Embed Size (px)
Citation preview

Die Umwandlung von N-Methylpolyborosilazanin amorphes Siliciumborcarbonitrid
Martin Jansena, * und Matthias Kroschelb
a Stuttgart, Max-Planck-Institut fuÈ r FestkoÈ rperforschungb Bonn, Institut fuÈ r Anorganische Chemie der UniversitaÈ t
Bei der Redaktion eingegangen am 7. Januar 2000.
InhaltsuÈ bersicht. Die Synthese der amorphen Keramik,SiBN3C`, die bis 1900 °C strukturell unveraÈndert bleibt, er-folgt aus dem EinkomponentenvorlaÈufer 1,1-Dichlor-N-(tri-chlorsilyl)-boranamin (TADB) uÈ ber die polymere Zwischen-stufe N-Methylpolyborosilazan. Der thermische Abbau despraÈkeramischen Polymers wurde mittels Thermischer Ana-
lyse, Infrarotspektroskopie und Kernresonanzspektroskopieuntersucht. Ûberraschend deutlich gliedert er sich in dreiAbschnitte, denen die Prozesse VervollstaÈndigung der Poly-kondensation des Polymers (200±350 °C), Fragmentierung/Pyrolyse (580±620 °C) und Eliminierung des Restwasser-stoffs (1000±1400 °C) zugeordnet werden koÈ nnen.
Conversion of N-Methylpolyborosilzane to Amorphous Siliconboroncarbonitride
Abstract. The amorphous ceramic `SiBN3C' has been synthe-sized starting from 1,1-dichloro-N-(trichlorosilyl)-boran-amine (TADB) with N-methylpolyborosilazane as a poly-meric intermediate. On heating the ceramic up to 1900 °C nomicrostructural changes occur. The thermal conversion ofthe preceramic polymer was investigated by thermal analysis,infrared spectroscopy and nuclear magnetic resonance spec-
troscopy. Surprisingly, the pyrolysis proceeds in three clearlydivided steps which can be assigned to completion of thepolycondensation (200±350 °C), fragmentation (580±620 °C)and elimination of residual hydrogen (1000±1400 °C).
Keywords: Amorphous materials; Ceramics; Siliconboron-carbonitrides; Preceramic polymers
1 Einleitung
Beginnend in den sechziger Jahren ist die Polymerrou-te als alternativer Zugang zu nitridischen und carbi-dischen Keramiken entwickelt worden. Dabei werdenuÈ ber anorganische Polymere als Zwischenstufen dieangestrebten Feststoffe durch Pyrolyse dargestellt [1].Ein wesentlicher Vorteil gegenuÈ ber den herkoÈ mm-lichen Syntheserouten liegt in der MoÈ glichkeit, dasPolymer zu formen [2] und so z. B. zu keramischen Fa-sern zu gelangen. Der Traum, aus einem praÈkera-mischen Polymer ein monolithisches Bauteil zu for-men, dessen Form nach der Pyrolyse erhalten bleibt,ist allerdings noch nicht in ErfuÈ llung gegangen. DerGrund hierfuÈ r ist im groûen Massenverlust bei der Py-rolyse zu sehen, der allen Polymeren gemeinsam istund zu makroskopischen Poren und Rissen im Werk-stuÈ ck fuÈ hrt.
Als besonders erfolgreich hat sich das Konzept her-ausgestellt, amorphe anorganische Netzwerke fuÈ r denEinsatz bei hohen Temperaturen heranzuziehen [3±6].Ein solcher Ansatz ist an sich nicht ohne weiteres
schluÈ ssig, da amorphe FestkoÈ rper stets thermodyna-misch instabil sind gegenuÈ ber der Kristallisation undderartige strukturelle Umwandlungen in einem Werk-stuÈ ck unter Belastung zu seiner ZerstoÈ ung fuÈ hren wuÈ r-den. Amorphe Netzwerke aus den Elementen Si, B, Nund C allerdings bleiben bis 1900 °C strukturell unve-raÈndert, was bisher ohne Beispiel ist. In der Kombina-tion aller unter Anwendungsaspekten relevantenStoffeigenschaften ist amorphes ,SiBN3C`, aus TADBals molekularem EinkomponentenvorlaÈufer uÈ ber dar-aus hergestelltes Polymer synthetisiert, allen anderenMaterialien uÈ berlegen [7]. Die AufklaÈrung der atoma-ren Struktur dieser amorphen Verbindungen ruÈ ckteschnell in den Blickpunkt des Interesses. Erste Struk-turinformationen wurden rasch gefunden und durchKombination unterschiedlicher Sonden waren weitereInformationen zugaÈnglich [8±10]. Die Zusammenset-zung der Keramik wurde zwischenzeitlich durch wei-tere Elementaranalysen zu SiBN2,3C0,8 eingegrenzt[11], jedoch verwenden wir weiterhin die vom Che-mical Abstract Service registrierte Formel SiBN3C(CAS-Nr. [159659-85-5]).
