Embed Size (px)
Citation preview

Untersuchungen zur Bildung von Furosin und
N-terminalen 2(1H)-Pyrazinonen
Dissertation
zur Erlangung des akademischen Grades
Doctor rerum naturalium
(Dr. rer. nat.)
vorgelegt
der Fakultät Mathematik und Naturwissenschaften
der Technischen Universität Dresden
von
staatl. geprüften Diplom-Lebensmittelchemiker René Krause
geboren am 23. Mai 1972 in Radeberg
Gutachter: Prof. Dr. rer. nat. T. Henle
Prof. Dr. rer. nat. W. Krause
Prof. Dr. rer. nat. L. W. Kroh
Eingereicht am: 4. November 2004
Tag der Disputation: 21. Januar 2005


„Ohne Spekulation gibt es keine neue Beobachtung.“
Charles Darwin (1809 - 1882)


Inhaltsverzeichnis
1 Einleitung und Zielstellung ..................................................................................................... 1
2 Theoretischer Teil ................................................................................................................... 3
2.1 Die Maillard-Reaktion ......................................................................................................... 3
2.1.1 Die Anfangsphase ..................................................................................................... 4
2.1.2 Die fortgeschrittene Phase......................................................................................... 8
2.1.3 Die finale Phase....................................................................................................... 10
2.1.4 Die Maillard-Reaktion in Relation zu anderen Reaktionen.................................... 11
2.1.5 Zur Bedeutung der Maillard-Reaktion in Lebensmitteln........................................ 12
2.1.6 Zur Bedeutung der Maillard-Reaktion in vivo ........................................................ 14
2.2 Analytik von Amadori-Produkten ...................................................................................... 17
2.2.1 Zur Bedeutung von Amadori-Produkten ................................................................. 17
2.2.2 Analytik von Furosin............................................................................................... 18
2.2.3 Zur molaren Ausbeute an Furosin........................................................................... 23
2.3 Reaktion von Proteinen mit reaktiven α-Dicarbonylverbindungen ................................... 26
2.3.1 Wichtige α-Dicarbonylverbindungen ..................................................................... 26
2.3.1.1 Glyoxal ............................................................................................................. 28
2.3.1.2 Methylglyoxal .................................................................................................. 30
2.3.2 Reaktion von α-Dicarbonylverbindungen mit Proteinen........................................ 31
2.3.3 Reaktion von α-Dicarbonylverbindungen mit freien Aminosäuren ....................... 35
2.3.4. Reaktion von α-Dicarbonylverbindungen am N-Terminus von Peptiden und
Proteinen........................................................................................................................... 37
2.4 Kenntnisse zu Verbindungen mit 2(1H)-Pyrazinon-Struktur............................................. 40
2.4.1 2(1H)-Pyrazinone in Lebensmitteln und Lebensmittelmodellsystemen ................. 40
2.4.2 2(1H)-Pyrazinone in vivo und als Pharmaka........................................................... 42
2.4.3 Naturstoffe mit 2(1H)-Pyrazinon-Struktur.............................................................. 43
3 Experimenteller Teil.............................................................................................................. 45
3.1 Chemikalien, Materialien und Geräte ................................................................................ 45
3.1.1 Chemikalien ............................................................................................................ 45

Inhaltsverzeichnis
3.1.2 Aminosäuren, Peptide und Proteine........................................................................ 47
3.1.3 Materialien und Hilfsmittel ..................................................................................... 48
3.1.4 Geräte ...................................................................................................................... 49
3.2.1 Hochdruckflüssigchromatographie ......................................................................... 50
3.2.2 Kopplung Hochdruckflüssigchromatographie Massenspektroskopie und direkte
Massenspektroskopie ....................................................................................................... 53
3.2.3 Aminosäureanalyse ................................................................................................. 54
3.2.4 Präparative Ionenaustauschchromatographie.......................................................... 55
3.2.5 Tüpfeltest................................................................................................................. 55
3.2.6 Dünnschichtchromatographie (DC) ........................................................................ 56
3.2.7 UV-VIS Spektroskopie ........................................................................................... 56
3.2.8 Fluoreszenzspektroskopie ....................................................................................... 57
3.2.9 Nuklearmagnetische Kernresonanz (NMR)-Spektroskopie.................................... 57
3.2.10 Elementaranalyse .................................................................................................. 58
3.3. Darstellung von Referenzmaterial zur Analytik der Amadori-Produkte........................... 58
3.3.1 Darstellung ausgewählter peptidgebundener Amadori-Produkte............................ 58
3.3.2 Darstellung von Nα-Hippuryl-Nε-carboxymethyl-L-lysin ...................................... 63
3.3.3 Darstellung von Nε-(1-Desoxy-D-fructos-1-yl)-L-lysin ......................................... 65
3.3.4 Darstellung von Pyridosin....................................................................................... 67
3.4 Studien zur Bildung der Hydrolyseprodukte...................................................................... 70
3.4.1 Hydrolyse der Amadori-Produkte ........................................................................... 70
3.4.2 Quantifizierung der Hydrolyseprodukte und Bestimmung der molaren Ausbeuten74
3.4.3 Bildung von Furosin ohne Salzsäurehydrolyse und Stabilität von Nε-Fructoselysin
in Salzsäure ...................................................................................................................... 75
3.5.1 Reaktion von ausgewählten Peptiden mit α-Dicarbonylverbindungen .................. 76
3.5.2 Darstellung von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin .................. 80
3.5.3 Darstellung von 2(1H)-Pyrazinon-Peptiden des Glyoxals ...................................... 83
3.5.4 Darstellung von N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin... 86
3.6 Analyse peptidgebundener 2(1H)-Pyrazinone nach Salzsäurehydrolyse........................... 88
3.6.1 Säurehydrolyse von 2(1H)-Pyrazinon-Peptiden...................................................... 88
3.6.2 Darstellung von 2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure und N1-Ethyl-
2(1H)-pyrazinon............................................................................................................... 90
3.6.3 Einfluss der Matrix auf die Säurehydrolyse von 2(1H)-Pyrazinon-Peptiden ......... 91

Inhaltsverzeichnis
3.7 Untersuchungen zur Reaktion von Insulin mit Glyoxal..................................................... 93
3.8 Untersuchungen zur Reaktion von Insulin mit Glyoxal nach Reduktion und Blockierung
der Sulfhydrylgruppen.............................................................................................................. 96
3.8.1 Reduktion und Blockierung der Sulfhydrylgruppen ............................................... 96
3.8.2 Untersuchungen nach enzymatischer Hydrolyse .................................................. 100
3.9 Untersuchungen zum Nachweis von N1-Alkyl-2(1H)-pyrazinonen nach
Salzsäurehydrolyse des glyoxal-modifizierten Insulins......................................................... 101
3.10 Vergleich der Reaktivität von Lysin, Arginin und dem N-Terminus unter den
Bedingungen der Inkubation von Insulin anhand von Peptiden ............................................ 104
4 Auswertung und Diskussion der Ergebnisse....................................................................... 107
4.1 Analytik von Amadori-Produkten .................................................................................... 107
4.1.1 Darstellung von Amadori-Produkten und Pyridosin als Referenzmaterial ........... 107
4.1.2 Bestimmung der molaren Ausbeuten der Hydrolyseprodukte .............................. 110
4.1.3 Betrachtungen zum Mechanismus der Bildung der Hydrolyseprodukte .............. 116
4.1.4 Untersuchungen in Hinblick auf die Bildung von Nε-Carboxymethyllysin.......... 119
4.1.5 Weitere Studien zur Bildung von Furosin............................................................. 121
4.2 Überblick zur Reaktivität von Peptiden und individuellen Aminosäureseitenketten
gegenüber α-Dicarbonylverbindungen .................................................................................. 123
4.3 Die Reaktion des N-Terminus von Peptiden mit Glyoxal................................................ 125
4.3.1 Die Reaktion von Gly-Ala-Phe mit Glyoxal ......................................................... 125
4.3.2 Identifikation des Reaktionsproduktes .................................................................. 127
4.3.3 Stabilität von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin .................... 130
4.3.4 Zum Mechanismus der Bildung von 2(1H)-Pyrazinon-Peptiden.......................... 131
4.4 Einflussfaktoren auf die Reaktion von Peptiden mit α-Dicarbonylverbindungen und die
Bildung von 2(1H)-Pyrazinon-Peptiden................................................................................. 132
4.4.1 Einfluss der Struktur des Peptides......................................................................... 132
4.4.2 Einfluss der Struktur der α-Dicarbonylverbindung .............................................. 134
4.4.3 Einfluss der Pufferionenart und Konzentration..................................................... 135
4.4.4 Einfluss des pH-Wertes......................................................................................... 137
4.5 Die Reaktion des N-Terminus von Peptiden mit Methylglyoxal..................................... 138
4.5.1 Die Reaktion von Gly-Ala-Phe mit Methylglyoxal .............................................. 138
4.5.2 Identifikation des Reaktionsproduktes .................................................................. 140
4.6 Die Reaktion des N-Terminus von Peptiden mit 3-Desoxyglucosulose .......................... 143

Inhaltsverzeichnis
4.7 Einordnung der Reaktivität des N-Terminus von Peptiden gegenüber
α-Dicarbonylverbindung........................................................................................................ 147
4.8 Analyse peptidgebundener 2(1H)-Pyrazinone nach Salzsäurehydrolyse......................... 149
4.8.1 Salzsäurehydrolyse von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin
........................................................................................................................................ 149
4.8.2 Identifikation der Hydrolyseprodukte von N-[2-(2-Oxo-2H-pyrazin-1-yl)-
propionyl]-phenylalanin................................................................................................. 151
4.8.3 Salzsäurehydrolyse von N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propionyl]-
phenylalanin ................................................................................................................... 154
4.8.4 Einfluss der Matrix auf das Hydrolyseverhalten................................................... 155
4.9 Untersuchungen zur Bildung von 2(1H)-Pyrazinonen am Protein Insulin ...................... 158
4.9.1 Inkubation von Insulin mit Glyoxal unter physiologischen Bedingungen............ 158
4.9.2 Charakterisierung der Reaktionsprodukte am intakten Insulin ............................. 159
4.9.3 Untersuchungen nach Reduktion und Blockierung der Sulfhydrylgruppen ......... 164
4.9.3.1 Charakterisierung der Reaktionsprodukte...................................................... 164
4.9.3.2 Vergleich der Reaktivität der N-Termini von A- und B-Kette ...................... 169
4.9.4 Charakterisierung der Reaktionsprodukte nach enzymatischer Hydrolyse........... 171
4.9.5 Untersuchungen zum Nachweis von N1-Alkyl-2(1H)-pyrazinonen nach
Salzsäurehydrolyse des glyoxal-modifizierten Insulins................................................. 173
4.9.6 Betrachtungen zur Reaktivität von Lysin und Arginin am Insulin ....................... 177
4.9.6 Zur möglichen physiologischen Relevanz der 2(1H)-Pyrazinon-Bildung am Insulin
........................................................................................................................................ 180
4.10 Zur möglichen Bildung von N-terminalen 2(1H)-Pyrazinonen in Lebensmitteln und
in vivo ..................................................................................................................................... 181
5 Zusammenfassung............................................................................................................... 185
6 Ausblick .............................................................................................................................. 189
7 Literaturverzeichnis............................................................................................................. 190

Abkürzungsverzeichnis
Abb. Abbildung
Ac Acetyl
AGE advanced glycation end-product
ALE advanced lipoxidation end-product
AU arbitrary unit
Boc tertiär-Butyloxycarbonyl
Bw Blindwert
Bz Benzoyl
BzG Hippursäure, Nα-Benzoylglycin
BzGFruK Nα-Hippuryl-Nε-(1-desoxy-D-fructos-1-yl)-L-lysin
BzGK Nα-Hippuryl-L-lysin
BzGCMK Nα-Hippuryl-Nε-carboxymethyl-L-lysin
BzGLctK Nα-Hippuryl-Nε-(1-desoxy-D-lactulos-1-yl)-L-lysin
BzGMltK Nα-Hippuryl-Nε-(1-desoxy-D-maltulos-1-yl)-L-lysin
BzGR Nα-Hippuryl-L-arginin
BzGTagK Nα-Hippuryl-Nε-(1-desoxy-D-tagatos-1-yl)-L-lysin
CEL Nε-(1-Carboxyethyl)-lysin
CMA N7-carboxymethylarginin
CMC S-Carboxymethylcystein
CML Nε-Carboxymethyllysin
COSY correlation spectroscopy
DAD Diodenarraydetektor
DEPT distortionless enhancement by polarization transfer
3-DG 3-Desoxyglucosulose
DMSO Dimethylsulfoxid
DOLD deoxyglucosulose-derived lysine dimer
DTT 1,4-Dithiothreitol
EAGLE either advanced glycation or lipoxidation end-product
EDTA Ethylendiamintetraessigsäure

Abkürzungsverzeichnis
em emissison - Emission
ESI-TOF-MS electrospray ionization – time of flight – mass spectrometry
et al. et alii (lat.) - und andere
ex excitation - Anregung
FruK Nε-(1-Desoxy-D-fructos-1-yl)-L-lysin
Glarg 5-(2-Imino-5-oxo-1-imidazolidinyl)-norvalin
GO Glyoxal
GOLD glyoxal-derived lysine dimer
HMBC heteronuclear multiple bond correlation
HPLC high pressure liquid chromatography
HSQC heteronuclear single quantum coherence
I Ionenstärke
LC-ESI-MS liquid chromatography - electrospray ionization – mass spectrometry
λem Emissionswellenlänge
λex Anregungswellenlänge
Lsg Lösung
MGO Methylglyoxal
MOLD methylglyoxal-derived lysine dimer
MOPS 3-Morpholino-propansulfonsäure
Mr relative monoisotopische Molekülmasse
m/z Masse/Ladung
NMR nuclear magnetic resonance - Kernmagnetische Resonanz
NP normal phase, Normalphase
PB phosphate buffer - Phosphatpuffer
PBS phosphate buffered saline - Phosphat gepufferte Kochsalzlösung
RP reversed phase, Umkehrphase
Tab. Tabelle
TAPSO N-[Tris(hydroxymethyl)-methyl]-3-amino-2-hydroxy-propansulfonsäure
TIC total ion current - Totalionenstrom
tR Retentionszeit
TRIS Tris(hydroxymethyl)-aminomethan
RAGE receptor for advanced glycation end-products
UV ultraviolett

1 Einleitung und Zielstellung
Insbesondere aufgrund des Einflusses auf den Genusswert ist die Maillard-Reaktion eine der
wichtigsten Reaktionen in gekochten, gebackenen, gerösteten oder anderweitig erhitzten
Lebensmitteln. Weiterhin findet sie mit langsamer Geschwindigkeit in biologischen Systemen
statt. In diesem Zusammenhang wird sie sowohl mit normalen Alterungsprozessen der
Proteine als auch mit unerwünschten physiologischen Veränderungen diskutiert.
Um die mit der Maillard-Reaktion einhergehenden Vorgänge zu verstehen und gezielt zu
beeinflussen, ist es notwendig die Reaktionswege zu kennen und die Produkte durch eine
geeignete Analytik zu erfassen. Die vorliegende Arbeit beschäftigt sich mit beiden
Teilaspekten. Im ersten Abschnitt werden Untersuchungen zur sicheren Interpretation der
Ergebnisse der Furosin-Analytik durchgeführt. Der zweite Teil widmet sich der Aufklärung
eines unter physiologischen Bedingungen bisher nicht beschriebenen Reaktionsweges sowie
der Charakterisierung und Analyse der neuen Produkte.
Amadori-Verbindungen des Lysins sind die ersten relativ stabilen Intermediate des sehr
komplexen Reaktionsgeschehens. Die oft angewendete Quantifizierung der Amadori-
Verbindungen nach Salzsäurehydrolyse anhand des Hydrolyseproduktes Furosin wurde
jedoch als wenig sicher angesehen. Insbesondere wurde eine gewisse Unsicherheit über die
Höhe der molaren Ausbeute an Furosin, welche die Berechnung der Lysin-Derivatisierung
erlaubt, diskutiert (Resmini et al., 1990, Van Boekel, 1998). Um diese Unsicherheit
auszuräumen, wurden zunächst die in Lebensmitteln und in physiologischen Proben aus
quantitativer Sicht wichtigen Amadori-Produkte des Lysins dargestellt. Anhand von
Hydrolyseexperimenten galt es schließlich die molaren Ausbeuten zu ermitteln und damit
eine sichere Berechnung der Lysin-Derivatisierung zu ermöglichen.
Der zweite Teil der Arbeit widmet sich Reaktionen von α-Dicarbonylverbindungen mit
Peptiden und Proteinen, welche in der fortgeschrittenen Phase der Maillard-Reaktion
stattfinden. Aus der Literatur war bekannt, dass α-Dicarbonylverbindungen bevorzugt mit den
Seitenketten von proteingebunden Arginin, Lysin und Cystein reagieren. Über Reaktionen mit
dem N-Terminus unter physiologischen Bedingungen (pH = 7,4, 37 °C) lagen demgegenüber
keine gesicherten Erkenntnisse vor. Ziel der weiteren Arbeiten war es deshalb, mögliche
Reaktionen der N-terminalen α-Aminogruppe mit physiologisch relevanten α-Dicarbonyl-
verbindungen näher zu untersuchen. Dazu wurden ausgewählte Peptide mit Glyoxal,
- 1 -

1 Einleitung und Zielstellung
Methylglyoxal oder 3-DG inkubiert. Dominierende Produkte konnten anschließend isoliert
und strukturell als 2(1H)-Pyrazinon-Peptide aufgeklärt werden. Es handelt sich dabei um eine
neue Klasse von advanced glycation end-products (AGEs), deren Bildung unter
physiologischen Bedingungen bisher nicht bekannt war. Um die Geschwindigkeit der Bildung
der N-terminalen 2(1H)-Pyrazinon-Struktur mit der von Reaktionen der bekannten
Reaktionspartner für α-Dicarbonylverbindungen gegenüberzustellen, erfolgten Inkubations-
experimente mit Tripeptiden sowie peptidgebundenen Arginin und Lysin. Schließlich stellte
sich die Frage ob die neue N-terminalen Derivatisierung ebenfalls am Protein stattfindet. Zur
Klärung wurde unter physiologischen Bedingungen mit Glyoxal umgesetztes Insulin direkt
und nach gestufter Fragmentierung näher untersucht. Im Weiteren galt es ein allgemein
anwendbares Verfahren zum Nachweis proteingebundener 2(1H)-Pyrazinone zu entwickeln
und zu prüfen.
- 2 -

2 Theoretischer Teil
2.1 Die Maillard-Reaktion
In dem Bestreben die in vivo stattfindende Proteinbiosynthese nachzuvollziehen setzte der
französische Mediziner und Chemiker Louis-Camille Maillard (1878 bis 1936) Glycin und
weitere Aminosäuren mit Glucose und später auch mit anderen Zuckern um. Dabei wurde er
auf eine besonders in der Wärme auftretende Bräunungsreaktion aufmerksam, welche mit der
Entwicklung von Kohlendioxid und der Bildung brauner Pigmente einhergeht (Maillard,
1911, 1912a, 1912b). Er beschäftigte sich im Folgenden intensiv mit dieser Reaktion und
diskutierte sie u. a. im Zusammenhang mit Bräunungsvorgängen bei Lebensmitteln (Maillard,
1917). Nicht zuletzt sah er als biochemisch orientierter Naturwissenschaftler schon damals
eine mögliche Relevanz für Veränderungen in vivo und studierte die Reaktion auch bei
physiologischer Temperatur (Maillard, 1916). Zu Leben und Werk von Louis-Camille
Maillard wird auf den Übersichtsartikel von Billaud & Adrian (2003) verwiesen.
Im Gegensatz zu anderen Namensreaktionen wird unter der nach Maillard benannten
Reaktion keine definierte chemische Reaktion verstanden, sondern eine komplexe
Reaktionsfolge zwischen Aminokomponenten wie Aminosäuren, Peptiden und Proteinen auf
der einen Seite sowie reduzierenden Zuckern und der Abbauprodukten auf der anderen Seite.
Die Phasen der Maillard-Reaktion
Um die im Verlauf der Maillard-Reaktion auftretenden Phänomene und die damit zu
assoziierenden Reaktionen systematisch zu betrachten, hat sich eine Einteilung der
Gesamtreaktionskaskade in drei Stadien (Hodge, 1953) bis heute als sinnvoll erwiesen. Da
das Reaktionsgeschehen sehr komplex ist, sind die Übergänge zwischen den Stadien jedoch
fließend und die Einteilung ist deshalb nicht als Dogma zu sehen.
Die frühe Phase ist gekennzeichnet durch die Kondensation der Amino- mit der
Zuckerkomponente zum Glycosylamin, welches nachfolgend einer Umlagerung zum
Amadori- oder Heyns-Produkt unterliegt. In diesem Stadium wird noch keine Farbbildung und
Absorption im nahen UV-Bereich beobachtet.
- 3 -

2 Theoretischer Teil
In der fortgeschrittenen Phase kommt es zum Abbau der Amadori- und Heyns-Produkte zu
zum Teil sehr reaktiven Intermediaten, welche in Folgereaktionen ein sehr vielfältiges
Produktspektrum liefern. Dies führt zu einer charakteristischen Zunahme der Absorption im
nahen UV-Bereich und dem Auftreten von Fluoreszenz. Reaktionsansätze in diesem Stadium
sind jedoch noch weitgehend farblos oder gelb gefärbt. Zusätzlich ist das Aroma von
geruchsaktiven Verbindungen wahrnehmbar und es wird die Freisetzung von Kohlendioxid
beobachtet.
Das Charakteristikum der finalen Phase ist das Auftreten von braunen Pigmenten, den
Melanoidinen. Weiterhin wird die Bildung von geruchsaktiven Verbindungen und
Kohlendioxid fortgesetzt.
2.1.1 Die Anfangsphase
Bildung von N-Glycosiden
Die ersten fassbaren Produkte der Reaktion zwischen der primären Aminogruppe einer
Aminokomponente und einem reduzierenden Zucker sind N-substituierten Glycosylamine,
welche auch als N-Glycoside bezeichnet werden. Die N-Glycoside von Aminosäuren und
Peptiden sind in freier Form nicht stabil und damit auch nicht isolierbar. Demgegenüber
können die N-Glycoside von Aminosäuren und Aldosen jedoch in Form ihrer Kalium-,
Magnesium-, Calcium- oder Schwermetall-Salze unter wasserfreien Bedingungen erhalten
werden (Weitzel et al., 1957, Beksan et al., 2003). Anhand von umfangreichen
Strukturuntersuchungen an N-Glycosiden von aliphatischen und aromatischen
Aminoverbindungen konnte gezeigt werden, dass die Kohlenhydratkomponente bevorzugt in
der cyclischen β-pyranoiden 4C1-Konformation vorliegt, acyclische Isomere vom Typ einer
Schiff Base konnten an Hand von IR-Untersuchungen ausgeschlossen werden (Paulsen &
Pflughaupt, 1980). Zu dem gleichen Ergebnis kamen Neglia et al. (1983), welche die
primären Glucose-Addukte mit RNase sowie mit n-Butylamin mittels NMR-Spektroskopie
untersuchten. Vom Primär-Addukt konnte jeweils nur das C-1 Atom des cyclischen
Glycosylamins detektiert werden. Ein C-Signal im Resonanz-Bereich der Schiff Basen wurde
dagegen nicht gefunden. Mossine et al. (1999) konnten mittels quantitativer NMR-
Untersuchungen von dem Glycosylamin N-Glucosyl-glycin zeigen, dass im anomeren
- 4 -

2 Theoretischer Teil
Gleichgewicht die β-Pyranose (87 %) gegenüber der α-Pyranose (12 %) dominiert. Die
acyclische „wahre“ Schiff Base wurde ebenfalls nicht detektiert.
Untersuchungen von Tannenbaum (1966) am Insulin-Glucose-System (molares Verhältnis
Insulin:Glucose = 9,4:1; aw = 0,74) zeigten eine höhere Stabilität von am Protein gebildeten
N-Glycosiden an. In der gleichen Weise ist das Vorkommen und die Quantifizierbarkeit eines
N-Glycosids am Hämoglobin, dem sogenannten pre-Hb AIc (= Hb AId), zu interpretieren
(Bolli et al., 1981, 1982, Higgins & Bunn, 1981).
HOO
H
HO
HOH
OH
OH
HOOH
H
HO
HOH
OH
OH
HOO
HO
OHOH
++ H2N R- H2N R
HOOH
H
HO
HOH
OH
OH
H2N R+
HOOH
H
HO
HOH
OH
HN R+
- H2O+ H2O
+ H+
- H+
HOO
H
HO
HOH
HN
OH
R
HOOH
HO
HOH
OH
HN R+
HOOH
HO
HOH
HN
OH
R
HOOH
HOO
HN
OH
R
+ H+- H+
NH
R
+ H+
- H+
Glucose Carbenium-Ion Carbinol-ammonium-Ion
Aminocarbenium-IonImmonium-Ion
Enaminol Glycosylamin
1-Amino-1-desoxyketose 1-Amino-1-desoxyketose (β-Pyranose)
Abb. 2.1.1-1: Reaktion von Glucose mit Aminosäuren - Bildung von Glycosylamin und Amadori-Produkt.
Die Bildung des N-Glycosids kann wie in Abb. 2.1.1-1 am Beispiel der Glucose gezeigt
formuliert werden (Simon & Kraus, 1970, Westphal & Kroh, 1985a). Die Reaktionssequenz
ist dabei grundsätzlich protonenkatalysiert. Im ersten Schritt führt die Protonierung des
- 5 -

2 Theoretischer Teil
endocyclischen Sauerstoffs zu einem Carbenium-Ion, an welches sich nachfolgend das Amin
unter der Bildung eines Carbinolammonium-Ions anlagert (SN2-Reaktion). Durch
Dehydratisierung entsteht eine protonierte Schiff Base (Immonium-Ion) bzw. das mesomere
Aminocarbenium-Ion. Unter Abspaltung eines Protons entsteht schließlich das cyclische
Glycosylamin. Die Reaktion kann auch als SN1-Reaktion oder bei Aminosäuren über einen
synchronen Mechanismus formuliert werden (Westphal & Kroh, 1985a).
Bildung von Amadori-Produkten
Die N-Glycoside der Aminosäuren sind wie bereits erwähnt instabil. Zum einen erfolgt leicht
die Hydrolyse unter Rückbildung der Ausgangsstoffe, und zum anderen kann es zur
sogenannten Amadori-Umlagerung kommen. Beide Prozesse erfolgen im Fall von
Aminosäuren durch die Säurefunktion autokatalytisch.
Der italienische Chemiker Mario Amadori (1886 bis 1941) beschäftigte sich näher mit den
Produkten der Umsetzung von Glucose mit p-Phenetidin (4-Ethoxyanilin) und weiteren
aromatischen Aminen. Nach dem Erhitzen ohne den Zusatz eines Lösungsmittels konnte er
zwei Verbindungen gleicher elementarer Zusammensetzung jedoch unterschiedlicher
Stabilität kristallin erhalten (Amadori, 1925). Das „instabile“ Isomer identifizierte Amadori
als N-Glycosid, was später von Kuhn & Dansi (1936) unter Verwendung von Toluidin als
Base bestätigt wurde. Die Struktur des „stabilen“ Isomers, Amadori vermutete eine Schiff
Base, konnte von Kuhn & Weygand (1937) als das Umlagerungsprodukt Isoglucosamin
(1-Amino-1-desoxyketose, Amadori-Produkt) identifiziert werden.
Es soll an dieser Stelle nicht unerwähnt bleiben, dass schon in früheren Arbeiten die
Umsetzung von aromatischen Aminen mit reduzierenden Zuckern beschrieben wurde
(Wrodnigg & Eder, 2001). Anhand der gewählten Reaktionsbedingungen ist anzunehmen,
dass Basilius Sorokin das Glycosylamin (Sorokin, 1888), Hugo Schiff (1834 bis 1915) die
1-Amino-1-desoxyketose (Schiff, 1871) und Robert Sachsse beide Produkte erhielt (Sachsse,
1871). Die Struktur der Produkte wurde jedoch zum damaligen Zeitpunkt noch nicht erkannt.
Weiterhin ist es bemerkenswert, dass die nach Amadori benannte Umlagerung bereits 1886
von Emil Fischer (1852 bis 1919) als Bestandteil der Bildung von Phenylglucosazon aus
Phenylhydrazin und Glucose beobachtet wurde. Um die Bedeutung ahnend vermutete Fischer
bereits: „Die Bildung des Isoglucosamins aus dem Phenylglucosazon ist der besondere Fall
einer allgemeineren Reaktion.“ (Fischer, 1886).
- 6 -

2 Theoretischer Teil
Der Mechanismus der Amadori-Umlagerung der N-Glycoside von Aldosen lässt sich wie in
Abb. 2.1.1-1 dargestellt beschreiben. Im ersten Schritt kommt es durch eine Protonierung der
endocyclischen Sauerstofffunktion des N-Glycosids zu einer Öffnung des halbacetalen Rings
und die beiden mesomeren Spezies das Immonium-Ion (protonierte Schiff Base) und das
Aminocarbenium-Ion können formuliert werden. Die Elektrophilie dieser Mesomeren führt zu
einer Acidifizierung des an 2-Position befindlichen Protons und fördert damit dessen
basekatalysierte Ablösung. Dieser Schritt wird als geschwindigkeitsbestimmend angesehen.
Das freigesetzte Enaminol tautomerisiert anschließend zur 1-Amino-1-desoxyketose, welche
bevorzugt als cyclisches Halbacetal vorliegt. Durch die Amadori-Umlagerung wird somit ein
Aldosylamin in ein Ketose-Derivat überführt (Simon & Kraus, 1970, Westphal & Kroh,
1985b).
Bildung von Heyns-Produkten
Die Umsetzung von Ketosen mit Aminosäuren führt wie bei den Aldosen über die Schiff Base
ebenfalls zu N-Glycosiden, im Fall der Fructose zum 2-Fructosylamin (Abb. 2.1.1-2).
HO
OH
HO
OOH
OH
HOO
HONHR
OH
OH
HOO
HO
NHR
OH
OH
+ H2N R
- H2N R
HO
NHR
HO
OOH
OH
+ H2O
- H2O
2-Fructosylamin
1-Amino-desoxymannose
Fructose
1-Amino-desoxyglucoseHO
NR
HO
OHOH
OH
OHHO OH
O
OH
NHR
*
*
Schiff Base
3-Keto-Umlagerungsprodukt
Abb. 2.1.1-2: Reaktion von Fructose mit Aminosäuren - Bildung von 2-Fructosylamin, Heyns- und 3-Keto-Umlagerungsprodukt.
Eingeleitet durch 1,2-Enolisierung der Schiff Base erfahren diese analog den Aldosylaminen
eine sogenannte Heyns-Umlagerung, welche zu den entsprechenden 2-Amino-
- 7 -

2 Theoretischer Teil
2-desoxyaldosen (Heyns-Produkten) führt (Heyns et al., 1952, 1956, 1957). Auf diese Weise
entsteht beispielsweise bei der Umsetzung von Fructose mit Glycin 2-N-Glycino-
2-desoxyglucose und erwartungsgemäß auch die epimere 2-N-Glycino-2-desoxymannose
(Heyns et al., 1957). Eine Besonderheit der Heyns-Umlagerung ist ihre Reversibilität (Heyns
& Breuer, 1958). Im Gegensatz dazu ist die Amadori-Umlagerung praktisch nicht reversibel.
Bei der Reaktion von Aminosäuren mit Fructose wird jedoch nicht nur die Bildung des
Heyns-Produktes beobachtet. Heyns et al. (1958) stellte unter Synthesebedingungen auch die
Bildung des Amadori-Produktes fest und führte dies zunächst auf das spezifische
Reaktionsvermögen der endständigen α-Hydroxyketon-Gruppe zurück. Eine vorherige
Isomerisierung der Fructose zu Glucose und Mannose im Sinne einer Lobry-de-Bruyn-
Alberda-van-Ekenstein-Umlagerung (De Bruyn & Van Ekenstein, 1895) und eine
anschließende Reaktion der Aldose zum Amadori-Produkt war nicht nachweisbar.
Entsprechend späterer Betrachtungen von Heyns et al. (1961) dürfte die Bildung des
Amadori-Produktes aus Aminosäuren und Fructose aus einer Sekundärreaktion des Heyns-
Produktes mit einer zweiten Aminosäure resultieren. Die Bildung des Amadori-Produktes
wurde ebenfalls bei der Umsetzung von Fructose mit humanen Serumalbumin (HSA) unter
physiologischen und damit sehr milden Bedingungen (wässrige phosphat-gepufferte Lösung,
pH = 7,4, 37 °C) beobachtet (McPherson et al., 1988).
Im Gegensatz zur Schiff Base aus Glucose und einem Amin, kann die Schiff Base der Fructose
auch einer 2,3-Enolisierung unterliegen und es kann zur Bildung isomerer 3-Keto-
Umlagerungsprodukte kommen (Abb. 2.1.1-2). Auf das Auftreten der 3-Keto-Umlagerungs-
produkte wurde von Suarez et al. (1989) nach Inkubation von bovinem Serumalbumin (BSA)
mit Fructose hingewiesen und nach Weenen et al. (1994) lässt sich nur über sie die Bildung
bestimmter Folgeprodukte wie 1-Desoxy- und 4-Desoxyglucosulose erklären.
2.1.2 Die fortgeschrittene Phase
Ausgangspunkt der fortgeschrittenen Phase ist der Abbau der Amadori- und Heyns-
Verbindungen sowie der 3-Keto-Umlagerungsprodukte zu den Desoxyhexodiulosen
(Desoxyosonen, DGs, siehe Abb. 2.1.2-1). Durch 1,2- bzw. 2,3-Enolisierung und
anschließende Eliminierung der Aminofunktion kommt es zur Bildung von 3-DG bzw. 1-DG.
Vorzugsweise bei Disacchariden bleibt nach 2,3-Enolisierung die Aminofunktion erhalten
und es wird das 1-Amino-4-desoxyoson (1-Amino-4-DG) gebildet (Weenen et al., 1994).
- 8 -

2 Theoretischer Teil
HOO
HONHR
OH
OH
Heyns-Produkt
OHHO OH
O
OH
NHR
*
*
3-Keto-Umlagerungsprodukt
HOO
HO
OHOH
NH
R
Amadori-Produkt
O
O
OH
OH
OH
3-DG
O
O
OH
OH
1-DG
O
O
OH
OH
NHR
1-Amino-4-DG
Abb. 2.1.2-1: Abbau von Amadori- und Heyns-Produkt sowie 3-Keto-Umlagerungsprodukt und Bildung der Desoxyosone (Weenen et al., 1994, modifiziert).
Die Desoxyosone sind sehr reaktiv und insbesondere über Enolisierungsreaktionen können sie
isomeriesieren, cyclisieren, dehydratisieren, sich an Redoxreaktionen beteiligen,
Spaltreaktionen wie der Retroaldolreaktion oder auch oxidativen Spaltungen sowie weiteren
Reaktionen unterliegen. Die daraus resultierenden Intermediate sind zum Teil wiederum
äußerst reaktiv und können ebenfalls die genannten Reaktionen eingehen. Es sind ferner
Reaktionen möglich die zur Knüpfung neuer C-C Bindungen führen, z. B. Reaktionen vom
Aldol-Typ. Weiterhin werden in diesem Stadium sowohl die zuvor freigesetzten
Aminogruppen als auch andere funktionelle Gruppen der Aminosäuren und Proteine,
insbesondere die Guanidinofunktion des Arginins, in die sehr komplexen Umsetzungen
einbezogen. Die Knüpfung neuer Bindungen und anschließende Cyclisierung führt dabei zur
Bildung einer Vielzahl von heterocyclischen Verbindungen (Ledl & Schleicher, 1990,
Weenen et al., 1994).
Eine gewisse Schlüsselrolle kommt in der fortgeschrittenen Phase insbesondere den
kürzerkettigen α-Dicarbonylverbindungen wie Glyoxal und Methylglyoxal zu (siehe
Abschnitt 2.3). Diese bewirken z. B. den raschen Strecker-Abbau von Aminosäuren (siehe
Abschnitt 2.3.3) und initiieren damit eine weitere bedeutende Reaktionskaskade die unter
anderen zu typischen Röstaromastoffen wie den Pyrazinen führt (Ledl & Schleicher, 1990).
- 9 -

2 Theoretischer Teil
Auf ausgewählte und für diese Arbeit relevante Intermediate und Reaktionsprodukte wird im
Abschnitt 2.3 näher eingegangen.
2.1.3 Die finale Phase
In der finalen Phase finden die im fortgeschrittenen Stadium eingeleiteten komplexen
Reaktionen ihre Fortsetzung und es kommt neben der vermehrten Bildung von
heterocyclischen Verbindungen zum Auftreten der sogenannten Melanoidine. Es handelt sich
dabei um braune und per Definition stickstoffhaltige Pigmente deren Molekulargewicht von
> 5000 bis zu Teil > 100000 reicht. Die Bildung ist zum einen ausschließlich aus
niedermolekularen Komponenten wie z. B. Glycin und Glucose möglich und zum anderen
können sie wie im Fall der Milch aus Proteinen und Zuckern entstehen. Im letzten Fall dürfte
die Bildung durch Anlagerung von Chromophoren an das Protein oder durch Bildung der
Chromophore direkt am Protein erfolgen (Ledl & Schleicher, 1990, Rizzi, 1997, Van Boekel,
1998).
Um die Struktur der Melanoidine näher zu untersuchen wurden bisher zahlreiche
niedermolekulare Komponenten miteinander in Anlehnung an die Versuche von Maillard
(Maillard, 1912a) umgesetzt. Unter den niedermolekularen Produkten, den sogenannten
Prämelanoidinen, konnten zahlreiche definierte z. T. farbige Verbindungen wie z. B. Blue-M1
(blue Maillard reaction intermediate-1) strukturell eindeutig aufgeklärt werden (Hayase et al.,
1999). Die Aufklärung der höhermolekularen Melanoidine ist auf Grund der strukturellen
Heterogenität schwierig. Mittels spektroskopischer Methoden konnten heterocyclische
Strukturelemente wie Pyrrol-, Indol-, Pyridin- und Furan-Ringe sowie eine große Anzahl
einfacher funktioneller Gruppen nachgewiesen werden (Ledl & Schleicher, 1990, Rizzi,
1997).
Cämmerer & Kroh (1995) schlossen aus der Analyse von Modellmelanoidinen, welche sie
aus verschiedenen Kohlenhydrate mit Aminosäuren erhielten, dass die elementare
Zusammensetzung und damit auch die Struktur von zahlreichen inneren Parametern, z. B. der
Art des Kohlenhydrates, und äußeren Faktoren wie z. B. der Umsetzung in Lösung oder
lösungsmittelfrei, der Temperatur und der Zeit abhängig ist. Die elementare
Zusammensetzung war jedoch von dem molaren Verhältnis Zucker zu Aminosäure unter
sonst gleichen Reaktionsbedingungen nahezu unabhängig.
- 10 -

2 Theoretischer Teil
Die experimentellen Befunde ließen insgesamt den Schluss zu, dass die Melanoidine keine
fundamentale und allgemeingültige Struktur im Sinn eines regulären Aufbaus aus sich
wiederholenden Einheiten besitzen. Für das Aminosäure-Zucker-System wurde eine
vergleichsweise variable Melanoidin-Grundstruktur vorgeschlagen, welche als
Strukturelemente Desoxyosone und durch Aldol-Kondensation gebildete oligomere
Desoxyosone enthält. Aminosäuren ermöglichen eine weitere Verknüpfung dieser Einheiten
z. B. in Form von Immonium-Brücken (>N+-RAminosäure) oder sie sind als Amadori-Produkt
gebunden (Cämmerer & Kroh, 1995, Cämmerer et al. 2002).
Die Struktur und die funktionellen Eigenschaften von Melanoidinen und auch von farbigen
Prämelanoidinen ist Gegenstand zahlreicher aktueller Untersuchungen. Hervorzuheben sind
hier insbesondere entscheidende Beiträge von der Arbeitsgruppe Hofmann (Hofmann, 1999,
Frank & Hofmann, 2000a, b, Lindenmeier et al., 2002).
2.1.4 Die Maillard-Reaktion in Relation zu anderen Reaktionen
In Lebensmitteln und in vivo finden neben der Maillard-Reaktion weitere
Bräunungsreaktionen statt, wobei mehrere Typen unterschieden werden (Friedman, 1996,
Høltermand, 1966).
Die Karamellisierung ist wie die Maillard-Reaktion eine nichtenzymatische Reaktion und
führt im fortgeschrittenen Stadium ebenfalls zu braunen Pigmenten und aromaaktiven
Verbindungen. Bei der Karamellisierung dienen jedoch ausschließlich Kohlenhydrate als
Ausgangsstoff. Die Umsetzungen sind zum Teil denen der Maillard-Reaktion ähnlich, auf
Grund der fehlenden Amin-Katalyse sind jedoch in der Regel drastischere Bedingungen wie
höhere Temperaturen und stärker saure oder basische pH-Werte erforderlich (Kroh, 1994).
Das bedeutet jedoch nicht, dass die Reaktionswege der Maillard-Reaktion immer dominieren.
Beim Erhitzen von Milch resultiert z. B. aus dem reinen Zuckerabbau mehr 3-Desoxy-
glucosulose als aus dem Abbau des Amadori-Produktes (Van Boekel, 1998). Einige
Reaktionen wie z. B. die Autoxidation von Glucose in Gegenwart von Luftsauerstoff finden
bereits unter physiologischen Bedingungen statt (Thornalley, 1985).
- 11 -

2 Theoretischer Teil
Der Ausgangspunkt der enzymatischen Bräunung sind die in pflanzlichen Material
vorkommenden phenolischen Verbindungen. Diese werden in einer durch Polyphenoloxidase
katalysierten Reaktion weiter hydroxyliert und zu Chinonen oxidiert. Anschließend
polymerisieren die Chinone zu höhermolekularen schwarzen Pigmenten und können damit
farbgebend wirken. Im Unterschied zur Maillard-Reaktion und der Karamellisierung ist die
enzymatische Bräunung an die Gegenwart von Sauerstoff geknüpft.
Als ein Subtyp der enzymatischen Bräunung sind die Interaktionen der Chinone mit
stickstoffhaltigen Verbindungen anzusehen, welche ebenfalls zur Bildung von dunkel
gefärbten Komponenten führt (Friedman, 1996).
Aufgrund der komplexen Zusammensetzung von Lebensmitteln und physiologischen
Systemen finden die einzelnen Bräunungsreaktionen parallel statt und beeinflussen einander.
Das resultierende Reaktionsgeschehen wird damit komplexer.
2.1.5 Zur Bedeutung der Maillard-Reaktion in Lebensmitteln
Durch die Maillard-Reaktion wird der Genusswert von erhitzten Lebensmitteln wesentlich
bestimmt. Die Ausbildung eines lebensmittel-typischen Koch-, Back-, Brat- oder Röst-
Aromas ist hierbei an erster Stelle zu nennen. Dafür sind die zahlreichen heterocyclischen
sowie nicht-cyclischen Verbindungen verantwortlich die ab der fortgeschrittenen Phase
gebildet werden. Weiterhin wird durch die Farbbildung insbesondere beim Backen und
Rösten eine charakteristische Bräunung erreicht (Ledl & Schleicher, 1990). Der Geschmack
wird z. B. durch die Bildung von Röstbitterstoffen beeinflusst (Von Reichenbach, 1844).
Diese Veränderungen sind in dem für das jeweilige Lebensmittel typischen Umfang
gewünscht und charakteristisch. Darüber hinausgehende Veränderungen können zum
off-flavor führen. Dies trifft auch auf Umsetzungen zu die für ein Lebensmittel nicht typisch
sind aber z. B. infolge längerer Lagerung auftreten (Ledl & Schleicher, 1990, Mauron, 1981).
Neben den organoleptischen Eigenschaften nimmt die Maillard-Reaktion auch direkten
Einfluss auf die nutritiven Eigenschaften. Hierbei spielt besonders die Verfügbarkeit der
essentiellen Aminosäuren eine große Rolle. Die chemische Modifikation einer Aminosäure
kann dazu führen, das diese vom Körper nicht mehr für die Proteinbiosynthese genutzt
- 12 -

2 Theoretischer Teil
werden kann. In diesem Zusammenhang schenkt man besonders Lysin Aufmerksamkeit
(siehe Abschnitt 2.2).
Zu einer Modifikation der anderen essentiellen Aminosäuren kommt es erst im Verlauf der
fortgeschrittenen Phase, wobei insbesondere Verluste bei den schwefelhaltigen Aminosäuren
zu verzeichnen sind. Daneben wird in diesem Stadium eine Abnahme der Verdaulichkeit des
gesamten Proteins beobachtet, welche die Verfügbarkeit sämtlicher Aminosäuren einschränkt
(Hurrell et al., 1983, Mauron, 1981, 1990, Möller et al., 1977).
Die Minderung der biologischen Wertigkeit des Proteins einzelner Lebensmittel ist beim
gesunden Erwachsenen und einer gemischten Kost in der Regel kein Problem. In einigen
Fällen wo nur eine sehr begrenzte Anzahl von Lebensmitteln als Ernährungsgrundlage dienen
wie z. B. bei der Säuglingsernährung ist jedoch erhöhte Aufmerksamkeit und die Anwendung
besonders schonender Technologien geboten (Hurrell, 1990).
Eng verknüpft mit der Stabilität und Haltbarkeit von Lebensmitteln sind antioxidativ
wirksame Stoffe. Beim Erhitzen von Lebensmittel kommt es zum Abbau von natürlichen
Antioxidationsmitteln. Gleichzeitig erfolgt aber auch eine Bildung von neuen antioxidativ
wirksamen Verbindungen im Zuge der Maillard-Reaktion wie dies z. B. bei
Tomatenprodukten gezeigt werden konnte (Anese et al., 2003, Giovanelli & Paradiso, 2002).
Antioxidativ wirksamen Maillard-Produkten wird auch in vivo eine positive Rolle
zugeschrieben werden. Van Chuyen et al. (1998) konnten beispielsweise nachweisen, dass
Bestandteile aus einem Peptid-Glucose-Reaktionsgemisch und aus Miso, einem „gebräunten“
Sojaprodukt, auch in vivo antioxidativ wirken. Die Aktivität gegenüber reaktiven
Sauerstoffspezies konnte insbesondere bei Peptid-Glucose-Reaktionsprodukten, Amadori-
Produkten, Modell-Melanoidinen und Melanoidinen aus verschiedenen Lebensmitteln wie
Kaffee und Miso gezeigt werden.
Die Produkte der Maillard-Reaktion werden kontrovers im Zusammenhang mit möglichen
mutagenen und karzinogenen Wirkungen diskutiert. In Modellansätzen und Lebensmitteln
konnten z. B. heterocyclische aromatische Amine identifiziert werden, welche sich in in vitro-
und in in vivo-Testsystemen als potente Mutagene und Karzinogene zeigten (Sugimura et al.,
1990). Gleichzeitig werden jedoch auch antimutagene und antikarzinogene Effekte durch
Bräunungsprodukte beobachtet, wobei insbesondere der antioxidativen Aktivität der
Melanoidine eine Schlüsselrolle zugeschrieben wird (Aeschbacher, 1990).
- 13 -

2 Theoretischer Teil
Die Maillard-Reaktion kann dazu benutzt werden die funktionellen Eigenschaften von
Lebensmittelproteinen unter ausschließlicher Verwendung natürlicher Lebensmittel-
bestandteile gezielt zu verändern. So kann z. B. durch Konjugation eines 34 kDa-Sojaproteins
mit Galactomannan die Löslichkeit insbesondere im isoelektrischen Bereich, die
Hitzestabilität sowie die emulgierenden Wirkung stark verbessert werden. Diese Modifikation
beseitigt gleichzeitig die Allergenität dieses Sojaproteins (Babiker et al., 1998).
2.1.6 Zur Bedeutung der Maillard-Reaktion in vivo
Die Maillard-Reaktion ist nicht auf Lebensmittel beschränkt, sondern sie findet ebenfalls
in vivo statt. In diesem Zusammenhang wird sie in Anlehnung an den englischen Begriff
„glycation“ oft als Glykierung bezeichnet. Zum Teil wird auch der Begriff Glycosylierung
gebraucht. Da es sich abgesehen von den anfangs gebildeten N-Glycosiden bei den Produkten
jedoch nicht um Glycoside handelt und zur Unterscheidung zu den enzymatisch gebildeten
„echten“ Glycosiden am Serin und Asparagin, ist die Verwendung des Begriffs Glykierung
jedoch geeigneter.
Die Proteinglykierung wurde in vivo als erstes beim Hämoglobins (Hb A) beobachtet. Unter
den verschiedenen Glycohämoglobinen (Hb AI) dominiert das sogennante Hb AIc. Als Ort der
Glykierung im Hb AIc konnte von Bookchin & Gallop (1968) die N-terminale
α-Aminogruppe der β-Kette identifiziert werden. Die Aufklärung der Struktur des
gebundenen Hexose-Restes als 1-Amino-1-desoxyfructose und damit als Amadori-Produkt
erfolgte durch Bunn et al. (1975, 1979) sowie Flückiger & Winterhalter (1976).
Zwischenzeitlich machten Rahbar et al. (1969) die interessante Beobachtung, dass der
Hb AIc-Anteil um den Faktor zwei bei Diabetikern erhöht ist. Damit wurde erhebliches
Interesse an in vivo stattfindenden Glykierungsprozessen geweckt. Im Folgenden wurde die
Glykierung sehr vieler Proteine aber auch von Aminophospholipiden (Ravandi et al., 1996)
in vivo beobachtet. Ein erhöhter Glykierungsstatus insbesondere bei Diabetes- und Urämie-
patienten erwies sich dabei als ein generelles Phänomen (Thornalley,1999, Henle & Miyata
2003).
Bunn & Higgins (1981) schrieben der in vivo stattfindenden Proteinglykierung evolutionäre
Bedeutung zu. Sie konnten die Reaktivität verschiedener Aldosen und Ketosen gegenüber
- 14 -

2 Theoretischer Teil
Hämoglobin mit dem Anteil der offenkettigen Form korrelieren und zeigten, dass Glucose
unter den getesteten Aldohexosen am langsamsten reagiert. Die Befunde wurden dahingehend
interpretiert, dass offenbar aus diesem Grund Glucose und nicht ein anderes Monosaccharid
als universeller Metabolit fungiert und somit das Vermögen der Saccharide zur Glykierung
von Proteinen ein Auswahlkriterium in der Evolution war.
Neben der Struktur des reduzierenden Zuckers hat weiterhin das Protein selbst einen
wesentlichen Einfluss auf die Geschwindigkeit der Amadori-Produkt-Bildung. Hierbei sind
insbesondere der pKa-Wert, die sogenannte Mikro-Umgebung und die Zugänglichkeit der
einzelnen Aminogruppen zu diskutieren. So kann der pKa-Wert erheblich durch benachbarte
Aminosäurereste beeinflusst werden. Bestimmte räumlich benachbarte Aminosäuren wie z. B.
Histidin bewirken weiterhin eine lokale Säure-Base-Katalyse oder sie fungieren als
kationische Bindungsstelle für Effektoren, welche dann katalytisch wirken (z. B. Phosphat).
Nicht zuletzt wirkt sich die Art des Lösungsmittels und das Ionenmilieu, z. B. die
Anwesenheit von Phosphat, aus. Im Ergebnis dessen wird die Bildung von Amadori-
Produkten in der Regel an ganz bestimmten individuellen Aminogruppen am Protein
beobachtet (Baynes et al., 1989). Im Fall des Hämoglobins ist der bevorzugte Reaktionsort die
N-terminale α-Aminogruppe der β-Kette, die anderen Aminogruppen sind deutlich weniger
reaktiv (Bunn et al., 1979).
Inzwischen wurde die Glykierung sehr vieler extra- und intrazellulärer Proteine in vivo
beschrieben. Neben der Bildung der frühen Maillard-Produkte kommt es in vivo ebenfalls zur
Bildung von Produkten der fortgeschrittenen Phase. Für diese wurde von Vlassara et al.
(1984) der Begriff AGEs („advanced glycosylation end-products“) eingeführt. Heute wird der
Begriff AGE als Abkürzung für „advanced glycation end-product“ gebraucht. Es werden
darunter stabile Endprodukte der Reaktion von aus Kohlenhydraten hervorgegangenen
Carbonylverbindungen mit Aminokomponenten verstanden. Da einige an der AGE-Bildung
beteiligte Carbonylverbindungen wie z. B. Glyoxal auch bei der Lipidoxidation entstehen,
werden nach Baynes (2000) diese Produkte auch als EAGLEs („either advanced glycation or
lipoxidation end-products“) bezeichnet. Weiterhin findet entsprechend der Begriff ALE
(„advanced lipoxidation end-product“) Anwendung (Requena et al., 1996).
- 15 -

2 Theoretischer Teil
Glykierung - Ursache oder Folge von Krankheiten?
Die Glykierung von Proteinen findet ständig statt und sie ist als ein normaler
Alterungsprozess der Proteine anzusehen. Da Glykierungsprozesse sehr oft mit einer
Änderung der Proteineigenschaften, der Struktur, der Halbwertszeit oder auch der Funktion
einhergehen, dürften sie von physiologischer und gegebenenfalls auch pathophysiologischer
Bedeutung sein (Thornalley, 1999).
Zur Anreicherung von Glykierungsprodukten kommt es z. B. wenn infolge von Diabetes
mellitus die Konzentration an Glucose und anderer glykierenden Agenzien erhöht ist, oder
wenn die Ausscheidung z. B. durch eine renale Dysfunktion vermindert ist. Weiterhin erfolgt
eine Akkumulation an besonders langlebigen Proteinen wie dem Linsenkristallin und
Kollagen (Thornalley, 1999).
Ein vermehrtes Auftreten von AGEs wird bei verschiedenen Krankheiten wie
diabetesbedingten Gefäßerkrankungen, diabetischer Katarakt, Arteriosklerose, chronischer
Niereninsuffizienz, dialysebedingter Amyloidose, neurodegenerativen Erkrankungen wie
Alzheimer sowie mit zunehmenden Alter beobachtet. Welche Rolle AGEs bei diesen
Prozessen zukommt, ob sie ursächlich ist oder ob die verstärkte Bildung und Akkumulation
nur eine Begleiterscheinung ist, wird sehr intensiv und kontrovers diskutiert (Bailey et al.,
1998, Baynes, 2000, 2001, Brownlee, 2000, 2001, Lyons, 2002, Miyata et al., 2000a, 2000b,
Raj et al., 2000, Singh et al., 2001, Stitt, 2001). In letzter Zeit schreiben einige Autoren auch
den mengenmäßig dominierenden proteingebundenen Amadori-Produkten eine patho-
physiologische Rolle zu (Salazar et al., 2000, Schalkwijk et al., 2002).
Nach Baynes (2000, 2001) sind AGEs nicht primär für die Geschwindigkeit des Alterns
verantwortlich, dies ist hauptsächlich die langsame kumulative Schädigung des Genoms. Es
ist jedoch denkbar, das Letzteres auch durch Glykierungsprozesse erfolgen kann.
Die Entstehung von Krankheiten ist multifaktoriell geprägt und Glykierungsprodukte können
deshalb kaum als eine alleinige Ursache angesehen werden. Die Bedeutung von
Glykierungsprodukten ist wahrscheinlich darin zu sehen, dass sie zum Fortschreiten
bestehender und zur Entwicklung neuer pathophysiologischer Prozesse beitragen können,
wenn sie auf einem bestimmten Niveau vorhanden und bestimmte weitere Faktoren gegeben
sind (Baynes, 2001, Stitt, 2001).
- 16 -

2 Theoretischer Teil
Dabei dürften nicht nur veränderte funktionelle Eigenschaften von intrazellulären Proteinen
eine Rolle spielen. Insbesondere bei Komplikationen in Folge von Diabetes mellitus werden
modifizierte extrazelluläre Matrixkomponenten als Ursache für gestörte Matrix-Matrix-,
Matrix-Zell- und Zell-Zell-Interaktionen diskutiert. Weiterhin erfolgt eine Bindung von AGE-
modifizierten Plasmaproteinen an AGE-Rezeptoren wie an RAGE („receptor for advanced
glycation end-products“). In dessen Folge soll es zu bestimmten Zellaktivierungsprozessen
kommen die zur Bildung reaktiver Sauerstoff-Spezies und zu pathophysiologischen
Veränderungen in der Genexpression führen (Brownlee, 2000, 2001). In ähnlicher Weise
werden auch bestimmte Fructoselysin bindende Proteine diskutiert (Salazar et al., 2000).
Neuere Befunde stellen jedoch nachteilige Zellaktivierungsprozesse als Folgen von AGE-
RAGE-Interaktionen in Frage (Valencia et al., 2004).
2.2 Analytik von Amadori-Produkten
2.2.1 Zur Bedeutung von Amadori-Produkten
Amadori-Produkte sind die ersten relativ stabilen Intermediate der Maillard-Reaktion und sie
werden deshalb in sehr vielen Lebensmitteln gefunden. Die Bildung ist besonders bei erhöhter
Temperatur, geringer Wasseraktivität (aw = 0,3 bis 0,7) oder längerer Lagerung begünstigt.
Eine hohe Stabilität und damit eine Anreicherung wird insbesondere bei verminderter
Wasseraktivität, z. B. in Milchtrockenerzeugnissen, Trockenfrüchten und Trockengemüse
beobachtet. Amadori-Produkte sind farb- und geruchlos und damit sensorisch nicht
wahrnehmbar. Sie sind jedoch wichtige Vorstufen für erwünschte oder auch nicht erwünschte
Aroma- und Farbstoffe. Damit sind sie aus lebensmitteltechnologischer Sicht als Parameter
zur Prozesslenkung interessant (Ciner-Doruk & Eichner, 1979, Schräder & Eichner, 1996).
Im Hinblick auf die Stabilität von Lebensmitteln dürfte die ausgesprochen hohe
Reduktionskraft bereits unter milden Bedingungen von Bedeutung sein, welche auch zur
analytischen Detektion genutzt werden kann. Die Desoxyfructosyl-Aminosäuren besitzen
z. B. ein Reduktionsvermögen gegenüber Kaliumhexacyanoferrat(III) in 0,1 M Natronlauge
bei 20 °C die von Glucose oder Fructose erst bei 100 °C erreicht wird. Das
Reduktionsvermögen der isomeren Glycosyl-Aminosäuren ist demgegenüber nur mit dem der
Monosaccharide vergleichbar (Abrams et al., 1955).
- 17 -

2 Theoretischer Teil
In Abhängigkeit von der Zusammensetzung der Lebensmittel bezüglich der Kohlenhydrat-
und Stickstoff-Fraktion werden die entsprechenden Amadori-Produkte von freien
Aminosäuren, Peptiden und Proteinen gefunden. Die Bildung von Amadori-Produkten an den
ε-Aminogruppen von proteingebundenen Lysin-Resten spielt bei den meisten Lebensmitteln
die quantitativ wichtigste Rolle. Da als Amadori-Produkt modifiziertes Lysin für die
Proteinbiosynthese nicht mehr genutzt werden kann und Lysin in vielen pflanzlichen
Proteinen (z. B. Getreideproteine) die limitierende essentielle Aminosäure ist, ist diese Lysin-
Modifizierung aus ernährungsphysiologischer Sicht nicht erwünscht. Infolgedessen war man
sehr früh bemüht das verfügbare und das nicht verfügbare (blockierte) Lysin zu bestimmen,
und auf Grundlage dessen technologische Prozesse zu verbessern. Unter den zahlreichen
Methoden die für diesen Zweck entwickelt wurden hat sich die Bestimmung von Furosin als
sehr vorteilhaft erwiesen (Finot et al., 1981) und sie ist neben der direkten
Fluordinitrobenzol-Methode das genaueste Verfahren (Hurrell et al., 1983).
2.2.2 Analytik von Furosin
Das Aminosäurederivat Furosin (Nε-(2-Furoylmethyl)-lysin) entsteht nicht direkt bei der
Maillard-Reaktion und ist damit nicht originär im Protein vorhanden, sondern es wird erst bei
der für die Aminosäureanalytik angewendeten Salzsäurehydrolyse aus Nε-Desoxyhexosyl-
Derivaten des Lysins gebildet (Abb. 2.2.2-1). Im Gegensatz dazu liefern die sich von
Pentosen ableitenden Amadori-Produkte, Heyns-Produkte und 3-Keto-Umlagerungsprodukte
bei der Salzsäurehydrolyse ohne vorherige Derivatisierung keine stabilen neuen Produkte.
Erbersdobler und Zucker (1966) fanden Furosin erstmals in salzsauren Hydrolysaten von
überhitztem Milchpulver und beschrieben es als unbekannte „Substanz X“, welche bei der
Ionenaustauschchromatographie nach Spackman et al. (1958) dem Arginin folgt.
Die unbekannte Verbindung wurde ebenfalls nach Hydrolyse eines Glucose-Lysin-
Reaktionsgemisches detektiert (Erbersdobler & Zucker, 1966), und schließlich konnte
Nε-Desoxyfructosyllysin als direkter Precursor identifiziert werden (Erbersdobler & Bock,
1967, Brüggemann & Erbersdobler, 1968). Heyns et al. (1968) sowie Finot et al. (1968)
isolierten anschließend die neue Verbindung aus Hydrolysaten von Nε-Desoxyfructosyllysin
und konnten sie strukturell als Nε-(2-Furoylmethyl)-lysin aufklären (Abb. 2.2.2-1). Die
- 18 -

2 Theoretischer Teil
Isolierung aus dem Hydrolysat eines Glucose-Casein-Erhitzungsansatzes mit
darauffolgendem strukturellen Nachweis erfolgte schließlich durch Nakai et al. (1975).
Als ein weiteres Folgeprodukt der Salzsäurehydrolyse von Nε-Desoxyfructosyllysin und
Nε-Desoxylactulosyllysin wurde Pyridosin (6-(5-Hydroxy-2-methyl-4-oxo-4H-pyridin-1-yl)-
L-norleucin, Abb. 2.2.2-1) detektiert (Finot et al. 1969). Ferner wurde eine Rückbildung von
Lysin beobachtet (Erbersdobler & Bock, 1967).
110 °C, 23 h
6 M HClHN
O
NH
O
HN
HO
NH2
O
O
O
H2N
HO
NH2
O
N
HO
NH2
O
O
HO
Nε-Desoxyfructosyllysin
Furosin
Lysin
Pyridosin
O
OH
OH
HOHO
Abb. 2.2.2-1: Salzsäurehydrolyse von Nε-Desoxyfructosyllysin.
Furosin wurde schnell als geeigneter Marker für die frühe Phase der Maillard-Reaktion
erkannt und insbesondere für die Beurteilung der Wärmebehandlung von Milchprodukten
sowie zur Verbesserung technologischer Prozesse herangezogen (Finot et al., 1981,
Erbersdobler, 1986). Die Bestimmung von Furosin und Lysin mittels Aminosäureanalyse
erlaubt weiterhin sowohl die Berechnung des biologisch verfügbaren als auch des nicht
verfügbaren Lysins (Bujard & Finot, 1978). Da zur Ermittlung des blockierten Lysins der
Ausgangsgehalt an Lysin nicht bekannt sein muss, ist dies ein Vorteil gegenüber anderen
Verfahren wie z. B. der Fluordinitrophenylierung. Weiterhin kommt es bei der Bestimmung
nicht zu Interferenzen durch die Anwesenheit des Glycosylamins und der im Gleichgewicht
befindlichen Schiff Base wie dies beim Natriumborhydrid-Verfahren zu beobachten ist
(Hurrell & Carpenter, 1981). Dies ist insbesondere bei erhitzter Milch von Interesse, da in
dieser das biologisch verfügbare Glycosylamin gegenüber dem Amadori-Produkt quantitativ
dominiert (Finot et al., 1977).
- 19 -

2 Theoretischer Teil
Methoden zur Bestimmung von Furosin
Furosin wurde und wird bei der Aminosäureanalyse mittels Kationenaustausch-
chromatographie und Nachsäulenderivatisierung mit Ninhydrin bestimmt, wobei sich die
Elution am Ende des Chromatogramms und damit die gute Abtrennung als vorteilhaft erwies
(Brandt & Erbersdobler, 1972, Henle et al., 1991). Schleicher & Wieland (1981) führten zur
Bestimmung die HPLC mit UV-Detektion bei 280 nm ein und konnten auf diese Weise die
Nachweisgrenze erheblich senken. Dies ermöglichte die nachweisstarke Untersuchung
biologischer Proben und verbesserte damit wesentlich die Kenntnisse um in vivo stattfindende
Glykierungsreaktionen (Schleicher et al., 1981). Im Folgenden wurden weitere Verfahren zur
Furosin-Bestimmung entwickelt, z. B. die Gaschromatographie (Büser & Erbersdobler,
1985), die Kapillarelektrophorese (Tirelli, 1998) sowie die HPLC mit massen-
spektroskopischer Detektion (Takemura et al., 1997). Die gaschromatographische Methode
erwies sich jedoch für die Routineanalytik als ungeeignet (Ruttkat & Erbersdobler, 1994).
Insbesondere bei Milchprodukten wird heute ein Ionenpaar-HPLC-Verfahren nach Resmini et
al. (1990) angewendet bei der das Hydrolysat vorher an einer C18-Phase gereinigt wird. Die
Nachweisgrenze wurde bei dem Verfahren nach Resmini et al. (1990) zu ca. 2 pmol ermittelt
(Knobloch, 1999), das bei der Kationenaustauschchromatographie beträgt ca. 200 pmol.
Furosin als Güteparameter bei Milchprodukten
Bei der Wärmebehandlung und Lagerung von Milch und Milchprodukten steigt der
Furosingehalt (nach Hydrolyse) in der Initialphase linear an. Die Bildung lässt sich mit einer
Kinetik pseudo-nullter Ordnung beschreiben (Claeys et al., 2001). Im fortgeschrittenen
Stadium der Maillard-Reaktion sinkt der Gehalt dagegen wieder. Furosin ist folglich ein sehr
geeigneter Indikator für den Wärmeeintrag bzw. die Lagerbedingungen in der frühen Phase
der Maillard-Reaktion, welche bei normal prozessierten Milchprodukten praktisch nicht
überschritten wird (Finot et al., 1981, Lopez-Fandino & Olano, 1999).
In Tabelle 2.2.2-1 sind die ermittelten Furosingehalte einiger wichtiger Milchprodukte
zusammengestellt. Die bei den Milchpulvern angegebenen Gehalte betreffen frische und sehr
schonend hergestellte Pulver. Nach Van Renterghem & De Block (1996) kann es bei
ungünstiger Lagerung, insbesondere leicht erhöhten Temperaturen, sehr schnell zum Anstieg
der Gehalte auf über 1000 mg / 100 g Protein kommen (12 d, 37 °C).
- 20 -
2 Theoretischer Teil
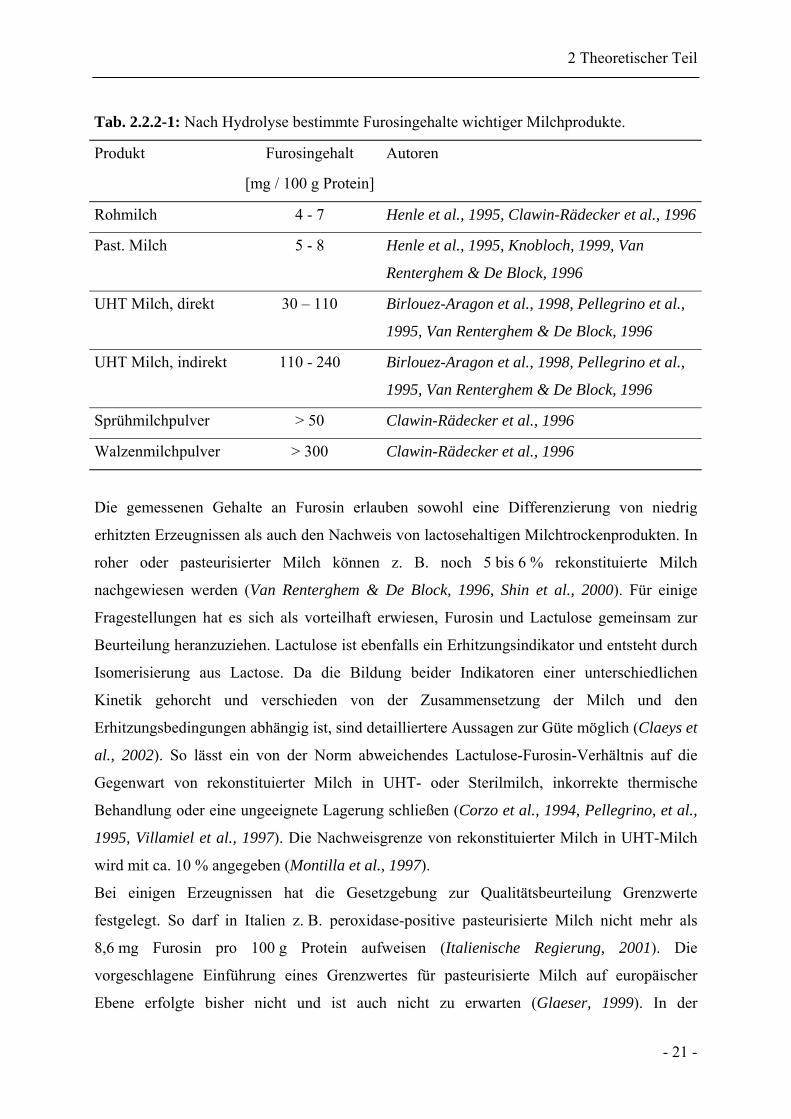
Tab. 2.2.2-1: Nach Hydrolyse bestimmte Furosingehalte wichtiger Milchprodukte.
Produkt Furosingehalt
[mg / 100 g Protein]
Autoren
Rohmilch 4 - 7 Henle et al., 1995, Clawin-Rädecker et al., 1996
Past. Milch 5 - 8 Henle et al., 1995, Knobloch, 1999, Van
Renterghem & De Block, 1996
UHT Milch, direkt 30 – 110 Birlouez-Aragon et al., 1998, Pellegrino et al.,
1995, Van Renterghem & De Block, 1996
UHT Milch, indirekt 110 - 240 Birlouez-Aragon et al., 1998, Pellegrino et al.,
1995, Van Renterghem & De Block, 1996
Sprühmilchpulver > 50 Clawin-Rädecker et al., 1996
Walzenmilchpulver > 300 Clawin-Rädecker et al., 1996
Die gemessenen Gehalte an Furosin erlauben sowohl eine Differenzierung von niedrig
erhitzten Erzeugnissen als auch den Nachweis von lactosehaltigen Milchtrockenprodukten. In
roher oder pasteurisierter Milch können z. B. noch 5 bis 6 % rekonstituierte Milch
nachgewiesen werden (Van Renterghem & De Block, 1996, Shin et al., 2000). Für einige
Fragestellungen hat es sich als vorteilhaft erwiesen, Furosin und Lactulose gemeinsam zur
Beurteilung heranzuziehen. Lactulose ist ebenfalls ein Erhitzungsindikator und entsteht durch
Isomerisierung aus Lactose. Da die Bildung beider Indikatoren einer unterschiedlichen
Kinetik gehorcht und verschieden von der Zusammensetzung der Milch und den
Erhitzungsbedingungen abhängig ist, sind detailliertere Aussagen zur Güte möglich (Claeys et
al., 2002). So lässt ein von der Norm abweichendes Lactulose-Furosin-Verhältnis auf die
Gegenwart von rekonstituierter Milch in UHT- oder Sterilmilch, inkorrekte thermische
Behandlung oder eine ungeeignete Lagerung schließen (Corzo et al., 1994, Pellegrino, et al.,
1995, Villamiel et al., 1997). Die Nachweisgrenze von rekonstituierter Milch in UHT-Milch
wird mit ca. 10 % angegeben (Montilla et al., 1997).
Bei einigen Erzeugnissen hat die Gesetzgebung zur Qualitätsbeurteilung Grenzwerte
festgelegt. So darf in Italien z. B. peroxidase-positive pasteurisierte Milch nicht mehr als
8,6 mg Furosin pro 100 g Protein aufweisen (Italienische Regierung, 2001). Die
vorgeschlagene Einführung eines Grenzwertes für pasteurisierte Milch auf europäischer
Ebene erfolgte bisher nicht und ist auch nicht zu erwarten (Glaeser, 1999). In der
- 21 -

2 Theoretischer Teil
Europäischen Gemeinschaft gilt jedoch ein Grenzwert von 10 mg Furosin pro 100 g Protein
für traditionellen Mozzarella im Sinne der Verordnung (EG) Nr. 2527/98 (Europäische
Kommission, 1998).
Furosin in anderen Lebensmitteln und in physiologischen Proben
Furosin wurde auch bei zahlreichen weiteren Lebensmittel als geeigneter Indikator für den
Fortschritt der frühen Maillard-Reaktion und damit als Güte- und Prozesslenkungsparameter
befunden, so z. B. bei Eiern (Hidalgo et al., 2000), gekochtem Schinken (Pompei &
Spagnolello, 1997a, 1997b), Gelee Royale (Marconi et al., 2002), Honig (Villamiel et al.,
2001), Käse (Villamiel et al., 2000), Marmelade (Rada-Mendoza et al., 2002), Teigwaren
(Acquistucci, 2000) und Tomatenprodukten (Hidalgo et al., 1998, Hidalgo & Pompei, 2000,
Giovanelli & Lavelli, 2002). Ein Feld von besonderer Bedeutung sind diätetische Erzeugnisse
wie Säuglings- und Kleinkindernahrungen (Evangelisti et al., 1994, Ferrer, et al., 2003,
Guerra-Hernandez et al., 1999, 2002, Rada-Mendoza et al., 2002) und Produkte für die
enterale Ernährung (Rufian-Henares et al., 2002).
Die Analyse von Furosin findet weiterhin bei biologischen Proben Anwendung. Dies
ermöglicht die Beurteilung der in vivo stattfindenden Proteinglykierung, z. B. zur
glykämischen Kontrolle und Lenkung (Takemura et al., 1997) oder bei Urämie (Floridi et al.,
1999). Sie kann jedoch auch genutzt werden um die Wirkung medizinischer Verfahren wie
z. B. der Hämo- und Peritoialdialyse zu bewerten (Floridi et al., 2002, Wu et al., 1995).
Ferner wird Furosin als definierter chemischer Marker in Studien zum Verbleib von alimentär
zugeführten Amadori-Produkten genutzt (Henle et al., 2000).
Neben Furosin sind auch die N-Furoylmethyl-Derivate anderer Aminosäuren bekannt, welche
analog dem Furosin bei der Salzsäurehydrolyse aus den entsprechenden freien
Nα-Desoxyhexosyl-Aminosäuren entstehen (Lipton et al., 1971). Einige Furoylmethyl-
Aminosäuren eignen sich ebenfalls als Erhitzungs- und Güteindikatoren. Die Anwendung
erfolgte insbesondere bei Honig (Sanz et al., 2003), Orangensaftpulver (Del Castillo et al.,
1998, 1999), Tomatenprodukten (Sanz et al., 2000) und Trockenfrüchten (Sanz et al., 2001).
- 22 -

