Embed Size (px)
Citation preview

Talanta. 1968. Vol. 15, PP. 379 10 384. Per@mon Press. Printed in Northern Ireland
ZUR POLAROGRAPHIE DER n-BUTYLZINNCHLORIDE
K. ISSLEIB, H. MATSCHINER und S. NAUMANN
Institut fti Anorganische Chemie der Universitat Halle, Saale, Deutsche Demokratische Republik
(Eingegangen am 15. September 1967. Angenommen am 10. Oktober 1967)
Zusammenfassung-Di-n-butylzinndichlorid (II) und Tri-n-butylzinn- chlorid (III) lassen sich neben Mono-n-butylzinntrichlorid (I) bei Anwesenheit von Diphenyldithiocarbamat gleichstrompolarographisch bestimmen. Die Untersuchungen werden in einer 5-%&en Natrium- acetattrihydrat-Methanoll6sung durchgefiihrt, urn die offensichtlich zwischen II bzw. III und Diphenyldithiocarbamat gebildeten, poiaro- graph&h aktivem “Dithiocarbamate” zu l&en und urn I als Acetat zu maskieren. In gewissen Grenzen besteht ProportionalitSit zwischen Wellenhiihe und Konzentration. In einem Organozinnchloridgemisch wird der Gehalt an I komplexometrisch ermittelt. Tetra-n-butylzinn ist in Methanol nahezu unliislich und stiirt die Bestimmungen nicht.
MONO-n-BVTYLZINNTRICHLORID (I), Di-n-butylzinnchlorid (II), Tri-n-butylzinn- chlorid (III) und Tetra-n-butylzinn (IV) lassen sich bekanntlich gravimetrisch, gas- chromatographisch,1-3 oszillopolarographisch* oder mit Hilfe der Gleich- bzw. Wechselstrompolarographies-lo quantitativ bestimmen. Versuche einer quantitativen Ermittlung von I-IV nebeneinander verliefen ergebnislos. So fiihrte die Gleichstrom- polarographie weder zu gut ausgebildeten Grenzstrbmen noch zu auswertbaren Polarogrammen. Die hier durch I verursachten hohen Maxima fiberdecken die Stufen von II und III.8-10 Auch oszillopolarographisch waren maximal nur zwei Komponenten, wenn die Konzentration einer dritten konstant gehalten wurde, nebeneinander zu bestimmen.4 Nach allem scheint eine polarographische Analytik von I-III nebeneinander nur dann miiglich zu sein, wenn eine der Komponenten durch Zugabe geeigneter Salze oder Komplexbildner stabilisiert bzw. komplexgebunden wird, was eine Trennung der polarographischen Stufen zur Folge haben wiirde.
Im Zusammenhang polarographischer Untersuchungenverschiedener substituierter Organozinn-dithiophosphinate bzw. -dithiophosphorsfiureester” waren gut ausge- prQte und in nichtw%Brigen Solventien charakteristische polarographische Stufen zu beobachten. Schwefelhaltige Komplexliganden wie Thiocarbons%uren, Dithiophos- phinsguren, Dithiocarbonate u.a. liefern in wiiI3riger Lijsung mit Organozinnhalogeni- den, hier vor allem I und II, offensichtlich unter Abspaltung von Halogenwasserstoff, die entsprechenden Reaktionsprodukte. Diese sind in Wasser unliislich, gut filtrierbar und teilweise zur selektiven Abtrennung verschiedener Organozinnhalogenide geeig- net. In MethanollGsungen, die Natriumacetat und Diphenyldithiocarbamat enthal- ten, ist eine gleichzeitige gleichstrompolarographische Bestimmung von II und III neben I miiglich. Natriumacetat verhindert, da13 I die ElektrodenvorgZinge von II und III beeinfluI3t. Gut auswertbare polarographische Stufen wurden hier aber nur unter Verwendung einer gesgttigten Kalomelelektrode als Gegenelektrode erhalten.
379

380 K. ISSLEIB, H. MATSCHINER und S. NAUMANN
Versuche, mit Bodenquecksilber als Gegenelektrode4~D*1’J fiihrten zu negativen Ergebnissen.
