Embed Size (px)
Citation preview

1
INSTITUT FÜR BIOCHEMIE
Biochemisches Grundpraktikum
für Biochemiker (Bachelor)
und Chemiker (Bachelor, Wahlpflicht)
Kurs Wintersemester 2017/2018
Betreuer: Dr. J. Stichel, M.Sc. O. Hennig, M.Sc. F. Ullm, C. Dammann

2
VORBEREITUNG DER VERSUCHE
Für die erfolgreiche Durchführung der einzelnen Versuche ist es notwendig, sich im Vorfeld des betreffenden Versuches mit den jeweiligen Vorschriften zu befassen.
GRUPPENARBEIT Die Versuche werden von einer Gruppe aus jeweils drei Kursteilnehmern durchgeführt. Bitte wechseln Sie sich bei der praktischen Versuchsdurchführung und beim Protokollieren ab, da sich viele Arbeitsgänge wiederholen.
ALLGEMEINE VERHALTENSREGELN
Alle Laborarbeiten bringen den Umgang mit Substanzen mit sich, die bei nicht sachgemäßer Handhabung zu Gesundheitsschäden führen können. Es ist deshalb darauf zu achten, dass bei allen Laborarbeiten die Sicherheit am Arbeitsplatz gewährleistet ist:
Bitte tragen Sie während des Aufenthaltes in den Praktikumsräumen einen Laborkittel, festes Schuhwerk sowie gegebenenfalls weitere Schutzausrüstung (Handschuhe/ Schutzbrillen)!
Im Labor darf nicht gegessen, getrunken oder geraucht werden!
Bitte unterlassen Sie ebenfalls das Abspielen von Tonträgern in den Laborräumen!
Bitte achten Sie auf Sauberkeit und Ordnung am Arbeitsplatz: Ein unordentlicher Arbeitsplatz führt leichter zu Unfällen, aber oftmals auch zum Misslingen des Experimentes!
Alle Gefäße, die Lösungen oder Chemikalien enthalten, sind sorgfältig zu beschriften!
Sämtliche Lösungen, insbesondere konzentrierte Säuren und Laugen sowie biologische Flüssigkeiten, dürfen nicht mit dem Mund pipettiert werden!
Jede Mitnahme von Chemikalien aus den Praktikumsräumen ist untersagt!
Alle Chemikalien dürfen nur in den zum Versuch notwendigen Mengen aus den Vorratsflaschen entnommen werden. Bitte füllen Sie nicht gebrauchte Lösungen nicht in die Vorratsgefäße zurück, Kontaminationsgefahr!
Bitte bedienen Sie kein Gerät ohne vorherige Einweisung! Die Schäden von Fehlbedienungen belaufen sich meist auf mehrere Tausend Euro!
Das Öffnen von unter Strom stehenden Elektrophoresekammern ist lebensgefährlich!
Bitte informieren Sie sich über die vorhandenen Sicherheitseinrichtungen (z. B. Feuerlöscher, Verbandskästen, Notbrause, Augendusche usw.)
Aus gegebenem Anlass möchten wir darauf hinweisen, dass das Telefonieren mit Mobiltelefonen nur außerhalb des Praktikumsraumes gestattet ist.

3
Haftungsregelungen Die ständig steigenden Reparaturkosten haben zu den folgenden Anweisungen des Institutsdirektors geführt:
Jeder Student muss für Glasbruch und sonstige selbst verschuldete Sachschäden in Höhe des Anschaffungspreises bzw. der Reparaturkosten selbst aufkommen.
Bei Gruppenpraktika haftet die Praktikumsgruppe, wobei alle Studenten einer Gruppe für die ausgehändigten Geräte quittieren.
Die Ausgabe der Praktikumsscheine erfolgt erst, wenn keine finanziellen Forderungen seitens des Institutes mehr bestehen.
BEENDIGUNG DER PRAKTISCHEN ARBEIT Bitte bewahren Sie Ihre Proben zunächst für eventuelle Nachmessungen auf. Entsorgen Sie sie erst, wenn sie wirklich nicht mehr benötigt werden. Nach Beendigung Ihrer Versuche hinterlassen Sie bitte den Arbeitsplatz sauber und aufgeräumt. Spülen Sie alle benutzten Hilfsmittel und Glasgeräte (Pipetten, Bechergläser, Vorratsflaschen etc.). Bitte schalten Sie alle elektrischen Messgeräte ordnungsgemäß aus.
Bitte lassen Sie ihr Laborheft vom Betreuer gegenzeichnen.

4
HINWEISE ZUR PROTOKOLLFÜHRUNG
Die Protokolle sind Gruppenprotokolle, d.h. es wird pro Versuch ein gemeinsam erarbeitetes Protokoll abgegeben und daher auch gemeinsam verantwortet. Die Experimente werden in einem Laborheft vorbereitet, die Pipettierschemata tabellarisch aufgestellt. Wichtig ist, dass alle Arbeitsvorgänge und Beobachtungen sofort im Laborheft notiert werden. Dies betrifft ermittelte Werte (z.B. Volumina, Ergebnisse, verwendete Geräte) als auch Pannen und Besonderheiten. In Verbindung mit der Praktikumsanleitung muss es dem Leser möglich sein, den Verlauf und das Ergebnis der Experimente nachvollziehen zu können. Laborhefte müssen leserlich geschrieben werden und dürfen nach Beendigung der Arbeiten nicht noch einmal abgeschrieben werden.
Das Protokoll wird mit Hilfe der Unterlagen (Laborheft, Chromatogramme, Tabellen, Messwerte usw.) zusammengestellt. Bitte behandeln Sie jeden Versuch einzeln, die einzelnen Protokolle werden möglicherweise von verschiedenen Betreuern korrigiert. Ein Protokoll ist für folgende Versuche anzufertigen:
Reinigung von Lysozym aus Hühnereiweiß und SDS-PAGE
Konzentrationsbestimmung von Proteinen
Saure und enzymatische Hydrolyse
Enzymkinetik
Die Protokolle aller Experimente sollen bitte spätestens 4 Wochen nach Beendigung des Praktikumskurses abgegeben werden. Alle Protokolle sind als Gruppenprotokolle anzufertigen. Jedes Protokoll muss zudem eine von den Studierenden unterschriebene Erklärung enthalten, dass die Arbeit selbstständig verfasst wurde. Alle Gruppenmitglieder sind für den Inhalt des Protokolls verantwortlich.
Erklärung
Hiermit versichern wir, dass wir das vorliegende Protokoll selbständig verfasst und keine anderen als die im Literaturverzeichnis angegebenen Quellen benutzt haben. Die Zeichnungen oder Abbildungen in dieser Arbeit sind von uns selbst erstellt worden oder mit einem entsprechenden Quellennachweis versehen. Leipzig, den <Datum>
<Vollständige, handschriftliche Unterschriften>
Sofern Sie eine korrigierte Version Ihres Protokolls anzufertigen haben, legen Sie der korrigierten Version bitte Ihre alte Version bei!
Für die Zusammenschrift der Protokolle wird folgende Gliederung vorgeschlagen, die sich nach den üblichen Vorschriften zur Anfertigung von wissenschaftlichen Arbeiten richtet. Folgendes Schema dient dabei als Orientierung:

5
Allgemeine Angaben / Deckblatt Namen der Praktikanten, Gruppennummer, ggf. email-Adresse(n) Studienrichtung Name/ Jahr des Praktikums (Grundpraktikum Biochemie, WS 2013/2014) Name des Versuches (Überschrift laut Skript) Einleitung Die Einleitung sollte nicht länger als eine halbe bis maximal eine Seite sein und zielgerichtet auf die Aufgabenstellung hinführen. Sie soll eine kurze theoretische Einführung und Aussagen zum Inhalt und Sinn des Versuches enthalten. Der theoretische Hintergrund der verwendeten Methoden soll kurz erläutert werden. Materialien und Methoden Materialien Die verwendeten Chemikalien, Enzyme, Puffer etc. sind anzugeben. Die verwendeten Konzentrationen müssen klar nachvollziehbar sein. Geräte Geräte müssen nur dann aufgeführt werden, wenn sie nicht unbedingt zur Grundausstattung eines Labors gehören (z.B. größere Zentrifugen mit Fabrikat, Rotortyp; Photometer). Dabei sollten Sie sich auf das Wesentliche beschränken, die Art der verwendeten Glasgeräte, Pipetten usw. muss nicht berücksichtigt werden. Methoden Hier reicht ein Verweis auf das Praktikumsskript, wenn die Versuche wie dort beschrieben durchgeführt wurden. Anzugeben sind jedoch die tatsächlichen Bedingungen bei der Durchführung sowie Abweichungen in den Methoden, so dass eine lückenlose Nachvollziehbarkeit gegeben ist. Ergebnisse Die Darstellung der Ergebnisse erfolgt wie am Ende jeden Versuchsteils beschrieben in Form von Tabellen und Abbildungen. Beachten Sie dabei:
Alle Messdaten sind übersichtlich anzuordnen, wenn möglich in Tabellenform!
Tabellen erhalten eine Überschrift, sollte eine Legende (z.B. für Abkürzungen) notwendig sein, steht diese unter der Tabelle. Sind Ergebnisse durch Berechnungen erhalten worden, muss stets ein Rechenbeispiel angegeben werden, das den kompletten Rechenweg enthält! Die Berechnung muss nachvollziehbar und mit der Angabe von Einheiten ausgeführt werden!
Abbildungen werden mit Legenden versehen, die eine präzise Zuordnung der aufgeführten Daten erlauben! Abbildungen erhalten zudem eine geeignete Bildunterschrift, die den Inhalt der Abbildung sinnvoll wiedergibt. Abbildungen erhaltene eine fortlaufende Nummerierung, auf die im Text verwiesen werden muss.
Die Achsen der Diagramme sind genau zu beschriften!
Eine ausführliche Diskussion der Ergebnisse erfolgt erst im sich anschließenden Ergebnisteil!
Diskussion

6
In der Diskussion (maximal eine Seite) erfolgt in einem zusammenhängenden Text eine kritische Bewertung der Daten. Dabei gehen Sie bitte auf die Fragen am Ende der jeweiligen Versuchbeschreibung ein. Literaturangaben Bitte nicht vergessen! Stil des Protokolls Alle Illustrationen (Abbildungen und Tabellen) müssen fortlaufend nummeriert, sowie mit Legenden (Abbildungen) bzw. Überschriften (Tabellen) versehen werden. Die Beschriftungen der Illustrationen müssen selbsterklärend sein. Sie müssen im fortlaufenden Text auf die Illustrationen hinweisen, damit der Leser weiß, wo die entsprechenden Daten gezeigt sind, und er so der Argumentation des Verfassers folgen kann. Bitte geben Sie für numerische Daten nur die Stellen an, die signifikant sind (nicht alle Stellen, die Ihnen der Rechner angibt)! Beispiele für die wissenschaftlich korrekte Beschriftung einer Abbildung und einer Tabelle:
Prüfliste für den Text
Ist der Text exakt und für jemanden verständlich, der den Versuch nicht gemacht hat?
Sind alle Rechnungen verständlich und nachvollziehbar beschrieben? Bitte geben Sie insbesondere alle Verdünnungsschritte bei der Berechnung von Enzymaktivitäten an (Lysozymversuch).
Sind alle Tabellen und Abbildungen im Text zitiert?
Sind die Seiten fortlaufend nummeriert?

7
VERSUCH 1 :
REINIGUNG VON LYSOZYM AUS HÜHNEREIWEISS
EINLEITUNG
Lysozym katalysiert die Hydrolyse von Zellwänden grampositiver Bakterien. Das Enzym
wurde 1922 von Alexander Fleming erstmals im Nasensekret entdeckt, nachdem er
festgestellt hatte, dass Nasenschleim Bakterienkulturen auflöst. Lysozym ist weit
verbreitet in Tieren und Pflanzen, in Speichel, Tränen, Milch, in den Sekreten der
Atemwege und des Verdauungstraktes, in Leukozyten und den Nieren von Säugetieren
sowie insbesondere im Hühnereiweiß.
Lysozym spaltet bevorzugt die -1,4-glykosidische Bindung zwischen N-
Acetylmuraminsäuren (NAM) und N-Acetylglucosaminen (NAG), welche in der Zellwand
vieler Mikroorganismen vorkommen. Dabei wird die Bindung zwischen dem
Kohlenstoffatom 1 und dem Brückensauerstoff gespalten, so dass Disaccharide aus NAG
und NAM entstehen, an denen noch Peptidseitenketten hängen (Abb. 1):
Abb. 1: Lysozym hydrolysiert die glykosidische Bindung zwischen N-Acetylmuraminsäure (NAM) und N-Acetylglucosamin (NAG).

8
Die Kristallstrukturanalyse des Lysozyms hat gezeigt, dass das aktive Zentrum einen
langen Spalt enthält, der sein normales Substrat, das langkettige Peptidoglykan der
Bakterienzellwand, aufnehmen kann.
Lysozym aus Hühnereiweiß ist das am gründlichsten untersuchte Lysozym und eines der
mechanistisch am besten verstandenen Enzyme. Es ist ein kleines, stark basisches
Protein (MW = 14,4 kDa), dessen einzige Polypeptidkette aus 129 Aminosäuren besteht
und in sich durch vier Disulfidbrücken quervernetzt ist.
Von allen Mikroorganimen ist Micrococcus lysodeikticus besonders empfindlich gegen
eine Verdauung von Lysozym. Die bakteriolytische Eigenschaft des Lysozyms ist die
Grundlage für eine Nachweismethode des Lysozyms. Hierbei wird das Enzym mit einer
trüben Suspension gefriergetrockneter Bakterien gemischt. Bei fortschreitender
Hydrolyse der Zellwände nimmt die Trübung der Suspension ab. Diese
Trübungsabnahme kann bei 450 nm in einem Spektralphotometer verfolgt werden.
EIGENSCHAFTEN VON LYSOZYM
Aufbau und physikalische Eigenschaften: Lysozym ist ein monomeres Protein (MW =
14,4 kDa). Die Absorption einer 1 mg/ml Lösung bei 280 nm (A280) liegt bei 2,48. Der
isoelektrische Punkt liegt bei pH 11.
Definition der Enzymaktivität: Eine Einheit (U) Lysozym ist definiert als die Menge
Enzym, welche unter optimalen Bedingungen eine Trübungsabnahme einer
Bakteriensuspension von 0,001 pro Minute (min) bei 450 nm, pH 7,0 und 25 °C
hervorruft. Die relative spezifische Aktivität und die Gesamtaktivität des Enzyms sind
folgendermaßen definiert:
Relative spezifische Aktivität =
TestansatzimProteinmg
10min /ΔA3
450
(1)
Gesamtaktivität = rel. spez. Aktivität mg Gesamtprotein (2)
PROBLEMSTELLUNG
Aus dem Eiklar von Hühnereiern soll das Enzym Lysozym gereinigt werden. Hierzu wer-
den pH-Präzipitation und eine Chromatographie an einer Kationenaustauschersäule
verwendet. Zur Bestimmung der enzymatischen Aktivität des Lysozyms werden als

9
Testverfahren die Trübungsmessung und als Testsubstrat Micrococcus lysodeikticus
Bakterien verwendet.
MATERIALIEN
Puffer und Lösungen
Testpuffer: 0,1 M Kaliumphosphatpuffer; pH 7,0
Säulenpuffer A: 0,1 M Ammoniumacetatpuffer; pH 4,5
Säulenpuffer B: 0,1 M Ammoniumacetatpuffer; pH 7,0
Säulenpuffer C: 0,1 M Ammoniumacetatpuffer; 0,025 M Na2CO3; pH 9,0
Säulenpuffer D: 0,1 M Ammoniumacetatpuffer; 0,25 M Na2CO3; pH 10,0
Proteinpuffer E: 0,1 M Ammoniumacetatpuffer; 0,25 M Na2CO3; pH 7,0
Bakteriensuspension: 0,3 mg/ml gefriergetrocknete Micrococcus lysodeikticus
Bakterien suspendiert in Testpuffer
Materialien Carboxymethylcellulose CM-52-Säule, in dest. Wasser,
Fotometer und Küvetten für UV- bzw. sichtbares Licht
PRAKTISCHE DURCHFÜHRUNG
Allgemeine Methoden
Nach jedem Reinigungsschritt werden Gesamtproteinkonzentration und Lysozymaktivität
bestimmt. Ein Aliquot von jedem Reinigungsschritt wird mittels SDS-PAGE-
Gelelektrophorese analysiert, um den Erfolg der Reinigung zu dokumentieren (siehe
Versuch 2).
a) Konzentrationsbestimmung des Enzyms
Die Proteinkonzentration in den bei der Reinigung erhaltenen Fraktionen wird durch
Absorptionsmessungen in Küvetten bei 280 nm ermittelt. Da die Zusammensetzungen
der Proteinlösungen zu diesem Zeitpunkt noch nicht bekannt sind, errechnen sich die
Proteinkonzentrationen zunächst aus der Näherungsformel für Proteingemische. Hierbei
wird eine Absorption von A280 = 1 in guter Näherung gleich einer Proteinkonzentration
von 1 mg/ml gesetzt. Für reine Proteinlösungen ist diese Näherung zu ungenau, hier
müssen die jeweiligen spezifischen Extinktionskoeffizienten berücksichtigt werden. Für
Lysozym liegt die Absorption einer 1 mg/ml Lösung bei 280 nm (A280) bei 2,48.

10
Verwenden Sie zur Proteinbestimmung in der Messküvette geeignete Vorverdünnungen
ihrer Aufarbeitungsstufen, nehmen Sie dazu Testpuffer als Verdünnungspuffer. Ihre
Referenzküvette enthält jeweils 2,0 ml Testpuffer als Vergleich. Achten Sie darauf, nur
solche Verdünnungen zu verwenden, bei denen Ihre bei 280 nm gemessenen
Absorptionen Werte von 0,1 weder unterschreiten noch Werte von 1,0 überschreiten.
Orientieren Sie sich dazu an den Empfehlungen zur Verdünnung für die
Proteinbestimmung am Ende der jeweiligen Aufarbeitungsstufe. Um das Ausmaß von
Messfehlern durch Verdünnung und Messung abzuschätzen, führen Sie drei
unabhängige Verdünnungen und die dazugehörigen Messungen durch. Tragen Sie alle
Verdünnungsfaktoren, die tatsächlich pipettierten Volumina und die dazugehörigen
Messwerte in ihr Laborheft ein. Um die Proteinkonzentration in der jeweiligen Fraktion zu
bestimmen, berücksichtigen sie alle Verdünnungsschritte und tragen den
Verdünnungsfaktor, die durchschnittlich gemessene Absorption bei 280 nm (A280) und
die daraus resultierende Proteinkonzentration in der Fraktion (mg/ml) in Tabelle 1 ein.
b) Enzymtest für Lysozym
Die Suspension der Micrococcus lysodeikticus Bakterien wird 20-30 Minuten bei 37°C
vorinkubiert und vor Gebrauch gut durchmischt (dieser Schritt wird von den
Praktikumsassistenten durchgeführt). 1,9 ml dieser Suspension werden in eine Küvette
(2 ml) gefüllt. Anschließend wird die Reaktion durch Zugabe von 100 µl Lysozymlösung
einer geeigneten Vorverdünnung (verwenden Sie dazu immer Testpuffer als
Verdünnungspuffer) gestartet (vorher kurz umrühren!) und die Abnahme der Absorption
bei 450 nm und Raumtemperatur in Abhängigkeit von der Zeit (2 min) verfolgt.
Verwenden Sie solche Enzymverdünnungen, welche eine Trübungsabnahme von
minimal 0,02 A450/min und maximal 0,08 A450/min verursachen. Um das Ausmaß von
Messfehlern durch Verdünnung und Messung abzuschätzen, führen Sie drei
unabhängige Verdünnungen und die dazugehörigen Messungen durch. Tragen Sie alle
Verdünnungsfaktoren, die tatsächlich pipettierten Volumina und die dazugehörigen
Messwerte in ihr Laborheft ein. Tragen Sie anschließend die durchschnittliche
Trübungsabnahme pro Minute (A450/min) in Tabelle 1 (wird am Praktikumstag
ausgegeben) ein und berechnen Sie die spezifische Aktivität (U/mg) und die
Gesamtaktivität (kU) wie oben unter „Eigenschaften von Lysozym“ angegeben.
Vergessen Sie bitte nicht, auch die Verdünnung in der Küvette in die Berechnung der
Verdünnungsschritte mit einzubeziehen! Vermeiden Sie eine Kontamination der
Stammlösung der Bakteriensuspension mit Spuren von Lysozym!