2 Theoretischer Teil
2.2.3 Zur molaren Ausbeute an Furosin
Wie bereits erwähnt wird bei der Salzsäurehydrolyse aus Nε-Desoxyhexosyl-Derivaten des
Lysins nicht nur Furosin sondern auch Pyridosin und Lysin gebildet, d. h. allein aus der
Quantifizierung des Furosins kann noch nicht auf den Gehalt an Amadori-Produkt
geschlossen werden. Dies schränkt die Aussagekraft der Methode zunächst erheblich ein. Zur
Berechnung des biologisch verfügbaren und des blockierten Lysins ist es erforderlich zu
wissen, wie viel des Amadori-Produkts in Furosin und Lysin überführt wird. Über die Höhe
der molaren Ausbeuten bestehen jedoch gewisse Unsicherheiten, welche einer Klärung
bedürfen (Resmini et al., 1990, Van Boekel, 1998).
In Tab. 2.2.3-1 sind die aus der Literatur bekannten molaren Ausbeuten an den
Hydrolyseprodukten zusammengefasst.
Tab. 2.2.3-1: Molare Ausbeuten der Hydrolyseprodukte nach Hydrolyse verschiedener
Amadori-Produkte mit Salzsäure.
Derivat Säure
[mol/l]
Furosin
[%]
Pyrid.1
[%]
Lysin
[%]
Kalibr.
Furosin
Autoren
For-Lys(Fru)-OH 6,00 20 10 50 Lys
For-Lys(Lct)-OH 7,75 29 11 44 Lys
Finot
& Mauron, 1972
~Lys(Lct)~
(Milchprodukte)
7,80 40 10 50 Arg Brandt
& Erbersdobler, 1972
~Lys(Lct)~
(Milchprodukte)
7,80 36 10 50 Arg Erbersdobler, 1986
~Lys(Lct)~
(Milchprodukte)
6,00 32 k.A.2 k.A.2 Lys Finot et al., 1977
H2N-Lys(Fru)OH 7,80 30 38 32 Furosin Steinig
& Montag, 1982
For-Lys(Fru)-OH 7,80 46 k.A.2 35 Furosin Szölgyenyi et al., 1989
~Lys(Lct)~
(Milchprodukte)
11,003 50 k.A.2 k.A.2 Furosin Watanabe et al., 1995
1 Pyridosin, 2 keine Angabe, 3 10 h, 110 °C
- 23 -

2 Theoretischer Teil
Wie Tab. 2.2.3-1 zu entnehmen ist, werden bei der Hydrolyse mit der für die
Aminosäureanalyse üblichen 6 M Salzsäure molare Ausbeuten für Furosin von 20 und 32 %
angegeben. Für die Hydrolyse mit 7,8 M Salzsäure bewegen sich die Angaben zwischen 29
und 46 %. Als eine entscheidende Ursache für die nicht unerheblichen Unterschiede ist die
Art der Kalibration und der verwendete Standard anzusehen. Da bis Anfang der Neunziger
Jahre des zwanzigsten Jahrhunderts kein Furosin-Standard kommerziell verfügbar war,
musste dieser entweder selbst als Referenz rein dargestellt werden oder es wurde
näherungsweise über die Kalibriergerade des Lysins oder Arginins ausgewertet.
Wenn auch gewisse Unklarheiten über die Höhe der molaren Ausbeuten der
Hydrolyseprodukte bestehen, so können einige wichtige Randbedingungen als gesichert
gelten. So sind die molaren Ausbeuten unter identischen Hydrolysebedingungen (Art und
Konzentration der Säure, Zeit, Temperatur) konstant. Die Art der Hydrolyse, ob unter
Rückfluss oder in verschlossenen Gefäßen, hat keinen Einfluss. Die Anwesenheit von
reduzierenden Zuckern oder das Protein-Salzsäure-Verhältnis wirkte sich gleichfalls nicht
nachteilig im getesteten Bereich aus (Finot, 1973). Die Konzentration der verwendeten
Salzsäure hat dagegen einen erheblichen Einfluss. Nach Henle et al. (1995) steigt die
Ausbeute an Furosin linear mit der Salzsäurekonzentration im Bereich von 4 bis 8 M (23 h,
110 °C).
Bei Verwendung höher konzentrierter Salzsäure ist eine weitere Steigerung der
Furosinbildung möglich. Dabei werden zwei Effekte beobachtet. So steigt am Anfang der
Hydrolyse die Furosinkonzentration rasch an, limitieren dürfte dabei neben der
Furosinbildung als solche die Freisetzung des Furosins aus dem Proteinverband sein.
Gleichzeitig findet ein nicht unerheblicher Abbau statt, so dass zu einer bestimmten Zeit ein
Maximalwert resultiert (Watanabe et al., 1995, Guerra-Hernandez & Corzo, 1996). Da das
Zeitfenster der maximalen Furosinbildung relativ klein ist, dürfte die Reproduzier- und
Vergleichbarkeit der Ergebnisse vermindert sein. Auf Grund dessen, der nur marginal
höheren Ausbeute und der arbeitstechnisch ungünstige Zeit von 10 h hat sich offenbar ein
Hydrolyseverfahren welches z. B. von Watanabe et al. (1995) beschrieben wurde nicht
durchgesetzt. Als Kompromiss wird heute in der Regel mit 8 M Salzsäure für 23 h bei 110 °C
hydrolysiert.
- 24 -

2 Theoretischer Teil
Um die Problematik über die Höhe der molaren Ausbeute zu umgehen, und trotzdem auf die
Lysin-Derivatisierung zu schließen, wurde von Schleicher & Wieland (1981) ein Hydrolysat
von Nε-Desoxyfructosyllysin als externer Standard zur Kalibration verwendet. Ähnlich ist das
Vorgehen von Takemura et al. (1997), welche der Probe zur Hydrolyse Nα-Formyl-
Nε-Desoxyfructosyl-d4-lysin als internen Standard zusetzten. Diese Vorgehensweisen sind im
Einzelfall geeignet, eine breite Anwendung ist jedoch an die kommerzielle Verfügbarkeit der
Standards geknüpft.
Furosin ist kommerziell erhältlich und kann somit zur Kalibration verwendet werden. Es ist
demzufolge sinnvoll käufliches Furosin als Standard zu verwenden und die molaren
Ausbeuten mit definierten Amadori-Produkten zu bestimmen. Anhand der so ermittelten
Überführungsfaktoren sollte dann eine breite und sichere Anwendung des Furosin-Verfahrens
zur Bestimmung der Lysin-Derivatisierung sowohl in Lebensmitteln als auch in biologischen
Proben möglich sein. Durch die Erweiterung dieses Hintergrundwissens dürfte gleichzeitig
die Verwendung von Furosin als Qualitätsparameter auf eine solidere Grundlage gestellt
werden.
Um dies zu erreichen, sind die in Lebensmitteln quantitativ wichtigen Amadori-Produkte als
Referenzsubstanzen darzustellen und gezielt zu hydrolysieren. Die Quantifizierung der
Hydrolyseprodukte mittels Aminosäureanalyse ermöglicht anschließend die Berechnung der
molaren Ausbeuten sowie der Überführungsfaktoren.
- 25 -

2 Theoretischer Teil
2.3 Reaktion von Proteinen mit reaktiven α-Dicarbonylverbindungen
2.3.1 Wichtige α-Dicarbonylverbindungen
Glyoxal Methylglyoxal
O
O
O
O
O
O
OH
OH
OH
3-DG
O
O
Diacetyl
Abb. 2.3.1-1: Wichtige α-Dicarbonylverbindungen.
α-Dicarbonylverbindungen wie z. B. Glyoxal, Methylglyoxal, 3-Desoxyglucosulose (3-DG)
treten sowohl in Lebensmitteln als auch in vivo als reaktive Intermediate auf. In
Lebensmitteln sind sie wichtige Precursoren für aromaaktive Substanzen und
Bräunungsprodukte, welche im Verlauf der Maillard-Reaktion entstehen. Sie haben damit
wesentlichen Einfluss auf dessen sensorischen und nutritiven Wert. Aus physiologischer Sicht
sind sie insbesondere wegen ihres proteinmodifizierenden Potentials von Interesse, und sie
gelten als Schlüsselverbindungen bei der Bildung von AGEs.
Weiterhin erwiesen sie sich in bestimmten Konzentrationen als cytotoxisch und wirkten im
Ames-Test ohne Aktivierung als mutagen. Als Ursache für die Mutagenität wird die
Reaktivität gegenüber den Purinbasen angesehen (Lee & Shibamoto, 2002).
Die Konzentrationen dieser Verbindungen ist in Lebensmitteln und in vivo in der Regel
gering, z. B. im Vergleich zur Plasmaglucosekonzentration (nüchtern) von ca. 5 mM (Lapolla
et al., 2002). Dies ist auf die hohe Reaktivität und auf in vivo stattfindende Entgiftungs-
prozesse zurückzuführen. Die im menschlichem Plasma gemessenen Gehalte bewegen sich
im µM-Bereich und sind bei bestimmten Krankheiten wie Urämie und Diabetes mellitus
erhöht (Tab. 2.3.1-1), wobei die Angaben offenbar methodisch bedingt voneinander
differieren. Die Ursache für die erhöhten Konzentrationen bei Nierenkranken und Diabetikern
ist sowohl in der vermehrten Bildung als auch in einer verminderten Entgiftung und
Elimination zu sehen.
- 26 -
2 Theoretischer Teil
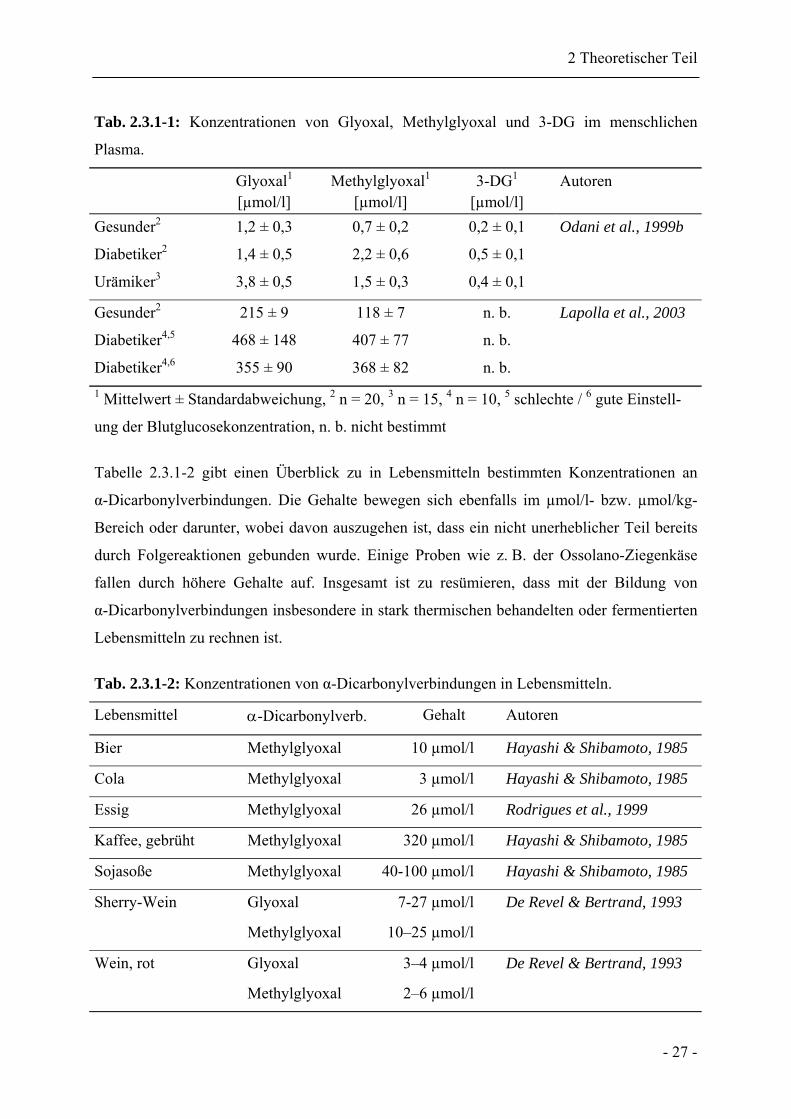
Tab. 2.3.1-1: Konzentrationen von Glyoxal, Methylglyoxal und 3-DG im menschlichen
Plasma.
Glyoxal1
[µmol/l] Methylglyoxal1
[µmol/l] 3-DG1
[µmol/l] Autoren
Gesunder2 1,2 ± 0,3 0,7 ± 0,2 0,2 ± 0,1 Odani et al., 1999b
Diabetiker2 1,4 ± 0,5 2,2 ± 0,6 0,5 ± 0,1
Urämiker3 3,8 ± 0,5 1,5 ± 0,3 0,4 ± 0,1
Gesunder2 215 ± 9 118 ± 7 n. b. Lapolla et al., 2003
Diabetiker4,5 468 ± 148 407 ± 77 n. b.
Diabetiker4,6 355 ± 90 368 ± 82 n. b. 1 Mittelwert ± Standardabweichung, 2 n = 20, 3 n = 15, 4 n = 10, 5 schlechte / 6 gute Einstell-
ung der Blutglucosekonzentration, n. b. nicht bestimmt
Tabelle 2.3.1-2 gibt einen Überblick zu in Lebensmitteln bestimmten Konzentrationen an
α-Dicarbonylverbindungen. Die Gehalte bewegen sich ebenfalls im µmol/l- bzw. µmol/kg-
Bereich oder darunter, wobei davon auszugehen ist, dass ein nicht unerheblicher Teil bereits
durch Folgereaktionen gebunden wurde. Einige Proben wie z. B. der Ossolano-Ziegenkäse
fallen durch höhere Gehalte auf. Insgesamt ist zu resümieren, dass mit der Bildung von
α-Dicarbonylverbindungen insbesondere in stark thermischen behandelten oder fermentierten
Lebensmitteln zu rechnen ist.
Tab. 2.3.1-2: Konzentrationen von α-Dicarbonylverbindungen in Lebensmitteln.
Lebensmittel α-Dicarbonylverb. Gehalt Autoren
Bier Methylglyoxal 10 µmol/l Hayashi & Shibamoto, 1985
Cola Methylglyoxal 3 µmol/l Hayashi & Shibamoto, 1985
Essig Methylglyoxal 26 µmol/l Rodrigues et al., 1999
Kaffee, gebrüht Methylglyoxal 320 µmol/l Hayashi & Shibamoto, 1985
Sojasoße Methylglyoxal 40-100 µmol/l Hayashi & Shibamoto, 1985
Sherry-Wein Glyoxal
Methylglyoxal
7-27 µmol/l
10–25 µmol/l
De Revel & Bertrand, 1993
Wein, rot Glyoxal
Methylglyoxal
3–4 µmol/l
2–6 µmol/l
De Revel & Bertrand, 1993
- 27 -

2 Theoretischer Teil
Lebensmittel α-Dicarbonylverb. Gehalt Autoren
Wein, weiß Glyoxal
Methylglyoxal
9 µmol/l
40 µmol/l
Barros et al., 1999
Butter Diacetyl 6 µmol/kg
330 µmol/kg
Rodrigues et al., 1999
Esteve et al., 2002
Emmentaler-Käse Diacetyl 1 µmol/kg Preininger & Grosch, 1994
Honig Glyoxal
Methylglyoxal
3-DG
29 µmol/kg
33 µmol/kg
1110 µmol/kg
Weigel et al., 2004
Joghurt Glyoxal
Methylglyoxal
Diacetyl
11–16 µmol/kg
8-18 µmol/kg
11-26 µmol/kg
Yamaguchi et al., 1994
Kaffeesahne 3-DG 3 µmol/l Rättich, 2004
Malzbier 3-DG 130 µmol/l Rättich, 2004
Ossolano-Ziegenkäse Diacetyl 52509 µmol/kg Zeppa et al., 2001
Parmesan,
direkt gesalzen
in Lake gesalzen
Methylglyoxal
Methylglyoxal
77 µmol/kg
9 µmol/kg
Lindsay & McDonald, 1992
2.3.1.1 Glyoxal
Glyoxal kann sowohl nichtenzymatisch als auch im Zuge enzymatischer Prozesse entstehen
(siehe Abb. 2.3.1.1-1). Auf nichtenzymatischen Weg kann die Bildung durch reinen
Zuckerabbau (Karamellisierung) oder amin-katalysiert erfolgen. Beim reinen Zuckerabbau
wird zunächst eine Fragmentierung (Retroaldol-Reaktion) der Glucose zu Glycolaldehyd
angenommen, welcher anschließend autoxidativ durch Sauerstoff zu Glyoxal oxidiert wird
(Thornalley, 1985). Es wird jedoch auch eine Retroaldol-Reaktion der Glucose diskutiert, die
direkt zu Glyoxal führt (Thornalley et al., 1999). Wesentlich schneller als der reine
Zuckerabbau erfolgt jedoch die Bildung von Glyoxal in Gegenwart einer Amino-Komponente
durch die Maillard-Reaktion (Thornalley et al., 1999). Hierbei kommt dem sogenannten
Namiki-Weg (Namiki & Hayashi, 1983) eine Schlüsselrolle zu. Dieser beinhaltet einen Abbau
- 28 -

2 Theoretischer Teil
des Glycosylamins durch Fragmentierung und Oxidation, wobei zur Reihenfolge dieser
Teilschritte und zu den Intermediaten in der Literatur konträre Angaben gemacht werden
(Namiki & Hayashi, 1983, Hofmann et al., 1999). Ferner wurde Glyoxal als ein
Oxidationsprodukt von Ascorbinsäure beschrieben (Osthoff, 1966). Ein weiterer Weg zur
Glyoxal-Bildung ist die Autoxidation von ungesättigten Fettsäuren (Loidl-Stahlhofen &
Spiteller, 1994).
Glyoxal
Glucose Glycosylamin Amadori-Produkt
Lipide
Serin
PyrazineOxazoleThiazole
DihydroxyimidazolidinylnorvalinCarboxymethyllysin
Hydroxyacetat
Ascorbat
Abb. 2.3.1.1-1: Bildung, Entgiftung und Reaktionen von Glyoxal (Literatur siehe Text).
Für die Bildung in vivo dürfte der enzymkatalysierte Abbau von Serin durch das Myelo-
peroxidase-Wasserstoffperoxid-Chlorid-System in Phagocyten von Bedeutung sein. Dieser
führt zunächst zur Bildung von Glycolaldehyd und findet bei Entzündungsprozessen statt
(Anderson et al., 1997). Zur Entgiftung ist in vivo das glutathionabhängige Glyoxalase-
System vorgesehen, welches das cytotoxisch wirkende Glyoxal in die inerte
Hydroxyessigsäure überführt. Hydroxyessigsäure wird anschließend über den Urin
ausgeschieden (Thornalley, 1990, 1998).
Sowohl Glyoxal als auch Methylglyoxal weisen eine sehr hohe Carbonylaktivität auf und
reagieren leicht mit Nucleophilen. Als bedeutsam werden insbesondere die Umsetzungen mit
freien Aminosäuren bei erhöhter Temperatur (Strecker-Abbau, siehe Abschnitt 2.3.3) sowie
die Reaktionen mit nucleophilen Seitenketten von proteingebunden Aminosäuren (siehe
Abschnitt 2.3.2) angesehen.
- 29 -

2 Theoretischer Teil
2.3.1.2 Methylglyoxal
Methylglyoxal wird ähnlich dem Glyoxal auf verschieden Wegen gebildet (Abb. 2.3.1.2-1).
Im Zuge der Maillard-Reaktion erfolgt die Bildung über das Amadori-Produkt, dessen Abbau
zum 1-DG und 3-DG und deren basekatalysierten Fragmentierungen (Retroaldolreaktion)
(Weenen, 1998). Es wird jedoch auch von der direkten Bildung aus dem Amadori-Produkt
durch Retroaldolreaktion berichtet (Hayashi & Namiki, 1986). Die Genese direkt aus Glucose
erfolgt ebenfalls durch basekatalysierte Fragmentierung (Thornalley et al., 1999). Im
Gegensatz zur nichtenzymatischen Glyoxal-Bildung sind diese Reaktionswege beim
Methylglyoxal nicht oxidativ.
Methylglyoxal
Glucose
Amadori-Produkt Aceton
Threonin
PyrazineOxazoleThiazole
MethylimidazolidinylnorvalinCarboxyethyllysin
D-Lactat
Desoxyosone
Triosephosphate
1,2-Propandiol
Abb. 2.3.1.2-1: Bildung, Entgiftung und Reaktionen von Methylglyoxal (Literatur siehe Text).
In vivo hat zusätzlich die Bildung aus Metaboliten des Stoffwechsels Bedeutung. Die größte
Rolle dürfte dabei der nichtenzymatischen Fragmentierung von Triosephosphaten zukommen,
welche als Intermediate der Glycolyse und des Polyol-Weges auftreten. Weiterhin wurde die
enzymatische Bildung im Katabolismus von Threonin und des Ketonkörpers Aceton
beschrieben. Die enzymatische Entgiftung erfolgt in erster Linie durch das
glutathionabhängige Glyoxalase-System und führt zur Bildung von D-Lactat. Weiterhin wird
Methylglyoxal durch die NADPH-abhängige Aldose-Reduktase zu 1,2-Propandiol umgesetzt
(Thornalley, 1990).
- 30 -

2 Theoretischer Teil
Methylglyoxal weist eine ähnlich hohe Reaktivität gegenüber primären Amino- sowie
Guanidino- und Sulfhydrylgruppen auf wie Glyoxal und es kommt folglich ebenfalls zur
Bildung von Folgeprodukten des Strecker-Abbaus und der Reaktion mit
Aminosäureseitenketten (siehe Abschnitt 2.3.3 und 2.3.2).
2.3.2 Reaktion von α-Dicarbonylverbindungen mit Proteinen
Auf Grund ihrer hohen Reaktivität kommt Verbindungen mit α-Dicarbonylstruktur bei der
posttranslationalen Modifikation von Proteinen im fortgeschrittenen Stadium der Maillard-
Reaktion eine Schlüsselrolle zu. Die Bildung von AGEs findet dabei insbesondere an den
nucleophilen Seitenketten von Lysin, Arginin und Cystein statt. Die Reaktion kann zu
monovalenten Derivaten führen, d. h. nur eine Aminosäureseitenkette wird modifiziert. Durch
Folgereaktionen entstehen jedoch auch vernetzende Aminosäurederivate, welche
intermolekular ausgebildet die Oligomerisierung von Proteinen bewirken (Thornalley et al.,
2003, Thorpe & Baynes, 2003).
Dem Nachweis und der Bestimmung von AGEs in vivo und in Lebensmitteln geht in der
Regel die Untersuchung von Modellansätzen möglicher Precursoren voraus. Die Isolierung
und strukturelle Aufklärung dort gebildeter Reaktionsprodukte ermöglichen anschließend die
Etablierung geeigneter Analyseverfahren und die gezielte Suche in komplexen Matrizes.
Nicht zuletzt auf Grund dieses systematischen Herangehens ist die Anzahl der identifizierten
und auch quantifizierten Verbindungen sehr groß und wächst ständig. Im Folgenden soll
deshalb vorzugsweise auf ausgewählte und für die vorliegende Arbeit relevante Produkte
Bezug genommen werden.
Nε-Carboxymethyllysin (CML, siehe Abb. 2.3.2-1) ist eines der analytisch bedeutendsten
Derivate. Die Bildung erfolgt hauptsächlich durch oxidativen Abbau von Amadori-Produkten
(Ahmed et al., 1986) und auf dem sogenannten „Namiki-Weg“ (Namiki & Hayashi, 1983) aus
Glycosylaminen (Glomb & Monnier, 1995). Weiterhin wurde CML als Produkt der Reaktion
von Lysin mit Glyoxal beschrieben (Al-Abed & Bucala, 1995). Das Strukturanaloge
Nε-(1-Carboxyethyl)-lysin (CEL) wird entsprechend bei der Reaktion von Lysin mit
Methylglyoxal gebildet (Ahmed et al., 1997). Beide Verbindungen sind salzsäurestabil und
wurden sowohl in Lebensmitteln als auch in physiologischen Proben quantifiziert (Ahmed et
al., 1997, Drusch et al., 1999, Odani et al., 1999a, Helling et al., 2002). Pyrralin
- 31 -

2 Theoretischer Teil
[ε-(2-Formyl-5-hydroxymethyl-pyrrol-1-yl)-norleucin] folgt aus der Reaktion von Lysin mit
3-DG und ist nicht salzsäurestabil. Die Quantifizierung erfolgte in Lebensmitteln und
biologischen Proben nach enzymatischer oder alkalischer Hydrolyse sowie immunologisch
(Hayase et al., 1989, Sengel et al. 1989, Förster & Henle, 2003). Aus der Reaktion von
Glyoxal, Methylglyoxal oder 3-DG mit jeweils zwei Lysin-Seitenketten und vermutlich unter
der Beteiligung von Formaldehyd resultieren die salzsäurestabilen vernetzenden Bislysyl-
imidazolium-Salze GOLD („glyoxal-derived lysine dimer“), MOLD („methylglyoxal-derived
lysine dimer“) und DOLD („deoxyglucosulose-derived lysine dimer“). GOLD und MOLD
wurden in biologischen Proben quantifiziert (Brinkmann et al., 1995, Wells-Knecht et al.,
1995, Skovsted et al., 1998, Odani et al., 1999a, Siegl, 2003). Pentosidin ist ebenfalls
salzsäurestabil und verknüpft die Seitenketten von Lysin und Arginin. Die Bestimmung
erfolgte sowohl in Lebensmittel als auch in physiologischen Flüssigkeiten und Geweben,
wobei oft die intrinsische Fluoreszenz (λex,max = 335 nm, λem,max = 385 nm) zur Detektion
genutzt wurde (Sell & Monnier, 1989, Henle et al., 1997, Odani et al., 1999a).
OHO
NH
H2N
OOH
OHO
NH
H2N
OOH
NHO O
OHO
H2N
N
N
OHO
H2N
OHO
H2N
+N
N
OHO
H2N
OHO
H2N
+
HO
OHHO
N
N
OHO
H2N
OHO
H2N
+
CML CEL Pyrralin GOLD MOLDDOLD Pentosidin
N
N NH
HN
OHO
H2N
+
OHO
H2N
Abb. 2.3.2-1: Ausgewählte Glykierungsprodukte des Lysins und Arginins (Literatur siehe Text).
Im Zusammenhang mit der Reaktion von Arginin mit Glyoxal werden zahlreiche Produkte
diskutiert, insbesondere 5-(4,5-Dihydroxy-2-imino-1-imidazolidinyl)-norvalin (Dihydroxy-
imidazolidin), 5-(2-Imino-5-oxo-1-imidazolidinyl)-norvalin (Glarg), N7-carboxymethyl-
arginin (CMA) und Nδ-(4,5-Dihydro-4-oxo-imidazol-2-yl)-ornithin (αNFC-1,
- 32 -

2 Theoretischer Teil
GO-Imidazolon) (siehe Abb. 2.3.2-2, Schwarzenbolz, 1997, Paul et al., 1998, Glomb & Lang,
2001). Die Vielfalt der Produkte und ihre zum Teil sehr begrenzte Stabilität gestalten die
Quantifizierung jedoch schwierig. CMA konnte bisher nach enzymatischer Hydrolyse im
menschlichen Blutserum bestimmt werden (Odani et al., 2001).
Als Produkte der Umsetzung von Arginin mit Methylglyoxal wurden u. a. Nδ-(4,5-Dihydro-
5-methyl-4-oxo-imidazol-2-yl)-ornithin (MGO-Imidazolon) und das blaue Fluorophor
Argpyrimidin (Nδ-(4,6-Dimethyl-5-hydroxy-pyrimidin-2-yl)-ornithin, λex, max = 320 nm,
λem, max = 382 nm) identifiziert (Henle et al., 1994a, Shipanova et al., 1997). Das
Hydroimidazolon-Derivat weist nur eine geringe Stabilität auf (Glomb & Lederer, 2002), und
konnte nach enzymatischer Hydrolyse in Laugengebäck bestimmt werden (Henle et al.,
1994a). Argpyrimidin wurde in Bier und zahlreichen biologischen Proben quantifiziert
(Glomb et al., 2001, Wilker et al., 2001). Aus der Reaktion von Arginin mit 3-DG resultiert
u. a. das Hydroimidazolon-Derivat Nδ-[4,5-Dihydro-4-oxo-5-(2,3,4-trihydroxybutyl)-
imidazol-2-yl]-ornithin (3-DG-Imidazolon, Imidazolon A). Die Detektion und Quantifizier-
ung erfolgte in Proben biologischer Herkunft (Hayase et al., 1995, Niwa et al., 1997).
H2NO
HO
HN
NHN
O
Glarg
H2NO
HO
HN
NHN
OH
OH
H2NO
HO
HN
NHHN
O
OH
H2NO
HO
HN
NN
OH
H2NO
HO
HN
N NH
O
HO
HOOH
H2NO
HO
HN
N NH
O
H2NO
HO
HN
N NH
O
Dihydroxy- imidazolidin
CMA MGO-Imidazolon
GO-Imidazolon
3-DG-Imidazolon
Argpyrimidin
Abb. 2.3.2-2: Ausgewählte Glykierungsprodukte des Arginins (Literatur siehe Text).
Die freie Sulfhydrylgruppe von Cystein reagiert mit α-Oxoaldehyden rasch und reversibel zu
Hemithioacetalen (siehe Abb. 2.3.2-3, Thornalley, 1998). Bei der Reaktion mit Glyoxal
wurde S-Carboxymethylcystein (CMC) als vergleichsweise stabiles Umlagerungsprodukt
beschrieben (Thorpe & Baynes, 2003).
- 33 -

2 Theoretischer Teil
S
O
OR
NH
OH
O
S
O
HOOH
NH2 O
Hemithioacetal CMC
Abb. 2.3.2-3: Ausgewählte Glykierungsprodukte des Cysteins (Literatur siehe Text).
Vergleichende Untersuchungen von verschiedenen Geweben und physiologischen
Flüssigkeiten zeigten, dass Hydroimidazolone in vivo die quantitativ bedeutsamsten der
untersuchten AGEs sind (siehe Tab. 2.3.2-1). Die Gehalte an CML und CEL sind deutlich
geringer und die der Bislysyl-imidazolium-Salze und des Pentosidins sind vergleichsweise
klein. Aus quantitativer Sicht ist Nε-Fructoselysin das dominierende Glykierungsprodukt. Die
Arginin-Derivatisierung ist jedoch vergleichbar und weist auf die hohe Reaktivität der
α-Oxoaldehyde insbesondere gegenüber Arginin hin (Thornalley et al., 2003).
Tab. 2.3.2-1: Plasmakonzentrationen von AGEs und Nε-Fructoselysin sowie Umfang der
Aminosäure-Derivatisierung bei normal gesunden Probanden (Thornalley et al., 2003).
Analyt Konzentration1
[nM]
Derivatisierung2
[mmol/mol Lys od. Arg]
CML 1109 0,021 ± 0,005
CEL 581 0,011 ± 0,005
GO-Imidazolon 962 0,06 ± 0,03
MG-Imidazolon 15540 0,92 ± 0,12
3-DG-Imidazolon 5922 0,35 ± 0,08
MOLD 42 0,0008 ± 0,0002
Argpyrimidin < 500 < 0,03
Pentosidin 474 0,006 ± 0,002 3
Nε-Fructoselysin 40504 0,77 ± 0,14 1 Mittelwert (n = 5), 2 Mittelwert ± Standardabweichung (n = 5), 3 mmol/mol Lys
- 34 -

2 Theoretischer Teil
2.3.3 Reaktion von α-Dicarbonylverbindungen mit freien Aminosäuren
Einer der bedeutendsten Reaktionswege im Zuge der Maillard-Reaktion ist eine von Adolf
Strecker (1822-1871) erstmals beschriebene Abbaureaktion von α-Aminosäuren durch
bestimmte Carbonylverbindungen (Strecker, 1862). Dabei wird die Aminosäuren simultan
decarboxyliert und desaminiert, und es resultiert der sich von der Aminosäure strukturell
ableitende und um die Carboxylgruppe verkürzte sogenannte Strecker-Aldehyd (Schönberg &
Moubacher, 1952). Diese als Strecker-Abbau bezeichnete Reaktion wird insbesondere beim
Erhitzen von Aminosäuren in der Gegenwart von 1,2-Di- oder 1,2,3-Tricarbonylverbindungen
beobachtet. In Abb. 2.3.3-1 sind zwei diskutierte Reaktionsmechanismen des Strecker-
Abbaus dargestellt.
O
OR2
R3
O
R2
R3 O
R1N
O
H
O
R2
R3
OR1N
O HH
OR1H2N
O HH
- H2O+ H2O+
O
R2
R3
OR1N
O HH
O
R2
R3
R1N
H-
H+O
R2
R3
R1N
H-
O
R2
R3
R1N
H
H
- CO2
H+- CO2
O
R2
R3
R1N
H
H
O
R2
R3
NH2H
O
HR1+
+ H2O- H2O
Aminosäureα-Dicarbonyl Schiff Base1 2
Azomethin-Anion
α-Aminoketon Strecker-Aldehyd
Abb. 2.3.3-1: Reaktion von α-Dicarbonylverbindungen mit Aminosäuren (Strecker-Abbau).
- 35 -

2 Theoretischer Teil
Der erste und essentielle Schritt beim Strecker-Abbau ist die säurekatalysierte Bildung einer
thermisch instabilen Schiff Base (Abb. 2.3.3-1, Schönberg et al, 1948). Daran schließt sich
eine Decarboxylierung an, wobei die undissozierte Säurefunktion der Ausgangspunkt ist (Van
Chuyen et al., 1972). Die Decarboxylierung und die damit einhergehende Umlagerung am
Stickstoff kann sowohl über einen ionischen Mechanismus (Baddar, 1949, 1950) als auch
über einen konzertierten Prozess mit einem cyclischen Übergangszustand (Fujimaki et al.,
1971, Rizzi, 1969) beschrieben werden. Im ersten Fall kommt es zur Bildung eines
mesomeriestabilisierten Azomethin-Anions, welches anschließend das freigesetzte Proton
oder ein Proton aus dem Medium aufnimmt. Die Tautomerisierung des resultierenden Enols
und die darauffolgende Hydrolyse der Azomethin-Bindung führt schließlich zur Freisetzung
eines α-Aminoketons und des Strecker-Aldehydes. Beim zweiten Mechanismus erfolgt die
Übertragung des Protons der Carboxylgruppe auf die Carbonylgruppe in einem konzertierten
Prozess. Denkbar wäre auch eine Übertragung des α-Protons der Aminosäure auf die
Carbonylfunktion, welche bei der Reaktion von Peptiden mit α-Dicarbonylverbindungen
postuliert wird (siehe 4.3.4). Da jedoch α-Aminoisobuttersäure (2-Methylalanin) Strecker-
aktiv ist (Fujimaki et al., 1971, Schönberg et al, 1948), sollte dieser Weg im Fall von
Aminosäuren nicht dominieren.
In der jüngeren Literatur werden beide Reaktionswege mit ähnlicher Häufigkeit angegeben
(Ho, 1996, Mottram et al., 2002, Rizzi et al., 1999). Konzertierte nichtionische Prozesse mit
cyclischem Übergangszustand sind z. B. für die Decarboxylierung von β-Oxocarbonsäuren
belegt (Westheimer & Jones, 1941). Nur der erste Mechanismus kann jedoch einige
Phänomene plausibel erklären. So sind bestimmte Carbonylverbindungen wie z. B.
p-Nitrobenzaldehyd Strecker-aktiv (Baddar, 1949), ohne das sie die Ausbildung eines
cyclischen Übergangszustandes strukturell zulassen. Weiterhin kann nur die Existenz des
Azomethin-Anions im Verlauf des ersten Mechanismus die Bildung von Oxazolidinen als
Folgeprodukte erklären (Ho & Hartman, 1982).
Die Produkte der Strecker-Reaktion und deren Intermediate sind sehr reaktiv und gehen
zahlreiche Folgereaktionen ein. Die dabei entstehenden Pyrazine, Thiazole, Thiazolidine,
Oxazole und Oxazolidine sind wesentlich am Aroma von gekochten, gebackenen und
gerösteten Lebensmitteln beteiligt. Die freigesetzten Strecker-Aldehyde sind als solche schon
aromaaktiv (Ho, 1996).
- 36 -

2 Theoretischer Teil
Der Strecker-Abbau von Aminosäuren durch Glyoxal, Methylglyoxal, Diacetyl und
3-Desoxyglucosulose in Lebensmittel erfolgt bei erhöhten Temperaturen wie 100 °C sehr
rasch. Bei niedrigen Temperaturen wie z. B. bei nur 50 °C ist diese Reaktion jedoch nicht
bevorzugt (Van Chuyen et al., 1972). Unter diesen vergleichsweise milden Bedingungen
kommt es z. B. bei der Reaktion von Alanin mit Glyoxal zur Bildung von
Nα-Carboxymethylalanin (Van Chuyen et al., 1973).
2.3.4. Reaktion von α-Dicarbonylverbindungen am N-Terminus von Peptiden
und Proteinen
Die Aminosäureseitenketten von Lysin, Arginin und Cystein werden als
Hauptreaktionspartner für α-Dicarbonylverbindungen am Protein angesehen und
entsprechende Umsetzungen sind und werden intensiv untersucht (siehe Abschnitt 2.3.2).
Reaktionen von α-Dicarbonylverbindungen am N-Terminus von Peptiden und Proteinen hat
man demgegenüber insbesondere unter physiologischen Bedingungen kaum Beachtung
geschenkt. Die Bildung einiger N-terminaler Modifikationen wurde jedoch unter nicht-
physiologischen oder synthetischen Bedingungen beschrieben (siehe Abb. 2.3.4-1).
Takahashi (1968) konnte bei der Umsetzung des Dipeptides Ala-Gly (1,2 mM) mit
Phenylglyoxal (82,2 mM) bei pH = 8,0 eine sehr rasche Desaminierung der α-Aminogruppe
und die Bildung des α-Ketoacyl-Peptides Pyruvylglycin zeigen (Abb. 2.3.4-1). Der Umsatz
an Peptid war mit ca. 94 % bei 25 °C nach 60 min nahezu vollständig. Die Reaktion wurde
mechanistisch über die Bildung der Schiff Base als Intermediat und eine sich anschließende
Transaminierung ähnlich dem Strecker-Abbau (siehe Abschnitt 2.3.3) erklärt (Takahashi,
1968). Im Unterschied zum Strecker-Abbau wird dabei die Säurefunktion durch die
Peptidbindung stabilisiert und eine peptidgebundene α-Ketosäure resultiert.
Ala-Gly reagierte ebenfalls sehr schnell mit Glyoxal, die präparative Ausbeute an
Pyruvylglycin war jedoch sehr viel geringer. Die Reaktion mit Glyoxal wurde jedoch
insgesamt als ähnlich der mit Phenylglyoxal gewertet und angenommen, dass die Reaktion
der α-Aminogruppe von Proteinen mit Glyoxalderivaten zu einer Ketoacyl-Modifikation des
N-Terminus führt (Takahashi, 1977a).
- 37 -
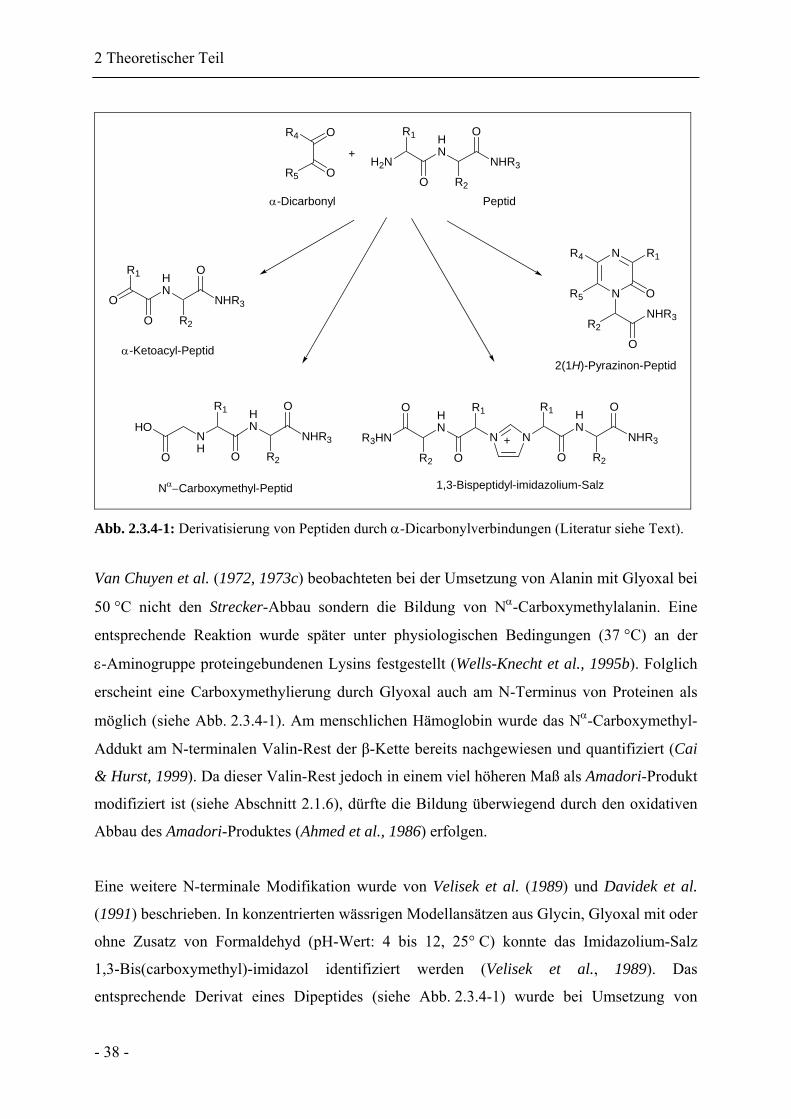
2 Theoretischer Teil
O
OR4
R5
+H2N
HN
NHR3
R1
O R2
O
O
HN
NHR3
R1
O R2
ON
N OR5
R4
R2NHR3
O
R1
NH
HN
NHR3
R1
O R2
O
HO
O
N N
HN
NHR3
R1
O R2
OHN
R3HN
R1
OR2
O
+
α-Ketoacyl-Peptid
Nα−Carboxymethyl-Peptid 1,3-Bispeptidyl-imidazolium-Salz
2(1H)-Pyrazinon-Peptid
Peptidα-Dicarbonyl
Abb. 2.3.4-1: Derivatisierung von Peptiden durch α-Dicarbonylverbindungen (Literatur siehe Text).
Van Chuyen et al. (1972, 1973c) beobachteten bei der Umsetzung von Alanin mit Glyoxal bei
50 °C nicht den Strecker-Abbau sondern die Bildung von Nα-Carboxymethylalanin. Eine
entsprechende Reaktion wurde später unter physiologischen Bedingungen (37 °C) an der
ε-Aminogruppe proteingebundenen Lysins festgestellt (Wells-Knecht et al., 1995b). Folglich
erscheint eine Carboxymethylierung durch Glyoxal auch am N-Terminus von Proteinen als
möglich (siehe Abb. 2.3.4-1). Am menschlichen Hämoglobin wurde das Nα-Carboxymethyl-
Addukt am N-terminalen Valin-Rest der β-Kette bereits nachgewiesen und quantifiziert (Cai
& Hurst, 1999). Da dieser Valin-Rest jedoch in einem viel höheren Maß als Amadori-Produkt
modifiziert ist (siehe Abschnitt 2.1.6), dürfte die Bildung überwiegend durch den oxidativen
Abbau des Amadori-Produktes (Ahmed et al., 1986) erfolgen.
Eine weitere N-terminale Modifikation wurde von Velisek et al. (1989) und Davidek et al.
(1991) beschrieben. In konzentrierten wässrigen Modellansätzen aus Glycin, Glyoxal mit oder
ohne Zusatz von Formaldehyd (pH-Wert: 4 bis 12, 25° C) konnte das Imidazolium-Salz
1,3-Bis(carboxymethyl)-imidazol identifiziert werden (Velisek et al., 1989). Das
entsprechende Derivat eines Dipeptides (siehe Abb. 2.3.4-1) wurde bei Umsetzung von
- 38 -

2 Theoretischer Teil
Diglycin mit Glyoxal und Formaldehyd in verdünnter Lösung bei pH = 4 und 80 °C erhalten
(Davidek et al. 1991). Bislysyl-Imidazolium-Salze wie GOLD und MOLD wurden wenig
später dargestellt und in physiologischen Proben quantifiziert (siehe Abschnitt 2.3.2).
Prey & Petershofer (1968) synthetisierten 2(1H)-Pyrazinon-Derivate (siehe Abb. 2.3.4-1) des
Diglycins durch Umsetzung von Diglycin mit überschüssigem Benzil, Glyoxal,
Methylglyoxal oder Diacetyl bei pH = 13. Da die dargestellten Derivate eine charakteristische
Absorption bei 320 bis 340 nm zeigten und diese UV-Bande auch in Zuckerfabrikationssäften
auftrat, wurde die Bildung von Verbindungen mit 2(1H)-Pyrazinon-Struktur bei der
Zuckerraffination vermutet.
Van Chuyen et al. (1973a, 1973b) erhitzten verschiedene Peptide mit Glyoxal in
konzentrierter Lösung bei 100 °C und pH = 5,0 für 1 h und beobachteten dabei eine intensive
Bräunung der Ansätze. Als niedermolekulare Reaktionsprodukte konnten 2(1H)-Pyrazinon-
Peptide, freie Aminosäuren und Strecker-Aldehyde identifiziert werden. Auffällig war, dass
in Erhitzungsansätzen mit Tripeptiden nicht nur die 2(1H)-Pyrazinon-Derivate der
Ausgangspeptide gefunden wurden, sondern auch die um die C-terminale Aminosäure
verkürzten 2(1H)-Pyrazinon-Dipeptide. Entsprechend waren in den Ansätzen der Tetrapeptide
die um die C-terminale Aminosäure verkürzten 2(1H)-Pyrazinon-Tripeptide und die um eine
weitere Aminosäure verkürzten 2(1H)-Pyrazinon-Dipeptide detektierbar. Es musste folglich
ein vom C-Terminus beginnender Abbau der 2(1H)-Pyrazinon-Peptide stattgefunden haben.
Aus Modellstudien zur Farbbildung wurde der Schluss gezogen, dass 2(1H)-Pyrazinone eine
die Bräunung fördernde Rolle spielen und dass dafür wahrscheinlich Reaktionen zwischen
den 2(1H)-Pyrazinonen und Zwischenprodukten die zu ihrer Entstehung führen
verantwortlich sind. Die Relevanz der beobachteten Reaktionen wurde für Peptid-Carbonyl-
Systeme wie Soja-Sauce und andere prozessierte Sojaprodukte bei der Farb- und
Aromabildung (Strecker-Aldehyde) gesehen. Weiterhin wurde der Einfluss auf den
Geschmack diskutiert. So schmeckt das Peptid Gly-L-Leu sehr bitter, das aus der Reaktion
mit Glyoxal resultierende 2(1H)-Pyrazinon-Peptid dagegen astringierend, ein wenig säuerlich
und später mild (Van Chuyen et al., 1973a).
- 39 -

2 Theoretischer Teil
2.4 Kenntnisse zu Verbindungen mit 2(1H)-Pyrazinon-Struktur
2.4.1 2(1H)-Pyrazinone in Lebensmitteln und Lebensmittelmodellsystemen
Da man sich nur wenig mit der Rolle von Peptiden bei der Maillard-Reaktion in Lebensmittel
beschäftigt hat, liegen nur einige wenige Studien zur Bildung von 2(1H)-Pyrazinonen vor. Im
wesentlichen sind dies die bereits erwähnten Untersuchungen von Van Chuyen et al. (1973a,
b), welche neben anderen Produkten 2(1H)-Pyrazinon-Peptide bei der Umsetzung von
Peptiden mit Glyoxal (pH = 5,0, 100 °C, 1 °h) erhielten (siehe Abschnitt 2.3.4). Bei späteren
Studien zur Bildung aromaaktiver Verbindungen wurden freie Aminosäuren oder Peptide mit
Glucose im Autoklaven bei pH = 5,2 und 180 °C für 2 h umgesetzt. Bei den peptid-haltigen
Ansätzen konnten N1-Alkyl-2(1H)-pyrazinone quantifiziert werden. Die Bildung kann über
den Abbau der Glucose zu α-Dicarbonylverbindungen, deren Reaktion mit dem Peptid zum
2(1H)-Pyrazinon-Peptid und eine sich anschließende Decarboxylierung erklärt werden (siehe
Abb. 2.4.1-1). Den N1-Alkyl-2(1H)-pyrazinone wurde ein einzigartiges röstiges Aroma
zugeschrieben. Aus freien Aminosäuren wurden keine 2(1H)-Pyrazinone gebildet, sie
fungierten als Precursoren für Pyrazine. Bei Di- und Tetrapeptiden dominierte die Bildung
von N1-Alkyl-2(1H)-pyrazinonen. Da diese Peptide nicht zu den freien Aminosäuren
abgebaut werden, fand kaum eine Bildung von Pyrazinen statt. Dipeptide sind hydrolytisch
stabil und Tetrapeptide werden bevorzugt in der Mitte gespalten. In Tripeptiden ist die zweite
Peptidbindung hydrolyselabil und sie lieferten folglich sowohl 2(1H)-Pyrazinone als auch
Pyrazine (Ho et al., 1992, Izzo & Ho, 1992, Izzo et al., 1993, Oh, et al., 1992).
O
OR4
R5
+
α-Dicarbonyl
N
N OR5
R4
R2NHR3
O
R1
Pyrazinon-Peptid
- H2O+ H2O
Peptid
H2N
HN O
R2NHR3
O
R1 N
N OR5
R4
R2OH
O
R1 N
N OR5
R4
R2
R1+ H2O- H2NR3 - CO2
Pyrazinon-Dipeptid N1-Alkyl-pyrazinon
Abb. 2.4.1-1: Bildung von N1-Alkyl-2(1H)-pyrazinonen (Van Chuyen et al, 1973b, Ho et al., 1992, Izzo & Ho, 1992, Izzo et al., 1993, Oh, et al., 1992, modifiziert).
Ein zweiter Bildungsweg für N1-Alkyl-2(1H)-pyrazinone konnte anhand von
Pyrolyseexperimenten (250 °C, 20 s) mit isotopenmarkiertem Glycin und Glucose
- 40 -

2 Theoretischer Teil
nachgewiesen werden. Für die Entstehung von N1-Alkyl-2(1H)-pyrazinonen wurde eine
mehrstufige Reaktion von einem Molekül Glyoxal oder Methylglyoxal mit insgesamt drei
Molekülen Glycin vorgeschlagen. Die Bildung über die Kondensation von zwei Molekülen
Glycin zum Diglycin und anschließende Reaktion entsprechend dem zuerst genannten Weg
konnte ebenfalls unter diesen Bedingungen postuliert werden (Keyhani & Yaylayan, 1996).
Eine für die freie Aminosäure Asparagin spezifische Bildung von 5,6-Dialkyl-5-methyl-
2(1H)-Pyrazinonen wurde bei Umsetzung von Asparagin mit verschiedenen Monosacchariden
in wässriger Lösung (200 °C, 1 h) beobachtet. Dabei findet zunächst eine Isomerisierung von
Asparagin über 3-Aminosuccinimid zu Isoasparagin statt (siehe Abb. 2.4.1-2). Letzteres
reagiert mit einer aus dem Kohlenhydratabbau stammenden α-Dicarbonylverbindung zu
einem 3-Carboxymethyl-2(1H)-pyrazinon, welches anschließend zum Endprodukt
decarboxyliert (Lawrence & Shu, 1995). Von der Decarboxylierung eines 3-Carboxymethyl-
2(1H)-pyrazinons zum 3-Methyl-Derivat wird ebenfalls in der synthetisch orientierten
Literatur berichtet (Taguchi et al., 1996).
O
OR1
R2
H2NHO O
NH2O
OHOH2N
H2N O
NH
O O
NH2
N
NH
OR2
R1
OH
ON
NH
OR2
R1
+Asn 3-Aminosuccinimid
3-Methyl-pyrazinon 3-Carboxymethyl-pyrazinon
Iso-Asn
- CO2
- 2 H2O
Abb. 2.4.1-2: Bildung von asparagin-spezifischen 3-Methyl-2(1H)-pyrazinonen (Lawrence & Shu, 1995).
Insgesamt kann resümiert werden, dass 2(1H)-Pyrazinone aus Modellexperimenten bei stark
erhöhter Temperatur bekannt sind. Der Nachweis in Lebensmitteln wurde jedoch bisher nicht
geführt.
- 41 -

2 Theoretischer Teil
2.4.2 2(1H)-Pyrazinone in vivo und als Pharmaka
Die Bildung von 2(1H)-Pyrazinonen im Zuge von in vivo stattfindenden Glykierungs-
prozessen oder in entsprechenden physiologischen Modellsystemen ist bisher nicht
beschrieben wurden. Es wird jedoch über die Entstehung eines 2(1H)-Pyrazinon-Derivates als
Abbauprodukt von bestimmten β-Lactam-Antibiotika berichtet. So konnte gezeigt werden,
dass Abbau und Aminolyse des Cephalosporins Cefaclor in Gegenwart von n-Propylamin zu
3-Phenyl-6-propylcarbamoyl-2(1H)-pyrazinon führt (siehe Abb. 2.4.2-1). Ein ähnliches
Konjugationsprodukt mit Polylysin wurde durch IgE-Antikörpern von bestimmten Patienten
mit Cefaclor-Allergie erkannt. Folglich können gewisse 2(1H)-Pyrazinon-Derivate als Hapten
bei der Entwicklung einer Aminocephalosporin-Allergie diskutiert werden (Venemalm, 2001).
S
N NH
HOOC
Cl
O
O
H2N N
NH
O
HN
O
H2N1.
2. pH = 7,4
3-Phenyl-6-propylcarbamoyl-2(1H)-pyrazinonCefaclor
Abb. 2.4.2-1: Bildung von 3-Phenyl-6-propylcarbamoyl-2(1H)-pyrazinon (Venemalm, 2001).
Neben diesem 2(1H)-Pyrazinon-Konjugat mit wahrscheinlich nachteiliger Wirkung gibt es
jedoch zahlreiche Verbindungen mit 2(1H)-Pyrazinon-Struktur, welche für gewünschte
pharmakologische Zwecke entwickelt wurden. Es handelt sich dabei insbesondere um
Peptidmimetica mit opioider (Okada, 1999) oder antithrombotischer Wirkung (Sanderson,
1999). Ein potenter und oral verfügbarer Thrombin-Inhibitor mit hoher Selektivität sowie sehr
guter Pharmakokinetik ist das in Abb. 2.4.2-2 dargestellte 2(1H)-Pyrazinon-Derivat
L-375,378 (Sanderson, 1999).
Bemerkenswert an der Struktur von L-375,378 ist die N1-Acetamid-Gruppierung am
2(1H)-Pyrazinon-Ring. Diese ist ebenfalls in 2(1H)-Pyrazinon-Peptiden vorhanden welche
sich von X-Glycyl-Peptiden ableiten (N-[2-(2-Oxo-2H-pyrazin-1-yl)-acetyl]-Peptide, siehe
Abb. 2.4.2-2).
Studien zur Biotransformation von L-375,378 und anderen Thrombin-Inhibitoren dieses Typs
weisen auf eine umfangreiche Metabolisierung des 2(1H)-Pyrazinon-Rings in vivo hin. Es
wurde sowohl die Hydroxylierung direkt am Ring als auch an der Methylgruppe beobachtet.
- 42 -

2 Theoretischer Teil
Weiterhin konnte die Konjugation der hydroxylierten Produkte mit Glutathion festgestellt
werden. Als Folge der metabolischen Aktivierung kam es jedoch auch zur Öffnung des
2(1H)-Pyrazinon-Rings und zur Umlagerung in ein Imidazol-Derivat oder zur Bildung eines
nicht-cyclischen Metaboliten. Eine kovalente Bindung an Plasmaproteine wurde ebenfalls
festgestellt (Riffel et al., 2000, Singh et al., 2003).
N
N O
NH
NH
O
N
H2N
L-375,378
N
N OR3
R1
NHR4
O
R2
N-[2-(2-Oxo-2H-pyrazin-1-yl)-acetyl]-Peptid
Abb. 2.4.2-2: Struktur von L-375,378 (Sanderson, 1999) und Struktur von N-[2-(2-Oxo-2H-pyrazin-1-yl)-acetyl]-Peptiden.
2.4.3 Naturstoffe mit 2(1H)-Pyrazinon-Struktur
Auf Grund ihrer biologischen Wirkung ist man bereits früh auf eine Reihe von
2(1H)-Pyrazinon-Derivaten insbesondere aus Aspergillus-Arten aufmerksam geworden. So
isolierte Dutscher (1947) den antibakteriell wirkenden Sekundärmetaboliten Aspergillussäure
(siehe Abb. 2.4.3-1) aus Aspergillus flavus und identifizierte ihn als 2(1H)-Pyrazinon-Derivat.
Da die ebenfalls gebildete Desoxyaspergillussäure keine antibakterielle Wirkung zeigt, wird
die Hydroxylierung am N1-Stickstoffatom als essentiell für diesen Effekt angesehen
(Dutscher, 1947).
Später wurden weitere 3,6-substituierte Derivate gefunden. Die Biogenese dieser
Verbindungen erfolgt aus zwei Aminosäure-Molekülen. Dabei werden die Kohlenstoffatome
1 und 2 der beteiligten Aminosäuren in den 2(1H)-Pyrazinon-Ring integriert und die
Aminosäure-Seitenketten bestimmen die Substituenten (MacDonald & Bishop, 1977). Das
vermutete Auftreten von Diketopiperazinen als Intermediate konnte nicht bestätigt werden
(Sammes, 1975).
- 43 -

2 Theoretischer Teil
N
NH
O
N
N O
OH
Aspergillussäure Desoxyaspergillussäure
Abb. 2.4.3-2: Struktur von Aspergillussäure und Desoxyaspergillussäure (Sammes, 1975).
In letzter Zeit führte die intensive Suche nach biologisch aktiven Stoffen im Bereich der
marinen Alkaloide zur Aufklärung von 3,6-Bis(indolyl)-2(1H)-pyrazinon- und 3-Benzyl-
5-phenyl-2(1H)-pyrazinon-Derivaten, welche sich vom Tryptophan bzw. Tyrosin ableiten
(Wright et al., 1992, Hirano et al., 2000). Ein besonders bemerkenswerter Vertreter ist
Dragmacidin D (siehe Abb. 2.4.3-2). Dragmacidin D wurde aus dem Tiefseeschwamm
Spongosoritis sp. isoliert und zeigt ein breites Spektrum an biologisch und pharmakologisch
interessanten Wirkungen (Wright, et al., 1992). So wird unter anderem von einer selektiven
Hemmung der neuronalen Stickstoffmonoxid-Synthase berichtet, welche ein therapeutischer
Ansatzpunkt für die Behandlung neurodegenerativer Erkrankungen wie z. B. der Alzheimer
Krankheit ist (Longley et al., 2000).
N
NH
O
HN
NH
HO
Br
NHN
NH2
Dragmacidin D
Abb. 2.4.3-2: Struktur von Dragmacidin D (Wright et al., 1992).
- 44 -

3 Experimenteller Teil
3.1 Chemikalien, Materialien und Geräte
3.1.1 Chemikalien
Verwendete Chemikalien unter Angabe von Bestellnummer, Reinheit und Verantwortlichen,
soweit im Text nicht anders vermerkt:
Acetonitril, chromasolv gradient grade, 34851, Riedel-de Haen, Seelze
Ameisensäure, 98-100 %, 33015, Riedel-de Haen, Seelze
Aminoguanidin, Bicarbonatsalz, A-7259, Sigma, Taufkirchen
Ammoniak 25 % p.a., 30501, Riedel-de Haen, Seelze
di-Ammoniumhydrogenphosphat, p.A., 30402, Riedel-de Haen, Seelze
Anionenaustauscher, Dowex 1X8, p.a., Chlorid-Form, 200 – 400 mesh, 44339, Fluka,
Taufkirchen
Borsäure, p.a., 31146, Riedel-de Haen, Seelze
Bromwasserstoffsäure, 48 %, p.a., 18730, Fluka, Taufkirchen
Citronensäure Monohydrat, p.a., 27490, Fluka, Taufkirchen
3-Desoxyglucosulose (3-DG), hergestellt nach Henle und Bachmann (1996), Diplomarbeit
J. Kahle, TU Dresden, Institut für Lebensmittelchemie
Dimethylformamid, puriss., > 99,5 % , 40228, Fluka, Taufkirchen
Chloroform, p.A., 25690, Fluka, Taufkirchen
Chloroform-d1, 99,9 Atom % D, 41,675-4, Aldrich, Taufkirchen
Deuteriumchlorid, 20 Gew.% in Deuteriumoxid, 99,5 Atom % D, 17,672-9, Aldrich,
Taufkirchen
Deuteriumoxid (D2O), 100,00 Atom % D, 19,170-1, Aldrich, Taufkirchen
Diacetyl, puriss., > 99,5 %, 31530, Fluka, Taufkirchen
Dimethylsulfoxid-d6, 99,5+ Atom % D, 17,594-3, Aldrich, Taufkirchen
Dithiothreitol, > 99,5 %, 43815, Fluka, Taufkirchen
Essigsäure, p.A., 33209, Riedel-de Haen, Seelze
Ethanol, purum, denaturiert, > 96 %, 02893, Fluka, Taufkirchen
Ethanol, absolut, denaturiert, > 99,8 %, 028404, Fluka, Taufkirchen
N-Ethylmorpholin, für Proteinsequenzierung, 04499, Fluka, Taufkirchen
- 45 -

3 Experimenteller Teil
Ethylendiamintetraessigsäure Dinatriumsalz Dihydrat, p.A., 03680, Fluka, Taufkirchen
Furosin, 98,5 % (HPLC), Neosystem, Strasbourg, Frankreich
D(+)-Glucose, wasserfrei, 49139, Fluka, Taufkirchen
Glyoxal, 40 % in Wasser, G-3140, Sigma, Taufkirchen
Glyoxylsäure Monohydrat, purum, > 97,0 %, 50710, Fluka, Taufkirchen
Iodacetamid, > 99 %, 57670, Fluka, Taufkirchen
Kaliumchlorid, p.a., 60130, Fluka, Taufkirchen
Kaliumdihydrogenphosphat, p.a., 60356, Fluka, Taufkirchen
Kationenaustauscher AG 50W-X8, H+-Form, 20 - 50 mesh , Bio-Rad Laboratories, München
Kationenaustauscher DOWEX 50X8-400, H+-Form, 200 - 400 mesh, 20308, Acros, Geel,
Belgien
Lactose Monohydrat, 107660, Merck, Darmstadt
Maltose Monohydrat, 105912, Merck, Darmstadt
Mercaptoethanol, > 99,0 %, 63689, Fluka, Taufkirchen
Methanol, chromasolv gradient grade, 65548, Riedel-de Haen, Seelze
Methanol-d4, 99,8+ Atom % D, 15,194-7, Aldrich, Taufkirchen
Methanol, p.a., 65543, Fluka, Taufkirchen
Methylglyoxal, 40 % in Wasser, M-0252, Sigma, Taufkirchen
3-Morpholinopropansulfonsäure (MOPS), > 99,5 %, 69948, Fluka, Taufkirchen
Natriumazid, p.a., > 99,5 %, 71289, Fluka, Taufkirchen
Natriumborhydrid, p.a., 71320, Fluka, Taufkirchen
Natriumchlorid, p.A., 106404, Merck, Darmstadt
di-Natriumhydrogenphosphat Dihydrat, z.A., 106580, Merck, Darmstadt
Natriumhydroxid, z.A., 106498, Merck, Darmstadt
Natrium-L-lactat, puriss., 71718, Fluka, Taufkirchen
Natriumsulfat, p.a., wasserfrei, 71962, Fluka, Taufkirchen
Ninhydrin, p.a., 33437, Riedel-de Haen, Seelze
Palladium auf Aktivkohle, 10 % Pd, puriss., 75990, Fluka, Taufkirchen
Phenol, p.a., 77610, Fluka, Taufkirchen
Pyridin, purum, > 99,0 %, 82703, Fluka, Taufkirchen
Salzsäure, 37%, ACS, 6011, J.T.Baker, Deventer, Holland (für Salzsäurehydrolysen)
Salzsäure, 37%, p.A., 84426, Fluka, Taufkirchen
Salzsäure, 37%, trace select, 84415, Fluka, Taufkirchen
- 46 -

3 Experimenteller Teil
Thioessigsäure, purum, > 95,0 %, 88620, Fluka, Taufkirchen
2,3,5-Triphenyl-2H-tetrazoliumchlorid (TTC), Feinchemie K.-H. Kallies KG, Sebnitz
Thymol, purum, > 99,0 %, 89330, Fluka, Taufkirchen
Tris-(carboxyethyl)phosphin-Hydrochlorid (TCEP HCl), 99 %, C-4706, Sigma, Taufkirchen
Tris-(hydroxymethyl)aminomethan (TRIS), > 99,8 %, 93349, Fluka, Taufkirchen
2-N-[Tris(hydroxymethyl)methyl]-ε-amino-2-hydroxypropansulfonsäure (TAPSO), > 99,0 %, 93357, Fluka, Taufkirchen
3.1.2 Aminosäuren, Peptide und Proteine
Es wurden ausschließlich L-Aminosäuren sowie Aminosäure-Derivate und Peptide von
L-konfiguierten Aminosäuren verwendet. Verwendete Aminosäuren, Peptide und Proteine
unter Angabe von Bestellnummer, Reinheit und Verantwortlichen, soweit im Text nicht
anders vermerkt:
Nα-Ac-Cys, 01039, Fluka, Taufkirchen
Nα-Ac-Lys, E-1120, Bachem, Weil am Rhein
Nα-Ac-Ser-Gly, G-1100, Bachem, Weil am Rhein
Nα-Ac-Trp, E-1220, Bachem, Weil am Rhein
Ala-Phe-Gly, H-1575, Bachem, Weil am Rhein
Arg-Phe-Ala × 2,0 CH3COOH × 0,5 H2O, Peptid-Gehalt: 75 %, H-1865, Bachem, Weil am
Rhein
Aminosäure-Standardlösung (Kalibration Aminosäureanalyse), AA-S-18, Sigma, Taufkirchen
Nα-Boc-Lys, A-1920, Bachem, Weil am Rhein
Nα-Bz-His, B-2254, Sigma, Taufkirchen
Nα-Bz-Met, E-1490, Bachem, Weil am Rhein
Carboxypeptidase B (EC 3.4.17.2), DFP treated from hog pankreas, 5 mg/ml, 133 U/mg
protein, 21942, Fluka, Taufkirchen
Endoproteinase Glu-C (EC 3.4.21.19), aus Staphylococcus aureus V8, 500-1000 U/mg,
45172, Fluka, Taufkirchen
Glu-Tyr, G-2000, Bachem, Weil am Rhein
Gly, E-3000, Bachem, Weil am Rhein
Glycinamid × 1,0 HCl, E-1950, Bachem, Weil am Rhein
- 47 -

3 Experimenteller Teil
Gly-Gly, G-2090, Bachem, Weil am Rhein
Gly-Ile, G-2135, Bachem, Weil am Rhein
Gly-Phe, G-2175, Bachem, Weil am Rhein
Gly-Gly-Gly, H-3350, Bachem, Weil am Rhein
Gly-Ala-Phe × 1,9 H2O, Bachem, Weil am Rhein
Gly-Ala-Tyr × 2,0 H2O, Peptid-Gehalt: 89 %, Bachem, Weil am Rhein
Gly-Phe-Ala × 1,0 H2O, H-3590, Bachem, Weil am Rhein
Hippursäure (BzG), 53280, Fluka, Taufkirchen
Nα-Hippuryl-arginin (BzGR), G-2265, Bachem, Weil am Rhein
Nα-Hippuryl-glycin (BzGG), G-2270, Bachem, Weil am Rhein
Nα-Hippuryl-lysin (BzGK), G-2275, Bachem, Weil am Rhein
Insulin aus Rinderpankreas, I-5500, Sigma, Taufkirchen
Leu-Asp, Gehalt: 95 %, G-3835, Bachem, Weil am Rhein
Lys × 1,0 HCl, E-2085, Bachem, Weil am Rhein
Nle, F-1915, Bachem, Weil am Rhein
Phe, E-2230, Bachem, Weil am Rhein
Phenylalaninamid, E-2235, Bachem, Weil am Rhein
Phe-Gly, G-2880, Bachem, Weil am Rhein
Phe-Val, G-2965, Bachem, Weil am Rhein
Phe-Gly-Gly, H-4525, Bachem, Weil am Rhein
3.1.3 Materialien und Hilfsmittel
Liste der verwendeten Materialien und Hilfsmittel, sofern an entsprechender Stelle nicht
detailliert beschrieben:
Einmal-Injektionskanülen Sterican®, (0,90 × 40) mm und (0,8 × 50) mm, Braun, Melsungen
Einmalspritzen, 1 ml, HSW Henke-Sass, Wolf GmbH, Tuttlingen
Einmalspritzen, 2 und 5 ml, Braun, Melsungen
Eppendorf-Gefäß, 1,5 und 2,0 ml, Eppendorf Nethler Hinz GmbH, Hamburg
Faltenfilter Selecta, Nr. 595½, 185 mm, Schleicher & Schuell, Dassel
Kartuschen Sep-Pak® C18, 360 mg, Nr. WAT 051910, Waters, Milford, USA
Kartusche Spe-edTM, SPE-ED C18-18 %, 1 g/6 ml, ASS-Chem, Bad Homburg
- 48 -

3 Experimenteller Teil
Membranfilter, Ø 45 mm, RC 0,45 µm, regenerierte Cellulose, Carl Roth GmbH + Co.,
Karlsruhe
Spritzenfilter Spartan, Ø 13 mm, RC 0,2 µm und 0,45 µm, regenerierte Cellulose, Schleicher
& Schuell, Dassel
3.1.4 Geräte
Liste der verwendeten Geräte, sofern an entsprechender Stelle nicht detailliert beschrieben:
Analysenwaage basia BP 121 S und BP 3100S, Sartorius, Göttingen
Brutschrank ICP 400, Memmert GmbH, Schwabach
Druckreaktor 4565M, 100 ml, Parr Instrument Company, Moline, Illinois, USA
Gefriertrocknungsanlage Alpha 1-2, Christ, Osterode
Gefriertrocknungsanlage Beta 1-8 K, Christ, Osterode
Heizplatte mit Magnetrührwerk IKAMAG® RH, Janke & Kunkel GmbH, Staufen
Hydrolyseröhrchen, 6 ml, 20 ml, Schott Glas, Mainz
Kolbenhubpipetten, 2 – 20 µl, 10 – 100 µl, 10 – 200 µl, 200 – 1000 µl, 1000 – 5000 µl,
Eppendorf Nethler Hinz GmbH, Hamburg
Minishaker MS 1, IKA Labortechnik, Staufen
NMR-Röhrchen Schott Professional, id. 4,20 mm, Art. Nr. 231700211, Schott Glas, Mainz
pH-Elektrode Mettler Toledo Inlab 422, Carl Roth GmbH + Co., Karlsruhe
pH-Meßgerät, WTW pH 526, WTW GmbH, Weilheim
Reinstwasseranlage Purelab plus purification system, USFilter, Ransbach-Baumbach
Reacti-BlockTM F, Pierce, Rockford, Illinois, USA
Reacti-ThermTM Heating/Stirring Module, Pierce, Rockford, Illinois, USA
Thermostat M12, Lauda, Lauda-Königshofen
Trockenschrank Modell 200, Memmert GmbH, Schwabach
Ultraschallbad Sonorex RK100, Bandelin electronic, Berlin
Vakuumrotationsverdampfer Laborata 4002, Heidolph, Schwabach
Zentrifugalverdampfer Speed Vac® SPD 111V mit Universal Vacuum System Plus UVS-
400A, Savant, Farmingdale, USA
Zentrifuge Centrifuge 5804 R, Eppendorf Nethler Hinz GmbH, Hamburg
Vakuumhydrolysegefäß, 8 ml, (10 × 150) mm, Pierce, Rockford, Illinois, USA
Vakuumpumpenstand PC 8, Vakuubrand GmbH + Co. KG, Wertheim
- 49 -

3 Experimenteller Teil
3.2 Analytische und präparative Methoden, Instrumente und Zubehör
3.2.1 Hochdruckflüssigchromatographie
HPLC-Anlagen
Merck Hitachi (Niederdruckgradienten-System)
bestehend aus: LC Kontrolleinheit L-5000, Gradientenpumpe L-6000, UV-VIS-Detektor
L-4200, Fluoreszenz-Detektor LaChrom L-7485 (Merck Hitachi, Darmstadt),
Autosampler, Säulenofen: Jetstream 2 Plus, 2 × UV-Detektor K2501 (Knauer, Berlin)
Knauer, analytisch (Niederdruckgradienten-System)
bestehend aus: Entgaser, Eluentendosier-System K1500, K1001 Pumpe mit 10 ml-
Pumpenkopf, dynamische Mischkammer, UV-Detektor K2501, Säulenofen: Jetstream 2 Plus,
Autosampler (Wissenschaftlicher Gerätebau, Dr. Ing. Herbert Knauer GmbH, Berlin)
Knauer, semipräparativ (Hochdruckgradienten-System)
bestehend aus: Entgaser, Eluentendosier-System K1500, 2 × K1001 Pumpe mit 50 ml-
Pumpenkopf, dynamische Mischkammer, UV-Detektor K2501, Säulenofen: Jetstream 2 Plus,
Autosampler (Knauer, Berlin)
Säulen
Säule SA1, Knauer (Berlin), Eurospher 100-C18
Knauer Vertex Column, 4,6 mm × 250 mm, Eurospher 100-C18, Partikelgröße 5 µm,
Porengröße 100 Å, mit integrierter Vorsäule, 4 mm × 5 mm, gleichen Trennmaterials
Säule SA2, Knauer (Berlin), Eurospher 100-C18
Knauer Vertex Column, 3,0 mm × 250 mm, Eurospher 100-C18, Partikelgröße 5 µm,
Porengröße 100 Å, mit integrierter Vorsäule, 3 mm × 5 mm, gleichen Trennmaterials
- 50 -

