
1
Biochemie/
Mikrobiologie
Ort: Praktikumsraum der Biochemie
MA Nord Ebene 0, Raum 428
Zeitplan: gemäß Gruppenverteilungsplan
Leitung: Prof. Dr. R. Erdmann
Der Teil Biochemie/Mikrobiologie des Biologiepraktikums findet an zwei
aufeinanderfolgenden Nachmittagen statt. Am ersten Praktikumstag werden die Versuche 1
bis 5 durchgeführt, die am zweiten Praktikumstag beendet und ausgewertet werden.
Daneben werden am zweiten Tag auch die Versuche 6 und 7 durchgeführt. Die Ergebnisse
der Versuche werden auf den dafür vorgesehenen Seiten der Praktikumsanleitung
protokolliert und anschließend dem Kursbetreuer vorgelegt.
Auflage 2014

2
VORBEMERKUNGEN ZUR SICHERHEIT IM LABORAT ORIUM
Im Praktikum gilt absolutes Rauchverbot. Es ist ebenfalls verboten, im Labor zu essen und
zu trinken. Außerdem ist zum Schutz der eigenen Kleidung ein Ki ttel (geschlossen!) zu
tragen.
Alle Lösungen, die in den Versuchen eingesetzt werden, müssen mit mechanischen
Pipettierhilfen (Eppendorf, Finnpipette) dosiert werden.
Im Labor vorhandene automatische Pipetten sind Präzisionsinstrumente und müssen mit
großer Sorgfalt behandelt werden. Die Handhabung dieser Pipetten ist ausführlich im
nächsten Kapitel dargestellt.
HANDHABUNG DER AUTOMATISCHEN PIPETTEN
Eine wichtige Voraussetzung für gutes Gelingen aller Versuche ist das richtige Pipettieren.
Die Dosierung von Flüssigkeiten im Mikroliter-Bereich wird mit mechanischen Pipettierhilfen
(Eppendorf-, und/oder Finn-Pipetten) durchgeführt. Beachten Sie die folgenden Regeln und
üben Sie den Umgang mit den Pipettierhilfen vor dem Versuch mit destilliertem Wasser.
FINN-PIPETTEN
Die Finn-Pipette ist ein volumetrisches Gerät zur genauen und sicheren Messung und Auf-
gabe von Flüssigkeiten. Je nach Modell können Volumina von 0,1 µl bis 5000 µl gemessen
und dosiert werden.
Die Finn-Pipette hat ein digitales Mikrometer, das das Volumen angibt. Das Volumen wird
durch Drehen der farblich geriffelten Einstellschraube eingestellt und ist innerhalb des
Einstellbereiches der Pipette kontinuierlich einstellbar. Das höchste Volumen steht auf der
Seite der Abwurfvorrichtung und entspricht der Nummer des Pipettenmodells.
Im Praktikum wird folgendes Pipettenmodelle verwendet:
Die Finn-Pipette wird mit Einwegpipettenspitzen aus Polypropylen benutzt.
Wichtig: Pipetten mit gefüllter Spitze niemals hinlegen!

3
Im Praktikum wird folgendes Pipettenmodelle verwendet:
Modell Einstellbereich (µl)
Rändelschraube gelb
20-100
Rändelschraube
blau
200-1000
Die Finn-Pipette wird mit Einwegpipettenspitzen aus Polypropylen benutzt. Die Einweg-
spritzen stellen die höchste Sicherheit für den Anwender dar und verhindern die Kontami-
nation zwischen den Proben. Um den Anwender vor einer Kontamination durch die Spitzen
zu schützen, verfügt die Finn-Pipette über einen eingebauten Spitzenabwerfer.
Einstellen des Volumens
Die Digitalanzeige besteht aus drei Ziffern und wird von oben nach unten abgelesen. Die drei
Ziffern geben das gewählte Volumen an.
Bei den blauen Pipetten bedeuten Ziffern Mikroliter
Das Volumen der Pipette wird durch Drehen der schwarzen, geriffelten Einstellschraube
eingestellt.
Diese Pipette wird auf 1,0 ml oder 1000 µl eingestellt.
Pipettierung
Die blaue Spitze auf den Schaft der Pipette aufstecken. Die Spitze dabei mit leichtem Drehen
fest andrücken, um absolute Dichtheit zu gewährleisten.
Anmerkung: Niemals Flüssigkeiten mit einer Finn-Pipette ohne Spitze aufnehmen!

4
Füllen
Den Druckknopf bis zum ersten Druckpunkt eindrücken (Abb. 2A). Die Pipette senkrecht
halten, und die Spitze in die Probenflüssigkeit ca. 1cm eintauchen.
Den Druckknopf langsam loslassen, um die Probe aufzusaugen (Abb. 2B). Eine Sekunde
warten, und dann die Spitze aus der Flüssigkeit herausnehmen.
Achtung: die Öffnung der Spitze nicht berühren!
Entleeren
Das Ende der Spitze in einem Winkel von 10 bis 40 Grad gegen die Innenwand des Gefäßes
halten. Den Druckknopf langsam bis zum ersten Druckpunkt herunterdrücken (Abb.2C). Eine
Sekunde warten. Den Druckknopf bis zum zweiten Druckpunkt herunterdrücken, um
restliche Flüssigkeit auszustoßen (Abb. 2D).
Die Spitze durch Drücken des Spitzenabwerfers abwerfen. Die Spitze muss nur gewechselt
werden, wenn eine andere Flüssigkeit aufgegeben oder die Volumeneinstellung geändert
wird.
Pipettenspitzen
Pipettenspitzen werden aus hochwertigem Polypropylen hergestellt. Die Spitzen sind farbig
und entsprechen der Farbe auf dem Druckknopf.Blaue oder gelbe Spitzen

5
Organisatorische Hinweise
• Die Zuteilung der Arbeitsplätze an die Studenten geschieht anhand der auf den
Praktikumskarten eingetragenen Nummern; jeder Student muss daher seine Nummer
kennen.
• An jedem Arbeitsplatz sind die für die Versuche benötigten Materialien und Geräte
aufgestellt.
• Jeder Student sollte einen wasserfesten Filzschreiber, ein Lineal und einen Arbeitskittel
mitbringen.
• Bei Bedarf, aber immer vor Verlassen des Praktikumsraums, sind die Hände zu Waschen und
zu Desinfizieren.
• Mäntel etc. können in den vor dem Kurssaal aufgestellten Spinden oder auf der Fensterbank
im Kurssaal abgelegt werden. Für die Spinde empfiehlt es sich ein Vorhängeschloss mitzubringen. Für abhanden gekommene Gegenstände wird keine Haftung übernommen.
• Gebrauchte Geräte (Pipettenspitzen, Kulturröhrchen) werden in den auf den Arbeitstischen
aufgestellten Abfallbehältern entsorgt. Objektträger und sonstige Glasabfälle werden in das
gekennzeichnete Becherglas gegeben.
• Die Ölimmersions-Objektive der Mikroskope sind nach der Benutzung mit einem mit Alkohol
benetzten Tuch vorsichtig zu reinigen.
• Vorsicht!! Bunsenbrenner!! (Haare, Kittel, Hände, Kittel, Steckdosen, Kabel)
• Bei Unklarheiten bitte rechtzeitig die Kursbetreuer um Rat fragen.
• Beimpfte Agarplatten nicht offen stehen lassen.
• Die am 1. Kurstag beimpften Agarplatten werden verschlossen und auf der Unterseite mit GruppenNr. (A1, A2, …D2), Name und Versuchstag beschriftet.
• Der Kursassistent legt die Aparplatten im Anschluss an alle Versuche in die entsprechenden
Brutschränke. Am 2. Kurstag liegen dann die Platten entsprechend der Gruppennummern
wieder aus.

