Embed Size (px)
Citation preview

R. Beckhaus, J. Oster, T. Wagner I003
Cyclische isomere vier- und funfgliedrige heterodinucleare Fischer-Carbenkomplexe - gebildet aus einem Vinylidentitanocen-Fragment und Metallcarbonylen Riidiger Beckhaus”, Jiirgen Oster und Trixie Wagner
Institut fur Anorganische Chemie der Technischen Hochschule Aachen, Professor-Pirlet-StraBe 1, D-52056 Aachen, Germany
Eingegangen am 22. Dezember 1993
Key Words: Metallocene complexes, bent / Titanium complexes / Vinylidene complexes / Transition metal complexes / Heterodinuclear complexes / Carbene complexes / Early-Late heterodinuclear complexes / Metallacycles / Oxatitanacyclobutane complexes / Oxatitanacyclopentene complexes
Cyclic Isomeric Four- and Five-Membered Heterodinculear Carbene Complexes - Formed from a Titanocene Vinylidene Fragment and Metal Carbonyls
The four-membered heterodimetallic carbene complexes Cp;TiC(=CH,)C(=ML,)d 16 [L,M: Cr(CO)5 (a], MO(CO)~
Fe(CO)* (i), Rh(acac)(CO) (i)] are easily prepared by trapping the vinylidenetitanocene [Cp,’Ti=C=CH2] fragment 6 with the corresponding metal carbonyls. The vinylidenetitano- cene 6 is generated in a favourable manner from Cp;- Ti(CH3)CH=CH2 (13) or Cp*(Fv)TiCH=CH2 (14) [Fv: C5(CH3)4CHZ] by a 1,3-H shift (a elimination) at room tempe- rature. The formation of 16 is characterized by a preferred suprafacial [2 +2] cycloaddition. Starting from Cp; - tiC(=CH2)CH2cH2 (5) or 14 and transition metal carbonyls unexpected isomeric five-membered titanacycles Cpz-
(b), W(CO)5 (c) , C P R ~ ( C ~ ) Z ( f ) , Mn2(CO)g(g), RedCOh (h),
kiCH=CH-C(=ML,)b 8 [ML, = a-c, MnCp(CO), (d), MnCp’(COf2 (e), f-i] are obtained at higher temperatures (70- 100°C). These oxatitanacyclopentenes 8 are formed also upon heating via the kinetic product 16 with high yield, as a result of a 1,2-hydrogen shift, proved for c, g, h. From the spectroscopic data an ql-vinylcarbene structure is deduced, exhibiting a cis- (16) or trans-configurated (8) L,M=C-C=C sequence. An X-ray structure analysis shows the importance of the acyl resonance form in 8a, typically observed for such heterodimetallic carbene complexes. The reversible libera- tion of the metal carbonyl is a remarkable property of com- plexes 16 in solution, whereas the thermodynamic product 8 shows the typical properties of Fischer carbene complexes.
Die a-Eliminierung kleiner Molekiile aus gesattigten Or- ganometallkomplexen 1 hat sich zu einem wirkungsvollen Verfahren zur Gewinnung von Metall-Ligand-Mehrfach- bindungssystemen 2 entwickelt. Dieses Reaktionsprinzip, wohletabliert fur die Synthese von Tantal-Carben-Komple- xen vom Schrock-Typ[’g21, findet zunehmend weitere Paral- lelen, wie z.B. zum Aufbau isolierbarer Komplexe rnit Zir- c~nium-[~] oder Mob-Kohlenstoff-Mehrfa~hbindungenl~]. Vielfach lassen sich a-H-Ubertragungsreaktionen auch zur Bildung intermediarer Metall-K~hlenstoff-[~] oder -Hete- roatom-Mehrfachbind~ngen[~~~] des Titans oder Zirconi- ums nutzen.
1 2
Eigene Untersuchungen zum Reaktionsverhalten von Vi- nylverbindungen elektronenarmer Ubergangsmetalle[8] er- brachten die spontane Umwandlung des Divinyltitanocens 3 zum Metallacyclobutan-Derivat 5[9,’01. Diese Reaktion ist durch die Ubertragung eines Wasserstoffatoms zwischen den Vinylgruppen unter Ausbildung des reaktivitatsbestim- menden Vinyliden-Ethylen-Intermediates 4 charakterisiert. In Verbindung rnit der bevorzugten C,-symmetrischen
Grundzustandsgeometrie des durch Thermolyse und Ethy- lenabspaltung aus 5 intermediar zuganglichen Vinylidenti- tanocens [Cp$Ti=C=CH2] 6L91 werden via 5-4 Cycloaddi- tionsprodukte rnit Cumulenen und Heterocumulenen unter Bildung von 7a-c gefunden“ ‘-12]. Enolisierbare Ketone reagieren stereo- und regioselektiv hingegen ausschlieDlich im Sinne einer 1,higmatropen H-Verschiebung zu Vinylti- tan~cen-enolaten[’~]. Unter analogen Reaktionsbedingun- gen, bestimmt durch die hohen Temperaturen zur Abspal- tung von Ethylen aus 5 (70- 12OoC), ergeben Umsetzungen rnit Metallcarbonylen unerwartete fiinfgliedrige dimetalli- sche Fischer-Carbenkomplexe 8[l l]. Diese uberraschende Differenzierung in der Bildung der Oxatitanacyclobutan- Ringe 7 einerseits und der Oxatitanacyclopenten-Derivate 8 andererseits veranlaBte uns, das Reaktionsverhalten von 6 gegeniiber Metallcarbonylen detaillierter zu untersuchen.
Im Gegensatz zu Cycloadditionen von Metallcarbonylen rnit Arinen[l41, Dienen[ls1, Olefinen[16] und Heterolefinen[17] an Metallocenfragmenten der Metalle der 4. Neben- gruppe[I8] (9-10) sind entsprechende Reaktionen von Schrock-Carben-Fragmenten rnit Metallcarbonylen (11-12) bisher kaum bekannt. In einigen Fallen wurden Zwischenprodukte vom Typ 12 teilweise als Intermediate spektroskopisch beobachtet[”] oder aufgrund von Folge-
Chem. Ber. 1994,127,1003- 1013 0 VCH Verlagsgesellschaft mbH, D-69451 Weinheim, 1994 0009-2940/94/0606- 1003 $10.00+.25/0
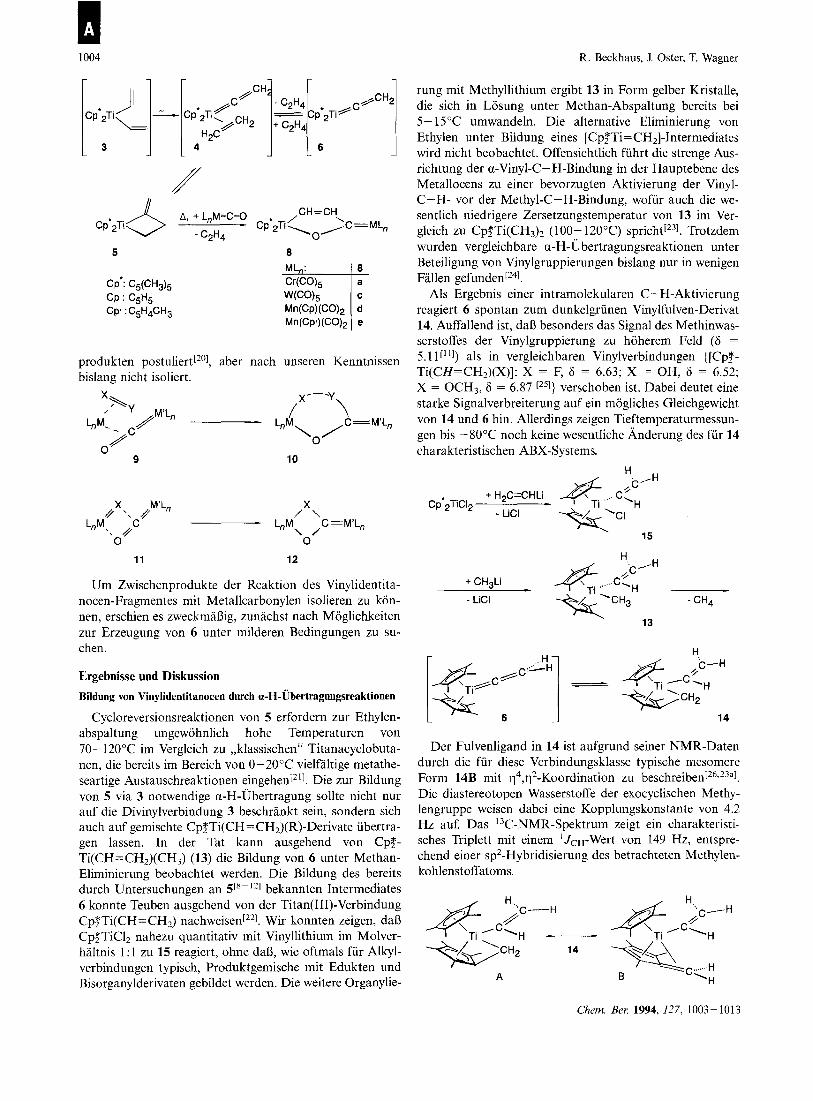
1004 R. Beckhaus, J. Oster, T. Wagner
CH-CH A, +L,M=C=O Cp II *Ti-\ ,/ ,C=ML" \
C2H4 0 Cp'2Ti
5 8
produkten postuliert[20], aber nach unseren Kenntnissen bislang nicht isoliert.
9 10
11 12
Um Zwischenprodukte der Reaktion des Vinylidentita- nocen-Fragmentes rnit Metallcarbonylen isolieren zu kon- nen, erschien es zweckmaDig, zunachst nach Moglichkeiten zur Erzeugung von 6 unter milderen Bedingungen zu su- chen.
Ergebnisse und Diskussion Bildung von Vinylidentitanocen durch a-H-Ubertragungsreaktionen
Cycloreversionsreaktionen von 5 erfordern zur Ethylen- abspaltung ungewohnlich hohe Temperaturen von 70- 120°C im Vergleich zu ,,klassischen" Titanacyclobuta- nen, die bereits im Bereich von 0-20°C vielfaltige metathe- seartige Austauschreaktionen eingehen[2']. Die zur Bildung von 5 via 3 notwendige a-H-Ubertragung sollte nicht nur auf die Divinylverbindung 3 beschrankt sein, sondern sich auch auf gemischte Cp2Ti(CH = CH2)(R)-Derivate ubertra- gen lassen. In der Tat kann ausgehend von Cp3- Ti(CH=CH2)(CH,) (13) die Bildung von 6 unter Methan- Eliminierung beobachtet werden. Die Bildung des bereits durch Untersuchungen an 5r8-l2l bekannten Intermediates 6 konnte Teuben ausgehend von der Titan(II1)-Verbindung Cp2Ti(CH=CH2) nachweisen[22]. Wir konnten zeigen, da13 CpZTiCl2 nahezu quantitativ mit Vinyllithium im Molver- haltnis 1 : 1 zu 15 reagiert, ohne daI3, wie oftmals fur Alkyl- verbindungen typisch, Produktgemische rnit Edukten und Bisorganylderivaten gebildet werden. Die weitere Organylie-
rung rnit Methyllithium ergibt 13 in Form gelber Kristalle, die sich in Losung unter Methan-Abspaltung bereits bei 5- 15°C umwandeln. Die alternative Eliminierung von Ethylen unter Bildung eines [Cp,*Ti=CH2]-Intermediates wird nicht beobachtet. Offensichtlich fuhrt die strenge Aus- richtung der a-Vinyl-C-H-Bindung in der Hauptebene des Metallocens zu einer bevorzugten Aktivierung der Vinyl- C-H- vor der Methyl-C-H-Bindung, wofur auch die we- sentlich niedrigere Zersetzungstemperatur von 13 im Ver- gleich zu Cp;Ti(CH3), (100- 120°C) s p r i ~ h t [ ~ ~ ] . Trotzdem wurden vergleichbare a-H-Ubertragungsreaktionen unter Beteiligung von Vinylgruppierungen bislang nur in wenigen Fallen g e f ~ n d e n [ ~ ~ ] .
