Embed Size (px)
Citation preview

200 H u i s g e n und K o c h
Die Kupplung aromatischer mit aliphatischen Diazoverbindungen
Von Rolf Huisgen und Hans- Joachim Koch
(Mitteilung aus dem Institut fur Organische Chemie der Universitiit Miinchen)
(Eingelaufen am 27. Oktober 1954)
(Mit 6 Figuren im Text)
A) E infuhrung Im Gegensatz zum Aryl-diazonium-kation entbehrt das Alkyl -
diazonium-ion der Mesomeriestabilisierung ; die Labilitat der Diazo- gruppe auI3ert sich in der raschen Abspaltung des Stickstoffmolekiils, wobei das zuruckbleibende Carbonium-ion mit dem Losungsmittel bzw . einem nucleophilen Agens weiterreagiert.
0 H,C-N IS -+ H3C0 + N,
t - H @ + H @ i.
0 0 Q E G 0 HZC-N N +--J HzC=N=N + + HzC-N=Nl
Ia Ib Ic
Entzieht man aber etwa dem Methyl-diazonium-kation ein Pro- ton, dann ermoglicht das freigelegte Elektronenpaar in I a der Diazo- gruppe die Entfaltung einer Mesomerie, die der des aromatischen Diazonium-ions entspricht. Eine solche Protonenabspaltung liegt tatsgchlich den bekannten Bildungswegen des Diazo-methans (I) aus Nitroso-acyl-methylaminenl), Nitroso-methylamino-isobutyl- methyl-keton 2, u. a. Verbindungen zugrunde.
Das freie Elektronenpaar am Kohlenstoff in den Grenzformeln I a und I c verleiht dem Diazo-methan und seinen Abkommlingen den Charakter von Basen und macht die Mehrzahl der Reaktionen dieser KorperMasse verstandlich. Die unter Stickstoff-Freisetzung von- statten gehende Reaktion der Diazo-alkane mit Sauren weist als ent- scheidende Stufe einen Protonen- Ubergang auf. Im obigen Formel- schema tritt diese Folge als Umkehr der Bildung des Diazo-methans aus dem Methyl-diazonium-ion in Erscheinung. Die Methylierung von Carbonsauren mit Diazo-methan gems8 I1 ist das bekannteste
l ) Die Synthesedes Diazomethans nach H.v.Pechmann ,Ber.dtsch.chem.Ges.28, 855 ( 1895) wurde im Lichte der neueren Untersuchungen uber Nitroso-acyl-amine von It. Huisg e n u. J. Re i ne r t s h o f e r , Liebigs Ann. Chem.575,174( 1952) betrachtet.
2, D. W. Adamson u. J. K e n n e r , J. chem. SOC. [London] 1935, 286; 1937, 1551.

Die Kupplung aromatischer mit aliphatischen Diazoverbindungen 201
Beispiel dieses Reaktionstypus ; neben den stochiometrischen exi- stieren auch saurekatalysierte Zerfallsreaktionen der Diazo-alkane.
8 0 0 0 I1 R-COOH + CH,-N, + [R-COO + CH,-NZ]
0 3% [R-COO + CH,"] ~ + R-COOCH,
Die Basizitiit der Diazo-alkane ist auch fur die zahlreichen Reak- tionen rnit Lewis - Sauren resp. Antibasen im Sinne von J. B j e r - rum 3, verantwortlich. Die Umsetzungen rnit Aldehyden, Ketonen, Saurechloriden, Benzopersaure, komplexbildenden Metallhalogeni- den, Nitrosyl-chlorid, a,P-ungesattigten Carbonyl- und Nitroverbin- dungen usf. werden stets eingeleitet von der Anlagerung des elektro- philen Agens an das freie Elektronenpaar des Diazo-alkans4). Der damit verbundene Verlust der Diazo-Mesomerie hat eine Entbindung von elementarem Stickstoff oder eine geeignete Stabilisierung durch RingschluB im Gefolge, wie mit zwei Beispielen illustriert sei.
N / \
CH2 N 0
+ 0 0 /N -N ('j
CHZ--N N ----f CHZ + CHz=CH-COOR CHZ-CH-COOR CHZ-CH-COOR
I n der Liste der elektrophilen Agentiera.hat nun auch das aroma- tische Diazonium-ion seinen Platz. Im Ubergang des Diazonium- kations in Diazo-cyanide, -sulfonate oder Azofarbstoffe bei der Be- handlung rnit Cyanid-, Sulfit- oder Phenolat-anion liegen Reaktio- nen rnit nucleophilen Partnern vor. Diese Reaktionsweise findet in einer Grenzformel des Diazonium-ions mit Elektronensextett am auBeren Stickstoff (111) eine formale Erklarung.
111
3) Angew. Chem. 63, 527 (1951). 4) Eine zusammenfassende Betrachtung dcr Seaktionen der Diazo-alkane
erscheint demnachst in Angew. Chem.

202 H u i s g e n und K o c h
Die nach diesen Uberlegungen vorauszusehende Reciktion der elektrophilen A4ryl-diazonium-ionen m i t d e n nucleophi len al iphat ischen Diazoverbindungen laSt sich ver~irkl ichen~) und bezuglich aller Reaktionsteilnehmer weitgehend variieren.
B) Diazo - e s sige s t e r a l s K u p p l u n g s k o m pone n t e Aus der eiskalten Losung von Diazo-essigester in Methanol wird
auf den Zusatz von p-Nitro-benzol-diazonium-chlorid hin ein Mol Stickstoff freigesetzt, wobei die Reaktionslosung zum Kristallbrei erstarrt. Bei dem in guter Ausbeute anfallenden blaflgelben Reak- tionsprodukt handelt es sich um das p-Nitro-phenyl-hydrazid-chlorid der Oxal-estersaure (IV). Der Bildungsweg 1aSt sich unschwer rekon- struieren : Auf die Azokupplung des Aryl-diazonium-ions gegen das nucleophile Zentrum des Diazo-essigesters folgt die AbstoSung des aliphatischen Diazo-stickstoffs nebst Eintritt des Chlor-anions in die Elektronenlucke des Carbonium-ions. Der Ubergang der Azoverbin- dung in die stabilere, tautomere Hydrazon-Form fuhrt zu IV.
/ S O , - - / \--N PIS + CH-COOR - S O , \ N S CH COOR \ / \ / C1” -PIS s c1- N n.
+ SO, ( ‘ 3 N-CH-COOR +NO,--/ \ S S-CH-COOR \-/ \ /
c‘1 c1
,COOR + NO,- < )--;UH-N=C \
C1 I v
Solche Abkommlinge der Saure-chloride, in denen sich der Car- bony1 -Sauerstoff gegen eine Hydrazon-Gruppe ersetzt findet, wurden unter Beschrankung auf die Oxal-estersaure-Reihe von D. A. B o w a c k und A. L a p w or t h 6, atus Aryl-diazonium-chlorid und x-Chlor-acetessigester unter Austritt eines Acetylrestes erhalten. Die Identitiit mit einem auf diesem Weg bereiteten Vergleichs- praparat sichert die Konstitution von IV.
Tab. 1 zeigt die bei der Reaktion des Diazo-essigesters mit ver- xchiedenen Diazonium-salzen isolierten Oxalester-aryl-hydrazid- chloride. Die Ausbeuten entsprechen denen schwer erfolgender Azo- kupplungen, bei denen der Zerfall des Aryl-diazonium-ions unter
j) Vorlaufige Mitteilung.: R. H u i s g e n , dngew. Chem. 65, 40 (1953); R. H u i s - g e n u. H. J. K o c h , Nsturwissenschaften 41, 16 (1954); R . H u i s g e n , Osteri. Chemiker Ztg. 55, 237 (1954).
6 , J . chem. SOC. [London] 87, 1854 (1905).

Aryl-
p-NO,-C,jH,- p-Br.C6H4- o-NO,-C,H,- P-Cl-COHd- 2,4-ClZ-C6H3- C6H5-
m-CH,-C,H,- p -c, ,H, -
p-CH,0-C6H,-
7, KinetikderAzokupplung: J. B. C o n a n t u . W. D. P e t e r s o n , J.Amer.chem. Soc. 52, 1220 (1930); H. Zol l inger , Helv. 36, 1730 (1953); Chem. Reviews 51, 347 (1952); R. H u i s g e n , W. R i e b l u. H. G. G e n t n e r , unverijffentlicht.
ubersicht bei J. F. B u n n e t t u. R. E. Z a h l e r , Chem. Reviews 49,294 (1951).
Hydrazid-chlorid Schmp.
191-193O
I Ausb. in Proc. d. T h
69 62 52 48 39 16 0 0 0
I 160-161 O
117,8-118° 146-147 O
92,5-93,5O 77-790
14*

204 H u i s g e n und K o c ' h
C) Azokupplungen m i t Diazo-ke tonen Die Diazo-ketone stehen dem Diazo-essigester in der Kupplungs-
Aktivitat nicht nach und liefern a-Ketosdure-arylhydrazid-halo- genide. Diazo-acetophenon wird bei der Azokupplung mit p-Nitro- benzol-diazonium-chlorid in eiskaltem Methanol in den Benzoyl- ameisensaure-Abkommling V mit 79-proc. Ausbeute ubergefiihrtg).
0 NO,--/ - CH-C--/ \
\ / \ / c1 s?;
Analog vollziehen sich die Reaktionen mit p-Chlor-benzol-diazo- nium-chlorid bzw. dem p-Nitro-benzol-diazonium-bromid. Zur konsti- tutionellen Sicherung wurden diese Hydrazid-halogenide auch, der Arbeitsweise von D. A. Bowack und A. Lapwor th6) entspre- chend, auf folgendem, hier nicht sonderlich ergiebigen Weg dar- gestellt :
0 ,,CO-C,H, ,CO-C6H; Ar-N N + CH, - Ar--NH-K=C,,
\ Cl'\ CO-CeH, CO-C6Hj
,,CO--C6H,
\ t Hal2 .cK,$ Ar-NH-X=C + C,B,-COOH - t HHal
VI
Unterwirft man das Kupplungsprodukt von p-Nitro-benzol-diazoniumsalz und Benzoyl-aceton der chlorierenden Spaltung, gelangt man unter Austritt voii Essigsiiure ebenfalls zu V; hier wird also ahnlich wie bei der Alkalibehandlung von Tri-acyl-methan-Derivaten der kleinere Acylrest bevorzugt abgelost.
Mit dem weniger aktiven unsubstituierten Benzol-diazonium- chlorid kuppelt Diazo-acetophenon nicht mehr ; ebenso weist schon die mehr als ein Mol betragende Stickstoff-Freisetzung bei der Reak- tion von P-Naphthalin-diazonium-salz mit Diazo-aceton auf die mangelnde Aktivitat der elektrophilen Komponente hin. Mit 2,4-Di- chlor-benzol-diazonium-chlorid dagegen laBt sich Diazo-aceton mit 75-proc. Ausbeute in das Brenztraubens&ure-(2,4-dichlor-phenyl)- hydrazid-chlorid (VII) uberfuhren. C. Bulow und P. W. Neberlo)
9, Die Kupplungsversuche rnit Diazo-acctophenon wurdcn von Herrn H. J. Schabel , Diplomarbeit Tiibingen 1952, durchgefuhrt.
10) Ber. dtsch. chem. Ges. 46, 2370 (1913).

Die Kupplung aromatischer mit aliphatischen Diazoverbindungen 205
erhielten die gleiche Verbindung durch Chlorierung von Benzol-azo- acetessigsaure unter Kernsubstitution und Carboxyl-Verdrangung.
Auch durch Verminderung der nucleophilen Aktivitat der ali- phatischen Diazoverbindung erreicht man die Grenze der Anwend- barkeit unserer Kupplungsreaktion. 1st schon der fjbergang vom Diazo-alkan zum Diazo-essigester oder Diazo-aceton von einer Basi- zitats-Schwachung begleitet, so bringt eine noch weitergehende Ein- bul3e an Basencharakter die Azokupplung ganz zum Erliegen
p-Yitro-diazo-acetophenon (VIII) u n d -propiophenon sind auch mit Halogen- oder Nitro-benzol-diazonium-salzen nicht zur Reaktion zu bringen ; selbst das extrem reaktive 2,4-Dinitro-benzol-diazo- nium-salzll) versagt. Die Resistenz dieser Diazo-ketone gegen Sauren geht mit der Kupplungstragheit Hand in Hand; fur beide Phanomene ist der Basizitatsverlust duroh induktiven Effekt der p-standigen Nitrogruppe verantwortlich. Das saurestabile Diazo- dihenzo y l -methan (IX) laat sich aus dem Versuch der Azokupplung ebenfalls unverandert zuruckerhalten. In diesem doppelseitig von Acylresten flankierten Diazomethan-System griindet sich der Rasi- zitatsschwund auf das Auftreten zweier Grsnzformeln desTyps IX b.
0 00 I
C6H,-C C,H,-C \o 0 \ @
/ C - x - N t - /C-X
C,H,-C C6H5-c , 0 IXa 0 IXb
D) Die Kupplungsreakt ion der Diary l -d iazo-methane Das aromatische Diazonium-ion setzt schon bei 0 O aus der metha-
nolischen Losung von Diphenyl-diazo-methan momentan den ali- phatischen Diazo-Stickstoff frei. Die aus der Reaktionslosung nach
11)K.H. Meyer, A. I r s c h i c k u. H. Schlosser , Ber. dtsch. chem. Ges.4'7, 1741 (1914).

