Embed Size (px)
Citation preview

VorlesungTheoretische Chemie ISommersemester 2015
Eckhard SpohrLehrstuhl fur Theoretische Chemie
Universitat Duisburg-EssenD-45141 Essen, Germany
26. Juni 2017


Inhaltsverzeichnis
I Einleitung und Motivation 1
1 Probleme der klassischen Physik zu Beginn des 20. Jahrhun-derts 21.1 Ubersicht . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21.2 Zusatzmaterial . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2.1 Atomspektren . . . . . . . . . . . . . . . . . . . . . . . 21.2.2 Schwarzer Strahler . . . . . . . . . . . . . . . . . . . . 41.2.3 Spezifische Warme . . . . . . . . . . . . . . . . . . . . 61.2.4 Photoelektrischer und Compton Effekt . . . . . . . . . 61.2.5 Dualitat . . . . . . . . . . . . . . . . . . . . . . . . . . 8
2 Was ist Theoretische Chemie? 92.1 Motivation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92.2 Teilgebiete der Theoretischen Chemie . . . . . . . . . . . . . . 92.3 Moderne theoretische Chemie . . . . . . . . . . . . . . . . . . 10
3 Klassische Teilchen und Wellen 133.1 Klassische Teilchen . . . . . . . . . . . . . . . . . . . . . . . . 133.2 Wellen (klassisch) . . . . . . . . . . . . . . . . . . . . . . . . . 14
4 Das Doppelspaltexperiment 16
II Quantenmechanik 20
5 Axiome der Quantenmechanik 215.1 Wellenfunktion und Aufenthaltswahrscheinlichkeitsdichte . . . 215.2 Hermitesche Operatoren und physikalische Observable . . . . . 22
5.2.1 Lineare Operatoren . . . . . . . . . . . . . . . . . . . . 225.2.2 Eigenfunktionen und Eigenwerte . . . . . . . . . . . . . 225.2.3 Operatoren . . . . . . . . . . . . . . . . . . . . . . . . 255.2.4 Kommutatoren . . . . . . . . . . . . . . . . . . . . . . 275.2.5 Dirac-Notation . . . . . . . . . . . . . . . . . . . . . . 285.2.6 Hermitesche Operatoren . . . . . . . . . . . . . . . . . 295.2.7 Operatordarstellungen . . . . . . . . . . . . . . . . . . 29
iii

5.3 Erwartungswerte . . . . . . . . . . . . . . . . . . . . . . . . . 315.4 Die Schrodingergleichung . . . . . . . . . . . . . . . . . . . . . 34
5.4.1 Zeitabhangige Schrodingergleichung . . . . . . . . . . . 345.4.2 Zeitunabhangige Schrodingergleichung . . . . . . . . . 36
5.5 Die Unscharferelation . . . . . . . . . . . . . . . . . . . . . . . 375.6 Zusammenfassung . . . . . . . . . . . . . . . . . . . . . . . . . 40
III Exakte Losungen der stationaren Schrodinger-gleichung 42
6 Eindimensionale Probleme 446.1 Das Teilchen im unendlich tiefen Kasten . . . . . . . . . . . . 45
6.1.1 Modell und Losung der Schrodingergleichung . . . . . . 456.1.2 Zustande des Teilchens im Kasten . . . . . . . . . . . . 496.1.3 Erwartungswerte und Varianzen fur das Teilchen im
Kasten . . . . . . . . . . . . . . . . . . . . . . . . . . . 546.1.4 Zusatzmaterial . . . . . . . . . . . . . . . . . . . . . . 57
6.2 Der harmonische Oszillator . . . . . . . . . . . . . . . . . . . . 586.2.1 Federmodell . . . . . . . . . . . . . . . . . . . . . . . . 586.2.2 Schrodingergleichung . . . . . . . . . . . . . . . . . . . 596.2.3 Losung der Schrodingergleichung . . . . . . . . . . . . 626.2.4 Form der Wellenfunktionen und Aufenthaltswahrschein-
lichkeitsdichte . . . . . . . . . . . . . . . . . . . . . . . 67
7 Zwei- und Dreidimensionale Probleme in kartesischen Koor-dinaten 727.1 Das Teilchen im zweidimensionalen Kasten . . . . . . . . . . . 727.2 Das Teilchen im dreidimensionalen Kasten . . . . . . . . . . . 807.3 Der harmonische Oszillator in 3 Dimensionen . . . . . . . . . . 847.4 Erweiterung auf mehr als ein Teilchen . . . . . . . . . . . . . . 88
8 Zentralkraft-Probleme 908.1 Einleitung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
8.1.1 Kugelkoordinaten . . . . . . . . . . . . . . . . . . . . . 918.1.2 Teilchen auf der Kugeloberfache . . . . . . . . . . . . . 948.1.3 Das Teilchen auf dem Ring . . . . . . . . . . . . . . . . 95
8.2 Der Drehimpuls . . . . . . . . . . . . . . . . . . . . . . . . . . 97
iv

8.3 Produktansatz der Schrodingergleichung in Kugelkoordinaten . 102
9 Das Wasserstoffatom 1109.1 Radiale Dichteverteilung . . . . . . . . . . . . . . . . . . . . . 1189.2 Entartung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
IV Mehrelektronenprobleme 121
10 Mehrelektronenprobleme ohne e-e-Wechselwirkung 12210.1 Allgemeine Losungen . . . . . . . . . . . . . . . . . . . . . . . 12210.2 Variationsprinzip . . . . . . . . . . . . . . . . . . . . . . . . . 12610.3 Grundzustand des He-Atoms . . . . . . . . . . . . . . . . . . . 128
11 Mehrelektronenatome 12911.1 Grundzustand des He-Atoms . . . . . . . . . . . . . . . . . . . 12911.2 Grundzustand des Li-Atoms . . . . . . . . . . . . . . . . . . . 13011.3 Der Spin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13111.4 Das Pauli-Prinzip . . . . . . . . . . . . . . . . . . . . . . . . . 13411.5 Die Eigenschaften von Atomen . . . . . . . . . . . . . . . . . . 13611.6 Drehimpulskopplung . . . . . . . . . . . . . . . . . . . . . . . 14011.7 Spin-Bahn-Kopplung und Hundsche Regeln . . . . . . . . . . . 14311.8 Zusammenfassung . . . . . . . . . . . . . . . . . . . . . . . . . 147
12 Molekule 14912.1 Die Born-Oppenheimer-Naherung . . . . . . . . . . . . . . . . 14912.2 Die Linear Combination of Atomic Orbital-Methode (LCAO) . 15212.3 Die Wellenfunktionen des H+
2 -Molekulions . . . . . . . . . . . 15712.4 Das Wasserstoffmolekul . . . . . . . . . . . . . . . . . . . . . . 15912.5 MO-Diagramme zweiatomiger Molekule . . . . . . . . . . . . . 159
v


“What I cannot create I do not understand”Richard Feynman
Ich bin Herrn Prof. Dr. Georg Jansen dankbar fur die Uberlassung seinesVorlesungsskriptes. Einige Beispiele und Darstellungen sind diesem Skriptentnommen. Der uberwiegende Teil des vorliegenden Skriptes lehnt sich engan P.W. Atkins and R. Friedman, “Molecular Quantum Mechanics”, Fourthedition, Oxford University Press, Oxford (2005, 2007) an. Ich danke HerrnDr. Klaus Kolster, Dr. Sergej Piskunovs und PD Dr. Holger Somnitz furFehlerkorrekturen und kritische Durchsicht des Skriptes sowie Frau HelgaFischer und Herrn Torsten de Montigny fur die Hilfe bei der Anfertigung derAbbildungen.


Teil I
Einleitung und Motivation
Inhaltsangabe
1 Probleme der klassischen Physik zu Beginn des 20. Jahr-hunderts 2
1.1 Ubersicht . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Zusatzmaterial . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2.1 Atomspektren . . . . . . . . . . . . . . . . . . . . . 2
1.2.2 Schwarzer Strahler . . . . . . . . . . . . . . . . . . 4
1.2.3 Spezifische Warme . . . . . . . . . . . . . . . . . . 6
1.2.4 Photoelektrischer und Compton Effekt . . . . . . . 6
1.2.5 Dualitat . . . . . . . . . . . . . . . . . . . . . . . . 8
2 Was ist Theoretische Chemie? 9
2.1 Motivation . . . . . . . . . . . . . . . . . . . . . . . . . . 9
2.2 Teilgebiete der Theoretischen Chemie . . . . . . . . . . . 9
2.3 Moderne theoretische Chemie . . . . . . . . . . . . . . . . 10
3 Klassische Teilchen und Wellen 13
3.1 Klassische Teilchen . . . . . . . . . . . . . . . . . . . . . . 13
3.2 Wellen (klassisch) . . . . . . . . . . . . . . . . . . . . . . . 14
4 Das Doppelspaltexperiment 16
1

1 Probleme der klassischen Physik zu Beginn
des 20. Jahrhunderts
1.1 Ubersicht
Slide 2 Ungeklarte experimentelle Probleme zu Beginn des 20. Jahrhun-derts
Um die Jahrhundertwende 1900 zeigte die klassische Physik (Mechanik,Thermodynamik, Elektrodynamik) mehr und mehr prinizipielle Unzulang-lichkeiten bei der Beschreibung von Eigenschaften auf atomarer Ebene.
• Atomspektren
• Strahlung des schwarzen Korpers
• Warmekapazitat bei niedrigen Temperaturen
• Photoelektrischer und Compton Effekt
• Dualitat der Materie
1.2 Zusatzmaterial
1.2.1 Atomspektren
Slide 3 Das Wasserstoffspektrum
This figure not shown due to copyright reasons! goto hyperlink
[http://www.astronomyknowhow.com/pics-res/hydrogen-spectra.jpg1]
Slide 41http://www.astronomyknowhow.com/pics-res/hydrogen-spectra.jpg
2

AtomspektrenDie von Atomen emittierte Strahlung ist nicht kontinuierlich, sondern be-
steht aus Spektrallinien. Die Balmerserie(1885), ν = RH
(1
22− 1
m2
)(Wel-
lenzahl ν = ν/c) beschreibt eine Serie von Spektrallinien im sichtbaren Licht.Die Rydberg-Konstante2 RH = 109737.32 cm−1 ist nach Johannes Rydberg3
benannt, der die Formel fur beliebige Serien (∗) ν = RH
(1
n2− 1
m2
)wie
z. B. die Lyman-Serie (n=1, UV), Paschen-Serie (n=3), Bracket-Serie (n=4)und Pfund-Serie (n=5) erweitert hat, die alle nach Wissenschaftlern benanntsind. Die Formel ist ein Spezialfall des Ritzschen4 Kombinationsprinzips,wonach alle beobachteten Spektrallinien als Termdifferenz ν = T1 − T2 ge-schrieben werden konnen.
Bohrsches AtommodellNiels Bohr (Nobelpreis 1922)a erklarte die Termformel (∗) durch dasBohrsche Atommodell (1913), wonach die erlaubten Energieniveausdes Wasserstoffatoms durch die Formel
En = − µe4
8h2ε20· 1
n2
beschrieben werden (Verknupfung von Strahlungstheorie und mecha-nischem Modell).Dabei heisst µ (1/µ = 1/mP + 1/me) reduzierte Masse, e ist die Ele-mentarladung, h ist die Plancksche Konstante, ε0 ist die Permittivitatdes Vakuums (“Dielektrizitatskonstante”) und n eine positive ganzeZahl.
ahttp://de.wikipedia.org/wiki/Niels Bohr
2http://de.wikipedia.org/wiki/Rydberg-Konstante3http://de.wikipedia.org/wiki/Johannes Rydberg4http://de.wikipedia.org/wiki/Walter Ritz
3

Kritik des Bohrschen Atommodells:Das Bohrsche Atommodell (und die Weiterentwicklung durch Ar-nold Sommerfelda) war zwar (naherungsweise) quantitativ korrekt,jedoch waren die Quantisierungsbedingungen fur die erlaubten Elek-tronenbahnen (ebenfalls ein nicht haltbares Konzept(↔ Unscharfere-lation)) ad hoc.Mit Hilfe der Quantenmechanik ergeben sich diese Quantisierungsbe-dingungen zwangslaufig!
ahttp://de.wikipedia.org/wiki/Arnold Sommerfeld
1.2.2 Schwarzer Strahler
Slide 5 Schwarzer Strahler
This figure not shown due to copyright reasons! goto hyperlink
[http://rugth30.phys.rug.nl/quantummechanics/ derived/black body.htm txt CAVITY.gif5]
Slide 65http://rugth30.phys.rug.nl/quantummechanics/ derived/black body.htm txt CAVITY.gif
4

Strahlung des schwarzen Korpers
• Stefan-Boltzmann-Gesetz M = σ · T 4
Stefan-Boltzmann-GesetzM : emittierte Leistung, dividiert durch die emittierende Flacheσ : Stefan-Boltzmann-Konstante;Bei 1000 K emittiert 1 cm2 eines schwarzen Strahlers ca. 6 Watt.
• Wiensches Verschiebungsgesetz λmax · T = const.
BeobachtungDas Wellenlangenmaximum der emittierten Strahlung nimmtmit zunehmender Temperatur ab, d.h. das Frequenzmaximumnimmt mit zunehmender Temperatur zu.
schwarz → rotgluhend → gelbgluhend → weißgluhend
• Rayleigh-Jeans-Gesetz ρ(λ) =8πkT
λ4
• “Ultraviolettkatastrophe”
InterpretationDie Energiedichte (also die Energie pro Volumeneinheit im Wel-lenlangenbereich λ bis λ+dλ) nimmt mit zunehmender Frequenzν (also abnehmendem λ) zu!Dieses Ergebnis wurde von Ehrenfest mit dem Namen “Ultra-violettkatastrophe” bezeichnet.
• Planck’sches Strahlungsgesetz (1900) ρ(λ) =8πhc
λ5
e−hc/λkT
1− e−hc/λkT
theoretische BegrundungEnergie wird in Einheiten von h · ν abgegeben
Planck (1858-1947) fuhrte die Naturkonstante h (PlanckscheKonstante) ad hoc ein, um die experimentellen Ergebnissezu erklaren. Seine Formel erklart die Schwarzkorperstrahlungvollstandig.
5

1.2.3 Spezifische Warme
Slide 7 spezifische Warme des Festkorpersbei niedrigen Temperaturen
• Gesetz von Dulong und Petit: Cv ≈ 3R
InterpretationJedes Atom verhalt sich wie ein klassischer Oszillator in 3 Di-mensionen und kann beliebige Betrage an Energie aufnehmen.
• Einstein: Cv = 3RfE(T ) mit fE(T ) =
{ΘE
T· eΘE/2T
1− eΘE/T
}2
InterpretationJedes Atom verhalt sich wie ein Oszillator, kann aber nur ange-regt werden, wenn die Anregungsenergie einen minimalen Wertubersteigt. Einstein nahm an, dass die Anregungsenergien furalle Oszillatoren gleich sind.Komplementaritat zur Planckschen Theorie!
• Debye: Cv = 3RfD(T ) mit fD(T ) = 3
(T
ΘD
)3 ∫ ΘD/T
0
x4ex
(ex − 1)2dx
InterpretationJedes Atom verhalt sich wie ein Oszillator, kann aber nurangeregt werden, wenn die Anregungsenergie einen mini-malen Wert ubersteigt. Im Ggs. zu Einstein nimmt Debyeeine Verteilung der charakteristischen Frequenzen (unddamit Anregungsenergien) an.
1.2.4 Photoelektrischer und Compton Effekt
Slide 8
6

Der Photoeffekt
This figure not shown due to copyright reasons! goto hyperlink
[https://www.llnl.gov/str/June05/gifs/Aufderheide3.jpg6]
[http://hyperphysics.phy-astr.gsu.edu/hbase/imgmod2/pelec.gif7]
Slide 9 Comptonstreuung
This figure not shown due to copyright reasons! goto hyperlink
[http://hyperphysics.phy-astr.gsu.edu/hbase/quantum/imgqua/compton.gif8]
Slide 10 Photoelektrischer und Compton Effekt
• Photoelektronen: EK = hν − Φ
Beobachtunglinearer Zusammenhang zwischen kinetischer Energie von Pho-toelektronen und der Frequenz des anregenden UV-LichtesEmission von Elektronen ist spontan (auch bei niedriger Inten-sitat), sobald die Strahlung eine Minimalfrequenz hat.
• Einstein verknupfte Planck’s Quantenhypothese mit dem Photoeffekt
Erklarung(Einstein 1905) Das elektromagnetische Feld ist quantisiert undbesteht aus Energiebundeln der Große hν
• G.N. Lewis pragte dafur den Begriff Photonen.
• Licht (Photonen) hat also Teilchencharakter
• relativistische Energie E =√m2c4 + c2p2
(Albert Einstein, 1879-1955, Nobelpreis 1921)a
m = 0⇒ E = p · cPhoton hat keine Ruhemasse, aber einen Impuls p und bewegtsich mit Lichtgeschwindigkeit.
ahttp://de.wikipedia.org/wiki/Einstein
6https://www.llnl.gov/str/June05/gifs/Aufderheide3.jpg7http://hyperphysics.phy-astr.gsu.edu/hbase/imgmod2/pelec.gif8http://hyperphysics.phy-astr.gsu.edu/hbase/quantum/imgqua/compton.gif
7

• fur Photonen: m = 0⇒ E = p · c = hν =hc
λ=⇒ λ =
h
poder p =
h
λ
• Photonen haben einen wellenlangenabhangigen Impuls.
• Experimentelle Bestatigung: Compton-Effekt
Bei der inelastischen Streuung von Photonen an Elektronen(im ursprunglichen Experiment (1923) in Graphit) andert sich
die Wellenlange der Photonen um δλ = 2λC sin2 1
2θ mit der
Compton-Wellenlange λC =h
mec. Diese Formel wird unter der
Annahme abgeleitet, dass Photonen einen linearen Impuls h/λbesitzen.
Die Quantenmechanik erklart diesen dualen Charakter. Pho-tonen haben einerseits Teilcheneigenschaften (z.B. einen linea-ren Impuls). Dies scheint ein Widerspruch zu zahlreichen Ex-perimenten, die den Wellencharakter des Lichtes untermauern.Die Quantenmechanik erklart diesen scheinbaren Widerspruchquantitativ, wohingegen die klassische Physik nicht einmal einequalitative Erklarung zu geben vermag.
1.2.5 Dualitat
Slide 11 Dualitat von Materie und StrahlungDie Synthese dieser Ideen und die Demonstration der engen Verknupfung
zwischen elektromagnetischer Strahlung und Materie begann mit Louis deBroglie (Nobelpreis 1929)9, der die Universalitat der de Broglie-Beziehung
λ =h
ppostulierte.
Dualitat⇒Dualitat, d.h. gleichzeitige Wellen- (λ) und Teilcheneigenschaften(Impuls p) von Materie und Strahlung!
9http://de.wikipedia.org/wiki/Louis-Victor de Broglie
8

2 Was ist Theoretische Chemie?
2.1 Motivation
Slide 12 Theoretische Chemie
Paul Adrian Maurice Dirac (1902-1984, Nobelpreis 1933)10 wird der Satz
zugeschrieben, dass “Once the laws of quantum mechanics are understood,
the rest is chemistry!”
=⇒ (Theoretische) Chemie ist also einfach(?) die Anwendung der Gesetzeder Quantenmechanik
Dirac wird ebenfalls der folgende Satz zugeschrieben: “The underlying physi-
cal laws for the mathematical theory of a large part of physics and the whole
of chemistry are thus completely known, and the difficulty is only that the
exact application of these laws leads to equations much too complicated to
be soluble.”
=⇒ Theoretische Chemie ist also doch nicht einfach ein Teilgebiet derIngenieurwissenschaften!
2.2 Teilgebiete der Theoretischen Chemie
Slide 13 Theoretische ChemieGrundlagen
• Die Losung der Gleichungen ist sogar so komplex, dass man haufig dieQuantenmechanik durch die klassische Mechanik ersetzen muss!
• Fur große Systeme mit einer großen Anzahl von Molekulen mussengemittelte Großen berechnet werden.
=⇒ Theoretische Chemie befasst sich mit
10http://de.wikipedia.org/wiki/Dirac
9

1. Quantenmechanik
2. Quantenchemie
3. klassische Mechanik
4. statistische Mechanik (Thermodynamik)
Slide 14 Teilgebiete der Theoretische Chemie
1. Theorie der Chemischen Bindung (z. B. Existenz von Molekulen, Geo-metrie, Bindungsenergien)
2. Theorie der Chemischen Reaktionen (z. B. Reaktionsdynamik, Reak-tionskinetik)
3. Theorie der Molekulspektroskopie (z. B. Wechselwirkung von Mo-lekulen mit elektromagnetischer Strahlung)
4. Theorie von Polymerstrukturen und -dynamik (z. B. Ionomere, Gas-diffusion, Scher- und Fließverhalten)
5. Theorie von Flussigkeiten (z. B. Solvatation, Relaxationsphanomene)
6. Theorie von Festkorpern (z. B. Transporteigenschaften, mechanischeEigenschaften)
7. Oberflachentheorie (Surface Science) (z. B. Adsorption, Katalyse, Elek-trochemie)
8. Mathematische Ordnungsstrukturen in der Chemie (z. B. Molekulstruk-tur und -topologie, Reaktionen, Organisation von Datenbanken)
9. . . .
1. – 7. uberlappen mit der (theoretischen) Physik
8. uberlappt mit Mathematik und Informatik
2.3 Moderne theoretische Chemie
Slide 15 10

Bedeutung heute
starke Zunahme in den letzten 30 Jahren
Theoretische Chemie ist Computerchemie:
Einsatz des Computers zur Losung chemischer Probleme
Ursachen dieser Entwicklung:
• Zuwachs an Rechenkapazitat (Moore’s Law)11
Moore’s Law“The complexity for minimum component costs has increased at arate of roughly a factor of two per year. Certainly over the short termthis rate can be expected to continue, if not to increase. Over thelonger term, the rate of increase is a bit more uncertain, althoughthere is no reason to believe it will not remain nearly constant for atleast 10 years. That means by 1975, the number of components perintegrated circuit for minimum cost will be 65,000.”G.E. Moore “Cramming more components onto integrated circuits”,Electronics Magazine, 19.4.1965.
• algorithmische Verbesserungen der Software
• verbesserte graphische Benutzerinterfaces (GUIs)
Slide 16 Moderne Theoretische ChemieIn weiten Bereichen der chemischen Forschung spielt die Unterstutzung
durch Rechnungen eine immer wichtigere Rolle. Dies gilt sowohl fur kleinereMolekule (z. B. Stabilitat und Struktur von Radikalen, chemische Verschie-bungen (NMR), Schwingungsspektroskopie) als insbesondere auch fur große-re Molekule wie Makromolekule und Proteine (z. B. Visualisierung von Struk-turen, Struktur-Wirkungsbeziehungen in der pharmazeutischen Forschung)und Molekulverbande (z. B. Docking, Materialsimulationen, also Simulatio-nen mit Umgebung, chemische Reaktionen) . Es geht weltweit 1/3 der Ka-pazitat von Supercomputern in chemische Anwendungen. Die Chemie liegt
11http://de.wikipedia.org/wiki/Moore’s Law
11

damit weit an der Spitze! Vor diesem Hintergrund muß sich auch die Ausbil-dung in Theoretischer Chemie muss verandert werden! Mehr Theorie / MehrComputation / Mehr Programmierung / Mehr Mathematik / Mehr Physik /Mehr Physikalische Chemie
12

3 Klassische Teilchen und Wellen
3.1 Klassische Teilchen
Slide 17 Klassische Teilchen
• Newton’s Bewegungsgleichungen
m · a = m · v = mdv
dt
= m · x = md2x
dt2= F
• oft ist F = −dV (x)
dx
• V = V (x) heißt Potentialfunktion
• Beispiele:
• Hookesches Federgesetz F = −kx V = 12kx2
• Bewegung im (konstanten) Gravitationsfeld der Erde F = −mg V =mgx (oft: z)
Slide 18 Verallgemeinerung
• n wechselwirkende Teilchen
x −→ xid
dx−→ ∂
∂xi
• Newtons Bewegungsgleichungen
mi · ai = mi · vi = midvidt
= mi · xi = mid2xidt2
= Fi = −∂V ({x1, . . . , xn})∂xi
• → Molekulardynamik
13

3.2 Wellen (klassisch)
Slide 19 Klassische Wellen
• Ortsabhangigkeit einer freien (ebenen) Welle:
φ(x) = cos kx oder φ(x) = sin kx
• aquivalent (mathematisch bequem)
φ(x) = cos kx+ i sin kx = eikx k = 2πλ
• Zeitabhangigkeit
φ(t) = cosωt− i sinωt = e−iωt ω = 2πν = 2πcν
• Gesamtwellenfunktion
ψ(x, t) = Aφ(x) · φ(t) = A︸︷︷︸Amplitude
·ei(kx−ωt)
kx− ωt heißt die Phase der Welle.
Bezug zu den “Materiewellen” atomarer Teilchen
Man ersetzt E = hν = ~ω und p =h
λ, und erhalt die Wellen-
funktion sich frei bewegender Teilchen als
ψ(x, t) = A · ei~ (p·x−E·t) ~ =
h
2π
Chemgapedia http://www.chemgapedia.de/...12
Slide 2012http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/1/pc/pc 11/pc 11 01/pc 11 01 01.vlu.html
14

Operatoren
• Man kann den Impuls aus der Wellenfunktion z.B. so gewinnen
~i
∂
∂xψ(x, t) =
~i· i~pψ(x, t) = pψ(x, t)
Operator~i
∂
∂xist ein Operator, durch dessen “Anwendung auf die Wel-
lenfunktion” wir die physikalische “Observable” des Impulses be-rechnen konnen.
Slide 21 Aufenthaltswahrscheinlichkeit
eine freie Welle ist im ganzen Raum ausgebreitet
Wo befindet sich das Teilchen?
Antwort:Irgendwo im Raum, mit uberall gleicher WahrscheinlichkeitW (x, y) = const.
Beobachtung: ψ∗(x, t) · ψ(x, t) = A2 = const.
⇒ Vermutung:W (x, t) = ψ∗(x, t)·ψ(x, t) konnte eine Wahrscheinlichkeitsdichtesein
15

4 Das Doppelspaltexperiment
Slide 22 Klassische Teilchen (Schrotkugeln)
Gewehr
Doppelspalt Auffangwand
Detektor
x
P1
P2
P12
Haufigkeitsverteilungen
Teilchen aus “Teilchenquelle” fliegen durch den Spalt
oder werden an einer Kante abgelenkt.
Man beobachtet die Verteilungen P1, P2, P12, wenn Spalt1, Spalt 2, oder beide Spalte offen sind.
Offensichtlich ist
1. P12 = P1 + P2
2. Es treffen nur ganze Kugeln auf.
Slide 23 Klassische Wellen (Wasserwellen)
16

