Embed Size (px)
Citation preview

DIE BEDEUTUNG DER BAKTERIEN DES
SOGENANNTEN ROTEN KOMPLEXES NACH
SOCRANSKY IN DER ÄTIOLOGIE
DER PARODONTITIS
-
EINE AKTUELLE LITERATURÜBERSICHT
Dissertation
zur Erlangung des akademischen Grades
„doctor medicinae dentariae“ (Dr. med. dent.)
vorgelegt dem Rat der Medizinischen Fakultät
der Friedrich- Schiller- Universität Jena
von Maja Anna-Böttcher M.Sc.
geboren am 15. 08.1972 in Greifswald

Gutachter
1. Prof. Dr. Wolfgang Pfister, Jena
2. Prof. Dr. Dr. Bernd W. Sigusch, Jena
3. PD Dr. Siegrun Eick, Bern/Schweiz
Tag der öffentlichen Verteidigung: 6.09.2016

Inhaltsverzeichnis
1 Zusammenfassung ............................................................................................................................ 1
2 Einleitung ......................................................................................................................................... 6
2.1 Parodontale Erkrankungen ....................................................................................................... 6
2.2 Bakterielle Komplexe nach Socransky .................................................................................... 8
2.3 Immuno-mikrobielle Pathogenese, Dysbiose und Wirtsreaktivität ........................................ 11
3 Aufgabenstellung ........................................................................................................................... 13
4 Material und Methode .................................................................................................................... 14
5 Ergebnisse und Diskussion ............................................................................................................ 17
5.1 Bakterielle Virulenzfaktoren .................................................................................................. 17
5.2 Porphyromonas gingivalis ...................................................................................................... 17
5.2.1 Klassifikation und Charakteristika ................................................................................. 17
5.2.2 Virulenzfaktoren von Porphyromonas gingivalis ........................................................... 20
5.3 Tannerella forsythia ................................................................................................................ 34
5.3.1 Klassifikation und Charakteristika ................................................................................. 34
5.3.2 Virulenzfaktoren von Tannerella forsythia ..................................................................... 35
5.4 Treponema denticola .............................................................................................................. 42
5.4.1 Klassifikation und Charakteristika ................................................................................. 42
5.4.2 Virulenzfaktoren ............................................................................................................. 43
5.5 Polymikrobielle Synergie, Biofilmbildung und Therapieansätze .......................................... 50
6 Literaturverzeichnis ....................................................................................................................... 53
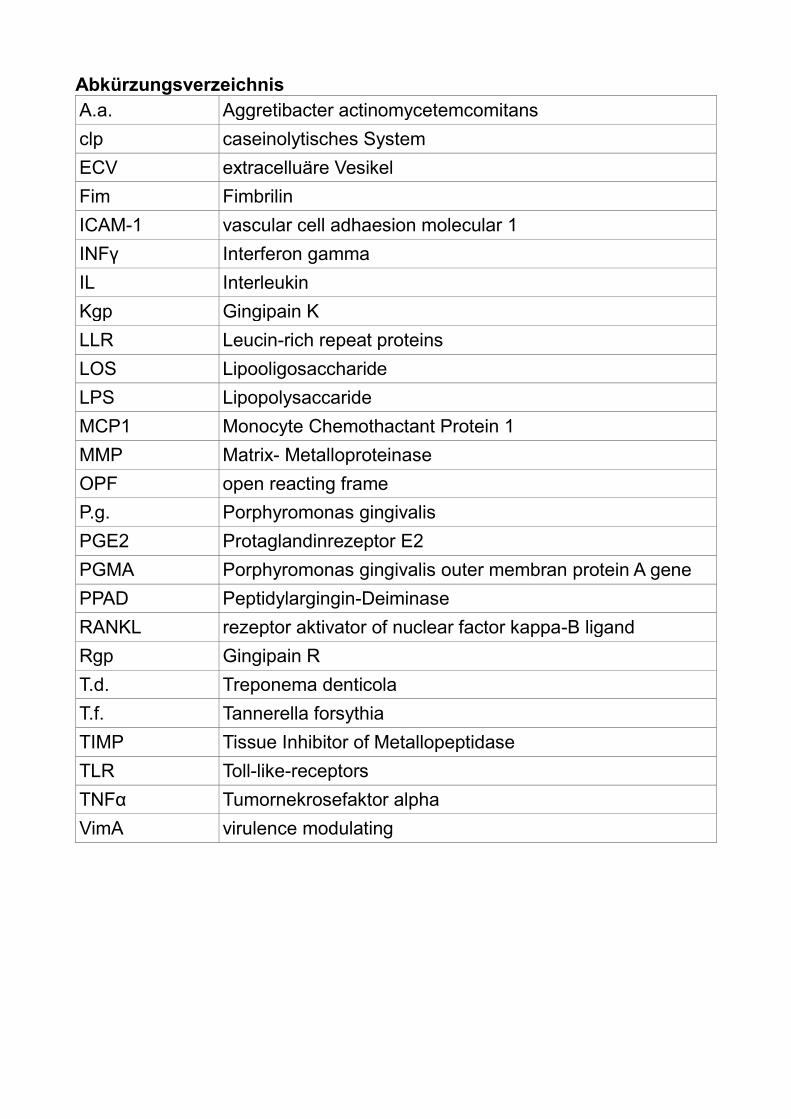
Abkürzungsverzeichnis
A.a. Aggretibacter actinomycetemcomitans
clp caseinolytisches System
ECV extracelluäre Vesikel
Fim Fimbrilin
ICAM-1 vascular cell adhaesion molecular 1
INFγ Interferon gamma
IL Interleukin
Kgp Gingipain K
LLR Leucin-rich repeat proteins
LOS Lipooligosaccharide
LPS Lipopolysaccaride
MCP1 Monocyte Chemothactant Protein 1
MMP Matrix- Metalloproteinase
OPF open reacting frame
P.g. Porphyromonas gingivalis
PGE2 Protaglandinrezeptor E2
PGMA Porphyromonas gingivalis outer membran protein A gene
PPAD Peptidylargingin-Deiminase
RANKL rezeptor aktivator of nuclear factor kappa-B ligand
Rgp Gingipain R
T.d. Treponema denticola
T.f. Tannerella forsythia
TIMP Tissue Inhibitor of Metallopeptidase
TLR Toll-like-receptors
TNFα Tumornekrosefaktor alpha
VimA virulence modulating

- 1 -
1 Zusammenfassung
Die chronische Parodontitis und ihre Folgeerscheinung, der Zahnverlust, stellen ein
schwerwiegendes gesundheitliches, wirtschaftliches und soziales Problem dar. Eine Gruppe
von Parodontopathogenen, welche als ‚Roter Komplex’ bezeichnet wird (Porphyromonas
gingivalis (P.g.), Tannerella forsythia (T.f.) und Treponema denticola (T.d.)), ist in ihrem
Auftreten mit der aggressiven und chronischen Verlaufsform der Parodontitis assoziiert. Ihre
Vertreter haben jedoch nicht nur einen Einfluss auf die Entstehung der Parodontitis sondern
werden auch bei anderen Erkrankungen nachgewiesen und sind möglicherweise in deren
Ätiologie kausal impliziert (z.B. Arteriosklerose, rheumathoide Arthritis, Osteoporose,
Alzheimer’s Erkrankung, Diabetes, ischaemische Herzerkrankungen und Erkrankungen des
Gastrointestinaltrakts). Die durch diese Mikoorganismen verursachte Problematik ist also
sehr weitreichend und erfordert eine umfassende Aufklärung der zugrundeliegenden
Pathogenitätsmechanismen.
Parodontale Gesundheit stellt einen Zustand dar, in dem sich antimikrobielle, pro-
inflammatorische Aktivitäten (welche die Infektion kontrollieren) und anti-inflammatorische
Regelkreise der Entzündungsresolution in einem dynamischen Gleichgewicht befinden.
Diese homeostatische Balance kann einerseits durch Veränderungen des Habitats (z.B.
immunologische, genetische oder von außen wirkende Umweltfaktoren) oder aber durch
das Wirken von ‚Keystone-Pathogenen’ gestört werden. Unter den Vertretern des ‚Roten
Komplexes’ nimmt P.g.die Rolle eines solchen Keystone-Pathogens ein. Trotz der
symbiotischen Beziehung, die die Mikrobiota im gesunden Zustand mit dem Wirt führt, so
besitzen ihre Vertreter dennoch Virulenzfaktoren, welche - wenn das Bakterium
mengenmäßig stark anwächst - eine andere Relevanz erlangen können als während des
symbiotischen Zustands. Diese Rolle der sog. ‚Pathobionten’ kommt im ‚Roten Komplex’
den Bakterien T.d. und T.f. zu, da sie auch in der gesunden Mikrobiota vorkommen, sich
unter bestimmten Umständen jedoch unverhältnismäßig stark vermehren und in Folge die
Pathogenese vorantreiben.
Porphyromonas gingivalis ist ein anaerober, asaccharolytischer und proteolytischer
Organismus, dessen Überleben und Wachstum von Abbauprodukten, die bei der
inflammatorischen Gewebszerstörung entstehen, abhängt. Während P.g. sich dem Angriff
durch das Immunsystem geschickt zu entziehen vermag, fördert das Bakterium gleichzeitig
das für das eigene Überleben so günstige inflammatorische Milieu. Die Vermutung, dass
P.g. seine Stellung als ‚Keystone-Pathogen’ durch Initiierung der Dysbiose (also der aus

- 2 -
Veränderung der mengenmäßigen Anteile verschiedener Spezies in der dentalen Plaque
resultierenden Verwandlung einer harmlosen symbiotischen in eine krankheitsassoziierte
Mikrobiota) erlangt, bestätigt sich auch im Tiermodell, wo P.g. die Zusammensetzung der
Mikrobiota zu verändern vermag, was zu pathogenen Veränderungen wie Knochenabbau
führt.
Obwohl der Hauptaspekt der pathogenen Aktivität von P.g. wohl in der Induktion der
Dysbiose liegt, so verfügt es doch über eine Vielzahl gut charakterisierter Virulenzfaktoren.
Es exprimiert z.B. drei Proteasen, Gingipaine genannt, die das umgebende Gewebe
proteolytisch angreifen, spezifisch Komponenten des Komplementsystems inaktivieren,
sowie bei der Zellinvasion und der Hämagglutination beteiligt sind. Stark virulent wirken
ebenfalls die Fimbrien, bakterielle Zellfilamente, die für Auto- und Koaggregation, die
Ausbildung von Biofilmen und die Zellinvasion wichtig sind. Die Kapselpolysaccharide
vermitteln einen Schutz vor dem Angriff des Immunsystems während die
Lipoppolysaccharide, normalerweise stark immunogen wirkende Oberflächenbestandteile,
nur eine außergewöhnlich schwache Reaktion des Immunsystems verursachen.
Auch bei Tannerella forsythia, einem ebenfalls anaerob lebenden, assaccharolytischen
Bakterium, können einer Reihe von Proteinen putativ pathogene Funktionen zugeordnet
werden. Beim Vergleich von Tannerella forsythia mit einer nicht-pathogenen Form von
Tannerella (BU 063, oral taxon 286) zeigte sich, dass letztere für die Faktoren Karilysin, PrtH
und BspA defizient ist, was deren Bedeutung als robuste Virulenzfaktoren untermauert. Das
Oberflächenprotein BspA spielt eine maßgebliche Rolle bei der Adhäsion an und Invasion
von Epithelzellen durch T.f., indem es spezifisch Aktin-Zytoskelett Rearrangements aktiviert.
Es induziert außerdem über den Rezeptor TLR2 (‚toll-like receptor 2’) die Freisetzung von
Chemokinen aus Epithelzellen und von pro-inflammatorischen Zytokinen aus Makrophagen.
Das Protein PrtH könnte durch seine proteolytische Wirkung die Ablösung des parodontalen
Gewebeverbands vom Zahnsubstrat unterstützen, jedoch werden aus manchen Patienten
auch Isolate ohne funktionelle PrtH isoliert, was die in-vivo Relevanz als Virulenzfaktor in
Frage stellt. Die Metalloproteinase Karilysin kann Tumornekrosefaktor alpha (Tnf-α), das
antibakterielle Peptid LL-37 sowie mehrere Komponenten des Komplementsystems
inaktivieren und hat daher ein sehr starkes virulentes Potential. Interessant ist ebenfalls der
charakteristische ‚S-Layer’, der die Autoaggregation beeinträchtigt und das Bakterium für
das Immunsystem schlechter erkennbar macht. Faktoren des Komplementsystems können
nicht binden, und der MAC (‚membrane attack complex’) kann nicht rekrutiert werden. Die
mangelhafte Immunabwehr erlaubt es T.f., TLR2 vermittelt Th2-Antworten zu induzieren und
dadurch zur Gewebe- und Knochenzerstörung beizutragen. Von T.f. exprimierte

- 3 -
Glycosidasen, Lipoproteine, Lipopolysaccharide und der Proteaseinhibitor Miropin sind
weitere Kandidaten, die aufgrund ihrer Funktionen als Virulenzfaktoren eine Rolle spielen
könnten.
Treponema denticola, ein Vertreter der oralen Spirochaeten, ist das einzige motile
Bakterium im ‚Roten Komplex’ und diese Eigenschaft ermöglicht es ihm, orale Epithelzellen
zu besiedeln. Im Tiermodell kann T.d. Knochenabbau induzieren, was eine ursächliche Rolle
der Virulenzfaktoren von T.d. bei der Gewebszerstörung im Rahmen der Parodontitis
nahelegt. Das Protein Msp kann Poren in der Zellmembran bilden und interagiert mit
zahlreichen Bindegewebsbestandteilen. Es kann wie BspA von T.f. in den
Aktinmetabolismus eingreifen und ist dadurch wahrscheinlich am Invasionspotential von T.d.
beteiligt. Außerdem kann es TLR2-vermittelt Makrophagen aktivieren und zur TNF-α
Ausschüttung anregen. TDE0471, ebenfalls ein Oberflächenantigen, verhindert die Bindung
des MAC an das Bakterium und schwächt daher die Immunantwort gegen das Bakterium.
TDE0214, ein c-di-GMP bindendes Protein, ist wichtig für Motilität, die Ausbildung von
Biofilmen und die Invasivität und wirkt außerdem immunstimulierend. Unter den
verschiedenen als Virulenzfaktoren von T.d. postulierten Lipoproteinen ist Dentilisin das am
besten untersuchte. Es beeinflusst interzelluläre Signalwege durch Abbau von
Signalmolekülen (IL-1b, IL-6, TNF-α und Mcp1) und moduliert dadurch Immunantworten. Es
greift die interzelluläre Matrix proteolytisch an und unterstützt dadurch die Penetration von
Epithelzellen. Außerdem bindet Dentilisin wie der RgpA-Kgp Komplex von P.g. an
Fibrinogen und bewirkt dessen Abbau, was Blutungsereignisse unterstützt. Dentilisin ist
außerdem an der Ko-aggregation von T.d. beteiligt. Ein wichtiger Aspekt der Virulenz ist
dabei auch, dass Dentilisin Komplementkomponenten, die zuvor durch das ebenfalls von
T.d. exprimierte FhbB gebunden wurden, spaltet und inaktiviert. Das T.d. Enzym Cystalisin
ist an einem Stoffwechselweg beteiligt, der in der Produktion von H2S resultiert und beteiligt
sich an der Pathogenese wohl durch Gewebeschädigung und Deregulation der Wirtsantwort.
Mit Hilfe von Oberflächen-Vesikeln können Virulenzfaktoren in tiefere Gewebeschichten
transportiert und dabei vor Abbau geschützt werden.
Wie oben dargelegt können einer großen Anzahl von Proteinen, die von Vertretern des
‚Roten Komplexes’ exprimiert werden, putativ pathogene Funktionen zugeordnet werden.
Jedoch sollte man beachten, dass unterschiedliche experimentelle Ansätze zur
Untersuchung gewählt wurden und es nicht immer möglich ist, die tatsächliche Bedeutung
der jeweils in vitro beobachteten Effekte für den Pathogeneseverlauf abzuschätzen.
Trotz der Vielzahl an Virulenzfaktoren und ihren strukturellen Unterschieden gibt es doch
augenscheinliche Gemeinsamkeiten betreffs der Angriffspunkte: Verschiedene Faktoren

- 4 -
sind z.B. an der Adhäsion und Invasion von Epithelzellen beteiligt, ein Charakteristikum, das
es den Bakterien erlaubt, in einem Reservoir zu ruhen, ohne vom Immunsystem angegriffen
zu werden. (Gingipaine und Fimbrien von P.g., S-layer und BspA von T.f. und Msp, Dentilisin,
LRR Proteine und TDE0214 von T.d.). Die meisten der gebildeten Proteasen besitzen das
Potential, umliegendes Gewebe proteolytisch anzugreifen, manchmal auch die
Blutungsneigung zu erhöhen. Andere Proteine wiederum haben Mechanismen entwickelt,
Komponenten des Komplementsystems unschädlich zu machen (die Gingipaine von P.g.,
das Karilysin von T.f. und TDE0471 und FhbB von T.d.). Außerdem induzieren zahlreiche
Faktoren die Zytokinexpression in Epithelzellen oder Zellen des Immunsystems (GroEL und
Fimbrien von P.g., BspA, PrtH, Karilysin, LPS und Lipoproteine von T.f. und Msp, OppA und
Dentilisin von T.d.), oft über den TLR2 (z.B. LPS von P.g.). Die immunologische Wirtsantwort
auf Parodontopathogene - vor allem auf ihre Oberflächenstrukturen, mit denen sie mit ihrer
Umwelt in Kontakt treten, und auf ihre metabolischen Produkte – ist eine kritische
Determinante im Fortschreiten der Parodontitis.
Neben diesen Gemeinsamkeiten sind auch mehrere Genprodukte der Vertreter des ‚Roten
Komplexes’ an der Ko-Aggregation untereinander und damit an der Entstehung des Biofilms
und der polymikrobiellen Synergie beteiligt (Fimbrien von P.g., LrrA und Dentilisin von T.d.)
Das Virulenzpotential der drei Mikroorganismen ist eng an ihre Lebensart im oralen Biofilm
gekoppelt. Kenntnisse darüber, welche Prozesse die Bildung von Biofilmen unterstützen
oder unterbrechen könnten hilfreich für den Eingriff in orale Pathogeneseabläufe sein.
Das Modell des ‚roten Komplexes’, in dem dessen drei Vertreter als Hauptverursacher der
Parodontitis dargestellt werden, erwuchs aus der Tatsache, dass ihre Bedeutung aufgrund
ihres besseren Wachstums in der Zellkultur überschätzt wurde. Heute nutzt man
metagenomische Ansätze um die Beteiligung verschiedener Spezies am Plaque genauer
zu definieren, mit dem Ergebnis, dass Hajishengallis 2012 das allgemeiner formulierte
Modell der polymikrobiellen Synergie und Dysbiose aufstellte. Die Entwicklung
problematischer Verlaufsformen der Parodontitis resultiert demgemäß aus der Dysbiose der
parodontalen Mikrobiota infolge der Zerstörung der Wirt-Mikroben Homöostase.
Im Rahmen dieser Arbeit wurden die Funktionen der verschiedenen Virulenzfaktoren von
P.g., T.f. und T.d. anhand aktueller Forschungsergebnisse erläutert und ihre mögliche
Bedeutung für die Pathogenese der Parodontitis in vivo abgeschätzt. Der Entwicklung neuer
therapeutischer Strategien liegen genaue Kenntnisse der Charakteristika und
Virulenzmechanismen der verursachenden Organismen zugrunde, und diese Arbeit soll
hierfür eine Grundlage bieten.

- 5 -
2 Einleitung
2.1 Parodontale Erkrankungen
In der Mundhöhle finden sich hunderte verschiedener Mikroorganismen, die mit dem
Wirtsorganismus im nicht krankmachenden ökologischen Gleichgewicht stehen. Etwa die
Hälfte hiervon ist bisher kultivierbar und folglich näher charakterisiert (Socransky et al., 1994;
Paster et al., 2001). Sie können nützlich, unbedeutsam (auch Kommensale oder Residente
genannt) oder schädlich sein (Wolf und Rateitschak, 2012). Opportunistische Keime, also
fakultativ pathogene Erreger, die nur im abwehrgeschwächten Wirt pathogen sind, finden
sich oft in der kommensalen Flora der Mundhöhle. Normalerweise schädigen sie den Wirt
nicht. Bei bestimmten Dispositionen wie z.B. Resistenzverminderung, Existenz von
Risikofaktoren oder Immunsuppression kann es jedoch zu einer selektiven Vermehrung von
Bakterien mit Virulenzfaktoren, und so zu einer opportunistischen Infektion wie der
Parodontitis kommen.
Die Gingivitis, welche sich auf das marginale suprakrestale Weichgewebe beschränkt,
manifestiert sich klinisch durch Blutung bei Sondierung des gingivalen Sulkus, in schweren
Fällen durch Rötung und Schwellung besonders im Bereich der Papillen (Wolf & Rateitschak
2012). Durch Veränderungen des oralen Mikrobioms, dem Überhandnehmen
parodontopathogener Bakterien in Zusammenhang mit einem veränderten, reduzierten
Immunstatus sowie der Präsenz von Risikofaktoren kann sich aus der Gingivitis eine
Parodontitis entwickeln: Die Entzündung der Gingiva greift dann auf die tieferen Strukturen
des Zahnhalteapparates über. Es kommt zur Desintegration des Kollagens und zum
Knochenabbau (Attachmentverlust). Das Saumepithel wandelt sich in ein Reservoir für
opportunistische pathogene Bakterien, welche ein Fortschreiten der Parodontitis fördern
können. Aufgrund der physiologischen Zusammenhänge können die Gingivitis und die
Parodontitis ineinander übergehen.
Die Parodontitis als bakteriell verursachte entzündliche Erkrankung aller Anteile des
marginale Parodontiums (d.h. der Gingiva, des Desmodontiums, der Wurzelzementes und
des Alveolarknochens) zeigt sich klinisch in der Bildung von Zahnfleischtaschen und dem
Auftreten von Attachmentverlust sowie von Knochenresorption. Durch fortschreitenden
Abbau des Stützgewebes kann dies bis zum Zahnverlust führen (Haunfelder et al. 1990).
Die Parodontitis verläuft in vier klinisch voneinander abgrenzbaren Phasen (Gaengler et al.
2005, Wolf und Rateitschak 2012):
Das erste Stadium beschreibt die „Erste Reaktion des gesunden Parodonts auf die Plaque“.
Plaquebakterien des marginalen Parodonts produzieren in ihrem Stoffwechsel Metaboliten,

- 6 -
die das Saumepithel veranlassen, Entzündungsmediatoren zu sezernieren. Freie
Nervenendigungen, der nicht unmittelbar von Blutgefäßen durchzogenen
Gewebestrukturen wie dem Saumepithel oder der freien und der anhaftenden Gingiva,
produzieren in Folge Neuropeptide und Histamin, die die lokale Gefäßreaktion
hochregulieren. Auch perivaskuläre Mastzellen setzen Histamin frei, welches das Endothel
veranlasst, den Entzündungsmediator Interleukin-8 (IL-8) in das Gefäß auszuschütten.
Das zweite Stadium beschreibt die „Aktivierung von Makrophagen und des
Serumproteinsystems“. Die in Stadium I beschriebene vaskuläre Reaktion bewirkt, dass
Serumproteine, z.B. Komplement, aus den postkapillären Venolen ins Bindegewebe
übertreten und eine lokale Entzündungsreaktion hervorrufen. Aktivierte Makrophagen
produzieren in der Folge Entzündungsmediatoren.
Das dritte Stadium der Pathogenese der Parodontitis umfasst das „Hochregulieren der
Aktivität der Entzündungszellen, das Ablösen des Saumepithels und die Bildung einer
gingivalen Tasche“. Das bindegewebige entzündliche Infiltrat wird jetzt von Lymphozyten
dominiert. Aktivierte T-Zellen koordinieren die Immunantwort auf die Plaque mittels
Zytokinen (IL-2 bis -6, IL-10 und -13, Tumornekrosefaktor alpha (TNFα), Interferon gamma
(INFγ)). Um die Erreger abzuwehren produzieren Plasmazellen Immunglobuline und
Zytokine. Aktivierte polymorphkernige neutrophile Granulozyten sezernieren diverse
Zytokine, Leukotriene (LTB4:Chemotaxis) und Matrix-Metallproteinasen (MMP), welche bei
der Destruktion des Bindegewebes und des Knochens eine zentrale Rolle einnehmen.
Aktivierte Fibroblasten produzieren statt Kollagen Matrix-Metallproteinasen und deren
Gegenspieler TIMP (Tissue Inhibitor of Metallopeptidase). Die Infiltratmenge wird größer.
Das vierte Stadium bildet den „Ersten Attachmentverlust des parodontalen Gewebes“. Im
infiltrierten Bindegewebe findet sich nun eine erhöhte Aktivität der Makrophagen sowie an
Mediatoren entzündlicher Prozesse. Immuninkompetente Zellen wie Fibroblasten
produzieren zahlreiche Zytokine (IL-1ß, IL-6, IL-8, TNFα) sowie Prostagladinrezeptor E2
(PGE2), MMP und TIMP. Plasmazellen dominieren das Infiltrat, was ein Kennzeichen für
eine fortgeschrittene parodontale Läsion ist (Thorbert-Mros et al. 2014). Die gestörte
Hämostase führt zum Abbau von Kollagen, Matrix und Knochen.
Epidemiologische Daten der deutschen Mundgesundheitsstudie (DMS IV (2007)) ergaben,
dass bei ca. 37 Mio. Erwachsenen eine moderate bis schwere Parodontitis nachgewiesen
werden kann, wobei zwischen 1997 und 2005 ein deutlicher Anstieg (bei 35- bis 44-jährigen:
32.2% auf 52.7% bei mittelschweren und 14.1% auf 20.5% bei schweren Parodontitiden) zu
verzeichnen war. Unverändert waren Zahnkaries (29,7 %) und Parodontitis (28,5 %) die
beiden Hauptursachen, die letztlich eine Zahnentfernung erforderlich machten; ab dem 40.

