
Medizinische UniversitätsklinikKnappschaftskrankenhaus Bochum
Morbus Hodgkin
Non-Hodgkin Lymphome
Plasmozytom
C. Teschendorf
Ruhr-Universität Bochum
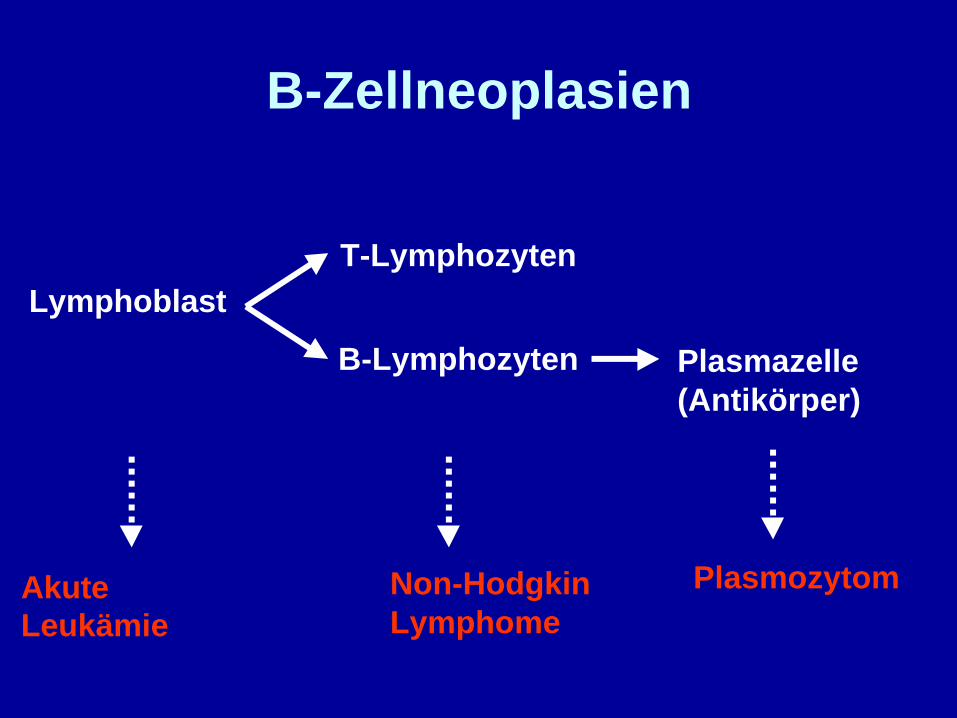
Lymphoblast
Plasmazelle(Antikörper)
Non-HodgkinLymphome
T-Lymphozyten
PlasmozytomAkuteLeukämie
B-Zellneoplasien
B-Lymphozyten

Plasmozytom

1845 Erstbeschreibung durch William MacIntyrekonsultiert Henry Bence Jones
1873 Rustitzky führt Begriff des Myeloms ein
1889 Kahler: Knochentumore und BJ-Protein verschiedene Facetten einer Krankheitsentität
Pasmozytom
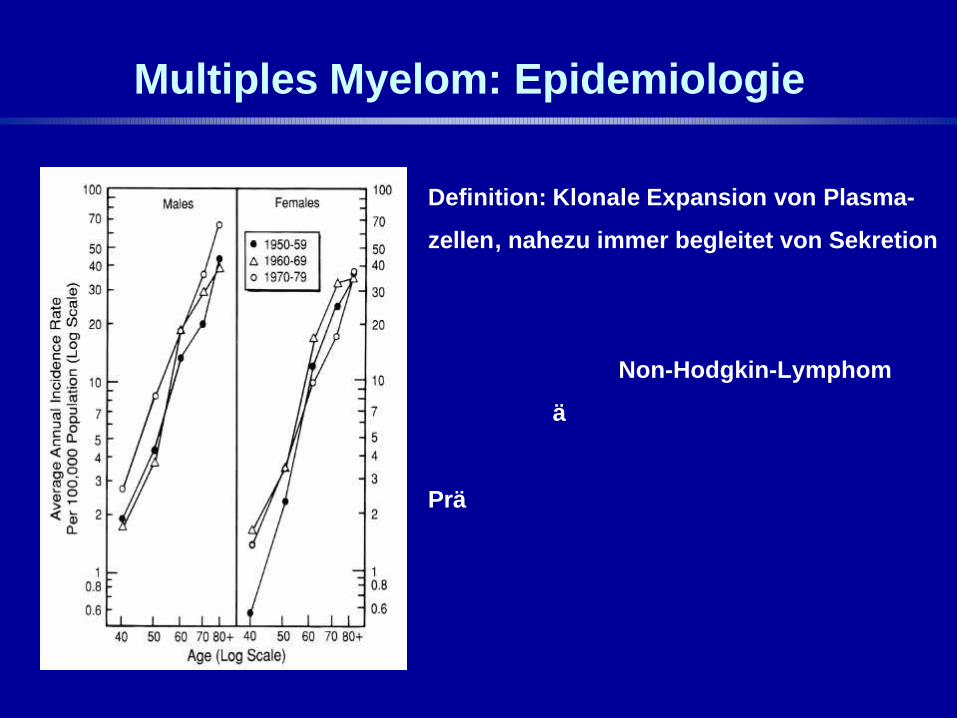
Multiples Myelom: Epidemiologie
Definition: Klonale Expansion von Plasma-
zellen, nahezu immer begleitet von Sekretion
Non-Hodgkin-Lymphom
ä
Prä
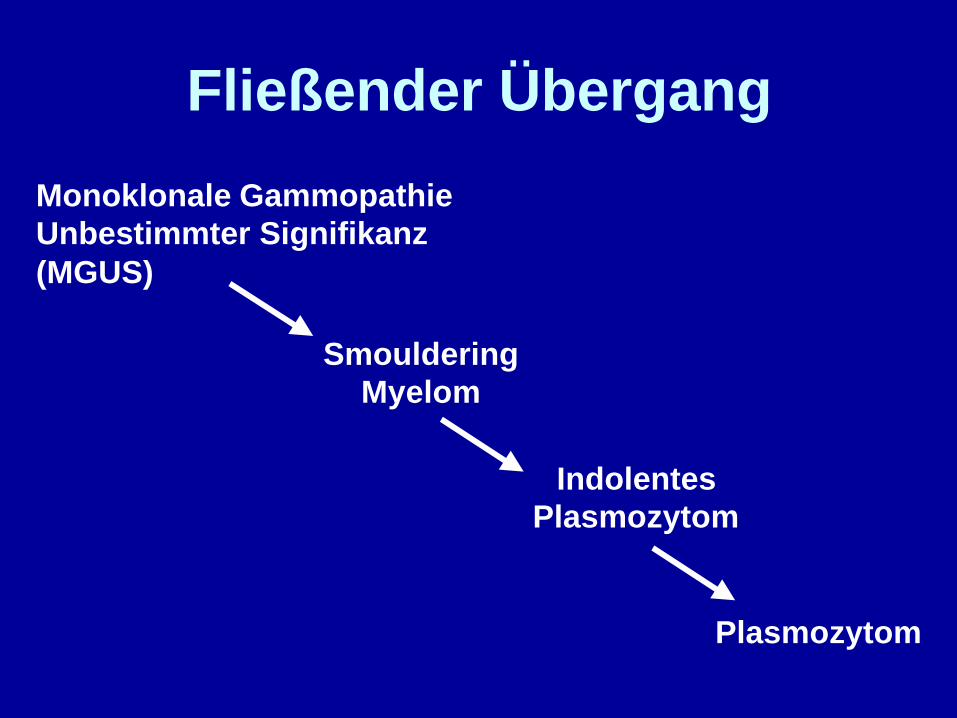
Monoklonale GammopathieUnbestimmter Signifikanz(MGUS)
SmoulderingMyelom
Plasmozytom
Fließender Übergang
IndolentesPlasmozytom
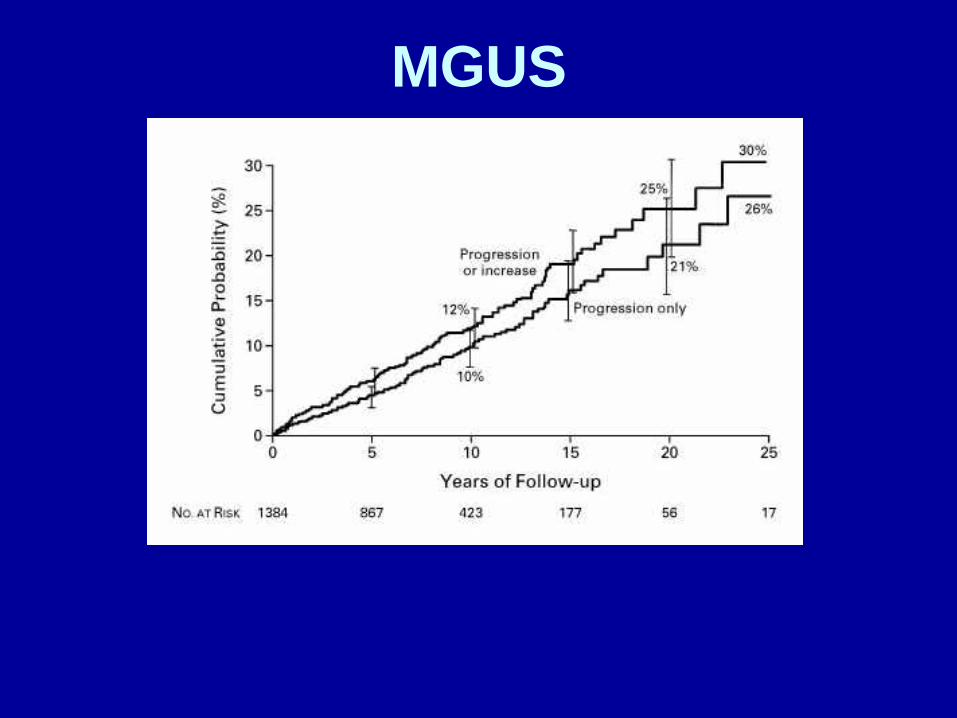
MGUS

