
1
BIOCHEMISCHES
PRAKTIKUM
für Toxikologen
Fassung vom Februar 2012
Universität Kaiserslautern
Fachbereich Chemie
Prof. Dr. W. E. Trommer

2
Inhaltsverzeichnis
Zeitplan zum Biochemie-Praktikum für Toxikologen zum WS 2011/12 .............................. 3
Bestimmung der katalytischen Konstanten und der Michaelis-Konstanten von
Chymotrypsin für N-Succinyl-alanyl-alanyl-prolyl-phenylalanin-4-nitroanilid (Suc-AAPF-
pNA) ............................................................................................................................. 4
Disk-Elektrophorese in Natriumdodecylsulfat (SDS-PAGE) ............................................. 12
Enzymatische Bestimmung von ADP (Adenosindiphosphat) ........................................ 20
Quantitative Proteinbestimmungsmethoden .................................................................... 24
Enzyme-Linked Immunosorbent Assay (ELISA) .............................................................. 30
Gezielte Mutagenese ...................................................................................................... 40
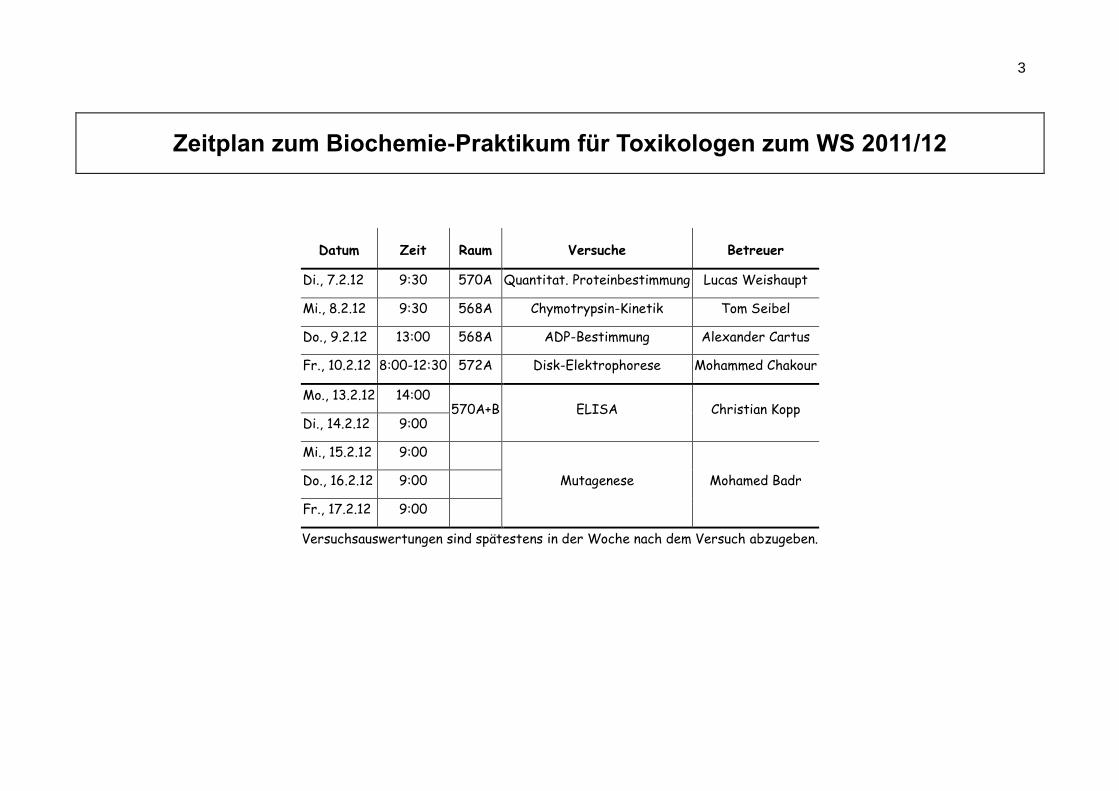
3
Zeitplan zum Biochemie-Praktikum für Toxikologen zum WS 2011/12
Datum Zeit Raum Versuche Betreuer
Di., 7.2.12 9:30 570A Quantitat. Proteinbestimmung Lucas Weishaupt
Mi., 8.2.12 9:30 568A Chymotrypsin-Kinetik Tom Seibel
Do., 9.2.12 13:00 568A ADP-Bestimmung Alexander Cartus
Fr., 10.2.12 8:00-12:30 572A Disk-Elektrophorese Mohammed Chakour
Mo., 13.2.12 14:00 570A+B ELISA Christian Kopp
Di., 14.2.12 9:00
Mi., 15.2.12 9:00
Mutagenese Mohamed Badr Do., 16.2.12 9:00
Fr., 17.2.12 9:00
Versuchsauswertungen sind spätestens in der Woche nach dem Versuch abzugeben.

4
Bestimmung der katalytischen Konstanten und der Michaelis-Konstanten von Chymotrypsin für
N-Succinyl-alanyl-alanyl-prolyl-phenylalanin-4-nitroanilid (Suc-AAPF-pNA)
Bestimmung der katalytischen Konstanten und der Michaelis-Konstanten von
Chymotrypsin für N-Succinyl-alanyl-alanyl-prolyl-phenylalanin-4-nitroanilid (Suc-AAPF-
pNA) ............................................................................................................................. 4
Graphische Bestimmung von KM und vmax ..................................................................... 6
Aufgabe ......................................................................................................................... 8
Herstellung der Lösungen .............................................................................................. 9
Testansatz ..................................................................................................................... 9
Auswertung .................................................................................................................. 10
Benötigte Angaben: ..................................................................................................... 10
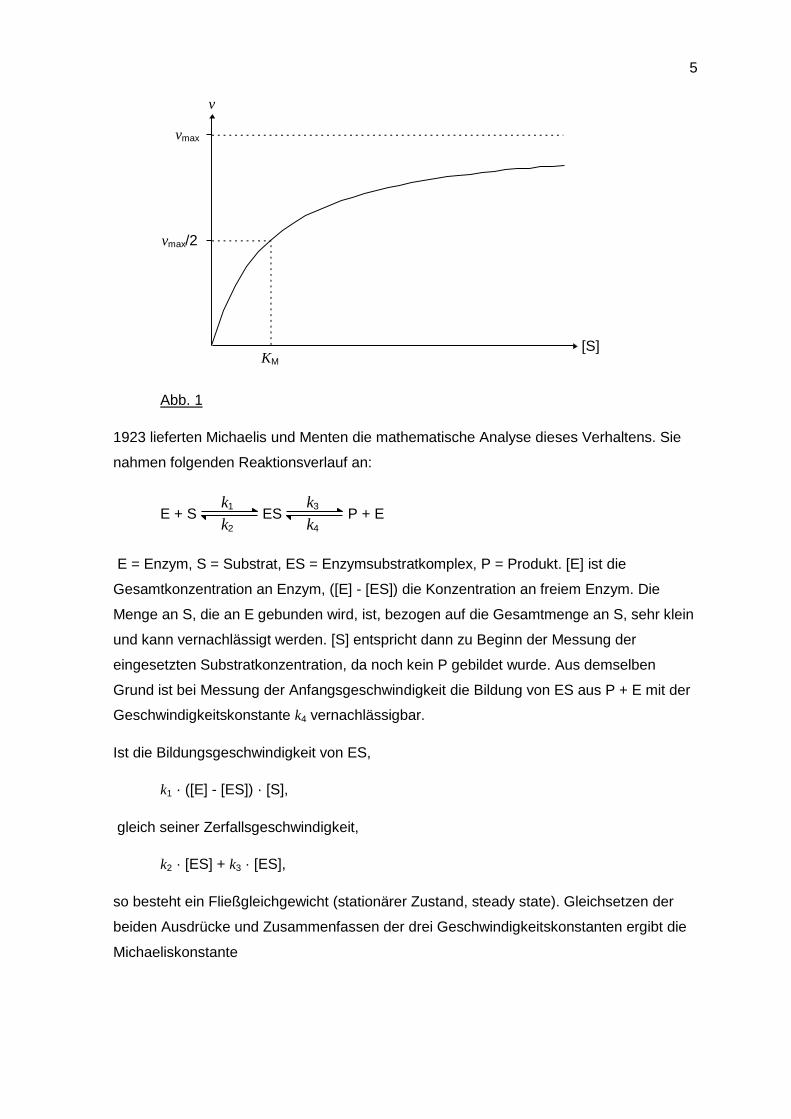
Die Reaktionsgeschwindigkeit einer enzymatisch katalysierten Reaktion hängt u. a. auch
von der Substratkonzentration ab. Bei der maximalen Umsatzgeschwindigkeit (vmax) ist
das Enzym vollständig mit Substrat abgesättigt, während bei geringeren Substrat-
konzentrationen nicht alle Enzymmoleküle abgesättigt sind.
5
KM
[S]
v
vmax
vmax/2
Abb. 1
1923 lieferten Michaelis und Menten die mathematische Analyse dieses Verhaltens. Sie
nahmen folgenden Reaktionsverlauf an:
E + S k2
k1 ES
k4
k3 P + E
E = Enzym, S = Substrat, ES = Enzymsubstratkomplex, P = Produkt. [E] ist die
Gesamtkonzentration an Enzym, ([E] - [ES]) die Konzentration an freiem Enzym. Die
Menge an S, die an E gebunden wird, ist, bezogen auf die Gesamtmenge an S, sehr klein
und kann vernachlässigt werden. [S] entspricht dann zu Beginn der Messung der
eingesetzten Substratkonzentration, da noch kein P gebildet wurde. Aus demselben
Grund ist bei Messung der Anfangsgeschwindigkeit die Bildung von ES aus P + E mit der
Geschwindigkeitskonstante k4 vernachlässigbar.
Ist die Bildungsgeschwindigkeit von ES,
k1 · ([E] - [ES]) · [S],
gleich seiner Zerfallsgeschwindigkeit,
k2 · [ES] + k3 · [ES],
so besteht ein Fließgleichgewicht (stationärer Zustand, steady state). Gleichsetzen der
beiden Ausdrücke und Zusammenfassen der drei Geschwindigkeitskonstanten ergibt die
Michaeliskonstante