- 7 -
Lebensjahr dominierte die Parodontitis als zahlenmäßiger Hauptgrund einer Zahnextraktion
(Glockmann et al 2007). Aktuelle Ergebnisse der V. Mundgesundheitsstudie werden laut
Institut der Deutschen Zahnärzte im Frühjahr 2016 erwartet.
2.2 Bakterielle Komplexe nach Socransky
Die Ende des 19. Jhs. beginnende Suche nach dem ätiologischen Agens, einem einzelnen
pathogenen Organismus, der gemäß der Koch’schen Postulate für die Entwicklung der
Parodontitis verantwortlich gemacht werden konnte, endete erfolglos. Auch die
‚Unspezifische Plaque Hypothese’, nach der die Menge aller Bakterien in der dentalen
Plaque die Krankheit verursacht, erwies sich als nicht zufriedenstellend. Auf der Basis der
spezifischen Plaquehypothese von Loesche (Loesche et al. 1972), nach der die Qualität der
Mikroorganismen der Mundflora für die Parodontitis verantwortlich ist, entwickelte Dr.
Sigmund Socransky schließlich aufgrund seiner Forschungsergebnisse 1998 (Socransky et
al. 1998) ein Modell, wonach verschiedenen Stadien der entzündlichen Veränderungen am
Parodontium das Vorkommen verschiedener Bakteriengruppen (sogenannter ‚Komplexe’)
zugeordnet werden kann. Es konnte im Fortschreiten des pathogenen Prozesses also eine
gewisse Besiedlungsabfolge mit unterschiedlichen und für die jeweilige Phase
charakteristischen Clustern von Pathogenen von gram-positiven hin zu gram-negativen
Organismen festgestellt werden (microbial shift disease). Die Beobachtung, dass die
entsprechenden Bakterien tatsächlich meist nicht alleine, sondern eben in diesen
charakteristischen Kombinationen an Entzündungsherden gefunden werden, unterstützt die
Hypothese, dass die Organismen der jeweiligen Komplexe während des pathogenen
Prozesses bei der Zerstörung parodontalen Gewebes miteinander kooperieren (Darveau et
al. 1997).
Die Gruppe um Socransky unterteilte parodontalpathogene Organismen in sechs
unterschiedliche mikrobiologische Komplexe (Socransky et al. 1998, 2002). Die genannten
Komplexe spiegeln bevorzugte Gemeinschaften von Mikroorganismen des subgingivalen
Biofilms bei parodontal Erkrankten wieder. Die Unterteilung der einzelnen Komplexe weist
auf eine engere Verwandtschaft der Organismen hin.
Die in der normalen Mundflora etablierten, grampositiven Keime wie Streptokokken und
Actinonmyceten stellen den gelben Komplex der parodontalpathogenen Organismen dar.
Sie werden von den sich im sauerstoffarmen Milieu des sich vertiefenden Sulkus von den

- 8 -
gramnegativen Anaerobiern verdrängt. Hierbei treten die Keime des grünen Komplexes
(drei Capnocytophaga-Spezies), Campylobacter concisus, Eikenella corrodens und vormals
Actinobacillus actinomycetemcomitans Serotyp B, neu: Aggregatibacter
actinomycetemcomitans (A.a.) auf. Letzterer wird in der Literatur aufgrund seiner hohen
Pathogenität als Leitkeim für die aggressive Parodontitis gehandelt (Fives-Taylor et al. 1999,
Fine et al. 2007, Amano 2010). Der violette Komplex der Acinomyceten reduziert sich zu
Gunsten des großen orangenen und des roten Komplexes. Dies gilt nicht nur für die
Relationen zueinander, sondern auch für die absoluten Zahlen (Socransky et al. 2002). Der
orangene Komplex enthält hauptsächlich potenziell pathogene Keime, die in geringer
Anzahl in jeder Mundhöhle zu finden sind und sich aufgrund der veränderten Pathologie
stark vermehren. Die höchste Pathogenität der hier eingeteilten parodontalpathogenen
Erreger weist der rote Komplex (Porphyromonas gingivalis (P.g.), Tanarella forsythia (T.f.)
und Treponema denticola (T.d.)) auf. So ist bei parodontal Erkrankten die Anwesenheit der
Mikroorganismen dieses Komplexes bei der klinischen Messung auffällig mit Bluten auf
Sondieren und Erhöhung der Sondierungstiefe verbunden (Haffajee et al. 1998, Holt et al.,
2000). Parameter sind Rötung der Gingiva, Blutung auf Sondierung, Suppuration und
Zusammensetzung der subgingivalen Besiedelung, welche eng mit der Sondierungstiefe
korreliert. Damit einhergehend sind das vermehrte Auftreten respektive die größere Anzahl
der Spezies des roten Komplexes wie auch des orangen Komplexes (Haffajee et al. 2004).
Spezies des roten Komplexes finden sich bei parodontal Erkrankten nicht nur an mehr
Messstellen, sondern auch in erheblich größerer Konzentration (Socransky et al. 1998).
Veränderungen der lokalen Gegebenheiten wie beispielsweise der Vertiefung von
parodontalen Taschen, führen zu einem sprunghaften Anstieg der Konzentration der
Spezies des orangenen und roten Komplexes, wohingegen Actinomycetes naeslundii
genospecies 2 von solchen Veränderung der Bedingungen unbeeindruckt bleiben
(Socransky et al. 1999).
Der Hauptunterschied zwischen supra- und subgingivaler Plaque von parodontal Erkrankten
und Gesunden spiegelt sich ebenfalls in den Verhältnissen und außerordentlich hohen
Spiegeln an Spezies des orangenen und roten Komplexes in der subgingivalen Plaque
wider (Ximenez-Fyvie et al. 2000).
Unter diesen verschiedenen von Socransky definierten Komplexen ist der ‚rote Komplex’ ein
Kollektiv aus Porphyromonas gingivalis, Tannerella forsythia und Treponema denticola, ein
‚später Besiedler’, der erst dann auftritt, wenn vorher durch die Besiedlung mit anderen
Mikroorganismen das entsprechende Umfeld geschaffen wurde (Bereitstellung von
Nährstoffen, anaerobes Milieu etc.). Er besteht aus Formen, die für parodontale Pathogene

- 9 -
gehalten werden, da ihr Vorhandensein mit aggressiven Infektionsverläufen assoziiert ist
(Socransky 1998). Untersucht wurden lange Zeit jedoch nur kultivierbare Spezies, und man
weiß, dass weitere Bakterien in die Gruppe der späten Besiedler fallen, die jedoch aufgrund
ihrer mangelnden Kultivierbarkeit noch nicht in dem Umfang untersucht und charakterisiert
werden konnten, wie es für die Vertreter des ‚Roten Komplex’ der Fall ist. Die Annahme,
dass die drei Pathogene des ‚Roten Komplex’ jedoch eine maßgebliche Rolle bei der
Entstehung der Parodontitis spielen, wird unterstützt durch Beobachtungen aus dem
Rattenmodell, wo Knochenabbau durch Ko-Infektion mit diesen Bakterien hervorgerufen
werden konnte, und darüber hinaus eine synergetische Wirkung der Parallelinfektion im
Vergleich mit Infektion mit den einzelnen Bakterien gezeigt wurde (Kesavalu et al. 2007).
Die Bakterien verfügen über zahlreiche Mechanismen, die Einfluss auf den
Krankheitsverlauf nehmen, und die als Virulenzfaktoren bezeichnet werden. Die Parodontitis
ist in ihrer Entstehung und Progression multifaktoriell bedingt, und während nur wenige
Virulenzfaktoren unmittelbar an der Zerstörung des Parodonts beteiligt sind, beeinflussen
viele die Wechselwirkung zwischen bakteriellem Stoffwechsel und immunologischer
Reaktion des Wirts. Diese indirekten Pathogenesefaktoren, die in das immunologische
Gleichgewicht eingreifen, sind wesentlich an der Entstehung der chronischen Parodontitis
beteiligt. Die Erkenntnis, dass letztendlich die multifaktoriell beeinflusste Immunantwort des
Wirts die Pathogenese der Parodontitis vorantreibt (Golub et al. 2006, Takashiba et al.2006)
führte dazu, dass sie 2008 als entzündliche und nicht länger als infektiöse Krankheit
klassifiziert wurde (Konsensuskonferenz der American Academy of Periodontology, AAP)

- 10 -
2.3 Immuno-mikrobielle Pathogenese, Dysbiose und Wirtsreaktivität
Die Mundhöhle ist einer der ökologisch komplexesten Bereiche des menschlichen Körpers
und ständig von von einer heterogenen Mischung an Mikroorganismen besiedelt, die
Lebensgemeinschaften wie Biofilme ausbilden. Das Immunsystem des Mundraumes ist
dem Kontakt mit Pathogenen und fremdartigen Substanzen angepasst und wird durch viele
komplexe immunologische Regelkreise kontrolliert. Bakterien bzw. bakterielle Faktoren wie
z.B. Lipopolysaccharide können in Epithelzellen die Ausschüttung von Zytokinen auslösen.
Es kommt in Folge zu einer Signalkaskade, die verschiedene Immunzellen und
Signalmoleküle umfasst (Takashiba et al. 2003). Dies führt zur lokalen Aktivierung des
Gefäßendothels, zur Infiltration weiterer Immunzellen und zur Progression der Entzündung
(Page 1991). Diesem Prozess entgegen wirken anti-entzündliche Zytokine, die die
Ausschüttung pro-inflammatorischer Zytokine hemmen und außerdem den Gewebeabbau
bremsen (Seymour et al. 2001, Silva et al. 2015).
In gesundem Gewebe herrscht eine Balance zwischen pro- bzw. antientzündlichen
Zytokinen und dem Entstehen einer Entzündungsantwort. Der durch mikrobielle Antigene
verursachte Reiz beruht also letztendlich auf einer Störung dieser Balance. Menge und
Aktivität der jeweiligen Zytokine sind jedoch direkt abhängig von individuellen Varianzen in
der immunologischen Kompetenz, die u.a. durch genetische Faktoren, z.B.
Genpolymorphismen an Interleukin-Loci, entstehen. Untersuchungen an Patienten mit der
aggressiven Form der Parodontitis zeigten, dass zahlreiche Genloci (bzw. Mutationen in
ihnen) an der Krankheitsentstehung beteiligt sein können und darüber hinaus wohl noch
eine große Anzahl weiterer, bislang nicht identifizierter krankheitsrelevanter Risikoallele
existiert (Vaithilingam et al. 2014). Auch andere endogene und exogene Faktoren wie
Immundefekte, Blut- und Stoffwechselerkrankungen und andere systemische Erkrankungen
(z.B. Diabetes), Mangelernährung, Stress oder Rauchen haben einen Einfluss auf die
Suszeptibilität und den Krankheitsverlauf, und zwar sowohl auf die immunvermittelte
Wirtsreaktivität als auch auf die pathogene Veränderung des dentalen Biofilms (Offenbacher
et al. 2008, Fullmer et al. 2009, Pöllänen et al. 2013, Camelo-Castillo et al. 2015).
Tatsächlich konnte gezeigt werden, dass in der großen Mehrzahl der Fälle (> 85%)
chronischer bzw. aggressiver Verlaufsformen der Parodontitis eine übersteigerte Reaktion
des oralen Immunsystems auf die Pathogene zugrunde liegt (man spricht hier von ‚high
respondern’). Interessanterweise konnte sogar dargestellt werden, dass sich unter anti-
inflammatorischer Therapie ein Wandel von pathologischer zurück zu gesunder Mikrobiota

- 11 -
vollzieht (Barthold et al. 2013, Hajishengallis 2014). In anderen Fällen können jedoch auch
Immundefekte für einen gewissen Prozentsatz an Parodontitisfällen verantwortlich gemacht
werden (Amaya et al. 2013).
Entgegen früherer Meinungen ist die Rückkehr zum gesunden Gewebezustand nach einer
Entzündung kein passiver sondern ein aktiver, stark regulierter Prozess, den man als
Resolution bezeichnet. Chronische Entzündungen wie die Parodontitis sind möglicherweise
generell durch ein Versagen der Entzündungsresolution und nicht durch die
inflammatorische Stimulation an sich bedingt (Serhan et al. 2007, Serhan et al. 2008,
Serhan 2008). Eine wichtige Stellung scheinen hier Makrophagen zu haben, indem sie
bestimmen, ob der inflammatorische Prozess sich chronisch entwickelt oder es zu einer
Entzündungsresolution kommt. (Zadeh et al. 1999, Hasturk 2012)
Scheitert die Resolution, so führt die Synthese weiterer Mediatoren zu Veränderungen des
Bindegewebes und des Knochen-Metabolismus, zum Beispiel werden Osteoklasten
aktiviert. Das den Zahn unterstützende Bindegewebe wird abgebaut, und es folgt die
Ablösung der Gewebeverbände vom Zahn.
Zusammenfassend handelt sich also bei der Parodontitis also um eine bakteriell stimulierte
destruktive Wirtsantwort, wobei der Entzündungsverlauf, die Entwicklung chronischer
Formen und die Progression von Gingivitis zu Parodontitis sowohl vom (sich durch den
mikrobiellen ‚shift’ und mit Entstehen der Dysbiose sowie durch polymikrobielle synergetisch
wirkende Virulenzfaktoren ändernden) mikrobiellen Reiz als auch von der ebenfalls
multifaktoriell bedingten Wirtsreaktivität abhängen. Die Erkenntnis, dass ein Versagen im
Prozess der Entzündungsresolution hier wohl eine wesentliche Rolle spielt, ist hinsichtlich
der Entwicklung neuer Therapieansätze sehr interessant, da Faktoren der Resolution als
neue Angriffspunkte genutzt werden können. Auch wenn weitere, bislang nicht identifizierte
Mikroorganismen an der Pathogenese beteiligt sein mögen, so wird die relevante Implikation
der drei Bakterien des ‚roten Komplexes’ in der Entstehung der Parodontitis jedoch durch
Experimente im Maus- und Rattenmodell deutlich dargestellt (Kesavalu et al. 2007).

- 12 -
3 Aufgabenstellung
Die Parodontitis stellt in steigendem Masse und weltweit eine Bedrohung für die
Zahngesundheit dar. Die medizinischen, aber auch gesellschaftlichen und wirtschaftlich
Folgen des Zahnverlusts sind immens, und stellen doch noch nicht den gesamten Umfang
des Problems dar: Die Pathogene des roten Komplexes, die in die Pathogenese der
Parodontitis impliziert sind, zeigen sich auch in erhöhtem Maße als mit anderen
Erkrankungen beim Menschen assoziiert: Arteriosklerose, rheumathoide Arthritis,
Osteoporose, Alzheimer Erkrankung, Diabetes und ischaemische Herzerkrankungen und
Erkrankungen des Gastrointestinaltrakts sind einige Beispiele (Olsen 2015, Otomo-Corgel
et al. 2102).
Zusammenhänge zwischen oraler und systemischer Gesundheit sind schon lange bekannt
und von immer mehr Erkrankungen erkennt man, dass sie auf einer pathologischen
Veränderung des Mikrobioms, der natürlichen mikrobiologischen Besiedlung verschiedener
Organsysteme, beruhen. Dies macht die Parodontitis auch zu einem Modell der
Pathogenese anderer Erkrankungen, über welche aus den Ergebnissen der
Parodontitisforschung ebenfalls Rückschlüsse gezogen werden können (Khocht et al.2014).
Um erfolgreiche neue Therapieansätze entwickeln zu können muss zunächst möglichst
umfassendes Wissen über die Pathogeneseabläufe, sowie die Virulenzfaktoren der in eine
Krankheit implizierten Mikroorganismen gewonnen werden. Im Rahmen dieser Arbeit soll
daher der derzeitige Kenntnisstand über Charakteristika und Virulenzfaktoren der Vertreter
des roten Komplexes, Porphyromonas gingivalis, Tannerella forsythia und Treponema
denticola, anhand aktueller Forschungsergebnisse zusammengefasst und diskutiert werden.

- 13 -
4 Material und Methode
Zur Beantwortung des Themas wurde eine systematische Literaturrecherche durchgeführt,
mit dem Ziel, anhand der verfügbaren und identifizierten Literatur eine aktuelle
Literaturübersicht zum gestellten Thema zu geben. Hierzu wurden die vorhandenen Quellen
zum aktuellen Zeitpunkt sowohl deutschlandweit als auch aus dem internationalen Raum
zusammengetragen.
Der Ablauf der systematischen Literaturrecherche gliederte sich in folgende Arbeitsschritte:
1. Bildung einer Suchstrategie einschließlich der Formulierung von differenzierten
Unterfragestellungen und Festlegung von Keywords (Suchbegriffe, Schlüsselwörter).
2. Die eigentliche Literatursuche erfolgt dabei über zwei Zugänge:
a.) Systematische Recherche in ausgewählten Datenbanken;
b.) Zusatzrecherche per Hand über Durchsicht der Referenzlisten bereits
gefundener Artikel nach neuen Literaturquellen („Reference Tracking“), sowie
ergänzende Recherche in spezifischen, bei der systematischen Recherche
nicht berücksichtigten Datenbanken.
3. Sichtung der Literatur und Auswahl der relevanten Literaturreferenzen mit Hilfe
definierter Ein- und Ausschlusskriterien
4. Bewertung der Literatur nach Evidenz
5. Dokumentation der Ergebnisse
Grundlage für die systematische Literatursuche bildet eine auf das Thema zugeschnittene
Suchstrategie. Zu dieser Suchfrage werden die jeweiligen Schlagworte (Keywords) für die
Recherche in den Datenbanken festgelegt. Im folgenden Abschnitt wird die Suchfrage der
entsprechenden Themas sowie die zugehörigen Keywords dargestellt.

- 14 -
Thema
Die Bedeutung der Bakterien des sogenannten roten Komplexes nach Socransky in
der Ätiologie der Parodontitis- Eine aktuelle Literaturübersicht
Dem Thema entsprechend werden Keywords abgeleitet. Zudem wurden themenrelevante
Artikel nach Schlagwörtern durchsucht und zur Vervollständigung in die Erarbeitung der
Keywords einbezogen. Diese wurden in deutscher und englischer Sprache formuliert:
Socransky
Roter Komplex
Porphyromonas gingivalis
Treponema denticola
Tannerella forsythia
Parodontitis
Pathogenität
Virulenzfaktor
Die Suchabfrage erfolgte datenbankspezifisch: die einzelnen Datenbanken bieten neben „
Limits“ (Einschränkungen) auch diverse Suchfilter an, mit deren Hilfe z.B. zeitliche und
sprachliche Merkmale eingestellt werden müssen. Die Datenbanken wurden, abhängig von
den Vorgaben und Möglichkeiten der jeweiligen Datenbank, wenn möglich nach Titel,
Abstract und Keyword durchsucht. Durch die Bool´sche Mengenoperation konnten
Schlagwörter miteinander verknüpt (AND) oder die Suchmenge mittels der Eingabe von OR
erweitert werden.
Die Auswahl adäquater Datenbanken erfolgte nach eingangs definierter Kriterien. Für die
vorliegende Studie waren ausschlaggebend das Fachgebiet, Evidenzgrad und
Verfügbarkeit. Gemäß der Leitfrage, möglichst evidenzbasierte Kriterien zu identifizieren,
erfolgte die Suche nach relevanten Studien zunächst in der Datenbank Cochrane Library, in
der Studien mit dem höchsten Evidenzniveau erfasst sind.
Zudem wurde in der Meta-Datenbank PubMed recherchiert um andere Publikationen mit
niedrigerem Evidenzniveau ebenfalls zu erfassen.

- 15 -
Generell wurde bei der Literatur teilweise eine zeitliche Einschränkung der letzten zehn
Jahre vorgenommen.
Nachfolgend werden die beiden verwendeten Datenbanken beschrieben.
Cochrane Library
Diese ist eine Datenbank der Cochrane Collaboration, einem intenationalen Netzwerk von
Wissenschaftlern für Wissenschaftler und Ärzte, in der systematische Übersichtsarbeiten
erstellt und für den Anwender zur Verfügung gestellt werden.
Es handelt sich um eine wissenschaftlich fundierte Informationsgrundlage, die es ermöglicht,
den aktuellen Stand der klinischen Forschung in kurzer Zeit objektiv beurteilen zu können.
Dies beruht auf der Tatsache, dass die Cochrane Library hinsichtlich der Evidenzbasiertheit
von Daten diejenigen Literatureferenzen zusammenfasst, die das höchste Evidenzniveau
aufweisen. Diese spiegeln sich beispielsweise in Meta-Analysen oder Reviews wider.
PubMed
Pubmed ist eine Meta-Datenbank und bibliographische Refenzdatenbank hauptsächlich im
englischsprachigen Raum. Es werden internationale medizinische Fachartikel,
insbesondere aus den vereinigten Staaten (National Library of Medicine, NLM) dokumentiert.
Pubmed bietet einen kostenfreien Zugang zu den Datenbanken Medline, Oldmedline (vor
1966) sowie Pubmed Central.
Neben der systematischen Datenbankrecherche wurde zusätzlich eine Handrecherche
vorgenommen. Hierfür wurde bei der Suche nach weiteren Referenzen systematisch in den
Literaturlisten der bisher gefundenen Artikel recherchiert („Reference Tracking“).

- 16 -
5 Ergebnisse und Diskussion
5.1 Bakterielle Virulenzfaktoren
Unter Virulenzfaktoren versteht man diejenigen spezifischen Eigenschaften oder
Bestandteile (struktureller oder metabolischer Natur) eines Pathogens, die zur
Krankheitsentstehung und zum Krankheitsverlauf beitragen. Bei der Parodontitis tragen
dabei einige dieser Virulenzfaktoren direkt zur Zerstörung von Parodont und Knochen bei.
Wie bereits erwähnt kann die pathogene Wirkung jedoch auch indirekt, durch Aktivierung
des Immunsystems, erfolgen. Viele der bakteriell exprimierten Faktoren, die zu einer
Aktivierung des Immunsystems führen, werden in der Literatur als Virulenzfaktoren
bezeichnet. Auch wenn prinzipiell die Parodontitis durch die Wirtsreaktivität vorangetrieben
wird, so sollte man dennoch beachten, dass nicht jeder potentiell immunstimulierende
Faktor unbedingt in die Krankheitsentstehung (und insbesondere in die chronische
Verlaufsform) involviert sein muss. Außerdem ist auch jeder Schutzmechanismus, durch
den sich das Bakterium dem Angriff durch das Wirtsimmunsystem widersetzt, ein
potentieller Virulenzfaktor, da er dem Bakterium die Möglichkeit vermittelt, den Mundraum
zu besiedeln und pathogene Veränderungen zu verursachen. In Folge werden also alle
diejenigen Faktoren beschrieben, die ein Virulenzpotential besitzen. Ob und wie stark jenes
jedoch ausgeprägt ist, ist oft noch Gegenstand andauernder Untersuchungen.
5.2 Porphyromonas gingivalis
5.2.1 Klassifikation und Charakteristika
Porphyromonas gingivalis (P.g.) ist ein gram-negativer, asaccharolytischer, obligat
anaerober Mikroorganismus. Das stäbchenförmige Bakterium ist nicht-motil und bildet keine
Sporen (Nakayama 2015). Es kann in vielen Bereichen des Mundraumes und auch in
gesunden Probanden nachgewiesen werden. Es finden sich bei P.g. viele verschiedene
Stämme und Genotypen und bei Patienten scheint jeweils ein Genotyp zu dominieren, ohne
dass verschiedenen Genotypen eindeutig unterschiedliche Pathogenität zugeordnet werden
kann. Bestimmte Kombinationen von Virulenzallelen sind jedoch mit Orten aktiver
Parodontitis und andere eher mit gesundem Parodontium assoziiert (siehe auch unten) und
in manchen Patienten werden auch besonders virulente Stämme gefunden (Yoshino et al.
2007). Durch horizontalen Gentransfer in dieser nicht-klonalen Population kommt es zu

- 17 -
neuen Allelkombinationen. Der hohe Grad an genetischer Diversifizierung bringt eine starke
Flexibilität hinsichtlich unterschiedlicher Umwelteinflüsse mit sich: P.g. kann so schnell auf
sich ändernde Umweltbedingungen reagieren, wobei der bestangepasste Stamm klonal
expandieren kann (Tribble et al. 2013).
Wie auch die anderen Mitglieder des ‚roten Komplexes’ so kann auch P.g. erst ‚spät’, d.h.
dann, wenn dafür durch andere Mikroorgansimen entsprechende Bedingungen geschaffen
wurden, orale Habitate besiedeln: Gesichert sein müssen zunächst die Bereitstellung von
notwendigen Nährstoffen durch metabolisch kompetente Arten, ein zunehmend anaerobes
Milieu und durch die Anhaftung anderer Bakterien entstehende Bindungsstellen (Lamont et
al. 1998).
Die Fähigkeit zur Auto- und Koaggregation stellt eine wichtige Voraussetzung für die
Besiedlung neuer Habitate dar und P.g. kann an Oberflächen binden und so ein Bestandteil
von Biofilmen werden. Die Adhärenz wird hierbei über Fimbrien (hitzebeständige Filamente,
die für die Bindung an eine Vielzahl von Substraten nötig sind) und Proteine der
Außenmembran vermittelt. Die Ausbildung einer Polysaccharidkapsel, wie sie einige
Stämme bilden, vermindert hingegen die Adhärenz (Wang et al. 2014). Die Besiedlung des
Mundraums erfolgt aber nicht nur durch Beteiligung am oralen Biofilm, sondern P.g. kann
auch lokal in parodontale Gewebe und in Epithelzellen eindringen.
Bereits in frühen Publikationen und bis heute wird Porphyromonas gingivalis in der Literatur
als eines der maßgeblichen Pathogene bei der Entstehung der Parodontitis beschrieben
(Slots et al. 1986, Lamont et al. 2000, Mysak et al. 2014). P.g. wird gemäß neueren
mechanistischen Modellen als ‚keystone’ (Brückenstein) Pathogen bezeichnet, womit seine
zentrale Rolle in der Ätiologie der Parodontitis beschrieben wird, welche es auch dann
ausüben kann, wenn es im Vergleich mit der gesamten Plaquemenge nur in geringer Anzahl
vorkommt (Hajishengallis und Lamont 2012). Während sich bei Pathobionten die
Pathogenität definitionsgemäß aus Habitatveränderungen (Umwelteinflüsse oder
Veränderungen der Immunabwehr) und damit meist einem zahlenmäßigen Anwachsen der
Population der Spezies ergibt (wie es z.B. bei Tannerella forsythia oder Treponema denticola
der Fall ist), verursachen ‚keystone Pathogene’ selbst den Zusammenbruch der
Homöostase - oder begünstigen ihn zumindest. Ihre Aktion erfolgt also zeitlich gesehen vor
der der Pathobionten (Hajishengallis 2014). Dysbiose, die Verschiebung des Mengenanteils
verschiedener Bakterien in der Mikrobiota zum Nachteil der Gesundheit, kann zum einen
durch Einflüsse von außen (also Veränderungen beim Wirt oder der Umwelt) aber auch
durch ‚keystone Pathogene’, die auch in geringer Anzahl das homöostatische Gleichgewicht
zum kippen bringen können, hervorgerufen werden (Hajishengallis 2014).

