Embed Size (px)
Citation preview

Anleitung zum Praktikum Strukturanalytik
WS 2018/19
Fakultät für Lebenswissenschaften
Institut für Biochemie
Professur für Biophysikalische Chemie

Inhalt -----------------------------------
Stationen/Kontakte
------------------------------------
X-ray I und II
X-ray III
FRET
MS I
MS II
NMR I
NMR II
IR
EPR

Fakultät Lebenswissenschaften Institut für Biochemie Professur für Biophysikalische Chemie Prof. Dr. T. Pompe
Praktikum Strukturanalytik für Biochemiker WS 2018/19
Zeitraum
Alle Praktika beginnen um 13:15 Uhr und enden ca. 16:30 Uhr, das MS-Praktikum endet ca.
17:00 Uhr.
Ausnahmen:
NMR II (Kernspintomographie) bei Herrn PD Dr. H. Busse beginnt um 14:30 Uhr.
Ort: Universitätsklinikum (UKL), Klinik für Diagnostische und Interventionelle Radiologie,
Treffpunkt Wartebereich vor der „Röntgen“-Anmeldung, Haupteingang UKL, Liebigstraße 20
(Haus 4), dann der Beschilderung folgen.
X-ray I und II sind zwei zweistündige Vorlesungen für alle Gruppen gemeinsam bei Prof.
Sträter, Fakultät für Chemie
Zeit: 15:00 bis 16:30 Uhr; Ort: Vorlesungsraum des Biomedizinisch-Biotechnologischen
Zentrums (BBZ), Deutscher Platz 5, Erdgeschoss
Praktikum Praktikumsleiter / Kontakt Ort
X-ray III Arbeitsgruppe Prof. Sträter BBZ, 1. OG, Deutscher Platz 5
An Klingel Dr. Weiße wählen
NMR I Herr Dr. Icker, Tel 36560 Institut für Organische Chemie Technikum-Analytikum, Linné-Str 3, Erdgeschoss, Raum 169/70
FRET Herr Riedel, Frau Dr. Franke,
Tel 36933
Institut für Biochemie, Johannisallee 21-23, Raum 035
MS I Frau Reppich-Sacher, Herr Dr. Stichel, Tel 38531 / 797
Institut für Biochemie, Stephanstrasse 24, Neubau Sonderlabore, 2. Etage
MS II Frau Dr. Popkova, Tel 15722
Herr Dr. Schiller, Tel 15733
Institut für Medizinische Physik und Biophysik, Härtelstrasse 16-18, 1. Etage, Raum 152.2
IR Frau Franz, Tel 36819 Institut für Pharmazie, Brüderstrasse 34, Raum 013
EPR Herr Prof. Pöppl, Frau Kultaeva, Tel 32608
Fakultät für Physik, Linnéstrasse 5,
Raum 111
3

X-ray I und II
Es handelt sich um zwei zweistündige Vorlesungen für alle Gruppen gemeinsam bei Prof. Sträter.
Unterlagen dazu finden Sie unter: www.uni-leipzig.de/~straeter/intern User: bbz Password: memento „Kurzer Abriss der Proteinkristallographie (xray1, xray2 Biochemie)“
4
X-ray III
5

Übung „X‐ray III“ im Praktikum „Biophysikalische Chemie“ für Biochemiker A. Kristallisation‐ Das Enzym Lysozym aus Hühnereiweiß (Fa. VWR, 1.05281.0001) soll kristallisiert werden. Als Kristallisationsmit‐tel (Fällungsmittel) dienen NaCl als auch Methyl‐Polyethylenglycol 5000 (MPEG‐5000). Die benötigten Lösun‐gen sind bereits vorhanden. Zusätzlich sind bereits vorhandene Lysozymkristalle der Vorgängergruppen am Mikroskop mit der Digitalkame‐ra zu dokumentieren. Die eigenen Kristalle wachsen bis zu drei Tage und sind anfänglich noch zu klein für eine optische Begutachtung. Beachten Sie bitte, dass die äußere Gestalt der Proteinkristalle schon Hinweise auf die innere Ordnung (Symmetrie) geben kann ‐ suchen Sie nach Symmetrie! Zur Verifizierung von Proteinkristallen gegenüber Salzkristallen werden ausgewählte Nachweismethoden ausprobiert (Bruchtest, Anfärbetest mit Proteinfarbstoffen). Dabei wird auch auf Handhabungsmethoden (Kryo‐Schleifen) für Proteinkristalle eingegan‐gen. Gearbeitet wird in Zweier‐Gruppen. Versuchsdurchführung A ‐ Lysozymlösung (Konzentration 30 mg/ml) in H2O ‐ 700 mM / 850 mM / 1000 mM / 1200 mM NaCl in 0,1 M NaAcetat (pH 4,5) Jede Gruppe pipettiert 4 (NaCl‐Konzentrationen) x 3 (Wiederholungen) = 12 Hängetropfen (je 4 µl Protein mit 4 µl Reservoirlösung mixen), entsprechend einer Kristallisations‐Halbplatte. Die Kristallisationsplatten werden anschließend bei 19 °C gelagert. Die Kristalle sind erst am nächsten Tag sichtbar. Versuchsdurchführung B (streak seeding) ‐ Lysozymlösungen (Konzentrationen 40 mg/ml; 24 mg/ml; 8 mg/ml) in 0,1 M NaAcetat (pH 4,5) ‐ 700 mM / 850 mM / 1000 mM / 1200 mM NaCl in 0,1 M NaAcetat (pH 4,5) und 25 % (w/v) MPEG‐5000 Kristalle einer Vorgängergruppe werden mechanisch zerkleinert. Die dabei entstehenden Kristallfragmente können als Kristallkeime in andere Kristallisationsansätze übertragen werden. Hierzu wird ein Haar durch die Suspension mit den zerstoßenen Kristallen und im Anschluss durch einen Wachstumstropfen gezogen. Alterna‐tiv streicht man mit dem Haar über einen intakten Kristall. An dessen Oberfläche befinden sich ebenfalls mikro‐skopisch kleine Kristalle, die als Kristallisationskeime im Wachstumstropfen verwendet werden können. Wachstumstropfen werden durch zusammenpipettieren von 4 (NaCl‐Konzentrationen) x 3 (Proteinkonzentrati‐onen) = 12 Hängetropfen (je 4 µl Protein mit 4 µl Reservoirlösung mixen) erhalten. Die Kristallisationsplatten werden anschließend bei 19 °C gelagert. Kristalle sollten bereits nach kurzer Zeit zu sehen sein.
B. Datensammlung‐ Ein nicht zu großer Proteinkristall (< 400 µm für maximale Kantenlänge) einer Vorgängergruppe ist für eine Messung im tiefgekühlten Zustand vorzubereiten. Dafür ist der Proteinkristall vorher sukzessive in einer mit ansteigender Konzentration eines Gefrierschutzmittels (25 v/v % Glycerol) versetzten Reservoirlösung zu inku‐bieren. Alternativ kann der Proteinkristall kurz durch einen Tropfen Paraffinöl gezogen werden, bevor er in den fl[ussigen Stickstoff getaucht wird. Beide Methoden verhindern unerwünschte Eisbildung, welche das Messer‐gebnis (Diffraktionsbild) erheblich verschlechtern. Ein exemplarisches Diffraktionsbild (30 sec ‐ 1 min Belichtungszeit bei Drehung des Kristalls um 1 ° entlang der sogenannten Spindelachse) wird begutachtet. Für die Messung eines Diffraktionsdatensatzes sind allerdings viele dutzende bis hunderte solcher Messaufnahmen notwendig.
6

C. Modellbau‐ Es soll die Struktur der menschlichen violetten sauren Phosphatase (HPAP) in Teilen bestimmt werden. Das Phasenproblem wurde mit der Methode des molekularen Ersatzes gelöst, wobei eine bekannte Struktur (hier die der saueren violetten Phosphatase des Schweins = Uteroferrin) in die Elementarzelle der zu bestimmenden Struktur „gedreht“ wurde. Dann konnte eine Elektronendichtekarte der zu bestimmenden Proteinstruktur berechnet werden. Mit Hilfe dieser Elektronendichtekarte kann die Struktur von HPAP ausgehend von der Struktur des Startmodells (Uteroferrin) gebaut werden. Linux! Das notwendige Programm ‚Coot‘ und die betreffenden Dateien sollten bereits eingeladen sein: Dateien: /HPAP/hpap.map Elektronendichtekarte der humanen PAP /HPAP/model.pdb Koordinatendatei des Uteroferrins nach molekularem Ersatz /HPAP/hpap.pdb Koordinatendatei der fertigen HPAP‐Struktur
Aufgaben: 1. Mutieren Sie 6 Aminosäurereste (6, 34, 60, 117, 121, 127) in der Modell‐Struktur, die laut Sequenzvergleich
(siehe Anlage) verschieden im Vergleich zur Zielstruktur sind, in die betreffenden richtigen Aminosäurereste.
2. Zentrieren Sie auf Cys142. Diese Aminosäure ist im Uteroferrin an einer Disulfidbrücke beteiligt. Ist diese Brücke auch in HPAP vorhanden (Elektronendichte)? 3. Verfolgen Sie die Kette nach Cys142 weiter C‐terminal. Bald kommt ein Bereich, in dem der Hauptkettenver‐lauf in HPAP anders verläuft als in Uteroferrin. Bevor hier die Seitenketten eingepasst werden können, muss zunächst der ganze Rest korrekt in die Dichte geschoben werden. Bauen Sie bis Asp147 oder auch weiter. Wel‐che Besonderheit weist Asp147 auf? Konturieren Sie dazu die Elektronendichte auf sigma größer 10 und mehr.
7

Sequenzvergleich Schwein (Uteroferrin) ‐ Mensch (HPAP)
Hinweis‐
Sequenznummern beziehen sich mit ihrer letzen Ziffer auf die entsprechende Position, so ist z.B. die Aminosäu‐
re 126 im Uteroferrin ein Arg (R), analog ist die Aminosäureposition 130 in HPAP ein Gln (Q).
8

Arbeiten in COOT Beachten Sie folgende Menüs: Display manager (Ein/Abschalten der einzelnen Objekte) Edit Map Colour hpap.map (Farbe der Elektronendichtekarte) Go to Atom (Anklickbare Aminosäuresequenz/Nummerneingabe mit Zentrierfunktion) Die Ansicht der Moleküle verändern Sie mit: Mausrad klicken auf ein Atom = Zentrieren auf das Atom und Aktualisierung der Elektronendichtekarte Rechte Maustaste gedrückt ziehen/schieben = Zoom Linke Maustaste gedrückt bewegen = dreidimensionale Rotation in allen drei Raumachsen Linke Maustaste auf Aminosäure im „Sequence View“‐Fenster klicken = Zentrieren auf die Aminosäure und Aktualisieren der Elektronendichtekarte Mausrad bewegen (VORSICHT!) = Konturierunglsevel der Elektronendichtekarte verändern (nach Betätigung wird Wert angezeigt und sollte wieder auf ca. sigma = 1 gebracht werden) Leertaste oder Shift+Leertaste = Springen der aktuellen Zentrierposistion von C� nach C�+1 oder nach C�‐1 Folgende Modellbauoptionen (rechtes Menü!) sollten Sie verwenden Mutate & Auto Fit: Mutieren und automatisches Anpassen der Seitenkette
Rotate/Translate Zone: Bewegen von kompletten Aminosäuren
(klicken Sie dazu nacheinander auf Hauptkettenamino‐ und Hauptkettencarboxylgruppe)
Edit Chi Angles: Verändern der Seitenkettenwinkel
Real Space Refine Zone: sehr wirkungsvolle Automatikbaufunktion für Aminosäurebereiche
Rigid Body Fit Zone: Automatikbauen ganzer Aminosäurebereiche (Anfang und Ende markieren)
9

FRET
10

Förster-Resonanz-Energietransfer (FRET)
Praktikum Strukturanalytik
i. Grundlagen
a. Förster-Resonanz-Energietransfer (FRET)
i. Prinzip
Für den Förster-Resonanz-Energietransfer werden zwei Moleküle (oder ein Molekül an verschiedenen Stellen) mit zwei Fluoreszenzfarbstoffen versehen, wobei des einen Emissionsspektrum mit dem Anregungsspektrum des anderen überlappt. Wenn die beiden Fluorophore in große räumliche Nähe kommen, sei es durch Proteinwechselwirkungen oder Konformationsänderungen, kann das eine Fluorophor (der Donor) die Energie von adsorbierten Photons direkt auf das andere Fluorophor (der Akzeptor) übertragen. Demnach wird Fluoreszenzlicht entsprechend des Emissionsspektrums des zweiten Fluorophors erzeugt. Der Energietransfer ist stark abstandsabhängig und geschieht nur über Distanzen von wenigen Nanometern. Deshalb eignet sich die Technik um Molekülstrukturen auf Nanometerskala zu detektieren.
ii. Fluoreszenzspektrometrie
Es können viele verschiedene Analysetechniken verwendet werden um den Energieübertrag aus FRET zu messen. Während meist ein Fluoreszenzmikroskop zur räumlichen Auflösung von FRET zum Einsatz kommt, soll in diesem Praktikumsversuch ein Fluoreszenzspektrometer eine qualitative Aussage über die Effizienz von FRET in einer Proteinlösung ermöglichen. Annähernd monochromatisches Licht wird durch einen ersten Filter erzeugt, der entsprechend der Anregungswellenlänge des ersten Fluorochroms gewählt ist. Nachdem das gefilterte Licht auf ein entsprechendes Fluorochrom getroffen ist, dieses angeregt hat, findet, je nachdem ob ein Fluorochrom der zweiten Art in der Nähe ist, FRET statt oder nicht. Für die Analyse der Häufigkeit von FRET wird das emittierte Licht durch einen zweiten Filter geleitet, der entsprechend der Emissionswellen
b. Untersuchung der Konformation von Fibronektin
i. Fibronektin als Bestandteil der extrazellulären Matrix
Fibronektin ist ein etwa 470kDa großes Glykoprotein der extrazellulären Matrix und dient da zum einen als strukturgebendes Element, zum anderen stellt es auch Bindungstellen für Zelladhäsionsrezeptoren und andere Matrixproteine bereit. Fibronektin ist ein Heterodimer aus zwei Untereinheiten, die wiederum aus einer Anzahl Domänen verschiedenen Typs (siehe Abbildung 1) bestehen.
Abbildung 1: Ein Fibronektinmonomer besteht aus Domänen dreier Typen, die Bindungstellen für andere Matrixproteine und Zelladhäsionsrezeptoren bereithalten. Über die Dissulfidbrücke sind zwei Monomere zu einem Heterodimer verknüpft. Quelle: (2)
In der extrazellulären Matrix kommt Fibronektin in unlöslichen, großen Aggregaten vor, die sich in fibrillärer Form zusammengelagert haben. Dieser Prozess geschieht unter Kraftausübung der Zelle, die über Zellrezeptoren an das Molekül gebunden ist und durch die anschließende Streckung des Moleküls werden kryptische Bindungstellen freigelegt, die eine Selbstassemblierung von Fibronektinmoleülen zu Fibrillen ermöglicht.
11

Als einzelnes Molekül ist Fibronektin löslich und wird zum Beispiel aus humanen Blutplasma gewonnen. Fibronektin kann verschiedene Konformationen annehmen, je nachdem ob es in Lösung, auf Oberflächen adsorbiert, oder in Fibrillen verbaut ist. Es gibt eine kompakte Konformation bei der die beiden Armes des Fibronektins übereinander gefaltet sind und die resultierende globuläre Form durch elektrostatische Wechselwirkungen zwischen den beiden Armen stabilisiert wird. Bei zunehmender Denaturierung, oder unter Kraftausübung der Zelle, wird die kompakte Konformation aufgelöst und Fibronektin liegt als langes, gestrecktes Molekül vor. Bei weiterer Dehnung werden Teile des Moleküls entfaltet und dabei Bindungsstellen freigelegt (siehe Abbildung 2).
Abbildung 2: Verlust der Struktur von Fibronektin unter denaturierenden Bedingungen. In Lösung nimmt Fibronektin eine kompakte Konformation an (A). Bei Denaturierung wird die kompakte Struktur aufgelöst und das Molekül ist eine lange Kette (B). Unter Fortschreitender Denaturierung werden Module entfaltet (C). Quelle: (1).
ii. Untersuchung von Fibronektin mittels FRET
Bei sehr starker Denaturierung von Fibronektin, zum Beispiel mittels 8M Guanidinhydrochlorid, liegen auf beiden Monomeren zwei Cysteine auf den Modulen III7 und III15 frei (siehe gelbe Module in Abbildung 2), die mittels eines funktionalisierten Fluoreszenzfarbstoffes markiert werden können. Im Versuch werden die mit Maleimiden versehenen Alexa Fluor 488 und Alexa Fluor 546 verwendet, bei denen unter großer räumlicher Nähe ein Energietransfer stattfinden kann. Abbildung 2 kann entnommen werden, dass die größte Wahrscheinlichkeit für einen Energietransfer in der kompakten Konformation von Fibronektin vorhanden ist und bei zunehmender Denaturierung ist eine Abnahme zu erwarten ist.
Im idealen Fall sind jeweils ein Modul III7 und III15 jeweils mit einem Donor-Fluorophor und die anderen mit einem Akzeptor-Molekül versehen. Da aber von einer großen Anzahl von Molekülen mit statistisch verteilten Fluorophoren ausgegangen wird, ist es unerheblich, ob die in jedem Fluorophorpaar FRET stattfinden kann.
Als Ziel des Versuchs sollen die Verhältnisse der Intensitäten nach erfolgtem Energiertransfers und der Fluoreszenz des Donors 𝑰𝐀 𝑰𝐃⁄ für verschieden stark denaturierende Bedingungen verglichen werden.
12

ii. Versuchsdurchführung
a. Fluoreszenzmarkierung des Fibronektins an freien Cysteinen
Vorbereitung der Farbstofflösungen
Vorbereitung von 1-10 mM Stammlösungen der Farbstoffe Alexa Flour 488 maleimide und Alexa Fluor 546 maleimide in DMSO (die Lösungen müssen lichtgeschützt aufbewahrt werden!)
Überprüfung der Konzentration durch Absorptionsspektroskopie (Eppendorf BioSpectrometer) über die Extinktionskoeffizienten der Farbstoffe:
Alexa Fluor 488: 𝑀 = 720,66 g mol⁄ , 𝜀488 nm
= 71 000 cm−1M−1
Alexa Fluor 546: 𝑀 = 1034,37 g mol⁄ , 𝜀546 nm
= 203 000 cm−1M−1
Dafür muss ein Teil der Farbstofflösungen abgenommen und verdünnt werden (etwa 200-1000 fach).
Berechnung der Konzentration: 𝐶Farbstoff =𝐴488/546
𝜀488/546∙𝑑 (Küvettendicke d=1cm)
Inkubation mit Alexa Flour 488 maleimide und Alexa Fluor 546 maleimide
Alle Lösungen in denen Fibronektin enthalten sind sollten keinen starken Scherkräften ausgesetzt werden, da Fibronektin sonst spontan ausfibrilliert.
Herstellung einer 8 M GndHCl-Lösung durch Lösen von 76,42 g GndHCl (𝑀 = 95,53 g mol⁄ ) in 100 ml Reinstwasser
Herstellung einer 1-2 µM (0.4-0.8 mg/ml) Fibronectinlösung (0.5 ml) in PBS
1:1 Mischung der Fibronektin-Lösung mit 8 M GndHCl
Hinzufügen der Farbstoff-Lösungen zur Proteinlösung (tropfenweise) jeweils bis zum ca. 5-fachen molaren Überschuss (Berechnung aus Fibronektinkonzentration) Bsp.: zu 0.5 ml einer 1 µm Fibronectinlösung werden je 2,5 µl von 1 mM Farbstofflösungen gegeben.
Reaktionsansatz nicht größer als 2,5 ml!
Ablauf der Reaktion wippend für 60 – 90 min bei Raumtemperatur oder über Nacht bei 4 °C (lichtgeschützt!)
Größenausschluss-Chromatographie
Abtrennung des ungebundenen Farbstoffs durch Größenausschluss-Chromatographie mit Sephadex PD 10-Säule
Äquilibrierung der Säule mit 25 ml PBS
Sobald das PBS durchgelaufen ist und die Säule trocken zu fallen droht wird der Reaktionsansatz aufgetragen
13

Gleichzeitig wird begonnen Fraktionen (ca. 5-10 Tropfen/Fraktion) aufzufangen und gegebenenfalls PBS auf die Säule nachgegeben
Bestimmung des Labelgrades und der Fibronektin-Konzentration
Messung der Absorption der Fraktionen bei 280 nm (A280), 494 nm (A494) und 554 nm (A554)
Konzentration der Farbstoffe und FN:
Alexa Fluor 488: d
AC
488
488488
, 488 = 71 000 cm-1 M-1, d = 1 cm
Alexa Fluor 546: d
AC
546
546546
, 546 = 203 000 cm-1 M-1, d = 1 cm
FN: d
AC
FN
FN
, FN = 563 200 cm-1 M-1, d = 1 cm
Labelgrad (LG): FN
Farbstoff
C
CLG
b. Messung des Förster-Resonanz-Energietransfer
Mit Hilfe des zweifach gefärbten Fibronektins kann die Konformationsänderung von Fibronektin bei fortschreitender Denaturierung untersucht werden.
In eine Mikrotiterplatte mit (flachem Boden) werden verschiedenen Volumina von 8M GndHCl-Lösung vorgelegt, sodass die GndHCl-Lösung in 0M, 1M,…, 4M vorliegt, jeweils mit der entsprechenden Menge PBS zum gleichen Volumen auffüllen (150 µl)
Im Anschluss wird eine bestimmte Menge (50 µl) an Fibronektinlösung hinzugegeben.
Die Messung der Fluoreszenzintensität erfolgt im Tecan Microplate Reader mit zwei verschiedenen Filtersets:
- Anregungswellenlänge bei 480 nm und Emissionswellenlänge bei 525 nm: Fluoreszenz des Donors mit Intensität ID
- Anregungswellenlänge bei 480 nm und Emissionswellenlänge von 585 nm: Anregung des Donors und Fluoreszenz des Akzeptors mit Intensität IA
c. FRET-Bleaching-Experiment
Mit Hilfe des in a. gelabelten Fibronektins kann das Adhäsionsverhalten auf hydrophilen bzw. hydrophoben
Oberflächen untersucht werden.
Herstellen von hydrophilen Oberflächen
Zur Herstellung von hydrophilen Oberflächen werden Deckgläschen mittels RCA gereinigt.
Die Deckgläser (DG) werden in einem Teflonkarussell für 30min in Millipore-Wasser Ultraschall-behandelt.
14

