Embed Size (px)
Citation preview

GATTOW und W]~DLA_~DT: Zur analytischen Untersuchung yon Bramlsteinen 15
Literatur 1 COVnSIER, J., J. t t v ~ u. 1~. PLATZE~: Anal. ehim. Act~ (Amsterdam) 13,
379 (1955); vgl. diese Z. 151, 376 (1956). -- 2 DUCI~.ET, L. u. P. SEGUIN: Vortrag beim XV. IUPAC-Kongrel~, Lissabon 1956. -- a LvK~, C. L. : AnMyt. Chemistry 27, 1150 (1955); vgl. diese Z. 151,310 (1956). - - 4 MO~IsoN, G. H., u. 1~. L. l~uP~: AnMy$. Chemistry 29, 892 (t957); vgl. dJese Z. 160, 394 (1958). - - 5 PO~L, F. A.: diese Z. 157, 6 (1957).
Doz. Dr.-Ing. F. A. Pomp, AE G-Forsehtmgs-ins~itut, Frankfurt am Main-Niederrad, GoldsteinstraBe 235
Aus dem Anorganiseh-chemischen Institut der Universit~t GSttingen
Zur analytischen Untersuchung yon Braunsteinen
Eine Methode zur Schnellbestimmung der Zusammensetzung yon M n 0 1 + ~ - P r o d u k t e n
Von
G. GATTOW und H.-G. WENDLAIN~DT
Mit 1 Text~bbildung
(Eingegangen am 28. November 1959)
W~hrend ffir die W e r t b e s t i m m u n g yon Brauns te in , worun te r die E r m i t t l u n g des Ante i l s Mn0~* in e iner gegebenen Probe ve r s t a nde n wird, eine Reihe yon Verfahren 1,~,7,1o zur Verf i igung stehen, die zum Teil auf die fr/ ihere Verwendung des Brauns te ins in der Chlor indust r ie zurf ickzuff ihren sind, f inden sich bemerkenswer terweise k a u m Hinweise ffir solche Methoden, die in einfacher, aber dennoch sicherer Weise eine B e s t i m m u n g des Mangan-Sauers toff -Verh~l tn isses ges ta t ten . Das m a g d a r a n liegen, dag die Bes t imm~ng dieses Verh'~ltnisses im t I i nb l i ck a u f die Beur te i lung yon Brauns t e inp roben in manchen Anwendungs- bere ichen eine geringe Rolle spielte, en tweder weft m a n an einer Charak- t e r i s ie rung des Mater ia ls dl t rch die s tSchiometr i sche Zusammense tznng weniger interessier~ war, oder aber dera r t ige Unte r sueh~ngen so r e l a t i~ wenig duregzuff ihren ha t t e , dab e twa der Ze i taufwand ffir e inen der- a r t igen Test unberf ieks ieht ig t ble iben konnte .
] )a bei der Bea rbe i tnng yon Problemen, wie sie sieh aus der Unter - suchung yon Zusammenhgngen zwisehen S t r u k t u r und s t6ehiometr i seher
* In alteren Arbeiten wurde angenommen, dab Braunstein die Zusammen- setzung ~inO 2 besitzt. Nach neuesten Untersuchungen wird diese Idealzusammen- setzung MnO 2 nur in den seltensten l~llen erreicht (z.B. fl-~In02), so da2 die fremdionenfreien ]~raunsteine Mlgemein als Nn01+ ~ eharakterisiert werden kSnnen (vgl. 3).

16 G. GATTOW lln(:]_ H.-G. WENDLANDT:
Zusammensetzung ergeben a, eine derartige Methode Voraussetzung ffir rationelles Arbeiten ist, haben wir uns mit dieser anMytischen Frage besch~ftigt, wor/iber naehstehend berichtet werden sell, nicht .zuletzt im Hinblick darauf, dal~ mit dem weiteren Vordringen yon synthetischem Braunstein die f~berprfifung des Mangan-Sauerstoff-Verhgltnisses sehon bei der I-Ierstellung kfinstlicher Braunsteine eine nicht zu unterschiitzende Hilfe bei der Kontrolle und Beurteilung der erzeugten Produkte bringen kSnnte.
