibenzo-1,4,7,10,13,16-hexathiahexadecan, wenn [Fe(CO),(C,H,S2),12- mit BrC,H,SC,H,SC,H,Br alkyliert und das gebildete
* V. Mitteilung s. Ref. 1. ** Herrn Prof. Dr. Dres. h. c. K&r1 Winnacker zu seinem 80. Geburtstag am 21.9.1983 gewidmet.
0022-328X/83/$03.00 0 1983 Elsevier Sequoia S.A.

316
[Fe(dhthd)CO] hydrolysiert wird. Die Umsetzung von dpttd2--Salzen mit [Mo(CO),] liefert [Mo(dpttd)(C0),12-, in dem der dpttd-Ligand nur dreiz&hnig wirkt; mit [Mo(CO),CI,] oder [Mo(CO),(PPh,),CI,] erhlilt man [Mo(dpttd)(CO),] mit dpttd als ftinfz;ihnigem Liganden. Dieser Komplex ergibt mit PMe, [Mo(dpttd)(CO)PMe,]. Die Reaktion des potentiell sechszahnigen dhthd-H, mit MO*‘-Carbonyl-Komplexen liefert nebeneinander [Mo(dhthd)CO] sowie [Mo(dhthd)(CO),]. Als neue Derivate von [Fe(dpttd)CO] wurden fernerhin das in den Benzolkernen vollstarrdig chlorierte
[Fe(Cl,-dpttd)CO] sowie der Methylhydrazin-Komplex [Fe(dpttd)(CH,N,H,)] erhalten.
Einleitung
Auf der Suche nach Modellverbindungen fur die aktiven Zentren redoxaktiver Metallenzyme besitzen Eisen- und Molybdan-Komplexe mit einer Ligandensph;ire von Schwefelatomen besondere Bedeutung wegen ihrer moglichen Beziehung zu Nitrogenasen und Hydrogenasen [2]. Unter diesem Gesichtspunkt untersuchen wir die Reaktivitat von Eisen- und Molybdan-Komplexen mit mehrzahnigen Thioether- thiol-Liganden. Ktirzlich haben wir tiber die Synthese von [Fe(dpttd)]-Komplexen berichtet, die als ftinfzahnigen Schwefel-Liganden das Dianion von dpttd-H, = 2,3,11,12-Dibenzo-1,4,7,10,13-pentathiatridecan enthalten [3].
Thioether-thiol-Verbindungen mit Alkylbrticken zwischen den Schwefelatomen,
wie z.B. 1,4,7,10_Tetrathiadecan (ttd-H,), konnen nach erprobten organischen Syn- thesemethoden in jeder beliebigen Kettenlange dargestellt werden [4]. Demgegentiber bereitet die Synthese von aromatischen, langerkettigen Thioether-thiol-Verbindun- gen, in denen ein Teil der Schwefelatome an Benzolkerne gebunden ist, nach klassischen Verfahren erhebliche Schwierigkeiten; z. B. lasst sich 2,3,8,9_Dibenzo- 1,4,7,10_tetrathiadecan (dttd-H,) ausgehend von o-Aminothiophenol nur in einer aufwendigen 5Stufensynthese in relativ geringen Ausbeuten erhalten [5].
Im Prinzip wesentlich einfacher kann dieser Ligand durch eine template-Reaktion nach Gl. 1 synthetisiert werden [6]:
[ Fe(S,C,H,),(CO),] 2- + BrC,H,Brz [Fe(dttd)(CO),]
Ebenso ist nach Gl. 2 der ftinfzal-mige Ligand dpttd2- zuganglich [3]:
[Fe(S2C,H,12 (CO),]
2-
+ SK&m), MeOH, RT Co , _2 Br- ) (2)

317
Bisher war es uns jedoch nicht gelungen, die Liganden aus den Komplexen such abzuspalten und in freier Form als dttd-H, bzw. dpttd-H, zu isolieren. Versuche,
durch Umsetzung von [Fe(dttd)(CO),] mit H,S oder Na,S in alkalischem Medium
unter verschiedenen, such drastischen, Reaktionsbedingungen eine Zersetzung zu Eisensulfid und dttd’- bzw. dttd-H, zu erzielen, schlugen fehl. Auch gegentiber Sauren schienen solche Komplexe relativ stabil zu sein; z. B. kann man [Fe(dpttd)CO] in konzentrierter Schwefelsaure auflosen und nach einigen Stunden mit H,O daraus unzersetzt wieder ausfallen. Freies dpttd-H, in reiner Form liess sich bislang such nicht auf dem fur dttd-H, eingeschlagenen Weg erhalten.
Der Zugang zu den freien Liganden ist jedoch unerlbslich, wenn ihre Koor- dinationseigenschaften gegentiber anderen Metallen untersucht werden sollen, an denen die Template-Synthesen nicht moglich sind. Auch beim Eisen selbst ist die Untersuchung der Eigenschaften freier Koordinationsstellen in Fragmenten wie [Fe(dttd)] nur moglich, wenn der freie Ligand eingesetzt werden kann.
Wie wir jetzt gefunden haben, lasst sich der dpttd-Ligand aus [Fe(dpttd)CO] mit verdiinnter Salzsaure in THF nach GI. 3 freisetzen. In der Folge zeigte sich, dass diese Methode such fur die Synthese des sechszahnigen Liganden dhthd-H, = 2,3,14,15-Dibenzo-1,4,7,10,13,16-hexathiahexadecan geeignet ist.
Ergebnisse und Diskussion
Synthese f&f- und sechsziihniger Thioether-thiol-Liganden sowie ihrer Fe”, Moo- und MO”-Komplexe
Die Hydrolyse von [Fe(dpttd)CO] (dpttd-H, = 2,3,11,12-Dibenzo-1,4,7,10,13- pentathiatridecan) in siedendem THF mit Salzslure gem&s Gl. 3,
[Fe(dpttd)CO] THF, HCI, 2.5 h Rtickfluss
-CO, -FeCI,
liefert eine klare gelbe Losung, aus der sich der ftinfzahnige S-Ligand dpttd-H, als farblose Festsubstanz in hohen Ausbeuten isolieren lasst.
