Embed Size (px)
Citation preview

Der Transkriptionsfaktor FKHR interagiert mit STAT-Faktoren und
verstärkt die IL-6-Signaltransduktion
Von der Medizinischen Fakultät der Rheinisch-Westfälischen Technischen Hochschule Aachen
zur Erlangung des akademischen Grades eines Doktors der Medizin genehmigte Dissertation
vorgelegt von
Florian Feld aus
Wuppertal
Berichter: Herr Universitätsprofessor Dr. rer. nat. Peter Claus Heinrich Herr Universitätsprofessor Dr. med. Günther Schmalzing Tag der mündlichen Prüfung: 11. Mai 2004 Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.


Veröffentlichungen wesentliche Teile der vorliegenden Arbeit wurden veröffentlicht in: Kortylewski, M.*, Feld, F.*, Krüger, K. D., Bahrenberg, G., Roth, R. A., Joost, H. G., Heinrich, P. C., Behrmann, I., Barthel, A. (2003) Akt modulates STAT3-mediated gene expression through a FKHR (FOXO1a)- dependent mechanism Journal of Biological Chemistry 278, 5242-5249 PMID: 12456685 * Both authors contributed equally to this publication. Heinrich, P.C., A. Barthel, I. Behrmann, F. Feld, J. Grötzinger, C. Haan, H. M. Hermanns, H.-G. Joost, I. M. Kerr, M. Kortylewski, U. Lehmann, G. Müller-Newen, S. Radtke, R. Roth, F. Schaper, and A. Timmermann. (2002) Regulation of interleukin-6-type cytokine signalling Cytokines in liver injury and repair: Falk Symposium 125 (A. M. Gressner, P. C. Heinrich, S. Matern) Kluwer Academic Publishers, Dordrecht, NL, pp. 221-237, ISBN: 0792387759


I
INHALTSVERZEICHNIS Inhaltsverzeichnis I
Verzeichnis der Abkürzungen V
1 Einleitung..............................................................................1
1.1 Zytokine ..................................................................................................................1
1.2 Interleukin-6 ............................................................................................................2
1.2.1 IL-6-Typ-Zytokine....................................................................................................2
1.2.2 Struktur, biologische Funktion und Nomenklatur von IL-6 ......................................3
1.2.3 Der Jak/STAT-Signaltransduktionsweg ..................................................................5
1.2.4 Regulation der IL-6-Signaltransduktion ..................................................................7
1.2.4.1 Die Tyrosinphosphatase SHP2...............................................................................7
1.2.4.2 SOCS-Proteine .......................................................................................................8
1.2.4.3 PIAS-Proteine .........................................................................................................9
1.2.4.4 Rezeptorvermittelte-Endozytose.............................................................................9
1.3 Die Ras / Raf / MAPK-Kaskade ............................................................................10
1.4 Forkhead box o (foxo) Transkriptionsfaktoren ......................................................11
1.4.1 Biologische Funktion und Nomenklatur ................................................................11
1.4.2 Regulation der foxo-Familienmitglieder ................................................................13
1.5 Zielsetzung der Arbeit...........................................................................................16
2 Material und Methoden ..................................................... 17
2.1 Verwendete Materialien ........................................................................................17
2.1.1 Chemikalien und Enzyme.....................................................................................17
2.1.2 Puffer und Medien ................................................................................................17
2.1.3 Verbrauchsmaterialien..........................................................................................17
2.2 Stimulanzien und Inhibitoren ................................................................................18
2.3 Antikörper .............................................................................................................19
2.4 Plasmide...............................................................................................................20
2.5 Eukaryontische Zellen und deren Kultivierung .....................................................23

II
2.6 Prokaryontische Zellen und deren Kultivierung ....................................................24
2.7 Herstellung kompetenter Bakterien ......................................................................24
2.8 Präparation, Modifikation und Analyse von Plasmid-DNA....................................25
2.8.1 Transformation von Bakterien...............................................................................25
2.8.2 Plasmid-Präparationen .........................................................................................25
2.8.3 Quantitative Bestimmung von Nukleinsäuren.......................................................25
2.8.4 Spaltung von DNA mit Restriktionsendonukleasen ..............................................26
2.8.5 Dephosphorylierung linearisierter DNA ................................................................26
2.8.6 Phenolextraktion...................................................................................................26
2.8.7 Elektrophoretische Analyse von Plasmid-DNA.....................................................27
2.8.8 Isolierung von DNA-Fragmenten ..........................................................................27
2.8.9 DNA-Ligation ........................................................................................................28
2.9 Transiente Transfektion von HepG2-Zellen..........................................................28
2.10 Proteinbestimmung nach Bradford .......................................................................29
2.11 Herstellung von Zell-Lysaten ................................................................................29
2.12 Immunpräzipitation ...............................................................................................29
2.13 SDS-Polyacrylamid-Gelelektrophorese ................................................................30
2.14 Proteintransfer auf eine PVDF-Membran durch das
„Western-Blot-semidry“-Verfahren........................................................................31
2.15 Detektion präzipitierter Proteine mit Hilfe eines Immunblots ................................33
2.16 Reportergen-Versuche .........................................................................................34
2.17 Ernte transfizierter Zellen für Reportergenuntersuchungen..................................34
2.17.1 Luziferase-Messung .............................................................................................35
2.17.2 β-Galaktosidase-Messung ....................................................................................35
2.18 Zellproliferationstest (XTT-Test) ...........................................................................36
2.19 Zellfärbungen für die Immunfluoreszenz ..............................................................36
2.20 Fluoreszenzmikroskopie .......................................................................................37
2.20.1 Geräte...................................................................................................................37
2.20.2 Bildaufnahme am LSM 510 ..................................................................................38

III
3 Ergebnisse......................................................................... 39
3.1 Hemmung der IL-6-Signaltransduktion in Hepatozyten
durch Wachstumsfaktoren ....................................................................................39
3.2 Nachweis einer funktionellen Interaktion zwischen STAT-Faktoren und FKHR ...44
3.2.1 Der Transkriptionsfaktor FKHR verstärkt
STAT3-abhängige Reportergenaktivität ...............................................................44
3.2.2 Die durch FKHR bewirkte transkriptionelle Steigerung ist STAT3-abhängig........48
3.2.3 Die funktionelle Interaktion mit FKHR ist spezifisch für STAT3 und STAT1.........49
3.2.4 FKHR vermindert die PIAS3-vermittelte STAT3-Inhibition ...................................53
3.2.5 Nachweis einer physikalischen Interaktion zwischen STAT3 und FKHR .............55
3.3 Auswirkungen von FKHR-modulierenden Effektoren
auf die IL-6-Signaltransduktion ............................................................................57
3.4 Die subzelluläre Lokalisation von FKHR und STAT3
in Abhängigkeit verschiedener Stimuli..................................................................62
3.5 Herstellung und erste Charakterisierung von HepG2-Transfektanten,
die stabil fluoreszierendes FKHR exprimieren......................................................64
4 Diskussion ......................................................................... 69
4.1 Interaktion zwischen FKHR und STAT-Faktoren..................................................69
4.2 Mögliche Mechanismen der Interaktion ................................................................70
4.2.1 FKHR könnte die Kernlokalisation der STAT-Faktoren begünstigen....................71
4.2.2 FKHR könnte als Vermittler zwischen STAT3
und weiteren Transkriptionsfaktoren fungieren.....................................................73
4.2.3 FKHR könnte die Aktivierung von STAT-Faktoren verstärken
oder deren Inaktivierung hemmen........................................................................74
4.2.4 FKHR könnte das STAT3-Dimer stabilisieren ......................................................74
4.3 Das IL-6-Signal scheint die hemmende Wirkung
von FKHR auf die Stoffwechselaktivität zu unterdrücken .....................................75

IV
5 Ausblick ............................................................................. 79
5.1 Generierung FKHR-defizienter Zellen ..................................................................79
5.2 Identifizierung der interagierenden Bereiche ........................................................80
5.3 Auswirkungen auf die Interaktion mit weiteren Proteinen.....................................81
5.4 in-vivo-Versuchsansätze.......................................................................................81
5.5 Studien zur DNA-Bindung von FKHR...................................................................82
5.6 Untersuchungen mit FKHR responsiven Promotoren...........................................83
6 Zusammenfassung ........................................................... 85
7 Literaturverzeichnis .......................................................... 87
8 Anhang ............................................................................. 103

V
Verzeichnis der Abkürzungen
Abb. Abbildung
AFX ALL1-fused-gene-from-chromosome-X
Ak Antikörper
APP Akut-Phase-Protein
APS Ammoniumperoxodisulfat
AS Aminosäure
ASV-16 avian sarcoma virus-16
ATP Adenosintriphosphat
Bad Bcl2/Bcl-XL-associated death promoter
Bcl2 B cell survival regulated by phospholipase Cγ2
Bim Bcl2-interacting mediator of cell death
bp Basenpaare
BSA bovines Serum Albumin
CBP CREB-binding-protein
CNTF ciliary neurotrophic factor
CREB cAMP response element binding protein
CT-1 Cardiotrophin-1
DMEM Dulbecco´s Modified Eagle Medium
DMSO Dimethylsulfoxid
DNA desoxyribonuleic acid
DTT 1,4-Dithio-DL-threitol (Cleland´s Reagenz)
E Extinktion
E. coli Escherichia coli
ECL enhanced chemoluminescence
EDTA ethylenediamine-tetraacetic acid
EGF epidermal growth factor
EMSA electrophoretic mobility shift assay
Epo Erythropoetin
ERK extracellular signal-regulated kinase
FCS fetal calf serum
FKHR forkhead related transcription factor

VI
FKHRL1 FKHR-like-1
foxo forkhead box o
FRET fluorescence resonance energy-transfer
G6P-ase Glukose-6-Phosphatase
GFP green fluorescent protein
GSK Glycogen-Synthase-Kinase-3
GST Glutathion S-Transferase
gp Glykoprotein
h Stunde
HEPES N-(2-Hydroxyethyl)-Piperazin-N´-2-Ethansulfonsäure
HGF hepatocyte growth factor
HNF hepatocyte nuclear factor
IFN Interferon
Ig Immunglobulin
IGFBP insulin-like growth factor binding protein
IL Interleukin
IRS Insulin-Rezeptor-Substrat
IRU / IRE insulin response unit / element
Jak Janus-Kinase
JH Jak homology
kDa Kilodalton
KIR kinase inhibitory region
LB Luria-Bertani
min Minute
Mr molare Masse
MAKK mitogen-activated protein kinase kinase
MAPK mitogen-activated protein kinase
NES nuclear export signal
NGF nerve growth factor
OD optical density
OSM Oncostatin M
PAA Polyacrylamid
PAGE Polyacrylamid-Gelelektrophorese

VII
PBS phosphate buffered saline
PCR polymerase chain reaction
PEPCK Phosphoenolpyruvat-Carboxykinase
PDK 3-phosphoinositidedependent protein kinase
PI3,4P2 Phosphatidylinositol-3,4-diphosphat
PI3,4,5P3 Phosphatidylinositol-3,4,5-triphosphat
PI3-Kinase Phosphatidylinositolphosphat-3-Kinase
PI4,5P2 Phosphatidylinositol-4,5-diphosphat
PI4P Phosphatidylinositol-4-phosphat
PIAS protein inhibitor of activated STAT
PKB Proteinkinase B
PTEN phosphatase and tension homology deleted on
chromosome ten
PVDF Polyvinylidendifluorid
pY Phosphotyrosin
R Rezeptor
RNA ribonucleic acid
sIL-6Rα soluble IL-6 receptor α
SDS sodium-dodecylsulfate
SGK serum-and-glucocorticoid-inducible-kinases
SH src homology
SHP SH2-domain-containing tyrosine phosphatase
SOCS suppressor of cytokine signalling
STAT signal transducer and activator of transcription
Tab. Tabelle
TCL total cell lysates
TEMED N, N, N´, N´- Tetramethyl-Ethylen-Diamin
TGF transforming growth factor
Tris Tris(hydroxy)aminomethan
U unit
Upm Umdrehungen pro Minute
YFP yellow fluorescent protein

VIII

1
1 Einleitung
1.1 Zytokine
Um die Integrität eines Individuums bei den unterschiedlichsten Um-
gebungsvariablen zu wahren, bedarf es eines hochkomplizierten Netz-
werks. Zum einen muss ein reger Informationsaustausch, zum anderen
eine optimale Steuerung von einzelnen Zellen sowie Zellverbänden ge-
währleistet sein. Der Körper bedient sich unterschiedlicher Mechanismen,
um diesen Aufgaben gerecht zu werden. Als bekanntestes Beispiel kann
man die bewusste Innervation von quergestreifter Muskulatur betrachten,
bei der ein Gedanke innerhalb von Bruchteilen einer Sekunde in eine aktive
Bewegung mündet.
Um solche leicht nachvollziehbaren Aktionen überhaupt durchführen zu
können, muss jedoch eine Vielzahl an Regelgrößen stimmen. Genannt
seien nur die notwendige Energiebereitstellung, ein adäquates Sauerstoff-
angebot und ein Abtransport anfallender Metabolite. Diese nicht bewusst
steuerbaren, vegetativen Funktionen erfüllt der Körper mittels spezieller
Transmitter. Diese Stoffe können von spezifischen Drüsen endokrin - über
die Blutbahn - abgegeben werden, wie bei der Glukagonfreisetzung zur
Glukoseutilisation. Auch parakrine – auf benachbarte Zellen zielende -
Wirkungsweisen wie die Stickstoffmonoxid (NO)-Synthese zur Arteriolen-
dilatation und damit vermehrter Sauerstoffbereitstellung sind unter-
stützende Mechanismen. Bei autokriner Stimulation ist die sezernierende
Zelle zugleich auch der Zielort.
Als eines der kompliziertesten und wichtigsten Systeme des menschlichen
Körpers kann man das Immunsystem ansehen. Dieses muss einerseits alle
eindringenden fremden Organismen vollständig beseitigen, andererseits
darf es sich keinen Fehler erlauben und eine Reaktion gegen körpereigene
Stoffe zulassen. Dafür ist ein besonders kompliziertes System an Signal-
übertragung notwendig, bei dem Zytokine die wichtigste Rolle spielen. So
sind sie in der Lage, die Proliferation und Entwicklung spezifischer Zell-
populationen zu fördern, die Sekretion unterschiedlicher von B-Lympho-

2
zyten gebildeter Antikörper zu steuern, aber auch bestimmte immuno-
logische Antworten zu unterdrücken.
Zytokine zählen zu den Gewebshormonen und wirken außer auf Leuko-
zyten auch auf viele andere Zellarten wie Fibroblasten, Hepatozyten und
Melanozyten. Zu den Zytokinen rechnet man Wachstumsfaktoren, Inter-
leukine, Interferone und Chemokine. Ihre Wirkung entfalten die Zytokine
über spezifische Rezeptoren, welche nach Rezeptor-Oligomerisierung eine
Signaltransduktionskaskade in Gang setzen. Zytokine können sowohl
autokrin wirken als auch para- und endokrin Effektorzellen ansprechen.
1.2 Interleukin-6
1.2.1 IL-6-Typ-Zytokine
Wie LIF (leukemia inhibitory factor), OSM (Oncostatin M), IL-11, CNTF
(ciliary neurotrophic factor), CLC (cardiotrophin-like cytokine), und CT-1
(Cardiotrophin-1) zählt IL-6 zu den langkettigen 4α−helix-bundle-Zytokinen
mit etwa 25 Aminosäuren in den α−Helices 1,2. Diese Zytokine weisen eine
ähnliche dreidimensionale Struktur auf. Außerdem haben alle Rezeptoren
dieser Botenstoffe die signaltransduzierende Rezeptoruntereinheit gp130
(ein Glykoprotein von 130 Kilodalton) gemeinsam. Auf Grund dieser
Ähnlichkeiten werden die genannten Zytokine auch als IL-6-Typ-Zytokine
bezeichnet.
Neben gp130 können die IL-6-Typ-Zytokin-Rezeptor-Komplexe eine
kürzere α-Rezeptor-Untereinheit (IL-6R, IL-11R, CNTFR) und/oder eine
zusätzliche signaltransduzierende Untereinheit (LIFR, OSMR) enthalten
(Abb. 1.2.1) 2,3. Eventuell sind noch weitere Proteine am Rezeptorkomplex
beteiligt, was durch den Balken mit dem Fragezeichen dargestellt wird.
Der IL-6Rα (gp80, ein 80 Kilodalton großes Glykoprotein) kommt physio-
logisch sowohl membrangebunden als auch löslich vor. Die lösliche Form
kann einerseits durch alternatives Spleißen der Prä-m-RNA als auch durch
limitierte Proteolyse (Shedding) entstehen.

3
gp130 / gp130 LIFR / gp130 OSMR / gp130
OSM CLC OSMCT-1LIF CNTFIL-11
?
gp130
gp130
gp130
gp130
gp130
gp130
gp130
gp130
LIFR
LIFR
LIFR
LIFR
LIFR
OSM
R
IL-6
gp130
gp130
IL-6R
IL-6R
IL-11R CN
TFR
CN
TFR
Abbildung 1.2.1: Die IL-6-Typ-Zytokine und ihre Rezeptorkomplexe.
1.2.2 Struktur, biologische Funktion und
Nomenklatur von IL-6
IL-6 besteht aus 184 Aminosäuren und weist ein Molekulargewicht von 26
kDa auf 4. Es wird posttranslational N- oder O-glykosyliert, wobei es etwa
dreifach aktiver als unglykosyliertes IL-6 ist 5. Zur Aufrechterhaltung der
Struktur sind zwei Cysteinbrücken wichtig.
Interleukin-6 ist eines der wichtigsten Zytokine bei der physiologischen
Antwort auf das Eindringen pathogener Organismen. Es wird auf Stimuli
wie Lipopolysaccharide (LPS), IL-1, TNFα und virale Nukleinsäuren von
einer Vielzahl von Zellen gebildet und induziert in der Leber Akut-Phase-
Proteine (APP) wie beispielsweise Serumamyloid A (SAA), C-reaktives
Protein (CRP) und α2-Makroglobulin 6-11.
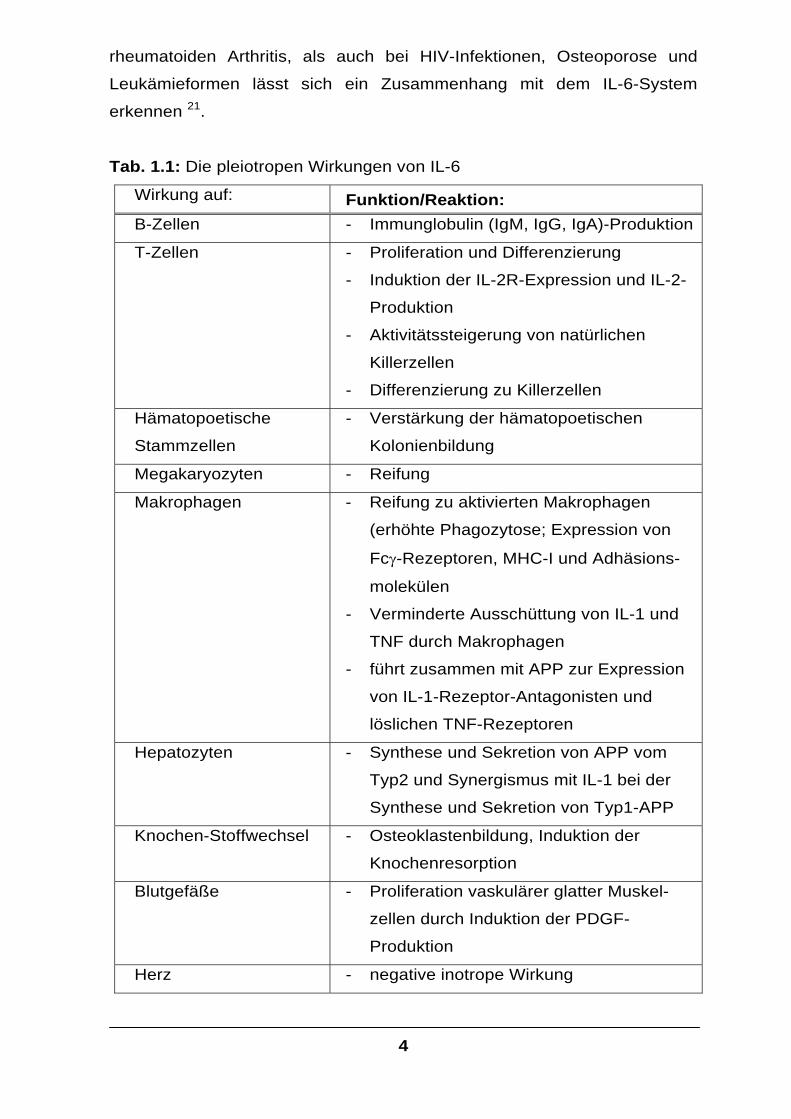
IL-6 wirkt auf eine Vielzahl von Zellen mit zum Teil antagonistischer Funk-
tion (Tabelle 1.1). Dem proinflammatorischen Effekt auf B- und T-Zellen
steht die antiinflammatorisch wirkende verminderte IL-1- und
TNFα−Sekretion gegenüber 12. Auf einige Zelllinien wirkt IL-6 antiproli-
ferativ 13,14, auf andere wachstumssteigernd 15,16 oder antiapoptotisch 17-20.
Bei einer Vielzahl von Krankheiten des menschlichen Organismus spielt IL-
6 eine wichtige Rolle. Sowohl bei Autoimmunerkrankungen wie der
4
rheumatoiden Arthritis, als auch bei HIV-Infektionen, Osteoporose und
Leukämieformen lässt sich ein Zusammenhang mit dem IL-6-System
erkennen 21.
Tab. 1.1: Die pleiotropen Wirkungen von IL-6
Wirkung auf: Funktion/Reaktion: B-Zellen - Immunglobulin (IgM, IgG, IgA)-Produktion
T-Zellen - Proliferation und Differenzierung
- Induktion der IL-2R-Expression und IL-2-
Produktion
- Aktivitätssteigerung von natürlichen
Killerzellen
- Differenzierung zu Killerzellen
Hämatopoetische
Stammzellen
- Verstärkung der hämatopoetischen
Kolonienbildung
Megakaryozyten - Reifung
Makrophagen - Reifung zu aktivierten Makrophagen
(erhöhte Phagozytose; Expression von
Fcγ-Rezeptoren, MHC-I und Adhäsions-
molekülen
- Verminderte Ausschüttung von IL-1 und
TNF durch Makrophagen
- führt zusammen mit APP zur Expression
von IL-1-Rezeptor-Antagonisten und
löslichen TNF-Rezeptoren
Hepatozyten - Synthese und Sekretion von APP vom
Typ2 und Synergismus mit IL-1 bei der
Synthese und Sekretion von Typ1-APP
Knochen-Stoffwechsel - Osteoklastenbildung, Induktion der
Knochenresorption
Blutgefäße - Proliferation vaskulärer glatter Muskel-
zellen durch Induktion der PDGF-
Produktion
Herz - negative inotrope Wirkung

5
Zentrales Nervensystem - Auslösen von Fieber durch die Induktion
von Prostaglandin E2 (PGE2)
- Neuronale Differenzierung
- Induktion der adrenocorticotropen
Hormonsynthese
Plazenta - Sekretion von chorionischem
Gonadotropin aus Trophoblasten
1.2.3 Der Jak/STAT-Signaltransduktionsweg
Nach Bindung von IL-6 an seinen α-Rezeptor (IL-6Rα, gp80) kommt es zur
Assoziation mit gp130. Zwei dieser Heterotrimere lagern sich zusammen
und bilden ein Heterohexamer, wie eine kürzlich erschienene Veröffent-
lichung zeigen konnte 22. Im Gegensatz zu Rezeptortyrosinkinasen besitzt
gp130 keine eigene Kinase-Domäne 1. Dafür sind sogenannte Janus-
Kinasen an die zytoplasmatische Domäne von gp130 assoziiert, von denen
vier bekannt sind: Jak1, Jak2, Jak3 und Tyk2. Eine besonders wichtige
Rolle für die gp130-Signaltransduktion kommt dabei Jak1 zu 23, was mit
Hilfe von Jak1-knock-out-Mäusen bestätigt werden konnte 24.
Janus-Kinasen interagieren mit den konservierten Membran-proximalen
Box1/Box2-Regionen der Zytokin-Rezeptoren 25-27. Dabei ist die Prolin-
reiche Box1 wie auch der Bereich zwischen Box1 und Box2 wichtig für die
Rezeptorbindung der Jaks 26,28. Die an hydrophoben Aminosäuren reiche
Box2 erhöht die Affinität der Bindungspartner zueinander 29.
Auf der Grundlage von Sequenzhomologien wurden die Janus-Kinasen in
sieben Jak-Homologie(JH)-Domänen unterteilt 30. Die C-terminal gelegene
JH1-Domäne ist eine Kinase-Domäne. Die hierzu homologe JH2-Domäne
wird wegen fehlender enzymatischer Aktivität als kinase-like-Domäne
bezeichnet und scheint an der Regulation der Kinase-Aktivität beteiligt zu
sein 31,32.
Es konnte gezeigt werden, dass N-terminal eine four-point-one, ezrin,
radixin, moesin (FERM) – Domäne liegt. Diese Domäne ist etwa 400
Aminosäuren lang und beinhaltet 19 hydrophobe Regionen 33. Sie setzt

6
sich aus drei Subdomänen, die gemeinsam eine Kleeblattform bilden,
zusammen: der Subdomäne F1, wobei die Aminosäuren 36 bis 112 eine
sogenannte Ubiquitin-like ß-grasp Domäne bilden, welche wahrscheinlich
mit ihrem vierten loop an der Rezeptorassoziation beteiligt ist 34, der
Subdomäne F2 mit einer Sekundärstruktur ähnlich dem acyl-coenzyme-A-
binding-protein, und der Subdomäne F3 mit einer Faltung wie phospho-
tyrosine-binding (PTB) oder pleckstrin-homology-Domänen 35.
Abbildung 1.2.2: Schematische Darstellung einer Jak 35.
Nachdem der initiale Schritt der IL-6-Signaltransduktion eine Aktivierung
der Janus-Kinasen durch gegenseitige Phosphorylierung bedingt, phospho-
rylieren diese fünf intrazelluläre Tyrosin-Reste (Y759, Y767, Y814, Y905
und Y915) der signaltransduzierenden Rezeptoruntereinheit gp130. Die
einzelnen Tyrosine scheinen unterschiedliche Signale im Zellinneren zu
generieren und für verschiedene Aufgaben zuständig zu sein 36. Dem
Membran-proximalen Y759 kommt für die Regulation eine besondere
Bedeutung zu (siehe 1.2.4.1 und 1.2.4.2) 37,38.
An die phosphorylierten Tyrosin-Reste können nun Proteine, die eine
passende SH2-Domäne aufweisen, binden 6,39,40. Proteine mit SH2-
Domänen sind unter anderem die STAT-Faktoren, die SOCS (suppressors
of cytokine signaling) feedback-Inhibitoren und die Tyrosinphosphatasen
SHPs (SH2 containing protein-tyrosine phosphatases).
STAT steht als Abkürzung für „signal transducer and activator of trans-
cription„ und beschreibt die Funktion dieser Transkriptionsfaktoren als
Bindeglied in der Zytokin-Signaltransduktion. Sie können mit ihrer SH2-
Domäne an Phosphotyrosin-Reste von gp130 binden. Dabei bindet STAT3
spezifisch an die Phosphotyrosin-Reste Y767 und Y814. Die Phospho-
tyrosin-Reste Y905 und Y915 können sowohl von STAT3, als auch von
STAT1 gebunden werden 40-42. Nach Phosphorylierung von Y701 im

7
STAT1 bzw. Y705 im STAT3 vermittelt die STAT coiled-coil-Domäne die
Bildung von Homo- oder Heterodimeren 43. Diese translozieren in den
Nukleus, wo sie mittels ihrer DNA-bindenden Domäne an spezifische
Basensequenzen der Doppelhelix (Enhancer) binden und über ihre Trans-
aktivierungsdomäne eine vermehrte oder verminderte Transkription IL-6-
induzierbarer Gene bewirken 6,44,45.
Abbildung 1.2.3: Schematische Darstellung eines STAT-Faktors 35.
1.2.4 Regulation der IL-6-Signaltransduktion
Der Jak/STAT-Signaltransduktionsweg wird durch eine Reihe von Faktoren
reguliert:
1.2.4.1 Die Tyrosinphosphatase SHP2
Die SHP2 ist ein ubiquitär exprimiertes Protein, welches bei der
Signaltransduktion von Zytokinen und Wachtumsfaktoren eine wichtige
Rolle spielt. Die SHP2 hat eine Größe von 65 kDa, besteht aus 585 Amino-
säuren und weist strukturell zwei N-terminal gelegene SH2-Domänen,
sowie eine, von Position 273 bis 510 reichende, Phospho-Tyrosin-
Phosphatasedomäne (PTP) auf 2,46,47.
Die SH2-Domänen dienen zum einen der Bindung an Proteine mit
phosphorylierten Tyrosin-Resten. So binden sie an Phosphotyrosine von
Rezeptorkomplexen wie dem gp130 38 oder PDGFR 48,49. Zum anderen
sind die SH2-Domänen an der Regulation der SHP2 beteiligt.
Die SHP2 kann sowohl mit gp130, als auch mit den Januskinasen Jak1 und
Jak2, jedoch nicht mit Jak3, assoziieren 50. Jak1 ist essentiell für die SHP2-
Aktivierung durch gp130 51.

8
Sie dient des weiteren als Adapterprotein zur Aktivierung der Ras / Raf -
Kaskade 47 und kann mit vielen anderen Proteinen wie IRS1 52 und Grb 53,54
interagieren.
Im IL-6-System scheint die SHP2 die Signaltransduktion zu unterdücken 37,
wobei der Assoziation mit dem Y759 der signaltransduzierenden Unter-
einheit gp130 eine besonders wichtige Rolle zukommt 36,37,55,56. Sie soll
unter anderem STAT-Faktoren dephosphorylieren 57. Die SHP1 ist in der
Lage Jak2 zu dephosphorylieren 58.
1.2.4.2 SOCS-Proteine
Eine weitere Familie von Inhibitoren der Signaltransduktion von Zytokinen
sind die suppressors of cytokine signaling (SOCS), Jak-binding protein
(JAB), STAT induced STAT inhibitor (SSI) oder cytokine-inducible SH2-
containing proteins (CIS). Diese Menge an Synonymen resultiert aus der
Tatsache, dass diese Proteine von verschiedenen Forschungsgruppen
relativ zeitgleich entdeckt wurden 59-61.
Bisher sind acht Mitglieder bekannt und kloniert worden (CIS, SOCS1-7).
Allen gemeinsam ist eine N-terminale variable Region, eine zentrale SH2-
Domäne und eine C-terminale, als SOCS-Box bezeichnete Region 62. Wie
schon aus der Nomenklatur hervorgeht, sind die Faktoren durch Zytokine
induzierbar und bilden damit einen negativen feedback-Mechanismus für
die Signaltransduktion von Zytokinen und verschiedenen Wachstums-
faktoren 59,61-63. Sie interagieren sowohl mit den Januskinasen als auch mit
den Rezeptoren 38,64-67.
Dabei hemmen die einzelnen Mitglieder der Familie unterschiedliche
Signaltransduktionswege. Für die Inhibierung des IL-6-Signals scheinen vor
allem SOCS1 und SOCS3 wichtig zu sein.
Die Jak1, Jak3 und Tyk2 werden durch SOCS1 gehemmt 60,61,65,67. Dem
Y759 der signaltransduzierenden Untereinheit gp130 wird in der Inhibierung
durch SOCS3 eine essenzielle Rolle zuteil 37,38.
Eine weitere Abschaltmöglichkeit von Signaltransduktionskaskaden wird in
der Markierung von Proteinen für ihre Degradation gesehen. Die SOCS-
Box interagiert mit Elongin B/C und könnte somit im Abbau über das
Proteasom eine Rolle spielen 68,69.

9
1.2.4.3 PIAS-Proteine
Im Gegensatz zu den bisher genannten Inhibitoren weisen die protein
inhibitors of activated STAT (PIAS) keine SH2-Domänen auf. Bisher
wurden fünf Homologe entdeckt, wobei die Interaktion zwischen PIAS und
STAT-Faktoren hochspezifisch ist und über den N-terminalen Bereich von
PIAS vermittelt wird 70. PIAS binden an phosphorylierte STAT-Faktoren:
PIAS1 nur an STAT1, PIAS3 nur an STAT3 71,72. Der genaue Inhibitions-
mechanismus ist noch nicht geklärt.
1.2.4.4 Rezeptorvermittelte-Endozytose
Ein weiterer Mechanismus das IL-6-Signals zu beenden ist die Rezeptor-
vermittelte Endozytose, die wahrscheinlich durch Clathrin-beschichtete
Vesikel erfolgt 73,74.

10
1.3 Die Ras / Raf / MAPK-Kaskade
Nach der IL-6-vermittelten Dimerisierung der IL-6-Rezeptor-Untereinheit
gp130 bindet die 65kDa große Protein-Tyrosin-Phosphatase SHP2 mit
einer SH2-Domäne an den Phosphotyrosin-Rest 759. Nach Phosphory-
lierung der SHP2 kann Grb2 (growth factor receptor bound protein)
binden 75. Grb2 ist über SH3-Domänen mit dem Guaninnucleotid-
Austausch-Faktor SOS (son of sevenless) verbunden 76. Dieser Komplex
ist in der Lage, GDP-Ras in die aktive Form GTP-Ras zu überführen 77,78.
Dieser Schritt leitet einerseits die Aktivierung der Ras/Raf/MAPK-Kaskade
ein, andererseits scheint GTP-Ras in vielen Zelllinien die PI3-Kinase
aktivieren zu können 79-82. Für HepG2-Zellen ist dieser Weg noch nicht
nachgewiesen.
Durch die Serin-/Threoninphosphorylierung von Raf wird diese Kinase
aktiviert und bewirkt durch Phosphorylierung von Erks, aus der Familie der
MAPKs, eine Aktivierung von verschiedenen Transkriptionsfaktoren (c-jun,
c-fos, c-myc, NF-IL-6), was in einer vermehrten Transkription Ras-induzier-
barer Gene resultiert 2,46,47,83,84.