Nach einem Spülvorgang in MilliQ-Wasser werden die DG erneut für 30min in Ethanol (vergällt) Ultraschall-behandelt.
Anschließend werden die Proben in MilliQ gespült und danach RCA gereinigt. Hierfür werden die DG in einem Gefäß aus Quarzglas in eine 60°C warme Lösung aus H2O2, NH3 und Millipore-Wasser im Verhältnis 1:1:5 getaucht. (! Nicht wärmer als 70°C !)
Nach zehn minütiger RCA-Reinigung werden die DG mit MilliQ-Wasser gespült und mit N2 trocken geblasen.
Herstellen von hydrophoben Oberflächen
Zur Herstellung von hydrophoben Oberflächen werden die zuvor gereinigten Deckgläschen mit einem
Silazan funktionalisiert.
Die gereinigten DG werden erhöht (z.B. auf zwei Objektträgern als Auflagefläche) in einer Glas-Petrischale gelagert. Anschließend werden mit Hilfe einer Spritze wenige Milliliter (2-4 ml) HMDS (Hexamethyldisilazan) auf den Grund der Petrischale gegeben und anschließend die Petrischale verschlossen.
Nach einer Inkubation der DG in dem Silazan über Nacht, werden diese mit Ethanol gespült und mit N2 getrocknet.
Fibronektinadhäsion auf hydrophilen/hydrophoben Oberflächen
Um das unterschiedliche Adhäsionsverhalten von Fibronektin auf Oberflächen mit verschiedener Hydrophobizität zu untersuchen, wird das FRET-gelabelte Fibronektin auf die hergestellten DG aufgebracht.
Die DG (bzw. die silazanierten DG) werden zur Aufnahme am LSM in eine Teflonhalterung gebracht und mit ca. 1-2 ml PBS benetzt.
Anschließend werden ca. 100 µl des gelabelten Fibronektins hinzugegeben und für 5 min inkubiert, bevor die Messung stattfinden kann.
Bleaching-Experiment
Die Konformation von Fibronektin ändert sich bei der Adhäsion auf Oberflächen je nach Hydrophobizität der Grenzfläche. Demnach sollte bei hydrophilen und hydrophoben Oberflächen ein unterschiedlich starker FRET erkennbar sein.
Nach der Einstellung des LSM auf die beiden Farbstoffe (Alexa Fluor 488 und AF 546) wird zunächst ein Bild der Fibronektin-funktionalisierten Oberfläche bei beiden Wellenlängen aufgenommen.
Anschließend wird der Kanal des FRET-Donors (AF 488) ausgeschaltet und die Laserintensität des FRET-Akzeptors (AF 546) auf 100% gesetzt.
Es erfolgt eine Bildserien-Aufnahme (100 Wiederholungen mit 1 sec Abstand) von einem festgelegten Bereich des Bildes.
Nach dem der FRET-Akzeptor nun geblichen wurde, erfolgt eine Bildaufnahme beider Farbstoffe mit den Parametern des ersten Bildes.
15

iii. Auswertung
Berechnung des Verhältnis der Fluoreszenzintensitäten 𝐼A 𝐼D⁄ für alle Ansätze
Auftragung des von 𝐼A 𝐼D⁄ gegen die Konzentration an denaturierender Substanz
Wie ist der Verlauf der Kurve zu erklären? Warum fällt 𝐼A 𝐼D⁄ nicht bis auf 0 ab?
Wie sieht die Konformation auf hydrophilen bzw. hydrophoben Oberflächen aus?
iv. Literaturhinweise
(1) Klotzsch, E., Smith, M. L., Kubow, K. E., Muntwyler, S., Little, W. C., Beyeler, F., Gourdon, D., Nelson, B. J. and Vogel, V. (2009). Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites, Proc. Natl. Acad. Sci. U. S. A., 106, pp. 18267-18272.
(2) Wierzbicka-Patynowski, I. and Schwarzbauer, J. E. (2003). The ins and outs of fibronectin matrix assembly, J. Cell. Sci., 116, pp. 3269-3276.
16

MS I
17

Universität Leipzig
Fakultät für Lebenswissenschaften Institut für Biochemie
AK Prof. Annette Beck-Sickinger Neubau Sonderlabore, 04103 Leipzig, Stephanstrasse 24 II. Etage
Praktikum Strukturanalytik Station MS I
betreut durch: Regina Reppich-Sacher; Dr. Jan Stichel
Tel.: 0341 9738531; 9736797
Mail: [email protected]; [email protected]
Massenspektrometrie
Die Massenspektrometrie ist eine analytische Methode zur Bestimmung der Molekülmasse von
Biomolekülen (z.B. Peptiden und Proteinen).
Das Massenspektrometer selbst besteht aus einer Ionenquelle, zur Erzeugung gasförmiger
Ionen, einem Massenanalysator, der die Ionen hinsichtlich ihres Masse/Ladungs-Quotienten
(m/z) auftrennt und einem Detektor, der das Massenspektrum liefert, aus dem abgelesen werden
kann, welche Ionen in welchen relativen Mengen gebildet worden sind.
Abb. 1 Allg. Prinzip eines Massenspektrometers
18

ESI-Massenspektrometrie
Die Electro Spray Ionisation (ESI)-Massenspektrometrie ist ein bevorzugtes
Ionisationverfahren zur Analyse von Biomolekülen, da es sehr schonend für das Analytmolekül
ist und kaum zu Fragmentationen führt. Bei der Elektrosprayionisation wird eine Analytlösung
durch eine Metallkapillare geleitet, an deren Spitze eine Spannung angelegt ist. Durch die
Spannung kommt es zur Bildung eines elektrischen Feldes zwischen der Kapillare und einer
Gegenelektrode. Das elektrische Feld durchdringt die Analytlösung und in ihr befindliche Ionen
bewegen sich elektrophoretisch auf die Gegenelektrode zu. Dabei bildet sich an der Spitze der
Kapillare ein Überschuss gleichartig geladener Ionen, die sich gegenseitig abstoßen und über
die Bildung eines Taylor-Kegels (Taylor-Cone) als feines Aerosol (etwa 10 µm Tropfengröße)
aus der Kapillare austreten. Ein neutrales Trägergas wie Stickstoff wird häufig benutzt um die
Vernebelung der Lösung und die Verdampfung des Lösungsmittels zu unterstützen. Aufgrund
der Verdampfung des Lösungsmittels verkleinert sich die Tropfengröße, während die Dichte
des elektrischen Feldes auf der Tropfen-Oberfläche zunimmt. Wenn der Radius der Tropfen
kleiner als das sogenannte Rayleigh-Limit wird, zerfallen die Tropfen aufgrund der Abstoßung
von gleichartigen Ladungen (Coulomb-Explosionen) in kleinere Tröpfchen.
IonSource Iontrap Spektrum
Abb. 2 Prinzip der ESI-MS
Für die Bildung freier Ionen in der Gasphase existieren mehrere Modellvorstellungen. Das
Charge Residue Model (CRM, Modell des geladenen Rückstands) geht davon aus, dass letztlich
winzige Tropfen von etwa 1 nm Durchmesser übrigbleiben, die nur ein ionisiertes
Analytmolekül enthalten. Beim Ion Evaporation Model (IEM, Ionenemissionsmodell) wird
angenommen, dass bereits aus größeren geladenen Tropfen freie Ionen in die Gasphase emittiert
formation multiply charged ions
19
werden. Die erzeugten Ionen werden durch die Potentialdifferenz zwischen Sprayerkapillare
und Orifice in das MS gelenkt.
Die Art der Spannung, die an der Kapillare angelegt wird, bestimmt die Ladung der Ionen, die
erzeugt werden. Durch eine positive Spannung werden positive geladene Ionen erzeugt und
durch eine negative Spannung negativ geladene Ionen. Bei der Elektrospray-Ionisierung
handelt es sich um eine sanfte Methode der Ionenerzeugung, bei der auch empfindliche
Moleküle und nicht kovalente Aggregate ionisiert werden können.
Typischerweise werden Quasimolekül-Ionen detektiert z.B. [M+H] + bei positiver Spannung.
Ein charakteristisches Phänomen der ESI-MS ist dabei die Bildung mehrfach geladener Ionen.
Signale in der Esi-MS
28
445.1
515.2
556.0
741.0
+MS, 0.2-6.4min #(5-148), Baseline subtracted(0.80)
0
1
2
3
4
5
8x10
Intens.
350 400 450 500 550 600 650 700 750 m/z
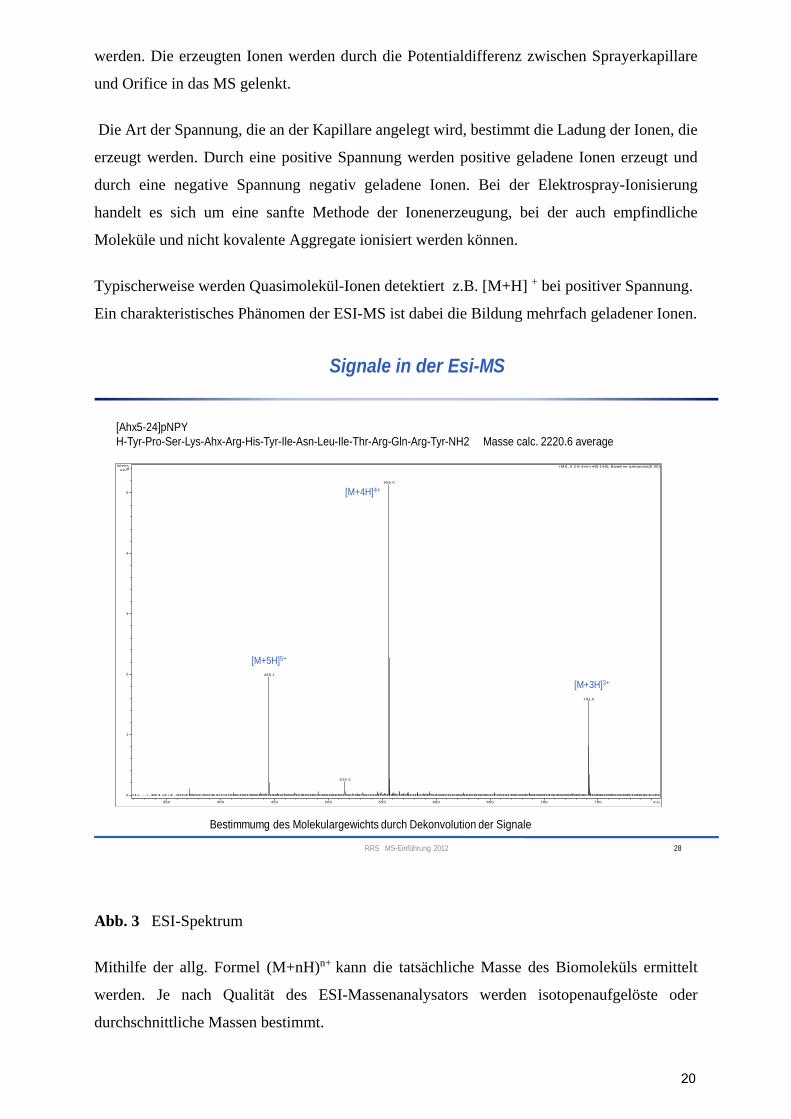
[Ahx5-24]pNPY
H-Tyr-Pro-Ser-Lys-Ahx-Arg-His-Tyr-Ile-Asn-Leu-Ile-Thr-Arg-Gln-Arg-Tyr-NH2 Masse calc. 2220.6 average
[M+5H]5+
[M+4H]4+
[M+3H]3+
RRS MS-Einführung 2012
Bestimmumg des Molekulargewichts durch Dekonvolution der Signale
Abb. 3 ESI-Spektrum
Mithilfe der allg. Formel (M+nH)n+ kann die tatsächliche Masse des Biomoleküls ermittelt
werden. Je nach Qualität des ESI-Massenanalysators werden isotopenaufgelöste oder
durchschnittliche Massen bestimmt.
20
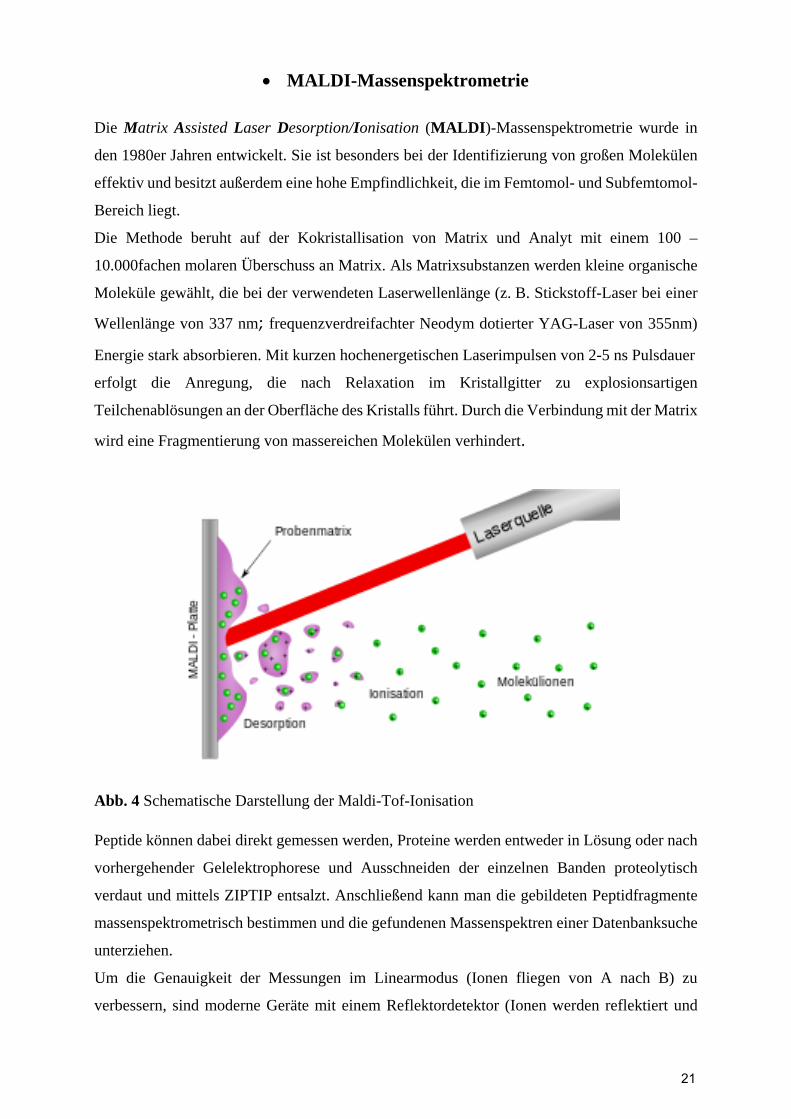
MALDI-Massenspektrometrie
Die Matrix Assisted Laser Desorption/Ionisation (MALDI)-Massenspektrometrie wurde in
den 1980er Jahren entwickelt. Sie ist besonders bei der Identifizierung von großen Molekülen
effektiv und besitzt außerdem eine hohe Empfindlichkeit, die im Femtomol- und Subfemtomol-
Bereich liegt.
Die Methode beruht auf der Kokristallisation von Matrix und Analyt mit einem 100 –
10.000fachen molaren Überschuss an Matrix. Als Matrixsubstanzen werden kleine organische
Moleküle gewählt, die bei der verwendeten Laserwellenlänge (z. B. Stickstoff-Laser bei einer
Wellenlänge von 337 nm; frequenzverdreifachter Neodym dotierter YAG-Laser von 355nm)
Energie stark absorbieren. Mit kurzen hochenergetischen Laserimpulsen von 2-5 ns Pulsdauer
erfolgt die Anregung, die nach Relaxation im Kristallgitter zu explosionsartigen
Teilchenablösungen an der Oberfläche des Kristalls führt. Durch die Verbindung mit der Matrix
wird eine Fragmentierung von massereichen Molekülen verhindert.
Abb. 4 Schematische Darstellung der Maldi-Tof-Ionisation Peptide können dabei direkt gemessen werden, Proteine werden entweder in Lösung oder nach
vorhergehender Gelelektrophorese und Ausschneiden der einzelnen Banden proteolytisch
verdaut und mittels ZIPTIP entsalzt. Anschließend kann man die gebildeten Peptidfragmente
massenspektrometrisch bestimmen und die gefundenen Massenspektren einer Datenbanksuche
unterziehen.
Um die Genauigkeit der Messungen im Linearmodus (Ionen fliegen von A nach B) zu
verbessern, sind moderne Geräte mit einem Reflektordetektor (Ionen werden reflektiert und
21
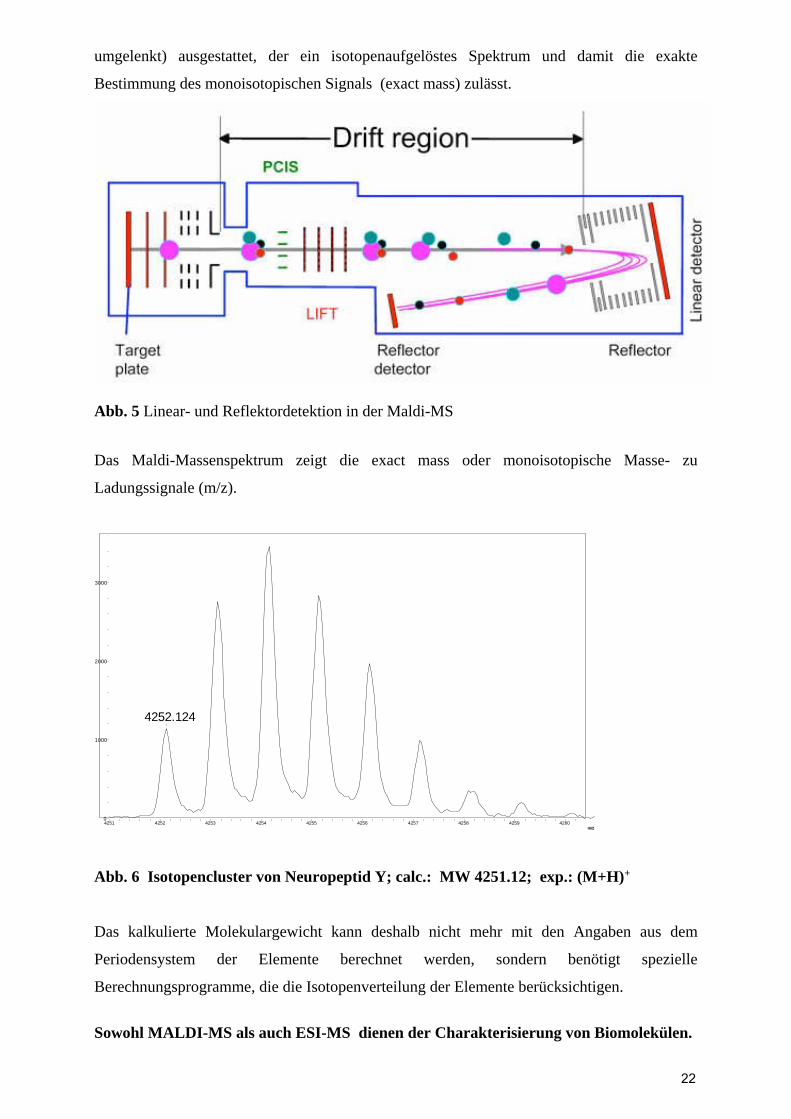
umgelenkt) ausgestattet, der ein isotopenaufgelöstes Spektrum und damit die exakte
Bestimmung des monoisotopischen Signals (exact mass) zulässt.
Abb. 5 Linear- und Reflektordetektion in der Maldi-MS Das Maldi-Massenspektrum zeigt die exact mass oder monoisotopische Masse- zu
Ladungssignale (m/z).
4252.124
0
1000
2000
3000
4251 4252 4253 4254 4255 4256 4257 4258 4259 4260
Abb. 6 Isotopencluster von Neuropeptid Y; calc.: MW 4251.12; exp.: (M+H)+
Das kalkulierte Molekulargewicht kann deshalb nicht mehr mit den Angaben aus dem
Periodensystem der Elemente berechnet werden, sondern benötigt spezielle
Berechnungsprogramme, die die Isotopenverteilung der Elemente berücksichtigen.
Sowohl MALDI-MS als auch ESI-MS dienen der Charakterisierung von Biomolekülen.
22