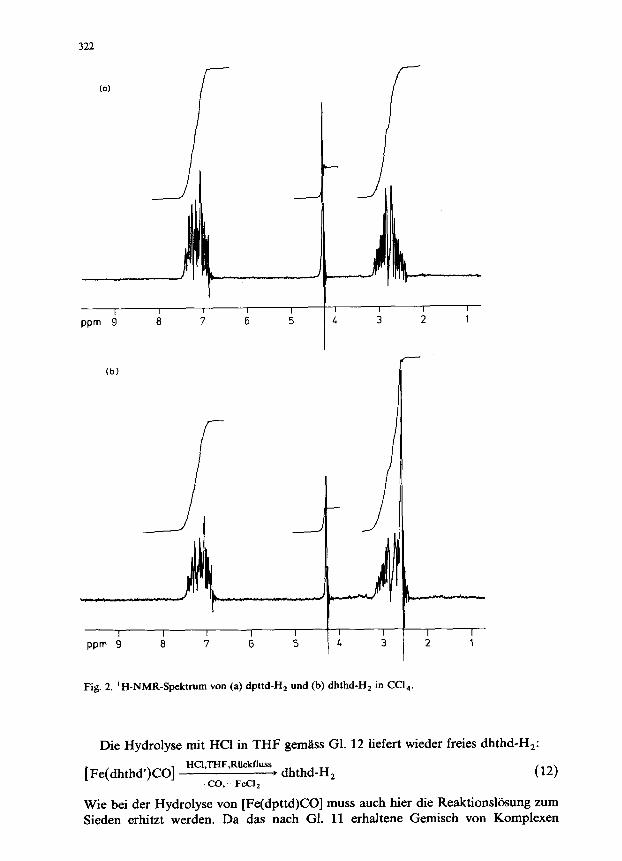
dpttd-H, ist in THF, Et,0 und Ccl, gut, in MeOH und EtOH nur massig lbslich; die spektroskopischen Daten sind in Tab. 3 zusammengefasst (vergl. such Fig. 2a). W&rend im Felddesorptions (FD)-Massenspektrum nur das Molektilion auftritt, werden im Elektronenstossionisierungs(EI)-Massenspektrumu.a. die in Tab. 1 angegebenen charakteristischen Fragmentionen beobachtet.
Die Isolierung des freien dpttd-H, ermoglichte nunmehr die Untersuchung seiner Koordinationseigenschaft such gegentiber Mo-Zentren. Wenn das Na-Salz dpttd-Na, mit Mo(CO), nach Gl. 4 umgesetzt wird,
Mo(CO), + dpttd2- EtOH THF Riickfluss
’ * 2 h,-CO
’ [Mo(dpttd’)(CO)$- (4)
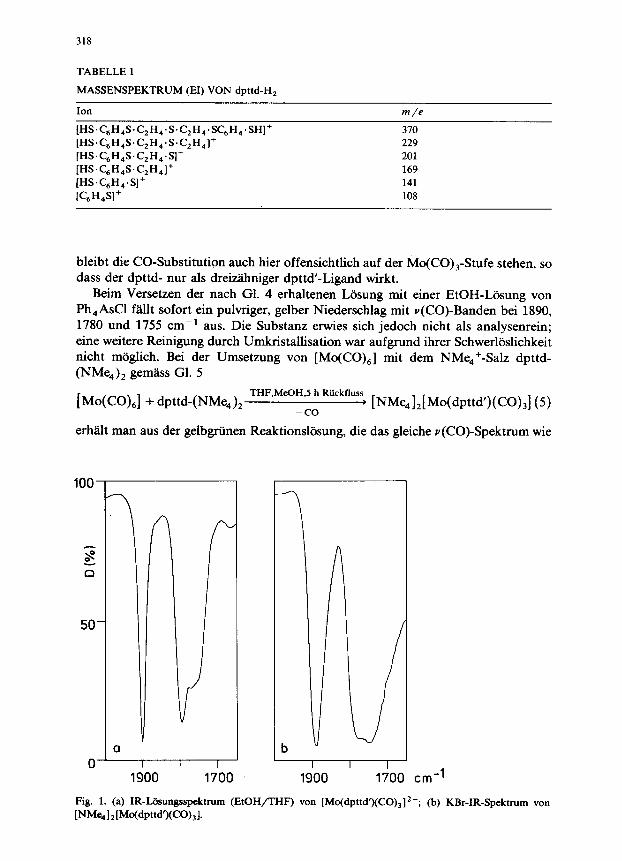
erh%lt man eine klare, gelbgrtine Losung; ihr IR-Spektrum weist das fur Mo(CO),- Komplexe typische v(CO)-Muster mit Banden bei 1900 und 1790 cm-’ sowie eine Schulter bei 1765 cm-’ auf (Fig. 1).
Wie bei der Umsetzung von Mo(CO), mit anderen mehrzahnigen S-Liganden [7]
318
TABELLE 1
MASSENSPEKTRUM (EI) VON dpttd-H,
IOll m/e
[HS.C6H,S.C2H4.S~C2H~.SChH~.SH]+ 370
[HS.C,H,S.C,H,.S.C,H.,]+ 229
[HS.C,H,S.C,H,.S]+ 201
[HS.C6H4S.C2H4]+ 169
[HS.C6H4.S]+ 141
GH,Sl+ 108
bleibt die CO-Substitution such hier offensichtlich auf der Mo(CO),-Stufe stehen, so dass der dpttd- nur als dreizahniger dpttd’-Ligand wirkt.
Beim Versetzen der nach Gl. 4 erhaltenen Losung mit einer EtOH-L&sung von Ph,AsCl f2illt sofort ein pulvriger, gelber Niederschlag mit v(CO)-Banden bei 1890, 1780 und 1755 cm-’ aus. Die Substanz erwies sich jedoch nicht als analysenrein; eine weitere Reinigung durch Umkristallisation war aufgrund ihrer Schwerloslichkeit nicht mbglich. Bei der Umsetzung von [Mo(CO),] mit dem NMe,+-Salz dpttd- (NMe,), gem&s Gl. 5
[Mo(CO),] + dpttd-(NMe,), THF’Meo,~~ Riickfl”ss, [NMe,],[Mo(dpttd’)(CO),] (5)
erh2lt man aus der gelbgrtinen Reaktionsliisung, die das gleiche v(CO)-Spektrum wie
loo-
-z o\ ii
so-
0-
:
cl
:
P
I 1 I
1900 1700
b
Fig. 1. (a) IR-Lihmgsspektrum (EtOH/THF) von [Mo(dpttd’)(CO),]2-; (b) KBr-IR-Spektrum van
[NM%1 Z 1 WWtd’XCO), 1.