WaÈhrend die Synthese des molekularen Einkom-ponentenvorlaÈufers TADB uÈ ber wohldefinierte undnachvollziehbare Schritte erfolgt [4], sind die VorgaÈn-ge, die bei der thermischen ÛberfuÈ hrung des Polymersin das rein anorganische Netzwerk ablaufen, noch un-
1634 Ó WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2000 0044±2313/00/6261634±1638 $ 17.50+.50/0 Z. Anorg. Allg. Chem. 2000, 626, 1634±1638
* Prof. Dr. M. Jansen,Max-Planck-Institut fuÈ r FestkoÈ rperforschung,Postfach 80 06 65,D-70506 Stuttgart,Fax: ++49(0)7 11-6 89 15 02E-mail: [email protected]

Die Umwandlung von N-Metylpolyborosilazan in amorphes Siliciumborcarbonitrid
verstanden. In der vorliegenden Arbeit wird die Ent-wicklung des FestkoÈ rpers aus dem geschmolzenen Po-lymer mittels thermischer Analyse und spektroskopi-scher Techniken (MS, IR, NMR) dokumentiert.
2 Experimentelles
2.1 Synthese von N-Methylpolyborosilazan
Das praÈkeramische Polymer N-Methylpolyborosilazan wirddurch Aminolyse des EinkomponentenvorlaÈufers TADBhergestellt (Abb. 1). Die Synthese dieses VorlaÈufers gehtvon 1,1,1,3,3,3-Hexamethyldisilazan aus. Durch zweifacheSilazanspaltung, erst mit Tetrachlorsilan und dann mitTrichlorboran, werden Silicium und Bor in das MolekuÈ l ein-gefuÈ hrt. TADB wird bei milden Bedingungen mit Mono-methylamin vernetzt. Alle Syntheseschritte sind bereits ananderer Stelle im Detail beschrieben [3]. Die Synthese, Pyro-lyse und Analyse des Polymers muss jeweils unter Schutzgas(N2, Ar, He) erfolgen.
2.2 Pyrolyse
FuÈ r die spektroskopischen Untersuchungen wurde das Poly-mer chargenweise bei verschiedenen Temperaturen (200,350, 400, 500, 600, 650, 700, 800, 900, 1000, 1400 °C) pyroly-siert und ex situ vermessen. Die Pyrolysen erfolgten in Bor-nitrid-Tiegeln in einem RoÈ hrenofen mit Quarzrohr (ober-halb von 1000 °C Korundrohr) unter stroÈ mendem Stickstoff.Die jeweiligen Temperaturen wurden mit 5 K/min ange-steuert und fuÈ r eine Stunde gehalten, die Proben danach mit5 K/min auf Raumtemperatur abgekuÈ hlt. Auûer der bei1400 °C getemperten Probe, die keinerlei Empfindlichkeitmehr gegenuÈ ber atmosphaÈrischen EinfluÈ ssen aufwies, erfolg-te auch nach der Pyrolyse die Handhabung der PraÈparateunter Schutzgas.
2.3 Thermische Analyse
Der thermische Abbau wurde mittels simultaner Thermo-analyse (DTA/TGA/MS) verfolgt. Die Untersuchung er-folgte mit einem Netzsch STA 429 in HeliumatmosphaÈre undKorundtiegeln mit einer Heizrate von 5 K/min. Zur Charak-terisierung fluÈ chtiger Abbauprodukte war ein BalzersQMG 421 Massenspektrometer uÈ ber eine heizbare Kapillareangeschlossen.