Als Ergebnis einer intramolekularen C- H-Aktivierung reagiert 6 spontan zum dunkelgrunen Vinylfulven-Derivat 14. Auffallend ist, daD besonders das Signal des Methinwas- serstoffes der Vinylgruppierung zu hoherem Feld (6 =
5.1 lC1 '1) als in vergleichbaren Vinylverbindungen {[Cpz- Ti(CH=CH2)(X)]: X = F, 6 = 6.63; X = OH, 6 = 6.52; X = OCH3, 6 = 6.87 [251) verschoben ist. Dabei deutet eine starke Signalverbreiterung auf ein mogliches Gleichgewicht von 14 und 6 hin. Allerdings zeigen Tieftemperaturmessun- gen bis -80°C noch keine wesentliche Anderung des fur 14 charakteristischen ABX-Systems.
H
+ H2C=CHLi c~'~TiCl ,
' 15
- CH4
+ CH3Li
- LiCl
13
Der Fulvenligand in 14 ist aufgrund seiner NMR-Daten durch die fur diese Verbindungsklasse typische mesomere Form 14B rnit q4,q2-Koordination zu bes~hreiben[*~~~~"] . Die diastereotopen Wasserstoffe der exocyclischen Methy- lengruppe weisen dabei eine Kopplungskonstante von 4.2 Hz auf. Das l3C-NMR-Spektrurn zeigt ein charakteristi- sches Triplett rnit einem 'JcH-Wert von 149 Hz, entspre- chend einer sp2-Hybridisierung des betrachteten Methylen- kohlenstoffatoms.
Chem. Ber. 1994,127, 1003-1013

Cyclische isomere vier- und fiinfgliedrige heterodinucleare Fischer-Carbenkomplexe 1005
Die Kopplungskonstante fur die a-CH-Vinylgruppierung ist auf einen Wert von 11 5 Hz erniedrigt. Unter Beriicksich- tigung der Hochfeldverschiebung des Methinsignals kann somit eine a-agostische Vinylgruppierung nicht ausge- schlossen ~ e r d e n l ~ ~ ] . Fur metallvermittelte reversible H- Ubertragungen unter direkter Beteiligung von M-H-Bin- dungen sind vielfiltige Beispiele bekannt. Das trifft einer- seits auf die ligandvermittelte ,,ReparaturFahigkeit" von Fulvenmetallocen-Verbindungen[26~28], aber auch auf viel- faltige anders geartete Metal l -a lken~l-[~~~] oder -alkyl-Ver- bindungen[2y1 zu. Nach unseren Kenntnissen sind reversible intramolekulare H-Ubertragungsreaktionen zwischen carb- anionischen Organylgruppen, die wahlweise vergleichbar ei- ner o-Organylverbindung (14A) oder als Carbenfragment (6) zu reagieren vermogen, bislang nahezu unbekanntL3O1. In dieser Arbeit wollen wir prufen, ob 13 oder auch 14 als potentielle Quellen fur 6 Beitrage zum Verstandnis der Bil- dung von 8 liefern.
Synthese heterodinuclearer Carbenkomplexe rnit Oxatitanacyclobutan-Teilstruktur
Versetzt man eine Losung von 13 in aliphatischen Koh- lenwasserstoffen mit einer aquimolaren Menge eines Hexa- carbonyls eines Metalls der 6 . Nebengruppe, so erfolgt oberhalb einer Temperatur von 0°C ein rascher Farbwech- sel der Losungen von gelb nach dunkelgriin. Dabei 1aSt sich eine Methan-Entwicklung nachweisen, verbunden rnit der iR-spektroskopisch zu verfolgenden Bildung von 16. Im Verlauf einer ein- bis zweistundigen Reaktionszeit fallen die Reaktionsprodukte 16a-c spontan aus, die in Form inten- siv gruner Kristalle rnit brillantem metallischen Oberfla- chenglanz isoliert werden konnen. In analoger Weise lassen sich die dinuclearen Carbenkomplexe 16f- j, ebenfalls mit Ausbeuten von 30- 55%, gewinnen.
+ L,M=C-0 13 c
- CH4
Im festen Zustand sind die so erhaltenen Verbindungen bis zu einer Temperatur von ca. 130°C stabil. Losungen von 16 sind ausgesprochen reaktiv und setzen bereits bei tiefen Temperaturen (-78 bis -20°C) das jeweilige Metallcarbo-
nyl wieder frei, besonders dann, wenn potentielle n-Akzep- torliganden (Ethylen, isocyanate), aber auch Feuchtigkeits- oder Sauerstoffspuren zugegen sind.
Im Unterschied zur funfgliedrigen Ringstruktur der er- wahnten heterodinuclearen Carbenkomplexe 8 zeigen die spektroskopischen Daten fur 16 eindeutig eine Oxatitanacy- clobutan-Teilstruktur. Als besonders charakteristisch erwei- sen sich dabei die im Unterschied zu 5 tieffeldverschobenen Protonenresonanzen der exocyclischen Methylengruppie- rungen in Form zweier Singuletts (s. Tab. 1).
Tab. 1. 'H-NMR-Daten (6-Werte) ausgewahlter Metallacyclen mit Oxatitanacyclobutan-Teilstrukturen (500 MHz, TMS) im Vergleich
zum Titanacyclobutan 5
7 21 (s)
7 17 (s)
7 21 (s)
7 01 (s)
7 Ol(s)
7 59 (s)
7 62 (s)
7 I7 (s)
1 2 2 178
I 19 179
191 1 5 5
I 4 4 185
177 I43
200 149
200 15')
2 3 8 161
Gleichzeitig zeigt sich ein Anwachsen der Unterschiede in den chemischen Verschiebungen der Protonen der exocy- clischen Methylengruppe in Abhangigkeit von der Natur der Substituenten am Oxatitanacyclobutan-Ring. Ein An- wachsen der A-Werte lauft dabei parallel rnit steigendem Elektronenangebot des jeweiligen Substituenten (X) in b- Position der Titanacyclen, so daS fur 16i und 16j und auch fur 7a und 7c die groDten Differenzen nachzuweisen sind.
Die IR-Spektren lassen fur die L,M(CO)5-Fragmente in 16a-c entsprechend der vorliegenden Punktgruppe C4, je zwei Streckschwingungen der Symmetrierasse A, sowie eine der Rasse E erkennen. Die vom Chrom zu Wolfram zuneh- mende Aufspaltung der E-Bande 1aSt sich rnit einer Storung der idealen C4,-Symmetrie im Titanacyclus durch das unge- sattigte Ca-Atom erklaren (s. Tab. 4)L3*1.
Die Massenspektren von 16 zeigen nur in vereinzelten Fallen den erwarteten Molekulpeak, wobei im ersten Frag- mentierungsschritt die vollstandige Abspaltung des Metall- carbonyls dominiert. Dieses Verhalten entspricht somit dem in Losung.
Chem. Ber. 1994, 127, 1003-1013

1006 R. Beckhaus, J. Oster, T. Wagner
Synthese heterodinuclearer Carben-Komplexe mit Oxatitanacyclopenten-Teilstruktur
Tab. 2. 'H-NMR-Daten (8-Werte) heterodinuclearer Carbenkomplexe rnit Oxatitanacyclopenten-Teilstruktur 8 (500 MHz, 25"C, TMS)
Erhitzt man 5 in Gegenwart von Hexacarbonylchrom oder -wolfram, von Cymantren oder Methylcymantren, so 1aDt sich unter Ethylen-Abspaltung bei Temperaturen von 90- 110°C die Bildung von 8a-e beobachten["l. Analoge Verbindungen entstehen auch ausgehend von 14, wobei aber bereits Reaktionstemperaturen von 50-70°C ausrei- chend sind. Auf diese Art und Weise konnen die Verbindun- gen 8a, c-f sowie 8i gewonnen werden. Die entsprechenden Umsetzungen mit Mo(CO)~ fiihren auch unter den genann- ten Reaktionsbedingungen zum thermolabilen Komplex Sb, dessen Isolierung in reiner Form bisher nicht gelungen ist.
Bei der Venvendung von Mn2(CO)10 oder Re2(CO)10 hin- gegen lassen sich unter analogen Reaktionsbedingungen zu- nachst ausgehend vom Vinylfulvenderivat 14 die dinucle- aren Carbenkomplexe mit Oxatitanacyclobutan-Strukturen 16g und h isolieren. Erst nach wesentlich Iangeren Reak- tionszeiten (14h, 70°C) erfolgt deren Urnwandlung in die Isomeren 8g und h. Die Tatsache, daB die Gewinnung von 16g ausgehend von 14 moglich ist, verdeutlicht, daD ein Gleichgewicht 14/6 bestehen muD. Gleichzeitig kann die primare Bildung von 16 als kinetisch kontrollierter Reak- tionsschritt aufgefaDt werden. Bestatigt wird diese An- nahme durch den Befund, daB ausgehend von den isolierten Verbindungen 16a-i eine Isomerisierung IR- und NMR- spektroskopisch (c, g quantitativ) ohne das Auftreten weite- rer Zwischenprodukte verfolgt werden kann. Die Isomeri- sierungen 16-8 sind stets rnit einem charakteristischen Farbwechsel von grun nach rot verbunden (16i-4 rot -+ rot braun).
H H
C=ML,
C-H Lg TI' c+ 'H + L,M=C=O
%'CH2 . 14 8
'H-NMR-spektroskopisch zeigt das Auftreten charakte- ristischer AB-Spektren die Bildung der Oxatitanacyclopen- ten-Ringe 8 rnit 'JHH-Werten von ca. 9 Hz an (Tab. 2).
Von groBem diagnostischen Wert fur die Strukturbe- schreibung sowohl von 16 als auch von 8 sind die 13C- NMR-Daten (Tab. 3), insbesondere die chemischen Ver- schiebungen der Carben-Kohlenstoffatome. Diese liegen im erwarteten Bereich fur Fischer-Carbenk~mplexe~~~], wobei aber im Vergleich rnit 0ffenen[~~1 oder carbocyclischen Fischer-Carbenk~mplexen[~~] zu hoherem Feld verschobene Werte gefunden werden. Besonders trifft dieses fur die Oxa- titanacyclobutane 16 verglichen mit den isomeren Oxatita- nacyclopenten-Komplexen 8 zu. Letzter Befund entspricht einerseits dem allgemeinen Trend, daD die Signale der p-C- Atome in Titanacyclobutanen zu hohem Feld verschoben
TiCH- =CH-C=X ' J , CP* *v Cp 2T~CH=CHC(=X)0
R X
7 01 (d)
7 07 (d)
7 20 (d)
6 72 (d)
6 73 (dj
6 82 (d)
7 06 (d)
7 25 (d)
7 17 (d)
7 46 (d)
7 42 (dj
7 45 (d)
7 17(d)
7 17 (d)
I 2 1 (d)
7 29(d)
7 28 (d)
7 51 (d)
~~
8 9 1 4 7
8 9 149
8 8 149
9 3 157
8 8 160
9 0 186
9 1 1 4 7
9 2 151
9 0 1 5 7
~ i n d [ ~ ~ , ~ ~ , ~ ~ l , andererseits werden Tieffeldverschiebungen der Carbenkohlenstoff-Signale rnit zunehmender RinggroRe z.B. auch beim Vergleich cyclischer 5- und 6-gliedriger Car- benkomplexe des Mangans nachgewie~en[~~]. Daruber hin- aus durfte auch eine polarere Struktur von 16 entsprechend einem verstarkten Acylcharakter, im Vergleich mit 8 zu hochfeldverschobenen 13C-Carben-Signalen beitragen (vgl. Diskussion der IR-Daten). Fur 16i wird eine charakteristi- sche Kopplung 'JRh-c von 52 Hz gefunden (Zr/Rh-Car- benkomplex lJRh-C 58.2 Hz["~]). Die Signale der titange- bundenen Kohlenstoffatome (Ti-C-) in den Oxatitanacy- clobutanen 16 sind im Vergleich rnit 5 zu hoherem Feld ver- schoben, wahrend die C-Atome der exo-Methylengruppe starker Tieffeld-absorbieren, so daB die genannten Signalla- gen sowohl in 16 als auch in 7a und 7c sich angleichen, was auf mogliche konjugative Wechselwirkungen in den Hetero- diensystemen hinweist. In Ubereinstimmung rnit dieser An- nahme wird in der Struktur von 7c ein dem Butadien ent- sprechender =C-C=-Abstand im Vierring von 1.477(4) A gefunden[l I]. Versuche, auch die Struktur von 16 diffrakto- metrisch belegen zu konnen, waren bislang nicht erfolg- reich.
