Embed Size (px)
Citation preview

Aus dem Institut für Pharmakologie, Toxikologie undPharmazie der Tierärztlichen Hochschule Hannover
_______________________________________________________
Untersuchung der Wirkung vonHypoxie auf das glutamaterge Systemals Tiermodell für die Ätiopathogenese
der Schizophrenie
INAUGURALDISSERTATION
Zur Erlangung des Grades eines Doktors der Veterinärmedizin
(Dr. med. vet.)durch die Tierärztliche Hochschule Hannover
Vorgelegt vonManfred Starke
aus Kalkar
Hannover 2002

Wissenschaftliche Betreuung: Prof. Dr. U. Ebert
1. Gutachter: Prof. Dr. U. Ebert
2. Gutachter: Prof. Dr. A. Tipold
Tag der mündlichen Prüfung: 22. November 2002

meinen Eltern und Annette,die immer zu mir gehalten haben


Inhaltsverzeichnis
1. Einleitung 9
2. Literaturübersicht 10
2.1. Schizophrenie 102.1.1. Ätiologie 102.1.2. Klinik – Klassifikation 112.1.3. Pathophysiologie – Erklärungshypothesen 112.1.4. Neurobiologie 122.1.5. Alterspräferenz 12
2.2. Das glutamaterge System 132.2.1. Glutamat-Rezeptoren 132.2.2. NMDA-Rezeptor 132.2.2.1. Aufbau und Vorkommen 132.2.2.2. NMDA-Rezeptor-Antagonisten 152.2.3. AMPA-Rezeptor 16
2.3. GABA-Rezeptor 162.3.1. GABAA-Rezeptor 17
2.4. Hypoxie – Ischämie 172.4.1. Effekte von Hypoxie und Ischämie auf die Zelle 172.4.2. Wirkung von Hypoxie und Ischämie auf das glutamaterge System 182.4.3. Wirkung von Hypoxie und Ischämie auf das GABAerge System 192.4.4. Wirkung von Hypoxie und Ischämie auf das Verhalten 20
2.5. Die Ratte als Tiermodell 20
2.6. Untersuchungsmethoden 212.6.1. Präpulsinhibition als Untersuchungsmethode für Verhaltensauffälligkeiten 212.6.2. In-Situ-Hybridisierung 222.6.3. Rezeptorautoradiographie 22
3. Eigene Untersuchungen 24
3.1. Material 243.1.1. Ratten 243.1.2. Geräte und Materialien 24
3.2. Methode 253.2.1. Versuche 253.2.1.1. Chronische Hypoxie (CH) 25

3.2.1.2. Kombinierte Hypoxie/Ischämie (HI) 263.2.1.3. Töten der Ratten - Gehirnentnahme und Lagerung 273.2.2. In-Situ-Hybridisierung 273.2.2.1. Das Anfertigen der Schnitte 273.2.2.2. Herstellung der benötigten Lösungen und Materialien 283.2.2.3. Versuchsablauf bei der In-Situ-Hybridisierung 293.2.2.3.1. Dehydrierung der Schnitte 293.2.2.3.2. In vitro – Transkription (Sonde) 303.2.2.3.3. Fraktionierung und Quantifizierung 313.2.2.3.4. Acetylierung 313.2.2.3.5. Hybridisierung 313.2.2.3.6. Inkubation 313.2.2.3.7. Posthybridisierung 323.2.2.3.7.1. Waschen 323.2.2.3.7.2. Dehydrierung 323.2.2.3.7.3. Exposition 323.2.3. Rezeptorautoradiographie 333.2.3.1. Das Anfertigen der Schnitte 333.2.3.2. Herstellung der benötigten Lösungen und Materialien 33
3.2.3.3. Versuchsablauf beim NMDA-Rezeptor - Ligand [3H]-MK-801 33
3.2.3.4. Versuchsablauf beim AMPA-Rezeptor - Ligand [3H]-AMPA 35
3.2.3.5. Versuchsablauf beim GABA-Rezeptor - Ligand [3H]-Muscimol 373.2.3.6. Entwicklung der belichteten Röntgen-Folien (RAR) 383.2.3.6.1. Herstellung der Lösungen 383.2.3.6.2. Entwicklung 383.2.4. Auswertung der Röntgenfolien (ISH und RAR) 383.2.5. Statistische Auswertung der Daten 393.2.6. Präpulsinhibition – Untersuchungen 393.2.6.1. Apparatur zur Auslösung und Messung der Schreckreaktion 393.2.6.2. Ablauf des Testprogrammes 403.2.6.3. Versuchsablauf 403.2.6.4. Auswertbare Parameter 40
3.3. Ergebnisse 423.3.1. Ergebnisse der Rezeptorautoradiographie: Rezeptorbindung 423.3.1.1. Präfrontaler Cortex – Gyrus Cinguli – (Cg1/3) 423.3.1.2. Infralimbischer Präfrontaler Cortex – (IL) 443.3.1.3. Lateraler Präfrontaler Cortex - (Fr1/2) 463.3.1.4. Nucleus Accumbens – (AcbC) 483.3.1.5. Striatum – (CPu) 503.3.1.6. Hippocampus: CA1 – Region 523.3.1.7. Hippocampus: CA2 – Region 543.3.1.8. Hippocampus: CA3a – Region 563.3.1.9. Hippocampus: CA3c – Region 583.3.1.10. Hippocampus: DG - Region 60

3.3.1.11. GABA: Präfrontaler Cortex - (Cg1/3, IL, Fr1/2) 623.3.1.12. GABA: Nucleus Accumbens - (AcbC) und Striatum - (CPu) 643.3.1.13. GABA: Hippocampus – (CA1, CA2, CA3a, CA3c, DG) 663.3.2. Ergebnisse der In-Situ-Hybridisierung (Rezeptordichte) 673.3.2.1. Präfrontaler Cortex – (Cg1/3) 683.3.2.2. Präfrontaler Cortex – Infralimbischer Cortex – (IL) 703.3.2.3. Lateraler Präfrontaler Cortex – (Fr1/2) 723.3.2.4. Nucleus Accumbens – (AcbC) 743.3.2.5. Striatum – (CPu) 763.3.2.6. Hippocampus: CA1 – Region 783.3.2.7. Hippocampus: CA2 – Region 803.3.2.8. Hippocampus: CA3a – Region 823.3.2.9. Hippocampus: CA3c – Region 843.3.2.10. Hippocampus: DG – Region 863.3.3. Ergebnisse der Messung der Schreckreaktion 883.3.3.1. Motorische Aktivität 883.3.3.2. Schreckreaktion und Habituation, Habituation in Prozent 883.3.3.3. Präpulsinhibition 883.3.3.4. Schreckreizhemmung in Prozent 89
4. Diskussion 93
4.1. Rezeptorautoradiographie 934.1.1. Vorinkubation bei NMDA-Rezeptoren 934.1.2. Rezeptoren 934.2. In-Situ-Hybridisierung 964.3. Präpulsinhibition 984.4. Schlussfolgerung 99
5. Zusammenfassung 101
6. Summary 104
7. Anhang 106
7.1. Einzeltierverzeichnis 106
7.2. Abkürzungsverzeichnis 110
7.3. Bildteil 112
8. Literaturverzeichnis 115


9
1. EinleitungSchizophrenie ist eine Erkrankung des Menschen, die in der Regel ab dem frühenErwachsenenalter auftreten kann. Mit einer Prävalenz von bis zu 1% ist sie nicht die häufigstepsychiatrische Erkrankung, ist aber aufgrund ihres meist schwerwiegenden Verlaufes ausmedizinischen, ethischen und wirtschaftlichen Gesichtspunkten bedeutsam. Momentan kann dieSchizophrenie nur mit begrenztem Erfolg und lediglich symptomatisch behandelt werden.Ferner wird häufig ein erneutes Ausbrechen der Erkrankung beobachtet.
Derzeit wird an der Ursache der Schizophrenie weltweit geforscht. Viele Erkenntnisse wurdenund werden gesammelt, aber ein großer Durchbruch steht noch aus. Aufgrund derunterschiedlichen Ausprägungsformen dieser Erkrankung und der komplexen Abläufe imGehirn ist es schwer, Zusammenhänge zwischen molekularbiologischen Veränderungen undVerhaltensveränderungen darzustellen.
Eine genetische Beteiligung an der Ätiologie der Schizophrenie wurde bereits nachgewiesen.Allerdings ist beispielsweise bei Zwillingsstudien lediglich eine 50%ige Konkordanz festgestelltworden. Es müssen folglich weitere Faktoren an der Entstehung der Schizophrenie beteiligtsein. Schwierige Umweltbedingungen, z.B. im sozialen Umfeld, als Kofaktoren zurBegünstigung der Schizophrenie, sind in den siebziger Jahren diskutiert und belegt worden.Derzeit werden zudem sowohl eine virale Beteiligung als auch Schwangerschafts- undGeburtskomplikationen von später an Schizophrenie erkrankten Patienten diskutiert.
Eine Hauptfolge von Schwangerschaftskomplikationen ist die Hypoxie bzw. die Ischämie.Verschiedene Gehirnregionen, wie der Präfrontale Cortex, das Striatum und der Hippocampus,werden als besonders empfindlich für Sauerstoffmangel erachtet.
Obwohl bereits viele Arbeiten zur Auswirkung von Sauerstoffmangel auf das Gehirnvorliegen, fehlen noch Untersuchungen, die den direkten Zusammenhang zwischenmolekularbiologischen Veränderungen und Verhaltensstörungen nach Hypoxie und Ischämiedarstellen.
In dieser Arbeit soll diese Frage erstmalig in einem geeigneten experimentellen Tiermodell(Gehnehmigungsnummer 164/98, siehe auch S. 23) untersucht werden. Hierfür wurde derZusammenhang zwischen Veränderungen an den Charakteristika von ionotropen Glutamat-und GABAA-Rezeptoren, zwei wichtigen Rezeptorklassen für die Neurotransmission imGehirn, und Verhaltensauffälligkeiten bei adulten Ratten untersucht, die in den erstenLebenstagen (vergleichbar dem noch ungeborenen humanen Fötus) eine Hypoxie und Ischämieerfahren hatten.

10
2. Literaturübersicht2.1. Schizophrenie2.1.1. ÄtiologieSchizophrenie ist eine schwere psychische Erkrankung, die Menschen geschlechtsübergreifend,in nahezu allen Altersgruppen und regional unabhängig ereilen kann. Die Prävalenz vonSchizophrenie wird mit ca. 1% angegeben (Häfner, 1995).Schizophrenie ist zwar medikamentös behandelbar, es kommt jedoch häufig zu Rückfällen, undeine Erkrankung an Schizophrenie hat häufig überdauernde psychische und sozialeBehinderungen zur Folge.
Die genaue Ätiologie der Schizophrenie ist noch immer ungeklärt (Henn, 1987). Schon seitdem frühen 20. Jahrhundert nimmt man eine genetische Disposition an (Günther-Genta et al.,1994). Aber auch weitere mögliche Ursachen werden diskutiert:Ein Ansatz der Ätiogeneseforschung ist die Suche nach Zusammenhängen von Schizophreniemit Schwangerschafts- und Geburtskomplikationen. Häufiger werden in der Vorgeschichteschizophrener Patienten Schwangerschafts- und Geburtskomplikationen, wie beispielsweiseFrühgeburt, Kaiserschnitt, verlängerte Geburtsdauer, Geburtsstillstände, gebärunfähigeKindslagen oder Asphyxie beschrieben (Parnas et al. 1982; McNeil, 1987; Lewis und Murray,1987; Owen et al., 1988; O'Callaghan et al., 1992;) Gemeinsamer Faktor dieserKomplikationen ist die perinatale Hypoxie oder Ischämie mit der Folge einerSauerstoffunterversorgung im Körper.Stress der schwangeren Mutter hat Einfluß auf das dopaminerge System des PräfrontalenCortex und erhöht dadurch die Summe der im Körper zirkulierenden Katecholamine. Währendder Schwangerschaft kann diese Katecholaminausschüttung zu einer Vasokonstriktion in derPlazenta führen. Damit ist der Fötus Hypoxie ausgesetzt und kann so geschädigt werden(Brixey et al., 1993).Auch andere Entwicklungsschäden des ungeborenen Kindes, vor allem des Gehirns, sind alsätiologische Ursachen der Schizophrenie in der Diskussion (DeLisi et al., 1987).Außerdem werden von manchen Autoren auch erbliche Autoimmunerkrankungen als Ursachevermutet (De Lisi et al., 1987; Brixey et al., 1993). Man nimmt an, dass der Körper postnatalzirkulierende Antikörper produziert, die gegen hirnspezifische Proteine gerichtet sind unddeshalb eine Gehirndysfunktion nach sich ziehen.Berichte über eine mögliche Rolle von Viren in der Ätiopathogenese der Schizophrenie werdenderzeit noch diskutiert. Vor allem die Borna-Virus-Infektion, aber auch der Befall mitRetroviren erweckt besonderes Interesse (Iwahashi et al., 1997; Yolken et al., 2000). Dergenaue Zeitpunkt der möglichen Infektion ist noch umstritten; sowohl prä- als auch perinataleInfektionswege sind in Betracht zu ziehen. Lipska und Weinberger (2000) konnten bei Rattenmit pränataler Bornavirus-Infektion vor allem Schäden im Hippocampus und im PräfrontalenCortex nachweisen.Auch der Befall von Influenza-A2 Viren bei Schwangeren im fünften Monat ist eingehenduntersucht worden. Eine jahreszeitliche Häufung der Geburtstermine bei Schizophrenenbesteht zwar, (Adams et al., 1993) – die Geburtstermine von Schizophrenen liegen gehäuft imHerbst und im Frühjahr - aber ein Zusammenhang zu Influenza A2-Viren konnte bisher nichthergestellt werden (Torrey, 1992).Einig sind sich dabei viele Autoren, dass der Einfluß von Hypoxie und viralen Infektionen aufdie Ätiologie der Schizophrenie dabei nicht die Rolle des genetischen Faktors beeinträchtigt. In

11
Wahrheit kann man sicher folgerichtig annehmen, dass sowohl genetische als auchUmweltfaktoren kombiniert zum Ausbruch dieser Krankheit führen (DeLisi et al., 1987; Brixeyet al., 1993)
2.1.2. Klinik - KlassifikationEs gibt bei der Schizophrenie zwei Formen von klinisch auffälligen Verhaltensveränderungen:eine Positiv- und eine Negativ-Symptomatik.Während sich die Positiv-Symptomatik durch eine produktiv psychotische Phase mitgesteigerter Aktivität, Wahn, Halluzinationen und Hypermotorik darstellt, zeichnet sich dieSchizophrenie bei der schlecht behandelbaren Negativ-Symptomatik durch Antriebslosigkeit,Aufmerksamkeitsstörungen, sozialem Rückzug und Affektstörungen aus. Der Verlauf derSchizophrenie kann akut oder chronisch sein. Bei chronischer Schizophrenie werden Stillständenach episodisch auftretenden akuten Schüben ebenso beschrieben wie Rückfälle nach langemStillstand. Mit zunehmendem Alter wird die Wahrscheinlichkeit eines Auftretens der erstenSymptome der Erkrankung immer unwahrscheinlicher (Henn, 1987; Farber et al., 1995).Obwohl seit einiger Zeit Antipsychotika zur Behandlung der akuten Positiv-Schizophreniesymptomatisch eingesetzt werden können, (z. B. Haloperidol oder Clozapin), ist mangelsKenntnis der Grundlagen eine ursächliche Therapie der Schizophrenie noch nicht in greifbarerNähe (Häfner, 1995).
2.1.3. Pathophysiologie - ErklärungshypothesenDer Neurotransmitter Dopamin scheint bei der Pathophysiologie der Schizophrenie eineentscheidende Rolle zu spielen, weil einige Hauptsymptome der Krankheit durch psychogeneStimulantien wie Amphetamin und Streß verschlimmert werden. Amphetamine und Streßbewirken beide eine Freisetzung von Dopamin bei Tieren (Brake et al., 1997). Es entstehen soSymptome, die denen der Positivsymptomatik der Schizophrenie gleichen.Allerdings lässt sich durch dieses Modell die Negativsymptomatik nicht erklären. Fernersprechen auch viele Patienten auf Dopaminantagonisten als Schizophrenie-Therapie nichtausreichend an.Die auf die Dopamin-Hypothese aufbauende Glutamathypothese der Schizophrenie wurde vonKim et al. (1980) begründet, aufbauend auf seine Befunde eines verminderten Glutamat-Spiegels im Liquor cerebrospinalis von Schizophrenie-Patienten, die jedoch nicht repliziertwerden konnten.
Phencycldin (PCP), ursprünglich als Anästhetikum verwendet, ist heute vor allem als Droge(„angel dust“) bekannt. PCP induziert einen psychotomimetischen Status, der derSchizophrenie sehr ähnelt. Im Gegensatz zur Amphetamin-induzierten Psychose, führt PCP zupositiven wie auch zu negativen Schizophrenie-ähnlichen Symptomen (Javitt und Zukin, 1991).Mit der durch Phencyclidin induzierten Psychose hat die experimentelle Schizophrenie-Forschung also ein Modell zur Verfügung, welches genauer die klinische Pathophysiologie derSchizophrenie widerspiegelt als die dopaminerge Stimulation (Thornberg und Saklad, 1996).PCP interagiert selektiv mit einer Bindungsstelle, die sich auf dem N-Methyl-D-Aspartat(NMDA) Rezeptor befindet (Javitt und Zukin, 1991). Die Besetzung dieses Rezeptors durchPCP induziert eine nicht kompetitive Hemmung von NMDA-Rezeptor-vermittelter Neuro-transmission. Andere NMDA-Antagonisten, wie z. B. Ketamin, induzieren bei im Verhältnis zuihrer Rezeptoraffinität vergleichbaren Dosierungen eben solche PCP-ähnlichen neurologischen

12
Verhaltenseffekte (Javitt und Zukin, 1991; Farber et al., 1995; Akbarian et al., 1996; Spandouet al., 1999).Diese Ergebnisse legen nahe, dass eine endogene Störung der NMDA-Rezeptor vermitteltenNeurotransmission bzw. eine Störung des glutamatergen Systems im Gehirn zur Pathogeneseder Schizophrenie beitragen könnte (Javitt und Zukin, 1991; Thornberg und Saklad, 1996).
2.1.4. NeurobiologieHistopathologische Veränderungen im Gehirn von Schizophrenie-Patienten sind vor allem imlimbischen System, im Dienzephalon und im Präfrontalen Cortex nachgewiesen worden(Weinberger, 1987). Viele Forscher haben aber auch eine Reduktion des Hippocampus-Volumens festgestellt (Stefanis et al., 1999; Weinberger, 1999). Diese Reduktion soll circa 4%betragen. Je geringer das Hippocampus-Volumen, umso früher kommt es zumKrankheitsausbruch. Außerdem scheinen diese Hippocampus-Abnormalitäten inSchizophrenie-Patienten besonders mit der postiven Symptomatik der Psychose assoziiert zusein (Stefanis et al., 1999). Der Hippocampus kann in das Ammonshorn und den Gyrusdentatus unterteilt werden. Der Anatom Rafael Lorente de No (1934) unterteilte dasHippocampus-Pyramidenzell-System in vier Untereinheiten, die noch heute so bezeichnetwerden: Cornu amonis (CA) 1-4. Die CA4-Region ist auch als Hilus des Gyrus dentatusbekannt (Nauta und Feirtag, 1986; Isaacson, 1987; Schmidt-Kastner und Freund, 1991).Weinberger (1999) konnte in der Region CA3-4 (Unterabteilungen des Ammonshorns)Änderungen bei Schizophrenie-Patienten feststellen: Die Expression von mRNA fürverschiedene Untereinheiten von nicht-NMDA-Glutamat-Rezeptoren war gesunken.Normalerweise steigt die Expression bei anderen psychischen Erkrankungen, wie z.B.Alkoholismus oder Alzheimer.Insgesamt sind aber die Daten über die normale Entwicklung, Zellbiologie und Funktion desHippocampus beim Menschen noch lückenhaft, so dass Schlussfolgerungen aus Unter-suchungen zu pathologischen Vorgängen im Hippocampus von schizophrenen Patienten mitVorsicht zu betrachten sind (Weinberger, 1999).Makroskopische Erweiterungen der 3. und 4. Hirnventrikel sind bei schizophrenen Patientenmittels MRI nachgewiesen worden (Cannon et al., 1989; Suddath et al., 1990; McGrath undMurray, 1995; Schmitt et al., 2001). Die Patienten mit vergrößerten lateralen Ventrikelnzeigten vermehrt negative Symptome und frühen Ausbruch der Krankheit. Dabei muß mannatürlich bedenken, dass die Patienten zum Teil unter Neuroleptika-Therapie standen undsomit die Positiv-Symptomatik unterdrückt sein kann (Pearlson et al., 1985).
2.1.5. AlterspräferenzÜber das Durchschnittsalter beim erstmaligen Auftreten der Schizophrenie liegenunterschiedliche Zahlen vor: Häfner (1995) gibt die Altersgruppe der 20 – 30-Jährigen alsbesonders anfällig an, laut Henn (1987) liegt das Durchschnittsalter bei 15-25 Jahren, Farber etal. (1995) konnten ein Durchschnittsalter von 19,9 Jahren feststellen.Die ersten Symptome können aber auch erst in viel späteren Jahren auftreten, besonders beiFrauen nach Beendigung des 40. Lebensjahres. Hierfür wird eine erhöhte Anfälligkeit vonFrauen durch den Abfall der Östrogenkonzentration in den Wechseljahren angenommen(Riecher-Rössler und Hafner, 1993).

13
2.2. Das glutamaterge System2.2.1. Glutamat-RezeptorenDer am häufigsten vorkommende kortikale Neurotransmitter ist Glutamat, gefolgt von GABA(McDonald und Johnston, 1990; Nakanishi, 1992; Carlsson et al., 1997;). Glutamat giltaußerdem als der am stärksten erregend wirkende Neurotransmitter (Cotman et al., 1987).Im Gehirn existieren fünf verschiedene Subtypen von Glutamat-Rezeptoren, die alle durchGlutamat aktiviert werden und nach für den jeweiligen Subtyp spezifischen Liganden benanntsind (Kim et al., 1998).Diese können in ionotrope Rezeptoren, N-Methyl-D-Aspartat (NMDA), 8-Amino-3-hydroxy-5-methyl-4-isoxazole-4-proprionat (AMPA) und Kainat (KA) und in mehrere metabotropeRezeptoren, z.B. L-2-amino-4-phosphonobuttersäure (L-AP4) oder 1S,3 R-trans-1-amino-cyclopethyl-1,3-dicarboxylate (trans-ACPD) unterteilt werden (McDonald und Johnston, 1990;Nakanishi, 1992; Kim et al., 1998). Die ionotropen Rezeptoren sind mit einem Ionenkanalverbunden. Dieser Ionenkanal ermöglicht im aktivierten Zustand den Ein- und Ausstrom vonKationen aus der Zelle und führt somit zu extra- und intrazellulären Veränderungen. Bei denmetabotropen Rezeptoren handelt es sich nicht um einen Kanal steuernden Rezeptor, sondernum einen Rezeptortyp, der an ein G-Protein gekoppelt ist. Bei diesen metabotropenGlutamatrezeptoren kann z.B. über die Phospholipase C ein spezielles Membranphospholipidgespalten werden (McDonald und Johnston, 1990).Die Verteilung der Glutamat-Bindungsstellen im Gehirn des Menschen verändert sich im Laufedes Lebens eines Individuums: im fetalen Hirn ist das Verteilungsmuster derGlutamatrezeptoren weiter gestreut, und auch eine quantitative Vermehrung dieser Rezeptorenist festzustellen (Barks et al., 1988). Die meisten Glutamatbindungsstellen im fetalen Hirnbefinden sich im Hippocampus, Nucleus caudatus/Putamen, Globus pallidus, subthalamischenNucleus und im reticulären Nucleus des Thalamus (Chaudieu et al., 1991). Eine maximaleExpression der Rezeptoren konnten Chaudieu et al. (1991) am 7. bis 14. postnatalen Tagausmachen. Gerade die Regionen mit hoher Glutamat-Rezeptor-Konzentration stehen imVerdacht, verwundbarer zu sein für neuronale Schädigungen durch Hypoxie oder Ischämie(Barks et al., 1988; Chaudieu et al., 1991). Carlsson et al. (1997) spekulieren sogar, dass einglutamaterges Defizit ein wichtiger pathogenetischer Faktor zumindest in einigen Fällen derSchizophrenie ist.
2.2.2. NMDA-Rezeptor2.2.2.1. Aufbau und VorkommenDer NMDA-Rezeptor ist einer von drei ionotropen Glutamat-Rezeptoren (Kim et al., 1998).An diesem Rezeptor bindet NMDA, ein Analog von Glutamat (Brewer und Cotman, 1989).Die wichtigste Funktion des NMDA-Rezeptors ist die Steuerung der GABAergen Neurone,um die tonische Hemmung beizubehalten (Farber et al., 1995).Der NMDA-Rezeptor ist mit einem Ionenkanal verbunden, der Na+ und Ca++ - Ionen denEintritt in die Zelle erlaubt (Espinoza und Parer, 1991). Dieser Rezeptor-Ionenkanal-Komplexspielt eine kritische Rolle in der neuronalen Entwicklung des Menschen, beim Lernen, Erinnernund bei der synaptischen Plastizität (Fritz et al., 1999a).Man kann zwei Hauptfamilien von NMDA-Rezeptor-Untereinheiten unterscheiden: diesogenannte NR1 und die NR2 – Familie (Cull-Candy et al., 1998; Kim et al., 1998). Eine NR1-Untereinheit und zwei bis drei NR2-Untereinheiten bilden den funktionellen Rezeptor (Waffordet al., 1993; Cull-Candy et al., 1998; Kim et al., 1998).

14
Die NR1-Untereinheiten-Familie besteht aus acht funktionellen Isoformen und einer nicht-funktionellen Isoform. Alle diese Varianten entstehen aus einem Gen (Cull-Candy et al., 1998).Die NR2-Untereinheiten-Familie besteht aus vier unterschiedlichen Formen: NR2A, NR2B,NR2C und NR2D. Alle diese Formen entstehen aus unterschiedlichen Genen (Cull-Candy etal., 1998). Von der NR2D-Untereinheit existieren zwei Varianten, wovon aber nur NR2D-2exprimiert wird (Wenzel et al., 1996).In neuesten Untersuchungen haben Sasaki et al. (2002) eine weitere Untereinheit des NMDA-Rezeptors charakterisieren können: NR3A. Diese Untereinheit scheint zu einem Abfall derCa++-Permeabilität und Mg++-Sensitivität zu führen. Die Expression von NR3A ist bisher inNeuronen von Nagern nachgewiesen worden (Sasaki et al., 2002).Die NR1-Untereinheit kodiert ein ca. 120 kDA großes Polypeptid, welches strukturelleÄhnlichkeit zu den Polypeptiden der AMPA- und KA-Rezeptoren von 22-26% aufweist(Moriyoshi et al., 1991; Kim et al., 1998). Die NR2-Untereinheiten bestehen aus größerenPolypeptiden (NR2A und NR2B mit z.B. ca. 165-170kDA). Sie sind zu 50-70% homologzueinander, aber nur zu 15-20% identisch zu der NR1-Untereinheit. Die vier Spezies der NR2-Untereinheit kann man in zwei Gruppen einteilen: (A, B) und (C, D) (Kim et al., 1998).Die NR1-Untereinheit scheint essentiell für die Funktion des Rezeptor-Kanal-Komplexes zusein, wohingegen die verschiedenen NR2-Untereinheiten die Funktion des NMDA-Rezeptorspotenzieren und modulieren (Sheng et al., 1994; Chen und Reiner, 1996; Kim et al., 1998).Der NMDA-Rezeptor besteht aus folgenden Bindungsstellen (Nakanishi, 1992; Hoffman et al.,1994; Fritz et al., 1999a; Fritz et al., 1999b):• Neurotransmitter-Bindungsregion mit Bindungsstellen für Agonisten (Glutamat) und
kompetetive Antagonisten (CPP)• regulatorische oder Co-Aktivator-Bindungsstelle (bindet Glycin)• Ionen-Kanal (im offenen Zustand bindet er die NMDA-Kanalblocker PCP und MK-801,
erlaubt Na+ und Ca++-Ionen den Eintritt in die Zelle)• eine spannungsabhängige Mg++-Bindungsstelle• inhibitorische Bindungsstelle (bindet Zn++)• Polyamin-Bindungsstelle (bindet Spermin oder Spermidin)Glycin ist für die Aktivierung des Rezeptors notwendig (Cotman et al., 1987). Auch derKationen-Kanal des NMDA-Rezeptors ist durch Glycin und auch Glutamat positiv modulierbar(Sakurai et al., 1991). Die NR1-NR2B - Rezeptoren sind sensibler gegenüber der Aktivierungdurch Glycin als zum Beispiel die NR1-NR2A - Rezeptoren (Zhong et al., 1996). DieBindungsaffinität des Rezeptors gegenüber Glutamat und Glycin hängt mit derZusammensetzung der verschiedenen Untereinheiten zusammen: so zum Beispiel hat derRezeptor NR1-NR2B die höchste Bindungsaffinität zu Glutamat (Laurie und Seeburg, 1994).Der NMDA-Rezeptor wird außerdem durch mikromolare Konzentrationen von Spermin,einem weiteren Polyamin, positiv moduliert. Liegt Spermin aber in höheren Konzentrationenvor, blockiert dieses den Rezeptor und den Ca++-Kanal (Ferchmin et al., 2000). Auch derMagnesium-Ionen-Spiegel der Zelle beeinflußt die Aktivierung des Rezeptors: bei sehrniedrigem Mg++-Gehalt der Zelle wird der NMDA-Rezeptor maximal aktiviert, durch hoheMg++-Spiegel gehemmt (Fritz et al., 1999b).
NMDA-Kanäle sind schon sehr früh im Gehirn auf sich noch entwickelnden neokortikalenNeuronen nachweisbar (Lo Turco et al., 1991). Die NMDA-Rezeptor-Verteilung ist ähnlichwie die AMPA-Rezeptor-Verteilung. Beide Rezeptoren sind im gesamten Gehirn zu finden,vor allem aber im Telenzephalon. Auch die CA1-Region des Hippocampus enthält sowohl

15
beim Menschen als auch bei der Ratte hohe NMDA-Rezeptor-Konzentrationen (Cotman et al.,1987). Die Zahl der NMDA-Rezeptoren ist im jugendlichen Rattenhirn größer als beierwachsenen Ratten. Im erwachsenen Rattenhirn findet man vermehrt Kainat-Rezeptoren(Hoffman et al., 1994).Rekombinante NR1-NR2-Kanäle zeigen vergleichbare Ca++-Permeabilität, weisen aberdeutliche Unterschiede im spannungs-abhängigen Mg++-Block auf (Monyer et al., 1994).Die Expression der verschiedenen NMDA-Rezeptor-Untereinheiten ist für jede Ent-wicklungsstufe in jeder Hirnregion der Ratte unterschiedlich: im cerebralen Kortex und imHippocampus (hier vor allem in CA1- und CA3-Pyramidenzellen) werden mRNAs, die NR2Cund NR2D kodieren nur ganz früh in der Entwicklung beobachtet. Normalerweise sind hier nurNR2A- und NR2B-mRNAs zu finden (Monyer et al., 1994; Perez-Velazquez und Zhang,1994). Die NR2D-mRNA nimmt während der postnatalen Entwicklung rapide ab (Wenzel etal., 1996). Der Peak in der Expression des Genes, welches NR1 kodiert, liegt hingegen beim10. postnatalen Tag (PND10) und im frühen Erwachsenenalter der Ratte (Pujic et al., 1993).Bei schizophrenen Human-Patienten hat man post mortem im Präfrontalen Cortex eine 53%igeSteigerung der Menge an NR2D-Untereinheit gefunden (Akbarian et al., 1996). ExperimentelleUntersuchungen an Tiermodellen, wie und warum es zur Steigerung der Menge dieserspeziellen NMDA-Rezeptor-Untereinheit gekommen ist, stehen noch aus.
2.2.2.2. NMDA-Rezeptor-AntagonistenDie NMDA-Rezeptor-Antagonisten binden am Rezeptor an unterschiedlichen Stellen undkönnen ihn so auf verschiedene Weise blockieren (Kornhuber et al., 1989; Fritz et al., 1999a).Sie blockieren entweder direkt den Ionen-Kanal (PCP (Phencyclidin), MK-801 ((+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate) und Ketamin) oder eine derregulatorischen Bindungsstellen des Rezeptors (CPP(3-(2-carboxypiperazin-4-yl)-1-phosphonic acid), ist Antagonist an der Glutamat-Bindungsstelle, D-Serin an der Glycin-Bindungsstelle) (Hoffmann et al., 1994; Farber et al., 1995; Fritz et al., 1999a; Spandou et al.,1999).MK-801 (seit kurzem auch Dizolcipin genannt) ist ein nicht-kompetitiver Antagonist, der imIonen-Kanal des NMDA-Rezeptors bindet. Interessanterweise kann MK-801 nur am offenenKanal binden, also erst nach physiologischer Aktivierung des NMDA-Rezeptorkomplexes(Hoffman et al., 1994). Die MK-801-Bindung wird daher bei Anwesenheit von physiologischenAgonisten, Koagonisten oder Modulatoren, wie NMDA, Glycin, Spermin oder Spermidinverstärkt (Sakurai et al., 1991; Subramaniam und McGonigle, 1991). Sakurai et al. (1991)weisen in ihren Untersuchungen nach, dass MK-801 in der quantitativen Autoradiographie alshochaffiner und selektiver Ligand für den NMDA-Rezeptor-Komplex eingesetzt werden kann.Die kompetitiven und die nicht-kompetitiven Antagonisten des NMDA-Rezeptors sind in derLage, eine neuroprotektive Wirkung zu entfalten (Arias et al., 1999), besitzen aberdosisabhängig auch neurotoxische und psychomimetische Aktivitäten: so zum Beispiel könnenalle diese genannten Antagonisten bei "normalen" Individuen, das heißt gesunden Patienten,eine Psychose mit negativen und positiven Symptomen der Schizophrenie erzeugen (Farber etal., 1995; Akbarian et al., 1996; Spandou et al., 1999). Auch bei der Ratte hat manexperimentelle Untersuchungen mit NMDA-Rezeptor-Antagonisten durchgeführt. Dieneurotoxische Aktivität von MK-801 war bei dieser Spezies stark altersabhängig: je älter dieRatte, umso sensitiver war sie. Dieses altersabhängige Profil (Anfang der Empfindlichkeit imfrühen Erwachsenenalter) bei der Ratte ist vergleichbar mit der gegenüber Ketamin induzierterPsychose oder Schizophrenie beim Menschen. Diese Befunde legten nahe, dass die NMDA-

16
Rezeptor-Hypofunktion (der Mechanismus, der der neurotoxischen und psychomimetischenAktivität der NMDA-Antagonisten zugrunde liegt) auch eine Rolle in der Schizophreniespielen könnte (Farber et al., 1995). Auch Olney und Farber (1995) nahmen bei derSchizophrenie eine Unterfunktion des NMDA-Rezeptors mit resultierender Zerstörung vonInterneuronen mit den Folgen mangelnder hemmender Neurotransmission und Zunahmeexzitatorischer Einflüsse an. Dies würde erst im frühen Erwachsenenalter, wenn entscheidendeNeuronenschaltkreise reifen, zu Fehlfunktionen führen, durch die eine Schizophrenie mittypischen verzögerten Symptomen ausgelöst wird.NMDA-Rezeptor-Antagonisten sind aber auch schon experimentell bei Ratten als neuro-protektive Drogen eingesetzt worden, vor allem in Situationen, wo Neuronen geschädigtwerden aufgrund von exzessiver Aktivität der NMDA-Rezeptoren (Farber et al., 1995).
2.2.3. AMPA-RezeptorDer AMPA-Rezeptor gehört wie der NMDA-Rezeptor zu den ionotropen Glutamat-Rezeptoren. Er ist mit einem Ionenkanal verbunden, der Kationen den Eintritt in die Zelleerlaubt (Espinoza und Parer, 1991; Nakanishi, 1992).Die Verteilung der AMPA-Rezeptoren ist mit der, der NMDA-Rezeptoren vergleichbar: inwelcher Region des Gehirns auch immer sich NMDA-Rezeptoren befinden, sind auch AMPA-Rezeptoren nachzuweisen (Cotman et al., 1987).Der AMPA-Rezeptor besitzt vier Glutamat-Rezeptor-Untereinheiten: GluR1 bis GluR4. Beieinem AMPA-Rezeptor mit GluR2-Untereinheiten kann bei Aktivierung wie bei NMDA-Rezeptoren, Ca++ in die Zelle strömen, ansonsten nicht (Gozal et al., 1999; Calabresi et al.,2000).Die AMPA-Rezeptor vermittelte Neurotoxizität zeigt ihren Höhepunkt am 10. postnatalen Tag(Romijn et al., 1991). Durch extrazelluläre Azidose kann sie im Gegensatz zur NMDA-Rezeptor vermittelten Neurotoxizität ansteigen (McDonald et al., 1998).AMPA-Antagonisten besitzen genauso wie NMDA-Antagonisten eine neuroprotektiveWirkung, die aber in verschiedenen Hirnregionen unterschiedlich ausgeprägt ist: So reduzierenAMPA-Rezeptor-Antagonisten experimentell an Zellkulturen nachweislich Neuronenschädenoder Neuronenverluste in der CA1-Region, wo NMDA-Antagonisten relativ unwirksam sind(McDonald et al., 1998).
2.3. GABA-RezeptorIm Gehirn besteht eine feinregulierte Balance zwischen dem exzitatorischen Glutamatsystemund dem inhibitorischen GABA-ergen System. GABA (γ-Amino-Buttersäure), der wichtigsteinhibitorische Neurotransmitter, bindet an 3 verschiedenen GABA-Rezeptoren im ZNS:GABAA-, GABAB- und GABAC-Rezeptoren (Van Eden et al., 1989; Starke, 1996; Bormann,2000).Der GABAB-Rezeptor ist nicht nur postsynaptisch, sondern auch präsynaptisch zu finden. Ersteht über einen G-Protein gekoppelten Mechanismus mit Ca++- und K+-Ionenkanälen inVerbindung (Watling, 1998).Der GABAC-Rezeptor ist wie der GABAA-Rezeptor mit einem Chlorid-Ionophor gekoppelt.Dieses moduliert die Öffnung von Ionenkanälen und damit den Chlorid-Ionenfluß. Bisherwurde er hauptsächlich in der Retina gefunden, man nimmt jedoch an, dass er auch inverschiedenen anderen Regionen des ZNS vorkommt (Watling, 1998).

17
2.3.1. GABAA-RezeptorDer GABAA-Rezeptor-Komplex ist eine pentamere Struktur, wobei fünf meist verschiedeneUntereinheiten so angeordnet sind, dass ein Chloridkanal entsteht (Olsen und Tobin, 1990;Cobas et al., 1991; Luhmann et al., 1993). Bisher wurden folgende Untereinheiten aus siebenFamilien entdeckt: 6 α-, 4 β-, 3 γ-, 1 δ-, 1 ε-, 1 π- und 3 ρ-Untereinheiten (Barnard et al.,1998). Whiting (1999) konnte auch Hinweise auf eine zusätzliche θ-Untereinheit geben.Demzufolge können theoretisch sehr viele GABAA-Rezeptoren mit unterschiedlichenKombinationen von Untereinheiten vorkommen. Es wurden bisher jedoch nur 15 verschiedeneUntertypen beschrieben, wobei der GABAA-Rezeptorkomplex aus zwei α-, zwei β- und einerweiteren Untereinheit zusammengesetzt ist (Cobas et al, 1991).Durch Bindung des Neurotransmitters GABA an den GABAA-Rezeptor, wird der Ionenkanalgeöffnet und die Chloridionen können in die Zelle einströmen (Segal, 1993).Die Ontogenese der GABAergen Innervation ist wie die anderen Neurotransmittersysteme imGehirn zum Zeitpunkt der Geburt noch nicht vollständig abgeschlossen. Der GABA-Gehalt imGehirn von neugeborenen Ratten beträgt nur ungefähr 50% des Gehaltes von erwachsenenRatten. Außerdem macht die Dichte der postsynaptischen GABAA-Rezeptoren bei acht Tagealten Ratten nur 25% derjenigen bei erwachsenen Tieren aus (Coyle und Enna, 1976).Der GABAA-Rezeptor besitzt folgende Bindungsstellen für Neurotransmitter undpharmakologische Modulatoren (Rothe et al., 1988; Olsen und Tobin, 1990; Möhler, 1992):• GABA-Bindungsstelle• Benzodiazepin-Bindungsstelle• Pikrotoxin-Bindungsstelle• Barbiturat-Bindungsstelle• Weitere Bindungsstellen (hier binden unter anderem einige Steroidhormone)Durch Bindung dieser Substanzen an den Rezeptor kann der durch GABA ausgelöste Chlorid-Ionenfluß moduliert werden, und damit auch die hemmende GABA-Wirkung verstärkt oderreduziert werden.So verstärken z.B. volle Benzodiazepinagonisten die GABA-Wirkung durch Erhöhung derÖffnungsfrequenz des Chlorid-Ionenkanals (Günther et al., 1995). Pikrotoxin und Tetrazol-Derivate reduzieren direkt den GABA-induzierten Chlorid-Ionenfluß, indem der Eintritt vonChlorid-Ionen in den Ionenkanal verhindert wird (Möhler, 1992; Sieghart, 1992). Barbiturateverstärken die Wirkung von GABA durch Verlängerung der Öffnungszeit des Ionenkanals.Auch Steroidhormone verstärken die Wirkung von GABA, allerdings durch die Verlängerungder Öffnungszeit des Ionenkanals (Sieghart, 1992).
2.4. Hypoxie - Ischämie2.4.1. Effekte von Hypoxie und Ischämie auf die ZelleHypoxie wird als verminderte bis unzureichende Sauerstoffversorgung der Körpergewebedefiniert. Unter Ischämie versteht man die Blutleere oder Minderdurchblutung eines Gewebesinfolge unzureichender oder fehlender arterieller Blutzufuhr (Boss, 1993).Ischämie setzt im allgemeinen größere Schäden im Gehirn als Hypoxie (Pearigen et al., 1996).Zerebrale Hypoxie und Ischämie führen zu verschiedenen zellulären pathologischenVeränderungen und können so in neuronalem Tod enden (Hoffman et al., 1994).Die Pathophysiologie der ischämischen Hirn-Läsion ist bis jetzt, auch beim Menschen, nochnicht vollständig verstanden. Nach der gegenwärtig vorherrschenden Hypothese führt eineischämische Verletzung zur Veränderung des zellulären Energie-Metabolismus, zur

18
intrazellulären Ca++-Akkumulation und zur Freisetzung exzitatorischer Aminosäuren (Bergerund Garnier, 2000). Auch Arias et al. (1999) und Toner und Stamford (1997) konnten zeigen,dass die extrazelluläre Konzenztration der Neurotransmitter, wie zum Beispiel Glutamat undAspartat, nach Ischämie stark erhöht ist.Ein zellulärer pathologischer Mechanismus, der mit ischämischer Neurodegeneration assoziiertist, ist Exzitotoxizität. Darunter versteht man das Absterben der Neurone aufgrund zu massiveroder zu langer Einwirkung von exzitatorischen Neurotransmittern, wie zum Beispiel Glutamatoder Aspartat (Arias et al., 1999; Johnston et al., 2001; Vannucci et al., 2001) und der darausresultierenden Erhöhung der intrazellulären Ca++-Konzentration. Diese Exzitotoxizität bei derPathogenese der Zellzerstörung spielt im sich entwickelnden Hirn eine größere Rolle als imerwachsenen Hirn (Johnston et al., 2001).Delivoria-Papadopoulos und Mishra (Mishra und Delivoria-Papadopoulos, 1999; Delivoria-Papadopoulos und Mishra, 2000) nehmen sogar an, dass der starke intrazelluläre Ca++-Anstiegzu einer Veränderung der Transkription spezifischer apoptotischer Gene führt, dies soEndonukleasen aktiviert und letztendlich zum programmierten Zelltod führt.Durch Hypoxie können in der Zelle außerdem freie Radikale freigesetzt werden. Diese freienRadikale können unter anderem zur Membran-Lipid-Peroxidierung und letztendlich zurZellmembran-Dysfunktion führen (Mishra und Delivoria-Papadopoulos, 1999). Eines der freienRadikale ist Stickstoffmonoxid (NO). NO+ ist die reduzierte Form des NO. NO hatneurotoxische Effekte. NO+ scheint dagegen neuroprotektive Effekte zu haben, in dem es denRedox-Zustand des NMDA-Rezeptor-Ionen-Kanals so verändert, dass sich der Kanal wenigeröffnet und deshalb weniger Ca++-Ionen in die Zelle gelangen (Groenendaal et al., 1997).Hypoxie und Ischämie führen gleichermaßen zu einer insuffizienten Adenosin 5'-Triphosphat-Produktion. Normalerweise wird bis zu 60% von diesem ATP genutzt, um das Ionen-Gleichgewicht in der Zelle zu halten, hauptsächlich indem Na+ aus der Zelle und K+ in die Zellegepumpt wird. Eine Reduzierung des ATP aufgrund von Hypoxie führt also zu schwerenStörungen des Ionen-Gleichgewichts. In dem Moment, wo Na+ physiologisch in die Zelleeintritt und K+ die Zelle verläßt, depolarisiert die Zellmembran und führt zu einem Spannungs-abhängigen Öffnen von Ca++-Kanälen und noch mehr Ca++-Einstrom in die Zelle (Espinoza undParer, 1991; Mishra und Delivoria-Papadopoulos, 1999).
2.4.2. Wirkung von Hypoxie und Ischämie auf das glutamaterge SystemDie Topographie der durch Hypoxie oder Ischämie verursachten Schäden im Gehirnkorrespondiert zur Verteilung der Glutamat-Rezeptoren (McDonald und Johnston, 1990). Sosind zum Beispiel Hippocampus-Neurone besonders sensitiv gegenüber zerebralen Schädendurch Ischämie. Man nimmt an, dass dies auf die hohe Dichte an Glutamat-Rezeptoren imHippocampus beruht (Barks et al., 1988; Zhao und Flavin, 2000; Liu et al., 2001). Innerhalbdes Hippocampus sind die CA1-Zellen sensitiver gegenüber Zellschäden durch Hypoxie oderIschämie als die CA3- oder DG-Region (Gyrus dentatus) des Hippocampus (Cheung et al.,2001).Exzitatorische Aminosäuren spielen eine wichtige Rolle bei neuronalen Schäden des Hirnsdurch Hypoxie und Ischämie: Die Glutamat- und Aspartat-Konzentrationen im extrazellulärenRaum des Gehirns steigen nach Hypoxie oder Ischämie an (Krajnc et al., 1996; Sebastião et al.,2001). Sowohl die alleinige in vivo als auch die in vitro Darreichung von Glutamat kannexperimentell zum Neuronen-Tod in Bezirken des Gehirns mit einer höheren Dichte vonGlutamat-Rezeptoren führen (Espinoza und Parer, 1991).

19
Die Hypoxie- bzw. Ischämie-bedingte Freisetzung von exzitatorischen Aminosäuren, aktiviertden NMDA-Rezeptor, und der intrazelluläre Spiegel von Na+ und Ca++-Ionen steigt an. Vorallem der Anstieg an Ca++-Ionen aktiviert wiederum Proteasen, Phospholipasen undStickstoffmonoxidsynthetasen und führt damit zur Bildung freier Radikale und zum Zelltod(Toner und Stamford, 1997; Fritz et al., 1999a; Xie et al., 1999; Yoshioka et al., 2000).Zerebrale Ischämie führt also zu Gewebeschädigung durch Überstimulation der neuronalenGlutamat-Rezeptoren. Aber auch Sauerstoffmangel-bedingte Membrandepolarisation führt zurpassiven Öffnung des NMDA-Rezeptor-Ionen-Komplexes (Johnston et al., 2001; Vannucci etal., 2001). Das pathologische Geschehen steigert sich zu einer verlängerten Öffnung derNMDA-Rezeptor-Ionen-Kanäle und kulminiert in abnorm hohen intrazellulären Ca++-Spiegeln(Arias et al., 1999; Fritz et al., 1999a; Delivoria-Papdopoulos und Mishra, 2000).Durch Gabe von NMDA-Rezeptor-Antagonisten vor den Insulten kann man die Hypoxie- oderIschämie-induzierten neuronalen Schäden mindern. Diese Befunde konnten experimentell beiRatten, neugeborenen Schweinen und Katzen nachgewiesen werden (Espinoza und Parer,1991; Gilland et al., 1998; Fritz et al, 1999a; Spandou et al., 1999).Zerebrale Hypoxie und Ischämie verändert den NMDA-Rezeptor auch in seinergrundsätzlichen Funktion, indem der Ionen-Kanal unspezifisch geöffnet wird (Hoffman et al.,1994). Wie und wo genau der Rezeptor durch den Sauerstoffmangel verändert wird, ist nochnicht genau bekannt: Fritz et al. (1999b) nehmen an, dass der Ionen-Kanal des NMDA-Rezeptors durch Hypoxie verändert wird. Simakajornboon et al. (2000) und Cheung et al.(2001) geben an, dass die NR1-Untereinheit des Rezeptors bei Hypoxie verstärktphosphoryliert vorliegt und somit die Funktion des NMDA-Rezeptors verändert ist.Liu et al. (2001) konnten eine Abnahme der NR2A-Untereinheit nach Sauerstoffmangel desGehirns nachweisen. Die außerdem nachgewiesene Phosphorylierung der NR2A-Untereinheitscheint ein wichtiger Weg, um den NMDA-Rezeptor zu regulieren.Takagi et al. (1997) fanden ebenfalls eine Phosphorylierung der NR2-Untereinheiten: imHippocampus liegen sowohl NR2A- als auch NR2B-Untereinheiten vermehrt phosphoryliertvor. Im Striatum und zerebralen Kortex hingegen, konnte nur eine vermehrte Phosphorylierungder NR2A-Untereinheit festgestellt werden. Diese Phosphorylierung potenziert die NMDA-Rezeptor-Aktivität.Ischämie führt außerdem zu einer Abnahme der Expression der NR2A- und NR2B-Untereinheiten im Hippocampus 24 Stunden nach der Läsion (Zhang et al., 1997). Außerdemist die Aktivität der AMPA- und der NMDA-Rezeptoren nach einem ischämischen Insultvermindert. Hsu et al. (1998) fanden diesen Abfall aber nur in der CA1- und DG-Region desHippocampus relativ früh, das heißt 24 Stunden nach dem Insult, während sich die Expressiondieser beiden Untereinheiten in der DG-Region des Hippocampus nach längerer Zeit wiedererholte.Gottlieb und Matute (1997) konnten zwei Tage nach dem ischämischen Insult eine mikroglialeReaktion im Hippocampus feststellen. Diese Reaktion hat ihren Höhepunkt am 4.-6. Tag nachdem Insult. Außerdem konnten sie zeigen, dass Astrozyten und Mikroglia vermehrt Proteineproduzieren. NR2A/B-Untereinheiten werden auf Zellen exprimiert, die morphologischeÄhnlichkeiten mit Astrozyten haben; und auch die NR1-Untereinheit steigt verstärkt an –zumindest zwischen dem 3. und 7. postischämischen Tag.
2.4.3. Wirkung von Hypoxie und Ischämie auf das GABAerge SystemHypoxie stört auch das GABAerge System. Es konnten höhere Konzentrationen von GABA inder Zerebrospinalflüssigkeit nach Sauerstoffmangel nachgewiesen werden. GABA als

20
inhibitorischer Neurotransmitter hemmt die Dopamin-Neuronen. Wenn die GABA-Konzentration niedrig ist, dann steigt die Dopamin-Ausschüttung, und wenn die GABA-Konzentration ansteigt - wie nach Hypoxie oder Ischämie - dann fällt die Dopamin-Konzentration im Hirn ab (Brixey et al., 1993).Hypoxie und Ischämie zerstören nachweislich GABAerge inhibitorische Interneurone (Romijn,1989; Romijn et al., 1992). NMDA-Rezeptor-Blocker können diese Zellverluste nach Hypoxievermindern (Romijn, 1989).Die GABA-Rezeptoren reagieren unterschiedlich sensibel auf den Sauerstoffmangel. So zumBeispiel kann man Ischämie-induzierte Änderungen am GABAA-Rezeptor-Ionenkanal sehen,die zu einer Schmälerung der inhibitorischen Effekte führen (Luhman et al., 1993).Außerdem haben Viapiano et al. (2001) gezeigt, dass die Bindungskapazität desNeurotransmitters GABA an den GABAA-Rezeptor nach Sauerstoffmangel des Gehirns um30% fällt. Dieser Abfall der Bindungskapazität konnte bis zum 10. Tag nach der Hypoxienachgewiesen werden, ist aber insgesamt reversibel (Viapiano et al., 2001).
2.4.4. Wirkung von Hypoxie und Ischämie auf das VerhaltenHypoxie und Ischämie bei der Geburt scheint bei Ratten auch akute und langfristige Effekte aufdas Verhalten zu haben:Ratten mit Anoxie bei der Geburt waren hyperaktiver (Iuvone et al., 1996; Brake et al., 2000)und zeigten ein, wenn auch subtiles, Defizit beim Erwerb des räumlichen Lernens (Iuvone etal., 1996; Boksa et al., 1998).Ein ischämischer Insult in neonatalen Ratten konnte außerdem längerfristige Defizite imgesamten Lernverhalten und im motorischen Verhalten des Tieres verursachen (Balduini et al.,2000).Weiterhin zeigten adulte Ratten mit perinataler Hypoxie eine Überreaktion aufwiederkehrenden Stress (El-Khodor und Boksa, 2000), gleichzeitig aber auch ein reduziertesAngst-geleitetes Verhalten (Hoeger et al., 2000).Hypoxie und Ischämie können anscheinend auch die chronobiologische Uhr von Rattenbeeinflussen (Antier et al., 1998b).
2.5. Die Ratte als TiermodellTier-Modelle sind wichtige Möglichkeiten, neue Theorien über Herkunft und Pathologie derSchizophrenie zu testen (Lipska und Weinberger, 2000). Da es keinen pathognomonischenMarker für das vielfältige Krankheitsbild der Schizophrenie gibt, muß man auf ein Tiermodellzurückgreifen, dass eine Konstellation von biologischen und Verhaltensphänomenenreproduziert, die bei der Schizophrenie relevant sind (Lipska und Weinberger, 2000).Das Ratten-Modell zur Untersuchung von neonataler zerebraler Hypoxie und Ischämie kannals vergleichbares Modell für den Menschen herhalten (Antier et al., 1998a; Ota et al., 1998).So ist zum Beispiel die Aminosäure-Sequenz der NR1-Untereinheit des NMDA-Rezeptors zu99% identisch zwischen Ratte, Maus und Mensch (Nakanishi, 1992).Der Ratten-Neocortex von 12-13 Tagen alten Ratten wird als vergleichbares Modell für dasmenschliche Hirn bei der Geburt angesehen, da Ratten zu einem frühen Zeitpunkt derHirnentwicklung geboren werden (Romijn et al., 1991).