319
die Losung von Gl. 4 aufweist, bei Ktihlung auf -24°C eine grtine, hochviskose
Substanz; sie lasst sich durch Waschen mit MeOH und Trocknen im HV in ein grtines Pulver iiberftihren und als [NMe,]z[Mo(dpttd’)(Co),l charakterisieren. Auch
dieses Salz ist gut nur in DMSO loslich. Der u(CO)-Bereich im KBr-IR-Spektrum dieses Salzes ist in Fig. 1 wiedergegeben. Im ‘H-NMR-Spektrum in DMSO-d, erscheinen die aromatischen Protonen als Multiplett bei 7.0 ppm, die NMe,+-Meth- ylgruppen als Singulett bei 3.1 ppm sowie die Protonen der Methylenbrticke als Multiplett zwischen 2-4 ppm.
Da der potentiell ftinfzahnige dpttd-Ligand bei der Reaktion mit Mo(CO), nur dreizahnig wirkt, wurden seine Koordinationseigenschaften such gegentiber MO”- Carbonyl-Verbindungen untersucht. Im Gegensatz zu Moo- weisen Mo”(CO)- Komplexe haufig siebenfache Koordination auf; ausserdem konnen ihre CO-Ligan- den verschiedentlich leicht substituiert werden, wie z.B. bei der Umsetzung von [Mo(dttd)(CO),] mit PMe, zu [Mo(dttd)(CO),PMe,].
Die Reaktionen mit Mo(CO),Cl, lieferten - wahrscheinlich wegen der beim L&en von [Mo(CO),Cl,] stets auftretenden Zersetzungsreaktionen - nur schwer reproduzierbare Ergebnisse. Bei den weiteren Experimenten wurde deswegen das weniger zersetzliche [Mo(CO),(PPh,),Cl,] als Ausgangssubstanz verwendet. Setzt man es, suspendiert in einem Losungsmittelgemisch aus Petrolether, MeOH und
( rjl S\Ma/s s’/ \‘s >
+ dpttd-No* PE,MeOH,THF
2. 3 h , RT
t 2 NaCl + 2 PPh3 + CO (6)
c-o co
THF (2/1/l), mit dpttd-Na, nach Gl. 6 urn, scheidet sich ein rotes Pulver ab. Es ist in THF, CH,Cl, und Aceton nur massig, in DMSO aber gut loslich und kann durch Umkristallisation aus DMSO/Aceton in eine rote kristalline Verbindung tiberftihrt werden, die elementaranalytisch sowie spektroskopisch als [Mo(dpttd)(CO),] char- akterisiert wurde.
[Mo(dpttd)(CO),] zeigt im KBr-IR-Spektrum zwei intensive v(CO)-Banden bei 1935 und 1880 cm-‘; sie weisen jeweils schwache Schultem bei 1950 und 1870 cm-’ auf, die wahrscheinlich auf Koordinationsisomere zurtickzuftihren sind. Im ‘H- NMR-Spektrum in DMSO-d, erscheinen die aromatischen Protonen aufgespalten in zwei Signalgruppen bei 7.2 ppm, die Signale der C,H,-Brticken erstrecken sich tiber den Bereich von 2-4 ppm. Das im freien Liganden beobachtete Aufspaltungsmuster sowie die chemischen Verschiebungen werden durch die Koordination vollig verandert; verantwortlich daftir ist vermutlich die Symmetrieerniedrigung bei der Koordination. Im FD-Massenspektrum wird das Molektilion (mit 96Mo) bei m/e 520 beobachtet.
Bemerkenswert ist die bei der Reaktion nach Gl. 6 bevorzugte Abspaltung von

320
PPh, aus [Mo(CO),(PPh,),CI,], die auf die im Vergleich zu CO besseren Austritts- eigenschaften von PPh, zurtickgeftihrt werden kann. Einen gleichen Reaktions- verlauf beobachtet man such bei der Umsetzung von [Fe(CO),(PPh,)I,] mit dttd-H,, die ausschliesslich zu [Fe(CO),dttd] ftihrt [l].
Wahrend sich z.B. [Mo(CO),dttd] leicht in [Mo(CO),(PR,)dttd] iiberftihren lass& konnte daraus bislang kein Monocarbonyl-Komplex wie [Mo(CO)(PR,),dttd] oder [Mo(CO)(dppe)dttd], dppe = 1,2-Bis(diphenylphosphino)ethan, erhalten werden; im Gegensatz dazu ist bei [Mo(dpttd)(CO),] eine solche Reaktion nach Gl. 7 leicht moglich.
bWWd)(CO),] F [Mo(dpttd)(PMe,)CO]
Rtihrt man eine Suspension von [Mo(dpttd)(CO),] in THF mit einem grossen PMe,-Uberschuss bei RT, erhalt man innerhalb von 15 min eine leuchtend rote Lbsung; im IR-Losungsspektrum tritt dabei eine scharfe v(CO)-Absorption bei 1810 cm-’ auf. Bei Ktihlung auf -78°C scheiden sich aus der Losung rotbraune Mikrokristalle ab, die als [Mo(dpttd)(PMe,)CO] (v(C0) (KBr) 1795 cm-‘) char- akterisiert wurden (Tab. 3). [Mo(dpttd)(PMe,)CO] ist gut loslich in CH,Cl,, CHCI, und THF.
[Mo(dpttd)(CO),] in analoger Weise mit o-Liganden wie Hydrazin oder Methyl- hydrazin zu den als potentielle Ausgangsverbindungen fur N,-Komplexe interes-
sierenden Hydrazin-Komplexen umzusetzen, gelang bisher nicht. In orientierenden Versuchen wurde bei der Umsetzung von [Mo(dpttd)(CO),] mit Methylhydrazin nur eine vollstandige CO-Abspaltung beobachtet, ohne dass sich in den resultierenden Produkten IR-spektroskopisch NH- oder gar N,-Banden nachweisen liessen.
[Mo(dpttd)(CO), ] stellt den ersten Mo’*(CO),-Komplex dar, in dem ausschliess- lich Thiolato- und Thioether-Schwefel-Atome als Koliganden fungieren. Seine relativ leichte Zuganglichkeit veranlasste uns, nunmehr die Synthese des zu dpttd-H, analogen sechszahnigen Liganden dhthd-H, = 2,3,14,15-Dibenzo-1,4,7,10,13,16- hexathiahexadecan zu versuchen, urn damit zu [Mo(dhthd)CO] zu gelangen.