Massenspektrometrie
Praktischer Teil
Identifizieren von Peptiden und Proteinen
- Teil Ia Messen am BRUKER-HCT-Gerät (Esi-MS) -
Abb.: I
1. Präparation:
a) Verdünnen Sie die beiden ausliegenden Peptidlösungen mit einem Acetonitril-Wasserbidest.-Ameisensäure-Gemisch bis zu einer Endkonzentration von ca 25 µM. Es werden etwa 50µl benötigt.
2. Messung:
a) Nachdem der Betreuer das Gerät gestartet und in den Meßbetrieb überführt hat, können Sie nun eine ausgewählte Probe über die Spritzenpumpe injizieren.
b) Nach Optimierung des Sprays und Wahl des Speicherortes für das aufgenommene
Spektrum, starten Sie die Aufnahme.
c) Nach 2-3 min kann die Messung beendet werden; Sie füllen die Spritze mit Spüllösung und spülen die Ionenfalle ausreichend.
d) Wenn das Spektrum frei von den Signalen Ihrer dosierten Probe ist, kann die nächste Probe injiziert werden.
* Alle Proben müssen frei von Trifluoressigsäure sein oder umgesalzen werden.
(200
23
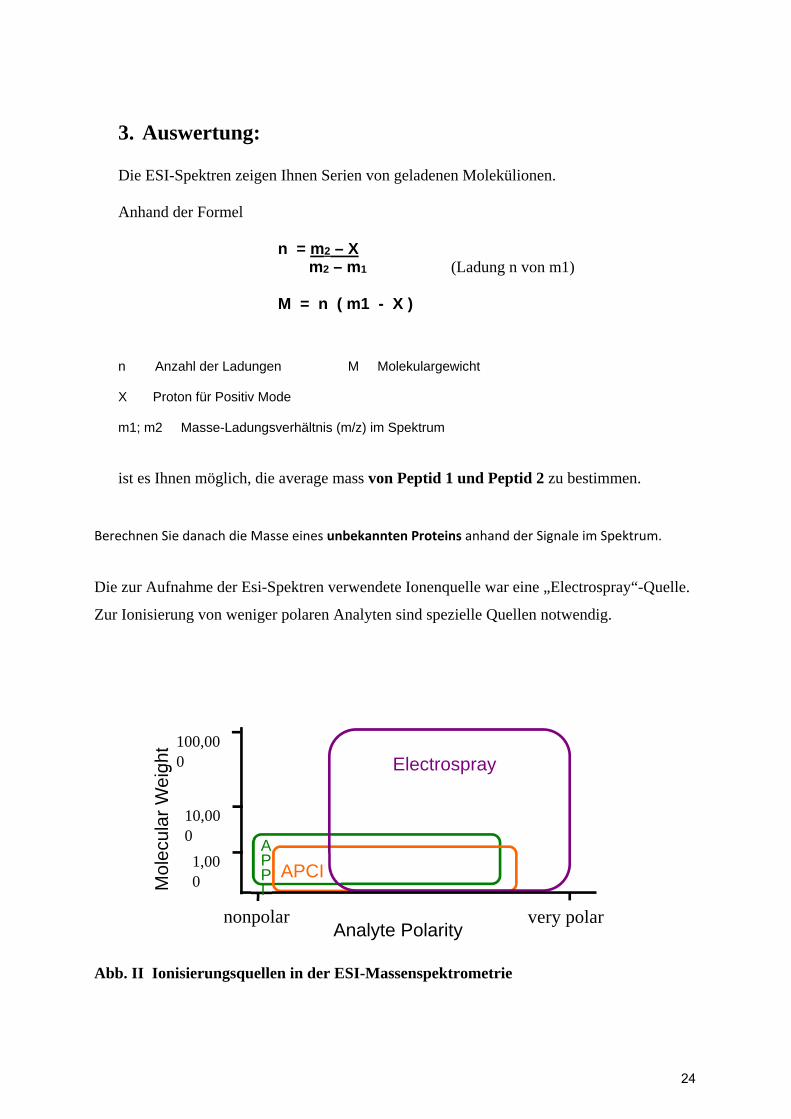
3. Auswertung:
Die ESI-Spektren zeigen Ihnen Serien von geladenen Molekülionen. Anhand der Formel n = m2 – X m2 – m1 (Ladung n von m1) M = n ( m1 - X ) n Anzahl der Ladungen M Molekulargewicht X Proton für Positiv Mode m1; m2 Masse-Ladungsverhältnis (m/z) im Spektrum
ist es Ihnen möglich, die average mass von Peptid 1 und Peptid 2 zu bestimmen.
Berechnen Sie danach die Masse eines unbekannten Proteins anhand der Signale im Spektrum.
Die zur Aufnahme der Esi-Spektren verwendete Ionenquelle war eine „Electrospray“-Quelle.
Zur Ionisierung von weniger polaren Analyten sind spezielle Quellen notwendig.
Abb. II Ionisierungsquellen in der ESI-Massenspektrometrie
Mo
lecu
lar
We
igh
t
Analyte Polarity very polar nonpolar
100,000
10,000
1,000
APCI
Electrospray
APPI
24

- Teil Ib Messen am Thermo Fisher-ORBITRAP ELITE-Gerät (Esi-MS) -
Abb. III Orbitrap-Massenspektrometer
1. Präparation:
Verdünnen Sie die ausliegende pNPY-Lösung mit einem Acetonitril-Wasserbidest.-Ameisensäure-Gemisch bis zu einer Endkonzentration von ca 0,25 µM. Es werden etwa 50µl benötigt.
2. Messung & Auswertung:
Nachdem der Betreuer die Software gestartet und in den Meßbetrieb überführt hat, können Sie nun die ausgewählte Probe über die Spritzenpumpe injizieren. Die in der Ionenquelle gebildeten Ionen passieren zunächst die normale Ionenfalle, werden in der C-Trap gebündelt und umgelenkt und gelangen in die Orbitrap. Dort bewegen sie sich in einem elektrischen Feld um eine spindelförmige Elektrode (Orbitrap) und aus der Frequenz mit der sie sich bewegen, kann das Masse zu Ladungsverhältnis nunmehr exact bestimmt werden. Die Prozessierungssoftware Thermo Xcalibur liefert nun die exact mass des gemessenen Peptids, exact mass calc. 4151.12.
25

- Teil IIa Messen am BRUKER-Microflex (Maldi-MS) -
Abb.: IV
1. Präparation:
a) Stellen Sie eine Matrixlösung aus alpha-Cyano-4-hydroxyzimtsäure nach ausliegender Vorschrift her.
b) Tragen Sie 3 Peptide (A, B, C) in geeigneter Verdünnung (ca.25µM) sowie die
Calibrierstandards (gelbes Label) mit frisch hergestellter Matrixlösung mittels Dried-Droplet-Verfahren (je 0.5µl) auf ein Edelstahltarget (klein) auf.
Alle Positionen der Substanzen auf den Platten sind zu notieren. Die Lösung zum Verdünnen ist 0.1%ige Trifluoressigsäure (TFA).
2. Messung:
Nach ausreichender Trocknung können Sie nun am Maldi-Tof-Gerät MICROFLEX die Peptide unter Anleitung mit einer geeigneten Meßmethode messen. Achten sie darauf, daß die monoisotopische Masse nur dann exakt bestimmt werden kann, wenn das Signal isotopenaufgelöst gemessen wurde.
3. Auswertung:
Ordnen Sie die gefundenen m/z-Werte in Dalton den folgenden Peptiden zu:
Tab. 1 Peptide A, B, C
Probenbezeichnung Masse calc. exact Masse calc. average
cj 2 1440.75 1441.67 cl 8 2219.24 2220.63
lH 1 2068.33 2069.60
26

- Teil IIb Messen am BRUKER-Ultraflex (Maldi-MS) –
Abb.: V
1. Präparation: Ein Protein wurde proteolytisch verdaut und anschließend über ZipTip aufgereinigt.
Tragen Sie diese Lösung in Originalkonzentration neben einem Calibrierspot (gelbes Label) auf das Target für das Ultraflexgerät mittels Dried-Droplet-Verfahren (je 0.5µl) auf. Präparieren Sie einen weiteren Spot mit Analyt & Matrix für ein MS_MS-Peptid oh 23 in geeigneter Konzentration (ca 25µM).
2. Messung und Auswertung:
a) Messen Sie unter Anleitung den tryptischen Verdau. Transferieren Sie das Spektrum des tryptisch verdauten Proteins in das Programm BIOTOOLS und starten Sie eine Datenbanksuche. Sie erhalten ein Mascot search result. Versuchen Sie, das ursprünglich eingesetzte Protein zu bestimmen.
b) Nehmen Sie das Spektrum eines Peptides auf. Führen Sie eine MS/MS-Messung
durch. Nachdem Sie das MS/MS-Spektrum aus dem Programm FLEX ANALYSIS nach BIOTOOLS transferiert haben, können Sie nun gemeinsam mit Ihrem Betreuer die MS/MS-Fragmente labeln und entscheiden, ob es sich um die unten vorgeschlagene Sequenz handelt.
H-Trp-Ala-Leu-Thr-His-Glu-Arg-Asn-Glu-Arg-Asn-Ser-Thr-OH
27

MS II
28

Praktikum Biophysikalische Chemie für den Studiengang Biochemie
Versuch MS II:
Lipidanalytik mittels MALDI-TOF Massenspektrometrie
und Dünnschichtchromatographie - Effekt der Ionenunterdrückung
Universität Leipzig, Medizinische Fakultät Institut für Medizinische Physik und Biophysik
Härtelstr. 16/18, D-04107 Leipzig
29

Biophysikalische Chemie (WS 2016/2017) - MS II
2
Lipidanalytik mittels MALDI-TOF Massenspektrometrie und Dünnschichtchromatographie
Kathrin Engel, Yulia Popkova, Jenny Schröter, Rosmarie Süß, und Jürgen Schiller Medizinische Fakultät, Institut für Medizinische Physik und Biophysik,
Härtelstr. 16/18, D-04107 Leipzig Tel.: 0341-9715733; Fax: 0341-9715709
e-mail: [email protected]
A. Allgemeine Einführung
1. Was sind Lipide und Phospholipide? Lipide spielen neben Proteinen und Kohlenhydraten eine gewaltige Rolle in der gesamten Biologie [1]. Obwohl sie in der Vergangenheit häufig etwas stiefmütterlich behandelt wur-den, kann man heute als gesichert annehmen, daß Lipide u.a. bei vielen Erkrankungen eine sehr wichtige Rolle spielen (z.B. bei der Arteriosklerose) [2]. Generell werden unter Lipiden vergleichsweise unpolare Verbindungen verstanden, die z.B. durch organische Lösungsmittel (in der Regel Hexan oder Chloroform) aus biologischen Proben extrahiert werden können. Lipide unterscheiden sich also von den meisten anderen Verbindungen dadurch, daß sie hydrophob bzw. lipophil ("fettliebend") sind. Eine Über-sicht der natürlich vorkommenden Lipide zeigt Abb. 1:
Abb. 1: Übersicht über die natürlich vorkommenden Lipide. Unter Lipiden werden in dem Schema ganz generell hydrophobe Verbindungen verstan-den. Bitte beachten Sie die große Variationsbreite der Lipide.
30

Biophysikalische Chemie (WS 2016/2017) - MS II
3
Wir werden uns im folgenden ganz überwiegend für die sogenannten "Phospholipide" (PL) und hier wiederum für die Glycerophospholipide interessieren. Diese Verbindungen ma-chen zwar im Gegensatz zu den klassischen Speicherfetten, d.h. den Triacylglycerolen (TAG) und auch dem Cholesterol nur einen mengenmäßig sehr geringen Anteil der Lipide im Organismus aus, besitzen aber dennoch überragende Bedeutung. So sind sie aufgrund ihres amphiphilen Charakters (sie besitzen einen polaren und einen apolaren Anteil) z.B. wesentlich am Aufbau von biologischen (Zell)-Membranen beteiligt. Darüber hinaus spie-len Phospholipide oder davon abgeleitete Verbindungen eine große Rolle als "second mes-senger Moleküle" bei der Signaltransduktion. Die Strukturen der wichtigsten Glycerophospholipide sind in Abb. 2 dargestellt:
Abb. 2: Übersicht über die wichtigsten Klassen von Glycerophospholipiden.
Dabei unterscheidet man in der Regel zwischen neutralen (PE und PC) und sauren Phospholipiden, d.h. Verbindungen, die bei annähernd neutralem pH-Wert (das Blut des gesunden Menschen besitzt einen pH-Wert von ca. 7.4) eine negative Ladung besitzen (PA, PS, PG, PI). Die Lipidzusammensetzung schwankt je nach Zell- oder Gewebetyp ganz be-trächtlich. Jedoch kann davon ausgegangen werden, daß PC in den meisten biologischen Gewebe den größten Teil der PL ausmacht [1]. Der in den Phospholipiden enthaltene Fettsäurerest kann in weiten Grenzen variieren, wo-bei sowohl die Kettenlänge, d.h. die Zahl der C-Atome als auch der Gehalt an Doppelbin-dungen (Sättigungsgrad) stark schwanken. Da die Synthese der PL jedoch immer aus Acetyl-Coenzym A erfolgt, besitzen alle physiologisch relevanten Phospholipide immer Fettsäurereste mit einer geraden Anzahl von C-Atomen. Einen Überblick über die wich-tigsten Fettsäuren gibt Tab. 1:
31

Biophysikalische Chemie (WS 2016/2017) - MS II
4
Tab. 1: Übersicht über die wichtigsten, physiologisch relevanten Fettsäuren, die Position der Doppelbindungen, ihr Trivialname und Molekulargewicht.
Die vollständige und eindeutige Bezeichnung eines Phospholipids lautet z.B. 1-Stearoyl-2-linoleoyl-sn-phosphatidylcholin. Zur Abkürzung verwendet man hier in der Regel auch Be-zeichnungen wie "SLPC" oder "PC 18:0/18:2". Die verwendeten Zahlen geben hierbei die Zahl der C-Atome bzw. die Zahl der Doppelbindungen (hinter dem Doppelpunkt) an. Na-türlich vorkommende Phospholipide besitzen in 1-Position in der Regel immer einen voll-ständig gesättigten Fettsäurerest, während sich in 2-Position normalerweise eine ungesät-tigte Fettsäure befindet. Von dieser Faustregel wollen wir im folgenden stets ausgehen, da mittels Massenspektrometrie (in der Regel) lediglich eine Unterscheidung hinsichtlich des Molekulargewichtes, aber nicht der entsprechenden Position eines bestimmten Fettsäure-restes möglich ist (s. unten) [3].
2. Lipidanalytik Die bei Lipiden etablierten Prozeduren sind - im Vergleich etwa zu den Proteinen - weit weniger genau definiert bzw. standardisiert [4]. Dennoch lassen sich auch hier unterschiedliche Schritte eindeutig unterscheiden: 2.1. Extraktion der Lipide In der Regel werden die Lipide zunächst aus den entsprechenden Köperflüssigkeiten (z.B. Blutplasma) oder Geweben (z.B. Leber) durch Anwendung organischer Lösungsmittel ext-rahiert, d.h. von den polareren Verbindungen wie z.B. Proteinen oder Kohlenhydraten ab-getrennt. Dazu werden Gemische organischer Lösungsmittel wie z.B. Chloro-
32

Biophysikalische Chemie (WS 2016/2017) - MS II
5
form/Methanol oder Hexan/Isopropanol eingesetzt. Am weitesten verbreitet ist dabei das System CHCl3/CH3OH: Chloroform ist mit Wasser nicht mischbar, wodurch sich ein 2-Phasensystem bildet (Abb. 3): In der unteren, organischen Physe sammeln sich die Lipide, während in der oberen, wäßrigen Phase (+CH3OH) die polareren Moleküle wie auch ein Großteil der Ionen verbleiben.
Abb. 3: Schematische Darstellung der Extraktion von Lipiden aus biologischen Materialien. Bitte be-achten Sie: CHCl3 hat eine größere Dichte als Wasser und findet sich deshalb immer "unten"! Aufgrund seiner Polarität ist das Methanol überwiegend in der H2O-Phase.
2.2. Trennung der erhaltenen Lipide in die einzelnen Unterklassen Für die meisten analytischen Anwendungen ist es notwendig die in dem erhaltenen Lipid-Rohextrakt vorhandenen Lipide in die einzelnen Klassen aufzutrennen, d.h. den Extrakt zu "fraktionieren". Dazu werden in der Regel chromatographische Verfahren wie z.B. die Dünnschichtchromatographie (TLC, "Thin-layer chromatography") [5] oder die Säulenchromatographie [3] eingesetzt. Die optimale Trennmethodik zu finden setzt hier große Erfahrung voraus und hängt in entscheidendem Maße von der Art des Lipidgemi-sches ab. Da wir lediglich die TLC einsetzen werden, soll nur auf dieses Verfahren näher eingegangen werden: Zur TLC werden in aller Regel mit Kieselgel beschichtete Kunststoff,- Aluminium,- oder Glasplatten als stationäre Phase verwendet. Die zu analysierenden Lipide werden in einem möglichst leicht flüchtigen, organischen Lösungsmittel wie z.B. Chloroform oder Hexan gelöst und anschließend mit einer Mikroliterspritze auf einer vorher markierten Linie auf die TLC-Platte aufgebracht. Nach dem Verdampfen des Lösungsmittels wird die so vorbe-reitete Platte in eine mit "Laufmittel" gesättigte Kammer gebracht. Steht die Platte in Kontakt mit dem Laufmittel, so beginnt das Laufmittel aufgrund der Ka-pillarkräfte in der Kieselgelschicht zu wandern. Dabei werden die einzelnen Komponenten des zu trennenden Lipidgemisches entsprechend ihrer Affinität zur stationären Phase unterschiedlich stark vom Laufmittel "mitgenommen". Dies führt dazu, daß unpolare Lipide (z.B. Triacylglycerole) unter identischen Bedingungen wesentlich weiter wandern als die viel polareren Phosphatidylcholine. Hat die Laufmittelfront fast den oberen Rand der Platte erreicht, wird die Chromatographie abgebrochen. Nach dem Trocknen der Platte werden die (farblosen) Lipide durch Besprü-hen mit einem Farbstoff sichtbar gemacht. Wir wollen dazu den Farbstoff Primulin (Struk-turformel in Abb. 4) einsetzen. Dieser Farbstoff bindet nicht-kovalent an die unpolaren
33

Biophysikalische Chemie (WS 2016/2017) - MS II
6
Reste von Lipiden, die anschließend unter UV-Licht sichtbar gemacht werden können (λmax der Emission ~ 365 nm) [6].
Abb. 4: Strukturformel des Fluoreszenzfarbstoffes "Primulin"
Der besondere Vorteil dieses Farbstoffes liegt darin, daß er bei Verwendung eines geeig-neten Lösungsmittels wieder ohne Zerstörung des Lipids abgelöst werden kann. Somit kön-nen die einzelnen Lipidklassen nach ihrer Trennung mittels TLC unmittelbar für die Mas-senspektrometrie verwendet werden. Ein wichtiger Begriff bei der TLC ist der sogen. "Rf-Wert" ("ratio of fronts"), der zur Cha-rakterisierung der einzelnen Fraktionen verwendet wird. Darunter versteht man den Quo-tienten aus der zurückgelegten Strecke der Substanz und der Strecke des Laufmittels (vgl. Abb. 5):
Abb. 5: Illustration zur Be-rechnung des Rf-Wertes an-hand einer schematisierten TLC-Platte
2.3. Quantitative Bestimmung der Phospholipide Hat man die einzelnen PL-Klassen voneinander getrennt, so erfolgt in der Regel eine Kon-zentrationsbestimmung der PL in den einzelnen Fraktionen. Der klassische Weg ist hier die Bestimmung des Phosphatgehaltes, der über biochemische Techniken sehr präzise und sen-sitiv erfolgen kann [7]. Dazu wird das entsprechende PL zunächst durch oxidierende Säuren vollständig in Phosphat überführt und dieses anschließend quantitativ bestimmt. Dann gilt: 1 Phosphat = 1 Phospholipidmolekül. Natürlich ist auch die sogen. "densitometrische" Auswertung der TLC-Platten direkt mög-lich. Dabei wird die Platte "eingescant" und digitalisiert. Die Quantifizierung erfolgt dann über die Intensitäten der einzelnen "Flecke".
34

Biophysikalische Chemie (WS 2016/2017) - MS II
7
Mit den bislang genannten Verfahren lassen sich zwar die einzelnen PL-Klassen quantitativ bestimmen, allerdings ist eine Bestimmung der Fettsäurezusammensetzung nur unter sehr großen Mühen möglich. In diesem Zusammenhang hat sich die Etablierung geeigneter mas-senspektrometrischer Techniken als außerordentlich hilfreich erwiesen. Die Mas-senspektrometrie ist außerdem eine extrem sensitive Technik und ermöglicht es mit nur sehr geringen Materialmengen auszukommen [8].
3. Massenspektrometrische Lipidanalytik Die Massenspektrometrie (MS) ist ein vergleichsweise altes analytisches Verfahren [8] und wurde bereits gegen Anfang des 20. Jahrhunderts entwickelt. Bei der MS wird die Masse von Ionen bestimmt. Dabei lassen sich generell zwei Schritte, die eigentliche Erzeugung von Ionen in der sogen. Ionenquelle und ihre anschließende Trennung bzw. die Detektion voneinander unterscheiden. Die klassische Methode zur Generierung von Ionen ist der Be-schuß der (zuvor verdampften) Probe mit Elektronen (Electron Impact, EI) in der Gasphase (Elektronenstoßionisation). Allerdings ist diese Technik durch zwei Randbedingungen er-heblich limitiert, da (a) die Substanz ohne Zersetzung zu verdampfen sein muß und (b) die gebildeten Ionen eine genügende Stabilität aufweisen müssen, um den Detektor erreichen zu können. Dabei werden - wenn die notwendige Ionisierungsenergie überschritten wird - Elektronen aus dem Analytmolekül "herausgeschlagen" und es entstehen positiv geladene Radikalkati-onen, die über ein ungepaartes Elektron verfügen. Da bewegte elektrische Ladungen in ei-nem Magnetfeld abgelenkt werden, kann auf diese Weise das entsprechende Molekularge-wicht bestimmt werden. Eine schematisierte Darstellung eines "klassischen" Mas-senspektrometers zeigt Abb. 6:
Abb. 6: Typische Anordnung bei der "klassischen" MS mit Elektronenstoß-Ionisation und Detektion der gebildeten Ionen in einem Magnetfeld.
35