6
Desinfektion und Sterilisation
Desinfektion: Ziel ist das Abtöten pathogener Mikroorganismen
Sterilisation: Ziel ist das Abtöten aller Bakterien (inkl. Sporen), Pilze und Protozoen,
sowie die Inaktivierung von Viren in einem Material.
Hygienische und chirurgische Händedesinfektion
Bei der ortständigen Besiedelung mit Bakterien handelt es sich um die residente gram-positive Flora, die bis in die Tiefen der Spaltlinien und entlang den Hautanhangsgebilden sitzt. Oberflächliche und immer wieder wechselnde Bakterien stellen die transiente Flora dar, die häufig auch gramnegativ sein kann.
Zur korrekten Desinfektion dürfen nur nach VAH-Liste (Verbund für angewandte Hygiene) bzw. Standardzulassung gemäß Arzneimittelgesetz § 36 zugelassene, alkoholische Hautdesinfektionsmittel verwendet werden.
Der Vorgang der hygienischen Händedesinfektion hat zum Ziel, die Übertragung von Krankheitserregern zu verhindern. Dies soll durch die Abtötung der transienten Flora erreicht werden. Durch die hygienische Händedesinfektion kann eine Keimübertragung von Mensch zu Mensch oder auch Arzt bzw. von Pflegepersonal auf den Patienten weitgehend vermieden werden. Sie ist ein wesentlicher Bestandteil der Prophylaxe von Hospitalinfektionen.
���� Die hygienische Händedesinfektion muss jedes Mal vor dem Verlassen des Praktikumsraumes durchgeführt werden!!!
Die chirurgische Händedesinfektion hat zum Ziel, neben der transienten auch möglichst die
gesamte residente Flora zu beseitigen. Sie wird vor operativen Eingriffen durchgeführt.
Durchführung der hygienischen Händedesinfektion:
• ausreichende Menge (mind. 3 ml) des Desinfektionsmittels mittels Ellenbogentechnik aus Desinfektionsmittelspender entnehmen
• Desinfektionsmittel über sämtliche Bereiche der trockenen Hände unter besonderer Berücksichtigung der Innen- und Außenflächen einschließlich Handgelenken, Flächen zwischen den Fingern, Fingerspitzen, Nagelfalze und Daumen einreiben
• für die Dauer der Einwirkzeit von 30 Sekunden feucht halten
Durchführung der chirurgischen Händedesinfektion:
• vor der chirurgischen Händedesinfektion werden Hände und Unterarme mit nach oben gerichteten Fingerspitzen und tieferliegenden Ellenbogen gewaschen (1 Min.)
• ausschließlich Nägel und Nagelfalzen sollen bei Bedarf mit weicher, desinfizierter Kunststoffbürste und hygienischen Handwaschpräparaten gereinigt werden
• trocknen der Hände mit einem keimarmen Einmal-Handtuch • einreiben der Hände und Unterarme nach Drei-Drittel-Regel mit einem
Hautdesinfektionsmittel (5 Min.), wobei die gesamte Hautoberfläche bis zum Ellenbogen 3 Minuten benetzt sein muss

7
1. Praktikumstag
1. Mikroorganismen in der Mund- und Rachenflora
Theoretischer Hintergrund und medizinische Relevanz:
Mundhöhle und Rachen sind von zahlreichen apathogenen Mikroorganismen besiedelt.
Strikt anaerobe Keime (Bakterien, die bei Zutritt von Sauerstoff in Oberflächenkulturen
nicht wachsen können) überwiegen mindestens um den Faktor 30 über aerobe Keime; da-
neben können Pilze und Protozoen in geringer Menge vorkommen. Unter den aeroben
Keimen überwiegen vergrünende Streptokokken, apathogene Neisserien (gramnegative
Kokken) und apathogene Corynebakterien (grampositive Stäbchen). Der Rachenflora
kommt eine wichtige Rolle bei der Entstehung der Zahnkaries zu, da es auf Zahnbelägen
aus nicht verdauten Kohlenhydraten zur Vermehrung von säureproduzierenden Strep-
tokokken kommt. Diese Keime sind in der Lage, hochmolekulare Kohlenhydrate zu pro-
duzieren, die sie vor einer Zerstörung durch das bakterizide Speichelenzym Lysozym
schützen. Unter dieser Schicht wird der Zahnschmelz demineralisiert. Daneben kommen
in geringer Anzahl häufig auch Keime vor, die bei massenhaftem Auftreten oder ein-
deutiger klinischer Symptomatik als Krankheitserreger gelten müssen (z.B. ß-
hämolysierende Streptokokken, Haemophilus influenzae). Es zeigt sich hier das Problem
einer quantitativen Beurteilung der Flora in physiologischer Weise mit Mikroorganismen
besiedelten Körperregionen.
Material: Blutagarplatte für jeden Teilnehmer eine Agarplatte steriler Abstrichtupfer Filzschreiber (Edding)
Bunsenbrenner
Impföse
Ausführung:
Eine Blutagarplatte wird auf der Rückseite mit den Initialen des Studenten und der
Gruppennummer als auch der Versuchsnummer beschriftet.
- Entnahme des Tonsillenabstrichs (Tupfer vorsichtig an die Tonsillen heranbringen und
unter drehenden Bewegungen abstreichen.
Wichtig: Berührung mit sonstigen Regionen der Mundhöhle vermeiden!
- mit Tupfer eine Hälfte der Blutagarplatte beimpfen.
- fraktioniertes Ausstreichen auf der Agarplatte siehe Abbildung Seite 9
- Die Platten werden verschlossen und ins Regal mit dem Deckel nach unten gelegt.
Der Betreuer legt die Platten anschließend für 2 Tage in den 37°C Brutschrank.
Am 2. Kurstag werden die Ergebnisse ausgewertet (Seite 27).

8
2. Differentialausstrich
Theoretischer Hintergrund:
Einzellige Mikroorganismen (Bakterien und Hefen) vermehren sich auf geeigneten
Nährmedien meist sehr rasch (Generationszeiten: wenige Minuten bis Stunden) durch
vegetative Teilung. Auf festen Nährböden (Agarplatten) kann eine einzelne Zelle in wenigen
Tagen zu einer Einzelkolonie mit ca.106-107 Zellen und mehreren mm Durchmesser
heranwachsen, die einen Klon identischer Zellen darstellt. Einzelkolonien verschiedener
Mikroorganismen unterscheiden sich häufig durch ihre Größe, Form und Farbe, so dass eine
Identifizierung möglich ist. Durch einen Verdünnungsausstrich auf einer geeigneten
Agarplatte ist es oft möglich, aus einem Gemisch verschiedener Mikroorganismen einzelne,
charakteristische Einzelkolonien zu gewinnen, mit deren Hilfe der Mikroorganismus weiter
charakterisiert und durch weitere Untersuchungen (z.B. der Stoffwechselleistungen) genau
identifiziert werden kann. Im folgenden Versuch sollen mehrere verschiedene Arten von
Mikroorganismen getrennt und ihr relativer Anteil in einer Mischkultur bestimmt werden.
Die auf dem Nährmedium gewachsenen Organismen werden am zweiten Praktikumstag
makroskopisch und mikroskopisch charakterisiert.
Medizinische Relevanz:
Die Gewinnung von Reinkulturen eines Erregers ist eine unabdingbare Voraussetzung für die
exakte Diagnostik von Infektionskrankheiten. In der Regel werden aus dem
Untersuchungsmaterial (Blut, Abstriche, Körperflüssigkeiten, Stuhlproben) zunächst
Anreicherungskulturen angelegt, aus denen sich durch einen Differentialausstrich auf einem
geeigneten Agar-Nährboden Einzelkolonien gewinnen lassen. Diese können als kleine
Reinkulturen für die weitere mikroskopische, biochemische und immunologische
Charakterisierung des Mikroorganismus verwendet werden.
Material: YPD-Agarplatte für jeden Teilnehmer eine Agarplatte
Reaktionsgefäß (Eppi) mit einer Mischkultur „M“
Impföse
Bunsenbrenner
Filsschreiber (Edding)
Vortex
Ausführung:
Die Mischkultur auf dem Vortex (Wirlmix) mischen.
Von der Mischkultur wird eine kleine Menge mit der sterilen Impfösen wie im folgenden
Schema beschrieben, (3-Ösen-Ausstrich nachfolgende Seite) gezeigt ausgestrichen. Das
fraktionierte Ausstreichen wird vom Kursbetreuer gezeigt. Die Platte wird verschlossen und
auf der Rückseite mit Gruppennummer, Name und Versuchsnummer beschriftet und mit
dem Deckel nach unten ins Regal gelegt.
Der Betreuer legt die Platten anschließend für 2 Tage in den 30°C Brutschrank.
Am 2. Kurstag werden die Ergebnisse ausgewertet (Seite 27). Dabei soll die relative Zahl der
verschiedenen Mikroorganismen bestimmt und von je einer repräsentativen Einzelkultur ein
mit Methylenblaufärbung gefärbtes Präparat mikroskopisch untersucht werden..

9
3-Ösen-Ausstrich
Um Bakterien zu isolieren werden diese auf YPD- Agar mit dem 3-Ösenausstrich vereinzelt
und das Wachstum der colony forming units (CFU) abgewartet. Mit der ausgeglühten und
abgekühlten Öse wird einmalig in das mit Keimen (M) beschriftete Eppendorfgefäß Probe
entnommen und im Zick-Zack vom Rand zur Mitte auf einem Drittel der Agar-Oberfläche
verteilt. Die Öse wird wieder ausgeglüht und nach Abkühlen zieht man sie einmal durch das
bereits beimpfte Feld und dann im Zick-Zack über das zweite Drittel des Nährbodens. Nach
Ausglühen wird in analoger Weise das letzte Drittel beimpft, nachdem man die Impföse einmal durch das zweite Drittel gezogen hat
Mit einer ausgeglühten und wieder erkalteten
Impföse etwas Material entnehmen.
Die Hälfte der Platte mit einer engen Zickzacklinie
beimpfen.
Öse ausglühen und erkalten lassen.
Kein neues Material entnehmen!
Impföse einmal durch den bereits beimpften
Bereich führen und ohne abzusetzen das nächste
Viertel in einer engen Zickzacklinie beimpfen.
Öse ausglühen und erkalten lassen.
Kein neues Material entnehmen!
Impföse einmal durch den zuletzt beimpften
Bereich führen und ohne abzusetzen das letzte
Viertel in einer engen Zickzacklinie beimpfen.

