Embed Size (px)
Citation preview

Einfluss der Lipidzusammensetzung der
Membran auf die Expression des
kdpFABC-Operons in Escherichia coli
Dissertation
zur Erlangung des Grades
Doktor der Naturwissenschaften (Dr. rer. nat.)
des Fachbereiches Biologie/Chemie
an der Universität Osnabrück
Maren Schniederberend
Osnabrück 2009

Inhaltsverzeichnis
2
Inhaltsverzeichnis Danksagung......................................... ..................................................................... 4 Abkürzungen........................................ ..................................................................... 5 Nomenklatur........................................ ...................................................................... 6 1. Einleitung ......................................... .............................................................. 7
1.1. K+-Transportsysteme von Escherichia coli ........................................................................... 7 1.2. Das Signaltransduktionssystem KdpD / KdpE von E. coli................................................... 9 1.3. Reiz und Reizaufnahme von KdpD in E. coli ....................................................................... 14 1.4. Die Zytoplasmamembran und ihre Phospholipide ....... ...................................................... 17 1.5. Einfluss der Membranzusammensetzung auf Membranprot eine durch die Interaktionen
mit Phospholipiden ................................. ............................................................................... 20 1.6. Aufgabenstellung ................................... ................................................................................ 21 2. Material und Methoden.............................. .................................................. 23
2.1. Materialien........................................ ....................................................................................... 23 2.2. Stämme, Plasmide und Oligonukleotide............... ............................................................... 24 2.3. Medien und Anzuchtverfahren........................ ...................................................................... 27 2.3.1. Mediumshift .............................................................................................................................. 28 2.3.2. Bestimmung der Lebendzellzahl .............................................................................................. 28 2.4. Molekularbiologische und genetische Methoden ....... ........................................................ 29 2.4.1. Plasmidisolierung ..................................................................................................................... 29 2.4.2. Isolierung genomischer DNA.................................................................................................... 29 2.4.3. DNA-Modifikation...................................................................................................................... 29 2.4.4. Elektrophoretische Auftrennung von DNA................................................................................ 30 2.4.5. Extraktion von DNA aus Agarosegelen .................................................................................... 30 2.4.6. Herstellung kompetenter Zellen ............................................................................................... 30 2.4.7. Transformation von kompetenten Zellen.................................................................................. 31 2.4.8. Polymerasekettenreaktion........................................................................................................ 31 2.4.9. DNA-Sequenzanalyse .............................................................................................................. 32 2.4.10. Isolierung von RNA................................................................................................................... 32 2.4.11. cDNA Synthese ........................................................................................................................ 32 2.4.12. Quantitative real-time RT-PCR................................................................................................. 32 2.4.13. P1-Phagenlysatpräparation und P1-Transduktion ................................................................... 33 2.5. Biochemische und analytische Methoden.............. ............................................................. 34 2.5.1. Herstellung von SDS-Proben aus ganzen Zellen..................................................................... 34 2.5.2. SDS-Polyacrylamidgelelektrophorese...................................................................................... 35 2.5.3. Immunoblot ............................................................................................................................... 35 2.5.4. Herstellung von multilamellaren Liposomen ............................................................................ 36 2.5.5. Präparation von invertierten Membranvesikeln........................................................................ 37 2.5.6. Reinigung von KdpD-6His ........................................................................................................ 37 2.5.7. Rekonstitution von KdpD-6His ................................................................................................. 37 2.5.8. Fusion von Membranvesikeln mit synthetischen Liposomen................................................... 39 2.5.9. Proteinbestimmung nach Lowry ............................................................................................... 39 2.5.10. Proteinphosphorylierung mit [γ−32P] ATP ................................................................................. 39 2.5.11. Lipid-Extraktion aus ganzen Zellen .......................................................................................... 40 2.5.12. Dünnschichtchromatographie................................................................................................... 41 2.5.13. Kopfgruppenanalyse der Phospholipide mittels 32P-Markierung.............................................. 41 2.5.14. Fettsäure-Extraktion aus ganzen Zellen................................................................................... 42 2.5.15. Fettsäureanalyse der Phospholipide mittels Gaschromatographie.......................................... 42 2.5.16. Flammenphotometrische Bestimmung des extrazellulären K+- und Na+-Gehaltes.................. 42 2.5.17. β-Galaktosidase-Aktivitätstest .................................................................................................. 43

Inhaltsverzeichnis
3
3. Ergebnisse ......................................... .......................................................... 44
3.1. Einfluss der K +-Limitation auf die Phospholipide von E. coli K12 Wildtyp ...................... 44 3.1.1. Totale Phospholipidzusammensetzung.................................................................................... 44 3.1.2. Neusynthetisiertes Cardiolipin nach K+-„downshift“ ................................................................. 45 3.1.3. Fettsäurezusammensetzung unter K+-Limitation ..................................................................... 46
3.2. Einfluss der K +-Limitation auf die Phospholipide von E. coli „Kalium-Stämmen“.......... 47 3.2.1. Totale Phospholipidzusammensetzung.................................................................................... 47 3.2.2. Neusynthetisiertes Cardiolipin nach K+-„downshift“ ................................................................. 48
3.3. Einfluss der K +-Limitation auf die Phospholipide von E. coli „Lipid-Stämmen“ ............. 50 3.4. Einfluss der K +-Limitation auf die Phospholipide von E. coli C43(DE3) / pEcb1............. 53 3.5. Cardiolipin-Anstieg unter K +-Limitation........................................ ....................................... 54 3.5.1. cls-Expression in verschiedenen E. coli Stämmen .................................................................. 54 3.5.2. Cardiolipin-Gehalt in einem ∆cls-Stamm.................................................................................. 56
3.6. Überprüfung eines gemeinsamen kch-cls Transkripts ....................................... ............... 58 3.7. Einfluss der Zusammensetzung der Zytoplasmamembran auf das Kdp-System ........... 60 3.7.1. Kinaseaktivität von KdpD-6His in Membran-Lipid-Fusionen.................................................... 60 3.7.2. Kinaseaktivität von KdpD-Derivaten in Membran-Lipid-Fusionen............................................ 62 3.7.3. kdpFABC-Expression unter K+-Limitation in Abhängigkeit von der Lipidzusammensetzung .. 63 3.8. Einfluss von Procain auf die kdpFABC-Expression ........................................ ................... 65 3.8.1. Q-RT-PCR in „Lipid-Stämmen“ ................................................................................................ 65 3.8.2. β-Galaktosidase-Aktivitäten von KdpD-Derivaten .................................................................... 67
3.9. Suche nach Kontaktstellen von KdpD mit Phospholipid en der Zytoplasmamembran... 68 3.10. Novobiocin-Resistenz in Abhängigkeit von KdpD...... ........................................................ 72 4. Diskussion......................................... ........................................................... 76
4.1. Einfluss der Lipidzusammensetzung auf das Kdp-Syste m ............................................... 76 4.2. Modell für den Anstieg von Cardiolipin ............. .................................................................. 81 4.3. Hypothesen zur Interaktion von KdpD................ ................................................................. 86 4.4. Ausblick........................................... ........................................................................................ 94 5. Zusammenfassung .................................... .................................................. 98 6. Summary ............................................ .......................................................... 99 7. Literaturverzeichnis............................... .................................................... 100 Eidesstattliche Erklärung.......................... ........................................................... 111

Danksagung
4
Danksagung Viele Menschen haben mich bei der Realisierung meiner Doktorarbeit unterstützt. Obwohl
diese Auflistung nicht vollständig sein kann, so möchte ich dennoch folgende Personen
hervorheben, die mich besonders unterstützt haben:
Herrn Professor K. Altendorf für die Möglichkeit an einem interessanten Thema zu
forschen und die guten Arbeitsbedingungen, die diese Arbeit erst möglich gemacht haben.
Besonders auch für sein Vertrauen, mich nach Houston in die Arbeitsgruppe von Professor
W. Dowhan zum Erlernen neuer Methoden und an zwei wunderbare Gordon-Konferenzen an
die amerikanische Ostküste zu entsenden, mit der Möglichkeit Kontakte zu knüpfen. Und
schließlich danke ich ihm für die kritische Durchsicht und die Begutachtung dieser Arbeit.
Herrn Professor G. Unden für die Begutachtung meiner Arbeit.
Dr. Petra Zimmann für ihre unermüdlichen Diskussionen, ihre Hilfsbereitschaft beim
Verfassen von meinem Manuskript, die kritische Durchsicht meiner Dissertation und ihrem
Verständnis für alles.
Monika, Knut, Kerstin und Eva für Eure tägliche Unterstützung als meine engsten
Mitstreiter im Labor 115/116, wenn Ihr alle leider auch nicht während meiner kompletten Zeit
dabei sein konntet, war es eine tolle Zeit mit Euch!
Allen ehemaligen und gegenwärtigen Mitarbeitern der Arbeitsgruppe Mikrobiologie für
das angenehme Arbeitsklima, die hilfreichen Ideen und die konstruktive Kritik. Insbesondere
seien hier meine Mitstreiter Dorthe, Inga und Heidi genannt.
Herrn Professor William Dowhan in Houston, Texas für die Aufnahme in seine Arbeits-
gruppe. Dabei gebührt ein besonderer Dank Dr. Mikhail „Misha“ Bogdanov für die Betreuung
und das Interesse an meinen Ergebnissen. Vielen Dank an beide auch für die Diskussionen
bezüglich des Manuskripts für meine Veröffentlichung.
Frau Professor Janet Wood in Guelph, Kanada für ihr Interesse am Fortschritt meiner
Arbeit und für Ihre konstruktiven Diskussionen.
Den Vorsitzenden der FAZIT- und der Hans Mühlenhoff-Stiftung für die finanzielle Unter-
stützung durch die Vergabe eines Stipendiums.
Meiner Schwester Tanja und ihrem Mann Ingo für die permanente Unterstützung und
Motivation während meiner Arbeit. Ohne sie hätte ich das alles nicht geschafft. Dazu hat
auch Geli einen wesentlichen Teil getragen. Danke!
Zu guter letzt bedanke ich mich bei all den lieben Menschen, die während der letzten
Jahre an meiner Seite waren. Ohne Euch hätte ich nur halb so viel Kraft und Spaß gehabt!

Abkürzungen
5
Abkürzungen AP Alkalische Phosphatase
Ap r Ampicillinresistenz
AS Aminosäure
bp Basenpaare
CA catalytic ATP-binding
cDNA komplementäre DNA
CL Cardiolipin
CMC “critical micelle concentration”
cpm “ Counts per minute”
CT “cycle threshold”
DC Dünnschichtchromatographie
DDM n-Dodecyl-β-Maltosid
DHp “ dimerization histidine phosphotransfer”
FAME Fettsäure Methyl Ester
HPt Histidin-enthaltende Phosphotransfer-Proteine
kdp „K+-dependent“
LDAO Lauryldimethylaminoxid
MV Membranvesikel
Ni2+-NTA Nickel-Nitrilotriessigsäure
ODx Optische Dichte der Wellenlänge x nm
OG Octylglycosid
PC Phosphatidylcholin
PE Phosphatidylethanolamin
PG Phosphatidylglycerol
Pi anorganisches Phosphat
Q-RT-PCR Quantitative real-time Reverse Transkriptase Polymerasekettenreaktion
rpm „rounds per minute“
RT Raumtemperatur
TAE Tris-Acetat-EDTA-Puffer
TG Tris/Glycerol
TM Transmembrandomäne
Usp “universal stress protein“
WT Wildtyp

Nomenklatur
6
Nomenklatur In der vorliegenden Arbeit werden Stämme von Escherichia coli mit einer Mutation in
den Genen, welche für Enzyme kodieren, die in die Biosynthese der Phospholipide
involviert sind, der Einfachheit halber als „Lipid-Stämme“ bezeichnet. Des Weiteren
werden Stämme mit einer Mutation in den Genen, welche für K+-Aufnahmesysteme
kodieren, der Einfachheit halber als „Kalium-Stämme“ bezeichnet.
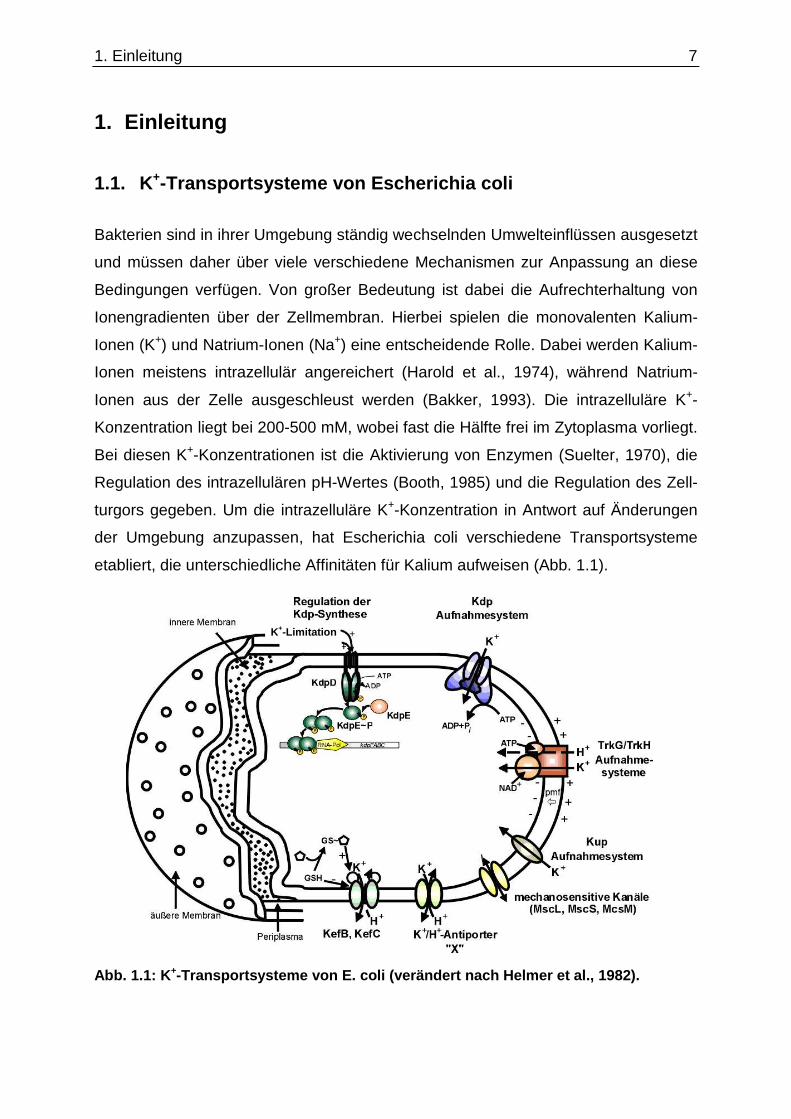
1. Einleitung
7
1. Einleitung
1.1. K+-Transportsysteme von Escherichia coli
Bakterien sind in ihrer Umgebung ständig wechselnden Umwelteinflüssen ausgesetzt
und müssen daher über viele verschiedene Mechanismen zur Anpassung an diese
Bedingungen verfügen. Von großer Bedeutung ist dabei die Aufrechterhaltung von
Ionengradienten über der Zellmembran. Hierbei spielen die monovalenten Kalium-
Ionen (K+) und Natrium-Ionen (Na+) eine entscheidende Rolle. Dabei werden Kalium-
Ionen meistens intrazellulär angereichert (Harold et al., 1974), während Natrium-
Ionen aus der Zelle ausgeschleust werden (Bakker, 1993). Die intrazelluläre K+-
Konzentration liegt bei 200-500 mM, wobei fast die Hälfte frei im Zytoplasma vorliegt.
Bei diesen K+-Konzentrationen ist die Aktivierung von Enzymen (Suelter, 1970), die
Regulation des intrazellulären pH-Wertes (Booth, 1985) und die Regulation des Zell-
turgors gegeben. Um die intrazelluläre K+-Konzentration in Antwort auf Änderungen
der Umgebung anzupassen, hat Escherichia coli verschiedene Transportsysteme
etabliert, die unterschiedliche Affinitäten für Kalium aufweisen (Abb. 1.1).
Abb. 1.1: K +-Transportsysteme von E. coli (verändert nach Helmer et al., 1982).
K+-Limitation

1. Einleitung
8
In E. coli sind bislang für den Export von K+ die KefB/KefC-Systeme (Bakker et al.,
1987; Booth et al., 1996) und ein K+/H+-Antiporter bekannt. Kalium kann aber auch
durch die mechanosensitiven Kanäle (Msc) aus der Zelle transportiert werden.
Dieses ist z.B. bei einem hypoosmotischen Schock der Fall, wenn K+-Ionen mit
anderen kleinen Molekülen über die unspezifischen Kanäle aus der Zelle exportiert
werden. Für die Aufnahme von Kalium bei einer Konzentration über 200 µM in der
Umgebung gibt es die niedrigaffinen TrkG/TrkH- und Kup-Systeme, deren Gene
konstitutiv exprimiert werden. Wenn diese Aufnahmesysteme bei niedrigeren K+-
Konzentrationen nicht mehr in der Lage sind, den Bedarf der Zelle an K+ aufrecht zu
erhalten (Epstein, 1992), synthetisiert E. coli ein Notfallsystem, um K+ aufzunehmen.
Dabei handelt es sich um den hochaffinen K+-Transporter KdpFABC, dessen
Genexpression über eine spezifische Induktion reguliert wird. Der KdpFABC-
Komplex ist eine K+-abhängige P-Typ ATPase und wird von dem kdpFABC-Operon
kodiert (Altendorf & Epstein, 1996).
Der hochaffine Transportkomplex besteht aus den vier Untereinheiten KdpF, KdpA,
KdpB und KdpC (Laimins et al., 1981; Hesse et al., 1984; Gaßel et al., 1998), die bei
der Aufnahme von K+-Ionen unterschiedliche Funktionen erfüllen. Es konnte gezeigt
werden, dass KdpA, welches mit zehn Transmembranhelices in der Zytoplasma-
membran verankert ist und ein Molekulargewicht von 59 kDa besitzt, eine Rolle bei
der Bindung und dem Transport von Kalium spielt (Buurmann et al., 1995). KdpA
stellt einen K+-Kanal vom MPM-Typ (membrane helix - P-loop - membrane helix) dar
(Durell et al., 2000). Die mit 72 kDa größte Untereinheit des Komplexes bildet KdpB,
welches die Membran vermutlich mit sieben Transmembrandomänen durchspannt
(Altendorf et al., 1998) und die notwendige Energie für den Transport liefert (Siebers
& Altendorf, 1989; Siebers & Altendorf, 1993). Dabei ist es unter den bakteriellen P-
Typ ATPasen einzigartig, dass die ATP-Hydrolyse und Substrattranslokation auf zwei
Untereinheiten verteilt ist (Bramkamp et al., 2007). Es wird vermutet, dass diese
beiden Vorgänge über die fünfte Transmembrandomäne von KdpB mit den hoch-
konservierten geladenen Aminosäuren D583 und K586 miteinander verbunden
werden (Bramkamp & Altendorf, 2003). Becker et al. (2007) konnten zeigen, dass
dieser Dipol für die Kopplung des K+-Ions und für den K+-Translokationsschritt
entscheidend ist. Die 20 kDa große KdpC-Untereinheit ist essentiell für den
Transport und sorgt für die korrekte Assemblierung des Komplexes (Gaßel, 1999).
Auch eine Funktion von KdpC bei der Energieübertragung von KdpB und KdpA wird

1. Einleitung
9
diskutiert (Gaßel & Altendorf, 2001). Des Weiteren wird vermutet, dass KdpC eine
katalytische Rolle bei dem Reaktionszyklus des KdpFABC-Komplexes spielt (Ahnert
et al., 2006). Möglicherweise könnte KdpC auch als katalytisches Chaperon fun-
gieren, welches durch Interaktion mit ATP die schwache ATP-Bindeaffinität der hoch-
konservierten Phosphorylierungsstelle von KdpB erhöht (Greie & Altendorf, 2007).
Der mit 3.1 kDa kleinsten Untereinheit des Komplexes, KdpF, wird eine Funktion bei
der Stabilisierung zugewiesen, wobei das Peptid für den Transport in vivo nicht
essentiell ist (Gaßel et al., 1999).
1.2. Das Signaltransduktionssystem KdpD / KdpE von E. coli Die Synthese des KdpFABC-Systems wird durch die membrangebundene Sensor-
kinase KdpD und den zytoplasmatischen Antwortregulator KdpE reguliert
(Walderhaug et al., 1992). Diese beiden Proteine stellen ein typisches Sensor-
kinase / Antwortregulator System dar. Solche Signaltransduktionssysteme sind bei
Prokaryoten und Eukaryoten weit verbreitet und weisen einen ähnlichen Mecha-
nismus der Reizwahrnehmung und Signalweiterleitung auf. Diese Systeme können
verschiedene externe Parameter wie Nährstofflimitation, Temperatur, Sauerstoffge-
halt, pH-Wert, Osmolalität, Anhäufung toxischer metabolischer Produkte und andere
Faktoren detektieren. Die Adaptation auf diese Variationen löst meist eine Änderung
der Genexpression oder der Motilität aus (Stock et al., 1990; Bourret et al., 1991;
Parkinson & Kofoid, 1992; Parkinson, 1993; Stock et al., 2000). Bei Bakterien
bestehen diese Systeme in der Regel aus zwei Proteinkomponenten (Abb.1.2). Die
Funktion des jeweiligen bakteriellen Zweikomponentensystems kann dabei ganz
unterschiedlich sein, denn diese Systeme werden sowohl für die Chemotaxis als
auch für den Metabolismus, die Pathogenität und den Transport benötigt (West &
Stock, 2001).
Abb. 1.2: Schematische Darstellung der Funktionswei se von Sensorkinase / Antwort-regulator Systemen anhand ihrer Domänenstruktur (ve rändert nach Laub & Goulian, 2007). Die direkt an der Phosphorylierungskaskade beteiligten Aminosäurereste Histidin und Aspartat sind durch die Buchstaben „H“ und „D“ gekennzeichnet.

1. Einleitung
10
Die bislang untersuchten Sensorkinasen sind überwiegend in der Zytoplasma-
membran verankert, wo sie einen oder mehrere Reize wahrnehmen können. Je
nachdem, ob sie den Reiz im Periplasma, in der Transmembranregion oder im
Zytoplasma wahrnehmen, können sie in verschiedene Gruppen eingeteilt werden
(Maschner et al., 2006). Ein typischer Antwortregulator liegt in löslicher Form im
Zytoplasma vor und kann so eine Zellantwort auslösen. Sensorkinasen und Antwort-
regulatoren sind in der Regel jeweils aus zwei funktionellen Domänen aufgebaut. Mit
Hilfe dieser Domänen findet die Reizwahrnehmung und Reizweiterleitung über eine
Phosphorylierung statt. Dabei besteht die Sensorkinase aus einer N-terminalen
“Input“-Domäne und einer C-terminalen “Transmitter“-Domäne. Der Antwortregulator
lässt sich in eine N-terminale “Receiver“-Domäne und eine oder mehrere C-terminale
“Output“-Domänen unterteilen (Parkinson, 1995). Nach der Wahrnehmung eines
Reizes durch die “Input“-Domäne der Sensorkinase kommt es zu einer Autophos-
phorylierung der Sensorkinase durch die γ-Phosphorylgruppe eines ATP-Moleküls an
einem hochkonservierten Histidinrest in der “Transmitter“-Domäne. Im Anschluss
erfolgt der Transfer dieser Phosphorylgruppe auf einen hochkonservierten Aspartat-
rest in der “Receiver“-Domäne des Antwortregulators. Der phosphorylierte Antwort-
regulator kann dann mittels seiner “Output“-Domäne die Adaptation der Zelle
bewirken (Stock et al., 1989; Parkinson & Kofoid, 1992; Stock et al., 2000). Dabei
führt meistens die Phosphorylierung des Antwortregulators zu einer Änderung der
Transkription spezifischer Gene (Laub & Goulian, 2007).
Die “Transmitter“-Domäne der Sensorkinase besteht aus zwei Subdomänen. Die
erste Subdomäne, welche den hochkonservierten Histidinrest beinhaltet, wurde als
“DHp“-Domäne (“dimerization histidine phosphotransfer“) bezeichnet. Aufgrund der
Phosphorylierung des Histidinrestes während der Signaltransduktion werden diese
Sensorkinasen auch Histidinkinasen genannt. Die andere Subdomäne, die “CA“-
Domäne (“catalytic ATP-binding“), enthält konservierte Sequenzmotive, die eine
Rolle bei der ATP-Bindung und bei der Translokation der Phosphorylgruppe zu
spielen scheinen (Tanaka et al., 1998).
Im Fall des Signaltransduktionssystems KdpD / KdpE kommt es unter K+-Limitation
(und bei hoher Osmolalität im deutlich geringeren Ausmaß) zur Autophos-
phorylierung von KdpD am hochkonservierten Histidinrest H673 in der “Transmitter“-
Domäne (Nakashima et al., 1992; Voelkner et al., 1993). Nach der Phosphorylierung

1. Einleitung
11
von KdpD erfolgt der Transfer der Phosphorylgruppe auf den Aspartatrest D52 in der
“Receiver“-Domäne des zytoplasmatischen Antwortregulators KdpE (Nakashima et
al., 1993; Jung et al., 1997; Lucassen, 1998). Diese Übertragung wird vermutlich
durch KdpE katalysiert, da KdpE in vitro auch durch Acetylphosphat phosphoryliert
wird (Lucassen, 1998). Weiterhin konnte gezeigt werden, dass die Phosphorylierung
von KdpE zur Bildung eines Homodimers führt (Lucassen, 1998). Dieses
phosphorylierte KdpE-Homodimer ist in der Lage, mit hoher Affinität stromaufwärts
an die kdpFABC-Promotorregion zu binden und die Transkription des kdpFABC-
Operons zu induzieren (Nakashima et al., 1993). Das Schema der Signaltransduktion
zwischen KdpD und KdpE ist in Abbildung 1.3 gezeigt.
Abb. 1.3: Modell der Signaltransduktion von KdpD / KdpE in E. coli (verändert nach Hamann, 2008). Dargestellt sind die Autophosphorylierung von KdpD und der Phospho-transfer von KdpD auf KdpE, durch welchen die kdpFABC-Expression induziert wird (links). Daneben ist auch gezeigt, wie KdpD phosphorylieres KdpE dephosphoryliert (rechts), wobei die Phosphorylgruppe als „P“ gekennzeichnet ist.
Die Sensorkinase KdpD (98.7 kDa) besteht aus einer zytoplasmatischen N-
terminalen und einer zytoplasmatischen C-terminalen Domäne, die durch vier Trans-
membrandomänen miteinander verbunden sind (Zimmann et al., 1995). Die mit
ungefähr 660 Aminosäuren ungewöhnlich große „Input“-Domäne von KdpD, welche

1. Einleitung
12
für die Reizwahrnehmung verantwortlich ist, wird aus der N-terminalen Domäne, den
vier Transmembrandomänen mit dem Arginincluster und ungefähr 140 Aminosäuren
der zytoplasmatischen C-terminalen Domäne gebildet (Abb. 1.4). Die anderen 230
Aminosäuren der zytoplasmatischen C-terminalen Domäne bilden eine für Histidin-
kinasen typische „Transmitter“-Domäne, welche die konservierten Sequenzmotive
“H“, “N“, “G1“, “F“ und “G2“ beinhaltet (Parkinson & Kofoid, 1992). In der “Input“-
Domäne fällt die 400 Aminosäuren große zytoplasmatische, hydrophile N-terminale
Domäne auf, welche bislang einzigartig unter den Sensorkinasen ist. In dieser N-
terminalen Domäne kommen zwei Sequenzmotive vor, die den “Walker A“- und
“Walker B“-Motiven ähneln, welche für eine ATP-Bindung charakteristisch sind. Bei
dem abgewandelten “Walker A“-Motiv “GXXXGXGKT“ liegt eine zusätzliche Amino-
säure zwischen den beiden ersten Glycinresten im Vergleich zum klassischen Motiv
“GXXGXGKT“ vor (Jung & Altendorf, 1998b).
Abb. 1.4: Schematische Darstellung der Domänen von KdpD aus E. coli (verändert nach Zimmann, 2007). Die Zahlen beziehen sich auf die Aminosäuresequenz. Der für die Signaltransduktion konservierte Histidinrest 673 ist durch das umkreiste “P” dargestellt. Das positiv geladene Arginincluster ist mit “+” eingezeichnet.
Aufgrund von Strukturmodellierungen wurde, neben den beiden “Walker“-ähnlichen
Motiven, eine weitere, potentielle ATP-bindende Domäne entdeckt. Diese Domäne
kommt in allen Mitgliedern der Familie der Usp-Proteine (“universal stress protein“)
vor, deren Synthese bei vielen wachstumshemmenden Stresssituationen erhöht ist
(Van Bogelen et al., 1990). Meist kommen in den Organismen viele verschiedene
Usp-Domänen vor, wobei diese entweder alleine oder mit mehreren anderen

1. Einleitung
13
Domänen ein Protein bilden (Siegele, 2005). Letzteres ist z. B. bei der Sensorkinase
KdpD von E. coli der Fall. Eine weitere in der „Input“-Domäne vorkommende Region
ist aufgrund der Sequenz möglicherweise eine GAF-Domäne, die zwischen dem
Arginincluster und einem „Coiled-coil“ Linker liegt, welcher die „Input“-Domäne mit
der „Transmitter“-Domäne verbindet. Aravind & Ponting (1997) vermuten, dass an
GAF-Domänen Liganden binden, welche eine Rolle bei der Reizwahrnehmung
spielen. Da die Aminosäuren 549-644 von KdpD Homologien zu den GAF-Domänen
aufweisen und eine Deletionsmutante zeigte, dass dieser Bereich für KdpD essentiell
ist (Hüging, 2006), könnte diese Domäne ebenfalls eine Rolle bei der Reizwahr-
nehmung bzw. bei der Signalkaskade spielen.
Für eine funktionelle Signalkaskade ist die korrekte Faltung der Sensorkinase und
des Antwortregulators von entscheidender Bedeutung. Da es sich bei KdpD um ein in
die Membran verankertes Protein handelt, ist der richtige Einbau in die Lipiddoppel-
schicht ebenfalls wichtig. Facey & Kuhn (2003) zeigten, dass die Insertion von KdpD
in die Zytoplasmamembran unabhängig von der Sec Translokase und YidC erfolgt.
Maier et al. (2008) konnten beobachten, dass eine amphiphile Region der N-
terminalen Domäne von KdpD (AS 22-48) für die Insertion in die Membran notwendig
zu sein scheint. Diese Aminosäuresequenz konnte von Signalerkennungspartikeln
(SRP) erkannt und folglich zur Membran gebracht werden (Maier et al., 2008). Diese
Beobachtung deckt sich mit der Theorie, dass die hydrophobe Aminosäuresequenz
an den Positionen 27-43 von KdpD für die Anlagerung der N-terminalen Domäne an
die Membran verantwortlich sein könnte (Zimmann, 1995). Des Weiteren ist es für die
Funktionalität von KdpD notwenig, dass das Protein im dimerisierten Zustand in der
Zytoplasmamembran vorliegt (Heermann et al., 1998). Dass der homooligomere
Zustand einer Sensorkinase häufig von entscheidender Bedeutung für die
Funktionalität ist, konnte bereits für viele Sensorkinasen gezeigt werden (Ninfa et al.,
1993; Swanson et al., 1993; Hidaka et al., 1997).
Wie viele Histidinkinasen ist auch KdpD bifunktional, da es in vitro nicht nur eine
Kinase- und Transferaktivität aufweist, sondern aufgrund seiner Phosphataseaktivität
auch in der Lage ist, phosphoryliertes KdpE zu dephosphorylieren (Jung et al.,
1997). Es konnte gezeigt werden, dass die Dephosphorylierung von KdpE~P
ausschließlich durch die Phosphataseaktivität der Sensorkinase KdpD erfolgt (Jung
et al., 1997). Brandon et al. (2000) stellten die Hypothese auf, dass die Initiation der

1. Einleitung
14
Signaltransduktion mittels KdpD durch die Inhibierung der phospho-KdpE spezi-
fischen Phosphataseaktivität herbeigeführt wird. Es wird vermutet, dass dieser
Wechsel zwischen Kinase- und Phosphataseaktivität von elektrostatischer Natur ist
(Jung & Altendorf, 1998).
1.3. Reiz und Reizaufnahme von KdpD in E. coli Obwohl die Bedingungen, unter denen die kdpFABC-Expression induziert wird,
bekannt sind, ist die Natur des Reizes immer noch ungeklärt. Während das
kdpFABC-Operon unter K+-limitierenden Bedingungen im Medium sehr effektiv und
kontinuierlich induziert wird, findet nur ein kleiner, transienter Anstieg der Expression
bei hoher Osmolalität statt (Hamann et al., 2008). Dabei ist es für die Induzierbarkeit
des Kdp-Systems von Bedeutung, durch welches Osmolyt die hohe Osmolalität im
Medium verursacht wird. Gowrishankar et al. (1985) konnten beobachten, dass eine
Erhöhung der Osmolalität im Medium und damit eine Verminderung des Turgor-
drucks nur dann eine kdpFABC-Expression auslöst, wenn es sich bei der osmotisch
wirksamen Substanz um ein Salz und nicht um einen Zucker handelt. In Salmonella
enterica konnte ebenfalls gezeigt werden, dass die Expression des kdpFABC-
Operons deutlich geringer durch Sucrose als durch NaCl ausfällt (Balaji et al., 2005).
Hamann et al. (2008) beobachteten, dass sich das Niveau und der Zeitraum der
kdpFABC-Expression unter K+-Limitation und hoher Osmolalität deutlich voneinander
unterscheiden. Unter K+-Limitation kam es zu einem tausendfachen Induktions-
verhältnis, welches schon Sekunden nach einem K+-„downshift“ gemessen wurde
und über den gesamten Zeitraum andauerte. Hingegen konnte durch Zugabe von
NaCl im Medium beobachtet werden, dass nur eine transiente Synthese des
KdpFABC-Systems mit einem hundertfachen Induktionsverhältnis erst Minuten nach
Zugabe des Salzes erfolgte. Wurde allerdings KCl als Salz zum Medium hinzugefügt,
so konnte keine kdpFABC-Expression ermittelt werden. Des Weiteren wurde bei
einem durch Zucker induzierten Stress nur eine transiente Expression mit einem
zehnfachen Induktionsverhältnis Minuten nach der Zugabe bestimmt. Aufgrund
dieser Ergebnisse vermuten Hamann et al. (2008), dass KdpD diese Reize unter-
schiedlich wahrnimmt. Heermann et al. (2009) konnten zeigen, dass das Stress-
protein UspC die Signalkaskade von KdpD / KdpE durch Interaktion mit der Usp-
Domäne von KdpD unter Salzstress stimuliert. Des Weiteren wurde von Jung &

1. Einleitung
15
Altendorf (2003) postuliert, dass KdpD wahrscheinlich eine Mischung verschiedener
Parameter wahrnimmt: Änderungen des Turgors, der Phospholipidzusammen-
setzung der Membran, der extra- und / oder intrazellulären K+-Konzentration, der
Osmolalität des Mediums, der Ionenstärke des Zytoplasmas und der internen ATP-
Konzentration. Allerdings zeigen die Ergebnisse von Hamann et al. (2008) deutlich,
dass weder die Änderung des Turgors noch die Änderung der Konzentration einiger
zytoplasmatisch gelöster Substanzen wie K+, ATP, Putrescine, Spermidine, Treha-
lose, Glutamat und Prolin der Stimulus für KdpD sein kann. Die Möglichkeit, dass der
K+-Gradient über der Zytoplasmamembran eine Rolle bei der Reizwahrnehmung
spielt, kann ebenfalls ausgeschlossen werden, da die kdpFABC-Expression von der
Anwesenheit anderer K+-Transportsysteme abhängig ist. Ein Wildtypstamm, welcher
die konstitutiven und niedrig affinen K+-Aufnahmesysteme Trk und Kup besitzt,
nimmt z. B. bei K+-Konzentrationen von 20 mM im Medium Kalium-Ionen über die
Trk- und Kup-Systeme auf, während der KdpFABC-Komplex nicht synthetisiert wird.
Hingegen synthetisiert unter denselben K+-Konzentrationen bereits ein Stamm, der in
den Genen dieser niedrig affinen K+-Aufnahmesystemen deletiert ist, den KdpFABC-
Komplex, um K+-Ionen aufnehmen zu können. Folglich kommt es im ersten Falle zu
keiner und im letzteren zu einer Induktion der kdpFABC-Expression, obwohl derselbe
K+-Gradient über der Zytoplasmamembran vorliegt (Abb. 1.5).
Abb. 1.5: kdpFABC-Expression in Abhängigkeit der niedrig affinen K +-Aufnahme-systeme in E. coli (verändert nach Hamann, 2008). In A ist ein Wildtypstamm dargestellt, der keine Mutation in den Genen der niedrig affinen K+-Aufnahmesystemen Trk und Kup aufweist, in B ein Stamm, der die Gene, welche für diese K+-Transportsysteme kodieren, deletiert hat. Obwohl der K+-Gradient über der Zytoplasmamembran in beiden Stämmen identisch ist, kommt es nur in einem der Stämme zur kdpFABC-Expression.
Bereits früher wurde die Hypothese aufgestellt, dass das positiv geladene Arginin-
cluster hinter der vierten Transmembranhelix mit den anionischen Phospholipiden
durch elektrostatische Wechselwirkung interagiert (Jung & Altendorf, 1998).
A) B)

1. Einleitung
16
Zimmann et al. (2007) konnten zeigen, dass einzelne Aminosäureaustausche nur in
diesem Arginincluster und der vierten Transmembrandomäne von KdpD zu einem
(semi-)konstitutiven Phänotyp in Bezug auf die kdpFABC-Expression unter K+-Limi-
tation führen, was vermutlich durch eine verringerte Phosphataseaktivität verursacht
wird. Deshalb ist es vorstellbar, dass solche Derivate von KdpD einen „locked-on“
Zustand annehmen (Zimmann et al., 2007). Des Weiteren konnte ein stimulierender
Einfluss von negativ geladenen Phospholipiden auf die Kinaseaktivität von KdpD in
vitro demonstriert werden (Stallkamp et al., 1999).
Darüber hinaus könnte der Aktivierung der Kinaseaktivität von KdpD eine Interaktion
mit einem anderen Protein zugrunde liegen. Für Mycobacterium tuberculosis konnte
von Steyn et al. (2003) gezeigt werden, dass die Lipoproteine LprF und LprJ mit dem
N-terminalen zytoplasmatischen Teil der Sensorkinase KdpD wechselwirken und so
die kdpFABC-Expression modulieren können.
Es gibt verschiedenen Hypothesen, welcher Bereich der Sensorkinase KdpD von
E. coli den Reiz wahrnimmt. Während von Heermann et al. (2003) der N-terminale
Bereich vorgeschlagen wird, wird von Rothenbücher et al. (2006) angenommen,
dass die zytoplasmatische C-terminale Domäne von KdpD als K+-Sensor fungiert.
Diesen Theorien liegen Ergebnisse verschiedener Konstrukte, die Teile der Sensor-
kinase deletiert haben, zugrunde. Heermann et al. (2003) zeigten mit einem
Konstrukt, dass nur die ersten 395 Aminosäuren (KdpD/1-395) von KdpD aus-
reichen, um die Bindung von KdpE an die DNA zu stabilisieren und somit die
Expression des kdpFABC-Operons zu induzieren, wobei die Phosphorylierung von
KdpE vermutlich durch niedermolekulare Phosphodonoren stattfindet. Die N-
terminale Domäne weist einen semi-konstitutiven Phänotyp in Bezug auf die
kdpFABC-Expression auf, obwohl die N-terminale Domäne aufgrund der fehlenden
Phosphorylierungsstelle im C-Terminus nicht in der Lage ist, das Kdp-System über
die normale Signalkaskade anzuschalten. In vitro konnte KdpD/1-395 weder
phosphoryliert noch der Phosphotransfer auf den Antwortregulator KdpE gezeigt
werden. Auch die Dephosphorylierung von KdpE~P durch KdpD/1-395 konnte in vitro
nicht gezeigt werden (Heermann et al., 2003a). Die N-terminale Domäne ist trotz der
fehlenden Transmembrandomänen an die Zytoplasmamembran assoziiert (Heer-
mann, 2001). Ein möglicher Grund für diese Anlagerung an die Membran könnte die
hydrophobe Aminosäuresequenz an den Positionen 27-43 von KdpD sein. Im

1. Einleitung
17
Gegensatz dazu wurde von Rothenbücher et al. (2006) beobachtet, dass ein aus den
Aminosäureresten 499-894 (C-terminale Domäne) bestehendes KdpD-Derivat eben-
falls wie das Konstrukt von Heermann et al. (2003) einen semi-konstitutiven Phäno-
typ aufweist und somit unter normalerweise nicht-kdpFABC-induzierenden
Bedingungen die Expression von kdpFABC induziert. Da jedoch die kdpFABC-
Expression im Fall der aus der C-TD bestehendes Derivat sehr niedrig ist, ist die
Aussage dieser Untersuchung der zytoplasmatischen C-TD von KdpD eine Funktion
als K+-Sensor zuzuschreiben (Rothenbücher et al., 2006) mehr als zweifelhaft. Diese
kritische Bewertung steht im Einklang mit der Beobachtung, dass die intrazelluläre
K+-Konzentration als Stimuli unwahrscheinlich ist (Malli & Epstein, 1998; Hamann et
al., 2008).
1.4. Die Zytoplasmamembran und ihre Phospholipide
Da gezeigt werden konnte, dass solubilisiertes KdpD in vitro keine Kinaseaktivität
aufweist (Zimmann, 1995), scheint die Zytoplasmamembran der Zelle eine wichtige
Rolle für die Funktionalität der Sensorkinase KdpD zu spielen. Die Zellhülle von
E. coli besteht aus drei verschiedenen Typen von Phospholipiden, welche über den
typischen „Kennedy Syntheseweg“ hergestellt werden (zum Überblick siehe Dowhan
et al., 2004). Das häufigste Phospholipid ist das zwitterionische Phosphatidylethanol-
amin (PE), die zwei anderen sind die anionischen Phospholipide Phosphatidyl-
glycerol (PG) und Cardiolipin (CL oder Diphosphatidylglycerol). Die hydrophilen Kopf-
gruppen der Phospholipide bilden die Oberfläche einer Lipiddoppelschicht, während
sich die hydrophoben Fettsäureketten im Inneren dieser Schicht befinden. Folglich
bestimmen die Kopfgruppen viele der strukturellen und chemischen Eigenschaften
der Oberfläche und die Fettsäuren die physikalischen Eigenschaften der Membran.
Das zwitterionische PE und das einfach negativ geladene PG besitzen zwei Fett-
säureketten, während das zweifach negativ geladene CL unter anderem aus vier
Fettsäureketten besteht, welche den hydrophoben Teil des Lipids bilden. Der Aufbau
der Phospholipide von E. coli ist in Abbildung 1.6 dargestellt. Die Phospholipide
haben verschiedene Fettsäureketten, welche innerhalb jedes Phospholipidtyps auch
unterschiedlich häufig vorkommen. Die häufigsten Fettsäuren von E. coli sind 16:0
und 16:1 cis 9 (Cronan, 1968). Die Zusammensetzung der Fettsäuren in der
Membran wird unter anderem durch die Temperatur beeinflusst. Die Erniedrigung der