Diagnosekriterien - WHO
Ø Plasmazellanteil des Knochenmarkes
Ø Höhe des Paraproteins im Blut, Urin
Ø Osteolysen

StadieneinteilungDurie und Salmon (1975)
NierenfunktionA bei Kreatinin < 2 mg/dl B bei Kreatinin ≥≥ 2 mg/dl
Stadium Befund
I Hb >10g/dlnormales Kalzium,
keine oder einzelne OsteolyseIgG < 5 g/dl, IgA < 3 g/dl, Bence Jones < 4 g/24h
II weder I noch III
III Hb < 8,5 g/dlHyperkalziämie
multiple OsteolysenIgG > 7 g/dl, IgA > 5 g/dl, Bence Jones > 12 g/24h
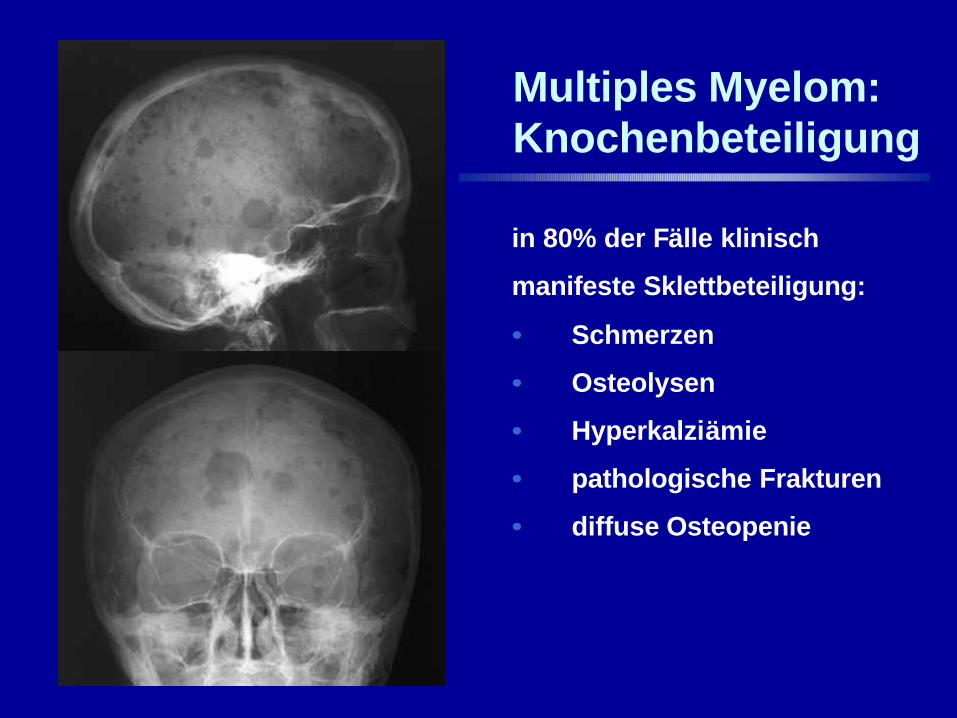
Multiples Myelom:Knochenbeteiligung
in 80% der Fälle klinisch
manifeste Sklettbeteiligung:
•• Schmerzen
•• Osteolysen
•• Hyperkalziämie
•• pathologische Frakturen
•• diffuse Osteopenie
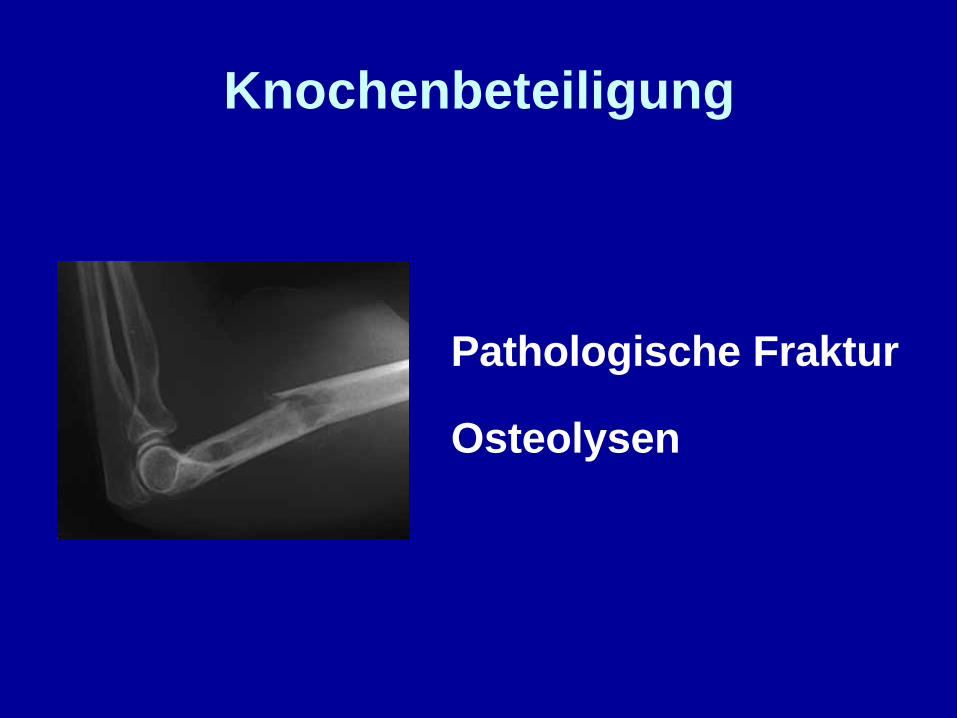
Knochenbeteiligung
Pathologische Fraktur
Osteolysen
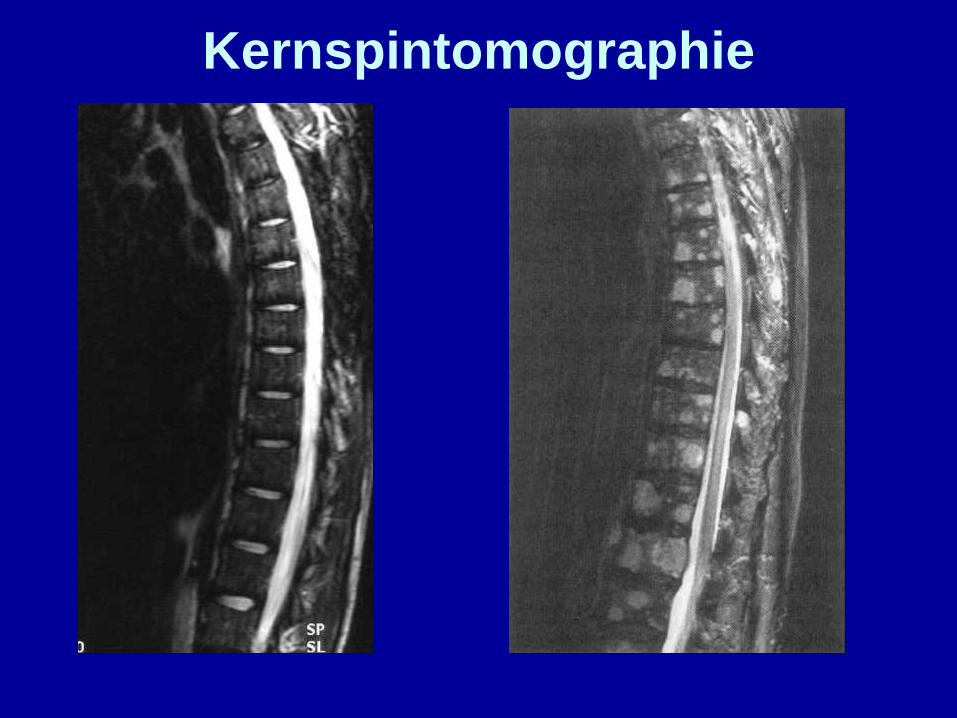
Kernspintomographie
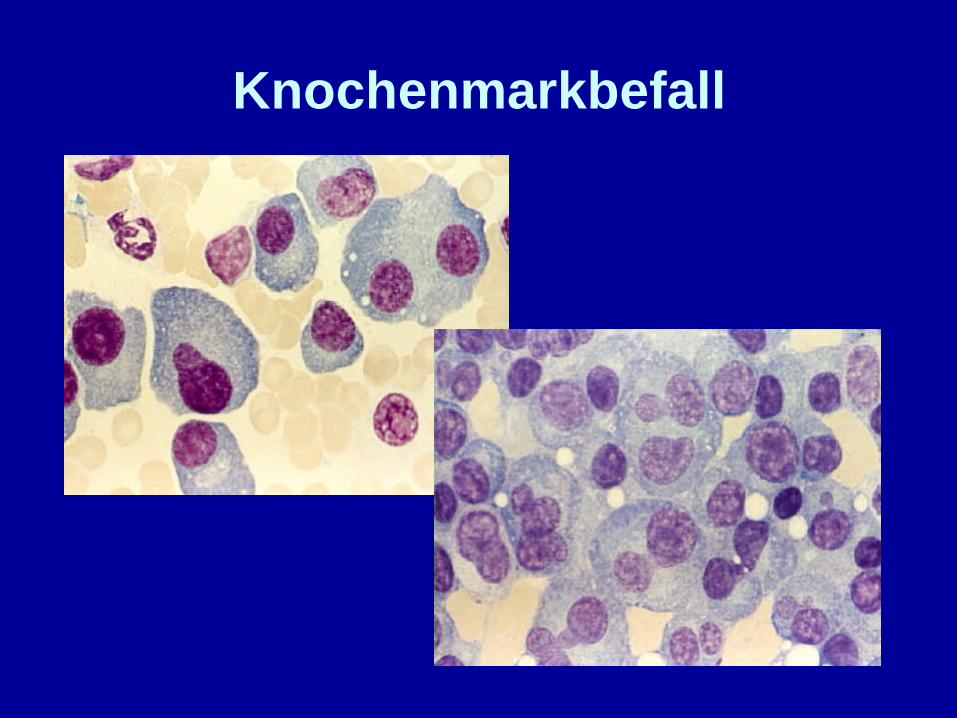
Knochenmarkbefall