2.4 Spektroskopische Untersuchungen
Vom Polymer und allen thermisch behandelten Proben wur-den FT-IR-Spektren mit einem Bruker IFS 113v Spektro-meter (KBr-Presslinge, 1 mg Probe in 500 mg trockenemKBr) aufgenommen.
Von fuÈ nf Proben (Polymer, 350, 650, 1000, 1400 °C) wur-den 11B- und 29Si-Magic-Angle-Spinning-Kernresonanz-Spektren (MAS-NMR) registriert. Es wurde ein 5 mm MAS-Probenkopf von Doty Scientific in einem Varian Unity 400Spektrometer verwendet. Die chemischen Verschiebungenwurden auf Diethylether-Trifluorboran bzw. Tetramethylsilanbezogen. Die Rotationsfrequenz betrug jeweils ca. 5 kHz.
3 Ergebnisse
3.1 Molekularer EinkomponentenvorlaÈuferund praÈkeramisches Polymer
Der EinkomponentenvorlaÈufer TADB enthaÈ lt eineSi±NH±B-BruÈ cke. Diese Gruppierung ist durch Dop-pelbindungsanteile stabilisiert [12] und bleibt auch beiTemperaturerhoÈ hung unveraÈndert erhalten. Es erfolgtkeine HCl-Elimination sondern Abspaltung von BCl3unter Bildung von Cl3Si±NH±BCl±NH±SiCl3 [13]. DieVernetzung mit Methylamin unter Dehydrohalogenie-rung zum praÈkeramischen Polymer erfolgt unterhalbvon ±60 °C in Pentan. Bei derart milden Bedingungenist eine Reaktion der Si±NH±B-BruÈ cke ausgeschlos-sen.
Im Polymer ist Silicium jeweils von vier und Bor je-weils von drei Stickstoffatomen umgeben, von denenjeweils genau eines als Si±NH±B-Gruppe vorliegt unddie anderen eine Methyl-Gruppe tragen. Diese Me-thyl-Stickstoffatome koÈ nnen endstaÈndig zusaÈtzlichWasserstoff tragen oder als BruÈ ckenfunktionen an einweiteres Silicium- oder Bor-Atom gebunden sein. Eshandelt sich damit um ein polymeres Borsilicium-imid(methylamid)(methylimid). Das VerhaÈltnis ausEndgruppen-NHMe und BruÈ cken-NMe laÈsst sich ausder quantitativen Analyse [3] ableiten. In [(Si±NH±B)-(±NMe±)x(±NHMe)y]n gilt 2 x + y = 5, daher ergibtsich fuÈ r eine typische Polymercharge der experi-mentell ermittelten Bruttozusammensetzung 17,9%(100 mi/Rmj) Silicium, 6,9% Bor, 38,6% Stickstoff,27,5% Kohlenstoff und 7,2% Wasserstoff x zu 1,7 undy zu 1,6. VerbruÈ ckende und endstaÈndige Methylamin-Gruppen liegen also etwa in gleichen Anteilen vor.Dieses VerhaÈltnis wird beim Tempern des Polymersdurch fortschreitende Polykondensation unter Abspal-tung von endstaÈndigem Methylamin und AusbildungverbruÈ ckender NMe-Gruppen kontinuierlich veraÈn-dert.
3.2 Thermische Analyse
Der thermische Abbau erfolgt unerwartet uÈ bersicht-lich in drei klar getrennten Stufen (Abb. 2). Zwischen200 und 500 °C (Massenverlust ca. 12%) wird vor al-lem Methylamin abgespalten. Es erfolgt somit eineVervollstaÈndigung der Kondensation. Durch den da-mit verbundenen Anstieg der mittleren Molmasse er-starrt das aufgeschmolzene Polymer in diesem Tem-peraturbereich. Die zweite Stufe bei 600 °C weist mitca. 28% den groÈ ûten Massenverlust auf. Die vorherr-
Z. Anorg. Allg. Chem. 2000, 626, 1634±1638 1635
Abb. 1 Synthese von N-Methylpolyborosilazan und ,SiBN3C`.