Zur weiteren Unterscheidung der Isomeren 8 und 16 kann zusatzlich zu den NMR-spektroskopischen Erkennt- nissen das Auftreten der Out-of-plane-CH-Deformations- schwingung der C = C-Doppelbindung herangezogen wer- den, die im Fall von 16a-c bei wesentlich hoheren Wellen- zahlen als fur 8a-c zu beobachten ist (Tab. 4).
Ansonsten zeigen die IR-Valenzschwingungen der CO- Liganden von 16 und 8 ein den jeweiligen Symmetrietypen entsprechendes Bandenmuster. In Carbenkomplexen homo- loger Metallcarbonyle treten fur die schwereren Metalle Verschiebungen zu groaeren Wellenzahlen auf. Der Nach- weis von nur funf Banden fur 16g,h und auch fur 8g,h zeigt, daB eine axiale Substitution (C4") bezuglich der M - M-Bindung der zweikernigen Decacarbonyle des Man- gans und Rheniums vorliegen muR. Die fur offene Fischer- Carben-Komplexe dominierende aquatoriale Substitution (C,) wiirde neun Banden erwarten 1 a ~ ~ e n [ ~ ~ ~ , ~ ~ , ~ ~ ] .
Eine axiale Substitution wurde auch fur das Cycloaddi- tionsprodukt von C P * ~ T ~ ( C ~ H ~ ) rnit Mn2(CO)lo und
Chem. Bec 1994, 127, 1003-1013
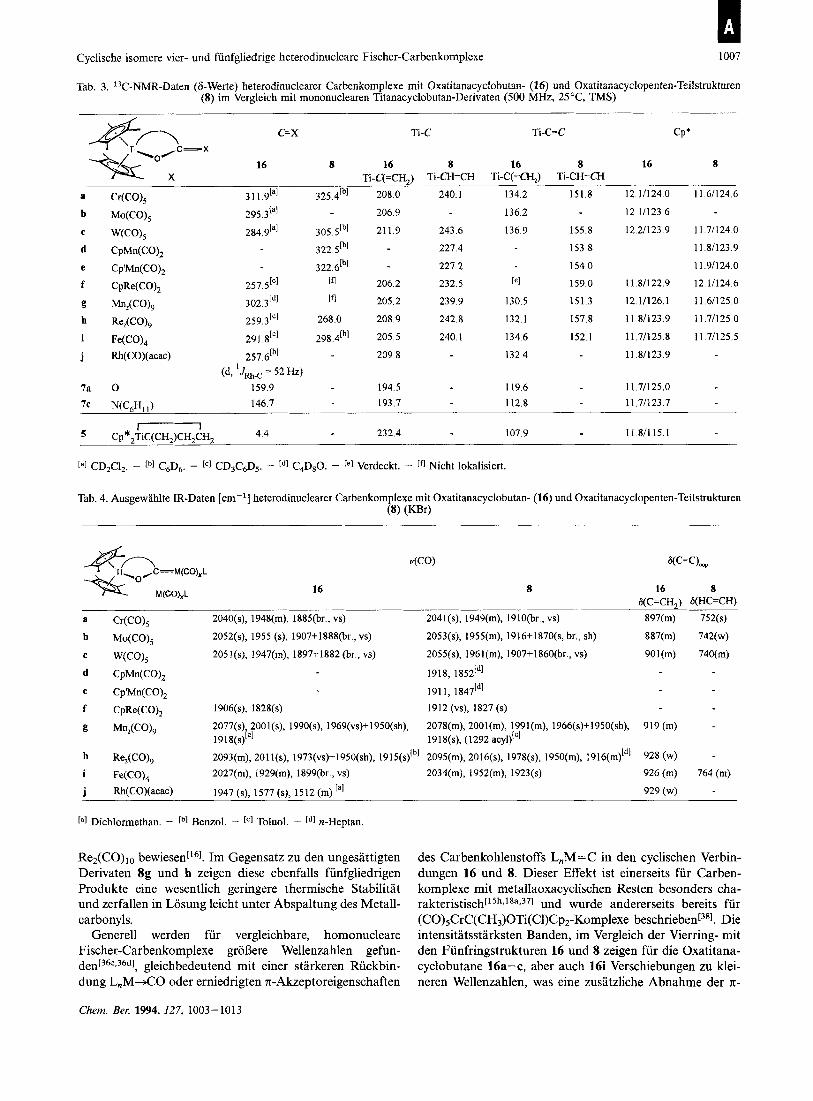
Cyclische isomere vier- und funfgliedrige heterodinucleare Fischer-Carbenkomplexe 1007
Tab. 3. 13C-NMR-Daten (6-Werte) heterodinuclearer Carbenkomplexe rnit Oxatitanacyclobutan- (16) und Oxatitanacyclopenten-Teilstrukturen (8) im Vergleich rnit mononuclearen Titanacyclobutan-Derivaten (500 MHz, 25 "C, TMS)
CP' Ti-C=C c=x Ti-C Ti\ ,C=X +/- O 16 8 16 8 16 8 16 8
F L X Ti-C(=CH,) Ti-CH=CH Ti-C(=CH,) Ti-CH=CH 12.1/124.0 11.6/124.6 a Cr(CO), 3 11 .91a1 325.4[b1 208.0 240.1 134.2 151.8
b
C
d
e
f
g h
i
i
?a 7c ~
295.3[a1
284.91a1
25 7.5"'
302.3'd1
259.3'"'
291 8"'
257.6[b1 (d, 'JmC = 52 Hz)
159.9 146.7
305, 5Ib1
322. 5Ib1
3 22. 6[b' [rl
Ifl
268.0
298.4Ib1
206.9
211.9
206.2
205.2
208.9
205.5
209.8
194.5 193.7
243.6
227 4
227 2
232.5
239 9
242 8
240.1
136.2
136 9
[el
130 5
132 1
134 6
132 4
119 6 112 8
155.8
153 8
154.0
159.0
151.3
157.8
152.1
12.U123.6
12.2h23.9
1 1 . W 22.9
12.1/126.1
11 N123.9
11.7/125.8
11.8/123.9
11.7/125.0 1 1.711 23.7
11.7/124 0
11.8/123.9
11.9/124.0
12 U124.6
11.6/125.0
11.7/125.0
11.7/125.5
~
4 4 232 4 107 9 11 8/115 1 m Cp*,TiC(CH,)CH,CH,
[a] CD2Cl2. - Lb] C6D+ - rC] CD3C6D5. - Ld] C4D80. - re] Verdeckt. - [fl Nicht lokalisiert.
Tab. 4. Ausgewahlte IR-Daten [cm-'1 heterodinuclearer Carbenkomplexe rnit Oxatitanacyclobutan- (16) und Oxatitanacyclopenten-Teilstrukturen (8) ( D r )
4CO) W=C),,
16 8 16 8 G(C=CH,) G(HC=CH)
2040(s), 1948(m), 1885(br., vs) 2041(s), 1949(m), 1910(br., vs) 897(m) 752(s)
2052(s), 1955 (s), 1907+1888(br., vs)
2051(s), 1947(m), 189711882 (br., vs)
1906(s), 1828(s)
2077(s), 2001(s), 1990(s), 1969(vs)+1950(sh), 191 8(s)[c1
2093(m), 201 l(s), 1973(vs)+195O(sh), 191 5(sfh1 2027(rn), 1929(m), 1899(br., vs)
j Rh(CO)(acac) 1947 (s). 1577 Is). 1512 (ml
2053(s), 1955(m), 1916+1870(s, br., sh)
2055(s), 1961(m), 1907+1860(br., vs)
1918, 1852[d1
1911, 18471d1 1912 (vs), 1827 (s) 2078(m), 2001(m), 199 1 (m), 1966(s)+1950(sh), 1918(s), (1292 acyl)'"]
2095(m), 2016(s), 1978(s), 1950(m), 1916(mfd' 2034(m), 195Z(m), 1923(s)
887(m) 742(w)
901(m) 740(rn)
919 (m)
928 (w)
926 (m) 764 (rn)
929 (w)
La] Dichlormethan. - rb] Benzol. - Toluol. - cd] n-Heptan.
Re2(CO)lo bewiesen[l6I. Im Gegensatz zu den ungesattigten Derivaten 8g und h zeigen diese ebenfalls funfgliedrigen Produkte eine wesentlich geringere thermische Stabilitat und zerfallen in Losung leicht unter Abspaltung des Metall- carbonyls.
Generell werden fur vergleichbare, homonucleare Fischer-Carbenkomplexe groI3ere Wellenzahlen gefun- den[36c,36d], gleichbedeutend rnit einer starkeren Ruckbin- dung L,M+CO oder erniedrigten n-Akzeptoreigenschaften
Chew. Ber. 1994, 127, 1003-1013
des Carbenkohlenstoffs L,M=C in den cyclischen Verbin- dungen 16 und 8. Dieser Effekt ist einerseits fur Carben- komplexe rnit metallaoxacyclischen Resten besonders cha- rakteri~tisch['~~~'*~,~~1 und wurde andererseits bereits fur (CO)5CrC(CH3)0Ti(C1)Cp2-Komplexe be~chrieben[~~]. Die intensitatsstarksten Banden, im Vergleich der Vierring- rnit den Funfringstrukturen 16 und 8 zeigen fur die Oxatitana- cyclobutane 16a-c, aber auch 16i Verschiebungen zu klei- neren Wellenzahlen, was eine zusatzliche Abnahme der n-
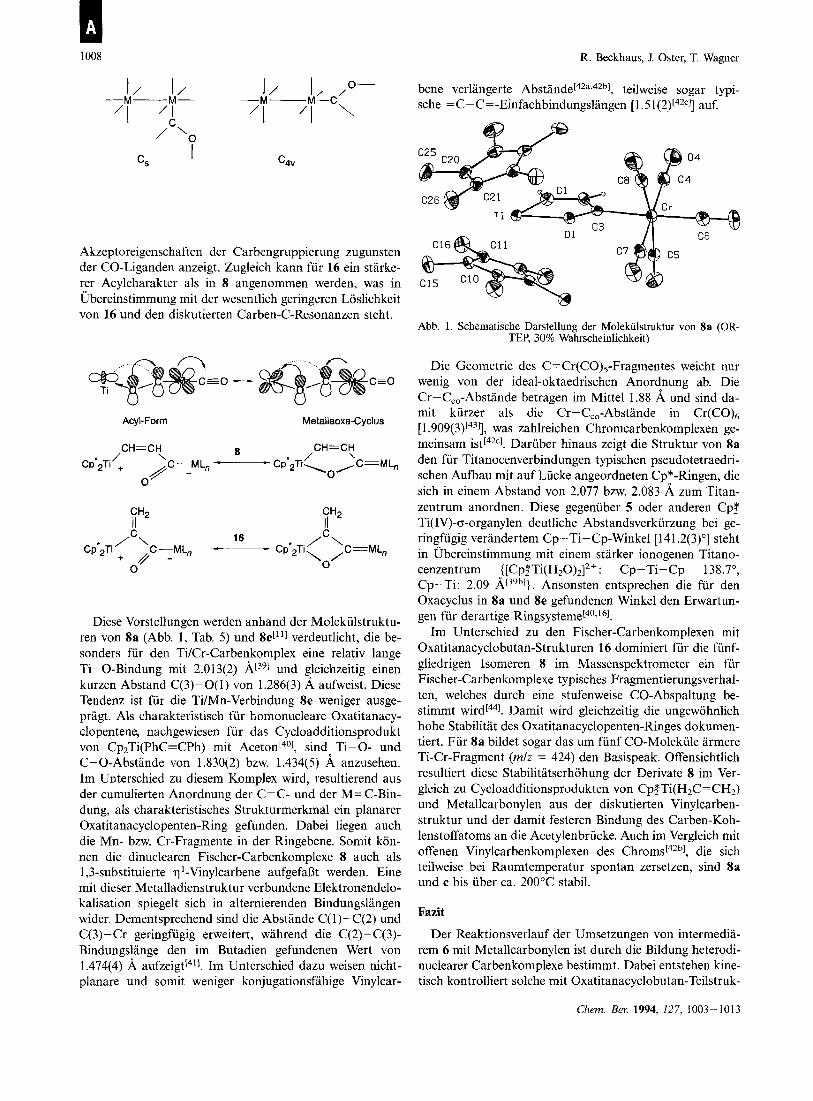
1008 R. Beckhaus, J. Oster, T. Wagner
bene verlangerte A b ~ t a n d e c ~ ~ ~ , ~ ~ ~ ] , teilweise sogar typi- sche = c- c= -Einfachbindungslangen [ 1.5 1 (2)[42c]] auf.