206 H u i s g e n und K o c h
kurzer Zeit kristallisierenden Verbindungen erweisen sich als halo- genfrei, enthalten aber eine der Wechselwirkung mit dem Losungs- mittel entstammende Methoxylgruppe. Es handelt sich um a--4ryl- azo-benzhydryl-methyZ-ather (XI), deren Bildung man sich folgender- maBen vorzustellen hat :
P l J n- n
Ar-N S -7 C-C,H, ---f Ar-X==X-C-C,H, K2
c1' CGH, CGH,
+CIS i, ,cn,oH d \
C1 OCH, [XII: OC,Hj]
Ar-X=S-C-C,H5 Ar-K=N-C-Cc,Ha
X CGH, XI CGHj
Auch hier wird auf Grund der groBeren nucleophilen Aktivitat des Chlor-anions das Arylazo-halogenid X als Zwischenprodukt ver- mutet ; gleich dem Stammkorper, dem Benzhydryl-chlorid, unter- liegt es der zu XI fuhrenden Solvolyse. Die in der GroBenordnung von SO-SO% d. Th. liegenden Ausbeuten an XI lassen sogar den SchluB zu, da13 die Bildung von X im Vergleich zur Solvolyse sehr rasch sein mul3. Im anderen Fall ware ein Absinken der Ausbeute durch Umsatz des entstehenden Halogenwasserstoffs mit Diphenyl- diazo-methan unvermeidbar. Fuhrt man die Azokupplung in Atha- no1 durch, werden in analoger Weise die Athyl-ather (XII) erhalten. Nicht minder glatt vollziehen sich die Kupplungen mit 9-Diazo- fluoren zu XIII.
/\ /\
\/A/\/ Ar-X=X OCH,
XI11
In diesen Kupplungsprodukten, die den Arylazo-Rest und die Athergruppe am gleichen Kohlenstoffatom tragen, haben wir es mit einer neuen Klasse von Azooerbindungen zu tun (Tab. 2). Auch im Diaryl-diazo-methan sollten die Aryl-Reste eine analoge Schwachung der nucleophilen Aktivitat des Diazo-methans auslosen wie die Car- bonylgruppe in Diazo-fettsaure-estem oder Diazo-ketonen. Die Er- fahrung lehrt jedoch - auBer dem Kupplungsvermogen ist hier die Saureempfindlichkeit anzufuhren -, daB die Angliederung des aro- matischen Kerns der Basizitat des zentralen Kohlenstoffatoms des Diazo-alkans wesentlich weniger Abbruch tut. Selbst mit dem reak-

Aryl- Ausbeute in Proc. d. Th. Schmp. Farbe
a-8r~l~zoo-ben~~ydryl-meth?(.18ther (XI) C6H6- 101-101,5° gelb p-NO,-C,H,- 134-135 ' rotorange
p-Br-C,H,- 136-137' 1 gelb P -c, OH,- 155,5-156° braunorange
p-CI-C,H,- 139-140' I gelb
9-Arylazo-fluoren-methylather (XIII)
70 56 54 66 65

208 H u i s g e n und K o c h
E) R e a k t i o n des p-Ni t ro-benzol -d iazonium-chlor ids m i t D i a z o - m e t h a n
Fur den Stammkorper der Reihe, das Diazo-methan selbst, 1aL3t das Fehlen jeglicher die Basizitat schwkchender Gruppen im Ein- klang mit der extremen Saureempfindlichkeit auch ein Maximum in der Befahigung zur Azokupplung voraussehen. Schon bei -100 vollzieht sich die Reaktion mit p-Nitro-benzol-diazonium-chlorid sehr stiirmisch ; das als Reaktionsprodukt zu erwartende p-Nitro- phenyl-hydrazid-chlorid der Ameisensaure (XVII) war jedoch zu- nachst nur in bescheidener Ausbeute isolierbar.
Im Gegensatz zu der oben beschriebenen Kupplung des Diazo- essigesters erweist sich hier die V e r e i n i g u n g des in t e rmed iaren C a r - b o n i u m - i o n s XVI mit d e m C h l o r - a n i o n als die empfindliche, die Aus- beute beeinflussende Stelle des Reaktionsablaufs. Mittels einer hohen Konzentration von Chlor-ionen lassen sich tatsachlich ander- weitige Reaktionswege ausschalten. Beim Eintropfen der atherisehen Diazomethan-Losung zur turbinierten, eiskalten, an Lithiumchlorid gesattigten methanolischen Losung des p-Nitro-benzol-diazonium- chlorids wird das kristallisierte Form-(p-nitro-phenyl-hydrazid)- clilorid (XVII) in nicht weniger als SO% d. Th. erhalten.
so,--, / >-X N + CH,-X -N -SO,-< >-S=N-CH,-S S 9 i' 0 +
\
--f SO,-( )--S=K-CH,
xv -NO,-< \ Y=S--CH,--C'I
-x2 d +cis
XVI H
/
\ / \ VH-S=C
XVII c1 - /-L
Im Gegensatz zu den Hydrazid-chloriden der Oxal-estersaure und der a-Ketosauren sind die der Ameisensaure instabil. XVII ist zwar im kristallisierten Zustand wochenlang haltbar. Die Losung in Methanol scheidet aber schon nach mehrtagigem Stehen Kristalle aus, die sich als p-Nitro-phenyl-hydrazin-hydrochlorid erweisen. Einer entsprechenden Reaktionsfolge unterliegt das im Benzolkcrn unsubstituierte Form-phenylhydrazid-chlorid schon im Verlauf einiger Minuten (S. 216). Wir sind geneigt, diese Reaktion mit dem Zerfall des hypothetischen Formy1;chlorids in Kohlenoxyd und Chlorwasserstoff in Parallele zii setzen.
H / r H?O
Ar--KH-S=C' -9 Br--SH--?iH,C'l I CO \ XVlll
c; 1

Die K u p p l u n g aromatischer m i t al iphat ischen Diazoverbindungen 209
Die Bedeutung der Chlor-ionen-Konzentration fur den Reaktions- erfolg weist auf die Polgereaktion des Carbonium-ions XVI als kriti- sche Stufe hin. Wie sieht nun die mit dem Eintritt des Chlor-ions konkurrierende Reaktion von XVI aus '2 Wir dachten zunachst an eine Umsetzung mit weiteren Molekeln Diazo-methan ; eine durch das aromatische Diazonium-ion gezundete Pol ymerisation in einer Ionenkette wiirde in der durch Borhalogenide oder Borsaure-ester katalysierten Polymethylen-Bildung aus Diazomethan eine Analogie besitzen12). Zwar fanden wir stets einen nichtfluchtigen Anteil im Reaktionsprodukt ; die kristallisierten Verbindungen stutzten aber unsere Vermutungen nicht.
Eine inverse Methodik vermindert die Chance von XVI, ein Chlor- ion einzufangen. In die eiskalte atherische Losung eines Diazo- methan-uberschusses lieBen wir langsam die Losung von p-Nitro- benzol-diazonium-chlorid in Methanol eintropfen. Aus dem kom- plexen Reaktionsprodukt , in welchem das Hydrazid-chlorid XVII fehlt, wurden nach Adsorption an Aluminiumoxyd und fraktio- nierter Kristallisation die Verbindungen der Tab. 3 isoliert.
Tab. 3 R e a k t i o n von p-Nitro-benzol-diazonium-chlorid
m i t i iberschi iss igem D i a z o m e t h a n
Ausbeute in -roc. d. Th.
Teilnahme Verb. Formel von x Mol Schmw. I r
C,H,O,N, 1 187-187,5' C8H702N3 156-157' C10H1102N3 1 1 1 102-103°
Die blaBgelbe Verbindung C,H,02N, ist formal das Ergebnis einer Reaktion der Komponenten im Verhaltnis 1 : 1 unter Abspaltung von Chlorwasserstoff. Am nachstliegenden erscheint eine Stabili- sierung des Primarproduktes XV der Azokupplung durch Ring- schluB zum 2-(p-Nitro-pheny1)-tetrazol (XIX). Zu einer solchen
N=N
NO,--/ \ Y--N \ XIX
12) H. Meerwein, Angew. Chem, 60. 78 (1928); G. G. Buckley u. X. N. R e y , J. chem. SOC. [London] 1952, 3701.
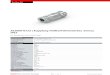
210 H u i s g e n und K o c h
Anlagerung des Diazomethans an die aromatische Diazogruppe wiirden sich in der Bildung von Pyrazol aus Acetylen13) sowie in der Addition des Diazomethans an die Dreifachbindung der Nitrile 14) Parallelen bieten. Die Darstellung des noch nicht beschriebe- nen Tetrazol-Abkommlings gelang durch Nitrierung des 2-Phenvl-
I3
t lo ? ,o k
5
retrozoJ __--- --- Z-Phenyl-tetruzo/-
-. . -2-Phenvl-tetrozol corbonsre
tetmzol-corbonsn -. -2-[p-Mtmphenyl-l
Ietmzol , - l-(p-hVrophenyld
250 300 350 m,u -
Fig. 1
tetrazolsly) ; XIX erwies sich mit unserem Reak- tionsprodukt aus Diazo- niethan als nicht iden- tisch.
Der spektrale Ver- gleich16) der Verbindung A (Tab. 3) mit verschie- denen Tetrazol-Derivaten (Fig. 1) lieB einen I-sub- stituierten Abkommiing vermuten; es handelt sich um I-(p-Nitro-pheny1)-te- trazol (XX), identisch mit einem nach M . F r e u n d und T. Paradiesl ' ) dar- gestellten Praparat.Dadie Tetrazol-Chemie keinen Anhaltspunkt fur eine Umlagerung von 1 -Aryl- in 2-Arvl-tetrazole bie-
tet, mu13 die Bildung von XX bei unserer Diazorkaktion auf eine primare Azokupplung des Diazomethans am Stickstoff zuriickgehen. Ein Blick auf die Grenzformeln des Diazomethans (S. 200) gestattet. es, das dupere Stickstoffatom als zweites basisches Zentrum anzuspre- chen. Obgleich eine Reaktion des Diazomethan-Stickstoffs mit, einem elektrophilen Agens bislang analogielos ist, vermogen wir nur mit folgendem Schema dem in 12-proc. Ausbeute auft,ret,enden Reaktionsprodukt Rechnung zu tragen.
13) H. v. P e c h m a n n , Bet. dtsch. chem. Ges. 31, 29.50 (1898); W. Hiickel u. J. D a t o w , Z.physik. Chem. Abt. A. 186, 159 (1940).
14) A. P e r a t o n e r u. E. A-lzzarello, Gazz. chim. ital. 38, I, 84 (1908); A. T a m - b u r o l l o u. A. M i l a z z o , ib. 38, 95 (1908); E. O l i v e r i - M a n d a l a , Gazz. chim. ital. 40, I, 123 (1910); F. A r n d t , H. Scholz u. E. F r o b e l , Liebigs Ann. Chem. 521, 95 (1936).
15) J. A. B l a d i n , Ber. dt,sch. chem. Ges. 18, 1544, 2907 (1885); 25, 1411 (189-7). DicKonstitution wurde vonE. B a m b e r g e r u n d P . de G r u y t , e r , Ber. dtsch. chem. Ges. 26, 2385 (1893) sowie von 0. W i d m a n n , ib. 26, 2618 (1893) richtig erkannt.
16) Die UV-Spektren dieser Arbeit wurden von Herrn Dr. H. Walz mit dem Unicam- Spe kt rophotometer SP 500 aufgenommen.
17) Ber. dtsch. chom. Ges. 34, 3120 (1901).

Die Kupplung arornatischer rnit aliphatischen Diazoverbindungen 21 1
Es ist einleuchtend, daB im Tetrazolring die bequemste Stabili- sierungsmoglichkeit des primaren Kations liegt. Von der Verbin- dung C der Tab. 3 wurde keine zur Konstitutionsermittlung aus-
KO,< >-$ N T X=X=CH, <? 6 I3
/ \-K=P\'-x=P\' CH, - /' c1 I b
HC=S 'K
__ + NO,-( > - N 4 /
xx
reichende Menge erhalten. Die Bruttoformel deutet auf die Reaktion von 1 Mol Diazoniumsalz mit 4 Molekeln Diazomethan hin.
Mit 24-proc. Ausbeute bilden die blaBgelben Blattchen der Ver- bindung C,H,O,N, das Hauptprodukt unserer inversen Reaktion des p-Nitro-benzol-diazonium-chlorids mit Diazomethan. 2 Mol Di- azomethan sind hier rnit dem Diazoniumsalz unter Abgabe von 2 Mol Stickstoff und 1 Mol Chlorwasserstoff zusammengetreten. Die UV-Absorption ist in Cyclohexan-Losung der des p-Nitranilins Bhn- lich (Fig. 2 ) , liegt jedoch kurzwelliger ; das Infrarot-Spektrum 18) gibt mit dem Auftreten einer fur die Nitrilgruppe typischen Bande bei 4,52 p einen wertvollen Anhaltspunkt.Die saure und die alkali- sche Verseifung fuhren zu N-Methyl-p-nitranilin, was uns von den beiden Formelmoglichkeiten XXI und XXII letztere bevorzugen ld3t. Mit der Konstitution eines iY-Methyl-AT-(p-nitro-pheny1)-cyan-
NO,--/ \ - G C K \ / NO,--/ \-NH-CH,-c -s \ /
XXI XXII CH,1
anzids (XXII) stehen auch das Fehlen derNH-Bande im Infrarot, die Unfahigkeit zur Nitrosamin-Bildung sowie die mit der Eliminie- rung des Cyanid-Restes verbundene Acetylierung zu N-Acetyl-N- methyl-p-nitranilin in Einklang. Das in der Lit. schon beschriebene N-(p-Nitropheny1)-cyanamid 19) (XXV) liefert als relativ starke SBure bei der Behandlung mit Diazomethan glatt das N-Methyl-Derivat, identisch mit unserem Kupplungsprodukt.
Auch bei unserer Kupplungsreaktion mit Diazomethan durfte XXII uber XXV als Zwischenstufe entstehen. Wie aber kommt es
la) Herrn Dr. U. S c h i e d t , Tubingen, danken wir fur die Aufnahme der IR-
19) P. P i e r r o n , Bull. SOC. chim. de France [3] 33, 69 (1905). Absorption. Die st-Schwingung des Cyanamids selbst liegt bei 4,48 ,u.

