Embed Size (px)
Citation preview

-- 76% -
Zur Kenntnis des Pinens 11. Versuche zur Herstellung der Homopinocampher- saure aus Pinonsaure. Uberfiihrung der Pinonsaure
in Tetrahydrocarvon vo11
L. Ruzieka und H. Trebler. (4. x. 20 I
Ein Korper vom Typus des Pinens konnte bisher weder aus monocyclischen noch anderen bicyclischen Verbindungen ge- wonnen werden. Es sind nur zwei Versuche in dieser Richtung bekannt geworden. Nach W. H. €'erkinjzml) entsteht bei der Brom- wasserstoffabspaltung aus dem Hydrobroniid I kein Nopinon (11).
co
. . I CH
I . 11.
(CEl&CUr
0. Starka) behauptet zwar, dass bei der Destillation des Calcium- salzes der Hexahydro-isophtalsaure ein Keton der Formel I11 ent- stehen solle, die von ihm beschriebenen Eigenschaften desselben sprechen jedoch fur das Vorliegen des isomeren inonocyclischeii l-Metliyl-3-cyclohexen-2-ons (IV).
1) Soc. 91, 1736 11907;. 2) 13. 45, 2369 (1912); schon die hci der hIolekularrefraktion gefunderie
Exaltation von 2,9 Einheiten ist sogar fur eiri ungesattigtes Keton reichlich und hei einem hicjclischen nach a 11 e 11 Erfahrungen ausgeschlossen. Eine rnomentarie Uromaufnnhme, wenn auch unter Uromwasserstoffentwickclung, be- obachtet man auch bei andern ungesattigten Ketooen. Der angegebene Snip. des Seinicarbazons 1780 stirnmt iiherein mit dem des Ketons IT. (s. Kotz und Steinhors~, A . 379, 10 119111).
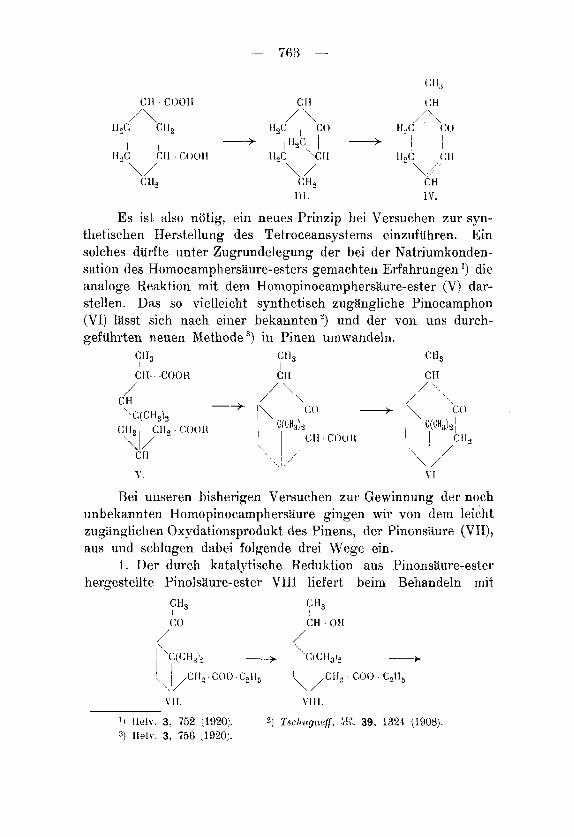
- 763 -
/\ I f & ; A\ CtI2 E!2c’i‘\c( J F 1 , C ( ’ ( J
1 ~~sc, 1 I i H,C ‘Cll II,C ( : H 1f2C CI1 COO11
\ / ’ \/ C:H1
I l l . I v. Es ist also niitig, ein neues Prinzip bei Versuchen zur syn-
thetischen Herstellung des Tetroceansystems einzufuhren. Ein solches durfte unter Zugrundelegung der bei der Natriumkonden- sation des HomocamphersBure-esters gemachten Erfahrungen ’) die analoge Reaktion mit dem Honiopinocamphersaure-ester (V) dar- stellen. Das so vielleicht synthetisch zugangliche Pinocainphon (VI) lasst sich nach einer bekannten2) und der von uns durch- gefuhrten neuen hlethode ’) in Pinen umwandeln.
CI1, Ctl, c H, I I I C H-C( )O R CH C H
/ \
1 1
\/’ C H c 11,
\ /’ / \
/
-+ ,( (:o + [\ co CH I \ \ ( :(CH,), CH, CH, COCII{ I C(CH,),I I(bH,& 1
c:li COC)I< 1 I Ctl, \ I /
\ I / ’ \ I / CII I\ I ’ /’ 1 V l
Bei unberen hisherigen Versuchen zur Gewinnung der noch unbekannten Homopinocampherslure gingen wir von dem leicht zugangliehen Oxydatioiisprodukt des Pinens, der Pinonsaure (VII), aus und schlugen dabei folgende drei Wege ein.
1. Der durch katalytische Reduktion aus Pinomlure-ester Iiergestellte Pinolsaure-ester VIIl liefert heim Behandeln mit
CH, I (:o
C H, I CH . Otl
11 Helv. 3. 752 (1‘320;. 21 Tseftugnrtf, ih‘. 39, 1324 (1908}. 3) H e l v . 3, 756 (1920;.

- 764 -
Cli, I C
CH, CHCl
1x. s Pliosphorpentachlorid hauptsachlich Pinocampholensaure-ester (For- me1 1X, wenn die noch nicht ganz sichergestellte Ansicht von Tiernnnn und Kerschbaum ') uber die Konstitution dieser Saure richtig ist) und nur wenig des gewunschten Chloresters X. Dieser sollte durch Umsetzm mit Kaliumcyanid und nachheriges Ver- seifen in Hornopinocamphersaure umgewandelt werden. Aber weder in den Chlorester noch in den analogen Jodester war es moglich, die Cyangruppe in nennenswerter Ausbeute einzufuhren.
2. Aus dem Cyanhydrin der Pinonsaure (XI) wollten wir durch Phosphorpentachlorid nach der ublichen Reaktion der a-Oxy- saurenitrile die Dehydro-homopinocampher-nitrilsaure XI1 her- stellen; die nebenbei entstehende Chlor-homopinocampher-nitril-
CH, &JH
/ 'CN t K \
i:(Cl13)2 __f
XI
CH3 I
C ,,I \
CH COO . C,H,
CH, , I c
xIr
CH, I i: // \
CH C N
C"3
C H CH,
/\ I
C li
s I \'. X V . CH(CH,), xv1.
I) I$. 33, 2668 (19001. lrn %usammenIinng rn i t den Ergebnissen unserer Methotie '2 ~ 1 ~ 1 1 die lii~nstitiition der I'iiiocam~~lioleiisBure nBher untersucht wetden.