11
Da Lysozym auch im menschlichen Speichel vorkommt, können Sie auch einen
entsprechenden Versuch zum Nachweis durchführen.
Isolierung von Lysozym aus Hühnereiweiß
a) Extraktion
Das Eiweiß von einem Hühnerei wird sorgfältig, ohne den Eidotter zu verletzen, von die-
sem getrennt. Das gesammelte Eiweiß (10 ml, Pasteurpipette) wird in einem Becherglas
mit einem doppelten Volumen an Testpuffer (20 ml) verdünnt und in einem schmalen
Becherglas für etwa 15 Sekunden in einem Küchenmixer homogenisiert. Anschließend
erfolgt eine Zentrifugation für 5 Minuten bei 4 °C und 13 000 U/min (den Überstand
weiterverwenden und Volumen messen nicht vergessen!). Ein Aliquot dieses
Rohextrakts von 1 ml wird zur Protein- und Aktivitätsbestimmung entnommen und (wie
alle weiteren Aliquots) im Eisbad aufbewahrt.
Empfohlene Gesamtverdünnung für die Proteinbestimmung 100-fach, für den
Aktivitätstest 500-1000-fach. Hinweis: Die empfohlenen Verdünnungen sind nur
Anhaltspunkte und können stark von Präparation zu Präparation variieren.
Nach Proteinbestimmung und Aktivitätstest werden alle Werte in Tabelle 1 eingetragen.
b) pH-Präzipitation
Zu dem klaren Extrakt wird unter pH-Kontrolle (pH-Glaselektrode in die Lösung eintau-
chen) langsam Eisessig (50% = 9M) getropft, bis der pH der Lösung einen Wert von 4,5
erreicht (Empfehlung: 50 µl-Schritte). Anschließend lässt man für 30 Minuten bei diesem
pH-Wert auf Eis rühren und zentrifugiert den entstandenen Niederschlag für 20 Minuten
bei 13 000 U/min und 4 °C ab. Der klare Überstand (er enthält das Lysozym) wird
vorsichtig dekantiert (Volumen messen!). Falls Sie nicht am nächsten Tag
weiterarbeiten, neutralisieren Sie die Probe mit 4M KOH auf pH von 7,5 und nehmen
anschließend ein Aliquot von 1ml zur Protein- und Aktivitätsbestimmung. Empfohlene
Gesamtverdünnung für die Proteinbestimmung 100-fach, für den Aktivitätstest 500-1000-
fach (Bei der Gesamtverdünnung auch die jeweiligen Verdünnungen in der Küvette
einbeziehen!).
c) Dialyse

12
Der Überstand der pH-Präzipitation wird in einen Dialyseschlauch (wird von den
Praktikumsassistenten vorbereitet) gefüllt und für mindestens 12 Stunden bei 4 °C gegen
5 l Säulenpuffer A über Nacht dialysiert (alle Gruppen gemeinsam). Danach wird die
Proteinlösung aus dem Schlauch entnommen und 5 Minuten lang bei 4 °C und 13 000
U/min zentrifugiert. Der klare Überstand wird dekantiert (Volumen messen!) und auf Eis
aufbewahrt sowie ein Aliquot von 1 ml für Proteinbestimmung und Aktivitätstest
entnommen. Empfohlene Gesamtverdünnung für die Proteinbestimmung: 100-fach, für
die Aktivitätsbestimmung: 500-1000-fach.
d) Chromatographie auf CM-Cellulose-Kationenaustauscher
Der CM-Cellulose-Kationenaustauscher wird wie im Anhang beschrieben am Vortag
vorbereitet. Danach wird die dialysierte Lösung mit einer Plaste-Pasteurpipette auf die
über Nacht in Säulenpuffer A äquilibrierte CM-Cellulose-Säule aufgetragen und mit dem
Sammeln des Durchflusses in einem Messzylinder (Durchfluss) begonnen. Mit
Säulenpuffer A wird solange gewaschen bis man 25 ml Säuleneluat gesammelt hat
(Falcon-Gefäß „Eluat A“). Der Flüssigkeitsmeniskus sollte gerade im Gel der Säule
verschwunden sein (Säule jedoch nicht trocken laufen lassen!). Anschließend wird das
Gelbett der Säule mit Puffer B zunächst überschichtet und das Vorratsgefäß mit Puffer B
gefüllt. Das entsprechende Eluat (wieder 25 ml) wird im Falcon-Gefäß „Eluat B“
gesammelt. Danach wird mit Säulenpuffer C (wie für Puffer B beschrieben) gewaschen
und es werden erneut 25 ml Eluat im Falcon-Gefäß „Eluat C“ gesammelt. Abschließend
wird mit Puffer D überschichtet und die Säule solange mit Puffer D gewaschen, bis man
25 ml Säuleneluat („Eluat D“) gesammelt hat. Dieses Eluat wird sofort mit Essigsäure auf
einen pH-Wert von 7,5 eingestellt, um die Probe zu neutralisieren. Ein jeweils
entnommenes Aliquot von 1 ml des Durchflusses und der Eluate A-D dient zur
Proteinbestimmung (empfohlene Gesamtverdünnung unverdünnt bis 25-fach). Bitte
mischen Sie die Eluate gründlich, bevor Sie das Aliquot entnehmen.
Zur Aktivitätsbestimmung können der Durchfluss sowie die Eluate A, B und C ohne
Vorverdünnung eingesetzt werden, Eluat D muss insgesamt ca. 500 bis 2.000-fach
verdünnt werden. (Bitte Verdünnungen in der Küvette berücksichtigen! Bei großen
Verdünnungen sollte mit 2 Verdünnungsschritten gearbeitet werden!)
Tipp: Es ist sinnvoll, bereits während der Elutionen mit der Bestimmung von
Proteinkonzentration und Lysozymaktivität zu beginnen.

13
e) Ammoniumsulfatfällung
Das mit Puffer D erhaltene und auf pH 7,5 eingestellte Eluat D wird bei 0 °C unter Eisküh-
lung (Eiswasser!) und 15 minütigem Rühren auf dem Magnetrührer portionsweise mit
soviel fein pulverisiertem Ammoniumsulfat versetzt, bis eine 70 %ige Sättigung mit
Ammoniumsulfat erreicht ist (dies entspricht ca. 440 g pro Liter Ausgangsvolumen von
Eluat D). Nach weiterem 15 minütigem Rühren wird das Proteinpräzipitat durch
Zentrifugation (für 20 Minuten im bei 13 000 U/min und 4 °C) vom Überstand abgetrennt.
Der Überstand wird möglichst vollständig vom Niederschlag dekantiert (Bitte testen Sie,
ob der Überstand tatsächlich kein Lysozym enthält!) Der Niederschlag wird dann in
einem möglichst geringen Volumen von Enzympuffer E aufgenommen. Verwenden Sie
zunächst nur 1 ml Puffer E und versuchen Sie den Niederschlag darin unter vorsichtigem,
schonenden Schwenken (Schaumbildung vermeiden) zu lösen. Geben Sie mehr Puffer
zu, falls das Präzipitat sich nicht vollständig löst, limitieren Sie die Puffermenge jedoch
auf maximal 2 ml. Nachdem sich der Niederschlag vollständig gelöst hat, muss das Eluat
noch einmal in einem Eppendorf-Tube kurz für 2 Minuten bei 4°C und 5 000 U/min in der
Tisch-Zentrifuge (Heraeus Biofuge fresco) zentrifugiert werden. Der klare Überstand wird
abgetrennt und dessen Volumen, Proteinkonzentration und Enzymaktivität bestimmt.
Empfohlene Gesamtverdünnung für die Proteinbestimmung 25- bis 100-fach, für den
Aktivitätstest 2000- bis 10 000-fach. Bitte auch hier wieder mit 2 Verdünnungsstufen
arbeiten, um den Fehler so gering wie möglich zu halten!
WICHTIG: Bitte bewahren Sie die Aliquots aller Reinigungsschritte für den
Versuch 2 (SDS-Polyacrylamid-Gelelektrophorese) auf.
Auswertung der Ergebnisse /Diskussion
Allgemeine Angaben zur Anfertigung des Protokolls finden Sie am Anfang des Skripts
(S. 4 – 6), detailliertere Informationen am Ende der Versuchsvorschrift 2 „SDS-
Gelelektrophorese“.
ANHANG

14
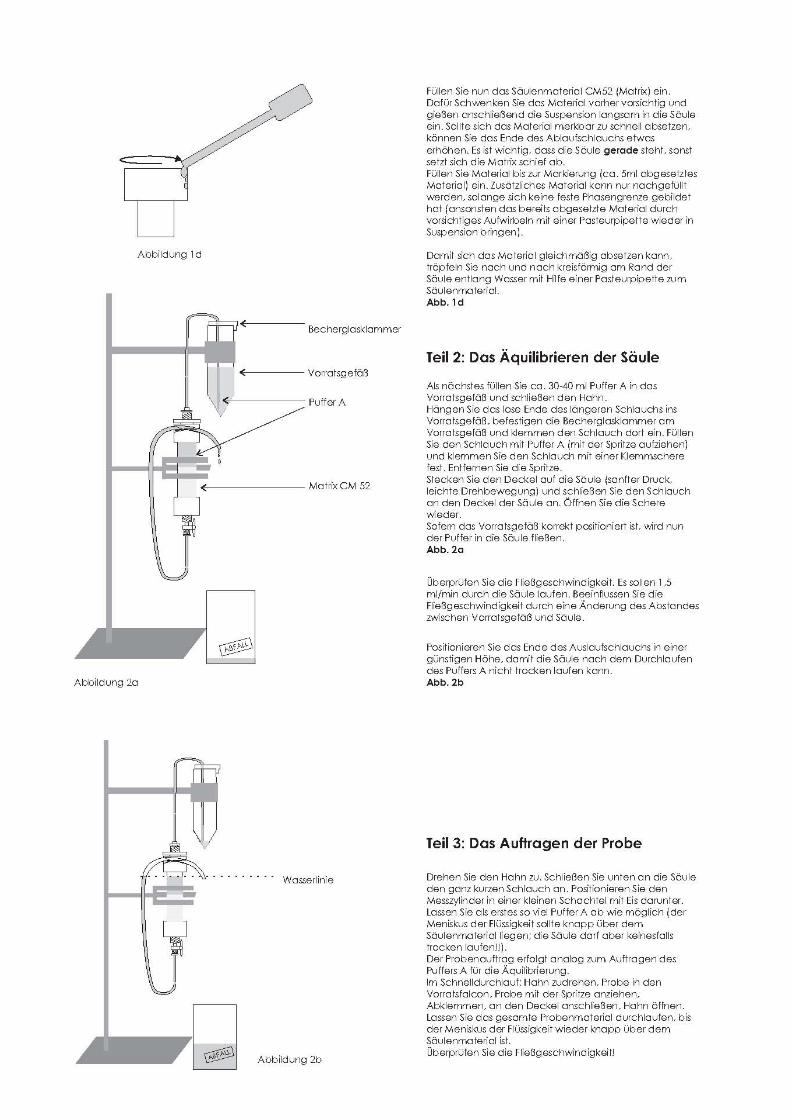
15

16
Veredelung entstehender Nebenprodukte der "Reinigung von
Lysozym aus Hühnereiweiß"
Einleitung
Biochemisches Arbeiten beinhaltet auch den verantwortungsvollen Umgang mit Chemikalien und Ressourcen. Ziel der Ausbildung ist es daher oftmals, organische Abfälle zeitnah und kostenneutral zu entsorgen. In der Praxis ist dies oft mit erheblichem Aufwand verbunden. Dass aber auch elegante Möglichkeiten der Altlastenbeseitigung zur Verfügung stehen, zeigt der Versuch "Veredelung entstehender Nebenprodukte der Reinigung von Lysozym aus Hühnereiweiß". In diesem Versuch ist es möglich, die entstandenen Abfallprodukte zu veredeln und auf diese Weise besonders interessanten biochemischen Abbauwegen zuzuführen.
Materialien und Methoden
Rohsubstrat: 5 Eigelb (aus Versuch: Reinigung von Lysozym aus Hühnereiweiß) Lipidpuffer: 400 ml Sahne (mind. 40% Lipidanteil) Chemikalien: 350 g Puderzucker
1-2 Päckchen Vanillezucker Fällungsmittel: 100 - 150 ml Ethanol
(R 11, S 7-16; M = 46,07 g/mol) Hilfsmittel: 1 Schneebesen .
1 große Schüssel Eis
Praktische Durchführung
Direkt nach der Präparation des Eigelbs muss dieses ohne Verzug und unter Vermeidung von Kontamination bzw. Kontakt mit Labormaterialien in ein sauberes Gefäß überführt werden. Bis zur Weiterverarbeitung bitte auf Eis aufbewahren! In einer großen Schüssel wird unter Rühren das Eigelb mit Lipidpuffer homogenisiert und weitere 5 Minuten cremig geschlagen. Portionsweise werden Puderzucker und Vanillezucker untergerührt, bis alle Kristalle gelöst sind. Der empfindlichste Schritt ist die Einstellung des Alkoholgehaltes, da es sich um eine hoch konzentrierte Stammlösung handelt (Handschuhe tragen, Alkohol nicht unverdünnt inkorporieren, Sicherheitsdatenblatt zu Rate ziehen)! Unter ständiger Geschmackskontrolle wird das Fällungsmittel über 30 min vorsichtig hinzugetropft, bis sich ein angenehmes Gefühl im Mundraum entfaltet. Die genaue Entsorgung sollte je nach persönlichen Vorlieben und Einstellungen variiert werden!
Auswertung der Ergebnisse
Sie können auch gerne andere Protokolle testen. Die Eierlikörverköstigung gestaltet sich in aller Regel abwechslungsreich und lustig, vor allem bei Miteinbeziehung der Assistenten.

17
VERSUCH 2 :
BIOCHEMISCHE TRENNVERFAHREN UND ANALYSEMETHODEN
SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) zur Analyse der Reinheit und
zur Bestimmung des Molekulargewichtes von Proteinen
EINLEITUNG
Die Elektrophoresetechnik ist ein sehr wichtiges analytisches Hilfsmittel in der Biochemie,
um Molekulargewicht und Reinheit von Makromolekülen (Peptide, Proteine,
Oligonukleotide, Nukleinsäuren) zu bestimmen. Die Methode beruht auf der Tatsache,
dass Moleküle wie DNA, RNA oder Proteine Ladungen tragen und daher in einem
elektrischen Feld wandern können. Als Trägermaterialien, an denen Elektrophorese
durchgeführt wird, fanden früher Cellulose (Papier) oder Stärke Verwendung, heute
werden hierfür Polyacrylamid oder Agarose benutzt.
SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE)
Als Trägermaterialien für die Gelelektrophorese eignen sich hydrophile Medien, welche
stabil und chemisch inert sind, und die Wanderung von kleinen Ionen in wässrigem Milieu
nicht behindern. All diese Eigenschaften vereinigt Polyacrylamid, welches man durch
Copolymerisation von Acrylamid und N,N'-Methylenbisacrylamid erhält (Abb. 2).
Abb. 2: Polymerisation und Vernetzung von Acrylamid und N,N’-Methylenbisacrylamid.

18
Der Vernetzungsgrad eines Polyacrylamid-Gels und damit seine Porengröße können
durch die Konzentrationen und das Verhältnis von Acrylamid zu N,N'-
Methylenbisacrylamid variiert werden. Die lineare Verknüpfung der Acrylamid-
Monomeren und die Quervernetzung mit N,N'-Methylen-bisacrylamid wird durch
radikalische Polymerisation mit Ammoniumperoxodisulfat (APS) gestartet.
Tetramethylethylendiamin (TEMED) wirkt dabei als Katalysator für die Bildung von
Sulfatradikalen. Die SDS-Gelelektrophorese von Proteinen verbindet die Methoden der
Gelfiltration und der Elektrophorese. Die Trennung erfolgt jedoch nur nach der Größe der
Proteine (relative Molekülmasse), wenn die Form und die Ladung der Moleküle einheitlich
ist. Diese Bedingungen werden ziemlich gut erfüllt, wenn die Tertiärstruktur der Proteine
durch Einwirkung des Detergens Natriumdodecylsulfat (SDS) zerstört wird. Die
Dodecylsulfatanionen binden mit ihren hydrophoben Kohlenwasserstoffketten an
hydrophobe Regionen eines Proteins, die ursprünglich im Inneren nativ gefalteter
Proteine lokalisiert waren. Dabei werden nicht kovalent verknüpfte Untereinheiten
dissoziiert und die geordnete Tertiärstruktur aufgelöst. Es werden etwa 1,4 g SDS/ g
Protein gebunden. Dadurch werden alle Proteine stark negativ geladen und die
Ladungsdichte (Ladung/Masse) wird für alle SDS-beladenen Proteine gleich. Die
Laufstrecke während der SDS-Gelelektrophorese hängt also nur noch von der Größe des
Proteins, d. h. von seiner relativen Molekülmasse ab. Die SDS-Polyacrylamid-
Gelelektrophorese ist eine der wichtigsten Routinemethoden der Biochemie.
Problemstellung
Mit Hilfe der SDS-Polyacrylamid-Gelelektrophorese soll der Erfolg der Reinigung des in
Versuch 1 präparierten Lysozyms dokumentiert werden. Als Referenz dient ein Gemisch
von Standardproteinen mit bekannten Molekülmassen (Proteinmarker) sowie ein
kommerziell erhältliches Lysozympräparat. Die Experimente werden mit einer Flachbett-
Elektrophoresekammer durchgeführt.