Biophysikalische Chemie (WS 2016/2017) - MS II
8
Diese Art der MS wird auch heute noch eingesetzt (insbesondere in Kombination mit der Gaschromatographie (GC)), da hier die Ionenausbeute im wesentlichen durch die Ionisie-rungsenergie bestimmter funktioneller Gruppen gegeben ist: Da z.B. die Ionisierungsener-gie von Fettsäuren durch die Carboxylgruppe bestimmt ist und diese in allen Fettsäuren identisch ist, können Fettsäuren mittels EI-MS quantitativ bestimmt werden. Dies ist ein wichtiger Unterschied zu Techniken wie z.B. ESI oder MALDI, da hier die Ionenausbeute mit der Azidität oder Basizität der einzelnen Verbindungen (in einem Gemsich) gewichtet ist. Die zugrunde liegenden physikalischen Gesetzmäßigkeiten lassen sich wie folgt wiederge-ben:
a) Elektrische Energie = Kinetische Energie:
z = Ladungszahl × Elementarladung
mUz2v
2vmUzEE 2
2
kinelek =⇒=⇒=
b) Kraft im Magnetfeld = Zentripetalkraft:
U2rB
zm
rBU2
mz
U2m
rzBm
Uz2m
rzB
:enGleichsetz
mrzBv
mrzBv
rvmvzBF
22
22
22
2
222
2
2222
2
=⇒=⇒
=⇒=
=⇒=⇒==
Bestimmt wird bei der MS also immer das Masse/Ladungs-(m/z)-Verhältnis. Geht man da-von aus, daß das Molekül nur eine einzige Ladung besitzt, dann entspricht dieses Verhältnis genau dem Molekulargewicht. Dabei ist es jedoch wichtig mit den Atommassen und nicht etwa mit den Isotopenmassen zu rechnen:
36

Biophysikalische Chemie (WS 2016/2017) - MS II
9
Tab. 2: Atomgewichte und mittlere Atomgewichte verschiedener, physio-logisch relevanter Elemente und ihrer verschiedenen Isotope
Mit dem genannten Verfahren sind jedoch nur Moleküle zu charakterisieren, die über eine ausreichende Flüchtigkeit verfügen und die ohne Zersetzung verdampft werden können. Dies machte die MS bis vor ca. 20 Jahren für die Analytik der meisten biologisch relevan-ten Moleküle ungeeignet. Erst Ende der 80er Jahre des 20. Jahrhunderts gelang es sogen. "Soft-Ionization"-Techniken zu etablieren, die diese Nachteile ausgleichen konnten. Von den beiden wichtigsten Verfahren, der "electrospray" (ESI) und der "MALDI"-Technik werden wir hier ausschließlich das MALDI-Verfahren zur Analytik von Lipiden einsetzen. Es soll noch darauf hingewiesen werden, daß sowohl die Erfindung von ESI wie auch von MALDI im Jahre 2002 mit dem Nobelpreis für Chemie geehrt wurde.
4. Was ist MALDI-TOF MS? MALDI-TOF MS ist die Abkürzung für "matrix-assisted laser desorption and ionization time-of-flight Massenspektrometrie" und wird heute - insbesondere für die Analytik von Proteinen - sehr häufig eingesetzt [9]. Das Verfahren nutzt einen Laser zur Verdampfung der Probe. Dabei werden vor allem Stickstoff-Laser eingesetzt, da sie relativ kostengünstig sind und im Betrieb recht reibungslos arbeiten. Diese Laser emittieren bei λ = 337 nm. Bei weitem nicht alle Proben absorbieren jedoch auch bei dieser Wellenlänge. Deshalb hat man
37

Biophysikalische Chemie (WS 2016/2017) - MS II
10
sich folgenden "Trick" einfallen lassen: Man setzt der zu untersuchenden Probe eine geeig-nete "Matrix" zu. Dies ist ein (in der Regel kleines, organisches) Molekül, das bei der an-gegebenen Wellenlänge die Energie des Lasers absorbiert. Dadurch wird die Probe "heiß" und verdampft. Analytmoleküle werden dabei "mitgerissen". Eine sehr vereinfachte Skizze des MALDI-Prozesses zeigt Abb. 7:
Abb. 7: Schematische Darstellung der Prozesse bei der MALDI-TOF MS (Ionen-er-zeugung und Detektion)
Durch Stöße zwischen der zu untersuchenden Substanz und der Matrix bzw. einem zuge-setzten Hilfsmedium (z.B. einer organischen Säure) bzw. von vorneherein enthaltenen Io-nen kommt es zur Bildung von Ionen, die zunächst in einem elektrischen Feld beschleunigt werden und anschließend in einem feldfreien Raum sich selbst überlassen werden. Dabei erfolgt eine Trennung gemäß des m/z-Verhältnisses: Leichte Ionen erreichen den Detektor schneller als schwerere und können auf diese Weise getrennt werden. Da die Massentren-nung sehr wesentlich von der freien Flugstrecke abhängig ist, erreicht man eine höhere Massenauflösung durch Verlängerung der Flugstrecke. Um dennoch möglichst kleine Ge-räte konstruieren zu können, wird eine spezieller "Umlenkspiegel" (Reflektor-Detektor) verwendet, der die Flugstrecke verlängert. In Abb. 8 ist das Positiv-Ionen MALDI-TOF-Massenspektrum von 1,2-Dipalmitoyl-phosphatidylcholin dargestellt, um einen Eindruck zu vermitteln wie Massenspektren von Lipiden aussehen und wie sie interpretiert werden müssen:
38

Biophysikalische Chemie (WS 2016/2017) - MS II
11
Abb. 8: Positiv-Ionen MALDI-TOF MS von DPPC (PC 2×16:0). Das Spektrum wurde mit einem µg Lipid und DHB als Matrix aufgenommen. Alle Peaks sind entsprechend des m/z-Verhältnisses gekennzeichnet. Die charakteristische Kopfgruppe von PC wie auch die Struktur von DHB sind ebenfalls gezeigt. "Th" ist die Abkürzung für "Thompson", die Ein-heit des m/z-Verhältnisses.
Das Molekulargewicht dieses Lipids im neutralen Zustand beträgt 733.6 g/mol. Man er-kennt 3 deutlich voneinander getrennte Peakgruppen, wobei das intensivste Signal jeweils bei m/z = 734.6, 756.6 bzw. 772.6 liegt. Dieses intensivste Signal entspricht der Verbin-dung, die ausschließlich die jeweils häufigsten Isotope enthält (1H, 12C, 14N, 16O, 23Na und 31P), während der Peak im Abstand von einem Dalton ein schwereres Isotop (wahrschein-lich 13C aufgrund seiner höchsten natürlichen Häufigkeit), der Peak im Abstand 2 Da zwei schwerere Isotope usw., enthält. Zur Vereinfachung sollen hier nur die jeweils intensivsten Peaks betrachtet werden. Diese Peaks entsprechen der Anlagerung unterschiedlicher Kationen, wodurch einfach geladene DPPC-Addukte mit charakteristischen Massendifferenzen im Vergleich zum neutralen DPPC entstehen. Die Anlagerung eines Protons entspricht einer Massendifferenz von 1 Da, die eines Natrium-Ions 23 Da und die Anlagerung eines Kalium-Ions schließlich 39 Da. Die Intensität der unterschiedlichen Addukte ist im wesentlichen durch den Kationen-Gehalt der verwendeten Lösungen bestimmt und somit gewissen Schwankungen unterworfen [10]. Der Peak bei m/z = 932.6 entspricht schließlich einem sogen. Cluster-Ion aus einem Mat-rix-Molekül (bei dem zwei Protonen gegen Natrium-Ionen ausgetauscht sind, d.h. MG = 198) und dem Protonaddukt des DPPC. Eine sehr ausführliche Arbeit zum gegenwärtigen Stand der Lipid- bzw. Phospholipidana-lytik mittels MALDI-TOF Massenspektrometrie findet sich in [11]. Ein vergleichbarer Re-view zur ESI-Problematik hingegen in [12].
39

Biophysikalische Chemie (WS 2016/2017) - MS II
12
B. Experimenteller Teil
Folgende Fragen sollen beantwortet werden: 1. Werden alle Lipide mit der gleichen MS-Empfindlichkeit
nachgewiesen? 2. Ist die Summe der Spektren der einzelnen Fraktionen gleich dem
Gemischspektrum? Bitte beachten Sie bei allen Arbeiten im Labor:
• Die zur Anwendung kommenden Chemikalien bzw. Lösungsmittel sind gesundheits-schädlich (dies gilt vor allem für Chloroform, das verdächtigt wird krebserregend zu sein!).
• Vermeiden Sie Berührungen mit ALLEN Chemikalien bzw. Lösungsmitteln. Dies gilt insbesondere für die Augen.
• Obwohl wir bei allen Experimenten nur mit sehr geringen Mengen an Chemikalien bzw. Lösungsmitteln arbeiten werden, sollten Sie darauf achten, sich den Lösungsmit-teldämpfen nur kurz auszusetzen. Das heißt: Bitte schließen Sie die entsprechenden Vorratsflaschen unmittelbar nach Entnahme wieder sorgfältig.
• Chloroform ist ein "aggressives" Lösungsmittel und darf NICHT in Kunststoffgefäße gefüllt werden! Aus dem gleichen Grund verbietet sich das Pipetieren mit Kunststoff-Pipetten ("Eppendorf-Pipetten"). Verwenden Sie ausschließlich Spritzen aus Glas (so-gen. "Hamilton"-Spritzen).
1. Lipidextraktion Untersucht werden sollen (a) eine Avocado, (b) ein Stück Rinderleber und c) ein Eigelb. Zur Entfernung polarer Bestandteile sowie zur Anreicherung der apolaren Bestandteile (Li-pide bzw. Phospholipide) ist dazu zunächst eine Extraktion mit organischen Lösungsmitteln erforderlich:
• Schneiden Sie die Avocado bzw. die Leber in möglichst kleine Stücke, das Eigelb wird homogenisiert.
• Wägen Sie jeweils ungefähr 0.5 g in ein Reagenzglas mit Schliff ein.
• Geben Sie 1 ml destilliertes Wasser, 1 ml Chloroform und 1 ml Methanol zu (Extrak-tion gemäß "Bligh & Dyer") [13].
• Vortexen Sie die Proben gründlich (ca. 1 min) lang.
40

Biophysikalische Chemie (WS 2016/2017) - MS II
13
• Füllen Sie die Probe in ein Zentrifugenröhrchen um und zentrifugieren Sie die Probe (20°C, 2500 U/min, 8 min) um die Phasentrennung zu beschleunigen.
• Entnehmen Sie mit einer Hamilton-Spritze (vorsichtig!) die untere (CHCl3) Phase und überführen Sie diese in ein anderes Gefäß.
2. Trennung der Lipidextrakte mittels TLC
Die Trennung der drei Lipid-Gesamtextrakte in die einzelnen Lipidklassen erfolgt an 10×10 cm HPTLC-("high performance thin-layer chromatography")-Kieselgel 60 Platten. Als "Laufmittel" verwenden Sie ein Gemisch aus Chloroform, Ethanol, Wasser und Triethylamin (30:35:7:35 v/v/v/v).
• Tragen Sie jeweils ca. 5 µl der drei Lipidextrakte im Abstand von ca. 1 cm vom unteren Rand und im Abstand von ca. 1,2 cm voneinander auf die TLC-Platte auf (Bitte achten Sie darauf, daß Sie die Beschichtung der Platte mit der Spritze nicht beschädigen).
• Neben den drei Lipidextrakten tragen Sie je 1 µl Stammlösung (10 mg/ml) der folgen-den Lipidstandards auf: PC 16:0/18:1 (a), PE 16:0/18:1 (b), und Triacylglycerol (TAG) 3×18:1 (Triolein) (c).
• Legen Sie die präparierte Platte in die mit dem Laufmittel gefüllte Entwicklungs-kammer. Die Trennung der Proben ist nach ca. 45 Minuten beendet.
• Nehmen Sie die Platte aus der Entwicklungskammer und trocknen Sie die Platte mit dem Fön.
• Besprühen Sie die Platte mit Primulin-Lösung (Direct Yellow 59) in Aceton/H2O (80:20 (v/v), 50 mg/l). Trocknen Sie die Platte.
• Unter dem UV-Betrachter (366 nm) erkennen Sie an den Stellen, wo der Farbstoff gebunden hat, violette Flecke. Markieren Sie diese Stellen indem Sie die Flecke mit Bleistift umkreisen. Durch Vergleich mit den Standards identifizieren Sie die jeweiligen Fraktionen in den Gewebeproben.
• Kratzen Sie die Spots von PC und PE des Leber-und Eigelbextraktes mit einem Spatel vorsichtig von der Platte ab und überführen Sie das Kieselgel in ein kleines Glasgefäß.
• Eluieren Sie die Lipide vom Kieselgel mit einem Gemisch aus 65 µl CHCl3, 65 µl Methanol und 65 µl 0.9%-iger wässriger NaCl-Lösung.
• Zentrifugieren Sie die Proben kurz um die Phasentrennung zu beschleunigen (5 Minu-ten bei 3500 U/min).
• Alle erhaltenen Proben werden in der Vakuumzentrifuge "Speed-Vak" zur Trockne eingedampft und mit 5 µl CHCl3 wieder aufgenommen.
41

Biophysikalische Chemie (WS 2016/2017) - MS II
14
3. MALDI Präparation
• Als Matrix für die MALDI-TOF MS wird 2,5-Dihydroxybenzosäure (DHB) verwendet. Stellen Sie sich bitte eine 0.5 M (77.06 mg/ml) Lösung von DHB in Methanol (CH3OH) her.
• Mischen Sie in einem kleinen Glasröhrchen 5 µl der auch bei der TLC als Standard verwendeten Lipidlösungen (1 mg/ml) mit 10 µl der Matrix-Lösung und mischen Sie sorgfältig (Vortexer).
• Tragen Sie auch die drei Lipid-Gesamtextrakte sowie die nach der Trennung erhaltenen Fraktionen auf.
Probe Position auf Target
Standard PC (POPC)
Standard PE (POPE)
Standard Triolein
Gesamtextrakt Leber
Gesamtextrakt Avocado
Gesamtextrakt Eigelb
PE-Fraktion Leber
PC-Fraktion Leber
PE-Fraktion Eigelb
PC-Fraktion Eigelb
4. Durchführung der MALDI-TOF MS • Nehmen Sie zunächst die Spektren der Standardlipide (Detektionsmodus: Positiv Ionen)
auf. Vergleich Sie die Anzahl der erhaltenen Peaks der einzelnen Verbindungen. Was fällt Ihnen beim Vergleich von PE/PC bzw. TAG/PC auf?
• Messen Sie nun die Gesamtlipidextrakte der Avocado, der Leber sowie des Eigelbs. Können Sie einzelne Peaks zuordnen? Detektieren Sie PE und PC (in der Leber und im Eigelb) mit vergleichbarer Empfindlichkeit?
• Betrachten Sie schließlich die Spektren der isolierten Fraktionen des Leber- und des Ei-gelbextraktes. Geben Sie die Fettsäurezusammensetzung von PC und PE durch Ver-gleich mit den bereitgestellten Massentabellen an.
42
Biophysikalische Chemie (WS 2016/2017) - MS II
15
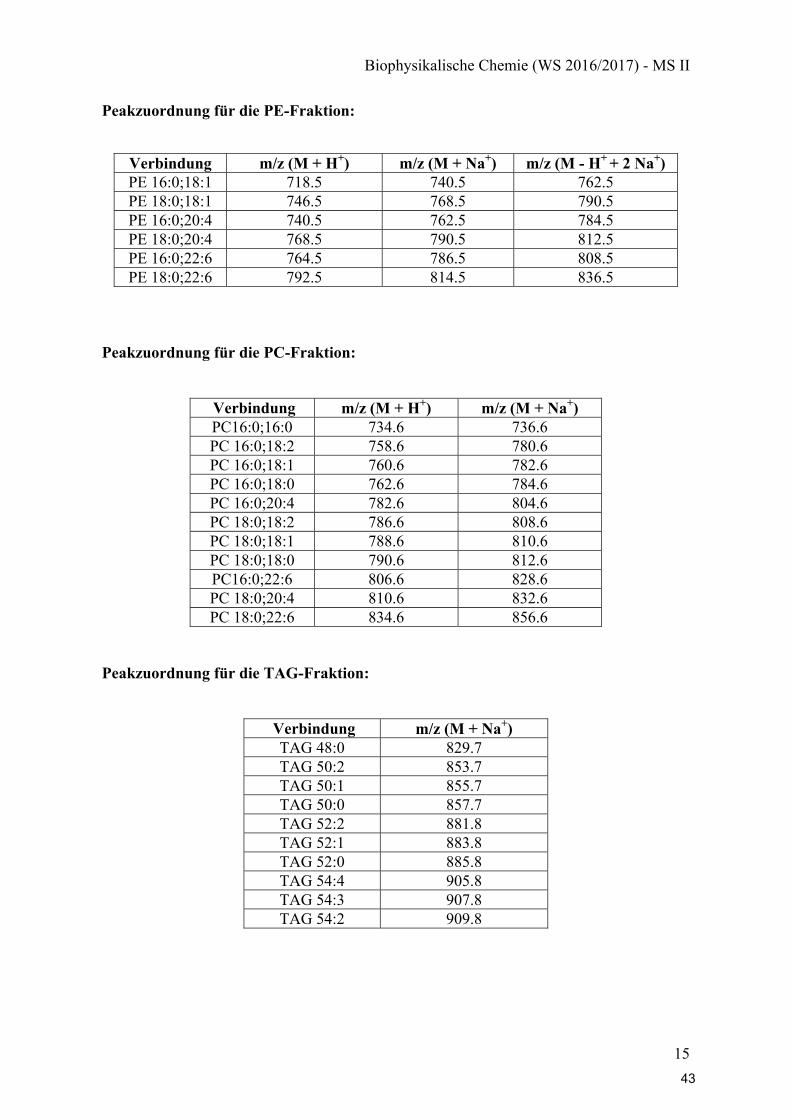
Peakzuordnung für die PE-Fraktion:
Verbindung m/z (M + H+) m/z (M + Na+) m/z (M - H+ + 2 Na+) PE 16:0;18:1 718.5 740.5 762.5 PE 18:0;18:1 746.5 768.5 790.5 PE 16:0;20:4 740.5 762.5 784.5 PE 18:0;20:4 768.5 790.5 812.5 PE 16:0;22:6 764.5 786.5 808.5 PE 18:0;22:6 792.5 814.5 836.5
Peakzuordnung für die PC-Fraktion:
Verbindung m/z (M + H+) m/z (M + Na+) PC16:0;16:0 734.6 736.6 PC 16:0;18:2 758.6 780.6 PC 16:0;18:1 760.6 782.6 PC 16:0;18:0 762.6 784.6 PC 16:0;20:4 782.6 804.6 PC 18:0;18:2 786.6 808.6 PC 18:0;18:1 788.6 810.6 PC 18:0;18:0 790.6 812.6 PC16:0;22:6 806.6 828.6 PC 18:0;20:4 810.6 832.6 PC 18:0;22:6 834.6 856.6
Peakzuordnung für die TAG-Fraktion:
Verbindung m/z (M + Na+) TAG 48:0 829.7 TAG 50:2 853.7 TAG 50:1 855.7 TAG 50:0 857.7 TAG 52:2 881.8 TAG 52:1 883.8 TAG 52:0 885.8 TAG 54:4 905.8 TAG 54:3 907.8 TAG 54:2 909.8
43

Biophysikalische Chemie (WS 2016/2017) - MS II
16
Literatur: [1] Stryer, L.: Biochemie, Vieweg, Braunschweig, 1987. [2] Libby, P.: Ateriosklerose als Entzündung. Spektrum der Wissenschaft. 2002, Heft 7,
48-59. [3] Lottspeich, F., Zorbas, H.: Bioanalytik, Spektrum Akademischer Verlag, Heidelberg,
1998. [4] www.cyberlipid.org [5] Touchstone; J.C.: Thin-layer chromatographic procedures for lipid separation. J.
Chromatogr. B 1995, 671: 169-195. [6] White, T., Bursten, S., Frederighi, D., Lewis, R.A., Nudelman, E.: High-resolution
separation and quantification of neutral lipid and phospholipid species in mammalian cells and sera by multi-one-dimensional thin-layer chromatography. Anal. Biochem. 1998, 10: 109–117.
[7] Chen, P. S., Toribara, T. Y., Wanner, H.: Microdetermination of phosphorus. Anal. Chem. 1956, 11: 1756–1758.
[8] Hesse, M., Meier, H., Zeeh, B.: Spektroskopische Methoden in der Organischen Chemie, Thieme, Stuttgart, 2005.
[9] Schiller, J., Arnold, K.: Mass spectrometry in structural biology. In: Encyclopedia of Analytical Chemistry (ed. by Meyers, R.A.), John Wiley & Sons Ltd., Chichester, 2000, 559-585.
[10] Schiller, J., Arnhold, J., Benard, S., Müller, M., Reichl, S., Arnold, K.: Lipid analysis by matrix-assisted-laser desorption and ionization mass spectrometry (MALDI-TOF-MS) - A methodological approach. Anal. Biochem. 1999, 267: 46-56.
[11] Schiller, J., Süß, R., Arnhold, J., Fuchs, B., Leßig, J., Müller, M., Petković, M., Zschörnig, O., Arnold, K.: Matrix-assisted laser desorption and ionization time-of-flight (MALDI-TOF) mass spectrometry in lipid and phospholipid research. Progr. Lipid Res. 2004, 43: 443-478.
[12] Pulfer, M., Murphy, R.C.: Electrospray mass spectrometry of phospholipids. Mass Spectrom. Rev. 2003, 22: 332-364.
[13] Bligh, E.G., Dyer, W.J.: A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 3: 911–917.
44