Akzeptoreigenschaften der Carbengruppierung zugunsten der CO-Liganden anzeigt. Zugleich kann fur 16 ein starke- rer Acylcharakter als in 8 angenommen werden, was in Ubereinstimmung mit der wesentlich geringeren Loslichkeit von 16 und den diskutierten Carben-C-Resonanzen steht.
r0 - CEO
Acyl-Form Metallaoxa-Cyclus
iH2 iH2
Diese Vorstellungen werden anhand der Molekiilstruktu- ren von 8a (Abb. 1, Tab. 5) und 8e[''] verdeutlicht, die be- sonders fur den Ti/Cr-Carbenkomplex eine relativ lange Ti-0-Bindung rnit 2.01 3(2) und gleichzeitig einen kurzen Abstand C(3)-0(1) von 1.286(3) A aufweist. Diese Tendenz ist fur die TiIMn-Verbindung 8e weniger ausge- pragt. Als charakteristisch fur homonucleare Oxatitanacy- clopentene, nachgewiesen fur das Cycloadditionsprodukt von Cp,Ti(PhC=CPh) rnit A~eton[~'], sind Ti-0- und C-0-Abstande von 1.830(2) bzw. 1.434(5) A anzusehen. Im Unterschied zu diesem Komplex wird, resultierend aus der cumulierten Anordnung der C=C- und der M=C-Bin- dung, als charakteristisches Strukturmerkmal ein planarer Oxatitanacyclopenten-Ring gefunden. Dabei liegen auch die Mn- bzw. Cr-Fragmente in der Ringebene. Somit kon- nen die dinuclearen Fischer-Carbenkomplexe 8 auch als 1,3-substituierte q '-Vinylcarbene aufgefaDt werden. Eine rnit dieser Metalladienstruktur verbundene Elektronendelo- kalisation spiegelt sich in alternierenden Bindungslangen wider. Dementsprechend sind die Abstande C( 1)-C(2) und C(3)-Cr geringfugig erweitert, wahrend die C(2)-C(3)- Bindungslange den im Butadien gefundenen Wert von 1.474(4) A a~fze ig t l~~] . Im Unterschied dazu weisen nicht- planare und somit weniger konjugationsfahige Vinylcar-
Abb. 1. Schematische Darstellung der Molekiilstruktur von 8a (OR- TEP, 30% Wahrscheinlichkeit)
Die Geometrie des C=Cr(CO)5-Fragmentes weicht nur wenig von der ideal-oktaedrischen Anordnung ab. Die Cr-C,,-Abstande betragen im Mittel 1.88 A und sind da- mit kurzer als die Cr-C,,-Abstande in Cr(C0)6 [ 1 .909(3)[431], was zahlreichen Chromcarbenkomplexen ge- meinsam ist[42cl. Daruber hinaus zeigt die Struktur von 8a den fur Titanocenverbindungen typischen pseudotetraedri- schen Aufbau mit auf Lucke angeordneten Cp*-Ringen, die sich in einem Abstand von 2.077 bnv. 2.083 A zum Titan- zentrum anordnen. Diese gegenuber 5 oder anderen Cp,* Ti(1V)-cr-organylen deutliche Abstandsverkiirzung bei ge- ringfugig verandertem Cp-Ti-Cp-Winkel [141.2(3)"] steht in Ubereinstimmung mit einem starker ionogenen Titano- cenzentrum {[CP?T~(H~O)~]~+: Cp-Ti-Cp 138.7", Cp-Ti: 2.09 Ansonsten entsprechen die fur den Oxacyclus in 8a und 8e gefundenen Winkel den Erwartun- gen fur derartige Ringsy~teme[~~.~~I .
Im Unterschied zu den Fischer-Carbenkomplexen mit Oxatitanacyclobutan-Strukturen 16 dominiert fur die funf- gliedrigen Isomeren 8 im Massenspektrometer ein fur Fischer-Carbenkomplexe typisches Fragmentierungsverhal- ten, welches durch eine stufenweise CO-Abspaltung be- stimmt wirdL41. Damit wird gleichzeitig die ungewohnlich hohe Stabilitat des Oxatitanacyclopenten-Ringes dokumen- tiert. Fur 8a bildet sogar das um funf CO-Molekule armere Ti-Cr-Fragment (mlz = 424) den Basispeak. Offensichtlich resultiert diese Stabilitatserhohung der Derivate 8 im Ver- gleich zu Cycloadditionsprodukten von Cp,*Ti(H2C=CH2) und Metallcarbonylen aus der diskutierten Vinylcarben- struktur und der damit festeren Bindung des Carben-Koh- lenstoffatoms an die Acetylenbrucke. Auch im Vergleich rnit offenen Vinylcarbenkomplexen des C h r o m ~ [ ~ ~ ~ ] , die sich teilweise bei Raumtemperatur spontan zersetzen, sind 8a und c bis uber ca. 200°C stabil.
Fazit
Der Reaktionsverlauf der Umsetzungen von intermedia- rem 6 rnit Metallcarbonylen ist durch die Bildung heterodi- nuclearer Carbenkomplexe bestimmt. Dabei entstehen kine- tisch kontrolliert solche rnit Oxatitanacyclobutan-Teilstruk-
Chew. Ber. 1994, 127, 1003-1013
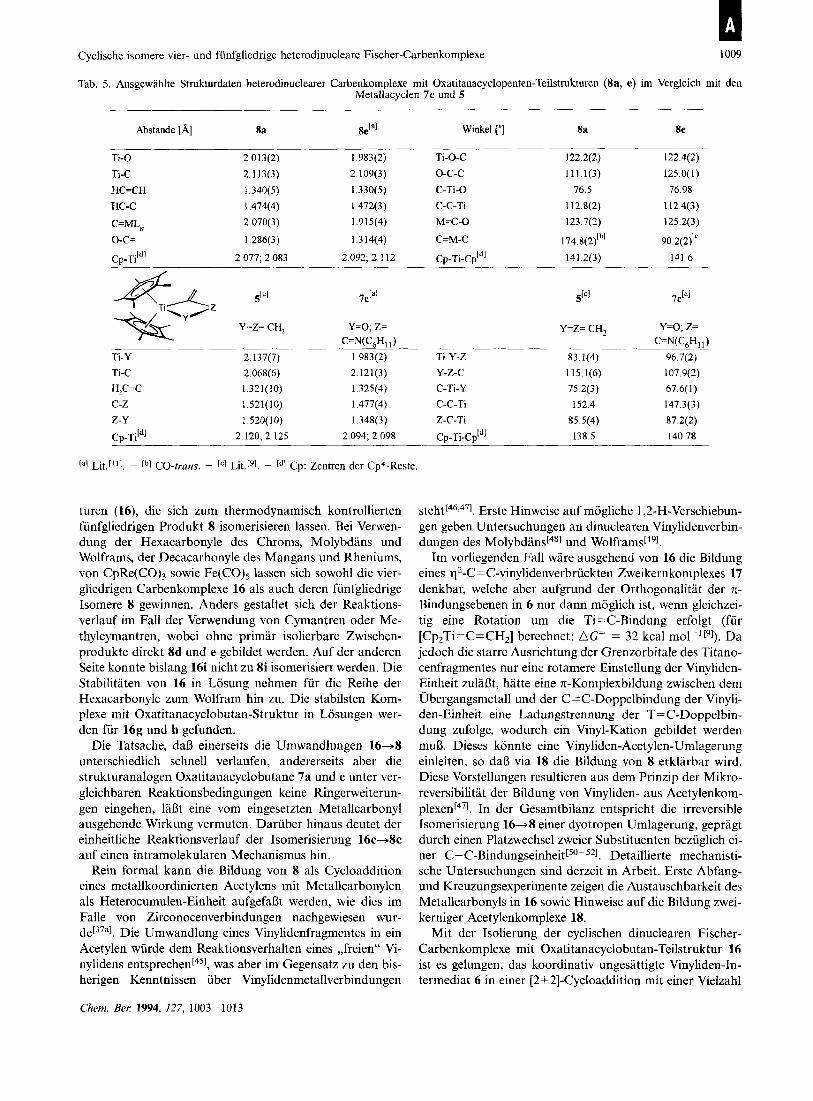
Cyclische isomere vier- und funfgliedrige heterodinucleare Fischer-Carbenkomplexe 1009
Tab. 5. Ausgewahlte Strukturdaten heterodinuclearer Carbenkomplexe rnit Oxatitanacyclopenten-Teilstrukturen @a, e) irn Vergleich rnit den Metallacyclen 7e und 5
Abstande [A] 8a 8 2 ' Winkel r] 8a 8e
TI-0 2 013(2) 1983(2) TI-0-C 122 2(2) 122 4(2)
TI-C 2 113(3) 2 109(3) 0-c-c 111 l(3) 125 0(1)
HC=CH 1 340(5) 1330(5) C-TI-0 76 5 76 98 HC-C 1474(4) 1472(3) C-C-TI I1 2 8(2) I12 4(3)
C=MLM 2 070(3) 1915(4) M=C-0 123 7(2) 125 2(3)
0-c= 1286(3) 1314(4) C=M-C 174 8(2fb1 90 2(2f1
Cp-Tiidl 2 077,2 083 2 092,2 112 Cp-Ti-Cpldl 141 2(3) 141 6
Y=Z= CH, Y=O, z= C=N(C,H, ,)
Ti-Y 2 137(7) 1.983(2) Ti-Y-Z 83.1(4) 96.7(2) Ti-C 2.068(6) 2.12 l(3) Y-2-C 1 15.1(6) 107.9(2) H,C-C 1.32 1 ( I 0) 1.325(4) C-Ti-Y 75 2(3) 67.6(1) C-Z 1.521(10) 1.477(4) C-C-Ti 152.4 147.3(3) 2-Y 1 520(10) 1.348(3) Z-C-Ti 85 5(4) 87 2(2) Cp-Ti'*' 2.120, 2.125 2.094, 2.098 Cp-Ti-Cp'*' 138.5 140.78
Lit.["]. - cbl CO-trans. - IC] Lit."]. - [*I Cp: Zentren der Cp*-Reste.
turen (16), die sich zum thermodynamisch kontrollierten funfgliedrigen Produkt 8 isomerisieren lassen. Bei Verwen- dung der Hexacarbonyle des Chroms, Molybdans und Wolframs, der Decacarbonyle des Mangans und Rheniums, von CpRe(CO)3 sowie Fe(CO), lassen sich sowohl die vier- gliedrigen Carbenkomplexe 16 als auch deren funfgliedrige Isomere 8 gewinnen. Anders gestaltet sich der Reaktions- verlauf im Fall der Verwendung von Cymantren oder Me- thylcymantren, wobei ohne primar isolierbare Zwischen- produkte direkt 8d und e gebildet werden. Auf der anderen Seite konnte bislang 16i nicht zu 8i isomerisiert werden. Die Stabilitaten von 16 in Losung nehmen fur die Reihe der Hexacarbonyle zum Wolfram hin zu. Die stabilsten Kom- plexe mit Oxatitanacyclobutan-Struktur in Losungen wer- den fur 16g und h gefunden.