Im folgenden sol1 zuntichst ausschliefilich fiber die polarographische Bestimmung von I-III berichtet werden. Eine Diskussion iiber den Mechanismus dieser elektro- chemischen Reaktion ist fi.ir spiitere Arbeiten vorgesehen.
EXPERIMENTELLER TEIL
Die polarographischen Untersuchungen werden mit dem Gleichstrompolarographen PTS 4 im Potentialbereich von -0,6 bis -1,6 V-Dtipfung 3, Empfindlichkeit 5 ,uA-durchgeftit. Als Geg- enelektrode dient eine ge&@te Kalomelelektrode (SCE). Die Tropfzeit der Kapillare betragt bei einer Behalterhiihe von 63 cm 5,2 sec/Tropfen. Als Grundelektrolyt wird eine 5- %ige Liisung von Natriumacetattrihydrat p.A. in Methanol p.A. verwendet. Als Komplexbilder fiir die Butylzinn- chloride dient eine O&%ige methanolische Diphenyldithiocarbamatl&ung. Das hierfiir verwendete Diphenyldithiocarbamat wird nach bekanntem Verfahre9 hergestellt. Die Untcrsuchungen werden in einem Kalousekgefg durchgeftihrt. Vor der polarographischen Untersuchung wird die M&z&e mehrmals mit Argon sekuriert und dann unter Inertgasatmosphtie Grundelektrolyt, jeweilige Substanzprobe und Komplexbildnerliisung zugegeben.
1. Bestimmung von Monobutylzinntrichlorid,(I), mit Komplexon II14Ja
Zehn ml einer sauren alkalischen oder alkoholischen Liisung, die I enthat, werden mit 30 ml Eisessig und 3 Tropfen einer OJ-%igen wlssrigen Brenzkatechinviolettl16sung versetzt. Bis zum Farbumschlag von Rot nach Blau wird festes Natriumacetat zugegeben. Ein ObcrschuR an Natriumacetat gestaltet den Titrationsverlauf gi”stig. Zu dieser Lasung 15iOt man eine 0,l his 0,Olm Lijsung von +omplexon III bis zum Farbumschlag von Blau nach Gelbrosa tropfen. Kurz vor Erreichen des Aquivalenzpunktes ist es erforderlich, nach Zugabe jedes Tropfens der Kom- plexonlijsung zu rtihren oder zu schiitteln. W&end der Titration ist auf Temperaturkonstanz (nicht iiber 20”) zu achten.
1 ml einer 0,lm Komplexonltisung = 28,22 mg Butylzinntrichlorid.
2. Bestimmung von Dibutylzinndichbrid,(ZZ), neben ButylzinntrichIorid,(Z), unter Verwendung von Thioglykolsiiure
Ein ml einer Petrol%therliisung von I und II wird mit 10 ml Petrolgther (Sdp. 50-60”) und 0,2ml Thioglykolsiiure versetzt. Nach Zugabe von 5-1Oml Wasser fiillt das Dibutylzinnthio-
lykolat aus. Diese wird nach 3 std. Stehen abfiltriert, mit wenig Wasser gewaschen und in 5 ml % ther und Methanol quantitativ gel&t. Fi.ir das Au&en des Niederschlages ist es giinstig, ein 25 ml M&kiilbchen zu verwenden. Der M&kolben wird mit Grundelektrolyt (l- %ige methanolischer Lithiumchloridlijsung) aufgefiillt und diese Liisung in einem Kalousekgefu polarographiert. Das Kalousekgefti wird auf 25 ml geeicht, um den w&end des Sekurierens verdunsteten Petrol&her durch Grundelektrolyt zu erg;inzen. Enthllt die bekannte Substanzprobe O-20% Dibutylzinn- dichlorid so ist eine Einwaage von 1 g Substanzgemisch auf 1OOml Petrolgther (Sdp.50-60”) zu empfehlen. Von dieser Liisung wird 1 ml, wie beschrieben, ftir die polarographische Untersuchung verwendet. Im Falle eirier geringeren Konzentration an II werden 2-5 ml der Petrolltherliisung eingesetzt.
