Embed Size (px)
Citation preview

Z. Anal. Chem. 284, 177-187 (1977) - �9 by Springer-Verlag 1977
Einsatz der ICP-Emissionsspektrometrie zur simultanen Multielementanalyse*
K. Ohls**, K. H. Koch und H. Grote
Hoesch Hfittenwerke AG, Postfach 902, D-4600 Dortmund 1
Eingegangen am 14. August 1976
Application of ICP Emission Spectrometry to Simultaneous Multielement Analysis. The inductively coupled plasma emission spectrometry is becoming a very important analytical tool in routine laboratories today. The application of ICP emission spectrometry in comparison to the analytical needs is described. The simultaneous determination of A1, B, Cr, Cu and Mn in waste water or steel solution is taken for example. The real limits of determination for several elements from steel samples depending on the applied ICP/spectrometer device and the working conditions are given. The most important parameters like tuning between generator, matchbox and coil, the kind of nebulizer, the sample flow gas rate and the observation height in the plasma are discussed.
Zusammenfassung. Die induktiv gekoppelte Plasma-Emissionsspektrometrie wird gegenw/irtig zu einem wichtigen analytischen Werkzeug in Routinelaboratorien. Der Einsatz der ICP-Emissionsspektrometrie wird an den analy- tischen Anforderungen gemessen. Die simultane Bestimmung yon A1, B, Cr, Cu und Mn in Abwasser oder L6sungen aus Stahlproben wird als Beispiel herangezogen. Die reellen Bestimmungsgrenzen ffir zahlreiche Ele- mente aus Stahl, die yon dem eingesetzten Apparat und den Arbeitsbedingungen abhfingen, werden mitgeteilt. Die wichtigsten Parameter wie Abstimmung zwischen Generator, Impedanzwandler und Spule, die Art des Zer- stfiubers, die Proben-Aerosolgasmenge und die Beobachtungsh6he im Plasma werden diskutiert.
Analyse yon Wasser, Stahl; Spektrometrie, ICP; Optimierung, Multielementanalyse.
1. Einleitung
Die quantitative Simultanbestimmung yon zahlrei- chen Elementen aus einer Probe ist ffir feste Stoffe ein gel6stes analytisches Problem. Dies geschieht mit Hilfe der R6ntgenfluorescenz- und Emissionsspektrometrie und war z.B. ftir den Bereich der Metallindustrie durch die Erm6glichung schneller Produktionsabl/iufe von grol3er wirtschaftlicher Bedeutung.
Die quantitative Simultanbestimmung yon zahl- reichen Elementen aus einer L6sung ist ein wesent- liches analytisches Problem, das bisher nur teilweise gel6st werden konnte. Neben der RFA yon L6sungen, die bisher nur auf bestimmte Ger~ite beschr/inkt blieb, ist wegen der erforderlichen Information die L6sungsemissionsspektralanalyse die Methode der Wahl. So hat es in den letzten Jahren zahlreiche Be-
* Auszugsweise vorgetragen auf dem First European ICP-Symposium Mfinchen, 3.-4.6. 1976.
** Korrespondenz-Anschrift.
schreibungen von Plasmen und speziellen Anregungs- techniken gegeben, deren Ziel die effektive Anregung (Ionisierung) yon Elementen (Atomen) aus L6sungen war. Prinzipiell lassen sich die bekanntesten Techniken in 4 Gruppen unterteilen, die alle gemeinsam von einer Probenl6sung ausgehen:
1. L6sung kommt mit den Elektroden in Kontakt, z.B. Eindampfen auf der Elektrode, Sprfihen durch eine Elektrode mit Bohrung oder die Radelektroden- technik.
2. L6sung ohne Kontakt mit den Elektroden, z.B. Zerstfiuben in einen Bogen.
3. Pr/iparation der L6sung auf Filterpapier und Analyse mit Hilfe der RFA oder Laser-Emissions- spektrographie.
4. Direktes Messen der L6sung, z.B. mit Hilfe der RFA.
Mit Ausnahme der RFA und der Kombination Radelektrode/Emissionsspektrometer handelt es sich dabei um spektrographische Techniken, die sehr arbeitsaufwendig sind.