10
Temperaturzonen bei einer Flamme am Beispiel einer Kerze

11
3. Gärversuch
Theoretischer Hintergrund:
Heterotrophe Mikroorganismen sind meist in der Lage, ihren gesamten Nährstoffbedarf aus
einfachen organischen Verbindungen zu bestreiten. Aus diesen können nicht nur die
komplexen Zellstrukturen aufgebaut sondern auch die für Wachstum und Vermehrung
benötigten Energien gewonnen werden. Besonders günstige C-Quellen sind verschiedene
Zucker. Zucker sind eine Untergruppe der Kohlenhydrate, sie enthalten Kohlenstoff (C) und
die Elemente des Wassers (H2O) im Verhältnis C6(H2O)6. Zucker sind entweder
Polyhydroxyaldehyde (Aldosen) oder Polyhydroxyketone (Ketosen). Für Mikroorganismen
sind die meisten Zucker hervorragende Kohlenstoff- und Energiequellen. Minimale
Strukturunterschiede können jedoch tiefgreifende Auswirkungen auf die biologische
Verwertbarkeit spezifischer Zucker haben. In den Mikrobenzellen können die Zucker unter
Gewinnung von ATP abgebaut werden (Gärung), dabei entstehen Gas (CO2) und
verschiedene organische Säuren (z.B. Milchsäure oder Essigsäure).Verschiedene
Mikroorganismen unterscheiden sich in ihrer Fähigkeit bestimmte Zucker zu vergären, so
dass diese Eigenschaft zur Charakterisierung eines unbekannten Organismus verwendet
werden kann. Im folgenden Versuch werden die beiden epimeren Einfachzucker
(Monosaccharide) Glucose und Galactose verwendet, die sich lediglich durch die räumliche
Anordnung (Konfiguration) der alkoholischen Hydroxylgruppe C-Atom 4 (Abb. 4) unterscheiden.
Daneben werden auch die aus zwei Glucoseeinheiten zusammengesetzten Doppelzucker
(Disaccharide) Maltose und Cellobiose eingesetzt, die sich nur durch die räumliche
Anordnung der Bindung zwischen den beiden Glucoseeinheiten (α−glycosidisch bzw. ß–
glycosidisch) unterscheiden, sonst jedoch völlig identisch sind (Abb.5). Diese scheinbar
geringfügigen Strukturunterschiede haben einen tiefgreifenden Einfluss auf die biologische
Wertigkeit dieser Zucker. Glucose (Traubenzucker) ist für fast alle Mikroorganismen eine
hervorragende C-Quelle. Galactose (ein Bestandteil des Milchzuckers) kann dagegen nicht
von allen Mikroorganismen verwertet werden. Ähnliches gilt für Maltose und Cellobiose.
Während viele Organismen aus Maltose (dem Grundbaustein der pflanzlichen und tierischen
Stärke) Glucose freisetzen können, vermögen nur einige wenige spezialisierte Pilze und
Protozoen Cellobiose (Grundbaustein der Cellulose) in die beiden Glucosemoleküle aufzuspalten.

12
In der Regel werden Einfachzucker wie Glucose und sein Epimer Galaktose gut verwertet.
Die aus zwei Zuckereinheiten zusammengesetzten Disaccharide Maltose (α−Glucosyl-(1-→
4)-Glucosid) und Cellobiose (ß-Glucosyl-(1-→ 4) -Glucosid) (vgl. Abb. 5) können dagegen
nicht von allen Mikroorganismen in die beiden Zuckereinheiten gespalten und verwertet
werden. Die Verwertung bestimmter C-Quellen lässt sich in einem einfachen Kulturversuch
überprüfen. Dabei wird in einem Kulturröhrchen ein nährstofffreies Basismedium(dieses
enthält einen Säureindikator) gegeben, mit der zu untersuchenden C-Quelle versetzt und
einer Kultur des Mikroorganismus beimpft. Zusätzlich enthält das Kulturmedium eine
Gasfalle (sog. Durham-Röhrchen), in der sich gebildetes CO2 fängt. Kann die zugesetzte C-
Quelle vergoren werden, führt die Säurebildung zum Farbumschlag des Indikators und in der
Falle fängt sich eine Gasblase.
Medizinische Relevanz:
Auch in der menschlichen Ernährung spielen Kohlenhydrate, besonders Polymere der
Glucose wie Stärke und Glykogen, sowie Galaktose als Bestandteil des Milchzuckers Lactose
(ß-Galaktosyl-1→4 Glucosid) eine wichtige Rolle. Von Gesunden wird die aus Lactose durch
das Verdauungsenzym Lactase freigesetzte Galaktose im Stoffwechsel zu Glucose
epimerisiert. Seltene angeborene Enzymdefekte in diesem Stoffwechselweg können jedoch
die Verwertung von Galaktose unmöglich machen und zum Krankheitsbild der Galaktosämie führen. Dabei staut sich die unverwertete Galaktose im Blut an und führt zu einer schweren,
unbehandelt tödlichen, Vergiftung des Gehirns. Bei einer rechtzeitigen Diagnose kann jedoch
durch eine frühzeitige Behandlung eine Schädigung der Kinder vollständig verhindert
werden.
13
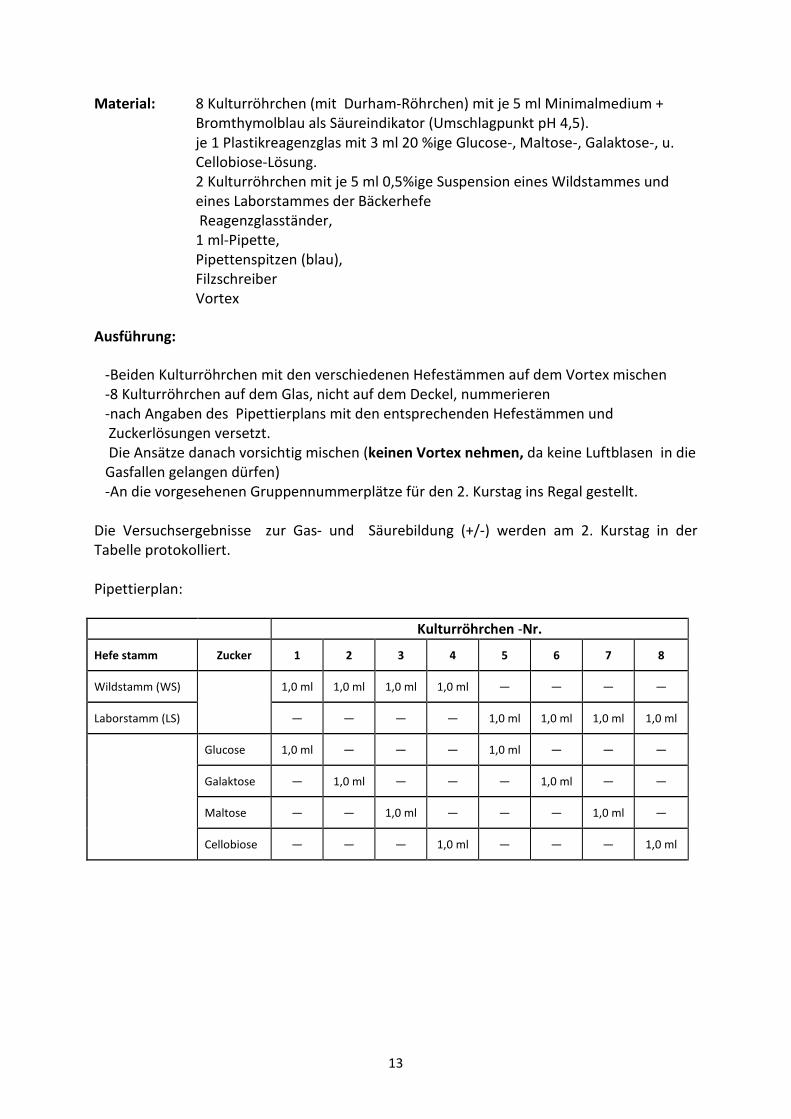
Material: 8 Kulturröhrchen (mit Durham-Röhrchen) mit je 5 ml Minimalmedium +
Bromthymolblau als Säureindikator (Umschlagpunkt pH 4,5).
je 1 Plastikreagenzglas mit 3 ml 20 %ige Glucose-, Maltose-, Galaktose-, u.
Cellobiose-Lösung.
2 Kulturröhrchen mit je 5 ml 0,5%ige Suspension eines Wildstammes und
eines Laborstammes der Bäckerhefe
Reagenzglasständer,
1 ml-Pipette,
Pipettenspitzen (blau),
Filzschreiber
Vortex
Ausführung:
-Beiden Kulturröhrchen mit den verschiedenen Hefestämmen auf dem Vortex mischen
-8 Kulturröhrchen auf dem Glas, nicht auf dem Deckel, nummerieren
-nach Angaben des Pipettierplans mit den entsprechenden Hefestämmen und
Zuckerlösungen versetzt.
Die Ansätze danach vorsichtig mischen (keinen Vortex nehmen, da keine Luftblasen in die
Gasfallen gelangen dürfen)
-An die vorgesehenen Gruppennummerplätze für den 2. Kurstag ins Regal gestellt.
Die Versuchsergebnisse zur Gas- und Säurebildung (+/-) werden am 2. Kurstag in der
Tabelle protokolliert.
Pipettierplan:
Kulturröhrchen -Nr.
Hefe stamm Zucker 1 2 3 4 5 6 7 8
Wildstamm (WS) 1,0 ml 1,0 ml 1,0 ml 1,0 ml — — — —
Laborstamm (LS) — — — — 1,0 ml 1,0 ml 1,0 ml 1,0 ml
Glucose 1,0 ml — — — 1,0 ml — — —
Galaktose — 1,0 ml — — — 1,0 ml — —
Maltose — — 1,0 ml — — — 1,0 ml —
Cellobiose — — — 1,0 ml — — — 1,0 ml
14
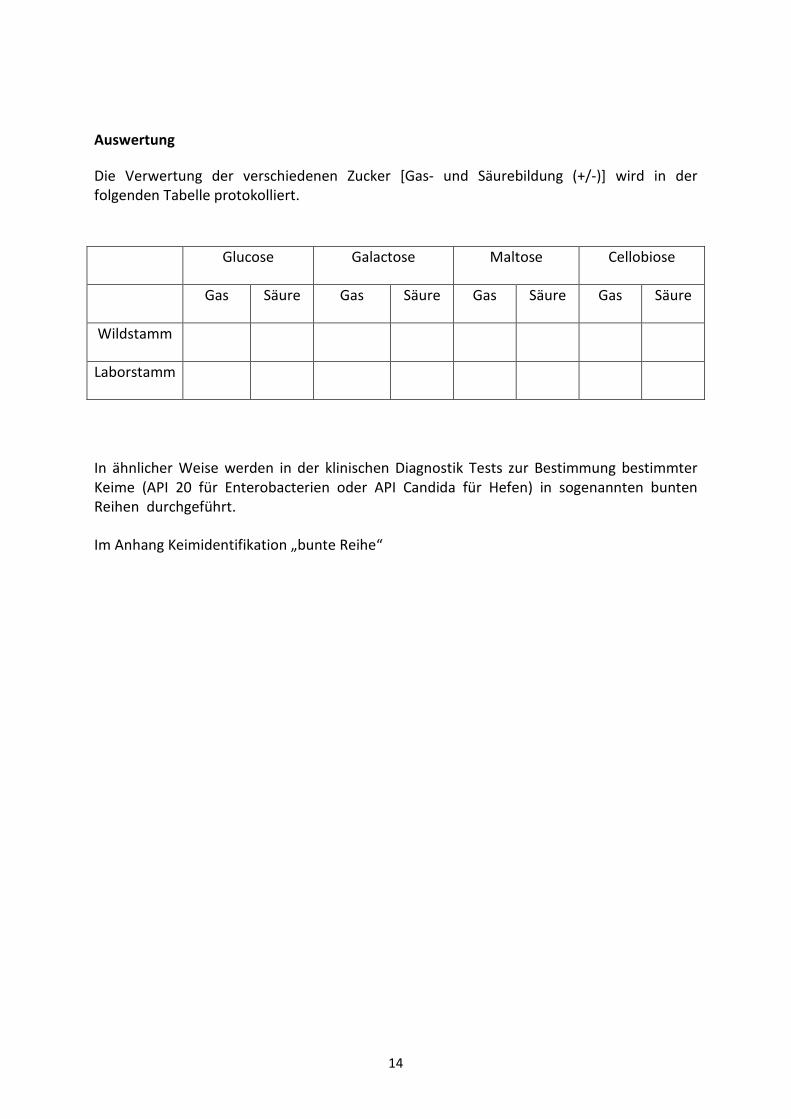
Auswertung
Die Verwertung der verschiedenen Zucker [Gas- und Säurebildung (+/-)] wird in der folgenden Tabelle protokolliert.
Glucose Galactose Maltose Cellobiose
Gas Säure Gas Säure Gas Säure Gas Säure
Wildstamm
Laborstamm
In ähnlicher Weise werden in der klinischen Diagnostik Tests zur Bestimmung bestimmter
Keime (API 20 für Enterobacterien oder API Candida für Hefen) in sogenannten bunten Reihen durchgeführt.
Im Anhang Keimidentifikation „bunte Reihe“