- 765 -
saure (analog der Formel XI) sollte durch Chlorwasserstoffab- spaltung in die gleiche Dehydrosaure XI1 ubergefuhrt werden. Wir hofften, dass bei der Wasser- bezw. Chlorwasserstoff-abspaltung hier die Umlagerung in den Cyclopentenring, wie sie heim Uber- gang der Pinolsaure in die Pinocampholensaure (VIII+IX) viel- leicht stattfindet, nicht eintreten wird, da bei der ungesattigten Saure in unserem Falle die der Pinocampholensaure analoge Lage der Doppelbindung nicht moglich ist oder aber verschoben werden musste.
Es erwies sich jedoch, dass der Dehydronitrilsaure die Konsti- tution einer aliphatischen Verbindung entsprechend der Formel XI11 zukommen muss, da die bei ihrer Verseifung entstehende Dicarbonsaure nach den Ergebnissen der katalytischen Reduktion zwei Doppelbindungen besitzt und aus der 2-Carboxyl-6-methyl- 2,6-heptadien-5-essigsaure (nach Formel XIV) oder ihren durch Verschiebung der 6-Doppelbindung resultierenden Isomeren be- steht '). Die gesattigte Dicarbonsaure murde als n-Methyl-P'-iso- propylpimelinsaure (XV) erkannt, denn sie liefert beim Ringschluss durch Destillation ihres Bleisalzes sowie diirch Kondensation des Esters mit Natrium als alleiniges Produkt Tetrahydrocarvon (XVI).
Eine derartige IJrnlagerung (XI+XIII) ist hei Pinolsaure- derivaten noch nicht beobachtet ; man kann sie der Bildung von a-Terpineol (XVIII) RUS Methylnopiiiol (XVII) an die Seite stellen').
CII, I
// \.
)" i \ /
'I'
HO . (:(CHJZ
S~II . S V I I I . XIX.
Es scheint danach, sowie hauptsachlich nach den Untersuchungen Wallnchs '), dass Pinen- und Pinonsiiurederivate mit einer am Vierring ,,semicyclisch" sitzenden Doppelbindung kaum bestandig sind und sich unter Ringumlagerung oder -aufspaltung stabili-
1) u h e r die Biltlung von Carvacrol aus diesel Saure vergl. Erp. Teil, S 778. a) Wullach, A . 356, 239 (1907); 360, 88 (1908). 3) Terpene uiicl Citinpher, Leipzig (1909) S. 253.

sieren. Eine Ausnahme wurde nur die Carvopinon- (XIX) oder Nitrosopinengruppe darstellen, deren Konstitution l) allerdings nicht sicher nachgewiesen ist. Fur die Versuche zur Gewinnung der Homopinocamphershre ergibt sich so immerhin die Richtlinie, Methoden zu vermeiden, wo eine solche Doppelbindung entstehen konnte.
3. Es wurde demzufolge versucht, Derivate der Pinonsaure zu gewinnen, die sich in die Homopinocampher-aldehydsaure (XX) iiberfuhren liessen, woraus dann durch Oxydation die Dicarbon- slure (V) zuganglich sein sollte. Aus Pinonssureester und Chlor-
CFI, C"3 CH, I I CH C-CH, OCH, C ~ C H COO. CzlIs
/' \crro / \ O H / \(j/ I\C(CFT,Iz "C(C ",I, I\C~C€l3),
\~/ CH, COO11 \/,CH, COO C,U, ( 1 / C H 2 COO . C.&
XXI XXII . xx methylather mit Zink2) konnte das Produkt XXI, das in diesem Sinne brauchbar ware, nicht erhalten werden. Dagegen liefert die Einwirkung von Chloressigester auf Pinonsiiureester in Gegen- wart von Natriurnamid ') den Glycidester der Formel XXII. Uber die Weiterverarbeitung dieses Esters sowie uber andersartige Dar- stellungsversuche der Homopinocamphersaure sol1 spater berichtet werden.
E x p e r ime n t e 11 e r T e il. Uber die Qewinnung dele r-Pinonsuure.
Zur Verwendung fur unsere Versuche kam nur die r-Pinon- saure, die wir irn wesentlichen nach der Methode von Bucyer4) herstellten. 600 gr Pinen vom Sdp. 155-156' (730 mm) wurden in 4 Liter Wasser unter Riihren mit der Turbine suspendiert und wahrend sechs Stunden eine Losung von 1400 gr Kaliumperman- ganat in 16 Liter Wasser zulaufen lassen, wobei die Temperatur
1) Vergl. die beiden vorhergehenden Fussnoten, sowie TValZacl~, A. 389,
2) Methode von Be'haZ, D. R. 1'. 180 202 (C 1907, I, 680). 3) Methode von Claisen, 13. 38, 693 (1905). 4 ) U. 29, 22 (1896).
185 (1912).