19
MATERIALIEN UND METHODEN
a, Lösungen, Puffer, Reagenzien
10x Elektrophoresepuffer: 250 mM Tris-Base; 1,9 M Glycin; 1 % (w/v) SDS
3x Probenpuffer: 6 % (w/v) SDS; 30% (v/v) Glycerin; 1,9 M ß-
Mercaptoethanol, 187,5 mM Tris-HCl-Puffer (pH 6,8);
0,2 mM Bromphenolblau
SDS-Sammelgel (3 %): 10 % (v/v) Acrylamid-Stock-Lösung (30 %); 0,125 M
Tris-HCl-Puffer (pH 6,8); 3,5 mM SDS; Bromphenolblau
Zugabe von 0,1 % (v/v) TEMED und 4,4 mM
Ammoniumperoxodisulfat
SDS-Trenngel (15 %): 50 % (v/v) Acrylamid-Stock-Lösung (30 %); 0,39 M
Tris-HCl-Puffer (pH 8,8); 1,75 mM SDS;
Zugabe von 0,05 % (v/v) TEMED und 2,2 mM
Ammoniumperoxodisulfat
Fixierlösung: 20 ml Methanol, 1 ml o-Phosphorsäure (85%), 79 ml
H2O
Waschlösung: 25 ml Methanol, 75 ml H2O
Färbelösung: Roti®-Blue-Färbelösung [Roth]
Proteinlösungen: Lysozym aus Hühnereiweiß
MW-Standard: Pageruler prestained protein ladder 10-180kDa
[ThermoFisher Scientific]

20
b, Herstellung des Polyacrylamid-Gels
Der Zusammenbau der Glasplatten für die Gelelektrophorese sowie die Herstellung und
das Gießen des Trenngels wird von den Praktikumsassistenten vorbereitet.
Nach erfolgter Polymerisation des Trenngels wird das Wasser entfernt, welches das
Trenngel überschichtet, um eine gleichmäßige Oberfläche zu garantieren.
Zur Herstellung des Sammelgels werden 5 ml Sammelgellösung in ein kleines
Becherglas pipettiert (Achtung: Handschuhe tragen!). Nach Zugabe von 50 µl 10 % APS-
Lösung und 5 µl TEMED wird kurz gemischt und die Gellösung sofort mit Hilfe einer
Pasteurpipette auf das Trenngel geschichtet. Es ist darauf zu achten, dass sich keine
Luftblasen bilden. Ist die Form gefüllt, steckt man vorsichtig den Kamm in das obere,
offene Ende der Form. Nach 20-30 Minuten ist die Polymerisation abgeschlossen
(Vergleichen Sie mit der restlichen, nicht in die Gelform gefüllten Lösung).
c) Vorbereitung der Elektrophorese
Beim Einsetzen der Gelgießform und der Befestigung des Gels mit zwei Klammern ist
darauf zu achten, dass nach dem Eintauchen der Gelform in den Puffer der
Anodenkammer keine Luftblasen am unteren Gelende hängen bleiben. Sind dennoch
welche vorhanden, werden diese mit Puffer weggespült. Jetzt wird der
Elektrophoresepuffer in den oberen Vorratsbehälter eingefüllt und schließlich der Kamm
so langsam und vorsichtig herausgezogen, dass immer ausreichend Puffer in die frei
werdenden Geltaschen fließen kann. Auch hier dürfen keine Luftblasen in den
Geltaschen vorhanden sein. Die vom Kamm erzeugten Geltaschen (in welche später die
Proben einpipettiert werden) sind vorsichtig und sorgfältig mit Hilfe einer Pipette mit
Elektrophoresepuffer zu spülen.
d) Denaturierung und Auftragen der Proben
Die jeweiligen Proteinlösungen werden so mit Wasser verdünnt, dass sie ca. 1,0 µg
Protein pro µl Lösung enthalten. (Bitte Beladungsplan ausfüllen und vom
Praktikumsassistenten gegenzeichnen lassen!) Anschließend werden in einem
Eppendorfgefäß jeweils zusammengemischt: 5 µl Probenpuffer und 15 µl der
Proteinverdünnung. Der Molekulargewichtsstandard muss nicht verdünnt werden und
enthält schon den Probenpuffer! Dann verschließt man die Gefäße, mischt am Vortex-
Mixer, zentrifugiert kurz an und inkubiert alle Gefäße für 5 Minuten bei 95 °C in einem
Heizblock und zentrifugiert wieder kurz an. (Der MG-Standard muss auch erhitzt werden!)
Die nun denaturierten Proteinproben werden mit einer Pipette so langsam in die
21
einzelnen Taschen des Gels einpipettiert, dass sich die Proben als schmale blaue
Banden direkt über dem Gel absetzen. Die einzelnen Proteinproben werden
folgendermaßen auf die Taschen des Gels verteilt:
Proteinprobe
Protein-
konzentration
[mg/ml]
Verdünnung
z. B. 1:20
tatsächliche
Verdünnung
z. B. 5+95
Spur 1 MG-Standard -
Spur 2 kommerzielles
Lysozym
1 mg/ml
Spur 3 Rohextrakt
Spur 4 pH-Präzipitation
Spur 5 Dialyse
Spur 6 Durchfluss
Spur 7 Eluat A
Spur 8 Eluat B
Spur 9 Eluat C
Spur 10 Eluat D
e) Elektrophorese
Man setzt den Deckel auf die Kammer, legt eine Spannung von 60 V an, bis die Proben
das Sammelgel passiert haben (ca. 30 min), erhöht dann die Spannung auf 160 V und
lässt die Elektrophorese so lange laufen, bis die Farbstoffbande des Markers
(Bromphenolblau) den unteren Rand des Gels erreicht hat. Dies ist in der Regel nach
etwa 45-60 Minuten der Fall.
f) Anfärben und Entfärben des Gels
Die Gelform wird nach Abschalten des Stroms aus der Elektrophoresekammer
genommen. Man entfernt dabei die vordere und hintere Platte der Gelform, legt das Gel
in eine Plastikschale und wäscht für 5 Minuten mit destilliertem Wasser. Danach wird
das Gel für 10-15 Minuten in Fixierlösung inkubiert und anschließend 3x mit destilliertem
Wasser gewaschen. Durch Zugabe von wenigen ml Roti®-Blue-Färbelösung erfolgt die
Färbung der Proteine. Ist der gewünschte Färbegrad erreicht (ca. 30 Minuten bei
Raumtemperatur), wird das Gel 3x mit dest. Wasser gewaschen, um überschüssigen

22
Farbstoff zu entfernen. Nach vollständiger Entfärbung werden die Gele mit Hilfe eines
Geldokumentationssystems fotografiert.
Achtung: Beim Umgang mit Polyacrylamidgelen sind Handschuhe zu tragen. Das Gel
ist sehr weich und reißt leicht, es ist daher mit äußerster Vorsicht zu behandeln!
Auswertung der Ergebnisse /Diskussion für die Versuche 1 und 2
Im Ergebnisteil sollten folgende Tabellen, Grafiken, Rechnungen etc. enthalten sein. Die
Form des Protokolls soll dabei der allgemeinen Form wissenschaftlicher Arbeiten (siehe
S. 5 -7) entsprechen.
1. Reinigungstabelle
Bitte füllen Sie die Reinigungstabelle für Lysozym aus. Die Berechnung der relativen
Aktivität und der Gesamtaktivität erfolgt wie in Gleichung 1 und 2 angegeben.
Berücksichtigen Sie bei diesen Berechnungen alle Verdünnungsschritte und geben Sie
für jede Rechnung (Proteinkonzentration, rel. spez. Aktivität usw.) je eine
Beispielrechnung an. Bitte beachten Sie, dass die Beispielrechnung alle Einheiten
enthalten muss!
2. SDS-PAGE
Im Diskussionsteil diskutieren Sie an Hand der Proteinmengen und der Aktivitätswerte
in der Aufreinigungstabelle den Reinigungserfolg und das Verfahren der
Ionenaustauschchromatographie. Beziehen Sie in ihre Bewertung auch das Ergebnis der
SDS-Gelelektrophorese aus Versuch 2 mit ein.
Bei der Diskussion Ihrer Ergebnisse sollten Sie u. a. auch auf folgende Fragen eingehen:
1. Welche Reinigungsschritte waren am erfolgreichsten?
2. Wieviel Protein bzw. Lysozymaktivität haben Sie auf die Chromatographiesäule
aufgetragen und wieviel Protein bzw. Lysozymaktivität haben Sie in den fünf
Elutionsschritten wieder im Eluat gefunden? Diskutieren Sie!
3. Betrachten Sie Proteinmenge und relative spezifische Aktivität vom ersten Schritt
der Reinigung bis zum Endprodukt! Was fällt auf?
4. Stellen Sie den Gesamtproteingehalt (in %) und die spezifische Aktivität (in U/mg)
grafisch dar!
23
5. Warum lassen sich bei der Reinigung mittels Ionenaustauschchromatographie
keine Lysozymaktivitäten in Eluat A, B und C nachweisen?
6. Welche Proteine könnten Sie im Durchfluss sowie den Eluaten A, B und C
gefunden haben? Welche Eigenschaften haben diese? Diskutieren Sie anhand
der Tabelle 1 „Zusammensetzung Hühnereiweiß“ und der Tabelle 2 „Prozentuale
Zusammensetzung der Hühnereiweißproteine“!
Tab. 1: Analyse isolierter Proteine aus dem Hühnereiweiß (aus Rhodes et al., 1957).
Tab. 2: Prozentuale Zusammensetzung der Hühnereiweißproteine.
7. Wie bewerten Sie den Anstieg der relativen spez. Aktivität von <500 U/mg bei den
ersten Reinigungsschritten auf ~5000 U/mg nach der Chromatographie?

24
8. Warum sehen Sie keine/ kaum Unterschiede in der relativen spezifischen Aktivität
zwischen Eluat D und dem Endprodukt nach der Ammonium-sulfatfällung?
Bitte bewerten Sie an Hand des SDS-Polyacrylamid-Gels in einer kurzen Diskussion
den Erfolg der einzelnen Reinigungsschritte sowie die Qualität des von Ihnen in
Versuch 1 gereinigten Lysozyms. Achten Sie bitte auch auf die richtige Beschriftung
von Abbildungen in wissenschaftlichen Arbeiten!

25
VERSUCH 3:
QUANTITATIVE BESTIMMUNG VON PROTEINEN
DURCH OPTISCHE MESSVERFAHREN
1. Konzentrationsbestimmung von Proteinen durch Messung der
UV-Absorption bei verschiedenen Wellenlängen
EINLEITUNG
Messung der Absorption. Für viele Versuche mit Proteinen in Lösung ist es notwendig,
ihre Konzentration zu kennen. Die Konzentration eines Proteins lässt sich am einfachsten
und genauesten durch Absorptionsspektroskopie im so genannten Aromatenbereich
(250-300 nm) bestimmen.
Die Absorption einer Lösung kann nicht direkt gemessen werden. Vielmehr wird die
Intensität des Lichts vor Durchtritt (I0) und nach Durchtritt (I) durch die Küvette verglichen.
Die Absorption A ergibt sich aus dem Verhältnis von I zu I0 gemäß Gleichung 3.
A = - log10 (I / I0) = c d (3)
Bitte beachten Sie, dass in Gleichung 3 der dekadische und nicht der natürliche
Logarithmus verwendet wird. Aufgrund des logarithmischen Zusammenhangs werden
bei einer Absorption von A = 1 (entsprechend -log (0,1)) 90% und bei A = 2 (entsprechend
-log (0,01)) bereits 99% des einfallenden Lichts absorbiert, d. h., es kommen nur 10%
bzw. 1% des einfallenden Lichts am Detektor an. Wenn Lösungen Absorptionswerte 1
aufweisen, sollten sie grundsätzlich verdünnt und die Messungen wiederholt werden.
Absorption durch Proteine. Die Absorption der Proteine im Bereich von 250 bis 300 nm
wird durch die drei aromatischen Aminosäuren Tyrosin (Tyr, Y), Tryptophan (Trp, W) und
Phenylalanin (Phe, F) sowie in geringem Ausmaß durch die Disulfidbrücken der Cystine
verursacht. Die Spektren von Phe, Tyr und Trp sind in Abb. 3 in verschiedenen
Maßstäben dargestellt. Tyr und Trp zeigen Maxima im Bereich von 274-280 nm, wobei
die Absorption von Trp besonders intensiv ist.
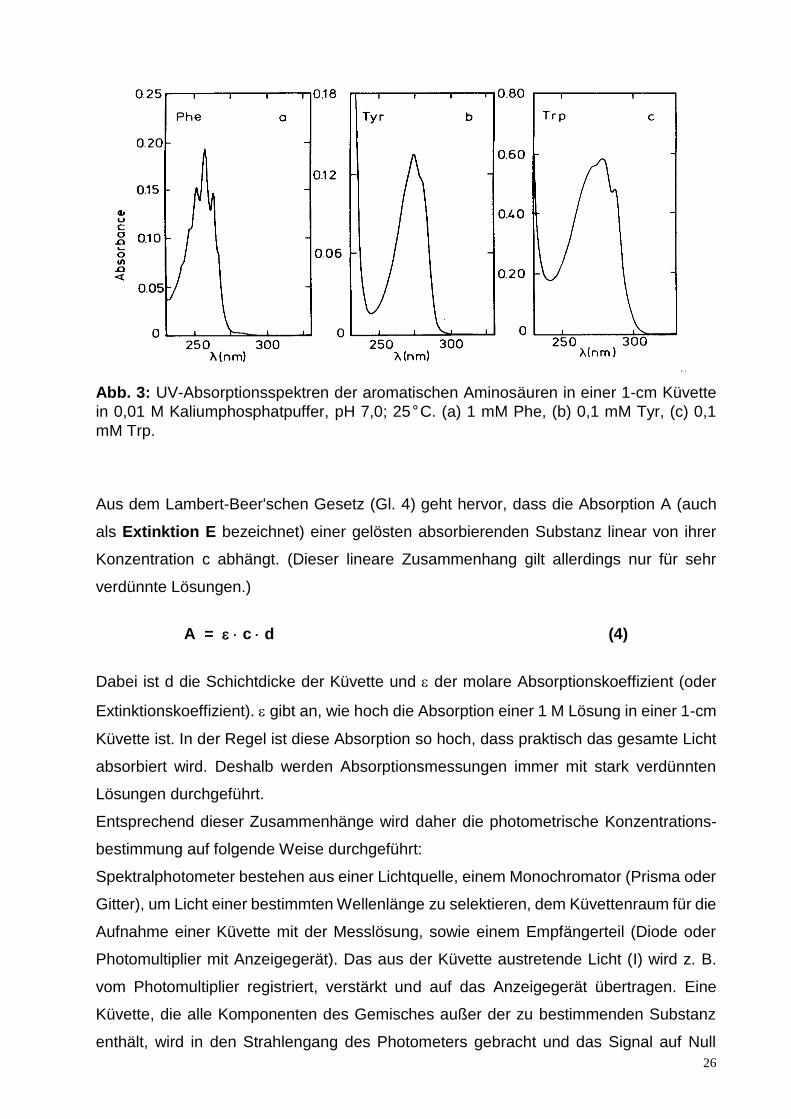
26
Abb. 3: UV-Absorptionsspektren der aromatischen Aminosäuren in einer 1-cm Küvette
in 0,01 M Kaliumphosphatpuffer, pH 7,0; 25°C. (a) 1 mM Phe, (b) 0,1 mM Tyr, (c) 0,1
mM Trp.
Aus dem Lambert-Beer'schen Gesetz (Gl. 4) geht hervor, dass die Absorption A (auch
als Extinktion E bezeichnet) einer gelösten absorbierenden Substanz linear von ihrer
Konzentration c abhängt. (Dieser lineare Zusammenhang gilt allerdings nur für sehr
verdünnte Lösungen.)
A = c d (4)
Dabei ist d die Schichtdicke der Küvette und der molare Absorptionskoeffizient (oder
Extinktionskoeffizient). gibt an, wie hoch die Absorption einer 1 M Lösung in einer 1-cm
Küvette ist. In der Regel ist diese Absorption so hoch, dass praktisch das gesamte Licht
absorbiert wird. Deshalb werden Absorptionsmessungen immer mit stark verdünnten
Lösungen durchgeführt.
Entsprechend dieser Zusammenhänge wird daher die photometrische Konzentrations-
bestimmung auf folgende Weise durchgeführt:
Spektralphotometer bestehen aus einer Lichtquelle, einem Monochromator (Prisma oder
Gitter), um Licht einer bestimmten Wellenlänge zu selektieren, dem Küvettenraum für die
Aufnahme einer Küvette mit der Messlösung, sowie einem Empfängerteil (Diode oder
Photomultiplier mit Anzeigegerät). Das aus der Küvette austretende Licht (I) wird z. B.
vom Photomultiplier registriert, verstärkt und auf das Anzeigegerät übertragen. Eine
Küvette, die alle Komponenten des Gemisches außer der zu bestimmenden Substanz
enthält, wird in den Strahlengang des Photometers gebracht und das Signal auf Null

27
abgeglichen (Referenzlösung). Anschließend wird eine ebenso vorbereitete Lösung in
den Strahlengang gebracht, die zusätzlich die zu bestimmende Substanz enthält
(Probelösung). Die angezeigte Extinktion der Probe ist dann direkt proportional zu der
gesuchten Konzentration. Aus dem Lambert-Beer'schen Gesetz folgt:
c = A / ( d) (siehe Gleichungen 3 und 4)
Zur Ermittlung der Konzentration der Probelösung muss natürlich der entsprechende
Verdünnungsfaktor der Probe einbezogen werden.
Ist bekannt, kann die Konzentration sofort berechnet werden. Andernfalls muss
zunächst mit Hilfe einer Eichreihe der Extinktionskoeffizient bestimmt werden. Dazu
werden Lösungen verschiedener Konzentrationen des gesuchten Stoffes hergestellt und
deren Extinktion bestimmt. Aus dem Anstieg der Geraden, die man erhält, wenn die
Extinktion gegen die Konzentration aufgetragen wird, lässt sich der Extinktions-
koeffizient berechnen. Oft verzichtet man jedoch auf eine Berechnung von und
entnimmt die gesuchte Konzentration direkt aus der Eichkurve.
Des Weiteren kann der molare Absorptionskoeffizient eines Proteins bei 280 nm mit
ausreichender Genauigkeit aus dem Gehalt an Tyrosin und Tryptophan sowie dem
Gehalt an Disulfidbrücken und ihren entsprechenden Absorptionskoeffizienten nach
Gleichung 5 berechnet werden:
280 [M-1cm-1] = 5 500 nTrp + 1 490 nTyr + 125 nSS (5)
280 ist der gesuchte molare Absorptionskoeffizient des Proteins, nTrp, nTyr und nSS
geben die Anzahl der Trp- und Tyr-Reste sowie der Disulfidbrücken wieder. Die molaren
Absorptionskoeffizienten von 5 500 M-1cm-1 für Trp, 1 490 M-1cm-1 für Tyr und 125 M-1cm-
1 für Disulfidbrücken in Gleichung 5 wurden aus der Analyse einer Vielzahl gut bekannter
Proteine erhalten.
Absorption durch Nukleinsäuren. Die in Gewebeextrakten vorkommenden
Nukleinsäuren, Nukleotide, Nukleoside, Purin- und Pyrimidinverbindungen absorbieren
ebenfalls im UV-Bereich. Die Absorptionsmaxima der einzelnen Basen liegen zwischen
250 und 280 nm. Polymere DNA zeigt daher eine relativ breite und sehr starke Absorption
mit einem Maximum bei 260 nm. Da die Proteine bei 280 nm eine ausgeprägte
Absorption zeigen, können aus Messungen bei 260 nm und 280 nm die Konzentrationen

