Embed Size (px)
Citation preview

UNIVERSITAT REGENSBURG
Institut fur Physikalische
und Theoretische Chemie
Prof. Dr. B. Dick
PRAKTIKUM PHYSIKALISCHE CHEMIE (TEIL I c)
(Spektroskopie)
Versuche UV 1, UV 2
Absorption im Sichtbaren und UV
1.1 Grundlagen
Im sichtbaren Spektralgebiet und im Gebiet des nahen UV findet man Absorptionen,die hauptsachlich Ubergangen zwischen elektronischen Energiezustanden der Atome oderMolekule entsprechen. So sind z. B. samtliche Farben und viele photochemische Reaktio-nen mit Ubergangen zwischen elektronischen Zustanden verbunden. Bei der Vielfalt derMolekule und der Unterschiedlichkeit der Elektronenstrukturen haben sich verschiedeneModelle zur Beschreibung der elektronischen Eigenschaften bewahrt.
0http://www-dick.chemie.uni-regensburg.de/studium/praktikum1c.html
1

1.1.1 Organische Molekule
Bei organischen Molekulen resultieren die hier zur Diskussion stehenden elektronischenMolekulzustande vorwiegend aus den 2s- und 2p- Zustanden der Kohlenstoffatome. Dieelektronische Wechselwirkung zwischen den Atomen des Molekuls laßt sich durch bin-dende σ-und π-Zustande sowie die zugehorigen antibindenden σ∗-und π∗-Zustande be-schreiben. In Verbindungen mit Heteroatomen (wie N, O, S oder Halogenen) gibt esauch elektronische Zustande, die nicht an den Bindungen beteiligt sind und mit einemfreien Elektronenpaar besetzt sind (nichtbindende Zustande, n–Zustande). Betrachtun-gen uber die Starke der Wechselwirkungen in den verschiedenen Zustanden ergeben eineinfaches, qualitativ gut verwendbares Energieniveauschema (s. Abb. 1.1) [1–4] .
Abbildung 1.1: Vereinfachtes Orbitalschema fur organische Molekule. n–Orbitale treten
nur in Molekulen auf, in denen sich Atome mit nichtbindenden Elek-
tronen befinden. Die Angabe uber die Besetzung bezieht sich auf den
Grundzustand des Molekuls. Besetzungsumlagerungen fuhren zu ange-
regten Zustanden.
Elektronische Ubergange konnen nur von besetzten zu nicht vollstandig besetzten Zu-standen stattfinden. Die im Grundzustand hochsten besetzten Molekulzustande heißenHOMO’s (highest occupied molecular orbitals), die niedrigsten nichtbesetzten Zustandeheißen LUMO’s (lowest unoccupied molecular orbitals). Viele Molekuleigenschaften wer-
2