3. Bestimmung von Dibutylzinndichlorid, (II), neben Monobutylzinntrichlorid, (Z), unter Verwendung von Diphenyldithocarbomat
Eid SuL&nzgemisch bekannter Zusammensetzung von II und I (vgl. Eichkurve) wird in einem Kalousekgefti mit Grundelektrolytl6sung (5- %ige methanolische Natriumacetattrihydratlasung) bis auf 25 ml aufgeftillt. Nach Sekurieren mit Argon werden 2 ml einer 0,4-x-igen methanolischen Diphenyldithiocarbamamatliisung zugegeben. Die Reihenfolge dieser Operation ist unbedingt einzu- halten. urn eine Umsetzune von I in das entsmechende Acetat zu pewtileisten und urn eine vorzeihge Komplexbildung & vermeiden; andeinfalls sind die polarogaphischen Stufen nicht zu trennen. Fti die Herstelhmg des Grundelektrolyten ist nur Natriumacetattrihydrat gee&net, da die Verwendung wasserfreien Natriumacetats nicht auswertbare Polarogramme liefert. Die Polarographie wird in einem Potentialbereich von -0,6 bis - 1,4 V und mit einer Empfindlichkeit von 2 ,uA durchgeftihrt.
Erfassungsgrenze: 0,0037-0,148 mglml II neben O-O,37 mg/ml I mit einer Genauigkeit f3 %.
4. Bestimmung von Dibutylzinndichlorid (II) und Tributylzinnchlorid (III) nebeneinander
Wie unter 3. beschrieben, wird ein bekanntes Substanzgemisch von II und III (vgl. Eichkurve) in 25 ml 5- %iger methanolischer NatriumacetattrihydratliSsung gel&t und mit 1 ml einer 0,Cproz.
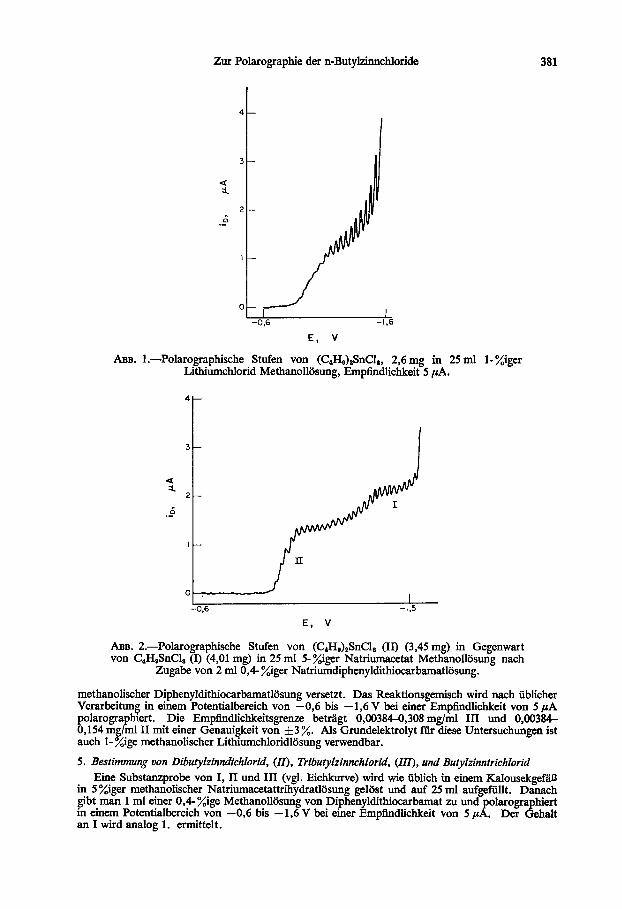
Zur Polarographie der n-Butylzhmchloride 381
E, V
ABB. l.-Polarographische Stufen von (C,H&SnCI,, 2,6 mg in 25 ml l-%iger Lithiumchlorid Methanolliisung, Emp6ndlichkeit 5 PA.
3-
a a.