212 H u i s g e n und K o c h
zur uberraschenden Ausbildung des Cyanamid-Systems Z Wenn man in Gegenwart einer hohenKonzentration am Chlor-anion das Hydra- ziti-chlorid XVII in guter Ausbeute erhalt, mu13 es auf der Stufe des Carbonium-ions XVI zur Verzweigung der Reaktionswege kommen. Wir nehmen an, da13 mit der Aufnahme des Anions eine innermole- k idare Unzlagerung dieses reaktiven Ions XVI konkurriert. Von der tautomeren Form XXIII des Carbonium-ions ausgehend, wandert der Nitranilino-Rest vom Stickstoff an den Kohlenstoff, wobei der Ubergangszustand mit dreigliedrigem Ring hier sogar im Gegensatz zu den meisten Carbonium-Umlagerungen mit einer ,,klassischen" Formel XXIV beschrieben werden kann. Mit der sich an die Um- lagerung anschliel3enden Abgabe eines Protons ist bereits die Cyan- amid-Gruppe fertig. Wir haben es hier mit einer neuartigeia Sextrtt- T'iiilagerung zu tun.
P -' - Cl-? SO,-- / \-N=X-CH, + + SO,- < FKH-N-CH + XVII
XVI /' XXIII \ /
/
XXIV
XXII --Ha +CH,N3 + NO,-< /' \-NH-C ?T
xxv
Es ist notwendig, noch eine zweite Moglichkeit zur Bildung von XXII in die Diskussion einzubeziehen. Das oben als Nebenprodukt isolierte 1-Nitrophenyl-tetrazol (XX) spaltet nach R. S t 0116 und F. H e n k e - S t a r k Z 0 ) in siedender Natronlauge Stickstoff ab und liefert glatt das N a t r i u m - s a l z des eben beschriebenen p - N i t r o - p h e n yl- c y a n a m i d s XXV. Fur diese Reaktion, sicherlich mit einer Protonen- abspsltung eingeleitet, ergibt sich zwangslaufig folgendes Schema :
HC- N, . \
:' N I' y: + 1-I :o
\ / --f KO,--/ )--N c N --f XXV SO2-< >-N--N,,
XX
Verschiedene Argumente sprecheii gegen einen genetischen Zu- samnienhang des Cyanamid-Derivats XXII und des Tetrazol-
"') .J. j m k t . Chem. 121 124, 290 (1930).

Die Kupplung arornatischer mit aliphatischen Diazoverbindungen 21 3
korpers X X bei unserer Diazoreaktion und lassen die Koinzidenz als zufallig erscheinen. Die 80-proc. Ausbeute am Hydrazid-chlorid XVII in Anwesenheit von Chlor-ion ist nur mit einer Azokupplung am Kohlenstoff des Diazomethans uber XV vereinbar, wiihrend eine Bildung von XXII aus X X eine mehr als 30-proc. Kupplung am Diazomethan-Stickstoff (S. 21 1) erfordern wurde. AuBerdem ist X X gegen Diazomethan stabil ; eine starkere Base als Diazomethan tritt aber im Reaktionsmedium gar nicht auf.
Eine unzweideutige Entscheidung zu Gunsten des Reaktions- weges uber die Carboniumumlagerung erzielten wir in Zusammen- arbeit mit K. Clusius und H. Hiirzeler2I) bei der Verfolgung des Reaktionsablaufs niit geeigneter Isotopen-Markierung. Wir iiber- fuhrten p-Nitranilin mit Athyl-nitrit, aus Natrium-nitrit mit 2,57 yo I5N bereitet, in das feste Diazoniumsalz und kuppelten rnit normalem , Jeichten" Diazomethan. Ein Blick auf die Formelfolge
xv ---r XVI -----f XXIII -+ XXIV -+ xxv --+ XXII
zeigt sofort, daB der markierte ,,auBere" Stickstoff des aromatischen Diazonium-ions im Zuge der Umlagerung in die Nitrilgruppe des Cyanamid-Derivats XXII gelangt. Nicht minder eindeutig ist, daB bei einer Bildung von XXII uber den Tetrazol-Abkommling X X das markierte Atom mit dem molekularen Stickstoff eliminiert wird, in XXII also gar nicht auftaucht.
Vorversuche hatten gezeigt, daB die saure Verseifung von XXII neben N-Methyl-p-nitranilin zu einer fast quantitativen Ausbeute an Ammoniak fuhrt, der Nitrilgruppe entstammend. Bei der Hydro- lyse des mit , ,schwerem" Diazonium-salz erhaltenen XXII wurden 97 yo des 15N-Gehaltes des eingesetzten schweren Natriumnitrits im freigesetztenAmmoniak aufgefunden; das N-Methyl-p-nitranilin zeigt das normale N-Isotopen-Verhaltnis. Fur die kleine Abweichung vom theoretischen N-Wert bedarf es nicht einmal der Annahme eines lsotopen- E f e k t s ; beim Abblasen des Ammoniaks aus schwach alkali- scher Losung 1aBt sich eine geringfiigige Verunreinigung mit ,,lei& tem" Methylamin, von der beginnenden nucleophilen Substitution des N-Methyl-p-nitranilins herriihrend, kaum vermeiden. Dieser Nachweis des markierten A t o m s in der C y a n g r u p p e gestattet es, den Bildungsweg uber X X auszuschlieBen, vermag dagegen die oben vorgeschlagene neue Sextett-Umlagerung zu stutzen.
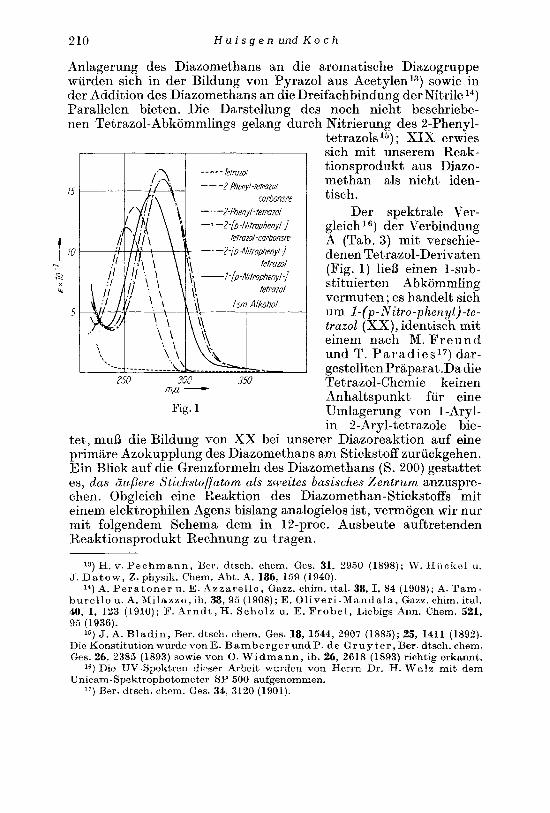
Es sei hier auf eine Besonderheit in der UV-Absorption des N-Methyl-p-nitra- nilins und des N-Methyl-p-nitrophenyl-cyanamids (XXII) hingewiesen, die zu- niichst den konstitutionellen Ruclrschlulj erschwerte. Bei Ahnlichkeit in Ex- tinktion und Habitus der Absorptionskurven tritt das Maximum beim N-Methyl- p-nitranilin in alkoholischer Losung bei 387 mp, beim Cyanamid-Abkommling
21) K. Clusius , H. H u r z e l e r , R. Huisgen u. H. J. K o c h , Nttturwissen- schaften 41, 213 (1954).
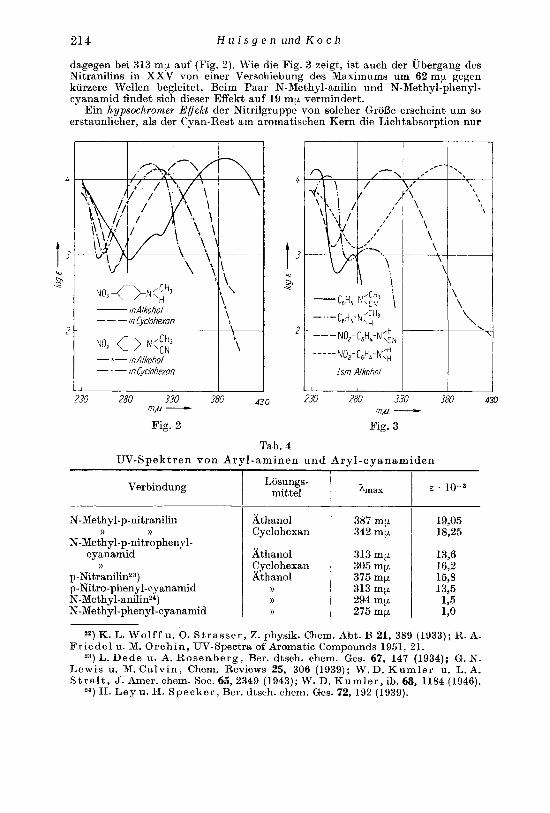
21 4 H u i s g e n und K o c h
dagegen bei 313 nip aiif (Fig. 2). U'ie die Fig. 3 zeigt, ist auch der Ubergang des Nitranilins in XXV von einer Verschiebung des Maximums um 6 2 m p gegen kurzere Wellen begleitet. Beim Paar N-Methyl-anilin und N-Methyl-phenyl- cyanamid findet sich dieser Effekt aiif 19 mp vermindert.
Ein hypsochromer Eflekt der Nitrilgruppe von solcher GroI3e erscheint um so erstaunlicher, als der Cyan-Rest am aromatischen Kern die Lichtabsorption nur
-x- in Alkohoj in Cycohexon
4
i 3
3
2
I 230 280 330 380 430 230 280
m,u - - Fig. 2 Fig. 3
Tab. 4 UV-Spektren v o n A r y l - a m i n e n u n d A r y l - c y a n a m i d e n
Verbindung Losungs- mittel
N-Methyl-p-nitranilin
S-Methyl-p-nitrophenyl- )) z
cyanamid ))
p-NitranilinZ3) p-Nitro-phenyl-cyanamid N-Methyl-anilinZ4) N-Methyl-phenyl-cyanamid
Athanol Cyclohexan
Athanol Cyclohexan Athano1
))
>)
))
h l a x
387 mp 342 mp
313 my 305 mp 375 mp 313 mp 294 mp 275 mp
E . 10-3
19,05 18,25
13,6 16,2 15,8 13,5
190 1,5
22) K. L. Wolff u. 0. S t r a s s e r , Z. physik. Chem. Abt. B 21, 389 (1933); R. A. F r i e d e l u. M. Orchin , W-Spectra of Aromatic Compounds 1951, 21.
23) L. D e d e u. A. R o s e n b e r g , Ber. dtsch. chem. Ges. 67, 147 (1934); G. N. Lewis u. M. Calv in , Chem. Reviews 25, 306 (1939); W.D. K u m l e r u. L. A. S t r a l t , J. Amer. chem. Soc. 65,2349 (1943); W. D. K u m l e r , ib. 68, 1184 (1946).
z4) H. L e y u . H. S p e c k e r , Ber. dtsch. chem. Ges. 72, 192 (1939).
Die Kupplung arornatischer rnit aliphatischen Diazoverbindungen 2 15
nenig oder gar im Sinne einer geringen Bathochromie zu beeinflussen pflegt, also ein sehr schwacher Chromophor zz) ist. Die Messung der Lichtabsorption in Cyclo- hexanlosung gibt den AufschluB, daB der spektrale Unterschied nur zum Teil auf den hypsochromen Effekt der Nitrilgruppe im Cyan-amid-System zuriick- geht. Die Cyanamid-Derivate zeigen eine wesentlich geringere Losungsmittel- abhangigkeit der Spektren als die Amine. Wie Fig. 2 und Tab. 4 zeigen, betragt der Unterschied in den Maxima des N-Methyl-p-nitranilins und X X I I hier nur mehr 37 mp. Die ungewohnlich starke Abhangigkeit der Lichtabsorption des p-Nitrani- lins vom Solvens wurde schon mehrfach diskutiertZs). Mit der Einfiihrung der Cyangruppe am Stickstoff, nicht aber durch Methylierung, geht diese Sonder- stellung des Nitranilins vollig verloren.
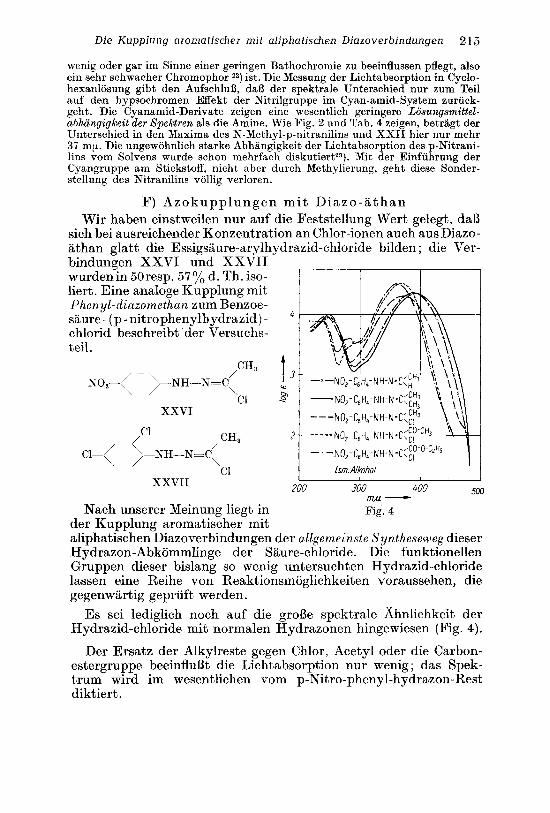
F) Azokupplungen mi t Diazo-a than Wir haben einstweilen nur auf die Feststellung Wert gelegt, daB
sich bei ausreichender Konzentration an Chlor-ionen auch aus Diazo- athan glatt die Essigsaure-arvlhydrazid-chloride bilden ; die Ver- bindunien XXVI und XXVII" wurdenin 50resp. 57% d. Th. iso- liert. Eine analoge Kupplung mit P h e n yl-diazomethan zum Benzoe- saure- (p-nitrophenylhydrazid) - chlorid beschreibt der Versuchs- teil .
,(-HA NO,--/ \-NH--Pu'=c
\ c1
\ / XXVI
,CH,
'C1
c1 /
Cl-/ ' \ /--NH--N=C
XXVII
Nach unserer Meinune lie& in
I Ism Alkohol
200 300 400 500 mN Fig. 4
der Kupplung aromati6heTmit aliphatischen Diazoverbindungen der allgemeinste S yntheseweg dieser Hydrazon-Abkommlinge der Saure-chloride. Die funktionellen Gruppen dieser bislang so wenig untersuchten Hydrazid-chloride lassen eine Reihe von Reaktionsmoglichkeiten voraussehen, die gegenwartig gepriift werden.
Es sei lediglich noch auf die grol3e spektrale Ahnlichkeit der Hydrazid-chloride mit normalen Hydrazonen hingewiesen (Fig. 4).
Der Ersatz der Alkylreste gegen Chlor, Acetyl oder die Carbon- estergruppe beeinfluat die Lichtabsorption nur wenig ; das Spek- trum wird im wesentlichen vom p-Nitro-phenyl-hydrazon-Rest diktiert .