3 Experimenteller Teil
Säule SA3, Polymer Laboratories GmbH, Darmstadt, PLRP-S
4,6 mm × 150 mm, PLRP-S, Partikelgröße 8 µm, Porengröße 300 Å, mit Vorsäule,
3 mm × 5 mm, gleichen Trennmaterials
Säule SA4, Polymer Laboratories GmbH, Darmstadt, PLRP-S
2,1 mm × 150 mm, PLRP-S, Partikelgröße 5 µm, Porengröße 300 Å, mit Vorsäule,
3 mm × 5 mm, gleichen Trennmaterials
Säule SP1, Knauer (Berlin), Eurospher 100-C18
Knauer Vertex Column, 8 mm × 250 mm, Eurospher 100-C18, Partikelgröße 10 µm,
Porengröße 100 Å, mit Vorsäule, 10 mm × 30 mm, gleichen Trennmaterials
Säule SP2, Knauer (Berlin), Eurospher 100-C18
Knauer Vertex Column, 16 mm × 250 mm, Eurospher 100-C18, Partikelgröße 15 bis 25 µm,
Porengröße 100 Å, mit Vorsäule, 16 mm × 30 mm, gleichen Trennmaterials
Eluenten-Systeme
Eluenten-System E1 (Phosphatpuffer-Methanol-System)
A: 10 mM Natriumphosphat-Puffer, pH = 7,0
1,78 g Dinatriummonohydrogenphosphat-Dihydrat in ca. 950 ml Reinstwasser lösen,
mit 12 M Salzsäure pH = 7,0 einstellen, in Maßkolben auf 1000 ml auffüllen,
membranfiltrieren.
B: Methanol, chromasolv
Eluenten-System E2 (Essigsäure-Methanol-System)
A: 50 mM Essigsäure
2,85 ml Essigsäure im Maßkolben mit Reinstwasser auf 1000 ml auffüllen,
membranfiltrieren.
B: 50 mM Essigsäure in Methanol, chromasolv
2,85 ml Essigsäure im Maßkolben mit Methanol auf 1000 ml auffüllen,
membranfiltrieren.
- 51 -

3 Experimenteller Teil
Eluenten-System E3 (Triethylamin-Essigsäure-Ameisensäure-Methanol-System)
A: 0,1 % Triethylamin, 0,05 % Essigsäure, 0,05 % Ameisensäure in Reinstwasser
1,0 ml Triethylamin, 0,5 ml Essigsäure (100 %), 0,5 ml Ameisensäure (100 %) im
Maßkolben mit Reinstwasser auf 1000 ml auffüllen, membranfiltrieren (pH = 3,73).
B: 0,1 % Triethylamin, 0,05 % Essigsäure, 0,05 % Ameisensäure in Methanol, chromasolv
1,0 ml Triethylamin, 0,5 ml Essigsäure (100 %), 0,5 ml Ameisensäure (100 %) im
Maßkolben mit Methanol (chromasolv) auf 1000 ml auffüllen, membranfiltrieren.
Eluenten-System E4 (Essigsäure-Methanol-System)
A: 0,1 % Essigsäure in Reinstwasser
1,0 ml Essigsäure (100 %) im Maßkolben mit Reinstwasser auf 1000 ml auffüllen,
membranfiltrieren.
B: 0,1 % Essigsäure in Methanol, chromasolv
1,0 ml Essigsäure (100 %) im Maßkolben mit Methanol (chromasolv) auf 1000 ml
auffüllen, membranfiltrieren.
Eluenten-System E5 (Ameisensäure-Methanol-System)
A: 0,1 % Ameisensäure in Reinstwasser (pH = 2,8 gemessen)
1,0 ml Ameisensäure (100 %) im Maßkolben mit Reinstwasser auf 1000 ml auffüllen,
membranfiltrieren.
B: 0,1 % Ameisensäure in Methanol, chromasolv
1,0 ml Ameisensäure (100 %) im Maßkolben mit Methanol (chromasolv) auf 1000 ml
auffüllen, membranfiltrieren.
Eluenten-System E6 (Ameisensäure-Acetonitril-System)
A: 0,1 % Ameisensäure in Reinstwasser (pH = 2,8 gemessen)
1,0 ml Ameisensäure (100 %) im Maßkolben mit Reinstwasser auf 1000 ml auffüllen,
membranfiltrieren.
B: 0,1 % Ameisensäure in Acetonitril, chromasolv
1,0 ml Ameisensäure (100 %) im Maßkolben mit Methanol (chromasolv) auf 1000 ml
auffüllen, membranfiltrieren.
- 52 -

3 Experimenteller Teil
3.2.2 Kopplung Hochdruckflüssigchromatographie Massenspektroskopie und
direkte Massenspektroskopie
HPLC-Anlage
Agilent 1100 series (Hochdruck Gradienten System)
bestehend aus: Entgaser, binäre Pumpe, Autosampler, Säulenthermostat, Diodenarraydetektor
(DAD), Fluoreszenzdetektor (Agilent Technologies, Palo Alto, USA)
Massenspektrometer
für ESI-TOF-MS (electrospray ionization – time of flight – mass spectrometry)
PerSeptive Biosystems Mariner Flugzeitmassenspektrometer mit ESI-Interface (Applied
Biosystems, Stafford, USA)
Kalibration: Bradykinin, Angiotensin I, Neurotensin
Direkte Analyse mittels ESI-MS:
Mariner Flugzeitmassenspektrometer (siehe oben),
oder: LC-MS-System: Esquire-LC (Hewlett Packard-Bruker, Bremen, Waldbronn, Germany)
Berechnung der relativen monoisotopischen Molekülmasse (Mr):
Es werden die Peaks mit dem geringsten m/z-Verhältnis (monoisotopischer Peak) von
prominenten mehrfach geladenen Ionen ausgewählt und Mr wird entsprechend den angegeben
Gleichungen berechnet.
Positiver Modus, [M+Hz]z+-Ionen: Mr = Mz × z – 1,0078 × z
Positiver Modus, [M+HnNam](m+n)+ -Ionen: Mr = Mz × (m + n) – 22,99 × m – 1,0078 × n
negativer Modus, [M-Hz]z--Ionen: Mr = Mz × z + 1,0078 × z
Mz Masse/Ladung (m/z)-Verhältnis
z Anzahl der Ladungen
m Anzahl der Natrium-Ionen
n Anzahl der Protonen
22,99 Masse von Natrium
1,0078 Masse eines Protons
- 53 -

3 Experimenteller Teil
3.2.3 Aminosäureanalyse
Gerät: Alpha Plus Aminosäureanalysator (LKB Biochrom, Cambridge, GB)
Säule: PEEK, (4,6 × 125) mm
Kationenaustauscher: Harz 5 µm, Serie 686, Analysentechnik K. Grüning, Olching
Fluss: 0,27 ml/min
Temperaturgradient: 55 bis 85 °C
Detektion: Nachsäulenderivatisierung mit Ninhydrin
Detektor: VIS-Photometer, λ = 570 nm, 440 nm (Prolin)
Puffer: siehe Tabelle 3.2.3-1
Methode: siehe Tabelle 3.2.3-2
Tab. 3.2.3-1: Puffer-Lösungen für die Aminosäureanalyse.
Puffer-Nr. Puffer Molarität [mol/l] pH-Wert
1 Natriumcitrat 0,2 2,20
2 Natriumcitrat 0,2 3,20
3 Natriumcitrat 0,2 4,25
5 Natriumcitrat 1,2 6,25
6 Natronlauge 0,4
Tab. 3.2.3-2: Trennprogramm der Aminosäureanalyse.
Puffer-Nr. Zeit [min] Temperatur [°C]
2 4 : 00 50
3 0 : 05 50
2 0 : 25 50
3 0 : 05 50
2 0 : 25 50
3 0 : 05 50
2 0 : 25 50
3 0 : 05 50
2 0 : 25 50
3 0 : 10 50
- 54 -

3 Experimenteller Teil
Puffer-Nr. Zeit [min] Temperatur [°C]
2 0 : 20 50
3 0 : 10 50
2 0 : 20 50
3 19 : 00 50
5 5 : 00 60
5 26 : 00 85
6 8 : 00 85
2 8 : 00 85
2 (20%) 8 : 00 85
2 11 : 00 50
3.2.4 Präparative Ionenaustauschchromatographie
BioLogic LP Niederdruckchromatographie-System Bio-Rad Laboratories, München
Econo-Column Flow-Adaptor, 15 mmm, 25 mm Bio-Rad Laboratories, München
Fraktionssammler Fraction Collector Model 2128 Bio-Rad Laboratories, München
Fraktionssammler RediFrac Pharmacia Biotech, Freiburg
Glassäule
Econo-Column (25 × 200) mm, (15 × 500) mm Bio-Rad Laboratories, München
Glassäule (25 × 120) mm, (25 × 200) mm Sigma, Steinheim
Schlauchpumpe P-1 Pharmacia Biotech, Freiburg
3.2.5 Tüpfeltest
DC-Platten: DC-Fertigplatten 10 × 20 cm, Schicht: 0,25 mm Kieselgel, Firma Machery-
Nagel, Düren
Auftragsmenge: 2 µl/Fraktion
Sprühreagenz: Ninhydrin-Lösung 0,1 %ig in Ethanol, 10 mg Ninhydrin in 10 ml
96 %igem Ethanol lösen
Sprühreagenz: 2,3,5-Triphenyl-2H-tetrazoliumchlorid (TTC)-Lösung 1 %ig, 100 mg TTC
in 10 ml 1 M Natronlauge lösen
- 55 -

3 Experimenteller Teil
Detektion: Trockenschrank bei ϑ = 90 °C (Ninhydrin)
Trockenschrank bei ϑ = 50 °C (TTC)
3.2.6 Dünnschichtchromatographie (DC)
DC-Platten:
Normalphase (NP): DC-Fertigplatten SIL G-25 UV254 , 10 × 20 cm, Schicht: 0,25 mm
Kieselgel mit Fluoreszenzindikator, Firma Machery-Nagel, Düren
Umkehrphase (RP): DC-Fertigplatten RP-18W UV254, 10 × 10 cm, Schicht: 0,25 mm
Kieselgel mit Fluoreszenzindikator, Firma Machery-Nagel, Düren
Kammer: DC-Automat: DC-MAT, Desaga, Wiesloch, alternativ: Glastrog-Kammer
Fließmittel: Acetonitril/Wasser/Essigsäure 80/20/20 (v/v/v)
Kammertyp: gesättigte Normalkammer
Konditionierung: 20 min (Kammersättigung)
Sprühreagenz: siehe Abschnitt 3.2.6
Detektion: siehe Abschnitt 3.2.6
Rf-Werte: BzGK (NP: 0,35, RP: 0,23), BzGCML (NP: 0,60), BzGFruK (NP: 0,29, RP: 0,19),
BzGLctK (NP: 0,20, RP: 0,11), BzGMltK (NP: 0,20, RP: 0,11), BzGTagK (NP: 0,32, RP:
0,21), FruK (NP: 0,17, RP: 0,07), Furosin (NP: 0,27, RP: 0,14), Lys (NP: 0,21, RP: 0,07),
Pyridosin (NP: 0,24, RP: 0,14)
3.2.7 UV-VIS Spektroskopie
Gerät: Spektralphotometer Specord S100, Carl Zeiss Jena
Messung: Integrationszeit: 41,0 ms
Akkumulation: 100 Spektren
Bereich: 189,97 bis 499,63 nm Extinktion
Weitere Parameter: automatische Dunkelmessung
Küvette: UV-Präzisions-Küvetten Suprasil 104-QS 10 mm aus Quarzglas
Hellma Optik GmbH, Jena
- 56 -

3 Experimenteller Teil
3.2.8 Fluoreszenzspektroskopie
Gerät: Fluoreszenz Spektrofluorimeter f-4500 mit Xenon-Lampe, Hitachi, Tokio, Japan
Küvette: Quarzküvette für die Fluoreszenzspektroskopie, 1,000 cm
Parameter für zweidimensionale Spektren:
λex = 300 bis 600 nm λem = 300 bis 600 nm
Scan: 12000 nm/min Contour interval: 50
3.2.9 Nuklearmagnetische Kernresonanz (NMR)-Spektroskopie
Die Aufnahme der NMR Spektren erfolgte durch Frau Annett Rudolph, Frau Dr. Margit
Grunar und Herrn Dr. Dieter Scheller am Institut für Organische Chemie der TU Dresden.
Die Messungen werden an einem NMR Spektrometer DRX 500 (Bruker, Reinstetten) mit
500 MHz für 1H-Experimente und 125 MHz für 13C-Experimente durchgeführt. Die
Zuordnung der Signale basiert auf den zweidimensionalen Experimenten 1H-1H COSY
(correlation spectroscopy), HSQC (heteronuclear single quantum coherence), HMBC
(heteronuclear multiple bond correlation) and DEPT (distortionless enhancement by
polarization transfer).
Die Angabe der chemischen Verschiebung δ in ppm erfolgt relativ zu den folgenden
Referenz-Signalen:
Chloroform (CDCl3): δH = 7,25 ppm, δC = 77,00 ppm (mittlere Linie),
(Lösungsmittel-Signale)
Deuteriumoxid (D2O): δH = 4,70 ppm (HDO intern),
δC = 0,00 ppm (TMS extern)
Dimethylsulfoxid-d6 (DMSO-d6): δH = 2,50 ppm, δC = 39,56 ppm (Lösungsmittel-Signale)
Methanol-d4: δH = 3,38 ppm, δC = 49,00 ppm (Lösungsmittel-Signale)
Das 1H-Spekrum wird weiterhin dazu benutzt die Ergebnisse der Elementaranalyse zu
interpretieren. Mit Hilfe der Flächenintegrale ausgewählter Peaks wird auf das molare
Verhältnis der Zielverbindung zu Lösungsmittelresten wie z. B. Essigsäure geschlossen.
- 57 -

3 Experimenteller Teil
3.2.10 Elementaranalyse
Die Ermittlung der elementaren Zusammensetzung der Präparate erfolgte durch Frau Anke
Peritz am Institut für Organische Chemie der TU Dresden. Die Bestimmung wird am
Elementar-Analysator Euro EA 3000 (Eurovector, Mailand, Italien) durchgeführt.
3.3. Darstellung von Referenzmaterial zur Analytik der Amadori-Produkte
3.3.1 Darstellung ausgewählter peptidgebundener Amadori-Produkte
Lösungen:
- 0,2 M N-Ethylmorpholin-Acetat-Puffer, pH = 8,0
1,25 ml N-Ethylmorpholin in ca. 40 ml Reinstwasser lösen, mit Essigsäure pH = 8,0
einstellen, im Maßkolben auf 50 ml auffüllen.
Synthese
Die Synthese von Nα-Hippuryl-Nε-(1-desoxy-D-fructos-1-yl)-L-lysin (Nα-Benzoylglycyl-Nε-
(1-desoxy-D-fructos-1-yl)-L-lysin, BzGFruK), Nα-Hippuryl-Nε-(1-desoxy-D-tagatos-1-yl)-
L-lysin (Nα-Benzoylglycyl-Nε-(1-desoxy-D-tagatos-1-yl)-L-lysin, BzGTagK), Nα-Hippuryl-
Nε-(1-desoxy-D-lactulos-1-yl)-L-lysin (Nα-Benzoylglycyl-Nε-(1-desoxy-D-lactulos-1-yl)-
L-lysin, BzGLctK) und Nα-Hippuryl-Nε-(1-desoxy-D-maltulos-1-yl)-L-lysin (Nα-Benzoyl-
glycyl-Nε-(1-desoxy-D-maltulos-1-yl)-L-lysin , BzGMltK) erfolgt in Anlehnung an Finot &
Mauron (1969).
Es werden 2,16 g (12,0 mmol) wasserfreie Glucose oder Galactose mit 0,62 g (2,0 mmol)
Nα-Hippuryl-L-lysin (BzGK) in 84 ml Methanol (wasserfrei) unter Rückfluss für 4 h erhitzt
(Finot & Mauron, 1969). Die Umsetzung von 1,44 g (4,0 mmol) Lactose- oder Maltose-
Monohydrat mit 0,62 g (2,0 mmol) BzGK erfolgt in einer Mischung aus 21 ml Methanol
(wasserfrei) und 9 ml Dimethylformamid (wasserfrei) unter Rückfluss für 3 h (Vinale et al.,
1999).
- 58 -

3 Experimenteller Teil
Isolierung der Amadori-Produkte der Monosaccharide
Der jeweilige Ansatz wird am Vakuumrotationsverdampfer unter reduziertem Druck bei
20 °C zur Trockene eingedampft und der Rückstand wird in insgesamt 9,0 ml 0,2 M
N-Ethylmorpholin-Acetat-Puffer pH = 8,0 gelöst. Die Lösung wird in ein Glasgefäß mit
Schraubdeckel überführt und der pH-Wert wird auf pH = 8,0 mit N-Ethylmorpholin
eingestellt. Nach Zugabe von 27 µl einer Lösung von Carboxypeptidase B (EC 3.4.17.2,
665 U/ml, Aktivität im Enzym-Substrat-Ansatz 2 U/ml) wird der Ansatz bei 25 °C und
gelegentlichem Umschwenken inkubiert. Die Kontrolle der Hydrolyse des nicht umgesetzten
BzGK zu Hippursäure erfolgt mittels RP-HPLC (siehe unten). Dazu werden 10 µl Ansatz
entnommen und 1 + 19 mit 10 mM Phosphatpuffer (Eluent A) verdünnt. Nach vollständiger
Hydrolyse (24 h) wird am Vakuumrotationsverdampfer unter reduziertem Druck bei 35 °C
zur Trockene eingedampft, der Rückstand wird in insgesamt 9,0 ml 10 mM Phosphatpuffer
(Eluent A) aufgenommen und die resultierende Lösung wird membranfiltriert (regenerierte
Cellulose, 0,45 µm).
Die Isolierung erfolgt mittels semipräparativer RP-HPLC unter Verwendung von zwei
Eluenten-Systemen (siehe unten). Nach dem ersten chromatographischen Schritt werden die
Fraktionen vereinigt und am Vakuumrotationsverdampfer unter reduziertem Druck bei 35 °C
auf ca. 2 ml eingeengt. Es wird mit 150 µl Essigsäure angesäuert und membranfiltriert
(regenerierte Cellulose, 0,45 µm). Die Fraktionen des zweiten chromatographischen Schrittes
werden vereinigt und am Vakuumrotationsverdampfer unter reduziertem Druck bei 35 °C auf
ca. 20 ml eingeengt und gefriergetrocknet. Die Präparate werden so oft wieder in
Reinstwasser gelöst und gefriergetrocknet, bis der Geruch nach Essigsäure nur noch schwach
wahrnehmbar ist (ca. 5 ×). Die Aufbewahrung der Lyophilisate erfolgt bei - 18 °C.
Isolierung der Amadori-Produkte der Disaccharide
Der jeweilige Synthese-Ansatz wird am Vakuumrotationsverdampfer unter reduziertem
Druck bei 20 °C eingeengt, der Rückstand wird in 10 ml Reinstwasser aufgenommen, die
Lösung wird mit 0,5 ml 6 M Salzsäure angesäuert und mittels eines Kateionenaustauschers
entzuckert.
Anlage: Schlauchpumpe P1 (siehe Abschnitt 3.2.4)
Sigma Glassäule (25 × 120) mm
- 59 -

3 Experimenteller Teil
Säulenbett: stark saurer Kationenaustauscher (AG 50W-X8, 20-50 mesh, H+-Form),
(25 × 120) mm
Konditionierung: 250 ml 6 M Salzsäure, Fluss: 0,5 ml/min
Reinstwasser bis pH-neutral, Fluss: 1,5 ml/min
Probenaufgabe: Probe, Fluss: 0,5 ml/min
20 ml Reinstwasser, Fluss: 0,5 ml/min
Waschen: 300 ml Reinstwasser, Fluss: 1,5 ml/min
Elution: 150 ml 2 M Ammoniak, Fluss: 1,5 ml/min
Das Eluat wird am Vakuumrotationsverdampfer unter reduziertem Druck bei 35 °C zur
Trockene eingeengt und der Rückstand wird in insgesamt 9,0 ml 0,2 M N-Ethylmorpholin-
Acetat-Puffer pH = 8,0 gelöst. Die enzymatische Hydrolyse und die weitere Aufarbeitung
erfolgen wie bei den Amadori-Produkten der Monosaccharide.
Analytische RP-HPLC
Proben: Enzym-Substrat-Ansatz, 1 + 19 mit Eluent A verdünnt
Reinheitskontrolle Präparate, Konzentration: 1 mg / 1 ml Eluent A
HPLC-Anlage: Merck Hitachi, siehe Abschnitt 3.2
Säule: SA1, Knauer Eurospher 100-C18, siehe Abschnitt 3.2
Eluenten-System: E1, Phosphatpuffer-Methanol-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 3 auf 7 % B in 25 min
Flussrate: 1,0 ml/min
Temperatur: 20 °C
Injektionsvolumen: 20 µl
Detektor: L4200 (Merck Hitachi), λ = 230 nm, siehe Abschnitt 3.2
Semipräparative RP-HPLC
1. Chromatographischer Schritt
HPLC-Anlage: Merck Hitachi, siehe Abschnitt 3.2
Säule: SP1, Knauer Eurospher 100-C18, siehe Abschnitt 3.2
Eluenten-System: E1, Phosphatpuffer-Methanol-System, siehe Abschnitt 3.2
- 60 -

3 Experimenteller Teil
Gradient: isokratisch, 10 % B 30 min
Flussrate: 1,5 ml/min
Temperatur: 20 °C
Injektionsvolumen: 200-500 µl
Detektor: L4200 (Merck Hitachi), λ = 280 nm, siehe Abschnitt 3.2
2. Chromatographischer Schritt
wie 1. Chromatographischer Schritt, geändert:
Eluenten-System: E2, Essigsäure-Methanol-System, siehe Abschnitt 3.2
Analytische Daten
Analytische Daten BzGFruK: ESI-MS, [M+H]+ m/z = 470,3, [M-H]- m/z = 468,2; 1H-NMR
(500 MHz, D2O, β-Pyranose) δ / ppm: 1,30 (2H, m, K-H4), 1,63 (1H, m, K-H3A), 1,63 (2H,
m, K-H5), 1,76 (1H, m, K-H3B), 2,98 (2H, t, K-H6), 3,17 (2H, s, H1'), 3,62 (1H, d, H3'), 3,64
(1H, dd, H6A'), 3,78 (1H, dd, H4'), 3,90 (1H, m, H5'), 3,90 (1H, m, H6B'), 4,00 (1H, d,
G-H2A), 4,05 (1H, d, G-H2B), 4,16 (1H, dd, K-H2), 7,44 (2H, t, Bz-Hm), 7,55 (1H, t,
Bz-Hp), 7,73 (2H, d, Bz-Ho), 13C-NMR (125 MHz, D2O, β-Pyranose) δ / ppm: 21,92 (t,
K-C4), 24,52 (t, K-C5), 30,84 (t, K-C3), 43,02 (t, G-C2), 48,11 (t, K-C6), 52,65 (t, C1'), 54,37
(d, K-C2), 63,77 (t, C6'), 68,77 (d, C5'), 69,16 (d, C4'), 69,43 (d, C3'), 95,27 (s, C2'), 127,10
(d, Bz-Co), 128,74 (d, Bz-Cm), 132,40 (d, Bz-Cp), 132,68 (s, Bz-Ci), 170,77 (s, G-C1),
171,05 (s, Bz-CO), 178,39 (s, K-C1); Elementaranalyse, C21H31N3O9
× 1,8 CH3COOH × 1,9 H2O (Mbrutto = 611,81 g/mol) berechnet: C = 48,29 %, H = 6,92 %,
N = 6,87 %, gefunden: C = 48,32 %, H = 7,06%, N = 6,95 %; weißer Feststoff,
chromatographische Reinheit: > 99 % (HPLC); Ausbeute: 672 mg (molare Ausbeute: 54,9 %)
Analytische Daten BzGTagK: ESI-MS, [M+H]+ m/z = 470,4, [M-H]- m/z = 468,1; 1H-NMR
(500 MHz, D2O, α-Pyranose) δ / ppm: 1,28 (2H, m, K-H4), 1,62 (1H, m, K-H3A), 1,62 (2H,
m, K-H5), 1,75 (1H, m, K-H3B), 2,95 (2H, m, K-H6), 3,09 (1H, d, H1A'), 3,23 (1H, d, H1B'),
3,51 (1H, m, H6A'), 3,71 (1H, m, H6B'), 3,76 (1H, m, H4'), 3,76 (1H, m, H5'), 3,81 (1H, d,
H3'), 3,99 (1H, d, G-H2A), 4,04 (1H, d, G-H2B), 4,14 (1H, dd, K-H2), 7,45 (2H, t, Bz-Hm),
7,54 (1H, t, Bz-Hp), 7,73 (2H, d, Bz-Ho), 13C-NMR (125 MHz, D2O, α-Pyranose) δ / ppm:
21,94 (t, K-C4), 24,38 (t, K-C5), 30,92 (t, K-C3), 43,06 (t, G-C2), 47,89 (t, K-C6), 52,58 (t,
- 61 -

3 Experimenteller Teil
C1'), 54,50 (d, K-C2), 62,58 (t, C6'), 65,67 (d, C5'), 70,54 (d, C4'), 71,76 (d, C3'), 95,24 (s,
C2'), 127,13 (d, Bz-Co), 128,76 (d, Bz-Cm), 132,42 (d, Bz-Cp), 132,71 (s, Bz-Ci), 170,74 (s,
G-C1), 171,02 (s, Bz-CO), 178,53 (s, K-C1); Elementaranalyse, C21H31N3O9
× 0,7 CH3COOH × 1,1H2O (Mbrutto = 531,39 g/mol) berechnet: C = 50,63 %, H = 6,83 %,
N = 7,91%, gefunden: C = 50,64 %, H = 6,89 %, N = 8,01 %; weißer Feststoff,
chromatographische Reinheit: > 99 % (HPLC); Ausbeute: 545 mg (molare Ausbeute: 51,2 %)
Analytische Daten BzGLctK: ESI-MS, [M+H]+ m/z = 633,5, [M-H]- m/z = 631,3; 1H-NMR
(500 MHz, D2O, β-Pyranose) δ / ppm: 1,28 (2H, m, K-H4), 1,60 (2H, m, K-H5), 1,61 (1H, m,
K-H3A), 1,74 (1H, m, K-H3B), 2,96 (2H, t, K-H6), 3,17 (2H, s, H1'), 3,48 (1H, dd, H2"),
3,56 (1H, m, H3"), 3,60 (1H, m, H5"), 3,67 (2H, m, H6"), 3,68 (1H, d, H6A'), 3,74 (1H, d,
H3'), 3,80 (1H, m, H4"), 3,88 (1H, d, H6B'), 3,98 (1H, d, G-H2A), 4,00 (1H, m, H4'), 4,03
(1H, d, G-H2B), 4,09 (1H, m, H5'), 4,14 (1H, dd, K-H2), 4,42 (1H, d, H1"), 7,43 (2H, t,
Bz-Hm), 7,53 (1H, t, Bz-Hp), 7,72 (2H, d, Bz-Ho), 13C-NMR (125 MHz, D2O, V) δ / ppm:
21,69 (t, K-C4), 24,27 (t, K-C5), 30,60 (t, K-C3), 42,78 (t, G-C2), 47,90 (t, K-C6), 52,49 (t,
C1'), 54,14 (d, K-C2), 60,81 (t, C6"), 63,14 (t, C6'), 66,20 (d, C5'), 67,90 (d, C3'), 68,30 (d,
C4"), 70,38 (d, C2"), 72,22 (d, C3"), 75,05 (d, C5"), 76,82 (d, C4'), 94,96 (s, C2'), 100,61 (d,
C1"), 125,50 (d, Bz-Cm), 126,87 (d, Bz-Co), 132,16 (d, Bz-Cp), 132,44 (s, Bz-Ci), 170,53 (s,
G-C1), 170,82 (s, Bz-CO), 178,22 (s, K-C1); Elementaranalyse, C27H42N3O14
× 1,2 CH3COOH × 1,1H2O (Mbrutto = 724,51 g/mol) berechnet: C = 48,74 %, H = 6,82 %,
N = 5,80 %, gefunden: C = 48,70 %, H = 6,79 %, N = 5,95 %; weißer Feststoff,
chromatographische Reinheit: > 99 % (HPLC); Ausbeute: 317 mg (molare Ausbeute: 21,9 %)
Analytische Daten BzGMltK: ESI-MS, [M+H]+ m/z = 633,5, [M-H]- m/z = 631,4; 1H-NMR
(500 MHz, D2O, β-Pyranose) δ / ppm: 1,28 (2H, m, K-H4), 1,60 (2H, m, K-H5), 1,61 (1H, m,
K-H3A), 1,74 (1H, m, K-H3B), 2,95 (2H, t, K-H6), 3,17 (2H, s, H1'), 3,29 (1H, dd, H3"),
3,45 (1H, dd, H5"), 3,60 (1H, d, H6A'), 3,66 (1H, m, H4"), 3,68 (2H, m, H6"), 3,70 (1H, dd,
H2"), 3,80 (1H, d, H3'), 3,84 (1H, dd, H4'), 3,89 (1H, d, H6B'), 3,99 (1H, d, G-H2A), 4,02
(1H, d, G-H2B), 4,05 (1H, m, H5'), 4,13 (1H, dd, K-H2), 5,10 (1H, dd, H1"), 7,43 (2H, t,
Bz-Hm), 7,53 (1H, t, Bz-Hp), 7,71 (2H, d, Bz-Ho); 13C-NMR (125 MHz, D2O, β-Pyranose) δ
/ ppm: 21,68 (t, K-C4), 24,25 (t, K-C5), 30,59 (t, K-C3), 42,77 (t, G-C2), 47,86 (t, K-C6),
52,33 (t, C1'), 54,14 (d, K-C2), 60,13 (t, C6"), 63,60 (t, C6'), 68,46 (d, C5'), 68,58 (d, C3'),
69,18 (d, C3"), 71,33 (d, C5"), 72,00 (d, C2"), 72,40 (d, C4"), 77,20 (d, C4'), 95,12 (s, C2'),
- 62 -

3 Experimenteller Teil
100,18 (d, C1"), 126,87 (d, Bz-Co), 128,50 (d, Bz-Cm), 132,16 (d, Bz-Cp), 132,43 (s, Bz-Ci),
170,52 (s, G-C1), 170,82 (s, Bz-CO), 178,22 (s, K-C1); Elementaranalyse,
C27H42N3O14 × 1,2 CH3COOH × 2,6 H2O (Mbrutto = 751,54 g/mol) berechnet: C = 46,99 %,
H = 6,97 %, N = 5,59 %, gefunden: C = 46,95 %, H = 6,82 %, N = 5,67 %; weißer Feststoff,
chromatographische Reinheit: > 99 % (HPLC); Ausbeute: 456 mg (molare Ausbeute: 30,3 %)
3.3.2 Darstellung von Nα-Hippuryl-Nε-carboxymethyl-L-lysin
Synthese
Die Synthese von Nα-Hippuryl-Nε-carboxymethyl-L-lysin (Nα-Benzoylglycyl-
Nε-carboxymethyl-L-lysin, BzGCML) erfolgt durch reduktive (hydrierende) Alkylierung von
Nα-Hippuryllysin mit Glyoxylsäure nach Liardon et al. (1987), modifiziert durch Helling
(2002). Für die Reinigung wurde ein Verfahren von Van Chuyen (1973c) adaptiert.
1,538 g (5,01 mmol) Nα-Hippuryllysin und 0,600 g (6,52 mmol) Glyoxylsäure-Monohydrat
werden in 20 ml Reinstwasser gelöst. Der pH-Wert wird mit 1,0 M Natronlauge (ca. 8,3 ml)
auf pH = 8,7 eingestellt. Unter Nachspülen mit Reinstwasser wird der Reaktionsansatz in ein
100 ml-Autoklaven-Gefäß überführt. Nach Zugabe von 52,0 mg Palladium auf Aktivkohle
(10 % Pd) wird der Autoklav verschlossen.
Die Umsetzung im Autoklaven (Druckreaktor Serie 4565M, Parr Instrument Company,
Moline, Illinois, USA) erfolgt nach dreimaligen Spülen der Apparatur mit Wasserstoff bei
einem Druck von 20 bar (Wasserstoff), unter Rühren, bei 23 °C und für 29 h. Anschließend
wird die Aktivkohle abfiltriert (Faltenfilter). Die Lösung wird membranfiltriert und am
Vakuumrotationsverdampfer unter reduziertem Druck bei 30 °C auf ca. 20 ml eingeengt.
Reinigung mittels Ionenaustauschchromatographie
Lösungen:
- 1 M Natronlauge (carbonatarm), 80 g Natriumhydroxid in 2000 l frisch gekochtem
und abgekühltem Reinstwasser unter Kühlung lösen.
- 0,5 M Essigsäure, 28,6 ml Essigsäure (Eisessig) im Maßkolben mit Reinstwasser auf
1000 ml auffüllen, membranfiltrieren, im Vakuum entgasen.
- 63 -

3 Experimenteller Teil
- 1,0 M Essigsäure, 57,2 ml Essigsäure (Eisessig) im Maßkolben mit Reinstwasser auf
1000 ml auffüllen, membranfiltrieren, im Vakuum entgasen.
- 1,5 M Essigsäure, 85,8 ml Essigsäure (Eisessig) im Maßkolben mit Reinstwasser auf
1000 ml auffüllen, membranfiltrieren, im Vakuum entgasen.
- weitere Lösungen und Reinstwasser im Vakuum entgasen.
Anlage: Bio-Rad-Anlage, BioLogic LP (siehe Abschnitt 3.2.4)
Fraktionssammler Bio-Rad Fraction Collector Model 2128 (siehe 3.2.4)
Bio-Rad Glassäule Econo-Column (25 × 200) mm (siehe 3.2.4)
Säulenbett: stark basischer Anionenaustauscher (DOWEX 1X8, 200-400 mesh,
Acetat-Form), (25 × 190) mm
Konditionierung: 2000 ml 1,0 M Natronlauge, Fluss: 1,0 ml/min
200 ml Reinstwasser, Fluss: 1,0 ml/min
300 ml 1,0 M Essigsäure, Fluss: 1,0 ml/min
500 ml Reinstwasser, Fluss: 1,0 ml/min
Probenaufgabe: 20 ml Probe, Fluss: 0,5 ml/min
20 ml Reinstwasser, Fluss: 0,5 ml/min
20 ml 0,1 M Essigsäure, Fluss: 0,5 ml/min
Elution: 300 ml 0,5 M Essigsäure, Fluss: 0,5 ml/min
300 ml 1,0 M Essigsäure, Fluss: 0,5 ml/min
300 ml 1,5 M Essigsäure, Fluss: 0,5 ml/min
Fraktionierung: ca. 6 ml (= 12 min) pro Fraktion
Regeneration: 1000 ml 1,0 M Salzsäure, Fluss: 1,0 ml/min
1000 ml Reinstwasser, Fluss: 1,0 ml/min
Zur Identifikation der Nα-Hippuryl-Nε-carboxymethyllysin enthaltenden Fraktionen wird
zunächst mittels Tüpfeltest und Ninhydrin-Detektion (siehe Abschnitt 3.2.5) und anschließend
mittels RP-HPLC (siehe Abschnitt 3.3.1) geprüft. Die Elution des Produktes erfolgt mit 1,0 M
Essigsäure. Die geeigneten Fraktionen werden vereinigt, am Vakuumrotationsverdampfer
unter reduziertem Druck bei 35 °C auf ca. 40 ml eingeengt und gefriergetrocknet. Das
Präparat wird so oft wieder in Reinstwasser gelöst und gefriergetrocknet, bis der Geruch nach
Essigsäure nur noch schwach wahrnehmbar ist (ca. 5 ×). Die Aufbewahrung des Lyophilisates
erfolgt bei – 18 °C.
- 64 -

3 Experimenteller Teil
Analytische Daten
Analytische Daten BzGCML: ESI-MS, [M+H]+ m/z = 366,2, [M-H]- m/z =364,1; 1H-NMR
(500 MHz, D2O) δ / ppm: 1,35 (2H, m, K-H4), 1,62 (1H, m, K-H3A), 1,69 (2H, m, K-H5),
1,84 (1H, m, K-H3B), 2,94 (2H, t, K-H6), 3,55 (2H, s, CM-H2), 4,01 (1H, d, G-H2A), 4,07
(1H, d, G-H2B), 4,31 (1H, dd, K-H2), 7,45 (2H, t, Bz-Hm), 7,55 (1H, t, Bz-Hp), 7,73 (2H, d,
Bz-Ho), 13C-NMR (125 MHz, D2O) δ / ppm: 23,66 (t, K-C4), 26,49 (t, K-C5), 31,78 (t,
K-C3), 44,58 (t, G-C2), 48,77 (t, K-C6), 50,47 (t, CM-C2), 54,58 (d, K-C2), 128,81 (d,
Bz-Co), 130,46 (d, Bz-Cm), 134,13 (d, Bz-Cp), 134,36 (s, Bz-Ci), 172,42 (s, G-C1), 172,77
(s, Bz-CO), 173,08 (s, CM-C1), 178,00 (s, K-C1); Elementaranalyse, C17H23N3O6
× 0,5 CH3COOH × 0,8 H2O (Mbrutto = 404,42 g/mol) berechnet: C = 53,46 %, H = 6,48 %,
N = 10,39 %, gefunden: C = 53,26 %, H = 6,38%, N = 10,41 %; weißer Feststoff,
chromatographische Reinheit: > 99 % (HPLC); Ausbeute: 1667 mg (molare Ausbeute:
82,4 %)
3.3.3 Darstellung von Nε-(1-Desoxy-D-fructos-1-yl)-L-lysin
Synthese
Die Synthese von Nε-Fructoselysin (Nε-(1-Desoxy-D-fructos-1-yl)-L-lysin, FruK) erfolgt in
Anlehnung an Reutter und Eichner (1989), wobei zur Vermeidung von Nebenprodukten von
Nα-Boc-Lysin ausgegangen wird.
Eine Lösung von 0,62 g (2,5 mmol) Nα-Boc-Lysin und 2,70 g (15,0 mmol) Glucose in
13,8 ml Reinstwasser wird mit 6,75 g Cellulose (mikrokristallin) zu einem Brei verarbeitet.
Die Mischung wird gefriergetrocknet und der resultierende Feststoff fein gemörsert. Zur
Einstellung des aw-Wertes wird im Exsikkator über einer gesättigten Lösung von
Calciumchlorid (aw = 0,35) für 14 d bei 20 °C aufbewahrt. Anschließend wird in einem
verschlossenen Schraubdeckelglas 12 d bei 40 °C inkubiert. Nachfolgend wird das
Reaktionsgemisch dreimal mit Reinstwasser extrahiert. Dazu wird jeweils mit 25 ml
Reinstwasser aufgeschlämmt, die Aufschlämmung zentrifugiert und der Überstand
abgenommen. Der vereinigte Extrakt wird filtriert (Faltenfilter) und mit 2,5 ml Essigsäure
angesäuert. Zur Abspaltung der Boc-Schutzgruppe und zur Entzuckerung mittels eines stark
sauren Kationenaustauscher wird wie unter 3.3.1 (Isolierung der Amadori-Produkte der
- 65 -

3 Experimenteller Teil
Disaccharide) angegeben verfahren. Um die Schutzgruppe vollständig zu entfernen, verbleibt
jedoch das Isolat nach dem Waschschritt über Nacht auf dem Austauscher und wird erst dann
eluiert. Das zur Trockene eingeengte Eluat wird in 10 ml 0,1 M Pyridinium-Formiat-Puffer,
pH = 3,0, aufgenommen und der pH-Wert wird mit Ameisensäure auf pH = 2,8 eingestellt.
Reinigung mittels Ionenaustauschchromatographie
Lösungen:
- 2 M Pyridin, 166 ml Pyridin (100 %) mit Reinstwasser im Maßkolben auf 1000 ml
auffüllen, membranfiltrieren, im Vakuum entgasen.
- 0,1 M Pyridinium-Formiat-Puffer, pH = 3,0, 8 ml Pyridin in ca. 950 ml Reinstwasser
lösen, mit Ameisensäure pH = 3,0 einstellen, im Maßkolben auf 1000 ml auffüllen,
membranfiltrieren, im Vakuum entgasen.
- 0,4 M Pyridinium-Acetat-Puffer, pH = 4,35, 33 ml Pyridin in ca. 950 ml Reinstwasser
lösen, mit Essigsäure pH = 4,35 einstellen, im Maßkolben auf 1000 ml auffüllen,
membranfiltrieren, im Vakuum entgasen.
- Reinstwasser, im Vakuum entgasen.
Anlage: Bio-Rad-Anlage, BioLogic LP (siehe Abschnitt 3.2.4)
Fraktionssammler Bio-Rad Fraction Collector Model 2128 (siehe
Abschnitt 3.2.4)
Bio-Rad Glassäule Econo-Column (15 × 500) mm (siehe 3.2.4)
Säulenbett: stark saurer Kationenaustauscher (DOWEX 50X 8-400, 200-400 mesh,
Pyridinium-Form), (15 × 480) mm
Konditionierung: 250 ml 4 M Salzsäure, Fluss: 0,17 ml/min
250 ml Reinstwasser, Fluss: 0,5 ml/min
250 ml 2 M Pyridin, Fluss: 0,17 ml/min
250 ml Reinstwasser, Fluss: 0,5 ml/min
250 ml 0,1 M Pyridinium-Formiat-Puffer, pH = 3,0, Fluss: 0,5 ml/min
Probenaufgabe: 10 ml Probe, Fluss: 0, 17 ml/min
20 ml 0,1 M Pyridinium-Formiat-Puffer, pH = 3,0, Fluss: 0,17 ml/min
Elution: 250 ml 0,4 M Pyridinium-Acetat-Puffer, pH = 4,35, Fluss: 0,17 ml/min
Fraktionierung: 5 ml (= 30 min) pro Fraktion
- 66 -

3 Experimenteller Teil
Die Reinheit der Fraktionen die im Tüpfeltest sowohl mit Ninhydrin als auch mit TTC eine
Rotfärbung zeigen (siehe Abschnitt 3.2.5) wird mittels Dünnschichtchromatographie (siehe
Abschnitt 3.2.6) und Aminosäureanalyse (siehe Abschnitt 3.2.3) überprüft. Die geeigneten
Fraktionen (Nr. 25 bis 36) werden vereinigt und am Vakuumrotationsverdampfer unter
reduziertem Druck bei 35 °C zur Trockene eingeengt. Der Rückstand wird in 5 ml trockenem
Methanol aufgenommen und es wird erneut zur Trockene eingeengt. Das zurückbleibende Öl
wird in 5 ml trockenem Methanol gelöst. Die Lösung wird unter intensiven Rühren in 25 ml
Ethylmethylketon eingetropft. Nach 16 h wird das Präzipitat mittels eines Büchnertrichters
abfiltriert und mit Ethylmethylketon gewaschen. Das Präparat wird im Vakuum (10 mbar)
getrocknet und anschließend bei - 18 C aufbewahrt.
Analytische Daten
Analytische Daten FruK: ESI-MS, [M+H]+ m/z = 309,2, [M-H]- m/z = 307,0;1H-NMR
(500 MHz, D2O, mit Deuteriumchlorid-Lösung pH = 5,1 eingestellt [pH-Wert gemessen,
Isotopeneffekt nicht korrigiert], β-Pyranose) δ / ppm: 1,35 (2H, m, K-H4),1,66 (2H, m,
K-H5), 1,79 (2H, m, K-H3), 3,03 (2H, dd, K-H6), 3,19 (2H, s, H1'), 3,61 (1H, d, H3'), 3,63
(1H, m, K-H2), 3,64 (1H, d, H6A'), 3,77 (1H, dd, H4'), 3,89 (1H, dd, H5'), 3,89 (1H, d, H6B'), 13C-NMR (125 MHz, D2O, β-Pyranose) δ / ppm: 21,34 (t, K-C4), 24,66 (t, K-C5), 29,68 (t,
K-C3), 47,83 (t, K-C6), 52,65 (t, C1'), 54,30 (d, K-C2), 63,75 (t, C6'), 68,73 (d, C5'), 69,10 (d,
C4'), 69,39 (d, C3'), 95,24 (s, C2'), 174,38 (s, K-C1); Elementaranalyse, C12H24N2O7
× 0,8 CH3COOH × 0,8CH3OH × 0,1 C2H5COCH3 (Mbrutto = 389,21 g/mol) berechnet:
C = 45,67 %, H = 8,08 %, N = 7,20 %, gefunden: C = 45,40 %, H = 7,99 %, N = 7,17 %;
schwach gelb gefärbter Feststoff, chromatographische Reinheit: > 99 % (Aminosäureanalyse);
Ausbeute: 399 mg (molare Ausbeute: 42,7 %)
3.3.4 Darstellung von Pyridosin
Synthese
Die Synthese von Pyridosin (6-(5-Hydroxy-2-methyl-4-oxo-4H-pyridin-1-yl)-L-norleucin
erfolgt in Anlehnung an Henle et al. (1994b).
1,8 g (10 mmol) Nα-Acetyllysin und 22 g (120 mmol) wasserfreie Glucose werden in 335 ml
absolutem Methanol (chromatosolv) für 4 h unter Rückfluss und Rühren umgesetzt.
- 67 -

3 Experimenteller Teil
Anschließend wird das Lösungsmittel am Vakuumrotationsverdampfer unter reduziertem
Druck bei ca. 20 °C entfernt, der Rückstand wird in 10 ml Reinstwasser gelöst und der Ansatz
wird wie unter 3.3.1 (Isolierung der Amadori-Produkte der Disaccharide) angegeben
entzuckert. Das Nα-Acetyl-Nε-(1-desoxy-D-fructos-1-yl)-L-lysin enthaltende Eluat wird am
Vakuumrotationsverdampfer unter reduziertem Druck bei ca. 35 °C zur Trockene eingeengt.
Der Rückstand wird in 1250 ml 8 M Salzsäure aufgenommen. Die Hydrolyse des Ansatzes
erfolgt durch Erhitzen unter Rückfluss und Rühren für 23 h. Anschließend wird die Salzsäure
im Wasserstrahlvakuum abdestilliert. Der Rückstand wird in 40 ml Reinstwasser gelöst, der
pH-Wert wird durch Zugabe von Trinatriumcitrat auf pH = 1,5 eingestellt und die Lösung
wird membranfiltriert.
Reinigung mittels Ionenaustauschchromatographie
Lösungen:
- 0,1 M Natriumcitrat-Puffer, pH = 3,0 (Aufgabepuffer), 4 g Natriumhydroxid in
ca. 900 ml Reinstwasser lösen, mit Citronensäure pH = 3,0 einstellen, auf 1000 ml
auffüllen, membranfiltrieren, im Vakuum entgasen.
- 0,3 M Natriumcitrat-Puffer, pH = 5,35 (Elutionspuffer), 12 g Natriumhydroxid in
ca. 900 ml Reinstwasser lösen, mit Citronensäure pH = 5,35 einstellen, auf 1000 ml
auffüllen, membranfiltrieren, im Vakuum entgasen.
- Reinstwasser, im Vakuum entgasen.
Anlage: Bio-Rad-Anlage, BioLogic LP (siehe Abschnitt 3.2.4)
Fraktionssammler Bio-Rad Fraction Collector Model 2128 (siehe 3.2.4)
Bio-Rad Glassäule Econo-Column (25 × 200) mm (siehe 3.2.4)
Säulenbett: stark saurer Kationenaustauscher (DOWEX 50X 8-400, 200-400 mesh,
Natrium-Form), (25 × 180) mm
Konditionierung: 250 ml 4 M Salzsäure, Fluss: 1,0 ml/min
Reinstwasser bis pH-neutral, Fluss: 2,0 ml/min
250 ml 2 M Natronlauge, Fluss: 1,0 ml/min
Reinstwasser bis pH-neutral, Fluss: 2,0 ml/min
250 ml Natriumcitrat-Puffer, pH = 3,0, Fluss: 1,0 ml/min
Probenaufgabe: 40 ml Probe, Fluss: 0,5 ml/min
- 68 -

3 Experimenteller Teil
Probenaufgabe: 100 ml 0,1 M Natriumcitrat-Puffer, pH = 3,0, Fluss: 0,5 ml/min
Elution: 250 ml 0,3 M Natriumcitrat-Puffer, pH = 5,35, Fluss: 0,5 ml/min
Fraktionierung: 8,5 ml (17 min) pro Fraktion
Die Reinheit der Fraktionen die im Tüpfeltest sowohl mit Ninhydrin als auch mit TTC eine
Rotfärbung zeigen (siehe Abschnitt 3.2.5) wird mittels Dünnschichtchromatographie (siehe
Abschnitt 3.2.6) und Aminosäureanalyse (siehe Abschnitt 3.2.3) überprüft. Die geeigneten
Fraktionen (Nr. 40 bis 50) werden vereinigt. Nach Verdünnen mit ca. 300 ml Reinstwasser
wird mit 6 M Salzsäure pH = 2 eingestellt und entsalzt.
Entsalzung mittels stark sauren Kationenaustauscher
Anlage: Schlauchpumpe P1 (siehe Abschnitt 3.2.4)
Sigma Glassäule (25 × 200) mm (siehe Abschnitt 3.2.4)
Säulenbett: stark saurer Kationenaustauscher (AG 50W-X8, 20-50 mesh, H+-Form),
(25 × 200) mm
Konditionierung: 250 ml 4 M Salzsäure, Fluss: 0,5 ml/min
Reinstwasser bis pH-neutral, Fluss: 1,5 ml/min
Probenaufgabe: Probe, Fluss: 0,5 ml/min
Waschen: 200 ml Reinstwasser, Fluss: 1,5 ml/min
400 ml 1 M Salzsäure, Fluss: 0,5 ml/min
Elution: 800 ml 4 M Salzsäure, Fluss: 1,5 ml/min
Fraktionierung: 12 ml (8 min) pro Fraktion
Die Fraktionen werden mittels Tüpfeltest mit Ninhydrin-Detektion (siehe Abschnitt 3.2.5)
und Aminosäureanalyse (siehe Abschnitt 3.2.3) überprüft. Die geeigneten Fraktionen (Nr. 8
bis 69) werden vereinigt und die Salzsäure wird im Wasserstrahlvakuum abdestilliert. Der
Rückstand wird in 10 ml Reinstwasser gelöst und die Lösung wird am
Vakuumrotationsverdampfer unter reduziertem Druck bei 35 °C zur Trockene eingeengt
(2 ×). Der Rückstand wird in 1 ml Reinstwasser gelöst und die Lösung wird auf eine
konditionierte RP-18 Kartusche (Spe-edTM, SPE-ED C18-18 %, 1 g/6 ml, Konditionierung:
5 ml Reinstwasser, 5 ml Methanol, 5 ml Reinstwasser) gegeben. Anschließend wird die
Kartusche mit 5 ml 0,001 M Salzsäure gespült. Das Eluat wird ab dem Beginn der
- 69 -

3 Experimenteller Teil
Probenaufgabe gesammelt und am Vakuumrotationsverdampfer unter reduziertem Druck bei
35 °C zur Trockene eingeengt. Der Rückstand wird in 5 ml Reinstwasser gelöst, die Lösung
wird membranfiltriert und gefriergetrocknet. Das Lyophilisat wird bei – 18 C aufbewahrt.
Analytische Daten
Analytische Daten Pyridosin: ESI-MS, [M+H]+ m/z = 255,2, [M-H]- m/z = 253,0; 1H-NMR
(500 MHz, DMSO-d6) δ / ppm: 1,38-1,49 (2 H, m, H-K4), 1,79-1,86 (2 H, m, H-K5), 1,87-
1,93 (2 H, m, H-K3), 2,48 (3 H, s, H-7´) 3,95 (1 H, t, H-K2), 4,18 (2 H, t, H-K6), 7,00 (1 H, s,
H-5´), 7,96 (1 H, s, H-2´), 13C-NMR (125 MHz, DMSO) δ / ppm: 18,31 (q, C-7´, 21,06 (t,
C-K4), 28,68 (t, C-K5), 29,17 (t, C-K3), 52,61 (d, C-K2), 55,39 (t, C-K6), 114,34 (d, C-5´),
131,12 (d, C-2´), 142,94 (s, C-3´), 148,48 (s, C-6´),159,42 (s, C-4´),171,98 (s, C-K1);
UV-Daten (0, 1 M Salzsäure): λmax = 245 nm, ε245 = 6655 l⋅mol-1⋅cm-1, λmax = 276 nm,
ε276 = 6086 l⋅mol-1⋅cm-1; Elementaranalyse, C12H18N2O4 × 2,0 HCl × 0,7 NaCl × 2,1 H2O
(Mbrutto = 405,95 g/mol) berechnet: C = 35,50 %, H = 6,01 %, N = 6,90 %, Cl = 23,58 %,
gefunden: C = 35,61 %, H = 6,04 %, N = 6,81 %, Cl = 23,70 %; gelbbrauner Feststoff,
chromatographische Reinheit: > 98 % (Aminosäureanalyse); Ausbeute: 250 mg (molare
Ausbeute: 6,2 %)
3.4 Studien zur Bildung der Hydrolyseprodukte
3.4.1 Hydrolyse der Amadori-Produkte
Lösungen
Amadori-Produkt-Stammlösungen
- Nα-Hippuryl-Derivat-Lösung (2 mg/ml), 50 mg BzGFruK, BzGTagK, BzGLctK oder
BzGMltK in 25 ml Reinstwasser im Maßkolben lösen.
- BzGFruK-Lösung (10 mg/ml), 20 mg BzGFruK in 2,000 ml Reinstwasser lösen
- Nε-Fructoselysin-Lösung (1 mg/ml), 25 mg FruK in 25 ml Reinstwasser im
Maßkolben lösen.
- 70 -

3 Experimenteller Teil
Salzsäure
- 12 M Salzsäure, „ACS Quality“ von J.T.Baker, Deventer, Niederlande
- 12 M Salzsäure, „trace select“ von Fluka, Taufenkirchen, Deutschland (nur für
Vakuumhydrolyse)
Salzsäurehydrolyse - Standard-Verfahren
Zusammensetzung der Hydrolyseansätze:
- Hydrolyse mit 6 M Salzsäure: 1000 µl Amadori-Produkt-Stammlösung (2 mg/ml) +
1000 µl 12 M Salzsäure (Amadori-Produkt-Salzsäure-Verhältnis: 1:1000, FruK:
1:2000)
- Hydrolyse mit 8 M Salzsäure: 1000 µl Amadori-Produkt-Stammlösung (2 mg/ml) +
2000 µl 12 M Salzsäure (Amadori-Produkt-Salzsäure-Verhältnis: 1:1500, FruK:
1:3000)
Zur Salzsäurehydrolyse wird die Amadori-Produkt-Stammlösung und die Salzsäure in ein
6 ml-Hydrolysegefäße pipettiert, der Ansatz wird gemischt, das Gefäß wird mit Stickstoff
gespült und fest verschlossenen. Die Hydrolyse erfolgt bei 110 °C für 23 h im Sandbad im
Trockenschrank. Zur Kontrolle der Dichtheit werden die Hydrolysegefäße vor und nach der
Hydrolyse gewogen. Nach der Hydrolyse werden jeweils 500 µl Hydrolysat in ein
1,5 ml-Eppendorf-Gefäß überführt und im Vakuumzentrifugalverdampfer zur Trockene
eingeengt. Der Rückstand wird in 1000 µl 0,2 M Natriumcitrat-Puffer (pH = 2,20, Puffer 1
siehe Abschnitt 3.2.3) gelöst, die Lösung wird zentrifugiert (6000 min-1, Zentrifuge
Eppendorf 5804 R) und membranfiltriert (0,2 µm, regenerierte Cellulose). Bei veränderter
Amadori-Produkt-Konzentration im Hydrolyseansatz (siehe unten) werden das zu
entnehmende Hydrolysat-Aliquot oder das Lösevolumen entsprechend angepasst. Es wird
jeweils eine Fünfachbestimmung beginnend mit fünf separaten Hydrolyseansätzen
durchgeführt.
Variation der Hydrolysebedingungen
Zusatz möglicher Effektoren
Es erfolgen Hydrolysen von BzGFruK und BzGTagK in 6 M Salzsäure (1:1000, siehe oben)
in Gegenwart von möglichen Effektoren. Die Höhe des Zusatzes beträgt jeweils 0,1 %
- 71 -

3 Experimenteller Teil
(1 mg/ml Gesamthydrolyseansatz) und 1,0 % (10 mg/ml). Es werden Glucose (wasserfrei),
Essigsäure, Citronensäure-Monohydrat, Natriumlactat, Natriumazid, Kaliumdihydrogen-
phosphat, Acetylcystein, Phenol, Thymol, Bromwasserstoffsäure (48 %ig), Mercaptoethanol
und Thioessigsäure geprüft.
Zusatz von Peptiden
Die Hydrolyse von BzGFruK erfolgt in Gegenwart von Dipeptiden (Peptid-Salzsäure-
Verhältnis: 1:125).
- Hydrolyse mit 6 M Salzsäure, 1000 µl BzGFruK-Stammlösung (2 mg/ml) + 1000 µl
12 M Salzsäure + 7 mg Glu-Tyr + 7 mg Leu-Asp
- Hydrolyse mit 8 M Salzsäure, 200 µl BzGFruK-Stammlösung (10 mg/ml) + 466 µl
Reinstwasser + 1334 µl 12 M Salzsäure + 7 mg Glu-Tyr + 7 mg Leu-Asp
Variation der Amadori-Produkt-Konzentration im Hydrolyseansatz
Die Hydrolyse von BzGFruK in 6 M und 8 M Salzsäure erfolgt bei ausgewählten Amadori-
Produkt-Salzsäure-Verhältnissen.
Hydrolyse mit 6 M Salzsäure
- Verhältnis 1:500, 400 µl Stammlösung (10 mg/ml) + 600 µl Reinstwasser + 1000 µl
12 M Salzsäure
- Verhältnis 1:10000, 250 µl Stammlösung (2 mg/ml) + 2250 µl Reinstwasser + 2500 µl
12 M Salzsäure
Hydrolyse mit 8 M Salzsäure
- Verhältnis 1:500, 400 µl Stammlösung (10 mg/ml) + 266 µl Reinstwasser + 1334 µl
12 M Salzsäure
- Verhältnis 1:1000, 200 µl Stammlösung (10 mg/ml) + 466 µl Reinstwasser + 1334 µl
12 M Salzsäure
- Verhältnis 1:10000, 250 µl Stammlösung (2 mg/ml) + 1417 µl Reinstwasser + 3333 µl
12 M Salzsäure
- 72 -

3 Experimenteller Teil
Hydrolyse mit 7,3 M Salzsäure
Die Hydrolyse von BzGFruK und BzGTagK erfolgt bei einem Amadori-Produkt-Salzsäure-
Verhältnis von 1:1275 mit 7,3 M Salzsäure (645 µl Amadori-Produkt-Stammlösung
[2 mg/ml] + 1000 µl 12 M Salzsäure).
Hydrolyse mit und ohne Spülen mit Stickstoff
Bei der Hydrolyse von BzGFruK in 6 M Salzsäure nach dem Standardverfahren wird der
Einfluss des Spülens mit Stickstoff geprüft.
Variation: vor dem Verschließen der Hydrolysegefäße
- Spülen des Gasraums mit Stickstoff (= Standardverfahren)
- kein Spülen des Gasraums mit Stickstoff
- Stickstoff durch Hydrolyseansatz blubbern lassen (1 oder 2 min)
Hydrolyse unter Vakuum
Die Hydrolyse von BzGFruK in 6 M und 8 M Salzsäure erfolgt in Vakuumhydrolysegefäßen
(Pierce) unter Verwendung von „Reacti-ThermTM Heating/Stirring Module“ und „Reacti-
BlockTM F“ (Pierce) für 23 h bei 110 °C.
Herstellung der Hydrolyseansätze wie Standardverfahren
Variation: vor dem Verschließen der Hydrolysegefäße
- keine zusätzliche Behandlung (Vergleich)
- Hydrolyseansatz auf -70 °C Kühlen, Gefäß evakuieren
(Endvakuum: 0,001 mbar, 1 min)
Hydrolyse nach Reduktion mit Natriumborhydrid
Das Amadori-Produkt wird mit Natriumborhydrid reduziert (molares Verhältnis: ca. 1:10, 1 h,
pH = 8,5) und anschließend wird mit 6 M Salzsäure hydrolysiert. Dazu werden 1000 µl
BzGFruK-Lösung (2 mg/ml) oder 1000 µl FruK-Lösung (1 mg/ml) in der folgenden Weise
behandelt:
- Zugabe von 1000 µl 100 mM Natriumphosphatpuffer (pH = 7,4, 500 µM EDTA),
- Zugabe von 1,2 mg Natriumborhydrid (resultierender pH-Wert: 8,5),
- 1 h Inkubation bei 23 C,
- Zugabe von 2000 µl 12 M Salzsäure,
- 73 -

3 Experimenteller Teil
- Standardhydrolyse (23 h, 110 °C).
Anschließend wird ein Aliquot von 500 µl entnommen und in der üblichen Weise (siehe oben:
Standard-Verfahren) aufgearbeitet und mittels Aminosäureanalyse untersucht.
3.4.2 Quantifizierung der Hydrolyseprodukte und Bestimmung der molaren
Ausbeuten
Die Quantifizierung von Furosin, Pyridosin, Lysin, CML und Glycin erfolgt mittels
Aminosäureanalyse (siehe Abschnitt 3.2.3) und externer Kalibration. Für die Kalibration von
Furosin sowie Lysin und Glycin werden käufliche Standards verwendet (siehe Abschnitt
3.1.2). Zur Pyridosin-Bestimmung wird das selbst dargestellte Präparat als Standard
verwendet. CML wird unter Verwendung eines salzsauren Hydrolysates von BzGCML mit
Glycin als interne Referenz kalibriert.
Zur Berechnung der molaren Ausbeuten der Hydrolyseprodukte der Nα-Hippuryl-Amadori-
Produkte wird Glycin als interne Referenz verwendet. Bei Nε-Fructoselysin wird der mittels
Elementaranalyse ermittelte Gehalt genutzt (siehe unten).
Formeln zur Berechnung der molaren Ausbeuten
%100⋅=Gly
xx n
nA (Nα-Hippuryl-Amadori-Produkte)
%100⋅=FruK
xx n
nA (Nε-Fructosyllysin)
Ax molare Ausbeute an Furosin, Pyridosin, Lysin oder CML [%]
nx Stoffmenge an Furosin, Pyridosin, Lysin oder CML [nmol/20µl]
nGly Stoffmenge an Glycin [nmol/20µl]
nFruK Ausgangsgehalt an Nε-Fructoselysin [nmol/20µl]
- 74 -

3 Experimenteller Teil
3.4.3 Bildung von Furosin ohne Salzsäurehydrolyse und Stabilität von
Nε-Fructoselysin in Salzsäure
Bildung von Furosin ohne Salzsäurehydrolyse
Die Untersuchung zur direkten Bildung von Furosin im Zuge der Maillard-Reaktion erfolgt in
Anlehnung an Hollnagel und Kroh (2000).
Dazu werden 36,0 mg (0,1 mmol) Maltose Monohydrat und 24,6 mg (0,1 mmol) Nα-Boc-
Lysin in 1000 µl Reinstwasser gelöst, die Lösung wird gefriergetrocknet und das
pulverförmiges Lyophilisat wird in verschlossenen Pyrex-Hydrolysegefäß bei 120 °C für
15 min im Sandbad im Trockenschrank erhitzt. Die rotbraun gefärbte Probe wird in 1000 µl
Reinstwasser gelöst. Zur Abspaltung der Boc-Gruppe werden zu einem Aliquot von 200 µl
18 µl 12 M Salzsäure hinzugefügt und anschließend wird über Nacht bei Raumtemperatur
inkubiert. Danach wird die Probe 1 + 9 mit 0,2 M Natriumcitrat-Puffer (pH = 2,20, Puffer 1
siehe Abschnitt 3.2.3) verdünnt, membranfiltriert (0,2 µm, regenerierte Cellulose) und mittels
Aminosäureanalyse untersucht (siehe Abschnitt 3.2.3).
Stabilität von Nε-Fructoselysin in Salzsäure
Nle-FruK-Stammlösung
6,1 mg (46,50 µmol) Norleucin (Nle) und 20,4 mg (52,27 µmol) Nε-Fructoselysin (Gehalt:
79 %) in 1000 µl Reinstwasser lösen.
Es wird auf die Stabilität von Nε-Fructoselysin in 6 M und 8 M Salzsäure unter nicht-
hydrolytischen Bedingungen mit Norleucin (Nle) als internen Standard geprüft.
Herstellung der Ansätze:
- Ansatz in 6 M Salzsäure: 200 µl Nle-FruK-Stammlösung + 200 µl 12 M Salzsäure
- Ansatz in 8 M Salzsäure: 200 µl Nle-FruK-Stammlösung + 400 µl 12 M Salzsäure
Die Inkubationen erfolgen bei 6 °C sowie 23 °C. Zur Untersuchung wird ein Aliquot von
20 µl (6 M-Ansätze) oder 30 µl (8 M-Ansätze) mit 0,2 M Natriumcitrat-Puffer (pH = 2,20,
Puffer 1 siehe Abschnitt 3.2.3) auf 1000 µl verdünnt, die Lösung wird membranfiltriert
(0,2 µm, regenerierte Cellulose) und mittels Aminosäureanalyse untersucht (siehe Abschnitt
3.2.3).
- 75 -

3 Experimenteller Teil
3.5 Studien zur Reaktion von Peptiden mit α-Dicarbonylverbindungen
3.5.1 Reaktion von ausgewählten Peptiden mit α-Dicarbonylverbindungen
Lösungen - Puffer
- 100 mM Phosphatpuffer (PB) (pH = 7,4, 1,25-fach)
2,23 g di-Natriumhydrogenphosphat-Dihydrat in ca. 90 ml Reinstwasser lösen, mit
Salzsäure pH = 7,4 einstellen und mit Reinstwasser auf 100 ml auffüllen.
- 50 mM Phosphatpuffer (PB) (pH = 7,4, 1,25-fach)
1,12 g di-Natriumhydrogenphosphat-Dihydrat in ca. 90 ml Reinstwasser lösen, mit
Salzsäure pH = 7,4 einstellen und mit Reinstwasser auf 100 ml auffüllen.
- 10 mM Phosphatpuffer (PB) (pH = 7,4, 1,25-fach)
0,223 g di-Natriumhydrogenphosphat-Dihydrat in ca. 90 ml Reinstwasser lösen, mit
Salzsäure pH = 7,4 einstellen und mit Reinstwasser auf 100 ml auffüllen.
- 100 mM Phosphatpuffer (PB) mit definiertem pH-Wert (1,25-fach)
1,12 g di-Natriumhydrogenphosphat-Dihydrat in ca. 40 ml Reinstwasser lösen, mit
Salzsäure (pH = 3,1, 4,0, 5,1, 6,1, 6,6, 7,1, 8,1, 8,5, 8,9) oder 4 M Natronlauge
(pH = 9,9, 11,1) den pH-Wert einstellen und mit Reinstwasser auf 50 ml auffüllen.
- Phosphatgepufferte Kochsalzlösung (PBS) (pH = 7,4, 1,25-fach)
184,2 mg di-Natriumhydrogenphosphat-Dihydrat, 29,3 mg Kaliumdihydrogen-
phosphat, 1000,1 mg Natriumchlorid und 25,2 mg Kaliumchlorid in 100 ml
Reinstwasser lösen.
- MOPS-Puffer (pH = 7,4, 1,25-fach)
2,62 g MOPS (3-Morpholino-propansulfonsäure) in ca. 90 ml Reinstwasser lösen, mit
4 M Natronlauge pH = 7,4 einstellen und mit Reinstwasser auf 100 ml auffüllen.
- TAPSO-Puffer (pH = 7,4, 1,25-fach)
3,24 g TAPSO (N-[Tris(hydroxymethyl)-methyl]-3-amino-2-hydroxy-propansulfon-
säure) in ca. 90 ml Reinstwasser lösen, mit 4 M Natronlauge pH = 7,4 einstellen und
mit Reinstwasser auf 100 ml auffüllen.
- TRIS-Puffer (pH = 7,4, 1,25-fach)
1,51 g TRIS (Tris(hydroxymethyl)-aminomethan) in ca. 90 ml Reinstwasser lösen, mit
Salzsäure pH = 7,4 einstellen und mit Reinstwasser auf 100 ml auffüllen.
- 76 -

3 Experimenteller Teil
- 100 mM und 200 mM Ameisensäure
377 µl Ameisensäure (100 %) werden mit Reinstwasser auf 100 ml (100 mM) oder
50 ml (200 mM) ergänzt.
Lösungen - α-Dicarbonylverbindungen
- Glyoxal (GO)-Lösung (5 mM, 5-fach)
143 µl Glyoxal-Lösung (40 %) mit Reinstwasser im Maßkolben auf 50 ml auffüllen.
- Methylglyoxal (MGO)-Lösung (5 mM, 5-fach)
188 µl Methylglyoxal-Lösung (40 %) mit Reinstwasser im Maßkolben auf 50 ml
auffüllen.
- 3-Desoxyglucosulose (3-DG)-Lösung (5 mM, 5-fach)
22,5 mg 3-DG in 5 ml Reinstwasser im Maßkolben lösen.
- Diacetyl-Lösung (5 mM, 5-fach)
109,3 µl Diacetyl-Lösung (>99 %) mit Reinstwasser im Maßkolben auf 50 ml
auffüllen.
Lösungen - Peptide
- Gly-Ala-Phe-Lösung (5 mM, 1,25-fach)
20,5 mg Gly-Ala-Phe x 1,9 H2O in 10 ml Puffer (1,25-fach) lösen.
- Phe-Gly-Gly-Lösung (5 mM, 1,25-fach)
17,5 mg Phe-Gly-Gly in 10 ml 100 mM (1,25-fach) PB lösen.
- Phe-Gly-Lösung (5 mM, 1,25-fach)
14,5 mg Phe-Gly (Gehalt: 95,9 %) in 10 ml 100 mM (1,25-fach) PB lösen.
- Ala-Phe-Gly-Lösung (5 mM, 1,25-fach)
18,4 mg Ala-Phe-Gly in 10 ml 100 mM (1,25-fach) PB lösen.
- Gly-Phe-Ala-Lösung (5 mM, 1,25-fach)
19,5 mg Gly-Phe-Ala in 10 ml 100 mM (1,25-fach) PB lösen.
- Phe-Lösung (5 mM, 1,25-fach)
10,6 mg Phe in 10 ml 100 mM (1,25-fach) PB lösen.
- Phenylalaninamid (F-NH2)-Lösung (5 mM, 1,25-fach)
5,1 mg in 5 ml 100 mM (1,25-fach) PB lösen.
- BGG-Lösung (5 mM, 1,25-fach)
14,8 mg BGG in 10 ml 100 mM (1,25-fach) PB lösen.
- 77 -

3 Experimenteller Teil
- BzGK-Lösung (5 mM, 1,25-facht)
19,2 mg BzGK in 10 ml 100 mM (1,25-fach) PB lösen.
- BzGR-Lösung (5 mM, 1,25-fach)
21,0 mg BzGR in 10 ml 100 mM (1,25-fach) PB lösen.
- AcSer-Gly-Lösung (5 mM, 1,25-fach)
12,8 mg AcSerGly in 10 ml 100 mM (1,25-fach) PB lösen.
- AcTrp-Lösung (5 mM, 1,25-fach)
15,4 mg AcTrp in 10 ml 100 mM (1,25-fach) PB lösen.
- AcCys-Lösung (5 mM, 1,25-fach)
10,2 mg AcCys in 10 ml 100 mM (1,25-fach) PB lösen.
- BzMet-Lösung (5 mM, 1,25-fach)
15,9 mg BzMet in 10 ml 100 mM (1,25-fach) PB lösen.
- BzHis-Lösung (5 mM, 1,25-fach)
16,2 mg BzHis in 10 ml 100 mM (1,25-fach) PB lösen.
- Gly-Ala-Phe-BzGR-Lösung (je 5 mM, 1,25-fach) (kompetitive Experimente)
20,5 mg Gly-Ala-Phe x 1,9 H2O sowie 21,0 mg BzGR in 10 ml Puffer (1,25-fach)
lösen.
- Glycinamid-Lösung (5 mM, 1,25-fach)
3,5 mg Glycinamid × 1,0 HCl × 0,8 H2O in 5 ml 100 mM (1,25-fach) PB lösen.
Durchführung
Die Inkubationsansätze werden wie in Tab. 3.5.1-1 angegeben angefertigt (jeweils dreifach)
und bei 60 °C für 72 h, bei 37 °C für 24 h sowie bei 37 °C für bis zu 14 d (ausgewählte
Ansätze) im Brutschrank inkubiert. Zum Abstoppen wird 1 + 1, 1 + 3 (Nα-Hippuryl-,
Nα-Benzoyl-Derivate) oder 1 + 9 (AcTrp) mit 100 mM Ameisensäure verdünnt
(resultierender pH = 2,8). Ansätze mit einem pH ≥ 9,0 werden mit 200 mM Ameisensäure
abgestoppt. Die Herstellung der 1 + 3 und 1 + 9 Verdünnungen erfolgt nach Abkühlen auf
Raumtemperatur mit einem Aliquot. Die Proben werden anschließend mittels RP-HPLC
analysiert. Der pH-Wert wird vor und nach der Inkubation in separaten Ansätzen kontrolliert.
- 78 -

3 Experimenteller Teil
Tab. 3.5.1-1: Zusammensetzung der Inkubationsansätze.
Probe Peptid-Lösung Puffer α-Dicarbonyl-Lösung Reinstwasser
Ansatz 400 µl - 100 µl -
Peptid-Bw* 400 µl - - 100 µl
Dicarbonyl-Bw* - 400 µl 100 µl -
*Bw: Blindwert
Der Umsatz an Peptid und die dazugehörige Standardabweichung wird wie folgt berechnet:
%100100[%] ×=ohne
mit
AAUmsatz
22
100 ⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟⎟
⎠
⎞⎜⎜⎝
⎛××=
ohne
ohne
mit
mit
ohne
mit
As
As
AAs
Amit Peakfläche (λ = 220 nm) des Peptidpeaks nach Inkubation mit der α-Dicarbonyl-
verbindung
Aohne Peakfläche (λ = 220 nm) des Peptidpeaks nach Inkubation ohne der α-Dicarbonyl-
verbindung (Peptid-Blindwert)
Analytische RP-HPLC
HPLC-Anlage: Merck Hitachi, siehe Abschnitt 3.2
Säule: SA1, Knauer Eurospher 100-C18, siehe Abschnitt 3.2
Nα-Hippuryl-Derivate:
Eluenten-System: E1, Phosphatpuffer-Methanol-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 6 auf 13 % B in 25 min (BzGK-Ansätze)
linear ansteigend, von 12 auf 19 % B in 25 min (BzGR-Ansätze)
Flussrate: 0,7 ml/min
Sonstige Derivate:
Eluenten-System: E3, Triethylamin-Essigsäure-Ameisensäure-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 12 auf 57 % B in 35 min (GO-Ansätze)
Flussrate: 0,5 ml/min (GO-Ansätze)
Gradient: linear ansteigend, von 25 auf 55 % B in 35 min (MGO-, 3-DG-Ansätze)
Flussrate: 0,7 ml/min (MGO-, 3-DG-Ansätze)
- 79 -