6
Kk k
kM
2 3E ES S
ES
1
(1)
Wenn k2 k3, kann k3 vernachlässigt werden, und KM k2/k1 = KD ist die Dissoziations-
konstante des Gleichgewichts E + S ES.
Eine Bestimmung von KM ist dann möglich, wenn [ES] bestimmt werden kann. Dies ist auf
direktem Weg sehr schwierig, aber die Reaktionsgeschwindigkeit ist proportional [ES]:
v = k3 · [ES] (2)
Aus Gleichung (1) wird [ES] errechnet:
ES
E S
SM
K
Dieser Wert wird in Gleichung (2) eingesetzt:
vk
K
3
M
E S
S
Wenn [S] so groß ist, dass die gesamte eingesetzte Enzymmenge als Enzymsubstrat-
komplex vorliegt ([E] = [ES]), ist die maximale Geschwindigkeit erreicht:
vmax = k3 · [E]
Daher:
vv
K
max
M
S
S
Dies ist die von Michaelis und Menten entwickelte Gleichung. Bei halbmaximaler
Geschwindigkeit wird KM = [S]. KM hat somit die Dimension einer Konzentration.
Graphische Bestimmung von KM und vmax
Die Michaelis-Menten-Gleichung kann in verschiedener Weise zur Bestimmung von vmax
und KM graphisch dargestellt werden:
a) v wird gegen [S] aufgetragen und ergibt eine Hyperbel. Für v = vmax/2 wird [S] = KM
(Abb. 1).
7
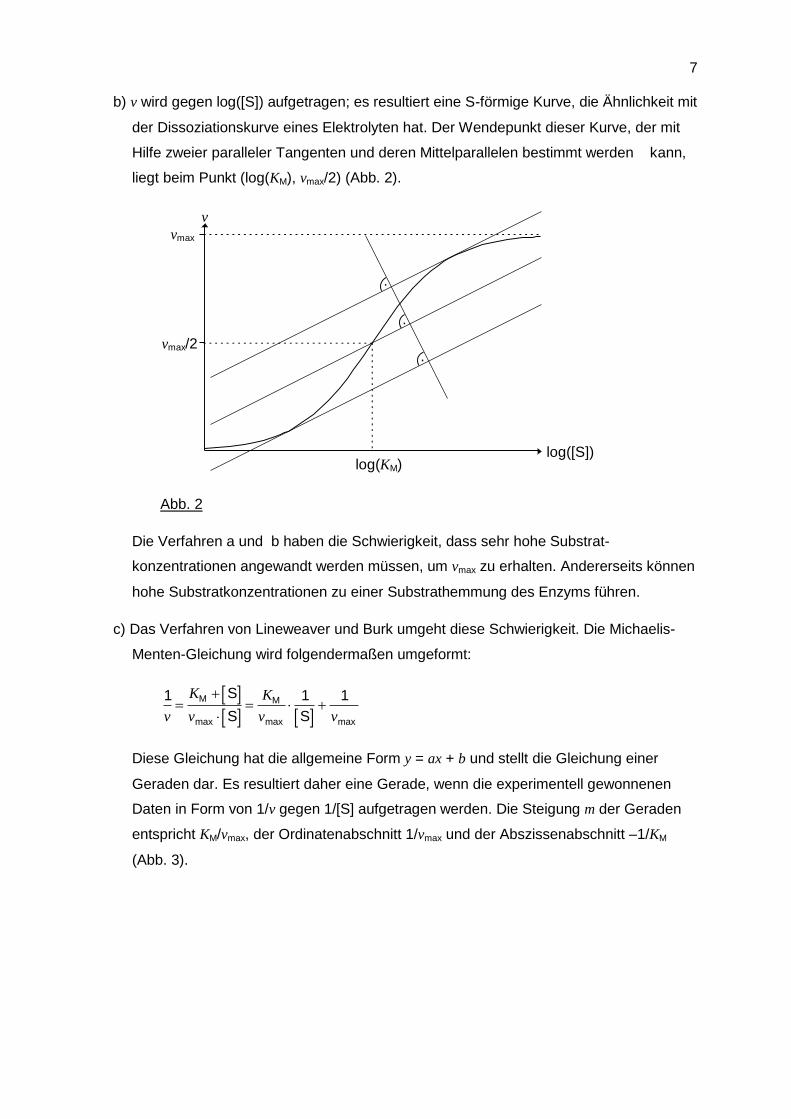
b) v wird gegen log([S]) aufgetragen; es resultiert eine S-förmige Kurve, die Ähnlichkeit mit
der Dissoziationskurve eines Elektrolyten hat. Der Wendepunkt dieser Kurve, der mit
Hilfe zweier paralleler Tangenten und deren Mittelparallelen bestimmt werden kann,
liegt beim Punkt (log(KM), vmax/2) (Abb. 2).
log([S])
.
.
.
log(KM)
v
vmax
vmax/2
Abb. 2
Die Verfahren a und b haben die Schwierigkeit, dass sehr hohe Substrat-
konzentrationen angewandt werden müssen, um vmax zu erhalten. Andererseits können
hohe Substratkonzentrationen zu einer Substrathemmung des Enzyms führen.
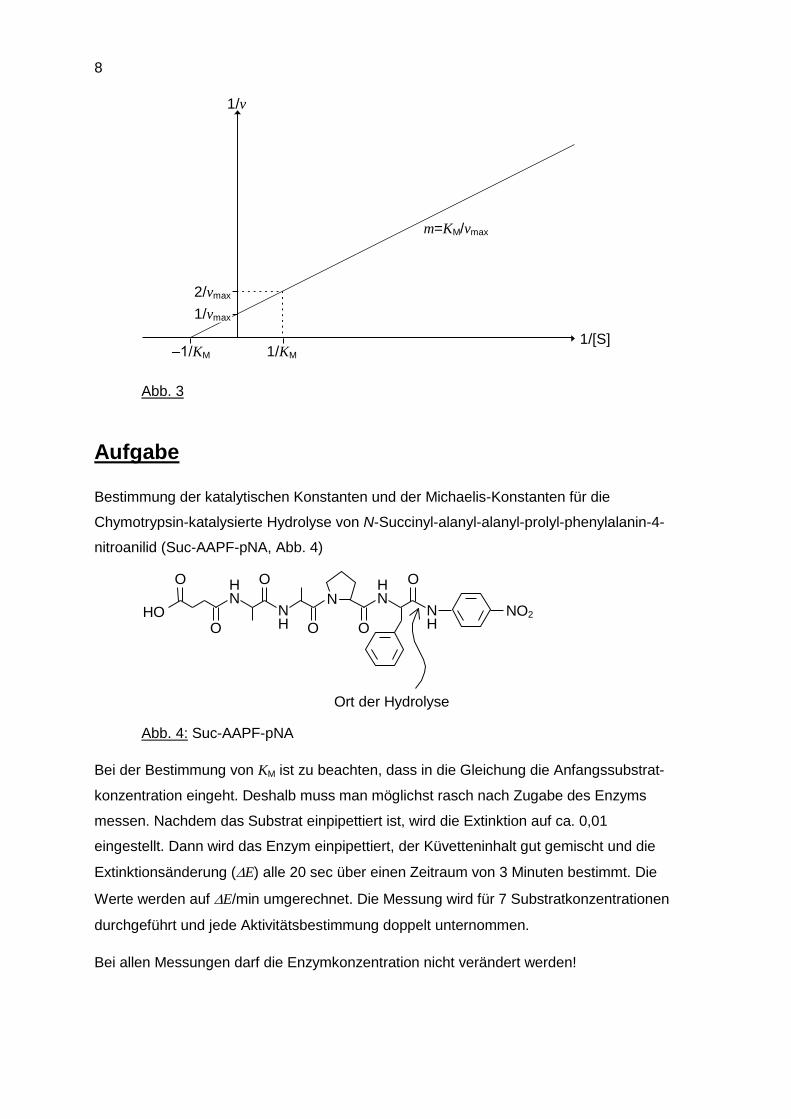
c) Das Verfahren von Lineweaver und Burk umgeht diese Schwierigkeit. Die Michaelis-
Menten-Gleichung wird folgendermaßen umgeformt:
1 1 1
v
K
v
K
v v
M
max
M
max max
S
S S
Diese Gleichung hat die allgemeine Form y = ax + b und stellt die Gleichung einer
Geraden dar. Es resultiert daher eine Gerade, wenn die experimentell gewonnenen
Daten in Form von 1/v gegen 1/[S] aufgetragen werden. Die Steigung m der Geraden
entspricht KM/vmax, der Ordinatenabschnitt 1/vmax und der Abszissenabschnitt –1/KM
(Abb. 3).
8
2/vmax
1/[S]–1/KM
m=KM/vmax
1/KM
1/v
1/vmax
Abb. 3
Aufgabe
Bestimmung der katalytischen Konstanten und der Michaelis-Konstanten für die
Chymotrypsin-katalysierte Hydrolyse von N-Succinyl-alanyl-alanyl-prolyl-phenylalanin-4-
nitroanilid (Suc-AAPF-pNA, Abb. 4)
O HN
HO
Ort der Hydrolyse
NO2
3O
O
NH O
NHN
O
O
NH
Abb. 4: Suc-AAPF-pNA
Bei der Bestimmung von KM ist zu beachten, dass in die Gleichung die Anfangssubstrat-
konzentration eingeht. Deshalb muss man möglichst rasch nach Zugabe des Enzyms
messen. Nachdem das Substrat einpipettiert ist, wird die Extinktion auf ca. 0,01
eingestellt. Dann wird das Enzym einpipettiert, der Küvetteninhalt gut gemischt und die
Extinktionsänderung (E) alle 20 sec über einen Zeitraum von 3 Minuten bestimmt. Die
Werte werden auf E/min umgerechnet. Die Messung wird für 7 Substratkonzentrationen
durchgeführt und jede Aktivitätsbestimmung doppelt unternommen.
Bei allen Messungen darf die Enzymkonzentration nicht verändert werden!

9
Herstellung der Lösungen
Molmassen: Tris: 121,14 g/mol; CaCl2: 147,02 g/mol; Suc-AAPF-pNA: 624,6 g/mol
Verdünnungspuffer (100 mM Tris, 10 mM CaCl2):
1,21 g Tris
147 mg CaCl2
pH 7,8 mit HCl
ad 100 ml Wasser
Substratstammlösung (1 mM):
3,1 mg Suc-AAPF-pNA
ad 5 ml Verdünnungspuffer
Enzymlösung:
2 mg Chymotrypsin ad 1 ml Verdünnungspuffer c = 2 mg/ml;
hiervon 250 l ad 1 ml Verdünnungspuffer c = 0,5 mg/ml;
hiervon 10 l ad 2 ml Verdünnungspuffer c = 0,0025 mg/ml
Pipettierschema:
Für die Messung ist es zweckmäßig, mit stärkster Verdünnung zu beginnen.
Testansatz
0,5 ml der verschiedenen, oben hergestellten Substratverdünnungen
0,1 ml Enzymlösung (0,0025 mg/ml)
Verdünnungspuffer [ml]
1,98 1,96 1,94 1,90 1,80 1,70 1,50
Substratstammlösung [ml]
0,02 0,04 0,06 0,10 0,20 0,30 0,50
Substratkonz. [mM] nach Verdünnen
0,01 0,02 0,03 0,05 0,10 0,15 0,25
Substratkonz. [mM] nach Enzymzugabe
0,0083 0,0167 0,0250 0,0417 0,0833 0,1250 0,2083

10
Auswertung
1) Bestimmen Sie die Anfangsreaktionsgeschwindigkeiten der jeweils vermessenen
Konzentrationen. Auftragung von E gegen die Zeit.
Ermitteln Sie graphisch die Anfangs-Steigung, indem Sie an den Anfang der
Messkurve eine Tangente anlegen.
(Die Messkurve ist eine Hyperbel, die entweder vom Computer an die Messdaten
gefittet oder von Hand näherungsweise eingezeichnet werden kann.)
2) Bestimmen Sie die spezifische Aktivität A für die vermessenen
Substratkonzentrationen nach:
3) Tragen Sie die spezifischen Aktivitäten gegen die Substratkonzentrationen auf
(Michaelis-Menten-Plot) und bestimmen Sie anhand des erhaltenen Graphen KM
und vmax.
4) Ermitteln Sie mit Hilfe der Auftragung nach Lineweaver-Burk ebenfalls KM und die
katalytische Konstante kcat.
Anhand der erhaltenen Geradengleichung lassen sich diese Konstanten
rechnerisch bestimmen.
5) Vergleichen Sie die von Ihnen ermittelten Werte von KM (aus graphischer und
rechnerischer Bestimmung)
6) Ergebnis und Fehlerdiskussion
Benötigte Angaben:
Das bei der Hydrolyse des Substrates freiwerdende 4-Nitroanilin wird in Abhängigkeit von
der Zeit bei 405 nm gemessen. Der molare Extinktionskoeffizient bei dieser Wellenlänge
beträgt 9 600 l mol–1 cm–1.
Die spezifische Aktivität A U/ mgmol Substrat
min mg Enzym
errechnet sich nach:
AE V
t d m
106

11
t = Zeit [min]
V = Volumen des Testansatzes in der Küvette [l]
d = Schichtdicke [cm]
m = Enzymmenge in Küvette [mg]
Bei den Hydrolasen, zu denen auch Trypsin und Chymotrypsin gehören, verwendet man
häufig anstelle von vmax die katalytische Konstante kcat:
kcat [1/s] = v max [U/mg] · MG [g/mol] / 60 000
MG = Molekulargewicht des Enzyms (hier: 23 000 g/mol)
60 000: Umrechnungsfaktor von mmol auf mol und von min auf s
In ein Diagramm sind einzutragen:
a) v gegen [S] und
b) v–1 gegen [S]–1,
wobei [S] = jeweilige Substratkonzentration in der Küvette. Anzugeben sind: KM [M],
vmax [U/mg] und kcat [s–1].

12
Disk-Elektrophorese in Natriumdodecylsulfat (SDS-PAGE)
Neben ihrer großen Bedeutung zur Reinheitskontrolle bei Proteinisolierungen ist die SDS-
Polyacrylamidgelelektrophorese (abgek.: SDS-PAGE) auch eine Methode zur
Molekulargewichtsabschätzung von Proteinen. Zur Trennung von Proteinen im Poly-
acrylamid-Gel wird hauptsächlich der Siebeffekt des Gels ausgenutzt. Um eine gute und
gleichsinnige Beweglichkeit der Proteinmoleküle im elektrischen Feld zu erreichen,
werden diese vor der Elektrophorese mit dem Detergenz Natriumdodecylsulfat (SDS)
beladen. Durch die Bindung von SDS werden Proteine zu Polyanionen, und das Protein
wird denaturiert. Da Proteine mit Disulfidbrücken in der SDS-PAGE häufig ein anomales
Verhalten zeigen, werden die Disulfidbrücken vor der Elektrophorese mit 2-Mercapto-
ethanol reduziert.
Die Vorinkubation der Proteine in SDS und 2-Mercaptoethanol schafft die Voraussetzung
für die Molekulargewichtsabschätzung im SDS-Gel, da jetzt die Beweglichkeit globulärer
Proteine bei gleicher Gelkonzentration (gleicher Porengröße) nur eine Funktion der Größe
des Moleküls ist.
Für die Molekulargewichtsbestimmung wird die relative Beweglichkeit von Proteinen mit
bekanntem Molekulargewicht in Bezug auf einen internen Standard (meist Bromphenol-
blau) verglichen und eine Eichkurve aufgestellt, indem der Logarithmus des Molekular-
gewichtes gegen den Rf-Wert (relative Beweglichkeit) aufgetragen wird. Aus dem Rf-Wert
des unbekannten Proteins kann somit dessen Molekulargewicht abgeschätzt werden.
Probleme: Die Beweglichkeit der Proteine im Polyacrylamid-Gel ist nicht allein von ihrem
Molekulargewicht abhängig, sondern zu einem erheblichen Teil auch von der jeweiligen
Form der denaturierten Eiweiße (globuläre Proteine wandern schneller als vollständig
entfaltete, lange Proteinfäden). Außerdem verändern bestimmte, natürlich vorkommende
Modifizierungen der Eiweiße, wie z. B. Glykosylierungen, deren Wanderungstendenz im
elektrischen Feld. Das SDS-Gel kann deshalb nur zur Abschätzung des Molekular-
gewichts dienen.
Das Trennmedium
Durch Kopolymerisation des monomeren Acrylsäureamids,

13
O
CCHCH2 NH2 ,
und eines quervernetzenden bifunktionellen Reagenzes, meist
N,N’-Methylenbisacrylsäureamid,
O
C CHCH CH2CH2 NH CH2 NH
O
C ,
wird ein dreidimensionales Netzwerk aufgebaut:
y. . .. . .
OC
CHCH2
NH2
OC
CHCH2
NH2
CH2
z
OC
CHCH2
NH2
w
. . .. . .
OC
CHCH2
NH2
OC
CHCH2
NH2 x
OC
CHCH2
NH2
Die Porengröße ist in einem recht weiten Bereich variabel, die Bisacrylamidkonzentration
kann in einem Bereich zwischen 2 % und 30 % der Acrylamid-Konzentration liegen. Bei
zu geringer Vernetzerkonzentration zerfließen die Gele, bei zu hoher werden sie brüchig.
Beispielsweise entspricht ein 7,5-%iges Gel einem Porendurchmesser von 50 Å und ein
30-%iges einem solchen von 20 Å. Zum Vergleich sei die räumliche Ausdehnung eines
Serumalbuminmoleküls angegeben: 40 Å × 150 Å. Die Polymerisation geschieht
radikalisch unter Zuhilfenahme von Radikalbildnern (hier: Ammoniumpersulfat).
Die Elektrophorese
Die Ausrüstung für eine Elektrophorese besteht aus zwei Teilen, einem Strom-
versorgungsteil (gewöhnlich ein Gleichspannungsgerät 0 - 550 V bzw. 0 - 200 mA) und
einer Elektrophoresekammer. Abb. 1 zeigt deren prinzipiellen Aufbau. Die Kammer
besteht aus einem oberen und unteren Pufferreservoir, die über die Gelplatte miteinander
verbunden sind. Da bei der SDS-Gelelektrophorese alle Proteine negativ geladen sind,
enthält das untere Reservoir die Anode, das obere die Kathode.