der Losung ohne Aussenkuhlung auf ca. 30' stieg. Es wurde iiber Nacht weiter geruhrt, dann durch Methylalkoholzusatz das noch vorhandene Kaliumpermanganat zerstort, vom Braunstein abfil- triert, der grosste Teil des Alkalis durch konzentrierte Salzsaure abgestumpft und am Dampfbade auf ca. vier Liter eingedampft. Nach dem Erkalten wurde von den ahgeschiedenen anorganischen Salzen abfiltriert ; ein Ausschutteln mit Ather von neutralen Pro- dukten, wie es Bneyel- empfiehlt, erwies sich als unnotig, da bei dieser Arbeitsweise fast keine entstehen. Nach dem Ansauern mit konzentrierter Salzsaure wurde die Hauptmenge der sauren Oxy- dationsprodukte durch eiiimaliges Ausschutteln mit Ather gewonnen und der Rest durch Extrahieren im Extraktionsapparate. Durch Einstellen der Rohsaure wahrend einer Woche (oder womoglich langer) in den Eisschrank und Abnutschen der Krystalle wird ein grosser Teil der vorhandenen r-Piuonsaure gewonnen, deren Menge bekanntlich je nach der Drehung des Pinens stark variieit. Nach einmaligem Umkrystallisieren aus Wasser ist die SLure ge- nugend rein fur die Weiterverarbeitung (Smp. ca. 103'). Urn aus dem fliissigen Hauptanteile der Rohsaure den Rest der Pinon- saure moglichst vollstandig zu gewinnen, ist es von Vorteil, nach Clem Entfernen des vorhandenen Wassers im Vakuum (bis looo), das Sauregemisch durch alkoholische Salzsaure zu verestern, am hesten nach der unten beschriebenen Methode. Die Destillation der Rohsaure (auch im abs. Vakuum) ist wegen hiebei eintretender starker Zersetzung zu verlustreich; ebenso ist auch die weitere Aufarbeitung nach Baeycr wenig empfehlenswert, da die gunstig- sten Bedingungen dabei nicht leicht einzuhalten sind. Durch Mischen der notigen Mengen 1-Pinonsaure-ester (aus franzosischem Terpentinol) und d-Pinonsaure-ester (aus amerikanischem 01) kann der r-Pinonslure-ester hergestellt werden und daraus lasst sich, wenn notig, durch Verseifung rnit alkoholischer Kalilauge die r-Pinonsaure gewjnnen.
Dnrstellung des Pinonsii.ure-uthylesters (VII, .
Nach Kneyer I ) entsteht aus der Pinonsaure heim Krhitzen rnit 50 proz. Schwefelsaure selir leicht das Ketolacton XXIII.

- 768 -
CB, I co
1 C(CII,\,-0
C[*, 1 >CO \dH-CH, XXIII.
Urn die Bedingungen ausfindig zu machen, bei denen sich Pinonsaure durch saure Agentien verestern lasst l), ohne diese Umlagerung auch nur teilweise befurchten zu mussen, erhitzten wir r-Pinonsaure mit 20 proz. Salzsaure einige Stunden am Wasser- bade und verdampften nachher vollstandig. Der Ruckstand er- starrt und besteht nach dem Smp. von 63' aus obigem Ketolacton. Auch beim Kochen von Pinonsaure mit gesattigter alkoholischer Salzsaure entsteht wenigstens teilweise das Lacton. Es zeigte sich dann, dass Pinonsiiure unter Einhaltung der fur die Herstellung des Llvulinesters ') ausgearbeiteten Bedingungen schon durch ganz schwache alkoholische Salzsaure glatt verestert werden kann.
60 gr Pinonslure wurden in 200 cm" absoluteni Alkohol ge- lost und rriit 20 cn13 gesattigter alkoholischer Salzsaure 'acht Stun- den gekocht, die Kalfte des Alkohols am Wasserbade abdestilliert und der Rest im Vakuum fast vollstandig abgesaugt. Nach dem Versetzen rnit Sodalosung wurde rnit Ather ausgeschuttelt und destilliert, wobei etwa 60 gr Athylester vom Sdp. ca. 100' ('1, mm) erhalten werden. Durch Einwirkung von Semicarbazid-acetat kann daraus das nach dem Umkrystallisieren aus wenig Alkohol bei 136' schmelzende Semicarbazon gewonnen werden.
0,1282 gr Subst. gaben 0,2726 gr CO, und 0,1027 gr H,O
C,,IJ,,O,N, Uer. C 57,95 13 8,590/0 Gef. ,, 58,Ol ,, 8,76 '10
Zum Vergleich wurde ein Pinonsaure-athylester hei Abwesen- lieit von anorganischer Slure hergestellt. 1,3 gr Pinonsaure wurde mit einer Losung von 0,3 gr Natrium in 5 cm3 absolutern Al- kohol und 2,5 gr khyljodid 5 Stunden gekocht. Der wie iiblich isolierte Ester lieferte ein nach Schmelzpunkt und Mischprobe rnit obigem identisches Semicarbazon.
l) Prvkin und Simonsen, SOC. 95, 1174 (1909) (C. 1909, 11. 803) stellten den Ester durch alkoholische Schwefelsaure her ohue Angahe dee Konzentration und Ausbeute. 2) Ruzicka, B. 50, 1366 (1917).

769 - -
Darstellung von Pinolsaure und ~ii2obuure-tithyleste7 (VIIQ.
Die Vorschrift von Tiemanii und iS'emmlel- ') zur Gewinnung dieser Slure durch Erhitzen von Pinonsaure rnit konzentriertern alkoholischen Kali (wir nahmen auf 16 gr PinonsLure 20 gr Atz- kali und 40 cm3 Alkohol) im Bombenrohr auf 180' ist fiir die Gewinnung grosserer Mengen Pinolsaure umstindlich; zudem betragt die Ausbeute nur hochstens 5Oo/o und ist auch ungewiss infolge der oft eintretenden merkwiirdigen Bildung der Pinocampholen- siiure (IX) bei der Destillation der Pinolsaure im Vakuum. Auch im absoluten Vakuum blieb uns die Halfte der Rohsaure im Des- tillierkolben als zahes Harz zuriick ; daneben erhielten wir imrner einen Vorlauf vom Sdp. 100-120' ( ' i s inni), bestehend aus un- reiner Pinocampholensaure ; die bei ca. 150' siedende Pinolsaure krystallisierte auch bei Yangerem Stehen nicht, w'slhrend Tiernann wenigstens teilweise Krystallisation erzielte. Durch den negativen Ausfall der Semicarbazonprobe uberzeugten wir uns von der Ab- wesenheit nennenswerter Mengen Pinonsilure.
Wir suchten daher eine ergiebigere Reduktionsmethode fur die Gewinnung der Pinolsaure ausfindig zu machen.
a) Natr iumamalgarn wirkt auf Pinonsaure in wbsriger Losung nach der fur die Reduktion der Lavulinsaure brauchbaren Vorschrift2) nur sehr langsam ein und in der so gewonnenen Pinolsaure war immer noch Pinonsaure durch Semicarbazid nach- weisbar.
b) M i t Nat r ium und Alkohol. 40 gr Pinonsaure wurden mit 100 cm3 absolutem Alkohol versetzt, 43 gr Natrium in grossen Stucken zugegeben und unter Erwarmen am Wasserbade im Laufe von zwei Stunden noch 450 cm3 Alkohol zugetropft. Nach der Auflosung des Natriums wurde der Alkohol rnit Wasserdampf abgeblasen, im Ruckstand unter Eiskuhlung mit konzentrierter Salzsaure die Natronlauge grosstenteils abgestumpft und die noch schwach alkalische Losung am Dampfbade konzentriert. Nach vorsichtigem Ansauern rnit starker Salzsaure wurde mit Ather ausgeschuttelt. Das im Vakuum scharf getrocknete zghe 0 1 kry- stallisierte nicht, auch konnte durch Behandeln rnit Losungsmit-
l) B. 30, 409 (1897); siehe auch B. 33, 2661 (1900). 2) H. E~*dmann, Anleituns zur Darst. orsan. Praparate, Stuttgart, S. 310
(1904). 4!)