216 H u i s g e n und K o c h
G) Die R e a k t i o n von Benzol -d iazonium-chlor id m i t D i a z o m e t h a n
Der komplexe Reaktionsverlauf und die Instabilitat des Form- phenylhydrazid-chlorids lassen es angezeigt erscheinen, die noch viele Unklarheiten bietende Kupplung der Stammkorper beider Reihen am SchluB zu besprechen.
Einleiten von gasformigem Diazomethan in die eisgekiihlte, me- tha,nolische Losung von Benzol-diazonium-chlorid fuhrt zu d. Th. an Phenylhydrazin-hydrochlorid. Bei weiteren Rupplungs- versuchen in Methanol-Ather konnten wir farblose Blattchen vom Schmp. 138-140O fassen, die sich beim Versuch des Umlosens in Phenylhydrazin-hydrochlorid umwandeln. In der in organischen Solventien loslichen Vorstufe vermuten wir das Form-phen ylh ydra- zid-chlorid (XXVIII), das rasch der S. 208 erwahnten Spaltungs- reaktion XVIII unterliegt.
191-192 150-151,5
169,5-170 O
179-180 O
Y l
XXVIII XXIX ‘x’ ‘ C6H5
Kicht basisch; IR: Konj. C=K Mit Saure + c ; IR: -CK, 3 NH Basisch Ful3n. 25 u. 26; IR : 2 NH
Als Nebenprodukte liefien sich in bescheidener Ausbeute u. a. zwei isomere Verbindungen C,,H,,N, fassen, die formal aus je 2 Mol der Komponenten unter Abgabe von 2 Mol Stickstoff und 2 Mol Chlor- wasserstoff entstehen. Wahrend das Isomere a (Tab. 5) in waBrigen Sauren nicht loslich ist, liefern die gelben Nadeln vom Schmp. 160 bis 151,5O (b von Tab. 5 ) mit Salzsaure ein leuchtendgelbes Hydro- chlorid, aus dem sich mit Alkali ein neues drittes Isomeres vom Schmp. 169,5-170 O freisetzen la&.
D. A. Bowack und A. L a p w o r t h 6 ) erzielten bei der Alkali- behandlung der Oxalester-hydrazid-chloride eine Kondensation zweier Molekeln unter Austritt von Chlorwasserstoff; die Reaktions- produkte wurden ohne Beweis als Diphenyl-dihydro-tetruzine an- gesprochen. Es lag nun nahe, in einer unserer isomeren Verbin- dungen C,,H,2N, das Ergebnis einer entsprechenden Reaktionsfolge
Tab. 5 Verbindungen C,,H,,N,
Bezeichn. I Schmp. I Bemerkungen I
a b
d 0
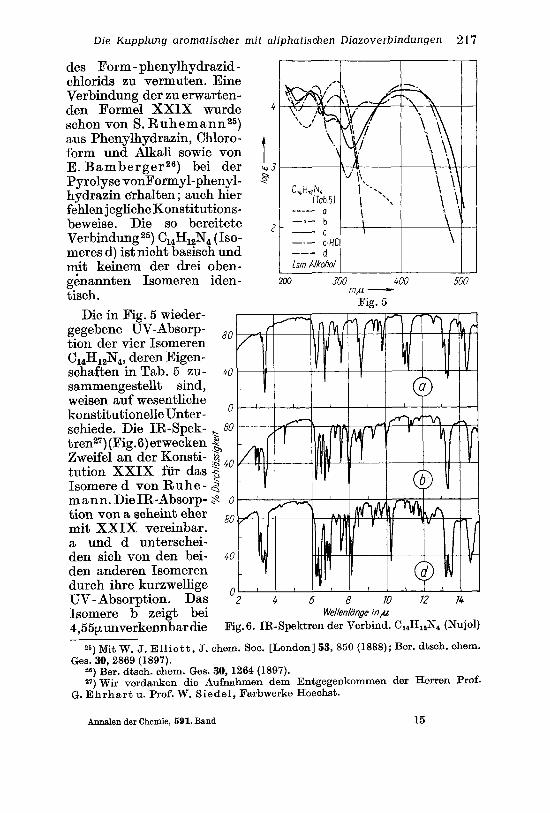
Die Kupplung aromatischer mit aliphatischen Diazoverbindungen 217
des Form - phenylhydrazid - chlorids zu vermuten. Eine Verbindung der zu erwarten- den Formel XXIX wurde 4 schon von S. Ruhemann25) aus Phenylhydrazin, Chloro- form und Alkali sowie von 1 E.BambergerZ6) bei der % 3 P yrolyse vonFormyl-phenyl- p hydrazin eyhalten; auch hier fehlen jegliche Konstitutions- beweise. Die so bereitete Verbindung 25) C14H184 (Iso- meres d) ist nicht basisch und mit keinem der drei oben- genannten Isomeren iden- tisch.
-.
\, ‘*\
\
500
Die in Fig. 5 wieder- gegebene UV-Absorp- tion der vier Isomeren C14H1&!4, deren Eigen- schaften in Tab. 5 zu- sammengestellt sind,
0 weisen auf wesentliche konstitutionelle Unter - schiede. Die IR-Spek- t-, 80 tren27) (Fig.6)erwecken % Zweifel an der Konsti- .$ 4o tution XXIX fur das 2 Isomere d von Ru he - 3 mann. DieIR-Absorp- S o tion von a scheint eher 8o mit XXIX vereinbar. a und d unterschei- den sich von den bei- 40 den anderen Isomeren durch ihre kurzwellige UV-Absorption. Das ‘2 4 6 8 JO 72 74 Isomere b zeigt bei Wellenlange i n p
4,55punverkennbar&e
Ges. 30, 2869 (1897).
40
Fig. 6. IR-Spektren der Verbind. C14H12N4 (Nujol)
25) Mit W. J. El l iot t , J. chem. SOC. [London] 53, 850 (1888); Ber. dtsch. chem.
28) Ber. dtsch. chem. Ges. 30, 1264 (1897). *’) Wir verdanken die Aufnahmen dem Entgegenkommen der Herren Prof.
G. Ehrhart u. Prof. W. Siedel, Farbwerke Hoechst.
Annalen der Chemie, 691. Band 15

21 8 H u i s g e n und K o c h
Valenzschwingung der Nitrilgruppe, daneben vermutlich drei ver- schiedene Typen von NH-Bindungen. Offensichtlich geht der Bildung von b die S. 21 2 beschriebene Sextett-Umlagerung zum Cyanamid voraus. Die Bruttoformel wiirde einem Dimeren des N-Phenyl-cyan- amids entsprechen ; allerdings ist bislang nur die Trimerisation dieses Cyanamid-Abkommlings zum Triphenylmelamin bekannt 28).
Die Umsetzung des Benzol-diazonium-chlorids mit Diazomethan bedarf weiterer Bearbeitung.
Der Deutschen Forschungsgemeinschaft schulden wir fur die Unterstutzung der Arbeit mit einer Sachbeihilfe Dank. Herrn H. Geyer und Frl. D. G r a f dauken wir fur die Ausfuhrung der Mikro-Analysen.
Beschreibung der Versuche
Die Aryl-diazonium-salze wurden wie ublich aus den Amin-hydrochloriden mit Athyl-nitrit in Eisessig oder abs. Alkohol bereitet und mit Ather oder, wenn Kristallisationsschwierigkeiten auftraten, mit Tetrahydrofuran vorsichtig aus- gefallt. Den Kupplungen mit aliphatischen Diazoverbindungen ging jeweils eine Gehaltsbestimmung des Aryl-diazonium-salzes voraus, die uber die Azokupplung mit p-Naphthol gravimetrisch oder photometrisch vorgenommen wurde.
Diazo-essigester a ls Kupplungskomponente Oxalsaure-athylester-(p-nitro-p,henyl)-hydrazid-chlorid (IV)
Eine Losung von 408 mg p-Nitro-benzol-diazonium-chlorid (2,20 mMol) in G ccm Methanol wurden unter Eiskuhlung mit 0,28 ccm Diazo-essigsaure-lthyl- ester (2,64 mMol) versetzt. Nach wenigen Minuten Stehen bei 0 0 wurde die Gas- entwicklung deutlich; nach etwa 10 Min. setzte die Kristallisation des blaagelben Hydrazid-chlorids ein. Innerhalb 4 Stunden wurden 46 ccm Stickstoff (red.) ent- wickelt, was 2,O mMol entspricht. Nach 15 Stunden wurde das zum Kristallbrei erstarrte Reaktionsgemisch abgesaugt und rnit wenig Methanol gewaschen. Es wurden 369 mg vom Schmp. 190--192° und aus der Mutterlauge weitere 13 mg vom Schmp. 185-189O isoliert. Ausbeute 64% d. Th. Umkristallisieren aus Methanol oder Athano1 fuhrte zu blaagelben, verfilzten Nadeln Tom Schmp. 191-193 ". Die Verbindung ist unzersetzt im Hochvakuum fluchtig.
CloHloO~N,C1 (271,66) Ber. C 44,21 H 3,71 N 15,47 C1 13,05 Gef. D 44,30 N 3,93 )) 15,11 1) 12,40
Das Hydrazid-chlorid reduzierte ammoniakalische Silberlosung nicht ; Er- warmen mit Salzsaure auf 100° verafidert den Schmp. nicht. Mit Kaliumhydroxyd in Methanol tritt Rotfarbung auf; nur wenn man sofort ansauert, lafit sich das unveranderte Hydrazid-chlorid wieder fassen.
Offensichtlich kommt rnit steigender Reaktionstemperatur die Solvolyse des Aryl-diazonium-salzes im Vergleich mit der Azokupplung starker zum Zug. Ein wie oben, nur bei 16", durchgefuhrter Kupplungsversuch lieferte 57% d. Th. am Hydrazid-chlorid; ein 40°-Versuch erbrachte gar nur 15% d. Th. Die Konkurrenz
28) F. A r n d t , Liebigs Ann. Chem. 384, 350 (1911).