1. Einleitung
18
Wachstumstemperatur führt in vielen Organismen zum Anstieg des Anteils der
ungesättigten Fettsäuren der Phospholipide (de Mendoza & Cronan, 1983). Dadurch
können Funktionen wie z.B. die Fluidität der Membran bei niedrigeren Temperaturen
aufrecht erhalten werden.
Abb. 1.6: Aufbau der Phospholipide. In A ist die Struktur der Phospholipide schematisch dargestellt (Quelle: www. bioteach.ubc.ca). In B sind die Strukturen der drei Hauptphospho-lipide PE, PG und CL von E. coli gezeigt (verändert nach Bogdanov et al., 2008). Das Rückgrad der Kopfgruppe ist fett hervorgehoben. Außerdem sind die mit „R“ gekenn-zeichneten Fettsäureketten in ihrer Länge und mit bzw. ohne Doppelbindungen variabel.
Die Phospholipidzusammensetzung von E. coli beträgt im Vollmedium circa
75 % PE, 20 % PG und 5 % CL (Cronan, 1968; Randle et al., 1969). Damit sind etwa
25 % der Lipide (PG und CL) negativ geladen. Außerdem konnten die oben ge-
nannten Arbeitsgruppen zeigen, dass in der stationären Wachstumsphase die Menge
von CL auf Kosten von PG um das 2-3 fache ansteigt, wodurch der Gehalt der
anionischen Phospholipide konstant bleibt (Cronan, 1968; Randle et al., 1969). Die
Phospholipide bilden in Gram-negativen Bakterien die Lipiddoppelschicht der
Zytoplasmamembran und die innere Schicht der äußeren Membran.
Die Biosynthese der drei Phospholipide von E. coli findet in 2-3 Schritten aus dem-
selben Vorstufenmolekül CDP-Diacylglycerol statt (Abb. 1.7). Das zwitterionische PE
wird durch die Herstellung von Phosphatidylserin (PS), welches als kurzzeitiges
Zwischenprodukt fungiert, synthetisiert. Die PS-Synthese erfolgt durch das Gen-
produkt des pssA-Gens und die PE-Synthese durch das des psd-Gens. Das
A) B)

1. Einleitung
19
anionische PG wird ebenfalls wie PE in zwei Schritten synthetisiert, während
Phosphatidylglycerol-3-Phosphat als Zwischenprodukt von PG fungiert. Das Gen-
produkt des pgsA-Gens katalysiert dabei den ersten Schritt, während das Enyzm mit
dem dazugehörigen Gen für die Synthese von PG bis jetzt noch unbekannt ist. Alle
Schritte dieses Syntheseweges in E. coli sind homolog zu dem Biosyntheseweg in
eukaryotischen Zellen mit Ausnahme der CL-Synthese. In E. coli wird CL produziert,
wenn das Enzym CL-Synthase (kodiert durch das cls-Gen) die Kondensation von
zwei PG Molekülen katalysiert und dabei Glycerol freisetzt.
Abb. 1.7: Biosyntheseweg der Phospholipide von E. coli (verändert nach Cronan, 2003). Dabei sind die Phospholipide rot, die Gene, deren Produkte den jeweiligen Schritt katalysieren, blau und benötigte zusätzliche bzw. freigesetzte Produkte grün dargestellt.
Des Weiteren sind die Auswirkungen von Mutationen in diesem Syntheseweg
drastisch und die Phänotypen der Stämme charakteristisch. Wenn eine Deletion des
psgA-Gens vorliegt, so dass kein PG mehr synthetisiert werden kann, kann ebenfalls
kein CL gebildet werden, was zu einer Phospholipidzusammensetzung ohne jegliche
anionische Lipide führt. Damit dieser Stamm ohne PG und CL überhaupt lebensfähig
ist, ist eine weitere Deletion des lpp-Gens essentiell, welches für das häufigste
Lipoprotein in E. coli kodiert (Shiba et al., 2004). Lpp ist in der aktiven Form mit der
Peptidoglycanschicht verbunden und fungiert so als Stabilisator und sorgt für die
Integrität der Zellhülle (Cascales et al., 2002). Um Lpp zu synthetisieren, benötigt die
Vorstufe (proLpp) den Diacylglyceryl-Rest von PG. In einem ∆pgsA lpp+ Stamm ist

1. Einleitung
20
dieser Syntheseschritt aufgrund der Abwesenheit von PG nicht möglich, so dass
proLpp größtenteils in der Zytoplasmamembran verbleibt und über den C-terminalen
Lysinrest das Peptidoglycan bindet, welches dadurch die innere mit der äußeren
Membran auf eine ungewöhnliche Weise verbindet, was letztendlich zur Zelllyse führt
(Shiba et al., 2004).
1.5. Einfluss der Membranzusammensetzung auf Membra nproteine durch die Interaktionen mit Phospholipiden
Stämme mit Mutationen in den Genen, welche für Enzyme kodieren, die in die
Biosynthese der Phospholipide involviert sind, resultieren in einer modifizierten
Phospholipidzusammensetzung. Dabei ist zu erwähnen, dass Änderungen in dieser
Zusammensetzung signifikante Effekte auf den gesamten Metabolismus der Zelle
haben. Die Bedeutung der Phospholipide konnte dadurch gezeigt werden, dass das
Fehlen des Hauptlipids PE zu Defekten essentieller Prozesse wie z.B. der Zellteilung
oder zu Konformationsänderungen von Membranproteinen führt (zum Überblick
siehe Dowhan et al., 2004). So konnte eine spezifische Rolle der Phospholipide für
die Topologie und die damit verbundene Aktivität für mehrere Membranproteine
gezeigt werden. Für die Lactose Permease LacY von E. coli konnte eine PE-ab-
hängige Topologie beobachtet werden (Xie et al., 2006). Die Abwesenheit von PE in
der Zytoplasmamembran führte neben einer Konformationsänderung des Proteins in
der Membran auch zum Verlust seiner Transportaktivität von Lactose und H+-Ionen
(Bogdanov et al., 1996). Die Topologie und die Transportaktivität der Permeasen
PheP und GabP werden ebenfalls von PE beeinflusst (Dowhan et al., 2004). Auch für
andere Bakterien konnten solche Interaktionen gezeigt werden. In Lactococcus lactis
interagiert der „multidrug-transporter“ LmrP mit den Kopfgruppen von PE
(Hakizimana et al., 2008). Bogdanov et al. (2008) stellten daher unter Einbezug der
„positive-inside rule“ (Von Heijne, 1986) die Theorie auf, dass PE die negative
Ladung von Aminosäureresten neutralisiert und dadurch die zytoplasmatische
Orientierung dieser Bereiche favorisiert wird, während in Abwesenheit von PE diese
Bereiche aufgrund ihrer stärker negativ wirkenden Ladungen und der „Proton motive
force“ (Pmf) in das Periplasma gelangen.
Für das Phospholipid CL konnte ebenfalls ein Zusammenhang mit verschiedenen
Membranproteinen beobachtet werden. Für die Glycosyltransferase MurG aus E. coli

1. Einleitung
21
konnte mittels einer Co-Reinigung gezeigt werden, dass eine erhöhte Bindeaffinität
zu CL besteht (Van den Brink-van der Laan et al., 2003). Des Weiteren konnte
beobachtet werden, dass CL die polare Lokalisation des Osmosensors ProP in
E. coli kontrolliert (Romantsov et al., 2008). Es konnte gezeigt werden, dass die
Zytoplasmamembran von E. coli und anderen Bakterien heterogen zusammen-
gesetzt ist (zum Überblick siehe Matsumoto et al., 2006). Domänen von CL konnten
mit einem spezifischen Farbstoff am Septum und an den Zellpolen lokalisiert werden
(Mileykovskaya & Dowhan, 2000). Es wird sogar vermutet, dass einige Membran-
proteine diese CL-Domänen stabilisieren (Matsumoto et al., 2006). Möglicherweise
könnte auch KdpD eine solche stabilisierende Funktion auf die CL-Domänen haben.
Ebenfalls konnte für das anionische PG ein Einfluss auf weitere Membranproteine
festgestellt werden. Für den Betaintransporter BetP aus Corynebacterium
glutamicum konnte beobachtet werden, dass neben der Osmolalität und der
Temperatur auch die Zusammensetzung der Phospholipide die Aktivität moduliert
(Özcan et al., 2007). Derivate von BetP wurden in ihrer inaktiven Konformation durch
das anionische PG stabilisiert (Schiller et al., 2006). Folglich sind Wechselwirkungen
zwischen den Kopfgruppen der Phospholipide und den Membranproteinen wahr-
scheinlich von der Ladung der jeweiligen Kopfgruppe abhängig.
1.6. Aufgabenstellung Der Schwerpunkt dieser Arbeit lag auf der Bestimmung der physiologischen
Zusammensetzung der Phospholipide von E. coli unter K+-Limitation. K+-limitierende
Wachstumsbedingungen sind am effektivsten, um eine permanente kdpFABC-
Expression zu erzeugen und stellen den Hauptreiz für die Sensorkinase KdpD dar
(Hamann et al., 2008). Wenn eine Änderung der Kopfgruppen in der exponentiellen
Wachstumsphase unter K+-Limitation beobachtet werden könnte, dann würden die
Reize, die das kdpFABC-Operon induzieren, auch einen Einfluss auf die Lipid-
zusammensetzung der Membran haben. Es konnte bereits gezeigt werden, dass bei
hoher Osmolalität im Medium der Gehalt von CL auf Kosten von PE bereits in der
exponentiellen Wachstumsphase ansteigt (Tsatskis et al., 2005). Folglich könnte
auch unter K+-Limitation im Medium der CL-Gehalt variieren. Ein Zusammenhang
zwischen der Lipidkomposition der Membran und der Aktivität der Sensorkinase
KdpD wäre demzufolge vorstellbar. Stallkamp et al. (1999) konnten bereits zeigen,

1. Einleitung
22
dass anionische Phospholipide einen stimulierenden Einfluss auf die Kinaseaktivität
der Sensorkinase KdpD haben.
Für diesen Ansatz sollte die Phospholipidzusammensetzung verschiedener E. coli
Stämme, die Neusynthese von CL und die Expression des dazugehörigen Gens
unter K+-Limitation bzw. direkt nach einem K+-„downshift“ sowie die Aktivität von
KdpD in verschiedenen Membranumgebungen untersucht werden, um ihre
Bedeutung für die kdpFABC-Expression zu ermitteln.

2. Material und Methoden
23
2. Material und Methoden
2.1. Materialien Für diese Arbeit wurden folgende Materialien verwendet:
Restriktionsenzyme New England Biolabs (Frankfurt)
T4 DNA-Ligase New England Biolabs (Frankfurt)
Taq DNA-Polymerase New England Biolabs (Frankfurt)
Phusion High-Fidelity DNA Polymerase New England Biolabs (Frankfurt)
“GeneRuler DNA Ladder Mix“ MBI Fermentas (St. Leon-Rot)
GelStar Lonza (Basel)
PeqGold Universal Agarose PeqLab (Erlangen)
“DNeasy-Tissue-Kit” Qiagen (Hilden)
“QIAprep-Spin-Miniprep Kit” Qiagen (Hilden)
“QIAquick-Gel-Extraction Kit” Qiagen (Hilden)
“RNeasy Mini Kit” Qiagen (Hilden)
DNAse I (RNase free) New England Biolabs (Frankfurt)
“RevertAid First Strand cDNA Synthesis
Kit” Fermentas (St. Leon-Rot)
IQ Sybr Green Supermix BioRad (München)
“PageRuler Prestained Protein Ladder“ MBI Fermentas (St. Leon-Rot)
Antiserum α-KdpD Zimmann et al. (1995)
Ziege-anti-(Kaninchen-IgG)-Antikörper-AP Rockland (Gilbertsville, Pennsylvania)
Protran Nitrocellulose-Membran Schleicher & Schüll (Dassel)
Filterpapier Schleicher & Schüll (Dassel)
Magermilchpulver Roth (Karlsruhe)
5-Brom-4-chlor-3-indoxylphosphat (BCIP) Biomol (Hamburg)
O-Nitrophenyl-β-D-galactopyranosid
(o-NPG) Sigma-Aldrich (Taufkirchen)

2. Material und Methoden
24
5-Brom-4-chlor-3-indoxyl-β-D-
galactopyranosid (X-Gal) Biomol (Hamburg)
32P-Phosphat Hartmann Analytic (Braunschweig)
[γ-32P] ATP Hartmann Analytic (Braunschweig)
Isopropyl-1-thio-β-D-Galactosid (IPTG) PeqLab (Erlangen)
„E. coli total lipid extract“ Avanti Polar Lipids (Alabaster, USA)
1,1´,2,2´-Tetraoleoyl Cardiolipin
(synthetisches CL) Avanti Polar Lipids (Alabaster, USA)
1,2-Dioleoyl-sn-Glycero-3-Phosphatidyl-
ethanolamin (synthetisches PE) Avanti Polar Lipids (Alabaster, USA)
1,2-Dioleoyl-sn-Glycero-3-Phosphatidyl-
cholin (synthetisches PC) Avanti Polar Lipids (Alabaster, USA)
L-α-Phosphatidylcholin (Egg PC) Sigma-Aldrich (Taufkirchen)
DC-Platten Sil G-25 (20 x 20 cm) Macherey-Nagel (Düren)
Molybdänblau Reagenz Sigma-Aldrich (Taufkirchen)
Ni2+-NTA Qiagen (Hilden)
n-Dodecyl-β-Maltosid (DDM) Glycon (Luckenwalde)
Bio-Beads AM-2 BioRad (München)
Alle hier nicht aufgeführten Materialien wurden von den Firmen Amersham
Biosciences (Freiburg), AppliChem (Darmstadt), Bayer (Leverkusen), Biomol
(Hamburg), BioRad (München), Biozym Diagnostics (Hess. Oldendorf), Fluka (Neu-
Ulm), Gibco/BRL (Eggenstein), Invitrogen (Karlsruhe), E. Merck (Darmstadt),
Riedel-de Häen (Seelze), Roche Diagnostics (Mannheim), Roth (Karlsruhe), Serva
(Heidelberg) und Sigma-Aldrich (Taufkirchen) im Reinheitsgrad “pro analysis“
bezogen.
2.2. Stämme, Plasmide und Oligonukleotide
Die im Rahmen dieser Arbeit verwendeten E. coli Stämme sind in der Tabelle 2.1
aufgelistet. Die verwendeten Plasmide sind in Tabelle 2.2 und die verwendeten
Oligonukleotide in Tabelle 2.3 aufgeführt.
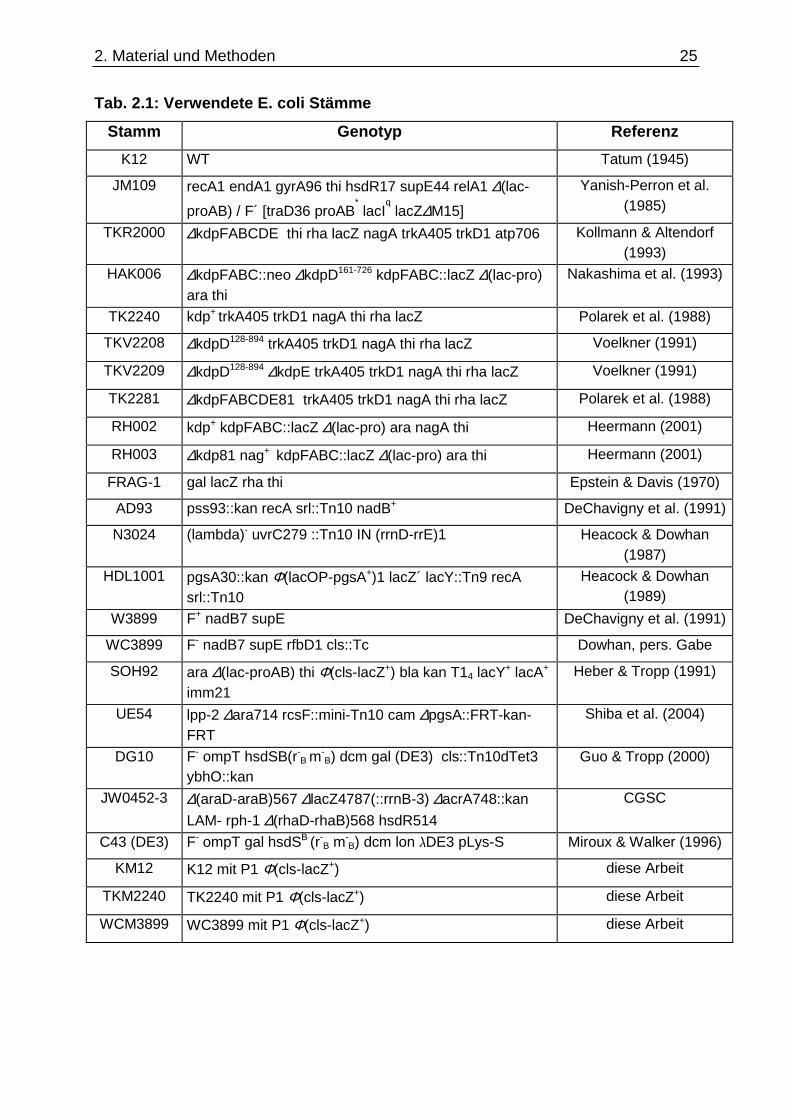
2. Material und Methoden
25
Tab. 2.1: Verwendete E. coli Stämme
Stamm Genotyp Referenz
K12 WT Tatum (1945)
JM109 recA1 endA1 gyrA96 thi hsdR17 supE44 relA1 ∆(lac-
proAB) / F´ [traD36 proAB* lacI
q lacZ∆M15]
Yanish-Perron et al. (1985)
TKR2000 ∆kdpFABCDE thi rha lacZ nagA trkA405 trkD1 atp706 Kollmann & Altendorf (1993)
HAK006 ∆kdpFABC::neo ∆kdpD161-726 kdpFABC::lacZ ∆(lac-pro) ara thi
Nakashima et al. (1993)
TK2240 kdp+ trkA405 trkD1 nagA thi rha lacZ Polarek et al. (1988)
TKV2208 ∆kdpD128-894 trkA405 trkD1 nagA thi rha lacZ Voelkner (1991)
TKV2209 ∆kdpD128-894 ∆kdpE trkA405 trkD1 nagA thi rha lacZ Voelkner (1991)
TK2281 ∆kdpFABCDE81 trkA405 trkD1 nagA thi rha lacZ Polarek et al. (1988)
RH002 kdp+ kdpFABC::lacZ ∆(lac-pro) ara nagA thi Heermann (2001)
RH003 ∆kdp81 nag+ kdpFABC::lacZ ∆(lac-pro) ara thi Heermann (2001)
FRAG-1 gal lacZ rha thi Epstein & Davis (1970)
AD93 pss93::kan recA srl::Tn10 nadB+ DeChavigny et al. (1991)
N3024 (lambda)- uvrC279 ::Tn10 IN (rrnD-rrE)1 Heacock & Dowhan (1987)
HDL1001 pgsA30::kan Φ(lacOP-pgsA+)1 lacZ´ lacY::Tn9 recA srl::Tn10
Heacock & Dowhan (1989)
W3899 F+ nadB7 supE DeChavigny et al. (1991)
WC3899 F- nadB7 supE rfbD1 cls::Tc Dowhan, pers. Gabe
SOH92 ara ∆(lac-proAB) thi Φ(cls-lacZ+) bla kan T14 lacY+ lacA+ imm21
Heber & Tropp (1991)
UE54 lpp-2 ∆ara714 rcsF::mini-Tn10 cam ∆pgsA::FRT-kan-FRT
Shiba et al. (2004)
DG10 F- ompT hsdSB(r-B m
-B) dcm gal (DE3) cls::Tn10dTet3
ybhO::kan Guo & Tropp (2000)
JW0452-3 ∆(araD-araB)567 ∆lacZ4787(::rrnB-3) ∆acrA748::kan
LAM- rph-1 ∆(rhaD-rhaB)568 hsdR514
CGSC
C43 (DE3) F- ompT gal hsdSB (r-B m-
B) dcm lon DE3 pLys-S Miroux & Walker (1996)
KM12 K12 mit P1 Φ(cls-lacZ+) diese Arbeit
TKM2240 TK2240 mit P1 Φ(cls-lacZ+) diese Arbeit
WCM3899 WC3899 mit P1 Φ(cls-lacZ+) diese Arbeit
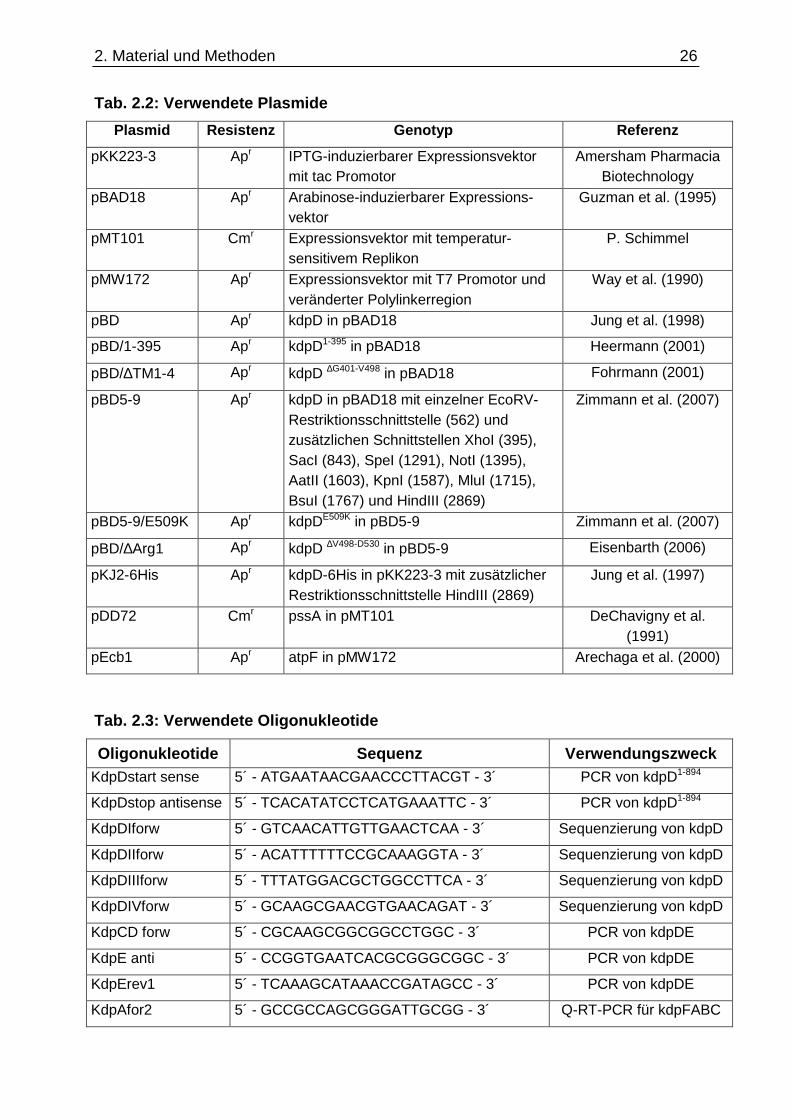
2. Material und Methoden
26
Tab. 2.2: Verwendete Plasmide
Plasmid Resistenz Genotyp Referenz
pKK223-3 Apr IPTG-induzierbarer Expressionsvektor mit tac Promotor
Amersham Pharmacia Biotechnology
pBAD18 Apr Arabinose-induzierbarer Expressions-vektor
Guzman et al. (1995)
pMT101 Cmr Expressionsvektor mit temperatur-sensitivem Replikon
P. Schimmel
pMW172 Apr Expressionsvektor mit T7 Promotor und veränderter Polylinkerregion
Way et al. (1990)
pBD Apr kdpD in pBAD18 Jung et al. (1998)
pBD/1-395 Apr kdpD1-395 in pBAD18 Heermann (2001)
pBD/∆TM1-4 Apr kdpD ∆G401-V498 in pBAD18 Fohrmann (2001)
pBD5-9 Apr kdpD in pBAD18 mit einzelner EcoRV-Restriktionsschnittstelle (562) und zusätzlichen Schnittstellen XhoI (395), SacI (843), SpeI (1291), NotI (1395), AatII (1603), KpnI (1587), MluI (1715), BsuI (1767) und HindIII (2869)
Zimmann et al. (2007)
pBD5-9/E509K Apr kdpDE509K in pBD5-9 Zimmann et al. (2007)
pBD/∆Arg1 Apr kdpD ∆V498-D530 in pBD5-9 Eisenbarth (2006)
pKJ2-6His Apr kdpD-6His in pKK223-3 mit zusätzlicher Restriktionsschnittstelle HindIII (2869)
Jung et al. (1997)
pDD72 Cmr pssA in pMT101 DeChavigny et al. (1991)
pEcb1 Apr atpF in pMW172 Arechaga et al. (2000)
Tab. 2.3: Verwendete Oligonukleotide
Oligonukleotide Sequenz Verwendungszweck KdpDstart sense 5´ - ATGAATAACGAACCCTTACGT - 3´ PCR von kdpD1-894
KdpDstop antisense 5´ - TCACATATCCTCATGAAATTC - 3´ PCR von kdpD1-894
KdpDIforw 5´ - GTCAACATTGTTGAACTCAA - 3´ Sequenzierung von kdpD
KdpDIIforw 5´ - ACATTTTTTCCGCAAAGGTA - 3´ Sequenzierung von kdpD
KdpDIIIforw 5´ - TTTATGGACGCTGGCCTTCA - 3´ Sequenzierung von kdpD
KdpDIVforw 5´ - GCAAGCGAACGTGAACAGAT - 3´ Sequenzierung von kdpD
KdpCD forw 5´ - CGCAAGCGGCGGCCTGGC - 3´ PCR von kdpDE
KdpE anti 5´ - CCGGTGAATCACGCGGGCGGC - 3´ PCR von kdpDE
KdpErev1 5´ - TCAAAGCATAAACCGATAGCC - 3´ PCR von kdpDE
KdpAfor2 5´ - GCCGCCAGCGGGATTGCGG - 3´ Q-RT-PCR für kdpFABC

2. Material und Methoden
27
KdpArev2 5´ - CTTCAACGGTATTCACAGCCTG - 3´ Q-RT-PCR für kdpFABC
GapAfor1 5´ - CTCCACTCACGGCCGTTTCG - 3´ Q-RT-PCR für gap
GapArev1 5´ - CTTCGCACCAGCGGTGATGTG -3´ Q-RT-PCR für gap
Clsfor2 5´ - ATGACAACCGTTTATACGTTG - 3´ PCR von cls1-486
Clsrev2 5´ - TTACAGCAACGGACTGAAGAA - 3´ PCR von cls1-486
Clsfor-35 5´ - TTTGCCAGATGAATCCATAT - 3´ Seq. der cls-lacZ Fusion
Clsfor+870 5´ - CAATATTATGCCGTTTGAAC - 3´ Seq. der cls-lacZ Fusion
LacZrev 5´ - CCGCTTCTGGTGCCGGAAACC - 3´ PCR der cls-lacZ Fusion
Cls-out at start 5´ - TCACCAACGTATAAACGGTTGTCAT - 3´ RT-PCR für cls-kch
Kch-out at end 5´ - CAGTAAAGAATCGGCGCAAAAATAG - 3´ RT-PCR für cls-kch
2.3. Medien und Anzuchtverfahren
Die Anzucht der in Tabelle 2.1 aufgeführten E. coli Stämme erfolgte in KML-Medium
(1 % KCl; 1 % Trypton; 0.5 % Hefeextrakt) nach Epstein & Kim (1971). Als Minimal-
medien wurden phosphat-gepufferte Medien, ebenfalls nach Epstein & Kim (1971),
eingesetzt, die 0.4 % Glucose als C-Quelle enthielten. Der E. coli Stamm AD93
wurde anstelle des phosphat-gepufferten Minimalmediums in GM56-Medium (Nes-
meyanova et al., 1991) kultiviert, welches durch verschiedene K+-Konzentrationen
modifiziert wurde. Die Bezeichnung der Minimalmedien erfolgte gemäß ihrer K+-Kon-
zentration in mM.
Bei Stämmen, die eine Deletion im pro- oder thi-Gen besaßen, wurde den Minimal-
medien zusätzlich 10 mg/l Prolin oder 1 mg/l Thiamin zugesetzt. Komponenten wie
Casaminosäuren (0.2 %), Bactopepton (0.145 %), Nicotinsäure (2 µg/ml) und Trypto-
phan (1 mM) wurden in den entsprechenden Konzentrationen bei Bedarf ebenfalls zu
den Minimalmedien zugesetzt. Andere Komponenten wie zum Beispiel IPTG
(0.7 mM oder 1 mM) oder MgCl2 (50 mM) wurden bei Bedarf zu allen Medien
hinzugefügt.
Zur Anzucht für die β-Galaktosidase-Aktivität wurden Minimalmedien mit ent-
sprechend angegebenen Konzentrationen von K+ verwendet. Nährböden wurden
durch Zugabe von 1.5 % Agar hergestellt. Für die Herstellung von Agarplatten mit
Minimalmedien wurde zuvor 3.8 % Agar mit 6 % NaCl versetzt und dreimal mit
H2OBidest gewaschen, um K+-Ionen aus dem Agar zu entfernen.

2. Material und Methoden
28
Antibiotika wurden in Konzentrationen von 100 µg/ml (Ampicillin, Carbenicillin,
Novobiocin, Erythromycin), 50 µg/ml (Kanamycin), 34 µg/ml (Chloramphenicol) bzw.
12.5 µg/ml (Tetrazyklin) zugesetzt. Die Anzucht der Zellen erfolgte aerob bei 37°C.
Die Optische Dichte wurde bei einer Wellenlänge von 600 nm in einem Shimadzu
UV-1202 Spektralphotometer (Shimadzu, Duisburg) bestimmt.
2.3.1. Mediumshift
Um die Einflüsse von schnellen Veränderungen der Wachstumsbedingungen (ins-
besondere kdpFABC-induzierende Bedingungen) zu ermitteln, wurden wachsende
Zellen nach dem Protokoll von Hamann et al. (2008) filtriert und in neuem Medium
resuspendiert. Dafür wurden zunächst Zellen über Nacht im 5 ml des ent-
sprechenden Mediums inkubiert und am nächsten Tag in 50 ml frisches Medium
überführt. Wenn die Zellen die exponentielle Wachstumsphase (OD600 von ~0.5)
erreicht hatten, wurden sie durch eine 0.45 µm Nitrocellulose-Membran vakuum-
filtriert. Die filtrierten Zellen wurden einmal mit Minimalmedium mit nominal keinen
K+-Ionen gewaschen. Anschließend wurden die Zellen mitsamt dem Filter in neues,
vorgewärmtes Medium mit der gewünschten K+-Konzentration und mit dem gleichen
Volumen überführt. Die Zellen wurden erneut im Wasserbad bei 37°C und 200 rpm
inkubiert, so dass sie weiterwachsen konnten. Diese Prozedur dauert zwischen 30
und 60 Sekunden. Der Moment, in dem der Filter mitsamt den Zellen in den neuen
Schüttelkolben überführt wurde, wurde als Zeitpunkt Null des Wachstums in dem
neuen Medium definiert. Abhängig von den gemessenen Parametern wurde die erste
Probe zwischen 30 und 60 Sekunden nach dem Zeitpunkt Null genommen. Als
Kontrolle wurden die Zellen in vorgewärmtes Medium mit derselben K+-Konzentration
wie zuvor überführt, um einen möglichen Einfluss der Filterprozedur auf die
gemessenen Parameter zu beobachten.
2.3.2. Bestimmung der Lebendzellzahl
Um das Resistenzverhalten verschiedener E. coli Stämme gegenüber dem
Antibiotikum Novobiocin zu untersuchen, wurden die Stämme über Nacht in den zu
testenden Flüssigmedien angezogen und am nächsten Tag mit frischem Medium so

2. Material und Methoden
29
verdünnt, dass die Bakterienkonzentration bei 1000 Keime / ml lag. Anschließend
wurde die Verdünnung der jeweiligen Übernachtkultur auf Selektionsplatten mit
verschiedenen Konzentrationen des Antibiotikums ausplattiert, so dass auf jeder
Agarplatte die gleiche Gesamtzellzahl von 100 Keimen ausplattiert wurde. Die
Selektionsplatten wurden dann für 1 - 2 Tage bei 37°C inkubiert und die Kolonien
ausgezählt. Für die Auswertung wurde jeweils die Anzahl der Kolonien auf Platten
ohne Antibiotikum als 100 % definiert, um den prozentualen Anteil lebender Zellen in
Abhängigkeit des Antibiotikums zu bestimmen.
2.4. Molekularbiologische und genetische Methoden
2.4.1. Plasmidisolierung
Die DNA von Plasmiden wurde aus 5 ml Übernachtkultur mittels des „QIAprep Spin
Miniprep Kits“ der Firma Qiagen (Hilden) nach Angaben des Herstellers isoliert.
2.4.2. Isolierung genomischer DNA
Genomische DNA wurde aus 1 ml Übernachtkultur mittels des “DNeasy Tissue-Kits“
der Firma Qiagen (Hilden) nach Angaben des Herstellers isoliert. Es wurde dabei
nach dem empfohlenen Protokoll für Gram-negative Bakterien gearbeitet.
2.4.3. DNA-Modifikation
Die Standard-DNA-Techniken wurden, falls nicht anders beschrieben, nach Maniatis
et al. (1989) und Ausubel et al. (1987) durchgeführt. Die in vitro-Veränderungen von
DNA-Molekülen wie Restriktionen und Ligationen wurden unter den vom jeweiligen
Hersteller empfohlenen Bedingungen durchgeführt. Linearisierte Vektoren wurden
zur Verhinderung einer Religation mit Alkalischer Phosphatase behandelt. Bei neu
konstruierten Plasmiden wurde die Korrektheit durch Kontrollrestriktionen mit ent-
sprechenden Restriktionsendonukleasen und DNA-Sequenzanalyse verifiziert.

2. Material und Methoden
30
2.4.4. Elektrophoretische Auftrennung von DNA
Die analytische und präparative Auftrennung von DNA-Fragmenten erfolgte mittels
Agarose-Gelelektrophorese. Dafür wurden Gele mit 0.7 - 1.5 % (w/v) Agarose in
TAE-Puffer (40 mM Tris; 40 mM Essigsäure; 1 mM EDTA) verwendet, die mit GelStar
(5 µl der 10.000-fachen Stocklösung in 50 ml TAE Puffer) versetzt waren. Ein
anderes Färbeverfahren war das Nachfärben der Agarosegele mit GelStar (5 µl in
50 ml TAE Puffer) für 1 Stunde bei Raumtemperatur auf einer Wippe. Vor dem Lauf
wurde zu den Proben 10 x DNA-Probenpuffer (50 % Glycerin; 0.1 M EDTA; 1 %
SDS; 0.1 % Bromphenolblau) gegeben. Zur Bestimmung der DNA-Fragment-Größen
diente der „GeneRuler DNA Ladder Mix“ der Firma MBI Fermentas (St. Leon-Rot).
Der Gellauf wurde in einer “Mini Sub DNA Cell“-Agarosegel-Laufkammer (BioRad,
München) bei konstant 100 V für 30 - 60 min durchgeführt. Die Detektion der
aufgetrennten DNA erfolgte auf einem UV-Transilluminator bei 304 nm und die Doku-
mentation der Gele mit einer Gel-Dokumentationsanlage von Herolab (Wiesloch).
2.4.5. Extraktion von DNA aus Agarosegelen
DNA-Fragmente wurden mittels des “QIAquick Gel Extraction Kits“ von Qiagen
(Hilden) nach Angaben des Herstellers aus Agarosegelen extrahiert.
2.4.6. Herstellung kompetenter Zellen
Die Transformation von E. coli Zellen mit Plasmid-DNA erfolgte nach einer modifi-
zierten RbCl-Methode (Promega Technical Manual, 1994). Zur Präparation von
kompetenten Zellen wurde eine Übernachtkultur 1 : 100 in frisches Medium überimpft
und aerob bei 37°C bis zu einer Optischen Dichte vo n 0.3 - 0.5 angezogen.
Anschließend wurden die Zellen bei 5.000 rpm für 10 min zentrifugiert und in dem
halben Volumen kalter Lösung A (10 mM MOPS, pH 7.0; 10 mM RbCl) resus-
pendiert. Nach erneuter zehnminütiger Zentrifugation bei 5.000 rpm wurden die
Zellen im gleichen Volumen kalter Lösung B (100 mM MOPS, pH 6.5; 50 mM CaCl2;
10 mM RbCl) resuspendiert und für 30 min auf Eis inkubiert. Nach einem weiteren
Zentrifugationsschritt wurden die Zellen in Lösung B (10 % des Ausgangsvolumens)
aufgenommen und dann direkt für Transformationen verwendet. Für eine längere

2. Material und Methoden
31
Aufbewahrung wurden die kompetenten Zellen mit 10 % Glycerin versetzt und nach
Schockgefrieren in flüssigem Stickstoff bei -80°C g elagert.
2.4.7. Transformation von kompetenten Zellen
Für die Transformation wurden 200 µl kompetente Zellen mit 100 - 200 ng Plasmid-
DNA (1 µl) oder einem kompletten Ligationsansatz mindestens eine Stunde auf Eis
inkubiert. Nach einem Hitzeschock (1.5 min bei 42°C ) und 10 minütiger Inkubation
auf Eis wurde zu jedem Ansatz 0.8 ml KML-Medium gegeben und für mindestens
eine Stunde aerob bei 37°C inkubiert. Anschließend wurden die Zellen auf geeig-
neten Selektionsnährböden ausplattiert. Bei Plasmidtransformationen wurden 100 µl
des Transformationsansatzes und bei Transformationen mit Ligationsansätzen wurde
der gesamte Ansatz ausplattiert. Es folgte eine Inkubation der Platten über Nacht bei
37°C.
2.4.8. Polymerasekettenreaktion
Die Amplifikation verschiedener Gene (kdpFABC, kdpDE oder cls) erfolgte mit der
Polymerasekettenreaktion (PCR). Als Matrize wurde chromosomale DNA oder
Plasmid-DNA eingesetzt und als Primer dienten jeweils zwei synthetisierte Oligo-
nukleotide, die beide komplementär zu gewünschten Abschnitten der Sequenz der
jeweiligen DNA-Fragmente bzw. in dem entsprechenden DNA-Abschnitt lokalisiert
sind. Die jeweils zwischen den Primern liegende Sequenz wurde mit der thermo-
stabilen Phusion-High-Fidelity DNA-Polymerase (für Konstruktionen) oder der eben-
falls thermostabilen Taq-DNA-Polymerase aus Thermus aquaticus (für Kontrollen)
amplifiziert. Im Einzelnen bestanden die PCR-Ansätze aus folgenden Komponenten:
10 ng Matrize, 15 pmol Primer sense und 15 pmol Primer antisense, 10 pmol dNTP,
1/10 des Endvolumens 10 x PCR-Puffer, 1/10 des Endvolumens Taq-Polymerase
und H2OBidest. Die Denaturierung der doppelsträngigen DNA erfolgte dabei pro
Reaktionszyklus für 0.8 min bei 96°C, das Anlagern der Primer für 0.8 min bei
Temperaturen entsprechend der Primer und die DNA-Amplifikation bei 72°C für
x Minuten (1000 bp / min) in Abhängigkeit der bp-Länge des gewünschten PCR-
Produktes. Nach 30 Zyklen wurde die Reaktion beendet und die Reaktionsansätze
bis zur Weiterbearbeitung bei 4°C gekühlt.

2. Material und Methoden
32
2.4.9. DNA-Sequenzanalyse
Die Sequenzierung von doppelsträngiger DNA erfolgte nach dem Prinzip des “Cycle
Sequencing”, welches auf dem nach Sanger et al. (1977) beschriebenen Ketten-
abbruchverfahren mit Didesoxynukleotiden basiert. Als Primer fanden verschiedene
synthetische Oligonukleotide (Operon, Köln) Verwendung, welche komplementär zu
gewünschten Abschnitten der Sequenz der jeweiligen DNA-Fragmente waren.
Die Sequenzierung der entsprechenden Plasmide oder PCR-Fragmente wurde durch
die Firma GATC Biotech AG (Konstanz) mit den passenden Primern durchgeführt
und mit der Software Chromas (Version 1.43) und SE Central (Clone Manager,
Version 8) ausgewertet.
2.4.10. Isolierung von RNA
Die Isolierung von RNA erfolgte aus dem Pellet von 1 ml Zellkultur (bei verschie-
denen Optischen Dichten) mittels des „RNeasy Mini Kits“ der Firma Qiagen (Hilden)
nach Angaben des Herstellers. Die Bestimmung der RNA-Konzentration erfolgte mit
dem BioPhotometer (Eppendorf, Hamburg). Zur weiteren Verwendung wurde die
isolierte RNA mit RNAse-freiem Wasser auf eine Konzentration von 20 µg/ml ein-
gestellt.
2.4.11. cDNA Synthese
Für die Synthese von cDNA wurde zunächst die isolierte RNA für 1 Stunde bei 37°C
und anschließend 10 min bei 70°C mit DNAse I (New E ngland Biolabs, Frankfurt)
inkubiert, um restliche Verunreinigungen von DNA zu entfernen. Anschließend
erfolgte die Synthese der cDNA mittels des „RevertAid First Strand cDNA Synthesis
Kits“ der Firma Fermentas (St. Leon-Rot) nach Angaben des Herstellers.
2.4.12. Quantitative real-time RT-PCR
Um die kdpFABC-Expression zu bestimmen, wurden die Primer kdpAfor2 und
kdpArev2 für die Q-RT-PCR genommen. Als interner Standard wurden die Primer

2. Material und Methoden
33
gapAfor1 und gapArev1 für das E. coli „housekeeping“-Gen gap benutzt. Die
spezifische Bindung der Primer für die Q-RT-PCR und die Amplifikation des
gewünschten PCR-Produktes, welche einen spezifischen 200 bp großen Teil von
kdpA bzw. gap vervielfältigt, wurde zuvor durch eine herkömmliche PCR in allen
Stämmen kontrolliert. Das Q-RT-PCR-Programm mit 2 min bei 95°C, der Zyklus
15 sec bei 95°C, 30 sec bei 62°C und 30 sec bei 72° C wurde für alle Primerpaare im
iCycler (BioRad, München) 40-mal durchgeführt und zum Schluss folgte einmalig
eine 5-minütige Inkubation bei 72°C.
Um Fehlerabweichungen zu vermeiden, wurde das Niveau der Expression von kdpA,
welches durch den Cycle threshold (CT) bestimmt wurde, gegen das Expressions-
niveau des „housekeeping“-Gens gap mit Hilfe eines Excel-Datenblattes der Firma
BioRad normalisiert. Für die statistische Auswertung wurden die Daten von mehreren
Messungen gemittelt. Die Induktion der kdpFABC-Expression ist in dem Verhältnis
der normalisierten Werte unter kdpFABC-induzierenden Bedingungen (0.1 mM K+)
dividiert durch die normalisierten Werte unter nicht-kdpFABC-induzierenden
Bedingungen (115 mM K+) angegeben. Es ist dabei anzumerken, dass, obwohl die
Induktionsverhältnisse aufgrund der extrem niedrigen Werte bei nicht-kdpFABC-
induzierenden Bedingungen schwankten, die allgemeine Größenordnung der CT-
Werte konstant war und deshalb nur die Tendenz der Induktionsverhältnisse
berücksichtigt wurde. Des Weiteren wurden Reaktionen ohne Zugabe einer cDNA als
Matrize als negative Kontrolle für die Aussagekräftigkeit der CT durchgeführt.
2.4.13. P1-Phagenlysatpräparation und P1-Transdukti on
Die Präparation von P1-Lysaten verschiedener E. coli Stämme und die
anschließende Transduktion der Phagen in E. coli Stämme wurden gemäß dem
Protokoll von Arber et al. (1960) durchgeführt. Zur Präparation des P1-Lysates
wurden über Nacht 5 ml Zellkultur des „Quellstammes“ kultiviert und am nächsten
Tag davon 100 µl in 5 ml frisches KML-Medium überimpft, bis eine OD600 von 0.5
erreicht worden war. Dann wurde je 1 ml Zellkultur mit 10 mM CaCl2 und
verschiedenen Volumina P1-Phagensuspension (0 µl, 1 µl, 5 µl, 20 µl, 50 µl, 100 µl,
200 µl, 500 µl) für 20 min bei 37°C inkubiert. Ansc hließend wurde zu jedem Ansatz
7 ml vorgewärmtes KML-Medium mit 7 mM CaCl2 hinzugefügt und die Kulturen für
3 - 6 h bei 37°C aerob inkubiert, bis eine totale L yse der Zellen erfolgt war. Es wurde

2. Material und Methoden
34
nur mit der Phagenkonzentration weitergearbeitet, welche die späteste Lyse erzielte,
um möglichst viele der neugebildeten Phagen zu erhalten. Das Lysat wurde mit
0.5 ml Chloroform versetzt, kurz gevortext und 10 min bei 5.000 rpm bei 4°C
zentrifugiert. Der das Phagenlysat enthaltende Überstand wurde erneut mit 0.5 ml
Chloroform versetzt, gevortext, zentrifugiert und bis zur Weiterverwendung bei 4°C
gelagert. Um ein genetisch homogenes Phagenlysat zu erhalten, wurde ein zweiter
Infektionszyklus durchgeführt, indem die beschriebene Prozedur wiederholt wurde.
Zur Transduktion der Lysate wurden E. coli Stämme, in die das jeweilige P1-Lysat
transduziert werden sollte, über Nacht in 5 ml KML-Medium angezogen und am
nächsten Tag in KML-Medium bis zu einer OD600 von 0.5 aerob bei 37°C kultiviert.
Anschließend wurden die Zellen von 1 ml Kultur durch Zentrifugation bei 5.000 rpm
geerntet, zweimal in dem gleichen Volumen Transduktionsmedium (100 mM NaCl;
10 mM CaCl2; 10 mM KCl; 1 mM MgCl2) gewaschen und so in Transduktionsmedium
resuspendiert, dass eine OD600 von ~ 0.1 eingestellt wurde. Danach wurden je 100 µl
der Zellsuspension mit verschiedenen Volumina des P1-Phagenlysats (0 µl, 1 µl,
5 µl, 20 µl, 100 µl) für 20 min bei 37°C ohne Schüt teln inkubiert. Anschließend
wurden die Ansätze mit 0.1 ml 1 M Natriumcitrat versetzt, um die Transduktion zu
beenden. Der gesamte Ansatz wurde mit 3 ml flüssigem Top-Agar (KML-Medium mit
0.6 % Agar) gemischt, auf entsprechenden Selektivnährböden ausplattiert und 1 - 2
Tage bei 37°C inkubiert. Die Transduktanten wurden anschließend auf ihren
korrekten Phänotyp getestet.
2.5. Biochemische und analytische Methoden
2.5.1. Herstellung von SDS-Proben aus ganzen Zellen
Jeweils 1 ml Zellsuspension einer OD600 von 0.5 - 2 wurde abzentrifugiert (5 min;
14.000 rpm) und das Pellet mit 2x SDS-Probenpuffer (50 mM Tris/HCl pH 6.8; 2 %
SDS; 10 % Glycerin; 5 % Mercaptoethanol; 0.1 % Bromphenolblau) auf eine Protein-
konzentration von 2 mg/ml eingestellt. Das Pellet wurde gründlich resuspendiert und
anschließend 5 min bei 60°C erhitzt.