14
Kathode
Anode
unteres Pufferreservoir
Glasplatten
Trenngel
oberes Pufferreservoir
Sammelgel
Abb. 1: Prizipieller Aufbau einer Vertikal-Elektrophoresekammer
In der Praxis verwendet man diskontinuierliche Gele und Puffer. Man spricht deshalb auch
von einer diskontinuierlichen SDS-Polyacrylamidgelelektrophorese (abgekürzt SDS-Disk-
Elektrophorese). Das obere Drittel des Gels besteht aus einem weitporigen Sammelgel
(engl. stacking gel), die unteren zwei Drittel des Gels bestehen aus einem engerporigen
Trenn- oder Laufgel (engl. running gel). Das Sammelgel hat die Aufgabe, die Probe,
sobald sie durch das Gel läuft, zu konzentrieren, sodass sie, sobald sie in das Laufgel ein-
dringt, eine extrem schmale Bande bildet. Im Laufgel findet dann die Trennung des
Proteingemisches statt. Die Wanderungsgeschwindigkeit von Proteinen im Sammelgel
entspricht der in freier Lösung. Der Reservoirpuffer ist ein Tris(hydroxymethyl)-
aminomethan/Glycin-Puffer (Tris/Glycin) pH 8,3, während die Sammel- und Trenngelpuf-
fer aus Tris/HCl bestehen, pH 6,8 bzw. 8,8. Der pH des Sammelgels ist also um zwei pH-
Einheiten niedriger als der des oberen Reservoirs. Bei pH 8,3 liegen nur ca. 5 % des
Glycins als Glycinat vor, das bedeutet, dass die Glycinationen eine relativ geringe effek-
tive Beweglichkeit besitzen. Nach Auftragen der Proteinproben wird das elektrische Feld
angelegt, und die Proteine wandern mit den Glycinationen in das Sammelgel. Die
Chloridionen im Gel fangen natürlich ebenfalls an zu wandern, so dass während der
Elektrophorese zusammen mit den Proteinen zwei verschiedene Ionenarten in
Trennrichtung wandern. Sie werden als Leitionen (Cl–) und als Folgeionen (Glycinat)
bezeichnet.
Zu Beginn der Trennung befinden sich die Leitionen (Cl–) in Sammel- und Trenngel,
während die Folgeionen nur in der Pufferlösung der Elektrodengefäße vorhanden sind.
Durch die Wahl des pH-Wertes von 6,8 für das Sammelgel ordnen sich die effektiven
Beweglichkeiten u (u = Beweglichkeit, = Dissoziationsgrad) folgendermaßen an:

15
uLeitionen > uProtein > uFolgeionen,
wobei Leitionen, Proteine und Folgeionen dasselbe Ladungsvorzeichen tragen, also zur
gleichen Elektrode wandern.
Im Sammelgel bei pH 6,8 wandert Glycinat etwa 100 mal langsamer als bei pH 8,8 im
Trenngel. Unterwirft man das ganze Gelsystem einem elektrischen Stromfluss, so eilen
die Leitionen infolge höherer Beweglichkeit den Proteinen und Folgeionen voraus und
lassen eine Zone geringerer Leitfähigkeit hinter sich.
Nun ist die spezifische Leitfähigkeit umgekehrt proportional zur Feldstärke. Diese Zone
gewinnt daher eine höhere Feldstärke, welche die Proteine und Folgeionen derart
beschleunigt, dass sie hinter den Leitionen mit gleicher Geschwindigkeit wandern und
sich dadurch ansammeln.
Es bewegen sich alle Ionenarten mit gleicher Geschwindigkeit, wenn die Produkte aus
Feldstärke und Beweglichkeit einander gleichen:
v = E · u
(v = Geschwindigkeit, E = Feldstärke, u = Beweglichkeit)
Es stellt sich ein regulierendes Gleichgewicht ein, das die Gleichheit der Produkte
aufrechterhält. Zwischen den Leit- und Folgeionen bildet sich daher eine Grenzschicht
aus, die gleichzeitig die Front zwischen Gebieten niedriger und hoher Feldstärke darstellt.
Man kann sie als eine von oben nach unten wandernde Schlierenfront im praktischen
Versuch wahrnehmen. Diese Grenzschicht wandert nun rasch durch das Sammelgel. Da
die Beweglichkeiten der Proteine zwischen denen der Leit- und Folgeionen liegen, werden
die Proteine von der wandernden Grenzschicht erfasst und zu einer schmalen,
hochkonzentrierten Proteinzone zusammengestaucht.
Erreicht nun die wandernde Proteinzone, eingezwängt zwischen Leit- und Folgeionen, die
Grenze zwischen Sammel- und Trenngel, so trifft sie dort zwei Diskontinuitäten an,
nämlich einen pH-Wechsel und einen Porengrößenwechsel. Der „neue“ pH-Wert im
Trenngel ist so gewählt, dass der Dissoziationsgrad der Folgeionen - und damit deren
Beweglichkeit - um ein Vielfaches zunimmt. Dadurch gewinnen die Folgeionen eine
Beweglichkeit, die der der Leitionen fast gleicht. Die Folgeionen überholen nun alle
Proteine und wandern direkt hinter den Leitionen den Proteinen voraus, die nun in einem
homogenen elektrischen Feld entsprechend ihrer Molekülgröße aufgetrennt werden.

16
Versuchsdurchführung
SDS-PAGE nach Smith, Methods in Molecular Biology, Volume1 (Proteins), 41-56.
Lösungen
Für alle Lösungen und Puffer entgastes destilliertes Wasser verwenden!!
Bemerkung: „Wasser ad 250 ml“ bedeutet, dass die Substanzen in ungefähr 150 ml dest.
Wasser gelöst und anschließend auf 250 ml Gesamtvolumen aufgefüllt werden.
Vorsicht: Acrylamid ist für Haut und Lunge sehr giftig. Abwiegen nur mit Handschuhen
und unter dem Abzug! Nicht mit dem Mund pipettieren! Die mit Acrylamid in Berührung
gekommenen Geräte sofort nach Gebrauch mit Wasser spülen!
2) Trenngelpuffer: SDS 1 g
Tris 45,5 g
HCl bis pH 8,8
Wasser ad 250 ml
Bemerkung: „Wasser ad 250 ml“ bedeutet hier, dass die Substanzen in ungefähr 200 ml
Wasser gelöst werden, der pH eingestellt und anschließend auf 250 ml Wasser aufgefüllt
wird.
Auf 2 l auffüllen. Es stellt sich ein pH von ca. 8,3 ein (Kontrolle!).
1) Acrylamidlösung: Acrylamid 73 g
Bisacrylamid 2 g
Wasser ad 250 ml
3) Sammelgelpuffer: SDS 1 g
Tris 15,5 g
HCl bis pH 6,8
Wasser ad 250 ml
4) Elektrophoresepuffer: Glycin 28,8 g
Tris 6 g
SDS 2 g

17
Bemerkung: Bromphenolblau schädigt die Membran der pH-Elektrode und darf deshalb
erst nach Einstellung des pH-Werts dazugegeben werden.
5) Ammoniumpersulfatlösung: Ammoniumpersulfat 1 g
Auf 10 ml Wasser auffüllen.
6) Trenngel (10 %): Acrylamidlösung 5 ml
Wasser 6,25 ml
Trenngelpuffer 3,75 ml
Gut entgasen!
Ammoniumpersulfatlsg. 25 l
TEMED 10 l
7) Sammelgel (4,5 %): Acrylamidlösung 0,75 ml
Wasser 3 ml
Sammelgelpuffer 1,25 ml
Gut entgasen!
Ammoniumpersulfatlsg. 15 l
TEMED 5 l
8) Probenpuffer (doppelt stark): SDS 0,92 g
Glycerin 4 g
Tris 0,3 g
Mercaptoethanol 2 ml
mit Wasser auf 7 ml
HCl bis pH 6,8
0,1 % Bromphenolblau 2 ml
mit Wasser auf 7 ml

18
9) Probenbereitung:
Proteinlösung wird 1:1 mit Probenpuffer versetzt. Die Lösungen können mit Wasser
verdünnt werden. Proteinmenge einer Auftragung: 5-25 g Protein in 10 l
Gesamtvolumen.
Arbeitsgang:
1) Zusammensetzen der sauberen, mit vergälltem Ethanol abgeriebenen Platten zur
Gelkammer.
2) Nach gründlichem Mischen der Trenngellösung (6) wird die Gelkammer bis 1 cm
unterhalb des Randes mit einer Pasteurpipette gefüllt und vorsichtig mit wasser-
gesättigtem Butanol überschichtet. Dadurch erhält man eine glatte Phasengrenze
zwischen Trenn- und Sammelgel.
3) Nach Beendigung der Polymerisation wird das Butanol mit einer Pasteurpipette
vorsichtig herausgesaugt und mit Wasser nachgespült. Die Sammelgellösung wird auf
das Trenngel aufgetragen und der Kamm vorsichtig in die Lösung eingeschoben.
4) Ist das Sammelgel polymerisiert, zieht man behutsam den Kamm heraus.
5) Die Gele werden mit den Glasplatten in die Elektrophoreseapparatur eingespannt und
diese mit Elektrophoresepuffer aufgefüllt.
6) Zur Elektrophorese werden 20 l der denaturierten Proben mit einer Mikropipette durch
den Elektrophoresepuffer hindurch in die Probentaschen eingebracht, ohne das
Sammelgel zu verletzen. Das Gel sollte insgesamt asymmetrisch beladen werden, da
sonst später keine Auswertung möglich ist.
7) Man setzt den Deckel auf und stellt eine konstante Spannung von 200V ein.
8) Nach erfolgter Elektrophorese und Abschalten der Spannungsquelle wird der
Elektrophoresepuffer dekantiert, und die Gelkammern werden ausgespannt. Das Gel
wird vorsichtig von den Platten abgelöst (nicht mit den Fingern anfassen!) und sofort in
Wasser eingetaucht.

19
9) Gefärbt wird das Gel mit PageBlue, einer Proteinfärbelösung von Fermentas nach
beiliegender Vorschrift. Zunächst wird das Gel in 100ml destilliertes Wasser gelegt und
1min bei höchster Leistung in der Mikrowelle erhitzt. Anschließend verbleibt das Gel für
5min auf dem Schüttler. Das Wasser wird entfernt und der Vorgang noch zwei weitere
Male wiederholt. Dieser erste Waschschritt dient zur Entfernung des SDS, da dieses
die Färbung stört. Anschließend wird das Gel mit genügend Färbelösung bedeckt und
erneut 30sec bei höchster Leistung in der Mikrowelle erhitzt und darauf hin 20min auf
dem Schüttler gefärbt. Danach wird die Färbelösung entfernt und das Gel mit
destilliertem Wasser von Farbresten befreit. Das Gel verbleibt über Nacht in
destilliertem Wasser auf dem Schüttler. Die Gele können so mehrere Tage gelagert
werden.
Aufgabe:
Bestimmung des Molekulargewichts der unbekannten Proteinproben.

20
Enzymatische Bestimmung von ADP (Adenosindiphosphat)
Einleitung zu den photometrischen Bestimmungen
Wird eine Küvette, die mit einer absorbierenden Flüssigkeit oder Lösung gefüllt ist, von
Licht durchstrahlt, dann bewirkt die Absorption des Küvetteninhalts eine
Intensitätsabnahme des Lichtstrahls. Diese ist von der Wellenlänge des Lichts, der
Konzentration der absorbierenden Probe, der Länge des Lichtweges durch die
Messlösung sowie einer Stoffkonstanten (dem Extinktionskoeffizienten ) abhängig. Die
Wellenlängen, bei denen ein Maximum hat (die Absorptionsmaxima) sind für
absorbierende Atomgruppierungen (Chromophore) wie z. B. Nitrogruppen,
Doppelbindungen, Aromaten u. v. a. charakteristisch.
Die Konzentrationsbestimmung durch Messung der Absorption einer monochromatischen
Strahlung nach Durchgang durch die Messlösung bezeichnet man als Photometrie.
Grundlage für die photometrischen Konzentrationsbestimmungen ist das Lambert-
Beersche Gesetz, nach dem sich die Konzentration einer Lichtenergie absorbierenden
Verbindung in verdünnter Lösung berechnen lässt:
cE
d
c = Konzentration (mol/l)
= molarer Extinktionskoeffizient (l mol–1 cm–1)
d = Schichtdicke der Küvette (cm)
E = Extinktion
Die Ableitung des Lambert-Beerschen Gesetzes ist den Lehrbüchern zu entnehmen.
In der Regel ist die Skala eines lichtelektrischen Messgerätes linear und logarithmisch
unterteilt. Die lineare Teilung zeigt das Verhältnis der austretenden zur eingestrahlten
Lichtintensität an (Durchlässigkeit, Transmission T ), auf der logarithmischen Teilung wird
die Extinktion E (negativer Logarithmus der Durchlässigkeit) angezeigt.