21
2.6. Untersuchungsmethoden2.6.1. Präpulsinhibition als Untersuchungsmethode für VerhaltensauffälligkeitenDie akustische Schreckreaktion (ASR) der Ratte wird durch plötzliche, intensive (> 85 dBSPL) Reize ausgelöst. Sie ist eine phylogenetisch alte, im Bereich der Säugetiere universellauftretende, motorische Antwort auf potentiell gefährliche Reize im Sinne einer willkürlichen,nicht unterdrückbaren, protektiven Reaktion. Sie besteht aus einer Kontraktion derGanzkörpermuskulatur, vor allem aber der Kopf- und Nackenmuskeln. Über ihren biologischenSinn wird noch spekuliert: zum einen schützt eine kontrahierte Muskulatur besser vor einemBiss oder Zugreifen mit Krallen, zum anderen wird durch eine erhöhte Muskelspannung dieLatenz einer etwaig folgenden Flucht- oder Angriffsreaktion verbessert (Fendt und Fanselow,1999). Die ASR kann durch unterschwellige Reize, die 30 bis 500 ms vor dem Schreckreizdargeboten werden und auch anderer Modalität sein können, gehemmt werden (Swerdlow etal., 1992). Dieses Phänomen wird Präpulsinhibition genannt (englisch: Prepulse Inhibition =PPI). Dem Phänomen der PPI liegt keine Konditionierung zugrunde. Schon bei der erstenReflexauslösung ist es vorhanden. Die PPI kann als Modell zur Erforschung der Filter-Funktionen des Gehirns herangezogen werden. Während der Verarbeitung eines Reizes (hierdes Präpulses) wird die Verarbeitung kurz danach eintreffender Reize (hier der Schreckreiz)abgeschwächt und so auch die motorische Reaktion reduziert (Swerdlow et al., 1992; Fendt etal., 2001; Meincke et al., 2001).Dieser sensomotorische Filter dient dazu, die Verarbeitung eines Reizes vor Störungen durchanderer, gleichzeitig eintreffender Reize zu schützen und ist bei einigen psychiatrischenErkrankungen, z.B. der Schizophrenie, der Parkinson Krankheit dem Tourette-Syndrom undder Huntington Krankheit, gestört (Swerdlow et al., 1992; Cadenhead et al., 1993; McDowdet al., 1993; Docherty und Grillon, 1995; Meincke et al., 2001). Dadurch könnte dieErforschung der neuronalen Grundlagen der Präpulsinhibition möglicherweise auch klinischeBedeutung für diese Erkrankung haben.Ein PPI-Defizit spiegelt also kein spezielles psychopathologisches Symptom oder Syndromwieder, sondern ist Ausdruck der Dysfunktion der die PPI regulierenden Hirnstrukturen. DieBeteiligung des anteromedialen Striatums wie auch des Hippocampus an der Regulation derPPI kann als gesichert gelten (Meincke et al., 2001; Swerdlow et al., 2001). Dem NucleusAccumbens - als Bindeglied zwischen Frontalhirn und limbischen Strukturen - kommt großeBedeutung in der Kontrolle von Verhalten und Kognition zu. Im Nucleus Accumbenskonvergieren glutamaterge Afferenzen aus Hippocampus, medialem Präfrontalen Cortex undAmygdala und dopaminerge aus dem ventralen tegmentalen Areal.In der negativen Beeinflussung der PPI gelten derzeit Glutamat-Rezeptor-Antagonisten, 5-HT(Serotonin) Rezeptor-Agonisten und Dopamin(D2)-Rezeptor-Agonisten als maßgebend (Kochund Robbins, 2001). Dopaminagonisten wie Apomorphin und D-Amphetamin bewirken insystemischer Applikation eine Reduktion der PPI, die durch Dopaminrezeptor-Antagonistenreversibel ist.Glutamat ist wichtiger Überträgerstoff kortikostriataler Verbindungen und spielt ebenfalls eineRolle in der Regulation der PPI. So wurde ein PPI-Defizit nach systemischer Anwendung vonNMDA-Antagonisten beobachtet, das sich nach Applikation von atypischen Neuroleptikawieder zurückbildete (Meincke et al., 2001).Die PPI kann sowohl bei gesunden als auch bei neuropsychiatrisch erkrankten Menschengemessen werden. In zahlreichen klinischen Fällen wird die PPI benutzt, um gestörteGehirnfunktionen zu identifizieren und zu verstehen und um möglichst zielgerichtet therapieren

22
zu können. Darüber hinaus ist PPI ein spezies-übergreifendes Phänomen, das sehr gut auch inVersuchstieren (Ratten, Mäuse) gemessen werden kann. Es ist hierbei wichtig, dass vieleCharakteristika der neuronalen Regulation der PPI, welche in Tierstudien entdeckt wurden, inStudien am Menschen bestätigt werden konnten. Ebenso finden sich aber auch Unterschiedezwischen den Spezies, z.B. Unterschiede in den Mustern neuronaler Regelkreise und derSubstrate, welche die PPI regeln. Die sensorimotorische Regulation (gating) des Schreck-Reflexes durch PPI kann spezies-übergreifend ausgelöst werden. Zum Auslösen gleicherReflexe können gleiche Stimuli benutzt werden (Olivier et al., 2001; Swerdlow et al., 2001).Bei der Schizophrenie liegt es nahe, ein Defizit der PPI zu erwarten. Daher stellt die Messungder PPI, auch bei Ratten, ein gutes, erprobtes Mittel dar, um die Verhaltens-Veränderungen zuuntersuchen und zu beurteilen.
2.6.2. In-Situ-HybridisierungDie In-Situ-Hybridisierung (ISH) ermöglicht ein Markieren von bestimmten, durch ihreNucleotidsequenz definierten DNA- und RNA-Strängen direkt im Gewebe (in situ). Mit Hilfeeines Plasmids wird eine radioaktiv-markierte mRNA (Sonde) gebildet, die dem Gegenstückder gesuchten Nucleotidsequenz entspricht. Diese kann auf das zu untersuchende Gewebeaufgetragen werden und bindet dort mit der gesuchten Nucleotidsequenz. Schließlich könnendie markierten Bereiche im Gewebe z.B. mit Hilfe einer strahlungs-sensitiven Folie, optischdargestellt werden (Ausubel et al., 1996).Die ISH ist heutzutage die Methode der Wahl, um die Verteilung und Dichte bestimmterNukleinsäuresequenzen in kleinen anatomischen Regionen oder Subregionen, z.B. des Gehirnszu studieren (Uhl, 1993). Sowohl DNA- als auch RNA-Sequenzen lassen sich direkt imGewebe (in situ) nachweisen. Hierfür kann man Zellausstriche, Zellkulturen oder wie in dieserArbeit, auf Objektträgern fixierte Gewebeschnitte verwenden. So lässt sich mit dieser Methodeder Nachweis von mRNAs erbringen (Schafer et al., 1993).
2.6.3. RezeptorautoradiographieDie Rezeptorautoradiographie ist eine Methode zur radiologischen Markierung vonRezeptoren. Sie ermöglicht die Messung der Dichte und Lokalisation von Rezeptoren anunfixierten Schnitten, welche nach standardisierten Versuchsprotokollen (Zilles und Schleicher,1995; Zilles et al., 1999) behandelt werden.Dabei werden radioaktive Liganden an die entsprechenden Rezeptoren gebunden. Schließlichkönnen diese gebundenen Liganden über Inkubation auf einer Röntgenfolie optisch dargestelltund quantifiziert werden. Nach der Auswertung der Folien kann nun eine quantitative Aussageüber den Rezeptor gemacht werden.Im Detail werden die unfixierten Gehirnschnitte in einer Magnesium- und Zink-freienPufferlösung gereinigt. Es befinden sich im Ionenkanal des NMDA-Rezeptors Bindungsstellensowohl für Magnesium- als auch für Zinkionen. Ein Vorhandensein solcher würde dasVersuchsergebnis beeinflussen.Bei der Hauptinkubation werden die Schnitte wieder in eine Pufferlösung mit radioaktivmarkierten Liganden gestellt. So können diese für einen definierten Zeitraum an dieRezeptoren binden. Schließlich werden in mehreren Waschungen die nicht gebundenenLiganden ausgewaschen. Da auch eine unspezifische Bindung im Gewebe stattfindet, wird einKontrollpräparat angefertigt. Es wird ebenso in eine Pufferlösung mit markierten Ligandengestellt, aber zusätzlich wird ein Kompetititor in 1000 facher Menge mit hinzugefügt (McCoyund Richfield, 1996a; McCoy und Richfield, 1996b). Der Kompetititor verdrängt den Liganden

23
von den Rezeptoren. Schließlich entsteht hier ein Kontrollbild mit einer unspezifischenBindung. Die unspezifische Bindung kann von der spezifischen subtrahiert werden. So kann dietatsächliche Rezeptordichte errechnet werden.

24
3. Eigene Untersuchungen3.1. Material3.1.1. RattenFür die Versuche wurden männliche Sprague Dawley Ratten verwendet. Das Alter der Tierewird aus Tabelle 1 ersichtlich. Das Gehirn der Ratte ist sehr gut charakterisiert und ermöglichtdadurch eine präzise Beschreibung und Auswertung einzelner Gehirnregionen.Die Ratten wurden im Tierhaus des Klinikum Mannheim für die Versuche gezüchtet. Die FirmaJanvier (Frankreich) lieferte die Zuchttiere.Die Tierversuche haben im Tierhaus des Klinikum Mannheim stattgefunden. Sie wurden durchdas Regierungspräsidium Karlsruhe (Aktenzeichen 35-9185.81/164/98) unter der Nummer164/98 genehmigt. Die molekularbiologischen Untersuchungen wurden im Labor desZentralinstituts für Seelische Gesundheit in Mannheim durchgeführt. 123 männlicheneugeborene Ratten gingen in die Versuche ein. Davon sind 24 Ratten noch während derlaufenden Versuche gestorben: 21 Tiere sind von ihren Müttern nach der Operation getötetworden, 3 Tiere sind während der Operation verstorben, die Daten von 3 Tieren im PPIVersuch konnten nicht verwendet werden. Insgesamt wurden von den 11 Tage alten Ratten 40Tiere molekularbiologisch untersucht; in den Verhaltensversuchen sind 56 Tiere bewertetworden.
Tabelle 1: Übersicht VersuchstiereVersuch . Gesamt Vers.-Tag 11. Tag getötet PPI
CH-V 25 Tiere 4.-8. 10 Tiere 15 TiereCH-K 24 Tiere 4.-8. 10 Tiere 14 TiereHI-V 22 Tiere 8. 10 Tiere 12 TiereHI-K 25 Tiere 8. 10 Tiere 15 Tiere(Vers.-Tag = Lebenstag, an welchem der Versuch stattgefunden hat; PPI = Präpulsinhibition,CH-V = chronische Hypoxie - Versuch; CH-K = chronische Hypoxie - Kontrolle; HI-V =Hypoxie / Ischämie - Versuch; HI-K = kombinierte Hypoxie/Ischämie – Kontrolle, siehe auch3.2.1.)
3.1.2. Geräte und MaterialienDie für die Versuche nötigen Geräte und Materialien sind in der nachfolgenden Tabelle 2aufgelistet.
Tabelle 2: Geräte und ReagenzienGeräte und Materialien Hersteller / Firma[3H]-Micro-scales; 0,111-4,07kBq/mg Amersham, EnglandAIS - Analytical Imaging Station Interfocus, DeutschlandAnexate® Roche, SchweizAntisedan® Pfizer, DeutschlandAutoradiographic[14C]Micro-Scales, Amersham, EnglandDextransulfat Sigma-Aldrich, DeutschlandDiethylpyrocarbonat - DEPC Roth, DeutschlandDomitor® Pfizer, Deutschland

25
Dormicum® Roche, SchweizDTT - 1,4-Dthiothreit Sigma-Aldrich, DeutschlandEDTA - Ethylenediaminetetraacetic acid Sigma-Aldrich, DeutschlandFormamid Merck, DeutschlandHCl RdH Laborchemikalien, DeutschlandHostamox® Pfizer, DeutschlandHyperfilm-[3H] Amersham, EnglandIlluminator, Model B ´95 Northern Light, USAKCl Mallinckrodt Baker, NiederlandeKH2PO4 Mallinckrodt Baker, NiederlandeKSCN Mallinckrodt Baker, NiederlandeKryostat Firma Laica CM 3000, DeutschlandNa2HPO4 Mallinckrodt Baker, NiederlandeNarcanti-vet® Janssen, DeutschlandNTP-Mix MBI Fermentas, DeutschlandObjektiv, Mikkor 55 mm Nikon, DeutschlandParaformaldehyd Sigma-Aldrich, Deutschlandpiezoelektrischen Akzelerometer Eigenbau Universität Tübingen, DeutschlandPolamivet® Intervet, DeutschlandQuick Spin Columns (G-50 Sephadex C.) Boehringer, DeutschlandRNase AWAY Molecular BioProducts inc., USARNase Inhibitor Roche, SchweizRNA – Polymerase MBI Fermentas, DeutschlandRöntgenentwickler Kodak, DeutschlandRöntgenfolie, BioMax MR Film Kodak, Deutschland[35S] – UTP Perkin-Elmer, USASauerstoffmeßgerät (GMH3690) Geisinger, DeutschlandSoundkarte Siggen, DeutschlandTEA Merck, DeutschlandTrocknungs-Perlen Roth, DeutschlandVicryl-Faden 0,4 metric Johnson und Johnson, USAyeast RNA (Hefe RNA – t-RNA) Roche, Schweiz
3.2. Methode3.2.1. Versuche3.2.1.1. Chronische Hypoxie (CH)25 Ratten im Alter von 4 Tagen wurden in Dreier- bis Fünfer-Gruppen in einen luftdichtabgeschlossenen Käfig gesperrt. Über fünf Tage, sechs Stunden je Tag, atmeten sie dort eineGasmischung aus 11% Sauerstoff und 89% Stickstoff. Das Muttertier war im Käfig anwesend

26
und hatte freien Zugang zu Futter und Wasser. Ebenso konnten die Jungtiere jederzeit vonihrer Mutter gesäugt werden.Die Kunststoff-Käfige waren 41 x 31 cm in der Grundfläche und 15 cm hoch. Der Gasfluß inden Versuchs-käfig betrug 5 Liter je Minute (+/- 0,5 L / min). Durch ein Einlaßventil wurdedas Gasgemisch in den Käfig eingeleitet und durch ein Auslaßventil konnte das überflüssigeGas wieder entweichen. Für die Kontrolle des Sauerstoffgehaltes wurde ein Sauerstoff-Messgerät benutzt. Die Abweichung des Meßgerätes betrug +/- 0,1 %, die Genauigkeit derGasmischbarkeit lag bei + 0,2 % bzw. - 0,3 %. Die Versuche fanden bei Raumtemperatur (ca.21°C) statt.26 Kontrolltiere wurden in einen ebensolchen Käfig gesperrt, unter gleichen Bedingungen undfür denselben Zeitraum, allerdings war dieser mit normaler Raumluft belüftet.Je 10 Versuchstiere und 10 Kontrolltiere wurden am elften postnatalen Tag getötet. Dieanderen 15 Versuchs- und 16 Kontrolltiere nahmen am geplanten Verhaltensexperiment teilund werden erst am 90. Tag getötet.
3.2.1.2. Kombinierte Hypoxie/Ischämie (HI)22 Ratten im Alter von acht Tagen wurden gewogen und entsprechend ihres Gewichtes (18-21g) mit dem in Tab. 3 angegebenen Gemisch aus einer Mischspritze subkutan narkotisiert.
Tab. 3: Dosierungsanleitung zur Narkose (Angaben in mg/kg KGW):
Methadon 1,0 (Polamivet®)
Midazolam 2,0 (Dormicum®)Medetomidin 0,15 (Domitor®)
Zur Antagonisierung:Naloxon 0,12 (Narcanti-vet®)
Flumazenil 0,2 (Anexate®)
Atipamezol 0,75 (Antisedan®)
Die Ratte wurde jeweils auf ein Wärmekissen (39°C) gelegt und fixiert. Die Haut wurdeventral in der Mitte des Halses durch einen ca. 5 mm langen Scherenschlag eröffnet. DerSchnitt verlief laterolateral. Nach Freipräparieren wurden beiderseitig der Trachea unter demMusculus sternocleidomastoideus die Arteria carotis communis sichtbar, die von dem Truncusvagosympaticus und der Vena jugularis interna isoliert werden. Die Arteria carotis communiswurde für eine Dauer von acht Minuten beidseits mit Aneurysmenklemmen abgeklemmt.Während dieser acht Minuten lagen die Tiere in einem gasdicht abgeschlossenen Käfig, der miteinem Gasgemisch aus 8% Sauerstoff und 92% Stickstoff belüftet wurde. Nach einerErholungsphase von 10 Minuten, während derer die Ratten Raumluft atmen konnten und dieAneurysmenklemmen gelöst wurden, wurde dieser Vorgang noch einmal wiederholt. ZumVersuchsende wurden die Klemmen gelöst und entfernt.Die Wunde wurde vernäht (0,4 metric Vicryl) und die Ratten mit einem Antidot aus ihrer
Narkose geweckt. Eine vorsorgliche Antibiose wurde mit Amoxicillin (Hostamox®, 13 mg/kg)subkutan gespritzt.

27
26 Kontrolltiere durchliefen das Versuchsschema ebenso, nur dass bei ihnen die Arteria carotiscommunis nicht abgeklemmt wurde. Die Operation wurde ansonsten genauso durchgeführt.Auch in diesem Teil der Experimente wurden je 10 Versuchs- und 10 Kontrolltiere am 11.postnatalen Tag getötet. Die anderen 12 Versuchs- und 16 Kontrolltiere wurden erst nach demVerhaltensexperiment am neunzigsten Lebenstag getötet.
3.2.1.3. Töten der Ratten – Gehirnentnahme und LagerungDie Ratten wurden am 11. Lebenstag, beziehungsweise am 90. Lebenstag getötet. Die Ratten,welche am 90. Lebenstag getötet werden, hatten zuvor an den Verhaltensversuchen (siehe3.2.6.) teilgenommen, welche am 85. Tag (± 4) stattfanden.Zum schmerzlosen Töten wurden die Ratten erst in Äther narkotisiert und schließlich mit einerGuillotine dekapitiert. Das Fell wurde vom Kopf entfernt und die Schädeldecke wurde miteiner Knochenzange entfernt. Das Gehirn wurde entnommen und in flüssigem Stickstoffschockgefroren. Um Läsionen zu vermeiden, wurde das Gehirn in ein Behältnis mit Isopentangelegt und dieses in den flüssigen Stickstoff gelegt, bis das Gehirn durchgefroren war. DasGehirn wurde in Aluminiumfolie und in Parafilm verpackt und wurde stoßfest in Röhrchen ineinem Gefrierfach bei -80°C gelagert.
3.2.2. In-Situ-Hybridisierung3.2.2.1. Das Anfertigen der SchnitteJe Gehirn wurden in drei verschiedenen Ebenen Schnitte in einer Schichtdicke von 20 µm fürdie In-Situ-Hybridisierung (ISH) mit dem Kryostat geschnitten. Diese Schnitte wurden aufbereits beschriftete Objektträger aufgezogen und mit 4%igem Paraformaldehyd 15 Minutenlang bei Raumtemperatur fixiert.Aus folgenden Ebenen wurden Schnitte erstellt: Präfrontaler Cortex, Striatum undHippocampus.In diesen Regionen wurden folgende Bereiche ausgewertet (Paxinos und Watson, 1986):• Präfrontaler Cortex: (3.70 mm rostral von Bregma), Region: 1. Fr1/2 = Frontaler Cortex;
2. Cg1/3 = Gyrus cinguli; 3. IL = Infralimbischer Cortex.• Striatum: (0.70 mm rostral vom Bregma), Regionen: 1. CPu = Nucleus caudatus +
Putamen = Striatum = Caudatus putamen; 2. AcbC = Nucleus Accumbens.• Hippocampus (-3.60 mm caudal von Bregma), Regionen: CA1; CA2; CA3a; CA3c; DG =
Hilus.Bei der Auswertung wurden sowohl die rechte als auch die linke Gehirnhälfte gemessen undausgewertet. Der Mittelwert hieraus ging in die Bewertung ein.
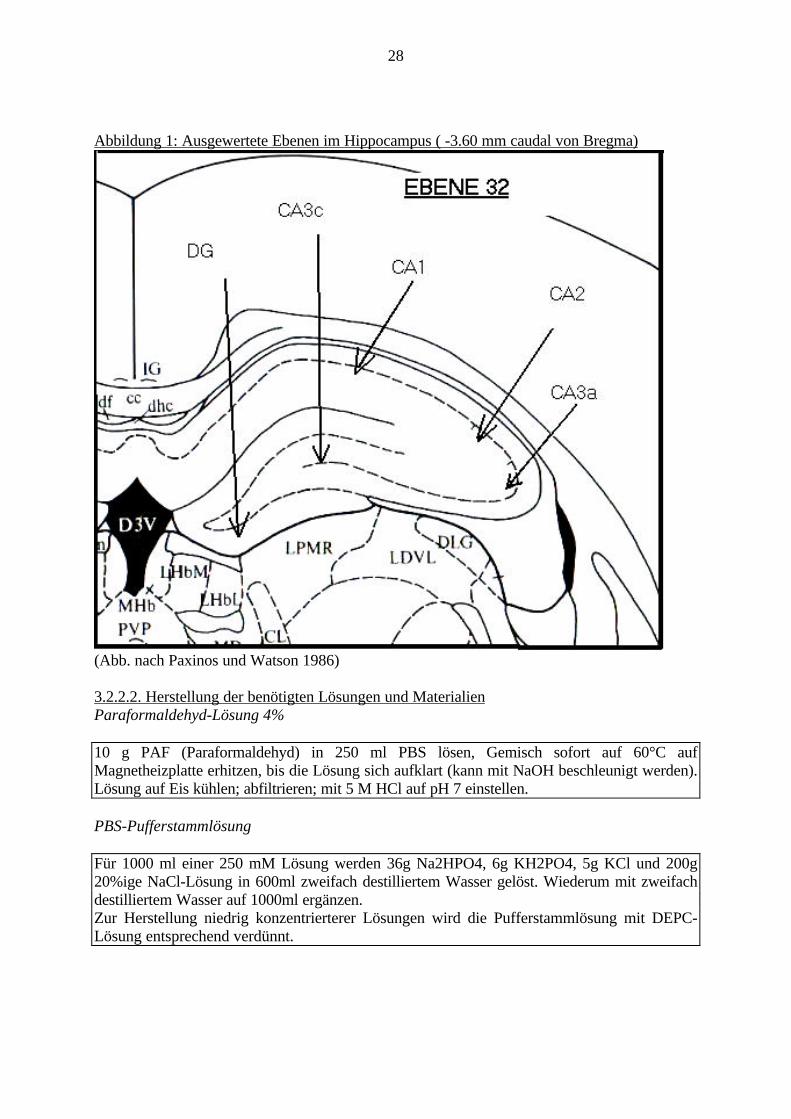
28
Abbildung 1: Ausgewertete Ebenen im Hippocampus ( -3.60 mm caudal von Bregma)
(Abb. nach Paxinos und Watson 1986)
3.2.2.2. Herstellung der benötigten Lösungen und MaterialienParaformaldehyd-Lösung 4%
PBS-Pufferstammlösung
Für 1000 ml einer 250 mM Lösung werden 36g Na2HPO4, 6g KH2PO4, 5g KCl und 200g20%ige NaCl-Lösung in 600ml zweifach destilliertem Wasser gelöst. Wiederum mit zweifachdestilliertem Wasser auf 1000ml ergänzen.Zur Herstellung niedrig konzentrierterer Lösungen wird die Pufferstammlösung mit DEPC-Lösung entsprechend verdünnt.
10 g PAF (Paraformaldehyd) in 250 ml PBS lösen, Gemisch sofort auf 60°C aufMagnetheizplatte erhitzen, bis die Lösung sich aufklart (kann mit NaOH beschleunigt werden).Lösung auf Eis kühlen; abfiltrieren; mit 5 M HCl auf pH 7 einstellen.
29
DEPC-Lösung
In 1 Liter zweifach destilliertes Wasser wird 1 ml Diethylpyrocarbonat (DEPC; dient derDeaktivierung von mRNAsen) - Konzentrat gegeben. Die Lösung wird autoklaviert.
Probenpuffer
Volumen Endonzentration100% Formamid (unautoklaviert) 12,5 ml 50%20 x SSC 2,5 ml 2 x0,5 M EDTA 62,5µl 1,25mM1,0 M TRIS pH 7,5 500 µl 20 mM50 x Denhardt´s solution 500 µl 1x50% Dextransulfat 5,0 ml 10%10% SDS 250µl 0,1%Mit DEPC-Lösung auf 25,0 ml auffüllen.Die oben genannten Substanzen (nicht das Formamid) werden zusammen gemischt und auf50°C erhitzt. Danach wird die Lösung autoklaviert. Nach dem Abkühlen wird das Formamidbeigemischt. Die fertige Lösung kann bei –20°C im Gefrierfach gelagert werden. (DieDextransulfat-Lösung kann in einer Menge von 2,5 g in 5,0 ml DEPC-Lösung bei 85°C gelöstund dann bei –20°C gelagert werden.)Details und Standard-Abläufe sind dem Buch „Sambrook, Fritsch, Maniatis (1989): MolecularCloning, A Laboratory manual“ entnommen.
20 x SSC-Stammlösung – (20mM)
3 M NaCl 175,32 g0,3 M Na-Citrat 88,2 gmit zweifach destilliertem Wasser auf 1000ml auffüllen.pH 7,0 mit HCl einstellen.
3.2.2.3. Versuchsablauf bei der ISHDie benutzten Gerätschaften wurden zuvor entweder im Autoklaven sterilisiert oder mitreinigenden bzw. RNAse deaktivierenden Reinigungsmitteln (RNase AWAY) behandelt.
3.2.2.3.1. Dehydrierung der SchnitteDie auf Objektträgern aufgetragenen Schnitte wurden nacheinander mit einer Reihe vonLösungen nach folgendem Protokoll behandelt:10 mM PBS 10 min10 mM PBS 10 min50% EtOH / DEPC 3 min75% EtOH / DEPC 3 min90% EtOH / DEPC 3 min100% EtOH / DEPC 5 min100% EtOH / DEPC 5 minLufttrocknen lassen
30
Die Schnitte können so behandelt bis zu einem Jahr bei –20°C in luftdicht verschlossenenBehältnissen aufbewahrt werden.
3.2.2.3.2. In vitro – Transkription (Sonde)Die Reagenzien wurden in der untenstehenden Reihenfolge in ein gekühltes (4°C) Eppendorf-Gefäß pipettiert.
Volumen Endkonzentration1. Polymerase-Puffer, 4,0 µl2. DTT (2,5M) 0,8 µl 0,1M3. DEPC-Lösung 4,0 µl4. NTP-Mix (10mM) 1,0 µl 0,5mM/Triphosphat5. Plasmid (200ng/µl) 0,5 µl (100 ng) 5 ng / µl6. RNAse Inhibitor, (40 U/µl) 1,0 µl 2 U / µl*7. RNA – Polymerase (20 U/µl) 3,5 µl (70 U) 3,5 U / µl*
8 [35
S] – UTP (40mCi/ml)** 5,0 µl (0,25 nmol) 200µCi* soll auch während des Pipettierens gekühlt werden** Vorsicht: radioaktiv
Die fertige Lösung wurde für 2 Stunden im Wasserbad bei 37°C inkubiert. Danach wurde dieLösung auf Eis gestellt und 50 µl DEPC-Lösung zugeben. Anschließend wurde das Gemischhomogenisiert.Je untersuchter Untereinheit des NMDA-Rezeptors stand ein Plasmid zur Verfügung.Es wurden jeweils zwei Stränge hergestellt: zum einen der Strang, welchen wir für dieAuswertung benötigen (antisense-Strang - AS), zum anderen der Ursprungsstrang (sense – S),welcher als Kontrolle sowohl während der Herstellung, als auch bei der Auswertung dienensollte. Je Plasmid wurde eine Probe mit einer T3- und eine Probe mit einer T7-RNA-Polymerase angesetzt.Je nachdem welches Plasmid benutzt wird, wurden unterschiedliche Polymerasen zur Bildungdes antisense-Stranges benutzt. Die genaue Aufteilung wird aus Tabelle 4 ersichtlich:
Tabelle 4: PolymerasenNR1 NR2-A NR2-B NR2-C NR2-D
anti-sense T7 T7 T3 T7 T3sense T3 T3 T7 T3 T7
Tabelle 5: Folgende Fragmente der Gesamt-Clone wurden benutzt:Gene GeneBank Fragment-Position Fragment-SizeRNR1-3-a U08265 2202-2721 520 bpRNR2A M91561 3774-4359 585 bpRNR2B NM008171 4030-4444 415 bpRNR2C M91562 2484-2862 378 bpRNR2D L31611 2157-2667 510 bp

31
3.2.2.3.3. Fraktionierung und QuantifizierungAus den beiden Gefäßen (AS und S pro Rezeptor-Subtyp bzw. Plasmid) wurde je 1,0 µlLösung in ein Szintillationsröhrchen (enthält Szintillationsflüssigkeit) gefüllt. Die restlicheLösung wurde auf Quick Spin Columns (G-50 Sephadex Columns) pipettiert und in einerZentrifuge für vier Minuten bei 1100x-g (2400 Umdrehungen) zentrifugiert. Das Eluat wurdeauf 200µl mit DEPC-Lösung aufgefüllt.Hiervon wurden wiederum je 1 µl in zwei Szintillationsröhrchen gegeben. Die vier Röhrchenwurden in dem Szintillationsgerät gemessen.
Je 10 Schnitte wurden 107 CPM (counts per minute) benötigt.Aus dem Ergebnis der Messung liess sich nun die Anzahl der Schnitte errechnen, welche mitdieser Probe inkubiert werden konnten.
3.2.2.3.4. AcetylierungZuerst wurde folgende Lösung durch Mischen der einzelnen Komponenten hergestellt:250 ml DEPC-Lösung1,25 g NaCl3,3 ml TEA (Triethanolamin)1 ml 5 M HCl (pH 8)625 µl 100 % Essigsäureanhydrid (unmittelbar vor Benutzung beifügen)
Die Küvette sollte vorgekühlt sein. Die Schnitte wurden 10 min bei Raumtemperatur in dieLösung gestellt. Ein Rührfisch diente zur besseren Benetzung der Schnitte.
Die Schnitte wurden anschließend zweimal mit 10mM PBS-Puffer für je 10 min beiRaumtemperatur gespült und nach dem Protokoll aus Tab. 4 dehydriert. Schnitte anschließendan der Luft trocknen lassen.
3.2.2.3.5. HybridisierungFür die Hybridisierung wurde das Plasmid in einer Hybridisierungslösung verdünnt und dieRNA durch Erhitzen aufgesplittet.Die Lösung wurde wie folgt hergestellt:900 µl Probenpuffer100 µl Hefe RNA – t-RNA40 µl 2,5 M DTT
1 x 107 cpm Plasmid-ProbeKomponenten zusammen in ein Eppendorf-Gefäß pipettieren und homogenisieren.
Die Probe wurde fünf Minuten lang bei 85°C im Wasserbad erhitzt (Aufsplitten der RNA).Anschließend wurde die Probe zum Abkühlen ins Eisbad gestellt.
3.2.2.3.6. InkubationJeder Objektträger wurde mit 100 µl der Plasmid-Probe beschichtet und mit einemDeckplättchen abgedeckt. Luftblasen unter dem Deckplättchen wurden hierbei vermieden.Danach wurden alle Objektträger waagerecht in einer Box gelagert.

32
Ein mit einer Lösung aus 20mM SSC und 50 %igem Formamid getränktes Tuch wurdeebenfalls mit in die Box eingebracht. Die Schnitte in der Box wurden 10 Stunden bei 55°Cinkubiert.
3.2.2.3.7. Posthybridisierung3.2.2.3.7.1. WaschenDer Waschvorgang diente dem Entfernen der Deckgläschen und dem Auswaschen derrestlichen Proben.Die Waschung sah wie folgt aus:
20 min 40mM SSC und anschließend die Deckgläser entfernen.5 min 40mM SSC bei Raumtemperatur5 min 20mM SSC
Zum Entfernen der nicht passenden, das heißt nicht gebunden vorliegenden RNA-Stränge,wurden im nächsten Schritt RNAsen beigegeben:30 min 200 ml RNAse A in 200 ml 15mM SSC bei 37°C
10 min 20mM SSC Raumtemperatur25 min 50% Formamid / 20mM SSC 55°C10 min 20mM SSC 55°C10 min 2mM SSC 55°C5 min 2mM SSC Raumtemperatur
3.2.2.3.7.2.Dehydrierung
Tabelle 6: Der Dehydrier-Vorgang wurde wie folgt durchgeführt:50% EtOH / DEPC 3 min75% EtOH / DEPC 3 min90% EtOH / DEPC 3 min100% EtOH / DEPC 5 min100% EtOH / DEPC 5 min
Die Schnitte wurden danach luftgetrocknet.
3.2.2.3.7.3.Exposition
Die Objektträger, wie auch ein [14C]-Standard-Streifen, (Autoradiographic[14C]Micro-Scales) wurden auf einem Karton festgeklebt. Darüber wurde eine Röntgenfolie gelegt undfünf Tage lang belichtet. Die Folie wurde für diesen Zeitraum in einer Röntgenkassettegelagert. Hiernach wurden die Filme in einem Röntgenentwickler entwickelt.
Die entwickelten Röntgenfilme wurden entsprechend gekennzeichnet, die abgebildeten Gehirnebeschriftet und in Klarsichtfolien gelagert.

33
3.2.3. Rezeptorautoradiographie (RAR)3.2.3.1. Das Anfertigen der SchnitteEs wurden wie bei der ISH drei Ebenen geschnitten und später untersucht. Die RAR- und dieISH-Schnitte wurden in einem Arbeitsgang geschnitten, um Materialverluste beim Anbringender Gehirne auf den Schneidkopf und dem Justieren bzw. Trimmen des Gerätes zu vermeiden.Die Schnittdicke betrug 16 µm.Die Objektträger mit den Schnitten wurden nach dem Schneiden mit einem kalten Fön ge-trocknet, und 12 Stunden auf Trocknungs-Perlen gelegt, um die Restfeuchtigkeit zu entfernen.In diesem Zustand konnten die Schnitte bis zu acht Tagen bei –20°C ohne weitere Fixation inverschlossenen Boxen gelagert werden.
3.2.3.2. Herstellung der benötigten Lösungen und MaterialienTris-HCl605,5 g Tris in 3000 ml zweifach destilliertem Wasser lösen300 ml Salzsäure (37%) über Nacht rührenmit Salzsäure auf pH 7,7 bei 25°C einstellenauf 5000 ml auffüllen
Aus 50 ml Tris-HCl-Stammpuffer und 1000 ml zweifach destilliertem Wasser werden 50 mMgebrauchsfertige Puffer-Lösung.
Tris-Acetat121,1 g Tris in 900 ml zweifach destilliertes Wasser lösenmit Essigsäure auf pH 7,2 bei 4°C einstellenauf 1000 ml auffüllen
Aus 50 ml Tris-Acetat-Stammpuffer und 1000 ml zweifach destilliertem Wasser wird 50 mMgebrauchsfertige Puffer-Lösung.
Tris-Citrat121,1 g Tris in 800 ml zweifach destilliertes Wasser lösenmit Citronensäure auf pH 7,0 bei 4°C einstellenauf 1000 ml auffüllen
Aus 50 ml Tris-Citrat-Stammpuffer und 1000 ml zweifach destilliertem Wasser werden 50 mMgebrauchsfertige Puffer-Lösung.
3.2.3.3. Versuchsablauf für RAR mit dem NMDA-Rezeptor - Ligand [3H]-MK-801Der Gesamtversuch war in eine Vorinkubation, eine Hauptinkubation, drei Waschvorgängeund die Trocknung aufgeteilt.
Die Vorinkubation wurde ausgeführt, um endogene Substanzen (z.B. Transmitter) und Ionenherauszuwaschen, welche im Gewebe an den untersuchten Rezeptoren binden können.Endogene Substanzen und Ionen könnten Bindungsstellen für den Liganden blockieren, und sodas Ergebnis beeinflussen. Für Magnesium- und auch für Zink-Ionen existiert eineBindungsstelle in dem NMDA-Ionenkanal. Daher wurden sämtliche Pufferlösungen mit

34
zweifach destilliertem Wasser hergestellt. Als Pufferlösung wird Tris-HCl verwendet. DieInkubation fand in handelsüblichen Kürvetten statt. Dauer der kurzen Inkubation war 15Minuten bei Zimmertemperatur (22°C), Dauer der langen Inkubation 180 Minuten bei 4°Ckaltem Wasser.
In der Hauptinkubation wurden die Schnitte mit dem jeweiligen tritiierten Liganden inkubiert,wobei der Ligand die Rezeptorbindungsstellen, aber bis zu einem gewissen Grad auchunspezifische Bindungsstellen im Gewebe markierte (Gesamtbindung = SB). Es war dahernötig, ein zweites Experiment an benachbarten Schnitten durchzuführen, um die unspezifischeBindung (UB) zu ermitteln. Dazu wurden die Schnitte mit dem tritiierten Liganden und einemnicht markierten Liganden (Kompetitor, in ungefähr 1000-fach höherer Konzentration als dermarkierte Ligand), der am gesuchten Rezeptor mit hoher Affinität bindet, zusammen inkubiert.Auf diesen Schnitten fand daher eine Kompetition zwischen dem tritiierten Liganden und demnicht markierten Liganden statt. Hierbei wurden die spezifischen Bindungsstellen durch denKompetitor blockiert und im Autoradiogramm wurden so nur die unspezifischenBindungsstellen durch den markierten Liganden dargestellt.In einem später folgenden Subtraktionsschritt der Computerauswertung wurde dieunspezifische Markierung von der Gesamtbindung abgezogen. Als Ergebnis erhielt man diespezifischen, durch den tritiierten Liganden markierten Rezeptorbindungsstellen. Je weniger dieunspezifische Bindung auftrat, desto spezifischer war der benutzte Ligand.Auch bei der Hauptinkubation diente Tris-HCl als Lösungspuffer.Glycin (30 µM) und Spermidin (50 µM) wurden ebenfalls beigesetzt. Die Hauptinkubationdauerte 60 min bei Zimmertemperatur.
Nach den Inkubationen war es nötig, die Schnitte durch Waschen von ungebundenemLiganden und von Puffersalzen, die für Artefakte auf der Filmemulsion verantwortlich seinkönnen, zu befreien. Daher wurde eine zweimalige Waschung der Schnitte in Pufferdurchgeführt, gefolgt von einer einmaligen Waschung der Schnitte in destilliertem Wasser,jeweils bei 4°C.Die Schnitte wurden mit einem kaltem Fön vorsichtig getrocknet und danach auf einenPappkarton aufgeklebt. Ein Tritium markierter Standardstreifen ([3H]-Micro-scales; 0,111-4,07kBq/mg) wurde ebenfalls beigelegt. Für die Dauer von fünf Wochen wurde dann auf die
markierten Schnitte ein Röntgenfilm (Hyperfilm-3H) aufgelegt.Nach fünf Wochen konnten die Röntgenfilme entwickelt und ausgewertet werden.
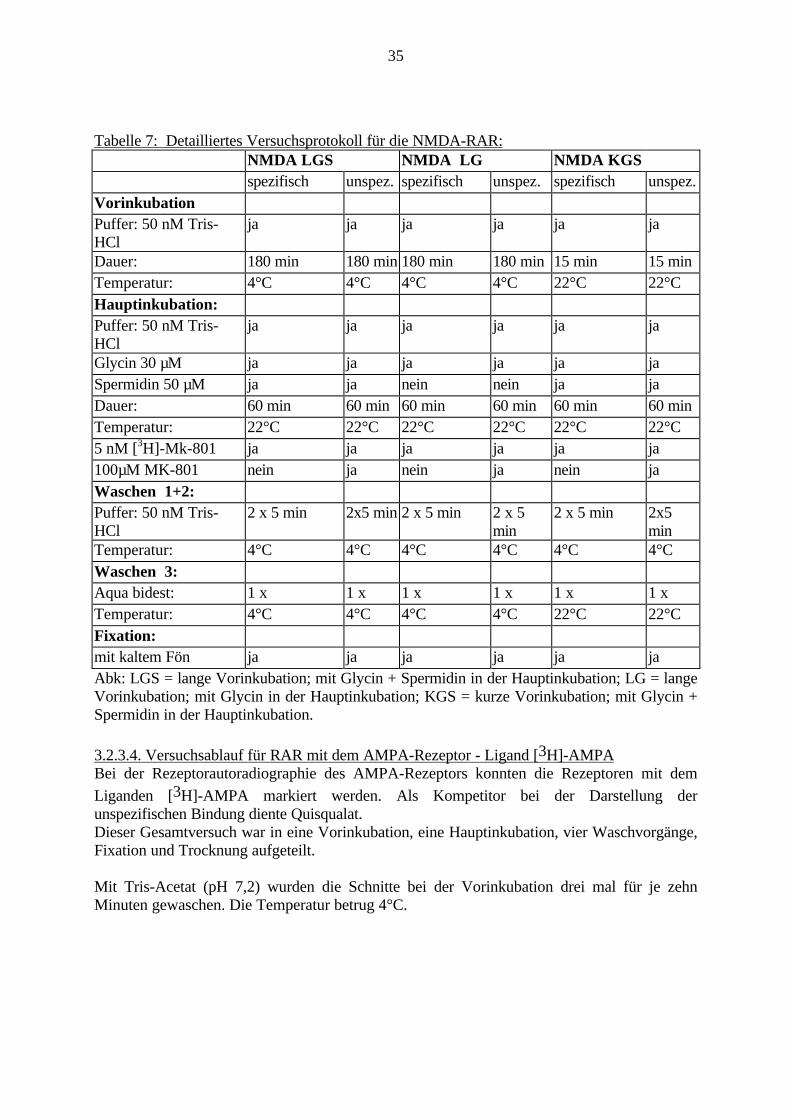
35
Tabelle 7: Detailliertes Versuchsprotokoll für die NMDA-RAR:NMDA LGS NMDA LG NMDA KGSspezifisch unspez. spezifisch unspez. spezifisch unspez.
VorinkubationPuffer: 50 nM Tris-HCl
ja ja ja ja ja ja
Dauer: 180 min 180 min 180 min 180 min 15 min 15 minTemperatur: 4°C 4°C 4°C 4°C 22°C 22°CHauptinkubation:Puffer: 50 nM Tris-HCl
ja ja ja ja ja ja
Glycin 30 µM ja ja ja ja ja jaSpermidin 50 µM ja ja nein nein ja jaDauer: 60 min 60 min 60 min 60 min 60 min 60 minTemperatur: 22°C 22°C 22°C 22°C 22°C 22°C5 nM [3H]-Mk-801 ja ja ja ja ja ja100µM MK-801 nein ja nein ja nein jaWaschen 1+2:Puffer: 50 nM Tris-HCl
2 x 5 min 2x5 min 2 x 5 min 2 x 5min
2 x 5 min 2x5min
Temperatur: 4°C 4°C 4°C 4°C 4°C 4°CWaschen 3:Aqua bidest: 1 x 1 x 1 x 1 x 1 x 1 xTemperatur: 4°C 4°C 4°C 4°C 22°C 22°CFixation:mit kaltem Fön ja ja ja ja ja jaAbk: LGS = lange Vorinkubation; mit Glycin + Spermidin in der Hauptinkubation; LG = langeVorinkubation; mit Glycin in der Hauptinkubation; KGS = kurze Vorinkubation; mit Glycin +Spermidin in der Hauptinkubation.
3.2.3.4. Versuchsablauf für RAR mit dem AMPA-Rezeptor - Ligand [3H]-AMPABei der Rezeptorautoradiographie des AMPA-Rezeptors konnten die Rezeptoren mit dem
Liganden [3H]-AMPA markiert werden. Als Kompetitor bei der Darstellung derunspezifischen Bindung diente Quisqualat.Dieser Gesamtversuch war in eine Vorinkubation, eine Hauptinkubation, vier Waschvorgänge,Fixation und Trocknung aufgeteilt.
Mit Tris-Acetat (pH 7,2) wurden die Schnitte bei der Vorinkubation drei mal für je zehnMinuten gewaschen. Die Temperatur betrug 4°C.
36
Bei der Hauptinkubation diente Tris-Acetat als Puffer. Dauer der Inkubation war 45 Minuten
bei einer Temperatur von 4°C. [3H]-AMPA (10 nM) diente als Ligand, Quisqualat (10 µM) alsKompetitor. Kaliumcyanid (100 mM) wurde ebenfalls zugegeben.
Wiederum wurden die Schnitte nach vollendeter Hauptinkubation dreimal in Tris-Acetat Pufferfür drei mal vier Sekunden gewaschen. Danach wurden sie zweimal für zwei Sekunden in eineFixationslösung (Aceton/Glutaraldehyd 100:2,5 ml) getaucht.Die Schnitte wurden danach vorsichtig kurz (zwei Sekunden) mit heißem Fön und schließlichmit kaltem Fön getrocknet und so fixiert.Auch diese Schnitte wurden auf einen Pappkarton aufgeklebt. Ein Tritium markierterStandardstreifen wurde beigelegt. Für die Dauer von vier Wochen wurde auf die markierten
Schnitte ein Röntgenfilm (Hyperfilm-[3H]) aufgelegt. Nach vier Wochen erfolgter Belichtungkonnten die Röntgenfilme entwickelt und ausgewertet werden.
Tabelle 8: Detailliertes Versuchsprotokoll für AMPA-RAR:AMPA AMPAspezifisch unspez.
Vorinkubation:Puffer: Tris-Acetat pH 7,2 Tris-Acetat pH 7,2Dauer: 3 x 10 min 3 x 10 minTemperatur: 4°C 4°CHauptinkubation:Puffer: Tris-Acetat pH 7,2 Tris-Acetat pH 7,2Dauer: 45 min 45 minTemperatur: 4°C 4°C10 µM Quisqualat nein ja 10 nM [3H]-AMPA ja ja100 mM KSCN ja jaWaschung 1-3:Puffer: Tris-Acetat pH 7,2 Tris-Acetat pH 7,2Dauer: 3 x 4 sec 3 x 4 secTemperatur: 4°C 4°CWaschung 4:Lösung: Aceton/Glutaraldehyd 100:2,5 ml;Dauer: 2 x 2 sec 2 x 2 secTrocknung: 2 sec mit heißem, dann mit kaltem Fön
37
3.2.3.5. Versuchsablauf beim GABA-Rezeptor - Ligand [3H]-Muscimol
Bei der Rezeptorautoradiographie des GABA-Rezeptors konnten der Ligand [3H]-Muscimolverwendet werden. Als Kompetitor bei der Darstellung der unspezifischen Bindung dienteGABA.Der Gesamtversuch wird in eine Vorinkubation, eine Hauptinkubation, drei Waschvorgängeund die Trocknung aufgeteilt.
In der Vorinkubation wurden die Schnitte drei mal für je fünf Minuten mit Tris-Citrat Puffer(ph 7,0) bei einer Temperatur von 4°C gewaschen.Auch bei der Hauptinkuabtion diente Tris-Citrat als Puffer. Dauer der Inkubation betrug hier40 Minuten bei einer Temperatur von 4°C.Anschließend wurden die Schnitte danach dreimal in Tris-Acetat Puffer für drei mal dreiSekunden gewaschen.Getrocknet wurden die Schnitte dieses Versuchs durch vorsichtiges Fönen.Auch diese Schnitte wurden anschließend auf einen Pappkarton aufgeklebt. Ein Tritiummarkierter Standardstreifen wurde beigelegt. Für die Dauer von fünf Wochen wurde auf die
markierten Schnitte ein Röntgenfilm (Hyperfilm-[3H]) aufgelegt. Schließlich konnten dieSchnitte nach der Entwicklung ausgewertet werden.
Tabelle 9: Der genaue Versuchsablauf sieht demnach wie folgt aus:GABAA GABAA
spezifisch unspez.Vorinkubation:Puffer: 50 nM Tris-Citrat pH 7 50 nM Tris-Citrat pH 7Dauer: 3 x 5 min 3 x 5 minTemperatur: 4°C 4°CHauptinkubation:Puffer: 50 nM Tris-Citrat pH 7 50 nM Tris-Citrat pH 7Dauer: 40 min 40 minTemperatur: 4°C 4°C3 nM [3H]-Muscimol ja jaKompetitor: 10 µM GABA nein jaWaschung:Puffer: 50 nM Tris-Citrat pH 7 50 nM Tris-Citrat pH 7Dauer: 3 x 3 sec 3 x 3 secTemperatur: 22°C 22°CTrocknung: mit kaltem Fön mit kaltem Fön

38
3.2.3.6. Entwicklung der belichteten Röntgen-Folien (RAR)3.2.3.6.1. Herstellung der Lösungen
Je 206 ml Entwickler- bzw. Fixiererlösung in 740 ml H2O lösen
3.2.3.6.2. EntwicklungDie Entwicklung der Röntgenfilme wurde in einem Dunkelraum bei Rotlicht nach folgendemProtokoll durchgeführt:1. Film auf Raumtemperatur erwärmen.2. Fotoschalen vorbereiten: Je eine Schale mit Entwickler, H2O und Fixierer vorbereiten.3. Entwickler filtrieren.4. Film für 5 min (mit Stoppuhr) in Entwickler einlegen (die Filmschicht – matt – nach oben).
Während den ersten 2 min, Fotoschale ständig schwenken.5. Film kurz in H2O abspülen.
6. Film 10 min fixieren (Filmschicht nach oben), die ersten 2 min schwenken.7. Film 30 min in fließendem Leitungswasser wässern.8. Film mit Wasser spülen.9. Trocknen.10. Aufbewahrung der Filme empfiehlt sich in Plastikhüllen.
Da die Filmschicht sehr empfindlich ist, durften die Filme nur am Rand angefaßt werden.
3.2.4. Auswertung der Röntgenfolien (ISH und RAR)Zum Erfassen und Speichern der Gehirne wurden die fertigen Röntgenfolien auf einenIlluminator gelegt und hiermit gleichmäßig ausgeleuchtet. In einer Entfernung von 39,5 cm warein Nikon Objektiv (Nikkor 55 mm) installiert, welches mit einem Computer verbunden ist. MitHilfe eines Programmes „AIS“ (Inter Active Systems) wurden die Bilder auf dem Bildschirmdargestellt und schließlich gespeichert. Auf jedem Röntgenfilm befand sich die Ablichtungeines Standard-Streifens, auf welchem festgelegte Einheiten von Radioaktivität dargestelltwaren. So konnte jeder Schattierung auf den Röntgenfollien ein entsprechender Wert in bq/gbei der ISH zugeordnet werden. Bei der RAR wurden die Standards an Gewebestandards vonhomogenisiertem Gehirn-Gewebe mit bekanntem Protein Gehalt kalibriert, um schließlich dieRezeptordichte in fmol/mg angeben zu können. Die Ergebnisse (in fmol) wurden multipliziertmit (KD+c)/c, um Bmax Werte zu erhalten. KD ist dabei die Dissoziationskonstante des
Liganden (5 nM für [3H]-MK-801). „c“ steht für die Inkubations-Konzentration des markiertenLiganden. „Bmax“ ist in dieser Formel die Gesamtzahl der Rezeptoren. Die
Dissoziationskonstanten sind für AMPA 10 nM, für GABA 3 nM und bei NMDA 5 nM (Zillesund Schleicher, 1999).
Beim Einscannen wurde mit jedem eingespeicherten Gehirn die zugehörige Standardkurvegesichert.Die entsprechenden Regionen der Gehirn-Darstellungen wurden abgespeichert undanschließend ausgewertet. Dabei wurde bei jeder Einzelmessung der Grauwert, die

39
ausgewertete Region, die Ebene des Schnittes, die Gehirnhälfte und die Tiernummer erfaßt.Auf die Auswahl der Gehirn-Areale wurde in Kapitel 3.2.2.1. bereits ausführlich eingegangen.Bei der Auswertung wurden sowohl die rechte als auch die linke Gehirnhälfte gemessen. DerMittelwert hieraus wurde für die weiteren Bewertungen verwendet.Diese Zahlenwerte sind im Anhang dargestellt und wurden mit dem SPSS-Coputerprogrammstatistisch ausgewertet. (SPSS Base 10.0 für Windows)
3.2.5. Statistische Auswertung der DatenDie statistische Auswertung der Daten erfolgte mit Hilfe des SPSS-Programmes. Die Auswahldes Tests wurde durch die Verteilung der Daten bestimmt. Es handelte sich, möglicherweisewegen der relativ geringen Tierzahlen, bei den erhaltenen Daten nicht um eineNormalverteilung (Gauss-Kurve). Daher konnten parametrische Tests wie der t-Test nichtverwendet werden. In diesem Fall kam der U-Test nach Mann-Whitney, ein nichtparametrischer Test für unabhängige Stichproben, zur Anwendung. Mit diesem Verfahrenkonnten die Daten der Kontroll- und Versuchsgruppen miteinander verglichen werden.
3.2.6. Präpulsinhibition - UntersuchungenDie Ratten, welche erst am 90. Tag getötet wurden, wurden unmittelbar vorher (Tag 85. ± 4d)für eine Messung der Schreckreaktion herangezogen.
3.2.6.1. Apparatur zur Auslösung und Messung der SchreckreaktionZur Messung der Amplitude der Akustischen-Schreckreaktion (ASR) wurden die Ratten ineinen Testkäfig (20x10x12 cm3) gesetzt, der auf einem piezoelektrischenBeschleunigungsmesser befestigt wurde. Dieser befand sich in einer Kammer (60x50x50 cm3),die mit schallabschwächendem Material ausgekleidet war.Bewegungen der Ratten verursachten eine Spannungsänderung der im Beschleunigungsmesserbefindlichen Piezofolie. Diese Spannungsänderung wurde elektronsch verstärkt und über einenAnalog/Digital-Wandler in den Computer gegeben.Zur Aufnahme wurde ein Softwareprogramm benutzt (DAPview), das die maximalenSpannungsschwankungen (peak-peak-Amplitude) in einem Zeitfenster 80 ms vor und nachdem Schreckreiz berechnete. Die ASR-Amplitude errechnete sich aus der Differenz zwischender maximalen Spannungsschwankung in dem Zeitfenster nach dem Schreckreiz und dermaximalen Spannungsschwankung in dem Zeitfenster vor dem Schreckreiz (motorischeSpontanaktivität).Die akustischen Schreckreize wurden über eine Soundkarte mit spezieller Software (BerndWaldmann) erzeugt, die durch selbstentwickelte Reizprogramme gesteuert wurden. DieseReize wurden über einen Stereo-Verstärker (WPA-600Pro) auf einen Lautsprecher gegeben,der 20 cm vom Testkäfig entfernt montiert war.