teln keine Krystallisation erzielt werden; die Semicarbazonprobe war gleichfalls negativ. Die Destillation im absoluten Vakuum verlief mit annahernd gleichem Ergebnis wie bei der nach Tie- m a w hergestellten Pinolsgure.
Zur Charakterisierung der nicht krystallisierenden destillierten Pinolsaure wurde eine kleine Probe rnit der berechneten Menge Kaliumpermanganat in wassriger Losung zur Pinonsaure oxydiert. Das beim Aufarbeiten erhaltene saure 61 erstarrte nicht, lieferte aber reichlich das bei 205' schmelzende Pinonsaure-semicarbazon (Mischprobe !).
Wird die rohe Pinolsgure nicht destilliert, sondern in den Ester ubergefuhrt, so erhalt man eine regelmassige, vom Zufall unabhlngige Ausbeute von ca. 60 Prozent Pinolsaureester, da sich letzterer beim Destillieren im absoluten Vakuum nicht zer- setzt. Zur konzentrierten alkoholischen Losung der Saure wurde ein kleiner uberschuss von Natriumathylatlosung zugesetzt und mit Athyljodid acht Stunden auf 190' im Bombenrohr erhitzt. Nach dem Entfernen des Alkohols und Versetzen mit Sodalosung wurde mit Ather ausgeschuttelt und destilliert. Der Pinolsaure- athylester ist ein diiqnflussiges farbloses 01 vom Sdp. ca looo ('/lo mm) (Analyse a). Auch aus destillierter Pinolsaure wurde der gleiche Ester erhalten (Analyse b).
a) b)
0,1990 gr Subst. yaben 0,4:901 pr CO, uiid 0,1780 gr II,O
0,1783 gr Subst. gaben 0,4397 y r CO, und 0,1671 yr H,O 0,1936 gr Subst. gabeii 0,4760 g r CO, u,nd 0,1779 gr H,O
Cl,Hz~03(VIII) Her. C 67,30 11 10,28'/0 Cef. a) ,, 67,19 10,000/0 Gef. 11) ,, 67.33, 67,08 ,, 10,FjO; 10,280lo
Vom Ester aus der undestillierten Pinolsaure wurde zur Charakterisierung eine Probe mit der berechneten Menge Chrom- saure in Eisessig am Wasserbade oxydiert. Bei der Aufarbeitung durch Absaugen des Eisessigs im Vakuum und Ubersattigen des Ruckstands rnit Sodalijsung konnte aus dem mit Ather ausge- schiittelten neutralen 01 das bei 136' schmelzende Semicarbazon des Pinonslure-athylesters erhalten werden (Mischprobe!).
c) Kata ly t i sche Redukt ion . Beim Schutteln von 60 gr Pinonsaure-athylester in Eisessiglosung rnit einem Zehntel der Gewichtsmenge Platinschwarz nach der Methode von WiZZstdittei* wird glatt in einigen Stunden eine Mol. Wasserstoff aufgenommen .

- 771 -
Die Eisessiglijsung wii*d mit vie1 Wasser versetzt, rnit Ather aus- geschuttelt, die atherkche Losung mit Sodalosung geschuttelt und das neutrale 01 destilliert; es ist nach dem Sdp. (ca. i00'. bei ' 1 3 mm) rnit dem oben beschriebenen Pinolsaure-athylester identisch. Eine Probe des Esters wurde nach vorheriger Behand- lung mit Seniicarbazid-acetat, wobei keine Spur Semicarbazon erhalten werden konnte, durch zehnstundiges Kochen mit alko- holischer Kalilauge verseift. Nach dern Abdestillieren des Alkohols wurde angesauert und mit Ather ausgeschuttelt. Die so gewonnene Pinolsaure krystallisiert sehr rasch vollstandig und schmilzt nach dem Umkrystallisieren aus Essigester und Petrolather bei 99 - 100' entsprechend den Angaben Tiemanns (1. c.). Die Ausbeuten an Ester und Saure sind nach dieser Methode annahernd quantitativ.
0,1049 gr Subst. gaben 0,2484 gr CO, und 0,0931 gr H,O C,oRl,03 (VIII, Pinolsaure) Ber. C 64,47 H 9,74010
Gef. ,, 64,60 ,, 9,930/0
Ohlorieruny des Pimolsuure-esters wnd 7,~mscctx ?nit Kaliumcyanid. Versuche, eine lirornierung des Pinolsaure-esters mit Phosphortribromid
durchzufiihren, hatter] wenig giinstige Resultate ergeben, indem dabei zum Teil phosphorhaltige Substanzen entstehen, die sich bei der Destillation zersetzeii rind schliesslich nur eici unscharf bei 100-1500 (*/3 mm) siedendes Produlrt von ca. 300/0 des theoretischen Bromgehaltes (ber. fur den Bromester analog der Pormel X) erhalten werden konnte.
Von den verschiedenen Chlorierungsversuchen sol1 nur fol- gender beschrieben werden. 54 gr des durch katalytische Reduk- tion gewonnenen Pinolsaure-athylesters wurden in 200 cm3 abso- luten Ather rnit 66 gr Phosphorpentachlorid (1 '14 Mol.) allmahlich unter Kuhlung rnit Kaltemischung versetzt, wobei darauf geachtet wurde, dass die Temperatur nicht uber Oo stieg. Nachdem das Phosphorpentachlorid aufgelost war und die Salzsaureentwickelung fast aufhorte, wurde noch zwei Stunden bei Zimmertemperatur stehen gelassen, das hellgefarbte Reaktionsgemisch auf Eis ge- gossen, gut durchgeschuttelt, die atherkche Losung rnit Soda- losung digeriert und rnit Natriumsulfat getrocknet. Beim Destil- lieren (0,3 mm) werden folgende Fraktionen erhalten :
1) 80- 90' ca. 25 gr 2) 90-120° ca. 10 gr 3) 120-130' ca. 15 gr