den durch die HOMO’s und LUMO’s gepragt. Es konnen die folgenden Ubergange auf-treten:
n → π∗-Ubergange findet man nur, wenn freie Elektronenpaare vorhanden sind. Diesebesetzen haufig die HOMO’s. Die Ubergange haben dann die geringste Ubergangsenergie(s. Abb. 1.1). n → π∗-Ubergange sind meistens sehr schwach (Grund: geringe Uber-lappung zwischen den n- und den π∗-Orbitalen, s. Uberlappungsregel, Abschnitt 1.1.3)Typische ε-Werte (ε = molarer dekadischer Extinktionskoeffizient) liegen zwischen 5und 200 l · mol−1cm−1 (Tab. 1.1). Die energetischen Lagen sowie die Starken der Ab-sorptionsbanden von n → π∗-Ubergangen hangen relativ stark von der Polaritat desLosungsmittels ab.
π → π∗-Ubergange findet man bei allen Molekulen, bei denen π-Elektronen an der Bin-dung beteiligt sind. Enthalt das Molekul keine freien Elektronenpaare, dann sind dieπ → π∗-Ubergange diejenigen mit geringster Ubergangsenergie (HOMO → LUMO). Dieenergetischen Lagen der Absorptionsbanden verschieben sich mit wachsender Konjuga-tionslange der Molekule zu kleineren Ubergangsenergien (s.Tab. 1.2). Erstrecken sichdie Absorptionsbanden in den sichtbaren Spektralbereich, dann erscheinen die Molekulefarbig. Die ε-Werte liegen in dem weiten ε-Bereich von 101 bis 105 l · mol−1cm−1, (Tab.1.1, Tab. 1.2, Tab. 1.3, Tab. 1.4 ) Im allgemeinen findet man pro Modell mehrereπ → π∗-Ubergange.
n → σ∗-Ubergange findet man bei gesattigten Molekulen mit Heteroatomen, die freieElektronenpaare besitzen. Die Ubergangsenergien liegen meistens uber 40 000 cm−1 (250nm), und die molaren dekadischen Extinktionskoeffizienten sind etwas hoher als die dern → π∗ Ubergange. (Verbindungen dieses Typs werden wegen ihrer Polaritat haufig alsLosungsmittel fur spektroskopische Untersuchungen eingesetzt. Siehe Tab. 1.5).
Tabelle 1.1: UV-Absorption einfacher chromophorerGruppen (nach [2, 4, 5]). n → π∗ Ubergange (λmax, εmax)zeigen eine relativ starke Losungsmittelabhangigkeit.
Chromophor Ubergang Beispiel λmax εmax
[nm] [l/mol · cm]C—H σ → σ∗ CH4 122 intensivC—C σ → σ∗ H3C—CH3 130 intensiv—O— n→ σ∗ H2O 167 1500
n→ σ∗ H3C—OH 183 200n→ σ∗ H5C2—O—C2H5 189 2000
—S— n→ σ∗ H3C—SH 235 180n→ σ∗ H3C—S—CH3 228 620n→ σ∗ H5C2—S—S—C2H5 250 380
3
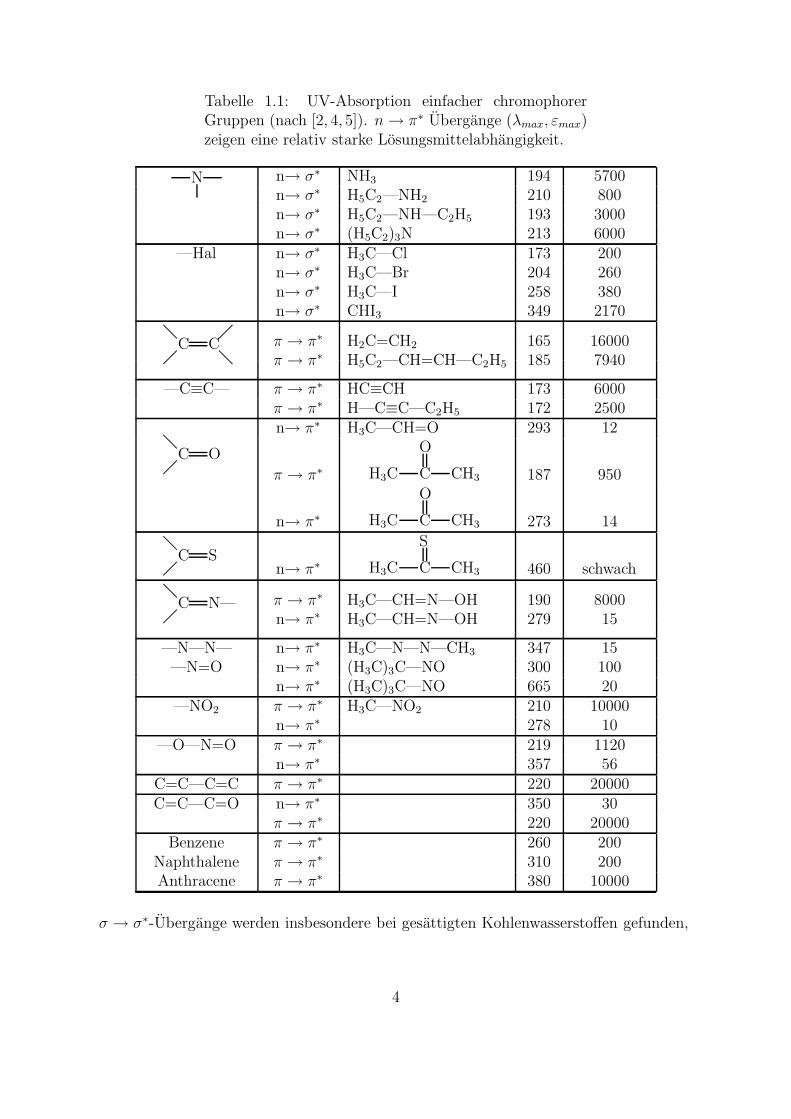
Tabelle 1.1: UV-Absorption einfacher chromophorerGruppen (nach [2, 4, 5]). n → π∗ Ubergange (λmax, εmax)zeigen eine relativ starke Losungsmittelabhangigkeit.
n→ σ∗ NH3 194 5700n→ σ∗ H5C2—NH2 210 800n→ σ∗ H5C2—NH—C2H5 193 3000
N
n→ σ∗ (H5C2)3N 213 6000—Hal n→ σ∗ H3C—Cl 173 200
n→ σ∗ H3C—Br 204 260n→ σ∗ H3C—I 258 380n→ σ∗ CHI3 349 2170
π → π∗ H2C=CH2 165 16000π → π∗ H5C2—CH=CH—C2H5 185 7940
C C@@
�� @@
��
—C≡C— π → π∗ HC≡CH 173 6000π → π∗ H—C≡C—C2H5 172 2500
C O@@
��
n→ π∗ H3C—CH=O 293 12
π → π∗ C
O
H3C CH3 187 950
n→ π∗ C
O
H3C CH3 273 14
C S@@
�� n→ π∗ C
S
H3C CH3 460 schwach
π → π∗ H3C—CH=N—OH 190 8000n→ π∗ H3C—CH=N—OH 279 15
C N—@@
��
—N—N— n→ π∗ H3C—N—N—CH3 347 15—N=O n→ π∗ (H3C)3C—NO 300 100
n→ π∗ (H3C)3C—NO 665 20—NO2 π → π∗ H3C—NO2 210 10000
n→ π∗ 278 10—O—N=O π → π∗ 219 1120
n→ π∗ 357 56C=C—C=C π → π∗ 220 20000C=C—C=O n→ π∗ 350 30
π → π∗ 220 20000Benzene π → π∗ 260 200
Naphthalene π → π∗ 310 200Anthracene π → π∗ 380 10000
σ → σ∗-Ubergange werden insbesondere bei gesattigten Kohlenwasserstoffen gefunden,
4