Multiples Myelom: Niereninsuffizienz
Klinik: Zeichen der Urämie (Übelkeit, Erbrechen, psychotische
Ö
•• Myelomniere, nephrotisches Syndrom: Niederschlag von
Eiweißzylindern in den Tubuli (Korrelation mit Leichtketten-
)
•• Hyperkalziämie: 30-40% der Patienten mit MM,
prä
•• Nephrotoxische Substanzen: NSAID, cave Konstrastmittel
•• Amyloidose

Multiples Myelom: Infektionen
Ätiologie:
•• Dysfunktion bei Antikörperantwort
•• oft zusätzlich T-Zell-, und NK-Zell-Defekt
•• Therapiefolge: CTX, Kortikosteroide
•• bevorzugt bakterielle Infektionen

Multiples Myelom: weitere Symptome
Allgemein: Schwäche, Müdigkeit, Gewichtsverlust
Neurologische Symptome:
•• spinale Kompression
•• periphere Polyneuropathie (selten), nahezu immer
ät:
•• selten (< 10% der Fälle, besonders IgA-Paraprotein)
Visusverschlechterung, zerebrale + pulmonale Symptome
•• Blutungsneigung
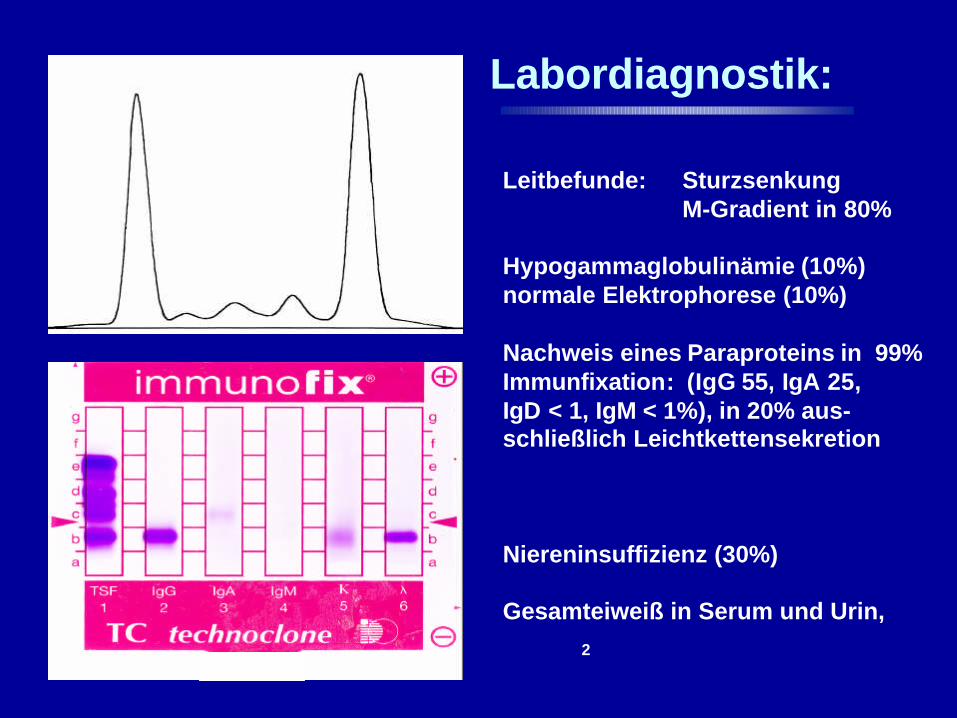
Labordiagnostik:
Leitbefunde: SturzsenkungM-Gradient in 80%
Hypogammaglobulinämie (10%)normale Elektrophorese (10%)
Nachweis eines Paraproteins in 99%Immunfixation: (IgG 55, IgA 25, IgD < 1, IgM < 1%), in 20% aus-schließlich Leichtkettensekretion
Niereninsuffizienz (30%)
Gesamteiweiß in Serum und Urin,
2

Prognosefaktoren
• Veränderungen des Erbguts
– Deletion Chromosom 13 oder 11
• Beta 2-Mikroglobulin, CRP
• Differenzierungsgrad
• Fortgeschrittenes Stadium
• Leichtkettenplasmozytom, IgA, IgD
• Nierenfunktionsstörung
• Alter des Patienten
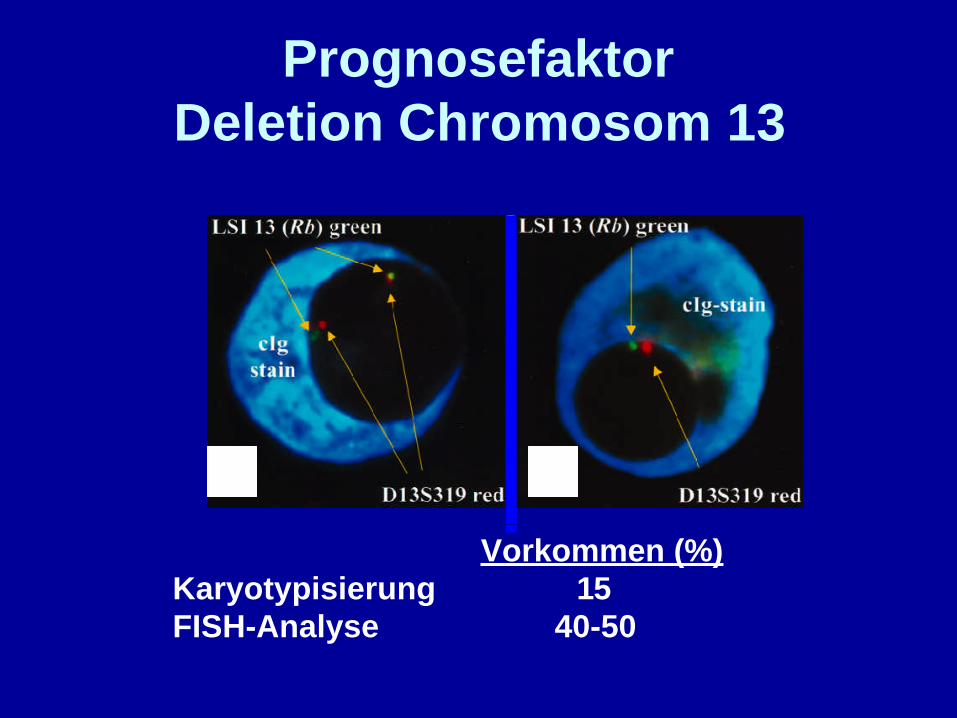
PrognosefaktorDeletion Chromosom 13
Vorkommen (%)Karyotypisierung 15FISH-Analyse 40-50
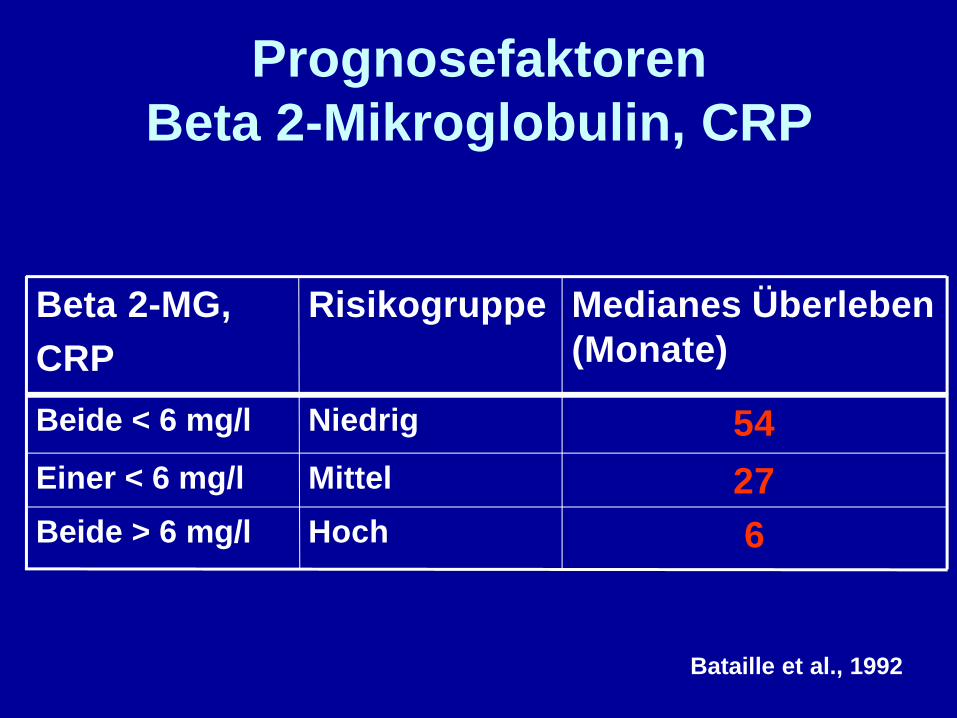
PrognosefaktorenBeta 2-Mikroglobulin, CRP
Bataille et al., 1992
6HochBeide > 6 mg/l
27MittelEiner < 6 mg/l
54NiedrigBeide < 6 mg/l
Medianes Überleben(Monate)
RisikogruppeBeta 2-MG,CRP

Bewährte Therapie
• Schmerztherapie
• Strahlentherapie, (Operation)
• Biphosphonate
• Standard Chemotherapie
• Hochdosistherapie mit Stammzelltransplantation

Therapie wann?
ØStadium II mit Fortschreiten, Stadium III
ØImmer:
ØNiereninsuffizienz
ØHyperkalzämie
ØSchmerzen
ØFrakturgefährdete Osteolysen