Die K u p p l u n g aromat ischer rnit al iphat ischen D i a z o v e r b i n d u n g e n 21 9
der bimolekularen Azokupplung und der unimolekularen Methanolyse 1aBt sich durch einen UberschuB Diazo-essigester zu Gunsten der Kupplung beeinflussen. So wurden etwa 1,34 g aktives p-Nitro-benzol-diazonium-chlorid in 16 ccm eis- kaltem Methanol mit 1,54 ccm Diazoessigester (100% OberschuD) versetzt. Die auf das Aryl-diazonium-salz bezogene Ausbeute betrug 69% d. Th. Die Aus- fiihrung der Azokupplung in mit Lithium-chlorid gesattigtem Methanol erbrachte keine Ausbeute-Steigerung.
Synthese des Vergleiehspraparatsa). 1,30 g p-Nitro-benzol-diazonium-chlorid wurden in 16 ccm Eiswasser gelost und langsam rnit einer Losung von 1,08 g a-Chlor-acetessigester und 0,86 g Natriumacetat in 20 ccm Athanol versetzt. Nach 5 Stunden Stehen bei Oo wurde die Kristallausscheidung durch Wasser- zusatz vervollstandigt. Wir erhielten 1,55 g am gelborange gefarbten Rohprodukt ; nach mehrfachem Umlosen unter Zusatz von Kohle lag der Schmp. bei 192-1930. Der Mischschmp. mit dem aus Diazo-essigester erbaltenen Produkt zeigte keine Depression.
Oxalester-p-nitro-phen yl-h ydrazid- bromid 1,0 g p-Nitranilin-hydrobromid wurde in 15 ccm abs. Alkohol bei 0 0 mit
0,8 ccm Athylnitrit diazotiert und ohne Ausfallung unmittelbar mit 0,s ccm Diazo-essigester gekuppelt. Die nach 4 Stunden abgeschiedenen hellbraunen Kristalle wurden abgesaugt und lieferten beim Umlosen aus Athanol oder Benzol zwei im Verhaltnis der Dimorphie stehende Kristallarten, namlich orangegelbe Polyeder neben blal3gelben Nadeln. Beide Formen zeigen gleiches Schmelz- verhalten. Schmp. 200-201 O.
C,,H,,O,N,Br (316,12) Ber. C 37,99 H 3,19 Gef. )) 37,93 )) 3,25
Mit Alkali tritt die obenerwahnte Rotfarbung auf. Beim Umgang mit dem Hydrazid-bromid ist Vorsicht am Platz; bei dem einen von uns verursachte es ein Hautekzem.
Beim Versuch der Kupplung des Diazo-essigesters mit p-Nitro-benzol-dia- zonium-fluoborat in Methanol, Acetonitril oder Pyridin entwich der gesamte Stickstoff der im OberschuB befindlichen aliphatischen Diazoverbindung. Aus der Reaktionslosung konnte jeweils das unveranderte Diazonium-fluoborat rnit Zers.- Punkt 141-1420 zuriickgewonnen werden. Es handelt sich also um eine kata- lytische Zersetzung des Diazo-essigesters, vielleicht von Spuren Borfluorid aus- gelost, die sich mit dem Fluoborat im Gleichgewicht befinden.
Oxalester-(p-brom- phen y l ) -h ydrazid-ehlorid 1,48 g aktives p-Brom-benzol-diazonium-chlorid (7,l mMol) wurden in 15 ccm
eiskaltem Methanol mit 1,35 ccm Diazo-essigester (123 mMol) zur Reaktion gebracht. Nach Stehen iiber Nacht wurde abgesaugt, mit Methanol gewaschen und getrocknet: 1,26 g cremefarbene SpieDe vom Schmp. 160-161 0; aus der Mutterlauge weitere 80 mg der gleichen Substanz. Das in 62% d. Th. erhaltene Hydrazid-chlorid kam aus Methanol in farblosen Lanzetten des oben angegebenen Schmo.
C,,H,,O,N,BrCl (305,57) Ber. C 39,30 H 3,30 Gef. )) 39,33 )) 3,47
Oxalester- (0-nitro- phen yl) -1Lydrazid-chlorid 1,02 g reines o-Nitro-benzol-diazonium-chlorid wurden wie oben in eiskaltern
Methanol mit der doppelt-molaren Menge Diazo-essigester umgesetzt, wobei nach einer Stunde die Abscheidung gelber, watteartig verfilzter Nadeln begann.
15*

i d 990 H u i s g e n und K o c h
Ausbeute 785 mg = 52% d. Th. Each Umlosen aus Methanol unter Kohlezusatz kanariengelbe Yadeln vom Schmp. 117,8--118°.
VloHl0O,N,CI (271,66) Ber. C 44,21 H 3,71 Gef. )) 44,52 )) 3,97
Oxulester-(p-c7Llor-phen?jl) -h ydrazid-chlorid Das mit 48-proc. Ausbeute isolierte Kupplungsprodukt des p-Chlor-benzol-
diazonium-chlorids bildet farblose Nadeln, die bei 146-1470 schmelzen. C,oHlo02N2Cl, (261,l l) Ber. C 46,OO H 3,86
Gef. )) 46,05 n 3,74
Oxalester-(2,4-dichlor-phen y1)-h ydruzid-chlorid Das trockene Diazonium-chlorid mit 1,36 g aktivem Gehalt wurde mit zwei
Aquivalenten Diazo-essigester wie iiblich in Methanol zur Reaktion gebracht. Nach Stehen iiber Nacht unter Aufarbeitung der Mutterlauge 748 mg, die bei 90-91 schmelzen. Aus Methanol elfenbeinfarbene Lanzetten vom Schmp. 92,5-93,5'.
C,,H,O,N,CI, (295,56) Ber. C 40,64 H 3,07 Gef. )) 41,07 )) 3,43
Oxalsaure-ath ylester-phenylh ydrazid-ehlorid Die bei 15O vorgenommene Reaktion von 1,70 g reinem Benzol-diazonium-
chlorid (12,l mMol) rnit 3,14 ccm Diazo-essigester (30 mMol) in 15 ccm Methanol lieferte innerhalb 14 Stunden 19,2 mMol Stickstoff. Die orangefarbene Losung roch nach Anisol, dem Ergebnis der Solvolyse. Kach dem Abziehen des Losungs- mittels i. V. ging iiberschiissiger Diazo-essigester bei 50°/12 iiber. Nach Auf- nehmen in Benzol wurde nacheinander mit 2n-Salzsaure, n-Natronlauge und Wasser ausgeschuttelt. Bei der Destillation des Riickstandes der Benzolphase ging gegen 170°/15 ein gelbes 01 iiber, das kristallin erstarrte. Nach Umlosen ails wenig Methanol 0,43 g bladgelbe Nadeln vom Schmp. 77-790. -
C,oH,102N2Cl (226,66) Ber. C 52,99 H 4,89 N 12,36 Gef. )) 52,99 )) 4,70 )) 12,15
Kupplungsversuche des Diazo-essigesters rnit p-Naphthalin-diazonium-chlorid, p-Methoxy-benzol- uad m-Methyl-benzol-diazonium-chlorid fielen negativ aus. Die langsame Gasentwicklung folgte dem Tempo der Solvolyse, der sich dann die Reaktion des Diazo-essigesters rnit dem freigesetzten Chlorwasserstoff anschlol3. Aus dem Versuch rnit dem Diazonium-salz der Naphthalin-Reihe wurde lediglich Serolin isoliert.
Azokupplungen m i t D iazo-ke tonen p-Nitro-benzol -diazonium-chlor id u n d D i a z o - a c e t o n
Aus einer eiskalten Losung von 1,0 g Diazonium-chlorid und 0,45 g frisch destilliertem D i a z o - a c e t ~ n ~ ~ ) in 20 ccm Methanol wurden beim Stehen iiber Nacht 152 ccm N, freigesetzt (6,s mMol). Nach dem Einengen kamen aus der braunen Losung 550 mg blal3gelber Nadeln vom Schmp. 219-224O (420/, d. Th.). Sach Umlosen aus Methanol wurde der Schmp. des Brenztraubensuure-(io-nitro- phenyl-)hl/drazid-chlorids bei 224-224,5 O gefunden.
C9H,0,hT,CI (241,63) Ber. C 44,73 H 3,34 Gef, )) 45,25 )) 3,32
29) F. A r n d t u. J. Amende, Ber. chch. chem. Ges. 61, 1124 (1928).

Die Kupplung aromatischer mit aljphatischen Diazoverbindungen
2,4-Dichlor -benzol -d iazonium-chlor id u n d D i a z o - a c e t o n
Aus 3,O g Diazonium-chlorid mit 13,5 mMol aktivem Gehalt und 1,28 g Diazo- aceton (15,2 mMol) in 10 ccm Methanol bei O o wurden innerhalb 3 Min. 15,5 mMol N, freigesetzt. Infolge der kleinen Losungsmittelmenge stieg die Losungs- temperatur trotz Eiskiihlung auf 400. Nach Absaugen und Waschen des Kristall- kuchens sowie Aufarbeitung der Mutterlauge wurden 2,70 g vom Schmp. 115 bis 116O erhalten. Aus Methanol farblose Nadeln, die bei 116-117" schmelzen. Das Praparat wurde durch Mischprobe mit einem nach C. Biilow und P. NeberlO) bereiteten Brenztraubensaure-(2,4-dichlor-phen2/1)-hydrazid-chlor~d (VII) iden- tifiziert. Auch das Vergleichspraparat schmolz bei 116-117 O; die Lit.-Angabelo) von 125O diirfte auf einem Versehen beruhen.
221
p-Ni t ro -benzo l -d i azon ium-ch lo r id u n d D i a z o - a c e t o p h e n o n @ )
Auf die Zugabe von 0,80 g reinem Diazo-acetophenon2@) zur kalten methano- lischen Losung von 1,0 g des aromatischen Diazonium-salzes setzte sofort die Entwicklung des Stickstoffs ein. Nach mehrstiindigem Stehen wurde der blaB- gelbe Kristallbrei abgesaugt und die Mutterla-uge eingeengt. Ausbeute 1,30 g verfilzter Nadeln = 79% d. Th. Nach mehrfachem Umlosen aus Aceton schmilzt das Benzoyl-ameisensaure-(p-nitro-phen~l)-hydrazid-clt.lorid (V) bei 244-245O u. Zers.
C14Hl,,03N3Cl (303,70) Ber. C 55,36 H 3,32 Gef. )) 55,25 )) 3,40
Die Substanz zeigt in konz. Schwefelsaure eine leuchtend orangerote Halo- chromie; beim Verdiinnen rnit Wasscr f&llt das unveranderte Hydrazid-chlorid wieder aus.
Ein Vergleichspraparat wurde durch Chlorolyse des Kupplungsproduktes aus p-Xitro-benzol-diazonium-salz und Dibenzoyl-methan erhalten. Dazu wurden 0,5 g des Diphenyl-triketon-p-nitro-phenyl-hydrazons und 0,2 g wasserfreies Katriumacetat in Eisessig gelost und bei Raumtemp. mit gasformigem Chlor behandelt. Nach Verdiinncn mit Wasser schied sich eine orangegelbe Verbindung aus, die nach Abpressen auf Ton rnit Ather digeriert wurde. Nach mehrfachem Umlosen aus Alkohol blal3gelbe Nadeln, die bei 243-244O schmolzen und mit dem Kupplungsprodukt des Diazo-acetophenons keine Depression gaben. Die Ausbeute betrug hier 30% d. Th. Auch aus dem p-Nitro-benzol-azo-benzoylaceton wurde das gleiche Hydrazid-chlorid bei der Behandlung mit Chlor in fast derselben Ausbeute erhalten.
Das p-Nitro-benzol-diazonium-bromid steht dem Chlorid im Kupplungs- vermogen gegeniiber Diazo-acetophenon nicht nach. Unter entsprechenden Be- dingungen wurden blaBgelbe Nadeln des Hydrazid-bromids vom Schmp. 241 bis 242O 11. Zers. mit 50-proc. Ausbeute erhalten.
C,,H,,O,K,Br (348,16) Ber. C 48,29 H 2,90 N 12,07 Gef. 1) 48,92 )) 3,03 )) 11,75
Das hier vorliegende Benzoyl-ameisensaure-(p-nitro-phen y1)-hydrazid-bromid wurde auch aus dem Diphenyl-triketon-p-nitrophenyl-hydrazon erhalten. 0,5 g dieses Hydrazons wurden in Eisessig in Gegenwart von Natriumacetat rnit 0,30 g Brom behandelt. Nach einstiindigem Erhitzen zum Sieden wurde mit Wasser- Benzol aufgearbeitet. Neben unverkndertem Ausgangsmaterial vom Schmp. 171 bis 172O wurden 86 mg einer schwerloslichen Verbindung vom Schmp. 238-241O isoliert, die nach Umlosen aus Alkohol bei 240-242O schmolzen.