Wenn gem&s Gl. 8 die tiefrote Losung von [Fe(S$,H,),(CO),]2- in MeOH mit dem Alkylierungsreagenz 1,8-Dichlor-3,6-dithiaoctan gertihrt wird,
[Fe(S2C,H,)2(CO)2]2-+ ClC,H,SC,H,SC,H,Cl s [Fe(dhthd)] (8)
fallt langsam eine ockerfarbene mikrokristalline Substanz aus; sie ist praktisch unloslich, weist im KBr-IR-Spektrum keine v(CO)-Absorptionen auf und wurde elementaranalytisch sowie durch Hydrolyse mit HCl als [Fe(dhthd)] identifiziert. Beim Versuch, gem&s Gl. 9
[ Fe(dhthd)] + CO2 [ Fe(dhthd’)CO] 0
mit CO eine Reaktion zu dem Monocarbonyl-Komplex [Fe(dhthd’)CO] zu erzielen, in dem der potentiell sechszalrnige Ligand dthtd2- nur als ftinfztihniger Ligand fungiert, wird such nach 5 h bei 50°C keine Reaktion beobachtet.
Die Hydrolyse von [Fe(dhthd)] mit Salzsaure in THF gem& Gl. 10 verlauft bei

321
RT rasch.
5
[Fe(dhthd)] HCI , THF, RT
- FeCI, -a 0 SH
57
(10)
HS
Die Suspension von [Fe(dhthd)] wandelt sich bereits nach 10 min in eine klare gelbe Losung urn, aus der sich der freie Ligand dhthd-H, in Form eines farblosen Pulvers isolieren lasst, das ‘H-NMR-, IR- und massenspektroskopisch charakterisiert wird. Im EI-Massenspektrum werden neben dem Molekiilion von dhthd-H, bei m/e 430
die in Tab. 2 angegebenen charakteristischen Fragmentionen beobachtet. Das ‘H-NMR-Spektrum in Ccl, (Fig. 2b) weist die Signale der aromatischen
Protonen als Multiplett bei 7.1 ppm und die der SH-Protonen als Singulett bei 4.3 ppm auf. W&hrend die Methylenprotonen der C-Atome 5,6,11 und 12 als sym-
metrisches 14Linienmultiplett bei 2.8 ppm erscheinen, liefern die Protonen der C-Atome 8 und 9 ein Singulett bei 2.6 ppm; das Intensitatsverhaltnis von aromatischen-, SH- sowie Methylenprotonen betragt 8/2/12.
Da die Darstellung von [Fe(dhthd)] nach Gl. 8 nur langsam ablauft und relativ geringe Ausbeuten liefert, wurde versucht, durch Verwendung von 1,8-Dibrom-3,6- dithiaoctan als Alkylierungsreagenz die Synthese zu optimieren. Dabei nimmt die Reaktion jedoch gem&ss Gl. 11 einen anderen Verlauf:
[Fe(S,C,H,),(C0)2]2-+ BrC,H,SC,H,SC,H,Br MeoH’THF’RT~ -CO, - 2Br
[Fe(dhthd’)CO] fNebenprodukt (11)
Aufgrund der schnelleren Alkylierung fallt namlich noch vor vollst;indiger Entcar-
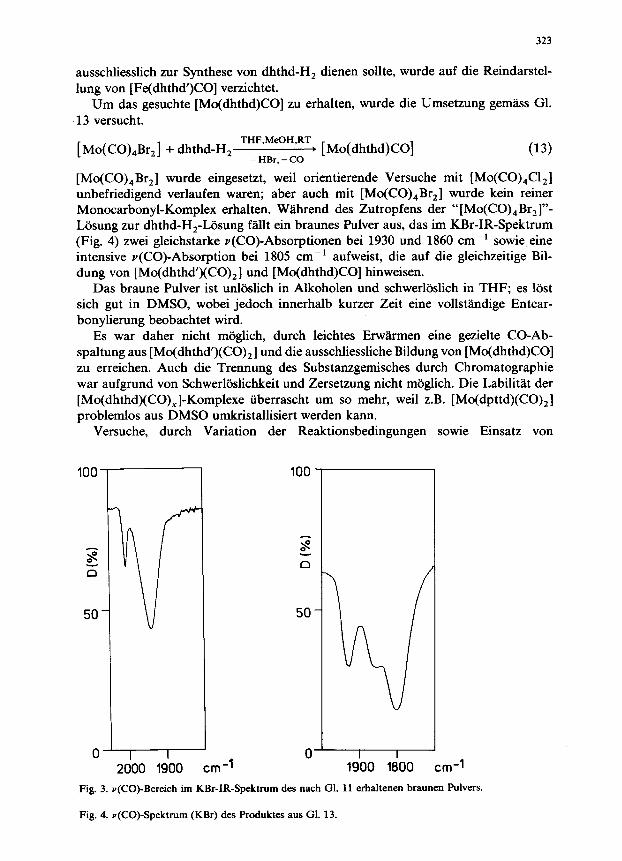
bonylierung schwerliisliches [Fe(dhthd’)CO] aus, das damit einer weiteren Reaktion entzogen ist. Es scheidet sich innerhalb von etwa 3 Stunden aus der tiefroten Reaktionsliisung als braunes Pulver ab, das im Gegensatz zu dem nach Gl. 8 erhaltenen Produkt im KBr-IR-Spektrum (Fig. 3) eine u(CO)-Absorption bei 1945 cm-’ aufweist. Die Schulter der Bande bei 1970 cm-’ und die schwache Y(CO)-Bande bei 2020 cm-’ weisen darauf hin, dass sich gleichzeitig such noch ein Fe(CO),- Komplex, wahrscheinlich [Fe(dhthd’)(CO),], bildet.
TABELLE 2
EI-MASSENSPEKTRUM VON dhthd-H,
[HSC,H,S.C,H,.S.C,H,.S.CzH.,+S.CbH$H]+ 430
.C,H, [HSC~H;S.C;H;.S .S.C2H4]+ 289
[HSC,H,S.C2H&C;H;.S]+ 261
[HSC,H,S.C,H,.S-C,H,]+ 229
[HSC6H.,S.C2H,.S]+ 201
[HSCsH,SC2H4]+ 169
lHSC&,Sl 141
Ion m/e
322
(0)
I I I I I I I I I wm 9 a 7 6 5 1 3 2 1
-
Fig. 2. ‘H-NMR-Spektrum von (a) dpttd-H, und (b) dhthd-H, in Ccl,.