Wellenerreger
Doppelspalt Auffangwand
Detektor
x
P1
P2
P12
Offensichtlich ist
1. P12 6= P1 + P2
Wellen zeigen Interferenzerscheinungen.
2. Die Intensitat kann jeden beliebigen Wert zwischen 0und der maximalen Intensitat annehmen.
Slide 24 Das Verhalten von Elektronen
17

Elektronenquellle
Doppelspalt Auffangwand
Detektor
x
P1
P2
P12
Offensichtlich ist
1. P12 6= P1 + P2
Elektronen zeigen Interferenzerscheinungen
2. Die Starke der Detektorimpulse ist immer gleich groß.
3. Selbst wenn die Elektronen einzeln nacheinander ankom-men, misst man stets ganze Elektronen, ihre Verteilungzeigt jedoch das Interferenzmuster
4. Es scheint, als ob die Elektronen mit sich selbst interfe-rieren.
Slide 25 Welle-Teilchen-Dualismus
• Das Elektron verhalt sich als Welle, soweit es die Statistik der Ereig-nisse betrifft.
• Andererseits verhalt sich das Elektron als Partikel, da bei jeder Mes-sung immer nur ein ganzes Teilchen im Detektor auftrifft.
18

Kopenhagener Interpretation (↔ Viel-Welten-Theorie)
– Kollaps der Wellenfunktion: Dem Teilchen stehen 2Wege offen, die es gleichzeitig benutzt ⇒ Interferenz
– Der Messprozess beeinflusst (stort) das Elektron und legtfest, welchen Weg es benutzt.
19

Teil II
Quantenmechanik
Inhaltsangabe
5 Axiome der Quantenmechanik 21
5.1 Wellenfunktion und Aufenthaltswahrscheinlichkeitsdichte . 21
5.2 Hermitesche Operatoren und physikalische Observable . . 22
5.2.1 Lineare Operatoren . . . . . . . . . . . . . . . . . . 22
5.2.2 Eigenfunktionen und Eigenwerte . . . . . . . . . . 22
5.2.3 Operatoren . . . . . . . . . . . . . . . . . . . . . . 25
5.2.4 Kommutatoren . . . . . . . . . . . . . . . . . . . . 27
5.2.5 Dirac-Notation . . . . . . . . . . . . . . . . . . . . 28
5.2.6 Hermitesche Operatoren . . . . . . . . . . . . . . . 29
5.2.7 Operatordarstellungen . . . . . . . . . . . . . . . . 29
5.3 Erwartungswerte . . . . . . . . . . . . . . . . . . . . . . . 31
5.4 Die Schrodingergleichung . . . . . . . . . . . . . . . . . . 34
5.4.1 Zeitabhangige Schrodingergleichung . . . . . . . . 34
5.4.2 Zeitunabhangige Schrodingergleichung . . . . . . . 36
5.5 Die Unscharferelation . . . . . . . . . . . . . . . . . . . . 37
5.6 Zusammenfassung . . . . . . . . . . . . . . . . . . . . . . 40
20

5 Axiome der Quantenmechanik
5.1 Wellenfunktion und Aufenthaltswahrscheinlichkeits-dichte
Slide 26 Axiom I
Die Grundlagen der Quantenmechanik konnen in Form von Axiomenbzw. Postulaten formuliert werden:
Postulat I:Der Zustand eines quantenmechanischen Systems ist vollstandigdurch eine Wellenfunktion
Ψ(r1, r2, . . . rN, t)
beschrieben. Die Funktion Ψ ist im Allgemeinen komplexwer-tiga.
ahttp://de.wikipedia.org/wiki/Komplexe Zahl
ri (oder ~ri) sind dabei die Koordinaten von Teilchen i, t die Zeit.
Wir werden sehen, dass die Wellenfunktion des Systems, also sein Zu-stand, haufig durch Quantenzahlen a, b, . . . charakterisiert werden kann.Die Zustande sind dann abzahlbar oder quantisiert, und konnen alsWellenfunktion Ψa,b,...(r1, r2, . . . rN, t) geschrieben werden. Fur ein ein-zelnes Teilchen ist die Wellenfunktion ψa,b,...(~r, t)
Slide 27 Bornsche InterpretationInterpretation als WahrscheinlichkeitsdichteDas Absolutquadrat der Wellenfunktion Ψ∗(~r, t)Ψ(~r, t) kann alsWahrscheinlichkeitsdichte p(r, t) interpretiert werden.
ψ∗(~r, t)ψ(~r, t)︸ ︷︷ ︸p(x,y,z,t)
dxdydz
ist dann die Wahrscheinlichkeit, ein Teilchen im infinitesimalenVolumenelement dV = dxdydz am Punkt ~r im Raum zur Zeit tzu finden, also zwischen x und x+ dx, y und y + dy und z undz + dz.
21

Offenbar ist dann
∞∫−∞
∞∫−∞
∞∫−∞
ψ∗(~r)ψ(~r)dxdydz = 1 die Wahrscheinlich-
keit, das Teilchen irgendwo im Raum zu finden.
5.2 Hermitesche Operatoren und physikalische Obser-vable
5.2.1 Lineare Operatoren
Slide 28 Lineare Operatoren
Ein Operator O wirkt auf eine Funktion f und erzeugt eine neue Funk-tion g:
g = Of
linearer OperatorEin linearer Operator hat die Eigenschaft
O(αf + βg) = αOf + βOg .
Operatoren in der Quantenmechanik sind lineare Operatoren.Multiplikation und Differentiation sind Beispiele fur lineareOperatoren. α und β sind Skalare (Zahlen), f und g sind Funk-tionen. Operatoren werden durch einˆcharakterisiert.
5.2.2 Eigenfunktionen und Eigenwerte
Slide 29 Eigenfunktionen und Eigenwerte IDefinition:Eine Funktion f ist Eigenfunktion zu einem Operator O, wenn
Of = αf
mit konstanten α. Die Konstante (Skalar) α heißt dann Eigen-wert.
Eigenfunktionen sind also Spezialfalle von Funktionen, die furjeden Operator charakteristisch sind.
22

Slide 30 Beispiel:
Gegeben sei die Funktion f(x) = cos(3x+ 5).
• sei O1 = ddx
O1f(x) = −3 sin(3x+ 5) =⇒ f(x) ist keine Eigenfunktion von O1.
• sei O2 = d2
dx2= O1O1
O2f(x) = ddx
ddxf(x) = d
dx[−3 sin(3x + 5)] = −9 cos(3x + 5) ist eine
Eigenfunktion von O2 zum Eigenwert -9.
• Ist exp(3x+ 5) eine Eigenfunktion von O1 oder O2?
O1 exp(3x+ 5) = 3 exp(3x+ 5)
O2 exp(3x+ 5) = ddx
3 exp(3x+ 5) = 9 exp(3x+ 5)
⇒ exp(3x + 5) ist eine Eigenfunktion von O1 (mit Eigenwert 3) und vonO2 (mit Eigenwert 9).
Slide 31 Eigenfunktionen und Eigenwerte II
wichtige Eigenschaften:
1. Die Menge aller Eigenfunktionen fn zu einem gegebenen Operator O(mit den entsprechenden Eigenwerten αn) bildet eine vollstandige Funk-tionenmenge.
Man sagt, dass die Funktionen dieser vollstandigen Funktionen-menge den Hilbertraum aufspannen. (Die Funktionen spielendie Rolle von Einheitsvektoren in diesem Raum, analog zum be-kannten dreidimensionalen Vektorraum.) Die Gesamtheit dieserFunktionen sowie aller moglichen Linearkombinationen darausnennt man Hilbert-Rauma.
ahttp://de.wikipedia.org/wiki/Hilbert-Raum
23

2. Eine Funktion, die uber dem gleichen Definitionsbereich definiert ist,kann nach diesen Funktionen entwickelt werden, d.h. eine Funktion gkann durch
g =∑n
cnfn
mit skalaren Koeffizienten cn dargestellt werden.
3. Die Menge der Eigenwerte {αn} nennt man auch das Eigenwertspek-trum des Operators O.
Slide 32 Eigenfunktionen und Eigenwerte III
Spezialfall: entartete Eigenwerte
Gibt es mehrere Eigenfunktionen des Operators O, z.B. fn undfm zum gleichen Eigenwert αn = αm = α (man spricht dannvon entarteten Eigenwerten), so ist jede Linearkombinationdieser Funktionen ebenfalls eine Eigenfunktion des OperatorsO.
Beweis:
Og = Ok∑
n=1
cnfn =k∑
n=1
cnOfn
=k∑
n=1
cnαfn = αk∑
n=1
cnfn = αg
Slide 33 Lineare Unabhangigkeit
24

DefinitionEine Funktionenmenge g1, g2, . . . gn heißt linear unabhangig,wenn es keinen Satz von Koeffizienten c1, c2, . . . cn gibt (außerdem trivialen Satz ci = 0∀i), fur den gilt:
n∑i=1
cigi = 0 .
Ein Satz von Funktionen, der nicht linear unabhangig ist, heißtlinear abhangig.
• Es ist moglich, aus n Basis(Eigen)funktionen eines Operators O einenSatz von n linear unabhangigen Funktionen zu erzeugen.
• Jede Funktion im Hilbertraum ist als Linearkombination des vollstandi-gen Funktionensatzes (=Basis) darstellbar.
5.2.3 Operatoren
Slide 34 Integrale uber Operatoren I
Es wurde weiter oben kurz eine “Analogie” zwischen Hilbertraum (derFunktionen) und dem (dreidimensionalen) Vektorraum angesprochen.
Das Analogon zum Skalarprodukt ~a ·~b = c sind Integrale uber Funktio-nen und/oder Operatoren der Form
I =
∫f ∗Ogdτ ,
wobei dτ ein verallgemeinertes Volumenelement ist (z.B. dτ = dxdydzfur Funktionen, die nur von einem Satz Koordinaten abhangen.
Da die Anwendung des Operators O auf g wieder eine Funktion, h,
ergibt, ist I =
∫f ∗Ogdτ =
∫f ∗hdτ ein Integral uber 2 Funktionen.
Slide 35
25

Integrale uber Operatoren II
• Fur den Operator O = 1 (Multiplikation mit 1) nennt man das Integral
S =
∫f ∗mfndτ das Uberlappungsintegral.
• Wenn S = 0 ist, klassifiziert man die Funktionen in Analogie zum drei-dimensionalen Vektorraum als orthogonal (analog zu zwei aufeinandersenkrechten Vektoren).
• Der Spezialfall n = m von S =
∫f ∗nfndτ heißt Normierungsintegral.
• Eine Funktion fn heißt (auf 1) normiert, wenn S =
∫f ∗nfndτ = 1
gilt. In der Regel kann man leicht einen Normierungsfaktor N finden,der eine Funktion fn normiert.
Slide 36 Beispiel: Normierungsfaktor
Sei f eine Funktion, wobei f(x) = sin(πx/L) im Definitionsbereich[0;L], ansonsten 0.
• Das Normierungsintegral lautet
S =
L∫0
f ∗fdx =
L∫0
N2 sin2(πx/L)dx =1
2LN2 !
= 1 .
⇒ N =
√2
L
Die normierte Funktion lautet also f =
(2
L
)1/2
sin(πx/L) (s. Ubungs-
aufgabe).
Slide 37
26

OrthonormalitatsbedingungEine Menge von Funktionen, die (a) normiert und (b) paarweiseorthogonal sind, genugt der Orthonormalitatsbedingung∫
f ∗mfndτ = δnm .
δnm heißt Kroneckerdelta, und hat den Wert 1 fur n = m, andernfalls0.
5.2.4 Kommutatoren
Slide 38 Kommutativitat
Zwei Operationen heißen kommutativ, wenn das Ergebnis un-abhangig von der Reihenfolge der Anwendung der Operationenist.(Genauer: Es muss noch angegeben werden, auf welche Menge man
sich bezieht.)
• Z.B. sind Addition und Multiplikation auf den Mengen der naturli-chen, ganzen, rationalen, reellen und komplexen Zahlen N,Z,Q,R,Ckommutativ.
Definition: KommutatorIm allgemeinen sind zwei Operatoren A und B nicht kommu-tativ. Man definiert den Kommutator [A, B] von A und Bals
[A, B] = AB − BA
Slide 39 Beispiel: Kommutator
Betrachten wir die Operatoren x und px := ~i
ddx
.
27

[x, px]f = (xpx − pxx)f
= x · ~i
∂f
∂x− ~i
∂(x · f)
∂x
= x · ~i
∂f
∂x− ~i
(f + x · ∂f
∂x
)(Kettenregel)
= −~if
=⇒ [x, px] = −~i
= i~
5.2.5 Dirac-Notation
Slide 40 Vereinfachung der Schreibweise: Dirac(“Bracket”)-Notation
Integrale des Typs I =
∫f ∗Ogdτ kommen in der Quantenmechanik so
haufig vor, dass eine vereinfachte, auf Dirac13 zuruckgehende, Notationsehr praktisch ist.
Diracsche “Bracket”-Notation∫fm∗Ofndτ =< m|O|n >
|n >:= fn heißt ket und ist eine Funktion.
< m| :=
∫f ∗mdτ heißt bra und ist ein linearer (Inte-
gral)Operator.
< m|O|n > nennt man bracket.
Wenn O = 1 ist, schreibt man vereinfacht < m|n >.
Per definitionem gilt < m|n >=< n|m >∗.< m|n >=
∫f∗mfndτ =
(∫fmf∗ndτ
)∗=(∫f∗nfmdτ
)∗= (< n|m >)∗
13http://de.wikipedia.org/wiki/Dirac
28

5.2.6 Hermitesche Operatoren
Slide 41 Hermitesche Operatoren
DefinitionEin Operator O heißt hermitesch, wenn fur zwei beliebigeFunktionen fn und fm∫
f ∗mOfndτ =
(∫f ∗nOfmdτ
)∗gilt.
In Diracschreibweise: < m|O|n >=< n|O|m >∗.
Eine alternative Definition lautet∫f ∗mOfndτ =
∫(Ofm)∗fndτ .
Slide 42 Axiom II
Postulat II: Observable
Physikalische Observable werden in der Quantenmechanikdurch hermitesche Operatoren reprasentiert, die die Kom-mutatorbeziehungen
[q, pq′ ] = i~δqq′ [q, q′] = 0 [pq, pq′ ] = 0
erfullen. Dabei stehen q und q′ jeweils fur x, y, z und pq undpq′ fur die zugehorigen linearen Impulse. i ist die imaginareEinheit, ~ = h/2π.
Hermitesche Operatoren haben reelle Eigenwerte. =⇒Messbare Großen sind reell!
5.2.7 Operatordarstellungen
Slide 43
29

Darstellungen
Ein großer Teil der Quantenmechanik kann mit solch abstrakten Ope-ratoren entwickelt werden. Die spezifische Wahl von Operatoren fureine Observable fuhrt zu spezifischen Darstellungen:
• OrtsdarstellungPositionsoperator: x→ x·Impulsoperator: px →
~i
∂
∂x
• Impulsdarstellung
Positionsoperator: x→ ~i
∂
∂pxImpulsoperator: px → px·
• Es gibt weitere Darstellungen, z.B. die Besetzungszahldarstellung. Wirwerden uns auf die Ortsdarstellung beschranken.
Slide 44 Konstruktion von Operatoren in der Quantenmechanik I
• Ortsoperator: x→ x·
• Impulsoperator: px → ~i
ddx
• Operator der kinetischen Energie T
in x-Richtung: Tklassisch =m
2(vx)
2 =(px)
2
2m→ − ~2
2m
d2
dx2
• in 3 Dimensionen: T = − ~2
2m
{∂2
∂x2+
∂2
∂y2+
∂2
∂z2
}= − ~2
2m∇2 = − ~2
2m∆
∇: Nabla-Operator, ∆: Laplace-Operator
Slide 45
30

Konstruktion von Operatoren in der Quantenmechanik II
• Operator der potentiellen Energie: V (x, y, z)→ V (x, y, z)·
• z.B. V (x, y, z) = − Ze2
4πε0√x2+y2+z2
· = V (r) = − Ze2
4πε0r·
fur die Coulombwechselwirkung eines Elektrons mit einem Kern derLadungszahl Z.
• Hamiltonoperator der Gesamtenergiez.B. des Wasserstoffatoms
H = − ~2
2m∇2 − e2
4πε0r·
Allgemeine VorschriftIn der Ortsdarstellung ersetzt man
1. x durch Multiplikation mit x·
2. px durch den Differentialoperator ~i∂∂x
3. und analog fur y und z.
5.3 Erwartungswerte
Slide 46 Matrixelemente
• Ausdrucke der Art
∫ψ∗Oφdτ = 〈ψ|O|φ〉 nennt man auch Matrixele-
mente des Operators O.
• Der Spezialfall∫ψ∗Oψdτ = 〈ψ|O|ψ〉 = 〈O〉ψ (ψ = φ)
heißt Erwartungswert des Operators O im Zustand ψ.
• Erwartungswerte von hermiteschen Operatoren sind reell:
31

Beweis:
〈O〉∗ =
(∫ψ∗Oψdτ
)∗=
∫ψOψ∗dτ
=
∫(Oψ∗)ψdτ
=
∫ψ∗Oψdτ
= 〈O〉
Slide 47 Axiom III
Postulat III:Wenn ein System durch eine Wellenfunktion ψ beschrieben ist,dann ist der Mittelwert einer physikalischen Große in ei-ner Serie von Messungen durch den Erwartungswert des zu-gehorigen Operators bestimmt.
Ist eine Wellenfunktion ψ eine Eigenfunktion des Operators O,dann gilt naturlich Oψ(~r, t) = akψ(~r, t).
=⇒ Ist ψ ein Eigenzustand des Operators O, dann wird bei jeder Mes-sung der gleiche Wert erhalten!
Postulat III’:Ist die Wellenfunktion ψ eine Eigenfunktion von O, so ist derErwartungswert einer Messung gleich dem Eigenwert des Ope-rators.
Slide 48 Axiom IV: Bornsche Interpretation
Postulat IV:Die Wahrscheinlichkeit, dass ein Teilchen im Volumenelementdτ = dx · dy · dz um den Punkt r zu finden, ist |ψ(r)|2dτ =ψ∗(r)ψ(r)dτ .
32

Fur Wellenfunktionen von Systemen aus n Teilchen ist
ψ∗(x1, y1, z1, x2, y2, z2, . . . , xn, yn, zn, t) ·ψ(x1, y1, z1, x2, y2, z2, . . . , xn, yn, zn, t) ·dx1dy1dz1 . . . dxndyndzn
die Wahrscheinlichkeit, das Teilchen 1 im Volumen dV1 = dx1 ·dy1 · dz1 um (x1, y1, z1) und gleichzeitig das Teilchen 2 im Volu-men dV2 = dx2 · dy2 · dz2 um (x2, y2, z2), etc., zu finden.
Slide 49 Normierung der WellenfunktionI
• Die Wahrscheinlichkeit, das Teilchen 1 irgendwo im Raum zu findenund gleichzeitig das Teilchen 2 irgendwo im Raum zu finden, usw.,muss gleich 1 sein.
• also mussen physikalisch sinnvolle Wellenfunktionen ψ normiert sein:
1 =
∞∫−∞
∞∫−∞
∞∫−∞
∞∫−∞
∞∫−∞
. . .
ψ∗(x1, y1, z1, x2, . . . , t)ψ(x1, y1, z1, x2, . . . , t)
dx1dy1dz1dx2 . . .
Slide 50 Normierung der WellenfunktionII
Gilt stattdessen
(∗) A =
∞∫−∞
∞∫−∞
∞∫−∞
∞∫−∞
∞∫−∞
. . .
ψ∗(x1, y1, z1, x2, . . . , t)ψ(x1, y1, z1, x2, . . . , t)
dx1dy1dz1dx2 . . .
mit 0 < A <∞ (∗∗), so ist φ =1√Aψ normiert.
33

• Funktionen, die (*) und (**) erfullen, heißen quadratintegrierbar oderquadratintegrabel oder normierbar.
• Die Gesamtheit aller quadratintegrablen Funktionen fur das n-Teilchensystemheißt der Hilbertraum fur das n-Teilchensystem.
• Bemerkung: ψ hangt im Allgemeinen von der Zeit t ab, die KonstanteA aber nicht.
Slide 51 Normierung der WellenfunktionII
• |ψ|2 ist eine Wahrscheinlichkeitsdichte
• |ψ|2dxdydz ist eine Wahrscheinlichkeit
5.4 Die Schrodingergleichung
5.4.1 Zeitabhangige Schrodingergleichung
Slide 52 Axiom V
Postulat V:Die zeitliche Entwicklung der Wellenfunktionψ(x1, y1, z1, x2, . . . , zn, t) wird durch die
zeitabhangige Schrodingergleichung i~∂ψ
∂t= Hψ
beschrieben.
• H ist der Hamiltonoperator, der im Allgemeinen explizit von der Zeitabhangig sein kann.
• Zeitabhangigkeit uber den Operator V der potentiellen Energie
• fur ein einzelnes Teilchen gilt
H = − ~2
2m
[∂2
∂x2+
∂2
∂y2+
∂2
∂z2
]+ V (x, y, z, t)
34

• in einer Dimension: H = − ~2
2m
∂2
∂x2+ V (x)
zeitabhangige Schrodingergleichung in einer Dimension
i~∂ψ(x,t)∂t
= − ~22m
∂2
∂x2ψ(x, t) + V (x)ψ(x, t)
Slide 53 Statt einer Herleitung
• Wir hatten weiter oben gesehen, dass eine Wellenfunktion fur atomareSysteme die Form
ψ(x, t) = A · ei~ (p·x−E·t)
besitzt.
• Fur ein freies Teilchen in einer Dimension ist E = Ekin, mit der kine-
tischen Energie Ekin = p2x2m
• i~∂ψ(x, t)
∂t= i~ ·
(−i~E
)ψ(x, t) = Eψ(x, t)
• − ~2
2m
∂2ψ(x, t)
∂x2+ V (x)ψ(x, t) =
−~2
2m
i2
~2p2xψ(x, t) + V (x)ψ(x, t)
= Ekinψ(x, t) + V (x)ψ(x, t) = Eψ(x, t)
=⇒ Schrodingergleichung ist erfullt.
Slide 54 Der Hamiltonoperator eines Vielteilchensystemsn-Teilchen-Operator
H =n∑i=1
− ~2
2m∆i + V ({~ri}, t)
wobei ∆i =∂2
∂x2i
+∂2
∂y2i
+∂2
∂z2i
der Laplace-Operator fur das i.
Teilchen ist.
35

Man beachte: Der Operator der kinetischen Energie entkoppelt (d.h.,ist eine einfache Summe uber Teilchen).
Alle Kopplungen im Vielteilchensystem stecken in der PotentialfunktionV ({~ri}, t).
5.4.2 Zeitunabhangige Schrodingergleichung
Slide 55 Separation der WellenfunktionOrtsfunktion & Zeitfunktion
• meistens ist die potentielle Energie nicht explizit zeitabhangig
• das System ist dann konservativ(wobei angenommen wurde, dass keine geschwindigkeitsabhangigen Wechsel-
wirkungsterme auftreten)
V (x1, y1, z1, x2, . . . zn, t) = V ({x}, t)→ V ({x})
• Dann kann man ψ(x1, y1, z1, x2, . . . zn, t) = ψ({x}, t) schreiben als
(∗) ψ({x}, t) = φ({x}) · χ(t)
• Man nennt dies einen Separationsansatz fur eine partielle Differential-gleichung. Die Schrodingergleichung ist eine solche.
Slide 56 Produktansatz
• Die Schrodingergleichung lautet mit (∗):
i~∂
∂t[φ({x}) · χ(t)] = H[φ({x}) · χ(t)]
=⇒ φ({x}) · i~ ∂∂tχ(t) = χ(t) · Hφ({x})
links wirkt kein Differentialoperator auf φ und rechts keiner auf χ.
Slide 57 36

Zeitunabhangige Schrodingergleichung
• Wir machen nun einen mathematisch unsauberen (aber gerechtfer-tigten) Trick, indem wir die Gleichung durch φ({x}) und χ(t) dividie-ren
=⇒ i~1
χ(t)· ∂∂tχ(t) =
1
φ({x})· Hφ({x})
oderi~∂χ(t)
∂t
χ(t)=
Hφ({x})φ({x})
R(t) = S({x})= E
= const.
Zeitunabhangige Schrodingergleichung
Hφ({x}) = Eφ({x})
5.5 Die Unscharferelation
Slide 58 Unscharfe
Nichtvertauschbare Operatoren bewirken, dass verschiedene Observa-blen nicht gleichzeitig exakte Werte annehmen konnen.
Messungen erzeugen Unscharfe, indem bei verschiedenen Messungendes gleichen Systems unterschiedliche Messwerte beobachtet werden(sei es durch meßtechnische Probleme (Ungenauigkeit) oder wie hierdurch prinzipielle Eigenschaften des Systems bedingt).
Diese Unscharfe wird (auch bei “klassischen” Messungen) durch dieVarianz
∆A2 = 〈(A− 〈A〉)2〉quantifiziert.
〈. . .〉 symbolisiert dabei einen Mittel- oder Erwartungswert.
Slide 59 37

Die Unscharferelation
Es gilt:
∆A2 = 〈(A− 〈A〉)2〉= 〈A2 − A〈A〉 − 〈A〉A+ 〈A〉〈A〉〉 (Ausmultiplizieren)
= 〈A2〉 − 〈A〉2 − 〈A〉2 + 〈A〉2 weil〈〈A〉〉 = 〈A〉= 〈A2〉 − 〈A〉2
∆A = {〈A2〉 − 〈A〉2}1/2 heißt Standardabweichung
Unscharferelation
Seien∆A = {〈A2〉 − 〈A〉2}1/2
und∆B = {〈B2〉 − 〈B〉2}1/2
Dann gilt ∆A∆B ≥ 1
2
∣∣∣〈[A, B]〉∣∣∣
Slide 60 Beweis der UnscharferelationI
• Seien 〈A〉 = 〈ψ|A|ψ〉 und 〈B〉 = 〈ψ|B|ψ〉.
• Operatoren fur die Verteilung von Einzelwerten von A und B sind dannδA = A− 〈A〉 und ˆδB = B − 〈B〉.
• Naturlich gilt [δA, ˆδB] = [A − 〈A〉, B − 〈B〉] = [A, B] =: iC, weil 〈A〉und 〈B〉 Skalare (Zahlen) sind.
• Man betrachtet nun fur reelles, ansonsten beliebiges α das Integral
I =
∫ ∣∣∣(αδA− i ˆδB)ψ∣∣∣2 dτ ≥ 0 .
Slide 61
38