222 H u i s g e n und K o c h
p - C h l o r - b e n z o 1 - d i a z o n i u m - c h 1 or i d u n d D i a z o - a c e t o p h e n o n Wahrend das Kupplungsprodukt des unsubstituierten Benzol-diazonium-
chlorids mit Diazo-acetophenon nicht gefaBt werden konnte, wurde das Benzoyl- ameisensaure-p-cWlor-phen yl-h ydrazid-chlorid in blaBgelben, verfilzten Nadeln er- halten, die nach Umlosen aus Athanol bei 194-196O schmelzen. Rote Halo- chromie in Schwefelsaure.
C,,Hl0ON,CI, (293,15) Ber. C 57,36 H 3,44 N 9,56 Gef. )) 57,33 )) 3,92 )) 9,20
Auch hier konnte ein Vergleichspraparat durch Chlorierung des Kupplungs- produktes von p-Chlor-benzol-diazonium-salz mit Dibenzoylmethan synthetisiert werden; neben Harzen fie1 eine geringe Menge des Hydrazid-chlorids rnit Schmp. 193-194O an.
K u p p l u n g s v e r s u c h e m i t p-Nitro-diazo-acetophenon Eine eiskalte Losung von 1,0 g p-Nitro-diazo-acet~phenon~~) in 30 ccm Metha-
nol wurde mit 0,97 g p-Nitro-benzol-diazonium-chlorid versetzt. Nach 16 Stunden hatte sich genau die dem aromatischen Diazonium-salz entsprechende Menge Stickstoff entwickelt. Aus der gelborange gefarbten Losung wurde das Diazo- keton mit dem Schmp. 94-95 O unverandert zuriickerhalten.
Auch mit 2,4-Dinitro-benzol-diazonium-sulfat lieB sich die Kupplung nicht erzwingen; wieder konnte das Diazo-keton aus der Zerfallslosung des aromati- schen Diazoniumsalzes zuriick isoliert werden. Das gleiche negative Ergebnis wurde bei den Kupplungsversuchen des bei 90-91 O schmelzenden p-Nitro-diazo- propiophenons mit p-Brom- und p-Nitro-benzol-diazonium-chlorid sowie mit 2,4-Dinitro-benzol-diazonium-sulfat erzielt. Ebenso wurde Dibenzoyl-diazo- methan31) aus Kupplungsversuchen mit dem p-Nitro- und 2,4-Dinitro-diazonium- salz unverandert wiedergewonnen.
Azokupplungen mit D i a r y l - d i a z o m e t h a n Benzo l -d i azon ium-ch lo r id u n d D i p h e n y l - d i a z o m e t h a n
Beim Versetzen einer eiskalten Losung von 1,0 g Benzol-diazonium-chlorid (7,l mMol) mit einer Losung von 1,40 g Diphenyl-dia~o-methan~~) (7,2 mMol) in 25 ccm Methanol entwich innerhalb weniger Minuten etwas mehr als die berechnete Menge Stickstoff. Nach dem Einengen schieden sich aus der dunkel- roten Losung 1,50 g gelber Polyeder vom Schmp. 97-990 aus. Nach Umlosen aus Methanol Schmp. 100,5--101,5°.
C,,H,,ON, (302,36) Ber. C 79,44 H 6,OO Gef. )) 79,33 )) 6,02
Die Ausbeute am cr-Benzol-azo-benzhydryl-methyl-uther (XI, Ar=C,H,) betrug 7076 d. Th.
In einem weiteren Kupplungsversuch wurde A’thanol als Losungsmittel ge- wahlt. Wenn die Solvolyse des intermediaren Halogenids sehr rasch erfolgt, sollte ein Pyridin-Zusatz das noch nicht umgesetzte Diphenyl-diazomethan vor dem Angriff dcs Halogen-Wasserstoffs schiitzen. Es ergab sich jedoch keine Ausbeute- steigerung.
30) B. E i s t e r t , Angew. Chem. 61, 186 (1949). 31) H. W i e l a n d u. S. Bloch , Ber. dtsch. chem. Ges. 37, 2526 (1904); 39, 1488
32) H. S t a u d i n g e r , E. A n t h e s u. F. P f c n n i n g e r , Bcr. dtsch. chem. Ges. 49, (1 906).
1932 (1916); Org. Syntheses 24, 53.

Die Kupplung arornatischer mit aliphatischen Diazoverbindungen 223
4,O g Diphenyl-diazomethan wurden in 50 ccm Athanol und 1,7 g Pyridin gelost und bei -5O mit 3 g trockenem Benzol-diazonium-chlorid (21,O Mol aktiver Gehalt) umgesetzt. Nach 45 Min. begann die Abscheidung hellgelber Kristalle aus der rotbraunen Liisung. Nach Stehen iiber Nacht wurde der Kristall- brei abgesaugt: 3,72 g (57% d. Th.) vom Schmp. 74-77O. Der a-Benzol-azo- henzhydryl-athyl-ather ist leicht loslich in den Alkoholen, Benzol und Essigester. Nach mehrfachem Umkristallisieren aus Petrolather liegt der Schmp. bei 80,5 bis 82O.
CziHzoONz (316,39) Rer. C 79,71 H 6,37 N 8,86 Gef. )) 79,78 )) 6,43 )) 9,24
Bei der Aufarbeitung der Mutterlauge wurde eine bescheidene Menge Tetra- phenyl-ketazin vom Schmp. 161-162 erhalten. Bei einem gleichartigen Kupp- lungsversuch wurde das Pyridin durch 2, l g Kaliumacetat ausgetauscht; die Ausbeute an der Benzol-azo-Verbindung betrug 51 % d. Th.
p -Ni t ro -benzo l -d i azon ium-ch lo r id u n d D i p h e n y l - d i a z o m e t h a n Beim Vereinigen der eiskalten Losungen von 9,0 g aktivem Diazonium-salz
(49 mMol) in 70 ccm Methanol und 9,4 g Diphenyl-diazomethan (48 mMol) in 15 ccm Methanol fuhrte die Reaktionswarme zu erheblicher Temperatursteigerung und stiirmischer Gasentwicklung. Schon nach wenigen Minuten begann die Kristallisation des Kupptungsprodukts, die durch 12-stundiges Stehen im Eis- schrank vervollstandigt wurde. Aus der weinroten Losung kamen 9,45 g rot- orange gefilrbte Tafeln, was einer 56-proc. Ausbeute a n ct-(p-Nitro-benzol-am)- henzhydryl-methylather entspricht. Der Rohschmp. von 124-126O stieg nach dem Umlosen aus Methanol auf 134-135O.
CzoHl,03N3 (347,36) Ber. C 69,15 H 4,93 N 12,lO CH,O 8,93 Gef. )) 69,26 )) 4,95 )) 11',96 D 9,15
p-Chlorbenzol -d iazonium-chlor id u n d D i p h e n y l - d i a z o m e t h a n Aquivalente Mengen der beiden Komponenten wurden in methanolischer
Losung bei -50 vereinigt. Aus 16,9 g Diphenyl-diazo-methan wurden 15,5 g gelbe rhombische Prismen erhalten (54% d. Th.). Nach mehrfachem Umlosen aus Aceton-Petroltither sowie aus Essigester wurde der Schmp. des a-(p-Chlor-benzol- uzo)-benzhydryl-methylathers bei 139-140° gefunden.
CzoH,,ONzCl (336,81) Ber. C 71,32 H 5,09 Gef. )) 71,lO )) 5,20
Bei der Reaktion von Diphenyl-diazomethan mit Nitroso-p-chlor-acetanil~d in benzolischer Losung, die zur Isomerisierung des Nitroso-acylamins 40 Stunden bei Raumtemp. stand, konnte kein kristallisiertes Kupplungsprodukt isoliert werden.
p -Brom-benzo l -d i azon ium-ch lo r id u n d D i p h e n y l - d i a z o m e t h a n Bei der heftigen Reaktion von 7,2 mMol des Diazonium-salzes und 7,O mMol
Diphenyl-diazomethan in 55 ccm Methanol wurden 8,3 mMol Stickstoff ent- wickelt. Nach dem Einengen kristallisierten aus der gelbbraunen Losung 1,75 g des gelben a(p-Brom-benzol-azo)-benzhydryl-methylathers (66% d. Th.), Schmp. 136-137O (aus Methanol).
C,,Hl,ONzBr (381,27) Ber. C 63,OO H 4,49 Gef. )) 62,72 )) 4,47

224 H u i s g e n und K o c h
p - X a p 11 t h a l i n - d i a z o n i u m - c h l o r i d u n d D i p he n y 1 - d i a z o m e t h a n Die wie iiblich durchgefiihrte Kupplung von 2,9 mMol der aromatischen und
3,6 mMol der aliphatischen Diazoverbindung in Methanol ergab unter Frei- setzung von 3,3 mMol Stickstoff 0,65 g braunorange gefarbter Kristalle (6576 d. Th.). Nach Umlosen aus Methanol schmolz der cc(P-Naphthalin-azo)-benzhydr?/l- methyliither bei 155,5--156O.
Cz,HzoONz (352,42) Ber. C 81,79 H 5 , i 2 Gef. )) 81,46 )) 5,70
p - B r o m - b e n z o 1 - d i a z o n i u m - c h 1 or i d u n d D i a z o - f lu o r en Die Iteaktion mit Dia~o-f luoren~~) in .Methanol wird wie oben durchgefiilirt
und lief bei 0" unter Entwicklung von 1,4 Aquivalenten Stickstoff ab. Aus kleineni Volumen Methanol kamen gelbe Polyeder vom Schmp. 112-114O in 60-proc. Ausbeute. Nach dem Umlosen liegt der Schmp. des 9- (p -Bro in -benzo l -azo ) -~uo~~~i - 9-methylathers (XIII, Ar=Br-C,H,) bei 115--115,5O.
C,oH,,ON,Br (379,25) Ber. C 63,33 H 4,OO Gef. )) 63,13 )) 4,18
p - N i t r o - b e n z o l - d i a z o n i u m - c h l o r i d u n d D i a z o - f l u o r e n 1,0 g Diazonium-salz in 35 ccm Methanol wurde bei 00 mit der Losung Ton
1,04 g Diazo-fluoren in 80 ccm Methanol und 20 ccm Ather vereinigt. Die Stick- stoffentwicklung war nach 30 Min. abgeschlossen. Unter Aufarbeitung der Mutter- lauge wurden 1,51 g gelber Nadelchen vom Schmp. 128-129O isoliert, einer Ausbeute von 81% d. Th. entsprechend. Nach Umlosen aus Methanol Schmp. des 9-(p-Nitro-benzol-azo)-9-methoxy-fluorens bei 138,4-13g0.
C,,H,,O,K, (345,34) Ber. C 69,55 H 4,38 Gef. )) 69,52 )) 4,36
H y d r i e r u n g d e s 0: (p ~ B r o m - b e n z o 1 - a z 0 ) ~ b e n z h y d r y 1 - m e t h y 1 a t h e r s 500 mg der Azoverbindung wurden in 50 ccm Methanol mit 30 mg Platinoxyd
katalytisch hydriert, bis nach 6 Stunden die Wasserstoffaufnahme trager wurde. Aus der eingeengten Losung wurden neben einer bescheidenen Menge farbloser, bei 243-245 0 schmelzender Kristalle nahezu 0,3 g farbloser Nadeln erhalten, die nach Umlosen aus Methanol bei 116,5-117,5 schmolzen.
C,,H,,N,Br (351,24) Ber. C 64,9i H 4,30 ?\' 7,98 Gef. )) 65,43 )) 4,24 )) 7,73
Es handelt sich um Benzophenon-p-brom-phen yl-hydrazmz, in der Mischprobe identisch mit einem authent. Prapnrat3*).
Bei einem zweiten, in Gegenwart einer grol3eren Menge Platinoxyd durch- gefuhrten Hydrieransatz wurde in guter Ausbeute Benzophenon-phenyl-hydrami isoliert, was einer Eliminierung des Bromatoms entspricht.
M e a k t i o n d e s a( p - N i t r o - b e n z o l - a z o ) - b e n z h y d r y 1 - m e t h y I a t h e r s m i t M i n e r a l s a u r e
0,3 g des Azo-athers wurden in 5 ccm Methanol mit 0, l ccm konz. Salzsaure 10 Stunden riickfluBgekocht. Nach Aufarbeitung rnit Wasser-Ather konnte neben verschmiertem Material 75 mg Substanz vom Schmp. 136-140 O gefaBt werden,
H. S t a u d i n g e r u. A. Gaulo , Ber. dtsch. chem. Ges. 49, 1951 (1916); A . Schonborg , W. I. Awad u. N. L a t i f , J. chem. SOC. [London] 1951. 1368.
3 4 ) L. Michael is , Rer. dtsch. chem. Grs. 26, 2190 (1893).

Die Kupplung aromatischer mit aliphatischen Diazoverbindungen 226
die sich nach Umlosen in derMischprobe m it Benzophenon-p-nitro-phenyE-hydrazon identisch erwiesen. Feine gelbe Nadeln vom Schmp. 151-152 O .
C i ~ H x N 3 0 ~ (317,331 Ber. C 71,91 H 4,76 Gef. )) 71,75 )) 4,85
Beim Versucli, den Azo-methyl-ather in siedendem k h a n o l in Gegenwart eiiier katalytischen Menge Schwefelsaure umzuathern, wurde das beschriebene p-Nitro-phenyl-hydrazon als einziges definiertes Produkt gefal3t. Schon die Ein- wirkung von gasformigem Chlorwasserstoff auf die kalte benzolische Losung fiihrte zur raschen Veranderung des Azoathers. Etwas sorgfaltiger wurde ein Zer- setzungsversuch mit konz. Schwefelsaure aufgearbeitet.
1,0 g Azo-iither losten sich beim UbergieBen mit 2 ccm konz. Schwefelsaure unter Gasentwicklung und Selbsterwarmung auf 50°. Nach 15-stundigem Stehen wurde mit Eis behandelt, in Benzol aufgenommen und mehrfach mit verd. Schwefelsaure ausgezogen. Die sauren Extrakte ergaben beim Alkalischmachen, Ausathern und Umkristallisieren des kherriickstandes aus Benzol-Petrolather eine bescheidene Menge gelber Nadeln vom Schmp. 145-1460, die sich als p - Y i t r a n i l i n erwiesen.
Die Neutralprodukte der Benzolphase wurden im Mikrokolbchen im Hoch- vakuum destilliert. Der gegen 70 iibergehende Anteil roch nach Nitrobenzol. Nur mit Reduktion und Azokupplung konnte dieser Stoff wahrscheinlich gemacht werden,nicht aber mit der Weiternitrierung zum m-Dinitro-benzol, dessen Kristalli- sation offensichtlich durch Begleitstoffe verhindert wurde. Aus der bis 1200/0,001 iibergehenden Fraktion schieden sich beim Erkalten farblose Blattchen vom Schmp. 69-70° aus, die nicht identifiziert werden konnten. Der rotbraune, olige Anteil dieser Fraktion wurde in Aceton aufgenommen und auf -70° gekiihlt. Die nach kurzer Zeit auskristallisierenden blal3gelben Nadeln losten sich bei Raum- temperatur wieder, wurden daher mit der Eintauchfritte aus der tiefgekiihlten Losung isoliert. Nach 2-maliger Wiederholung des Prozesses erhielt man 160 mg farbloser Kristalle, die nach Umlosen aus Petrolather bei 4 4 , 5 4 6 O schmolzen und mit Benzophenon keine Depression ergaben. Auch das 2,4-Dinitro-phenyl- hydrazon ergab mit einem bei 238,50 schmelzenden Vergleichspraparat keine Sc1imp.-Erniedrigung.
Die zwischen 120 und 2200/0,001 iibergehende Fraktion kristallisierte teilweise beim Erkalten. Beim Digerieren mit Petrolather blieben gelbe Nadeln zuriick, die nach mehrfachem Umlosen aus Methanol bei 153-154O schmolzen und sich mit Benzophenon-p-nitro-phenyl-hydrazon in der Mischprobe identisch erwiesen.
Kupp lung von p-Nitro-benzol-diazonium-chlorid m i t Phenyl-diazo -me t han
1,7 g Diazonium-salz in 12,5 ccm mit Lithiumchlorid gesattigtem Methanol wurden bei - 5 O mit der methanolischen Losung von 1,0 g Phenyl-diazo-methan tropfenweise versetzt. Die lebhafte Stickstoffentwicklung war bald abgeschlossen. Aufarbeitung mit Wasser-Ather. Aus der eingeengten Losung kristallisierten rot- braune Tafeln mit positiver Beilstein-Probe. Der Schmp. lag nach Umkristalli- sieren aus Methanol unter Kohlezusatz bei 191--191,5O.
C,3Hlo0,”,Cl (275,69) Ber. C 56,63 H 3,66 Gef. )) 57,02 )) 3,85