Die Hydrolyse mit HCl in THF gem&s Gl. 12 liefert wieder freies dhthd-H,:
[Fe(dhthd’)CO] Hc~oF~F~~~ dhthd-H, 2
(12)
Wie bei der Hydrolyse von [Fe(dpttd)CO] muss such hier die Reaktionsliisung zum Sieden erhitzt werden. Da das nach Gl. 11 erhaltene Gemisch von Komplexen
323
ausschliesslich zur Synthese von dhthd-H, dienen sollte, wurde auf die Reindarstel-
lung von [Fe(dhthd’)CO] verzichtet. Urn das gesuchte [Mo(dhthd)CO] zu erhalten, wurde die Umsetzung gem&s Gl.
13 versucht.
[Mo(CO),Br,] + dhthd-H, T~~~r~~~* [Mo(dhthd)CO] 03)
[Mo(CO),Br,] wurde eingesetzt, weil orientierende Versuche mit [Mo(CO),Cl,] unbefriedigend verlaufen waren; aber such mit [Mo(CO),Br,] wurde kein reiner Monocarbonyl-Komplex erhalten. W&end des Zutropfens der “[Mo(CO),Br,]“- Losung zur dhthd-H,-Liisung fallt ein braunes Pulver aus, das im KBr-IR-Spektrum (Fig. 4) zwei gleichstarke v(CO)-Absorptionen bei 1930 und 1860 cm-’ sowie,eine intensive Y(CO)-Absorption bei 1805 cm-’ aufweist, die auf die gleichzeitige Bil- dung von [Mo(dhthd’)(CO),] und [Mo(dhthd)CO] hinweisen.
Das braune Pulver ist unloslich in Alkoholen und schwerloslich in THF; es lost sich gut in DMSO, wobei jedoch innerhalb kurzer Zeit eine vollstandige Entcar- bonylierung beobachtet wit-d.
Es war daher nicht moglich, durch leichtes Erwkmen eine gezielte CO-Ab-
spaltung aus [Mo(dhthd’)(CO),] und die ausschliessliche Bildung von [Mo(dhthd)CO] zu erreichen. Auch die Trennung des Substanzgemisches durch Chromatographie war aufgrund von Schwerlijslichkeit und Zersetzung nicht moglich. Die Labilitat der [Mo(dhthd)(CO),]-Komplexe tiberrascht urn so mehr, weil z.B. [Mo(dpttd)(CO),] problemlos aus DMSO umkristallisiert werden kann.
Versuche, durch Variation der Reaktionsbedingungen
100
gi 2;
50
0
v .
100 -
‘;; o\
0
50-
0- 2000 1900 cm”
sowie Einsatz von
v I I
1900 1800 cm-1
Fig. 3. v(CO)-Bereich im KBr-IR-Spektrum des nach Gl. 11 erhaltenen braunen Pulvers.
Fig. 4. v(CO)-Spektrum (KBr) des Produktes aus Gl. 13.

324
[Mo(CO),(PPh,),Cl,] die ausschliessliche Bildung von entweder [Mo(dhthd’)(CO), ] oder [Mo(dhthd)CO] zu erreichen, verliefen bislang ebenfalls erfolglos.
Synthese von [Fe(dpttd)CO]-Derivaten Wie wir ktirzlich berichtet haben, scheiterten die Versuche, [Fe(dpttd)N,H,] . THF
zu einem N,-Komplex zu oxidieren, bereits daran, dass es extrem schwerloslich ist und sich in keinem geeigneten Losungsmittel ohne Zersetzung l&en llsst. Methyl- substitution in den Benzolkernen des dpttd-Liganden von [Fe(dpttd)CO] ftihrt zu keinen besseren Liislichkeitseigenschaften, wie sich durch die Synthese von 2,3,11,12-Di(tetramethylbenzo)-1,4,7,lO,l3-pentathiatridecanato-carbonyl-eisen(II) zeigen liess; auf weitere Versuche mit diesem Komplex wurde daher verzichtet [3].
Urn zu prtifen, ob sich eventuell durch Halogensubstituenten in den Benzolkernen die Loslichkeit solcher Komplexe gtinstig beeinflussen lasst, haben wir deshalb gem&s Gl. 14:
[Fe(S,C6CI,)rK0)2] *- t S(C2H,Br,, THF, MeOH. RT
-Ew-, -co (14)
I co
such das chlorsubstituierte Analoge von [Fe(dpttd)CO], [Fe(Cl,-dpttd)CO], 2,3,11,12-~i(tetrachlorbenzo)-1,4,7,lO,l3-pentathiatridecanato-carbonyl-eisen(II), dargestellt. Es ist jedoch such nicht besser lbslich als [Fe(dpttd)CO], so dass keine weiteren Umsetzungen damit durchgeftihrt wurden. Das mikrokristalline, ockerfar- bene [Fe(Cl,-dpttd)CO] weist im KBr-IR-Spektrum eine v(CO)-Bande bei 1965 cm-’ auf; im EI-Massenspektrum wird das isotopenverbreiterte Signal der ent- carbonylierten Spezies [Fe(Cl,-dpttd)]+ bei m/e 696-704 beobachtet.
Auf der Suche nach weiteren Verbindungen, die als potentielle Vorstufen fur Distickstoff-Komplexe dienen sollten, konnte nunmehr in einer tiberraschend einfachen Reaktion gem&s Gl. 15 [Fe(dpttd)(N,H,CH,)] synthetisiert werden.
[Fe(Wtd){P(OPh)~)] - N2H3CH3’PE [Fe(dpttd)(N,H,CH,)] - P(oPh),
(15)
Rtihrt man [Fe(dpttd){P(OPh), }] in Methylhydrazin bei RT, so ist praktisch ausser einer schwachen Rotfarbung des Methylhydrazins keine Reaktion zu beobachten; such nach ErwSirmen auf ca. 45°C liegt der rotbraune Phosphit-Komplex immer noch ungelbst in N,H,CH, vor. Versetzt man diese Suspension,jedoch mit Petrol- ether, PE, urn gem&s Gl. 16 im Gleichgewicht befindliches P(OPh),
[ Fe(dpttd) { P(OPh),}] + N,H,CH 3 + [ Fe(dpttd)N,H,CH,] + P(OPh), (16)
aus der Methylhydrazin-Phase zu entfernen, so erhalt man durch kraftiges Rtihren bei RT schon nach wenigen Minuten eine klare, tiefrote Methylhydrazin-Phase, die mit der farblosen Petrolether-Phase tiberschichtet ist. Abdekantieren des Petrolethers und erneute Zugabe von frischem Petrolether ergeben schliesslich eine Methylhy- drazin-Lbsung, aus der sich bei 50°C durch langsame Zugabe von MeOH gelbe Kristalle von [Fe(dpttd)(N,H,CH,)] ausfallen lassen.