- 772 -
Die Fraktion 1 ist fast chlorfrei 'und besteht wahrscheinlich aus dem PinocampholensBure-ester ') (IX) ; in der Fraktion 3 ist nach einer Chlorbestimmung zu etwa zwei Drittel der gesuchte Chlorester X enthalten
C,,t~,,O,Cl (Xj Ber. CI i.5,l O/o Gef. ,, 10,70!0
Beim Erhitzen des Chlorproduktes mit Kaliuincyanid in schwach wasserhaltiger alkoholischer Losung wird hauptsachlich Halogenwasserstoff abgespalten, was aus dem starken Sinken des Siedepunktes beim Reaktionsprodukte folgt, ohne dass eine stick- stoffhaltige Fraktion erhalten wurde.
Wir stellten dann noch durch Kochen des Chloresters in alkoholischer Losung mit Natriumjodid den Jodester her. Da sich derselbe beim Destillieren teilweise zersetzt, wurde das Rohpro- dukt in wassrig-alkoholischer Losung mit Kaliumcyanid gekocht, wobei wieder in erster Linie Jodwasserstoff abgespalten wird und nur in kleiner Menge eine bei 110-130' (I/:! mm) siedende stick- stoffhaltige Fraktion erhalten wurde, die fur eine weitere Unter- suchung ungenugend war.
Cyadydr in der. Pinomawe (XI).
340 gr Kaliumcyanid wurden in 600 em3 Wasser gelost, auf -loo abgekuhlt und allmahlich eine Aufschlemmung von 125 gr Pinonsaure in 300 om3 Wasser zugegeben. Nachdem Losung ein- getreten war, wurde unter andauernder Eiskuhlung im Laufe von zwei Tagen 500 cm3 konzentrierter Salzsaure portionsweise ein- getragen in dem Masse, dass keine Erwarmung uber ca. 5' statt- findet. Die braungefiirbte Losung wurde dann mit konzentrierter Salzskiure angesauert und einige Ma1 rnit Ather ausgeschuttelt, wobei die Losung, um eine leichtere Trennung zu ermirglichen, von suspendierten geringen Harzmengen abfiltriert werden muss. Die mit Natriumsulfat getrocknete atherische Losung wird am Wasserbade konzentriert und schliesslich im Vakuum vollstandig abgesaugt. Zur weiteren Reinigung wird das 01 in Chloroform aufgenommen, nochmals rnit Natriumsulfat getrocknet und das Chloroform durch Evakuieren vollig entfernt. Das so gewoniiene
1 ) Derselbe sol1 noch naher untersuctit werdeu

- 773 -
rohe zahflussige, hellbraune Cyanhydrin (mindestens 90"/0 Aus- beute) wurde fur die weitere Verarbeitung direkt verwendet. Es wurden Proben von zwei verschiedenen Darstellungen zur Ge- wichtskonstanz im Vakuum getrocknet und durch Stickstoffbe- stimmungen der Prozentgehalt des Rohprodukts an Pinonsaure- cyanhydrin ermittelt.
C,,H,,03N (XI) ljer. N 6.64 O/o
Gef. ,, 5,32 und 5,99O/o
entsprechend ca. 80 untl 90 O / o Cyanhydrin.
Bei langem Stehen krystallisiert das Cyanhydrin grossten- teils ; nach dem Abpressen auf Ton wurden die farblosen Krystalle aus Eenzol uinkrystallisiert und zeigen einen Smp. von ca. 94" nach vorhergehendem Sintern. Zur Gewichtskonstanz wurde im absoluten Vakuum bei gewohnlicher Temperatur getrocknet.
0,1610 gr Subst. gaben 9,0 cm3 N, (200,725 mm)
CllHl,O,N (XI) Ber. N 6,64 O/o
Gef. ,, 6,21 O/o
Dieses Cyanhydrin enthalt wohl noch geringe Mengen Pi- nonsaure.
Verseifung des Pinonsaure-cyanhydrins.
a) Durch Erwarmen m i t wassr igem Alkali wird, wie zu erwaiqen war, die Blausaure unter Regenerierung der Pinonsaure wieder abgespalten.
b) Eine Probe wurde rnit 20proz. S a l z s a u r e zwolf Stunden eekocht, dann zur Trockne verdampft und der Ruckstand rnit ?. Ather ausgezogen. Aus der konzentrierten Losung scheidet sich das Ketolacton XXIII vom Srnp. und Mischprohe 63O ab.
c) Durch zwolfstundiges Kochen rnit der zehnfachen Menge 10 proz. absoluter a lkohol i scher Sa lzsaure , Absaugen des Lo- sungsmittels im Vakuum, Ausschutteln rnit Ather des rnit Sodali5sung versetzten Kiickstandes und Destillieren des so erhaltenen 61s bei 1 mm wird neben etwas Pinonsaure-athylester vom Sdp. ca. l l O o eine bei ca. 14.0' siedende Hauptfraktion erhalten, die nach dem Erstarren bei 63' schmilzt und durch die Mischprobe rnit dem Ketolacton XXIII identifiziert werden konnte.