2. Material und Methoden
35
2.5.2. SDS-Polyacrylamidgelelektrophorese
Die elektrophoretische Auftrennung von Proteinen erfolgte mittels SDS-PAGE nach
Laemmli (1970). Dazu wurden 0.75 mm dicke Flachgele der Größe 7 x 10 cm
verwendet. Die Acrylamidkonzentration betrug im Sammelgel 4.9 % und im Trenngel
9 %. Die SDS-Gele wurden mit Hilfe von Rotiphorese-Fertiglösung (30 % (w/v)
Acrylamid; 0.8 % (w/v) Bisacrylamid) (Roth, Karlsruhe) hergestellt. Die Proteinproben
wurden vor dem Lauf mit SDS-Probenpuffer versetzt, so dass eine Endkonzentration
von 50 mM Tris/HCl, pH 6.8, 10 % Glycerol (v/v), 2 % SDS (w/v), 5 % β-Mercapto-
ethanol (v/v) und 0.1 % Bromphenolblau (w/v) erzielt wurde. Als Proteinstandard
wurde “PageRuler Prestained Protein Ladder“ verwendet. Der SDS-Laufpuffer be-
stand aus 0.025 M Tris, 0.19 M Glycin und 0.1 % SDS. Der Gellauf wurde in einer
“Mini-Protean“-Laufanlage (BioRad, München) für Minigele bei konstant 200 V durch-
geführt.
Die aufgetrennten Proteine wurden anschließend mit Serva Blau G-250 (Coomassie-
Blau) nach Weber & Osborn (1969) detektiert, wobei die Färbelösung zur Fixierung
der Proteine neben 0.25 % (w/v) Coomassie Brilliant Blue und 50 % (v/v) Methanol
zusätzlich 10 % TCA (w/v) enthielt. Die Entfärbung der Gele erfolgte in 5 % Methanol
(v/v) und 7.5 % Essigsäure (v/v).
2.5.3. Immunoblot
Der Elektrotransfer von Proteinen aus SDS-Gelen auf Nitrocellulose (Porengröße
0.45 µm) erfolgte mit Hilfe einer “Trans-Blot SD semi dry“-Anlage (BioRad, München)
für 20 min bei 20 V. Dafür wurde, nach Angaben des Herstellers, ein Puffer mit
125 mM Tris/HCl, pH 8.3; 192 mM Glycin und 20 % Methanol (v/v) verwendet.
Anschließend wurde die Nitrocellulose für eine Stunde bei 37°C in Puffer A (0.9 %
NaCl (w/v); 50 mM Tris/HCl, pH 7.4) + 5 % Magermilch (w/v) inkubiert, um die
Membran mit Protein abzusättigen und somit eine unspezifische Bindung der
Antikörper auf der Blotmembran zu verhindern. Mit Hilfe des Antiserums αKdpD
(1 : 2.500 verdünnt in Puffer A + 5 % Magermilch) konnte KdpD nachgewiesen
werden (Zimmann, 1995). Nach der Behandlung mit dem ersten Antikörper (über
Nacht bei 4°C) wurde die Membran dreimal für 10 min in Puffer A gewaschen.

2. Material und Methoden
36
Daraufhin wurde die Membran für 10 min mit Puffer A und 5 % Magermilch
gequencht. Anschließend erfolgte die Bindung des zweiten Antikörpers für 1 Stunde
bei RT. Dazu wurde, gemäß dem Protokoll nach Zimmann (1995), Ziege-anti-
(Kaninchen-IgG) konjugiert mit Alkalischer Phosphatase für die Detektion von KdpD
verwendet. Vor der Detektion wurde die Membran wieder dreimal für 15 min mit
Puffer A gewaschen. Die Entwicklung des Immunoblots erfolgte durch Inkubation in
Substratlösung (45 mM Natriumcarbonat, pH 9.5; 0.01 % Nitro-Blue-Tetrazolium
(w/v); 0.045 % 5-Bromo-4-chloro-3-indolylphosphat (w/v)).
2.5.4. Herstellung von multilamellaren Liposomen
Die Herstellung von multilamellaren Liposomen erfolgte in Anlehnung an das
Protokoll von Knol et al. (1998). Es wurde zunächst „E. coli total lipid extract“ von
Avanti aufgereinigt und anschließend mit Phosphatidylcholin (PC) aus Eiern (EggPC)
in einem Verhältnis von 3 : 1 (w/w) gemischt (Hänelt, pers. Gabe). Alternativ wurden
auch Liposomen bestehend aus synthetischen Lipiden (100 % CL bzw. 50 % PE und
50 % PC) für die Fusion mit Membranvesikeln hergestellt. Die Lipide wurden in
einem Rotationsverdampfer bei Raumtemperatur getrocknet. Der Lipidfilm wurde in
dem gleichen Volumen 96 %-iges Ethanol resuspendiert und die Lipide erneut einge-
dampft. Die Lipide wurden dann so in 50 mM KPi-Puffer, pH 7.0 aufgenommen, dass
eine Konzentration von 10 mg/ml eingestellt wurde. Die homogene Lösung wurde mit
dem Ultraschallgerät Sonifer B-15 (Branson) 4 - 6 mal für 15 sec kontinuierlich unter
Stickstoffgas und im Eiswasser beschallt, wobei zwischen den Wiederholungen
immer eine 45 sec Pause eingehalten wurde. Anschließend wurde die Liposomen-
lösung (kleine Vesikel) in flüssigem Stickstoff eingefroren und langsam wieder bei
Raumtemperatur aufgetaut. Dieser Einfrier-Auftau-Schritt wurde insgesamt dreimal
durchgeführt, um große multilamellare Liposomen zu erhalten, die heterogen in ihrer
Größe und Anzahl der Lamellaren waren. Die Liposomen wurden bis zum Gebrauch
in flüssigem Stickstoff gelagert. Vor dem Gebrauch wurden die Liposomen 11-mal
durch einen Polycarbonat-Filter mit einer Porengröße von 400 nm extrudiert, um eine
homogene Lösung an unilamellaren Liposomen zu erhalten. Die Überprüfung der
richtigen Formation der Leerliposomen erfolgte im SLM-Aminco 8100 Spektrofluoro-
meter (SLM-Aminco) nach Elferink et al. (1992).

2. Material und Methoden
37
2.5.5. Präparation von invertierten Membranvesikeln
Invertierte Membranvesikel wurden nach dem Protokoll von Siebers & Altendorf
(1988) hergestellt. In Abwandlung zu diesem Protokoll wurde ein Aufschlusspuffer
verwendet, der nicht Hepes/Tris, sondern Tris/HCl, pH 7.5 enthielt. Aufgeschlossen
wurden die Zellen am Aufschlussgerät Basic-Z (Düse: 0,18 mm, Aufschlussdruck:
1,35 kbar). Nicht aufgeschlossene Zellen und Zelltrümmer wurden durch einen
niedertourigen Zentrifugationsschritt für 10 min bei 10.000 rpm und 4 °C entfernt. Die
invertierten Membranvesikel wurden bei 42.000 rpm in einer Beckman L-80 XP
Ultrazentrifuge pelletiert, einmal in niederionischem Puffer (1 mM Tris/HCl, pH 7.5;
3 mM EDTA, pH 7.5; 0.5 mM PMSF) gewaschen und anschließend in TG-Puffer
(50 mM Tris/HCl, pH 7.5; 10 % Glycerol (v/v)) aufgenommen. Die Lagerung der
Vesikel erfolgte in flüssigem Stickstoff.
2.5.6. Reinigung von KdpD-6His
Die Reinigung von KdpD-6His erfolgte mittels Ni-NTA Affinitätschromatographie im
Batchverfahren nach dem Protokoll von Jung et. al (1997). Aus invertierten
Membranvesikeln (Endkonzentration 5 mg/ml) wurde KdpD-6His in Gegenwart von
0.6 M NaCl durch schrittweise Zugabe von Lauryldimethylaminoxid (LDAO) bis zu
einer Endkonzentration von 2 % solubilisiert. Nach 30 min Rühren auf Eis wurde der
Solubilisationsansatz zentrifugiert (70.000 rpm, 1 h, 4°C). Der Überstand wurde
anschließend zu der gewaschenen und equilibrierten Ni-NTA Agarose (Qiagen,
Hilden) gegeben und für 30 min bei 4°C auf einer Wi ppe inkubiert. Die Reinigung von
KdpD-6His erfolgte durch dreimaliges Waschen mit Equilibrierungspuffer (50 mM
Tris/HCl, pH 7.5; 0.04 % Dodecylmaltosid; 10 mM Imidazol; 0.5 M NaCl; 10 mM β-
Mercaptoethanol; 10 % Glycerin). KdpD-6His wurde mit Equilibrierungspuffer mit
einer Konzentration von 100 mM bzw. 250 mM Imidazol eluiert. Die Reinigung wurde
mittels Immunoblot und Proteinbestimmung überprüft.
2.5.7. Rekonstitution von KdpD-6His
Die Rekonstitution von KdpD-6His erfolgte gemäß dem Protokoll von Jung et al.
(1997). Die Liposomen wurden wie unter 2.5.8 beschrieben hergestellt und vor der

2. Material und Methoden
38
Rekonstitution wurde 0.64 % Triton X-100 hinzugefügt. Das gereinigte KdpD-6His
wurde im Verhältnis Lipid zu Protein 25 : 1 (w/w) hinzugefügt und unter Rühren bei
RT für 10 min inkubiert. Zur Entfernung des Detergenz wurden Bio-Beads im
Verhältnis von 5 : 1 (w/w) zum Detergenz eingesetzt. Die Bio-Beads wurden zuvor
mit Methanol und H2OBidest nach dem Protokoll von Holloway (1973) gewaschen und
in H2OBidest bei 4°C gelagert. Nach einer Stunde Inkubation bei RT auf der Wippe
erfolgte die erneute Zugabe derselben Menge an Bio-Beads. Es folgte eine
Inkubation über Nacht bei 4°C. Dann wurde die doppe lte Menge an Bio-Beads hinzu-
gefügt und erneut für 1 h bei RT inkubiert. Zum Entfernen der Bio-Beads wurden die
Proteoliposomen über Polyproylensäulen (Qiagen, Hilden) gegeben und der Durch-
fluss aufgefangen. Die Bio-Beads wurden mit TG-Puffer gewaschen, um den Verlust
von Proteoliposomen zu minimieren. Der Durchfluss wurde zentrifugiert (70.000 rpm,
1 h, 4°C), das Pellet in TG-Puffer resuspendiert un d erneut wie zuvor zentrifugiert.
Die Proteoliposomen wurden in TG-Puffer aufgenommen und in flüssigem Stickstoff
gelagert.
Alternativ wurde die Rekonstitution von KdpD-6His gemäß dem Protokoll von Knol et
al. (1998) durchgeführt. Die wesentlichen Unterschiede zu dem herkömmlichen
Rekonstitutionsprotokoll für KdpD bestehen darin, dass das Verhältnis von Lipid zu
Protein 100 : 1 beträgt und dass andere Mengen der Bio-Beads (jedes Mal Zugabe
von 40 mg/ml) und andere Zeitintervalle (15 min, 15 min, 30 min, über Nacht,
1 Stunde) verwendet wurden. Außerdem wurden die Liposomen jeweils so titriert,
dass sie partiell solubilisiert waren, welches eine Tritonkonzentration von nur 0.18 %
zur Folge hatte.
Eine weitere Rekonstitutionsmethode erfolgte durch Verdünnen des Detergenz unter
die CMC („critical micelle concentration“) direkt nach der Reinigung von KdpD-6His
nach dem Protokoll von Heuberger et al. (2001). Dazu erfolgte zunächst bei der
Standardreinigung von KdpD (siehe 2.5.6) ein Detergenzwechsel von DDM
(Dodecylmaltosid) zu OG (Octylglycosid) auf der Ni-NTA, da das Detergenz DDM
nicht so verdünnt werden kann, dass es unterhalb seiner CMC liegt, weil es einen
sehr kleinen CMC-Wert (0.17 mM) aufweist. Hingegen kann OG so verdünnt werden,
dass es unterhalb seiner im Vergleich zu DDM höheren CMC (25 mM) liegt. Die an
die Ni-NTA gebundene Probe wurde mit dem 10-fachen Säulenvolumen mit OG-
Puffer (1.25 %) gewaschen, um das restliche Detergenz DDM vom gebundenen
Protein zu entfernen. Die Elution von KdpD-6His von der Ni-NTA erfolgte mit 100 mM

2. Material und Methoden
39
Imidazol in Anwesenheit von OG. Die Rekonstitution erfolgte sofort nach der Protein-
bestimmung, da die Micellen aus OG instabil sind. Dazu wurde OG durch die Zugabe
von OG-freiem Puffer und Lipid so verdünnt, dass die OG-Konzentration unter der
CMC von 25 mM lag, um die Bildung von Liposomen mit zufällig eingebautem KdpD
zu erzielen.
2.5.8. Fusion von Membranvesikeln mit synthetischen Liposomen
Um die Lipidzusammensetzung bzw. den anionischen Anteil an Phospholipiden zu
variieren, wurden multilamellare Liposomen aus synthetischen Lipiden in ver-
schiedenen Verhältnissen mit invertierten Membranvesikeln gemischt. Dafür wurden
zum Einen Liposomen bestehend aus 100 % anionischem CL und zum Anderen
Liposomen bestehend aus 100 % zwitterionischen Lipiden (50 % PE und 50 % PC)
hergestellt und benutzt. Die Fusion dieser Liposomen mit den Membranvesikeln
erfolgten in Anlehnung an das Protokoll von Özcan et al. (2007). Abweichend zu der
in der Literatur beschriebenen Methode wurde ohne die Zugabe von PEG und ohne
Extrusion gearbeitet, da beobachtet werden konnte, dass die Kinaseaktivität von
KdpD-6His nach dem Extrudieren bereits in den invertierten Membranvesikeln
reduziert war.
2.5.9. Proteinbestimmung nach Lowry
Proteinbestimmungen wurden in Abwandlung des Protokolls von Lowry et al. (1951)
nach Peterson et al. (1977) durchgeführt. Als Standardprotein für die Erstellung von
Eichgeraden wurde BSA (2 mg/ml) in den Konzentrationen von 0 µg bis 25 µg ver-
wendet. Die Messung der Extinktion erfolgte im Shimadzu UV-1202 Spektralphoto-
meter (Shimadzu, Duisburg) bei einer Wellenlänge von 750 nm.
2.5.10. Proteinphosphorylierung mit [ γγγγ−−−−32P] ATP
Für die Messung der Kinaseaktivität von KdpD-6His wurden invertierte Membran-
vesikel aus dem Stamm TKR2000 (atp-) verwendet. Das radioaktive Phosphat kann
folglich nicht durch die ATPase abgebaut werden. Die invertierten Membranvesikel
wurden mit TG-Puffer auf eine Endkonzentration von 2 mg/ml eingestellt. Die Kinase-

2. Material und Methoden
40
aktivität wurde in Phosphorylierungspuffer (25 mM Tris/HCl, pH 7.5; 2 mM DTT;
0.01 mM MgCl2; 0.5 M NaCl; 5 % Glycerin) bei Raumtemperatur gemessen. Die
Reaktion wurde durch Zugabe von 20 µM [γ-32P]-ATP (2.38 Ci/mmol) gestartet. Nach
verschiedenen Zeitpunkten wurden Proben entnommen und die Reaktion durch
Zugabe von 2 x SDS Probenpuffer im Verhältnis 1 : 2 abgestoppt. Die Proben
wurden durch SDS-Polyacrylamidgelelektrophorese aufgetrennt (11 % Gel; 200 V;
60 min). Als Standard wurden der ,,Page Ruler Prestained Protein Ladder“ und
0.702 pmol [γ-32P]-ATP eingesetzt. Die SDS-Gele wurden anschließend für 60 min
bei 80°C getrocknet und über Nacht auf einem ,,Stor age Phosphor Screen“
exponiert. Die phosphorylierten Proteine konnten mittels „Phosphoimager Storm 820“
(Molecular Dynamics) detektiert werden. Die Quantifizierung erfolgte mit Hilfe der
Software ,,Image Quant 5.2“ von Molecular Dynamics.
2.5.11. Lipid-Extraktion aus ganzen Zellen
Um die Kopfgruppenzusammensetzung der Phospholipide bestimmen zu können,
wurden zunächst die Lipide aus ganzen Zellen extrahiert. Das Protokoll erfolgte in
Anlehnung an Wikström et al. (2004). Dafür wurden Zellen des jeweiligen Stammes
über Nacht im entsprechenden Minimalmedium mit 25 µCi/ml 32P-Orthophosphat
kultiviert und am nächsten Tag in frisches, radioaktives Medium derselben K+-
Konzentration überimpft. Wenn die Zellen die frühe exponentielle Wachstumsphase
(OD600 ~0.5) erreicht hatten, wurden Zellen von 5 ml Kultur durch Zentrifugation
(1 min, 13.000 rpm) vom Medium getrennt und bis zur Weiterverarbeitung bei 4°C
gelagert.
Um die Einflüsse von schnellen Veränderungen der Wachstumsbedingungen
(insbesondere kdpFABC-induzierenden Bedingungen) zu ermitteln, wurden wach-
sende Zellen wie unter 2.3.1 beschrieben filtriert und in neues, ebenfalls radioaktiv-
markiertes Medium resuspendiert. Anschließend wurden zu verschiedenen Zeit-
punkten der Wachstumskurve Proben genommen.
Die polaren Lipide wurden mittels Chloroform/Methanol extrahiert, wie bei Tsatskis et
al. (2005) beschrieben. Das Pellet wurde in 0.2 ml Puffer N (0.5 N NaCl; 0.1 N HCl)
resuspendiert und für 5 min gevortext. Danach wurde 0.6 ml Chloroform-Methanol
(3 : 1) zu der Suspension hinzugefügt und für mindestens 30 min gevortext. Ab-
schließend wurde erneut 0.2 ml Puffer N dazugegeben, für 10 min gevortext und

2. Material und Methoden
41
1 min bei 13.000 rpm zentrifugiert, so dass sich die Suspension in zwei Phasen
trennte. Die obere Phase wurde verworfen und die extrahierten Lipide der unteren
Phase in ein neues Eppendorfgefäß überführt, um Verunreinigungen durch den
ebenfalls radioaktiven Überstand zu vermeiden.
2.5.12. Dünnschichtchromatographie
Damit für die Quantifizierung der Dünnschichtchromatographie (DC) sicher gestellt
wurde, dass jede Spur die gleiche Menge an Radioaktivität enthielt, wurden die
flüssigen Proben zunächst im Szintillationszähler Tri-Carb 2300 TR (Packard
Bioscience) analysiert. Dafür wurden je 5 µl der Probe mit 4 ml Szintillationsflüssig-
keit versetzt und die Radioaktivität mit einem Programm zur Detektion von 32P be-
stimmt. Diese Werte geben die gemittelte Radioaktivität pro Minute in „counts per
minute“ (cpm) für eine 10-minütige Messung an. Die Kalibrierung des Szintillations-
zählers fand anhand interner Eichstandards des Herstellers statt.
Die DC wurde gemäß des Protokolls von Tsatskis et al. (2005) durchgeführt. Für die
Auftrennung der Phospholipide wurden entsprechende Kieselgel-Platten verwendet.
Es wurde jeweils eine maximale Radioaktivität von 8.000 - 10.000 cpm (4 - 5 nCi) pro
Spur aufgetragen. Damit ebenfalls das aufgetragene Volumen der Proben bei
gleicher Radioaktivität identisch war, wurden die Proben - falls nötig - mit ent-
sprechenden Mengen Chloroform gemischt. Als Laufmittel wurde Chloroform-
Methanol-Essigsäure im Verhältnis 68 : 25 : 8 verwendet. Nach Auftrennung und
entsprechender Trocknung der DC-Platten wurden diese über Nacht unter einem
„Storage Phosphor Screen“ exponiert.
2.5.13. Kopfgruppenanalyse der Phospholipide mittel s 32P-Markierung
Die radioaktiv-markierten Phospholipide konnten mittels „Phosphoimager Storm 820“
(Molecular Dynamics) detektiert werden. Die Quantifizierung erfolgte mit Hilfe der
Software ,,Image Quant 5.2“ von Molecular Dynamics. Der Gehalt jedes Lipids wurde
in mol % angegeben und zuvor an den jeweiligen PO4-Gehalt des Lipids angepasst.
Als biochemische Kontrolle wurde nicht-radioaktiv-markiertes Phospholipid-Extrakt
mittels Dünnschichtchromatographie aufgetrennt. Die DC-Platte wurde mit Molybdän-
blau Reagenz angesprüht, um die Phospholipide zu detektieren. Die Spots der

2. Material und Methoden
42
Phospholipide wurden klassifiziert, indem parallel verschiedene synthetische Lipide
ebenfalls auf die DC-Platte aufgetragen wurden.
2.5.14. Fettsäure-Extraktion aus ganzen Zellen
Zellen des E. coli Stammes K12 wurden in Minimalmedien mit verschiedenen K+-
Konzentrationen kultiviert und bei verschiedenen Optischen Dichten durch Zentrifu-
gation geerntet und für die Fettsäure-Extraktion verwendet. Das Protokoll für die
Extraktion von Fettsäure Methyl Estern (FAMEs) aus ganzen Zellen erfolgte in An-
lehnung an Sasser (1990). Dabei fand nach Zellernte eine Verseifung durch eine
Natronlauge statt, gefolgt von einer Methylierung der Fettsäuren, damit diese flüchtig
werden. Die Fettsäuren wurden anschließend mittels eines Hexan-Ether-Gemisches
extrahiert und mit Natronlauge gewaschen.
2.5.15. Fettsäureanalyse der Phospholipide mittels Gaschromatographie
Die Identifizierung der FAMEs wurde mittels Gaschromatographie-Massenspektro-
metrie (GC-MS) nach Lipski & Altendorf (1997) bestimmt. Die Auswertung der Werte
erfolgte mittels eines mitgeführten Standards (9 : 0 Fettsäure-Leiter). Dazu wurden
zunächst die gemessenen RT-Werte des Standards zu den Fettsäuren im Standard
zugeordnet. Anhand einer eigenen ECL-Datenbank (A. Lipski, Universität Osna-
brück) wurden dann die umgerechneten Werte der Proben (von RT auf ECL) auto-
matisch zu einer bestimmten Fettsäure zugeordnet. Die Auswertung der nicht
identifizierten Fettsäuren erfolgte anhand des jeweiligen Massenspektrums mit Hilfe
der Software „Enhanced ChemStation“ (Agilent Technologies) durch den Vergleich
mit einer anderen Fettsäure-Datenbank („FAME“). Die Zuordnung erfolgte über
typische Merkmale verschiedener Fettsäuren. Der Anteil jeder Fettsäure wurde in %
von der gesamten Fettsäurezusammensetzung angegeben.
2.5.16. Flammenphotometrische Bestimmung des extraz ellulären K +- und Na +-
Gehaltes
Die flammenphotometrische Bestimmung des extrazellulären Kalium- oder
Natriumgehaltes wurde nach dem Protokoll von Bakker et al. (1993) durchgeführt.

2. Material und Methoden
43
1 ml der zu testenden Zellsuspension wurde 5 min bei 13.000 rpm zentrifugiert und
vom Überstand des Zentrifugates 750 µl abgenommen. Bei Kaliumgehalten über
100 µM wurde die Probe mit H2OBidest verdünnt. Die gegebenenfalls verdünnte Probe
wurde 1 : 1 mit einer 10 mM CsCl / 2.5 % TCA-Lösung gemischt und der Kalium-
bzw. Natriumgehalt am Flammenphotometer der Marke Eppendorf ELEX 6361 mit
Hilfe von Standardlösungen bestimmt.
2.5.17. ββββ-Galaktosidase-Aktivitätstest
Die Expression von kdpFABC oder cls in vivo wurde mittels des Reportergens lacZ
(kdpFABC- bzw. cls-Promotor/lacZ-Genfusion) nachgewiesen. Die mit dem β-
Galaktosidase-Aktivitätstest nach Miller (1972) bestimmte Aktivität der vom lacZ-Gen
kodierten β-Galaktosidase diente als indirektes Maß für die Expression von kdpFABC
bzw. cls. Für den Test wurden Zellen des Stammes E. coli HAK006 (kdpFABC-lacZ
∆kdpD161-726) mit einem entsprechenden Plasmid (kodierend für KdpD-Derivate) in
Minimalmedien nach Epstein & Kim (1971) bis zu einer OD600 von etwa 1.0
angezogen. Für den Test der cls-Expression wurden Zellen des Stammes E. coli
SOH92 (cls-lacZ) in Minimalmedien nach Epstein & Kim (1971) angezogen und bei
verschiedenen Optischen Dichten der Wachstumskurve wurden Proben für den β-
Galaktosidase-Aktivitätstest genommen.
Anschließend wurde 1 ml jeder Zellkultur für 10 min bei 13.000 rpm zentrifugiert und
in 1 ml Phosphatpuffer (200 mM, pH 7.0) resuspendiert. Der Zellaufschluss erfolgte
durch Zugabe von 100 µl Chloroform und 50 µl SDS (0.1 % (w/v)). Zur Bestimmung
der Aktivität wurden 200 µl ortho-Nitrophenyl-β-D-Galaktosid (4 mg/ml) zu den
Proben gegeben. Die Reaktionen wurden nach 4 min durch Zugabe von 500 µl
Natriumcarbonat (1 M) gestoppt. Zelltrümmer konnten durch Zentrifugation bei
13.000 rpm für 10 min entfernt werden. Die Intensität der durch die Reaktion hervor-
gerufenen Färbung wurde photometrisch bei einer Absorption von 420 nm bestimmt.
Die Aktivität konnte mit Hilfe folgender Formel berechnet werden:
Aktivität [units] = (A420 x 1000) / (t x V x OD600)
Dabei ist A420 als die Absorption der Reaktionslösung, OD600 als die Optische Dichte
der Zellsuspension, t als die Reaktionszeit [min] und V als das Reaktionsvolumen
[ml] definiert.
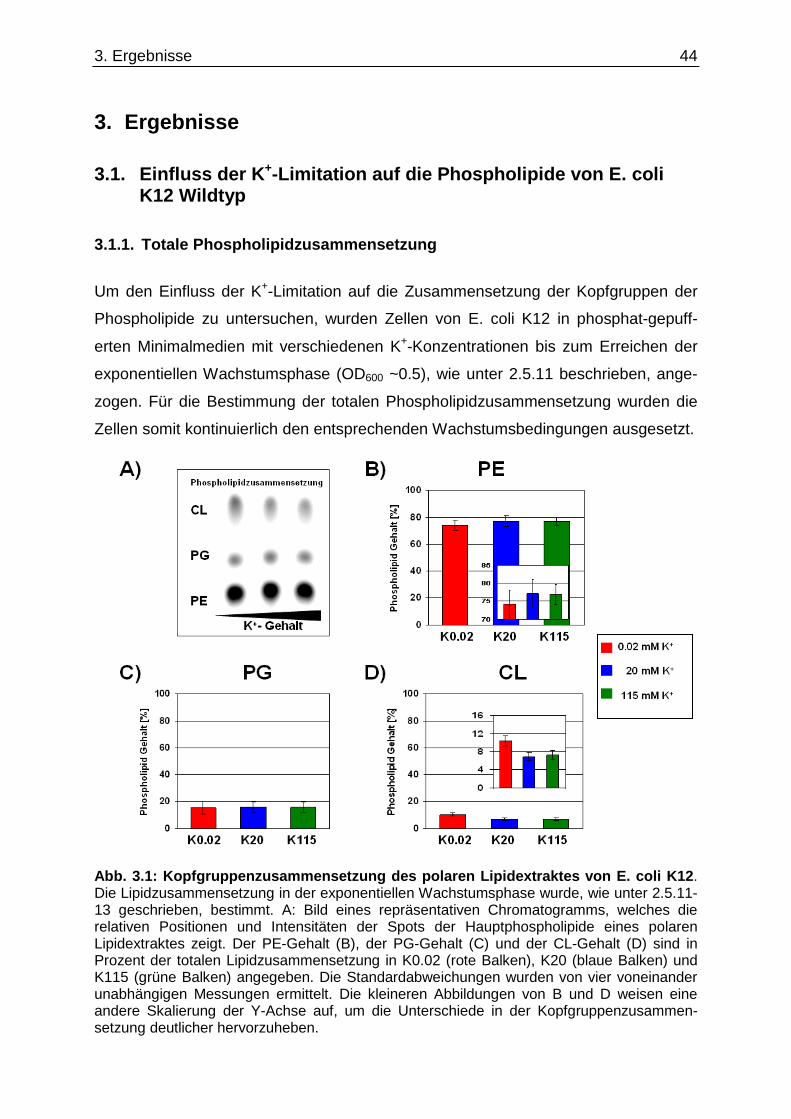
3. Ergebnisse
44
3. Ergebnisse
3.1. Einfluss der K +-Limitation auf die Phospholipide von E. coli K12 Wildtyp
3.1.1. Totale Phospholipidzusammensetzung
Um den Einfluss der K+-Limitation auf die Zusammensetzung der Kopfgruppen der
Phospholipide zu untersuchen, wurden Zellen von E. coli K12 in phosphat-gepuff-
erten Minimalmedien mit verschiedenen K+-Konzentrationen bis zum Erreichen der
exponentiellen Wachstumsphase (OD600 ~0.5), wie unter 2.5.11 beschrieben, ange-
zogen. Für die Bestimmung der totalen Phospholipidzusammensetzung wurden die
Zellen somit kontinuierlich den entsprechenden Wachstumsbedingungen ausgesetzt.
Abb. 3.1: Kopfgruppenzusammensetzung des polaren Li pidextraktes von E. coli K12. Die Lipidzusammensetzung in der exponentiellen Wachstumsphase wurde, wie unter 2.5.11-13 geschrieben, bestimmt. A: Bild eines repräsentativen Chromatogramms, welches die relativen Positionen und Intensitäten der Spots der Hauptphospholipide eines polaren Lipidextraktes zeigt. Der PE-Gehalt (B), der PG-Gehalt (C) und der CL-Gehalt (D) sind in Prozent der totalen Lipidzusammensetzung in K0.02 (rote Balken), K20 (blaue Balken) und K115 (grüne Balken) angegeben. Die Standardabweichungen wurden von vier voneinander unabhängigen Messungen ermittelt. Die kleineren Abbildungen von B und D weisen eine andere Skalierung der Y-Achse auf, um die Unterschiede in der Kopfgruppenzusammen-setzung deutlicher hervorzuheben.
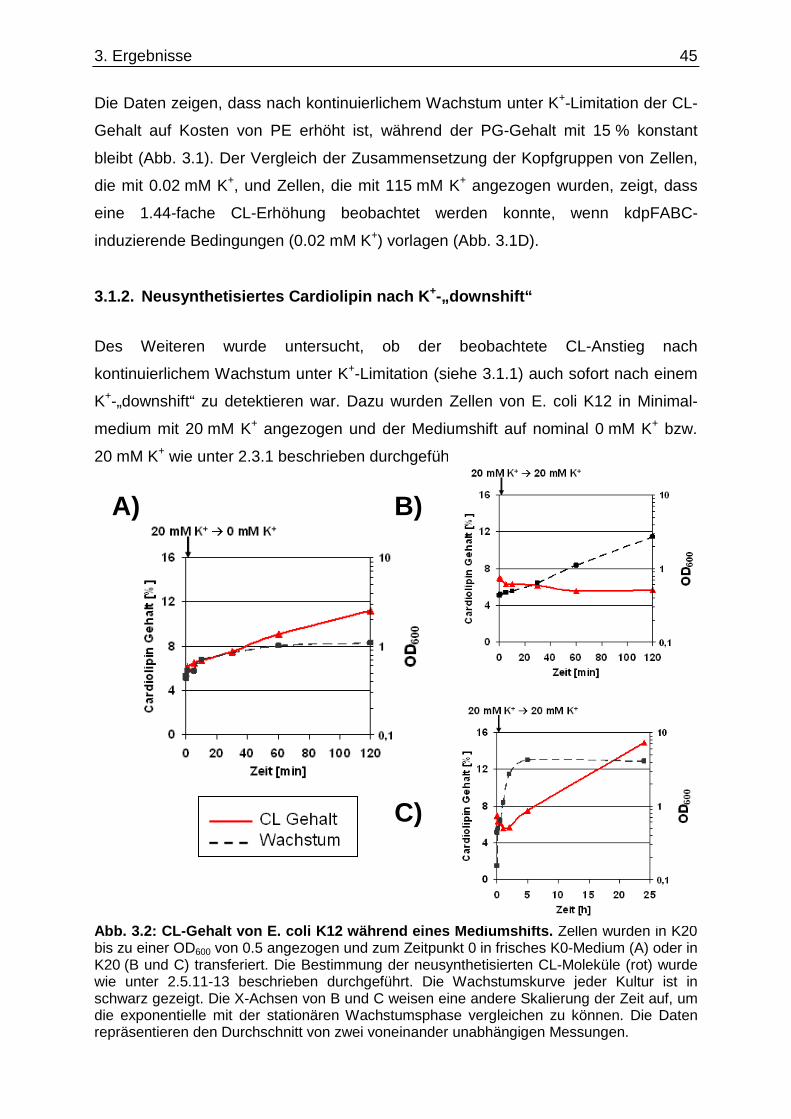
3. Ergebnisse
45
Die Daten zeigen, dass nach kontinuierlichem Wachstum unter K+-Limitation der CL-
Gehalt auf Kosten von PE erhöht ist, während der PG-Gehalt mit 15 % konstant
bleibt (Abb. 3.1). Der Vergleich der Zusammensetzung der Kopfgruppen von Zellen,
die mit 0.02 mM K+, und Zellen, die mit 115 mM K+ angezogen wurden, zeigt, dass
eine 1.44-fache CL-Erhöhung beobachtet werden konnte, wenn kdpFABC-
induzierende Bedingungen (0.02 mM K+) vorlagen (Abb. 3.1D).
3.1.2. Neusynthetisiertes Cardiolipin nach K +-„downshift“
Des Weiteren wurde untersucht, ob der beobachtete CL-Anstieg nach
kontinuierlichem Wachstum unter K+-Limitation (siehe 3.1.1) auch sofort nach einem
K+-„downshift“ zu detektieren war. Dazu wurden Zellen von E. coli K12 in Minimal-
medium mit 20 mM K+ angezogen und der Mediumshift auf nominal 0 mM K+ bzw.
20 mM K+ wie unter 2.3.1 beschrieben durchgeführt.
Abb. 3.2: CL-Gehalt von E. coli K12 während eines Mediumshifts. Zellen wurden in K20 bis zu einer OD600 von 0.5 angezogen und zum Zeitpunkt 0 in frisches K0-Medium (A) oder in K20 (B und C) transferiert. Die Bestimmung der neusynthetisierten CL-Moleküle (rot) wurde wie unter 2.5.11-13 beschrieben durchgeführt. Die Wachstumskurve jeder Kultur ist in schwarz gezeigt. Die X-Achsen von B und C weisen eine andere Skalierung der Zeit auf, um die exponentielle mit der stationären Wachstumsphase vergleichen zu können. Die Daten repräsentieren den Durchschnitt von zwei voneinander unabhängigen Messungen.
A) B)
C)