226 H u i s g e n und K o c h
In einem Kupplungsversuch ohne Zusatz von Lithiumchlorid wurde das Benzoesaure-p-nitro-phenyl-hydrazid-chlorid gar nicht aufgefunden; die hier auftre- tenden gelben Nadeln von Schmp. 80-81O harren noch der weiteren Bearbeitung.
Reakt ion des p-Ni t ro-benzol -d iazonium-chlor ids mi t Diazomethan
In einem Vorversuch wurde in die Losung von 3 g des Aryldiazoniumsalzes in 30 ccm Methanol bei 00 gasformiges Diazomethan (1,2 Aquivalente) eingeleitet. Aus der dunkelroten Losung schieden sich orangerote Kristalle aus, die aus siedendem n-Butanol umkristallisiert wurden: 56 mg vom Schmp. 230-232 O. Die Substanz (Gef. 42,12; H 3,03; N 21,05) gab mit Natronlauge eine blaue Farb- reaktion und wurde nicht naher untersucht. Der dunkle olige Ruckstand der Reaktionslosung wurde mehrfach mit Petrolather ausgekocht ; der Eindampf- ruckstand der Extrakte lie13 sich aus Benzol umkristallisieren. I m besten Versuch wurden 0,19 g gelber Nadeln vom Schmp. 140,5-141,5O erhalten, die sich als Form-p-nitro-phenyl-h ydrazid-chlorid erwiesen. Eine glatte Bildung dieses Hydra- zid-chlorids wurde erst bei der K u p p l u n g i n m i t L i t h i u m c h l o r i d g e s a t t i g - t e m M e t h a n o l erzielt.
In eine eiskalte Losung von 5,8 g p-Nitro-benzol-diazonium-chlorid (31 mMol) in 80 ccm Methanol, die 12 g wasserfreies Lithiumchlorid gelost enthielten, lielj man unter Ruhren innerhalb 30 Min. bei -4O 2,16 g Diazomethan (52 mMol) in 80 ccm Ather eintropfen. Das Reaktionsgemisch wurde noch weitere 30 Min. mecha- nisch geriihrt und iiber Nacht stehengelassen. Die abgeschiedenen gelbbraunenKri- stalle erwiesen sich als 0,80g unveriindertes Aryl-diazonium-salz, das sich offensicht- lich durch die Ausfallung mit Ather der Reaktion entzogen hatte. Der Eindampf- ruckstand der rotbraunen Reaktionslosung wurde aus Methanol fraktiouiert, kristallisiert. Auf 2,55 g gelbbrauner Nadeln vom Schmp. 141,5-142° folgten zwei weitere Fraktionen vom Sahmp. 123-1270. Bei dem in 81% d. Th. Roh- ausbeute angefallenen Stoff handelt; ea sich um das Form-p-nitro-phenyl-hydrazid- chlorid, das aus Methanol umgelost wurde: Gelbe Nadeln vom Schmp. 141-142 O.
C,H,O,N’,Cl (199,60) Ber. C 42,12 H 3,03 ?; 21,05 Gef. )) 42,53 )) 3,12 )) 20,44
Bus der methanolischen Losung des Form-p-nitro-phenyl-hydrazid-chlorids schieden sich nach 4-tiigigem Stehen Kristalle von hoherem Schmp. aus, die durch Ausfallen aus alkoholischer Losung mit CCl, gereinigt wurden. I n den farblosen Blattchen vom Schmp. 196-197 O lag p-Nitro-phenylhydrazin-h ydro- chlorid vor.
C,H,O,N,Cl (189,60) Ber. C 38,Ol H 4,25 N 22,16 Gef. )) 38,02 x 3,83 )) 21,42
Das mit Aceton erhaltene gelbe Hydrazon schmolz bei 147-148 ”.
K u p p l u n g m i t u b e r s c h u s s i g e m D i a z o - m e t h a n
Mit einer inversen Methode, Eintropfen der Aryl-diazoniumsalz-Losung in einen UberschuB verdiinnter Diazomethan-Losung wurden eine grolere Zahl von Versuchen ausgefuhrt, Ton denen nur einer ausfuhrlich wiedergegeben sei.
Eine Losung von 10,O g p-Ritro-benzol-diazonium-chlorid (95-proc., 51 mMol) in 100 ccm eiskaltem Methanol wurde innerhalb 50 Min. in die auf -loo bis -3O

Die Kupplung aromatischer mit aliphatischen Diazoverbindungen 227
gekiihlte, .mechanisch geriihrte Losung von 11,3 g Diazomethan (270 mMol) in 350 ccm Ather und 100 ccm Methanol einfliel3en gelassen. Jeder einfallende Tropfen loste sofort Stickstoffentwicklung aus. Nach Stehen iiber Nacht im Kiihhchrank hatte sich aus der orangegelben Losung ein korniger Niederschlag ausgeschieden, der beim Umlosen aus Methanol-Aceton 20 mg gelbe Nadeln vom Schmp. 183-185O lieferte (Stoff A der Tab. 3). Die iitherisch-methanolische Reaktionslosung wurde mit vie1 Wasser gewaschen und anschlieBend vom Losungsmittel befreit. Nach Aufnehmen in Benzol wurde an einer 80 cm langen Aluminiumoxyd-Siiule adsorbiert und mit dem gleichen Losungsmittel ent- wickelt und eluiert. Der Durchlauf wurde in 80-ccm- Quantitiiten aufgefangen, wobei jeweils nach Abdampfen des Losungsmittels die Rohschmelzpunkte ge- nommen wurden. I n der dritten bis sechsten Eluat-Fraktion fanden sich 2,3 g vom Stoff B (Tab. 3) mit einem Rohschmp. von 131--1410. Mehrfaches Umlosen aus Essigester fiihrte zu blaBgelben, bei 155,5-157 O schmelzenden SpieBen.
C,H,O,N, (177,15) Ber. C 54,24 H 3,98 N23,72 Gef. )) 54,45 )) 3,85 )) 23,39
Molgew. nach R a s t (Campher) 180, 183.
Die siebente bis zehnte Eluat-Fraktion ergab oberhalb 240° u. Zers. schmel- zende kristallisierte Anteile, deren Reinigung nicht gelang.
Eine planmiiDige Variation der Versuchsbedingungen zeigte einen iiberraschend groBen EinfluB des Losungsmittels. Ein Methanol-Ather-Gemisch von 1 : 1,7 Vo1.- teilen bot die optimalen Bedingungen fur die Bildung von B. Mit hoherem Ather- gehalt (1:7) erreichte die Ausbeute a n Stoff A mit 12% d. Th. ein Maximum, wiihrend die a n B absank. Bei einem Versuch in Methanol-Ather 3 : 1 konnte keinerlei kristallisiertes Produkt erhalten werden. Tab. 6 faBt die Resultate zu- sammen.
Tab. 6 R e a k t i o n v o n p-Nitro-benzol-diazonium-chlorid m i t i i b e r -
sch i i ss igem D i a z o m e t h a n
1 : 7,5 1 : 6,2 1 : 2,6 1 : 1,75 1 : 1,75 1: 1,6 1 : 0,3
87 46 76 93 85
111 31
420 46
136 440 405 340 150
12 10 12 3 1
23 24
I nur o’2 rote ie3 Schmieren
Der Versuch mit dem Losungsmittelverhiiltnis 1 : 6,2 sei kurz skizziert: 1,85 g aktives Diazonium-chlorid in 30 ccm Methanol wurden innerhalb 12 Min. bei -4O in die Losung von 420 mg Diazomethan in 185 ccm &,her eingeruhrt. Nach mehrstiindigem Stehen hatten sich an der Kolbenwand braune, mit 01 durchsetzte Kristalle abgesetzt ; beim Digerieren mit kaltem Benzol blieben 220 mg voni Schmp. 174-1770 zuriick (Stoff A der Tab. 3). MehrfachesUmlosenaus Methanol- Aceton unter Kohlezusatz fiihrte zu gelben Nadeln vom Schmp. 187-187,5O u. Zers.
C,H,O,N, (191,15) Ber. C43,98 H 2,64 N 36,64 Gef. )) 44,03 1) 2,25 )) 36,07

225 H u i s g e n und K o c h
Die abdekantierte, methanolisch-atherische Reaktionslosung wurde wie obeu aufgearbeitet. Das erste und zweite benzolische Eluat der Aluminiumoxyd- Adsorption ergaben 25 mg der Substanz C (Tab. 3), die aus Petrolather in gelben Prismen vom Schmp. 102-103° kam.
C,,H,,O,K, (205,21) Ber. C 58,52 H 5,40 X 20,48 Gef. 1) 58,72 )) 5,21 )) 19,85
Molgew. nach Rast (Campher) 204. Die UV-Absorption des noch nicht geklarten Stoffes C zeigt ein Maximum bei
297 in;). (log z=4,2), eine Schulter bei 390 mi* und eine Vorbande bei 445 in?.
K o n s t i t u t i o n s e r m i t t l u n g d e r V e r b i n d u n g A (C,H,O,N,) Die Synthese des 2 ( p - Nitro-pheny1)-tetrazols (XIX) wurde durch Decarboxylie-
rung der 5-Carbonsa~re'~) angestrebt ; alleVersuche fuhrten zu Verharzung, obwolil sich die 2-Phenyl-tetrazol-5-carbonsaure leicht thermisch decarboxylieren lieI.315). Am dem oligen 2-Phenyl-tetrazol erhielten wir rnit rauch. Salpetersaure ein Produkt, das wir aus Analogiegrunden -- die UV-Spektren (Fig. 1) stutzen diesen SchluB - als Bz-p-Kitro-Derivat ansprechen. Aus Methanol farblose Xadeln vom Schmp. 143-144O.
C,H,O,PI', (191,15) Ber. C 43,98 H 3,64 N 36,64 Gef. )) 44,24 )) 2,48 )) 35,99
Das 1 (p-iVitro-pheny1)-tetrazoZ( XX)findet sich rnit einem S~hmp.205~in der Lite- ratur17) beschrieben. Bei der Nacharbeitung erhielten wir ein Praparat, das nach vielfachem Umlosen aus Methanol bei 190° u. Zers. schmolz. Die Ursache des Unterschieds ist nicht klar. Das Praparat vom Schmp. 190° erwies sich mit dcm Stoff A in Mischschmp. und UV-Absorption identisch.
K o n s t i t u t i o n s e r mi t t l u n g d e r Ver b i n d u n g B (C,H70,X,) Sunre Verseifung. 248 mg Substanz wurden rnit je 1 ccm Wasser, Eisessig und
konz. Schwefelsaure 6 Stunden auf dem Wasserbad erwarmt. Die rotbraune Losung wurde rnit wenig Wasser in die Destillationsapparatur zur Ammoniak- Bestimmung gespult und mit Natronlauge auf den Phenolphtalein-Umschlag ein- gestellt. Nach Abtreiben des Ammoniaks mit Wasserdampf wurde die vorgelegte n/lO-Salzsaure zur Entfernung kleiner Mengen N-Methyl-p-nitranilin mit reinem Ather ausgezogen. Die Rucktitration der nichtverbrauchten Saure wies anf 23,l mg Ammoniak hin, was 97 % d. Th. entspricht. Vorversuche hatten ergeben. da13 unter diesen Bedingungen aus N-Methyl-p-nitranilin im schwachalkalischen Medium 2-4% d. Th. Methylamin freigesetzt und abgeblasen werden.
Bus dem Ruckstand der Wasserdampf-Destillation wurden nach den1 Erkalten 166 mg Rohkristallisat erhalten; nach Umlosen aus Essigester unter Kohlezusatz 136 mg gelber Prismen (64% d. Th.) vom Schmp. 149,5-150°. Die Mischprobe ergab Identitat mit einem authent. Yraparat von N-Methyl-p-nitranilin, das durch Methylierung von p-Toluol-sulfonyl-p-nitranilid und anschlieoende Ver- seifung mit 80-proc. Schwefelsaure erhalten wurde.
C,H,O,N, (152,15) Ber. C 55,25 H 5,30 N 18,41 Gef. )) 55,51 )) 5,36 )) 18,32
Die Behandlung des Stoffes B mit 20-proc. methanolischer Kalilauge fuhrte ebenfalls zur Freisetzung von Ammoniak. Erst nach Chromatographie a n rlluminiumoxyd wurde hier N-Methyl-p-nitranilin in maBiger Ausbeute erhalten.
Scetylierung. Kach 2-stundigem Erwarmen mit Acetanliydrid auf 100O wurde der Stoff B unverandert ziiruckerhalten. Daraufhin wurden scharfere Be-