- 18 -
In einem Mausmodell der Parodontitis kann P.g. in ansonsten keimfreien Tieren keinen
Knochenabbau auslösen, im Beisein weiterer oraler Bakterien die Mikrobiota jedoch soweit
verändern, dass der Knochenabbau vorangetrieben wird (Hajishengallis et al. 2011). Diese
Aktivität wird teilweise seinem atypischen Lipopolysaccharid (LPS) zugeschrieben, welches
nicht Toll-like Receptor 4 (TLR4) sondern TLR2 aktiviert und TLR4 in Abhängigkeit vom
lokalen Entzündungsstatus sogar inhibiert, was zu einer lokalen Schwächung des
Immunsystems führt (Su et al. 2015). P.g. hat jedoch noch weitere Mechanismen entwickelt,
um selbst dem Immunsystem zu entgehen und gleichzeitig das für sich und andere
pathogene Keime (u.a. des roten Komplex) günstige inflammatorische Milieu zu fördern
(Singhrao et al. 2015). Es verursacht z.B. die Ausschüttung hoher Mengen
proinflammatorischer Zytokine wie IL-1b und IL-6, unterdrückt aber gleichzeitig die
Aktivierung des Komplementsystems und von TLRs (Singhrao et al. 2015). Das
inflammatorische Exudat bietet eine Quelle essentieller Nährstoffe für P.g. da es z.B. Peptide
und verwertbares Eisen aus haemolytischen Vorgängen, das im Metabolismus von P.g. eine
große Rolle spielt, enthält (Hajishengallis 2011).
Die Mechanismen, die immunmodulatorisch und auf das Komplementsystem wirken und
proteolytisch das Gewebe angreifen, sowie die Besiedlung oraler Epithelzellen erlauben,
und der nachteilige Einfluss von P.g. auf die Zusammensetzung der symbiontischen
Gemeinschaft der oralen Mikrobiota, der schließlich zum ‚microbial shift’ bzw. der Dysbiose
führt, stellen zum derzeitigen Wissensstand die Hauptmerkmale der Virulenz von
Porphyromonas gingivalis dar. Im Folgenden werden nun die Virulenzfaktoren von P.g.
beschrieben und ihre Funktion erklärt.

- 19 -
5.2.2 Virulenzfaktoren von Porphyromonas gingivalis
Da die Virulenz über die Interaktion mit der Umwelt vermittelt wird, gelten in erster Linie
Proteine, die an der Zelloberfläche gebunden sind, die also als erste mit der Umwelt in
Kontakt treten, sowie sekretierte Proteine als Virulenzfaktoren von Bakterien. Da die gut
untersuchten Gingipain-Proteasen und ihre Biosynthese stark mit verschiedenen Aspekten
der Virulenz von P.g. in Zusammenhang stehen, werden hier zunächst die Proteasen von
P.g. und dann erst strukturgebende Elemente wie Kapsel-Polysaccharide,
Lipopolysaccharide, Fimbrien etc. behandelt.
Proteasen und andere Enzyme
Proteasen spielen als Virulenzfaktoren eine große Rolle. Sie werden von den Bakterien
sekretiert und können durch Spaltung unterschiedlicher Substrate stark gewebsschädigend
wirken. Als asaccharolytischer Organismus ist P.g. auf ein komplexes System zum Abbau
von Fremdproteinen angewiesen und verfügt daher über ein breites Arsenal an proteolytisch
wirkenden Proteinen.
Gingipaine (Proteolyse, Zellinvasion, Haemagglutination + Haemolyse)
Die Proteine Gingipain R (Rgp-A und Rgp-B) und Gingipain K (Kgp), die Proteine spezifisch
hinter Arginin- bzw Lysinresten spalten, entfalten ihren Einfluss auf die Virulenz von P.g.
durch direkte und indirekte Mechanismen in jedem Stadium der Parodontitis (Bindung und
Besiedlung des Habitats, Erwerb von Nährstoffen, Umgehung der Immunabwehr des Wirtes,
Penetration von Geweben und Vermehrung). In erster Linie geschieht dies dadurch, dass
sie zahlreiche an der Virulenz beteiligte bakterielle Proteine prozessieren, die zum Beispiel
am Fimbrienaufbau beteiligt sind oder auf der Zelloberfläche exprimiert werden. Sie greifen
den Wirtsorganismus proteolytisch an, werden durch dessen Proteasen aber kaum inhibiert.
Hierbei werden zum einen Nährstoffe für das Bakterium freigesetzt, gleichzeitig aber auch
wichtige, normalerweise eng kontrollierte Signaltransduktionswege des Wirtes dysreguliert
(Kadowaki et al. 2000‚ Fitzpatrick et al. 2009).
Gingipaine spielen zunächst in der Zell- und Gewebepenetration eine entscheidende Rolle
und entsprechende Deletionsmutanten zeigen ein stark verringertes Penetrationspotential
(Andrian et al. 2004). Das lysin spezifische Gingipain K (Kgp) baut sehr effektiv Fibrinogen

- 20 -
und Fibrin ab (und trägt dadurch zur Blutungstendenz bei). Die Arginin spezifische Protease
Gingipain A (Rgp-A) aktiviert Gerinnungsfaktoren und degradiert Fibrinogen/Fibrin durch
seine für Phospholipide und Fibrinogen hochaffine Adhäsions/Hämagglutinations-Domäne
(effizienter als Rgp-B). P.g. attenuiert außerdem durch proteolytische Effekte der Gingipaine
den PI3K/Akt Signalweg was zur Dysregulation PI3K/Akt-abhängiger Zellfunktionen und in
Folge zur Zerstörung von Epithelialbarrieren führt (Nakayama 2015).
Auf der Bakterienzelloberfläche bilden Rgp-A und Kgp große, multifunktionale Komplexe,
die Funktionen wie Proteolyse, die Gewinnung von Hämoglobin, Aktivierung von
Blutplättchen, Agglutination von roten Blutkörperchen, Hämolyse und Adhäsion an die
extrazelluläre Matrix bewerkstelligen (Olsen et al. 2014). Dies ist in Einklang mit
Beobachtungen aus dem Mausmodell der Parodontitis, wo RgpA und Kgp, nicht jedoch Rgp-
B, eine maßgebliche Rolle im Pathogeneseprozess der Parodontitis spielen (Pathirana et
al. 2007). Gingipaine können in solchen - durch Lipopolysaccharide (LPS) an die Oberfläche
gebundenen - Formen vorkommen, jedoch auch abgespalten und in die Umgebung
freigesetzt werden (Olsen et al. 2014).
Gingipaine aktivieren das Kininsystems (Kininogen/ Kallikrein/ Kinin-System (Rapala-Kozik
et al. 2011), indem die sich auf der Bakterienoberfläche bildenden (und oben beschriebenen)
Adhäsionspatches (bestehend aus Gingipainen mit Hämagglutinin/Adhäsin-Domänen)
Bestandteile des Kininsystems binden. Auch Fibronektin und Laminin (Glykoproteine der
extrazellulären Matrix) werden gebunden und abgebaut (Lantz et al. 1991, Pike et al. 1996).
Gingipaine stimulieren die Expression von Matrix-Metalloproteinasen (MMPs) in
Fibroblasten und aktivieren sekretierte latente MMPs, welche das parodontale Gewebe
zerstören können (Imamura et al. 2003). Potempa et al. (2000) beschreiben den großen
Einfluss von Gingipainen auf den Kontrollmechanismus der MMPs auf der Ebene der Gen-
und Zymogenaktivierung.
Gingipaine sind auch am Abbau des Makrophagen-Oberflächenrezeptors CD14 beteiligt,
was die LPS-vermittelte Leukozytenaktivierung inhibiert, und dadurch die Besiedlung durch
P.g. erleichtert (Imamura et al. 2003). Gingipaine stören außerdem die Funktion neutrophiler
Granulozyten und führen zur Inaktivierung von Tumornekrosefaktor-α (TNF-α) und der
Modifizierung von Elastase (Jagels et al. 1996, Calkins et al. 1998, Abrahamson et al. 1997).
Die Multifunktionalität der Gingipaine erklärt den in seiner Virulenz völlig abgeschwächten
Phänotyp der Gingipain-negativen Mutanten von Porphyromonas gingivalis (Palm et al.
2015).
Unter Verwendung eines modifizierten Zellinvasionsassays konnte demonstriert werden,
dass verschiedene Subtypen innerhalb einer Population von P.g. Stämmen Unterschiede in

- 21 -
ihrer Fähigkeit, in orale Epithelzellen einzudringen, aufweisen (Suwannakul et al. 2010). Der
hochinvasive Subtyp dringt hierbei 10-30x so effizient ein wie der schlecht invasive Subtyp.
Eine Analyse der Gingipain Aktivität dieser Subtypen ergab, dass bei der hochinvasiven Art
die zellassoziierte Rgp-Aktivität reduziert war.
Klinisch manifestiert sich die Aktivität der Gingipaine in der Ausbildung von Ödemen durch
Erhöhung der Gefäßpermeabilität (Kallikrein/Kinin Signalwegaktivierung durch Rgp),
Infiltration von neutrophilen Immunzellen (Aktivierung des Komplementsystems durch Rgp)
und Blutung (Abbau von Fibrinogen/Fibrin durch Kgp) (Imamura et al. 2003, Travis et al.
1997).
Neben den Gingipainen selbst sind für die Virulenz von P.g. natürlich auch in die
Gingipainsynthese involvierte Faktoren maßgeblich. Die Acetyltransferase VimA (virulence
modulating) von P.g. beispielsweise spielt eine Rolle in der Resistenz gegenüber oxidativem
Stress, was wichtig ist bezüglich der harschen inflammatorischen Lebensbedingungen in
der parodontalen Tasche (Aruni et al. 2013). VimA ist außerdem mit für den pigmentierten
Phaenotyp von P.g. verantwortlich (Vanterpool et al. 2005). VimA moduliert die Gingipain
Biogenese und hat daher indirekten Einfluss auf dessen virulente Aktivität. Es beeinflusst
außerdem die Glykosylierung und damit Verankerung von Gingipainen und spielt auch bei
der Sialinisierung, im Acetyl-Coenzym A Transfer, im Lipid A Metabolismus und in anderen
Bereichen der Proteinbiosynthese eine Rolle. Die posttranslationale Modifikation
verschiedener Oberflächenbestandteile stellt ein Schlüsselelement bei der Modulation des
pathogenen Potentials von P.g. dar. Der Reifungsprozess der Gingipaine steht mit der
Kohlehydrat-Biosynthese in Verbindung und wird neben VimA durch weitere Proteine (VimE,
VimF, Por, PorR, Sov und Rfa) reguliert (Aruni et al. 2013). Das ‚Por’- Sekretionssystem aus
PorT, Sov, Pg27 sowie weiteren P.g. Proteinen vermittelt den Transport und die Sekretion
von Gingipainen und auch von anderen P.g.-Proteinen, die ebenfalls verwandte Export-
Domänen besitzen (Sato et al. 2010). Es gibt weitere Proteine mit bislang wenig geklärter
Funktion, die ebenfalls an der Funktion von Gingipainen beteiligt sind , z.B. PG534 (Saiki et
al. 2010).
Trypsin-like proteases
Neben den Gingipainen wurden bei P.g. mehrere weitere Proteasen mit trypsin-artiger
Aktivität nachgewiesen. Trypsin verdaut Proteine der extrazellulären Matrix und kann daher
einen Gewebeverband schädigen, indem es die Zelladhäsion beeinträchtigt. Trotzdem
spielen diese Proteine bei der Entstehung der Parodontitis keine entscheidende Rolle, da

- 22 -
sie durch Wirtsfaktoren weitgehend inaktiviert werden.
PPAD (P.g. Peptidylarginin-Deiminase)
Als Citrullinierung bezeichnet man einen Vorgang der post-translationalen Modifikation bei
der Argininreste in Proteinen deiminiert werden. Normalerweise finden sich diese Enzyme
nur bei Vertrebraten, doch einzigartig unter Bakterien kodiert auch P.g. für eine
Peptidylarginin-Deiminase (PPAD), die allerdings zu denen der höheren Eukaryonten
genetisch keine Verwandschaft aufweist. Ihre Funktion wird bei der Implikation von P.g. in
die Rheumatoide Arthritis diskutiert, da hier Citrullinierungsprozesse pathologisch verändert
sind. PPAD modifiziert andere P.g.-Proteine, jedoch auch sich selbst und Wirtsproteine
(Bielecka et al. 2014, Goulas et al. 2015).
Dipeptidylpeptidase IV (DPP-IV)
Aminosäuren werden von P.g. als Energie- und Kohlenstoffquelle genutzt und sie werden in
erster Linie in Form von Dipeptiden eingebaut, welche durch Dipeptidylpeptidasen (DPP)
zur Verfügung gestellt werden. Die am besten charakterisierte DPP von P.g., DPP-IV, hat
selbst keine Gelatinase oder Kollagenase-Aktivität gegenüber humanem Kollagen, kann
jedoch MMP’s (Matrix-Metalloproteinasen) des Wirtsorganismus (MMP1 = Kollagenase,
MMP2 = Gelatinase,) die eine große Rolle in späten Stadien der Parodontitis spielen,
aktivieren. Es konnte eine Korrelation zwischen der Fähigkeit von P.g. Stämmen, Biofilme
auszubilden, und der DPP-IV Aktivität festgestellt werden (Clais et al. 2014). In einem
Mausmodell der subkutanen Abszessbildung stellten sich die biofilmbildenden Isolate mit
hoher DPP-IV Aktivität als pathogener heraus als die nicht-biofilmbildende Stämme mit
niedriger DPP-IV Aktivität, die keine Abszesse bildeten. Der Zusammenhang zwischen
DPP-IV Aktivität und der Bildung von Biofilmen ist jedoch nicht geklärt. Beide Faktoren
werden jedoch als mögliche Ansätze von Therapien diskutiert (Krauss et al. 2000).
Mittlerweile wurden weitere DPP (DDP-7, DPP-11) von P.g. identifiziert und charakterisiert
(Banbula et al. 2001, Ohara-Nemoto 2011).
GroEL
Das GroEL Protein induziert bei Parodontitispatienten hohe Antikörper-Titer und wurde

- 23 -
daher in seiner Funktion näher untersucht. Es handelt sich um ein Protein aus der Hsp60
Familie, die an Proteinfaltungsprozessen beteiligt sind und schnell auf Stressreaktionen
reagieren (heat-shock Proteine). Es zeigte sich, dass Inkubation von humanen parodontalen
Ligamentzellen mit rekombinantem GroEL zur Ausschüttung von Zytokinen (IL-6, IL-8) führt,
die mit vermehrter Knochenresorption in Zusammenhang stehen. Außerdem fördert GroEL
bedingt durch Zytoskelett-Reorganisation die Zellmigration. Es wird vermutet, dass GroEL
durch die Aktivierung des Faktors RANKL, an der Osteoklastogenese beteidigt ist. Im
Rattenmodell zeigten sich verstärkte Entzündungsreaktionen und Knochenschwund nach
einer GroEL Inokulation. Forscher vermuten, dass die Immunantwort auf Stress-assoziierte
Proteine eine Entzündungsantwort hervorruft, die schließlich zu einer Chronifizierung führen
kann (Lin et al. 2014).
Kapsel / Polysaccharide und Lipopolysaccharide
Bakterielle Zelloberflächenglykane wie Polysaccharide und Lipopolysaccharide der Kapsel
beeinflussen den Prozess der Wirts-Immunantwort auf den pathogenen Reiz und werden
daher als Schlüsselelemente der Virulenz definiert. P.g. exprimiert zumindest drei
Oberflächenglykane, O-LPS, A-LPS und K-Antigen. K-Antigen Kapseln sind phagozytose-
resistent, während nicht kapselbildende Stämme besser in Epithelzellen eindringen und dort
replizieren können (Laine et al. 1998, Irshad et al. 2012). Die bakterielle Kapsel verhindert
den Zugang des Immunsystems zum Bakterium und kann die Anwesenheit des Bakteriums
auch maskieren. Ein P.g. Phänotyp ohne Kapsel induziert wesentlich höhere Mengen an
inflammatorischen Zytokinen, was die Kapselbildung zu einem Faktor der Unterwanderung
des Immunsystems macht (Brunner et al. 2010). Andererseits zeigten sich in einem
Tiermodell kapselbildende Stämme virulenter als nicht-kapselbildende (Van Steenbergen et
al. 1987).
Lipopolysaccharide (LPS), strukturgebende komplexe Glykanstrukturen auf der
Bakterienoberfläche, die durch Lipide in der Zelle verankert sind, wirken normalerweise
stark immunogen und die ‚innate immunity’ (natürliche, nicht-adaptive Immunität) der
Wirtsorganismen reagiert spezifisch auf Kontakt mit bakteriellen LPS (toll-like receptors
(TLR)-Aktivierung). Bei P.g. finden sich wie oben erwähnt O- und A-Typ LPS, die auch die
Serotyp-Spezifität der P.g.-Stämme determinieren (Sims et al. 2001). Bei P.g. scheint es sich
jedoch um atypische LPS zu handeln, die hauptsächlich TLR2, aber auch TLR4 Rezeptoren
stimulieren (auch Fimbrien, Lipoprotein PG1828 und Phosphoceramide sind potentielle

- 24 -
Kandidaten in P.g., die eine solche Antwort hervorrufen können) und in Makrophagen nur
eine im Vergleich zur Stimulation mit Eschiarichia coli LPS transiente Immunreaktion
hervorrufen (Holden et al. 2014, Sellers et al. 2015). Die von den Immunzellen produzierten
Zytokine haben einen schädigenden Effekt auf den Alveolarknochen und sind am
Knochenabbau beteiligt, während sie keinerlei Effekt auf das Weiterbestehen der P.g.
Infektion haben (Burns et al. 2006, Liang et al. 2011). Das LPS von P.g. kann in dendritischen
Zellen Reifung und antigenpräsentierende Funktionen initiieren und es regt die Bildung von
Osteoklasten an, indem es TLR2-abhängig RANKL Expression induziert (Kassem et al.
2015).
Eine in vivo-Relevanz der Beobachtung, dass P.g. LPS in gingivalen Fibroblasten TLR4
Signalwege stimuliert, hat sich hingegen bislang noch nicht nachweisen lassen können
(Wang et al. 2000).
Adhärenz /Adhäsionsmoleküle
Fimbrien
Fimbrien sind hitzebeständige Filamente von etwa 5 nm Durchmesser und 0,3 bis 3 μm
Länge, die aus 41 bis 49 kDa großen Fimbrillin-Untereinheiten bestehen und für P.g. den
wichtigsten Faktor für die Adhärenz darstellen. Durch sie ist die Bindung am Substrate wie
Epithelzellen, Speichelmoleküle, Gerinnungsfaktoren wie Fibrinogen, das extrazelluläre
Matrixprotein Fibronektin, Laktoferrin aber auch an andere Bakterien (Auto- sowie
Koaggregation) möglich.
Der fim-Gencluster besteht aus 7 Genen: FIMX, Porpyromonas gingivalis outer membran
protein A gene (PGMA) und fimA-E. Das strukturelle Molekül ist hierbei FimA (auch Fimbrillin
genannt) und die P.g.-Fimbrien bestehen ausschließlich aus FimA-Molekülen. Es ist bekannt,
dass die P.g.-Stämme ATCC 33277 und 381 (33277) aberrant lange Fimbrien (über einige
µm in der Länge) exprimieren, was auf FimB Mangel zurückzuführen ist. Tatsächlich
scheinen die Proteine FIMX, PGMA und FimB bis FimE nur regulatorisch auf die
Fimbrienbildung einzuwirken – und zwar sowohl bezüglich der Menge als auch der Länge -
ohne für die Ausbildung der Fimbrien jedoch essentiell notwendig zu sein (Nishiyama et al.
2007, Wang et al. 2007, Nagano et al. 2010). Es zeigte sich für verschiedene fimA-
Genotypen, dass in gesunden Probanden häufig Genotyp I und V gefunden wird, während
bei Parodontitis-Patienten die Typen II und IV dominieren, was eine starke Implikation der
Funktion der Fimbrien in die Pathogenese der Parodontitis belegt (Amano et al. 2000, 2010).
Auf der Oberfläche von P.g. werden auch kürzere Fimbrien, welche aus dem Protein Mfa

- 25 -
(auch als MFA1 bezeichnet) bestehen, exprimiert (Amano et al. 2010), welche nur in
Abwesenheit der längeren Fimbrien sichtbar gemacht werden können, und die so zuvor
unentdeckt geblieben waren. Beide Typen scheinen bei der Entwicklung der Parodontitis
eine Rolle zu spielen (Amano et al. 2004)
P.g. ist in der Lage, mit verschiedenen oralen gram-positiven und-negativen Spezies zu ko-
aggregieren (Whittaker et al. 1996). Die Länge der Fimbrien definiert, dass sie die ersten
Bestandteile der Bakterienzelle sind, die mit anderen Bakterien und auch mit Wirtszellen
interagieren. In der Literatur wird berichtet, dass die langen Fimbrien von P.g. die Ko-
adhäsion mit einer Anzahl oraler Mikroorganismen vermitteln: Actinomyces viscosus,
Treponema denticola (Dentilisin), Streptococcus gordonii und Streptococcus oralis (beide
über das zentrale Glykolyse-Molekül Glycerinaldehyd-3-phosphat-Dehydrogenase /
GAPDH) (Hashimoto et al. 2003, Park et al. 2005, Goulbourne et al. 1991, Maeda et al.
2004, Amano et al 2010). Kurze Fimbrien von P.g. ko-adhärieren mit Streptococcus gondii
mittels Adhäsin-Rezeptor-Interaktion und mit Streptococcus-SspA und -SspB
Oberflächenproteinen. (Kuboniwa et al. 2010). Lange Fimbrien scheinen sowohl bei der
Ausbildung des Biofilms als auch bei seiner Reifung eine Rolle zu spielen, während kurze
Fimbrien und Kgp (das lysin-spezifische Gingipain) suppressive und regulatorische Rollen
während der Biofilmentwicklung haben (Kuboniwa et al. 2009). Darüber hinaus steuert Rgp
(das Arginin-spezifische Gingipain) die Morphologie und das Biovolumen der Mikrokolonie.
Gemeinsam scheinen diese Moleküle in einer koordinierten Art und Weise zu handeln, um
die Entwicklung des reifen Biofilm zu regulieren.
TLRs (T-cell lymphotropic receptors, Rezeptoren auf der Oberfläche von T-Zellen) co-
clustern mit CD14, CD36, CD55 (den Zerfall beschleunigender Faktor), Komplement
Rezeptor 3 (CR3; CD11b/CD18), CXC-Chemokin-Rezeptor 4 (CXCR4) und
Wachstumsdifferenzierungsfaktor 5 (GDF5) (Hajishengallis et al. 2006, Asai et al. 2001). Bei
langen Fimbrien wurde gezeigt, dass sie nuclear factor-kappaB (NF-kB) mittels TLR2 und
CD14 stimulieren, was zu einer Induktion von Zytokinen führt, die - wie auch TNF-α, IL-1β,
IL-8 und IL-6 - an der Knochenresorption beteidigt sind (Hajshengallis et al. 2006, Asai et al.
2001, Ogawa et al. 2002, Davey et al. 2008, Harokopakis et al. 2006). Obwohl CD14 ein
wichtiger Co-Rezeptor für die Aktivierung von Epithelzellen durch lange Fimbrien ist, wurde
festgestellt, dass die Cytokin-Reaktionen (IL-6, IL-8, Granulozyten-Makrophagen-Kolonie-
stimulierenden Faktor, und TNF-α) von primären gingivalen Epithelzellen auf P.g. moderat
sind. Ein Effekt der durch den Mangel an membranassoziiertem CD14 auf diesen Zellen
zurückzuführen ist (Hajishengallis et al. 2006, Eskan et al. 2007). Im Gegensatz dazu
reagieren Monozyten mit CD14 stark auf lange Fimbrien und sekretieren große Mengen von

- 26 -
IL-6, IL-8 und TNF-α (Eskan et al. 2007). Lange Fimbrien induzieren auch IL-1β, IL-8,
monocyte chemoattractant protein-1 (MCP-1), vascular cell adhesion molecule-1 ( ICAM-1)
und E-Selectin in menschlichen Endothelzellen der Aorta (Takahashi et al. 2006). Obwohl
lange Fimbrien nur eine von mehreren pro-inflammatorischen Molekülen von P.g. sind,
zeigten Mutanten ohne lange Fimbrien im Vergleich deutlich schwächere Zytokin-Antworten,
was möglicherweise auf die Tatsache zurückzuführen ist, dass die langen Fimbrien wohl
zuerst mit den Wirtszellrezeptoren interagieren, was die unmittelbare Aktivierung
intrazellulären Signalkaskaden zur Folge hat (Kato et al. 2007).
Andere Studien zeigen, dass kurze Fimbrien stark in Wechselwirkung mit CD14 und TLR2
(aber nicht TLR4) stehen. Sie induzieren die Expression von Zytokinen (IL-1α, IL-β, IL-6 und
TNF-α) sowohl in menschlichen Monozyten-Zelllinien als auch in Makrophagen der Maus
( Hamada et al. 2002, Hiramine et al. 2003).
Lange Fimbrien interagieren außerdem mit CXCR4, einem Ko-Rezeptor für die TLR2
Aktivierung in menschlichen Monozyten und Makrophagen der Maus. Sie induzieren die
CXCR4-vermittelte Aktivierung der cAMP-abhängigen Proteinkinase A, die wiederum die
TLR2-induzierte NF-κB-Aktivierung hemmt (Hajishengallis et al. 2008, Pierce et al. 2009).
Die Bedeutung der Fimbrien wird deutlich in einem Experiment, in dem durch Immunisierung
von Ratten alleine mit isolierten Fimbrien eine pathogene Wirkung der P.g.-Infektion
unterdrückt wurde. P.g. Mutanten, die nicht zur Ausbildung von Fimbrien in der Lage sind,
zeigen außerdem einen drastisch verminderten Pathogenitätsphänotyp hinsichtlich der
Zerstörung des Alveolarknochens. Ein P.g. Stamm der keine Fimbrien exprimiert zeigt
außerdem eine signifikant bessere Bildung von Fibronektin (Evans et al. 1992, Malek et al.
1994, Puschmann 2004).
Hämagglutinine (Hämagglutination und Hämolyse)
Auf der Oberfläche von P.g. werden mehrere Proteine mit hämagglutinierender Funktion
exprimiert, die ebenfalls an der Zelladhäsion beteiligt sind und auch den Prozess der
Kolonie- bzw Biofilmbildung durch Quervernetzung mit humanen Zellbestandteilen
unterstützen (Lamont und Jenkinson 1998).
Eisen spielt beim Wachstum und der Virulenz von Porphyromonas gingivalis eine
entscheidende Rolle. Da Eisen in Form von Hämin einen essentiellen Nahrungsbestandteil
von P.g. darstellt, hat das Bakterium Mechanismen entwickelt, um an die Oberfläche von
Erythrozyten zu binden und Hämin daraus freizusetzen. Wie oben beschrieben besitzen