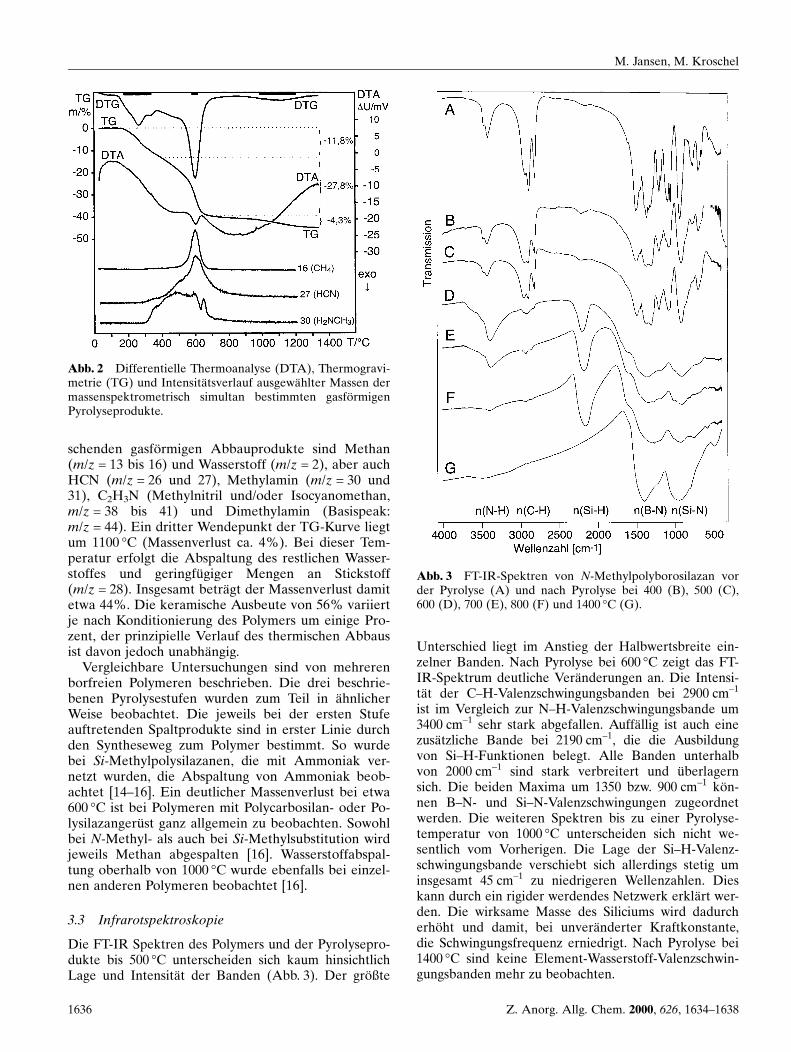
schenden gasfoÈ rmigen Abbauprodukte sind Methan(m/z = 13 bis 16) und Wasserstoff (m/z = 2), aber auchHCN (m/z = 26 und 27), Methylamin (m/z = 30 und31), C2H3N (Methylnitril und/oder Isocyanomethan,m/z = 38 bis 41) und Dimethylamin (Basispeak:m/z = 44). Ein dritter Wendepunkt der TG-Kurve liegtum 1100 °C (Massenverlust ca. 4%). Bei dieser Tem-peratur erfolgt die Abspaltung des restlichen Wasser-stoffes und geringfuÈ giger Mengen an Stickstoff(m/z = 28). Insgesamt betraÈgt der Massenverlust damitetwa 44%. Die keramische Ausbeute von 56% variiertje nach Konditionierung des Polymers um einige Pro-zent, der prinzipielle Verlauf des thermischen Abbausist davon jedoch unabhaÈngig.
Vergleichbare Untersuchungen sind von mehrerenborfreien Polymeren beschrieben. Die drei beschrie-benen Pyrolysestufen wurden zum Teil in aÈhnlicherWeise beobachtet. Die jeweils bei der ersten Stufeauftretenden Spaltprodukte sind in erster Linie durchden Syntheseweg zum Polymer bestimmt. So wurdebei Si-Methylpolysilazanen, die mit Ammoniak ver-netzt wurden, die Abspaltung von Ammoniak beob-achtet [14±16]. Ein deutlicher Massenverlust bei etwa600 °C ist bei Polymeren mit Polycarbosilan- oder Po-lysilazangeruÈ st ganz allgemein zu beobachten. Sowohlbei N-Methyl- als auch bei Si-Methylsubstitution wirdjeweils Methan abgespalten [16]. Wasserstoffabspal-tung oberhalb von 1000 °C wurde ebenfalls bei einzel-nen anderen Polymeren beobachtet [16].