3. Ergebnisse
46
Wie Abbildung 3.2A zeigt, wurde der CL-Anstieg von Zellen auch sofort nach einem
Shift (30 sec) in Medium mit nominal keinen K+-Ionen beobachtet, wobei dieser An-
stieg die Proportion von neusynthetisiertem CL widerspiegelt. Eine solche Änderung
der Zusammensetzung der Kopfgruppen wurde hingegen nicht ermittelt, wenn die
Zellen in frisches Medium mit derselben K+-Konzentration überführt wurden (Abb.
3.2B). Unabhängig von einem Wechsel des Mediums, stieg der CL-Gehalt auf
Kosten von PG an, wenn die Zellen die stationäre Wachstumsphase erreicht hatten
(Abb. 3.2C). Dieser Effekt in der stationären Wachstumsphase konnte bereits früher
beobachtet werden (Cronan, 1968; Randle et al., 1969).
3.1.3. Fettsäurezusammensetzung unter K +-Limitation
Da bereits gezeigt werden konnte, dass sich unter K+-Limitation die Zusammen-
setzung der Kopfgruppen der Phospholipide der Zellhülle von E. coli ändert (siehe
3.1.1-2), wurde untersucht, ob sich auch die Zusammensetzung der Fettsäureketten
der Phospholipide unter K+-Limitation ändert. Die Zusammensetzung der Fettsäure-
ketten wurde mittels Gaschromatographie bestimmt. Dazu wurden Flüssigkulturen
von E. coli K12 in Minimalmedium mit 0.1 mM K+ oder 115 mM K+ angezogen und
nach Zellernte bei verschiedenen Optischen Dichten die Fettsäuren extrahiert und
analysiert.
Tab. 3.1: Fettsäurezusammensetzung der Phospholipid e von E. coli K12.
Zellen wurden in K0.1 oder in K115 angezogen. Der Gehalt jeder Fettsäure ist in Prozent angegeben und wurde wie unter 2.5.14-15 beschrieben bestimmt. Die Daten repräsentieren den Durchschnitt von zwei voneinander unabhängigen Messungen. Die Werte für die Fettsäurezusammensetzung sind in Tabelle 3.1 dargestellt. Die
prozentualen Anteile der häufigsten Fettsäuren von E. coli in K0.1 sind 16:0 mit
38.8 % (bzw. 36.4 % in K115) und 16:1 cis 9 mit 29.8 % (bzw. 34.8 % in K115).
Fettsäure [%] in K0.1 K115
12:0 2.6 3.1 14:0 6.5 7.4 16:0 38.8 36.4 16:1 cis 9 29.8 34.8 18:0 0 0 18:1 cis 11 11.0 10.4 17:0 cyclo 9-10 6.1 3.7 19:0 cyclo 11-12 0 0 Andere 5.2 4.2
3. Ergebnisse
47
Sowohl in K0.1 als auch in K115 sind die prozentualen Anteile der Fettsäuren
vergleichbar mit denen in Vollmedium ermittelten Werten (Cronan, 1968).
3.2. Einfluss der K +-Limitation auf die Phospholipide von E. coli „Kalium-Stämmen“
3.2.1. Totale Phospholipidzusammensetzung
Um den Einfluss von verschiedenen K+-Transportern auf die Zusammensetzung der
Kopfgruppen zu untersuchen, wurden entsprechende „Kalium-Stämme“ verwendet.
Der E. coli Stamm TK2240 (kdp+ trk– kup–) wurde getestet, um den Einfluss der
niedrig affinen K+-Aufnahmesysteme Trk und Kup zu untersuchen. Der Stamm
RH003 hingegen ist für die Gene, die für das komplette KdpFABCDE-System
kodieren, deletiert. Der Stamm HAK006, der unter anderem für die Konstruktion von
RH003 verwendet wurde, weist nur eine Teildeletion des kdpD-Gens in den Basen-
paaren auf, welche für die Aminosäuren 161-726 von KdpD kodieren. Als Referenz
wurden die gewonnenen Ergebnisse aus 3.1.1 von E. coli K12 genommen. Dieser
Vergleich ist besonders im Falle von TK2240 wichtig, da die kdpFABC-Expression
davon abhängig ist, ob auch andere K+-Transportsysteme wie Trk und Kup in der
Zelle vorhanden sind (siehe Abb. 1.5). Die Stämme wurden in verschiedenen Mini-
malmedien mit 32P-Orthophosphat angezogen und die Zusammensetzung der Kopf-
gruppen nach kontinuierlichem Wachstum während der exponentiellen Wachstums-
phase (OD600 ~0.5) bestimmt.
Tab. 3.2: Zusammensetzung der Kopfgruppen verschied ener E. coli „Kalium-Stämme“.
Kopfgruppengehalt [%] in K0.1 Stamm Relevanter
Genotyp PE PG CL
Anteil anionischer Phospholipide
K12 trk+ kdp+ 75.1 (77.3) 15.1 (16.5) 9.8 (6.2) 24.9 (22.7)
TK2240 trk- kdp+ 77.3 14.1 8.6 22.7
RH003 trk+ kdp- 78.8 17.3 3.9 21.2
HAK006 trk+ kdpD- 78.6 17.9 3.5 21.4
Die Zusammensetzung der Kopfgruppen ist angegeben in Prozent und wurde wie unter 2.5.11-13 beschrieben bestimmt. Die Daten repräsentieren den Durchschnitt von zwei - drei voneinander unabhängigen Messungen. Der Anteil anionischer Lipide ist durch die Summe von PG und CL angegeben. Bei den Werten in Klammern handelt es sich um die Werte des Referenzstammes K12 in K115.
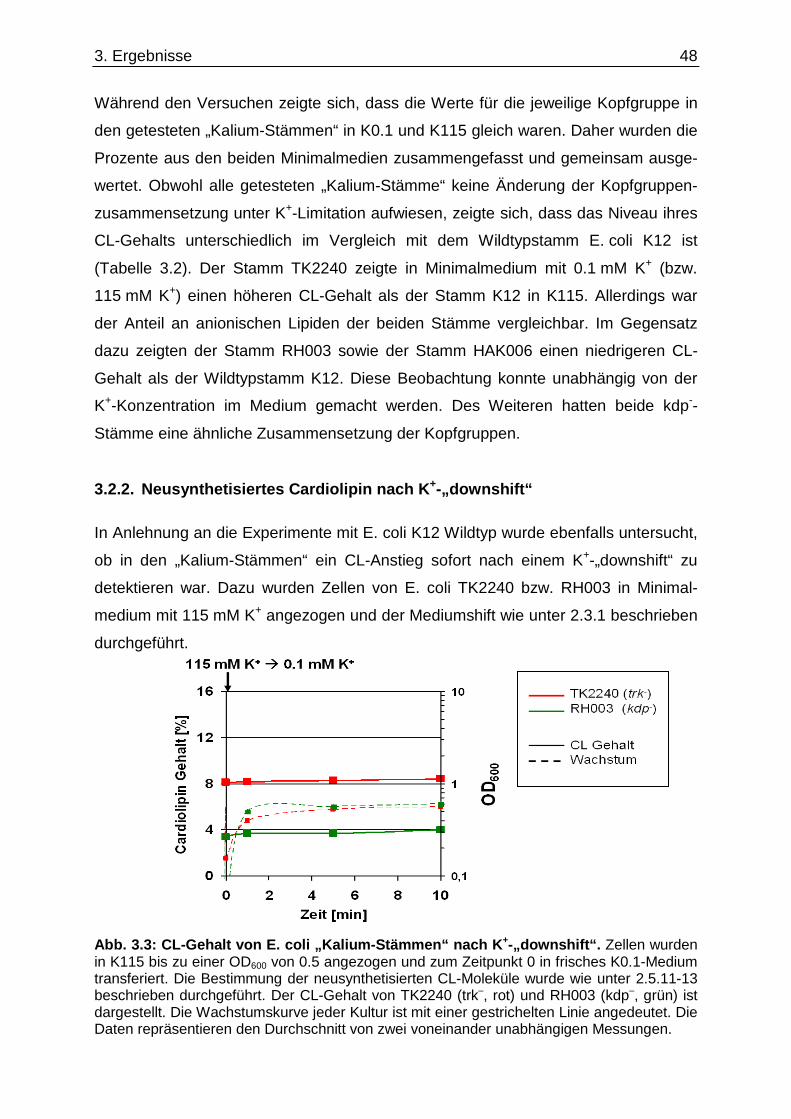
3. Ergebnisse
48
Während den Versuchen zeigte sich, dass die Werte für die jeweilige Kopfgruppe in
den getesteten „Kalium-Stämmen“ in K0.1 und K115 gleich waren. Daher wurden die
Prozente aus den beiden Minimalmedien zusammengefasst und gemeinsam ausge-
wertet. Obwohl alle getesteten „Kalium-Stämme“ keine Änderung der Kopfgruppen-
zusammensetzung unter K+-Limitation aufwiesen, zeigte sich, dass das Niveau ihres
CL-Gehalts unterschiedlich im Vergleich mit dem Wildtypstamm E. coli K12 ist
(Tabelle 3.2). Der Stamm TK2240 zeigte in Minimalmedium mit 0.1 mM K+ (bzw.
115 mM K+) einen höheren CL-Gehalt als der Stamm K12 in K115. Allerdings war
der Anteil an anionischen Lipiden der beiden Stämme vergleichbar. Im Gegensatz
dazu zeigten der Stamm RH003 sowie der Stamm HAK006 einen niedrigeren CL-
Gehalt als der Wildtypstamm K12. Diese Beobachtung konnte unabhängig von der
K+-Konzentration im Medium gemacht werden. Des Weiteren hatten beide kdp--
Stämme eine ähnliche Zusammensetzung der Kopfgruppen.
3.2.2. Neusynthetisiertes Cardiolipin nach K +-„downshift“
In Anlehnung an die Experimente mit E. coli K12 Wildtyp wurde ebenfalls untersucht,
ob in den „Kalium-Stämmen“ ein CL-Anstieg sofort nach einem K+-„downshift“ zu
detektieren war. Dazu wurden Zellen von E. coli TK2240 bzw. RH003 in Minimal-
medium mit 115 mM K+ angezogen und der Mediumshift wie unter 2.3.1 beschrieben
durchgeführt.
Abb. 3.3: CL-Gehalt von E. coli „Kalium-Stämmen“ nach K +-„downshift“. Zellen wurden in K115 bis zu einer OD600 von 0.5 angezogen und zum Zeitpunkt 0 in frisches K0.1-Medium transferiert. Die Bestimmung der neusynthetisierten CL-Moleküle wurde wie unter 2.5.11-13 beschrieben durchgeführt. Der CL-Gehalt von TK2240 (trk–, rot) und RH003 (kdp–, grün) ist dargestellt. Die Wachstumskurve jeder Kultur ist mit einer gestrichelten Linie angedeutet. Die Daten repräsentieren den Durchschnitt von zwei voneinander unabhängigen Messungen.
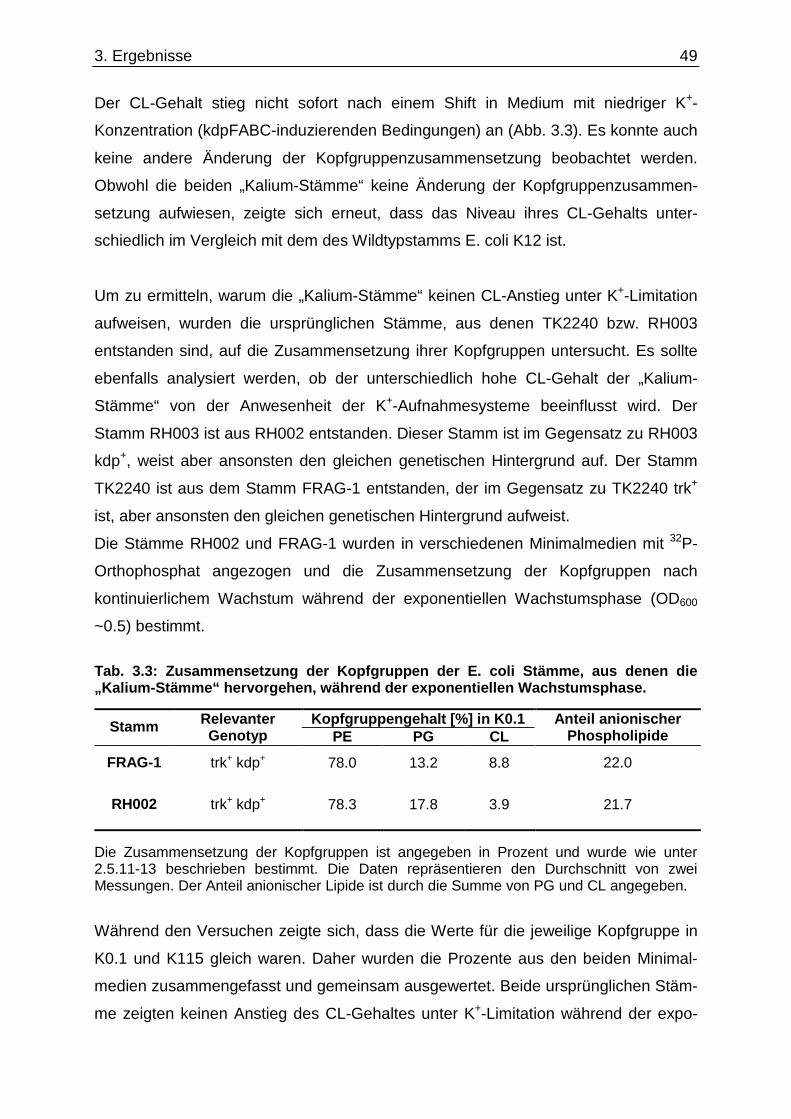
3. Ergebnisse
49
Der CL-Gehalt stieg nicht sofort nach einem Shift in Medium mit niedriger K+-
Konzentration (kdpFABC-induzierenden Bedingungen) an (Abb. 3.3). Es konnte auch
keine andere Änderung der Kopfgruppenzusammensetzung beobachtet werden.
Obwohl die beiden „Kalium-Stämme“ keine Änderung der Kopfgruppenzusammen-
setzung aufwiesen, zeigte sich erneut, dass das Niveau ihres CL-Gehalts unter-
schiedlich im Vergleich mit dem des Wildtypstamms E. coli K12 ist.
Um zu ermitteln, warum die „Kalium-Stämme“ keinen CL-Anstieg unter K+-Limitation
aufweisen, wurden die ursprünglichen Stämme, aus denen TK2240 bzw. RH003
entstanden sind, auf die Zusammensetzung ihrer Kopfgruppen untersucht. Es sollte
ebenfalls analysiert werden, ob der unterschiedlich hohe CL-Gehalt der „Kalium-
Stämme“ von der Anwesenheit der K+-Aufnahmesysteme beeinflusst wird. Der
Stamm RH003 ist aus RH002 entstanden. Dieser Stamm ist im Gegensatz zu RH003
kdp+, weist aber ansonsten den gleichen genetischen Hintergrund auf. Der Stamm
TK2240 ist aus dem Stamm FRAG-1 entstanden, der im Gegensatz zu TK2240 trk+
ist, aber ansonsten den gleichen genetischen Hintergrund aufweist.
Die Stämme RH002 und FRAG-1 wurden in verschiedenen Minimalmedien mit 32P-
Orthophosphat angezogen und die Zusammensetzung der Kopfgruppen nach
kontinuierlichem Wachstum während der exponentiellen Wachstumsphase (OD600
~0.5) bestimmt.
Tab. 3.3: Zusammensetzung der Kopfgruppen der E. coli Stämme, aus denen die „Kalium-Stämme“ hervorgehen, während der exponentie llen Wachstumsphase.
Kopfgruppengehalt [%] in K0.1 Stamm Relevanter Genotyp PE PG CL
Anteil anionischer Phospholipide
FRAG-1 trk+ kdp+ 78.0 13.2 8.8 22.0
RH002 trk+ kdp+ 78.3 17.8 3.9 21.7
Die Zusammensetzung der Kopfgruppen ist angegeben in Prozent und wurde wie unter 2.5.11-13 beschrieben bestimmt. Die Daten repräsentieren den Durchschnitt von zwei Messungen. Der Anteil anionischer Lipide ist durch die Summe von PG und CL angegeben.
Während den Versuchen zeigte sich, dass die Werte für die jeweilige Kopfgruppe in
K0.1 und K115 gleich waren. Daher wurden die Prozente aus den beiden Minimal-
medien zusammengefasst und gemeinsam ausgewertet. Beide ursprünglichen Stäm-
me zeigten keinen Anstieg des CL-Gehaltes unter K+-Limitation während der expo-

3. Ergebnisse
50
nentiellen Wachstumsphase und einen vergleichbaren Anteil an CL im Vergleich mit
den jeweiligen Stämmen, die aus ihnen konstruiert wurden (Tabelle 3.2 vgl. Tabelle
3.3). Der Stamm FRAG-1 (bzw. TK2240) hat mit 8.8 % (8.6 %) einen höheren CL-
Gehalt als der Stamm RH002 (RH003 und HAK006) mit 3.9 % (bzw. 3.5 %).
3.3. Einfluss der K +-Limitation auf die Phospholipide von E. coli „Lipid-Stämmen“
Um den Einfluss der K+-Limitation auf die Zusammensetzung der Kopfgruppen von
„Lipid-Stämmen“ zu untersuchen, wurden entsprechende Stämme analysiert. Solche
„Lipid-Stämme“ mit einer veränderten nativen Zusammensetzung der Kopfgruppen
sind bereits detailliert charakterisiert worden (zum Überblick siehe Dowhan et al.,
2004). Auch die Zusammensetzung der Kopfgruppen, der in der vorliegenden Arbeit
untersuchten Stämme UE54, WC3899, HDL1001 und AD93, wurde bereits für das
Wachstum in Vollmedium beschrieben. In dieser Arbeit wurde das Wachstum unter
K+-Limitation untersucht, da bislang für diese Wachstumsbedingung keine Werte
vorlagen. Als ein Extrem wurde der Stamm AD93, der nur anionische Phospholipide
enthält, und als ein anderes Extrem der Stamm UE54, der nur zwitterionische
Phospholipide enthält, getestet.
Zunächst wurde ein biochemischer Nachweis erbracht, dass es sich bei den
verschiedenen E. coli „Lipid-Stämmen““ jeweils um die in der Literatur für Vollmedium
beschriebene Phospholipidzusammensetzung handelt. Dafür wurden Zellen von den
Stämmen AD93 und K12 in Minimalmedium mit 115 mM K+ und in KML-Medium
angezogen und die Lipide extrahiert. Neben den Extrakten der Lipide der verschie-
denen Stämme wurde ein synthetisches Gemisch von PE, PG und CL auf die DC-
Platte aufgetragen, um die Phospholipide identifizieren zu können. Anschließend
wurde die DC-Platte mit einem speziellen Farbstoff für Phospholipide (Molybdänblau)
angesprüht (siehe Abb. 3.4A). Des Weiteren wurden alle untersuchten „Lipid-
Stämme“ in den verschiedenen Minimalmedien mit radioaktivem 32P-Orthophosphat
angezogen, die Lipide extrahiert und die Zusammensetzung der Kopfgruppen
bestimmt (siehe 2.5.11-13), um diese untereinander vergleichen zu können.
Mit dem Vergleich von Abbildung 3.4A mit 3.4B konnten die radioaktiven Spots den
jeweiligen Phospholipiden zugeordnet werden. Mit dieser Methode konnte bestätigt
werden, dass alle untersuchten Stämme die beschriebenen und keine weiteren

3. Ergebnisse
51
Phospholipide aufweisen. Des Weiteren wurde eine dem Vollmedium ähnliche
Tendenz der prozentualen Anteile der Kopfgruppen bei der Zusammensetzung der
Phospholipide des jeweiligen Stammes anhand der Intensitäten der Spots ange-
deutet. Die genaue Quantifizierung der Zusammensetzung der Kopfgruppen erfolgte
wie unter 2.5.13 beschrieben.
Abb. 3.4: Identifizierung der Phospholipide. Der biochemische Nachweis der Zusammen-setzung der Kopfgruppen ist in A und im Vergleich der radioaktive Nachweis von [32P]-eingebauten Phospholipiden in B nach Auftrennung mittels 1-Dimensionaler Dünnschicht-chromatographie dargestellt. Die Besonderheiten in der Zusammensetzung des jeweiligen Stammes sind durch die roten bzw. grünen Kreise gekennzeichnet, je nachdem, ob es sich um den Anteil von anionischen oder zwitterionischen Phospholipiden handelt. Die Daten repräsentieren den Durchschnitt von zwei - drei voneinander unabhängigen Messungen.
Die Zusammensetzung der Kopfgruppen der Phospholipide der einzelnen Stämme
ist in Tabelle 3.4 dargestellt. Während den Versuchen zeigte sich, dass die Werte für
die jeweilige Kopfgruppe der „Lipid-Stämme“ in K0.1 und K115 gleich waren. Daher
wurden die Prozente aus den beiden Minimalmedien zusammengefasst und gemein-
sam ausgewertet. Unter K+-Limitation zeigte der Stamm AD93, der eine Deletion des
pssA-Gens beinhaltet, eine Zusammensetzung der Kopfgruppen ohne PE, welche in
eine ausschließliche Zusammensetzung aus anionischen Kopfgruppen, haupt-
sächlich aus PG und CL bestehend, resultiert. Die Transformation des Stammes mit
dem Plasmid pDD72, welches das pssA-Gen kodiert, führte zu einer Zusammen-
setzung der Kopfgruppen, die vergleichbar mit der des Wildtypstamms K12 ist.
CL
PG
PE
Start
Stamm K12 HDL1001 AD93 W3899 DG10 IPTG + - WC3899 UE54 pDD72 - +
CL
PG
PE
synthetische AD93 Lipide K12
A) B)
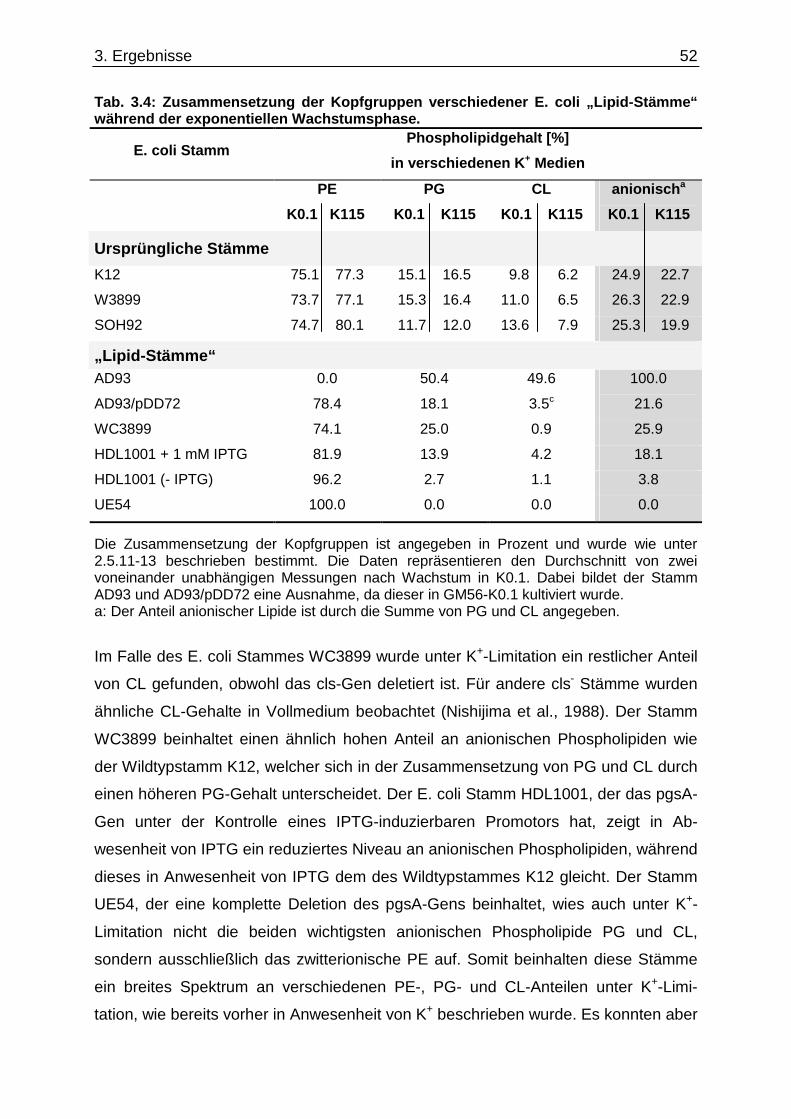
3. Ergebnisse
52
Tab. 3.4: Zusammensetzung der Kopfgruppen verschied ener E. coli „Lipid-Stämme“ während der exponentiellen Wachstumsphase.
E. coli Stamm Phospholipidgehalt [%]
in verschiedenen K + Medien
PE
K0.1 K115
PG
K0.1 K115
CL
K0.1 K115
anionisch a
K0.1 K115
Ursprüngliche Stämme
K12 75.1 77.3 15.1 16.5 9.8 6.2 24.9 22.7
W3899 73.7 77.1 15.3 16.4 11.0 6.5 26.3 22.9
SOH92 74.7 80.1 11.7 12.0 13.6 7.9 25.3 19.9
„Lipid-Stämme“ AD93 0.0 50.4 49.6 100.0
AD93/pDD72 78.4 18.1 3.5c 21.6
WC3899 74.1 25.0 0.9 25.9
HDL1001 + 1 mM IPTG 81.9 13.9 4.2 18.1
HDL1001 (- IPTG) 96.2 2.7 1.1 3.8
UE54 100.0 0.0 0.0 0.0
Die Zusammensetzung der Kopfgruppen ist angegeben in Prozent und wurde wie unter 2.5.11-13 beschrieben bestimmt. Die Daten repräsentieren den Durchschnitt von zwei voneinander unabhängigen Messungen nach Wachstum in K0.1. Dabei bildet der Stamm AD93 und AD93/pDD72 eine Ausnahme, da dieser in GM56-K0.1 kultiviert wurde. a: Der Anteil anionischer Lipide ist durch die Summe von PG und CL angegeben.
Im Falle des E. coli Stammes WC3899 wurde unter K+-Limitation ein restlicher Anteil
von CL gefunden, obwohl das cls-Gen deletiert ist. Für andere cls- Stämme wurden
ähnliche CL-Gehalte in Vollmedium beobachtet (Nishijima et al., 1988). Der Stamm
WC3899 beinhaltet einen ähnlich hohen Anteil an anionischen Phospholipiden wie
der Wildtypstamm K12, welcher sich in der Zusammensetzung von PG und CL durch
einen höheren PG-Gehalt unterscheidet. Der E. coli Stamm HDL1001, der das pgsA-
Gen unter der Kontrolle eines IPTG-induzierbaren Promotors hat, zeigt in Ab-
wesenheit von IPTG ein reduziertes Niveau an anionischen Phospholipiden, während
dieses in Anwesenheit von IPTG dem des Wildtypstammes K12 gleicht. Der Stamm
UE54, der eine komplette Deletion des pgsA-Gens beinhaltet, wies auch unter K+-
Limitation nicht die beiden wichtigsten anionischen Phospholipide PG und CL,
sondern ausschließlich das zwitterionische PE auf. Somit beinhalten diese Stämme
ein breites Spektrum an verschiedenen PE-, PG- und CL-Anteilen unter K+-Limi-
tation, wie bereits vorher in Anwesenheit von K+ beschrieben wurde. Es konnten aber

3. Ergebnisse
53
keine signifikanten Änderungen in der Zusammensetzung der Kopfgruppen während
der exponentiellen Wachstumsphase unter K+-Limitation festgestellt werden.
Im Gegensatz dazu zeigte der Stamm SOH92, der eine cls-lacZ Fusion zum
Nachweis der cls-Expression aufweist, einen CL-Anstieg unter K+-Limitation. Dieser
Stamm beinhaltet jedoch keine Mutation in den Genen, welche für Enzyme, die in die
Biosynthese der Phospholipide involviert sind, kodieren, und eine regulatorische
Auswirkung auf die Phospholipidzusammensetzung haben.
3.4. Einfluss der K +-Limitation auf die Phospholipide von E. coli C43(DE3) / pEcb1
Um einen genaueren Aufschluss über die Wechselwirkung von KdpD und den
Phospholipiden in der Zytoplasmamembran zu erlangen, wurde der E. coli Stamm
C43(DE3), welcher bei Überproduktion der b-Untereinheit der ATP-Synthase eine
Proliferation der Zytoplasmamembran aufweist (Arechaga et al., 2000), auf seine
Zusammensetzung der Phospholipide untersucht. Die Kopfgruppen der Phospho-
lipide im Stamm C43(DE3) und C43(DE3) / pEcb1, kodierend für die b-Untereinheit
der ATP-Synthase, wurden nach Kultivierung in verschiedenen Minimalmedien mit 32P-Orthophosphat bestimmt.
Tab. 3.5: Zusammensetzung der Kopfgruppen von E. coli C43(DE3) und C43(DE3) / pEcb1 während der exponentiellen Wachstu msphase.
Kopfgruppengehalt [%] in K0.1 Stamm Relevanter Genotyp PE PG CL
Anteil anionischer Phospholipide
C43(DE3) WT 78.1 19.0 2.9 21.9
C43(DE3)/pEcb1 atpF ↑ 79.4 14.6 6.0 20.6
Die Zusammensetzung der Kopfgruppen der exponentiellen Wachstumsphase ist angegeben in Prozent und wurde wie unter 2.5.11-13 beschrieben bestimmt. Die Daten repräsentieren den Durchschnitt von zwei voneinander unabhängigen Messungen. Der Anteil anionischer Lipide ist durch die Summe von PG und CL angegeben.
Die Zusammensetzung der Kopfgruppen ist in Tabelle 3.5 dargestellt. Während den
Versuchen zeigte sich, dass die Werte für die jeweilige Kopfgruppe in K0.1 und K115
gleich waren. Daher wurden die Prozente aus den beiden Minimalmedien zu-
sammengefasst und gemeinsam ausgewertet. Sowohl bei C43(DE3) als auch bei
C43(DE3) / pEcb1 konnte keine Änderung der Kopfgruppen während der exponen-
tiellen Wachstumsphase unter K+-Limitation im Vergleich zur Kultivierung mit hohen

3. Ergebnisse
54
K+-Konzentrationen beobachtet werden. Beim Vergleich von C43(DE3) mit
C43(DE3) / pEcb1 hingegen führte die Proliferation der Zytoplasmamembran zu
einer veränderten Zusammensetzung der Kopfgruppen. Der Stamm C43(DE3) hat
mit 2.9 % ein niedrigeres CL-Niveau, aber mit 21.9 % einen höheren Anteil
anionischer Phospholipide als C43(DE3) / pEcb1, wobei diese Beobachtung unab-
hängig von der K+-Konzentration im Medium war. Der CL-Anstieg infolge der
Proliferation der Zytoplasmamembran wurde bereits für Vollmedium beobachtet
(Arechaga et al., 2000).
3.5. Cardiolipin-Anstieg unter K +-Limitation
3.5.1. cls-Expression in verschiedenen E. coli Stämmen
Die unter 3.1.1 beobachtete Erhöhung des CL-Gehalts unter K+-Limitation könnte auf
eine erhöhte cls-Expression oder auf eine erhöhte Enzymaktivität der CL-Synthase
unter K+-Limitation zurückzuführen sein. Deswegen wurde der Einfluss der K+-
Limitation auf die Induktion der cls-Expression untersucht, indem Zellen von E. coli
SOH92 in phosphat-gepufferten Minimalmedien mit verschiedenen K+-Konzen-
trationen kultiviert wurden. Mit diesem Stamm kann die cls-Expression indirekt
anhand der chromosomalen cls-lacZ Fusion mittels eines β-Galaktosidase-Aktivitäts-
test bestimmt werden, da das Gen der CL-Synthase an ein Reportergen gekoppelt ist
(vgl. auch Heber & Tropp, 1991). Ansonsten hat der Stamm keine Beeinträchtigung
in der Synthese der K+-Transportsysteme oder bei der Biosynthese der Phospho-
lipide. Der Stamm SOH92 ist vom CL-Gehalt (8 – 13 % in Abhängigkeit der K+-
Konzentration) und vom Gehalt an negativen Phospholipiden (20 – 25 %) vergleich-
bar mit dem Wildtypstamm E. coli K12 (genaue Werte siehe Tabelle 3.4). Zu
verschiedenen Zeitpunkten während des kontinuierlichen Wachstums der Kulturen in
K0.1 bzw. K115 wurden Proben für den β-Galaktosidase-Aktivitätstest entnommen.
In Abbildung 3.5 wird deutlich, dass zu jedem Zeitpunkt der Wachstumskurve der
Zellen in Medium mit 0.1 mM K+ die cls-Expression höher war als in Medium mit
115 mM K+. Während der exponentiellen Wachstumsphase waren die β-Galakto-
sidase-Aktivitäten in beiden Minimalmedien höher als in der stationären Wachstums-
phase der jeweiligen Kultur. Das Induktionsverhältnis (K0.1 / K115) für die cls-
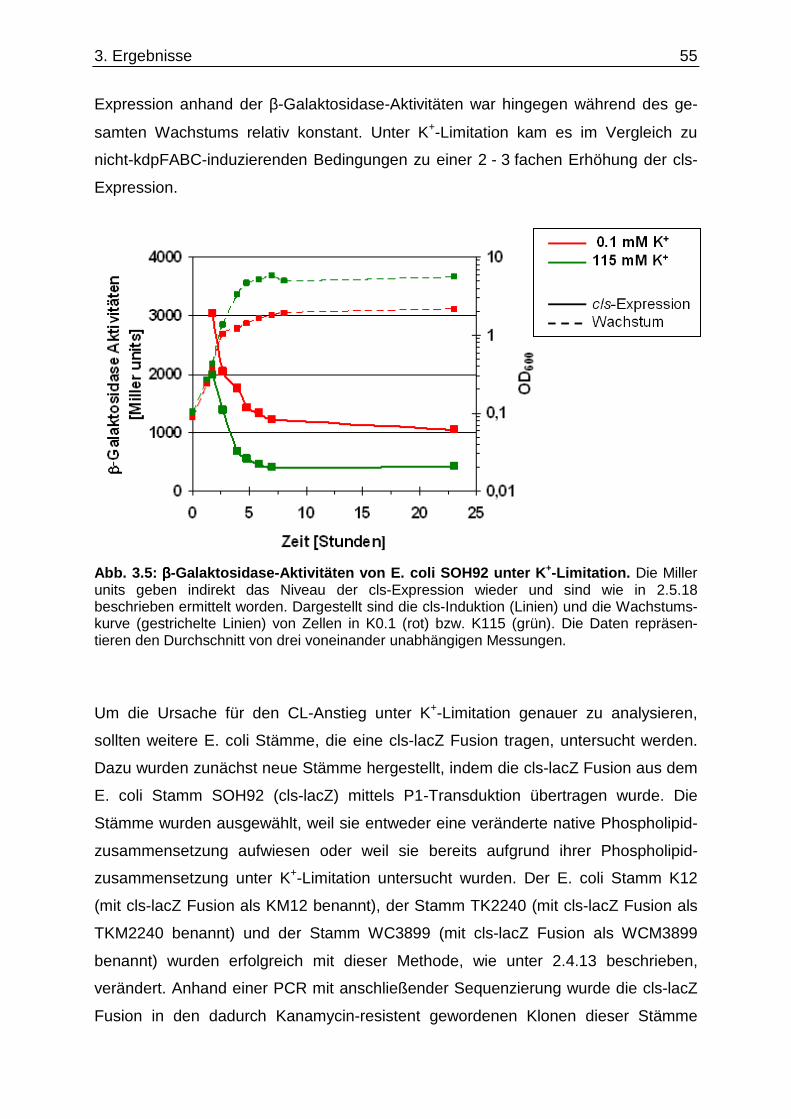
3. Ergebnisse
55
Expression anhand der β-Galaktosidase-Aktivitäten war hingegen während des ge-
samten Wachstums relativ konstant. Unter K+-Limitation kam es im Vergleich zu
nicht-kdpFABC-induzierenden Bedingungen zu einer 2 - 3 fachen Erhöhung der cls-
Expression.
Abb. 3.5: ββββ-Galaktosidase-Aktivitäten von E. coli SOH92 unter K +-Limitation. Die Miller units geben indirekt das Niveau der cls-Expression wieder und sind wie in 2.5.18 beschrieben ermittelt worden. Dargestellt sind die cls-Induktion (Linien) und die Wachstums-kurve (gestrichelte Linien) von Zellen in K0.1 (rot) bzw. K115 (grün). Die Daten repräsen-tieren den Durchschnitt von drei voneinander unabhängigen Messungen.
Um die Ursache für den CL-Anstieg unter K+-Limitation genauer zu analysieren,
sollten weitere E. coli Stämme, die eine cls-lacZ Fusion tragen, untersucht werden.
Dazu wurden zunächst neue Stämme hergestellt, indem die cls-lacZ Fusion aus dem
E. coli Stamm SOH92 (cls-lacZ) mittels P1-Transduktion übertragen wurde. Die
Stämme wurden ausgewählt, weil sie entweder eine veränderte native Phospholipid-
zusammensetzung aufwiesen oder weil sie bereits aufgrund ihrer Phospholipid-
zusammensetzung unter K+-Limitation untersucht wurden. Der E. coli Stamm K12
(mit cls-lacZ Fusion als KM12 benannt), der Stamm TK2240 (mit cls-lacZ Fusion als
TKM2240 benannt) und der Stamm WC3899 (mit cls-lacZ Fusion als WCM3899
benannt) wurden erfolgreich mit dieser Methode, wie unter 2.4.13 beschrieben,
verändert. Anhand einer PCR mit anschließender Sequenzierung wurde die cls-lacZ
Fusion in den dadurch Kanamycin-resistent gewordenen Klonen dieser Stämme
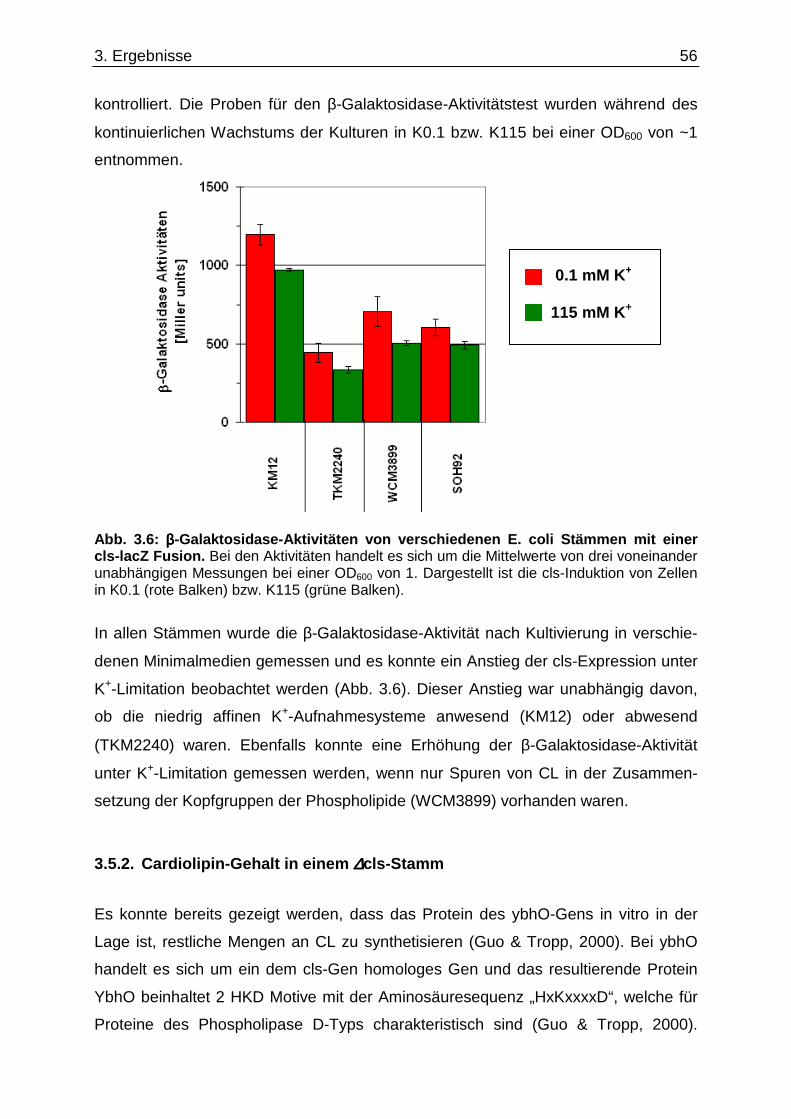
3. Ergebnisse
56
0.1 mM K+ 115 mM K+
kontrolliert. Die Proben für den β-Galaktosidase-Aktivitätstest wurden während des
kontinuierlichen Wachstums der Kulturen in K0.1 bzw. K115 bei einer OD600 von ~1
entnommen.
Abb. 3.6: ββββ-Galaktosidase-Aktivitäten von verschiedenen E. coli Stämmen mit einer cls-lacZ Fusion. Bei den Aktivitäten handelt es sich um die Mittelwerte von drei voneinander unabhängigen Messungen bei einer OD600 von 1. Dargestellt ist die cls-Induktion von Zellen in K0.1 (rote Balken) bzw. K115 (grüne Balken).
In allen Stämmen wurde die β-Galaktosidase-Aktivität nach Kultivierung in verschie-
denen Minimalmedien gemessen und es konnte ein Anstieg der cls-Expression unter
K+-Limitation beobachtet werden (Abb. 3.6). Dieser Anstieg war unabhängig davon,
ob die niedrig affinen K+-Aufnahmesysteme anwesend (KM12) oder abwesend
(TKM2240) waren. Ebenfalls konnte eine Erhöhung der β-Galaktosidase-Aktivität
unter K+-Limitation gemessen werden, wenn nur Spuren von CL in der Zusammen-
setzung der Kopfgruppen der Phospholipide (WCM3899) vorhanden waren.
3.5.2. Cardiolipin-Gehalt in einem ∆∆∆∆cls-Stamm
Es konnte bereits gezeigt werden, dass das Protein des ybhO-Gens in vitro in der
Lage ist, restliche Mengen an CL zu synthetisieren (Guo & Tropp, 2000). Bei ybhO
handelt es sich um ein dem cls-Gen homologes Gen und das resultierende Protein
YbhO beinhaltet 2 HKD Motive mit der Aminosäuresequenz „HxKxxxxD“, welche für
Proteine des Phospholipase D-Typs charakteristisch sind (Guo & Tropp, 2000).
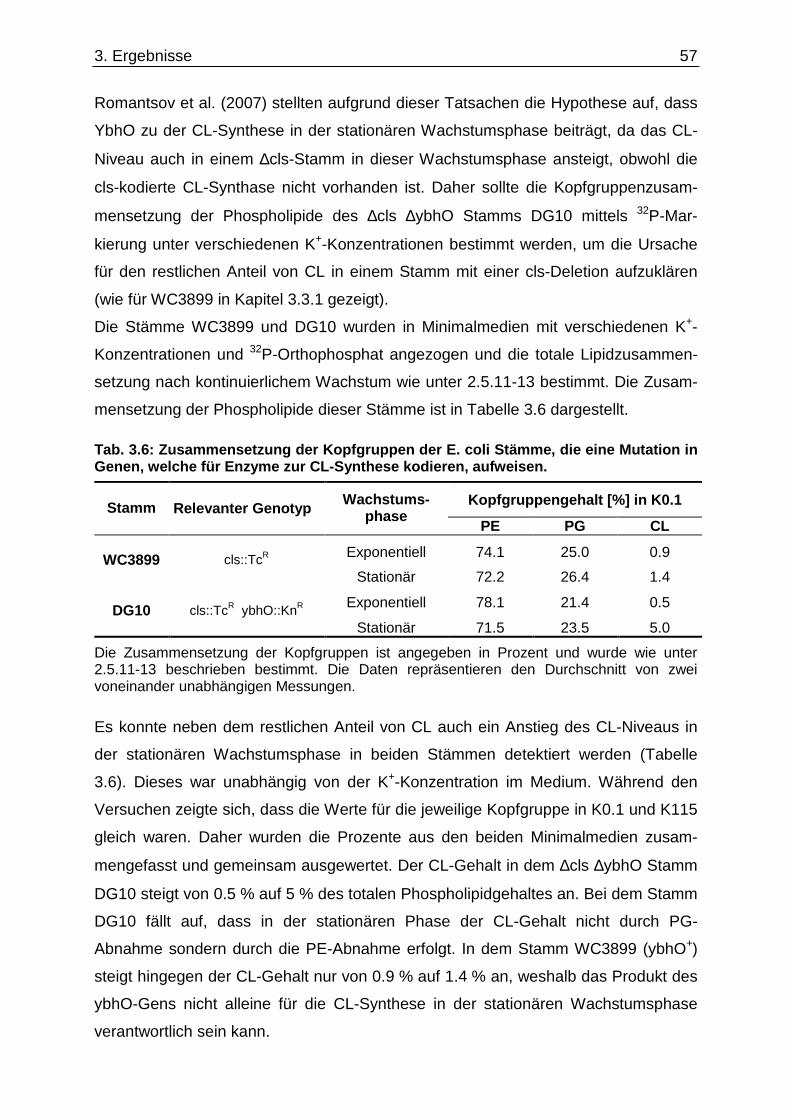
3. Ergebnisse
57
Romantsov et al. (2007) stellten aufgrund dieser Tatsachen die Hypothese auf, dass
YbhO zu der CL-Synthese in der stationären Wachstumsphase beiträgt, da das CL-
Niveau auch in einem ∆cls-Stamm in dieser Wachstumsphase ansteigt, obwohl die
cls-kodierte CL-Synthase nicht vorhanden ist. Daher sollte die Kopfgruppenzusam-
mensetzung der Phospholipide des ∆cls ∆ybhO Stamms DG10 mittels 32P-Mar-
kierung unter verschiedenen K+-Konzentrationen bestimmt werden, um die Ursache
für den restlichen Anteil von CL in einem Stamm mit einer cls-Deletion aufzuklären
(wie für WC3899 in Kapitel 3.3.1 gezeigt).
Die Stämme WC3899 und DG10 wurden in Minimalmedien mit verschiedenen K+-
Konzentrationen und 32P-Orthophosphat angezogen und die totale Lipidzusammen-
setzung nach kontinuierlichem Wachstum wie unter 2.5.11-13 bestimmt. Die Zusam-
mensetzung der Phospholipide dieser Stämme ist in Tabelle 3.6 dargestellt.
Tab. 3.6: Zusammensetzung der Kopfgruppen der E. coli Stämme, die eine Mutation in Genen, welche für Enzyme zur CL-Synthese kodieren, aufweisen.
Relevanter Genotyp Kopfgruppengehalt [%] in K0.1 Stamm
Wachstums-phase
PE PG CL
Exponentiell 74.1 25.0 0.9 WC3899 cls::TcR Stationär 72.2 26.4 1.4
Exponentiell 78.1 21.4 0.5 DG10 cls::TcR ybhO::KnR Stationär 71.5 23.5 5.0
Die Zusammensetzung der Kopfgruppen ist angegeben in Prozent und wurde wie unter 2.5.11-13 beschrieben bestimmt. Die Daten repräsentieren den Durchschnitt von zwei voneinander unabhängigen Messungen.
Es konnte neben dem restlichen Anteil von CL auch ein Anstieg des CL-Niveaus in
der stationären Wachstumsphase in beiden Stämmen detektiert werden (Tabelle
3.6). Dieses war unabhängig von der K+-Konzentration im Medium. Während den
Versuchen zeigte sich, dass die Werte für die jeweilige Kopfgruppe in K0.1 und K115
gleich waren. Daher wurden die Prozente aus den beiden Minimalmedien zusam-
mengefasst und gemeinsam ausgewertet. Der CL-Gehalt in dem ∆cls ∆ybhO Stamm
DG10 steigt von 0.5 % auf 5 % des totalen Phospholipidgehaltes an. Bei dem Stamm
DG10 fällt auf, dass in der stationären Phase der CL-Gehalt nicht durch PG-
Abnahme sondern durch die PE-Abnahme erfolgt. In dem Stamm WC3899 (ybhO+)
steigt hingegen der CL-Gehalt nur von 0.9 % auf 1.4 % an, weshalb das Produkt des
ybhO-Gens nicht alleine für die CL-Synthese in der stationären Wachstumsphase
verantwortlich sein kann.