- 27 -
manche Gingipaine funktionelle Hämagglutinations-Domänen und damit
hämagglutinierende Eigenschaften. P.g. besitzt jedoch auch weitere Hämagglutinine, die
durch die Gene hagA, hagB und hagC codiert werden. Deren Deletion schwächt den HA
Phaenotyp von P.g., inhibiert ihn aber, wegen der Redundanz dieser Aktivität in
verschiedenen Proteinen, nicht (Lepine und Progluske- Fox 1996). Auch
Lipopolysaccharide und Fimbrienproteine können für einen Hämagglutinationsphänotyp
verantwortlich sein (Ogawa und Hamada 1994). Hämagglutination und proteolytische
Prozesse scheinen durch Ko-expression in engem Zusammenhang miteinander und zu
Adhärenz und Fimbrienaktivität zu stehen (Lamont 1998).
Invasion und Besiedlung von Epithelzellen
Nach Bindung an die Zellmembran wird P.g. über lipid rafts internalisiert und gelangt in frühe
Phagosomen. Es muss dann von P.g. ein Prozess der zellulären Autophagie initiiert werden,
da P.g. sonst in Phagosomen abgebaut wird. (Belanger et al. 2006, Mysak et al. 2014). Im
Laufe des Autophagieprozesses entstehen für P.g. optimale Ernährungs- und
Wachstumsbedingungen in der Epithelzelle (Belanger et al 2006). Es ist noch unklar, durch
welche Mechnismen und bakterielle Faktoren dieser Prozess abläuft.
Integrine sind für die Ausbildung der epithelialen Barriere essentiell, weil sie durch
sogenannte ‘focal adhesions’, lokale Adhäsionspunkte, die physikalische Verbindung
zwischen dem intrazellulären Zytoskelett und der extrazellulären Matrix vermitteln. Sie
beeinflussen außerdem u.a. Prozesse der Zellproliferation und Motilität aber auch der
Wundheilung und Regeneration (Gumbiner 1996). Bei diesen Ereignissen spielen die
Proteine Paxillin und Focal Adhesion Kinase (FAK) eine wichtige Rolle und die
Phosphorylierung von FAK ist ein zentraler Regulator bei der Integrin-vermittelten
Zellmigration (Schlaepfer et al. 1999). Paxillin findet sich konzentriert an ‘focal adhesions’
und spielt eine Rolle bei der Ausbildung des Aktin-Zytoskeletts (Nakamura et al. 2000). Das
Eindringen von P.g. in Epithelzellen führt zur Herabregulierung von Paxillin und FAK, was
zu einer Beeinträchtigung der Zellfunktion und Beeinflussung des Aktin-Zytoskeletts führt.
Gingivale Fibroblasten und Epithelzellen, welche mit P.g. infiziert sind, zeigen eine
reduzierte Adhäsion an die extrazelluläre Matrix. Veränderungen in der Morphologie von
einer ausgebreiteten zu einer abgerundeten Form und die damit beeinträchtigte Motilität
nach Inkubation mit P.g. wurden bereits von mehreren Gruppen beschrieben, was auf oben
beschriebene zytoskelettale Veränderungen zurückzuführen ist (Yilmaz et al. 2003). Durch

- 28 -
P.g. induzierte Apoptosisvorgänge resultieren allerdings ebenfalls in einem Abbau von Aktin
und führen zu einem Kollaps des Zytoskeletts. Zhang et al. (2013) berichten über die
Invasion von P.g. in Osteoblasten mittels Bindung der α5β1-Fimbrien an Integrin auf
Osteoblasten und anschließender Kondensation von peripherem Aktin. In den infizierten
Zellen wurde über den Signaltransduktionsweg der JNK-Kinase Apoptose induziert, ein
Prozess, der für die Bakterien jedoch nachteilig ist (Zhang et al. 2013). Auch Gingipaine
können die Zellintegrität modifizieren (Chen et al. 2001, Sheets et al. 2005, Tamai et al.
2005, Kinane et al. 2012).
Am Invasionsprozess sind auch Fimbrienproteine beteiligt (s.o.). Fimbrien erleichtern über
zellassoziiertes Fibronektin und dem an Fibronektin bindenden α5β1-Integrin die bakterielle
Invasion von mehreren humanen epithelialen Zelllinien und die Persistenz von P.g. an
intrazellulären Orten in vitro, was den Erreger vor der Erkennung durch das Immunsystem
schützen und seiner weiteren Ausbreitung in benachbarte Gewebe dienlich sein kann
( Yilmaz et al. 2002, 2003, 2004, Tsuda et al. 2008).
Verschiedene Subtypen von P.g. zeigen Unterschiede in ihrer Fähigkeit, in orale
Epithelzellen einzudringen (Suwannakul et al. 2010). Nach Veening et al. (2008) resultiert
diese epigenetische Veränderung in der Entstehung von phänotypischen Merkmalen, die
sich als Unterarten innerhalb einer isogenen Bevölkerung manifestiert, in der eine solche
Variation einen Mechanismus bietet, um eine schnelle Auswertung der ökologischen
Chancen oder die Reaktion auf Belastungen zu manifestieren ("bet-hedging"). Eine Analyse
ergab eine Reihe von Genen, die in die Resistenz gegenüber oxidativem Stress und in den
Eisentransport involviert sind, und die entscheidend für das Eindringen und das Überleben
in Epithelzellen sein könnten. P.g. exprimiert außerdem eine Reihe von Proteinen, die zum
sogenannten ‘caseinolytischen System’ (Clp) gehören. ClpC und ClpXP scheinen dabei für
das Eindringen in Wirtsepithelzellen notwendg zu sein, ClpB spielt hierbei hingegen keine
Rolle (Capestani et al. 2008)
Auto- und Ko-aggregation, Ausbildung von Biofilmen
In die Bindung an andere Vertreter der gleichen Spezies (Autoaggregation) und Vertreter
anderer Spezies (Ko-Aggregation) sind bei Porphyromonas gingivalis in erster Linie die
Fimbrien involviert. Die langen Fimbrien initiieren dabei die Entstehung des Biofilms,
während sie die Reifung des Biofilms nicht vorantreiben. Kurze Fimbrien und das Gingipain
Kgp haben suppressive und regulatorische Rollen während der Entwicklung des Biofilms.

- 29 -
Das Gingipain Rgp hingegen bestimmt die Morphologie der Kolonie in einem
Versuchsansatz und auch deren Volumen. Diese Faktoren scheinen gemeinsam an der
Regulation der Biofilmentwicklung beteiligt zu sein (Kuboniwa et al. 2009).
An der Ko-Aggregation mit anderen Bakterien sind auf der Seite von P.g. ebenfalls Fimbrien
und Gingipaine wichtig (Haraguchi et al. 2014). Bei der Ko-Aggregation von P.g. mit T.d. ist
das Protein Dentilisin beteiligt, und an der Ko-Aggregation mit Vertretern anderer Spezies,
die nicht zum roten Komplex gehören, sind auf deren Seite zahlreiche weitere Faktoren
bekannt, die am Aggregationsprozess beteiligt sind (Sano et al. 2014).
Interaktion mit dem Komplementsystem
Das Komplementsystem als Bestandteil der natürlichen Immunität des Wirtsorganismus
(innate immunity) ist ein schnell aktivierbares Verteidigungssystem gegen eindringende
Mikroben. Die Aktivierung des Komplementsystems resultiert in einer Art von Kettenreaktion
von proteolytischen Vorgängen, die jeweils auf den nächsten Faktor aktivierend wirken. In
diesem Prozess werden die Mikroorganismen von Komplementbestandteilen bedeckt
(opsonisiert) und letztendlich werden Immunzellen dadurch herbeigelockt und aktiviert. Die
Eindringlinge werden phagozytiert und lysiert (Markiewski und Lambris 2007).
Das Komplementsystem rekrutiert und aktiviert Zellen des Immunsystems und ist damit ein
Schlüsselfaktor bei der Pathogenabwehr. Es steht mit der Entwicklung der Parodontitis in
engem Zusammenhang: Überreaktion (durch z.B. Gendefekte) oder Dysregulation (durch
pathogene Faktoren) tragen zum Entzündungsverlauf maßgeblich bei und parodontale
Entzündung und lokale Aktivierung des Komplementsystems korrelieren in histologischen
und klinischen Untersuchungen.
Es existieren drei unterschiedliche Signalwege bei der Komplementaktivierung, sie alle
enden jedoch (nach Aktivierung von signalweg-abhängigen Konvertasen) zunächst am
gleichen zentralen Schlüssel-Punkt, dem C3 Faktor. Dieses so zentrale Protein wird jedoch
direkt durch alle drei P.g. Gingipaine angegriffen (Popadiak et al. 2007). Durch Gingipain-
vermittelten C3-Abbau inhibiert P.g. maßgeblich dessen Aktivierung, und zwar unabhängig
davon, welcher der Signalwege genutzt wird. Die Opsonisierung und Bindung des MAC
(membrane attack complex) an der Bakterienoberfläche wird also durch Gingipaine
unterdrückt (Popadiak 2007). Außer C3 degradiert P.g. auch die Komplementfaktoren C1,
C4 und C5 und fängt das C4b Protein ab (Wang et al. 2010). Während der massive Abbau
und die Inaktivierung von Komplementbestandteilen dem ohnehin komplement-resistenten

- 30 -
Bakterium wenig Vorteile bringt, verbessert es jedoch die Lebensbedingungen für andere,
vielleicht komplement-sensitivere Bakterien (Hussain 2015).
Ein wichtiger Bestandteil der innate immunity sind außerdem sogenannte ‚pattern
recognition receptors’ (PRR) wie z.B. die Familie der ‚toll-like receptors’ (TLR, s.o.). Sie
detektieren konservierte mikrobielle Strukturen wie z.B. die Lipopolysaccharide der gram-
negativen Bakterien und reagieren auf den Kontakt (Medzhitov 2007). In engem
Zusammenspiel mit den TLRs vermittelt das Komplementsystem auch zwischen der
natürlichen und der adaptiven Immunität. Um eine persistierende oder chronische Infektion
zu etablieren besteht für Mikroorganismen eine wichtige Evolutionsstrategie darin, einen
Weg finden, dieses System zu überlisten oder sogar für die eigenen Zwecke auszunutzen
(Krauss et al. 2010).
Ein anderer Bestandteil der Komplementkaskade ist der C5a-Rezeptor, der teilweise im
Zusammenspiel mit dem TLR2 wichtige inflammatorische und antimikrobielle Funktionen
vermittelt. P.g. besitzt selbst eine konvertaseartige Aktivität, kann C5a daher selbst aus
dessen Vorläufer generieren. Dies erscheint biologisch gesehen kontraproduktiv, da C5a ein
sehr potenter Aktivator des Komplementsystems ist (Wang et al. 2010). Das durch P.g.
hergestellte C5a aktiviert jedoch lokal den C5aR und stimuliert dadurch intrazelluläre Ca+-
Signalwege, die synergistisch die ansonsten schwachen cAMP Antworten der P.g.-
induzierten TLR2 Aktivierung verstärken. Die erhöhte cAMP Produktion wirkt wiederum auf
die Proteinkinase A (PKA), die infolge die Glykogen-Synthase Kinase 3b inaktiviert und
daher NO-vermittelten Angriff auf P.g. verhindert (Hajishengallis et al. 2008, Wang et al
2010). Ebenso befremdlich erscheint auf den ersten Blick die präferentielle Nutzung des
TLR2 Rezeptors durch P.g. obwohl TLR4 normalerweise bakterielle LPS erkennt (Wang et
al. 2010). Allerdings hat P.g. Wege gefunden, um TLR4 auf Umwegen zu inhibieren und nur
ein gewisser Teil der TLR2 Mechanistik wird inhibiert, so dass Signalwege, die für das
Wachstum des Bakteriums förderlich sind unterstützt werden, während solche, die ihm
schaden, inhibiert werden (Coats et al. 2009, Hajishengallis et al. 2012). TLR2- sowie C5aR-
defiziente Mäuse zeigen nach P.g. Infektion nicht den Knochenabbau-Phänotyp des P.g.
Wildtyp, was in vivo zeigt, dass dieser Pathogeneseprozess auf dem Zusammenspiel von
TLR2 und C5aR beruht (Raby et al. 2011). Von besonderem Interesse dabei ist die
spezifische Inhibition von IL-12p70, das durch TLR2 induziert wird, während durch die
Interaktion von C5aR und TLR2 weiterhin die Bildung proinflammatorischer und dem
Knochenabbau dienlicher Zytokine (IL-1b, IL-6 und TNF-α) angeregt wird. In vivo kann P.g.
erwiesenermaßen einer Elimination durch IL-12p70-abhängige Mechanismen entgehen,
während es gleichzeitig den Knochenabbau begünstigt, und damit gleichzeitig destruktive

- 31 -
Entzündungsprozesse fördern und trotzdem für das eigene Überleben sorgen kann (Raby
et al. 2011). Dieser ausgeklügelte Mechanismus ist für P.g. von Vorteil, denn eine
generalisierte Immunsuppression herbeizuführen wäre nicht im Interesse von P.g.. Die
Attacke durch Leukozyten könnte so vielleicht verhindert werden, andererseits könnte das
Bakterium durch fehlende Exudatbestandteile schlicht verhungern: als asaccharolytischer
Organismus braucht es das proteinreiche, inflammatorische Milieu um zu gedeihen
(Hajishengallis und Lambris 2012).
Die langen Fimbrien von P.g. sind ebenfalls an der Subversion des Komplementsystems
beteiligt, indem sie mit CR3 interagieren (Haijshengallis et al. 2007). Die CR3-Aktivierung
vermittelt wie oben beschrieben das Zusammenspiel von TLR2 und IL-12p70, und damit
fördert dieser Prozess das Überleben von P.g. in vitro und in vivo (Hajishengallis et al. 2011).
Die gleiche Gruppe zeigte auch, dass P. gingivalis die zellvermittelte Immunität unterdrückt,
indem es die Menge von ausgeschüttetem Interferon (IFN)-γ, einem Aktivator der
zellvermittelten Immunität, herabreguliert (Hajishengallis et al 2011). P.g. manipuliert also
die Bestandteile des Komplemetsystems in seinem Sinne um der Immunerkennung zu
entgehen, die ökologische Nische erfolgreich zu besiedeln und die lokale Mikrobiota zu
verändern.
Vesikel
Porphyromonas gingivalis kann Vesikel (extrazelluläre Vesikel, ECV) von der äußeren
Membran absondern, die aufgrund ihrer geringen Größe (20 – 150 nm) leicht das
umliegende Gewebe penetrieren und so auch zu einer lokalen Gewebszerstörung führen
können (Kamaguchi et al. 2003, Mayrand et al. 1988). Vesikel können als Vehikel für
toxische Substanzen und zahlreiche proteolytische Enzyme dienen (Mayrand et al. 1988).
Es wurde gezeigt, dass ca. 80% der wichtigsten toxischen Substanzen von P.g. in die
extrazellulären Vesikel inkorporiert werden können (Tsuda et al. 2005, Grenier et al. 1987).
Insgesamt konnten 151 Proteine von P.g. in ECVs nachgewiesen werden (Veith et al. 2014).
Pham et al. (2002) fanden in den äußeren Membranvesikeln von P.g. neben Gingipain große
Mengen an Cysteinproteasen.
Vesikel können auch die spezifische Immunantwort behindern, indem sie mit
Bakterienzellen um Antikörper-Bindungsstellen konkurrieren, und sind an der Ko-
aggregation mit Eubacterium saburreum und Capnocytophaga ochracea beteiligt (Grenier

- 32 -
et al. 1987).
Von ECVs wird außerdem die Ausschüttung von Matrixmetalloproteinase-1 (MMP-1) sowie
von Tissue inhibitor of metalloproteinase-1 (TIMP-1) aus humanen parodontalen
Ligamentfibroblasten angeregt, was zur Auflösung der extrazellulären Matrix führen kann
(Sang et al. 2014). ECVs führen außerdem zu einer Herabregulierung von CD-14
Rezeptoren, was wiederum die Hyporeaktivität von Makrophagen auf LPS-Stimulation
bewirkt, und die Immunantwort auf P.g. so schwächt (Duncan et al.. 2004).
Die Vesikel können auch Gingipaine enthalten und Inkubation mit ECVs kann experimentell
die Ablösung des oralen Plattenepithels bewirken (Nakao et al 2014).
Serotypen K1 und K2
Destruktive Parodontitis ist assoziiert mit einer Th1-Th17 Immunantwort und der Aktivierung
von RANKL–spezifischen Osteoklasten (RANKL (receptor aktivator of nuclear factor kappa-
B ligand) ist ein in die Osteoklastogenese involvierter Faktor). Die Serotypen K1 und K2 von
Porphyromonas gingivalis induzieren eine besonders starke Th1-Th17 Immunantwort und
man kann bei ihnen eine im Gegensatz zu anderen Serotypen verstärkte RANKL Produktion
nachweisen. Vermittelt durch die Th17-assoziierte RANKL Produktion induzieren sie eine
starke Osteoklasten Aktivierung, was wiederum die Basis für eine stärkere
Knochenresorption und damit eine Erklärung für die schwerere Verlaufsform der K1- und
K2-assoziierten Parodontitis darstellt (Vernal et al. 2014).
Effekt von Porphyromonas gingivalis auf die Wundheilung
In verschiedenen Studien konnte kürzlich gezeigt werden, dass P.g. die Wundheilung
deutlich verlangsamt. (Bhattacharya et al. 2014, Laheij et al. 2013). Es konnte gezeigt
werden, dass Zellzyklus-assoziierte Gene, die notwendig für die Zellteilung sind, sowie
Integrin-b-3 und -6, die wichtig für die Zellmigration sind, während dieses Prozesses
herabreguliert werden. Dieser Nachweis deckt sich mit der früheren Beobachtung, dass P.g.
die Wiederherstellung von gesundem oralen Gewebe durch Interferenz mit Zellmigration
und Zellwachstum behindert (Amano et al. 2007, Furata et al. 2009). Die am beschriebenen
Prozess beteiligten Faktoren sind bislang nicht eindeutig identifiziert worden, sie sind jedoch
mit der Zellwand von P.g. verbunden, können ins Medium abgegeben werden und sind

- 33 -
hitzeresistent. Es wird daher spekuliert, dass es sich um die Fimbrien, die
kapselassoziierten Polysaccharide oder die LPSs von P.g. handelt (Laheij et al. 2013).
5.3 Tannerella forsythia
5.3.1 Klassifikation und Charakteristika
Tannerella forsythia (T.f.), von der Gestalt her ein spindelförmiges Stäbchen, ist ein
gramnegatives Bakterium mit anaerobem Metabolismus, das in die Familie der Cytophaga
Bacteroides eingeordnet wurde. Es besitzt keine Fimbrien und ist unbeweglich.
Es wurde 1986 isoliert und als Bacteroides forsythus bezeichnet, 2006 jedoch basierend
auf 16s RNA Sequenzen reklassifiziert und in Tannerella forsythia umbenannt (Sakamoto et
al. 2002, Maiden et al. 2003, Tanner und Izard 2006, Posch et al. 2012)
T.f. kann nach den Definitionen von Socransky (Socransky 1979) als Pathogen klassifiziert
werden, da es bei Parodontitis in (relativ zum gesunden Status) erhöhter Zahl vorzufinden
ist (Sharma et al. 1998, Bird et al. 2001, Yoo et al. 2007), da es Hinweise auf eine
Wirtsantwort gegen seine Antigene gibt, es im Tierversuch Krankheitssymptome
hervorrufen kann (Takemoto et al. 1997, Sharma et al. 2005, Kesavalu et al. 2007) und es
Virulenzfaktoren exprimiert die möglicherweise am Pathogeneseprozess beteiligt sind
(Sharma et al. 2010, Posch et al. 2012).
T.f. besiedelt subgingivale Plaque Biofilme und besitzt eine einzigartige Glycoprotein Glucan
Oberflächenschicht mit S Struktur. Die virulenten Hauptmerkmale von Tannerella forsythia
sind Anheftung an und Invasion von Epithelzellen, der proteolytische und pro-apoptotische
Angriff von Epithelzellen sowie die Freisetzung pro-inflammatorischer Zytokine.
5.3.2 Virulenzfaktoren von Tannerella forsythia
Der S-layer der Zelloberfläche
‚S’-Oberflächenschichten (sogenannte ‚S-layers’) bestehen aus wasserunlöslichen
Proteinen, die die intrinsische Fähigkeit zur Selbstanordnung besitzen und auf der
Oberfläche von Bakterien Netzstrukturen ausbilden. Bei Tannerella forsythia besteht der S-
layer aus zwei Glycoproteinen, TfsA und TfsB (mit einem berechneten Molekulargewicht

- 34 -
von 135 bzw 154 kDa, das durch posttranslationale Modifikationen tatsächlich jedoch höher
ist, die Angaben belaufen sich auf 200/210 bzw 230/270 kDa), wobei sich bedingt durch die
Anwesenheit zweier unterschiedlicher Proteine eine einzigartige Struktur ergibt (Kerusou
1988, Lee 2006). Sie ist durch sogenannte ‚rough polysaccharides’ in der Bakterienzelle
verankert. Der S-layer bietet einen Selektionsvorteil für T.f. indem er einen Schutzmantel
darstellt, als molekulare Sortierstation und Ionenfalle dient und Zelladhäsion,
Zelloberflächenerkennung und das Anbinden von Phagen vermittelt (Beveridge et al. 1997,
Sabet et al. 2003, Sara und Sleytr 2000).
Tannerella forsythia besitzt, einzigartig unter gram-negativen Prokaryoten, Glycan Core
Strukturen auf der Oberfläche, die O-glycosidisch mit dem S-layer sowie mit anderen
Glykoproteinen verbunden sind (Lee et al. 2006). Die O-Glycosylierung findet man ebenfalls
bei mehreren anderen T.f.-Proteinen und Posch et al. (2011) postulieren hier ein
Virulenzmerkmal. Ein bestimmtes Motiv in diesem Glycan Core vermag Th17-vermittelte
Immunantworten (die Anlockung neutrophiler Zellen) zu supprimieren und so die
Effektorfunktionen dendritischer Zellen zu modulieren. Ein Bakterienstamm ohne dieses
Motiv induziert im Vergleich zum Wildtyp-Stamm vermehrt IL-23 und IL-6 Ausschüttung aus
Makrophagen (Settem 2013).
Der Glycosylierungsstatus bakterieller Proteine beeinflusst auch das Potential von T.f. zur
Autoaggregation. Der ORF (open reading frame) TF0022 codiert für ein Protein, das die
Expression von Genen, die in Glycosylierungsprozesse involviert sind, stimuliert. Mutanten
ohne diesen ORF zeigen eine verstärkte Neigung zur Autoaggregation, wahrscheinlich
durch Beeinflussung der post-translationalen Modifikation von Zelloberflächenkomponenten,
so dass auch hier eine virulente Komponente möglich ist (Niwa et al. 2011).
Orale Bakterien, unter anderem auch die beiden anderen Vertreter des roten Komplexes,
haben verschiedene Wege entwickelt, dem Angriff durch Faktoren des Komplementsystems
der natürlichen Immunabwehr im Wirtsserum zu entgehen (Serumresistenz), z.B. den
Proteaseverdau von Komplementproteinen, die Rekrutierung von Faktoren, die die
Komplementaktivierung inhibieren sowie die polysaccharidvermittelte
Komplementinhibierung (Shimotahira et al. 2013). Es konnte gezeigt werden, dass der S-
layer ebenfalls eine Rolle bei der Erkennung durch Komplementfaktoren spielt. Eine
Mutante mit defektem S-layer bindet große Mengen des Faktors c3b und zeigt
Wachstumsstörungen in Anwesenheit von Serumproteinen (FCS, fetal calf serum). Durch
den S-layer kann c3b nicht an die Zelloberfläche binden und T.f. kann in Anwesenheit von
Serumproteinen besser wachsen als die S-layer-defiziente Mutante, die durch c3b-Bindung

- 35 -
den MAC (membrane attack complex) rekrutiert, was zu einer Zellwandperforation der
Bakterien führt (Shimotahira et al. 2013).
Auch eine andere Arbeitsgruppe zeigte die Relevanz des S-layer bei der bakteriellen
Unterwanderung des Immunsystems durch mRNA Expressionsprofile in stimulierten
Effektorzellen. Eine T.f. Mutante ohne S-layer vermag in diesem experimentellen Ansatz die
Ausschüttung hoher Mengen an proinflammatorischen Botenstoffen zu induzieren, während
T.f. eine im Vergleich hierzu wesentlich verzögerte Immunantwort hervorruft (Sekot et al.
2011).
Die Daten legen nahe, dass der S-layer in der Form, wie er sich durch evolutionäre
Anpassung entwickelt hat, und -vermittelt durch das Glucan Core- dazu beiträgt, dass T.f.
der Kontrolle durch Mechanismen der natürlichen, angeborenen Immunität (innate immunity)
entgeht und dadurch die Immunantwort des Wirtsorganismus attenuiert. Das durch die
mangelhafte immunzellvermittelte Elimination bedingte Verbleiben im Wirtsorganismus
erlaubt es Tannerella forsythia, TLR2 und Th2 Antworten zu induzieren und dadurch zur
Gewebe- und Knochenzerstörung beizutragen (Settem 2013). Im Gegensatz zu
Porphyromonas gingivalis, welches aktiv das Immunsystem vor Ort schwächt und damit
aktiv auch anderen Organismen die Möglichkeit zu verstärktem Wachstum vermittelt (s.o.),
handelt es sich hier jedoch um passive Mechanismen.
Sabet et al. (2001) zeigten hämagglutinierende Eigenschaften von T.f., bei der sie eine
Abhängigkeit vom 210 kDa S-layer Protein demonstrierten. Diese Beobachtung konnte
jedoch von einer anderen Arbeitsgruppe nicht reproduziert werden und wird daher
kontrovers diskutiert (Sabet et al. 2001, Murakami et al. 2002, Sakakibara et al. 2007).
T.f. kann sich an Epithelzellen der Mundhöhle anheften und in sie eindringen (Sabet et al.
2001), und in der Tat lässt sich das Bakterium in Epithelialzellen nachweisen (Rudney et al.
2005, Colombo et al. 2007). Dabei werden anti-apoptotische Mechanismen in Gang gesetzt,
die den bakteriell vermittelten progammierten Zelltod blockieren. Die Zellen bieten so dem
Bakterium ein Reservoir, in dem es der Immunantwort entgehen kann und so bei der
Neubesiedlung nach erfolgter Therapie eine Rolle spielt (Inagaki 2006). 2001 postulierten
Sabet et al., dass dieser Mechanismus der Zellinvasion durch den S-layer vermittelt wird,
da er durch Präinkubation mit Antiseren gegen S-layer Proteine blockiert wurde. Die
Funktionen des S-layer als äußerste Zellschicht und damit Kontaktoberfläche mit der
Umwelt lassen sich nicht von den Funktionen der dort gebundenen Proteine und
anheftenden Moleküle separieren. Die Änderung der Struktur des S-layer kann auch
Änderungen in der Bindungsmöglichkeit anderer bakterieller Bestandteile mit sich bringen.