3 Experimenteller Teil
Gly-Ala-Phe-Ansätze zur Umsatzbestimmung:
Eluenten-System: E5, Ameisensäure-Methanol-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 15 auf 43 % B in 30 min
Flussrate: 0,7 ml/min
Temperatur: 20 °C
Injektionsvolumen: 20 µl
Detektor: L4200 (Merck Hitachi), λ = 220 nm, alternativ K2501 (Knauer),
λ1 = 220 nm, siehe Abschnitt 3.2
K2501 (Knauer), λ2 = 320 nm, siehe Abschnitt 3.2
LaChrom L-7485 (Merck Hitachi), λex = 320 nm, λem = 400 nm, siehe
Abschnitt 3.2
3.5.2 Darstellung von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin
Darstellung
Um das Reaktionsprodukt der Umsetzung von Gly-Ala-Phe mit Glyoxal strukturell
aufzuklären, wurde ein semipräparativer Ansatz angefertigt und das Produkt mittels
semipräparativer RP-HPLC isoliert.
Lösungen
- Natriumphosphatpuffer (PB) pH = 7,4 (100 mM, einfach)
1,78 g di-Natriumhydrogenphosphat-Dihydrat in ca. 90 ml Reinstwasser lösen, mit
1 M Salzsäure pH-Wert einstellen und mit Reinstwasser auf 100 ml auffüllen.
235 mg (0,72 mmol) Gly-Ala-Phe × 1,9 H2O werden in 100 ml 100 mM Natriumphosphat-
puffer (pH = 7,4, einfach konzentriert) gelöst und mit insgesamt 0,80 mmol Glyoxal bei 37 °C
für 7 d inkubiert. Die Zugabe des Glyoxals (40 %ig) erfolgt gestaffelt in Portion zu 9,1 µl
(0,08 mmol) zweimal täglich in den ersten fünf Tagen. Der pH-Wert wird täglich geprüft und
falls erforderlich mit 4 M Natronlauge wieder auf pH = 7,4 eingestellt. Nach der Inkubation
wird die Lösung unter reduziertem Druck am Vakuumrotationsverdampfer auf ca. 10 ml
eingeengt, mit 1,0 ml Essigsäure angesäuert, membranfiltriert (0,45 µm, regenerierte
Cellulose) und mittels semipräparativer RP-HPLC fraktioniert.
- 80 -

3 Experimenteller Teil
Semipräparative RP-HPLC
HPLC-Anlage: Knauer, semipräparativ , siehe Abschnitt 3.2
Säule: SP2, Knauer Eurospher 100-C18, siehe Abschnitt 3.2
Eluenten-System: E4, Essigsäure-Methanol-System, siehe Abschnitt 3.2
Gradient: isokratisch, 30 % B 30 min
Flussrate: 6,0 ml/min
Temperatur: 20 °C
Injektionsvolumen: 500 - 1000 µl
Detektor: K2501 (Knauer), λ = 320 nm, siehe Abschnitt 3.2
Die Produkt-Fraktionen werden vereinigt, am Vakuumrotationsverdampfer unter reduziertem
Druck bei 35 °C auf ca. 20 ml eingeengt und gefriergetrocknet. Das Präparat wird so oft
wieder in Reinstwasser gelöst und gefriergetrocknet, bis der Geruch nach Essigsäure nur noch
schwach wahrnehmbar ist (ca. 5 ×). Die Aufbewahrung des Lyophilisates erfolgt bei – 18 °C.
Analytische Daten N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin: ESI-MS, [M+H]+
m/z = 315,7, [M-H]- m/z = 313,7; 1H-NMR (500 MHz, DMSO-d6) δ / ppm: 1,45 (3H, d, 3J = 7,2 Hz, A-H3), 2,87 (1H, dd, 2J = 13,3 Hz, 3J = 6,5 Hz, F-H3A), 3,02 (1H, dd, 2J = 13,3 Hz, 3J = 4,9 Hz, F-H3B), 4,08 (1H, dd, 3J = 4,9 Hz, 3J = 6,5 Hz, F-H2), 5,38 (1H, q, 3J = 7,2 Hz, A-H2), 7,04 (2H, m, F-H5,5´), 7,07 - 7,10 (2H, m, F-H6,6´), 7,07 - 7,10 (1H, m,
F-H7), 7,30 (1H, d 3J = 4,5 Hz, P-H5), 7,52 (1H, dd, 3J = 4,5Hz, 5J = 1,0 Hz, P-H6), 7,81 (1H,
d, 3J = 7,1Hz, A-NH-F), 7,99 (1H, d, 5J = 1,0 Hz, P-H3), 13C-NMR (125 MHz, DMSO-d6)
δ / ppm: 16,29 (q, A-C3), 37,14 (t, F-C3), 52,50 (d, A-C2), 55,50 (d, F-C2), 122,97 (d, P-C5),
125,65 (d, F-C7), 127,40 (d, P-C6), 127,63 (d, F-C6,6´), 129,35 (d, F-C5,5´), 138,77 (s,
F-C4), 147,56 (d, P-C3), 155,03 (s, P-C2), 167,50 (s, A-C1), 172,71 (s, F-C1); UV- and
Fluoreszenz Daten (100 mM PB, pH = 7,0): λUVmax = 322 nm, ε322 = 4900 l⋅mol-1⋅cm-1,
λex, max = 330 nm, λem, max = 395 nm; Elementaranalyse, C16H17N3O4 × 0,05 CH3COOH
× 0,45 H2O (Mbrutto = 326,43 g/mol) berechnet: C = 59,24 %, H = 5,59 %, N = 12,87 %,
gefunden: C = 59,75 %, H = 5,49 %, N = 12,86 %; Ausbeute: 112 mg (molare Ausbeute:
47,9 %); weißer Feststoff, chromatographische Reinheit: > 99 % (HPLC)
- 81 -

3 Experimenteller Teil
Untersuchungen zur Stabilität von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin -
Stabilität bei ausgewählten pH-Werten
Lösungen
- Phosphatpuffer (PB) mit pH = 2,0, 4,0, 7,4 oder 9,1 (100 mM, einfach)
1,78 g di-Natriumhydrogenphosphat-Dihydrat in ca. 90 ml Reinstwasser lösen, mit
Salzsäure pH-Wert einstellen und mit Reinstwasser auf 100 ml auffüllen.
- N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin-Lösung (5 mM)
1,6 mg Präparat in 1000 µl 100 mM PB definierten pH-Wertes lösen.
Zur Untersuchung der Stabilität bei verschiedenen pH-Werten werden je 450 µl N-[2-(2-Oxo-
2H-pyrazin-1-yl)-propyl]-phenylalanin-Lösung (5 mM) bei 60 °C für 72 h im Brutschrank
inkubiert. Anschließend wird 1 + 1 mit 100 mM Ameisensäure verdünnt und mittels HPLC
untersucht (siehe Abschnitt 3.5.1). Es wird je pH-Wert eine Doppelbestimmung durchgeführt.
Untersuchungen zur Stabilität von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin -
Stabilität gegenüber Aminoguanidin (AMG)
Lösungen
- 100 mM Phosphatpuffer (PB) (pH = 7,4, 1,25-fach), siehe Abschnitt 3.5.1
- Aminoguanidin (AMG)-Lösung (5 mM, 5-fach)
17,0 mg Aminoguanidin Bicarbonat in 1000 µl Reinstwasser lösen.
- Glyoxal-Lösung (5 mM, 5-fach), siehe Abschnitt 3.5.1
- N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin-Lösung (5 mM)
4,1 mg Präparat in 2000 µl 100 mM PB (pH = 7,4, 1,25-fach) lösen.
Die Inkubationsansätze werden wie in Tab. 3.5.2-1 angegeben angefertigt (jeweils zweifach)
und bei 60 °C für 72 h im Brutschrank inkubiert. Anschließend wird 1 + 1 mit 100 mM
Ameisensäure verdünnt und mittels HPLC untersucht (siehe Abschnitt 3.5.1). Der Umsatz an
2(1H)-Pyrazinon-Peptid wird wie unter 3.5.1 angegeben berechnet.
- 82 -
3 Experimenteller Teil
Tab. 3.5.2-1: Zusammensetzung der Inkubationsansätze.
Probe Pyrazinon-Lsg Puffer AMG-Lsg Glyoxal-Lsg Reinstwasser
Ansatz 400 µl - 100 µl - -
Pyrazinon-Bw* 400 µl - - - 100 µl
AMG-Bw* - 400 µl 100 µl - -
Triazin-Ansatz - 300 µl 100 µl 100 µl -
*Bw: Blindwert, AMG: Aminoguanidin
3.5.3 Darstellung von 2(1H)-Pyrazinon-Peptiden des Glyoxals
Um die Reaktionsprodukte der Umsetzung von weiteren Peptiden mit Glyoxal strukturell
aufzuklären, wurden entsprechende semipräparative Ansätze angefertigt und die Produkte
mittels Extraktion oder semipräparativer RP-HPLC isoliert.
3-Methyl-2-(2-oxo-2H-pyrazin-1-yl)-valeriansäure und 2-(2-Oxo-2H-pyrazin-1-yl)-2-
phenyl-essigsäure
377 mg (2,0 mmol) Gly-Ile und 335 mg (1,5 mmol) Gly-Phe werden jeweils in 25 ml
100 mM Natriumphosphatpuffer (pH = 7,4, einfach konzentriert, siehe Abschnitt 3.5.2) gelöst
und mit insgesamt 2,1 mmol (Gly-Ile-Ansatz) oder 1,6 mmol (Gly-Phe-Ansatz) Glyoxal bei
37 °C für 7 d inkubiert. Die Zugabe des Glyoxals (40 %ig) erfolgt gestaffelt in Portion zu
23,9 µl (0,21 mmol, Gly-Ile-Ansatz) oder 18,0 µl (0,16 mmol, Gly-Phe-Ansatz) zweimal
täglich in den ersten fünf Tagen. Der pH-Wert wird täglich geprüft und falls erforderlich mit
1 M Natronlauge wieder auf pH = 7,4 eingestellt.
Zur Extraktion des 2(1H)-Pyrazinon-Dipeptides wird der pH-Wert des Ansatzes mit 2 M
Kaliumhydrogensulfat-Lösung (ca. 2,4 ml) auf pH = 2,0 eingestellt, und es wird mit 3 × 30 ml
Ethylacetat extrahiert. Das Ausgangsdipeptid verbleibt dabei in der wässrigen Phase. Die
vereinigten Extrakte werden zweimal mit je 10 ml Reinstwasser gewaschen und anschließend
über Natriumsulfat getrocknet. Nach der Abtrennung des Natriumsulfats durch Filtration über
einen Faltenfilter wird das Ethylacetat am Vakuumrotationsverdampfer (30 °C, 78 mbar)
entfernt. Der Rückstand wird in 20 ml Methanol aufgenommen und es wird am
Vakuumrotationsverdampfer wieder zur Trockene eingeengt. Schließlich wird für 5 h im
Vakuum (25 °C, 0,05 mbar) getrocknet.
- 83 -

3 Experimenteller Teil
Analytische Daten 3-Methyl-2-(2-oxo-2H-pyrazin-1-yl)-valeriansäure: ESI-MS, [M+H]+
m/z = 211,10; 1H-NMR (500 MHz, DMSO-d6) δ / ppm: 0,85 (3H, m, I-H5), 0,85 (3H, m,
I-H6), 1,30 (2H, m, I-H4), 1,85 (1H, m, I-H3), 5,02 (1H, d, 3J = 9,8Hz, I-H2), 7,36 (1H, d, 3J = 4,5 Hz, P-H5), 7,67 (1H, d, 3J = 4,5 Hz, 5J = 1,1 Hz, P-H6), 8,04 (1H, d, 5J = 1,1 Hz
P-H3), 13C-NMR (125 MHz, DMSO-d6) δ / ppm: 11,31 (q, I-C5), 15,54 (q, I-C6), 24,74 (t,
I-C4), 36,47 (d, I-C3), 61,43 (d, I-C2), 123,11 (d, P-C5), 128,36 (d, P-C6), 148,08 (d, P-C3),
155,28 (s, P-C2), 170,22 (s, I-C1), UV- and Fluoreszenz Daten: (HPLC-DAD bzw.
Fluoreszenzdetektor, HPLC-Eluent Wasser-Methanol mit 0,1 % (v/v) Ameisensäure):
λUVmax = 326 nm, λem,max = 396 nm bei λex = 320 nm; Ausbeute: 226 mg (molare Ausbeute:
54 %); dunkelbrauner Feststoff, chromatographische Reinheit: > 92 % (HPLC)
Analytische Daten 2-(2-Oxo-2H-pyrazin-1-yl)-2-phenyl-essigsäure: ESI-MS, [M+H]+
m/z = 245,07; 1H-NMR (500 MHz, DMSO-d6) δ / ppm: 3,39 (1H, d, 2J = 6,4Hz, F-H3A), 3,48
(1H, d, 2J = 6,4Hz, F-H3B), 5,40 (1H, dd, 3J = 11,3Hz, 3J = 4,9Hz, F-H2), 7,10 (2H, m, F-
H5,5´), 7,17 (1H, m, F-H7), 7,18 (1H, d, 3J = 4,4Hz, P-H5), 7,23 (2H, m, F-H6,6´), 7,47 (1H,
d, 3J = 4,4Hz, P-H6), 7,92 (1H, d, 5J = 0,8Hz, P-H3), 13C-NMR (125 MHz, DMSO-d6)
δ / ppm: 34,13 (t, F-C3), 61,21 (d, F-C2), 122,61 (d, P-C5), 126,72 (d, F-C7), 128,46 (d,
F-C6,6´), 128,77 (d, F-C5,5´), 129,63 (d, P-C6), 136,53 (s, F-C4), 148,13 (d, P-C3), 155,14
(s, P-C2), 169,64 (s, F-C1), UV- and Fluoreszenz Daten: (HPLC-DAD bzw.
Fluoreszenzdetektor, HPLC-Eluent Wasser-Methanol mit 0,1 % (v/v) Ameisensäure):
λUVmax = 324 nm, λem,max = 395 nm bei λex = 320 nm; Ausbeute: 295 mg (molare Ausbeute:
82 %); dunkelbraunes Öl, chromatographische Reinheit: > 90 % (HPLC)
N-[2-(2-Oxo-2H-pyrazin-1-yl)-2-phenyl-acetyl]-alanin
389 mg (1,25 mmol) Gly-Phe-Ala × 1,0 H2O werden in 200 ml 100 mM Natriumphosphat-
puffer (pH = 7,4, 1,25-fach konzentriert, siehe Abschnitt 3.5.1) gelöst und mit 1,25 mmol
Glyoxal (50 ml 5 mM Glyoxal-Lösung [5-fach, siehe Abschnitt 3.5.1]) bei 37 °C für 5 d
inkubiert. Der pH-Wert wird täglich geprüft und falls erforderlich mit 1 M Natronlauge
wieder auf pH = 7,4 eingestellt. Nach der Inkubation wird die Lösung unter reduziertem
Druck am Vakuumrotationsverdampfer auf ca. 25 ml eingeengt, mit 2,5 ml Essigsäure
angesäuert, membranfiltriert (0,45 µm, regenerierte Cellulose) und mittels semipräparativer
RP-HPLC fraktioniert. Die Anwendung des bei den 2(1H)-Pyrazinon-Dipeptiden
- 84 -

3 Experimenteller Teil
angewendeten Extraktionsverfahrens ist nicht möglich, da das Ausgangstripeptid ebenfalls
extrahierbar ist. Die semipräparative RP-HPLC und die Aufarbeitung der Fraktionen wird wie
in Abschnitt 3.5.2 angegeben durchgeführt.
Analytische Daten N-[2-(2-Oxo-2H-pyrazin-1-yl)-2-phenyl-acetyl]-alanin: ESI-MS, [M+H]+
m/z = 316,09; 1H-NMR (500 MHz, DMSO-d6) δ / ppm: 1,25 (3H, d, 3J = 7,2 Hz, A-H3), 3,28
(1H, dd, 2J = 14,6 Hz, 3J = 12,0 Hz, F-H3A), 3,42 (1H, dd, 3J = 14,6 Hz, 3J = 4,5 Hz, F-H3B),
4,08 (1H, q, A-H2), 5,86 (1H, dd, 3J = 12,0Hz, 3J = 4,5Hz, F-H2), 7,13 (1H, m, F-H7), 7,22
(2H, m, F-H5,5´), 7,22 (2H, m, F-H6,6´), 7,25 (1H, d, 3J = 4,5 Hz, P-H5), 7,81 (1H, dd, 3J = 4,5 Hz, 5J = 1,0 Hz, P-H6), 7,84 (1H, d, 5J = 1,0 Hz, P-H3), 8,59 (1H, d, 3J = 7,2 Hz,
A-NH-F), 13C-NMR (125 MHz, DMSO-d6) δ / ppm: 17,94 (q, A-C3), 35,55 (t, F-C3), 49,10
(d, A-C2), 57,21 (d, F-C2), 122,27 (d, P-C5), 126,68 (d, F-C7), 128,29 (d, F-C6,6´), 128,71
(d, P-C6), 128,76 (d, F-C5,5´), 136,25 (s, F-C4), 147,38 (d, P-C3), 155,27 (s, P-C2), 167,26
(s, F-C1), 174,20 (s, A-C1), UV- and Fluoreszenz Daten: (HPLC-DAD bzw.
Fluoreszenzdetektor, HPLC-Eluent Wasser-Methanol mit 0,1 % (v/v) Ameisensäure):
λUVmax = 326 nm, λem,max = 397 nm bei λex = 320 nm; Ausbeute: 193 mg (molare Ausbeute:
46 %); schwach gelb gefärbter Feststoff, chromatographische Reinheit: > 98 % (HPLC)
N-[2-(2-Oxo-2H-pyrazin-1-yl)-acetyl]-glycin
946 mg (5,0 mmol) Gly-Gly-Gly werden in 40 ml 100 mM Natriumphosphatpuffer (pH = 7,4,
einfach konzentriert, siehe Abschnitt 3.5.2) gelöst und mit insgesamt 5,0 mmol Glyoxal bei
37 °C für 7 d inkubiert. Die Zugabe des Glyoxals (40 %ig) erfolgt gestaffelt in Portion zu
114,3 µl (0,5 mmol) zweimal täglich in den ersten fünf Tagen. Der pH-Wert wird täglich
geprüft und falls erforderlich mit 1 M Natronlauge wieder auf pH = 7,4 eingestellt. Nach der
Inkubation wird die Lösung unter reduziertem Druck am Vakuumrotationsverdampfer auf
ca. 15 ml eingeengt, mit 1,5 ml Essigsäure angesäuert, membranfiltriert (0,45 µm,
regenerierte Cellulose) und mittels semipräparativer RP-HPLC fraktioniert.
Semipräparative RP-HPLC und Aufarbeitung der Fraktionen
siehe Abschnitt 3.5.2, geändert:
Gradient: isokratisch, 100 % A, 30 min
- 85 -

3 Experimenteller Teil
Analytische Daten N-[2-(2-Oxo-2H-pyrazin-1-yl)-acetyl]-glycin: ESI-MS, [M+H]+
m/z = 212,08; 1H-NMR (500 MHz, Methanol-d4) δ / ppm: 3,94 (1H, s, G3-H2), 4,80 (1H, s,
G2-H2), 7,47 (1H, d, 3J = 4,4Hz, P-H5), 7,61 (1H, dd, 5J = 1,0Hz, 3J = 4,4Hz, P-H6), 8,13
(1H, s, 5J = 1,0Hz, P-H3), UV-Daten: (0,01 mg/ml in 0,1 % (v/v) Ameisensäure):
λUVmax = 319 nm; Elementaranalyse, berechnet: C = 45,50 %, H = 4,30 %, N = 19,9 %,
gefunden: C = 36,18 %, H = 4,17 %, N = 15,91 %, Gehalt auf C-Basis = 79,5 %; Ausbeute:
770 mg (molare Ausbeute: 58 % [Gehalt auf C-Basis berücksichtigt]); schwach gelb gefärbter
Feststoff, chromatographische Reinheit: > 97 % (HPLC)
3.5.4 Darstellung von N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-
phenylalanin
Um das Hauptreaktionsprodukt der Umsetzung von Gly-Ala-Phe mit Methylglyoxal
strukturell aufzuklären, wurde ein semipräparativer Ansatz angefertigt und das Produkt
mittels semipräparativer RP-HPLC isoliert.
470 mg (1,44 mmol) Gly-Ala-Phe × 1,9 H2O werden in 200 ml 100 mM Natriumphosphat-
puffer (pH = 7,4) gelöst und mit insgesamt 1,60 mmol Methylglyoxal bei 37 °C für 12 d
inkubiert. Die Zugabe des Methylglyoxals (40 %ig) erfolgt gestaffelt in Portion zu 24,6 µl
(0,16 mmol) einmal täglich in den ersten zehn Tagen. Der pH-Wert wird täglich geprüft und
falls erforderlich mit 4 M Natronlauge wieder auf pH = 7,4 eingestellt. Nach der Inkubation
wird die Lösung unter reduziertem Druck am Vakuumrotationsverdampfer bei 35 °C auf
ca. 20 ml eingeengt, mit Ameisensäure auf pH = 4,7 angesäuert, membranfiltriert (0,45 µm,
regenerierte Cellulose) und mittels semipräparativer RP-HPLC fraktioniert.
Semipräparative RP-HPLC
HPLC-Anlage: Merck Hitachi, siehe Abschnitt 3.2
Säule: SP1, Knauer Eurospher 100-C18, siehe Abschnitt 3.2
Eluenten-System: E4, Essigsäure-Methanol-System, siehe Abschnitt 3.2
Gradient: isokratisch, 35 % B, 30 min
Flussrate: 2,1 ml/min
Temperatur: 20 °C
- 86 -

3 Experimenteller Teil
Injektionsvolumen: 400 µl
Detektor: K2501 (Knauer), λ1 = 220 nm, siehe Abschnitt 3.2
K2501 (Knauer), λ2 = 320 nm, siehe Abschnitt 3.2
LaChrom L-7485 (Merck Hitachi), λex = 320 nm, λem = 400 nm, siehe
Abschnitt 3.2
Das Hauptreaktionsprodukt wird anhand der Absorption bei λ = 320 nm und der Fluoreszenz
gesammelt. Die Fraktionen werden vereinigt, am Vakuumrotationsverdampfer unter
reduziertem Druck bei 35 °C auf ca. 20 ml eingeengt und gefriergetrocknet. Das Präparat wird
so oft wieder in Reinstwasser gelöst und gefriergetrocknet, bis der Geruch nach Essigsäure
nur noch schwach wahrnehmbar ist (ca. 5 ×). Die Aufbewahrung des Lyophilisates erfolgt bei
- 18 °C.
Analytische Daten N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin: ESI-MS,
[M+H]+ m/z = 330,1, [M-H]- m/z = 328,2; 1H-NMR (500 MHz, DMSO-d6) δ / ppm: 1,61
(3H, d, 3J = 7,2 Hz, A-H3), 2,32 (1H, s, P-5Me-H), 3,02 (1H, dd, 2J = 14,0 Hz, 3J = 8,9 Hz,
F-H3A), 3,27 (1H, dd, 2J = 14,0Hz, 3J = 8,9 Hz, F-H3B), 4,73 (1H, dd, 3J = 8,9 Hz, 3J = 4,9 Hz , F-H2), 5,51 (1H, q, 3J = 7,2 Hz , A-H2), 7,19-7,26 (2H, m, F-H5,5´), 7,19-7,26
(2H, m, F-H6,6´), 7,19-7,26 (1H, m, F-H7), 7,36 (1H , s, P-H6), 8,06 (1H , s, P-H3), 13C-NMR (125 MHz, DMSO) δ / ppm: 16,45 (q, A-C3), 19,51 (q, P-C5Me), 38,27 (t, F-C3),
54,67 (d, A-C2), 55,20 (d, F-C2), 124,74 (d, P-C6), 127,73 (d, F-C7), 129,43 (d, F-C6,6´),
130,37 (d, F-C5,5´), 134,06 (s, P-C5), 138,17 (s, F-C4), 147,72 (d, P-C3), 156,63 (s, P-C2),
170,87 (s, A-C1), 174,32 (s, F-C1); UV- and Fluoreszenz Daten (100 mM PB, pH = 7,0):
λUVmax = 336 nm, ε336 = 4600 l⋅mol-1⋅cm-1, λex, max = 340 nm, λem, max = 405 nm; Elementar-
analyse, C17H19N3O4 × 0,13 CH3COOH × 0,50 H2O (Mbrutto = 346,17 g/mol) berechnet:
C = 59,87 %, H = 5,97 %, N = 12,14 %, gefunden: C = 59,72 %, H = 5,90 %, N = 12,17 %;
Ausbeute: 29 mg (molare Ausbeute: 5,8 %); weißer Feststoff, chromatographische Reinheit:
> 99 % (HPLC)
- 87 -

3 Experimenteller Teil
3.6 Analyse peptidgebundener 2(1H)-Pyrazinone nach Salzsäurehydrolyse
3.6.1 Säurehydrolyse von 2(1H)-Pyrazinon-Peptiden
Lösungen
- N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin -Lösung
3,4 mg N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin × 0,05 Essigsäure
× 0,45 Wasser (Mbrutto = 326,43 g/mol) in 3000 µl Reinstwasser lösen.
- N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin -Lösung
3,6 mg N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin × 0,13 Essig-
säure × 0,50 Wasser (Mbrutto = 346,17 g/mol) in 3000 µl Reinstwasser lösen.
- Citrat-Puffer (2 M, pH = 9,0)
4,2 g (20,0 mmol) Citronensäure Monohydrat in ca. 5 ml Reinstwasser lösen, mit 4 M
Natronlauge pH = 9,0 einstellen und mit Reinstwasser auf 10 ml auffüllen.
Salzsäurehydrolyse
Es werden je 900 µl 2(1H)-Pyrazinon-Peptid-Lösung und 900 µl 12 M Salzsäure (Baker, ACS
quality) in ein 6 ml-Hydrolysegefäße pipettiert, der Ansatz wird gemischt, das Gefäß wird mit
Stickstoff gespült und fest verschlossen. Die Hydrolyse erfolgt bei 110 °C im Sandbad im
Trockenschrank für 16 h, 23 h oder 48 h.
Aufarbeitung - Zentrifugalverdampfer
143 µl (ca. 250 nmol) N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin-Hydrolysat bzw.
144 µl (ca. 250 nmol) N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin-
Hydrolysat werden je in Eppendorf-Gefäß pipettiert und im Zentrifugalverdampfer zur
Trockene eingeengt. Die Proben werden in 500 µl Citrat-Puffer (0,2 M, pH = 2,2, Puffer 1
Tab. 3.2.3-1) im Ultraschallbad gelöst und anschließend membranfiltriert (regenerierte
Cellulose; 0,2 µm). Es werden je 20 µl (ca. 10 nmol) am Aminosäureanalysator (3.2.3) sowie
mittels RP-HPLC untersucht.
- 88 -

3 Experimenteller Teil
Aufarbeitung – Abpuffern der Säure mit Citrat-Puffer
143 µl (ca. 250 nmol) N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin-Hydrolysat bzw.
144 µl (ca. 250 nmol) N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin-
Hydrolysat werden je in ein Eppendorf-Gefäß pipettiert. Die Proben werden jeweils mit dem
gleichen Volumen Citratpuffer (2 M, pH = 9) versetzt, mit Reinstwasser auf 500 µl ergänzt
(resultierender pH = 1 bis 2) und membranfiltriert (regenerierte Cellulose; 0,2 µm). Es werden
je 20 µl (ca. 10 nmol) am Aminosäureanalysator (3.2.3) sowie mittels RP-HPLC untersucht.
Die aufgearbeiteten Lösungen der 16 h-Hydrolysate werden weiterhin mittels LC-ESI-MS
analysiert.
Aufarbeitung – Extraktion der Abbauprodukte im Alkalischen
500 µl Hydrolysat werden mit 4 M Natronlauge alkalisch gemacht (ca. pH = 12).
Anschließend wird drei mal mit je 300 µl Chloroform (p.a.) extrahiert. Die organischen
Phasen werden mittels Pipette abgenommen und vereinigt. Der so erhaltene Extrakt wird im
Stickstoffstrom zur Trockene eingeengt. Dabei wird nur so lang als notwendig mit Stickstoff
gespült. Der Rückstand wird anschließend sofort in 500 µl Fließmittel (0,1 % Ameisensäure
in Reinstwasser (v/v)) aufgenommen. Die Isolierung wird mittels HPLC kontrolliert.
RP-HPLC
HPLC-Anlage: Merck Hitachi, siehe Abschnitt 3.2
Säule: SA1, Knauer Eurospher 100-C18, siehe Abschnitt 3.2
Eluenten-System: E5, Ameisensäure-Methanol-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 8 auf 22 % B in 25 min (Pyrazinon-Hydrolysate)
linear ansteigend, von 15 auf 42 % B in 35 min (5-Methyl-pyrazinon-
Hydrolysate)
Flussrate: 0,7 ml/min
Temperatur: 20 °C
Injektionsvolumen: 20 µl
Detektor: K2501 (Knauer), λ1 = 220 nm, siehe Abschnitt 3.2
K2501 (Knauer), λ2 = 320 nm, siehe Abschnitt 3.2
LaChrom L-7485 (Merck Hitachi), λex = 320 nm, λem = 400 nm, siehe
Abschnitt 3.2
- 89 -

3 Experimenteller Teil
LC-ESI-MS
Proben: 16 h-Hydrolysate
HPLC-Anlage: Agilent 1100 series, siehe Abschnitt 3.2.2
Säule: SA1, Knauer Eurospher 100-C18, siehe Abschnitt 3.2.1
Eluenten-System: E5, Ameisensäure-Methanol-System, siehe Abschnitt 3.2.1
Gradient: linear ansteigend, von 8 auf 34 % B in 25 min (Pyrazinon-Hydrolysate)
linear ansteigend, von 15 auf 42 % B in 35 min (5-Methyl-pyrazinon-
Hydrolysate)
Flussrate: 0,3 ml/min
Temperatur: 20 °C
Injektionsvolumen: 100 µl
Detektor: DAD (Agilent 1100 series), λ1 = 220 nm, λ1 = 320 nm, s. Absch. 3.2.2
ESI-TOF-MS (Applied Biosystems), siehe Abschnitt 3.2.2
3.6.2 Darstellung von 2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure und N1-Ethyl-
2(1H)-pyrazinon
Zur Aufklärung der Hydrolyseprodukte von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-
phenylalanin wird 2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure synthetisiert und anschließend
mit Salzsäure behandelt. Das resultierende Produkt N1-Ethyl-2(1H)-pyrazinon wird im
semipräparativen Maßstab in chromatographisch reiner Form gewonnen.
730,7 mg (5,0 mmol) Gly-Ala werden in 70 ml 100 mM Natriumphosphatpuffer (pH = 7,4,
siehe Abschnitt 3.5.1) gelöst und mit insgesamt 571 µl (5,0 mmol) Glyoxal bei 37 °C für 8 d
inkubiert. Die Zugabe des Glyoxals (40 %ig) erfolgt gestaffelt in Portion zu 57,1 µl
(0,5 mmol) zweimal täglich in den ersten fünf Tagen. Der pH-Wert wird täglich geprüft und
falls erforderlich mit 1 M Natronlauge wieder auf pH = 7,4 eingestellt.
Nach der Inkubation wird der Ansatz (ca. 70 ml) unter Kühlung mit 70 ml 12 M Salzsäure
p. a. gemischt und auf 20-ml-Hydrolysegefäße verteilt. Nach dem Spülen mit Stickstoff
werden die Gefäße fest verschlossen und die Hydrolyse erfolgt bei 110 °C für 48 h im
Sandbad im Trockenschrank.
- 90 -

3 Experimenteller Teil
Der pH-Wert des Hydrolysates (ca. 140 ml) wird unter Eiswasserkühlung mit 4 M
Natronlauge auf pH = 11 bis 12 eingestellt. Die Lösung wird 3 × mit je 50 ml Chloroform
(reinst) im Schütteltrichter extrahiert. Die vereinigten organischen Phasen werden mit 50 ml
Reinstwasser gewaschen und anschließend über Natriumsulfat (wasserfrei) getrocknet. Der
Extrakt wird am Vakuumrotationsverdampfer (ca. 180 mbar, 25 °C) fast bis zur Trockne
eingeengt. Restliches Chloroform wird anschließend mit Stickstoff abgeblasen. Das Präparat
wird bei – 18 °C im Gefrierschrank aufbewahrt.
RP-HPLC
Proben: - 2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure-Ansatz
- N1-Ethyl-2(1H)-pyrazinon-Präparat
Durchführung: siehe Abschnitt 3.6.1, geändert:
Gradient: linear ansteigend, von 8 auf 22 % B in 25 min
Analytische Daten N1-Ethyl-2(1H)-pyrazinon: ESI-MS, [M+H]+ m/z = 125,09; 1H-NMR
(500 MHz, CDCl3) δ / ppm: 1,36 (3H, t, 3J = 7,26 Hz, Et-H2), 3,92 (2H, q, 3J = 7,26 Hz,
Et-H1), 7,30 (1H, d, 3J = 4,4 Hz, P-H5), 8,11 (1H, d, 5J = 1,0 Hz, P-H3), 7,09 (1H, dd, 3J = 4,4 Hz, 5J = 1,0 Hz, P-H6), 13C-NMR (125 MHz, CDCl3) δ / ppm: 13,98 (q, Et-C2),
44,45 (t, Et-C1), 123,95 (d, P-C5), 128,03 (d, P-C6), 149,58 (d, P-C3), 156,04 (s, P-C2); UV-
and Fluoreszenz Daten (0,01 mg / ml 0,1 % (v/v) Ameisensäure): λUVmax = 318 nm,
ε336 = 3535 l⋅mol-1⋅cm-1, λex, max = 360 nm, λem, max = 420 nm; Ausbeute: 17,6 mg (molare
Ausbeute: 2,0 %); brauner, schwach grüner Feststoff, chromatographische Reinheit: > 98 %
(HPLC)
3.6.3 Einfluss der Matrix auf die Säurehydrolyse von 2(1H)-Pyrazinon-Peptiden
Lösungen
- N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin–Lösung (1 mg / ml)
10 mg N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin × 0,05 Essigsäure
× 0,45 Wasser (Mbrutto = 326,43 g / mol) in 10 ml Reinstwasser lösen.
- Citrat-Puffer (2 M, pH = 9,0)
- 91 -

3 Experimenteller Teil
Durchführung
Entsprechend Tab. 3.6.3-1 werden die Zusätze in 6 ml Hydrolysegefäße eingewogen und mit
1,000 ml 2(1H)-Pyrazinon-Lösung oder Reinstwasser sowie mit je 1,000 ml 12 M Salzsäure
versetzt (Endkonzentration der Salzsäure: 6 M). Die Ansätze werden gemischt, die Gefäße
werden mit Stickstoff gespült und fest verschlossen. Die Hydrolyse erfolgt bei 110 °C mit den
angegebenen Zeiten im Sandbad im Trockenschrank.
Tab. 3.6.3-1: Zusammensetzung der zu Hydrolyse eingesetzten Lösungen.
Probe Zusammensetzung Zeit [h]
Pyrazinon 1 ml Pyrazinon-Lsg 16, 23, 48
Pyrazinon + Glucose 10 1 ml Pyrazinon-Lsg + 10 mg Glucose 16, 23, 48
Pyrazinon + Natriumazid 1 ml Pyrazinon-Lsg + 10 mg Natriumazid 16, 23, 48
Pyrazinon + Mercaptoethanol 1 ml Pyrazinon-Lsg + 10 mg Mercaptoethanol 16, 23, 48
Pyrazinon + Cornflakes 1 ml Pyrazinon-Lsg + 10 mg Cornflakes 16, 23, 48
Cornflakes 1 ml Reinstwasser + 10 mg Cornflakes 16, 23, 48
Pyrazinon + Insulin 1 ml Pyrazinon-Lsg + 2 mg Insulin 23
Pyrazinon + Glucose 1 1 ml Pyrazinon-Lsg + 1 mg Glucose 16
Pyrazinon + Glucose 1 +
Mercaptoethanol
1 ml Pyrazinon-Lsg + 1 mg Glucose +
10 mg Mercaptoethanol
16
RP-HPLC der Hydrolysate
Von den Hydrolysaten wird jeweils 1 ml abgenommen, es wird mit 6 M Natronlauge pH = 2
bis 4 eingestellt, die Lösung wird membranfiltriert (regenerierte Cellulose, 0,45 µm) und
anschließend mittels RP-HPLC untersucht.
Durchführung der RP-HPLC:
siehe Abschnitt 3.6.1, geändert: Injektionsvolumen: 50 µl
- 92 -

3 Experimenteller Teil
3.7 Untersuchungen zur Reaktion von Insulin mit Glyoxal
Inkubation von Insulin mit Glyoxal
Lösungen
- Insulin-Lösung (0,017 mM, 10-fach)
2,0 mg Rinderinsulin in 2000 µl 2 mM Salzsäure lösen.
- Glyoxal-Lösung (0,05 mM, 10-fach)
57,2 µl Glyoxal-Lösung (40 %) im Maßkolben mit Reinstwasser auf 100 ml ergänzen
und ein Aliquot dieser Lösung 1 + 9 mit Reinstwasser verdünnen.
- Phosphatgepufferte Kochsalzlösung (PBS) (pH = 7,4, 1,25-fach), siehe 3.5.1
- 10 mM Phosphatpuffer (PB) (pH = 7,4, 1,25-fach), siehe 3.5.1
Durchführung
Die Inkubationsansätze werden wie in Tab. 3.7-1 angegeben angefertigt (jeweils dreifach) und
bei 37 °C für 15 d im Brutschrank inkubiert. Anschließend werden die Ansätze entnommen,
auf Raumtemperatur abgekühlt und zum Abstoppen der Reaktion werden 33 µl 10 %ige
Ameisensäure (2,65 M) je 1000 µl Ansatz zugegeben (pH = 2,9, gemessen). Ansätze, die im
Anschluss reduziert werden sollen, dürfen nicht abgestoppt werden. Nach Membranfiltration
mittels Spritzenfilter (regenerierte Cellulose, 0,2 µm, Cellulosemischester sind ungeeignet)
werden die Proben unverdünnt mittels HPLC analysiert. Der pH-Wert wird vor und nach der
Inkubation in separaten Ansätzen kontrolliert.
Tab. 3.7-1: Inkubationsansätze von Insulin mit Glyoxal
Probe Puffer Insulin-Lösung Glyoxal-Lösung Reinstwasser
Ansatz 800 µl 100 µl 100 µl -
Insulin-Bw* 800 µl 100 µl - 100 µl
Glyoxal-Bw* 800 µl - 100 µl 100 µl
* Bw: Blindwert
- 93 -

3 Experimenteller Teil
UV-Spektroskopie von Insulin
Im Hinblick auf mögliche Einflüsse des Puffers auf die Struktur von Insulin wird das
UV-Spektrum von Insulin in PBS und in 10 mM Phosphatpuffer aufgenommen.
Lösungen
- PBS (1,25-fach) und 10 mM Phosphatpuffer (PB, 1,25-fach), siehe Abschnitt 3.5.1
- PBS (einfach) zum Verdünnen
8,00 ml PBS (1,25-fach) + 1,00 ml 2 mM Salzsäure + 1,00 ml Reinstwasser
- 10 mM Phosphatpuffer (PB, einfach) zum Verdünnen
8,00 ml 10 mM PB (1,25-fach) + 1,00 ml 2 mM Salzsäure + 1,00 ml Reinstwasser
- 0,1000 mg/ml Insulin in PBS
800 µl PBS (1,25-fach) + 100 µl Insulin-Lösung (0,017 mM, 10-fach, in 2 mM Salz-
säure) + 100 µl Reinstwasser
- 0,0500 und 0,0125 mg/ml Insulin in PBS
500 µl bzw. 125µl der 0,1000 mg/ml Insulin-Lösung in PBS werden mit PBS
(einfach) auf 1000 µl ergänzt.
- 0,1000 mg/ml, 0,0500 und 0,0125 mg/ml Insulin in 10 mM Phosphatpuffer
Die Herstellung erfolgt analog den Lösungen in PBS.
Durchführung
siehe Abschnitt 3.2.8, Lösung in Referenzküvette: jeweiliger Puffer (einfach) zum Verdünnen
Charakterisierung der Inkubationsansätze mittels RP-HPLC
Proben: Glyoxal-Insulin-Inkubationsansätze, siehe oben
HPLC-Anlage: Merck Hitachi, siehe Abschnitt 3.2
Säule: Säule SA3, Polymer Laboratories, PLRP-S, siehe Abschnitt 3.2
Eluenten-System: E6, Ameisensäure-Acetonitril-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 5 auf 50 % B in 30 min
Flussrate: 1,0 ml/min
Temperatur: 20 °C
Injektionsvolumen: 100 µl
- 94 -

3 Experimenteller Teil
Detektor: K2501 (Knauer), λ1 = 220 nm, siehe Abschnitt 3.2
K2501 (Knauer), λ2 = 320 nm, siehe Abschnitt 3.2
LaChrom L-7485 (Merck Hitachi), λex = 320 nm, λem = 400 nm, siehe
Abschnitt 3.2
Der Umsatz an Insulin und die dazugehörige Standardabweichung wird wie folgt berechnet:
%100100[%] ×=ohne
mit
AAUmsatz
22
100 ⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟⎟
⎠
⎞⎜⎜⎝
⎛××=
ohne
ohne
mit
mit
ohne
mit
As
As
AAs
Amit Peakfläche (λ = 220 nm) des Insulin-Peaks nach Inkubation mit Glyoxal
Aohne Peakfläche (λ = 220 nm) des Insulin-Peaks nach Inkubation ohne Glyoxal (Blindwert)
Charakterisierung der Inkubationsansätze mittels RP-HPLC und LC-ESI-MS
Proben: Glyoxal-Insulin-Inkubationsansätze, siehe oben
HPLC-Anlage: Agilent 1100 series, siehe Abschnitt 3.2.2
Säule: Säule SA3, Polymer Laboratories, PLRP-S, siehe Abschnitt 3.2
Eluenten-System: E6, Ameisensäure-Acetonitril-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 5 auf 50 % B in 30 min
Flussrate: 1,0 ml/min
Temperatur: 20 °C
Injektionsvolumen: 500 µl
Detektor: DAD (Agilent 1100 series), λ1 = 220 nm, λ1 = 320 nm, s. Absch. 3.2.2
ESI-TOF-MS (Applied Biosystems), Flussrate zum Massenspektrometer
ca. 0,5 ml/min, siehe Abschnitt 3.2.2
Untersuchungen mittels Aminosäureanalyse
Lösungen, siehe oben Inkubation von Insulin mit Glyoxal
- 95 -

3 Experimenteller Teil
Probenvorbereitung
Es wird ein Inkubationsansatz bestehend aus 16,0 ml PBS, 2,0 ml Insulin-Lösung (0,017 mM,
10-fach, in 2 mM Salzsäure) und 2,0 ml Glyoxal-Lösung (0,05 mM, 10-fach) angefertigt. Die
Inkubation erfolgt bei 37 °C für 15 d im Brutschrank. Anschließend wird mit 66 µl
Ameisensäure (100 %ig) angesäuert und die Probe wie in Abschnitt 3.8.1 beschrieben
angereichert. Zum Vergleich wird in der gleichen Weise ein Ansatz von Insulin ohne
Glyoxal-Zugabe (dafür Reinstwasser) angefertigt, inkubiert und angereichert. Die erhaltenen
Eluate werden im Spitzkolben am Rotationsverdampfer (30 °C, 12 mbar) zur Trockene
eingeengt.
Die trockenen Rückstände werden in je 2,0 ml 6 M Salzsäure gelöst, die Lösungen werden in
6 ml-Hydrolysegefäße überführt, die Gefäße werden mit Stickstoff gespült und fest
verschlossen. Die Hydrolyse erfolgt für 23 h bei 110 °C im Sandbad im Trockenschrank. Im
Anschluss werden je 1,5 ml Hydrolysat im Zentrifugalverdampfer zur Trockne eingeengt.
Weiterhin wird Insulin direkt zur Salzsäurehydrolyse eingesetzt. Dazu werden 2,0 mg Insulin
mit 2,0 ml 6 M Salzsäure versetzt und wie oben beschrieben hydrolysiert und getrocknet.
Anschließend werden die Proben in je 300 µl 0,2 M Citratpuffer (pH = 2,2, Puffer 1, siehe
Abschnitt 3.2.3) im Ultraschallbad gelöst. Nach Verdünnung mit 0,2 M Citratpuffer werden
20 µl (1 + 2 Verdünnung) oder 40 µl (1 + 9 Verdünnung) mittels Aminosäureanalyse
analysiert (siehe Abschnitt 3.2.3).
3.8 Untersuchungen zur Reaktion von Insulin mit Glyoxal nach Reduktion
und Blockierung der Sulfhydrylgruppen
3.8.1 Reduktion und Blockierung der Sulfhydrylgruppen
Um die beiden Ketten des Insulins zu trennen, werden die Disulfidbrücken mit Dithiothreitol
(DTT) reduziert und die freigesetzten Sulfhydrylgruppen durch Umsetzung mit Iodacetamid
blockiert. Die Durchführung erfolgt entsprechend einem modifizierten Protokoll von Gray
(1990).
Lösungen
- Dithiothreitol (DTT)-Lösung (30 mM, 25-fach)
18,5 mg 1,4-Dithiothreitol in 160 µl Reinstwasser frisch vor Gebrauch lösen.
- 96 -

3 Experimenteller Teil
- Tris-(carboxyethyl)phosphin (TCEP)-Lösung (30 mM, 25-fach)
34,4 mg Tris-(carboxyethyl)phosphin-Hydrochlorid in 160 µl Reinstwasser frisch vor
Gebrauch lösen.
- Natrium-EDTA-Lösung (2 mM, 10-fach)
74,5 mg Di-Natrium-EDTA-dihydrat in 10 ml Reinstwasser lösen.
- Tris-Acetat-Puffer (0,5 M, pH = 8,0)
1,515 g Tris-(hydroxymethyl)aminomethan in ca. 20 ml Reinstwasser lösen, 2,5 ml
2 mM Natrium-EDTA-Lösung (10-fach) hinzugeben, mit Essigsäure pH = 8,0
einstellen und mit Reinstwasser auf 25 ml ergänzen. Unmittelbar vor Gebrauch
5 - 10 min Stickstoff durch die Lösung blubbern lassen.
- Citronensäure-Lösung (0,5 M)
1,05 g Citronensäure-Monohydrat in 10 ml Reinstwasser lösen.
Reduktion
240 µl einer nicht abgestoppten Insulin-Glyoxal-Inkubation werden mit 10 µl DTT- bzw.
TCEP-Lösung (jeweils 30 mM, 25-fach) gemischt. Zur vollständigen Reduktion wird 50 min
(DTT) bzw. 20 h (TCEP) bei Raumtemperatur im Dunklen aufbewahrt.
Blockierung mit Iodacetamid
100 mg Iodacetamid und 200 µl Tris-Acetat-Puffer werden in ein Eppendorf-Gefäß (1,5 ml)
geben. Das Iodacetamid wird unter gelegentlichem Schütteln im Trockenschrank bei 60 °C
innerhalb von 10 min gelöst. Nach kurzem Abkühlen (ca. 30 °C) werden 250 µl der zuvor
reduzierten Probe mit einer Spritze (feine Nadel) direkt in die Iodacetamid-Lösung
eingespritzt. Es wird intensiv gemischt und nach 35 s wird die Reaktion mit 400 µl 0,5 M
Citronensäure abgestoppt.
Anreicherung an einer C18-Phase
Für einige Folgeuntersuchungen ist eine Anreicherung als auch die Abtrennung der
überschüssigen Reagenzien erforderlich. Die Festphasenextraktion erfolgt entsprechend den
Angaben in Tab. 3.8.1-1. Falls notwendig, wird der pH-Wert der aufzugebenden Lösung vor
der Probenaufgabe mit 10 %iger Ameisensäure auf pH < 3 gebracht.
Zur guten Handhabbarkeit wird auf die Kartusche (Sep-Pak® C18-Kartuschen, 360 mg,
Waters, WAT051910) eine 2 ml-Einwegspritze ohne Stempel gesteckt. Durch das Einbringen
- 97 -

3 Experimenteller Teil
einer gekürzte 200 µl-Eppendorfspitze (gelb) in den Auslauf der Kartusche wird die Flussrate
reproduzierbar gestaltet, sie beträgt ca. 0,6 ml/min. Nach Aufgabe der Elutionslösung werden
die ersten 0,5 ml Eluat verworfen. Das darauffolgende Eluat (ca. 1,5 ml) wird aufgefangen.
Tab. 3.8.1-1: Protokoll zur Anreicherung.
Schritt Lösung Volumen [ml]
Konditionierung Acetonitril
0,1 % Ameisensäure (v/v)
2,0
2,0
Probeaufgabe Probelösung, pH < 3 1,0 bis 20,0
Waschen 0,1 % Ameisensäure (v/v) 4,0
Elution 0,1 % Ameisensäure in 50 % Acetonitril (v/v) 2,0
Analyse mittels RP-HPLC
Proben: - Proben, reduziert und blockiert, ohne Anreicherung (siehe oben) für
Reaktivitätsvergleich A- vs. B-Kette
- Proben, reduziert und blockiert, mit Anreicherung (siehe oben)
Trennsystem und Parameter siehe Abschnitt 3.7, geändert:
Gradient: linear ansteigend, von 15 auf 40 % B in 30 min
Injektionsvolumen: 100 µl oder 500 µl
Der Gesamtumsatz je Kette wird wie folgt berechnet:
%100100[%] ×=ohne
mit
AAKettejetzGesamtumsa
Amit Peakfläche (λ = 220 nm) des Peaks der jeweiligen Kette nach Inkubation mit Glyoxal
Aohne Peakfläche (λ = 220 nm) des Peaks der jeweiligen Kette nach Inkubation ohne
Glyoxal (Blindwert)
Der Umsatz zur pyrazinon-modifizierten Kette wird wie folgt berechnet:
%100[%] ×++
=−NPorgPyr
Pyr
AAAA
KettePyrazinonzurUmsatz
- 98 -

3 Experimenteller Teil
APyr Peakfläche (λ = 220 nm) des Peaks der pyrazinon-modifizierten Kette
Aorg Peakfläche (λ = 220 nm) des Peaks der orginären (nicht umgesetzten) Kette
ANP Peakfläche (λ = 220 nm) eines Nebenproduktpeaks (nur bei B-Kette)
(APyr, ARes, ANP innerhalb eines Chromatogramms bestimmt)
Analyse mittels LC-ESI-MS
Lösungen
siehe oben und Abschnitt 3.7
Inkubation
Es wird ein Inkubationsansatz bestehend aus 16,0 ml PBS, 2,0 ml Insulin-Lösung (0,017 mM,
10-fach, in 2 mM Salzsäure) und 2,0 ml Glyoxal-Lösung (0,05 mM, 10-fach) angefertigt. Die
Inkubation erfolgt bei 37 °C für 15 d im Brutschrank.
Probenvorbereitung - Verfahren mit Anreicherung
20 ml des Inkubationsansatzes werden mit 833 µl DTT-Lösung (30 mM, 25-fach) gemischt.
Die Reduktion erfolgt bei Raumtemperatur im Dunklen für 30 min. Zur Alkylierung werden
3,0 g Iodacetamid und 6,0 ml Tris-Acetat-Puffer in einen Spitzkolben (50 ml) gegeben. Das
Iodacetamid wird unter gelegentlichem Umschwenken im Trockenschrank bei 60 °C
innerhalb von 10 min gelöst. Nach kurzem Abkühlen (ca. 30 °C) wird die zuvor reduzierte
Probe in die Iodacetamid-Lösung gegeben. Es wird intensiv gemischt und nach 35 s wird die
Reaktion mit 12,0 ml 0,5 M Citronensäure abgestoppt. Anschließend erfolgt sofort die oben
beschriebene Anreicherung. Das Eluat (1,5 ml) wird für die Analyse mittels LC-ESI-MS
eingesetzt.
Probenvorbereitung - Verfahren ohne Anreicherung
Die Proben werden wie oben beschrieben reduziert und blockiert. Die Derivatisierung wird
jedoch mit 576 µl Inkubationsansatz, 24 µl DTT-Lösung und 200 mg Iodacetamid in 400 µl
Tris-Acetat-Puffer durchgeführt. Zum Abstoppen werden 40 µl Ameisensäure (100 %ig)
verwendet. Die Proben werden anschließend sofort intensiv geschüttelt und mittels
LC-ESI-MS analysiert. Pro LC-ESI-MS-Lauf sind vier Derivatisierungen vorzunehmen.
- 99 -

3 Experimenteller Teil
LC-ESI-MS
Proben: Reduziert und blockiert, Eluat nach Anreicherung, siehe oben
Trennsystem und Parameter: siehe Abschnitt 3.7, geändert:
Gradient: linear ansteigend, von 15 auf 40 % B in 30 min
Injektionsvolumen: 500 µl
Proben: Proben, reduziert und blockiert, ohne Anreicherung, siehe oben
Trennsystem und Parameter: siehe Abschnitt 3.7, geändert:
Gradient: isokratisch 5 % B, 25 min, Flussrate: 1,0 ml/min, anschließend
linear ansteigend, von 5 auf 42 % B in 30 min, Flussrate: 0,5 ml/min
Injektionsvolumen: 4000 µl
3.8.2 Untersuchungen nach enzymatischer Hydrolyse
Lösungen
- Natriumchlorid-Lösung (0,2 M)
116,9 mg Natriumchlorid in 10 ml Reinstwasser lösen.
- di-Ammoniumhydrogenphosphat-Puffer (1 M)
1,32 g di-Ammoniumhydrogenphosphat in 10 ml Reinstwasser lösen.
- Endoproteinase Glu-C-Lösung (ca. 1 U/µl)
1,0 mg Endoproteinase Glu-C aus Staphylococcus aureus V8 (E.C. 3.4.21.19) in
500 µl Natriumchlorid-Lösung (0,2 M) rekonstituieren. Aufbewahrung kurzfristig bei
5 - 8 °C, längerfristig bei – 18 °C.
- weitere Lösungen, siehe Abschnitt 3.7 und 3.8.1
Probenvorbereitung
Es wird ein 20 ml-Inkubationsansatz von Insulin mit Glyoxal wie in Abschnitt 3.8.1
beschrieben angefertigt, inkubiert, reduziert, blockiert und angereichert. Bei der Anreicherung
werden 1,5 ml Eluat erhaltenen. Zur enzymatischen Hydrolyse werden davon 750 µl mit
400 µl di-Ammoniumhydrogenphosphat-Puffer (1 M) gemischt. Die Hydrolyse erfolgt nach
Zugabe von 50 µl Endoproteinase Glu-C-Lösung (ca. 1 U/µl) mit leichter Bewegung im
Minishaker bei Raumtemperatur für 20 h. Zum Abstoppen werden 120 µl Ameisensäure
- 100 -

3 Experimenteller Teil
(100 %ig) zugegeben. Nach Membranfiltration (regenerierte Cellulose, 0,45 µm) wird mittels
LC-ESI-MS analysiert.
LC-ESI-MS
Proben: siehe oben
HPLC-Anlage: Agilent 1100 series, siehe Abschnitt 3.2.2
Säule: Säule SA4, Polymer Laboratories, PLRP-S, siehe Abschnitt 3.2
Eluenten-System: E6, Ameisensäure-Acetonitril-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 10 auf 25 % B in 30 min
Flussrate: 0,2 ml/min
Temperatur: 20 °C
Injektionsvolumen: 100 µl
Detektor: DAD (Agilent 1100 series), λ1 = 220 nm, λ1 = 320 nm, s. Absch. 3.2.2
ESI-TOF-MS (Applied Biosystems), siehe Abschnitt 3.2.2
3.9 Untersuchungen zum Nachweis von N1-Alkyl-2(1H)-pyrazinonen nach
Salzsäurehydrolyse des glyoxal-modifizierten Insulins
Darstellung von N1-(2-Methyl-butyl)-2(1H)-pyrazinon und 3-Benzyl-N1-isobutyl-
2(1H)-pyrazinon
Die nach Salzsäurehydrolyse von glyoxal-modifizierten Insulin erwarteten N1-Alkyl-
2(1H)-pyrazinone werden im analytischen Maßstab als Vergleichssubstanzen dargestellt.
Dazu werden die Dipeptide Gly-Ile und Phe-Val mit Glyoxal zu den entsprechenden
2(1H)-Pyrazinon-Dipeptiden umgesetzt. Deren Behandlung mit Salzsäure liefert schließlich
die N1-Alkyl-2(1H)-pyrazinone.
Lösungen
- Phosphatpuffer (100 mM, 1,66-fach)
2,97 g di-Natriumhydrogenphosphat-Dihydrat in ca. 90 ml Reinstwasser lösen, mit
konzentrierter Salzsäure pH = 7,4 einstellen und mit Reinstwasser im Maßkolben auf
100 ml auffüllen.
- 101 -

3 Experimenteller Teil
- Phe-Val-Lösung (10 mM, 1,66-fach)
8,8 mg Phe-Val in 2 ml Phosphatpuffer (100 mM, 1,66-fach) lösen.
- Gly-Ile-Lösung (10 mM, 1,66-fach)
6,5 mg Phe-Val in 2 ml Phosphatpuffer (100 mM, 1,66-fach) lösen.
- Glyoxal-Lösung (10 mM, 2,5-fach)
143 µl Glyoxal-Lösung (40%) mit Reinstwasser auf 50 ml ergänzen.
Durchführung
900 µl Phe-Val- und 900 µl Gly-Ile-Lösung werden jeweils mit insgesamt 600 µl Glyoxal-
Lösung (10 mM, 2,5-fach) versetzt und bei 37 °C für 3 d im Brutschrank inkubiert. Die
Zugabe der Glyoxal-Lösung erfolgt gestaffelt in Portion zu 300 µl an den ersten beiden
Tagen. Anschließend wird der Umsatz an Peptid mittels RP-HPLC kontrolliert. In der
gleichen Weise wird eine Blindprobe aus 900 µl Peptid-Lösung und insgesamt 600 µl
Reinstwasser inkubiert.
Jeweils 1,5 ml der inkubierten Lösungen werden im Hydrolyseröhrchen mit 1,5 ml 12 M
Salzsäure (p.a.) gemischt, die Gefäße werden mit Stickstoff gespült und fest verschlossen. Die
Hydrolyse erfolgt bei 110 °C für 48 Stunden im Sandbad im Trockenschrank. Anschließend
wird 1,0 ml Hydrolysat entnommen, mit 6 M Natronlauge wird pH = 2 bis 4 eingestellt, die
Lösung wird membranfiltriert (regenerierte Cellulose, 0,45 µm) und mittels RP-HPLC und
LC-ESI-MS untersucht.
RP-HPLC
HPLC-Anlage: Merck Hitachi, siehe Abschnitt 3.2
Säule: Säule SA1, Knauer (Berlin), Eurospher 100-C18, siehe Abschnitt 3.2
Eluenten-System: E5, Ameisensäure-Methanol-System, siehe Abschnitt 3.2
Gradient:
Glyoxal-Gly-Ile-Ansatz: linear ansteigend, von 8 auf 34 % B in 25 min, anschließend:
linear ansteigend auf 68 % B in 5 min, isokrat. 68 % B 5 min
Glyoxal-Phe-Val-Ansatz: linear ansteigend, von 21 auf 64 % B in 25 min, anschließend:
linear ansteigend auf 68 % B in 5 min, isokrat. 68 % B 5 min
Proben nach Hydrolyse: linear ansteigend, von 43 auf 68 % B in 30 min, anschließend:
isokratisch 68 % B 5 min
Flussrate: 0,7 ml/min
- 102 -

3 Experimenteller Teil
Temperatur: 20 °C
Injektionsvolumen: 50 µl
Detektor: K2501 (Knauer), λ1 = 220 nm, siehe Abschnitt 3.2
K2501 (Knauer), λ2 = 320 nm, siehe Abschnitt 3.2
LaChrom L-7485 (Merck Hitachi), λex = 320 nm, λem = 400 nm, siehe
Abschnitt 3.2
LC-ESI-MS
HPLC-Anlage: Agilent 1100 series, siehe Abschnitt 3.2.2
Säule: Säule SA2, Knauer (Berlin), Eurospher 100-C18, siehe Abschnitt 3.2
Eluenten-System: E5, Ameisensäure-Methanol-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 43 auf 68 % B in 30 min, anschließend isokrat.
68 % B 5 min
Flussrate: 0,3 ml/min
Temperatur: 20 °C
Injektionsvolumen: 100 µl
Detektor: DAD (Agilent 1100 series), λ1 = 220 nm, λ1 = 320 nm, s. Absch. 3.2.2
ESI-TOF-MS (Applied Biosystems), siehe Abschnitt 3.2.2
Untersuchungen am glyoxal-modifizierten Insulin
Lösungen
siehe Abschnitt 3.7 und 3.8.1
Inkubation, Anreicherung, Salzsäurehydrolyse, Probenvorbereitung
Es wird ein Inkubationsansatz bestehend aus 80,0 ml PBS, 10,0 ml Insulin-Lösung
(0,017 mM, 10-fach, in 2 mM Salzsäure) und 10,0 ml Glyoxal-Lösung (0,05 mM, 10-fach)
angefertigt. Die Inkubation erfolgt bei 37 °C für 15 d im Brutschrank. Anschließend werden
vier 20 ml-Portionen entnommen. Jede Portion (= 2 mg Insulin) wird mit 66 µl Ameisensäure
(100 %ig) angesäuert (pH < 3,0) und an je einer Kartusche wie in Abschnitt 3.8.1 beschrieben
angereichert.
- 103 -

3 Experimenteller Teil
Die Eluate von jeweils zwei Anreicherungen (= 4 mg Insulin) werden im Spitzkolben
vereinigt. Die beiden resultierenden Ansätze werden am Rotationsverdampfer (30 °C,
12 mbar) auf ca. 500 µl eingeengt. Das Volumen der Rückstände wird mit einer Eppendorf-
Pipette bestimmt und jeweils auf 1000 µl mit Reinstwasser ergänzt. Zu jedem Ansatz werden
1000 µl 12 M Salzsäure gegeben (Salzsäure-Endkonzentration im Ansatz: 6 M). Einem
Ansatz werden 30 µl (33,6 mg) Mercaptoethanol hinzugefügt. Die Gefäße werden mit
Stickstoff gespült und fest verschlossen. Die Hydrolyse erfolgt bei 110 °C für 48 h (ohne
Mercaptoethanol) oder 23 h (mit Mercaptoethanol) im Sandbad im Trockenschrank.
Anschließend werden je 1,0 ml Hydrolysat entnommen, mit 6 M Natronlauge wird pH = 2 bis
4 eingestellt, die Lösungen werden membranfiltriert (regenerierte Cellulose, 0,45 µm) und
mittels RP-HPLC und LC-ESI-MS untersucht.
RP-HPLC und LC-ESI-MS
Anlagen und Trennparameter: siehe oben (Proben nach Salzsäurebehandlung der
2(1H)-Dipeptid-Pyrazinone), geändert:
Injektionsvolumen: 100 µl
3.10 Vergleich der Reaktivität von Lysin, Arginin und dem N-Terminus
unter den Bedingungen der Inkubation von Insulin anhand von Peptiden
Nα-Hippuryllysin (BzGK), Nα-Hippurylarginin (BzGR) und das Tripeptid Gly-Ala-Tyr
werden jeweils mit Glyoxal unter den Bedingungen der Inkubation von Insulin (Peptid:
17 µM, Glyoxal: 50 µM) als auch bei höherer Konzentration (Peptid und Glyoxal: je 5 mM)
inkubiert.
Lösungen
- Phosphatgepufferte Kochsalzlösung (PBS) (pH = 7,4, 1,25-fach), siehe Abschnitt
3.5.1
- Glyoxal-Lösung (0,05 mM, 10-fach), siehe Abschnitt 3.7
- Glyoxal-Lösung (5 mM, 5-fach), siehe Abschnitt 3.5.1
- BzGK-Lösung (0,017 mM, 10-fach)
5,2 mg BzGK in 100 ml Reinstwasser im Maßkolben lösen.
- 104 -

3 Experimenteller Teil
- BzGK-Lösung (5 mM, 1,25-facht)
19,2 mg BzGK in 10 ml PBS (1,25-fach) im Maßkolben lösen.
- BzGR-Lösung (0,017 mM, 10-fach)
5,7 mg BzGR in 100 ml Reinstwasser im Maßkolben lösen.
- BzGR-Lösung (5 mM, 1,25-fach)
21,0 mg BzGR in 10 ml PBS (1,25-fach) im Maßkolben lösen.
- Gly-Ala-Tyr-Lösung (0,017 mM, 10-fach)
5,9 mg Gly-Ala-Tyr × 2 H2O in 100 ml Reinstwasser im Maßkolben lösen.
- Gly-Ala-Tyr-Lösung (5 mM, 1,25-fach)
20,6 mg Gly-Ala-Tyr x 2 H2O in 10 ml PBS (1,25-fach) im Maßkolben lösen.
Durchführung
Die Inkubationsansätze werden wie in Tab. 3.10-1 und Tab. 3.10-2 angegeben angefertigt
(jeweils dreifach) und bei 37 °C für 15 d im Brutschrank inkubiert. Zum Abstoppen werden je
Ansatz 33 µl 10 %ige (2,65 M) Ameisensäure zugesetzt. Die 5 mM-Ansätze von Gly-Ala-Tyr
werden 1 + 1 mit 10 mM Phosphatpuffer (HPLC-Eluent A) und die 5 mM-Ansätze der
Nα-Hippuryl-Derivate 1 + 3 mit 0,1 % Ameisensäure (HPLC-Eluent A) verdünnt. Um die
mögliche Wirkung der Ameisensäure auf das Produktspektrum zu prüfen, wird diese beim
Abstoppen und Verdünnen durch Reinstwasser ersetzt. Der pH-Wert wird vor und nach der
Inkubation in separaten Ansätzen geprüft. Die Analyse der Proben erfolgt mittels RP-HPLC.
Der Umsatz an Peptid und die dazugehörige Standardabweichung wird wie in Abschnitt 3.5.1
angegeben berechnet.
Tab. 3.10-1: Zusammensetzung der Inkubationsansätze (Peptid 17 µM, Glyoxal: 50 µM).
Probe Peptid-Lösung
(17 µM, 10-fach)
PBS Glyoxal-Lösung
(50 µM, 10-fach)
Reinstwasser
Ansatz 100 µl 800 µl 100 µl -
Peptid-Blindwert 100 µl 800 µl - 100 µl
- 105 -

3 Experimenteller Teil
Tab. 3.10-2: Zusammensetzung der Inkubationsansätze (Peptid 5 mM, Glyoxal: 5 mM).
Probe Peptid in PBS
(5 mM, 1,25-fach)
Glyoxal-Lösung
(5 mM, 5-fach)
Reinstwasser
Ansatz 800 µl 200 µl -
Peptid-Blindwert 800 µl - 200 µl
Analytische RP-HPLC
HPLC-Anlage: Merck Hitachi, siehe Abschnitt 3.2
Säule: SA1, Knauer Eurospher 100-C18, siehe Abschnitt 3.2
Ansätze der Nα-Hippuryl-Derivate:
Eluenten-System: E1, Phosphatpuffer-Methanol-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 6 auf 13 % B in 25 min (BzGK)
linear ansteigend, von 12 auf 19 % B in 25 min (BzGR)
Gly-Ala-Tyr-Ansätze:
Eluenten-System: E5, Ameisensäure-Methanol-System, siehe Abschnitt 3.2
Gradient: linear ansteigend, von 13 auf 72 % B in 35 min
Flussrate: 0,7 ml/min
Temperatur: 20 °C
Injektionsvolumen: 20 µl (5 mM-Ansätze)
100 µl (17 µM-Ansätze)
Detektor: K2501 (Knauer), λ1 = 220 nm, siehe Abschnitt 3.2
K2501 (Knauer), λ2 = 320 nm, siehe Abschnitt 3.2
LaChrom L-7485 (Merck Hitachi), λex = 320 nm, λem = 400 nm, siehe
Abschnitt 3.2
- 106 -

4 Auswertung und Diskussion der Ergebnisse
4.1 Analytik von Amadori-Produkten
Die Bestimmung von Furosin ist eine geeignete und oft angewendete Methode zur
Beurteilung der Wärmebehandlung und des nutritiven Wertes von Lebensmitteln,
insbesondere bei Milchprodukten. Ferner erlaubt die Anwendung der Furosin-Analytik
Rückschlüsse auf in vivo stattfindende Glykierungsprozesse (siehe Abschnitt 2.2.2). Die
Unsicherheit über die Höhe der molaren Ausbeute an Furosin bei der Salzsäurehydrolyse der
Amadori-Produkte des Lysins wurde jedoch immer wieder als Kritikpunkt angeführt (Resmini
et al., 1990, Van Boekel, 1998). Um diese Unsicherheit auszuräumen wurden die in
Lebensmitteln und in vivo relevanten Amadori-Produkte Nε-Fructosyl-, Nε-Tagatosyl-,
Nε-Lactulosyl-, und Nε-Maltulosyl-lysin als peptidgebundene Derivate und Nε-Fructosyl-lysin
in freier Form dargestellt. Die gezielte Hydrolyse und die Quantifizierung der
Hydrolyseprodukte erlaubten anschließend die Bestimmung der molaren Ausbeuten sowie der
sogenannten Überführungsfaktoren auf Basis von Furosin. Letztere ermöglichen jetzt die
sichere Korrelation der Ergebnisse der Furosin-Analytik mit der Lysin-Derivatisierung und
damit exakte Aussagen zum Fortschritt nichtenzymatischer Glykierungsreaktionen in
Lebensmitteln und in vivo.
4.1.1 Darstellung von Amadori-Produkten und Pyridosin als Referenzmaterial
Die ausgewählten Amadori-Produkte des Lysins wurden als Nα-Hippuryl-Derivate (siehe
Abb. 4.1.1-1) dargestellt. Nε-Fructosyllysin wurde weiterhin als freies Derivat gewonnen. Der
Hippursäure-Rest erwies sich auf Grund der UV-Absorption bei 230 nm und der Retention
auf RP-Material (Smith & Thornalley, 1992) sowohl für die Analytik als auch für die
semipräparative Isolierung mittels HPLC als vorteilhaft.
Die Synthese der Hippuryl-Amadori-Produkte der Monosaccharide erfolgte durch Umsetzung
von Nα-Hippuryllysin mit dem entsprechenden Saccharid in siedendem Methanol unter
Rückfluss (Finot & Mauron, 1969). Dabei wurden laut HPLC-Analyse Umsätze zum
Amadori-Produkt von 90 bis 95 % erreicht. Bei den Amadori-Produkten der Disaccharide
- 107 -

4 Auswertung und Diskussion der Ergebnisse
waren die Bildungsraten mit ca. 50 % unter diesen Bedingungen deutlich geringer. Deshalb
erfolgte deren Darstellung in einer Mischung aus Methanol und Dimethylformamid (Vinale et
al., 1999). Der auf diese Weise erzielte höhere Umsatz von ca. 95 % beruht vermutlich auf
der besseren Löslichkeit der Disaccharide in dem Lösungsmittelgemisch und der höheren
Reaktionstemperatur beim Kochen unter Rückfluss.
HOO
HO
HO
OH
O
OH
OH
HOHO
Nα-Hippuryl-Nε-fructosyllysin
OHO
HO OH
O
OHO
OH
OH
HO
HO
O
HO OOH
HO O
OH
OHHO
R =
R =R =
R = R =
Nα-Hippuryllysin
Nα-Hippuryl-Nε-tagatosyllysin
Nα-Hippuryl-Nε-maltulosyllysin
Nα-Hippuryl-Nε-lactulosyllysin
HN
NH
OH
O
OO
NHR
H
Abb. 4.1.1-1: Struktur von Nα-Hippuryllysin und der dargestellten Hippuryl-Amadori-Produkte.
Mittels analytischer RP-HPLC und unter Verwendung eines phosphat-gepufferten und
methanolhaltigen Eluenten-Systems konnte eine sehr gute Trennung von Nα-Hippuryllysin
und dem jeweiligen Syntheseprodukt erreicht werden (siehe Abb. 4.1.1-2). Die Trennung war
jedoch stark von der Flussrate abhängig, so dass mit der semipräparativen Säule und
vermindertem linearen Fluss keine Trennung gegeben war. Die Trennung im
semipräparativen Maßstab konnte erst durch eine vorherige Behandlung des Syntheseansatzes
mit Carboxypeptidase B (EC 3.4.17.2) erzielt werden. Carboxypeptidase B ist eine
Exopeptidase, welche C-terminal gebundenes Arginin oder Lysin von Peptiden abspaltet
- 108 -

4 Auswertung und Diskussion der Ergebnisse
(Ambler, 1972). Im Syntheseansatz führte die Behandlung mit Carboxypeptidase B folglich
zur Hydrolyse von nicht umgesetzten Nα-Hippuryllysin zu Hippursäure und Lysin. Das als
Amadori-Produkt modifizierte Nα-Hippuryllysin wurde demgegenüber nicht angegriffen
(siehe Abb. 4.1.1-2). Zur Gewinnung der Syntheseprodukte wurden die mit
Carboxypeptidase B behandelten Ansätze zunächst unter Verwendung des trennstarken
phosphat-gepufferten und methanolhaltigen Eluenten-Systems isoliert und anschließend in
einem zweiten chromatographischen Schritt mit wässriger Essigsäure und Methanol als
Eluenten entsalzt.
0 10 20 30 4
0,00
0,25
0,50
0,75
1,00
1,25
0
Abso
rptio
n (λ
= 2
30 n
m) [
AU]
BzG
BzGFruK
BzGK
B
A
Zeit [min]
Abb. 4.1.1-2: HPLC-Analyse eines Syntheseansatzes von Nα-Hippuryl-Nε-fructosyllysin (BzGFruK) vor (A) und nach (B) Behandlung mit Carboxypeptidase B. Hippursäure (BzG), Nα-Hippuryllysin (BzGK). (Durchführung: siehe Abschnitt 3.3.1).
Die Synthese von freiem Nε-Fructosyllysin wurde in Anlehnung an Reutter & Eichner (1989)
durchgeführt. Es wurde jedoch Nα-Boc-lysin statt Lysin mit Glucose umgesetzt. Dadurch
konnte die Bildung von Nα-Fructosyllysin und Nα,Nε-Difructosyllysin vermieden und die
chromatographische Reinigung vereinfacht werden. Die Abspaltung der Boc-Schutzgruppe
erfolgte auf schonende Weise an einem stark sauren Kationenaustauscher.
- 109 -