28
von DNA und Protein simultan in einer Probe bestimmt und störende Absorptionen von
z. B. DNA korrigiert werden (DNA absorbiert bei 260 nm etwa 50-fach stärker als
Proteine).
Methode nach Warburg und Christian. Da nach einem Aufschluss von Zellen neben
Proteinlösungen auch größere Mengen an Nukleinsäuren und Nukleotiden vorhanden
sind, müssen die bei A280 gemessenen Werte korrigiert werden, da die Basen der
Nukleinsäuren ebenfalls bei 280 nm absorbieren. Warburg und Christian ermittelten
daher einen Wert bei 260 nm (A260), der mit dem bei A280 gemessenen Wert in folgende
Beziehung gesetzt werden kann:
Proteinkonzentration (mg/ml) = 1,55 ∙ A280 – 0,76 ∙ A260 (6)
Diese Gleichung kann bis zu einem Anteil von 20% Nukleinsäuren in der Lösung
angewendet werden.
Methode nach Whitaker und Granum. Die Differenz in der Absorption zwischen 235 nm
und 280 nm kann verwendet werden, um die Konzentration von Proteinen in Gegenwart
von Nukleinsäuren zu bestimmen. Für Proteine ist der Absorptionskoeffizient bei 235 nm
etwa 2,8 mal größer als der Absorptionskoeffizient bei 280 nm. Diese hohe Absorption
bei 235 nm rührt von der Peptidbindung her. Daher kann die Differenz zwischen der
Absorption bei 235 nm und bei 280 nm als Maß für die vorhandenen Peptidbindungen
eines Proteins und damit als Grundlage für dessen Konzentrationsbestimmung dienen.
Ein besonderer Vorteil dieser Methode ist die Tatsache, dass Nukleinsäuren in geringer
Konzentration die Messungen nicht stören, da sie bei 235 nm und 280 nm eine
annähernd gleiche Absorption besitzen.
Proteinkonzentration (mg/ml) = (A235 - A280) / 2,51 (7)

29
Materialien
Puffer und Lösungen
Lysozymlösung: Lysozymlösung unbekannter Konzentration in 0,1 M
Kaliumphosphatpuffer, pH 7,0
Messpuffer: 0,1 M Kaliumphosphatpuffer, pH 7,0
Hilfsmittel: Fotometer und Küvetten für UV-Bestimmung
Problemstellung
Am Beispiel des Lysozyms sollen mehrere Proteinbestimmungsmethoden miteinander
verglichen werden, die auf der Messung der UV-Absorption beruhen.
Praktische Durchführung
Messung der Absorption der Proteinlösungen
In der Küvette werden 1,92 ml Messpuffer vorgelegt. Damit wird im Gerät die
Absorption bei 280 nm (bzw. bei 260 nm und 235 nm) auf Null abgeglichen. Dann
werden in die Küvette genau 80 µl der Proteinlösung (Lysozym) gegeben. Der Inhalt
der Küvette wird mit einem Plastikrührer vorsichtig vermischt (Blasenbildung vermeiden,
da diese die Messung verfälschen würden!). Anschließend wird die Küvette wieder in den
Küvettenhalter des Photometers gestellt und es werden bei 280 nm (bzw. bei 260 nm
und 235 nm) die Absorptionen gemessen. Der gesamte Messvorgang wird danach
zweimal erneut durchgeführt, zunächst mit 1,86 ml Messpuffer und 140 µl
Proteinlösung, dann mit 1,8 ml Messpuffer und 200 µl Proteinlösung in der Küvette.
Protein-
lösung
Ver-
dünnungs-
faktor
A280
gemessen
A280
absolut
(Verdünnungs-
faktor
einbeziehen!)
A260
gemessen
A260
absolut
(Verdünnungs-
faktor
einbeziehen!)
A235
gemessen
A235
absolut
(Verdünnungs-
faktor
einbeziehen!)
Messung 1
Messung 2
Messung 3

30
Proteinlösung
Mittelwert
A280
absolut
Mittelwert
A260
absolut
Mittelwert
A235
absolut
Unbekannte
Lysozymlösung
Problemstellung A
Für das Enzym Lysozym soll zunächst der molare Absorptionskoeffizient bei 280 nm (
280 [M-1 cm-1]) berechnet und dann die Konzentration der unbekannten Lysozymlösung
über das Lambert-Beer'sche Gesetz bestimmt werden.
a) Berechnung des Absorptionskoeffizienten
Bitte berechnen Sie mit Hilfe von Gl. 5 den Absorptionskoeffizienten 280 [M-1cm-1] für
Lysozym. Hühnereiweiß-Lysozym besteht aus 129 Aminosäuren (MW: 14,4 kDa, d.h. 1
mol Lysozym = 14`400 g) mit einem Gehalt von 4,7 % Trp und 2,3 % Tyr, außerdem sind
4 Disulfidbrücken vorhanden.
b) Berechnung der Lysozymkonzentration
Berechnen Sie aus den Messungen jeweils die absolute Absorption der Lysozym-
stammlösung für die Wellenlängen 260 nm und 280 nm (jeweilige Verdünnungen
einbeziehen!), und mit dem in (a) bestimmten Wert für 280 die molare Konzentration von
Lysozym in der Stammlösung aus c = A280 / (280 d). Aus den Absorptionswerten bei 260
nm und 280 nm lässt sich ferner das Verhältnis A280/A260 bestimmen. Es sollte größer als
1 sein, solange keine Nukleinsäuren in der Messlösung vorhanden sind.
Mittelwert
A260
absolut
Mittelwert
A280
absolut
A280/ A260
Protein-
konzentration
[mg/ml]
Unbekannte
Lysozymlösung
31
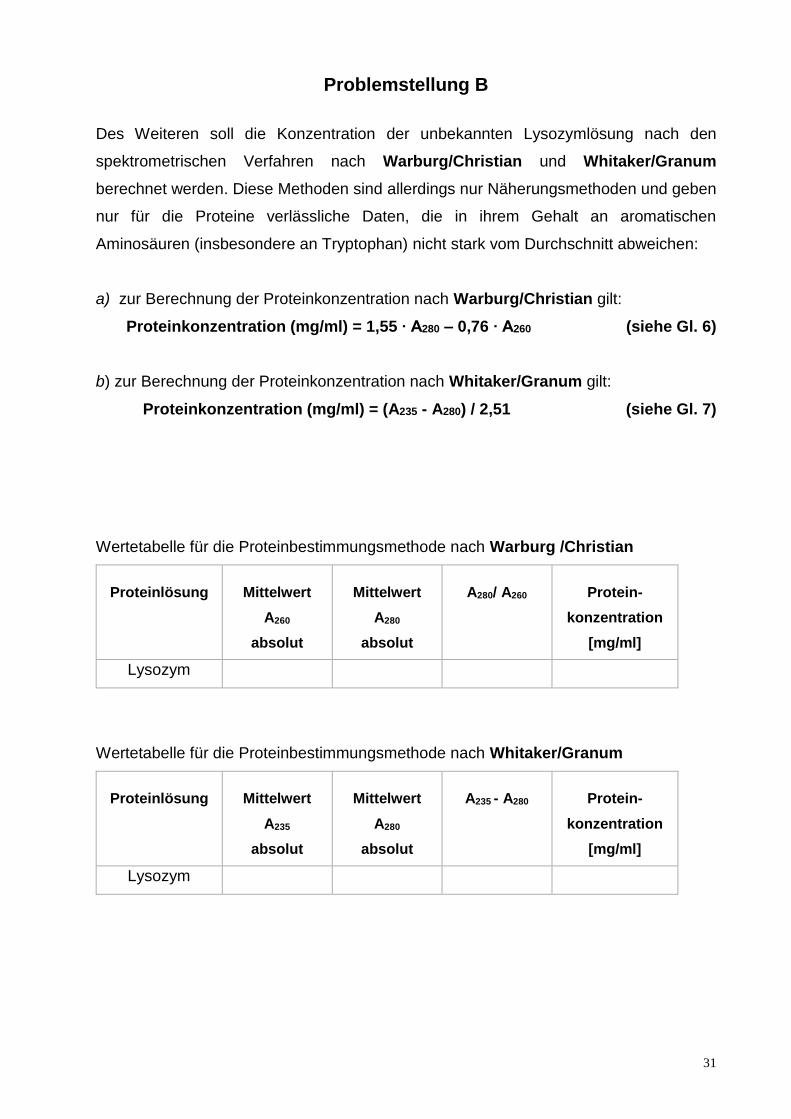
Problemstellung B
Des Weiteren soll die Konzentration der unbekannten Lysozymlösung nach den
spektrometrischen Verfahren nach Warburg/Christian und Whitaker/Granum
berechnet werden. Diese Methoden sind allerdings nur Näherungsmethoden und geben
nur für die Proteine verlässliche Daten, die in ihrem Gehalt an aromatischen
Aminosäuren (insbesondere an Tryptophan) nicht stark vom Durchschnitt abweichen:
a) zur Berechnung der Proteinkonzentration nach Warburg/Christian gilt:
Proteinkonzentration (mg/ml) = 1,55 ∙ A280 – 0,76 ∙ A260 (siehe Gl. 6)
b) zur Berechnung der Proteinkonzentration nach Whitaker/Granum gilt:
Proteinkonzentration (mg/ml) = (A235 - A280) / 2,51 (siehe Gl. 7)
Wertetabelle für die Proteinbestimmungsmethode nach Warburg /Christian
Proteinlösung
Mittelwert
A260
absolut
Mittelwert
A280
absolut
A280/ A260
Protein-
konzentration
[mg/ml]
Lysozym
Wertetabelle für die Proteinbestimmungsmethode nach Whitaker/Granum
Proteinlösung
Mittelwert
A235
absolut
Mittelwert
A280
absolut
A235 - A280
Protein-
konzentration
[mg/ml]
Lysozym

32
Problemstellung C
In Versuch 1 „Reinigung von Lysozym aus Hühnereiweiß“ haben Sie zur Bestimmung
der Proteinkonzentration ein Näherungsverfahren angewendet und eine Absorption von
A280 = 1 gleich einer Proteinkonzentration von 1 mg/ml gesetzt. Für reine Protein-
lösungen ist dieses Näherungsverfahren jedoch zu ungenau, da die jeweiligen
spezifischen Absorptionskoeffizienten nicht berücksichtigt wurden.
Für Lysozym würde dabei die Absorption einer 1 mg/ml Lösung bei 280 nm einen Wert
von 2,48 aufweisen.
Bestimmen Sie die Proteinkonzentration der unbekannten Lysozymlösung nach der
Näherungsmethode A280, 1 mg/ml = 1 und nach der für Lysozym spezifischen Korrelation
A280, 1 mg/ml = 2,48.

33
VERSUCH 3:
QUANTITATIVE BESTIMMUNG VON PROTEINEN
DURCH OPTISCHE MESSVERFAHREN
2. Konzentrationsbestimmung von Proteinen durch colorimetrische Verfahren
EINLEITUNG
Die zuvor behandelte direkte UV-spektrometrische Konzentrationsbestimmung von
Proteinen ist in der Regel nur für gereinigte Proteine geeignet. Zellextrakte oder nur
teilweise gereinigte Proteinfraktionen enthalten noch viele Coenzyme, Ionen, Nukleotide
und Nukleinsäuren, die alle sehr stark im UV-Bereich absorbieren und die
Proteinabsorption überdecken. In solchen Fällen können Proteine nur mit
colorimetrischen Verfahren nachgewiesen und quantifiziert werden. In diesen Färbe-
verfahren werden die gelösten Proteine zunächst mit einem Reagenz umgesetzt, wobei
das Reaktionsprodukt im Bereich des sichtbaren Lichts absorbiert. Die Konzentration des
angefärbten Proteins wird über eine Eichgerade bestimmt, die in parallelen Ansätzen
unter identischen Bedingungen mit Proben eines Eichproteins bekannter Konzentration
erstellt wurde.
Bei den Nachweismethoden nach Biuret, Folin, BCA, Lowry oder Bradford ist die
BRADFORD-Methode aufgrund ihrer hohen Empfindlichkeit und ihrer geringen
Störanfälligkeit eine der am häufigsten in der Laborpraxis angewendeten
colorimetrischen Proteinbestimungsmethoden:
Die bei der Bindung des kommerziell erhältlichen, synthetischen Farbstoffs Coomassie
Brilliant Blue G-250 an Proteine auftretende Verschiebung des Absorptionsmaximums
von 465 zu 595 nm wird zur einfachen, raschen und sensitiven Bestimmung von
Proteinen genutzt.
Abb. 4: Chemische Formel des Farbstoffs Coomassie Brilliantblau G 250.

34
Materialien
Lysozymlösung: Lysozymlösung unbekannter Konzentration in 0,1 M
Kaliumphosphatpuffer, pH 7,0
Messpuffer: 0,1 M Kaliumphosphatpuffer, pH 7,0
Hilfsmittel: Fotometer und Küvetten für sichtbares Licht
Proteinlösungen: Lysozym (20 mg/ml) in Kaliumphosphatpuffer, pH 7,0
Assay-Reagenz: BRADFORD-Reagenz [Bio-Rad], bestehend aus
Farbstoff G250, Phosphorsäure und Methanol
Praktische Durchführung
Bei der indirekten Proteinbestimmungsmethode nach BRADFORD wird zunächst eine
Eichkurve mit Lysozym bekannter Konzentration aufgenommen, aus der dann die
Proteinmenge der unbekannten Lysozymprobe bestimmt werden kann.
a) Herstellung einer Verdünnungsreihe des Proteins Lysozym
Für die Verdünnungsreihe werden die in untenstehender Tabelle zu berechnenden
Mengen der Lysozym-Stammlösung (20 mg/ml; muss vorher noch 1:25 verdünnt werden)
mit den entsprechenden Mengen an Messpuffer in 2,0 ml Eppendorfreaktionsgefäße
zusammenpipettiert und mit dem Vortex-Mixer sorgfältig gemischt. Bitte beachten Sie,
dass jeder Testansatz ein Volumen von 40 µl besitzt.
Des Weiteren wird die in Versuch 3.1 untersuchte Lysozymlösung unbekannter
Konzentration 1:5 mit Messpuffer verdünnt (Dreifachbestimmung!), jeweils 40 µl werden
anschließend in ein 2 ml Eppendorfreaktionsgefäß pipettiert.
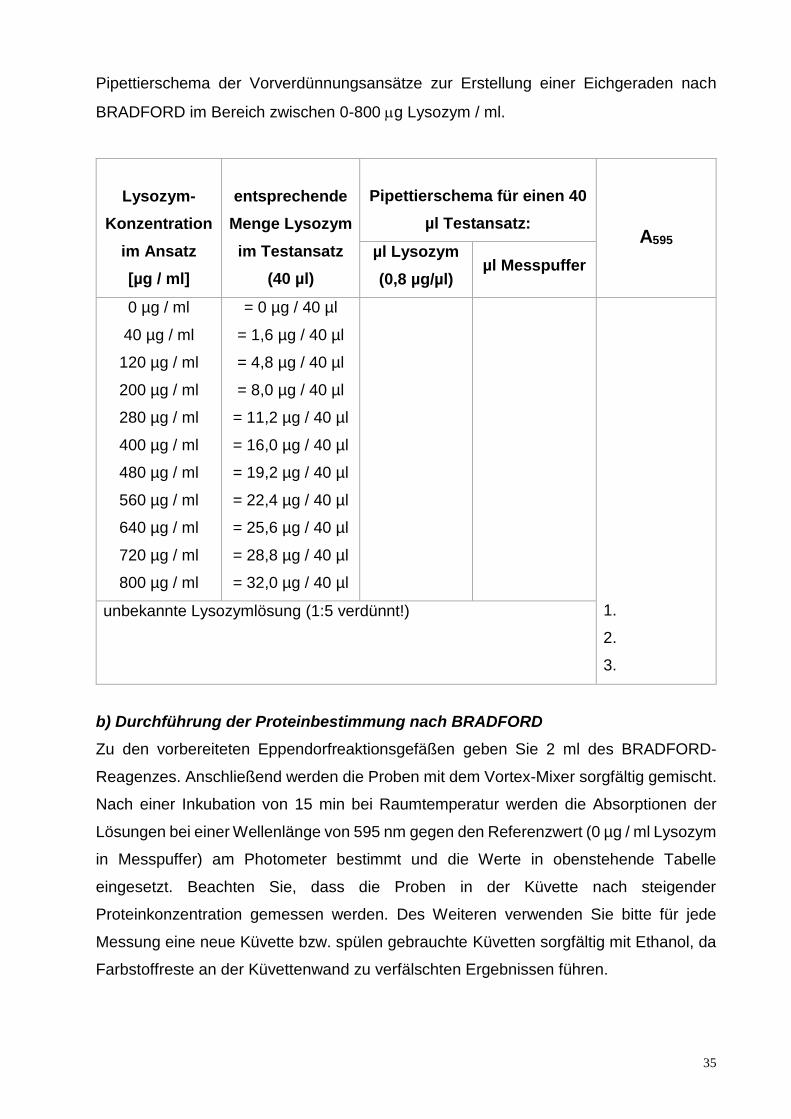
35
Pipettierschema der Vorverdünnungsansätze zur Erstellung einer Eichgeraden nach
BRADFORD im Bereich zwischen 0-800 g Lysozym / ml.
Lysozym-
Konzentration
im Ansatz
[µg / ml]
entsprechende
Menge Lysozym
im Testansatz
(40 µl)
Pipettierschema für einen 40
µl Testansatz:
A595 µl Lysozym
(0,8 µg/µl) µl Messpuffer
0 µg / ml
40 µg / ml
120 µg / ml
200 µg / ml
280 µg / ml
400 µg / ml
480 µg / ml
560 µg / ml
640 µg / ml
720 µg / ml
800 µg / ml
= 0 µg / 40 µl
= 1,6 µg / 40 µl
= 4,8 µg / 40 µl
= 8,0 µg / 40 µl
= 11,2 µg / 40 µl
= 16,0 µg / 40 µl
= 19,2 µg / 40 µl
= 22,4 µg / 40 µl
= 25,6 µg / 40 µl
= 28,8 µg / 40 µl
= 32,0 µg / 40 µl
1.
2.
3.
unbekannte Lysozymlösung (1:5 verdünnt!)
b) Durchführung der Proteinbestimmung nach BRADFORD
Zu den vorbereiteten Eppendorfreaktionsgefäßen geben Sie 2 ml des BRADFORD-
Reagenzes. Anschließend werden die Proben mit dem Vortex-Mixer sorgfältig gemischt.
Nach einer Inkubation von 15 min bei Raumtemperatur werden die Absorptionen der
Lösungen bei einer Wellenlänge von 595 nm gegen den Referenzwert (0 µg / ml Lysozym
in Messpuffer) am Photometer bestimmt und die Werte in obenstehende Tabelle
eingesetzt. Beachten Sie, dass die Proben in der Küvette nach steigender
Proteinkonzentration gemessen werden. Des Weiteren verwenden Sie bitte für jede
Messung eine neue Küvette bzw. spülen gebrauchte Küvetten sorgfältig mit Ethanol, da
Farbstoffreste an der Küvettenwand zu verfälschten Ergebnissen führen.