11
1.4 Forkhead box o (foxo)
Transkriptionsfaktoren
1.4.1 Biologische Funktion und Nomenklatur
Die Familie der forkhead box o (foxo)-Tanskriptionsfaktoren umfasst
verschiedene sogenannte winged-helix-Proteine. Dieser Name ergibt sich
aus der Tertiärstruktur, da die Faktoren mit gewundenen Helices an die
DNA binden.
DNA -Bindungsdomäne Transaktivierungsdomäne
T
C
SSNES
N
Abbildung 1.4.1: Schematische Darstellung eines forkhead-Transkriptionsfaktors
Die einzelnen Mitglieder weisen untereinander und mit ihrem Orthologen
DAF-16 von Caenorhabditis elegans, einem Helminthen aus dem Stamm
der Nematoden, Homologien von 85% in der DNA-Bindungsdomäne auf.
Beim Menschen wurden drei dieser Familie zugehörige Faktoren identi-
fiziert. Sie spielen in der Entwicklung von Neoplasien eine Rolle:
forkhead related transcription factor (FKHR, entspricht foxo1A), FKHR-like1
(FKHRL1, entspricht foxo3A) und ALL1-fused-gene-from-chromosome-X
(AFX, entspricht foxo4).
Allen gemeinsam ist ein konservierter N-Terminus, welcher von der DNA-
Bindungsdomäne gefolgt wird. C-terminal findet sich die Transaktivierungs-
domäne (Abbildung 1.4.1). Des weiteren verfügen alle über drei phosphory-
lierbare Reste, denen bei der Regulation eine essentielle Bedeutung
zukommt. Es handelt sich um einen N-terminal gelegenen Threonin-Rest
sowie zwei Serin-Reste, wobei einer in der DNA-Bindungsdomäne, der

12
andere in der Transaktivierungsdomäne nahe bei einem nuclear export
signal (NES) (Abbildung 1.4.1).
Bei der Entstehung vom alveolären Typ des Rhabdomyosarkoms sowie
beim kindlichen Ewing-Sarkom konnten Fusionsproteine aus der Trans-
aktivierungsdomäne von FKHR mit anderen Transkriptionsfaktoren wie
PAX3 und PAX7 beobachtet werden 85-88. Auch andere maligne Neoplasien
wie bestimmte Leukämieformen lassen sich auf Defekte von Mitgliedern der
Familie zurückführen 89.
In dephosphoryliertem Zustand sind die forkhead-Faktoren im Zellkern
lokalisiert und als Transkriptionsfaktoren aktiv. Dort binden sie an Promo-
toren verschiedener Zielgene und bewirken eine vermehrte Expression.
Sowohl nach Überexpression von AFX 90, als auch von FKHR 91 und
FKHRL1 92 konnte eine Wachstumshemmung durch erhöhte p27Kip-
Konzentrationen nachgewiesen werden. Als weiteres Zellzyklus-suppri-
mierendes Protein ließ sich das proapoptotische Bim aus der Familie der
Bcl2-Proteine bei Anwesenheit von FKHRL1 verstärkt exprimieren 93. Die
Transkription von Cyclinen des D-Typs wird durch FKHR unterdrückt 94.
Auch Schlüsselenzyme im Glukosestoffwechsel unterliegen der Kontrolle
der forkhead-Transkriptionsfaktoren. So binden die foxo-Familienmitglieder
an die insulin response unit / element (IRU /IRE), die in dem Promotor-
bereich der Gene der Glukose-6-Phosphatase 95 sowie der Phosphoenol-
pyruvat-Carboxykinase und des insulin-like growth factor binding protein 1
(IGFBP1) 96 zu finden sind. Dabei ist für DAF-16 eine direkte Interaktion mit
Transkriptionskoaktivatoren nachgewiesen worden. Das Orthologe von
Caenorhabditis elegans ist in der Lage, p300/CREB-binding-protein (CBP)
sowie steroid receptor coactivator (SRC) an den IGFBP-1-Promotor zu
rekrutieren. Dafür ist das C-terminale Ende von DAF-16 essentiell. Es
interagiert mit der sogenannten KIX-Domäne und der E1A-bindenden
Domäne von CBP 97.

13
1.4.2 Regulation der foxo-Familienmitglieder
Das Heterodimer der PI3-Kinase besteht aus einer in fünf Isoformen
vorkommenden regulatorischen und einer in zwei Isoformen ubiquitär
exprimierten katalytischen Untereinheit von 110kDa 98. Die regulatorischen
Untereinheiten kann man nach ihren Molekulargewichten in drei Gruppen
einteilen: p50, p55, p85 98. Sie enthalten eine SH3- und zwei SH2-Domä-
nen 99.
Ein virales Onkogen des ASV-16-Virus bildet eine konstitutiv aktive Form
der katalytischen Untereinheit der PI3-K und führt in kultivierten Hühner-
embryoblasten zu erhöhten Phosphatidylinositol-3,4-diphosphat (PI3,4P2)-,
Phosphatidylinositol-3,4,5-triphosphat (PI3,4,5P3)-Spiegeln und aktiviertem
Akt 100.
Verschiedene Tyrosinkinase-Rezeptoren dienen als Signaltransduktoren,
um ein extrazelluläres Signal in ein intrazelluläres umzusetzen. Ein von
vielen Wachstumsfaktoren wie EGF, NGF, HGF, Insulin, aber auch GM-
CSF, Interleukin-3, und -5 101,102,103,104 gemeinsam genutzter Weg führt zur
Aktivierung der heterodimeren Phosphatidylinositol-3-Kinase (PI3-K), die
die Substrate Phosphatidylinositol-4-phosphat (PI4P) sowie Phosphatidyl-
inositol-4,5-diphosphat (PI4,5P2) an der dritten Position phosphoryliert und
so Phosphatidylinositol-3,4-diphosphat (PI3,4P2) und Phosphatidylinositol-
3,4,5-triphosphat (PI3,4,5P3) bildet 101,102,103,104 (Abbildung1.4.2).
Die Aktivierung der PI3-Kinase kann auf verschiedenen Wegen erfolgen.
Einerseits besteht die Möglichkeit, durch direkte Tyrosinphosphorylierung
der katalytischen Untereinheit durch Rezeptortyrosinkinasen eine Akti-
vierung zu erzielen 99 105. Andererseits können Adapterproteine wie Insulin-
Rezeptor-Substrat (IRS) zwischengeschaltet sein 106. Auch existiert ein
Weg in Form einer Phosphorylierung der katalytischen Untereinheit bei
Aktivierung von Ras 82. Über diese Wege sind unter anderem verschiedene
Wachstumsfaktoren in der Lage, die PI3-Kinase zu aktivieren.
Ein funktioneller Antagonist der PI3-K ist die Phosphatase PTEN. Sie
katalysiert die Abspaltung des Phosphatrestes an der dritten Position der
Phosphatidylinositole und hat als Tumorsuppressor eine wichtige Funktion.
So konnte gezeigt werden, dass in PTEN-defizienten Mäusen stark er-

14
niedrigte p27Kip-Spiegel vorliegen, und es zu multipler neoplastischer
Tumorbildung kommt 91,107-110.
Die in der Zellmembran verankerten und phosphorylierten Phosphoinositol-
Lipide rekrutieren die in drei Isoformen vorhandene Proteinkinase B (PKB) /
Akt mittels ihrer Plekstrin-homologen Domäne an die Plasmamembran, wo
die Kinase an T308 und S473 phosphoryliert und damit aktiviert wird 111,112
(Abbildung 1.4.2). Diesen Schritt katalysiert für das Threonin die so-
genannte Phosphatidylinositol-(3,4,5)-Phosphate dependent Kinase 1
(PDK1), für das Serin die kürzlich entdeckte PDK2 113,114.
Eine weitere, von PDKs aktivierte Gruppe von Kinasen sind die serum-and-
glucocorticoid-inducible-kinases (SGKs), welche nach Stimulation mit
Glukokortikoiden und Wachstumsfaktoren vermehrt gebildet werden. Auch
sie werden durch eine Plekstrin-homologe Domäne an die Plasmamembran
rekrutiert. Wie bei PKB / Akt sind zwei Phosphorylierungsstellen für ihre
Aktivierung wichtig. Die erste, T256, wird ebenfalls von der PDK1 phospho-
ryliert, für die zweite, S422, konnte die entsprechende Kinase bislang nicht
identifiziert werden 115.
Sowohl die PKB / Akt als auch die SGKs translozieren nach ihrer Akti-
vierung in den Nukleus und fungieren als Serin-/ Threonin-Kinasen. Sie
phosphorylieren die Mitglieder der foxo-Familie an den beschriebenen
Resten. Dabei weist PKB / Akt eine höhere Spezifität für die zweite Stelle
(bei FKHR: S256), SGKs für die dritte (bei FKHR: S315) auf. Das erste
Threonin (bei FKHR: T32) wird von beiden mit gleicher Spezifität phospho-
ryliert 116.
Die PKB / Akt phosphoryliert neben den foxo-Transkriptionfaktoren noch
weitere Proteine wie Glycogen-Synthase-Kinase-3 (GSK-3) und Bad 117,118.
Phosphorylierte foxo-Transkriptionsfaktoren sind inaktiv und werden aus
dem Nukleus transportiert. Im Zytosol werden sie an 14-3-3-Proteine
gebunden. Diese stellen eine große Gruppe konservierter regulatorischer
Proteine dar, die in der Lage sind, verschiedene Proteine wie Enzyme,
Rezeptoren oder Transkriptionsfaktoren zu binden. 14-3-3-Proteine kom-
men in allen eukaryontischen Zellen vor und erfüllen unterschiedliche
Aufgaben. Dephosphorylierte foxo-Transkriptionsfaktoren werden von den
14-3-3-Proteinen nicht gebunden. Für die Bindung an diese Proteine sind
die ersten beiden Phosphorylierungsstellen wichtig, die dritte, welche in der

15
Nähe des nuclear export signal (NES) liegt, scheint für den nukleären
Export verantwortlich zu sein 116.
Mutiert man die beschriebenen Reste zu nicht phosphorylierbaren
Alaninen, so sind die foxo-Familienmitglieder fast ausschließlich im Zellkern
lokalisiert und konstitutiv aktiv. Eine Bindung an 14-3-3-Proteine ist nicht
möglich.
Die Transkriptionsfaktoren können zusätzlich jedoch auf einem bisher
unbekannten weiteren Weg inaktiviert werden, wie Studien mit dem PI3-K-
Inhibitor LY294002 und Wortmannin zeigen. So lässt sich durch Zugabe
von LY294002 zu dem normalen Kulturmedium auch die Aktivität von
mutiertem, nicht phosphorylierungsfähigem, DAF-16 weiter steigern 119.
PTEN
PI3-Kinase-Signaltransduktion
PI3 K
\/\/\
\/\/
PI(4,5)P2PI(3,4,5)P3
PKB / Akt
FKHR
LY 294002
Wortmannin
14 – 3 - 3
FKHRP
P
P
T24
S256
S319
FKHRP
P
P
T24
S256
S319
SGK
PDKs
WachstumsfaktorenIL-3, IL-5und andere Mediatoren
IGFBP
PEPCK
G6P-ase
P27
bim
Glukokortikoide
Abbildung 1.4.2: Schematische Darstellung der Regulation und Funktion von FKHR.
Die einzelnen Proteine, Inhibitoren und durch FKHR regulierten Genprodukte werden im Text unter 1.4.2 näher beschrieben. Aktive Proteine sind in grüner, inaktive oder inhibierende Proteine oder Substanzen in roter Farbe dargestellt.

16
1.5 Zielsetzung der Arbeit
In Folge verschiedener Prozesse, wie einer partiellen Hepatektomie,
Infekten, Traumen oder auch chemischen Noxen, setzen regenerative
Vorgänge in der Leber ein. Durch Proliferation ist die menschliche Leber in
der Lage, einen Substanzdefekt von bis zu 80% wieder auszugleichen 120.
Dafür verantwortlich sind vor allem erhöhte growth hormone-(GH)-Spiegel.
Man findet nach Leberresektionen einen Anstieg der Plasmakonzentration
von IL-6, TNF, EGF, IGF und HGF 121-125. Es kommt neben einer Zunahme
der Mitoserate jedoch auch zu einer Abnahme der Sekretion von IL-6-
induzierbaren Proteinen, wie den Akut-Phase-Proteinen (APP). Es konnte
gezeigt werden, dass die APP-Produktion von Hepatozyten nach Stimu-
lation mit Wachstumsfaktoren vermindert ist 126-129.
Eine mögliche Erklärung hierfür ist eine Hemmung der IL-6-Signaltrans-
duktion nach Induktion von SOCS3 durch verschiedene Wachstums-
faktoren und Zytokine 63,64,130. Neben den Wachstumsfaktoren ist jedoch
auch Cortisol in der Lage, die APP-Synthese negativ zu beeinflussen 12.
Welche intrazellulären Vorgänge für die beschriebenen Phänomene
Verantwortung tragen, wurde bisher jedoch nicht hinreichend geklärt.
Zielsetzung dieser Arbeit war es deshalb zu untersuchen, welche Mecha-
nismen der verminderten APP-Produktion unter Stimulation mit Wachs-
tumsfaktoren und Cortisol zu Grunde liegen.

17
2 Material und Methoden
2.1 Verwendete Materialien
2.1.1 Chemikalien und Enzyme
Alle Chemikalien wurden in p. a. -Qualität eingesetzt und von den Firmen
AGS (Heidelberg), BioRad (München), Roche (Mannheim), Fluka (Neu-
Ulm), Merck (Darmstadt), New England Biolabs (Schwalbach), Amersham
(Freiburg), Qiagen (Hilden), Sigma (Deisenhofen) und Whatman
(Maidstone, England) bezogen. In der Methodenbeschreibung wird näher
auf die verwendeten Chemikalien eingegangen. Alle Enzyme wurden, falls
im Text nicht anders angegeben, von der Firma Roche bezogen.
2.1.2 Puffer und Medien
Alle Puffer und Medien wurden in wässriger Lösung angesetzt.
Die Zusammensetzung der verwendeten Puffer und Medien wird in der
Methodenbeschreibung näher erläutert.
2.1.3 Verbrauchsmaterialien
Die eingesetzten Verbrauchsmaterialien stammten von den Firmen DuPont
(Dreireich), Eppendorf (Hamburg), Millipore (Eschborn), Sartorius
(Göttingen), Serva (Heidelberg), Sigma (Deisenhofen) und Whatman
(Maidstone, England).

18
2.2 Stimulanzien und Inhibitoren
IL-6 rekombinantes humanes IL-6 wurde nach der Methode von
Arcone et al. 131 hergestellt und freundlicherweise von A.
Küster zur Verfügung gestellt; die spezifische Aktivität be-
trug 2 x 106 BSF2 (B-cell stimulatory factor-2) U/mg Pro-
tein.
sIL-6Rα rekombinanter humaner sIL-6Rα wurde nach der Methode
von Weiergräber et al. 132 von A. Küster produziert.
Insulin rekombinantes Insulin wurde von der Firma Roche
(Penzberg) bezogen und in 1 µM Konzentration eingesetzt.
TGFß R&D Systems (Wiesbaden) lieferte TGFß. 10 Einheiten /
ml wurden dem Zellkulturmedium zugesetzt.
IFN γ Interferon γ lieferte das Unternehmen PeproTech
(Montreal, Quebec). 1000 Einheiten / ml wurden dem Zell-
kulturmedium zugesetzt.
Epo Erythropoetin wurde von J. Burg und K.-H. Sellinger (Firma
Roche, Penzberg) zur Verfügung gestellt. Bei der Stimu-
lation wurden 5 Einheiten / ml verwendet.
EGF epidermal growth factor wurde von M. Kortylewski
(Aachen) zur Verfügung gestellt und in einer Konzentration
von 50 ng/ml dem Medium zugesetzt.
HGF hepatocyte growth factor wurde von W. Birchmeier (Berlin)
zur Verfügung gestellt und in einer Konzentration von 3300
Einheiten / ml Medium eingesetzt.
NGF nerve groth factor wurde von Boehringer (Ingelheim am
Rhein) zur Verfügung gestellt. Bei der Stimulation kamen
50 ng/ml Medium zum Einsatz.
Dexamethason wurde von der Firma Merck (Darmstadt) bezogen und in 2
µM Konzentration verwendet.

19
Wortmannin Das von dem Unternehmen Calbiochem (San Diego,
Californien) gelieferte Wortmannin kam als 1 µM Lösung
zum Einsatz.
LY294002 Ebenfalls von Calbiochem (San Diego, Kalifornien)
bezogenes LY294002 wurde in 50 µM Konzentration ver-
wendet.
Zellkulturmaterialien:
DMEM; Flüssigmedium mit 4,5 g Glukose und
Glutamax Gibco, Eggenstein
DMEM/NUT-MIX-F12 Flüssigmedium Gibco, Eggenstein
Penicillin/Streptomycin Gibco, Eggenstein
Trypsin / EDTA-Lösung:
0,05 % Trypsin / 0,02 % EDTA Biochrom, Berlin
FCS: mykoplasmenfreies fetal-calf-serum Cytogen, Berlin
2.3 Antikörper
polyklonales anti-STAT3 Kaninchenserum
(C20, Santa Cruz Biotechnology, Kalifornien)
monoklonales anti-STAT3 IgG1 der Maus
(610189, Transduction Laboratory, NJ, USA)
polyklonales anti-FKHR Kaninchenserum
(H128 Santa Cruz Biotechnology, Kalifornien)
polyklonales anti-FKHR Kaninchenserum
(wurde von A. Barthel durch GST-FKHR gereinigt)
TRITC-konjugierter Schwein-anti-Kaninchen-Antikörper
(R0156, Dako, Glostrup, Dänemark)
FITC-konjugierter Ziege-anti-Maus-Antikörper
(F0479, Dako, Glostrup, Dänemark)

20
2.4 Plasmide
pSVL eukaryontischer Expressionsvektor, enthält den
späten SV40 Promoter, AmpR (Pharmacia, Freiburg)
pCAGGS eukaryontischer Expressionsvektor, enthält den
Promoter des cytomegalo-Virus, AmpR (zur Ver-
fügung gestellt von K. Nakajima und T. Hirano,
Osaka in Japan)
pRC/CMV eukaryontischer Expressionsvektor, enthält den
Promoter des cytomegalo-Virus, AmpR (Pharmacia,
Freiburg)
pCDNA3.1 eukaryontischer Expressionsvektor, enthält den PCMV
Promoter, AmpR (Invitrogen, Carlsbad, Kalifornien)
pEF-GFP-C1 eukaryontischer Expressionsvektor, enthält den PCMV
Promoter, KanR und die kodierende Sequenz für das
green fluorescent protein (GFP) (Clontech, Palo Alto)
pEF-YFP eukaryontischer Expressionsvektor, enthält den PCMV
Promoter, AmpR und die kodierende Sequenz für das
yellow fluorescent protein (YFP) (zur Verfügung ge-
stellt von U. Sommer und S. Haan, RWTH-Aachen)
pSVL-STAT3 enthält die murine STAT3-cDNA (zur Verfügung
gestellt von J. Sasse, RWTH-Aachen)
pCAGGS-STAT1 enthält die kodierende Sequenz der humanen
STAT1-cDNA (zur Verfügung gestellt von T. Hirano,
Osaka in Japan)
pECE-STAT5b trägt die für STAT5b kodierende cDNA, enthält den
späten SV40 Promoter, AmpR

21
pSVL∆EcoRI-Epo-gp130
enthält die Erbinformation einer Rezeptorchimäre,
welche extrazellulär aus einem Teil des Erythro-
poetinrezeptors, transmembranär und intrazellulär
aus Box1/Box2 des gp130 und einer STAT5 Bin-
dungsstelle besteht. AmpR. (kloniert von Petra May
und Claudia Gerhartz) 133
pCAGGS-STAT3D enthält eine mutierte murine STAT3-cDNA, die für
ein Protein kodiert, das nicht in der Lage ist, an die
DNA zu binden (zur Verfügung gestellt von K.
Nakajima und T. Hirano, Osaka in Japan)
pCAGGS-STAT3F enthält eine mutierte murine STAT3-cDNA, die für
ein Protein kodiert, das nicht in der Lage ist, zu di-
merisieren (zur Verfügung gestellt von K. Nakajima
und T. Hirano, Osaka in Japan)
pCDNA-FKHR enthält die kodierende Sequenz des humanen FKHR
(kloniert von A. Barthel, RWTH-Aachen)
pEF-GFP-FKHR enthält 5’ vom GFP die kodierende Sequenz von
FKHR (kloniert von A. Barthel, RWTH-Aachen)
pEF-GFP-FKHR 1A enthält die kodierende Sequenz des humanen
FKHR, wobei T24 gegen Alanin ausgetauscht wurde
(kloniert von A. Barthel, RWTH-Aachen)
pEF-GFP-FKHR 2A enthält die kodierende Sequenz des humanen
FKHR, wobei T24 und S256 gegen Alanin ausge-
tauscht wurden (kloniert von A. Barthel, RWTH-
Aachen)
pEF-GFP-FKHR 3A enthält die kodierende Sequenz des humanen
FKHR, wobei T24, S256 und S319 gegen Alanin
ausgetauscht wurden (kloniert von A. Barthel,
RWTH-Aachen)
pEF-YFP-FKHR enthält 5’ vom YFP die kodierende Sequenz von
FKHR (wurde im Rahmen dieser Dissertation klo-
niert)

22
pCMV-PIAS3 enthält die kodierende Sequenz von PIAS3 (zur
Verfügung gestellt von K. Shuai, K.C.L.A., Los
Angeles)
pGl3α2M-215Luc enthält den Promotorbereich –215 bis +8 vom α2-
Macroglobulin-Gen fusioniert mit der kodierenden
Sequenz der Luziferase
pGl3hACT-359Luc enthält den Promotorbereich –359 bis +25 vom
humanen α1-Anitchymotrypsin-Gen fusioniert mit
der kodierenden Sequenz der Luziferase
pGL3-IRE-tk-Luc STAT1-responsives Element des IRF-1-Promotors
mit nachgeschaltetem Luziferase-Gen, enthält den
späten SV40-Promotor, AmpR (kloniert von P. May)
pGL3-ß-Casein-tk-Luc STAT5-Bindungselement des ß-Casein-Promotors
mit nachgeschaltetem Luziferase-Gen, enthält den
späten SV40-Promotor, AmpR (kloniert von P. May)
pUC19-SIE-tk-Luc drei identische, STAT1 und STAT3 responsive
Elemente in Serie mit nachgeschaltetem Luziferase-
Gen, AmpR (zur Verfügung gestellt von H. Gascan,
Angers, Frankreich)
pCH110 Expressionsvektor für die β-Galaktosidase
(Pharmacia, Freiburg)

23
2.5 Eukaryontische Zellen und deren
Kultivierung
Es wurde ausschließlich mit adhärent wachsenden humanen Heptomzellen
HepG2 (ATCC, CRL 1651) gearbeitet.
Die Zellen wurden bei 37°C in einer wassergesättigten Atmosphäre mit 5%
CO2 kultiviert. Die Medien enthielten, falls nicht anders angegeben, 10%
fötales Kälberserum, 60 mg/ml Penicillin und 100 mg/ml Streptomycin. Zur
Weiterkultivierung wurden die adhärent wachsenden HepG2-Zellen auf
einer konfluent bewachsenen Petrischale zweimal mit PBS gewaschen und
mit Trypsin/EDTA abgelöst. Die Zellen wurden in einer 1:2- bis 1:20-
Verdünnung in neuem Medium aufgenommen.
Für die Langzeitaufbewahrung wurden die von der Platte gelösten Zellen
mit 10% DMSO-haltigem Medium versetzt und in Kryoröhrchen aliquotiert.
Zur langsamen Abkühlung der Zellen erfolgte eine Lagerung für 24 h bei
-80°C, bevor die Zellen zur längeren Aufbewahrung in flüssigen Stickstoff
gestellt wurden. Zum schnellen Auftauen wurden die Zellen bei 37°C
inkubiert und das DMSO-haltige Medium mit 10 ml kaltem Medium ver-
dünnt. Die Zellen wurden bei 950 Upm bei 4°C abzentrifugiert, in neuem
Medium aufgenommen und in einer Petrischale ausgesät.
Die Kultivierung der von den permanenten Zelllinien abgeleiteten tran-
sienten Transfektanten erfolgte in gleicher Weise.
PBS:
0,2 M NaCl
2,5 mM KCl
8 mM Na2HPO4
1,5 mM KH2PO4
pH 7,4

24
2.6 Prokaryontische Zellen und deren
Kultivierung
Zur Transformation mit rekombinanten Plasmiden wurde der E. coli-Stamm
JM-83 verwendet. Die mit den verschiedenen Plasmiden transformierten
Bakterien wurden in LB-Medium unter Zugabe von Ampicillin: 100 µg/ml
bzw. Kanamycin: 40 µg/ml oder Neomycin 0,6 mg/ml entsprechend der
plasmidvermittelten Antibiotikaresistenz bei 37°C in einem Schüttelkolben
(bei 200 Upm) kultiviert. Die Stammhaltung erfolgte in Form eines Gefrier-
vorrates in LB-Medium mit 20% Glycerin bei -80°C.
LB-Medium:
1 0 g/l NaCl
1 0 g/l Bactotrypton (Difco)
5 g/l Yeast Extract (Difco)
stationäre Kultur:
1 5 g/l Agar (in LB-Medium)
2.7 Herstellung kompetenter Bakterien
500 ml LB-Medium wurden mit Bakterien aus einer 50 ml Übernachtkultur
zu einer OD600 von 0,02 angeimpft und unter Schütteln bei 37°C inkubiert.
Nach Erreichen einer OD600 von 0,3 musste die Kultur in einem
Eis/Wasserbad schnell abgekühlt, die Bakterien bei 4°C abzentrifugiert
(6000 Upm, 10 min) und das Sediment in 125 ml einer eiskalten 50 mM
CaCl2-Lösung resuspendiert werden. Nach erneutem Zentrifugieren wurde
das Sediment in 25 ml 50 mM CaCl2/10%-Glyzerin-Lösung aufgenommen
und in Aliquots von je 500µl in flüssigem Stickstoff schockgefroren und bei
-80°C gelagert. Auf diese Weise vorbehandelte Zellen konnten über
mehrere Monate zur Transformation verwendet werden.

25
2.8 Präparation, Modifikation und Analyse
von Plasmid-DNA
2.8.1 Transformation von Bakterien
10 µl eines Ligationsansatzes oder 10 ng einer Plasmid-DNA wurden mit
100 µl einer Suspension kompetenter Bakterien für 10 min auf Eis inkubiert
und danach für 90 sec auf 42°C erwärmt. Nach kurzer Abkühlung auf Eis
und Zugabe von 800 µl LB-Medium erfolgte eine Inkubation der Bakterien
für 1 h bei 37°C. Die Zellen wurden durch Zentrifugation (2 min bei 10000
Upm) sedimentiert, das Medium bis auf 100 µl abgenommen und die
Bakterien auf Agarplatten, denen das entsprechende Antibiotikum zu-
gesetzt wurde, ausgestrichen. Nach einer Inkubation bei 37°C für 12-16
Stunden konnten die gewachsenen Kolonien gezählt und analysiert wer-
den.
2.8.2 Plasmid-Präparationen
Plasmid-Präparationen erfolgten nach den Herstellerangaben der Firma
QIAGEN (Hilden).
2.8.3 Quantitative Bestimmung von
Nukleinsäuren
Die quantitative Bestimmung der DNA erfolgte photometrisch in einem
UV/Vis-Spektrophotometer (Pharmacia, Freiburg) über die optical density
OD260. Eine Extinktion E260 = 1,0 entspricht 50 µg doppelsträngiger (ds)
DNA bzw. 20 µg einzelsträngiger (ss) DNA. Die Reinheit der Nukleinsäure-
präparationen wurde mit Hilfe des OD260 / OD280 Quotienten ermittelt.
Dieser sollte bei 1,8-2,0 liegen 134.

26
2.8.4 Spaltung von DNA mit
Restriktionsendonukleasen
Die Restriktion von Plasmid-DNA erfolgte nach Standardvorschrift 134. Die
Restriktionsbedingungen wurden entsprechend den Empfehlungen der
Hersteller gewählt. In der Regel betrug die Inkubationszeit 1-3 h. Die
vollständige Restriktion der DNA wurde mit Hilfe analytischer Agarosegele
überprüft.
2.8.5 Dephosphorylierung linearisierter DNA
Um die Wahrscheinlichkeit der Religation eines linearisierten Plasmids zu
verringern und so die Effizienz der gewünschten Insertion einer Fremd-
DNA zu erhöhen, wurde die endständige 5’-Phosphatgruppe des line-
arisierten Vektors entfernt. 100-200 ng DNA wurden mit einer Einheit
alkalischer Phosphatase 30 min bei 37°C inkubiert. Zum Entfernen des
Enzyms erfolgte anschließend die Aufreinigung über ein Agarosegel oder
mittels Phenolextraktion.
2.8.6 Phenolextraktion
Der zu reinigende Ansatz wurde mit H2O auf 400 µl aufgefüllt, es wurden
20 µl 2 M Tris-Base und 400 µl Wasser-gesättigtes Phenol zugegeben.
Nach gründlichem Mischen und 2 minütiger Zentrifugation bei 14000 Upm,
wurde die obere wässrige Phase abgenommen und in ein frisches Eppen-
dorfreaktionsgefäß überführt.
Nun erfolgte die Zugabe von je 400 µl Phenol/Chloroform bzw. Chloroform,
und es wurde jeweils wie oben beschrieben verfahren.
Zum Fällen der DNA wurde schließlich die wässrige Lösung mit 2 µl
Glycogen, 10 µl 8 M LiCl und 400 µl Isopropanol versetzt, gemischt und
eine Stunde bei -20°C inkubiert. Nach 10 minütiger Zenrifugation bei 14000
Upm wurde der Überstand verworfen, der Bodensatz zweimal mit 70%igem
Ethanol gewaschen, bei 37°C getrocknet und schließlich in 40 µl H2O
resuspendiert.

27
2.8.7 Elektrophoretische Analyse von Plasmid-
DNA
Nach Verdau mit geeigneten Restriktionsendonukleasen erfolgte die
Analyse der DNA-Fragmente mittels Elektrophorese. Dazu wurde die DNA-
Lösung mit 0,1 Volumen eines 10-fach DNA-Probenpuffers vermischt. Im
Agarosegel aufgetragen, konnten die DNA-Fragmente im elektrischen Feld
nach ihrer Größe aufgetrennt werden. Dieses Gelsystem wurde benutzt,
um DNA-Fragmente von 300-8000 bp aufzutrennen. Die Gelmatrix bestand
aus 1% Agarose in 1xTAE und 0,1 ng/ml Ethidiumbromid. Der Einsatz von
Ethidiumbromid ermöglichte es, die DNA-Banden im UV-Licht sichtbar zu
machen.
10 x DNA Probenpuffer:
2 5 % Ficoll’sche Lösung
0,4 % Bromphenolblau
0,4 % Xylencyanolblau
50 x TAE:
2 M Tris-Base
1 M Eisessig
50 mM EDTA pH 8,0
Laufpuffer:
1 x TAE
2.8.8 Isolierung von DNA-Fragmenten
Die durch den Verdau der Plasmide entstandenen DNA-Fragmente wurden
auf Agarosegelen aufgetrennt, das gewünschte Fragment ausgeschnitten
und mit Hilfe des „Gel-Extraktion-Kit“ der Firma Qiagen (Hilden) isoliert. Die
Konzentration des isolierten Fragments konnte durch Abschätzung der
Helligkeit der Bande auf einem analytischen Agarosegel ermittelt werden.

28
2.8.9 DNA-Ligation
Die Ligation doppelsträngiger DNA-Moleküle erfolgte mit dem Enzym T4-
DNA-Ligase. In der Regel wurden 10 ng des geschnittenen Vektors und ein
5-facher molarer Überschuß an gereinigtem Fragment für den Ligations-
ansatz eingesetzt. Die Inkubationszeit lag bei 14-16 Stunden bei 14°C im
Wasserbad.
2.9 Transiente Transfektion von HepG2-
Zellen
Für Reportergen-Assays wurden HepG2-Zellen transient mit der Calcium-
Phosphat-Methode transfiziert. Dazu wurden 24 h vor der Transfektion die
konfluent bewachsenen Schalen 1 zu 8 bis 1 zu 10 gesplittet. Das
DMEM/NUT-MIX-F12 Medium wurde 30 min vor der Transfektion durch
DMEM ersetzt. Die zur Transfektion verwendete Calcium-Phosphat-DNA-
Lösung setzte sich aus 25 µg DNA, 75 µl 2 M CaCl2 und 600 µl 2 x HBS in
12 ml DMEM zusammen. Die Zellen wurden nach einer Inkubationszeit von
18 h in DMEM/NUT-MIX-F12 weiterkultiviert.
2 x HBS:
1 0 g/l HEPES
1 6 g/l NaCl
0,74 g/l KCl
0,25 g/l NaH2PO4xH2O
2 g/l Glukose
Der pH-Wert der Lösung wurde mit NaOH auf exakt 7,05 eingestellt.