21
Viele Substanzen, die farblos oder nur schwach gefärbt sind, können durch Zugabe eines
Reagenzes, mit dem sie unter Bildung stark gefärbter Produkte reagieren, bestimmt
werden. Dabei werden die Reaktionsbedingungen so standardisiert, dass die Menge der
zu bestimmenden Verbindung direkt proportional der gemessenen Extinktion ist.
c F E
c' = Gewichtsmenge der zu bestimmenden Verbindung im Standardtest
F = Proportionalitätsfaktor
Unter diesen Bedingungen wird eine Eichkurve mit bekannten Mengen der zu
bestimmenden Substanz aufgenommen und E gegen c' aufgetragen. Die Kurve sollte,
wenn von den gemessenen Extinktionswerten die Extinktion des Blindwertes (Extinktion
des Ansatzes ohne Analysensubstanz) subtrahiert wird, durch den Koordinatenursprung
gehen und eine Gerade ergeben, deren Steigung F entspricht.
Der ermittelte Proportionalitätsfaktor F dient zur Gehaltsbestimmung bei Lösungen
unbekannter Konzentration.
Aufgabe
In einer ADP-Lösung unbekannter Konzentration ist der Gehalt an ADP (Abb. 1) auf
enzymatischem Wege zu bestimmen.
O
O
ONa
P O
O
O
Na
Na
P O CH2
HO
O
OH
N
N
NH2
N
N
Abb. 1: ADP, Trinatriumsalz (MG 493,15)
Prinzip der enzymatischen Methode
Zur enzymatischen Bestimmung von ADP werden folgende Enzymreaktionen benutzt:
ADP + PEP PK
ATP + Pyruvat (1)

22
Pyruvat + NADH + H+ LDH
Lactat + NAD+ (2)
PEP = Phosphoenolpyruvat
PK = Pyruvatkinase
ATP = Adenosintriphosphat
NAD+ (NADH) = Nicotinamid-Adenin-Dinucleotid, oxidierte (bzw. reduzierte) Form
LDH = Lactatdehydrogenase
Durch Koppeln der Reaktion (1) mit der Indikatorreaktion (2), einem
Dehydrogenasesystem, kann ADP durch einen optischen Test gemessen werden. Da die
Gleichgewichte beider Reaktionen ganz auf der rechten Seite liegen, ist eine quantitative
Umsetzung von ADP in ATP und von Pyruvat in Lactat gewährleistet.
Reagenzien (M = mol/l)
0,1 M Tris-Puffer, mit HCl auf pH 7,5 eingestellt
PEP-Lösung in Wasser (2,5 mg / 0,4 ml)
NADH-Lösung in Wasser (4,5 mg / 0,6 ml)
0,5 M Magnesiumsulfat-Lösung
1 M KCl-Lösung
PK-Lösung in Puffer (ca. 0,1 mg / ml)
LDH-Lösung in Puffer (ca. 0,1 mg / ml)
Berechnung des Versuchsergebnisses
Aus den Reaktionsgleichungen (1) und (2) ist ersichtlich, dass pro mol umgesetztes ADP
1 mol NADH verbraucht wird. Die verbrauchte NADH-Menge kann aus der Extinktions-
änderung der Inkubationslösung berechnet werden:
340 = 6,3 · 103 l mol–1 cm–1
365 = 3,4 · 103 l mol–1 cm–1
Durchführung der Bestimmung
In eine Küvette (Schichtdicke 1 cm) werden nacheinander eingefüllt:
720 l Tris-Puffer

23
40 l MgSO4-Lösung
40 l 1 M KCl-Lösung
16 l PEP-Lösung
24 l NADH-Lösung
80 l der zu untersuchenden ADP-Lösung
40 l LDH-Lösung
Es wird gründlich gemischt und die Extinktion bei 365 nm oder 340 nm über 3 min
verfolgt. Wenn sie konstant bleibt, wird abgelesen, es werden 40 l der PK-Lösung
zugegeben, und es wird umgemischt. Ist die Extinktion erneut konstant, wird E365
bestimmt und daraus die ADP-Konzentration berechnet. Sollte die Menge zu groß oder
zu klein sein, um eine Extinktionsänderung von ca. 0,1 - 0,2 zu erreichen, dann sind die
Menge der ADP-Lösung und entsprechend die Puffermenge zu verändern; das Gesamt-
volumen des Ansatzes sollte dabei konstant bleiben. Es sollen mindestens drei Bestim-
mungen ausgeführt werden.
Tabelle zur Auswertung:
Ergebnis: ADP-Gehalt aufgrund der enzymatischen Bestimmung: ...................
ADP-Lsg [ml] E365 umgesetzte NADH-Menge
ADP-Menge im Ansatz
ADP-Konz. in der Lsg

24
Quantitative Proteinbestimmungsmethoden
Die quantitative Bestimmung von Proteinen gehört wohl zu den wichtigsten Methoden in
der allgemeinen Biochemie. In der Literatur ist eine Vielzahl verschiedener Bestimmungs-
verfahren beschrieben, die jedoch fast alle mit bestimmten Vor- und Nachteilen behaftet
sind.
Es soll hier dringend darauf hingewiesen werden, dass im Allgemeinen mit keinem der
Bestimmungsverfahren absolute Proteinwerte erhalten werden können. In den meisten
Fällen werden zur Bestimmung der unbekannten Proteinkonzentrationen andere, leicht
zugängliche Proteine (wie z. B. Rinderserumalbumin, BSA), als Standard verwendet. Die
über solche Methoden erhaltenen Proteinwerte sind also nur als relative Werte zu dem
jeweiligen Standardprotein zu verstehen.
Absolute Proteinbestimmung kann also nur dann durchgeführt werden, wenn zum einen
das zu untersuchende Protein als Standard verwendet werden kann (nach Lyophilisieren
und genauem Auswiegen kann eine Standardkurve ermittelt werden) oder zum anderen
bei bekanntem Extinktionskoeffizienten des untersuchten Proteins die UV-Absorption bei
einer definierten Wellenlänge bestimmt werden kann.
1. Proteinbestimmung mit BCA-Test
Einleitung
Der BCA-Test ist ein Proteinbestimmungs-Assay der Firma Uptima, der sich durch hohe
Empfindlichkeit und geringe Störanfälligkeit auszeichnet. Er ist kompatibel mit vielen
ionischen und nicht-ionischen Detergenzien. Der Test ist einfacher und schneller
durchzuführen als die Proteinbestimmungsmethode nach Lowry. Die Empfindlichkeit
erreicht 5 µg/ml im erweitertem Protokoll. Außerdem kann der Test in angepasster Form
in Microtiterplatten durchgeführt werden.
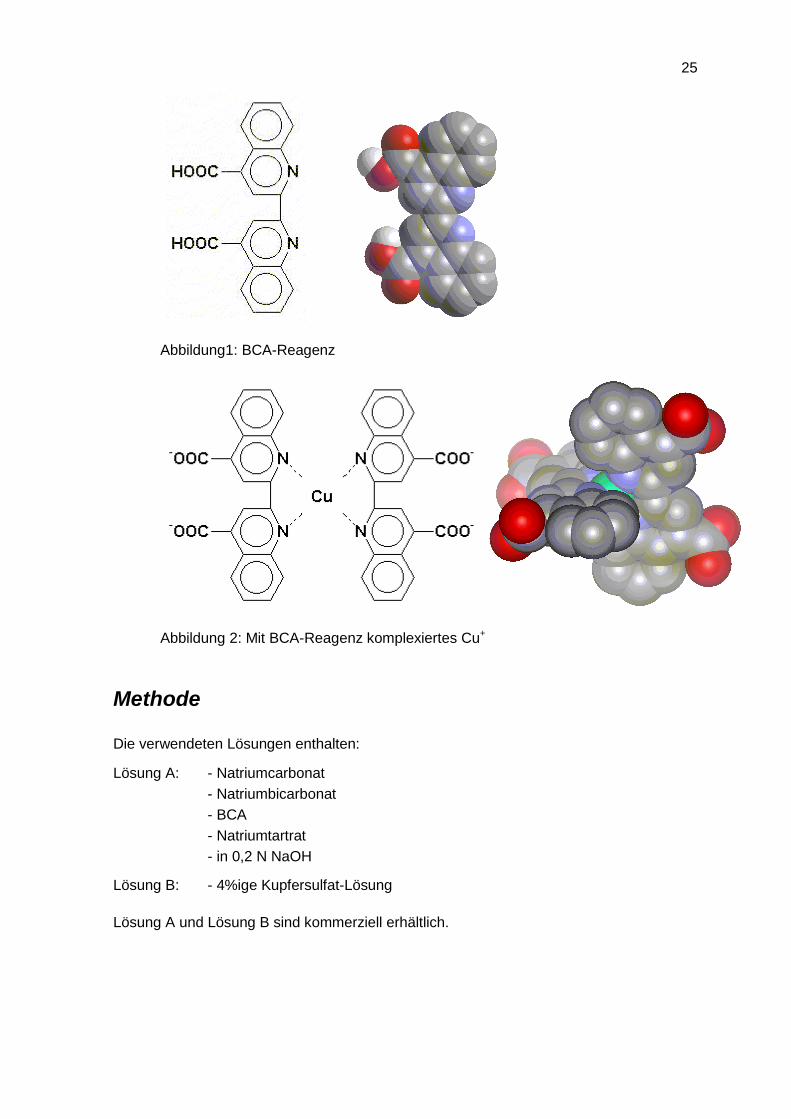
Bicinchoninsäure (2,2’-Bichinolin-4,4’-dicarbonsäure, bicinchoninic acid, BCA) ist ein Cu+-
Reagenz. Die Farbbildung wird auf die vier Aminosäuren Cystein, Cystin, Tryptophan und
Tyrosin zurückgeführt. In einer Biuret-Reaktion reduziert das Protein Cu2+ in alkalischem
Medium zu Cu+. Anschließend wird das Kupfer(I)-Ion von zwei Molekülen BCA
komplexiert.
25
Abbildung1: BCA-Reagenz
Abbildung 2: Mit BCA-Reagenz komplexiertes Cu+
Methode
Die verwendeten Lösungen enthalten:
Lösung A: - Natriumcarbonat
- Natriumbicarbonat
- BCA
- Natriumtartrat
- in 0,2 N NaOH
Lösung B: - 4%ige Kupfersulfat-Lösung
Lösung A und Lösung B sind kommerziell erhältlich.
26
Herstellung der Eichgeraden
Es wird eine BSA-Lösung der Konzentration c = 250 µg/ml unter Verwendung einer
Stammlösung (SL) der Konzentration c = 2 mg/ml hergestellt.
Eine Verdünnung von 100 µl SL (2 mg/ml) 200 µg BSA mit 700 µl Wasser ergibt eine
BSA-Lösung von c = 250 µg/ml. Diese Lösung wird zum Erstellen der Eichgerade
verwendet.
Herstellung des BCA-Reagenzes
BCA-Lösung A und BCA-Lösung B werden im Verhältnis 50:1 gemischt. Zum Beispiel
29,4 ml Lösung A und 0,6 ml Lösung B.
Das so angesetzte Reagenz kann nur einen Tag lang verwendet werden; immer so viel
Reagenz ansetzen, dass es auch für die zu bestimmenden Proben ausreicht.
Pipettierschema für die Eichgerade
Die Eichgerade sowie die Proben werden in Eppendorf-Tubes doppelt angesetzt.
Eppi-Nr. BSA-Lösung [µl] H2O [µl] BCA-Reagenz [µl] µg BSA/Probe
1 (Blank) 0 100 900 0
2 10 90 900 2,5
3 20 80 900 5
4 30 70 900 7,5
5 40 60 900 10
6 50 50 900 12,5
7 60 40 900 15
8 70 30 900 17,5
Am günstigsten legt man das Wasser vor und pipettiert anschliessend die BSA-Lösung
zu.

27
Pipettierschema für die zu bestimmende Proteinprobe
Eppi-Nr. Protein-Lsg. [µl] H2O [µl] BCA-Reagenz [µl]
1 (Blank) 0 100 900
2 20 80 900
3 40 60 900
4 60 40 900
Vermessung
Erst wenn alle Proben (d.h. Eichgerade und Proteinproben) bereit sind wird das BCA-
Reagenz zugegeben. Sofort danach erfolgt eine 30-minütige Inkubation im 60 °C warmen
Wasserbad. Danach werden die Proben auf Eis abgekühlt und sofort im UV/Vis-
Spektrometer bei einer Wellenlänge von 562 nm vermessen.
2. Konzentrationsbestimmung nach Bradford
Theorie
Die Bestimmung der Proteinkonzentration nach Bradford ist, ähnlich wie die Methode
nach Lowry, ein Verfahren, das auf einer Farbreaktion basiert. Die Nachweismethode
findet im stark sauren Medium statt, das Coomassie Brilliant Blue G-250, einen
Triarylmethanfarbstoff enthält. Durch Bindung des Farbstoffes an das Protein erfolgt ein
spezifischer Farbwechsel von einem Aborptionsmaximum von 465 nm zu 595 nm. Die
Intensität des Farbkomplexes ist direkt vom Proteingehalt abhängig.
Der Farbkomplex ist nach zwei Minuten gut sichtbar und für ca. eine Stunde stabil.
Diese Proteinbestimmungsmethode ist schnell durchführbar, gut zu reproduzieren und
sehr sensitiv. Sie wird durch Pufferchemikalien und reduzierende Stoffe kaum gestört,
versagt allerdings, wenn in der Proteinlösung Substanzen enthalten sind, die in stark
phosphorsauren Lösungen grobflockige Niederschläge bilden (z. B. Detergenzien wie
Desoxycholat o. ä.). Störungen des Testes werden auch durch Natriumdodecylsulfat
(SDS) und Triton X-100 verursacht.
Der Nachteil der Methode liegt, wie bei allen farb-chemischen Nachweismethoden darin,
dass die Proteinprobe wegen Denaturierung nicht mehr wiederverwendet werden kann.

28
Aufgabe
Ermittlung der Konzentration einer unbekannten Proteinlösung.
Reagenzien
Bradford-Reagenz
0,1 g Coomassie Brilliant Blue G-250 werden in 50 ml 50-%igem Ethanol (v/v) gelöst.
Dann werden 100 ml 85-%ige Phosphorsäure zugegeben und mit dest. Wasser auf
250 ml aufgefüllt.
Vor Gebrauch wird ein Volumen des Reagenz mit 4 Volumen dest. Wasser gemischt und
filtriert.
Modifizierung nach M. Holtzhauer (1988)
Albuminlösung als Standard für die Eichgerade
10 mg Rinderserumalbumin (BSA) werden in einem 10-ml-Messkolben eingewogen und
mit dest. Wasser bis zur Marke aufgefüllt. Die Einwaage muss genau bekannt sein. Lösen
und Mischen durch vorsichtiges Drehen des Kolbens - nicht schütteln!
Durchführung
Erstellen der Eichgerade
Nach folgendem Schema wird der Albuminstandard (1 mg/ml) in einzelne Reagenzgläser
pipettiert.
2 × 0 l (Blank), 2 × 5 l, 2 × 7 l, 2 × 10 l, 2 × 12 l, 2 × 15 l (Dupletts)
Daraufhin werden in jedes Reagenzglas 1 ml Bradford-Reagenz hinzugefügt und auf
einem Whirlmix durchmischt.
Nach ca. 2 Minuten ist der Farbkomplex stabil. Dann erfolgt die Extinktionsmessung in
Kunststoffküvetten bei 595 nm. (Eppendorf-Photometer: 578 nm oder 623 nm)
Konzentrationsbestimmung der unbekannten Proteinlösung
2 × 3 l, 2 × 5 l, 2 × 7 l der Proteinlösung in einzelne Reagenzgläser pipettieren.
Proben mit jeweils 1 ml Bradford-Reagenz versetzen und analog zur Vorschrift der
Eichgeraden vermessen.