2-
.-D
I-
-0,6 -I,5
E, V
A~B. 2.-Polarographische Stufen von (C4Hl)$nClB (II) (3,45 mg) in Gegenwart von CdH9SnCl, (I) (4,Ol mg) in 25 ml 5- Xiger Natriumacetat Methanolliisung nach
Zugabe von 2 ml 0,4- %iger Natriumdiphenyldithiocarbamatl6sung.
methanolischer Diphenyldithiocarbamatlosung versetzt. Das Reaktionsgemisch wird nach iiblicher Verarbeitung in einem Potentialbereich von -0,6 bis -1,6 V bei einer Emptindlichkeit von 5 PA polarographiert. Die Empfmdlichkeitsgrenze betriigt 0,00384-0,308 mglml III und 0,00384- 0,154 mg/ml II mit einer Genauigkeit von A3 %. Als Grundelektrolyt ftir diese Untersuchungen ist such l- %ige methanolischer Lithiumchloridlijsung verwendbar.
5. Bestimmung van Dibutylzinndichlorid, (ZZ), Tributylzinnchlorid, (ZZZ), und Butykinntrichlorid
Eine Substanzprobe von I, II und III (vgl. Eichkurve) wird wie tiblich in einem Kalousekgefs in S%iger methanol&her Natriumacetattrihydratlratliisung gel&t und auf 25 ml aufgeftillt. Danach gibt man 1 ml einer 0,4- %ige Methanolli%ung von Diphenyldithiocarbamat zu und polarographiert in einem Potentialbereich von -0,6 bis -1,6 V bei einer Emptindlichkeit von 5 PA. Der Gehalt an I wird analog 1. ermittelt.
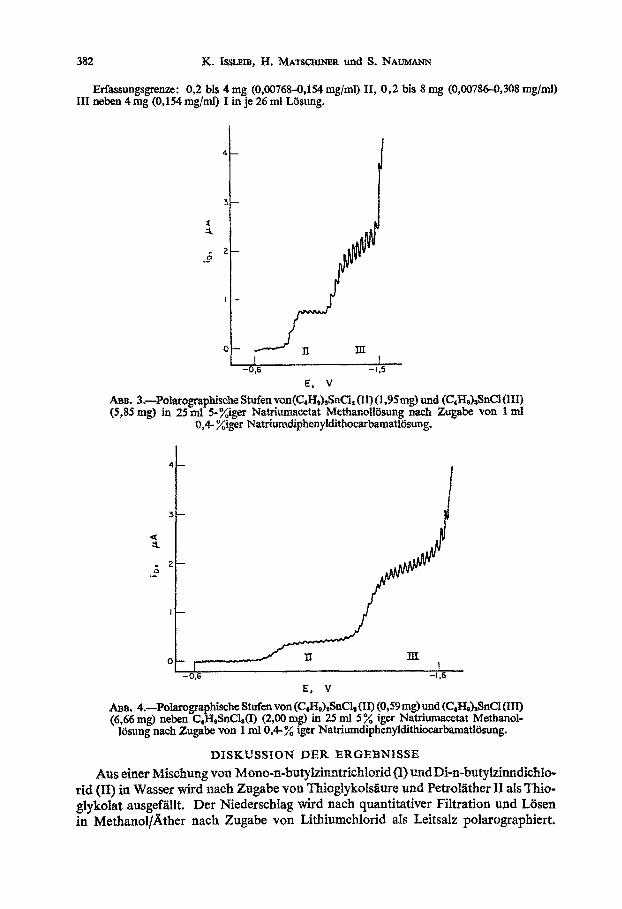
382 K. Iswim, H. MATSCHINER und S. NAUMANN
Erfassungsgrenze: 0,2 his 4 mg (0,00768-0,154 mglml) II, 0,2 bis 8 mg (0,00786-0,308 q/ml) III n&en 4 mg (0,154 mg/m.l) I in je 26 ml L6sung.
J-
:.
L 2- .a
O-
I -0,6 -I,5
fL v
ABB. 3.-potarograpbische Stufen von(C~H~)~Sn~~ (II) (i,9Smg) und (CpfIalsSnCl (III1 (5,85 mg) in 25 ml 5-Qger Nat~~a~~t Me~a~II~su~g nach Zugabe van 1 ml
O& %iger Natri~~p~~nyldis~g.
4-
3-
or I
-0,6 -I,6
E, v
&m. 4.-Polarographische Stufen von (GH&SnCl, (II) (0,59mg) )mdtC,HJ&%KYl (III) (6,66 mg) neben C,HpSnCl,(I) (2,OO ms> in 25 ml 53 iger N_aFmmacetat Methanol-
)&ung nach Zugabe von 1 ml 0,4- % iger Natriumdlphenyl&tiocarbamatlBsung.