Biphosphonate

Bisphosphonate Präparate
Präparat Dosis Dauer Intervall
Clodronat 1600 mg po tgl.
(Bonefos, Ostac®) 1500 mg iv 4h 4 Wochen
Pamidronat (Aredia®) 90 mg iv 4h 4 Wochen
Zoledronat (Zometa®) 4 mg iv 15 min 4 Wochen
Ibandronat (Bondronat®) (4 mg iv 30 min 4 Wochen)
Normaler Knochenumbau
•
•
•
•
Mundy GR, 1999
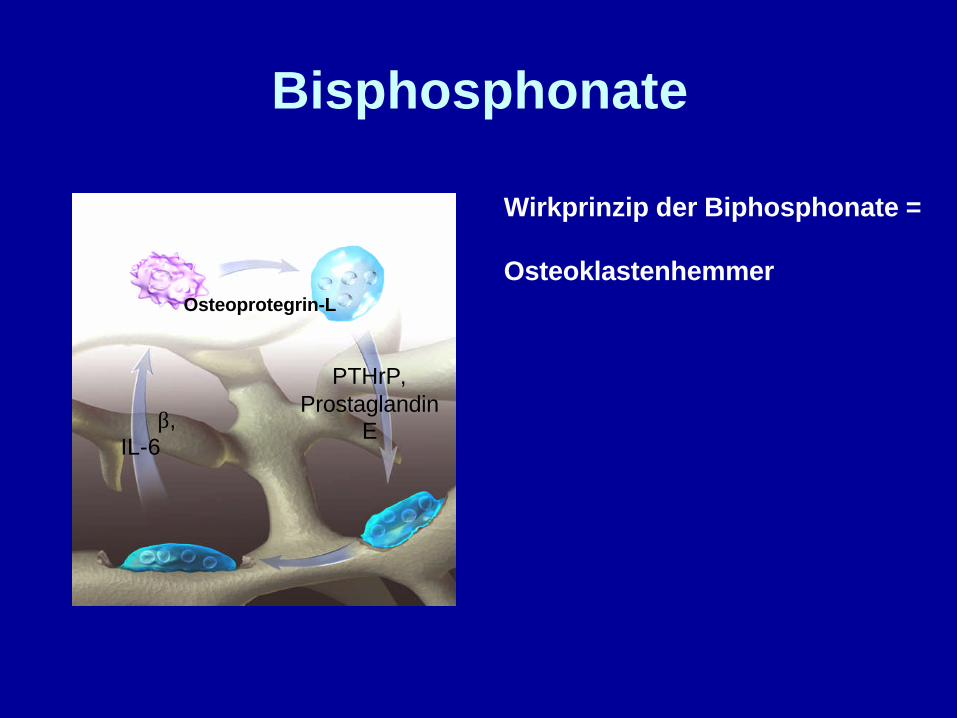
Bisphosphonate
β,IL-6
PTHrP,Prostaglandin
E
Osteoprotegrin-L
Wirkprinzip der Biphosphonate =
Osteoklastenhemmer
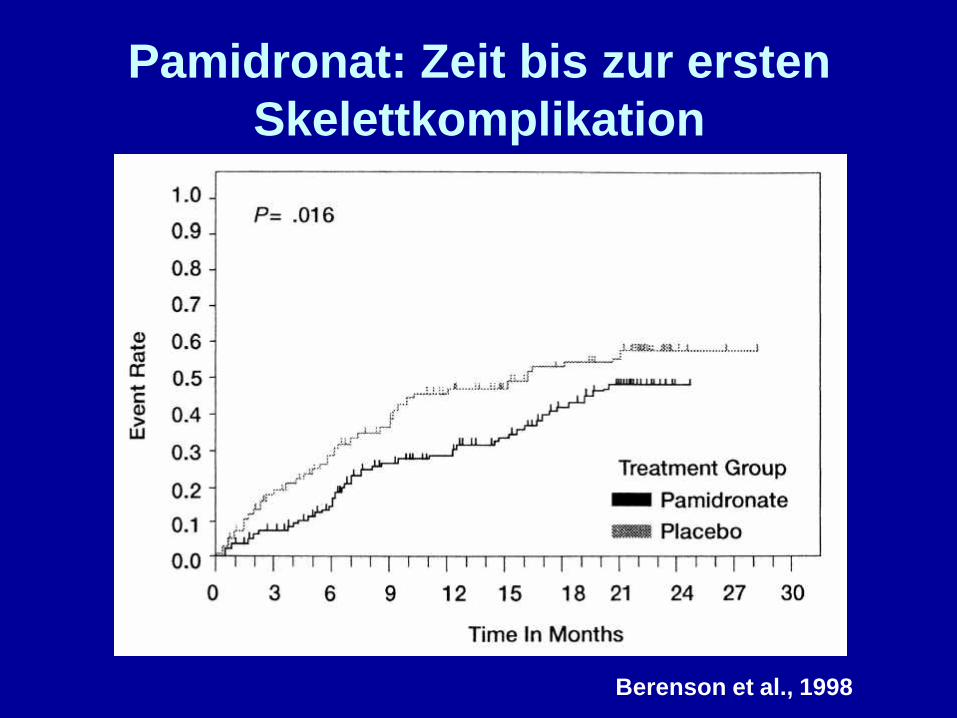
Pamidronat: Zeit bis zur erstenSkelettkomplikation
Berenson et al., 1998

Standard Chemotherapie

Alexanian-ProtokollMelphalanPrednisolon
VAD-ProtokollVinristinAdriamycinDexamethason
ID-ProtokollIdarubicinDexamethason
Standard Chemotherapie

Analyse von 6633 behandelten Patienten
Kombination (VAD)
Alexanian vs Kombinationstherapien