Tabelle 1.2: Absorptionspeaklagen einiger Polyine (nach [2, 6]): Hauptmaxima von kon-
jugierten Polyinen Me(C ≡ C)nMe
λmax εmax λmax εmaxn
[nm] [l/mol · cm] [nm] [l/mol · cm]
2 - - 250 160
3 207 135000 306 120
4 234 281000 354 105
5 260,5 352000 394 120
6 284 445000 - -
Tabelle 1.3: Absorptionspeaklagen einiger Polyene (nach [2, 6]): Langwelligste Absorp-
tion in konjugierten all-trans-Polyenen R − (CH = CH)n − R
R = CH31 R = C6H5
2
λmax εmax λmax εmaxn
[nm] [l/mol · cm] [nm] [l/mol · cm]
1 174 24000 306 24000
2 227 24000 334 48000
3 275 30200 358 75000
4 310 76500 384 86000
5 342 122000 403 94000
6 380 146000 420 1130001 aufgenommen in Petrolether bzw. Ether2 aufgenommen in Benzol
5
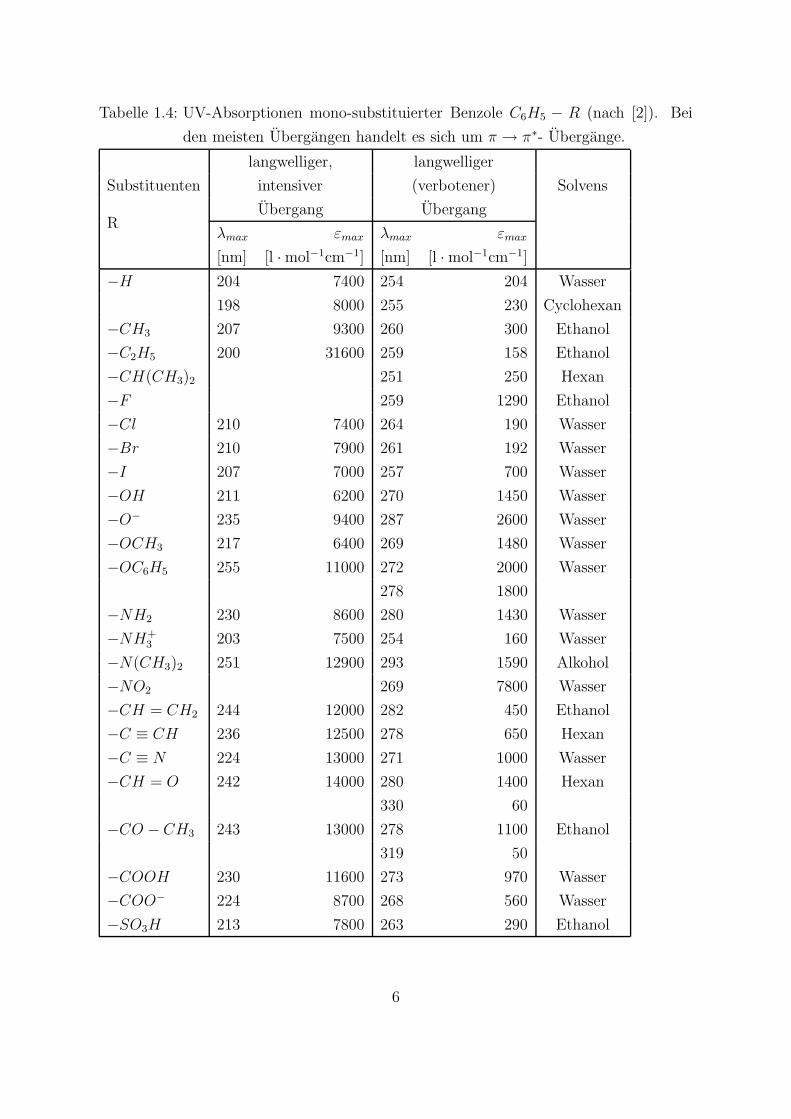
Tabelle 1.4: UV-Absorptionen mono-substituierter Benzole C6H5 − R (nach [2]). Bei
den meisten Ubergangen handelt es sich um π → π∗- Ubergange.
langwelliger, langwelliger
Substituenten intensiver (verbotener) Solvens
Ubergang UbergangR
λmax εmax λmax εmax
[nm] [l · mol−1cm−1] [nm] [l · mol−1cm−1]
−H 204 7400 254 204 Wasser
198 8000 255 230 Cyclohexan
−CH3 207 9300 260 300 Ethanol
−C2H5 200 31600 259 158 Ethanol
−CH(CH3)2 251 250 Hexan
−F 259 1290 Ethanol
−Cl 210 7400 264 190 Wasser
−Br 210 7900 261 192 Wasser
−I 207 7000 257 700 Wasser
−OH 211 6200 270 1450 Wasser
−O− 235 9400 287 2600 Wasser
−OCH3 217 6400 269 1480 Wasser
−OC6H5 255 11000 272 2000 Wasser
278 1800
−NH2 230 8600 280 1430 Wasser
−NH+3 203 7500 254 160 Wasser
−N(CH3)2 251 12900 293 1590 Alkohol
−NO2 269 7800 Wasser
−CH = CH2 244 12000 282 450 Ethanol
−C ≡ CH 236 12500 278 650 Hexan
−C ≡ N 224 13000 271 1000 Wasser
−CH = O 242 14000 280 1400 Hexan
330 60
−CO − CH3 243 13000 278 1100 Ethanol
319 50
−COOH 230 11600 273 970 Wasser
−COO− 224 8700 268 560 Wasser
−SO3H 213 7800 263 290 Ethanol
6