4 Auswertung und Diskussion der Ergebnisse
Weiterhin wurden Nα-Hippuryl-Nε-carboxymethylhippuryllysin als Standard zur
Quantifizierung von CML sowie Pyridosin als Standard gewonnen. Alle dargestellten
Verbindungen wurden in chromatographisch reiner Form erhalten. Die massen- und NMR-
spektroskopischen Daten sowie die Ergebnisse der Elementaranalysen der Syntheseprodukte
(siehe Abschnitt 3.3) decken sich mit den erwarteten bzw. den zum Teil aus der Literatur
bekannten Werten (Kroh et al., 1992, Olano et al., 1992, Mossine et al., 1994, Feather &
Mossine, 1998). Die Präparate wurden damit für die folgenden Hydrolysestudien als geeignet
angesehen.
4.1.2 Bestimmung der molaren Ausbeuten der Hydrolyseprodukte
Die Hydrolyse der dargestellten Amadori-Produkte erfolgte unter den für die
Aminosäureanalytik standardisierten Bedingungen in 6 M Salzsäure für 23 h bei 110 °C und
bei einem Präparat-Säure-Verhältnis von 1:1000. Ferner wurde in der für die Furosin-Analyse
oft benutzten 8 M Salzsäure hydrolysiert. Für die Quantifizierung der Hydrolyseprodukte
wurde die Kationenaustauschchromatographie mit Nachsäulenderivatisierung durch
Ninhydrin (Aminosäureanalysator) verwendet, welche die gleichzeitige Bestimmung aller
Hydrolyseprodukte ermöglicht (siehe Abb. 4.1.2-1).
Da Amadori-Produkt und Glycin in den Hippuryl-Derivaten im molaren Verhältnis 1:1
vorliegen, wurde dieser Vorteil genutzt und Glycin als interner Standard zur Berechnung der
molaren Ausbeuten verwendet. Dies sollte sich positiv sowohl auf die Richtigkeit als auch auf
die Präzession der Ergebnisse auswirken.
Abbildung 4.1.2-1 zeigt typische Aminosäurechromatogramme von Hydrolysaten von
Nα-Hippuryl-Nε-fructosyllysin und Nα-Hippuryl-Nε-tagatosyllysin. Schon visuell wird
deutlich, dass sich die Gehalte an Pyridosin, Lysin und Furosin in den beiden Hydrolysaten
deutlich unterscheiden und damit die Hydrolyseprodukte in unterschiedlichem Umfang
gebildet werden. So ist im Vergleich zu Nε-Fructosyllysin bei Nε-Tagatosyllysin der Umfang
der Bildung von Furosin und Pyridosin höher und die Rückbildung von Lysin erniedrigt (6 M
Salzsäure, siehe Abb. 4.1.2-1). Weiterhin ist auffällig, dass in beiden Hydrolysaten geringe
Mengen an CML detektierbar sind (siehe auch Abschnitt 4.1.4).
- 110 -

4 Auswertung und Diskussion der Ergebnisse
0 20 40 60
0,00
0,25
0,50
0,75
1,00Ammoniak
Lys
FurosinPyridosin
CML
Gly
BzGTagK
BzGFruK
Abso
rptio
n (λ
= 5
70 n
m) [
AU
]
Zeit [min]
Abb. 4.1.2-1: Aminosäureanalyse von Hydrolysaten (6 M Salzsäure) von Nα-Hippuryl-Nε-fructosyllysin (BzGFruK) und Nα-Hippuryl-Nε-tagatosyllysin (BzGTagK). (Durchführung: siehe Abschnitt 3.4.1).
Für Fructosyl-, Lactulosyl- und Maltulosyllysin ist ein ähnliches Hydrolyseverhalten
festzustellen (Abb. 4.1.2-2, Tab. 4.1.2-1). Nach Hydrolyse mit 6 M Salzsäure wurden molare
Ausbeuten an Furosin zwischen 30 und 35 % bestimmt. Die anscheinend leicht verminderte
Bildung von Furosin aus freiem Fructoselysin dürfte auf die unterschiedliche Art der
Berechnung zurückzuführen sein. Die Berechnung der Ausgangskonzentration erfolgte bei
diesem Derivat anhand der Elementaranalyse. Die bestimmten Ausbeuten an Furosin bei
Hydrolyse in 6 M Salzsäure stimmen sehr gut mit der von Finot et al. (1977) für
lactulosyliertes Milchprotein ermittelten Ausbeute von 32 % überein. Bei Hydrolyse mit 8 M
Salzsäure liefern Fructosyl-, Lactulosyl- und Maltulosyllysin mit 46, 50 und 51 %
erwartungsgemäß höhere Ausbeuten an Furosin. Die ermittelte Ausbeute bei Fructosyllysin
deckt sich nahezu mit der von Szölgyenyi et al. (1989) ermittelten Ausbeute von 46 % bei
Hydrolyse mit 7,8 M Salzsäure.
- 111 -

4 Auswertung und Diskussion der Ergebnisse
Verglichen mit Fructosyl-, Lactulosyl- und Maltulosyllysin, welche an der Amino-Gruppe
einen Fructosyl-Rest aufweisen, zeigt Tagatosyllysin ein anderes Hydrolyseverhalten. Aus
Tagatosyllysin wird bei Hydrolyse mit 6 M Salzsäure mit 42 % signifikant (P < 0,01) mehr
Furosin gebildet. Die von Finot et al. (1981) vermutete erhöhte Bildung von Furosin aus
Tagatosyllysin wurde damit abgesichert und quantifiziert. Im Gegensatz zu den Fructosyl-
Amadori-Produkten bewirkt beim Tagatosyllysin die Erhöhung der Salzsäurekonzentration
auf 8 M keine Erhöhung der molaren Ausbeute an Furosin, so dass bei dieser Konzentration
Tagatosyllysin eine signifikant (P < 0,01) geringere molare Ausbeute liefert.
FruK BzGFruK BzGLctK BzGMltK BzGTagK0
10
20
30
40
50po
nn
nn
mm m
m
Amadori-Produkt
Mol
are
Aus
beut
e an
Fur
osin
[%]
Abb. 4.1.2-2: Molare Ausbeute an Furosin bei Hydrolyse verschiedener Amadori-Produkte in 6 M (graue Balken) oder 8 M Salzsäure (schwarze Balken). Mittelwert ± Standardabweichung (n = 5, f = 4). P < 0,01: m vs n, m vs o, n vs p. (Durchführung: siehe Abschnitt 3.4.1, 3.4.2).
Bei der Bildung von Pyridosin verhalten sich die Fructosyl-Amadori-Produkte wiederum
ähnlich, in 6 M Salzsäure werden jeweils ca. 16 % Pyridosin gebildet (Tab. 4.1.2-1). Die
Bildung aus Tagatosyllysin ist mit 36 % demgegenüber signifikant erhöht. Aus früheren
Arbeiten ist bekannt, dass bei langer Lagerung von UHT-Milch das Pyridosin-zu-Furosin-
Verhältnis steigt (Möller, et al. 1977). Eine Hydrolyse der Lactose, die Reaktion der
freigesetzten Galactose zum Tagatoselysin und die vermehrte Bildung von Pyridosin aus
Tagatosyllysin dürfte dieses Phänomen mit erklären.
- 112 -

4 Auswertung und Diskussion der Ergebnisse
Anhand der bestimmten molaren Ausbeuten an Furosin kann das als Amadori-Produkt
modifizierte Lysin berechnet werden. Die dazu notwendigen Konversionsfaktoren sind in
Tab. 4.1.2-2 zusammengefasst. Die Berechnung ist einfach, solange nur ein Typ von
Amadori-Produkt vorliegt. In Milchprodukten in denen die Lactose hydrolysiert wurde liegen
jedoch Nε-Fructosyl- und Nε-Tagatosyllysin nebeneinander vor. Um auch in diesen
Lebensmitteln das nicht verfügbare Lysin zu bestimmen, müssen beide Amadori-Produkte die
gleiche molare Ausbeute an Furosin liefern. Die dazu erforderliche Salzsäurekonzentration
wurde zunächst anhand der mit beiden Nα-Hippuryl-Amadori-Produkten mit 6 M und 8 M
Salzsäure bestimmten Ausbeuten zu 7,3 M berechnet (Gleichungssystem) und anschließend
experimentell bestätigt (Tab. 4.1.2-1). Zur Bestimmung des nicht verfügbaren Lysins in
lactose-hydrolysierten Milchprodukten kann folglich die Hydrolyse mit 7,3 M Salzsäure
empfohlen werden.
Tab. 4.1.2-1: Molare Ausbeuten an Hydrolyseprodukten bei Hydrolyse verschiedener
Amadori-Verbindungen in 6 M, 8 M oder 7,3 M Salzsäure für 23 h bei 110 °C.
Molare Ausbeute [%] * Salzsäure-
konzentration
Amadori-
Verbindung Lysin Furosin Pyridosin CML
6,0 M FruK 56,1 ± 1,4m 30,0 ± 1,2m 14,9 ± 0,3m 1,5 ± 0,1
BzGFruK 56,2 ± 0,4m 32,4 ± 0,5m 15,9 ± 0,2m 1,6 ± 0,1
BzGLctK 57,9 ± 1,5m 33,8 ± 1,4m 15,9 ± 0,9m 1,0 ± 0,2
BzGMltK 56,0 ± 0,6m 34,6 ± 0,5m 15,5 ± 0,2m 1,1 ± 0,1
BzGTagK 28,6 ± 0,2 42,1 ± 0,4 35,6 ± 0,3 2,3 ± 0,1
8,0 M FruK 42,5 ± 0,9m 43,4 ± 1,1 18,6 ± 1,1m 5,0 ± 0,3
BzGFruK 41,7 ± 0,8m 46,1 ± 1,9m 15,9 ± 0,8m 5,9 ± 0,1
BzGLctK 40,5 ± 0,6m 50,2 ± 1,3m 18,4 ± 0,7m 2,5 ± 0,2
BzGMltK 39,4 ± 0,4m 51,0 ± 0,7m 17,5 ± 0,2m 3,5 ± 0,2
BzGTagK 32,2 ± 0,5 41,9 ± 1,7 32,0 ± 1,1 4,1 ± 0,2
7,3 M BzGFruK 48,3 ± 1,2m 42,8 ± 1,8 14,3 ± 0,8m 3,1 ± 0,3
BzGTagK 29,7 ± 1,1 42,1 ± 1,1 35,3 ± 0,7 2,7 ± 0,1
* Mittelwert ± Standardabweichung (n = 5, f = 4), m P < 0,01 vs BzGTagK
- 113 -

4 Auswertung und Diskussion der Ergebnisse
Tab. 4.1.2-2: Überführungsfaktoren zur Berechnung des als Amadori-Produkt modifizierten
Lysins.
Überführungsfaktor bei Hydrolyse mit Salzsäure Amadori-Verbindung
6,0 M 8,0 M 7,3 M
Fructosyl- 3,1 2,2 2,4
Lactulosyl-, Maltulosyl- 2,9 2,0 n. b.
Tagatosyl- 2,4 2,4 2,4
n. b. nicht bestimmt
Prüfung von möglichen Einflussfaktoren auf die Hydrolyse
Um zu prüfen, ob sich bei der Hydrolyse die Gegenwart bestimmter
Lebensmittelinhaltsstoffe, Bestandteile von Inkubationsansätzen oder Verfahrensweisen auf
die molare Ausbeute an Furosin auswirken, wurden entsprechende Hydrolyseexperimente
durchgeführt (siehe Abschnitt 3.4.1).
Dazu wurde die Standard-Hydrolyse von Nα-Hippuryl-Nε-fructosyllysin und Nα-Hippuryl-
Nε-tagatosyllysin (6 M und 8 M Salzsäure, Protein-Salzsäure-Verhältnis 1:1000) wie folgt
modifiziert:
- Zusatz von Glucose (bis 10 mg/ml Hydrolyseansatz)
- Zusatz von Peptiden (7 mg/ml, Protein-Salzsäure-Verhältnis 1:125)
- Variation des Protein-Salzsäure-Verhältnisses von 1:500 bis 1:10000
- Zusatz möglicher Effektoren (bis 10 mg/ml), z. B. organische Säuren, Antioxidations-
mittel wie Acetylcystein, Oxidationsmittel wie Natriumazid
- Hydrolyse ohne vorheriges Spülen des Gefäßes mit Stickstoff
- Hydrolyse nachdem Stickstoff direkt durch Hydrolyseansatz geleitet wurde (bis 2 min)
Vom Natriumazid-Zusatz abgesehen zeigte keiner der getesteten Parameter einen
signifikanten (P < 0,05) Einfluss auf die molare Ausbeute an Furosin. Dies spricht sehr für die
Verlässlichkeit der Methode. So wirkte sich insbesondere die Gegenwart von reduzierenden
Zuckern nicht nachteilig auf die Richtigkeit und Präzession der Ergebnisse aus. Das gleiche
gilt für einen verminderten Salzsäureüberschuss von nur 1:125, welcher von Resmini et al.
(1990) zur Furosin-Analytik eingeführt wurde. Weiterhin zeigten die Art des Spülens der
Hydrolysegefäße mit Stickstoff oder ein Verzicht darauf keine Wirkung, so dass dieser
- 114 -

4 Auswertung und Diskussion der Ergebnisse
Arbeitsschritt bei der reinen Furosin-Analytik streng genommen nicht notwendig ist. In dieser
Weise ist auch die Feststellung von Finot (1973) zu interpretieren, dass die Hydrolyse im
Kolben unter Rückfluss oder in abgeschmolzenen Ampullen jeweils zum gleichen Ergebnis
führt. Von den geprüften weiteren Zusätzen erwies sich lediglich die Anwesenheit größerer
Mengen von Natriumazid (10 mg/ml Hydrolyseansatz) als ungünstig. Dadurch kam es zu
einer Verminderung der Ausbeute an Furosin (10 % in 6 M Salzsäure) und zur vermehrten
Bildung von CML (10 % in 6 M Salzsäure). Eine geringere Konzentration an Natriumazid
(1 mg/ml Hydrolyseansatz) zeigte keinen Effekt.
Wie bereits in früheren Studien (Finot et al., 1972, Henle et al., 1995) festgestellt wurde, ist
die molare Ausbeute an Furosin stark von der verwendeten Salzsäurekonzentration abhängig.
Die vorliegenden Untersuchungen zeigten jedoch auch, dass dies im untersuchten
Konzentrationsbereich nicht für Tagatosyllysin gilt. Auch in Hinblick auf die Bildung von
Pyridosin und Lysin wurde im Hydrolyseverhalten eine Abhängigkeit von der Art des
Amadori-Produktes beobachtet. Insgesamt betrachtet zeigten damit Amadori-Produkte,
welche einen Fructosyl-Rest enthalten, ein ähnliches Verhalten, das sich signifikant von dem
des Tagatosyllysins unterschied.
Um das blockierte Lysin auch in Proben zu bestimmen, die sowohl Fructosyl- als auch
Tagatosyllysin enthalten, wie z. B. bei Milchprodukten in denen die Lactose hydrolysiert
wurde, hat sich die Hydrolyse mit 7,3 M Salzsäure als geeignet erwiesen. Bei dieser
Konzentration erfolgt die Bildung von Furosin aus beiden Amadori-Produkten im gleichen
Umfang. Insbesondere die Hydrolyse in Gegenwart von überschüssig vorhandenem Zucker
oder Protein (Peptiden) zeigte keinen Einfluss auf die Höhe der molaren Ausbeute an Furosin,
so dass sich folglich diese Lebensmittelbestandteile nicht negativ auf die Ergebnisse
auswirken.
Zusammenfassend ist festzustellen, dass anhand der durchgeführten Hydrolyseexperimente
das Hintergrundwissen um die Furosin-Bestimmung und ihre Anwendbarkeit erweitert
werden konnte. Insbesondere wurden die molaren Ausbeuten an Furosin und die damit in
Zusammenhang stehenden Überführungsfaktoren bestimmt. Anhand der ermittelten
Überführungsfaktoren ist nun erstmals eine sichere Bestimmung der Lysin-Derivatisierung
auf der Basis der Analytik von Furosin möglich. Dies erlaubt exakte Aussagen zum
Fortschritt der frühen Maillard-Reaktion sowohl in Lebensmittel als auch in vivo.
- 115 -

4 Auswertung und Diskussion der Ergebnisse
4.1.3 Betrachtungen zum Mechanismus der Bildung der Hydrolyseprodukte
Die bei der Salzsäurehydrolyse der Amadori-Produkte festgestellten Phänomene werfen
zwangsläufig die Frage nach den Ursachen auf. Dabei erscheinen insbesondere die
Hintergründe für die Bildung von drei Haupthydrolyseprodukten, die beobachtete
Abhängigkeit der molaren Ausbeuten von der Salzsäurekonzentration und der Art des
Amadori-Produktes interessant. Obwohl z. B. die beiden erstgenannten Beobachtungen bereits
mehrere Jahrzehnte bekannt sind, wurden die Ursachen bisher kaum diskutiert. Grundlegend
für die Betrachtungen ist der Mechanismus der Bildung der Hydrolyseprodukte. Dieser wurde
jedoch ebenfalls noch nicht im Detail und unter Hydrolysebedingungen untersucht.
Anhand von bekannten Reaktionen von Zuckern und Amadori-Produkten unter Säurekatalyse
(Anet, 1962, 1964) und Untersuchungen zum Abbau von Amadori-Produkten bei der
Thermolyse im Neutralen (Huber & Ledl, 1990) wird angenommen, dass die Bildung von
Furosin durch eine 2,3-Enolisierung des Amadori-Produktes eingeleitet wird und intermediär
das 1-Amino-1,4-didesoxyoson auftritt (Abb. 4.1.3-1). Die Rückbildung von Lysin ist über
eine 1,2-Enolisierung und anschließende Eliminierung des Lysins erklärbar (Anet, 1960). Für
die Bildung von Pyridosin sind zwei Wege diskutierbar. Die erste Möglichkeit beinhaltet eine
direkte Dehydratisierung der Furanose-Form des Amadori-Produktes zu einem
2-Aminomethyl-furan-Derivat, welches anschließend einer Umlagerung zum Pyridosin
unterliegt (Finot et al., 1969a). Weiterhin ist die Bildung über das 1-Amino-1,4-didesoxyoson
formulierbar (Abb. 4.1.3-1).
Die in Abb. 4.1.3-1 angegebenen Reaktionswege geben zunächst den Fakt wieder, dass es
sich bei den Hydrolyseprodukten um Endprodukte handelt und damit keines ein Intermediat
bei der Bildung eines anderen ist. Eine Schlüsselstellung in Hinblick auf die Anteile bei der
Produktbildung dürfte das spezifische Vermögen der Amadori-Produkte zur 1,2- und
2,3-Enolisierung sowie die Neigung der enolischen Intermediate zur Dehydratisierung
einnehmen. Neben diesen von der Art des Amadori-Produktes abhängigen Faktoren wird
insbesondere bei der Dehydratisierung die Salzsäurekonzentration von Bedeutung sein.
Hierbei sind die Protonierungsgleichgewichte an den Hydroxylgruppen und am Stickstoff zu
betrachten. Für die Dehydratisierung ist die Protonierung der jeweiligen Hydroxylgruppe
Voraussetzung und folglich sollte dieser Prozess mit steigender Säurekonzentration
beschleunigt werden. Gleichzeitig dürfte entsprechend dem angegebenen Mechanismus eine
Protonierung am Stickstoff der Dehydratisierung des 1,2-Enols und damit der Lysinbildung
- 116 -

4 Auswertung und Diskussion der Ergebnisse
entgegenwirken. Weiterhin ist anzunehmen, dass die protonierte Aminofunktion aus
elektrostatischen Gründen die Protonierung an der Hydroxylgruppe an Position 3 mehr
behindert als an Position 4. In der Summe kann dies als Erklärung für die vermehrte Furosin-
Bildung bei höheren und die vermehrte Lysinbildung bei niedrigeren Säurekonzentrationen
bei den Fructoysl-Amadori-Produkten angesehen werden. Als Ursache für die sehr ähnliche
molaren Ausbeuten bei den Fructoysl-Amadori-Produkten ist eine vergleichsweise schnelle
Hydrolyse von Nε-Lactulosyl- und Nε-Maltulosyllysin zum Nε-Fructosyllysin anzusehen.
NHR
HO
O
OH
OH
OH
NHR
OH
OH
OH
OH
OH
NHR
O
OH
OH
OH
NHR
O
OH
OH
O
NHR
OO
Furosin
- H2O - 2 H2O
Amadori-Produkt
2,3-Enol 1-Amino-2,3-didesoxyoson
NR
O
Lysin
Pyridosin
O
HO OH
O
OH
HO HOOHNHR
NHR
OH
Amadori-Produkt(Furanose-Form)
1,2-Enol
NHR
HO
OH
OH
OH
OH
NH
OH
OH
OH
OH
R O
O
OH
OH
OH
3-DG
H2N R- H2O
+ H2O+
Aminomethyl-furan
- 2 H2O - H2O
- 2 H2O
NHR
H2O
OH
OH
OH
OH
+
_ +
- H++ H+
NHR
O
OH2
OH
OH
OH
H
+- H+
+ H+
Abb. 4.1.3-1: Überblick zum Bildungsmechanismus von Lysin, Furosin und Pyridosin aus Nε-Fructosyllysin (Literatur siehe Text).
Die tendenziell höheren Werte für Furosin bei den Disaccharid-Amadori-Produkten sind
vermutlich auf eine gewisse Unvollständigkeit der eben genannten Hydrolyse und die
- 117 -

4 Auswertung und Diskussion der Ergebnisse
leichtere Eliminierbarkeit des Glycosyl-Restes gegenüber der Hydroxylgruppe vom
Kohlenstoffatom 4 im Fructosyl-Rest zurückzuführen (Hollnagel & Kroh, 2000).
Da sich Nε-Fructosyllysin und Nε-Tagatosyllysin „nur“ in der Stellung der Hydroxylgruppe an
Position 4 unterscheiden, ist diese strukturelle Differenz als Ursache für das unterschiedliche
Hydrolyseverhalten verantwortlich. Bei Nε-Tagatosyllysin ist die stark erhöhte Pyridosin-
Bildung auffällig. Da Pyridosin nach Finot et al. (1969a) direkt aus der furanoiden Form des
Amadori-Produktes hervorgehen soll, wäre es denkbar, dass dieses Konformer beim
Nε-Tagatosyllysin im Vergleich zum Nε-Fructosyllysin im erhöhten Maß vorhanden ist.
Entsprechend den bei Raumtemperatur in Deuteriumoxid und schwach saurem pH-Wert
mittels 13C-NMR Spektroskopie ermittelten Anomer-Anteilen ist dies aber nicht der Fall
(Tab. 4.1.3-1). Die Daten zeigen jedoch, dass sich die beiden Amadori-Produkte sehr deutlich
in ihrer anomeren Zusammensetzung unterscheiden. Die Anomer-Anteile bei den Fructosyl-
Amadori-Produkten sind ähnlich, es dominiert jeweils die β-Pyranose. Zu den Konformeren-
Anteilen unter Hydrolysebedingungen kann keine sichere Aussage getroffen werden, da die
Bestimmung mittels 13C-NMR Spektroskopie in 6 M Salzsäure bereits bei Raumtemperatur
nicht möglich ist. Aus der Literatur ist bekannt, dass die Anomer-Anteile stark vom
Lösungsmittel abhängig sind (Tjan et al., 1974, Jakas & Horvat, 1996). Weiterhin dürfte die
Temperatur von Einfluss sein (Funcke & Klemer, 1976). Für D-Fructose, welche eine nahezu
identische anomere Zusammensetzung aufweist wie Nε-Fructosyllysin, wurde z. B. bereits im
Temperatur-Bereich von 0 bis 80 °C in Wasser eine sehr starke Abhängigkeit in der Weise
beobachtet, dass mit steigender Temperatur die Anteile der beiden furanoiden Formen auf
Kosten der β-Pyranose zunehmen (Tsuzuki et al., 1950, Schneider et al., 1985). Zu einer
Abhängigkeit vom pH-Wert liegen für wässrige Lösungen konträre Angaben vor. Während
eine solche von Altena et al. (1981) im Bereich von pH = 3,5 bis 9,5 festgestellt wurde,
konnte diese von Röper et al. (1983) im Bereich von pH = 0,9 bis 11,9 nicht bestätigt werden.
Zusammenfassend ist festzustellen, dass sich die Anomer-Anteile unter Hydrolyse-
bedingungen von denen unter moderaten Bedingungen mit Sicherheit unterscheiden werden.
Der Umfang der Verschiebung in der anomeren Zusammensetzung und damit die Plausibilität
der postulierte Bildung von Pyridosin aus der Furanose-Form kann jedoch ohne die
entsprechenden Daten nicht beurteilt werden.
Die Bildung von Pyridosin ist weiterhin aus dem 1-Amino-1,4-didesoxyoson zu diskutieren.
Sofern unter den Hydrolysebedingungen die cis,trans-Isomerie des als Enol entstehenden
1-Amino-1,4-didesoxyoson keine signifikante Rolle spielt, sollte bei Nε-Tagatosyllysin und
- 118 -

4 Auswertung und Diskussion der Ergebnisse
Nε-Fructosyllysin das Furosin-zu-Pyridosin-Verhältnis gleich sein. Ein solches vom Amadori-
Produkt unabhängiges Verhältnis wurde jedoch nicht festgestellt und folglich sollte dieser
Bildungsweg nicht der einzige sein.
Tab. 4.1.3-1: Anomer-Anteile der dargestellten Hippuryl-Amadori-Produkte in Deuterium-
oxid-Lösung (pH = 4 bis 5 [gemessener pH-Wert, nicht korrigiert], ϑ = 20 °C).
Amadori- Anomer-Anteil [%] 1,2
Produkt α-Pyranose β-Pyranose α-Furanose β-Furanose
BzGFruK 5 (4) 71 (70) 12 (13) 12 (13) 3
BzGTagK 70 (67) 23 (21) 7 (9) - (3) 4
BzGLctK - (2) 73 (65) 14 (18) 13 (15) 5
BzGMltK - (2) 70 (64) 16 (18) 14 (16) 4
1 Bestimmt mittels 13C-NMR Spektroskopie anhand der Signalintensität ausgewählter
gleichartiger C-Atome der messbaren Anomere. 2 Literaturangaben in Klammern: 3 Mossine
et al. (1994). 4 Feather & Mossine (1998). 5 Olano et al. (1992).
Insgesamt betrachtet können die diskutierten Reaktionswege die Hydrolyse der Amadori-
Produkte im Wesentlichen erklären. Einige Phänomene können jedoch zur Zeit noch nicht
vollständig auf molekularer Ebene interpretiert werden. Zur weiteren Klärung sind
insbesondere Kenntnisse zur Struktur der Amadori-Produkte unter Hydrolysebedingungen
sowie zum Mechanismus der Produktbildung erforderlich.
4.1.4 Untersuchungen in Hinblick auf die Bildung von Nε-Carboxymethyllysin
In den Hydrolysaten der Amadori-Produkte wurden neben Lysin, Furosin und Pyridosin in
geringen Mengen CML detektiert, die molare Ausbeute wurde zu 1 bis 2 % bestimmt. CML
ist als ein oxidatives Abbauprodukt von Fructosyllysin bekannt (Ahmed et al., 1986). Um eine
Verunreinigung der Präparate der Hippuryl-Amadori-Produkte auszuschließen, wurden diese
mittels RP-HPLC auf das Vorhandensein von Nα-Hippuryl-Nε-carboxymethyllysin geprüft.
Das Nε-Fructosyllysin-Präparat wurde mittels Aminosäureanalyse untersucht. Es konnte
weder Nα-Hippuryl-Nε-carboxymethyllysin bzw. CML in den Präparaten nachgewiesen
- 119 -

4 Auswertung und Diskussion der Ergebnisse
werden. Weiterhin konnte CML nicht detektiert werden, wenn vor der Salzsäurehydrolyse das
Amadori-Produkt mit Natriumborhydrid reduziert wurde. Folglich muss der oxidative Abbau
während der Hydrolyse erfolgen. Darauf weist auch die vermehrte CML-Bildung bei
Hydrolyse in Gegenwart von Natriumazid hin (siehe Abschnitt 4.1.2), welches in saurer
Lösung als Oxidationsmittel wirkt.
Um die Wirkung von Luftsauerstoff als Oxidationsmittel zu minimieren, wurde die Hydrolyse
unter Vakuum (p < 0,001 mbar) durchgeführt (Abb. 4.1.4-1).
A B C0,0
0,3
0,6
0,9
1,2
1,5
1,8n
m
po
n
m
Art der Hydrolyse
Mol
are
Ausb
eute
[%]
Abb. 4.1.4-1: Molare Ausbeute an CML bei Hydrolyse von Nα-Hippuryl-Nε-fructosyllysin in 6 M Salzsäure: Standardhydrolyse in Hydrolysegefäß nach Spülen mit Stickstoff (A), Hydrolyse in Vakuumhydrolysegefäß, ohne Spülen mit Stickstoff unter Normaldruck (B), Hydrolyse in Vakuumhydrolysegefäß, nach Evakuierung (p < 0,001 mbar) (C). Hydrolyse mit Salzsäure „ACS quality“ von J. T. Baker (graue Balken) und „trace select“ von Fluka (schwarze Balken). Mittelwert ± Standardabweichung (n = 5, f = 4). P < 0,01: m vs o, n vs p. (Durchführung: siehe Abschnitt 3.4.1).
Im Vergleich zur Standardhydrolyse führte die Hydrolyse unter Vakuum zu einer
signifikanten (P < 0,01) Verringerung der CML-Bildung auf etwa die Hälfte. Gewisse Spuren
von Chlor in der Salzsäure sind durch die Vakuum-Behandlung vermutlich nicht vollständig
entfernbar und als Ursache für die restliche CML-Bildung anzusehen. Da Spuren von
Schwermetallen den oxidativen Abbau katalysieren können, fand eine weitere Salzsäure
- 120 -

4 Auswertung und Diskussion der Ergebnisse
höchster analytischer Qualität Verwendung. Diese bewirkte jedoch keine signifikant
niedrigere Bildung von CML. Eine vollständige Unterdrückung der CML-Bildung war nur
möglich, wenn vor der Hydrolyse das Amadori-Produkt mit Natriumborhydrid reduziert
wurde. Die Bestimmung von CML in Lebensmitteln nach einer solchen Reduktion wurde
z. B. von Drusch et al. (1999) beschrieben.
Zusammenfassend ist festzustellen, dass unter Standardhydrolysebedingungen eine geringe
CML-Bildung stattfindet. In Hinblick auf die Bestimmung von Furosin ist dieser Abbau
jedoch aus quantitativen Gründen ohne Belang. Da in Lebensmittel und in vivo das Amadori-
Produkt gegenüber CML um den Faktor 200 bis 1000 dominiert, ist zur Bestimmung von
CML der oxidative Abbau des Amadori-Produktes während der Hydrolyse unbedingt zu
vermeiden. Um dies zu erreichen, hat sich unter Modellbedingungen die vorherige Reduktion
des Amadori-Produktes mit Natriumborhydrid als geeignet erwiesen.
4.1.5 Weitere Studien zur Bildung von Furosin
Bisher wurde Furosin ausschließlich als Produkt der Salzsäurehydrolyse von Amadori-
Produkten betrachtet. Huber & Ledl (1990) berichteten jedoch auch von der thermisch
induzierten Bildung eines 2-Aminoacetylfuran-Derivates aus Salzen des N-Fructosyl-
propylamins in wässriger Lösung im Zuge der Maillard-Reaktion. Weiterhin konnten
Hollnagel & Kroh (2000) nach Umsetzung von Lysin und Maltose unter ähnlichen
Bedingungen Furosin detektieren. In Anlehnung an diese Experimente wurde das Lyophilisat
einer wässrigen Lösung von Nε-Boc-Lysin und Maltose für 15 min bei 120 °C erhitzt. Nach
Abspaltung der Schutzgruppe konnte mittels Aminosäureanalyse eindeutig die Bildung von
Furosin nachgewiesen und die obigen Befunde bestätigt werden. Der Umsatz zu Furosin
wurde zu ca. 0,5 % bestimmt. Über eine Bildung von Pyridosin kann auf Grund von
Interferenzen durch die erforderliche große Injektionsmenge keine sicher Aussage getroffen
werden. Huber (1989) konnte keine Bildung eines dem Pyridosin ähnlichen Derivates aus
N-Fructosylpropylamin beobachten.
Um die Stabilität von Amadori-Produkten in Salzsäure zu untersuchen, wurde freies
Nε-Fructosyllysin in 6 M und 8 M Salzsäure bei 23 °C inkubiert und zu definierten Zeiten
wurde die Lösung mittels Aminosäureanalyse untersucht. Nε-Fructosyllysin zeigte unter den
- 121 -

4 Auswertung und Diskussion der Ergebnisse
gewählten Inkubationsbedingungen eine ausgesprochen hohe Stabilität. Erst nach 14 d
Inkubation in 8 M Salzsäure konnte eine geringfügige Furosin-Bildung beobachtet werden. In
6 M Salzsäure war demgegenüber zu diesem Zeitpunkt noch kein Abbau detektierbar. Damit
zeigt Nε-Fructosyllysin in 6 M und 8 M Salzsäure bei 23 °C eine bemerkenswert hohe
Stabilität. Strukturanalytische Untersuchungen der Amadori-Produkte in Salzsäure zur
weiteren Klärung der Reaktionswege bei der Hydrolyse sollten somit prinzipiell möglich sein.
Aus messtechnischen Gründen sind jedoch 13C-NMR-spektroskopische Untersuchungen in
Salzsäure beispielsweise zur Bestimmung der Anteile der einzelnen Anomeren nicht
realisierbar.
- 122 -

4 Auswertung und Diskussion der Ergebnisse
4.2 Überblick zur Reaktivität von Peptiden und individuellen
Aminosäureseitenketten gegenüber α-Dicarbonylverbindungen
Obwohl die in vivo stattfindende Glykierung von Proteinen zuerst am N-Terminus des
Hämoglobins festgestellt wurde (Bunn et al., 1975, 1979, Flückiger & Winterhalter, 1976),
schenkte man später Reaktionen an der α-Aminogruppe von Proteinen und Peptiden kaum
Beachtung. Autoren, welche sich aus protein- oder lebensmittelchemischer Sicht mit
Umsetzungen von α-Dicarbonylverbindungen am N-Terminus befassten, kamen zum Teil zu
konträren Ergebnissen (Takahashi, 1968, 1977a, Van Chuyen et al., 1973a, b). Auf Grund der
gewählten Bedingungen lassen diese Untersuchungen auch keine Rückschlüsse auf in vivo
stattfindende Prozesse zu (siehe Abschnitt 2.3.4).
Um Reaktionen am N-Terminus in der fortgeschrittenen Phase der Maillard-Reaktion zu
untersuchen, wurden im Rahmen dieser Arbeit kurzkettige Peptide mit in vivo und in
Lebensmitteln relevanten α-Dicarbonylverbindungen unter physiologischen Bedingungen
bezüglich des pH-Wertes (pH = 7,4) und der Temperatur (ϑ = 37 °C) umgesetzt. Die Peptide
und Aminosäure-Derivate wurden dabei in Hinblick auf die Analytik mittels RP-HPLC und
UV-Detektion bei λ = 220 nm ausgewählt. Da der pH-Wert wesentlichen Einfuß auf die
Kinetik von Amino-Carbonyl-Reaktionen hat, erfolgten die Inkubationen in phosphat-
gepufferter Lösung. Die Pufferionenkonzentration wurde so gewählt, dass die
Konstanthaltung des pH-Wertes (∆pH ≤ 0,1) über den untersuchten Inkubationszeitraum
gewährleistet war.
Als Einstieg wurden ausgewählte Peptide ohne nucleophile Seitenketten, z. B. Gly-Ala-Phe,
Gly-Phe-Ala, Phe-Gly-Gly, mit Glyoxal, Methylglyoxal oder 3-DG im equimolaren
Verhältnis (je 5 mM) für 3 d bei leicht erhöhter Temperatur von 60 °C umgesetzt. Um die
Reaktivität beurteilen zu können, wurden die bekannten Reaktionspartner für α-Dicarbonyl-
verbindungen Lysin und Arginin sowie weitere potentiell reaktive Aminosäuren als
Nα-geschützte Derivate unter den gleichen Bedingungen inkubiert.
Die Umsätze wurden mittels RP-HPLC anhand der Abnahme der Peakfläche des
Ausgangspeptides bestimmt. Anhand der so ermittelten Umsätze (siehe Abb. 4.2-1) ist auf
eine hohe Reaktivität von Peptiden wie Gly-Ala-Phe und Phe-Gly-Gly zu schließen.
Nα-Hippurylarginin war erwartungsgemäß ebenfalls reaktiv. Weiterhin waren Umsätze mit
Cystein und Methionin festzustellen. Als Reaktionsprodukte des Cysteins sind insbesondere
- 123 -

4 Auswertung und Diskussion der Ergebnisse
Thiohemiacetale und Thiohemiketale zu vermuten (Thornalley, 1998). Demgegenüber konnte
mit Nα-Hippuryllysin, Nα-Acetyl-Tryptophan, Nα-Ac-Ser-Gly, Nα-Benzoyl-Histidin,
Nα-Hippurylglycin keine Reaktion beobachtet werden. Da CML und CEL als Produkte der
Lysin-Derivatisierung durch Glyoxal bzw. Methylglyoxal bekannt sind (Al-Abed & Bucala,
1995, Ahmed et al., 1997), war die geringe Reaktivität von Lysin nicht zu erwarten. In
Hinblick auf die Untersuchungen von Saito et al. (1986) gilt das gleiche für Tryptophan.
Anhand der Inkubationen mit Nα-Hippurylglycin ist festzustellen, dass der Peptidstickstoff
ebenfalls nicht reaktiv ist.
GAF GFA AFG FGG F AcC BzGR BzMet0
20
40
60
80
100
Um
satz
[%]
Peptid, Aminosäure-Derivat
Abb. 4.2-1: Umsatz an Peptid nach Umsetzung mit Glyoxal (hellgraue Balken), Methylglyoxal (mittelgraue Balken) oder 3-DG (schwarze Balken). (Peptid und α-Dicarbonylverbindung je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 60 °C, 3 d, Durchführung: 3.5.1).
Wie Abb. 4.2-1 weiterhin zu entnehmen ist, zeigen die α-Dicarbonylverbindungen gegenüber
den Tripeptiden eine abgestufte Reaktivität, wobei die Reaktivität in der Reihenfolge vom
3-DG über Methylglyoxal zum Glyoxal hin zunimmt. Dieser Befund steht offenbar in
Zusammenhang mit der Elektrophilie der Carbonylgruppen, da diese auf Grund von
Substituenteneffekten in der gleichen Weise steigt. Weiterhin wird die Neigung zur
Hydratisierung von Bedeutung sein. So ist beim Methylglyoxal bevorzugt die
Aldehydfunktion hydratisiert (Thornalley, 1996) und als reaktive Funktion verbleibt damit
nur die weniger elektrophile Ketogruppe. Beim Glyoxal ist demgegenüber bei Bildung des
Monohydrates immer noch eine reaktivere Aldehydfunktion vorhanden. Bei 3-DG dürfte die
- 124 -
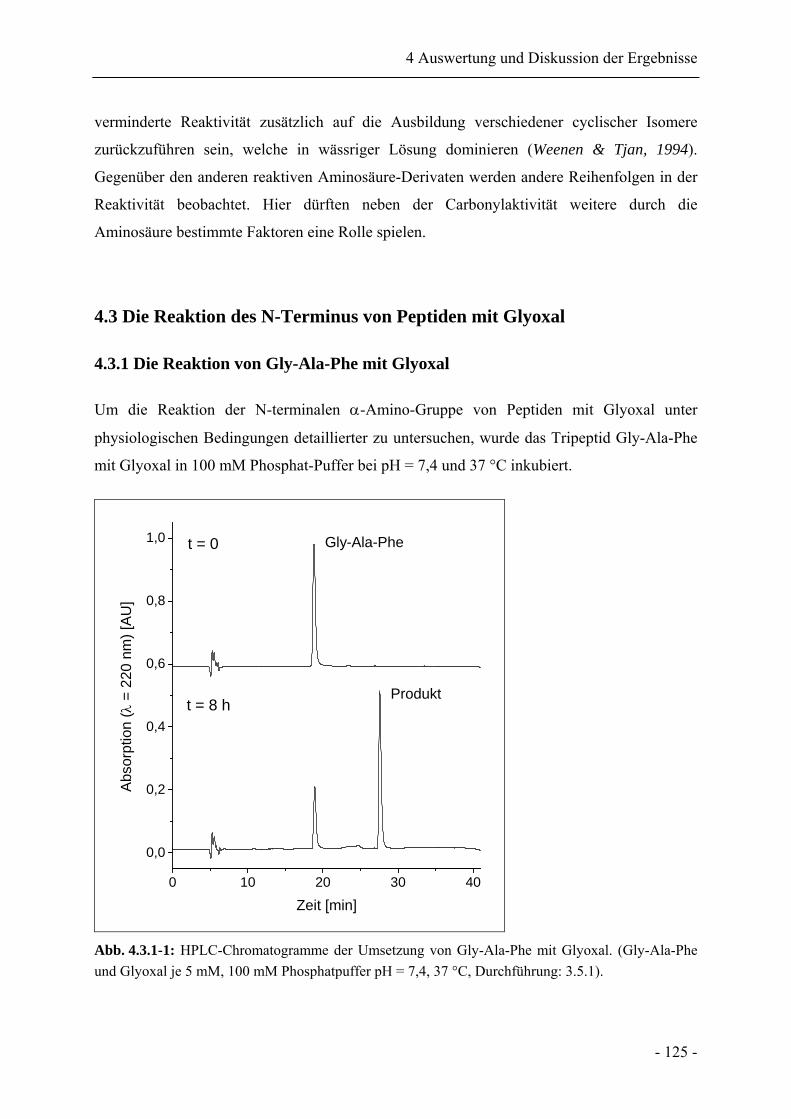
4 Auswertung und Diskussion der Ergebnisse
verminderte Reaktivität zusätzlich auf die Ausbildung verschiedener cyclischer Isomere
zurückzuführen sein, welche in wässriger Lösung dominieren (Weenen & Tjan, 1994).
Gegenüber den anderen reaktiven Aminosäure-Derivaten werden andere Reihenfolgen in der
Reaktivität beobachtet. Hier dürften neben der Carbonylaktivität weitere durch die
Aminosäure bestimmte Faktoren eine Rolle spielen.
4.3 Die Reaktion des N-Terminus von Peptiden mit Glyoxal
4.3.1 Die Reaktion von Gly-Ala-Phe mit Glyoxal
Um die Reaktion der N-terminalen α-Amino-Gruppe von Peptiden mit Glyoxal unter
physiologischen Bedingungen detaillierter zu untersuchen, wurde das Tripeptid Gly-Ala-Phe
mit Glyoxal in 100 mM Phosphat-Puffer bei pH = 7,4 und 37 °C inkubiert.
0 10 20 30 40
0,0
0,2
0,4
0,6
0,8
1,0 Gly-Ala-Phe
Produktt = 8 h
t = 0
Abs
orpt
ion
(λ =
220
nm
) [AU
]
Zeit [min]
Abb. 4.3.1-1: HPLC-Chromatogramme der Umsetzung von Gly-Ala-Phe mit Glyoxal. (Gly-Ala-Phe und Glyoxal je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 37 °C, Durchführung: 3.5.1).
- 125 -
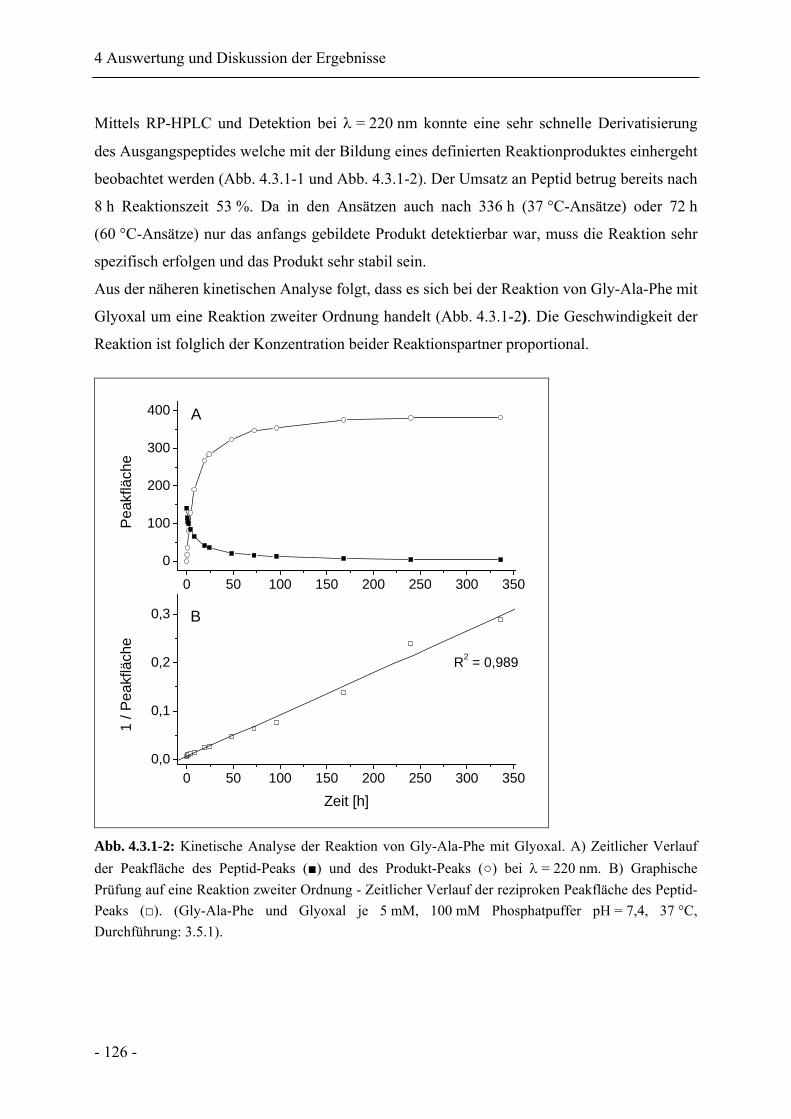
4 Auswertung und Diskussion der Ergebnisse
Mittels RP-HPLC und Detektion bei λ = 220 nm konnte eine sehr schnelle Derivatisierung
des Ausgangspeptides welche mit der Bildung eines definierten Reaktionproduktes einhergeht
beobachtet werden (Abb. 4.3.1-1 und Abb. 4.3.1-2). Der Umsatz an Peptid betrug bereits nach
8 h Reaktionszeit 53 %. Da in den Ansätzen auch nach 336 h (37 °C-Ansätze) oder 72 h
(60 °C-Ansätze) nur das anfangs gebildete Produkt detektierbar war, muss die Reaktion sehr
spezifisch erfolgen und das Produkt sehr stabil sein.
Aus der näheren kinetischen Analyse folgt, dass es sich bei der Reaktion von Gly-Ala-Phe mit
Glyoxal um eine Reaktion zweiter Ordnung handelt (Abb. 4.3.1-2). Die Geschwindigkeit der
Reaktion ist folglich der Konzentration beider Reaktionspartner proportional.
0 50 100 150 200 250 300 3500,0
0,1
0,2
0,3
0 50 100 150 200 250 300 350
0
100
200
300
400
B
R2 = 0,989
1 / P
eakf
läch
e
Zeit [h]
A
Pea
kflä
che
Abb. 4.3.1-2: Kinetische Analyse der Reaktion von Gly-Ala-Phe mit Glyoxal. A) Zeitlicher Verlauf der Peakfläche des Peptid-Peaks (■) und des Produkt-Peaks (○) bei λ = 220 nm. B) Graphische Prüfung auf eine Reaktion zweiter Ordnung - Zeitlicher Verlauf der reziproken Peakfläche des Peptid-Peaks (□). (Gly-Ala-Phe und Glyoxal je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 37 °C, Durchführung: 3.5.1).
- 126 -

4 Auswertung und Diskussion der Ergebnisse
4.3.2 Identifikation des Reaktionsproduktes
Zur Aufklärung der Struktur wurden 112 mg der neuen Verbindung aus einem
semipräparativen Inkubationsansatz mittels RP-HPLC in chromatographisch reiner Form
isoliert. Das UV-Spektrum des Isolates bei pH = 7,0 weist neben der peptidspezifischen
Absorption bei λ = 210 nm eine charakteristische Bande mit einem Maximum bei λ = 322 nm
auf. Bei der daraufhin durchgeführten Prüfung auf Fluoreszenz erwies sich die isolierte
Verbindung als fluoreszierend mit λex, max = 330 nm und λem, max = 395 nm (siehe
Abb. 4.3.2-1). Durch Messung bei pH = 2,4 (100 mM Ameisensäure) oder pH = 8,3 (100 mM
Borat-Puffer) kommt es weder zu einer signifikanten Verschiebung des Maximums im
UV-Spektrum (λ = 322 nm) noch zu einer Veränderung des molaren Extinktionskoeffizienten
(ε322nm). Das gleiche gilt für das Maximum im Fluoreszenzspektrum und die Intensität der
Fluoreszenzemission. Folglich ist anzunehmen, das der Chromophor und Fluorophor keine
leicht ionisierbaren funktionellen Gruppen aufweist.
Mittels direkter ESI-MS Analyse konnte die relative monoisotopische Masse des Produktes
zu Mr = 314,7 bestimmt werden. Anhand dessen ist von einer Reaktion von einem Molekül
Peptid mit einem Molekül Glyoxal unter Elimination von zwei Molekülen Wasser
auszugehen.
200 300 400 5000,0
0,2
0,4
0,6
0,8
1,0
1,2
200 300 400 500 600
A
Ext
inkt
ion
Wellenlänge [nm]
B
Rel
ativ
e Fl
uore
szen
zem
issi
on
Wellenlänge [nm]
Abb. 4.3.2-1: Absorptionsspektrum (A) und Fluoreszenzemissionsspektrum (λex, max = 330 nm, B) des Glyoxal-Gly-Ala-Phe-Reaktionsproduktes. Aufgenommen in 100 mM Phosphat-Puffer pH = 7,0.
- 127 -

4 Auswertung und Diskussion der Ergebnisse
Mittels ein- und zweidimensionale NMR Spektroskopie (1H, 13C, 1H-1H COSY, DEPT,
HSQC, HMBC) konnte die in Abb. 4.3.2-2 gezeigte Verbindung als Reaktionsprodukt
identifiziert werden. Die spektroskopischen Daten der 1H- und 13C-NMR Experimente sind in
Abschnitt 3.5.2 zusammengefasst.
In den Spektren werden neben den Signalen von Alanin und Phenylalanin zusätzliche Signale
im olefinischen Bereich detektiert. Bemerkenswert ist die Tieffeld-Verschiebung des Signals
des Alanin Methinprotons von δ = 4,37 ppm im Ausgangspeptid nach δ = 5,38 ppm im
Produkt, welche die Bildung eines neuen elektronenziehenden Substituenten anzeigt. Im
HMBC Spektrum korreliert dieses Proton mit dem C-2- und dem C-6-Atom des neuen
Substituenten und zeigt die Verknüpfung über das benachbarte Stickstoff-Atom an. Aus dem 1H-1H COSY und HSQC Spektren ist die Nachbarschaft der CH-5 Methingruppe zur CH-6
Methingruppe zu entnehmen. Das H-3 Methinproton erscheint als kleines Dublett und im
HMBC Spektrum sind Kreuzpeaks zu C-5 und zum quartären C-2 sowie eine schwache
Korrelation zu C-6 festzustellen. In der Summe der Ergebnisse konnte damit das eine
2(1H)-Pyrazinon-Struktur tragende Peptid N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-
phenylalanin (Abb. 4.3.2-2) als Reaktionsprodukt aufgeklärt werden.
H
N
NO
CH3
H
H
H1
3
6HN
OOH
O
N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin
Abb. 4.3.2-2: Strukturformel von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin. Die Pfeile zeigen charakteristische im HMBC Spektrum beobachtete Konnektivitäten an.
Die gefundene chemische Struktur deckt sich mit dem Ergebnis der massenspektroskopischen
Analyse. Die Bildung eines 2(1H)-Pyrazinon-Rings erklärt weiterhin die charakteristische
Absorption im UV oberhalb von 300 nm (λmax = 322 nm). Für das in der Struktur ähnliche
N1-Methyl-2(1H)-pyrazinon wurde ein UV-Maximum mit praktisch identischer Lage
- 128 -

4 Auswertung und Diskussion der Ergebnisse
(λmax = 323 nm, wässrige Lösung, pH = 7,0) festgestellt (Mason, 1957). Eine UV-Bande
oberhalb 300 nm ist ein allgemeines Charakteristikum für Pyrazin-Derivate und auf einem
n → π Übergang zurückzuführen (Halverson & Hirt, 1951, Mason, 1959). Der pKa-Wert des
Produktes dürfte ähnlich niedrig sein wie der von N1-Methyl-2(1H)-pyrazinon (pKa = - 0,04,
Albert & Phillips, 1956) und für die Konstanz der Lage der UV-Bande sowie die Konstanz
des Fluoreszenzspektrums im untersuchten pH-Bereich (pH = 2,4 bis 8,3) verantwortlich sein.
N
N O
OH
O
N
N O
OH
O
N
N O
NH
O
OH
O
N
N O
NH
O
OH
O
N-[2-(2-Oxo-2H-pyrazin-1-yl)-acetyl]-glycin
N-[2-(2-Oxo-2H-pyrazin-1-yl)-2-phenyl-acetyl]-alanin2-(2-Oxo-2H-pyrazin-1-yl)-2-phenyl-essigsäure
3-Methyl-2-(2-oxo-2H-pyrazin-1-yl)-valeriansäure
Abb. 4.3.2-3: Strukturformeln von dargestellten 2(1H)-Pyrazinon-Peptiden
Im Folgenden wurden Inkubationsansätze bei 37 °C und pH = 7,4 von weiteren Peptiden
(z. B. Gly-Phe-Ala, Gly-Gly-Gly, Phe-Gly-Gly, Phe-Gly, Gly-Ile, Gly-Phe) und Glyoxal auf
die Bildung von entsprechenden 2(1H)-Pyrazinon-Peptiden geprüft. Ausgewählte
2(1H)-Pyrazinon-Peptide wurden dabei semipräparativ gewonnen (Abb. 4.3.2-3). Die Analyse
der Inkubationsansätze erfolgte im ersten Schritt mittels RP-HPLC. Zur spezifischen
Detektion wurde mit Hilfe von zwei UV-Detektoren bei λ = 220 nm und λ = 320 nm sowie
mittels Fluoreszenzdetektor (λex = 320 nm, λem = 400 nm) gemessen. Die Ermittlung der
Masse der Produkte erfolgte mittels LC-ESI-MS. Diese Experimente zeigten, dass es bei der
Umsetzung der geprüften Tripeptide mit Glyoxal sehr schnell und spezifisch zur Bildung der
entsprechenden 2(1H)-Pyrazinon-Peptide kommt. Dipeptide reagierten langsamer und die
Bildung des 2(1H)-Pyrazinon-Peptides war bei einigen Dipeptiden ebenfalls sehr spezifisch,
z. B. bei Phe-Gly. Im Fall von Phe-Val wurde jedoch ein sehr heterogenes Produktspektrum
- 129 -

4 Auswertung und Diskussion der Ergebnisse
und kaum 2(1H)-Pyrazinon-Bildung beobachtet. Die Produkte der Reaktion von Gly-Phe-Ala,
Gly-Gly-Gly, Gly-Ile und Gly-Phe mit Glyoxal wurden im semipräparativen Maßstab isoliert
und die Struktur mittels ein- und zweidimensionalen NMR-Spektroskopie sowie ESI-MS
zweifelsfrei als die in Abb. 4.3.2-3 gezeigten 2(1H)-Pyrazinon-Peptide identifiziert (siehe
Abschnitt 3.5.3). Insgesamt betrachtet ist damit die Reaktion von Peptiden und Glyoxal zu
2(1H)-Pyrazinon-Peptiden unter physiologischen Bedingungen als ein allgemein zu
beobachtendes Phänomen anzusehen.
Bei den identifizierten 2(1H)-Pyrazinon-Peptiden handelt es sich um Derivate die neben
anderen Produkten bereits von Prey & Petershofer (1968) unter Synthesebedingungen
(pH = 13, hoher Überschuss an Glyoxal) und Van Chuyen et al. (1973a, b) beim Erhitzen
(pH = 5,0, 100 °C, 1 h) erhalten wurden (siehe Abschnitt 2.3.4). Mit den vorliegenden
Untersuchungen konnte nun gezeigt werden, dass die Bildung von 2(1H)-Pyrazinon-Peptiden
aus Peptiden und Glyoxal bereits unter physiologischen und damit sehr milden Bedingungen
erfolgt. Dabei ist besonders die ausgesprochen schnelle Reaktion und die spezifische Bildung
nur eines Reaktionsproduktes (bei Tripeptiden) hervorzuheben. Auf Grund der von Prey &
Petershofer (1968) und Van Chuyen et al. (1973a, b) angewendeten drastischen Bedingungen
war dieses Ergebnis nicht und vor allem nicht mit dieser Deutlichkeit zu erwarten. Eher war
demgegenüber die Bildung von α-Ketoacyl-Peptiden zu vermuten. Takahashi (1977a)
beschrieb deren Entstehung unter relativ milden Bedingungen (pH = 8,0, 25 °C, 1 h). Mittels
der angewendeten Analytik waren jedoch α-Ketoacyl-Peptide sowie andere mögliche
Reaktionsprodukte wie 1,3-Bispeptidyl-imidazolium-Salze (Davidek et al., 1991) und
Nα-Carboxymethyl-Peptide (Van Chuyen et al., 1972, 1973c) nicht nachweisbar.
4.3.3 Stabilität von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin
Um die Stabilität von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin sowohl im leicht
Basischen als auch im Sauren beurteilen zu können, wurden Inkubations-Experimente im
pH-Bereich von 2,0 bis 9,0 (60 °C, 3 d) durchgeführt. Das 2(1H)-Pyrazinon-Peptid zeigte sich
dabei als sehr stabil. Lediglich bei pH = 9,0 war ein kleiner zusätzlicher später eluierender
Peak feststellbar. Der Abbau war jedoch sehr gering (< 3 %).
- 130 -

4 Auswertung und Diskussion der Ergebnisse
Die Reversibilität der 2(1H)-Pyrazinon-Bildungsreaktion wurde durch Inkubation von
N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin (5 mM) in Gegenwart des α-Dicarbonyl-
reagenzes Aminoguanidin (25 mM, pH = 7,4, 60 °C, 3 d) geprüft. Mittels HPLC wurde ein
Abbau des 2(1H)-Pyrazinon-Peptides von ca. 9 % bestimmt. Ein zusätzlicher im
Chromatogramm auftretender Peak konnte dem 1,2,4-Triazin-Derivat zugeordnet werden,
welches bei der Reaktion von Aminoguanidin mit Glyoxal gebildet wird. Die Bildung von
2(1H)-Pyrazinon-Peptiden ist damit reversibel. Die nur geringe Rückbildung in Gegenwart
eines fünffachen molaren Überschusses an Aminoguanidin belegt aber auch die hohe
Stabilität der N-terminalen 2(1H)-Pyrazinon-Struktur.
Im Rahmen der durchgeführten Stabilitätsuntersuchungen ist dem 2(1H)-Pyrazinon-Tripeptid
damit insgesamt eine hohe Stabilität zuzuschreiben.
4.3.4 Zum Mechanismus der Bildung von 2(1H)-Pyrazinon-Peptiden
Der Bildungsweg von 2(1H)-Pyrazinon-Peptiden kann wie in Abb. 4.3.4-1 dargestellt
formuliert werden.
O
OR4
R5
+
α-Dicarbonyl
NHR3NH
H2N
H
O R2
O
R1 NHR3NH
O R2
OO
NR4
R5
R1
H
NHR3NH
O R2
OOH
NR4
R5
R1N
HN O
R5
R4
R2NHR3
O
R1
O
N
N OR5
R4
R2NHR3
O
R1
2(1H)-Pyrazinon-Peptid
- H2O+ H2O
+ H2O- H2O
Schiff Base
Enaminolα-Aminoketon
Peptid
Abb. 4.3.4-1: Postulierter Bildungsmechanismus von 2(1H)-Pyrazinon-Peptiden.
Im ersten Schritt erfolgt eine Protonen-katalysierte Bildung einer Schiff Base. Anschließend
findet ähnlich dem Strecker-Abbau (siehe Abschnitt 2.3.3) eine Umlagerung am Azomethin-
- 131 -

4 Auswertung und Diskussion der Ergebnisse
Stickstoffatom statt. Es handelt sich dabei um eine Tautomerisierung eines α-Iminoketons in
ein Enaminol eines Imins. Dabei kann eine Übernahme des α-Protons der N-terminalen
Aminosäure durch die Ketogruppe postuliert werden kann. Die Abgabe und Aufnahme eines
Protons an bzw. aus dem Medium wäre ebenfalls denkbar (nicht gezeigt). Anschließend
unterliegt das gebildete Enaminol einer Tautomerisierung zum α-Aminoketon und es folgt
eine intramolekulare nucleophile Addition des Peptid-Stickstoffes an die Ketofunktion. Nach
Wasserabspaltung kommt es schließlich zur Ausbildung eines konjugierten cyclischen
Enamin-Imino-Keton-Systems, welches als 2(1H)-Pyrazinon bezeichnet wird.
Die Annahme, dass ein Enaminol eines Imins als Intermediat auftritt wird durch die
Isolierung und Identifizierung solcher Derivate in Form von N-(2-(3-Desoxy-ascorb-
3-ylimino)-acetyl)-Aminosäuren erhärtet (siehe Abb. 4.3.4-2). Diese Produkte wurden nach
Umsetzung von Dehydroascorbinsäure (DHA) mit verschiedenen Dipeptiden bei 75 °C für
30 min in Methanol erhalten (Sakurai & Ishii, 1988, Sakurai et al., 1988).
O
HO
OH
O
O
O H2N
HN O
OH
O
HH
OH
HO
OH
O
O
N
HN O
OH
O
+- H2O+ H2O
H
Gly-Ala DAIA-AlaDHA
Abb. 4.3.4-2: Reaktion von Dehydroascorbinsäure (DHA) mit Gly-Ala und Bildung von N-(2-(3-Desoxy-ascorb-3-ylimino)-acetyl)-Ala (DAIA-Ala) (Sakurai & Ishii, 1988, Sakurai et al., 1988).
4.4 Einflussfaktoren auf die Reaktion von Peptiden mit
α-Dicarbonylverbindungen und die Bildung von 2(1H)-Pyrazinon-Peptiden
4.4.1 Einfluss der Struktur des Peptides
Zur Beurteilung des Einflusses der Primärstruktur des Peptides auf die 2(1H)-Pyrazinon-
Bildung wurden Inkubationsexperimente mit weiteren Peptiden und Aminosäure-Derivaten
durchgeführt (siehe Abb. 4.4.1-1). Die Peptide reagierten dabei spezifisch zum
- 132 -

4 Auswertung und Diskussion der Ergebnisse
2(1H)-Pyrazinon-Peptid, wobei sich jedoch eine deutliche Abhängigkeit der Primärstruktur
auf die Reaktion zeigte. Während das Ausmaß der 2(1H)-Pyrazinon-Bildung bei Gly-Phe-Ala
praktisch identisch wie bei Gly-Ala-Phe war, wurden bei Ala-Phe-Gly ein deutlich niedriger
und bei Phe-Gly-Gly ein deutlich höherer Umsatz gemessen (Abb. 4.4.1-1). Um diese
gemessenen Reaktivitäten zu diskutieren, wurden die pKa-Werte der N-terminalen
α-Aminogruppen mit der Software Solaris V 4.67 (siehe Literaturverzeichnis: Advanced
Chemistry Development [ACD]) berechnet. Da die pKa-Werte der N-terminalen
α-Aminogruppen von Gly-Phe-Ala (pKa = 7,67, ACD) und Gly-Ala-Phe (pKa = 7,66, ACD)
praktisch identische sind, Ala-Phe-Gly (pKa = 8,12, ACD) den höchsten und Phe-Gly-Gly
(pKa = 7,39, ACD) den niedrigsten pKa-Werte aufweist, bestimmen diese offensichtlich
wesentlich die Reaktionsgeschwindigkeit und vermutlich auch die Gleichgewichtslage. Für
die Kinetik der 2(1H)-Pyrazinon-Bildungsreaktion bedeutet dies, das vermutlich der erste und
einleitende Reaktionsschritt, die Bildung der Schiff Base (siehe Abschnitt 4.3.4), der
geschwindigkeitsbestimmende Schritt ist und die folgenden intramolekularen Schritte
vergleichsweise schnell erfolgen.
GAF GFA AFG FGG FG F FG F-NH20
20
40
60
80
100
*
*
Um
satz
[%]
Peptid, Aminosäure-Derivat
Abb. 4.4.1-1: Umsatz an Peptid nach Umsetzung mit Glyoxal nach 24 h oder 2 h (*). (Peptid und Glyoxal je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 37 °C, Durchführung: 3.5.1).
Das Dipeptid Phe-Gly zeigte eine deutlich geringere Reaktivität als das in der N-terminalen
Sequenz gleiche Tripeptid Phe-Gly-Gly. Auch hier sind die unterschiedlichen pKa-Werte als
Ursache anzusehen. Der pKa-Wert der α–Aminogruppe von Peptiden sinkt im allgemeinen
- 133 -

4 Auswertung und Diskussion der Ergebnisse
mit zunehmender Kettenlänge, so dass der pKa-Wert von Phe-Gly (pKa = 7,53 [25 °C],
Agoston et al.,1999, pKa = 7,55, ACD) höher ist als der von Phe-Gly-Gly (pKa = 7,39, ACD).
Die 2(1H)-Pyrazinon-Bildung aus Säureamiden ist ebenfalls möglich. Phenylalaninamid
(F-NH2) reagiert dabei deutlich schneller als Phe-Gly. Dies dürfte sowohl auf den niedrigeren
pKa-Wert der α-Aminogruppe von Phenylalaninamid (pKa = 7,26 [25 °C], Dallavalle et
al.,1989, pKa = 7,50, ACD) als auch auf eine bessere sterische Zugänglichkeit und Reaktivität
der Amidfunktion des Phenylalaninamids zurückzuführen sein. Aufgrund des Fehlens der
Amidfunktion war aus Phenylalanin erwartungsgemäß keine 2(1H)-Pyrazinon-Bildung zu
beobachten. Da auch kein sonstiger Abbau von Phenylalanin feststellbar war, findet unter den
angewendeten physiologischen Bedingungen ebenfalls kein Strecker-Abbau zum
Phenylacetaldehyd (siehe Abschnitt 2.3.3) und keine Bildung von
Nα-Carboxymethylphenylalanin (siehe Abschnitt 2.3.4) statt. Für diese Reaktionen sind die
Reaktionsbedingungen damit zu mild bzw. die Reaktionszeit ist zu kurz.
4.4.2 Einfluss der Struktur der α-Dicarbonylverbindung
Zur Abschätzung der Reaktivität wichtiger α-Dicarbonylverbindungen gegenüber der
α-Aminogruppe von Peptiden wurden diese mit Gly-Ala-Phe inkubiert. Dabei zeigte Glyoxal
auf Grund seiner hohen Carbonylaktivität die höchste Reaktivität (Abb. 4.4.2-1). Die anderen
α-Dicarbonylverbindung waren ebenfalls reaktiv. Als Ursache für die geringeren Umsätze bei
diesen Derivaten sind die verminderten Carbonylaktivitäten und zusätzlich im Fall von 3-DG
die Ausbildung cyclischer Formen anzusehen (siehe Abschnitt 4.2). Unterschiede waren
weiterhin im Produktspektrum festzustellen. Während die Reaktion mit Glyoxal spezifisch
zum 2(1H)-Pyrazinon-Peptid führte, wurden bei der Umsetzung mit Methylglyoxal mehrere
Produkte detektiert, wobei das 2(1H)-Pyrazinon-Peptid das Hauptprodukt war (siehe
Abschnitt 4.5.1). Bei der Reaktion von Diacetyl mit dem gewählten Peptid wurde
demgegenüber ein sehr komplexes Produktspektrum gebildet und der Umfang der
2(1H)-Pyrazinon-Bildung war gering.
- 134 -

4 Auswertung und Diskussion der Ergebnisse
GO MGO 3-DG Diacetyl0
20
40
60
80U
msa
tz a
n P
eptid
[%]
α-Dicarbonylverbindung
Abb. 4.4.2-1: Umsatz an Gly-Ala-Phe nach Reaktion mit Glyoxal, Methylglyoxal, 3-DG oder Diacetyl. Mittelwert ± Standardabweichung (n = 3). (Peptid und α-Dicarbonylverbindung je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 37 °C, 24 h, Durchführung: 3.5.1).
4.4.3 Einfluss der Pufferionenart und Konzentration
Um den Einfluss des Puffers auf die 2(1H)-Pyrazinon-Bildung näher zu untersuchen, wurde
die Inkubation von Gly-Ala-Phe mit Glyoxal in verschiedenen Puffer-Lösungen
vorgenommen. Phosphat-Puffer wurde als Natriumphosphat-Puffer mit einer Konzentration
von 100 mM (Ionenstärke I = 285 mM), 50 mM (I = 143 mM) und 10 mM (I = 29 mM) sowie
als phosphatgepufferte Kochsalz-Lösung (PBS, cPhosphat = 10 mM, I = 167 mM) getestet.
Während die Umsätze im 100 mM und 50 mM Natriumphosphat-Puffer praktisch identisch
waren, wurde mit dem 10 mM Natriumphosphat-Puffer ein signifikant niedriger Umsatz
erzielt (Abb. 4.4.3-1). Die Reaktion in PBS führt zu einem Umsatz, der denen im 100 mM
und 50 mM Natriumphosphat-Puffer vergleichbar war. PBS enthält ebenfalls 10 mM
Phosphat, er weist jedoch eine deutlich höhere Ionenstärke auf. Nach Martell et al. (1998)
bewirkt im unteren Konzentrationsbereich eine Erhöhung der Ionenstärke eine leichte
Absenkung des pKa-Wertes der α-Aminogruppe, so dass in einem Puffer höherer Ionenstärke
der Anteil an unprotonierten und damit reaktiven α-Aminogruppen größer ist. Folglich dürfte
die Reaktionsgeschwindigkeit im untersuchten Konzentrationsbereich nicht primär durch die
Phosphatkonzentration sondern vielmehr durch die Ionenstärke beeinflusst sein. Die
2(1H)-Pyrazinon-Bildung unterliegt damit keiner konzentrationsabhängigen katalytischen
- 135 -
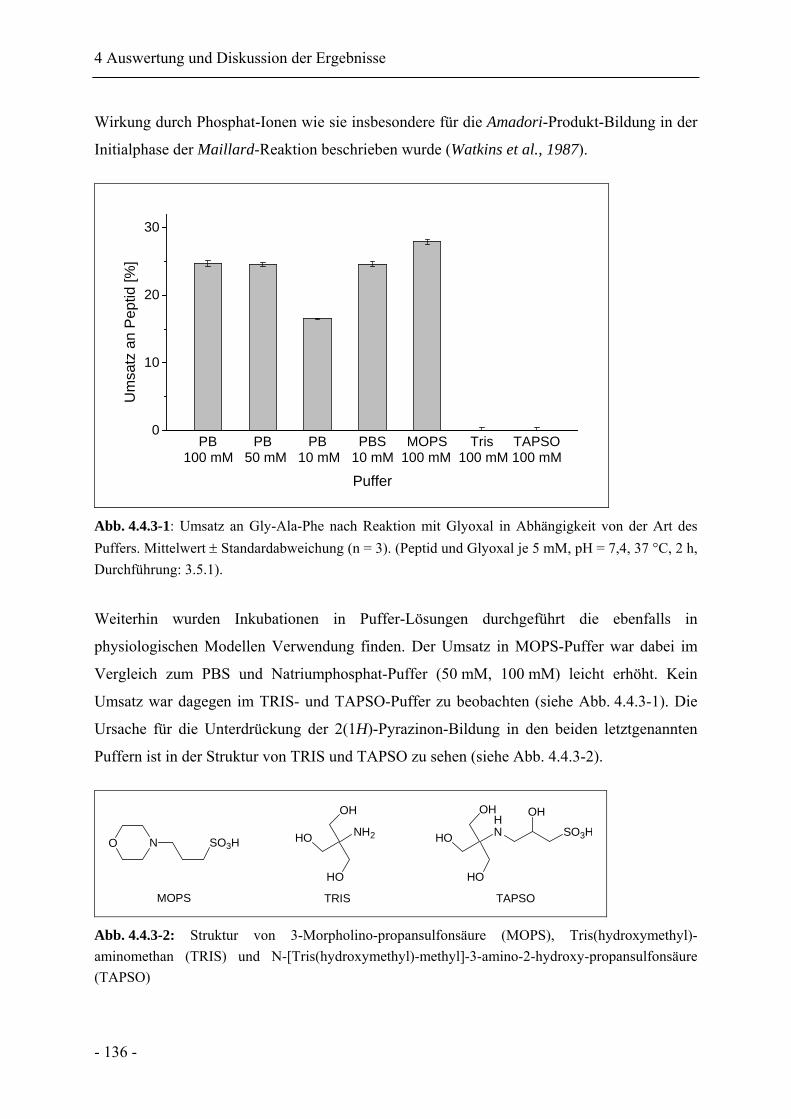
4 Auswertung und Diskussion der Ergebnisse
Wirkung durch Phosphat-Ionen wie sie insbesondere für die Amadori-Produkt-Bildung in der
Initialphase der Maillard-Reaktion beschrieben wurde (Watkins et al., 1987).
PB PB PB PBS MOPS Tris TAPSO0
10
20
30
100 mM 50 mM 10 mM 10 mM 100 mM 100 mM 100 mM
Um
satz
an
Pep
tid [%
]
Puffer
Abb. 4.4.3-1: Umsatz an Gly-Ala-Phe nach Reaktion mit Glyoxal in Abhängigkeit von der Art des Puffers. Mittelwert ± Standardabweichung (n = 3). (Peptid und Glyoxal je 5 mM, pH = 7,4, 37 °C, 2 h, Durchführung: 3.5.1).
Weiterhin wurden Inkubationen in Puffer-Lösungen durchgeführt die ebenfalls in
physiologischen Modellen Verwendung finden. Der Umsatz in MOPS-Puffer war dabei im
Vergleich zum PBS und Natriumphosphat-Puffer (50 mM, 100 mM) leicht erhöht. Kein
Umsatz war dagegen im TRIS- und TAPSO-Puffer zu beobachten (siehe Abb. 4.4.3-1). Die
Ursache für die Unterdrückung der 2(1H)-Pyrazinon-Bildung in den beiden letztgenannten
Puffern ist in der Struktur von TRIS und TAPSO zu sehen (siehe Abb. 4.4.3-2).
HN SO3HHO
OH
HO
OH
NH2HO
OH
HO
NO SO3H
MOPS TRIS TAPSO
Abb. 4.4.3-2: Struktur von 3-Morpholino-propansulfonsäure (MOPS), Tris(hydroxymethyl)-aminomethan (TRIS) und N-[Tris(hydroxymethyl)-methyl]-3-amino-2-hydroxy-propansulfonsäure (TAPSO)
- 136 -
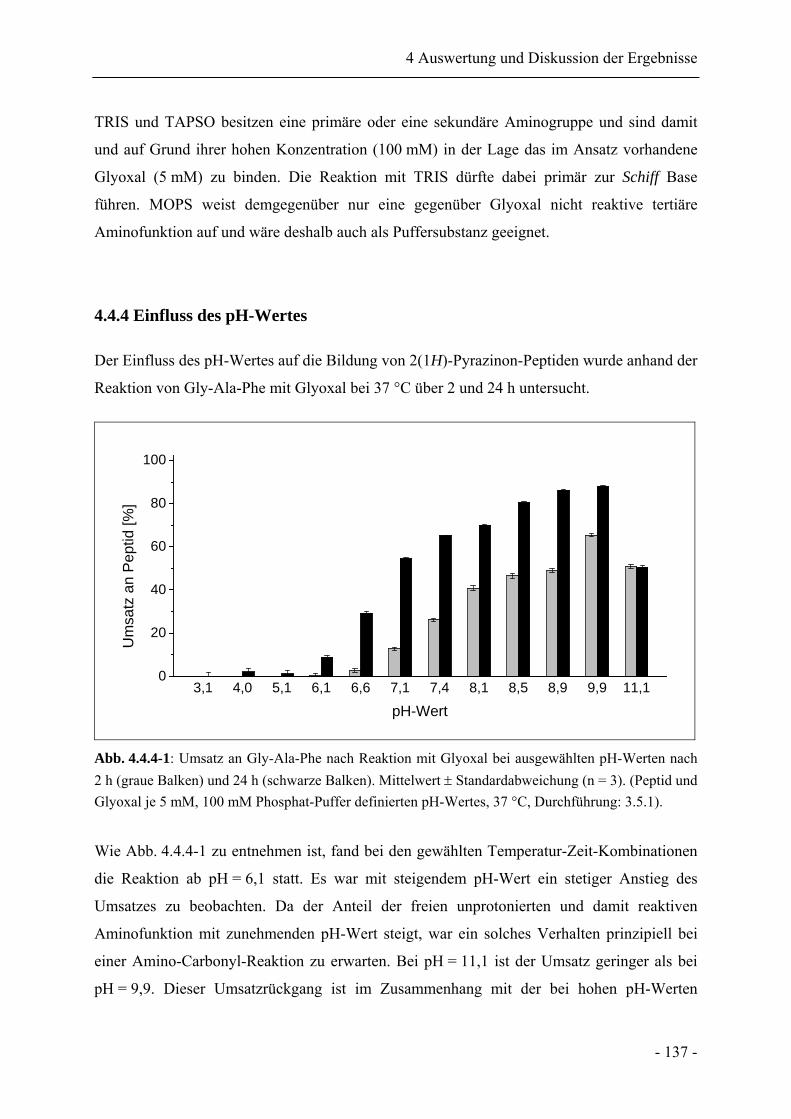
4 Auswertung und Diskussion der Ergebnisse
TRIS und TAPSO besitzen eine primäre oder eine sekundäre Aminogruppe und sind damit
und auf Grund ihrer hohen Konzentration (100 mM) in der Lage das im Ansatz vorhandene
Glyoxal (5 mM) zu binden. Die Reaktion mit TRIS dürfte dabei primär zur Schiff Base
führen. MOPS weist demgegenüber nur eine gegenüber Glyoxal nicht reaktive tertiäre
Aminofunktion auf und wäre deshalb auch als Puffersubstanz geeignet.
4.4.4 Einfluss des pH-Wertes
Der Einfluss des pH-Wertes auf die Bildung von 2(1H)-Pyrazinon-Peptiden wurde anhand der
Reaktion von Gly-Ala-Phe mit Glyoxal bei 37 °C über 2 und 24 h untersucht.
3,1 4,0 5,1 6,1 6,6 7,1 7,4 8,1 8,5 8,9 9,9 11,10
20
40
60
80
100
Um
satz
an
Pep
tid [%
]
pH-Wert
Abb. 4.4.4-1: Umsatz an Gly-Ala-Phe nach Reaktion mit Glyoxal bei ausgewählten pH-Werten nach 2 h (graue Balken) und 24 h (schwarze Balken). Mittelwert ± Standardabweichung (n = 3). (Peptid und Glyoxal je 5 mM, 100 mM Phosphat-Puffer definierten pH-Wertes, 37 °C, Durchführung: 3.5.1).
Wie Abb. 4.4.4-1 zu entnehmen ist, fand bei den gewählten Temperatur-Zeit-Kombinationen
die Reaktion ab pH = 6,1 statt. Es war mit steigendem pH-Wert ein stetiger Anstieg des
Umsatzes zu beobachten. Da der Anteil der freien unprotonierten und damit reaktiven
Aminofunktion mit zunehmenden pH-Wert steigt, war ein solches Verhalten prinzipiell bei
einer Amino-Carbonyl-Reaktion zu erwarten. Bei pH = 11,1 ist der Umsatz geringer als bei
pH = 9,9. Dieser Umsatzrückgang ist im Zusammenhang mit der bei hohen pH-Werten
- 137 -