- 36 -
Alle Funktionen, die dem S-layer zugschrieben werden, hängen folglich auch von den dort
sich befindlichen und im folgenden beschriebenen Zellbausteinen ab. So weiß man
mittlerweile, dass die Funktion der Anheftung zwar einen intakten S-layer erfordert
(Sakakibara et al. 2007), das Eindringen in Wirtszellen jedoch durch das BspA Protein (das
sich ebenfalls an der Zelloberfläche und damit am S-layer befindet) vermittelt wird (Inagaki
et al. 2006).
BspA
BspA ist ein Oberflächenantigen, welches sowohl an der Zelloberfläche gebunden
vorkommt als auch sekretiert wird. Es bindet an extrazelluläres Fibrinogen und Fibronectin,
die im Aufbau der extrazellulären Matrix und als ‘clotting factors’ eine Rolle spielen. Es wirkt
außerdem immunogen und bei Parodontitis Patienten werden Antikörper gegen BspA
gefunden (Sharma et al. 1998).
BspA ist für Anheftung an und die Invasion von Epithelzellen nötig (Honma et al. 2001). Die
Bindung von BspA an die Wirtszelle führt zur Aktivierung von PI3K und Rac1 (GTPasen der
Rho-Familie) und in Folge zu Veränderungen im Aktin Cytoskelett, welche bei vielen
Pathogenen eine Rolle bei der Internalisierung spielen (Inagaki et al. 2006). Weitere
Untersuchungen liefern Hinweise darauf, dass die Internalisierung sowohl über das Molekül
Clathrin als auch über sogenannte ‘lipid rafts’ erfolgen kann. (Mishima and Sharma 2011)
Wie bereits erwähnt bietet der Prozess der Internalisierung dem Bakterium die Möglichkeit,
der Immunkontrolle zu entgehen und in einem Reservoir zu ruhen, aus dem es auch nach
erfolgter Therapie wieder hervortreten kann (Inagaki et al. 2006)
BspA spielt außerdem eine Rolle im Entzündungsprozess, indem es die Freisetzung sowohl
von Chemokinen aus Epithelzellen als auch von pro-inflammatorischen Zytokinen aus
Makrophagen induziert. Dies geschieht durch die BspA-vermittelte Aktivierung des toll-like
receptors 2 (TLR2), wobei der leucin-rich repeat (LRR) von BspA an TLR2 bindet und als
Ligand für diesen Rezeptor zu wirken scheint. (Hajishengallis et al. 2002, Akira et al. 2004,
Onishi et al. 2008, Sharma et al. 2010). Infolge der Bindung und Aktivierung kommt es im
experimentellen Ansatz von Onishi et al. (2008) zur Ausschüttung von IL-8 aus humanen
gingivalen Epithelzellen, in früheren Publikationen war jedoch von einer nur sehr geringen
IL-8 Ausschüttung aus Primärkulturen von humanen gingivalen Epithelzellen berichtet
worden (Han et al. 2000). Im Mausmodell führt Infektion mit T.f. zu parodontalem
Knochenverlust (Sharma et al 2005). Mäuse ohne funktionellen TLR2 (TLR-2-/-) bzw mit

- 37 -
Stat-6 Defizienz (Stat-6 -/-) zeigten hingegen deutlich eingeschränkte Suszeptibilität für T.f.
–induzierten Knochenabbau sowie abgeschwächte Th2 Antworten, was darauf schließen
lässt, dass Infektion mit T.f. zu einer durch TLR2-signaling vermittelten knochendestruktiven
Th2-Zell Antwort führt (Myneni et al. 2011). Die BspA-vermittelte TLR2-Aktivierung stellt
folglich ein starkes Virulenzmerkmal von T.f. dar.
Miropin
Miropin, ein ebenfalls von T.f. exprimiertes Serpin (= Familie von Serin-protease Inhibitoren),
vermag ein weites Spektrum von Proteasen zu inhibieren. Es entwickelte sich wohl in
Anpassung an die stark proteolytischen Lebensbedingungen im Mundraum und könnte
neben einer putativen Funktion im Stoffwechsel des Bakteriums dadurch, dass es T.f. vor
der destruktiven Aktivität neutrophiler Serinproteasen schützt, ein Virulenzpotential besitzen
(Ksiazek et al. 2015).
Proteasen
Da T.f. als asaccharolytischer Organismus nicht in der Lage ist, Kohlenhydrate abzubauen,
muss es Proteinbestandteile der Wirtszellen verstoffwechseln und nutzt dazu verschiedene
Proteasen. Bedingt durch ihre biologische Funktion greifen die bakteriellen Proteasen
jedoch auch das umliegende epitheliale Gewebe des Wirtsorganismus, sowie das
Immunsystem an, da Faktoren der natürlichen / angeborenen Immunität (innate immunity)
durch Abbau neutralisiert werden können.
Die in vivo Pathogenesefunktion einer bereits 1995 publizierten ‘trypsin-like serine protease’
Aktivität (Grenier 1995) eines 81kDa Proteins der Zelloberfläche ist ebenfalls als gering zu
bewerten, da sie nur kleinere Proteinsubstrate spalten kann.
Die PrtH Cystein-Protease mit Hämolysin Aktivität wurde erstmals 1997 von Saito et al.
(1997) beschrieben. Das für PrtH codierende Gen prtH wurde während einer Studie zur
Funktionalität verschiedener genetischer Fragmente von T.f. isoliert. Ein Klon mit dem prtH-
tragenden Genabschnitt führte in Eschiarichia coli zu Hydrolyse von Milcheiweißen. In der
Sequenz wurde ein Gen identifiziert, welches für ein putatives Protein mit Ähnlichkeiten zu
Cysteinproteasen kodiert, PrtH. In einer anderen Studie wurde ein Protein charakterisiert,
das an der Ablösung von Zellen aus dem Verband beteiligt ist und als forsythia detaching

- 38 -
factor (Fdf, codiert durch das Gen fdf) bezeichnet wurde. Es zeigte sich, dass es sich bei
prtH um einen Teil des fdf Gens handelte (Pei et al. 2009). In der Literatur wird aber nach
wie vor häufig der Terminus PrtH verwendet, obwohl er streng genommen nur für einen Teil
des Fdf Vollängenproteins steht. PrtH zeigt strukturelle Ähnlichkeiten bzw.
Sequenzhomologien mit Caspasen und den Gingipainen von Porphyromonas gingivalis.
Die PrtH Protease (Saito et al. 1997) kann größere Proteinsubstrate spalten und damit
potentiell auch zur Ablösung von Epithelzellen aus dem Gewebe führen, was mit der
Freisetzung von Chemokinen (IL-8) einhergeht. Beeinträchtigung der Zelladhäsion ist als
ein maßgeblicher Faktor zu bewerten, der den Prozess der Ablösung des parodontalen
Gewebeverbands vom Zahnsubstrat unterstützt und damit am Fortschritt der
Paradontalerkrankung und in letzter Konsequenz am Zahnverlust beteiligt ist. Auch wenn
die biologischen Aktivitäten den Verdacht nahelegen, dass eine ursächliche Verbindung zur
Entwicklung der Parodontitis besteht, und dies auch lange Zeit postuliert wurde, so scheint
PrtH in vivo für ihre Enstehung zumindest nicht notwendig zu sein, da in einem gewissen
Prozentsatz von Patienten Isolate ohne funktionelle PrtH gefunden wurden (van der Reijden
et al. 2006)
Die kürzlich identifizierte und charakterisierte Metalloproteinase Karilysin scheint hingegen
ein stark virulentes Potential zu besitzen (Karim et al. 2010). Karilysin besitzt die Fähigkeit
zur Autoprozession und vermag weiterhin eine Vielzahl anderer Proteine, z.B. Fibrinogen,
Fibronektin, Elastin u.a., zu spalten (Karim et al. 2010). Außerdem führt es sehr effektiv zur
Ablösung des Immunfaktors TNF-α von der Oberfläche von Makrophagen. TNF- α wird
teilweise von Karilysin neutralisiert, der Rest des in die Umgebung abgegebenen TNF- α
behält jedoch seine biologische Aktivität und kann sowohl Apoptosis als auch autokrine
Signalkaskaden proinflammatorischer Genexpression auslösen (Bryzek et al. 2014). Bereits
im Jahre 2000 war gezeigt worden, dass T.f., T.f.-Lysate sowie der Zellkulturüberstand von
T.f. die Apoptosis-vermittelte Zell-Lyse von HL-60 Zellen und anderen Zellen humanen
Ursprungs herbeiführen (Arakawa et al. 2000). Diese Beobachtungen könnten auf
Karilysinaktivität zurückzuführen sein.
Weiterhin kann Karilysin sowohl das humangenerierte, antibakterielle Peptid LL-37, welches
eine bedeutsame Rolle bei der Abwehr der Besiedlung durch Pathogene im Mundraum
spielt, spalten und dadurch inaktivieren als auch alle Komponenten des
Komplementsystems inaktivieren, eine Funktion auf der wohl die Serumresistenz von T.f.
beruht. (Koziel et al. 2010, Jusko et al. 2012)

- 39 -
Glycosidasen
Tannerella forsythia exprimiert eine Reihe Glycosidasen (SiaH1 und NanH Sialidasen, sowie
alpha-D-glucosidase and N-acetyl-beta-glucosaminidase, (Moncla. 1990, Ishikura et al.
2003, Hughes 2003, Thompson et al. 2009) die während der Pathgenese verschiedene
Rollen spielen. Ihre Aktivität liegt im Abbau von Oligosacchariden und Proteoglucanen,
wodurch potentiell das Endothel der Mundhöhle angegriffen warden kann. Währrend des
Abbauprozesses können neue Oberflächenepitope entstehen, die dem Bakterium selbst
oder anderen bakteriellen Mitbesiedlern die Anheftung und Koloniebildung erlauben. Die
glycosidische Aktivität führt auch zur Anhäufung eines toxischen Abfallprodukts
(Methylglyoxal), das seinerseits zur Gewebeschädigung beitragen kann (Sharma et al. 2000)
Das Enzym α-I-fucosidase , eine kürzlich identifizierte 51-kDa Glycosidase von T.f., scheint
keine extrazelluläre Wirkung zu haben, sondern am Zuckermatabolismus im Periplasma
beteiligt zu sein und somit nur eine indirekte Virulenzfunktion zu haben (Megson et al. 2015).
Lipopolysaccharide
Bei den meisten gram-negativen Bakterien befinden sich aus strukturgebenden und
funktionellen Gründen Lipopolysaccharide (LPS) in der Außenhülle. Sie wirken
immunstimulierend, indem sie von Monozyten und Makrophagen erkannt werden und
dienen so dem Wirtsorganismus zum Erkennen und Bekämpfen der Infektion durch gram-
negative Bakterien. In Folge des Kontakts mit LPS werden eine Reihe inflammatorischer
Botenstoffe (z.B. TNFα , IL-6, IL-1a) ausgeschüttet, die wiederum das Immunsystem dazu
anregen, gegen das eindringende Pathogen vorzugehen (Posch et al. 2013). Die
Lipopolysaccharidstruktur von T.f. wurde durch Massenspektroskopie teilweise geklärt. Das
aufgereinigte Lipopolysaccharid vom R-Typus (‚rough-type’) induziert in Macrophagen -
jedoch kofaktorabhängig - die Produktion der proinflammatorischen Cytokine IL-1 , IL-6 und
TNF- α (Posch et al. 2013).

- 40 -
Lipoproteine
Die Lipoprotein-Fraktion einer T.f. Aufreinigung vermochte es, unter experimentellen
Bedingungen sowohl IL-6 Ausschüttung aus einer humanen gingivalen Fibroblasten-Zellinie
als auch TNF-α Ausschüttung aus Th-Zellen auszulösen. Sie induzierte weiterhin TLR2-
abhängig CD25-Expression auf der Oberfläche von CD14-positiven Zellen sowie Apoptosis
(Hasebe et al. 2004, Bodet und Grenier 2010). Auf diesen Beobachtungen beruht die
Einordnung der Lipoproteine als Virulenzfaktoren von T.f., ohne dass diese Lipoproteine
näher charakterisiert wurden. Aufgrund der redundanten Effektorfunktionen könnten hier
BspA, Karilysin und Lipopolysaccharide der Zellhülle eine Rolle in der Lipoproteinfraktion
spielen. Die Autoren argumentieren zumindest, dass die beobachteten Effekte nicht
aufgrund von Lipopolysacchariden in der Aufarbeitung stammen. Karilysin war zum
Zeitpunkt der Studie noch nicht identifiziert worden (Hasebe et al. 2004).
5.4 Treponema denticola
5.4.1 Klassifikation und Charakteristika
Treponema denticola (T.d.) gehört zur Familie der Spirochaetaceae. Die oralen
Spirochaeten gehören der Gattung Treponema an, es gibt daneben die Gattungen Borrelia
und Spirochaeta. T.d. ist ein gram-negatives, von der Form her schraubenförmig
gekrümmtes, motiles Stäbchen. Die Fortbewegung erfolgt dabei durch Rotation um die
Längsachse und die Motilität stellt einen wichtigen Virulenzfaktor von T.d. dar, da sie an der
Invasion humaner Epithelzellen beteiligt ist. In einem murinen Parodontitismodell führt
Inokluation mit T.d. zu Besiedlung der Mundhöhle des Tieres, Induktion einer spezifischen
Immunantwort und der Herbeiführung von signifikantem Knochenabbau, was eine
ursächliche Rolle von T.d. bei der Gewebszerstörung im Rahmen der Parodontitis nahelegt
(Kesavalu et al. 2007). Wie Porphyromonas gingivalis und Tannerella forsythia kann auch
Treponema denticola an Epithelzellen binden und in sie eindringen.

- 41 -
5.4.2 Virulenzfaktoren
Oberflächenantigene
Die an der Oberfläche von Bakterien vorkommenden Proteine spielen eine Rolle bei der
Interaktion mit dem Umfeld, insbesondere bei der Zell-Interaktion (mit anderen Bakterien
oder Wirtszellen), und sind auch gleichzeitig die Angriffspunkte der antibakteriellen
Immunabwehr. Bei den Bakterien des Roten Komplexes handelt es sich hier spezifischer
Weise um extrazelluläre Proteasen, deren Substrate sehr vielfältiger Natur sind.
Msp
Msp ist das von der Molekülzahl häufigste Protein auf der Oberfläche von T.d. und besteht
aus drei Domänen, wobei die zentrale Domäne, anders als die hoch konservierten C- und
N-Termini, zwischen den verschiedenen Stämmen einen hohen Grad an Variabilität zeigen.
In Tieren, die mit T.d. immunisierten wurden, finden sich verschiedene Serotypen in
Abhängigkeit vom verwendeten T.d. Stamm und die variable Region ist hierfür verantwortlich
(Fenno et al. 1997, Edwards et al. 2005, Lee et al. 2005, Capone et al. 2008). Es besitzt die
Fähigkeit, Poren auszubilden (porin) und tritt über sog. ‚loops’ mit einer Vielzahl
extrazellulärer Proteine wie Laminin, Fibronectin, Keratin, Kollagen, Fibrinogen,
Hyaluronsäure und Heparin in Kontakt, was die Besiedlung von Wirtsgeweben unterstützt
(Fenno and McBride 1998, Edwards et al 2005). Msp kann, wie auch T.f. BspA, in den
Aktinmetabolismus eingreifen und dadurch in aktinabhängige Prozesse eingreifen. Dies
wiederum beeinträchtigt die Chemotaxis neutrophiler Immunzellen und die Aktivität von
Phagozyten. (Batista da Silva et al. 2004, Puthengady et al. 2006, Amin et al. 2007, Jobin
et al. 2007, Magalhaes et al. 2008). In primären gingivalen Epithelzellen löste Msp keine IL-
8 Induktion aus, welches normalerweise neutrophile Immunzellen zum Infektionsherd lockt
(Brissette et al. 2008). Durch Umgehung dieser Immunaktivierung könnte die Persistenz von
T.d. verbessert werden.
Msp kann TLR-2 abhängig Makrophagen aktivieren und hier TNF-α Produktion auslösen
(Rosen et al. 2012, Anand et al. 2013).

- 42 -
Td92
Ein weiteres Oberflächenprotein ist Td92, welches wohl die Bindung an Epithelzellen
vermittelt, da ein Antiserum gegen Td92 diese Bindung verhindert (Jun et al. 2008). Chin et
al. (2013) berichten, dass die Interferon -dominante Zytokin- Reaktion bei chronischen
Parodontitispatienten beeinträchtigt war, und der Td92 -induzierte Interferon-Spiegel-
negativ mit der Zahnzerstörung bei Patienten assoziiert war. Die Studienergebnisse von Kim
et al. (2012) legen nahe, dass Td92 die Bildung von Osteoclasten durch die Regulierung
der RANKL und die Osteoprotegerin- Produktion über eine Prostaglandin E2 -abhängigen
Mechanismus fördert.
TDE0471
Die putative Neuraminidase (Sialidase) TDE0471, eine oberflächenassoziierte Exo-
Neuraminidase, entfernt Sialic Acid von humanen Serumproteinen. Dies dient Bakterien
einerseits zur Nahrungsaufnahme, und andererseits wirkt die Anheftung von Wirtsproteinen
an der Bakterienzelloberfläche dem Angriff durch das Immunsystem entgegen. TDE0471
verhindert die Bindung von MAC (membrane attack complexes) an die
Bakterienzelloberfläche. Untersuchungen im Tiermodell ergaben, dass TDE0471-defiziente
T.d. Mutanten in BALB/C Mäusen weniger virulent sind, als T.d.. Dieser Unterschied ist
jedoch in mutanten Mäusen mit Komplementdefekten nicht gegeben, was darauf hinweißt
dass der virulente Phänotyp durch Unschädlichmachung des Komplementsystems entsteht
(Kurniyati et al. 2013).
TDE0214
Bei TDE0214 handelt es sich um ein PilZ-like c-di-GMP bindendes Protein, das an der c-di-
GMP-Signalkette beteiligt ist, und dessen Funktion wichtig ist für die Motilität des Bakteriums
und seine Fähigkeit, Biofilme auszubilden. In in-vivo Mausmodellen zeigte eine Mutante
ohne TDE0214 eine verminderte Invasivität des Bakteriums, eine weniger starke Fähigkeit,
Abszesse zu bilden und rief eine weniger starke Immunantwort hervor, was die Funktion
dieses Faktors bei der Virulenz unterstreicht.

- 43 -
Trypsin-like Protease Aktivität
Die Oligopeptidase B OpdB, die möglicherweise in zwei verschiedenen Formen von zwei
verschiedenen Genloci exprimiert wird, verleiht T.d. eine trypsin-like Proteaseaktivität (Lee
and Fenno 2004).
Lipoproteine
Zu den Oberflächenproteinen gehören viele Lipoproteine. T.d. kodiert für 166 putative
Lipoproteine, die zu verschiedenen Proteinfamilien gehören, jedoch alle N-terminal
acetyliert und dadurch in der Membran verankert sind (Veith et al. 2009). Die Expression
der entsprechenden Gene variiert in Abhängigkeit vom Wachstumsverhalten (Suspension
vs. Biofilm), was die Bedeutung dieser Proteine für das Bakterium zeigt (Mitchell et al. 2010).
OppA
Das Lipoprotein OppA bindet z.B. lösliche Proteine wie Plasminogen und Fibrinogen, nicht
jedoch unlösliche Proteine oder Wirtszellen. Es hat adhäsive Eigenschaften und führt
möglicherweise zur Bestückung der Bakterienzelloberfläche mit Wirtszellproteinen, was die
Immunerkennung verzögern oder verhindern kann (Fenno et al. 2000).
FhbB
Das Lipoprotein FhbB bindet Proteine der Faktor-H (FH)-Familie, welche regulatorisch in
das Komplementsystem eingreifen. Das gebundene FH Protein wird dann durch Dentilisin
gespalten und inaktiviert. Die genaue Rolle dieser Funktion bei der Virulenz von T.d. muss
noch geklärt werden. Es liegt nahe, dass die Komplementfunktionen beeinträchtigt werden,
aber FhbB könnte auch bei der Zellbindung und -invasion beteiligt sein (McDowell et al.
2007, 2009).

- 44 -
Dentilisin
Ein weiteres Lipoprotein ist die Protease Dentilisin (auch PrtP, chymotrypsin-like protease,
subtilisin-like protease). Sie kommt in einem Komplex mit zwei weiteren Proteinen vor,
PrcA1 und PrcA2, und trägt auf mehreren Wegen zur Virulenz von T.d. bei. Zum einen
beeinflusst das Protein interzelluläre Signalwege des Wirtsorganismus, indem es
Signalmoleküle wie IL-1b, IL-6, TNF-a und Mcp1 (Monocyte chemoattractant protein 1)
proteolytisch abbaut und Immunantworten dadurch moduliert werden (Miyamoto et al. 2006,
Okuda et al. 2007, Jo et al. 2014). Außerdem greift es die interzelluläre Matrix proteolytisch
an und durch den Abbau interzellulärer Adhäsionsmoleküle wird scheinbar auch die
Penetration von Epithelzellen durch T.d. begünstigt (Chi et al. 2003). Dentilisin spaltet
möglicherweise auch andere Oberflächenproteine wie Msp und interagiert mit den Fimbrien
von P.g. und ist an der Co-Aggregation dieser Bakterien beteiligt (Sano et al. 2014). Wie
auch der RgpA-Kgp Komplex von P.g., so bindet auch der Dentilisin-PrcA-Komplex von T.d.
an Fibrinogen und bewirkt dessen Abbau, wodurch Effekte wie Blutungsneigung und
Verzögerungen in der Reparatur der Schadstellen erklärt werden können (Bamford et al.
2007).
Leucin-rich-repeat Proteine
LrrA ist ein Protein der Klasse der LLR Proteine (leucin-rich repeat proteins), welches die
Bindung von T.d. an T.f. (nicht jedoch an P.g.) und an Epithelzellen vermittelt. So wird die
Expression weiterer putativer LRR-Proteine bei T.d. anhand von Sequenzanalysen vermutet.
Bei P.g. spielen LRR Proteine eine Rolle im Prozess der Zellpenetration epithelialer Zellen
und der Ausbildung des Biofilms. Auch bei T.f. spielt das LRR-Protein BspA bei der
Zellinvasion eine Rolle und als Virulenzfaktor in einem murinen Modell des Knochenabbaus
(Sharma et al. 1998, 2005, Capestany et al. 2006, Inagaki et al. 2006, Dashper et al. 2009).
Dies legt die Vermutung nahe, dass auch bei T.d. LRR-Proteine eine Rolle als
Virulenzfaktoren spielen mögen.

- 45 -
Cystalisin
Die metabolischen Endprodukte von T.d., u.a. Acetat, Lactat, Pyruvat, Hydrogensulfid (H2S)
und Ammoniak können sowohl die biologische Zusammensetzung des Biofilms
beeinflussen als auch das Wirtsgewebe angreifen und Immunantworten modulieren
(Hespell und Canele Parola 1971, Carlsson 1997, Kuramitsu et al. 2007). Die flüchtige aber
extrem toxische Schwefelverbindung Hydrogensulfid z.B. entsteht bei der
Cysteinfermentation von T.d. in einem metabolischen Prozess, der auch das neu entdeckte
Protein Cystalysin involviert (Chu et al. 2002, 2008). Cystalysin-vermittelte H2S-Produktion
führt zur Zersetzung der Membranen von Erythrozyten, was die hämolytische Aktivität von
T.d. erklärt, und hat sowohl pro- als auch anti-inflammatorische Effekte, was zu einer
Deregulation der Wirtsimmunantwort beitragen mag (Chu et al. 1995, Chen et al. 2010).
Lipooligosaccharide
Treponemen gehören gemäß aktuellen Klassifizierungen weder zu den gram-positiven noch
zu den gram negativen Bakterien. Daher besitzen sie auch nicht die für gram-negative
Bakterien typischen, immunstimulierenden Lipopolysaccharide (LPS) in der Außenhülle.
Stattdessen besitzten Treponemen Lipooligosaccharide (LOS), die funktionelle
Ähnlichkeiten zu LPS haben. LOS besitzen ein proinflammatorisches Potential und binden
an Proteine der extrazellulären Matrix, an Mucosazellen und an andere orale Bakterien, was
die proentzündliche Funktion weiter verstärken könnte (Grenier et al. 2013). TLR (toll-like
receptors), Oberflächenrezeptoren, die der Erkennung von konservierten
Bakterienstrukturen dienen, leiten entsprechende Signale weiter, die zur Expression
proinflammatorischer Gene führen. Kommt es bei diesem Prozess zu einer Deregulation,
kann die für die Parodontitis typische Gewebeschädigung auftreten (s.o.). Auch von P.g. und
T.f. ist es bekannt, dass sie in TLR-vermittelte Signaltransduktionswege eingreifen. Bei T.d.
tritt TLR Stimulierung hauptsächlich durch LOS auf, welche in einer Vielzahl von Zellen pro-
inflammatorische Botenstoffe induzieren (Tanabe et al. 2008).
Oberflächen-Vesikel
Die runden Vesikel die an der Oberfläche von T.d. entstehen, enthalten Adhäsine, Toxine
und proteolytische Proteine und werden daher als starke Virulenzfaktoren eingeordnet. Sie

- 46 -
sind an Funktionen wie bakterieller Aggregation und Zellinvasion beteiligt, sie sind
zytotoxisch und können die Wirts-Immunantwort modulieren (Wensink und Withold 1981,
Kuehn und Ketsy 2005). Als Transportvesikel können sie wenig stabile Signalmoleküle an
weiter entfernte Stellen transportieren, dabei vor Abbau schützen und können dabei besser
in Gewebe eindringen als das Bakterium selbst (Li Z et al. 1996, Mashburn und Whiteley
2005, Kuehn und Kesty 2005). Die Vesikel werden in der Monokultur aber auch bei Ko-
Kultivierung mit Porphyromonas gingivalis beobachtet, eine in-vivo Relevanz ist also
wahrscheinlich (Dashper et al. 2011). Sowohl LOS als auch Msp finden sich in den Vesikeln
und beide können in Makrophagen ein Immuntoleranzverhalten auslösen, was dem
Bakterium einen Überlebensvorteil sichert (Nussbaum et al. 2009).
Motilität und Chemotaxis
Auch die Beweglichkeit und die Chemotaxis von T.d. können zur Virulenz des Bakteriums
beitragen. Ein spezieller Flagellen-Aufbau erlaubt Treponema, sich auch unter sehr
viscosen Bedingungen zu bewegen, was einen Selektionsvorteil darstellt (Fenno und
McBride 1998). Durch Chemotaxis kann das Bakterium auf sich ändernde
Umweltbedingungen reagieren. Umweltreize werden dabei durch Chemotaxis-Proteine
durch die Zellhülle geleitet und wirken über weitere Proteine auf den Flagellenmotor und die
Richtung der Flagellenrotation (Sim et al. 2005). Anziehend auf T.d. wirken dabei Glukose,
Serum und Albumin, Stoffe, die auch in Zusammenhang mit Gewebezerstörung stehen, was
darauf hinweist, dass sich T.d. zu Orten hinbewegt, and denen bereits Gewebeschädigung
aufgetreten ist (Umemoto et al. 2001, Ruby et al. 2008). Die Möglichkeit der Chemotaxis
hängt natürlich mit einem intakten Bewegungsapparat zusammen. Interessant in Bezug auf
die Virulenz ist dabei die Beobachtung, dass sowohl Mutanten ohne Flagellen als auch
Mutanten mit Störungen in Chemotaxis-Signaltransduktionswegen Störungen bzw.
Versagen der Fähigkeit der extrazellulären Penetration von Geweben zeigen (Li H et al.
1996, Lux et al. 2002, Kataoka et al. 1997, Li et al. 1999). Internalisierung von Bakterien
durch Epithelzellen bieten den Mikroorganismen die Möglichkeit, vom Nahrungsvorrat zu
profitieren und gleichzeitig teilweise der Immunantwort und auch dem Angriff durch z.B.
pharmazeutische Antibiotika zu entgehen, weshalb dieser Aufenthaltsort ein Reservoir für
die Latenz der Organismen darstellt (Colombo et al. 2007, Johnson et al. 2008).