Beweis der UnscharferelationII
I =
∫ {(αδA− i ˆδB)ψ
}∗ {(αδA− i ˆδB)ψ
}dτ
=
∫ψ∗(αδA+ i ˆδB)(αδA− i ˆδB)ψdτ (Hermitizitat)
= 〈(αδA+ i ˆδB)(αδA− i ˆδB)〉 (Erwartungswert)
= α2〈(δA)2〉+ 〈( ˆδB)2〉−iα〈δA ˆδB − ˆδBδA〉 (Ausmultiplizieren)
= α2〈(δA)2〉+ 〈( ˆδB)2〉+ α〈C〉≥ 0
Slide 62 Beweis der UnscharferelationIII
0 ≤ I = α2〈(δA)2〉+ 〈( ˆδB)2〉+ α〈C〉
= 〈(δA)2〉
(α +
〈C〉2〈(δA)2〉
)2
+〈( ˆδB)2〉 − 〈C〉2
4〈(δA)2〉(quadr. Erganzung)
gilt fur beliebiges α, also insbesondere auch furdasjenige α, das den ersten Term verschwinden laßt
=⇒ I = 〈( ˆδB)2〉 − 〈C〉2
4〈(δA)2〉≥ 0
=⇒ 〈(δA)2〉〈( ˆδB)2〉 ≥ 1
4〈C〉
2
Slide 63
39

Beweis der UnscharferelationIV
〈(δA)2〉 = 〈(A− 〈A〉)2〉= 〈A2 − 2A〈A〉+ 〈A〉2〉= 〈A2〉 − 2〈A〉〈A〉+ 〈A〉2
= 〈A2〉 − 〈A〉2
〈(δA)2〉 ist also die mittlere quadratische Abweichung von A von seinemMittelwert.
Analoges gilt fur B. mit ∆A =
√δA
2, ∆B =
√ˆδB
2
=⇒ ∆A∆B ≥ 12|〈C〉|
5.6 Zusammenfassung
Slide 64 Weitere Eigenschaften der Wellenfunktion• Die Schrodingergleichung ist eine DGL 2. Ordnung bzgl. der
Koordinaten der Teilchen
⇒ ψ({x}) muss uberall stetig sein
⇒ ∂
∂xiψ({x}) muss stetig an allen Stellen sein, an denen die po-
tentielle Energie stetig ist.
• Naturlich muss ψ eindeutig sein (genau genommen: ψ∗ψ musseindeutig sein).
• die Wellenfunktion darf nicht uber einen endlichen Bereich un-endlich groß werden
Slide 65
40
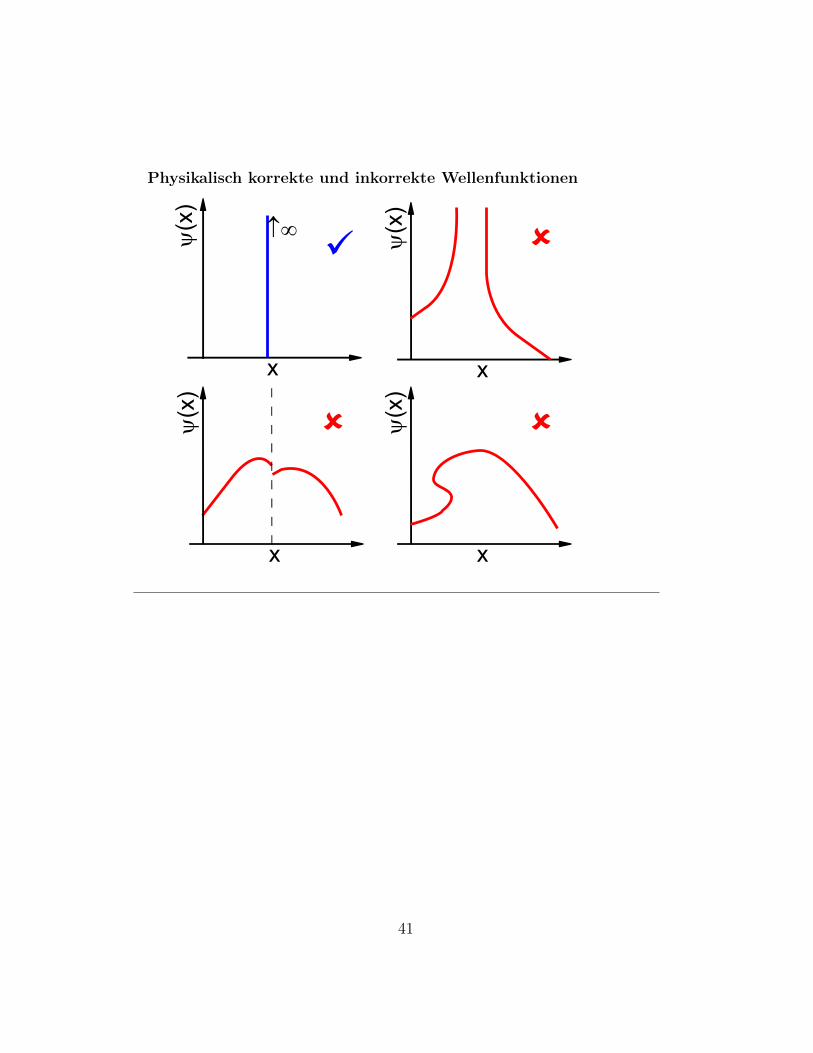
Physikalisch korrekte und inkorrekte Wellenfunktionen
y(x)
x
y(x)
x
y(x)
x
y(x)
x
41

Teil III
Exakte Losungen derstationarenSchrodingergleichung
Inhaltsangabe
6 Eindimensionale Probleme 44
6.1 Das Teilchen im unendlich tiefen Kasten . . . . . . . . . . 45
6.1.1 Modell und Losung der Schrodingergleichung . . . 45
6.1.2 Zustande des Teilchens im Kasten . . . . . . . . . 49
6.1.3 Erwartungswerte und Varianzen fur das Teilchenim Kasten . . . . . . . . . . . . . . . . . . . . . . . 54
6.1.4 Zusatzmaterial . . . . . . . . . . . . . . . . . . . . 57
6.2 Der harmonische Oszillator . . . . . . . . . . . . . . . . . 58
6.2.1 Federmodell . . . . . . . . . . . . . . . . . . . . . . 58
6.2.2 Schrodingergleichung . . . . . . . . . . . . . . . . . 59
6.2.3 Losung der Schrodingergleichung . . . . . . . . . . 62
6.2.4 Form der Wellenfunktionen und Aufenthaltswahr-scheinlichkeitsdichte . . . . . . . . . . . . . . . . . 67
7 Zwei- und Dreidimensionale Probleme in kartesischenKoordinaten 72
7.1 Das Teilchen im zweidimensionalen Kasten . . . . . . . . 72
7.2 Das Teilchen im dreidimensionalen Kasten . . . . . . . . . 80
7.3 Der harmonische Oszillator in 3 Dimensionen . . . . . . . 84
7.4 Erweiterung auf mehr als ein Teilchen . . . . . . . . . . . 88
8 Zentralkraft-Probleme 90
8.1 Einleitung . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
42

8.1.1 Kugelkoordinaten . . . . . . . . . . . . . . . . . . . 91
8.1.2 Teilchen auf der Kugeloberfache . . . . . . . . . . 94
8.1.3 Das Teilchen auf dem Ring . . . . . . . . . . . . . 95
8.2 Der Drehimpuls . . . . . . . . . . . . . . . . . . . . . . . . 97
8.3 Produktansatz der Schrodingergleichung in Kugelkoordi-naten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102
9 Das Wasserstoffatom 110
9.1 Radiale Dichteverteilung . . . . . . . . . . . . . . . . . . . 118
9.2 Entartung . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
43

6 Eindimensionale Probleme
Slide 66 Die Schrodingergleichung in einer Dimension
• wir betrachten Wellenfunktionen, die nur von einer Variablen abhangen
d.h. die Bewegung eines einzigen Teilchens ist eingeschrankt auf eineRaumrichtung (x-Achse).
1D-SGL: Hψ(x) = Eψ(x)(− ~2
2m
d2
dx2+ V (x)
)ψ(x) = Eψ(x)
44

6.1 Das Teilchen im unendlich tiefen Kasten
6.1.1 Modell und Losung der Schrodingergleichung
Slide 67 Das Teilchen im unendlich tiefen Kasten
• Das Teilchen soll sich zwischen x = 0 und x = L frei bewegen konnen
(frei bedeutet: kraftefrei: ⇒ V = const.wahle oBdA: V = 0)
• aber nicht außerhalb dieses “Kastens” der Lange L gelangen konnen.
(außerhalb des Kastens ist die potentielle Energie also “unendlich groß”)
Systemskizze
x
V(x)=0 V(x)V(x)
0 L
Slide 68 Losungsweg
• Unterteilung des Definitionsbereiches in 3 Bereiche
1. Fur x ≤ 0 gilt V (x)→∞.
2. Fur 0 < x < L gilt V (x) = 0.
3. Fur x ≥ L gilt wie fur Bereich 1 V (x)→∞.
Slide 6945

Bereiche 1 und 3
• Wir sind nur an endlichen Energien E des Teilchens interessiert.
⇒(− ~2
2m
d2
dx2+ V (x)
)ψ(x) = Eψ(x)
kann nur durch ψ(x) = 0 fur x ≤ 0 und x ≥ L erfullt sein.
Dann gilt naturlich auch ρ(x) = ψ∗(x)ψ(x) = 0 fur x ≤ 0 und x ≥ L,d.h. das Teilchen kann sich nicht außerhalb des Bereiches 0 < x < Laufhalten.
• die Aufenthaltswahrscheinlichkeit außerhalb des “Potentialtopfes” ver-schwindet also, in Ubereinstimmung mit der Problemstellung.
Slide 70 Bereich 2
Fur 0 < x < L gilt V (x) = 0.
Somit lautet die zu losende Differentialgleichung (DGL)
− ~2
2m
d2
dx2ψ(x) = Eψ(x).
Die Energie E muss immer großer oder gleich 0 sein,da E immer ≥ Vmin = 0 ist, also Ekin ≥ 0!
• gesucht: Funktion ψ(x), deren 2. Ableitung proportional zum Negativenihrer selbst ist.
• Wir wissen:
d2
dx2sin(ax) = −a2 sin(ax)
d2
dx2cos(ax) = −a2 cos(ax)
⇒ Losungsansatz ψ(x) = A sin(ax) +B cos(ax)
(A, B und a sind noch festzulegen!).
Slide 71 46

Eigenwerte
d2
dx2ψ =
d2
dx2[A sin(ax) +B cos(ax)]
= Ad2
dx2sin(ax) +B
d2
dx2cos(ax)
= A · (−a2) · sin(ax) +B · (−a2) · cos(ax)
= −a2[A sin(ax) +B cos(ax)] = −a2ψ(x)
Somit ist−~2
2m
d2
dx2ψ(x) =
~2a2
2mψ(x) = Eψ(x)
⇒ E =~2a2
2m
A, B, und a mussen noch durch die Randbedingungen festgelegt werden.
Slide 72 Randbedingungen
(i) ψ(0) = ψ(L)!= 0
da die Wellenfunktion im Topf stetig in die Wellenfunktion außerhalbdes Topfes ubergehen muss.
(ii) Die Normierungsbedingung
∞∫−∞
ψ∗(x)ψ(x)dx =
L∫0
ψ∗(x)ψ(x)dx!= 1
47

Anschlußbedingungen
– aus (i) folgt an der Stelle x = 0:
ψ(0) = A sin(0 · a)︸ ︷︷ ︸0
+B cos(0 · a)︸ ︷︷ ︸1
!= 0 ⇒ B=0
– damit gilt an der Stelle x = L: ψ(L) = A sin(L · a)!= 0
– A 6= 0, da sonst ψ(x) = 0 auch im Potentialkasten
⇒ sin(L · a)!= 0
– analog, n 6= 0, da sonst uberall ψ(x) = 0:
– Zur Erinnerung: sin(nπ) = 0 fur n = 1, 2, 3, . . .
– Man kann also die Bedingung ψ(L) = 0 immer dann
erfullen, wenn L · a = n · π ist, also wenn a =nπ
L
– kleinstes n ist n = 1
– negatives n nicht moglich, da ψ−n = −ψn ware(identisches |ψ|2!)
Slide 73 Energieeigenwerte
• zulassige Energiewerte E =~2a2
2m⇒ En =
~2π2
2mL2· n2 n = 1, 2, 3, . . .
Nur wenn E einen der Eigenwerte En annimmt, hat die Schrodinger-gleichung fur das Teilchen im Kasten eine physikalisch sinnvolle (d.h.mit den Randbedingungen vertragliche) Losung
Offenbar ist E “gequantelt”!
Slide 7448

Normierung
(iii)
∫ L
0
ψ∗(x)ψ(x)dx = A∗A
∫ L
0
sin2(ax)dx!= 1
|A|2∫ L
0
sin2(nπx
L)dx = |A|2L
2!= 1
⇒ Wir wahlen A =
√2
L
• Man hatte durchaus die Freiheit, A = −√
2
Lzu wahlen.
oder A = i
√2
L
oder A = eiα√
2
Lmit beliebigem reellem α
• Diese Wahlfreiheit besteht, weil nur das Betragsquadrat der Wellen-funktion physikalische Bedeutung hat.
6.1.2 Zustande des Teilchens im Kasten
Slide 75 Zustande
Zusammenfassend gilt also fur n = 1, 2, 3, . . .
Energiezustande En =~2π2
2mL2n2 =
h2
8mL2n2
Wellenfunktionen ψn(x) =
√2
Lsin(nπL· x)
• Es existieren unendlich viele Eigenwerte und (normierte) Eigenfunktio-nen fur das Teilchen im Kasten.
49

• Die Grundzustandsenergie (n=1) lautet E1 =~2π2
2mL2=
h2
8mL2
• Die Energien der angeregten Zustande En = E1 · n2 , n = 2, 3, . . .
• n heißt Quantenzahl
Slide 76 Nullpunktsenergie
• Die minimale Energie des Grundzustands des Teilchens im Kasten istimmer vorhanden.
• Sie kann nicht konvertiert werden.
• Dieser unveranderliche minimale Energiebeitrag heißt auch Nullpunkt-senergie.
Slide 77 Spektrum der Zustande
• Anregungsenergien zwischen zweiaufeinanderfolgenden Zustanden
∆En = En+1 − En = E1 · (2n+ 1)
• Die Energien der angeregten Zustandewachsen quadratisch mit der Quanten-zahl n
• die Anregungsenergien zwischen benach-barten Zustanden linear
Slide 78
50

Massenabhangigkeit
1. E1 ∝1
m
damit sind alle Energien umgekehrt pro-portional zur Masse des Teilchens
⇒ auch ∆E ∝ 1
m
Slide 79 Großenabhangigkeit
2. E1 ∝1
L2
alle Energien sind umgekehrt proportio-nal zum Quadrat der Lange des Kastens
⇒ auch ∆E ∝ 1
L2
Slide 80 Quasikontinuum
Großenabschatzung: L = 2 A(≈Atom), m = me
51

→ ∆E1 ≈ 4.5 · 10−18J → ν ≈ 225000 cm−1
(mit ∆E = hcν; vglbar der Rydbergkonstanten (Ry = 1.097·105cm−1 =1.097 · 107m−1)!
Betrachten wir den Grenzfall
• großer Massen
L = 2 A, m = 1 g→ ∆E1 ≈ 1.8 · 10−33J → ν ≈ 9 · 10−11 cm−1!
• oder großer Kastenlangen
L = 1 cm, m = me
→ ∆E1 ≈ 4.2 · 10−45J → ν ≈ 2.1 · 10−22 cm−1!
Dann rucken die Energien der Zustande also sehr dicht zusammen.
→ Es sieht so aus, als waren (fast) beliebige kontinuierliche Variationender Energie moglich. Die Energieniveaus sind quasikontinuierlich wiein der klassischen Physik
Slide 81 Wellenfunktionen und Wahrscheinlichkeitsdichten
• Grundzustandswellenfunktion und zugehorige Wahrscheinlichkeitsdich-te
ψ1(x) =
√2
Lsin(πxL
)ρ1(x) = |ψ1(x)|2 =
2
Lsin2
(πxL
)• Wellenfunktionen der angeregten Zustande und zugehorige Wahrschein-
lichkeitsdichten fur n = 2, 3, . . .
ψn(x) =
√2
Lsin(πxL· n)
ρ1(x) = |ψ1(x)|2 =2
Lsin2
(πxL· n)
52

Slide 82 Wellenfunktionen und Aufenthaltswahrscheinlichkeiten
• die Zahl der Knoten (Nulldurchgange von ψ(x)) nimmt mit zunehmen-dem n zu (wie n− 1).
• Fur sehr hohe Quantenzahlen gibt es sehr viele Knoten.
Slide 83 Bohr’sches Korrespondenzprinzip
Fur großes n gilt:
• Man wird in der Regel den Zu-stand des Systems nur als ei-ne Uberlagerung von Zustandenmit ahnlichem n finden.
• Sind viele dieser Zustande uber-lagert, ergibt sich eine Wahr-scheinlichkeitsdichte ρ ≈ const.
• wie in der klassischen Physik
Dies ist ein Beispiel fur das Bohr’sche Korrespondenzprinzip zwischen Quan-tenmechanik und klassischer Mechanik.
Slide 84 Alternative Darstellung
53

Wellenfunktionen oder Aufenthaltswahrscheinlichkeitsdichten werden im Po-tentialtopf bei Energie E eingezeichnet (s. z.B. Atkins).
6.1.3 Erwartungswerte und Varianzen fur das Teilchen im Kasten
Slide 85 Grundzustand des Teilchens im KastenEnergieerwartungswert
• Der Energieerwartungswert im GZ ist
〈E〉ψ1= 〈ψ1
∗|H|ψ1〉 =
L∫0
ψ1∗Hψ1dx
=
L∫0
ψ1∗E1ψ1dx
= E1
L∫0
ψ1∗ψ1dx = E1
• In einem Eigenzustand des Operators (hier H) ist naturlich der Erwar-tungswert des Operators gleich dem Eigenwert!
Slide 86 54

Grundzustand des Teilchens im KastenErwartungswert des Ortes
• Der Ortserwartungswert im GZ (n = 1) ist
〈x〉ψ1= 〈ψ1
∗|x|ψ1〉 =
(√2
L
)2 L∫0
sin(πx
L)x sin(
πx
L)dx
=2
L
L∫0
x sin2(πxL
)dx =
L
2(s. Ubungsaufgabe)Rechenweg
• Der zustandsgemittelte Aufenthaltsort des Teilchens ist also in der Mittedes Potentialkastens!
(nicht unerwartet!)
Slide 87 Grundzustand des Teilchens im KastenErwartungswert des Impulses
• Der Impulserwartungswert im GZ ist
〈px〉ψ1= 〈ψ1
∗|~i
∂
∂x|ψ1〉 =
2
L
L∫0
sin(πx
L)~i
∂
∂xsin(
πx
L)dx
=2
L
L∫0
sin(πxL
) ~πiL
cos(πxL
)dx = 0
aus Symmetriegrunden
• Der zustandsgemittelte Impuls des Teilchens ist also 0.
(MaW: das Teilchen bewegt sich “genauso oft” nach links wie nachrechts!)
(ebenfalls nicht unerwartet!)
Slide 88 55

Grundzustand des Teilchens im KastenVarianzen
Um diese Mittelwerte ergeben sich Schwankungen der Messung von Ortoder Impuls (Ort und Impuls sind, da [x, H] 6= 0 und [px, H] 6= 0, nichtgleichzeitig mit der Energie scharf messbar) in Form von Standardab-weichungen und Varianzen
• Varianzen (∆A)2 = 〈A2〉 − 〈A〉2
• Standardabweichungen σA =√
(∆A)2 =
√〈A2〉 − 〈A〉
2
• Standardabweichung des Ortes σx =√
(∆x)2 =√〈x2〉 − 〈x〉2︸︷︷︸
schon berechnet
• Standardabweichung des Impulses σpx =√
(∆px)2 =√〈px2〉 − 〈px〉2︸ ︷︷ ︸
schon berechnet
Slide 89 Orts-Impuls-Unscharfe im Grundzustand
• 〈x2〉 = 〈ψ1|x2|ψ1〉 = L2
(1
3− 1
2π2
)⇒ σx =
L
π√
12
√π2 − 6
• 〈p2x〉 = 〈ψ1|p2
x|ψ1〉 =~2π2
L2
• ⇒ σpx =~πL
=⇒ σx · σpx = ~√π2 − 6
12
• da
√π2 − 6
12≈ 0.568, ist auf jeden Fall σx · σpx >
1
2~
Die Heisenberg’sche Unscharferelation ist also erfullt.
56

6.1.4 Zusatzmaterial
Slide 90 Integral∫ L
0x sin2(πx
L)dx
〈x〉 =2
L
L∫0
x sin2(πxL
)dx =
2L
π2
π∫0
y sin2 ydy
Substitution: y = πx/L⇒ dx = dxdy
dy = Lπ
dy
=2L
π2
π∫0
y · 1
2[1− cos(2y)]dx Additionstheorem
=2L
π2
[y2
4− 1
8cos(2y)− 1
4y sin(2y)
]∣∣∣∣y=π
y=0
partielle Integration
=2L
π2
π2
4− 1
8cos(2π)︸ ︷︷ ︸
=1
−π4
sin(2π)︸ ︷︷ ︸=0
−02
4+
1
8cos(0)︸ ︷︷ ︸
=1
+π
4sin(0)︸ ︷︷ ︸
=0
=
2L
π2
π2
4=L
2
57

6.2 Der harmonische Oszillator
6.2.1 Federmodell
Slide 91 Modell
• Eine Masse m sei durch eine Feder mit einer Wand verbunden.
• Auslenkungen aus der Ruhelage (x = 0), in der die Feder entspannt ist,seien nur entlang einer Achse (der x-Achse) moglich.
Slide 92 Federkraft und Hookesches Gesetz
• Fur (kleine) Auslenkungen ist die Ruckstellkraft
~F = −k · ~x.
k heißt Federkonstante
• Krafte sind die negativen Ableitungen der potentiellen Energie
~F = −~∇V .
Slide 93 Wechselwirkungspotential
• Da die Kraft nur von x abhangen soll (und damit das Potential), sieht
man leicht, daß V (x, y, z) = V (x) =1
2kx2
• denn ~∇V (x) =
∂V (x)∂x
∂V (x)∂y
∂V (x)∂z
=
kx00
= kx~ex = k~x
Slide 94 58

Hamilton-Funktion und Hamilton-Operator
• Die kinetische Energie der Masse m ist T =~p2
2m=px
2
2m
• die klassische Hamiltonfunktion (also die Gesamtenergie) lautet
H(px, x) = T + V =px
2
2m+
1
2kx2
• der entsprechende Hamiltonoperator in der Quantenmechanik lautet
H = T + V =p2x
2m+
1
2kx2
oder explizit H = − ~2
2m
d2
dx2+
1
2kx2
6.2.2 Schrodingergleichung
Slide 95 Schrodingergleichung des Harmonischen Oszillators
Hψ(x) = Eψ(x)⇐⇒ − ~2
2m
d2ψ(x)
dx2+
1
2kx2ψ(x) = Eψ(x)
man definiert a2 :=~√k ·m
und multipliziert alle Terme mit2m
~2· a2
und erhalt
−a2 d2ψ(x)
dx2+x2
a2ψ(x) =
2a2mE
~2ψ(x) = εψ(x)
mit der Definition ε :=2a2mE
~2=
2
~
√m
k· E
Slide 96
59

Variablentransformation ξ = x/a
Man definiert eine neue Variable ξ =x
a
dann istd
dξ= a · d
dx
undd2
dξ2= a2 · d2
dx2
Damit erhalt man schließlich
d2
dξ2ψ(ξ) + (ε− ξ2)ψ(ξ) = 0
Slide 97 Ansatze zur Losung der Schrodingergleichung des harmonischenOszillators
Man kann diese Gleichung auf mehrere Arten losen.
1. klassisches Verfahren durch Losen der Differentialgleichung mit Produkt-und Potenzreihenansatz
=⇒ Eigenfunktionen und Eigenwerte
2. algebraisches Verfahren mit Erzeugungs- und Vernichtungsoperatoren
=⇒ Eigenwerte und Matrixelemente
Die Kenntnis von Eigenfunktion und Eigenwert ist aquivalent zur Kennt-nis von Eigenwert und allen Matrixelementen. (Beide Techniken lieferndie gleiche Einsicht in das Problem.)
Slide 98
60

Eigenwerte
• Man erhalt als Eigenwerte fur ε die Werteε = 2n+ 1, n = 0, 1, 2, . . .
und mit den Definitionen
a2 :=~√k ·m
und ε :=2a2mE
~2=
2
~
√m
kE erhalt man
E = ~ω(n+
1
2
)= ~√k
m
(n+
1
2
), n = 0, 1, 2, . . .
Slide 99 Eigenfunktionen
ψn(x) = NnHn(ξ)e−ξ2/2 mit ξ = x/a = x
4√m·k√~ =
√mω~ x
ψn(x) = NnHn(x/a)e−(x/a)2/2 = NnHn
(√mω
~x
)e−
mω2~ x
2
mit den Hermiteschen Polynomen
Hn(ξ) = (−1)neξ2 dn
dξne−ξ
2
und den Normierungsfaktoren
Nn =(m · ω
~π
)1/4(
1
2nn!
)1/2
Slide 100 Hermitesche Polynome
61

H0(y) = 1
H1(y) = 2y
H2(y) = 4y2 − 2
H3(y) = 8y3 − 12y
H4(y) = 16y4 − 48y2 + 12
H5(y) = 32y5 − 160y3 + 120y
H6(y) = 64y6 − 480y4 + 720y2 − 120
H7(y) = 128y7 − 1344y5 + 3360y3 − 1680y
H8(y) = 256y8 − 3584y6 + 13440y4 − 13440y2 + 1680
6.2.3 Losung der Schrodingergleichung
Slide 101 Losung der SchrodingergleichungAsymptotischer Losungsansatz
Schrodingergleichung
d2
dξ2ψ(ξ) + (ε− ξ2)ψ(ξ) = 0
Fur sehr große Werte von ξ gilt ξ2 � ε und damit asymptotisch (alsofur ξ → ±∞)
d2ψ
dξ2− ξ2ψ(ξ) = 0
Diese Differentialgleichung hat die allgemeine Losung
ψ(ξ) = Ae−12ξ2 +Be+ 1
2ξ2
Damit ψ(ξ) normierbar bleibt, muss ψ(ξ) → 0 fur ξ → ±∞ =⇒B = 0
Slide 102 62