15
4. Antibiotika/ Resistenzbestimmung
Theoretischer Hintergrund und medizinische Relevanz:
Antibiotika sind sehr komplexe organische Verbindungen, die von einigen Mikroorgansimen
(Bes. Streptomyceten und Pilzen) in die Umgebung ausgeschieden werden und die in
anderen Mikroorganismen grundlegende Lebensprozesse (z.B. Replikation, Genexpression
oder die Zellwandbiosynthese) blockieren. Etwaige Nahrungskonkurrenten werden dadurch
abgetötet zumindest in ihrem Wachstum gehemmt. Penicillin (Abb.6), das von einigen
filamentösen Pilzen der Gattung Penicillium sezerniert wird, hemmt spezifisch eine
essentielle Reaktion in der Biosynthese des Mureins der Zellwand. Das Außenskelett
wachsender Bakterien verliert dadurch an Festigkeit und die Bakterienzellen werden durch
osmotische Lyse zerstört. Da Murein ausschließlich in den Zellwänden von Prokaryonten
vorkommt, ist dieses Antibiotikum spezifisch nur gegen Bakterien wirksam und kann zur Therapie bei bakteriellen Infektionen eingesetzt werden.
Der wichtigste Strukturbestandteil des Penicillins ist der sog.ß-Lactamring (vgl. Abb. 6)der für
die Wirkung auf die Mureinsynthese verantwortlich ist. Viele an sich penicillinempfindliche
Bakterien können durch Bildung des Enzymes ß-Lactamase (=Penicillinase), das den ß-
Lactamring hydrolytisch spaltet, gegenüber dem Antibiotikum resistent werden, jedoch kann
das native Penicillinmolekül durch chemische Strukturveränderungen gegenüber ß-
Lactamase resistent gemacht werden. Die pharmazeutische Industrie bietet eine Reihe von
substituierten Penicillinen (darunter auch Oxacillin) an, die durch Lactamase nicht mehr
gespalten werden können und deshalb erfolgreich gegen Penicillin-resistente Bakterien eingesetzt werden.
Eine antibiotische Therapie verspricht aber nur Erfolg, wenn das Antibiotikum gegen den
Krankheitserreger wirksam ist, bzw. keine Resistenz vorliegt. Die Wirksamkeit von
Antibiotika kann mit verschiedenen Methoden überprüft werden. Die am häufigsten verbreitete Methode ist der Agardiffusionstest

16
Abb.7.
Bei dem ein mit dem zu testenden Antibiotikum getränkter Filterpapierstreifen auf eine mit
dem Keim beimpfte Agarplatte gelegt wird. Aus der Größe der Hemmhöfe lassen sich
Rückschlüsse auf die Empfindlichkeit des Bakteriums ziehen. Die theoretischen Grundlagen
dieses Verfahrens sowie weitere Methoden der Resistenzbestimmung werden im Kursus der Medizinischen Mikrobiologie (5. Semester) behandelt.
Staphylococcus epidermidis ist ein Bestandteil der normalen Hautflora, der aber bei
bestimmen Patienten auch Infektionen hervorrufen kann. Dies ist auf seine Fähigkeit an
Polymeren zu binden zurückzuführen Im Krankenhaus aber kann er bei abwehrgeschwächten
Menschen bei Unsauberkeit eine Ursache für schwere Erkrankungen (sog. nonosomiale Infektion)
sein.
Im folgenden Versuch sollen zwei S.epidermis-Isolate (genetisch minimal veränderte
Stämme) auf Ihre Empfindlichkeit gegen Penicillin G und Oxacillin (einem Penicillinase-festen Penicillinderivat) hin untersucht werden.

17
Material: Flüssigkultur von 2 Staphylococcus epidermidis-Stämmen (Staph 1 und 2)
2 Reagenzgläser mit 2 ml physiolog. NaCl (steril)
Wattetupfer
2 Müller-Hinton-Agarplatten
Penicillin (P)- und Oxacillin (Ox)-Testblättchen
Pinzette
Ausführung
� von beiden Bakterienkulturen werden Verdünnungen hergestellt, indem man 100µl
der zuvor gevortexten Kultur in ein Röhrchen mit 2 ml physiolog. NaCl pipettiert
� aus jeder Verdünnung wird mit einem Wattetupfer eine Müller-Hinton-Agarplatte
gleichmässig mit Bakterien beimpft
� mit einer Pinzette wird je ein Penicillin (P)- und Oxacillin (Ox)-Testblättchen mit der
Pinzette aufgelegt und leicht angedrückt
� Platten mit dem Deckel nach oben ins Regal legen
� die Müller-Hinton-Agarplatten werden bei 37°C über Nacht bebrütet
Auswertung:
Die Hemmhofdurchmesser werden ausgemessen und anhand folgender Tabelle
interpretiert:
empfindlich (E) resistent (R)
Penicillin 10 U ≥ 29 mm ≤ 28 mm
Oxacillin 1 µg ≥ 18 mm ≤ 17 mm
Staph-1 Staph-2
Penicillin 10 U
Oxacillin 1 µg