Hochdosistherapie mitStammzelltransplantation
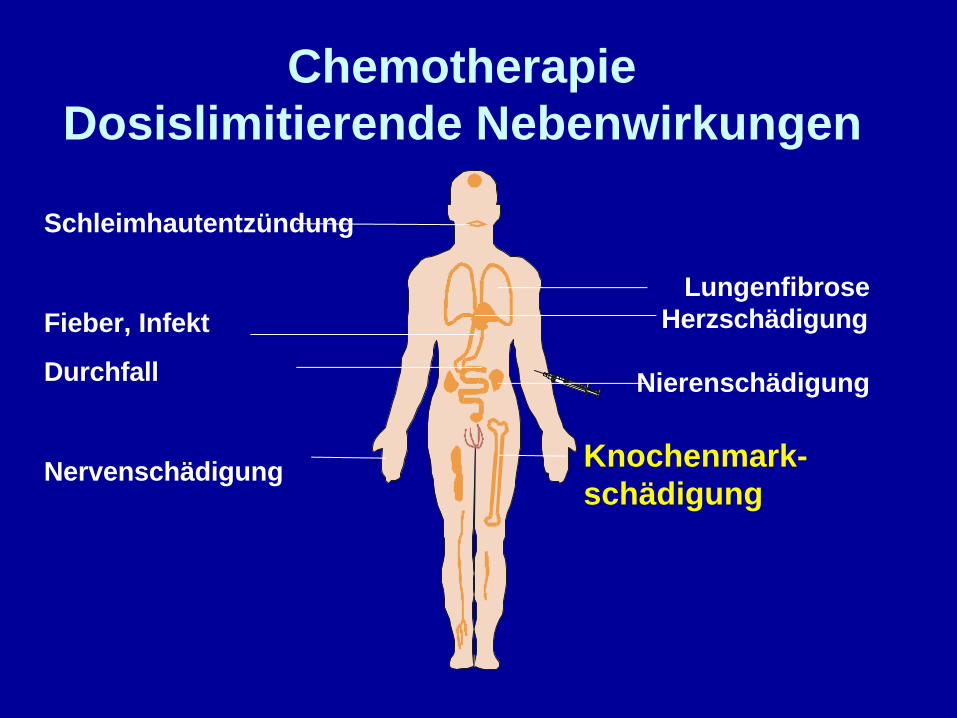
Schleimhautentzündung
Fieber, Infekt
Durchfall
Nervenschädigung
LungenfibroseHerzschädigung
Nierenschädigung
Knochenmark-schädigung
ChemotherapieDosislimitierende Nebenwirkungen
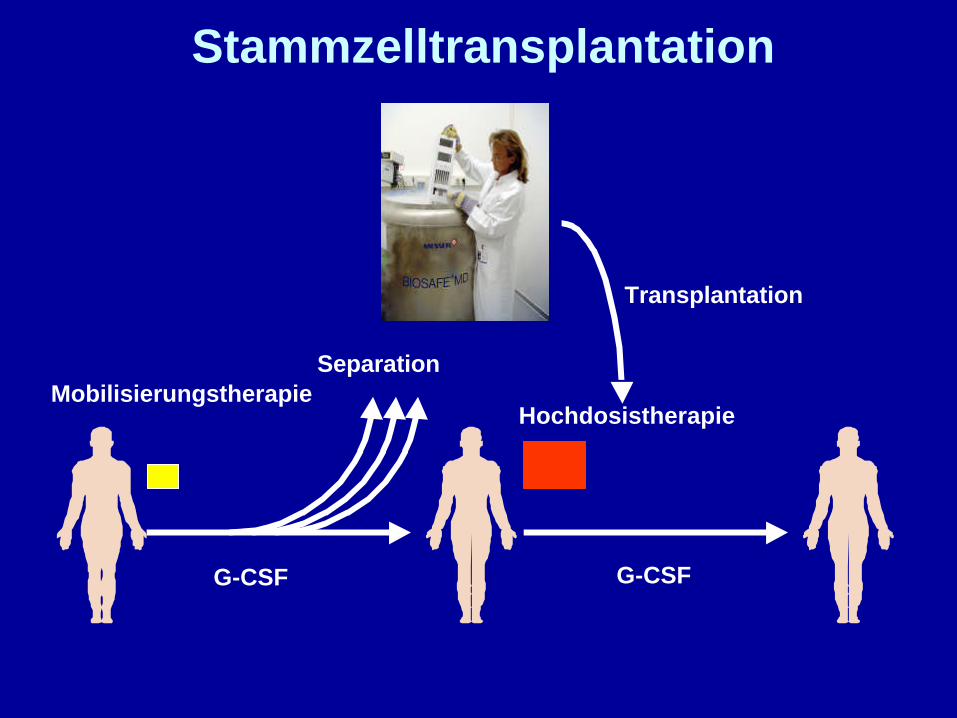
Stammzelltransplantation
G-CSF G-CSF
MobilisierungstherapieHochdosistherapie
Transplantation
Separation
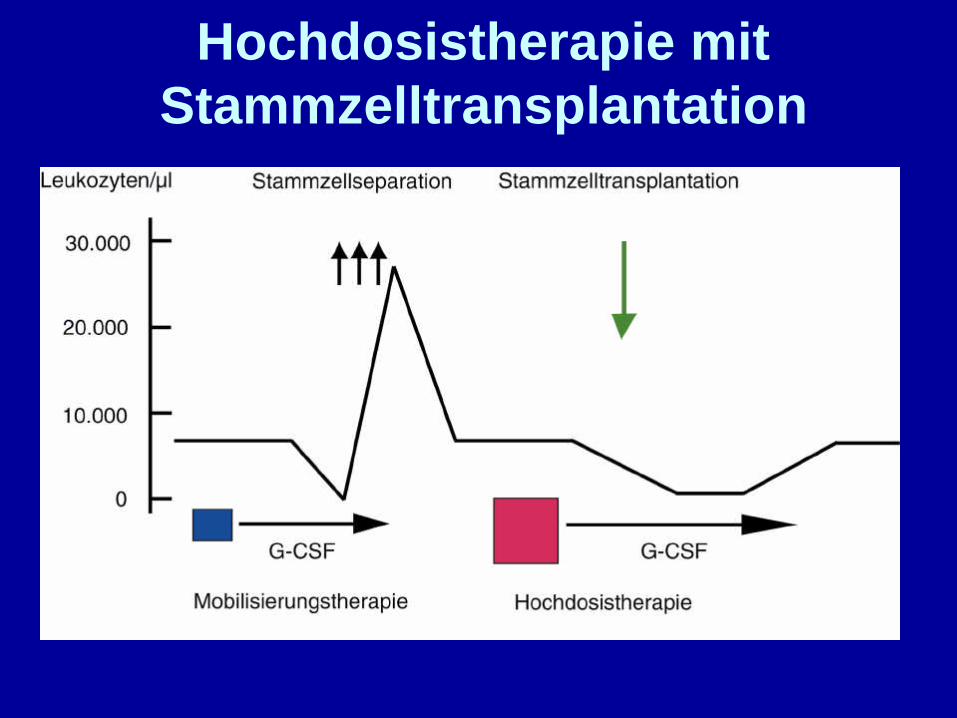
Hochdosistherapie mitStammzelltransplantation
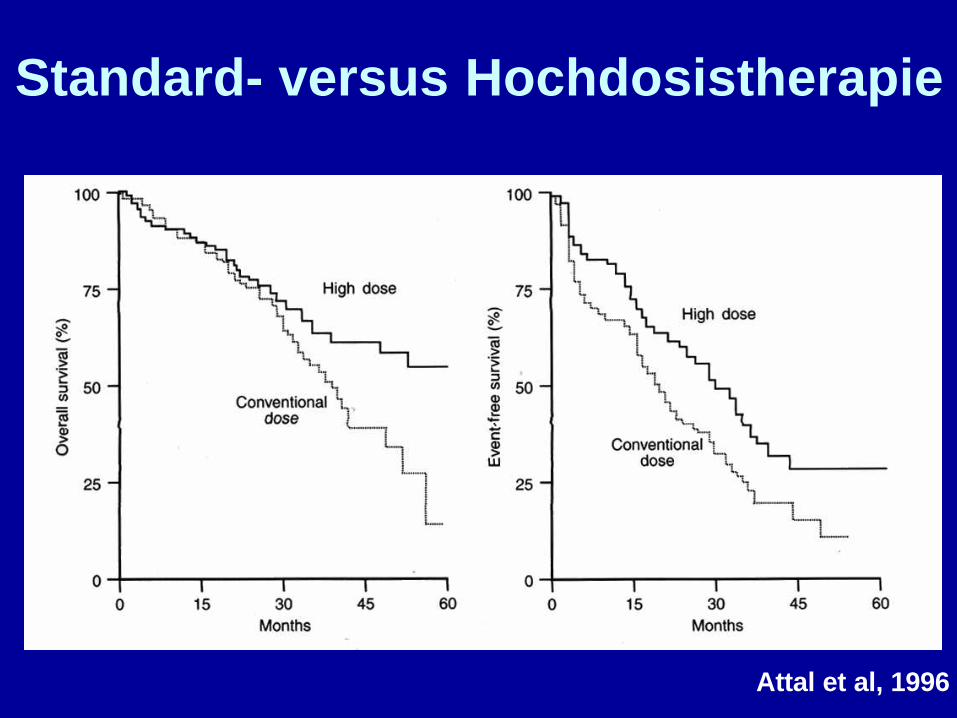
Attal et al, 1996
Standard- versus Hochdosistherapie

αα

Neue Therapeutika
• Thalidomid
• Bortezomib (Velcade®)
• Lenalidomid

Morbus Hodgkin

MORBUS HODGKIN
Ø häufigste maligne Erkrankung des
Ø Erstbeschreibung 1832 durch den Engländer Thomas
Ø pathologisches Merkmal = große (Sternberg-Reed-Zellen )
Ø überschießende Entzündungsreaktion des Organismusmit Bildung eines Granulationsgewebes
Krankheitsmanifestationen

EPIDEMIOLOGIE DESMORBUS HODGKIN
Ø ca. 1% aller malignen Erkrankungen
Ø Inzidenz: 2-3/100.000/Jahr
Ø bimodale Altersverteilung (Haupterkrankungsgipfel20-30 Jahre, zweiter Gipfel nach dem 60. Lebensjahr)
Ø Männer doppelt so häufig wie Frauen betroffen
Ø Ätiologie unbekannt (Viren? genetische Einflüsse?
Epstein-Barr-Virus?)

Histologische KLASSIFIKATIONØ lymphozytenreicher Subtyp
Häufigkeit < 5%, Hauptlokalisation: zervikaleLymphknoten, sehr günstige Prognose
Ø noduläre Sklerose
Häufigkeit 60-70%, Hauptlokalisation: Lymphknoten, günstige Prognose
Ø Mischtyp
Häufigkeit 20-30%, keine topographischePräferenz, mäßige Prognose
Ø lymphozytenarmer Typ
Häufigkeit < 5%, Hauptlokalisation: Lymphknoten, schlechte

KLINIK DES MORBUS HODGKINØ Allgemeinsymptome (allgemeines Krankheitsgefühl,
Gewichtsverlust, Fieber (Sonderform = Pel-Ebstein-Fieber),Nachtschweiß, Hautjucken)
Ø Leitbefund = schmerzlose Lymphadenopathie,
Ø Splenomegalie (35% der Patienten).
Ø Hepatomegalie
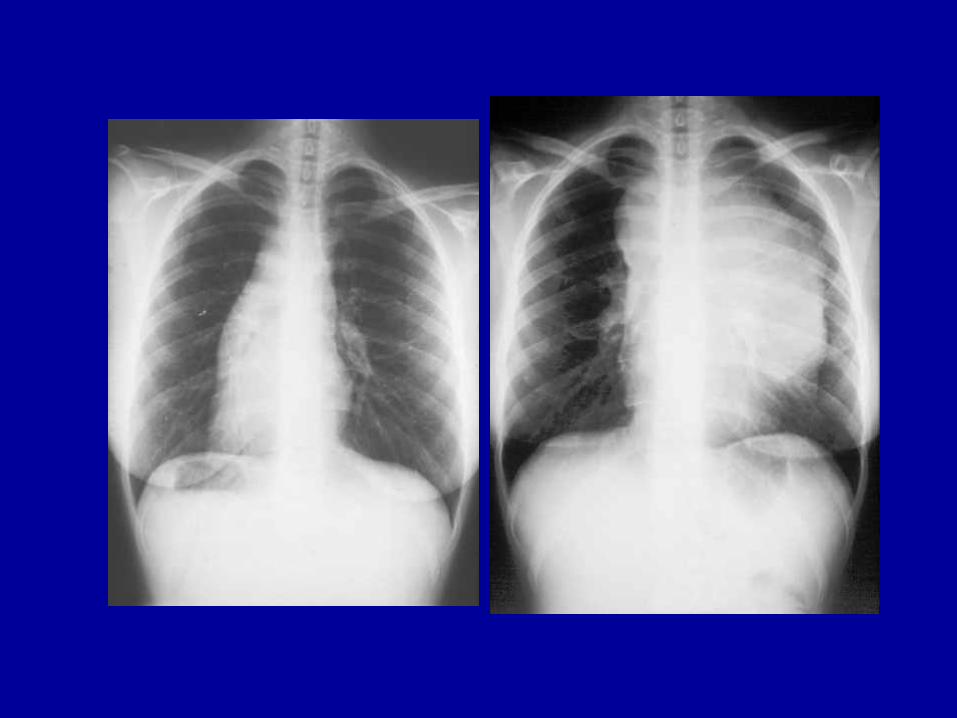
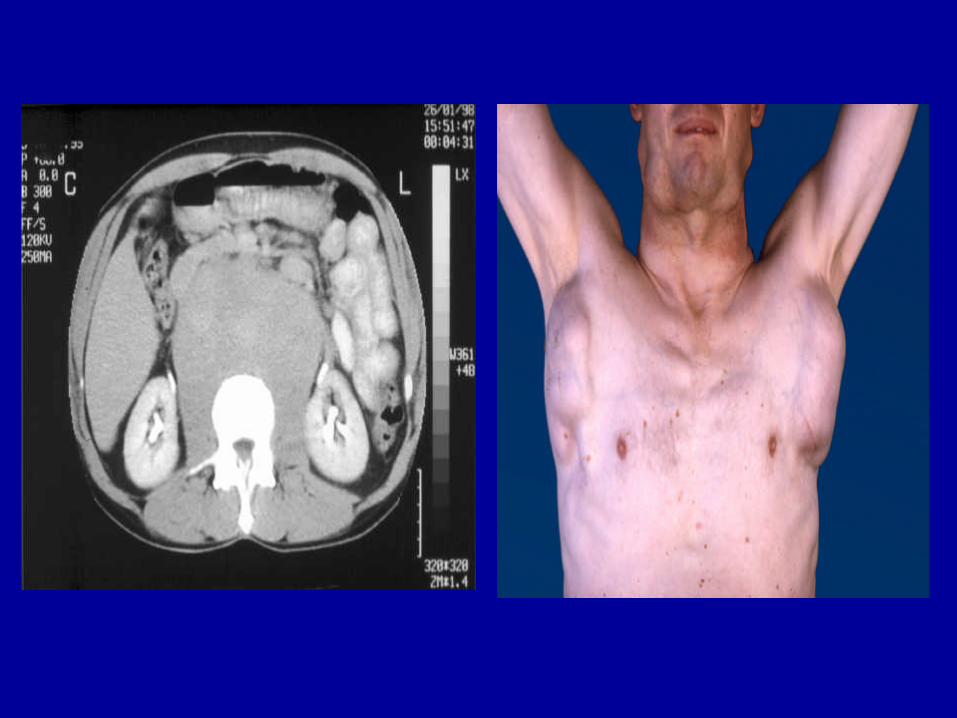
Ø Symptome durch andere extranodale Krankheitsmanifestationen(Lunge und Pleura (10-20%), Knochen (7%), Knochenmark (3%),ZNS, Perikard, Haut u.a.)