178 Z. Anal. Chem., Band 284 (1977)
Spectrometer
I I,c~ I ~ A r
Aerosolgas
Abb. 1. Schema der Versuchsanordnung
Generator
27MHz
2j 5 kw
Der grundlegende Unterschied im Vergleic'h zur Analyse fester Stoffe ist jedoch die Forderung, dab die v o n d e r L6sung ausgehenden Methoden in der
R e g e l um den Faktor 102 besser in bezug auf die Bestimmungsgrenze sein miissen. Es ist noch darauf einzugehen, dal3 der durch das L6sen entstehende Verdfinnungsfaktor ausgeglichen werden muB.
Somit ist die bisher erfolgreiche Entwicklung der von Greenfield [9] und Fassel [7] beschriebenen, induktiv gekoppelten Plasmaflamme als Anregung von L6sungen in Verbindung mit einem leistungs- f~ihigen Spektrometer erkl/irlich. Durch die Kombina- tion ICP/Spektrometer/Datenverarbeitungssystem er- gaben sich extrem kleine Nachweisgrenzen, die zu der Hoffnung berechtigten, in praktisch allen Gehalts- bereichen analysieren zu k6nnen.
Die gegenw~irtige Aufgabe besteht nun darin, das prinzipiell beschriebene System der ICP-Anregung so zu stabilisieren, dab es sich ffir die Routineanalyse eignet.
Die Aufgaben in grogen Routinelaboratorien sind in bezug auf die L6sungsanalyse von zweifacher Art; es sind entweder Proben zu untersuchen, die in flfissiger Form angeliefert werden, wie z.B. Wasser, Abwasser, Beizlaugen, Absorptionsl6sungen u .a . oder feste Stoffe, die in L6sung gebracht werden mfissen. Die Anzahl dieser Proben kann eine betrfichtliche H6he erreichen, wenn man bedenkt, dab ein Teil der Proben, die fiber die RFA und Emissionsspektrometrie laufen, zu Kontrollzwecken bzw. zur Sicherstellung der Rich- tigkeit chemisch analysiert werden mug.
In praktisch allen Proben sind mehrere Elemente nebeneinander zu bestimmen. Insofern stellte der Ein- satz der Atomabsorptionsspektrometrie vor einigen Jahren zwar gegenfiber der Photometrie und besonders der Volumetrie oder Gravimetrie einen erheblichen Fortschritt in bezug auf den Zeitaufwand dar, war aber eben nicht die Optimall6sung. Der zu erwartende
rationelle Effekt der simultanen Multielementanalyse sollte zu einer schnellen Verbreitung der Anwendung der ICP-Emissionsspektrometrie ffihren. Hier sei auf die ausffihrliche Literaturbetrachtung durch Boumans [4] verwiesen. Wesentlich ist dabei auch, dab sich bestimmte Elementkombinationen, die bisher zu er- heblichen analytischen Schwierigkeiten ffihrten, wie z.B. Nb/Ta, Ce/La, Se/Te oder B/A1, problemlos ermitteln lassen.
2. Prinzip der ICP-Emissionsspektrometrie
Das gesamte Apparatesystem, das zur simultanen Multielementanalyse ben6tigt wird, besteht prinzipiell aus einer Generator/Brennerkombination, einem Mehrkanal-Emissionsspektrometer und einem Rech- ner zur Steuerung der Parametereinstellung und Da- tenauswertung. Dies ist schematisch in Abbildung 1 dargestellt.
Wenn z. B. Argon in einem Rohr, auf das durch eine Spule induktiv eine bestimmte HF-Leistung fibertragen wird und an dem ein definiertes Magnetfeld existiert, geziindet wird, so bleibt durch fortw/ihrende Ionisation eine Plasmaflamme brennen. Durch ein bestimmtes Rohrsystem, das in der Regel aus einem fiul3eren Rohr, einem inneren Rohr und einer zentrisch angeordneten Capillare besteht, und die Kopplung mit einer bestimmten HF-Leistung fiber eine Spule bei einem konstanten Magnetfeld lfil3t sich ein stabiles, ftir die L6sungs- anregung geeignetes Plasma erzeugen. Das Charakteristische der ICP ist die spezielle Anderung der Flammenform, wenn durch die zentrische Capillare ein Gasstrom eingeblasen wird. Es bildet sich ein durchgehendes Loch im Plasma; in der N0.he der Capillaren6ffnung entsteht eine Herz- oder Pfann- kuchenform.