DESKWSSION DER ERGEBNISSE
Aus &er Mischung van Moue-u-bu~~~~chlo~d (I) ~d~~-~-bu~lzi~dich~~ rid err) in Wasser wird nach Zugabe van ~~oglyko~s~~~e und Petrol&her II als Ihio- glykolat ausgef2illt. Der Niederschlag wird nach quantitativer Filtration und L&en in Methhanol/Ather nach Zugabe von Lithiumchlorid als Leitsalz polarographiert.

Zur Poiarographie der n-Butykimchbride 383
Man erhSilt fur II eine gut auswertbare Stufe mit dem Halbstufenpotential El,, = -0,924 V gegen SCE. Zwischen der Stufenhohe und der Konzentration im Bereich von 0 bis 2,77 mg II/25 ml Liisung besteht Proportiona~t~t.
Im Falle hoherer Konzentrationen von II erfolgt starke Adsorption an der Quecksilberoberfhiche, es besteht also hier keine Proportionalitat zwischen der Wellenhohe und der Konzentration des Depolarisators.
Das Filtrat der Mischung enthalt I als Hydrolysat, das in zwei Stufen an der Quecksilbertropfelektrode reduziert wird. Es sind zwei gut ausgepragte Wellen zu beobachten, deren erste der Konzentration von I proportional ist. Obwohl sich der Gehalt von I einer L&sung auf diese Weise ermitteln last, ist es vorteilhafter, I neben II komplexometrisch mit Komplexon III quantitativ zu bestimmen.4J2J3 Stijrungen fiir diese Titration treten durch zu geringe Eisessigkonzentration-sie sollte 60% betragen-und durch Zinn(IV)-ionen, die hier miterfal3t werden, auf. Die Erfassungsgrenze dieser Methode erlaubt, 6 mg I neben 2 g II quantitativ nach- zuweisen.
Weit gtinstiger verlauft die gleichstrompolarographische Bestimmung von II in einem Gemisch mit I unter Verwendung von Diphenyldithiocarbamat statt Thioglykolsaure und bei Anwesenheit von Natriumacetat. Letzteres fiihrt das leicht
lp hydrolysierbare I in das stabilere Acetat tiber, was eine Verschiebung des Haibstu- fenpotentials von I zu negativeren Potentialen zur Foige hat. II reagiert mit Natrium- diphenyldit~ocarbamat unter Komplexbildung ve~ut~ch zu dem entsprechenden Dithiocarbamat, dessen polarographische Stufe sich nun deutlich von der des Mono-n-butylzinntrichlorids unterscheidet (vgl. Abb. 2). Die Gehaltsermittlung von I wird such hier durch komplexometrische Titration vorgenommen.13 Nach dieser Methode waren 0,l bis 4 mg II in 27 ml neben 0 bis 10 mg I in 27 ml mit einer Genauigkeit von f 3 % nachzuweisen.
Auch II und Tri-n-butyl~nnchlorid (III) iassen sich in 5-%iger methano~scher Natriumacetatliisung als Grundelektrolyt nach Zugabe von Diphenyldithiocarbamat im gewissen Konzentrationsbereich leicht polarographisch nebeneinander bestimmen.
Bei gleicher Konzentration verhalten sich die Hiihen der polarographischen Stufen von II und III wie 29 (vgl. Abb. 3). In den Grenzen von 0,l bis 4 mg II in 26 ml und 0 ,I bis 8 mg III in 26 ml Grundelek~olyt besteht Proportiona~t~t zwischen der Wellenhijhe und der Konzentration von II und III. Es kijnnen such II und III neben I polarographisch bestimmt werden, da in Natriumacetat/Methanol und Natriumdiphenyldithiocarbamat die Abscheidungspotentiale der Reaktionsprodukte von II und III wesentlich positivere Werte aufweisen als das des Acetats von I (vgl. Abb. 4). Die vor den Stufen des II und III evti. auftretenden Wellen werden durch Hydrolyseprodukte von I verursacht und stiiren keineswegs die Auswe~ung der Polarogramme. Enthalt das Methanol sehr wenig Wasser, so werden diese Stufen nicht beobachtet. Neben 4 mg I lassen sich nach diesem Verfahren noch 4 mg II und 8 mg III je 26 ml Gesamtliisung bestimmen. Bis zu dieser Grenzkonzentration von I-III resultiert such hier die gleiche zuvorgenannte Proportionalitit zwischen Wellen- hijhe und Konzentration. In einem Gemisch von I-III wird der Gehalt von I wie tiblich komplexome~isch ermittelt. Tetra-n-buty~nn (IV) stijrt infolge weitgehender Unioslichkeit in Methanol die Best&mung von I-III niche.