3. Ergebnisse
58
3.6. Überprüfung eines gemeinsamen kch-cls Transkripts
Bei der Betrachtung der Lage des cls-Gens im Chromosom fiel auf, dass direkt
neben dem cls-Gen das kch-Gen lokalisiert ist. Das kch-Gen kodiert für einen funk-
tionalen K+-Kanal (Kuo et al., 2003). Dies ist erwähnenswert, da zytoplasmatisches
K+ eine Rolle für die cls-Expression und für die Aktivität der CL-Synthase spielt.
Obwohl diese zwei Gene vor ihrem Transkriptionsstart eine eigene Promotorsequenz
haben, wäre es denkbar, dass sie ebenfalls gemeinsam über ein kch-cls mRNA
Transkript exprimiert und/oder reguliert werden. Dem cls-Gen geht eine Shine-
Dalgarno-Sequenz und ein σ70-Promotor voraus (Ivanisevic et al., 1995). Es wird
vermutet, dass das kch-Gen über einen wahrscheinlich σ54-abhängigen Promotor
exprimiert wird, der durch Computeranalyse identifiziert wurde (Reitzer & Schneider,
2001). Jedoch ist der Abstand zwischen der vermeintlich σ54 bindenden Stelle und
dem Translationsstart für kch mit 264 bp größer als für alle anderen bekannten σ54-
abhängigen Promotoren, die Abstände zwischen 34 und 136 bp aufweisen (Reitzer &
Schneider, 2001). Dieser potentielle σ54-Promotor war nicht in der Lage kch zu über-
exprimieren, wodurch es nicht möglich war, die Funktion von Kch genauer zu unter-
suchen (Kuo et al., 2003). Es wäre trotzdem denkbar, dass zusätzlich beide Gene
gemeinsam über ein Transkript exprimiert und evtl. auch gemeinsam reguliert
werden, da für beide Genprodukte K+ eine entscheidende Rolle spielt (Abb. 3.7).
Abb. 3.7: Schema einer möglichen, gemeinsamen Trans kription von kch und cls. P: Promotor.
Um eine gemeinsame Expression der beiden Gene nachzuweisen, wurde eine
Reverse Transkriptase PCR durchgeführt. Dafür wurde zunächst, wie in 2.4.10-11
beschrieben, RNA aus Zellen von E. coli K12 isoliert und mittels eines Reversen
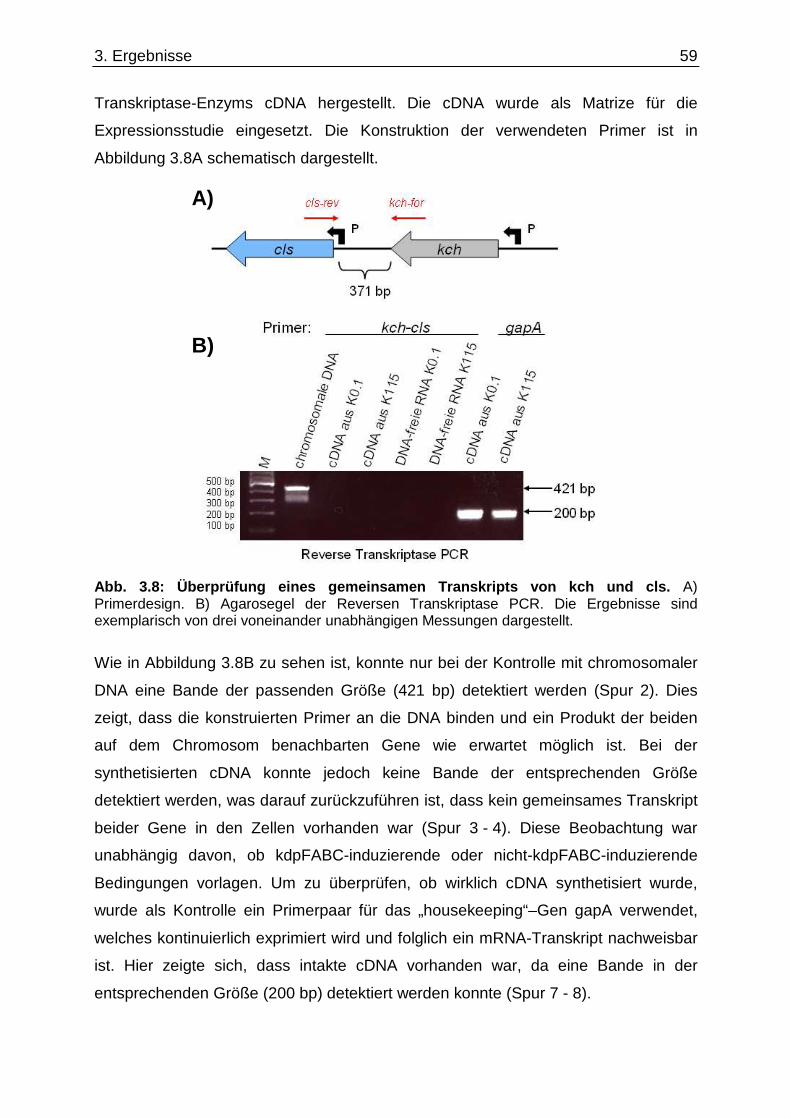
3. Ergebnisse
59
Transkriptase-Enzyms cDNA hergestellt. Die cDNA wurde als Matrize für die
Expressionsstudie eingesetzt. Die Konstruktion der verwendeten Primer ist in
Abbildung 3.8A schematisch dargestellt.
Abb. 3.8: Überprüfung eines gemeinsamen Transkripts von kch und cls. A) Primerdesign. B) Agarosegel der Reversen Transkriptase PCR. Die Ergebnisse sind exemplarisch von drei voneinander unabhängigen Messungen dargestellt.
Wie in Abbildung 3.8B zu sehen ist, konnte nur bei der Kontrolle mit chromosomaler
DNA eine Bande der passenden Größe (421 bp) detektiert werden (Spur 2). Dies
zeigt, dass die konstruierten Primer an die DNA binden und ein Produkt der beiden
auf dem Chromosom benachbarten Gene wie erwartet möglich ist. Bei der
synthetisierten cDNA konnte jedoch keine Bande der entsprechenden Größe
detektiert werden, was darauf zurückzuführen ist, dass kein gemeinsames Transkript
beider Gene in den Zellen vorhanden war (Spur 3 - 4). Diese Beobachtung war
unabhängig davon, ob kdpFABC-induzierende oder nicht-kdpFABC-induzierende
Bedingungen vorlagen. Um zu überprüfen, ob wirklich cDNA synthetisiert wurde,
wurde als Kontrolle ein Primerpaar für das „housekeeping“–Gen gapA verwendet,
welches kontinuierlich exprimiert wird und folglich ein mRNA-Transkript nachweisbar
ist. Hier zeigte sich, dass intakte cDNA vorhanden war, da eine Bande in der
entsprechenden Größe (200 bp) detektiert werden konnte (Spur 7 - 8).
A) B)

3. Ergebnisse
60
3.7. Einfluss der Zusammensetzung der Zytoplasmamem bran auf das Kdp-System
3.7.1. Kinaseaktivität von KdpD-6His in Membran-Lip id-Fusionen
Stallkamp et al. (1999) konnten zeigen, dass die Aktivierung der Sensorkinase KdpD
durch die Anwesenheit von 50 % anionischer Phospholipide in Liposomen beein-
flusst werden kann. Dieser Einfluss sollte genauer untersucht werden, indem die
Menge an negativ geladenen Phospholipiden variiert wurde. Da in invertierten Mem-
branvesikel von TRK2000 / pKJ2-6His mit [γ-32P]-ATP Kinaseaktivität von KdpD-6His
gemessen werden kann, sollte auf dieser Ebene die Autophosphorylierung von KdpD
untersucht werden, um das Protein in seiner nativen Membranumgebung zu be-
lassen.
Zunächst wurden die „Lipid-Stämme“ mit einer veränderten nativen Phospholipid-
zusammensetzung mit dem Plasmid pKJ2-6His transformiert. Anschließend wurden
Membranvesikel präpariert, um mit diesen eine Aussage über den Einfluss des
Anteils an anionischen Phospholipiden auf die Kinaseaktivität treffen zu können. Es
konnte für die Stämme UE54 / pKJ2-6His (100 % PE), WC3899 / pKJ2-6His (75 %
PE, 24 % PG, 1 % CL) und HDL1001 / pKJ2-6His (mit 1 mM IPTG: 82 % PE, 14 %
PG, 4 % CL bzw. ohne IPTG: 96 % PE, 3 % PG, 1 % CL) allerdings keine Kinase-
aktivität von KdpD-His6 gemessen werden (Daten nicht gezeigt). Dieses ist
vermutlich darauf zurückzuführen, dass die Stämme nicht atp- sind und somit das
radioaktiv-markierte ATP über die ATPase abgebaut werden kann.
Daraufhin wurden die Membranvesikel von TRK2000/pKJ2-6His, in denen Kinase-
aktivitäten gemessen werden konnten, in verschiedenen Verhältnissen mit Lipo-
somen aus synthetischen Lipiden (50% PE / 50% PC oder 100% CL) gemischt, um
den Anteil an anionischen Phospholipiden zu variieren. Diese Methode wurde bereits
für andere Proteine angewandt (Özcan et al., 2007). Ein Vorteil dabei ist, dass das
Protein in seiner natürlichen Membranumgebung bleibt und nicht durch die
Reinigung und anschließende Rekonstitution möglicherweise funktionell beein-
trächtigt wird.
Mit einem Immunoblot wurde überprüft, dass vergleichbare KdpD-Proteinmengen bei
allen Fusionen von Membranvesikeln mit synthetischen Liposomen eingesetzt
wurden. In den Autoradiogrammen waren neben dem phosphorylierten KdpD-His6
Monomer (KdpD-6His~P) auch das phosphorylierte Dimer und andere phos-
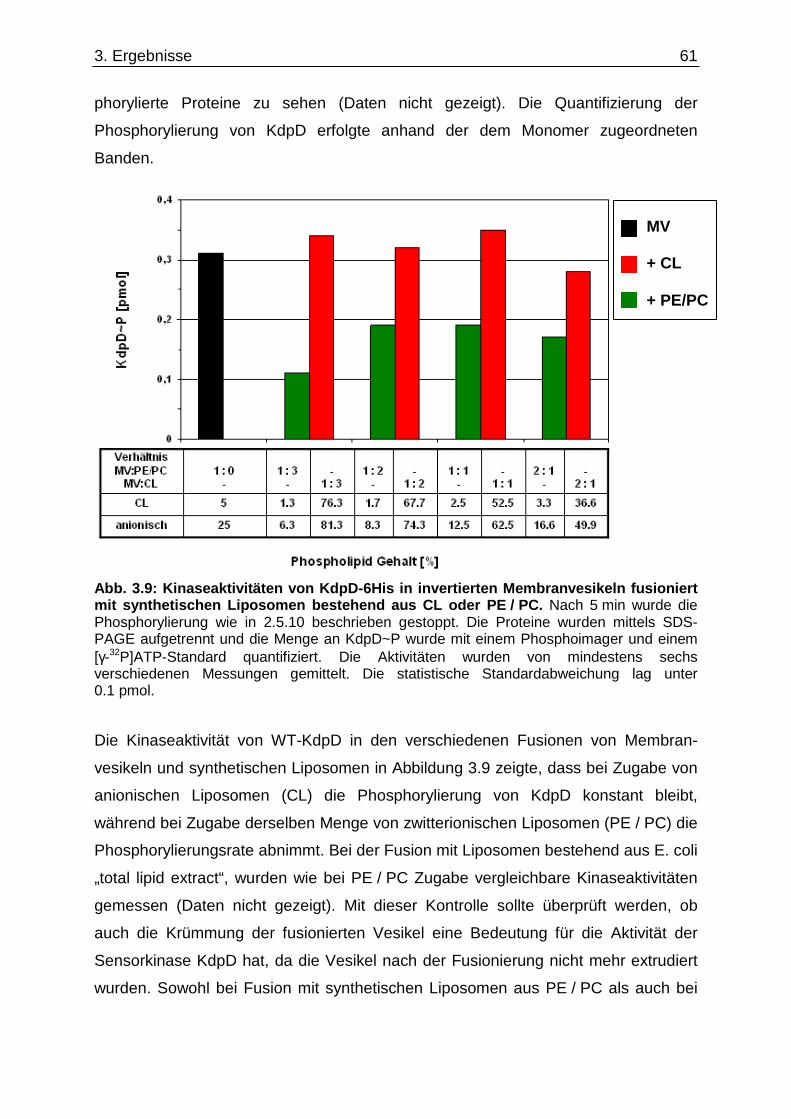
3. Ergebnisse
61
phorylierte Proteine zu sehen (Daten nicht gezeigt). Die Quantifizierung der
Phosphorylierung von KdpD erfolgte anhand der dem Monomer zugeordneten
Banden.
Abb. 3.9: Kinaseaktivitäten von KdpD-6His in invertierten Mem branvesikeln fusioniert mit synthetischen Liposomen bestehend aus CL oder P E / PC. Nach 5 min wurde die Phosphorylierung wie in 2.5.10 beschrieben gestoppt. Die Proteine wurden mittels SDS-PAGE aufgetrennt und die Menge an KdpD~P wurde mit einem Phosphoimager und einem [γ-32P]ATP-Standard quantifiziert. Die Aktivitäten wurden von mindestens sechs verschiedenen Messungen gemittelt. Die statistische Standardabweichung lag unter 0.1 pmol.
Die Kinaseaktivität von WT-KdpD in den verschiedenen Fusionen von Membran-
vesikeln und synthetischen Liposomen in Abbildung 3.9 zeigte, dass bei Zugabe von
anionischen Liposomen (CL) die Phosphorylierung von KdpD konstant bleibt,
während bei Zugabe derselben Menge von zwitterionischen Liposomen (PE / PC) die
Phosphorylierungsrate abnimmt. Bei der Fusion mit Liposomen bestehend aus E. coli
„total lipid extract“, wurden wie bei PE / PC Zugabe vergleichbare Kinaseaktivitäten
gemessen (Daten nicht gezeigt). Mit dieser Kontrolle sollte überprüft werden, ob
auch die Krümmung der fusionierten Vesikel eine Bedeutung für die Aktivität der
Sensorkinase KdpD hat, da die Vesikel nach der Fusionierung nicht mehr extrudiert
wurden. Sowohl bei Fusion mit synthetischen Liposomen aus PE / PC als auch bei
MV + CL + PE/PC
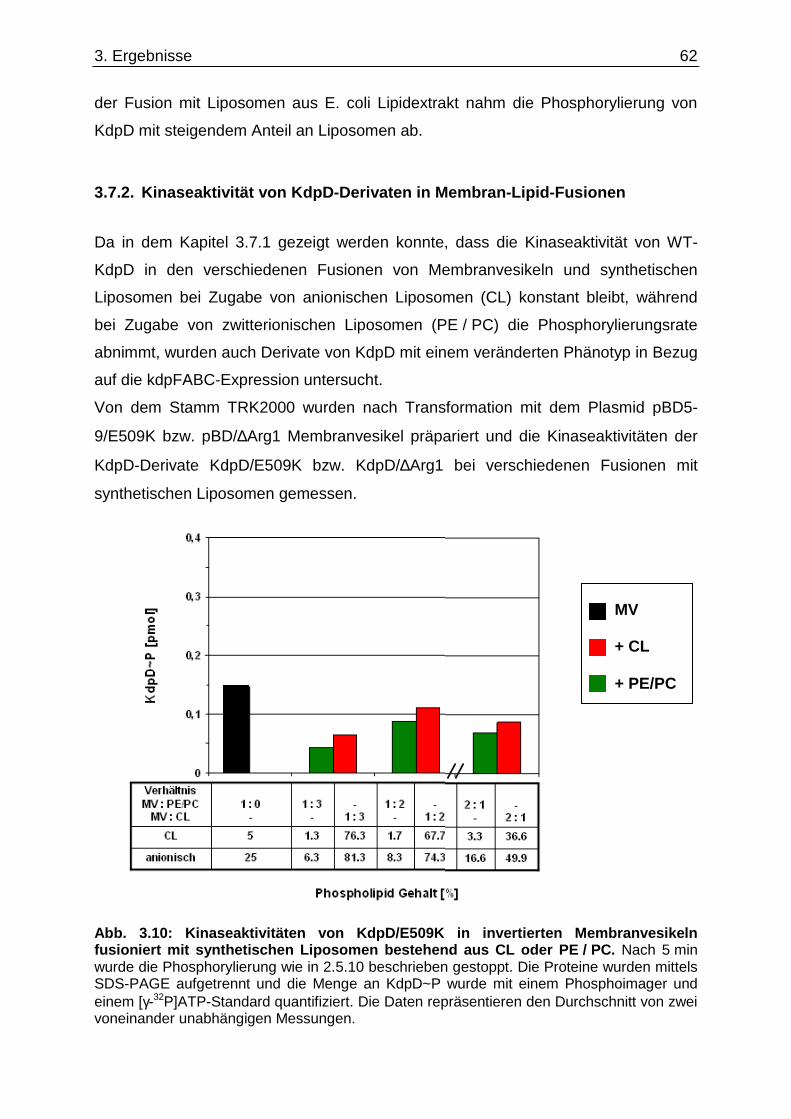
3. Ergebnisse
62
der Fusion mit Liposomen aus E. coli Lipidextrakt nahm die Phosphorylierung von
KdpD mit steigendem Anteil an Liposomen ab.
3.7.2. Kinaseaktivität von KdpD-Derivaten in Membra n-Lipid-Fusionen
Da in dem Kapitel 3.7.1 gezeigt werden konnte, dass die Kinaseaktivität von WT-
KdpD in den verschiedenen Fusionen von Membranvesikeln und synthetischen
Liposomen bei Zugabe von anionischen Liposomen (CL) konstant bleibt, während
bei Zugabe von zwitterionischen Liposomen (PE / PC) die Phosphorylierungsrate
abnimmt, wurden auch Derivate von KdpD mit einem veränderten Phänotyp in Bezug
auf die kdpFABC-Expression untersucht.
Von dem Stamm TRK2000 wurden nach Transformation mit dem Plasmid pBD5-
9/E509K bzw. pBD/∆Arg1 Membranvesikel präpariert und die Kinaseaktivitäten der
KdpD-Derivate KdpD/E509K bzw. KdpD/∆Arg1 bei verschiedenen Fusionen mit
synthetischen Liposomen gemessen.
Abb. 3.10: Kinaseaktivitäten von KdpD/E509K in invertierten Me mbranvesikeln fusioniert mit synthetischen Liposomen bestehend au s CL oder PE / PC. Nach 5 min wurde die Phosphorylierung wie in 2.5.10 beschrieben gestoppt. Die Proteine wurden mittels SDS-PAGE aufgetrennt und die Menge an KdpD~P wurde mit einem Phosphoimager und einem [γ-32P]ATP-Standard quantifiziert. Die Daten repräsentieren den Durchschnitt von zwei voneinander unabhängigen Messungen.
MV + CL + PE/PC

3. Ergebnisse
63
Die semi-konstitutive Mutante KdpD/E509K zeigte eine geringere Aktivität im
Vergleich zum Wildtyp-KdpD, aber eine ähnliche Tendenz bei Zugabe von
synthetischen Liposomen. Die Zugabe von anionischen Liposomen führte ebenfalls
zu einem konstanteren Niveau der Kinaseaktivität (Abb. 3.10 vgl. Abb. 3.9). Die K+
insensitive Mutante KdpD/∆Arg1 wies keine Kinaseaktivität auf, unabhängig davon in
welchem Verhältnis synthetische Liposomen mit den Membranvesikeln fusioniert
wurden (Daten nicht gezeigt). Es konnten somit keine unterschiedlichen Einflüsse auf
die Phosphorylierung der Sensorkinase KdpD im Vergleich von Wildtyp und den
Derivaten in Abhängigkeit des Anteils der anionischen Phospholipide gezeigt
werden.
3.7.3. kdpFABC-Expression unter K +-Limitation in Abhängigkeit von der Lipid-
zusammensetzung
Basierend auf den bisher gezeigten Ergebnissen ist es vorstellbar, dass eine
Korrelation zwischen der Aktivität der membrangebundenen Sensorkinase KdpD und
der Zusammensetzung der Kopfgruppen existiert. Um diese Beziehung aufzuklären,
wurden „Lipid-Stämme“ mit einer veränderten nativen Zusammensetzung der Kopf-
gruppen auf die kdpFABC-Expression untersucht. Dafür wurden Zellen der jeweiligen
Stämme in Minimalmedien mit verschiedenen K+-Konzentrationen angezogen und
die RNA wie unter 2.4.10 beschrieben für die Q-RT-PCR extrahiert. Anschließend
wurde mittels einer Reversen Transkriptase aus der DNAse-verdauten RNA cDNA
hergestellt, welche als Matrize für die Q-RT-PCR eingesetzt wurde. Um die
kdpFABC-Expression zu ermitteln, wurde ein Primerpaar in kdpA und als Kontrolle
ein Primerpaar in gap benutzt. Das Produkt jeder PCR betrug 200 bp.
Diese Methode ermöglicht es, die kdpFABC-Expression direkter zu messen als im
Vergleich zu der bereits beschriebenen KdpFABC-Detektion mittels der Immunoblot-
Technik von ganzen Zellen (Stallkamp et al., 1999). Es wurden Proben von Zellen
unter kdpFABC-induzierenden Bedingungen (0.1 mM K+) und nicht-kdpFABC-
induzierenden Bedingungen (115 mM K+) untersucht. Zuvor wurden die Primer,
welche für die Q-RT-PCR benutzt werden sollten, anhand isolierter chromosomaler
DNA aller untersuchten Stämme und dem Q-RT-PCR-Protokoll mittels einer
standardisierten PCR kontrolliert.
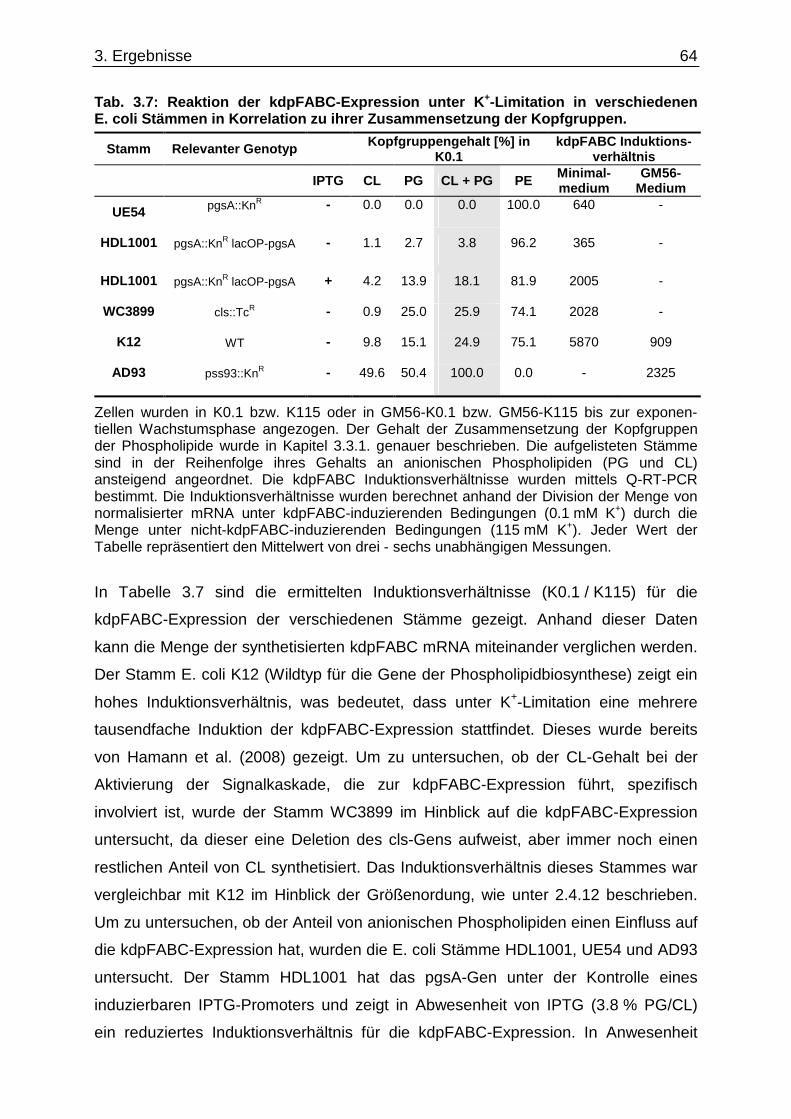
3. Ergebnisse
64
Tab. 3.7: Reaktion der kdpFABC-Expression unter K +-Limitation in verschiedenen E. coli Stämmen in Korrelation zu ihrer Zusammensetzung de r Kopfgruppen.
Stamm Relevanter Genotyp Kopfgruppengehalt [%] in K0.1
kdpFABC Induktions-verhältnis
IPTG CL PG CL + PG PE Minimal- medium
GM56- Medium
UE54 pgsA::KnR - 0.0 0.0 0.0 100.0 640 -
HDL1001 pgsA::KnR lacOP-pgsA - 1.1 2.7 3.8 96.2 365 -
HDL1001 pgsA::KnR lacOP-pgsA + 4.2 13.9 18.1 81.9 2005 -
WC3899 cls::TcR - 0.9 25.0 25.9 74.1 2028 -
K12 WT - 9.8 15.1 24.9 75.1 5870 909
AD93 pss93::KnR - 49.6 50.4 100.0 0.0 - 2325
Zellen wurden in K0.1 bzw. K115 oder in GM56-K0.1 bzw. GM56-K115 bis zur exponen-tiellen Wachstumsphase angezogen. Der Gehalt der Zusammensetzung der Kopfgruppen der Phospholipide wurde in Kapitel 3.3.1. genauer beschrieben. Die aufgelisteten Stämme sind in der Reihenfolge ihres Gehalts an anionischen Phospholipiden (PG und CL) ansteigend angeordnet. Die kdpFABC Induktionsverhältnisse wurden mittels Q-RT-PCR bestimmt. Die Induktionsverhältnisse wurden berechnet anhand der Division der Menge von normalisierter mRNA unter kdpFABC-induzierenden Bedingungen (0.1 mM K+) durch die Menge unter nicht-kdpFABC-induzierenden Bedingungen (115 mM K+). Jeder Wert der Tabelle repräsentiert den Mittelwert von drei - sechs unabhängigen Messungen.
In Tabelle 3.7 sind die ermittelten Induktionsverhältnisse (K0.1 / K115) für die
kdpFABC-Expression der verschiedenen Stämme gezeigt. Anhand dieser Daten
kann die Menge der synthetisierten kdpFABC mRNA miteinander verglichen werden.
Der Stamm E. coli K12 (Wildtyp für die Gene der Phospholipidbiosynthese) zeigt ein
hohes Induktionsverhältnis, was bedeutet, dass unter K+-Limitation eine mehrere
tausendfache Induktion der kdpFABC-Expression stattfindet. Dieses wurde bereits
von Hamann et al. (2008) gezeigt. Um zu untersuchen, ob der CL-Gehalt bei der
Aktivierung der Signalkaskade, die zur kdpFABC-Expression führt, spezifisch
involviert ist, wurde der Stamm WC3899 im Hinblick auf die kdpFABC-Expression
untersucht, da dieser eine Deletion des cls-Gens aufweist, aber immer noch einen
restlichen Anteil von CL synthetisiert. Das Induktionsverhältnis dieses Stammes war
vergleichbar mit K12 im Hinblick der Größenordung, wie unter 2.4.12 beschrieben.
Um zu untersuchen, ob der Anteil von anionischen Phospholipiden einen Einfluss auf
die kdpFABC-Expression hat, wurden die E. coli Stämme HDL1001, UE54 und AD93
untersucht. Der Stamm HDL1001 hat das pgsA-Gen unter der Kontrolle eines
induzierbaren IPTG-Promoters und zeigt in Abwesenheit von IPTG (3.8 % PG/CL)
ein reduziertes Induktionsverhältnis für die kdpFABC-Expression. In Anwesenheit

3. Ergebnisse
65
von 1 mM IPTG, welches zu einem beinahe restaurierten PG- und CL-Gehalt führte,
konnte ein höheres Induktionsverhältnis in dem Bereich des Wildtypstammes K12
beobachtet werden. Des Weiteren wurde die kdpFABC-Expression unter K+-
Limitation des Stammes UE54, der eine Deletion des pgsA-Gen hat, was in einer
Membran ohne die anionischen Phospholipide PG und CL resultiert, untersucht.
Dieser Stamm zeigte ein niedriges Induktionsverhältnis für die kdpFABC-Expression
wie HDL1001 ohne IPTG, was den Einfluss des Gesamtanteils der anionischen
Phospholipide andeutet. Der Stamm AD93 ist aufgrund des Fehlens des zwitter-
ionischen Phospholipids PE das gegenteilige Extrem von UE54, was zu einer
Zytoplasmamembran bestehend aus 100 % anionischen Phospholipiden führt. Die
Q-RT-PCR Daten dieses Stammes zeigen ein 10-fach höheres Induktionsverhältnis
für die kdpFABC-Expression gemessen in GM56-Medium im Vergleich zu K12, was
ebenfalls den stimulierenden Einfluss der anionischen Phospholipide auf die
kdpFABC-Expression bestätigt. Für den Stamm C43(DE3) konnte mit einer ausge-
stülpten Zytoplasmamembran (C43(DE3) / pEcb1), was zu einem leichten CL-
Anstieg führte, ein höheres Induktionsverhältnis für die kdpFABC-Expression
beobachtet werden (Daten nicht gezeigt).
3.8. Einfluss von Procain auf die kdpFABC-Expression
3.8.1. Q-RT-PCR in „Lipid-Stämmen“
Des Weiteren sollte die Wirkung von Procain auf die kdpFABC-Expression in
Stämmen mit einer veränderten nativen Phospholipidzusammensetzung untersucht
werden. Procain lagert sich in die Zytoplasmamembran ein und bewirkt dadurch eine
Änderung der Membranspannung, welche möglicherweise von KdpD wahrge-
nommen wird. Procain könnte aber auch die Aktivität oder den Einbau der Sensor-
kinase KdpD bzw. des Transportkomplexes KdpFABC in die Zytoplasmamembran
beeinflussen.
In diesem Ansatz sollte die kdpFABC-Expression mit Hilfe von Q-RT-PCR in den
„Lipid-Stämmen“ mit einer veränderten nativen Phospholipidzusammensetzung und
chromosomal-kodiertem Wildtyp-kdpD ermittelt werden. Dafür wurden zunächst die
Stämme AD93 (∆PE), UE54 (∆PG∆CL), WC3899 (∆CL) und K12 (native Lipid-
zusammensetzung) in Minimalmedium mit 0.1 mM K+ bzw. 115 mM K+ angezogen.
Bei einer OD600 0.4 erfolgte die Zugabe von 20 mM Procain. Für die Q-RT-PCR
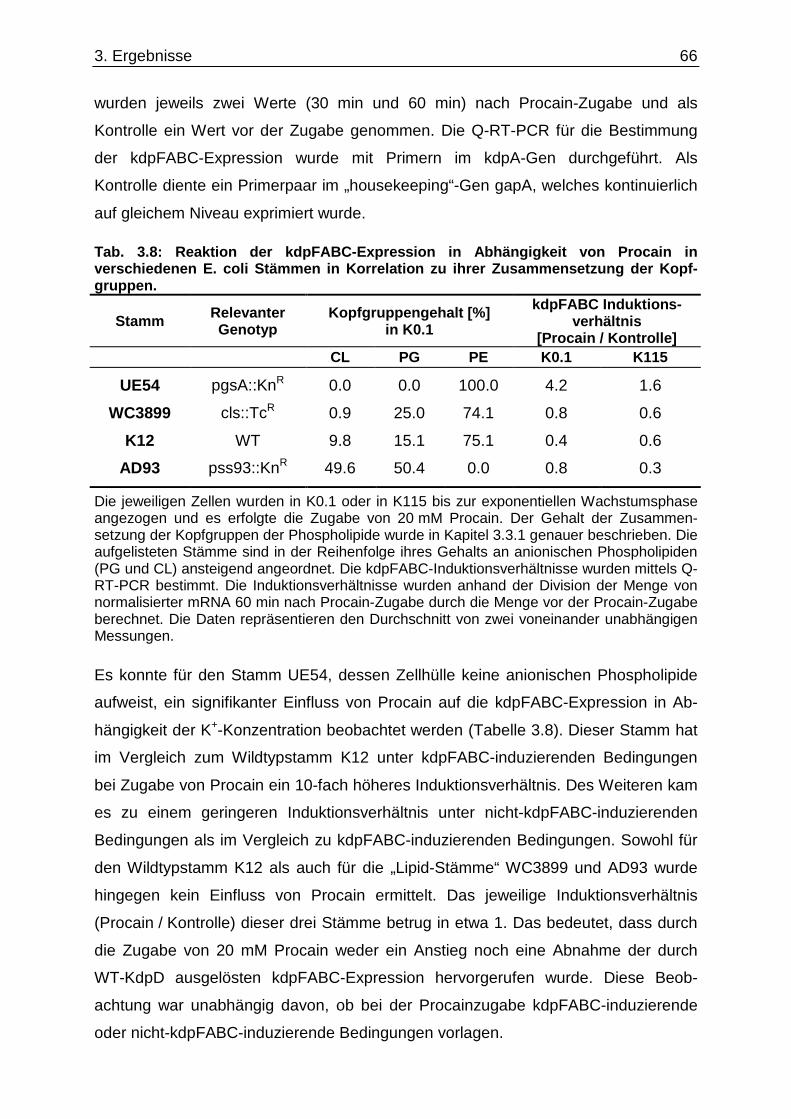
3. Ergebnisse
66
wurden jeweils zwei Werte (30 min und 60 min) nach Procain-Zugabe und als
Kontrolle ein Wert vor der Zugabe genommen. Die Q-RT-PCR für die Bestimmung
der kdpFABC-Expression wurde mit Primern im kdpA-Gen durchgeführt. Als
Kontrolle diente ein Primerpaar im „housekeeping“-Gen gapA, welches kontinuierlich
auf gleichem Niveau exprimiert wurde.
Tab. 3.8: Reaktion der kdpFABC-Expression in Abhängigkeit von Procain in verschiedenen E. coli Stämmen in Korrelation zu ihrer Zusammensetzung de r Kopf-gruppen.
Stamm Relevanter Genotyp
Kopfgruppengehalt [%] in K0.1
kdpFABC Induktions-verhältnis
[Procain / Kontrolle] CL PG PE K0.1 K115
UE54 pgsA::KnR 0.0 0.0 100.0 4.2 1.6
WC3899 cls::TcR 0.9 25.0 74.1 0.8 0.6
K12 WT 9.8 15.1 75.1 0.4 0.6
AD93 pss93::KnR 49.6 50.4 0.0 0.8 0.3
Die jeweiligen Zellen wurden in K0.1 oder in K115 bis zur exponentiellen Wachstumsphase angezogen und es erfolgte die Zugabe von 20 mM Procain. Der Gehalt der Zusammen-setzung der Kopfgruppen der Phospholipide wurde in Kapitel 3.3.1 genauer beschrieben. Die aufgelisteten Stämme sind in der Reihenfolge ihres Gehalts an anionischen Phospholipiden (PG und CL) ansteigend angeordnet. Die kdpFABC-Induktionsverhältnisse wurden mittels Q-RT-PCR bestimmt. Die Induktionsverhältnisse wurden anhand der Division der Menge von normalisierter mRNA 60 min nach Procain-Zugabe durch die Menge vor der Procain-Zugabe berechnet. Die Daten repräsentieren den Durchschnitt von zwei voneinander unabhängigen Messungen. Es konnte für den Stamm UE54, dessen Zellhülle keine anionischen Phospholipide
aufweist, ein signifikanter Einfluss von Procain auf die kdpFABC-Expression in Ab-
hängigkeit der K+-Konzentration beobachtet werden (Tabelle 3.8). Dieser Stamm hat
im Vergleich zum Wildtypstamm K12 unter kdpFABC-induzierenden Bedingungen
bei Zugabe von Procain ein 10-fach höheres Induktionsverhältnis. Des Weiteren kam
es zu einem geringeren Induktionsverhältnis unter nicht-kdpFABC-induzierenden
Bedingungen als im Vergleich zu kdpFABC-induzierenden Bedingungen. Sowohl für
den Wildtypstamm K12 als auch für die „Lipid-Stämme“ WC3899 und AD93 wurde
hingegen kein Einfluss von Procain ermittelt. Das jeweilige Induktionsverhältnis
(Procain / Kontrolle) dieser drei Stämme betrug in etwa 1. Das bedeutet, dass durch
die Zugabe von 20 mM Procain weder ein Anstieg noch eine Abnahme der durch
WT-KdpD ausgelösten kdpFABC-Expression hervorgerufen wurde. Diese Beob-
achtung war unabhängig davon, ob bei der Procainzugabe kdpFABC-induzierende
oder nicht-kdpFABC-induzierende Bedingungen vorlagen.
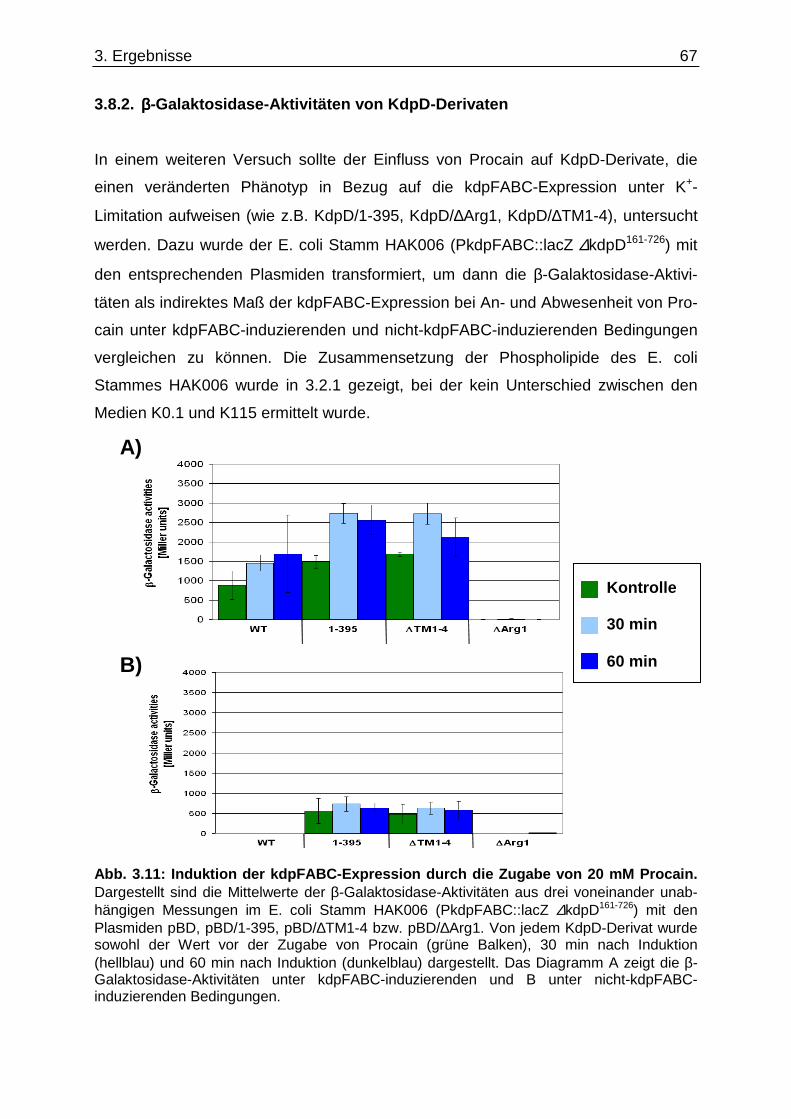
3. Ergebnisse
67
3.8.2. ββββ-Galaktosidase-Aktivitäten von KdpD-Derivaten
In einem weiteren Versuch sollte der Einfluss von Procain auf KdpD-Derivate, die
einen veränderten Phänotyp in Bezug auf die kdpFABC-Expression unter K+-
Limitation aufweisen (wie z.B. KdpD/1-395, KdpD/∆Arg1, KdpD/∆TM1-4), untersucht
werden. Dazu wurde der E. coli Stamm HAK006 (PkdpFABC::lacZ ∆kdpD161-726) mit
den entsprechenden Plasmiden transformiert, um dann die β-Galaktosidase-Aktivi-
täten als indirektes Maß der kdpFABC-Expression bei An- und Abwesenheit von Pro-
cain unter kdpFABC-induzierenden und nicht-kdpFABC-induzierenden Bedingungen
vergleichen zu können. Die Zusammensetzung der Phospholipide des E. coli
Stammes HAK006 wurde in 3.2.1 gezeigt, bei der kein Unterschied zwischen den
Medien K0.1 und K115 ermittelt wurde.
Abb. 3.11: Induktion der kdpFABC-Expression durch die Zugabe von 20 mM Procain. Dargestellt sind die Mittelwerte der β-Galaktosidase-Aktivitäten aus drei voneinander unab-hängigen Messungen im E. coli Stamm HAK006 (PkdpFABC::lacZ ∆kdpD161-726) mit den Plasmiden pBD, pBD/1-395, pBD/∆TM1-4 bzw. pBD/∆Arg1. Von jedem KdpD-Derivat wurde sowohl der Wert vor der Zugabe von Procain (grüne Balken), 30 min nach Induktion (hellblau) und 60 min nach Induktion (dunkelblau) dargestellt. Das Diagramm A zeigt die β-Galaktosidase-Aktivitäten unter kdpFABC-induzierenden und B unter nicht-kdpFABC-induzierenden Bedingungen.
Kontrolle 30 min 60 min
A) B)