Tabelle 1.5: Absorption gesattigter Verbindungen (nach [4]).
Verbindung λmax εmax Ubergang
[nm] [l · mol−1cm−1]
CH4 125 - σ → σ∗
CH3 − CH3 135 - σ → σ∗
H2O 167 7000 n → σ∗
CH3OH 183 500 n → σ∗
CH3 − O − CH3 185 - n → σ∗
CH3NH2 213 - n → σ∗
CH3Cl 173 - n → σ∗
CH3 258 - n → σ∗
CH3(CH2)5SH 224 126 n → σ∗
wobei die Ubergangsenergien vorwiegend im Vakuum-UV-Gebiet liegen (unter ≈ 190 nm).Diese Ubergange konnen nur mit speziellen Spektralphotometern (evakuierter Strahlen-gang) beobachtet werden. Die ε-Werte liegen in den gleichen Großenordnungen wie dieder π → π∗-Ubergange (Tab. 1.1, Tab. 1.5).
σ → π∗-Ubergange. Diese relativ schwachen hochenergetischen Ubergange liegen mei-stens unter den sehr starken π → π∗-und σ → σ∗-Ubergangen verborgen.
In Abb. 1.2 sind die Energiebereiche der oben diskutierten Elektronenubergange in uber-sichtlicher Form zusammengestellt.Die Beschreibung der elektronischen Molekulzustande durch σ - ,π- und n–Zustandehat den Charakter einer Abschirmfeldnaherung. Jedes Elektron
”bewegt“ sich im Mo-
lekul unabhangig von den anderen (Einelektronennaherung). Es sind z.B. die explizitenWechselwirkungen der Elektronen untereinander, die Spins der Elektronen sowie dieWechselwirkungen mit den Molekulschwingungen vernachlassigt.
1.1.2 Ubergangsmetallkomplexe
Elektronische Ubergange in Ubergangsmetallkomplexen fuhren sehr oft zu charakteri-stischen Farben. Cr3+-Zentren bewirken z.B. die rote Farbe des Rubin oder die gruneFarbe des Smaragd. Die entsprechenden Absorptionsbanden lassen sich im wesentlichendurch Ubergange zwischen den durch das elektrische Feld der Liganden aufgespaltetend–Zustanden erklaren. Die theoretische Beschreibung erfolgt im Rahmen der Liganden-feldtheorie [1, 7].In vielen Ubergangsmetallkomplexen findet man eine oktaedrische (Oh–Symmetrie) odereine nahezu oktaedrische Koordination der Liganden um das Zentralion. Die d–Elektro-
7
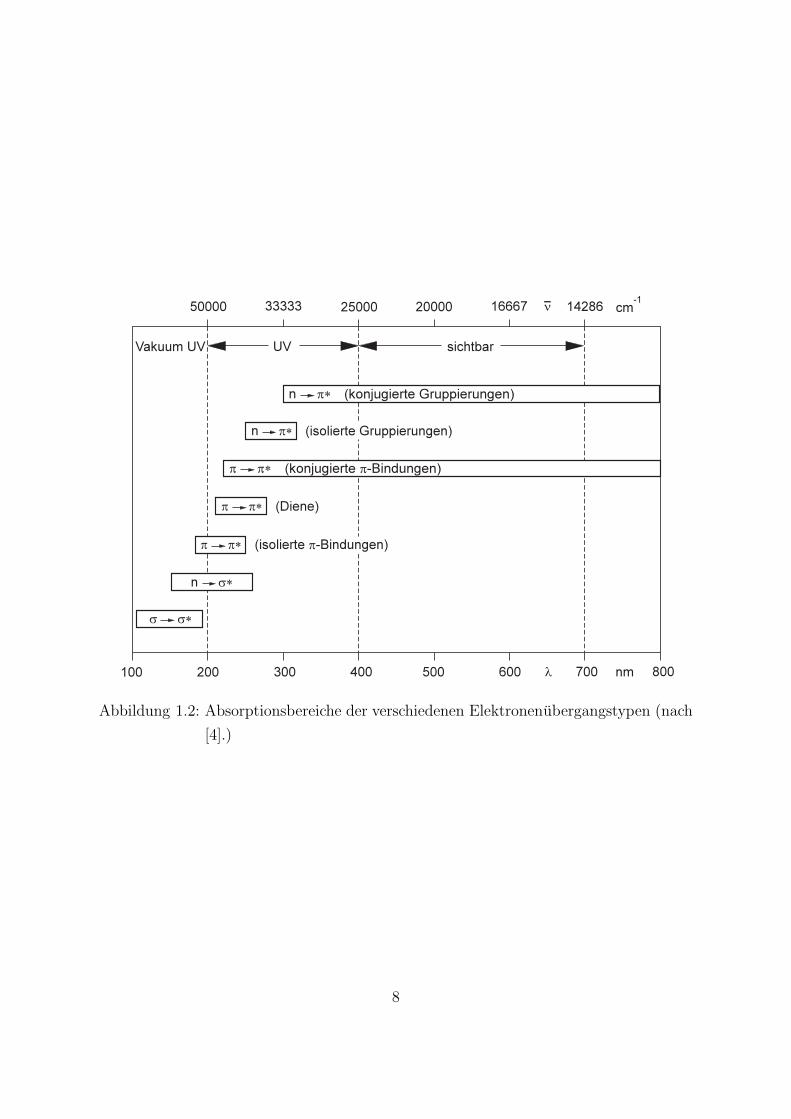
Abbildung 1.2: Absorptionsbereiche der verschiedenen Elektronenubergangstypen (nach
[4].)
8

nenzustande, die im freien Ion funffach entartet sind, spalten in der Oh–Symmetrie ineinen dreifach und einen zweifach entarteten Zustand auf (s. Abb. 1.3) . Die Großeder Aufspaltung wird mit ∆ ≡ 10 Dq bezeichnet. Der Wert des FeldstarkeparametersDq laßt sich experimentell bestimmen. Er hangt unter anderem stark von der Art derLiganden ab. Diese lassen sich nach wachsender Ligandenfeldstarke in der spektroche-mischen Reihe ordnen (Tab. 1.6, Tab. 1.7).
Abbildung 1.3: Aufspaltung der d–Elektronenzustande in einem oktaedrischen Feld.
Den Zustanden des Komplexes werden die gruppentheoretischen Sym-
bole eg bzw. t2g zugeordnet.
Tabelle 1.6: Spektrochemische Serie: Ordnung der Liganden nach steigender Liganden-
feldstarke ( Dq –Parameter). Je weiter rechts ein Ligand steht, desto starker
ist sein Ligandenfeld. Am Zentralion koordinierende Atome sind unterstri-
chen.
I− < Br− < Cl− ≈ SCN− ≈ N−3 < (C2H5O)2PS−
2 < F− < (C2H5)2NCS−2 <
< (NH2)2CO < OH− < (COO)2−2 ≈ H2O < NCS− < NH2CH2COO− <
< NCSHg+ ≈ NH3 ≈ C5H5N < NH2 · CH2 · CH2 · NH2 ≈ SO2−3 < NH2OH <
< NO−2 < H− ≈ CH−
3 < CN−
Es zeigt sich, daß die Einelektronen-Naherung, die zu dem in Abb. 1.3 dargestelltenEnergieniveaudiagramm fuhrt, im allgemeinen zu grob fur eine Beschreibung der expe-rimentellen Spektren ist. Die Ursache hierfur liegt insbesondere darin, daß dabei die
9