NMR I
45

Praktikumsskript NMR
Strukturaufklärung bzw. -bestätigung mittels NMR-Spektroskopie
Einleitung Strukturanalytik im Wandel der Zeit Ziel von Strukturaufklärung bzw. -bestätigung ist die Beschreibung einer Substanz mit Hilfe einer Formel, wobei 2 Arten von Formeln bedeutsam sind:
a) Summenformel Sie gibt die qualitative und quantitative Zusammensetzung einer Verbindung wider. b) Strukturformel Für die chemische Struktur spielt das Kriterium der Isomerie eine entscheidende Rolle, wobei man zwischen Konstitutions-, Konfigurations- und Konformations-isomerie unterscheidet.
Voraussetzung für Strukturuntersuchung: Reinheit der Substanz Reinigungsoperationen: u.a. Destillation, Umkristallisation, Sublimation Chromatographische Methoden (DC, GC, SC, HPLC)
Früher: Strukturaufklärung auf chemischem Wege Beurteilung der Substanz nach äußerer Erscheinung Vorproben (Bestimmung physikalische Konstanten) Nachweisreaktionen funktioneller Gruppen Identifizierung über Derivate Unabhängige Synthese
Heute: Strukturbestimmung mit physikalischen Methoden
mehr Informationen mit geringerem Zeitaufwand wesentliche Methoden sind u.a.: UV/Vis-Spektroskopie IR-Spektroskopie NMR-Spektroskopie Massenspektrometrie
46

Eine besondere Bedeutung kommt dabei heute der NMR-Spektroskopie und Massenspektrometrie zu.
Die NMR-Spektroskopie ist die einzige Methode zur Strukturbestimmung, mit deren Hilfe sowohl Aussagen zu Konstitution als auch Konfiguration und
Konformation gemacht werden können.
400 MHz NMR-Spektrometer der Firma Bruker (Magnet links, Konsole rechts) In der NMR-Spektroskopie benutzt man dazu für die Strukturaufklärung hochauflösende Fourier-Transform-NMR-Spektrometer entsprechender Konfiguration mit einem supraleitenden Magneten.
47

Praktikumsablauf
1) Jede Praktikumsgruppe wählt aus einem Angebot von Wirk- oder auch Naturstoffen eine Verbindung aus, von der zunächst Informationen zusammen getragen und dann die NMR-Spektren (mindestens 1H und 13C; meist dazu auch APT, H,H-COSY, HSQC und HMBC) im automatischen Routinebetrieb aufgenommen werden (Dauer mindestens 3 Stunden).
Angebotsliste von Wirkstoffen für den praktischen Teil:
Ibuprofen Thalidomid Coffein Theophyllin Sulfadiazin Sulfacetamid Trimethoprim Indometacin Atropinsulfat
Andererseits kann aber bei Bedarf auch aus einem breiten Angebot von Naturstoffen (Terpene, Steroide, Flavonoide etc.) ausgewählt werden.
2) Während des eigentlichen Messvorgangs erfolgt in der Zwischenzeit am
Beispiel eines außer Betrieb genommenen Gerätes die Erläuterung der apparativen Ausstattung mit Aufbau und Funktionsweise eines NMR-Spektrometers.
Supraleitender Magnet Probenkopf
48

Konsole (Electronics Cabinet) eines Varian-NMR-Spektrometers
3) Die NMR-Analytik organischer Verbindungen wird bevorzugt in Lösung ausgeführt. Es folgen daher Erläuterungen zur Probenvorbereitung (NMR-Röhrchen, notwendige Probenmenge, Wahl eines geeigneten Lösungsmittels etc.) und Hinweise auf die für die Messung notwendigen Vorarbeiten am Spektrometer (Tuning/Matching; Lock; Shim).
Tuning/Matching
Deuterium-Locksignal
49
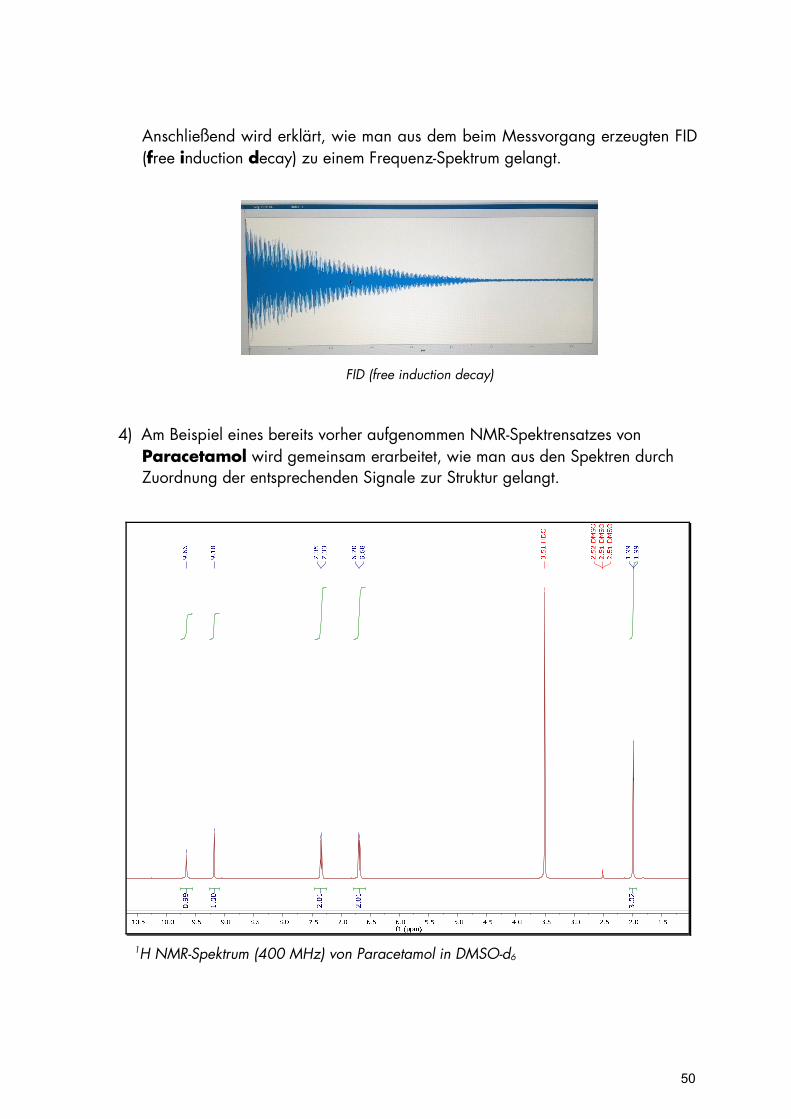
Anschließend wird erklärt, wie man aus dem beim Messvorgang erzeugten FID (free induction decay) zu einem Frequenz-Spektrum gelangt.
FID (free induction decay)
4) Am Beispiel eines bereits vorher aufgenommen NMR-Spektrensatzes von Paracetamol wird gemeinsam erarbeitet, wie man aus den Spektren durch Zuordnung der entsprechenden Signale zur Struktur gelangt.
1H NMR-Spektrum (400 MHz) von Paracetamol in DMSO-d6
50

Die Aufnahme des 1H NMR-Spektrums einer Verbindung bedarf des geringsten Aufwandes aller NMR-Messungen. Häufig dauern Probenvorbereitung und Vorbereitung der Messung länger als der eigentliche Messprozess selbst. Das erzeugte Spektrum liefert im Wesentlichen Informationen zu 3 Kriterien: die Lage der Signale (chemische Verschiebung δ)
Dabei erscheinen in Abhängigkeit von der chemischen Umgebung Protonen von ali-phatischen/alicyclischen Verbindungen in einem Bereich von 0 - 4 ppm, Protonen von Olefinen im Bereich von 4 - 6 ppm und Protonen von Aromaten im Bereich von 6 - 8 ppm. Aldehydprotonen werden typischerweise bei etwa 9 - 10.5 ppm beobachtet. In diesem Bereich registriert man häufig auch NH- und OH-Protonen, wobei das Auftret-en dieser Signale stark vom verwendeten Lösungsmittelabhängig ist, und sie infolge von H/D-Austausch oftmals auch verbreitert sind oder ganz verschwinden. XH-Proto-nen, die Bestandteil von starken Wasserstoffbrückenbindungen sind, können deshalb aber auch weit oberhalb der üblichen 10 ppm (bis ca. 20 ppm) registriert werden.
die Intensität der Signale Die über das entsprechende Signal integrierte Fläche ist dabei direkt proportional zur Zahl der gebundenen Protonen und somit wichtig für die Summenformel.
die Spin-Kopplung Sie gibt Auskunft über die Nachbarschaft von Protonen und damit die Bindungsver-hältnisse; man unterscheidet dabei bevorzugt Kopplungen über 2 (geminale) und über 3 (vicinale) Bindungen. In Abhängigkeit von der Struktur der Substanz beobachtet man manchmal aber auch Fernkopplungen über 4 und mehr Bindungen. Das Maß für die Kopplung ist die sogenannte Kopplungskonstante (J), deren Größe ebenfalls wichtige Strukturinformationen liefert.
Aufgrund der viel geringeren Empfindlichkeit des 13C Kerns gegenüber 1H benötigt man für die Messung von 13C NMR-Spektren mehr Substanz, und man nimmt diese bevorzugt auch als sogenannte protonenentkoppelte Spektren auf. Das bedeutet, man verzichtet bewusst auf zusätzliche Informationen über Kopplung und Signalintensität, erreicht aber dadurch schneller ein günstiges Signal-Rausch-Verhältnis. Vorteilhaft wirkt sich in diesem Zusammenhang auch aus, dass man damit für jedes unterschiedlich gebundene C-Atom nur ein Signal erhält. Außerdem erleichtert der viel größere Verschiebungsbereich (ca. 200 ppm gegenüber ca. 10 ppm im 1H) die Zuordnung.
51
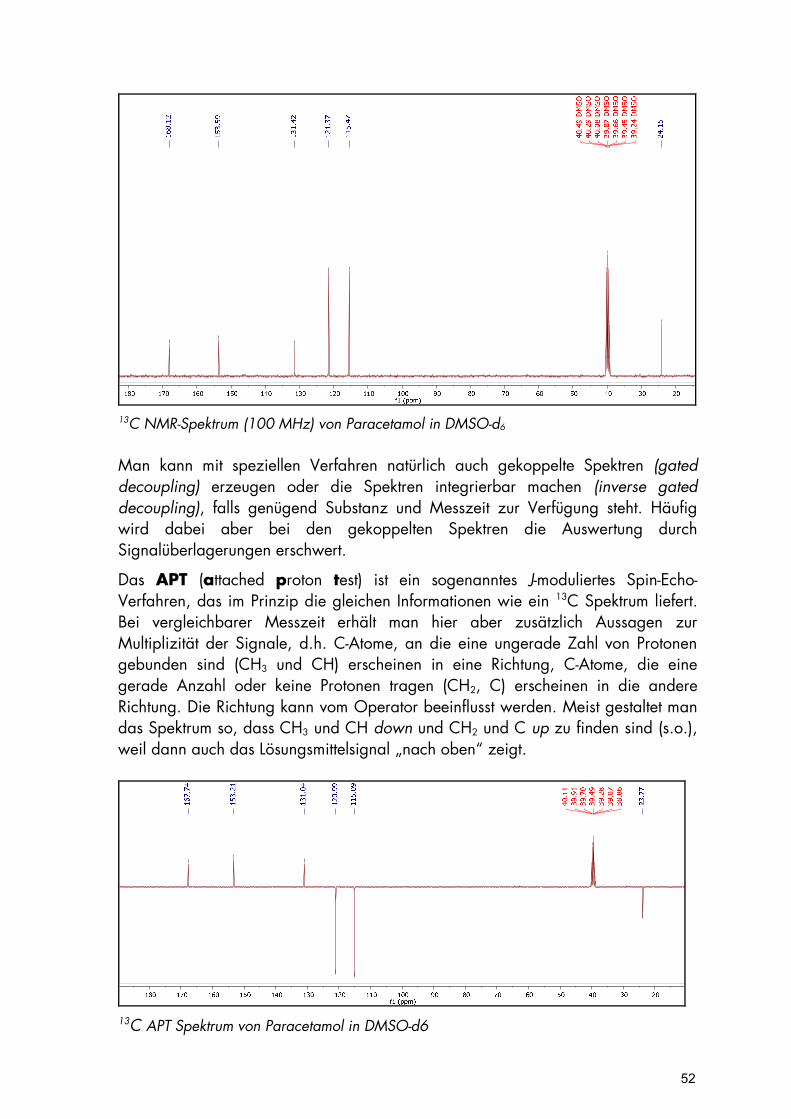
13C NMR-Spektrum (100 MHz) von Paracetamol in DMSO-d6 Man kann mit speziellen Verfahren natürlich auch gekoppelte Spektren (gated decoupling) erzeugen oder die Spektren integrierbar machen (inverse gated decoupling), falls genügend Substanz und Messzeit zur Verfügung steht. Häufig wird dabei aber bei den gekoppelten Spektren die Auswertung durch Signalüberlagerungen erschwert.
Das APT (attached proton test) ist ein sogenanntes J-moduliertes Spin-Echo-Verfahren, das im Prinzip die gleichen Informationen wie ein 13C Spektrum liefert. Bei vergleichbarer Messzeit erhält man hier aber zusätzlich Aussagen zur Multiplizität der Signale, d.h. C-Atome, an die eine ungerade Zahl von Protonen gebunden sind (CH3 und CH) erscheinen in eine Richtung, C-Atome, die eine gerade Anzahl oder keine Protonen tragen (CH2, C) erscheinen in die andere Richtung. Die Richtung kann vom Operator beeinflusst werden. Meist gestaltet man das Spektrum so, dass CH3 und CH down und CH2 und C up zu finden sind (s.o.), weil dann auch das Lösungsmittelsignal „nach oben“ zeigt.
13C APT Spektrum von Paracetamol in DMSO-d6
52
Schwierig ist die Unterscheidung allerdings bei Verwendung von D2O als Lösungsmittel, weil es hier aufgrund des Fehlens von C-Atomen im Spektrum gar kein Lösungsmittelsignal gibt. Erleichtert wird die Auswertung von APT Spektren aber noch dadurch, dass CH3-Gruppen im Vergleich zu CH stärker abgeschirmt werden, ebenso wie CH2 gegenüber C. Da das APT Spektrum von der direkten 13C-1H-Kopplung (Kopplungen über eine Bindung mit einer Kopplungskonstante von ca. 120 bis 160 Hz) abhängt, kann es aber bei der Auswertung des Spektrums bei mangelnder Erfahrung zu Fehlinterpretationen kommen: Ist die direkte 13C-1H-Kopplungskonstante, wie im Falle von Acetylenen, Aldehyden oder auch einer Vielzahl von Heterocyclen, wesentlich größer (ca. 180 bis 250 Hz), erscheinen die Signale mit einer „falschen“ Phase oder sie verschwinden gänzlich.
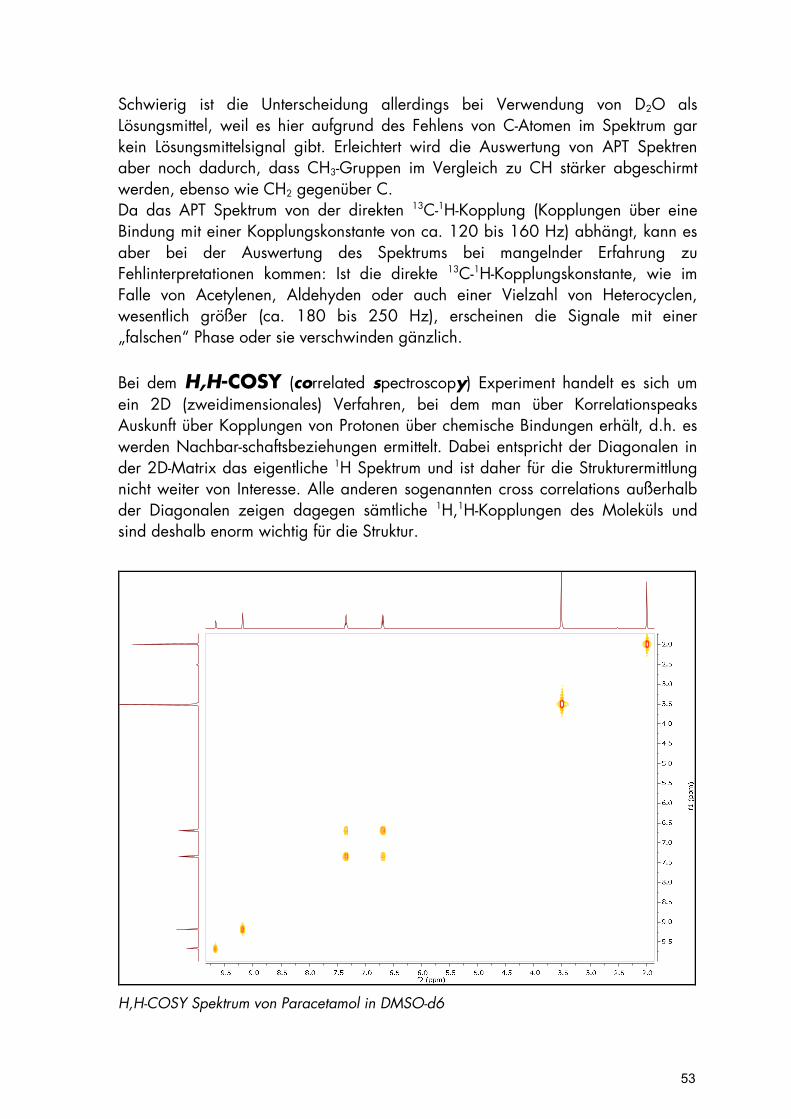
Bei dem H,H-COSY (correlated spectroscopy) Experiment handelt es sich um ein 2D (zweidimensionales) Verfahren, bei dem man über Korrelationspeaks Auskunft über Kopplungen von Protonen über chemische Bindungen erhält, d.h. es werden Nachbar-schaftsbeziehungen ermittelt. Dabei entspricht der Diagonalen in der 2D-Matrix das eigentliche 1H Spektrum und ist daher für die Strukturermittlung nicht weiter von Interesse. Alle anderen sogenannten cross correlations außerhalb der Diagonalen zeigen dagegen sämtliche 1H,1H-Kopplungen des Moleküls und sind deshalb enorm wichtig für die Struktur.
H,H-COSY Spektrum von Paracetamol in DMSO-d6
53

Vom COSY gibt es aufgrund seiner Bedeutung eine Vielzahl von Spielarten (u.a. COSY-45, COSY-90, long-range-COSY, DQ-COSY, TOCSY etc.) auf die hier nicht weiter eingegangen werden soll, die aber alle wichtige Informationen in Abhängigkeit von der Struktur der vermessenen Verbindung geben können. Außerdem kann man mit diesem Experiment auch andere Kerne (z.B. 19F, 31P) mit 1H oder auch untereinander korrelieren.
Erwähnt sei in diesem Zusammenhang aber noch das NOESY-Experiment, das Auskunft über dipolare Wechselwirkungen von Protonen „durch den Raum“ (und nicht über Bindungen) gibt und somit Abstandsmessungen von Protonen ermöglicht, die für die Ermittlung der Stereostruktur einer Substanz besonders bedeutsam sind.
Das HSQC- (heteronuclear single quantum coherence) Experiment ist auch ein 2D Verfahren und beruht auf der direkten Kopplung von C-Atomen mit Protonen über eine Bindung (C-H; mittlere Kopplungskonstante ca. 140 Hz). Dem Spektrum kann man deshalb Korrelationen entnehmen, die vom entsprechenden C-Atom zu den direkt daran gebundenen Protonen führen. In seiner editierten Form kann man auch noch zusätzlich die Multiplizität der C-Atome ermitteln. Das Experiment ist deshalb für die Strukturbestimmung von extremer Bedeutung. Findet man z.B. ausgehend von einem C-Signal Korrelation zu zwei verschiedenen Protonen, lässt das Rückschlüsse auf eine CH2-Gruppe mit diastereotopen Protonen in einer chiralen Umgebung zu, und man weiß dann sofort, dass es sich um eine optisch aktive Verbindung handelt.
Edited-HSQC Spektrum von Paracetamol in DMSO-d6
54
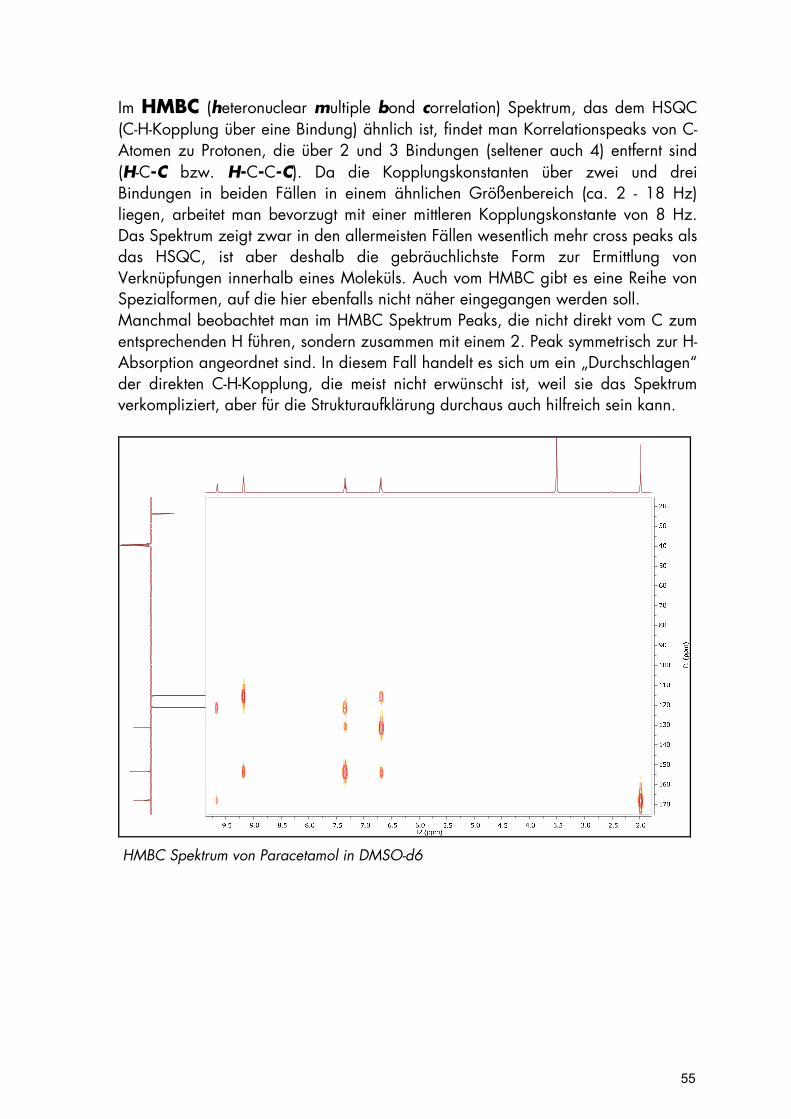
Im HMBC (heteronuclear multiple bond correlation) Spektrum, das dem HSQC (C-H-Kopplung über eine Bindung) ähnlich ist, findet man Korrelationspeaks von C-Atomen zu Protonen, die über 2 und 3 Bindungen (seltener auch 4) entfernt sind (H-C-C bzw. H-C-C-C). Da die Kopplungskonstanten über zwei und drei Bindungen in beiden Fällen in einem ähnlichen Größenbereich (ca. 2 - 18 Hz) liegen, arbeitet man bevorzugt mit einer mittleren Kopplungskonstante von 8 Hz. Das Spektrum zeigt zwar in den allermeisten Fällen wesentlich mehr cross peaks als das HSQC, ist aber deshalb die gebräuchlichste Form zur Ermittlung von Verknüpfungen innerhalb eines Moleküls. Auch vom HMBC gibt es eine Reihe von Spezialformen, auf die hier ebenfalls nicht näher eingegangen werden soll. Manchmal beobachtet man im HMBC Spektrum Peaks, die nicht direkt vom C zum entsprechenden H führen, sondern zusammen mit einem 2. Peak symmetrisch zur H-Absorption angeordnet sind. In diesem Fall handelt es sich um ein „Durchschlagen“ der direkten C-H-Kopplung, die meist nicht erwünscht ist, weil sie das Spektrum verkompliziert, aber für die Strukturaufklärung durchaus auch hilfreich sein kann.
HMBC Spektrum von Paracetamol in DMSO-d6
55