Das Semicarbazon des dabei gewonnenen Pinonsaure-athyl- esters schmolz nach dem Uinkrystallisieren aus wenig Alkohol bei 141O. Die Mischung mit dem oben beschriebenen bei 136' schmel- zenden Semicarbazon des Pinonsaure-athylesters, der durch direkte Veresterung der Pinonsaure hergestellt war, zeigte den Smp. von etwa 138O- 139'. Die beiden Sernicarbazone sind vielleicht stereo- isomer.
0.1519 gr Subst. gaben 0,3214 gr C O , iind 0.1229 gr H,O 0,1130 gr Subst. gaben 0,2*389 gr CO, u n d 0,0915 gr H,O
CI3H,,O,N, Ber. C 57,93 H 8,59 O/o . Gef . , 57,73; 57,68 ,l 9,05; 9,060/0
Es wird also auch durch Salzsaure aus dem Cyanhydrin Blausaure abgespalten.
Daystellung des Athylestem von Pinonsir'ui.e-cya?zkydl.in (nach Formel XI).
Zu 10,s gr rohen Pinonsaure-cyanhydrin in 50 cm3 absolutem Ather wurde eine Losung von 20 gr Thionylchlorid in der gleichen Menge Ather unter Kuhlung mit Kaltemischung tropfenweise zu- gesetzt. Nach mehrstundigem Stehen bei Zimmertemperatur wurde sechs Stunden am Wasserbade gekocht, dann 10 cm3 absoluten Alkohol zugegeben und nochmals aufgekocht. Aufgearbeitet wurde durch Schutteln mit Eiswasser und Sodalosung (beim Ansauern derselben tritt keine Abscheidung ein). Das in Ather geloste neu- trale Produkt siedet neben einern geringen Vorlauf von Pinon- saure-athylester in der Hauptsache bei 155-165' (0,7 mm). Das dickflussige 01 wurde nochmals destilliert und eine Mittelfraktioii vom Sdp. 150-152O (0,4 mm) analysiert. Es besteht aus dem Cyanhydrin des Pinonsaure-athylesters.
0,2445 gr Subst. gaben 0,5819 gr CO, irnd 0,19RO gr f1,O 0,2715 gr Subst. gaben 13,9 cm3 9, (80,720 mni)
C,,H,,O,S Ber C 65,27 H 8,79 N 5,85 O/o
Gel'. ,, 64,94 9,06 ,) 5,89'/0
Bei einem Versuche, wo das Thionylchlorid ohne Kuhlung zugesetzt wurde, erhielt man eine geringe Ausbeute an Cyan- hydrinester, daneben aber eine grossere Menge Pinonsaure-athyl- ester. Um die Hlauslureabspaltung zu vermeiden, muss also bei ganz geliiiden Bedingungen gearbeitet werden.

- 775 -
Ein analoger Versuch in Gegenwart voii Chinolin bei tiefer Temperatur durchgefuhrt ergab ein undestillierbares schwefelhal- tiges Keaktionsprodukt.
Beim Kochen des analysierten Cyanhydrinesters rnit alko- holischem Kali wird Pinonsaure erhalten, wodurch seine Koii- stitution bewiesen ist.
Herstellung cles 2-~gan-6-rnethyl-2,6-heptadi~n-n"-essigsau~e- tithylestew ( X I I a .
94,5 gr des rohen Pinonsaurecyanhydrins wurden in 300 cm3 Chloroform unter Kiihlung mit Kaltemischung allmahlich mit 260 gr gepulvertem Phosphorpentachlorid versetzt, wobei darauf geachtet wurde, dass die Temperatur des Gemisches unter +5" blieb. Nach jedem Zusatz erfolgt heftige Salzsaureentwickelung. Die rotviolette Losung wurde iiber Nacht im Eiskeller stehen gelassen und dann langsam unt er Einhaltung obiger Vorsichts- massregeln 500 cm3 absoluten Alkohols eingetragen. Aufgearbeitet wurde durch langeres kraftiges Durchschutteln mit Eisstiicken und mehrmaliges Ausziehen der wassrigen Losung rnit Chloro- form. Die Chloroformlosung wurde rnit Sodalosung digeriert, wobei nur geringe Mengen saurer Substanzen durch Ansaiuern der Letz- teren gewonnen werden, die nicht weiter untersucht wurden. Die neutralen Anteile wurden der Destillation im Vakuum unterworfen und zwar entfernte man zuerst durch Erhitzen bis auf 150' (1 2 mm) die Phosphorsaureester. Durch dreimaliges Fraktionieren des Restes im absoluten Vakuum wurden etwa 20 gr Pinonsaure- athylester erhalten vom Sdp. ca. 100" (l1.1 mm), der einmal als Semicarbazon vom Smp. 136O und dann auch durch Analyse identifiziert wurde.
0,1455 gr Subst. gaben 0,3611 gr CO, und 0,1259 gr 11,O
L ~ ~ F J ~ ~ O , (VII) Ber. C 67,94 11 9,43 ('/o Cef. ,, 67,70 9,680/0
Neben einer Zwischenfraktion von etwa 10 gr (Sdp. 100-120") siedet die Hauptmenge als gelbliches 01 (40 gr) unscharf bei 120-150" ('/3 mm) und konnte auch durch mehrmaliges Destil- lieren nicht in scharf gesonderte Anteile getrennt werden. Alle Fraktionen erwiesen sich als chlor- und stickstoffhaltig und ent- farben mornentan Rrom in Schwefelkohlenstofflosung. Die hochste

- 776 -
Fraktion ist zudem phosphorhaltig, wie auch der Destillations- ruckstand.
Da es sich in einem Vorversuche erwies, dass zur vollstan- digen Chlorabspaltung die Behandlung mit Dimethylanilin unge- eignet ist, wurden die gesamten bei 120-150' siedenden Frak- tionen mit 60 gr Chinolin in 100 cm' Xylol zwolf Stunden ge- kocht. Nach den1 Erkalten wurde init verdiinnter Salzsaure das Chinolin entfernt, die Xylolbsung durch Ather verdiinnt (zur bessern Trennung) und mit Sodaliisung ausgeschuttelt ; in der Letzteren waren nur Spuren saurer Substanzen enthalten. Die Hauptmenge der neutralen Produkte (= 32 gr) siedet von 120 his 130' bei ' 1 3 mm, ist chlorfrei und besteht aus dem in der Auf- schrift genannten Korper, der nicht ganz rein erhalten werden konnte. Ein mittlerer Anteil wurde nach zweimaliger Fraktionie- rung analysiert. Das 0 1 ist schwach gelb gefgrbt.
0,2034 gr Subst. gaben 0,5212 gr CO, urrd 0,1608 gr lI,O 0,1966 gr Subst. gaben 0,5035 gr CO, rind 0,1544 gr 13,O 0,2380 gr Subst. gaben 12,s crns K2 (140, 729 J I I ~ I )
(:lStllSOSN (SIII) Rer. C 70,58 I1 8,60 K 6,33 O/o
Gef. 69,92; 6937 ,, 8,m; a,m ,, 5,80"/0
Es wurderi aucli Versuclie uiit,crnommen, die l~insetziing des Pinonsbure- c~anhydr ins mit I'hosphortribromid durchxufiihren, da hei clieser Reaktioii uud hufarbeitung weniger von den starkeu aiiorganischen Sanreii entsteheri. dir Anlass zur Urnlagerung geben konnten. Nest? Arheitsweise erwies sich jedorli als ungunstig, d a eine schlechte busbeute erhalten wiirde und nebeiibei iiielir phosphorhnltige Serhinduiigen entstchcn, die sicli bei der Destillation xersetzeir.
huch die Clilorierung des Cyanbydriiis des PinonsBure-athglcster~ lmt keine Vorteile vor der hnwendung des t'iriorisiiuI.e-~:!'anli~driiis.
Herstellung der Ester der 2-(%ir boxyl-~-~nethyl-~,6'-he;ptccdii.,2- 5-essigsicure (bezw. der Isomeren, Formel XIV).
32 gr des Cyanesters XI11 wurden mit 60 cm' 50proz. Kalilauge und Alkohol bis zur homogenen Losung versetzt und zwei Tage am Wasserbade gekocht. Da nach dem Abdanipfen des Alkohols und mehrstiindigem Erhitzen auf 100' der 'Ruckstand immer noch Ammoniak entwickelt, wurde mit noch 50 cm3 50- proz. Kalilauge versetzt und weitere zwei Tage gekocht, worauf die Verseifung beendigt war. Nach dem Ansauern und Ausschiit-