Miiglichkeiten und Voraussetzungen fiir die Bestimmung des Mangan- Sauerstoff-u
Zur Ermit t lung des Mangan-Sauerstoff-Verh/iltnisses muI~ zuss zur Bestimmung des aktiven Sauerstoffs mit einer zweiten Analyse der Gesamtmangangehalt festgestellt werden. Fiir die Bestimmung des akt iven* Sauerstoffs kann man eine der bek~nnten Methodenl,e,v, ~~ heranziehen. Dabei sollte die Methode bevorzugt werden, die bei gleicher Sieherheit und vergleichbarem Zeitaufwand die M6glichkeit bietet, den GesamtmangangehMt im Anschlug an die Best immung des aktiven Sauerstoffs durchzuf/ihren. Es ist naheliegend, dab nicht nut Arbeits- aufw~nd, sondern auch eine zusi~tzliche FehlerqueUe durch Fortfall der Einwaage ausgeschaltet wird, zumal die Kombinat ion beider Bestim- mungen mit besonderer Einfachheit erfolgt, wenn man, wie bereits P~IBIL u. 01I~AL~ 9 ~ndeuteten, die Bestimmung des Mn 2+ ira AnschluB an die Reduktion chelatometrisch durchf~hrt.
Unter diesem Gesichtspunkt scheiden naturgem/~6 einige Methoden zur Bestimmung des ~ktiven Sauerstoffs aus, wie z.B. die Eisen(II)- sulfatmethodeX, ~ oder die neuerdings yon KRxJzovg, SIMON u. Z:~KA s vorgeschlagene tIydrochinonmethode, w~hrend andere, wie die OxM~t- methode 1,a, nur bedingt brauchbar sind. Bei der OxalaSmethode liegen die Schwierigkeiten nicht bei tier Methode selbst. Diese hat im Gegenteil den Vorzug, unempfindlich gegen St6rungen dureh Fe ~+ zu sein, sondern bei tier chelatometrischen Mn-Bestimmung, da man hier zusi~tzlich das fiber die KMnO4-Riiektitration eingebrachte Mn ~+ mit erfaBt und ge- sondert in Reehnung stellen m/il~te.
Es ersehien deshalb angebraeht, sieh mit der KJ-IVfethode zu befassen, obgleich diese Methode zur Bestimmung des akt iven Sauerstoffs weder als Sehiedsverfahren noch sonst eir~drucksvoll erw/ihnt wirdi,~, i~
Unter den St6rmlgen dureh Fremdionen bei der K J - ~ e t h o d e zur Erfassung des akt iven Sauerstoffs ist besonders Fe ~+ zu nennen. Hierauf sell an sp/~terer Stelle ausffihrlich eingegangen werden. Die weiteren Ausffihrungen beziehen sich deshMb zuns auf die Beschreibung der
* Unte r ~ktlvem Sauerstoff soil die fiber die Zusammensetzung ~r vorliegende O-~r x im Br~unstein 5{rtO 1 + ~ verst&nden werden.

Zur analytischen Untersuchung yon Braunsteinen 17
Arbe i t en mi t P roben yon syn the t i schem Brauns te in , bei denen nur solehe F r e m d i o n e n (z. B. Spuren yon Na +, K +, Nl~a + ) i m Gi t te r e ingebau t sind, die keine Beeinflussung in ana ly t i scher t I in s i eh t a~siiben.
Die Bestimmung des aI~tiven Sauersto/]s mit der KJ-Methode
Vorauszuschicken is t zun~chst , dab n icht nur be im Arbe i t en mi t der KJ-Methode~ sondern ganz al lgemein bei al len Methoden es sehr wiin- schenswert ist , m i t e iner hohen AuflSsegesehwindigkei t zu arbei ten. Eine mSgl ichst feine Ver te ihlng der Brauns te inprobe un te r kr~f t igem Schfit- teln, dessen W i r k l m g durch Glasperlen un te r s t i i t z t wird, b r ing t eine e rs taunl ieh gute W i r k u n g - - aueh bei der Oxa la tmethode . Dies is t b ier u m so wiehtiger , als bei l~ngerer AuflSsezei t die s tSrende a~ toka ta ly t i s ehe Zerse tzung der Oxals~ure m e r k b a r das Ergebnis f~lschen kann, selbst wenn man, wie KATZ, CLARKE U. Nu 6 festgestel l t haben, un t e r CO 2- A tmosphe re arbe i te t . Der Vol ls t~ndigkei t ha lber sei erw~hnt , dab die Vorbehand lung und die Modif ikat ion des vor l iegenden Brauns te ins in die LSsegesehwindigkei t eingehen, ohne dal~ aus d iesem Z usa mme nha ng fiir die Ana lyse und S t r u k t u r p rak t i seh Nutzen gezogen werden k6nnte . Bei ungi ins t igen L6sungsvorausse tzungen a rbe i t e t man zweekm~Big mi~ E inwaagen zwischen 10--30 rag, wodureh bemerkenswer te Er le iehte- rungen erziel t werden. W i t haben auch aug diesem Grunde die Methode au f derar t ige Subs tanzmengen abgestel] t . Sie kann jedoeh aueh analog bei grSi~eren E inwaagen (etwa 200 rag) durehgeff ihr t werden.