40
3.2.6.2. Ablauf des Testprogrammes• 5 Minuten Eingewöhnung ohne Reize• 10 Minuten Schreckreize (100 dB; weißes Rauschen; 20 ms)• je 5 Blöcke:
1. Schreckreiz allein2.-4. verschiedene Präpulse (60, 70 und 80 dB; 10 kHz, 20 ms; Anfang 70 ms vor dem Schreckreiz) mit Schreckreiz5.-7. verschiedene Präpulse allein8. ohne Reiz
• alles in zufälliger Reihenfolge.
3.2.6.3. VersuchsablaufDie Ratte wurde in den Versuchskäfig gesetzt, und nach 5 Minuten Eingewöhnungszeitwurden zehn Schreckreize (100 dB SPL, weißes Rauschen, 20 ms) präsentiert. Danach wurden5 Blöcke mit den folgenden acht Reiztypen in zufälliger Reihenfolge gegeben(Interstimulusintervall: 20 Sekunden): Schreckreiz allein (100 dB SPL, weißes Rauschen, 20ms), Schreckreiz mit verschiedenen Präpulsen (60, 70 bzw. 80 dB, 10 kHz, 20 ms incl. 0,4 msAnstiegs- und Abfallflanke, Reizanfang 70 ms vor dem Schreckreiz), die Präpulse allein undkein Reiz. Zur Auswertung wurden die Reaktionsamplituden auf die unterschiedlichenReiztypen gemittelt. Als Maß für die Präpulsinhibition diente die prozentuale Abschwächungder Reaktionsamplitude nach Schreckreiz mit Präpuls gegenüber der Reaktion auf einenSchreckreiz allein. Zur Kontrolle wurde überprüft, dass auf den Präpuls, bzw. auf die Situationohne Reiz, keine ASR erfolgte.
3.2.6.4. Auswertbare ParameterAuswertbare Parameter der Schreckreaktionshemmung in dieser Arbeit:PRÄ: Ist die normale, motorische Aktivität vor Versuchsbeginn, also vor den Schreckreizen.Sie dient als Kontrolle, daher werden hier Werte im Bereich um Null erwartet.NIX: Ist die normale, motorische Aktivität während des Versuches, die regelmäßig zwischenden Schreckreizen gemessen wurde. Sie dient ebenso wie die PRÄ als Kontrolle. Daherwurden auch hier ebenso wie bei der PRÄ Werte im Bereich um Null erwartet.HAB: Habituation ist die Phase der Gewöhnung an die Schreckreize, die hier ohne Präpulseangeboten werden. Die Gewöhnung an die Schreckreize wurde hiermit gemessen.HAB%: Habituation in Prozent ist der Mittelwert (MW) aus den ersten fünf HAB-Werten imVerhältnis zu den zweiten fünf HAB-Werten ([MW HAB 6-10 – MW HAB 1-5] / [MW HAB1-5 / 100]) in Prozent. Hieraus lässt sich die Gewöhnung an die Schreckreize beurteilen. Beischizophrenen Patienten ist die Gewöhnungsfähigkeit mitunter eingeschränkt. (Werte tief imnegativen Bereich lassen auf eine besonders starke Gewöhnung schließen.)SR: Reaktion auf den Schreckreiz; hier ohne vorherigen Präpuls gemessen und während desVersuches. Auch diese dienten der Kontrolle.P60, P70, P80: Präpuls bei P60, P70 und P80: Hiermit wurden die Präpulse bei 60, 70 und 80dB ohne anschließenden Schreckreiz ausgeführt und die Reaktionen der Ratten gemessen. DiePräpulse sind Töne, die unterhalb der Reizschwelle liegen sollen. Diese Messung stellt eineKontrolle dar. Die Werte werden im Bereich um Null erwartet.P60+SR, P70+SR, P80+SR: Schreckreizhemmung durch einen Präpuls der keineSchreckreaktion auslöst. Die Lautstärke der Präpulse liegt wie zuvor in der Kontrolle bei 60,70 und 80 dB, Der Effekt von Präpulsen vor dem Schreckreiz ist im Normalfall eine