Die Tatsache, daB einerseits die Umwandiungen 16-43 unterschiedlich schnell verlaufen, andererseits aber die strukturanalogen Oxatitanacyclobutane 7a und c unter ver- gleichbaren Reaktionsbedingungen keine Ringerweiterun- gen eingehen, laBt eine vom eingesetzten Metallcarbonyl ausgehende Wirkung vermuten. Dariiber hinaus deutet der einheitliche Reaktionsverlauf der Isomerisierung 16c+8c auf einen intramolekularen Mechanismus hin.
Rein formal kann die Bildung von 8 als Cycloaddition eines metallkoordinierten Acetylens rnit Metallcarbonylen als Heterocumulen-Einheit aufgefaBt werden, wie dies im Falle von Zirconocenverbindungen nachgewiesen wur- de[37aI. Die Umwandlung eines Vinylidenfragmentes in ein Acetylen wurde dem Reaktionsverhalten eines ,,freien" Vi- nylidens entspre~hen[~~I, was aber im Gegensatz zu den bis- herigen Kenntnissen uber Vinylidenmetallverbindungen
~ t e h t [ ~ ~ , ~ ~ ] . Erste Hinweise auf mogliche 1,2-H-Verschiebun- gen geben Untersuchungen an dinuclearen Vinylidenverbin- dungen des M o l y b d a n ~ [ ~ ~ ] und Wolf ram~[~~] .
Im vorliegenden Fall ware ausgehend von 16 die Bildung eines q2-C = C-vinylidenverbruckten Zweikernkomplexes 17 denkbar, welche aber aufgrund der Orthogonalitat der x- Bindungsebenen in 6 nur dann moglich ist, wenn gleichzei- tig eine Rotation um die Ti=C-Bindung erfolgt (fur [Cp2Ti=C=CH2] berechnet: AG* = 32 kcal m ~ l - ' [ ~ I ) . Da jedoch die starre Ausrichtung der Grenzorbitale des Titano- cenfragmentes nur eine rotamere Einstellung der Vinyliden- Einheit zulaDt, hatte eine x-Komplexbildung zwischen dem Ubergangsmetall und der C = C-Doppelbindung der Vinyli- den-Einheit eine Ladungstrennung der T=C-Doppelbin- dung zufolge, wodurch ein Vinyl-Kation gebildet werden muB. Dieses konnte eine Vinyliden-Acetylen-Umlagerung einleiten, so dal3 via 18 die Bildung von 8 erklarbar wird. Diese Vorstellungen resultieren aus dem Prinzip der Mikro- reversibilitat der Bildung von Vinyliden- aus Acetylenkom- p l e ~ e n [ ~ ~ ] . In der Gesamtbilanz entspricht die irreversible lsomerisierung 16-8 einer dyotropen Umlagerung, gepragt durch einen Platzwechsel zweier Substituenten beziiglich ei- ner C-C-Bindung~einheit[~~-~*I. Detaillierte mechanisti- sche Untersuchungen sind derzeit in Arbeit. Erste Abfang- und Kreuzungsexperimente zeigen die Austauschbarkeit des Metallcarbonyls in 16 sowie Hinweise auf die Bildung zwei- kerniger Acetylenkomplexe 18.
Mit der Isolierung der cyclischen dinuclearen Fischer- Carbenkomplexe rnit Oxatitanacyclobutan-Teilstruktur 16 ist es gelungen, das koordinativ ungesattigte Vinyliden-In- termediat 6 in einer [2+2]-Cycloaddition rnit einer Vielzahl
Chem. Bel: 1994, 127, 1003-1013
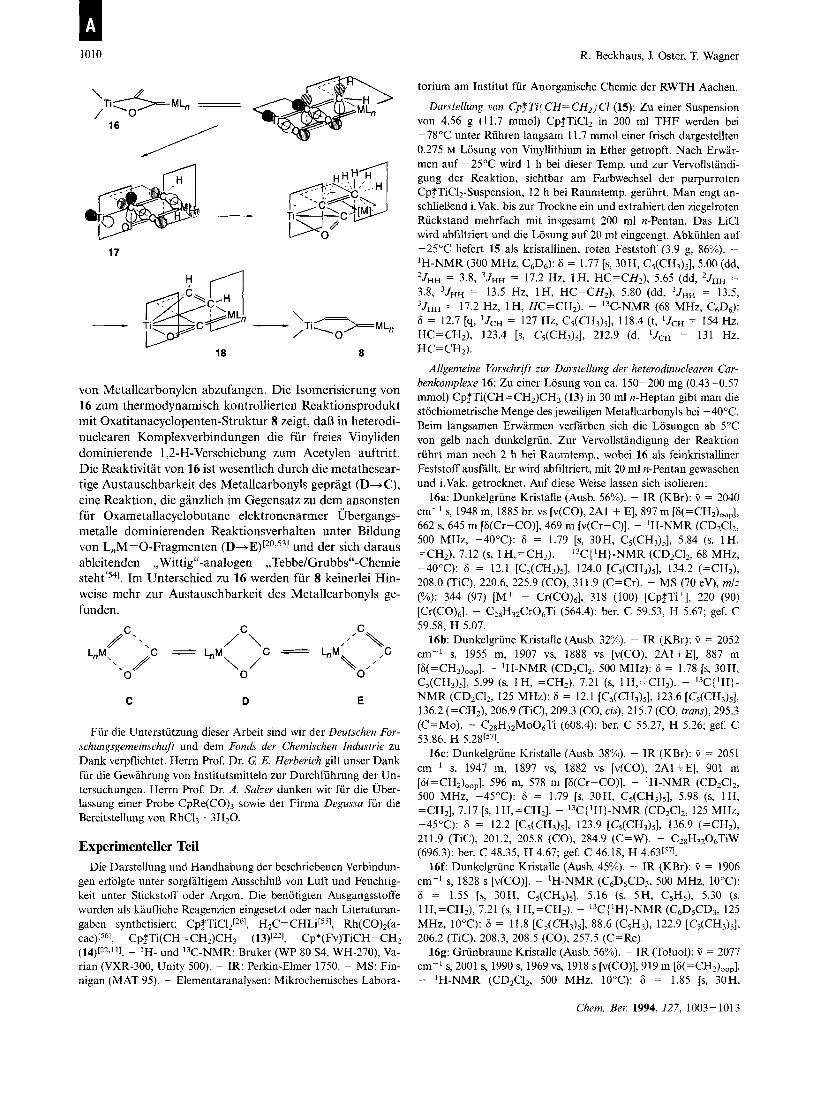
1010 R. Beckhaus, J. Oster, T. Wagner
17
18 8
von Metallcarbonylen abzufangen. Die Isomerisierung von 16 zum thermodynamisch kontrollierten Reaktionsprodukt mit Oxatitanacyclopenten-Struktur 8 zeigt, da13 in heterodi- nuclearen Komplexverbindungen die fur freies Vinyliden dominierende 1 ,ZH-Verschiebung zum Acetylen auftritt. Die Reaktivitat von 16 ist wesentlich durch die metathesear- tige Austauschbarkeit des Metallcarbonyls gepragt (D-C), eine Reaktion, die ganzlich im Gegensatz zu dem ansonsten fur Oxarnetallacyclobutane elektronenarmer Ubergangs- metalle dominierenden Reaktionsverhalten unter Bildung von L,M=O-Fragmenten (D-3E)[20,531 und der sich daraus ableitenden ,,Wittig"-analogen ,,Tebbe/Grubbs"-Chemie ~ t e h t [ ~ ~ ] . Im Unterschied zu 16 werden fur 8 keinerlei Hin- weise mehr zur Austauschbarkeit des Metallcarbonyls ge- funden.
n P
C D E
Fur die Unterstutzung dieser Arbeit sind wir der Deutschen For- schungsgemeinschufi und dem Fonds der Chemischen Industrie zu Dank verpflichtet. Herrn Prof. Dr. G E. Herberich gilt unser Dank fur die Gewahrung von Institutsmitteln zur Durchfuhrung der Un- tersuchungen. Herrn Prof. Dr. A. Sulzer danken wir fur die Uber- lassung einer Probe c ~ R e ( C 0 ) ~ sowie der Firma Degussu fur die Bereitstellung von RhC1, . 3H20.
Experimenteller Teil Die Darstellung und Handhabung der beschriebenen Verbindun-
gen erfolgte unter sorgfaltigem AusschluR von Luft und Feuchtig- keit unter Stickstoff oder Argon. Die benotigten Ausgangsstoffe wurden als kaufliche Reagenzien eingesetzt oder nach Literaturan- gaben synthetisiert; Cp,*TiC12[261, H2C=CHLi[551, Rh(CO)2(a- c~c) [ ,~] , Cp,*Ti(CH=CH2)CH3 (13)[22], Cp*(Fv)TiCH=CH2 (14)[22,11]. - 'H- und I3C-NMR: Bruker (WP 80 S4, WH-270), Va- rian (VXR-300, Unity 500). - IR: Perkin-Elmer 1750. - MS: Fin- nigan (MAT 95). - Elementaranalysen: Mikrochemisches Labora-
torium am Institut fur Anorganische Chemie der RWTH Aachen.
Darstellung von Cp,* Ti(CH= CH2) CI (15): Zu einer Suspension von 4.56 g (11.7 mmol) CptTiC12 in 200 ml THF werden bei -78°C unter Riihren langsam 11.7 mmol einer frisch dargestellten 0.275 M Losung von Vinyllithium in Ether getropft. Nach Erwar- men auf -25°C wird 1 h bei dieser Temp. und zur Vervollstandi- gung der Reaktion, sichtbar am Farbwechsel der purpurroten Cp,*TiCI2-Suspension, 12 h bei Raumtemp. geruhrt. Man engt an- schliel3end i.Vak. bis zur Trockne ein und extrahiert den ziegelroten Ruckstand mehrfach mit insgesamt 200 ml n-Pentan. Das LiCl wird abfiltriert und die Losung auf 20 ml eingeengt. Abkuhlen auf -25°C liefert 15 als kristallinen, roten Feststoff (3.9 g, 86%). - 'H-NMR (300 MHz, C6D6): 6 = 1.77 [s, 30H, C,(CH,),], 5.00 (dd, 2 J ~ ~ 3.8, 3 J ~ ~ = 17.2 HZ, IH, HC=CH2), 5.65 (dd, 2 J ~ ~ = 3.8, 3 J ~ ~ = 13.5 Hz, IH, HC=CH2), 5.80 (dd, 3JHH = 13.5, ,JHH 17.2 Hz, IH, HC=CHz). - I3C-NMR (68 MHz, c6D6): 6 = 12.7 [q, ' JCH = 127 Hz, C5(CH3)5], 118.4 (t, ' J c H = 154 Hz, HC=CH2), 123.4 [s, C,(CH,),], 212.9 (d, 'JCH = 131 Hz, HC=CH2).