29
2.10 Proteinbestimmung nach Bradford
Zum quantitativen Nachweis gelöster Proteine wurde ein Test der Firma
BioRad (München) verwendet. Das Prinzip der Bestimmung basiert auf der
von Bradford 135 beschriebenen Methode. Für die Kalibrationsgerade bzw.
zur Probenbestimmung wurde eine Verdünnung von 0-12 µg BSA bzw. 5 µl
der Testlösung in 800 µl Wasser und 200 µl der BioRad-Gebrauchslösung
hergestellt. Die Proben wurden gemischt und 5 min bei RT inkubiert. Die
Proteinkonzentration konnte mit Hilfe der OD595-Messung bestimmt werden.
2.11 Herstellung von Zell-Lysaten
Zu lysierende HepG2-Zellen wurden zunächst zweimal mit kaltem
PBS/1mM Vanadat gewaschen und mit einem Gummischaber von der
Platte gelöst. Die Zellen wurden abzentrifugiert (2 min, 1200 Upm) und in
300 µl IP-Puffer (Brij-Lysispuffer: 20 mM Tris/HCl pH 7,4; 150 mM NaCl; 10
mM NaF; 1% Brij’97; 1 mM Na-Orthovanadat; 0,5 mM EDTA; 5 mg/ml
Aprotinin; 1 mg/ml Leupeptin) aufgenommen. Nach einer Inkubation von 30
min auf Eis wurde 1 min geschüttelt und zentrifugiert (5 min, 10000 Upm).
Die Überstände wurden in neue Eppendorf-Reaktionsgefäße überführt.
2.12 Immunpräzipitation
Zelllysate wurden auf gleiche Proteinmengen und Flüssigkeitsvolumina
eingestellt und mit 1 µg Antikörper über Nacht bei 4°C inkubiert. Zur
Immunpräzipitation von STAT3 wurde der polyclonale C20-Antikörper
(Santa Cruz Biotech.) und zur Immunpräzipitation von FKHR FKHR-
Antiserum, welches von A. Barthel zur Verfügung gestellt wurde, verwen-
det. Anschließend wurden die Immunkomplexe mit Hilfe von Protein-A-
Sepharose (Pharmacia) präzipitiert. Nach dreimaligem Waschen wurden
die Immunkomplexe mit Lämmli-Puffer 136 von der Sepharose gelöst, mit
Hilfe einer SDS-Polyacrylamid-Gelelektrophorese der Masse nach auf-
getrennt und anschließend durch Western-blotting auf eine PVDF-Membran

30
übertragen. Kopräzipitierte Proteine konnten durch Immundetektion nach-
gewiesen werden.
2.13 SDS-Polyacrylamid-Gelelektrophorese
Zur Auftrennung von Proteinen wurde, wenn nicht anders angegeben, eine
diskontinuierliche SDS-Polyacrylamidgel-Elektrophorese (7,5% PPA) nach
Laemmli durchgeführt 136.
Die elektrophoretische Auftrennung von Proteinen unter denaturierenden
und reduzierenden Bedingungen erfolgt proportional zur Masse. Mer-
captoethanol bewirkt die Reduktion von Disulfidbrücken zu den ent-
sprechenden Thiolen. Die hier angewendete Methode der diskontinuier-
lichen Elektrophorese nach Laemmli erfolgte in einer vertikalen Flach-
gelkammer von Biometra nach Herstellerangaben.
Trenngel:
7 ml aqua dest.
3,5 ml Acrylamidlösung
3,5 ml 4-fach Trenngelpuffer
1 8 0 µl 20% APS
4,5 µl TEMED
Sammelgel:
4 ml aqua dest.
0,6 ml Acrylamidlösung
1,5 ml 4-fach Sammelgelpuffer
5 0 µl 20% APS
3 µl TEMED
Acrylamidlösung:
3 0 % Acrylamid
0,8 % Bisacrylamid

31
4-fach Trenngelpuffer:
1,5 M Tris/HCl, pH8,8
0,4 % SDS
4-fach Sammelgelpuffer:
0,5 M Tris/HCl, pH6,8
0,4 % SDS
Elektrodenpuffer:
2 5 mM Tris-Base
1 9 2 mM Glycin
0,1 % SDS
Die Elektrophorese erfolgte für 16 h bei konstanter Spannung von 60 V
oder 2 h bei konstanter Stromstärke von 35 mA.
2.14 Proteintransfer auf eine PVDF-
Membran durch das
„Western-Blot-semidry“-Verfahren
Die elektrophoretisch aufgetrennten Proteine wurden zur Detektion auf eine
PVDF-Membran übertragen. Dazu diente das „Western-Blot-semidry“-
Verfahren. Es wurden insgesamt 15 3MM Whatman-Papiere und die
PVDF-Membran genau auf die Gelgröße zugeschnitten. Die PVDF-Memb-
ran musste für 5-7 sec in 100% Methanol getaucht, weitere 5 min gewäs-
sert und dann für 15 min in Anoden-Puffer-II äquilibriert werden. Das Gel
wurde für mindestens 5 min in Kathoden-Puffer äquilibriert. Nach kurzem
Tränken der Whatman-Filter mit den unten angegebenen Puffern erfolgte
der Aufbau des Blots:

32
1. Graphitplatte, bildet Anode
2. 6 Whatman-Filter getränkt mit Anoden-Puffer-I
3. 3 Whatman-Filter getränkt mit Anoden-Puffer-II
4. PVDF-Membran äquilibriert in Anoden-Puffer-II
5. Polyacrylamidgel äquilibriert in Kathoden-Puffer
6. 6 Whatman-Filter getränkt mit Kathoden-Puffer
7. Graphitplatte, bildet Kathode
Die oben aufgeführten Komponenten wurden luftblasenfrei aufeinander-
gelegt. Die Übertragung der Proteine auf die Membran erfolgte bei 0,8
mA/cm2 für 1h bei einem Andruckgewicht von 1 kg.
Anodenpuffer-I:
0,3 M Tris-Base
2 0 % Methanol
Anodenpuffer-II:
0,025 M Tris-Base
2 0 % Methanol
Kathodenpuffer:
0,04 M E-Aminocapronsäure
2 0 % Methanol

33
2.15 Detektion präzipitierter Proteine mit
Hilfe eines Immunblots
Nach dem Transfer der Proteine auf eine PVDF-Membran wurden die
freien Bindungsstellen 10 min mit 10% BSA in TBS-N blockiert, die Memb-
ran zweimal für 15 min mit TBS-N gespült und 1 h bei RT mit einer 1:1000
Verdünnung eines entsprechenden Antikörpers in TBS-N inkubiert. Nach
zwei kurzen Spülschritten und dreimaligem Waschen für 1x15 min und
2x10 min erfolgte die Inkubation mit dem Peroxidase-konjugierten Anti-
körper in einer Verdünnung von 1:1000 in TBS-N für 30 min bei RT. Das
überschüssige Antiserum wurde durch zweimaliges Spülen und erneute
Waschschritte für 2 x 15 min mit TBS-N entfernt. Die Proteine konnten in
einer anschließenden ECL-Reaktion (Amersham, Braunschweig) detektiert
werden.
TBS-N:
2,42 g / l Tris-Base
8 g / l NaCl
3,81 ml / l 1 M HCl, pH 7,6
0,1 %v/ v Nonidet P-40

34
2.16 Reportergen-Versuche
Zur Analyse genregulatorischer Eigenschaften von Proteinen wurden
Reportergenexperimente durchgeführt. Ein Reportergen-Konstrukt enthält
neben einer Promotorsequenz das kodierende Gen für das Reporter-
protein. Humane Hepatomzellen (HepG2) wurden mit dem entsprechenden
Expressionskonstrukt transient transfiziert. Indirekt konnte dann über die
Messung der Reporterenzym-Aktivität die Expressionstärke des Reporter-
gens bestimmt werden. Die Genauigkeit der in Reportergensystemen
gemessenen Aktivitäten wird durch verschiedene Faktoren, wie z.B.
Unterschiede von Zellkulturschale zu Zellkulturschale, Anzahl und Zustand
der kultivierten Zellen, Transfektionseffizienz, Zell-Lyse und Pipettier-
ungenauigkeiten bestimmt. Deshalb wurde neben dem experimentellen
Reportergenplasmid noch ein weiteres Kontrollexpressionskonstrukt, das
eine konstitutive Expression der β-Galaktosidase (pCH110, Phamacia)
bewirkt, kotransfiziert.
2.17 Ernte transfizierter Zellen für
Reportergenuntersuchungen
Mit der Calcium-Phosphat-Methode transifizierte HepG2-Zellen wurden
einen Tag nach der Transfektion für weitere 16 h mit (stimulierte Zellen)
oder ohne (unstimulierte Zellen) Zytokin bzw. Wachstumsfaktoren weiter-
kultiviert. Anschließend wurde das Kulturmedium entfernt, und die Zellen
wurden mit PBS gewaschen und lysiert. Zur Zelllyse wurde der Reporter
Lysis Buffer der Firma Promega (Madison, USA), der für Luziferase- und β-
Galaktosidase-Reporterenzym-Versuche geeignet ist, nach
Herstellervorschriften verwendet.

35
2.17.1 Luziferase-Messung
Die Zell-Lyse der HepG2-Zellen wurde mit einem Kit (Luciferase Assay
System with Reporter Lysis Buffer) der Firma Promega nach modifizierten
Herstellervorschriften durchgeführt. 10 µl der aus der Zell-Lyse erhaltenen
Zellextrakte wurden in einer lichtundurchlässigen 96er-Lochplatte mit 50 µl
Assay Reagent des Kits versetzt, woduch die von der Luziferase kata-
lysierte Reaktion gestartet wurde. Die Luziferase des Leuchtkäfers,
Photinus pyralis (firefly luciferase) oxidiert ihr Substrat Luciferin zu
Oxyluciferin, und bei dieser Reaktion wird Licht emittiert. Normalerweise
verläuft die Reaktion über das Enzymintermediat Luciferyl-AMP und klingt
recht schnell ab. Wird jedoch Coenzym A (CoA) zugegeben, so entsteht
anstatt Luciferyl-AMP Luciferyl-CoA, die Reaktion erreicht nach wenigen
Sekunden ihr Maximum und bleibt über einen Zeitraum von 60 Sekunden
stabil. Mit dem Gerät „Microlumat 96 P“ der Firma EG&G Berthold (Bad
Wildbad) wurde die Lumineszenz gemessen. Die erhaltenen Werte wurden
mit den Werten für die Transfektionseffizienz (β-Galaktosidase-Aktivität)
korrigiert. Aus mindestens dreifach unabhängig voneinander durchge-
führten Versuchen wurden die Mittelwerte und Standartabweichungen
berechnet. Sofern nicht anders angegeben, wurden alle Messwerte und
Standartabweichungen durch den ersten basalen Wert geteilt und dieser so
auf eins normiert.
2.17.2 β-Galaktosidase-Messung
Um die Transfektionseffizienz zu überprüfen, wurde das Kontrollreporter-
plasmid pCH 110 (Pharmacia) stets kotransfiziert. Die β-Galaktosidase-
Aktivität wurde bestimmt, indem 50 µl der Zell-Lysate mit 500 µl β-Galakto-
sidase-Puffer und 100 µl ONPG (1 mg/ml o-Nitrophenylgalactosid), chro-
mogenes Substrat der β-Galaktosidase, bei
37 °C inkubiert wurde. Sobald eine leichte Gelbfärbung der Proben fest-
zustellen war, wurde die Reaktion mit 250 µl 1M Na2CO3 gestoppt und die
OD der Proben bei 420 nm bestimmt.

36
Die β-Galaktosidase-Aktivität errechnet sich dann wie folgt:
Aktivität=(OD420 x 6000) / (Inkubationszeit in min)
β-Galaktosidase-Puffer:
6 0 mM Na2HPO4
4 0 mM NaH2PO4
1 mM KCl
1 mM MgCl2
3 8 6 µl β-Mercaptoethanol pro 100 ml Lösung
2.18 Zellproliferationstest (XTT-Test)
Stabil transfizierte HepG2-Zellen wurden zweimal in PBS gewaschen und
in DMEM/NUT-MIX-F12 aufgenommen. Je 2x104 Zellen in 100 µl Medium
wurden in eine 96er-Lochplatte ausgesät. Zum Stimulieren wurden die
Zellen mit Konzentrationen von 800 bis 0 Einheiten IL-6/ml und sIL-6Rα der
gleichen Konzentration inkubiert. Nach drei Tagen erfolgte die kolori-
metrische Quantifizierung der metabolisch aktiven Zellen mit Hilfe des Cell
Proliferaiton Kit II (XTT) (Boehringer Mannheim, Mannheim). Die Um-
setzung von Tetrazoliumsalz XTT wurde nach 4-12 h Inkubation der
HepG2-Zellen mit dem XTT-Reaktiongemisch spektralphotometrisch bei
450 nm (Referenzwellenlänge 650 nm) im Mikrotiterplatten-Reader gemes-
sen.
2.19 Zellfärbungen für die Immunfluoreszenz
Um endogene Proteine im Mikroskop sichtbar zu machen, wurden die
HepG2-Zellen über Nacht auf 12 mm cover slips kultiviert. Darauf folgte
eine Inkubation in Medium ohne FCS für sechs Stunden. Nach der sich
anschließenden Stimulation von 15 min mit IL-6 und sIL-6Rα bzw. Insulin
wurden die Zellen mit PBS2+ gewaschen und mittels 2% Paraformaldehyd
20 min fixiert. Da intrazelluläre Proteine im Zentrum des Interesses stan-

37
den, musste die Zellmembran permeabilisiert werden, damit die Antikörper
ins Zytosol gelangen konnten. Dieses erfolgte durch eine fünf-minütige
Inkubation in PBS2+T. Nach Einbringen der Zellen für fünf Minuten in 50mM
NH4Cl, folgte ein weiterer Waschschritt und eine 30-minütige Absättigung
der unspezifischen Bindungen durch 1% BSA, welches in PBS2+T gelöst
war. Anschließend gab man für die gleiche Zeit den Primärantikörper, in
PBS2+T 0,1% BSA gelöst und 1:100 verdünnt, hinzu. Zur Anwendung kam
der monoklonale Maus anti-STAT3 Antikörper (610189, Transduction) und
polyklonales anti-FKHR (H-128, Santa Cruz Biotechnology) Kaninchen-
serum. Nach den folgenden drei Waschschritten mit PBS2+T 0,1% BSA
wurde mit dem Sekundärantikörper, in PBS2+T 0,1% BSA gelöst und 1:100
verdünnt, für 30 Minuten inkubiert. Dabei richtete sich ein TRITC-makierter
Antikörper gegen ein Epitop von Kaninchen-Antikörpern (R0156, Dako,
Glostrup, Dänemark) und ein an FITC gebundener Antikörper gegen ein
Epitop von Mausantikörpern (F0479, Dako, Glostrup, Dänemark). Nach
weiteren Waschvorgängen wurden die Cover Slips mit der bewachsenen
Seite auf einem Objektträger mit Moviol fixiert.
2.20 Fluoreszenzmikroskopie
Fluorochrome zeichnen sich insofern aus, als dass man sie mittels eines
engen Wellenlängenspektrums anregen kann, und sie dann langwelligeres
Licht emittieren.
2.20.1 Geräte
Die mikroskopische Untersuchung der Zellpräparate erfolgte an einem
konfokalen Laser Scanning Mikroskop Typ LSM 510 der Firma Carl Zeiss
Mikroskopie, Jena.
Eine spektrometrische Auswertung einzelner Zellbereiche wurde durch ein
direkt mit dem LSM 510 gekoppeltes modulares Dioden-Array-Spektro-
meter Typ MCS 521vis der Firma Carl Zeiss Mikroskopie, Jena, ermöglicht.

38
2.20.2 Bildaufnahme am LSM 510
Durch Anregung im Wellenlängenbereich zwischen 450 bis 490 nm (Filter-
satz ”FITC”) und im Bereich zwischen 540 und 552 nm (Filtersatz ”TRITC”)
mittels einer Quecksilberdampflampe konnten im okularvisuellen Arbeits-
modus Zellen ausgewählt werden, welche die gewünschte Fluoreszenz-
markierung aufwiesen.
Im konfokalen Arbeitsmodus wird die Probe mit Laserlicht angeregt. Es
kamen die Wellenlängen 488 nm (Argon-Laser) und 543 nm (Helium-Neon-
Laser) zum Einsatz. Dabei wurden das Fluoreszeinisothiocyanat (FITC) mit
488 nm und das tetramethyl rhodamine isothiocyanate (TRITC) mit 543 nm
angeregt.
Verschiedene Filterkombinationen ermöglichten die selektive Detektion des
Emissionslichtes der verschiedenen Fluorophore. Für FITC kam der
Bandpassfilter Typ BP 505-530 (Bandbreite 505 bis 530 nm) zum Einsatz.
Die TRITC-Fluoreszenz wurde mit einem Langpassfilter Typ LP 560
(Durchlaß >560 nm) abgenommen. Die konfokalen Lochblenden ließen sich
auf jede Emmissionswellenlänge der Probe einstellen und sicherten somit
einen möglichst hohen Grad an Auflösung und geringer optischer
Schnittdicke.
Die Handhabung des konfokalen Mikroskops erfolgte gemäß den Vor-
schriften im Operating Manual (Release 2.5) der Firma Carl Zeiss.
Für die Bildaufnahmen wurde die Laserleistung des Argon-Lasers im
Bereich zwischen 25% und 55% variiert. Die Laserblende wurde im Bereich
4% bis 90% eingestellt. Die Auflösung betrug entweder 512x512 Bildpunkte
oder 1024x1024 Bildpunkte. Die Tiefenschärfe der Bilder wurde konstant im
12 Bit-Modus belassen. Die Aufnahmegeschwindigkeit wurde in Abhängig-
keit von der Aufnahmeart jeweils einem optimalen Signal zu Rausch-
Verhältnis angepasst.

39
3 Ergebnisse
3.1 Hemmung der IL-6-Signaltransduktion
in Hepatozyten durch
Wachstumsfaktoren
Wie in der Einleitung (siehe 1.5) beschrieben, kommt es bei HepG2-Zellen
unter Stimulation mit Wachstumsfaktoren oder Cortisol zu einer ver-
minderten Produktion IL-6-induzierbarer Proteine.
FCS (fetal calf serum) ist, wie der Name schon sagt, das von fetalen
Kälbern gewonnene Serum. Da in der fetalen Phase das Organwachstum
im Vordergrund steht, werden in dieser Zeit eine Fülle von Wachstums-
faktoren benötigt. Das in den Versuchen verwendete FCS stellt somit ein
nicht genau definiertes Gemisch verschiedenster löslicher, nicht korpus-
kulärer Blutbestandteile da.
Um die Auswirkung von FCS auf die IL-6-Signaltransduktion und damit die
Produktion von Akut-Phase-Proteinen (APP) nachzuvollziehen, wurden
verschiedene Versuchsreihen durchgeführt.
In dem ersten Experiment wurden humane HepG2-Hepatom-Zellen mit
einem Plasmid transfiziert, das den Promotor des Gens für alpha-2-
Makroglobulin, einem APP der Ratte, mit nachgeschaltetem Luziferase-
Gen, enthält. Zur Transfektionskontrolle wurde ein die ß-Galaktosidase
kodierendes Plasmid kotransfiziert. Diese beiden Konstrukte wurden auch,
sofern nicht anders angegeben, in den folgenden Versuchen eingesetzt.

40
0123456789
10 %
FCS
7,5 %
FCS
5 % FCS
2,5 %
FCS
1,25 %
FCS
0 % FCS
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6Rmit IL-6/sIL-6R
Abbildung 3.1.1: Abnehmende Mengen von FCS im Kulturmedium bewirken eine steigende IL-6-induzierbare Luziferase Aktivität.
HepG2-Zellen wurden mit insgesamt 25 µg Plasmid mittels Calciumphosphat-Methode transfiziert. Dabei wurden je 5 µg Plasmid, welches für die β-Galaktosidase kodiert, sowie ein Expressionskonstrukt mit dem alpha-2-Makroglobulin Promotor mit nachgeschaltetem Luziferase-Gen kotransfiziert. Für die verbleibenden 15 µg wurden Leervektoren verwendet. Das Medium, in dem die Transfektion durchgeführt wurde, war DMEM und enthielt 5% FCS. Nach 24 Stunden wurde das Medium gegen DMEM-F12 ausgetauscht und die Zellen für 16 Stunden wie angegeben stimuliert. Die weißen Balken geben die Aktivität ohne, die schwarzen die Aktivität mit IL-6-Stimulus wieder (160 Einheiten IL-6 und 1µg löslicher IL-6 Rezeptor pro ml). Als Abgleich für unterschiedliche Transfektionseffizienzen wurden alle gemessenen Luziferasewerte durch die zugehörigen ß-Galaktosidasewerte geteilt. Alle Versuche wurden unabhängig voneinander mindestens dreifach durchgeführt, der Mittelwert berechnet und die Standardabweichung gebildet. Alle Mittelwerte und Standardabweichungen wurden durch den, in der Reihe ersten basalen Mittelwert geteilt, und dieser so auf eins normiert.
Bei dem Versuch (Abbildung 3.1.1) konnte gezeigt werden, dass mit
sinkender FCS-Konzentration die gemessene Luziferase Aktivität stieg.
Dies gilt vor allem für die Inkubation mit IL-6. Zehnprozentiges FCS im
Medium bewirkt eine Halbierung der Luziferase Aktivität nach Stimulation
im Vergleich zu FCS-freiem Medium. In Abwesenheit von IL-6 betrug die
Abnahme nur etwa 15%. Somit ergibt sich bei Zugabe von FCS eine
deutliche Abnahme der Induzierbarkeit dieses IL-6-responsiven Genes,
was bedeutet, dass FCS hemmend auf die Signaltransduktion des IL-6
wirkt.
Die Tatsache, dass der Effekt bei unstimulierten Zellen gering ist und auch
die ß-Galaktosidaseaktivität nicht signifikant vermindert wurde (Daten nicht
gezeigt), macht eine generelle Hemmung der RNA-, beziehungsweise
Proteinbiosynthese unwahrscheinlich.
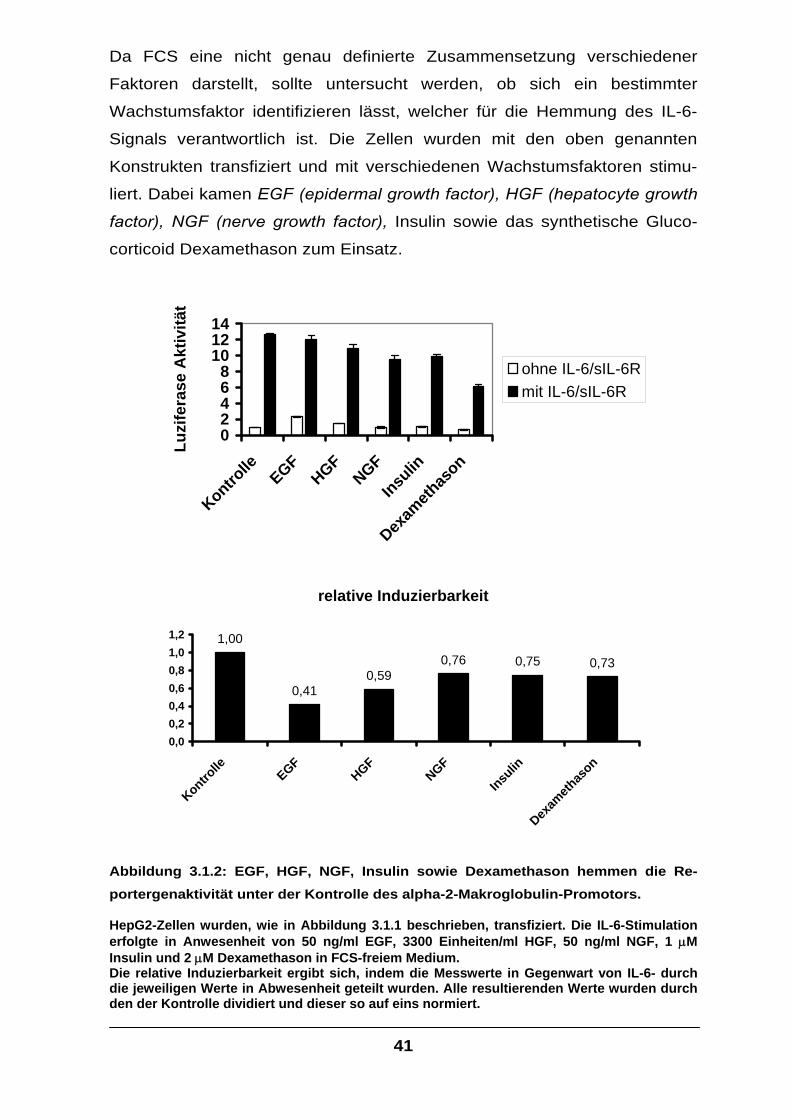
41
Da FCS eine nicht genau definierte Zusammensetzung verschiedener
Faktoren darstellt, sollte untersucht werden, ob sich ein bestimmter
Wachstumsfaktor identifizieren lässt, welcher für die Hemmung des IL-6-
Signals verantwortlich ist. Die Zellen wurden mit den oben genannten
Konstrukten transfiziert und mit verschiedenen Wachstumsfaktoren stimu-
liert. Dabei kamen EGF (epidermal growth factor), HGF (hepatocyte growth
factor), NGF (nerve growth factor), Insulin sowie das synthetische Gluco-
corticoid Dexamethason zum Einsatz.
02468
101214
Kontrolle EGF
HGFNGF
Insulin
Dexam
ethas
on
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6Rmit IL-6/sIL-6R
relative Induzierbarkeit
1,00
0,410,59
0,76 0,75 0,73
0,00,20,40,60,81,01,2
Kontrolle EGF
HGFNGF
Insulin
Dexam
ethas
on
Abbildung 3.1.2: EGF, HGF, NGF, Insulin sowie Dexamethason hemmen die Re-portergenaktivität unter der Kontrolle des alpha-2-Makroglobulin-Promotors.
HepG2-Zellen wurden, wie in Abbildung 3.1.1 beschrieben, transfiziert. Die IL-6-Stimulation erfolgte in Anwesenheit von 50 ng/ml EGF, 3300 Einheiten/ml HGF, 50 ng/ml NGF, 1 µM Insulin und 2 µM Dexamethason in FCS-freiem Medium. Die relative Induzierbarkeit ergibt sich, indem die Messwerte in Gegenwart von IL-6- durch die jeweiligen Werte in Abwesenheit geteilt wurden. Alle resultierenden Werte wurden durch den der Kontrolle dividiert und dieser so auf eins normiert.

42
Wie in Abbildung 3.1.2 gezeigt, stellte sich bei allen getesteten Substanzen
eine leichte Abnahme der IL-6-induzierten Luziferase Aktivität ein. Die
basale Aktivität nahm in Gegenwart der Wachtumsfaktoren deutlich zu.
Stellt man ein Verhältnis zwischen den jeweiligen mit IL-6 stimulierten und
unstimulierten Proben auf, so ergibt sich wie beim vorangegangenen
Versuch (Abbildung 3.1.1) ebenfalls eine Verringerung der Induzierbarkeit
um durchschnittlich etwa 30% (Abbildung 3.1.2).
Für Insulin wurde bereits eine Hemmung der IL-6-Signaltransduktion
beschrieben 127. Insulin, wie auch sein Rezeptor, besitzt große Ähnlichkeit
mit anderen Wachstumsfaktoren beziehungsweise ihren Rezeptoren,
woraus für einige auch der Name insulin-like-growth-factor (IGF) resultiert.
Außerdem handelt es sich bei Insulin um ein anaboles Hormon, welches
die Glukose-Utilisation der Zelle fördert und somit wichtig für Wachstum
und Proliferation ist.
Wie in der Einleitung beschrieben, transduziert Insulin, wie auch viele
andere Wachstumsfaktoren, sein Signal über eine Rezeptortyrosinkinase.
Insulin-Bindung bedingt eine Tyrosin-Phosphorylierung des Rezeptors und
in der Folge eine Aktivierung der PI3-Kinase, welche über Zwischenstufen
zur Phosphorylierung und Aktivierung einer weiteren Kinase, der Protein-
kinaseB führt. Ein synthetischer Inhibitor der PI3-Kinase ist Wortmannin.
Von Interesse war nun, ob die Hemmung der IL-6-Signaltransduktion durch
Insulin durch eine Inhibierung der PI3-Kinase mittels Wortmannin aufzu-
heben ist. Dabei wurden die Zellen analog zu den vorangegangenen
Versuchen transfiziert und entweder gar nicht, nur mit Insulin oder mit
Insulin und Wortmannin stimuliert (Abbildung 3.1.3).

43
0
5
10
15
20
25
Kontrolle Insulin Insulin +Wortmannin
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6R
mit IL-6/sIL-6R
Abbildung 3.1.3: Der inhibitorische Effekt von Insulin auf die IL-6-Signaltransduktion lässt sich mit Hilfe von Wortmannin komplett unterdrücken.
HepG2-Zellen wurden, wie in Abbilung 3.1.1 beschrieben, transfiziert und mit IL-6 und sIL-6R stimuliert. Insulin lag 1 µM und Wortmannin 100 nM im Kulturmedium vor.
Dabei stellte sich bei Insulin-Inkubation die auch aus den Vorversuchen zu
erwartende Minderung der Luziferase Aktivität ein (Abbildung 3.1.2 und
3.1.3). Es konnte weiterhin gezeigt werden, dass aus einer Behandlung mit
Insulin und Wortmannin im Vergleich zu normalem Kulturmedium unter IL-
6-Stimulation sogar eine höhere Luziferase Aktivität resultiert. Die PI3-
Kinase soll in vielen Zelllinien über die Ras/Raf/MAPK-Kaskade ebenfalls
bei einem IL-6-Stimulus aktiviert werden 79-82,137. Da Wortmannin die PI3-
Kinase inhibiert, wäre eine Erklärungsmöglichkeit, dass die PI3-Kinase
direkt oder indirekt für den hemmenden Effekt von Insulin auf die IL-6-
Signaltransduktion verantwortlich sein könnte.
Die PI3-Kinase phosphoryliert Phosphatidylinositole der Zellmembran.
Davon abhängige Kinasen, die phosphatidylinositolphoshate-dependent-
kinases (PDK), spielen in der weiteren Signaltransduktions-Kaskade der
PI3-Kinase eine wesentliche Rolle. Ein Effektorprotein, welches der PDK1
als Substrat dient, ist PKB/Akt. Es wird in phosphorylierter Form aktiv,
transloziert in den Zellkern und phosphoryliert bestimmte Proteine.
In weiteren Versuchen konnte PKB/Akt als eine Kinase, deren Phospho-
rylierung und Aktivierung für die Unterdrückung des IL-6-Signals ausreicht,
identifiziert werden (Daten nicht gezeigt, vgl. 138).