Losungsansatz fur die Schrodingergleichung
Die asymptotische Losung ψ(ξ) ∼ e−12ξ2 verwendet man als Ansatz fur
eine allgemeine Losung fur alle ξ, nicht nur fur sehr große.
ψ(ξ) = NH(ξ)e−12ξ2
ist ein Produkt aus
Normierungskonstante N (spater festzulegen),
einer unbekannten “Korrektur”Funktion H(ξ) (als nachstes zu bestim-men)
und der bereits bekannten asympotischen Losung ψ(ξ)
Slide 103 Ableitungen des Losungsansatzes ψ = NH(ξ)ψ(ξ)
Differenziert man ψ(ξ) nach der Kettenregel, so erhalt man
dψ(ξ)
dξ= N
d
dξ
(e−
12ξ2 ·H(ξ)
)= N
(−ξe−
12ξ2 ·H(ξ) + e−
12ξ2 dH(ξ)
dξ
)
und
d2ψ(ξ)
dξ2= N
((ξ2 − 1)e−
12ξ2H(ξ)− 2ξe−
12ξ2 dH(ξ)
dξ
+e−12ξ2 d2H(ξ)
dξ2
)
Slide 104
63

Differentialgleichung fur H(ξ)
Einsetzen in die SGLd2
dξ2ψ(ξ) + (ε− ξ2)ψ(ξ) = 0
und Multiplikation von links mit(
e12ξ2)/N ergibt
(ξ2 − 1)H(ξ)− 2ξdH(ξ)
dξ+
d2H(ξ)
dξ2+ (ε− ξ2)H(ξ) = 0
oder
H ′′(ξ)− 2ξH ′(ξ) + (ε− 1)H(ξ) = 0
Slide 105 Potenzreihenansatz fur H(ξ)I
Gleichungen der Form
H ′′(ξ)− 2ξH ′(ξ) + (ε− 1)H(ξ) = 0
lost man durch Potenzreihenentwicklung
Ansatz H(ξ) =∞∑j=0
ajξj
⇒ H ′(ξ) =∞∑j=0
j · ajξj−1
⇒ H ′′(ξ) =∞∑j=0
j(j − 1) · ajξj−2
Einsetzen:
∞∑j=0
j(j − 1) · ajξj−2 − 2ξ∞∑j=0
j · ajξj−1
︸ ︷︷ ︸ξ...ξj−1→ξj
+(ε− 1)∞∑j=0
ajξj = 0
Slide 106 64

Potenzreihenansatz fur H(ξ)II
∞∑j=0
j(j − 1) · ajξj−2
︸ ︷︷ ︸⇓
−2∞∑j=0
j · ajξj + (ε− 1)∞∑j=0
ajξj = 0
∞∑j=2
j(j − 1) · ajξj−2
︸ ︷︷ ︸mit k + 2 := j
∞∑k=0
(k + 2)(k + 2− 1) · ak+2ξk+2−2
︸ ︷︷ ︸und nach Umbenennen k → j
∞∑j=0
{(j + 2)(j + 1)aj+2 + (ε− 1− 2j)aj} ξj = 0
Slide 107 Potenzreihenansatz fur H(ξ)III Rekursionsformel
∞∑j=0
{(j + 2)(j + 1)aj+2 + (ε− 1− 2j)aj} ξj = 0
Diese Gleichung kann nur dann fur alle ξ gelten, wenn jede einzelnePotenz von ξ verschwindet.
⇒ (j + 2)(j + 1)aj+2 = (2j + 1− ε)aj
Dies ist eine Rekursionsformel fur die aj
• kennt man a0, sind alle weiteren a2, a4, a6, . . . festgelegt.
• kennt man a1, sind alle weiteren a3, a5, a7, . . . festgelegt.
a0 und a1 kann man prinzipiell frei wahlen, da man spater noch denNormierungsfaktor bestimmen muss.
Slide 108 65

Abbruchbedingung
Man kann nun zeigen (tun wir aber nicht), dass i.A. fur einen beliebigenWert von ε die Funktion H(ξ) so schnell wachst, dass das Produkt
H(ξ)e−12ξ2 fur ξ → ±∞ nicht gegen Null strebt.
=⇒ Die Wellenfunktion ist dann nicht normierbar.
Fur normierbare Losungen muss die Potenzreihe fur H(ξ) also bei ir-gendeiner maximalen Potenz n (also bei ξn) abbrechen.
also: an+2!= 0
⇒ 2n+ 1− ε(n+ 2)(n+ 1)
!= 0
=⇒ 2n+ 1− ε = 0
oder εn = 2n+ 1
Slide 109 Quantenzahl n
εn = 2n+ 1
Es fallt wieder automatisch eine Quantenzahl n an, die die verschiede-nen moglichen Losungen (fur die maximalen endlichen Potenzen vonξ) durchzahlt.
ε war definiert als ε =2
~
√m
kE
Die Energieniveaus des harmonischen Oszillators lauten dann
En = ~√k
m
(n+
1
2
)
Slide 110
66

Frequenz des Oszillators
En = ~ω(n+
1
2
)= hν
(n+
1
2
)
Frequenz: ν =1
2π
√k
m
Kreisfrequenz: ω = 2πν =
√k
m
Frequenz des quantenmechanischen Oszillators ist dieselbe wie die desklassischen Oszillators
Die Energie des klassischen Oszillators hangt direkt von der Amplitude(maximale Auslenkung) der Schwingung ab und kann kontinuierlichvariieren
Die Energie des quantenmechanischen Oszillators ist wieder gequantelt.
6.2.4 Form der Wellenfunktionen und Aufenthaltswahrscheinlich-keitsdichte
Slide 111 Wellenfunktionen
ψ(ξ) = NnHn(ξ)e−12ξ2
mit
67

Hn(ξ) =n∑j=0
ajξj
a(n)j+2 =
2(j − n)
(j + 2)(j + 1)a
(n)j
wenn n gerade, dann verschwinden
alle aj fur ungerades j
wenn n ungerade, dann verschwinden
alle aj fur gerades j
Slide 112 Grundzustand
ψn(x) = NnHn(ξ)e−ξ2/2
mit ξ = x/a = x4√m·k√~ =
√mω~ x
und Nn =(m · ω
~π
)1/4(
1
2nn!
)1/2
• EGZ = E0 =1
2~ω
ψ0(x) =(m · ω
~π
)1/4
· 1 · 1 · e−12mω~ x2
=(m · ω
~π
)1/4
e−12mω~ x2
=
√1√π
(4
√m · ω~
)· e−
12mω~ x2
Slide 113
68
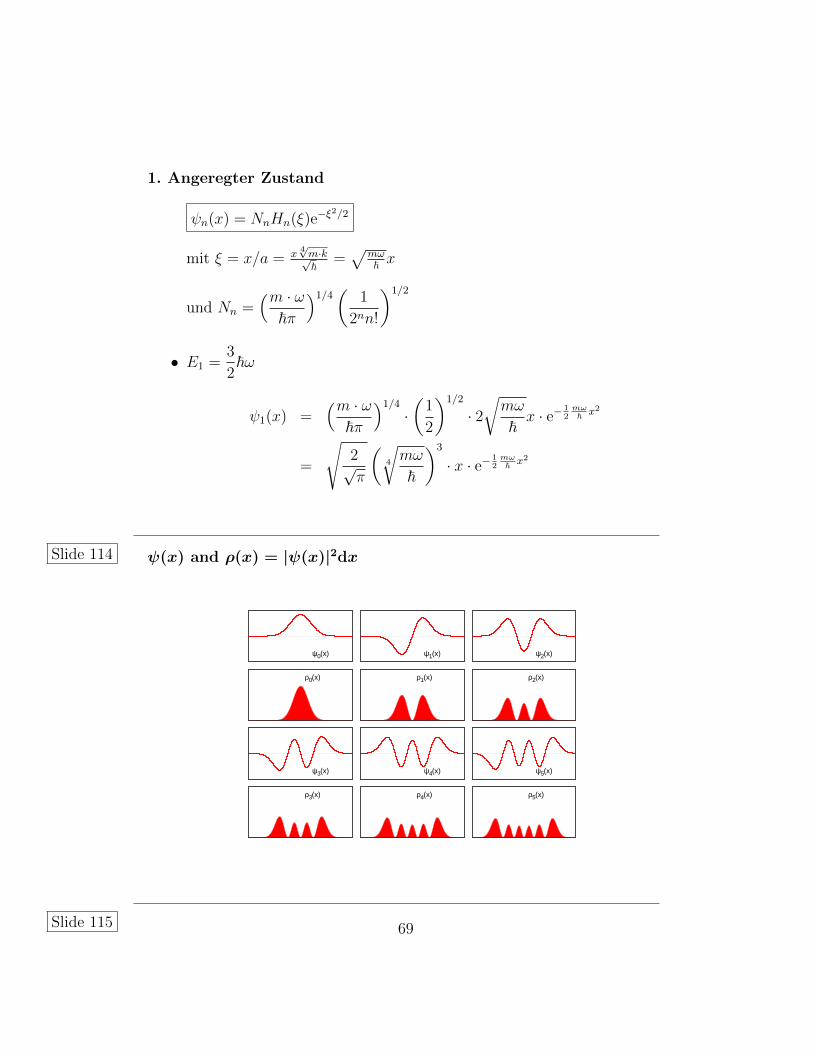
1. Angeregter Zustand
ψn(x) = NnHn(ξ)e−ξ2/2
mit ξ = x/a = x4√m·k√~ =
√mω~ x
und Nn =(m · ω
~π
)1/4(
1
2nn!
)1/2
• E1 =3
2~ω
ψ1(x) =(m · ω
~π
)1/4
·(
1
2
)1/2
· 2√mω
~x · e−
12mω~ x2
=
√2√π
(4
√mω
~
)3
· x · e−12mω~ x2
Slide 114 ψ(x) and ρ(x) = |ψ(x)|2dx
ψ0(x) ψ1(x) ψ2(x)
ρ0(x) ρ1(x) ρ2(x)
ψ3(x) ψ4(x) ψ5(x)
ρ3(x) ρ4(x) ρ5(x)
Slide 115 69

Darstellung im “Topf der potentiellen Energie”
Slide 116 Eigenschaften I• Zahl der Knoten(=Nulldurchgange der Wellenfunktion) = Quantenzahln
• Die niedrigste Energie 12~ω > 0 Nullpunktsenergie
Ein quantenmechanischer Oszillator ist niemals in Ruhe! Er“schwingt immer um seine Gleichgewichtslage”!
Slide 117 EigenschaftenII
70

• Ein klassischer Oszillator durfte sich, bei den gegebenen Energien, nichtim Bereich außerhalb der gelben Parabel aufhalten. (Hier ware wegenEn = T + V und V > En die kinetische Energie T negativ)!
• Ein quantenmechanischer Oszillator darf sich in diesem klassisch “ver-botenen” Bereich aufhalten. Die Wellenfunktion hat eine endliche Am-plitude und Aufenthaltswahrscheinlichkeitsdichte in diesem Bereich.
TunneleffektDieses Hinein’tunneln’ in klassisch verbotene Bereiche ist einallgemeines Phanomen der QuantenmechanikDer Name dafur ist
Tunneleffekt
Slide 118 Unscharferelation
• Die Orts-Impulsunscharfe√
∆x2∆p2x ist im Grundzustand des harmo-
nischen Oszillators gegeben durch√
∆x2∆p2x =
~2
.
• Es gilt also das Gleichheitszeichen in der Unscharferelation
Zustand minimaler quantenmechanischer UnscharfeDer GZ des HO ist derjenige quantenmechanische Zustand mitder minimal moglichen Orts-Impuls-Unscharfe.
Slide 119 Der harmonische Oszillator:Ein Modell fur zweitatomige Molekule
• Alle obigen Resultate gelten auch fur zweiatomige Schwinger
• x ist dann die Differenz zwischen aktuellem Atomabstand und demGleichgewichtsabstand re: x = x2 − x1 − re
• Dazu muss m→ µ =m1 ·m2
m1 +m2
ersetzt werden.
• µ heißt reduzierte Masse.
71

7 Zwei- und Dreidimensionale Probleme in
kartesischen Koordinaten
7.1 Das Teilchen im zweidimensionalen Kasten
Slide 120 Das Teilchen im Kasten
• Das Teilchen soll sich zwischen x = 0 und x = Lx und y = 0 undy = Ly frei bewegen konnen
(frei bedeutet: kraftefrei: ⇒ V = const.wahle oBdA: V = 0)
• aber nicht außerhalb dieses “Kastens” der Dimension (Flache) Lx · Lygelangen konnen.
(außerhalb des Kastens ist die potentielle Energie also “unendlich groß”)
Systemskizze
Slide 121 Die Schrodingergleichung im zweidimensionalen Kasten
• − ~2
2m
[∂2
∂x2+
∂2
∂y2
]ψ(x, y) = Eψ(x, y)
72

fur 0 ≤ x ≤ Lx und 0 ≤ y ≤ Ly.
• ψ(x, y) = 0 fur alle anderen Werte von (x, y).
• Losung: Produktansatz ψ(x, y) = X (x) · Y(y)
− ~2
2m
[∂2
∂x2+
∂2
∂y2
]X (x) · Y(y) = E · X (x) · Y(y)
−Y(y)~2
2m
∂2X (x)
∂x2−X (x)
~2
2m
∂2Y(y)
∂y2= E · X (x) · Y(y)∣∣∣∣· 1
X (x) · Y(y)
− ~2
2m
1
X (x)
∂2X (x)
∂x2− ~2
2m
1
Y(y)
∂2Y(y)
∂y2= E
Slide 122 Faktorisierung
− ~2
2m
1
X (x)
∂2X (x)
∂x2︸ ︷︷ ︸⇓
− ~2
2m
1
Y(y)
∂2Y(y)
∂y2︸ ︷︷ ︸⇓
= E︸︷︷︸⇓
Fkt. nur von x Fkt. nur von y Konstante
• Die Summe einer Funktion, die nur von x abhangt, und einer anderenFunktion, die nur von y abhangt, kann nur dann konstant sein, wennjede der beiden Funktionen fur sich konstant ist.
− ~2
2m
1
X (x)
∂2X (x)
∂x2= Ex
− ~2
2m
1
Y(y)
∂2Y(y)
∂y2= Ey
Slide 123
73

2 Schrodingergleichungen in einer Dimension
− ~2
2m
∂2X (x)
∂x2= ExX (x)
− ~2
2m
∂2Y(y)
∂y2= EyY(y)
Ex + Ey = E
• Die Losungen fur diese beiden Gleichungen kennen wir aber schon!
Es sind die gleichen Losungen wie die des Teilchens im eindimensiona-len Kasten.
Slide 124 Losungen der eindimensionalen Gleichung
=⇒
E(j)x =
~2π2
2mLx2 · j
2 j = 1, 2, 3, . . .
E(k)y =
~2π2
2mLy2 · k
2 k = 1, 2, 3, . . .
j und k sind unabhangig voneinander
Xj(x) =
√2
Lxsin
(jπ
Lx· x)
Yk(y) =
√2
Lysin
(kπ
Ly· y)
Slide 125
74

Energieeigenwerte und Eigenfunktionen des zweidimensionalen Pro-blems
Ej,k =~2π2
2mLx2 · j
2 +~2π2
2mLy2 · k
2
=~2π2
2m
[j2
Lx2 +
k2
Ly2
]j, k = 1, 2, 3, . . .
ψj,k(x, y) =
√4
Lx · Ly· sin
(jπ
Lx· x)· sin
(kπ
Ly· y)
• Die Losungen enthalten zwei Quantenzahlen j und k!
• Die Losungen hangen von der Form des Kastens (via Lx und Ly) ab!
• Es ist denkbar, dass 2 unterschiedliche Kombinationen (j, k) den glei-chen Energiewert liefern!
Dieses Phanomen nennt man (Energie)Entartung
Slide 126 Grundzustand im quadratischen Kasten
• Betrachten wir als Spezialfall einen quadratischen Kasten mit Lx =Ly = L.
• Die Energieeigenwerte lauten dann
E =~2π2
2m
[j2
L2+k2
L2
]=
~2π2
2mL2
[j2 + k2
]= ε[j2 + k2]
• Der Grundzustand (1, 1) ist gegeben durch E1,1 = 2ε, da j, k ≥ 1 seinmussen.
• Der Grundzustand ist nicht entartet, d.h., es gibt nur eine Kombinationvon j und k (beide = 1).
75

• Die Wellenfunktion des Grundzustandes ist ψ1,1(x, y) =
√4
L2·sin
(πL· x)·
sin(πL· y)
Slide 127 Angeregte Zustande im quadratischen Kasten
E = ε[j2 + k2]
• Der 1. angeregte Zustand ist gegeben durch E = 5ε.
• Er ist zweifach entartet.
• Energie des Zustandes (1, 2): j = 1, k = 2 =⇒ E1,2 = ε[1 + 4] = 5ε
• Wellenfunktion ψ1,2(x, y) =
√4
L2· sin
(πL· x)· sin
(2π
L· y)
• Energie des Zustandes (2, 1): j = 2, k = 1 =⇒ E2,1 = ε[4 + 1] = 5ε
• Wellenfunktion ψ2,1(x, y) =
√4
L2· sin
(2π
L· x)· sin
(πL· y)
Slide 128 Entartung
• Man erkennt leicht, dass alle Zustande, in denen j = k ist, nicht odereinfach entartet sind.
• Alle anderen Zustande, fur die j 6= k ist, sind zweifach entartet.
Slide 129
76

Wellenfunktionen im quadratischen Kasten
• Es gibt Knotenlinien.
• Die Wellenfunktionen zu entarteten Zustanden lassen sich durch Rota-tion ineinander uberfuhren.
Slide 130 Rechteckiger Kasten
• Der allgemeine Fall ist der, fur den Lx 6= Ly ist.
• Der Grundzustand ist immer durch ψ1,1 und E1,1 gegeben.
• Die Reihenfolge der angeregten Zustande hangt von der Form des Ka-stens ab.
Slide 131 Energieniveaus im Rechteck-Kasten mit Ly = 2Lx
E =~2π2
2m
[j2
L2x
+k2
L2y
]=
~2π2
2m
[j2
L2x
+k2
4L2x
]=
~2π2
8mLx2
[4j2 + k2
]= ε
[4j2 + k2
]
77

j k 4j2 + k2
1 1 51 2 81 3 131 4 201 5 292 1 172 2 202 3 25
. . .
• Anregung in y-Richtung ist leichter.
• Die Zustande (j, k) = (2, 2) und (1, 4) sind entartet. Man nennt dies auchzufallige Entartung.
• Es gibt weniger entartete Energieniveaus als fur den quadratischen Kasten.
• Entartung ist eine Konsequenz der Symmetrie. Die hier beobachtete zufalli-ge Entartung ist die Konsequenz einer “versteckten” Symmetrie.
Slide 132 Wellenfunktionen im Rechteck-Kasten mit Ly = 2Lx
Slide 133 Zufallige Entartung von ψ1,4 und ψ2,2
78

• Versteckte Symmetrie: Man kann durch Rotation der Halften der Wel-lenfunktion um 90◦ die beiden Wellenfunktionen ineinander uberfuhren.
(Eine Knotenlinie geht mitten durch den Kasten, und die Wellenfunk-tion verschwindet auch an den Randern.)
79

7.2 Das Teilchen im dreidimensionalen Kasten
Slide 134 Das Teilchen im Kasten
• Das Teilchen soll sich zwischen x = 0 und x = Lx und y = 0 undy = Ly und z = 0 und z = Lz frei bewegen konnen
(frei bedeutet: kraftefrei: ⇒ V = const.wahle oBdA: V = 0)
• aber nicht außerhalb dieses “Kastens” der Dimension (Volumen) Lx ·Ly · Lz gelangen konnen.
(außerhalb des Quaders ist die potentielle Energie also “unendlich groß”)
Systemskizze
Slide 135 Die Schrodingergleichung im dreidimensionalen Kasten
• − ~2
2m
[∂2
∂x2+
∂2
∂y2+
∂2
∂z2
]ψ(x, y, z) = Eψ(x, y, z)
fur 0 ≤ x ≤ Lx, 0 ≤ y ≤ Ly und 0 ≤ z ≤ Lz.
• ψ(x, y, z) = 0 fur alle anderen Werte von (x, y, z).
80

• Losung: Produktansatz ψ(x, y, z) = X (x) · Y(y) · Z(z)
− ~2
2m
[∂2
∂x2+
∂2
∂y2+
∂2
∂z2
]XYZ = E · XYZ
−YZ ~2
2m
∂2X∂x2
−XZ ~2
2m
∂2Y∂y2−XY ~2
2m
∂2Z∂z2
= E · XYZ∣∣∣∣· 1
XYZ
− ~2
2m
1
X∂2X∂x2
− ~2
2m
1
Y∂2Y∂y2− ~2
2m
1
Z∂2Z∂z2
= E
Slide 136 Faktorisierung
− ~2
2m
1
X∂2X∂x2︸ ︷︷ ︸
⇓
− ~2
2m
1
Y∂2Y∂y2︸ ︷︷ ︸
⇓
− ~2
2m
1
Z∂2Z∂z2︸ ︷︷ ︸
⇓
= E
Fkt. nur von x Fkt. nur von y Fkt. nur von z
• Die Summe dreier Funktionen, von denen eine nur von x, eine nur von y undeine nur von z abhangt, kann nur dann konstant sein, wenn jede der dreiFunktionen fur sich konstant ist.
− ~2
2m
1
X∂2X∂x2
= Ex
− ~2
2m
1
Y∂2Y∂y2
= Ey
− ~2
2m
1
Z∂2Z∂z2
= Ez
Slide 137 Energieeigenwerte und Eigenfunktionen des dreidimensionalen Pro-blems
81

Die Losungen sind bekannt, und konnen analog zu denen in einer undzwei Dimensionen konstruiert werden:
Ej,k,l =~2π2
2mLx2 · j
2 +~2π2
2mLy2 · k
2 +~2π2
2mLZ2 · l
2
=~2π2
2m
[j2
Lx2 +
k2
Ly2 +
l2
Lz2
]j, k, l = 1, 2, 3, . . .
ψj,k,l(x, y, z) =
√8
LxLyLz· sin
(jπx
Lx
)sin
(kπy
Ly
)sin
(lπz
Lz
)
• Die Losungen enthalten drei Quantenzahlen j, k, und l!
• Die Losungen hangen von der Form des Quaders (via Lx, Ly und Lz)ab!
• Es ist denkbar, dass mehrere unterschiedliche Kombinationen (j, k, l)den gleichen Energiewert liefern!
Slide 138 Energieniveaus im Wurfel Lx = Ly = Lz = L
E =~2π2
2mL2
[j2 + k2 + l2
]= ε
[j2 + k2 + l2
]j k l j2 + k2 + l2
1 1 1 3
1 1 2 61 2 1 62 1 1 6
1 2 2 92 1 2 92 2 1 9
. . .1 2 3 141 3 2 142 1 3 142 3 1 143 1 2 143 2 1 14
. . .
82

j k l j2 + k2 + l2
. . .4 4 3 41 (3 Permutationen)6 2 1 41 (6 Permutationen)
. . .
• Aufgrund der kubischen Symmetrie beobachtet man regelmaßig Entartung.
• Man beobachtet unterschiedliche Entartungsgrade g.
• z.B. ist g = 1 fur den Grundzustand.
• g = 3 fur die Zustande E = 6ε und E = 9ε.
• g = 6 fur den Zustand E = 14ε.
• g = 9 fur den Zustand E = 41ε! (normale und zufallige Entartung)
83

7.3 Der harmonische Oszillator in 3 Dimensionen
Slide 139 Potentielle EnergieI
• Bei der Losung des Harmonischen Oszillators in einer Dimension war
die potentielle Energie der Masse an der Feder durch Epot(x) =1
2kx2
gegeben.
Die Feder konnte sich nur in x-Richtung bewegen.
• Man kann dies auf 3 Dimensionen verallgemeinern.
Dazu nimmt man an, dass die Feder an einem Punkt fixiert ist, und inalle Raumrichtungen ausgelenkt werden kann.
Slide 140 Potentielle EnergieII
Die potentielle Energie lautet allgemein (wieder mit der Annahme, dassdas Hookesche Gesetz erfullt ist)
V (x, y, z) =1
2kxx
2 +1
2kyy
2 +1
2kzz
2
Diese Gleichung nimmt an, dass die Federkraft in unterschiedlichenRichtungen unterschiedlich stark wirkt.
Nimmt man dagegen an, dass kx = ky = kz = k, dann gilt
V (x, y, z)1
2(x2 + y2 + z2) = V (r) =
1
2kr2
wobei r2 = x2 +y2 +z2 das Abstandsquadrat der Masse von der Gleich-gewichtslage im Raum ist.
Slide 141
84

Hamilton Operator
H = − ~2
2m
[∂2
∂x2+
∂2
∂y2+
∂2
∂z2
]+
1
2kr2
H = − ~2
2m
[∂2
∂x2+
∂2
∂y2+
∂2
∂z2
]+
1
2k(x2 + y2 + z2)
=
[− ~2
2m
∂2
∂x2+
1
2kx2
]+
[− ~2
2m
∂2
∂y2+
1
2ky2
]+
[− ~2
2m
∂2
∂z2+
1
2kz2
]
• H zerfallt aufgrund der speziellen Form der potentiellen Energie (∝ r2)in die Summe aus drei harmonischen Oszillatoren.
Slide 142 Schrodingergleichung
• Man kann wieder einen Produktansatz machen: ψ(x, y, z) = X (x)Y(y)Z(z).
• Die Schrodingergleichung lautet dann
Hψ(x, y, z) =
[− ~2
2m
∂2
∂x2+
1
2kx2
]XYZ
+
[− ~2
2m
∂2
∂y2+
1
2ky2
]XYZ
+
[− ~2
2m
∂2
∂z2+
1
2kz2
]XYZ
= E · XYZ
• Man dividiert wieder durch XYZ und erhalt die
Slide 143
85

Faktorisierte Schrodingergleichung
1
XYZHXYZ =
[− 1
X~2
2m
∂2X∂x2
+1
2kx2
]+
[− 1
Y~2
2m
∂2Y∂y2
+1
2ky2
]+
[− 1
Z~2
2m
∂2Z∂z2
+1
2kz2
]= E
• Wieder ist die Summe von drei Funktionen, von denen jede von genaueiner unabhangigen Variable abhangt, eine Konstante.
⇒ Jeder der Klammerausdrucke fur sich muss wieder konstant sein.
Slide 144 Eindimensionale Schrodingergleichung
• Man erhalt also, z.B. fur die x-Richtung[− 1
X~2
2m
∂2X∂x2
+1
2kx2
]= Ex
Multiplikation mit X[− ~2
2m
∂2X∂x2
+1
2kx2X
]= Ex · X
• Dies ist wieder die Schrodingergleichung eines harmonischen Oszilla-tors.
• Analoges gilt fur die y und die z-Richtung
• Das Problem des kugelsymmetrischen Harmonischen Oszillators kannalso in drei eindimensionale harmonische Oszillatorprobleme transfor-miert werden.
Slide 14586