DIAGNOSE DES MORBUS HODGKIN
Ø Diagnosesicherung immer histologisch durchgroßzügige Lymphknotenbiopsie (Biopsie), nach Möglichkeit nicht ausLeistenlymphknoten
Ø zytologische Lymphknotenuntersuchung nichtausreichend!

STAGING-UNTERSUCHUNGEN
Ø Anamnese (B-Symptomatik?)
Ø körperliche Untersuchung (Lymphknotenstatus,Hepatosplenomegalie)
Ø Laboruntersuchungen (BSG, Blutbild, ,,
Retentionswerte, Serumeiweißelektrophorese,

B-SYMPTOMATIK
Ø Nachtschweiß
Ø Fieber (> 38 °C) unklarer Genese
Ø Gewichtsverlust > 10 % in 6 Monaten
Computertomographie Abdomen

STAGING-UNTERSUCHUNGEN
Ø Röntgen LungeØ Ultraschall Bauch, HalsØ CT (MRT) von Hals, Thorax und Abdomen/BeckenØ BeckenkammbiopsieØ SkelettszintigrammØ Organbiopsie (Leber)Ø Galliumszintigraphie, PET (Positronenemissions-
tomographie)
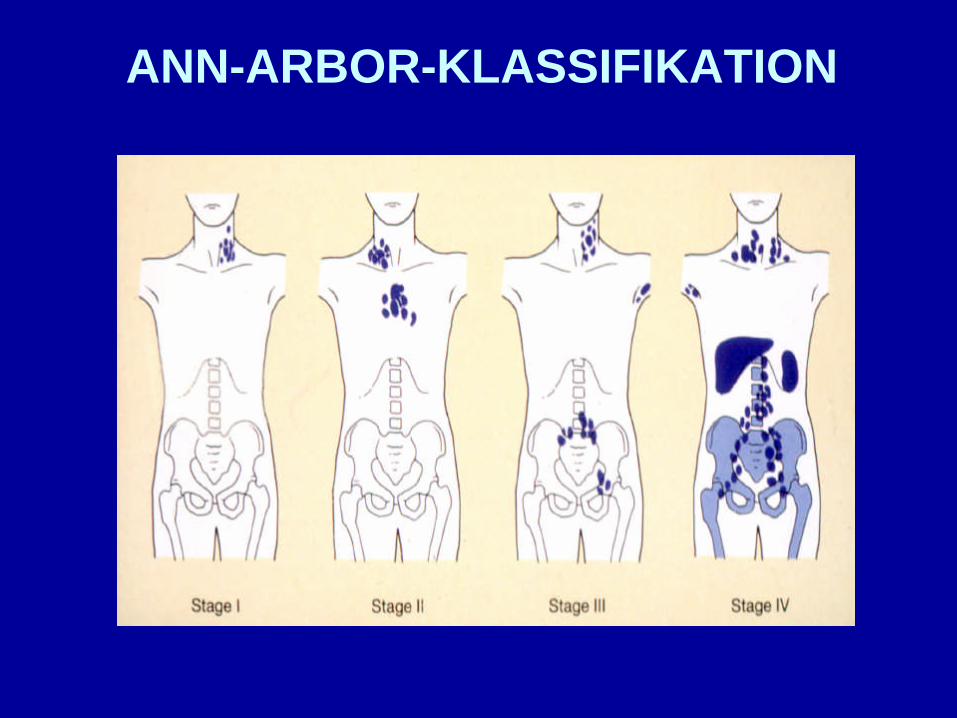
ANN-ARBOR-KLASSIFIKATION

ANN-ARBOR-KLASSIFIKATION
Zusätze
A/B B-Symptomatik
E Extranodalbefall
S Milzbefall

TherapiezielHeilung

PROGNOSEGRUPPEN(DEUTSCHE HODGKIN-STUDIENGRUPPE)
Ø Frühe oder lokalisierte StadienCS I/II ohne Risikofaktoren
Ø Intermediäre StadienCS IA/IIA mit einem oder mehreren RisikofaktorenCS IIB mit BSG > 30 mm und/oder > 3 befallenen
Lymphknotenarealen
Ø Fortgeschrittene StadienCS IIB mit mediastinalem Bulk, E-Befall oder massiverMilzbeteiligungCS IIIA mit einem oder mehreren Risikofaktoren

Deutsche Hodgkin Lymphom StudiengruppeUniversität Köln
Ø HD 13 für limitierte Stadien
Ø HD 14 für intermediäre Stadien
Ø HD 15 für fortgeschrittene Stadien
Ø LPHD für lymphozytenreiche Typ (IA)
Ø HD-R2 Rezidiv
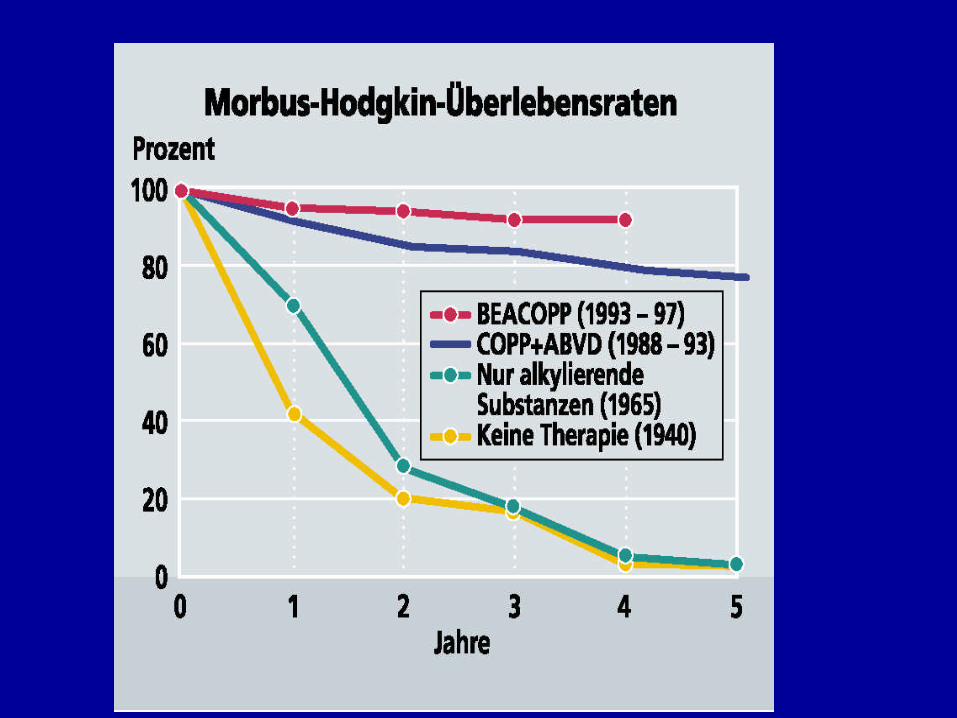

LANGZEITNEBENWIRKUNGENVON ZYTOSTATIKA
Ø Zweitneoplasien (nach 15 Jahren 11%), solideTumoren, MDS und AML, Non-Hodgkin-Lymphome
Ø Infertilität (Azoospermie bzw. Amenorrhoe nach 6Kursen MOPP 80%, nach ABVD nur 30%)
Ø Anthrazyklin-induzierte Kardiomyopathie
Ø Lungenfibrose (Bleomycin)
Ø Femurkopfnekrose (Steroide!, ca. 2% der Patienten)

Non Hodgkin-Lymphom

MALIGNER LYMPHOME
Ø Hodgkin-LymphomInzidenz 2-3/100.000/Jahr
Ø Non-Hodgkin-LymphomeInzidenz 3-6/100.000/Jahr

NHL: Diagnostik
Ø Histologie (Immunhistochemie)Ø ImmunphänotypisierungØ Zytogenetik

NHL: Symptome derErkrankung
ØLymphknotenvergrößerungen(tastbar)ØExtranodalbefall - spezifische
Symptome (Knochen, NNH, Lunge,Haut ...)Øseltener
• Fieber, Nachtschweiß,Gewichtsverlust
• Schmerzen