ArbeitsvorsehriIt (vgl. KOr,T~OFFT). 10--30 nag Substanz werden auf • 0,05 mg abgewogen und in einen mit Schliff verseheaen 500 ml-Erlenmeyer-Kolben gebraeht, in den kurz zuvor etwa 10 G]asperlen (~ 1 mm), 70--80 ml Wasser, 20--30 ml 4 n Sehwefel- oder Phosphorsg, are und 4real je etwa 0,5 g NaKCO a unter besoaders gutem Umschiitteln gegeben wurden. Durch die letzte Mal~nahme soil der Sauer- stoffgeha]t im Kolbea hinreichend klein gehalten werden.
Man gibt dann 1--2 g K J hinzu und verschlie~t. Wenn man nicht bei Rotlieht arbeiten karm, amhtillt man den Kolben mit einem dichten sehwarzen Tuch und schiittelt bis zur LSsung der Probe. Dies ist je nach Probe in 1--5 rain erreicht. Anschliel~end titriert man sofort in iiblicher Weise mit 0,1 n Na2S2Os-LSsung gegen St~trke unter Verwendung einer Mikrobiirette. Beim Offnen des Kolbens ist Vorsicht geboten, da infolge des COe-Uberdruekes leicht Jodverluste dutch Mit- reiBen auftreten kSrmen. Wir benutzen desha]b gelegentIieh einen Aufsatz, bei dem des gebildete CO 2 regulierbar dureh eine mit Wasser geffillte Vorlage gdeite~ werden kann (G~raufsatz). Das Ablassen des lJberdruckes kann so auf 20--30 see verzSgert werden. Jodverluste werden vermieden.
Anmerkungen zur KJ-Methode. I~ach P$I~IL u. Ci~rALtK 9 soll bei der KJ- ]~{ethode zwischen Aeiditat and dem Fehler (gemeint ist der Absolutbetrag der Abweichung des gefundenen Wertes yon der Vorgabe) Korrelation bestehen, derart, dab mit s~eigender Aciditat der Fehler einsinnig negativ w~chs$.
Wir habea diesen ]~efund weder bei der Verwendung yon Schwefelsaure, noeh bei der Verwendung yon Phosphors~ure best~tigen k5nnen. Mit Essigs~ture finden wir zwar geringe negative Abweichungen, aber diese Abweiehungen weisen keinen Gang auf (vgl. Tab. 1). Ein Zusammenhang zwischen einsimfig negativer Abwei-
Z. analy~. Chem., Bd. 174 2

18 G. GATTOW und H.-G. WE~DLANDT:
chung und tier Aciditi~t ist jedenfalls nicht verstandlich, es sei denn, dab man Verunreinigungen in der S~ure annimmt, die sekund~tr einen Jodverlust bringen.
Wir k6nnen auch die Bemerkung yon P~mL u. Mitarb2 nicht best~tigen, dab man mit Phosphors~ure nicht arbeiten k6nne, da die Reduktion yon Mn02 im phosphorsauren Medium unvollst~ndig sei.
L~ Ubereinstimmung mit KOLWHOF~ ~ u.a. stellen wir im Gegenteil fest, dab man in phosphorsaurer LSsung einwandfreie Resultate erhMt (vgl. Tab. 1). Dieses Ergebnis verdient besondere Beachtung im Hinblick auf die Tarnung yon Fe a+ (s. S. 22).
Tabelle 1. Einflufi yon Sdureart und Sgurekonzentration au~ die l~eduktion yon .MnO~ +
(Einw~ge etwa 10--60 mg)
S~urekonzentrat ion
a) 10 ml 4 n Si~ure -~ 90 ml It20 b) 20 ml 4 n S~ure ~- 80 ml H20 c) 40 ml 4 n Si~ure ~- 60 ml It20 d) 60 ml 4 n S~ure -~ 40 ml H20
Prozent O bei Yerwendung yon
4 n tt.~SO4 4 n ttaPO4
15,68 15,67 15,65 15,65 15,66 15,68 15,65 15,66
4 n CHaCOOH
15,52 15,53 15,51
Erklgrung zur Tab. 1. Bezugsgr5Be ist der mit 20 ml 4 n Schwefels~ure gewonnene Wert, der auch nach der Oxalatmethode kontrolliert wurde, und der das Mittel aus 25 Einzelbestimmungen repr~sentiert. Der mittlere Fehler (~M betri~gt -+-0,025. Mit der 3 a-Regel ergibt sich das Interval1 15,57--15,73. Mit grSBter Wahrschein- lichkeit darf angenommen werden, dab bei Wiederholung der Messungen ein Ergeb- his gefundenwird, d as innerhalb dieses Intervalls liegt. Da andererseits ulle Ergebnisse der unter verschiedenen Bedingungen durchgeffihrten Versuche - - bis auf die unter Verwendung yon Essigs~ure durchgefiihrten - - in dem abgesteckten Spielraum liegen, daft n~herungsweise gefolgert werden, dab die Wasserstoffionenkonzen- tration unter den gegebenen und fiir die Anwendung des Verfahrens wichtigen ]~edingungen keinen EinfluB auf die Reduktion yon Braunstein hat.