Den Gasstrom im/iul3eren Rohr nennt man Kfihlgas, den- jenigen im inneren Rohr Brenngas, entsprechend den ursprfing- lichen Aufgaben. Damit die Wirkung des Ktihlgases erh6ht wird, ist das innere Rohr oben tulpenf6rmig ausgebildet, so dab ein 1 mm weiter Ringschlitz entsteht, in dem der Gas- strom beschleunigt wird.
Leitet man durch die Capillare einen Aerosolgasstrom, so wird in Abhfingigkeit yon der Gasgeschwindigkeit eine relativ lange Verweilzeit der Aerosolpartikel im Plasma erreicht. Durch die Form des Plasmas ist das Einbringen bzw. Ein- dringen des Aerosolgases gewfihrleistet.
Die Erzeugung des Aerosols und die Trfigergas-Durch- flul3menge (sample flow gas) sind yon besonderer Bedeutung.
Fiir die Leistungserzeugung und die Kopplung sind unter- schiedliche Systeme beschrieben worden. Die fibertragenen Leistungen schwanken zwischen einigen Hundert Watt und mehr als 10 kW.
Der Frequenzbereich der eingesetzten Generatoren liegt zwischen 6 und 55 MHz.
Inzwischen haben sich zwei unterschiedliche Ar- beitsweisen herauskristallisiert:
A. das Arbeiten mit konstanter Frequenz (27,12 MHz) und variabler Leistung (600-2000 W),
B. das Arbeiten mit stabilisierter Leistung (z.B. 1000 W) und freilaufender Frequenz (50-55 MHz).

K. Ohls et al. : ICP-Emissionsspektrometrie zur Multielementanalyse 179
Zwei weitere Varianten ergeben sich durch die Verwendung von Argon als Kfihlgas und als Brenngas oder nur als Brenn- gas mit Stickstoff als Kfihlgas. Der letztere Fall erfordert eine wesentlich h6here Leistungsfibertragung, weil durch die Ionisation von N2 ein erheblicher Anteil der Leistung ver- braucht wird.
Obwohl mit derartigen Hochleistungsplasmen die lfingsten praktischen Erfahrungen vorliegen (Green- field, England, und Watson, Sfidafrika), haben wir uns bisher nur mit dem yon Fassel u. a. [8] beschriebe- nen Ar-/Ar-Plasma beschfiftigt. Die bier beschriebe- nen Ergebnisse wurden mit einem Generator mit kristallstabilisierter Frequenz (27,12 MHz) bei 1,5 KW Ausgangsleistung erarbeitet.
Kfihlgas und Brenngas sind vereinigt, weil das Kfhlgas (10,81 Ar/min) auch ffir die Unterhaltung des Plasmas ausreicht. Der eingesetzte Brenner (torch) bzw. die Dimensionen des Quarzrohrsystems wurden nicht ver/indert. Wir haben lediglich eine justierbare Haltevorrichtung nach einer Idee von Souilliart u. Robin [13] gebaut, so dab Einzelrohre mit einer Capil- lare kombiniert werden k6nnen und ihr Auswechseln erleichtert wird (Abb. 2).
Die Zerstfiuberkammer wurde von Scott [12] fiber- nommen, weil wir meinen, dab sie ftir die pneu- matische Zerstfiubung eine optimale Gr613e hat. Die wesentliche Anderung liegt in der Verwendung des seit Jahren in der Flammenphotometrie bew/ihrten Zerst/iubers yon Eppendorf (Abb.2). Dieser Zer- stfiuber erfordert einen relativ hohen Vordruck (1,5 1 Ar/min), ist jedoch in der Lage, zeitkonstant ein sehr feines Aerosol zu liefern. Der L6sungsverbrauch liegt bei etwa 1 ml/min (sample flow), wovon etwa 0,005- 0,01 ml/min (0,5 - 1 ~) tatsfichlich in die Flam- me gelangen.