44
Es galt herauszufinden, wie Akt die IL-6-Signaltransduktion zu hemmen
vermag. Insulin und anderen Wachstumsfaktoren ist gemeinsam, dass sie
über eine Aktivierung der PI3-Kinase PKB/Akt phosphorylieren. In Ab-
bildung 3.1.2 wurde gezeigt, dass auch Dexamethason zu einer Unter-
drückung des IL-6-Signals und damit zu einer Synthesehemmung von
APPs befähigt ist. Cortisol wirkt über verschiedene Mechanismen auf
zelluläre Funktionen. Eine Möglichkeit ist die Aktivierung von serum- and
glucocorticoid-inducible kinases (SGK), welche in ihren Kinasedomänen
starke Homologien zu PKB/Akt aufweisen. Für Akt sind als bekannte
Substrate die Glykogen-Synthase-Kinase-3 (GSK3), Bad sowie FKHR zu
nennen. Eines dieser Proteine könnte möglicherweise mit Proteinen der IL-
6-Signaltransduktions-Kaskade interagieren und so die APP-Produktion
beeinflussen.
Ein Protein, welches einer regulatorischen Kontrolle sowohl der Protein-
kinase B, als auch der SGK untersteht 115, ist der forkhead transcription
factor (FKHR). So konzentrierte sich die Aufmerksamkeit zunächst auf
dieses Protein, ein Familienmitglied der forkhead box, class O (foxo)
Transkriptionsfaktoren.
3.2 Nachweis einer funktionellen Interaktion
zwischen STAT-Faktoren und FKHR
3.2.1 Der Transkriptionsfaktor FKHR verstärkt
STAT3-abhängige Reportergenaktivität
Um herauszufinden, ob ein Zusammenhang zwischen der beobachteten
PKB/Akt-Phosphorylierung und der Abnahme des IL-6-Signals auf eine
Interaktion von STAT3 mit FKHR zurückzuführen ist, wurde eine Versuchs-
reihe mit Konstrukten des Wildtyps dieses Transkriptionsfaktors durch-
geführt.
Dabei wurden neben den für die Reportergene kodierenden Vektoren
steigende Mengen von FKHR-cDNA transfiziert. Falls die Signaltrans-
duktionshemmung des IL-6 durch aktiviertes Akt auf eine Phosphorylierung
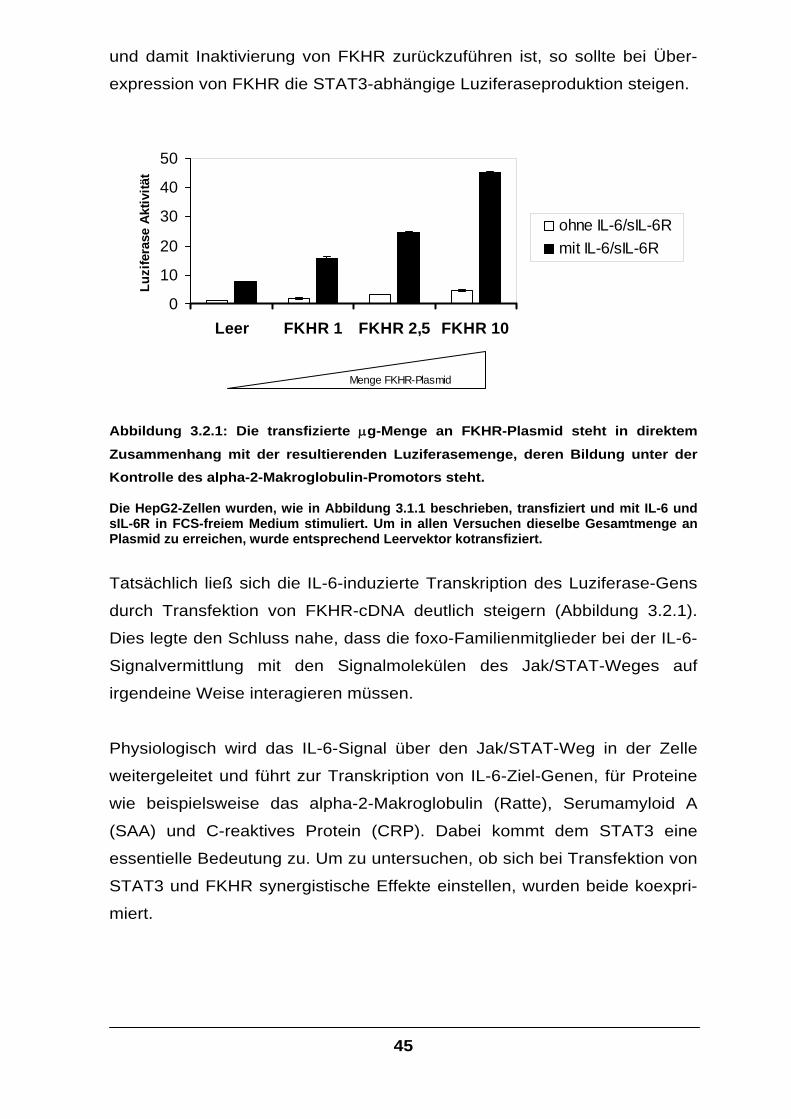
45
und damit Inaktivierung von FKHR zurückzuführen ist, so sollte bei Über-
expression von FKHR die STAT3-abhängige Luziferaseproduktion steigen.
0
10
20
30
40
50
Leer FKHR 1 FKHR 2,5 FKHR 10
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6Rmit IL-6/sIL-6R
Menge FKHR-Plasmid
Abbildung 3.2.1: Die transfizierte µg-Menge an FKHR-Plasmid steht in direktem
Zusammenhang mit der resultierenden Luziferasemenge, deren Bildung unter der Kontrolle des alpha-2-Makroglobulin-Promotors steht.
Die HepG2-Zellen wurden, wie in Abbildung 3.1.1 beschrieben, transfiziert und mit IL-6 und sIL-6R in FCS-freiem Medium stimuliert. Um in allen Versuchen dieselbe Gesamtmenge an Plasmid zu erreichen, wurde entsprechend Leervektor kotransfiziert.
Tatsächlich ließ sich die IL-6-induzierte Transkription des Luziferase-Gens
durch Transfektion von FKHR-cDNA deutlich steigern (Abbildung 3.2.1).
Dies legte den Schluss nahe, dass die foxo-Familienmitglieder bei der IL-6-
Signalvermittlung mit den Signalmolekülen des Jak/STAT-Weges auf
irgendeine Weise interagieren müssen.
Physiologisch wird das IL-6-Signal über den Jak/STAT-Weg in der Zelle
weitergeleitet und führt zur Transkription von IL-6-Ziel-Genen, für Proteine
wie beispielsweise das alpha-2-Makroglobulin (Ratte), Serumamyloid A
(SAA) und C-reaktives Protein (CRP). Dabei kommt dem STAT3 eine
essentielle Bedeutung zu. Um zu untersuchen, ob sich bei Transfektion von
STAT3 und FKHR synergistische Effekte einstellen, wurden beide koexpri-
miert.
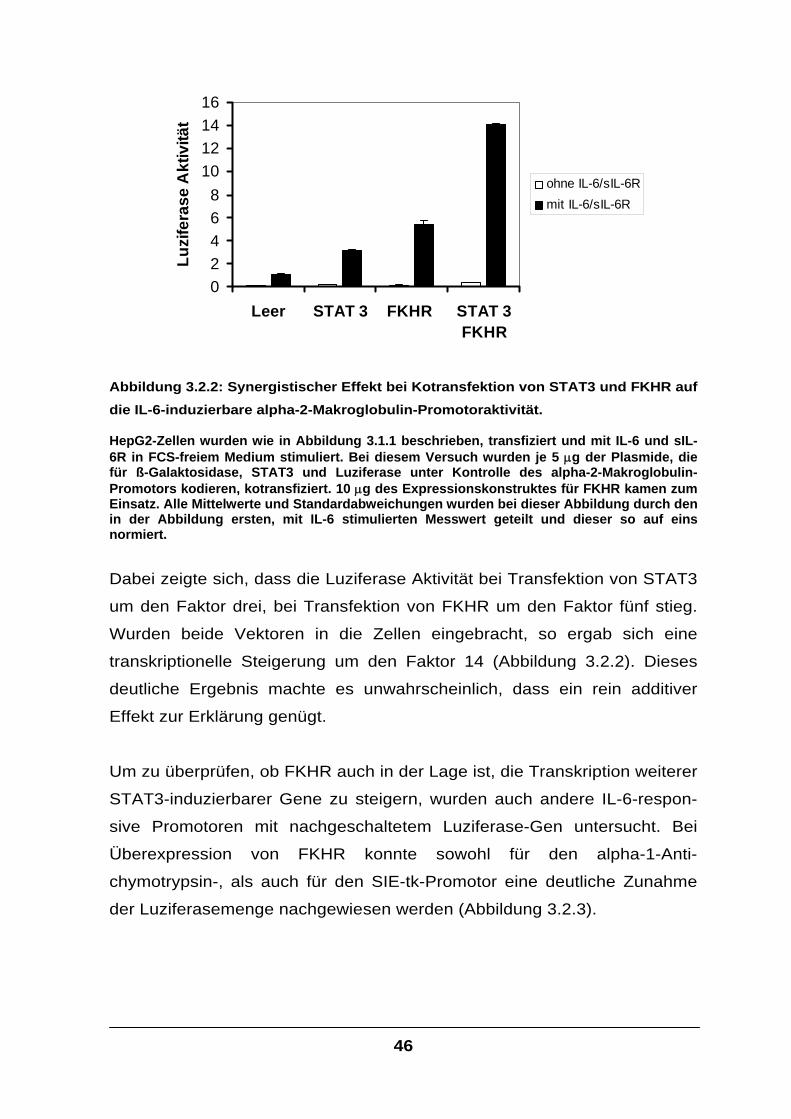
46
02468
10121416
Leer STAT 3 FKHR STAT 3FKHR
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6Rmit IL-6/sIL-6R
Abbildung 3.2.2: Synergistischer Effekt bei Kotransfektion von STAT3 und FKHR auf die IL-6-induzierbare alpha-2-Makroglobulin-Promotoraktivität.
HepG2-Zellen wurden wie in Abbildung 3.1.1 beschrieben, transfiziert und mit IL-6 und sIL-6R in FCS-freiem Medium stimuliert. Bei diesem Versuch wurden je 5 µg der Plasmide, die für ß-Galaktosidase, STAT3 und Luziferase unter Kontrolle des alpha-2-Makroglobulin-Promotors kodieren, kotransfiziert. 10 µg des Expressionskonstruktes für FKHR kamen zum Einsatz. Alle Mittelwerte und Standardabweichungen wurden bei dieser Abbildung durch den in der Abbildung ersten, mit IL-6 stimulierten Messwert geteilt und dieser so auf eins normiert.
Dabei zeigte sich, dass die Luziferase Aktivität bei Transfektion von STAT3
um den Faktor drei, bei Transfektion von FKHR um den Faktor fünf stieg.
Wurden beide Vektoren in die Zellen eingebracht, so ergab sich eine
transkriptionelle Steigerung um den Faktor 14 (Abbildung 3.2.2). Dieses
deutliche Ergebnis machte es unwahrscheinlich, dass ein rein additiver
Effekt zur Erklärung genügt.
Um zu überprüfen, ob FKHR auch in der Lage ist, die Transkription weiterer
STAT3-induzierbarer Gene zu steigern, wurden auch andere IL-6-respon-
sive Promotoren mit nachgeschaltetem Luziferase-Gen untersucht. Bei
Überexpression von FKHR konnte sowohl für den alpha-1-Anti-
chymotrypsin-, als auch für den SIE-tk-Promotor eine deutliche Zunahme
der Luziferasemenge nachgewiesen werden (Abbildung 3.2.3).
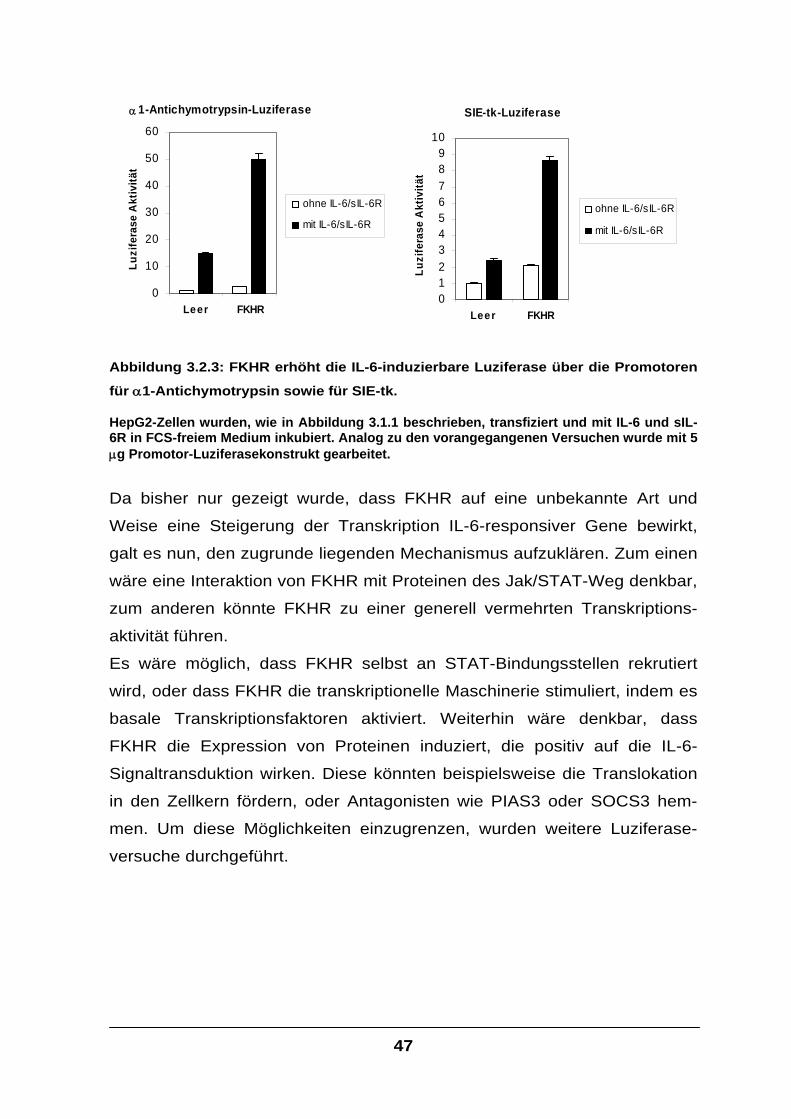
47
Abbildung 3.2.3: FKHR erhöht die IL-6-induzierbare Luziferase über die Promotoren
für α1-Antichymotrypsin sowie für SIE-tk.
HepG2-Zellen wurden, wie in Abbildung 3.1.1 beschrieben, transfiziert und mit IL-6 und sIL-6R in FCS-freiem Medium inkubiert. Analog zu den vorangegangenen Versuchen wurde mit 5 µg Promotor-Luziferasekonstrukt gearbeitet.
Da bisher nur gezeigt wurde, dass FKHR auf eine unbekannte Art und
Weise eine Steigerung der Transkription IL-6-responsiver Gene bewirkt,
galt es nun, den zugrunde liegenden Mechanismus aufzuklären. Zum einen
wäre eine Interaktion von FKHR mit Proteinen des Jak/STAT-Weg denkbar,
zum anderen könnte FKHR zu einer generell vermehrten Transkriptions-
aktivität führen.
Es wäre möglich, dass FKHR selbst an STAT-Bindungsstellen rekrutiert
wird, oder dass FKHR die transkriptionelle Maschinerie stimuliert, indem es
basale Transkriptionsfaktoren aktiviert. Weiterhin wäre denkbar, dass
FKHR die Expression von Proteinen induziert, die positiv auf die IL-6-
Signaltransduktion wirken. Diese könnten beispielsweise die Translokation
in den Zellkern fördern, oder Antagonisten wie PIAS3 oder SOCS3 hem-
men. Um diese Möglichkeiten einzugrenzen, wurden weitere Luziferase-
versuche durchgeführt.
SIE-tk-Luziferase
0123456789
10
Leer FKHR
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6R
mit IL-6/sIL-6R
α 1-Antichymotrypsin-Luziferase
0
10
20
30
40
50
60
Leer FKHR
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6R
mit IL-6/sIL-6R
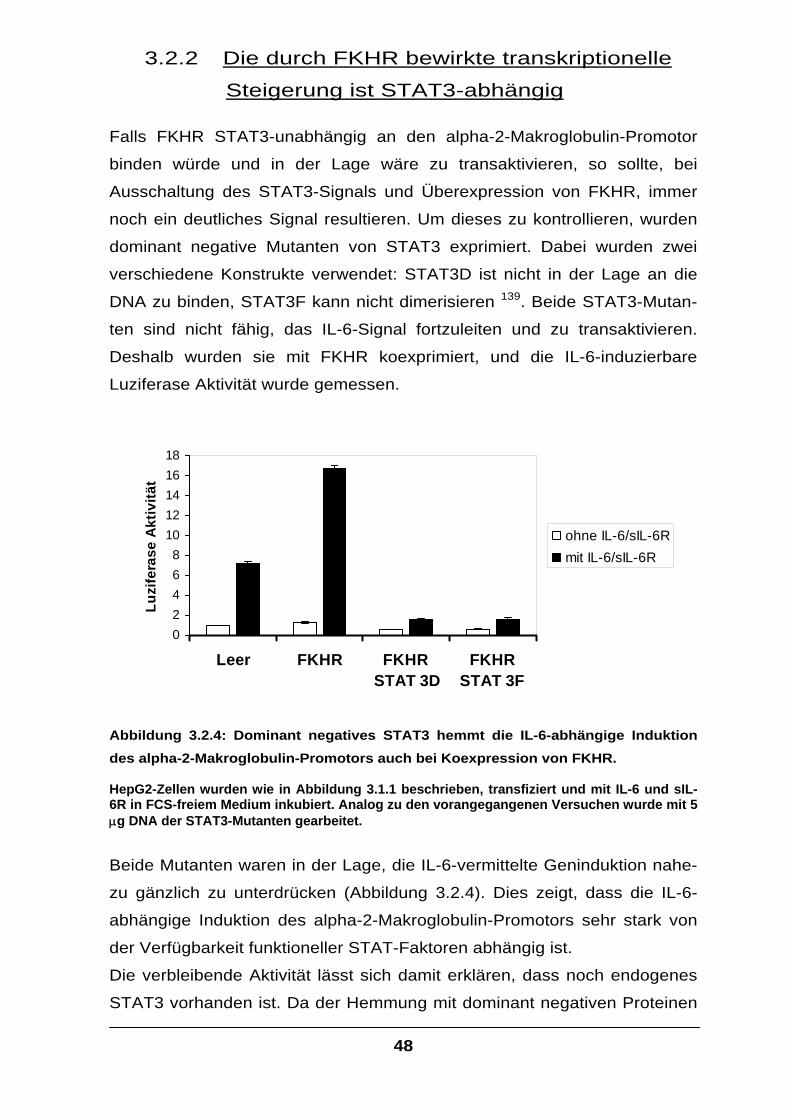
48
3.2.2 Die durch FKHR bewirkte transkriptionelle
Steigerung ist STAT3-abhängig
Falls FKHR STAT3-unabhängig an den alpha-2-Makroglobulin-Promotor
binden würde und in der Lage wäre zu transaktivieren, so sollte, bei
Ausschaltung des STAT3-Signals und Überexpression von FKHR, immer
noch ein deutliches Signal resultieren. Um dieses zu kontrollieren, wurden
dominant negative Mutanten von STAT3 exprimiert. Dabei wurden zwei
verschiedene Konstrukte verwendet: STAT3D ist nicht in der Lage an die
DNA zu binden, STAT3F kann nicht dimerisieren 139. Beide STAT3-Mutan-
ten sind nicht fähig, das IL-6-Signal fortzuleiten und zu transaktivieren.
Deshalb wurden sie mit FKHR koexprimiert, und die IL-6-induzierbare
Luziferase Aktivität wurde gemessen.
02468
1012141618
Leer FKHR FKHR STAT 3D
FKHR STAT 3F
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6Rmit IL-6/sIL-6R
Abbildung 3.2.4: Dominant negatives STAT3 hemmt die IL-6-abhängige Induktion des alpha-2-Makroglobulin-Promotors auch bei Koexpression von FKHR.
HepG2-Zellen wurden wie in Abbildung 3.1.1 beschrieben, transfiziert und mit IL-6 und sIL-6R in FCS-freiem Medium inkubiert. Analog zu den vorangegangenen Versuchen wurde mit 5 µg DNA der STAT3-Mutanten gearbeitet.
Beide Mutanten waren in der Lage, die IL-6-vermittelte Geninduktion nahe-
zu gänzlich zu unterdrücken (Abbildung 3.2.4). Dies zeigt, dass die IL-6-
abhängige Induktion des alpha-2-Makroglobulin-Promotors sehr stark von
der Verfügbarkeit funktioneller STAT-Faktoren abhängig ist.
Die verbleibende Aktivität lässt sich damit erklären, dass noch endogenes
STAT3 vorhanden ist. Da der Hemmung mit dominant negativen Proteinen
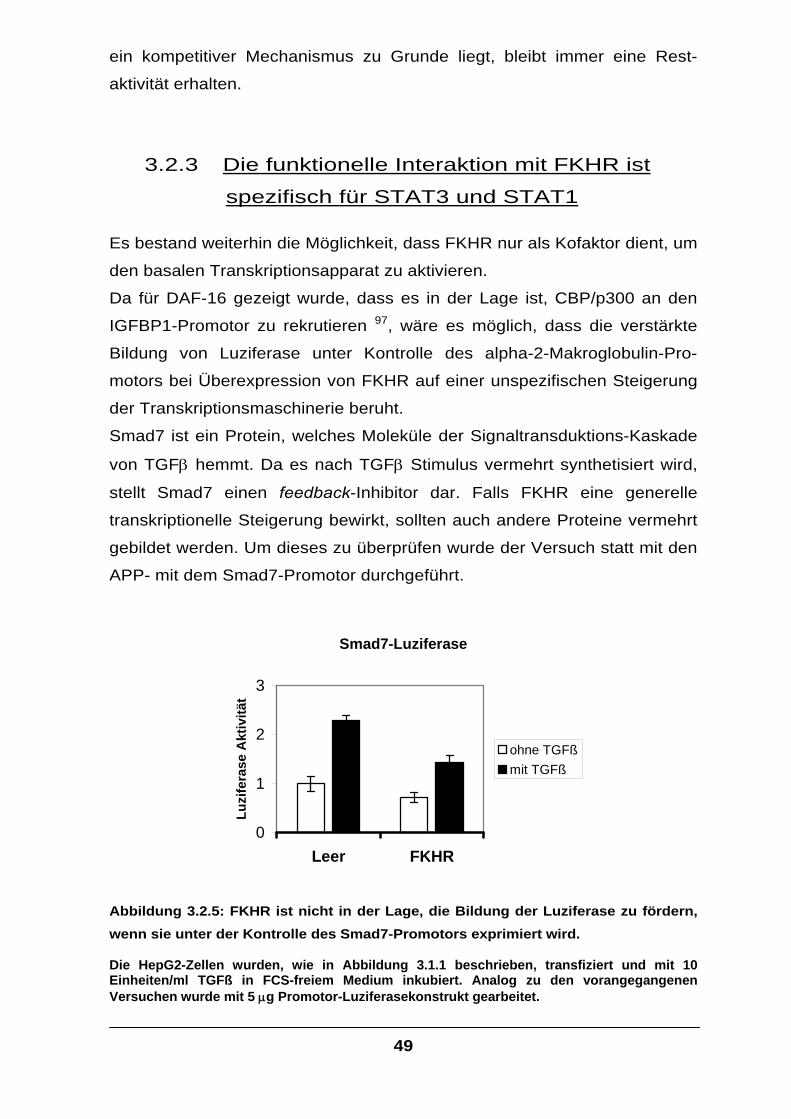
49
ein kompetitiver Mechanismus zu Grunde liegt, bleibt immer eine Rest-
aktivität erhalten.
3.2.3 Die funktionelle Interaktion mit FKHR ist
spezifisch für STAT3 und STAT1
Es bestand weiterhin die Möglichkeit, dass FKHR nur als Kofaktor dient, um
den basalen Transkriptionsapparat zu aktivieren.
Da für DAF-16 gezeigt wurde, dass es in der Lage ist, CBP/p300 an den
IGFBP1-Promotor zu rekrutieren 97, wäre es möglich, dass die verstärkte
Bildung von Luziferase unter Kontrolle des alpha-2-Makroglobulin-Pro-
motors bei Überexpression von FKHR auf einer unspezifischen Steigerung
der Transkriptionsmaschinerie beruht.
Smad7 ist ein Protein, welches Moleküle der Signaltransduktions-Kaskade
von TGFβ hemmt. Da es nach TGFβ Stimulus vermehrt synthetisiert wird,
stellt Smad7 einen feedback-Inhibitor dar. Falls FKHR eine generelle
transkriptionelle Steigerung bewirkt, sollten auch andere Proteine vermehrt
gebildet werden. Um dieses zu überprüfen wurde der Versuch statt mit den
APP- mit dem Smad7-Promotor durchgeführt.
Smad7-Luziferase
0
1
2
3
Leer FKHR
Luzi
fera
se A
ktiv
ität
ohne TGFßmit TGFß
Abbildung 3.2.5: FKHR ist nicht in der Lage, die Bildung der Luziferase zu fördern, wenn sie unter der Kontrolle des Smad7-Promotors exprimiert wird.
Die HepG2-Zellen wurden, wie in Abbildung 3.1.1 beschrieben, transfiziert und mit 10 Einheiten/ml TGFß in FCS-freiem Medium inkubiert. Analog zu den vorangegangenen Versuchen wurde mit 5 µg Promotor-Luziferasekonstrukt gearbeitet.
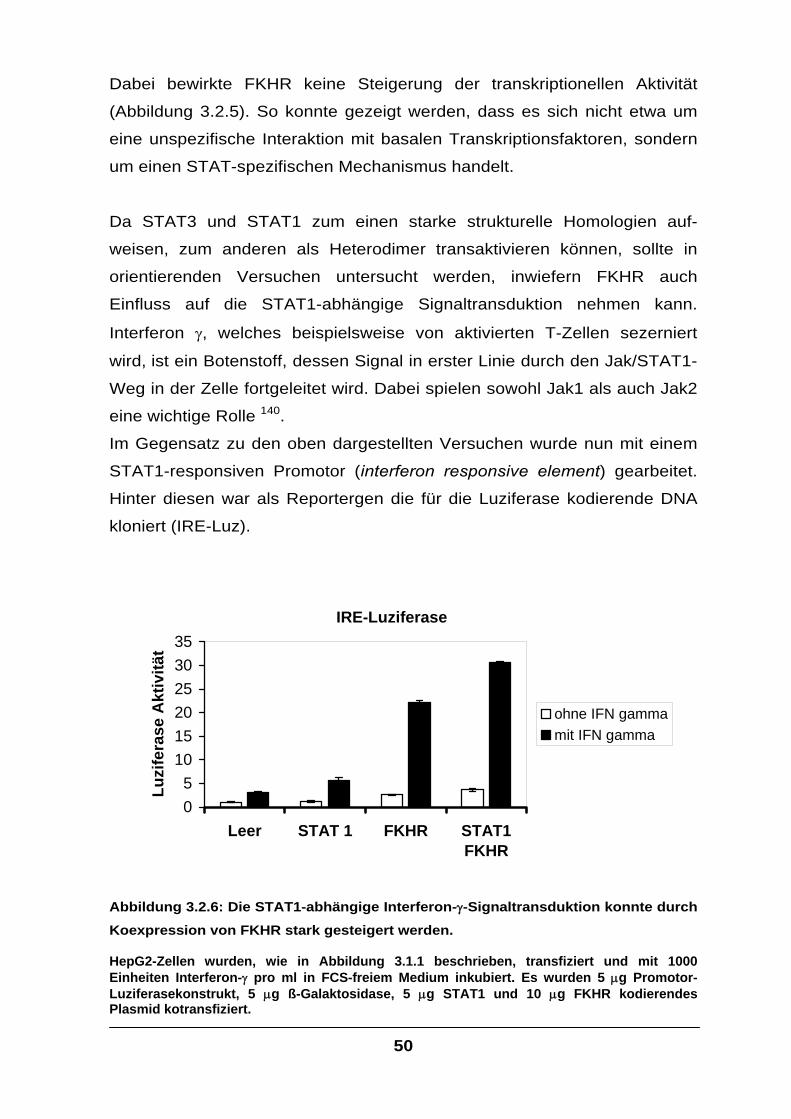
50
Dabei bewirkte FKHR keine Steigerung der transkriptionellen Aktivität
(Abbildung 3.2.5). So konnte gezeigt werden, dass es sich nicht etwa um
eine unspezifische Interaktion mit basalen Transkriptionsfaktoren, sondern
um einen STAT-spezifischen Mechanismus handelt.
Da STAT3 und STAT1 zum einen starke strukturelle Homologien auf-
weisen, zum anderen als Heterodimer transaktivieren können, sollte in
orientierenden Versuchen untersucht werden, inwiefern FKHR auch
Einfluss auf die STAT1-abhängige Signaltransduktion nehmen kann.
Interferon γ, welches beispielsweise von aktivierten T-Zellen sezerniert
wird, ist ein Botenstoff, dessen Signal in erster Linie durch den Jak/STAT1-
Weg in der Zelle fortgeleitet wird. Dabei spielen sowohl Jak1 als auch Jak2
eine wichtige Rolle 140.
Im Gegensatz zu den oben dargestellten Versuchen wurde nun mit einem
STAT1-responsiven Promotor (interferon responsive element) gearbeitet.
Hinter diesen war als Reportergen die für die Luziferase kodierende DNA
kloniert (IRE-Luz).
IRE-Luziferase
05
101520253035
Leer STAT 1 FKHR STAT1FKHR
Luzi
fera
se A
ktiv
ität
ohne IFN gammamit IFN gamma
Abbildung 3.2.6: Die STAT1-abhängige Interferon-γ-Signaltransduktion konnte durch
Koexpression von FKHR stark gesteigert werden.
HepG2-Zellen wurden, wie in Abbildung 3.1.1 beschrieben, transfiziert und mit 1000 Einheiten Interferon-γ pro ml in FCS-freiem Medium inkubiert. Es wurden 5 µg Promotor-Luziferasekonstrukt, 5 µg ß-Galaktosidase, 5 µg STAT1 und 10 µg FKHR kodierendes Plasmid kotransfiziert.

51
Nach Transfektion von FKHR war ein deutlicher Anstieg der Luziferase zu
verzeichnen (Abbildung 3.2.6). Im Verhältnis zu den Kontrolltransfektionen
mit Leervektoren, wurde unstimuliert etwa zweieinhalb mal mehr, nach
IFNγ-Gabe das achtfache an Luziferase gebildet.
Wurden STAT1 und FKHR kotransfiziert, war die Luziferasebildung in
unstimuliertem Zustand um den Faktor drei angestiegen im Vergleich zu
nur mit Leervektor transfizierten Zellen. Nach Stimulation war eine zehn-
fach höhere Luziferase Aktivität zu messen.
So zeigte sich, ähnlich zu den Ergebnissen mit STAT3 nach IL-6-Stimu-
lation (Abbildung 3.2.2), ein additiver Effekt der Transkriptionsfaktoren
FKHR und STAT1 nach IFNγ-Stimulation.
Es sollte im folgenden untersucht werden, ob auch weitere STAT-Faktoren
wie STAT5 mit FKHR interagieren, und FKHR auch in der Lage ist, die
vermehrte Transkription STAT5-responsiver Gene zu bewirken.
Prolaktin, ein Peptidhormon, bindet extrazellulär an seinen Rezeptor.
Daraufhin kommt es zur Rezeptordimerisierung, Aktivierung von Jak2 und
einer Phosphorylierung von Tyrosin-Resten des Rezeptors. An diese lagert
sich STAT5 an, dimerisiert, transloziert in den Zellkern und aktiviert STAT5-
responsive Gene wie beispielsweise das β–Casein-Gen 141-144.
In Analogie zu den vorangegangenen Versuchen kam nun ein Konstrukt
zum Einsatz, bei dem das Reportergen der Luziferase unter den Einfluss
des β–Casein-Promotors gestellt war.
HepG2-Zellen exprimieren nur sehr wenig Prolaktinrezeptoren.
Um die Bedingungen zu den vorangegangenen Versuchen möglichst
konstant zu halten und HepG2-Zellen verwenden zu können, wurde
deshalb zusätzlich mit einem Expressionskonstrukt gearbeitet, welches für
eine Rezeptorchimäre codiert, die extrazellulär aus dem Erythropoetin-
Rezeptor, transmembranär und intrazellulär aus einem Teil des gp130 mit
einer zusätzlichen STAT5 Bindungsstelle besteht.
Nach Stimulation mit Erythropoetin resultiert so intrazellulär eine STAT5
Aktivierung, wie sonst nach Prolaktinstimulation.
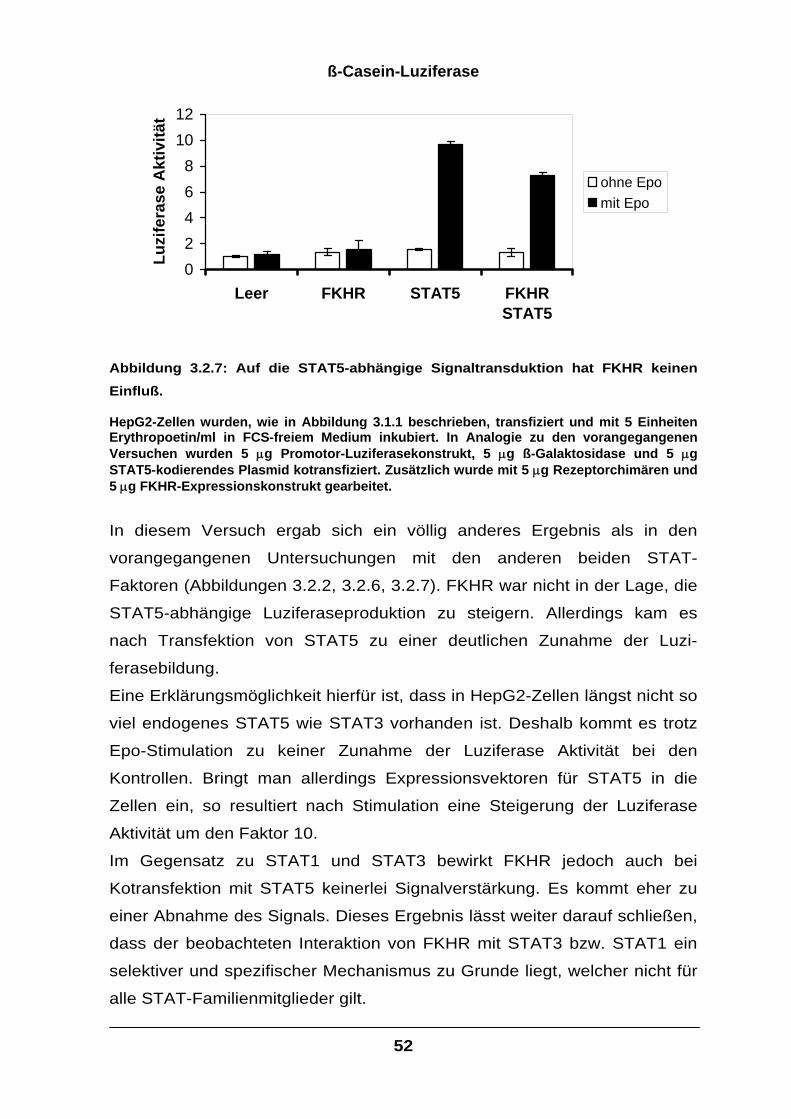
52
ß-Casein-Luziferase
02468
1012
Leer FKHR STAT5 FKHR STAT5
Luzi
fera
se A
ktiv
ität
ohne Epomit Epo
Abbildung 3.2.7: Auf die STAT5-abhängige Signaltransduktion hat FKHR keinen Einfluß.
HepG2-Zellen wurden, wie in Abbildung 3.1.1 beschrieben, transfiziert und mit 5 Einheiten Erythropoetin/ml in FCS-freiem Medium inkubiert. In Analogie zu den vorangegangenen Versuchen wurden 5 µg Promotor-Luziferasekonstrukt, 5 µg ß-Galaktosidase und 5 µg STAT5-kodierendes Plasmid kotransfiziert. Zusätzlich wurde mit 5 µg Rezeptorchimären und 5 µg FKHR-Expressionskonstrukt gearbeitet.
In diesem Versuch ergab sich ein völlig anderes Ergebnis als in den
vorangegangenen Untersuchungen mit den anderen beiden STAT-
Faktoren (Abbildungen 3.2.2, 3.2.6, 3.2.7). FKHR war nicht in der Lage, die
STAT5-abhängige Luziferaseproduktion zu steigern. Allerdings kam es
nach Transfektion von STAT5 zu einer deutlichen Zunahme der Luzi-
ferasebildung.
Eine Erklärungsmöglichkeit hierfür ist, dass in HepG2-Zellen längst nicht so
viel endogenes STAT5 wie STAT3 vorhanden ist. Deshalb kommt es trotz
Epo-Stimulation zu keiner Zunahme der Luziferase Aktivität bei den
Kontrollen. Bringt man allerdings Expressionsvektoren für STAT5 in die
Zellen ein, so resultiert nach Stimulation eine Steigerung der Luziferase
Aktivität um den Faktor 10.
Im Gegensatz zu STAT1 und STAT3 bewirkt FKHR jedoch auch bei
Kotransfektion mit STAT5 keinerlei Signalverstärkung. Es kommt eher zu
einer Abnahme des Signals. Dieses Ergebnis lässt weiter darauf schließen,
dass der beobachteten Interaktion von FKHR mit STAT3 bzw. STAT1 ein
selektiver und spezifischer Mechanismus zu Grunde liegt, welcher nicht für
alle STAT-Familienmitglieder gilt.
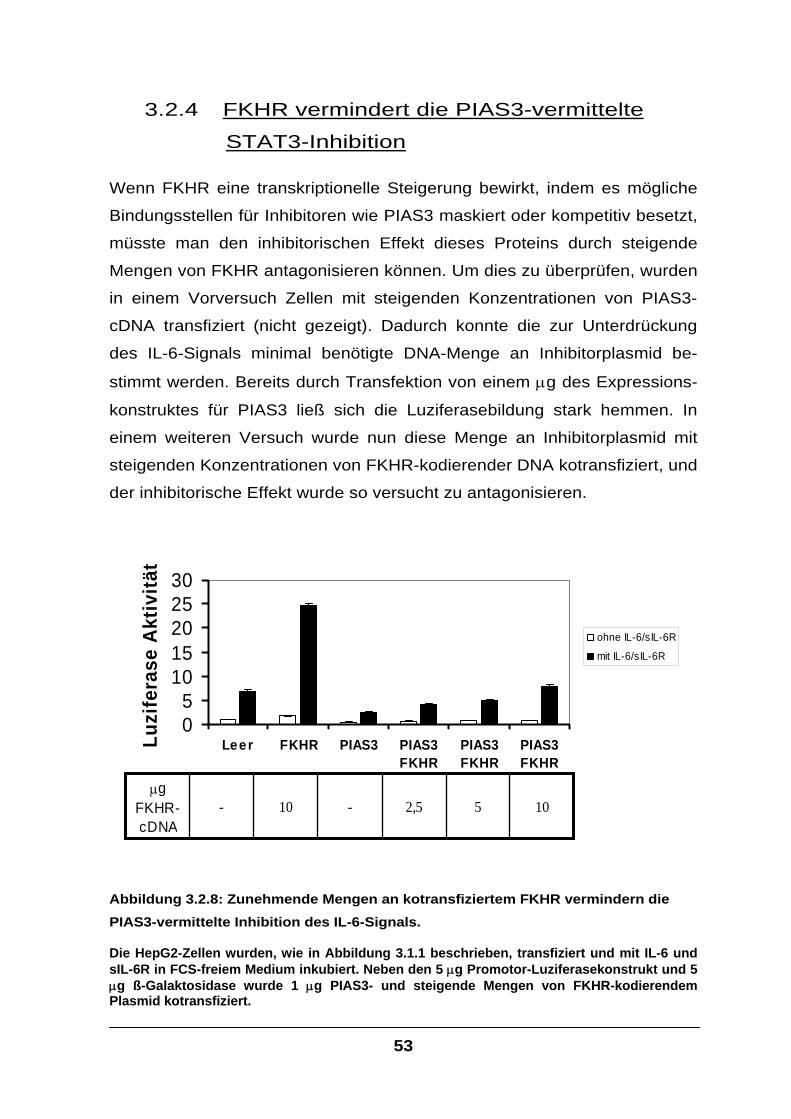
53
3.2.4 FKHR vermindert die PIAS3-vermittelte
STAT3-Inhibition
Wenn FKHR eine transkriptionelle Steigerung bewirkt, indem es mögliche
Bindungsstellen für Inhibitoren wie PIAS3 maskiert oder kompetitiv besetzt,
müsste man den inhibitorischen Effekt dieses Proteins durch steigende
Mengen von FKHR antagonisieren können. Um dies zu überprüfen, wurden
in einem Vorversuch Zellen mit steigenden Konzentrationen von PIAS3-
cDNA transfiziert (nicht gezeigt). Dadurch konnte die zur Unterdrückung
des IL-6-Signals minimal benötigte DNA-Menge an Inhibitorplasmid be-
stimmt werden. Bereits durch Transfektion von einem µg des Expressions-
konstruktes für PIAS3 ließ sich die Luziferasebildung stark hemmen. In
einem weiteren Versuch wurde nun diese Menge an Inhibitorplasmid mit
steigenden Konzentrationen von FKHR-kodierender DNA kotransfiziert, und
der inhibitorische Effekt wurde so versucht zu antagonisieren.
05
1015202530
Leer FKHR PIAS3 PIAS3FKHR
PIAS3FKHR
PIAS3FKHR
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6R
mit IL-6/sIL-6R
1052,5-10-µg
FKHR-cDNA
Abbildung 3.2.8: Zunehmende Mengen an kotransfiziertem FKHR vermindern die PIAS3-vermittelte Inhibition des IL-6-Signals.
Die HepG2-Zellen wurden, wie in Abbildung 3.1.1 beschrieben, transfiziert und mit IL-6 und sIL-6R in FCS-freiem Medium inkubiert. Neben den 5 µg Promotor-Luziferasekonstrukt und 5 µg ß-Galaktosidase wurde 1 µg PIAS3- und steigende Mengen von FKHR-kodierendem Plasmid kotransfiziert.