KLASSIFIKATION DERNON-HODGKIN-LYMPHOME
Ø Kiel-Klassifikation (Lennert et al, 1978)
Ø Working-Formulation (Rosenberg et al,1982)
Ø REAL-Klassifikation (Harris et al, 1994)
Ø WHO-Klassifikation (1997)

KLASSIFIKATION DERNON-HODGKIN-LYMPHOME
ØHochmaligne/AggressiveLymphome
ØNiedrigmaligne/IndolenteLymphome

KLASSIFIKATION DER NON-HODGKIN-LYMPHOME DERB-ZELL-REIHE (NIEDRIG-MALIGNE LYMPHOME)
Kiel-Klassifikation REAL-Klassifikation
Chronische lymphatische Leukämie vom B-Zell-chronische lymphatischeLeukämieB-Zell-Typ (B-CLL) LeukämieLymphoplasmozytoides Immunozytom B-CLL mit plasmazellulärerDifferenzierungLymphozytisches Lymphom Kleinzelliges lymphozytischesLymphomLymphoplasmozytisches Immunozytom Immunozytom (Morbus Waldenström)
Extranodale Marginalzonen-LymphomeMonozytoide Lymphome, einschließlich Nodale Marginalzonen-Lymphome(mono-Marginalzonen-Lymphome zytoid)
Splenisches Marginalzonen-LymphomHaarzellenleukämie HaarzellenleukämiePlasmozytisches Lymphom Plasmozytom, multiples MyelomZentroblastisch-zentrozytisches Keimzentrumslymphom,
Mantelzell-Lymphom

KLASSIFIKATION DER NON-HODGKIN-LYMPHOME DERB-ZELL-REIHE (HOCH-MALIGNE LYMPHOME)
Kiel-Klassifikation REAL-Klassifikation
Zentroblastisches Lymphom Diffuses großzelliges B-Zell-Lymphom
B-immunoblastisches Lymphom Diffuses großzelliges B-Zell-Lymphom
Großzelliges anaplastisches B-Zell- Diffuses großzelliges B-Zell-LymphomLymphom (Ki-1+) Mediastinales B-Zell-Lymphom
Burkitt-Lymphom Burkitt-LymphomBurkitt-ähnliches B-Zell-Lymphom
Lymphoblastisches Lymphom Vorläuferzell-B-lymphoblastisches

Merkmale niedrigmaligner Lymphome
1. Langsam progredienter Verlauf mitMöglichkeit spontaner Remissionen
2. Lebenserwartung mehrere Jahre
3. Mäßige Empfindlichkeit gegenüberkonventioneller Chemotherapie ohneHeilungschance
4. Gute Empfindlichkeit gegenüberRadiotherapie in lokalisierten Stadien

Merkmale hochmaligner Lymphome
1. Rasch progredienter Verlauf
2. Lebenserwartung ohne Behandlung mehrere
3. Mäßige bis gute Empfindlichkeit gegenüberkonventioneller Chemotherapie, kurativeTherapieoption
4. Gute Empfindlichkeit gegenüberRadiotherapie als adjuvante Maßnahme
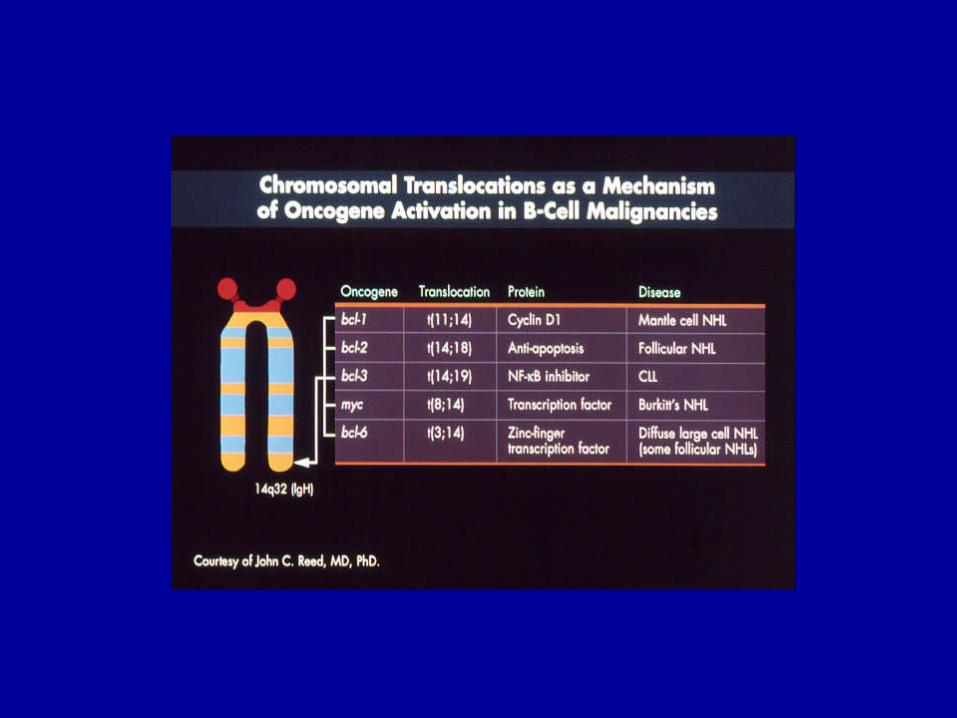

Parameter Günstig Ungünstig
Alter < 60 J. > 60 J.Allgemeinzustand < 2 > 2Extranodalbefall < 2 > 2LDH normal erhöhtAnn Arbor Stadium I, II III, IV
Hochmaligne NHL:Internationale Prognostische Index (IPI)

Non-Hodgkin-LymphomeTherapeutische Strategien
Ø Indolente Lymphome („Niedrig-maligne“)–– In niedrigen Stadien lokale Radiatio - sonst
systemisch
Ø Aggressive Lymphome („Hoch-maligne“)– primär kurativ– immer systemisch
Ø Sehr aggressive Lymphome (ALL-ähnlich)– Primär kurativ– ALL-ähnliche Mehrphasenprotokolle, .

Standard-Chemotherapie NHL:R-CHOP
Rituximab 375 mg/m2 i.v. d 1
Cyclophosphamid 750 mg/m2 i.v. d 1Doxorubicin 50 mg/m2 i.v. d 1Vincristin 1.4 mg/m2 i.v. d 1Prednis(ol)on 100 mg p.o. d 1-5
Wiederholung: 21-tägig
McKelvey et al. 1976
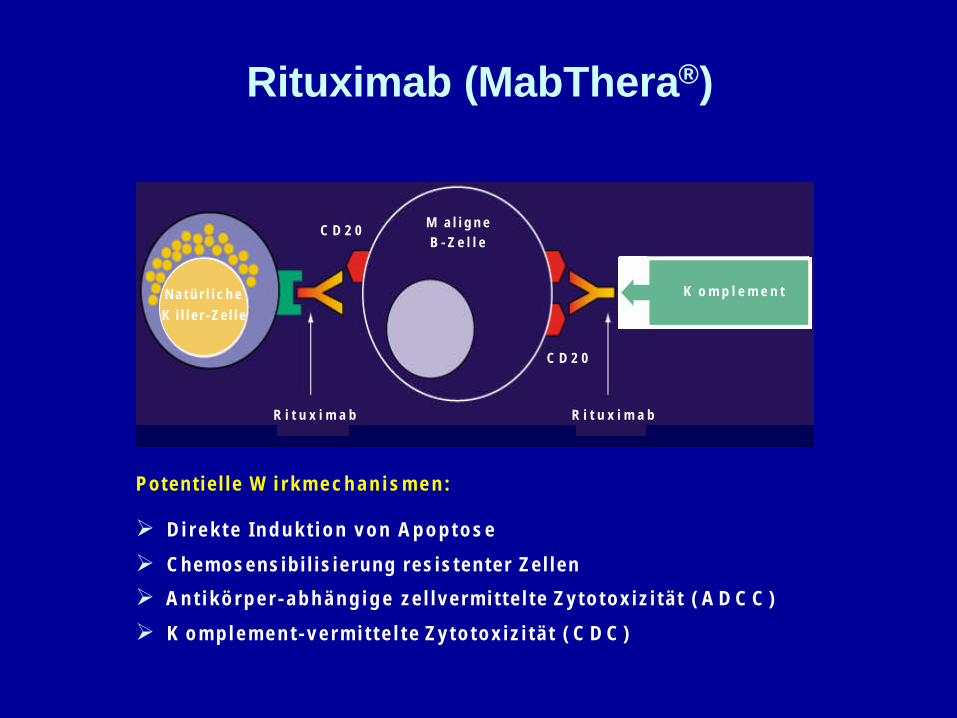
Poten t i e l l e W i r k m e c h a n i s m e n :
Ø D i r e k t e I n d u k t i o n v o n A p o p t o s e
Ø C h e m o s e n s i b i l i s i e r u n g r e s i s t e n t e r Z e l l e n
Ø A n t i k ö r p e r - a b h ä n g i g e z e l l v e r m i t t e l t e Z y t o t o x i z i t ä t ( A D C C )
Ø K o m p l e m e n t - v e r m i t t e l t e Z y t o t o x i z i t ä t ( C D C )
K o m p l e m e n t
R i t u x i m a bR i t u x i m a b
M a l i g n eB - Z e l l e
C D 2 0
C D 2 0
N a t ü r l i c h eK i l l e r - Z e l l e
K o m p l e m e n t
Rituximab (MabThera®)
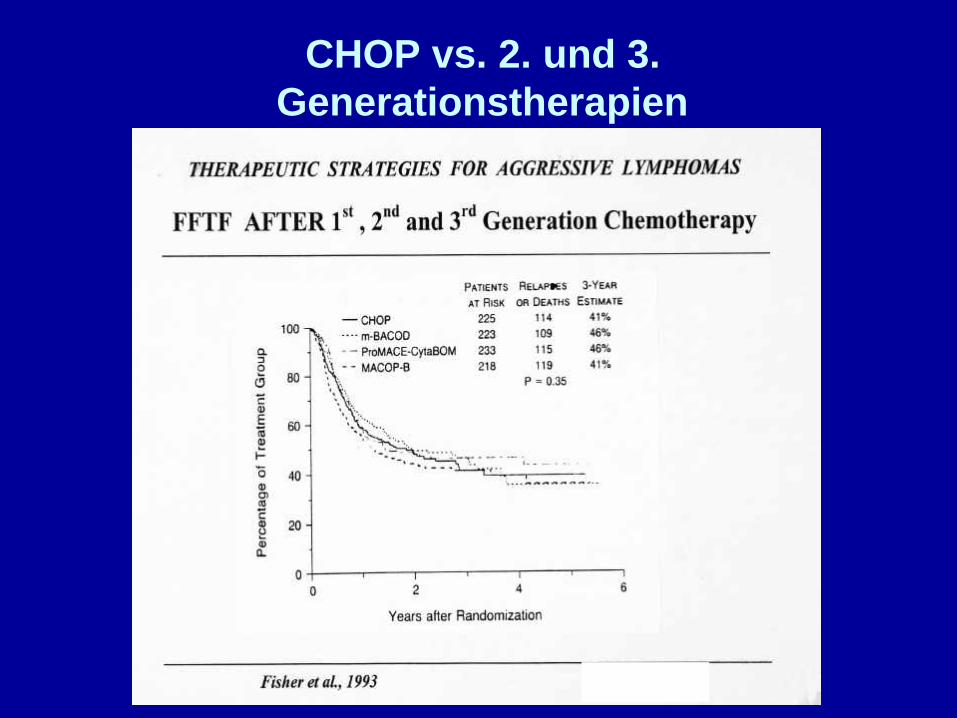
CHOP vs. 2. und 3.Generationstherapien