- 777 -
teln mit Ather wird die Dicarbonsaure XIV als zahe gelbliche Rfasse erhalten, die stickstofffrei ist und auch bei langem Stehen nicht krystallisierte.
Dia thyles te r . Zur konzentrierten alkoholischen Losung der rohen Dicarbonsaure wurde eine Losung von zwei Mol. Na- trium in Alkohol zugegeben, wobei das schwer losliche Natrium- salz abgeschieden wird. Nach achtstundigem Erhitzen im Bomben- rohr rnit uberschussigem Athyljodid auf 120' wurde das Losungs- mittel verdampft, mit Sodalosung versetzt und rnit Ather aus- geschuttelt. Aus der Sodalosung kann durch Ansauern etwas unveresterte Saure zuruckgewonnen werden. Das neutrale Produkt siedet bei 110-120' ('/s mm) als dunnflussiges, farbloses 01. Eine Xttelfraktion vom Sdp. 115' wurde analysiert.
0,1797 gr Suhst. gaben 0,4420 gr CO, und 0,1508 g r H,O 0,2282 gr Subst. gaben 0,5597 gr CO, und 0,1719 gr 11,O
CI5Hz4Oe (XIY) Ber. C 67Jl 11 9,020/0 Gef. ,, 67,iO; 66,91 )1 9,17; 9,400/0
Dimethyles te r . 30 gr der rohen Dicarbonsaure XIV wer- den in einer eiskalten Losung von 17 gr Stangenkali in 40 cm' Wasser mit 35 gr Dimethylsulfat zwolf Stunden geschuttelt. Auch anfangs tritt keine wesentliche Erwarmung ein. Es wird mit etwas Sodalosung versetzt und mit Ather ausgeschuttelt. Aus der alka- lischen Losung kann man durch Ansauern einige Gramm saurer Substanzen (wohl z. T. Estersaure) gewinnen, die durch erneute Behandlung mit Dimethylsulfat in den neutralen Ester uberfuhrbar sind. Der gesamte neutrale Anteil wurde im absoluten Vakuum des- tilliert, wobei die Hauptmenge als farbloses, dunnflussiges 01 bei 1 10-120' ( '13 mm) siedet. Nach zweimaliger Fraktionierung wurde eine Mittelfraktion vom Sdp. 115' ('/3 mm) analysiert ; dieselbe erwies sich als der Dimethylester der Saure XIV.
0,2034 gr Subst. gaben 0,4843 gr CO, iiiid 0,1663 gr 1120
C,sH,004 Eer. C 64,96 H 8,400/0 Gef. ,, 64,97 8,600/0
Beim Verseifen der beiden reinen Ester durch zwanzigstun- diges Kochen rnit alkoholischem Kali, Verdampfen zur Trockne, Ansauern und Ausziehen mit Ather konnte keine krystallisierte Saure erhalten werden.

XXIT.
-
CH, 1 C
\/ fY I
CH /\
CH, CH,
XST.
-+
2 gr der rohen Dicarbonsaure XIV wurden mit der drei- faclien Menge Essigsaureanhydrid im Bombenrolire zehn Stunden auf etwa 180" erhitzt und dann der Destillation im Vakuum un- terworfen. Durch Zersetzung des intermedcar entstehenden An- hydrids der Dicarbonsaure (XXIV) bildet sich hiebei wohl das zweifach ungesattigte Keton XXV, das durch Enolisierung sofort in Carvacrol (XXVI) ubergeht. Letzteres wird unter den Versuchs- bedingungen acetyliert. Das bei der Destillation erhaltene ober- halb 100" (12 mm) siedende neutrale 01 (etwa 0,5 gr), das als Carvacryl-acetat betrachtet wurde, verseiften wir durcli 15-stiin- diges Kochen mit alkoholischem Kali. Nach dem Verdampfen des Alkohols wurde der Ruckstand angesauert und das, abge- scliiedene 01 rnit Ather ausgeschiittelt. Es siedet nach dem Trocknea uher Pottasche bei etwa 110' (12 mm), zeigt Carvacrolgeruch und gibt mit Eisenchlorid in alkoholischer Lijsung eine Grunfarbung. Durch 20-stundiges Erhitzen mit Phenylisocyanat auf 100' und Waschen des in der Kalte emtarrenden Reaktionsproduktes rnit Petrolather werden feine Ngdelchen erhalten, die direkt somie iiacli deni Umkrystallisieren aus Essigester und Petrolather bei 136-137" schmelzen und nach der Mischprobe rnit dem gleich schmelzenden Phenylurethan des Carvacrols ') identisch sind.
Diese Reaktion stellt eine neue Gewinnungsart eines Phe- 1101s dar.