- 47 -
Transposasen
Neben Proteinen, die direkt oder indirekt als Virulenzfaktoren wirken, kann auch die
genetische Variabilität, mit deren Hilfe das Bakterium sich an die Umwelt anpassen kann,
als potentiell virulent eingestuft werden. Bei der Evolution der Spirochäten spielte
horizontaler Gentransfer mit anderen Organismen eine große Rolle was die Bereitschaft des
Organismus zur genetischen Adaption zeigt (Dashper et al. 2011). T.d. besitzt
beispielsweise 35 Gensequenzen mit Homologien zu Transposasen, Enzymen, welche in
der Lage sind, Genabschnitte aus dem Genom herauszuspalten und – an anderer Stelle –
wieder einzufügen. 25 dieser Gene sind heraufreguliert, wenn das Bakterium im Biofilm
eingebunden ist, was eine Vielzahl chromosomaler Rearrangements nahelegt. Auch
horizontaler Gentransfer durch Bakteriophagen (φtd1) findet statt, was zu einem Import von
virulenzassoziierten Genen führen mag. Beides stellen Mechanismen der genetischen
Diversifizierung dar, welche der Vereinfachung der Umweltanpassung sowie der
Genregulation dienlich sein können (Mitchell 2010).
Toxin/Antitoxinsysteme
Bakterielle Toxin-Antitoxinsysteme, von denen in T.d. 33 anhand von Sequenzanalysen
postuliert wurden, können an bakterieller Resistenz sowie der Persistenz von Biofilmen
beteiligt sein und damit als Virulenzfaktoren verstanden werden (Kim et al. 2009, Makarova
et al. 2009, Jayaraman 2008).

- 48 -
5.5 Polymikrobielle Synergie, Biofilmbildung und Therapieansätze
Die Virulenzfaktoren der Vertreter des ‚Roten Komplex’ wirken nicht nur alleine bzw. im
Kontext des Bakteriums, sondern auch synergistisch. Im oralen Biofilm kommt es unter den
Bakterien durch Zell-Zell Kontakt und lösliche Botenstoffe zu zahlreichen Interaktionen, z.B.
zum sogenannten ‚cross-feeding’, einem Prozess, in dem ein Bakterium die Abbauprodukte
des anderen verstoffwechselt, gemeinsam werden Aktivitäten wie Genexpression reguliert,
Nährsoffe aquiriert und DNA ausgetauscht. Andererseits stehen die Mikroben aber auch im
Wettstreit um Nährstoffe. Von polymikrobieller Synergie spricht man dann, wenn Bakterien
in der Anwesenheit bestimmter anderer Mikroben besser wachsen und erfolgreicher
Krankheitssymptome auslösen können (Kuboniwa und Lamont 2010, Hajishengallis und
Lamont 2012).
Auf der Zahnoberfläche binden Bakterien in charakteristischer Reihenfolge, was zu einer
aus verschiedenen Schichten bestehenden, komplexen Mikrobiota führt. Tiefer liegende
Schichten bieten dabei ideale Lebensbedingungen für Anaerobier, die hier expandieren.
Eine Spezies, die sich in diesem Biofilm ansiedeln möchte, muss nun an andere
Komponenten binden und unter den herrschenden Lebensbedingungen (Sauerstoffgehalt,
Nährstoffe) gedeihen können. Die Anheftung kann auf Wirtsoberflächen, aber auch durch
Auto- und Koaggregation vonstattengehen. Die Anordnung im Biofilm ermöglicht den
metabolischen Austausch und die Ausführung synergistischer Strategien und ist daher von
Vorteil für die Bakterien (Hansen et al. 2007).
Synergistische Effekte bei Mischinfektionen mit Organismen des roten Komplexes wurden
bereits beschrieben: P.g. und T.d. wachsen in einer Mischkultur deutlich besser als in
Monokultur, und durch die Anwesenheit des jeweils anderen Bakteriums werden bekannte
Virulenzfaktoren hochreguliert (Tan et al. 2014). In Tierversuchen wurde gezeigt, dass
während P.g. alleine keine Parodontitis-assoziierten Pathogenese-Phaenotypen
hervorrufen kann, sie es doch vermag, eine vorhandene Mikrobiota in die Dysbiose zu
führen (Kesavalu et al. 2007, Suzuki et al. 2013). In vitro zeigen die LPS von P.g., T.d. und
T.f. synergistische Effekte bei der Stimulation von Immunantworten (Bodet et al. 2010) Ein
interessantes weiteres Beispiel für die mikrobielle Synergie ist die gut untersuchte Bindung
von Streptococcus gordonii und P.g. Eine Interaktion die P.g. von einer nicht pathogenen in
eine pathogene Form transferieren kann (Kuboniwa und Lamont 2010).
P.g. bindet an frühe Besiedler des Biofilms und kann als eine Art Brücke fungieren, da es
auch an späte Besiedler bindet (Kolenbrander et al. 2002, Haffajee et al. 2008) Es ist

- 49 -
weiterhin bekannt, dass Vertreter des ‚Roten Komplexes’ auch mit anderen Spezies Biofilme
ausbilden, z.B. T.d. mit Fusobacterium nucleatum oder die oben beschriebene Interaktion
von P.g. mit Streptococcus gordonii (Kuboniwa und Lamont 2010, Honma et al. 2015).
Weitere Interaktionen sind bekannt. Die Untersuchung von Kooperation im Biofilm ist
teilweise aber auch noch deskriptiver Natur, und es fehlt die Identifikation von
zugrundeliegenden Mechanismen und Virulenzfaktoren (Suzuki et al. 2013).
Mehr noch als sich auf die biochemischen Eigenschaften einzelner im pathogenen
Bakterienverbund vorkommender Keime zu konzentrieren, sieht man heute im
Pathogeneseprozess das polymikrobiell synergistische Zusammenspiel der
Mikroorganismen im Verband als essentiell an. Immunsuppression, Beeinflussung
gegenseitiger Genexpression sowie der gemeinschaftlich koordinierte Abbau von
Makromolekülen und DNA- Austausch spielen hierbei eine Rolle. Dabei ist weniger das
Vorkommen spezifischer Pathogene entscheidend als vielmehr die Besetzung von
Schlüsselfunktionen des Pathogeneseprozesses durch hierfür geeignete Mikroorganismen
(Hajishengallis und Lamont 2012).
Das Modell des ‚roten Komplexes’, in dem dessen drei Vertreter als Hauptverursacher der
Parodontitis dargestellt werden, erwuchs aus der Tatsache, dass ihre Bedeutung aufgrund
ihres besseren Wachstums in der Zellkultur überschätzt wurde. Heute nutzt man
metagenomische Ansätze um die Beteiligung verschiedener Spezies am Plaque genauer
zu definieren, und es zeigte sich, dass auch Vertreter der Gattungen Anaeroglobus, Bulleidia,
Desulfobulbus, Filifactor, Mogibacterium, Phocaeicola, Schwartzia, TM7, Atopobium,
Eubacterium und Peptostreptococcus zu den ‚späten Besiedlern’ zu zählen sind. Die
Untersuchung der Einflüsse dieser Bakterien auf den Pathogeneseverlauf steht noch in
ihren Anfängen. Hajishengallis formulierte daher 2012 das allgemeiner gehaltene Modell
der ‚polymikrobiellen Synergie und Dysbiose’. Demmzufolge die Entwicklung
problematischer Verlaufsformen der Parodontitis aus der Dysbiose der parodontalen
Mikrobiota infolge der Zerstörung der Wirt-Mikroben Homöostase resultiert.
Die pathologischen Mechanismen, die der Parodontitis zugrunde liegen, werden im Verlauf
der Forschungen besser verstanden, und es eröffnen sich daraus neue therapeutische und
auch präventive Ansatzmöglichkeiten. Probiotika und antiinflammatorische Medikamente
werden beispielsweise verwendet, um die Mikrobiota in ihrer Zusammensetzung zu
beeinflussen (Takashiba et al. 1999, Anusha et al. 2015). Auch photodynamische Therapie
oder peptidbasierte Therapeutika kommen zum Einsatz (Moreira et al. 2015, Guevara et al.
2013). Polysaccharide können möglicherweise Biofilme zur Auflösung bringen, und auch
die Hemmung bestimmter Bestandteile des Komplementsystems (C3) hat sich im

- 50 -
Tiermodell bewährt (Renudueles et al. 2013, Maekawa et al. 2014).

- 51 -
6 Literaturverzeichnis
Abrahamson MM, Kikstrom J, Potempa S, Renvert A. 1997. Modification of cystatin C
activity by bacterial proteinases and neutrophil elastase in periodontitis. Mol Pathol, 50:291-
297.
Akira S, Takeda K. 2004. Toll-like receptor signalling. Nat Rev Immunol, 4:499–511.
Amano A. 2007. Disruption of epithelial barrier and impairment of cellular function by
Porphyromonas gingivalis. Front Biosci, 12:3965-74.
Amano A . 2010. Bacterial adhesins to host components in periodontitis. Periodontol 2000,
52(1):12-37.
Amano A. Host-parasite interactions in periodontitis: microbial pathogenicity and innate
immunity. 2010. Periodontol 2000, 54(1):9-14
Amano A, Nakagawa I, Okahashi N, Hamada N. 2004. Variations of Porphyromonas
gingivalis fimbriae in relation to microbial pathogenesis. J Periodontal Res, 39(2):136-42.
Amano A, Kuboniwa M, Nakagawa I, Akiyama S, Morisaki I, Hamada S. 2000. Prevalence
of specific genotypes of Porphyromonas gingivalis fimA and periodontal health status. J Dent
Res, 79(9):1664-8.
Amaya MP, Criado L, Blanco B, Gómez M, Torres O, Flórez L, González CI, Flórez O. 2013.
Polymorphisms of pro-inflammatory cytokine genes and the risk for acute suppurative or
chronic nonsuppurative apical periodontitis in a Colombian population. Int Endod J,
46(1):71-8.
Amin M, Grove DA, Kapus A, Glogauer M, Ellen RP. 2007. An actin-stabilizing peptide
conjugate deduced from the major outer sheath protein of the bacterium Treponema
denticola. Cell Motil Cytoskeleton, 64(9):662-74.
Anand A, Luthra A, Edmond ME, Ledoyt M, Caimano MJ, Radolf JD. 2013. The major outer
sheath protein (Msp) of Treponema denticola has a bipartite domain architecture and exists

- 52 -
as periplasmic and outer membrane-spanning conformers. J Bacteriol,195(9):2060-71.
Andrian E, Grenier D, Rouabhia M. 2004. In vitro models of tissue penetration and
destruction by Porphyromonas gingivalis. Infect Immun. 2004 Aug;72(8):4689-98.
Anusha RL, Umar D, Basheer B, Baroudi K. 2015. The magic of magic bugs in oral cavity:
Probiotics. J Adv Pharm Technol Res. 6(2):43-7
Arakawa S, Nakajima T, Ishikura H, Ichinose S, Ishikawa I, Tsuchida N. 2000 Novel
apoptosis-inducing activity in Bacteroides forsythus: a comparative study with three
serotypes of Actinobacillus actinomycetemcomitans. Infect Immun, 68(8):4611-5.
Aruni AW, Robles A, Fletcher HM. 2013. VimA mediates multiple functions that control
virulence in Porphyromonas gingivalis. Mol Oral Microbiol, 28(3):167-80.
Asai Y1, Ohyama Y, Gen K, Ogawa T. 2001. Bacterial fimbriae and their peptides activate
human gingival epithelial cells through Toll-like receptor 2. Infect Immun, 69(12):7387-95.
Batista da Silva AP, Lee W, Bajenova E, McCulloch CA, Ellen RP. 2004. The major outer
sheath protein of Treponema denticola inhibits the binding step of collagen phagocytosis in
fibroblasts. Cell Microbiol, 6(5):485-98.
Bamford CV, Fenno JC, Jenkinson HF, Dymock D. 2007. The chymotrypsin-like protease
complex of Treponema denticola ATCC 35405 mediates fibrinogen adherence and
degradation. Infect Immun, 75(9):4364-72.
Banbula A, Yen J, Oleksy A, Mak P, Bugno M, Travis J, Potempa J. 2001. Porphyromonas
gingivalis DPP-7 represents a novel type of dipeptidylpeptidase. J Biol Chem, 276(9):6299-
305.
Bartold PM, Van Dyke TE. 2013. Periodontitis: a host-mediated disruption of microbial
homeostasis. Unlearning learned concepts. Periodontol 2000, 62(1):203-17.
Beall CJ, Campbell AG, Dayeh DM, Griffen AL, Podar M, Leys EJ. 2014. Single cell
genomics of uncultured, health-associated Tannerella BU063 (Oral Taxon 286) and

- 53 -
comparison to the closely related pathogen Tannerella forsythia. PLoS One, 9(2):e89398.
Bélanger M, Rodrigues PH, Dunn WA Jr, Progulske-Fox A. 2006. Autophagy: a highway for
Porphyromonas gingivalis in endothelial cells. Autophagy, 2(3):165-70.
Beveridge, T. J., Pouwels, P. H., Sara, M. & 22 other authors. 1997. Functions of S-layers.
FEMS Microbiol Rev, 20:99–149.
Bhattacharya R, Xu F, Dong G, Li S, Tian C, Ponugoti B, Graves DT. 2014. Effect of bacteria
on the wound healing behavior of oral epithelial cells. PLoS One, 9(2):e89475.
Bian J, Liu X, Cheng YQ, Li C. 2013. Inactivation of cyclic Di-GMP binding protein TDE0214
affects the motility, biofilm formation, and virulence of Treponema denticola. J Bacteriol.
195(17):3897-905.
Bielecka E, Scavenius C, Kantyka T, Jusko M, Mizgalska D, Szmigielski B, Potempa B,
Enghild JJ, Prossnitz ER, Blom AM, Potempa J. 2014. Peptidyl arginine deiminase from
Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J Biol Chem,
289(47):32481-7.
Bird PS, Shakibaie F, Gemmell E, Polak B, Seymour GJ. 2001. Immune response to
Bacteroides forsythus in a murine model. Oral Microbiol Immunol,16(5):311-5.
Bodet C, Grenier D. 2010. Synergistic effects of lipopolysaccharides from periodontopathic
bacteria on pro-inflammatory cytokine production in an ex vivo whole blood model. Mol Oral
Microbiol, 25(2):102-11.
Brissette CA, Pham TT, Coats SR, Darveau RP, Lukehart SA. 2008. Treponema denticola
does not induce production of common innate immune mediators from primary gingival
epithelial cells. Oral Microbiol Immunol, 23(6):474-81.
Brunner J, Scheres N, El Idrissi NB, Deng DM, Laine ML, van Winkelhoff AJ, Crielaard W.
2010. The capsule of Porphyromonas gingivalis reduces the immune response of human
gingival fibroblasts. BMC Microbiol, 10:5.

- 54 -
Bryzek D, Ksiazek M, Bielecka E, Karim AY, Potempa B, Staniec D, Koziel J, Potempa J. A
pathogenic trace of Tannerella forsythia - shedding of soluble fully active tumor necrosis
factor α from the macrophage surface by karilysin. 2014. Mol Oral Microbiol, 29(6):294-306.
Burns E, Bachrach G, Shapira L, Nussbaum G. 2006.Cutting Edge: TLR2 is required for the
innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and
TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol, 177(12):8296-300.
Calkins CC, Platt K, Potempa J, Travis J. 1998. Inactivation of tumor necrosis factor a by
proteinases (gingipains) from the periodontal pathogen, Porphyromonas gingivalis.
Implications of immune evasion. J Biol Chem. 273:6611-6614.
Camelo-Castillo AJ, Mira A, Pico A, Nibali L, Henderson B, Donos N, Tomás I. 2015.
Subgingival microbiota in health compared to periodontitis and the influence of smoking.
Front Microbiol, 24(6):119.
Capestany CA, Tribble GD, Maeda K, Demuth DR, Lamont RJ. 2008. Role of the Clp system
in stress tolerance, biofilm formation, and intracellular invasion in Porphyromonas gingivalis.
J Bacteriol, 190(4):1436-46.
Capestany CA, Kuboniwa M, Jung IY, Park Y, Tribble GD, Lamont RJ 2006. Role of the
Porphyromonas gingivalis InlJ protein in homotypic and heterotypic biofilm development.
Infect Immun, 74(5):3002-5.
Capone R, Wang HT, Ning Y, Sweier DG, Lopatin DE, Fenno JC. 2008. Human serum
antibodies recognize Treponema denticola Msp and PrtP protease complex proteins. Oral
Microbiol Immunol, 23(2):165-9.
Carlsson J. 1997. Bacterial metabolism in dental biofilms. Adv Dent Res,11(1):75-80.
Chen T, Nakayama K, Belliveau L, Duncan MJ. 2001. Porphyromonas gingivalis gingipains
and adhesion to epithelial cells. Infect Immun, 69(5):3048-56.
Chen W, Kajiya M, Giro G, Ouhara K, Mackler HE, Mawardi H, Boisvert H, Duncan MJ, Sato
K, Kawai T. 2010. Bacteria-derived hydrogen sulfide promotes IL-8 production from epithelial

- 55 -
cells. Biochem Biophys Res Commun, 391(1):645-50.
Chi B, Qi M, Kuramitsu HK. 2003. Role of dentilisin in Treponema denticola epithelial cell
layer penetration. Res Microbiol, 154(9):637-43.
Chu L, Burgum A, Kolodrubetz D, Holt SC. 1995. The 46-kilodalton-hemolysin gene from
Treponema denticola encodes a novel hemolysin homologous to aminotransferases. Infect
Immun. 63(11):4448-55.
Chu L, Dong Z, Xu X, Cochran DL, Ebersole JL. 2002. Role of glutathione metabolism of
Treponema denticola in bacterial growth and virulence expression. Infect Immun,
70(3):1113-20.
Chu L, Lai Y, Xu X, Eddy S, Yang S, Song L, Kolodrubetz D. 2008. A 52-kDa leucyl
aminopeptidase from treponema denticola is a cysteinylglycinase that mediates the second
step of glutathione metabolism. J Biol Chem, 283(28):19351-8.
Clais S, Boulet G, Kerstens M, Horemans T, Teughels W, Quirynen M, Lanckacker E, De
Meester I, Lambeir AM, Delputte P, Maes L, Cos P. 2014. Importance of biofilm formation
and dipeptidyl peptidase IV for the pathogenicity of clinical Porphyromonas gingivalis
isolates. Pathog Dis, 70(3):408-13.
Coats SR, To TT, Jain S, Braham PH, Darveau RP. 2009. Porphyromonas gingivalis
resistance to polymyxin B is determined by the lipid A 4'-phosphatase, PGN_0524. Int J Oral
Sci, 1(3):126-35.
Colombo AV, da Silva CM, Haffajee A, Colombo AP. 2007.Dentification of intracellular oral
species within human crevicular epithelial cells from subjects with chronic periodontitis by
fluorescence in situ hybridization. J Periodontal Res, 42(3):236-43.
Colombo APV, Boches SK, Cotton SL, Goodson JM, Kent R, Haffajee AD, Socransky SS,
Hasturk H, Van Dyke TE, Paster BJ. 2009. Comparisons of subgingival microbial profiles of
refractory periodontitis, severe periodontitis and periodontal health using the human oral
microbe identification microarray (HOMIM). J Periodontol, 80(9):1421-1432.

- 56 -
Darveau RP, Tanner A, Page RC. 1997. The microbial challenge in periodontitis. Periodontol
2000.14:12-32.
Dashper SG, Seers CA, Tan KH, Reynolds EC. 2011. Virulence factors of the oral spirochete
Treponema denticola. J Dent Res, 90(6):691-703.
Dashper SG, Ang CS, Veith PD, Mitchell HL, Lo AW, Seers CA, Walsh KA, Slakeski N, Chen
D, Lissel JP, Butler CA, O'Brien-Simpson NM, Barr IG, Reynolds EC. 2009. Response of
Porphyromonas gingivalis to heme limitation in continuous culture. J Bacteriol, 191(3):1044-
55.
Davey M, Liu X, Ukai T, Jain V, Gudino C, Gibson FC 3rd, Golenbock D, Visintin A, Genco
CA. 2008. Bacterial fimbriae stimulate proinflammatory activation in the endothelium through
distinct TLRs. J Immunol, 180(4):2187-95.
Duncan L, Yoshioka M, Chandad F, Grenier D. 2004. Loss of lipopolysaccharide receptor
CD14 from the surface of human macrophage-like cells mediated by Porphyromonas
gingivalis outer membrane vesicles. Microb Pathog, 36(6):319-25.
Edwards AM1, Jenkinson HF, Woodward MJ, Dymock D. 2005. Binding properties and
adhesion-mediating regions of the major sheath protein of Treponema denticola ATCC
35405. Infect Immun, 73(5):2891-8.
Eskan MA, Hajishengallis G, Kinane DF. 2007. Differential activation of human gingival
epithelial cells and monocytes by Porphyromonas gingivalis fimbriae. Infect Immun,
75(2):892-8.
Evans RT, Klausen B, Sojar HT, Bedt GS, Sfintescu, Ramamurthy NS, Golub M, Genco RJ.
1992. Immunization with Porphyromonas (Bacteroides) gingivalis fimbriae protects against
periodontal destruction. Infect Immun, 60:2926-2935.
Fenno JC, McBride BC. 1998. Virulence factors of oral treponemes. Anaerobe, 4(1):1-17.
Fenno JC, Tamura M, Hannam PM, Wong GW, Chan RA, McBride BC. 2000. Identification
of a Treponema denticola OppA homologue that binds host proteins present in the
subgingival environment. Infect Immun, 68(4):1884-92.

- 57 -
Fenno JC, Wong GW, Hannam PM, Müller KH, Leung WK, McBride BC. 1997. Conservation
of msp, the gene encoding the major outer membrane protein of oral Treponema spp. J
Bacteriol, 79(4):1082-9.
Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, Nasri C, McKiernan M, Gunsolley
J. 2007. Aggregatibacter actinomycetemcomitans and its relationship to initiation of
localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents.
J Clin Microbiol. 45(12):3859-69
Fitzpatrick RE, Wijeyewickrema LC, Pike RN. 2009. The gingipains: scissors and glue of the
periodontal pathogen, Porphyromonas gingivalis. Future Microbiol, 4(4):471-87.
Fives-Taylor PM, Meyer DH, Mintz KP, Brissette C. 1999. Virulence factors of Actinobacillus
actinomycetemcomitans. Periodontol 2000. 20:136-67.
Fullmer SC, Preshaw PM, Heasman PA, Kumar PS. 2009. Smoking cessation alters subgingival microbial recolonization. J Dent Res, 88(6):524.
Furuta N, Takeuchi H, Amano A. 2009. Entry of Porphyromonas gingivalis outer membrane
vesicles into epithelial cells causes cellular functional impairment. Infect Immun,
77(11):4761-70.
Gängler P, Hoffmann T, Willershausen B, Schwenzer N, Ehrenfeld M, Hrsg. 2005.
Konservierende Zahnheilkunde und Parodontologie, Zweite Aufl. Stuttgart, New York;
Thieme Verlag, S.263-264.
Glockmann, E., Panzner, K.-D., Huhn, P., Sigusch, B. W., Glockmann, K. 2007.
Ursachen des Zahnverlustes in Deutschland. – Dokumentation einer bundesweiten
Erhebung IDZ-Information Nr. 2/11.
Golub LM, Payne JB, Reinhardt RA, Nieman G. 2006. Can systemic diseases co-induce
(not just exacerbate) periodontitis? A hypothetical "two-hit" model. J Dent Res, (2):102-5.
Goulas T, Mizgalska D, Garcia-Ferrer I, Kantyka T, Guevara T, Szmigielski B, Sroka A, Millán
C, Usón I, Veillard F, Potempa B, Mydel P, Solà M, Potempa J, Gomis-Rüth FX. 2015.
Structure and mechanism of a bacterial host-protein citrullinating virulence factor,

- 58 -
Porphyromonas gingivalis peptidylarginine deiminase. Sci Rep, 5:11969.
Goulbourne PA1, Ellen RP. 1991. Evidence that Porphyromonas (Bacteroides) gingivalis
fimbriae function in adhesion to Actinomyces viscosus. J Bacteriol, 173(17):5266-74.
Grenier D. 1995. Characterization of the trypsin-like activity of Bacteroides forsythus.
Microbiology, 141:921–926.
Grenier D. 2013. Binding properties of Treponema denticola lipooligosaccharide. J Oral
Microbiol, 16:5.
Grenier D, Mayrand D. 1987. Functional characterization of extracellular vesicles produced
by Bacteroides gingivalis. Infect Immun, 55(1):111-7.
Guevara T, Ksiazek M, Skottrup PD, Cerdà-Costa N, Trillo-Muyo S, de Diego I, Riise E,
Potempa J, Gomis-Rüth FX. 2013. Structure of the catalytic domain of the Tannerella
forsythia matrix metallopeptidase karilysin in complex with a tetrapeptidic inhibitor. Acta
Crystallogr Sect F Struct Biol Cryst Commun. 69(Pt 5):472-6.
Gumbiner BM. 1996. Cell adhesion: the molecular basis of tissue architecture and
morphogenesis. Cell, 84(3):345-57.
Haffajee AD, Socransky SS, Patel MR, Song X. 2008. Microbial complexes in supragingival
plaque. Oral Microbiol Immunol. 23(3):196-205.
Haffajee AD, Bogren A, Hasturk H, Feres M, Lope, NJ, Socransky SS. 2004. Subgingival
microbiota of chronic periodontitis subjects from different geographic locations. J Clin
Periodontol, 11:996–1002.
Haffajee AD, Cugini MA, Tanner A, Pollack RP, Smith C, Kent RL Jr, Socransky SS. 1998.
Subgingival microbiota in healthy, well-mainted elder and periodontitis subjects. J Clin
Periodontol, 25(5):346-53.
Hajishengallis G 2014. Immunomicrobial pathogenesis of periodontitis: keystones,
pathobionts, and host response. Trends Immunol, ;35(1):3-11.