Eigenwerte und Eigenfunktionen des Harmonischen Oszillators indrei Dimensionen
Ej,k,l = ~ω(j +
1
2
)+ ~ω
(k +
1
2
)+ ~ω
(l +
1
2
)= ~ω
(j + k + l +
3
2
)j, k, l = 0, 1, 2, . . .
ψj,k,l = NjklHj
(xa
)Hk
(ya
)Hl
(za
)e−(x2+y2+z2)/2a2
Hj, Hk, Hl sind wieder Hermitesche Polynomen, und a und ω sind wiezuvor definiert. Njkl ist ein Normierungsfaktor.
• Die Nullpunktsenergien sind additiv!
Slide 146 Energieeigenwerte E = ~ω(j + k + l + 3
2
)und Entartung
j k l j + k + l + 32
Permutationen
0 0 0 3/2 1
1 0 0 5/2 3
2 0 0 7/2 31 1 0 7/2 3
3 0 0 9/2 32 1 0 9/2 61 1 1 9/2 1
4 0 0 11/2 33 1 0 11/2 62 2 0 11/2 32 1 1 11/2 3
. . .
• Entartungsgrad g steigt rasch mit der Energie an.
• g(3/2) = 1 (GZ)
• g(5/2) = 3
87

• g(7/2) = 6
• g(9/2) = 10
• g(11/2) = 15
Slide 147 Symmetrie und Entartung
• Man nennt einen Energieeigenwert der Schrodingergleichung n-fachentartet, wenn er durch n unterschiedliche Kombinationen der zugehori-gen Quantenzahlen realisiert werden kann.
• Weist das zu losende Problem und damit der zugehorige Hamilton-operator eine oder mehrere Symmetrien auf, so finden sich in seinemEnergiespektrum entartete Eigenwerte.
• Der Umkehrschluß (Entartung ⇒ Symmetrie) gilt nicht. Es kann zuzufalligen Entartungen kommen.
7.4 Erweiterung auf mehr als ein Teilchen
Slide 148 Schrodingergleichung fur 1 Teilchen
• Produktansatz: ψ(x, y, z) = X (x)Y(y)Z(z).
• Schrodingergleichung:
Hψ(x, y, z) =
[− ~2
2m
∂2
∂x2+
1
2kx2
]XYZ
+
[− ~2
2m
∂2
∂y2+
1
2ky2
]XYZ
+
[− ~2
2m
∂2
∂z2+
1
2kz2
]XYZ
= E · XYZ
• E = Ex + Ey + Ez = ~ω[(j + 1
2) + (k + 1
2) + (l + 1
2)]
Slide 149 88

Schrodingergleichung fur 2 Teilchen
• Produktansatz: ψ(x1, y1, z1, x2, . . .) = X1(x1)Y1(y1)Z1(z1)X2(x2)Y2(y2)Z2(z2).
• Schrodingergleichung:
Hψ(x1, y1, z1, x2, y2, z2)
=
[− ~2
2m
∂2
∂x21
+1
2kx2
1
]X1Y1Z1X2Y2Z2
+
[− ~2
2m
∂2
∂y21
+1
2ky2
1
]X1Y1Z1X2Y2Z2
+
[− ~2
2m
∂2
∂z21
+1
2kz2
1
]X1Y1Z1X2Y2Z2
+ . . .
+
[− ~2
2m
∂2
∂z22
+1
2kz2
2
]X1Y1Z1X2Y2Z2
= E · X1Y1Z1X2Y2Z2
Slide 150 Schrodingergleichung fur 2 Teilchen
• E = Ex1 + Ey1 + Ez1 + Ex2 + Ey2 + Ez2
• E = ~ω[(j1 + 1
2) + (k1 + 1
2) + (l1 + 1
2) +
(j2 + 12) + (k2 + 1
2) + (l2 + 1
2)]
Slide 151 Schrodingergleichung fur N Teilchen
E = Ex1 + Ey1 + Ez1 + Ex2 + Ey2 + Ez2 + Ex3 + . . .
E = ~ω[(j1 + 1
2) + (k1 + 1
2) + (l1 + 1
2)+
(j2 + 12) + (k2 + 1
2) + (l2 + 1
2) + (j3 + 1
2) + . . .
]E =
k=3N∑k=1
~ω(nk+1
2) mit nk den Quantenzahlen fur den k. Freiheitsgrad
89

8 Zentralkraft-Probleme
8.1 Einleitung
Slide 152 Der dreidimensionale harmonische Oszillatorin kartesischen Koordinaten
• Die potentielle Energie des symmetrischen (kx = ky = kz = k) Hamilton-Operators des dreidimensionalen harmonischen Oszillators laßt sichaufgrund seiner speziellen Form auch als abstandsabhangige potentielleEnergie schreiben:
H(x, y, z) = − ~2
2m
[∂2
∂x2+
∂2
∂y2+
∂2
∂z2
]+
1
2k(x2 + y2 + z2
)︸ ︷︷ ︸r2
Kann man den kinetischen Energie-Anteil vielleicht ebenfalls als Funk-tion von r und zwei weiteren Winkelkoordinaten formulieren?
Slide 153 Der dreidimensionale harmonische Oszillatorin Kugelkoordinaten
• Kann man den kinetischen Energie-Anteil vielleicht ebenfalls als Funktion
von r und zwei weiteren Winkelkoordinaten formulieren?
Etwa in der Form?
H(r, ϑ, φ) = − ~2
2m∆(r, ϑ, φ) +
1
2kr2
• Klassisch ist diese Zerlegung der kinetischen Energie in einen Radial-anteil und einen Drehimpulsanteil moglich.
• Man zerlegt dabei die kinetische Energie im kartesischen Koordinaten-system in zwei Komponenten, namlich die der Radialbewegung weg voneinem (wahlbaren) Ursprungspunkt und die einer Drehbewegung um ei-ne Achse durch den Ursprungspunkt
Slide 154 90

Beispiel: Planetenbewegung
• Die Bewegung der Planeten um die Sonne (oder des Mondes um die Er-de) ist eine Drehbewegung auf einer elliptischen Bahn, wobei die Sonne(bzw. die Erde) in einem Brennpunkt stehen. (Kepler’sche Gesetze)14
• Die Bewegung findet (wenn man von außeren storenden Einflussen ab-sieht), in einer Ebene statt.
(Ebene der Ekliptik ≈ 23.5◦ fur die Erde)
• Durch diverse (zeitlich veranderliche) Storungen (z.B. durch die Kon-stellation anderer Planeten) variiert die elliptische Bahn der Erde umdie Sonne langfristig (Radialbewegung).
8.1.1 Kugelkoordinaten
Slide 155 Kugelkoordinaten
• Azimutwinkel “Theta”θ oder ϑ(→ Breitengrad)
• Winkel “Phi”φ oder ϕ(→ Langengrad)
14http://de.wikipedia.org/wiki/Johannes Kepler
91

• z = r cosϑ
• ρ = r sinϑ
• x = r sinϑ cosϕ
• y = r sinϑ sinϕ
• Die Ersetzung {x, y, z} → {r, ϑ, ϕ} nennt man eine Koordinatentrans-formation.
Slide 156 Laplace-Operator in KugelkoordinatenKoordinatentransformation
• Annahme: Koordinatentransformationx = x(r, ϑ, ϕ)
y = y(r, ϑ, ϕ)
z = z(r, ϑ, ϕ)
• Weiterhin gebe es eine Rucktransformationr = r(x, y, z)
ϑ = ϑ(x, y, z)
ϕ = ϕ(x, y, z)
• Dann gilt fur eine Funktion f(r, ϑ, φ)(∂f
∂x
)=
(∂f
∂r
)(∂r
∂x
)+
(∂f
∂ϑ
)(∂ϑ
∂x
)+
(∂f
∂ϕ
)(∂ϕ
∂x
)
Slide 157
92

Differentialoperatoren(∂∂x
)und
(∂2
∂x2
)In Operatorschreibweise(
∂
∂x
)f =
[(∂r
∂x
)(∂
∂r
)+
(∂ϑ
∂x
)(∂
∂ϑ
)+
(∂ϕ
∂x
)(∂
∂ϕ
)]f
Also (∂
∂x
)=
[(∂r
∂x
)(∂
∂r
)+
(∂ϑ
∂x
)(∂
∂ϑ
)+
(∂ϕ
∂x
)(∂
∂ϕ
)]und (
∂2
∂x2
)=
[(∂r
∂x
)(∂
∂r
)+
(∂ϑ
∂x
)(∂
∂ϑ
)+
(∂ϕ
∂x
)(∂
∂ϕ
)][(
∂r
∂x
)(∂
∂r
)+
(∂ϑ
∂x
)(∂
∂ϑ
)+
(∂ϕ
∂x
)(∂
∂ϕ
)]
Slide 158 Laplace-Operator in Kugelkoordinaten
• . . . analog fur y und z
• =⇒ . . . =⇒ . . .
∆ =
(∂2
∂x2
)+
(∂2
∂y2
)+
(∂2
∂z2
)=
(∂2
∂r2
)+
2
r
(∂
∂r
)+
1
r2
1
sinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)+
1
r2
1
sin2 ϑ
(∂2
∂φ2
)=:
(∂2
∂r2
)+
2
r
(∂
∂r
)+
Λ2
r2
mit dem Legendreschen Operator Λ = Λ(ϑ, ϕ)
Slide 159 93

Hamilton-Operator in Kugelkoordinaten
H = V (r)− ~2
2m
[(∂2
∂r2
)+
2
r
(∂
∂r
)]− ~2Λ2
2mr2
• mr2 ist das Tragheitsmoment der Massem im Abstand r vom Ursprung.
• L2 = −~2Λ2 ist der Drehimpulsoperator.
• Der winkelabhangige Term im Operator der kinetischen Energie hatalso wie in der klassischen Mechanik die Struktur
1
2
Drehimpuls2
Tragheitsmoment.
8.1.2 Teilchen auf der Kugeloberfache
Slide 160 Das Teilchen auf der Kugeloberflache
H = V (r)− ~2
2m
[(∂2
∂r2
)+
2
r
(∂
∂r
)]− ~2Λ2
2mr2
• Was passiert, wenn wir den “Aktionsradius” des Teilchens auf die Ku-geloberflache (Radius R) begrenzen?
(homo sapiens sapiens vor der Erfindung der Schaufel und des Flug-zeugs)
1.
(∂
∂r
)= 0!
2. V (R) = 0, sonst V (r)→∞
=⇒ Die Wellenfunktion in R-Richtung ist “uninteressant”!
Slide 161
94

Schrodingergleichung des Teilchens auf der Kugel
Hψ(ϑ, ϕ;R) = − ~2
2mR2
[1
sinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)+
1
sin2 ϑ
(∂2
∂ϕ2
)]ψ(ϑ, ϕ;R)
R ist ein Parameter.
• Die Losung dieser Differentialgleichung fuhrt auf die sogenannten Ku-gelflachenfunktionen Ylm (s.u.).
8.1.3 Das Teilchen auf dem Ring
Slide 162 Das Teilchen auf dem Ring(auf dem “Kugelaquator”)
• Was passiert, wenn wir weiterhin den “Aktionsradius” des Teilchensauf den Aquator beschranken?
1. ϑ wird konstant sein: ϑ = 90◦ ⇒ sinϑ = 1
2.
(∂
∂ϑ
)= 0!
3. Formal wird aus der Variable ϑ wieder ein Parameter.
• Die Schrodingergleichung des Teilchens auf dem “Ring” lautet
Hψ(ϕ;R, ϑ) = − ~2
2mR2
(∂2
∂ϕ2
)ψ(ϕ;R, ϑ)
allgemein, auf einem Breitengrad:
= − ~2
2msin2 ϑR2
(∂2
∂ϕ2
)ψ(ϕ;R, ϑ)
Slide 163 95

Losung der Schrodingergleichung
− ~2
2mR2
∂2ψ(ϕ)
∂ϕ2= Eψ(ϕ)
∂2ψ(ϕ)
∂ϕ2= −2mR2E
~2ψ(ϕ)
• Diese Losung ist formal die Gleiche wie fur das Teilchen im eindimen-sionalen Kasten.
Diesmal ist jedoch die unabhangige Variable keine kartesische Koordi-nate, sondern eine periodische Winkelvariable.
Die formale Losung lautet
ψ(ϕ) = a sin(mlϕ) + b cos(mlϕ)
= Aeimlϕ
a, b sind reell, A i.A. komplex, i ist die imaginare Einheit, ml ist eben-falls reell.
Slide 164 Quantisierungsbedingung fur ml
• Um die Schrodingergleichung zu erfullen, muss naturlich gelten
ml2 =
2mR2E
~2oder E =
~2
2mR2ml
2
• Da die Variable ϕ periodisch ist, muss man fordern
ψ(ϕ+ 2π) = ψ(ϕ) ,
oder
eiml(ϕ+2π) = eimlϕ · ei2mlπ = eimlϕ
um physikalisch eindeutige Losungen zu erhalten.
• Also ei2mlπ = 1 =⇒ ml ist eine ganze Zahl.
• Daraus ergibt sich als Quantisierungsbedingung fur ml
ml = 0,±1,±2, . . .
Slide 165 96

Teilchen auf dem RingZusammenfassung
Wellenfunktionen
ψml = Aeimlϕ
Energieeigenwerte
Emi =~2
2mR2ml
2
Quantenzahlen
mi = 0,±1,±2,±3 . . .
BemerkungObwohl die Differentialgleichung identisch mit der des Teilchens imKasten ist, sind die Randbedingungen unterschiedlich!Dies fuhrt dann auch zu unterschiedlichen erlaubten Quantenzahlen(ml = 0).
8.2 Der Drehimpuls
Slide 166 Drehimpuls (klassisch)
• Der Drehimpuls ~L ist ein Vektor.
Er ist das Vektorprodukt aus Orts- und Impulsvektor.
~L = ~r × ~p
~L =
y · pz − z · pyz · px − x · pzx · py − y · px
• Sind die Krafte auf ein Teilchen radialsymmetrisch, so ist der Drehim-
puls eine Konstante der Bewegung (Erhaltungsgroße).
• Der Drehimpuls ist mit der Winkelgeschwindigkeit ~ω und dem Tragheits-tensor I uber
~L = I~ω
verknupft.
Slide 167 97

Ubergang zur Quantenmechanik
• x→ x = x · px → px =~i
(∂
∂x
)
~L =
y · ~i∂∂z− z · ~
i∂∂y
z · ~i∂∂x− x · ~
i∂∂z
x · ~i∂∂y− y · ~
i∂∂x
=
~i
y ∂∂z− z ∂
∂y
z ∂∂x− x ∂
∂z
x ∂∂y− y ∂
∂x
Operator des Quadrats des Drehimpulses:
L2 = L2x + L2
y + L2z
Slide 168 Eigenschaften des Drehimpulsoperators:Kommutatoren der Komponenten
[Lx, Ly
]=
(LxLy − LyLx
)=
(~i
)2([y∂
∂z− z ∂
∂y
] [z∂
∂x− x ∂
∂z
])−
(~i
)2([z∂
∂x− x ∂
∂z
] [y∂
∂z− z ∂
∂y
])=
(~i
)2(y ∂∂zz ∂∂x− y ∂
∂zx ∂∂z− z ∂
∂yz ∂∂x
+ z ∂∂yx ∂∂z
)−
(~i
)2(z ∂∂xy ∂∂z− z ∂
∂xz ∂∂y− x ∂
∂zy ∂∂z
+ x ∂∂zz ∂∂y
)∂∂z
x = x ∂∂z
=(~i
)2(y ∂∂zz ∂∂x
+ z ∂∂yx ∂∂z− z ∂
∂xy ∂∂z− x ∂
∂zz ∂∂y
)
Slide 169 98

Eigenschaften des DrehimpulsoperatorsKommutatoren der Komponenten
[Lx, Ly
]=
(~i
)2(y ∂∂zz ∂∂x
+ z ∂∂yx ∂∂z− z ∂
∂xy ∂∂z− x ∂
∂zz ∂∂y
)Benutze ∂
∂zz = 1 + z ∂
∂z
=
(~i
)2(y∂
∂x+ yz
∂
∂z
∂
∂x+ zx
∂
∂y
∂
∂z
)−
(~i
)2(zy
∂
∂x
∂
∂z+ x
∂
∂y+xz
∂
∂z
∂
∂y
)=
~i· ~i
(y∂
∂x− x ∂
∂y
)= i~Lz
Slide 170 Kommutatoren der Drehimpulsoperatoren
• Kommutatoren[Lx, Ly
]= i~Lz
[Ly, Lz
]= i~Lx
[Lz, Lx
]= i~Ly
[Lx, L
2]
= 0[Ly, L
2]
= 0[Lz, L
2]
= 0
(Beweise in den Ubungen)
Konsequenzen
– Ein beliebiger Zustand des Systems kann immer nur einEigenzustand fur eine der 3 Drehimpulskomponenten sein.
– Die Zustande des Systems sind gleichzeitig Eigenzustandedes Absolutbetragsquadrates des Drehimpulses, L2.
– Also haben L2 und (beispielsweise) Lz gemeinsame Eigen-funktionen.
Slide 171 99

Der Operator des Quadrates des Drehimpulses
• Mit den Definitionen der kartesischen Komponenten des DrehimpulsesLx, Ly, Lz kann man den Operator L2 des Quadrates des Drehimpulsesausdrucken als
L2 = L2x + L2
y + L2z
• Eine etwas langwierige Rechnung zeigt, dass man L2 auch durch
L2 = −~2
[1
sinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)+
1
sin2 ϑ
(∂2
∂φ2
)]ausdrucken kann (s.o., ohne Beweis).
Slide 172 Die Drehimpulsoperatoren in KugelkoordinatenUnter Verwendung der Koordinatentransformationsgleichungen fur Ku-
gelkoordinaten lassen sich auch die Komponenten des Drehimpulsoperatorsin Kugelkoordinaten angeben:
Lx =~i
[− sinϕ
(∂
∂ϑ
)− cotϑ cosϕ
(∂
∂ϕ
)]Ly =
~i
[cosϕ
(∂
∂ϑ
)− cotϑ sinϕ
(∂
∂ϕ
)]Lz =
~i
(∂
∂ϕ
)
Anmerkung: Lz und das Teilchen auf dem Ring besitzen die gleichenEigenvektoren!
Slide 173 Eigenwertgleichung des Drehimpulses
• Die Schrodingergleichung des Teilchens auf der Kugel
Hψ(ϑ, ϕ;R) = − ~2
2mR2
[1
sinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)+
1
sin2 ϑ
(∂2
∂ϕ2
)]ψ(ϑ, ϕ;R)
= Erotψ(ϑ, ϕ;R)
100

laßt sich auch schreiben als
Hψ(ϑ, ϕ) =L2
2mR2ψ(ϑ, ϕ) = − ~2
2mR2Λ2ψ(ϑ, ϕ) = Erotψ(ϑ, ϕ)
da R nur parametrisch auftaucht (d.h. R ist ein konstanter Parameter,keine Variable).
• Letztendlich muss man also die Eigenwertgleichung fur das Absolut-quadrat des Drehimpulses, bzw. des Operators Λ2, losen:
Slide 174 Eigenwertgleichung fur Drehimpuls und Legendre-OperatorI
L2ψ = (Erot · 2mR2) ψ
bzw.
Λ2ψ(ϑ, ϕ) =[
1sinϑ
(∂∂ϑ
)sinϑ
(∂∂ϑ
)+ 1
sin2 ϑ
(∂2
∂ϕ2
)]ψ(ϑ, ϕ)
= −Erot · 2mR2
~2ψ(ϑ, ϕ) = kψ(ϑ, ϕ)
Diese Gleichung (mit einem noch zu bestimmenden Eigenwert k) lasstsich durch Faktorisierung gemaß ψ = Θ(ϑ) · Φ(ϕ) faktorisieren undlosen.
Slide 175 Eigenwertgleichung fur ΛLosungsweg
• Faktorisierung ψ = Θ · Φ
• Losung der Gleichung fur Φ (“Teilchen auf dem Ring”) liefert eineQuantenzahl ml (s.o.).
• Variablensubstitution z = cosϑ, Θ(ϑ)→ P (z) (Legendre-Funktionen)
101

• Faktorisierung von P (z) (analog zum harmonischen Oszillator)P (z) = (1− z2)|ml|/2G(z)
• Potenzreihenansatz fur G(z)
• Losung G(z) fuhrt wieder auf eine Rekursionsformel, die aus Normie-rungsgrunden abbrechen muss (→ Quantenzahl l)
k = l · (l + 1) mit l = 0, 1, 2, . . . und |ml| ≤ l
Slide 176 Losungen
• Λ2ψ = −l · (l + 1)ψ
• L2ψ = ~2l · (l + 1)ψ
• Lzψ = ~mlψ
• Anmerkung: ml = −l,−l + 1, . . . , l − 1, l ⇒ 〈Lz2〉 ist immer kleinerals 〈L2〉 (außer fur l = 0).
Dies bedeutet, dass der Drehimpulsvektor immer eine Komponente inder xy-Ebene hat (→ “Prazession” von Spins in der NMR-Spektroskopie).
8.3 Produktansatz der Schrodingergleichung in Kugel-koordinaten
Slide 177 Separation der Schrodingergleichung in Kugelkoordinaten
• Die Schrodingergleichung eines Teilchens in einem Zentralfeldpotentiallautet also
Hψ(r, ϑ, ϕ) =
[− ~2
2m
{(∂2
∂r2
)+
2
r
(∂
∂r
)}− ~2Λ2
2mr2+ V (r)
]ψ(r, ϑ, ϕ)
= Eψ(r, ϑ, ϕ)
102

• mit
Λ2 =1
sinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)+
1
sin2 ϑ
(∂2
∂φ2
)
Slide 178• Macht man den Ansatz ψ(r, ϑ, ϕ) = R(r) ·Θ(ϑ) ·Φ(ϕ) und multipliziert
mit2mr2
~2RΘΦ,
• so erhalt man
r2
R
[−{(
∂2
∂r2
)+
2
r
(∂
∂r
)}+
2mV (r)
~2
]R
− 1
ΘΦΛ2(ϑ, ϕ)ΘΦ =
2mr2E
~2
• oder
r2
R
[−{(
∂2
∂r2
)+
2
r
(∂
∂r
)}+
2m(V (r)− E)
~2
]R
=1
ΘΦΛ2(ϑ, ϕ)ΘΦ
Slide 179r2
R
[−{(
∂2
∂r2
)+
2
r
(∂
∂r
)}+
2m(V (r)− E)
~2
]R
=1
ΘΦΛ2(ϑ, ϕ)ΘΦ
Dies ist wieder ein Ausdruck, dessen linke Seite nur von r und dessenrechte Seite nur von ϑ und ϕ abhangt.
• Dies ist nur moglich, wenn beide Seiten gleich einer Konstanten sind,die wir k nennen.
• Zunachst betrachtet man die rechte Seite.
Slide 180 103

Faktorisierung der Drehimpulsgleichung
Λ2ΘΦ = kΘΦ[1
sinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)+
1
sin2 ϑ
(∂2
∂φ2
)]ΘΦ = kΘΦ
Multiplikation mitsinϑ2
ΘΦergibt
1
Θsinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)Θ +
1
Φ
(∂2
∂φ2
)Φ = k sin2 ϑ
oder1
Θsinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)Θ− k sin2 ϑ
+1
Φ
(∂2
∂φ2
)Φ = 0
Slide 181 1
Θsinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)Θ− k sin2 ϑ
+1
Φ
(∂2
∂φ2
)Φ = 0
• Wieder ist die Summe eines Ausdrucks, der nur von ϑ abhangt, undeines Ausdrucks, der nur von ϕ abhangt, eine Konstante, die wir −m2
l
nennen.
⇒ Beide Ausdrucke mussen jeweils konstant sein.
Slide 182 Die Φ-Gleichung
1
Φ
(∂2
∂ϕ2
)= −m2
l(∂2
∂ϕ2
)Φ = −m2
l Φ
104

• Diese Gleichung hat Losungen
Φml =1√2π
eimlφ
mit ml = 0,±1,±2, . . .
Slide 183 Die Θ-Gleichung
1
Θsinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)Θ− k sin2 ϑ
+1
Φ
(∂2
∂φ2
)Φ︸ ︷︷ ︸
=−ml2
= 0
[Multiplikation mit Θ, Division durch sin2 ϑ]
1
sinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)Θ− m2
l
sin2 ϑΘ = kΘ
1
sinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)Θ =
(k +
m2l
sin2 ϑ
)Θ
Slide 184 Losungen der Θ-Gleichung
1
sinϑ
(∂
∂ϑ
)sinϑ
(∂
∂ϑ
)Θ =
(k +
m2l
sin2 ϑ
)Θ
Die Losungen der DGL lauten
Θl ml(ϑ) =
[(2l + 1
2
)(l − |ml|)!(l + |ml|)!
]1/2
P|ml|l (cosϑ)
mit Eigenwerten
k = l(l + 1) l = 0, 1, 2, . . . und |ml| ≤ l
105

• P |ml|l heißen assoziierte Legendresche Polynome.
Slide 185 Kugelflachenfunktionen
• Das Produkt aus Θl ml(ϑ) und Φml(ϕ) heißt Kugelflachenfunktion Ylm(wobei das Subskript l in ml der Ubersichtlichkeit halber weggelassenwird).
englisch:“spherical harmonics”
Slide 186 Kugelflachenfunktionen
l ml Ylm(ϑ, ϕ)
0 0√
14π
(→ s-Funktion)
1 0√
34π
cosϑ (→ pz-Funktion)
1 ±1 ∓√
38π
sinϑe±iϕ (→ p-Funktionen)
2 0√
516π
(cos2 ϑ− 1)
2 ±1 ∓√
158π
cosϑ sinϑe±iϕ (d-Funktionen)
2 ±2√
1532π
sin2 ϑe±2iϕ
Slide 187 p-Orbitale
• Die Funktion Y10(ϑ, ϕ) laßt sich mit der Definition der Kugelkoordina-ten auch schreiben als
Y10 =
√3
4π
z
r
• Dies ist fur die Funktionen Y1±1 nicht moglich.
106

• Wir konnen aber Linearkombinationen bilden, z. B.
ψpx =1√2
(Y1−1 − Y11)
=1√2
√3
8π
(sinϑe−iϕ + sinϑe+iϕ
)=
1√2
√3
8πsinϑ · 2 cosϕ =
√3
4πsinϑ cosϕ =
√3
4π
x
r
Slide 188 py-Orbital
• Analog bilden wir
ψpy =i√2
(Y1−1 + Y11)
=i√2
√3
8π
(sinϑe−iϕ − sinϑe+iϕ
)=
1√2
√3
8πsinϑ · 2 sinϕ =
√3
4πsinϑ sinϕ =
√3
4π
y
r
• pz ist eine Eigenfunktion zu L2 und Lz.
• px und py sind nur Eigenfunktion zu L2, aber nicht zu Lz.
Slide 189 Radialgleichung
Die Schrodingergleichung lautete:
r2
R
[−{(
∂2
∂r2
)+
2
r
(∂
∂r
)}+
2m(V (r)− E)
~2
]R
=1
ΘΦΛ2(ϑ, ϕ)ΘΦ︸ ︷︷ ︸
= −l(l + 1)
107