779 - -
Darstellung der a-Metlayl-~'-isopl-o~~yl~inaelii~-este~~ f X l 7 .
Die beiden ungesattigten Ester XIV wurden in athylalkoho- lischer bezw. methylalkoholischer Losung mit Wasserstoff in Ge- genwart von ein Zehntel des Gewichts Platinschwarz der Re- duktion unterworfen, wobei in einigen Stunden zmei Mol. Wasser- stoff aufgenommen werden. Die gesattigten Ester wurden zur Reinigung im Vakuum destilliert.
Der D i a t h y 1 e s t e r ist ein dunnflussiges farbloses 01 vom Sdp. i10-l2O0 ( ' 1 3 mm); eine Mittelfraktion vom Sdp. ca. 115' wurde analysiert.
0.1344 gr Subst. gaben 0,3273 gr CO, und 0,1200 gr 11,O
C,,H,80, (XV) Ber. C 66,18 H 10,29O/o Get. ,, 66,43 10,OOo/o
Der D i m e t h y l e s t e r siedet bei 105-1i5° ( '19 mm) als dunnflussiges farbloses 01; eine Analysenfraktion u-urde bei ca. 108' entnommen.
0,1621 gr Suhst. %ahen 0,3784 gr CU, uncl 0,1410 gr I t & ) C13112404 (analog XV) Rer. C 63,93 11 9,83 o/o
Gef. ll 63$9 ,, 9,73 o/o
Durch Verseifung der beiden Ester nach dem fur die un- gesattigten Ester XIV angegebenen Verfahren wurde eine zahe amorphe Masse gewonnen, welche. die gesattigte Saure darstellt und auch bei inonatelangem Steheii keine Neigung zur Krystal- lisation zeigte.
Gewimung des Tctrahydl-ocarvons (XU) aids dem a-Methyl-P'-iso- prop ylpimelin-estw.
7,4 gr des Dimethylesters wurden mit 0,s gr gepulvertein Natrium in 20 cm3 Xylol funf Stunden irn &bade gekocht, nach dem Erkalten mit verdunnter Salzsaure durchgeschuttelt, die Xy- Iollosung abgetrennt und die wassrige 1,osuiig mi t Ather ausge- zogen. Die vereinigte Xylol-Atherlosung wurde mit Natriumsulfat getrocknet und die Losungsmittel vollstandig entferiit (zum Scliluss durch langeres Erhitzen im Vakuum auf looo)>. Der Ketoester, dessen Anwesenheit aus der starken Eisenchloridreaktion des

- 780 -
Ruckstandes (= 6,s gr) folgt, wurde nicht isoliert, da er sich auch bei oftmaligem Ausziehen rnit 10 proz. Natronlauge nicht voll- standig gewinnen l'asst. Es wurde daher der ganze Ruckstand cler Verseifung (sowohl sauer wie alkalisch) unterworfen.
Bei einem Versuch wurde rnit der funffachen Menge 10proz. alkoholischer Kalilauge 15 Stunden gekocht, mit Wasser bis zum Aufhoren der Triibung versetzt, ausgeathert und der Extrakt nach dem Trocknen rnit Natriumsulfat im Vakuum destilliert.
Bei einem andern Versuch wurde der rohe Ketoester niit konzentrierter Salzsaure 8 Stunden am Ruckflusskuhler erhitzt, mit Ather ausgezogen und nach dem Trocknen wie oben im Va- kuum destilliert.
In beiden Fallen wird eine bei 80-100° (12 mm) siedende Fraktion von starkem Geruch nach Kiimmel und Pfeffermiinz erhalten. Daraus lasst sich das r-Tetrahydrocarvon isolieren, das die von Wallack ') beschriebenen Eigenschaften besitzt. Die ge- samte Menge wurde ins Semicarbazon verwandelt, das schon iiach etwa einer halben Stunde vollst'andig gebildet ist. Es schmilzt in rohem Zustande sehr unscharf und nach zweimaligem Umkrystal- lisieren aus Alkohol werden Krystalle vom Smp. 170-171° er- halten; die leichter loslichen Anteile schmelzen tiefer (etwa zwischeii 140 und 160°), wie auch Wallnch beobachtete.
Das ganze Semicarbazon wurde durch Kochen rnit 20-proz. Salzs'aure im De'stillierkolben gespalten, wobei das sehr rasch re- generierte Tetrahydrocarvon mit den Wasserdiimpfen ubergeht. Es wurde rnit Ather ausgezogen, mit Calciumchlorid getrocknet und im Vakuum destilliert. Man erhalt so aus der angewandten Menge Ester etwa 1 gr eines scharf bei SOo (11 mm) siedenden, farblosen, diinnflussigen Ols, das den oben beschriebenen Geruch auf w eist.
0,1171 gr Siibst. gaben 0,3336 ~r CO, i i i i c l 0,1249 gr H,O
~ l o H 1 8 0 (XVI) Hrr. C 77,85 H 11,77 Yo Get. ,, 77,72 ,, 11,93 O/o
0 xi m. 0,5 gr Tetrahydrocarvon wurde in methylalkoholischer Losung niit einer Lissung yon 1 gr Hydroxylaminchlorhydrat in 1 cm' Wasser versetzt und mit 1,2 gr Natriumbicarbonat drei Stun-
1) Zusamme~~gesteIlt in Terpeue und Campher (1909), s. 375. Yemic:ir- haLon Srrip. 1740, Oxim 1050.

781 - -
den gekocht. Nach dem Abdampfen des Methylalkohols scheiden sich beim Erkalten Krptalle ab, die in den organischen Losungs- mitteln leicht loslich sind und aus wassrigem Alkohol in Form feiner Nadelchen vom Smp. 103-104° (nach dem Trocknen) er- halten wurden.
0,1568 gr Suhst. gahen 0,4049 g r CO, mid 0,1589 gr II,O
ClOH,,ON Ber. C 70,94 H 11,320,'o Gef. 70,45 ,, 11,340/0
D i e D e s t i l l a t i o n d e s B l e i s a l z e s der a-Methyl-P'-iso- propylpimelinsaure (hergestellt durch Auflosen der rohen Saure in Ammoniak, Vertreiben des Uberschusses an Letzterem am Was- serbade, Fallen mit Bleiacetat und Trocknen des Niederschlags bei 100°) im Kohlendioxydstrom liefert ebenfalls Tetrahydrocarvon.
Die Ausfuhrung dieser Brbeit war durch Mittel gefordert. die dern einen von t ins von der vrrn't Hnff-Stzftu?~g dry A-gl Akadeinre der Wzssm- s( htrttrw Amsterdam gewahrt wurden, wofnr auch hier bestens yedankt sel
h n m e r k u n g
Zurich, Chemisches Institut der Eidg. Techn. Hochschule.
Uber die Polymerisation der 1,2 - Cyclohexenone von
L. Ruzieka. (4. x. 20.)
Bei vereinzelteii 1,2 - Cyclohexenonen wurde die Reobach- tung gemacht, dass durch die Einwirkung alkalischer Reagentien
CH, CI1, I I
CII C
I . 11. I11