18
5. Wachstum und Nachweis von Bakteriophagen (Bakterienviren)
Theoretischer Hintergrund:
Viren sind sehr kleine (> 100nm) Partikel, die an der Grenze zwischen unbelebten
Makromolekülkomplexen und sehr einfachen lebenden Organismen stehen. Sie bestehen
praktisch nur aus einem Innenkörper, einem DNA-oder RNA-Molekül, das ihre gesamte
genetische Information enthält, und einer Schutzhülle aus Proteinen (Kapsid). Viren haben
keinen eigenen Stoffwechsel und können daher Nährstoffe weder aufnehmen noch
verwerten. Auf der anderen Seite besitzt jedes Viruspartikel (Virion) die vollständige
genetische Information für seine Vermehrung. Eine Fortpflanzung findet jedoch nur statt,
wenn das Virus in eine geeignete Wirtszelle eindringen und ihren Stoffwechsel auf die
Produktion von Viruspartikeln umprogrammieren kann. Die infizierte Wirtszelle produziert
bis zu mehreren hundert neue Viruspartikel und geht in der Regel selbst dabei zugrunde.
Viren sind auf bestimmte Wirtszellen spezialisiert. So vermehren sich z. B. bakterielle Viren,
die als Bakteriophagen oder Phagen bezeichnet werden, nur in geeigneten Bakterienzellen,
wobei meistens nur eine oder wenige verwandte Bakterienarten von einem bestimmten
Phagen infiziert werden können. So infiziert z.B. der hier verwendete Bakteriophage T4
spezifisch bestimmte Stämme des Enterobakteriums Escherichia coli. Der Nachweis von
Bakteriophagen lässt sich am einfachsten aufgrund ihrer Infektiosität für das Wirtsbakterium
führen. Dazu wird ein kleines Volumen einer Phagensuspension mit einer großen Zahl von
Wirtsbakterien gemischt. Die Mischung wird gleichmäßig auf einem Nähragar verteilt, so
dass die Bakterien zu einer homogenen Zellschicht („Zellrasen“) auswachsen. In diesem
Rasen bilden diejenigen Bakterien, die mit einem Phagen infiziert wurden, ein infektiöses
Zentrum, von dem aus benachbarte Zellen ebenfalls infiziert und zerstört werden. Da
Bakteriophagen sich nur in wachsenden Zellen vermehren können, wiederholt sich der
Infektionsprozess nur solange, bis die Wirtszellen das Wachstum aus Nährstoffmangel
einstellen. Zu diesem Zeitpunkt ist im Bereich des infektiösen Zentrums der Bakterienrasen
zerstört. Es hat sich ein mehrere mm großes kreisrundes Loch („Plaque“) gebildet. Bei
geeigneter Verdünnung der Phagensuspension ist jeder Plaque von einem einzelnen
Viruspartikel verursacht, so dass sich aus der Zahl der Plaques die Anzahl der infektiösen
Partikel in der Suspension (= „Virustiter“) errechnen lässt.
Medizinische Relevanz:
In der Medizin haben Bakteriophagen nur eine untergeordnete Bedeutung. Versuche,
bakteriell bedingte Infektionskrankheiten durch spezifische gegen Erreger gerichtete Phagen
zu bekämpfen, erwiesen sich als erfolglos. Dagegen spielen Infektionen durch animalische
Viren (z.B. Grippe, Hepatitis, Pocken, Masern, Röteln, Windpocken, sowie AIDS) in der
medizinischen Praxis eine ungeheure Rolle. Auch bei diesen Viruskrankheiten führt die
Infektion zu einer lytischen Zerstörung der Wirtszelle, die je nachdem betroffenen Organe zu
den charakteristischen Krankheitssymtomen führen. Da es sich bei Viren um intracelluläre
Parasiten handelt, die den Stoffwechsel der Wirtszelle nutzen, ist eine Behandlung mit Antibiotika zwecklos und gefährlich.
Problem der Entwicklung antiviraler Medikamente
Da Viren beziehungsweise Virionen im Gegensatz zu Bakterien keine Zellen sind, können sie
auch nicht wie solche abgetötet werden. Es ist lediglich möglich, eine virale Infektion und die

19
Virusvermehrung durch Virostatika zu be- oder zu verhindern. Besonders die biochemischen
Vermehrungsabläufe können von Virusart zu Virusart sehr unterschiedlich sein, was die
Findung eines hemmenden oder unterbindenden Wirkstoffes erschwert.
Da die Vermehrung der Viren im Inneren von normalen Zellen stattfindet und sich dort sehr
eng an die zentralen biochemischen Zellmechanismen ankoppelt, müssen die in Frage
kommenden antiviralen Wirkstoffe entweder • das Eindringen der Virionen in die Wirtszellen verhindern,
• in den Zellstoffwechsel zum Nachteil der Virusvermehrung eingreifen,
• oder nach einer möglichen Virusvermehrung in den Zellen das Austreten der neuen Viren aus
den Zellen unterbinden.
Andererseits müssen diese gesuchten Wirkstoffe jedoch auch für den Körperstoffwechsel,
den Zellverband und/oder den internen Zellstoffwechsel insgesamt verträglich sein, da sonst
nicht nur beispielsweise die Virusvermehrung in den Zellen zum Erliegen kommt, sondern
schlimmstenfalls auch das (Zell-)Leben des gesamten behandelten Organismus.
Weil diese Bedingungen sehr schwer zu vereinbaren sind, sind die bisher entwickelten
antiviralen Medikamente auch oft mit schweren Nebenwirkungsrisiken verbunden. Es
handelt sich um eine Gratwanderung, welche die Medizin bislang meist vor eine unlösbare
Aufgabe stellt.
Verschärft wird die Entwicklung von effektiven antiviralen Medikamenten außerdem durch
die Resistenzentwicklung von Seiten der zu bekämpfenden Viren gegenüber einem einmal
gefundenen, brauchbaren Wirkstoff, zu der sie auf Grund ihres extrem schnell ablaufenden
Vermehrungszyklus und der biochemischen Eigenart dieser Replikation gut in der Lage sind.
Der Virentiter Der Titer ist in der Biologie und Medizin ein Maß für eine Konzentration, z. B. eines
Antikörpers, Antigens oder eines Virus. Er wird dadurch bestimmt, dass die Probe
fortlaufend verdünnt wird und mit den Verdünnungen ein bestimmter Test auf den zu
bestimmenden Stoff (z. B. Elisa/ Immunoassay) durchgeführt wird. Die weitestgehende
Verdünnung, bei der noch eine Reaktion nachweisbar ist, wird als Titer angegeben. Die
Ermittlung des Titers war früher eine übliche Methode, um eine Immunität nach einer
Impfung oder den Anstieg der Konzentration von Antikörpern während einer akuten
Infektionskrankheit zu beurteilen.
In der Regel wird – zum Beispiel ein Blutserum – in Zweierstufen verdünnt, d. h.
Verdünnungen von 1:2, 1:4, 1:8, 1:16, 1:32 usw. hergestellt. Die Verdünnungen gibt man
dann z. B. auf Zellkulturen, die dann mit einem Virus infiziert werden. Die höchste
Verdünnungsstufe, bei der noch eine Infektion der Zellen vollständig verhindert wird (also
noch ausreichend Antikörper vorhanden sind), wird als Titer angegeben. Ein Titer von 1:1024
gibt also eine höhere Konzentration an als 1:128, da trotz höherer Verdünnung noch eine
positive Reaktion des Tests festzustellen war.
Die Angabe des Titers ist heute aufgrund moderner und einfacherer Verfahren zur
Antikörper- oder Antigenbestimmung ungebräuchlich geworden. Nur bei wenigen
Krankheitserregern ist eine Verdünnungsreihe noch notwendig, wenn ein Neutralisationstest
oder eine Komplementbindungsreaktion durchgeführt werden muss. Auch bei der
manchmal noch gebräuchlichen Bestimmung des Rötelntiters haben doch andere Verfahren
zur Feststellung einer Immunität den Titer weitgehend verdrängt. Aber selbst wenn heute in
der Serologie Antikörperkonzentrationen in ng/ml oder IE/ml angegeben werden, wird der
Begriff „Titer“ für diese Angaben fälschlicherweise verwendet.