Multiplikation mitR(r)
r2ergibt die Radialgleichung[
−{(
∂2
∂r2
)+
2
r
(∂
∂r
)}+
2m(V (r)− E)
~2
]R(r)
= − l(l + 1)
r2R(r)
Slide 190 RadialgleichungII
[−{(
∂2
∂r2
)+
2
r
(∂
∂r
)}+
2m(V (r)− E)
~2
]R(r) = −
l(l + 1)
r2R(r)
=⇒ [Umstellen]
−{(
∂2
∂r2
)+
2
r
(∂
∂r
)}R(r) +
[2mV (r)
~2+l(l + 1)
r2
]R(r) =
2mE
~2R(r)
• Wir machen uns zunutze, dass gilt
(rR)′′ = (R+ rR′)′ = (R′ +R′ + rR′′) = 2R′ + rR′′
=1
r(r2R′)′
[R′ :=
(∂R
∂r
)](Beweis in Ubungen)
so dass der Term mit der geschweiften Klammer durch1
r· (rR)′′ ersetzt
werden kann.
Slide 191 RadialgleichungIII
Man definiert dann u := r ·R(r) und erhalt aus
−{(
∂2
∂r2
)+
2
r
(∂
∂r
)}R(r) +[
2mV (r)
~2+l(l + 1)
r2
]R(r) =
2mE
~2R(r)
zunachst
−1
r
(∂2
∂r2
)u(r) +
[2mV (r)
~2+l(l + 1)
r2
]R(r) =
2mE
~2R(r)
und schließlich nach Multiplikation mit r
108

−(∂2
∂r2
)u(r) +
[2mV (r)
~2+l(l + 1)
r2
]u(r) =
2mE
~2u(r)
• Bemerkung: Die Drehimpulsquantenzahl l taucht hier als Parameter derDifferentialgleichung auf. Die Energie hangt im Allgemeinen auch von l ab(vgl. jedoch die Energiewerte des H-Atoms (s.u.)).
109

9 Das Wasserstoffatom
Slide 192 Radiale Schrodingergleichung
• Ersetzt man V (r) = − Ze2
4πε0rin
−(∂2
∂r2
)u(r) +
[2mV (r)
~2+l(l + 1)
r2
]u(r) =
2mE
~2u(r)
so erhalt man −(∂2
∂r2
)u(r) +
[− 2mZe2
4πε0r~2+l(l + 1)
r2
]u(r) =
2mE
~2u(r)
• Fur das H-Atom ist naturlich Z = 1. Die Gleichung gilt jedoch fur alle1-Elektronen-Ionen (z.B. He+, Li++).
• Anmerkung: Dies ist eine eindimensionale Schrodingergleichung mit demeffektiven Potential
Veff = − Ze2
4πε0r+l(l + 1)~2
2mr2
• Der Drehimpulsterm spielt dabei die Rolle eines Zentrifugal”potentials”.
Slide 193 Reduzierte Variable
• Zur Eliminierung der physikalischen Konstanten fuhrt man ein
a0 =4πε0~2
me2=
ε0h2
πme2
Bohrscher Atomradius (Dimension: Lange)
sowie
R∞ =me4
64π3ε20~3c=
me4
8ε20h3c
Rydberg-Konstante (Dimension: cm−1)
• a0 = 0.52918 A = 52.918 pm
• R∞ = 109677.6 cm−1
Slide 194 110

Substitution der radialen SGL
• Man definiert ρ =r
a0und ε = E
hcR∞und erhalt aus
−{(
∂2
∂r2
)+ 2
r
(∂∂r
)}R(r) +
[− 2mZe2
4πε0~2r + l(l+1)r2
]R(r) =
2mE~2 R(r)
− 1r2
(∂∂r
)r2(∂∂r
)R(r) +
[− 2mZe2
4πε0~2r + l(l+1)r2
]R(r) =
2mE~2 R(r)
nach Einsetzen
− 1ρ2
(∂∂ρ
)ρ2(∂∂ρ
)R(ρ) +
[−2Z
ρ + l(l+1)ρ2
]R(ρ) = εR(ρ)
Slide 195 Asymptotische BetrachtungI
• Fur ρ→∞ kann man die Terme2Z
ρund
l(l + 1)
ρ2vernachlassigen.
• Fur ρ → 0 wiederum dominiert der Drehimpulsterml(l + 1)
ρ2(wenn l > 0).
Der Term divergiert nach +∞.
• Wenn l = 0 ist, dann dominiert der Coulombterm2Z
ρund divergiert nach
−∞.
• Wir vermuten weiterhin (richtigerweise), dass ε < 0.
Dies ist plausibel, da V (r) < 0 fur alle r!
Slide 196 Asymptotische BetrachtungII
• Wir betrachten nur die sogenannten gebundenen Zustande (d.h. die normier-baren Losungen der SGL).
• Als asymptotische Losung ergibt sichRas(ρ) = ρl exp(−
√−ερ).
Slide 197 111

Transformierte Schrodingergleichung und Potenzreihenansatz
• Mit dem Ansatz R(ρ) = f(ρ) ·Ras(ρ) = f(ρ) · ρl exp(−√−ερ)
ergibt sich die Differentialgleichung fur f(ρ) als
d2f(ρ)dρ2
= 2(l+1ρ −
√−ε)
df(ρ)dρ + 2
ρ
(Z −√−ε(l + 1)
)f(ρ) = 0 ,
die man wieder mit einem Potenzreihenansatz
f(ρ) =∞∑k=0
Akρk
lost.
Slide 198 Rekursionsformel
• Man erhalt (ohne Beweis) wieder eine Rekursionsformel fur die Koeffizienten
Ak =
(2(l + k)
√−ε− 2Z
k(k − 1) + Z(l + 1)kAk−1
)• Wieder ware die Funktionen R(r) nicht normierbar, wenn nicht die Potenz-
reihe abbrechen wurde, d.h. fur irgendein k gilt:
Ak = 0
• Daraus ergibt sich Ak = 2(l + k)√−ε− 2Z = 0 bzw.
√−ε =
Z
l + kl = 0, 1, 2, . . . , k = 1, 2, . . .
Slide 199 Losung der Schrodingergleichung des H-Atoms
εn = −Z2
n2n = l + 1, l + 2, l + 3, . . .
oder (da l ≥ 0)
En = −hcR∞εn = −hcR∞Z2
n2n = 1, 2, 3, . . .
• Die zugehorigen Wellenfunktionen lauten
ψnlm(r, ϑ, ϕ) = Rnl(r)Ylm(ϑ, ϕ)
n = 1, 2, 3, . . . l = 0, 1, 2, . . . m = −l,−l + 1, . . . , 0, 1, . . . , l − 1, l
Slide 200 112

Radiale Wellenfunktion
Rnl(r) = −
√4Z3
n4a30
(n− l − 1)!
[(n+ l)!]3
(2Zr
na0
)le− Zrna0L2l+1
n+l
(2Zr
na0
)
• Die Lqs(x) sind die sogenannten assoziierten Laguerre-Polynome (die durch
die angegebene Rekursionsformel fur die Ak und eine spezielle Wahl von A0
gegeben sind).
• Die Wellenfunktionen des H-Atoms nennt man Orbitale.
• (“orbit” → “orbital”)
Slide 201 Radialfunktionen des H-Atoms
n l Orbital Rnl(ρ) ·(a0Z
)3/21 0 1s 2 · e−ρ/2
2 0 2s√
1/8 (2− ρ)e−ρ/2
2 1 2p√
1/24 ρe−ρ/2
3 0 3s√
1/243 (6− 6ρ+ ρ2)e−ρ/2
3 1 3p√
1/486 (4− ρ)ρe−ρ/2
3 2 3d√
1/2430 ρ2e−ρ/2
• Anmerkung: ρ = (2Z/na0) · r ist abhangig von n!! =⇒Fur großere n fallt die Wellenfunktion langsamer ab, die Orbitale sind also“großer”!
Slide 202
113
Energiediagramm
2 4 6 8 10 12 14 ρ/a0
n=1
n=2n=3
n=4
Kontinuumungebundene Zustaendenicht auf 1 normierbareLoesungen
(100) 1s
(200) 2s (211) (21-1) (210) 2pz
Slide 203 Termschema des H-Atoms
• Zustande En ∝ 1n2
• E2s − E1s = −hcR∞(
14 − 1
)= 0.75hcR∞
• E3s − E1s = −hcR∞(
19 − 1
)= 0.88hcR∞
• E4s − E1s = −hcR∞(
116 − 1
)= 0.9375hcR∞
• E5s − E1s = −hcR∞(
125 − 1
)= 0.96hcR∞
• . . . Lyman-Serie
• E3s − E2s = −hcR∞(
19 −
14
)= 0.138hcR∞
• E4s − E2s = −hcR∞(
116 −
14
)= 0.1875hcR∞
• E5s − E2s = −hcR∞(
125 −
14
)= 0.21hcR∞
• E6s − E2s = −hcR∞(
136 −
14
)= 0.222hcR∞
• . . . Balmer-Serie
Slide 204 114

Skizzen der radialen Wellenfunktion Rnl(r)
Anmerkungen:
• Die y-Achse besitzt unterschiedliche Skalierung!
• Beachten Sie die x-Achsenskalierung aufgrund der Definition von ρ = (2Z/na0)·r.
Slide 205 OrbitaleWinkelanteil
• Ylm(cosϑ, ϕ) sind Kugelflachenfunktionen, also Funktionen, die auf der Ober-flache der Einheitskugel definiert sind (Definitionsbereich 0 ≤ ϑ < π und0 ≤ ϕ < 2π).
• eine ubliche Datstellung dieser Funktionen ist, dass der Funktionswert ent-lang der Richtung, die durch (ϑ, ϕ) auf der Einheitskugel gegeben ist, auf-getragen wird. Die durch diese Punkte definierte Oberflache charakterisiertdas Orbital.
Slide 206 s-Orbitale
• s-Orbitale sind kugelsymmetrisch.
• Die Funktion Y00 ist eine Konstante auf der Kugeloberflache.
• Der Graph dieser Funktion ist demzufolge eine Kugel.
115

Slide 207 p-Orbitale
• p-Orbitale (∝ cos(ϑ) oder ∝ sin(ϑ)) sind positiv in einem Halbraum, negativim anderen Halbraum.
• Die Funktion Y10 besitzt demzufolge eine Knotenebene.
• Die Funktion Y10 = pz besitzt zwei Bereiche unterschiedlichen Vorzeichens
pz py px
• px und py-Orbitale sehen gleich aus, sind nur um 90◦ in die x- bzw. y-Richtung gedreht
Slide 208 d-Orbitale
• d-Orbitale sind gemischte Polynome 2. Grades in cosϑ und sinϑ.
• Die Funktion Y2m besitzt demzufolge zwei Knotenebenen.
• Die Funktion Y20 = dz2 besitzt 3 Bereiche unterschiedlichen Vorzeichens.
116

+
–
+
dxy, dxz, dyz, und dx2−y2 Orbitalesind Linearkombinationen aus denY2m,m = −2,−1, 1, 2.Sie sehen gleich aus, besitzen jedochunterschiedliche Vorzugsrichtungenim Raum.
Slide 209 d-Orbitale
• dz2 = Y20 ∝ (3 cos2 ϑ− 1)Zylindersymmetrie um die z-Achse positive Bereiche entlang der positivenund negativen z-Achse negativer Bereich in einem Ring in der xy-Ebene
• dxy =1√2
(Y22 − Y2−2) ∝ sin2 ϑ sinϕ cosϕ liegt in der xy-Ebene positive
Bereiche entlang der Hauptdiagonale x = y (45◦ and 225◦) negative Bereicheentlang der Nebendiagonale x = −y (135◦ and 315◦)
• dx2−y2 =1√2
(Y22 + Y2−2) ∝ sin2 ϑ(cos2 ϕ− sin2 ϕ) liegt in der xy-Ebene po-
sitive Bereiche entlang der x-Achse (0◦ and 180◦) negative Bereiche entlangder y-Achse (90◦ and 270◦)
Slide 210 d-Orbitale
• dxz =1√2
(Y21 − Y2−1) ∝ sinϑ cosϑ cosϕ liegt in der xz-Ebene positive
Bereiche entlang der Hauptdiagonale x = z (45◦ and 225◦) negative Bereicheentlang der Nebendiagonale x = −z (135◦ and 315◦)
• dyz =1√2
(Y21 + Y2−1) ∝ sinϑ cosϑ cosϕ liegt in der yz-Ebene positive
Bereiche entlang der Hauptdiagonale y = z (45◦ and 225◦) negative Bereicheentlang der Nebendiagonale y = −z (135◦ and 315◦)
117

9.1 Radiale Dichteverteilung
Slide 211 Matrixelemente
Durch die Koordinatentransformation (x, y, z) → (r, ϑ, ϕ) andert sich nichtnur die Form des Laplace-Operators ∆, sondern auch die Integrationsgrenzenund das differentielle Volumenelement.
∞∫−∞
dx
∞∫−∞
dy
∞∫−∞
dz →∞∫
0
r2dr
π∫0
sinϑdϑ
2π∫0
dϕ
also dx dy dz → r2dr sinϑdϑ dϕ = r2dr dcosϑ dϕ
Slide 212 Radiale VerteilungsfunktionI
• Die Wellenfunktion ψnlm = Rnl(r)Ylm(ϑ, ϕ) ist naturlich in jedem Punkt(r, ϑ, ϕ) und damit auch fur jeden Punkt (x, y, z) bekannt.
• Es ist aber haufig wichtig, die Wahrscheinlichkeit zu kennen, das Teilchenin einer Kugelschale um den Ursprung, also in einer Schale zwischen r undr + dr, unabhangig von der Orientierung zu finden.
• Diese Wahrscheinlichkeit P (r)dr ist gegeben durch
P (r)dr =
∫Ober-flache
|ψnlm|2dτ =
π∫0
2π∫0
|Rnl|2|Ylm|2r2 sinϑdr dϑ dϕ
• Da die Ylm auf 1 normiert sind, gilt
π∫0
2π∫0
|Ylm|2 sinϑdϑ dϕ = 1
Slide 213
118

Radiale VerteilungsfunktionII
• Daher gilt
P (r)dr =
π∫0
2π∫0
|Rnl|2|Ylm|2r2 sinϑdr dϑ dϕ = |Rnl(r)|2r2dr
• P (r) = |Rnl(r)|2r2 heißt radiale Verteilungsfunktion.
• Multipliziert man P (r) mit dr, so erhalt man die Wahrscheinlichkeit, dasElektron in einer Kugelschale zwischen r und r + dr zu finden.
• Anmerkung: Die radiale Verteilungsfunktion ist in allen Orbitalen am Ur-sprung = 0, d.h., die Wahrscheinlichkeit, das Elektron am Ursprung zu fin-den, ist verschwindend gering.
Slide 214 Skizzen der radialen Verteilungsfunktion
Anmerkungen:
• Die radiale Verteilungsfunktion ist fur r = 0 immer Null!
• Die Funktion hat n− l − 1 Knoten zwischen r = 0 und r =∞.
9.2 Entartung
Slide 215
119

Quantenzahlen
• Drei Quantenzahlen bestimmen die Eigenzustande des H-Atoms.
• Die Hauptquantenzahl n bestimmt die Energieeigenwerte (also die Eigenwer-te des Hamiltonoperators H.
En = −hcR∞Z2
n2
• Die Nebenquantenzahl l bestimmt die Eigenwerte des DrehimpulsoperatorsL2.
• Die magnetische Quantenzahl m bestimmt die Eigenwerte der z-Komponentedes Drehimpulses Lz.
• Die drei Operatoren L2, Lz, H kommutieren paarweise und besitzen gemein-same Eigenfunktionen, namlich die Wassserstofforbitale ψnlm(r, ϑ, ϕ).
Slide 216 Entartung
• Zu jeder Hauptquantenzahl n gibt es n verschiedene Drehimpulsquanten-zahlen l = 0, . . . n− 1.
• Zu jeder Drehimpulsquantenzahl l gibt es 2l + 1 verschiedene magnetischeQuantenzahlen m, namlich −l,−l + 1, . . . , l − 1, l.
• Die Gesamtzahl der Zustande zu gegebenem n ist also
g(n) =n−1∑l=0
(2l + 1) = 2n−1∑l=0
l +n−1∑l=0
1
= 2n(n− 1)
2+ n = n(n− 1) + n = n2
• Das H-Atom besitzt n2 entartete Energiezustande.
• Die Entartung bei verschiedenen l-Zustanden bei gegebenem n im H-Atomist “zufallig” (sie ist Ausdruck einer hoheren Symmetrie des Coulombpoten-tials).
120

Teil IV
Mehrelektronenprobleme
Inhaltsangabe
10 Mehrelektronenprobleme ohne e-e-Wechselwirkung 122
10.1 Allgemeine Losungen . . . . . . . . . . . . . . . . . . . . . 122
10.2 Variationsprinzip . . . . . . . . . . . . . . . . . . . . . . . 126
10.3 Grundzustand des He-Atoms . . . . . . . . . . . . . . . . 128
11 Mehrelektronenatome 129
11.1 Grundzustand des He-Atoms . . . . . . . . . . . . . . . . 129
11.2 Grundzustand des Li-Atoms . . . . . . . . . . . . . . . . . 130
11.3 Der Spin . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131
11.4 Das Pauli-Prinzip . . . . . . . . . . . . . . . . . . . . . . . 134
11.5 Die Eigenschaften von Atomen . . . . . . . . . . . . . . . 136
11.6 Drehimpulskopplung . . . . . . . . . . . . . . . . . . . . . 140
11.7 Spin-Bahn-Kopplung und Hundsche Regeln . . . . . . . . 143
11.8 Zusammenfassung . . . . . . . . . . . . . . . . . . . . . . 147
12 Molekule 149
12.1 Die Born-Oppenheimer-Naherung . . . . . . . . . . . . . . 149
12.2 Die Linear Combination of Atomic Orbital-Methode (LCAO)152
12.3 Die Wellenfunktionen des H+2 -Molekulions . . . . . . . . . 157
12.4 Das Wasserstoffmolekul . . . . . . . . . . . . . . . . . . . 159
12.5 MO-Diagramme zweiatomiger Molekule . . . . . . . . . . 159
121

10 Mehrelektronenprobleme ohne e-e-Wechselwirkung
10.1 Allgemeine Losungen
Slide 217 Hamiltonoperator fur N Elektronen
• allgemeine Form:
H = −N∑i=1
~2
2m∆i +
N∑i=1
Vext(~ri) +
N∑i=1
N∑j=i+1︸ ︷︷ ︸
′′j>i′′
e2
4πε0rij
• Vext(~ri) ist die potentielle Energie des i. Elektrons, also z. B. die Coulomb-Energie dieses Elektrons im Feld von n Kernen k.
Vext(~ri) = −n∑k=1
Zke2
4πε0rik
Slide 218 Vernachlassigung der interelektronischen Wechselwirkung
• Eine (schlechte) Naherung ist es, den Term
N∑i<j
e2
4πε0rij
zu vernachlassigen.
• Dann kann man offensichtlich
H0 = −N∑i=1
~2
2m∆i +
N∑i=1
Vext(~ri) =N∑i=1
h(i)
schreiben.
H0 ist also eine Summe aus Einelektronenoperatoren h(i) mit
h(i) = − ~2
2m∆i + Vext(~ri)
Slide 219122

Einelektronenhamiltonoperatoren h(i)
• Jeder dieser Einelektronenoperatoren sieht (bis auf den Laufindex i) wie derandere aus.
• Er konnte z. B. die Form des Hamiltonoperators fur
– das Teilchen im Kasten
– den harmonischer Oszillator
– oder ein Atom mit Kernladungszahl Z (H-”ahnliches” Atom)
haben.
Slide 220 Wellenfunktionen des Mehrteilchenproblems
• Nehmen wir an, wir haben das Einteilchenproblem gelost,
z. B. fur N Elektronen im Kasten
h(i)ψni(~ri) =
[− ~2
2m∆i + Vext(~ri)
]ψni(~ri) = eniψni(~ri)
ni = 1, 2, . . .∞
oder fur N Elektronen im Zentralpotential eines Atomkerns mit Kernla-dungszahl Z
h(i)ψnilimi(~ri) =
[− ~2
2m∆i + Vext(~ri)
]ψnilimi(~ri)
= eniliψnilimi(~ri)
Slide 221 Wellenfunktionen des Mehrteilchenproblems
• Dann sind mogliche Losungen des Mehrteilchenproblems
H0Ψ(~r1, ~r2, . . . , ~rN ) = EΨ(~r1, ~r2, . . . , ~rN )
gegeben durch
123

Ψ(~r1, . . . , ~rN ) = ψn1l1m1(~r1) · ψn2l2m2(~r2) · . . . · ψnN lNmN (~rN )
=N∏i=1
ψnilimi(~ri)
mit
E = en1l1 + en2l2 + . . .+ enN lN
=N∑i=0
enili
Slide 222 Produktwellenfunktion
• Die Faktorisierung der Wellenfunktion ist immer dann moglich, wenn derHamiltonoperator eine Summe aus Termen ist.
• Dann gilt auch die Additivitat der Energien.
• Man nennt die Wellenfunktion auch eine Produktfunktion (manchmal auchdas Hartree-Produkt, s.u.).
Slide 223 Beispiel: He-Atom ohne Elektron-Elektron-Wechselwirkung
H0 = h(1) + h(2)
h(i) = − ~2
2m∆i −
2e2
4πε0ri.
• Schauen wir uns unterschiedliche Elektronenkonfigurationen an:
• 1s2 beide Elektronen im 1s-Orbital.
• 1s 2s Elektronen 1 im 1s-OrbitalElektron 2 im 2s-Orbital.
Slide 224
124

Energie der Elektronenkonfiguration 1s2
H0Ψ1s2(~r1, ~r2) = (h(1) + h(2))ψ1s(~r1) · ψ1s(~r2)
= (h(1)ψ1s(~r1))ψ1s(~r2) + ψ1s(~r1)(h(2)ψ1s(~r2))
= (e1sψ1s(~r1))ψ1s(~r2) + ψ1s(~r1)(e1sψ1s(~r2))
= (e1s + e1s)︸ ︷︷ ︸E1s2
ψ1s(~r1)ψ1s(~r2)
= E1s2Ψ1s2(~r1, ~r2)
Slide 225 Energie der Elektronenkonfiguration 1s 2s
H0Ψ1s 2s(~r1, ~r2) = (h(1) + h(2))ψ1s(~r1) · ψ2s(~r2)
= (h(1)ψ1s(~r1))ψ2s(~r2) + ψ1s(~r1)(h(2)ψ2s(~r2))
= (e1sψ1s(~r1))ψ2s(~r2) + ψ1s(~r1)(e2sψ2s(~r2))
= (e1s + e2s)︸ ︷︷ ︸E1s 2s
ψ1s(~r1)ψ2s(~r2)
= E1s 2sΨ1s 2s(~r1, ~r2)
ahnlich ist (Elektron 2 im 1s-Zustand, 1 im 2s-Zustand)
H0Ψ2s 1s(~r1, ~r2) = (h(1) + h(2))ψ2s(~r1) · ψ1s(~r2)
= (e2s + e1s)︸ ︷︷ ︸E2s 1s
ψ2s(~r1)ψ1s(~r2)
= E2s 1sΨ2s 1s(~r1, ~r2)
Slide 226 Entartung
• Offensichtlich gilt E1s 2s = E2s 1s, da die Anregung vom 1. Elektron von 1sin 2s oder vom 2. Elektron aquivalent ist.
125

• Die Elektronen verhalten sich physikalisch gleich.
• Es liegt also im 1s 2s-Zustand eine Entartung vor.
• Die allgemeine mathematische Losung fur die Elektronenkonfiguration 1s 2s(fur ein beliebiges Elektron im 1s-Zustand und das andere Elektron im 2s-Zustand) lautet also
H0 [aΨ1s 2s(~r1, ~r2) + bΨ2s 1s(~r1, ~r2)]
= E1s 2s [aΨ1s 2s(~r1, ~r2) + bΨ2s 1s(~r1, ~r2)]
mit beliebigem a und b.
• Sinngemaß gilt Ahnliches fur andere Konfigurationen des Pseudo-He-Atomssowie fur Konfigurationen von Pseudo-Atomen mit mehr als zwei Elektronen.
10.2 Variationsprinzip
Slide 227 Variationsprinzip
Von allen Wellenfunktionen beschreibtdiejenige den Grundzustand eines Sy-stems (am Besten), die den niedrigstenErwartungswert der Energie liefert.
Slide 228 Beweis des VariationsprinzipsI
• Der Beweis beruht auf der Tatsache, dass jede Funktion mit gleichem Defi-nitionsbereich nach den Eigenfunktionen eines hermiteschen Operators ent-wickelt werden kann. Man benutzt die Eigenfunktionen des Hamiltonoperatorund zeigt so leicht, dass jede Funktion einen hoheren Energieerwartungswertbesitzt als der wahre Grundzustand.
• Sei f({x}) eine Funktion auf dem Definitionsbereich {x}.
• Der Definitionsbereich {x} kann ein- oder mehrdimensional sein.
126

• Kenne ich die Eigenfunktionen ψi({x}), i = 1, 2, . . . eines Hermiteschen Ope-rators, die auf dem gleichen Definitionsbereich definiert sind, so kann ich fschreiben als
f({x}) = c0ψ0 + c1ψ1 + c2ψ2 + . . . =
∞∑i=0
ciψi({x})
Slide 229 Beweis des VariationsprinzipsII
• Wir wahlen als Hermiteschen Operator den Hamiltonoperator H des Sy-stems.
• Wir ordnen die Wellenfunktionen in der ublichen Weise, so dass ψ0 dieGrundzustandsenergie von H ist.
• Der Erwartungswert der Energie im Zustand f lautet
〈H〉f = 〈f |H|f〉 · 〈f |f〉−1
〈f |f〉 · 〈H〉f = 〈∞∑i=0
ciψi|H|∞∑j=0
cjψj〉
=∞∑i=0
∞∑j=0
ci∗cj〈ψi|H|ψj〉 =
∞∑i=0
∞∑j=0
ci∗cj〈ψi|Ej |ψj〉
=∞∑i=0
∞∑j=0
ci∗cjEj〈ψi|ψj〉 =
∞∑i=0
∞∑j=0
ci∗cjEjδij
=∞∑i=0
|ci|2Ei ≥ E0
∞∑i=0
|ci|2 = E0〈f |f〉 q.e.d
Slide 230 Konsequenzen aus dem Variationsprinzip
• 〈H〉f ≥ E0
• Selbst wenn wir den GZ nicht kennen, konnen wir eine echte obere Grenzefur den Energiewert des GZ angeben, indem wir den Energieerwartungswertfur eine wie auch immer ausgewahlte Funktion berechnen.
127