36
Auswertung der Ergebnisse / Hinweise zum Verfassen des Protokolls
„Proteinbestimmungsmethoden“
Am Beispiel des Lysozyms sollen mehrere Proteinbestimmungsmethoden miteinander
verglichen werden. Vergleichen und diskutieren Sie die erhaltenen Messergebnisse der
unterschiedlichen Methoden.
1. Integrieren Sie Ihre Originaldaten für die UV-Absorptionsmessung!
2. Berechnen Sie die Proteinkonzentration auf der Basis der verschiedenen
Korrelationen! Geben Sie Gleichung und Rechenweg mit an!
3. Proteinbestimmung nach Bradford
Erstellen Sie mit Hilfe der gemessenen Absorptionswerte für die Lysozym-Lösungen eine
Eichkurve und ermitteln Sie die Konzentration der unbekannten Lysozymlösung.
Beachten Sie dabei den Vorverdünnungsfaktor!
4. Stellen Sie die Daten aller Versuche sowohl in Tabellenform als auch grafisch dar!

37
Diskussion
Vergleichen Sie die erhaltenen Daten und diskutieren Sie Vor- und Nachteile der
verwendeten Proteinbestimmungsmethoden. Welche Werte sind am genauesten/ am
ungenauesten und warum?
Warum müssen die Ergebnisse aus der Anwendung des Lambert-Beer’schen-Gesetzes
bzw. aus der Korrelation A280 (2,48) = 1 mg/ml einfach genauer sein als die Ergebnisse
aus den Korrelationen nach Warburg/Christian und Whitaker/Granum?
Tipp: Berechnen Sie dazu den experimentell bestimmten Extinktionskoeffizienten für
Lysozym, der sich aus der Korrelation A280 (2,48) = 1 mg/ml ergibt und vergleichen Sie
diesen mit Ihrem berechneten Extinktionskoeffizienten.

38

39
VERSUCH 4:
KOHLENHYDRATE
Saure und enzymatische Hydrolyse von Glykogen und Dextran sowie
Durchführung einer enzymatischen Glukosebestimmung
EINLEITUNG
Kohlenhydrate. Unter dem Begriff „Kohlenhydrate“ werden sehr viele Naturstoffe
zusammengefasst, welche als Baumaterial oder Betriebsstoffe für sämtliche Lebewesen
von Bedeutung sind. Während Cellulose, Hemicellulose und Pektine der Pflanzenzelle
als Baustoffe dienen, sind Stärke und Zucker (Saccharose, Fruktose, Laktose) als
Nahrung für Mensch und Tier wichtig. Im Organismus des Säugers ist die D-Glukose das
Substrat der Glykolyse. Dieser Zucker steht seinerseits mit dem in Leber und Muskulatur
gespeicherten Reservepolysaccharid, dem Glykogen, im Gleichgewicht. Zudem sind
Zucker am Aufbau der verschiedenartigsten Biomoleküle beteiligt: Pentosen finden sich
in den Nukleinsäuren, Ribose in der Ribonukleinsäure, 2-Desoxyribose in der DNA. Die
Grundsubstanz des Bindegewebes, Schleimstoffe und Blutgruppensubstanzen enthalten
Zuckerderivate, wie Aminozucker und Uronsäuren. In konjugierter Form kommen Zucker
zudem in Glykoproteinen und Glykolipiden vor. Schließlich ist zu erwähnen, dass die
Kopplung körperfremder und körpereigener Giftstoffe an Glukuronsäure der vom
Säugerorganismus am häufigsten angewandte Entgiftungsvorgang ist.
Während der Skelettmuskel und insbesondere der Herzmuskel auch Fettsäuren und
Acetoacetat als Energiequelle des oxidativen Stoffwechsels nutzen können, dient
Glukose in allen Geweben als primäre Energiequelle, welche den Gewebezellen ständig
aus dem Blut zur Verfügung steht. Aus der Nahrung aufgenommene Glukose, die nicht
sofort zur Deckung des Energiebedarfes metabolisiert wird, kann vor allem in der Leber
sowie in der Muskulatur als Glykogen in polymerer Form gespeichert werden, welches
gegenüber der monomeren Glukose sehr unterschiedliche physikalische, chemische und
biologische Eigenschaften zeigt.
Eigenschaften von Glykogen. Glykogen ist das wichtigste Reservepolysaccharid
tierischer Muskel- und Leberzellen. So können z. B. bis zu 18 % des Lebergewichtes aus
Glykogen bestehen. Glykogen ist ein Polymer aus D-Glukose mit α (14) glykosidischen
Bindungen, die auf je 12-18 Monosaccharid-Einheiten eine Verzweigungsstelle mit 16-
Verknüpfungen enthält (MG: 5-15106). Aufgrund dieser stark verzweigten Struktur wird

40
ein rascher Abbau des Glykogens durch die gleichzeitige Freisetzung der Glukose an
den Enden aller Verzweigungen möglich.
Abb. 5: Aufbau von Glykogen.
Die Spaltung der glykosidischen Bindungen im Glykogen verläuft durch saure oder
enzymatisch katalysierte Hydrolyse; in vivo vor allem durch Phosphorolyse
(Glykogenphosphorylase). Während bei der Säurehydrolyse über die intermediäre
Bildung verschiedener Oligosaccharide sämtliche 14- und 16-Bindungen gelöst
werden, wirken die Glykogen-abbauenden Enzyme spezifischer. Von einem bestimmten
Enzym werden immer nur ganz bestimmte Verknüpfungsmuster gespalten. Durch
Phosphorylasen werden bestimmte Sauerstoffbrücken unter Einlagerung von Phosphat
gespalten, wobei z. B. in Leber und Muskel aus Glykogen Glukose-1-Phosphat entsteht.
Während die Kohlenhydrate im Intermediärstoffwechsel ausschließlich phosphorolytisch
abgebaut werden, erfolgt die Spaltung bei der Verdauung durch hydrolytische Enzyme.
Bei den polysaccharidspaltenden Hydrolasen muss streng zwischen α-Glukosidasen und
β-Glukosidasen unterschieden werden. Erstere spalten nur α-glykosidische Bindungen,
z.B. Amylase (Substrat: Stärke, Glykogen), letztere greifen nur β-Glukoside an, z. B.
Cellulase (Substrat: Cellulose). Da der Säuger nur α-Glukosidasen besitzt, kann er
Stärke, nicht aber Cellulose spalten. Diese „Lücke“ in der Enzymausrüstung wird
allerdings teilweise dadurch kompensiert, dass Darmbakterien über β-Glukosidasen
verfügen. Die α-Amylase des menschlichen Speichels spaltet 14-, aber nicht 16-
Bindungen im Innern eines Glykogenmoleküls (Endoamylase), so dass es
zwischenzeitlich zur Bildung von noch relativ hochmolekularen Dextrinen kommt. Bei
anhaltendem Einwirken des Enzyms entstehen schließlich Maltose, Isomaltose und
Glukose.

41
Das Ausmaß der anschließend durchgeführten sauren und enzymatische Hydrolyse
(mittels α-Amylase) kann durch einen Vergleich beider Methoden in Abhängigkeit von der
Hydrolysedauer bestimmt werden. Als Referenzsubstanz dient das
Speicherpolysaccharid Dextran, dessen Glukoseeinheiten fast ausschließlich α-16-
Bindungen enthalten.
MATERIALIEN
Puffer und Lösungen
Glucose (Sigma-Aldrich)
(Glukosestandard 83 µg/ml)
(Glukoselösung f. Eichkurve 2mg/ml)
Dinitrosalicylat-Reagenz (1%, VWR)
Na-Phosphatpuffer (5 mM NaCl, 20 mM Na-Phosphat, pH 6,9)
Peroxidase 172 U/ml (Sigma-Aldrich)
Glukoseoxidase 810 U/ml (Sigma-Aldrich)
o-Dianisidin 4,75 mg/ml (Sigma-Aldrich)
Praktische Durchführung
a) Glykogenhydrolyse und Quantifizierung der Produkte
Erstellen einer Eichkurve
Zum Erstellen der Eichkurve erhalten Sie eine Glukosestammlösung (Konzentration von
2 mg/ml). 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75 und 2 mg Glukose werden in je 1 ml dest.
Wasser gelöst. (Verdünnen Sie dafür die Glukosestammlösung entsprechend
untenstehender Tabelle in 1,5 ml Eppendorf-Tubes!).
Nr. 0 1 2 3 4 5 6 7 8
Glukose (mg/ml)
0
0,25
0,5
0,75
1
1,25
1,5
1,75
2
Glukose (2 mg/ml)
0 125 µl 1 ml
Wasser 1 ml 875 µl -
Davon werden in einem Reagenzglas je 0,5 ml (Leerwert 0 mit 0,5 ml dest. Wasser
anstelle von Glukose) mit 0,5 ml Dinitrosalicylat-Reagenz versetzt. Nach 5 minütiger
42
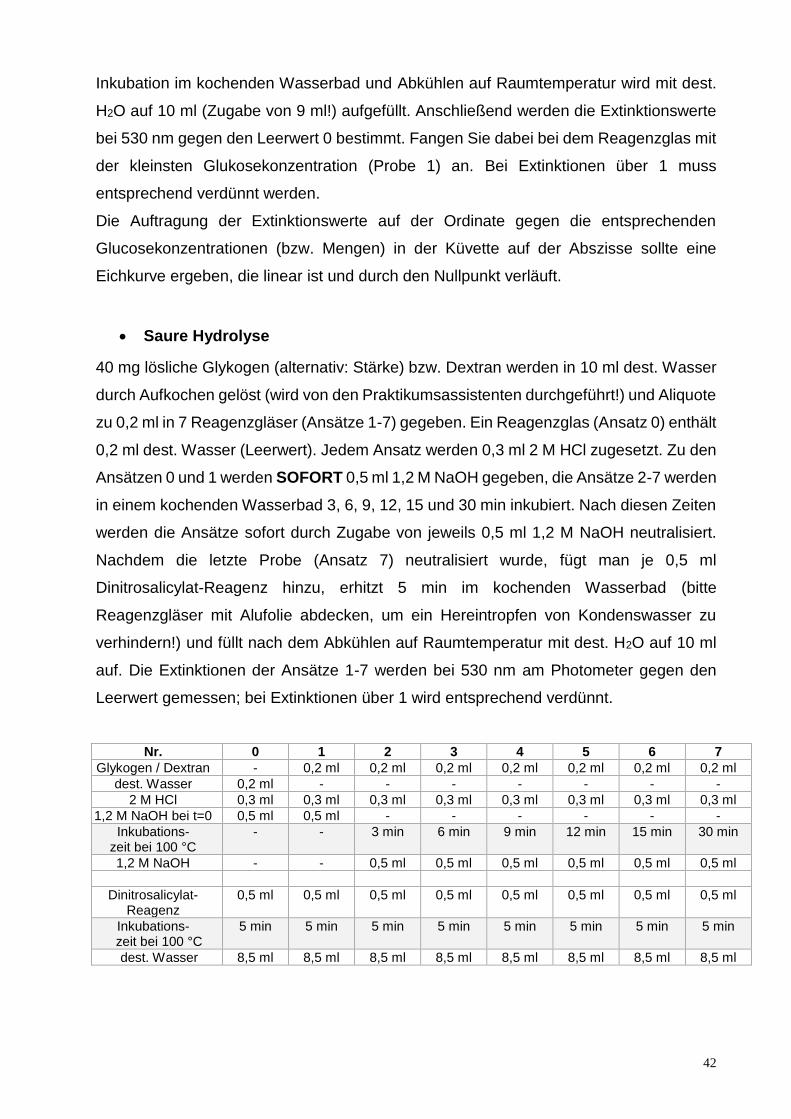
Inkubation im kochenden Wasserbad und Abkühlen auf Raumtemperatur wird mit dest.
H2O auf 10 ml (Zugabe von 9 ml!) aufgefüllt. Anschließend werden die Extinktionswerte
bei 530 nm gegen den Leerwert 0 bestimmt. Fangen Sie dabei bei dem Reagenzglas mit
der kleinsten Glukosekonzentration (Probe 1) an. Bei Extinktionen über 1 muss
entsprechend verdünnt werden.
Die Auftragung der Extinktionswerte auf der Ordinate gegen die entsprechenden
Glucosekonzentrationen (bzw. Mengen) in der Küvette auf der Abszisse sollte eine
Eichkurve ergeben, die linear ist und durch den Nullpunkt verläuft.
Saure Hydrolyse
40 mg lösliche Glykogen (alternativ: Stärke) bzw. Dextran werden in 10 ml dest. Wasser
durch Aufkochen gelöst (wird von den Praktikumsassistenten durchgeführt!) und Aliquote
zu 0,2 ml in 7 Reagenzgläser (Ansätze 1-7) gegeben. Ein Reagenzglas (Ansatz 0) enthält
0,2 ml dest. Wasser (Leerwert). Jedem Ansatz werden 0,3 ml 2 M HCl zugesetzt. Zu den
Ansätzen 0 und 1 werden SOFORT 0,5 ml 1,2 M NaOH gegeben, die Ansätze 2-7 werden
in einem kochenden Wasserbad 3, 6, 9, 12, 15 und 30 min inkubiert. Nach diesen Zeiten
werden die Ansätze sofort durch Zugabe von jeweils 0,5 ml 1,2 M NaOH neutralisiert.
Nachdem die letzte Probe (Ansatz 7) neutralisiert wurde, fügt man je 0,5 ml
Dinitrosalicylat-Reagenz hinzu, erhitzt 5 min im kochenden Wasserbad (bitte
Reagenzgläser mit Alufolie abdecken, um ein Hereintropfen von Kondenswasser zu
verhindern!) und füllt nach dem Abkühlen auf Raumtemperatur mit dest. H2O auf 10 ml
auf. Die Extinktionen der Ansätze 1-7 werden bei 530 nm am Photometer gegen den
Leerwert gemessen; bei Extinktionen über 1 wird entsprechend verdünnt.
Nr. 0 1 2 3 4 5 6 7
Glykogen / Dextran - 0,2 ml 0,2 ml 0,2 ml 0,2 ml 0,2 ml 0,2 ml 0,2 ml
dest. Wasser 0,2 ml - - - - - - -
2 M HCl 0,3 ml 0,3 ml 0,3 ml 0,3 ml 0,3 ml 0,3 ml 0,3 ml 0,3 ml
1,2 M NaOH bei t=0 0,5 ml 0,5 ml - - - - - -
Inkubations- zeit bei 100 °C
- - 3 min 6 min 9 min 12 min 15 min 30 min
1,2 M NaOH - - 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml
Dinitrosalicylat-Reagenz
0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml
Inkubations- zeit bei 100 °C
5 min 5 min 5 min 5 min 5 min 5 min 5 min 5 min
dest. Wasser 8,5 ml 8,5 ml 8,5 ml 8,5 ml 8,5 ml 8,5 ml 8,5 ml 8,5 ml
43
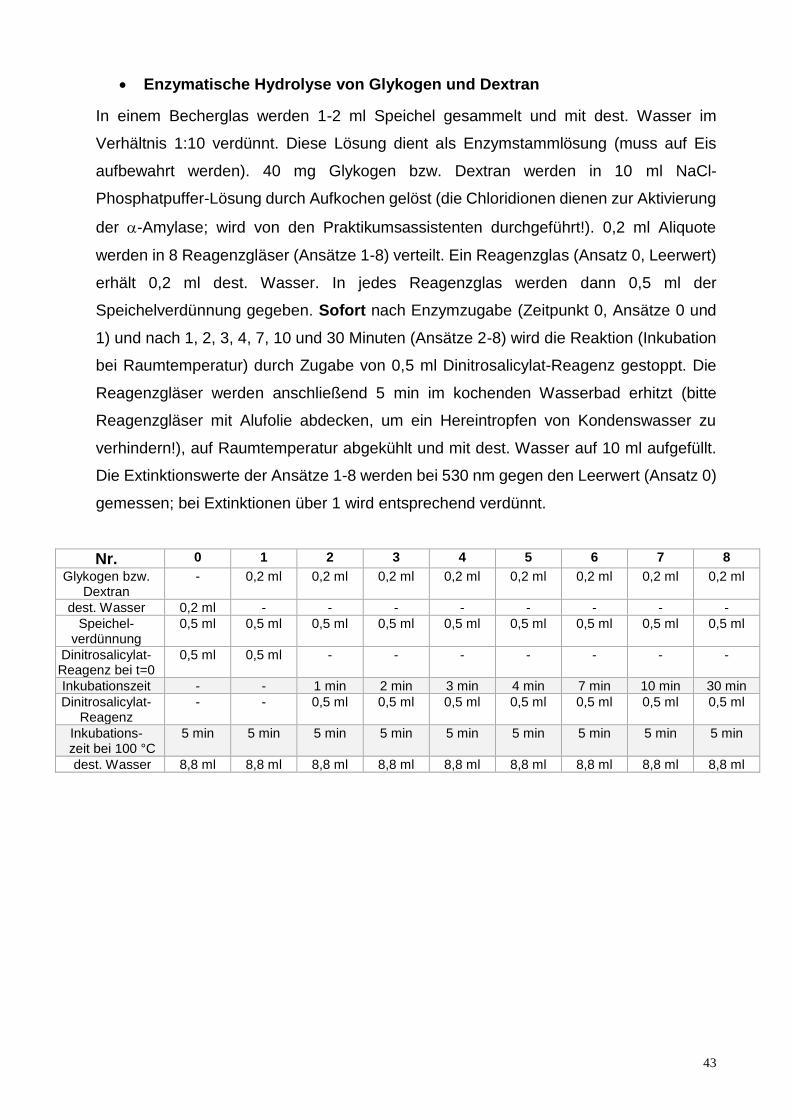
Enzymatische Hydrolyse von Glykogen und Dextran
In einem Becherglas werden 1-2 ml Speichel gesammelt und mit dest. Wasser im
Verhältnis 1:10 verdünnt. Diese Lösung dient als Enzymstammlösung (muss auf Eis
aufbewahrt werden). 40 mg Glykogen bzw. Dextran werden in 10 ml NaCl-
Phosphatpuffer-Lösung durch Aufkochen gelöst (die Chloridionen dienen zur Aktivierung
der -Amylase; wird von den Praktikumsassistenten durchgeführt!). 0,2 ml Aliquote
werden in 8 Reagenzgläser (Ansätze 1-8) verteilt. Ein Reagenzglas (Ansatz 0, Leerwert)
erhält 0,2 ml dest. Wasser. In jedes Reagenzglas werden dann 0,5 ml der
Speichelverdünnung gegeben. Sofort nach Enzymzugabe (Zeitpunkt 0, Ansätze 0 und
1) und nach 1, 2, 3, 4, 7, 10 und 30 Minuten (Ansätze 2-8) wird die Reaktion (Inkubation
bei Raumtemperatur) durch Zugabe von 0,5 ml Dinitrosalicylat-Reagenz gestoppt. Die
Reagenzgläser werden anschließend 5 min im kochenden Wasserbad erhitzt (bitte
Reagenzgläser mit Alufolie abdecken, um ein Hereintropfen von Kondenswasser zu
verhindern!), auf Raumtemperatur abgekühlt und mit dest. Wasser auf 10 ml aufgefüllt.
Die Extinktionswerte der Ansätze 1-8 werden bei 530 nm gegen den Leerwert (Ansatz 0)
gemessen; bei Extinktionen über 1 wird entsprechend verdünnt.
Nr. 0 1 2 3 4 5 6 7 8
Glykogen bzw. Dextran
- 0,2 ml 0,2 ml 0,2 ml 0,2 ml 0,2 ml 0,2 ml 0,2 ml 0,2 ml
dest. Wasser 0,2 ml - - - - - - - -
Speichel-verdünnung
0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml
Dinitrosalicylat-Reagenz bei t=0
0,5 ml 0,5 ml - - - - - - -
Inkubationszeit - - 1 min 2 min 3 min 4 min 7 min 10 min 30 min
Dinitrosalicylat-Reagenz
- - 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml 0,5 ml
Inkubations- zeit bei 100 °C
5 min 5 min 5 min 5 min 5 min 5 min 5 min 5 min 5 min
dest. Wasser 8,8 ml 8,8 ml 8,8 ml 8,8 ml 8,8 ml 8,8 ml 8,8 ml 8,8 ml 8,8 ml