325
[Fe(dpttd)(N,H,CH,)] ist praktisch unlijslich in allen gebrluchlichen Lijsungs-
mitteln. Bei Versuchen, es z.B. in MeOH, THF oder Et,0 zu l&en, wandeln sich die gelben Kristalle binnen weniger Minuten in ein braunes Pulver urn. Es weist
elementaranalytisch keinen Stickstoff auf und stellt wahrscheinlich [Fe(dpttd)], dar. Mit DMSO wird kurzfristig eine Lijsung erhalten, aus der sich aber nach ca. 30-60 Sekunden ein braungriines Pulver, vermutlich [Fe(dpttd)DMSO], abscheidet. LGslich ist [Fe(dpttd)N,H,CH,] nur in Methylhydrazin; es liess sich deshalb nur IR- und massenspektroskopisch sowie elementaranalytisch charakterisieren.
Im KBr-IR-Spektrum werden drei v(NH)-Banden bei 3260, 3180 und 3130 cm-’ sowie eine S(NH,)-Absorption bei 1590 cm -’ beobachtet. Das Molekiilion von [Fe(dpttd)(N,H,CH,)] lgsst sich such im FD-Massenspektrum nicht beobachten; 15-Masseneinheiten tiefer bei m/e 455 erscheint jedoch ein Signal, das der ent- methylierten Spezies [Fe(dpttd)(N,H,)]+ zugeordnet werden kann. Bei m/e 424 erscheint das ftir [Fe.(dpttd)]-Komplexe typische [Fe(dpttd)]+-Fragmention. Methyl- hydrazin-Lbsungen von [Fe(dpttd)(N,H,CH,)] weisen beim Erw;irmen von 0 auf 40°C einen reversiblen Farbwechsel von orange nach griin auf; sowohl aus der orangen wie aus der gfinen LBsung werden beim Versetzen mit MeOH gelbe Kristalle ausgeschieden.
Wenn man die Synthese von [Fe(dpttd)N,H,CH,] gem&s Gl. 15 bei 20°C durchfiihrt und anschliessend bei 5-10°C mit MeOH ausf&llt, erh%lt man griine Kristalle. Sie weisen zwar die praktisch gleichen elementaranalytischen Daten wie die gelben Kristalle, im KBr-IR-Spektrum jedoch fiinf v(NH)-Banden bei 3280, 3250, 3200, 3160 und 3100 cm-’ sowie eine 6(NH,)-Bande bei 1600 cm-’ auf. Wahrscheinlich handelt es sich bei den griinen Kristallen urn ein metastabiles Isomeres. Nach Aufliisen in Methylhydrazin und Au&lien mit MeOH bei 50°C erh< man wiederum die gelben Kristalle.
Oxidationsversuche an [Fe(dpttd)N,H,CH,] in DMSO sowie in Methylhydrazin ergaben keine Hinweise auf die Bildung von N,-Komplexen.
Experimentelles
Alle Reaktionen wurden unter Stickstoff in absoluten LBsungsmitteln durchgeftihrt und - soweit mijglich - IR-spektroskopisch verfolgt. IR-Spektren wurden mit dem Spektrometer IMR 16 der Fa. Zeiss, Massenspektren auf dem MAT 212 der Fa. Varian und ‘H-NMR-Spektren auf einem JNM-PMX 60 der Fa. Jeol aufgenommen.
o-Benzoldithiol [8], Tetrachlor-o-benzoldithiol [9], Bis[/%bromethyl)sulfid [lo], 1,8-Dichlor-3,6-dithiaoctan[ll], 1,8-Dibrom-3,6_dithiaoctan[12], [Mo(CO),Cl,] [13],
[Mo(CO,)Br,] [14], [Mo(CO),(PPh,),Cl,] [13] und [Fe(dpttd)CO] [3] wurden nach der angegebenen Literatur dargestellt.
Synthesen
dpttd-H, 2 g (4.4 mmol) [Fe(dpttd)CO] werden in 200 ml THF mit 1.8 ml konz. HCl
solange zum Sieden erhitzt (ca. 2.5 h), bis eine klare, gelbe Lbsung vorliegt. Das THF wird abkondensiert und der Riickstand viermal mit je 10 ml Et,0 extrahiert. Die vereinigten Et,O-Extrakte werden iiber Na,SO, getrocknet und filtriert.

326
Abkondensieren des Et,0 ergibt ein gelbbraunes 01, das in 15 ml Ccl, aufgenom- men wird, wobei schwarzbraune Verunreinigungen ungelost zurtickbleiben. Die Ccl,-Losung wird tiber SiO,/60 filtriert, das 3 X mit je 10 ml Ccl, nachgewaschen wird. Abkondensieren des Ccl, und Umkristallisation (20/- 78°C) des zurtickbleibenden farblosen 01s aus Et,0 ergeben 1.3 g dpttd-H, (80% d.Th. bez.
auf [Fe(dpttd)CO]). Fp. 22°C. Elementaranalyse: Gef.: C, 52.03; H, 4.99; S, 42.6. C,,H,,S, (370.3) ber.: C,
51.85; H, 4.86; S, 43.29%.
0.5 g (1.35 mmol) dpttd-H, werden mit 1.3 ml 20%iger (CH,),NOH-Losung und 0.36 g (1.35 mmol) Mo(CO), in 20 ml THF/MeOH (l/3) 5 h unter Rtickfluss erhitzt. Bei anschliessender Ktihlung der grtingelben Losung auf -24°C scheidet sich eine grtine, viskose Substanz ab. Die tiberstehende Losung wird abdekantiert und die viskose Substanz mit MeOH und Et,0 gewaschen. Nach Trocknen im HV erhalt man als pulverige, grtine Festsubstanz 230 mg [NMeJJMo(dpttd’)(Co),l. (24% d. Th. bez. auf dpttd-Hz)
Elementaranalyse: Gef.: C, 45.85; H, 5.96; N, 3.96. C,,H,O,S,N,Mo (696.76) ber.: C, 46.54; H, 5.79; N, 4.02%.