Struktur und Zuordnung der entsprechenden NMR-Signale von Paracetamol in DMSO-d6
56

5) Im Anschluss daran wird gezeigt, welche weiteren modernen Hilfsmittel heute bei der Strukturbestimmung in Anspruch genommen werden können, wie z.B. die Suche in Datenbanken (SciFinder; SDBS) und die vorherige Berechnung der Spektren mit Hilfe von Inkrementensystemen, sowie Modelling und Geometrie- bzw. Energieoptimierung der Strukturen mit der entsprechenden Software (ChemOffice).
SciFinder
What is SciFinder?
SciFinder® is a research discovery application that provides integrated access to the world's most comprehensive and authoritative source of references, substances and reactions in chemistry and related sciences.
Spectral Database for Organic Compounds, SDBS
https://sdbs.db.aist.go.jp
ChemOffice
Umfangreiches Software-Paket zur Darstellung chemischer Strukturen mit vielen Optionen (Spektrensimulation NMR, MS; Stoffdatenvorhersage…; 3D-Strukturen)
6) Nach Abschluss der Messungen werden die Spektren der jeweils gewählten Substanz bearbeitet und ausgedruckt. Die Spektren bilden dann die Grundlage für die vollständige Strukturzuordnung, die dann gegebenenfalls von interessierten Studenten in Heimarbeit vorgenommen werden kann.
57

NMR II
58

Praktikum Strukturanalytik – Bachelorstudiengang Biochemie Nuclear Magnetic Resonance II (NMR-Bildgebung)
Klinik und Poliklinik für Diagnostische und Interventionelle Radiologie
Universitätsklinikum Leipzig AöR, Liebigstr. 20 (Operatives Zentrum), 04109 Leipzig Grundlagen – Kernmagnetische Resonanz
– Kernspin und magnetisches Moment
– Magnetisches Moment im äußeren Magnetfeld: Larmorpräzession
– Kernspin ½: Wasserstoff
– Energieniveaus und Besetzungszahlen
– Spin-Ensemble: Longitudinal- und Transversalmagnetisierung
– Detektion des NMR-Signals
– Hochfrequenz (HF)-Anregung: 90°-Puls
– Transversale und longitudinale Relaxation
– Relaxationszeiten biologischer Gewebe: T1 und T2
– NMR-Experiment: Spin-Echo
– Experimentelle Parameter: Echozeit (TE) und Repetitionszeit (TR)
– Bildkontrast: T1-Wichtung, T2-Wichtung, Protonendichte-Wichtung
Praktischer Teil: Klinisches 1,5-Tesla Ganzkörper-System
– Sicherheitsaspekte
– HF-Kabine und Kontrollraum
– Systemaufbau
– Magnetfelder: Grundfeld, Anregung, Gradienten, Abschirmung, Homogenisierung (Shim)
– Sende- und Empfangsspulen
– Bedienkonsole und Bedienoberfläche
– MR-Protokolle und Pulssequenzen
– Spin-Echo-Sequenz und grundlegende Messparameter
– Ablauf einer MR-Untersuchung
• Patientenvorbereitung und Spulenanbringung
• Aufnahme der Bilddaten
• Betrachtung und Bewertung der MR-Bilder
– Besonderheiten der NMR-Bildgebung (MR-Tomographie)
– Anwendungsbeispiele
_____________________________________________________
Kontakt: [email protected] • (0341) 97-17413
59

IR
60

Strukturanalytik: Infrarot-Spektroskopie (IR)
1. Einführung
Diese Art der Molekülspektroskopie macht sich die Anregung von Energiezuständen
im Molekül zunutze, welche durch Bestrahlung mit infrarotem Licht im
Wellenlängenbereich von 800 nm bis 1 mm erreicht werden.
Diese Technik kann sowohl zur Strukturaufklärung unbekannter Substanzen, zum
Vergleich mit Referenzsubstanzen als auch zur quantitativen Analyse genutzt werden.
Voraussetzung für die IR-Aktivität einer Bindung ist ein ausgeprägtes Dipolmoment.
Durch die Bestrahlung mit infrarotem Licht werden Schwingungszustände im Molekül
angeregt die je nach Art von Atom und Bindung unterschiedliche Frequenzbereiche
absorbieren. Gemessen wird die Transmission (%) des Lichts in Korrelation zum
Frequenzbereich (Wellenzahl: cm-1). Die auftretenden mechanischen Schwingungen
lassen sich in 2 Arten unterteilen (siehe Abb.).
Jede Bindungs- und Schwingungsart besitzt charakteristische Absorptionsmuster.
Daraus resultiert für jedes Molekül ein einzigartiges Spektrum.
61

Tab.: Schwingungsdaten häufiger Molekülgruppen
Bindung Wellenzahlbereich Art der Schwingung
v 2850–3200 Valenzschwinung
δ 1400 Deformationsschwingung
1650 Valenzschwinung
2200-2500 Valenzschwinung
v 3200-3600 Valenzschwinung, bei H-
Brückenbindung breite Bande
v
Carboxy
2500-3000
1700
v 2200-2260 Valenzschwinung
v 3100-3500 Valenzschwinung
v <1500 Hal.: beliebiges Halogen
2. Qualitative Analyse
Jede Gruppe erhält 3 unbekannte Arznei-/Hilfsstoffe. Diese werden spektroskopisch
untersucht (Messanweisungen siehe 5.) und mit den Referenzspektren aus der
Datenbank (Ordner: Referenzspektren P5) verglichen und zugeordnet (siehe Tab.).
Nach der Identifikation suchen sie sich ein Spektrum aus und vergleichen es mit den
Spekten, welche sich auf der frei zugänglichen SDBS (Spectral Database for Organic
Compounds) finden lassen.
62
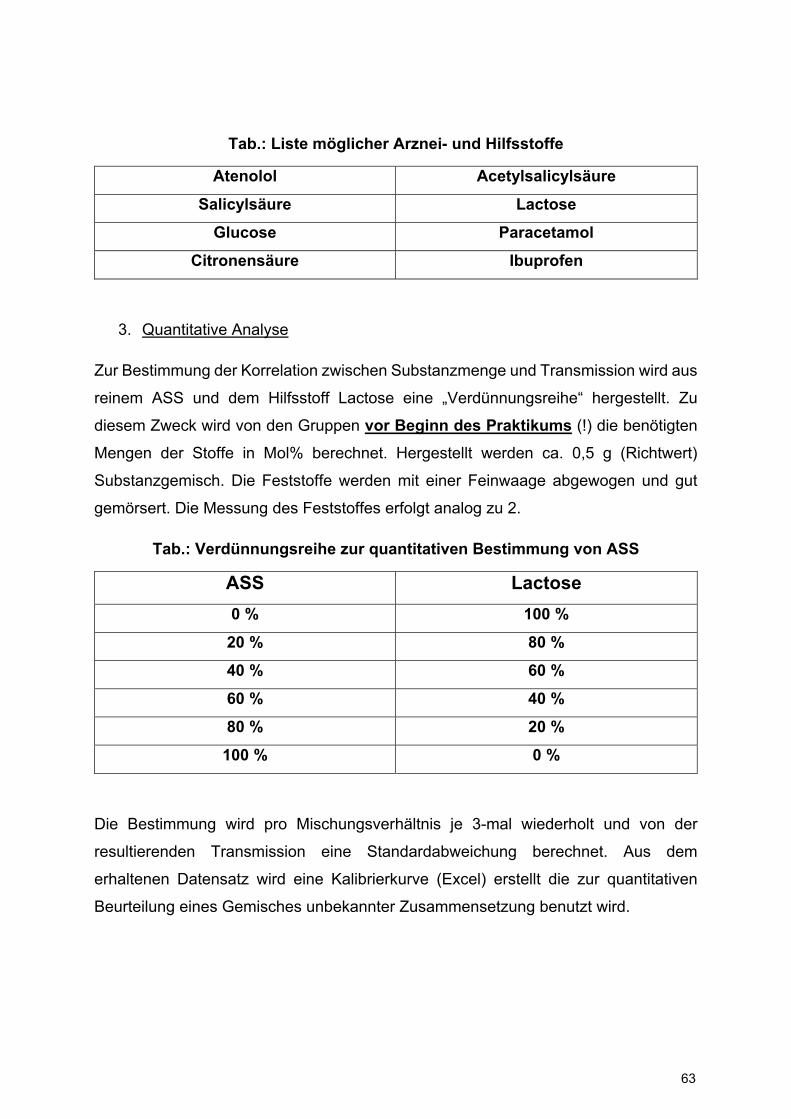
Tab.: Liste möglicher Arznei- und Hilfsstoffe
Atenolol Acetylsalicylsäure
Salicylsäure Lactose
Glucose Paracetamol
Citronensäure Ibuprofen
3. Quantitative Analyse
Zur Bestimmung der Korrelation zwischen Substanzmenge und Transmission wird aus
reinem ASS und dem Hilfsstoff Lactose eine „Verdünnungsreihe“ hergestellt. Zu
diesem Zweck wird von den Gruppen vor Beginn des Praktikums (!) die benötigten
Mengen der Stoffe in Mol% berechnet. Hergestellt werden ca. 0,5 g (Richtwert)
Substanzgemisch. Die Feststoffe werden mit einer Feinwaage abgewogen und gut
gemörsert. Die Messung des Feststoffes erfolgt analog zu 2.
Tab.: Verdünnungsreihe zur quantitativen Bestimmung von ASS
ASS Lactose
0 % 100 %
20 % 80 %
40 % 60 %
60 % 40 %
80 % 20 %
100 % 0 %
Die Bestimmung wird pro Mischungsverhältnis je 3-mal wiederholt und von der
resultierenden Transmission eine Standardabweichung berechnet. Aus dem
erhaltenen Datensatz wird eine Kalibrierkurve (Excel) erstellt die zur quantitativen
Beurteilung eines Gemisches unbekannter Zusammensetzung benutzt wird.
63

4. Kontrollfragen
Informieren sie sich über das Funktionsprinzip der ATR-Infrarotspektroskopie.
Inwiefern unterscheidet sie sich von konventionellen Methoden der IR?
Wo sehen sie die Grenzen dieser Messtechnik?
Mit welcher analytischen Methode kann man IR-inaktive Bindungen sichtbar machen?
Inwieweit lässt sich die Genauigkeit der Quantifizierung beurteilen? Nehmen sie dazu
ihre eigenen Daten aus 3. Zu Hilfe.
Was ist die sogenannte Fourier-Transformation (FT)? Welche Vorteile ergeben sich
aus dieser Technik?
5. Arbeitsanweisung
- Öffnen sie das Programm „Spektrum“ von PerkinElmer® (Einloggdaten sind
beim Assistenten zu erfragen)
- Bestücken sie die „Andrückvorrichtung“ des Messgerätes mit dem großen
Stempel, schieben diese über das Messfenster und drehen sie den Stempel
vorsichtig nach unten (Vorsicht: Andruckmesser sollte nie 150
überschreiten!!!)
- Drücken sie im Programmfenster auf „Untergrund“ und warten sie bis die
Messung (siehe Ladebalken) abgeschlossen ist
64
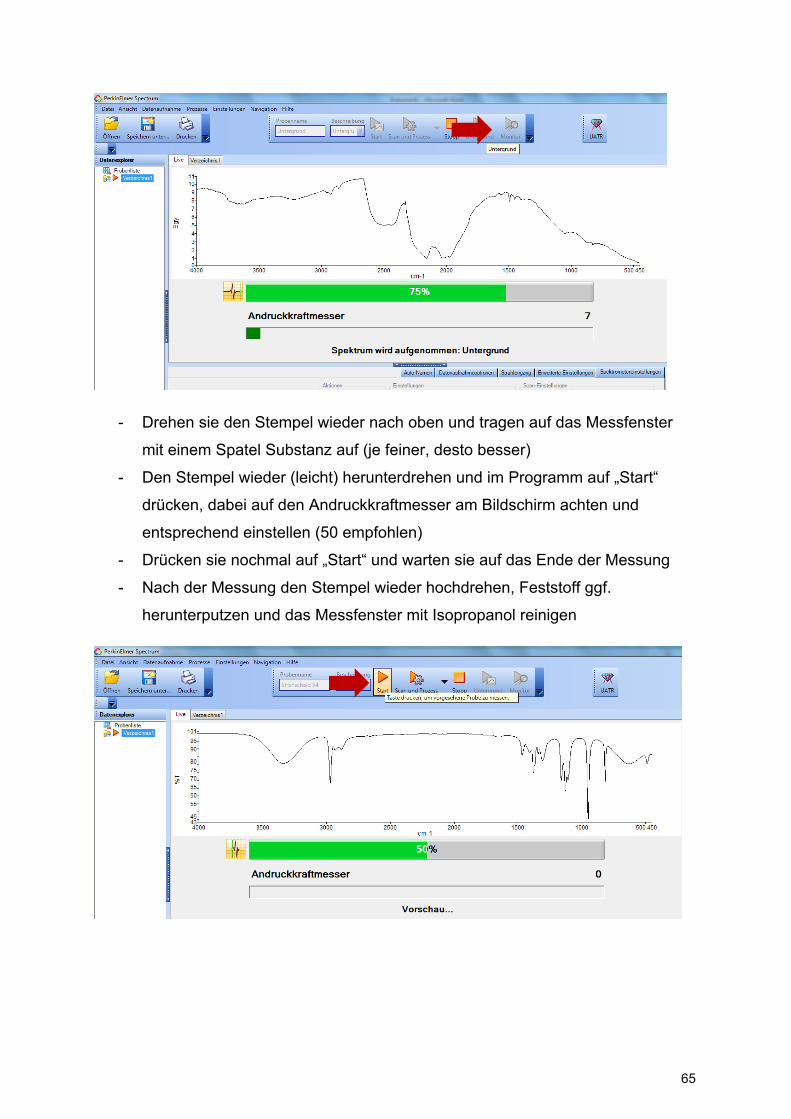
- Drehen sie den Stempel wieder nach oben und tragen auf das Messfenster
mit einem Spatel Substanz auf (je feiner, desto besser)
- Den Stempel wieder (leicht) herunterdrehen und im Programm auf „Start“
drücken, dabei auf den Andruckkraftmesser am Bildschirm achten und
entsprechend einstellen (50 empfohlen)
- Drücken sie nochmal auf „Start“ und warten sie auf das Ende der Messung
- Nach der Messung den Stempel wieder hochdrehen, Feststoff ggf.
herunterputzen und das Messfenster mit Isopropanol reinigen
65

- Ihr Messergebnis ist links im Bildschirm unter „Verzeichnis“ einseh- und
bearbeitsbar
- Zur Öffnung der Datenbank klicken sie auf „Öffnen“ und laden aus dem
Dateiordner „Referenzspetren P5“ die Arznei- und Hilfstoffe aus 2. in das
Programm
66