~Tir haben besondere Versuche ausgeffihrt, um zu prfifen,ob der AusschluB yon Luftsauerstoff und Licht bei der beschriebenen jodometrischen Bestimmung tatsi~chlich notwendig ist 5.
Die ])urchffihrung der Versuche entsprach der oben gegebenen Arbeitsweise, wobei lediglich Braunsteinpr~parate fortgelassen wurden. Daffir wurde aber bei einer Reihe von Versuchen eine ~quivalente Menge Mn u+ vorgegeben, um einen etwaigen katalytischen EinfluB des Mn ~+ zu erfassen. Das Schtitteln der l%eaktions- gef~Be wurde ebenfalls wie bei der Analyse durchgeffihrt.
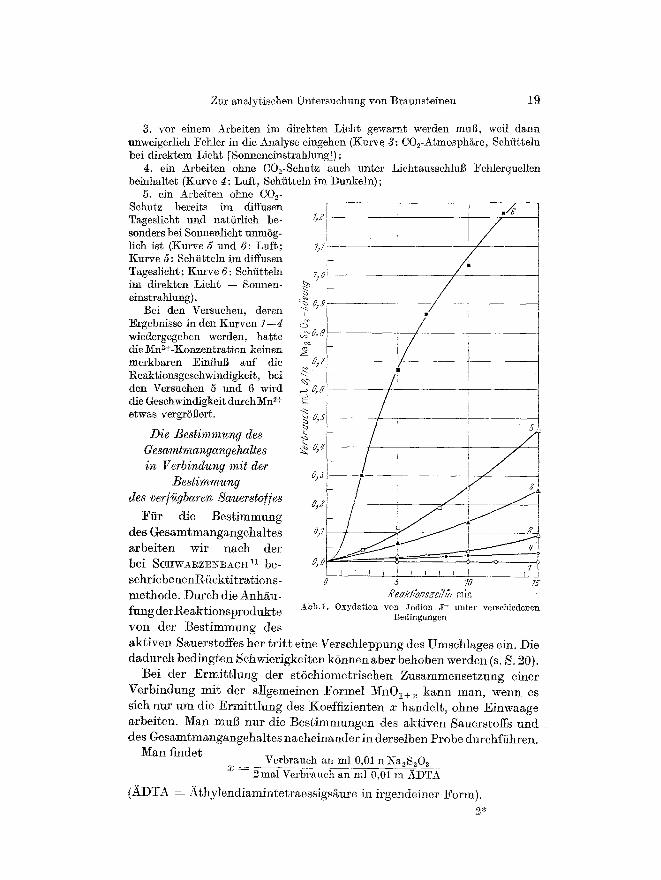
Abb. 1 zeigt die Geschwindigkeit des photochemisch begSnstigten Umsatzes yon J - unter den Bedimgungen der Analyse
hu 2 J- -+-H20-+- O ~ J 2 ~ - 2 O H - .
Man sieht, duB 1. bei Einhaltung der Vorschrift keine Bildung yon J2 als StSrreaktion m6glich
ist (Kurve 1: CO2-Atmosph~re, Schfittehl unter LichtausschluB); 2. bei Tageslicht (diffusem Licht) bei l~ngerem Schii~teln eine merkbure J2-
Bildung eintritt, was man damit erkl~ren kann, dab die einfache Verdr~ngung yon Luft dutch CO s natiirlich nicht so ausreichend sein kann, du~ jegliche Nebenreak- tion unterbunden wird (Kurve 2: CO.~-Atmosph~re, Schiitteln bei diffusem Tages- licht) ;
Zur analytischen Untersuchung yon Braunsteinen 19
3. vor einem Arbeiten im direkten Licht gewa.rnt werden muff, weil dann unweigerlich Fehler in die AnMyse eingehen (Kurve 3: C02-Atmosph&re, Schiitteln bei direktem Lieht [Solmeneinstrahlung]);
4. ein Arbeiten ohne C02-Schutz aueh unter LichtausschluB Fehlerquellen beinhalte$ (Xurve g: Luft, Sehfitteln im Dunkeln);
5. ein Arbeiten olme COe- Schutz bereits im diffusen Tageslicht und natiirlich be- sonders bei Sonnenlicht unmSg- lich ist (Kurve 5 und 6: Luft; Kurve 5: Sehiitteln im diffusen Tageslieht; Kurve 6: Sehfitteln im direkten Licht -- Sonnen- einstrahlung).