370 mg (1 mmol) dpttd-H, werden in 5 ml MeOH/Sml THF gel&t und mit einer Losung aus 50 mg Na (2 mmol) in 5 ml MeOH metalliert. Nach Zugabe von 10 ml Petrolether wird die klare Losung mit 780 mg (1 mmol) [Mo(CO),(PPh,),Cl,] versetzt und bei RT gertihrt. Die zunachst gelbe Suspension wandelt sich innerhalb von 3 h in eine orangerote urn. Der Feststoff wird abfiltriert, mit MeOH und Et *O gewaschen und im Hochvakuum getrocknet. Man erhalt 400 mg orangerotes [Mo(dpttd)(CO),] (78% d. Th. bez. auf dpttd-Hz). Umkristallisation erfolgt durch Versetzen einer bei 20°C gesattigten DMSO-LBsung mit der doppelten Menge Aceton; bei Ktihlung auf -24°C kristallisiert analysenreines [Mo(dpttd)(CO),] aus.
Elementaranalyse: Gef.: C, 41.41; H, 3.24. Molmasse: 520 (massenspektrosko- pisch (FD) bez. auf 96 Mo). ‘1s 16 2 5 H 0 S MO (520.51) ber.: C, 41.53; H, 3.10%.
100 mg (0.19 mmol) [Mo(dpttd)(CO),] und 0.5 ml PMe, werden in 10 ml THF 1.5 h bei RT gertihrt. Nach Filtration der leuchtend roten Losung iiber 5 g Kieselgel 60 wird auf -78°C gektihlt. Das auskristallisierte [Mo(dpttd)CO(PMe,)] wird abfiltriert, zweimal mit Et ,O gewaschen und 6 h im HV getrocknet. Ausbeute 60 mg (55% d. Th. bez. auf [Mo(dpttd)(CO),]).
Elementaranalyse: Gef.: C, 42.38. H, 4.61. Molmasse: 568 (massenspektrosko- pisch, FD). C,,H,,OS,PMo (568.57) ber.: C, 42.25; H, 4.43%.
dhthd-H, (a) [Fe(S,C,H,),(CO), J2 -. 850 mg (6 mmol) o-Benzoldithiol in 50 ml MeOH
werden mit einer L&sung aus 280 mg (12 mmol) Na in 30 ml MeOH metalliert. Nach Zugabe von 0.6 g (3 mmol) FeCl, .4H,O wird 10 mm bei RT gertihrt und anschliessend 2 h CO eingeleitet. Man erhalt eine klare, tiefrote Losung mit 3 mm01
P+GVVLd2(Co)212-.

327
(b) [Fe(dhthd)J,. 3 mm01 [Fe(SJ,H,),(CO),]2- in 80 ml MeOH werden mit 660 mg (3 mmol) 1,8-Dichlor-3,6-dithiaoctan in 60 ml MeOH versetzt und 2-3 d bei RT gertihrt. Die aus der tiefroten Losung ausgefallenen ockerfarbenen Mikrokris-
talle werden abfiltriert, mit MeOH sowie Et,0 gewaschen und 8 h im HV getrock-
net. Ausbeute: 300 mg [Fe(dhthd)],. (20% d. Th. Bez. auf [Fe(S,C,H,),(CO),]*-). Elementaranalyse: Gef.: C, 44.34; H, 4.30. C,,H,,&Fe (484.54) ber.: C, 44.62;
H, 4.16%. (c) [Fe(dhthd’)CO]. Zu 3 mmol [Fe(S&,H,),(CO),]*- in 80 ml MeOH werden
bei RT innerhalb von 10 min 980 mg 1,8-Dibrom-3,6-dithiaoctan in 20 ml THF/ZO ml MeOH getropft. Nach 3.5 h Ri.ihren wird das ausgefallene braune Pulver abfiltriert, mit MeOH und Et,0 gewaschen und 5 h im HV getrocknet. Ausbeute: 1.1 g braunes Pulver, das vorwiegend aus [Fe(dhthd’)CO] besteht. (ca. 70% d. Th.
bez. auf [Fe(!S&H,)2(CO)2]2-). (d) dhthd-H,. 2.4 g (- 4.5 mmol) [Fe(dhthd)] bzw. [Fe(dhthd’)CO] werden in
150 ml THF mit 2 ml konz. HCl etwa 1 h bis zur Bildung einer klaren gelben Lijsung zum Sieden erhitzt. Nach Abkondensieren des THF wird der gelbbraune, viskose Rtickstand ftinfmal mit je 20 ml Et,0 extrahiert. Die vereinigten gelben Et,O-Ex- trakte werden tiber Na,SO, getrocknet und tiber 5 g Kieselgel filtriert; das SiO, wird zweimal mit je 10 ml Et *O nachgewaschen. Die nunmehr farblose Et ,O-Losung wird auf -78°C gekiihlt. Wahrend 1 d fallt farbloses dhthd-H, aus, das bei - 30°C filtriert und anschliessend bei RT im HV getrocknet wird. Ausbeute: 1.1 g dhthd-H, (Fp. 59°C) (51% d. Th. bez. auf den Fe-Komplex). Elementaranalyse: Gef.: C, 50.18; H, 5.18; S, 42.7. Molmasse: 430 (massenspektrometrisch). C,,H,,S, (430.54) ber.: C, 50.21; H, 5.11; S, 44.67%.
Versuch zur Synthese von [Mo(dhthd)CO] Zu 115 mg (0.27 mmol) dhthd-H, in 70 ml MeOH/lO ml THF werden bei 50°C
innerhalb von 10 min 100 mg (0.27 mmol) Mo(CO),Br, in 20 ml MeOH getropft. Das ausgefallene braune Pulver wird abfiltriert mit MeOH und Et ,O gewaschen und im HV getrocknet. Ausbeute: 120 mg. Elementaranalyse: Gef.:, C, 39.22; H, 3.09. C,,H,,S,OMo (552.6) ber.: C, 41.29; H, 3.64%.
[Fe(Cl,-dpttd)CO] Zii 65 mg (1.2 mmol) MeONa in 30 ml THF/MeOH (l/l) werden 150 mg (0.54
mmol) Tetrachlor-o-benzoldithiol gegeben. Unter gleichzeitiger CO-Einleitung werden 55 mg (0.27 mmol) FeCl, .4H,O zugeftigt; nach weiterem 60 min CO- Einleiten wird in die klare tiefrote Liisung innerhalb von 3 min eine Liisung von 65 mg (0.27 mmol) Bis-(&bromethyl)sulfid in 4 ml THF/MeOH (l/l) getropft. W&rend 2-stiindigem Rtihren unter CO fallen aus der L&sung feine, hellbraune Kristaile aus, die abfiltriert und nach Waschen mit MeOH und Et ,O im HV getrocknet werden. Man erh< 90 mg mikrokristallines [Fe(Cl,-dpttd)CO] (46% d. Th. bez. auf FeCl, . H,O).