Der grol3e Einflul3 des Aerosols auf die spektrale Intensit/it und die Abhfingigkeit des Streulichtanteiles vom Zerstfiubergasdruck sowie die zu beachtende An- saugh6he und eine Diskussion des Aerosoltransportes sind von Kranz [10] ausf/ihrlich beschrieben worden.
Es sind auch bereits Untersuchungen fiber die An- regungstemperaturen, die Gasgeschwindigkeitsprofile oder die Partikelverteilung im Plasma ver6ffentlicht worden, wobei speziell auf die theoretischen Arbeiten yon Barnes [1] zu verweisen ist. Es ist anzunehmen, dab unter den bier gew/ihlten Bedingungen eine Anregungs- temperatur von etwa 8000 K erreicht wird.
Ffir den Anwender ergibt sich aus diesen Unter- suchungen der wesentliche Hinweis, dab ftir alle Atome in bezug auf ihre Ionisation eine optimale H6he im Plasma existiert, so dab der Beobachtungsh6he (Licht- abnahme zum Spektrometer) eine besondere Bedeu- tung zukommt.
F f r die Wahl des optischen Indikationssystems gibt es verschiedene Uberlegungen. Generell 1/il3t sich bis
Ar
Probe Kclmmer
[CP Brenner
notch Scott ZerstSuber Eppendorf Abb. 2. Brenner und Zerstfiubersystem
heute der Prozel3 im Plasma noch nicht beschreiben, so dab sich auch nicht voraussagen lfil3t, welche Spektral- linien geeignet oder sogar st6rungsfrei sind. Dies mul3 experimentell ermittelt werden. Allgemein lfil3t sich bisher nur sagen, dab Atomlinien mit zunehmendem Ionisierungspotential an Intensitfit verlieren, wfihrend diejenige von Ionenlinien langsam ansteigt.
Dadurch wird das Einsetzen von Spektrometern mit festinstallierten Spalten und Detektoren nicht sofort ffr alle Probleme ideal sein. Eine volle Aus- nutzung der ICP-Anregung ist jedoch nur dann gege- ben, wenn hochleistungsf~ihige Spektrometer einge- setzt werden. Dies hat seinen Grund in der Umsetzung sehr geringer Absolutmengen, denn man sollte wegen der Konstanz des Zerst/iubers nur mit maximal 1 ~igen L6sungen arbeiten, d.h. 10 mg/ml. Ffir das Beispiel einer L6sung aus Reineisen mit 0,001 ~ Cr wfirde das bedeuten, dab 0,1 ~tg Cr/ml enthalten ist, wovon etwa 1 ng in das Plasma gelangen. Das Spektro- meter mul3 also m6glichst wenig Lichtverlust garan- tieren und mit allen M6glichkeiten einer genauen Verst/irkung der erzielbaren Spannung ausgestattet sein.
Weitere Anforderungen an das Gerfit sind die Ver- wendung eines holographischen Gitters bzw. eines Gitters mit hoher Strichanzahl in einer Anordnung, die nur geringes Streulicht erzeugt, die Brennweite von minimal I m und eine optimale Dispersion. Das Aufl6sungsverm6gen sollte 0,02 nm nicht fiberschrei-

180 Z. Anal. Chem., Band 284 (1977)
ten, weil praktisch alle Spektrallinien angeregt werden - also in der Regel linienre{che Spektren vorliegen - und ein Zusammenhang zwischen Aufl6sungsverm6- gen und Nachweisgrenze besteht.