29
Falls die bestimmte Extinktion außerhalb des Bereiches der Eichgeraden liegt, muss die
Stammproteinlösung verdünnt eingesetzt werden.
Achtung: Bei dieser Proteinbestimmungsmethode sind die Eichgeraden nur bis zu einem
maximalen Proteingehalt von 15 g pro Probe proportional zur Proteinmenge, danach
knickt die Eichgerade ab.
Auswertung
Ermittelte Extinktionswerte für die verschiedenen BSA-Gehalte (g) in einem Diagramm
auftragen. Mittels linearer Regression kann daraufhin aus den gemessenen
Extinktionswerten der unbekannten Proteinlösung deren Konzentration bestimmt werden.
Literatur:
M. M. Bradford, (1976) Anal. Biochem. 72, 248-254
Holtzhauer M. (1988) Biochemische Labormethoden, Arbeitsvorschriften und Tabellen,
Springer-Verlag, Berlin, Heidelberg, New York, 5-6

30
Enzyme-Linked Immunosorbent Assay (ELISA)
1. Einleitung ................................................................................................................. 30
2. Versuchsbeschreibung ............................................................................................ 35
3. Versuchsdurchführung ............................................................................................. 36
3.1 Reagenzien und Chemikalien ............................................................................. 36
3.2 Versuchsablauf ................................................................................................... 37
3.3 Bestückung der Mikrotiterplatte .......................................................................... 38
4. Auswertung .............................................................................................................. 39
5. Literatur ................................................................................................................... 39
1. Einleitung
Die intakte Oberfläche des Körpers stellt eine wirksame Barriere gegenüber den meisten
Mikroorganismen (Viren, Bakterien, Pilzen, Parasiten) dar. Um dennoch eingedrungene
Mikroben unschädlich zu machen, verfügen Wirbeltiere über ein Abwehrsystem aus
Molekülen und Zellen, das Immunsystem.
Alle Zellen des Immunsystems stammen von pluripotenten Stammzellen ab, die sich im
Laufe ihrer Entwicklung in zwei Zellinien, die myeloische und die lymphatische,
differenzieren. Die myeloische Reihe besteht zum gößten Teil aus Phagozyten, welche in
der Lage sind, Fremdorganismen zu endocytieren und zu verdauen. Die Zellen der
lymphatischen Reihe (Lymphocyten) differenzieren, je nach Umgebung (micro-
environment) in der sie heranreifen, in T- und B-Zellen. T-Zellen entwickeln sich dabei im
Thymus, während B-Zellen bei Säugetieren im Knochenmark (bone marrow) entstehen.

31
Neben der zellvermittelten Immunreaktion spielen auch lösliche Faktoren bei der
Immunantwort eine große Rolle (humorale Immunantwort). Den Hauptträger dieser
Abwehr bilden die Immunglobuline oder Antikörper. Diese werden - nach einem Kontakt
des lymphatischen Systems mit fremden immunogenen Molekülen - von Plasmazellen,
die sich aus B-Zellen entwickeln, gebildet. Moleküle, die im Organismus die Bildung
dieser Antikörper induzieren, nennt man Antigene.
Die Antikörper erkennen das infektiöse Agens spezifisch, das heißt, ein bestimmter
Antikörper erkennt nur sein zugehöriges Antigen. Die Bindung erfolgt dabei nicht an das
gesamte Antigen, sondern nur an eine bestimmte Stelle, die Antigendeterminante
(Epitop).
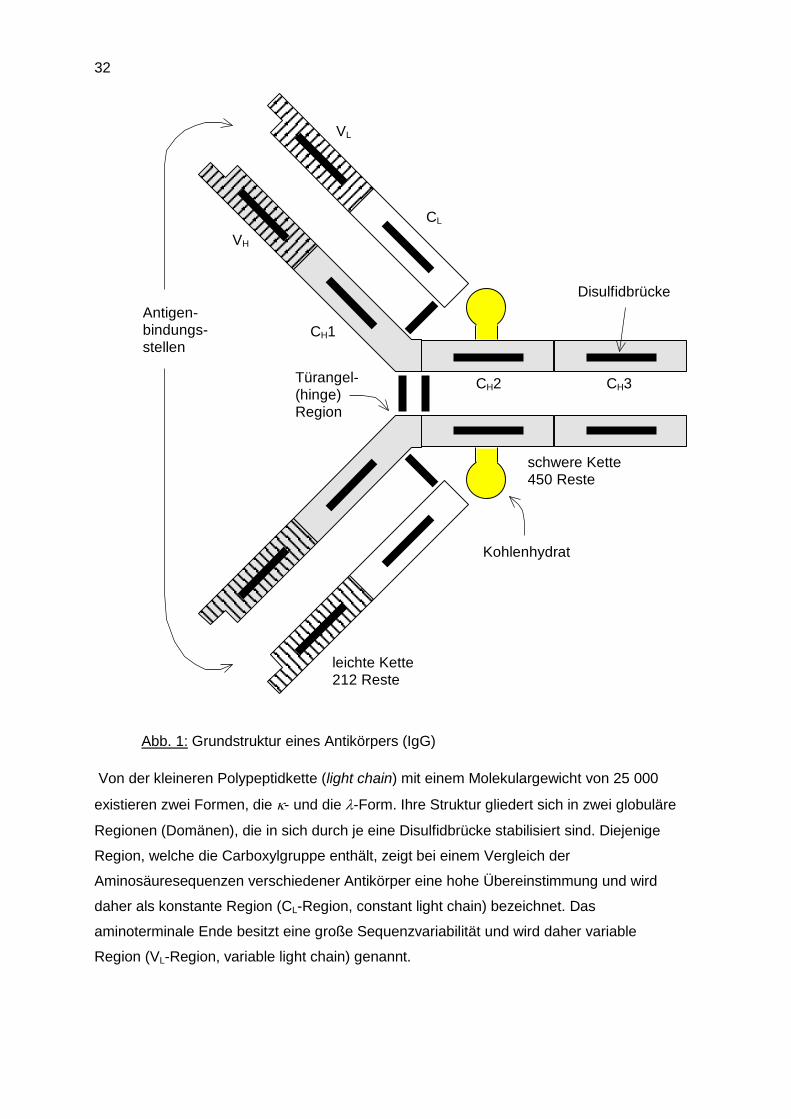
Die Grundstruktur aller Antikörper besteht aus je zwei identischen leichten und schweren
Polypeptidketten, die über Disulfidbrücken miteinander verbunden sind (Abb. 1).
32
Kohlenhydrat
Disulfidbrücke
schwere Kette450 Reste
leichte Kette
212 Reste
CH3CH2
CH1
VH
CL
VL
Türangel-(hinge)
Region
Antigen-
bindungs-stellen
Abb. 1: Grundstruktur eines Antikörpers (IgG)
Von der kleineren Polypeptidkette (light chain) mit einem Molekulargewicht von 25 000
existieren zwei Formen, die - und die -Form. Ihre Struktur gliedert sich in zwei globuläre
Regionen (Domänen), die in sich durch je eine Disulfidbrücke stabilisiert sind. Diejenige
Region, welche die Carboxylgruppe enthält, zeigt bei einem Vergleich der
Aminosäuresequenzen verschiedener Antikörper eine hohe Übereinstimmung und wird
daher als konstante Region (CL-Region, constant light chain) bezeichnet. Das
aminoterminale Ende besitzt eine große Sequenzvariabilität und wird daher variable
Region (VL-Region, variable light chain) genannt.

33
Die schwere Polypeptidkette (heavy chain) existiert in fünf Formen () mit einem
Molgewicht von 50.000-77.000. Jede dieser Formen ist mit einem Leichtkettentyp frei
kombinierbar, wodurch die fünf Immunglobulinklassen (IgA, IgD, IgE, IgG bzw. IgM)
entstehen. Variationen in der Schwerkettenstruktur innerhalb einer Klasse (z. B. 1, 2, 3
u. 4) führt zur Bildung von Subklassen (entsprechend: IgG1, IgG2, IgG3, bzw. IgG4).
Die Struktur der schweren Ketten ist denen der Leichtkette sehr ähnlich. Bedingt durch die
größere Molmasse besitzen sie jedoch neben der variablen Region (VH-Region, variable
heavy chain) drei konstante Domänen (CH1, CH2, CH3). Der Kohlenhydratanteil, den alle
Antikörper besitzen, ist an die CH2-Region gebunden.
Die pflanzliche Proteinase Papain spaltet Immunglobuline in der Region zwischen den
CH1- und CH2-Domänen, der sogenannten Türangelregion (hinge region), wodurch zwei
identische Fab-Fragmente und ein Fc-Fragment entstehen (Abb. 2). Ein weiteres Enzym
für die Fragmentierung von Antikörpern ist Pepsin, welches zwei größere Fragmente
erzeugt, das F(ab')2-Fragment, das die über die Hinge-Region miteinander verbundenen
Fab-Regionen umfasst, und das pFc'- Fragment, welches den CH3-Domänen des
Antikörpermoleküls entspricht.
Mit diesen Fragmenten konnte gezeigt werden, dass die Antigen-Antikörper-Bindungsorte
in den variablen Bereichen (VL, VH) des Antikörpers liegen, während der Fc-Teil die
Bindung des Immunglobulins an verschiedene Zellen des Immunsystems, sowie
Komplementfaktoren vermittelt. Die hohe Beweglichkeit der Hinge-Region erlaubt eine
Änderung des Abstandes der Antigenbindungsorte, wodurch diese unabhängig
voneinander verfügbar sind.
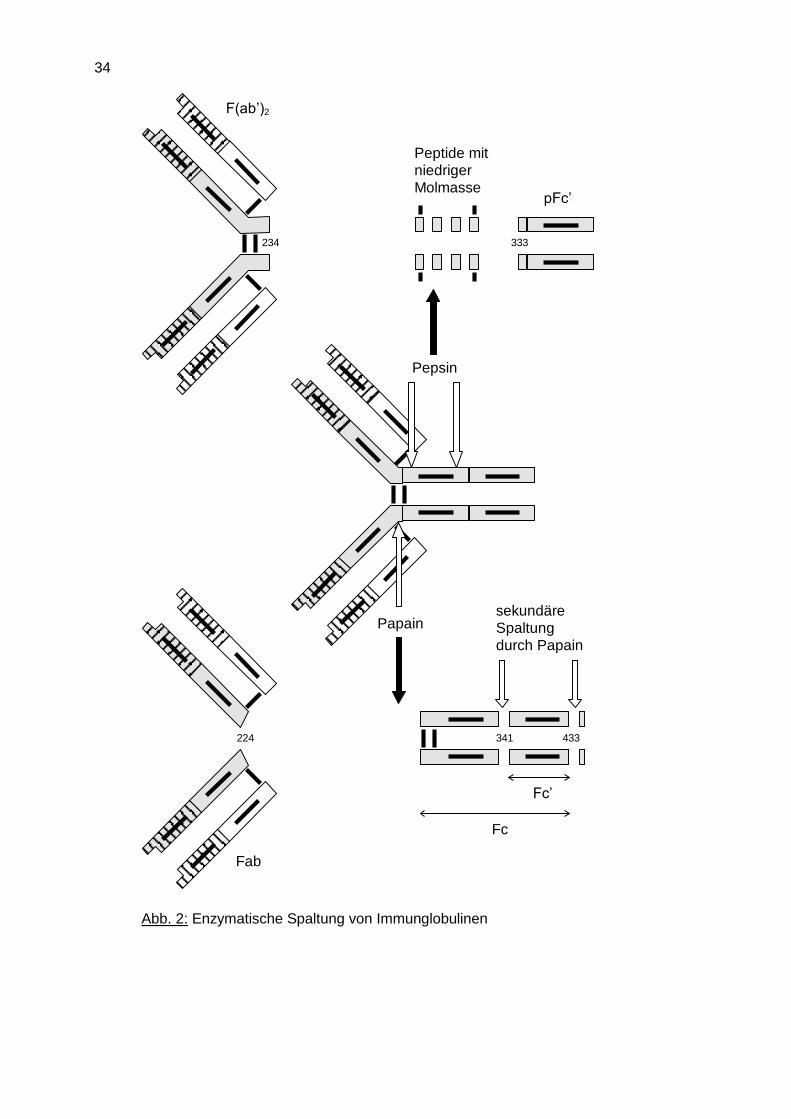
34
F(ab’)2
Peptide mitniedriger
Molmasse
sekundäre
Spaltung
durch Papain
Papain
Pepsin
pFc’
333234
Fab
Fc’
Fc
433341224
Abb. 2: Enzymatische Spaltung von Immunglobulinen
35
2. Versuchsbeschreibung
Zur Bestimmung von Antikörpertitern gegen bestimmte Antigene existiert mittlerweile eine
Vielzahl von Methoden, wobei an dieser Stelle nur der RIA (radioimmunoassay) und der
ELISA (enzyme-linked immunosorbent assay) genannt seien. Beide Assays sind sehr
einfach in der Durchführung, doch sehr sensitiv und zuverlässig, was zu deren weit
verbreiteter Anwendung geführt hat. Die Grundlage beider Methoden beruht auf der
Tatsache, dass bestimmte Kunststoffoberflächen (z. B. Polystyrol, Polypropylen,
Polycarbonat, Polyvinylchlorid) geringe Mengen der meisten Proteine fest binden können.
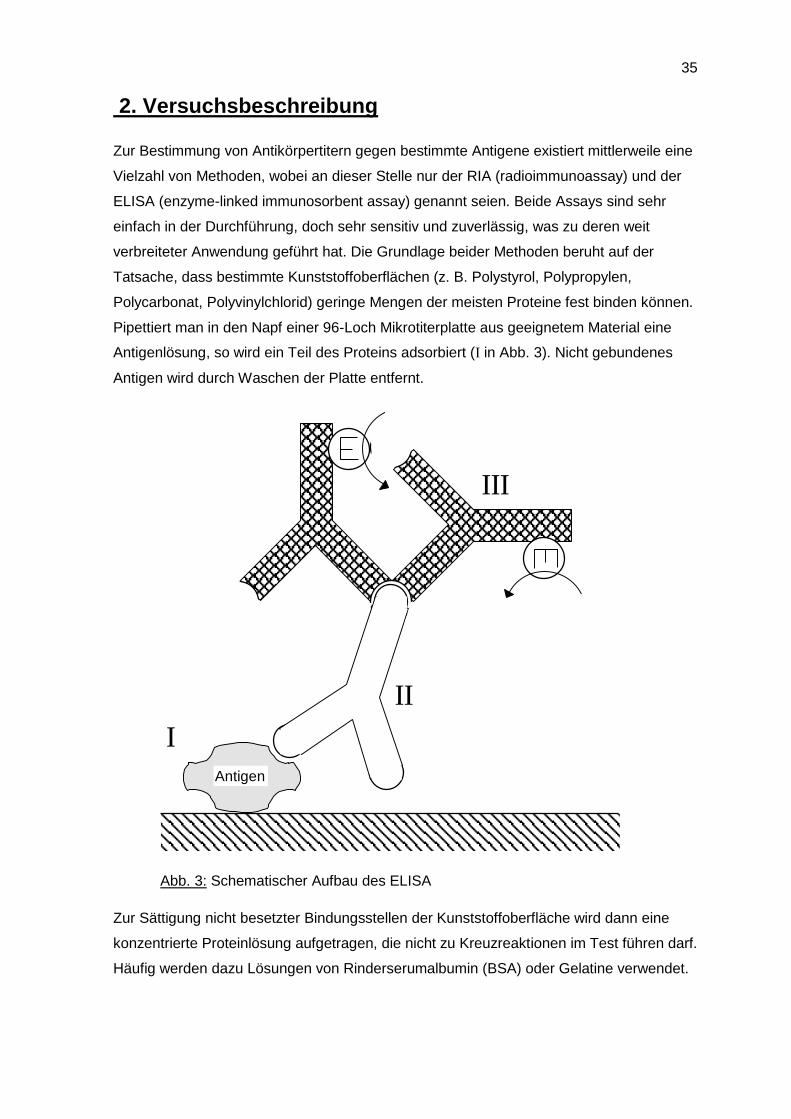
Pipettiert man in den Napf einer 96-Loch Mikrotiterplatte aus geeignetem Material eine
Antigenlösung, so wird ein Teil des Proteins adsorbiert (I in Abb. 3). Nicht gebundenes
Antigen wird durch Waschen der Platte entfernt.
I
II
III
Antigen
Abb. 3: Schematischer Aufbau des ELISA
Zur Sättigung nicht besetzter Bindungsstellen der Kunststoffoberfläche wird dann eine
konzentrierte Proteinlösung aufgetragen, die nicht zu Kreuzreaktionen im Test führen darf.
Häufig werden dazu Lösungen von Rinderserumalbumin (BSA) oder Gelatine verwendet.