Tabelle 1.7: Dq –Werte fur einige Cr3+-Komplexe [8], DMSO = OS(CH3)2; urea =
(NH2)2CO; ox = (COO)2−2 ; acac = (CH3 · CO · CH · CO · CH3)
−; en =
NH2 − CH2 − CH2 − NH2
Oktaedrische Komplexe Dq –Werte[cm−1]
[CrCl6]3− 1380
[CrF6]3− 1490
[Cr(DMSO)6]3+ 1580
[Cr(urea)6]3+ 1620
[Cr(NO3)6]3− 1700
[Cr(ox)3]3− 1740
[Cr(H2O)6]3+ 1740
[Cr(NCS)6]3− 1770
[Cr(acac)3]0 1790
[Cr(NH3)6]3+ 2160
[Cr(en)3]3+ 2190
[Cr(CN)6]3− 2660
Wechselwirkung der Elektronen untereinander nicht vollstandig berucksichtigt ist. Ausquantenmechanischen Rechnungen unter Einschluß der Elektronenwechselwirkung erhaltman das Orgeldiagramm fur oktaedrische Chromkomplexe (s. Abb. 1.4) ([1, 7]).
Elektronische Ubergange zwischen den aus den d-Zustanden resultierenden Termen sindrelativ schwach. Die ε-Werte liegen fur Ubergange zwischen Termen mit gleichem Spin(spin-erlaubte Ubergange z.B. Quartett-Quartett-Ubergange) in der Großenordnung von100 l · mol−1cm−1 und fur Ubergange zwischen Termen mit verschiedenem Spin (spin-verbotene Ubergange oder Interkombinationsubergange,z.B. Quartett–Dublett–Ubergange)unter 10 l · mol−1cm−1 (bei Cr3+-Komplexen).
Erlauterung zur Abb. 1.4Aus diesem Diagramm Abb. 1.4 kann man fur Komplexe mit verschiedenen Liganden,sofern deren Dq –Werte bekannt sind, die Termenergien ablesen. Der Grundzustandliegt in der Abszisse. Als Beispiel ist die Konstruktion des Energieniveauschemas fur[Cr(CN)6]
3− dargestellt. Nach Tab. 1.7 gilt fur diesen Komplex Dq = 2660 cm−1. DieSchnittpunkte zwischen der Kurvenschar und der vertikalen Linie durch den Abszissen-wert Dq = 2660 cm−1 entsprechen den Termenergien des Komplexes [Cr(CN)6]
3−. Nachquantenmechanischen Rechnungen hat der energetische Abstand zwischen dem Grundzu-stand 4A2g und dem angeregten Zustand 4T2g in oktaedrischen Cr3+-Komplexen immerden Wert 10 Dq . 4A2g ↔4 T2g, ist der niedrigste spin-erlaubte Ubergang. Der entspre-chende Absorptionspeak ist im allgemeinen sehr breit.
10
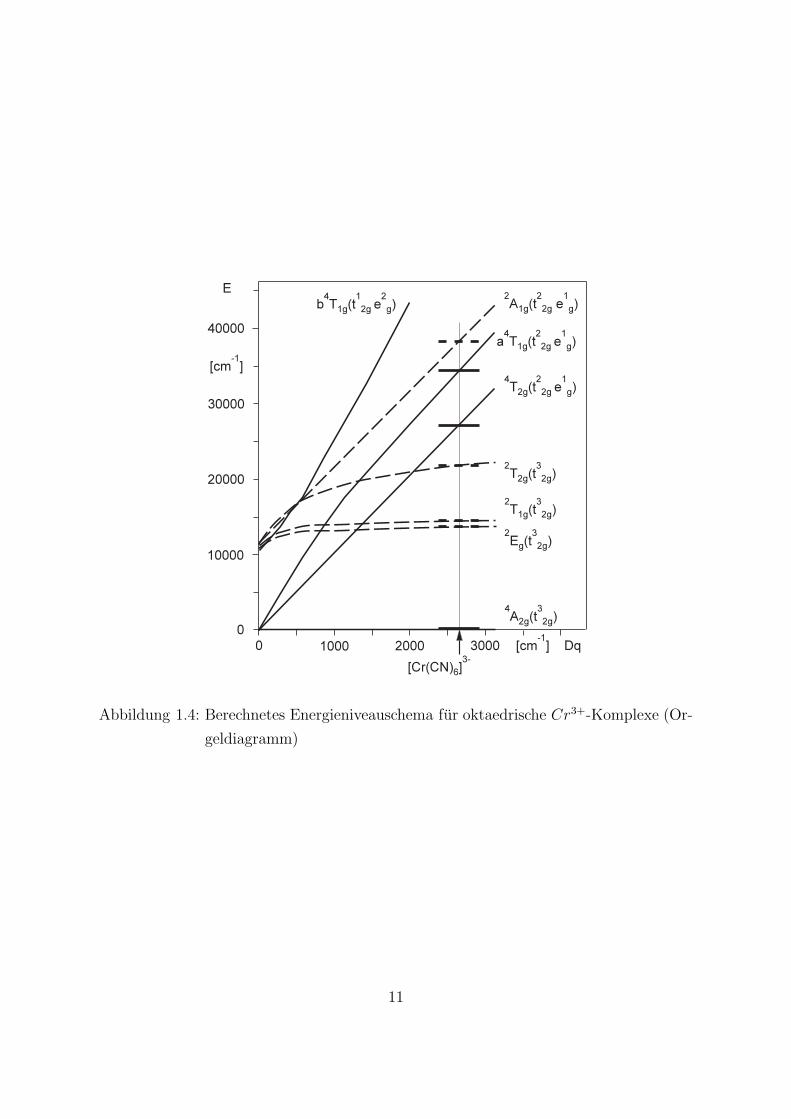
Abbildung 1.4: Berechnetes Energieniveauschema fur oktaedrische Cr3+-Komplexe (Or-
geldiagramm)
11