54
Bei dem Versuch war mit zunehmender Menge an kotransfiziertem FKHR-
kodierendem Plasmid eine gewisse transkriptionelle Steigerung zu be-
obachten (Abbildung 3.2.8). Es konnte bei Kotransfektion von 10 µg FKHR
kodierender DNA die annähernd gleiche Luziferase Aktivität wie bei den
Kontrollen beobachtet werden, wobei die Menge an Expressionsvektor für
FKHR um den Faktor zehn größer war als die von PIAS3. Mit diesen
Ergebnissen lässt sich ein Antagonismus von FKHR und PIAS3 weder
beweisen, noch sicher ausschließen.
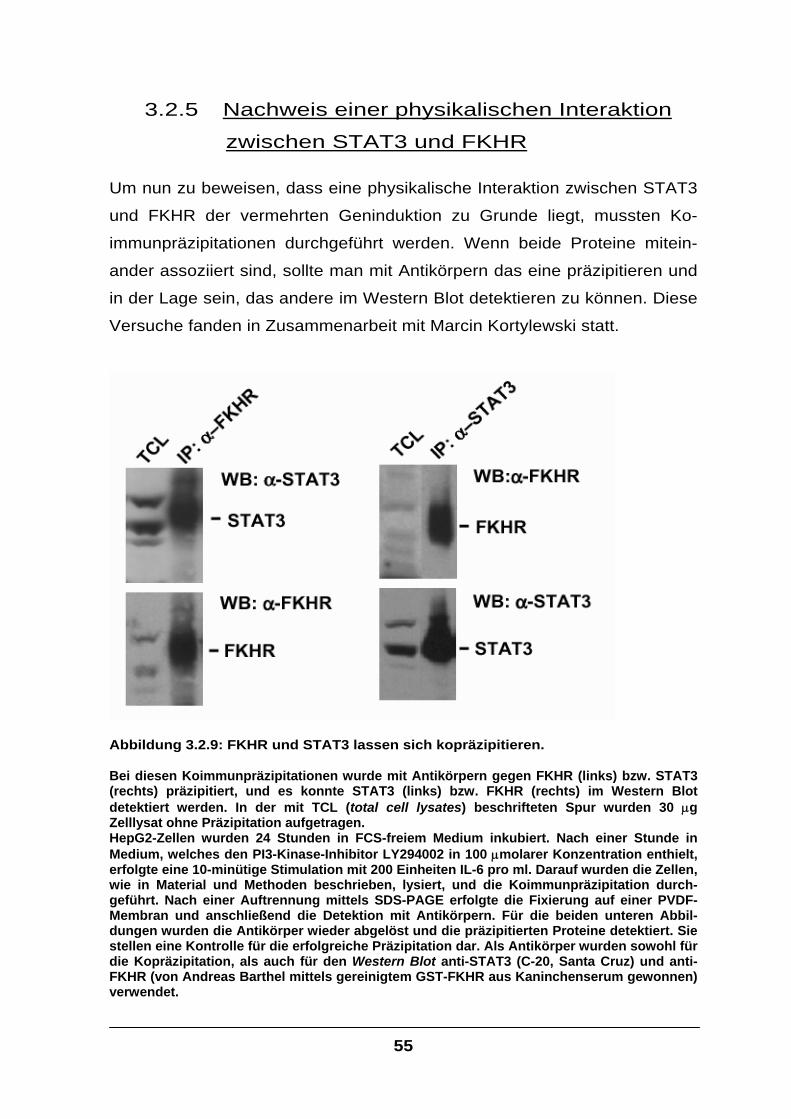
55
3.2.5 Nachweis einer physikalischen Interaktion
zwischen STAT3 und FKHR
Um nun zu beweisen, dass eine physikalische Interaktion zwischen STAT3
und FKHR der vermehrten Geninduktion zu Grunde liegt, mussten Ko-
immunpräzipitationen durchgeführt werden. Wenn beide Proteine mitein-
ander assoziiert sind, sollte man mit Antikörpern das eine präzipitieren und
in der Lage sein, das andere im Western Blot detektieren zu können. Diese
Versuche fanden in Zusammenarbeit mit Marcin Kortylewski statt.
Abbildung 3.2.9: FKHR und STAT3 lassen sich kopräzipitieren.
Bei diesen Koimmunpräzipitationen wurde mit Antikörpern gegen FKHR (links) bzw. STAT3 (rechts) präzipitiert, und es konnte STAT3 (links) bzw. FKHR (rechts) im Western Blot detektiert werden. In der mit TCL (total cell lysates) beschrifteten Spur wurden 30 µg Zelllysat ohne Präzipitation aufgetragen. HepG2-Zellen wurden 24 Stunden in FCS-freiem Medium inkubiert. Nach einer Stunde in Medium, welches den PI3-Kinase-Inhibitor LY294002 in 100 µmolarer Konzentration enthielt, erfolgte eine 10-minütige Stimulation mit 200 Einheiten IL-6 pro ml. Darauf wurden die Zellen, wie in Material und Methoden beschrieben, lysiert, und die Koimmunpräzipitation durch-geführt. Nach einer Auftrennung mittels SDS-PAGE erfolgte die Fixierung auf einer PVDF-Membran und anschließend die Detektion mit Antikörpern. Für die beiden unteren Abbil-dungen wurden die Antikörper wieder abgelöst und die präzipitierten Proteine detektiert. Sie stellen eine Kontrolle für die erfolgreiche Präzipitation dar. Als Antikörper wurden sowohl für die Kopräzipitation, als auch für den Western Blot anti-STAT3 (C-20, Santa Cruz) und anti-FKHR (von Andreas Barthel mittels gereinigtem GST-FKHR aus Kaninchenserum gewonnen) verwendet.

56
Sowohl bei der Koimmunpräzipitation mit Antikörpern gegen STAT3 als
auch im analogen Versuch mit anti-FKHR ließ sich das jeweils andere
Protein im Western Blot detektieren. Die Kontrollen mit ProteinA-Sepharose
und irrelevanten Antikörpern waren negativ (nicht gezeigt).
Dies beweist, dass STAT3 und FKHR physikalisch miteinander interagieren
können. Ob sie direkt oder unter Vermittlung anderer Faktoren miteinander
assoziiert sind, konnte mit diesem Versuch allerdings nicht gezeigt werden.
Überraschenderweise resultierte aus diesem Experiment ebenfalls, dass
sie auch ohne IL-6-Stimulation miteinander im Komplex vorliegen (nicht
gezeigt). Dieses könnte eine mögliche Begründung dafür sein könnte, dass
überexprimiertes FKHR auch in unstimulierten HepG2-Zellen die Luziferase
Aktivität unter der Kontrolle des alpha-2-Makroglobulin-Promotors erhöht
(Abbildung 3.2.1).

57
3.3 Auswirkungen von FKHR-
modulierenden Effektoren auf die
IL-6-Signaltransduktion
Für die Regulation von FKHR spielt die PI3-Kinase eine sehr wichtige
Rolle. Es konnte gezeigt werden, dass FKHR mit STAT3 interagiert und die
Transkription IL-6-induzierbarer Gene fördert.
Wie bereits in der Einleitung (siehe 1.3) erwähnt, kommt es nach IL-6-
Stimulus über den Komplex aus SHP2/Grb2/SOS zu einer Aktivierung von
Ras. In vielen Zelllinien soll GTP-Ras in der Lage sein, die PI3-Kinase zu
aktivieren 79-82,137.
Der Literatur nach könnte das unter normalen Bedingungen gemessene
IL-6-Signal somit einen Zustand von zumindest partieller PI3-Kinase-Akti-
vierung und somit einer teilweisen Inaktivierung von FKHR darstellen.
Um die Auswirkung der PI3-Kinase und ihren untergeordneten Effektoren
auf die IL-6-Signaltransduktion weiter untersuchen zu können, musste
ausgeschlossen werden, dass das endogene FKHR einen limitierenden
Faktor darstellen kann. So wäre es denkbar, dass bereits bei geringer PI3-
Kinase-Aktivierung der größte Teil des endogenen FKHRs inaktiviert wird
und gemessene Effekte nicht mehr auf die Interaktion von STAT3 und
FKHR zurückzuführen sind.
Deshalb wurde in den folgenden Versuchen die FKHR-Konzentration durch
Überexpression von FKHR angehoben und verschiedene Aktivatoren und
Inhibitoren der PI3-Kinase untersucht. Dadurch sollten sich auch die
Unterschiede der Luziferasewerte nach den verschiedenen Stimulationen
besser erkennen lassen.
Falls die IL-6-Signaltransduktion von dephosphoryliertem FKHR-abhängig
ist, sollte sich eine deutliche Abnahme der unter der Kontrolle des alpha-2-
Makroglobulin-Promotors stehenden Luziferase Aktivität bei Aktivierung der
PI3-Kinase verzeichnen lassen.
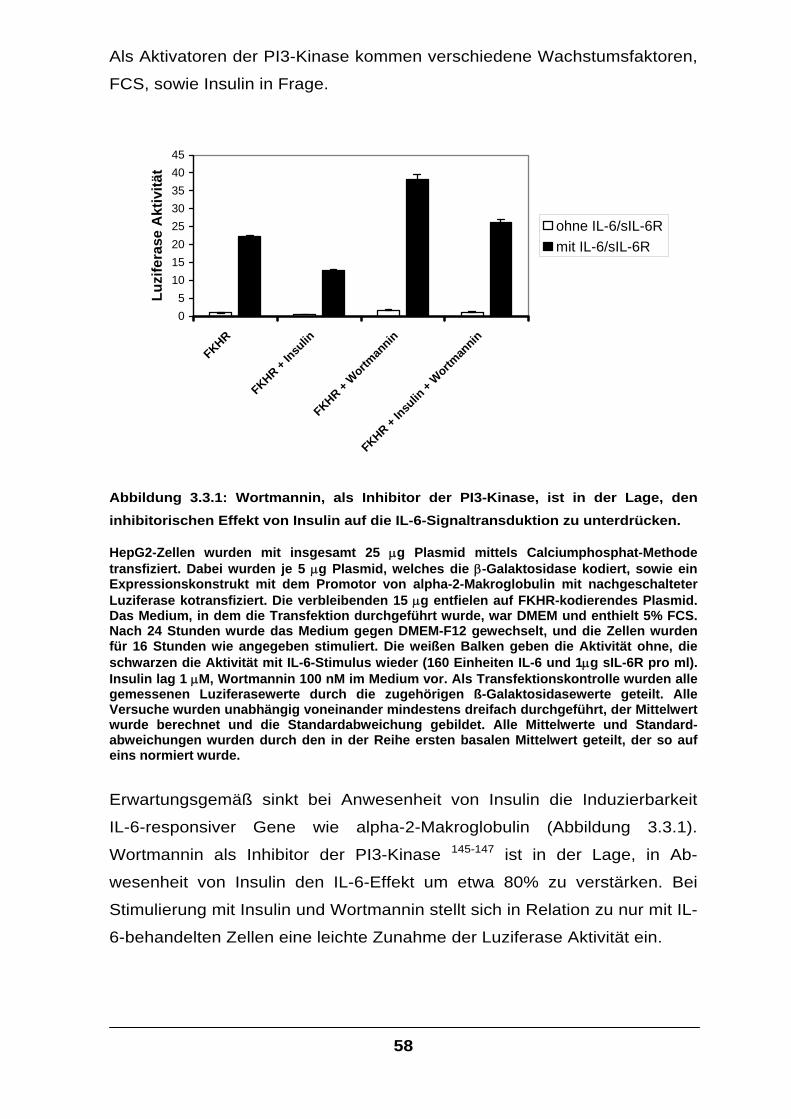
58
Als Aktivatoren der PI3-Kinase kommen verschiedene Wachstumsfaktoren,
FCS, sowie Insulin in Frage.
05
1015202530354045
FKHR
FKHR + Insu
lin
FKHR + Wortm
annin
FKHR + Insu
lin + W
ortman
nin
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6Rmit IL-6/sIL-6R
Abbildung 3.3.1: Wortmannin, als Inhibitor der PI3-Kinase, ist in der Lage, den inhibitorischen Effekt von Insulin auf die IL-6-Signaltransduktion zu unterdrücken.
HepG2-Zellen wurden mit insgesamt 25 µg Plasmid mittels Calciumphosphat-Methode transfiziert. Dabei wurden je 5 µg Plasmid, welches die β-Galaktosidase kodiert, sowie ein Expressionskonstrukt mit dem Promotor von alpha-2-Makroglobulin mit nachgeschalteter Luziferase kotransfiziert. Die verbleibenden 15 µg entfielen auf FKHR-kodierendes Plasmid. Das Medium, in dem die Transfektion durchgeführt wurde, war DMEM und enthielt 5% FCS. Nach 24 Stunden wurde das Medium gegen DMEM-F12 gewechselt, und die Zellen wurden für 16 Stunden wie angegeben stimuliert. Die weißen Balken geben die Aktivität ohne, die schwarzen die Aktivität mit IL-6-Stimulus wieder (160 Einheiten IL-6 und 1µg sIL-6R pro ml). Insulin lag 1 µM, Wortmannin 100 nM im Medium vor. Als Transfektionskontrolle wurden alle gemessenen Luziferasewerte durch die zugehörigen ß-Galaktosidasewerte geteilt. Alle Versuche wurden unabhängig voneinander mindestens dreifach durchgeführt, der Mittelwert wurde berechnet und die Standardabweichung gebildet. Alle Mittelwerte und Standard-abweichungen wurden durch den in der Reihe ersten basalen Mittelwert geteilt, der so auf eins normiert wurde.
Erwartungsgemäß sinkt bei Anwesenheit von Insulin die Induzierbarkeit
IL-6-responsiver Gene wie alpha-2-Makroglobulin (Abbildung 3.3.1).
Wortmannin als Inhibitor der PI3-Kinase 145-147 ist in der Lage, in Ab-
wesenheit von Insulin den IL-6-Effekt um etwa 80% zu verstärken. Bei
Stimulierung mit Insulin und Wortmannin stellt sich in Relation zu nur mit IL-
6-behandelten Zellen eine leichte Zunahme der Luziferase Aktivität ein.
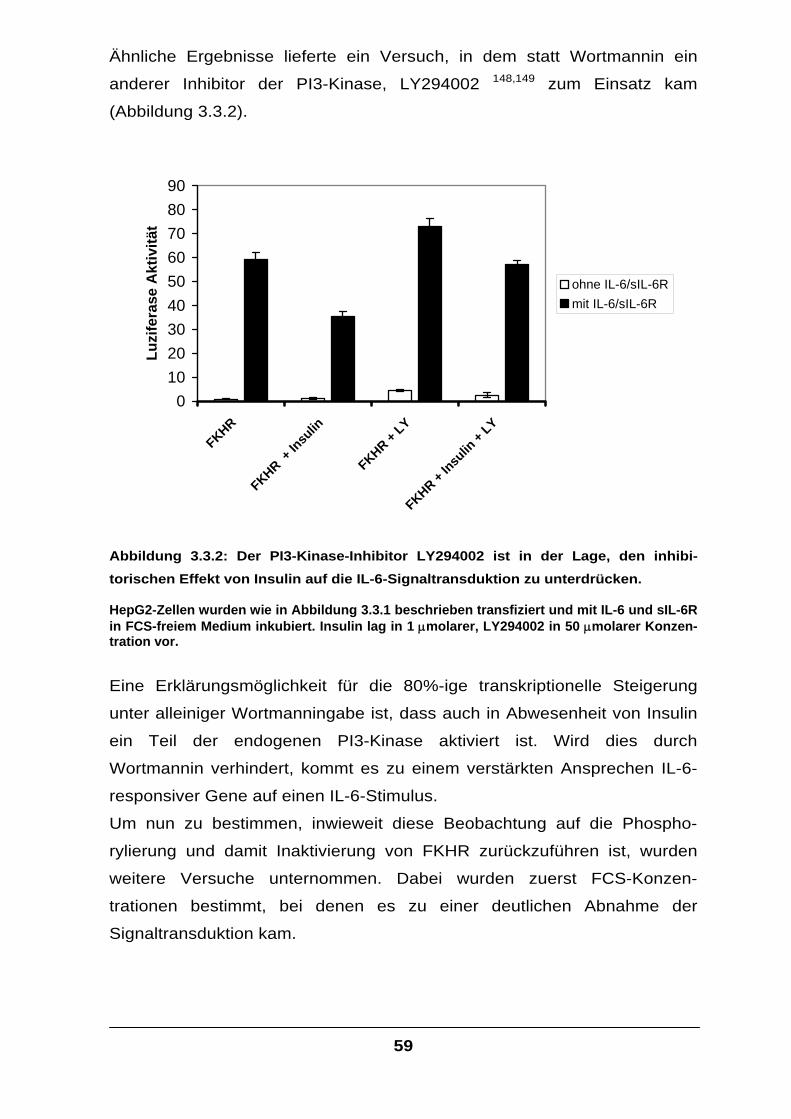
59
Ähnliche Ergebnisse lieferte ein Versuch, in dem statt Wortmannin ein
anderer Inhibitor der PI3-Kinase, LY294002 148,149 zum Einsatz kam
(Abbildung 3.3.2).
0102030405060708090
FKHR
FKHR + In
sulin
FKHR + LY
FKHR + Insu
lin +
LY
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6Rmit IL-6/sIL-6R
Abbildung 3.3.2: Der PI3-Kinase-Inhibitor LY294002 ist in der Lage, den inhibi-torischen Effekt von Insulin auf die IL-6-Signaltransduktion zu unterdrücken.
HepG2-Zellen wurden wie in Abbildung 3.3.1 beschrieben transfiziert und mit IL-6 und sIL-6R in FCS-freiem Medium inkubiert. Insulin lag in 1 µmolarer, LY294002 in 50 µmolarer Konzen-tration vor.
Eine Erklärungsmöglichkeit für die 80%-ige transkriptionelle Steigerung
unter alleiniger Wortmanningabe ist, dass auch in Abwesenheit von Insulin
ein Teil der endogenen PI3-Kinase aktiviert ist. Wird dies durch
Wortmannin verhindert, kommt es zu einem verstärkten Ansprechen IL-6-
responsiver Gene auf einen IL-6-Stimulus.
Um nun zu bestimmen, inwieweit diese Beobachtung auf die Phospho-
rylierung und damit Inaktivierung von FKHR zurückzuführen ist, wurden
weitere Versuche unternommen. Dabei wurden zuerst FCS-Konzen-
trationen bestimmt, bei denen es zu einer deutlichen Abnahme der
Signaltransduktion kam.
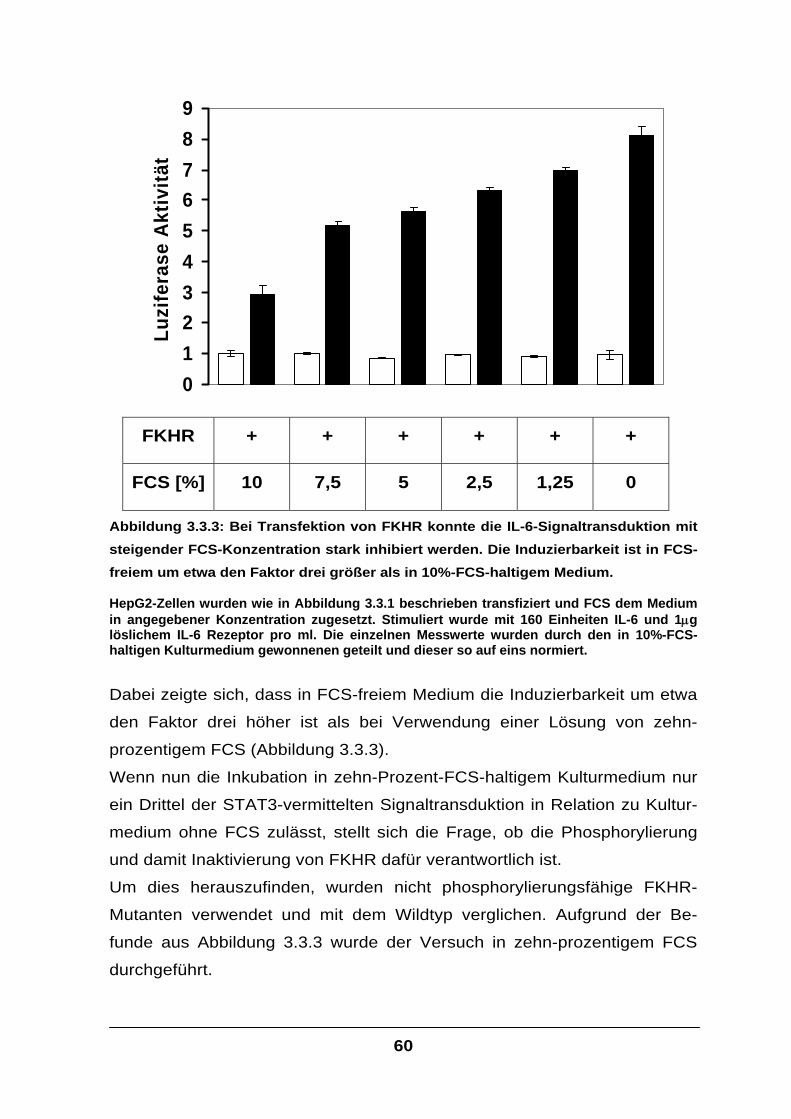
60
0123456789
Luzi
fera
se A
ktiv
ität
FKHR + + + + + +
FCS [%] 10 7,5 5 2,5 1,25 0
Abbildung 3.3.3: Bei Transfektion von FKHR konnte die IL-6-Signaltransduktion mit steigender FCS-Konzentration stark inhibiert werden. Die Induzierbarkeit ist in FCS-freiem um etwa den Faktor drei größer als in 10%-FCS-haltigem Medium.
HepG2-Zellen wurden wie in Abbildung 3.3.1 beschrieben transfiziert und FCS dem Medium in angegebener Konzentration zugesetzt. Stimuliert wurde mit 160 Einheiten IL-6 und 1µg löslichem IL-6 Rezeptor pro ml. Die einzelnen Messwerte wurden durch den in 10%-FCS-haltigen Kulturmedium gewonnenen geteilt und dieser so auf eins normiert.
Dabei zeigte sich, dass in FCS-freiem Medium die Induzierbarkeit um etwa
den Faktor drei höher ist als bei Verwendung einer Lösung von zehn-
prozentigem FCS (Abbildung 3.3.3).
Wenn nun die Inkubation in zehn-Prozent-FCS-haltigem Kulturmedium nur
ein Drittel der STAT3-vermittelten Signaltransduktion in Relation zu Kultur-
medium ohne FCS zulässt, stellt sich die Frage, ob die Phosphorylierung
und damit Inaktivierung von FKHR dafür verantwortlich ist.
Um dies herauszufinden, wurden nicht phosphorylierungsfähige FKHR-
Mutanten verwendet und mit dem Wildtyp verglichen. Aufgrund der Be-
funde aus Abbildung 3.3.3 wurde der Versuch in zehn-prozentigem FCS
durchgeführt.
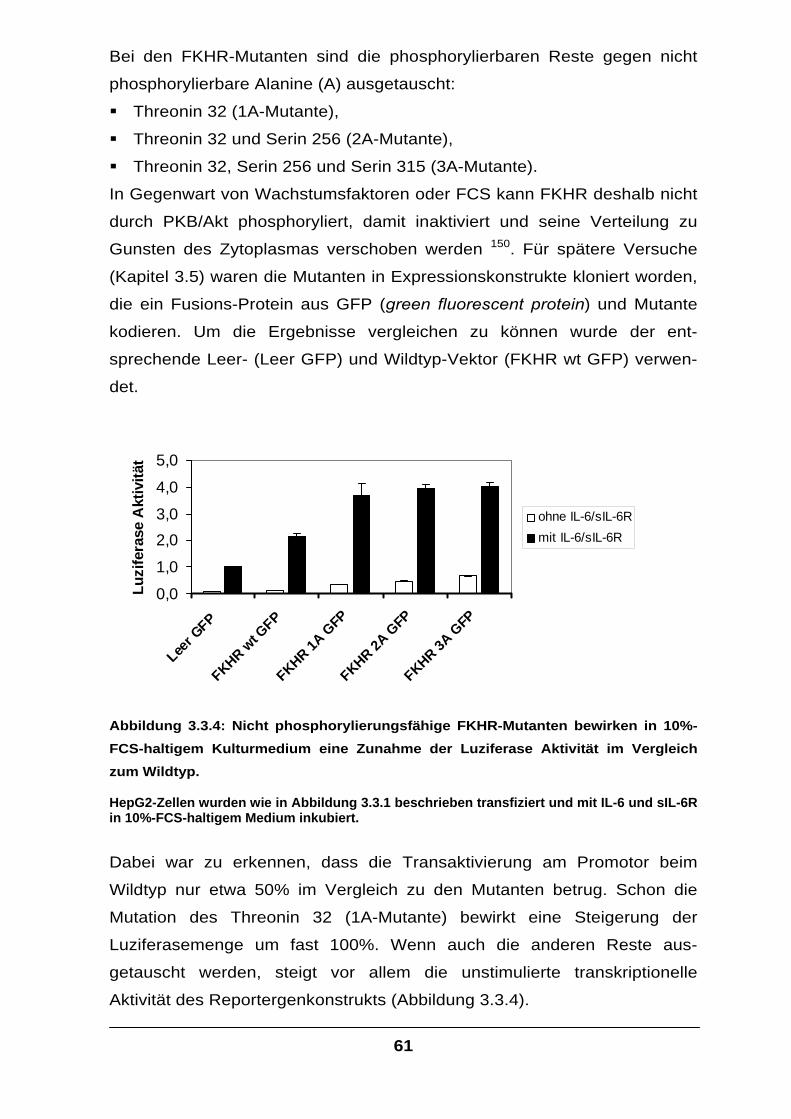
61
Bei den FKHR-Mutanten sind die phosphorylierbaren Reste gegen nicht
phosphorylierbare Alanine (A) ausgetauscht:
Threonin 32 (1A-Mutante),
Threonin 32 und Serin 256 (2A-Mutante),
Threonin 32, Serin 256 und Serin 315 (3A-Mutante).
In Gegenwart von Wachstumsfaktoren oder FCS kann FKHR deshalb nicht
durch PKB/Akt phosphoryliert, damit inaktiviert und seine Verteilung zu
Gunsten des Zytoplasmas verschoben werden 150. Für spätere Versuche
(Kapitel 3.5) waren die Mutanten in Expressionskonstrukte kloniert worden,
die ein Fusions-Protein aus GFP (green fluorescent protein) und Mutante
kodieren. Um die Ergebnisse vergleichen zu können wurde der ent-
sprechende Leer- (Leer GFP) und Wildtyp-Vektor (FKHR wt GFP) verwen-
det.
0,0
1,0
2,03,0
4,0
5,0
Leer G
FP
FKHR wt G
FP
FKHR 1A G
FP
FKHR 2A G
FP
FKHR 3A G
FP
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6Rmit IL-6/sIL-6R
Abbildung 3.3.4: Nicht phosphorylierungsfähige FKHR-Mutanten bewirken in 10%-FCS-haltigem Kulturmedium eine Zunahme der Luziferase Aktivität im Vergleich zum Wildtyp.
HepG2-Zellen wurden wie in Abbildung 3.3.1 beschrieben transfiziert und mit IL-6 und sIL-6R in 10%-FCS-haltigem Medium inkubiert.
Dabei war zu erkennen, dass die Transaktivierung am Promotor beim
Wildtyp nur etwa 50% im Vergleich zu den Mutanten betrug. Schon die
Mutation des Threonin 32 (1A-Mutante) bewirkt eine Steigerung der
Luziferasemenge um fast 100%. Wenn auch die anderen Reste aus-
getauscht werden, steigt vor allem die unstimulierte transkriptionelle
Aktivität des Reportergenkonstrukts (Abbildung 3.3.4).