Allgemeine Vorschrift zur Darstellung der heterodinuclearen Car- benkomplexe 16: Zu einer Losung von ca. 150-200 mg (0.43-0.57 mmol) Cp,*Ti(CH=CH2)CH3 (13) in 30 ml n-Heptan gibt man die stochiometrische Menge des jeweiligen Metallcarbonyls bei -40°C. Beim langsamen Erwarmen verfarben sich die Losungen ab 5°C von gelb nach dunkelgrun. Zur Vervollstandigung der Reaktion riihrt man noch 2 h bei Raumtemp., wobei 16 als feinkristalliner Feststoff ausfallt. Er wird abfiltriert, mit 20 ml n-Pentan gewaschen und i.Vak. getrocknet. Auf diese Weise lassen sich isolieren:
16a: Dunkelgrune Kristalle (Ausb. 56%). - IR (KBr): 0 = 2040 cm-' s, 1948 m, 1885 br. vs [v(CO), 2A1 + El, 897 m [6(=CH2)oop], 662 s, 645 m [&(Cr-CO)], 469 m [v(Cr-C)]. - 'H-NMR (CD2CI2, 500 MHz, -40°C): 6 = 1.79 [s, 30H, C,(CH3),], 5.84 ( s , IH, =CH2), 7.12 (s, lH,=CHZ). - '3C{'H}-NMR (CDZC12, 68 MHz, -40°C): 6 = 12.1 [C,(CH,),], 124.0 [C,(CH,),], 134.2 (=CH2), 208.0 (TIC), 220.6, 225.9 (CO), 311.9 (C=Cr). - MS (70 eV), mlz (%): 344 (97) [M+ - Cr(CO),], 318 (100) [Cp,*Ti'], 220 (90) [cr(co),]. - C28H32Cr06Ti (564.4): ber. C 59.53, H 5.67; gef. C 59.58, H 5.07.
16b: Dunkelgrune Kristalle (Ausb. 32%). - IR (KBr): 0 = 2052 cm-I s, 1955 m, 1907 vs, 1888 vs [v(CO), 2AI+E], 887 m [6(=CH2),,,]. - 'H-NMR (CDzC12, 500 MHz): 6 = 1.78 [s, 30H, C,(CH3)5], 5.99 (s, 1 H, =CH2), 7.21 ( s , 1 H,=CH,). - I3C{'H}- NMR (CDzC12, 125 MHz): 6 = 12.1 [Cj(CH,),], 123.6 [C.j(CH,),], 136.2 (=CH,), 206.9 (Tic), 209.3 (CO, cis), 215.7 (CO, trans), 295.3 (C=Mo). - C28H32M006Ti (608.4): ber. C 55.27, H 5.26; gef. C 53.86, H 5.28[571.
16c: Dunkelgrune Kristalle (Ausb. 38%). - IR (KBr): F = 2051 cm-' s, 1947 m, 1897 vs, 1882 vs [v(CO), 2Al+E], 901 m [6(=CH2),,,], 596 m, 578 m [6(Cr-C0)]. - 'H-NMR (CD2Cl2, 500 MHz, -45°C): 6 = 1.79 [s, 30H, Cs(CH,),], 5.98 (s, IH, =CH2], 7.17 [s, 1H,=CH2]. - '3C{'H}-NMR (CD2C12, 125 MHz, -45°C): 6 = 12.2 [C5(CH&], 123.9 [C,(CH,),], 136.9 (=CH,), 211.9 (TIC), 201.2, 205.8 (CO), 284.9 (C=W). - C28H3206TiW (696.3): ber. C 48.35, H 4.67; gef. C 46.18, H 4.63r5'1.
16f: Dunkelgrune Kristalle (Ausb. 45%). - IR (KBr): 5 = 1906 cm-' S, 1828 S [V(Co)]. - 'H-NMR (C6D5CD3, 500 MHz, 10°C): 6 = 1.55 [s , 30H, C5(CH&], 5.16 (s, 5H, C5H5), 5.30 (s, 1 H,=CHZ), 7.21 (s, 1 H,=CH2). - l3C('H}-NMR (CsD5CD3, 125 MHz, 10°C): 6 = 11.8 [C,(CH3)5], 88.6 (C5H5), 122.9 [C,(CH,),], 206.2 (Tic), 208.3, 208.5 (CO), 257.5 (C=Re).
16g: Grunbraune Kristalle (Ausb. 56%). - IR (Toluol): F = 2077 cm-' s, 2001 s, 1990 s, 1969 vs, 1918 s [v(CO)], 919 m [6(=CH2),,,]. - 'H-NMR (CD2C12, 500 MHz, 10°C): 6 = 1.85 [s, 30H,
Chem. Ber. 1994, 127, 1003-I013

Cyclische isomere vier- und funfgliedrige heterodinucleare Fischer-Carbenkomplexe B 1011
C,(CH,),], 5.57 (s, IH, =CH2), 7.01 (s, lH , =CH2). - 'H-NMR ([DSITHF, 500 MHz): 6 = 1.87 [s, 30H, C5(CH3)5], 5.63 (s, 1H, =CHz), 7.01 (s, lH,=CHJ. - I3C{'H}-NMR ([DslTHF, 125 MHz): 6 = 12.1 [C,(CH,),], 126.1 [Cs(CH,),], 130.5 (=CH2), 205.2 (TIC), 226.3 (CO), 302.3 (C=Mn). - C32H32Mn201~Ti (733.8): ber. C 52.32, H 4.36; gef. C 50.55, H 4.47[571.
16h: Violette Kristalle (Ausb. 38%). - IR (Toluol): 0 = 2093 cm-' m, 2011 s, 1973 vs, 1950 sh, 1915 s [v(CO)], 928 m [6(=CHz)oop]. - 'H-NMR (C6D5CD3, 500 MHz): 6 = 1.43 [s, 30H, C,(CH,),), 5.24 (s , IH, =CH3, 7.01 (s, IH,=CH2). - I3C('H}-NMR (C~DSCD~, 125 MHz): 6 = 11.8 [C,(CH&], 123.9 [C5(CH3)5], 132.1 (=CH,), 201.4, 202.0 (CO), 208.9 (Tic), 259.3 (C= Re).
16i: Rote Kristalle (Ausb. 37%). - IR (KBr): F = 2027 cm-' m, 1929 m, 1899 vs [v(CO)], 926 m [6(=CH2),,,]. - 'H-NMR (C6DsCD3, 500 MHz): 6 = 1.49 [s, 30H, C,(CH,),], 5.59 (S,
1 H,=CHz), 7.59 (s, I H,=CHz). - I3C('H}-NMR (CsD5CD3, 125 MHz): 6 = 11.7 [C5(CH3)5], 125.8 [C,(CH3)5], 134.6 (=CH2), 205.5 (TIC), 218.9, 219.1 (CO), 291.8 (C=Fe). - C27H32Fe05Ti (540.3): ber. 59.99, H 5.92; gef. C 57.60, H 6.33[571.
16: Schwarze Kristalle (Ausb. 37%). - IR (CH2CI2): F = 1947 cm-' s [v(CO), Rh-CO], 1577 s, 1512 m [v(CO), acac], 929 w
Cs(CH3),], 1.97 (s, 6H, CH3-acac), 5.43 (s, I H, CH-acac), 5.62 (d, [6(=CH,),,,]. - 'H-NMR (C6D6, 500 MHz): 6 = 1.59 [s, 30H,
* J H H = 0.9 Hz, IH,=CH2), 7.62 (d, 2JHH = 0.9 Hz, 1H,=CH2). - 13C{'H}-NMR (CsD5CD3, 125 MHz): 6 = 11.8 [C,(CH,),], 27.4 (CH3-acac), 28.1 (CH,-acac), 100.4 (CH-acac), 123.9 [C5(CH3),], 132.4 (=CH2), 185.0 (CCH3-acac), 188.3 (CCH,-acac), 194.0 (d, 'JKhC = 92 Hz, RhCO), 209.8 (TIC), 257.6 (d, ' J R h C = 52 Hz, C=Rh). - C29H3904RhTi (601.9): ber. C 57.82, H 6.48; gef. C 56.50, H 6.56[571.
Allgemeine Vorschr$t zur Darstellung der heterodinuclearen Carbenkomplexe 8: Eine Losung von ca. 200 mg (571 mmol) Cp,*Ti(CH=CH2)CH3 (13) in 20 ml n-Heptan wird 12 h bei Raum- temp. geriihrt. Die dabei entstdndene grune Losung von Cp* (Fv)Ti(CH=CH,) (14) wird mit der stochiometrischen Menge des jeweiligen Metallcarbonyls versetzt. Man erhitzt auf 5O-7O0C, wo- bei die Farbe im Verlauf von 20 h nach rot umschlagt. Es wird auf ca. 5 ml bis zur beginnenden Kristallisation eingeengt. Abtrennung und Trocknung i.Vak. liefert 8 als roten Feststoff. Zur Reinigung kann aus Toluol umkristallisiert werden. Auf diese Weise lassen sich isolieren:
8a: Rote Kristalle (Ausb. 54%). - IR (KBr): F = 2041 cm-' s, 1949 m, 1910 vs [v(CO), 2A1 + El, 752 s [6(CH=CH),,,]. - 'H- NMR (C6D6, 270 MHZ): 6 = 1.47 [s, 30H, CS(CH3)5], 7.01 (d, ,JHH = 8.9 HZ, IH,=CH), 7.46 (d, 3 J ~ ~ = 8.9 HZ, IH, HC=). - I3C{'H}-NMR (C6D6, 67.9 MHz): 6 = 11.6 [CS(CH3)5], 124.6 [C5(CH3),], 151.8 (=CH-C=Cr), 240.1 (Tic), (CO), 322.6 (C=Cr). - MS (70 eV), mlz ("10): 564 (10) [M+], 536 (3) [M+ -
4 CO], 424 (100) [M+ - 5 CO]. - C28H32Cr06Ti (564.4): ber. C 59.53, H 5.67; gef. C 59.32, H 5.53.
8b: Nicht rein isoliert. - IR (KBr): 0 = 2053 cm-' s, 1955 m,
CO], 508 ( 5 ) [M+ - 2 CO], 480 (4) [M+ - 3 CO], 352 (10) [M+ -
1916 vs [v(CO), 2AI + El, 742 w [G(CH=CH),,,]. - 'H-NMR (C6D6, 80 MHZ): 6 = 1.49 [S, 30H, C,(CH3)5], 7.07 (d, = 8 Hz, 1 H,=CH), 7.42 (d, 3 J ~ ~ = 8 Hz, 1 H, HC=). - 'H-NMR (CD2C12, 80 MHz): 6 = 1.90 [S, 30H, C,(CH,),], 7.24 (d, 3 J ~ ~ =
9 Hz, 1 H,=CH), 7.59 (d, 3 J H i r = 9 Hz, 1 H, HC=). 8c: Rote Kristalle (Ausb. 59%). - IR (KBr): 0 = 2055 cm-' s,
1961 m, 1907 + 1860 vs [v(CO), 2A1 + El, 740 m [S(CH=CH),,,]. - 'H-NMR (C6D6, 270 MHz): 6 = 1.49 [s, 30H, Cs(CH,),], 7.20 (d, 3 J ~ ~ = 8.8 Hz, 1 H,=CH), 7.45 (d, 3 J f 1 ~ = 8.8 Hz, 1 H, HC=). - I3C{'H}-NMR (C6D6, 67.9 MHz): 6 = 11.7 [C5(CH3)5], 124.6
[C,(CH,),], 155.8 (=GI-C=W), 201.5, 205.7 (CO), 243.6 (TIC), 305.5 (C=W). - MS (70 eV), mlz: 697 [M+]. - C28H3206TiW (696.3): ber. C 48.25, H 4.67; gef. C 48.35, H 4.68.