1.1.3 Auswahlregeln und Ubergangsintensitaten fur optische
Ubergange
Die Aussage, ein Ubergang sei”verboten“, bezieht sich meistens auf eine bestimmte
Naherung. In der Natur fuhren jedoch verschiedene Einflusse (wie Symmetrieernied-rigung, Spin-Bahn-Kopplung, Beimischung anderer Elektronenzustande usw.) sehr oftdazu, daß die ”verbotenen” Ubergange dennoch beobachtet werden konnen, allerdingshaufig nur mit sehr kleiner Intensitat. Je mehr dieser ”Verbote” bestehen, desto kleinerist der ε-Wert. Wichtige Regeln sind:
Laporte-Verbot
Besitzt das System ein Symmetriezentrum (Inversionszentrum), so bezeichnet man einenElektronen-Zustand als gerade (g), wenn seine Wellenfunktion bei Spiegelung am Inver-sionszentrum ihr Vorzeichen nicht andert (gerade Paritat). Anderenfalls spricht man voneinem ungeraden (u) Zustand (mit ungerader Paritat). Nach Laporte sind Elektronen-Ubergange zwischen Zustanden gleicher Paritat verboten.Beispiel:Atomare p-Funktionen haben u-Paritat, d-Funktionen haben g-Paritat. Nach Laportesind p ↔ p-Ubergange und d ↔ d-Ubergange paritatsverboten; p ↔ d-Ubergange sindparitatserlaubt.
Spin-Verbot
Elektronen-Ubergange zwischen Zustanden mit verschiedenem Spin sind verboten (In-terkombinationsverbot).
Uberlappungs-Regel
Elektronenubergange zwischen lokalisierten n-Zustanden (n- Elektronen sind im allge-meinen als Elektronenpaare an den Heteroatomen lokalisiert) und delokalisierten π∗-Zustanden sind verboten [4].
Um den Begriff des”Verbots“ eines Elektronenuberganges mit der relativ einfach meß-
baren Große ε in Beziehung zu setzen, ist in Abb. 1.5 ein hypothetisches Spektrumdargestellt. Es ist zu erkennen, daß sich die molaren dekadischen Extinktionskoeffizien-ten (fur verschiedene Ubergange) uber viele Zehnerpotenzen erstrecken konnen.
12
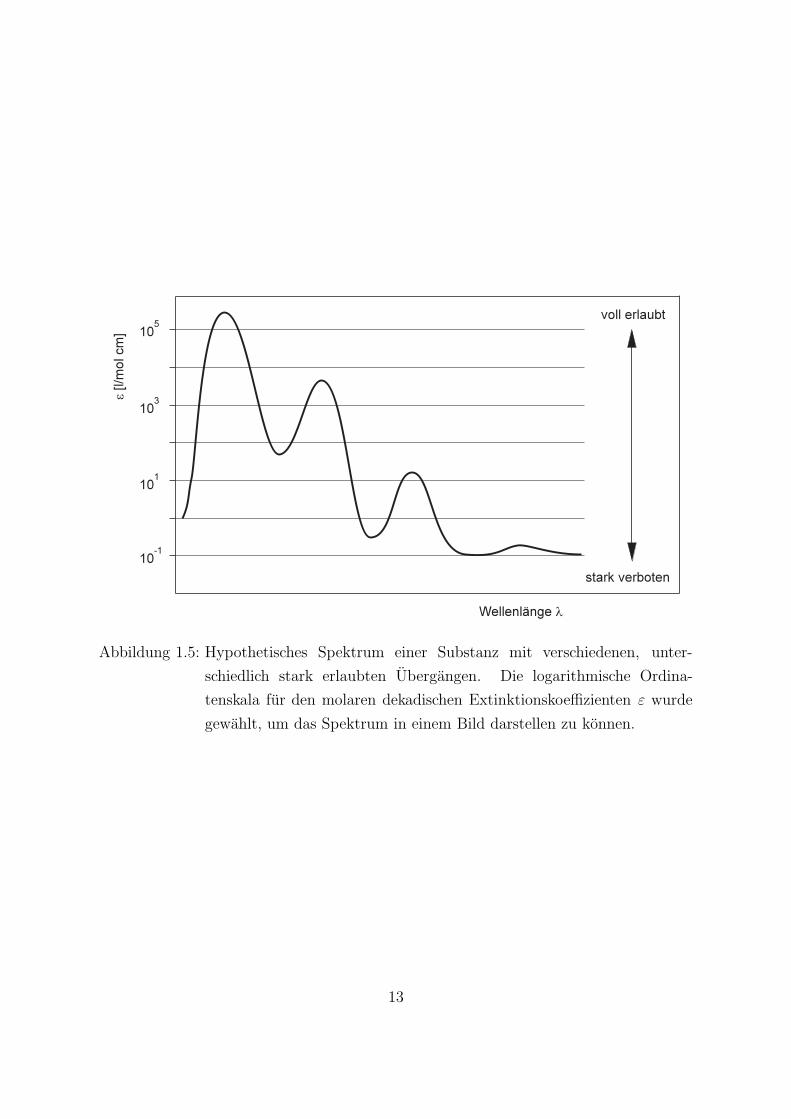
Abbildung 1.5: Hypothetisches Spektrum einer Substanz mit verschiedenen, unter-
schiedlich stark erlaubten Ubergangen. Die logarithmische Ordina-
tenskala fur den molaren dekadischen Extinktionskoeffizienten ε wurde
gewahlt, um das Spektrum in einem Bild darstellen zu konnen.
13