44
b) Enzymatische Glukosebestimmung
Die enzymatische Bestimmung der Glukose erfolgt nach folgendem Schema:
Das für die Glukosebestimmung benötigte Glukosereagenz wird nach folgender
Vorschrift hergestellt:
9,79 ml 0,1 M Na-Phosphatpuffer, pH 7,0 + 10 µl Peroxidase (POD) + 100 µl
Glukoseoxidase (GOD) + 100 µl o-Dianisidin.
Für diesen Versuch werden 2 ml Glukosereagenz mit 0,1 ml der unbekannten
Glukoselösung (vorher 1:10 verdünnen) bzw. dem Leerwert (0,1 ml H2O) und dem
Standard (0,1 ml Glukose-Standardlösung mit einer Konzentration von 83 g/ml) für 90
Minuten bei Raumtemperatur inkubiert (direktes Sonnenlicht vermeiden). Die Reaktionen
werden durch Zugabe von 2 ml 8 M H2SO4 beendet. Es wird gut gemischt und danach
die Extinktion bei 530 nm gegen den Leerwert gemessen.

45
Auswertung der Ergebnisse / Hinweise zum Verfassen des Protokolls
„Kohlenhydrate“
1. Glykogenhydrolyse:
Glukoseeichkurve: Tragen Sie die gemessenen Extinktionen (y-Achse) gegen die
Glukosekonzentrationen (Angabe in mg/ml, mg Glukose/Reaktionsansatz und
Glukosekonzentration in mM) (x-Achse) auf.
Werte der sauren bzw. enzymatischen Hydrolyse:
Protokollieren Sie den Verlauf der sauren und enzymatischen Glykogenhydrolyse
als auch der enzymatischen Dextranhydrolyse.
Tragen Sie des Weiteren die gemessenen Extinktionen (y-Achse) gegen die Zeit (x-
Achse) auf, verwenden Sie für alle drei Kurven ein und dasselbe Diagramm!
Ermitteln Sie aus dem Plateau der Kurven die Werte mit der höchsten Extinktion
(Endpunkt) und bestimmen Sie über die Glukoseeichkurve die bei der Hydrolyse
gebildete Menge Glukose!
Verwenden Sie den Wert von Ansatz 8 der sauren Hydrolyse als Wert der
maximalen Umsetzung (100 % = vollständige Spaltung des Glykogens in Glukose)
und berechnen Sie für die enzymatische Hydrolyse den prozentualen Wert der
maximalen Umsetzung!

46
Wieviel Glukose hätte maximal freigesetzt werden können? Vergleichen Sie diesen
Wert mit der von Ihnen bestimmten Menge an freigesetzter Glukose bei der sauren
Hydrolyse!
2. Enzymatische Glukosebestimmung:
Bestimmen Sie den Glukose-Gehalt der unbekannten Probe (µg/ml). Berechnen Sie
die Molarität der unbekannten Glukoselösung (Angabe in mM; Molekulargewicht
Glukose: 180,2). Bitte geben Sie den Rechenweg mit an!
Diskussion
Diskutieren Sie Ihre Daten!
Hätten die Maximalwerte der enzymatischen Hydrolyse die Maximalwerte der
sauren Hydrolyse übersteigen können, wenn Sie den Speichel nicht vorher 1:20
verdünnt hätten? Wie würde oben stehendes Diagramm aussehen, wenn Sie
unverdünnten Speichel bei der enzymatischen Hydrolyse eingesetzt hätten?
Diskutieren Sie die Ergebnisse für die Hydrolyse mit Dextran!

47
VERSUCH 5:
ENZYMKINETIK AM BEISPIEL DER ALKALISCHEN PHOSPHATASE
EINLEITUNG
Enzyme
Enzyme sind Proteine mit katalytischen Eigenschaften. Sie sind in der Lage, chemische
Reaktionen in biologischen Systemen Tausend- bis Milliarden-fach zu beschleunigen,
ohne dabei selbst verbraucht zu werden. Dies bedeutet, dass ein einzelnes
Enzymmolekül in der Lage ist, Millionen von Substratmolekülen pro Minute
umzuwandeln. Viele Stoffwechselvorgänge in unserem Körper würden ohne die
hocheffektiven Enzyme praktisch nicht ablaufen können, d.h. Reaktionen, die nur
Millisekunden brauchen, würden ohne Enzyme Jahre benötigen.
Enzyme besitzen eine hochspezifische Bindungsstelle für ihr Substrat, das aktive
Zentrum. Durch die optimale Bindung des Substrates an das aktive Zentrum des
Enzymes werden die Substrate so orientiert, dass sie die für die Bildung des notwendigen
Übergangszustandes optimale Lage einnehmen. Enzyme verringern dabei zum einen die
Aktivierungsenergie, die für die Bildung des Übergangszustandes notwendig ist und sind
zum anderen auch für die Bildung und Stabilisierung des Übergangszustandes
verantwortlich.
Die Kinetik enzymkatalysierter Reaktionen, der Michaelis-Menten Formalismus
Um eine einfache Beziehung zwischen der Anfangsgeschwindigkeit v0 einer enzymkataly-
sierten Reaktion und der eingesetzten Substratkonzentration [S] zu erhalten, geht man von
einer sehr einfachen Reaktionsgleichung aus, wobei zunächst das Substrat S an das
Enzym E bindet und so den Enzym-Substrat-Komplex ES bildet. In diesem Komplex läuft
die katalysierte Reaktion ab und das Produkt P dissoziiert vom Enzym ab. Diese
Reaktionsgleichung ist in Gl. 1 wiedergegeben.
k1 k2 E + S ES E + P
k-1 (1)

48
Wenn die Bedingungen berücksichtigt werden, unter denen eine enzymkatalysierte
Reaktion gemessen wird, ergibt sich eine einfache Beziehung zwischen der
Anfangsgeschwindigkeit v0 und der eingesetzten Substratkonzentration [S], nämlich das
Michaelis-Menten-Modell. Die Michaelis-Menten-Gleichung (Gl. 2) erklärt die
charakteristische hyperbolische Abhängigkeit der Enzymaktivität von der
Substratkonzentration und liefert Größen, die die Wirksamkeit des Enzyms quantitativ
charakterisieren.
V0 = vmax [S] / (KM + [S]) (2)
vmax ist die Maximalgeschwindigkeit der Reaktion bei sehr hoher Substratkonzentration,
wenn praktisch das gesamte Enzym als Enzym-Substrat-Komplex vorliegt. Die Michaelis-
Konstante KM kennzeichnet die Affinität eines Enzyms zu seinem Substrat. Die Michaelis-
Konstante entspricht derjenigen Substratkonzentration, bei der die halbmaximale
Geschwindigkeit erreicht ist.
Abb. 1: Schematische Darstellung der Kinetik einer enzymkatalysierten Reaktion, die der Michaelis-Menten-Beziehung folgt.
Näherungswerte für KM und vmax können sehr leicht aus den experimentellen Daten abge-
schätzt werden. KM entspricht der Substratkonzentration bei v0 = vmax /2, vmax ist der
Grenzwert, dem die gemessenen Werte für v0 zustreben.
Der schematische Verlauf einer enzymkatalysierten Reaktion, die der Michaelis-Menten
Beziehung folgt, ist in Abb. 1 dargestellt.
49
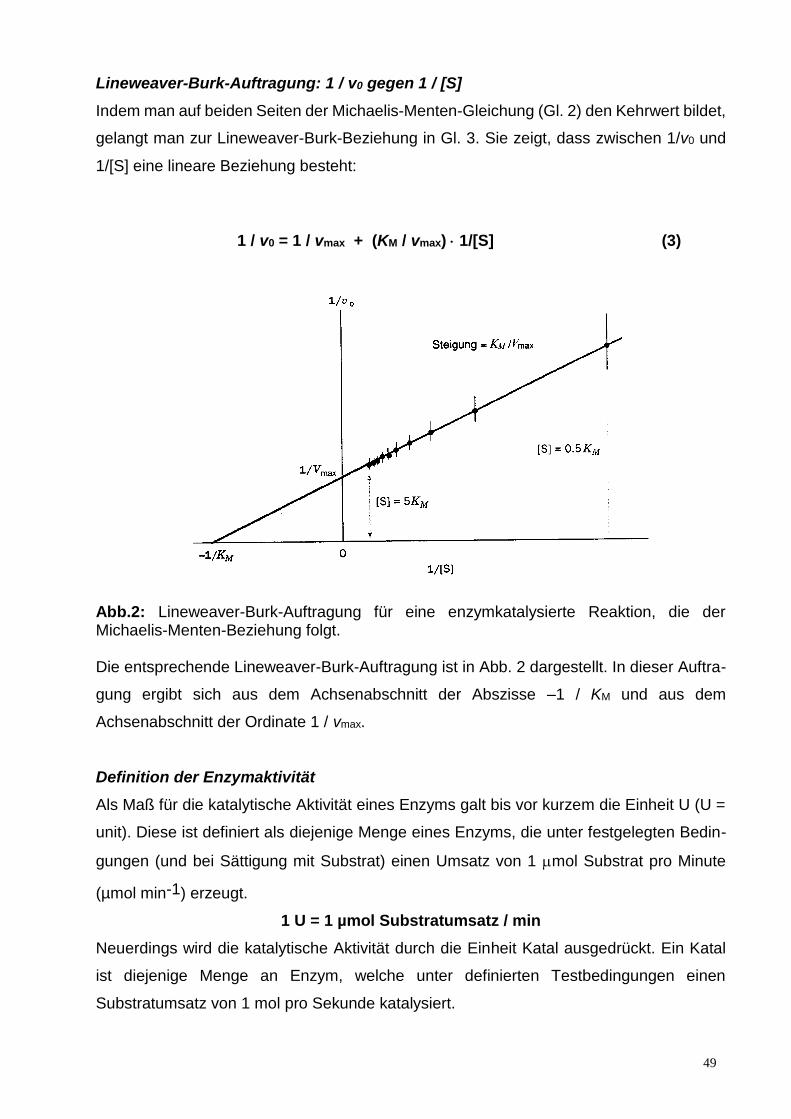
Lineweaver-Burk-Auftragung: 1 / v0 gegen 1 / [S]
Indem man auf beiden Seiten der Michaelis-Menten-Gleichung (Gl. 2) den Kehrwert bildet,
gelangt man zur Lineweaver-Burk-Beziehung in Gl. 3. Sie zeigt, dass zwischen 1/v0 und
1/[S] eine lineare Beziehung besteht:
1 / v0 = 1 / vmax + (KM / vmax) 1/[S] (3)
Abb.2: Lineweaver-Burk-Auftragung für eine enzymkatalysierte Reaktion, die der Michaelis-Menten-Beziehung folgt. Die entsprechende Lineweaver-Burk-Auftragung ist in Abb. 2 dargestellt. In dieser Auftra-
gung ergibt sich aus dem Achsenabschnitt der Abszisse –1 / KM und aus dem
Achsenabschnitt der Ordinate 1 / vmax.
Definition der Enzymaktivität
Als Maß für die katalytische Aktivität eines Enzyms galt bis vor kurzem die Einheit U (U =
unit). Diese ist definiert als diejenige Menge eines Enzyms, die unter festgelegten Bedin-
gungen (und bei Sättigung mit Substrat) einen Umsatz von 1 mol Substrat pro Minute
(µmol min-1) erzeugt.
1 U = 1 µmol Substratumsatz / min
Neuerdings wird die katalytische Aktivität durch die Einheit Katal ausgedrückt. Ein Katal
ist diejenige Menge an Enzym, welche unter definierten Testbedingungen einen
Substratumsatz von 1 mol pro Sekunde katalysiert.

50
Die spezifische Aktivität gibt an, wieviel Aktivitätseinheiten in einem mg Enzym enthalten
sind.
1 U/mg = 1 µmol/(minmg)
Die spezifische Aktivität eines Proteins wird unter Substratsättigung gemessen. Unter
diesen Bedingungen ist die Anfangsgeschwindigkeit v0 eine lineare Funktion der
Enzymkonzentration [E].
Die Wechselzahl eines Enzyms (turnover number; kcat)
Die Wechselzahl (turnover number, kcat) eines Enzyms gibt an, wieviele Substratmoleküle
durch ein Enzymmolekül (bzw. ein aktives Zentrum) pro Minute (oder pro Sekunde)
umgesetzt werden. Für Enzyme, die dem Michaelis-Menten-Formalismus gehorchen, ist
die Wechselzahl identisch mit der Geschwindigkeitskonstanten k2 (vgl. Gl. 1). Sie kann
sehr einfach aus vmax berechnet werden (Gl. 4 und 5):
vmax = k2 [E] (4)
k2 = vmax / [E] (5)
Die Alkalische Phosphatase
Die alkalische Phosphatase (AP) ist ein in der Natur weit verbreitetes Enzym, welches in
tierischen Geweben, Pflanzen, Bakterien und Hefen vorkommt. Dieses Enzym katalysiert
die Spaltung einer Vielzahl von Phosphatestern (Ester von primären und sekundären
Alkoholen, von Zuckeralkoholen, zyklischen Alkoholen, Phenolen, Aminen und
Nukleosidmonophosphaten AMP, GMP, UMP, CMP). Allerdings werden Phosphodiester,
wie sie z.B. bei der DNA oder RNA vorkommen, nicht gespalten. An z. B. Proteinen kann
sie Phosphatreste von phosphoryliertem Serin, Threonin oder Tyrosin abspalten.
Die AP kommt vor allem in Osteoblasten des Knochenmarks vor und besitzt eine große
Bedeutung bei der Knochenneubildung. Medizinische Bedeutung hat die Bestimmung der
alkalischen Phosphatase im Serum, da ein erhöhter Spiegel der AP auf Knochentumore
hinweist. Die AP besitzt daher eine große Rolle als Tumormarker.
Die „Alkalische Phosphatase“ bekam ihren Namen, da ihr Aktivitätsoptimum im alkalischen
pH-Bereich liegt. Die Alkalische Phosphatase ist ein Dimer und spaltet relativ unspezifisch
durch Hydrolyse Phosphatester entsprechend der Gleichung:

51
AP
ROPO32- + H2O ROH + HPO4
2- (6)
Das Gleichgewicht der dargestellten Reaktion liegt weit auf der rechten Seite.
Die von der alkalischen Phosphatase katalysierte Reaktion kann sehr einfach durch die
Verwendung des synthetischen Substrats para-Nitrophenylphosphat (pNPP) verfolgt
werden (Gl. 7):
(7)
pNPP zeigt bei 405 nm eine vernachlässigbar kleine Absorption. Die Abspaltung des
Phosphatrestes führt jedoch zur Bildung eines p-Nitrophenolat-Anions, das sich im
alkalischen Milieu photometrisch gut quantifizieren lässt, da es eine charakteristische
gelbe Farbe zeigt und damit eine ausgeprägte Absorption bei 405 nm aufweist
(Extinktionskoeffizient: 405 = 18 500 M-1cm-1). Die hydrolytische Spaltung von pNPP,
welche durch die AP katalysiert wird, kann somit direkt photometrisch an Hand der
Zunahme der Absorption bei 405 nm verfolgt werden und bietet eine einfache Möglichkeit,
die enzymatische Aktivität der AP zu bestimmen.
PROBLEMSTELLUNG
Im vorliegenden Versuch soll die Enzymkinetik der Alkalischen Phosphatase untersucht
werden. Dazu sollen die für ein Michaelis-Menten-Diagramm notwendigen Daten
gemessen werden (d. h. die Hydrolyse von pNPP bei verschiedenen Konzentrationen an
pNPP). Aus diesen Daten sollen die kinetischen Konstanten vmax und KM, sowie die spezi-
fische Aktivität und die Wechselzahl (kcat) bestimmt werden.
In der praktischen Durchführung gliedert sich der Versuch in zwei Teile. Im ersten Teil wird
zunächst die Kinetik der Hydrolyse von pNPP als Funktion des pH-Werts untersucht, um
das pH-Optimum der AP zu finden. Im zweiten Teil wird unter optimalen pH-Bedingungen
die Hydrolyse bei unterschiedlichen Substratkonzentrationen gemessen, um Daten für die
Michaelis-Menten Auftragung zu erhalten.
bas. pH