36
In die so vorbehandelte Mikrotiterplatte wird dann die zu testende Lösung einpipettiert.
Falls in der Probe antigenspezifischer Antikörper vorhanden ist, bindet dieser an das an
die Festphase fixierte Antigen (II in Abb. 3). Alles nicht gebundene Material wird in einem
anschließenden Waschvorgang entfernt. Um den gebundenen Antikörper detektieren zu
können, wird die Platte dann mit einer Lösung von markiertem Anti-Antikörper behandelt
(III in Abb. 3).
Im Praktikumsversuch soll ein His-Tag markiertes Protein (Gelonin) aus
Expressionsversuchen immunologisch nachgewiesen werden. Es wird deshalb eine
monoclonale Anti-His-Tag Antikörperlösung verwendet. Die Markierung dieses zweiten
Antikörpers kann auf unterschiedliche Art und Weise erfolgen. Beim RIA ist der
Sekundärantikörper radioaktiv markiert, beim ELISA ist er mit einem Enzym (E in Abb. 3)
gekoppelt.
Nach einem letzten Waschvorgang wird das gebundene Enzym-Anti-Maus-Ig-Konjugat
durch eine Farbreaktion nachgewiesen, bzw. beim RIA an die Festphase gebundene
Radioaktivität gemessen. Je nach Anwendung können verschiedene Enzyme zur
Markierung des Anti- Antikörpers verwendet werden. Häufig werden Peroxidase(POD)-
und alkalische Phosphatase(AP)-Konjugate benutzt.
3. Versuchsdurchführung
3.1 Reagenzien und Chemikalien
Testprotein (Antigen) Rekombinantes Gelonin mit His-Tag (20 µg/ml)
Gelonin aus Samen von Gelonium multiflorum (20 µg/ml)
Kontrollen
Negativkontrollen: 20 µg/ml Cytochrom C
Positivkontrolle: 20 µg/ml rekombinantes Gelonin
Primärantikörper Monoklonale, antigenspezifischer Antikörper:
Mouse Anti-His Antikörper
Verdünnung: 1:2000 mit PBS-Tween
Enzymkonjugat Alkalische Phosphatase markierte, polyklonale Antikörper gegen Maus-Immunglobuline:
Anti-mouse lgG Antikörper (AP)
Verdünnung: 1:1000 mit PBS-Tween

37
Coating-Puffer Lösung A: 100 ml 0,2 M Na2CO3 (2,12 g ad 100 ml)
Lösung B: 100 ml 0,2 M NaHCO3 (1,68 g ad 100 ml)
Gebrauchslösung : 8,5 ml Lösung A + 4 ml Lösung B
pH 10,6 (pH-Kontrolle)
ad 50 ml mit bidest. Wasser
Waschpuffer PBS-Tween Na2HPO4 0,92 g
NaH2PO4 * H2O 0,35 g
NaCl 8,18 g
pH 7,2
ad 1 l mit deionisiertem Wasser
100 ml zur Seite stellen (= PBS-Puffer)
zu den restlichen 900 ml:
Tween 20 0,45 ml
Blockierlösung 1% BSA in PBS
250 mg BSA in 25 ml PBS
Substratpuffer (AP) NaN3 0,2 g (0,02 %)
MgCl2 * 6 H2O 0,1 g
DEA (Diethanolamin) 97 ml
pH 9,8
ad 1 l bidest. Wasser
Substratlösung (AP) 15 mg p-Nitrophenylphosphat ad 15 ml Substratpuffer
3.2 Versuchsablauf
a) Das Antigen wird im Coating-Puffer auf eine Konzentration von 20 g/ml eingestellt. Je
Napf werden 100 µl dieser Lösung eingebracht. Die Bindung an die feste Phase erfolgt
über Nacht bei 4 °C.
b) Die Näpfe der Platte werden durch Ausschlagen geleert. Pro Napf werden 200 µl der
Blockierlösung eingefüllt. Die Inkubation erfolgt bei Raumtemperatur für 90 min.
c) 2-maliges Waschen mit jeweils 200 µl PBS-Tween-Puffer. Ausschlagen der Näpfe.
d) Je Napf werden 100 l Primärantikörperlösung eingebracht. Die Inkubation erfolgt bei
Raumtemperatur für 1 h (siehe „Aufteilung der Mikrotiterplatte“, Abschn. 3.3)
e) 3 × Waschen. Näpfe ausschlagen.

38
f) Das Enzymkonjugat wird auf eine geeignete Arbeitskonzentration gebracht, d. h. ca.
1000 × verdünnt (10 l Enzymkonjugatlsg. + 10 ml PBS-Tween) und 100 l pro Napf
eingebracht. Die Inkubation erfolgt bei Raumtemperatur für 1 h.
g) 4 × Waschen. Näpfe ausschlagen.
h) 100 l Substratlösung zugeben. Die Entwicklung erfolgt bei Raumtemperatur. Die
Platte wird nach 10 min mit Hilfe eines EIA-Readers bei 414 nm vermessen.
3.3 Bestückung der Mikrotiterplatte
a) Blank:
Für die Messung benötigt der EIA-Reader eine Spalte der Platte als Blank. In die Spalte 1
dürfen deshalb keine Proben eingebracht werden. Stattdessen wird PBS einpipettiert. Alle
anderen Schritte wie Blockieren, Konjugatzugabe usw. erfolgen wie beschrieben.
b) Negativkontrollen:
Um falsche positive Ergebnisse auszuschließen, müssen in jedem ELISA
Negativkontrollen durchgeführt werden. Zu diesem Zweck werden 2 Spalten der Platte
nicht mit Antigen beschichtet. Das weitere Vorgehen erfolgt wieder wie beschrieben. Als
weitere Negativkontrolle wird in einer antigenbeschichteten Spalte ein Antikörper, der
nicht mit dem Antigen reagiert, als Probe eingebracht.
Die Ursachen für falschpositive Ergebnisse können sein:
Unspezifische Adsorption des Enzymkonjugats an das Plastikmaterial,
Kreuzreaktionen des Enzymkonjugats,
c) Positivkontrollen:
Um sicherzustellen, dass der ELISA richtig durchgeführt wurde, sollte immer eine Spalte
der Platte mit einer Probe bestückt werden, von der bekannt ist, dass sie für das
verwendete Antigen spezifische Antikörper enthält. Diese Löcher müssen sich beim
Entwickeln auf jeden Fall färben.

39
4. Auswertung
Die Auswertung erfolgt zunächst optisch, indem man die Löcher notiert, die deutlich
stärker gefärbt sind als die Negativkontrollen. Die Messwerte des EIA-Readers werden
statistisch ausgewertet.
Dazu werden die Negativkontrollen gemittelt und die Standardabweichung berechnet. Für
jede Probe wird ebenfalls der Mittelwert aus den Messwerten gebildet. Eine Probe ist
dann positiv, wenn dieser Mittelwert größer ist als die Summe aus dem Mittelwert der
Negativkontrollen und der zweifachen Standardabweichung.
5. Literatur
Harlow, E., Lane, D.,
Antibodies. A Laboratory Manual,
Cold Spring Harbor Laboratory, New York, 1988.
Kap. 14, Immunoassays, S. 553-612.
Goding, J. W.,
Monoclonal Antibodies: Principles and Practice, 2nd ed.,
Academic Press, London, 1986.
Kap. 3.10, Screening Assays, S. 76-89.
Peters, J. H., Baumgarten, H., (Hrsg.),
Monoklonale Antikörper. Herstellung und Charakterisierung, 2. Auflage,
Springer-Verlag, Berlin, Heidelberg, 1990.
Kap. 10, Nachweis von monoklonalen Antikörpern, S. 317-458.

40
Gezielte Mutagenese
Gezielte Mutagenese ...................................................................................................... 40
1. Grundlagen .............................................................................................................. 41
1.1. Der Ort der Mutation und mutagene Oligonukleotide ......................................... 41
1.2. LacZ / -Galactosidase ...................................................................................... 41
2. DNA Isolierung nach der „QiaPrep“ Methode ........................................................... 45
2.1. Das QIAprep Prinzip .......................................................................................... 45
2.2. Alkalische Lyse von Bakterien ........................................................................... 45
2.3. DNA Adsorption an die QIAprep-Membran ........................................................ 46
2.4. Waschen und Elution der Plasmid DNA ............................................................. 46
2.5. DNA Ausbeute ................................................................................................... 46
3. Miniprep-Protokoll für den „QIAprep spin kit“ unter Verwendung einer Tischzentrifuge47
4. Ortsgerichtete Mutagenese ...................................................................................... 50
4.1. Einführung ......................................................................................................... 50
4.2. DNA-Polymerase ............................................................................................... 50
4.3. Verdau mittels DpnI Restriktionsendonuklease .................................................. 51
4.4. Primer-Design ................................................................................................... 51
4.5. Überblick ........................................................................................................... 52
4.6. Durchführung der PCR-Reaktion ....................................................................... 53
4.7. Durchführung der Restriktion ............................................................................. 53
5. Agarose-Gelelektrophorese ..................................................................................... 54
5.1. Durchführung der Gelelektrophorese: ................................................................ 55
6. Transformation ......................................................................................................... 56
7. Vorbereiten der Agar-Platten für Blue-White-Screening ........................................... 57