1.2 Aufbau des Spektralphotometers
Ein Schema des optischen Aufbaus des Spektralphotometers (UV 200 bzw. 210 von Shi-madzu) zeigt die Abb. 1.6. Mit dem Umklappspiegel M1 kann das Licht der Wolfram-Faden-Lampe W (Spektralbereich: ca. 350 nm - 850 nm) oder das der Deuterium-LampeD2 (Spektralbereich: ca. 200 nm–350 nm) gewahlt werden. Die Filter F1 bis F5, dienenzur Blockierung hoherer Ordnungen (Gittermonochromator!) und zur Streulichtvermin-derung. Der Wechsel der Filter erfolgt automatisch durch mechanische Kopplung mitdem Wellenlangenvorschub, und zwar passend zum jeweiligen Spektralbereich. Das Lichtgelangt durch den Eintrittsspalt S1 in den Monochromator, wird mit den Spiegeln M2
und M3 auf das Gitter G gefuhrt und dort spektral zerlegt reflektiert.
Abbildung 1.6: Schematischer Aufbau eines Zweistrahl-Spektralphotometers zur Mes-
sung der Absorption im UV- bzw. im sichtbaren Spektralgebiet.
Uber die Spiegel M4 und M5 verlaßt je nach Gittereinstellung das gewunschte mono-chromatische Licht den Monochromator durch den Austrittsspalt S2. Anschließend wirdmit Hilfe der Spiegelanordnung M6, M8 und M10 und der sich drehenden Spiegelsek-torscheibe M7 der Lichtstrahl so zerlegt, daß er abwechselnd entweder uber den SpiegelM8 auf die Referenzkuvette oder uber den Spiegel M10 auf die Probenkuvette gefuhrtwird. Hinter den Kuvetten wird das Licht durch eine entsprechende Spiegelanordnung(M9, M12, M13 und M11) wieder so zusammengefuhrt, daß abwechselnd Licht entwederaus dem Referenzstrahlengang oder aus dem Probenstrahlengang den Detektor (Photo-multiplier Pm) trifft. Die beiden rotierenden Spiegelsektorscheiben M7 und M12 mussenmit hoher Prazision mit genau der gleichen Umdrehungszahl und genau korreliert mit-einander laufen. Bei der elektronischen Verarbeitung der vom Multiplier geliefertenStrome werden die beiden Signale aus dem Proben- bzw. Referenzstrahlengang getrenntverstarkt. Je nach Einstellung des Gerates wird dann auf elektronischem Wege dasVerhaltnis der beiden Signale (Transmission) oder der negative dekadische Logarithmusdieses Verhaltnisses (Extinktion) gebildet und anschließend auf dem Schreiber ausgege-ben.
14

1.3 Losungsmittel
Nimmt man Absorptionsspektren von Substanzen im gelosten Zustand auf, ist vomLosungsmittel zu fordern, daß es im zu untersuchenden Spektralgebiet nicht absorbiert.Die Kenntnis der Absorption von Losungsmitteln ist also eine Voraussetzung zur er-folgreichen Registrierung von Spektren. Von einigen gebrauchlichen Losungsmitteln istin der folgenden Tabelle die Durchlassigkeitsgrenze im UV angegeben; das ist die Wel-lenlange, bis zu der das Losungsmittel (vom Sichtbaren aus) eingesetzt werden kann.Die angegebenen Grenzen gelten fur Losungsmittel, die fur die UV-Spektroskopie spezi-ell gereinigt sind (siehe z.B. Tab. 1.5).
Tabelle 1.8: UV-Durchlassigkeitsgrenzen fur verschiedene Losungsmittel
Petrolether Tetrachlorkohlenstoff 257 nm
Ligroin Schwefelkohlenstoff 340 nm
n-Hexan Methanol
n-Heptan 195nm Athanol200 nm
n-Octan Diathylether 200 nm
Cyclohexan Tetrahydrofuran 274 nm
Decalin 210 nm Dioxan 240 nm
Benzol 270 nm Wasser 185 nm
Methylenchlorid
Chloroform240 nm
1.4 Experimentelle Hinweise
1. Das Spektralphotometer muß zu Beginn einer Meßreihe entsprechend der auslie-genden Kalibriervorschrift justiert werden.
2. Die Schreiberanzeige muß entsprechend der Anzeige des Spektralphotometers (z.B.Extinktionswerte) bei maximaler Transmission (keine Probe) und minimaler Trans-mission (mit dem Shutterblock unterbrochener Strahlengang) kalibriert werden.
3. Beim Verlassen des sichtbaren Spektralbereichs in Richtung zum UV-Bereich mußbei ca. 350 nm von der Wolframfadenlampe auf die D2-Lampe umgestellt werden.Dazu mussen der Schreiber und das Spektralphotometer angehalten werden.
4. Die Referenzkuvette wird immer mit dem Losungsmittel gefullt, in dem die zuvermessende Substanz gelost wurde.
15

5. Die nicht mehr benotigten Losungen werden in die dafur vorgesehenen Abfallka-nister geschuttet.
6. Eine einwandfreie Absorptionsmessung ist nur moglich, wenn auf peinliche Sau-berkeit der Kuvetten geachtet wird. Die Kuvetten durfen nicht an den optischenFlachen beruhrt werden! Die Reinigung der Kuvetten erfolgt mit destilliertemWasser und anschließend mit Aceton. Gegebenenfalls konnen auch die jeweiligenLosungsmittel verwendet werden. 1 cm– und 2 cm–Kuvetten werden in der Zen-trifuge getrocknet. Ausnahme: Die 1mm-Kuvetten durfen nicht in der Zentrifugegetrocknet werden! Bruchgefahr! Verwenden Sie zum Trocknen den Fon. Zerbro-chene Kuvetten mussen vom dafur verantwortlichen Studenten ersetzt werden.
7. ProtokollfuhrungDie gesamte Protokollfuhrung erfolgt auf dem Schreiberpapier. Es mussen ne-ben den vermessenen Substanzen, Losungsmitteln und Kuvettendicken samtlicheveranderbaren Geratefunktionen notiert werden. Die Spektren sind in der Reihen-folge der Aufnahme durchzunumerieren.
1.5 Aufgabenstellungen
1.5.1 Das Lambert-Beersche Gesetz lautet:
T = I/I0 = 10−ε·c·d
E = ε · c · d
T = Transmission, E = Extinktion, ε = dekadischer molarer Extinktionskoeffizient[l · mol−1cm−1], c = Konzentration des absorbierenden Stoffes [mol/l], d = Dicke derabsorbierenden Schicht [cm]. Uberprufen Sie die Gultigkeit dieses Gesetzes fur zweiSysteme:
K2CrO4-Losung
Variieren Sie
1. die Konzentration [mol/l] in den Schritten c1 = 4 · 10−4, c2 = 2, 5 · 10−4, c3 =1 · 10−4, c4 = 2, 5 · 10−5 bei konstanter Schichtdicke d = 1 cm,
2. die Schichtdicke in den Schritten d1 = 0, 1 cm, d2 = 1 cm, d3 = 2 cm bei konstanterKonzentration c = 2 · 10−4 mol/l.
Tragen Sie die gemessenen Extinktionswerte E fur das langwellige Extinktionsmaximumnach Gleichung E = ε · c · d als Funktion des Produktes (c · d) auf (2 Meßkurven,graphische Fehlerbetrachtung).Hinweis: Losungsmittel 5 · 10−3 n NaOH
16