52
MATERIALIEN
Puffer und Lösungen
Testpuffer 1-8: 1 M Tris/HCl Puffer, pH 6; 7; 7,5; 8; 8,5; 9; 9,5 und 10 Substratlösung: 10 mM p-Nitrophenylphosphat (BoehringerMannheim;
Bestell-Nr. 107 891) in H2O (vor Licht geschützt und eisgekühlt aufbewahren, über Nacht einfrieren)
Enzym: Alkalische Phosphatase, 5 mg/ml aus Kälberdarm
(AppliChem, Darmstadt), 24,2 U/mg, in Testpuffer pH 8,5 gelöst MW (pro Untereinheit): 57 300 Da
PRAKTISCHE DURCHFÜHRUNG
a) Durchführung der einzelnen Enzymtests
Für alle Proben der Pipettierschemata I und II werden zunächst Testpuffer, H2O, pNPP in
die Küvette gegeben und mit dem Plastikrührspatel gemischt. Danach wird die
Hydrolysereaktion durch Zugabe von jeweils 100 µl einer geeignet verdünnten
Enzymlösung (Alkalische Phosphatase, Verdünnung von Praktikumsassistenten erfragen!
[üblicherweise 1:200 in H2O]) gestartet. Nach einer Messzeit von 2 Minuten kann die
Reaktionsgeschwindigkeit als ΔE405/min abgelesen werden (Auf Linearität der Reaktion
achten!). Jede Messung wird als Doppel- ggf. als Dreifachbestimmung durchgeführt, die
Ergebnisse werden in die vorbereitete Tabelle eingetragen.
b) pH-Abhängigkeit der Aktivität der alkalischen Phosphatase
Jedes Enzym besitzt einen mehr oder weniger engen pH-Bereich, in welchem es optimal
arbeiten, d.h. seine höchste spezifische Aktivität entfalten kann. Optimale
Enzymgeschwindigkeiten werden überwiegend im Bereich von pH 6-9 beobachtet.
In diesem Versuchsteil wird die Aktivität der AP bei acht verschiedenen pH-Werten
zwischen pH 6 und pH 10 gemessen. Dazu werden verschiedene Puffer verwendet. Die
Konzentrationen an Enzym und Substrat sind in allen Ansätzen gleich. Entsprechend dem
folgenden Pipettierschema I werden zunächst pNPP-Lösungen bei verschiedenen pH-
Werten hergestellt.
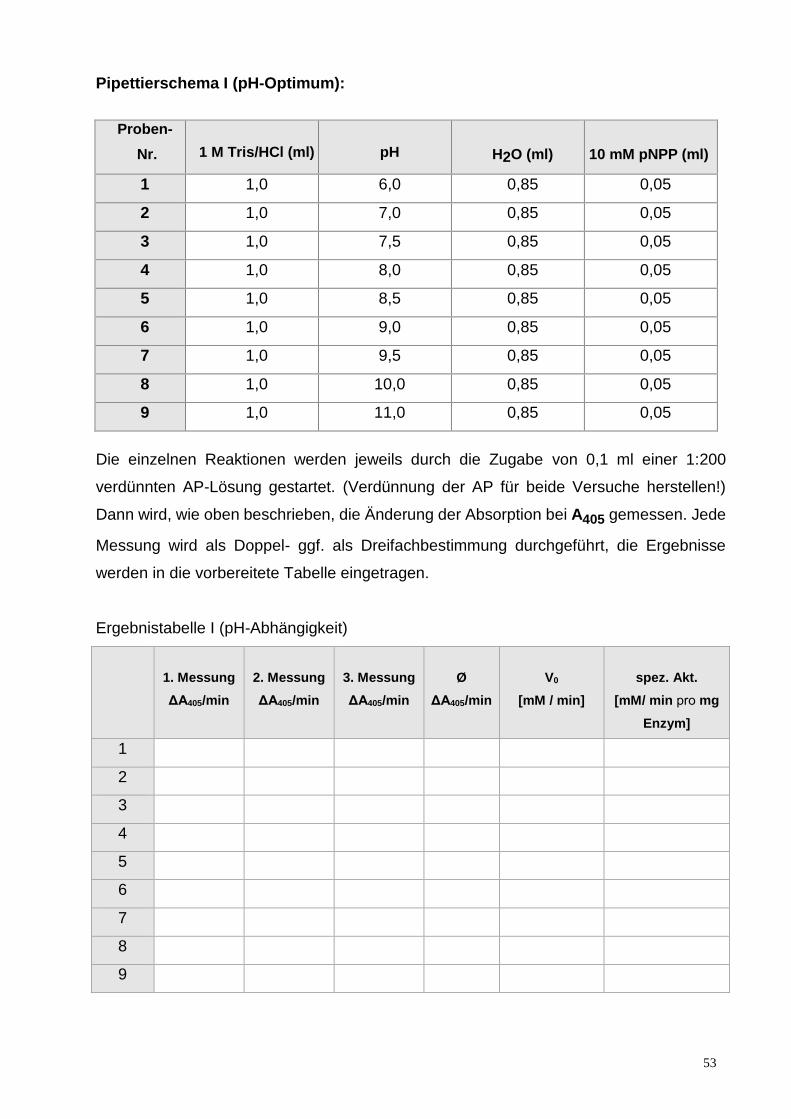
53
Pipettierschema I (pH-Optimum):
Proben-
Nr.
1 M Tris/HCl (ml)
pH
H2O (ml)
10 mM pNPP (ml)
1 1,0 6,0 0,85 0,05
2 1,0 7,0 0,85 0,05
3 1,0 7,5 0,85 0,05
4 1,0 8,0 0,85 0,05
5 1,0 8,5 0,85 0,05
6 1,0 9,0 0,85 0,05
7 1,0 9,5 0,85 0,05
8 1,0 10,0 0,85 0,05
9 1,0 11,0 0,85 0,05
Die einzelnen Reaktionen werden jeweils durch die Zugabe von 0,1 ml einer 1:200
verdünnten AP-Lösung gestartet. (Verdünnung der AP für beide Versuche herstellen!)
Dann wird, wie oben beschrieben, die Änderung der Absorption bei A405 gemessen. Jede
Messung wird als Doppel- ggf. als Dreifachbestimmung durchgeführt, die Ergebnisse
werden in die vorbereitete Tabelle eingetragen.
Ergebnistabelle I (pH-Abhängigkeit)
1. Messung
ΔA405/min
2. Messung
ΔA405/min
3. Messung
ΔA405/min
Ø
ΔA405/min
V0
[mM / min]
spez. Akt.
[mM/ min pro mg
Enzym]
1
2
3
4
5
6
7
8
9
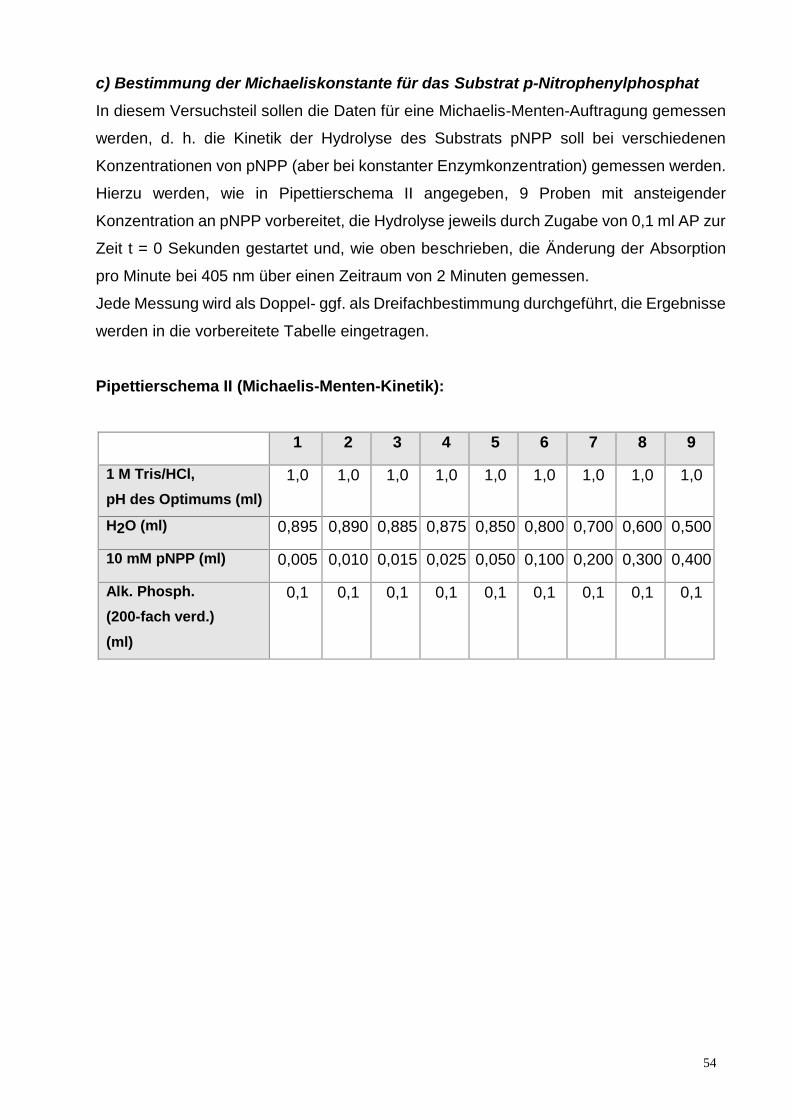
54
c) Bestimmung der Michaeliskonstante für das Substrat p-Nitrophenylphosphat
In diesem Versuchsteil sollen die Daten für eine Michaelis-Menten-Auftragung gemessen
werden, d. h. die Kinetik der Hydrolyse des Substrats pNPP soll bei verschiedenen
Konzentrationen von pNPP (aber bei konstanter Enzymkonzentration) gemessen werden.
Hierzu werden, wie in Pipettierschema II angegeben, 9 Proben mit ansteigender
Konzentration an pNPP vorbereitet, die Hydrolyse jeweils durch Zugabe von 0,1 ml AP zur
Zeit t = 0 Sekunden gestartet und, wie oben beschrieben, die Änderung der Absorption
pro Minute bei 405 nm über einen Zeitraum von 2 Minuten gemessen.
Jede Messung wird als Doppel- ggf. als Dreifachbestimmung durchgeführt, die Ergebnisse
werden in die vorbereitete Tabelle eingetragen.
Pipettierschema II (Michaelis-Menten-Kinetik):
1 2 3 4 5 6 7 8 9
1 M Tris/HCl,
pH des Optimums (ml)
1,0 1,0 1,0 1,0 1,0 1,0 1,0 1,0 1,0
H2O (ml) 0,895 0,890 0,885 0,875 0,850 0,800 0,700 0,600 0,500
10 mM pNPP (ml) 0,005 0,010 0,015 0,025 0,050 0,100 0,200 0,300 0,400
Alk. Phosph.
(200-fach verd.)
(ml)
0,1 0,1 0,1 0,1 0,1 0,1 0,1 0,1 0,1
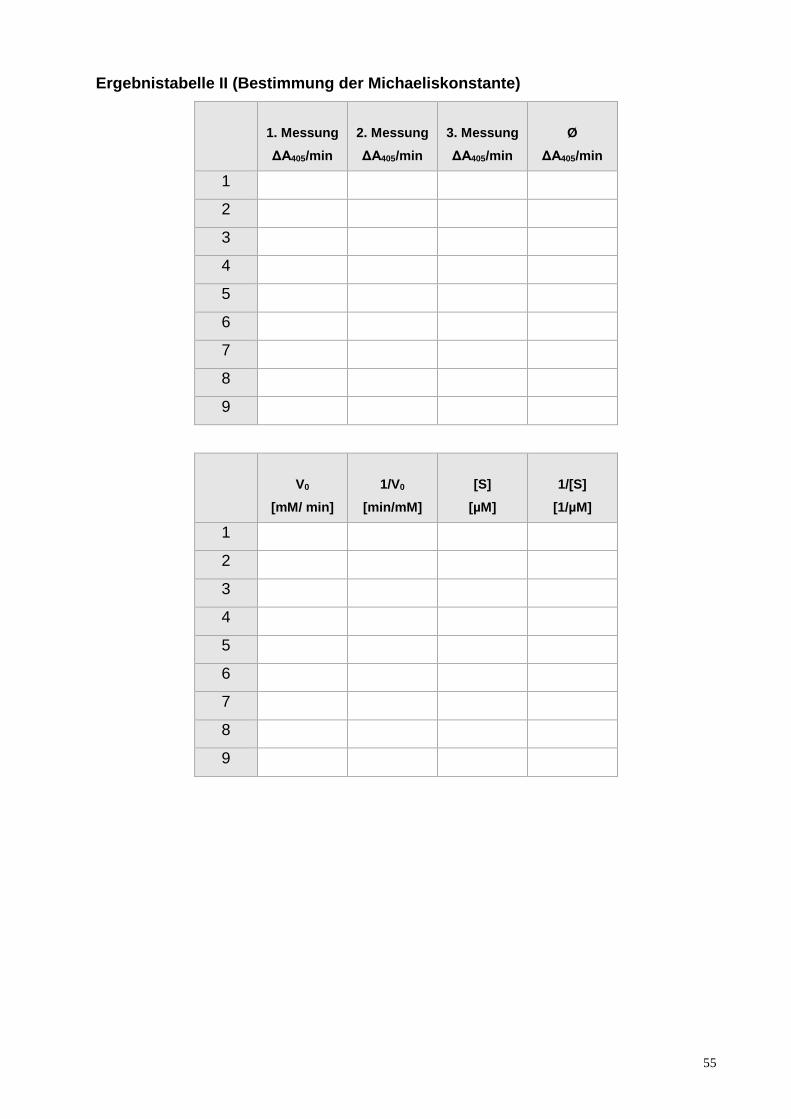
55
Ergebnistabelle II (Bestimmung der Michaeliskonstante)
1. Messung
ΔA405/min
2. Messung
ΔA405/min
3. Messung
ΔA405/min
Ø
ΔA405/min
1
2
3
4
5
6
7
8
9
V0
[mM/ min]
1/V0
[min/mM]
[S]
[µM]
1/[S]
[1/µM]
1
2
3
4
5
6
7
8
9

56
AUSWERTUNG DER ERGEBNISSE
a) Bestimmung des pH-Optimums der AP
Bestimmen Sie aus den Werten ΔA405 /min mittels des Lambert-Beerschen Gesetzes die
Anfangsgeschwindigkeit in mM /min und unter Berücksichtigung der Enzymverdünnung
die spezifische Aktivität des Enzyms (in mM / min mg Enzym) und tragen Sie diese Werte
in die Ergebnistabelle „pH-Abhängigkeit“ ein. Geben Sie je eine Beispielrechnung für die
Berechnung der Initialgeschwindigkeit sowie der spezifischen Aktivität an. Tragen Sie die
spezifische Aktivität gegen den pH-Wert auf. Wo liegt das pH-Optimum der AP?
b) Bestimmung der Michaeliskonstante für das Substrat p-Nitrophenylphosphat
Tragen Sie bitte Ihre Daten in die Ergebnistabelle „Bestimmung der Michaeliskonstanten“
ein und berechnen Sie die jeweilige Initialgeschwindigkeit in mM / min bei den jeweiligen
Substratkonzentrationen (Bitte je eine Beispielrechnung angeben). Tragen Sie diese
anschließend als Funktion der Substratkonzentration im Michaelis-Menten Diagramm auf.
Aus diesem Diagramm können Sie erste Werte für vmax und KM abschätzen.
Tragen Sie dann Geschwindigkeit und Substratkonzentration entsprechend Lineweaver-
Burk doppelt reziprok auf und bestimmen Sie daraus KM und vmax. Bestimmen Sie
außerdem aus dem Wert für vmax die Wechselzahl (kcat) für die AP (Molekulargewicht für
das Enzymdimer: 114 600 Da).

57
BERECHNUNGSMODUS und HILFESTELLUNG
für die Bestimmung der Enzymaktivität der Alkalischen Phosphatase
1) Bestimmung der Initialgeschwindigkeiten v0
Basis hierfür ist das Lambert-Beer’sche Gesetz: ΔE = ε dc
umgeformt: Δd
Ec
dabei sind Δc: die Konzentrationsänderung in mol/l ΔE: die Extinktionsänderung / min ε: der molare Extinktionskoeffizient für pNPP in M-1 · cm-1 (= l ·mol-1 ·cm-1) d: die Schichtdicke der Küvette in cm
Δc v0
v0 d
E
min/405
cm
cmM
min cml
cmmol
min
min
M
Beispiel:
ΔE405 / min = 0,08
ε405 = 18.500 M-1 · cm-1
d = 1 cm
v0 d
E
min/405
cm
cmM
min500.18
08,0
= 0,0000043 · M / min
= min/0043,0 mM
Die Initialgeschwindigkeit v0 gibt an, dass im Falle des o.g. Beispiels 0,0043 µmol pNPP-
Substrat pro Minute in einem Volumen von 1 ml durch die entsprechende Enzymmenge
umgesetzt wurden.

58
2) Bestimmung der spez. Aktivität der Alkalischen Phosphatase
Die spez. Aktivität gibt die Enzymaktivität pro mg Protein an, d.h. wieviel
Aktivitätseinheiten in einem mg Enzym enthalten sind. Das bedeutet, dass sich aus der
Initialgeschwindigkeit v0 unter Berücksichtigung der Enzymverdünnung die spez. Aktivität
des Enzyms in [M / min · mg Enzym] berechnen lässt.
mgml
molin
Enzymmg
vAktivitätspez
min.
0
Beispiel:
z.B. Ausgangskonzentration der AP: 5 mg/ml
Verdünnung 1:200: 0,025 mg/ml
Verdünnung in Küvette nochmals 1: 20
(100 µl Enzymlösung / 2 ml Ansatz): [E] = 0,00125 mg / ml
Enzymmenge in 2mL =0,0025mg
mg
mM
mgEnzym
vAktivitätspez
min0025,0
0043,0.
0
mgml
mol
mg
mM
min72,1
min72,1

59
3) Bestimmung der Wechselzahl der Alkalischen Phosphatase
Die Wechselzahl (kcat) eines Enzyms gibt an, wieviele Substratmoleküle durch ein
Enzymmolekül (bzw. ein aktives Zentrum) pro Minute (oder pro Sekunde) unter der
Voraussetzung der Substratsättigung umgesetzt werden.
][2
max
E
Vkcatk [E] = Enzymkonzentration (Enzymmenge)
Berechnung von [E]:
z.B. Ausgangskonzentration der AP: 5 mg/ml
Verdünnung 1:200: 0,025 mg/ml
Verdünnung in Küvette nochmals 1: 20
(100 µl Enzymlösung / 2 ml Ansatz): [E] = 0,00125 mg / ml
Umrechnung mg/ml in M:
MG AP: 114600 Da
d.h. 1 molAP = 114600 g
d.h., eine 1 molare Lösung Alkalische Phosphatase (1 M) enthält 114600 g / l.
1MAP = 114600 g / l
? = 0,00125 mg / ml
= 1,09·10-5 mM
min1009,1
01,0
][2
5
max
mM
mM
E
Vk
= 917 min-1
= 15 sec -1
Die Alkalische Phosphatase setzt unter den gegebenen Bedingungen 917 Moleküle
pNPP pro Minute (bzw. 15 Moleküle pro Sekunde) um. Da die AP als Dimer aktiv ist,
ergibt sich für 1 katalytisches Zentrum ein Umsatz von 458 Molekülen pro Minute.