3.3 Infrarotspektroskopie
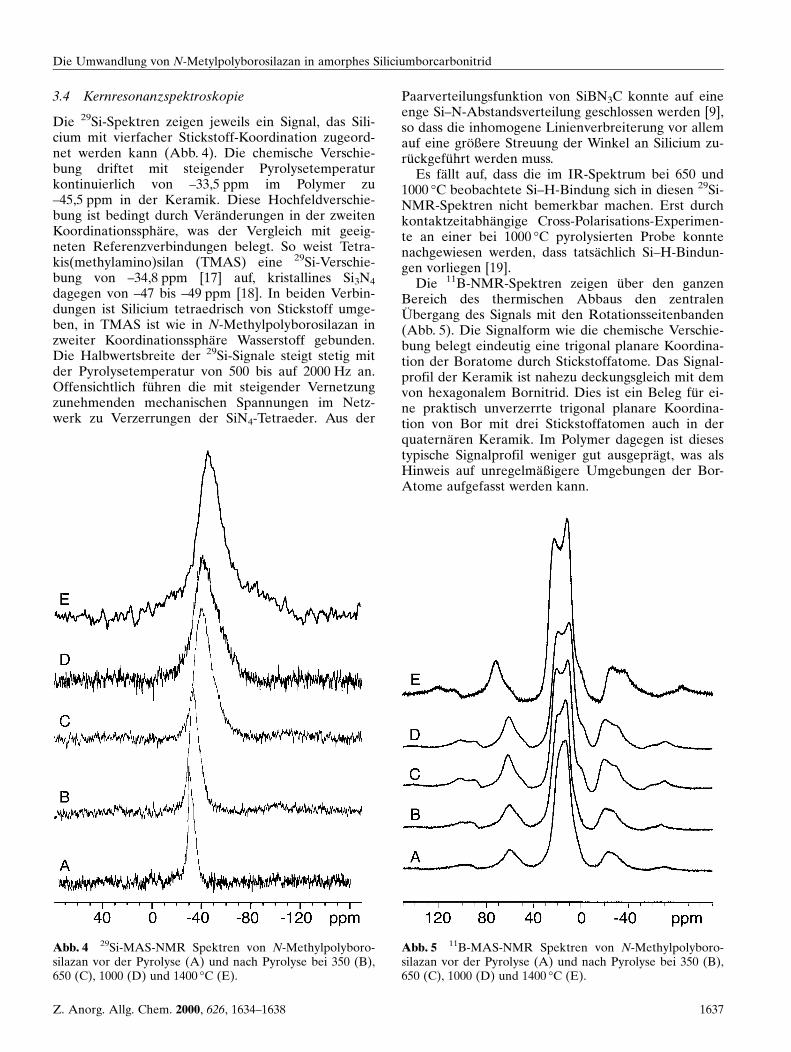
Die FT-IR Spektren des Polymers und der Pyrolysepro-dukte bis 500 °C unterscheiden sich kaum hinsichtlichLage und IntensitaÈ t der Banden (Abb. 3). Der groÈ ûte
Unterschied liegt im Anstieg der Halbwertsbreite ein-zelner Banden. Nach Pyrolyse bei 600 °C zeigt das FT-IR-Spektrum deutliche VeraÈnderungen an. Die Intensi-taÈ t der C±H-Valenzschwingungsbanden bei 2900 cm±1
ist im Vergleich zur N±H-Valenzschwingungsbande um3400 cm±1 sehr stark abgefallen. AuffaÈ llig ist auch einezusaÈtzliche Bande bei 2190 cm±1, die die Ausbildungvon Si±H-Funktionen belegt. Alle Banden unterhalbvon 2000 cm±1 sind stark verbreitert und uÈ berlagernsich. Die beiden Maxima um 1350 bzw. 900 cm±1 koÈ n-nen B±N- und Si±N-Valenzschwingungen zugeordnetwerden. Die weiteren Spektren bis zu einer Pyrolyse-temperatur von 1000 °C unterscheiden sich nicht we-sentlich vom Vorherigen. Die Lage der Si±H-Valenz-schwingungsbande verschiebt sich allerdings stetig uminsgesamt 45 cm±1 zu niedrigeren Wellenzahlen. Dieskann durch ein rigider werdendes Netzwerk erklaÈrt wer-den. Die wirksame Masse des Siliciums wird dadurcherhoÈ ht und damit, bei unveraÈnderter Kraftkonstante,die Schwingungsfrequenz erniedrigt. Nach Pyrolyse bei1400 °C sind keine Element-Wasserstoff-Valenzschwin-gungsbanden mehr zu beobachten.