41
1. Grundlagen
Bei der ortsspezifischen oder gezielten Mutagenese (im engl. site-directed mutagenesis)
wird mit Hilfe der rekombinanten Gentechnik eine gezielte Veränderung der DNA
ermöglicht. Es können damit gezielt einzelne Nukleinbasen eines Gens ausgetauscht
oder auch ganze Gene entfernt werden. Dieses Verfahren ist eine inzwischen weit
verbreitete Methode in der Molekularbiologie, welches von der Veränderung eines Gens
auf einem Plasmid bis hin zur Knockout-Maus reicht
1.1. Der Ort der Mutation und mutagene Oligonukleotide
Bevor eine zielgerichtete Mutation durchgeführt werden kann, müssen folgende
Voraussetzungen erfüllt sein:
Das interessierende Gen muss in einem geeigneten Vector (Bacteriophage oder
Plasmid) vorliegen.
Die Stelle, in welche die gewünscht Mutation in das Gen bzw. das Protein
eingeführt werden soll, muss bekannt sein.
Ebenso muss bekannt sein, welche Base entfernt und welche eingefügt werden
soll um das gewünschte Ergebnis zu erzielen.
1.2. LacZ / -Galactosidase
Das lacZ-Gen kodiert das Enzym -Galactosidase. Die -Galactosidase aus E. coli
hydrolysiert das Disaccharid Lactose in die beiden einfachen Zucker Galactose und
Glucose.
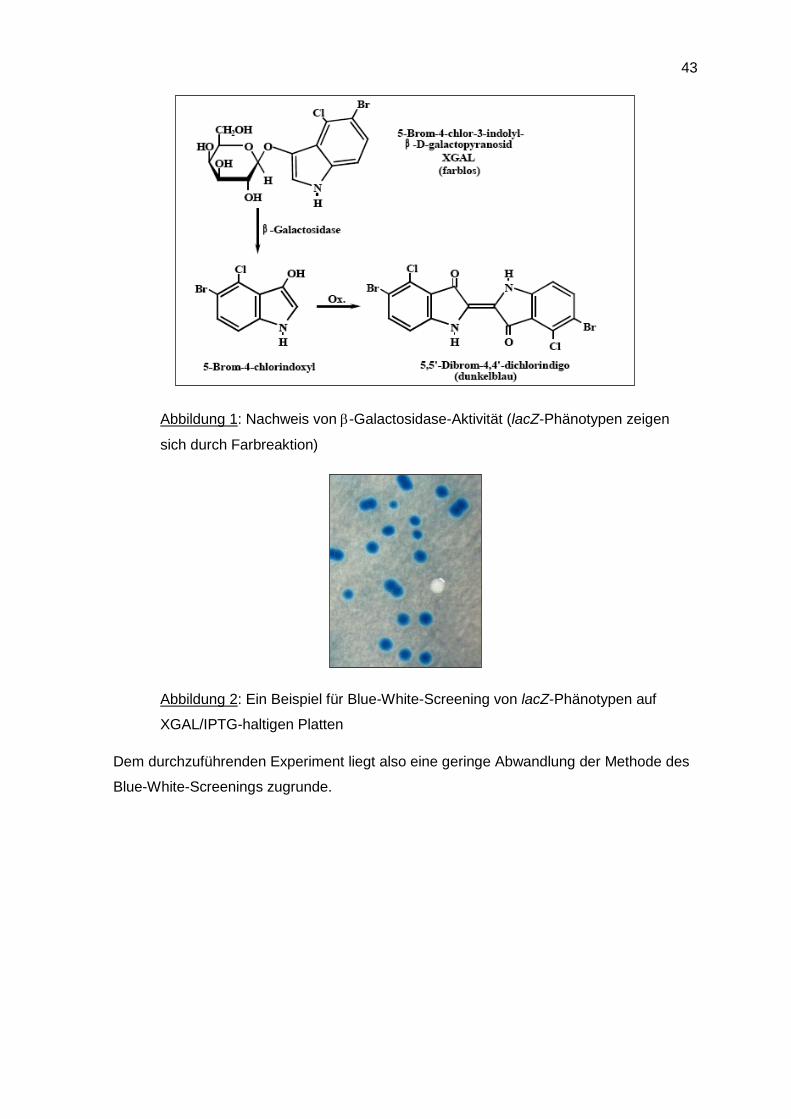
Eine weitere Anwendung von -Galactosidase ist das sogenannte blue-white-screening,
bei dem die farblose Verbindung 5-Brom-4-chlor-3-indolyl-b-Dgalactopyranosid (XGAL)
als Substratanalogon verwendet wird. Hierbei wird XGAL von der -Galactosidase erkannt
und in Galactose und eine Indoxyl-Verbindung gespalten. Diese wird durch Luftoxidation
in den dunkelblauen Farbstoff 5-Brom-4-chlorindigo überführt (siehe Abbildung 1).
XGAL und -Galactosidase-Aktivität werden seit Jahren in der Molekular- und Mikrobiolo-
gie sehr effektiv für in vivo Screening eingesetzt. Bei der Kultivierung von E. coli mit einer
aktiven -Galactosidase auf XGAL-haltigen Festmedien nehmen die Kolonien die dunkel-
blaue Färbung durch den gebildeten Farbstoff an.

42
Die klassische Anwendung der Blau-Weiß-Selektion liegt in der Detektion von insertierter
(eingefügter) DNA in das lacZ-Gen. Eines der ursprünglichen Probleme bei Klonierungs-
arbeiten war, die wenigen „neuen“ Zellen zu finden, die das gewünschte Fremd-DNA-
Stück („insert“) enthalten und aus Tausenden oder Millionen nicht veränderten Zellen
sicher herauszupicken. Dieses Problem wurde von Joachim Messing (University of
California and Minnesota) gelöst. Messing konstruierte Vektoren (Plasmide und
Bacteriophagen), bei denen das interessierende fremde DNA-Fragment innerhalb des
normalen lacZ-Genabschnitts ein-kloniert werden konnte.
Erfolgt die Klonierung des Inserts erfolgreich in die -Galactosidase-kodierende Sequenz,
wird von der betroffenen Zelle keine intakte -Galactosidase mehr exprimiert. Diese
Zellen wachsen auf XGAL-haltigen Medien und bleiben weiß. Alle Zellen, die kein DNA-
Insert in ihrem lacZ-Gen tragen, färben sich aufgrund der -Galactosidase-Aktivität
dunkelblau. Das Problem, die „richtige“ Zelle (mit der klonierten DNA) zu finden wurde
somit trivial: Man pickt eine weiße Kolonie aus den blauen Kolonien heraus.
In unserem Experiment liegt ein defektes lacZ-Gen, einkloniert im Plasmid pWhitescript
(siehe Abbildung 4), vor. Dieses defekte Gen wird nun durch gezielte Mutagenese wieder
in ein aktives Gen zurückverwandelt. Die Mutation innerhalb des lacZ-Gens von
pWhitescript verursacht einen Austausch von C mit T innerhalb der Protein-kodierenden
Sequenz wodurch aus einem Glutamin-Codon (CAA) ein Stop-Codon (TAA) an der
Aminosäurenposition 9 der -Galactosidase entsteht. Diese Substitutionsmutation
verhindert, bedingt durch einen vorzeitigen Abbruch der Proteinsynthese, die Expression
des intakten Enzyms. Wird diese Mutation wieder revidiert, erhalten wir ein neues Plasmid
(pBluescriptIISK) mit aktiver -Galactosidase. Im lacZ-Gen vom pBluescriptIISK ist das T
also erfolgreich durch C ersetzt worden, wodurch wieder die Aminosäure Nummer 9
Glutamin exprimiert wird.
43
Abbildung 1: Nachweis von -Galactosidase-Aktivität (lacZ-Phänotypen zeigen
sich durch Farbreaktion)
Abbildung 2: Ein Beispiel für Blue-White-Screening von lacZ-Phänotypen auf
XGAL/IPTG-haltigen Platten
Dem durchzuführenden Experiment liegt also eine geringe Abwandlung der Methode des
Blue-White-Screenings zugrunde.

44
Kolonien eines geeigneten E. coli Stammes, welcher das Plasmid pWhitescript enthält,
sind auf XGAL-haltigen Platten farblos bis weiß. Das Ziel ist, aus dem veränderten Stop-
Codon (TAA), durch ortsspezifische Mutation wieder einen Codon für Glutamin (CAA)
herzustellen. Das bedeutet, dass eine einzige der rund 3000 Basen des Plasmids
ausgetauscht werden muss. Bei erfolgreich durchgeführter Mutation und anschließender
Transformation wird das inaktive lacZ--Gen und die damit verbundene inaktive -
Galactosidase wieder in ein aktives lacZ+-Gen und damit in eine aktive -Galactosidase
überführt. Diese Oligonukleotid-vermittelte Mutage-nese erzeugt aus den vorher
ungefärbten Kolonien wieder blaue Kolonien eines lacZ+-Stamms.
Zur Amplifikation steht ein synthetisches Oligonukleotid („QC primer1“) zur Verfügung.
Dieser Primer ist zur Plasmid-DNA-Sequenz in der Umgebung von Codon 9
komplementär (siehe Abbildung 3). Direkt im Codon 9 enthält der Primer eine
Fehlpaarung welche zur Mutation am Zielcodon (fett dargestellt in Abbildung 3) führt: TAA
wird durch CAA ersetzt und die ursprüngliche „Wildtyp“-Sequenz lacZ+ sowie die
Enzymaktivität werden wieder her-gestellt.
Abbildung 3: Mutation von pWhitescript Stop-Codon 9 (lacZ-) in Gln9 (lacZ+)

45
Abbildung 4: Plasmidkarte von pWhitescriptä
2. DNA Isolierung nach der „QiaPrep“ Methode
2.1. Das QIAprep Prinzip
Das QIAprep Minipreparationsverfahren besteht aus zwei grundlegenden Schritten:
Alkalische Lyse der Bakterien
Adsorption der DNA an die QIAprep Membran, Waschen und Elution der Plasmid-
DNA.
2.2. Alkalische Lyse von Bakterien
Das QIAprep-Verfahren verwendet eine modifizierte Methode der alkalischen Lyse von
Birn-boim und Doly. Hierbei werden zuerst die Bakterien unter alkalischen Bedingungen
lysiert. Das Lysat wird im darauf folgenden Schritt gleichzeitig neutralisiert und auf eine,
für die Reinigung an der QIAprep Silicagel-Membran notwendig hohe Salzkonzentration,
eingestellt.

46
2.3. DNA Adsorption an die QIAprep-Membran
QIAprep Säulchen verwenden ein spezielle Silicagel-Membran, welche:
unter hohen Salzkonzentrationen die selektive Adsorption
unter geringen Salzkonzentration die Desorption
der Plasmid-DNA ermöglichen.
Die Kombination des optimierten Puffersystem für die Lyse und dieser speziellen
Membran stellt sicher, dass nur Plasmid-DNA in den Säulchen absorbiert wird. RNA,
zelluläre Proteine und niedermolekulare Stoffwechselverbindungen hingegen binden nicht
an der Membran.
2.4. Waschen und Elution der Plasmid DNA
Salze werden effizient durch einen kurzen Waschschritt mit Puffer PE von der Membran
ent-fernt. Danach kann die hochreine Plasmid-DNA mit Puffer von der QIAprep-Säule
eluiert werden. Diese DNA kann dann in einer Vielzahl verschiedener Anwendungen
eingesetzt werden, wie z. B. Restriktionsverdau, PCR oder Sequenzierungsreaktionen.
2.5. DNA Ausbeute
Die Plasmid-Ausbeute variiert:
in Abhängigkeit mit der Anzahl der Plasmidmoleküle pro Zelle
mit der Größe des Plasmids
durch Zellwachstumsfaktoren
durch das Elutionsvolumen und der Elutionsdauer.
So können 1,5 ml Übernachtkultur können zwischen 5 und 15 mg Plasmid-DNA liefern.

47
3. Miniprep-Protokoll für den „QIAprep spin kit“ unter
Verwendung einer Tischzentrifuge
1. Animpfen einer Übernachtkultur von DH5a mit dem pWhitescript-Plasmid :
10 ml LBAmp-Medium (100 μl/ml Ampicillin) werden mit einer Einzelkolonie des DH5a
pWhitescript angeimpft (steril arbeiten!).
Die Kultur wird über Nacht bei 37°C mit 300 rpm in einem Rotationsschüttler inkubiert.
2. Bakterienernte durch Zentrifugation:
Es werden jeweils 2 ml der Kultur in ein Eppi überführt und die Zellen bei 13.000 rpm
für 30 Sekunden abzentrifugiert. Die überstehende Flüssigkeit wird in einem Gefäß
gesam-melt, später autoklaviert und verworfen.
3. Resuspendieren des Zellpellets:
Jedes Zellpellet wird in 250 µl Puffer P1 resuspendiert. Es sollten anschließend keine
Zellklumpen mehr sichtbar sein.
4. Lyse des Zellpellets:
In jede der resuspendierten Zell-Lösungen wird nun 250 µl Puffer P2 zugegeben.
Die Flüssigkeiten durch vorsichtiges Invertieren der Eppis mischen. Nicht vortexen, da
dies zur Fragmentierung der genomischen DNA führt.
Die Lösung sollte viskos und leicht durchsichtig werden. Die Lyse darf nicht länger als
fünf Minuten stehen bleiben.
Anmerkung zur Lyse des Zellpellets:
Nach der Ernte und Resuspension weden die Bakterienzellen in NaOH / SDS (Buffer P2)
in der Gegenwart von RNase A lysiert. SDS solubilisiert die Phospholipide und Proteine
der Zellmembranen, was zur Auflösung der Zellwand führt. Das Cytosol wird
ausgeschüttet und die Alkalien denaturieren die Proteine wie auch chromosomale und
Plasmid-DNA. Bleiben die Plasmidmoleküle den alkalischen Bedingungen zu lange
ausgesetzt, wird die DNA irreversibel denaturiert. Diese denaturierte Form ist resistent
gegenüber Restriktionsverdau und läuft schneller im Agarosegel als die native Form.

48
Heftiges Schütteln oder Rühren des Lyse-Ansatzes muss vermieden werden, um zu
vermeiden, dass die chromosomale DNA in kleinere Fragmente zerbricht. Unter den
herrschenden Bedingungen könnten diese Fragmente nicht von der Plasmid-DNA
chemisch unterschieden werden. Die Fragmentierung würde also dazu führen, dass die
Plasmide mit anderen DNA-Bruchstücken verunreinigt werden.
5. Herstellung geeigneter Bedingungen zur Adsorption:
Jedem Ansatz werden 350 µl Puffer N3 zugegeben und das Eppi sofort vorsichtig
inver-tiert um die Lösungen zu durchmischen und um lokale Präzipitationen zu
vermeiden.
Die Lösung sollte eine zähflüssige Konsistenz annehmen.
Anmerkung zu den Adsorptionsbedingungen:
Das Lysat wird durch den Zusatz von Essigsäure neutralisiert und gleichzeitig wird eine
hohe Salzkonzentration erzeugt, die für die Adsorption an die Silicagel-Membran
notwendig ist.
Die hohe Salzkonzentration fällt denaturierte Proteine, Zelltrümmer, SDS und
chromosomale DNA aus, während die kleineren Plasmidmoleküle in Lösung bleiben. Es
ist sehr wichtig, dass die Lösungen gut durchmischt werden, um die Fällung zu
vervollständigen.
Die chromosomale DNA ist an die Zellwand gebunden und fällt als unlöslicher Komplex,
der aus Salzen, Detergenzien und Proteinen besteht, aus.
6. Zentrifugation:
Die Zentrifugation wird für 10 min. bei maximaler Geschwindigkeit durchgeführt um
die unlöslichen Bestandteile abzutrennen. Es sollte sich ein kompaktes, weißes
Pellet bilden. Die Plasmid-DNA bleibt im Überstand gelöst.
7. Adsorption der Plasmid-DNA an die Säulenmembran:
Für jede Probe eine QIAprep-Säule in ein 2-ml Sammelröhrchen setzen.
Den Überstand der Zentrifugation in eine QIAprep-Säule pipettieren.
Die Zentrifugation erfolgt für 30 Sekunden mit höchster Geschwindigkeit. Der Durch-
fluss wird verworfen.