Bei den Versuehen, deren Ergebnisse in den Kurven 1--4 wiedergegeben werden, barge die Nm2+-Konzentration keinen merkbaren Einfluff auf die Reaktionsgeschwindigkeit, bei den Versuchen 5 und 6 wird die Gesehwindigkeit durchMn ~+ etwas vergrSBel~.
Die Bestimmung des Gesamtmangangehaltes in Verbindung mit der
Bestimmung des ve~']iigbaren Sauersto/]es
Fi i r die Bes t immung des Gesamtmangangeha l t e s a rbe i t en wir nach der b e i SCJ~WARZENtI&CI-[ 11 be- sch r i ebeneng / i ck t i t r a t ions - methode . Dureh die Anhgu- lung d e r R e a k t i o n s p r o d u k t e yon der Bes t immung des
L
;J <- L / '
k / -
. o / ! /
k / ': . / /-+
0 5- 7O 15 Rea/(f/'o/Tsx#/'/~ m~n
Abb.1. Oxydation yon Jodion 5- unter versehiedet~en Bedingungen
ak t i ven Sauerstoffes her t r i t t eine Versehleppnng des Umsehlages ein. Die dadureh beding~en Schwier igkei ten k6nnen a b e t behoben werden (s. S. 20).
Bei der E r m i t t l u n g der s tSehiometr i sehen Zusammense tzung einer Verb indung mi t der a l lgemeinen F o r m e l Mn01+ . k a n n man, wenn es sich nur u m die E r m i t t l u n g des Koeff iz ienten x ha.ndelt, ohne Einwaage arbei ten . ~{an muff n u t die Bes t immungen des ak t iven Sauerstoffs und des GesamtmangangehMtes naehe inander in derselben Probe dnrchffihren.
Man f indet Verbraueh an ml 0,01 n Na2S20~
X ~ - 2 real Verbraueh an ml 0,01 in ]4DTA
(•DTA = 2&thylendiamintetra.essigsgure in i rgendeiner Form) .
2*

20 G. GATTOW und tt.-G. WENDLANDT:
D e r Vor t e i l des A r b e i t e n s ohne E i n w a a g e l i eg t n i ch t n u r in de r E i n -
s p a r u n g y o n Arbe i t s ze i t , s onde rn - - was w e s e n t l i e h e r se in d / i r f te - - m a n
g e w i n n t e inen h 6 h e r e n G r a d an G e n a u i g k e i t , da m a n die m i t d e n E i n -
w a a g e n v e r b u n d e n e n F e h l e r m S g l i e h k e i t e n , i n sbesonde re b e i m A r b e i t e n i m H a l b m i k r o m a B s t a b , aus seha l t e t .
Arbeitsvorschrift . Es wird yon einer LSsuag ausgegaagea, wie sie naeh der Titrat ion des Jods mit Thiosulfat bei der Bes~immung des aktiven Sauerstoffs naeh der KJ-Nethode vorliegt (Substanzmenge 10--20 mg).
Man versetzt die LSsung mit etwa 0,1--0,2 g Itydi-oxylammoniumchlorid und erw~rmt kurz bis auf etwa 80~ Darm gibt man soviel 0,0t m )kDTA-LSsung hinzu, daft eia etwa 5--10~ l~berschuB vorhanden ist. Uater gutem Schtitteln wird mit ~ n Natronlauge neutralisiert. Bei riehtiger Durchfiihruag wird die Probe nut ~uBerst schwaeh gelb verfSrbt. Bei unsaehgem~gem Arbeiten kSnaea dunkel- braune F~irbungen auftreten, iasbesondere, wean man zu weir ins alkalisehe Milieu hineingekommen ist. Man sSuert dam1 wieder mit einigen Tropfen Sehwefels~ure an..