Elementaranalyse: Gef.: C, 27.89; H, 1.34. C,,H,OS,Cl,Fe (727.99) ber.: C, 28.05; H, 1.11%.
Eine Suspension von 1 g (1.36 mmol) [Fe(dpttd){P(OPh),}] in 6 ml Methylhy- drazin wird durch kraftiges Rtihren bei RT ftinfmal mit je 10 ml Petrolether

328
TABELLE 3
AUSGEWAHLTE SPEKTROSKOPISCHE DATEN
Verbindung IR
(cm-‘)
dpttd-H, v(SH) 2520’
dhthd-H, v(SH) 2520 ’
[NMe.,]2[Mo(dpttd’)(CO)s] v(C0) 1900 a 1790 mit Schuher bei 1765
]Mo(dpttdXCO), 1 r(C0) 1935 d 1880
v(C0) 1795 LI v(C0) 1810 c
[Fe@&,-dpttd)CO] v(C0) 1965 ’
v(NH) 3260 ’
3180
3130
‘H-NMR MS (ppm) rel. TMS (m/e)
7.1 b ( &Ar),m) [dpttd-Ha]+ 4.3 (&SH),s) 370
2.8 (G(CH,).m) (EI/FD) 7.1 h (S(Ar),m) [dhthd-Ha] +
4.3 (8(SH).s) 430
2.8 (S(CH,),m) (EI/FD) 2.6 (G(CH,),s) 7.0 c ( 8(Ar),m)
2-4 (F(CH,)) 3.1 (a(CH,),s) 7.2 ’ (a(Ar),m) lMo(WWW,l+ 2-4 (8(CH,)) 520
(FD) 7.2 d ( 8 (ArXm) [Mo(dpttd)PMe,(CO)]+ 1.8-3.8 (S(CH,)) 568
1.75 (a(CH,),d) (FD) _ [Fe(CI,-dpttd)]+
696-704
(EI)
Fe(W4W2Hdl+ 455
(FD)
a In KBr b In Ccl, ’ In DMSO-d, d In CD&I, e In THF; s = Singulett, d = Dublett, m = Multiplett.
extrahiert, wobei die Suspension in eine Losung tibergeht. Nach der letzten Extrak- tion wird die klare, tiefrote Methylhydrazinlosung auf 45°C erwarmt und unter Riihren langsam mit 20 ml MeOH versetzt, wobei gelbes, kristallines [Fe(dpttd)(N,H,CH,)] ausfallt, das bei -30°C abfiltriert und mit kaltem MeOH sowie Et,0 gewaschen wird. Nach Trocknen im HV (zunachst bei - 10°C und schliesslich bei RT) erhalt man 0.47 g gelbes [Fe(dpttd)(N,H,CH,)] (74% d. Th. bez.
auf [Fe(dpttd){P(OPh),}]). Elementaranalyse: Gef.: C, 43.15; H, 4.63; N, 5.98. C,,H,,N,S,Fe’(470.49) ber.:
C, 43.39; H, 4.71; N, 5.95%.
Dank
Diese Arbeit wurde von der Deutschen Forschungsgemeinschaft, dem Verband der Chemischen Industrie - Fonds der Chemischen Industrie - und durch eine Spende der Dr. Otto Rijhm Stiftung untersttitzt, woftir wir such an dieser Stelle herzlich danken mijchten.
Literatur
1 D. Sellmann, G. Lanzrath, G. Huttner, L. Zsolnai, C. Kruger und K.H. Claus, Z. Naturforsch, B, 38
(1983) 961. 2 VergI. dazu z.B. A.W. Addison, W.R. Cullen, D. Dolphin und B.R. James (Eds.), Biological Aspects of
Inorganic Chemistry, J. Wiley and Sons, New York, 1977.

329
3 D. Sellmann und U. Kleine-Kleffmann, J. Organomet. Chem., 247 (1983) 307.
4 J. Bradshaw, J. Heterocyl. Chem., 11 (1974) 45.
5 D. Sellmann und E. Bbhlen, Z. Naturforsch. B, 37 (1982) 1026.
6 D. Sellmann, H.E. Jonk und H.R. Pfeil, J. Organomet. Chem., 191 (1980) 171.
7 D. Sellmann und J. Schwarz, J. Organomet. Chem., 241 (1983) 343.
8 I. Degani und R. Fochi, Synthesis, 7 (1976) 741.
9 (a) Josef Pikl, Polychlorbenzenedithiols, U.S. Patent 2, 842, 578 Jul. 8. 1958; (b) M.J. Baker-Hawkes,
E. Billing und Harry B. Gray, J. Am. Chem. Sot., 88 (1966) 4870; (c) J.A. McCleverty, N.M. Atherton,
J. Locke, E.J. Wharton und C.J. Winscom, J. Am. Chem. Sot., 89 (1967) 6082.
10 W. Steinkopf, J. Herold und J. Stiihr, Ber. Deut. Chem. Ges., 53 (1920) 1007.
11 (a) G.M. Benett und E.M. Whincop, J. Chem. Sot., 119 (1921) 1860; (b) C. Price und R. Roberts, J.
Org. Chem., 12 (1947) 265.
12 Analog zu den in lla bzw. 10 angegebenen Vorschriften.
13 R. Colton und J.B. Tomkins, Aust. J. Chem., 19 (1966) 1143.
14 J.A. Bowden und R. Colton, Aust. J. Chem., 21 (1968) 2657.


![Metallkomplexe mit biologisch wichtigen Liganden, LXVIII ... · R. Bergs, K. Sünkel, W. Beck 2429 Notiz / Note Metallkomplexe mit biologisch wichtigen Liganden, LXVIII[1] Metallorganische](https://img.pdfslide.org/doc/110x75/5d66bbe188c9937b6e8bd214/metallkomplexe-mit-biologisch-wichtigen-liganden-lxviii-r-bergs-k-suenkel.jpg)




![FaktenzumRauchen - dkfz.de · Dibenzo[c,g]carbazol Benzo[b]furan 2B 2B 2B 2B 2B N-Nitrosamine N-Nitrosodimethylamin N-Nitrosomethylethylamin N-Nitrosodiethylamin N-Nitrosodi-n-propylamin](https://img.pdfslide.org/doc/110x75/5d611c4c88c993d14a8b5aab/faktenzumrauchen-dkfzde-dibenzocgcarbazol-benzobfuran-2b-2b-2b-2b-2b.jpg)





