Die Moglichkeit einer gleichzeitigen Gehaltsermittlung von IV in I-IV beschrankt sich auf den Chlors~ureaufschlu115 und anschliel3ende komplexometrische Titration

384 K. ISSLEIB, H. IvI.&rscm~aa und S. NAUMANN
der Zinn(IV)-ionen. IV 1 3% sich darauf hin nach der Restmethode quantitativ bestimmen.
Anerkennung-Die Untersuchungen wurden auf Anregung des VEB EK Bitterfeld durchgeftihrt; wir danken dem Betrieb ftir finanxielle und apparative Untersttitxung.
Sammary-The determination of di-n-butyltin(IV) dichloride and tri- n-butyltin(IV) chloride in the presence of mono-n-butyltin(IV) tri- chloride with sodium diphenyldithiocarbamate by d.c. polarography is described. The investigations were made in solutions containing 5% of sodium acetate trihvdrate in methanol. which dissolves the dithio- carbamates of the di-I;-butyl and tri-n-b&y1 compounds and shifts the reduction wave of the mono-n-butyl compound to more negative potentials. Within certain limits the wave height is proportional to the concentration. In a mixture of the organotin chlorides, mono-n- butyltin trichloride is determined complexometrically. Tetrabutyltin is practically insoluble in methanol and does not interfere in the determinations.
1. 2.
::
:* 7: 8. 9.
10.
::: 13. 14. 15. 16.
R&&-Gn d&it le dosage du dichlorure de di-n-butyl&ain(IV) et du chlorure de tri-n-butyletain(IV) en presence de trichlorure de mono-n-butyl&ain(IV) au moyen de diphenyldithiocarbamate de sodium par polarographie en courant continu. I_es recherches ont et6 effect&es dam des solutions contenant 5% da&ate de sodium trihydrate en methanol, ce qui dissout les dithiocarbamates des composes di-n-nutyl et tri-n-butyl et deplace la vague de reduction du compose mono-n-butyl vers des potentiels plus negatifs. Dam certaines limites, la hauteur de vague est proportionnelle 51 la concentration. Dans un melange des chlorures organostanniques, on dose le trichlorure de mono-n-butylbtain complexometriquement. Le tetrabutyletain est pratiquement tmsoluble en methanol et ne &ne pas dans les dosages.
LITERATUR
H. GeiOler und H. Kriegsmann, Z. Chem., 1964,4,354. R. D. Steinmeyer, A. F. Fentiman und E. J. Kahler, Anal. Chem., 1965,37,520. H. Jehring und H. Mehner, Z. Anal. Chem., 1967,224,136. U. Rothermund, Diplomarbeit TH Leuna-Merseburg, 1964. G. Costa, Gazz. Chim. Ital., 1950, 80,42. H. Jehring und H. Mehner, Z. Chem., 1963,3,34. Idem, ibid., 1964,4,273. Idem, ibid., 1963,3,472. P. Rdske, Diplomarbeit TH Leuna-Merseburg, 1964. J. Gerstenberger, D@lomarbeit TH Leuna-Merseburg, 1963. K. Issleib und W. Mertens, unveriiffentlichte Ergebnisse. K. Btiger, Z. Lebensm. Untersuch. Forsch., 1961,114,1. J. Efer, D. Quaas und W. Spichale, Z. Chem., 1965, 5,390. J. Gasparic und A. Gee, J. Chromatog., 1962, 8, 393. R. Geyer und H. J. Seudlitx, Z. Chem., 1964,4,468. Methoden der organische Chemie (Houben-Weyl), 4.Aufl., Bd. IX, S. 826. G. Thieme, Stuttgart, 1962.





