8d: Orangegelbe Kristalle (Ausb. 55%). - IR (n-Pentan): 0 = 1918, 1852 cm-' [ ~ ( c o ) ] . - 'H-NMR (C6Dbr270 MHz): 6 = 1.57 [S, 30H, C5(CH3)5], 4.56 (S, C5H5), 6.72 (d, 3 J ~ ~ = 9.3 HZ, lH,=CH), 7.17 (d, 3 J ~ ~ = 9.3 Hz, IH, HC=). - I3C-NMR (C6D6, 125 MHZ): 6 = 11.8 [q, 'JCH = 126 HZ, C S ( ~ T ) ~ ] , 86.5 (d, 'JCtr = 175 Hz, C5H5), 123.9 [S, C5(CH3)5], 153.8 (d, ' J C H = 156 Hz, CH-C=Mn), 227.4 (d, ' J C H = 143 Hz, TIC), 236.3 (s, CO), 322.5 (s, C=Mn). - MS (70 eV), mlz (%): 548 (100) [M+], 492 (20) [M+ - 2 CO]. - C30H37Mn03Ti (548.5): ber. C 65.63, H 6.75; gef. C 66.04, H 6.85.
8e: Ockerbrauner Feststoff (Ausb. 64%). - IR (n-Pentan): 0 =
1911, 1847 cm-' [V(CO)]. - 'H-NMR (C6D6, 500 MHZ): 6 = 1.60 [s, 30H, C,(CH,),], 1.96 [S, 3H, C5H4(CH3)], 4.42, 4.55 [t, J E ~ I ~ = 1.9 Hz, je 2H, C5H4(CH3)], 6.73 (d, 3JHH = 8.8 Hz, IH,=CH), 7.17 (d, ?JHH = 8.8 Hz, I H, HC=). - '3Cf'Hf-NMR (C6D6, 125 MHz): 6 = 11.9 [C5(CH3)5], 14.5 [C5H4CH3Ir 85.8, 88.2, 101.2 (C5H4CH3), 124.0 [C5(CH3)5], 154.0 ( = a - C = M n ) , 227.2 (TIC), 236.8 (CO), 322.6 (C=Mn). - MS (70 eV), m/z ("/o): 563 (25) [M+], 506 (100) [M+ - 2 CO]. - C31H39Mn03Ti (562.5): ber. C 66.13, H 6.93; gef. C 66.31, H 7.04.
8f: Ockerbrauner Feststoff (Ausb. 42%). - IR (CH2C12): 0 = 1912 cm-' vs, 1827 s [v(CO)]. - 'H-NMR (CD2CI2, 300 MHz): 6 = 1.86 [s, 30H, C5(CH3)5], 5.15 (CsHs), 6.82 (d, 3 J ~ ~ = 9.0 Hz, 1 H,=CH), 7.21 (d, 3JHH = 9.0 Hz, 1 H, HC=). - I3C{'H)-NMR (CDZC12, 75 MHz): 6 = 12.1 [C5(CH3)5], 87.9 (C5H5), 124.6 [C5(CH3),], 159.0 ( = a - C = R e ) , 206.8, 208.2 (CO), 232.5 (Tic), (C=Re) nicht lokalisiert.
8g: Rote Kristalle (Ausb. 36%). - IR (Toluol): F = 2078 cm-' m, 2001 m, 1991 m, 1966 s, 1918 m [v(CO)]. - 'H-NMR (C6D6, 500 MHz): 6 = 1.47 [S, 30H, C5(CH3)5], 7.06 (d, 3 J ~ ~ = 9.1 Hz, lH,=CH), 7.29 (d, 3JHH = 9.1 Hz, IH, HC=). - 13C{'H}-NMR
(=CH-C=Mn), 225.6, 226.5 (CO), 239.9 (TIC), (C=Mn) nicht lo- kalisiert.
8h: Rote Kristalle. - IR (Toluol): F = 2095 cm-l m, 2016 ni, 1978 s, 1950 m, 1916 m [v(CO)]. - 'H-NMR (CD2CI2, 80 MHz):
(C6D.5, 125 MHz): 6 = 11.6 [C,(m3)5], 125.0 [C,(CH,),], 151.3
6 = 1.91 [S, 30H, C5(CH3)5], 7.22 (d, 3 J ~ ~ = 9.1 HZ, IH,=CH), 7.74 (d, 3 J ~ ~ 9.1 Hz, 1 H, HC=). - 'H-NMR (C6D6,500 MHz): 6 = 1.51 [s, 30H, C5(CH3),], 7.25 (d, ,JHH = 9.2 Hz, IH,=CH), 7.28 (d, 3JHH = 9.2 HZ, 1 H, ffc=). - "C{'H}-NMR (C6D6, 125
(=CH-C=Re), 200.5, 201.0 (CO), 242.8 (Tic), 268.0 (C=Re). - MS (70 eV), mlz: 698 [M+].
8i: IR (KBr): 0 = 2034 cm-' m, 1952 m, 1923 s [v(CO)], 764 m
MHz): 6 = 11.7 [C5(CH3)5], 125.0 [C,(CH,),], 157.8
[G(CH=CH),,]. - 'H-NMR (C6D6,500 MHz): 6 = 1.57 [s, 30H, C5(CH3)5], 7.17 (d, 3 J ~ ~ = 9.0 Hz, 1H,=CH), 7.51 (d, 3 J k 1 ~ ~ = 9.0 Hz, 1H, HC=). - I3C{'H}-NMR (C6D6, 125 MHz): 6 = 11.7 [C5(CH3)5], 125.5 [C5(CH3),], 152.1 (=CH-C=Fe), 219.0 (CO), 240.1 (TIC), 298.4 (C=Fe).
Daten zur Kristallstrukturnnalyse von 8a: Summenformel C28H32Cr06Ti, Molmasse 564.46 g/mol. Ein Einkristall der GroBe 0.3 X 0.4 X 0.6 mm3 wurde bei - 10°C auf einem Vierkreisdiffrak- tometer CAD4 der Firma Enraf-Nonius vermessen (Mo-K,). Raumgruppe: P2Jn (Nr. 14) mit Z = 4; Zelldimensionen: a = 9.541(5), b = 20.132(7), c = 14.092(5) A, p = 97.95(3)", V = 2681(3) A3. Dber. = 1.399 glcm3, p = 7.3 cm-'. Es wurden 4840 Reflexe im Bereich 3" < 0 < 25" gemessen. Fur die Verfeinerung (333 Parameter) wurden 2905 symmetrieunabhangige Reflexe mit I > 30(4 verwendet. Die Strukturlos~ng[~~] erfolgte mittels Patter- son-Synthese und Differenz-Fourier-Methoden. Alle Nicht-H-
Chem. Ber. 1994,127, 1003-1013

1012 R. Beckhaus, J. Oster, T. Wagner
Atome wurden anisotrop, die H-Atome am Fiinfring isotrop verfei- nert. Alle anderen Wasserstoffe wurden bei der Verfeinerung mitge- fiihrt [d,--" = 0.98 A, Biso(H) = 1.3 Biso(C)]. Gewichtungsfaktor l/02(Fo), R = 0.044, R, = 0.062, GOF = 2.378. Keine Absorp- tionskorrektur. Die Restelektronendichte betrug 0.47 1 ec'/A3[59].
['I [ I a ] R. R. Schrock, J Organornet. Chem. 1976,122,209-225. - [ Ib ] C. D. Wood. S. J. McLain, R. R. Schrock. J Am. Chem. SOC. 1979, 101, 3210-3222. [2a] R. R. Schrock, Science 1983, 219, 13-18. - LZb] R. R. Schrock, Ace. Chem. Res. 1979, 12, 98-104.
C31 M. D. Frvzuk. S. S. H. Mao, M. J. Zaworotko, L. R. MacGilliv- ray, J Am. Chem. Soc 1993, 115, 5336-5339.
E41 J. K. Cockcroft, V. C. Gibson, J. A. K. Howard, A. D. Poole, U. Siemeling, C. Wilson, 1 Chem. Soc, Chem. Commun.
r S ] [5a1 N. A. Petasis, D.-K. Fui, J. Am. Chem. Soc. 1993, 115, 7208-7214. - [5b] N. A. Petasis, D.-K. Fui, Organometallics 1993,12,3776-3780. - [''I N. A. Petasis, I. Akritopoulou, Syn- left 1992, 6655667. - Lsd] N. A. Petasis, E. 1. Bzowej, J Org. Chem. 1992, 57, 1327-1330. - I? DeShong, P. J. Rybczyn- ski, J Org Chem. 1991, 56, 3207-3210. - Lsfl N. A. Petasis, E. I. Bzowej, .L Am. Chem. Soc. 1990, 112, 6392-6394.
Ti=Si: E. Hengge, M. Weinberger, J Organomet. Chem. 1993,443, 167- 173. - Wb] Ti=N: C. C. Cummins, C. P. Schal- ler, G. D. Van Duyne, P. T. Wolczanski, A. W. E. Chan, R. Hoffmann, 1. Am. Chem. Soc. 1991,113,2985-2994.
c71 [7a] Zr=N: P. J. Walsh, F. J. Hollander, R. G. Bergman, Organo- metallics 1993, 12, 3705-3723. - [7b1 Zr=P: J. Ho, Z. Hou, R. J. Drake, D. W. Stephan, Organometallics 1993, 12, 3145-3157. - [7cl Zr=P: 2. Hou, D. W. Stephan, J Am. Chem. Soc. 1992, 114, 10088-10089. - [7d1 Zr=O, Zr=S: M. J. Carney, P. J. Walsh, F. J. Hollander, R. G. Bergman, Organometallics 1992, 11, 761-777. -- [7e1 Zr=O: M. J. Carney, P. J. Walsh, F. J. Hol- lander, R. G. Bergman, J Am. Chem. Soc. 1989, 111, 8751 -8753. R. Beckhaus, Zur Chemie von Vinylverbindungen elektronenar- mer Ubergangsmetalle, Habilitation Thesis, Fakultat fur Natur- wissenschaften, Technische Hochschule Leuna-Merseburg, 1989. 1991: Mathematisch-Naturwissenschaftliche Fakultat der
1992, 1668-1670.
L61
RWTH Aachen, 1993. r91 R. Beckhaus, S. Flatau, S. I. Troyanov, P. Hofmann, Chem. Ber.
[ ' O ] R. Beckhaus. K.-H. Thiele. D. Strohl, J Orfanomet. Chem. 1992, 125, 291 -299.
1989,369, 43-54. -
(''1 R. Beckhaus, 1. StrauB, T. Wagner, P. Kiprof, Angew. Chem. 1993, 105, 281-283; Angew. Chem. Int. Ed. Engl. 1993, 32, 264-266.
[I2 ] R. Beckhaus in Organometallics in Organic Synthesis (OSM4) (Hrsg.: D. Enders, H.-J. Gais, W. Keim), Vieweg Verlag, Braun- schweig, 1993, S. 131.
[ I3 ] R. Beckhaus, I. StrauD, T. Wagner, J Organomet. Chem. 1994, 464, 155-161.
[14] [I4'] G. Erker, U. Dorf, R. Lecht, M. T. Ashby, M. Aulbach, R. Schlund, C. Kriiger, R. Mynott, Organometallics 1989, 8, 2037-2044. - G. Erker, U. Dorf, R. Mynott, Y-H. Tsay, C. Kriiger, Angew. Cliem. 1985, 97, 572-514; Angew. Chem. Znt. Ed. Engl. 1985, 24, 584-586. - [I4'] G. Erker, R. Lecht, J Or anomet. Chem. 1986, 311, 45-55.
1991, ZO, 3559--3568. - G. Erker, F. Sosna, Organometal- lics 1990, 9, 1949-1953. - ["'I G. Erker, B. Menjon, Chem. Ber. 1990, 123, 1327-1329. - [lsd] G. Erker, F. Sosna, R. Pfaff, R. Noe, C. Sarter, A. Kraft, C. Kru er, R. Zwettler, J Organo- met. Chem. 1990, 394, 99-112. - lFse1 G. Erker, R. Lecht, Y.- H. Tsay, C. Kriiger, Chem. Ber. 1987,120, 1763- 1765. - [ " f ~ G. Erker, R. Lecht, J. L. Petersen, H. Bonnemann, Organometallics 1987. 6. 1962-1967. - ["gl G. Erker. R. Lecht, R. Schlund,
[Is] 4 G. Erker, R. Pfaff, C. Kriiger, S. Werner, Organometallics
K. Angermund, C. Kruger, Angew. Chem. 1987, 99, 708-710; Annew. Chem. Int. Ed. End. 1987, 26, 666-668. - [Ish] G. Er- ke< U. Dorf, R. Benn, R.-D. Reinhardt, J Am. Chem. SOC.