41
Verringerung der Schreckreaktion der Ratte. Je lauter die Präpulse werden, desto geringerwird die folgende Reaktion des Versuchstieres erwartet. Also je stärker der Präpuls, destogeringer die Schreckreaktion unter physiologischen Bedingungen. Bei schizophrenen Patientenkann in diesem Versuch eine verringerte Präpulsinhibition des Schreckreizes auftreten.PPI60, PPI70, PPI80: Schreckreizhemmung in Prozent bei 60, 70 und 80 dB. Sie errechnetsich aus dem Schreckreiz „SR“ und „Schreckreiz mit Präpuls“ wie folgt: ([SR – P80+SR] /[SR / 100]). Ist der Wert positiv, so hat eine Schreckreizhemmung (PPI) stattgefunden. Ist derWert negativ, dann hat keine PPI stattgefunden ( in dem Fall war P80+SR größer als SR, alsohat der Präpuls im Vergleich der beiden Werte keinen Effekt gehabt). Um diesen Wert bei denVersuchstieren dreht sich der Versuch.
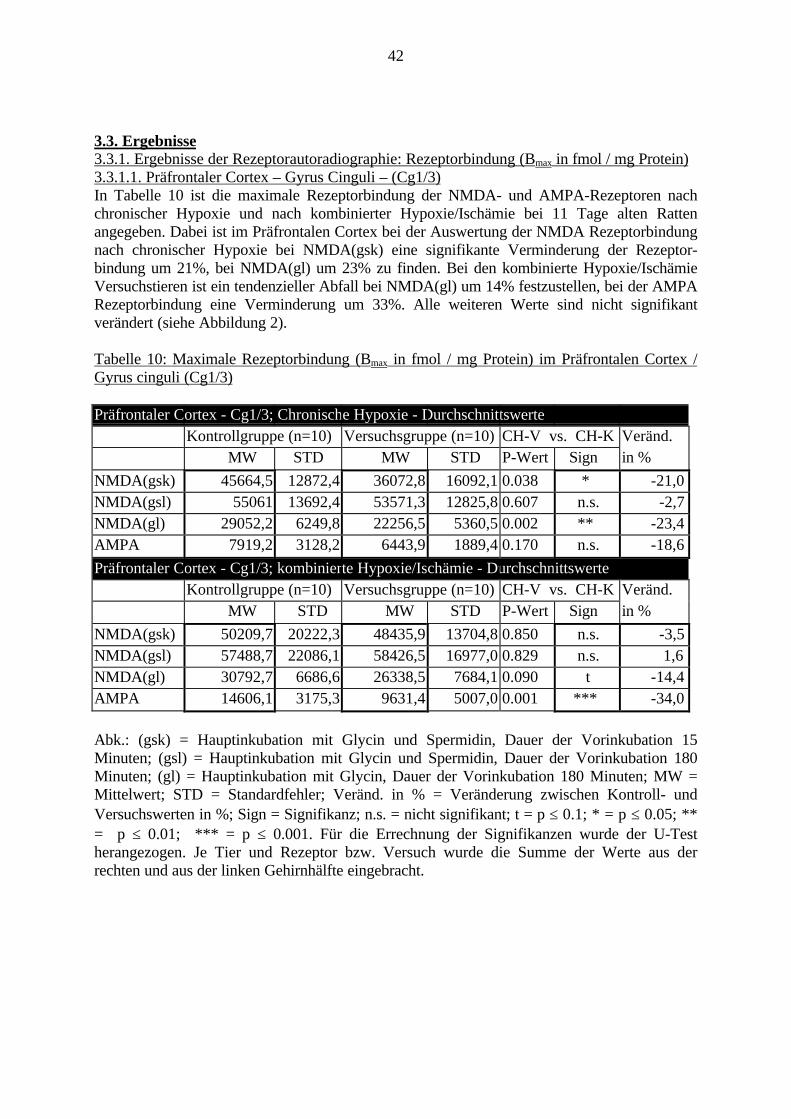
42
3.3. Ergebnisse3.3.1. Ergebnisse der Rezeptorautoradiographie: Rezeptorbindung (Bmax in fmol / mg Protein)3.3.1.1. Präfrontaler Cortex – Gyrus Cinguli – (Cg1/3)In Tabelle 10 ist die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptoren nachchronischer Hypoxie und nach kombinierter Hypoxie/Ischämie bei 11 Tage alten Rattenangegeben. Dabei ist im Präfrontalen Cortex bei der Auswertung der NMDA Rezeptorbindungnach chronischer Hypoxie bei NMDA(gsk) eine signifikante Verminderung der Rezeptor-bindung um 21%, bei NMDA(gl) um 23% zu finden. Bei den kombinierte Hypoxie/IschämieVersuchstieren ist ein tendenzieller Abfall bei NMDA(gl) um 14% festzustellen, bei der AMPARezeptorbindung eine Verminderung um 33%. Alle weiteren Werte sind nicht signifikantverändert (siehe Abbildung 2).
Tabelle 10: Maximale Rezeptorbindung (Bmax in fmol / mg Protein) im Präfrontalen Cortex /Gyrus cinguli (Cg1/3)
Präfrontaler Cortex - Cg1/3; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 45664,5 12872,4 36072,8 16092,1 0.038 * -21,0NMDA(gsl) 55061 13692,4 53571,3 12825,8 0.607 n.s. -2,7NMDA(gl) 29052,2 6249,8 22256,5 5360,5 0.002 ** -23,4AMPA 7919,2 3128,2 6443,9 1889,4 0.170 n.s. -18,6
Präfrontaler Cortex - Cg1/3; kombinierte Hypoxie/Ischämie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 50209,7 20222,3 48435,9 13704,8 0.850 n.s. -3,5NMDA(gsl) 57488,7 22086,1 58426,5 16977,0 0.829 n.s. 1,6NMDA(gl) 30792,7 6686,6 26338,5 7684,1 0.090 t -14,4AMPA 14606,1 3175,3 9631,4 5007,0 0.001 *** -34,0
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1; * = p ≤ 0.05; **= p ≤ 0.01; *** = p ≤ 0.001. Für die Errechnung der Signifikanzen wurde der U-Testherangezogen. Je Tier und Rezeptor bzw. Versuch wurde die Summe der Werte aus derrechten und aus der linken Gehirnhälfte eingebracht.
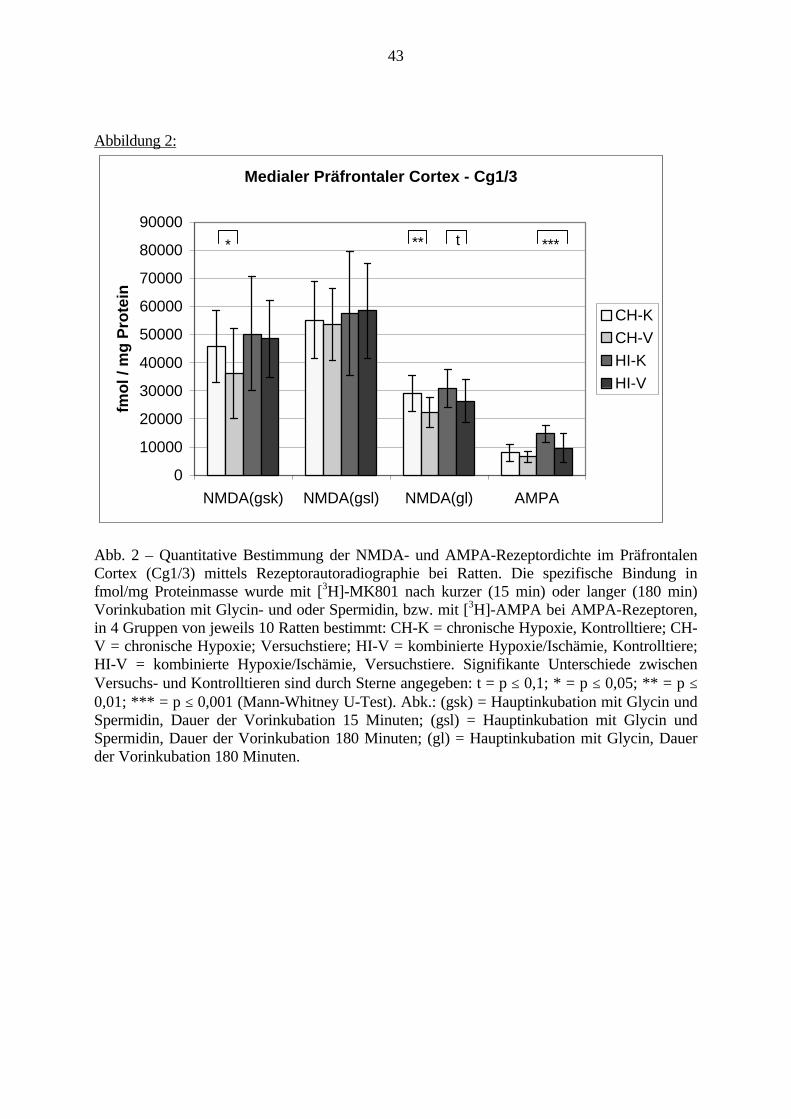
43
Abbildung 2:
Abb. 2 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im PräfrontalenCortex (Cg1/3) mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung infmol/mg Proteinmasse wurde mit [3H]-MK801 nach kurzer (15 min) oder langer (180 min)Vorinkubation mit Glycin- und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren,in 4 Gruppen von jeweils 10 Ratten bestimmt: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere;HI-V = kombinierte Hypoxie/Ischämie, Versuchstiere. Signifikante Unterschiede zwischenVersuchs- und Kontrolltieren sind durch Sterne angegeben: t = p ≤ 0,1; * = p ≤ 0,05; ** = p ≤0,01; *** = p ≤ 0,001 (Mann-Whitney U-Test). Abk.: (gsk) = Hauptinkubation mit Glycin undSpermidin, Dauer der Vorinkubation 15 Minuten; (gsl) = Hauptinkubation mit Glycin undSpermidin, Dauer der Vorinkubation 180 Minuten; (gl) = Hauptinkubation mit Glycin, Dauerder Vorinkubation 180 Minuten.
Medialer Präfrontaler Cortex - Cg1/3
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-K
CH-V
HI-K
HI-V
* ** t ***
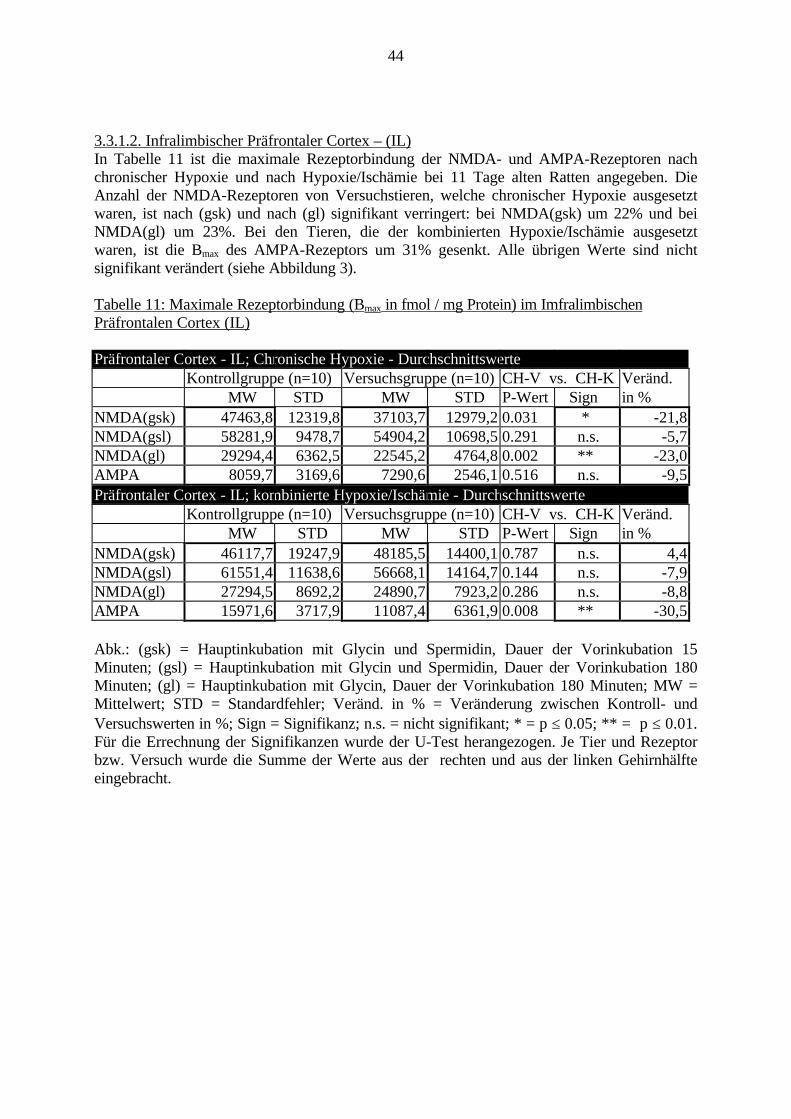
44
3.3.1.2. Infralimbischer Präfrontaler Cortex – (IL)In Tabelle 11 ist die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptoren nachchronischer Hypoxie und nach Hypoxie/Ischämie bei 11 Tage alten Ratten angegeben. DieAnzahl der NMDA-Rezeptoren von Versuchstieren, welche chronischer Hypoxie ausgesetztwaren, ist nach (gsk) und nach (gl) signifikant verringert: bei NMDA(gsk) um 22% und beiNMDA(gl) um 23%. Bei den Tieren, die der kombinierten Hypoxie/Ischämie ausgesetztwaren, ist die Bmax des AMPA-Rezeptors um 31% gesenkt. Alle übrigen Werte sind nichtsignifikant verändert (siehe Abbildung 3).
Tabelle 11: Maximale Rezeptorbindung (Bmax in fmol / mg Protein) im ImfralimbischenPräfrontalen Cortex (IL)
Präfrontaler Cortex - IL; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 47463,8 12319,8 37103,7 12979,2 0.031 * -21,8NMDA(gsl) 58281,9 9478,7 54904,2 10698,5 0.291 n.s. -5,7NMDA(gl) 29294,4 6362,5 22545,2 4764,8 0.002 ** -23,0AMPA 8059,7 3169,6 7290,6 2546,1 0.516 n.s. -9,5Präfrontaler Cortex - IL; kombinierte Hypoxie/Ischämie - Durchschnittswerte
Kontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 46117,7 19247,9 48185,5 14400,1 0.787 n.s. 4,4NMDA(gsl) 61551,4 11638,6 56668,1 14164,7 0.144 n.s. -7,9NMDA(gl) 27294,5 8692,2 24890,7 7923,2 0.286 n.s. -8,8AMPA 15971,6 3717,9 11087,4 6361,9 0.008 ** -30,5
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; * = p ≤ 0.05; ** = p ≤ 0.01.Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptorbzw. Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
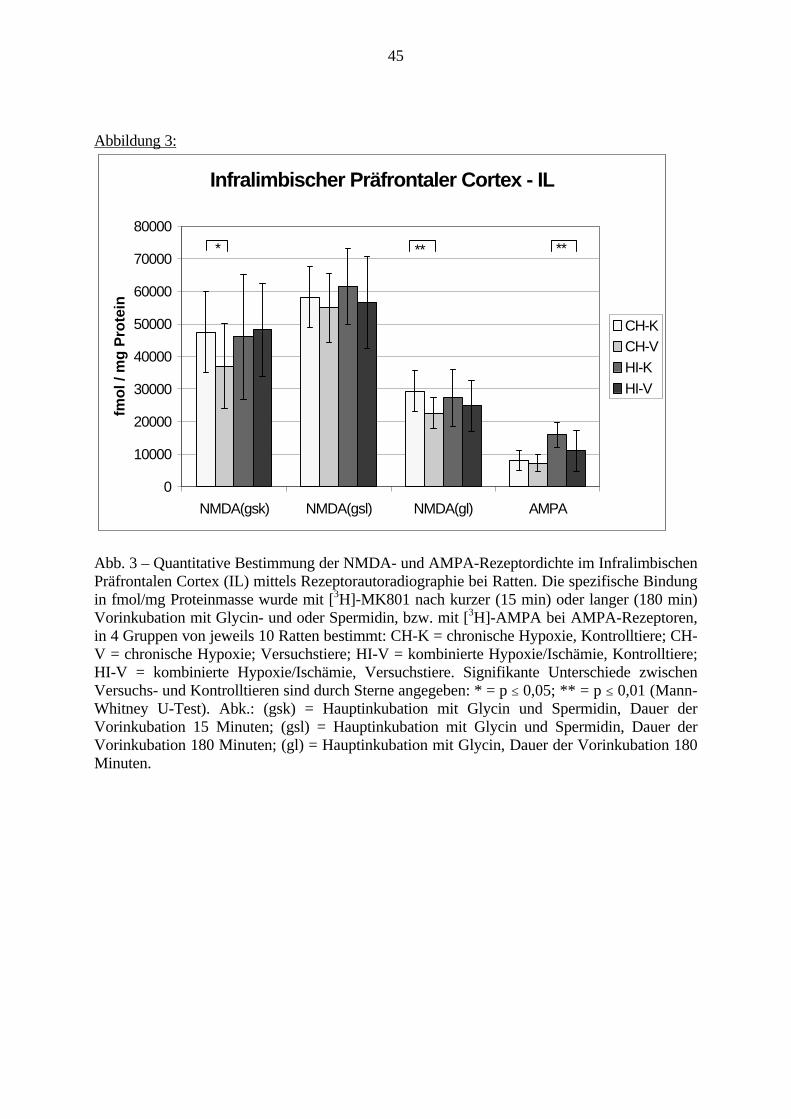
45
Abbildung 3:
Abb. 3 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im InfralimbischenPräfrontalen Cortex (IL) mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindungin fmol/mg Proteinmasse wurde mit [3H]-MK801 nach kurzer (15 min) oder langer (180 min)Vorinkubation mit Glycin- und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren,in 4 Gruppen von jeweils 10 Ratten bestimmt: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere;HI-V = kombinierte Hypoxie/Ischämie, Versuchstiere. Signifikante Unterschiede zwischenVersuchs- und Kontrolltieren sind durch Sterne angegeben: * = p ≤ 0,05; ** = p ≤ 0,01 (Mann-Whitney U-Test). Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer derVorinkubation 15 Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer derVorinkubation 180 Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180Minuten.
Infralimbischer Präfrontaler Cortex - IL
0
10000
20000
30000
40000
50000
60000
70000
80000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-KCH-VHI-KHI-V
* ** **
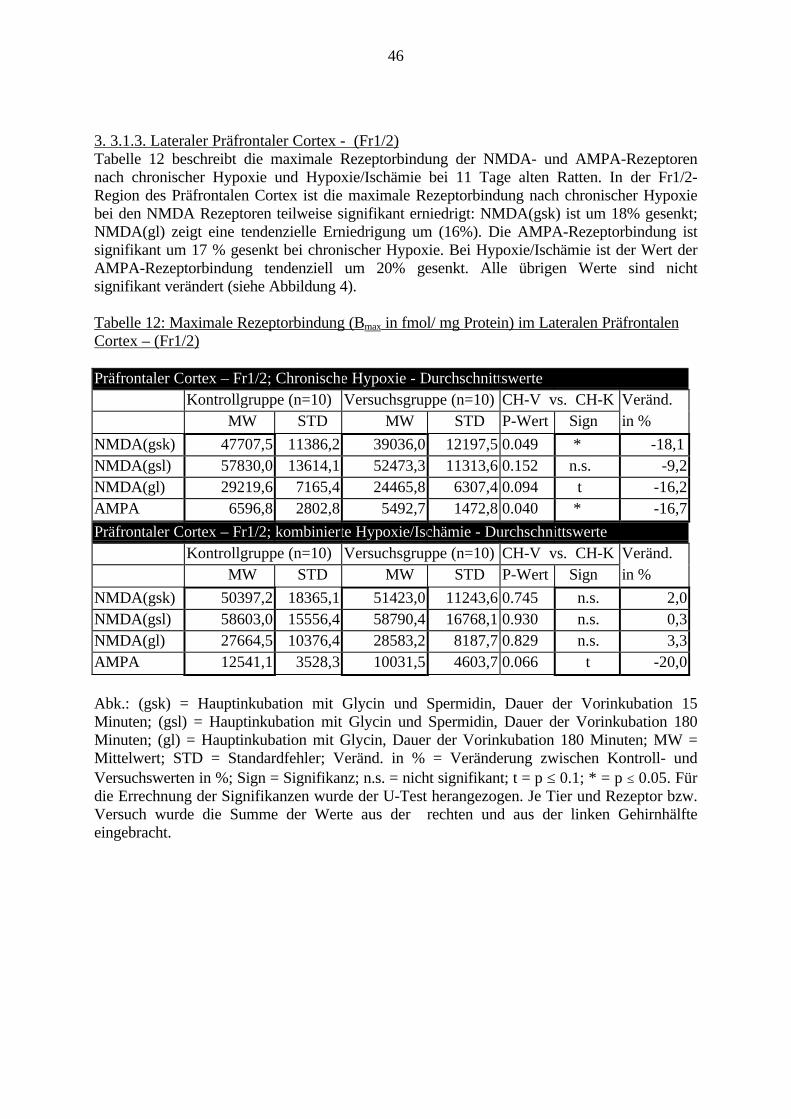
46
3. 3.1.3. Lateraler Präfrontaler Cortex - (Fr1/2)Tabelle 12 beschreibt die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptorennach chronischer Hypoxie und Hypoxie/Ischämie bei 11 Tage alten Ratten. In der Fr1/2-Region des Präfrontalen Cortex ist die maximale Rezeptorbindung nach chronischer Hypoxiebei den NMDA Rezeptoren teilweise signifikant erniedrigt: NMDA(gsk) ist um 18% gesenkt;NMDA(gl) zeigt eine tendenzielle Erniedrigung um (16%). Die AMPA-Rezeptorbindung istsignifikant um 17 % gesenkt bei chronischer Hypoxie. Bei Hypoxie/Ischämie ist der Wert derAMPA-Rezeptorbindung tendenziell um 20% gesenkt. Alle übrigen Werte sind nichtsignifikant verändert (siehe Abbildung 4).
Tabelle 12: Maximale Rezeptorbindung (Bmax in fmol/ mg Protein) im Lateralen PräfrontalenCortex – (Fr1/2)
Präfrontaler Cortex – Fr1/2; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 47707,5 11386,2 39036,0 12197,5 0.049 * -18,1NMDA(gsl) 57830,0 13614,1 52473,3 11313,6 0.152 n.s. -9,2NMDA(gl) 29219,6 7165,4 24465,8 6307,4 0.094 t -16,2AMPA 6596,8 2802,8 5492,7 1472,8 0.040 * -16,7
Präfrontaler Cortex – Fr1/2; kombinierte Hypoxie/Ischämie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 50397,2 18365,1 51423,0 11243,6 0.745 n.s. 2,0NMDA(gsl) 58603,0 15556,4 58790,4 16768,1 0.930 n.s. 0,3NMDA(gl) 27664,5 10376,4 28583,2 8187,7 0.829 n.s. 3,3AMPA 12541,1 3528,3 10031,5 4603,7 0.066 t -20,0
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1; * = p ≤ 0.05. Fürdie Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw.Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
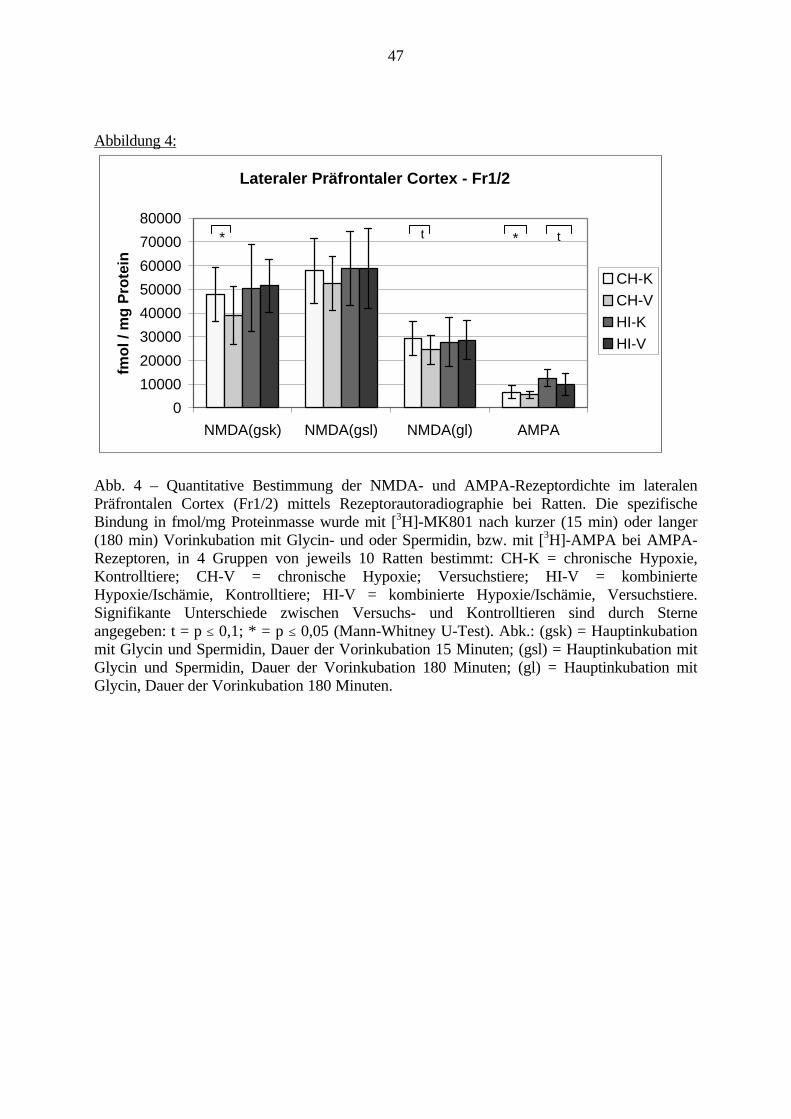
47
Abbildung 4:
Abb. 4 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im lateralenPräfrontalen Cortex (Fr1/2) mittels Rezeptorautoradiographie bei Ratten. Die spezifischeBindung in fmol/mg Proteinmasse wurde mit [3H]-MK801 nach kurzer (15 min) oder langer(180 min) Vorinkubation mit Glycin- und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren, in 4 Gruppen von jeweils 10 Ratten bestimmt: CH-K = chronische Hypoxie,Kontrolltiere; CH-V = chronische Hypoxie; Versuchstiere; HI-V = kombinierteHypoxie/Ischämie, Kontrolltiere; HI-V = kombinierte Hypoxie/Ischämie, Versuchstiere.Signifikante Unterschiede zwischen Versuchs- und Kontrolltieren sind durch Sterneangegeben: t = p ≤ 0,1; * = p ≤ 0,05 (Mann-Whitney U-Test). Abk.: (gsk) = Hauptinkubationmit Glycin und Spermidin, Dauer der Vorinkubation 15 Minuten; (gsl) = Hauptinkubation mitGlycin und Spermidin, Dauer der Vorinkubation 180 Minuten; (gl) = Hauptinkubation mitGlycin, Dauer der Vorinkubation 180 Minuten.
Lateraler Präfrontaler Cortex - Fr1/2
0
10000
20000
30000
40000
50000
60000
70000
80000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-K
CH-V
HI-K
HI-V
* t * t
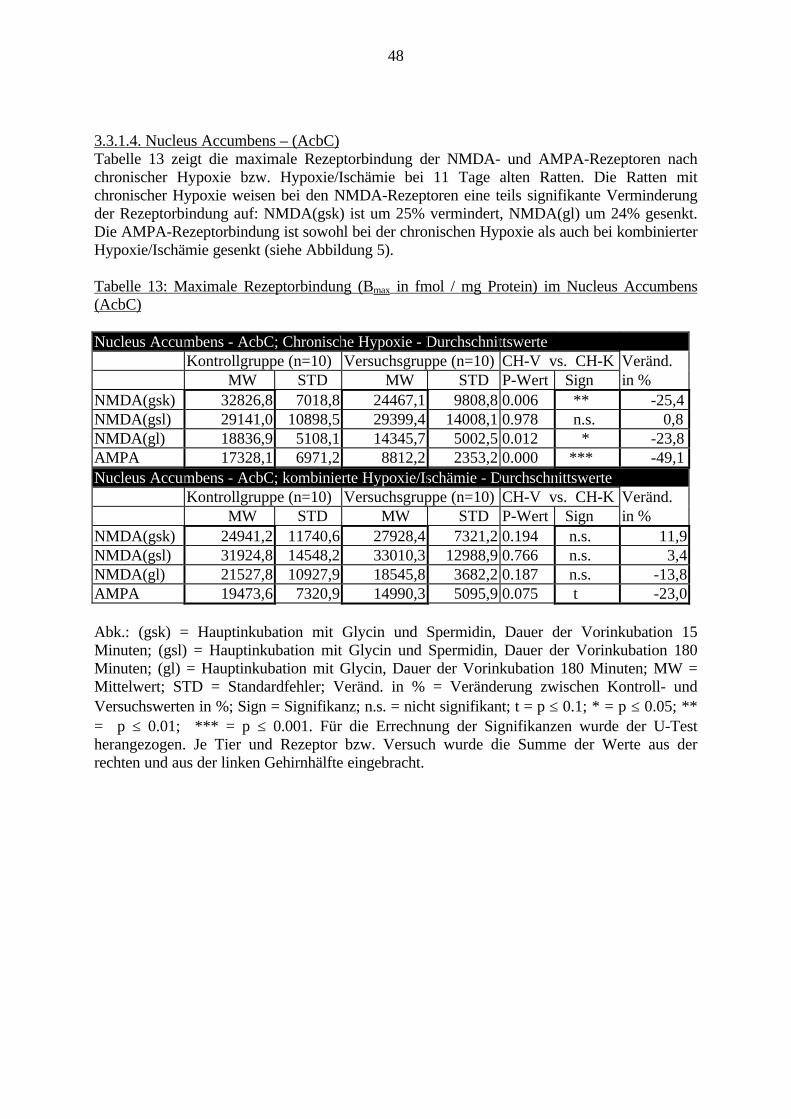
48
3.3.1.4. Nucleus Accumbens – (AcbC)Tabelle 13 zeigt die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptoren nachchronischer Hypoxie bzw. Hypoxie/Ischämie bei 11 Tage alten Ratten. Die Ratten mitchronischer Hypoxie weisen bei den NMDA-Rezeptoren eine teils signifikante Verminderungder Rezeptorbindung auf: NMDA(gsk) ist um 25% vermindert, NMDA(gl) um 24% gesenkt.Die AMPA-Rezeptorbindung ist sowohl bei der chronischen Hypoxie als auch bei kombinierterHypoxie/Ischämie gesenkt (siehe Abbildung 5).
Tabelle 13: Maximale Rezeptorbindung (Bmax in fmol / mg Protein) im Nucleus Accumbens(AcbC)
Nucleus Accumbens - AcbC; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 32826,8 7018,8 24467,1 9808,8 0.006 ** -25,4NMDA(gsl) 29141,0 10898,5 29399,4 14008,1 0.978 n.s. 0,8NMDA(gl) 18836,9 5108,1 14345,7 5002,5 0.012 * -23,8AMPA 17328,1 6971,2 8812,2 2353,2 0.000 *** -49,1Nucleus Accumbens - AcbC; kombinierte Hypoxie/Ischämie - Durchschnittswerte
Kontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 24941,2 11740,6 27928,4 7321,2 0.194 n.s. 11,9NMDA(gsl) 31924,8 14548,2 33010,3 12988,9 0.766 n.s. 3,4NMDA(gl) 21527,8 10927,9 18545,8 3682,2 0.187 n.s. -13,8AMPA 19473,6 7320,9 14990,3 5095,9 0.075 t -23,0
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1; * = p ≤ 0.05; **= p ≤ 0.01; *** = p ≤ 0.001. Für die Errechnung der Signifikanzen wurde der U-Testherangezogen. Je Tier und Rezeptor bzw. Versuch wurde die Summe der Werte aus derrechten und aus der linken Gehirnhälfte eingebracht.
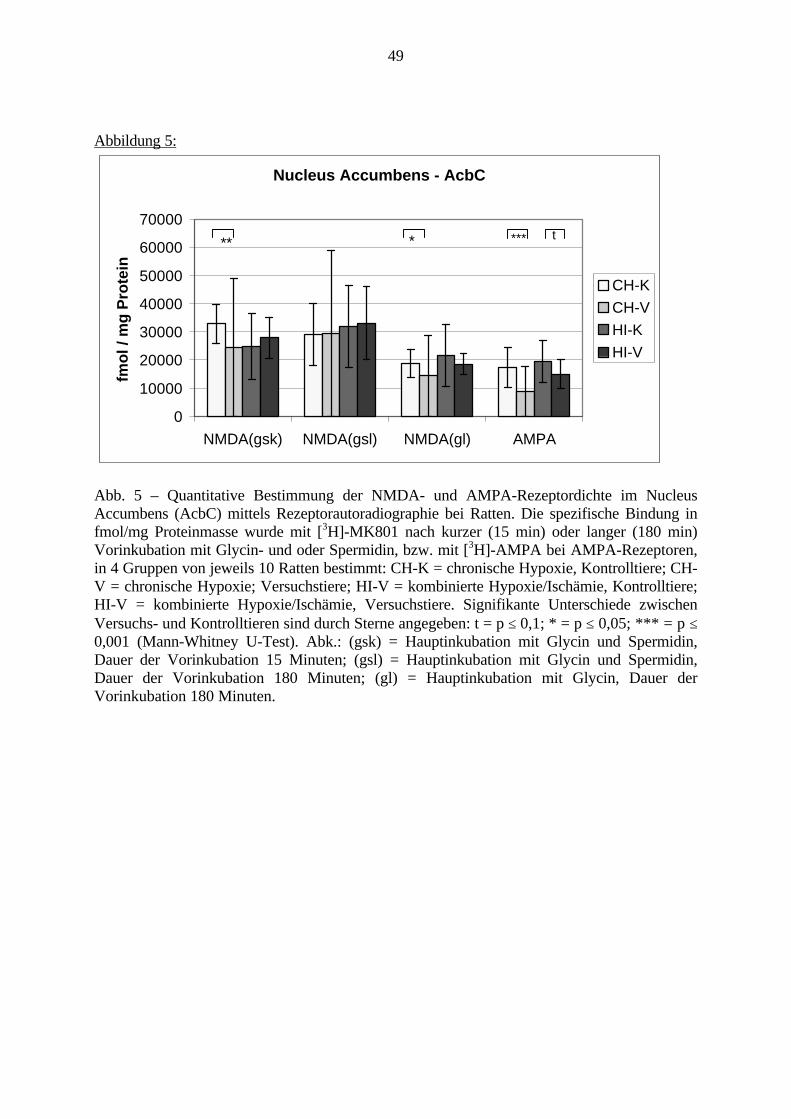
49
Abbildung 5:
Abb. 5 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im NucleusAccumbens (AcbC) mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung infmol/mg Proteinmasse wurde mit [3H]-MK801 nach kurzer (15 min) oder langer (180 min)Vorinkubation mit Glycin- und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren,in 4 Gruppen von jeweils 10 Ratten bestimmt: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere;HI-V = kombinierte Hypoxie/Ischämie, Versuchstiere. Signifikante Unterschiede zwischenVersuchs- und Kontrolltieren sind durch Sterne angegeben: t = p ≤ 0,1; * = p ≤ 0,05; *** = p ≤0,001 (Mann-Whitney U-Test). Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin,Dauer der Vorinkubation 15 Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin,Dauer der Vorinkubation 180 Minuten; (gl) = Hauptinkubation mit Glycin, Dauer derVorinkubation 180 Minuten.
Nucleus Accumbens - AcbC
0
10000
20000
30000
40000
50000
60000
70000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-K
CH-V
HI-K
HI-V
** * *** t
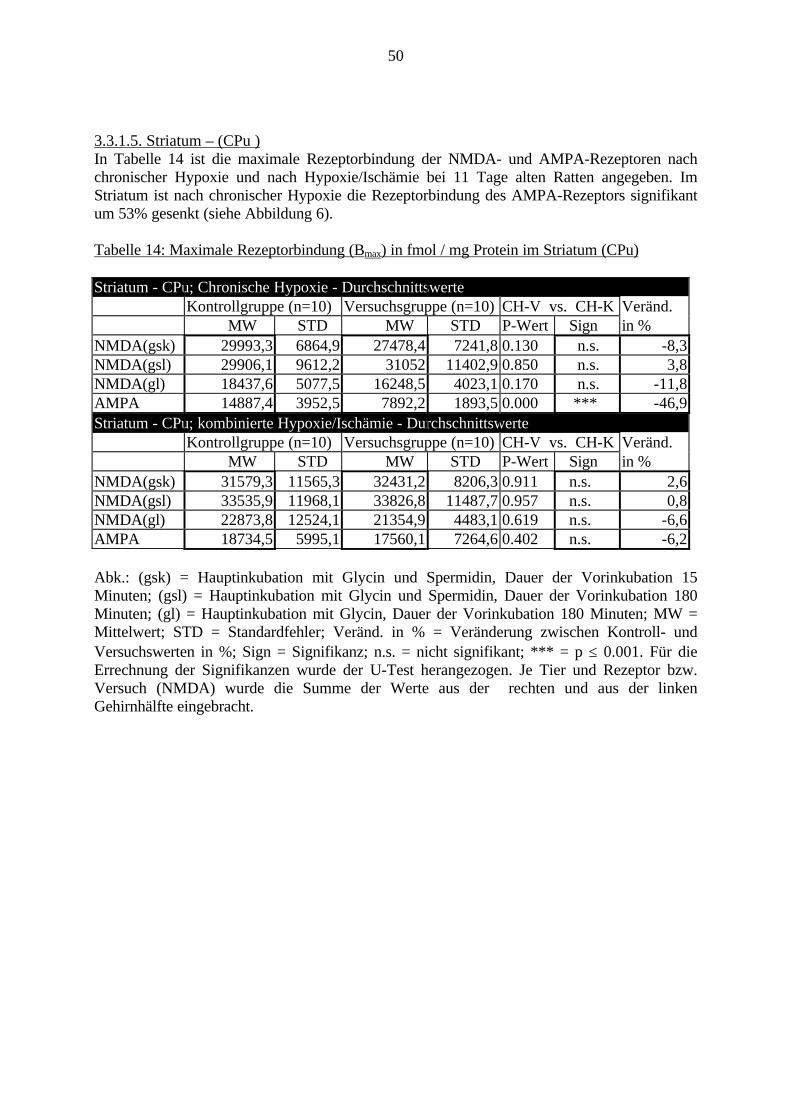
50
3.3.1.5. Striatum – (CPu )In Tabelle 14 ist die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptoren nachchronischer Hypoxie und nach Hypoxie/Ischämie bei 11 Tage alten Ratten angegeben. ImStriatum ist nach chronischer Hypoxie die Rezeptorbindung des AMPA-Rezeptors signifikantum 53% gesenkt (siehe Abbildung 6).
Tabelle 14: Maximale Rezeptorbindung (Bmax) in fmol / mg Protein im Striatum (CPu)
Striatum - CPu; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 29993,3 6864,9 27478,4 7241,8 0.130 n.s. -8,3NMDA(gsl) 29906,1 9612,2 31052 11402,9 0.850 n.s. 3,8NMDA(gl) 18437,6 5077,5 16248,5 4023,1 0.170 n.s. -11,8AMPA 14887,4 3952,5 7892,2 1893,5 0.000 *** -46,9Striatum - CPu; kombinierte Hypoxie/Ischämie - Durchschnittswerte
Kontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 31579,3 11565,3 32431,2 8206,3 0.911 n.s. 2,6NMDA(gsl) 33535,9 11968,1 33826,8 11487,7 0.957 n.s. 0,8NMDA(gl) 22873,8 12524,1 21354,9 4483,1 0.619 n.s. -6,6AMPA 18734,5 5995,1 17560,1 7264,6 0.402 n.s. -6,2
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; *** = p ≤ 0.001. Für dieErrechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw.Versuch (NMDA) wurde die Summe der Werte aus der rechten und aus der linkenGehirnhälfte eingebracht.
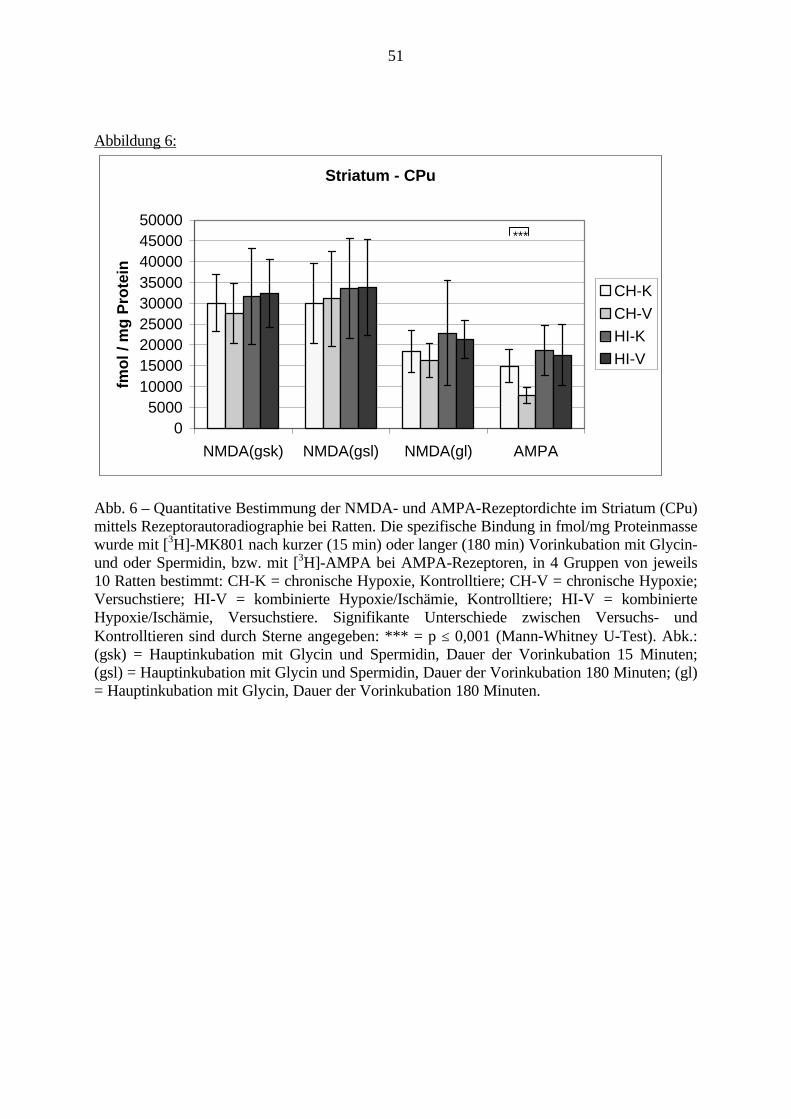
51
Abbildung 6:
Abb. 6 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im Striatum (CPu)mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung in fmol/mg Proteinmassewurde mit [3H]-MK801 nach kurzer (15 min) oder langer (180 min) Vorinkubation mit Glycin-und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren, in 4 Gruppen von jeweils10 Ratten bestimmt: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie;Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V = kombinierteHypoxie/Ischämie, Versuchstiere. Signifikante Unterschiede zwischen Versuchs- undKontrolltieren sind durch Sterne angegeben: *** = p ≤ 0,001 (Mann-Whitney U-Test). Abk.:(gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15 Minuten;(gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180 Minuten; (gl)= Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten.
Striatum - CPu
05000
100001500020000250003000035000400004500050000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-K
CH-V
HI-K
HI-V
***
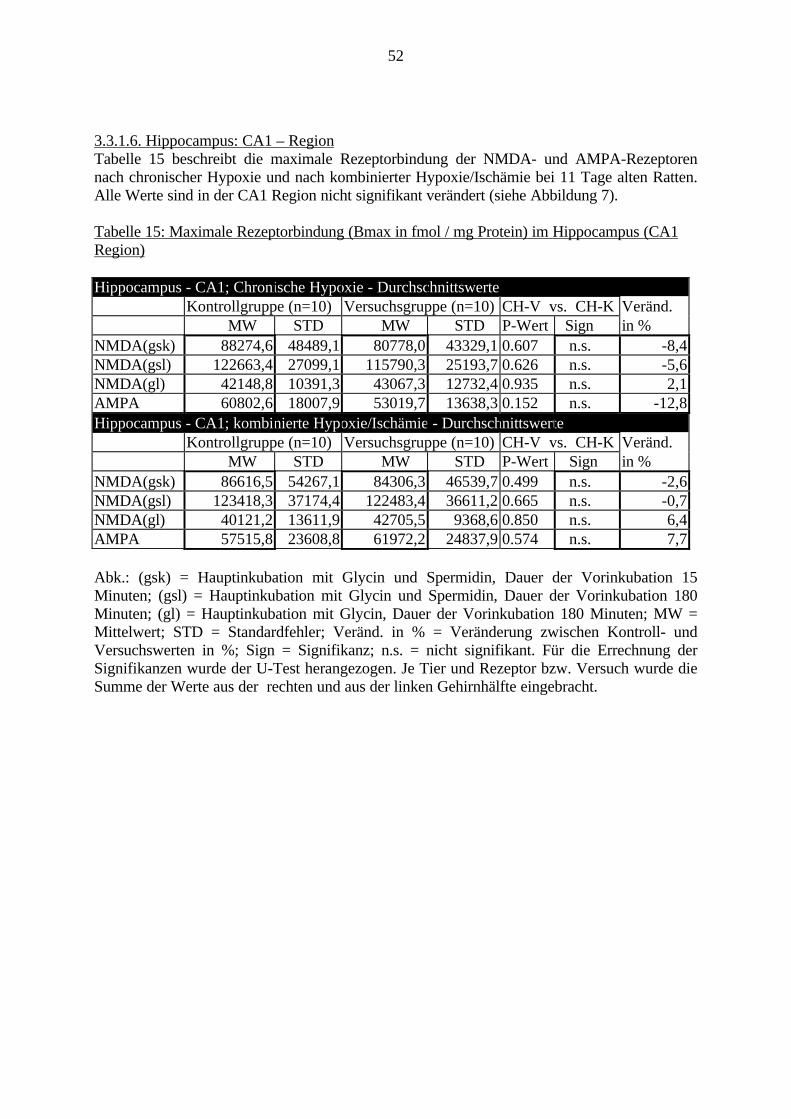
52
3.3.1.6. Hippocampus: CA1 – RegionTabelle 15 beschreibt die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptorennach chronischer Hypoxie und nach kombinierter Hypoxie/Ischämie bei 11 Tage alten Ratten.Alle Werte sind in der CA1 Region nicht signifikant verändert (siehe Abbildung 7).
Tabelle 15: Maximale Rezeptorbindung (Bmax in fmol / mg Protein) im Hippocampus (CA1Region)
Hippocampus - CA1; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 88274,6 48489,1 80778,0 43329,1 0.607 n.s. -8,4NMDA(gsl) 122663,4 27099,1 115790,3 25193,7 0.626 n.s. -5,6NMDA(gl) 42148,8 10391,3 43067,3 12732,4 0.935 n.s. 2,1AMPA 60802,6 18007,9 53019,7 13638,3 0.152 n.s. -12,8Hippocampus - CA1; kombinierte Hypoxie/Ischämie - Durchschnittswerte
Kontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 86616,5 54267,1 84306,3 46539,7 0.499 n.s. -2,6NMDA(gsl) 123418,3 37174,4 122483,4 36611,2 0.665 n.s. -0,7NMDA(gl) 40121,2 13611,9 42705,5 9368,6 0.850 n.s. 6,4AMPA 57515,8 23608,8 61972,2 24837,9 0.574 n.s. 7,7
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant. Für die Errechnung derSignifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw. Versuch wurde dieSumme der Werte aus der rechten und aus der linken Gehirnhälfte eingebracht.
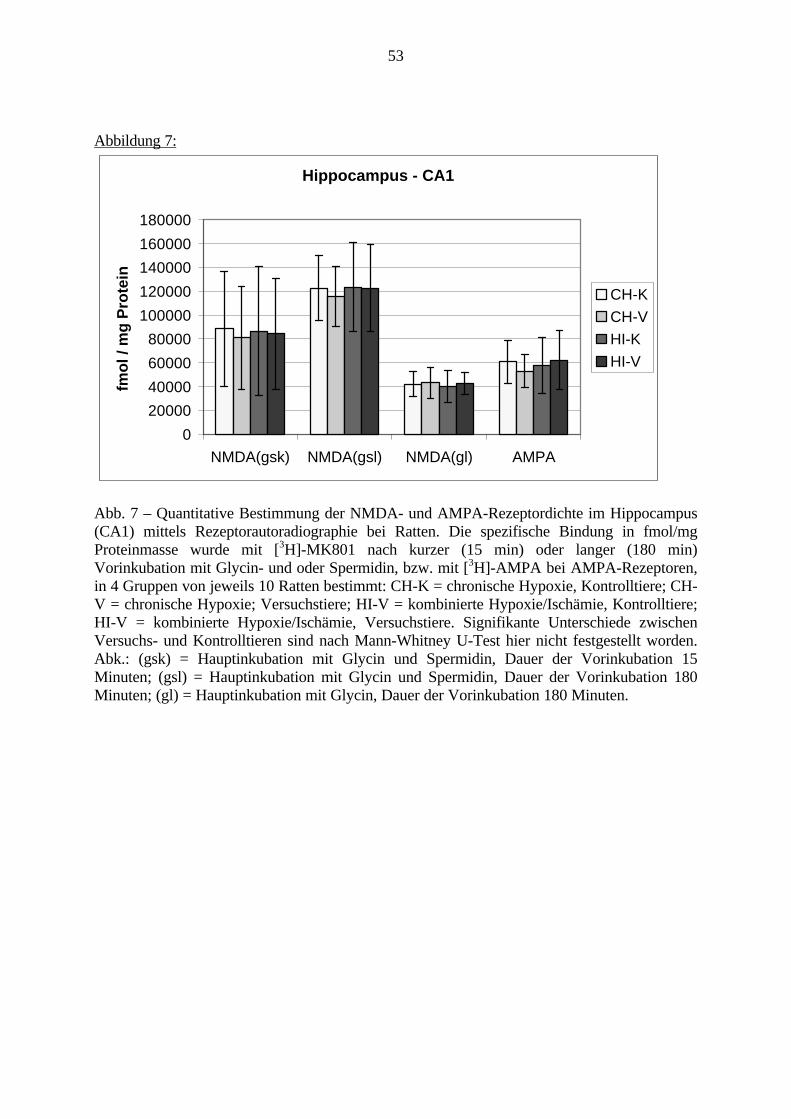
53
Abbildung 7:
Abb. 7 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im Hippocampus(CA1) mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung in fmol/mgProteinmasse wurde mit [3H]-MK801 nach kurzer (15 min) oder langer (180 min)Vorinkubation mit Glycin- und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren,in 4 Gruppen von jeweils 10 Ratten bestimmt: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere;HI-V = kombinierte Hypoxie/Ischämie, Versuchstiere. Signifikante Unterschiede zwischenVersuchs- und Kontrolltieren sind nach Mann-Whitney U-Test hier nicht festgestellt worden.Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten.
Hippocampus - CA1
0
20000
40000
60000
80000
100000
120000
140000
160000
180000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-K
CH-V
HI-K
HI-V
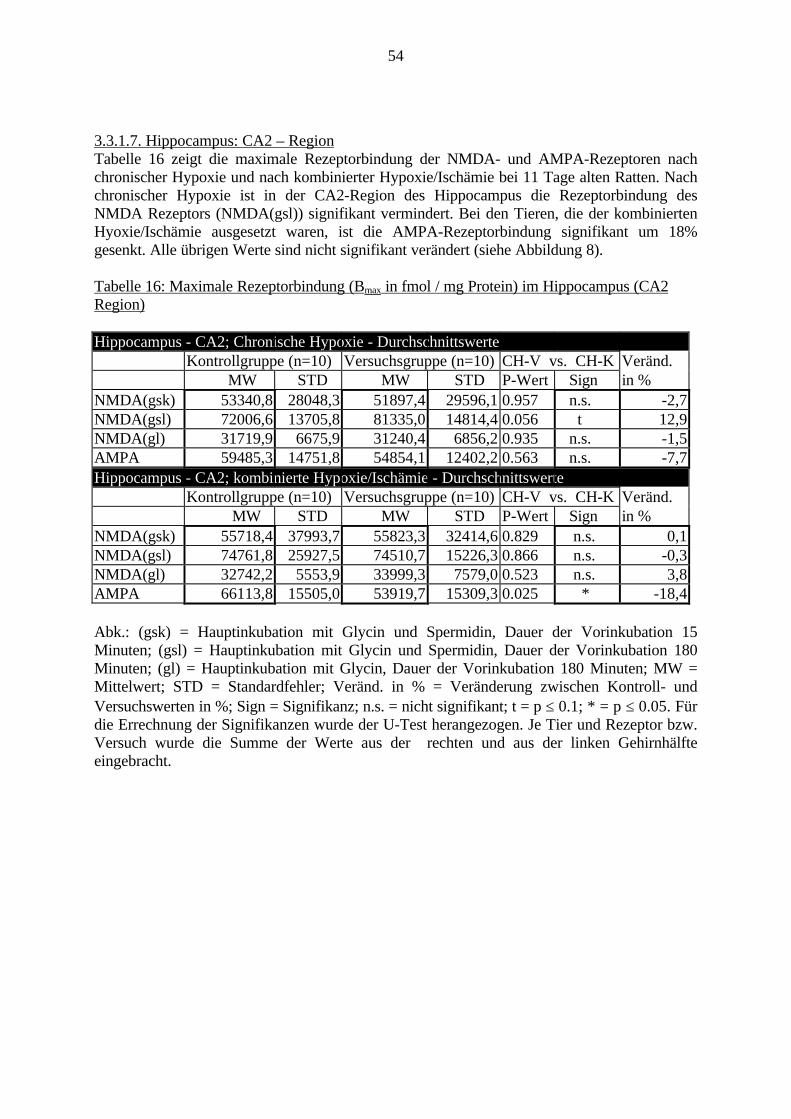
54
3.3.1.7. Hippocampus: CA2 – RegionTabelle 16 zeigt die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptoren nachchronischer Hypoxie und nach kombinierter Hypoxie/Ischämie bei 11 Tage alten Ratten. Nachchronischer Hypoxie ist in der CA2-Region des Hippocampus die Rezeptorbindung desNMDA Rezeptors (NMDA(gsl)) signifikant vermindert. Bei den Tieren, die der kombiniertenHyoxie/Ischämie ausgesetzt waren, ist die AMPA-Rezeptorbindung signifikant um 18%gesenkt. Alle übrigen Werte sind nicht signifikant verändert (siehe Abbildung 8).
Tabelle 16: Maximale Rezeptorbindung (Bmax in fmol / mg Protein) im Hippocampus (CA2Region)
Hippocampus - CA2; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 53340,8 28048,3 51897,4 29596,1 0.957 n.s. -2,7NMDA(gsl) 72006,6 13705,8 81335,0 14814,4 0.056 t 12,9NMDA(gl) 31719,9 6675,9 31240,4 6856,2 0.935 n.s. -1,5AMPA 59485,3 14751,8 54854,1 12402,2 0.563 n.s. -7,7Hippocampus - CA2; kombinierte Hypoxie/Ischämie - Durchschnittswerte
Kontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 55718,4 37993,7 55823,3 32414,6 0.829 n.s. 0,1NMDA(gsl) 74761,8 25927,5 74510,7 15226,3 0.866 n.s. -0,3NMDA(gl) 32742,2 5553,9 33999,3 7579,0 0.523 n.s. 3,8AMPA 66113,8 15505,0 53919,7 15309,3 0.025 * -18,4
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1; * = p ≤ 0.05. Fürdie Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw.Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
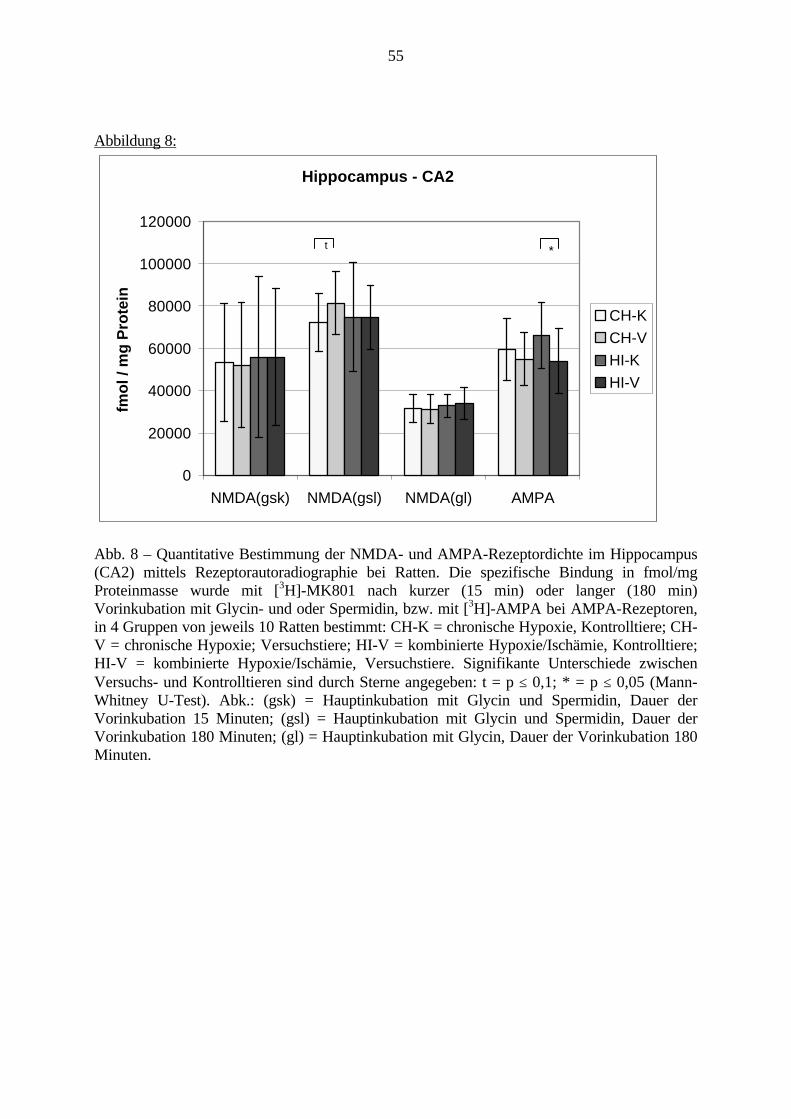
55
Abbildung 8:
Abb. 8 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im Hippocampus(CA2) mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung in fmol/mgProteinmasse wurde mit [3H]-MK801 nach kurzer (15 min) oder langer (180 min)Vorinkubation mit Glycin- und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren,in 4 Gruppen von jeweils 10 Ratten bestimmt: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere;HI-V = kombinierte Hypoxie/Ischämie, Versuchstiere. Signifikante Unterschiede zwischenVersuchs- und Kontrolltieren sind durch Sterne angegeben: t = p ≤ 0,1; * = p ≤ 0,05 (Mann-Whitney U-Test). Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer derVorinkubation 15 Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer derVorinkubation 180 Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180Minuten.
Hippocampus - CA2
0
20000
40000
60000
80000
100000
120000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-K
CH-V
HI-K
HI-V
t *
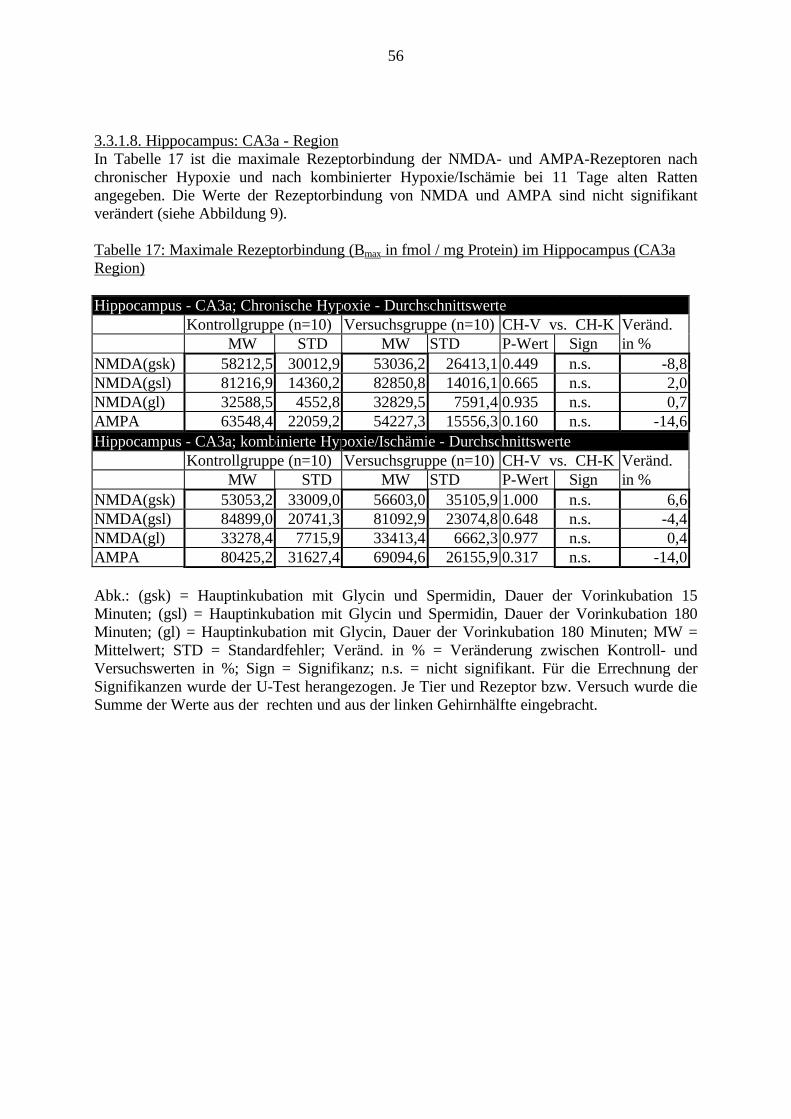
56
3.3.1.8. Hippocampus: CA3a - RegionIn Tabelle 17 ist die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptoren nachchronischer Hypoxie und nach kombinierter Hypoxie/Ischämie bei 11 Tage alten Rattenangegeben. Die Werte der Rezeptorbindung von NMDA und AMPA sind nicht signifikantverändert (siehe Abbildung 9).
Tabelle 17: Maximale Rezeptorbindung (Bmax in fmol / mg Protein) im Hippocampus (CA3aRegion)
Hippocampus - CA3a; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 58212,5 30012,9 53036,2 26413,1 0.449 n.s. -8,8NMDA(gsl) 81216,9 14360,2 82850,8 14016,1 0.665 n.s. 2,0NMDA(gl) 32588,5 4552,8 32829,5 7591,4 0.935 n.s. 0,7AMPA 63548,4 22059,2 54227,3 15556,3 0.160 n.s. -14,6Hippocampus - CA3a; kombinierte Hypoxie/Ischämie - Durchschnittswerte
Kontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 53053,2 33009,0 56603,0 35105,9 1.000 n.s. 6,6NMDA(gsl) 84899,0 20741,3 81092,9 23074,8 0.648 n.s. -4,4NMDA(gl) 33278,4 7715,9 33413,4 6662,3 0.977 n.s. 0,4AMPA 80425,2 31627,4 69094,6 26155,9 0.317 n.s. -14,0
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant. Für die Errechnung derSignifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw. Versuch wurde dieSumme der Werte aus der rechten und aus der linken Gehirnhälfte eingebracht.
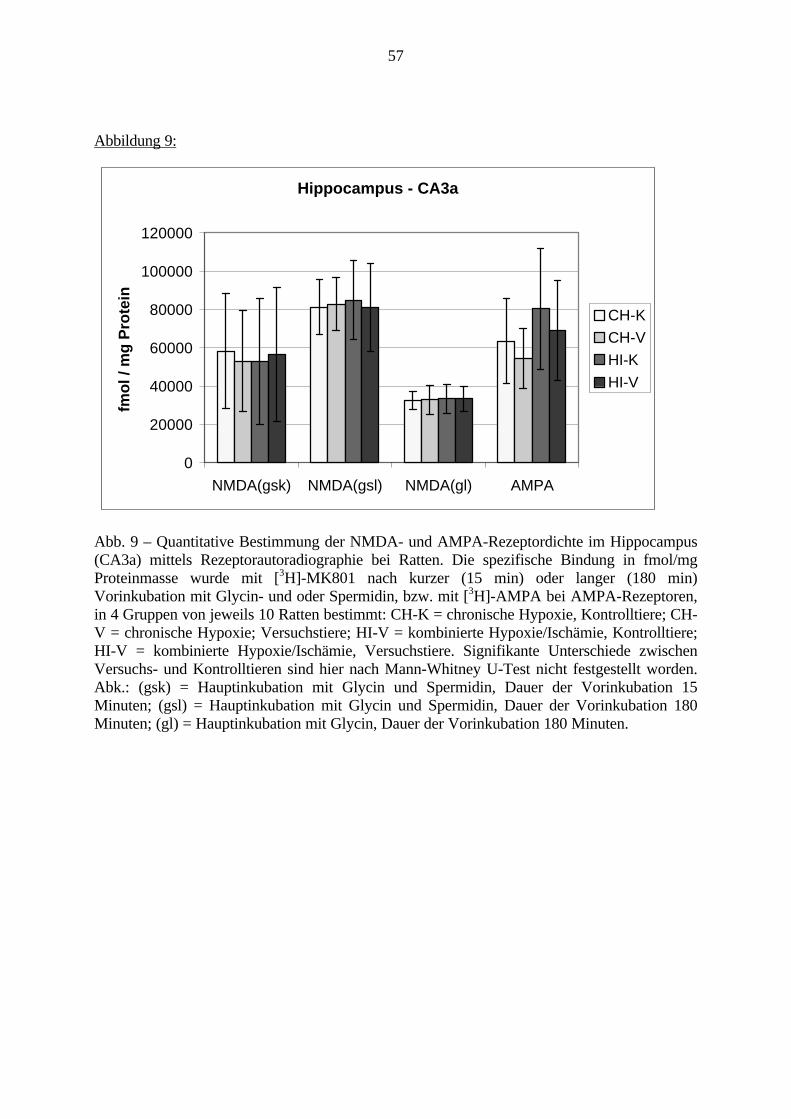
57
Abbildung 9:
Abb. 9 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im Hippocampus(CA3a) mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung in fmol/mgProteinmasse wurde mit [3H]-MK801 nach kurzer (15 min) oder langer (180 min)Vorinkubation mit Glycin- und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren,in 4 Gruppen von jeweils 10 Ratten bestimmt: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere;HI-V = kombinierte Hypoxie/Ischämie, Versuchstiere. Signifikante Unterschiede zwischenVersuchs- und Kontrolltieren sind hier nach Mann-Whitney U-Test nicht festgestellt worden.Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten.
Hippocampus - CA3a
0
20000
40000
60000
80000
100000
120000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-K
CH-V
HI-K
HI-V
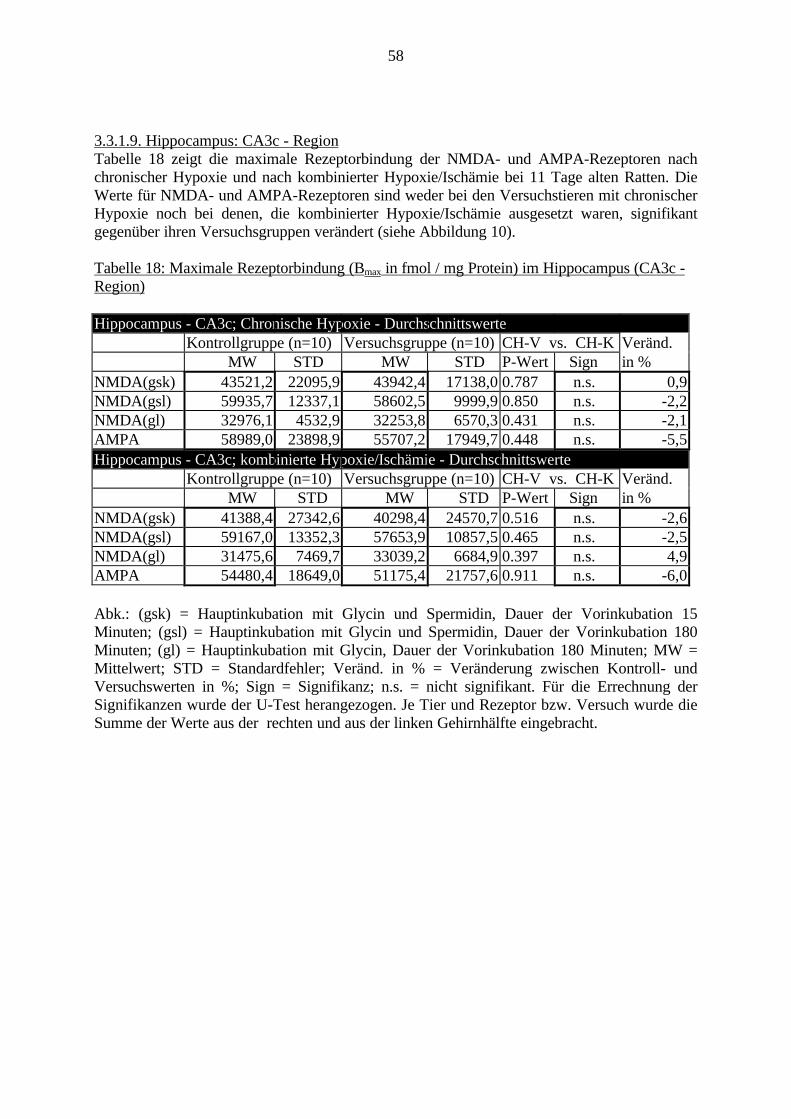
58
3.3.1.9. Hippocampus: CA3c - RegionTabelle 18 zeigt die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptoren nachchronischer Hypoxie und nach kombinierter Hypoxie/Ischämie bei 11 Tage alten Ratten. DieWerte für NMDA- und AMPA-Rezeptoren sind weder bei den Versuchstieren mit chronischerHypoxie noch bei denen, die kombinierter Hypoxie/Ischämie ausgesetzt waren, signifikantgegenüber ihren Versuchsgruppen verändert (siehe Abbildung 10).
Tabelle 18: Maximale Rezeptorbindung (Bmax in fmol / mg Protein) im Hippocampus (CA3c -Region)
Hippocampus - CA3c; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 43521,2 22095,9 43942,4 17138,0 0.787 n.s. 0,9NMDA(gsl) 59935,7 12337,1 58602,5 9999,9 0.850 n.s. -2,2NMDA(gl) 32976,1 4532,9 32253,8 6570,3 0.431 n.s. -2,1AMPA 58989,0 23898,9 55707,2 17949,7 0.448 n.s. -5,5Hippocampus - CA3c; kombinierte Hypoxie/Ischämie - Durchschnittswerte
Kontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 41388,4 27342,6 40298,4 24570,7 0.516 n.s. -2,6NMDA(gsl) 59167,0 13352,3 57653,9 10857,5 0.465 n.s. -2,5NMDA(gl) 31475,6 7469,7 33039,2 6684,9 0.397 n.s. 4,9AMPA 54480,4 18649,0 51175,4 21757,6 0.911 n.s. -6,0
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant. Für die Errechnung derSignifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw. Versuch wurde dieSumme der Werte aus der rechten und aus der linken Gehirnhälfte eingebracht.
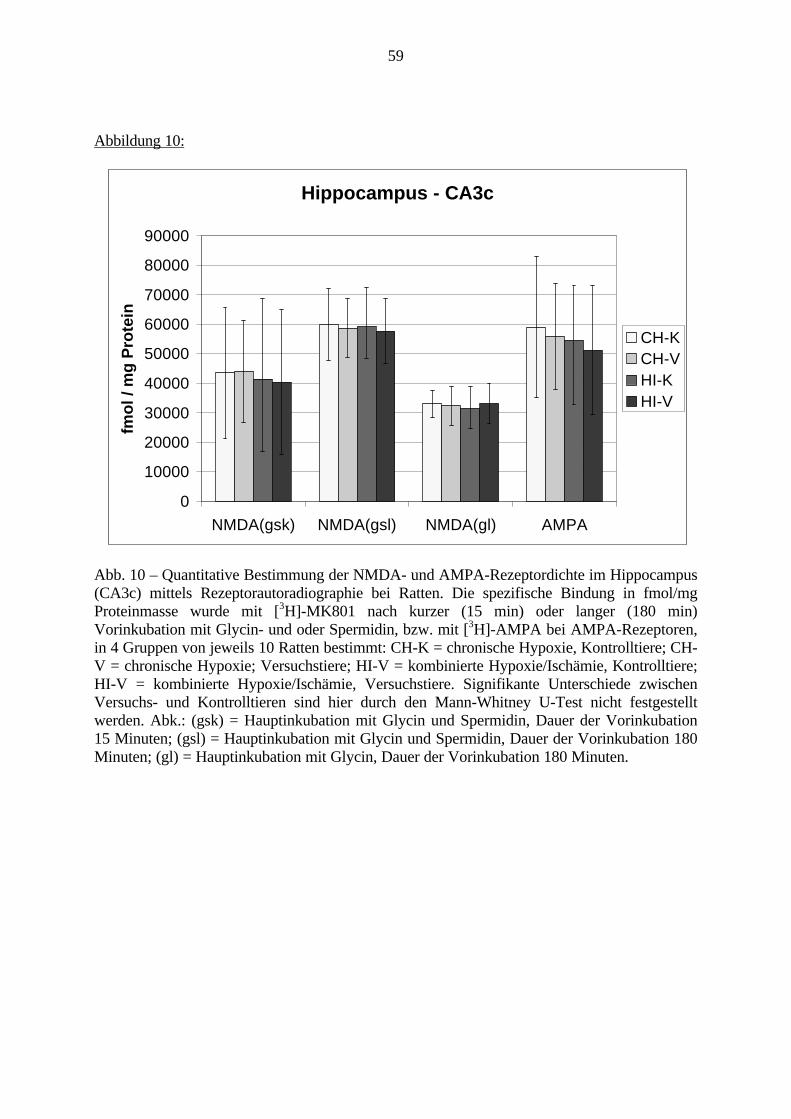
59
Abbildung 10:
Abb. 10 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im Hippocampus(CA3c) mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung in fmol/mgProteinmasse wurde mit [3H]-MK801 nach kurzer (15 min) oder langer (180 min)Vorinkubation mit Glycin- und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren,in 4 Gruppen von jeweils 10 Ratten bestimmt: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere;HI-V = kombinierte Hypoxie/Ischämie, Versuchstiere. Signifikante Unterschiede zwischenVersuchs- und Kontrolltieren sind hier durch den Mann-Whitney U-Test nicht festgestelltwerden. Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation15 Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten.
Hippocampus - CA3c
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-KCH-VHI-KHI-V
60
3.3.1.10. Hippocampus: DG - RegionTabelle 19 beschreibt die maximale Rezeptorbindung der NMDA- und AMPA-Rezeptorennach chronischer Hypoxie und nach kombinierter Hypoxie/Ischämie bei 11 Tage alten Ratten.In der DG-Region des Hippocampus ist weder bei chronischer Hypoxie noch bei kombinierterHypoxie/Ischämie die Rezeptorbindung der NMDA- und AMPA-Rezeptoren signifikantverändert (siehe Abbildung 11).
Tabelle 19: Maximale Rezeptorbindung (Bmax in fmol / Protein) im Hippocampus (DG –Region)
Hippocampus - DG; Chronische Hypoxie - DurchschnittswerteKontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 53817,1 26276,6 52781,9 26662,7 0.787 n.s. -1,9NMDA(gsl) 67881,6 12044,6 73742,4 13456,4 0.213 n.s. 8,6NMDA(gl) 32799,4 6581,3 32371,7 7095,0 0.829 n.s. -1,3AMPA 40116,9 14595,1 43271,6 12402,6 0.761 n.s. 7,8Hippocampus - DG; kombinierte Hypoxie/Ischämie - Durchschnittswerte
Kontrollgruppe (n=10) Versuchsgruppe (n=10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
NMDA(gsk) 47387,9 25766,6 51866,1 27118,4 0.766 n.s. 9,4NMDA(gsl) 72273,1 17023,7 71587,2 11196,8 0.888 n.s. -0,9NMDA(gl) 29198,7 8370,8 32706,2 6457,1 0.273 n.s. 12,0AMPA 40616,0 18723,2 38882,3 13786,8 0.757 n.s. -4,2
Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 15Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten; MW =Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- undVersuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant. Für die Errechnung derSignifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw. Versuch wurde dieSumme der Werte aus der rechten und aus der linken Gehirnhälfte eingebracht.
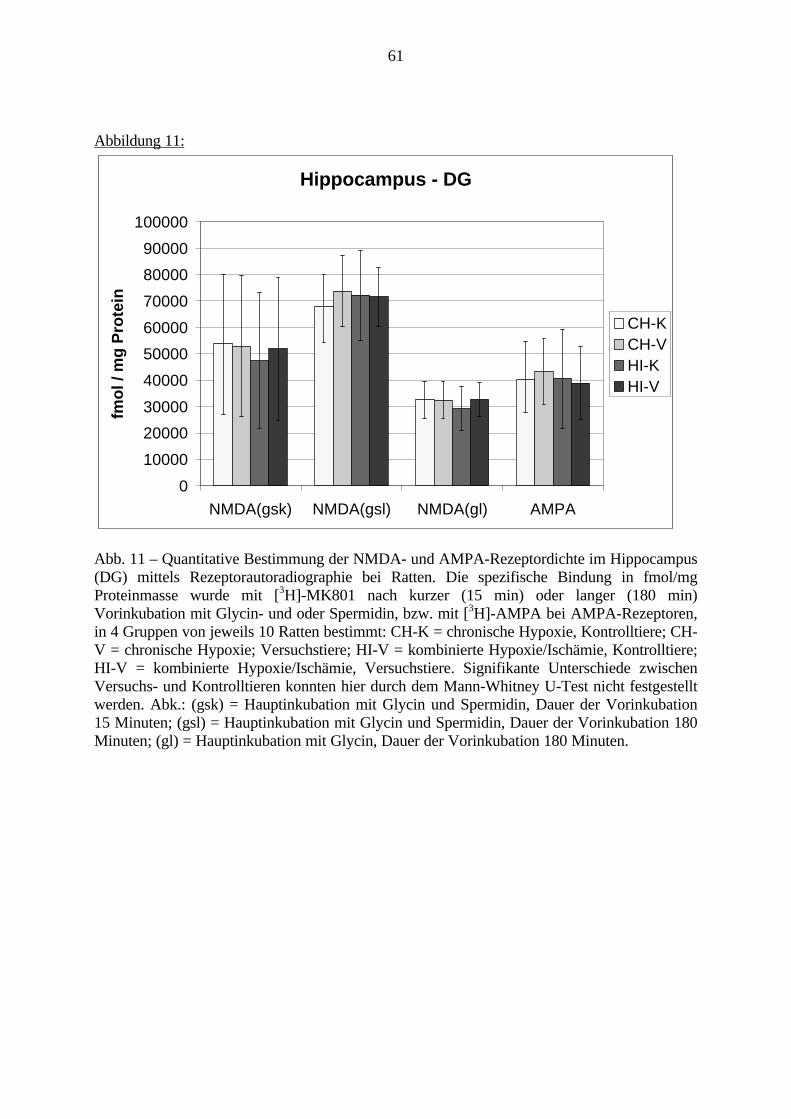
61
Abbildung 11:
Abb. 11 – Quantitative Bestimmung der NMDA- und AMPA-Rezeptordichte im Hippocampus(DG) mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung in fmol/mgProteinmasse wurde mit [3H]-MK801 nach kurzer (15 min) oder langer (180 min)Vorinkubation mit Glycin- und oder Spermidin, bzw. mit [3H]-AMPA bei AMPA-Rezeptoren,in 4 Gruppen von jeweils 10 Ratten bestimmt: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere;HI-V = kombinierte Hypoxie/Ischämie, Versuchstiere. Signifikante Unterschiede zwischenVersuchs- und Kontrolltieren konnten hier durch dem Mann-Whitney U-Test nicht festgestelltwerden. Abk.: (gsk) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation15 Minuten; (gsl) = Hauptinkubation mit Glycin und Spermidin, Dauer der Vorinkubation 180Minuten; (gl) = Hauptinkubation mit Glycin, Dauer der Vorinkubation 180 Minuten.
Hippocampus - DG
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000
NMDA(gsk) NMDA(gsl) NMDA(gl) AMPA
fmo
l / m
g P
rote
in
CH-KCH-VHI-KHI-V
62
3.3.1.11. GABA: Präfrontaler Cortex: Cg1/3 – Region, IL – Region, Fr1/2 - RegionIn Tabelle 20 ist die maximale Rezeptorbindung der GABAA-Rezeptoren nach chronischerHypoxie und nach kombinierter Hypoxie/Ischämie bei 11 Tage alten Ratten angegeben. Nachstatistischem U-Test haben sich die Daten nicht signifikant verändert (siehe Abbildung 12).
Tabelle 20: Maximale Rezeptorbindung (Bmax in fmol / Protein) im Striatum an GABAA
Rezeptoren
Striatum - Cg1/3; IL; Fr1/2 - Mittelwerte in fmol / mg Protein Kontrollwerte (n = 10) Versuchswerte (n = 10) CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
Cg1/3 - CH 3309,7 1558,3 3131,0 1476,8 0.706 n.s. -5,3Cg1/3 - HI 3656,3 2308,3 3473,2 1393,3 0.974 n.s. -5,0IL - CH 4407,0 1426,5 3609,9 1189,3 0.152 n.s. -18,0IL - HI 3234,5 1915,5 3036,7 1111,1 0.934 n.s. -6,1Fr1/2 - CH 3594,6 1566,1 3336,7 1720,3 0.588 n.s. -7,1Fr1/2 - HI 4730,9 2320,7 3466,6 1122,8 0.121 n.s. -26,7
Abk.: CH = chronische Hypoxie; HI = kombinierte Hypoxie/Ischämie. MW = Mittelwert; STD= Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- und Versuchswerten in %;Sign = Signifikanz; n.s. = nicht signifikant. Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw. Versuch wurde die Summe der Werte aus derrechten und aus der linken Gehirnhälfte eingebracht.
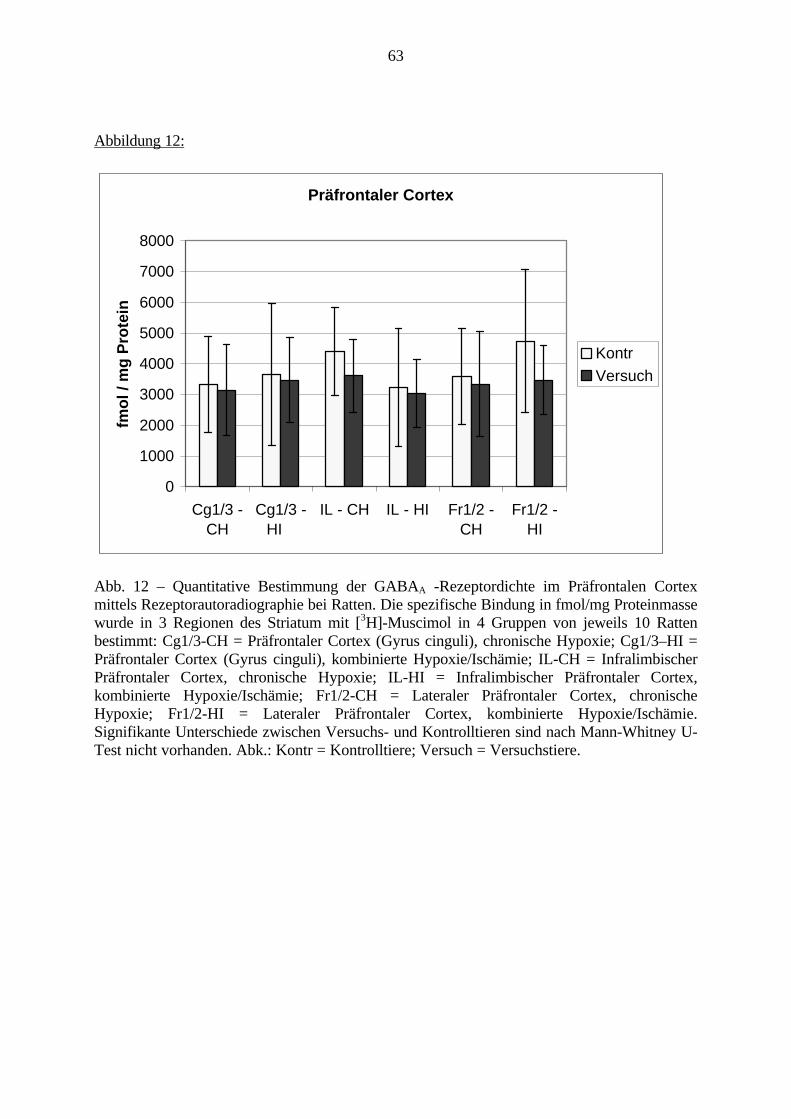
63
Abbildung 12:
Abb. 12 – Quantitative Bestimmung der GABAA -Rezeptordichte im Präfrontalen Cortexmittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung in fmol/mg Proteinmassewurde in 3 Regionen des Striatum mit [3H]-Muscimol in 4 Gruppen von jeweils 10 Rattenbestimmt: Cg1/3-CH = Präfrontaler Cortex (Gyrus cinguli), chronische Hypoxie; Cg1/3–HI =Präfrontaler Cortex (Gyrus cinguli), kombinierte Hypoxie/Ischämie; IL-CH = InfralimbischerPräfrontaler Cortex, chronische Hypoxie; IL-HI = Infralimbischer Präfrontaler Cortex,kombinierte Hypoxie/Ischämie; Fr1/2-CH = Lateraler Präfrontaler Cortex, chronischeHypoxie; Fr1/2-HI = Lateraler Präfrontaler Cortex, kombinierte Hypoxie/Ischämie.Signifikante Unterschiede zwischen Versuchs- und Kontrolltieren sind nach Mann-Whitney U-Test nicht vorhanden. Abk.: Kontr = Kontrolltiere; Versuch = Versuchstiere.
Präfrontaler Cortex
0
1000
2000
3000
4000
5000
6000
7000
8000
Cg1/3 -CH
Cg1/3 -HI
IL - CH IL - HI Fr1/2 -CH
Fr1/2 -HI
fmo
l / m
g P
rote
in
Kontr
Versuch
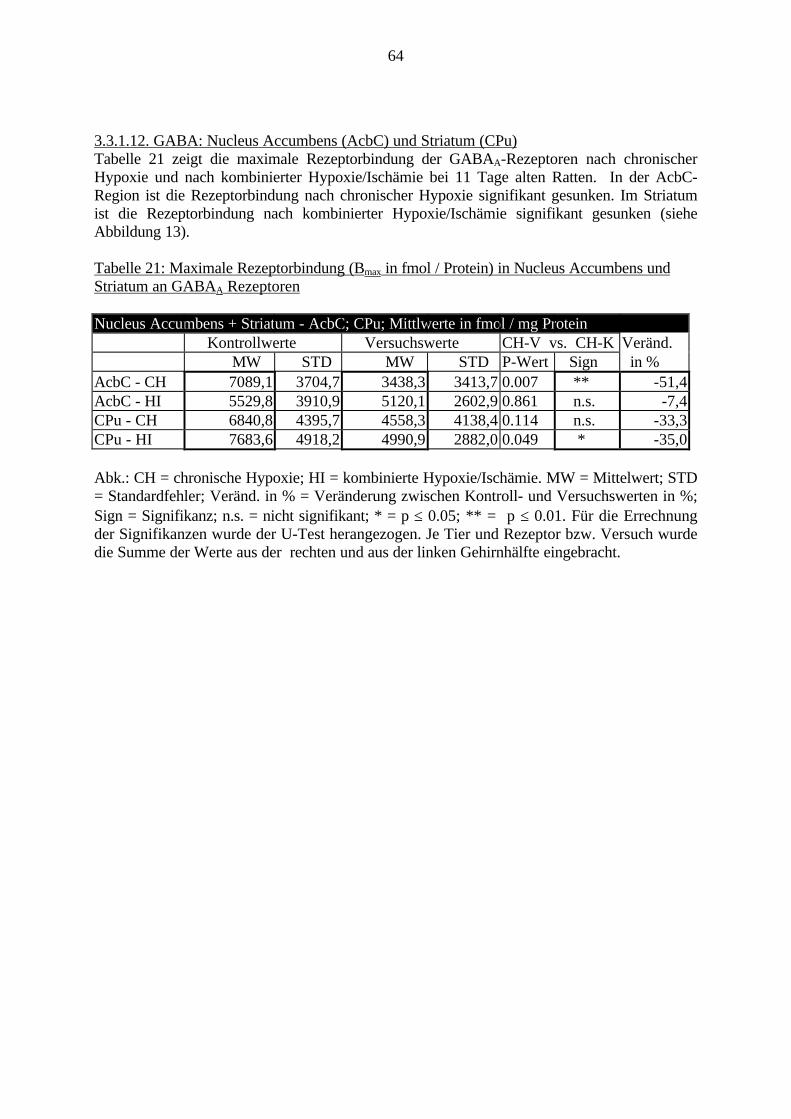
64
3.3.1.12. GABA: Nucleus Accumbens (AcbC) und Striatum (CPu)Tabelle 21 zeigt die maximale Rezeptorbindung der GABAA-Rezeptoren nach chronischerHypoxie und nach kombinierter Hypoxie/Ischämie bei 11 Tage alten Ratten. In der AcbC-Region ist die Rezeptorbindung nach chronischer Hypoxie signifikant gesunken. Im Striatumist die Rezeptorbindung nach kombinierter Hypoxie/Ischämie signifikant gesunken (sieheAbbildung 13).
Tabelle 21: Maximale Rezeptorbindung (Bmax in fmol / Protein) in Nucleus Accumbens undStriatum an GABAA Rezeptoren
Nucleus Accumbens + Striatum - AcbC; CPu; Mittlwerte in fmol / mg Protein Kontrollwerte Versuchswerte CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
AcbC - CH 7089,1 3704,7 3438,3 3413,7 0.007 ** -51,4AcbC - HI 5529,8 3910,9 5120,1 2602,9 0.861 n.s. -7,4CPu - CH 6840,8 4395,7 4558,3 4138,4 0.114 n.s. -33,3CPu - HI 7683,6 4918,2 4990,9 2882,0 0.049 * -35,0
Abk.: CH = chronische Hypoxie; HI = kombinierte Hypoxie/Ischämie. MW = Mittelwert; STD= Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- und Versuchswerten in %;Sign = Signifikanz; n.s. = nicht signifikant; * = p ≤ 0.05; ** = p ≤ 0.01. Für die Errechnungder Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw. Versuch wurdedie Summe der Werte aus der rechten und aus der linken Gehirnhälfte eingebracht.
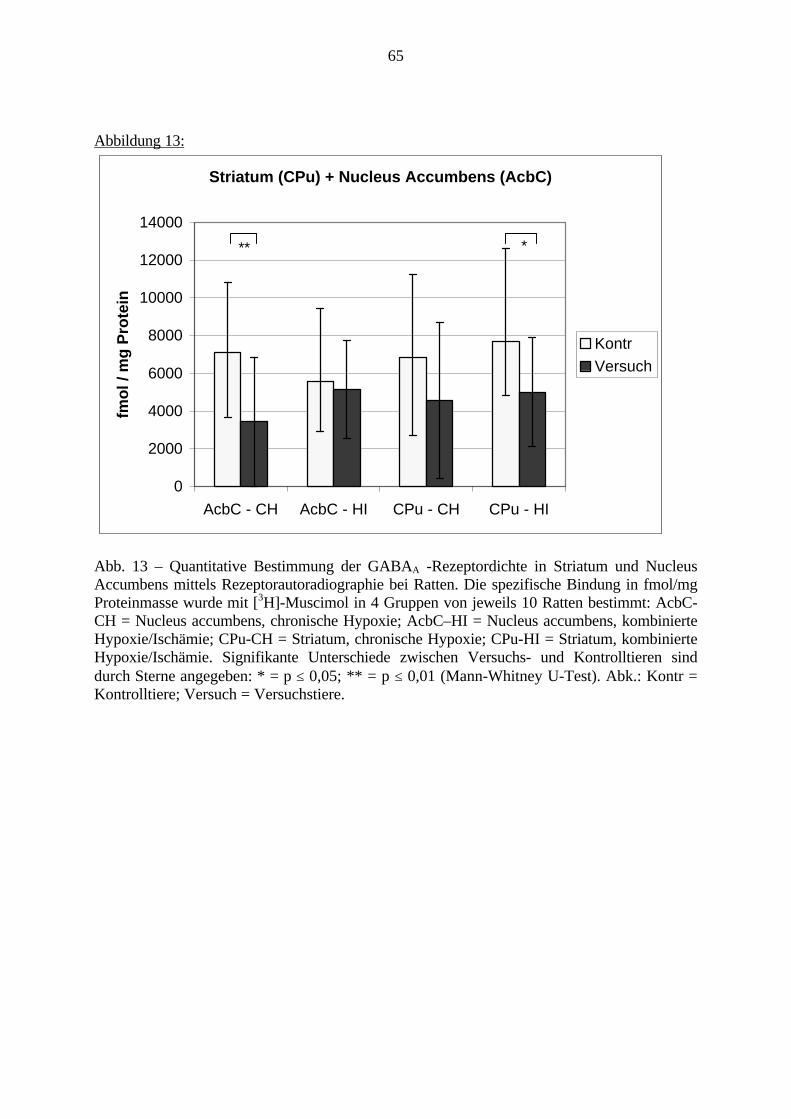
65
Abbildung 13:
Abb. 13 – Quantitative Bestimmung der GABAA -Rezeptordichte in Striatum und NucleusAccumbens mittels Rezeptorautoradiographie bei Ratten. Die spezifische Bindung in fmol/mgProteinmasse wurde mit [3H]-Muscimol in 4 Gruppen von jeweils 10 Ratten bestimmt: AcbC-CH = Nucleus accumbens, chronische Hypoxie; AcbC–HI = Nucleus accumbens, kombinierteHypoxie/Ischämie; CPu-CH = Striatum, chronische Hypoxie; CPu-HI = Striatum, kombinierteHypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- und Kontrolltieren sinddurch Sterne angegeben: * = p ≤ 0,05; ** = p ≤ 0,01 (Mann-Whitney U-Test). Abk.: Kontr =Kontrolltiere; Versuch = Versuchstiere.
Striatum (CPu) + Nucleus Accumbens (AcbC)
0
2000
4000
6000
8000
10000
12000
14000
AcbC - CH AcbC - HI CPu - CH CPu - HI
fmo
l / m
g P
rote
in
Kontr
Versuch
** *
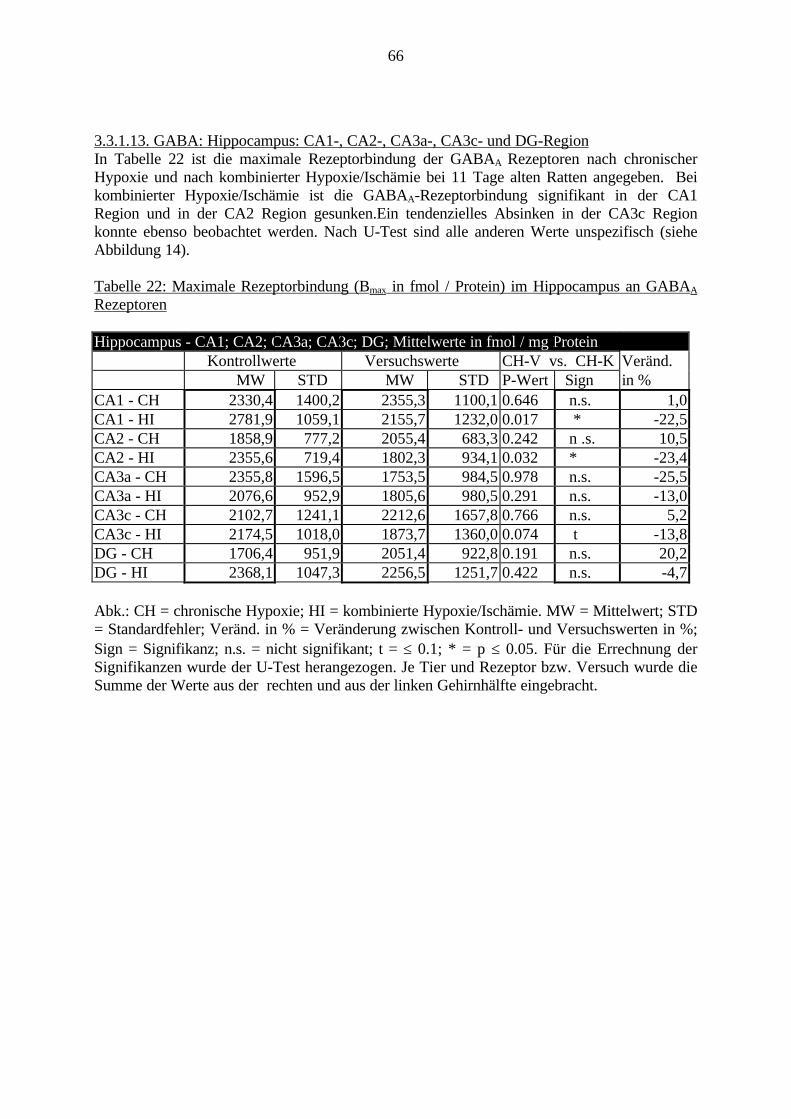
66
3.3.1.13. GABA: Hippocampus: CA1-, CA2-, CA3a-, CA3c- und DG-RegionIn Tabelle 22 ist die maximale Rezeptorbindung der GABAA Rezeptoren nach chronischerHypoxie und nach kombinierter Hypoxie/Ischämie bei 11 Tage alten Ratten angegeben. Beikombinierter Hypoxie/Ischämie ist die GABAA-Rezeptorbindung signifikant in der CA1Region und in der CA2 Region gesunken.Ein tendenzielles Absinken in der CA3c Regionkonnte ebenso beobachtet werden. Nach U-Test sind alle anderen Werte unspezifisch (sieheAbbildung 14).
Tabelle 22: Maximale Rezeptorbindung (Bmax in fmol / Protein) im Hippocampus an GABAA
Rezeptoren
Hippocampus - CA1; CA2; CA3a; CA3c; DG; Mittelwerte in fmol / mg Protein Kontrollwerte Versuchswerte CH-V vs. CH-K Veränd. MW STD MW STD P-Wert Sign in %
CA1 - CH 2330,4 1400,2 2355,3 1100,1 0.646 n.s. 1,0CA1 - HI 2781,9 1059,1 2155,7 1232,0 0.017 * -22,5CA2 - CH 1858,9 777,2 2055,4 683,3 0.242 n .s. 10,5CA2 - HI 2355,6 719,4 1802,3 934,1 0.032 * -23,4CA3a - CH 2355,8 1596,5 1753,5 984,5 0.978 n.s. -25,5CA3a - HI 2076,6 952,9 1805,6 980,5 0.291 n.s. -13,0CA3c - CH 2102,7 1241,1 2212,6 1657,8 0.766 n.s. 5,2CA3c - HI 2174,5 1018,0 1873,7 1360,0 0.074 t -13,8DG - CH 1706,4 951,9 2051,4 922,8 0.191 n.s. 20,2DG - HI 2368,1 1047,3 2256,5 1251,7 0.422 n.s. -4,7
Abk.: CH = chronische Hypoxie; HI = kombinierte Hypoxie/Ischämie. MW = Mittelwert; STD= Standardfehler; Veränd. in % = Veränderung zwischen Kontroll- und Versuchswerten in %;Sign = Signifikanz; n.s. = nicht signifikant; t = ≤ 0.1; * = p ≤ 0.05. Für die Errechnung derSignifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw. Versuch wurde dieSumme der Werte aus der rechten und aus der linken Gehirnhälfte eingebracht.
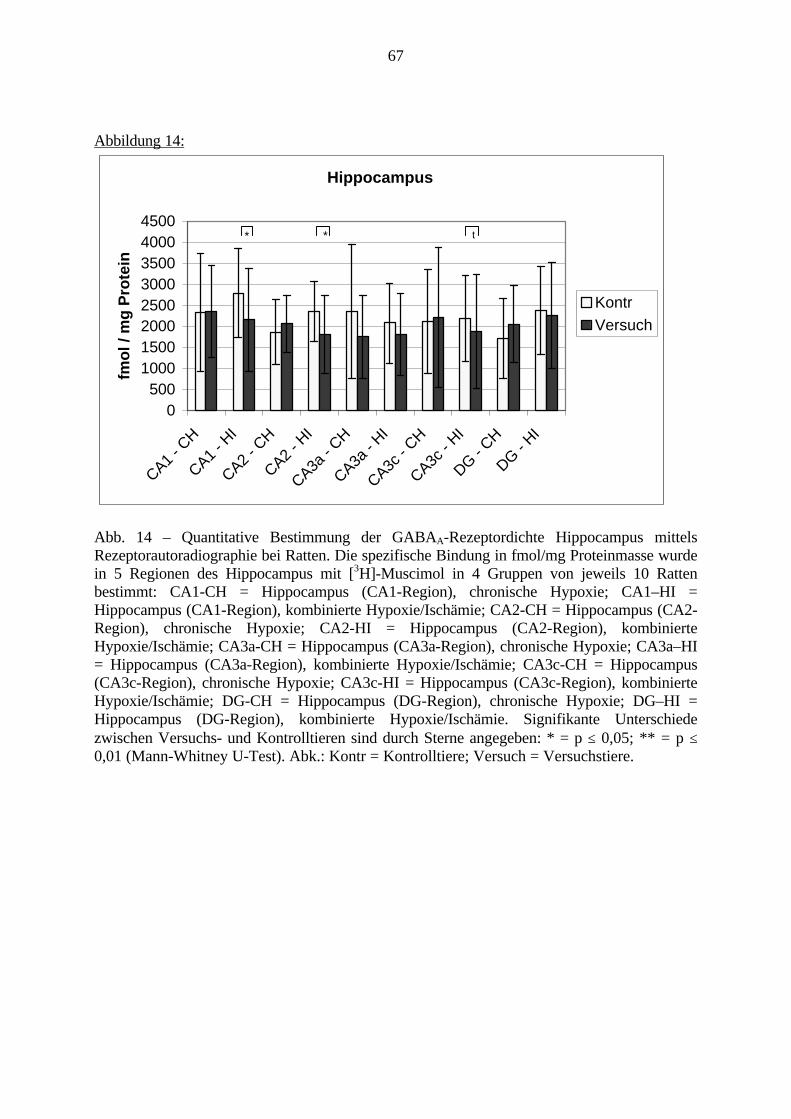
67
Abbildung 14:
Abb. 14 – Quantitative Bestimmung der GABAA-Rezeptordichte Hippocampus mittelsRezeptorautoradiographie bei Ratten. Die spezifische Bindung in fmol/mg Proteinmasse wurdein 5 Regionen des Hippocampus mit [3H]-Muscimol in 4 Gruppen von jeweils 10 Rattenbestimmt: CA1-CH = Hippocampus (CA1-Region), chronische Hypoxie; CA1–HI =Hippocampus (CA1-Region), kombinierte Hypoxie/Ischämie; CA2-CH = Hippocampus (CA2-Region), chronische Hypoxie; CA2-HI = Hippocampus (CA2-Region), kombinierteHypoxie/Ischämie; CA3a-CH = Hippocampus (CA3a-Region), chronische Hypoxie; CA3a–HI= Hippocampus (CA3a-Region), kombinierte Hypoxie/Ischämie; CA3c-CH = Hippocampus(CA3c-Region), chronische Hypoxie; CA3c-HI = Hippocampus (CA3c-Region), kombinierteHypoxie/Ischämie; DG-CH = Hippocampus (DG-Region), chronische Hypoxie; DG–HI =Hippocampus (DG-Region), kombinierte Hypoxie/Ischämie. Signifikante Unterschiedezwischen Versuchs- und Kontrolltieren sind durch Sterne angegeben: * = p ≤ 0,05; ** = p ≤0,01 (Mann-Whitney U-Test). Abk.: Kontr = Kontrolltiere; Versuch = Versuchstiere.
Hippocampus
0500
10001500200025003000350040004500
CA1 - C
H
CA1 - H
I
CA2 - C
H
CA2 - H
I
CA3a -
CH
CA3a -
HI
CA3c -
CH
CA3c -
HI
DG - CH
DG - HI
fmo
l / m
g P
rote
in
Kontr
Versuch
* * t
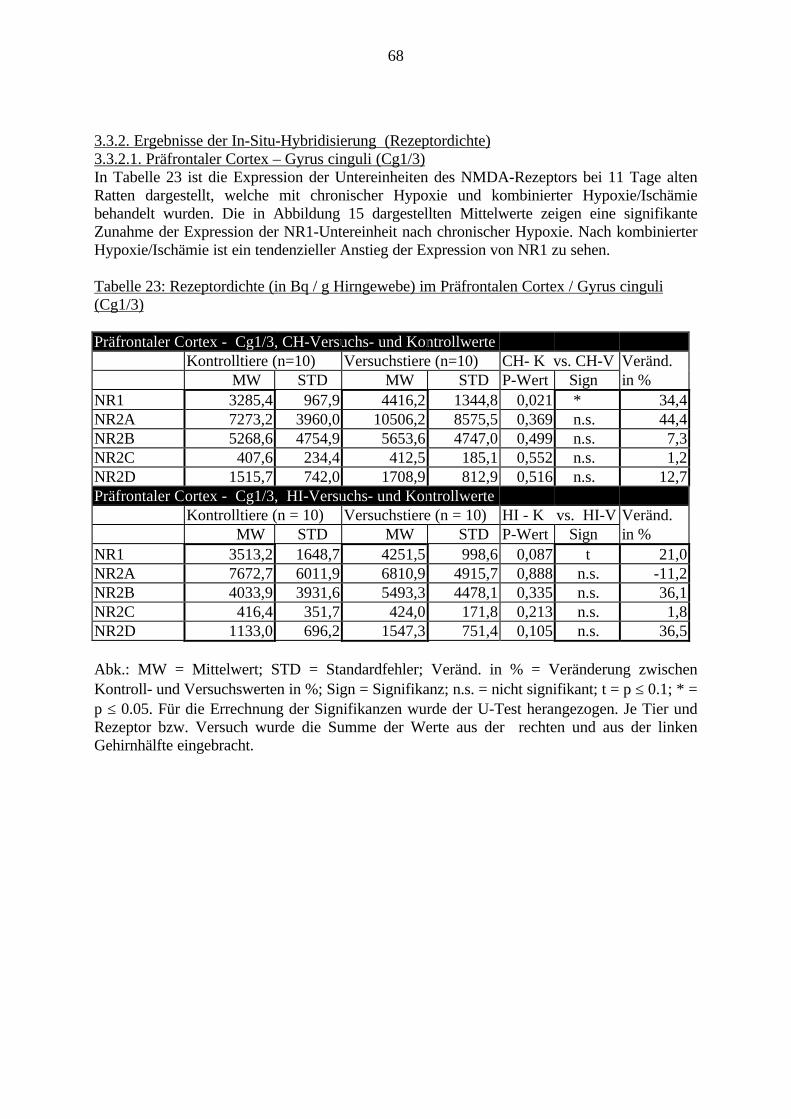
68
3.3.2. Ergebnisse der In-Situ-Hybridisierung (Rezeptordichte)3.3.2.1. Präfrontaler Cortex – Gyrus cinguli (Cg1/3)In Tabelle 23 ist die Expression der Untereinheiten des NMDA-Rezeptors bei 11 Tage altenRatten dargestellt, welche mit chronischer Hypoxie und kombinierter Hypoxie/Ischämiebehandelt wurden. Die in Abbildung 15 dargestellten Mittelwerte zeigen eine signifikanteZunahme der Expression der NR1-Untereinheit nach chronischer Hypoxie. Nach kombinierterHypoxie/Ischämie ist ein tendenzieller Anstieg der Expression von NR1 zu sehen.
Tabelle 23: Rezeptordichte (in Bq / g Hirngewebe) im Präfrontalen Cortex / Gyrus cinguli(Cg1/3)
Präfrontaler Cortex - Cg1/3, CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH- K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 3285,4 967,9 4416,2 1344,8 0,021 * 34,4NR2A 7273,2 3960,0 10506,2 8575,5 0,369 n.s. 44,4NR2B 5268,6 4754,9 5653,6 4747,0 0,499 n.s. 7,3NR2C 407,6 234,4 412,5 185,1 0,552 n.s. 1,2NR2D 1515,7 742,0 1708,9 812,9 0,516 n.s. 12,7Präfrontaler Cortex - Cg1/3, HI-Versuchs- und Kontrollwerte
Kontrolltiere (n = 10) Versuchstiere (n = 10) HI - K vs. HI-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 3513,2 1648,7 4251,5 998,6 0,087 t 21,0NR2A 7672,7 6011,9 6810,9 4915,7 0,888 n.s. -11,2NR2B 4033,9 3931,6 5493,3 4478,1 0,335 n.s. 36,1NR2C 416,4 351,7 424,0 171,8 0,213 n.s. 1,8NR2D 1133,0 696,2 1547,3 751,4 0,105 n.s. 36,5
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1; * =p ≤ 0.05. Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier undRezeptor bzw. Versuch wurde die Summe der Werte aus der rechten und aus der linkenGehirnhälfte eingebracht.
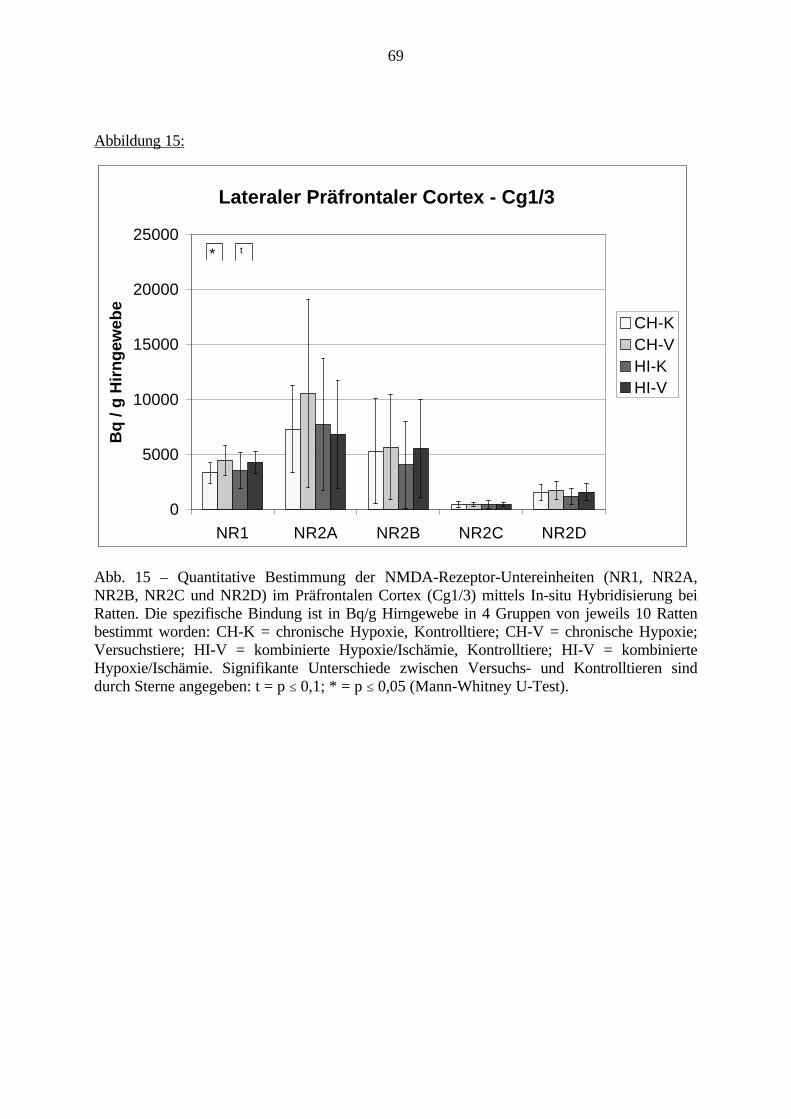
69
Abbildung 15:
Abb. 15 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) im Präfrontalen Cortex (Cg1/3) mittels In-situ Hybridisierung beiRatten. Die spezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen von jeweils 10 Rattenbestimmt worden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie;Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V = kombinierteHypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- und Kontrolltieren sinddurch Sterne angegeben: t = p ≤ 0,1; * = p ≤ 0,05 (Mann-Whitney U-Test).
Lateraler Präfrontaler Cortex - Cg1/3
0
5000
10000
15000
20000
25000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-KCH-VHI-KHI-V
* t
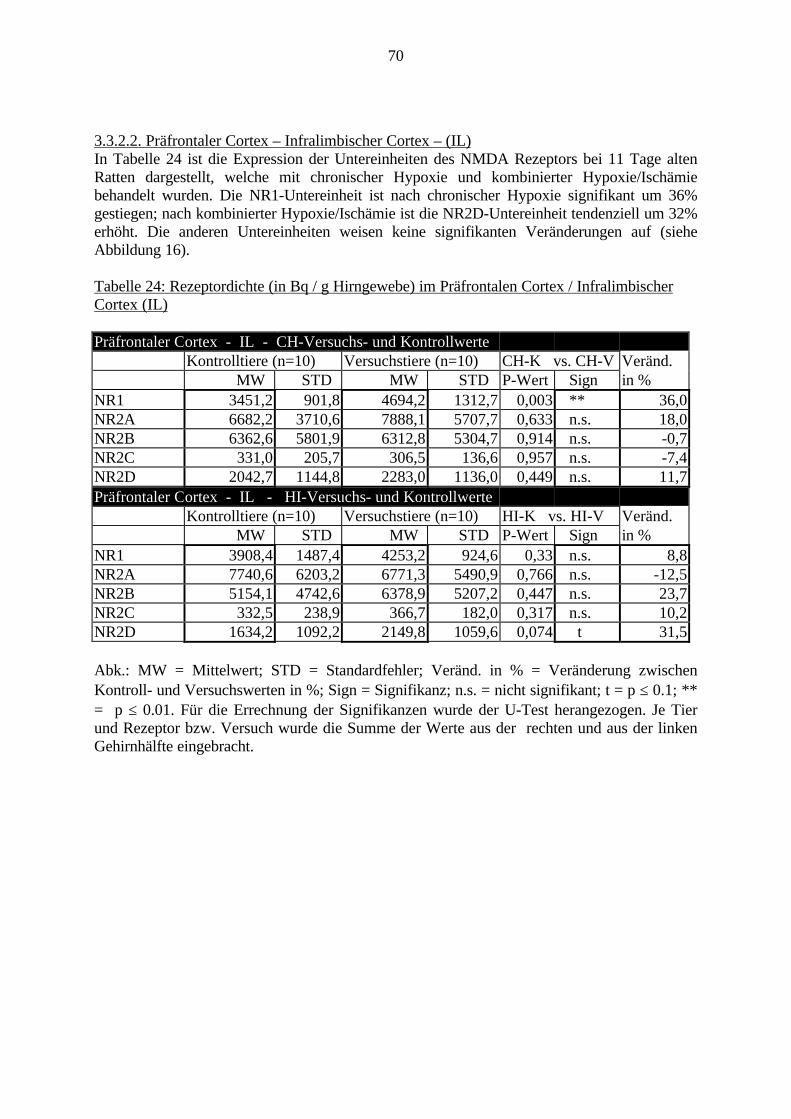
70
3.3.2.2. Präfrontaler Cortex – Infralimbischer Cortex – (IL)In Tabelle 24 ist die Expression der Untereinheiten des NMDA Rezeptors bei 11 Tage altenRatten dargestellt, welche mit chronischer Hypoxie und kombinierter Hypoxie/Ischämiebehandelt wurden. Die NR1-Untereinheit ist nach chronischer Hypoxie signifikant um 36%gestiegen; nach kombinierter Hypoxie/Ischämie ist die NR2D-Untereinheit tendenziell um 32%erhöht. Die anderen Untereinheiten weisen keine signifikanten Veränderungen auf (sieheAbbildung 16).
Tabelle 24: Rezeptordichte (in Bq / g Hirngewebe) im Präfrontalen Cortex / InfralimbischerCortex (IL)
Präfrontaler Cortex - IL - CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH-K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 3451,2 901,8 4694,2 1312,7 0,003 ** 36,0NR2A 6682,2 3710,6 7888,1 5707,7 0,633 n.s. 18,0NR2B 6362,6 5801,9 6312,8 5304,7 0,914 n.s. -0,7NR2C 331,0 205,7 306,5 136,6 0,957 n.s. -7,4NR2D 2042,7 1144,8 2283,0 1136,0 0,449 n.s. 11,7Präfrontaler Cortex - IL - HI-Versuchs- und Kontrollwerte
Kontrolltiere (n=10) Versuchstiere (n=10) HI-K vs. HI-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 3908,4 1487,4 4253,2 924,6 0,33 n.s. 8,8NR2A 7740,6 6203,2 6771,3 5490,9 0,766 n.s. -12,5NR2B 5154,1 4742,6 6378,9 5207,2 0,447 n.s. 23,7NR2C 332,5 238,9 366,7 182,0 0,317 n.s. 10,2NR2D 1634,2 1092,2 2149,8 1059,6 0,074 t 31,5
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1; **= p ≤ 0.01. Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tierund Rezeptor bzw. Versuch wurde die Summe der Werte aus der rechten und aus der linkenGehirnhälfte eingebracht.
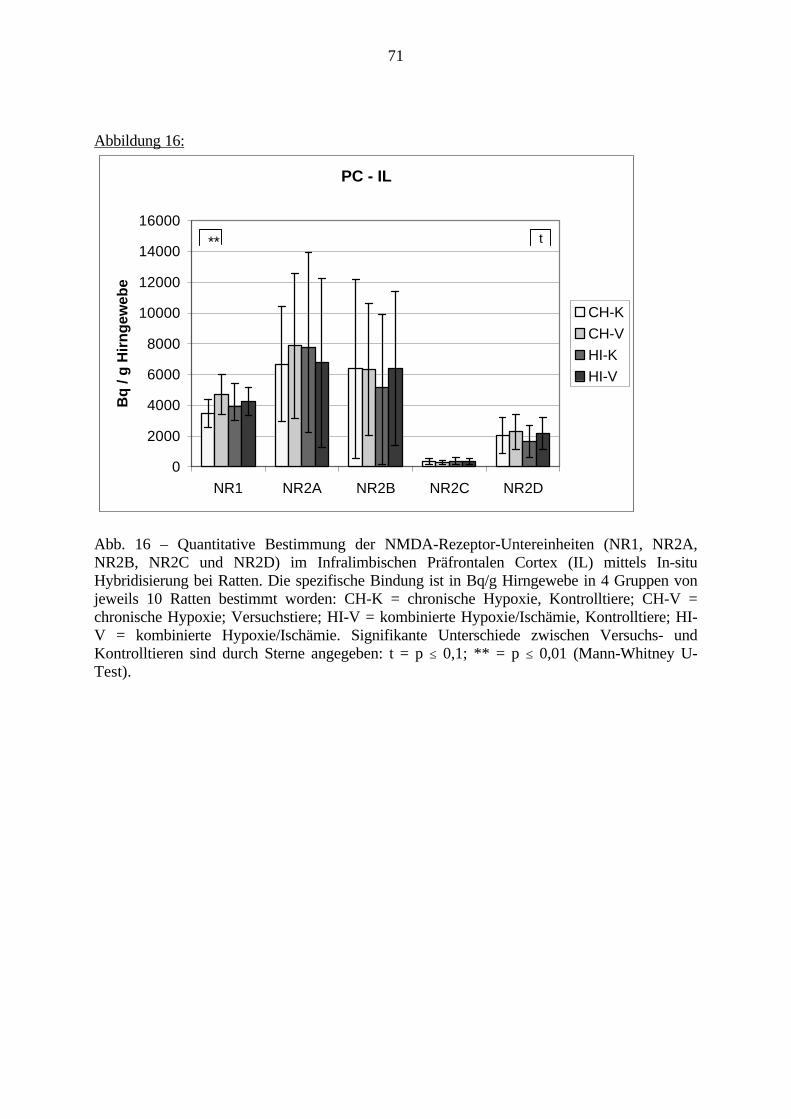
71
Abbildung 16:
Abb. 16 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) im Infralimbischen Präfrontalen Cortex (IL) mittels In-situHybridisierung bei Ratten. Die spezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen vonjeweils 10 Ratten bestimmt worden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V =chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V = kombinierte Hypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- undKontrolltieren sind durch Sterne angegeben: t = p ≤ 0,1; ** = p ≤ 0,01 (Mann-Whitney U-Test).
PC - IL
0
2000
4000
6000
8000
10000
12000
14000
16000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-K
CH-V
HI-K
HI-V
** t
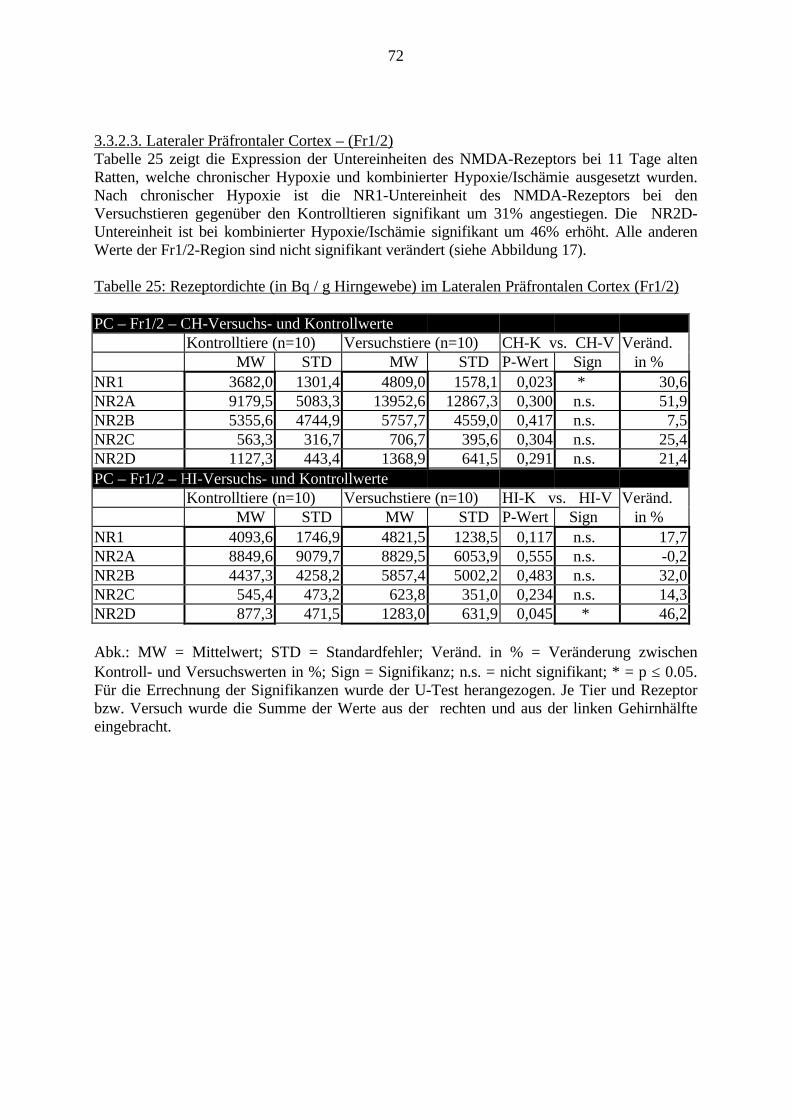
72
3.3.2.3. Lateraler Präfrontaler Cortex – (Fr1/2)Tabelle 25 zeigt die Expression der Untereinheiten des NMDA-Rezeptors bei 11 Tage altenRatten, welche chronischer Hypoxie und kombinierter Hypoxie/Ischämie ausgesetzt wurden.Nach chronischer Hypoxie ist die NR1-Untereinheit des NMDA-Rezeptors bei denVersuchstieren gegenüber den Kontrolltieren signifikant um 31% angestiegen. Die NR2D-Untereinheit ist bei kombinierter Hypoxie/Ischämie signifikant um 46% erhöht. Alle anderenWerte der Fr1/2-Region sind nicht signifikant verändert (siehe Abbildung 17).
Tabelle 25: Rezeptordichte (in Bq / g Hirngewebe) im Lateralen Präfrontalen Cortex (Fr1/2)
PC – Fr1/2 – CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH-K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 3682,0 1301,4 4809,0 1578,1 0,023 * 30,6NR2A 9179,5 5083,3 13952,6 12867,3 0,300 n.s. 51,9NR2B 5355,6 4744,9 5757,7 4559,0 0,417 n.s. 7,5NR2C 563,3 316,7 706,7 395,6 0,304 n.s. 25,4NR2D 1127,3 443,4 1368,9 641,5 0,291 n.s. 21,4PC – Fr1/2 – HI-Versuchs- und Kontrollwerte
Kontrolltiere (n=10) Versuchstiere (n=10) HI-K vs. HI-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 4093,6 1746,9 4821,5 1238,5 0,117 n.s. 17,7NR2A 8849,6 9079,7 8829,5 6053,9 0,555 n.s. -0,2NR2B 4437,3 4258,2 5857,4 5002,2 0,483 n.s. 32,0NR2C 545,4 473,2 623,8 351,0 0,234 n.s. 14,3NR2D 877,3 471,5 1283,0 631,9 0,045 * 46,2
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; * = p ≤ 0.05.Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptorbzw. Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
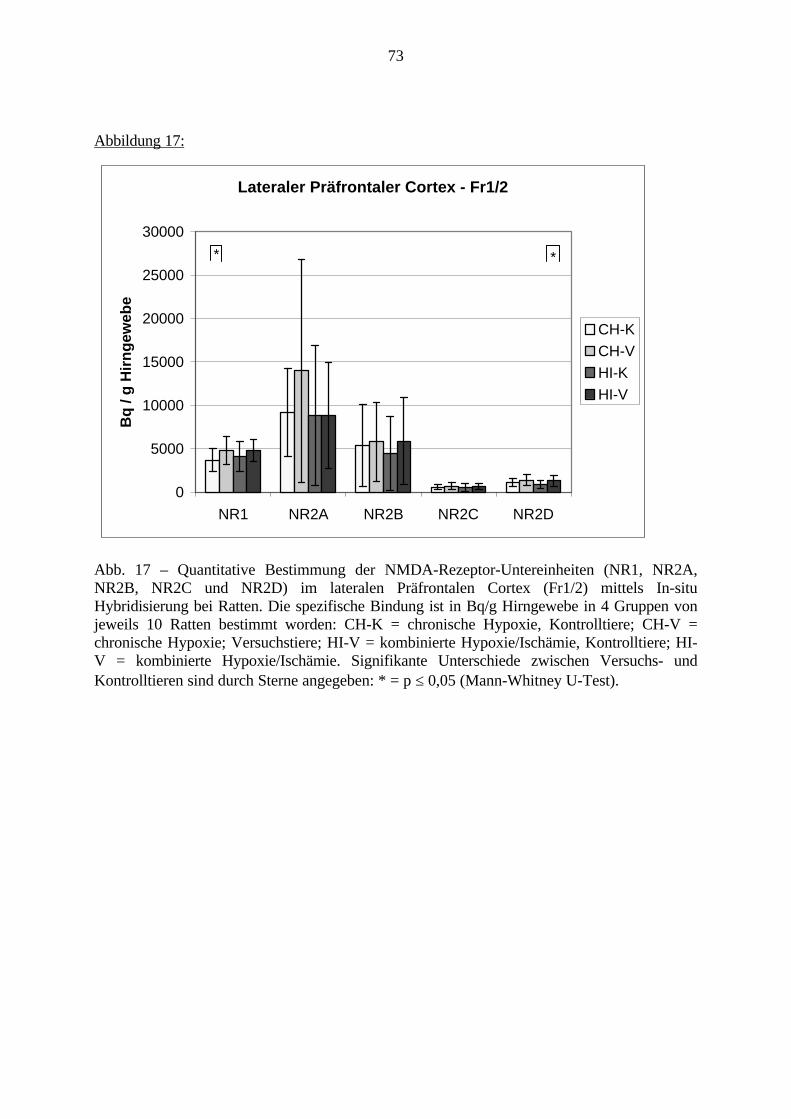
73
Abbildung 17:
Abb. 17 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) im lateralen Präfrontalen Cortex (Fr1/2) mittels In-situHybridisierung bei Ratten. Die spezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen vonjeweils 10 Ratten bestimmt worden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V =chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V = kombinierte Hypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- undKontrolltieren sind durch Sterne angegeben: * = p ≤ 0,05 (Mann-Whitney U-Test).
Lateraler Präfrontaler Cortex - Fr1/2
0
5000
10000
15000
20000
25000
30000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-K
CH-V
HI-K
HI-V
* *
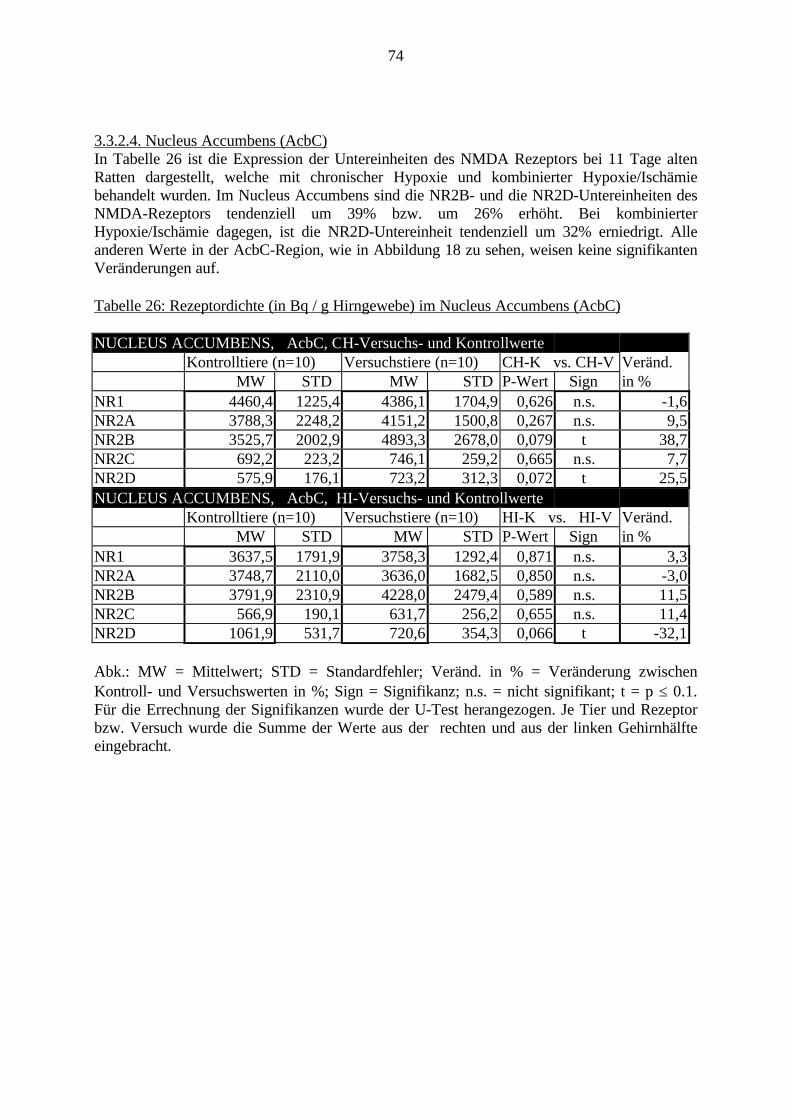
74
3.3.2.4. Nucleus Accumbens (AcbC)In Tabelle 26 ist die Expression der Untereinheiten des NMDA Rezeptors bei 11 Tage altenRatten dargestellt, welche mit chronischer Hypoxie und kombinierter Hypoxie/Ischämiebehandelt wurden. Im Nucleus Accumbens sind die NR2B- und die NR2D-Untereinheiten desNMDA-Rezeptors tendenziell um 39% bzw. um 26% erhöht. Bei kombinierterHypoxie/Ischämie dagegen, ist die NR2D-Untereinheit tendenziell um 32% erniedrigt. Alleanderen Werte in der AcbC-Region, wie in Abbildung 18 zu sehen, weisen keine signifikantenVeränderungen auf.
Tabelle 26: Rezeptordichte (in Bq / g Hirngewebe) im Nucleus Accumbens (AcbC)
NUCLEUS ACCUMBENS, AcbC, CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH-K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 4460,4 1225,4 4386,1 1704,9 0,626 n.s. -1,6NR2A 3788,3 2248,2 4151,2 1500,8 0,267 n.s. 9,5NR2B 3525,7 2002,9 4893,3 2678,0 0,079 t 38,7NR2C 692,2 223,2 746,1 259,2 0,665 n.s. 7,7NR2D 575,9 176,1 723,2 312,3 0,072 t 25,5NUCLEUS ACCUMBENS, AcbC, HI-Versuchs- und Kontrollwerte
Kontrolltiere (n=10) Versuchstiere (n=10) HI-K vs. HI-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 3637,5 1791,9 3758,3 1292,4 0,871 n.s. 3,3NR2A 3748,7 2110,0 3636,0 1682,5 0,850 n.s. -3,0NR2B 3791,9 2310,9 4228,0 2479,4 0,589 n.s. 11,5NR2C 566,9 190,1 631,7 256,2 0,655 n.s. 11,4NR2D 1061,9 531,7 720,6 354,3 0,066 t -32,1
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1.Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptorbzw. Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
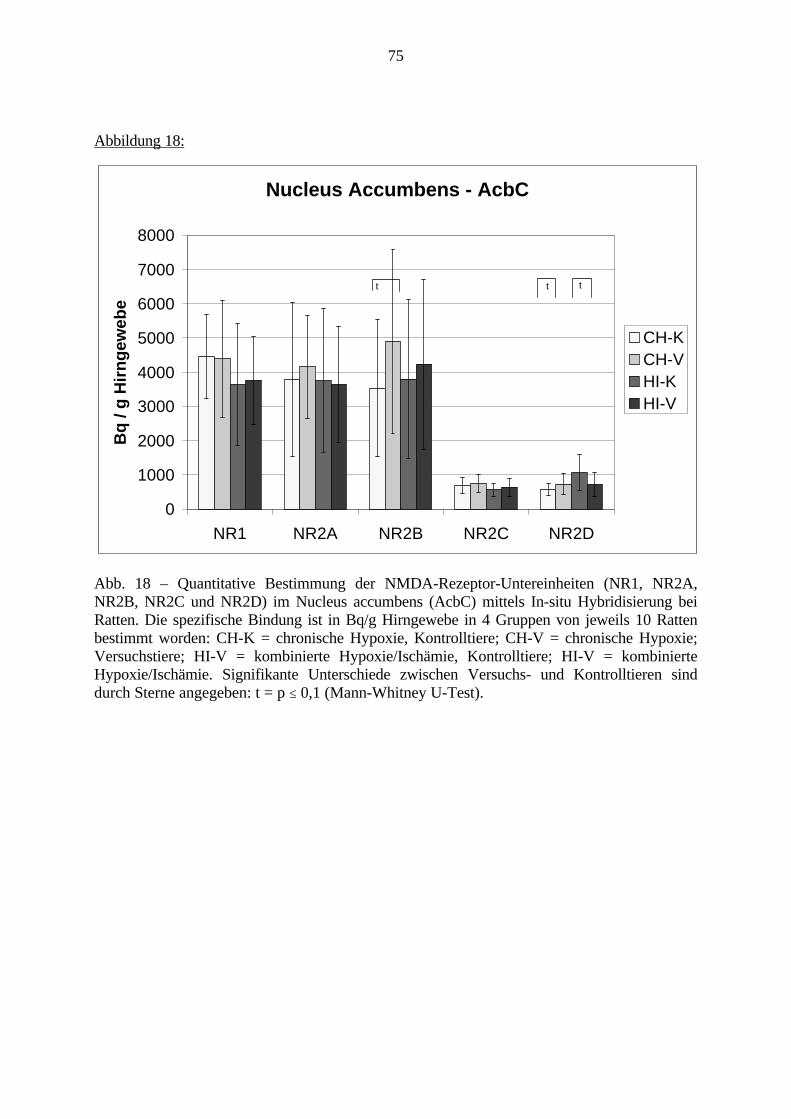
75
Abbildung 18:
Abb. 18 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) im Nucleus accumbens (AcbC) mittels In-situ Hybridisierung beiRatten. Die spezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen von jeweils 10 Rattenbestimmt worden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie;Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V = kombinierteHypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- und Kontrolltieren sinddurch Sterne angegeben: t = p ≤ 0,1 (Mann-Whitney U-Test).
Nucleus Accumbens - AcbC
0
1000
2000
3000
4000
5000
6000
7000
8000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-KCH-VHI-KHI-V
t t t
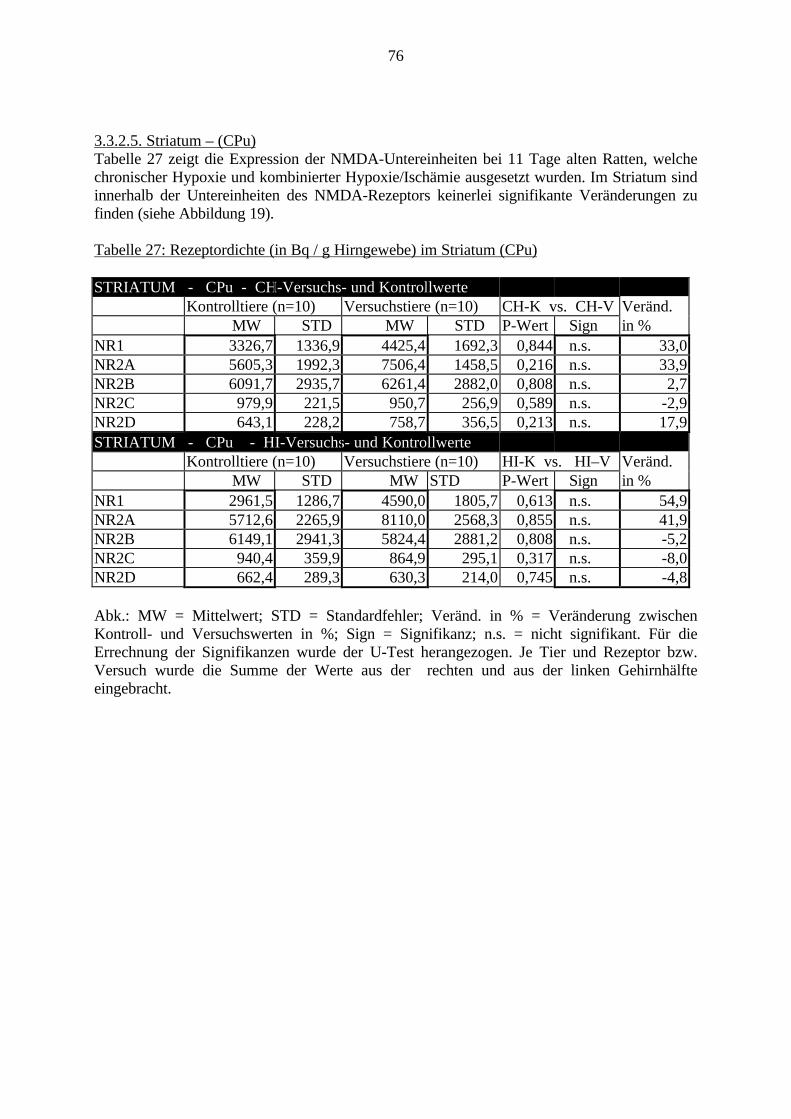
76
3.3.2.5. Striatum – (CPu)Tabelle 27 zeigt die Expression der NMDA-Untereinheiten bei 11 Tage alten Ratten, welchechronischer Hypoxie und kombinierter Hypoxie/Ischämie ausgesetzt wurden. Im Striatum sindinnerhalb der Untereinheiten des NMDA-Rezeptors keinerlei signifikante Veränderungen zufinden (siehe Abbildung 19).
Tabelle 27: Rezeptordichte (in Bq / g Hirngewebe) im Striatum (CPu)
STRIATUM - CPu - CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH-K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 3326,7 1336,9 4425,4 1692,3 0,844 n.s. 33,0NR2A 5605,3 1992,3 7506,4 1458,5 0,216 n.s. 33,9NR2B 6091,7 2935,7 6261,4 2882,0 0,808 n.s. 2,7NR2C 979,9 221,5 950,7 256,9 0,589 n.s. -2,9NR2D 643,1 228,2 758,7 356,5 0,213 n.s. 17,9STRIATUM - CPu - HI-Versuchs- und Kontrollwerte
Kontrolltiere (n=10) Versuchstiere (n=10) HI-K vs. HI–V Veränd. MW STD MW STD P-Wert Sign in %
NR1 2961,5 1286,7 4590,0 1805,7 0,613 n.s. 54,9NR2A 5712,6 2265,9 8110,0 2568,3 0,855 n.s. 41,9NR2B 6149,1 2941,3 5824,4 2881,2 0,808 n.s. -5,2NR2C 940,4 359,9 864,9 295,1 0,317 n.s. -8,0NR2D 662,4 289,3 630,3 214,0 0,745 n.s. -4,8
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant. Für dieErrechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptor bzw.Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
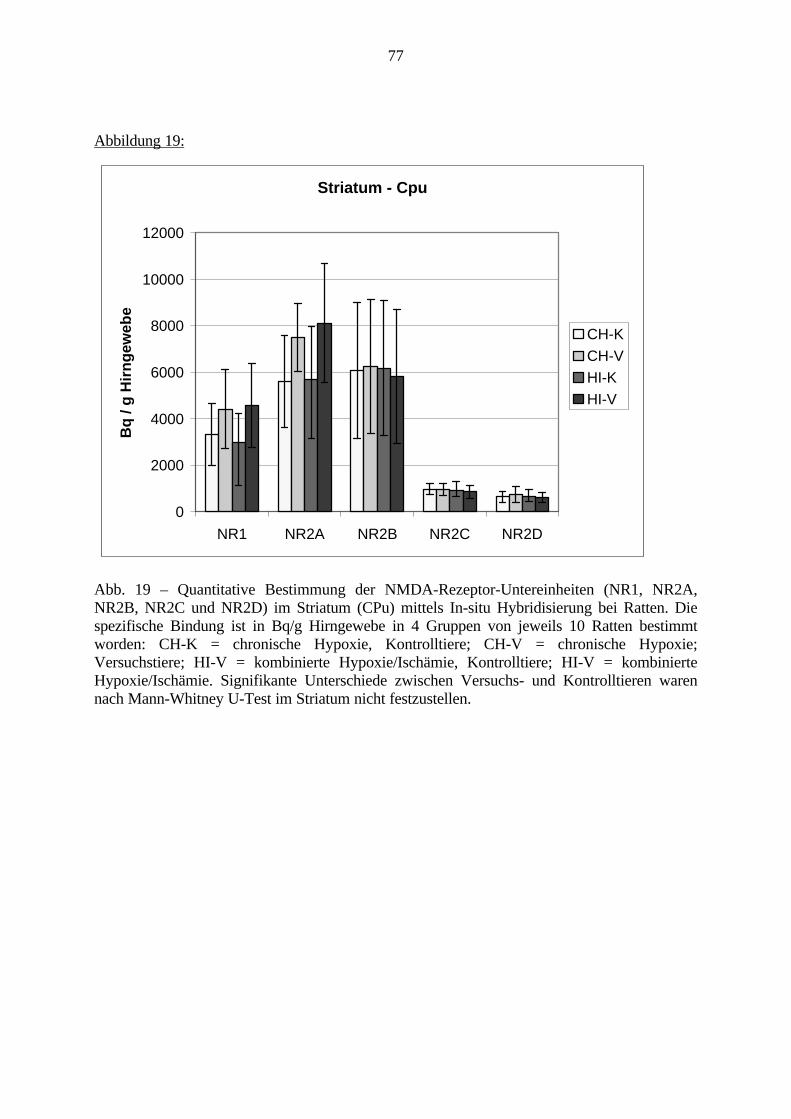
77
Abbildung 19:
Abb. 19 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) im Striatum (CPu) mittels In-situ Hybridisierung bei Ratten. Diespezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen von jeweils 10 Ratten bestimmtworden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronische Hypoxie;Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V = kombinierteHypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- und Kontrolltieren warennach Mann-Whitney U-Test im Striatum nicht festzustellen.
Striatum - Cpu
0
2000
4000
6000
8000
10000
12000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-K
CH-V
HI-K
HI-V
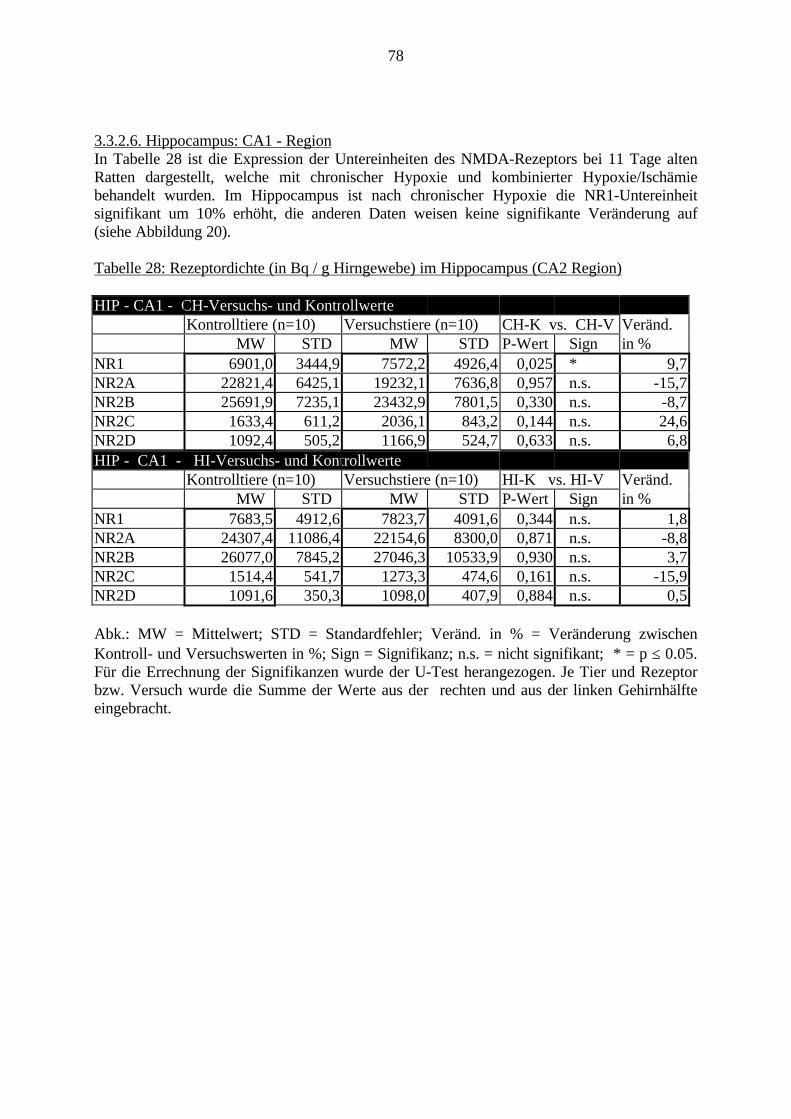
78
3.3.2.6. Hippocampus: CA1 - RegionIn Tabelle 28 ist die Expression der Untereinheiten des NMDA-Rezeptors bei 11 Tage altenRatten dargestellt, welche mit chronischer Hypoxie und kombinierter Hypoxie/Ischämiebehandelt wurden. Im Hippocampus ist nach chronischer Hypoxie die NR1-Untereinheitsignifikant um 10% erhöht, die anderen Daten weisen keine signifikante Veränderung auf(siehe Abbildung 20).
Tabelle 28: Rezeptordichte (in Bq / g Hirngewebe) im Hippocampus (CA2 Region)
HIP - CA1 - CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH-K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 6901,0 3444,9 7572,2 4926,4 0,025 * 9,7NR2A 22821,4 6425,1 19232,1 7636,8 0,957 n.s. -15,7NR2B 25691,9 7235,1 23432,9 7801,5 0,330 n.s. -8,7NR2C 1633,4 611,2 2036,1 843,2 0,144 n.s. 24,6NR2D 1092,4 505,2 1166,9 524,7 0,633 n.s. 6,8HIP - CA1 - HI-Versuchs- und Kontrollwerte
Kontrolltiere (n=10) Versuchstiere (n=10) HI-K vs. HI-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 7683,5 4912,6 7823,7 4091,6 0,344 n.s. 1,8NR2A 24307,4 11086,4 22154,6 8300,0 0,871 n.s. -8,8NR2B 26077,0 7845,2 27046,3 10533,9 0,930 n.s. 3,7NR2C 1514,4 541,7 1273,3 474,6 0,161 n.s. -15,9NR2D 1091,6 350,3 1098,0 407,9 0,884 n.s. 0,5
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; * = p ≤ 0.05.Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptorbzw. Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
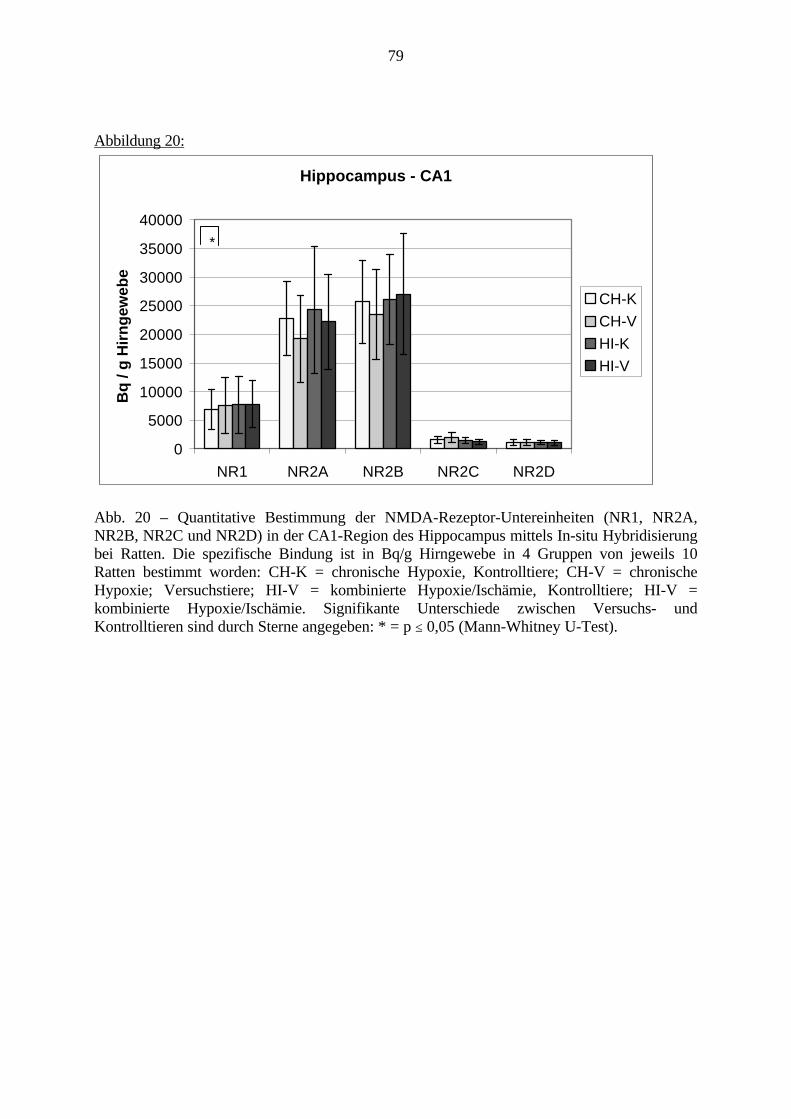
79
Abbildung 20:
Abb. 20 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) in der CA1-Region des Hippocampus mittels In-situ Hybridisierungbei Ratten. Die spezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen von jeweils 10Ratten bestimmt worden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronischeHypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V =kombinierte Hypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- undKontrolltieren sind durch Sterne angegeben: * = p ≤ 0,05 (Mann-Whitney U-Test).
Hippocampus - CA1
0
5000
10000
15000
20000
25000
30000
35000
40000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-K
CH-V
HI-K
HI-V
*
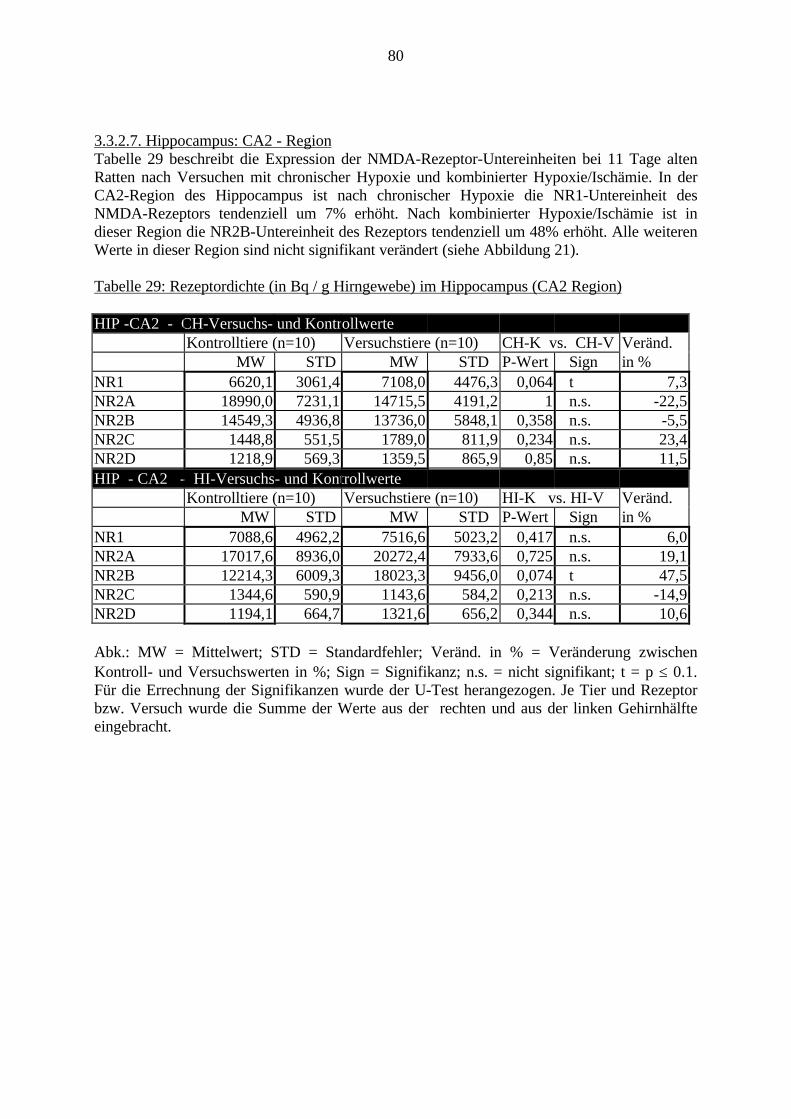
80
3.3.2.7. Hippocampus: CA2 - RegionTabelle 29 beschreibt die Expression der NMDA-Rezeptor-Untereinheiten bei 11 Tage altenRatten nach Versuchen mit chronischer Hypoxie und kombinierter Hypoxie/Ischämie. In derCA2-Region des Hippocampus ist nach chronischer Hypoxie die NR1-Untereinheit desNMDA-Rezeptors tendenziell um 7% erhöht. Nach kombinierter Hypoxie/Ischämie ist indieser Region die NR2B-Untereinheit des Rezeptors tendenziell um 48% erhöht. Alle weiterenWerte in dieser Region sind nicht signifikant verändert (siehe Abbildung 21).
Tabelle 29: Rezeptordichte (in Bq / g Hirngewebe) im Hippocampus (CA2 Region)
HIP -CA2 - CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH-K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 6620,1 3061,4 7108,0 4476,3 0,064 t 7,3NR2A 18990,0 7231,1 14715,5 4191,2 1 n.s. -22,5NR2B 14549,3 4936,8 13736,0 5848,1 0,358 n.s. -5,5NR2C 1448,8 551,5 1789,0 811,9 0,234 n.s. 23,4NR2D 1218,9 569,3 1359,5 865,9 0,85 n.s. 11,5HIP - CA2 - HI-Versuchs- und Kontrollwerte
Kontrolltiere (n=10) Versuchstiere (n=10) HI-K vs. HI-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 7088,6 4962,2 7516,6 5023,2 0,417 n.s. 6,0NR2A 17017,6 8936,0 20272,4 7933,6 0,725 n.s. 19,1NR2B 12214,3 6009,3 18023,3 9456,0 0,074 t 47,5NR2C 1344,6 590,9 1143,6 584,2 0,213 n.s. -14,9NR2D 1194,1 664,7 1321,6 656,2 0,344 n.s. 10,6
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1.Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptorbzw. Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
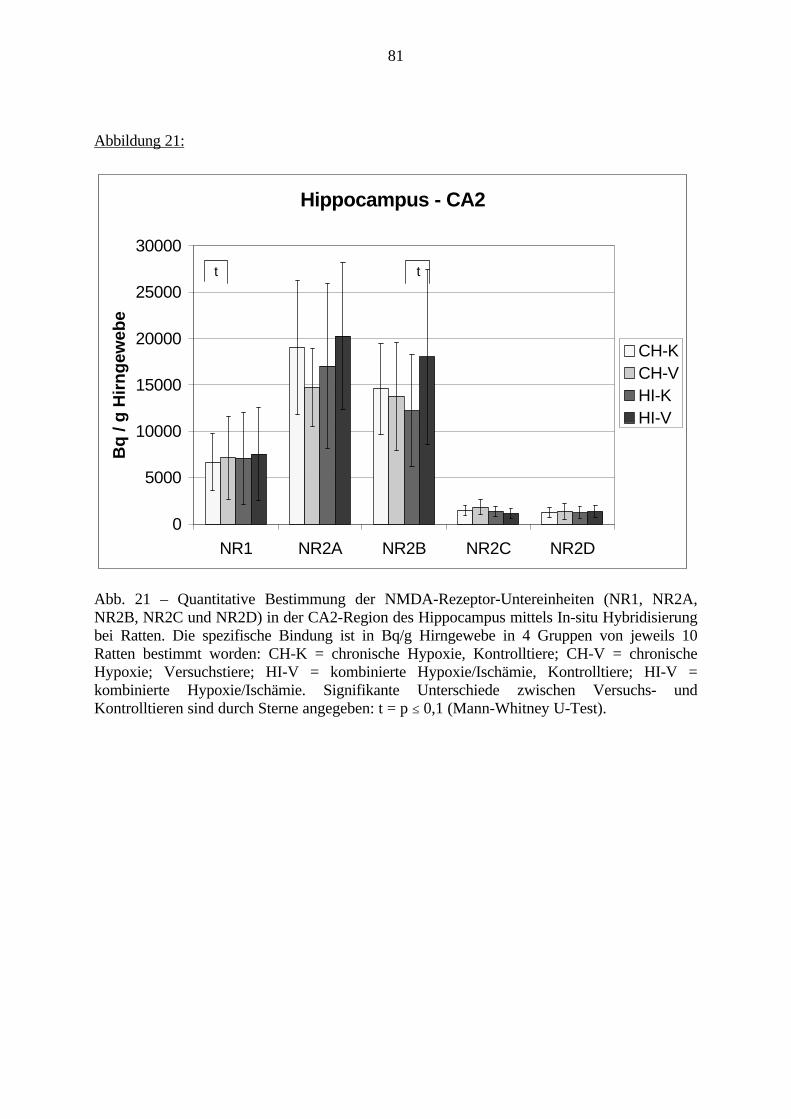
81
Abbildung 21:
Abb. 21 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) in der CA2-Region des Hippocampus mittels In-situ Hybridisierungbei Ratten. Die spezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen von jeweils 10Ratten bestimmt worden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronischeHypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V =kombinierte Hypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- undKontrolltieren sind durch Sterne angegeben: t = p ≤ 0,1 (Mann-Whitney U-Test).
Hippocampus - CA2
0
5000
10000
15000
20000
25000
30000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-KCH-VHI-KHI-V
t t
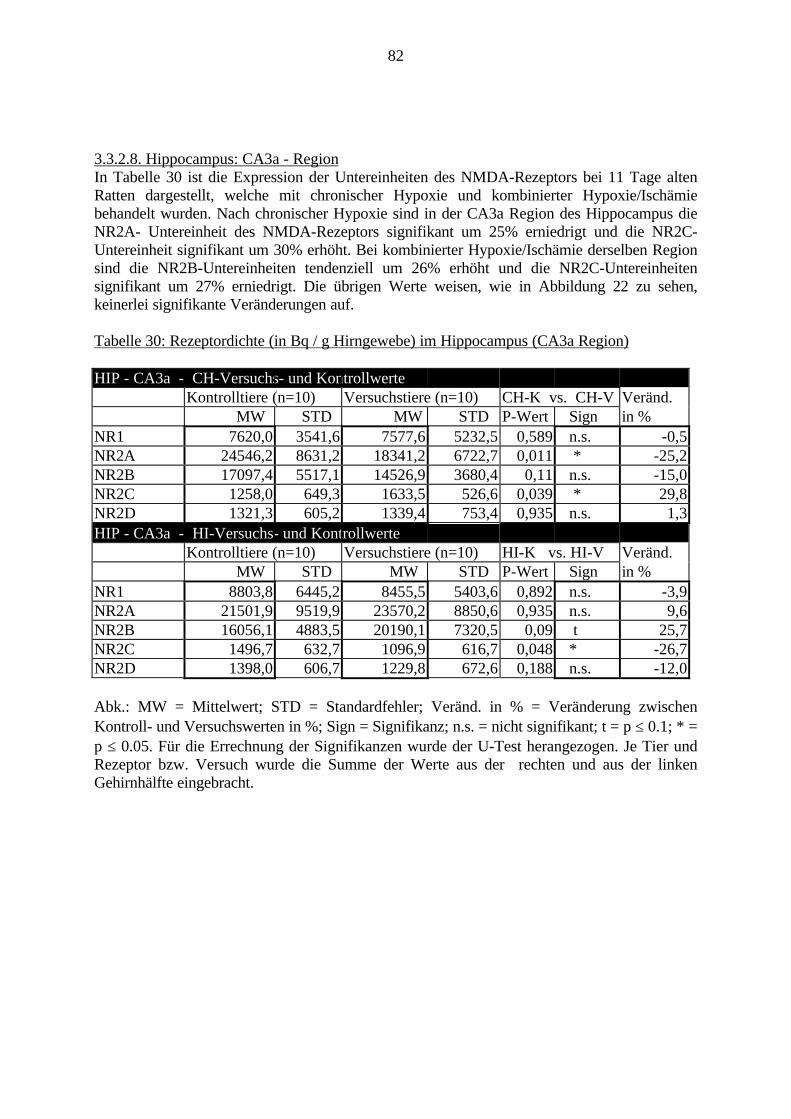
82
3.3.2.8. Hippocampus: CA3a - RegionIn Tabelle 30 ist die Expression der Untereinheiten des NMDA-Rezeptors bei 11 Tage altenRatten dargestellt, welche mit chronischer Hypoxie und kombinierter Hypoxie/Ischämiebehandelt wurden. Nach chronischer Hypoxie sind in der CA3a Region des Hippocampus dieNR2A- Untereinheit des NMDA-Rezeptors signifikant um 25% erniedrigt und die NR2C-Untereinheit signifikant um 30% erhöht. Bei kombinierter Hypoxie/Ischämie derselben Regionsind die NR2B-Untereinheiten tendenziell um 26% erhöht und die NR2C-Untereinheitensignifikant um 27% erniedrigt. Die übrigen Werte weisen, wie in Abbildung 22 zu sehen,keinerlei signifikante Veränderungen auf.
Tabelle 30: Rezeptordichte (in Bq / g Hirngewebe) im Hippocampus (CA3a Region)
HIP - CA3a - CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH-K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 7620,0 3541,6 7577,6 5232,5 0,589 n.s. -0,5NR2A 24546,2 8631,2 18341,2 6722,7 0,011 * -25,2NR2B 17097,4 5517,1 14526,9 3680,4 0,11 n.s. -15,0NR2C 1258,0 649,3 1633,5 526,6 0,039 * 29,8NR2D 1321,3 605,2 1339,4 753,4 0,935 n.s. 1,3HIP - CA3a - HI-Versuchs- und Kontrollwerte
Kontrolltiere (n=10) Versuchstiere (n=10) HI-K vs. HI-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 8803,8 6445,2 8455,5 5403,6 0,892 n.s. -3,9NR2A 21501,9 9519,9 23570,2 8850,6 0,935 n.s. 9,6NR2B 16056,1 4883,5 20190,1 7320,5 0,09 t 25,7NR2C 1496,7 632,7 1096,9 616,7 0,048 * -26,7NR2D 1398,0 606,7 1229,8 672,6 0,188 n.s. -12,0
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1; * =p ≤ 0.05. Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier undRezeptor bzw. Versuch wurde die Summe der Werte aus der rechten und aus der linkenGehirnhälfte eingebracht.
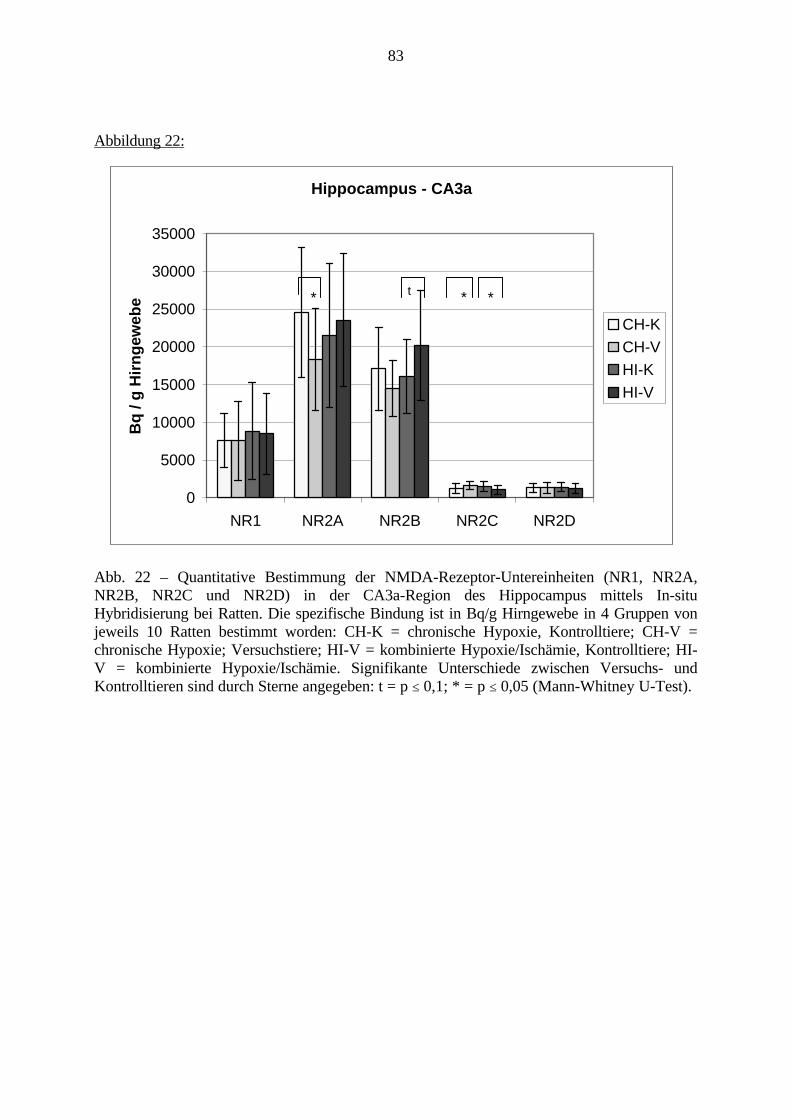
83
Abbildung 22:
Abb. 22 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) in der CA3a-Region des Hippocampus mittels In-situHybridisierung bei Ratten. Die spezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen vonjeweils 10 Ratten bestimmt worden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V =chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V = kombinierte Hypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- undKontrolltieren sind durch Sterne angegeben: t = p ≤ 0,1; * = p ≤ 0,05 (Mann-Whitney U-Test).
Hippocampus - CA3a
0
5000
10000
15000
20000
25000
30000
35000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-K
CH-V
HI-K
HI-V
* * *t
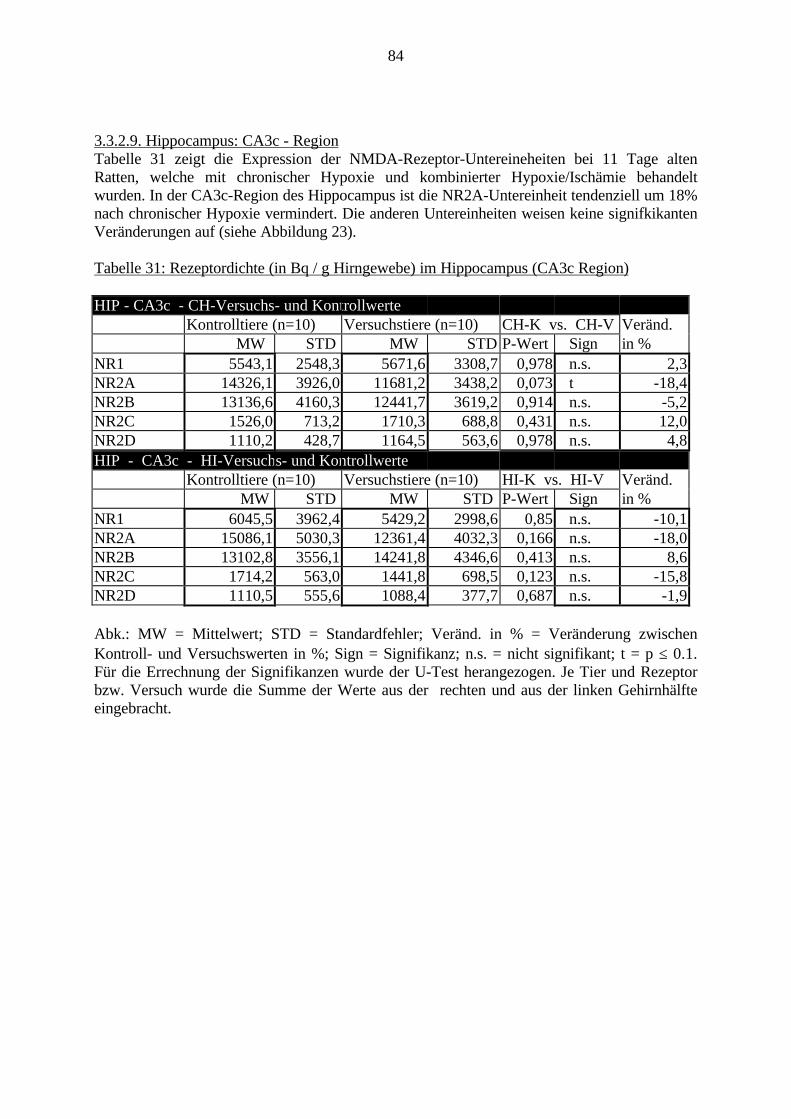
84
3.3.2.9. Hippocampus: CA3c - RegionTabelle 31 zeigt die Expression der NMDA-Rezeptor-Untereineheiten bei 11 Tage altenRatten, welche mit chronischer Hypoxie und kombinierter Hypoxie/Ischämie behandeltwurden. In der CA3c-Region des Hippocampus ist die NR2A-Untereinheit tendenziell um 18%nach chronischer Hypoxie vermindert. Die anderen Untereinheiten weisen keine signifkikantenVeränderungen auf (siehe Abbildung 23).
Tabelle 31: Rezeptordichte (in Bq / g Hirngewebe) im Hippocampus (CA3c Region)
HIP - CA3c - CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH-K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 5543,1 2548,3 5671,6 3308,7 0,978 n.s. 2,3NR2A 14326,1 3926,0 11681,2 3438,2 0,073 t -18,4NR2B 13136,6 4160,3 12441,7 3619,2 0,914 n.s. -5,2NR2C 1526,0 713,2 1710,3 688,8 0,431 n.s. 12,0NR2D 1110,2 428,7 1164,5 563,6 0,978 n.s. 4,8HIP - CA3c - HI-Versuchs- und Kontrollwerte
Kontrolltiere (n=10) Versuchstiere (n=10) HI-K vs. HI-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 6045,5 3962,4 5429,2 2998,6 0,85 n.s. -10,1NR2A 15086,1 5030,3 12361,4 4032,3 0,166 n.s. -18,0NR2B 13102,8 3556,1 14241,8 4346,6 0,413 n.s. 8,6NR2C 1714,2 563,0 1441,8 698,5 0,123 n.s. -15,8NR2D 1110,5 555,6 1088,4 377,7 0,687 n.s. -1,9
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1.Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptorbzw. Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
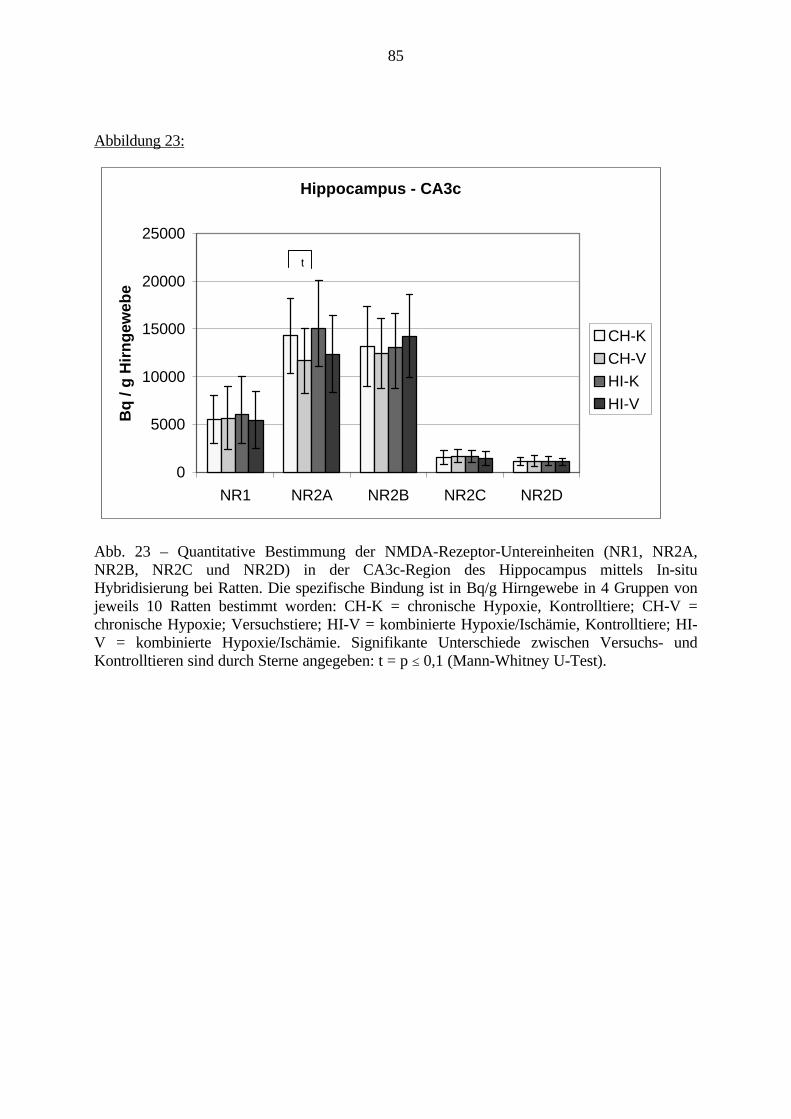
85
Abbildung 23:
Abb. 23 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) in der CA3c-Region des Hippocampus mittels In-situHybridisierung bei Ratten. Die spezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen vonjeweils 10 Ratten bestimmt worden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V =chronische Hypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V = kombinierte Hypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- undKontrolltieren sind durch Sterne angegeben: t = p ≤ 0,1 (Mann-Whitney U-Test).
Hippocampus - CA3c
0
5000
10000
15000
20000
25000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-K
CH-V
HI-K
HI-V
t
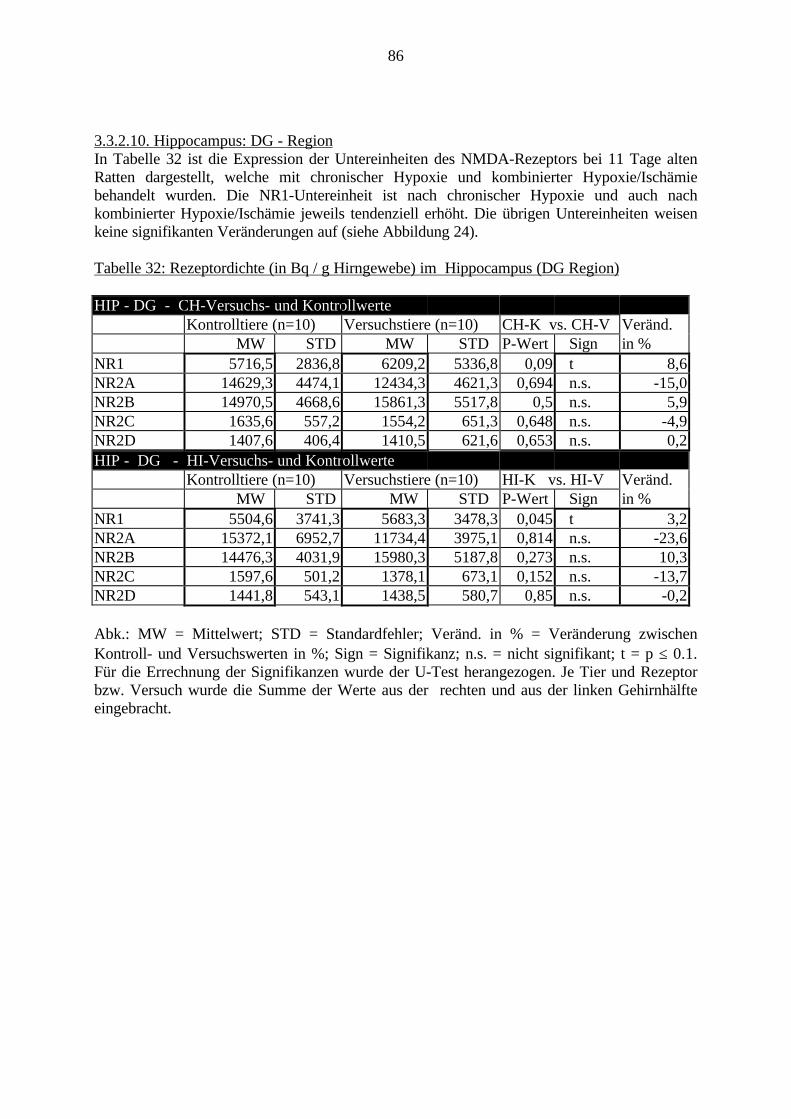
86
3.3.2.10. Hippocampus: DG - RegionIn Tabelle 32 ist die Expression der Untereinheiten des NMDA-Rezeptors bei 11 Tage altenRatten dargestellt, welche mit chronischer Hypoxie und kombinierter Hypoxie/Ischämiebehandelt wurden. Die NR1-Untereinheit ist nach chronischer Hypoxie und auch nachkombinierter Hypoxie/Ischämie jeweils tendenziell erhöht. Die übrigen Untereinheiten weisenkeine signifikanten Veränderungen auf (siehe Abbildung 24).
Tabelle 32: Rezeptordichte (in Bq / g Hirngewebe) im Hippocampus (DG Region)
HIP - DG - CH-Versuchs- und KontrollwerteKontrolltiere (n=10) Versuchstiere (n=10) CH-K vs. CH-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 5716,5 2836,8 6209,2 5336,8 0,09 t 8,6NR2A 14629,3 4474,1 12434,3 4621,3 0,694 n.s. -15,0NR2B 14970,5 4668,6 15861,3 5517,8 0,5 n.s. 5,9NR2C 1635,6 557,2 1554,2 651,3 0,648 n.s. -4,9NR2D 1407,6 406,4 1410,5 621,6 0,653 n.s. 0,2HIP - DG - HI-Versuchs- und Kontrollwerte
Kontrolltiere (n=10) Versuchstiere (n=10) HI-K vs. HI-V Veränd. MW STD MW STD P-Wert Sign in %
NR1 5504,6 3741,3 5683,3 3478,3 0,045 t 3,2NR2A 15372,1 6952,7 11734,4 3975,1 0,814 n.s. -23,6NR2B 14476,3 4031,9 15980,3 5187,8 0,273 n.s. 10,3NR2C 1597,6 501,2 1378,1 673,1 0,152 n.s. -13,7NR2D 1441,8 543,1 1438,5 580,7 0,85 n.s. -0,2
Abk.: MW = Mittelwert; STD = Standardfehler; Veränd. in % = Veränderung zwischenKontroll- und Versuchswerten in %; Sign = Signifikanz; n.s. = nicht signifikant; t = p ≤ 0.1.Für die Errechnung der Signifikanzen wurde der U-Test herangezogen. Je Tier und Rezeptorbzw. Versuch wurde die Summe der Werte aus der rechten und aus der linken Gehirnhälfteeingebracht.
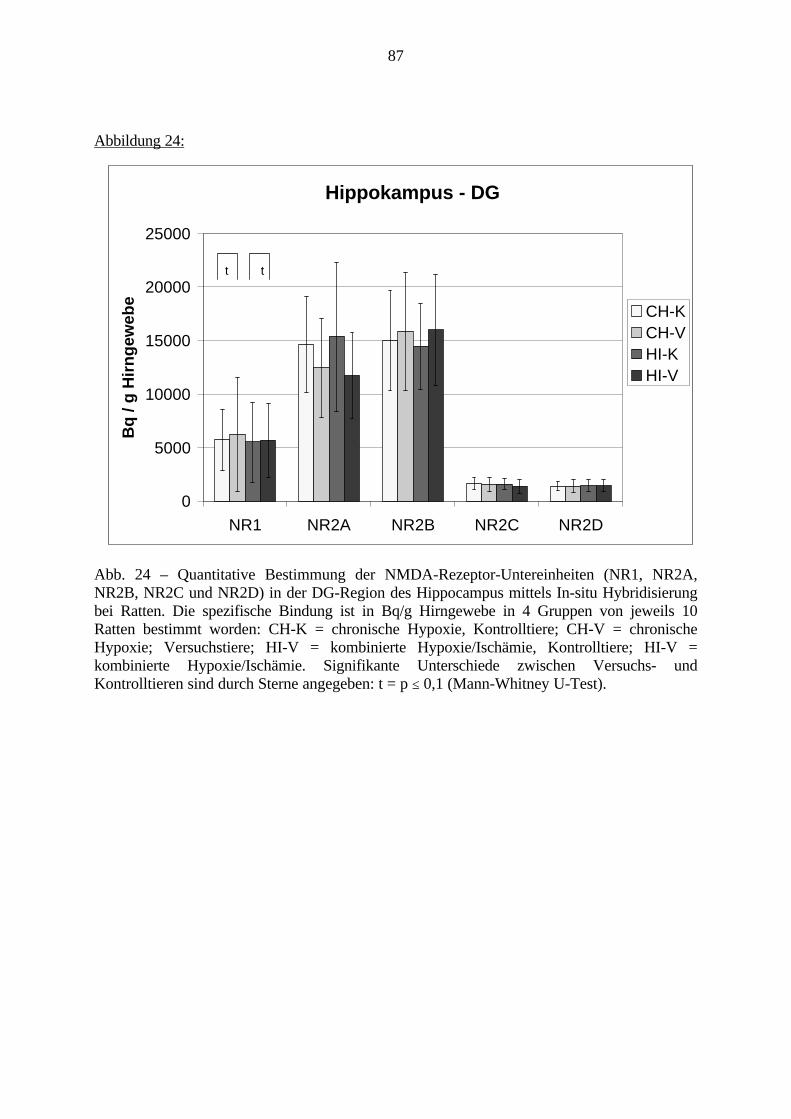
87
Abbildung 24:
Abb. 24 – Quantitative Bestimmung der NMDA-Rezeptor-Untereinheiten (NR1, NR2A,NR2B, NR2C und NR2D) in der DG-Region des Hippocampus mittels In-situ Hybridisierungbei Ratten. Die spezifische Bindung ist in Bq/g Hirngewebe in 4 Gruppen von jeweils 10Ratten bestimmt worden: CH-K = chronische Hypoxie, Kontrolltiere; CH-V = chronischeHypoxie; Versuchstiere; HI-V = kombinierte Hypoxie/Ischämie, Kontrolltiere; HI-V =kombinierte Hypoxie/Ischämie. Signifikante Unterschiede zwischen Versuchs- undKontrolltieren sind durch Sterne angegeben: t = p ≤ 0,1 (Mann-Whitney U-Test).
Hippokampus - DG
0
5000
10000
15000
20000
25000
NR1 NR2A NR2B NR2C NR2D
Bq
/ g
Hir
ng
eweb
e
CH-KCH-VHI-KHI-V
t t
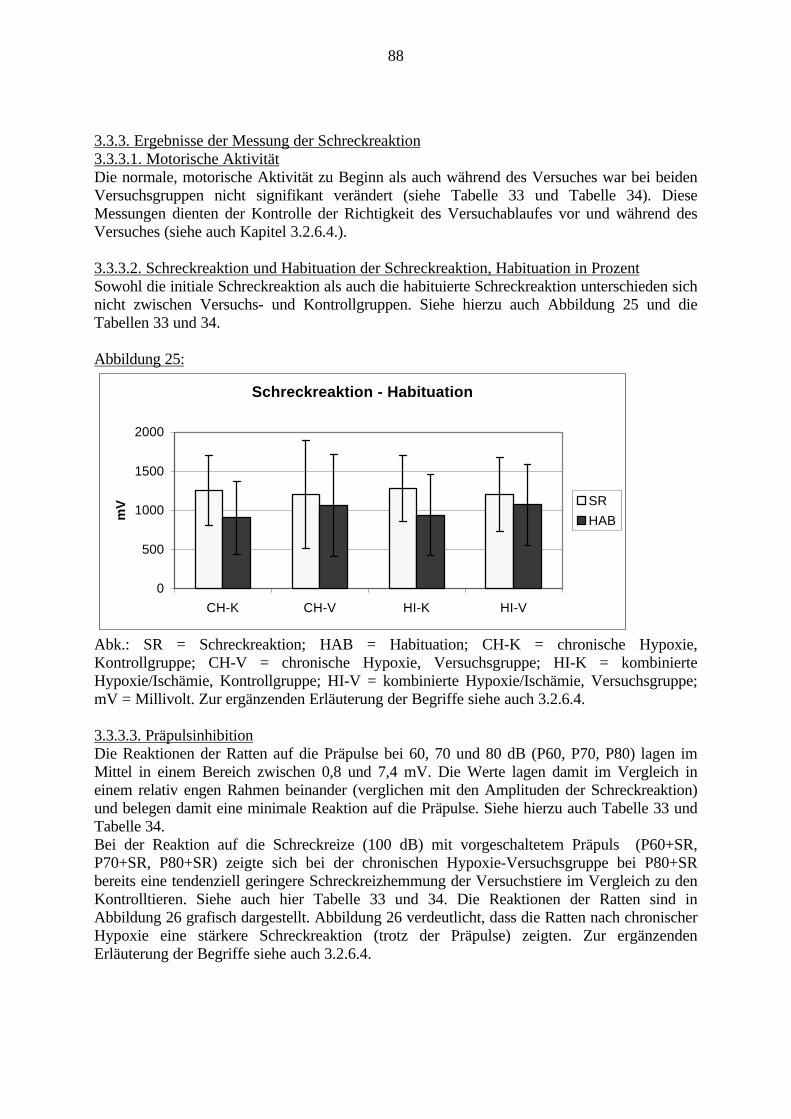
88
3.3.3. Ergebnisse der Messung der Schreckreaktion3.3.3.1. Motorische AktivitätDie normale, motorische Aktivität zu Beginn als auch während des Versuches war bei beidenVersuchsgruppen nicht signifikant verändert (siehe Tabelle 33 und Tabelle 34). DieseMessungen dienten der Kontrolle der Richtigkeit des Versuchablaufes vor und während desVersuches (siehe auch Kapitel 3.2.6.4.).
3.3.3.2. Schreckreaktion und Habituation der Schreckreaktion, Habituation in ProzentSowohl die initiale Schreckreaktion als auch die habituierte Schreckreaktion unterschieden sichnicht zwischen Versuchs- und Kontrollgruppen. Siehe hierzu auch Abbildung 25 und dieTabellen 33 und 34.
Abbildung 25:
Abk.: SR = Schreckreaktion; HAB = Habituation; CH-K = chronische Hypoxie,Kontrollgruppe; CH-V = chronische Hypoxie, Versuchsgruppe; HI-K = kombinierteHypoxie/Ischämie, Kontrollgruppe; HI-V = kombinierte Hypoxie/Ischämie, Versuchsgruppe;mV = Millivolt. Zur ergänzenden Erläuterung der Begriffe siehe auch 3.2.6.4.
3.3.3.3. PräpulsinhibitionDie Reaktionen der Ratten auf die Präpulse bei 60, 70 und 80 dB (P60, P70, P80) lagen imMittel in einem Bereich zwischen 0,8 und 7,4 mV. Die Werte lagen damit im Vergleich ineinem relativ engen Rahmen beinander (verglichen mit den Amplituden der Schreckreaktion)und belegen damit eine minimale Reaktion auf die Präpulse. Siehe hierzu auch Tabelle 33 undTabelle 34.Bei der Reaktion auf die Schreckreize (100 dB) mit vorgeschaltetem Präpuls (P60+SR,P70+SR, P80+SR) zeigte sich bei der chronischen Hypoxie-Versuchsgruppe bei P80+SRbereits eine tendenziell geringere Schreckreizhemmung der Versuchstiere im Vergleich zu denKontrolltieren. Siehe auch hier Tabelle 33 und 34. Die Reaktionen der Ratten sind inAbbildung 26 grafisch dargestellt. Abbildung 26 verdeutlicht, dass die Ratten nach chronischerHypoxie eine stärkere Schreckreaktion (trotz der Präpulse) zeigten. Zur ergänzendenErläuterung der Begriffe siehe auch 3.2.6.4.
Schreckreaktion - Habituation
0
500
1000
1500
2000
CH-K CH-V HI-K HI-V
mV SR
HAB
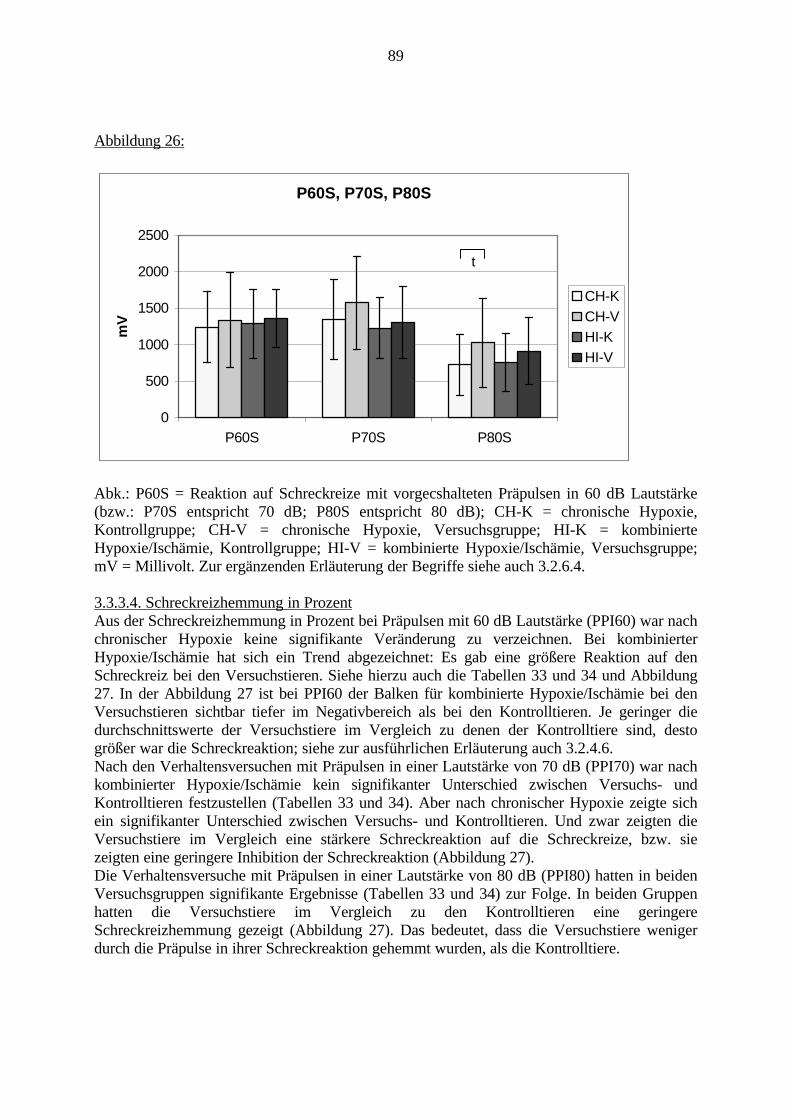
89
Abbildung 26:
Abk.: P60S = Reaktion auf Schreckreize mit vorgecshalteten Präpulsen in 60 dB Lautstärke(bzw.: P70S entspricht 70 dB; P80S entspricht 80 dB); CH-K = chronische Hypoxie,Kontrollgruppe; CH-V = chronische Hypoxie, Versuchsgruppe; HI-K = kombinierteHypoxie/Ischämie, Kontrollgruppe; HI-V = kombinierte Hypoxie/Ischämie, Versuchsgruppe;mV = Millivolt. Zur ergänzenden Erläuterung der Begriffe siehe auch 3.2.6.4.
3.3.3.4. Schreckreizhemmung in ProzentAus der Schreckreizhemmung in Prozent bei Präpulsen mit 60 dB Lautstärke (PPI60) war nachchronischer Hypoxie keine signifikante Veränderung zu verzeichnen. Bei kombinierterHypoxie/Ischämie hat sich ein Trend abgezeichnet: Es gab eine größere Reaktion auf denSchreckreiz bei den Versuchstieren. Siehe hierzu auch die Tabellen 33 und 34 und Abbildung27. In der Abbildung 27 ist bei PPI60 der Balken für kombinierte Hypoxie/Ischämie bei denVersuchstieren sichtbar tiefer im Negativbereich als bei den Kontrolltieren. Je geringer diedurchschnittswerte der Versuchstiere im Vergleich zu denen der Kontrolltiere sind, destogrößer war die Schreckreaktion; siehe zur ausführlichen Erläuterung auch 3.2.4.6.Nach den Verhaltensversuchen mit Präpulsen in einer Lautstärke von 70 dB (PPI70) war nachkombinierter Hypoxie/Ischämie kein signifikanter Unterschied zwischen Versuchs- undKontrolltieren festzustellen (Tabellen 33 und 34). Aber nach chronischer Hypoxie zeigte sichein signifikanter Unterschied zwischen Versuchs- und Kontrolltieren. Und zwar zeigten dieVersuchstiere im Vergleich eine stärkere Schreckreaktion auf die Schreckreize, bzw. siezeigten eine geringere Inhibition der Schreckreaktion (Abbildung 27).Die Verhaltensversuche mit Präpulsen in einer Lautstärke von 80 dB (PPI80) hatten in beidenVersuchsgruppen signifikante Ergebnisse (Tabellen 33 und 34) zur Folge. In beiden Gruppenhatten die Versuchstiere im Vergleich zu den Kontrolltieren eine geringereSchreckreizhemmung gezeigt (Abbildung 27). Das bedeutet, dass die Versuchstiere wenigerdurch die Präpulse in ihrer Schreckreaktion gehemmt wurden, als die Kontrolltiere.
P60S, P70S, P80S
0
500
1000
1500
2000
2500
P60S P70S P80S
mV
CH-KCH-VHI-KHI-V
t
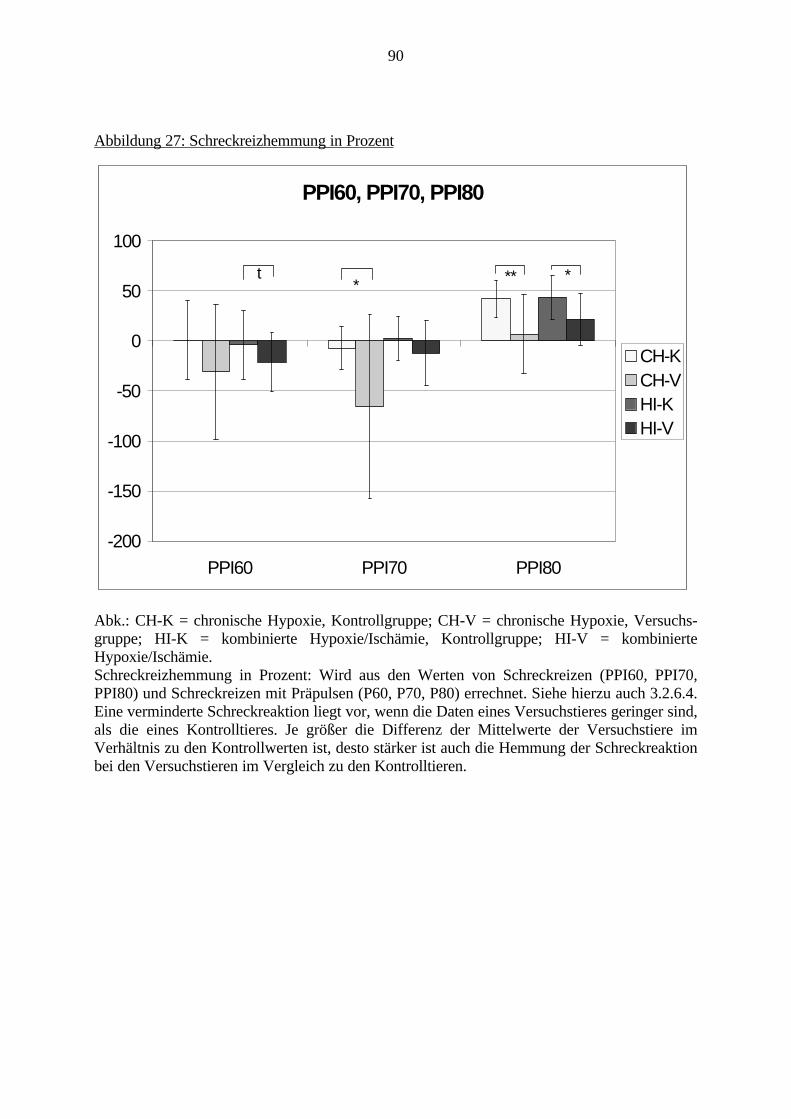
90
Abbildung 27: Schreckreizhemmung in Prozent
Abk.: CH-K = chronische Hypoxie, Kontrollgruppe; CH-V = chronische Hypoxie, Versuchs-gruppe; HI-K = kombinierte Hypoxie/Ischämie, Kontrollgruppe; HI-V = kombinierteHypoxie/Ischämie.Schreckreizhemmung in Prozent: Wird aus den Werten von Schreckreizen (PPI60, PPI70,PPI80) und Schreckreizen mit Präpulsen (P60, P70, P80) errechnet. Siehe hierzu auch 3.2.6.4.Eine verminderte Schreckreaktion liegt vor, wenn die Daten eines Versuchstieres geringer sind,als die eines Kontrolltieres. Je größer die Differenz der Mittelwerte der Versuchstiere imVerhältnis zu den Kontrollwerten ist, desto stärker ist auch die Hemmung der Schreckreaktionbei den Versuchstieren im Vergleich zu den Kontrolltieren.
PPI60, PPI70, PPI80
-200
-150
-100
-50
0
50
100
PPI60 PPI70 PPI80
CH-KCH-VHI-KHI-V
** **
t
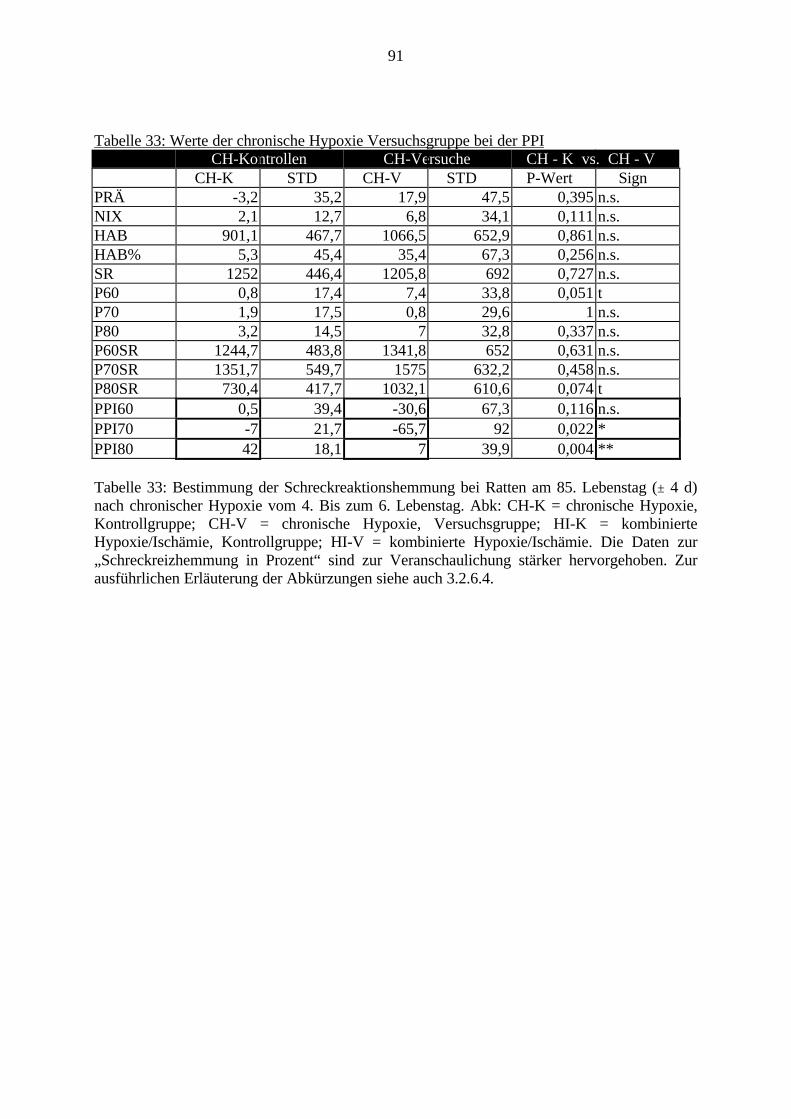
91
Tabelle 33: Werte der chronische Hypoxie Versuchsgruppe bei der PPI CH-Kontrollen CH-Versuche CH - K vs. CH - V CH-K STD CH-V STD P-Wert Sign
PRÄ -3,2 35,2 17,9 47,5 0,395 n.s.NIX 2,1 12,7 6,8 34,1 0,111 n.s.HAB 901,1 467,7 1066,5 652,9 0,861 n.s.HAB% 5,3 45,4 35,4 67,3 0,256 n.s.SR 1252 446,4 1205,8 692 0,727 n.s.P60 0,8 17,4 7,4 33,8 0,051 tP70 1,9 17,5 0,8 29,6 1 n.s.P80 3,2 14,5 7 32,8 0,337 n.s.P60SR 1244,7 483,8 1341,8 652 0,631 n.s.P70SR 1351,7 549,7 1575 632,2 0,458 n.s.P80SR 730,4 417,7 1032,1 610,6 0,074 tPPI60 0,5 39,4 -30,6 67,3 0,116 n.s.PPI70 -7 21,7 -65,7 92 0,022 *PPI80 42 18,1 7 39,9 0,004 **
Tabelle 33: Bestimmung der Schreckreaktionshemmung bei Ratten am 85. Lebenstag (± 4 d)nach chronischer Hypoxie vom 4. Bis zum 6. Lebenstag. Abk: CH-K = chronische Hypoxie,Kontrollgruppe; CH-V = chronische Hypoxie, Versuchsgruppe; HI-K = kombinierteHypoxie/Ischämie, Kontrollgruppe; HI-V = kombinierte Hypoxie/Ischämie. Die Daten zur„Schreckreizhemmung in Prozent“ sind zur Veranschaulichung stärker hervorgehoben. Zurausführlichen Erläuterung der Abkürzungen siehe auch 3.2.6.4.
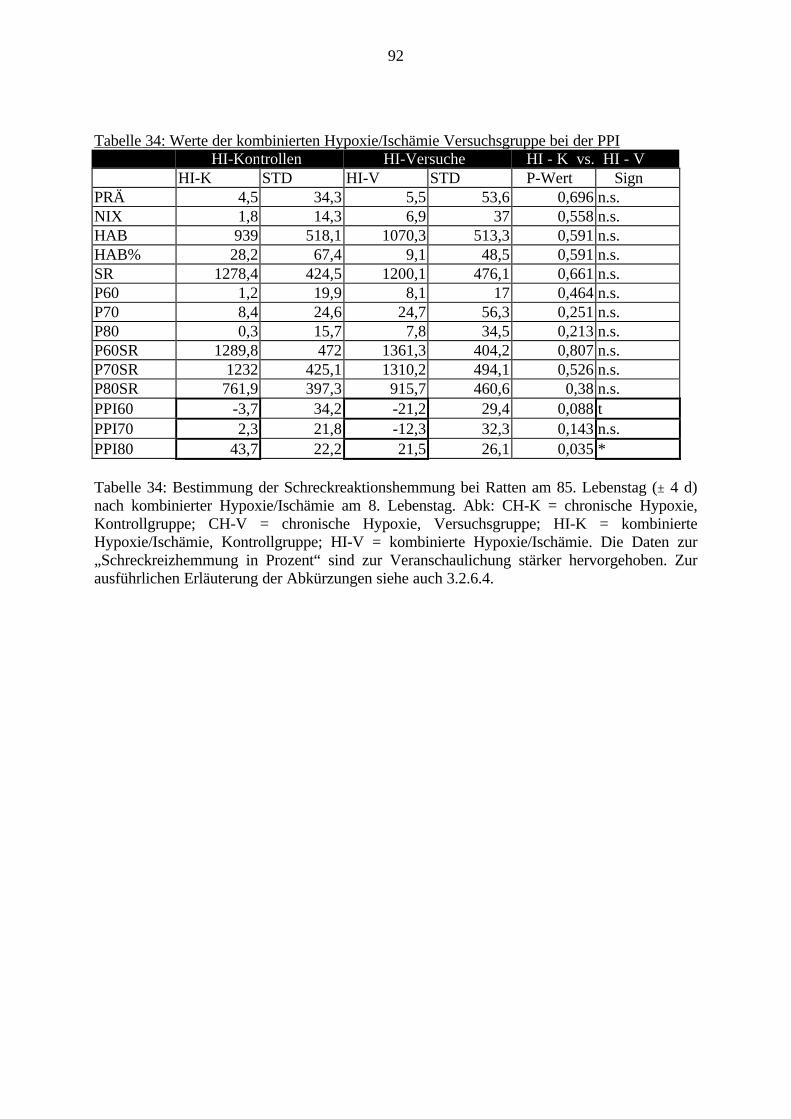
92
Tabelle 34: Werte der kombinierten Hypoxie/Ischämie Versuchsgruppe bei der PPI HI-Kontrollen HI-Versuche HI - K vs. HI - VHI-K STD HI-V STD P-Wert Sign
PRÄ 4,5 34,3 5,5 53,6 0,696 n.s.NIX 1,8 14,3 6,9 37 0,558 n.s.HAB 939 518,1 1070,3 513,3 0,591 n.s.HAB% 28,2 67,4 9,1 48,5 0,591 n.s.SR 1278,4 424,5 1200,1 476,1 0,661 n.s.P60 1,2 19,9 8,1 17 0,464 n.s.P70 8,4 24,6 24,7 56,3 0,251 n.s.P80 0,3 15,7 7,8 34,5 0,213 n.s.P60SR 1289,8 472 1361,3 404,2 0,807 n.s.P70SR 1232 425,1 1310,2 494,1 0,526 n.s.P80SR 761,9 397,3 915,7 460,6 0,38 n.s.PPI60 -3,7 34,2 -21,2 29,4 0,088 tPPI70 2,3 21,8 -12,3 32,3 0,143 n.s.PPI80 43,7 22,2 21,5 26,1 0,035 *
Tabelle 34: Bestimmung der Schreckreaktionshemmung bei Ratten am 85. Lebenstag (± 4 d)nach kombinierter Hypoxie/Ischämie am 8. Lebenstag. Abk: CH-K = chronische Hypoxie,Kontrollgruppe; CH-V = chronische Hypoxie, Versuchsgruppe; HI-K = kombinierteHypoxie/Ischämie, Kontrollgruppe; HI-V = kombinierte Hypoxie/Ischämie. Die Daten zur„Schreckreizhemmung in Prozent“ sind zur Veranschaulichung stärker hervorgehoben. Zurausführlichen Erläuterung der Abkürzungen siehe auch 3.2.6.4.