20
Material: 2 Petrischalen mit LB-Agar
2 Plastikreagenzgläser mit Weichagar im 50°C Wasserbad
Reaktionsgefäß (Eppi) mit Phagensuspension „P“
Reaktionsgefäß (Eppi) mit 0,9ml Natriumchloridlösung „NaCl“
Reaktionsgefäß (Eppi) mit E.coli Kultur „E“
0,1 ml bzw. 100 µl Pipette
gelbe Pipettenspitzen
Filzschreiber (Edding)
Ausführung: Beide LB-Platten mit dem Edding am Rand beschriften, (GruppenNr., verdünnt
oder unverdünnt)
Phagen und E.coli Lösungen vortexen
100 µl der gemischten Phagenlösung in das Eppi mit den 0,9ml
Natriumchloridlösung geben (entstandene Verdünnung 1:10 der
Ursprungsphagenlösung
Zügiges Arbeiten ist nun wichtig, da der Weichagar schnell erkaltet und sich ansonsten
ungleichmäßig verfestigt
100 µµµµl der unverdünnten Phagenlösung und 100 µ µ µ µl E.coli-Kultur in den
Weichagar (am Wasserbad) pipettieren
3x schwenken und den mit Phagen und E.coli versetzten Weichagar auf die
entsprechende LB-Platte gießen und gleichmäßig verteilen
100 µ µ µ µl der verdünnten Phagenlösung und 100 µ µ µ µl E.colil-Kultur in den
Weichagar (am Wasserbad) pipettieren
3x schwenken und den mit Phagen und E.coli versetzten Weichagar auf die
entsprechende LB-Platte gießen und gleichmäßig verteilen
Beide Platten mit dem Deckel nach oben am Arbeitsplatz stehen lassen.
Auswertung:
Am 2. Kurstag werden die entstandenen Plaques ausgezählt (Phagenanzahl/ml) und
derDurchschnittswert aller Arbeitsgruppen der unverdünnten Phagensuspension
bestimmt.
Anhand der auf beiden Platten ausgezählten Plaques wird die Anzahl der in der Suspension
enthaltenen Phagen errechnet. (Verdünnungsfaktor beachten!)
Ergebnis:
Die Anzahl der Phagen in eine ml Suspension ist:__________ Phagenpartikel/ml

21
2. Praktikumstag
Laufzettel: 1.Wachstumskurve von E.coli - alle 30 min „Trübung „ im Photometer messen
-Werte im Graphen eintragen
- nach 90 min. anhand es Graphen kontrollieren, ob die Verdopplungsrate bestimmt werden
kann (Berechnung oder im Graphen ablesen
2. Objektträger von den verschiedenen Keimen (direkt nach der Wachstumskurve anfertigen) - Zwei Klone vom Rachenabstrich (Versuch 1) und 3 bis 4 der verschiedenen im
Differentialausstrich (Versuch 2) gefundenen Kolonien auf je einen Objektträger geben,
Anmerkung: Objektträger braucht zum Trocknen Zeit.
-eigene Objekte mikroskopieren und es in den entsprechenden Tabellen protokollieren.
-als Vergleich die bereitliegenden Objekte anschauen.
3. Gärversuch auswerten -Farbveränderung und Gasbildung protokollieren und evtl. quantifizieren.
4. Antibiotika-Versuch -Hemmhöfe ausmessen (Angabe im Skript in Millimetern)
5. Bakteriophagen-Versuch -Plaques (Löcher) zählen
-Phagentiter/ml berechnen (Achtung Verdünnung beachten)
-Werte an die Tafel schreiben
22
6. Wachstumskurve von E.coli
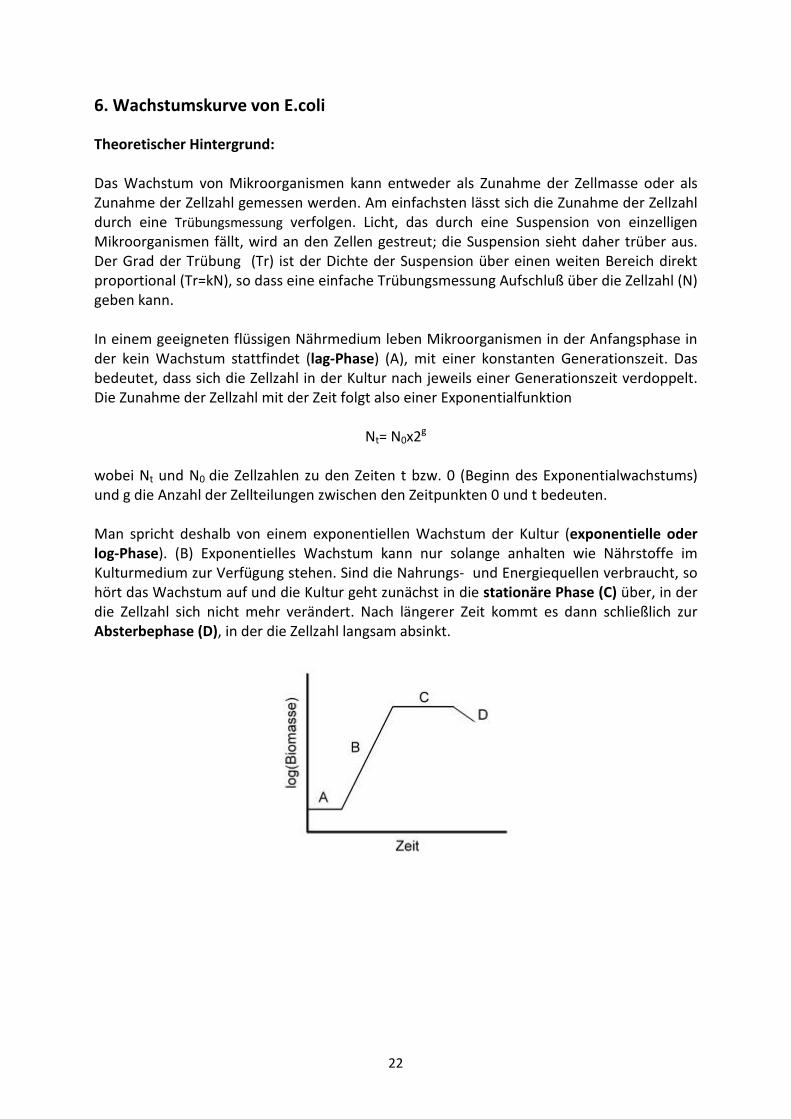
Theoretischer Hintergrund: Das Wachstum von Mikroorganismen kann entweder als Zunahme der Zellmasse oder als
Zunahme der Zellzahl gemessen werden. Am einfachsten lässt sich die Zunahme der Zellzahl
durch eine Trübungsmessung verfolgen. Licht, das durch eine Suspension von einzelligen
Mikroorganismen fällt, wird an den Zellen gestreut; die Suspension sieht daher trüber aus.
Der Grad der Trübung (Tr) ist der Dichte der Suspension über einen weiten Bereich direkt
proportional (Tr=kN), so dass eine einfache Trübungsmessung Aufschluß über die Zellzahl (N)
geben kann.
In einem geeigneten flüssigen Nährmedium leben Mikroorganismen in der Anfangsphase in
der kein Wachstum stattfindet (lag-Phase) (A), mit einer konstanten Generationszeit. Das
bedeutet, dass sich die Zellzahl in der Kultur nach jeweils einer Generationszeit verdoppelt.
Die Zunahme der Zellzahl mit der Zeit folgt also einer Exponentialfunktion
Nt= N0x2g
wobei Nt und N0 die Zellzahlen zu den Zeiten t bzw. 0 (Beginn des Exponentialwachstums)
und g die Anzahl der Zellteilungen zwischen den Zeitpunkten 0 und t bedeuten.
Man spricht deshalb von einem exponentiellen Wachstum der Kultur (exponentielle oder log-Phase). (B) Exponentielles Wachstum kann nur solange anhalten wie Nährstoffe im
Kulturmedium zur Verfügung stehen. Sind die Nahrungs- und Energiequellen verbraucht, so
hört das Wachstum auf und die Kultur geht zunächst in die stationäre Phase (C) über, in der
die Zellzahl sich nicht mehr verändert. Nach längerer Zeit kommt es dann schließlich zur
Absterbephase (D), in der die Zellzahl langsam absinkt.
23
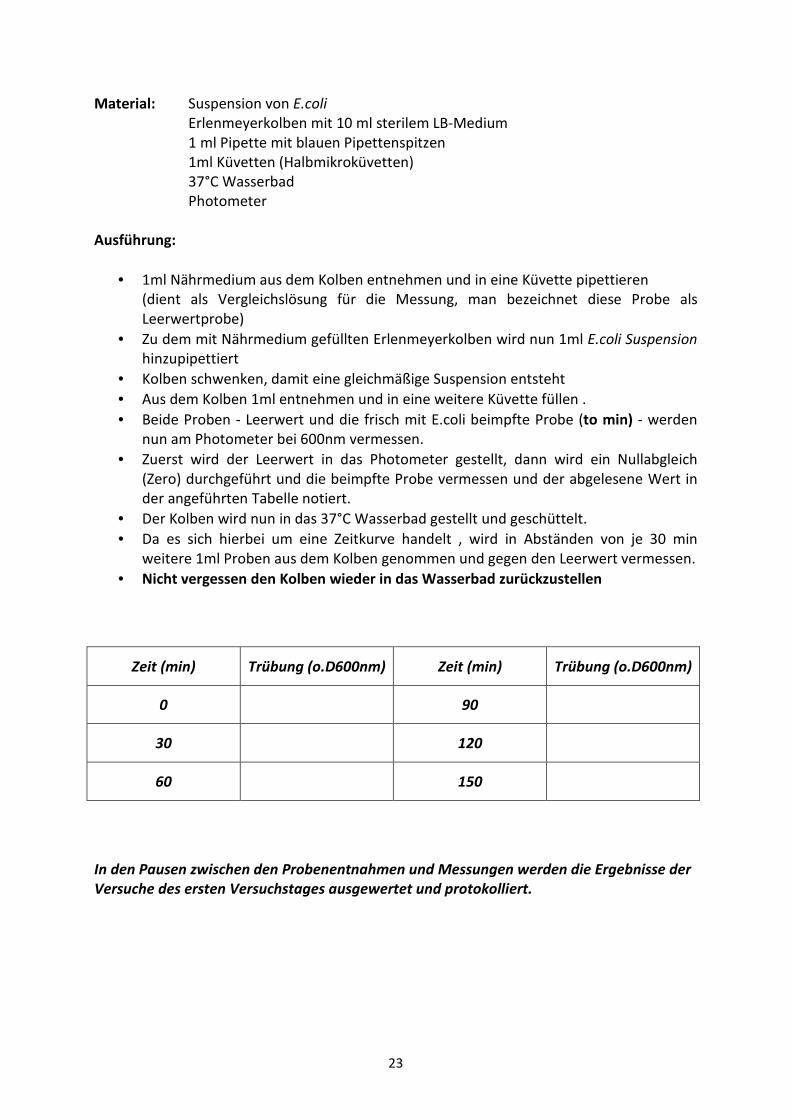
Material: Suspension von E.coli
Erlenmeyerkolben mit 10 ml sterilem LB-Medium
1 ml Pipette mit blauen Pipettenspitzen
1ml Küvetten (Halbmikroküvetten)
37°C Wasserbad
Photometer
Ausführung:
• 1ml Nährmedium aus dem Kolben entnehmen und in eine Küvette pipettieren
(dient als Vergleichslösung für die Messung, man bezeichnet diese Probe als
Leerwertprobe)
• Zu dem mit Nährmedium gefüllten Erlenmeyerkolben wird nun 1ml E.coli Suspension
hinzupipettiert
• Kolben schwenken, damit eine gleichmäßige Suspension entsteht
• Aus dem Kolben 1ml entnehmen und in eine weitere Küvette füllen .
• Beide Proben - Leerwert und die frisch mit E.coli beimpfte Probe (to min) - werden
nun am Photometer bei 600nm vermessen.
• Zuerst wird der Leerwert in das Photometer gestellt, dann wird ein Nullabgleich
(Zero) durchgeführt und die beimpfte Probe vermessen und der abgelesene Wert in
der angeführten Tabelle notiert.
• Der Kolben wird nun in das 37°C Wasserbad gestellt und geschüttelt.
• Da es sich hierbei um eine Zeitkurve handelt , wird in Abständen von je 30 min
weitere 1ml Proben aus dem Kolben genommen und gegen den Leerwert vermessen.
• Nicht vergessen den Kolben wieder in das Wasserbad zurückzustellen
Zeit (min) Trübung (o.D600nm) Zeit (min) Trübung (o.D600nm)
0 90
30 120
60 150
In den Pausen zwischen den Probenentnahmen und Messungen werden die Ergebnisse der
Versuche des ersten Versuchstages ausgewertet und protokolliert.