EPR
67

strukturanalytik: epr praktikum 1EPR
EPR / MQF, Institut für Experimentalphysik II, Universität Leipzig
−
Strukturanalytik: EPR1 Praktikum 1 EPR – engl. Electron ParamagneticResonance, auch ESR – Electron SpinResonance
Abbildung 1: Proteinvermessung mitSpinlabels. Aus
Die Elektronenspinresonanz ist ein Messverfahren zur Untersu-chung von paramagnetischen Zentren in Materie. Sie wird in derPhysik, Chemie, Biologie und vielen angrenzenden Bereichen zurStukturaufklärung eingesetzt. Für biochemische Anwendungen sindbeispielsweise Entfernungsverteilungen zwischen paramagnetischenZentren relevant (Abb. 1). Diese können im Bereich von 1,8 bis 6 nmin Membranproteinen und bis zu 10 nm in deuterierten löslichenProteinen mit der sog. DEER-Technik untersucht werden. Die Anzahlvon paramagnetischen Zentren und deren relative Orientierung istdamit charakterisierbar.2 DEER setzt keine Kristallisation voraus
2 Razzaghi, S., Brooks, E. K., Bordi-gnon, E., Hubbell, W. L., Yulikov, M.,Jeschke, G., ChemBioChem 2013, 14, 14,1883
und ist nicht im Hinblick auf die Größe der Proteine oder Prote-inkomplexe beschränkt. Diamagnetische Proteine können durchseitenorientierende Spinlabels untersucht werden.3
3 Jeschke, G., Annual review of physicalchemistry 2012, 63, 419
Dieses Praktikum wird Ihnen einen Überblick über die verschie-denen EPR-Messtechniken und ihren Einsatz in biochemischenUntersuchungen geben. Sie lernen ein Messverfahren der EPR-Spektroskopie praktisch kennen und charakterisieren Proben inverschiedenen Konzentrationen und Lösungsmitteln nach den g-und A-Werten.
Grundlagen der EPR-Spektroskopie
Abbildung 2: J. K. Sawoiski (1907 –1976), russ. Physiker
Die EPR ist eine relativ alte Messmethode, deren theoretische Be-schreibung mit dem berühmten Stern-Gerlach-Experiment von1921 zusammenhängt. Mit Etablierung der Radar- und Funk-Bauteil-verwandten Röhrentechnik wurde bereits 1944 an derU Kasan durch J. K. Sawoiski die heute bekannte Form der sog.CW-EPR4 eingeführt. Grundlage ist die Aufspaltung der Zeeman-
4 CW – continous wave, das herkömm-liche Messverfahren mit kontinuierlicheingestrahlter Mikrowelle
Energieniveaus eines paramagnetischen Stoffes unter dem Einflusseines Magnetfeldes. Mit fortschreitender elektronischer Entwick-lung wurden auch in der EPR die aus der NMR bekannten, vielfäl-tigen gepulsten Messungen eingeführt und finden heute insbeson-dere für die Biochemie breite Verwendung. Im Gegensatz zur NMRist in der EPR die CW-Methode noch immer weit verbreitet undliefert wichtige Forschungsergebnisse. Die folgenden Kapitel sollenin beide Techniken einführen.
Elektronen-Zeeman-Effekt: CW-EPR
mS = + 12 −
mS = − 12
−
∆E
B
E
Abbildung 3: Ein aufgezeichnetes EPR-Spektrum der Zeeman-Aufspaltungder Elektronenspin-EnergieniveausmS = ± 1
2 in Abhängigkeit des Magnet-feldes B.
Ungepaarte Elektronen, wie sie in organischen Radikalen, Farbzen-tren in Kristallen, Übergangsmetallen und weiteren Stoffen vorkom-men, sind paramagnetische Zentren, die sich unter dem Einflusseines Magnetfeldes B gemäß ihres Elektronenspins ausrichten. Sys-teme mit einem ungepaarten Elektron können hier die Energienvon Niveaus der Quantenzahlen mS = ± 1
2 annehmen (Abb. 3).
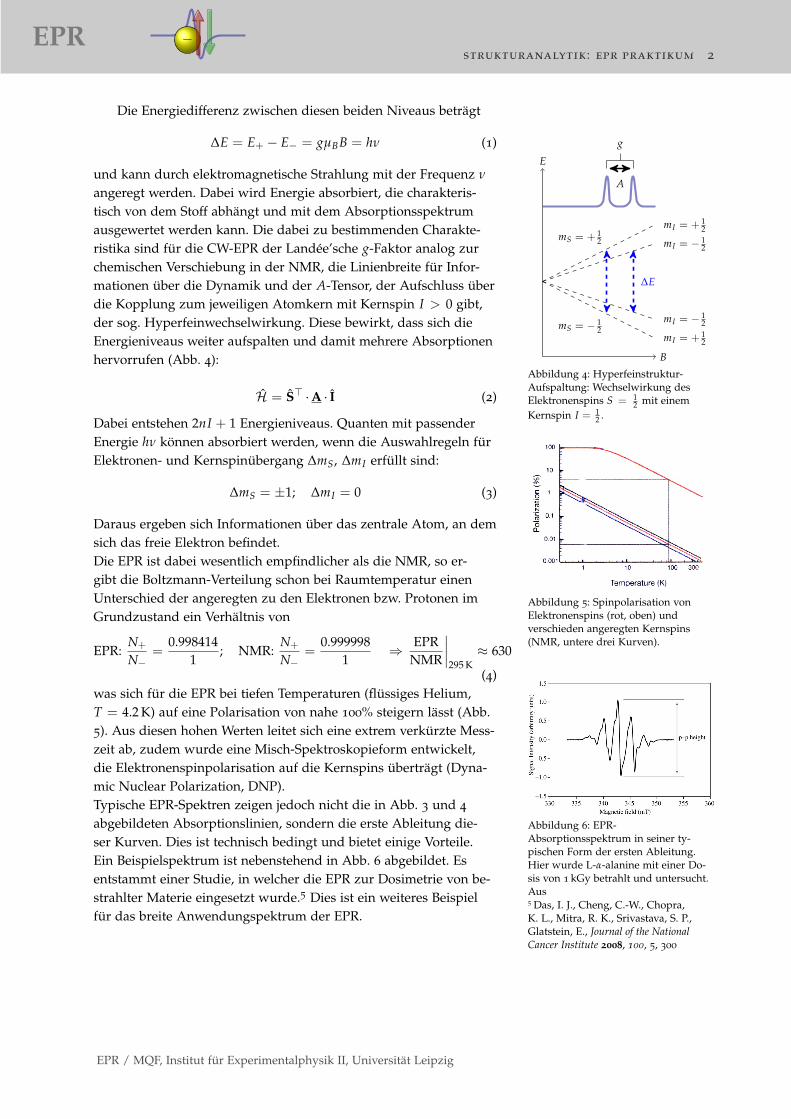
strukturanalytik: epr praktikum 2EPR
EPR / MQF, Institut für Experimentalphysik II, Universität Leipzig
−
Die Energiedifferenz zwischen diesen beiden Niveaus beträgt
∆E = E+ − E− = gµBB = hν (1)
und kann durch elektromagnetische Strahlung mit der Frequenz ν
angeregt werden. Dabei wird Energie absorbiert, die charakteris-tisch von dem Stoff abhängt und mit dem Absorptionsspektrumausgewertet werden kann. Die dabei zu bestimmenden Charakte-ristika sind für die CW-EPR der Landée’sche g-Faktor analog zurchemischen Verschiebung in der NMR, die Linienbreite für Infor-mationen über die Dynamik und der A-Tensor, der Aufschluss überdie Kopplung zum jeweiligen Atomkern mit Kernspin I > 0 gibt,der sog. Hyperfeinwechselwirkung. Diese bewirkt, dass sich dieEnergieniveaus weiter aufspalten und damit mehrere Absorptionenhervorrufen (Abb. 4):
mI = + 12
mI = − 12
mI = + 12
mI = − 12
mS = + 12
mS = − 12
∆E
B
E
g
A
Abbildung 4: Hyperfeinstruktur-Aufspaltung: Wechselwirkung desElektronenspins S = 1
2 mit einemKernspin I = 1
2 .
H = S> · A · I (2)
Dabei entstehen 2nI + 1 Energieniveaus. Quanten mit passenderEnergie hν können absorbiert werden, wenn die Auswahlregeln fürElektronen- und Kernspinübergang ∆mS, ∆mI erfüllt sind:
∆mS = ±1; ∆mI = 0 (3)
Daraus ergeben sich Informationen über das zentrale Atom, an demsich das freie Elektron befindet.
Abbildung 5: Spinpolarisation vonElektronenspins (rot, oben) undverschieden angeregten Kernspins(NMR, untere drei Kurven).
Die EPR ist dabei wesentlich empfindlicher als die NMR, so er-gibt die Boltzmann-Verteilung schon bei Raumtemperatur einenUnterschied der angeregten zu den Elektronen bzw. Protonen imGrundzustand ein Verhältnis von
EPR:N+
N−=
0.9984141
; NMR:N+
N−=
0.9999981
⇒ EPRNMR
∣∣∣∣295 K
≈ 630
(4)was sich für die EPR bei tiefen Temperaturen (flüssiges Helium,T = 4.2 K) auf eine Polarisation von nahe 100% steigern lässt (Abb.5). Aus diesen hohen Werten leitet sich eine extrem verkürzte Mess-zeit ab, zudem wurde eine Misch-Spektroskopieform entwickelt,die Elektronenspinpolarisation auf die Kernspins überträgt (Dyna-mic Nuclear Polarization, DNP).
confirmed in subsequent studies (Kuroda andMiyagawa, 1982; Muto and Iwasaki, 1973), but anumber of authors have speculated that thisradical may not be the only one present in theobserved spectrum (Arber et al., 1991; Ciesielskiand Wielopolski, 1994; Kuroda and Miyagawa,1982; Miyagawa and Gordy, 1960; Simmons, 1962).A comprehensive spectroscopic study by Sagstuenet al. (1997) presented evidence for the existence oftwo further radicals: a hydrogen abstraction radical
present in significant amounts, and a minorityradical tentatively assigned the equilibriumstructure
The central feature of the observed room temp-erature EPR spectrum is estimated to compriseapproximately 55% of the deamination radical,35 % of the hydrogen-abstraction radical, and 10%of the minority radical (Heydari et al., 2002).
The deamination radical is thought to be pro-duced via a reductive pathway involving theprotonated anion (Muto and Iwasaki, 1973):
CH!CH3"!NH3#"!COOH$†" # e$ #H#
$! CH!CH3"!NH3#"!COOH$"; !5:1"
CH!CH3"!NH3#"!COOH$†"
$! †CH!CH3"COOH#NH3: !5:2"
The hydrogen-abstraction radical is thought tobe produced via an oxidative pathway (Sagstuenet al., 1997), possibly involving decarboxylation andsubsequent hydrogen abstraction.
Direct evidence for more than one alanine-derived radical has been presented by Wieser et al.(1993), after prolonged exposure of irradiatedalanine to uv, and at very high absorbed doses(1.6 MGy) by Desrosiers et al. (1995) using spin-trapping techniques.
The principles of EPR spectroscopy are welldescribed in the literature (e.g., Abragam and
Bleany, 1970). The alignment of magnetic momentsof the unpaired electrons in an external magneticfield can be reversed (electron spin flip) by resonantabsorption of energy from incident microwave radi-ation. The amount of the absorbed energy is a func-tion of the applied magnetic field and the magneticmoment of the electron (electron Zeeman inter-action), as well as the magnetic moments of neigh-boring nuclei (hyperfine interaction and nuclearZeeman interaction). The hyperfine interactionbetween the electron and nuclear magneticmoments leads to the splitting of the resonancepeak in the EPR spectrum. The resonance con-dition in the case of dominant electron–Zeemaninteraction can be described by Eq. (5.3), whichgives the relationship of the microwave frequencyand the magnetic flux density at the center of theEPR spectrum,
hn % gbB; !5:3"
where h is Planck’s constant, n frequency of appliedmicrowave field, g Lande g-factor (for free elec-trons, g ! 2.0023), b the Bohr magneton (9.27 &10224 J T21), and B is magnetic flux density of theexternal magnetic field.
The EPR spectrum of L-a-alanine irradiated with60Co radiation is shown in Fig. 5.1. Electron para-magnetic resonance spectra are customarily pre-sented in terms of the first derivative of themicrowave absorption intensity as a function of themagnetic field strength. The peak-to-peak ampli-tude of the central feature is generally used fordosimetry, although it is possible to use the doubleintegral in certain circumstances (Ahlers andSchneider, 1991). The increase in peak-to-peak
Figure 5.1. EPR spectrum of L-a-alanine irradiated to a dose ofapproximately 1 kGy. Modulation amplitude ! 0.3 mT;microwave power ! 2 mW.
DOSIMETRY SYSTEMS FOR USE IN RADIATION PROCESSING
30
at UB
Leipzig on Novem
ber 29, 2011http://jicru.oxfordjournals.org/
Dow
nloaded from
Abbildung 6: EPR-Absorptionsspektrum in seiner ty-pischen Form der ersten Ableitung.Hier wurde L-α-alanine mit einer Do-sis von 1 kGy betrahlt und untersucht.Aus
Typische EPR-Spektren zeigen jedoch nicht die in Abb. 3 und 4abgebildeten Absorptionslinien, sondern die erste Ableitung die-ser Kurven. Dies ist technisch bedingt und bietet einige Vorteile.Ein Beispielspektrum ist nebenstehend in Abb. 6 abgebildet. Esentstammt einer Studie, in welcher die EPR zur Dosimetrie von be-strahlter Materie eingesetzt wurde.5 Dies ist ein weiteres Beispiel 5 Das, I. J., Cheng, C.-W., Chopra,
K. L., Mitra, R. K., Srivastava, S. P.,Glatstein, E., Journal of the NationalCancer Institute 2008, 100, 5, 300
für das breite Anwendungspektrum der EPR.

strukturanalytik: epr praktikum 3
Gepulste EPR-Techniken
Gepulste EPR-Verfahren wie das eingangs beschriebene DEER6 6 DEER – Double Electron-ElectronResonance, nutzt die Wechselwirkungzwischen zwei ungepaarten Elektronen
verwenden im Gegensatz zur CW-EPR nur kurze Sequenzen vonMikrowellen. Diese, genannt Pulse, beeinflussen bestimmte Spins,welche wiederum Auswirkung auf ihre Umgebung haben können.Sobald vom Senden der Pulse auf Empfang umgeschaltet werdenkann (dies hängt von vielen physikalischen und technischen Li-mitierungen ab) beobachtet man die zeitliche Veränderung derMagnetisierung im konstanten Magnetfeld. Dies gibt Informationenüber umgebende Kerne oder weitere ungepaarte Elektronen.
z
ωL
π
π/2
tMW
π2
π
τ τ
Abbildung 7: Oben: einfachste Puls-Folge, das Hahn-Echo. Unten: Orien-tierung der Spins durch die beidenPulse.
Die einfachste Pulsfolge ist das Hahn-Echo (Abb. 7). Ein π/2-Pulsorientiert alle zunächst entlang des Magnetfeldes B in der z-Achseausgerichteten Spins in die xy-Ebene durch eine Drehung um 90◦.Sogleich beginnen alle Spins mit ihrer jeweiligen LamorfrequenzωL = 2πν in dieser Ebene zu präzedieren, d.h. sie drehen sich umdie z-Achse. Dabei ist B die Feldstärke, bei der mit der CW-EPRmit Frequenz ν das entsprechende Signal angeregt wird. Die Spinslaufen entsprechend dem ωL unterschiedlich schnell auseinander.Nun folgt nach einer Zeit τ ein π-Puls, der die Orientierung um180◦ ändert, sodass alle Spins wieder zusammenlaufen, weil dievormals schnellsten Spins nun am weitesten hinten liegen (sichaber noch immer in dieselbe Richtung drehen). Die Zeit τ, nachder das Zusammenlaufen aller Spins beobachtet werden kann, istcharakteristisch für jede Probe und stellt somit den Messparameterdar.Neben dieser Pulsfolge existieren viele weitere, die im Praktikumvorgestellt werden. Beispielhaft seien HYSCORE7 (regt auf zwei un- 7 Hyperfine Sublevel Correlation
terschiedlichen Mikrowellenfrequenzen benachbarte Spins an unduntersucht deren Wechselwirkung), ENDOR8 (zusätzlich zum Mi- 8 Electron Nuclear Double Resonance
krowellenpuls wird ein NMR-Radiopuls auf die Probe eingestrahltund die Auswirkung auf den Elektronenspin beobachtet) und dasbereits besprochenene DEER (eine besondere Pulsfolge regt auchzwei Elektronenspins an und untersucht den Einfluss des zweitenSpins auf den ersten) genannt. Auf letzteres wird im kommendenAbsatz näher eingegangen.
EPR
EPR / MQF, Institut für Experimentalphysik II, Universität Leipzig
−
Proteinstrukturaufklärung mit SDSL
The results of the simulations (Figure 6) for the Gd3+ tagsare in good agreement with the experimental distance distribu-tions in terms of both the positions of the maxima and theirwidths. The maximum of the distance distribution ofGd3+-Gd3+ ions in the samples tagged with 3MDPA ispredicted to be shorter by 0.2 nm than for the samples taggedwith 4MMDPA for both proteins. This small difference has notbeen detected experimentally. The maximum of the Gd3+-Gd3+
distance distribution is predicted to be at 3.3 nm for τC14, whichis close to the measured value of 3.4 nm. For p75ICD, themaximum of the calculated Gd3+-Gd3+ distance distributionis 3.2 nm. This distance reduces to 2.9 nm when one of theGd3+ ions is assumed to be immobilized by coordination toAsp397, leading to better agreement with the experimentalvalues. Assistance by Asp397 in coordinating the Gd3+ ionbound to C416 is supported by the ENDOR and NMR results.The simulations show distance distributions with a width at half-height of about 0.8 nm, in close agreement with the experimentalresults. The width of the distance distribution is only littleaffected by immobilization of one of the Gd3+ ions. In the caseof the MTSL-labeled proteins, the maximum of the calculated
distance distribution (2.9 nm) was longer than the experimentalvalue (2.5 nm) for both proteins.
Discussion
Our results demonstrate the attraction of Gd3+ spin labelingas a tool for nanometer-scale distance determinations in proteinsby high-magnetic-field DEER measurements. The unique high-field EPR spectral properties of Gd3+ greatly enhance thesensitivity, correspondingly reducing the quantity of samplerequired. Labeled proteins at concentrations below 100 µM andin volumes of about 2 µL (corresponding to total amounts ofless than 200 pmol) can be routinely measured and analyzedfor distances up to 4 nm with typical measurement times of8-12 h. Larger distances will require larger concentrations orlonger accumulation times. The sensitivity is more than an orderof magnitude larger compared with standard X-band DEERmeasurements with nitroxides. The modulation depth of DEERtraces of Gd3+-Gd3+ pairs is significantly lower than in pairsof S ) 1/2 spins because of the low probability of finding apair of spins with MS ) (1/2.28 As long as the temperature is
Figure 6. Distributions of Gd3+-Gd3+ and nitrogen-nitrogen distances in p75ICD (PDB code 1NGR) and τC14 (PDB code 2AYA). Distances were calculatedby randomly generating rotamers of the bonds connecting the paramagnetic centers with the CR atom of the tagged cysteine residues, excluding conformationsthat clash with protein atoms in the NMR structures. (a) Distance distributions calculated for p75ICD. The different curves were calculated for the tagsdescribed in the figure. The dotted-dashed curves were calculated by assuming complete immobilization of one of the Gd3+ ions by coordination to the tag(attached to C416) and D397. Lower panel: Distance distributions calculated for τC14 for the tags described in the figure. (b) Cartoon of the NMR structureof p75ICD tagged with two 4MMDPA-Gd3+ complexes. The conformer chosen shows the Gd3+ ion coordinated to the tag as well as to D397. The Gd3+-Gd3+
distance is indicated for a random conformer of the 4MMDPA tag attached to C379. (c) Cartoon of the NMR structure of τC14 tagged with two 3MDPA-Gd3+ complexes. The Gd3+-Gd3+ distance is indicated for randomly chosen conformers of the tags attached to C528 and C553.
9046 J. AM. CHEM. SOC. 9 VOL. 132, NO. 26, 2010
A R T I C L E S Potapov et al.
Abbildung 8: Gd3+-SDSL an demProtein p75ICD (PDB code 1NGR).Aus
Site Directed Spin Labeling (SDSL) wird eine Methode genannt, beider Proteine durch eine site-directed mutagenesis verändert werden(Abb. 8). Es werden die Wechselwirkungen der Spinlabel unter-sucht, um Rückschlüsse auf Abstands- und Orientierungsverhält-nisse in dem Protein zu ziehen. Dazu können Linienbreitenmodellein der CW-EPR oder DEER eingesetzt werden.9
9 Potapov, A., Yagi, H., Huber, T.,Jergic, S., Dixon, N. E., Otting, G.,Goldfarb, D., Journal of the AmericanChemical Society 2010, 132, 26, 9040