93
4. Diskussion4.1. Rezeptorautoradiographie4.1.1. Vorinkubation bei NMDA-RezeptorenBei der Rezeptorautoradiographie in dieser Untersuchung fällt auf, dass unabhängig von derVorbehandlung die höchsten Bmax -Werte (in absoluten Zahlen) bei der Verwendung vonGlycin und Spermidin nach der längeren Vorinkubationszeit festzustellen waren. Bei alleinigemZusatz von Glycin – trotz langer Vorinkubation mit dem Puffer – oder bei kürzererVorinkubationszeit waren alle Werte, unabhängig von der Region, deutlich niedriger.Das heißt, dass die Vorinkubationszeit anscheinend keinen so großen Einfluß auf dieBindungskapazität des Liganden hat. Die Vorinkubation mit dem Puffer ist dazu gedacht, allestörenden Elektrolyte und Transmitter im Hirn zu entfernen, wofür in der vorliegendenUntersuchung 15 Minuten offensichtlich ausreichten.Man kann aus den vorliegenden Daten vermuten, dass bei Anwesenheit von sowohl Glycin alsauch Spermidin die besten Koaktivator-Effekte erzielt werden können.
4.1.2. RezeptorenDie absoluten Zahlen der Bmax-Werte des NMDA-Rezeptors der Kontrolltiere dieser Arbeitstimmen nicht mit den in der Literatur gefundenen absoluten Zahlen überein. Die absolutenZahlen dieser Werte sind aber auch in der Literatur sehr unterschiedlich. Dabei muß manbedenken, dass die verschiedenen Autoren andere Untersuchungsmethoden als in dervorliegenden Arbeit benutzten oder die untersuchten Ratten im Alter nicht mit den hieruntersuchten Tieren übereinstimmten:Pichiule et al. (1996) (Bmax des NMDA-Rezeptors im Hippocampus ca. 3800 fmol/mg Protein)und Zhang et al. (1995) (Bmax des NMDA-Rezeptors im Hippocampus ca. 30 fmol/mg Protein)zum Beispiel untersuchten Membranfraktionen, das heißt man kann nicht davon ausgehen, dassauch wirklich alle NMDA-Rezeptoren noch intakt waren zum Zeitpunkt der Untersuchung.Der Marker MK-801 bindet aber nur am offenen Ionenkanal des NMDA-Rezeptors, benötigtalso einen funktionstüchtigen Rezeptor. Die Werte von Sakurai et al. (1991), die einen Bmax-Wert des NMDA-Rezeptors im Hippocampus von ca. 199 bis 1090 fmol/mg Protein angeben,(je nach Region des Hippocampus) sind mit der hier vorliegenden Untersuchung auch nicht zuvergleichen, da diese Autoren junge, erwachsene Ratten mit einem Körpergewicht von 175-250 g untersuchten. Das Verteilungsmuster und auch die Quantität der Glutamat-Rezeptorenverändern sich aber im Laufe des Lebens eines Individuums: im fetalen Hirn ist dasVerteilungsmuster der Glutamatrezeptoren weiter gestreut, und auch eine quantitativeVermehrung dieser Rezeptoren ist festzustellen (Barks et al., 1988). Eine maximale Expressionder Rezeptoren konnten Chaudieu et al. (1991) am 7. bis 14. postnatalen Tag der Ratteausmachen, also genau im Untersuchungszeitraum der hier vorliegenden Arbeit. Dass Sakuraiet al. (1991) also weitaus niedrigere Werte nachweisen konnten, kann nicht verwundern.Außerdem inkubierten sie die Schnitte nicht mit dem Co-Aktivator Glycin bzw. Spermidin, derfür die Aktivierung des Kanals und damit Öffnung des Ionenkanals notwendig ist (Cotman etal., 1987).
Durch diese Inkubation der Schnitte in dieser Arbeit mit Glycin und Spermidin,nachgewiesenen Co-Aktivatoren von NMDA-Rezeptoren (Cotman et al., 1987; Ferchmin etal., 2000), kann man die absoluten Zahlen der hier vorliegenden Bmax-Werte von NMDA-,AMPA- und GABAA-Rezeptoren nicht direkt miteinander vergleichen. Die NMDA-