Die neutralisierte LSsung wird mit etwa 100--150 ml Wasser verdfinnt. Man f/igg naeh Vorsehrift NHa-NH~CI-Puffer (p~ ~ 10) hinzu und ti~riert den JkDTA- t3bersehug mit eiaer Zinkl6sung unter Verwendtmg einer Mikrobtirette gegen Erioehromsehwarz T zuriiek, d.h. man gitriert yon Blau auf l~ot. Als Endpunkt w/~hlt man zweekm/~13igerweise nieht den ers~en Bereich des Umschlagsintervalls, soadern man nimmt den Bereich, in dem der Indicator sichtbar ins Rote nmschl/~gt, da dieser Punkt im allgemeinea bei den Bedinguagen der vorliegenden Analyse sch~rfer erkennbar ist.
Die Titerstellung der ZinksulfatISsung nimmt man unter den Bedingungen der tIauptanalyse vor, indem man etwa zu einer eben austitrierten Probe eraeut 20 ml einer eingestellten 0,01 m ADTA-LSsung zugibt und dan~ mit der unbekannten ZiaklSsung auf den beschriebeaen Endpunkt zur Ermit t lung des Titers der Zink- 15sung zuriiektitriert. Naeh derselbert Methode 1/~gt sieh im tibrigen -- werm der Titer der ZinklSsung erst eimnal festgestellt ist -- die Sch/~rfe des Umschlagpunktes jeder weiteren Titration leieht koatrollieren, indem man beispielsweise naeh Been- diguag der Titration wieder 10 ml~DTA-LSsung vorlegt und mit Zinksulfat zuriick- titriert. Dieses Koutrollverfahren empfiehlt sieh besonders durra, wenn bei der vor- ausgegangenen Sauerstoffbestimmung stark abweichende Bedingungen vorgelegen haben. Man sollte dana den I)bersehug an JkDTA jeweils mit dem den Bedingungen des Systems entspreehenden Zinkfaktor zuriiekreehnea, den man leicht aus den Koatrollversuchen ermittelt.
Es hat sieh als zweekm~gig herausgestellt, die Zinkl6sung etwa doppelt so stark einzustellen als die )kDTA-LSsung, da man so einen sch/~rferen Umschlag erzie]t. Fiir die Verringerung des Einflusses des unvermeidbarea Titerfehlers ist es wesent- lich, dab nur ein kleiaer Ubersehul3 bei der erstert Zugabe an ~ D T A genommen wird. Je kleiaer dieser r des~o geringer der EirdtuI~ des bei der Erfassung dieses ~bersehusses auftreteadea FehIers.
Sollte der Umschlagspunkt sehwer zu erkennen sein, was z.B. darm tier Fall sein kann, wenn man mehr als 5-- 10 mg Nn -~+ zur Titration bringt, dann empfiehlt es sieh, die Probe naeh tier Neutralisation mit Natronlauge zu teilen, die Teile, deren Volumen man nicht zu kenaen braueht, getrermt zu verdiinuen, mit Puffer zu versetzen und in der ~ngegebenen Weise einzeln zu titrieren. Man titriert den zweiten Teil der Probe sofort auf die Umsehlagsfarbe des ersteu Teils der Probe; hierdureh wird dem Einflug eines mSgliehea Titrierfehlers wesentlieh entgegengewirkt. Diese visuelle Itilfe empfiehl~ sieh aueh bei der oben besehriebenen Titration und Titer- stellung.

Zur analytischen Untersuchung yon Braunsteinen 21
Legt man Wert auf den Wassergehalt der Probe, dann geht man yon einer Einwaage aus. Man errechnet den MnO- und O-Gehalt der Probe in Prozenten aus, der Wassergehalt ist dann die Differenz der Summe der Prozente zu ttundert*.
Beleganalysen
Das beschriebene Verfahren wurde an fiber 200 synthetischen Braunsteinproben verschiedener Darstellung erprobt. Vergleichsuntersuchungen zu glteren iMethoden der Untersuchung auf st0chiometrische Zusammensetzung (vgl. z.B.4), bei der yon einer Probe der Sauerstoffgehalt nach der OxaIatmethode und von einer weiteren Probe der Gesamtm~nga.ngeh~lt n~ch der Pyrophosphatmethode bestimmt wurde, ergaben eine sehr gute ~bereinstimmung, wie die Zahlen in Tab. 2 zeigen.
Eine weitere Bestgtigung der l~ichtigkeit der mit der neuen Methode erhaltenen Werte ergab sich durch den Vergleich dieser Werte mit den Ergebnissen struktur- analytischer Untersuchungen a. So wurde yon mehreren Proben Mn20 s (= MnOl+~ mit x ~ 0,500) folgende stSchiome~risehe Zusammensetzung 1 -k x ermittelt: Probe a 1,503; Probe b 1,502; Probe c J[,500; Probe d 1,499; Probe e 1,500.