CHOP
Langzeitremissionen ca. 40 %
McKelvey et al., 1976
Aggressive LymphomeStandardtherapie
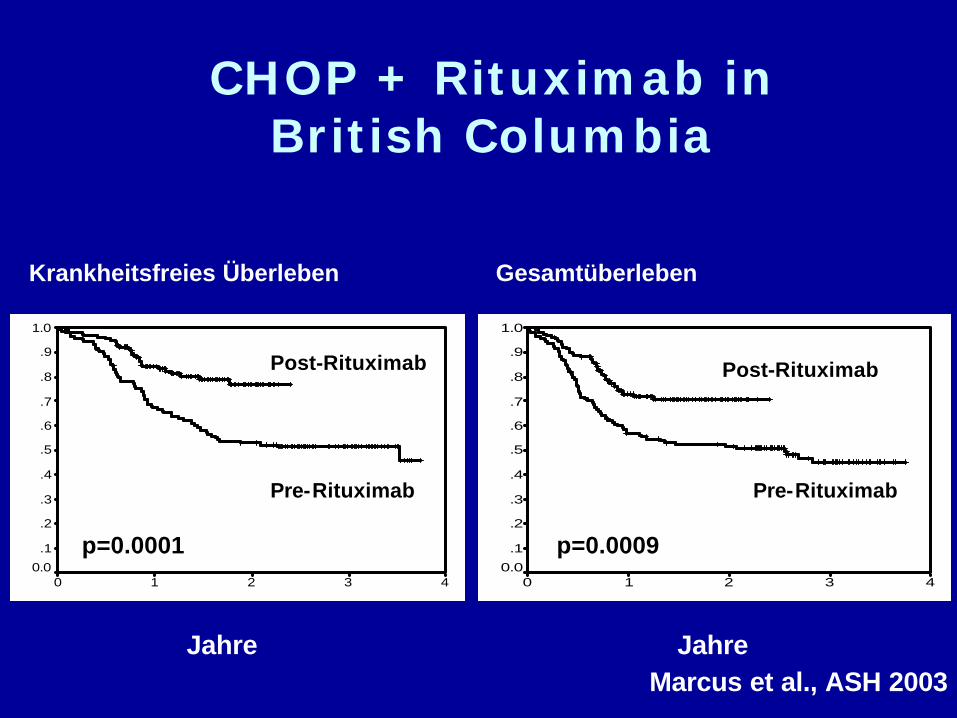
Jahre
CHOP + Rituximab inBritish Columbia
43210
1.0
.9
.8
.7
.6
.5
.4
.3
.2
.1
0.0
Post-Rituximab
Pre-Rituximab
p=0.000143210
1.0
.9
.8
.7
.6
.5
.4
.3
.2
.1
0.0
p=0.0009
Post-Rituximab
Pre-Rituximab
JahreMarcus et al., ASH 2003
GesamtüberlebenKrankheitsfreies Überleben
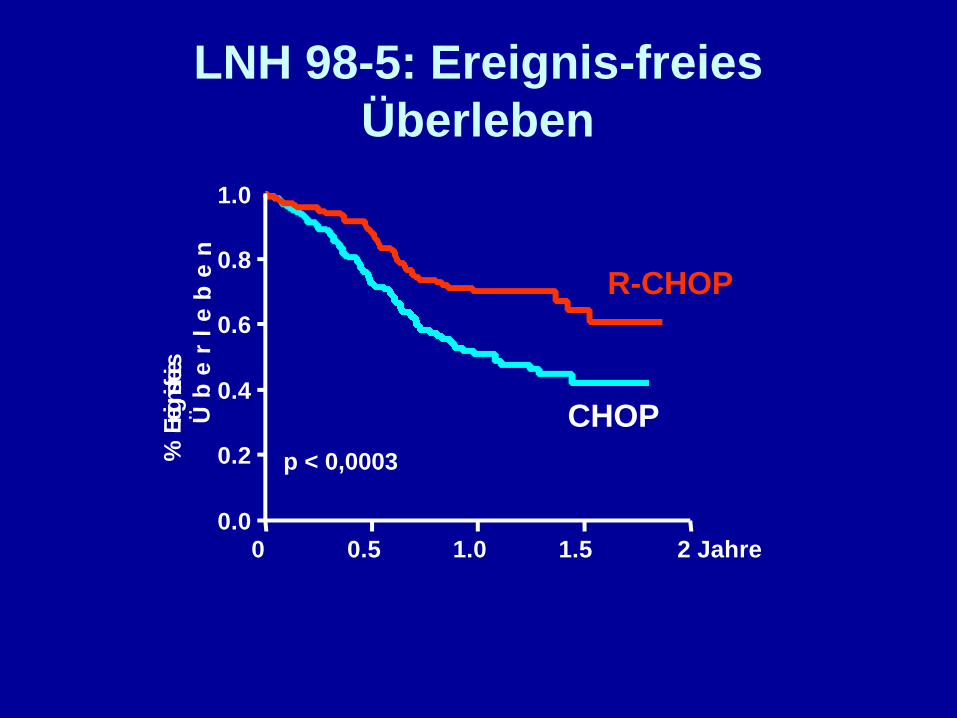
0 0.5 1.0 1.5 2 Jahre0.0
0.2
0.4
0.6
0.8
1.0%
Ere
ignisfre
ies
Üb
er
le
be
n
R-CHOP
CHOPp < 0,0003
LNH 98-5: Ereignis-freiesÜberleben

Burkitt-Lymphom: Therapie
Ø Operation mit Entfernung großer Tumoranteile
Ø Heilung durch intensive ChemotherapieØ „Hölzer –Protokoll“Ø Rituximab großer Fortschritt
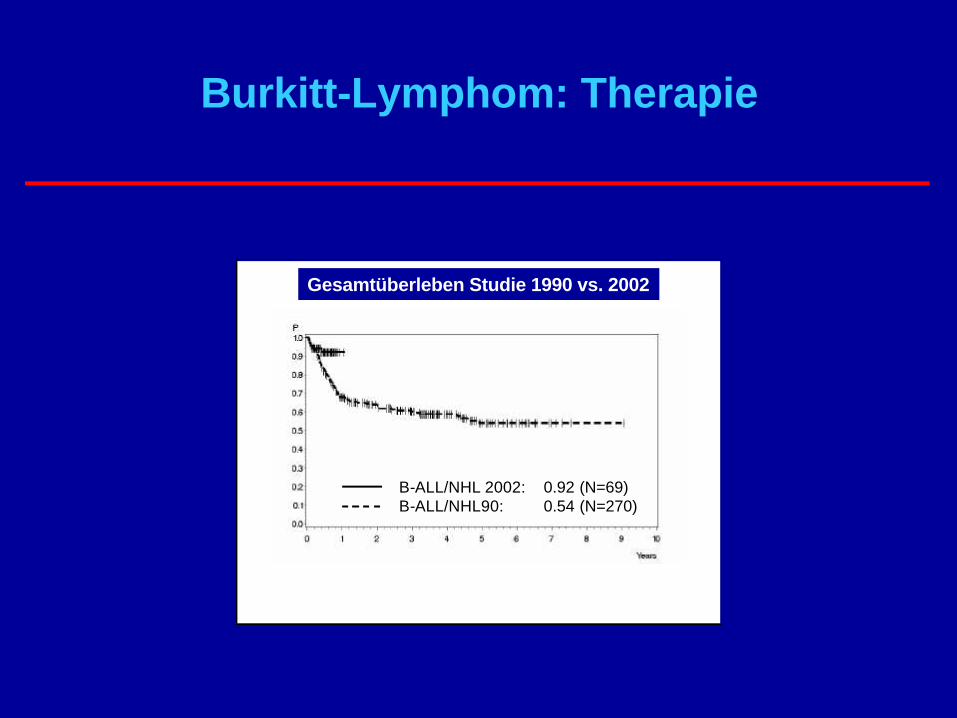
Burkitt-Lymphom: Therapie
Overall Survival GMALL in Adult B-ALL/NHL
B-ALL/NHL 2002: 0.92 (N=69)B-ALL/NHL90: 0.54 (N=270)
Gesamtüberleben Studie 1990 vs. 2002

Recommended

