94
Rezeptoren sind maximal aktiviert worden, das heißt der markierende Ligand MK-801 konntemaximal binden und somit vermutlich auch eine maximale Anzahl der intakten NMDA-Rezeptoren im Schnitt markieren. Eine zusätzliche Aktivierung der AMPA- oder GABAA-Rezeptoren, damit auch diese Rezeptoren maximal markiert werden konnten, ist nichtdurchgeführt worden. Zusammenfassend muß man also bei der Auswertung der absolutenRezeptorzahlen diese Information mit in Betracht ziehen. Die Auswertung, ob die Rezeptorennach Sauerstoffmangel in der Zahl abnehmen, ist davon jedoch nicht betroffen, da die Werte jamit Kontrolltieren verglichen wurden.
Generell ist festzustellen, dass durch Hypoxie die Bindungskapazität aller Rezeptoren inverschiedenen Hirnregionen abgenommen hat.Die Bmax des NMDA-Rezeptors ist bei Tieren, die eine chronische Hypoxie erfahren hatten,signifikant im Präfrontalen Cortex (sowohl in der Region Cg 1/3 (21-23%) als auch Fr 1/2 (16-18%) als auch IL (22-23%)) und im Nucleus Accumbens (25%) gesunken. Dagegen war dieBindungskapazität bei Tieren, die einer kombinierten Ischämie/Hypoxie unterzogen wurden, imPräfrontalen Cortex nicht verändert.Damit konnte die Aussage von Pearigen et al. (1996), dass Ischämie im allgemeinen größereSchäden im Hirn setzt als Hypoxie, nicht bestätigt werden. Auch die erwarteten größerenLäsionen im deutlich empfindlicheren Hippocampus (Zhao und Flavin, 2000; Barks et al.,1988) konnten nach beiden Methoden der hypoxischen Schädigung nicht nachgewiesenwerden. Allerdings ist mein Ischämie-Modell sehr mild, während die chronische Hypoxiehäufiger wiederholt wurde. Bei den Ischämie-Versuchen waren bei den Ratten zweimal füracht Minuten die Arteria carotis communis beidseits abgeklemmt worden und die Tierekonnten dabei nur 8% Sauerstoff atmen. Bei der chronischen Hypoxie dagegen, konnten dieRatten über fünf Tage, jeweils sechs Stunden lang nur 11% Sauerstoff atmen.Die MK-801-Bindung an den NMDA-Rezeptor ist bei Anwesenheit der ModulatorenSpermidin und Glycin normalerweise bei intakten Rezeptoren verstärkt (Sakurai et al., 1991;Subramaniam und McGonigle, 1991). Eine Senkung der Bindungskapazität nach chronischerHypoxie im Präfrontalen Cortex und im Nucleus Accumbens trotz Inkubation der Schnitte mitGlycin und /oder Spermidin gibt also einen Hinweis darauf, dass die NMDA-Rezeptoren inihrer Funktion gestört sein müssen. Denn interessanterweise kann MK-801 nur am offenenIonen-Kanal des NMDA-Rezeptors binden (Hoffman et al., 1994) und die Öffnung des Kanalsbedingt einen in der Funktion ungestörten Rezeptor.Zerebrale Hypoxie führt zu einer vermehrten Ausschüttung von Glutamat und damit zu einerÜbererregung des NMDA-Rezeptors. Diese Exzitotoxizität gipfelt im Ca++-Ionen-Einstrom indie Zelle und führt letztendlich zum Zelltod und damit auch zur Zerstörung der normalenFunktionalität des NMDA-Rezeptors (Fritz et al., 1999a; Yoshioka et al., 2000). DiesesPhänomen dürfte auch in dieser Untersuchung als Erklärung dienen, warum dieBindungskapazität des NMDA-Rezeptors nach Hypoxie abfällt. Das Fehlen vonVeränderungen beim kombinierten ischämisch/hypoxischen Insult könnte auf eine geringereEmpfindlichkeit gegenüber einer kurzzeitig gesetzten Sauertoffunterversorgung des Hirns beijuvenilen Tieren hindeuten. Kurzzeitig bedeutet nach dem Protokoll der vorliegenden Arbeitzweimal für jeweils acht Minuten, wobei die chronische Hypoxie fünf Tage lang für sechsStunden, das heißt also insgesamt für 30 Stunden, durchlaufen wurde.Dieser NMDA-Rezeptorabfall im Präfrontalen Cortex einige Tage nach Hypoxie könnte imspäteren Entwicklungsstadium zu einer NMDA-Rezeptor-Hypofunktion führen, die später