60
Versuch 6:
Kompetitive Hemmung
EINLEITUNG
Die kompetitive Hemmung von Enzymen spielt eine wichtige Rolle bei der
Stoffwechselregulation. Der Hemmstoff ist dabei häufig dem Substrat strukturell ähnlich
und bindet wie das Substrat im aktiven Zentrum. Substrat und Inhibitor haben also eine
zumindest teilweise überlappende Bindungsstelle, um die beide konkurrieren.
Analog zur Bildung des Enzym-Substrat-Komplexes führt die Bindung des Inhibitors an
das Enzym zur Bildung des Enzym-Inhibitor-Komplexes. Ausgehend von der Michaelis-
Menten-Gleichung lässt sich die Gleichgewichtskonstante des Enzym-Inhibitor-
Komplexes (KI) wie folgt ableiten:
𝐾𝐼 =[𝐸][𝐼]
[𝐸𝐼]
Setzt man dies in die Michaelis-Menten-Gleichung ein, so erhält man für die
Reaktionsgeschwindigkeit in Gegenwart eines Inhibitors:
𝑣 =𝑣𝑚𝑎𝑥 ∙ [𝑆]
𝐾𝑀 (1 +[𝐼]𝐾𝐼) + [𝑆]
Transformiert in die doppelt reziproke Form (Lineweaver-Burk-Darstellung) ergibt sich:
1
𝑣=𝐾𝑀 (1 +
[𝐼]𝐾𝐼) + [𝑆]
𝑣𝑚𝑎𝑥 ∙ [𝑆]=
1
𝑣𝑚𝑎𝑥+𝐾𝑀 (1 +
[𝐼]𝐾𝐼)
𝑣𝑚𝑎𝑥 ∙ [𝑆]
Vmax wird durch die Bindung des Inhibitors nicht verändert. Der scheinbare KM-Wert des
Enzyms vergrößert sich jedoch um den Faktor (1 +[𝐼]
𝐾𝐼). Für eine konstante
Substratkonzentration [S] ergibt sich ein linearer Zusammenhang zwischen 1/v und der
Inhibitorkonzentration [I].

61
PROBLEMSTELLUNG
Im vorliegenden Versuch soll untersucht werden, inwiefern die Aktivität der alkalischen
Phosphatase durch ihr Produkt Phosphat gehemmt wird. Die Gleichgewichtskonstante des
Enzym-Inhibitor-Komplexes soll bestimmt werden.
MATERIALIEN
Puffer und Lösungen
Testpuffer: 1 M Tris/HCl Puffer, pH 9,5 Substratlösung: 10 mM p-Nitrophenylphosphat in H2O (vor Licht geschützt
und eisgekühlt aufbewahren, über Nacht einfrieren) Inhibitorlösung: 100 mM K2HPO4 in Testpuffer
Enzym: Alkalische Phosphatase, 5 mg/ml aus Kälberdarm
(AppliChem, Darmstadt), 24,2 U/mg, in Testpuffer pH 9,5 gelöst MW (pro Untereinheit): 57 300 Da
PRAKTISCHE DURCHFÜHRUNG
a) Bestimmung der Michaeliskonstante für das Substrat pNPP in Gegenwart von
anorganischem Phosphat
In diesem Versuchsteil sollen die Daten für eine Michaelis-Menten-Auftragung gemessen
werden, d. h. die Kinetik der Hydrolyse des Substrats pNPP soll bei verschiedenen
Konzentrationen von pNPP (aber bei konstanter Inhibitor- und Enzymkonzentration)
gemessen werden. Hierzu werden, wie in Pipettierschema I angegeben, 9 Proben mit
ansteigender Konzentration an pNPP vorbereitet, die Hydrolyse jeweils durch Zugabe von
0,1 ml AP zur Zeit t = 0 Sekunden gestartet und, wie oben beschrieben, die Änderung der
Absorption pro Minute bei 405 nm über einen Zeitraum von 2 Minuten gemessen.
Jede Messung wird als Doppel- ggf. als Dreifachbestimmung durchgeführt, die Ergebnisse
werden in die vorbereitete Tabelle eingetragen.
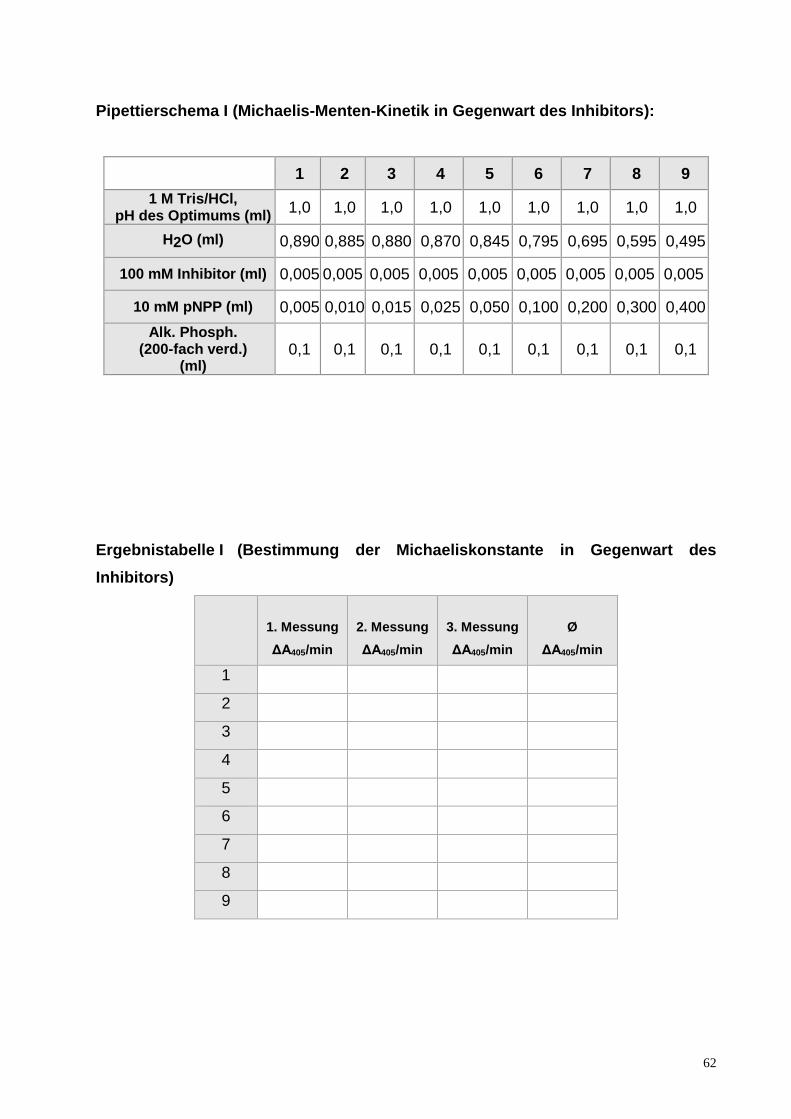
62
Pipettierschema I (Michaelis-Menten-Kinetik in Gegenwart des Inhibitors):
1 2 3 4 5 6 7 8 9
1 M Tris/HCl, pH des Optimums (ml)
1,0 1,0 1,0 1,0 1,0 1,0 1,0 1,0 1,0
H2O (ml) 0,890 0,885 0,880 0,870 0,845 0,795 0,695 0,595 0,495
100 mM Inhibitor (ml) 0,005 0,005 0,005 0,005 0,005 0,005 0,005 0,005 0,005
10 mM pNPP (ml) 0,005 0,010 0,015 0,025 0,050 0,100 0,200 0,300 0,400
Alk. Phosph. (200-fach verd.)
(ml) 0,1 0,1 0,1 0,1 0,1 0,1 0,1 0,1 0,1
Ergebnistabelle I (Bestimmung der Michaeliskonstante in Gegenwart des
Inhibitors)
1. Messung
ΔA405/min
2. Messung
ΔA405/min
3. Messung
ΔA405/min
Ø
ΔA405/min
1
2
3
4
5
6
7
8
9

63
V0
[mM/ min]
1/V0
[min/mM]
[S]
[µM]
1/[S]
[1/µM]
1
2
3
4
5
6
7
8
9
b) Bestimmung der Inhibitorkonstante für anorganisches Phosphat
In diesem Versuchsteil sollen die Daten für eine Enzyminhibition gemessen werden, d. h.
die Kinetik der Hydrolyse des Substrats pNPP soll bei verschiedenen Konzentrationen von
anorganischem Phosphat (aber bei konstanter Substrat- und Enzymkonzentration)
gemessen werden. Hierzu werden, wie in Pipettierschema II angegeben, 8 Proben mit
ansteigender Konzentration an Phosphat vorbereitet, die Hydrolyse von pNPP jeweils
durch Zugabe von 0,1 ml AP zur Zeit t = 0 Sekunden gestartet und, wie oben beschrieben,
die Änderung der Absorption pro Minute bei 405 nm über einen Zeitraum von 2 Minuten
gemessen.
Jede Messung wird als Doppel- ggf. als Dreifachbestimmung durchgeführt, die Ergebnisse
werden in die vorbereitete Tabelle eingetragen.
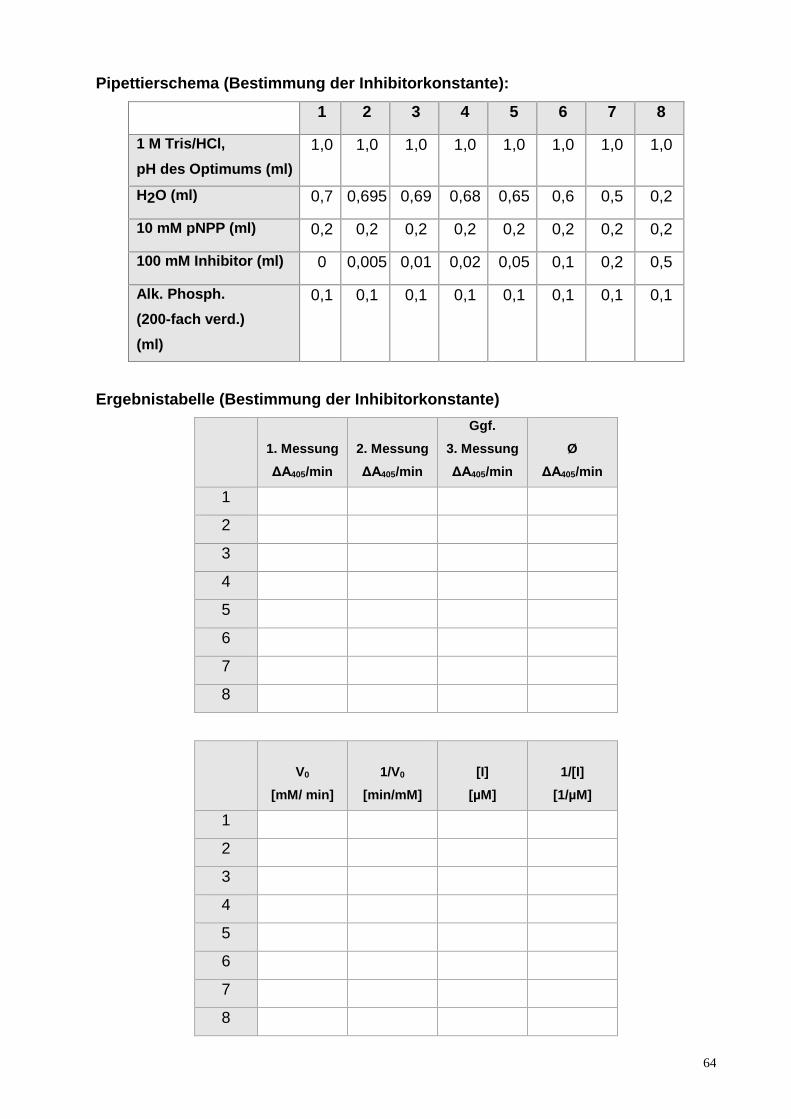
64
Pipettierschema (Bestimmung der Inhibitorkonstante):
1 2 3 4 5 6 7 8
1 M Tris/HCl,
pH des Optimums (ml)
1,0 1,0 1,0 1,0 1,0 1,0 1,0 1,0
H2O (ml) 0,7 0,695 0,69 0,68 0,65 0,6 0,5 0,2
10 mM pNPP (ml) 0,2 0,2 0,2 0,2 0,2 0,2 0,2 0,2
100 mM Inhibitor (ml) 0 0,005 0,01 0,02 0,05 0,1 0,2 0,5
Alk. Phosph.
(200-fach verd.)
(ml)
0,1 0,1 0,1 0,1 0,1 0,1 0,1 0,1
Ergebnistabelle (Bestimmung der Inhibitorkonstante)
1. Messung
ΔA405/min
2. Messung
ΔA405/min
Ggf.
3. Messung
ΔA405/min
Ø
ΔA405/min
1
2
3
4
5
6
7
8
V0
[mM/ min]
1/V0
[min/mM]
[I]
[µM]
1/[I]
[1/µM]
1
2
3
4
5
6
7
8

65
AUSWERTUNG DER ERGEBNISSE
a) Bestimmung der Michealis-Menten-Parameter in Gegenwart eines Inhibitors
Tragen Sie bitte Ihre Daten in die Ergebnistabelle „Bestimmung der Michaeliskonstanten
in Gegenwart eines Inhibitors“ ein und berechnen Sie die jeweilige Initialgeschwindigkeit
in mM / min bei den jeweiligen Substratkonzentrationen (Bitte je eine Beispielrechnung
angeben). Tragen Sie diese anschließend als Funktion der Substratkonzentration im
Michaelis-Menten Diagramm auf. Aus diesem Diagramm können Sie erste Werte für vmax
und KM abschätzen.
Tragen Sie dann Geschwindigkeit und Substratkonzentration entsprechend Lineweaver-
Burk doppelt reziprok auf und bestimmen Sie daraus KM und vmax in Gegenwart des
Inhibitors. Bestimmen Sie außerdem aus dem Wert für vmax die Wechselzahl (kcat) für die
AP in Gegenwart des Inhibitors (Molekulargewicht für das Enzymdimer: 114 600 Da).
b) Bestimmung der Inhibitorkonstante für anorganisches Phosphat
Tragen Sie bitte Ihre Daten in die Ergebnistabelle „Bestimmung der Inhibitorkonstante“ ein
und berechnen Sie die jeweilige Initialgeschwindigkeit in mM / min bei den jeweiligen
Substratkonzentrationen (Bitte je eine Beispielrechnung angeben). Tragen Sie diese
anschließend reziprok als Funktion der Inhibitorkonzentration im Diagramm auf. Aus
diesem Diagramm können Sie erste Werte für KI abschätzen.
c) Bewertung des Einflusses des Inhibitors auf die enzymatische Reaktion
Bewerten Sie ausgehend von den erhaltenen Daten welche enzymkinetischen Parameter
in Gegenwart des Inhibitors auf welche Art und Weise beeinflusst werden. Um welchen
Typ Hemmung handelt es sich (kompetitiv, nichtkompetitiv, unkompetitiv?)

66
VERSUCH FÜR ZU HAUSE:
Isolierung von DNA aus Tomaten
EINLEITUNG
Für die Isolierung von DNA existieren sehr viele unterschiedliche Protokolle, die für
verschiedene Zellen und Gewebe, für Plasmid- oder genomische DNA im kleinen oder
großen Maßstab entwickelt wurden. Die folgende Methode ist eine sehr „grobe“ Methode,
um DNA und RNA aus pflanzlichem Gewebe zu isolieren. Sie wird natürlich so nicht im
Labor angewendet, zeigt aber die Grundprinzipien der DNA-Extraktion aus Geweben auf.
Ferner eignet sich diese Methode hervorragend für das Experimentieren zu Hause oder
in der Schule.
Das Gewebe wird zunächst mechanisch, die Zell- und Kernmembranen durch die
Tensidwirkungen eines Haushaltsreinigers zerstört. Die Zellreste werden herausgefiltert
und die Nukleinsäuren gelangen durch die groben Poren eines Kaffeefilters ins Filtrat.
Außer einer halben Tomate (oder Zwiebel) werden noch folgende Stoffe benötigt:
5 ml Spülmittel, 1,5 g Kochsalz, 2 Tropfen flüssiges Waschmittel, kalter Alkohol aus der
Apotheke (z.B. über Nacht im Gefrierfach aufbewahrt)
Praktische Durchführung
1. Zu 50 ml Wasser werden Kochsalz und Spülmittel
gegeben. Gut Umrühren, damit sich das Salz löst!
2. Die Tomate wird in kleine Stücke geschnitten und die Stücke in die salzige
Spülmittellösung gegeben. Das Spülmittel bricht die Zellmembranen auf, so dass
die DNA aus den Zellen und Zellkernen entlassen wird.
3. Die Mischung muss nun mindestens 15 Minuten stehenbleiben. Man kann sie
auch erhitzen (auf ca. 60°C), denn Wärme beschleunigt den Auflöseprozess und
zerstört zugleich die sogenannten DNasen (das sind Enzyme, die die DNA abbauen).
4. Jetzt wird die Mischung im Mörser püriert (oder gemixt – aber nicht zu lange, sonst
wird die DNA zerstört).

67
5. Anschließend wird die Mischung filtriert – zu Hause kann man dazu einen
Kaffeefilter verwenden. Dabei bleiben die festen Zellwandbestandteile im Filter
zurück und werden so von der gelösten DNA und den Proteinen getrennt.
6. Etwa 10ml des Filtrats werden in ein hohes, schmales Glas (z.B. Reagenzglas)
gegeben. Dazu kommen 2 bis 3 Tropfen Flüssigwaschmittel. Wieder gut
mischen! Im Waschmittel sind Proteasen enthalten, Enzyme, die die Proteine in der
Lösung abbauen. Dazu sind nur winzige Mengen Enzym nötig.
7. Zum Schluss wird die Lösung mit dem gleichen Volumen kalten Alkohols
überschichtet – Dabei wird der Alkohol vorsichtig an der Wand des Gefäßes
entlang hineingegossen. Man erhält zwei Flüssigkeitsschichten: oben den Alkohol
und unten den Tomatenextrakt.
8. Innerhalb kurzer Zeit reichert sich die DNA an der Grenze zwischen beiden
Schichten an. DNA löst sich nicht in Alkohol und wird daher als weißer Schleim
sichtbar. Mit Hilfe eines Holzstäbchens kann man die DNA herausfischen.
Literatur Pingoud, A., Urbanke, C. (1997). Arbeitsmethoden der Biochemie. Walter de Gruyter,
Berlin, New York
Rhodes, M., Azari, P. und Feeney, R. (1957). Analysis, Fractionation, and Purification of
Egg White Proteins with Cellulose-Cation Exchanger. J. Biol. Chem. 230(1): 399-408.
Lottspeich, F., Zorbas, H. (1998). Bioanalytik. Spektrum, Akad. Verl., Heidelberg, Berlin
Die biochemischen Grundlagen der hier beschriebenen Versuche können in diversen
Lehrbüchern (Stryer, Voet etc.) nachgelesen werden.





