• Je besser wir diese Funktion wahlen, desto naher kommen wir (von oben)an die wahre Grundzustandsenergie heran.
• Man kann auf diesem Prinzip Algorithmen entwickeln, um moglichst guteNaherungen zu finden
=⇒ Basis der Quantenchemie
• Berechnet man mit einem Variationsverfahren einen Energieerwartungswert,der niedriger als die wahre Grundzustandsenergie liegt, so liegt entwederein Rechenfehler vor oder die theoretische Beschreibung berucksichtigt einewesentliche Eigenschaft des Systems nicht oder unzureichend!
10.3 Grundzustand des He-Atoms
Slide 231 He-Energie ohne e-e-WW
• Der experimentelle Wert der Grundzustandsenergie des He-Atoms liegt bei−79 eV = −2.9065 Hartree.
• 79 eV an Energie mussen aufgebracht werden, um das He-Atom in ein He++
(ein α-Teilchen) und 2 Elektronen in unendlichem Abstand voneinander zudissoziieren.
• E1s2 = 2 ·(−hcR∞Z2
)= −8EH = −108.8eV
• Die berechnete Energie ist niedriger als die experimentelle Energie
⇒ Widerspruch zum Variationsprinzip
• Der Grund fur den Widerspruch ist naurlich leicht gefunden:Vernachlassigung der Elektron-Elektron-Wechselwirkung!
128

11 Mehrelektronenatome
11.1 Grundzustand des He-Atoms
Slide 232 Bessere Naherung: Verwendung von Ψ1s2 als Grundzustandswellen-funktion
• Die exakte Wellenfunktion des He-Grundzustandes ist nicht bekannt.
• Nehmen wir an, dass die oben berechnete Wellenfunktion Ψ1s2 den Grund-zustand hinreichend gut beschreibt,
(dass also die Wellenfunktion durch die Elektron-Elektron-WW nicht starkbeeinflusst wird).
• Dies ist wieder eine Naherung, die zumindest besser als die totale Ver-nachlassigung der e-e-Wechselwirkung sein sollte.
• Dazu muss man den Erwartungswert 〈Ψ1s2 |H|Ψ1s2〉 berechnen.
Slide 233 Energieerwartungswert im Grundzustand Ψ1s2
〈H〉 = 〈h(1) + h(2) +e2
4πε0r12〉
= 〈ψ1s(1)|〈ψ1s(2)|h(1)|ψ1s(1)〉|ψ1s(2)〉+ 〈ψ1s(1)|〈ψ1s(2)|h(2)|ψ1s(1)〉|ψ1s(2)〉
+ 〈ψ1s(1)|〈ψ1s(2)| e2
4πε0r12|ψ1s(1)〉|ψ1s(2)〉
= e1s + e1s + 〈ψ1s(1)|〈ψ1s(2)| e2
4πε0r12|ψ1s(1)〉|ψ1s(2)〉
= −108.8 + 〈ψ1s(1)|〈ψ1s(2)| e2
4πε0r12|ψ1s(1)〉|ψ1s(2)〉
= −108.8eV + 34eV = −74.8eV > E0 = −79eV
=⇒ Kein Widerspruch zum Variationstheorem!
• (Das hier skizzierte Verfahren ist ein Beispiel fur die Anwendung der Storungs-theorie (1. Ordnung)).
Slide 234 129

Coulombintegral J1s 1s
• Das Integral
J1s 1s = 〈Ψ1s2 |e2
4πε0r12|Ψ1s2〉
=
∫ ∫ ∫dx1dy1dz1
∫ ∫ ∫dx2dy2dz2
e2
4πε0r12|Ψ|2
=: (1s 1s|1s 1s)
heißt Coulombintegral
• Coulombintegrale sind zentrale Großen, die in der ab initio Quantenchemieberechnet werden mussen (milliardenfach!).
• Fazit: Physikalisch ist (scheint?) alles in Ordnung!
11.2 Grundzustand des Li-Atoms
Slide 235 Li-Energie im (hypothetischen) Zustand Ψ1s3
• Experimentelle Energie des Li-Atoms: −203.4 eV
• Wir nehmen an, dass die Wellenfunktion des Li-Atoms durch
Ψ1s3 = ψ1s(1) · ψ1s(2) · ψ1s(3)
beschrieben werden kann,
dass sich also alle Elektronen im niedrigsten Orbital befinden.
• Wir berechnen wieder den Energieerwartungswert
〈H〉 = 〈h(1) + h(2) + h(3) +e2
4πε0
(1
r12+
1
r13+
1
r23
)〉
fur das Li-Atom ist Z=3
= 3 ∗ (−9 ∗ 13.6)eV + 3 ∗ 51eV
= −214.2eV < E0 = −203.4eV
Slide 236 130

Fazit
• Eine schlechte Wellenfunktion liefert eine Energie, die niedriger als die ex-perimentelle Energie ist.
=⇒ Widerspruch zum Variationstheorem!
• Es liegt der Verdacht nahe, dass in der Beschreibung der elektronischenEigenschaften etwas fehlt!
Elektronenspin
11.3 Der Spin
Slide 237 Der Stern-Gerlach-Versuch
• Beobachtung: In einem inhomogenen Magnetfeld spaltet ein Atomstrahl ausAg-Atomen ([Kr]4d105s1(2S1/2)) in zwei Teilstrahlen auf.
• die 5s-Elektronen des Ag-Atoms sollten keinen Drehimpuls, also auch keinmagnetisches Moment besitzen!
• Interpretation: Elektronen besitzen aufgrund eines inneren Drehimpulsesein magnetisches Moment.
• Man nennt den inneren Drehimpuls Elektronenspin.
• Der Spin ist durch zwei mogliche Einstellungen bzgl. einer raumfesten Achsecharakterisiert.
spin-up = ↑ = α-Spinspin-down = ↓ = β-Spin
• Die Spin-Einstellung stellt einen zuatzlichen Freiheitsgrad fur ein Elektrondar, die man als “zusatzliche Koordinate” σ (mit nur 2 moglichen Werten)auffassen kann.
Slide 238
131

Koordinaten und Spinorbitale
• Die Spinkoordinate σ, die nur zwei Werte (α und β) annehmen kann, musszu den Ortskoordinaten hinzugefugt werden.
~x = {~r, σ} =
{{~r, α}{~r, β}
• Orbitale (=Einelektronenwellenfunktionen) hangen nicht nur von ~r, sondernvon ~x ab
χ(~x) = χ(~r, σ) =
{ψ(~r)|α〉ψ(~r)|β〉
• Die χ(~x) heißen Spinorbitale, wahrend man die bisher betrachteten ψ(~r) reinraumliche Orbitale oder Raumorbitale nennt.
Slide 239 Spinquantenzahlen
• Der Spin verhalt sich “kinematisch” wie ein Bahndrehimpuls.
⇒ Er wird durch zwei Quantenzahlen bestimmt.
• Die Quantenzahl s charakterisiert den Betrag des Spins.
• Die Quantenzahl ms charakterisiert die Spineinstellung bzgl. einer raumli-chen Achse (z.B. der z-Achse).
Slide 240 Eigenwerte
• Die Wirkung der Operatoren auf die Dirac’schen “kets” laßt sich in folgenderWeise beschreiben:
s2|α〉 = ~2s(s+ 1)|α〉 =3
4~2|α〉 → s =
1
2
s2|β〉 = ~2s(s+ 1)|β〉 =3
4~2|β〉
sz|α〉 = ~ms|α〉 =1
2~|α〉 → ms = +
1
2
sz|β〉 = ~ms|β〉 = −1
2~|β〉 → ms = −1
2
132

• Anmerkung: Das Wirkung der Spinoperatoren auf die Eigenfunktionen istanalog zu der der Bahndrehimpulsoperatoren, jedoch mit halbzahligen Quan-tenzahlen!
Slide 241 SpinorbitaleI
• Spinorbitale sind Produkte aus Raumorbitalen und einer “Spinfunktion”,die nicht explizit definiert ist, fur die aber die Wirkung von Operatoren unddie Werte von Matrixelementen bekannt sind.
• Vernachlassigt man die Spin-Bahn-Kopplung (s.u.), dann sieht der Hamil-tonoperator mit Spin genauso aus wie der bisher ohne Spin betrachtete Ha-miltonoperator fur Elektronen.
• Spinorbitale fur das H-Atom:
χnlm(~x) = χnlm(~r, σ) =
{ψlmn(~r) |α〉ψlmn(~r) |β〉
Slide 242 SpinorbitaleII
• Wenn der Hamiltonoperator nicht vom Spin abhangt sind α- und β-Spinorbitaleentartet:
Hψlmn(~r)|α〉 = Enlψlmn(~r)|α〉Hψlmn(~r)|β〉 = Enlψlmn(~r)|β〉
• Fur Spinorbitale gilt
s2χ(~x) = s2ψ(~r)|α〉 = ψ(~r)s2|α〉= ψ(~r)~2s(s+ 1)|α〉 = ~2s(s+ 1)ψ(~r)|α〉= ~2s(s+ 1)χ(~r)
szχ(~r) = ~msχ(~x)analog
s2 und sz “wirken nicht” auf den raumlichen Anteil des Spinorbitals.
133

11.4 Das Pauli-Prinzip
Slide 243 Ununterscheidbare Teilchen
• Elemenarteilchen (also auch Elektronen, Protonen, etc.) sind untereinanderununterscheidbar!
• Das bedeutet
1. Der Hamiltonoperator muss symmetrisch in den Teilchenkoordinatensein. (Kein Elektron darf “ausgezeichnet” sein.)
2. Die Aufenthaltswahrscheinlichkeitsdichte |Ψ|2 der Vielteilchenwellen-funktion muss invariant (unveranderlich) sein bzgl. Vertauschung vonTeilchenkoordinaten.
=⇒ Ψ(~x1, ~x2, . . . , ~x i, . . . , ~x
j, . . . , ~xN )
= ±Ψ(~x1, ~x2, . . . , ~x j, . . . , ~x
i, . . . , ~xN )
• Relativistischen Uberlegungen fuhren dazu, dass Elektronen als Fermionen(Teilchen mit halbzahligem Spin) antisymmetrische Wellenfunktionen habenmussen.
Slide 244 Das Pauli-Prinzip
Die Wellenfunktion von Elektronen ist antisymmetrischbezuglich der Vertauschung der Ortskoordinaten und Spinein-stellungen jeweils zweier beliebiger Elektronen.
• Die Mehrelektronenwellenfunktion Ψ hangt nicht, wie weiter oben angenom-men, nur von den ~ri ab, sondern von den ~xi.
Ψ(~r1, ~r2, . . . , ~rN ) −→ Ψ(~x1, ~x2, . . . , ~xN )
Damit ist ausgeschlossen, dass ein Spinorbital “zweifach besetzt” ist, denn
Ψ(~x1, ~x2) = χ1s(~r1, α(1))χ1s(~r2, α(2))
= ψ1s(~r1)|α(1)〉ψ1s(~r2)|α(2)〉= ψ1s(~r2)|α(2)〉ψ1s(~r1)|α(1)〉= Ψ(~x2, ~x1) 6= −Ψ(~x1, ~x2)
Slide 245 134

Konsequenzen
• Man kann jedoch leicht Wellenfunktionen konstruieren, deren “Raumorbita-le” zweifach besetzt sind, namlich mit Elektronen verschiedener Spineinstel-lung.
z.B.
Ψ(~x1, ~x2) = ψ1s(~r1)|α(1)〉ψ1s(~r2)|β(2)〉−ψ1s(~r1)|β(1)〉ψ1s(~r2)|α(2)〉
y Ψ(~x2, ~x1) = ψ1s(~r2)|α(2)〉ψ1s(~r1)|β(1)〉−ψ1s(~r2)|β(2)〉ψ1s(~r1)|α(1)〉
= −Ψ(~x1, ~x2)
• Dies kann man auch in Form einer Determinante schreiben
Ψ(~x1, ~x2) =
∣∣∣∣ χ1s(~r1, α(1)) χ1s(~r1, β(1))χ1s(~r2, α(2)) χ1s(~r2, β(2))
∣∣∣∣=
∣∣∣∣ ψ1s(~r1)|α(1)〉 ψ1s(~r1)|β(1)〉ψ1s(~r2)|α(2)〉 ψ1s(~r2)|β(2)〉
∣∣∣∣
Slide 246 Slater-Determinanten
• In dieser Form kann das Rezept zur Konstruktion von Mehrelektronenwel-lenfunktionen verallgemeinert werden.
Konstruiert man aus einem Satz von Spinorbitalen eine sogenannte Slater-Determinante
Ψ(~x1, ~x2, . . . , ~xN ) =1√N !
∣∣∣∣∣∣∣∣∣χ1(~x1) χ2(~x1) . . . χN (~x1)χ1(~x2) χ2(~x2) . . . χN (~x2)...
.... . .
...χ1(~xN ) χ2(~xN ) . . . χN (~xN )
∣∣∣∣∣∣∣∣∣so erfullt diese automatisch das Pauli-Prinzip.
Slide 247
135

Antisymmetrisierungsoperator
• andere Schreibweise fur Slaterdeterminanten:Ψ(~x1, ~x2, . . . , ~xN ) = |χ1 χ2 χ3 . . . χN 〉 oderΨ(~x1, ~x2, . . . , ~xN ) = Aχ1(~x1) · χ2(~x2) · . . . · χN (~xN )
• A heißt Antisymmetrisierungsoperator.A erzeugt eine Slaterdeterminante aus einem Produkt von Orbitalen.
• fur zwei Teilchen
A =1
2!(1− P12)
• fur drei Teilchen
A =1
3!(1− P12 − P13 − P23 + P12P23 + P13P23)
• Pij heißt Permutationsoperator (Transpositionsoperator)PijΨ(~x1, ~x2, . . . , ~xi, . . . , ~xj , . . . , ~xN ) = Ψ(~x1, ~x2, . . . , ~xj , . . . , ~xi, . . . , ~xN )
Slide 248 Nicht-wechselwirkende Elektronen
Die Eigenfunktionen von Hamiltonoperatoren fur nicht-wechselwirkendeElektronen konnen immer als Slater-Determinante geschrieben werden.
H|χ1 χ2 χ3 . . . χN 〉 = E|χ1 χ2 χ3 . . . χN 〉E = e1 + e2 + . . .+ eN
• Anmerkungen
1. |χ1 χ2 . . . χi . . . χi . . . , χN 〉 = 0(Zwei Zeilen der Determinante sind identisch!)
2. Will man zusatzliche Forderungen wie die nach “guten” Quantenzah-len fur den Gesamtspin erfullen, muss man oft Linearkombinationenmehrerer (entarteter) Slater-Determinanten bilden.
11.5 Die Eigenschaften von Atomen
Slide 249
136

Li-AtomII
• Das Pauli-Prinzip sagt uns, dass nur 2 Elektronen im 1s-Zustand sein konnen,und dass das 3. Elektron offensichtlich im 2s-Zustand sein muss.
• Entsprechend gilt fur die Gesamtenergie nun statt
〈H〉 = 〈h(1) + h(2) + h(3) +e2
4πε0
(1
r12+
1
r13+
1
r23
)〉
= 3 ∗ (−9 ∗ 13.6)eV + 3 ∗ 51eV = −214.2eV
vielmehr
〈H〉 = 2 ∗ (−9 ∗ 13.6)− 9 ∗ 13.6
22eV + Vee
= −200.0eV > E0 = −203.4eV
⇒ Kein Widerspruch zum Variationsprinzip.
Slide 250 Orbitale und Elektronenkonfiguration
• Wir hatten weiter oben gesehen, dass die Elektron-Elektron-Abstoßung von2 Elektronen im 1s-Orbital des He-Atoms ca. 34 eV betragt, im 1s-Orbitaldes Li-Atoms ca. 51 eV.
• Die abstoßende Wechselwirkung ist ∝ Z, weil starker geladene Kerne dieElektronen im Mittel starker anziehen, so dass auch die Abstoßung zunimmt.
⇒ Offensichtlich ist der Erwartungswert 〈 e2
4πε0r〉 abhangig von den Einelek-
tronenorbitalen.
• Die Elektron-Elektron-Abstoßung hebt die Entwartung der Zustande zu un-terschiedlichem Drehimpuls l (H-Atom) auf!
• Die Zustande der Atome sind daher nicht nur wesentlich durch die Haupt-quantenzahlen, sondern durch durch die Elektronenkonfiguration (und damitden Drehimpuls) charakterisiert.
Slide 251
137

Mehrelektronenatome ohne Elektron-Elektron-Wechselwirkung
• Pseudo-He 1s2 1s ��
• Pseudo-C 1s22s2p3
2s, 2p � � � �
1s ��
• Pseudo-N 1s22s22p3
2s, 2p �� � � �
1s ��
• Hier wurde angenommen, dass die Elektronen zunachst einfach auf die Raum-orbitale verteilt werden (s. Hundsche Regeln).
Slide 252 Mehrelektronenatome mit Elektron-Elektron-Wechselwirkung
• Durch die Elektron-Elektron-Wechselwirkung andern sich die Gesamtener-gien
• Die Orbitalenergien andern sich und spalten (teilweise) auf.
• Anmerkung: Die Gesamtenergien sind bei Berucksichtigung der e-e-WWnicht die Summe der Orbitalenergien (Vermeidung von Doppelzahlungen).
• C 1s22s22p2
2p � �2s ��
1s ��Elektronenkonfiguration des C-Atoms andert sich.
• N 1s22s22p3
2p � � �2s ��
1s ��Elektronenkonfiguration des N-Atoms bleibt gleich.
Slide 253
138
Konfigurationsenergien
• Die relative Energie der Orbitale zum Drehimpuls l hangt vom “Fullgrad”der Schale ab, also von der Anzahl der Elektronen, und damit von der Kern-ladungszahl Z.
• Durch die Elektron-Elektron-Abstoßung liegen die s-Orbitale niedriger alsdie p-Orbitale zu gegebener Hauptquantenzahl, die p-Orbitale niedriger alsdie d-Orbitale.
• Dies kann soweit gehen, dass fur bestimme Werte von Z, die 4s-Orbitalenergieniedriger liegt als die 3d-Orbitalenergie. → d-Elemente (Ubergangsmetalle)
• Berechnung erfolgt z.B. uber die Methode des Selbstkonsistenten Feldes(Hartree-Fock-Verfahren, ThC II).
Slide 254 Konfigurationsenergien
Slide 255 Ionisierungsenergien
139
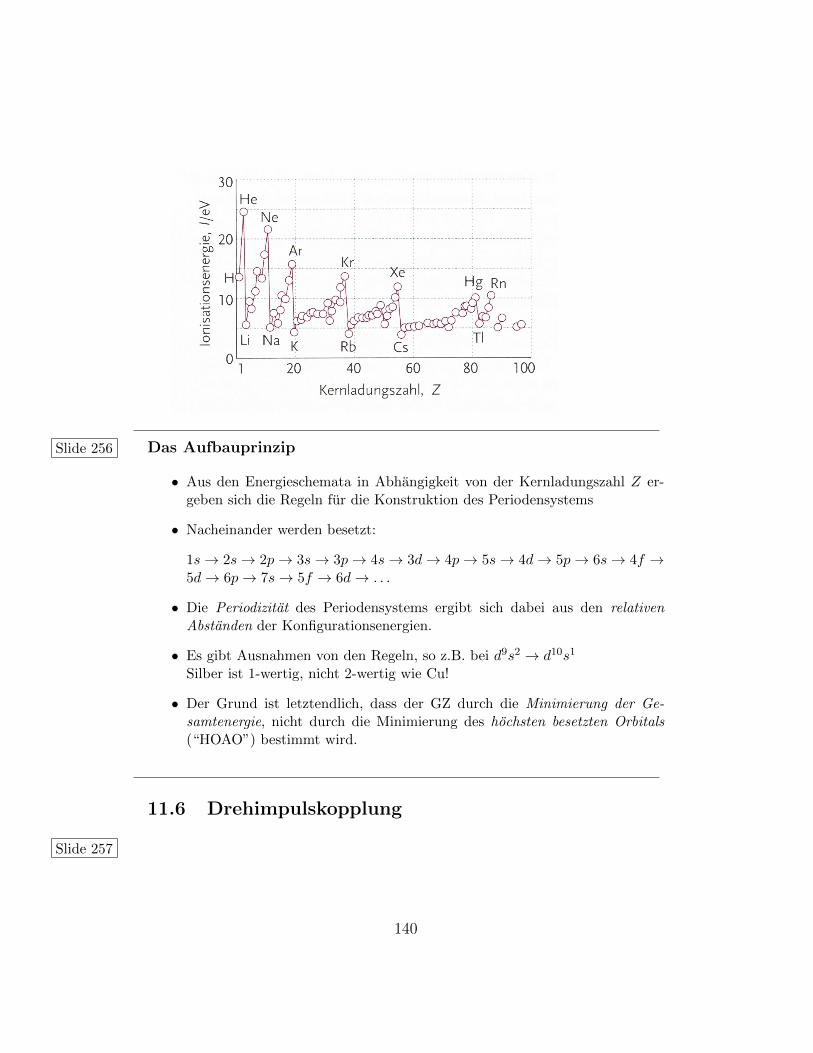
Slide 256 Das Aufbauprinzip
• Aus den Energieschemata in Abhangigkeit von der Kernladungszahl Z er-geben sich die Regeln fur die Konstruktion des Periodensystems
• Nacheinander werden besetzt:
1s→ 2s→ 2p→ 3s→ 3p→ 4s→ 3d→ 4p→ 5s→ 4d→ 5p→ 6s→ 4f →5d→ 6p→ 7s→ 5f → 6d→ . . .
• Die Periodizitat des Periodensystems ergibt sich dabei aus den relativenAbstanden der Konfigurationsenergien.
• Es gibt Ausnahmen von den Regeln, so z.B. bei d9s2 → d10s1
Silber ist 1-wertig, nicht 2-wertig wie Cu!
• Der Grund ist letztendlich, dass der GZ durch die Minimierung der Ge-samtenergie, nicht durch die Minimierung des hochsten besetzten Orbitals(“HOAO”) bestimmt wird.
11.6 Drehimpulskopplung
Slide 257
140

AdditionsregelnI
• Aufgrund der Ununterscheidbarkeit der Elektronen kann weder der Bahn-drehimpuls li noch der Spin si eines Elektrons i eine Observable sein!
• Betrachten wir ein Beispiel mit zwei Elektronen in p-Orbitalen (li = 1)
• Zustande:
l1 m1 l2 m2 M = m1 +m2 L?
1 1 1 1 2 21 1 1 0 11 1 1 -1 01 0 1 1 11 0 1 0 01 0 1 -1 -11 -1 1 1 01 -1 1 0 -11 -1 1 -1 -2 2
Slide 258 AdditionsregelnII
• Zustande:
l1 m1 l2 m2 M = m1 +m2 L?
1 1 1 1 2 21 1 1 0 11 1 1 -1 01 0 1 1 11 0 1 0 01 0 1 -1 -11 -1 1 1 01 -1 1 0 -11 -1 1 -1 -2 2
• 2 Zustande (M = ±2) sind nur moglich, wenn der Gesamtdrehimpuls L denWert 2 annimmt (|M | ≤ L!).
• Es gibt 3 Zustande mit M = 0!
• Es gibt je 2 Zustande mit M = +1 und M = −1!
Slide 259
141

Addition von Bahndrehimpulsen
• Der Gesamtbahnimpuls und seine z-Komponente sind durch folgende Ope-ratoren bestimmt:
L = l1 + l2 + . . .+ lN
Lz = l1,z + l2.z + . . .+ lN,z
Regel Zwei Bahndrehimpulse werden vektoriell addiert!Der Betrag (die Quantenzahl L) kann ganzzahlige Werte zwi-schen |~l1 +~l2| und |~l1 −~l2| annehmen.
• Im Beispiel ergibt dies die Zustande L = 2 (5-fach entartet), L = 1 (3-fachentartet) und L = 0 (1-fach entartet).
⇒ insgesamt 9 Zustande des Gesamtdrehimpulses (= Zahl der Ausgangs-zustande)
• Zustande zu verschiedenem Gesamtbahndrehimpuls L klassifiziert man mitGroßbuchstaben.L = 0, 1, 2, 3, . . . → S, P,D, F, . . .
Slide 260 Addition von Spins
• Fur Spins gilt analog
Regel Zwei Spins werden vektoriell addiert!Der Betrag (die Quantenzahl S) kann ganzzahlige Werte zwi-schen |~s1 + ~s2| und |~s1 − ~s2| annehmen.
• Kopplung zweier Spins kann nur auf die Gesamtspins S = 1 und S = 0fuhren.
• Gesamtspinzustande S charakterisiert man entsprechend ihrer Multiplizitat(=Zahl der MS-Zustande).
• Zustande mit S = 0 heißen Singuletts.
• Zustande mit S = 1 heißen Tripletts.
• Zustande mit S = 12 (nur ein ungepaarter Spin) heißen Dubletts.
Slide 261 142

Addition von mehreren Drehimpulsen
• Die Kopplung von mehr als zwei Spins (oder Bahndrehimpulsen) kann mansich so vorstellen, dass zunachst 2 Spins gekoppelt werden, dann das Ergebnismit dem 3., usw.
• Mehr als 2 Drehimpulse konnen entsprechend zu Gesamtdrehimpulszustandenmit hoherem L oder S fuhren.
• Die entsprechenden Wellenfunktionen sind Linearkombinationen von Pro-dukten von Orbitalen.
• Die entsprechenden Koeffizienten heißen Clebsch-Gordon-Koeffizienten.
• Der Gesamtspin ist wieder ein Drehimpuls, ebenso wie der Gesamtbahndre-himpuls. Beide konnen wieder miteinander koppeln (=wechselwirken), unddabei einen Gesamtdrehimpuls ~J bilden.
11.7 Spin-Bahn-Kopplung und Hundsche Regeln
Slide 262 Spin-Bahn-Kopplung
• Der innere Drehimpuls (Spin) der geladenen Elektronen bewirkt ein magne-tisches Moment, mit dem der Bahndrehimpuls der Elektronen wechselwirkt.
• Es gibt also einen Zusatzterm im Hamilton-Operator, der bisher nicht beruck-sichtigt wurde.
• Man kann zeigen, dass dieser Term die Form einer Kopplung zwischen Ge-samtspin S und Gesamtbahndrehimpuls L annimmt.
• Man nennt diesen Effekt daher Spin-Bahn-Kopplung (Spin-Orbit-Coupling).
HSO =Ze2
8πε0m2er
3c2~L · ~S = ξ(r) ~L · ~S
• Verglichen mit der Kern-Elektron-Anziehung und der e-e-Abstoßung ist HSO
klein.
⇒ EHSO ist eine Storung der Konfigurationsenergie.
Slide 263143