95
auftretende Zeichen einer Schizophrenie erklärt. Diese Hypothese als Erklärungsversuch derSchizophrenie stellten schon Olney und Farber (1995) auf.Der Abfall der Bindungskapazität des NMDA-Rezeptors nur im Präfrontalen Cortex und imNucleus Accumbens aber nicht im Hippocampus der untersuchten Ratten paßt nicht zurHypothese, dass gerade die Regionen mit hoher Glutamat-Rezeptor-Konzentration imVerdacht stehen, verwundbarer zu sein für neuronale Schädigungen durch Hypoxie oderIschämie (Barks et al., 1988; Chaudieu et al., 1991). Die meisten Glutamatbindungsstellen imfetalen Rattenhirn befinden sich im Hippocampus, Nucleus caudatus/Putamen, Globus pallidus,subthalamischen Nucleus und im reticulären Nucleus des Thalamus (Chaudieu et al., 1991).Anscheinend ist der Hippocampus juveniler Ratten unempfindlicher gegenüber hypoxischerSchädigung als der Präfrontale Cortex.
Die Bindungskapazität des AMPA-Rezeptors ist auch nach Hyopxie in einigen Hirnregionenzurückgegangen. Während nach chronischer Hypoxie ein Abfall der Bmax für AMPA nur inTeilen des Präfrontalen Cortex (17% in der Fr1/2-Region), im Nucleus Accumbens (49%) undim Striatum (53%) stattfand, konnte nach kombinierter Ischämie/Hypoxie eine generelleAbnahme der AMPA-Rezeptorbindung im gesamten Präfrontalen Cortex beobachtet werden(Cg1/3: 34%, IL: 31%, Fr1/2: 20%). Der Hippocampus war (mit Ausnahme der CA2 Regionnach kombinierter Hypoxie/Ischämie (18%)) wiederum nicht betroffen.Die Regionen Nucleus Accumbens und Striatum zeigten mit Abstand den höchstenprozentualen Abfall an Bindungskapazität der AMPA-Rezeptoren. Anscheinend sind dieseRegionen besonders anfällig für eine hypoxische Schädigung.Die Bindungskapazitäten sowohl der AMPA- als auch der NMDA-Rezeptoren sind imPräfrontalen Cortex und im Nucleus Accumbens nach chronischer Hypoxie gesunken. Dieslässt auf eine verstärkte Empfindlichkeit des juvenilen Rattenhirns gegenüber wiederholterhypoxischer Schädigung in diesen Regionen schließen. Die kombinierte Hypoxie/Ischämiezeigte nur beim AMPA-Rezeptor eine Abnahme der Bindungskapazität im PräfrontalenKortex; der NMDA-Rezeptor ließ dort keine Veränderung der Bmax erkennen. Es fällt auf, dassdas juvenile Rattenhirn im Bezug auf die sinkende Bindungskapazität des NMDA-Rezeptorsempfindlicher auf die chronische Hypoxie als auf eine kurz einwirkende Ischämie reagiert. DerHippocampus scheint insgesamt unempfindlicher gegenüber hypoxischer Schädigung zu sein.
Die Bindungskapazität des GABAA-Rezeptors hat sich nach Hypoxie nur leicht verändert.Während Hypoxie, egal wie verursacht, zu keiner veränderten Bmax im Präfrontalen Cortexführte, fand sich eine Abnahme der GABAA-Rezeptorbindung im Nucleus Accumbens (nachchronischer Hypoxie (51,5%)) und im Striatum (nach kombinerter Ischämie/Hypoxie (35%).Interessanterweise war auch in den Regionen CA1 und CA2 des Hippocampus nachkombinierter Ischämie/Hypoxie eine Abnahme der Bindung um jeweils 23% zu verzeichnen.Diese Daten bestätigen die Aussage von Brixey et al. (1993), dass zerebrale Hypoxie auch dasGABA-System schädigt. Auch die Feststellung von Viapiano et al. (2001), die eine 30%igeSenkung der Bindungskapazität von GABA am GABAA-Rezeptor nach zerebralemSauerstoffmangel nachgewiesen hatten, konnte in der vorliegenden Arbeit bestätigt werden.Diese Veränderungen werden voraussichtlich durch Ischämie bedingte Änderungen am GABA-Rezeptor-Ionenkanal verursacht (Luhman et al., 1993). Anscheinend ist der NucleusAccumbens eine sehr vulnerable Region für eine hypoxische Schädigung des GABAA-Rezeptors. Auch hier scheint chronische Hypoxie mehr Schäden zu verursachen, als diekombinierte kurzzeitige Hypoxie/Ischämie. Mein Ischämie-Modell scheint sehr mild, während

96
die chronische Hypoxie häufiger wiederholt wurde und deshalb anscheinend mehr Läsionensetzen konnte.
4.2. In-Situ-HybridisierungDie Expression der mRNA der NR1-Untereinheit ist im Präfrontalen Cortex (Regionen Fr 1/2(31%), Cg 1/3 (34%) und IL (36%)) und in der CA1-Region des Hippocampus (10%)signifikant bei juvenilen Ratten nach chronischer Hypoxie angestiegen. Ein Hinweis auf einenAnstieg war im Präfrontalen Cortex nach Hypoxie/Ischämie in der Region Cg 1/3 (21%), imHippocampus CA2 (7%) bei chronischer Hypoxie und in der Region DG bei chronischerHypoxie (9%) und bei kombinierter Hypoxie/Ischämie (3%) zu sehen.Die Aussage von Gottlieb und Matute (1997), die eine Steigerung der Expression der NR1-Untereinheiten in der CA1-Region des Hippocampus vom dritten bis siebten postischämischenTag nachweisen konnten, können aufgrund der Daten dieser Untersuchung nicht bestätigtwerden. In der vorliegenden Untersuchung konnte nur im Gyrus dentatus des Hippocampus beiIschämie eine Steigerung der Expression beobachtet werden. In der CA1-Region desHippocampus war die Expression der NR1-Untereinheit nach chronischer Hypoxie, nicht nachkombinierter Ischämie/Hypoxie gesteigert. Allerdings ist mein Ischämie-Modell im Gegensatzzur Untersuchung von Gottlieb und Matute (1997) sehr mild, während die chronische Hypoxiehäufiger wiederholt wurde.Außerdem scheinen aber noch andere Regionen des Hirns empfindlich zu reagieren, wie zumBeispiel der Präfrontale Cortex. Hier konnten die höchsten prozentualen Zunahmen derExpression dieser Untereinheit beobachtet werden. Die Zunahme der mRNA der NR1-Untereinheit könnte als "Gegenregulation" des Körpers verstanden werden, der versucht, diezerstörten oder nicht mehr voll funktionierenden NMDA-Rezeptoren zu reparieren und zuersetzen. Hierzu passt auch die herabgesetzte NMDA-Rezeptorbindungskapazität imPräfrontalen Cortex.
Die mRNA der NR2A-Untereinheit ist im Bereich der CA3a Region des Hippocampus beichronischer Hypoxie signifikant gefallen (25%). Einen Hinweis auf einen Abfall dieser mRNAist in der CA3c-Region des Hippocampus bei chronischer Hypoxie (18%) sichtbar.Die Aussagen von Liu et al. (2001) und Zhang et al. (1997), die einen Abfall der Expressionder NR2A-Untereinheit im gesamten Hippocampus nach Ischämie nachweisen konnten,können aufgrund dieser Daten nicht bestätigt werden. In der vorliegenden Untersuchung findetman diesen Abfall nur nach chronischer Hypoxie. Auch hier muß mein mildes Modell derIschämie als Erklärungsversuch in Betracht gezogen werden, weshalb das juvenile Rattenhirnvulnerabler auf chronische Hypoxie als auf kombinierte Hypoxie/Ischämie reagiert. Hsu et al.(1998) konnten diesen Abfall der Expression der NR2A-Untereinheit in anderen Regionen desHippocampus beobachten (CA1- und DG-Region des Hippocampus) als in dieserUntersuchung. Die Aussage von Hsu et al. (1998), dass direkt nach dem postischämischenInsult ein Abfall der Expression der NR2A-Untereinheit in diesen Regionen besteht, der amTag 3 schon wieder fast normal sei, kann als Erklärung dienen, warum in der vorliegendenUntersuchung nur sehr vereinzelte Veränderungen in der Expression der NR2A-Untereinheitzu sehen waren. Die molekularbiologischen Untersuchungen fanden genau 3 Tage nach demInsult statt, so dass ein eventueller Abfall der Expression dieser Untereinheit dann aufgrund desUntersuchungsprotokolls schon nicht mehr nachzuweisen war. Die Steigerung des mRNA-Gehaltes der NR2A-Untereinheit in der CA1-Region des Hippocampus, die Gottlieb undMatute (1997) nachweisen konnten, konnte aufgrund meiner Daten nicht bestätigt werden.

97
Die Expression der NR2B-Untereinheit ist im Nucleus Accumbens bei chronischer Hypoxienur schwach gestiegen (39%), des Weiteren kann man Hinweise auf einen Anstieg imHippocampus in den Regionen CA2 (47%) und CA3a (26%) nach kombinierterHypoxie/Ischämie sehen.Das Ergebnis der Studie von Gottlieb und Matute (1997), die eine maximale Expression vonNR2B nach Ischämie in der CA1-Region des Hippocampus beobachten konnten, kannaufgrund der hier vorliegenden Daten nicht bestätigt werden. Auch die Ergebnisse von Zhanget al. (1997) und Hsu et al. (1998), die einen Abfall der mRNA der NR2B-Untereinheit imHippocampus feststellen konnten, konnten in der vorliegenden Untersuchung nicht beobachtetwerden.Die Zunahme der Expression der NR2B-Untereinheit kann sicherlich wieder als sekundäreReparaturleistung des Körpers verstanden werden. Im Hinblick darauf, dass die NR2B-Untereinheit für eine verbesserte Leitfähigkeit des NMDA-Rezeptors zuständig sein soll(Reyes et al., 1998), das heißt der Ionenkanal des Rezeptorkomplexes öffnet länger, könnteman annehmen, dass der Körper versucht, die Abnahme des NMDA-Rezeptors dahingehend zuregulieren, dass die nachfolgend synthetisierten Rezeptoren besonders leitfähig sind und so eineMinderzahl des Rezeptors ausgeglichen werden soll.
Die mRNA der NR2C-Untereinheit ist im Hippocampus in der CA3a-Region bei chronischerHypoxie signifikant gestiegen (30%); in derselben Region bei der kombiniertenHypoxie/Ischämie ist die mRNA dieser Untereinheit signifikant gefallen (27%).Die Aussage von Perez-Velazquez und Zhang (1994), die erstmalig nach Hypoxie imerwachsenen Hippocampus mRNA der NR2C-Untereinheit gefunden haben, kann mit denvorliegenden Daten nicht bestätigt werden, denn die hier untersuchten Ratten befinden sichnoch in der Entwicklung des Hirns. Erst im Laufe des frühen Erwachsenenalters kann mandavon ausgehen, dass NR2C im Hippocampus nicht mehr nachzuweisen ist.Anscheinend sind die Schädigungen, die mit der chronischen Hypoxie und mit der Ischämie imHippocampus an der NR2C-Untereinheit gesetzt wurden, unterschiedlich gewichtet.Auffallend ist die Wirkung von kombinierter Hypoxie/Ischämie und chronischer Hypoxie aufdie NR2B-Untereinheiten in der CA3a-Region des Hippocampus. Bei chronischer Hypoxiesteigt die NR2C-Untereinheit schwach an. Dagegen fällt sie bei der kombiniertenIschämie/Hypoxie ab. Dieses Phänomen könnte als Kompensationsversuch des Hirnes erklärtwerden. An dieser Stelle sollte in Betracht gezogen werden, dass möglicherweise dieunterschiedliche Qualität von chronischer Hypoxie und kombinierter kurzzeitigerHypoxie/Ischämie verschiedenartige Effekte auf das Hirn haben konnte.
Die Expression der NR2D-Untereinheit ist im Präfrontalen Cortex in der Region Fr1/2 beiHypoxie/Ischämie signifikant gestiegen (46%). Einen Hinweis auf einen Anstieg kann man inder IL-Region (32%) des Präfrontalen Cortex bei kombinierter Hypoxie/Ischämie und imNucleus Accumbens (26%) bei chronischer Hypoxie beobachten. Außerdem ist ein Hinweis aufeinen Abfall der Expression im Nucleus Accumbens bei kombinierter Hypoxie/Ischämie zusehen (32%). Die Beobachtung von Akbarian et al. (1996), der bei humanen Schizophrenie-Patienten einen Anstieg der NR2D-Untereinheit im Präfrontalen Cortex gefunden hat, konntein der vorliegenden Arbeit nach kombinierter Hypoxie/Ischämie bei der Ratte beobachtetwerden.

98
Zusammenfassend kann man sagen, dass das Gehirn der juvenilen Ratten auf dieSauerstoffunterversorgung im Hinblick auf die unterschiedlichen Untereinheiten verschiedenreagiert hat. Die NR1-Untereinheit ist gestiegen, die Expression der verschiedenen NR2-Untereinheiten ist entweder gestiegen oder gefallen. Dies könnte dahingehend verstandenwerden, dass der Körper zwar eine Reparaturleistung versucht, dass aber dieZusammensetzung der NMDA-Rezeptoren nach dem Insult sich verändert hat. Vor allem dieNR2A-Untereinheiten scheinen in bestimmten Hirnregionen durch andere Untereinheitenersetzt worden zu sein.
4.3. PräpulsinhibitionDie Messung der Schreckreaktion hat dargestellt, dass die Ratten nach chronischer Hypoxieeine deutliche Verringerung ihrer Schreckreaktionshemmung (bei 70 bzw. bei 80 dBPräpulsen) im Vergleich zu den Kontrolltieren hatten. Besonders auffällig ist hierbei, dass erstmit zunehmender Lautstärke des Präpulses eine signifikante Verminderung derSchreckreaktionshemmung einsetzt (PPI70) und sich das Defizit bei noch lauterem Präpuls(PPI80) noch einmal erhöht. Das bedeutet, dass die Versuchstiere im Vergleich zu denKontrolltieren nach chronischer Hypoxie weniger sensitiv auf den Präpuls reagieren.Die Versuchstiere zeigten im Vergleich zu den Kontrolltieren nach Hypoxie und Ischämie nachleisen Präpulsen (PPI60) und nach stärkeren Präpulsen (PPI80) eine Verminderung ihrerSchreckreaktionshemmung. Das bedeutet, der Effekt der Präpulse ist bei den Versuchstierengeringer als bei den Kontrolltieren. Bei „normalen“, d.h. gesunden Ratten, würde man aufSchreckreize mit vorhergehenden Präpulsen eine Verminderung der Schreckreaktion, d.h. eineSchreckreaktionshemmung, erwarten. Ebenso wie die Ratten mit chronischer Hypoxieerschienen die Tiere, die kombinierter Hypoxie/Ischämie ausgesetzt waren, weniger beeinflußtvon dem Effekt der Präpulse.Die Ergebnisse von Meincke et al. (2001) und Swerdlow et al. (2001), die eine vermindertePräpulsinhibition und auch eine vermutliche Beteiligung des Hippocampus und des NucleusAccumbens bei Ratten nachgewiesen haben, können durch die Daten dieser Arbeit nur zumTeil bestätigt werden. Denn die massivsten molekularbiologischen Veränderungen bei denRatten dieser Arbeit befanden sich im Präfrontalen Cortex und im Nucleus Accumbens. ImNucleus Accumbens konvergieren glutamaterge Afferenzen aus Hippocampus, medialemPräfrontalen Cortex und Amygdala und dopaminerge Afferenzen aus dem ventralentegmentalen Areal (Swerdlow et al., 2001).Dennoch bieten die Ergebnisse eine Möglichkeit, bereits jetzt einen Vergleich anzustellen.Auch Swerdlow et al. (2001) konnten nachweisen, dass die PPI-Regulation bei Ratten unteranderem im Präfrontalen Cortex stattfindet. Außerdem könnte die PPI-Regulation in Rattendurchaus als Modell für den Menschen dienen, könnte aber nicht direkt so auf andere Speziesübertragen werden (Swerdlow et al., 2001).Es konnten in dieser Arbeit im Nucleus Accumbens und im Präfrontalen Cortex, beidessensitive Gebiete, was die Reizverarbeitung bezogen auf die Verarbeitung der Schreckreizeangeht, bei den 11 Tage alten Tieren massive Veränderungen an den Rezeptoren nachgewiesenwerden. Daher liegt bereits zum jetzigen Zeitpunkt die Vermutung nahe, dass hier ein kausalerZusammenhang bestehen könnte. Voraussetzung hierfür wäre natürlich eine mangelhafteReparation der Läsionen, die durch Hypoxie und Ischämie und durch chronische Hypoxieverursacht wurden. Eine Bestätigung dieser Vermutung kann die noch ausstehendemolekularbiologische Untersuchung bei adulten Ratten erbringen, die im juvenilen Alter nachdem Protokoll der vorliegenden Arbeit geschädigt wurden.

99
Die Bedeutung dieser Versuchsergebnisse liegt derzeit bei der Erkenntnis, dass chronischeHypoxie und kombinierte Hypoxie/Ischämie im jungen Alter der Ratte einen Effekt auf dasVerhalten im Erwachsenenalter haben können. Es ist naheliegend, diese Beobachtung auf denMenschen zu übertragen. In dieser Arbeit ist bereits erwähnt worden (siehe hierzu 2.5.), dasseinige Bausteine der Rezeptoren sich bei Mensch und Ratte sehr ähneln. Die Ratte scheint eingutes Modell dafür zu sein, die Empfindlichkeit des menschlichen fetalen Gehirnes in Bezugauf chronische Hypoxie und kombinierte Hypoxie/Ischämie und die Möglichkeit vonLangzeitschäden zu untersuchen.
4.4. SchlussfolgerungBei Schizophrenen werden gehäuft und in Kombination Schwangerschafts- und Geburts-komplikationen in der Vorgeschichte gefunden. Gemeinsamer Faktor dieser Komplikationen istHypoxie oder Ischämie. Erst im frühen Erwachsenenalter kommt es zum Auftreten ersterSymptome.Ein Sauerstoffmangel im Gehirn der juvenilen Ratten bewirkt eine Senkung der Bindungs-kapazität der Liganden an dem NMDA-Rezeptor. Der Präfrontale Cortex ist in dieserUntersuchung am stärksten davon betroffen, aber auch andere Hirnregionen zeigen eineverminderte Bmax. Durch die gesenkte Bindung, kommt es in vivo zu einer Unterfunktion diesesRezeptors. Beim Menschen nimmt man an, dass solche Veränderungen erst im frühenErwachsenenalter, wenn entscheidende Neuronenschaltkreise reifen, zu Fehlfunktionen undzum Auftreten von schizophrenen Symptomen führen. Eines dieser Symptome konnte in dervorliegenden Arbeit am Tiermodell der juvenilen hypoxischen Ratte durch dieVerhaltensversuche am 85. Tag nachgestellt werden: die Defizite bei der Hemmung derakustischen Schreckreaktion durch einen Präpuls zeigten deutlich eine Fehlfunktion der diePräpulsinhibition regulierenden Hirnstrukturen. Eine mögliche Erklärung für dieses Defizitkönnte die Hypofunktion des NMDA-Rezeptors durch die juvenile Hypoxie darstellen. Dermolekularbiologische Nachweis der permanent gesenkten Bindungskapazität für NMDA, unddamit der Hypofunktion des Rezeptors im adulten Tier, steht allerdings noch aus und sollte inweiteren Untersuchungen angegangen werden.Auch der AMPA-Rezeptor zeigte eine gesenkte Bindungskapazität nach Hypoxie im juvenilenAlter. Diese Hypofunktion mag sicherlich wie auch die Hypofunktion des NMDA-Rezeptorszu dem später beobachteten Defizit der Präpulsinhibition beigetragen haben.Außerdem ist eine Senkung der Bindungskapazität des GABAA-Rezeptors in der RegionNucleus Accumbens bei chronischer Hypoxie und in den Regionen Striatum, CA1, CA2 undCA3c des Hippocampus bei kombinierter Hypoxie/Ischämie festzustellen. Dies kann zu einerSchmälerung der inhibitorischen Effekte führen. Ob dieser Abfall der Bindungskapazitätwirklich reversibel ist, wie Viapiano et al. (2001) festgestellt haben, wird voraussichtlich diezum jetzigen Zeitpunkt noch laufende molekularbiologische Untersuchung der nach demVerhaltensversuch getöteten Ratten zeigen. Gleichzeitig muß man bei der Auswertung dieserGABAA-Rezeptor-Werte bedenken, dass die Ratten unter anderem mit einemBenzodiazepinagonisten (Midazolam) narkotisiert wurden. Benzodiazepinagonisten erhöhennämlich die Bindungskapazität von GABA am Rezeptor und verstärken damit die GABA-Wirkung durch eine Erhöhung der Öffnungsfrequenz des Ionenkanals (Günther et al., 1995).Da aber sowohl die Versuchs- als auch die Kontrolltiere narkotisiert wurden, kann man davonausgehen, dass die Ergebnisse der Bindungskapazität des GABAA-Rezeptors davonunbeeinflußt sind.

100
Die Expression der NR1-Untereinheit des NMDA-Rezeptors nimmt nach Sauerstoffmangel desjuvenilen Rattenhirns zu. Auffallend ist, dass überall dort, wo im Präfrontalen Cortex die Bmax
des NMDA-Rezeptors signifikant gefallen ist, die mRNA der NR1-Untereinheit signifikantgestiegen ist. Diesen Zusammenhang könnte man als einen Kompensationsversuch des Gehirnsverstehen. Sowohl die Zusammensetzung der einzelnen Untereinheiten der NMDA-Rezeptorenals auch die Anzahl der Rezeptoren im Hirn ist bei der Geburt noch nicht abgeschlossen. Soliegt zum Beispiel der maximale Peak in der Expression des Genes, welches NR1 kodiert, beim10. postnatalen Tag und im frühen Erwachsenenalter der Ratte (Pujic et al., 1993). Ob dieser Kompensationsversuch nun ausgereicht hat, dass nach einigen Wochen wiedergenügend neue und funktionstüchtige NMDA-Rezeptoren im Präfrontalen Cortex vorliegen,darf angezweifelt werden. Einen Hinweis darauf, ob nun tatsächlich eine vollständige,funktionelle Reparation stattgefunden hat, geben die Verhaltensversuche. Die Messung der PPIzeigt deutliche Defizite in den die Reizverarbeitung regulierenden Hirnregionen und lässt aufeine länger anhaltende Schädigung schließen. Die noch ausstehende molekularbiologischeUntersuchung der Tiere aus den PPI-Versuchen könnte eine weitere Bestätigung für dieseHypothese erbringen.
Auch bei den hier untersuchten Ratten ist eine vermehrte Expression der NR2D-Untereinheitim Präfrontalen Cortex, wie bei humanen Schizophrenie-Patienten, gefunden worden (Akbarianet al., 1996). Dies ist ein weiterer Hinweis darauf, dass Sauerstoffmangel im juvenilen Hirn zurÄtiopathogenese der Schizophrenie beitragen könnte.
In dieser Untersuchung fällt auf, dass das juvenile Rattenhirn im Bezug auf die sinkendeBindungskapazität des NMDA-Rezeptors und auf die Steigerung der Expression der NR1-Untereinheit empfindlicher auf die chronische Hypoxie, als auf eine kurz einwirkende Ischämiereagiert. Dieser Effekt ist sicherlich im Hinblick auf die Prävention von Folgeschäden durchSauerstoffmangel bei der Schwangerschaft und bei der Geburt beachtenswert.
Die Versuchsergebnisse legen dar, dass Hypoxie oder Ischämie bei jungen Ratten meßbareSchäden an NMDA-, AMPA- und GABA-Rezeptoren verursachen können, die auch bis zumErwachsenenalter nicht ausgeglichen werden können. Die Ergebnisse zeigen, dass chronischeHypoxie bzw. die Ischämie als Ursache sowohl für akute molekularbiologische Veränderungenim Hirn als auch für die späteren Störungen in der Reizverarbeitung anzusehen sind. DieVerhaltensauffälligkeiten zusammen mit den molekularbiologischen Veränderungen legen nahe,dass auch die Ratte als Labortier und Tiermodell an erworbenen psychischen Krankheiten, wiez. B. der Schizophrenie, erkranken kann. Der Schluß, dass sicherlich auch andere Haustieredavon betroffen sein können, liegt nahe. Diese Diagnosestellung ist natürlich klinisch beiHaustieren extrem schwierig zu stellen und wird im Normalfall sicher selten medikamentelltherapiert, sondern eher durch Euthanasie behoben.Dieses Tiermodell erscheint geeignet, um den Zusammenhang von Verhaltensstörungen undVeränderungen an Rezeptoren experimentell zu untersuchen. Und es ist geeignet, ummögliche Therapien der Schizophrenie und Prävention von Folgeschäden durchSauerstoffmangel bei der Schwangerschaft und bei der Geburt experimentell zu testen.

101
5. Zusammenfassung
Manfred Starke
Untersuchung der Wirkung von Hypoxie auf das glutamaterge System als Tiermodellfür die Ätiopathogenese der Schizophrenie
Bei der Schizophrenie kommen Schwangerschafts- und Geburtskomplikationen gehäuft in derVorgeschichte der Patienten vor. Besonders das Auftreten hypoxie-assoziierterKomplikationen weist auf eine mögliche Rolle von Hypoxie oder Ischämie in derPathophysiologie der Erkrankung hin. In dieser Arbeit wurde der Einfluß von Hypoxie undIschämie auf glutamaterge Rezeptoren am Tiermodell untersucht, da bei der Schizophrenie einglutamaterges Defizit angenommen wird. Anhand von Messungen der Reaktionsinhibitiondurch Schreckreize mit vorhergehenden Präpulsen, einem Verhalten, das bei schizophrenenPatienten verändert ist, soll durch das Anführen von Zusammenhängen zwischen Hypoxie undIschämie mit dem späteren Auftreten von Verhaltensänderungen an Ratten ein verbessertesModell für die Beziehung von Molekularbiologie zu Verhalten erstellt und getestet werden.96 Ratten wurden Hypoxie bzw. Ischämie ausgesetzt, um sie anschließend aufmolekularbiologische Veränderungen bzw. Verhaltensabweichungen zu untersuchen. Die Tierewurden in 4 Gruppen aufgeteilt. Gruppe 1 wurde vom 4. bis zum 8. postnatalen Lebenstag6h/d in einem luftdichten Käfig mit 11% O2 / 89% N2 (Chronische Hypoxie = CH) behandelt.Gruppe 2 diente als Kontrollgruppe hierzu. Der entsprechende Käfig war identisch aufgebaut,aber es wurde Luft eingeleitet. Den Tieren der Gruppe 3 wurde am 8. postnatalen Tag inNarkose 2 mal im Abstand von 10 min für je 8 min beidseitig die Arteria carotis communisabgeklemmt (kombinierte Hypoxie/Ischämie = HI). Während dieser Abklemmphase wurden dieTiere in ein gasdichtes Behältnis mit 8% O2 / 92% N2 gelegt. Gruppe 4 diente alsKontrollgruppe hierzu. Die Tiere wurden ebenso in Narkose operiert, aber die Carotidenwurden nicht abgeklemmt.Am 11. postnatalen Tag wurden je Gruppe 10 Tiere getötet, die Gehirne freipräpariert und inIsopentan und flüssigem Stickstoff schockgefroren. Aus folgenden Ebenen der Gehirne wurdenin einem Kryostaten dünne Schnitte hergestellt (16µm für die Rezeptorautoradiographie =RAR, 20µm für die In-Situ-Hybridisierung = ISH) und auf Objektträgern fixiert: PräfrontalerCortex (PC), Striatum (ST) und Hippocampus (HIP). Innerhalb dieser Ebenen wurdenfolgende Regionen untersucht: PC: Fr1/2, Cg1/3 und IL; ST: CPu und AcbC; HIP: CA1, CA2,CA3a, CA3c und DG. Die eine Hälfte der Präparate wurde mittels Rezeptorautoradiographie(RAR) untersucht. Drei verschiedene Rezeptoren wurden dabei mit folgenden Ligandenradioaktiv markiert: der NMDA-Rezeptor mit [3H]-MK801, der AMPA-Rezeptor mit [3H]-AMPA und der GABA-A-Rezeptor mit [3H]-Muscimol. Dem Liganden MK801 wurde zurVerstärkung der Bindung Spermidin bzw. Glycin während der Hauptinkubation zugemischt.Die andere Hälfte der Präparate wurde mittels In-Situ-Hybridisierung untersucht, wobei mitHilfe von mit 35S markierten Sonden die mRNAs folgender NMDA-Rezeptor-Untereinheitenmarkiert werden: NR1, NR2A, NR2B, NR2C und NR2D. Anschließend wurden diemarkierten Präparate auf strahlungssensitive Folien gelegt und bei NMDA und GABA 5Wochen, bei AMPA 4 Wochen und bei der ISH 5 Tage inkubiert. Die Folien wurdenentwickelt, die Abbildungen darauf abfotografiert und digital gespeichert. Schließlich konntenmit einem Auswertungsprogramm die beschriebenen Regionen der Rattengehirne auf ihre

102
stattgefundene Bindung untersucht werden. Die überlebenden Tiere wurden am 85. postnatalenTag (± 4d) in einem Verhaltensversuch auf ihre Schreckreaktionshemmung (PPI) mit Präpulsen(20ms) bei 60, 70 und 80 dB, einem Inter-Stimulus Intervall (50ms) und Schreckreizen (100dB, 20ms) untersucht.Es war eine verminderte Rezeptoranzahl bezüglich NMDA-, AMPA- und GABAA-Rezeptorenin den untersuchten Ebenen sowohl nach chronischer Hypoxie als auch nach kombinierterHypoxie/Ischämie festzustellen. Ein Anstieg an NMDA-Rezeptoren war nach chronischerHypoxie in der CA2 Region des Hippocampus festzustellen. Es kann desweiteren einevermehrte Expression der mRNA der NR1-Untereinheiten nach chronischer Hypoxie (PC:Cg1/3, Fr1/2, IL; HIP: CA1, CA2, DG), der NR2B-Untereinheit (Nucleus Accumbens =AcbC), der NR2C-Untereinheiten (HIP: CA3a) und der NR2D-Untereinheiten (AcbC)festgestellt werden. Eine Verminderung ist feststellbar bei den NR2A-Untereinheiten imHippocampus (CA3A; CA3c). Nach kombinierter Hypoxie/Ischämie ist eine Heraufregulationder NR1-Untereinheiten im Präfrontalen Cortex (Fr1/2) und im Hippocampus (DG)feststellbar; eine Heraufregulation zeigt sich bei den NR2B-Untereinheiten im Hippocampus(CA2; CA3a) und bei den NR2D-Untereinheiten im Präfrontalen Cortex (Cg1/3; IL). EineVerminderung der mRNA der NR2A-Untereinheiten ist im Hippocampus (CA3a, CA3c) nachchronischer Hypoxie festzustellen; nach kombinierter Hypoxie/Ischämie sind die NR2D-Untereinheiten im Nucleus Accumbens und die NR2C-Untereinheiten CA3a-Region desHippocampus erniedrigt. Die Effekte durch chronische Hypoxie schienen größer zu sein als dieEffekte durch kombinierte Hypoxie/Ischämie. Bei der Messung der PPI bei 56 Versuchstierenam 85. Lebenstag (± 4d) hat sich eine deutliche Erniedrigung der Hemmung derSchreckreaktion in beiden Versuchsgruppen gezeigt.Das Absinken der Rezeptorzahlen bei NMDA-, AMPA- und GABAA-Rezeptoren ist einHinweis auf die erfolgte Schädigung durch chronische Hypoxie bzw. Ischämie und ein Belegfür die Auswirkung von kombinierter Hypoxie/Ischämie bzw. chronischer Hypoxie. DieHeraufregulation der NR1- und der NR2B-Untereinheiten könnte als Kompensationsversuchverstanden werden. Dabei weist die Abnahme der NMDA-Rezeptorzahl auf eineUnterfunktion dieses Rezeptors hin, wie sie auch in der Pathophysiologie der Schizophrenieangenommen wird. Der Anstieg der leicht erregbaren NR2B-Untereinheit könnte alsVerschiebung der Verhältnisse zugunsten leichter reagierender Untereinheiten verstandenwerden. Die uneinheitliche Entwicklung der NR2C-Untereinheit könnte sowohl dieunterschiedliche Qualität der Auswirkung von chronischer Hypoxie und kombinierterHypoxie/Ischämie widerspiegeln, als auch die unterschiedliche Empfindlichkeit derGehirnareale. Der Anstieg der NR2D-Untereinheiten, wobei wie bei schizophrenen Patientenvermehrt NR2D-Untereinheiten im Präfrontalen Cortex gefunden wurden, kann als weiteresIndiz verstanden werden, dass bei der Äthiopathogenese der Schizophrenie chronischeHypoxie bzw. kombinierte Hypoxie/ Ischämie beteiligt sein können.Offensichtlich besteht ein Zusammenhang zwischen chronischer Hypoxie bzw. Hypoxie/Ischämie und morphologischen und quantitativen Veränderungen an NMDA-, AMPA- undGABAA-Rezeptoren. Es liegt nahe, die gemessenen PPI-Defizite als Folge der erwähntenVeränderungen anzusehen.Zusammengefaßt zeigen die Ergebnisse eine besondere Rolle von Hypoxie und Ischämie ineinem vulnerablen Stadium der Gehirnentwicklung und weisen auf eine mögliche Beteiligungan der Pathophysiologie der Schizophrenie hin.

103
Die Versuchsergebnisse legen dar, dass Hypoxie oder Ischämie bei jungen Ratten meßbareSchäden an NMDA-, AMPA- und GABAA-Rezeptoren verursachen können, die auch bis zumErwachsenenalter nicht ausgeglichen werden können. Diese Ergebnisse zeigen, dass chronischeHypoxie bzw. die Ischämie als Ursache sowohl für akute molekularbiologische Veränderungenim Hirn als auch für die späteren Störungen in der Reizverarbeitung anzusehen sind. DieseErgebnisse werden auch durch die Literatur bestätigt. Daher erscheint dieses Tiermodell alsgeeignet, um den Zusammenhang von Verhaltensstörungen und Veränderungen an Rezeptorenexperimentell zu untersuchen. Und es ist geeignet, um mögliche Therapien der Schizophrenieund Prävention von Folgeschäden durch Sauerstoffmangel bei der Schwangerschaft und bei derGeburt experimentell an diesem Modell zu testen.

104
6. Summary
Manfred Starke
Investigation of the effect of hypoxia on the glutamatergic system as an animal model forthe etiopathogenesis of schizophrenia
In schizophrenia, pregnancy and birth complications occur at an increased rate in the casehistory of the patients. Especially the occurrence of hypoxia-associated complications points toa possible role of hypoxia or ischemia in the pathophysiology of the disease. The influence ofhypoxia and ischemia on the glutamatergic receptors was investigated in an animal model studyas a glutamatergic deficit is assumed in schizophrenia. By means of behavior tests, indicatingdeviations with schizophrenia, further associations of hypoxia and ischemia with thesubsequent occurrence of disturbance of behavior will be demonstrated with rats to developand characterize an improved model of the relation between molecularbiology and behavior.96 rats were exposed to chronic hypoxia or combined hypoxia/ischemia in order to examinethem subsequently for molecular-biological changes or behavioral deviations. The animals weredivided into 4 groups. Group 1 was ventilated, from the 4th to the 8th postnatal day of life,6hrs/d, in an airtight cage with 11% O2 / 89% N2 (chronic hypoxia = CH); group 2 serving ascontrol group. The corresponding cage was of identical design, but ventilated with air. Theanimals of group 3 had, on the 8th postnatal day, under anesthesia, 2x at an interval of 10 min,for 8 min each time, their arteria carotis communis clamped off on both sides (combinedhypoxia/ischemia = HI). During this clamp-off phase, the animals were placed in a gas-tightcontainer with 8% O2 / 92%N2, group 4 serving as control group. The animals were likewiseoperated on under anesthesia, but the carotid arteries were not clamped off.On the 11th postnatal day, 10 animals out of each group were killed, their brains exposed andquick-frozen in isopentane and liquid nitrogen. From the following planes of the brains, in acryostat, thin slices were cut (16µm for the “receptor autoradiography” = RAR, 20µm for the“in situ hybridization” = ISH) and fixed on object slides: pre-frontal cortex (PC), striatum (ST)and hippocampus (HIP). Within these planes, the following regions were examined: PC: Fr1/2,Cg1/3 and IL; ST: CPu and AcbC; HIP: CA1, CA2, CA3a, CA3c and DG. One half of thepreparations was examined by means of receptor autoradiography. In the process, threedifferent receptors were marked radioactively with the following ligands: the NMDA receptorwith [3H]-MK801, the AMPA-receptor with [3H]-AMPA and the GABAA-receptor with [3H]-muscimol. The ligand MK801 was mixed to spermidine or glycine during the main incubationfor binding enhanced. The other half of the preparations was examined by means of in situhybridization; in the process, the mRNAs of the following NMDA receptor subunits werelabeled by 35S: NR1, NR2A, NR2B, NR2C and NR2D. Next, the labeled preparations wereplaced onto radiation-sensitive film sheets and incubated for 5 weeks in the case of NMDA andGABAA, 4 weeks in the case of AMPA and 5 days in the case of ISH. The film sheets weredeveloped, the images on them photographed and digitally stored. Finally, the describedregions of the rat brains were examined for their binding with the aid of an evaluation program.The surviving animals were examined, on the 85th postnatal day (± 4d), for their prepulseinhibition (PPI) with prepulses (20ms) at 60, 70 and 80 dB, an inter-stimulus interval (50ms)and pulses at 100 dB (20ms).

105
A reduced number of NMDA, AMPA and GABAA receptors was noted after chronicalhypoxia and combined hypoxia/ischemia in the examined areas. Moreover, an increasedexpression of the mRNA of the NR1 subunit in CA2 region of hippocampus was noted. Anincreased expression of the mRNAs of the NR1 subunits after chronical hypoxia was noted(PC: Cg1/3, Fr1/2, IL; HIP: CA1, CA2, DG) as well as an increase of the NR2B subunits(AcbC), the NR2C subunits (HIP: CA3a) and the NR2D subunits(AcbC). A reduction of themRNA of the NR2A subunit (HIP: CA3a, CA3c) can be registered in other regions. Followingcombined hypoxia/ischemia an increase of the NR1 subunits in the prefrontal cortex (Fr1/2)and the hippocampus (DG) was found. Also, an increase of the NR2B subunits in hippocampus(CA2, CA3a) and an increase of the NR2D subunits in the prefrontal cortex (Cg1/3, IL) wasobserved. A decrease of the mRNA of the NR2A subunits in the hippocampus (CA3a, CA3c)following chronical hypoxia was found. Following combined hypoxia/ischemia the NR2Dsubunits in the nucleus accumbens and the NR2C subunits of the CA3a region of thehippocampus were decreased. The effects by chronical hypoxia seem to be larger than theeffects by combined hypoxia/ischemia. When measuring the PPI in 56 experimental animals onthe 85th day of life (± 4d), a significant reduction of the PPI in both experimental groups wasnoted.The reduction of receptor numbers in the case of NMDA, AMPA and GABAA receptors is anindication of the damage and impact of chronic hypoxia or combined hypoxia/ischemia.Upregulation of the NR1 and NR2B subunits could be interpreted as a compensatory attempt.In the process, the reduction of the NMDA receptor number suggests a hypofunction of thisreceptor, which is also assumed in the pathophysiology of schizophrenia. The increase of theeasily excitable NR2B subunit could be seen as a shift of conditions in favor of more readilyresponding units. The non-uniform development of the NR2C subunit could reflect both thediverse quality and effect of chronic hypoxia and combined hypoxia/ischemia and the diversesensitivities of the brain regions. Increase of the NR2D subunits can be interpreted as a furtherindication that chronic hypoxia or combined hypoxia/ischemia could be involved in theetiopathogenesis of schizophrenia.This study indicates a connection between chronic hypoxia or combined hypoxia/ischemia andmorphologic and quantitative changes at the NMDA, AMPA- and GABAA receptors. It seemslogical to consider the measured PPI deficits as a result of these changes.In summary, the findings demonstrate a critical role of hypoxia and ischemia in a vulnerablephase of brain development and indicate a possible involvement in the pathophysiology ofschizophrenia.The results of these tests demonstrate that hypoxia and/or ischemia are able to cause significantdamage to NMDA-, AMPA- and GABAA-receptors that can not be repaired until adulthood.These results suggest that chronic hypoxia respective combined hypoxia/ischemia are the causefor the acute molecularbiological changes in the young rat brain as well as for the laterdisruption in the sensorimotor gating in adult rats. These results are in accordance to otherinvestigations.Therefore, this animal model seems to be a useful tool to investigate the connection ofdisorders in behaviour and molecularbiological changes of receptors. It seems also to behelpful to establish new strategies for the treatment of schizophrenia and the prevention of theeffects of oxygene lack during pregnancy and during birth.
106
7. Anhang7.1.Einzeltierverzeichnis
Tabelle 30: Übersicht Versuchstiere – 1Nr. V M-Nr. * +D +T10 CH-V 2 05.01.01 16.01.01 11.11 CH-V 2 05.01.01 16.01.01 11.12 CH-V 2 05.01.01 16.01.01 11.13 CH-V 2 05.01.01 16.01.01 11.14 CH-V 7 09.03.01 20.03.01 11.15 CH-V 7 09.03.01 20.03.01 11.16 CH-V 7 09.03.01 20.03.01 11.17 CH-V 7 09.03.01 20.03.01 11.18 CH-V 8 10.03.01 21.03.01 11.19 CH-V 8 10.03.01 21.03.01 11.Nr. V M-Nr. * +D +T20 CH-K 1 06.01.01 17.01.01 11.21 CH-K 1 06.01.01 17.01.01 11.22 CH-K 1 06.01.01 17.01.01 11.23 CH-K 1 06.01.01 17.01.01 11.24 CH-K 4 18.03.01 29.03.03 11.25 CH-K 4 18.03.01 29.03.03 11.26 CH-K 4 18.03.01 29.03.03 11.27 CH-K 2 18.03.01 29.03.03 11.28 CH-K 2 18.03.01 29.03.03 11.29 CH-K 2 18.03.01 29.03.03 11.Nr: V M-Nr. * +D +T30 HI-V 1 06.01.01 17.01.01 11.31 HI-V 1 06.01.01 17.01.01 11.32 HI-V 1 06.01.01 17.01.01 11.33 HI-V 1 06.01.01 17.01.01 11.34 HI-V 4 24.12.00 05.01.01 11.35 HI-V 4 24.12.00 05.01.01 11.36 HI-V 4 24.12.00 05.01.01 11.37 HI-V 4 24.12.00 05.01.01 11.38 HI-V 9 10.03.01 21.03.01 11.39 HI-V 9 10.03.01 21.03.01 11.
107
Nr. V M-Nr. * +D +T40 HI-K 4 24.12.00 05.01.01 11.41 HI-K 6 12.11.00 23.11.00 11.42 HI-K 6 12.11.00 23.11.00 11.43 HI-K 6 12.11.00 23.11.00 11.44 HI-K 1 18.03.01 29.03.03 11.45 HI-K 1 18.03.01 29.03.03 11.46 HI-K 1 18.03.01 29.03.03 11.47 HI-K 1 12.11.00 23.11.00 11.48 HI-K 1 12.11.00 23.11.00 11.49 HI-K 1 12.11.00 23.11.00 11.
Abk.: Nr. = Nummer des Versuchstieres; V = durchgeführter Versuch; M-Nr. = Muttertier-Nummer; * = Geburtstag der Versuchs-Ratte; +D = Datum, zu welchem die Ratte getötetwurde; +T = Lebenstag, an welchem die Ratte getötet wurde.
108
Tabelle 31: Übersicht Versuchstiere – 2Nr. V M-Nr. * PPI-D PPI-T +D +T
50 CH-V 7 09.03.01 05.06.01 88. 07.06.01 90.51 CH-V 7 09.03.01 05.06.01 88. 07.06.01 90.52 CH-V 7 09.03.01 05.06.01 88. 07.06.01 90.53 CH-V 7 09.03.01 05.06.01 88. 07.06.01 90.54 CH-V 7 09.03.01 05.06.01 88. 07.06.01 90.55 CH-V 8 13.03.01 05.06.01 84. 11.06.01 90.56 CH-V 8 13.03.01 05.06.01 84. 11.06.01 90.57 CH-V 8 13.03.01 05.06.01 84. 11.06.01 90.58 CH-V 8 13.03.01 05.06.01 84. 11.06.01 90.59 CH-V 8 13.03.01 05.06.01 84. 11.06.01 90.59-b CH-V 1 05.11.01 30.01.02 86. 03.02.02 90.59-c CH-V 1 05.11.01 30.01.02 86. 03.02.02 90.59-d CH-V 1 05.11.01 30.01.02 86. 03.02.02 90.59-e CH-V 1 05.11.01 30.01.02 86. 03.02.02 90.59-f CH-V 1 05.11.01 30.01.02 86. 03.02.02 90.Nr. V M-Nr. * PPI-D PPI-T +D +T
60 CH-K 2 12.03.01 05.06.01 85. 10.06.01 90.61-J CH-K 7 30.05.01 21.08.01 83. 28.08.01 90.63 CH-K 1 28.05.01 21.08.01 81. 30.08.01 90.64-A CH-K 7 07.11.01 30.01.02 84. 05.02.02 90.64-B CH-K 7 07.11.01 30.01.02 84. 05.02.02 90.64-C CH-K 7 07.11.01 30.01.02 84. 05.02.02 90.64-D CH-K 7 07.11.01 30.01.02 84. 05.02.02 90.67 CH-K 1 06.08.01 30.10.01 85. 04.11.01 90.68-A CH-K 2 05.11.01 30.01.02 86. 04.02.02 90.68-B CH-K 2 05.11.01 30.01.02 86. 04.02.02 90.68-C CH-K 2 05.11.01 30.01.02 86. 04.02.02 90.68-D CH-K 2 05.11.01 30.01.02 86. 04.02.02 90.69-a CH-K 7 06.08.01 30.10.01 85. 04.11.01 90.69-b CH-K 7 06.08.01 30.10.01 85. 04.11.01 90.
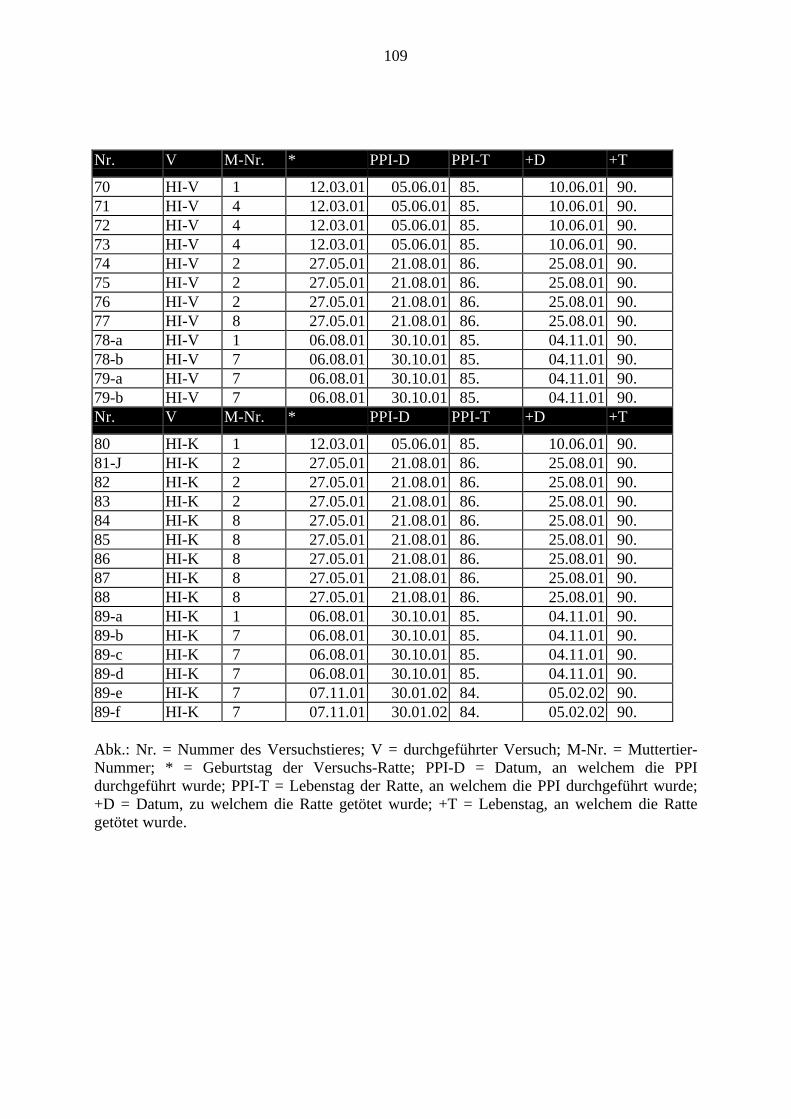
109
Nr. V M-Nr. * PPI-D PPI-T +D +T
70 HI-V 1 12.03.01 05.06.01 85. 10.06.01 90.71 HI-V 4 12.03.01 05.06.01 85. 10.06.01 90.72 HI-V 4 12.03.01 05.06.01 85. 10.06.01 90.73 HI-V 4 12.03.01 05.06.01 85. 10.06.01 90.74 HI-V 2 27.05.01 21.08.01 86. 25.08.01 90.75 HI-V 2 27.05.01 21.08.01 86. 25.08.01 90.76 HI-V 2 27.05.01 21.08.01 86. 25.08.01 90.77 HI-V 8 27.05.01 21.08.01 86. 25.08.01 90.78-a HI-V 1 06.08.01 30.10.01 85. 04.11.01 90.78-b HI-V 7 06.08.01 30.10.01 85. 04.11.01 90.79-a HI-V 7 06.08.01 30.10.01 85. 04.11.01 90.79-b HI-V 7 06.08.01 30.10.01 85. 04.11.01 90.Nr. V M-Nr. * PPI-D PPI-T +D +T
80 HI-K 1 12.03.01 05.06.01 85. 10.06.01 90.81-J HI-K 2 27.05.01 21.08.01 86. 25.08.01 90.82 HI-K 2 27.05.01 21.08.01 86. 25.08.01 90.83 HI-K 2 27.05.01 21.08.01 86. 25.08.01 90.84 HI-K 8 27.05.01 21.08.01 86. 25.08.01 90.85 HI-K 8 27.05.01 21.08.01 86. 25.08.01 90.86 HI-K 8 27.05.01 21.08.01 86. 25.08.01 90.87 HI-K 8 27.05.01 21.08.01 86. 25.08.01 90.88 HI-K 8 27.05.01 21.08.01 86. 25.08.01 90.89-a HI-K 1 06.08.01 30.10.01 85. 04.11.01 90.89-b HI-K 7 06.08.01 30.10.01 85. 04.11.01 90.89-c HI-K 7 06.08.01 30.10.01 85. 04.11.01 90.89-d HI-K 7 06.08.01 30.10.01 85. 04.11.01 90.89-e HI-K 7 07.11.01 30.01.02 84. 05.02.02 90.89-f HI-K 7 07.11.01 30.01.02 84. 05.02.02 90.
Abk.: Nr. = Nummer des Versuchstieres; V = durchgeführter Versuch; M-Nr. = Muttertier-Nummer; * = Geburtstag der Versuchs-Ratte; PPI-D = Datum, an welchem die PPIdurchgeführt wurde; PPI-T = Lebenstag der Ratte, an welchem die PPI durchgeführt wurde;+D = Datum, zu welchem die Ratte getötet wurde; +T = Lebenstag, an welchem die Rattegetötet wurde.