- 59 -
Hajishengallis G, Harokopakis E. 2007. Porphyromonas gingivalis interactions with
complement receptor 3 (CR3): innate immunity or immune evasion? Front Biosci,12:4547-
57.
Hajishengallis G, Lambris JD. 2012. Complement and dysbiosis in periodontal disease.
Immunobiology, 217(11):1111-6.
Hajishengallis G, Lamont RJ 2012. Beyond the red complex and into more complexity: the
polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral
Microbiol, 27(6):409-19.
Hajishengallis G, Chavakis T, Hajishengallis E, Lambris JD. 2014. Neutrophil homeostasis
and inflammation: novel paradigms from studying periodontitis. J Leukoc Biol, pii:
jlb.3VMR1014-468R.
Hajishengallis G, Wang M, Bagby GJ, Nelson S. 2008. Importance of TLR2 in early innate
immune response to acute pulmonary infection with Porphyromonasgingivalis in mice. J
Immunol, 181(6):4141-9.
Hajishengallis G, Martin M, Sojar HT, Sharma A, Schifferle RE, DeNardin E, Russell MW,
Genco RJ. 2002. Dependence of bacterial protein adhesins on toll-like receptors for
proinflammatory cytokine induction. Clin. Diagn. Lab. Immunol, 9:403–401.
Hajishengallis G, Tapping RI, Harokopakis E, Nishiyama S, Ratti P, Schifferle RE, Lyle EA,
Triantafilou M, Triantafilou K, Yoshimura F. 2006. Differential interactions of fimbriae and
lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred
pattern recognition apparatus. Cell Microbiol, 8(10):1557-70.
Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam
A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species
orchestrates inflammatory periodontal disease through the commensal microbiota and
complement. Cell Host Microbe, 10(5):497-506.
Hamada N, Watanabe K, Arai M, Hiramine H, Umemoto T. 2002. Cytokine production
induced by a 67-kDa fimbrial protein from Porphyromonas gingivalis. Oral Microbiol Immunol,

- 60 -
17(3):197-200.
Han YW, Shi W, Huang GT, Kinder Haake S, Park NH, Kuramitsu H. 2000. Interactions
between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum
adheres to and invades epithelial cells. Genco RJ Infect Immun, 68(6):3140-6.
Hansen SK, Rainey PB, Haagensen JA, Molin S. 2007. Evolution of species interactions in
a biofilm community. Nature. 445(7127):533-6.
Haraguchi A, Miura M, Fujise O, Hamachi T, Nishimura F. 2014. Porphyromonas gingivalis
gingipain is involved in the detachment and aggregation of Aggregatibacter
actinomycetemcomitans biofilm. Mol Oral Microbiol, 29(3):131-43.
Harokopakis E, Albzreh MH, Martin MH, Hajishengallis G. 2006. TLR2 transmodulates
monocyte adhesion and transmigration via Rac1- and PI3K-mediated inside-out signaling in
response to Porphyromonas gingivalis fimbriae. J Immunol, 176(12):7645-56.
Hasebe A, Yoshimura A, Into T, Kataoka H,Tanaka S,Arakawa S, Ishikura H,
Golenbock DT, Sugaya T, Tsuchida N, Kawanami M, Hara Y, Shibata K . 2004.
Biological Activities of Bacteroides forsythus Lipoproteins and Their Possible Pathological
Roles in Periodontal Disease. Infect Immun, 72(3):1318–1325.
Hashimoto M, Ogawa S, Asai Y, Takai Y, Ogawa T 2003. Binding of Porphyromonas
gingivalis fimbriae to Treponema denticola dentilisin. FEMS Microbiol Lett, 226(2):267-71.
Hasturk H, Kantarci A, Van Dyke TE. 2012. Oral inflammatory diseases and systemic
inflammation: role oft he macrophage. Front Immunol, 16;3:118.
Haunfelder D. 1990. Praxis der Zahnheilkunde Bd.4 Parodontologie 2. Aufl. Urban u.
Schwarzenberg. München-Wien-Baltimore.
Hespell RB, Canale-Parola E. 1971. Amino acid and glucose fermentation by Treponema
denticola. Arch Mikrobiol, 78(3):234-51.
Hiramine H, Watanabe K, Hamada N, Umemoto T. 2003. Porphyromonas gingivalis 67-kDa

- 61 -
fimbriae induced cytokine production and osteoclast differentiation utilizing TLR2. FEMS
Microbiol Lett., 229(1):49-55.
Holden JA, Attard TJ, Laughton KM, Mansell A, O'Brien-Simpson NM, Reynolds EC. 2014.
Porphyromonas gingivalis lipopolysaccharide weakly activates M1 and M2 polarized mouse
macrophages but induces inflammatory cytokines. Infect Immun, 82(10):4190-203.
Holt, SC and Ebersole JL. 2005. Porphyromonas gingivalis, Treponema denticola, and
Tannerella forsythia: the "red complex", a prototype polybacterial pathogenic consortium in
periodontitis. Periodontol 2000, 38:72-122.
Honma, K., Kuramitsu, H. K., Genco, R. J. & Sharma, A. 2001. Development of a gene
inactivation system for Bacteroides forsythus: construction and characterization of a BspA
mutant. Infect Immun, 69:4686–4690.
Honma K, Ruscitto A, Frey AM, Stafford GP, Sharma A. 2015. Sialic acid transporter NanT
participates in Tannerella forsythia biofilm formation and survival on epithelial cells.Microb
Pathog. pii: S0882-4010(15)00143-6.
Hughes CV, Malki G, Loo CY, Tanner AC, Ganeshkumar N. 2003. Cloning and expression
of alpha-D-glucosidase and N-acetyl-beta-glucosaminidase from the periodontal pathogen,
Tannerella forsythensis (Bacteroides forsythus). Oral Microbiol Immunol,18(5):309-12.
Hussain M, Stover CM, Dupont A. 2015. P. gingivalis in Periodontal Disease and
Atherosclerosis - Scenes of Action for Antimicrobial Peptides andComplement. Front
Immunol, 6:45.
Imamura T, Travis J, Potempa J 2003. The biphasic virulence activities of gingipains:
activation and inactivation of host proteins. Curr Protein Pept Sci, 4(6):443-50.
Inagaki S, Onishi S, Kuramitsu HK, Sharma A. 2006. Porphyromonas gingivalis vesicles
enhance attachment, and the leucine-rich repeat BspA protein is required for invasion of
epithelial cells by "Tannerella forsythia". Infect Immunol, 74(9):5023-8.
Irshad M, van der Reijden WA, Crielaard W, Laine ML. 2012. In vitro invasion and survival

- 62 -
of Porphyromonas gingivalis in gingival fibroblasts; role of the capsule. Arch Immunol Ther
Exp (Warsz), 60(6):469-76.
Ishikura H, Arakawa S, Nakajima T, Tsuchida N, Ishikawa I. 2003. Cloning of the Tannerella
forsythensis (Bacteroides forsythus) siaHI gene and purification of the sialidase enzyme. J
Med Microbiol, 52(Pt 12):1101-7.
Jagels MA, Travis J, Potempa J, Pike R, Hugli TE. 1996. Proteolytic inactivation of the
leukozyte C5a receptor by proteinases derived from Porphyromonas gingivalis. Infect
Immun, 64:1984-1991.
Jayaraman R. 2008. Bacterial persistence: some new insights into an old phenomenon.
J. Biosci, 33(5):795-805.
Jo AR, Baek KJ, Shin JE, Choi Y. 2014. Mechanisms of IL-8 suppression by Treponema
denticola in gingival epithelial cells. Immunol Cell Biol, 92(2):139-47.
Jobin MC, Virdee I, McCulloch CA, Ellen RP. 2007. Activation of MAPK in fibroblasts by
Treponema denticola major outer sheath protein. Biochem Biophys Res Commun,
356(1):213-8.
Johnson JD, Chen R, Lenton PA, Zhang G, Hinrichs JE, Rudney JD. 2008. Persistence of
extracrevicular bacterial reservoirs after treatment of aggressive periodontitis. J Periodontol.
79(12):2305-12.
Jun HK, Lee SH, Lee HR, Choi BK. 2012. Integrin α5β1 activates the NLRP3 inflammasome
by direct interaction with a bacterial surface protein. Immunity, 36(5):755-68.
Jusko M, Potempa J, Karim AY, Ksiazek M, Riesbeck K, Garred P, Eick S, Blom AM. 2012.
A metalloproteinase karilysin present in the majority of Tannerella forsythia isolates inhibits
all pathways of the complement system. J Immunol, 188(5):2338-49.
Kadowaki T1, Nakayama K, Okamoto K, Abe N, Baba A, Shi Y, Ratnayake DB, Yamamoto
K.2000.Porphyromonasgingivalis proteinases as virulence determinants in progression of
periodontal diseases. J Biochem, 128(2):153-9.

- 63 -
Kamaguchi A, Nakayama K, Ichiyama S, Nakamura R, Watanabe T, Ohta M, Baba H,
Ohyama T. 2003. Effect of Porphyromonas gingivalis vesicles on coaggregation of
Staphylococcus aureus to oral microorganisms. Curr Microbiol, 47(6):485-91.
Karim AY, Kulczycka M, Kantyka T, Dubin G, Jabaiah A, Daugherty PS, Thogersen IB,
Enghild JJ, Nguyen KA, Potempa J. 2010. A novel matrix metalloprotease-like
enzyme(karilysin) of the periodontal pathogen Tannerella forsythia ATCC 43037. Biol
Chem,391(1):105-17.
Kassem A, Henning P, Lundberg P, Souza PP, Lindholm C, Lerner UH. 2015.
Porphyromonas gingivalis Stimulates Bone Resorption by Enhancing RANKL (Receptor
Activator of NF-κB Ligand) through Activation of Toll-like Receptor 2 in Osteoblasts. J Biol
Chem. 290(33):20147-58.
Kataoka M, Li H, Arakawa S, Kuramitsu H. 1997. Characterization of a methyl-accepting
chemotaxis protein gene, dmcA, from the oral spirochete Treponemadenticola. Infect Immun,
65(10):4011-6.
Kato T, Kawai S, Nakano K, Inaba H, Kuboniwa M, Nakagawa I, Tsuda K, Omori H, Ooshima
T, Yoshimori T, Amano A. 2007. Virulence of Porphyromonas gingivalis is altered by
substitution of fimbria gene with different genotype. Cell Microbiol, 9(3):753-65.
Kerusuo E. 1988. Ultrastructure oft he cell envelope of Bacteroides forsythus strain ATCC
43037T. Oral Microbiol Immunol, 3(3):134-7.
Kesavalu L, Sathishkumar S, Bakthavatchalu V, Matthews C, Dawson D, Steffen M,
Ebersole JL . 2007. Rat model of polymicrobial infection, immunity, and alveolar bone
resorption in periodontal disease. Infect Immun, 75(4):1704-12.
Khocht A, Albandar JM. 2014. Aggressive forms of periodontitis secondary to systemic
disorders. Periodontol 2000, 65(1):134-48.
Kinane JA, Benakanakere MR, Zhao J, Hosur KB, Kinane DF. 2012. Porphyromonas
gingivalis influences actin degradation within epithelial cells during invasion and apoptosis.
Cell Microbiol, 14(7):1085-96.

- 64 -
Kim M, Jun HK, Choi BK, Cha JH, Yoo YJ. 2012. Td92, an outer membrane protein of
Treponema denticola, induces osteoclastogenesis via prostaglandin E(2)-mediated
RANKL/osteoprotegerin regulation. J Periodontal Res, 45(6):772-9.
Kim TS, Kang NW, Lee SB, Eickholz P, Pretzl B, Kim CK. 2009. Differences in subgingival
microflora of Korean and German periodontal patients. Arch Oral Biol, 54(3):223-9.
Kolenbrander P, Andersen RA, Blehert DS, Egland PG, Foster JS, Palmer RJ. 2002.
Communication among Oral Bacteria.Microbiol Mol Biol Rev. 66(3): 486–505.
Koziel J, Karim AY, Przybyszewska K, Ksiazek M, Rapala-Kozik M, Nguyen KA, Potempa
J. 2010. Proteolytic inactivation of LL-37 by karilysin, a novel virulence mechanism of
Tannerella forsythia. J Innate Immun, 2(3):288-93.
Krauss JL, Potempa J, Lambris JD, Hajishengallis G. 2010. Complementary Tolls in the
periodontium: how periodontal bacteria modify complement and Toll-like receptor responses
to prevail in the host. Periodontol 2000, 52(1):141-62.
Ksiazek M, Mizgalska D, Enghild JJ, Scavenius C, Thogersen IB, Potempa J. 2015. Miropin,
a novel bacterial serpin from the periodontopathogen Tannerella forsythia, inhibits a broad
range of proteases by using different peptide bonds within the reactive center loop. J Biol
Chem, 2;290(1):658-70.
Kuboniwa M, Lamont RJ. 2010. Subgingival biofilm formation. Periodontol 2000. 52(1):38-
52.
Kuboniwa M, Tribble GD, Hendrickson EL, Amano A, Lamont RJ, Hackett M. 2010. Insights
into the virulence of oral biofilms: discoveries from proteomics. Expert Rev Proteomics,
9(3):311-23.
Kuboniwa M, Amano A, Hashino E, Yamamoto Y, Inaba H, Hamada N, Nakayama K, Tribble
GD, Lamont RJ, Shizukuishi S. 2009. Distinct roles of long/short fimbriae and gingipains in
homotypic biofilm development by Porphyromonasgingivalis. BMC Microbiol, 9:105.

- 65 -
Kuramitsu HK, He X, Lux R, Anderson MH, Shi W. 2007. Interspecies interactions within oral
microbial communities. Microbiol Mol Biol Rev,71(4):653-70.
Kurniyati K, Zhang W, Zhang K, Li C. 2013. A surface-exposed neuraminidase affects
complement resistance and virulence of the oral spirochaete Treponema denticola. Mol
Microbiol, 89(5):842-56.
Kuboniwa M, Lamont RJ. 2010. Subgingival biofilm formation. Periodontol 2000, 52(1):
38–52.
Kuehn MJ, Kesty NC. 2005. Bacterial outer membrane vesicles and the host-pathogen
interaction. Genes Dev,19(22):2645-55.
Laheij AM, de Soet JJ, Veerman EC, Bolscher JG, van Loveren C. 2013. The influence of
oral bacteria on epithelial cell migration in vitro. Mediators Inflamm. 154532.
Laine ML, van Winkelhoff AJ. 1998. Virulence of six capsular serotypes
of Porphyromonas gingivalis in a mouse model. Oral Microbiol Immunol, 13(5):322-5.
Lamont RJ, Jenkinson HF. 1998. Life below the gum line: pathogenic mechanisms of
Porphyromonas gingivalis. Microbiol Mol Biol Rev. 62(4):1244-63.
Lamont RJ, Jenkinson HF. 2000. Subgingival colonization by Porphyromonas gingivalis.
Oral Microbiol Immunol, 15(6):341-9.
Lantz MS, Allen RD, Duck LW, Switalski LM, Hook M. 1991. Porphyromonas gingivalis
surface components bind and degrade connective tissue proteins. Periodont Res, 26:283-
285.
Lee SY, Fenno JC. 2004. Expression of Treponema denticola oligopeptidase B in
Escherichia coli. Curr Microbiol, 48(5):379-82.
Lee SH, Kim KK, Choi BK. 2005. Upregulation of intercellular adhesion molecule 1 and
proinflammatory cytokines by the major surface proteins of Treponema maltophilum and
Treponema lecithinolyticum, the phylogenetic group IV oral spirochetes associated with
periodontitis and endodontic infections. nfect Immun, 73(1):268-76.

- 66 -
Lee SW, Sabet M, Um HS, Yang J, Kim HC, Zhu W. 2006. Identification and characterization
of the genes encoding a unique surface (S-) layer of Tannerella forsythia.
Gene, 371(1):102-11.
Lépine G, Progulske-Fox A. 1996. Duplication and differential expression of hemagglutinin
genes in Porphyromonas gingivalis. Oral Microbiol Immunol, 11(2):65-78.
Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, Li F, Tzekou A, Lambris JD,
Hajishengallis G. 2011. The C5a receptor impairs IL-12-dependent clearance of
Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol,
186(2):869-77.
Li H, Kuramitsu HK. 1996. Development of a gene transfer system in Treponema denticola
by electroporation. Oral Microbiol Immunol, 11(3):161-5.
Li H, Arakawa S, Deng QD, Kuramitsu H. 1996. Characterization of a novel methyl-accepting
chemotaxis gene, dmcB, from the oral spirochete Treponemadenticola. Infect Immun,
67(2):694-9.
Li Z, Clarke AJ, Beveridge TJ. 1996. A major autolysin of Pseudomonas aeruginosa:
subcellular distribution, potential role in cell growth and division and secretion in surface
membrane vesicles. J Bacteriol, 178(9):2479-88.
Lin FY, Hsiao FP, Huang CY, Shih CM, Tsao NW, Tsai CS, Yang SF, Chang NC, Hung SL,
Lin YW. 2014. Porphyromonas gingivalis GroEL induces osteoclastogenesis of periodontal
ligament cells and enhances alveolar bone resorption in rats. PLoS One. 9(7):e102450.
Loesche, WJ, Bretz WA, Kerschensteiner D, Stoll J, Socransky SS, Hujoel P, Lopatin DE.
1990. Development of a diagnostic test for anaerobic periodontal infections based on plaque
hydrolysis of benzoyl-dl-arginine-naphthylamide. J Clin Microbiol, 28:1551–1559.
Lux R, Sim JH, Tsai JP, Shi W. 2002. Construction and characterization of a cheA mutant of
Treponema denticola. J Bacteriol, 184(11):3130-4.
Maeda K, Nagata H, Kuboniwa M, Kataoka K, Nishida N, Tanaka M, Shizukuishi S. 2004.

- 67 -
Characterization of binding of Streptococcus oralis glyceraldehyde-3-phosphate
dehydrogenase to Porphyromonas gingivalis major fimbriae. Infect Immun, 72(9):5475-7.
Maekawa T, Hajishengallis G. 2014. Topical treatment with probiotic Lactobacillus brevis
CD2 inhibits experimental periodontal inflammation and bone loss. J Periodontal Res.
49(6):785-91.
Magalhães MA, Sun CX, Glogauer M, Ellen RP. 2008.The major outer sheath protein of
Treponema denticola selectively inhibits Rac1 activation in murine neutrophils.Cell
Microbiol,10(2):344-54.
Maiden MF, Cohee P, Tanner AC. 2003. Proposal to conserve the adjectival form of the
specific epithet in the reclassification of Bacteroides forsythus Tanner et al. 1986 to the
genus Tannerella Sakamoto et al. 2002 as Tannerella forsythia corrig., gen. nov., comb. nov.
Request for an Opinion. Int J Syst Evol Microbiol,53(Pt 6):2111-2.
Makarova KS, Wolf YI, Koonin EV. 2009. Comprehensive comparative-genomic analysis of
type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes.
Biol Direct, 4:19.
Malek R, Fisher JG, Caleca A, Stinson M, van Oss CJ, Lee JY, Cho MI, Genco RJ, Evans
RT, Dyer DW. 1994. Inactivation of the Porphyromonas gingivalis fimA gene blocks
periodontal damage in gnotobiotic rats. J Bacteriol, 176(4):1052-9.
Markiewski MM, Lambris JD. 2007. The role of complement in inflammatory diseases from
behind the scenes into the spotlight. Am J Pathol, 171(3):715-27.
Mashburn LM, Whiteley M. 2005. Membrane vesicles traffic signals and facilitate group
activities in a prokaryote. Nature, 437(7057):422-5.
Mayrand D, Holt SC. 1988. Biology of asaccharolytic black-pigmented Bacteroides species.
Microbiol Rev, 52(1):134-52.
McDowell JV, Frederick J, Stamm L, Marconi RT. 2007. Identification of the gene encoding
the FhbB protein of Treponema denticola, a highly unique factor H-like protein 1 binding

- 68 -
protein. Infect Immun, 75(2):1050-4.
McDowell JV, Huang B, Fenno JC, Marconi RT, Reynolds EC. 2009. Analysis of a unique
interaction between the complement regulatory protein factor H and the periodontal
pathogen Treponema denticola. Infect Immun, 77(4):1417-25.
Medzhitov R 2007. TLR-mediated innate immune recognition. Semin Immunol, 19(1):1-2.
Megson ZA, Koerdt A, Schuster H, Ludwig R, Janesch B, Frey A, Naylor K, Wilson I,
Stafford GP, Messner P, Schäffer C. Characterization of an α-l-fucosidase from the
periodontal pathogen Tannerella forsythia. Virulence, 6(3):282-92.
Meyle J, Chapple I 2015. Molecular aspects of the pathogenesis of periodontitis. Periodontol
2000, 69(1):7-17.
Mishima E, Sharma A.2011. Tannerella forsythia invasion in oral epithelial cells requires
phosphoinositide 3-kinase activation and clathrin-mediated endocytosis. Microbiology,
157(Pt 8):2382-91.
Mitchell HL, Dashper SG, Catmull DV, Paolini RA, Cleal SM, Slakeski N, Tan KH, Reynolds
EC. 2010. Treponema denticola biofilm-induced expression of a bacteriophage, toxin-
antitoxin systems and transposases. Microbiology, 156(Pt 3):774-88.
Miyamoto M, Ishihara K, Okuda K. 2006. The Treponema denticola surface protease
dentilisin degrades interleukin-1 beta (IL-1 beta), IL-6, and tumor necrosis factor alpha.
Infect Immun, 74(4):2462-7.
Moncla BJ, Braham P, Hillier S L. 1990. Sialidase (neuraminidase) activity among gram-
negative anaerobic and capnophilic bacteria. J Clin Microbiol, 28:422–425.
Moreira MS, de FreitasArchilla JR, Lascala CA, Ramalho KM, Gutknecht N, Marques MM.
2015. Post-Treatment Apical Periodontitis Successfully Treated with Antimicrobial
Photodynamic Therapy Via Sinus Tract and Laser Phototherapy: Report of Two
Cases.Photomed Laser Surg.
Murakami Y, Higuchi N, Nakamura H, Yoshimura F, Oppenheim FG. 2002. Bacteroides

- 69 -
forsythus hemagglutinin is inhibited by N-acetylneuraminyllactose. Oral Microbiol Immunol,
17(2):125-8.
Myneni SR, Settem RP, Connell TD, Keegan AD, Gaffen SL, Sharma A. 2011.TLR2
signaling and Th2 responses drive Tannerella forsythia-induced periodontal bone loss. J.
Immunol, 187:501–509.
Mysak J, Podzimek S, Sommerova P, Lyuya-Mi Y, Bartova J, Janatova T, Prochazkova J,
Duskova J. 2014. Porphyromonas gingivalis: major periodontopathic pathogen overview. J
Immunol Res, 476068.
Nagano K, Hasegawa Y, Murakami Y, Nishiyama S, Yoshimura F. 2010. FimB regulates
FimA fimbriation in Porphyromonas gingivalis. J Dent Res, 89(9):903-8.
Nakamura K, Yano H, Uchida H, Hashimoto S, Schaefer E, Sabe H. 2000. Tyrosine
phosphorylation of paxillin alpha is involved in temporospatial regulation of paxillin-
containing focal adhesion formation and F-actin organization in motile cells. J Biol Chem,
275(35):27155-64.
Nakao R, Takashiba S, Kosono S, Yoshida M, Watanabe H, Ohnishi M, Senpuku H. 2014.
Effect of Porphyromonas gingivalis outer membrane vesicles on gingipain-mediated
detachment of cultured oral epithelial cells and immune responses. Microbes Infect,16(1):6-
16.
Nakayama K. 2015, Porphyromonas gingivalis and related bacteria: from colonial
pigmentation to the type IX secretion system and gliding motility. J Periodontal Res, 50(1):1-
8.
Nishiyama S, Murakami Y, Nagata H, Shizukuishi S, Kawagishi I, Yoshimura F. 2007.
Involvement of minor components associated with the FimA fimbriae of Porphyromonas
gingivalis in adhesive functions. Microbiology, 153(Pt 6):1916-25.
Niwa D, Nishikawa K, Nakamura H. 2011. A hybrid two-component system of Tannerella
forsythia affects autoaggregation and post-translational modification of surface proteins.
FEMS Microbiol Lett, 318(2):189-96.

- 70 -
Nussbaum G, Ben-Adi S, Genzler T, Sela M, Rosen G. 2009. Involvement of Toll-like
receptors 2 and 4 in the innate immune response to Treponema denticola and its outer
sheath components. Infect Immun, 77(9):3939-47.
Offenbacher S, Barros SP, Beck JD. 2008. Rethinking periodontal inflammation.
J. Periodontol, 79(8 Suppl):1577-84.
Ogawa T, Hamada S. 1994. Hemagglutinating and chemotactic properties of synthetic
peptide segments of fimbrial protein from Porphyromonas gingivalis. Infect. Immun, 62:
3305–3310.
Ogawa T, Asai Y, Hashimoto M, Uchida H. 2002. Bacterial fimbriae activate human
peripheral blood monocytes utilizing TLR2, CD14 and CD11a/CD18 as cellular receptors.
Eur J Immunol, 32(9):2543-50.
Ohara-Nemoto Y, Shimoyama Y, Kimura S, Kon A, Haraga H, Ono T, Nemoto TK. 2011.
Asp- and Glu-specific novel dipeptidyl peptidase 11 of Porphyromonas gingivalis ensures
utilization of proteinaceous energy sources. J. Biol. Chem. 286, 38115-38127.
Okuda T, Kimizuka R, Miyamoto M, Kato T, Yamada S, Okuda K, Ishihara K. 2007.
Treponema denticola induces interleukin-8 and macrophage chemoattractant protein 1
production in human umbilical vein epithelial cells. Microbes Infect, 9(7):907-13.
Olsen I. 2015. From the acta prize lecture 2014: the periodontal-systemic connection seen
from a microbiological standpoint. Acta Odontol Scand, 73(8):563-8.
Onishi S, Honma K, Liang S, Stathopoulou P, Kinane D, Hajishengallis G, Sharma A. 2008.
Toll-like receptor 2-mediated interleukin-8 expression in gingival epithelial cells by the
Tannerella forsythia leucine-rich repeat protein BspA. Infect. Immun, 76:198–205.
Otomo-Corgel J, Pucher JJ, Rethman MP, Reynolds MA. 2012. State of the science: chronic
periodontitis and systemic health. J Evid Based Dent Pract, 12(3 Suppl):20-8.
Page RC. 1991. The role of inflammatory mediators in the pathogenesis of periodontal
disease. J Periodontol Res.