M. Jansen, M. Kroschel
1636 Z. Anorg. Allg. Chem. 2000, 626, 1634±1638
Abb. 2 Differentielle Thermoanalyse (DTA), Thermogravi-metrie (TG) und IntensitaÈ tsverlauf ausgewaÈhlter Massen dermassenspektrometrisch simultan bestimmten gasfoÈ rmigenPyrolyseprodukte.
Abb. 3 FT-IR-Spektren von N-Methylpolyborosilazan vorder Pyrolyse (A) und nach Pyrolyse bei 400 (B), 500 (C),600 (D), 700 (E), 800 (F) und 1400 °C (G).
Die Umwandlung von N-Metylpolyborosilazan in amorphes Siliciumborcarbonitrid
3.4 Kernresonanzspektroskopie
Die 29Si-Spektren zeigen jeweils ein Signal, das Sili-cium mit vierfacher Stickstoff-Koordination zugeord-net werden kann (Abb. 4). Die chemische Verschie-bung driftet mit steigender Pyrolysetemperaturkontinuierlich von ±33,5 ppm im Polymer zu±45,5 ppm in der Keramik. Diese Hochfeldverschie-bung ist bedingt durch VeraÈnderungen in der zweitenKoordinationssphaÈre, was der Vergleich mit geeig-neten Referenzverbindungen belegt. So weist Tetra-kis(methylamino)silan (TMAS) eine 29Si-Verschie-bung von ±34,8 ppm [17] auf, kristallines Si3N4
dagegen von ±47 bis ±49 ppm [18]. In beiden Verbin-dungen ist Silicium tetraedrisch von Stickstoff umge-ben, in TMAS ist wie in N-Methylpolyborosilazan inzweiter KoordinationssphaÈre Wasserstoff gebunden.Die Halbwertsbreite der 29Si-Signale steigt stetig mitder Pyrolysetemperatur von 500 bis auf 2000 Hz an.Offensichtlich fuÈ hren die mit steigender Vernetzungzunehmenden mechanischen Spannungen im Netz-werk zu Verzerrungen der SiN4-Tetraeder. Aus der
Paarverteilungsfunktion von SiBN3C konnte auf eineenge Si±N-Abstandsverteilung geschlossen werden [9],so dass die inhomogene Linienverbreiterung vor allemauf eine groÈ ûere Streuung der Winkel an Silicium zu-ruÈ ckgefuÈ hrt werden muss.
Es faÈ llt auf, dass die im IR-Spektrum bei 650 und1000 °C beobachtete Si±H-Bindung sich in diesen 29Si-NMR-Spektren nicht bemerkbar machen. Erst durchkontaktzeitabhaÈngige Cross-Polarisations-Experimen-te an einer bei 1000 °C pyrolysierten Probe konntenachgewiesen werden, dass tatsaÈchlich Si±H-Bindun-gen vorliegen [19].
Die 11B-NMR-Spektren zeigen uÈ ber den ganzenBereich des thermischen Abbaus den zentralenÛbergang des Signals mit den Rotationsseitenbanden(Abb. 5). Die Signalform wie die chemische Verschie-bung belegt eindeutig eine trigonal planare Koordina-tion der Boratome durch Stickstoffatome. Das Signal-profil der Keramik ist nahezu deckungsgleich mit demvon hexagonalem Bornitrid. Dies ist ein Beleg fuÈ r ei-ne praktisch unverzerrte trigonal planare Koordina-tion von Bor mit drei Stickstoffatomen auch in derquaternaÈren Keramik. Im Polymer dagegen ist diesestypische Signalprofil weniger gut ausgepraÈgt, was alsHinweis auf unregelmaÈûigere Umgebungen der Bor-Atome aufgefasst werden kann.