4 Auswertung und Diskussion der Ergebnisse
stattfindenden Disproportionierung des Glyoxals zu Hydroxyessigsäure und Glyoxylsäure
(Cannizzaro-Reaktion) zu sehen. In den Reaktionsansätzen war ausschließlich das als
2(1H)-Pyrazinon modifizierte Peptid als Produkt zu detektieren, so dass die gemessenen
Umsätze der Bildungsrate des 2(1H)-Pyrazinons entsprechen. Es kann somit auf eine hohe
Spezifität der Reaktion in einem weiten pH-Bereich geschlossen werden.
Auf Grund der Ergebnisse sollten mit Basen behandelte Lebensmittel wie z. B. Laugengebäck
oder aufgeschlossenes Kakaopulver für eine 2(1H)-Pyrazinon-Bildung prädestiniert sein. Um
Anhaltspunkte über die 2(1H)-Pyrazinon-Bildung in den mengenmäßig dominierenden
Lebensmitteln mit schwach saurem pH-Wert zu erhalten, müssten insbesondere Experimente
bei technologisch relevanteren höheren Temperaturen durchgeführt werden.
Die Ergebnisse zeigen ferner, dass das Abstoppen der Inkubationen mit 100 mM oder
200 mM Ameisensäure, was zu einer Absenkung des pH-Wertes auf pH = 3 bis 4 führt, für
die Analytik sehr gut geeignet ist.
4.5 Die Reaktion des N-Terminus von Peptiden mit Methylglyoxal
4.5.1 Die Reaktion von Gly-Ala-Phe mit Methylglyoxal
Die Reaktion von Peptiden mit Methylglyoxal wurde näher anhand des Peptides Gly-Ala-Phe
unter physiologischen Bedingungen (pH = 7,4, 37 °C) untersucht. Wie bereits bei den
Inkubationen mit Glyoxal konnte eine sehr schnelle Derivatisierung des Peptides festgestellt
werden, z. B. 50 % Umsatz nach 8 h. Gegenüber den Umsetzungen mit Glyoxal war das
Produktspektrum jedoch komplexer (siehe Abb. 4.5.1-1). Der Hauptproduktpeak zeigte
jedoch die für 2(1H)-Pyrazinone typische Absorption bei λ = 320 nm sowie Fluoreszenz
(λex = 320 nm, λem = 400 nm) und es konnte die Dominanz eines solchen Produktes vermutet
werden.
- 138 -

4 Auswertung und Diskussion der Ergebnisse
0 10 20 30 40
0,0
0,2
0,4
0,6Fluoreszenzex. 320 nmem. 400 nm
Gly-Ala-Phe
Produkt
UV 320 nm
UV 220 nm
Abs
orpt
ion,
Flu
ores
zenz
[AU
]
Zeit [min]
Abb. 4.5.1-1: HPLC-Chromatogramme der Umsetzung von Gly-Ala-Phe mit Methylglyoxal. (Peptid und Methylglyoxal je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 37 °C, 8 h, Durchführung: 3.5.1).
Die kinetischen Betrachtungen zur Reaktion mit Methylglyoxal weisen ebenfalls auf ein
komplizierteres Reaktionsgeschehen hin (Abb. 4.5.1-2). Bis zu einer Reaktionszeit von 96 h
ist bei der Reaktion mit Methylglyoxal der zeitliche Verlauf der Peptid-Derivatisierung der
Umsetzung mit Glyoxal (siehe Abb. 4.3.1-2) ähnlich. Anschließend ist jedoch ein annähernd
linearer Abbau festzustellen. Bei Betrachtung bis zu 240 h lässt sich die Reaktion dennoch gut
als Reaktion zweiter Ordnung beschreiben (Abb. 4.5.1-2, B). Die Produkt-Bildungskurve
weist wie bei Glyoxal einen asymptotischen Verlauf auf. Der Umfang der Bildung des
Hauptproduktes ist jedoch deutlich geringer und die Produkt-Bildung stagniert bereits nach
48 h bis 72 h. Da nach 336 h eine leichte Abnahme an Produkt festzustellen ist, unterliegt
dieses offenbar Folgereaktionen.
- 139 -
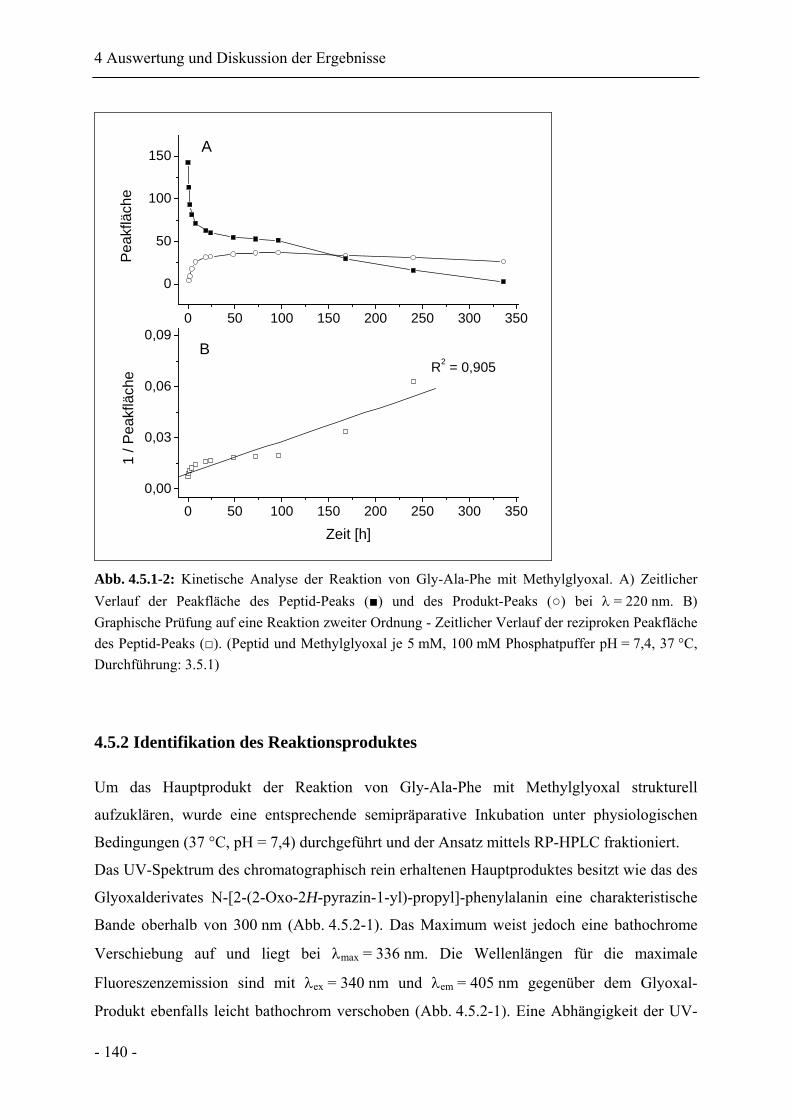
4 Auswertung und Diskussion der Ergebnisse
0 50 100 150 200 250 300 3500,00
0,03
0,06
0,090 50 100 150 200 250 300 350
0
50
100
150
BR2 = 0,905
1 / P
eakf
läch
e
Zeit [h]
AP
eakf
läch
e
Abb. 4.5.1-2: Kinetische Analyse der Reaktion von Gly-Ala-Phe mit Methylglyoxal. A) Zeitlicher Verlauf der Peakfläche des Peptid-Peaks (■) und des Produkt-Peaks (○) bei λ = 220 nm. B) Graphische Prüfung auf eine Reaktion zweiter Ordnung - Zeitlicher Verlauf der reziproken Peakfläche des Peptid-Peaks (□). (Peptid und Methylglyoxal je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 37 °C, Durchführung: 3.5.1)
4.5.2 Identifikation des Reaktionsproduktes
Um das Hauptprodukt der Reaktion von Gly-Ala-Phe mit Methylglyoxal strukturell
aufzuklären, wurde eine entsprechende semipräparative Inkubation unter physiologischen
Bedingungen (37 °C, pH = 7,4) durchgeführt und der Ansatz mittels RP-HPLC fraktioniert.
Das UV-Spektrum des chromatographisch rein erhaltenen Hauptproduktes besitzt wie das des
Glyoxalderivates N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin eine charakteristische
Bande oberhalb von 300 nm (Abb. 4.5.2-1). Das Maximum weist jedoch eine bathochrome
Verschiebung auf und liegt bei λmax = 336 nm. Die Wellenlängen für die maximale
Fluoreszenzemission sind mit λex = 340 nm und λem = 405 nm gegenüber dem Glyoxal-
Produkt ebenfalls leicht bathochrom verschoben (Abb. 4.5.2-1). Eine Abhängigkeit der UV-
- 140 -

4 Auswertung und Diskussion der Ergebnisse
und Fluoreszenz-Charakteristika vom pH-Wert war im untersuchten Bereich von pH = 2,4
(100 mM Ameisensäure) bis pH = 8,3 (100 mM Borat-Puffer) nicht festzustellen. Damit ist
anzunehmen, das der Chromophor und Fluorophor keine leicht ionisierbaren funktionellen
Gruppen aufweist.
Mittels direkter ESI-MS Analyse konnte die relative monoisotopische Masse des Produktes
zu Mr = 329,1 bestimmt werden. Wie bei der Reaktion mit Glyoxal ist folglich von einer
Reaktion von einem Molekül Peptid mit einem Molekül Methylglyoxal unter Elimination von
zwei Molekülen Wasser auszugehen.
200 300 400 5000,0
0,2
0,4
0,6
0,8
1,0
1,2
200 300 400 500 600
A
Extin
ktio
n
Wellenlänge [nm]
BR
elat
ive
Fluo
resz
enze
mis
sion
Wellenlänge [nm]
Abb. 4.5.2-1: Absorptionsspektrum (A) und Fluoreszenzemissionsspektrum (λex, max = 330 nm, B) des Methylglyoxal-Gly-Ala-Phe-Reaktionsproduktes. Aufgenommen in 100 mM Phosphat-Puffer pH = 7,0.
Die chemische Struktur des Reaktionsproduktes konnte anhand der entsprechenden ein- und
zweidimensionale NMR-Experimente (1H, 13C, 1H-1H COSY, DEPT, HSQC, HMBC) als
N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin (Abb. 4.5.2-2) aufgeklärt
werden und ist konsistent mit der massenspektroskopischen Analyse. Es handelt sich damit
wie das Produkt der Umsetzung mit Glyoxal um ein 2(1H)-Pyrazinon-Derivat.
Bei der Struktur-Aufklärung stellte sich insbesondere die Frage, ob sich die vom
Methylglyoxal stammende Methylgruppe an Position 5 oder 6 am 2(1H)-Pyrazinon-Ring
- 141 -

4 Auswertung und Diskussion der Ergebnisse
befindet. Aufschluss gaben hierzu vor allem die im HMBC-Spektrum beobachteten
Kopplungen (Abb. 4.5.2-2). So zeigte das Methinproton vom Alanin nur zum C6-Atom des
2(1H)-Pyrazinon-Rings und nicht zum C5-Atom eine Kopplung. Die Protonen der Ring-
Methylgruppe koppelten sowohl mit C5 als auch mit C6. Anhand dessen war eindeutig
festzustellen, dass sich die Methylgruppe an Position 5 befindet.
H
N
NO
CH3
H
H3C
H1
3
6HN
OOH
O
4
N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin
Abb. 4.5.2-2: Strukturformel von N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin. Die Pfeile zeigen charakteristische im HMBC Spektrum beobachtete Konnektivitäten an.
Da das 5-Methyl-2(1H)-pyrazinon das Hauptprodukt ist und keine Hinweise auf das isomere
6-Methyl-Derivat gefunden wurden, muss die Reaktion im hohen Maß regioselektiv sein. Bei
der Bildungsreaktion (siehe Abschnitt 4.3.4) wird dementsprechend im ersten Schritt die
Ketogruppe und nicht die Aldehydfunktion des Methylglyoxals durch die primäre
Aminogruppe angegriffen. Eine Aldehydfunktion ist im allgemeinen reaktiver als eine
Ketogruppe. Im Fall von Methylglyoxal liegt die Aldehydgruppe in wässriger Lösung jedoch
überwiegend hydratisiert und damit weniger reaktiv vor (Thornalley,1996), so das es offenbar
aufgrund dessen zu der beobachteten Regioselektivität kommt.
Die selektive Bildung von 5-substituierten 2(1H)-Pyrazinon-Derivaten ist nicht nur auf die
angewendeten milden physiologischen Bedingungen begrenzt. Bei Umsetzungen von
Methylglyoxal mit N-(Benzyloxy)-glycinamid (Ohkanda, et al., 1996) oder Phenylglyoxal mit
Glycinamid (Sato, 1978) unter synthetischen Bedingungen im stark alkalischen Milieu
wurden ebenfalls eine solche Regioselektivität festgestellt.
- 142 -

4 Auswertung und Diskussion der Ergebnisse
4.6 Die Reaktion des N-Terminus von Peptiden mit 3-Desoxyglucosulose
Als weiterer potenzieller Reaktionspartner für die α-Aminogruppe von Peptiden wurde die
α-Dicarbonylverbindung 3-Desoxyglucosulose in die Studien einbezogen. Wie bei den
kürzerkettigen α-Dicarbonylverbindungen war in den Inkubationsansätzen mittels RP-HPLC
die Bildung eines Hauptproduktes zu detektieren, welches Absorption bei λ = 320 nm zeigt
(Abb. 4.6-1).
0 10 20 30 400,0
0,2
0,4
0,6
0,8
1,0
UV 220 nm
UV 320 nm
HauptproduktGly-Ala-Phe
Abso
rptio
n [A
U]
Zeit [min]
Abb. 4.6-1: HPLC-Chromatogramme der Umsetzung von Gly-Ala-Phe mit 3-DG. (Peptid und 3-DG je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 60 °C, 3 d, Durchführung: 3.5.1).
Im Vergleich zu den zuvor untersuchten α-Dicarbonylverbindungen erfolgt die Reaktion mit
3-DG jedoch deutlich langsamer. Nach 24 h war z. B. ein Umsatz von ca. 10 % festzustellen
(pH = 7,4, 37 °C). Der zeitliche Verlauf der Derivatisierung des Peptides (Abb. 4.6-2) weist
ähnlich wie bei Methylglyoxal auf ein komplexeres Reaktionsgeschehen mit zunehmender
Reaktionszeit hin. Auf Grund dessen ist die Reaktion nicht mehr mit einem einfachen
Geschwindigkeits-Zeit-Gesetz sinnvoll zu beschreiben. Die Auswertung in Hinblick auf eine
- 143 -

4 Auswertung und Diskussion der Ergebnisse
Reaktion zweiter Ordnung liefert z. B. lediglich ein Bestimmtheitsmaß von R2 = 0,84 (Daten
bis 240 h) oder R2 = 0,67 (Daten bis 336 h). Hinzu kommt, dass nach 240 h erst ein Umsatz
von 58 % erreicht wird und bei Umsätzen bis 50 % und einem analytischen Fehler von ± 5 %
die Bestimmung der Reaktionsordnung nicht sicher möglich ist (Labuza, 1984). Insbesondere
ab einer Reaktionszeit von 168 h entspricht die Bildung des Hauptproduktes nicht mehr der
Peptid-Derivatisierung. Im Gegensatz zur Umsetzung mit Methylglyoxal kommt es jedoch im
untersuchten Zeitfenster bis 336 h nicht zur Stagnation der Bildung des Hauptproduktes.
0 50 100 150 200 250 300 350
0.000
0.005
0.010
0.015
0 50 100 150 200 250 300 350
0
50
100
150
200
R2 = 0,844
B
1 / P
eakf
läch
e
Zeit [h]
A
Pea
kflä
che
Abb. 4.6-2: Kinetische Analyse der Reaktion von Gly-Ala-Phe mit 3-DG. A) Zeitlicher Verlauf der Peakfläche des Peptid-Peaks (■) und des Produkt-Peaks (○) bei λ = 220 nm. B) Graphische Prüfung auf eine Reaktion zweiter Ordnung - Zeitlicher Verlauf der reziproken Peakfläche des Peptid-Peaks (□). (Peptid und 3-DG je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 37 °C, Durchführung: 3.5.1)
Das UV-Spektrum des Hauptproduktes weist eine Bande oberhalb von 300 nm mit einem
Maximum von λUV, max = 334 nm auf. Weiterhin fluoresziert die Verbindung mit
λex, max = 337 nm und λem, max = 402 nm (siehe Abb. 4.6.-3). Beide Merkmale wurden bereits
bei N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin (siehe Abschnitt 4.3.2) und
- 144 -

4 Auswertung und Diskussion der Ergebnisse
N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin (siehe Abschnitt 4.5.2)
festgestellt und sind charakteristisch für 2(1H)-Pyrazinone. Es ist folglich davon auszugehen,
dass es bei der Reaktion der N-Termini von Peptiden mit 3-Desoxyglucosulose ebenfalls zur
Bildung von 2(1H)-Pyrazinon-Peptiden kommt. Die weitere Charakterisierung und der
spektroskopische Nachweis der Struktur dieser Produkte sollten Gegenstand weiterer
Untersuchungen sein.
200 300 400
0,0
0,1
0,2
0,3
0,4
200 300 400 500 600
A
Extin
ktio
n
Wellenlänge [nm]
B
Rel
ativ
e Fl
uore
szen
zem
issi
on
Wellenlänge [nm]
Abb. 4.6-3: Absorptionsspektrum (A) und Fluoreszenzemissionsspektrum (λex, max = 337 nm, B) des Hauptproduktes der Reaktion von Gly-Ala-Phe mit 3-DG. Peak bei HPLC mit tR = 31 min (siehe Abb. 4.6.1-1) gesammelt und Spektren in HPLC-Eluent (Eluenten-System E3, ca. 30 % B, siehe Abschnitt 3.2.1) aufgenommen.
Wie bereits erwähnt erfolgte sowohl die Derivatisierung von Gly-Ala-Phe als auch von
anderen Peptiden (siehe auch Abb. 4.2-1) durch 3-Desoxyglucosulose langsamer als durch
Glyoxal und Methylglyoxal. Die in vivo gemessenen Gehalte an Glyoxal, Methylglyoxal und
3-Desoxyglucosulose bewegen sich im µmol/kg-Bereich und in der gleichen Größenordnung
(siehe Tab. 2.3.1-1). Die in Lebensmitteln bestimmten Gehalte an Glyoxal und Methylglyoxal
bewegen sich ebenfalls in diesem Bereich (siehe Tab. 2.3.1-2). Über die Gehalte von
3-Desoxyglucosulose in Lebensmitteln ist demgegenüber vergleichsweise wenig bekannt.
Weigel et al. (2004) bestimmten relativ hohe Gehalte von 1110 µmol/kg (Median) in Honig.
Von Rättich (2004) wurden Gehalte an 3-Desoxyglucosulose in Kaffeesahne, Kondensmilch
und Kakaomilch von ca. 3 µmol/l und in Malzbier von ca. 130 µmol/l ermittelt. Damit sind in
- 145 -

4 Auswertung und Diskussion der Ergebnisse
Lebensmitteln Gehalte an 3-Desoxyglucosulose messbar die auch für Glyoxal und
Methylglyoxal gefunden werden. Bei der Beurteilung der bestimmten Gehalte an
α-Dicarbonylverbindungen ist zu berücksichtigen, dass bei der Bestimmung nur die freien
und ggf. die leicht reversibel gebundenen α-Dicarbonylverbindungen erfasst werden.
α-Dicarbonylverbindungen gehen aufgrund ihrer hohen Reaktivität nach der Bildung schnell
Folgereaktionen ein. Die bestimmten Gehalte geben deshalb nur begrenzt Auskunft über die
insgesamt auftretenden Mengen. Zusätzlich hat die Probenvorbereitung zur Bestimmung der
Gehalte einen erheblichen Einfluss auf die Ergebnisse (Lal et al., 1997).
Auf Grund von Modelluntersuchungen von Hofmann (1999) ist das Auftreten von
3-Desoxyglucosulose insbesondere bei stärker erhitzten Lebensmitteln zu erwarten. Weiterhin
ist diesen Untersuchungen zu entnehmen, dass unter den Bedingungen der Maillard-Reaktion
3-Desoxyglucosulose aus quantitativer Sicht bedeutender ist als Glyoxal und Methylglyoxal.
In gewissem Umfang ist dies auch vom Bildungsweg der 3-Desoxyglucosulose zu erwarten.
3-Desoxyglucosulose ist ein direktes Abbauprodukt des Amadori-Produktes (Ledl &
Schleicher, 1990) und Amadori-Produkte sind die quantitativ wichtigsten Verbindungen der
Maillard-Reaktion in Lebensmittel und in vivo (Henle, 2003). Die Bildung von
3-Desoxyglucosulose erfolgt ferner auch direkt aus dem Zucker ohne Aminkatalyse (Anet,
1964) und in vivo zusätzlich aus Fructose-3-phosphat (Szwergold et al., 1990). Fructose-
3-phosphat entsteht bei dem enzymatischen Abbau von Fructoselysin durch das deglykierende
Enzym Fructosamin-3-kinase (Delpierre et al., 2002). Als Folgeprodukte der 3-Desoxy-
glucosulose sind z. B. Hydroxymethylfurfural in Lebensmitteln (Van Boekel, 1998, Sanz et
al., 2003) sowie Pyrralin in Lebensmitteln und in vivo von Bedeutung (Hayase, et al. 1989,
Förster & Henle, 2003). In Hinblick auf die Quantität der Reaktionsprodukte der einzelnen
α-Dicarbonylverbindungen ist damit insgesamt betrachtet zu vermuten, dass die etwas
geringere Reaktivität der 3-Desoxyglucosulose durch ihre höhere Bildungsrate ausgeglichen
wird. Somit könnten N-terminale 2(1H)-Pyrazinone des Glyoxals und Methylglyoxals auf
Grund der hohen Reaktivität und 2(1H)-Pyrazinone der 3-Desoxyglucosulose auf Grund der
hohen Quantität der Ausgangsdicarbonylverbindungen sowohl in Lebensmittel als auch
in vivo von Bedeutung sein.
- 146 -

4 Auswertung und Diskussion der Ergebnisse
4.7 Einordnung der Reaktivität des N-Terminus von Peptiden gegenüber
α-Dicarbonylverbindung
In den vorangegangenen Inkubationsexperimenten wurde unter den gewählten
physiologischen Modell-Bedingungen eine hohe Reaktivität der α-Aminogruppe von
Peptiden gegenüber α-Dicarbonylverbindungen festgestellt. In der Literatur werden die
nucleophilen Seitenketten von Arginin und Lysin als die Hauptreaktionspartner für
α-Dicarbonylverbindungen an Proteinen angesehen, wobei Arginin ein höheres Reaktions-
vermögen zugeschrieben wird (siehe Abschnitt 2.3.2, Nakaya, et al. 1967, Takahashi,
1977a, b).
Um die Reaktivität der α-Aminogruppe von Peptiden gegenüber den bekannten
Reaktionspartnern beurteilen zu können, wurden ausgewählte Peptide mit Glyoxal,
Methylglyoxal oder 3-Desoxyglucosulose (3-DG) inkubiert (siehe Abb. 4.7-1 und 4.7-2).
Dabei dienten Gly-Ala-Phe (GAF), Nα-Hippuryllysin (BzGK) und Nα-Hippurylarginin
(BzGR) als Modellpeptide für den N-Terminus, für proteingebundenes Lysin bzw.
proteingebundenes Arginin. Es wurden sowohl separate Ansätze, ein Peptid plus eine
α-Dicarbonylverbindung, als auch kompetitive Ansätze, Gly-Ala-Phe plus Nα-Hippuryl-
arginin plus eine α-Dicarbonylverbindung, angefertigt. Die Konzentration jedes Reaktions-
partners betrug 5 mM und es wurde in 100 mM Phosphatpuffer bei 37 °C für 24 h inkubiert.
0
20
40
60
80
100
separate gemeinsame Inkubationen Inkubation
BzGK BzGR GAF BzGR GAF
Der
ivat
isie
rung
[%]
Abb. 4.7-1: Umsatz an Peptid nach Inkubation mit Glyoxal. (Peptide und Glyoxal je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 37 °C, 24 h, Durchführung: 3.5.1).
- 147 -

4 Auswertung und Diskussion der Ergebnisse
Bei den Umsetzungen mit Glyoxal zeigten Nα-Hippurylarginin und Gly-Ala-Phe sowohl bei
separater Inkubation als auch unter kompetiven Bedingungen jeweils eine nahezu gleiche
Reaktivität (siehe Abb. 4.7-1). Eine Lysin-Derivatisierung konnte unter den gewählten
Bedingungen nicht festgestellt werden. Die Inkubation in PBS über 15 d und unter sonst
identischen Bedingungen führte jedoch zu einer gewissen Bildung von Nα-Hippuryl-
Nε-carboxymethyllysin, der Umsatz zum CML-Derivat betrug ca. 0,5 %.
Bei den Inkubationen mit Methylglyoxal zeigte Nα-Hippurylarginin eine deutlich höheres
(separate Inkubationen) oder leicht erhöhtes (gemeinsame Inkubation) Reaktionsvermögen als
Gly-Ala-Phe (siehe Abb. 4.7-2). Mit Nα-Hippuryllysin konnte keine Reaktion beobachtet
werden. Gegenüber 3-DG zeigten Nα-Hippurylarginin und Gly-Ala-Phe sowohl bei separater
als auch bei gemeinsamer Inkubation eine ähnliche Reaktivität (siehe Abb. 4.7-2).
Nα-Hippuryllysin erwies sich wiederum als nicht reaktiv.
0
20
40
60
80
100
separate gemeinsame Inkubationen Inkubation
BzGK BzGR GAF BzGR GAF
Der
ivat
isie
rung
[%]
0
20
40
60
80
100
separate gemeinsame Inkubationen Inkubation
BzGK BzGR GAF BzGR GAF
Der
ivat
isie
rung
[%]
Abb. 4.7-2: Umsatz an Peptid nach Inkubation mit Methylglyoxal (links) oder 3-DG (rechts). (Peptide und α-Dicarbonylverbindung je 5 mM, 100 mM Phosphatpuffer pH = 7,4, 37 °C, 24 h, Durchführung: 3.5.1).
Die Untersuchungen belegen für das gewählte Modellsystem eine ähnliche bis nahezu gleiche
Reaktivität des N-Terminus von Peptiden und der Guanidino-Funktion des Arginins
gegenüber Glyoxal, Methylglyoxal und 3-DG. Lysin zeigte sich demgegenüber als nicht
reaktiv. In vivo als auch in Lebensmitteln wurden bereits zahlreiche Reaktionsprodukte der
untersuchten α-Dicarbonylverbindungen mit Arginin nachgewiesen sowie quantifiziert (siehe
Abschnitt 2.3.2). Folglich ist anzunehmen, dass es ebenfalls zur Derivatisierung der
- 148 -

4 Auswertung und Diskussion der Ergebnisse
N-terminalen α-Aminogruppe und der Bildung von 2(1H)-Pyrazinonen in diesen Systemen
kommt.
Als ein weiteres Indiz für diese These ist die charakteristische Fluoreszenz der untersuchten
.8 Analyse peptidgebundener 2(1H)-Pyrazinone nach Salzsäurehydrolyse
4.8.1 Salzsäurehydrolyse von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenyl-
Um peptid- und proteingebundene 2(1H)-Pyrazinone in Lebensmitteln und in in vivo-
ie
2(1H)-Pyrazinone zu werten (λex, max = 330 nm bis 340 nm, λem, max = 395 nm bis 405 nm).
Mit dem Fortschreiten der Maillard-Reaktion wird im allgemeinen eine Zunahme der
Fluoreszenz beobachtet. Die Messung dieser Maillard-spezifischen Fluoreszenz wird oft
genutzt um den Fortschritt von Glykierungsreaktionen zu beurteilen. Über die Struktur der
verantwortlichen Fluorophore ist jedoch nur zum Teil bekannt (Kollias et al., 1998, Avendano
et al., 1999, Birlouez-Aragon, et al. 2001). Es ist deshalb zu vermuten, dass ein Teil der in
in vivo und in thermisch behandelten Lebensmitteln beobachteten Fluoreszenz auf die
Bildung von 2(1H)-Pyrazinonen zurückzuführen ist. Weitere Studien sollten sich deshalb mit
dem Nachweis von 2(1H)-Pyrazinonen in in vivo-Proben und in Lebensmitteln befassen.
4
alanin
Materialien spezifisch nachweisen und bestimmen zu können, ist eine reproduzierbare
Freisetzung der 2(1H)-Pyrazinon-Struktur oder eines Folgeproduktes aus dem Peptid- bzw.
Proteinverband notwendig. Ob dies durch eine Säurehydrolyse möglich ist, wurde anhand des
2(1H)-Pyrazinon-Peptides N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin geprüft.
Wie dem in Abb. 4.8.1-1 gezeigten Aminosäurechromatogramm zu entnehmen ist, sind d
Gehalte an Glycin und Alanin in einem Hydrolysat von N-[2-(2-Oxo-2H-pyrazin-1-yl)-
propionyl]-phenylalanin deutlich geringer als der Gehalt an Phenylalanin. Die Hydrolyse der
2(1H)-Pyrazinon-Struktur zu den Ausgangsaminosäuren ist folglich nicht vollständig. Damit
ist zu vermuten, dass der 2(1H)-Pyrazinon-Ring zu einem nicht unerheblichen Anteil intakt
geblieben ist oder dass ein Abbau zu anderen mit Ninhydrin nicht anfärbbaren Verbindungen
stattgefunden hat. Weiterhin ist eine zeitliche Abhängigkeit des Abbaus während der
Hydrolyse festzustellen. So steigt die Wiederfindung an Glycin (bezogen auf Phenylalanin)
- 149 -

4 Auswertung und Diskussion der Ergebnisse
mit 15,5 % (16 h Hydrolysezeit), 21,0 % (23 h) oder 26,0 % (48 h) und die von Alanin mit
29,6 % (16 h), 35,0 % (23 h) oder 40,2 % (48 h) mit zunehmender Hydrolysezeit.
0 10 20 30 40 50 600,0
0,1
0,2
0,3
0,4
Ammoniak
Phe
AlaGlyA
bsor
ptio
n (λ
= 5
70 n
m) [
AU
]
Zeit [min]
Abb. 4.8.1-1: Aminosäurechromatogramm eines Salzsäurehydrolysates von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin. (6 M Salzsäure, 110 °C, 23 h, Durchführung: 3.2.3, 3.6.1).
Um im UV absorbierende sowie fluoreszierende Abbauprodukte zu detektieren, wurden die
Hydrolysate mittels RP-HPLC untersucht. Dazu wurde zunächst wie zur Aminosäureanalyse
üblich die Salzsäure aus den Hydrolysaten im Vakuum am Zentrifugalverdampfer entfernt.
Anschließend wurden die Proben in HPLC-Eluent gelöst. Die HPLC-Chromatogramme der
auf diese Weise aufbereiteten Proben unterschieden sich jedoch wesentlich von den HPLC-
Chromatogrammen bei denen der Salzsäureüberschuss durch Zugabe von Citrat-Puffer
beseitigt wurde (siehe Abb. 4.8.1-2).
In den mit Citrat-Puffer behandelten Proben waren zwei zusätzliche Peaks detektierbar,
welche die für 2(1H)-Pyrazinone charakteristische Absorption bei λ = 320 nm sowie
Fluoreszenz zeigten. Es war somit davon auszugehen, dass es sich um Abbauprodukte mit
2(1H)-Pyrazinon-Struktur handelt, welche im Zentrifugalverdampfer flüchtig sind oder einen
weiteren Abbau unterliegen. Bei den folgenden Untersuchungen wurde deshalb der
Salzsäureüberschuss durch Zugabe von Citrat-Puffer entfernt.
- 150 -
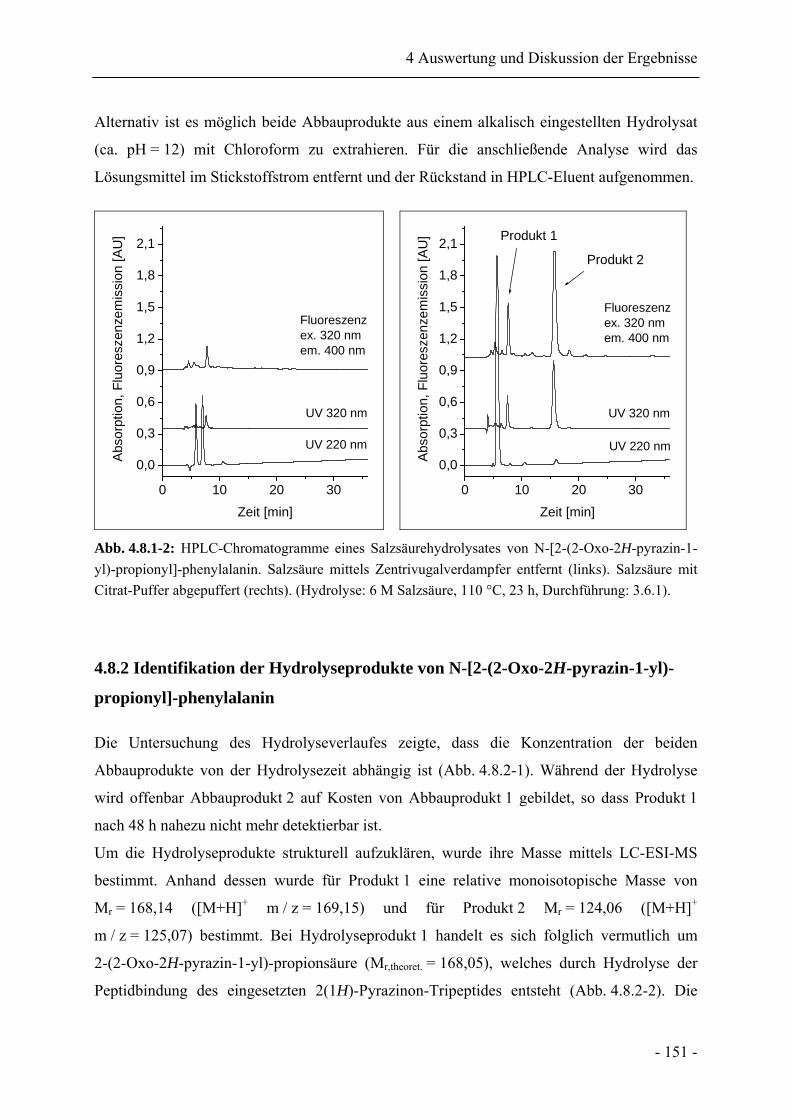
4 Auswertung und Diskussion der Ergebnisse
Alternativ ist es möglich beide Abbauprodukte aus einem alkalisch eingestellten Hydrolysat
(ca. pH = 12) mit Chloroform zu extrahieren. Für die anschließende Analyse wird das
Lösungsmittel im Stickstoffstrom entfernt und der Rückstand in HPLC-Eluent aufgenommen.
0 10 20 30
0,0
0,3
0,6
0,9
1,2
1,5
1,8
2,1
Fluoreszenzex. 320 nmem. 400 nm
UV 320 nm
UV 220 nm
Abs
orpt
ion,
Flu
ores
zenz
emis
sion
[AU
]
Zeit [min]
0 10 20 30
0,0
0,3
0,6
0,9
1,2
1,5
1,8
2,1 Produkt 1
Produkt 2
Fluoreszenzex. 320 nmem. 400 nm
UV 320 nm
UV 220 nm
Abs
orpt
ion,
Flu
ores
zenz
emis
sion
[AU
]
Zeit [min]
Abb. 4.8.1-2: HPLC-Chromatogramme eines Salzsäurehydrolysates von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin. Salzsäure mittels Zentrivugalverdampfer entfernt (links). Salzsäure mit Citrat-Puffer abgepuffert (rechts). (Hydrolyse: 6 M Salzsäure, 110 °C, 23 h, Durchführung: 3.6.1).
4.8.2 Identifikation der Hydrolyseprodukte von N-[2-(2-Oxo-2H-pyrazin-1-yl)-
propionyl]-phenylalanin
Die Untersuchung des Hydrolyseverlaufes zeigte, dass die Konzentration der beiden
Abbauprodukte von der Hydrolysezeit abhängig ist (Abb. 4.8.2-1). Während der Hydrolyse
wird offenbar Abbauprodukt 2 auf Kosten von Abbauprodukt 1 gebildet, so dass Produkt 1
nach 48 h nahezu nicht mehr detektierbar ist.
Um die Hydrolyseprodukte strukturell aufzuklären, wurde ihre Masse mittels LC-ESI-MS
bestimmt. Anhand dessen wurde für Produkt 1 eine relative monoisotopische Masse von
Mr = 168,14 ([M+H]+ m / z = 169,15) und für Produkt 2 Mr = 124,06 ([M+H]+
m / z = 125,07) bestimmt. Bei Hydrolyseprodukt 1 handelt es sich folglich vermutlich um
2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure (Mr,theoret. = 168,05), welches durch Hydrolyse der
Peptidbindung des eingesetzten 2(1H)-Pyrazinon-Tripeptides entsteht (Abb. 4.8.2-2). Die
- 151 -
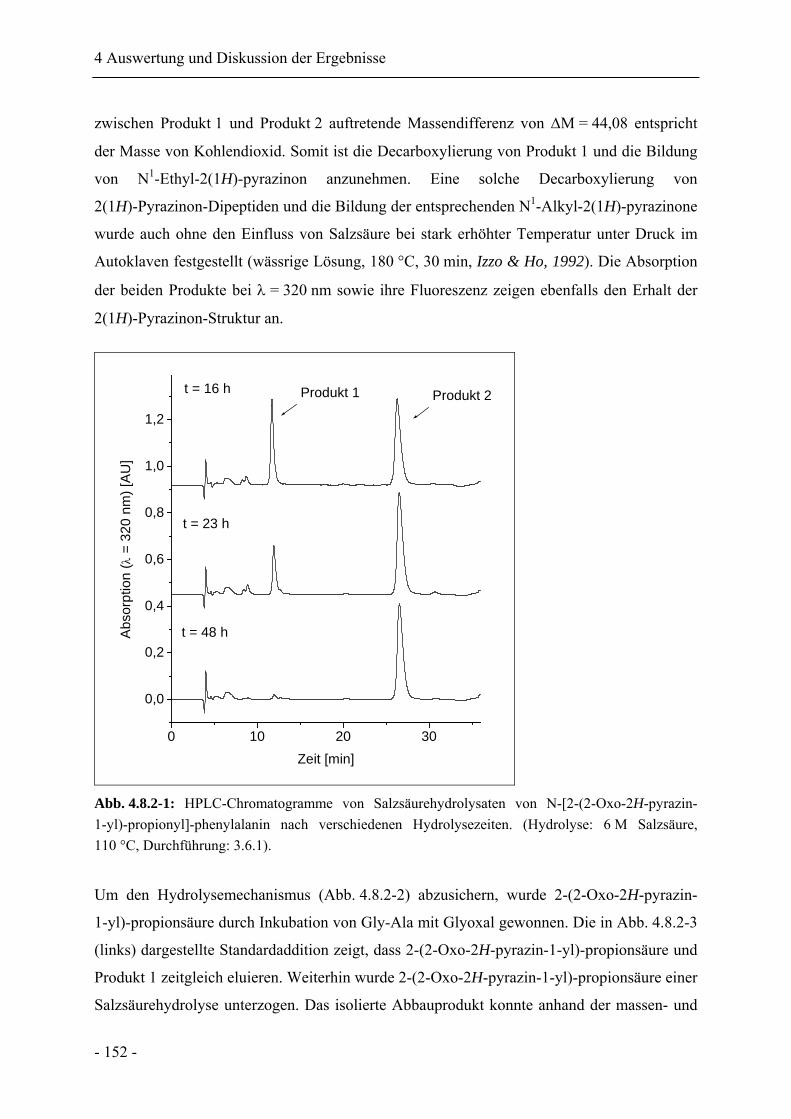
4 Auswertung und Diskussion der Ergebnisse
zwischen Produkt 1 und Produkt 2 auftretende Massendifferenz von ∆M = 44,08 entspricht
der Masse von Kohlendioxid. Somit ist die Decarboxylierung von Produkt 1 und die Bildung
von N1-Ethyl-2(1H)-pyrazinon anzunehmen. Eine solche Decarboxylierung von
2(1H)-Pyrazinon-Dipeptiden und die Bildung der entsprechenden N1-Alkyl-2(1H)-pyrazinone
wurde auch ohne den Einfluss von Salzsäure bei stark erhöhter Temperatur unter Druck im
Autoklaven festgestellt (wässrige Lösung, 180 °C, 30 min, Izzo & Ho, 1992). Die Absorption
der beiden Produkte bei λ = 320 nm sowie ihre Fluoreszenz zeigen ebenfalls den Erhalt der
2(1H)-Pyrazinon-Struktur an.
0 10 20 30
0,0
0,2
0,4
0,6
0,8
1,0
1,2
Produkt 2Produkt 1t = 16 h
t = 23 h
t = 48 hAbs
orpt
ion
(λ =
320
nm
) [A
U]
Zeit [min]
Abb. 4.8.2-1: HPLC-Chromatogramme von Salzsäurehydrolysaten von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin nach verschiedenen Hydrolysezeiten. (Hydrolyse: 6 M Salzsäure, 110 °C, Durchführung: 3.6.1).
Um den Hydrolysemechanismus (Abb. 4.8.2-2) abzusichern, wurde 2-(2-Oxo-2H-pyrazin-
1-yl)-propionsäure durch Inkubation von Gly-Ala mit Glyoxal gewonnen. Die in Abb. 4.8.2-3
(links) dargestellte Standardaddition zeigt, dass 2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure und
Produkt 1 zeitgleich eluieren. Weiterhin wurde 2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure einer
Salzsäurehydrolyse unterzogen. Das isolierte Abbauprodukt konnte anhand der massen- und
- 152 -

4 Auswertung und Diskussion der Ergebnisse
NMR-spektroskopischen Daten zweifelsfrei als das erwartete N1-Ethyl-2(1H)-pyrazinon
aufgeklärt werden (siehe Abschnitt 3.6.2). Die Zuspritzung des semipräparativ gewonnenen
N1-Ethyl-2(1H)-pyrazinon zum Hydrolysat von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-
phenylalanin (Abb. 4.8.2-3) zeigt die Identität des N1-Ethyl-2(1H)-pyrazinons mit Produkt 2
an.
NHOH
O
O
N
N
O
OH
O
N
N
O N
N
O
N1-Ethyl-2(1H)-pyrazinon(1-Ethyl-2H-oxo-2H-pyrazin)
2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure
N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin
- Phe - CO2
Abb. 4.8.2-2: Salzsäurehydrolyse von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin.
0 10 20 30
0,0
0,2
0,4
0,6
0,8
1,0
C
B
A Produkt 1
Abso
rptio
n (λ
= 3
20 n
m) [
AU
]
Zeit [min]
0 10 20 30
0,0
0,2
0,4
0,6
F
E
D Produkt 2
Abso
rptio
n (λ
= 3
20 n
m) [
AU
]
Zeit [min]
Abb. 4.8.2-3: HPLC-Chromatogramme zur Identifikation der Abbauprodukte im Hydrolysat von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin. Links: Standardaddition (A) von 2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure (B) zu Hydrolysat von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin (C). Rechts: Standardaddition (D) von N1-Ethyl-2(1H)-pyrazinon (E) zu Hydrolysat von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin (F).
Um zu prüfen ob es bei der Hydrolyse von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-
phenylalanin auch zur Bildung des am Ringstickstoffatom nicht substituierten
- 153 -

4 Auswertung und Diskussion der Ergebnisse
2(1H)-Pyrazinon kommt, wurde diese Verbindung im analytischen Maßstab durch Inkubation
von Glycinamid mit Glyoxal gewonnen. Die Bildung wurde mittels LC-ESI-MS ([M+H]+
m / z = 97,05, Mr = 96,04, Mr, theoretisch = 96,03) bestätigt. 2(1H)-Pyrazinon eluierte bei der
HPLC bei tR = 8,5 min (Trennbedingungen wie in Abb. 4.8.2-3) und zeigte Absorption bei
λ = 320 nm sowie Fluoreszenz. In den Hydrolysaten von N-[2-(2-Oxo-2H-pyrazin-1-yl)-
propionyl]-phenylalanin sowie von 2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure konnte dieser
Peak nicht detektiert werden, auch nicht mittels massenspektroskopischer Detektion. Folglich
findet bei der Hydrolyse kein Abbau zu 2(1H)-Pyrazinon statt.
Insgesamt ist damit festzustellen, dass die bestimmten Massen der Hydrolyseprodukte, der
strukturelle Nachweis von N1-Ethyl-2(1H)-pyrazinon als Folgeprodukt von 2-(2-Oxo-
2H-pyrazin-1-yl)-propionsäure sowie die gezeigten Standardadditionen eindeutig den in
Abb. 4.8.2-2 gezeigten Hydrolysemechanismus belegen.
4.8.3 Salzsäurehydrolyse von N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propionyl]-
phenylalanin
Bei der Hydrolyse von N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin war
ein ähnliches Verhalten wie bei der von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-
phenylalanin festzustellen. Anhand der für 2(1H)-Pyrazinone charakteristischen Absorption
bei λ = 320 nm sowie Fluoreszenz ist darauf zu schließen, dass drei Abbauprodukte mit
2(1H)-Pyrazinon-Struktur gebildet werden (Abb. 4.8.3-1). Für das dominierende
Abbauprodukt 2 konnte dies anhand der Masse bewiesen werden. Es wurde eine relative
monoisotopische Masse von Mr = 138,07 ([M+H]+ m / z = 139,08) bestimmt. In Analogie zur
Hydrolyse von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin handelt es sich
folglich um N1-Ethyl-5-methyl-2(1H)-pyrazinon (Mr,theoret. = 138,08). Die Massen von
Abbauprodukt 1 und 3 waren auf Grund der geringen Bildung nicht mittels LC-ESI-MS
bestimmbar. Eine strukturelle Zuordnung ist damit nicht möglich. Es ist jedoch anzunehmen,
dass es sich bei einem dieser Produkte um das 2(1H)-Pyrazinon-Dipeptid handelt.
Für die gemeinsame Analytik von 2(1H)-Pyrazinon-Peptiden, welche sich vom Glyoxal oder
Methylglyoxal ableiten, ist jeweils das Abbauprodukt 2 (N1-Alkyl-2(1H)-pyrazinon) ein
geeigneter Analyt. Auf Grund des unterschiedlichen zeitlichen Verlaufs der Bildung der
N1-Alkyl-2(1H)-pyrazinone bei den Hydrolysen (Abb. 4.8.2-1 und Abb. 4.8.3-1) ist eine
- 154 -

4 Auswertung und Diskussion der Ergebnisse
Hydrolysezeit von 23 h zu wählen. Bei dieser Hydrolysezeit war sowohl das sich vom
Glyoxal als auch das sich vom Methylglyoxal ableitende N1-Alkyl-2(1H)-pyrazinon im
ausreichenden Maß detektierbar.
0 10 20 30
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
Produkt 3
Produkt 2
Produkt 1
UV 220 nm
UV 320 nm
Fluoreszenz ex. 320 nmem. 400 nm
Abs
orpt
ion,
Flu
ores
zem
issi
on [A
U]
Zeit [min]
0 10 20 30
0,0
0,1
0,2
0,3
0,4 Produkt 2
Produkt 3Produkt 1
t = 48h
t = 23 h
t = 16 h
Abso
rptio
n (λ
= 3
20 n
m) [
AU]
Zeit [min]
Abb. 4.8.3-1: HPLC-Chromatogramme von Salzsäurehydrolysaten von N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin. Links: Mit verschiedenen Detektoren aufgenommen (Hydrolysezeit: 16 h). Rechts: Nach ausgewählten Hydrolysezeiten. (Hydrolyse: 6 M Salzsäure, 110 °C, Durchführung: 3.6.1).
Zusammenfassend ist festzustellen, dass N-[2-(5-Methyl-2-oxo-2H-pyrazin-1-yl)-propionyl]-
phenylalanin einem Abbau unterliegt wie er schon für N-[2-(2-Oxo-2H-pyrazin-1-yl)-
propionyl]-phenylalanin und 2-(2-Oxo-2H-pyrazin-1-yl)-propionsäure beobachtet wurde
(siehe Abschnitt 4.8.2). Dieser Abbau von pyrazinon-modifizierten N-Termini über das
2(1H)-Pyrazinon-Dipeptid zum N1-Alkyl-2(1H)-pyrazinon dürfte somit allgemeine Gültigkeit
besitzen.
4.8.4 Einfluss der Matrix auf das Hydrolyseverhalten
Um einen möglichen Einfluss der Matrix auf den Abbau von 2(1H)-Pyrazinon-Peptiden
während der Säurehydrolyse zu überprüfen, wurde N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-
phenylalanin in Gegenwart von ausgewählten Zusätzen hydrolysiert und das Hydrolysat
mittels RP-HPLC untersucht. Dabei wurde insbesondere geprüft, ob die Zusätze den Abbau
- 155 -

4 Auswertung und Diskussion der Ergebnisse
des 2(1H)-Pyrazinon-Tripeptides über das 2(1H)-Pyrazinon-Dipeptid (Produkt 1) zum
N1-Alkyl-2(1H)-pyrazinon (Produkt 2) beeinflussen.
Entsprechend den Chromatogrammen des Hydrolysates ohne Zusätze und denen des
Hydrolysates unter Zusatz von Insulin (Abb. 4.8.4-1), fand in Gegenwart des Proteins die
Hydrolyse ungehindert statt.
0 10 20 30
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4 Produkt 2Produkt 1
UV 220 nm
UV 320 nm
Fluoreszenzex. 320 nmem. 400 nm
Abso
rptio
n, F
luor
esze
nzem
issi
on [A
U]
Zeit [min]
0 10 20 30
0.0
0.2
0.4
0.6
0.8
1.0
1.2 Produkt 2Produkt 1
UV 220 nm
UV 320 nm
Fluoreszenzex. 320 nmem. 400 nm
Abso
rptio
n, F
luor
esze
nzem
issi
on [A
U]
Zeit [min]
Abb. 4.8.4-1: HPLC-Chromatogramme von Hydrolysaten von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin. Links: ohne Zusatz (0,5 mg 2(1H)-Pyrazinon-Peptid pro 1 ml Hydrolyse-ansatz). Rechts: Zusatz von Insulin (0,5 mg 2(1H)-Pyrazinon-Peptid, 1 mg Insulin pro 1 ml Hydrolyseansatz). (Hydrolysezeit: 16 h, Durchführung: 3.6.3).
Stark störend wirkte sich demgegenüber der Zusatz von Glucose aus. Dieser unterdrückte den
Abbau zu Produkt 1 und 2 vollständig, so dass beide Hydrolyseprodukte nicht detektierbar
waren (Abb. 4.8.4-2). Die gleiche Beobachtung wurde bei Zusatz von zucker- und
stärkehaltigen Cornflakes oder bei Zusatz des Oxidationsmittels Natriumazid gemacht. Bei
Hydrolyse in Anwesenheit von Glucose konnte der störende Einfluss durch Zugabe von
Mercaptoethanol beseitigt werden (Abb. 4.8.4-2). Die Stabilität der 2(1H)-Pyrazinon-Struktur
weist damit eine gewisse Ähnlichkeit mit der des Tryptophans auf. Tryptophan wird bei der
Salzsäurehydrolyse ebenfalls durch Oxidation abgebaut, wobei schon Spuren von Sauerstoff
und Chlor oder auch Cystin als Oxidationsmittel ausreichen (Olcott & Fraenkel-Conrat,
1947). Dieser Abbau kann durch Zusatz von Mercaptoethanol oder auch anderer Thiole und
sorgfältige Entfernung von Sauerstoff und Chlor unterdrückt werden (Ng, 1987). Weiterhin
wird Tryptophan auch bei Hydrolyse in Anwesenheit von Kohlenhydraten zerstört (Gortner
- 156 -
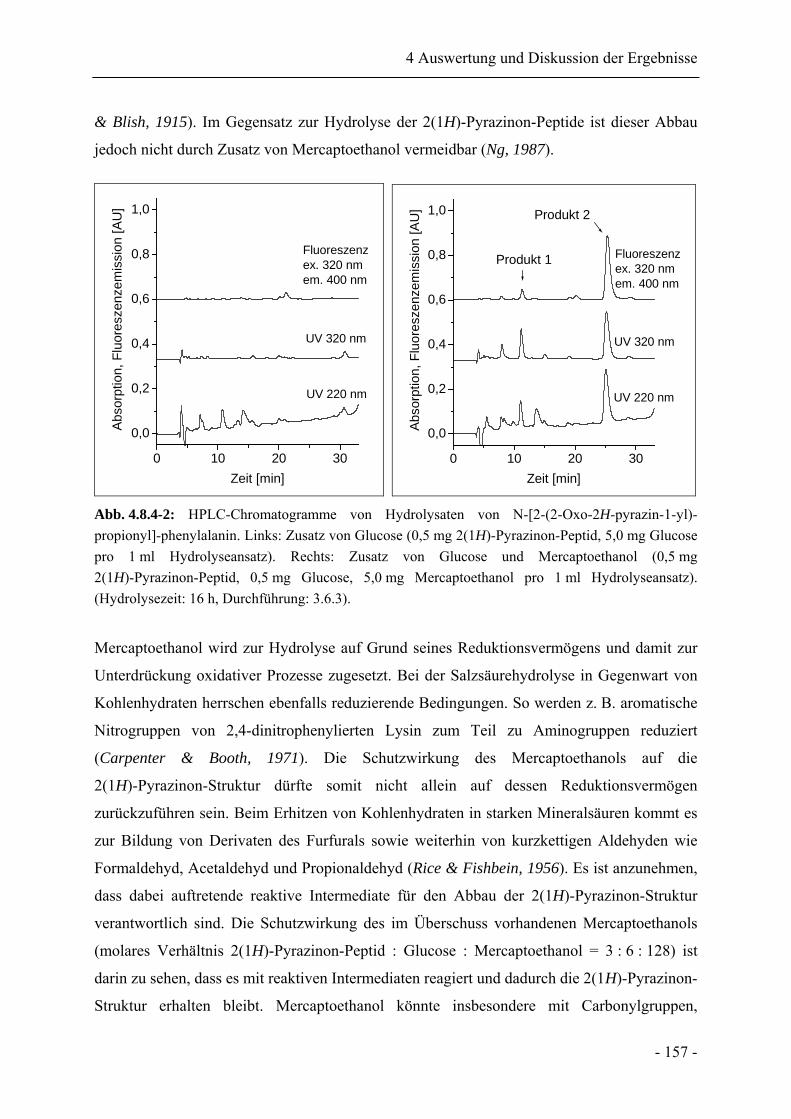
4 Auswertung und Diskussion der Ergebnisse
& Blish, 1915). Im Gegensatz zur Hydrolyse der 2(1H)-Pyrazinon-Peptide ist dieser Abbau
jedoch nicht durch Zusatz von Mercaptoethanol vermeidbar (Ng, 1987).
0 10 20 30
0,0
0,2
0,4
0,6
0,8
1,0
UV 220 nm
UV 320 nm
Fluoreszenzex. 320 nmem. 400 nm
Abso
rptio
n, F
luor
esze
nzem
issi
on [A
U]
Zeit [min]
0 10 20 30
0,0
0,2
0,4
0,6
0,8
1,0 Produkt 2
Produkt 1
UV 220 nm
UV 320 nm
Fluoreszenzex. 320 nmem. 400 nm
Abso
rptio
n, F
luor
esze
nzem
issi
on [A
U]
Zeit [min]
Abb. 4.8.4-2: HPLC-Chromatogramme von Hydrolysaten von N-[2-(2-Oxo-2H-pyrazin-1-yl)-propionyl]-phenylalanin. Links: Zusatz von Glucose (0,5 mg 2(1H)-Pyrazinon-Peptid, 5,0 mg Glucose pro 1 ml Hydrolyseansatz). Rechts: Zusatz von Glucose und Mercaptoethanol (0,5 mg 2(1H)-Pyrazinon-Peptid, 0,5 mg Glucose, 5,0 mg Mercaptoethanol pro 1 ml Hydrolyseansatz). (Hydrolysezeit: 16 h, Durchführung: 3.6.3).
Mercaptoethanol wird zur Hydrolyse auf Grund seines Reduktionsvermögens und damit zur
Unterdrückung oxidativer Prozesse zugesetzt. Bei der Salzsäurehydrolyse in Gegenwart von
Kohlenhydraten herrschen ebenfalls reduzierende Bedingungen. So werden z. B. aromatische
Nitrogruppen von 2,4-dinitrophenylierten Lysin zum Teil zu Aminogruppen reduziert
(Carpenter & Booth, 1971). Die Schutzwirkung des Mercaptoethanols auf die
2(1H)-Pyrazinon-Struktur dürfte somit nicht allein auf dessen Reduktionsvermögen
zurückzuführen sein. Beim Erhitzen von Kohlenhydraten in starken Mineralsäuren kommt es
zur Bildung von Derivaten des Furfurals sowie weiterhin von kurzkettigen Aldehyden wie
Formaldehyd, Acetaldehyd und Propionaldehyd (Rice & Fishbein, 1956). Es ist anzunehmen,
dass dabei auftretende reaktive Intermediate für den Abbau der 2(1H)-Pyrazinon-Struktur
verantwortlich sind. Die Schutzwirkung des im Überschuss vorhandenen Mercaptoethanols
(molares Verhältnis 2(1H)-Pyrazinon-Peptid : Glucose : Mercaptoethanol = 3 : 6 : 128) ist
darin zu sehen, dass es mit reaktiven Intermediaten reagiert und dadurch die 2(1H)-Pyrazinon-
Struktur erhalten bleibt. Mercaptoethanol könnte insbesondere mit Carbonylgruppen,
- 157 -

4 Auswertung und Diskussion der Ergebnisse
vinylogen Carbonylgruppen und Carbeniumionen reagieren sowie Redoxprozesse
beeinflussen. Auf Grund einer solchen Scavenger-Funktion werden Thiole ebenfalls bei der
Peptidsynthese wie z. B. bei der Abspaltung von Schutzgruppen eingesetzt (Bodanszky,
1993).
Zusammenfassend kann festgestellt werden, dass der Abbau von 2(1H)-Pyrazinon-Peptiden
zum Teil stark von der Matrix abhängig ist. Insbesondere Zucker aber auch Oxidationsmittel
wie Natriumazid stören den Abbau zu den entsprechenden N1-Alkyl-2(1H)-pyrazinonen.
Demgegenüber zeigte die Gegenwart von Protein keine Wirkung. Der nachteilige Einfluss
von Zucker konnte durch den Zusatz von Mercaptoethanol unterbunden werden. Für die
Analytik von pyrazinon-modifizierten Komponenten in kohlenhydrathaltigen Matrices sollte
der Zusatz von Mercaptoethanol folglich die gewünschte Bildung der N1-Alkyl-
2(1H)-pyrazinone ermöglichen.
4.9 Untersuchungen zur Bildung von 2(1H)-Pyrazinonen am Protein Insulin
4.9.1 Inkubation von Insulin mit Glyoxal unter physiologischen Bedingungen
Anhand von Umsetzungen von kurzkettigen Peptiden mit α-Dicarbonylverbindungen wie
Glyoxal und Methylglyoxal konnte gezeigt werden, dass es unter physiologischen
Bedingungen zu einer schnellen Derivatisierung des N-Terminus zum 2(1H)-Pyrazinon
kommt. Um zu prüfen ob diese Reaktion ebenfalls an einem Protein stattfindet, wurde die
Reaktion von bovinem Insulin mit Glyoxal untersucht. Dazu wurde Insulin (17 µM) mit
Glyoxal (50 µM) in PBS („phosphate-buffered saline“, 10 mM Phosphat, 0,8 % [w/v]
Natriumchlorid) bei pH = 37 °C für 15 d inkubiert. Die gewählte Pufferionen-Konzentration
erwies sich dabei als ausreichend und gewährleistete die Konstanz des pH-Wertes über den
Inkubationszeitraum (∆pH < 0,2).
Insulin wurde für die vorliegenden Modell-Untersuchungen gewählt, da es im monomeren
Zustand ein kleines und damit übersichtliches Protein ist (Molekulargewicht bovines Insulin
MW = 5733). Es ist aus zwei über zwei Disulfid-Brücken verknüpften Ketten aufgebaut und
besitzt damit zwei N-Termini als potentielle Orte der Bildung von 2(1H)-Pyrazinonen
(Abb. 4.9.1-1). Am N-Terminus der A-Kette befindet sich die Sequenz Gly-Ile und am
N-Terminus der B-Kette die Sequenz Phe-Val. Entsprechend der Aminosäuresequenz des
- 158 -

4 Auswertung und Diskussion der Ergebnisse
Insulins kommen der Arginin-Rest Arg-B22 und der Lysin-Rest Lys-B29 innerhalb der
B-Kette als weitere mögliche Reaktionsorte in Betracht.
Da sich bei den vorangegangenen Untersuchungen Lysin als nicht reaktiv zeigte (siehe
Abschnitt 4.7) wurde ein dreifacher molarer Überschuss an Glyoxal gegenüber Insulin
gewählt, so dass pro vermuteten Reaktionsort ein Molekül Glyoxal vorhanden ist. Um
vorzugsweise die spezifischsten und damit die wichtigsten Reaktionen zu erfassen, wurde bei
vergleichsweise niedrigen Konzentrationen inkubiert.
Gly-Ile-Val-Glu-Gln-Cys-Cys-Ala-Ser-Val-Cys-Ser-Leu-Tyr-Gln-Leu-Glu-Asn-Tyr-Cys-Asn
Phe-Val-Asn-Gln-His-Leu-Cys-Gly-Ser-His-Leu-Val-Glu-Ala-Leu-Tyr-Leu-Val-Cys-Gly
Ala-Lys-Pro-Thr-Tyr-Phe-Phe-Gly-Arg-Glu
S SA-Kette
B-Kette
2 31 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 2021
30 29 28 27 26 25 24 23 22 21
SS
SS
Abb. 4.9.1-1: Primärstruktur von Rinderinsulin (Ryle et al., 1955). ↓: Spaltstelle von Endoproteinase Glu-C (siehe Abschnitt 4.9.4).
4.9.2 Charakterisierung der Reaktionsprodukte am intakten Insulin
Analyse mittels RP-HPLC
Abb. 4.9.2-1 zeigt Chromatogramme der HPLC-Analyse eines Inkubationsansatzes von
Insulin mit Glyoxal nach 15 d Reaktionszeit. Neben dem Peak des originären Insulins waren
die Peaks von mindestens vier Folgeprodukten detektierbar. Die direkt dem originären Insulin
folgende Komponente trat ebenfalls bei der Inkubation von Insulin ohne Glyoxal auf, so dass
es sich folglich um ein Autolyseprodukt handeln muss. Durch massenspektroskopische
Analyse (siehe unten) konnte es als desaminiertes Insulin identifiziert werden. Anhand der
Peakflächen bei λ = 220 nm wurde der Umfang der Desaminierung zu ca. 10 % bestimmt.
Desaminierungen sind beim Insulin häufig zu beobachten (Dotsikas und Loukas, 2002).
Insulin weist insgesamt sechs Asparagin- und Glutamin-Reste als mögliche
- 159 -

4 Auswertung und Diskussion der Ergebnisse
Desaminierungsstellen auf, von denen der Asparagin-Rest Asn-A21 am leichtesten
desaminiert werden soll (Sundby, 1962).
15,0 17,5 20,0 22,5 25,0
0,0
0,2
0,4
0,6
0,8
1,0Insulin,pyrazinon-modifiziertInsulin,
desaminiert
Insulin,originär
UV 320 nm
UV 220 nm
Fluoreszenzex. 320 nmem. 400 nm
Abs
orpt
ion,
Flu
ores
zenz
emis
sion
[AU
]
Zeit [min]
Abb. 4.9.2-1: HPLC-Chromatogramme der Umsetzung von bovinem Insulin mit Glyoxal. (17 µM Insulin, 50 µM Glyoxal, PBS pH = 7,4, 37 °C, 15 d, Durchführung: 3.7).
Weiterhin waren mindestens drei auf die Inkubation mit Glyoxal zurückführbare Produkte
festzustellen. Sie wiesen die für 2(1H)-Pyrazinone typische Absorption bei λ = 320 nm sowie
Fluoreszenz auf. Die detaillierte Untersuchung mittels LC-ESI-MS zeigte die Bildung von
monopyrazinon-modifizierten Insulin-Komponenten an (siehe unten).
Einfluss des Phosphatpuffers auf die Reaktion
Um den Einfluss des verwendeten Puffers auf die Reaktion zu untersuchen, wurde die
Inkubation ebenfalls in 10 mM Phosphatpuffer durchgeführt. Dieser enthielt im Unterschied
zum PBS kein Natriumchlorid und ebenfalls kein Kaliumchlorid. Der allein durch Glyoxal
bedingte Umsatz an Insulin war in PBS mit (32,1 ± 2,0) % (Mittelwert ±
Standardabweichung, n = 3) höher als der in 10 mM Phosphatpuffer von (22,0 ± 1,6) %
- 160 -

4 Auswertung und Diskussion der Ergebnisse
(n = 3). Der zusätzlich durch Desaminierung hervorgerufene Umsatz war dagegen in beiden
Puffern praktisch gleich und wurde zu (9,8 ± 0,4) % (n = 3, PBS) und (10,1 ± 0,4) % (n = 3,
10 mM Phosphatpuffer) bestimmt. Die Reaktion von Insulin mit Glyoxal in PBS bewirkte
insbesondere die vermehrte Bildung der am stärksten fluoreszierenden pyrazinon-
modifizierten Komponente. Wie bereits bei den Umsetzungen von Gly-Ala-Phe mit Glyoxal
gezeigt werden konnte (siehe Abschnitt 4.4.3), spielt offenbar auch bei der Bildung von
2(1H)-Pyrazinonen am Protein die Ionenstärke eine erhebliche Rolle. Dabei ist insbesondere
die Wirkung auf die pKa-Werte der α-Aminogruppen zu diskutieren (siehe Abschnitt 4.4.3).
Weiterhin könnte sich das komplexe Assoziationsverhalten des Insulins (Kaarsholm &
Ludvigsen, 1995) oder andere strukturelle Änderungen auf die Zugänglichkeit der N-Termini
und damit auf die Reaktivität auswirken. Dass die Struktur des Insulins von der Art des
Puffers abhängig ist, verdeutlichen bereits die in Abb. 4.9.2-2 gezeigten UV-Spektren. Wie
ersichtlich ist, wird sowohl die Lage des dominierenden Maximums als auch dessen Intensität
stark vom Puffer beeinflusst. Anhand dessen kann auf eine ungleiche Zugänglichkeit oder
Anregbarkeit der Chromophore und damit auf eine unterschiedliche Struktur geschlossen
werden.
200 250 300 350 4000,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
200 250 300 350 4000,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
Ext
inkt
ion
Wellenlänge [nm]
0,1000 mg/ml PB
0,0500 mg/ml PB
0,0125 mg/ml PB
0,1000 mg/ml PBS
0,0500 mg/ml PBS
0,0125 mg/ml PBS
Wellenlänge [nm]
Abb. 4.9.2-2: UV-Spektren von Insulin in PBS (links) und in 10 mM Phosphatpuffer (PB, rechts). (Durchführung: 3.7).
- 161 -

4 Auswertung und Diskussion der Ergebnisse
Analyse mittels LC-ESI-MS
Um die Peaks anhand ihrer Masse bestimmten Reaktionsprodukte zuordnen zu können,
wurden die Inkubationsansätze mittels LC-ESI-MS untersucht. Die Messungen erfolgten im
positiven Modus. Zur Detektion diente ein Flugzeitmassenspektrometer. Für die Berechnung
der relativen monoisotopischen Molmassen (Mr) wurde der Peak mit dem geringsten
m/z-Verhältnis von dominanten mehrfach geladene Molekülionen ausgewählt und Mr anhand
der Ladung des Molekülions berechnet (siehe Abschnitt 3.2.2, Tab. 4.9.2-1).
Abb. 4.9.2-3 zeigt das TIC-Chromatogramm sowie die beiden UV-Spuren des
Inkubationsansatzes von Insulin mit Glyoxal. Anhand der bestimmten Molmasse kann Insulin
dem zuerst eluierenden Peak A zugeordnet werden (Tab. 4.9.2-1). Die beiden darauffolgenden
Komponenten B und C sind Autolyseprodukte, wobei die detektierten Massenzunahmen
jeweils eine Desaminierung (∆Mr, theoret = 1,00) des Insulins anzeigen. Auf Grund der
Trennung der Produkte erfolgt die Desaminierung offensichtlich an verschiedenen Positionen.
Peak D, E und F sind bei λ = 320 nm (Abb. 4.9.2-3) sowie anhand ihrer Fluoreszenz
(Abb. 4.9.2-1) detektierbar. Damit war die Bildung von pyrazinon-modifizierten Derivaten zu
vermuten, welche es mittels massenspektroskopischer Analyse abzusichern galt. Aus der
Bildung der 2(1H)-Pyrazinon-Struktur resultiert eine Massenzunahme von ∆Mr, theoret = 21,99
(monoisotopisch). Gegenüber dem originären Insulin war eine solche Massenzunahme bei
den Komponenten D und E festzustellen und es handelt sich somit um monopyrazinon-
modifizierte Insulin-Derivate. Bei Komponente F handelt es sich ebenfalls um mono-
pyrazinon-modifiziertes Insulin, welches zusätzlich eine Desaminierung aufweist. Die
Bildung von 2(1H)-Pyrazinonen am intakten Insulin konnte damit massenspektroskopisch
abgesichert werden. Die Bildung einer N-terminalen 2(1H)-Pyrazinon-Struktur ist damit nicht
nur am N-Terminus von kurzkettigen Peptiden möglich sondern sie findet ebenfalls an dem
gewählten Protein statt.
Das Vorhandensein eines Insulin-Derivates mit Mr = 5653,5 und unbekannter Struktur
(Peak D) lässt auf weitere zur Zeit noch nicht aufgeklärte Reaktionen schließen.
- 162 -