Ffir verschiedene Proben MnsO ~ (= MnOl+~ mit x = 0,333) wurde foIgende Zusammensetzung bestimmt: Probe / 1,334; Probe g 1,333; Probe h 1,335; Probe i 1,333.
Fehlerbreite des Verfahrens. Nach unseren Versuchen betr/igt der mit t - lere Fehler des Mittelwertes ~_ 0,0021. Un te r Einbeziehung der 3~-Grenze
Tabelle 2. Bestimmung der stSchio- metrischen Zusammensetzung 1 q- x
verschiedener Braunsteinproben Mn01 +
werte yon (lq-x) t efunden
a) nach der Oxalat- b) nach dem in methode und Pyrophos- dieser Arbeit
phatme~hode beschriebenel~ Verfahren
Probe 1 1,932 1,936
Probe 2 1,914 1,914
Probe 3 1,874= 1,870
Probe 4= 1,887 1,889
Probe 5 1,84=1 1,836
k a n n m a n deshalb sagen, dab es die Methode gestat tet ,
1,934 1,935 1,919 1,915 1,868 1,871 1,885 1,883 1,838 1,841
den Ko- effizienten x auf • 0,007 sicher zu bes t immen (A x/x ~ 0~3 bis :~0,4~ ).
I m al lgemeinen wird diese Schranke ausreiehend sein. Eine darfiber- h inausgehende Genauigkei t zu verlangen, scheint auch in Anbe t rach t der sonstigen Fehlerm6glichkei ten n icht gerechtfertigt.
Die analyt ische Fehlerbet rachtung, bei der wir un te r Auswer tung der bei den Ti t ra t ionen gewonnenen Er fahrungen vorgingen, zeigte, dab
beim Arbei ten ohne Einwaage ebenfalls eine der obigen Angabe ent- sprechende Sch~rfe erwar te t werden darf. Aus der Feh]erbe t rachtung ist yon Bedeutung, daI3 ein Fehler bei der Bes t immung des Sauer- stoffs mi t etwa 4real gr6Berem Gewicht eingeht als ein Fehler bei der 5~anganbes~imlnnng. Dies war ffir ires AnlaB, sich in der beschrie- benen Weise nochmals eingehend mi t der Sauers toffbes t immung zu befassen.
* Dieses gilt natiir]ich nur, wmm der Fremdionengehalt versehwindend klein is{.

22 GATTOW und W E N ' D L A l g D T : Zur analytisehen Untersuchung yon Braunsteinen
Anwendung der Methode auf teehnisehen Braunstein
Sofern in dem Braunstein keine st6renden Kationen enthalten sind, ist die Methode ohne weiteres anwendbar. St6rend wirken bei der K J - Bestimmung alle mehrwertigen Kationen, die unter den angegebenen Bedingungen mit Jodid reagieren, und selbstversti~ndlieh bei der Mangan- best immung alle diejenigen, die mit J~DTA Umsetzungen ergeben. Bei der KJ-Methode dfirften im wesentlichen Fea+-Ionen Ms Gitterbestand- teile des Braunsteins ftir St6rungen in Frage kommen. Aber auch f/Jr die Manganbestimmung ist Eisen unangenehm, da sieh nut kleinste Mengen dutch bekannte Tarnungsmittel unseh~dlieh machen lassen. Die St6run- gen bei der KJ-Methode dutch Eisen lassen sieh dutch Anwendung van Phosphors~m'e umgehen; wie Tab. 3 zeigt, ist es m6glich, Fe a+ bei den teehnisch in Frage kommenden EisengehMten des Braunsteins einwand- frei zu maskieren.
Tabelle 3. Vorgelegt: 5 mg ~e~+; 0,4 g K J + 2 g NaHCO~, 5 rain Schii~teln x ml 4 n Si~ure -~ y ml H~O; Verbraueh: ml 0,1 n Na2S203-LSsung
y
10 90 20 80 40 60 60 40
Da die Titrat ion
4 n H~804
0,03 0,06 0,1t 0,17
mit ~iDTA bei
4 n II~P04 4 n CKaOOH,
0 0 0 0 0 0 0 0
Gegenwart yon Phosphors~ure unschiirfer als bei Anwendung yon Schwefels/ture verli~uft, empfiehlt es sich, in solchen F/tllen das Mangan aus saurem Medium (HNOa oder H2SO~) dutch 0xydat ion mit festem N a B r 0 3 in der Sied~hitze aus- zufs fiber eine Glasfritte (G 4) abzunutschen, auszuwasehen, mit Salzs~ure aufznl6sen und dann nach Verkochen des Chlors und Neu- tralisation in bekannter Weise mit XDTA zu titrieren. Mit dieser Methode hat man auch gleichzeitig die Behinderung der XDTA-Titrat ion dutch Fremdkat ionen umgangen, da die Ausffi.llung des M~ngans unter den gegebenen Bedingungen spezifisch ist.