110
7.2. AbkürzungsverzeichnisAlle Abkürzungen, die in dieser Arbeit verwendet werden, sind nachfolgend alphabetischgeordnet aufgeführt:
* : Geburtstag der Versuchs-Ratte+D: Datum, zu welchem die Ratte getötet wurde+T: Lebenstag, zu welchem die Ratte getötet wurde5-HT: SerotoninA.: ArterieAcbC: Nucleus AccumbensAMPA: 8-amino-3-hydroxy-5-methyl-4-isoxazole-4-proprionateAS: Antisense-StrangASR: Allgemeine SchreckreaktionBmax: Rezeptorbindungbq: Bequerelbzw.: beziehungsweiseCa: CalciumCg1/3: Gyrus cinguliCH: chronische HypoxieCH-V: chronische Hypoxie - VersuchCH-K: chronische Hypoxie - Kontrollecpm: Lichtblitze pro MinuteCpu: Caudatus putamenCPP: 3-(2-carboxypiperazin-4-yl)-1-phosphonic aciddB: DezibelDG: Gyrus dentatus – Region des Ammonshorns (siehe auch Abbildung 1)DNA: DesoxyribonukleinsäureFr1/2: Frontaler Cortexd.h.: das heißtGABA: γ-Amino-ButtersäureGluR: Glutamat-Rezeptorgsl: Glycin, Sperimidin, lang: 180 min Vorinkubation; in der Hauptinkubation
werden Glycin und Spermidin beigemischtgl: Glycin, lang: 180 min Vorinkubation; in der Hauptinkubation wird Glycin
beigemischt, kein SpermidinHAB: HabituationHAB%: Habituation in %HI: kombinierte Hypoxie/IschämieHI-V: kombinierte Hypoxie/Ischämie - VersuchHI-K: kombinierte Hypoxie/Ischämie - KontrolleHIP: HippocampusIL: Infralimbischer CortexISH: In-Situ-Hybridisierungk: kurz: Vorinkubation 15 min; in der Hauptinkubation werden Glycin und
Spermidin beigischtKA: Kainat

111
kDA: Kilo-DaltonkHz: Kilo-HertzM-Nr.: Muttertier-NummerM.: MuskelMg: MagnesiumMK-801: (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleatemRNA: messenger-RibonukleinsäureMRI: Magnet-Resonanz-Imagingms: MillisekundenmV: MillivoltMW: Mittelwertn.s. nicht signifikantNa: NatriumNIX: normale, motorische Aktivität während des VersuchesNMDA: N-Methyl-D-AspartatNO: StickstoffmonoxidNr.: Nummer des VersuchstieresP60: Präpuls bei 60 dBP70: Präpuls bei 70 dBP80: Präpuls bei 80 dBP60SR: Präpuls mit darauf folgendem Schreckreiz bei 60 dBP70SR: Präpuls mit darauf folgendem Schreckreiz bei 70 dBP80SR: Präpuls mit darauf folgendem Schreckreiz bei 80 dBPAF: ParaformaldehydPC: Präfrontaler CortexPCP: PhencyclidinPCR: Polymerase-Ketten-Reaktionpnd: postnataler TagPRÄ: normale, motorische Aktivität vor VersuchsbeginnPPI60: Schreckreizhemmung (bei 60 dB gemessen) in ProzentPPI70: Schreckreizhemmung (bei 70 dB gemessen) in ProzentPPI80: Schreckreizhemmung (bei 80 dB gemessen) in ProzentPPI-D: Datum, an welchem die PPI durchgeführt wurdePPI-T: Lebenstag der Ratte, an welchem die PPI durchgeführt wurdeRAR: RezeptorautoradiographieRNA: RibonukleinsäureS: Sense-Strang (bei der ISH)STD: StandardabweichungSR: SchreckreizST: Striatumt: Trend, t ≤ 0.1TEA: TriethanolaminV.: VeneV: durchgeführter VersuchVers.-Tag: Lebenstag, an welchem der Versuch stattgefunden hat (siehe Tabelle 1)Zn: Zink
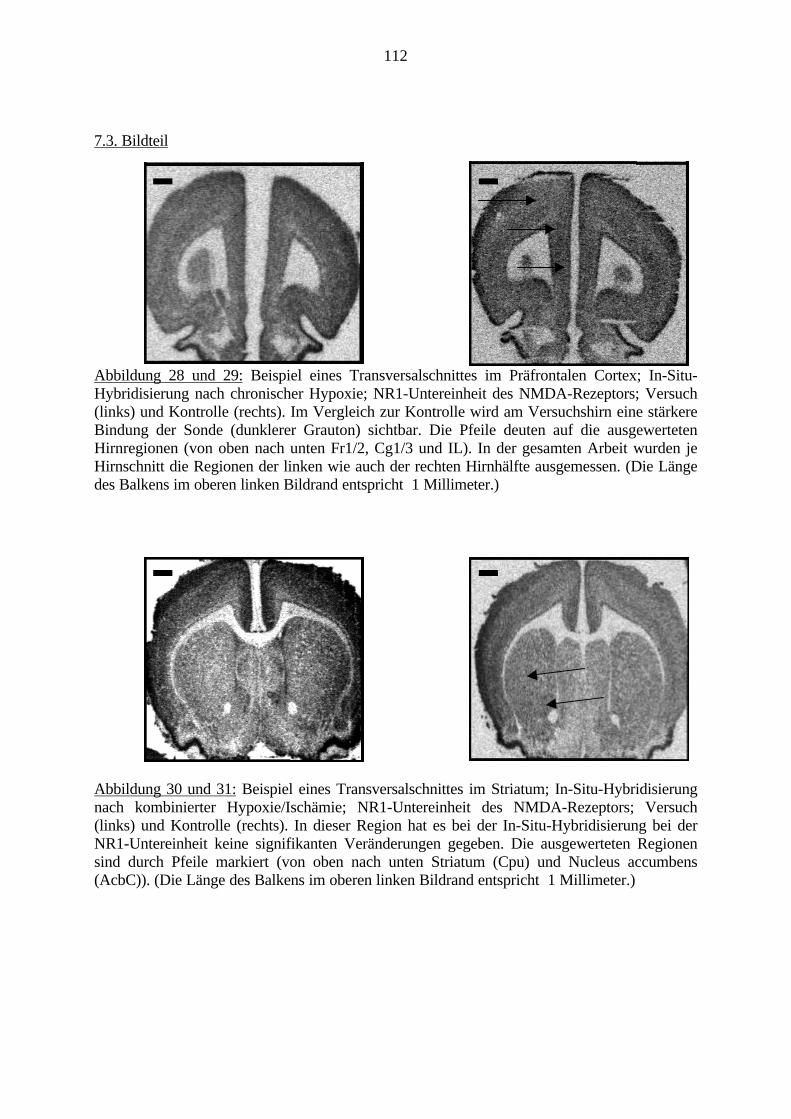
112
7.3. Bildteil
Abbildung 28 und 29: Beispiel eines Transversalschnittes im Präfrontalen Cortex; In-Situ-Hybridisierung nach chronischer Hypoxie; NR1-Untereinheit des NMDA-Rezeptors; Versuch(links) und Kontrolle (rechts). Im Vergleich zur Kontrolle wird am Versuchshirn eine stärkereBindung der Sonde (dunklerer Grauton) sichtbar. Die Pfeile deuten auf die ausgewertetenHirnregionen (von oben nach unten Fr1/2, Cg1/3 und IL). In der gesamten Arbeit wurden jeHirnschnitt die Regionen der linken wie auch der rechten Hirnhälfte ausgemessen. (Die Längedes Balkens im oberen linken Bildrand entspricht 1 Millimeter.)
Abbildung 30 und 31: Beispiel eines Transversalschnittes im Striatum; In-Situ-Hybridisierungnach kombinierter Hypoxie/Ischämie; NR1-Untereinheit des NMDA-Rezeptors; Versuch(links) und Kontrolle (rechts). In dieser Region hat es bei der In-Situ-Hybridisierung bei derNR1-Untereinheit keine signifikanten Veränderungen gegeben. Die ausgewerteten Regionensind durch Pfeile markiert (von oben nach unten Striatum (Cpu) und Nucleus accumbens(AcbC)). (Die Länge des Balkens im oberen linken Bildrand entspricht 1 Millimeter.)
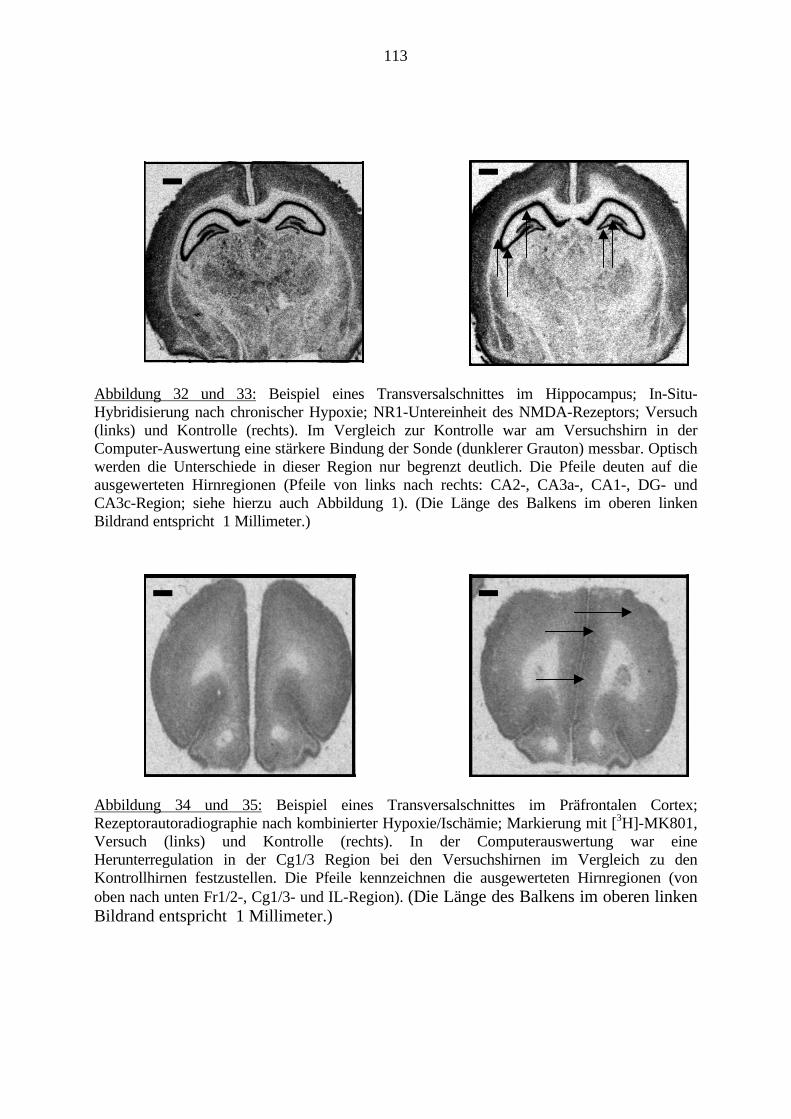
113
Abbildung 32 und 33: Beispiel eines Transversalschnittes im Hippocampus; In-Situ-Hybridisierung nach chronischer Hypoxie; NR1-Untereinheit des NMDA-Rezeptors; Versuch(links) und Kontrolle (rechts). Im Vergleich zur Kontrolle war am Versuchshirn in derComputer-Auswertung eine stärkere Bindung der Sonde (dunklerer Grauton) messbar. Optischwerden die Unterschiede in dieser Region nur begrenzt deutlich. Die Pfeile deuten auf dieausgewerteten Hirnregionen (Pfeile von links nach rechts: CA2-, CA3a-, CA1-, DG- undCA3c-Region; siehe hierzu auch Abbildung 1). (Die Länge des Balkens im oberen linkenBildrand entspricht 1 Millimeter.)
Abbildung 34 und 35: Beispiel eines Transversalschnittes im Präfrontalen Cortex;Rezeptorautoradiographie nach kombinierter Hypoxie/Ischämie; Markierung mit [3H]-MK801,Versuch (links) und Kontrolle (rechts). In der Computerauswertung war eineHerunterregulation in der Cg1/3 Region bei den Versuchshirnen im Vergleich zu denKontrollhirnen festzustellen. Die Pfeile kennzeichnen die ausgewerteten Hirnregionen (vonoben nach unten Fr1/2-, Cg1/3- und IL-Region). (Die Länge des Balkens im oberen linkenBildrand entspricht 1 Millimeter.)
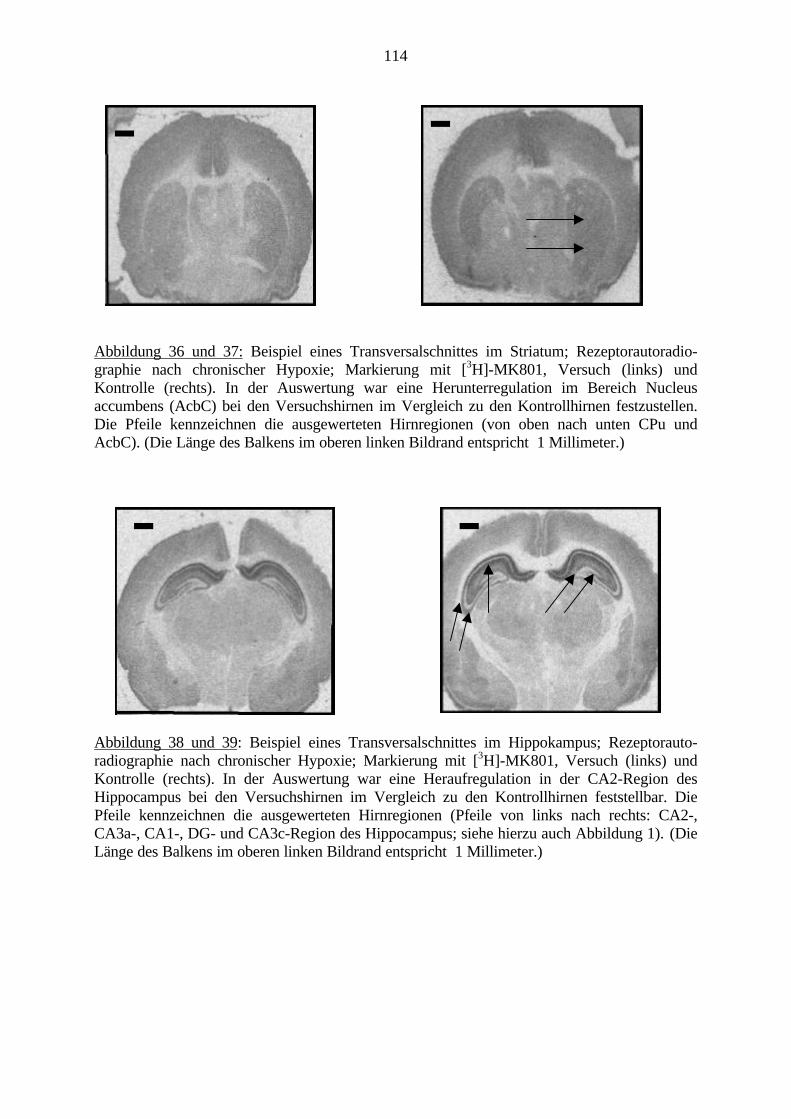
114
Abbildung 36 und 37: Beispiel eines Transversalschnittes im Striatum; Rezeptorautoradio-graphie nach chronischer Hypoxie; Markierung mit [3H]-MK801, Versuch (links) undKontrolle (rechts). In der Auswertung war eine Herunterregulation im Bereich Nucleusaccumbens (AcbC) bei den Versuchshirnen im Vergleich zu den Kontrollhirnen festzustellen.Die Pfeile kennzeichnen die ausgewerteten Hirnregionen (von oben nach unten CPu undAcbC). (Die Länge des Balkens im oberen linken Bildrand entspricht 1 Millimeter.)
Abbildung 38 und 39: Beispiel eines Transversalschnittes im Hippokampus; Rezeptorauto-radiographie nach chronischer Hypoxie; Markierung mit [3H]-MK801, Versuch (links) undKontrolle (rechts). In der Auswertung war eine Heraufregulation in der CA2-Region desHippocampus bei den Versuchshirnen im Vergleich zu den Kontrollhirnen feststellbar. DiePfeile kennzeichnen die ausgewerteten Hirnregionen (Pfeile von links nach rechts: CA2-,CA3a-, CA1-, DG- und CA3c-Region des Hippocampus; siehe hierzu auch Abbildung 1). (DieLänge des Balkens im oberen linken Bildrand entspricht 1 Millimeter.)

115
8. Literaturverzeichnis*Adams, W.; R.E. Kendell; E.H. Hare; P. Munk-Jorgensen (1993):Epidemiological evidence that maternal influenca contributes to the aetiology of schizophrenia.An analysis of Scottish, English, and Danish data.Br J Psychiatry 163: 522-534.
Akbarian, S.; N.J. Sucher; D. Bradley; A. Tafazzoli; D. Trinh; W.P. Hetrick; S.G. Potkin; C.A.Sandman; W.E. Bunney, Jr.; E.G. Jones (1996):Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex ofschizophrenics.The Journal of Neuroscience 16: 19-30.
Antier, D.; B.L. Zhang; F. Maillet; S. Akoka; L. Pourcelot; F. Sannajust F (1998a):Effects of neonatal focal cerebral hypoxia-ischemia on sleep-waking pattern, EcoG powerspectra and locomotor activity in the adult rat.Brain Research 807: 29-37.
Antier, D.; B.L. Zhang; D. Poisson; L. Pourcelot; F. Sannajust (1998b):Influence of neonatal focal cerebral hypoxia-ischemia on cardiovascular and neurobehavioralfunctions in adult Wistar rats.Neuroscience Letters 250: 57-60.
Arias, R.L.; J.R. Tasse; M.R. Bowlby (1999):Neuroprotective interaction effects of NMDA and AMPA receptor antagonist an in vitromodel of cerebral ischemia.Brain Research 816: 299-308.
Ausubel F.M.; R. Breut; R.E. Kongston; D.D. Moore; J.G. Seidmann; J.A. Smith; K. Struhl(1996):In Situ Hybridization and Immunhistochemistry. In: Current Protocols in Molecular Biology.John Wiley and Sons Inc. Chapter 14, Vol. 2: 11-28.
Balduini, W.; V. De Angelis; E. Mazzoni; M. Cimino (2000):Long-lasting behavioral alterations following a hypoxic/ischemic brain injury in neonatal rats.Brain Research 859: 318-325.
Barks, J.D.; F.S. Silverstein; K. Sims; J.T. Greenamyre; M.V. Johnston MV (1988):Glutamate recognition sites in human fetal brain.Neuroscience Letters 84: 31-35.
*Zeitschriftentitel-Abkürzungen gemäß:1. List of journals indexed in Index Medicus, Bethesda, Md.: Nat. Library of Medicine, 19892. Serial Sources for the BIOSIS Data Base. Philadelphia, Pa.: BioScience Information Services, 1988

116
Barnard, E.A.; P. Skolnick, R.W. Olsen; H. Möhler; W. Sieghart; G. Briggio; C. Braestrup;A.E. Bateson; S.Z. Langer (1998):International Union of Pharmacology. XV. Subtypes of gamma-aminobutyric acidA receptors:classification on the basis of subunit structure and receptor function.Pharmacol. Rev. 50: 291-313.
Berger, R.; Y. Garnier (2000):Perinatal brain injury.J. Perinat. Med. 28: 261-285.
Boksa, P.; D. Wilson; J. Rochford (1998):Responses to stress and novelty in adult rats born vaginally, by cesarean section or by cesareansection with acute anoxia.Biology of the Neonate 74: 48-59.
Bormann, J. (2000):The „ABC“ of GABA receptors.Trends Pharmacol. Sci. 21: 16-19.
Boss, N. (1993):Roche Lexikon Medizin, 3. Auflage, Urban & Schwarzenberg Verlag, München.
Brake, W.G.; P. Boksa; A. Gratton (1997):Effects of perinatal anoxia on the acute locomotor response to repeated amphetamineadministration in adult rats.Psychopharmacology 133: 389-395.
Brake, W.G.; R.M. Sullivan; A. Gratton A (2000):Perinatal distress leads to lateralized medial prefrontal cortical dopamine hypofunction in adultrats.Journal of Neurocscience 20: 5538-5543.
Brewer, G.J.; C.W. Cotman (1989):NMDA receptor regulation of neuronal morphology in cultured hippocampal neurons.Neuroscience Letters 99: 268-273.
Brixey, S.N.; B.J. Gallager III; J.A. McFallls Jr.; L.F. Parmelee (1993):Gestational and neonatal factors in the etiology of schizophrenia.Journal of Clinical Psychology 49: 447-456.
Cadenhead, K.S.; M.A. Geyer; D.L. Braff (1993):Impaired startle prepulse inhibition and habituation in patients with schizotypal personalitydisorder.Am J Psychiatry 150: 1862-1867.

117
Calabresi, P.; D. Centonze; G. Bernardi (2000):Cellular factors controlling neuronal vulnerability in the brain.Nuerology 55: 1249-1255.
Cannon; T.D.; S.A. Mednick; J. Parnas (1989):Genetic and perinatal determinants of structural brain deficits in schizophrenia.Arch. Gen. Psychiatry 46: 883-889.
Carlsson, A.; L.O. Hansson; N. Waters; M.L. Carlsson (1997):Neurotransmitter aberrations in schizophrenia: new perspectives and therapeutic implications.Life Sciences 61: 75-94.
Chaudieu, I.; H. Mount; R. Quirion; P. Boksa (1991):Transient postnatal increases in excitatory amino acid binding sites in rat ventral mesencepalon.Neuroscience Letters 133: 267-270.
Chen, Q.; A. Reiner (1996):Cellular distribution of the NMDA receptor NR2A / 2B subunits in the rat striatum.Brain Research 743: 346-352.
Cheung, H.H.; L. Teves; M.C. Wallace; J.W. Gurd (2001):Increased phosphorylation of the NR1 subunit of the NMDA receptor following cerebralischemia.Journal of Neurochemistry 78: 1179-1182.
Cobas, A.; A. Farien; G. Alvarez-Bolado; M.P. Sanchez (1991):Prenatal development of the intrinsic neurons of the rat neocortex: a comparative study of thedistribution of GABA-immunoreactive cells and the GABA-A receptor.Neuroscience 40: 375-397.
Cotman, C.W.; D.T. Monaghan; O.P. Ottersen; J. Storm-Mathisen (1987):Anatomical organization of excitatory amino acid receptors and their pathways.TINS 10: 273-280.
Coyle, J.T.; S.J. Enna (1976):Neurochemical aspects of the ontogenesis of GABAergic neurons in the rat brain.Brain Res. 11: 119-133.
Cull-Candy, S.G.; S.G. Brickley; C. Misra; D. Feldmeyer; A. Momiyama; M. Farrant (1998):NMDA receptor diversity in the cerebellum: identification of subunits contributing tofunctional receptors.Neuropharmacology 37: 1369-1380.

118
DeLisi, L.E.; O. Abramsky; B.M. Angrist; B. Bondy; K. Davison; H. Helmchen; J.H.Henderson; G. Huber; B. Jacobsen; J.G. Knight; T.F. McNeil; E.F. Torrey; D.A.J.Tyrell(1987):Nongenetic etiological factors. Group report.In: Biological Perspectives of Schizophrenia, Hrsg: Helmchen, H.; F.A. Henn, John Wiley &Sons Limited, Dahlem Konferenzen: 162-184.
Delivoria-Papadopoulos, M.; O.P. Mishra (2000):Mechanisms of perinatal cerebral injury in fetus and newborn.Annals of the New York Academy of Sciences 900: 159-168.
Docherty, N.M.; C. Grillon (1995):Affective reactivity of language and the startle response in schizophrenia.Biol Psychiatry. 38: 68-70.
El-Khodor, B.F.; P. Boksa (2000):Transient birth hypoxia increases behavioral responses to repeated stress in the adult rat.Behav Brain Res. 107: 171-175.
Espinoza, M.I.; J.T. Parer (1991):Mechanisms of asphyxial brain damage, and possible pharmacologic interventions, in the fetus.American Journal of Obstetrics and Gynecology 164: 1582-1591.
Farber, N.B.; D.F. Wozniak; M.T. Price; J.H. Labruyere; H. St. Peter; J.W. Olney (1995):Age-specific neurotoxicity in the rat associated with NMDA receptor blockade: potentialrelevance to schizophrenia?Biological Psychiatry 38: 788-796.
Fendt, M.; M.S. Fanselow (1999):The neuroanatomical and neurochemical basis of conditioned fear.Neurosci Biobehav Rev. 23: 743-760.
Fendt, M.; L. Li; J.S.Yeomans (2001):Brain stem circuits mediating prepulse inhibition of the startle reflex.Psychopharmacology 156: 216-224.
Ferchmin, P.A.; D. Perez; M. Biello (2000):Spermine is neuroprotective against anoxia and N-methyl-D-aspartate in hippocampal slices.Brain Research 859: 273-279.
Fritz, K.I.; F. Groenendaal; C. Andersen; S.T. Ohnishi; O.P. Mishra; M. Delivoria-Papadopoulos (1999a):Deleterious brain cell membrane effects after NMDA receptor antagonist administration tonewborn piglets.Brain Research 816: 438-445.

119
Fritz; K.I.; O.P. Mishra; M. Delivoria-Papadopoulos (1999b):Mg2+dependent modification of the N-methyl-D-aspartate receptor following graded hypoxiain the cerebral cortex of newborn piglets.Neuroscience 92: 685-692.
Gilland, E.; M. Puka-Sundvall; L. Hillered; H. Hagberg (1998):Mitochondrial function and energy metabolism after hypoxia-ischemia in the immature ratbrain: involvement of NMDA-receptors.Journal of Cerebral Blood Flow and Metabolism 18: 297-304.
Gottlieb, M.; C. Matute (1997):Expression of ionotropic glutamate receptor subunits in glial cells of the hippocampal CA1area following transient forebrain ischemia.Journal of Cerebral Blood Flow and Metabolism 17: 290-300.
Gozal, D.; Y.D. Xue; N. Simakajornboon (1999):Hypoxia induces c-fos protein expression in NMDA but not AMPA glutamat receptor labeledneurons within the nucleus tractus solitaris of the conscious rat.Neuroscience Letters 262: 93-96.
Groenendaal, F.; O.P. Mishra; J.E. McGowan; D.J. Hoffman; M. Delivoria-Papadopoulos(1997):Function of cell membranes in cerebral cortical tissue of newborn piglets after hypoxia andinhibition of nitric oxide synthase.Pedriatric Research 42: 174-179.
Günther-Genta, F.; P. Bovet; P. Holfeld (1994):Obstetric complications and schizophrenia. A case-control study.British Journal of Psychiatry 164: 165-170.
Günther, U.; J. Benson; D. Benke; J.M. Fritschy; G. Reyes; H. Möhler; B. Lüscher (1995):Benzodiazepine-insensitive mice generated by target disruption of the gamma2 subunit-gene ofgamma-aminobutyric acid type A receptors.Proc.Natl.Acad.Sci.USA 92: 7749-7753.
Häfner, H. (Hrsg.) (1995):Was ist Schizophrenie?Gustav Fischer Verlag.
Henn, F.A. (1987):Neurotransmitter Abnormalities in Schizophrenia.In: Helmchen, H.; F.A. Henn, John Wiley & Sons Limited (Hrsg.): Biological Perspectives ofSchizophrenia, Dahlem Konferenzen: 299-309.

120
Hoeger, H.; M. Engelmann; G. Bernert; R. Seidl; H. Bubna-Littitz; W. Mosgoeller; B. Lubec;G. Lubec (2000):Long term neurological and behavioral effects of graded perinatal asphyxia in the rat.Life Sciences 66: 947-962.
Hoffman, D.J.; J.E. McGowan; P.J. Marro; O.P. Mishra; M. Delivoria-Papadopoulos (1994):Hypoxia induced modification of the N-methyl-D-aspartate receptor in the brain of thenewborn piglet.Neuroscience Letters 167: 156-160.
Hsu, J.C.; Y. Zhang; N. Takagi; J.W. Gurd; M.C. Wallace; L. Zhang; J.H. Eubanks (1998):Decreased expression anf functionality of NMDA receptor complexes persist in the CA1, butnot in the dentate gyrus after transient cerebral ischemia.Journal of Cerebral Blood Flow and Metabolism 18: 768-775.
Isaacson, R.L. (1987):In: Encyclopedia of Neuroscience: Adelmann,G. (Hrsg.).Birkhäuser-Verlag: 492-495.
Iuvone, L.; M.C. Geloso; E. Dell'Anna (1996):Changes in open field behavior, spatial memory, and hippocampal parvalbuminimmunoreactivity following enrichment in rats exposed to neonatal anoxia.Experimental Neurology 139: 25-33.
Iwahashi, K.; M. Watanabe; K. Nakamura; H. Suwaki; T. Nakaya; Y. Nakamura;H. Takahashi; K. Ikuta (1997):Clinical investigation of the relationship between Borna disease virus (BDV) infection andschizophrenia in 67 patients in Japan.Acta Psychiatr Scand 96: 412-415.
Javitt, D.C.; S.R. Zukin (1991):Recent advances in the phencyclidine model of schizophrenia.American Journal of Psychiatry 148: 1301-1308.
Johnston, M.V.; W.H.Trescher; A. Ishida; W. Nakajima (2001):Neurobiology of hypoxic-ischemic injury in the developing brain.Pediatric Research 49: 735-741.
Kim J.S.; H.H. Kornhuber; Schmid-Burgk, B. Holzmüller (1980):Low cerebrospinal fluid glutamate in schizophrenie patients and a new hypothesis ofschizophrenia.Neuroscience Letters 20: 379-382.
Kim, W.T.; M.F. Kuo; O.P. Mishra; M. Delivoria-Papadopoulos (1998):Distribution and expression of the subunits of N-methyl-D-aspartate (NMDA) receptors;NR1,NR2A, and NR2B in hypoxic newborn piglet brains.Brain Research 799: 49-54.

121
Koch, M.; T.W. Robbins (2001):Special issue on the psychopharmacology of prepulse inhibition: basic and clinical studies.Psychopharmacology 156: 115-116.
Kornhuber, J.; F. Mack-Burkhardt; P. Riederer; G.F. Hebenstreit; G.P. Reynolds;H.B. Andrews; H. Beckmann (1989):[3H]MK-801 binding sites in postmortem brain regions of schizophrenic patients.Journal of Neural Transmission 77: 231-236.
Krajnc, D.; N.H. Neff; M. Hadjiconstantinou (1996):Glutamate, glutamine and glutamine synthetase on the neonatal rat brain following hypoxia.Brain Research 707: 134-137.
Laurie, D.J.; P.H. Seeburg (1994):Ligand affinities at recombinant N-methyl-D-aspartate receptors depend on subunitcomposition.European Journal of Pharmacology 268: 335-345.
Lewis, S.W.; R.M. Murray (1987):Obststric complications, neurodevelopmental deviance, and risk of schizophrenia.J. Psychiat. Res. 21: 413-421.
Lipska, B.K.; D.R. Weinberger (2000):To model a psychiatric disorder in animals: Schizophrenia as a reality test.Neuropsychopharmacology 23: 223-239.
Liu, Y.; G. Zhang; C. Gao; X. Hou (2001):NMDA receptor activation results in tyrosine phosphorylation of NMDA receptor subunit2A(NR2A) and interaction of Pyk2 and Src with NR2A after transient cerebral ischemia andreperfusion.Brain Research 909: 51-58.
Lo Turco, J.J.; M.G. Blanton; A.R. Kriegstein (1991):Initial expression and endogenous activation of NMDA channels in early neocorticaldevelopment.The Journal of Neuroscience 11: 792-799.
Lorente de No, R. (1934):Studies on the structure of the cerebral cortex. II. Continuation of the study of the ammonicsystem.J. Psychol. Neurol. 46: 113-177.
Luhmann, H.J.; T. Kral; U. Heinemann (1993):Influence of hypoxia on excitation and GABAergic inhibition in mature and developing ratneocortex.Exp. Brain Res. 97: 209-224.

122
McCoy, L.; E. K. Richfield. (1996a):Washable endogenous substances and regional heterogeneity in agonist enhanced [3H]MK-801binding in rat brain.Brain Research 710:103-111.
McCoy, L.; E. K. Richfield. (1996b):Chronic antipsychotic treatment alters glycine-stimulated NMDA receptor binding in rat brain.Neuroscience Letters 213:137-141.
McDonald, J.W.; T. Bhattacharyya; S.L. Sensi; D. Lobner; H.S. Ying; L.M. Canzoniero; D.W.Choi (1998):Extracellular acidity potentiates AMPA receptor-mediated cortical neuronal death.Journal of Neuroscience 18: 6290-6299.
McDonald, J.; M.V. Johnston (1990):Physiological and pathophysiological roles of excitatory amino acids during central nervoussystem development.Brain Research reviews 15: 41-70.
McDowd, J.M.; D.L. Filion; M.J. Harris; D.L. Braff (1993):Sensory gating and inhibitory function in late-life schizophrenia.Schizophr Bull. 19: 733-746.
McGrath, J.; R. Murray (1995):Risk factors for schizophrenia: from conception to birth.In: Hirsch, S.R.; D.R. Weinberger: Schizophrenia. Blackwell science 187-205.
McNeil, T.F. (1987):Perinatal influences in the development of schizophrenia.In: Helmchen H, Henn FA (eds.): Biological perspectives of schizophrenia. John Wiley & sonsLimited. S. Bernhard: Dahlem Konferenzen: 125-138.
Meincke, U.; Gouzoulis-Mayfrank; H. Saß (2001):Der Startle-Reflex in der Schizophrenieforschung.Nervenarzt, 72: 844-852.
Mishra, O.P.; M. Delivoria-Papadopoulos (1999):Cellular mechanisms of hypoxic injury in the developing brain.Brain Research Bulletin 48: 233-238.
Möhler, H. (1992):GABAergic synaptic transmission. Regulation by drugs.Drug Res. 42: 211-214.

123
Monyer, H.; N. Burnashev; D.J. Laurie; B. Sakmann; P.H. Seeburg (1994):Developmental and regional expression in the rat brain and functional properties of fourNMDA receptors.Neuron 12: 529-540.
Moriyoshi, K.; M. Masu; T. Ishii; R. Shigemoto; N. Mizuno; S. Nakanishi (1991):Molecular cloning and characterization of the rat NMDA receptor.Nature 354: 31-37.
Nakanishi, S (1992):Molecular diversity of glutamate receptors and implications for brain function.Science 258: 597-603.
Nauta, W.J.H.; M. Feirtag (1986):Fundamental Neuroanatomy.W.H.Freeman and Company Verlag: 272-278.
O'Callaghan, E.; T. Gibson; H.A. Colohan; P. Buckley; D.G. Walshe; C. Larkin;J.L. Waddington (1992):Risk of schizophrenia in adults born after obstetric complications and their association withearly onset of illness: a controlled study.BMJ 305: 1256-1259.
Olivier, B.; C. Leahy; T. Mullen; R. Paylor; V.E. Groppi; Z. Sarnyai (2001):The DBA/2J strain and prepulse inhibition of startle: a Model system to test antipsychotics?Psychopharmacology 156: 284-290.
Olney, J.W.; Farber, N.B. (1995):Glutamate receptor dysfunction and schizophrenia.Archives of General Psychiatry 52: 998-1007.
Olsen, R.W. und A.J. Tobin (1990):Molecular biology of GABA-A reeptors.FASEB 4: 1469-1480.
Ota, A.; T. Ikeda; K. Abe; H. Sameshima; X.Y. Xia; Y.X. Xia; T. Ikenoue (1998):Hypoxic-ischemic tolerance phenomenon observed in neonatal rat brain.American Journal of Obstetrics and Gynecology 179: 1075-1078.
Owen, M.J.; S.W. Lewis; R.M. Murray (1988):Obstetric complications and schizophrenia: a computed tomographic study.Psychological Medicine 18: 331-339.
Parnas, J.; F. Schulsinger; T.W. Teasdale; H. Schulsinger; P.M. Feldman; S.A. Mednick(1982):Perinatal complications and clinical outcome within the schizophrenia spectrum.Brit. J. Psychiat. 140: 416-420.

124
Paxinos, G.; C. Watson (1986):The Rat Brain in Stereotaxic Coordinates.Academic Press, Sydney.
Pearigen, P.; R. Gwinn; R.P. Simon (1996):The effects in vivo of hypoxia on brain injury.Brain Research 725: 184-191.
Pearlson, G.D.; D.J. Garbacz; P.J. Moberg; H.S. Ahn; J.R. DePaulo (1985):Symptomatic, familial, perinatal, and social correltes of computerized axial tomography (CAT)changes in schizophrenics and bipolars.The Journal of Nervous and Mental Disease 173: 42-50.
Perez-Velazquez, J.L.; L. Zhang (1994):In vitro hypoxia induces expression of the NR2C subunit of the NMDA receptor in rat cortexand hippocampus.Journal of Neurochemistry 53: 1171-1173.
Pichiule, P.; J.C. Chavez; J. Boero; A. Arregui (1996):Chronic hypoxia induces modification of the N-methyl-D-aspartate receptor in rat brain.Neuroscience Letters 218: 83-86.
Pujic, Z.; I. Matsumoto; P.A. Wilce (1993):Expression of the gene coding for the NR1 subunit of the NMDA receptor during rat braindevelopment.Neuroscience Letters 162: 67-70.
Reyes, M.; A. Reyes; T. Opitz; M.A. Kapin; P.K. Stanton (1998):Eliprodil, a non-competitive, NR2B-selective NMDA antagonist, protects pyramidal neuronsin hippocampal slices from hypoxic/ischemic damage.Brain Res. 782: 212-218.
Riecher-Rössler, A.; H. Hafner (1993):Schizophrenia and oestrogens - is there an association?Eur Arch Psychiatry Clin Neurosci. 242: 323-328.
Romijn, H.J. (1989):Preferential loss of GABAergic neurons in hypoxia-exposed neocortex slab cultures isattenuated by the NMDA receptor blocker D-2-amino-7-phosphonoheptanoate.Brain Research 501: 100-104.
Romijn, H.J.; M.A. Hofman; A. Gramsbergen (1991):At what age is the developing cerebral cortex of the rat comparable to that of the full-termnewborn human baby.Early Human Development 26: 61-67.

125
Romijn, H.J., A.W.J.W. Janszen; M.J.D. van Voorst, R.M. Buijs, R. Balazs; D.F. Swaab(1992):Perinatal hypoxic ischemic encephalopathy affects the proportion of GABA-immunoreactiveneurons in the cerebral cortex of the rat.Brain Research 592: 17-28.
Rothe, T.; H. Middleton-Price, V. Bigl (1988):The ontogeny of GABA receptors and glutamic acid decarboxylase in regions of the rat brain.Neuropharmacology 27: 661-667.
Sakurai, S.Y.; J.H.J. Cha; J.B. Penney; A.B. Young (1991):Regional distribution and properties of [H]MK-801 binding sites determined by quantitativeautoradiography in rat brain.Neuroscience 40: 533-543.
Sambrook, J.; e. F. Fritsch; T. Maniatis (1989):Molecular Cloning, A Labarotory manual, 2. Auflage.Cold Spring Harbor Laboratory Press.
Sasaki, Y.F.; Rothe, T.; Premkumar, L.S.; Das, S.; Cui, J.; Talantova, M.V.; Wong, H.K.;Chan, S.F.; Zhang, D.; Nakanishi, N.; Sucher N.J.; Lipton, S.A. (2002):Characterization and comparison of the NR3A subunit of the NMDA receptor in recombinantsystems and primary cortical neurons.J Neurophysiol, 87: 2052-2063.
Schafer, M.K.-H., J.P. Herman; S.J. Watson (1993):In situ hybridization histochemistry.In: Imaging Drug Action in the Brain, hrsg. V. D.D. London, CRC Press, Boca Rhadon [u.a.]:337-378.
Schmidt-Kastner, R.; T.F. Freund (1991):Selective vulnerability of the hippocampus in brain ischemia.Neuroscience 40: 599-636.
Schmitt, A.; W. Weber-Fahr; A. Jatzko; H. Tost; F.A. Henn; D.F. Braus (2001):Aktueller Überblick über strukturelle Magnetresonanztomographie bei Schizophrenie.Fortschr Neurol Psychiat: 69105-69115.
Sebastião, A.M.; A. de Mendoca; T. Moreira; J.A. Ribeiro (2001):Activation of synaptic NMDA receptors by action potential-dependent release of transmitterduring hypoxia impairs recovery of synaptic transmission on reoxygenation.The Journal of Neuroscience 21: 8564-8571.
Segal, M. (1993):GABA induces a unique rise of Ca in cultured rat hippocampal neurons.Hippocampus 3: 229-238.

126
Sheng, M.; J. Cummings; L.a. Roldean; Y.N. Jan; L.Y. Jan (1994):Changing subunit composition of heteromeric NMDA receptors during development of ratcortex.Nature 368: 144-147.
Sieghart, W. (1992):GABAA receptors: ligand-gated Cl- ion channels modulated by multiple drug-binding sites.Trends Pharmacol. Sci. 13: 446-450.
Simakajornboon, N.; E. Gozal; Y.M. Gozal; D. Gozal (2000):Hypoxia induces activation of a N-methyl-D-aspartate glutamate receptor-Protein-kinase Cpathway in the dorsocaudal brainstem of the conscious rat.Neuroscience Letters 278: 17-20.
Spandou, E.; G. Karkavelas; V. Soubashi; P. Avgovstides-Savvopoulou; T. Loizidis;O. Guiba-Tziampiri (1999):Effect of ketamine on hypoxic-ischemic brain damage in newborn rats.Brain Research 819: 1-7.
Starke, K. (1996):Grundlagen der Pharmakologie des Nervensystems.In: W. Forthe, D. Henschler, W. Rummel und K.Starke (Hrsg.):Allgemeine und spezielle Pharmakologie und Toxikologie.Spektrum Akademischer Verlag GmbH, Heidelberg/Berlin/Oxford: 103-133.
Stefanis, N.; S. Frangon; J. Yakeley; T. Sharma; P. O´Connel; K. Morgan; T. Sigmudsson; M.Taylor; R. Murray (1999):Hippocampal volume reduction in schizophrenia: effects of genetic risk and pregnancy andbirth complications.Biological Psychiatry 46: 697-702.
Subramaniam, S.; P. McGonigle (1991):Quantitative autoradiographic characterization of the binding of (+)-5-methyl-10, 11-dihydro-5H-dibenzo[a,d]cyclohepten-5,1-imine ([3H]MK-801) in rat brain: regional effects ofpolyamines.Journal of Pharmacology and Experimental Therapeutics 256: 811-819.
Suddath, R.L.; G.W. Christison; E.F. Torrey; M.F. Casanova; D.R. Weinberger (1990):Anatomical abnormalities in the brains of monozygotic twins discordant for schizophrenia.The New England Journal of Medicine 322: 789-794.
Swerdlow, N.R.; S.B. Caine; M.A. Geyer (1992):Regionally selective effects of intracerebral dopamine infusion on sensorimotor gating of thestartle reflex in rats.Psychopharmacology 108: 189-195.

127
Swerdlow, N.R.; M.A. Geyer; D.L. Braff (2001):Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge andfuture challenges.Psychopharmacology 156: 194-215.
Takagi, N.; K. Shinno; L. Teves; N. Bissoon; M.C. Wallace; J.W. Gurd (1997):Transient ischemia differentially increases tyrosine phosphorylation of NMDA receptorsubunits 2A and 2B.Journal of Neurochemistry: 1060-1065.
Thornberg, S.A.; S.R. Saklad (1996):A review of NMDA receptors and the phencyclidine model of schizophrenia.Pharmacotherapy 16: 82-93.
Toner, C.C.; J.A. Stamford (1997):Characteristics of the NMDA receptor modulating hypoxia / hypoglycaemia-induced rat striataldopamine release in vitro.European Journal of Pharmacology 340: 133-143.
Torrey, E.F. (1992):Are we overestimating the genetic contribution to schizophrenia?Schizophr Bull. 18: 159-170.
Uhl, G.R. (1993):A review of in situ hybridization technique: relevance to combined immunocytochemicalstudies.In: Immunhistochemistry II, hrsg. V. A.C. Cuello, IBRO, Chichester, 1993: 281-300.
Van Eden, C.G.; L. Mrzljak; P. Voorn; H.B.M. Uylings (1989):Prenatal development of GABA-ergic neurons in the neocortex of the rat.The Journal of Comparative Neurology 289: 213-227.
Vannucci, R.C.; R.M. Brucklacher; S.J. Vannucci (2001):Intracellular calcium accumulation during the evolution of hypoxic-ischemic brain damage inthe immature rat.Developmental Brain Research 126: 117-120.
Viapiano, M.S.; A.M. Mitridate de Novara, S. Fiszer de Plazas; C.E. Bozzini (2001):Prolonged exposure to hypobaric hypoxia transiently reduces GABA(A) receptor number inmice cerebral cortex.Brain Res. 894: 31-36.
Wafford, K.A.; C.J. Bain; B. Le Bourdelles; P.J. Whiting; J.A. Kemp (1993):Preferential co-assembly of recombinant NMDA receptors composed of three differentsubunits.Neuro Report 4: 1347-1349.

128
Watling, K.J. (1998):The RBI Handbook of Receptor Classification and Signal Transduction.3rd Edition, Sigma-Aldrich Research Biochemicals Incorporated.
Weinberger, D.R. (1987):Implications of normal brain development for the pathogenesis of schizophrenia.Archives of General Psychiatry 44: 660-669.
Weinberger, D.R. (1999):Cell biology of the hippocampal formation in schizophrenia.Biol Psychiatry 45: 395-402.
Wenzel, A.; M. Villa; H. Mohler; D. Benke (1996):Developmental and regional expression of NMDA receptor subtypes containing the NR2Dsubunit in rat brain.Journal of Neurochemistry 66: 1240-1248.
Whiting, P.J.; T.P. Bonnert; R.M. McKernan; S. Farrar; B. Le Bourdelles; R.P. Heavens; D.W.Smith; L. Hewson; M.R. Rigby; D.J. Sirinathsinghji; S.A. Thompson; K.A. Wafford (1999):Molecular and functional diversity of the expanding GABAA receptor gene family.Ann N Y Acad Sci. 868: 645-53.
Xie, J.; G. Lu; Y. Hou (1999):Role of excitatory amino acids in hypoxic preconditioning.Biological Signals and Receptors 8: 267-274.
Yolken, R.H.; H. Karlsson; F. Yee; N.L. Johnston-Wilson; E.F. Torrey (2000):Endogenous retroviruses and schizophrenia.Brain Research, Brain Research Review 31: 193-199.
Yoshioka, A.; Y. Yamaya; S. Saiki; M. Kanemoto; G. Hirose; J. Beesley; D. Pleasure (2000):Non-N-methyl-D-aspartate glutamate receptors mediate oxygen-glucose deprivation-inducedoligodendroglial injury.Brain Research 854: 207-215.
Zhang, L.; J.C. Hsu; N. Takagi; J.W. Gurd; M.C. Wallace; J.H. Eubanks (1997):Transient global ischemia alters NMDA receptor expression in rat hippocampus: correlationwith decreased immunoreactive protein levels of the NR2A/B subunits, and an altered NMDAreceptor functionality.Journal of Neurochemistry: 1983-1994.
Zhao, G.; M.P. Flavin (2000):Differential sensitivity of rat hippocampal and cortical astrocytes to oxygen-glucosedeprivation injury.Neuroscience Letters 285: 177-180.

129
Zhong, J.; V.K. Gribkoff; P.B. Molinoff (1996):Use of subunit specific antisense oligodeoxynucleotides to define developmental changes in theproperties of N-methyl-D-aspartate receptors.Molecular Pharmacology 50: 631-638.
Zilles, K.; A. Schleicher. (1995):Correlative imaging of transmitter receptor distributions in human cortex.pp 277-307, in Stumpf W.E., Solomon H.F. (eds.), Autoradiography and correlative imaging,Academic Press, San Diego
Zilles K.; Qu M.S.; Kohling R.; Speckmann E.J. (1999):Ionotropic glutamate and GABA receptors in human epileptic neocortical tissue: quantitativein vitro receptor autoradiography.Neuroscience. 94:1051-1061.

130
Danksagung
Abschließend möchte ich allen danken, die zum Gelingen dieser Arbeit beigetragen haben.
Mein Dank gilt meiner Betreuerin am Zentralinstitut für Seelische Gesundheit (ZI) inMannheim, Frau Dr. A. Schmitt für die Überlassung der abwechslungsreichen, umfassendenThematik, für Ihre Ratschläge und die Möglichkeit, selbstständig zu arbeiten, ebenso wie fürdie Einarbeitung in die Methode der Rezeptorautoradiographie. Ferner danke ich ihr für ihreRatschläge beim Umgang mit dem SPSS-Statistik-Programm. Ihr und Herrn Dr. M. Zink giltmein besonderer Dank für das zur Verfügung Stellen der Plasmide.
Danken möchte ich Herrn Prof. Löscher für die sehr freundliche Vermittlung meiner Arbeit anHerrn Prof. Dr. U. Ebert.
Herrn Prof. Dr. U. Ebert danke ich sehr herzlich, dass er sich dieser externen Arbeitangenommen und immer Zeit gefunden hat, sie mit seiner sympathischen Art immerkonstruktiv zu betreuen.
Herrn PD Dr. Markus Fendt danke ich für seine Mühe, mehrfach seine PPI Apparatur nachMannheim zu bringen, dort aufzubauen und mit mir die Versuche durchzuführen. Sehr herzlichdanke ich ihm für die zur Verfügung gestellten Daten und ganz besonders für seine freundlicheUnterstützung durch viele hilfreiche Ratschläge.
Für die Einarbeitung in die In-Situ-Hybridisierung und für die stete Bereitschaft meine Fragenausführlich zu diskutieren, danke ich sehr herzlich Frau Brigitte May, Frau Bettina Müller undFrau Simone Keck.
Den Tierpflegern im Klinikum Mannheim, Frau Petra Prochatzka und Frau Martina Bohrer,möchte ich für ihr kompetentes Engagement bei der Pflege und Zucht der Tiere danken; ihnenund Frau Viktoria Skude gilt besonderer Dank, für ihre hilfreichen Ratschläge zurVorbereitung und Organisation meiner Versuche, im Besonderen aber für das zur VerfügungStellen von Gerätschaften und für viele lustige Geschichten.
Den Mitarbeitern des Labors im ZI in Mannheim und allen anderen Mitarbeitern, die mich mitRatschlägen oder Aufmunterungen unterstützt haben, möchte ich an dieser Stelle herzlichdanken. Ich habe mich in diesem Kreis immer sehr wohl gefühlt.
Frau Reinhard von der biostatistischen Abteilung des ZI Mannheim danke ich für ihrEngagement, mir statistische Problemstellungen zu erklären.
Herrn PhD. W. Gunia von der Firma Interfocus danke ich für das geduldige Eingehen aufmeine vielen telefonischen Fragen zum AIS-Auswertungs Programm.





