Z. Anorg. Allg. Chem. 2000, 626, 1634±1638 1637
Abb. 4 29Si-MAS-NMR Spektren von N-Methylpolyboro-silazan vor der Pyrolyse (A) und nach Pyrolyse bei 350 (B),650 (C), 1000 (D) und 1400 °C (E).
Abb. 5 11B-MAS-NMR Spektren von N-Methylpolyboro-silazan vor der Pyrolyse (A) und nach Pyrolyse bei 350 (B),650 (C), 1000 (D) und 1400 °C (E).

4 Zusammenfassung
Der thermische Abbau von N-Methylpolyborosilazanzu SiBN3C gliedert sich deutlicher als bei anderenpraÈkeramischen Polymeren in drei getrennte Schritte.Der erste Schritt erfolgt im Temperaturbereich von200±350 °C. IR- und NMR-Spektren zeigen hier auûereiner Zunahme der Halbwertsbreiten keine signifikan-te VeraÈnderung. Es schreitet die Kondensation unterAbspaltung von Methylamin und Ausbildung einerNMe-BruÈ cke zwischen Silicium- und/oder Boratomenvoran. Es entstehen dadurch keine andersartigenfunktionellen Gruppen, nur die relative HaÈufigkeitder unterschiedlichen Aminfunktionen veraÈndert sich,und die mittlere MolekuÈ lmasse steigt an. Die Abspal-tung von Methylamin ist massenspektrometrisch be-legt, und der Anstieg der Halbwertsbreiten erklaÈrtsich aus der fortschreitenden Vernetzung, die zuneh-mende Spannungen im Netzwerk und damit eine staÈr-kere Streuung der Koordinationsgeometrien hervor-ruft.
Der zweite Pyrolyseschritt erfolgt zwischen 580 und620 °C. In diesem Temperaturbereich erfolgen wenigergut nachvollziehbare chemische Reaktionen, eskommt verstaÈrkt zu thermisch induzieren Bindungs-bruÈ chen. Die Fragmente saÈttigen sich teilweise ab, sodass vollstaÈndige MolekuÈ le wie Methan, HCN undMethylnitril detektiert werden. Die IR-Spektren zei-gen, dass nach diesem Pyrolyseschritt nur noch einBruchteil der C±H-Bindungen vorhanden ist. Die da-mit verbundene Ønderung der StickstoffumgebungschlaÈgt sich im 29Si-Spektrum als deutliche Hochfeld-verschiebung um 8 ppm nieder.
Oberhalb von 1000 °C erfolgt im letzten Pyrolyse-schritt die Abspaltung des letzten enthaltenen Wasser-stoffs, vor allem aus N±H- und Si±H-Funktionen. Dieschlieûlich erhaltene Keramik ist wasserstofffrei undhat die ungefaÈhre Zusammensetzung SiBN3C. Hervor-zuheben ist, dass bei der Pyrolyse keine Abspaltungvon Bor- oder Silicium-Komponenten erfolgt, das Ver-haÈ ltnis von Si : B bleibt damit vom MolekuÈ l bis zurKeramik unveraÈndert bei 1 : 1. Der Stickstoff-Gehaltstellt sich passend dazu ein, waÈhrend der Kohlenstoff-gehalt in gewissen Grenzen variabel zu sein scheint.So ist es durch Pyrolyse in Ammoniak-AtmosphaÈremoÈ glich, aus demselben Polymer die kohlenstofffreieKeramik Si3B3N7 herzustellen [3].
Bor ist in der Keramik trigonal planar von Stick-stoff umgeben. Silicium liegt stets in tetraedrischerStickstoffumgebung vor. Die N±Si±N-Bindungswinkelweisen eine groÈ ûere Streuung auf als die entsprechen-den mit Bor als Zentralatom. Dies ist im Einklang mitBerechnungen nach denen eine Verzerrung der Bin-
dungswinkel an N und Si in diesem GeruÈ st den ge-ringsten Energieaufwand erfordert [20, 21]. Es gibtkeine Anhaltspunkte dafuÈ r, dass die im VorlaÈufermo-lekuÈ l vorgegebene Si±N±B-BruÈ cke bei der Polymerisa-tion oder Pyrolyse abgebaut wird.