3. Ergebnisse
68
Es konnte gezeigt werden, dass die Zugabe von 20 mM Procain die kdpFABC-
Expression der semi-konstitutiven Mutanten KdpD/1-395 und KdpD/∆TM1-4 signi-
fikant beeinflusst (Abb. 3.11). Unter kdpFABC-induzierenden Bedingungen stieg die
kdpFABC-Expression dieser KdpD-Derivate durch die Zugabe von Procain an (Abb.
3.11A). Die K+ insensitive Mutante KdpD/∆Arg1 hingegen induzierte auch durch die
Zugabe von Procain keine signifikante kdpFABC-Expression, da die Werte der β-
Galaktosidase-Aktivitäten um 5 Miller units lagen, unabhängig ob kdpFABC-indu-
zierende oder nicht-kdpFABC-induzierende Bedingungen vorlagen. Die Expression
des Wildtyp-KdpD wurde unabhängig ob kdpFABC-induzierende oder nicht-
kdpFABC-induzierende Bedingungen vorlagen ebenfalls durch die Einlagerung von
Procain in die Membran nicht signifkant beeinträchtigt. Des Weiteren führte Procain
dazu, dass die bereits vorhandene kdpFABC-Expression der semi-konstitutiven
Mutanten KdpD/1-395 und KdpD/∆TM1-4 in Minimalmedium mit 115 mM K+ ebenfalls
anstieg (Abb. 3.11B). Somit wurde für KdpD-Proteine mit einem semi-konstitutiven
Phänotyp eine Steigerung der Aktivität von KdpD in Abhängigkeit der Anwesenheit
von Procain in der Membran beobachtet.
3.9. Suche nach Kontaktstellen von KdpD mit Phospho lipiden der Zytoplasmamembran
Für die Untersuchung der Interaktion von Phospholipiden mit der Sensorkinase KdpD
wurde eine Co-Reinigung durchgeführt. Dazu wurden Zellen des Stammes
TKR2000/pKJ2-6His in Vollmedium angezogen, invertierte Membranvesikel
präpariert und KdpD-6His wie unter 2.5.6 beschrieben gereinigt. Als Detergenz für
die Solubilisierung des membrangebundenen Proteins wurde LDAO verwendet.
Anschließend wurden die Phospholipide mittels einer Chloroform-Methanol-
Extraktion (wie unter 2.5.11 beschrieben) vom solubilisierten Protein entfernt. Das
Extrakt wurde auf eine DC-Platte aufgetragen, um die Phospholipide mit einem
speziellen Laufmittel aufzutrennen und schließlich mit Molybdänblau anzufärben. Um
die Phospholipide identifizieren zu können, wurde als Kontrolle ein synthetisches
Gemisch aus PE, PG und CL ebenfalls auf die DC-Platte aufgetragen.
Wie in der Abbildung 3.12 zu erkennen ist, reicht die eingesetzte Menge von
gereinigtem KdpD nicht aus, um die prozentuale Verteilung der Phospholipide genau
bestimmen zu können. Es wurde eine ähnliche Verteilung der Phospholipide im
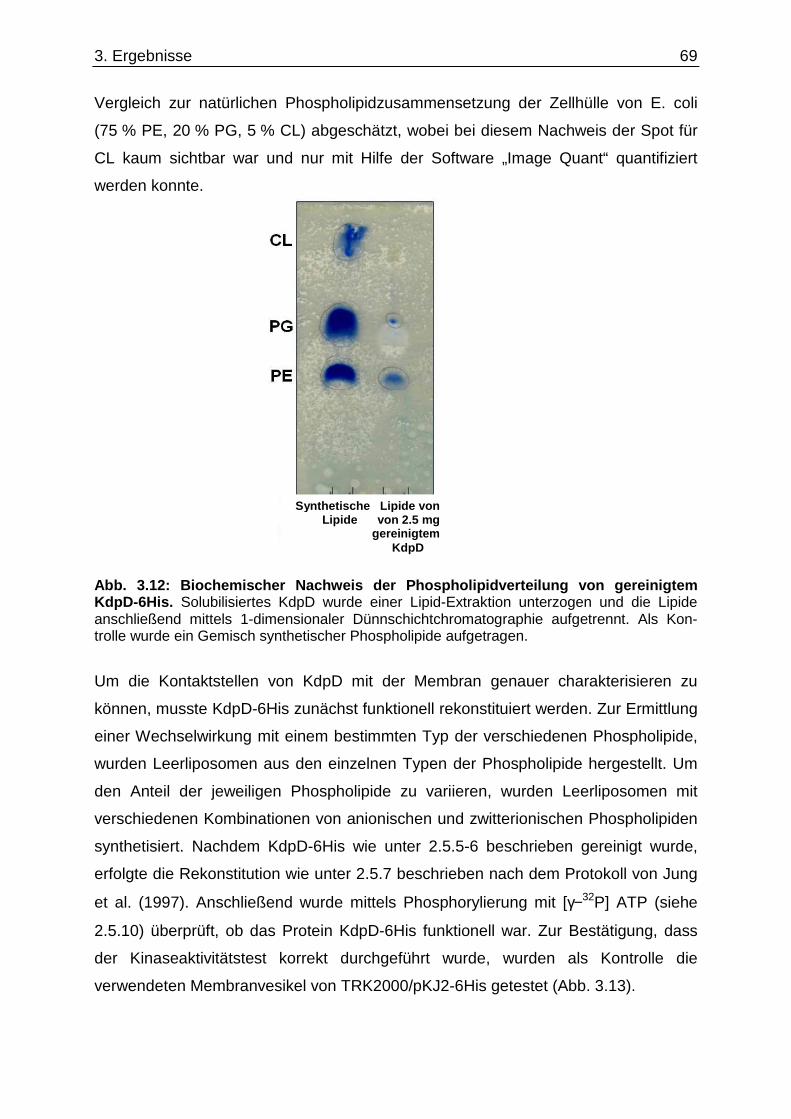
3. Ergebnisse
69
Vergleich zur natürlichen Phospholipidzusammensetzung der Zellhülle von E. coli
(75 % PE, 20 % PG, 5 % CL) abgeschätzt, wobei bei diesem Nachweis der Spot für
CL kaum sichtbar war und nur mit Hilfe der Software „Image Quant“ quantifiziert
werden konnte.
Abb. 3.12: Biochemischer Nachweis der Phospholipidv erteilung von gereinigtem KdpD-6His. Solubilisiertes KdpD wurde einer Lipid-Extraktion unterzogen und die Lipide anschließend mittels 1-dimensionaler Dünnschichtchromatographie aufgetrennt. Als Kon-trolle wurde ein Gemisch synthetischer Phospholipide aufgetragen.
Um die Kontaktstellen von KdpD mit der Membran genauer charakterisieren zu
können, musste KdpD-6His zunächst funktionell rekonstituiert werden. Zur Ermittlung
einer Wechselwirkung mit einem bestimmten Typ der verschiedenen Phospholipide,
wurden Leerliposomen aus den einzelnen Typen der Phospholipide hergestellt. Um
den Anteil der jeweiligen Phospholipide zu variieren, wurden Leerliposomen mit
verschiedenen Kombinationen von anionischen und zwitterionischen Phospholipiden
synthetisiert. Nachdem KdpD-6His wie unter 2.5.5-6 beschrieben gereinigt wurde,
erfolgte die Rekonstitution wie unter 2.5.7 beschrieben nach dem Protokoll von Jung
et al. (1997). Anschließend wurde mittels Phosphorylierung mit [γ−32P] ATP (siehe
2.5.10) überprüft, ob das Protein KdpD-6His funktionell war. Zur Bestätigung, dass
der Kinaseaktivitätstest korrekt durchgeführt wurde, wurden als Kontrolle die
verwendeten Membranvesikel von TRK2000/pKJ2-6His getestet (Abb. 3.13).
Synthetische Lipid e von Lipide von 2.5 mg gereinigtem KdpD

3. Ergebnisse
70
Abb. 3.13: Autoradiogramm der Kinaseaktivitäten. Als Kontrolle wurden Membranvesikel von TRK2000/pKJ2-6His und als Proteoliposomen mit rekonstituiertem KdpD-6His nach der Methode von Jung et al. (1997) exemplarisch Liposomen aus 100% synthetischem CL dargestellt.
Es konnte allerdings nur bei der Kontrolle eine Phosphorylierung von KdpD detektiert
werden. Nach der Rekonstitution von KdpD-His6 konnte in allen Proteoliposomen
unabhängig von der Lipidzusammensetzung der Liposomen keine Kinaseaktivität
gemessen werden (Abb. 3.13). In Kapitel 3.7.1 konnte bereits gezeigt werden, dass
KdpD-6His in seiner natürlichen Membranumgebung aktiv war. Da unklar war, ob die
nicht vorhandene Kinaseaktivität der rekonstituierten Sensorkinase KdpD auf die
verwendeten Liposomen oder die Rekonstitutionsmethode zurückzuführen war,
wurden zunächst die hergestellten Leerliposomen überprüft. Die richtige Formation
der Liposomen konnte mittels DiSC3-Fluorimeter-Messung bestätigt werden (Daten
nicht gezeigt). Für weitere Versuche wurden dann Liposomen verwendet, die aus
E. coli : EggPC im Verhältnis 3:1 bestehen. Diese Liposomen wurden ebenfalls auf
ihre richtige Formation überprüft. Des Weiteren war es möglich, in diese ein
gewünschtes Zielprotein so zu rekonstituieren, dass es funktionell war (Hänelt, pers.
Mitteilung). Damit sollte ausgeschlossen werden, dass die synthetischen Liposomen
einen möglicherweise negativen Einfluss auf die Funktionalität von KdpD haben. Die
Rekonstitution in diese Liposomen erfolgte zunächst wieder nach dem Protokoll von
Jung et al. (1997). Auch mit diesen Liposomen konnte für KdpD-6His keine Aktivität
gemessen werden (Daten nicht gezeigt). Daraufhin wurden weitere Bedingungen für
die Rekonstitution getestet, um ein funktionelles Protein zu erhalten. Es wurde

3. Ergebnisse
71
A) B)
zunächst ein Protokoll für die Rekonstitution von KdpD-6His angewandt, das für viele
Membranproteine benutzt wird (Knol et al., 1998). Zur Abschätzung der Einbaurate
von KdpD-6His in die Liposomen wurde ein Coomassie-Gel angefertigt. Auf dem mit
Coomassie gefärbten SDS-Gel konnte im Vergleich zum gereinigten KdpD-Protein
nur eine Einbaurate von 0 – 10 % des KdpD-6His in die E. coli : EggPC Liposomen
(3:1) ermittelt werden (Abb. 3.14 B), während nach der Standardmethode für KdpD
von Jung et al. (1997) circa 50 % KdpD-6His in die Liposomen eingebaut zu sein
schien (Abb. 3.14 A).
Abb. 3.14: Coomassie-Gele zur Bestimmung der Einbau rate bei der Rekonstitution von KdpD-His6. Aufgetragen wurde dieselbe Proteinmenge sowohl von gereinigtem als auch von rekonstituiertem Protein nach der Methode von A: Jung et al. (1997) und B: Knol et al. (1998). Die Ergebnisse repräsentieren exemplarisch die Einbauraten von voneinander unabhängigen Versuchsansätzen mit verschiedenen Liposomen.
Das in die E. coli : EggPC Liposomen (3:1) rekonstituierte KdpD-6His wurde einem
Kinaseaktivitätstest unterzogen. Es konnte aber kein phosphoryliertes KdpD detek-
tiert werden (Daten nicht gezeigt). Mit dieser Methode war parallel ein anderes Mem-
branprotein rekonstituiert worden, welches eine bessere Einbaurate (80 %) und vor
allem seine Funktionalität nach der Rekonstitution aufzeigte (Hänelt, pers. Mit-
teilung). Es ist möglich, dass KdpD-6His nicht korrekt in die Membran der Liposomen
eingebaut wurde oder dass das Protein bereits nach der Reinigung nicht mehr aktiv
war. Dies würde den völligen Kinaseaktivitätsverlust erklären. Da auch die Wieder-
holung dieser Rekonstitutionsmethode kein funktionelles KdpD Protein nach der
Rekonstitution brachte, wurde eine alternative Rekonstitutionsmethode durch Ver-
dünnen des Detergenz unter die CMC direkt nach der Reinigung von KdpD-6His

3. Ergebnisse
72
nach dem Protokoll von Heuberger et al. (2001) durchgeführt. Das Coomassie-Gel
und der Immunoblot mit einem Antikörper gegen KdpD zeigten jedoch, dass die
Effizienz der Reinigung von KdpD-6His schlechter als gewöhnlich (ohne Detergenz-
wechsel von DDM zu OG) war (Daten nicht gezeigt). Die Einbaurate von KdpD in die
Liposomen lag mit dieser Methode bei 0 - 10 % und es konnte ebenfalls keine
Phosphorylierung von KdpD detektiert werden (Daten nicht gezeigt).
Für die beiden neu getesteten Rekonstitutionsmethoden wurde auch überprüft, ob
ein Verlust des Proteins beim Extrudieren für die Herstellung uniformer Proteolipo-
somen durch eine unspezifische Anlagerung des Proteins an dem Polycarbonat-
Filter stattfand, da folglich das Protein möglicherweise falsch in oder nur an die
Membran der Liposomen assoziiert war. Der Kinaseaktivitätstest zeigte jedoch, dass
auch ohne das Extrudieren keine Phosphorylierung mit den oben beschriebenen
Rekonstitutionsmethoden ermittelt werden konnte (Daten nicht gezeigt).
3.10. Novobiocin-Resistenz in Abhängigkeit von KdpD Es konnte gezeigt werden, dass ein kdp- Stamm im Gegensatz zu einem kdp+
Stamm eine erhöhte Resistenz gegenüber dem Antibiotikum Novobiocin hat (Zhou et
al., 2003). Des Weiteren wurde das cls-Gen früher auch als nov-Gen bezeichnet, da
ein ∆cls Stamm sensitiver als ein cls+ Stamm gegenüber Novobiocin ist (Tropp et al.,
1995). Aufgrund dieser beiden Beobachtungen sollte der Zusammenhang genauer
untersucht werden. Dazu sollte die Resistenz gegenüber Novobiocin eines kdp+
Stammes mit der eines kdp- Stammes unter kdpFABC-induzierenden und nicht-
kdpFABC-induzierenden Bedingungen verglichen werden. Der Antibiotikatest von
Zhou et al. (2003) erfolgte nur in Vollmedium, also unter nicht-kdpFABC-indu-
zierenden Bedingungen. Das bedeutet für den in diesem Versuch verwendeten kdp+
Stamm, dass kein KdpFABC-Komplex synthetisiert wird und KdpD / KdpE nur in
geringen Mengen in der Zelle vorkommt, während im kdp- Stamm sowohl kein
KdpFABC als auch kein KdpD / KdpE gebildet wird. Da bei dem in der Literatur
beschriebenen Versuch allerdings ein Unterschied in der Novobiocin-Resistenz
zwischen den beiden Stämmen beschrieben wurde, müsste die Resistenz gegenüber
Novobiocin mit der membrangebundenen Sensorkinase KdpD zusammenhängen.
Folglich wurden die „Kalium-Stämme“ TK2240 und TKV2208 getestet, da beide
Stämme aufgrund der Abwesenheit der niedrig affinen K+-Aufnahmesysteme Trk und
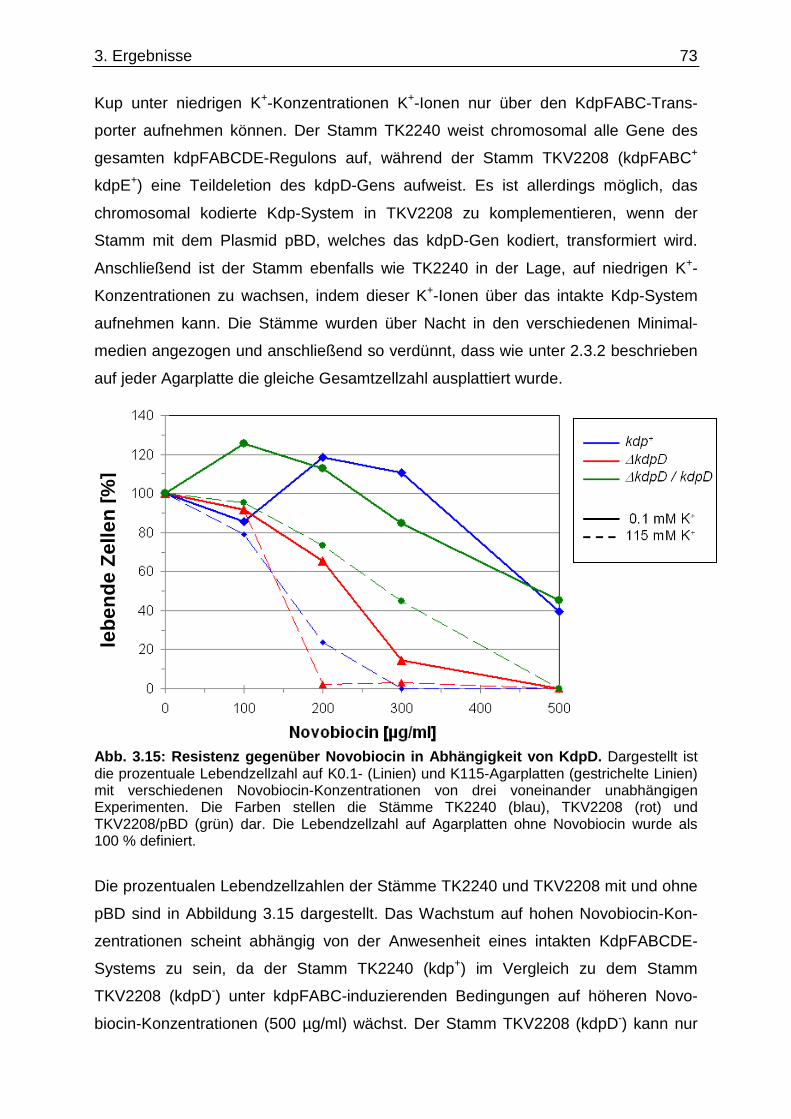
3. Ergebnisse
73
Kup unter niedrigen K+-Konzentrationen K+-Ionen nur über den KdpFABC-Trans-
porter aufnehmen können. Der Stamm TK2240 weist chromosomal alle Gene des
gesamten kdpFABCDE-Regulons auf, während der Stamm TKV2208 (kdpFABC+
kdpE+) eine Teildeletion des kdpD-Gens aufweist. Es ist allerdings möglich, das
chromosomal kodierte Kdp-System in TKV2208 zu komplementieren, wenn der
Stamm mit dem Plasmid pBD, welches das kdpD-Gen kodiert, transformiert wird.
Anschließend ist der Stamm ebenfalls wie TK2240 in der Lage, auf niedrigen K+-
Konzentrationen zu wachsen, indem dieser K+-Ionen über das intakte Kdp-System
aufnehmen kann. Die Stämme wurden über Nacht in den verschiedenen Minimal-
medien angezogen und anschließend so verdünnt, dass wie unter 2.3.2 beschrieben
auf jeder Agarplatte die gleiche Gesamtzellzahl ausplattiert wurde.
Abb. 3.15: Resistenz gegenüber Novobiocin in Abhäng igkeit von KdpD. Dargestellt ist die prozentuale Lebendzellzahl auf K0.1- (Linien) und K115-Agarplatten (gestrichelte Linien) mit verschiedenen Novobiocin-Konzentrationen von drei voneinander unabhängigen Experimenten. Die Farben stellen die Stämme TK2240 (blau), TKV2208 (rot) und TKV2208/pBD (grün) dar. Die Lebendzellzahl auf Agarplatten ohne Novobiocin wurde als 100 % definiert.
Die prozentualen Lebendzellzahlen der Stämme TK2240 und TKV2208 mit und ohne
pBD sind in Abbildung 3.15 dargestellt. Das Wachstum auf hohen Novobiocin-Kon-
zentrationen scheint abhängig von der Anwesenheit eines intakten KdpFABCDE-
Systems zu sein, da der Stamm TK2240 (kdp+) im Vergleich zu dem Stamm
TKV2208 (kdpD-) unter kdpFABC-induzierenden Bedingungen auf höheren Novo-
biocin-Konzentrationen (500 µg/ml) wächst. Der Stamm TKV2208 (kdpD-) kann nur
lebe
nde
Zel
len
[%]
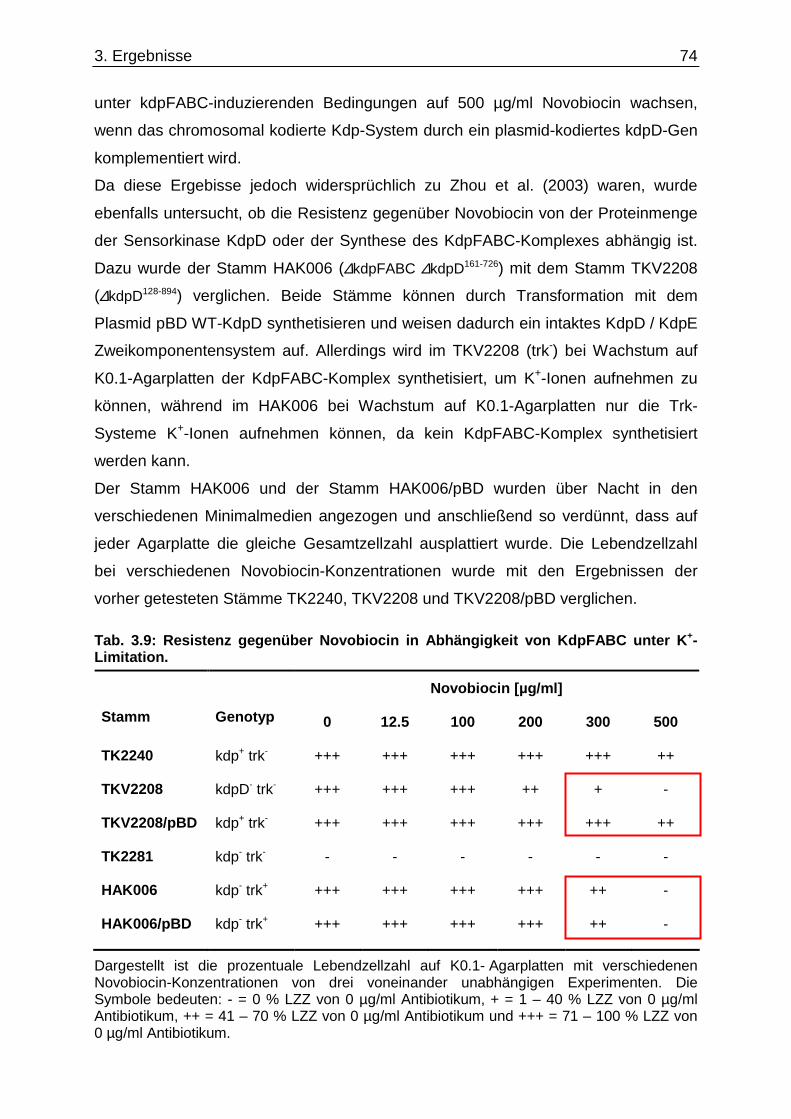
3. Ergebnisse
74
unter kdpFABC-induzierenden Bedingungen auf 500 µg/ml Novobiocin wachsen,
wenn das chromosomal kodierte Kdp-System durch ein plasmid-kodiertes kdpD-Gen
komplementiert wird.
Da diese Ergebisse jedoch widersprüchlich zu Zhou et al. (2003) waren, wurde
ebenfalls untersucht, ob die Resistenz gegenüber Novobiocin von der Proteinmenge
der Sensorkinase KdpD oder der Synthese des KdpFABC-Komplexes abhängig ist.
Dazu wurde der Stamm HAK006 (∆kdpFABC ∆kdpD161-726) mit dem Stamm TKV2208
(∆kdpD128-894) verglichen. Beide Stämme können durch Transformation mit dem
Plasmid pBD WT-KdpD synthetisieren und weisen dadurch ein intaktes KdpD / KdpE
Zweikomponentensystem auf. Allerdings wird im TKV2208 (trk-) bei Wachstum auf
K0.1-Agarplatten der KdpFABC-Komplex synthetisiert, um K+-Ionen aufnehmen zu
können, während im HAK006 bei Wachstum auf K0.1-Agarplatten nur die Trk-
Systeme K+-Ionen aufnehmen können, da kein KdpFABC-Komplex synthetisiert
werden kann.
Der Stamm HAK006 und der Stamm HAK006/pBD wurden über Nacht in den
verschiedenen Minimalmedien angezogen und anschließend so verdünnt, dass auf
jeder Agarplatte die gleiche Gesamtzellzahl ausplattiert wurde. Die Lebendzellzahl
bei verschiedenen Novobiocin-Konzentrationen wurde mit den Ergebnissen der
vorher getesteten Stämme TK2240, TKV2208 und TKV2208/pBD verglichen.
Tab. 3.9: Resistenz gegenüber Novobiocin in Abhängi gkeit von KdpFABC unter K +-Limitation.
Dargestellt ist die prozentuale Lebendzellzahl auf K0.1- Agarplatten mit verschiedenen Novobiocin-Konzentrationen von drei voneinander unabhängigen Experimenten. Die Symbole bedeuten: - = 0 % LZZ von 0 µg/ml Antibiotikum, + = 1 – 40 % LZZ von 0 µg/ml Antibiotikum, ++ = 41 – 70 % LZZ von 0 µg/ml Antibiotikum und +++ = 71 – 100 % LZZ von 0 µg/ml Antibiotikum.
Novobiocin [µg/ml]
Stamm
Genotyp 0 12.5 100 200 300 500
TK2240 kdp+ trk- +++ +++ +++ +++ +++ ++
TKV2208 kdpD- trk- +++ +++ +++ ++ + -
TKV2208/pBD kdp+ trk- +++ +++ +++ +++ +++ ++
TK2281 kdp- trk- - - - - - -
HAK006 kdp- trk+ +++ +++ +++ +++ ++ -
HAK006/pBD kdp- trk+ +++ +++ +++ +++ ++ -

3. Ergebnisse
75
Der Stamm HAK006 zeigt nach Transformation mit pBD keine Erhöhung der
Resistenz gegenüber Novobiocin (Tabelle 3.9). Dieses ist gegensätzlich zum
TKV2208/pBD, der eine Erhöhung der Novobiocin-Resistenz unter kdpFABC-indu-
zierenden Bedingungen aufweist. Folglich scheint ein intaktes KdpFABCDE-System
bzw. speziell die Synthese von KdpFABC eine Rolle bei der Novobiocin-Resistenz zu
spielen. Diese Beobachtung widerspricht der von Zhou et al. (2003) aufgestellten
Behauptung, dass ein kdp- Stamm im Gegensatz zu einem kdp+ Stamm eine erhöhte
Resistenz gegenüber Novobiocin aufweist.
Die Beobachtungen der Novobiocin-Resistenz in Abhängigkeit des Kdp-Systems
scheinen spezifisch für Novobiocin zu sein, da bei Erythromycin, einem anderen Anti-
biotikum, das ebenfalls durch das „multi-drug“ Effluxsystem AcrAB aus der Zelle
geschleust wird, kein Unterschied bei dem Wachstum auf verschiedenen Erythro-
mycin-Konzentrationen zwischen den Stämmen TK2240 und TKV2208 ermittelt
werden konnte (Daten nicht gezeigt). Des Weiteren konnte ein Wachstum der
Stämme nur bei Konzentrationen von unter 100 µg/ml Erythromycin beobachtet
werden.

4. Diskussion
76
4. Diskussion
4.1. Einfluss der Lipidzusammensetzung auf das Kdp- System
Es ist bekannt, dass die Lipidzusammensetzung der Zytoplasmamembran von
Bakterien mit der Osmolalität im Medium und mit der Wachstumsphase variiert. In
E. coli steigt der CL-Gehalt auf Kosten von PE in der exponentiellen Wachstums-
phase in Medium mit hoher Osmolalität unabhängig vom Osmolyt um das 2-3 fache
an (Tsatskis et al., 2005). In der stationären Wachstumsphase hingegen steigt der
CL-Gehalt auf Kosten von PG um das 2-3 fache an (Cronan, 1968; Randle et al.,
1969). Hierbei kommt es zu keinem Nettoanstieg des anionischen Phospholipid-
gehaltes. Allerdings ist der Abbau von CL langsamer als der von PG (Kanemasa et
al., 1967), was zu der Vermutung führt, dass CL in wachsenden E. coli Zellen stabiler
ist als PG. Dies bezüglich wurde von Cronan (1968) vorgeschlagen, dass eine solche
Änderung in dem Anteil der anionischen Phospholipide in der stationären
Wachstumsphase möglicherweise ein Schutzmechanismus ist, um die anionischen
Phospholipide gegen Abbau zu schützen.
In dieser Arbeit wurden die Änderungen der Phospholipidzusammensetzung der
Membran unter K+-limitierenden Bedingungen untersucht, um den Einfluss auf die
kdpFABC-Expression in E. coli zu analysieren. Es konnte zum ersten Mal gezeigt
werden, dass sich die Zusammensetzung der Kopfgruppen der Phospholipide von
E. coli K12 ändert, indem der Anteil an CL auf Kosten von PE nicht nur während der
exponentiellen Wachstumsphase nach kontinuierlichem Wachstum unter K+-
Limitation, sondern auch direkt nach einem K+-„downshift“ ansteigt. Bei einem K+-
„downshift“ ist der CL-Anstieg nur auf neusynthetisierte CL-Moleküle zurückzuführen,
während bei kontinulierlichem Wachstum unter K+-Limitation der höhere CL-Gehalt
aus bereits vorhandenen und neusynthetisierten CL-Moleküle resultiert. Für die
Messungen der Phospholipidzusammensetzung nach einem K+-„downshift“ ist dabei
erwähnenswert, dass die Translokation der Phospholipide von der inneren zur
äußeren Membran von E. coli innerhalb von Sekunden stattfindet (Donohue-Rolfe et
al., 1980). Des Weiteren unterscheidet sich die Verteilung der Phospholipide in den
beiden Bilayern der Zellhülle. Es konnte für E. coli und Salmonella typhimurium
beobachtet werden, dass die innere Schicht der äußeren Membran mehr PE und
weniger PG und CL als die Zytoplasmamembran beinhaltet (Osborn et al., 1972;

4. Diskussion
77
Lugtenberg & Peters, 1976). Folglich sollte der ständig steigende CL-Anstieg nach
einem K+-„downshift“ zu einer Änderung des CL-Gehalts in der Zytoplasmamembran
führen, da das neusynthetisierte CL zunächst vollständig in die Zytoplasmamembran
eingebaut wird und davon nur ein kleiner Teil in die innere Schicht der äußeren
Membran verlagert wird.
Interessanterweise konnte der in E. coli K12 beobachtet CL-Anstieg unter K+-
Limitation während der exponentiellen Wachstumsphase jedoch nicht in den Kalium-
oder „Lipid-Stämmen“ gemessen werden. Das unterschiedliche CL-Niveau und der
bis zur stationären Wachstumsphase konstante CL-Gehalt in den „Kalium-Stämmen“
konnten auf den Stammhintergrund bzw. auf andere Mutationen in den „Kalium-
Stämmen“ und nicht auf die Abwesenheit des Kdp-Systems oder der niedrig affinen
K+-Aufnahmesysteme Trk und Kup zurückgeführt werden. In der vorliegenden Arbeit
wurde ebenfalls gezeigt, dass Stämme aus denen die „Kalium-Stämme“ konstruiert
wurden, das gleiche CL-Niveau und einen bis zur stationären Wachstumsphase
konstanten CL-Gehalt aufwiesen. In den „Lipid-Stämmen“ konnte kein CL-Anstieg
unter K+-Limitation während der exponentiellen Wachstumsphase beobachtet
werden. Jedoch zeigte der Stamm W3899, der für die Konstruktion der „Lipid-
Stämme“ WC3899 und AD93 verwendet wurde, und keine Mutation in den Genen
aufweist, welche für Enzyme kodieren, die in die Biosynthese der Phospholipide
involviert sind, einen CL-Anstieg unter K+-Limitation im Gegensatz zu seinen
Derivaten (siehe Tabelle 3.4). Diese Beobachtung lässt vermuten, dass die genetisch
bedingte Veränderung der ursprünglichen Kopfgruppenzusammensetzung aufgrund
der Mutationen in den entsprechenden Genen der Stämme WC3899 und AD93 den
CL-Anstieg unter K+-Limitation beeinflusst. Des Weiteren konnte der CL-Anstieg nur
beobachtet werden, wenn das pssA-Gen auf dem Chromosom wie im ursprünglichen
Stamm W3899 und nicht wie bei AD93 / pDD72 auf einem Plasmid kodiert ist (siehe
Tabelle 3.4). Diese Beobachtung führt zu der Hypothese, dass das Expressions-
niveau des pssA-Gens möglicherweise einen Effekt auf die Synthese von CL unter
K+-Limitation hat.
Für einige Sensorkinase / Antwortregulator Systeme konnte bereits beobachtet
werden, dass die Zusammensetzung der Phospholipide einen Einfluss auf die
Aktivierung dieser Systeme hat. Der Cpx-Zweikomponenten-Signaltransduktionsweg
zum Beispiel wurde in E. coli Stämmen aktiviert, die kein PE mehr in der Membran

4. Diskussion
78
aufwiesen (Mileykovskaya & Dowhan, 1997). Im Falle des KdpD / KdpE Systems
konnte mit der vorliegenden Arbeit gezeigt werden, dass die Kinaseaktivität der
Sensorkinase KdpD in seiner natürlichen Membranumgebung durch die Zugabe von
CL in vitro stimuliert wurde, während die gleiche Menge PE / PC zu einer niedrigeren
Aktivität führte. Des Weiteren lassen die Q-RT-PCR-Daten dieser Arbeit darauf
schließen, dass in vivo eher der gesamte Anteil anionischer Phospholipide als die
Höhe des CL-Gehalts einen Einfluss auf die kdpFABC-Expression hat. Der Stamm
WC3899 (∆cls) zum Beispiel hat ein vergleichbares Induktionsverhältnis für die
kdpFABC-Expression wie der Wildtypstamm K12, obwohl sich nur die Zusammen-
setzung der anionischen Phospholipide aufgrund des niedrigeren CL-Gehaltes unter-
scheidet, nicht jedoch die Höhe des anionischen Anteils. Eine Korrelation zwischen
dem Induktionsverhältnis für die kdpFABC-Expression und dem totalen Anteil an
anionischen Phospholipiden (siehe Tabelle 3.7) ist durch die Beobachtung unter-
stützt, dass Stämme mit einem höheren anionischen Phospholipidanteil (18-25 %,
wie in den Stämmen K12, WC3899 und HDL1001 + IPTG) ein höheres Induk-
tionsverhältnis für die kdpFABC-Expression aufweisen, als Stämme mit einem
niedrigeren anionischen Phospholipidanteil (0-4 %, wie in den Stämmen UE54 und
HDL1001 - IPTG). Allerdings ist die Nettoladung der Oberfläche der Membran, wel-
che durch die Phospholipidzusammensetzung beeinflusst wird, nicht ausschließlich
für die kdpFABC-Expression verantwortlich. Dieses ist auf die Beobachtung zurück-
zuführen, dass in dem E. coli Stamm UE54 immer noch kdpFABC exprimiert wird,
obwohl die Membran keine anionischen Phospholipide PG und CL aufweist. Die
restliche Aktivität könnte auf die Akkumulation der Vorstufenmoleküle Phosphatidyl-
säure (PA) und CDP-Diacylglycerol in diesem Stamm zurückzuführen sein, welche
der Membranoberfläche einige negative Ladungen bereitstellen (Tsatskis et al.,
2005). Die Beobachtung, dass die höchste kdpFABC-Expression in dem Stamm, be-
stehend aus einer Membran mit ausschließlich anionischen Phospholipiden (AD93),
gemessen wurde, unterstützt die Theorie, dass anionische Phospholipide einen
stimulierenden Effekt auf die kdpFABC-Expression haben. Auch die Proliferation der
Zytoplasmamembran und der damit verbundene CL-Anstieg scheinen einen Einfluss
auf die Aktivität der Sensorkinase KdpD zu haben. Dies zeigte sich dadurch, dass die
bei der Überproduktion der b-Untereinheit der ATP-Synthase verursachte Ausstül-
pung der Membran ebenfalls in einer Änderung der kdpFABC-Expression resultierte.
Die beschriebene Beobachtung ist vermutlich auf das angestiegene Verhältnis von

4. Diskussion
79
anionischen zu zwitterionischen Phospholipiden zurückzuführen, welche sich mit den
vorherigen Hypothesen deckt.
Auch die ermittelte Kinaseaktivität der Sensorkinase KdpD bei Fusion von Membran-
vesikeln mit synthetisch hergestellten Liposomen zeigte, dass die Autophospho-
rylierungsrate von KdpD nicht nur durch die Anwesenheit von anionischen Phospho-
lipiden, sondern auch durch den prozentualen Anteil von negativ geladenen
Phosphoipiden beeinflusst wird. Da bei dem Versuch die identische Menge von
anionischen oder zwitterionschen Liposomen bzw. Liposomen aus „E. coli total
extract“ zugegeben wurde, kann diese Beobachtung auf den spezifischen Effekt von
negativ geladenen Phospholipiden zurückgeführt werden. Der Einfluss des prozen-
tualen Anteils von negativ geladenen Phospholipiden auf die Kinaseaktivität von
KdpD (siehe 3.7.1) deckt sich mit der kdpFABC-Expression, die in Stämmen mit
einer veränderten nativen Phospholipidzusammensetzung gemessenen wurde. Mit
den beiden Versuchsansätzen konnten folglich die Daten von Stallkamp et al. (1999)
untermauert und detaillierter beschrieben werden, da der Anteil von anionischen
Phospholipiden variiert wurde. Bei den Fusionen der Membranvesikel könnte neben
dem Einfluss der negativen Liposomen auch ein krümmungsbedingter Effekt eine
Rolle spielen. Die Phosphorylierung sowohl von Wildtyp-KdpD als auch vom Derivat
KdpD/E509K nahm ab, je höher der Anteil der zwitterionischen bzw. E. coli Lipo-
somen im Verhältnis zu den Membranvesikeln war. Da die fusionierten Vesikel
anschließend nicht extrudiert wurden, könnte es zu Vesikeln mit unterschiedlichen
Größen gekommen sein. Eine Theorie wäre, dass je höher der Anteil an zuge-
gebenen Liposomen ist, desto größer wird der Durchmesser der fusionierten Vesikel.
Folglich wären die Vesikel im Verhältnis von Membranvesikeln zu Liposomen mit
1 : 0 am kleinsten, gefolgt von denen im Verhältnis von 1 : 1, dann 1 : 2 bzw. 2 : 1
und am größten wären die Vesikel im Fusionsverhältnis von 1 : 3. Das würde
bedeuten, dass die Größe bzw. die Krümmung der Membran der Vesikel einen
Einfluss auf die Autophosphorylierung von KdpD haben könnte. Je kleiner die Vesikel
sind, desto größer ist die Krümmung der Membran. In den kleineren (nativen)
Membranvesikeln, extrudiert auf 400nm Durchmesser, konnte eine höhere Aktivität
gemessen werden, wenn sie nicht mit Liposomen fusioniert wurden. Ramamurthi et
al. (2009) konnten für B. subtilis sogar zeigen, dass die Verteilung von Proteinen in
der Zelle aufgrund der Krümmung der Membran zu erfolgen scheint.

4. Diskussion
80
Die geringere Phosphorylierungsrate von KdpD/E509K bei diesem Versuch ist
vermutlich auf den niedrigeren KdpD-Anteil in den Membranvesikeln zurückzuführen,
da ein anderer Vektor für die Überexpression verwendet wurde. Bei pBD5-9/E509K
handelt es sich um ein Plasmid mit einem Arabinose-induzierbaren Promotor, der
allerdings mit Glucose reprimiert wurde, während es sich bei pKJ2-6His um ein
Plasmid mit einem starken tac-Promotor handelt. Folglich ist das Expressionsniveau
von WT-KdpD deutlich höher als bei dem KdpD-Derivat, was die Ursache für die
unterschiedliche Höhe der Aktivitäten beider Proteine zu sein scheint.
In diesem Zusammenhang ist zu erwähnen, dass es sich bei den eingesetzten
Membranvesikeln um invertierte Membranvesikel handelt. Das bedeutet, dass die
KdpD-Proteine in umgekehrter Orientierung in den Vesikeln vorlagen, damit die
eigentlich zytoplasmatischen Domänen mit dem konserviertem Histidinrest an den
Außenseite der Proteoliposomen lagen, um für das radioaktiv-markierte ATP
zugänglich zu sein. Folglich handelt es sich bei der Krümmung in allen gemessenen
Proben nicht um den nativen Zustand in der Zelle, sondern um die entgegengesetzte
Richtung. Diese Orientierung spielt auch bei der Rekonstitution von KdpD-6His in
Liposomen eine Rolle, da die unterschiedliche Krümmung in einer schlechteren
Einbaurate resultieren könnte. Möglicherweise konnte das Protein nicht funktionell in
die Liposomen rekonstituiert werden, da das Protein bereits in der Zelle durch die
Überproduktion teilweise an der Zytoplasmamembran aggregiert vorliegen könnte.
Dafür spräche auch die Beobachtung, dass invertierte Membranvesikel nach dem
Extrudieren deutlich niedrigere Phosphorylierungsraten aufwiesen. Das aggregierte
Protein könnte demzufolge bei der Formierung gleich großer Vesikel an dem Poly-
carbonat-Filter unspezifisch binden. Bislang brachten weitere Versuchsansätze,
KdpD-6His funktionell zu rekonstituieren, keine messbare Aktivität des Proteins
(Kipschull, pers. Mitteilung).
Bei der Einlagerung von membranverändernden Substanzen in die Zytoplasma-
membran konnte durch die Zugabe von Procain eine signifikante Änderung der
kdpFABC-Expression, induziert durch WT-KdpD / KdpE, gemessen werden, aller-
dings nur wenn kdpFABC-induzierende Bedingungen vorlagen und die Zytoplasma-
membran ausschließlich aus zwitterionischem PE bestand. Dieses Ergebnis schränkt
die Beobachtung von Jung et al. (2000) ein, die zeigen konnten, dass Procain keinen
Einfluss auf die Phosphorylierung von WT-KdpD in Zellen mit einer nativen Phospho-

4. Diskussion
81
lipidzusammensetzung hat. Demzufolge hat Procain nur eine Auswirkung auf die
Aktivität des Kdp-Systems, wenn anionische Phospholipide in der Zellhülle wie im
Stamm UE54 fehlen. Des Weiteren konnte für Derivate von KdpD mit einem
veränderten Phänotyp hinsichtlich der kdpFABC-Expression durch die Zugabe von
Procain eine Erhöhung der vorhandenen Aktivität ermittelt werden. Dieses deckt sich
mit der Beobachtung von Sugiura et al. (1994), die zeigen konnten, dass semi-
konstitutive Derivate eine erhöhte kdpFABC-Expression bei Zugabe von Procain
aufwiesen. Demzufolge scheinen solche interkalierenden Substanzen die Aktivität
oder den Einbau der Sensorkinase KdpD bzw. des Transportkomplexes KdpFABC in
die Zytoplasmamembran zu beeinflussen. Somit sprechen die mittels Procain
gewonnenen Ergebnisse ebenso wie die Kinaseaktivitäten der fusionierten Vesikel
für die Hypothese, dass KdpD die Änderung der Membranspannung wahrnimmt bzw.
die Aktivität von KdpD durch die Krümmung der Membran beeinflusst wird.
4.2. Modell für den Anstieg von Cardiolipin Der Effekt von zytoplasmatischem K+ auf die Aktivität der CL-Synthase wurde in
früheren Studien analysiert, in denen gezeigt werden konnte, dass K+-Phosphat als
ein Aktivator für die CL-Synthase fungiert (Hiraoka et al., 1991). Außerdem konnte in
der vorliegenden Arbeit experimentell gezeigt werden, dass unter K+-Limitation die
Menge an CL anstieg. Für diese Beobachtung in der exponentiellen Wachstums-
phase gibt es mindestens zwei mögliche Erklärungen: Einerseits könnte eine höhere
Aktivität der CL-Synthase und andererseits eine höhere Expression des cls-Gens
und damit eine größere Menge an CL-Synthase Molekülen die Ursache sein. Die
Ergebnisse des Stammes mit der cls-lacZ transkriptionalen Fusion passen zu der
zweiten Erklärung. Das deutet darauf hin, dass die CL-Synthese auf genetischer
Ebene reguliert wird, wodurch die Ergebnisse von Heber & Tropp (1991) bestätigt
werden. Unter K+-Limitation kommt es vermutlich durch einen bislang unbekannten
Faktor zu einer Erhöhung der cls-Expression. In Bezug auf den CL-Anstieg während
der stationären Wachstumsphase widerspricht die Vermutung der genetischen
Regulation der Beobachtung, dass der CL-Gehalt in dieser Wachstumsphase auf-
grund der höheren Enzymaktivität der CL-Synthase ansteigt (Hiraoka et al., 1993).
Des Weiteren konnte gezeigt werden, dass die CL-Synthase durch CL inhibiert
wurde, welches eines der beiden Produkte dieser katalysierten Reaktion ist (Ragolia
& Tropp, 1994). Das verdeutlicht, dass die Synthese von CL auch auf dem

4. Diskussion
82
enzymatischen Niveau reguliert wird und unterstützt das Modell einer Regulierung
auf beiden Ebenen (Abb. 4.1).
Abb. 4.1: Schema einer möglichen Regulation des CL- Niveaus in E. coli. OM: Äußere Membran, CM: Zytoplasmamembran, P: Promotor.
Diese mehrfache Regulierung könnte es der Zelle ermöglichen, ihren CL- und/oder
ihren anionischen Phospholipidgehalt genauer zu steuern und dabei negative
Einflüsse auf essentielle Prozesse und Funktionen in der Zelle zu vermeiden.
Romantsov et al. (2007) vermuten, dass der von Tsatskis et al. (2005) beobachtete
CL-Anstieg auf Kosten von PE unter hohen Osmolalitäten auf den Anstieg der PG-
Synthese und der PG-Konversion zu CL zurückzuführen ist, obwohl das detektierte
Niveau von PG unverändert war. Diese Hypothese könnte ebenfalls den in dieser
Arbeit gezeigten CL-Anstieg unter K+-Limitation erklären. Auch der ermittelte Anstieg
der cls-Expression unter K+-Limitation untermauert diese Hypothese, da folglich mehr
CL-Synthase synthetisiert wird um CL zu bilden und somit für den CL-Anstieg
verantwortlich zu sein scheint. Schließlich gibt es bislang keine experimentellen
Hinweise dafür, dass die CL-Synthase CL auch über einen PG-unabhängigen Weg
synthetisieren kann.
Bezugnehmend auf die cls-Expression ist es erwähnenswert, dass in einem Stamm
mit einer cls-Deletion der restliche Anteil von CL ansteigt, wenn die Zellen die
stationäre Wachstumsphase erreichen (Nishijima et al., 1988). Es wurde von
Nishijima und Mitarbeitern vermutet, dass CL in solchen Stämmen durch einen
alternativen Syntheseweg unter Gebrauch des pssA-Genproduktes hergestellt wird.