62
So konnte gezeigt werden, dass die Phosphorylierung von FKHR für die
IL-6-Signaltransduktion eine wichtige Rolle spielt. Die in Abbildung 3.3.1 bis
3.3.3 dargestellte Abnahme des IL-6-induzierten Signals in Anwesenheit
von FCS oder Insulin könnte auf eine Phosphorylierung und damit Inakti-
vierung von FKHR zurückzuführen sein.
3.4 Die subzelluläre Lokalisation von FKHR
und STAT3 in Abhängigkeit
verschiedener Stimuli
Um nachvollziehen zu können, wie die Transkriptionsfaktoren STAT3 und
FKHR in Abhängigkeit verschiedener Stimuli innerhalb der Zelle verteilt
sind, wurden Immunfluoreszenzstudien durchgeführt. Dabei wird ein gegen
das anzufärbende Protein gerichteter Antikörper mit permeabilisierten und
fixierten Zellen inkubiert. Ein zweiter Antikörper richtet sich gegen den
ersten und trägt einen Farbstoff, welcher bei Anregung durch eine definierte
Wellenlänge ein spezifisches Lichtspektrum emittiert. Dieses wird registriert
und ermöglicht so, die zelluläre Lokalisation des Proteins zu bestimmen.
Durch unterschiedliche Absorptions- und Emissionsmaxima der ver-
schiedenen Fluoreszenzstoffe können mehrere Proteine gleichzeitig
angefärbt und detektiert werden.
In diesem Versuch kamen Antikörper gegen STAT3 und FKHR zum
Einsatz. Es sollte die subzelluläre Lokalisation beider Proteine nach
verschiedenen Stimuli untersucht werden. In diesen Versuchen wurden
ausschließlich native, nicht transfizierte Zellen verwendet, um Artefakte zu
minimieren.
Bei den hier gezeigten Färbungen wird FKHR in roter und STAT3 in grüner
Farbe wiedergegeben.
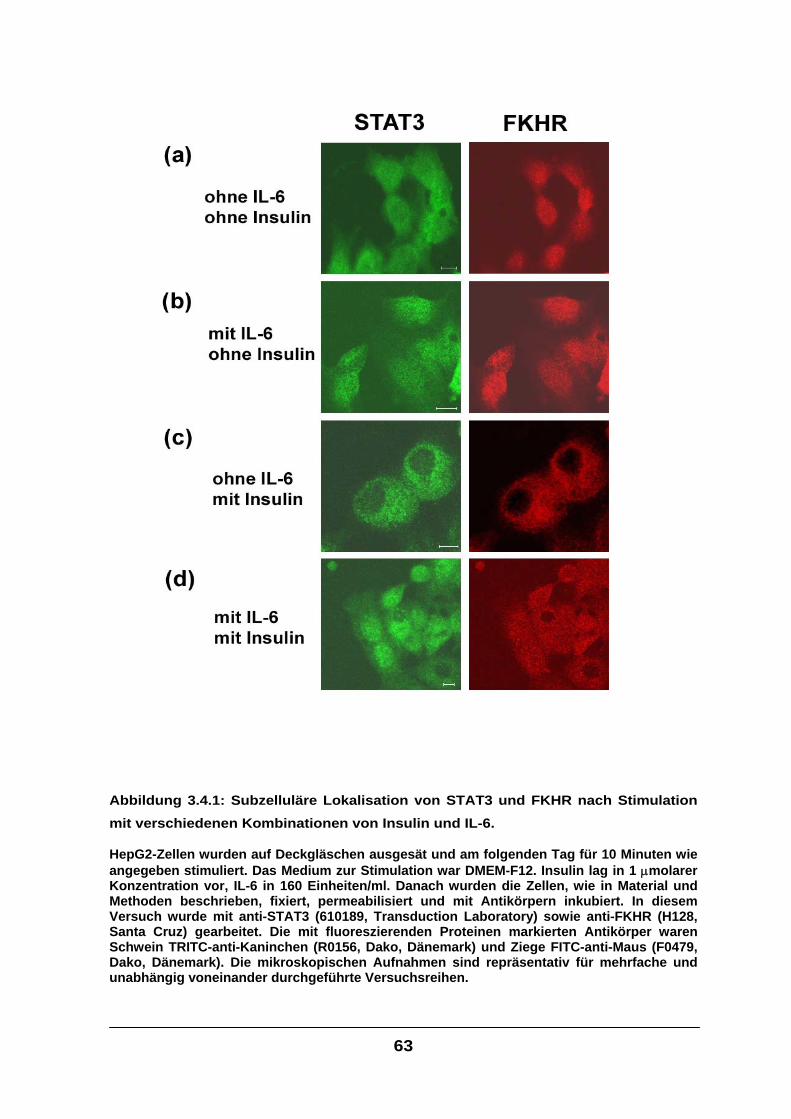
63
Abbildung 3.4.1: Subzelluläre Lokalisation von STAT3 und FKHR nach Stimulation mit verschiedenen Kombinationen von Insulin und IL-6.
HepG2-Zellen wurden auf Deckgläschen ausgesät und am folgenden Tag für 10 Minuten wie angegeben stimuliert. Das Medium zur Stimulation war DMEM-F12. Insulin lag in 1 µmolarer Konzentration vor, IL-6 in 160 Einheiten/ml. Danach wurden die Zellen, wie in Material und Methoden beschrieben, fixiert, permeabilisiert und mit Antikörpern inkubiert. In diesem Versuch wurde mit anti-STAT3 (610189, Transduction Laboratory) sowie anti-FKHR (H128, Santa Cruz) gearbeitet. Die mit fluoreszierenden Proteinen markierten Antikörper waren Schwein TRITC-anti-Kaninchen (R0156, Dako, Dänemark) und Ziege FITC-anti-Maus (F0479, Dako, Dänemark). Die mikroskopischen Aufnahmen sind repräsentativ für mehrfache und unabhängig voneinander durchgeführte Versuchsreihen.

64
In unstimuliertem Zustand liegt STAT3 relativ diffus in der Zelle vor, jedoch
findet sich tendenziell eher eine Anhäufung im Zytoplasma. FKHR stellt
sich deutlich im Nukleus dar (Abbildung 3.4.1 a).
Bei einem Stimulus mit IL-6 transloziert STAT3 in den Zellkern, wo sich
auch FKHR befindet (Abbildung 3.4.1 b). Dabei weisen intranukleäre
Strukturen Helligkeitsmaxima-Übereinstimmungen für beide Proteine auf.
Werden die Zellen nur mit Insulin behandelt, so resultiert eine andere
intrazelluläre Verteilung der Proteine: Sowohl STAT3, als auch FKHR sind
im Zytoplasma und sind nicht mehr im Zellkern anzutreffen (Abbildung 3.4.1
c). Dabei liegt STAT3 längst nicht mehr so diffus wie in unstimulierten
Zellen (Abbildung 3.4.1 a), sondern deutlich abgegrenzt im Zytoplasma vor.
Bei Inkubation mit Insulin und IL-6 ist STAT3 im Nukleus lokalisiert. FKHR
weist eine diffuse, jedoch eher zytoplasmatische Lokalisation auf (Ab-
bildung 3.4.1 d).
Diese mikroskopischen Befunde zeigen also, dass nach IL-6-Stimualtion
eine deutliche Kolokalisation beider Transkriptionsfaktoren im Zellkern
nachzuweisen ist.
3.5 Herstellung und erste Charakterisierung
von HepG2-Transfektanten, die stabil
fluoreszierendes FKHR exprimieren
In den bisher im Rahmen dieser Dissertationsarbeit gezeigten Versuchen
wurde der verstärkende Effekt von FKHR auf den IL-6 / gp130 / Jak /
STAT3-Weg entdeckt. Um orientierend zu untersuchen, ob sich ebenfalls
synergistische oder gar antagonistische Effekte auf die FKHR-vermittelte
Proteinbiosynthese unter IL-6-Einfluss einstellen, wurden stabil transfizierte
HepG2-Zellen generiert. Dabei wurde ein Konstrukt verwendet, welches für
ein Fusionsprotein aus FKHR und einem fluoreszierenden Protein kodiert.
Dieses diente zum einen als Transfektionskontrolle, zum anderen sollten
spätere Versuche ermöglichen, die intrazelluläre Lokalisation und Be-
wegung in Reaktion auf verschiedene Stimuli in vivo zu untersuchen.
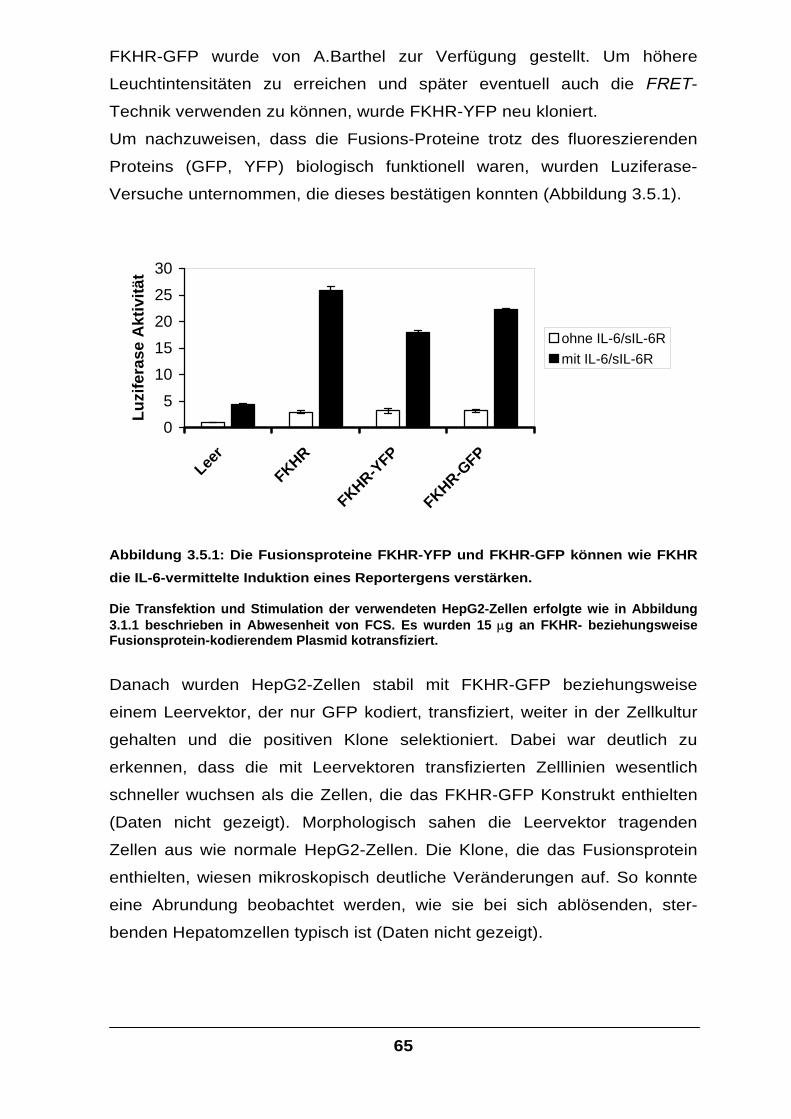
65
FKHR-GFP wurde von A.Barthel zur Verfügung gestellt. Um höhere
Leuchtintensitäten zu erreichen und später eventuell auch die FRET-
Technik verwenden zu können, wurde FKHR-YFP neu kloniert.
Um nachzuweisen, dass die Fusions-Proteine trotz des fluoreszierenden
Proteins (GFP, YFP) biologisch funktionell waren, wurden Luziferase-
Versuche unternommen, die dieses bestätigen konnten (Abbildung 3.5.1).
05
101520
2530
Leer
FKHR
FKHR-YFP
FKHR-GFP
Luzi
fera
se A
ktiv
ität
ohne IL-6/sIL-6Rmit IL-6/sIL-6R
Abbildung 3.5.1: Die Fusionsproteine FKHR-YFP und FKHR-GFP können wie FKHR die IL-6-vermittelte Induktion eines Reportergens verstärken.
Die Transfektion und Stimulation der verwendeten HepG2-Zellen erfolgte wie in Abbildung 3.1.1 beschrieben in Abwesenheit von FCS. Es wurden 15 µg an FKHR- beziehungsweise Fusionsprotein-kodierendem Plasmid kotransfiziert.
Danach wurden HepG2-Zellen stabil mit FKHR-GFP beziehungsweise
einem Leervektor, der nur GFP kodiert, transfiziert, weiter in der Zellkultur
gehalten und die positiven Klone selektioniert. Dabei war deutlich zu
erkennen, dass die mit Leervektoren transfizierten Zelllinien wesentlich
schneller wuchsen als die Zellen, die das FKHR-GFP Konstrukt enthielten
(Daten nicht gezeigt). Morphologisch sahen die Leervektor tragenden
Zellen aus wie normale HepG2-Zellen. Die Klone, die das Fusionsprotein
enthielten, wiesen mikroskopisch deutliche Veränderungen auf. So konnte
eine Abrundung beobachtet werden, wie sie bei sich ablösenden, ster-
benden Hepatomzellen typisch ist (Daten nicht gezeigt).
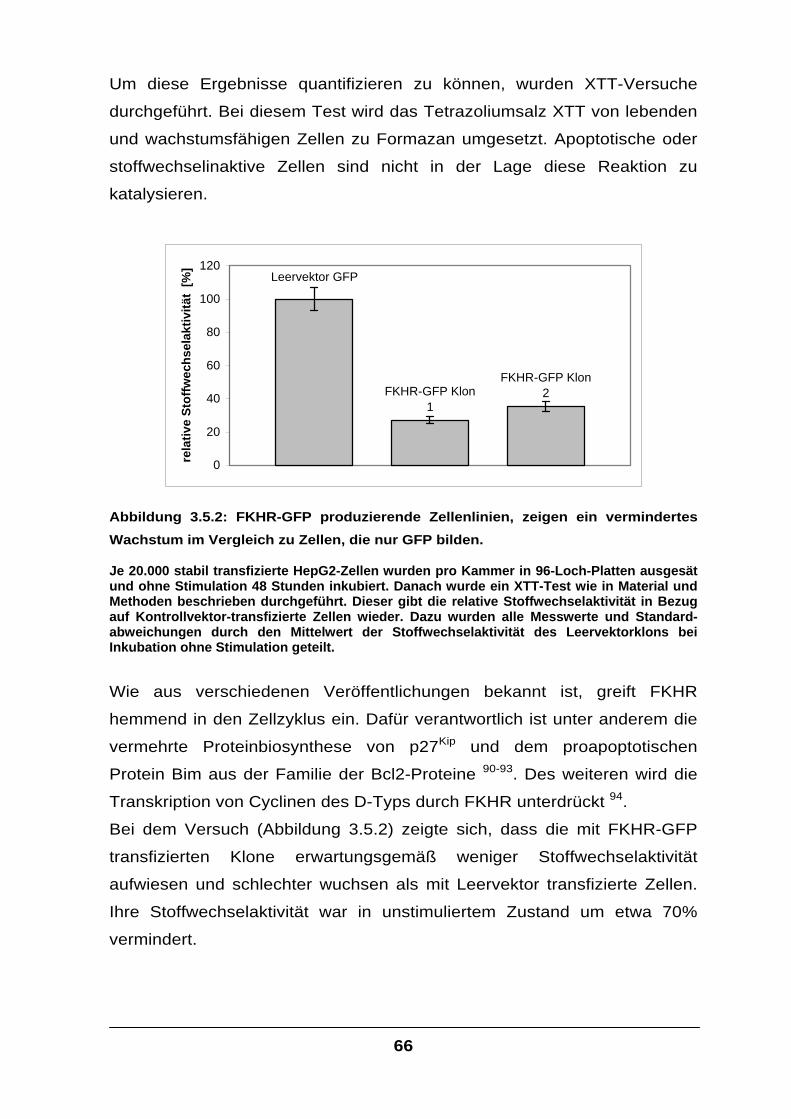
66
Um diese Ergebnisse quantifizieren zu können, wurden XTT-Versuche
durchgeführt. Bei diesem Test wird das Tetrazoliumsalz XTT von lebenden
und wachstumsfähigen Zellen zu Formazan umgesetzt. Apoptotische oder
stoffwechselinaktive Zellen sind nicht in der Lage diese Reaktion zu
katalysieren.
FKHR-GFP Klon 1
FKHR-GFP Klon 2
Leervektor GFP
0
20
40
60
80
100
120
rela
tive
Stof
fwec
hsel
aktiv
ität
[%]
Abbildung 3.5.2: FKHR-GFP produzierende Zellenlinien, zeigen ein vermindertes Wachstum im Vergleich zu Zellen, die nur GFP bilden.
Je 20.000 stabil transfizierte HepG2-Zellen wurden pro Kammer in 96-Loch-Platten ausgesät und ohne Stimulation 48 Stunden inkubiert. Danach wurde ein XTT-Test wie in Material und Methoden beschrieben durchgeführt. Dieser gibt die relative Stoffwechselaktivität in Bezug auf Kontrollvektor-transfizierte Zellen wieder. Dazu wurden alle Messwerte und Standard-abweichungen durch den Mittelwert der Stoffwechselaktivität des Leervektorklons bei Inkubation ohne Stimulation geteilt.
Wie aus verschiedenen Veröffentlichungen bekannt ist, greift FKHR
hemmend in den Zellzyklus ein. Dafür verantwortlich ist unter anderem die
vermehrte Proteinbiosynthese von p27Kip und dem proapoptotischen
Protein Bim aus der Familie der Bcl2-Proteine 90-93. Des weiteren wird die
Transkription von Cyclinen des D-Typs durch FKHR unterdrückt 94.
Bei dem Versuch (Abbildung 3.5.2) zeigte sich, dass die mit FKHR-GFP
transfizierten Klone erwartungsgemäß weniger Stoffwechselaktivität
aufwiesen und schlechter wuchsen als mit Leervektor transfizierte Zellen.
Ihre Stoffwechselaktivität war in unstimuliertem Zustand um etwa 70%
vermindert.
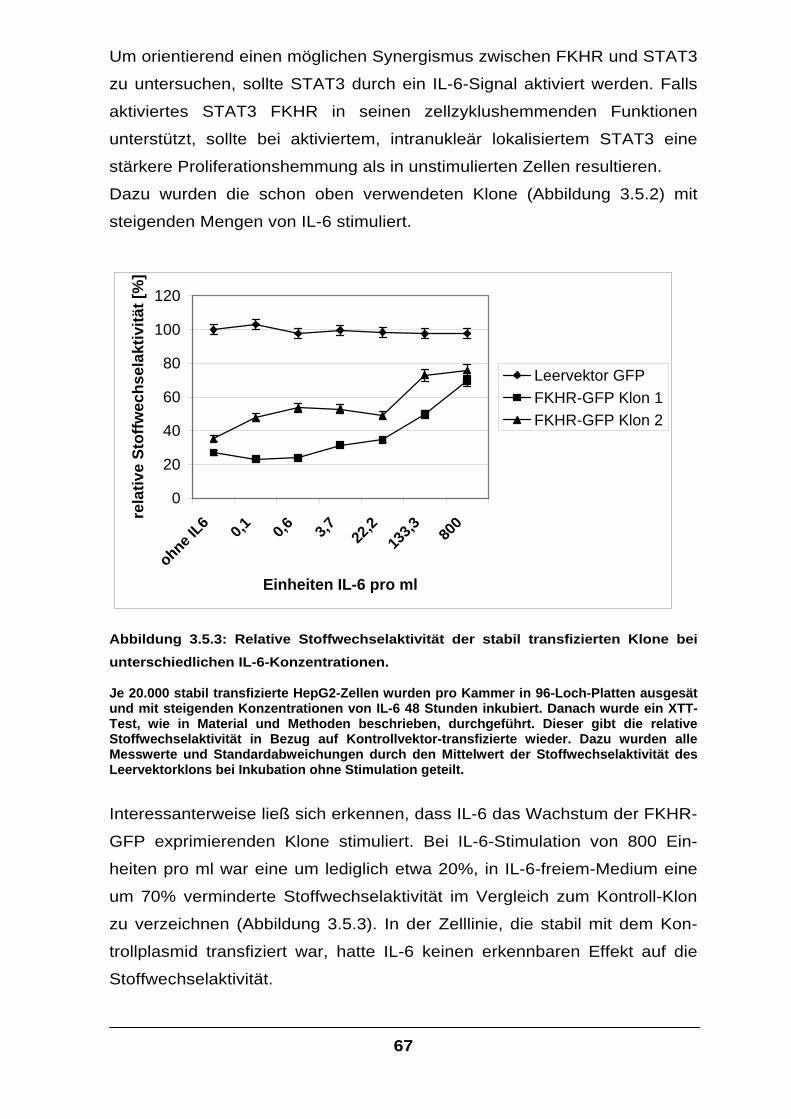
67
Um orientierend einen möglichen Synergismus zwischen FKHR und STAT3
zu untersuchen, sollte STAT3 durch ein IL-6-Signal aktiviert werden. Falls
aktiviertes STAT3 FKHR in seinen zellzyklushemmenden Funktionen
unterstützt, sollte bei aktiviertem, intranukleär lokalisiertem STAT3 eine
stärkere Proliferationshemmung als in unstimulierten Zellen resultieren.
Dazu wurden die schon oben verwendeten Klone (Abbildung 3.5.2) mit
steigenden Mengen von IL-6 stimuliert.
0
20
40
60
80
100
120
ohne IL6 0,1 0,6 3,7 22
,213
3,3 800
Einheiten IL-6 pro ml
rela
tive
Stof
fwec
hsel
aktiv
ität [
%]
Leervektor GFPFKHR-GFP Klon 1FKHR-GFP Klon 2
Abbildung 3.5.3: Relative Stoffwechselaktivität der stabil transfizierten Klone bei unterschiedlichen IL-6-Konzentrationen.
Je 20.000 stabil transfizierte HepG2-Zellen wurden pro Kammer in 96-Loch-Platten ausgesät und mit steigenden Konzentrationen von IL-6 48 Stunden inkubiert. Danach wurde ein XTT-Test, wie in Material und Methoden beschrieben, durchgeführt. Dieser gibt die relative Stoffwechselaktivität in Bezug auf Kontrollvektor-transfizierte wieder. Dazu wurden alle Messwerte und Standardabweichungen durch den Mittelwert der Stoffwechselaktivität des Leervektorklons bei Inkubation ohne Stimulation geteilt.
Interessanterweise ließ sich erkennen, dass IL-6 das Wachstum der FKHR-
GFP exprimierenden Klone stimuliert. Bei IL-6-Stimulation von 800 Ein-
heiten pro ml war eine um lediglich etwa 20%, in IL-6-freiem-Medium eine
um 70% verminderte Stoffwechselaktivität im Vergleich zum Kontroll-Klon
zu verzeichnen (Abbildung 3.5.3). In der Zelllinie, die stabil mit dem Kon-
trollplasmid transfiziert war, hatte IL-6 keinen erkennbaren Effekt auf die
Stoffwechselaktivität.

68
Dieser Versuch konnte keinen bilateralen Synergismus zwischen STAT3
und FKHR nachweisen. So scheint aktiviertes STAT3 nicht in der Lage zu
sein, die FKHR-vermittelte Wachstumshemmung in HepG2-Zellen zu
unterstützen. Allerdings wirft dieses Experiment auch einige Fragen auf, die
in der Diskussion weiter thematisiert werden (siehe Kapitel 4.3).

69
4 Diskussion
4.1 Interaktion zwischen FKHR und STAT-
Faktoren
Im Rahmen dieser Arbeit konnte erstmals nachgewiesen werden, dass
FKHR, ein Protein aus der Familie der foxo-Transkriptionsfaktoren, mit
STAT-Faktoren interagiert.
In Abbildung 3.2.2 ist der deutliche Synergismus zwischen FKHR und
STAT3 bezüglich der IL-6-Signaltransduktion dargestellt. Dabei bewirkt die
Koexpression von FKHR und STAT3 eine Transkriptions-Steigerung um
den Faktor 14 im Vergleich zu Zellen, die nur mit Leervektor transfiziert
worden waren. Gemessen wurde dies mit einem alpha-2-Makroglobulin-
Promotor-Luziferase-Konstrukt. Die alleinige Überexpression von STAT3
erhöhte die Luziferase Aktivität nur um den Faktor 3, die von FKHR um den
Faktor 5. FKHR und STAT3 interagieren bei ihrem Zusammenspiel physi-
kalisch miteinander, was mit Koimmunpräzipitationen nachgewiesen wurde
(Abbbildung 3.2.9).
Analog zu STAT3 zeigt Abbildung 3.2.6 die Interaktion mit STAT1, wohin-
gegen für STAT5 kein Effekt gemessen werden konnte (Abbildung 3.2.7).
Bei der TGFß-Signaltransduktion konnte durch Überexpression von FKHR
keine gesteigerte Smad7-Promotor-Aktivierung beobachtet werden (Ab-
bildung 3.2.5). Auch die gemessenen ß-Galaktosidasewerte waren bei
Versuchsreihen mit und ohne FKHR-Transfektion ähnlich, was eine gene-
relle Steigerung der Transkriptionsmaschinerie durch FKHR noch unwahr-
scheinlicher macht.
Dem beobachteten Zusammenspiel von FKHR und STAT3 beziehungs-
weise STAT1 liegt also ein spezifischer Mechanismus zu Grunde, welcher
nur für einige STAT-Familienmitglieder zu gelten scheint.
Dass foxo-Familienmitglieder mit anderen Transkriptionsfaktoren inter-
agieren und auch selbst in der Lage sind zu transaktivieren, konnte bereits
in verschiedenen Veröffentlichungen gezeigt werden 90,92,93,95,96,97,151.

70
In jüngster Zeit ließ sich außerdem eine Inhibition von hepatocyte nuclear
factor 4 (HNF-4), ein Mitglied der Steroid- und Thyroidrezeptoren, durch
aktives, also dephosphoryliertes FKHR nachweisen 152. Auch dieser
Interaktion liegt eine direkte Assoziation beider Proteine zugrunde.
Für verschiedene STAT-Faktoren wurde ebenfalls ein Zusammenspiel mit
anderen DNA-bindenden Proteinen beschrieben. So ist eine Kooperation
zwischen STAT1 und Sp1 bekannt 153. STAT5 soll mit dem Glucokortikoid-
rezeptor interagieren können 154. Für STAT3 wurde für eine Vielzahl von
Genen eine Assoziation mit dem Transkriptionsfaktor c-Jun beobachtet 2,35.
Vor kurzem konnte gezeigt werden, dass für die Leberregeneration nach
Verletzung ein Komplex aus hepatocyte nuclear factor 1 (HNF-1), STAT3
und AP-1 (c-Fos/c-Jun) verantwortlich ist. Dabei verstärken die IL-6-
aktivierten STAT3-Faktoren die Transkription HNF-1-abhängiger Gene, wie
beispielsweise das insulin-like growth factor binding protein 1(IGFBP-1) 155.
4.2 Mögliche Mechanismen der Interaktion
Ursprünglich entdeckt wurde FKHR in dem alveolären Typ des Rhabdo-
myosarkoms. Dort liegt es als Fusionsprotein mit PAX3 vor 85.
Es konnte gezeigt werden, dass das C-terminale Ende des FKHR für die
stark gesteigerte transkriptionelle Aktivität der Chimäre verantwortlich
ist 156,157,158. Durch den Einsatz von Deletionsmutanten wurde bewiesen,
dass auch für den Synergismus mit STAT3 der C-terminale Anteil von
FKHR essentiell ist 138.
Wie die dargestellte Koimmunpräzipitation (Abbildung 3.2.9) belegt, liegt
eine Assoziation der Proteine STAT3 und FKHR vor. Der Nachweis, ob
beide Transkriptionsfaktoren direkt oder unter Vermittlung weiterer Proteine
miteinander assoziiert sind, steht noch aus.
Über den genauen Mechanismus, wie FKHR die IL-6-abhängige, STAT3
vermittelte transkriptionelle Steigerung bewirkt, lässt sich zur Zeit noch
keine Aussage treffen. Es sind verschiedene Möglichkeiten zu diskutieren.

71
4.2.1 FKHR könnte die Kernlokalisation der
STAT-Faktoren begünstigen
Eine Möglichkeit, wie FKHR die STAT3-vermittelte, IL-6-induzierte
Transkription responsiver Gene steigern könnte, wäre eine Förderung der
Kernlokalisation von STAT3. Um dies zu erreichen, existieren zwei Mög-
lichkeiten. Es könnte einerseits der nukleäre Export gehemmt werden,
indem beispielsweise das STAT3-Dimer durch anwesendes FKHR ver-
mehrt intranukleär lokalisiert bleibt. Andererseits könnte FKHR zytoplas-
matisch mit STAT3 assoziieren und auf dem Weg in den Zellkern, bei-
spielsweise durch Interaktion mit Transportproteinen, unterstützend wirken
und so den Import fördern.
Vermehrt intranukleär lokalisierte STAT-Faktoren wären so in der Lage zu
transaktivieren und den beobachteten Luziferaseanstieg zu bewirken.
Abbildung 3.4.1 gibt Aufschluss über die Lokalisationen der beiden Prote-
ine in Abhängigkeit verschiedener Stimuli.
In unstimuliertem Zustand liegt STAT3 relativ diffus in der Zelle vor, jedoch
findet sich tendenziell eher eine Anhäufung im Zytoplasma. FKHR stellt
sich deutlich im Zellkern dar (Abbildung 3.4.1 a). In den Luziferasepro-
motorstudien zeigte sich bei unstimulierten, mit FKHR transfizierten Zellen
eine Zunahme der basalen Luziferasemenge (Abbildung 3.2.1 und 3.2.2).
Auch bei der Koimmunpräzipitation waren beide Proteine in unstimuliertem
Zustand bereits assoziiert. Eine mögliche Erklärung könnte sein, dass das
nukleär lokalisiertes FKHR in der Lage ist, STAT3 dort zu stabilisieren und
vor dem Export zu bewahren.
Bei einem Stimulus mit IL-6 transloziert der größte Teil des STAT3 in den
Nukleus, wo es sich zusammen mit FKHR darstellen lässt (Abbildung 3.4.1
b). Dabei weisen intranukleäre Strukturen Helligkeitsmaxima-Überein-
stimmungen für beide Proteine auf. In den Reportergenversuchen ließ sich
bei Anwesenheit von FKHR eine deutliche Transkriptions-Steigerung
beobachten (Abbildungen 3.2.1 und 3.2.2). Auch in der Koimmun-
präzipitation waren nach IL-6-Stimulation die präzipitierten Proteinmengen
erheblich größer, wie der Western Blot nachweisen konnte (nicht gezeigt).

72
Zusammengenommen wäre eine Interpretationsmöglichkeit eine vermehrte
intranukleäre Assoziation der beiden Transkriptionsfaktoren nach IL-6-
Stimulation.
Werden die Zellen nur mit Insulin behandelt, so findet sich eine andere
intrazelluläre Verteilung der Proteine: Sowohl STAT3 als auch FKHR
befinden sich im Zytoplasma und sind nicht mehr im Zellkern anzutreffen
(Abbildung 3.4.1 c). Dabei stellt sich STAT3 längst nicht mehr so diffus wie
in unstimulierten Zellen (Abbildung 3.4.1 a), sondern deutlich im Zyto-
plasma dar. In den Luziferaseversuchen resultiert nach Insulinstimulation
eine Abnahme der transkriptionellen Aktivität am alpha-2-Makroglobulin-
Promotor um etwa 25% bei Abwesenheit von IL-6 (Abbildung 3.1.2 und
3.1.3). Als Erklärungsmodell könnte dienen, dass FKHR nicht mehr intra-
nukleär lokalisiert ist und dadurch auch nicht mehr die Lokalisation von
STAT3 im Zellkern unterstützen kann.
Bei Inkubation mit Insulin und IL-6 resultiert für STAT3 eine intranukleäre
Lokalisation. FKHR weist eine diffuse, jedoch eher zytoplasmatische
Lokalisation auf (Abbildung 3.4.1 d). In den Luziferaseversuchen zeigte sich
eine deutliche Verringerung der Luziferase Aktivität im Vergleich zu nur
IL-6-behandelten Zellen (Abbildung 3.1.3). In Analogie zu den schon
erwähnten Erklärungsansätzen lassen auch diese Ergebnisse auf eine, für
die transkriptionelle Aktivität essentielle intranukleäre Interaktion schließen.
Ob beide Proteine schon vor der Stimulation zytoplasmatisch assoziiert
sind, kann nicht eindeutig beurteilt werden. Im Vergleich IL-6-stimulierter
Zellen mit und ohne Insulin kommt es durch Inkubation mit Insulin zum
nukleären Export von einem großen Teil des FKHR, und die Assoziation mit
STAT3 scheint weitgehend verloren zu gehen. Die mikroskopischen
Befunde zeigen weiterhin, dass nach IL-6-Stimualtion in Abwesenheit von
Insulin eine deutliche Kolokalisation beider Transkriptionsfaktoren im
Zellkern nachzuweisen ist.
Eine klare Aussage lassen diese Ergebnisse nicht zu. Allerdings scheint in
Abwesenheit von IL-6 durch einen Insulinstimulus sowohl FKHR als auch
STAT3 vermehrt im Zytoplasma lokalisiert zu sein. Dies spricht für die

73
These, dass FKHR die Kernlokalisation von STAT3 fördert, da andernfalls
FKHR im Zytoplasma und STAT3 - wie bei unstimulierten Zellen - weiterhin
diffus in der Zelle vorliegen würde.
4.2.2 FKHR könnte als Vermittler zwischen
STAT3 und weiteren Transkriptionsfaktoren
fungieren
Dass FKHR unabhängig von STAT3 eine vermehrte Transkription IL-6-
responsiver Gene bewirken kann, konnte ausgeschlossen werden, da
dominant negative STAT3-Faktoren dann die IL-6-Signaltransduktion nicht
nahezu vollständig unterdrücken könnten (Abbildung 3.2.4). Außerdem hat
FKHR auch einen verstärkenden Effekt auf die Transkription von Genen
unter Kontrolle des SIE-Promotors (Abbildung 3.2.3). Dort finden sich aber
keine potentiellen FKHR-Bindungsstellen, was eine STAT3-abhängige
Promotorbindung wahrscheinlich macht.
Des Weiteren handelt es sich nicht um eine unselektive Steigerung der
Aktivität der Transkriptionsmaschinerie, da dann auch andere Proteine
vermehrt gebildet würden. Für die Smad7- und ß-Caseinpromotoren wurde
gezeigt, dass FKHR keine Luziferase Aktivitätserhöhung bewirkt (Ab-
bildungen 3.2.5 und 3.2.7).
Als offene Frage bleibt jedoch, ob FKHR eventuell nur als Vermittler
zwischen den STAT-Faktoren einerseits und weiteren Transkriptions-
koaktivatoren andererseits fungiert.
Sowohl für DAF16, dem Orthologen aus Caenorhabditis elegans 97, als
auch für STAT3 und STAT1 159 wurde beschrieben, dass sie in der Lage
sind, p300/CREP-binding-protein (CBP) an Promotorbereiche zu rekru-
tieren.
Dabei wäre es möglich, dass auch FKHR weitere Transkriptionskoaktiva-
toren verstärkt zum STAT3 bringt oder einmal gebundene stabilisiert. In der
Vergangenheit wurde beispielsweise eine Assoziation von STAT3 und c-
Jun beschrieben 160.