strukturanalytik: epr praktikum 4
Strukturaufklärung mit CW-EPR
am Beispiel von Colicin E1 mit einem Nitroxid-Radikal Spinlabel
298
- C r O x
A. P. TODD ET AL.
+ C r O x - C r O x + C r O x
m
+Jk-+-- I 1 I
i n --fn I I 1
I
Fig. 2. (a) EPR spectra of MTSSL-labeled colicin mutants 398- 406 in aqueous solution in the absence and presence of 50 mM CrOx. The vertical lines pass through the outermost hyperfine extrema of 404. The scan range is 100 G. (b) EPR spectra of
Spin Label Mobility Varies With Position The spin labeled mutant spectra in aqueous solu-
tion exhibit a variety of lineshapes depending upon the label and position along the peptide, as can be seen in Figures 2 and 3. Because of the small sample volume required by the loop-gap resonator (<5 ~ l ) , each spectrum required on the order of only 50 pmol of protein. The most useful qualitative information that can be readily extracted from such spectra is the relative mobility of the label. Consider first the spectra for the MTSSL-labeled mutants of the hy- drophilic group in Figure 2a (left column). It can be seen that three of the spectra (398, 400, 404) are distinctive from their neighbors in the sequence by
406 - b
PDSSL-labeled colicin mutants 398-406 in aqueous solution in the absence and presence of 50 mM CrOx. Arrows above 398 denote outermost hyperfine extrema of mobile (m) and motionally restricted (r) components. The scan range is 100 G.
having widely spaced hyperfine extrema (see verti- cal lines), indicating that the label is strongly im- mobilized and buried in the protein interior. In con- trast, the six other spectra have sharper spectral features and much more closely spaced hyperfine ex- trema. This indicates more rapid motion and sug- gests that these labels project from the protein sur- face in contact with water. On this qualitative basis, all of the spectra in Figure 2a may be classified as either mobile or motionally restricted. These results are summarized in the second column of Table I. A similar classification can be made of the spectra for PDSSL-labeled mutants of this sequence (Fig. 2b), which is summarized in the same table. The spectra
Abbildung 9: Nitroxid-Radikal gela-belte Colicin E1-Mutanten 398–406 inwässriger Lösung ohne und mit 50 mMCr(Ox)3−
3 untersucht mit CW-EPR. (r)kennzeichnet starre und (m) mobilePositionen der Label. Aus
Zur Modifizierung des Proteins Colicin E1 wird an einer gewünsch-ten Stelle eine Aminosäure durch Cystein ersetzt. Dabei werdendie natürlichen Cystein-Bausteine durch Glycin ausgetauscht undder Einbau durch Gen-Sequenzierung und einen Aktivitätstest mit[35S
]Cystein kontrolliert. Die damit gemessenen EPR-Spektren wei-
sen verschiedene Signallagen und -schärfen auf, die auf mobile undstarre (restricted mobility) Positionen hindeuten (Abb. 9). Auchdurch Zugabe von 50 mM Trioxalatchromat(III)-(Cr(Ox)3−
3 )-Lösungkann eine Veränderung je nach Labelposition erkennbar werden,das Signal verschwindet immer genau dann, wenn das Label anPositionen sitzt, die für die wässrige Lösung zugänglich sind.10
10 Todd, A. P., Cong, J., Levinthal, F.,Levinthal, C., Hubell, W. L., Proteins:Structure, Function, and Bioinformatics1989, 6, 3, 294
Aus den Spektren können auch direkt Informationen über denAbstand zwischen den Labeln gewonnen werden. So ist die dipo-lare Wechselwirkung, die zu einer Signalaufspaltung von 2B führt,proportional zum Abstand r−3 der Label:
2B ∝g2µ2
B(3 cos2 θ − 1
)
r3 (5)
In ungeordneten Systemen tritt zudem eine Linienverbreiterungauf, da verschiedene Winkel θ in der Lösung vorliegen:
M (B) = ∑r
P (r) D (r, B) (6)
Wird diese Wahrscheinlichkeitsverteilung M(B) mit der Abstands-verteilung P(r) und der Linienbreitenfunktion D(r, B) in Gl. 5 ein-gesetzt und über alle Positionen gemittelt, ist die Berechnung desmittleren Abstandes der Label aus der Linienbreite möglich:11
11 Rabenstein, M. D., Shin, Y.-K.,Proceedings of the National Academy ofSciences 1995, 92, 18, 8239
〈r〉 = 3
√1, 125g2µ2
B〈2B〉 (7)
Gd3+ Complexes as Potential Spin Labels for High Field Pulsed EPR DistanceMeasurements
Arnold M. Raitsimring,† Chidambaram Gunanathan,‡ Alexey Potapov,§ Irena Efremenko,‡Jan M. L. Martin,‡ David Milstein,*,‡ and Daniella Goldfarb*,§
Department of Chemistry, UniVersity of Arizona, Tucson, Arizona 85721, and Departments of Organic Chemistryand Chemical Physics, Weizmann Institute of Science, RehoVot 76100, Israel
Received August 4, 2007; E-mail: [email protected]; [email protected]
Distance measurements between spin labels by means of pulseEPR have attracted considerable attention because of the applica-tions to biological systems.1,2 These determine the dipolar interactionbetween two electron spins, which for two S ) 1/2 spins is
In eq 1, g1 and g2 are the g values, r is the interspin distance, andθ is the angle between r and the external magnetic field, B0. Theaccessible distance range, 1.5-6.0 nm, allows one to obtainstructural information on biomolecules such as proteins, DNA andRNA, and their complexes.3,4 Two nitroxide spin labels (NSL) areintroduced in the biomolecule, and the measurements are usuallycarried out at X-band frequencies (∼9.5 GHz) on samples of ∼50µL of 0.1-0.2 mM solutions (∼12 h accumulation time). Distancemeasurements, however, are not limited to organic radicals, andmeasurements involving metal ions were reported as well.5-7In principle, high field (HF)/high microwave frequency (HM-
WF)8,9 measurements can offer improvement in sensitivity overX-band. However, due to the g anisotropy, the width of the NSLEPR spectrum increases at HF, leading to a decrease in the spectraldensity and of the flip probability of the pumped spins. Therefore,excluding cases where the relative orientation of the g frames ofthe NSLs is of prime interest, HF measurements provide onlymoderate gain (if any).For HF distance measurements, a different class of spin labels,
the spectrum of which narrows with increasing B0, such as highspin half integer systems (HSS) like Gd3+ (S ) 7/2) may beattractive. While the width of the entire spectrum of these ions isdetermined by the crystal field interaction (cfi), which is magneticfield independent, the sub-spectrum of the central |-1/2⟩ f |1/2⟩transition (CT) narrows as B0 increases. Here we present first resultson the synthesis and pulse double electron-electron resonance(DEER) measurements between two Gd3+ ions connected via a rigidlinker, shown in Scheme 1.The bis-Gd3+ complex comprises two units of a pyridine-based
tetracarboxylate gadolinium complex, developed as an MRI contrastagent.10 Double Sonogashira coupling of the pyridine tetracarboxy-late ester 1 and 1,4-diethynylbenzene (2) provided the correspondingocta-ester11 (Scheme 1). Deprotection of the octa-ester11 andsubsequent complexation of the octa-acid with excess of an aqueoussolution of GdCl3‚3H2O gave the desired bis-Gd3+ complex 5 in86% yield. The paramagnetic complex 5 was characterized by IR,MS (ESI), HRMS, and magnetic moment (p ) 15.63(2) µB)analyses.11
The DEER measurements were carried out at Ka- (33.78 and29.6 GHz) and W- (94.9 GHZ) bands.12,13 The corresponding echo-detected (ED) EPR spectra are shown in Figure 1b. The width ofthe W-band CT is about 0.4 of that at Ka-band, as expected.14The broad background the CT is superimposed on is due to all
other transitions, and their relative intensities are a function of thetemperature and spectrometer frequency. The relative populations,P(MS), of the MS ) (1/2 energy levels for the frequencies andtemperatures at which the experiments were carried out are listedin Table 1. The four-pulse,15 time domain DEER traces, shown inFigure 2a, exhibit a steep initial decay and shallow but clear
† University of Arizona.‡ Department of Organic Chemistry, Weizmann Institute of Science.§ Department of Chemical Physics, Weizmann Institute of Science.
νDD(θ,r) )g1g2#
2µ04πhr3
(3 cos2 θ - 1) (1)
Scheme 1. Synthesis of the Bis-Gd3+ Complexa
a Conditions: (a) trans-PdCl2(PPh3)2, CuI, dry iPr2NH, rt, 40 h, 67%;(b) CF3CO2H/CH2Cl2, rt, 12 h, 82%; (c) pH 5-7, GdCl3‚3H2O, rt, 5 h,86%.
Figure 1. (a) The geometry of the bis-Gd3+ complex with a total chargeof -2 and 14 unpaired electrons, optimized at the PBE0/sdd level of theorywith D2h symmetry constrain.11 (b) Two-pulse ED-EPR spectra of the bis-Gd3+ complex recorded at Ka-band (∼1.5 mM, 13 K), W-band (0.2 mM,10, 25 K). The positions of the pump and observed pulses are shown onthe figure.
Published on Web 10/27/2007
14138 9 J. AM. CHEM. SOC. 2007, 129, 14138-14139 10.1021/ja075544g CCC: $37.00 © 2007 American Chemical Society
Abbildung 10: Gd3+-Label. Aus
Typische Spinlabels in der EPR sind Nitroxidradikale oder Gadolinium-(Gd3+)-Komplexe, wie nebenstehend gezeigt. Dabei werden Ni-troxidradikale bevorzugt verwendet, wenn auch Informationenüber die Orientierung gewonnen werden sollen, während typischeGadolinium-Label nicht orientierungsselektiv sind und damit bes-sere Aussagen über die Abstandsverteilung liefern können.12 Mit 12 Raitsimring, A. M., Gunanathan, C.,
Potapov, A., Efremenko, I., Martin,J. M., Milstein, D., Goldfarb, D., Journalof the American Chemical Society 2007,129, 46, 14138
Gadolinium lässt sich die Intensität um eine Größenordnung stei-gern, sodass weniger Substanzbedarf besteht und die Messzeit sinkt(7 – 10 Std.). Spinlabel sollen die Eigenschaften des Proteins nichtändern.
EPR
EPR / MQF, Institut für Experimentalphysik II, Universität Leipzig
−
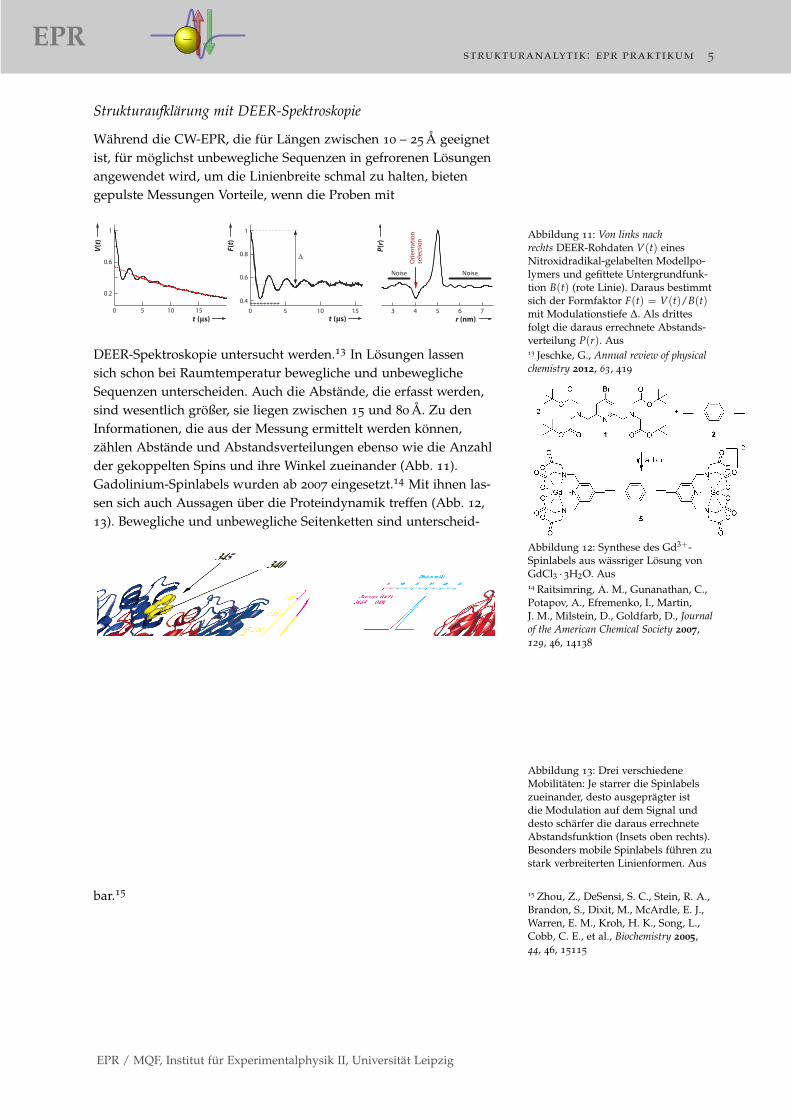
strukturanalytik: epr praktikum 5
Strukturaufklärung mit DEER-Spektroskopie
Während die CW-EPR, die für Längen zwischen 10 – 25 Å geeignetist, für möglichst unbewegliche Sequenzen in gefrorenen Lösungenangewendet wird, um die Linienbreite schmal zu halten, bietengepulste Messungen Vorteile, wenn die Proben mit
EPR
EPR / MQF, Institut für Experimentalphysik II, Universität Leipzig
−
PC63CH19-Jeschke ARI 27 February 2012 17:17
O
O NO
O
ONO
•
•a
b
e
c
f
d
g
0 5 10 15
0.2
0.6
1
0 5 10 150.4
0.6
0.8
1
–3 –2.5 –2 –1.5 –1
–25
–20
–15
3 4 5 6 7 3 4 5 6 7
3 4 5 6 7
α = 0.001
α = 0.001α = 1 α = 1
α = 1,000
α = 1,000
∆
2H NoiseNoise
Noise Noise
Orie
ntat
ion
sele
ctio
n
Hex Hex Hex Hex
Hex Hex Hex Hex
V(t)
F(t)
t (µs) t (µs) r (nm)
P(r)
r (nm)r (nm)log r
log h
Figure 3Data analysis and artifacts. (a) Structure of model compound 2. (b) Primary four-pulse DEER data V(t) of 2 (black line) and fittedbackground function B(t) (red line). The dotted segments are extrapolated. (c) Form factor F (t) = V (t)/B(t) with modulation depth !.The purple dashed line denotes the part accessible with tmax = 4 µs. (d ) Distance distribution P(r) obtained by approximate Paketransformation of F(t). Black bars denote noise artifacts, and the maroon arrow an orientation selection artifact. (e) Tikhonovregularization L curve with three selected regularization parameters α. ( f ) Distance distributions obtained by Tikhonov regularizationwith constraint P (r) > 0. Undersmoothing (red ) causes unrealistic peak splittings; oversmoothing (blue) causes artificial broadening. Atthe L-curve corner ( green), P(r) is most realistic. ( g) Best experimental distance distribution ( green), artifacts owing to missing nuclearmodulation averaging (black line, red bar), and loss of shape when restricting data to tmax = 4 µs ( purple).
Equation 2. Most notably, a growing dipolar oscillation is introduced near tmax where the pumppulse approaches the final refocusing pulse. At the cost of sensitivity, this end artifact can besuppressed by decreasing excitation bandwidths, by increasing the frequency difference betweenpump and observer pulses, or by increasing the difference between τ 2 and tmax. For standardX-band conditions (32-ns observer pulses, 12-ns pump pulse, 65-MHz frequency difference), theend artifact is usually negligible.
2.3.4. Combination frequencies for multiple spins. If more than two paramagnetic centersare situated in a protein or protein complex, the pump pulse may excite more than one spin Bi neara given A spin. The form factor F(t) then contains products of the pair form factors fi (t) for theindividual A-Bi spin pairs (25). Thus, products of cosine functions of the dipolar frequencies ωee,i
occur, which correspond to sum and difference combinations of these frequencies. The relative
426 Jeschke
Ann
u. R
ev. P
hys.
Che
m. 2
012.
63:4
19-4
46. D
ownl
oade
d fr
om w
ww
.ann
ualre
view
s.org
by W
IB60
91 -
Uni
vers
itaet
sklin
ikum
Lei
pzig
on
07/1
1/14
. For
per
sona
l use
onl
y.
Abbildung 11: Von links nachrechts DEER-Rohdaten V(t) einesNitroxidradikal-gelabelten Modellpo-lymers und gefittete Untergrundfunk-tion B(t) (rote Linie). Daraus bestimmtsich der Formfaktor F(t) = V(t)/B(t)mit Modulationstiefe ∆. Als drittesfolgt die daraus errechnete Abstands-verteilung P(r). Aus
DEER-Spektroskopie untersucht werden.13 In Lösungen lassen 13 Jeschke, G., Annual review of physicalchemistry 2012, 63, 419sich schon bei Raumtemperatur bewegliche und unbewegliche
Sequenzen unterscheiden. Auch die Abstände, die erfasst werden,sind wesentlich größer, sie liegen zwischen 15 und 80 Å. Zu denInformationen, die aus der Messung ermittelt werden können,zählen Abstände und Abstandsverteilungen ebenso wie die Anzahlder gekoppelten Spins und ihre Winkel zueinander (Abb. 11).
Gd3+ Complexes as Potential Spin Labels for High Field Pulsed EPR DistanceMeasurements
Arnold M. Raitsimring,† Chidambaram Gunanathan,‡ Alexey Potapov,§ Irena Efremenko,‡Jan M. L. Martin,‡ David Milstein,*,‡ and Daniella Goldfarb*,§
Department of Chemistry, UniVersity of Arizona, Tucson, Arizona 85721, and Departments of Organic Chemistryand Chemical Physics, Weizmann Institute of Science, RehoVot 76100, Israel
Received August 4, 2007; E-mail: [email protected]; [email protected]
Distance measurements between spin labels by means of pulseEPR have attracted considerable attention because of the applica-tions to biological systems.1,2 These determine the dipolar interactionbetween two electron spins, which for two S ) 1/2 spins is
In eq 1, g1 and g2 are the g values, r is the interspin distance, andθ is the angle between r and the external magnetic field, B0. Theaccessible distance range, 1.5-6.0 nm, allows one to obtainstructural information on biomolecules such as proteins, DNA andRNA, and their complexes.3,4 Two nitroxide spin labels (NSL) areintroduced in the biomolecule, and the measurements are usuallycarried out at X-band frequencies (∼9.5 GHz) on samples of ∼50µL of 0.1-0.2 mM solutions (∼12 h accumulation time). Distancemeasurements, however, are not limited to organic radicals, andmeasurements involving metal ions were reported as well.5-7In principle, high field (HF)/high microwave frequency (HM-
WF)8,9 measurements can offer improvement in sensitivity overX-band. However, due to the g anisotropy, the width of the NSLEPR spectrum increases at HF, leading to a decrease in the spectraldensity and of the flip probability of the pumped spins. Therefore,excluding cases where the relative orientation of the g frames ofthe NSLs is of prime interest, HF measurements provide onlymoderate gain (if any).For HF distance measurements, a different class of spin labels,
the spectrum of which narrows with increasing B0, such as highspin half integer systems (HSS) like Gd3+ (S ) 7/2) may beattractive. While the width of the entire spectrum of these ions isdetermined by the crystal field interaction (cfi), which is magneticfield independent, the sub-spectrum of the central |-1/2⟩ f |1/2⟩transition (CT) narrows as B0 increases. Here we present first resultson the synthesis and pulse double electron-electron resonance(DEER) measurements between two Gd3+ ions connected via a rigidlinker, shown in Scheme 1.The bis-Gd3+ complex comprises two units of a pyridine-based
tetracarboxylate gadolinium complex, developed as an MRI contrastagent.10 Double Sonogashira coupling of the pyridine tetracarboxy-late ester 1 and 1,4-diethynylbenzene (2) provided the correspondingocta-ester11 (Scheme 1). Deprotection of the octa-ester11 andsubsequent complexation of the octa-acid with excess of an aqueoussolution of GdCl3‚3H2O gave the desired bis-Gd3+ complex 5 in86% yield. The paramagnetic complex 5 was characterized by IR,MS (ESI), HRMS, and magnetic moment (p ) 15.63(2) µB)analyses.11
The DEER measurements were carried out at Ka- (33.78 and29.6 GHz) and W- (94.9 GHZ) bands.12,13 The corresponding echo-detected (ED) EPR spectra are shown in Figure 1b. The width ofthe W-band CT is about 0.4 of that at Ka-band, as expected.14The broad background the CT is superimposed on is due to all
other transitions, and their relative intensities are a function of thetemperature and spectrometer frequency. The relative populations,P(MS), of the MS ) (1/2 energy levels for the frequencies andtemperatures at which the experiments were carried out are listedin Table 1. The four-pulse,15 time domain DEER traces, shown inFigure 2a, exhibit a steep initial decay and shallow but clear
† University of Arizona.‡ Department of Organic Chemistry, Weizmann Institute of Science.§ Department of Chemical Physics, Weizmann Institute of Science.
νDD(θ,r) )g1g2#
2µ04πhr3
(3 cos2 θ - 1) (1)
Scheme 1. Synthesis of the Bis-Gd3+ Complexa
a Conditions: (a) trans-PdCl2(PPh3)2, CuI, dry iPr2NH, rt, 40 h, 67%;(b) CF3CO2H/CH2Cl2, rt, 12 h, 82%; (c) pH 5-7, GdCl3‚3H2O, rt, 5 h,86%.
Figure 1. (a) The geometry of the bis-Gd3+ complex with a total chargeof -2 and 14 unpaired electrons, optimized at the PBE0/sdd level of theorywith D2h symmetry constrain.11 (b) Two-pulse ED-EPR spectra of the bis-Gd3+ complex recorded at Ka-band (∼1.5 mM, 13 K), W-band (0.2 mM,10, 25 K). The positions of the pump and observed pulses are shown onthe figure.
Published on Web 10/27/2007
14138 9 J. AM. CHEM. SOC. 2007, 129, 14138-14139 10.1021/ja075544g CCC: $37.00 © 2007 American Chemical Society
Abbildung 12: Synthese des Gd3+-Spinlabels aus wässriger Lösung vonGdCl3 · 3H2O. Aus
Gadolinium-Spinlabels wurden ab 2007 eingesetzt.14 Mit ihnen las-
14 Raitsimring, A. M., Gunanathan, C.,Potapov, A., Efremenko, I., Martin,J. M., Milstein, D., Goldfarb, D., Journalof the American Chemical Society 2007,129, 46, 14138
sen sich auch Aussagen über die Proteindynamik treffen (Abb. 12,13). Bewegliche und unbewegliche Seitenketten sind unterscheid-
Also shown in Figure 10 are the predicted averagedistances between the unpaired electrons of the two spinlabels from molecular modeling of the R1 side chain at eachsite based on the static crystal structure. The results of thesemodeling experiments, which were carried out to ap-proximate contributions from the dimensions and flexibilityof the R1 side chain as described in Materials and Methods(see refs 46-49 for additional discussion), are in reasonableagreement with the interprobe distances that were obtainedfrom the experimental measurements. When combined withthe data in Figure 7, which showed that residues 337-348
form a continuous R-helix, these long-range distance con-straints show that the spatial arrangement of these two helicesin the cdb3 dimer is very similar, if not identical, to theirspatial arrangement in the crystal structure at pH 4.8.Intersubunit Distances at Additional Sites. Previous studies
have provided compelling evidence that the Stokes radiusof cdb3 reversibly increases from 55 to 66 Å as the pH israised in the range of 6 to 10 with the most dramatic increase(∼80%) occurring between pH 8 and 10 (3, 19, 38). Basedon the crystal structure, it has been hypothesized that cdb3could form elongated structures without an overall changein secondary structure, by rotations about residues G305 orE291 (3). To test whether cdb3 exhibited a significantlyelongated structure at neutral pH relative to the static crystalstructure, 11 different sites on the side of the compactperipheral domain of each monomer that is proximal to thedimer interface were chosen for measurement of interprobedistances as shown in Figure 11. Following expression,purification, and spin labeling of each of these 11 singlecysteine mutants, EPR or DEER experiments were carried
FIGURE 9: DEER characterization of intersubunit distances between residues 340-345 in the dimerization arm. The upper left panel showsthe structure and location of residues 340-345 in the crystal structure (yellow) on the ribbon diagram of the two subunits of the cdb3 dimer(blue and red). The scaled echo amplitude (shown as dots, normalized to 1) is plotted as a function of time in a 4-pulse DEER experimentwith the R1 side chain at positions 340 (upper right), 342 (lower left), and 344 (lower right). The solid line is the best fit of the scaled echoamplitude versus time using DeerPackage 2002.1 as described in Materials and Methods. The insets in the upper right of each DEERspectrum show the average interprobe distance and the distribution of distances from fitting the experimental data. Tabular data for residues341 and 343 (DEER data not shown) are listed in Table 1.
Table 1: Interresidue Distances in the Dimerization Arm of Cdb3
residue no.: 339 340 341 342 343 344 345R (Å) 14.7a 34.6 31.9 24.9 34.0 37.0 35.5σ (Å) 0.4a 0.8 3.6 1.1 2.3 4.2 6.6a The values for R and σ at residue 339 were obtained by fitting the
CW-EPR data to a tether-in-a-cone model (26). The values for R andσ at positions 340 through 345 were determined by fitting thecorresponding DEER data to a single Gaussian distribution of interspindistances as described in Materials and Methods.
Solution Structure of Cdb3 Biochemistry, Vol. 44, No. 46, 2005 15121
Abbildung 13: Drei verschiedeneMobilitäten: Je starrer die Spinlabelszueinander, desto ausgeprägter istdie Modulation auf dem Signal unddesto schärfer die daraus errechneteAbstandsfunktion (Insets oben rechts).Besonders mobile Spinlabels führen zustark verbreiterten Linienformen. Aus
bar.15 15 Zhou, Z., DeSensi, S. C., Stein, R. A.,Brandon, S., Dixit, M., McArdle, E. J.,Warren, E. M., Kroh, H. K., Song, L.,Cobb, C. E., et al., Biochemistry 2005,44, 46, 15115
strukturanalytik: epr praktikum 6
Praktikumsaufgaben
Aufgaben
EPR / MQF, Institut für Experimentalphysik II, Universität Leipzig
−
1. Beschriften Sie alle Bestandteile des Spektrometers! 4! Bitte bringen Sie einen Taschen-rechner zum Praktikum mit. Schutz-kleidung ist nicht erforderlich. KeinUmgang mit offenen Lösungsmit-teln, keine Gefährdung durch starkeMagnetfelder außerhalb der Magneten.
Sicherheitshinweise beachten!
2. Bestimmen Sie den g- und A (MHz)-Wert aus dem folgendenSpektrum von 0.01 M Acetylacetonato-Komplex (Cu(acac)2) inCHCl3: 63,65Cu2+ mit
Kernspin beider Isotope I = 32 ,
Elektronenkonfiguration 3d9 → S =12
Experimentelle Bedingungen:T = 295 K,ν = 9.47 GHz,Mikrowellenleistung: 1,3 mW
Benötigte Gleichungen:A [MHz] = 28 · (g/ge) · A [mT]A
[cm−1] = 3, 34× 10−5 · A [MHz]
Nützliche Konstanten:ge = 2, 002319µB = 9, 274× 10−24 J
Th = 6, 6261× 10−34 Js
280 290 300 310 320 330 340 350 360B (mT)
Inte
nsit
ät(B
.E.)
3. Praxisaufgabe Spektroskopieren Sie das in Aufgabe 2 gezeigte Cu(acac)2 in verschiedenen Lösungsmit-teln (CHCl3, C5H5N, THF, C8H10) und vergleichen Sie die g- und A-Werte!
4. Praxisaufgabe Spektroskopieren Sie Di-tert-Butylnitroxid (DTBN) in drei Konzentrationen (10−4, 10−3, 10−2)gelöst in Toluol. Welchen Elektronen- und Kernspin kann man aus den Spektren ableiten? Um welchesIsotop des Stickstoffes handelt es sich? Ordnen Sie die drei Konzentrationen zu!





