4. Diskussion
83
Jedoch waren Zellen mit einer pssA-Deletion in einem Stamm mit einer cls-Deletion
nicht lebensfähig (Nishijima et al., 1988). Bislang konnte kein Enzym identifiziert
werden, welches den restlichen Anteil von CL in einem Stamm mit einer cls-Deletion
synthetisiert. In E. coli gibt es zwei zum cls-Gen homologe Gene ybhO und ymdC,
diese konnten CL in vitro synthetisieren (Guo & Tropp, 2000). Romantsov et al.
(2007) stellten daher die Hypothese auf, dass YbhO für den CL-Anstieg in der
stationären Phase verantwortlich ist. Aufgrund der Ergebnisse der vorliegenden
Arbeit kann diese Vermutung jedoch widerlegt werden, da auch in dem ∆cls ∆ybhO
Stamm DG10 der CL-Gehalt in der stationären Wachstumsphase von etwa 0.5 % auf
5 % des totalen Phospholipidgehaltes ansteigt. Folglich ist YbhO nicht bzw. nicht
alleine für den CL-Anstieg in der stationären Phase verantwortlich, da auch in dem
∆ybhO Stamm diese Veränderung in der Kopfgruppenzusammensetzung auftrat. Bei
dem Stamm DG10 fällt allerdings auf, dass in der stationären Phase der CL-Gehalt
nicht durch die sonst übliche PG-, sondern durch eine PE-Abnahme erfolgte. Dieses
liegt vermutlich an der cls-Deletion, da PG nicht mehr wie sonst in CL umgewandelt
werden konnte und somit während des Wachstums akkumulierte. Dieses spricht für
die aufgestellte Theorie, dass es sich bei der CL-Synthese möglicherweise um eine
Nebenreaktion des pssA-Gens handelt, wodurch auch der PE-Abfall bei zunehmen-
dem CL-Anteil zu erklären wäre.
Abb. 4.2: Alternative Wege für die CL-Synthese von E. coli. Dabei ist der vom Gen-produkt des pssA-Gens abhängige Weg für die CL-Synthese grün und der vom Genprodukt des pgsA-Gens abhängige Weg rot eingezeichnet.
Eine andere Theorie ist, dass die CL-Synthese durch YbhO ebenfalls wie die durch
die CL-Synthase von der Anwesenheit des Produktes des pgsA-Gens abhängig sein
könnte (Abb. 4.2). Das erklärt sich daraus, dass der Stamm UE54, der keine

4. Diskussion
84
anionischen Lipide aufgrund seiner pgsA-Deletion besitzt, auch in der stationären
Wachstumsphase keinen CL-Anstieg unter K+-Limitation aufwies (Daten nicht
gezeigt), obwohl er keine ybhO-Deletion besitzt. Folglich scheinen verschiedene
Mechanismen zu existieren, welche die Transkription des cls-Gens und folglich die
CL-Synthese in der exponentiellen und in der stationären Wachstumsphase auf
genetischer Ebene regulieren.
Auch die beiden Reize von KdpD, K+-Limitation und hohe Osmolalität, scheinen den
CL-Gehalt unterschiedlich zu beeinflussen. Der Vergleich des CL-Gehalts unter K+-
Limitation und hoher Osmolalität zeigt, dass es unter beiden kdpFABC-induzierenden
Bedingungen während der exponentiellen Wachstumsphase zu einem Anstieg des
anionischen Phospholipids kommt. Die Höhe des CL-Anstiegs ist allerdings unter-
schiedlich, denn während es unter hohen Osmolalitäten zu einem 2-3 fachen Anstieg
kommt (Tsatskis et al., 2005), konnte unter K+-Limitation nur eine 1.44-fache
Erhöhung gemessen werden (siehe 3.1.1). Ein weiterer Unterschied der beiden
Wachstumsbedingungen ist die Höhe des Induktionsverhältnisses der kdpFABC-
Expression, denn während bei hoher Osmolalität kdpFABC nur transient und auf
einem niedrigeren Niveau exprimiert wird, kommt es unter K+-Limitation zu einer
kontinuierlichen und wesentlich höheren Induktion des Systems (Hamann et al.,
2008). Anhand dieser beiden Stresssituationen für die Zelle kann folglich kein
gemeinsames Regulationsmodell für den CL-Anstieg in der exponentiellen Wachs-
tumsphase postuliert werden.
Des Weiteren fiel bei den Induktionsverhältnissen für die kdpFABC-Expression auf,
dass der Stamm K12 in dem GM56-Medium ein vermindertes Induktionsverhältnis für
die kdpFABC-Expression im Gegensatz zum Induktionsverhältnis in Minimalmedium
nach Epstein und Kim aufwies. Die stärkere Induktion der kdpFABC-Expression in
K12 könnte möglicherweise auf die unterschiedliche Natrium-Konzentration in den
phosphat-gepufferten Medien K0.1 und K115 zurückzuführen sein. Da aber in GM56
mit 0.1 mM K+ bzw. 115 mM K+ die Na+-Konzentration mit 78 mM konstant ist, zeigt
sich, dass diese Induktion definitiv nur auf die K+-Limitation zurückzuführen ist. Das
würde bedeuten, dass bei einer höheren Na+-Konzentration (wie z.B. in GM56-
Medium im Vergleich zu dem Minimalmedium nach Epstein und Kim) die Synthese
des KdpFABC-Komplexes stärker induziert wird, weil durch die Na+-Ionen eine

4. Diskussion
85
geringere K+-Verfügbarkeit suggeriert wird (Hamann et al., 2008). Aufgrund der
höheren kdpFABC-Expression in GM56 mit 115 mM K+ verringert sich folglich das
Induktionsverhältnis. Ebenfalls könnte auch die unterschiedliche Höhe der Ionen-
stärke dabei eine Rolle spielen. Während das phosphat-gepufferte Minimalmedium
sowohl mit 0.1 mM K+ als auch mit 115 mM K+ eine Gesamtionenstärke von 115 mM
aufweist, ist die Ionenstärke in GM56-K0.1 und GM56-K115 mit 78.1 mM bzw.
183 mM unterschiedlich. Ähnliche Daten für unterschiedliche Expressionen unter
verschiedenen Osmolalitäten konnten für Salmonella enterica beobachtet werden
(Balaji et al., 2005).
Im Hinblick auf die Induktion der Expression unter K+-Limitation werden die
Promotoren des cls-Gens und des kdpFABC-Operons von E. coli hochreguliert bzw.
aktiviert. Deshalb wurde ein Alignment der Sequenzen beider Promotoren durch-
geführt (Daten nicht gezeigt). Es wurde untersucht, ob sich ähnliche Sequenzen vor
dem jeweiligen Transkriptionsstart befinden, die evtl. auf eine ähnliche Regulation
(z.B. Aktivierung) hinweisen. Vor dem Transkriptionsstart (+1) des kdpFABC-Operons
konnte eine 23 bp lange Bindestelle (-72 bis -50) für dimerisiertes KdpE~P identi-
fiziert werden (Nakashima et al., 1993). Der 11 bp große Abstand zur -35 Region ist
typisch für Aktivatoren wie KdpE, welche bei einer größeren Entfernung der Binde-
stelle zur -35 Region nicht mehr mit der RNA-Polymerase interagieren und bei einem
kleineren Abstand zur -35 Region diese blockieren können. Die KdpE-Bindesequenz
war allerdings nicht vor dem Transkriptionsstart von cls zu finden. Auch bei Berück-
sichtigung der DNA-Windung, bei der nur ein Teil der Basen mit den entsprechenden
Abständen identisch sein muss, konnte keine passende Sequenz identifiziert werden.
Dieses deutet auf keine ähnliche Regulation der Expression von cls und kdpFABC
unter K+-Limitation hin. Auch sonst konnten keine Homologien der Sequenzen der
beiden Promotoren beobachtet werden. Außerdem zeigten beide Gene bzw.
Operons deutliche Unterschiede in der Höhe ihres Induktionsverhältnisses bezüglich
niedriger und hoher K+-Konzentrationen im Medium. Während es für die cls-Ex-
pression zu einem mit 2 - 3 kleinen Induktionsverhältnis kommt, wird das kdpFABC-
Expression ~6000-fach induziert. Dieses lässt vermuten, dass verschiedene Mecha-
nismen für die Regulation der cls- und kdpFABC-Expression unter K+-Limitation
existieren.

4. Diskussion
86
Zusammenfassend ist noch offen, warum die Zelle in der exponentiellen Wachstums-
phase unter K+-Limitation eine Änderung der Zusammensetzung der Kopfgruppen
der Phospholipide vornimmt. Vermutlich ist der CL-Anstieg ebenso wie unter hoher
Osmolalität oder in der stationären Wachstumsphase ein Schutzmechanismus auf
solch einen Stress, um die Phospholipide gegen den Abbau zu schützen, da CL
stabiler als PG ist. Ebenfalls wird in Stresssituationen die Zusammensetzung der
Fettsäuren variiert, indem der Anteil an 17:0 cyclo 9-10 auf Kosten von 16:1 cis 9
erhöht wird (Cronan, 1968). Dabei wird die Doppelbindung am 9. C-Atom von 16:1
cis 9 zu einer zyklischen Verbindung zwischen dem 9. und 10. C-Atom in 17:0 cyclo
9-10 umgewandelt. Diese Verschiebung bei der Zusammensetzung der Fettsäuren
konnte in der vorliegenden Arbeit auch erstmalig unter K+-Limitation beobachtet
werden. Die kleinen Schwankungen des prozentualen Anteils von 16:1 cis 9 und 17:0
cyclo 9-10 zwischen K0.1 und K115 sind damit zu erklären, dass die Zellen unter K+-
Limitation aufgrund des Kaliummangels schneller die stationäre Wachstumsphase
erreichen. Diese Änderung der Fettsäurezusammensetzung hat keinen Einfluss auf
die Beschaffenheit der Lipiddoppelschicht der Zytoplasmamembran, da beide Fett-
säuren die gleichen chemischen Eigenschaften aufweisen. Somit konnte gezeigt
werden, dass sich unter K+-Limitation nur die Zusammensetzung der Kopfgruppen
und nicht die der Fettsäureketten der Phospholipide ändert. Folglich wird nur die
Oberfläche der Lipiddoppelschicht unter K+-Limitation verändert, indem der Anteil an
negativen Ladungen (CL-Anstieg durch PE-Abnahme) erhöht wird.
4.3. Hypothesen zur Interaktion von KdpD Die Ergebnisse der vorliegenden Arbeit zeigen, dass eine Korrelation zwischen dem
Gehalt an anionischen Phospholipiden und dem Signalstatus der membran-
gebundenen Sensorkinase KdpD sowohl in vitro als auch in vivo besteht. Für den K+-
Kanal KcsA aus Streptomyces lividans konnte gezeigt werden, dass die Anwesenheit
von anionischen Phospholipiden für die Aktivierung bzw. Öffnung des Kanals jedoch
nicht für die Tetramerisierung von KcsA essentiell ist (Alvis et al., 2003). Ähnlich
wäre für KdpD in E. coli vorstellbar, dass die Bildung eines Homodimers auch ohne
die Anwesenheit von negativ geladenen Phospholipiden stattfindet. Folglich würde es
erst zu einer Aktivierung der Sensorkinase durch eine Wechselwirkung mit den
anionischen Phospholipiden kommen, wenn diese unter kdpFABC-induzierenden Be-
dingungen vermehrt in die Membranumgebung der Sensorkinase gelangen. Für die

4. Diskussion
87
chemische Wechselwirkung der nicht-integralen Membranbereiche von KdpD mit den
Phospholipiden der Zytoplasmamembran sind verschiedene Möglichkeiten denkbar.
Zum Einen ist vorstellbar, dass die N-terminale Domäne von KdpD aufgrund der
hydrophoben Aminosäuresequenz an der Position 27-43 mit der Membran
interagiert. Heermann et al. (2003) konnten zeigen, dass ein KdpD-Derivat ohne
Transmembrandomänen und C-terminale Domäne an die Zytoplasmamembran
assoziiert vorliegt. Maier et al. (2008) beobachteten sogar, dass die amphiphile
Region der N-terminalen Domäne von KdpD (AS 22-48) für die Insertion in die
Membran notwendig zu sein scheint, da diese Aminosäuresequenz von Signal-
erkennungspartikeln erkannt wurde, welche für den Transport zur Membran ver-
antwortlich sind (Maier et al., 2008). Zum Anderen wäre es denkbar, dass das positiv
geladene Arginincluster hinter der vierten Transmembrandomäne mit den Kopf-
gruppen der negativ geladenen Phospholipide an der Oberfläche der Zytoplasma-
membran wechselwirkt (Abb. 4.3). Dafür spräche auch die helikale Anordnung der
Aminosäurereste in dem Arginincluster, da mit einem Computerprogramm sechs der
acht positiven Reste auf einer Seite der Helix lokalisiert wurden (Andreeßen, 2007).
Solche Interaktionen der nicht-integralen Membranbereiche von KdpD könnten
widerum zu einer Aktivierung der Signalkaskade von KdpD / KdpE bzw. zu einem
Wechsel zwischen Kinase- und Phosphataseaktivität von KdpD führen.
Abb. 4.3: Topologie-Modell der Sensorkinase KdpD (v erändert nach Zimmann et al., 1995). Dargestellt sind mögliche Protein-Lipid-Interaktionen. Die Zahlen beziehen sich auf die Aminosäuresequenz. Das positiv geladene Arginincluster ist mit „++++“ gekennzeichnet.
Eine Interaktion des Argininclusters mit den anionischen Phospholipiden wird auch
für die RCK-Domäne des K+ Kanals MjK2 von Methanococcus jannaschii vermutet
(Ptak et al., 2005). Des Weiteren konnten Schmidt et al. (2006) zeigen, dass die
Funktion eines spannungsabhängigen K+ Kanals von negativ geladenen Phospho-
lipiden abhängig ist, da diese eine stabilisierende Interaktion mit den positiv gela-
denen Argininresten des Spannungssensors bilden. Die Position der α-Helix nach
401 498
475 466
446444
428
423
27 43
P
H673 N 1
C 894
Periplasma
Zytoplasmamembran
Zytoplasma
? ?

4. Diskussion
88
den Transmembrandomänen von OpuA aus Lactococcus lactis wird sowohl von dem
PG-Gehalt der Membran als auch von der Ionenstärke reguliert (Biemans-Oldehinkel
et al., 2006), da beobachtet wurde, dass bei einem höheren PG-Gehalt die Aktivität
des Proteins ansteigt. Frühere Studien unserer Arbeitsgruppe konnten auch die
Bedeutung des Argininclusters für die Aktivität der Sensorkinase KdpD zeigen.
Derivate von KdpD, denen die vier Transmembrandomänen und das dahinter liegen-
de Arginincluster fehlen, zeigten keine Aktivität unter allen getesteten Bedingungen
(Puppe et al., 1996). Im Gegensatz dazu konnte bei Derivaten, denen nur die vier
Transmembrandomänen fehlten, das Arginincluster jedoch vorhanden war, ein semi-
konstitutiver Phänotyp in Bezug auf die kdpFABC-Expression unter K+-Limitation
beobachtet werden (Heermann et al., 2003a). Derivate von KdpD mit einer Deletion
des Argininclusters (Aminosäure 498 - 530 bzw. 499 - 513) konnten jedoch weder
phosphoryliert werden, noch waren diese in der Lage das Kdp-System zu
komplementieren (Eisenbarth, 2006; Andreeßen, 2007). Des Weiteren wurden durch
ungerichtete Mutagenese ausschließlich Derivate von KdpD mit einem einzelnen
Aminosäureaustausch und einem semi-konstitutiven Phänotyp in der α-Helix loka-
lisiert, die den vierten Transmembrandurchgang und das dahinter liegende Arginin-
cluster bildet (Sugiura et al., 1994; Brandon et al., 2000; Zimmann et al., 2007).
Diese Ergebnisse untermauern die Hypothese, dass solche Derivate von KdpD
wahrscheinlich durch Änderung der Ladungen in diesem Bereich eine „locked-on“
Konformation annehmen (Zimmann et al., 2007). Eine vom Wildtyp abweichende
Aktivität der Sensorkinase durch einen derartigen starren Zustand könnte möglicher-
weise durch eine veränderte Protein-Lipid-Interaktion bewirkt werden. Ein solcher
Effekt wurde bereits für den Antennenkomplex II in dem Purpurbakterium
Rhodobacter sphaeroides beobachtet, bei dem ein einzelner Aminosäureaustausch
die Protein-Lipid-Interaktion verändert (Kwa et al., 2008). Außerdem kann durch
einen einzelnen Aminosäureaustausch die Orientierung der α-Helices zueinander
verändert werden, wie für ProP in E. coli gezeigt wurde (Tsatskis et al., 2008).
Anstelle einer direkten Interaktion mit Phospholipiden wäre es auch möglich, dass
KdpD einen bislang unbekannten Interaktionspartner hat, wie für KdpD aus of
Mycobacterium tuberculosis gezeigt wurde (Steyn et al., 2003). Die Lipoproteine
LprF und LprJ aus M. tuberculosis wechselwirken mit dem N-terminalen zytoplasma-
tischen Teil der Sensorkinase KdpD und können so die kdpFABC-Expression modu-
lieren (Steyn et al., 2003). Bei einem Vergleich der Aminosäuresequenz zwischen
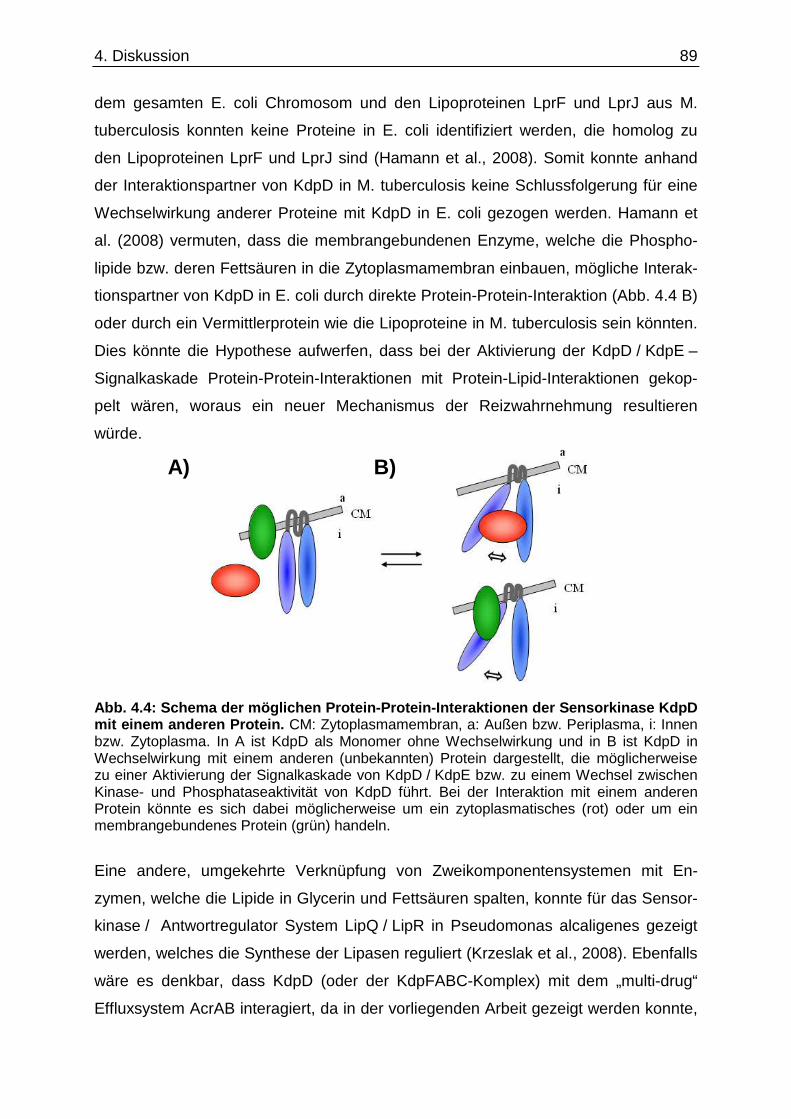
4. Diskussion
89
A) B)
dem gesamten E. coli Chromosom und den Lipoproteinen LprF und LprJ aus M.
tuberculosis konnten keine Proteine in E. coli identifiziert werden, die homolog zu
den Lipoproteinen LprF und LprJ sind (Hamann et al., 2008). Somit konnte anhand
der Interaktionspartner von KdpD in M. tuberculosis keine Schlussfolgerung für eine
Wechselwirkung anderer Proteine mit KdpD in E. coli gezogen werden. Hamann et
al. (2008) vermuten, dass die membrangebundenen Enzyme, welche die Phospho-
lipide bzw. deren Fettsäuren in die Zytoplasmamembran einbauen, mögliche Interak-
tionspartner von KdpD in E. coli durch direkte Protein-Protein-Interaktion (Abb. 4.4 B)
oder durch ein Vermittlerprotein wie die Lipoproteine in M. tuberculosis sein könnten.
Dies könnte die Hypothese aufwerfen, dass bei der Aktivierung der KdpD / KdpE –
Signalkaskade Protein-Protein-Interaktionen mit Protein-Lipid-Interaktionen gekop-
pelt wären, woraus ein neuer Mechanismus der Reizwahrnehmung resultieren
würde.
Abb. 4.4: Schema der möglichen Protein-Protein-Inte raktionen der Sensorkinase KdpD mit einem anderen Protein. CM: Zytoplasmamembran, a: Außen bzw. Periplasma, i: Innen bzw. Zytoplasma. In A ist KdpD als Monomer ohne Wechselwirkung und in B ist KdpD in Wechselwirkung mit einem anderen (unbekannten) Protein dargestellt, die möglicherweise zu einer Aktivierung der Signalkaskade von KdpD / KdpE bzw. zu einem Wechsel zwischen Kinase- und Phosphataseaktivität von KdpD führt. Bei der Interaktion mit einem anderen Protein könnte es sich dabei möglicherweise um ein zytoplasmatisches (rot) oder um ein membrangebundenes Protein (grün) handeln.
Eine andere, umgekehrte Verknüpfung von Zweikomponentensystemen mit En-
zymen, welche die Lipide in Glycerin und Fettsäuren spalten, konnte für das Sensor-
kinase / Antwortregulator System LipQ / LipR in Pseudomonas alcaligenes gezeigt
werden, welches die Synthese der Lipasen reguliert (Krzeslak et al., 2008). Ebenfalls
wäre es denkbar, dass KdpD (oder der KdpFABC-Komplex) mit dem „multi-drug“
Effluxsystem AcrAB interagiert, da in der vorliegenden Arbeit gezeigt werden konnte,

4. Diskussion
90
dass ein intaktes KdpFABCDE-System für das Wachstum auf Agarplatten mit
höheren Konzentrationen des Antibiotikums Novobiocin, welches durch AcrAB aus
der Zelle geschleust wird (Ma et al., 1995), notwenig zu sein scheint (siehe 3.10).
Auch Lebendzellzahlen von mehr als 100 % bei niedrigeren Novobiocin-Konzen-
trationen im Vergleich zu Agarplatten ohne Antibiotikum sprechen für eine Abhängig-
keit von KdpD, da dieser Effekt nur bei einem funktionsfähigen Kdp-System bzw. bei
Überproduktion der Sensorkinase KdpD unter kdpFABC-induzierenden Bedingungen
(0.1 mM K+) beobachtet werden konnte. Es wäre möglich, dass durch die größere
Menge von KdpD in der Zytoplasmamembran eine Interaktion mit AcrAB beeinflusst
wird bzw. vermehrt auftritt und daraus womöglich eine höhere Resistenz gegenüber
Novobiocin resultiert.
Für eine Interaktion des KdpFABC-Komplexes mit AcrAB sprächen hingegen die
Daten unter nicht-kdpFABC-induzierende Bedingungen (Daten nicht gezeigt). Im
Vollmedium KML herrschen aufgrund der Kaliumkonzentration von ~144 mM K+
ebenso wie im Minimalmedium K115 mit 115 mM K+ nicht-kdpFABC-induzierende
Bedingungen und folglich wird kein KdpFABC-Komplex synthetisiert. Unter diesen
Wachstumsbedingungen liegen die regulatorischen Proteine KdpD / KdpE jedoch nur
in einem kdp+ Stamm in einer geringen Menge vor. Da allerdings unter nicht-
kdpFABC-induzierende Bedingungen in der vorliegenden Arbeit kein signifikanter
Unterschied zwischen einem kdp+ und kdp- Stamm für die Resistenz gegenüber
Novobiocin beobachtet werden konnte, scheint die Höhe der Novobiocin-Resistenz
von dem Vorhandensein des KdpFABC-Komplexes beeinflusst zu werden. Dieses
Ergebnis widerspricht den Beobachtungen von Zhou et al. (2003), die einen Unter-
schied in dem Resistenzverhalten zwischen einem kdp+ und kdp- Stamm im
Vollmedium ermittelten. Des Weiteren klassifizierten Zhou et al. (2003) einen kdp-
Stamm im Gegensatz zu einem kdp+ Stamm als resistenter gegenüber Novobiocin.
Die vorliegene Arbeit zeigt jedoch, dass genau der umgekehrte Fall eintritt und ein
intaktes KdpFABCDE-System für das Wachstum auf höheren Novobiocin-Konzen-
trationen notwenig zu sein scheint. Die widersprüchlichen Ergebnisse von Zhou et al.
(2003) sind möglicherweise damit zu erklären, dass sie in ihrer Veröffentlichung nicht
genauer auf ihren Versuchsansatz eingegangen sind und sich auch nur am Rande
mit dem Kdp-System beschäftigt haben, da sie alle Sensorkinase / Antwortregulator
Systeme von E. coli untersuchten.

4. Diskussion
91
Damit der Stamm TKV2208 (kdpD- trk-) überhaupt auf Agarplatten mit 0.1 mM K+
wachsen kann, muss der Stamm den KdpFABC-Komplex synthetisieren, um K+-
Ionen in die Zelle transportieren zu können. Mittels Q-RT-PCR konnte in diesem
Stamm die kdpFABC-Expression nachgewiesen werden, obwohl das für die Sensor-
kinase kodierende kdpD-Gen deletiert war (Zimmann, pers. Mitteilung). Somit könnte
die kdpFABC-Expression durch eine KdpD-unabhängige Phosphorylierung des
Antwortregulators KdpE induziert werden. Dabei könnte KdpE durch nieder-
molekularen Phosphodonoren oder durch eine andere Sensorkinase („cross-talk“)
phosphoryliert werden. Ein solcher Mechanismus wurde oft in Organismen beob-
achtet, die aufgrund einer Mutation nicht mehr in der Lage waren, die normale
Signalkaskade zur Aktivierung einer spezifischen Antwort zu benutzen (Laub &
Goulian, 2007). In E. coli wurden alle Zweikomponentensysteme bezüglich ihrer
funktionellen Eigenschaften (Autophosphorylierung und Phosphotransfer) und
möglicher „cross-talk“ Reaktionen in vitro untersucht (Yamamoto et al., 2005). Dabei
konnte in vitro gezeigt werden, dass einige Sensorkinasen die Antwortregulatoren
anderer Systeme phosphorylieren, wobei die Sensorkinase KdpD nur in der Lage ist
den eigenen Antwortregulator KdpE zu phosphorylieren. Jedoch konnten Yamamoto
et al. (2005) zeigen, dass UhpB neben KdpD die einzige Sensorkinase von E. coli ist,
die KdpE in vitro phosphorylieren kann. Die aus drei Komponenten bestehende
UhpABC Signalkaskade reguliert die Aufnahme von Glucose-6-Phosphat über den
Transporter UhpT (Verhamme et al., 2001). Folglich wurde für die Aufklärung der
Reizweiterleitung von KdpD auf KdpE je ein Alignment der Aminosäuresequenz der
Sensorkinasen UhpB und KdpD bzw. der Antwortregulatoren UhpA und KdpE durch-
geführt. Jedoch zeigten beide Alignments keine signifikanten Homologiebereiche, die
einen Aufschluss über die strukturelle Wechselwirkung von KdpD (bzw. UhpB) mit
KdpE bringen könnten (Daten nicht gezeigt).
Die hypothetische Wechselwirkung mit AcrAB könnte auch eine für das Kdp-System
regulierende Funktion haben. Eine Regulation der Resistenzen gegenüber
verschiedenen Antibiotika durch solche Sensorkinase / Antwortregulator Systeme ist
bereits für verschiedene Bakterien bekannt (Matsushita & Janda, 2002). In E. coli
konnte beobachtet werden, dass das Zweikomponentensystem VanS / VanR die
Resistenz gegenüber Vancomycin reguliert (Silva et al., 1998). Hirakawa et al.
(2003a) untersuchten alle 32 Antwortregulatoren von E. coli, wobei sie für KdpE

4. Diskussion
92
keinen Einfluss auf die Resistenz gegenüber den getesteten β-Lactam-Antibiotika
feststellen. Bereits zuvor hatte diese Arbeitsgruppe veröffentlicht, dass die Über-
produktion einiger Antwortregulatoren, darunter auch KdpE, oft nur die Resistenz
gegenüber bestimmten Antibiotika, welche über den „multi-drug“ Effluxsystem AcrAB
ausgeschleust werden, beeinflusst (Hirakawa et al., 2003b). Es konnte für über-
produziertes KdpE eine 4-fach höhere Resistenz gegenüber Kanamycin, jedoch
keine Änderung gegenüber Novobiocin bestimmt werden (Hirakawa et al., 2003b).
Ein weiterer Hinweis auf eine globalere Regulierung durch die Sensorkinase KdpD
könnte sein, dass das Kdp-System nicht nur in E. coli, sondern auch in vielen
anderen Mikroorganismen existiert. Folglich hätte die konstitutiv synthetisierte Sen-
sorkinase KdpD zwei grundverschiedene Funktionen. Eine Funktion ist die Regula-
tion des KdpFABC-Systems, um die K+-Homeostase bei extremen Bedingungen
durch die Aufnahme von K+ über den KdpFABC-Komplex in der Umgebung aufrecht
zu erhalten. Die andere Funktion könnte sein, weitere Prozesse durch die Interaktion
mit einem anderen Protein unter nicht-kdpFABC-induzierenden Bedingungen zu
regulieren. Diese Theorie ist denkbar, da die Anordnung der Gene kdpA, kdpB und
kdpC relativ konserviert ist, während die regulatorischen Gene kdpD / kdpE in den
verschiedenen Organismen unterschiedlich angeordnet sind oder nur teilweise
vorhanden sind (Ballal et al., 2007). In dem Archaeon Halobacterium salinarium wird
ein KdpFABC-Transporter unter K+-Limitation gebildet, der K+-Ionen in die Zelle
transportiert und so ein Wachstum unter extremen Bedingungen in Abhängigkeit von
der Anwesenheit der Produkte des kdpFABCcat3-Operons ermöglicht (Strahl &
Greie, 2008). Allerdings fehlen in diesem halophilen Organismus die zu kdpD und
kdpE homologen regulatorischen Gene. Jedoch könnte das zusätzlich chromosomal-
kodierte cat3-Gen ebenfalls eine Rolle bei der Transkription spielen. In dem
thermoacidophilen Bakterium Alicyclobacillus acidocaldarius hingegen wurde in zwei
Regionen eine zu KdpD homologe Sensorkinase lokalisiert. Die eine Region befand
sich im Operon kdpZFABCN, wobei kdpN für einen Homodimer der N-terminalen
Domäne von KdpD kodiert und die andere in dem kdpHE-Operon, wobei kdpH die
Transmembrandomänen und die C-terminale Domäne von KdpD kodiert
(Schleußinger et al., 2006). In dem Stickstoff-fixierenden Cyanobakterium Anabena
sp. L-31 konnten zwei Kopien der Gene kdpAGBC im Genom lokalisiert werden,
wobei nur in einem der beiden Operone (kdp1) eine verkürzte Form der N-terminalen

4. Diskussion
93
Domäne von KdpD vorhanden war und ein kdpE homologes Gen weder in dem
einen noch in dem anderen Operon vorhanden war (Ballal & Apte, 2005). Allerdings
scheint die Expression von nur einem der beiden Operone (kdp2) unter K+-Limitation
induziert zu werden. In Sinorhizobium meliloti konnte neben den zu E. coli
homologen niedrig affinen K+-Aufnahmesystemen ebenfalls ein Kdp-System anhand
von Sequenzhomologien ermittelt werden, dass ebenfalls nur unter K+-Limitation und
hoher Osmolalität K+ in die Zelle transportiert (Domingeuz-Ferreras et al., 2009).
Es ist wichtig zu erwähnen, dass die bakterielle Membran eine heterogene Verteilung
der Lipide besitzt (zum Überblick siehe Matsumoto et al., 2006). In E. coli ist das
anionische Phospholipid CL am Septum und an den Zellpolen angereichert, wodurch
CL-reiche Domänen vorliegen (Mileykovskaya & Dowhan, 2000). Huang et al. (2006)
vermuten, dass diese Clusterung ein krümmungsbedingtes Phänomen ist. Bezug-
nehmend auf die heterogene Verteilung der Lipide konnten mehrere Membran-
proteine am Septum und/oder an den Polen der Bakterienzelle lokalisiert werden (Lai
et al., 2004). Diese Lokalisation deutet an, dass diese Membranproteine die Nähe
anionischer Phospholipide bevorzugen. Des Weiteren wurde vermutet, dass Mem-
branproteine mit einer hohen Affinität für CL die Domänen aus CL stabilisieren (zum
Überblick siehe Matsumoto et al., 2006) oder dass CL die polare Lokalisation dieser
Proteine fördert, wie für den Osmosensor ProP in E. coli vorgeschlagen wurde
(Romantsov et al., 2007). Es ist auch für KdpD vorstellbar, dass dieses Protein in
diesen CL-reichen Domänen geclustert vorkommt oder sogar mit CL interagiert.
Mikrodomänen, die CL und KdpD enthalten, könnten somit erklären, warum ein
Anstieg von CL unter K+-Limitation nicht unbedingt essentiell ist, wie in einigen
Stämmen beobachtet wurde. Bisherige Daten mit KdpD-GFP lassen jedoch keine
Clusterung von KdpD vermuten (Feuerbaum, 2008). In dem Stamm UE54, der kein
PG und CL aufweist, konnte Phosphatidylsäure und ein neu identifiziertes an-
ionisches Lipid, N-acyl PE, ebenfalls an den Polen der Zelle angereichert, gefunden
werden (Mileykovskaya et al., 2009). Eine erhöhte Bindeaffinität zu CL konnte für die
Glycosyltransferase MurG mittels einer Co-Reinigung gezeigt werden (Van den
Brink-Van der Laan et al., 2003). Mit diesem Ansatz konnten für KdpD jedoch keine
Rückschlüsse auf eine spezifische Interaktion von Phospholipiden mit KdpD
herausgefunden werden (siehe 3.9). Dieses liegt vermutlich daran, dass es sich im
Gegensatz zu der mit vier Transmembrandomänen in der Zytoplasmamembran

4. Diskussion
94
verankerten Sensorkinase KdpD bei MurG nur um ein an die Membran assoziiertes
Protein handelt. Demzufolge ist die Zugabe eines Detergenz, welches für die
Stabilität von Membranproteinen in Lösung notwendig ist, bei der Co-Reinigung von
MurG nicht essentiell. Bei der Solubilisierung von KdpD hingegen könnte das Deter-
genz mit einer höheren Affinität die verschieden Phospholipidtypen unterschiedlich
stark vom Protein verdrängen. Dadurch würde sich auch die Frage stellen, ob die
Quantifizierung der Phospholipide vom gewählten Detergenz abhängig ist und wie es
sich bei anderen Membranproteinen verhält, um von einer wirklich spezifischen
Wechselwirkung sprechen zu können.
Eine andere Begründung für die verschiedenen Aktivitäten von KdpD in den „Lipid-
Stämmen“ und folglich eine veränderte native Lipidzusammensetzung haben, könnte
sein, dass jeder Typ der Phospholipide zu einem anderen lateralen Packungsdruck
führt. De Kruijff (1997) vermutet, dass nicht-bilayer-bevorzugende Lipide wie PE
einen höheren lateralen Packungsdruck auf Membranproteine ausüben, um diese in
eine für sie bessere Konformation zu bringen, als es bilayer-bevorzugende Lipide wie
CL und PG tun. Wikström et al. (2009) stellen die Hypothese auf, dass der laterale
Packungsdruck auf die Größe der Kopfgruppen der Phospholipide zurückzuführen
ist, wobei kleinere Kopfgruppen eine höhere Krümmung der Membran verursachen
und mehr Vorteile für viele Membranproteine als größere Kopfgruppen (niedrigere
Krümmung) darstellen.
4.4. Ausblick
In dieser Arbeit konnte gezeigt werden, dass der Anteil von anionischen Phospho-
lipiden in der Zellhülle von E. coli die kdpFABC-Expression beeinflusst. Des Weiteren
könnte die heterogene Verteilung der Lipide ein Parameter sein, der für den Signal-
status von KdpD von Bedeutung ist. Daher wird zurzeit die Lokalisation von KdpD in
der Zytoplasmamembran mit KdpD-GFP Fusionen untersucht (Zimmann, pers.
Mitteilung), die möglicherweise mit der Clusterung von CL übereinstimmt und auf
eine Interaktion mit dem Phospholipid hindeuten könnte.
Zur Untersuchung der Wechselwirkungen der nicht-integralen Membranbereiche von
KdpD mit den Phospholipiden der Zytoplasmamembran (siehe Abb. 4.3) bietet sich
folgende experimentelle Vorgehensweise an: Zunächst muss KdpD in Liposomen
rekonstituiert werden, in denen eine Fettsäure einen TID-Rest (Diazirin) trägt. Dieser

4. Diskussion
95
geht bei Bestrahlung mit UV-Licht kovalente Bindungen mit Proteinen in der nahen
Umgebung ein. Um die Kontaktstellen zwischen dem Protein und der Membran zu
lokalisieren, kann die Position dieser Markierung mit Hilfe der MALDI-Analyse und
des Edman-Abbaus bestimmt werden. Solch ein Ansatz wurde bereits für die Identi-
fizierung von Kontaktstellen anderer Proteine erfolgreich angewandt (Von Ballmoos
et al., 2002).
Alternativ könnte auch die BIAcore Technik („biomolecular interaction analysis“) mit
KdpD Peptiden angewandt werden. Dafür müssten Liposomen bestehend aus einem
Typ Phospholipid (also nur PE, PG oder CL) auf spezielle Chips gezogen werden.
Die Messung der Peptidbindung würde mittels SPR („surface plasmon resonance“)
erfolgen, wobei KdpD-Peptide in verschiedenen Puffern über die Chips fließen
würden, um verschiedene Bedingungen wie kdpFABC-induzierend und nicht-
kdpFABC-induzierend zu untersuchen. Mit dieser Methode können Informationen
über die Kinetik und die Affinität der Bindung von Phospholipiden an KdpD ge-
wonnen werden.
Außerdem wäre es interessant zu überprüfen, ob anionische Phospholipide einen
Einfluss auf die Topologie von KdpD haben und womöglich dadurch die Aktivität der
Sensorkinase beeinflussen, wie für die Lactose Permease LacY von E. coli
beobachtet werden konnte (Dowhan et al., 2004). Dazu müssten zunächst die „Lipid-
Stämme“ mit einer veränderten nativen Phospholipidzusammensetzung mittels P1-
Transduktion so verändert werden, dass sie ∆lacZ oder ∆phoA sind, um an-
schließend mit Plasmiden, die eine kdpD-lacZ oder ein kdpD-phoA Fusion kodieren,
transformiert werden zu können. Dabei wäre es sinnvoll Positionen von KdpD zu
untersuchen, die beim Wildtyp in der Nähe der Zytoplasmamembran lokalisiert sind.
Somit sollten die Fusionen an Aminosäuren in den periplasmatischen Loops
zwischen den Transmembrandomänen 1 - 2 bzw. 3 - 4 liegen. Des Weiteren sollten
Positionen für die Fusion kurz vor der ersten und hinter der vierten Trans-
membrandomäne bzw. im Loop zwischen den Transmembrandomänen 2 - 3 sowie in
dem N- und C-terminalen zytoplasmatischen Bereich gewählt werden. Mit dem
jeweiligen Enzymaktivitätstest könnten dann die verschiedenen Konstrukte auf ihre
Zugänglichkeit untersucht werden. Anhand der unterschiedlichen Enzymaktivitäten
kann die getestete Position der Fusion lokalisiert werden, da die alkalische Phos-
phatase im Zytoplasma und die β-Galaktosidase im Periplasma inaktiv ist. Die

4. Diskussion
96
Topologie von KdpD in einem Stamm mit einer nativen Phospholipidzusammen-
setzung wurde unter anderem mit dieser Methode analysiert (Zimmann et al., 1995).
Dabei fiel jedoch auf, dass die synthetisierten Mengen der verschiedenen Fusions-
proteine unterschiedlich und zudem auch noch instabil waren (Zimmann et al., 1995).
Solch eine Topologiestudie könnte unter Berücksichtigung dieser Problematik die
Funktionalität von KdpD in den „Lipid-Stämmen“, wie in der vorliegenden Arbeit
gezeigt, ergänzen.
Es wäre ebenfalls denkbar, dass KdpD mit anderen Proteinen wechselwirkt. Dieses
könnte anhand des „Bacterial two-hybrid systems“ für potentielle Kandidaten
überprüft werden (Karimova et al., 1998). Es wurde von Hamann et al. (2008) die
Hypothese vorgeschlagen, dass KdpD mit den membrangebundenen Phospholipid-
synthasen PlsB und PssA interagieren könnte, da diese in einem Komplex an den
Septen lokalisiert sind (Gully & Bouveret, 2006). Ebenfalls könnte mit dieser Methode
eine potentielle Wechselwirkung mit dem „multi-drug“ Effluxsystem AcrAB anhand
der gezeigten Ergebnisse in der vorliegenden Arbeit untersucht werden. Für die
globalere Suche nach Interaktionspartnern von KdpD können in vivo Crosslinks
mittels HPINE („His-Protein-Interaction-Experiment“) nach Herzberg et al. (2007)
durchgeführt werden. Zu diesem Zweck wird das Zielprotein mit einen His-Tag
versehen. Zu der Bakterienkultur wird Formaldehyd gegeben, wodurch die momen-
tanen Protein-Protein-Interaktionen fixiert werden. Nach einer kurzen Inkubationszeit
werden die Zellen geerntet und aufgeschlossen. Das Protein kann dann zusammen
mit seinen potentiellen Partnern über seinen His-Tag mittels Ni2+-NTA-Affinitäts-
chromatographie aufgereinigt werden. Nach der Auflösung der Crosslinks können die
Interaktionspartner durch Massenspektrometrie identifiziert werden. Erste Versuche
waren bislang jedoch noch nicht erfolgreich (Zimmann, pers. Mitteilung).
Aufgrund der Beobachtung von Hirakawa et al. (2003b), dass überproduziertes KdpE
in Zellen zu einer erhöhten Resistenz gegenüber Kanamycin führt, könnten weitere
Versuche unter diesem Aspekt durchgeführt werden. Möglicherweise könnten
entsprechende Plattentests ein Zusammenhang zwischen dem Kdp-System und dem
Effluxsystem AcrAB genauer verifizieren. Es könnten an die vorliegende Arbeit
angelehnte Versuche durchgeführt werden.

4. Diskussion
97
Die oben beschriebenen möglichen Interaktionsstudien von KdpD mit den Phospho-
lipiden der Zytoplasmamembran, intra- oder intermolekular mit sich selbst oder mit
anderen Proteinen sowie die Studie möglicher Konformationsänderungen sind für die
Erforschung des genaueren Mechanismus der Reizwahrnehmung durch das
KdpD / KdpE - System bzw. der Signalweiterleitung innerhalb dieses Systems von
großer Bedeutung.