49
8. Waschen der gebundenen Plasmid-DNA:
Jeweils 75 µl Puffer PE in die QIAprep-Säule pipettieren und 1 min bei maximaler
Geschwindigkeit zentrifugieren.
Den Durchfluss verwerfen.
Die Säule erneut unter gleichen Bedingungen zentrifugieren um Reste des
Waschpuffers vollständig zu entfernen.
9. Elution der Plasmid-DNA:
Die QIAprep-Säule in ein 1,5 ml Eppi setzten.
Zur Elution werden 50 µl Puffer auf die Mitte der QIAprep-Membran pipettiert. Nach
einer Minute Inkubationszeit bei Raumtemperatur wird mittels Zentrifugation (1 min,
vmax) die Plasmid-DNA eluiert.
Die Säule wird verworfen.
10. Bestimmung der DNA-Konzentration im Eluat:
Die Konzentrationsbestimmung der Plasmid-Lösung (1:100 verdünnt) erfolgt bei
einer Extinktion von 260 nm im Photometer. Zum Messen wird eine Quarzküvette
verwendet.
Es werden 5 µl des Eluats mit 495 µl Wasser (bidest.) verdünnt um die
Konzentration zu bestimmen. Vor der eigentlichen Messung muss der Blank-Wert
bestimmt werden.
11. Berechnung der Konzentration:
Es wird die Konzentration der gewonnenen Plasmid-DNA bestimmt (1 OD260 = 50
μg
doppelsträngige DNA/ml). Den Verdünnungsfaktor berücksichtigen!
Die Konzentration der isolieren Plasmid-DNA wird auf 40 ng/µl eingestellt.

50
4. Ortsgerichtete Mutagenese
4.1. Einführung
Ortsspezifische Mutation ist eine wertvolle Technik zur Untersuchung von Struktur- und
Funktionsbeziehungen in Proteinen. Zur Durchführung einer solchen Mutation wird in der
Regel einzelsträngige DNA (ssDNA) als Template verwendet.
Mittels des QuikChange ortsgerichteter Mutagenese Kit™ von Stratagene wird die direkte
Einführung einer ortspezifischen Mutation in ein doppelsträngiges Plasmid ermöglicht.
Dies erspart die Umklonierung in Bakteriophagen und die Herstellung von ssDNA. Hinzu
kommt, dass dieses QuikChange-Verfahren keine speziellen Vektoren benötigt und auch
nicht an bestimme Restriktionsstellen geknüpft ist. Dieses schnelle Verfahren erzeugt die
gewünschten Mutanten in wenigen Schritten mit einer Effizienz von mehr als 80 %.
4.2. DNA-Polymerase
Das QuikChange-Verfahren kann verwendet werden:
um Punktmutationen zu erzeugen
um einzelne oder mehrere Aminosäuren auszutauschen, einzufügen oder zu
entfernen
um Restriktionsstellen einzufügen oder zu entfernen
Die Methode wird mittels Polymerase-Kettenreaktion (PCR) im Thermocycler unter
Verwen-dung einer DNA-Polymerase (Taq- oder Pfu-DNA-Polymerase) durchgeführt.
Die DNA-Polymerase repliziert beide Plasmidstränge mit hoher Geschwindigkeit unter
Ver-wendung der mutagenen Oligonukleotide als Primer (siehe Abbildung 3). Diese sind
zu beiden Strängen des Templats komplementär und werden während der Temperatur-
zyklen von der Polymerase an ihrem 3’-Ende verlängert. Dadurch entsteht, ausgehend
von den Primern, ein mutiertes Plasmid mit zwei gestaffelten Einzelstrangbrüchen am 5’-
Ende.

51
4.3. Verdau mittels DpnI Restriktionsendonuklease
Nach der Amplifikation wird das PCR-Produkt mit dem Restriktionsenzym DpnI behandelt
um restliche, unerwünschte Plasmid-DNA zu entfernen. Die DpnI Endonuklease (Zielse-
quenz: 5’-Gm6ATC-3’) schneidet nur methylierte DNA spezifisch. Somit kann das Enzym
zwischen der isolierten Plasmid-DNA und der in-vitro synthetisierten, mutagenen DNA
unterscheiden. In vivo hergestellte DNA, welche beispielsweise aus E. coli-Stämmen
isoliert wird, ist methyliert und wird deswegen von DpnI abgebaut.
Der doppelstrangbrüchige Vektor mit der gewünschten Mutation wird anschließend in
kompetente DH-Zellen transformiert.
4.4. Primer-Design
Mutagene Primer führen eine spezifische experimentelle Mutation ein. Die mutagenen
Oligo-nukleotide müssen individuell für die jeweils gewünschte Mutation entworfen
werden.
Folgende Überlegungen sollten dem Primer-Design zugrunde liegen:
1. Beide mutagenen Primer müssen die gewünschte Mutation enthalten und sich an die
Zielsequenzen auf den komplementären Strängen des Plasmids anlagern.
2. Die Primer sollten 25 bis 45 Basen lang sein und die Schmelztemperatur (Tm)
mindes-tens 78° C betragen.
Folgende Formel wird üblicherweise für die Berechnung der Tm verwendet:
Tm = 81.5 + 0.41(%GC) - 675/N - %mismatch
Wobei N die Primerlänge in Basenpaaren bedeutet.
3. Die gewünschte Mutation (Deletion oder Insertion) sollte in der Mitte der Primer
liegen,
flankiert von 10 bis 15 Basen der korrekten (komplementären) Sequenz auf beiden
Seiten.
4. Der GC-Gehalt der Primer sollte mindestens 40 % betragen und an beiden Enden
der Primer sollte mindestens eine G- oder C-Base lokalisiert sein.

52
4.5. Überblick
Die schwarzen Punkte kennzeichnen das Gen im Plasmid
mit der Zielsequenz für die Mutation.
Im ersten PCR-Schritt wird die Plasmid-DNA zu Einzel-
strängen denaturiert
Nach einer Absenkung der Temperatur erfolgt die Anlage-
rung (annealing) der mutagenen Oligonukleotide an die
komplementäre Stelle des Einzelstranges.
Im finalen PCR-Schritt verlängert (elongation) die DNA-
Polymerase die Primer und insertiert die Mutation in zirku-
läre Stränge mit Einzelstrangbrüchen.
Diese drei Schritte werden so oft wiederholt, bis genügend
PCR-Produkt vorhanden ist.
Die parentalen, nichtmutierte Template-DNA wird mittels
der Restriktionsendonuklease DpnI verdaut.
Nur der doppelstrangbrüchige Vektor mit der insertierten
Mutation liegt jetzt noch vor.
Dann erfolgt die Transformation der doppelstrangbrüchi-
gen dsDNA in kompetente DH5a-Zellen.
In den Zellen werden die Strangbrüche im mutierten
Plasmid repariert.

53
4.6. Durchführung der PCR-Reaktion
Folgende Reagenzien werden, der Reihenfolge entsprechend, auf Eis in ein PCR-
Reaktions-gefäß pipettiert:
32,50 µl Steriles Wasser
6,00 µl 10 x Puffer Y
1,00 µl MgCl2
2,50 µl pWhitescript ( = 100 ng)
2,00 µl Primer 1 ( = 10 nmol)
2,00 µl Primer 2 ( = 10 nmol)
2,00 µl dNTP-Mix (2,5 mM je Nukleotid)
2,00 µl Taq-Polymerase (ca. 2 U/µl)
50,00 µl Gesamtvolumen
Der Thermocycler nutzt folgendes Programm zur Amplifikation:
Segment Zyklen Temp. [°C] Dauer
1 1 95 3 min
95 30 sec
2 28 51 1 min
72 3 min
3 1 72 7 min
4 1 4 hold
4.7. Durchführung der Restriktion
Es werden 25 µl des amplifizierten PCR-Produktes in ein neues Reaktionsgefäß
überführt. Dann werden 2,0 µl der Restriktionsendonuklease DpnI zupipettiert und der
Reaktionsansatz bei 37 °C für eine Stunde im Thermocycler inkubiert.

54
5. Agarose-Gelelektrophorese
!
Ethidiumbromid (EtBr) ist sehr giftig, reizend, kanzerogen und mutagen!
Beim Umgang damit sind Schutzhandschuhe zu tragen! Alle Geräte, die mit der
Lösung in Berührung gekommen sind, müssen als kontaminiert betrachtet werden.
Der Umgang mit diesem Stoff ist nur an den speziell ausgewiesenen Arbeitsplätzen
erlaubt.
Die Elektrophorese mit einem Agarosegel als Trennmaterial ist eine Standardmethode,
die zur Trennung, Identifizierung und Reinigung von DNA-Molekülen dient. Die DNA wird
mittels dem interkalierenden Fluoreszenzfarbstoff Ethidiumbromid im ultraviolettem Licht
sichtbar.
Die Wanderungsgeschwindigkeit der Nukleinsäuren im Gel hängt von deren Konformation
ab. Die überspiralisierte (supercoiled) Form wandert am schnellsten durch die Poren des
Gels. Dann folgt offene zirkulare DNA (Einzelstrangbruch) und letztendlich die lineare
DNA-Konformation (Doppelstrangbruch). Die Laufstrecke dieser Form steht in einem
direkten Verhältnis zu ihrer Länge in bp. Ebenfalls elektrophoretisch aufgetrennt wird ein
Marker mit definierten Bandengrößen. Durch einen Vergleich dieser Banden (hier: l-
Phagen-DNA mit Hind III verdaut) kann die Größe des Plasmids bestimmt werden.
Mittels Gelelektrophorese wird überprüft, ob die Amplifikation erfolgreich war. Hierzu wird
ein Teil des PCR-Produkts, Template-DNA als Vergleichsprobe sowie ein Marker auf das
Gel geladen.

55
Abbildung 5: 0,9 % Agarosegel. (Bahn 1: l-Marker; Bahn 2: Template-DNA;
Bahnen 3+4: PCR-Produkt)
5.1. Durchführung der Gelelektrophorese:
Herstellung der Trenngellösung (0,9 %):
Folgende Zutaten werden in einem Erlenmeyerkolben vermischt:
1. 0,63 g Agarose einwiegen.
2. Mittels einer Messpipette 7 ml TBE-Puffer (10fach konzentriert) zugeben.
3. 63 ml Wasser auf der Waage zugeben.
Die Gellösung wird aufgekocht (Heizplatte oder Mikrowelle) bis die Agarose komplett
gelöst ist.
Gießen und Beladen des Gels:
1. Die offenen Seiten der Gelkammer werden sorgfältig mit Klebeband abgeklebt, wobei
auf korrekten Sitz (speziell an den Stegen) zu achten ist, um ein Auslaufen der Gel-
lösung zu verhindern.

56
2. Die Trenngellösung wird möglichst blasenfrei in die zuvor abgeklebte Kammer einge-
füllt und der Probenkamm in die Lösung gesteckt. Eventuell vorhandene Luftblasen
werden mittels einer Pipettenspitze vorsichtig an den Rand geführt.
3. Nachdem das Gel ausgehärtet ist (ca. 30 min.) wird es in die Elektrophoresekammer
gelegt. Die Apparatur wird bis knapp über den oberen Rand des Gels mit Laufpuffer
(1 x TBE) befüllt. Nun können die Proben in die Geltaschen eingebracht werden.
4. 20 μl des PCR-Produktes werden in ein 1,5 ml Eppendorf-Tube pipettiert, 4 μl Lade-
Puffer zugesetzt und kurz gemischt (Vortexer).
5. Um die an der Wand haftenden Tropfen am Boden des Tubes zu sammeln wird die
Probe wenige Sekunden zentrifugiert.
6. Die Proben (PCR-Produkt, Template-DNA, Marker) werden in jeweils eine Geltasche
pipettiert.
7. Die Kammer mit dem Deckel verschließen. Am Netzgerät eine konstante Spannung
von 100 V einstellen und die Elektrophorese starten.
8. Zwei Farbstoffe im Lade-Puffer zeigen die momentane Lauffront an. Nach ca. einer
Stunde kann die Elektrophorese gestoppt und das Gel im EtBr-Bad angefärbt
werden. Die Aufnahme erfolgt unter UV-Licht.
Durch einen Vergleich mit den Markerbanden wird die Längen der Plasmidfragmente be-
stimmt.
6. Transformation
Das PCR-Produkt wird mittels Temperaturschock in kompetente DH-Zellen transformiert.
Diese Zellen wurden durch Waschvorgänge mit verschiedenen Metallsalzlösungen so
modifi-ziert, dass sie durch einen Temperaturschock Plasmid-DNA ins Zellinnere
schleusen können. Trotz dieser chemischen Behandlung nimmt von etwa einer Million
Zellen nur eine einzige die fremde DNA auf.
Transformation der mutierten Plasmid-DNA:
1. Die kompetenten Zellen werden auf Eis aufgetaut. Ein Wasserbad auf 42 °C
vorgeheizt.

57
2. Zu der Zellsuspension wird der komplette Rest des DpnI-verdauten PCR-Produktes
zugegeben und kurz auf dem Vortexer vermischt.
3. Die Zellen werden für 60 Minuten auf Eis inkubiert.
4. Der Hitzeschock der Zellen erfolgt durch die Inkubation für 3 Minuten bei 42 °C im
Wasserbad. Anschließend sofort wieder auf Eis stellen.
5. Die transformierten Zellen werden komplett in 1 ml Flüssigmedium pipettiert und für 1
Stunde bei 37 °C im Schüttler inkubiert.
7. Vorbereiten der Agar-Platten für Blue-White-Screening
Während der Transformation werden die Agarplatten für das Screening der
Transformanten vorbereitet. Nacheinander wird zuerst 40 μl IPTG-Lösung und dann 40 μl
XGAL-Lösung mit einem Trigalski-Spatel (bzw. einer gebogenen Pasteur-Pipette) auf LB-
Ampicillin-Platten ausgestrichen, bis die Flüssigkeit jeweils in das Medium eingezogen ist.
Nach vollendeter Inkubation werden von der Kultur 50, 100 und 200 µl mit einem
Trigalski-Spatel auf den vorbereiteten Agarplatten ausgestrichen.
Die Platten werden über Nacht bei 37° C in einem Brutschrank inkubiert, wobei die Petri-
schalen auf dem Deckel liegen (Beschriftung auf der Unterseite nicht vergessen).
Am nächsten Tag wird überprüft, ob und welche Kolonien gewachsen sind.
Recommended

