(NH4)FeIII(SO4)2 (A) / (NH4)SCN (B)-Losung
Vermessen Sie die Losungen mit folgenden (NH4)FeIII(SO4)2 ·12H2O-Konzentrationen:c = 10−2, 5 · 10−3, 4 · 10−3, 3 · 10−3, 2 · 10−3 und 10−3mol/l. Tragen Sie den Wert deslangwelligen Extinktionsmaximums uber der Konzentration auf.Hinweise:
1. Losungsmittel 0, 2 n H2SO4
2. Das Konzentrationsverhaltnis (A:B) der Stammlosung betragt 1:4.
3. Fur die Konzentrationen c = 10−2 und 5 · 10−3 mol/l mussen 1 mm-Kuvettenund fur c = 10−3 mol/l eine 2 cm–Kuvette verwendet werden. Fur die ubrigenKonzentrationen werden 1 cm–Kuvetten verwendet.
1.5.2 Losungsspektren organischer Molekule
Nehmen Sie die Absorptionsspektren von folgenden Substanzen auf (Konzentrationenin mol/l): Benzol (7 · 10−3), Benzaldehyd (5 · 10−2 und 1 · 10−4), p-Xylol (2 · 10−3),1,4-Diphenyl-1,3-Butadien (2 · 10−5) , Aceton (1 · 10−1).Hinweis:
1. Kuvettendicke 1 cm
2. Losungsmittel: Hexan
3. Die Spektren sind bis 200 nm(=50 000 cm−1) aufzunehmen.
1.5.3 Losungsspektrum eines Cr3+-Komplexes
Nehmen Sie das Absorptionsspektrum eines oktaedrischen Cr3+ -Komplexes auf. (DieKonzentration ist auf dem Meßkolben angegeben.)
1.6 Auswertung
Die Auswertung soll nach dem ausgeteilten Auswertungsschema erfolgen.
1. Diskutieren Sie, ob das Lambert-Beersche Gesetz fur beide Losungen (gem. 1.5.1)gilt. Falls das nicht der Fall ist, geben Sie eine Begrundung an. Anderenfalls be-stimmen Sie jeweils aus der Steigung der Meßkurve den ε-Wert des entsprechendenUberganges. Geben Sie Ihr Ergebnis mit Meßfehler an.
2. Bestimmen Sie fur alle Absorptionsubergange der organischen Substanzen die ener-getischen Lagen der Absorptionsubergange und die ε-Werte der Maxima. Ord-nen Sie die Absorptionsbanden dem Einelektronen-Obergangstyp (n → π∗, π →π∗, σ → σ∗ usw.) zu.
17

3. Um welches Ligandensystem handelt es sich bei dem vermessenen Cr3+-Komplex?Beachten Sie dazu die Tabelle mit den Dq –Werten (Tab. 1.7). KlassifizierenSie ferner alle auftretenden Ubergange gruppentheoretisch unter Verwendung desOrgeldiagramms (Abb. 1.4). Bestimmen Sie die zugehorigen ε–Werte.
18
Tabelle 1.9: Auswerteschema I — Spektren organischer Molekule
Substanz λmax νmax εmax λ ε Zuordnung der Uber- Begrundungen /
[nm] [cm−1] [l/mol cm] [nm] [l/mol · cm] gange zwischen den Bemerkungen
exp. exp. exp. Lit. Lit. Einelektronen-MOs
19
Tabelle 1.10: Auswerteschema II — Losungsspektrum eines Cr3+– Komplexes
λmax[nm] νmax[cm−1] εmax ∆ν1/2εmax Zuordnung Begrundung /
[l/mol cm] Bemerkungen
Dq-Wert:
Ligandsystem:
20

Literaturverzeichnis
[1] B. Dick oder H. Yersin: Vorlesung”Physikalische Chemie II“
[2] M. Hesse, H. Meier, B. Zeeh:”Spektroskopische Methoden in der organischen
Chemie“, G. Thieme Verlag, Stuttgart 1984.
[3] R. Demuth, F. Kober:”Grundlagen der Spektroskopie“, Diesterweg Salle, Verlag
Sauerlander 1977, S. 65 ff.
[4] H. G. O. Becker:”Einfuhrung in die Photochemie“, G. Thieme Verlag, Stuttgart
1983, Kap. 1 und 2.
[5] N. J. Turro:”Modern Molecular Photochemistry“, The Benjamin/Cummings Pu-
bl. Co., Menlo Park, California 1978.
[6] D. H. Williams, I. Fleming:”Spektroskopische Methoden in der organischen Che-
mie“, G. Thieme Verlag, Stuttgart 1971.
[7] H. L. Schlafer, G. Gliemann:”Einfuhrung in die Ligandenfeldtheorie“, Akad. Ver-
lagsges. Frankfurt/Main 1967.
[8] A. B. P. Lever:”Inorganic Electronic Spectroscopy“, Elsevier, Amsterdam 1984,
S. 419.
21





























![Vorexperimente zur koh˜arent ˜uberh ˜ohten Raman-Streuung ... · B Spektroskopie des Zustands E2§ ... Stimulated Raman Scattering, SRS, [2{4,7,9{11]). Dabei wird in Molek˜ulen](https://img.pdfslide.org/doc/110x75/5e2007194fe028629f270f1e/vorexperimente-zur-kohoearent-oeuberh-oeohten-raman-streuung-b-spektroskopie.jpg)