Wir danken der Deutschen Forschungsgemeinschaft(SFB 408, Anorganische FestkoÈ rper ohne Translationssym-metrie) fuÈ r finanzielle FoÈ rderung und Herrn Dr. W. Hoff-bauer fuÈ r die Aufnahme der MAS-NMR-Spektren.
Literatur
[1] Bayer A.G., G. Winter, W. Verbeek, M. Mansmann, US-Pat. 3892583, 1. 7. 1975, [C. A. 1974, 81, P 126134 n].
[2] P. Chantrell, E. P. Popper, in: Special Ceramics, E. P.Popper (Herausgeber), Academic Press, New York1964, S. 87.
[3] H.-P. Baldus, O. Wagner, M. Jansen, Mat. Res. Soc.Symp. Proc. 1992, 271, 821.
[4] H.-P. Baldus, M. Jansen, Angew. Chem. 1997, 109, 338;Angew. Chem., Int. Ed. Engl. 1997, 36, 328.
[5] M. Jansen, H. JuÈ ngermann, Curr. Opin. Solid State &Mater. Sci. 1997, 2, 150.
[6] F. Aldinger, M. Weinmann, J. Bill, Pure Appl. Chem.1998, 70, 439.
[7] P. Baldus, M. Jansen, D. Sporn, Science, 1999, 285, 699.[8] U. MuÈ ller, W. Hoffbauer, M. Jansen, Chem. Mater., im
Druck.[9] D. Heinemann, W. Assenmacher, W. Mader, M. Kro-
schel, M. Jansen, J. Mater. Res. 1999, 14, 3746.[10] R. Franke, St. Bender, H. JuÈ ngermann, M. Kroschel,
M. Jansen, J. Electron Spectrosc. Relat. Phenom. 1999,101±103, 641.
[11] S. Mann, D. Geilenberg, J. A. C. Broekaert, M. Jansen,J. Anal. At. Spectrom. 1997, 12, 975.
[12] M. MuÈ hlhaÈuser, M. Gastreich, C. M. Marian, H. JuÈ nger-mann, M. Jansen, J. Phys. Chem. 1996, 100, 16551.
[13] O. Wagner, Diplomarbeit, Univ. Bonn 1991.[14] D. Bahloul, M. Pereira, P. Goursat, J. Am. Ceram. Soc.
1993, 76, 1156.[15] H. N. Han, D. A. Lindquist, J. S. Haggerty, D. Seyferth,
Chem. Mater. 1992, 4, 705.[16] M. Narisawa, M. Shimoda, K. Okamura, M. Sugimoto,
T. Seguchi, Bull. Chem. Soc. Jpn. 1995, 68, 1098.[17] I. Mokros, M. Jansen, Monatsh. Chem. 1996, 127, 117.[18] E. A. Leone, S. Curran, M. E. Kotun, J. Am. Ceram.
Soc. 1996, 79, 513.[19] G. Jeschke, M. Kroschel, M. Jansen, J. Non-Cryst. Solids
1999, 260, 216.[20] P. Kroll, R. Hoffmann, Angew. Chem. 1998, 110, 2616;
Angew. Chem., Int. Ed. Engl. 1998, 37, 2527.[21] A. Hannemann, J. C. Schoen, M. Jansen, ªModelling of
Si±B±N-ceramicsº in Proceedings of the 9. Workshopon ªNichtkristalline und partiell kristalline Materialienºof the DGK, Wolfersdorf 1999, B. MuÈ ller (Heraus-geber), Jena.
M. Jansen, M. Kroschel
1638 Z. Anorg. Allg. Chem. 2000, 626, 1634±1638















![10 Elektrischer Strom 2013.ppt [Kompatibilitätsmodus]...- im Elektromotor, Umwandlung in mechanische Arbeit - im Akku, Umwandlung in chemische Energie - im Widerstand, Umwandlung](https://img.pdfslide.org/doc/110x75/5f01e8147e708231d4019e8d/10-elektrischer-strom-2013ppt-kompatibilittsmodus-im-elektromotor-umwandlung.jpg)