5. Zusammenfassung
98
5. Zusammenfassung In dieser Arbeit wurde untersucht, ob K+-Limitation die Zusammensetzung der Phos-
pholipide der Membran in Escherichia coli beeinflusst. Es konnte gezeigt werden,
dass es unter K+-limitierenden Bedingungen im Medium zu einem Anstieg des
Cardiolipin (CL) Gehaltes auf Kosten des zwitterionischen Phospholipids Phos-
phatidylethanolamin (PE) während der exponentiellen Wachstumsphase kommt.
Durch den CL-Anstieg auf Kosten von PE steigt ebenfalls der Anteil an anionischen
Phospholipiden (PG und CL) an. Der Anstieg von CL konnte auf die Erhöhung der
transkriptionalen Aktivität des cls-Gens zurückgeführt werden, welches für die CL-
Synthase kodiert. Im Gegensatz zu den Kopfgruppen änderte sich die Fettsäure-
zusammensetzung der Phospholipide nicht signifikant unter K+-Limitation. Dieses
Phänomen tritt vermutlich auf, um die Lipide gegen den Abbau zu schützen.
Des Weiteren konnte gezeigt werden, dass die Kinaseaktivität von KdpD in seiner
nativen Membranumgebung durch Fusion mit Liposomen aus anionischen Phospho-
lipiden stimuliert wird, während diese durch Fusion mit Liposomen aus zwitter-
ionischen Phospholipiden reduziert wird. Es konnte ebenfalls gezeigt werden, dass
die Höhe des gesamten Anteils anionischer Phospholipide neben der Kinaseaktivität
von KdpD auch die kdpFABC-Expression beeinflusst.
Die in die Zytoplasmamembran interkalierende Substanz Procain zeigte einen
signifikanten Einfluss auf die kdpFABC-Expression in Abhängigkeit von der Phospho-
lipidzusammensetzung der Membran. Für Derivate von KdpD konnte eine Erhöhung
der bereits vorhandenen Aktivität, aber kein neuer Phänotyp der Sensorkinase
ermittelt werden. Aufgrund dieser Ergebnisse ist es vorstellbar, dass die Sensor-
kinase KdpD Änderungen der Membranspannung wahrnimmt. Des Weiteren
sprechen die Kinaseaktivitäten der fusionierten Membranvesikel für die Hypothese,
dass die Aktivität von KdpD durch die Krümmung der Membran beeinflusst wird.
Aufgrund der Beobachtung, dass die Resistenz gegenüber hohen Novobiocin-
Konzentrationen von dem Vorhandensein eines intakten KdpFABCDE-Systems
abhängig zu sein scheint, könnte auch eine Interaktion mit dem „multi-drug“ Efflux-
system AcrAB möglich sein.

6. Summary
99
6. Summary In this study it was investigated whether K+ limitation affects membrane phospholipid
composition in Escherichia coli. Our measurements revealed that there is an
increase in the cardiolipin (CL) content during the exponential growth phase at the
expense of the zwitterionic phospholipid phosphatidylethanolamine (PE) under K+
limitation. In addition, the CL increase at the expense of PE results in a higher
anionic phospholipid content (PG and CL). It could be shown, that the increase of CL
is the result of the increased transcriptional activity of the cls gene coding for the CL
synthase. In contrast to the head groups there was no significant change in the fatty
acid composition of the phospholipids under K+ limitation. Probably, this phenomenon
occurs to protect the lipids against degradation.
Furthermore, the kinase activity of KdpD is stimulated in its native membrane
environment by fusion with liposomes containing anionic CL while the addition of the
same amount of zwitterionic PE/PC liposomes results in a reduced kinase activity. In
addition, it could be observed, that the content of anionic phospholipids affects
beside the kinase activity of KdpD also the kdpFABC expression.
In addition, the cytoplasmic membrane intercalating reagent procaine had a
significant influence on kdpFABC expression depending on the phospholipid
composition of the membrane. For derivatives of KdpD an increase of already
existing activity, but no new phenotype for the sensor kinase could be observed.
Regarding these results, it is possible that the sensor kinase KdpD senses changes
of the membrane strain. Furthermore, the kinase activities of the fused vesicles lead
to the assumption that the activity of KdpD is influenced by the curvature of the
membrane.
Due to the fact that the resistance against high novobiocin concentrations seems to
depend on the presence of an intact KdpFABCDE system, an interaction with the
multi-drug efflux system AcrAB could be envisaged.

7. Literaturverzeichnis
100
7. Literaturverzeichnis Ahnert, F., R. Schmid, K. Altendorf, and J. C. Grei e. 2006. ATP binding properties of the soluble part of the KdpC subunit from the Escherichia coli K(+)-transporting KdpFABC P-type ATPase. Biochem. 45: 11038-11046. Altendorf, K., and W. Epstein. 1996. The Kdp-ATPase of Escherichia coli, pp 403-420. In A.G. Lee (ed.), Biomembranes, Vol 5 (ATPases), London, JAI Press. Altendorf, K., M. Gaßel, W. Puppe, T. Möllenkamp, A . Zeeck, C. Boddien, K. Fendler, E. Bamberg, and S. Drosse. 1998. Structure and function of the Kdp-ATPase of Escherichia coli. Acta Physiol. Scand. Suppl. 643: 137-146. Alvis, S. J., I. M. Williamson, J. M. East, and A. G. Lee. 2003. Interactions of anionic phospholipids and phosphatidylethanolamine with the potassium channel KcsA. Biophys. J. 85: 3828-3838. Andreeßen, B. 2007. Einfluss des C-terminalen Bereichs der Sensordomäne auf die Reizperzeption der Histidinkinase KdpD von Escherichia coli. Masterarbeit. Universität Osnabrück. Aravind, L., and C. P. Ponting. 1997. The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem Sci 22: 458-459. Arber, W. 1960. Transduction of chromosomal genes and episomes in Escherichia coli. Virology 11: 273-288. Arechaga, I., B. Miroux, S. Karrasch, R. Huijbregts , B. de Kruijff, M. J. Runswick, and J. E. Walker. 2000. Characterisation of new intracellular membranes in Escherichia coli accompanying large scale over-production of the b subunit of F1F0 ATP synthase. FEBS Letters 482: 215-219. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moo re, J. G. Seidman, J. A. Smith, and K. Struhl. 1987. Current Protocols in Molecular Biology. Greene Publishing Associates and Wiley Interscience. John Wiley and Sons. New York. Bakker, E. P. 1993. Cell K+ & K+-transport systems in prokaryotes, Alkali cation transport systems in prokaryotes (Bakker, E. P. ed.), CRC Press, 205-224, Boca Raton, Florida. Bakker, E. P., A. Borchard, M. Michels, K. Altendor f, and A. Siebers. 1987. High affinity potassium uptake system in Bacillus acidocaldarius showing immunological cross-reactivity with the Kdp system from Escherichia coli. J. Bacteriol. 147: 820-826. Balaji, B., K. O´Connor, J. R. Lucas, J. M. Anderso n, and L. N. Csonka. 2005. Timing of induction of osmotically controlled genes in Salmonella enterica serovar typhimurium determined with quantitative real-time reverse transcription-PCR. Appl. Environ. Microbiol. 71: 8273-8283. Ballal, A., and S. K. Apte. 2005. Differential expression of the two kdp operons in the nitrogen-fixing cyanobacterium Anabaena sp. Strain L-31. Appl. Environ. Microbiol. 71: 5297-5303. Ballal, B., B. Basu, and S. K. Apte. 2007. The Kdp-ATPase system and its regulation. J. Biosci. 32: 559-568. Becker, D., K. Fendler, K. Altendorf, and J.-C. Gre ie. 2007. The conserved dipole in transmembrane helix 5 of KdpB in the Escherichia coli KdpFABC P-type ATPase is crucial for coupling and the electrogenic K+-translocation step. Biochemistry 46: 13920-13928. Bogdanov M., J. Sun, H. R. Kaback, and W. Dowhan. 1996. A phospholipid acts as a chaperone in assembly of a membrane transport protein. J. Biol. Chem. 271: 11615–11618.

7. Literaturverzeichnis
101
Bogdanov, M., J. Xie, P. Heacock. W. Dowhan. 2008. To flip or not to flip: lipid-protein charge interactions are a determinat of final membrane protein topology. J. Cell. Biol. 182: 925-935. Bogdanov, M., E. Mileykovskaya, and W. Dowhan. 2008. Lipids in the assembly of membrane proteins and organization of protein supercomplexes: Implications for lipid-linked disorders. Subcell. Biochem. 49: 197-239. Booth, I. R. 1985. Regulation of cytoplasmic pH in bacteria. Microbiol. Rev. 49: 359-378. Booth, I. R., M. A. Jones, D. McLaggan, Y. Nikolaev , L. S. Ness, C. M. Wood, S. Miller, S. Tötemeyer, and G. P. Ferguson. 1996. Bacteriol ion channels, Handbook of Biological Physics Vol. 2 (Koning, W. N.; Kaback, H. R.; Lolkema, J. S.; eds.) 693-729, Elsevier Science, Amsterdam. Bourret, R. B., K. A. Borkovich, and M. I. Simon. 1991. Signal transduction pathways involving protein phosphorylation in prokaryotes. Annu. Rev. Biochem. 60: 401-441. Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248-254. Bramkamp, M., and K. Altendorf. 2003. Mutational analysis of charged residues in the putative KdpB-TM5 domain of the Kdp-ATPase of Escherichia coli. Ann. N.Y. Acad. Sci. 986: 351-353. Bramkamp, M., K. Altendorf, and J.-C. Greie. 2007. Common pattern and unique features of P-type ATPases: a comparative view on the KdpFABC complex from Escherichia coli. Mol. Membr. Biol. 24: 375-386. Brandon, L., S. Dorus, W. Epstein, K. Altendorf, an d K. Jung. 2000. Modulation of KdpD phosphatase implicated in the physiological expression of the Kdp ATPase of Escherichia coli. Mol. Microbiol. 38: 1086-1092. Buurman, E. T., K.-T. Kim, and W. Epstein. 1995. Genetic evidence of two sequentially occupied K+ binding sites in the Kdp transport ATPase. J. Biol. Chem. 270: 6678-6685. Cascales, E., A. Bernadac, M. Gavioli, J.-C. Lazzar oni, and R. Lloubes. 2002. Pal lipoprotein of Escherichia coli plays a major role in outer membrane integrity. J. Bacteriol. 184: 754-759. Cronan, J. E. 1968. Phospholipid alterations during growth of Escherichia coli. J. Bacteriol. 95: 2054-2061. Cronan, J. E. 2003. Bacterial membrane lipids: where do we stand? Annu. Rev. Microbiol. 57: 203-224. DeChavigny, A., P. N. Heacock, and W. Dowhan. 1991. Sequence and inactivation of the pss gene of Escherichia coli - Phosphatidylethanolamine may not be essential for cell viability. J. Biol. Chem. 266: 5323-5332. De Kruijff, B. 1997. Biomembranes. Lipids beyond the bilayer. Nature 386: 129-130. De Mendoza, D., A. Klages Ulrich, and J. E. Cronan. 1983. Thermal regulation of membrane fluidity in Escherichia coli: Effects of overproduction of beta-ketoacyl carrier protein synthase I. J. Biol. Chem. 258: 2098-2101. Dominguez-Ferreras, A., S. Munoz, J. Olivares, M. J Soto, and J. Sanjuan. 2009. Role of potassium uptake systems in Sinorhizobium meliloti osmoadaptatation and symbiotic performance. J. Bacteriol. In press. Donohue-Rolfe, A. M. and M. Schaechter. 1980. Translocation of phospholipids from the inner to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. 77: 1867-1871. Dowhan, W., E. Mileykovskaya, and M. Bogdanov. 2004. Diversity and versatility of lipid-protein interactions revealed by molecular genetic approaches, Biochim. Biophys. Acta 1666: 19-39.

7. Literaturverzeichnis
102
Durell, S. R., E. P. Bakker, and H. R. Guy . 2000. Does the KdpA subunit from the high affinity K+-translocating P-type kdp-ATPase have a structure similar to that of K+ channels? Biophys. J. 78: 188-199. Eisenbarth, M. 2006. Die Sensorkinase KdpD aus Escherichia col: a) Funktionalitätsstudien KdpD-CTD b) Konstruktion und Charakterisierung von KdpD/∆Arg. Großpraktikumsbericht. Universität Osnabrück. Elferink, M. G., J. G. de Wit, R. Demel, A. J. Drie ssen, and W. N. Konings. 1992. Functional reconstitution of membrane proteins in monolayer liposomes from bipolar lipids of Sulfolobus acidocaldarius. J. Chem. Biochem. 267: 1375-1381. Epstein, W. 1992. Kdp, a bacterial P-type ATPase whose expression and activity are regulated by turgor pressure. Acta Physiol. Scand. 146: 193-199. Epstein, W., and B. S. Kim. 1971. Potassium transport loci in Escherichia coli K-12. J. Bacteriol. 108: 639-644. Epstein, W., and M. Davis. 1970. Potassium-dependent mutants of Escherichia coli. J. Bacteriol. 101: 836-843. Epstein, W., V. Whitelaw, and J. Hesse. 1978. A K+ transport ATPase in Escherichia coli. J. Biol. Chem. 253: 6666-6668. Facey, S. J., and A. Kuhn. 2003. The sensor protein KdpD inserts into the Escherichia coli membrane independent of the Sec translocase and YidC. Eur. J. Biochem. 270: 1724-1734. Feuerbaum, E.-A. 2008. In vivo Lokalisations- und Interaktionsstudien der Sensorkinase KdpD aus Escherichia coli. Dissertation. Universität Osnabrück. Fohrmann, A. 2001. Bedeutung der Transmembrandomänen für die Reizaufnahme durch die Sensorkinase KdpD aus Escherichia coli. Diplomarbeit. Universität Osnabrück. Gaßel, M. 1999. Charakterisierung, Reinigung und Rekonstitution des Kdp-Komplexes aus Escherichia coli unter besonderer Berücksichtigung der Untereinheiten KdpC und KdpF sowie Untersuchungen zur Identifikation der Plekomakrolidbindestelle von P- und V-ATPasen. Dissertation. Universität Osnabrück. Gaßel, M., and K. Altendorf. 2001. Analysis of KdpC of the K(+)-transporting KdpFABC complex of Escherichia coli. Eur. J. Biochem. 268: 1772-1781. Gaßel, M., T. Möllenkamp, W. Puppe, and K. Altendor f. 1999. The KdpF subunit is part of the K+-translocating Kdp complex of Escherichia coli and is responsible for the stabilization of the complex in vitro. J. Biol. Chem. 274: 37901-37907. Gaßel, M., A. Siebers, W. Epstein, and K. Altendorf . 1998. Assembly of the Kdp complex, the multi-subunit K+-transport ATPase of Escherichia coli. Biochim. Biophys. Acta 1415: 77-84. Gowrishankar, J. 1985. Identification of osmoresponsive genes in Escherichia coli: evidence for participation of potassium and proline transport systems in osmoregulation. J. Bacteriol. 164: 434-445. Greie, J.-C., and K. Altendorf. 2007. The K+-translocating KdpFABC complex from Escherichia coli: A P-type ATPase with unique features. J. Bioenerg. Biomembr. 39: 397-402. Gully, D., and E. Bouveret. 2006. A protein network for phospholipid synthesis uncovered by a variant of the tandem affinity purification method in Escherichia coli. Proteomics 6: 282-293. Gunasekera, T. S., L. N. Csonka, and O. Paliy. 2008. Genome-wide transcriptional responses of Escherichia coli K-12 to continuous osmotic and heat stresses. J. Bacteriol. 190: 3712-3720. Guo, D., and B. E. Tropp. 2000. A second Escherichia coli protein with CL synthase activity. Biochim. Biophys. Acta 1483: 263-274.

7. Literaturverzeichnis
103
Hakizimana, P., M. Masureel, B. Gbaguidi, J.-M. Ruy sschaert, and C. Govaerts. 2008. Interactions between phosphatidylethanolamine headgroup and LmrP, a multidrug transporter: A conserved mechanism for proton gradient sensing? J. Biol. Chem. 283: 9369-9376. Hamann, K. 2008. Systematic Analysis of stimulus perception of the sensor kinase KdpD of Escherichia coli. Dissertation. Universität Osnabrück. Hamann, K., P. Zimmann, and K. Altendorf. 2008. Reduction of turgor is not the stimulus for the sensor kinase KdpD of Escherichia coli. J. Bacteriol. 190: 2360-2367. Harold, F., K. Altendorf, and H. Hirata. 1974. Probing membrane transport mechanisms with ionophores. Ann. NY Acad. Sci. 235: 149-160. Heacock P. N., and W. Dowhan. 1987. Construction of a lethal mutation in the synthesis of the major acidic phospholipids of Escherichia coli. J. Biol. Chem. 262: 13044-13049. Heacock, P. N. and W. Dowhan. 1989. Alteration of the phospholipid composition of Escherichia coli through genetic manipulation. J. Biol. Chem. 264: 14972-14977. Heber, S., and B. E. Tropp. 1991. Genetic regulation of cardiolipin synthase in Escherichia coli. Biochim. Biophys. Acta 1129: 1-12. Heermann, R. 2001. Funktionelle und strukturelle Eigenschaften der Sensorkinase KdpD aus Escherichia coli unter besonderer Berücksichtigung der N-terminalen Domäne. Dissertation. Universität Osnabrück. Heermann, R., K. Altendorf, and K. Jung. 1998. The turgor sensor KdpD of Escherichia coli is a homodimer. Biochim. Biophys. Acta 1415: 114-124. Heermann, R., K. Altendorf, and K. Jung. 2003. The N-terminal input domain of the sensor kinase KdpD of Escherichia coli stabilizes the interaction between the cognate response regulator KdpE and the corresponding DNA-binding site. J. Biol. Chem. 278: 51277-51284. Heermann, R., A. Fohrmann, K. Altendorf, and K. Jun g. 2003. The transmembrane domains of the sensor kinase KdpD of Escherichia coli are not essential for sensing K+ limitation. Mol. Microbiol. 47: 839-848. Heermann, R., A. Weber, B. Mayer, M. Ott, E. Hauser , G. Gabriel, T. Pirch, and K. Jung . 2009. The universal stress protein UspC scaffolds the KdpD/KdpE signaling cascade of Escherichia coli under salt stress. J. Mol. Biol. 386: 134-148. Heitkamp, T., R. Kalinowski, B. Böttcher, M. Börsch , K. Altendorf, and J. C. Greie. 2008. K+-translocating KdpFABC P-type ATPase from Escherichia coli acts as a functional and structural dimer. Biochemistry. 47: 3564-3575. Helmer, G. L., L. A. Laimins, and W. Epstein, W. 1982. Mechanisms of potassium transport in bacteria, In: Membranes and transport (Martonosi, A.N., ed.), Plenum Publishing Corp., New York. 2: 123-128. Herzberg, C., L. A. F. Weidinger, B. Dörrbecker, S. Hübner, J. Stülke, and F. M. Commichau . 2007. SPINE: A method for the rapid detection and analysis of protein–protein interactions in vivo. Proteomics. 7: 4032-4035. Hesse, J. E., L. Wieczorek, K. Altendorf, A. S. Rei cin, E. Dorus, and W. Epstein. 1984. Sequence homology between two membrane transport ATPases, the Kdp-ATPase of Escherichia coli and the Ca2+-ATPase of sarcoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 81: 4746-4750. Heuberger, E. H. M. L., E. Smits, and B. Poolman. 2001. Xyloside transport by XylP, a member of the galactoside-pentoside-hexuronide family. J. Biol. Chem. 276: 34465-34472.

7. Literaturverzeichnis
104
Hidaka, Y., H. Park, and M. Inouye. 1997. Demonstration of dimer formation of the cytoplasmatic domain of a transmembrane osmosensor protein, EnvZ, of Escherichia coli using Ni-histidine tag affinity chromatography. FEBS Letts. 400: 238-242. Hirakawa, H., K. Nishino, J. Yamada, T. Hirata, and A. Yamaguchi. 2003a. β-Lactam resistance modulated by the overexpression of response regulators of two-component signal transduction systems in Escherichia coli. J. Antimicrob. Chemother. 52: 576-582. Hirakawa, H., K. Nishino, T. Hirata, and A. Yamaguc hi. 2003b. Comprehensive studies of drug resistance mediated by overexpression of response regulators of two-component signal transduction systems in Escherichia coli. J. Bacteriol. 185: 1851-1856. Hiraoka, S., H. Matsuzaki, and I. Shibuya. 1993. Active increase in cardiolipin synthesis in the stationary growth phase and its physiological significance in Escherichia coli. FEBS Lett. 336: 221-224. Hiraoka, S., K. Nukui, N. Uetake, A. Ohta, and I. S hibuya. 1991. Amplification and substantial purification of cardiolipin synthase of Escherichia coli. J. Biochem. 110: 443-449. Holloway, P. W. 1973. A simple procedure for removal of Triton X-100 from protein samples. Anal. Biochem. 53: 304-308. Huang, K. C., R. Mukhopadhyay, and N. S. Wingreen. 2006. A curvature-mediated mechanism for localization of lipids to bacterial poles. PLOS 2: 1357-1364. Hüging, K. 2006. Charakterisierung von Mutanten im C-terminalen Bereich der Sensorkinase KdpD aus Escherichia coli. Bachelorarbeit. Universität Osnabrück. Ivanisevic, R., M. Milic, D. Ajdic, J. Rakonjac, an d D. Savic. 1995. Nucleotide sequence, mutational analysis, transcriptional start site, and product analysis of nov, the gene which affects Escherichia coli K-12 resistance to the gyrase inhibitor novobiocin. J. Bacteriol. 177: 1766-1771. Jung, K., and K. Altendorf. 1998a. Individual substitutions of clustered arginine residues of the sensor kinase KdpD of Escherichia coli modulate the ratio of kinase to phosphatase activity. J. Biol. Chem. 273: 26415-26420. Jung, K., and K. Altendorf. 1998b. Truncation of amino acids 12-128 causes deregulation of the phosphatase activity of the sensor kinase KdpD of Escherichia coli. J. Biol. Chem. 273: 17406-17410. Jung, K., and K. Altendorf. 2003. Stimulus perception and signal transduction by the KdpD/KdpE system of Escherichia coli, pp. 53-58. In P. Dürre, and B. Friedrich (ed.), Regulatory networks in prokaryotes, Wymondham, GB, Horizon Scientific Press. Jung, K., B. Tjaden, and K. Altendorf. 1997. Purification, reconstitution and characterization of KdpD, the turgor sensor of Escherichia coli. J. Biol. Chem. 272: 10847-10852. Jung, K., M. Veen, and K. Altendorf. 2000. K+ and ionic strength directly influence the autophosphorylation activity of the putative turgor sensor KdpD of Escherichia coli. J. Biol. Chem. 275: 40142-40147. Jung, K., R. Heermann, M. Meyer, and K. Altendorf. 1998. Effect of cysteine replacements on the properties of the turgor sensor KdpD of Escherichia coli, Biochim. Biophys. Acta 1372: 311-322. Laimins, L. A., D. B. Rhoads, and W. Epstein. 1981. Osmotic control of kdp operon expression in Escherichia coli. Proc. Natl. Acad. Sci. USA 78: 464-468. Lucassen, M. 1998. Regulation des Kdp-Systems aus Escherichia coli: Biochemische Charakterisierung des Antwortregulators KdpE und Nachweis von Konformationsänderungen im Zuge der Aktivierung. Dissertation. Universität Osnabrück. Lugtenberg, E. J. J., and R. Peters. 1976. Distribution of lipids in cytoplasmic and outer membranes of Escherichia coli K12. Biochim. Biophys. Acta 441: 38-47.

7. Literaturverzeichnis
105
Kanemasa, Y., Y. Akamatsu, and S. Nojima. 1967. Composition and turnover of the phospholipids in Escherichia coli. Biochim. Biophys. Acta. 144: 382-390. Karimova, G., J. Pidoux, A. Ullmann, and D. Ladant. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. USA 95: 5752-5756. Knol, J., K. Sjollema, and B. Poolman. 1998. Detergent-mediated reconstitution of membrane proteins. Biochemistry 37: 16410-16415. Kollmann, R., and K. Altendorf. 1993. ATP-driven potassium transport in right-side-out vesicles via the Kdp system of Escherichia coli. Biochim. Biophys. Acta 1143: 62-66. Krzeslak, J., G. Gerritse, R. van Merkerk, R. H. Co ol, and W. J. Quax. 2008. Lipase expression in Pseudomonas alcaligenes is under the control of a two-component regulatory system. Appl. Environ. Microbiol. 74: 1402-1411. Kuo, M. M., Y. Saimi, and C. Kung. 2003. Gain-of-function mutations indicate that Escherichia coli Kch forms a functional K+ conduit in vivo. EMBO J. 22: 4049-4058. Kusters, R., W. Dowhan, and B. de Kruijff. 1991. Negatively charged phospholipids restore prePhoE translocation across phosphatidylglycerol-depleted Escherichia coli inner membranes. J. Biol. Chem. 266: 8659-8662. Kwa, L. G., D. Wegmann, B. Brügger, F. T. Wieland, G. Wanner, and P. Braun. 2008. Mutation of a single residue, β-glutamate-20, alters protein-lipid interactions of light harvesting complex II. Mol. Microbiol. 67: 63-77. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685. Lai, E.-M., U. Nair, N. D. Phadke, and J. R. Maddoc k. 2004. Proteomic screening and identification of differentially distribution membrane proteins in Escherichia coli. Mol. Microbiol. 52: 1029-1044. Laub, M. T., and M. Goulian. 2007. Specificity in two-component signal transduction pathways. Annu. Rev. Genet. 41: 121-145. Lipski, A., and K. Altendorf. 1997. Identification of heterotrophic bacteria isolated from ammonia-supplied experimental biofilters. System. Appl. Microbiol. 20: 448–457. Ma D., D. N. Cook, M. Alberti, N. G. Pon, H. Nikaid o, and J. E. Hearst. 1995. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol. Microbiol. 16:45-55. Maier, K. S., S. Hubich, H. Liebhart, S. Krauss, A. Kuhn, and S. J. Facey. 2008. An amphiphilic region in the cytoplasmic domain of KdpD is recognized by the signal recognition particle and targeted to the Escherichia coli membrane. Mol. Microbiol. 68: 1471-1484. Malli, R. and W. Epstein. 1998. Expression of the Kdp ATPase is consistent with regulation by turgor. J. Bacteriol. 180: 5102-5108. Maniatis, T.; E. F. Fritsch, and J. Sambrook. 1989. Molecular Cloning. A laboratory manual. Cold Spring Harbor Laboratory Press. Cold Spring Harbor. New York. Mascher, T., J. D. Helmann, and G. Unden. 2006. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol. Mol. Biol. Rev. 70: 910-938. Matsumoto, K., J. Kusaka, A. Nishibori, and H. Hara . 2006. Lipid domains in bacterial membranes. Mol. Microbiol. 61: 1110-1117. Matsushita, M., and K. D. Janda. 2002. Histidine kinases as targets for new antimicrobial agents. Bioorg. Med. Chem. 10: 855-867.

7. Literaturverzeichnis
106
Mileykovskaya, E. and W. Dowhan. 2000. Visualization of phospholipid domains in Escherichia coli by using the Cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J. Bacteriol. 182: 1172-1175. Mileykovskaya, E., and W. Dowhan. 1997. The Cpx two-component signal transduction pathway is activated in Escherichia coli mutant strains lacking phosphatidylethanolamine. J. Bacteriol. 179: 1029-1034. Mileykovskaya, E., A. C. Ryan, X. Mo, C.-C. Lin, K. I. Khalaf, W. Dowhan, and T. A. Garrett. 2009. Phosphatidic acid and N-acylphosphatidylethanolamine form membrane domains in Escherichia coli mutant lacking cardiolipin and phosphatidylglycerol. J. Bio. Chem. 284: 2990-3000. Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Habor Laboratory Press. Cold Spring Habor. New York. Miroux, B., and J. E. Walker. 1996. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Biol. Mol. 260: 289-298. Nakashima, K., A. Sugiura, K. Kanamaru, and T. Mizu no. 1993. Signal transduction between the two regulatory components involved in the regulation of the kdpABC operon in Escherichia coli: Phosphorylation-dependent functioning of the positive regulator, KdpE. Mol. Microbiol. 7: 109-116. Nakashima, K., A. Sugiura, H. Momoi, and T. Mizuno. 1992. Phosphotransfer signal transduction between two regulatory factors involved in the osmoregulated kdp operon in Escherichia coli. Mol. Microbiol. 6: 1777-1784. Nesmeyanova, M. A., I. M. Tsfasman, A. L. Karamyshe v, and N. E. Suzina. 1991. Secretion of the overproduced periplasmic PhoA protein into the medium and accumulation of its precursor in phoA-transformed Escherichia coli strains: involvement of outer membrane vesicles. World J. Microbiol. Biotechnol. 7: 394-406. Ninfa, E. G., M. R. Atkinson, E. S. Kamberov, and A . J. Ninfa. 1993. Mechanism of autophosphorylation of Escherichia coli nitrogen regulator II (NRII or NtrB): trans-phosphorylation between subunits. J. Bacteriol. 175: 7024-7032. Nishijima, S., Y. Asami, N. Uetake, S. Yamagoe, A. Ohta, and I. Shibuya. 1988. Disruption of the Escherichia coli cls gene responsible for cardiolipin synthesis. J. Bacteriol. 170: 775-780. Osborn, M. J., J. E. Gander, E. Parisi, and J. Cars on. 1972. Mechanism of assembly of the outer membrane of Salmonella typhimurium. Isolation and characterization of cytoplasmic and outer membrane. J. Biol. Chem. 247: 3962-3972. Özcan, N., C. S. Ejsing, A. Shevchenko, A. Lipski, S. Morbach, and R. Krämer. 2007. Osmolality, temperature, and membrane lipid composition modulate the activity of betaine transporter BetP in Corynebacterium glutamicum. J. Bacteriol. 189: 7485-7496. Parkinson, J. S., and E. C. Kofoid. 1992. Communication modules in bacterial signaling proteins. Annu. Rev. Genet. 26: 71-112. Parkinson, J. S. 1993. Signal tranduction schemes of bacteria. Cell 73: 857-871. Parkinson, J. S. 1995. Genetic approaches for signaling pathways and proteins, in: Two-component signal transduction (Ed. Hoch, J. A. & Silhavy, T. J.), 9-23, American Society for Microbiology, Washington, D. C. Peterson, G. L. 1977. A simplification of the protein assay method of Lowry et al., which is more generally applicable. Anal. Biochem. 83: 346-356. Polarek, J. W., G. Williams, and W. Epstein. 1992. The products of kdpDE operon are required for expression of the Kdp-ATPase of Escherichia coli. J. Bacteriol. 174: 2145-2151.

7. Literaturverzeichnis
107
Polarek, J. W., M. O. Walderhaug, and W. Epstein. 1988. Genetics of Kdp, the K+ transport ATPase of Escherichia coli. Methods Enzymol. 157: 655-667. Promega. 1994. Technical manual. Altered sites II in vitro mutagenesis systems. Ptak, C. P., L. G. Cuello, and E. Perozo. 2005. Electrostatic interaction of a K+ channel RCK domain with charged membrane surfaces. Biochemistry 44: 62-71. Puppe, W., P. Zimmann, K. Jung, M. Lucassen, and K. Altendorf. 1996. Characterization of truncated forms of the KdpD protein, the sensor kinase of the K+-translocating Kdp system of Escherichia coli. J. Biol. Chem. 271: 25027-25034. Quigley, B. 2007. The N-terminal sequence of Escherichia coli CL synthase: function(s) of the conserved residues. Dissertation. City University of New York. Raetz, C. R., and W. Dowhan . 1990. Biosynthesis and function of phospholipids in Escherichia coli. J. Biol. Chem. 265: 1235-1238. Ragolia, L. and B. E. Tropp. 1994. The effects of phosphoglycerides on Escherichia coli cardiolipin synthase. Biochim. Biophys. Acta 1214: 323-332. Ramamurthi, K. S., S. Lecuyer, H. A. Stone, and R. Losick. 2009. Geometric cue for protein localization in a bacterium. Science 323: 1354-1357. Randle, C. L., P. W. Albro, and J. C. Dittmer. 1969. The phosphoglyceride composition of Gram-negative bacteria and the changes in composition during growth. Biochim. Biophys. Acta 187: 214-220. Reitzer, L. and B. L. Schneider. 2001. Metabolic context and possible physiological themes of sigma54-dependent genes in Escherichia coli. Microbiol. Mol. Biol. Rev. 65: 422-444. Romantsov, T., S. Helbig, D. E. Culham, C. Gill, L. Stalker, and J. M. Wood. 2007. Cardiolipin promotes polar localization of osmosensory transporter ProP in Escherichia coli. Mol. Microbiol. 64: 1455-1465. Romantsov, T., L. Stalker, D. E. Culham, and J. M. Wood. 2008. Cardiolipin controls the osmotic stress response and the subcellular location of transporter ProP in Escherichia coli. J. Biol. Chem. 283: 12314-12323. Rothenbücher, M. C., S. J. Facey, D. Kiefer, M. Kos smann, and A. Kuhn. 2006. The cytoplasmic C-terminal domain of the Escherichia coli KdpD protein funtions as a K+ sensor. J. Bacteriol. 188: 1950-1958. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA Sequencing with chain-terminating inhibitors. Proc. Nat. Acad. Sci. USA 74: 5463-5467. Sasser, M. 1990. Identification of bacteria through fatty acid analysis. In Methods in Phytobacteriology ed. Klement, Z., Rudolph, K. and Sands, D.C. pp. 199–204. Budapest : Akademiai Kiado. Schiller, D., V. Ott, R. Krämer, and S. Morbach. 2006. Influence of membrane composition on osmosensing by the betaine carrier BetB from Corynebacterium glutamicum. J. Biol. Chem. 281: 7737-7746. Schleußinger, E., R. Schmid, and E. P. Bakker. 2006. New type of kdp region with a split sensor-kinase kdpD gene located within two divergent kdp operons from the thermoacidophilic bacterium Alicyclobacillus acidocaldarius. Biochim. Biophys. Acta 1759: 437-441. Schmidt, D., Q.-X. Jiang, and R. MacKinnon. 2006. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature 444: 775-779. Shiba, Y., Y. Yokoyama, Y. Aono, T. Kiuchi, J. Kusa ka, K. Matsumoto, and H. Hara. 2004. Activation of the Rcs signal transduction system is responsible for the thermosensitive growth defect

7. Literaturverzeichnis
108
of an Escherichia coli mutant lacking phosphatidyl-glycerol and cardiolipin. J. Bacteriol. 186: 6526-6535. Siebers, A., and K. Altendorf. 1988. The K+-translocating Kdp-ATPase from Escherichia coli. Purification, enzymatic properties and production of complex- and subunit-specific antisera. Eur. J. Biochem. 178:131-140. Siebers, A., and K. Altendorf. 1989. Characterization of the phosphorylated intermediate of the K+-translocating Kdp-ATPase from Escherichia coli. J. Biol. Chem. 264: 5831-5838. Siebers, A., and K. Altendorf. 1993. K+-translocating Kdp-ATPases and other bacterial P-type ATPases. In: Alkali cation transport systems in prokaryotes (Bakker, E.P., ed.), pp. 225-252, CRC Press, Boca Raton, Florida. Siegele, D. A. 2005. Universal stress proteins in Escherichia coli. J. Bacteriol. 187: 6253-6254. Silva, J. C.; A. Haldimann, M. K. Prahalad, C. T. W alsh, and B. L. Wanner . 1998. In vivo characterization of the type A and B vancomycin-resistant enterococci (VRE) VanRS two-component systems in Escherichia coli: A nonpathogenic model for studying the VRE signal transduction pathways. Proc. Natl. Acad. Sci. 95: 11951-11956. Stallkamp, I., W. Dowhan, K. Altendorf, and K. Jung . 1999. Negatively charged phospholipids influence the activity of the sensor kinase KdpD of Escherichia coli. Arch. Microbiol. 172: 295-302. Steyn, A. J., J. Joseph, and B. R. Bloom. 2003. Interaction of the sensor module of Mycobacterium tuberculosis H37Rv KdpD with members of the Lpr family. Mol. Microbiol. 47:1075-1089. Stock, J. B., A. J. Ninfa, and A. M. Stock. 1989. Protein phosphorylation and regulation of adaptive response in bacteria. Microbiol. Rev. 53: 789-795. Stock, A. M., V. L. Robinson, and P. N. Goudreau. 2000. Two-component signal transduction. Annu. Rev. Biochem. 69: 183-215. Stock, J. B., A. M. Stock, and J. M. Mottonen. 1990. Signal transduction in bacteria. Nature 344: 395-400. Strahl, H. and J. C. Greie. 2008. The extremely halophilic archaeon Halobacterium salinarum R1 responds to potassium limitation by expression of the K+-transporting KdpFABC P-type ATPase and by a decrease in intracellular K+. Extremophiles 12: 741-751. Sugiura, A., K. Nakashima, K. Tanaka, and T. Mizuno . 1992. Clarification of the structural and functional features of the osmoregulated kdp operon of Escherichia coli. Mol. Microbiol. 6:1769-1776. Suelter, C. H. 1970. Enzymes activated by monovalent cations. Science 168: 789-795. Sugiura, A., K. Hirokawa, K. Nakashima, and T. Mizu no. 1994. Signal-sensing mechanisms of the putative osmosensor KdpD in Escherichia coli. Mol. Microbiol. 14:929-938. Swanson, R. V., L. A. Alex, and M. I. Simon. 1994. Histidine and aspartate phosphorylation: two- component systems and the limits of homology. TIBS 19: 485-490. Swanson, R. V., R. B. Bourret, and M. I. Simon . 1993. Intermolecular complementation of the kinase activity of CheA. Mol. Microbiol. 8: 435-441. Tanaka, T., S. K. Saha, R. I. Tomomori, R. Ishima, D. Liu, K. I. Tong, H. Park, R. Dutta, L. Qin, M. B. Swindells, T. Yamazaki, A. M. Ono, M. Kainosho, M. Inouye, and M. Ikura. 1998. NMR structure of the histidine kinase domain of the E. coli osmosensor EnvZ. Nature 396: 88-92. Tatum, E. L. 1945. X-ray induced mutant strains of Escherichia coli. PNAS USA 31:215-219. Tropp, B. E. 1997. Cardiolipin synthase from Escherichia coli. Biochim. Biophys. Acta 1348:192-200.

7. Literaturverzeichnis
109
Tropp, B. E., L. Ragolia, W. Xia, W. Dowhan, R. Mil kman, K. E. Rudd, R. Ivanisevic, and D. J. Savic. 1995. Identity of the Escherichia coli cls and nov genes. J. Bacteriol. 177: 5155–5157. Tsatskis, Y., J. Khambati, M. Dobson, M. Bogdanov, W. Dowhan, and J. M. Wood. 2005. The osmotic activation of transporter ProP is tuned by both its C-terminal coiled-coil and osmotically induced changes in phospholipid composition. J. Biol. Chem. 280:41387-41394. Tsatskis, Y., S. C. Kwok, E. Becker, C. Gill, M. N. Smith, R. A. B. Keates, R. S. Hodges, and J. M .Wood. 2008. Core residue replacements cause coiled-coiled orientation switching in vitro and in vivo: structure-function correlations for osmosensory transporter ProP. Biochem. 47: 60-72. Van Bogelen, R. A., M E. Hutton, and F. C. Neidhard t. 1990. Gene-protein database of Escherichia coli K-12. edition 3. Electrophoresis 11: 1131-1166. Van den Brink-Van der Laan, E., J.-W. Boots, R. E. Spelbrink, G. M. Kool, E. Breukink, J. A. Killian, and B. de Kruijff. 2003. Membrane interaction of the glycosyltransferase MurG: a special role for cardiolipin. J. Bacteriol. 185: 3773-3779. Verhamme, D. T., J. C. Arents, P. W. Postma, W. Cri elaard, and K. J. Hellingwerf. 2001. Glucose-6-phosphate-dependet phosphoryl flow through the Uhp two-component regulatory system. Microbiology. 147: 3345-3352. Voelkner, P. 1991. Molekularbiologische und biochemische Untersuchungen der Regulationsproteine KdpD und KdpE des Kdp-Systems von Escherichia coli, Diplomarbeit, Universität Osnabrück. Voelkner, P., W. Puppe, and K. Altendorf. 1993. Characterisation of the KdpD protein, the sensor kinase of the K+-translocating system of Escherichia coli. Eur. J. Biochem. 217: 1019-1026. von Ballmoos, C., Y. Appoldt, J. Brunner, T. Granie r, A. Vasella, and P. Dimroth. 2002. Membrane topography of the coupling ion binding site in Na+-translocating F1Fo ATP synthase. J. Biol. Chem. 277: 3504-3510. Von Heijne, G. 1986. The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. EMBO J. 5: 3021-3027. Walderhaug, M. O., J. W. Polarek, P. Voelkner, J. M . Daniel, J. E. Hesse, K. Altendorf, and W. Epstein. 1992. KdpD and KdpE, proteins that control expression of the kdpABC operon, are members of the two-component sensor-effector class of regulators. J. Bacteriol. 174: 2152-2159. Weber, K., and M. Osborn. 1969. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 244: 4406-4412. West, A. H.; and A. M. Stock. 2001. Histidine kinases and response regulator proteins in two-component signalling systems. Trends Biochem. Sci. 26: 369-376. Wikström, M., A. A. Kelly, A. Georgiev, H. M. Eriks son, M. R. Klement, M. Bogdanov, W. Dowhan, and A. Wieslander. 2009. Lipid-engineered Escherichia coli membranes reveal critical lipid headgroup size for protein function. J. Biol. Chem. 284:954-965. Wikström, M., J. Xie, M. Bogdanov, E. Mileykovskaya , P. Heacock, A. Wieslander, and W. Dowhan. 2004. Monoglucosyldiacylglycerol, a foreign lipid, can substitute for phosphatidylethanolamine in essential membrane-associated functions in Escherichia coli. J. Biol. Chem. 279:10484-10493. Xie, J., M. Bogdanov, P. N. Heacock, and W. Dowhan. 2006. Phosphatidylethanolamine and monoglucosyldiacylglycerol are interchangeable in supporting topogenesis and function of the polytopic membrane protein lactose permease. J. Biol. Chem. 281: 19172-19178. Yamamoto, K., K. Hirao, T. Oshima, H. Aiba, R. Utsu mi, and A. Ishihama. 2005. Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J. Biol. Chem. 280:1448-1456.

7. Literaturverzeichnis
110
Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13m18 and pUC19 vectors. Gene 33: 103-119. Zhou, L., X.-H. Lei, B. R. Bochner, and B. L. Wanne r. 2003. Phenotype microarray analysis of Escherichia coli K-12 mutants with deletions of all two-component systems. J. Bacteriol. 185: 4956-4972. Zimmann, P. 1995. Molekulare Analyse des Regulationsproteins KdpD des Kdp-Systems von Escherichia coli. Dissertation. Universität Osnabrück. Zimmann, P., A. Steinbrügge, M. Schniederberend, K. Jung, and K. Altendorf. 2007. The extension of the fourth transmembrane helix of the sensor kinase KdpD of Escherichia coli is involved in sensing. J. Bacteriol. 189:7326-7334. Zimmann, P., W. Puppe, and K. Altendorf. 1995. Membrane topology analysis of the sensor kinase KdpD of Escherichia coli. J. Biol. Chem. 270:28282-28288.

Eidesstattliche Erklärung
111
Eidesstattliche Erklärung Ich erkläre hiermit, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und
ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Die aus
anderen Quellen direkt oder indirekt übernommenen Daten und Konzepte sind unter
Angabe der Quelle gekennzeichnet. Bei der Auswahl und Auswertung folgenden
Materials haben mir die nachstehend aufgeführten Personen in der jeweils beschrie-
benen Weise entgeltlich / unentgeltlich geholfen. Weitere Personen waren an der
inhaltlichen materiellen Erstellung der vorliegenden Arbeit nicht beteiligt.
Insbesondere habe ich hierfür nicht die entgeltliche Hilfe von Vermittlungs- bzw.
Beratungsdiensten (Promotionsberater oder andere Personen) in Anspruch ge-
nommen. Niemand hat von mir unmittelbar oder mittelbar geldwerte Leistungen für
Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation
stehen. Die Arbeit wurde bisher weder im In- noch im Ausland in gleicher oder
ähnlicher Form einer anderen Prüfungsbehörde vorgelegt.
................................................... ................................................................. (Ort, Datum) (Unterschrift)





