Die Fokussierung des emittierten Lichtes auf den Eintrittsspalt kann zu Schwierigkeiten ffihren. Es wurde gemessen, daB Beeinflussungen relativ grog sein k6nnen und sich wfihrend einer Megperiode findern. Das Arbeiten mit direkter Abbildung bzw. Ausleuch- ten des gesamten Gitters hat sich bew/ihrt, die erreich- baren Intensit/iten werden gr6ger. Hier ist der generelle Unterschied zwischen der Routineanalytik und dem Auffinden der besten Nachweisgrenze angesprochen. Ftir Routineanalysen ist die h6chstm6gliche Intensitfit (Z/ihlrate) anzustreben, um statistisch gesicherte Re- sultate zu erhalten. Im anderen Fall spielt ein Inten- sitfitsverlust kaum eine Rolle, weil letztlich nur das Verhfiltnis Signal/Untergrund zu ermitteln ist.
Das Spektrometer/Rechnersystem sollte mit der M6glichkeit ausgestattet sein, Untergrundmessungen und -korrekturen ausffihren zu k6nnen. Das ist leicht gefordert, denn wo soll man spezielle Spalte im Be- reich des Spektrums setzen. Die apparative Entwick- lung sollte dahingehen, daB z.B. mit lichtempfind- lichen Dioden an beiden Seiten neben einer Analysen- linie gemessen werden kann.
Der dynamische Mel3bereich des Systems sollte mindestens 10 s oder besser 106 betragen, da normaler- weise der Konzentrationsbereich von ng/ml bis mg/ml erfaBt werden kann.
Als Resultat lfiBt sich formulieren, dab es fiir spezielle Probleme der Multielementanalyse erfor- derlich ist, ein Simultanspektrometer einzusetzen. Dann mtissen Aufgabenstellung und Umfang definiert bekannt sein. Die Verwendung eines Vakuumspektro- meters hat den Vorteil, dab auch mit Spektrallinien im nahen UV gearbeitet werden kann, wenn das Plasma direkt vor der Eintrittslinse des Spektrometers an- geordnet wird. Bis zu Wellenl/ingen von 180 nm lassen sich noch ausreichende Intensit/iten messen [3].
Variabler w/ire dann die Kombination ICP/Sequenz- spektrometer, wobei zwischen den einzelnen Element- bestimmungen die Parameter entsprechend optimiert werden k6nnen.
Da sich mit der ICP-Spektrometrie analytische Pro- bleme werden 16sen lassen, die heute nur mit grogen Schwierigkeiten oder iiberhaupt nicht 16sbar sind, sollte die ICP-Anregung auch mit einem variablen Monochromatorger/it hoher Gtite kombiniert werden, um problemorientierte Spektrallinien frei w/ihlen zu k6nnen.
In allen F/illen 1/il3t sich durch die Anwendung der ICP-Spektrometrie ein rationeller Effekt erreichen. Vielf~iltig wird die Anwendung, wenn die Versuche zum Einbringen von Gasen, Metalld/impfen, Partikel-
aerosolen oder Pulvern [6] erfolgreich weitergeftihrt werden.
3. Apparative Bedingungen
Ftir die geplante Durchftihrung simultaner Multi- elementanalysen stehen uns folgende Apparaturen zur Verffigung, die mit den angegebenen Bedingungen be- trieben werden:
Generator Henry Radio, Los Angeles, Modell 2000 PGC 27,12 MHz, kristallstabilisiert 2500 W 1000-1500 W
Frequenz maximale Leistung Arbeitsleistung Leistungsverlust
am Generator ICP-Anregung
Impedanzwandler Spule
Quarzbrenner
Argongasstrom Zfindung
H6henverstellung des Brenners
Zerst/iuber
Zerst~iuberkammer Aerosolgas Kontrolle der
Gasstr6mung
Spektrometer
Brennweite Gitter Wellenl/ingenbereich Aufl6sungsverm6gen Profilierung Analysenlinien
0 - 5 W Plasmaspec (Prototyp) Kontron, Mfinchen Henry Radio, Los Angeles Kupferrohr,
3 mm Innen-0 4 mm AuBen-0
Windungen: 1,5 23 mm Innen-0 27 mm AuBen-0