Erwartungswerte des Spin-Bahn-Kopplungsoperators
• Zustande sind jetzt durch “gute Quantenzahlen” L, S, J , und mJ charakte-risiert (Gesamtdrehimpuls J).
ESO = 〈L, S; J,MJ |HSO|L, S; J,MJ〉 =
Z4e4
(4πε0~c)2hcR∞
{J(J + 1)− L(L+ 1)− S(S + 1)
2n3L(L+ 1/2)(L+ 1)
}• Die Energie ESO ist klein, aber sie spaltet die Zustande zu gegebenem L, S
und J auf.
• Dies fuhrt zur beobachtbaren Feinstruktur der Atomspektren.
• Zustande zu festem L, S, J nennt man Terme
• Der Grundzustand eines Vielelektronenatoms ist also keine Konfiguration,sondern ein Term.
Slide 264 Hundsche Regeln
• Die Hundschen Regeln beschreiben die Reihenfolge der Terme und legeninsbesondere fest, wie man den Grundzustand eines Atoms findet.
• 1. Hundsche RegelDer Term mit maximaler Spin-Multiplizitat hat die niedrigste Energie!
• 2. Hundsche RegelBei gegebener Spin-Multiplizitat hat der Term mit dem hochstem L-Wert die niedrigste Energie
• 3. Hundsche RegelFur Atome mit weniger als halb gefullten Schalen hat der Zustand mitdem kleinsten J-Wert die niedrigste Energie.
Slide 265
144

Beispiel: Grundzustand des KohlenstoffatomsI
• Die Elektronenkonfiguration ist 1s22s22p2.
• Die Orbitaldrehimpulse li sind (0,0,0,0,1,1).
• Als praktische Regel gilt: Volle Schalen oder Unterschalen brauchen nichtbetrachtet zu werden
• Also betrachten wir die Subkonfiguration 2p2 mit l1 = l2 = 1 und s1 = s2 =1/2
• L kann nach den Regeln der Drehimpulskopplung die Werte 0, 1, 2 (S, P,D)annehmen.
• S kann nach den Regeln der Drehimpulskopplung die Werte 0 oder 1 anneh-men
• 1. Hundsche Regel: S = 1⇒ Triplett.
• 2. Hundsche Regel: L maximal. Demnach wurde man D erwarten.
Slide 266 Beispiel: Grundzustand des KohlenstoffatomsII
• Jedoch ist die Spinfunktion in Triplett-Funktionen stets symmetrisch bzgl.Vertauschung der zwei Elektronen. Ebenso ist die D-Funktion symmetrisch,wahrend die P -Funktion antisymmetrisch bzgl. Vertauschung der zwei Elek-tronen ist.
• 3D ist also aufgrund des Pauli-Prinzips nicht moglich, wohl aber 3P .
• 3. Hundsche Regel: fur 2p2 (weniger als halb besetzt) soll J minimal sein.L = S = 1 =⇒ J = 0, 1, 2. Also ist J = 0 der gesuchte Zustand.
• Der Grundzustand des C-Atoms ist 3P0.
Slide 267
145

Hundsche Regeln“verbesserte” Formulierung nach Haken / Wolf
• 1. Hundsche RegelDer Term mit maximaler Spin-Multiplizitat hat die niedrigste Energie!Elektronen mit gleichem l werden so aufgefullt, dass der GesamtspinS =
∑si maximal wird.
• 2. Hundsche RegelUnter Beachtung des Pauli-Prinzips werden die Elektronen mit gleicheml so auf die ml verteilt, dass |Lz| = |
∑ml~| = mL~ ein Maximum wird.
L ist dann gleich mL.
• 3. Hundsche RegelFur Atome mit weniger als halb gefullten Schalen hat der Zustand mitdem kleinsten J-Wert die niedrigste Energie.
Slide 268 Beispiel: Die Na-D-LinieI
• Das Na-Atom hat die Elektronenkonfiguration 1s22s22p63s.
• Die teilweise besetzte Schale hat ein Elektron im l = 0 und s = 1/2-Zustand.Es ist nur j = 1/2 moglich!
• Der Grundzustand ist demzufolge 2S1/2 (“Duplett-S-Einhalb”).
• Anregungen des Na-Atoms erfolgen in 4s bzw. 4p bzw. 4d-Orbitale. Die4s-Orbitale sind die energetisch niedrigsten.
• Fur die Abregung (“Emission von Licht”) sind die Ubergangswahrschein-lichkeiten (bei weitem) am großten fur den Ubergang aus dem 4p-Zustand.
• Begrundung: Photonen besitzen einen Spin von S = 1 (sie sind Boso-nen). Drehimpulserhaltung erfordert, dass das Photon den Drehimpuls “mit-nimmt”.
⇒ Auswahlregel ∆L = 1
Slide 269
146

Beispiel: Die Na-D-LinieII
• Die 4p-Konfiguration des Na-Atoms besteht aus einem L = 1 Elektron mitSpin S = 1/2. Mogliche Werte des Gesamtdrehimpulses J sind also J = 3/2oder J = 1/2.
• Die Natrium-Doppellinie (D-Linie) setzt sich zusammen aus den Ubergangen2P1/2 → 2S1/2 und 2P3/2 → 2S1/2
11.8 Zusammenfassung
Slide 270 Zusammenfassung Atomphysik
• Das bisher vorgeschlagene Schema ist anwendbar, wenn die Spin-Bahn-Kopp-lungsenergie klein im Vergleich zur e-e-Abstoßung ist.
• Dann koppeln zunachst die li zu L und die si zu S (Russell-Saunders-Kopplung), die dann zu J koppeln.
• Fur schwere Kerne (hohes Kernladungszahl Z) koppeln die individuellenli starker an die si zu einem ji, die dann wieder zu einem J koppeln (jj-Kopplung).
Slide 271 Virialtheorem
• Fur alle Atome gilt
〈T 〉 = −12〈V 〉
als Konsequenz des allgemeinen Virialtheorems:
147

Falls V (λ~r1, λ~r2, . . . , λ~rN ) = λνV (~r1, ~r2, . . . , ~rN ), so gilt
〈T 〉 =ν
2〈V 〉
• Coulomb-Wechselwirkung: V ∝ 1r y ν = −1
• Harmonischer Oszillator: V ∝ r2 y ν = +2
148

12 Molekule
Slide 272 Vorbemerkungen
• Da Atome viele ununterscheidbare Elektronen besitzen, sind ihre Zustandedurch interelektronische Coulomb- und Austausch-Wechselwirkungen be-stimmt.
• Je 2 Elektronen konnen in einem Raumorbital existieren. (jedes in einemunterschiedlichen Spinorbital).
• Dies ist analog bei Molekulen der Fall. Hier nennt man die entsprechendenEinelektronenzustande Molekulorbitale.
• Wir hatten beim Atom die Bewegung des Kerns immer vernachlassigt, daman diese kinematisch exakt abseparieren kann.
• Bei Molekulen spielt die Kernbewegung eine großere Rolle (Schwingungen).Die Separation ist nicht exakt moglich.
• Im dynamischen Molekul bewegen sich sehr leichte Massen (Elektronen) und103 − 104 mal schwerere Massen (Kerne).
12.1 Die Born-Oppenheimer-Naherung
Slide 273 Ein einfaches Molekul mit einem Elektron: H+2
~RAB = ~RB − ~RA
~reA = ~re − ~RA
~reB = ~re − ~RB
149

H = TA + TB + VAB + Te + VeA + VeB
= − ~2
2mA∆A −
~2
2mB∆B +
ZAZBe2
4πε0RAB
− ~2
2me∆e −
ZAe2
4πε0reA− ZBe
2
4πε0reB
Ti: kinetische Energieoperatoren
Slide 274 Elektronischer Hamiltonoperator
• Der Gesamt-Hamilton-Operator
H = − ~2
2mA∆A −
~2
2mB∆B +
ZAZBe2
4πε0RAB
− ~2
2me∆e −
ZAe2
4πε0reA− ZBe
2
4πε0reB
enthalt Terme, die das Elektron im Feld der Kerne beschreiben
Hel(~RA, ~RB) = − ~2
2me∆e −
ZAe2
4πε0reA− ZBe
2
4πε0reB
• der elektronische Hamiltonoperator Hel hangt implizit uber reA und reB vonRA und RB ab.
Slide 275 Elektronische Schrodingergleichung
• In der Born-Oppenheimer-Naherung lost man zunachst die elektronischeSchrodingergleichung
Hel(~RA, ~RB)φel(~re; ~RA, ~RB) = Eel(~RA, ~RB)φel(~re; ~RA, ~RB)
bei festgehaltenem ~RA und ~RB
• Dadurch hangt Eel und φel parametrisch von den Koordinaten der Kerneab.
Slide 276150

Schrodingergleichung der Kerne
• mit der Definition EBO(~RA, ~RB) = Eel(~RA, ~RB) + VAB
• gelangt man zur SGL fur die Kernbewegung[TA + TB + EBO)(~RA, ~RB)
]φnuc(~RA, ~RB)
= Eφnuc(~RA, ~RB)
• Die Gesamtwellenfunktion ist dann
φ(~re, ~RA, ~RB) = φel(~re; ~RA, ~RB) · φnuc(~RA, ~RB)
• Der suggerierte Separationsansatz ist nicht exakt, aber haufig ausreichend.
• Eigentlich musste die Gesamtwellenfunktion in eine Summe aus Produktendieser Art entwickelt werden, also etwa
φ(~re, ~RA, ~RB) =∑i
φi,el(~re; ~RA, ~RB) · φi,nuc(~RA, ~RB)
Slide 277 Anmerkungen
• Physikalische Motivation: Die Elektronen sind so leicht, dass sie den Kerneninstantan folgen konnen.
• Eel(~RA, ~RB) hangt eigentlich nur von den internen Koordinaten ab, also hierz. B. nur von RAB.
• Dies kann man durch Separation von Massenschwerpunkts- und Rotations-bewegung berucksichtigen.
• In der Nahe von Req gilt EBO(RAB) = 12kAB(RAB −Req)2.
→ harmonischer Oszillator als Modell fur schwingende chemische Bindungen
151

12.2 Die Linear Combination of Atomic Orbital-Methode(LCAO)
Slide 278 Das einfachste Molekul H+2
• Die gesuchte Wellenfunktion des einen Elektrons im H+2 nennt man Mo-
lekulorbital (MO).
• MOs sind analog zu AOs Einelektronenwellenfunktionen.
• Der minimale Ansatz fur die Wellenfunktion ist
ψ = c1ψ1 + c2ψ2
mit ψ1 und ψ2 den Atomorbitalen (hier 1s-Orbitale) der beiden H-Atomean deren jeweiligen Ort.
• ψ ist eine Linearkombination von Atomorbitalen (LCAO).
Slide 279 Variationsprinzip
• In mathematischer Formulierung bedeutet das Variationsprinzip, dass
E = 〈E〉 =〈ψ|H|ψ〉〈ψ|ψ〉
≥ E0
sein muss.
• Wir erhalten also die optimalen Koeffizienten c1 und c2 (diejenigen, die diebestmogliche Energie mit dem obigen LCAO-Ansatz ergeben) durch Mini-mierung des Energieerwartungswertes.
Slide 280
152

Energieerwartungswert
E =〈c1ψ1 + c2ψ2|H|c1ψ1 + c2ψ2〉〈c1ψ1 + c2ψ2|c1ψ1 + c2ψ2〉
E =c∗1c1〈ψ1|H|ψ1〉+ c∗1c2〈ψ1|H|ψ2〉+ c∗2c1〈ψ2|H|ψ1〉+ c∗2c2〈ψ2|H|ψ2〉
c∗1c1〈ψ1|ψ1〉+ c∗1c2〈ψ1|ψ2〉+ c∗2c1〈ψ2|ψ1〉+ c∗2c2〈ψ2|ψ2〉
• Man definiert Sij = 〈ψi|ψj〉 (Uberlappungsintegrale) und Hij = 〈ψi|H|ψj〉(Matrixelemente des Hamiltonoperators)
• Wenn die Atomorbitale normiert sind, ist Sii = 1 (aber nicht Sij = 0)!
• Fur reelle Atomorbitale gilt Sij = S∗ji = Sji und Hij = H∗ji = Hji.
E =c∗1c1H11 + c∗1c2H12 + c∗2c1H12 + c∗2c2H22
c∗1c1S11 + c∗1c2S12 + c∗2c1S12 + c∗2c2S22
Slide 281 Energieminimierung
• E ist minimal, wenn alle
(∂E
∂ci
)= 0 fur alle ci (oder, analog, wenn alle(
∂E
∂c∗i
)= 0).
• Wir bilden die Ableitungen
(∂E
∂c∗i
).
• Anstatt nun
E =c∗1c1H11 + c∗1c2H12 + c∗2c1H12 + c∗2c2H22
c∗1c1S11 + c∗1c2S12 + c∗2c1S12 + c∗2c2S22
nach der Quotientenregel abzuleiten, und dann = 0 zu setzen, kann manauch die Gleichung
E · [c∗1c1S11 + c∗1c2S12 + c∗2c1S12 + c∗2c2S22]
= [c∗1c1H11 + c∗1c2H12 + c∗2c1H12 + c∗2c2H22]
rechts und links ableiten (implizite Differentiation).
Slide 282 153

Ableitungen
∂
∂c∗1
∣∣∣∣ E · [c∗1c1S11 + c∗1c2S12 + c∗2c1S12 + c∗2c2S22]
∂
∂c∗1
∣∣∣∣ = [c∗1c1H11 + c∗1c2H12 + c∗2c1H12 + c∗2c2H22]
⇒ E [c1S11 + c2S12] = [c1H11 + c2H12]
analog: Ableiten nach c∗2
E [c1S12 + c2S22] = [c1H12 + c2H22]
oder nach Umsortieren in c1 und c2-Terme
(H11 − ES11)c1 + (H12 − ES12)c2 = 0
(H12 − ES12)c1 + (H22 − ES22)c2 = 0
Slide 283 Sakulardeterminante des Gleichungssystems
• Das Gleichungssystem (H11 − ES11)c1 + (H12 − ES12)c2 = 0
(H12 − ES12)c1 + (H22 − ES22)c2 = 0
hat genau dann eine nichttriviale Losung (d.h. nicht: c1 = c2 = 0), wenn dieSakulardeterminante∣∣∣∣ (H11 − ES11) (H12 − ES12)
(H12 − ES12) (H22 − ES22)
∣∣∣∣ != 0
verschwindet.
• allgemein: det(Hij − SijE) = 0
Slide 284
154

Energieeigenwerte
• Fur H+2 gilt H11 = H22 und S11 = S22 = 1.
• Man erhalt
(H11 − E)2 − (H12 − ES12)2 = 0
⇒ (H11 − E) = ±(H12 − ES12)
H11 ∓H12 = E ∓ ES12
⇒ E± =H11 ±H12
1± S12
∣∣∣∣±H11S12 ∓H11S12
1± S12
= H11 ±H12 −H11S12
1± S12
= H11 ±β
1± S12
β = H12 −H11S12 heißt reduziertes Resonanzintegral.
Slide 285 Resonanzintegral
E± = H11 ±β
1± S12
= H11 ±H12 −H11S12
1± S12
=H11 · (1± S12)± (H12 −H11S12)
1± S12
=H11 ±H12
1± S12=α± β∗
1± S12
• β∗ heißt Resonanzintegral.
• Grenzwertbetrachtung: S12, β∗ → 0 =⇒ keine Bindung
• Energieerniedrigung des bindenden Orbitals ist kleiner als die Energieerhohungdes antibindenden Orbitals
Slide 286
155

AbstandsabhangigkeitS12 = S12(RHH) β∗ = β∗(RHH)
S12 = 〈ψ1s(~r − ~R1)|ψ1s(~r − ~R2)〉= 〈ψ1s(~r − ~R1)|ψ1s(~r − ~R1 − ~R)〉
• die r-Abhangigkeit verschwindet durch die Integration, die R1-Abhangigkeitaufgrund der freien Wahlbarkeit des Koordinatenursprungs.
• die Winkelabhangigkeit verschwindet, weil der Raum isotrop ist ⇒ S12 =S12(R)
• Analog werden H11 und H12 (α und β∗) ebenfalls nur noch von R abhangen.
• der exponentielle Abfall von ψ1s fuhrt zu einem exponentiellen Abfall derIntegrale fur große R.
Slide 287 Eigenvektoren
• Nacheinander setzt man E+ = H11 +β
1 + S12und E− = H11 −
β
1− S12in
das Gleichungssystem
(H11 − ES11)c1 + (H12 − ES12)c2 = 0
(H12 − ES12)c1 + (H22 − ES22)c2 = 0
ein und erhalt dann fur jeden Energieeigenwert die Koeffizienten c1 und c2
(d.h. den Eigenvektor bzw. Eigenfunktion) (→ Ubungsaufgabe).
• Man erhalt
E+ =H11 +H12
1 + S12= H11 +
β
1 + S12EF: ψ+ = ψ1 + ψ2
E− =H11 −H12
1− S12= H11 −
β
1− S12EF: ψ− = ψ1 − ψ2
Slide 288
156
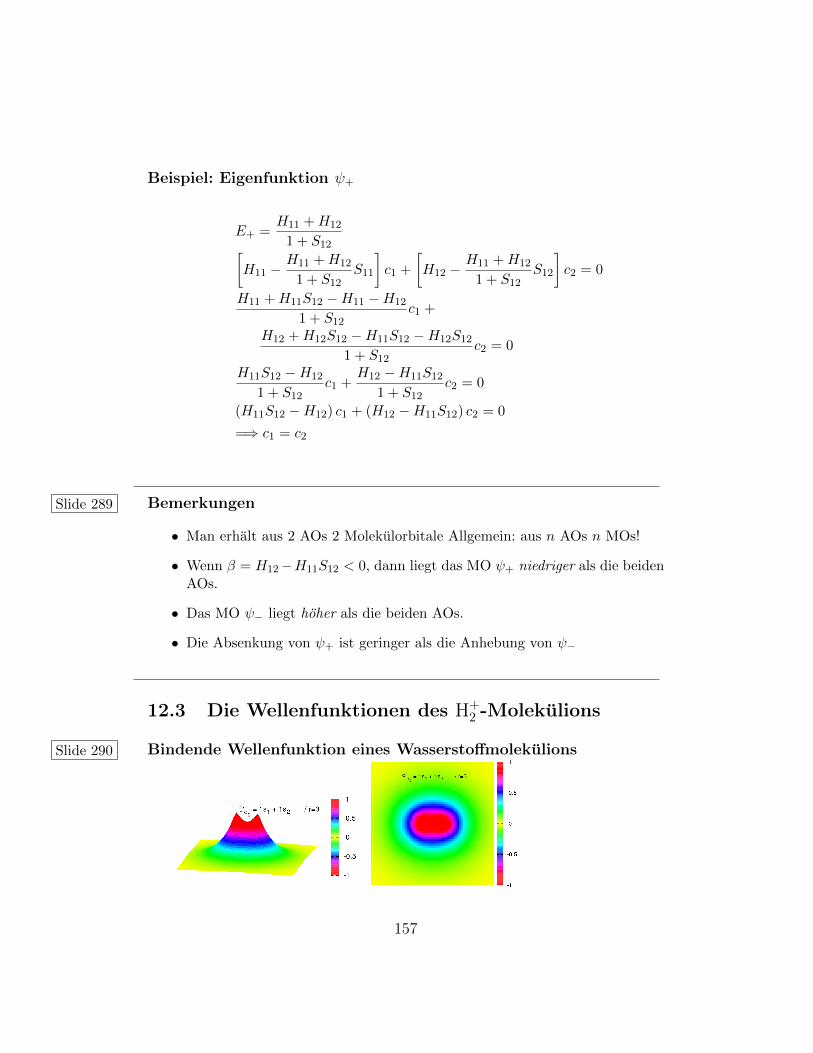
Beispiel: Eigenfunktion ψ+
E+ =H11 +H12
1 + S12[H11 −
H11 +H12
1 + S12S11
]c1 +
[H12 −
H11 +H12
1 + S12S12
]c2 = 0
H11 +H11S12 −H11 −H12
1 + S12c1 +
H12 +H12S12 −H11S12 −H12S12
1 + S12c2 = 0
H11S12 −H12
1 + S12c1 +
H12 −H11S12
1 + S12c2 = 0
(H11S12 −H12) c1 + (H12 −H11S12) c2 = 0
=⇒ c1 = c2
Slide 289 Bemerkungen
• Man erhalt aus 2 AOs 2 Molekulorbitale Allgemein: aus n AOs n MOs!
• Wenn β = H12−H11S12 < 0, dann liegt das MO ψ+ niedriger als die beidenAOs.
• Das MO ψ− liegt hoher als die beiden AOs.
• Die Absenkung von ψ+ ist geringer als die Anhebung von ψ−
12.3 Die Wellenfunktionen des H+2 -Molekulions
Slide 290 Bindende Wellenfunktion eines Wasserstoffmolekulions
157
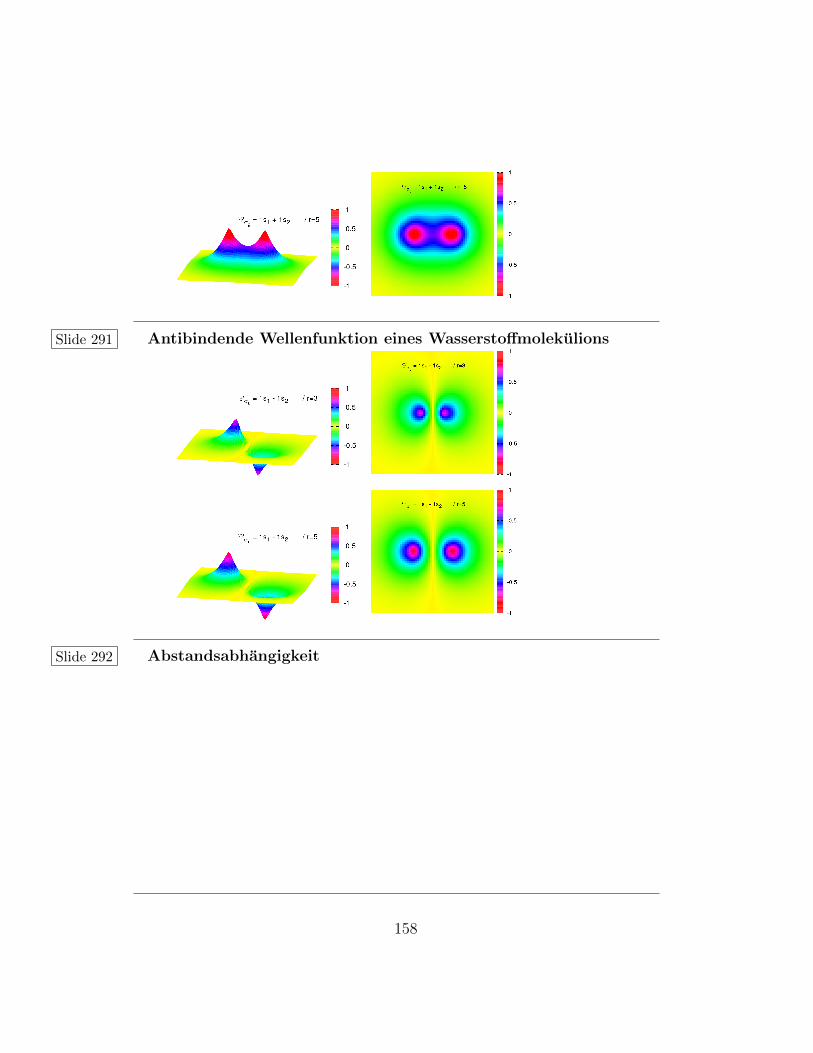
Slide 291 Antibindende Wellenfunktion eines Wasserstoffmolekulions
Slide 292 Abstandsabhangigkeit
158

12.4 Das Wasserstoffmolekul
Slide 293 Hamiltonoperator
• Im Rahmen der Born-Oppenheimer-Naherung hat der elektronische Hamil-tonoperator fur die Elektronen 1 und 2 im Feld der Kerne A und B dieForm
H = T1 + T2 + VA1 + VA2 + VB1 + VB2 + V12
= T1 + VA1 + VB1
+ T2 + VA2 + VB2
+ V12
• Ohne den Elektron-Elektron-Wechselwirkungsterm V12 = e2
4πε0r12hatte er
die Form einer Summe von 2 Hamiltonoperatoren H1 und H2 fur H+2 :
H = H1 + H2
Slide 294 Slater-Determinante
• Wir wollen die Orbitale des H+2 ψ+ = ψσ als Naherungen fur die Orbitale
des H2 benutzen.
• Analog zu den Vielteilchenatomen erstellen wir die Grundzustandswellen-funktion des H2 als eine Slaterdeterminante von Spinorbitalen.
• Diese generieren wir, indem wir von unten Elektronen maximal paarweise indie Molekulorbitale einfugen und diese antisymmetrisieren
• Die Slaterdeterminante lautet dann
Ψσ2 =1√2
∣∣∣∣ ψσ(~r1)|α1〉 ψσ(~r1)|β1〉ψσ(~r2)|α2〉 ψσ(~r2)|β2〉
∣∣∣∣• Energie Eσ2 = 2Eσ + 〈V12〉 mit Eσ = H11 + β/(1− S12)
(oder Vernachlassigung von V12).
12.5 MO-Diagramme zweiatomiger Molekule
Slide 295 159

Regeln fur die Kombination von AtomorbitalenDie durch die Elektron-Elektron-Wechselwirkungen modifizierten Atomorbitale
und Atomorbitalenergien bilden zusammen mit dem LCAO-Konzept die Grundlageder Molekulorbital-Diagramme.
Regeln:
1. Man braucht nur AOs zu kombinieren, deren Uberlappung nicht Null ergibt(z. B. aus Symmetriegrunden).
2. Man braucht nur energetisch ahnliche AOs (in erster Naherung) zu verwen-den.
3. Man besetzt die Orbitale von unten nach oben.
Slide 296 MO-Diagramm fur H2
• Energieabsenkung
• bindende Wechselwirkung → chemische Bindung
Slide 297 MO-Diagramm fur He2
160

• Energieerhohung (das obere Orbital ist starker erhoht als das untere abge-senkt ist)
• antibindende Wechselwirkung → keine chemische Bindung
Slide 298 MO-Diagramm fur ein heteropolares Molekul MX
• Energieabsenkung eines Elektrons ist klein, des anderen Elektrons groß
• bindende Wechselwirkung → chemische Bindung
• Bindender Zustand dem niedrigeren Orbital ahnlicher.
161





