Zusammenfassung
Es wird eine, dureh. Kombinat ion zweier bekannter, auf jodometrischer und ehel~tometrischer Grundlage stehender Einzelverfahren entwiekelte Methode besehrieben, mit der es im I-Ialbmikrom~i]stab m6glieh ist, die st6ehiometrische Zusammensetzung einer Brannstein-Probe in etwa 10 rain, unter Verzieht auf jegliehe Einwaage, mit einer Genauigkeit yon •176 zu bestimmen. Die Methode wird im Hinblick auf die Unter- suehung natfirlieher und synthetiseher Braunsteine diskutiert.
Herrn Professor Dr. O. GL~MS~R gilt unser Dank fiir wertvolle Diskussion. Der I)eutschen Forschungsgemeinsehaf~ danken wir fiir d~e zur Verfiigung gestellten fIilfsmittel.

SACKINDRA KUMAR DATTA: Organic Reagents in Inorganic Analysis. X 23
Literatur 1 Analyse der Metalle, Bd. I Schiedsverfahren, Bd. II Betriebsanalysen, Berlin-
G6ttingen-Heidelberg: Springer 1953. -- 2 B~RL-LvNG]~: Chemisch-teehnische Unter- suchungsmethoden, 8. Anti., Bd. II/1, S. 784ff. -- 3 GLEMSER, O., U. G. GATTOW: Z. anorg, allg. Chem., im Druck. -- 4 G ~ I s ] ~ , 0., u. g . M]~ISIEK : J. prakt. Chem. (4) 5,219 (1958). -- 5 Vgl. z. B. Gmelins Handbuch der anorganischen Chemic, System Nr.8 (Jod), Berlin: V erlag Chemie, 1933, S. 324-- 334. -- 6 KATZ, M. J., R. C. C5~KE u. W. F. lkTu Analyt. Chemistry 28,507 (1956) ; vgl. diese Z. 155, 63 (1957). -- 7 XO~THOF~, I. M. : Die Praxis der MaBanalyse, Tell II. Berlin: Springer 1931. -- s Y~]~JzovA, V., V. Snv~o~ u. J. Z:~]~A: Collect. czeehoslov, chem. Commun. 24, 293 (1959); vgl. diese Z. 165, 291 (1959). -- 9 Pf~I]~IL, R., u. J. C~ALI~: Collect. ezechoslov, chem. Commun. 20, 562 (1955); vgl. diese Z. 148, 220 (1955/56). -- 10 RurP, E.: Arch. Pharmaz. Ber. dtseh, pharmaz. Ges. 254, 135 (1916); vgl. diese Z. 56, 114 (1917); vgl. auch BmTz-BI~,Tz: Ausfiihrung quantitativer Analysen, III. Auti., Leipzig 1940, S. 213. -- n SC~W.~ZEN~Aeg, G. : Diekomplexometrische Titration. Stuttgart: F. Enke-Verlag 1955.
Dr. G. GA~TOW, Anorganisch-chemisches Institut der Universitat GSttingen, ttospitalstr. 8--10
Victoria (Government) College, Cooehbehar, West Bengal, India
Use of Organic Reagents in Inorganic Analysis Part X*
Determination of Thorium with Maleanilic Acids
By SACHINDRA KUlVIAR ])ATTA
With 1 Figure in the Text
(Received May 19, 1959)
Maleanilie acids are found to produce somewhat selective precipi ta t ion of tho r ium and zirconium, which are more or less quan t i t a t ive ~. The present paper describes the appl icat ion of these acids in the analysis of tho r ium gravimetrical ly. These maleanil ie acids have been prepared from anil ine, its chloro and n i t ro derivatives, toluidines, anisidines, naphthy lamines , m-phenylened iamine and tol idine. Among these, the acids obta ined from 2,5-dichloroaniline, o- and p-toluidines, o- and p-anisidines, 1- and 2-naphthylamines , o-tolidine and m-phenylenedi- amine proved useful for the de te rmina t ion of thor ium.
Experimental Reagents and Apparatus
Reagents. 1% solution of the reagents in 500/0 alcohol were prepared with the aid of heat.
Thorium and other Solutions. Stock solutions of pure thorium, cerium, and lan- thanum nitrates (reagent grade) were prepared and their earth contents determined
* Part IX: See Z. analyt. Chem. 168, 418 (1959).