K. Mashima. K. Jvodoi. A. Ohvshi. H. Takavd. J Chem. Soc.. 1984, 106, 7649-7650.
&em. Commun. 1986, 1145- 1 i46. [ I 7 ] G. Erker, M. Mena. U. Hoffrnann, B. Menjon, J. L. Petersen.
Or anometallics 1991, 10, 291 -298. cLsJ [Isd 4 G. Erker, Angew. Chem. 1989,101,411-426; Angew. Chem.
Int. Ed. Engl. 1989, 28, 397-412. - 1988. 7. 2451-2463.
G. Erker, Polyhedron
[I9] E. V'Anslyn, B. D. Santarsiero, R. H. Grubbs, Organometallics 1988, 7 , 2137-2145.
['O] G. Proulx, R. G. Bergman, J Am. Chem. Soc. 1993, 115, 9802-9803.
12'] J. Feldman, R. R. Schrock, Prog. lnorg Chem. 1991, 39, 1-74. G. A. Luinstra, J. H. Teuben, Organometallics 1992, 11,
[231 [23a1 C. McDade, J. C. Green, J. E. Bercaw, Organometallics 1982,1, 1629-1634. - [23b] C. P. Gibson, D. S. Bern, J Organo- met. Chem. 1991, 414, 23-32. - [23c1 S. H. Bertz, G. Dabbagh, C. P. Gibson, Organometallics 1988, 7, 563- 565.
LZ4] [24a1 A. van Asselt, B. J. Burger, V. C. Gibson, J. E. Bercaw, J Am. Chem. Sot. 1986, 108, 5347-5349. - [24b1 G. Parkin, E. Bunel, B. J. Burger, M. S. Trimmer, A. van Asselt, J. E. Bercaw, J Mol. Catal. 1987, 41, 21 -39. - [24c] W. Bell, D. M. Haddle- ton, A. McCamley, M. G. Partridge, R. N. Perutz, H. Willner, J Am. Chem. Soc. 1990, 112, 9212-9226.
LZs] R. Beckhaus, J. Sang, J. Oster, T. Wagner, J Organomet. Chem., im Druck. J. E. Bercaw, J Am. Chem. Soc. 1974, 96, 5087-5095.
M. Brookhart, M. L. H. Green, J Organomet. Chem. 1983, 250, 395-408. - Zum Vergleich P-agostische Vinylsysteme in Cp,Zr-Komplexen: 'J(CB-H) 104- 112 Hz: [27b1 I. Hyla-Kry- spin, R. Gleiter, C. Kriiger, R. Zwettler, G. Erker, Organonzetal- lies 1990, 9, 517-523. - [27c1 G. Erker, W. Fromberg, K. Anger- mund, R. Schlund, C. Kriiger, J Chem. Soc., Chem. Commun. 1986, 372-374. - 'J(CB-H) 96-97 Hz: [27d1 U. Rosenthal, A. Ohff, M. Michalik, H. Gorls, V. V. Burlakov, V. B. Shur, Angew. Chem. 1993, 105, 1228-1230; Angew. Chem. Int. Ed. Engl. 1993,32, 1193-1195.
[281 S. T. Carter, W. Clegg, V. C. Gibson, T. P. Kee, R. D. Sanner, Organometallicv 1989, 8, 253-255.
[291 Siehe z.B. R. M. Bullock, C. E. L. Headford, K. M. Hennessy, S. E. Kegley, J. R. Norton, J Am. Chem. Soc. 1989, 111, 3897 - 3908.
L3O] R. Beckhaus, I. StrauB, T. Wagner, unveroffentlicht. L3'1 K. H. Dotz, H. Fischer, P. Hofmann, F. R. Kreissl, U. Schubert,
K. Weiss, Transition Metal Carbeize Complexes, Verlag Che- mie, 1983.
L3'1 G. M. Bodner, S. B. Kahl, K. Bork, B. N. Storhoff, J. E. Wuller, L. J. Todd, Znorg Chem. 1973, 12, 1071-1074.
[331 [33al J.-A. M. Andersen, S. J. Archer, J. R. Moss, M. L. Niven, Inorg. Chim. Acta 1993, 206, 187-192. - [33b1 N. A. Bailey, D. A. Dunn, C. N. Foxcroft, G. R. Harrison, M. J. Winter, S. Woodward, J Chem. Sue., Dulton Trans. 1988, 1449-1456. - [33c] N. A. Bailey. P. L. Chell. C. P. Manuel. A. MukhoDadhyay,
1793- 1801.
D. Rogers, H. E. Tabbron, M. J. Winter, J Chem. Soc, Daitoi Trans. 1983. 2397-2403.
[341 J. W. F. L. Seetz, B. J. J. v. de Heisteeg, G. Schat, 0. S. Akker-
[351 J.-A. M. Garner, A. Irving, J. R. Moss, Organonzetallics 1990,
[361 [36al G. Huttner, D. Regler, Chem. Ber. 1972, 10.5, 1230-1244. - [36b1 E. W. Post, K. L. Watters, Inorg. Chim. Acta 1978, 26, 29-36. - [36c1 E. 0. Fischer, E. Ofmaus, Chem. Ber. 1969, 102, 2449-2455. - [36d1 E. 0. Fischer, P. Rustemeyer, J Organornet. Chem. 1982, 225, 265-277. - M. Y Darensbourg, D. J. Darensbourg, Inorg. Chem. 1970, 9, 32-39.
[371 [37al R. Beckhaus, K.-H. Thiele, J Organomet. Chem. 1989, 368, 315-322. - [37b1 P. Fu, M. A. Khan, K. M. Nicholas, Organo- metallics 1991, 10, 382-384. - [37cl P. T. Barger, J. E. Bercaw, Organometallics 1984, 3, 278-284.
PX] E. 0. Fischer, S. Fontana, .l Organornet. Chem. 1972, 40,
[391 Kurze TiTO-Bindung z.B. 1.750(2) A, mittIFre Ti-0-Bindung 1.855(2) A [3ya] lange Ti-0-Bindung 2.11 A [39bl. - [39a1 J. C. Huffman, K. G. Moloy, J. A. Marsella, K. G. Caulton, J Am. Chem. Soc. 1980, 102, 3009-3014. - [39bl U. Thewalt, B. Ho- nold, J Organomet. Chem. 1988, 348, 291-303.
I4O] V. B. Shur, V. V. Burlakov, A. I. Yanovsky, P. V. Petrovsky, Y. T. Struchkov, M. E. Volpin, J Organornet. Chem. 1985, 297,
L4'1 K. P. C. Vollhardt, Organische Chemie, 1. Aufl., VCH Verlagsge- sellschaft mbH, Weinheim, 1990.
I4,1 [42al M. Sabat, M. F. Gross, M. G. Finn, Organometallics 1992, 11, 745-751. - [42bl K. H. Dotz, W. Kuhn, K. Ackermann, 2.
man, F. Bickelhaupt, J Mol. Catal. 1985, 28, 71 -83.
9, 2836-2840.
159-162.
51-59.
Chem. Ber. 1994, 127, 1003-1013

Cyclische isomere vier- und funfgliedrige heterodinucleare Fischer-Carbenkomplexe 1013
Nutuvforsch., Teil B, 1983, 38, 1351-1356. - [42c] G. Huttner, S. Lange, Chem. Ber. 1970, 103, 3149-3158.
[431 A. Whitaker, J. W. Jeffery, Actu Crystalogr 1967,23, 977-984. [441 r44al J. A. Connor, E. M. Jones, 1 Orgunomet. Chem. 1971, 31,
389-393. - LUhl J. W. Wilson, E. 0. Fischer, .I Organomet. Chem. 1973, 57, C63-C66. - J. Muller, J. A. Connor, Chem. Ber. 1969, 102, 1148-1160.
[451 [45al M. M. Gallo, T. P. Hamilton, H. F. Schaefer 111, J Am. Chem. SOC. 1990, 112, 8714-8719. - [451 D. Siilze, H. Schwarz, Chem. Phys. Lett. 1989, 156, 397-400. - [45cl G. Franking, Chem. Phys. Lett. 1983, 100, 484-487.
[461 [46al M. I. Bruce, Chem. Rev. 1991, 91, 197-257. - [46b1 H. Wer- ner, Nuchr. Chem. Tech. Lab. 1992, 40, 435-444. - [46c1 M. I. Bruce. A. G. Swincer. Adv. Orpanomet. Chem. 1983, 22, 59-128. - [46d1 N. M. 'Kostic, R-F. Fenske, Organometullics
I4'I J. Silvestre. R. Hoffmann. Helv. Chim. Actu 1985. 68. 1982, I , 974-982.
I ,
1461 - 1506: [481 [48A] N. M. Doherty, C. Elschenbroich, H.-J. Kneuper, S. A. R.
Knox, J Chem. Soc., Chem. Commun. 1985, 170-171. - [48hl S. F. T. Froom, M. Green, R. J. Mercer, K. R. Nagle, A. G. Orpen, S. Schwiegk, J. Chem. Soc., Chem. Commun. 1986, 1666-1668.
[491 J. Levisalles, H. Rudler, Y Jeannin, F. Dahan, J. Orgunornet. Chem. 1979, 178, C8-CI2.
G. Erker, R. Petrenz, Organometullics 1992,11, 1646-1655. - G. Erker, R. Petrenz, 1 Chem. Soc., Chem. Commun. 1989, 345-346.
["I ['la] G. N. Sastry, E. D. Jemmis, J. Orgunornet. Chem. 1990, 388. 289-299. - [51h] G. Erker. K. Kroou. Chem. Ber. 1982.
1 1 1
115; 2437-3446. L S 2 ] M. T. Reetz. Adv. Orcanomet. Chem. 1977. 16. 33-65. cS31 [53a] B. Schiott, K. AT Jorgensen J Chem.' So;., Dalton Trans.
1993, 337-344. - LS3'] L. L. Whinnery Jr., L. M. Henling, J. E. Bercaw, J. Am. Chem. SOC. 1991,113, 7375-7582.
rS41 [54d] S. H. Pine G. S. Shen, H. Hoang, Synthesis 1991, 165- 167. - [54bl H. U: ReiBig, Nachr. Chem. Techn. Lab. 1986, 34,
[551 C. S. Johnson Jr., M. A. Weiner, J. S. Waugh, D. Seyferth, J. Am. Chem. SOC. 1961,83, 1306-1307.
r S 6 ] F. Bonati, G. Wilkinson, J Chem. SOC. 1964, 3156-3160. P7I Der Kohlenstoffwert fallt fur 16 aufgrund unvollstandiger Ver-
brennung, offensichtlich bedingt durch bevorzugte MC-Bil-
562-565.
dung, zu niedrig aus. L5'l B. A. Frenz. 1978. The ENRAF-NONIUS CADI-SDP - a Re-
al-Time System for Concurrent X-Ray Data Collection and Cry- stal Structure Determination. In Computing in Crystallography (Hrsg.: H. Schenk, R. Olthof-Hazekamp, H. van Koningsveld, G. C. Bassi), Delft University, SDP-PLUS, Version 1.1 (1984) and VAXSDP, Version 2.2 (1985).
[591 Weitere Einzelheiten zur Kristallstrukturuntersuchung konnen beim Fachinformationszentrum Karlsruhe, Gesellschaft fur wissenschaftlich-technische Information mbH, D-76344 Eggen- stein-Leopoldshafen, unter Angabe der Hinterlegungsnummer CSD-400535 angefordert werden.
[416/93]
Chem. Ber. 1994, 127, 1003-1013





