Abstand zwischen den Windungen: 2,2 mm
InduktivitS.t der Spule: 0,14 ~H Kombinierter Brenn- und Kfihlgas-
strom im Aul3enrohr mit tangen- tialem Eingang
AuBenrohr: 18 mm Innen- 0 (20 mm lfinger als Innenrohr)
Innenrohr (Tulpe): 14 mm Innen-0 Capillare: 1,5 mm Loch-0
(5 mm unterhalb Innenrohr) 10,8 l/rain Funkenfibertragung durch Tesla-
Spule
8 - 36 mm fiber der Spule (Digital- anzeige auf 0,04 mm genau)
Eppendorf (Netheler & Hinz, Hamburg) Typ A, Capillare 1,98 mm
nach Scott u. a. [12] 1,5 - 3 1 Ar/min
getrennte Druckreduzierung ffir Brenn-/Kfihlgasversorgung und Aerosolgas; konstante Einstellung fiber Porter Instrumente Typ B 250-6 bzw. B 125-50
Labtester V 25 mit Auswertegerfit CRT 100
l m 2160 Linien/mm 160- 430 nm 0,02 nm Hg-Lampe fiber Hg 296,73 nm Se 196,03 nm Sb 206,84 nm As 228,81 nm B 249,67 nm
K. Ohls et al. : ICP-Emissionsspektrometrie zur Multielementanalyse 181
P 253,56 nm Mn 257,61 nm Fe 259,94 nm Si 288,16nm Cu 324,75 nm Ti 334,94 nm Ni 341,47 nm Ca 393,37 nm La 394,91 nm A1 396,15 nm W 400,88 nm Nb 407,97 nm Cr 425,43 nm
4. Experimentelle Untersuchungen
Bei der Durchffihrung der Versuche konnten wir uns auf die grundlegenden Arbeiten des AMES Laboratory [5] stfitzen. Es erschien uns wenig sinnvoll, bestimmte Erfahrungen jahrelanger Arbeiten kontrollieren oder findern zu wollen, zumal wir die gleichen Dimensionen des Brenners, der Spule usw. sowie einen ~ihnlichen Generator gew~ihlt hatten. Somit haben wir weder die Frequenz noch die Leistung des Generators variiert. Bei einem Ar/Ar-Plasma und gegebener Anordnung f~hrt nach Barnes eine Ver/inderung der Frequenz von 15 auf 70 MHz lediglich zu einer Temperatur- verfinderung von 10000 K auf 9000 K [1]. Im AMES Laboratory [11] konnte gezeigt werden, dab bei aus- gewfihlten Spektrallinien auch eine Leistungsfinderung um einige 100 W keinen wesentlichen EinfluB auf die Intensit~it hat, wenn die Kopplung nicht gest6rt wird.
Wesentlichen Einflul3 hat die Resonanzabstimmung zwischen Impedanzwandler und Generator. Dazu geh6rt auch die Kopplung fiber die Spule. Die in Form yon abgestrahlter Wfirme verlorene Leistung kann
durch eine gute Abstimmung in Grenzen gehalten werden. Bei einer Ausgangsleistung von 1500 W am Generator kann geschfitzt werden, dab etwa 700- 900 W tats~ichlich am Brenner vorliegen.
Die weiteren Gr6Ben, die die Intensitfitsmessung stark beeinftussen, sind die Art des Zerst/iubers, die Zerstfiuberkammer und die Aerosolgasmenge, von der die umgesetzte Menge der Probel6sung und letzt- lich die Verweilzeit im Plasma abhfingen, sowie die Beobachtungsh6he im Plasma, d.h. der Bereich der Plasmaflamme, aus dem das emittierte Licht zum Spektrometer entnommen wird. Bei gegebenem Zer- st~iuber und feststehender Kammer haben wir daher im wesentlichen die Abh/ingigkeiten zwischen relativer Intensit/it und Beobachtungsh6he bei den einzelnen Elementen untersucht, wobei entweder die Aerosol- gasmenge oder die Beobachtungsh6he variiert wurden.
Nachdem mit 0,1 N salzsauren, w/iBrigen L6sungen jeweils eines Elementes die Profilierung geprfift und das Verh/iltnis Signal/Untergrund gemessen wurden, erfolgten alle weiteren Untersuchungen an einer 0,1 N salzsauren L6sung, die alle interessierenden Elemente enthielt, mit Hilfe der simultanen Multielementana- lyse, d.h. alle Daten wurden unter den gegebenen Versuchsbedingungen gleichzeitig gemessen und aus- gedruckt.