Die Kupplung aromatischer mit aliphatischen Diazoverbindungen 229
dingungcn gewahlt. 100mg B wurden mit 2ccm Acetanhydrid und einem Tropfen konz. Schwefelsaure 6 Stnnden riickfluBgekocht. Nach Schiitteln mit einer Spatelspitze Calciumcarbonat wurde i. V. zur Trockne gebracht, und mit Benzol aufgenommen. Nach wiederholtem Umkristallisieren aus Essigester unt.er Kohlezusatz farblose Prismen vom Schmp. 150,5-151 O,
C,H,,O,N, (194,19) Ber. C 55,66 H 5,19 Gef. u 55,44 )) 5,28
Der Mischschmp. bewies die 1dent.itat rnit N - Acet yl-iV-methyl-p-nitraiiilin.
S p n t hese d e s K - Met h y 1 - p - n i t r o - p h e n y l - c y a n a m i d s (XXII) Die Einwirkung von Bromcyan auf p-Nitranilin in wal3rigem Alkohol1*) fiihrte
zu einem mit dem Harnstoffabkommling stark verunreinigten Praparat von p-Nitro-phenyl-eyanamid (XXV). Gunstiger ist die Stickstoff-Abspaltung aus 1-(p-Nitro-pheny1)-tetrazol ( X X ) mit siedender verd. Natronlauge20). Das orange- farbene Natriumsalz gab .mit verd. Salzsaure das nahezu farblose p-Nitro-phenyl- cyanamid, das wegen seiner Neigung zur Trimerisation 35) rasch aus Methanol umgelost wurde. 165 mg p-Nitro-phenyl-cyanamid wurden in 10 ccm Ather nnd 2 ccm Methanol gelost und mit 120 mg Diazomethan in 7 ccm Ather umgesetzt. Schon nach 3 Min. begann unter Stickstoffentwicklung die Ausscheidung der bla13gelben Kristalle des N-Methyl-Deriwats, die rnit einem Schmp. 155-156O sofort rein anfielen und mit dem Stoff B in der Mischprobe keine Depression gaben. Eine zweite Fraktion aus der Mutterlauge mit Schmp. 140-152O erhohte die Ausbeute auf 70% d. Th. I n dem oberhalb 250° schmelzenden Restanteil lag vermutlich das trimere X X V Tor.
V e r f o lg u n g d e r R e a k t i o n d e s I5N - m a r k i e r t e n p - N i t r o - b e n z o 1 - d i a z o - n i u m - c h 1 or i d s m i t ii b e r s c h ii s s ig e m D i a z o m e t h a n
Die Uberfiihrung des p-Nitranilins in das wasserfreie Diazonium-chlorid wird normalerweise durch Eintragen des Amin-hydrochlorids in einen Uberschul3 Athylnitrit in abs. .Alkohol erzielt. Die Kostbarkeit des markierten Natrinm- nitrits zwang zu rationellerem Einsatz des Salpetrigsaure-esters. Vorversuche er- gaben, daO bei kleiner Nitritkonzentration nur in stark saurer Losung die Weiter- kupplung zur Diazo-amino-Verbindung in Grenzen gehalten wird. Das portions- weise Eintragen des p-Nitranilinium-chlorids in die salzsaure alkoholische Losung des Athyl-nitrits mu13 man beenden, sobald eine gelinde Triibung auftritt, von p,p’-Dinitro-diazoamino-benzol herriihrend. Beim Ausfallen mit abs. Ather er- hielten wir 70--80% d. Th. am festen Diazoniumsalz, bezogen auf das eingesetzte Athyl-nitrit. 1 g Natriumnitrit mit 2,57 Proc. 15N, von Herrn Prof. K. Clus ius freundlich iiberlassen, wurde wie iiblich in Athylnitrit iibergefiihrt, das gar nicht isoliert, sondern gleich in der methanolischen Losung kondensiert und mit dem Amin-hydrochlorid umgesetzt wurde. Die Reaktion mit Diazomethan in Metha- nol-Ather vollzog sich planmaBig und lieferte 195 mg rohes N-Methyl-p-nitro- phenyl-cyanamid, aus dem 163 mg eines bei 146-149O schmelzenden Produkts erhalten wurden. Die Hydrolyse mit Wasser-Eisessig-Schwefelsaure und an- schlieI3ende Wasserdampfdestillation aus schwachalkalischem Medium lieferte aus 163 mg Cyanamid-Derivat 47 mg rohes Ammonchlorid (95% d. Th.). Aus dem Riickstand wurden 119 mg rohes N-Methyl-p-nitranilin isoliert; Umlosen aus Essigester gab 90 mg vom Schrnp. 149-150°.
Die spektroskopische Isotopen-Anal yse des aus dem Ammonchlorid mit Hypo- chlorit freigesetzten Stickstoff wurde von I(. Clus ius und H. HiirzelerZ1) vor- genommen und ergab 2,50-Proc. 15K. Da gewohnlicher Stickstoff 0,37 Proc. 15K enthalt, wurde somit 97 % des bei der Diazotierung eingesetzten Nitrit-Stickstoffs

230 H u i s g e n und K o c h
in der Nitrilgruppe von XXII aufgefunden. Das N-Methyl-p-nitranilin wurde mit Jodwasserstoff reduziert und nach K j e d a h l aufgeschlossen. Der den beiden K- Atomen des Nitranilin-Abkommlings entstammende Stickstoff wies 0,36 Proc. I5?T auf, was innerhalb der Fehlergrenzen mit dem Isotopengemisch des normalen Ytickstoffs ubereinstimmt.
D i a z o - a t h a n a l s K u p p l u n g s k o m p o n e n t e E ssi g s a u r e ~ (p - n i t r o - p h e n y 1 - h y d r a z i d ) - c h lor i d (XXVI)
I n eine Losung von 2,22 g aktivem p - Nitro - benzol- diazonium - chlorid (12,O mMol) und 10 g Lithiumchlorid in 90 ccm Methanol wurden zwischen -13 und -60 1 , l g Diazo-athanS5) (19,6 mMol) in 50 ccm Ather innerhalb 20.,Min. eingetragen. Nach 15 Stunden Stehen im Kiihlschrank wurde rnit Wasser-Ather aufgearbeitet. Einengen der atherischen Losung fiihrte zu drei Fraktionen mit insgesamt 1,3 g Hydrazid-chlorid (50% d. Th.), das nach mehrfachem Umlosen aus Benzol bei 138,5--139,5O schmolz.
C,H,O,S,Cl (213,62) Ber. C44,98 H 3,77 N 19,67 Gef. )) 45,323 )) 3,79 )) 19,38
E s s i g s a u r e - ( 2,4 - d i c h l o r - p h e n y 1 - h y d r a z i d ) ~ c h l or i d (XXVII) Wie oben wurden 2,61 g aktives 2,4-Dichlor-benzol-diazonium-chlorid in
methanolischer Lithiumchlorid-Losung mit Diazo-athan in Ather umgesetzt und wie ublich aufgearbeitet. Man erhielt 1,70 g Rohkristallisat (57% d. Th.) vom Schmp. 53-55 0. Die Reinigung gelang durch Tieftemperatur-Kristallisation aus Aceton. Farblose Kristalle vom Schmp. 57-58O.
C,H;S,Cl, (237,52) Bex. C 40,45 H 2,97 Gef. )) 40,54 )) 2,81
R e a k t i o n v o n Benzol-diazonium-chlor id m i t D i a z o m e t h a n
Selbst eine grodere Zahl von Versuchen ermoglichte es uns bislang nicht, die optimalen Bedingungen fur die Bildung der einzelnen Reaktionsprodukte anzu- geben. Schon kleine Anderungen in den Konzentrationen, in der Zusammen- setzung des Losungsmittels oder der Reaktionstemperatur vermochten das Reak- tionsbild erheblich zu verandern. Es seien daher lediglich einige Versuche heraus- gegriffen und skizziert.
1. I n eine Losung von 4,O g Benzol-diazonium-chlorid (28,5 mMol) in 30 ccni Methanol wurden bei 00 2,O g Diazomethan (47 mMol) eingeleitet, die aus einer toluolischen Losung bei 50-70° im Stickstoff-Strom ausgetrieben wurden. Aus der violettblauen Losung schied sich nach einigem Stehen bei O o ein dicker Kristallbrei aus. Nach Waschen mit eiskaltem Methanol 2,4 g glanzender Blgtt- chen, die nach Umlosen aus Athano1 vollig farblos sind und bei 234-236O u. Zers. schmolzen. Die Substanz reduziert Fehling und ammoniakalische Silberlosung ; rnit Natronlauge der charakteristische Geruch des Phenylhydrazins.
C,H,N,Cl (144,60) Ber. C 49,233 H 6,27 N 19,37 Gef. )) 50,43 )) 6,29 )) 19,03
Ausbeute am Phenyl-hydrazonium-chlorid 59% d. Th. Ein zweiter, bei -40° durchgefiihrter Versuch lieferte 60% d. Th. am gleichen Produkt 3 6 ) .
3 3 ) D. W. Adamson u. J. K e n n e r , J. chem. SOC. [London] 1937, 1651. 36) Diese beiden Versuche wurden von H e i n H. J. Schabelg) durchgefuhrt.

Die Kupplurig aromatischer mit aliphatischen Diazoverbindungen 231
2. Wir lieden eine Losung von 1, l g Diazomethan (26,2 mMol) in 43 ccm Ather innerhalb 45 Min. in die mechanisch geriihrte Losung von 17,9 mMol Benzol- diazonium-chlorid in 80 ccm mit Lithiumchlorid gesattigten Methanols bei -15O eintropfen; die lebhafte Gasentwicklung zeigte den raschen Reaktionsablauf an. Nach EingieBen in Wasser wurde in Ather aufgenommen. Der Ruckstand der Ather-Losung kristallisierte beim Anreiben rnit eiskaltem Methanol in farblosen Blattchen vom Schmp. 138-140° (0,80 g). Beim Versuch des Umkristallisierens aus heiBem Methanol-Benzol wurden lediglich die glanzenden Schuppen des Phenyl-hydrazonium-chlorids vom Schmp. 223-2240 u. Zers. erhalten. In dem uber die atherische Phase isolierten Primarprodukt vermuten wir das Form- p h e n yl-h ydrazid-chlorid.
3. Bei einem gleichartigen Ansatz blieb die Reaktionslosung vor der Auf- arbeitung iiber Nacht im Kiihlschrank stehen. Eine gelbe kristalline Fallung wurde abgesaugt, rnit Wasser gewaschen und getrocknet. 175 mg vom Schmp. 178-183O. Hulsen-Extraktion mit Petrolather und Umlosen aus Methanol lied den Schmp. auf 191-192O steigen (Stoff a der Tab. 5). Kein Reduktions- vermogen, unloslich in heiIjer verd. Salzsaure.
Ci4HizN4 (236727) Ber. C 71,16 H 5,12 N 23,72 Gef. )) 70,58 )) 4,88 )) 23,18
Mo1.-Gew. nach R a s t (Campher) 231, 240.
4. Eine Losung von 3,O g Diazonium-chlorid (21,4 mMol) in 65 ccm mit Lithiumchlorid gesattigtem Methanol wurde bei -5O innerhalb 26 Min. mit 1,3 g Diazomethan (31 mMol) in 50 ccm Ather versetzt. Wahrend der Eintropf- zeit wurden portionsweise 2,2 g feingepulvertes Calciumcarbonat (22 mMol) zugesetzt. Nach Stehen iiber Nacht wurde rnit Wasser-Ather aufgearbeitet. Nach Vertreiben des Athers kristallisierte der Ruckstand und wurde auf Ton abgepredt. 400 mg gelbe Nadeln vom Schmp. 142-148O (8% d. Th.). Nach Umkristallisieren aus Benzol Schmp. 151--151,5O. (Stoff b der Tab. 5.)
Ci4HizN4 (236927) Ber. C 71,16 H 5,12 N23,72 Gef. )) 71,32 )) 5,31 )) 23,57
Mo1.-Gew. nach R a s t (Exalton) 233, 241.
Die Verbindung ist basisch; aus der Losung in heider 2n-Salzsaure schied sich beim Erkalten ein leuchtendgelbes Chlorhydrat vom Zers-Punkt 245-246O aus, das sich iiberraschenderweise von einer isomeren Base ableitet. Die mit Ammoniak freigemachte Base lieB sich aus Toluol oder aim Aceton-Benzol zu einem gelben Nadelfilz vom Schmp. 169,5-170° umkristallisieren (Verbindung d der Tab. 5). Da die Base unangenehm zu reinigen ist, kristallisiert man zweckmaRig das Hydrochlorid mehrfach aus Salzsaure um. Die Zersetzlichkeit in geschmolzenem Campher oder Exalton erlaubte eine Mo1.-Gew.-Bestimmung nicht.
Ci4H,,N, (236927) Ber. C 71,16 H5,12 N23.72 Gef. )) 70,64 )) 5,12 )) 22 68