4 Auswertung und Diskussion der Ergebnisse
15,0 17,5 20,0 22,5 25,0
0
2
4
6
0,0
0,2
0,4
0,6
0,8
1,0
0
5
10
15
20
25
30
35
E
F
D
UV 320 nm
Abso
rptio
n [m
AU]
Zeit [min]
F
EDCB
AUV 220 nm
Abs
orpt
ion
[AU
]
F
EDCB
ATIC
TIC
[cou
nts
x 10
00]
Abb. 4.9.2-3: HPLC-Chromatogramme (MS- und UV-Detektion) der Umsetzung von bovinem Insulin mit Glyoxal. (17 µM Insulin, 50 µM Glyoxal, PBS pH = 7,4, 37 °C, 15 d, Durchführung: 3.7).
- 163 -

4 Auswertung und Diskussion der Ergebnisse
Tab. 4.9.2-1: Bestimmte relative monoisotopische Molmassen und deren Zuordnung.
Peak m / z Ion Mr Mr ∆Mr* Zuordnung Mr, theo.
A 1146,85
1433,29
MH55+
MH44+
5729,61
5729,61
5729,61 0,00 Insulin 5729,56
B 1147,08
1433,66
MH55+
MH44+
5730,36
5730,61
5730,49 0,88 Insulin, desaminiert 5730,57
C 1147,08
1433,66
MH55+
MH44+
5730,37
5730,59
5730,48 0,87 Insulin, desaminiert 5730,57
D 1151,28 MH55+ 5751,36 5751,33 21,72 Insulin, monopyrazinon- 5751,55
1438,83 MH44+ 5751,29 modifiziert
D 1414,38 MH44+ 5653,49 5653,49 - 76,12 ?
E 1151,26
1438,85
MH55+
MH44+
5751,26
5751,37
5751,32 21,71 Insulin, monopyrazinon-
modifiziert
5751,55
F
1151,51
1439,24
MH55+
MH44+
5752,51
5752,93
5752,72 23,11 Insulin, monopyrazinon-
modifiziert, desaminiert
5752,56
* Massendifferenz zur gemessenen relativen monoisotopischen Molmasse des Insulins
4.9.3 Untersuchungen nach Reduktion und Blockierung der Sulfhydrylgruppen
4.9.3.1 Charakterisierung der Reaktionsprodukte
Aus der Anfangsphase der Maillard-Reaktion ist bekannt, das bestimmte Aminogruppen am
Protein eine erhöhte Reaktivität aufweisen. Zum Beispiel findet in vivo die Bildung des
Amadori-Produktes am Hämoglobin bevorzugt an der α-Aminogruppe des N-terminalen
Valin-Restes der β-Kette statt (Bunn et al., 1979). Humanes Serumalbumin wird in vivo
demgegenüber bevorzugt an der ε-Aminogruppe eines Lysin-Restes (Lys-525) durch Glucose
modifiziert (Garlick & Mazer, 1983). Da Insulin zwei N-Termini besitzt, stellte sich die Frage
ob eine solche Spezifität auch bei der Bildung von 2(1H)-Pyrazinonen zu beobachten ist. Um
die Orte der Derivatisierung aufzuklären, wurden die beiden Kette durch Reduktion der
Disulfid-Brücken mit Dithiothreitol (DTT) getrennt und die freigesetzten Ketten durch
Umsetzung mit Iodacetamid stabilisiert. Der Nachweis der 2(1H)-Pyrazinon-Struktur an
kleineren Fragmenten dient ebenfalls zur weiteren Absicherung von deren Bildung.
- 164 -

4 Auswertung und Diskussion der Ergebnisse
Analyse mittels RP-HPLC und LC-ESI-MS
Abb. 4.9.3.1-1 zeigt die Chromatogramme der RP-HPLC eines Inkubationsansatzes von
Insulin mit Glyoxal nachdem die Disulfid-Brücken reduziert und die Sulfhydrylgruppen
blockiert wurden. Neben den Peaks der nicht durch Glyoxal modifizierten Ketten werden
weitere Peaks detektiert. Zwei Komponenten zeigen die für die 2(1H)-Pyrazinon-Struktur
charakteristische Absorption bei λ = 320 nm und Fluoreszenz.
10 15 20 25 30
0,0
0,2
0,4
0,6
Fluoreszenz,ex. 320 nm,em. 400 nm
UV 320 nm
UV 220 nm
A-Kette,pyrazinon-modifiziert
B-Kette,pyrazinon-modifiziert
B-Kette
A-Kette
Abso
rptio
n, F
luor
esze
nz [A
U]
Zeit [min]
Abb. 4.9.3.1-1: HPLC-Chromatogramme der Umsetzung von bovinem Insulin mit Glyoxal nach Reduktion und Blockierung der Sulfhydrylgruppen. (17 µM Insulin, 50 µM Glyoxal, PBS pH = 7,4, 37 °C, 15 d, Durchführung: 3.8.1).
Im Unterschied zu den Untersuchungen mittels RP-HPLC war für die Analyse mittels
LC-ESI-MS eine Anreicherung der reduzierten und blockierten Insulin-Derivate mittels
Festphasenextraktion an einer C18-Phase erforderlich. Dies ermöglichte eine eindeutigere
massenspektroskopische Detektion und überführte durch die Abtrennung überschüssiger
- 165 -

4 Auswertung und Diskussion der Ergebnisse
Reagenzien gleichzeitig die Proben in eine stabilere Form. Alternativ war ein modifiziertes
Derivatisierungsprotokoll, welches zu einem geringeren Endvolumen führt, und die Injektion
großer Probenmengen möglich. In diesem Fall diente das verwendete HPLC-System zur
Anreicherung.
Abb. 4.9.3.1-2 zeigt das TIC-Chromatogramm sowie die beiden UV-Spuren eines
entsprechend aufgearbeiteten Inkubationsansatzes von Insulin mit Glyoxal. In Tab. 4.9.3.1-1
sind die häufigsten Molekülionen und deren Zuordnungen zu bestimmten Derivaten
zusammengefasst. Das bei λ = 320 nm erhaltene Chromatogramm ist durch zwei dominante
Peaks geprägt. Anhand der bestimmten relativen Molmassen lassen sich diese Peaks eindeutig
der pyrazinon-modifizierten A-Kette (Peak C) und der pyrazinon-modifizierten B-Kette
(Peak I) zuordnen. Die Bildung der 2(1H)-Pyrazinon-Struktur an beiden Ketten ist damit
zweifelsfrei belegt.
Für die korrekte Interpretation der Spektren waren insbesondere bei diesen LC-ESI-MS
Analysen ein erhöhtes Vorkommen von Natrium-Addukten von Bedeutung. So führt die
Bildung eines Natrium-Adduktes zu einer identischen Massenzunahme wie durch
2(1H)-Pyrazinon-Bildung. Zum Beispiel wird für das MNaH2+-Molekülion der originären
A-Kette (m/z = 1295,19) die gleiche Masse erhalten für das MH22+-Molekülion der
pyrazinon-modifizierten A-Kette (m/z = 1295,24, siehe Tab. 4.9.3.1-1). Da die originäre
A-Kette und die modifizierte A-Kette durch die Chromatographie getrennt werden, kann das
MNaH2+-Addukt der originären A-Kette das Vorhandensein der pyrazinon-modifizierten
A-Kette jedoch nicht vortäuschen. Wenn die originäre A-Kette mit in Peak C enthalten wäre,
müsste weiterhin die Masse des MH22+-Molekülions der originären A-Kette (m/z = 1284,18)
detektierbar sein. Diese wurde jedoch nicht gemessen. Zusätzlich war in Peak C das
MNa22+-Molekülion der modifizierten A-Kette messbar (m/z = 1316,90). Ein Peak mit diesem
m/z-Verhältnis kann nur aus der modifizierten A-Kette und nicht aus originären A-Kette
resultieren. Ferner ist Peak C anhand der Absorption bei λ = 320 nm sowie mittels
Fluoreszenz-Detektion erfassbar, so dass insgesamt die 2(1H)-Pyrazinon-Bildung gesichert
ist.
Neben den Peaks der originären und der pyrazinon-modifizierten Ketten waren weitere
Komponenten detektierbar. Es handelt sich dabei um desaminierte Produkte (Peak B, E und
F) sowie um unbekannte Produkte der B-Kette (Peak G und H).
- 166 -

4 Auswertung und Diskussion der Ergebnisse
10 15 20 250
1
2
3
0,0
0,1
0,2
0,3
0,0
0,5
1,0
1,5
2,0
2,5
3,0
UV 320 nm
I
C
Abso
rptio
n [m
AU]
Zeit [min]
IHGFE
D
B
A
UV 220 nm
TICAb
sorp
tion
[AU
]
IHGF
C
E
D
B
A
TIC
[cou
nts
x 10
0000
]
Abb. 4.9.3.1-2: HPLC-Chromatogramme (MS- und UV-Detektion) der Umsetzung von bovinem Insulin mit Glyoxal nach Reduktion und Blockierung der Sulfhydrylgruppen sowie Anreicherung. (Inkubation: 17 µM Insulin, 50 µM Glyoxal, PBS pH = 7,4, 37 °C, 15 d, Durchführung: 3.8.1).
- 167 -

4 Auswertung und Diskussion der Ergebnisse
Tab. 4.9.3.1-1: Bestimmte relative monoisotopische Molmassen und deren Zuordnung.
Peak m / z Ion Mr Mr ∆ Mr* Zuordnung Mr, theo.
A 1284,18
1295,19
856,47
MH22+
MNaH2+
MH34+
2566,35
2566,38
2566,37
2566,37 0,00 A-Kette 2566,03
B 1284,71
1295,67
1306,67
MH22+
MNaH2+
MNa22+
2567,40
2567,35
2567,35
2567,37 1,00 A-Kette,
desaminiert
2567,04
C 1295,24
1306,24
1316,90
MH22+
MNaH2+
MNa22
2588,48
2588,48
2587,82
2588,26 21,89 A-Kette,
pyrazinon- modifiziert
2588,02
D 703,46
879,061
1171,74
1757,07
MH55+
MH44+
MH33+
MH22+
3512,23
3512,21
3512,18
3512,11
3512,18 0,00 B-Kette 3511,71
E 703,66
879,32
1172,08
MH55+
MH44+
MH33+
3513,25
3513,25
3513,22
3513,24 1,06 B-Kette,
desaminiert
3512,71
F 703,65
879,31
1172,08
1757,59
MH55+
MH44+
MH33+
MH22+
3513,20
3513,19
3513,22
3513,17
3512,20 1,02 B-Kette,
desaminiert
3512,71
G 860,29
1146,71
1719,56
MH44+
MH33+
MH22+
3437,13
3437,11
3437,10
3437,11 - 75,07 B-Kette,
?
H 886,21
1181,27
1771,43
MH44+
MH33+
MH22+
3540,80
3540,80
3540,85
3540,82 28,64 B-Kette,
?
I 884,55
1179,06
1768,11
MH44+
MH33+
MH22+
3534,17
3534,16
3534,21
3534,17 21,99 B-Kette,
pyrazinon- modifiziert
3533,69
* Massendifferenz zur gemessenen relativen monoisotopischen Molmasse der jeweiligen Kette
- 168 -

4 Auswertung und Diskussion der Ergebnisse
4.9.3.2 Vergleich der Reaktivität der N-Termini von A- und B-Kette
Um zu klären welche der beiden Ketten die reaktivere ist, wurden reduzierte und blockierte
Proben von Inkubationsansätzen (17 µM Insulin, ohne oder mit 50 µM Glyoxal, PBS
pH = 7,4, 37 °C, 15 d) mittels RP-HPLC untersucht und die Peakflächen bei λ = 220 nm zur
Auswertung herangezogen.
Der Gesamtumsatz je Kette wurde anhand der Peakflächen nach der Inkubation mit und ohne
Glyoxal ermittelt. Der Gesamtumsatz an der A-Kette wurde zu (5,1 ± 1,2) % (Mittelwert ±
Standardabweichung, n = 3) und der an der B-Kette zu (25,9 ± 1,6) % (n = 3) bestimmt. Die
B-Kette ist damit deutlich reaktiver als die A-Kette. Das Ergebnis stimmt gut mit dem
ermittelten Gesamtumsatz des Insulins von (32,1 ± 2,0) % überein (siehe Abschnitt 4.9.2).
Die Autolyse des Insulins von insgesamt ca. 10 % ist in den Ergebnissen nicht enthalten.
Der Umsatz zur pyrazinon-modifizierten A-Kette wurde zu (2,0 ± 0,4) % (Mittelwert ±
Standardabweichung, n = 3) und zur pyrazinon-modifizierten B-Kette zu (5,9 ± 0,6) % (n = 3)
berechnet. Damit ist die 2(1H)-Pyrazinon-Bildung am N-Terminus der B-Kette (Phe-Val)
etwa um den Faktor drei gegenüber der am N-Terminus der A-Kette (Gly-Ile) bevorzugt.
Bei der B-Kette ist der Unterschied zwischen dem Gesamtumsatz und dem Umsatz zur
pyrazinon-modifizierten B-Kette besonders markant. Folglich findet die Bildung weiterer
Derivate statt. Diese sind z. B. als Peak G und H detektierbar (siehe Abschnitt 4.9.3.1). Ihre
Struktur war nicht Gegenstand dieser Arbeit, die Aufklärung sollte jedoch das Ziel weiterer
Untersuchungen sein.
Zur Erklärung der bevorzugten 2(1H)-Pyrazinon-Modifizierung am N-Terminus der B-Kette
sind insbesondere die pKa-Werte der beiden α-Aminogruppen zu betrachten. Wie bereits bei
den Umsetzungen von Peptiden verschiedener Sequenz mit Glyoxal diskutiert (siehe
Abschnitt 4.4.1), nimmt die Geschwindigkeit der 2(1H)-Pyrazinon-Bildung mit zunehmenden
Anteil an unprotonierter Aminogruppe zu. Daher ist der niedrigere pKa-Wert der
α-Aminogruppe des N-Terminus der B-Kette (pKa = 7,1) im Vergleich zu dem der A-Kette
(pKa = 8,4, Gao et al., 1995) für die beobachtete Selektivität als Ursache anzusehen. Als
weiteres Kriterium ist die Zugänglichkeit der beiden Termini zu diskutieren. Bei der
verwendeten Konzentration von 17 µM liegt Insulin als Dimer vor (Kaarsholm & Ludvigsen,
1995). Entsprechend der Struktur im Kristall (Blundell et al., 1972) und der Struktur in
Lösung (Kaarsholm & Ludvigsen, 1995) befindet sich keiner der beiden N-Termini im dimer-
bildenden Bereich des Moleküls. In Lösung liegt der N-Terminus der A-Kette zwar relativ
- 169 -

4 Auswertung und Diskussion der Ergebnisse
nah am Rumpf, jedoch an der Oberfläche des Moleküls. Der N-Terminus der B-Kette ragt
demgegenüber leicht vom Rumpf weg und die Konformation ist etwas flexibler
(Abb. 4.9.3.2-1, Kaarsholm & Ludvigsen, 1995). Insgesamt betrachtet ist damit anzunehmen,
dass beide Reaktionsorte für Glyoxal gut zugänglich sind.
Phe-B1
Gly-A1
Arg-B22
Lys-B29
Abb. 4.9.3.2-1: Struktur von B9(Asp)-Humaninsulin in Lösung, welche nahezu mit der Struktur von Humaninsulin in Lösung und der T-Konformation im 2Zn-Kristall identisch ist (Jørgensen et al., 1992).
Eine höhere Reaktivität der N-terminalen α-Aminogruppe der B-Kette gegenüber der von der
A-Kette wurde ebenfalls bei der Reaktion von Insulin mit Glucose festgestellt. Dieses
Ergebnis wurde sowohl bei der Umsetzung in einem trockenen System bei einer relativen
Gleichgewichtsfeuchte von 70 % und 37 °C (Schwartz & Lea, 1952, Amaya-F et al., 1976) als
auch in Lösung in Gegenwart von Natriumcyanoborhydrid bei pH = 7,4 und 37 °C erhalten
(O’Harte et al., 1996, 2000). Die Ursache der unterschiedlichen Reaktivität wurde von
Amaya-F et al. (1976) und O’Harte et al. (1996, 2000) nicht diskutiert. Lediglich Schwartz &
Lea (1952) vermuteten eine unterschiedliche Zugänglichkeit der Reaktionsorte als Ursache.
Insulin wurde in zahlreichen weiteren Untersuchungen mit für Aminogruppen spezifischen
Reagenzien wie N-Hydroxysuccinimidylestern (Lindsay & Shall, 1971, Hashimoto et al.,
- 170 -

4 Auswertung und Diskussion der Ergebnisse
1989), Diketen (Lindsay & Shall, 1969), weiteren aktivierten Säurederivaten (Tsai et al.,
1997) sowie Aldehyden (Mei et al., 1999) umgesetzt. Diesen Arbeiten ist zu entnehmen, dass
die Reihenfolge der Reaktivität der drei primären Aminogruppen von dem modifizierenden
Reagenz, dem Lösungsmittel, dem pH-Wert, dem Insulin-zu-Reagenz-Verhältnis und
gegebenenfalls von zugesetzten Effektoren abhängig ist. So ist z. B. die selektive
Carbamoylierung der α-Aminogruppe der A-Kette und der ε-Aminogruppe des Lysinrestes
möglich ohne die α-Aminogruppe der B-Kette zu modifizieren (Tsai et al., 1997). Eine
spezifisch Derivatisierung der α-Aminogruppe der B-Kette ist ebenfalls möglich. Dies konnte
beispielsweise in 54 %igen wässrigen Isopropanol bei pH = 6,8 in Gegenwart von 1,5 M
Natriumsalicyclat durch reduktive Alkylierung mit cis-9-Hexadecenal und Natriumcyano-
borhydrid erreicht werden (Mei et al., 1999). Als Ursache für spezifische Derivatisierungen
sind damit die oben genannten inneren und äußeren Einflussfaktoren anzusehen.
4.9.4 Charakterisierung der Reaktionsprodukte nach enzymatischer Hydrolyse
Um den Nachweis der 2(1H)-Pyrazinon-Bildung an kleineren Peptidfragmenten zu
ermöglichen, wurde in Anlehnung an die Arbeit von O´Harte et al. (1996) eine enzymatische
Fragmentierung des glyoxal-modifizierten Insulins (17 µM Insulin, 50 µM Glyoxal, PBS
pH = 7,4, 37 °C, 15 d) mit Endoproteinase Glu-C (E.C. 3.4.21.19) vorgenommen.
Endoproteinase Glu-C spaltet bei pH = 8,0 spezifisch Peptidbindungen, die sich C-terminal an
Glutaminsäure in der Aminosäuresequenz befinden (Drapeau et al., 1972). Dementsprechend
sind aus reduziertem und blockiertem Insulin drei Fragmente der A-Kette (A1-4, A5-17, A18-
21) und drei Fragmente der B-Kette (B1-13, B14-21, B22-30) zu erwarten (siehe
Abb. 4.9.1-1). Abb. 4.9.4-1 zeigt das mittels LC-ESI-MS im positiven Modus erhaltene
TIC-Chromatogramm der enzymatisch hydrolysierten Probe.
Das pyrazinon-modifizierte N-terminale Fragment der B-Kette (PyrB1-13) konnte eindeutig
anhand seiner Masse (MH22+ m/z = 781,26, Mr = 1560,50, Mr, theoret. = 1560,71) identifiziert
werden (Abb. 4.9.4-2). Das Vorhandensein der 2(1H)-Pyrazinon-Struktur wird weiterhin
durch das UV-Spektrum belegt, welches das für 2(1H)-Pyrazinone charakteristische
Maximum bei λmax = 324 nm aufweist (Abb. 4.9.4-2).
Ein pyrazinon-modifiziertes N-terminales Fragment der A-Kette war nicht nachweisbar. Als
Ursache ist die geringere 2(1H)-Pyrazinon Bildung an der A-Kette und die offenbar etwas
- 171 -
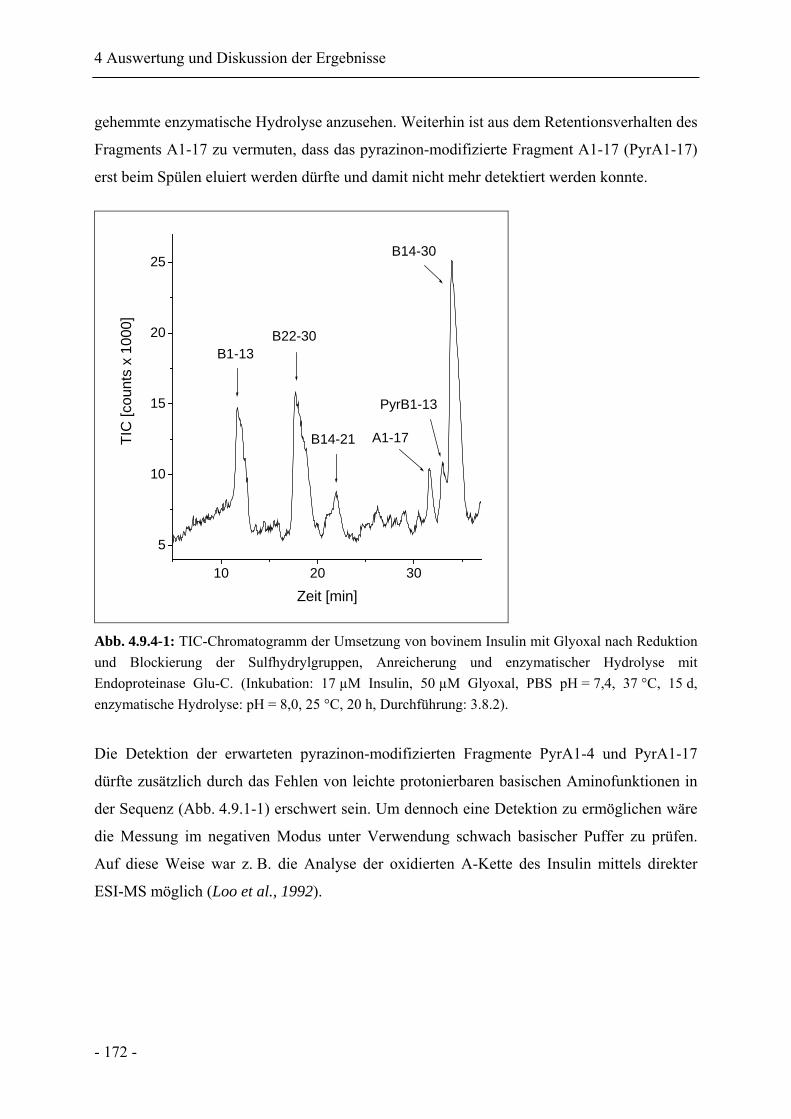
4 Auswertung und Diskussion der Ergebnisse
gehemmte enzymatische Hydrolyse anzusehen. Weiterhin ist aus dem Retentionsverhalten des
Fragments A1-17 zu vermuten, dass das pyrazinon-modifizierte Fragment A1-17 (PyrA1-17)
erst beim Spülen eluiert werden dürfte und damit nicht mehr detektiert werden konnte.
10 20 30
5
10
15
20
25B14-30
A1-17
PyrB1-13
B22-30
B14-21
B1-13
TIC
[cou
nts
x 10
00]
Zeit [min]
Abb. 4.9.4-1: TIC-Chromatogramm der Umsetzung von bovinem Insulin mit Glyoxal nach Reduktion und Blockierung der Sulfhydrylgruppen, Anreicherung und enzymatischer Hydrolyse mit Endoproteinase Glu-C. (Inkubation: 17 µM Insulin, 50 µM Glyoxal, PBS pH = 7,4, 37 °C, 15 d, enzymatische Hydrolyse: pH = 8,0, 25 °C, 20 h, Durchführung: 3.8.2).
Die Detektion der erwarteten pyrazinon-modifizierten Fragmente PyrA1-4 und PyrA1-17
dürfte zusätzlich durch das Fehlen von leichte protonierbaren basischen Aminofunktionen in
der Sequenz (Abb. 4.9.1-1) erschwert sein. Um dennoch eine Detektion zu ermöglichen wäre
die Messung im negativen Modus unter Verwendung schwach basischer Puffer zu prüfen.
Auf diese Weise war z. B. die Analyse der oxidierten A-Kette des Insulin mittels direkter
ESI-MS möglich (Loo et al., 1992).
- 172 -

4 Auswertung und Diskussion der Ergebnisse
770 775 780 785 7900
50
100
150
200
250MH2
2+
m/z = 781,26
Inte
nsitä
t [co
unts
]
m / z
200 250 300 350 4000
50
100
150
200
250
300
324 nm
Abs
orpt
ion
[mA
U]
Wellenlänge [nm]
Abb. 4.9.4-2: Massenspektrum (links) und UV-Spektrum (DAD) (rechts) des pyrazinon-modifizierten Fragments der B-Kette (PyrB1-13). Durchführung: 3.8.2).
4.9.5 Untersuchungen zum Nachweis von N1-Alkyl-2(1H)-pyrazinonen nach
Salzsäurehydrolyse des glyoxal-modifizierten Insulins
Für die Analyse von peptid- und proteingebundenen 2(1H)-Pyrazinonen wurde ein allgemein
anwendbares Verfahren entwickelt (siehe Abschnitt 4.8). Dieses sieht im ersten Schritt die
Hydrolyse des Proteins mit Salzsäure ggf. unter Zusatz von Mercaptoethanol vor. Dabei
werden N-terminale 2(1H)-Pyrazinone als N1-Alkyl-2(1H)-pyrazinone freigesetzt. Der
Nachweis der N1-Alkyl-2(1H)-pyrazinone erfolgt anschließend mittels RP-HPLC mit UV-
und Fluoreszenz-Detektion sowie mittels LC-ESI-MS. Um die 2(1H)-Pyrazinon-Bildung am
Insulin zusätzlich abzusichern und die Eignung der Methode zu beurteilt, wurde das
entwickelte Verfahren am glyoxal-modifizierten Insulin (17 µM Insulin, 50 µM Glyoxal, PBS
pH = 7,4, 37 °C, 15 d) angewendet.
Darstellung der erwarteten N1-Alkyl-2(1H)-pyrazinone
Für die Anwendung des Verfahrens wurden zunächst die erwarteten N1-Alkyl-
2(1H)-pyrazinone der A-Kette (N1-(2-Methyl-butyl)-2(1H)-pyrazinon, A2, Abb. 4.9.5-1) und
der B-Kette (3-Benzyl-N1-isobutyl-2(1H)-pyrazinon, B2) im analytischen Maßstab als
- 173 -

4 Auswertung und Diskussion der Ergebnisse
Vergleichssubstanzen gewonnen. Wie in Abb. 4.9.5-1 gezeigt ist wurden dazu im ersten
Schritt die 2(1H)-Pyrazinon-Dipeptide aus den Dipeptiden Gly-Ile und Phe-Val (N-terminale
Sequenz der A- bzw. B-Kette) jeweils durch Umsetzung mit Glyoxal dargestellt. Durch
Behandlung der Ansätze mit Salzsäure (6 M, 48 h, 110 °C) erfolgte anschließend die
Freisetzung der gewünschten N1-Alkyl-2(1H)-pyrazinone.
3-Benzyl-N1-isobutyl-2(1H)-pyrazinon (B2)
N1-(2-Methyl-butyl)-2(1H)-pyrazinon (A2)
N
N
O
OH
O
N
N
OHN
H2N
O
OH
O
O
O
N
N
ON
N
O
OH
O
HN
H2N
O
OH
O
O
O
2-(3-Benzyl-2-oxo-2H-pyrazin-1-yl)-3-methyl-buttersäure (B1)
2-(2-oxo-2H-pyrazin-1-yl)-3-methyl-pentansäure (A1)
Gly-Ile(A-Kette)
Phe-Val(B-Kette)
Glyoxal
Glyoxal
+
+(HCl), ∆
(HCl), ∆
- 2 H2O
- 2 H2O
- CO2
- CO2
Abb. 4.9.5-1: Reaktionswege zur Darstellung von N1-(2-Methyl-butyl)-2(1H)-pyrazinon (A2) und 3-Benzyl-N1-isobutyl-2(1H)-pyrazinon (B2).
Abb. 4.9.5-2 zeigt die HPLC-Chromatogramme der erhaltenen Lösungen nach der salzsauren
Behandlung der Glyoxal-Dipeptid-Ansätze. Die N1-Alkyl-2(1H)-pyrazinone wurden anhand
ihrer Absorption bei λ = 320 nm und der Fluoreszenz identifiziert. Wie aus Abb. 4.9.5-2
weiterhin zu entnehmen ist, konnte N1-(2-Methyl-butyl)-2(1H)-pyrazinon (A2) in relativ guter
Ausbeute erhalten werden. Auf Grund der geringen Bildung des 2(1H)-Pyrazinon-Dipeptides
aus Phe-Val und Glyoxal (B1) war der Gesamtumsatz zum 3-Benzyl-N1-isobutyl-
2(1H)-pyrazinon (B2) demgegenüber relativ gering. Der Nachweis des N1-Alkyl-
2(1H)-pyrazinons mittels LC-ESI-MS gelang deshalb nur bei N1-(2-Methyl-butyl)-
2(1H)-pyrazinon (MH+ m/z = 167,15, Mr = 166,14, Mr, theoret. = 166,12).
- 174 -

4 Auswertung und Diskussion der Ergebnisse
0 10 20 30 40 50 60
0,00
0,25
0,50
0,75
1,00
1,25
1,50
UV 220 nm
UV 320 nm
Fluoreszenzex. 320 nmem. 400 nm
A2
Abs
orpt
ion,
Flu
ores
zenz
[AU
]
Zeit [min]
0 10 20 30 40 50 60
0,00
0,25
0,50
0,75
1,00
1,25
1,50 B2
UV 220 nm
UV 320 nm
Fluoreszenzex. 320 nmem. 400 nm
Abs
orpt
ion,
Flu
ores
zenz
[AU
]
Zeit [min]
Abb. 4.9.5-2: HPLC-Chromatogramme der mit Salzsäure behandelten Ansätze zur Darstellung von N1-(2-Methyl-butyl)-2(1H)-pyrazinon (Peak A2) (links) und von 3-Benzyl-N1-isobutyl-2(1H)-pyrazinon (Peak B2) (rechts). (Durchführung: 3.9).
Analyse von glyoxal-modifiziertem Insulin
Für die Analyse wurde das glyoxal-modifizierte Insulin zunächst an einer C18-Phase
angereichert und anschließend mit Salzsäure in Gegenwart von Mercaptoethanol hydrolysiert.
Die Hydrolyse in Gegenwart von Mercaptoethanol erwies sich dabei für die Bildung der
gewünschten Hydrolyseprodukte als erforderlich. Als dominierender Peak im bei λ = 320 nm
erhaltenen Chromatogramm des Hydrolysates wurde das 2(1H)-Pyrazinon-Dipeptid der
B-Kette 2-(3-Benzyl-2-oxo-2H)-pyrazin-1-yl)-3-methyl-buttersäure (Peak B1, siehe
Abb. 4.9.5-1) anhand der Masse (MH+ m/z = 287,21, Mr = 286,20, Mr, theoret. = 286,13)
eindeutig identifiziert (Abb. 4.9.5-3). Mit geringerer Intensität wurde das N1-Alkyl-
2(1H)-pyrazinon der pyrazinon-modifizierten B-Kette 3-Benzyl-N1-isobutyl-2(1H)-pyrazinon
(Peak B2) detektiert (siehe Abb. 4.9.5-3, LC-ESI-MS: MH+ m/z = 243,21, Mr = 243,20,
Mr, theoret. = 242,15). Die geringere Peakfläche des N1-Alkyl-2(1H)-pyrazinons (Peak B2)
gegenüber dem 2(1H)-Pyrazinon-Dipeptid (Peak B1) ist auf die kurze Zeit der Salzsäure-
Behandlung von 23 h zurückzuführen. Nach 48 h würde Peak B2 dominieren. Das N1-Alkyl-
2(1H)-pyrazinon der pyrazinon-modifizierten A-Kette N1-(2-Methyl-butyl)-2(1H)-pyrazinon
- 175 -

4 Auswertung und Diskussion der Ergebnisse
war mittels RP-HPLC nicht sicher und mit LC-ESI-MS nicht nachweisbar. Als primäre
Ursache hierfür ist die geringere 2(1H)-Pyrazinon-Modifizierung der A-Kette anzusehen.
0 10 20 30 40
0
10
20
B1
B2
Abs
orpt
ion
(λ =
320
nm
) [m
AU
]
Zeit [min]
275 280 285 290 295
0
50
100
150
200
250 MH+
m/z = 287,21
Inte
nsitä
t [co
unts
]
m / z
Abb. 4.9.5-3: HPLC-Chromatogramm von einem Salzsäurehydrolysat des glyoxal-modifizierten Insulins mit Peak B1 (2-(3-Benzyl-2-oxo-2H)-pyrazin-1-yl)-3-methyl-buttersäure) und Peak B2 (3-Benzyl-N1-isobutyl-2(1H)-pyrazinon) (links). Massenspektrum der bei 22,4 min eluierenden Verbindung (Peak B1) (rechts). (Inkubation: 17 µM Insulin, 50 µM Glyoxal, PBS pH = 7,4, 37 °C, 15 d, Hydrolyse mit Zusatz von Mercaptoethanol, Durchführung: 3.9).
Insgesamt betrachtet ermöglichte die zuvor entwickelte Analysenmethode den sicheren
Nachweis (inklusive Massenspektrum) der 2(1H)-Pyrazinon-Bildung an der B-Kette des
Insulins anhand der Hydrolyseprodukte. Die Methode ist damit prinzipiell zum Nachweis
proteingebundener 2(1H)-Pyrazinone geeignet. Im Vergleich zum Nachweis am intakten oder
partiell fragmentierten Insulin mittels LC-ESI-MS ist die erforderliche Menge an Protein
jedoch deutlich größer. Weitere Untersuchungen sollten deshalb darauf zielen die
Nachweisgrenze zu senken. Hierzu könnten die Analyse mittels LC-ESI-MS und Messung im
negativen Modus sowie die GC-MS als Alternativmethoden geprüft werden. Weiterhin sollten
die Vorgänge während der Hydrolyse näher untersucht werden. In Hinblick auf eine
nachweisstarke Analytik ist insbesondere die Höhe der molaren Ausbeute an N1-Alkyl-
2(1H)-pyrazinon aus dem proteingebunden 2(1H)-Pyrazinon, der Einfluss der Matrix auf die
- 176 -

4 Auswertung und Diskussion der Ergebnisse
molare Ausbeute und ob durch geeignete Zusätze oder Verfahrensweisen die molare Ausbeute
erhöht werden kann von Interesse.
4.9.6 Betrachtungen zur Reaktivität von Lysin und Arginin am Insulin
Bei den zuvor diskutierten Untersuchungen am glyoxal-modifizierten Insulin konnte
eindeutig und erstmals die Bildung der 2(1H)-Pyrazinon-Struktur am Protein unter
physiologischen Bedingungen nachgewiesen werden. Damit stellt sich die Frage, ob die
bekannten Reaktionsorte für Glyoxal also die Seitenketten von Lysin und Arginin ebenfalls
reagieren. Um dies zu klären, wurden die Massenspektren der LC-ESI-MS Untersuchungen
auf mögliche Arginin- und Lysin-Derivate (siehe Abschnitt 2.3.2) geprüft. Produkte wie
5-(4,5-Dihydroxy-2-imino-1-imidazolidinyl)-norvalin (Dihydroxyimidazolidin, Abb. 2.3.2-2,
∆Mr = +58,0), 5-(2-Imino-5-oxo-1-imidazolidinyl)-norvalin (Glarg, Abb. 2.3.2-2,
∆Mr = +40,0), N7-carboxymethylarginin (CMA, Abb. 2.3.2-2, ∆Mr = +58,0) und CML
(Abb. 2.3.2-1, ∆Mr = +58,0) konnten jedoch nicht detektiert werden. Weiterhin wurde eine
Aminosäureanalyse des glyoxal-modifizierten Insulins durchgeführt. Es war jedoch nur ein
tendenzieller und statistisch nicht signifikanter Abbau von Arginin und kein Abbau von Lysin
festzustellen. Spezifische Produkte des Arginins oder CML konnten nicht nachgewiesen
werden. Die Aminosäuresequenz des Insulins enthält als weitere basische Aminosäure
zweimal Histidin. Ein Abbau war mittels Aminosäureanalyse nicht feststellbar. Zu dem
gleichen Ergebnis kam Takahashi (1977a, b). Dieser beobachtete unter ähnlichen
Bedingungen ebenfalls keine Derivatisierung von Nα-Acetyl-histidin oder proteingebundenem
Histidin durch Glyoxal.
Um zu prüfen, welche Reaktionen mit 50 µM Glyoxal prinzipiell zu erwarten sind, wurden
entsprechende Modellexperimente mit 17 µM Gly-Ala-Tyr, 17 µM Nα-Hippuryllysin oder
17 µM Nα-Hippurylarginin unter den Bedingungen der Inkubation des Insulins (PBS,
pH = 7,4, 37 °C, 15 d) durchgeführt. Anhand der Analyse mittels HPLC konnte keine Bildung
von Nα-Hippuryl-Nε-carboxymethyllysin oder anderer Lysin-Derivate beobachtet werden. Als
primäre Ursache für die geringe Reaktivität des Lysins ist der hohe pKa-Wert der
ε-Aminogruppe anzusehen. Die ε-Aminogruppe des Lysins im Insulin weist z. B. einen
pKa-Wert von 11,1 auf (Gao, 1995). Folglich ist diese Aminogruppe bei pH = 7,4 vollständig
protoniert und damit nicht reaktiv. Bei dem Ansatz mit Nα-Hippurylarginin mit Glyoxal war
- 177 -
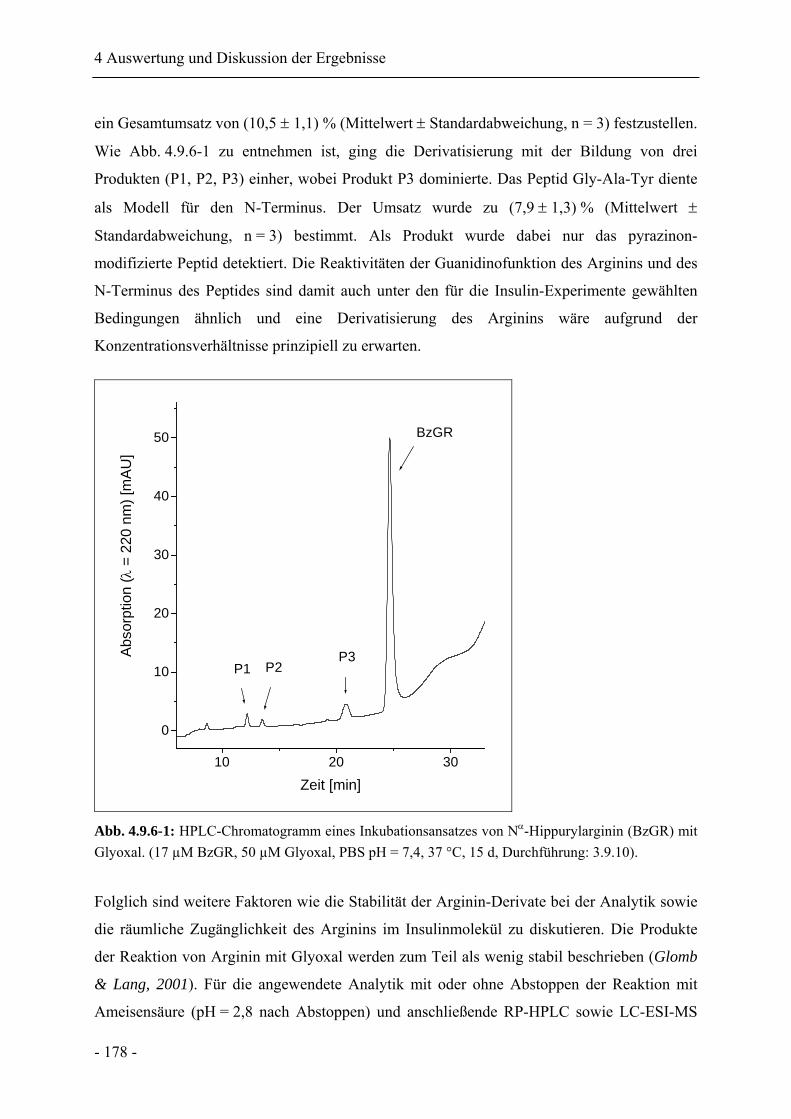
4 Auswertung und Diskussion der Ergebnisse
ein Gesamtumsatz von (10,5 ± 1,1) % (Mittelwert ± Standardabweichung, n = 3) festzustellen.
Wie Abb. 4.9.6-1 zu entnehmen ist, ging die Derivatisierung mit der Bildung von drei
Produkten (P1, P2, P3) einher, wobei Produkt P3 dominierte. Das Peptid Gly-Ala-Tyr diente
als Modell für den N-Terminus. Der Umsatz wurde zu (7,9 ± 1,3) % (Mittelwert ±
Standardabweichung, n = 3) bestimmt. Als Produkt wurde dabei nur das pyrazinon-
modifizierte Peptid detektiert. Die Reaktivitäten der Guanidinofunktion des Arginins und des
N-Terminus des Peptides sind damit auch unter den für die Insulin-Experimente gewählten
Bedingungen ähnlich und eine Derivatisierung des Arginins wäre aufgrund der
Konzentrationsverhältnisse prinzipiell zu erwarten.
10 20 30
0
10
20
30
40
50 BzGR
P3P2P1
Abs
orpt
ion
(λ =
220
nm
) [m
AU]
Zeit [min]
Abb. 4.9.6-1: HPLC-Chromatogramm eines Inkubationsansatzes von Nα-Hippurylarginin (BzGR) mit Glyoxal. (17 µM BzGR, 50 µM Glyoxal, PBS pH = 7,4, 37 °C, 15 d, Durchführung: 3.9.10).
Folglich sind weitere Faktoren wie die Stabilität der Arginin-Derivate bei der Analytik sowie
die räumliche Zugänglichkeit des Arginins im Insulinmolekül zu diskutieren. Die Produkte
der Reaktion von Arginin mit Glyoxal werden zum Teil als wenig stabil beschrieben (Glomb
& Lang, 2001). Für die angewendete Analytik mit oder ohne Abstoppen der Reaktion mit
Ameisensäure (pH = 2,8 nach Abstoppen) und anschließende RP-HPLC sowie LC-ESI-MS
- 178 -

4 Auswertung und Diskussion der Ergebnisse
jeweils mit 0,1 % Ameisensäure im Eluenten sollte die Stabilität jedoch gegeben sein. Wie
Abb. 4.9.6-2 zeigt, wurden mittels RP-HPLC identische Produktspektren und Peakflächen
erhalten, wenn ein Ansatz von Nα-Hippurylarginin und Glyoxal ohne Abstoppen oder nach
Abstoppen analysiert wird.
10 20 30
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7 P3
P2
P1
BzGR
B
A
Abso
rptio
n (λ
= 2
20 n
m) [
AU
]
Zeit [min]
Abb. 4.9.6-2: HPLC-Chromatogramme eines Inkubationsansatzes von Nα-Hippurylarginin (BzGR) mit Glyoxal. Injektion in das HPLC-System direkt nach der Inkubation (A) und nach Abstoppen (pH = 2,8) und 12 h Aufbewahrung bei Raumtemperatur (B). (Inkubation: 5 mM BzGR, 5 mM Glyoxal, PBS pH = 7,4, 37 °C, 15 d, Durchführung: 3.9.10).
Nakaya et al. (1967) untersuchten unter proteinchemischen Aspekten die Reaktion von
Insulin (0,5 mM) mit Glyoxal (z. B. 30 mM) bei pH = 9,2, (15 ± 5)° C und einer
Reaktionszeit von 3 h. Die Arginin-Derivatisierung erwies sich dabei als stark vom Zustand
des Proteins abhängig. So war der Arginin-Abbau beim nativen Insulin (ca. 4 % bei 30 mM
Glyoxal) stets deutlich kleiner als beim denaturierten Protein (ca. 36 % bei 30 mM Glyoxal).
Entsprechend der Struktur des Insulins im 2Zn-Kristall (Blundell et al., 1972) und der
Struktur in Lösung (Jørgensen et al., 1992) bildet die Guanidinofunktion des Arginins
- 179 -

4 Auswertung und Diskussion der Ergebnisse
(Arg-B22) eine Salzbrücke mit der C-terminalen Carboxylat-Gruppe von Asn-A22 aus. Dies
dürfte sowohl die Zugänglichkeit als auch die Nucleophilie der Guanidinogruppe herabsetzen.
Insgesamt betrachtet erscheint es damit als sehr wahrscheinlich, dass der Arginin-Rest Arg-22
für eine Reaktion mit Glyoxal unter den gewählten Inkubationsbedingungen nicht verfügbar
war und damit eine selektive Umsetzung an den N-Termini erfolgte.
4.9.6 Zur möglichen physiologischen Relevanz der 2(1H)-Pyrazinon-Bildung am
Insulin ist ein Peptidhormon mit großer physiologischer Bedeutung insbesondere für den
tiden mit Glyoxal unter physiologischen
roteinen kann sich die Glykierung auf die funktionellen
Insulin
Kohlenhydrat- und Lipidstoffwechsel aber auch für die Proteinbiosynthese. Die in vitro
beobachtete Bildung der 2(1H)-Pyrazinon-Struktur an den beiden N-Termini wirft die Frage
auf, ob diese Reaktion auch in vivo am Insulin oder anderen Proteinen oder Peptiden
stattfindet. Wenn ja, stellt sich die Frage nach der Bedeutung und den möglichen
physiologischen Folgen dieser Derivatisierung.
Da die Inkubationen von Insulin oder Pep
Bedingungen (pH = 7,4, 37 °C) und bei relativ niedrigen Konzentrationen (Insulin- und
Gly-Ala-Tyr-Ansätze) erfolgten, ist die 2(1H)-Pyrazinon-Bildung in vivo prinzipiell zu
vermuten. Dafür spricht auch die in vivo stattfindende Glykierung von Insulin durch Glucose
oder phosphorylierte Monosaccharide. So beträgt der Anteil des glykierten Insulins am
gesamten zirkulierenden Insulin im menschlichen diabetischen Plasma (Typ 2 Diabetiker)
ca. 9 % (Hunter et al., 2003). Bei Nicht-Diabetikern waren demgegenüber glykiertes Insulins
nicht nachweisbar (McKillop et al., 1999). Die Glykierung selbst findet nicht im Plasma statt,
sondern während der Synthese und Speicherung in den β-Zellen des Pankreas (Abdel-Wahab
et al., 1996). Aufgrund des intensiven Glucose-Metabolismus in den β-Zellen ist das Milieu
in diesem Zelltyp für intrazelluläre Glykierungen geradezu prädestiniert (Boyd et al., 2000).
Infolgedessen ist auch mit der Freisetzung von α-Dicarbonylverbindungen und der Bildung
von 2(1H)-Pyrazinonen zu rechnen.
Insbesondere bei biologisch aktiven P
Eigenschaften auswirken. So zeigte das an der α-Aminogruppe der B-Kette glykierte Insulin
eine gegenüber dem originären Insulin verminderte biologische Aktivität (O’Harte, 2000,
Boyd et al., 2000, Hunter et al., 2003). Im in vitro-Modell wirkte das monoglykierte Insulin
- 180 -

4 Auswertung und Diskussion der Ergebnisse
auf Muskelzellen beispielsweise signifikant weniger stimulierend auf den Glucosetransport
und Metabolismus. Die Hormon-Wirkung in vivo wurde an der Maus geprüft. Dabei zeigte
das monoglykierte Insulin ein signifikant vermindertes Vermögen den Plasmaglucosespiegel
zu erniedrigen und damit die Plasmaglucose-Homöostase zu regulieren (Boyd et al., 2000).
Da die Bindung des monoglykierten Insulins an den Insulin-Rezeptor und die metabolische
Elimination im Vergleich zum nativen Insulin unverändert waren, wurde ein
postrezeptorischer Effekt als Ursache für die beeinträchtigte Hormonwirkung angesehen
(Hunter et al., 2003). Auf Grund der Zirkulation glykierten Insulins bei Typ 2 Diabetes und
dessen reduzierter biologischer Aktivität wurde schließlich vermutet, dass die Glykierung von
Insulin mit für die bei Diabetes mellitus Typ 2 charakteristische Glucoseintoleranz und
Insulinresistenz verantwortlich ist (McKillop et al., 1999, 2001, Boyd et al., 2000, Hunter et
al., 2003).
Entsprechend der Untersuchungen am glykierten Insulin (O’Harte, 2000, Boyd et al., 2000,
.10 Zur möglichen Bildung von N-terminalen 2(1H)-Pyrazinonen in
Die Bildung von N-terminalen 2(1H)-Pyrazinonen konnte sowohl an Peptiden als auch an
Hunter et al., 2003) könnte auch das an der B-Kette pyrazinon-modifizierte Insulin eine
verminderte biologische Aktivität aufweisen. Da die N-terminale Aminogruppe der A-Kette
(Gly-A1) und die darauffolgende Peptidbindung zwischen Gly-A1 und Ile-A2 für die
Rezeptorbindung von Bedeutung sind (Nakagawa & Tager, 1992, Olsen et al., 1998), dürfte
dies auch für das an der A-Kette pyrazinon-modifizierte Insulin zutreffen. Insgesamt
betrachtet wäre es somit interessant auf das Vorkommen von pyrazinon-modifizierten Insulin
in vivo sowie dessen biologische Aktivität zu prüfen.
4
Lebensmitteln und in vivo
dem Protein Insulin nachgewiesen werden (Abschnitt 4.3, 4.5, 4.9). Als potente Precursoren
erwiesen sich dabei die α-Dicarbonylverbindungen Glyoxal, Methylglyoxal und
3-Desoxyglucosulose (Abschnitt 4.4.2). Die Bildungsreaktion erfolgt bereits bei
physiologischer Temperatur in einem weiten pH-Bereich von pH = 6 bis 11 (Abschnitt 4.4.4).
Weiterhin wurde jeweils eine nahezu gleiche Geschwindigkeit der 2(1H)-Pyrazinon-Bildung
und der Derivatisierung der Guanidinofunktion des Arginins durch Glyoxal, Methylglyoxal
oder 3-Desoxyglucosulose beobachtet. Die ε-Aminogruppe des Lysins reagierte
- 181 -

4 Auswertung und Diskussion der Ergebnisse
demgegenüber nicht (Abschnitt 4.7). Aus Lebensmitteln und in vivo sind bereits zahlreiche
Reaktionsprodukte von Glyoxal, Methylglyoxal oder 3-Desoxyglucosulose mit den
Aminosäureseitenketten von Lysin oder Arginin bekannt (Abschnitt 2.3.2). Da die
Precursoren N-terminaler 2(1H)-Pyrazinone, α-Dicarbonylverbindungen sowie Peptide und
Proteine, sowohl in Lebensmitteln als auch in vivo vorhanden sind, ist folglich insgesamt
betrachtet fest anzunehmen, dass eine Bildung von 2(1H)-Pyrazinonen in Lebensmitteln und
in vivo stattfindet.
Als Indiz für die Bildung von 2(1H)-Pyrazinonen kann die im fortgeschrittenen Stadium der
illard-spezifischen Fluoreszenz wird ebenfalls in vivo beobachtet. Die
2(1H)-Pyrazinone einen Teil der beobachteten Fluoreszenz erklären können. Besonders
Maillard-Reaktion auftretende Fluoreszenz gewertet werden. In Lebensmitteln und
entsprechenden Modellsystemen wird durch eine Wärmebehandlung eine Zunahme der
Fluoreszenzintensität beobachtet. Auf Basis dessen konnte Birlouez-Aragon et al. (1998,
2001) ein auf Fluoreszenzmessung beruhendes Analysenverfahren zur Beurteilung der
Wärmebehandlung etabliert werden. Die Anwendung erfolgte z. B. bei Milch, Cornflakes,
frittiertem Lachs und gerösteten Sojabohnen. Die maximalen Anregungs- und Emissions-
wellenlängen der Maillard-spezifischen Fluoreszenz waren dabei vom Lebensmittel abhängig
und bewegten sich für die Anregung im Bereich von λex, max = 320 nm bis 350 nm und für die
Emission im Bereich von λem, max = 395 nm bis 440 nm. Über die Struktur der
verantwortlichen individuellen Fluorophore konnten jedoch keine Angaben getroffen werden.
Da die identifizierten N-terminalen 2(1H)-Pyrazinone mit einer maximalen Anregungs-
wellenlänge von λex, max = 330 bis 340 nm und einer maximalen Emissionswellenlänge von
λem, max = 395 nm bis 405 nm ebenfalls fluoreszieren, könnte ein Teil der in Lebensmitteln
beobachteten Fluoreszenz auf das Vorkommen dieser N-terminalen Modifikation
zurückzuführen sein.
Das Auftreten der Ma
Intensität nimmt mit steigendem Alter der Proteine zu und tritt verstärkt bei Diabetes mellitus
oder Urämie auf. Auf Grund dessen wird die Fluoreszenz ähnlich wie bei Lebensmitteln als
Summenparameter für den AGE-Status genutzt (Yanagisawa et al., 1998, Henle et al., 1999).
Die Messung der Fluoreszenz erfolgt dabei oft mit der Wellenlängenkombination des
Pentosidins mit λex = 335 nm und λem = 385 nm oder bei höheren Wellenlängen, z. B. mit
λex = 370 nm und λem = 440 nm (Sell & Monnier, 1989, Yanagisawa et al., 1998, Avendano et
al., 1999). Abgesehen vom Pentosidin ist über die Struktur der verantwortlichen individuellen
Fluorophore wenig bekannt. Es ist wiederum zu vermuten, dass N-terminale
- 182 -

4 Auswertung und Diskussion der Ergebnisse
wahrscheinlich erscheint dies bei den sogenannten AGE-Peptiden. AGE-Peptide sind
fluoreszierende Peptide mit einer Masse bis 10 kDa die im Blut zirkulieren. Die Gehalte im
Serum und im Urin sind insbesondere bei Diabetikern mit Nierenfehlfunktion erhöht
(Yanagisawa et al., 1998). Durch Hämodialyse werden diese Peptide aus dem Blut entfernt
(Henle et al., 1999, Floridi et al., 2002). Hämodialysate solcher Patienten wären deshalb ein
geeigneter Ausgangspunkt um auf die in vivo-Bildung N-terminale 2(1H)-Pyrazinone zu
prüfen. Die gegenüber Gesunden erhöhten Gehalte an α-Dicarbonylverbindungen im Blut
(Odani et al., 1999b) lassen ebenfalls die 2(1H)-Pyrazinon-Bildung bei Urämikern vermuten.
Auf Grund der hohen Reaktivitäten von Glyoxal und Methylglyoxal sowie der vermuteten
quantitativen Bedeutung der 3-Desoxyglucosulose sind sowohl in Lebensmitteln als auch
in vivo die N-terminalen 2(1H)-Pyrazinone aller drei α-Dicarbonylverbindungen zu erwarten
(siehe Abschnitt 4.6). Weitere α-Dicarbonylverbindungen wie z. B. 1-Desoxyglucosulose
könnten ebenfalls potente Precursoren sein. Eine hohe Anzahl möglicher 2(1H)-Pyrazinone
resultiert aus der N-terminalen Sequenz der Peptide oder Proteine. An der 2(1H)-Pyrazinon-
Bildung sind die erste und zweite Aminosäure vom N-Terminus beginnend betroffen. Es gibt
insgesamt 342 Möglichkeiten, zwei Aminosäuren der neunzehn für die 2(1H)-Pyrazinon-
Bildung in Frage kommenden Aminosäuren (alle proteinogenen Aminosäuren außer Prolin)
zu variieren. Folglich sind bereits 342 2(1H)-Pyrazinon-Dipeptide des Glyoxals formulierbar.
In Hinblick auf den Nachweis einer möglichen 2(1H)-Pyrazinon-Bildung in Lebensmitteln
und in physiologischen Proben wäre es deshalb sinnvoll, gezielt auf diese an bestimmten
ausgewählten Proteinen oder Peptiden zu prüfen. Die Analyse könnte wie der Nachweis am
Insulin mittels RP-HPLC mit UV- und Fluoreszenz-Detektion sowie LC-MS erfolgen. Um
den Nachweis spezifisch zu gestalten, sollte die Analyse ebenfalls nach geeigneter
enzymatischer Hydrolyse durchgeführt werden.
- 183 -


5 Zusammenfassung
Zur Bildung von Furosin aus Amadori-Verbindungen des Lysins
Furosin entsteht bei der Salzsäurehydrolyse aus den Amadori-Produkten des Lysins und wird
als Marker für den Fortschritt der frühen Maillard-Reaktion, zur Beurteilung von
lebensmitteltechnologischen Prozessen sowie zur Berechnung des verfügbaren und des nicht
verfügbaren Lysins verwendet. Für die Nutzung von Furosin als Qualitätsparameter ist die
reproduzierbare und konstante Bildung während der Salzsäurehydrolyse entscheidend. Dies
wird in der Literatur jedoch kontrovers diskutiert. Im ersten Abschnitt dieser Arbeit galt es
deshalb die molaren Ausbeuten an Furosin und den anderen Hydrolyseprodukten zu
bestimmen und damit eine sichere Interpretation der Ergebnisse zu ermöglichen.
1 Es wurden sowohl die in Lebensmitteln als auch die in vivo quantitativ wichtigen Amadori-
Produkte des Lysins in Form der Nα-Hippuryl-Derivate Nα-Hippuryl-Nε-fructosyl-lysin,
Nα-Hippuryl-Nε-tagatosyl-lysin, Nα-Hippuryl-Nε-lactulosyl-lysin und Nα-Hippuryl-
Nε-maltulosyl-lysin dargestellt. Die chromatographische Isolierung mittels RP-HPLC konnte
dabei durch eine vorgelagerte enzymatische Hydrolyse von nicht umgesetztem
Nα-Hippuryllysin effizient gestaltet werden. Weiterhin wurde freies Nε-Fructosyl-lysin sowie
Pyridosin als Standard gewonnen. Die Absicherung der erforderlichen Reinheit der
Verbindungen erfolgte chromatographisch. Ihre Identität konnte mittels Massenspektroskopie
sowie durch ein- und zweidimensionale NMR-Experimente sichergestellt werden.
2 Die Hydrolyse der Amadori-Verbindungen erfolgte mit der für die Aminosäureanalytik
üblichen Salzsäurekonzentration von 6 M sowie mit der bei der Furosin-Bestimmung oft
verwendeten Salzsäurekonzentration von 8 M. Die Quantifizierung der Hydrolyseprodukte
Furosin, Pyridosin, Lysin und CML in den Hydrolysaten konnte mittels Kationenaustausch-
chromatographie und Nachsäulenderivatisierung mit Ninhydrin erreicht werden.
3 Die Fructosyl-Verbindungen Nε-Fructosyl-lysin, Nε-Lactulosyl-lysin und Nε-Maltulosyl-
lysin zeigten ein ähnliches Hydrolyseverhalten, welches sich deutlich von dem von
Nε-Tagatosyl-lysin unterschied. Nach Hydrolyse mit 6 M Salzsäure waren bei den Fructosyl-
- 185 -

5 Zusammenfassung
Amadori-Produkten molare Ausbeuten an Furosin zwischen 32 und 35 % feststellbar. Die
Furosin-Bildung aus Nε-Tagatosyl-lysin war demgegenüber mit 42 % signifikant höher. Bei
Hydrolyse mit 8 M Salzsäure wurden bei den Fructosyl-Amadori-Produkten deutlich höhere
molare Ausbeuten zwischen 46 und 51 % bestimmt. Die Bildung aus Nε-Tagatosyl-lysin blieb
bei dieser Konzentration hingegen unverändert bei 42 %. Für die Bestimmung der Lysin-
Derivatisierung bei Proben, welche sowohl Nε-Fructosyl-lysin als auch Nε-Tagatosyl-lysin
enthalten, wie z. B. lactose-hydrolysierte Milchprodukte, wird deshalb die Hydrolyse mit
7,3 M Salzsäure empfohlen. Bei dieser Konzentration liefern beide Amadori-Produkte die
gleiche Menge an Furosin.
4 Die auf Basis der molaren Ausbeuten ermittelten Überführungsfaktoren ermöglichen nun
erstmals eine sichere Bestimmung der Lysin-Derivatisierung auf der Basis der Analytik von
Furosin. Damit sind jetzt exakte Aussagen zum Fortschritt nichtenzymatischer
Glykierungsreaktionen sowohl in Lebensmittel als auch in vivo möglich.
Zur Bildung von N-terminalen 2(1H)-Pyrazinonen
Aufgrund der physiologischen Relevanz von in vivo stattfindenden Glykierungsreaktionen ist
die Untersuchung der Reaktion von Proteinen mit α-Dicarbonylverbindung ein intensiv
untersuchtes Gebiet. Dabei befasste man sich bisher vorzugsweise mit Umsetzungen an den
nucleophilen Seitenketten von Lysin und Arginin. Mögliche Reaktionen am N-Terminus
wurden demgegenüber unter physiologischen Bedingungen (pH = 7,4 und 37 °C) bislang
nicht betrachtet und sollten deshalb der Gegenstand von weiteren Untersuchungen sein.
5 Die Inkubation des Tripeptides Gly-Ala-Phe mit Glyoxal unter physiologischen
Bedingungen führte zu einer sehr schnellen Derivatisierung des Peptides und der
gleichzeitigen Bildung eines definierten Produktes. Nach Isolierung mittels semipräparativer
RP-HPLC konnte das Reaktionsprodukt durch Massenspektroskopie und NMR-Analyse
zweifelsfrei als N-[2-(2-Oxo-2H-pyrazin-1-yl)-propyl]-phenylalanin identifiziert werden. Die
Verbindung erwies sich in einem weiten pH-Bereich, bei erhöhter Temperatur sowie in
Gegenwart eines Überschusses des α-Dicarbonyl-Reagenzes Aminoguanidin als stabil.
- 186 -

5 Zusammenfassung
6 Bei der Reaktion anderer ausgewählter Peptide, wie Gly-Phe-Ala, Phe-Gly-Gly,
Gly-Gly-Gly, Phe-Gly, Gly-Ile und Gly-Phe, mit Glyoxal war ebenfalls die spezifische
Bildung der entsprechenden 2(1H)-Pyrazinon-Peptide festzustellen. Dabei zeigte sich, dass
die Geschwindigkeit der Reaktion von der Primärstruktur des Peptides, vom pH-Wert und der
Ionenstärke abhängig ist. Die Inkubation von Gly-Ala-Phe mit anderen
α-Dicarbonylverbindung wie Methylglyoxal und 3-Desoxyglucosulose führte ebenfalls zu
den entsprechenden 2(1H)-Pyrazinon-Peptiden sowie weiteren Produkten. Die Reaktion war
damit weniger spezifisch.
7 Nach Inkubation von Gly-Ala-Phe mit Methylglyoxal wurde das Hauptreaktionsprodukt in
chromatographisch reiner Form isoliert und strukturell eindeutig als N-[2-(5-Methyl-2-oxo-
2H-pyrazin-1-yl)-propyl]-phenylalanin aufgeklärt. Die Reaktion mit Methylglyoxal führt
damit regioselektiv zum 5-Methyl-2(1H)-pyrazinon-Derivat.
8 Die identifizierten 2(1H)-Pyrazinon-Derivate sind durch eine charakteristische Bande im
UV oberhalb 300 nm (λUV, max = 322 nm bis 336 nm) und die Eigenschaft zu fluoreszieren
(λex, max = 330 nm bis 340 nm, λem, max = 395 nm bis 405 nm) gekennzeichnet. Dies ermöglicht
die gezielte Analyse mittels RP-HPLC mit UV- und Fluoreszenz-Detektion. Eine
Abhängigkeit der UV- und Fluoreszenz-Charakteristika vom pH-Wert (pH = 2,4 bis 8,3) war
nicht feststellbar.
9 Um die Bedeutung der N-terminalen 2(1H)-Pyrazinon-Bildung beurteilen zu können,
wurden vergleichende Inkubationsexperimente mit peptidgebundenem Lysin und Arginin
sowie Gly-Ala-Phe durchgeführt. Dabei wurde eine ähnliche bis nahezu identische Reaktivität
des N-Terminus von Gly-Ala-Phe und Nα-Hippurylarginin gegenüber Glyoxal, Methylglyoxal
und 3-DG festgestellt. Nα-Hippuryllysin zeigte unter den gewählten Bedingungen hingegen
keine Reaktion. Die Bildung von 2(1H)-Pyrazinonen dürfte damit sowohl in vivo als auch in
Lebensmitteln von Bedeutung sein.
10 Zum Nachweis der 2(1H)-Pyrazinon-Bildung an einem Protein wurde Insulin mit Glyoxal
unter physiologischen Bedingungen in verdünnter Lösung inkubiert. Die Untersuchungen
erfolgten mittels RP-HPLC mit UV- und Fluoreszenz-Detektion sowie mittels LC-ESI-MS
direkt nach der Inkubation am intakten Insulin, am Insulin nach Separierung der Ketten sowie
- 187 -

5 Zusammenfassung
nach enzymatischer Hydrolyse der separierten Ketten. Anhand dessen konnte die Bildung der
2(1H)-Pyrazinon-Struktur sowohl an der A-Kette als auch an der B-Kette eindeutig bewiesen
werden. Dabei wurde eine bevorzugte Derivatisierung des N-Terminus der B-Kette gegenüber
dem der A-Kette festgestellt.
11 Die Reaktion von Insulin mit Glyoxal unter physiologischen Bedingungen in verdünnter
Lösung wurde ebenfalls in Hinblick auf eine Derivatisierung weiterer potentieller
Reaktionsorte geprüft. Modifikationen der Seitenketten des Argininrestes Arg-B22 und des
Lysinrestes Lys-B29 waren durch Anwendung von LC-ESI-MS und Aminosäureanalyse nicht
nachweisbar. Bei Modellumsetzungen von Nα-Hippurylarginin und Nα-Hippuryllysin jeweils
mit Glyoxal unter identischen Bedingungen fanden demgegenüber Reaktionen am Arginin
statt. Nα-Hippuryllysin reagierte nicht. Als Ursache für die verminderte Reaktivität des
proteingebundenen Argininrestes Arg-B22 ist damit die schlechte Zugänglichkeit der
Guanidino-Gruppe in der Raumstruktur des Insulins anzusehen.
12 Es wurde eine Analysenstrategie zum Nachweis peptid- und proteingebundener
2(1H)-Pyrazinone entwickelt. Diese beinhaltet im ersten Schritt die Freisetzung der
N-terminalen 2(1H)-Pyrazinon-Struktur als N1-Alkyl-2(1H)-pyrazinon durch Hydrolyse mit
Salzsäure. Daran schließt sich der Nachweis mittels RP-HPLC mit UV- und Fluoreszenz-
Detektion sowie mittels LC-ESI-MS an. Das Verfahren konnte erfolgreich an glyoxal-
modifizierten Insulin angewendet werden.
Zusammenfassend ist festzustellen, dass die Bildung von 2(1H)-Pyrazinonen am N-Terminus
von Peptiden und am Protein Insulin durch Reaktion mit α-Dicarbonylverbindungen unter
physiologischen Bedingungen aufgeklärt und bewiesen werden konnte. In den verwendeten
Modell-Systemen zeigte der N-Terminus eine ähnliche Reaktivität wie peptidgebundenes
Arginin. Aufgrund dessen und der durchgeführten Untersuchungen zur Stabilität ist fest
davon auszugehen, dass es sich bei Verbindungen mit 2(1H)-Pyrazinon-Struktur um eine neue
Klasse von AGEs (advanced glycation end-products) mit Bedeutung in physiologischen
Systemen als auch in Lebensmitteln handelt.
- 188 -

6 Ausblick
Die Ergebnisse der im ersten Abschnitt dieser Arbeit durchgeführten Studien zur Analytik
von Amadori-Produkten erlauben eine sichere Bestimmung von Furosin und daraus ableitbare
Berechnung der Lysin-Derivatisierung. Insbesondere bei stark fructosehaltigen Backwaren
wie z. B. bestimmte Diabetikerbackwaren oder Honiggebäcken ist eine dominante
Derivatisierung des Lysins als Heyns-Produkt sowie 3-Keto-Umlagerungsprodukt zu
vermuten. Aufgrund des Fehlens einer geeigneten Analysenmethode für diese Verbindungen
liegen jedoch über diese Derivatisierungen kaum Erkenntnisse vor. Die Etablierung einer
entsprechenden Analytik ist demzufolge erstrebenswert und sollte Gegenstand zukünftiger
Untersuchungen sein.
Anhand der Studien zur Reaktion von Peptiden oder Insulin mit α-Dicarbonylverbindungen
ist davon auszugehen, dass die Bildung proteingebundener N-terminaler 2(1H)-Pyrazinone
ebenfalls sowohl in vivo als auch in Lebensmitteln stattfindet. Weitere Untersuchungen
sollten sich deshalb mit dem Nachweis dieser Derivatisierung in entsprechenden Proben
befassen. Für die chemische Analytik könnte dabei insbesondere die charakteristische
Fluoreszenz der 2(1H)-Pyrazinone genutzt werden. Wenn die 2(1H)-Pyrazinon-Modifizierung
bestimmter Proteine wie z. B. von Insulin vermutet wird, sollte durch die Verwendung
monoklonaler Antikörper eine sehr spezifische und nachweisstarke Detektion möglich sein.
Weitere Studien sollten sich mit physiologische Aspekten befassen. Dabei könnte in Hinblick
auf eine alimentäre Aufnahme zunächst die Verdau- und Resorbierbarkeit von pyrazinon-
modifizierten Proteinen sowie die Art der Metabolisierung und Elimination der
2(1H)-Pyrazinon-Struktur geprüft werden. Im Zusammenhang mit der möglichen Bildung von
2(1H)-Pyrazinonen in vivo sind insbesondere mögliche Auswirkungen auf die Funktion der
modifizierten Proteine zu betrachtet.
- 189 -

7 Literaturverzeichnis
Abdel-Wahab, Y.H.A., O’Harte, F.P.M., Ratcliff, H., McClenaghan, N.H., Barett, C.R., Flatt, P.R.
(1996). Glycation of insulin in the islets of Langerhans of normal and diabetic animals.
Diabetes 45, 1489-1496
Abrams, A., Lowy, P.H., Borsook, H. (1955). Preparation of 1-Amino-1-deoxy-2-ketohexosees from
aldohexoses and α-amino acids. I. Journal of the American Chemical Society 77, 4794-4796
Acquistucci, R. (2000). Influence of Maillard reaction on protein modification and colour
development in pasta. Comparison of different drying conditions. Lebensmittel-Wissenschaft
und -Technologie 33, 48-52
Advanced Chemistry Development (ACD). Software Solaris V 4.67 (© 1994-2004 ACD). In:
American Chemical Society. Scifinder Scholar, Version 2004
Aeschbacher, H.U. (1990). Anticarcinogenic effect of browning reaction products. In: Finot, P.A.,
Aeschbacher, H.U., Hurrell, R.F., Liardon, R. (Ed.), The Maillard reaction in food processing,
human nutrition and physiology. Basel: Birkhäuser Verlag, Advances in Life Sciences, 245-258
Agoston, C., Janlowska, T., Sovago, I.. (1999). Potentiometric and NMR studies on palladium (II)
complexes of oligoglycines and related ligands with non-coordinating side chains. Journal of
the Chemical Society - Dalton Transactions 16-18, 3295-3302
Ahmed, M.U., Thorpe, S.R., Baynes, J.W. (1986). Identification of Nε-carboxymethyllysine as a
degradation product of fructoselysine in glycated protein. Journal of Biological Chemistry 261,
4889-4894
Ahmed, M.U., Brinkmann Frye, E., Degenhardt, T.P., Thorpe, S.R., Baynes, J.W. (1997).
Nε-carboxyethyllysine, a product of the chemical modification of proteins by methylglyoxal,
increases with age in human lens proteins. Biochemical Journal 324, 565-570
Al-Abed, Y., Bucala, R. (1995). Nε-carboxymethyllysine formation by direct addition of glyoxal to
lysine during the Maillard reaction. Bioorganic & Medicinal Chemistry Letters 5, 2161-2162
Albert, A., Phillips, J.N. (1956). 264. Ionization constants of heterocyclic substances. Part II.
Hydroxy-derivatives of nitrogenous six-membered ring-compounds. Journal of the Chemical
Society, 1294-1304
Altena, J.H., Van De Ouweland, G.A.M., Teunis, C.J., Tjan, S.B. (1981). Analysis of the 220-MHz,
P.M.R. spectra of some products of the Amadori and Heyns rearrangements. Carbohydrate
Research 92, 37-49
- 190 -

7 Literaturverzeichnis
Amadori, M. (1925). Prodotti di condensazione tra glucosio e p-fenetidina. Atti Rendiconti Accademia
Nazionale dei Lincei [Roma] 2 (6.Ser.), 337-342
Amaya-F, J., Lee, T.-C., Chichester, C.O. (1976). Biological inactivation of proteins by the Maillard
reaction: Effect of mild heat on the tertiary structure of insulin. Journal of Agricultural and
Food Chemistry 24, 465-467
Ambler, R.P. (1972). Enzymatic hydrolysis with carboxypeptidases. Methods in Enzymology 25, 143-
154
Anderson, M.M., Hazen, S.L., Hsu, F.F., Heinecke, J.W. (1997). Human neutrophils employ the
myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into
glycolaldehyde, 2-hydroxypropanal and acrolein. Journal of Clinical Investigation 99, 424-432
Anese, M, Calligaris, S., Nicoli, M.C., Massini, R. (2003). Influence of total solids concentration and
temperature on the changes in redox potential of tomato pastes. International Journal of Food
Science and Technology 38, 55-61
Anet, E.F.L.J. (1962). Formation of furan compounds from sugars. Chemistry and Industry, 262
Anet, E.F.L.J. (1964). 3-Deoxyglucosuloses (3-Deoxyglucosones) and the degradation of
carbohydrates. Advances in Carbohydrate Chemistry 19, 181-218
Anet, E.F.L.J. (1970). Proton exchange during the conversion of hexose into 5-hydroxymethyl-
2-furaldehyde. Australian Journal of Chemistry 23, 2383-2385
Avendano, G.F., Agarwal, R.K., Bashey, R.I., Lyons, M.M., Soni, B.J,. Jyothirmayi, G.N., Regan, T.J.
(1999). Effects of glucose intolerance on myocardial function and collagen-linked glycation.
Diabetes 48, 1443-1447
Babiker, El Fadil E., Hiroyuki, A., Matsudomi, N., Iwata, H., Ogawa, T., Bando, N., Kato, A. (1998).
Effect of polysaccaride conjugation or transglutaminase treatment on the allergenicity and
functional properties of soy protein. Journal of Agricultural and Food Chemistry 46, 866-871
Baddar, F. G. (1949). The electronic interpretation of the Strecker degradation. Journal of the
Chemical Society, S163-S167
Bailey, A.J., Paul, R.G., Knott, L. (1998). Mechanism of maturation and aging of collagen.
Mechanisms of Aging and Development 106, 1-56
Baddar, F.G. (1950). Triad prototropic systems. Part I. The mobility of the azomethine system during
decarboxylation. Journal of the Chemical Society, 136-139
Barros, A., Rodrigues, J.A., Almeida, P.J., Oliva-Teles, M.T. (1999). Determination of glyoxal,
methylglyoxal, and diacetyl in selected beer and wine, by HPLC with UV spectrophotometric
detection, after derivatization with o-phenylenediamine. Journal of Liquid Chromatography &
Related Technology 22, 2061-2069
- 191 -