74
4.2.3 FKHR könnte die Aktivierung von STAT-
Faktoren verstärken oder deren
Inaktivierung hemmen
Eine Hemmung von Inhibitoren der STAT3-Signaltransduktion, wie PIAS3
oder SOCS3, wäre ein denkbarer Mechanismus, wie FKHR die IL-6-
Signaltransduktion positiv beeinflussen könnte. Es wäre möglich, dass
FKHR am STAT3 Bindungsstellen für inhibitorische Proteine kompetitiv
blockiert und so in der Lage ist, die STAT-Aktivierung zu verlängern.
Wie in Abbildung 3.2.8 zu erkennen ist, stellt sich bei einer Koexpression
von PIAS3 und FKHR eine Steigerung der Transkriptions-Aktivität ein.
Diese ist umso größer je mehr FKHR kotransfiziert wird.
Nicht auszuschließen ist auch, dass FKHR auf andere inhibitorische
Proteine wie SHP2 oder SOCS3 hemmend wirkt. Ebenso ist denkbar, dass
es durch FKHR zu einer verstärkten Aktivierung von STAT3 kommt. So
könnte FKHR beispielsweise STAT3 vermehrt zum Rezeptor oder den
Januskinasen rekrutieren und so die Phosphorylierung und Dimerisierung
der STAT-Faktoren fördern.
4.2.4 FKHR könnte das STAT3-Dimer
stabilisieren
Eine weitere Möglichkeit, wie FKHR das IL-6-Signal positiv beeinflussen
könnte, wäre eine Stabilisierung der aktivierten STAT-Dimere. So könnten
die STAT3- und STAT1-Faktoren als Homo- oder Heterodimer zusammen-
gehalten werden. Durch eine Oligomerisierung mit FKHR und eventuell
weiteren Proteinen könnten Bindungsstellen für inhibitorische Proteine
besetzt / maskiert werden.

75
4.3 Das IL-6-Signal scheint die hemmende
Wirkung von FKHR auf die
Stoffwechselaktivität zu unterdrücken
Merkwürdigerweise fand sich in der Immunfluoreszenz bei Stimulation mit
Insulin und IL-6 für FKHR eine diffuse Verteilung im Zytoplasma und im
Zellkern, wohingegen die alleinige Gabe von Insulin zu einer klar zyto-
plasmatischen Lokalisation von FKHR führte (Abbildung 3.4.1).
Eine mögliche Begründung hierfür könnte sein, dass nicht nur FKHR die
STAT3-Dimere stabilisiert, sondern dass durch Interaktion von STAT3 und
FKHR auch FKHR vermehrt nukleär lokalisiert bleibt. Durch eine Hetero-
Oligomerisierung von FKHR und STAT-Faktoren könnten Phosphory-
lierungsstellen von FKHR maskiert werden und das Protein so vor dem
nukleären Export bewahrt werden. In diesem Fall sollte eine verstärkte
Transkription FKHR-abhängiger Gene resultieren. Wie schon in der Ein-
leitung erwähnt, wird FKHR für die vermehrte Bildung einer Vielzahl
proapoptotischer / proliferationshemmender Proteine verantwortlich ge-
macht. So konnte nach Überexpression der foxo-Familienmitglieder AFX,
FKHR und FKHRL1 eine Wachstumsinhibition von Hepatozyten durch
erhöhte p27Kip-Konzentrationen nachgewiesen werden 90-92. Als weiteres,
den Zellzyklus unterdrückendes Protein, ließ sich das proapoptotisch
wirkende Bim aus der Familie der Bcl2-Proteine bei Anwesenheit von
FKHRL1 verstärkt exprimieren 93. Die Transkription von Cyclinen des D-
Typs wird durch FKHR unterdrückt 94.
Zellen, die intranukleär erhöhte FKHR-Konzentrationen aufweisen, sollten
deshalb eine geringere Stoffwechselaktivität besitzen als Zellen, in denen
das FKHR zytoplasmatisch lokalisiert ist.
Für IL-6 werden in der Literatur unterschiedliche Funktionen auf die Proli-
feration verschiedener Zelltypen beschrieben. So konnte einerseits gezeigt
werden, dass IL-6 proliferative und antiapoptotische Signale sowohl in
Zelllinien des multiplen Myeloms, wie auch in Hep3B-Hepatomzellen
vermittelt 18,161,162. Ein in jüngster Zeit erschienener Artikel macht dafür die
Phosphorylierung und Inaktivierung von FKHR verantwortlich 163.

76
Andererseits wurde für maligne Melanomzellen eine IL-6-vermittelte
Wachstumsinhibition nachgewiesen 14,164,165.
Im XTT-Test (Abbildung 3.5.3) konnte bei IL-6-Gabe jedoch keine vermin-
derte Proliferation des HepG2-Kontroll-Klons beobachtet werden. Die
Zellen, die fluoreszierendes FKHR exprimieren, wuchsen in Anwesenheit
von IL-6 sogar besser.
Zumindest für die antiproliferativen Eigenschaften von FKHR ließ sich so
kein Synergismus von FKHR und Proteinen des IL-6-induzierten Jak /
STAT-Weges nachweisen. Würde STAT3 das foxo-Familienmitglied
unterstützen, so sollte insbesondere bei hohen Konzentrationen von IL-6
eine stärkere Wachstumsinhibition resultieren.
Eine mögliche Erklärung für dieses Ergebnis liefert die in der Einleitung
beschriebene Aktivierung der PI3-Kinase über den SHP2 / Ras / Raf-Weg
(siehe Kapitel 1-3). Nach diesem Modell würde ein IL-6-Stimulus neben der
STAT3-Rekrutierung zu einer Aktivierung der PI3-Kinase führen. Diese
könnte dann über Zwischenschritte Akt aktivieren, welches wiederum in der
Lage wäre, FKHR zu phosphorylieren und damit zu inaktivieren. So könnte
ein IL-6-Stimulus zu einem nukleären Export und zur Hemmung der
Wirkung von FKHR führen 150,166. Dieser Mechanismus wird auch in der
Literatur für den antiapoptotischen Effekt des IL-6 in Zelllinien des multiplen
Myeloms und Hep3B Hepatomzellen angeführt 18,161,162 (s.o.).
Für die in den durchgeführten Versuchen verwendeten HepG2-Zellen ließ
sich diese These jedoch nicht bestätigen. So konnte nach IL-6-Inkubation
weder eine verstärkte Phosphorylierung von PKB/Akt im Western Blot
(nicht gezeigt), noch eine zytoplasmatische Häufung von FKHR in der
Immunfluoreszenz nachgewiesen werden (Abbildung 3.4.1 b). Auch in der
Literatur ist dieser Weg bisher nicht für HepG2-Zellen beschrieben worden.
Demnach muss für die stoffwechselfördernde Wirkung des IL-6 ein anderer
Mechanismus verantwortlich sein, den es weiterhin zu identifizieren gilt.
Denkbar wäre eine Hemmung der Apoptose oder auch eine Förderung der
Proliferation.

77
In HepG2-Zellen wurde gezeigt, dass OnkostatinM und IL-6 eine Zell-
zyklushemmung durch erhöhte p27kip Konzentrationen bewirken 167, was
einen Widerspruch zu den Beobachtungen darstellt (Abbildung 3.5.3). Auf
die Proliferation und Stoffwechselaktivität des nur GFP exprimierenden
Klons schien IL-6 keinen Einfluß zu haben.
In den Immunfluoreszenzstudien (Abbildung 3.4.1) konnte auf einen reinen
IL-6-Stimulus keine deutliche Verhältnisverschiebung des FKHRs vom
Zellkern zu Gunsten des Zytoplasmas beobachtet werden. Ob der hohe
endogene STAT3-Gehalt der HepG2-Zellen und das Postulat, dass akti-
viertes STAT3 eventuell FKHR im Kern halten kann, eine Erklärung bieten,
bleibt Gegenstand weiterer Untersuchungen.

78

79
5 Ausblick Im Rahmen dieser Dissertationsarbeit konnte erstmals gezeigt werden,
dass die Transkriptionsfaktoren STAT3 und FKHR miteinander inter-
agieren. Den Beweis hierfür brachten Ergebnisse aus Luziferaseversuchen
und Koimmunpräzipitationen. Wie der genaue Mechanismus der Wechsel-
wirkung ist, bleibt jedoch weiterhin ungeklärt.
In Kapitel 4 wurden bereits mehrere Möglichkeiten diskutiert wie FKHR das
STAT3-Signal verstärken könnte. Allerdings haben die aufgezeigten
Ansätze zum heutigen Zeitpunkt rein spekulativen Charakter. Um den
genannten Postulaten nachzugehen und sie zu beweisen oder zu wider-
legen, bedarf es weiterer Experimente.
Im folgenden möchte ich einen Ausblick geben, wie diese Versuche
aussehen könnten und kurz umreißen, auf welche Fragen man sich
Antworten erhoffen kann.
5.1 Generierung FKHR-defizienter Zellen
Experimente mit FKHR-defizienten Zellen sind von großem Interesse, da
mit Hilfe solcher Zelllinien der Einfluss von FKHR auf die IL-6-Signaltrans-
duktion überprüft werden könnte. Da die, in den beschriebenen Versuchen
verwendeten HepG2-Zellen endogenes FKHR enthalten, lässt sich keine
Aussage treffen, ob der foxo-Transkriptionsfaktor FKHR essentiell für die
Induktion STAT3-abhängiger Gene ist. Um die Bedingungen möglichst
konstant zu halten, wäre es optimal, HepG2-Zellen so zu verändern, dass
das vorhandene FKHR nur noch in minimaler Konzentration vorliegt. Um
dieses Ziel zu erreichen, gibt es verschiedene Möglichkeiten. Einerseits
kann man anti-sense-DNA-Konstrukte verwenden. Die nach Transfektion
daraus gebildete anti-sense-mRNA hybridisiert mit der endogenen FKHR-
mRNA und blockiert die Translation. Andererseits und als neuere Methode
kommt auch eine siRNA Transfektion in Frage. Hier kommt es zur Zer-
störung der FKHR-mRNA.

80
Wenn die Interaktionsbereiche von FKHR und STAT3 besser verstanden
sind, lassen sich dominant negative Transkriptionsfaktoren herstellen. Mit
diesen könnten durch kompetetive Hemmung die Auswirkungen des einen
Partners auf die Signaltransduktion des Anderen weiter untersucht werden.
Auch Studien mit Zellen von FKHR knock-out-Tieren könnten interessante
Resultate liefern. Tiere genetisch so zu manipulierten wäre jedoch mit sehr
hohem Aufwand verbunden.
5.2 Identifizierung der interagierenden
Bereiche
Da für FKHR und STAT3 eine physikalische Assoziation nachgewiesen
werden konnte, wäre es nötig zu analysieren, welche Regionen und
Aminosäure-Reste für die Interaktion verantwortlich sind. Die Lösung der
Tertiärstruktur des Komplexes würde natürlich das beste Verständnis der
Wechselwirkung erlauben. Da dies ein nur mit sehr großem Aufwand zu
erreichendes Ziel ist, könnten zunächst die Aminosäure-Reste identifiziert
werden, welche die Interaktion vermitteln. Durch gezielte Punktmutationen
ließen sich die dabei wichtigen Bereiche ermitteln. In Abschnitt 4.1 der
Diskussion wurde bereits vorgeschlagen, dass die Interaktion am C-
Terminus des FKHR stattfindet.
Auch durch den Einsatz von Chimären ließen sich verantwortliche Bereiche
eingrenzen. Als Beispiel sind Konstrukte zu nennen, die für definierte
Domänen von STAT3 und STAT5 kodieren.
Wenn die Aminosäure-Reste identifiziert sind und Mutanten vorliegen,
welche nicht mehr in der Lage sind zu assoziieren wäre dies ein wichtiger
Schritt zum Verständnis des Interaktions-Mechnismus.

81
5.3 Auswirkungen auf die Interaktion mit
weiteren Proteinen
Auch weitere Koimmunpräzipitationen könnten Aufschluss über einige
interessante Fragestellungen geben. Sowohl für STAT-Faktoren 153,160,168,
als auch für FKHR 90,92,93,95-97,151 konnten Interaktionen mit verschiedenen
Transkriptionskoaktivatoren nachgewiesen werden. Mit solchen Versuchen
ließe sich der gegenseitige Einfluss auf die Bindung dieser Faktoren
studieren. So ist fraglich, in welchem Maße die einzelnen Proteine mitein-
ander assoziieren und sich dabei gegenseitig behindern oder gar ver-
stärken und zu einer Bindung anderer Proteine beitragen können.
Ebenfalls ist von Interesse, ob FKHR schon bei der Rezeptoraktivierung
eine Rolle spielt. Um dies herauszufinden könnte man Koimmunprä-
zipitationen mit Jaks durchführen.
Auch die Frage ob FKHR die STAT-Dimere stabilisiert und vor negativ
wirkenden Regulatoren wie PIAS3 oder SOCS3 schützt, bleibt zu beant-
worten. Versuche bezüglich der Kinetik können hierfür Anhaltspunkte
liefern. Mit solchen Experimenten lässt sich klären, ob bei Anwesenheit von
FKHR die STAT-Faktoren länger aktiv bleiben oder ein höheres Akivitäts-
Maximum erreichen.
5.4 in-vivo-Versuchsansätze
Im Laufe dieser Arbeit wurden bereits stabil transfizierte Zellen generiert,
die ein Fusionsprotein aus FKHR und einem fluoreszierenden Protein
exprimieren. Mit Hilfe dieser Zelllinien lassen sich Studien mit lebenden
Zellen durchführen. Dabei könnte mit Hilfe der konfokalen laser-scanning
Mikroskopie untersucht werden, wie FKHR in nativen Zellen auf ver-
schiedene Stimuli reagiert. Insbesondere wäre dabei auch die zeitliche
Komponente von Interesse.

82
Da auch STAT3 in der Arbeitsgruppe als fluoreszierendes Protein vorliegt,
könnte man zum einen eventuell über die FRET-Technik die Interaktion mit
FKHR weiter spezifizieren, veranschaulichen und verifizieren. Zum anderen
lässt dies ein Verfahren zu, Punktmutanten auf Funktionalität zu überprüfen
und so die interagierenden Bereiche zu identifizieren.
Durch die Generierung von mit beiden Konstrukten stabil transfizierten
Zellen könnten weitere Experimente unbeantwortete Fragen klären. Da
STAT3 und FKHR aber mit dem fluoreszierenden Protein gekoppelt sind
und durch Transfektion auch eine Überexpression vorliegt, handelt es sich
um ein artifizielles System.
Trotzdem lassen Studien mit solchen lebenden Zellen eindeutigerere
Ergebnisse als Versuche mit fixierten Zellen erhoffen. Insbesonders
bezüglich der zeitlichen Lokalisation von Proteinen in Abhängigkeit von
verschiedenen Stimulanzien bieten solche Zelllinien Vorteile. Eine Voraus-
setzung dafür ist, dass beide Proteine funktionell sind, was in Vorversuchen
gezeigt werden muss. Für die Steigerung der Reportergenaktivität unter
Kontrolle des alpha-2-Makroglobulin-Promotors ist dieses für die FKHR-
Expressionskonstrukte bereits geschehen (Abbildung 3.5.1). Im Falle des
STAT3 konnten Ulrike Sommer und Serge Haan eine Funktionalität des
fluoreszierenden Fusionsproteins ebenfalls nachweisen.
5.5 Studien zur DNA-Bindung von FKHR
Ob FKHR an die DNA binden muss, um die transkriptionelle Steigerung zu
bewirken, ist noch nicht hinreichend geklärt und bedarf weiterer Studien.
Hierzu bieten sich Versuche mit mutieren Promotoren von Reportergen-
Expressionsvektoren an.

83
5.6 Untersuchungen mit FKHR responsiven
Promotoren
Wie in Kapitel 3.5 gezeigt, wuchsen stabil mit FKHR transfizierte HepG2-
Zellen erwartungsgemäß schlechter als die Kontrollen. Unter IL-6-Stimu-
lation war jedoch eine höhere Proliferation nachweisbar als bei unstimu-
lierten. Ob durch das IL-6-Signal oder durch aktivierte STAT-Faktoren
eventuell eine Inhibierung von FKHR erfolgt bleibt zu beweisen.
Dafür wären Versuche mit FKHR-abhängigen Promotoren viel ver-
sprechend. Mit Hilfe solcher Studien ließe sich zeigen, ob es zu einer
bilateralen transkriptionellen Steigerung responsiver Gene kommt oder ob
der IL-6-Signaltransduktionsweg FKHR bei der Ausübung seiner bio-
logischen Funktion hemmt. Für FKHR konnte eine verstärkte Transkription
STAT3-induzierbarer Gene nachgewiesen werden. Ob STAT3 in der Lage
ist, die FKHR-abhängige Proteinbiosynthese zu unterstützen oder sie eher
unterdrückt, bleibt bis zur Durchführung dieser Experimente unklar.
So bleiben viele interessante Versuche durchzuführen, die Antworten auf
die verschiedenen Fragen geben können.

84

85
6 Zusammenfassung Die Tatsache, dass Wachstumsfaktoren in der Lage sind, Hepatozyten zur
Proliferation anzuregen, es jedoch gleichzeitig zu einer verminderten
Synthese von Akut-Phase-Proteinen kommt, wurde in verschiedenen
Veröffentlichungen beschrieben 127-129. Im Rahmen der vorgelegten Arbeit
wird eine mögliche Begründung für diese Beobachtungen aufgezeigt.
Ein wichtiger Signaltransduktionsweg von Wachstumsfaktoren verläuft über
eine Aktivierung der PI3-Kinase und der ProteinkinaseB. Das IL-6-Signal
wird in erster Linie über Januskinasen und STAT-Faktoren innerhalb der
Zelle weitergeleitet.
In dieser Arbeit wurde erstmals der Transkriptionsfaktor FKHR als ein
Bindeglied zwischen dem PI3-Kinase/PKB-Signalweg und der IL-6/gp130/
Jak/STAT-Kaskade identifiziert.
Es konnte gezeigt werden, dass die Transkriptionsfaktoren STAT3 und
FKHR synergistisch zusammen wirken und dass FKHR in der Lage ist, die
STAT3 vermittelte, IL-6-induzierte Transkription responsiver Gene signi-
fikant zu steigern. Eine STAT3-unabhängige Geninduktion am alpha-2-
Makroglobulin-Promotor durch FKHR wurde ausgeschlossen. Es konnte
eine physikalische Interaktion von FKHR und STAT3 nachgewiesen
werden. In Immunfluoreszenzstudien ließ sich insbesondere nach einem
IL-6-Stimulus eine deutliche Kolokalisation beider Proteine im Zellkern
erkennen. Die Wechselwirkung zwischen dem foxo-Transkriptionsfaktor
FKHR und bestimmten Faktoren der STAT Familie ist spezifisch. Neben
STAT3 wurde auch für STAT1, nicht jedoch für STAT5 oder Smad7 ein
synergistischer Effekt beobachtet.
Wachstumsfaktoren aktivieren über die PI3-Kinase die ProteinkinaseB,
welche durch Serin/Threonin Phosphorylierung den nukleären Export von
FKHR einleitet. Es ist gezeigt worden, dass die Stimulation von HepG2-
Zellen mit Insulin oder FCS in einer Abnahme der Akut-Phase-Protein-
Synthese und Sekretion resultiert. Dem Rückschluss Rechnung tragend
bewirkt die Hemmung der PI3-Kinase ebenso wie nicht phospho-
rylierungsfähige Mutanten von FKHR trotz FCS- oder Insulin-haltiger
Medien starke transkriptionelle Induktionen von Akut-Phase-Proteinen.

86

87
7 Literaturverzeichnis 1. Bazan, J.F. (1990). Haemopoietic receptors and helical cytokines. Immunol Today 11, 350-354. 2. Heinrich, P.C., Behrmann, I., Müller-Newen, G., Schaper, F. & Graeve, L. (1998). Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334 ( Pt 2), 297-314. 3. Senaldi, G., Varnum, B.C., Sarmiento, U., Starnes, C., Lile, J., Scully, S., Guo, J.,
Elliott, G., McNinch, J., Shaklee, C.L., Freeman, D., Manu, F., Simonet, W.S., Boone, T. & Chang, M.S. (1999).
Novel neurotrophin-1/B cell-stimulating factor-3: a cytokine of the IL-6 family. Proc Natl Acad Sci U S A 96, 11458-11463. 4. Hirano, T., Yasukawa, K., Harada, H., Taga, T., Watanabe, Y., Matsuda, T.,
Kashiwamura, S., Nakajima, K., Koyama, K., Iwamatsu, A., Tsunasawa, S., Sakiyama, F., Matsui, H., Takahara, Y., Taniguchi, T. & Kishimoto, T. (1986).
Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin.
Nature 324, 73-76. 5. Dufhues, G. (1991). Postranslationale Modifikation von humanem IL-6: Charakterisierung der
Kohlenhydrat-Seitenketten, ihre Lokalisation und Funktion. Institut für Biochemie der RWTH Aachen, Dissertation. 6. Heinrich, P.C., Castell, J.V. & Andus, T. (1990). Interleukin-6 and the acute phase response. Biochem J 265, 621-636. 7. Gauldie, J., Richards, C., Harnish, D., Lansdorp, P. & Baumann, H. (1987). Interferon beta 2/B-cell stimulatory factor type 2 shares identity with monocyte-
derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells.
Proc Natl Acad Sci U S A 84, 7251-7255. 8. Baumann, H. & Schendel, P. (1991). Interleukin-11 regulates the hepatic expression of the same plasma protein genes as
interleukin-6. J Biol Chem 266, 20424-20427. 9. Andus, T., Geiger, T., Hirano, T., Kishimoto, T., Tran-Thi, T.A., Decker, K. &
Heinrich, P.C. (1988). Regulation of synthesis and secretion of major rat acute-phase proteins by
recombinant human interleukin-6 (BSF-2/IL-6) in hepatocyte primary cultures. Eur J Biochem 173, 287-293. 10. Castell, J.V., Gomez-Lechon, M.J., David, M., Hirano, T., Kishimoto, T. &
Heinrich, P.C. (1988). Recombinant human interleukin-6 (IL-6/BSF-2/HSF) regulates the synthesis of acute
phase proteins in human hepatocytes. FEBS Lett 232, 347-350.

88
11. Andus, T., Geiger, T., Hirano, T., Northoff, H., Ganter, U., Bauer, J., Kishimoto, T.
& Heinrich, P.C. (1987). Recombinant human B cell stimulatory factor 2 (BSF-2/IFN-beta 2) regulates beta-
fibrinogen and albumin mRNA levels in Fao-9 cells. FEBS Lett 221, 18-22. 12. Tilg, H., Dinarello, C.A. & Mier, J.W. (1997). IL-6 and APPs: anti-inflammatory and immunosuppressive mediators. Immunol Today 18, 428-432. 13. Mori, S., Murakami-Mori, K. & Bonavida, B. (1999). Interleukin-6 induces G1 arrest through induction of p27(Kip1), a cyclin-dependent
kinase inhibitor, and neuron-like morphology in LNCaP prostate tumor cells. Biochem Biophys Res Commun 257, 609-614. 14. Kortylewski, M., Heinrich, P.C., Mackiewicz, A., Schniertshauer, U., Klingmuller,
U., Nakajima, K., Hirano, T., Horn, F. & Behrmann, I. (1999). Interleukin-6 and oncostatin M-induced growth inhibition of human A375 melanoma
cells is STAT-dependent and involves upregulation of the cyclin-dependent kinase inhibitor p27/Kip1.
Oncogene 18, 3742-3753. 15. Anderson, K.C., Jones, R.M., Morimoto, C., Leavitt, P. & Barut, B.A. (1989). Response patterns of purified myeloma cells to hematopoietic growth factors. Blood 73, 1915-1924. 16. Kawano, M., Hirano, T., Matsuda, T., Taga, T., Horii, Y., Iwato, K., Asaoku, H.,
Tang, B., Tanabe, O., Tanaka, H., Kuramoto, A. & Kishimoto, T. (1988). Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature 332, 83-85. 17. Kunioku, H., Inoue, K. & Tomida, M. (2001). Interleukin-6 protects rat PC12 cells from serum deprivation or chemotherapeutic
agents through the phosphatidylinositol 3-kinase and STAT3 pathways. Neurosci Lett 309, 13-16. 18. Kuo, M.L., Chuang, S.E., Lin, M.T. & Yang, S.Y. (2001). The involvement of PI 3-K/Akt-dependent up-regulation of Mcl-1 in the prevention of
apoptosis of Hep3B cells by interleukin-6. Oncogene 20, 677-685. 19. Hardin, J., MacLeod, S., Grigorieva, I., Chang, R., Barlogie, B., Xiao, H. &
Epstein, J. (1994). Interleukin-6 prevents dexamethasone-induced myeloma cell death. Blood 84, 3063-3070. 20. Lichtenstein, A., Tu, Y., Fady, C., Vescio, R. & Berenson, J. (1995). Interleukin-6 inhibits apoptosis of malignant plasma cells. Cell Immunol 162, 248-255. 21. Akira, S., Taga, T. & Kishimoto, T. (1993). Interleukin-6 in biology and medicine. Adv Immunol 54, 1-78. 22. Boulanger, M.J., Chow, D.C., Brevnova, E.E. & Garcia, K.C. (2003). Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130
complex. Science 300, 2101-2104.

89
23. Guschin, D., Rogers, N., Briscoe, J., Witthuhn, B., Watling, D., Horn, F.,
Pellegrini, S., Yasukawa, K., Heinrich, P., Stark, G.R. & Kerr, I.M. (1995). A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction
pathway in response to interleukin-6. Embo J 14, 1421-1429. 24. Rodig, S.J., Meraz, M.A., White, J.M., Lampe, P.A., Riley, J.K., Arthur, C.D., King,
K.L., Sheehan, K.C., Yin, L., Pennica, D., Johnson, E.M., Jr. & Schreiber, R.D. (1998).
Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses.
Cell 93, 373-383. 25. Tanner, J.W., Chen, W., Young, R.L., Longmore, G.D. & Shaw, A.S. (1995). The conserved box 1 motif of cytokine receptors is required for association with JAK
kinases. J Biol Chem 270, 6523-6530. 26. Murakami, M., Narazaki, M., Hibi, M., Yawata, H., Yasukawa, K., Hamaguchi, M.,
Taga, T. & Kishimoto, T. (1991). Critical cytoplasmic region of the interleukin 6 signal transducer gp130 is conserved in
the cytokine receptor family. Proc Natl Acad Sci U S A 88, 11349-11353. 27. Pellegrini, S. & Dusanter-Fourt, I. (1997). The structure, regulation and function of the Janus kinases (JAKs) and the signal
transducers and activators of transcription (STATs). Eur J Biochem 248, 615-633. 28. Haan, C., Hermanns, H.M., Heinrich, P.C. & Behrmann, I. (2000). A single amino acid substitution (Trp(666)-->Ala) in the interbox1/2 region of the
interleukin-6 signal transducer gp130 abrogates binding of JAK1, and dominantly impairs signal transduction.
Biochem J 349, 261-266. 29. Haan, C., Heinrich, P.C. & Behrmann, I. (2002). Structural requirements of the interleukin-6 signal transducer gp130 for its interaction
with Janus kinase 1: the receptor is crucial for kinase activation. Biochem J 361, 105-111. 30. Harpur, A.G., Andres, A.C., Ziemiecki, A., Aston, R.R. & Wilks, A.F. (1992). JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene 7, 1347-1353. 31. Yeh, T.C., Dondi, E., Uze, G. & Pellegrini, S. (2000). A dual role for the kinase-like domain of the tyrosine kinase Tyk2 in interferon-alpha
signaling. Proc Natl Acad Sci U S A 97, 8991-8996. 32. Saharinen, P., Takaluoma, K. & Silvennoinen, O. (2000). Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol 20, 3387-3395. 33. Hilkens, C.M., Is'harc, H., Lillemeier, B.F., Strobl, B., Bates, P.A., Behrmann, I. &
Kerr, I.M. (2001). A region encompassing the FERM domain of Jak1 is necessary for binding to the
cytokine receptor gp130. FEBS Lett 505, 87-91.

90
34. Haan, C., Is'harc, H., Hermanns, H.M., Schmitz-Van De Leur, H., Kerr, I.M.,
Heinrich, P.C., Grotzinger, J. & Behrmann, I. (2001). Mapping of a region within the N terminus of Jak1 involved in cytokine receptor
interaction. J Biol Chem 276, 37451-37458. 35. Heinrich, P.C., Behrmann, I., Haan, S., Hermanns, H.M., Muller-Newen, G. &
Schaper, F. (2003). Principles of IL-6-type cytokine signalling and its regulation. Biochem J Pt. 36. Schmitz, J., Dahmen, H., Grimm, C., Gendo, C., Müller-Newen, G., Heinrich, P.C.
& Schaper, F. (2000). The cytoplasmic tyrosine motifs in full-length glycoprotein 130 have different roles in
IL-6 signal transduction. J Immunol 164, 848-854. 37. Lehmann, U., Schmitz, J., Weissenbach, M., Sobota, R.M., Hortner, M.,
Friederichs, K., Behrmann, I., Tsiaris, W., Sasaki, A., Schneider-Mergener, J., Yoshimura, A., Neel, B.G., Heinrich, P.C. & Schaper, F. (2003).
SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130.
J Biol Chem 278, 661-671. 38. Schmitz, J., Weissenbach, M., Haan, S., Heinrich, P.C. & Schaper, F. (2000). SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the
SHP2 recruitment site of gp130. J Biol Chem 275, 12848-12856. 39. Heim, M.H., Kerr, I.M., Stark, G.R. & Darnell, J.E., Jr. (1995). Contribution of STAT SH2 groups to specific interferon signaling by the Jak-STAT
pathway. Science 267, 1347-1349. 40. Hemmann, U., Gerhartz, C., Heesel, B., Sasse, J., Kurapkat, G., Grotzinger, J.,
Wollmer, A., Zhong, Z., Darnell, J.E., Jr., Graeve, L., Heinrich, P.C. & Horn, F. (1996).
Differential activation of acute phase response factor/Stat3 and Stat1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. II. Src homology SH2 domains define the specificity of stat factor activation.
J Biol Chem 271, 12999-13007. 41. Gerhartz, C., Heesel, B., Sasse, J., Hemmann, U., Landgraf, C., Schneider-
Mergener, J., Horn, F., Heinrich, P.C. & Graeve, L. (1996). Differential activation of acute phase response factor/STAT3 and STAT1 via the
cytoplasmic domain of the interleukin 6 signal transducer gp130. I. Definition of a novel phosphotyrosine motif mediating STAT1 activation.
J Biol Chem 271, 12991-12998. 42. Stahl, N., Farruggella, T.J., Boulton, T.G., Zhong, Z., Darnell, J.E., Jr. &
Yancopoulos, G.D. (1995). Choice of STATs and other substrates specified by modular tyrosine-based motifs in
cytokine receptors. Science 267, 1349-1353. 43. Zhong, Z., Wen, Z. & Darnell, J.E., Jr. (1994). Stat3: a STAT family member activated by tyrosine phosphorylation in response to
epidermal growth factor and interleukin-6. Science 264, 95-98.