24
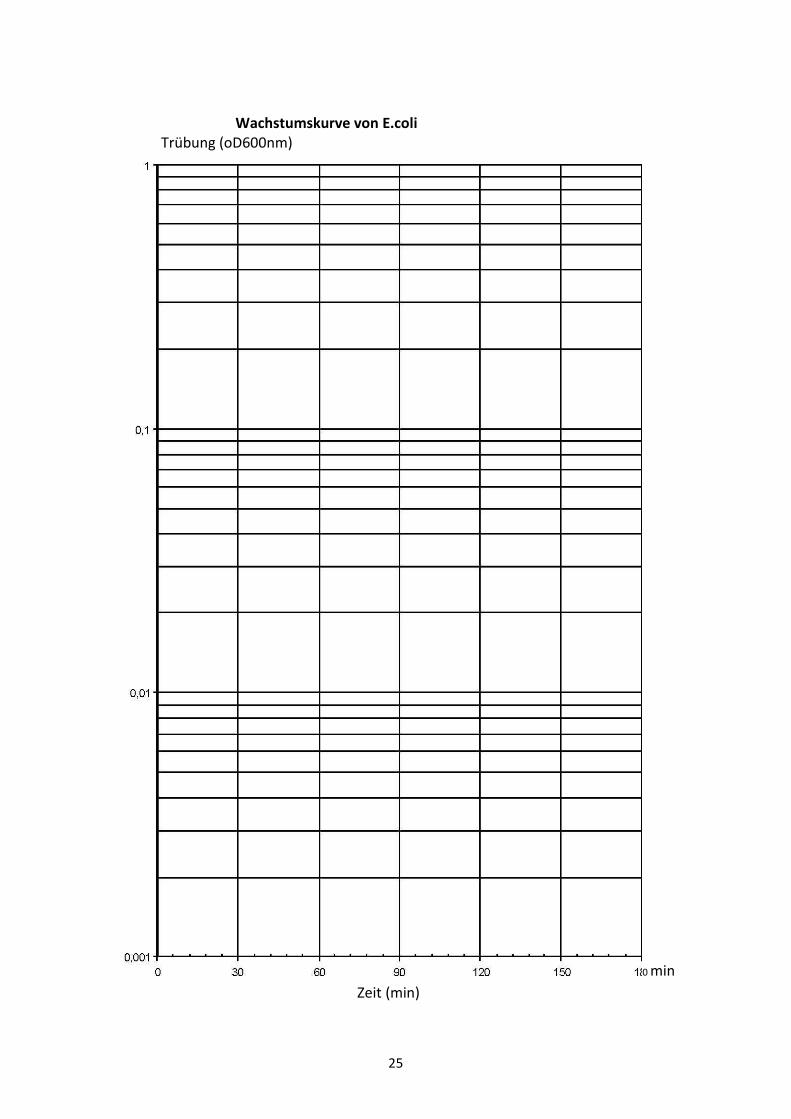
Auswertung
Die gemessenen Trübungswerte werden in das vorgegebene halblogarithmische
Koordinatensystem eingezeichnet und die Punkte durch eine Kurve verbunden. Aus dem
linearen Abschnitt der Wachstumskurve kann die Verdopplungszeit (tD), die benötigt wird
um die Zellzahl in der Kultur zu verdoppeln und die der Generationszeit entspricht, direkt
abgelesen oder nach der Formel tD =���2(2−1)
log �2−����1 berechnet,
wobei t1; t2 und N1; N2 die Zeiten bzw. Zellzahlen zu Beginn und Ende des exponentiellen
Wachstums bedeuten:
Ergebnis:
Unter den verwendeten Versuchsbedingungen beträgt die Generationszeit der Bakterien
____________ min.
25
Wachstumskurve von E.coli Trübung (oD600nm)
0 min
Zeit (min)

26
7. Mikroskopischer Nachweis von Bakterien (Methylenblau-Färbung) Mithilfe dieser Färbemethoden werden 2 bis 3 unterschiedliche Kolonien von Hautkeimen
(Versuch 1) und 3 bis 4 der verschiedenen im Differentialausstrich (Versuch 2) gefundenen
Kolonien untersucht. Die Ergebnisse der Färbung werden wie in der nachfolgenden Tabelle
bei der Auswertung nach morphologischen und mikroskopischen Kriterien von Versuch 1 und Versuch 2 protokolliert.
Material: Objektträger,
schwarze Kachel
Glasschreiber zur Nummerierung der Objektträger
phys. NaCl in Tropfflaschen
Methylenblaulösung
Filterblock
Ausführung:
Herstellung eines fixierten Präparates mit Kolonien einer Agarplatte:
• Objektträger auf die schwarze Kachel legen und mit dem Glasschreiber die Nummer
des Präparates auf den Objektträger kratzen
• einen kleinen Tropfen NaCl in die Mitte des Objektträger geben
• Impföse ausglühen und auskühlen lassen
• mit der erkalteten Impföse eine Kolonie auf der Agarplatte leicht „antippen“
• Material im NaCl großflächig verteilen und komplett lufttrocknen lassen, bis kein
Flüssigkeitsfilm auf dem Objektträger mehr sichtbar ist (damit mit dem nächsten
Schritt die Bakterien nicht gekocht werden!!)
• Hitzefixierung: Objektträger mit der „Bakterienseite“ nach oben (Pinzette,
Schutzbrille) gleichmäßig der Länge nach durch die Flamme ziehen, diesesn Vorgang
3mal wiederholen
• Hitzefixierte Präparate auf die Färbebank am Waschbecken legen.
Methylenblaulösung auf das Präparat für 1 min tropfen und nach 1 min unter
fließendem Wasser
• Die überschüssige Farblösung unter fließendem Wasser abspülen
• Präparat in den Filterblock legen und abtrocknen.
Mikroskopische Untersuchung der Präparate
Auf das gefärbte Präparat einen Tropfen Immersionsöl geben und mit dem
Ölimmersionsobjektiv (100fache Vergrößerung) bei offener Blende und
hochgestelltem Kondensor mikroskopieren.
Beurteilung der Bakterienform (Kokken, Stäbchen, Größe) und ihrer Lagerung (Ketten,
Haufen).
Nach dem Mikroskopieren wird das Objektiv mit 70%igem Alkohol gereinigt.
Die selbst angefertigten Präparate werden in dem an den Waschbecken stehenden
Plastikbechergläsern entsorgt und die Beispielpräparate zurück in den Objektträgerhalter
gelegt.
27
Auswertung der Versuche
Zu Versuch 1 (Rachenflora)
Bestimmen Sie die Anzahl der Kolonien. Protokollieren Sie die Morphologie der Kolonien
(Größe, Form, Farbe) und führen Sie mit den Mikroorganismen aus charakteristischen
Kolonien eine Methylenblaufärbung durch. Protokollieren Sie die Morphologie der gefunden
Mikroorganismen und versuchen Sie, sie aufgrund ihrer Form (Stäbchen, Kokken) und
Lagebeziehung (Haufen, Ketten) zu charakterisieren.
Charakterisieren Sie maximal drei Keime nach makroskopischen und mikroskopischen
Kriterien.
Koloniemorphologie (Durchmesser, Farbe,
Oberfläche glänzend oder
matt, Geruch etc.
Mikroskopie Vorbereitung der Proben
siehe Aufgabenteil 7
Methylenblaufärbung
Hautkeim 1
Hautkeim 2
Hautkeim 3
Zu Versuch 2 (Differentialausstrich)
Bestimmen Sie die Morphologie der gefundenen Keime nach Größe, Form und Farbe. Führen
Sie mit den Mikroorganismen aus charakteristischen Kolonien eine Methylenblaufärbung
durch. Protokollieren Sie die Morphologie der gefunden Mikroorganismen und versuchen
Sie, sie aufgrund ihrer Form (Stäbchen, Kokken) und Lagebeziehung (Haufen, Ketten) und Größe zu charakterisieren.
Charakterisieren Sie 3 Keime nach makroskopischen und mikroskopischen Kriterien und
nehmen Sie noch 2 Keime von unterschiedlicher Größe auf einen Objektträger und
mikroskopieren Sie diese.
Koloniemorphologie (Durchmesser, Farbe, Oberfläche
glänzend oder matt, Geruch etc.
Mikroskopie Vorbereitung der Proben siehe
Aufgabenteil 7
Keim 1
Keim 2
Keim3