7 Literaturverzeichnis
Baynes, J.W. (2000). From life to death - the struggle between chemistry and biology during aging:
the Maillard reaction as an amplifierer of genomic damage. Biogerontology 1, 235-246
Baynes, J.W. (2001). The role of AGEs in aging: causation or correlation. Experimental Gerontology
36, 1527-1537
Baynes, J.W., Watkins, N.G., Fisher, C.I., Hull, C.J., Patrick, J.S., Ahmed, M.U., Dunn, J.A., Thorpe,
S.R. (1989). The Amadori product on protein: Structure and reactions. In: Baynes, J.W.,
Monnier, V.M. (Ed.), The Maillard reaction in aging, diabetes and nutrition. New York: Alan R.
Liss, Inc., 43-67
Beksan, E., Schieberle, P., Robert, S., Blank, I., Fay, L. B., Schlichtherle-Cerny, H., Hofmann, T.
(2003). Synthesis and sensory characterization of novel umami-tasting glutamate
glycoconjugates. Journal of Agricultural and Food Chemistry 51, 5428-5436
Billaud, C., Adrian, J. (2003). Louis-Camille Maillard, 1878-1936. Food Reviews international 19,
345-374
Birlouez-Aragon, I., Nicolas, M., Metais, A., Marchond, N., Grenier, J., Calvo, D. (1998). A rapid
fluorimetric method to estimate the heat treatment of liquid milk. International Dairy Journal 8,
771-777
Birlouez-Aragon, I., Leclere, J., Quedraogo, C.L., Birlouez, E., Grongnet, J.-F. (2001). The FAST
method, a rapid approach of the nutritional quality of heat-treated foods. Nahrung/Food 45,
201-205
Blundell, T.L., Dodson, G.G., Hodgkin, D.M.C., Mercola, D.A. (1972). Insulin: The structure in the
crystal and its reflection in chemistry and biology. Advances in Protein Chemistry 26, 279-402
Bodanszky, M. (1993). Peptide chemistry. Berlin: Springer-Verlag, 159
Bolli, G., Compagnucci, P., Cartechini, M.G., De Feo, P., Santeusanio, F., Brunetti, P. (1981).
Analysis of short-term changes in reversibly and irreversibly glycosylated haemoglobin AI:
Relevance to diabetes mellitus. Diabetologia 21, 70-72
Bolli G., Cartechini, M.G., Compagnucci, P., De Feo, P., Santeusanio, F., Brunetti, P. (1982). Kinetic
behaviour of the glucose-hemoglobin labile adduct in normal and diabetic erythrocytes. Diabete
& Metabolisme (Paris) 21, 21-27
Bookchin R.M., Gallop P.M. (1968). Structure of hemoglobin AIc: Nature of the N-terminal beta
chain blocking group. Biochemical and Biophysical Research Communications 32, 86-93
Boyd A.C., Abdel-Wahab, Y.H.A., McKillop, A.M., McNulty, H., Barett, C.R., O’Harte, F.P.M.,
Flatt, P.R. (2000). Impaired ability of glycated insulin to regulate plasma glucose and stimulate
glucose transport and metabolism in mouse abdominal muscle. Biochimica et Biophysica Acta
1523, 128-134
- 192 -

7 Literaturverzeichnis
Brandt, A., Erbersdobler, H.F. (1972). Zur Bestimmung von Furosin in Nahrungs- und Futtermitteln.
Landwirtschaftliche Forschung, Sonderheft, 28/II, 115-119
Brinkmann, E., Wells-Knecht, K.J., Thorpe, S.R., Baynes, J.W. (1995). Characterization of an
imidazolium compound formed by the reaction of methylglyoxal and Nα-hippuryllysine.
Journal of the Chemical Society, Perkin Transactions 1, 2817-2818
Bujard, E., Finot, P.A. (1978). Mesure de la disponibilite et du blocage de la lysine dans les laits
industriels. Annales de la Nutrition et de l'Alimentation 32, 291-305
Bunn, H.F., Haney, D.N., Gabbay, K.H., Gallop, P.M. (1975). Further identification of the nature and
the linkage of the carbohydrate in hemoglobin AIc. Biochemical and Biophysical Research
Communications 36, 103-109
Bunn, H.F., Shapiro, R., McManus, M., Garrick, L., McDonald, M.J., Gallop, P.M., Gabbay, K.H.
(1979). Structural heterogeneity of human hemoglobin A due to nonenzymatic glycosylation.
Journal of Biological Chemistry 254, 3892-3898
Bunn, H.F., Higgins, P.J. (1981). Reaction of monosaccharides with proteins: Possible evolutionary
significance. Science 213, 222-224
Büser, W., Erbersdobler, H.F. (1985). Determination of furosine by gas-liquid chromatography.
Journal of Chromatography 346, 363-368
Brownlee, M. (2000). Negative consequences of glycation. Metabolism 49 Suppl. 1, 9-13
Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature 414,
813-820
Brüggemann, J., Erbersdobler, H.F. (1968). Fructoselysin als wichtiges Reaktionsprodukt von Lysin
mit Glucose bei Hitzeschädigung von Lebens- und Futtermitteln. Zeitschrift für Lebensmittel-
Untersuchung und -Forschung 137, 137-143
Cai, J., Hurst, H.E. (1999). Identification and quantitation of N-(carboxymethyl)valine adduct in
hemoglobin by gas chromatography/mass spectrometry. Journal of Mass Spectrometry 34, 537-
543
Cämmerer, B., Jalyschko, W., Kroh, L.W. (2002). Intact carbohydrate structures as part of the
melanoidin skeleton. Journal of Agricultural and Food Chemistry 50, 2083-2087
Cämmerer, B., Kroh, L.W. (1995). Investigation of the influence of reaction conditions on the
elementary composition of melanoidins. Food Chemistry 53, 55-59
Carpenter, K.J., Booth, V.H. (1971). Damage to lysine in food processing: Its measurement and its
significance. Nutrition Abstracts & Reviews 43, 423-451
Ciner-Doruk, M., Eichner, K. (1979). Bildung und Stabilität von Amadori-Verbindungen in wasser-
armen Lebensmitteln. Zeitschrift für Lebensmittel-Untersuchung und -Forschung 168, 9-20
- 193 -

7 Literaturverzeichnis
Claeys, W.L., Ludikhuyze, L.R., Hendrickx, M.E. (2001). Formation kinetics of hydroxymethyl-
furfural, lactulose and furosine in milk heated under isothermal and non-isothermal conditions.
Journal of Dairy Research 68, 287-301
Claeys, W.L., Van Loey, A.M., Hendrickx, M.E. (2002). Intrinsic time temperature integrators for
heat treatment of milk. Trends in Food Science & Technology 13, 293-311
Clawin-Rädecker, I, Koehn, A., Schlimme, E. (1996). Furosin: Hitzeindikator in Milch und
Frischkäse. Deutsche Milchwirtschaft 47, 902-905
Corzo, N., Delgado, T., Troyano, E., Olano, A. (1994). Ratio of lactulose to furosine as indicator of
quality of commercial milks. Journal of Food Protection 57, 737-739
Dallavalle, E., Fisicaro, E., Corradini, R., Marchelli, R. (1989). Complex formation equilibria of
L-Amino-acid amides with copper (II) in aquous solution. Helvetica Chimica Acta 72, 1479-
1486
Davidek, T., Velisek, J., Davidek, J., Pech, P. (1991). Glycylglycyl-derived 1,3-disubstituted
imidazole in nonenzymatic browning reactions. Journal of Agricultural and Food Chemistry 39,
1374-1377
De Bruyn, C.A.L., Van Ekenstein, W.A. (1895). Einwirkung von Alkalien auf Kohlenhydrate.
Wechselseitige Umwandlung von Glucose, Fructose und Mannose in einander. Berichte der
Deutschen Chemischen Gesellschaft 28, 3078-3082
Del Castillo, M.D., Corzo, N., Polo, M.C., Pueyo, E., Olano, A. (1998). Changes in the amino acid
composition of dehydrated orange juice during accelerated nonenzymatic browning. Journal of
Agricultural and Food Chemistry 46, 277-280
Del Castillo, M.D., Corzo, N., Olano, A. (1999). Early stages of Maillard reaction in dehydrated
orange juice. Journal of Agricultural and Food Chemistry 47, 4388-4390
Delpierre, G., Collard, F., Fortpied, J., Van Schaftingen, E. (2002). Fructosamine 3-kinase is involved
in an intracellular deglycation pathway in human erythrocytes. Biochemical Journal 365, 801-
808
De Revel, G., Bertrand, A. (1993). A method for the detection of carbonyl compounds in wine:
Glyoxal and methylglyoxal. Journal of the Science of Food and Agriculture 61, 267-272
Dotsikas, Y., Loukas, Y.L. (2002): Kinetic degradation study of insulin complexed with methyl-beta
cyclodextrin. Confirmation of complexation with electrospray mass spectrometry and 1H NMR.
Journal of Pharmaceutical and Biomedical Analysis 29, 487-494
Drapeau, G.R., Boily, Y., Houmard, J. (1972). Purification and properties of an extracellular protease
of staphylococcus aureus. Journal of Biological Chemistry 247, 6720-6726
Drusch, S., Faist, V., Erbersdobler, H.F. (1999). Determination of Nε-carboxymethyllysine in milk
products by a modified reversed-phase HPLC method. Food Chemistry 65, 547-553
- 194 -

7 Literaturverzeichnis
Dutscher, I.D. (1947). Aspergillic acid: An antibiotic substance produced by aspergillus flavus.
Journal of Biological Chemistry 171, 320-338
Erbersdobler, H.F. (1986). Twenty years of furosine - better knowledge about the biological
significance of Maillard-reaction in food and nutrition. In: Fujimaki, M., Namiki, M., Kato, H.
(Ed.), Amino-carbonyl reactions in food and biological systems. Amsterdam: Elsevier,
Developments in Food Science 13, 481-491
Erbersdobler, H.F., Zucker H. (1966). Untersuchungen zum Gehalt an Lysin und verfügbarem Lysin
in Trockenmagermilch. Milchwissenschaft 21, 564-568
Erbersdobler, H.F., Bock, G. (1967). Untersuchungen zur Identifizierung einer in Hydrolysaten
hitzegeschädigter Proteine nachweisbaren ninhydrin-positiven Substanz. Naturwissenschaften
54, 648
Esteve, M.J., Frigola, A., Rodrigo, M.C., Rodrigo, M. (2002). Use of polarography as a quality-control
method for determining diacetyl in citrus and vegetable juices, yoghurt and butter. Food
Additives and Contaminants 19, 519-523
Europäische Kommission (1998). Verordnung (EG) Nr. 2527/98 der Kommission vom 25.11.1998.
Amtsblatt der Europäischen Gemeinschaft L317, 14-17
Evangelisti, F., Calcagno, C., Zunin, P. (1994). Relationship between blocked lysine and carbohydrate
composition of infant formulas. Journal of Food Science 59, 335-337
Feather, M.S., Mossine, V.V. (1998). Correlation between structure and reactivity of Amadori
compounds: The reactivity of acyclic forms. In: O´Brien, J., Nursten, H.E., Crabbe, M.J.C.,
Ames, J.M. (Ed.), The Maillard reaction in foods and medicine. Cambridge: The Royal Society
Chemistry, 37-42
Ferrer, E., Alegria, A., Farre, R., Abellan, P., Romero, F., Clemente, G. (2003). Evolution of available
lysine and furosine contents in milk-based infant formulas throughout the shelf-life storage
period. Journal of the Science of Food and Agriculture 83, 465-472
Finot, P.A. (1973). Non-enzymic browning. In: Porter, J.W.G., Rolls, B.A. (Ed.), Proteins in human
nutrition. London: Academic Press, 501-514
Finot, P.A., Bricout, J., Viani, R., Mauron, J. (1968). Identification of a new lysine derivative obtained
upon acid hydrolysis of heated milk. Experientia 24, 1097-1099
Finot, P.A., Viani, R., Bricout, J., Mauron, J. (1969a). Detection and identification of pyridosine, a
second lysine derivative obtained upon acid hydrolysis of heated milk. Experientia 25, 134-135
Finot, P.A., Mauron, J. (1969b). Le blocage de la lysine par la reaction de Maillard. I. Synthese de
N-(desoxy-1-D-fructosyl-1) et N-(desoxy-1-D-lactulosyl-1) de la lysines. Helvetica Chimica
Acta 52, 1488-1494
- 195 -

7 Literaturverzeichnis
Finot, P.A., Mauron, J. (1972). Le blocage de la lysine par la reaction de Maillard. II. Proprietes
chimiques des derives N-(desoxy-1-D-fructosyl-1) et N-(desoxy-1-D-lactulosyl-1) de la lysine.
Helvetica Chimica Acta 55, 1153-1164
Finot, P.A., Bujard, E., Mottu, F., Mauron, J. (1977). Availability of the true Schiff´s base of lysine.
Chemical evaluation of the Schiff´s base between lysine and lactose in milk. In: Friedman, M.
(Ed.), Protein Crosslinking. Nutritional and medical consequences, New York: Plenum press,
343-365
Finot, P.A., Deutsch, R., Bujard, E. (1981). The extent of the Maillard reaction during the processing
of milk. In: Eriksson, C. (Ed.), Maillard reactions in food. Chemical, physiological and
technological aspects. Oxford: Pergamon Press, Progress in Food and Nutrition Science 5, 159-
176
Fischer, E. (1886). Ueber Isoglucosamin. Berichte der Deutschen Chemischen Gesellschaft 19, 1920-
1924
Floridi, A., Trizza, V., Paolotti, P., Lucarelli, C. (1999). Analytical strategy for the assessment of the
protein glycation status in uremic patients by high-performance liquid chromatography. Journal
of Chromatography A 846, 65-71
Floridi, A., Antolini, F., Galli, F., Fagugli, R.M., Floridi, E., Buoncristiani, U. (2002). Daily
haemodialysis improves indices of protein glycation. Nephrology, Dialysis, Transplantation 17,
871-878
Flückiger, R., Winterhalter, K.H. (1976). In vitro synthesis of hemoglobin AIc. FEBS Letters 71, 356-
360
Förster, A., Henle, T. (2003). Glycation in food and metabolic transit of dietary AGEs (advanced
glycation end-products): Studies on the urinary excretion of pyrraline. Biochemical Society
Transactions 33, 1383-1385
Frank, O., Hofmann, T. (2000a). On the influence of the carbohydrate moiety on chromophore
formation during food-related Maillardreactions of pentoses, hexoses, and disaccharides.
Helvetica Chimica Acta 83, 3246-3261
Frank, O., Hofmann, T. (2000b). Characterization of key chromophores formed by nonenzymatic
browning of hexoses and L-alanine by using the color activity concept. Journal of Agricultural
and Food Chemistry 48, 6303-6311
Friedman, M. (1996). Food browning and its prevention: An overview. Journal of Agricultural and
Food Chemistry 44, 631-653
Fujimaki, M., Van Chuyen, N., Kurata, T. (1971). Studies on the decarboxylation of amino acids with
glyoxal. Agricultural and Biological Chemistry 35, 2043-2049
- 196 -

7 Literaturverzeichnis
Funcke, W., Klemer, A. (1976). 13C-N.M.R.-Untersuchungen an Mutarotations-Gleichgewichten von
D-Fructose und 1-Amino-1-desoxy-D-fructose-Derivaten (Amadoriverbindungen).
Carbohydrate Research 50, 9-13
Gao, J., Mrksich, M., Gomez, F.A., Whitesides, G.M. (1995). Using capillary electrophoresis to
follow the acetylation of the amino groups of insulin and to estimate their basicities. Analytical
chemistry 67, 3093-3100, Korrektur: Analytical Chemistry 68, 2287
Garlick, R.L., Mazer, J.S. (1983). The principal site of nonenzymatic glycosylation of human serum
albumin in vivo. Journal of Biological Chemistry 258, 6142-6146
Giovanelli, G., Lavelli, V. (2002). Evaluation of heat and oxidative damage during storage of
processed tomato products. I. Study of heat damage indices. Journal of the Science of Food and
Agriculture 82, 1263-1267
Giovanelli, G., Paradiso, A. (2002). Stability of dried and intermediate moisture tomato pulp during
storage. Journal of Agricultural and Food Chemistry 50, 7277-7281
Glaeser, H. (1999). Persönliche Mitteilung. Milchkonferenz 1999, 23./24. September, Kiel
Glomb, M.A., Monnier V.M. (1995). Mechanism of protein modification by glyoxal and
glycolaldehyde, reactive intermediates of the Maillard reaction. Journal of Biological Chemistry
270, 10017-10026
Glomb, M.A., Lang, G. (2001). Isolation and characterization of glyoxal-arginine modifications.
Journal of Agricultural and Food Chemistry 49, 1493-1501
Glomb, M.A., Lederer, M.O., Bunzel, M. (2002). Lebensmittelchemie 2001. Nachrichten aus der
Chemie 50, 358-363
Glomb, M.A., Rösch, D., Nagaraj, R.H. (2001). Nδ-(5-Hydroxy-4,6-dimethylpyrimidine-2-yl)-L-
ornithine, a novel Methylglyoxal-Arginine modification in beer. Journal of Agricultural and
Food Chemistry 49, 366-372
Gortner, R.A., Blish, M.J. (1915). On the origin of the humin formed by the acid hydrolysis of
proteins. Journal of the American Chemical Society 37, 1630-1636
Gray, W.R. (1997). Disulfide bonds between cysteine residues. In: Creighton, T.E. (Ed.), Protein
structure. A practical approach. Oxford: IRL Press, 164-186
Guerra-Hernandez, E., Corzo, N. (1996). Furosine determination in baby cereals by ion-pair reversed-
phase liquid chromatography. Cereal Chemistry 73, 729-731
Guerra-Hernandez, E., Corzo, N., Garcia-Villanova, B. (1999). Maillard reaction evaluation by
furosine determination during infant cereal processing. Journal of Cereal Science 29, 171-176
Guerra-Hernandez, E., Leon, C., Corzo, N., Garcia-Villanova, B., Romera, J.M. (2002). Chemical
changes in powdered infant formulas during storage. International Journal of Dairy Technology
55, 171-176
- 197 -

7 Literaturverzeichnis
Halverson, F., Hirt, R.C. (1951). Near ultraviolett solution spectra of the diazines. Journal of Chemical
Physics 19, 711-718
Hashimoto, M., Takada, K., Kiso, Y., Muranishi, (1989). Synthesis of palmitoyl derivatives of insulin
and their biological activities. Pharmaceutical Research 6, 171-176
Hayase, F., Nagaraj, R.H., Miyata, S., Njoroge, F.G., Monnier, V.M. (1989). Aging of proteins:
Immunological detection of a glucose-derived pyrrole formed during Maillard reaction in vivo.
Journal of Biological Chemistry 263, 3758-3764
Hayase, F., Konishi, Y., Kato, H. (1995). Identification of the modified structure of arginine residues
in proteins with 3-deoxyglucosone, a Maillard reaction intermediate. Bioscience, Biotechnology,
and Biochemistry 59, 1407-1411
Hayase, F., Takahashi, Y., Tominaga, S., Miura, M., Gomyo, T., Kato, H. (1999). Identification of
blue pigment formed in a D-xylose-glycine reaction system. Bioscience, Biotechnology, and
Biochemistry 63, 1512-1514
Hayashi, T., Shibamoto, T. (1985). Analysis of methylglyoxal in foods and beverages. Journal of
Agricultural and Food Chemistry 33, 1090-1093
Hayashi, T., Namiki, M. (1986). Role of sugar fragmentation in the Maillard reaction. In: Fujimaki,
M., Namiki, M., Kato, H. (Ed.), Amino-carbonyl reactions in food and biological systems.
Amsterdam: Elservier, Developments in Food Science 13, 29-38
Helling, R. (2002). Persönliche Mitteilung
Helling, R., Richter, S., Henle, T. (2002). Bestimmung des Glykosylierungsproduktes
Nε-Carboxyethyllysin in Lebensmitteln. Lebensmittelchemie 56, 35
Henle, T. (2003). AGEs in foods: Do they play a role in uremia? Kidney International 63, Suppl. 84,
S145-S147
Henle T., Walter H., Krause I., Klostermeyer H. (1991). Efficient determination of individual Maillard
compounds in heat-treated milk products by amino acid analysis. International Dairy Journal 1,
125-135
Henle, T., Walter, A.W., Haeßner, R., Klostermeyer, H. (1994a). Detection and identification of a
protein-bound imidazolone resulting from the reaction of arginine residues and methylglyoxal.
Zeitschrift für Lebensmittel-Untersuchung und –Forschung 199, 55-58
Henle, T., Walter, A.W., Klostermeyer, H. (1994b). A simple method for the preparation of furosine
and pyridosine reference material. Zeitschrift für Lebensmittel-Untersuchung und -Forschung
198, 66-67
Henle, T., Zehetner, G., Klostermeyer, H. (1995). Fast and sensitive determination of furosine.
Zeitschrift für Lebensmittel-Untersuchung und –Forschung 200, 235-237
- 198 -

7 Literaturverzeichnis
Henle, T., Bachmann, A. (1996). Synthesis of pyrraline reference material. Zeitschrift für
Lebensmittel-Untersuchung und -Forschung 202, 72-74
Henle, T., Schwarzenbolz, U., Klostermeyer, H. (1997). Detection and quantification of pentosidine in
foods. Zeitschrift für Lebensmittel-Untersuchung und -Forschung A 204, 95-98
Henle, T., Deppisch, R., Beck, W., Hergesell, O., Hänisch, G.M., Ritz, E. (1999). Advanced glycated
end-products (AGE) during haemodialysis treatmeant: Discrepant results with different
methodologies reflecting the heterogeneity of AGE compounds. Nephrology, Dialysis,
Transplantation 14, 1968-1975
Henle, T., Schwenger, V., Ritz, E. (2000). Preliminary studies on the renal handling of lactuloselysine
from milk products. Czech Journal of Food Sciences 18, 101-102
Henle, T., Miyata, T. (2003). Advanced glycation end products in uremia. Advances in Renal
Replacment Therapy 10, 321-331
Heyns, K., Koch, W. (1952). Über die Bildung eines Aminozuckers aus d-Fructose und Ammoniak.
Zeitschrift für Naturforschung 7b, 486-488
Heyns, K., Paulsen, H., Breuer, H. (1956). Umsetzung von Fructose mit Aminosäuren zu
Glucosaminosäuren. Angewandte Chemie 68, 334-335
Heyns, K., Breuer, H., Paulsen, H. (1957). Darstellung und Verhalten der 2-N-Aminosäure-2-desoxy-
glucosen („Glucose-Aminosäuren“) aus Glycin, Alanien Leucin und Fructose. Chemische
Berichte 90, 1374-1386
Heyns, K., Breuer, H. (1958). Darstellung und Verhalten weiterer N-substituierter 2-Amino-2-desoxy-
aldosen aus Fructose und Aminosäuren. Chemische Berichte 91, 2750-2762
Heyns, K., Paulsen, H., Schroeder, H. (1961). Umsetzung von Ketohexosen mit sekundären
Aminosäuren und sekundären Aminen. Tetrahedron 13, 247-257
Heyns, K., Heukeshoven, J., Brose, K.-H. (1968). Abbau von Fructose-aminosäuren zu
N-(2-Furoylmethyl)-aminosäuren. Zwischenprodukte von Bräunungsreaktionen. Angewandte
Chemie 80, 627
Hidalgo, A., Rossi, M., Pompei, C. (1995). Furosine as a freshness parameter of shell eggs. Journal of
Agricultural and Food Chemistry 43, 1673-1677
Hidalgo, A., Pompei, C., Zambuto, R. (1998). Heat damage evaluation during tomato products
processing. Journal of Agricultural and Food Chemistry 46, 4387-4390
Hidalgo, A., Pompei, C. (2000). Hydroxymethylfurfural and furosine reaction kinetics in tomato
products. Journal of Agricultural and Food Chemistry 48, 78-82
Higgins, P.J., Bunn, H.F. (1981). Kinetic analysis of the nonenzymatic glycosylation of hemoglobin.
Journal of Biological Chemistry 256, 5204-5208
- 199 -

7 Literaturverzeichnis
Hirano, K., Kubota, T., Tsuda, M., Watanabe, K., Fromont, J., Kobajashi, J. (2000). Ma´edamines A
and B, cytotoxic bromotyrosine alkaloids with a unique 2(1H)pyrazinone ring from sponge
Suberea sp.. Tetrahedron 56, 8107-8110
Ho, C.-T. (1996). Thermal generation of Maillard aromas. In: Ikan, R. (Ed.), The Maillard reaction:
Consequences for the chemical and live science. Chichester, U.K.: John Wily & Sons Ltd, 27-
53
Ho, C.-T., Hartman, G.J. (1982). Formation of oxazolines and oxazoles in the Strecker degradation of
DL-alanine and DL-cysteine with butanedione. Journal of Agricultural and Food Chemistry 30,
793-794
Ho, C.-T., Oh, Y.-C., Zhang, Y., Shu, C.-K. (1992). Peptides as flavor precursors in model Maillard
reactions. In: Teranishi, R., Takeoka, G. R., Güntert, M. (Ed.), Flavor precursors: Thermal and
enzymatic conversions. Washington, D. C.: American Chemical Society, ACS Symposium
Series 490, 193-202
Hodge, J.E (1953). Dehydrated foods. Chemistry of browning reactions in model systems. Journal of
Agricultural and Food Chemistry 1, 928-943
Hofmann, T. (1999). Quantitative studies on the role of browning precursors in the Maillard reaction
of pentoses and hexoses with L-alanine. European Food Research and Technology 209, 113-
121
Hofmann, T., Bors, W., Stettmaier, K. (1999). Studies on radical intermediates in the early stage of the
nonenzymatic browning reaction of carbohydrates and amino acids. Journal of Agricultural and
Food Chemistry 47, 379-390
Hollnagel, A., Kroh, L.W. (2000). Degradation of oligosaccharides in nonenzymatic browning by
formation of α-dicarbonyl compounds via a „peeling off“ mechanism. Journal of Agricultural
and Food Chemistry 48, 6219-6226
Høltermand, A. (1966). The browning reaction. Die Stärke 18, 319-328
Huber, B. (1989). Untersuchungen zur Maillard-Reaktion. Isolierung und Identifizierung von
reaktiven Zwischenprodukten. Dissertation, Technische Universität München
Huber, B., Ledl, F. (1990). Formation of 1-amino-1,4-dideoxy-2,3-hexodiuloses and 2-aminoacetyl-
furans in the Maillard reaction. Carbohydrate Research 204, 215-220
Hunter, S.J., Boyd A.C., O’Harte, F.P.M., McKillop, A.M., Wiggam, M.I., Mooney, M.H.,
McCluskey, J.T., Lindsay, J.R., Ennis, C.N., Gamble, R., Sheridan, B., Barett, C.R., McNulty,
H., Bell, P.M., Flatt, P.R. (2003). Demonstration of glycated insulin in human diabetic plasma
and decreased biological activity assessed by euglycemic-hyperinsulinemic clamp technique in
humans. Diabetes 52, 492-498
- 200 -

7 Literaturverzeichnis
Hurrell, R.F., Carpenter, K.J. (1981). The estimation of available lysine in foodstuffs after Maillard
reactions. In: Eriksson, C. (Ed.), Maillard reactions in food. Chemical, physiological and
technological aspects. Progress in Food and Nutrition Science 5, Oxford: Pergamon Press, 159-
176
Hurrell, R.F., Finot, P.A., Ford, J.E. (1983). Storage of milk powders under adverse conditions. I. Loss
of lysine and of other essential amino acids as determined by chemical and microbiological
methods. The British Journal of Nutrition 49, 343-354
Hurrell, R.F. (1990). Influence of the Maillard reaction on the nutritional value of foods. In: Finot,
P.A., Aeschbacher, H.U., Hurrell, R.F., Liardon, R. (Ed.), The Maillard reaction in food
processing, human nutrition and physiology. Basel: Birkhäuser Verlag, Advances in life
Sciences, 245-258
Italienische Regierung (2000). Ministero delle politische agricole e forestali decreto 15. dicembre
2000. Fissazione dei valori massimi di furosina nei formaggi freschi a pasta filata e nel latte
(crudo e pastorizzato perossidasi-positivo). Gazzetta Ufficiale RI n. 31, 07.02.2001
Izzo, H.V., Ho, C.-T. (1992). Peptide-specific Maillard-reaction products: A new pathway for flavor
chemistry. Trends in Food Science & Technology 3, 253-256
Izzo, H.V., Yu, T.-H., Ho, C.-T. (1993). Flavor generation from the Maillard reaction of peptides and
proteins. In: Schreier, P., Winterhalter, P. (Ed.), Progress in flavour precursor studies: Analysis,
generation, biotechnology. Carol Stream: Allured publishing cooperation, 314-328
Jakas, A., Horvat, S. (1996). Synthesis and 13C NMR investigation of novel Amadori compounds
(1-amino-1-deoxy-D-fructose derivatives) related to the opioid peptide, leucine-enkephalin.
Journal of the Chemical Society, Perkin Transactions 2, 789-794
Jones, R.G. (1949). Pyrazines and related compounds. I. A new synthesis of hydroxypyrazines. The
journal of the American Chemical Society 71, 78-81
Jørgensen, A.M.M., Kristensen, S.M, Led J.J., Balschmidt, P. (1992). Three-dimensional solution
structure of an insulin dimer. A study of the B9(Asp) mutant of human insulin using nuclear
magnetic resonance, distance geometry and restrained molecular dynamics. Journal of
Molecular Biology 227, 1146-1163
Kaarsholm, N.C., Ludvigsen, S. (1995). The high resolution solution structure of the insulin monomer
determined by NMR. Receptor 5, 1-8
Keyhani, A., Yaylayan, V.A. (1996). Elucidation of the mechanism of pyrazinone formation in glycine
model systems using labeled sugars and amino acids. Journal of Agricultural and Food
Chemistry 44, 2511-2516
Knobloch, A. (1999). Furosin als Qualitätsparameter für Milch und Milchprodukte. Diplomarbeit,
Technische Universität Dresden
- 201 -

7 Literaturverzeichnis
Kollias, N., Gillies, R., Moran, M., Kochevar, I.E., Anderson, R.R. (1998). Endogenous skin
fluorescence includes bands that may serve as quantitative markers of aging and photoaging.
Journal of Investigative Dermatology 111, 776-780
Kuhn, R., Dansi, A (1936). Über eine molekulare Umlagerung von N-Glucosiden. Berichte der
Deutschen Chemischen Geselschaft 69, 1745-1754
Kuhn, R., Weygand, F. (1937). Die Amadori-Umlagerung. Berichte der Deutschen Chemischen
Geselschaft 70. 769-772
Kroh, L.W., Schröder, R., Mügge, C., Westphal, G., Baltes, W. (1992). Thermisch induzierter Abbau
von Disaccharid-Amadori-Verbindungen in quasi wasserfreiem Reaktionsmilieu. Zeitschrift für
Lebensmittel-Untersuchung und -Forschung 194, 216-221
Kroh, L.W. (1994). Caramelization in food and beverages. Food Chemistry 51, 373-379
Labuza, T.P. (1984). Application of chemical kinetics to deterioration of foods. Journal of Chemical
Education 61, 348-358
Lal, S., Kappler, F., Walker, M., Orchard, T.J., Beisswenger, P.J., Szwergold, B.S., Brown, T.R.
(1997). Quantitation of 3-deoxyglucosone levels in human plasma. Archives of Biochemistry
and Biophysics 342, 254-260
Lapolla, A., Tonani, R., Fedele, D., Garbeglio, M., Senesi, A., Seraglia, R., Favretto, D., Traldi, P.
(2002). Non-enzymatic glycation of IgG: An in vivo study. Hormone and Metabolic Research
34, 260-264
Lapolla, A., Flamini, R., Vedova A.D., Senesi, A., Reitano, R., Fedele, D., Basso, E., Seraglia, R.,
Traldi, P. (2003). Glyoxal and methylglyoxal levels in diabetic patients: Quantitative
determination by a new GC/MS method. Clinical Chemistry and Laboratory Medicine 41,
1166–1173
Ledl, F., Schleicher, E. (1990). Die Maillard-Reaktion in Lebensmitteln und im menschlichen Körper -
neue Ergebnisse zu Chemie, Biochemie und Medizin. Angewandte Chemie 102, 597-626
Lee, K.-G., Shibamoto, T. (2002). Toxicology and antioxidant activities of non-enzymatic browning
reaction products: Review. Food Reviews International 18, 151-175
Liardon, R., De Weck-Gaudard, D., Philippossian, G., Finot, P.-A. (1987). Identification of
Nε-carboxymethyllysine: A new Maillard reaction product, in rat urine. Journal of Agricultural
and Food Chemistry 35, 427-431
Lindenmeier, M., Faist, V., Hofmann, T. (2002). Structural and functional characterization of pronyl-
lysine, a novel protein modification in bread crust melanoidins showing in vitro antioxidative
and phase I/II enzyme modulating activity. Journal of Agricultural and Food Chemistry 50,
6997-7006
Lindsay, D.G., Shall, S. (1969). Acetoacetylation of insulin. Biochemical Journal 115, 587-595
- 202 -

7 Literaturverzeichnis
Lindsay, D.G., Shall, S. (1971). The acetylation of insulin. Biochemical Journal 121, 737-745
Lindsay, R.C., McDonald, S.T. (1992). Browning in stirred-cured, directly-salted Parmesan cheese
linked to α-dicarbonyl production by wild lactobacillus contaminants. Marschall Italian
Specialty Cheese Seminars, 1-5
Lipton, S.H., Finegold, H., Dutky, R.C. (1971). N-(2-Furacyl)glycine - a furan amino acid derived
from fructose glycine. Journal of Agricultural and Food Chemistry 19, 125-128
Loidl-Stahlhofen, A., Spiteller, G. (1994). α-Hydroxyaldehydes, products of lipid peroxidation.
Biochimica et Biophysica Acta 1211, 156-160
Longley, R.E., Isbrucker, R.A., Wright, A.E. (2000). Use of imidazole and indole compounds as
inhibitors of nitric oxide synthase. United States Patent 6087363
Loo, J.A., Loo, R.R.O., Light, K.J., Edmonds, C.G., Smith, R.D.S. (1992). Multiply charged negative
ions by elektospray ionization of polypeptides and proteins. Analytical Chemistry 64, 81-88
Lopez-Fandino, R., Olano, A. (1999). Review: Selected indicators of the quality of thermal processed
milk. Food Science and Technology International 5, 121-137
Lyons, T. J. (2002). Glycation, carbonyl stress, EAGLEs, and the vascular complications of diabetes.
Seminars in Vascular Medicine 2, 175-189
MacDonald, J.C., Bishop, G.G. (1977). Biosynthesis of arglecin and related compounds. Canadian
Journal of Biochemistry 55, 165-172
Maillard, L.-C. (1911). Condensation des acides amines en prescence de la glycerine;
Cycloglycylglycine et polypeptides. Comptes Rendus Hebdomadaires des Séances de
l'Académie des Sciences 153, 1078-1080
Maillard, L.-C. (1912a). Action des acides amines sur les sucres: Formation des melanoidines par voie
methodique. Comptes Rendus Hebdomadaires des Séances de l’Académie des Sciences 154, 66-
68
Maillard, L.-C. (1912b). Formation d’humus et de combustibles mineraux sans intervention de
l’oxygene atmospherique, des microorganismes, des hautes temeratures, ou des fortes pressions.
Comptes Rendus Hebdomadaires des Séances de l’Académie des Sciences 155, 1554-1556
Maillard, L.-C. (1916). Syntheses des matieres humiques par action des acides amines sur les sucres
reducteurs. Annales de Chimie (Paris) 5, 258-317
Maillard, L.-C. (1917). Identite des matieres humiques de synthese avec les matieres humiques
naturelles. Annales de Chimie (Paris) 7, 113-152
Marconi, E., Caboni, M.F., Messia, M.C., Panfili, G. (2002). Furosine: A suitable marker for assessing
the freshness of royal jelly. Journal of Agricultural and Food Chemistry 50, 2825-2829
- 203 -

7 Literaturverzeichnis
Mason, S.F. (1957). 246. The electronic spectra of N-hetereoaromatic systems. Part II. Substituted
monocyclic azones. Journal of the Chemical Society, 1247-1253
Mason, S.F. (1959). 1002. The tautomerism of N-hetereoaromatic hydroxy-compounds. Part II.
Ultraviolett spectra. Journal of the Chemical Society, 5010-5017
Mauron, J. (1981). The Maillard reaction in food: A critical review from the nutritional standpoint. In:
Eriksson, C. (Ed.), Maillard reactions in food. Chemical, physiological and technological
aspects. Progress in Food and Nutrition Science 5, Oxford: Pergamon press, 5-35
Mauron, J. (1990). Influence of processing on protein quality. Journal of Nutritional Science and
Vitaminology 36, S57-S69
McKillop, A.M., McGurk, C., McCluskey, J.T., Harriott, P., Bell, P.M., Flatt, P.R., O’Harte, F.P.M.
(1999). Detection of glycated insulin in plasma of type 2 diabetic subjects. Digestion 60, 507
McKillop, A.M., Mooney, M.H., Harriott, P., Flatt, P.R., O’Harte, F.P.M. (2001). Evaluation of
glycated insulin in diabetic animals using immunocytochemistry and radioimmunoassay.
Biochemical and Biophysical Research Communications 286, 524-528
McPherson, J.D., Shilton, B.H., Walton, D.J. (1988). Role of fructose in glycation and cross-linking of
proteins. Biochemistry 27, 1901-1907
Mei, H., Yu, C., Chan, K. (1999). NB1-C16-Insulin: Site-specific synthesis, purification, and
biological activity. Pharmaceutical Research 16, 1680-1686
Miyata, T., Kurokawa, T., VanYpersele DeStrihou, C. (2000a). Relevance of oxidative and carbonyl
stress to long-term uremic complications. Kidney International 58 (suppl. 76), 120-125
Miyata, T., Kurokawa, T., VanYpersele DeStrihou, C. (2000b). Advanced glycation and lipoxidation
end products: Role of reactive carbonyl compounds generated during carbohydrate and lipid
metabolism. Journal of the American Society of Nephrology 11, 1744-1752
Möller, A.B., Andrews, A.T., Cheeseman, G.C. (1977). Chemical changes in ultra-heat-treated milk
during storage. II. Lactuloselysine and fructoselysine formation by the Maillard reaction.
Journal of Dairy Research 44, 267-275
Montilla, A., Calvo, M.M., Santa-Maria, G., Corzo, N., Olano, A. (1997). Correlation between
lactulose and furosine in UHT-heated milk. Journal of Food Protection 59, 1061-1064
Mossine, V.V., Glinsky, G.V., Feather, M.S. (1994). The preparation and characterization of some
Amadori compounds (1-amino-1-deoxy-D-fructose derivatives) derived from a series of
aliphatic ω-amino acids. Carbohydrate Research 262, 257-270
Mossine, V.V., Linetsky, M., Glinsky, G.V., Ortwerth, B.J., Feather, M.S. (1999). Superoxide free
radical generation by Amadori compounds: The role of acyclic forms and metal ions. Chemical
Research in Toxicology 12, 230-236
- 204 -

7 Literaturverzeichnis
Mottram, D.S., Wedzicha, B.L., Dodson, A.T. (2002). Acrylamid is formed in the Maillard reaction.
Nature 419, 448-449
Nakagawa, S.H., Tager, H.S. (1992). Importance of aliphatic side-chain structure at positions 2 and 3
of the insulin A chain in insulin-receptor interactions. Biochemistry 31, 3204-3214
Nakai, T., Ohta, T., Hatsumi, M., Horikoshi, S. (1975). Isolation of Nε-(2-furoylmethyl)-L-lysine
(furosine) from a acid hydrolyzate of casein heated with glucose. Agricultural and Biological
Chemistry 39, 2421-2422
Nakaya, K., Horinishi, H., Shibata, K. (1967). States of amino acid residues in proteins. XIV. Glyoxal
as a reagent for discrimination of arginine residues. Journal of Biochemistry 61, 345-351
Namiki, M., Hayashi, T. (1983). A new mechanism of the Maillard reaction involving sugar
fragmentation and free radical formation. In: Waller, G.R., Feather, M.S. (Ed.), The Maillard
reaction in foods and nutrition. ACS Symposium Series 215, 21-46
Neglia, C.I., Cohen, H.J., Garber, A.R., Ellis, P.D., Thorpe, S.R., Baynes J.W. (1983). 13C NMR
investigation of nonenzymatic glucosylation of protein. Model studies using RNase A. Journal
of Biological Chemistry 258, 14279-14283.
Ng, L.T., Pascaud, A., Pascaud, M. (1987). Hydrochloric acid hydrolysis of proteins and
determination of tryptophan by reversed-phase high-performance liquid chromatography.
Analytical Biochemistry 167, 47-52
Niwa, T., Katsuzaki, T., Ishizaki, Y., Hayase, F., Miyazaki, T., Uematsu, T., Tatemichi, N., Takei, Y.
(1997). Imidazolone, a novel advanced glycation end product, is present at high levels in
kidneys of rats with streptozotocin-induced diabetes. FEBS Letters 407, 297-302
Odani, H., Matsumoto, Y., Shinzato,. T, Usami, J., Maeda, K. (1999a). Mass spectrometric study on
the protein chemical modification of uremic patients in advanced Maillard reaction. Journal of
Chromatography B 731, 131-140
Odani, H., Shinzato, T., Matsumoto, Y., Usami, J., Maeda, K. (1999b). Increase in three
α,β-dicarbonyl compound levels in human uremic plasma: Specific in vivo determination of
intermediates in advanced Maillard reaction. Biochemical and Biophysical Research
Communications 256, 89-93
Odani, H., Iijima, K., Nakata, M., Miyata, S., Kusunoki, H., Yasuda, Y., Yoshiyuki, H., Irie, S.,
Maeda, K., Fujimoto, D. (2001). Identification of Nω-carboxymethylarginine, a new advanced
glycation endproduct in serum proteins of diabetic patients: Possibility of a new marker of aging
and diabetes. Biochemical and Biophysical Research Communications 285, 1232-1236
Oh, Y.-C., Shu, C.-K., Ho, C.-T. (1992). Formation of novel 2(1H)-pyrazinones as peptide-specific
Maillard-reaction products. Journal of Agricultural and Food Chemistry 40, 118-121
- 205 -

7 Literaturverzeichnis
O’Harte, F.P.M., Højrup, P., Barett, C.R., Flatt, P.R. (1996). Identification of the site of glycation of
human insulin. Peptides 17, 1323-1330
O’Harte, F.P.M., Boyd, A.C., McKillop, A.M., Abdel-Wahab, Y.H.A., McNulty, H., Barett, C.R.,
Conlon, J.M., Højrup, P., Flatt, P.R. (2000). Structure, antihyperglycemic activity and cellular
actions of a novel diglycated human insulin. Peptides 21, 1519-1526
Ohkanda, J., Kumasaka, T., Takasu, A., Hasegawa, T., Katoh, A. (1996). N-hydroxyamide-containing
heterocycles. Part 7. Preparation and photochemical behavior of 1-benzyloxy-
2(1H)-pyrazinones. Heterocycles 43, 883-889
Okada, Y., Fukumizu, A., Takahashi, M., Yamazaki, J., Yokoi, T., Tsuda, Y., Bryant, S.D., Lazarus,
L.H. (1999). Amino acids and peptides. LVI. Synthesis of pyrazinone ring-containing opioid
mimetics and examination of their opioid receptor binding activity. Tetrahedron 55, 14391-
14406
Olano, A., Santa-Maria, G., Corzo, N., Calvo, M.M., Martinez-Castro, I., Jimeno, M.L. (1992).
Determination of free carbohydrates and Amadori compounds formed at the early stage of non-
enzymic browning. Food Chemistry 43, 351-358
Olcott, H.S., Fraenkel-Conrat, H. (1947). Formation and loss of cysteine during acid hydrolysis of
proteins. Role of tryptophan. Journal of Biological Chemistry 171, 583-593
Olsen, H.B., Ludvigsen, S., Kaarsholm, N.C. (1998). The relationship between insulin bioactivity and
structure in the NH2-terminal A-chain helix. Journal of Molecular Biology 284, 477-488
Osthoff, D. (1966). Dissertation, Universität Frankfurt / M., zit. in: Velisek, J., Davidek, J., Cuhrova,
J., Kubelka, V. (1976). Volatile heterocyclic compounds in the reaction of glyoxal with glycine.
Journal of Agricultural and Food Chemistry 24, 3-7
Paul, R.G., Avery, N.C., Slatter, D.A., Sims, T.J., Bailey, A.J. (1998). Isolation and characterization of
advanced glycation end products derived from the in vitro reaction of ribose and collagen.
Biochemical Journal 330, 1241-1248
Paulsen, H., Pflughaupt, K.-W. (1980). In: Pigmann, W., Horton, D. (Ed.), The carbohydrates:
Chemistry and biochemistry, Band 1B, New York: Academic press, 881-927, zit. in: Westphal,
G., Kroh, L. (1985a)
Pellegrino, L., De Noni, I., Resmini, P. (1995). Coupling of lactulose and furosine indices for quality
evaluation of sterilized milk. International Dairy Journal 5, 647-659
Pompei, C., Spagnolello, A. (1997a). Furosine as an index of heat treatment intensity in meat
products: Its application to cooked ham. Meat Science 46, 139-146
Pompei, C., Spagnolello, A. (1997b). Influence of tumbling and dextrose on furosine content in
cooked ham. Journal of Agricultural and Food Chemistry 45, 4080-4083
- 206 -

7 Literaturverzeichnis
Pötzsch, W.R., Fischer, A., Müller, W. (1988). Lexikon bedeutender Chemiker. Leipzig:
Bibliographisches Institut
Preininger, M., Grosch, W. (1994). Evaluation of key odorants of the volatiles of Emmentaler cheese
by calculation of the odour activity values. Lebensmittel-Wissenschaft und -Technologie 33,
237-244
Prey, V., Petershofer, G. (1968). Zur Kenntnis von Melanoidinvorprodukten. Zeitschrift für die
Zuckerindustrie 18, 63-65
Rada-Mendoza, M., Olano, A., Villamiel, M. (2002). Furosine as indicator of Maillard reaction in
jams and fruit-based infant foods. Journal of Agricultural and Food Chemistry 50, 4141-4145
Rahbar, S., Blumenfeldt, O., Ranney, H.M. (1969). Studies of an unusual hemoglobin in patients with
diabetes mellitus. Biochemical and Biophysical Research Communications 36, 838-843
Raj, D.S.C., Choudhury, D., Welburne, T.C., Levi, M. (2000). Advanced glycation end products: A
nephrologist´s perspective. American Journal of Kidney Diseases 35, 365-380
Rättich, S. (2004). Quantitative Bestimmung von α-Dicarbonylen in Milchprodukten. Diplomarbeit,
Technische Universität Dresden
Ravandi, A., Kuksis, A., Marai, L., Myher, J. J., Steiner, G., Lewisa, G., Kamido, H. (1996). Isolation
and identification of glycated aminophospholipids from red cells and plasma of diabetic blood.
FEBS Letters 381, 77-81
Requena, J.R., Fu, M.X., Ahmed, M.U., Jenkins, A.J., Lyons, T.J., Thorpe, S.R. (1996). Lipoxidation
products as biomarkers of oxidative damage to proteins during lipid peroxidation reactions.
Nephrology, Dialysis, Transplantation 11, 48-53
Resmini, P., Pellegrino, L., Batelli, G. (1990). Accurate quantification of furosine in milk and dairy
products by a direct HPLC method. Italian Journal of Food Science 2, 177-183
Rice, F.A.H., Fishbein, L. (1956). Effect of aqueous sulfuric acid on sugars. II. Spectrometric studies
on the hexoses; identification of the ether-soluble products formed. Journal of the American
Chemical Society 78, 3731-3734
Riffel, K.R., Song, H., Gu, X., Yan, K., Lo, M.-W. (2000). Simultaneous determination of a novel
thrombin inhibitor and its two metabolites in human plasma by liquid chromatography/tandem
mass spectrometry. Journal of Pharmaceutical and Biomedical Analysis 23, 607-616
Rizzi, G.P. (1969). The formation of tetramethylpyrazine and 2-isopropyl-4,5-dimethyl-3-oxazoline in
the Strecker degradation of DL-valine with 2,3-butandione. Journal of Organic Chemistry 34,
2002-2004
Rizzi, G.P. (1997). Chemical structure of colored Maillard reaction products. Food Reviews
International 13, 1-28
- 207 -

7 Literaturverzeichnis
Rizzi, G.P. (1999). The Strecker degradation and its contribution to food flavor. In: Teranishi, R.,
Wick, E.L., Hornstein, I. (Ed.), Flavor chemistry: Thirty years of progress, New York: Kluwer
Academic / Plenum Publishers, 335-343
Rodrigues, J.A., Barros, A.A. , Rodrigues, P.G. (1999). Differential pulse polarographic determination
of alpha-dicarbonyl compounds in foodstuffs after derivatization with o-phenylenediamine.
Journal of Agricultural and Food Chemistry 47, 3219-3222
Röper, H., Röper, S., Heyns, K., Meyer, B. (1983). N.M.R. spectroscopy of N-(1-Deoxy-D-fructos-
1-yl)-L-amino acids (“fructose-amino acids”). Carbohydrate Research 116, 183-195
Rufian-Henares, J.A., Garcia-Villanova, B., Guerra-Hernandez, E. (2002). Furosine content, loss of
o-phthaldialdehyde reactivity, fluorescence and colour in stored enteral formulas. International
Journal of Dairy Technology 55, 121-126
Ruttkat, A., Erbersdobler, H.F. (1994). Degradation of furosine during heptafluorobutyric anhydride
derivatization for gas chromatographic determination. Journal of Chromatography A 678, 103-
107
Ryle, A.P., Sanger, F., Smith, L.F., Kitai, R. (1955). The disulfide bonds of insulin. Biochemical
Journal 60, 541-556
Sachsse, R. (1871). Ueber einige stickstoffhaltige Verbindungen des Milchzuckers. Berichte der
Deutschen Chemischen Geselschaft 4, 834-836
Saito, G., Okitani, A., Hayase, F., Kato, H. (1986). Characterization of trytophan derivatives from the
reaction of Nα-acetyl-tryptophan with carbonyl compounds. Agricultural and Biological
Chemistry 50, 2315-2323
Sakurai, H., Ishii, K. (1988). Structural analysis of ninhydrin-positive substance produced by the
reaction of dehydroascorbic acid with glycylleucine. Bulletin of the College of Agriculture and
Veterinary Science, Nihon University 45, 50-59
Sakurai, H., Ito, T., Ishii, K. (1988). Structure and ninhydrin reaction mechanism of ninhydrin-positive
substances produced by the reaction of dehydroascorbic acid with glycyl dipeptides. Bulletin of
the College of Agriculture and Veterinary Science, Nihon University 45, 59-70
Salazar, R., Brandt, R., Kellermann, J., Krantz, S. (2000). Purification and characterization of a
200 kDa fructosyllysine-specific binding protein from cell membranes of U937 cells.
Glycoconjugate Journal 17, 713-716
Sanderson, P.E.J. (1999). Small, noncovalent serine protease inhibitors. Medical Research Reviews 19,
179-197
Sanz, M.L., Del Castillo, M.D., Corzo, N., Olano, A. (2000). Presence of 2-furoylmethyl derivatives in
hydrolysates of processed tomato products. Journal of Agricultural and Food Chemistry 48,
468-471
- 208 -

7 Literaturverzeichnis
Sanz, M.L., Del Castillo, M.D., Corzo, N., Olano, A. (2001). Formation of Amadori compounds in
dehydrated fruits. Journal of Agricultural and Food Chemistry 49, 5228-5231
Sanz, M.L., Del Castillo, M.D., Corzo, N., Olano, A. (2003). 2-Furoylmethyl amino acids and
hydroxymethylfurfural as indicators of honey quality. Journal of Agricultural and Food
Chemistry 51, 4278-4283.
Sammes, P.G. (1975). Naturally occuring 2,5-dioxopiperazines and related compounds. Fortschritte
der Chemie Organischer Naturstoffe 32, 51-118
Sato, N. (1978). Studies on pyrazines. 4. (1). The synthesis of 2-hydroxy-6-phenylpyrazine and its
derivatives. Journal of Heterocyclic Chemistry 15, 665-670
Schalkwijk, C.G., Lieuw-a-Fa, M., Van Hinsberg, V.W.M., Stehouwer, C.D.A. (2002).
Pathophysiological role of Amadori-glycated proteins in diabetic microangiopathy. Seminars in
Vascular Medicine 2, 191-197
Schleicher, E., Scheller, L., Wieland, O.H. (1981). Quantitation of lysine-bound glucose of normal and
diabetic eryothrocyte membranes by HPLC analysis of furosine (ε-N(2-furoylmethyl)-L-lysin).
Biochemical and Biophysical Research Communications 99, 1011-1019
Schleicher, E., Wieland, O.H. (1981). Specific quantitation by HPLC of protein (lysine) bound glucose
in human serum albumin and other glycosylated proteins. Journal of Clinical Chemistry and
Clinical Biochemistry 19, 81-87
Schneider, B., Lichtenthaler, F.W., Steinle, G., Schiweck, H. (1985). Studies on ketoses. 1.
Distribution of furanoid and pyranoid tautomers of D-fructose in water, dimethyl sulfoxide, and
pyridine via proton NMR intensities of anomeric hydroxy groups in [D6]DMSO. Liebig's
Annalen der Chemie, 2443-2453
Schönberg, A., Moubacher, B. (1952). The Strecker degradation of α-amino acids. Chemical Reviews
50, 261-277
Schönberg, A., Moubacher, R., Mostafa, A. (1948). Degradation of α-amino acids to aldehydes and
ketones by interaction with carbonyl compounds. Journal of the Chemical Society, 176-182
Schräder, I., Eichner, K. (1996). Veränderungen von Inhaltsstoffen bei der Verarbeitung von Tomaten.
Zeitschrift für Lebensmittel-Untersuchung und -Forschung 202, 474-480
Schwartz, H.M., Lea, C.H. (1952). The reaction between proteins and reducing sugars in the dry state.
Relative reactivity of the α- and ε-amino groups of insulin. Biochemical Journal 50, 713-716
Schwarzenbolz, U., Henle, T., Haeßner, R., Klostermeyer, H. (1997). On the reaction of glyoxal with
proteins. Zeitschrift für Lebensmittel-Untersuchung und -Forschung A 205, 121-124
- 209 -

7 Literaturverzeichnis
Sell, D.R., Monnier, V.M. (1989). Structure elucidation of a senescence cross-link from human
extracellular matrix – implication of pentoses in the aging process. Journal of Biological
Chemistry 264, 21597-21602
Sengl, M., Ledl, F., Severin, T. (1989). Maillard-Reaktion von Rinderserumalbumin mit Glucose.
Hochleistung-Flüssigkeitschromatographischer Nachweis des 2-Formyl-5-(hydroxymethyl)-
pyrrol-1-norleucins nach alkalischer Hydrolyse. Journal of Chromatography 463, 119-125
Shin, P.K., Ahn, Y.T., Kim, H.U. (2000). Studies on the assay of furosine in domestic milk and milk
products. Journal of Animal Science and Technology (Korea) 42, 325-330
Shu, C.K., Lawrence, B.M. (1995). 3-Methyl-2(1H)-pyrazinones, the asparagine-specific Maillard
products formed from asparagine and monosaccharides. Journal of Agricultural and Food
Chemistry 43, 779-781
Siegl, T. (2003). Studien zur Quervernetzung von Milchproteinen und zur Bildung individueller
Crosslink-Aminosäuren. Dissertation, Technische Universität Dresden
Simon, H., Kraus, A. (1970). Mechanistische Untersuchungen über Glykosylamine, Zuckerhydrazone,
Amadori-Umlagerungsprodukte und Osazone. Fortschritte der chemischen Forschung 14, 430-
471
Singh, R., Barden, A., Mori, T., Beilin, L. (2001). Advanced glycation end-products: A review.
Diabetologia 44, 129-146
Singh, R., Elipe, M.V.S., Pearson, P.G., Arison, B.H., Wong, B.K., White, R., Yu, X., Burgey, C.S.,
Lin, J.H., Baillie, T.A. (2003). Metabolic activation of a pyrazinone-containing thrombin
inhibitor. Evidence for novel biotransformation involving pyrazinone ring oxidation,
rearrangement, and covalent binding to proteins. Chemical Research in Toxicology 16, 198-207
Skovsted, I.C., Christensen, M., Breinholt, J., Mortensen, S.B. (1998). Characterization of a novel
AGE-compound derived from lysine and 3-deoxyglucosulose. Cellular and Molecular Biology
44, 1159-1163
Smith, P.R., Thornalley, P.J. (1992). Preparation and characterization of Nε-(1-deoxy-D-fructosyl-
1-yl)-hippuryl-lysine. Carbohydrate Research 223, 293-298
Sorokin, B. (1888). Mitteilungen aus dem agronomischen Laboratorium der Universität Kasan. 1. Die
Anilide und Toluide der Glycosen. Journal für praktische Chemie 37, 291-317
Spackman, D.H., Stein, W.H., Moore, S. (1958). An automatic recording apparatus for use in the
chromatography of amino acids. Analytical Chemistry 30, 1185-1190
Stitt, A.W. (2001). Advanced glycation: An important pathological event in diabetic and age related
ocular disease. British Journal of Ophthalmology 85, 746-753
Strecker, A. (1862). Notiz über eine eigentümliche Oxydation durch Alloxan. Annalen der Chemie
und Pharmacie 123, 363-365
- 210 -

7 Literaturverzeichnis
Suarez, G., Rajaram, R., Oronsky, A.L., Gawinowicz, M.A. (1989). Nonenzymatic glycation of bovine
serum albumin by fructose (fructation). Comparison with the Maillard reaction initiated by
glucose. Journal of Biological Chemistry 264, 3674-3679
Sugimura,T., Takayama, S., Ohgaki, H., Wakabayashi, K., Nagao, M. (1990). Mutagens and
carcinogens formed by cooking meat and fish: Heterocyclic amines. In: Finot, P.A.,
Aeschbacher, H.U., Hurrell, R.F., Liardon, R. (Ed.), The Maillard reaction in food processing,
human nutrition and physiology. Basel: Birkhäuser Verlag, Advances in Life Sciences, 245-258
Sundby, F. (1962). Separation and characterization of acid-induced insulin transformation products by
paper electrophoresis in 7 M urea. Journal of Biological Chemistry 237, 3406-3411
Szölgyenyi, G.P., Winsauer, K.J.B., Deutsch, E. (1989). Determination of furosine as a measure for
irreversibly bound glucose in human fibrinogen. Monatshefte für Chemie / Chemical Monthly
120, 1147-1158
Szwergold, B.S., Kappler, F., Brown, T.R. (1990). Identification of fructose-3-phosphate in the lens of
diabetic rats. Science 247, 451-454
Taguchi, H., Yokoi, T., Okada, Y. (1996). Amino acids and peptides. XLVI. Introduction of carboxyl
function into pyrazinone by using newly developed procedure for pyrazinone ring formation:
Observation of immediate decarboxylation. Chemical & Pharmaceutical Bulletin 44, 2037-2041
Takahashi, K. (1968). The reaction of phenylglyoxal with arginine residues in proteins. Journal of
Biological Chemistry 243, 6171-6179
Takahashi, K. (1977a). The reaction of phenylglyoxal and related reagents with amino acids. Journal
of Biochemistry (Tokyo) 81, 395-402
Takahashi, K. (1977b). Further studies on the reaction of phenylglyoxal and related reagents with
proteins. Journal of Biochemistry (Tokyo) 81, 403-414
Takemura, A., Muramatsu, S., Kobayashi, N., Nakagawa, A., Kitazawa, E., Uematsu, T. (1997).
Determination of furosine in hair by liquid chromatography-tandem mass spectrometry.
Biomedical Chromatography 11, 61-62
Tannenbaum, S.R. (1966). Protein carbonyl browning systems: A study of the reaction between
glucose and insulin. Journal of Food Science 31, 53-57
Thornalley, P.J. (1985). Monosaccharide autoxidation in health and disease. Environmental Health
Perspectives 64, 297-307
Thornalley, P.J. (1990): The glyoxalase system: New developments towards functional
characterization of a metabolic pathway fundamental to biological life. Biochemical Journal
269, 1-11
- 211 -

7 Literaturverzeichnis
Thornalley, P.J. (1996). Pharmacology of methylglyoxal: Formation, modification of proteins and
nucleic acids, and enzymatic detoxification - a role in pathogenesis and antiproliferative
chemotherapy. General Pharmacology 27, 565-573
Thornalley, P.J. (1998). Glutathione-dependent detoxification of α-oxoaldehydes by glyoxalase
system: Involvement in disease mechanisms and antiproliferative activity of glyoxalase I
inhibitors. Chemico-Biological Interactions 111-112, 137-151
Thornalley, P.J. (1999). The clinical significance of glycation. Clinical Laboratory 45, 263-273
Thornalley, P.J., Langborg, A., Minhas, H.S. (1999). Formation of glyoxal, methylglyoxal and
3-deoxyglucosone in the glycation of proteins by glucose. Biochemical Journal 344, 109-116
Thornalley, P.J., Battah, S., Ahmed, N., Karachalias, N., Agalou, S., Babaei-Jaddi, R., Dawnay, A.
(2003). Quantitative screening of advanced glycation endproducts in cellular and extracellular
proteins by tandem mass spectrometry. Biochemical Journal 375, 581-592
Thorpe, S.R., Baynes, J.W. (2003). Maillard reaction products in tissue proteins: New products and
new perspectives. Amino Acids 25, 275-281
Tirelli, A. (1998). Improved method for the determination of furosine in food by capillary
electrophoresis. Journal of Food Protection 61, 1400-1404
Tjan, S.B., Van De Ouweland, G.A.M. (1974). PMR investigation into the structure of some
N-substituted 1-amino-1-deoxy-D-fructoses (Amadori rearrangement products). Evidence for a
preferential conformation in solution. Tetrahedron 30, 2891-2897
Tomita, I., Shimoi, K., Suzuki, M., Sano, M., Nakamura, Y., Kinae, N., Boriboon, P., Hashizume, T.
(1991). Changes in the levels of lipid peroxidation and mutagenicity of fish after sun-drying and
broiling. Journal of the Food Hygienic Society of Japan 32, 543-547
Tsai, Y.J., Rottero, A., Chow, D.D., Hwang, K.J., Lee, V.H.L., Zhu, G., Chan, K.K. (1997). Synthesis
and purification of NB1-Palmitoyl insulin. Journal of Pharmaceutical Sciences 72, 1264-1268
Tsuzuki, Y., Yamazaki, J., Kagami, K. (1950). The specific rotation of fructose. Journal of the
American Chemical Society 72, 1071-1073
Valencia, J.V., Mone, M., Koehne, C., Rediske, J., Hughes, T. E. (2004). Binding of receptor for
advanced glycation end products (RAGE) ligands is not sufficient to induce inflammatory
signals: Lack of activity of endotoxin-free albumin-derived advanced glycation end products.
Diabetologia 47, 844-852
Van Boekel, M.A.J.S. (1998). Effect of heating on Maillard reactions in milk. Food Chemistry 62,
403-414
Van Chuyen, N., Kurata, T., Fujimaki, M. (1972). Studies on the Strecker degradation of alanine with
glyoxal. Agricultural and Biological Chemistry 36, 1199-1207
- 212 -

7 Literaturverzeichnis
Van Chuyen, N., Kurata, T., Fujimaki, M. (1973a). Studies on the reaction of dipeptides with glyoxal.
Agricultural and Biological Chemistry 37, 327-334
Van Chuyen, N., Kurata, T., Fujimaki, M. (1973b). Formation of N-[2(3-allkylpyrazin-2-on-1-yl)-
acyl]amino acids or -peptides on heating tri- or tetrapeptides with glyoxal. Agricultural and
Biological Chemistry 37, 1613-1618
Van Chuyen, N., Kurata, T., Fujimaki, M. (1973c). Formation of N-carboxymethyl amino acids from
the reaction of α-amino acids with glyoxal. Agricultural and Biological Chemistry 37, 2209-
2210
Van Chuyen, N., Iijichi, K., Umetsu, H., Moteki, K. (1998). Antioxidative properties of products from
amino acids or peptides in the reaction with glucose. In: Shahidi, F., Ho, C.-T., Van Chuyen, N.
(Ed.), Process-induced chemical changes in food. New York: Plenum press, 201-212
Van Renterghem, R., De Block, J. (1996). Furosine in consumption milk and milk powders.
International Dairy Journal 6, 371-382
Velisek, J., Davidek, T., Davidek, J., Trska, P., Kvasnicka, F., Velcova, K. (1989). New imidazoles
formed in nonenzymatic browning reactions. Journal of Food Science 54, 1544-1546
Venemalm, L. (2001). Pyrazinone conjugates as potential cephalosporin allergens. Bioorganic &
Medicinal Chemistry Letters 11, 1869-1870
Villamiel, M., Arias, M., Corzo, N., Olano, A. (1997). Use of different thermal indices to assess the
quality of pasteurized milks. Zeitschrift für Lebensmittel-Untersuchung und -Forschung A 208,
169-171
Villamiel, M., Arias, M., Corzo, N., Olano, A. (2000). Survey of the furosine content in cheeses
marketed in Spain. Journal of Food Protection 63, 974-975
Villamiel, M., Del Castillo, M.D., Corzo, N., Olano, A. (2001). Presence of furosine in honeys.
Journal of the Science of Food and Agriculture 81, 790-793
Vinale, F., Monti, S.M., Panunzi, B., Fogliano, V. (1999). Convenient synthesis of lactuloselysine and
its use for LC-MS analysis in milk-like model systems. Journal of Agricultural and Food
Chemistry 47, 4700-4706
Vlassara, H., Brownlee, M., Cerami, A. (1984). Accumulation of diabetic rat peripheral nerve myelin
by macrophages increases with the presence of advanced glycosylation endproducts. Journal of
Experimental Medicine 160, 197-207
Von Reichenbach, C.-L. (1844). Ueber die Röstung organischer Körper. Annalen der Chemie und
Pharmacie 49, 1-17
Watanabe, K., Nakamura, F., Suyama, K. (1995). Analysis of furosine as an indicator of lysine residue
glycation in milk protein by HPLC. Animal Science and Technology (Japan) 66, 293-298
- 213 -

7 Literaturverzeichnis
Watkins, N.G., Neglia-Fisher, C.I., Dyer, D.G., Thorpe, S.R., Baynes, J.W. (1987). Effect of
phosphate on the kinetics and specificity of glycation of protein. Journal of Biological
Chemistry 262, 7207-7212
Weigel, K.U., Opitz, T., Henle, T. (2004). Studies on the occurrence and formation of 1,2-dicarbonyls
in honey. European Food Research and Technology 218, 147-151
Weitzel, G., Geyer, H.-U., Fretzendorf, A.-M. (1957). Darstellung und Stabilität der Salze von
Aminosäure-N-Glycosiden. Chemische Berichte 90, 1153-1161
Wells-Knecht, K.J., Brinkmann, E., Baynes, J.W. (1995a). Characterization of an imidazolium salt
formed from glyoxal and Nα-hippuryllysine: A model for Maillard reaction crosslinks in
proteins. Journal of Organic Chemistry 60, 6246-6247
Wells-Knecht, K.J., Zyzak, D.V., Litchfield, J.E., Thorpe, S.R., Baynes, J.W. (1995b). Mechanism of
autoxidative glycosylation: Identification of glyoxal and arabinose as intermediates in the
autoxidative modification of proteins by glucose. Biochemistry 34, 3702-3709
Weenen, H. (1998). Reactive intermediates and carbohydrate fragmentation in Maillard chemistry.
Food Chemistry 62, 393-401
Weenen, H., Tjan, S.B. (1994). 3-Deoxyglucosone as flavor precursor. In: Maarse, H., Van der Heij,
D.G. (Ed.), Trends in flavor research. Amsterdam: Elsevier Science B.V., 327-337
Weenen, H., Tjan, S.B., De Valois, P.J., Bouter, N., Pos, A., Vonk, H. (1994). Mechanism of pyrazine
formation. In: Parliment, T.H., Morello, M.J., McGorrin, R.J. (Ed.), Thermally generated
flavors. Maillard, microwave, and extrusion process. Washington, DC: American Chemical
Society, 142-157
Westheimer, F.H., Jones, W.A. (1941). The effect of solvent on some reaction rates. Journal of the
American Chemical Society 63, 3283-3286
Westphal, G., Kroh, L. (1985a): Zum Mechanismus der „frühen Phase“ der Maillard-Reaktion. 1.
Mitteilung Einfluß der Struktur des Kohlenhydrates und der Aminosäure auf die Bildung des
N-Glycosids. Nahrung 29, 757-764
Westphal, G., Kroh, L. (1985b): Zum Mechanismus der „frühen Phase“ der Maillard-Reaktion. 2.
Mitteilung Folgereaktionen von N-Glycosiden. Nahrung 29, 765-775
Weygand, F. (1940). Über N-Glycoside. II. Amadori-Umlagerungen. Berichte der Deutschen
Chemischen Geselschaft 73, 1259-1278
Wilker, S.C., Chellan, P., Arnold, B.M., Nagaraj, R.H. (2001). Chromatographic quantification of
argpyrimidine, a methylglyoxal-derived product in tissue proteins: Comparison with
pentosidine. Analytical Biochemistry 290, 353-358
- 214 -

7 Literaturverzeichnis
Wright, A.E., Pomponi, S.A., Cross, S.S., McCarthy, P. (1992). A new bis(indole) alkaloid from a
deep-water marine sponge of the genus spongosorites. Journal of Organic Chemistry 57, 4772-
4775
Wrodnigg, T.M., Eder, B. (2001). The Amadori and Heyns rearrangements: Landmarks in the history
of carbohydrate chemistry or unrecognised synthetic opportunities? Topics in Currant
Chemistry 215, 115-152
Wu, Y.C., Monnier, V., Friedlander, M. (1995). Reliable determination of furosine in human serum
and dialysate proteins by high-performance liquid chromatography. Journal of
Chromatography B 667, 328-332
Yamaguchi, Y., Ishida, J., Xuan-Xuan, Z., Nakamura, M., Yoshitake, T. (1994). Determination of
glyoxal, methylglyoxal, diacetyl and 2,3-pentanedione in fermented foods by high-performance
liquid chromatography with fluorescence detection. Journal of Liquid Chromatography 17, 203-
211
Yanagisawa, K., Makita, Z., Shiroshita, K., Ueda, T., Fusegawa, T., Kuwajima, S., Takeuchi, M.,
Koike, T. (1998). Specific Assay for advanced glycation end products in blood and urine of
diabetic patients. Metabolism 47, 1548-1553
Zeppa, G., Conterno, L., Gerbi, V. (2001). Determination of organic acids, sugars, diacetyl, and
acetoin in cheese by high-performance liquid chromatography. Journal of Agricultural and
Food Chemistry 49, 2722-2726
- 215 -


Erklärung
Hiermit versichere ich, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und
ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe; die aus fremden
Quellen direkt oder indirekt übernommenen Gedanken sind als solche kenntlich gemacht. Die
Arbeit wurde bisher weder im Inland noch im Ausland in gleicher oder ähnlicher Form einer
anderen Prüfungsbehörde vorgelegt.
Die Dissertation wurde am Institut für Lebensmittelchemie der Technischen Universität
Dresden unter der wissenschaftlichen Betreuung von Herrn Prof. Dr. rer. nat. Dr.-Ing. habil.
Thomas Henle angefertigt.
Erfolglose Promotionsverfahren haben durch mich bis jetzt nicht stattgefunden.
Die Promotionsordnung der Fakultät Mathematik und Naturwissenschaften der Technischen
Universität Dresden in der aktuell gültigen Fassung erkenne ich in allen Teilen an.
René Krause


Danksagung
Mein ganz besonderer Dank gilt Herrn Prof. Dr. Thomas Henle, Direktor am Institut für
Lebensmittelchemie der TU Dresden, für das entgegengebrachte Vertrauen, die Möglichkeit
ein so interessantes Thema bearbeiten zu dürfen sowie die stetige Unterstützung und das
ausgezeichnete freundschaftliche Verhältnis.
Herrn Prof. Dr. Wolfgang Krause, emeritierter Professor am Institut für Lebensmittelchemie
der TU Dresden, und Herrn Prof. Dr. Lothar W. Kroh, Direktor am Institut für
Lebensmittelchemie der TU Berlin, danke ich für die freundliche Übernahme der Korreferate.
Bei Frau DLC Kerstin Knoll, Frau DLC Janina Kühn und Frau DLC Ilka Penndorf möchte ich
mich für die sehr engagierte Mitarbeit im Rahmen ihrer Diplomarbeiten bedanken. Frau Dipl.-
Ing. (FH) Karla Schlosser danke ich für die Durchführung der Aminosäureanalysen und Dr.
Uwe Schwarzenbolz für die Messungen mittels LC-ESI-MS. Bei Frau Annett Rudolf, Herrn
Dr. Dieter Scheller, Frau Dr. Margit Gruner, Herrn Dr. Herbert Kroschwitz und Frau Anke
Peritz vom Institut für Organische Chemie gilt mein Dank für die Aufnahme von NMR- und
MS-Spektren sowie die Durchführung von Elementaranalysen.
Allen Doktoranden, Mitarbeitern, Diplomanden und Studenten des Institutes für
Lebensmittelchemie der TU Dresden danke ich für das freundschaftliche Umfeld und die
angenehme Arbeitsatmosphäre. Ganz besonders möchte ich mich bei meinem Mitstreiter und
sehr guten Freund Dr. Thomas Siegl sowie meinen beiden Diplomandinnen Frau Evelyn
Schwarzer und Frau Katja Wolf bedanken.
Nicht zuletzt danke ich meiner Familie und Freunden durch deren Unterstützung das Gelingen
dieser Arbeit mit ermöglicht wurde.





