- 71 -
Palm E, Khalaf H, Bengtsson T. 2015. Suppression of inflammatory responses of human
gingival fibroblasts by gingipains from Porphyromonasgingivalis. Mol Oral Microbiol,
30(1):74-85.
Park Y, Simionato MR, Sekiya K, Murakami Y, James D, Chen W, Hackett M, Yoshimura F,
Demuth DR, Lamont RJ. 2005. Short fimbriae of Porphyromonas gingivalis and their role in
coadhesion with Streptococcus gordonii. Infect Immun, 73(7):3983-9.
Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanuos VA, Sahasrabudhe A,
Dewihirst FE. 2001. Bacterial diversity in human subgingival plaque. J Bacteriol, 183:3770-
3783.
Pathirana RD, O'Brien-Simpson NM, Brammar GC, Slakeski N, Reynolds EC. 2007. Kgp
and RgpB, but not RgpA, are important for Porphyromonas gingivalis virulence in the murine
periodontitis model. Infect Immun, 75(3):1436-42
Pei J, Grishin NV. 2009. Prediction of a caspase-like fold in Tannerella forsythia virulence
factor PrtH. Cell Cycle, 8(9):1453-5.
Pham K, Feik D, Hammond BF, Rams TE, Whitaker EJ. 2002. Aggregation of human
platelets by gingipain-R from Porphyromonas gingivalis cells and membrane vesicles.
Platelets, 13(1):21-30.
Pierce DL, Nishiyama S, Liang S, Wang M, Triantafilou M, Triantafilou K, Yoshimura F,
Demuth DR, Hajishengallis G. 2009. Host adhesive activities and virulence of novel fimbrial
proteins of Porphyromonas gingivalis. Infect Immun, 77(8):3294-301.
Pike RN, Potempa J, McGraw W, Coetzer THT, Travis J. 1996. Characterization of the
binding activities of proteinase-adhesin complexes from Porphyromonas gingivalis.
J. Bacteriol, 178:2876-2882
Pöllänen MT, Paino A, Ihalin R. 2013. Environmental stimuli shape biofilm formation and the
virulence of periodontal pathogens. cInt J Mol Sci, 14(8):17221-37.

- 72 -
Popadiak K, Potempa J, Riesbeck K, Blom AM. 2007. Biphasic effect of gingipains from
Porphyromonas gingivalis on the human complement system. J Immunol, 178(11):7242-50.
Posch G, Pabst M, Brecker L, Altmann F, Messner P, Schäffer C. 2011. Characterization and
scope of S-layer protein O-glycosylation in Tannerella forsythia. J Biol Chem,
286(44):38714-24.
Posch G, Sekot G,† Friedrich V, Megson ZA, Koerdt A, Messner P, Schäffer C. 2012.
Glycobiology Aspects of the Periodontal Pathogen Tannerella forsythia. Biomolecules,
2(4): 467–482.
Potempa J, Banbula A, Travis J. 2000. Role of bacterial proteinases in matrix destruction
and modulation of host responses. Periodontol 2000, 24:153-92.
Puthengady TB, Sun CX, Bajenova E, Ellen RP, Glogauer M. 2006. Modulation of human
neutrophil functions in vitro by Treponema denticola major outer sheath protein. Infect
Immun, 74(3):1954-7.
Ruddy MJ, Shen F, Smith JB, Sharma A, Gaffen SL. 2004. Interleukin-17 regulates
expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation
and neutrophil recruitment. J. Leukoc. Biol, 76:135-144.
Puschmann Ch. 2003. Untersuchungen zur Wirksamkeit verschiedener Antibiotika auf
Porphyromonas gingivalis in oralen Epithelzellen [Dissertation]. Jena: Friedrich-Schiller-
Universität.
Raby AC, Holst B, Davies J, Colmont C, Laumonnier Y, Coles B, Shah S, Hall J, Topley N,
Köhl J, Morgan BP, Labéta MO. 2011. TLR activation enhances C5a-induced pro-
inflammatory responses by negatively modulating the second C5a receptor, C5L2. Eur J
Immunol, 41(9):2741-52.
Rapala-Kozik M, Bras G, Chruscicka B, Karkowska-Kuleta J, Sroka A, Herwald H, Nguyen
KA, Eick S, Potempa J, Kozik A. 2011. Adsorption of components of the plasma kinin-
forming system on the surface of Porphyromonas gingivalis involves gingipains as the major
docking platforms. Infect Immun, 79(2):797-805.

- 73 -
Rendueles O, Kaplan JB, Ghigo JM. 2013. Antibiofilm polysaccharides. Environ Microbiol.
15(2):334-46.
Rosen G, Sela MN, Bachrach G. 2012. The antibacterial activity of LL-37 against Treponema
denticola is dentilisin protease independent and facilitated by the major outer sheath protein
virulence factor. Infect Immun, 80(3):1107-14.
Ruby JD, Lux R, Shi W, Charon NW, Dasanayake A. 2008. Effect of glucose on Treponema
denticola cell behavior. Oral Microbiol Immunol, 23(3):234-8.
Rudney JD, Chen R, Sedgewick GJ. 2005. Actinobacillus actinomycetemcomitans,
Porphyromonas gingivalis, and Tannerella forsythensis are components of a polymicrobial
intracellular flora within human buccal cells. J Dent Res, 84(1):59-63.
Sabet M, Lee SW, Nauman RK, Sims T, Um HS. 2003.The surface (S-) layer is a virulence
factor of Bacteroides forsythus. Microbiology, 149(Pt 12):3617-27.
Saiki K, Konishi K. 2010. Identification of a novel Porphyromonas gingivalis outer membrane
protein, PG534, required for the production of active gingipains. FEMS Microbiol Lett,
310(2):168-74.
Saito T., Ishihara K., Kato T. & Okuda, K. 1997. Cloning, expression, and sequencing of a
protease gene from Bacteroides forsythus ATCC 43037 in Escherichia coli. Infect Immun,
65:4888–4891.
Sakakibara J, Nagano K, Murakami Y, Higuchi N, Nakamura H, Shimozato K, Yoshimura F.
2007. Loss of adherence ability to human gingival epithelial cells in S-layer protein-deficient
mutants of Tannerellaforsythensis. Microbiology, 153(Pt 3):866-76.
Sakamoto M, Suzuki M, Umeda M, Ishikawa I, Benno Y. 2002. Reclassification of
Bacteroides forsythus (Tanner et al. 1986) as Tannerella forsythensis corrig., gen. nov.,
comb. Nov. Int J Syst Evol Microbiol, 52(Pt 3):841-9.
Sang SG, Rong H, Wang JB, Xie YQ. 2014. Effects of Porphyromonas gingivalis
extracellular vesicles on human periodontal ligament fibroblasts. Int J Clin Exp Med,

- 74 -
7(2):379-83.
Sano Y, Okamoto-Shibayama K, Tanaka K, Ito R, Shintani S, Yakushiji M, Ishihara K. 2014.
Dentilisin involvement in coaggregation between Treponema denticola and Tannerella
forsythia. Anaerobe, ;30:45-50.
Sara M, Sleytr UB. 2000. S-Layer proteins. J. Bacteriol, 182:859–868.
Sato K, Naito M, Yukitake H, Hirakawa H, Shoji M, McBride MJ, Rhodes RG, Nakayama K
2010. A protein secretion system linked to bacteroidete gliding motility and pathogenesis.
Proc Natl Acad Sci U S A. 107(1):276-81.
Schlaepfer DD, Hauck CR, Sieg DJ. 1999. Signaling through focal adhesion kinase. Prog
Biophys Mol Bioll, 71(3-4):435-78.
Sellers RM, Payne JB, Yu F, LeVan TD, Walker C, Mikuls TR. 2015. TLR4 Asp299Gly
polymorphism may be protective against chronic periodontitis. J Periodontal Res.
Settem RP, Honma K, Nakajima T, Phansopa C, Roy S, Stafford GP, Sharma A . 2013. A
bacterial glycan core linked to surface (S)-layer proteins modulates host immunity through
Th17 suppression. Mucosal Immunol, 6(2):415-26.
Sekot G, Posch G, Messner P, Matejka M, Rausch-Fan X, Andrukhov O, Schäffer C. 2011.
Potential of the Tannerella forsythia S-layer to delay the immune response. J Dent Res,
90(1):109-14.
Serhan CN. 2008. Controlling the resolution of acute inflammation: a new genus of dual anti-
inflammatory and proresolving mediators. J Periodontol, 79(8 Suppl):1520-6.
Serhan CN, Chiang N, Van Dyke TE. 2008. Resolving inflammation: dual anti-inflammatory
and pro-resolution lipid mediators. Nat Rev Immunol, 8(5):349-61
Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, Perretti M, Rossi AG,
Wallace JL. 2007. Resolution of inflammation: state oft he art, definitions and terms .ASEB
J, 21(2):325-32.

- 75 -
Seymour GJ. 1991.Importance of the host response in the periodontium. J Clin Periodontol,
18(6):421-6.
Seymour GJ, Gemmell E. 2001. Cytokines in periodontal disease: where to from here? Acta
Odontol Scand, 59(3):167-73.
Sharma A. 2010. Virulence mechanisms of Tannerella forsythia. Periodontol 2000,
54(1):106-16.
Sharma A, Inagaki S, Honma K., Sfintescu C, Baker PJ, Evans R.T. 2005. Tannerella
forsythia-induced alveolar bone loss in mice involves leucine-rich-repeat BspA protein. J.
Dent. Res, 84:462–467.
Sharma A, Novak EK, Sojar HT, Swank RT, Kuramitsu HK, Genco RJ 2000. Porphyromonas
gingivalis platelet aggregation activity: outer membrane vesicles are potent activators of
murine platelets. Oral Microbiol Immunol, 15(6):393-6.
Sharma A, Sojar HT, Glurich I, Honma K, Kuramitsu HK, Genco RJ. 1998. Cloning,
expression, and sequencing of a cell surface antigen containing a leucine-rich repeat motif
from Bacteroides forsythus ATCC 43037. Infect Immun, 66(12):5703-10.
Sheets SM, Potempa J, Travis J, Casiano CA, Fletcher HM. 2005. Gingipains from
Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in
endothelial cells. Infect Immun, 73(3):1543-52.
Shimotahira N, Oogai Y, Kawada-Matsuo M, Yamada S, Fukutsuji K, Nagano K, Yoshimura
F, Noguchi K, Komatsuzawa H. 2013. The surface layer of Tannerella forsythia contributes
to serum resistance and oral bacterial coaggregation. Infect Immun, 81(4):1198-206.
Silva N, Abusleme L, Bravo D, Dutzan N, Garcia-Sesnich J, Vernal R, Hernández M,
Gamonal J. 2015. Host response mechanisms in periodontal diseases. J Appl Oral Sci.
23(3):329-55.
Sim JH, Shi W, Lux R. 2005.Protein-protein interactions in the chemotaxis signalling
pathway of Treponema denticola. Microbiology, 151(Pt 6):1801-7.

- 76 -
Sims TJ, Schifferle RE, Ali RW, Skaug N, Page RC. 2001. Immunoglobulin G response of
periodontitis patients to Porphyromonas gingivalis capsular carbohydrate and
lipopolysaccharide antigens. Oral Microbiol Immunol, 16(4):193-201.
Slots J. 1986. Bacterial specificity in adult periodontitis. A summary of recent work. J Clin
Periodontol, 13(10):912-7.
Socransky SS. 1979. Criteria for the infectious agents in dental caries and periodontal
disease. J Clin Periodontol, 6(7):16-21.
Socransky SS, Haffajee AD. 1994. Evidence of bacterial etiology: a historical
perspective.Periodontol 2000, 5:7-25.
Socransky SS, Smith C, Haffajee AD. 2002. Subgingival microbial profiles in refractory
periodontal disease J Clin Periodontol, 29(3):260-8.
Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL Jr. 1998. Microbial complexes in
subgingival plaque. J Clin Periodontol, 25(2):134-44.
Socransky SS, Haffajee AD, Ximenez-Fyvie LA, Ferres M, Mager D. 1999. Ecological
considerations in the treatment of Actinobacillus actinomycetemcomitans and
Porphyromonas gingivalis periodontal infections. Periodontol 2000, 20:341-62.
Su H, Yan X, Dong Z, Chen W, Lin ZT, Hu QG. 2015. Differential roles of Porphyromonas
gingivalis lipopolysaccharide and Escherichia coli lipopolysaccharide in maturation and
antigen-presenting functions of dentritic cells. Eur Rev Med Pharmacol Sci, 19(13):2482-92.
Suwannakul S, Stafford GP, Whawell SA, Douglas CW. 2010. Identification of bistable
populations of Porphyromonas gingivalis that differ in epithelial cell invasion. Microbiology,
156(Pt 10):3052-64.
Takashiba S, Naruishi K. 2006. Gene polymorphisms in periodontal health and disease.
Periodontol 2000, 40:94-106.
Takashiba S, Naruishi K, Murayama Y .2003. Perspective of cytokine regulation for

- 77 -
periodontal treatment: fibroblast biology. J Periodontol, 74(1):103-10.
Takashiba S, Ohyama H, Oyaizu K, Kogoe-Kato N, Murayama Y. 1999. HLA genetics for
diagnosis of susceptibility to early-onset periodontitis. J Periodontal Res. 34(7):374-8
Takahashi Y, Davey M, Yumoto H, Gibson FC 3rd, Genco CA. 2006. Fimbria-dependent
activation of pro-inflammatory molecules in Porphyromonas gingivalis infected human aortic
endothelial cells. Cell Microbiol, 8(5):738-57.
Takemoto T, Kurihara H, Dahlen G. 1997. Characterization of Bacteroides forsythus isolates.
J Clin Microbiol, 35(6):1378-81.
Tamai R, Asai Y, Ogawa T. 2005. Requirement for intercellular adhesion molecule 1 and
caveolae in invasion of human oral epithelial cells by Porphyromonas gingivalis. Infect
Immun, 73(10):6290-8.
Tan KH, Seers CA, Dashper SG, Mitchell HL, Pyke JS, Meuric V, Slakeski N, Cleal SM,
Chambers JL, McConville MJ, Reynolds EC. 2014. Porphyromonas gingivalis and
Treponema denticola exhibit metabolic symbioses. PLoS Pathog. 10(3):e1003955.
Tanabe S, Bodet C, Grenier D. 2008. Treponema denticola lipooligosaccharide activates
gingival fibroblasts and upregulates inflammatory mediator production. J Cell Physiol,
216(3):727.
Tanner AC, Izard J. 2006. Tannerella forsythia, a periodontal pathogen entering the genomic
era. Periodontol 2000,42:88-113.
Terheyden H, Stadlinger B, Sanz M, Garbe AI, Meyle J. 2014. Inflammatory reaction -
communication of cells. Clin Oral Implants Res. 25(4):399-407.
Thompson H, Homer KA, Rao S, Booth V, Hosie AH. 2009. An orthologue of Bacteroides
fragilis NanH is the principal sialidase in Tannerella forsythia. J Bacteriol, 191(11):3623-8.
Thorbert-Mros S, Larsson L, Berglundh T. 2014. Cellular composition of long-standing
gingivitis and periodontitis lesions. J Periodontal Res, 50(4):535-43.

- 78 -
Travis J, Pike R, Imamura T, Potempa J. 1997. Porphyromonas gingivalis proteinases as
virulence factors in the development of periodontitis. J Periodontal Res, 32(1 Pt 2):120-5.
Tribble GD, Kerr JE, Wang BY. 2013. Genetic diversity in the oral pathogen Porphyromonas
gingivalis: molecular mechanisms and biological consequences. Future Microbiol, 8(5):607-
20.
Tsuda K, Amano A, Umebayashi K, Inaba H, Nakagawa I, Nakanishi Y, Yoshimori T. 2007.
Molecular dissection of internalization of Porphyromonas gingivalis by cells using
fluorescent beads coated with bacterial membrane vesicle. Cell Struct Funct, 30(2):81-91.
Tsuda K, Furuta N, Inaba H, Kawai S, Hanada K, Yoshimori T, Amano A. 2008. Functional
analysis of alpha5beta1 integrin and lipid rafts in invasion of epithelial cells by
Porphyromonasgingivalis using fluorescent beads coated with bacterial membrane vesicles.
Cell Struct Funct, 33(1):123-32.
Umemoto T, Jinno T, Taiji Y, Ogawa T. 2001. Chemotaxis of oral treponemes toward sera
and albumin of rabbit. Microbiol Immunol. 45(8):571-7.
Vaithilingam RD, Safii SH, Baharuddin NA, Ng CC, Cheong SC, Bartold PM, Schaefer AS,
Loos BG. 2014. Moving into a new era of periodontal genetic studies: relevance of large
case-control samples using severe phenotypes for genome-wide association studies. J
Periodontal Res, 49(6):683-95.
Takashiba S, Naruishi K. 2006. Gene polymorphisms in periodontal health and disease.
Periodontol 2000, 40:94-106.
Van der Reijden WA, Bosch-Tijhof CJ, Strooker H, van Winkelhoff AJ. 2006. prtH in
Tannerella forsythensis is not associated with periodontitis. J Periodontol,77(4):586-90.
Van Steenbergen TJ, Delemarre FG, Namavar F, De Graaff J. 1987. Differences in virulence
within the species Bacteroides gingivalis. Antonie Van Leeuwenhoek, 53(4):233-44.
Veening JW, Stewart EJ, Berngruber TW, Taddei F, Kuipers OP, Hamoen LW. 2008. Bet-
hedging and epigenetic inheritance in bacterial cell development. Proc Natl Acad Sci U S A,

- 79 -
105(11):4393-8.
Veith PD, Dashper SG, O'Brien-Simpson NM, Paolini RA, Orth R, Walsh KA, Reynolds EC.
2009. Major proteins and antigens of Treponema denticola. Biochim Biophys Acta,
1794(10):1421-32.
Veith PD, Chen YY, Gorasia DG, Chen D, Glew MD, O'Brien-Simpson NM, Cecil JD, Holden
JA, Reynolds EC. 2014. Porphyromonas gingivalis outer membrane vesicles exclusively
contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence
factors. J Proteome Res, 13(5):2420-32.
Vernal R, Díaz-Zúñiga J, Melgar-Rodríguez S, Pujol M, Diaz-Guerra E, Silva A, Sanz M,
Garcia-Sanz JA. 2014. Activation of RANKL-induced osteoclasts and memory T
lymphocytes by Porphyromonas gingivalis is serotype dependant. J Clin Periodontol,
41(5):451-9.
Wang PL, Azuma Y, Shinohara M, Ohura K. 2000. Toll-like receptor 4-mediated signal
pathway induced by Porphyromonas gingivalis lipopolysaccharide in human gingival
fibroblasts. Biochem Biophys Res Commun, 273(3):1161-7.
Wang YL, Pan KQ, Sun Y, Deng J. 2014. Effect of lipopolysaccharide on the expression of
ALP,BSP,DSPP in rat dental pulp cells. Shanghai Kou Qiang Yi Xue, 23(4):431-5.
Wang M, Shakhatreh MA, James D, Liang S, Nishiyama S, Yoshimura F, Demuth DR,
Hajishengallis G. 2007. Fimbrial proteins of porphyromonas gingivalis mediate in vivo
virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J
Immunol, 179(4):2349-58.
Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, Triantafilou M, Triantafilou K,
Lambris JD, Hajishengallis G. 2010. Microbial hijacking of complement-toll-like receptor
crosstalk. Sci Signal, 3(109):ra11.
Wensink J, Witholt B. 1981. Outer-membrane vesicles released by normally growing
Escherichia coli contain very little lipoprotein. Eur J Biochem, 116(2):331-5.

- 80 -
Whittaker CJ, Clemans DL, Kolenbrander PE. 1996. Insertional inactivation of an
intrageneric coaggregation-relevant adhesin locus from Streptococcus gordonii DL1
(Challis). Infect Immun, 64(10):4137-42.
Wolf F, Rateitschak H. 2012. Farbatlanten der Zahnmedizin 1- Parodontologie. Thieme.
Ximinez-Fyvie LA, Haffajee AD, Socransky SS. 2000. Comparison of the micrbiota of supra-
and subgingival plaque in health and periodontitis. J Clin Periodontol, 27(9):648-57.
Yilmaz O, Watanabe K, Lamont RJ. 2002. Involvement of integrins in fimbriae-mediated
binding and invasion by Porphyromonas gingivalis. Cell Microbiol, 4(5):305-14.
Yilmaz O, Jungas T, Verbeke P, Ojcius DM 2004. Activation of the phosphatidylinositol 3-
kinase/Akt pathway contributes to survival of primary epithelial cells infected with the
periodontal pathogen Porphyromonas gingivalis. Infect Immun, 72(7):3743-51.
Yilmaz O, Young PA, Lamont RJ, Kenny GE. 2003. Gingival epithelial cell signalling and
cytoskeletal responses to Porphyromonas gingivalis invasion. Microbiology, 149(Pt 9):2417-
26.
Yoshino T, Laine ML, van Winkelhoff AJ, Dahlén G. 2007. Genotype variation and capsular
serotypes of Porphyromonas gingivalis from chronic periodontitis and periodontal
abscesses. FEMS Microbiol Lett, 270(1):75-81.
Yoo JY, Kim HC, Zhu W, Kim SM, Sabet M, Handfield M, Hillman J, Progulske-Fox A, Lee
SW. 2007. Identification of Tannerella forsythia antigens specifically expressed in patients
with periodontal disease. FEMS Microbiol Lett, 275(2):344-52.
Zadeh HH, Nichols FC, Miyasaki KT. 1999. The role of the cell-mediated immune
response to Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in
periodontitis. Periodontol 2000, 20:239-88.
Zhang W, Ju J, Rigney T, Tribble G . 2013. Integrin α5β1-fimbriae binding and actin
rearrangement are essential for Porphyromonas gingivalis invasion of osteoblasts and
subsequent activation of the JNK pathway. BMC Microbiol, 13:5.

- 81 -
Danksagung
Bedanken möchte ich mich bei Herrn Universitätsprofessor Dr. med. Wolfgang Pfister für
die Möglichkeit, die Überlassung des Themas, seinem Fachwissen und seiner freien Zeit,
die er für mich opferte.
Bedanken möchte ich mich auch bei meinen Eltern, die mir stets mit Rat und Tat zur Seite
stehen.
Ganz besonderem Dank gilt meinem Mann, dafür dass er für unsere Kinder und mich der
beste Vater und Ehemann der Welt ist und unseren Kindern, die dafür sorgen, dass unsere
Welt bunt bleibt.

- 82 -
Lebenslauf
Maja Anna- Böttcher
15.8.1972 in Greifswald
Neunkircher Str. 131
66113 Saarbrücken
Tel: 0681- 49488
Eltern
Sigrun Böttcher, geb. Lembke Fotografenmeisterin
Dr. Volker Böttcher Zahnarzt, Arzt für Mund-, Kiefer- und Gesichtschirurgie
Familienstand
verheiratet mit Walter Anna Dipl. Ingenieurinformatik
Kinder: Noah Anna, 17.7.2000
Laurin Anna, 19.10.2004
Charlotte Anna, 5.9.2007
Schule/ Studium
1979-1983 Grundschule Schafbrücke, Saarbrücken
1983-1990 Deutsch- Französisches- Gymnasium, Saarbrücken
1990-1991 Realschule Ludwigspark, Saarbrücken
1991-1993 Wirtschaftsschulen Saarbrücken, Fachoberschule Wirtschaft
1993-1996 Kaufmännisches Bildungszentrum Saarbrücken, Halberg
1993-1996 Abendgymnasium Saarbrücken
1996-2002 Studium der Zahnmedizin, Universitätsklinikum Homburg
8/2002 Approbation Zahnmedizin
12/2009 Anerkennung der Weiterbildung zur Fachzahnärztin für
Oralchirurgie
9/2013 Ernennung zum Master of Science (M.Sc.) „Parodontologie und
Implantattherapie“ der DIU/DGParo

- 83 -
Berufsausbildung und Berufspraxis
1992-1993 Praktikum Allgemeiner Finanzdienst GmbH
1993-1996 Ausbildung zur Arzthelferin
2002-2007 Zahnärztin der Klinik für Mund- Kiefer- und Gesichtschirurgie
der Universitätskliniken Homburg/Saar
2010-2012 Zulassung und Teilhaber in der Gemeinschaftspraxis
Dr. C. Assaf, Dr. V. Böttcher, M.Anna-Böttcher, Saarbrücken
2013-heute Teilhaber in der Gemeinschaftspraxis
Dr. Assaf, A.-K.Böttcher und M. Anna-Böttcher M.Sc., Saarbrücken

- 84 -
Ehrenwörtliche Erklärung
Hiermit erkläre ich, dass mir die Promotionsordnung der Medizinischen Fakultät der
FriedrichSchiller-Universität bekannt ist,
ich die Dissertation selbst angefertigt habe und alle von mir benutzten Hilfsmittel,
persönlichen Mitteilungen und Quellen in meiner Arbeit angegeben sind,
mich folgende Personen bei der Auswahl und Auswertung des Materials sowie bei der
Herstellung des Manuskripts unterstützt haben:
Prof. Dr. med. habil. W. Pfister
die Hilfe eines Promotionsberaters nicht in Anspruch genommen wurde
und dass Dritte weder unmittelbar noch mittelbar geldwerte Leistungen von mir für Arbeiten
erhalten haben, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen,
dass ich die Dissertation noch nicht als Prüfungsarbeit für eine staatliche oder andere
wissenschaftliche Prüfung eingereicht habe
und dass ich die gleiche, eine in wesentlichen Teilen ähnliche oder eine andere Abhandlung
nicht bei einer anderen Hochschule als Dissertation eingereicht habe.
Saarbrücken, den Maja Anna-Böttcher





