28
29
Glossar
Absterbephase Endphase des Wachstums von Mikroorgansimen
in der der Zelltod gegenüber der Zellteilung überwiegt
Aldose Einfachzucker mit einer Aldehydgruppe (-CH=O) als wichtigste funktionelle Gruppe
Agar
Aus einer Alge gewonnenes Polysaccharid, das
bei hoher Temperatur flüssig ist und beim
Abkühlen geliert. Das Gel kann durch Einschluss
der unterschiedlichen Nährlösungen feste
Nährböden für Mikroorganismen bilden.
Antibiotika
Von Mikroorganismen gebildete und
ausgeschiedene hochkomplexe organische
Verbindungen, die durch eine spezifische
Hemmung zentraler Lebensprozesse andere
Mikroorganismen abtöten oder im Wachstum
hemmen kann,
apathogen ncht krankheitserregend
Bakteriophagen für Bakterien spezifische Viren
bakterizid abtötende Wirkung von Desinfektionsmitteln auf
Bakterien
Blutagar Agarnährboden mit eingeschlossenen roten
Blutkörperchen
denaturieren Zerstörung der nativen Raumstruktur (besonders
von Proteinen)
Desinfektion Abtötung aller Erreger übertragbarer
Infektionskrankheiten
Disaccharid Aus zwei über eine glykosidische Bindung
verknüpfte Einfachzucker zusammengesetzt zu einem Doppelzucker
Einzelkolonie
Auf einem festen Nährboden (Agarplatte) durch vegetative Vermehrung aus einer einzelnen Zelle
gewachsener Klon identischer Mikroorganismen.
Eine Einzelkolonie stellt eine Reinkultur dar.
Enterobakterien
In der Darmflora vorkommende, meist
apathogene Bakterien, vorwiegend
gramnegative Stäbchen. Zu ihnen gehören
allerdings auch verschiedene Krankheitserreger.
Enzym Katalytisch wirksames Protein, das spezifische
biochemische Reaktionen zu beschleunigen
vermag.
epimere Zucker
Zucker, deren Konfiguration nur in einem
einzigen asymmetrischen Zentrum
unterschiedlich ist, wie z.B. Glucose und
Galaktose
30
Galaktosämie angeborene Stoffwechselkrankheit, bei der
Galaktose nicht abgebaut werden kann.
Galaktose Hexose, Bestandteil des Milchzuckers Laktose
Generationszeit Die Zeit, in der sich die Mikroorganismen
verdoppeln
Genexpression
Umsetzung der im Genom gespeicherten
genetischen Information in spezifische
funktionelle Genprodukte: Proteine (durch
Transkription und Translation) oder rRNA bzw.
tRNA (nur Transkription)
Glucose = Traubenzucker, wichtigstes Kohlenhydrat im
Stoffwechsel
Heterotroph(er) Stoffwechsel Baustoffwechsel, der vorgefundene organische
Moleküle als Kohlenstoffquelle verwendet.
Ketose
Einfachzucker mit einer Ketogruppe
als wichtigste funktionelle Gruppe
ß-Lactamring
Lactame sind intramolekulare Säureamide. Der
viergliedrige ß-Lactamring ist wesentlich für den
antibiotischen Wirkungsmechanismus der sog. ß-
Lactamantibiotika (Penicillin und Cephalosporin)
LB-Medium Von den Biologen Luria und Burrows
entwickeltes optimales Nährmedium für E.coli
log-Phase/ exponentielle Phase Phase der Wachastumskurve, in der sich die Zellen mit konstanter Generationszeit
exponentiell teilen
Membran, biologisch Biologische Membranen bestehen aus durch
eingelagerte Proteine stabilisierte
Lipiddoppelschichten
Minimalmedium Nährmedium, das lediglich die einfachsten für
das Wachstum von Mikroorganismen
unentbehrliche Nährstoffe enthält
Monosaccharide Einfachzucker
Müller-Hinton Agar Von Müller und Hinton entwickeltes
Optimalmedium für den Antibiotika-
Diffusionstest
Murein
Charakteristisches netzartiges Bauelement der
Bakterienzellwand aus langen
Kohlenhydratketten, die durch kurze
Peptideketten quervernetzt sind. Bei der
Mureinsynthese wird die Ausbildung der
Peptidvernetzung durch Penicillin spezifisch blockiert.
31
parasitisch, parasitierend von (oft auch) anderen Organismen lebend
pathogen krankheitsauslösend
Penicillin
Von Schimmelpilzen der Gattung Penicillium
gebildetes Antibiotikum, das spezifisch die
Biosynthese von Murein in der
Bakterienzellwand hemmt.
Polymer Durch Aneinanderreihung von ähnlichen
Grundbausteinen (Monomeren) aufgebautes
Makromolekül
Proteine wichtigste Klasse von biologischen Polymeren;
aus Aminosäuren aufgebaut
Replikation DNA-Synthese
Resistenz Unempfindlichkeit gegenüber äußeren
Einflüssen z.B. Antibiotika oder anderen
Medikamenten
saprophytisch von abgestorbenem Zellmaterial lebend
stationäre Phase Dritte Phase der Wachstumskurve, in der die
Zellzahl konstant bleibt, weil sich
Zellvermehrung und Zelltod die Waage halten.
Sterilisation Abtötung aller vermehrungsfähigen
Mikroorganismen
Titer Durch eine Verdünnungsreihe bestimmte
Konzentration
Viren
Subzelluläre, sehr kleine (<0.3m) Lebensformen
an der Grenze zwischen unbelebten Makromolekülkomplexen und sehr primitiven
Mikroorgansimen, die aus einer Nukleinsäure
(DNAoder RNA) und einigen Proteinen bestehen.
virulent hochgradig pathogen
Virion/ Viruspartikel
Infektiöse Viruseinheit. Aufgebaut aus einem
Innenkörper aus DNAoder RNA und dem aus
Kapsomerenproteinen zusammengesetztes
Kapsid
Wachstum von Mikroorganismen
Da sich Mikrooganismen bei Verdoppelung ihrer
Zellmasse in zwei Tochterzellen teilen, ist für sie
Wachstum gleichbedeutend mit Zellvermehrung
von Mikroorganismen
Wachstumskurve
Auftragung der Anzahl von Mikroorganismen
gegen die Zeit. Eine Wachstumskurve umfasst
vier Phasen: 1.lag-Phase, 2.exponentielle Phase,
3.stationäre Phase und die Absterbephase
YPD-Agar Optimalmedium für das Wachstum von
Mikroorganismen auf der Basis von Yeast (=Hefe), Pepton und Dextrose

32
Keimidentifikation
Die Diagnostik mikrobieller Kontaminanten kann – je nach Aufgabenstellung – auf unterschiedlichem Niveau ausgeführt werden. Zur primären Charakterisierung dienen neben einer Beurteilung des makroskopisch erkennbaren Wachstumsverhaltens einfache Reaktionen, wie Gram-Färbung, Oxidase-Test,… sowie mikroskopische Methoden. Hierzu steht ein leistungsfähiges Mikroskop zur Verfügung, welches auch fluoreszenzanalytische Messungen erlaubt.
Präzisionsbestimmungen bakterieller Keime werden über das API-System durchgeführt, das auf der Basis zahlreicher, spezifischer Stoffwechselvorgänge arbeitet („bunte Reihe“) und rechnergestützt einen großen Vergleichsdatenbestand zur exakten Diagnose heranzieht.
Recommended