91
44. Lütticken, C., Wegenka, U.M., Yuan, J., Buschmann, J., Schindler, C., Ziemiecki,
A., Harpur, A.G., Wilks, A.F., Yasukawa, K., Taga, T., Kishimoto, T., Barbieri, G., Pellegrini, S., Sendtner, M., Heinrich, P.C. & Horn, F. (1994).
Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130.
Science 263, 89-92. 45. Stahl, N., Boulton, T.G., Farruggella, T., Ip, N.Y., Davis, S., Witthuhn, B.A.,
Quelle, F.W., Silvennoinen, O., Barbieri, G., Pellegrini, S., Ihle, J.N. & Yancopoulos, G.D. (1994).
Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components.
Science 263, 92-95. 46. Freeman, R.M., Jr., Plutzky, J. & Neel, B.G. (1992). Identification of a human src homology 2-containing protein-tyrosine-phosphatase: a
putative homolog of Drosophila corkscrew. Proc Natl Acad Sci U S A 89, 11239-11243. 47. Perkins, L.A., Larsen, I. & Perrimon, N. (1992). corkscrew encodes a putative protein tyrosine phosphatase that functions to
transduce the terminal signal from the receptor tyrosine kinase torso. Cell 70, 225-236. 48. Kazlauskas, A., Feng, G.S., Pawson, T. & Valius, M. (1993). The 64-kDa protein that associates with the platelet-derived growth factor receptor
beta subunit via Tyr-1009 is the SH2-containing phosphotyrosine phosphatase Syp. Proc Natl Acad Sci U S A 90, 6939-6943. 49. Lechleider, R.J., Sugimoto, S., Bennett, A.M., Kashishian, A.S., Cooper, J.A.,
Shoelson, S.E., Walsh, C.T. & Neel, B.G. (1993). Activation of the SH2-containing phosphotyrosine phosphatase SH-PTP2 by its
binding site, phosphotyrosine 1009, on the human platelet-derived growth factor receptor.
J Biol Chem 268, 21478-21481. 50. Yin, T., Shen, R., Feng, G.S. & Yang, Y.C. (1997). Molecular characterization of specific interactions between SHP-2 phosphatase and
JAK tyrosine kinases. J Biol Chem 272, 1032-1037. 51. Schaper, F., Gendo, C., Eck, M., Schmitz, J., Grimm, C., Anhuf, D., Kerr, I.M. &
Heinrich, P.C. (1998). Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal
transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression.
Biochem J 335 ( Pt 3), 557-565. 52. Pluskey, S., Wandless, T.J., Walsh, C.T. & Shoelson, S.E. (1995). Potent stimulation of SH-PTP2 phosphatase activity by simultaneous occupancy of
both SH2 domains. J Biol Chem 270, 2897-2900. 53. Schiemann, W.P., Bartoe, J.L. & Nathanson, N.M. (1997). Box 3-independent signaling mechanisms are involved in leukemia inhibitory factor
receptor alpha- and gp130-mediated stimulation of mitogen-activated protein kinase. Evidence for participation of multiple signaling pathways which converge at Ras.
J Biol Chem 272, 16631-16636.

92
54. Li, W., Nishimura, R., Kashishian, A., Batzer, A.G., Kim, W.J., Cooper, J.A. &
Schlessinger, J. (1994). A new function for a phosphotyrosine phosphatase: linking GRB2-Sos to a receptor
tyrosine kinase. Mol Cell Biol 14, 509-517. 55. Nicholson, S.E., De Souza, D., Fabri, L.J., Corbin, J., Willson, T.A., Zhang, J.G.,
Silva, A., Asimakis, M., Farley, A., Nash, A.D., Metcalf, D., Hilton, D.J., Nicola, N.A. & Baca, M. (2000).
Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130.
Proc Natl Acad Sci U S A 97, 6493-6498. 56. Stahl, N., Farruggella, T.J., Boulton, T.G., Zhong, Z., Darnell, J.E., Jr. &
Yancopoulos, G.D. (1995). Choice of STATs and other substrates specified by modular tyrosine- based motifs in
cytokine receptors. Science 267, 1349-1353. 57. Shuai, K. (1999). The STAT family of proteins in cytokine signaling. Prog Biophys Mol Biol 71, 405-422. 58. Jiao, H., Berrada, K., Yang, W., Tabrizi, M., Platanias, L.C. & Yi, T. (1996). Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-
containing protein tyrosine phosphatase SHP-1. Mol Cell Biol 16, 6985-6992. 59. Starr, R., Willson, T.A., Viney, E.M., Murray, L.J., Rayner, J.R., Jenkins, B.J.,
Gonda, T.J., Alexander, W.S., Metcalf, D., Nicola, N.A. & Hilton, D.J. (1997). A family of cytokine-inducible inhibitors of signalling. Nature 387, 917-921. 60. Endo, T.A., Masuhara, M., Yokouchi, M., Suzuki, R., Sakamoto, H., Mitsui, K.,
Matsumoto, A., Tanimura, S., Ohtsubo, M., Misawa, H., Miyazaki, T., Leonor, N., Taniguchi, T., Fujita, T., Kanakura, Y., Komiya, S. & Yoshimura, A. (1997).
A new protein containing an SH2 domain that inhibits JAK kinases. Nature 387, 921-924. 61. Naka, T., Narazaki, M., Hirata, M., Matsumoto, T., Minamoto, S., Aono, A.,
Nishimoto, N., Kajita, T., Taga, T., Yoshizaki, K., Akira, S. & Kishimoto, T. (1997). Structure and function of a new STAT-induced STAT inhibitor. Nature 387, 924-929. 62. Hilton, D.J., Richardson, R.T., Alexander, W.S., Viney, E.M., Willson, T.A.,
Sprigg, N.S., Starr, R., Nicholson, S.E., Metcalf, D. & Nicola, N.A. (1998). Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A 95, 114-119. 63. Adams, T.E., Hansen, J.A., Starr, R., Nicola, N.A., Hilton, D.J. & Billestrup, N.
(1998). Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a
novel inhibitor of cytokine receptor signaling. J Biol Chem 273, 1285-1287. 64. Sasaki, A., Yasukawa, H., Suzuki, A., Kamizono, S., Syoda, T., Kinjyo, I., Sasaki,
M., Johnston, J.A. & Yoshimura, A. (1999). Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by
binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 4, 339-351.

93
65. Yasukawa, H., Misawa, H., Sakamoto, H., Masuhara, M., Sasaki, A., Wakioka, T.,
Ohtsuka, S., Imaizumi, T., Matsuda, T., Ihle, J.N. & Yoshimura, A. (1999). The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in
the activation loop. Embo J 18, 1309-1320. 66. Narazaki, M., Fujimoto, M., Matsumoto, T., Morita, Y., Saito, H., Kajita, T.,
Yoshizaki, K., Naka, T. & Kishimoto, T. (1998). Three distinct domains of SSI-1/SOCS-1/JAB protein are required for its suppression
of interleukin 6 signaling. Proc Natl Acad Sci U S A 95, 13130-13134. 67. Nicholson, S.E., Willson, T.A., Farley, A., Starr, R., Zhang, J.G., Baca, M.,
Alexander, W.S., Metcalf, D., Hilton, D.J. & Nicola, N.A. (1999). Mutational analyses of the SOCS proteins suggest a dual domain requirement but
distinct mechanisms for inhibition of LIF and IL-6 signal transduction. Embo J 18, 375-385. 68. Zhang, J.G., Farley, A., Nicholson, S.E., Willson, T.A., Zugaro, L.M., Simpson,
R.J., Moritz, R.L., Cary, D., Richardson, R., Hausmann, G., Kile, B.J., Kent, S.B., Alexander, W.S., Metcalf, D., Hilton, D.J., Nicola, N.A. & Baca, M. (1999).
The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation.
Proc Natl Acad Sci U S A 96, 2071-2076. 69. Kamura, T., Sato, S., Haque, D., Liu, L., Kaelin, W.G., Jr., Conaway, R.C. &
Conaway, J.W. (1998). The Elongin BC complex interacts with the conserved SOCS-box motif present in
members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev 12, 3872-3881. 70. Liao, J., Fu, Y. & Shuai, K. (2000). Distinct roles of the NH2- and COOH-terminal domains of the protein inhibitor of
activated signal transducer and activator of transcription (STAT) 1 (PIAS1) in cytokine-induced PIAS1-Stat1 interaction.
Proc Natl Acad Sci U S A 97, 5267-5272. 71. Chung, C.D., Liao, J., Liu, B., Rao, X., Jay, P., Berta, P. & Shuai, K. (1997). Specific inhibition of Stat3 signal transduction by PIAS3. Science 278, 1803-1805. 72. Liu, B., Liao, J., Rao, X., Kushner, S.A., Chung, C.D., Chang, D.D. & Shuai, K.
(1998). Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci U S A 95, 10626-10631. 73. Zohlnhofer, D., Graeve, L., Rose-John, S., Schooltink, H., Dittrich, E. & Heinrich,
P.C. (1992). The hepatic interleukin-6 receptor. Down-regulation of the interleukin-6 binding
subunit (gp80) by its ligand. FEBS Lett 306, 219-222. 74. Dittrich, E., Rose-John, S., Gerhartz, C., Mullberg, J., Stoyan, T., Yasukawa, K.,
Heinrich, P.C. & Graeve, L. (1994). Identification of a region within the cytoplasmic domain of the interleukin-6 (IL-6)
signal transducer gp130 important for ligand-induced endocytosis of the IL-6 receptor. J Biol Chem 269, 19014-19020.

94
75. Skolnik, E.Y., Batzer, A., Li, N., Lee, C.H., Lowenstein, E., Mohammadi, M.,
Margolis, B. & Schlessinger, J. (1993). The function of GRB2 in linking the insulin receptor to Ras signaling pathways. Science 260, 1953-1955. 76. Li, N., Batzer, A., Daly, R., Yajnik, V., Skolnik, E., Chardin, P., Bar-Sagi, D.,
Margolis, B. & Schlessinger, J. (1993). Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine
kinases to Ras signalling. Nature 363, 85-88. 77. Buday, L. & Downward, J. (1993). Epidermal growth factor regulates the exchange rate of guanine nucleotides on
p21ras in fibroblasts. Mol Cell Biol 13, 1903-1910. 78. Buday, L. & Downward, J. (1993). Epidermal growth factor regulates p21ras through the formation of a complex of
receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 73, 611-620. 79. Rodriguez-Viciana, P., Warne, P.H., Vanhaesebroeck, B., Waterfield, M.D. &
Downward, J. (1996). Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. Embo J 15, 2442-2451. 80. Rodriguez-Viciana, P., Marte, B.M., Warne, P.H. & Downward, J. (1996). Phosphatidylinositol 3' kinase: one of the effectors of Ras. Philos Trans R Soc Lond B Biol Sci 351, 225-231; discussion 231-222. 81. Rodriguez-Viciana, P., Warne, P.H., Khwaja, A., Marte, B.M., Pappin, D., Das, P.,
Waterfield, M.D., Ridley, A. & Downward, J. (1997). Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin
cytoskeleton by Ras. Cell 89, 457-467. 82. Scita, G., Tenca, P., Frittoli, E., Tocchetti, A., Innocenti, M., Giardina, G. & Di
Fiore, P.P. (2000). Signaling from Ras to Rac and beyond: not just a matter of GEFs. Embo J 19, 2393-2398. 83. Schaeffer, H.J. & Weber, M.J. (1999). Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19, 2435-2444. 84. Lu, W., Gong, D., Bar-Sagi, D. & Cole, P.A. (2001). Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine
phosphorylation of SHP-2 in cell signaling. Mol Cell 8, 759-769. 85. Bennicelli, J.L., Fredericks, W.J., Wilson, R.B., Rauscher, F.J., 3rd & Barr, F.G.
(1995). Wild type PAX3 protein and the PAX3-FKHR fusion protein of alveolar
rhabdomyosarcoma contain potent, structurally distinct transcriptional activation domains.
Oncogene 11, 119-130.

95
86. Biegel, J.A., Nycum, L.M., Valentine, V., Barr, F.G. & Shapiro, D.N. (1995). Detection of the t(2;13)(q35;q14) and PAX3-FKHR fusion in alveolar
rhabdomyosarcoma by fluorescence in situ hybridization. Genes Chromosomes Cancer 12, 186-192. 87. Downing, J.R., Khandekar, A., Shurtleff, S.A., Head, D.R., Parham, D.M.,
Webber, B.L., Pappo, A.S., Hulshof, M.G., Conn, W.P. & Shapiro, D.N. (1995). Multiplex RT-PCR assay for the differential diagnosis of alveolar rhabdomyosarcoma
and Ewing's sarcoma. Am J Pathol 146, 626-634. 88. Anderson, J., Ramsay, A., Gould, S. & Pritchard-Jones, K. (2001). Pax3-fkhr induces morphological change and enhances cellular proliferation and
invasion in rhabdomyosarcoma. Am J Pathol 159, 1089-1096. 89. Borkhardt, A., Repp, R., Haas, O.A., Leis, T., Harbott, J., Kreuder, J.,
Hammermann, J., Henn, T. & Lampert, F. (1997). Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias
with a t(X;11)(q13;q23). Oncogene 14, 195-202. 90. Medema, R.H., Kops, G.J., Bos, J.L. & Burgering, B.M. (2000). AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB
through p27kip1. Nature 404, 782-787. 91. Nakamura, N., Ramaswamy, S., Vazquez, F., Signoretti, S., Loda, M. & Sellers,
W.R. (2000). Forkhead transcription factors are critical effectors of cell death and cell cycle arrest
downstream of PTEN. Mol Cell Biol 20, 8969-8982. 92. Dijkers, P.F., Medema, R.H., Pals, C., Banerji, L., Thomas, N.S., Lam, E.W.,
Burgering, B.M., Raaijmakers, J.A., Lammers, J.W., Koenderman, L. & Coffer, P.J. (2000).
Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1).
Mol Cell Biol 20, 9138-9148. 93. Dijkers, P.F., Medema, R.H., Lammers, J.W., Koenderman, L. & Coffer, P.J.
(2000). Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead
transcription factor FKHR-L1. Curr Biol 10, 1201-1204. 94. Ramaswamy (2002). A novel mechanism of gene regulation and tumor suppression by the transcription
factor FKHR. Cancer Cell Vol 2. 95. Schmoll, D., Walker, K.S., Alessi, D.R., Grempler, R., Burchell, A., Guo, S.,
Walther, R. & Unterman, T.G. (2000). Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and
the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity.
J Biol Chem 275, 36324-36333.

96
96. Hall, R.K., Yamasaki, T., Kucera, T., Waltner-Law, M., O'Brien, R. & Granner,
D.K. (2000). Regulation of phosphoenolpyruvate carboxykinase and insulin-like growth factor-
binding protein-1 gene expression by insulin. The role of winged helix/forkhead proteins.
J Biol Chem 275, 30169-30175. 97. Nasrin, N., Ogg, S., Cahill, C.M., Biggs, W., Nui, S., Dore, J., Calvo, D., Shi, Y.,
Ruvkun, G. & Alexander-Bridges, M.C. (2000). DAF-16 recruits the CREB-binding protein coactivator complex to the insulin-like
growth factor binding protein 1 promoter in HepG2 cells. Proc Natl Acad Sci U S A 97, 10412-10417. 98. Funaki, M., Katagiri, H., Inukai, K., Kikuchi, M. & Asano, T. (2000). Structure and function of phosphatidylinositol-3,4 kinase. Cell Signal 12, 135-142. 99. Schlessinger, J. (1994). SH2/SH3 signaling proteins. Curr Opin Genet Dev 4, 25-30. 100. Chang, H.W., Aoki, M., Fruman, D., Auger, K.R., Bellacosa, A., Tsichlis, P.N.,
Cantley, L.C., Roberts, T.M. & Vogt, P.K. (1997). Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-
kinase. Science 276, 1848-1850. 101. Carter, A.N. & Downes, C.P. (1992). Phosphatidylinositol 3-kinase is activated by nerve growth factor and epidermal
growth factor in PC12 cells. J Biol Chem 267, 14563-14567. 102. de Groot, R.P., Coffer, P.J. & Koenderman, L. (1998). Regulation of proliferation, differentiation and survival by the IL- 3/IL-5/GM-CSF
receptor family. Cell Signal 10, 619-628. 103. Arcaro, A., Zvelebil, M.J., Wallasch, C., Ullrich, A., Waterfield, M.D. & Domin, J.
(2000). Class II phosphoinositide 3-kinases are downstream targets of activated polypeptide
growth factor receptors. Mol Cell Biol 20, 3817-3830. 104. Tacchini, L., Dansi, P., Matteucci, E. & Desiderio, M.A. (2001). Hepatocyte growth factor signalling stimulates hypoxia inducible factor-1 (HIF-1)
activity in HepG2 hepatoma cells. Carcinogenesis 22, 1363-1371. 105. Zhang, W., Johnson, J.D. & Rutter, W.J. (1993). Association and phosphorylation-dependent dissociation of proteins in the insulin
receptor complex. Proc Natl Acad Sci U S A 90, 11317-11321. 106. Virkamaki, A., Ueki, K. & Kahn, C.R. (1999). Protein-protein interaction in insulin signaling and the molecular mechanisms of
insulin resistance. J Clin Invest 103, 931-943.

97
107. Brunet, A., Bonni, A., Zigmond, M.J., Lin, M.Z., Juo, P., Hu, L.S., Anderson, M.J.,
Arden, K.C., Blenis, J. & Greenberg, M.E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription
factor. Cell 96, 857-868. 108. Pennisi, E. (1997). New tumor suppressor found--twice. Science 275, 1876-1878. 109. Maehama, T. & Dixon, J.E. (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273, 13375-13378. 110. Myers, M.P., Pass, I., Batty, I.H., Van der Kaay, J., Stolarov, J.P., Hemmings,
B.A., Wigler, M.H., Downes, C.P. & Tonks, N.K. (1998). The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A 95, 13513-13518. 111. Burgering, B.M. & Coffer, P.J. (1995). Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 376, 599-602. 112. Franke, T.F., Yang, S.I., Chan, T.O., Datta, K., Kazlauskas, A., Morrison, D.K.,
Kaplan, D.R. & Tsichlis, P.N. (1995). The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-
activated phosphatidylinositol 3-kinase. Cell 81, 727-736. 113. Hresko, R.C., Murata, H. & Mueckler, M. (2003). Phosphoinositide-dependent kinase-2 is a distinct protein kinase enriched in a novel
cytoskeletal fraction associated with adipocyte plasma membranes. J Biol Chem 278, 21615-21622. 114. Kops, G.J. & Burgering, B.M. (1999). Forkhead transcription factors: new insights into protein kinase B (c- akt) signaling. J Mol Med 77, 656-665. 115. Brunet, A., Park, J., Tran, H., Hu, L.S., Hemmings, B.A. & Greenberg, M.E.
(2001). Protein kinase SGK mediates survival signals by phosphorylating the forkhead
transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 21, 952-965. 116. Rena, G., Prescott, A.R., Guo, S., Cohen, P. & Unterman, T.G. (2001). Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in
regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem J 354, 605-612. 117. Cross, D.A., Alessi, D.R., Cohen, P., Andjelkovich, M. & Hemmings, B.A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785-789. 118. Scheid, M.P. & Duronio, V. (1998). Dissociation of cytokine-induced phosphorylation of Bad and activation of PKB/akt:
involvement of MEK upstream of Bad phosphorylation. Proc Natl Acad Sci U S A 95, 7439-7444.

98
119. Cahill, C.M., Tzivion, G., Nasrin, N., Ogg, S., Dore, J., Ruvkun, G. & Alexander-
Bridges, M. (2001). Phosphatidylinositol 3-kinase signaling inhibits DAF-16 DNA binding and function via
14-3-3-dependent and 14-3-3-independent pathways. J Biol Chem 276, 13402-13410. 120. Schumpelick, V., Bleese, N.M. & Mommsen, U. (1999). Chirurgie. Leerbuck ENKE Verlag 4. Auflage, Seite 966. 121. Milland, J., Tsykin, A., Thomas, T., Aldred, A.R., Cole, T. & Schreiber, G. (1990). Gene expression in regenerating and acute-phase rat liver. Am J Physiol 259, G340-347. 122. Kraizer, Y., Mawasi, N., Seagal, J., Paizi, M., Assy, N. & Spira, G. (2001). Vascular endothelial growth factor and angiopoietin in liver regeneration. Biochem Biophys Res Commun 287, 209-215. 123. de Jong, K.P., von Geusau, B.A., Rottier, C.A., Bijzet, J., Limburg, P.C., de Vries,
E.G., Fidler, V. & Slooff, M.J. (2001). Serum response of hepatocyte growth factor, insulin-like growth factor-I, interleukin-6,
and acute phase proteins in patients with colorectal liver metastases treated with partial hepatectomy or cryosurgery.
J Hepatol 34, 422-427. 124. Fausto, N. (2000). Liver regeneration. J Hepatol 32, 19-31. 125. Iwai, M., Cui, T.X., Kitamura, H., Saito, M. & Shimazu, T. (2001). Increased secretion of tumour necrosis factor and interleukin 6 from isolated,
perfused liver of rats after partial hepatectomy. Cytokine 13, 60-64. 126. Campos, S.P. & Baumann, H. (1992). Insulin is a prominent modulator of the cytokine-stimulated expression of acute-phase
plasma protein genes. Mol Cell Biol 12, 1789-1797. 127. Campos, S.P., Wang, Y. & Baumann, H. (1996). Insulin modulates STAT3 protein activation and gene transcription in hepatic cells. J Biol Chem 271, 24418-24424. 128. Derfalvi, B., Igaz, P., Fulop, K.A., Szalai, C. & Falus, A. (2000). Interleukin-6-induced production of type II acute phase proteins and expression of
junB gene are downregulated by human recombinant growth hormone in vitro. Cell Biol Int 24, 109-114. 129. Wang, Y., Ripperger, J., Fey, G.H., Samols, D., Kordula, T., Wetzler, M., Van
Etten, R.A. & Baumann, H. (1999). Modulation of hepatic acute phase gene expression by epidermal growth factor and
Src protein tyrosine kinases in murine and human hepatic cells. Hepatology 30, 682-697. 130. Campbell, J.S., Prichard, L., Schaper, F., Schmitz, J., Stephenson-Famy, A.,
Rosenfeld, M.E., Argast, G.M., Heinrich, P.C. & Fausto, N. (2001). Expression of suppressors of cytokine signaling during liver regeneration. J Clin Invest 107, 1285-1292.

99
131. Arcone, R., Pucci, P., Zappacosta, F., Fontaine, V., Malorni, A., Marino, G. &
Ciliberto, G. (1991). Single-step purification and structural characterization of human interleukin-6
produced in Escherichia coli from a T7 RNA polymerase expression vector. Eur J Biochem 198, 541-547. 132. Weiergräber, O., Hemmann, U., Küster, A., Müller-Newen, G., Schneider, J.,
Rose-John, S., Kurschat, P., Brakenhoff, J.P., Hart, M.H., Stabel, S. & Heinrich, P.C. (1995).
Soluble human interleukin-6 receptor. Expression in insect cells, purification and characterization.
Eur J Biochem 234, 661-669. 133. May, P., Gerhartz, C., Heesel, B., Welte, T., Doppler, W., Graeve, L., Horn, F. &
Heinrich, P.C. (1996). Comparative study on the phosphotyrosine motifs of different cytokine receptors
involved in STAT5 activation. FEBS Lett 394, 221-226. 134. Maniatis, T., Frisch, E.F. and Sambrook, J. (1982). Molecular Cloning: A Labatory Manual. Cold Spring Harbor (New York): Cold Spring Harbor Laborator. 135. Bradford, M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein
utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248-54. 136. Laemmli, U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, (259), p. 680-5. 137. Hideshima, T., Nakamura, N., Chauhan, D. & Anderson, K.C. (2001). Biologic sequelae of interleukin-6 induced PI3-K/Akt signaling in multiple myeloma. Oncogene 20, 5991-6000. 138. Kortylewski, M., Feld, F., Kruger, K.D., Bahrenberg, G., Roth, R.A., Joost, H.G.,
Heinrich, P.C., Behrmann, I. & Barthel, A. (2002). Akt modulates STAT3-mediated gene expression through a FKHR (FOXO1a)-
dependent mechanism. J Biol Chem 26, 26. 139. Nakajima, K., Yamanaka, Y., Nakae, K., Kojima, H., Ichiba, M., Kiuchi, N.,
Kitaoka, T., Fukada, T., Hibi, M. & Hirano, T. (1996). A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1
leukemia cells. Embo J 15, 3651-3658. 140. Shuai, K., Horvath, C.M., Huang, L.H., Qureshi, S.A., Cowburn, D. & Darnell, J.E.,
Jr. (1994). Interferon activation of the transcription factor Stat91 involves dimerization through
SH2-phosphotyrosyl peptide interactions. Cell 76, 821-828. 141. Clevenger, C.V. & Medaglia, M.V. (1994). The protein tyrosine kinase P59fyn is associated with prolactin (PRL) receptor and is
activated by PRL stimulation of T-lymphocytes. Mol Endocrinol 8, 674-681.

100
142. Lebrun, J.J., Ali, S., Sofer, L., Ullrich, A. & Kelly, P.A. (1994). Prolactin-induced proliferation of Nb2 cells involves tyrosine phosphorylation of the
prolactin receptor and its associated tyrosine kinase JAK2. J Biol Chem 269, 14021-14026. 143. Schmitt-Ney, M., Doppler, W., Ball, R.K. & Groner, B. (1991). Beta-casein gene promoter activity is regulated by the hormone-mediated relief of
transcriptional repression and a mammary-gland-specific nuclear factor. Mol Cell Biol 11, 3745-3755. 144. Rosen, J.M., Wyszomierski, S.L. & Hadsell, D. (1999). Regulation of milk protein gene expression. Annu Rev Nutr 19, 407-436. 145. Powis, G., Bonjouklian, R., Berggren, M.M., Gallegos, A., Abraham, R.,
Ashendel, C., Zalkow, L., Matter, W.F., Dodge, J., Grindey, G. & Vlahos, C.J. (1994).
Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res 54, 2419-2423. 146. Nakanishi, S., Kakita, S., Takahashi, I., Kawahara, K., Tsukuda, E., Sano, T.,
Yamada, K., Yoshida, M., Kase, H. & Matsuda, Y. (1992). Wortmannin, a microbial product inhibitor of myosin light chain kinase. J Biol Chem 267, 2157-2163. 147. Wymann, M.P., Bulgarelli-Leva, G., Zvelebil, M.J., Pirola, L., Vanhaesebroeck,
B., Waterfield, M.D. & Panayotou, G. (1996). Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-
802, a residue involved in the phosphate transfer reaction. Mol Cell Biol 16, 1722-1733. 148. Vlahos, C.J., Matter, W.F., Brown, R.F., Traynor-Kaplan, A.E., Heyworth, P.G.,
Prossnitz, E.R., Ye, R.D., Marder, P., Schelm, J.A., Rothfuss, K.J., Serlin, B.S. & Simpson, R.J. (1995).
Investigation of neutrophil signal transduction using a specific inhibitor of phosphatidylinositol 3-kinase.
J Immunol 154, 2413-2422. 149. Vlahos, C.J., Matter, W.F., Hui, K.Y. & Brown, R.F. (1994). A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-
benzopyran-4-one (LY294002). J Biol Chem 269, 5241-5248. 150. Biggs, W.H., 3rd, Meisenhelder, J., Hunter, T., Cavenee, W.K. & Arden, K.C.
(1999). Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the
winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A 96, 7421-7426. 151. von Groote-Bidlingmaier, F., Schmoll, D., Orth, H.M., Joost, H.G., Becker, W. &
Barthel, A. (2003). DYRK1 is a co-activator of FKHR (FOXO1a)-dependent glucose-6- phosphatase
gene expression. Biochem Biophys Res Commun 300, 764-769. 152. Hirota, K., Daitoku, H., Matsuzaki, H., Araya, N., Yamagata, K., Asada, S.,
Sugaya, T. & Fukamizu, A. (2003). HNF-4 is a novel downstream target of insulin via FKHR as a signal- regulated
transcriptional inhibitor. J Biol Chem 7, 7.

101
153. Look, D.C., Pelletier, M.R., Tidwell, R.M., Roswit, W.T. & Holtzman, M.J. (1995). Stat1 depends on transcriptional synergy with Sp1. J Biol Chem 270, 30264-30267. 154. Stocklin, E., Wissler, M., Gouilleux, F. & Groner, B. (1996). Functional interactions between Stat5 and the glucocorticoid receptor. Nature 383, 726-728. 155. Leu, J.I., Crissey, M.A., Leu, J.P., Ciliberto, G. & Taub, R. (2001). Interleukin-6-induced STAT3 and AP-1 amplify hepatocyte nuclear factor 1-mediated
transactivation of hepatic genes, an adaptive response to liver injury. Mol Cell Biol 21, 414-424. 156. Fredericks, W.J., Galili, N., Mukhopadhyay, S., Rovera, G., Bennicelli, J., Barr,
F.G. & Rauscher, F.J., 3rd (1995). The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar
rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol 15, 1522-1535. 157. Galili, N., Davis, R.J., Fredericks, W.J., Mukhopadhyay, S., Rauscher, F.J., 3rd,
Emanuel, B.S., Rovera, G. & Barr, F.G. (1993). Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar
rhabdomyosarcoma. Nat Genet 5, 230-235. 158. Lam, P.Y., Sublett, J.E., Hollenbach, A.D. & Roussel, M.F. (1999). The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3
homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol Cell Biol 19, 594-601. 159. Paulson, M., Pisharody, S., Pan, L., Guadagno, S., Mui, A.L. & Levy, D.E. (1999). Stat protein transactivation domains recruit p300/CBP through widely divergent
sequences. J Biol Chem 274, 25343-25349. 160. Schaefer, T.S., Sanders, L.K. & Nathans, D. (1995). Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc Natl Acad Sci U S A 92, 9097-9101. 161. Tu, Y., Gardner, A. & Lichtenstein, A. (2000). The phosphatidylinositol 3-kinase/AKT kinase pathway in multiple myeloma plasma
cells: roles in cytokine-dependent survival and proliferative responses. Cancer Res 60, 6763-6770. 162. Chen, R.H., Chang, M.C., Su, Y.H., Tsai, Y.T. & Kuo, M.L. (1999). Interleukin-6 inhibits transforming growth factor-beta-induced apoptosis through the
phosphatidylinositol 3-kinase/Akt and signal transducers and activators of transcription 3 pathways.
J Biol Chem 274, 23013-23019. 163. G-Amlak, M., Uddin, S., Mahmud, D., Damacela, I., Lavelle, D., Ahmed, M., van
Besien, K. & Wickrema, A. (2002). Regulation of myeloma cell growth through Akt/Gsk3/forkhead signaling pathway. Biochem Biophys Res Commun 297, 760-764. 164. Kortylewski, M., Heinrich, P.C., Mackiewicz, A. & Behrmann, I. (2001). Cytokine-mediated growth inhibition of human melanoma cells. Adv Exp Med Biol 495, 169-172.

102
165. Lazar-Molnar, E., Hegyesi, H., Pallinger, E., Kovacs, P., Toth, S., Fitzsimons, C.,
Cricco, G., Martin, G., Bergoc, R., Darvas, Z., Rivera, E.S. & Falus, A. (2002). Inhibition of human primary melanoma cell proliferation by histamine is enhanced by
interleukin-6. Eur J Clin Invest 32, 743-749. 166. Rena, G., Guo, S., Cichy, S.C., Unterman, T.G. & Cohen, P. (1999). Phosphorylation of the transcription factor forkhead family member FKHR by protein
kinase B. J Biol Chem 274, 17179-17183. 167. Klausen, P., Pedersen, L., Jurlander, J. & Baumann, H. (2000). Oncostatin M and interleukin 6 inhibit cell cycle progression by prevention of p27kip1
degradation in HepG2 cells. Oncogene 19, 3675-3683. 168. Shuai, K. (2000). Modulation of STAT signaling by STAT-interacting proteins. Oncogene 19, 2638-2644.

103
8 Anhang
Danksagung
Bei Herrn Prof. Dr. P. C. Heinrich bedanke ich mich für die freundliche
Aufnahme in seinen Arbeitskreis, sein Interesse am Fortgang dieser Arbeit
und besonders die Zeit, die er sich für persönliche Gespräche und Pro-
bleme nahm.
Ganz besonders danke ich Frau Priv. Doz I. Behrmann, für die hervor-
ragende Betreuung, die guten Ideen, die vielen Diskussionen und die
freundliche Atmosphäre. Ein weiterer großer Verdienst von ihr liegt in der
Durchsicht und Korrektur des Manuskriptes mit vielen hilfreichen Anre-
gungen.
Marlies Kauffmann, Marcin Kortylewski und Andreas Barthel danke ich für
die fachliche Unterstützung.
Jens Greiser danke ich für die vielen Gespräche und die entgegen-
gebrachte Empathie.
Mein außerordentlicher Dank gilt vor allem den Menschen, die mich im
Laufe der Arbeit unterstützt haben und es mir erst ermöglichten, diese
Dissertation anzunehmen. Dabei sind insbesondere meine Eltern und Frau
Jenny Baschke zu nennen.

104

105
Curriculum vitae Name : Florian Feld Geburtsdatum : 31. August 1976 Geburtsort : Wuppertal Wohnort : Roermonder Str. 112 52072 Aachen Telefon : 0241 - 9967537 Eltern : Dr. rer. nat. Hans Feld, Studiendirektor am Städt. Gymnasium Gevelsberg i.R. Ehefrau Inge Feld, geborene Drünkler, Oberstudienrätin am Städt. Gymnasium Ennepetal i.R. Staatsangehörigkeit : deutsch Ausbildung : 8 / 1982 bis 6 / 1986 Städt. Gemeinschaftsgrundschule "Am Strückerberg" 8 / 1986 bis 6 / 1995 Städt. Gymnasium Gevelsberg mit Erlangen der allgemeinen Hochschulreife 8 / 1995 bis 10 / 1996 Zivildienst 10 / 1996 bis 3 / 2000 Sudium der Medizin an der RWTH – Aachen Physikum September 1998 mit der Note befriedigend (2,66) Erstes Staatsexamen April 2000 mit der Note gut (2,0) 4 / 2000 bis 4 / 2001 molekularbiologische Promotionsarbeit am Institut für Biochemie 4 / 2001 bis 11 / 2004 Sudium der Medizin an der RWTH – Aachen Zweites Staatsexamen September 2002 mit der Note gut (1,66) Drittes Staatsexamen am 21. November 2003 mit der Note sehr gut und Abschluss des Studiums mit der Note sehr gut (1,49)








![BOXON STAT - boxonbulk.de · [NAME] BAGS TECHNICAL INFORMATION SICHERE LAGERUNG OHNE RISIKEN Die Big Bags der Reihe Boxon STAT werden speziell für den Schutz vor statischer Elektrizität](https://img.pdfslide.org/doc/110x75/5e0317c7d9e2ea2f2041b969/boxon-stat-name-bags-technical-information-sichere-lagerung-ohne-risiken-die.jpg)



