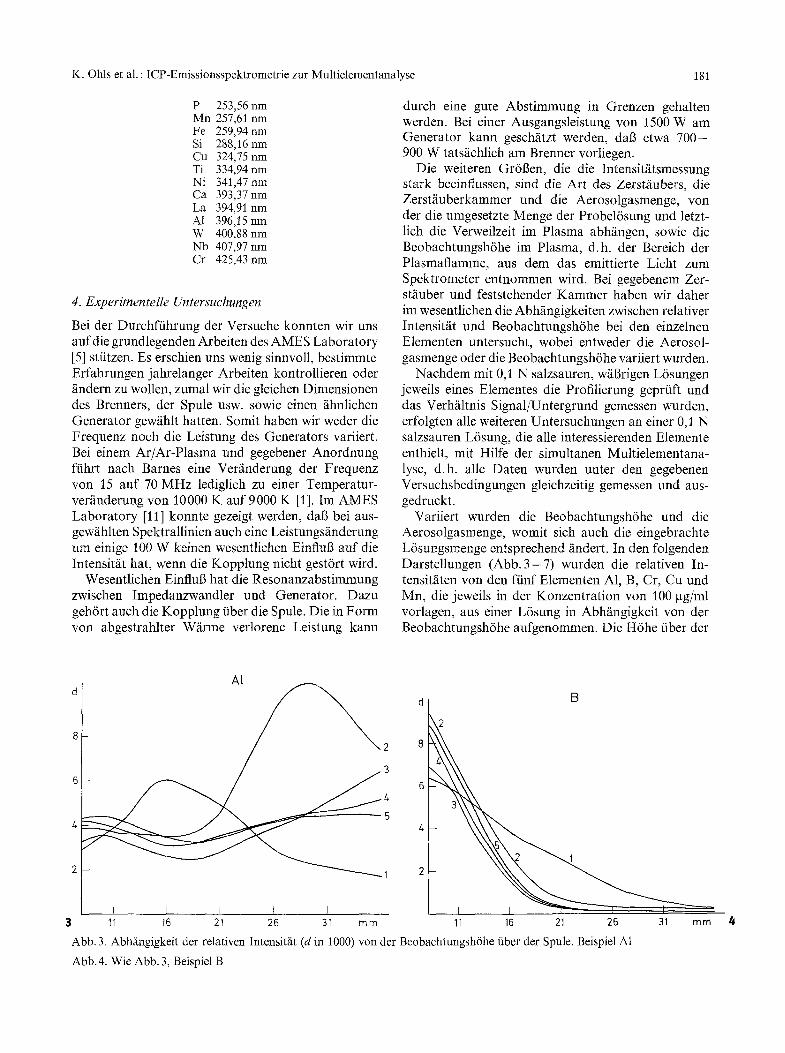
Variiert wurden die Beobachtungsh6he und die Aerosolgasmenge, womit sich auch die eingebrachte L6sungsmenge entsprechend findert. In den folgenden Darstellungen (Abb. 3 -7 ) wurden die relativen In- tensitfiten von den ffinf Elementen A1, B, Cr, Cu und Mn, die jeweils in der Konzentration von 100 gg/ml vorlagen, aus einer L6sung in Abh/ingigkeit yon der Beobachtungsh6he aufgenommen. Die H6he fiber der
A[
d B
2 8
3
6
~ 5 4
1 2 2 ~ ~
I I I ( I I I 11 16 21 26 31 mm 11 16 21 26 31 mm 4
Abb. 3. AbhS~ngigkeit der relativen Intensitfit (d in 1000) vonder Beobachtungsh6he fiber der Spule. Beispiel Al
Abb. 4. Wie Abb. 3, Beispiel B
182 Z. Anal. Chem., Band 284 (1977)
d
10
3
5 2
I I I I I I 11 16 21 26 31 mm
Or
4 - 3
1
I I I I I I 11 16 21 26 31 mm
d
6
M n
5
I I / 1 I 11 16 21 26 31 mm
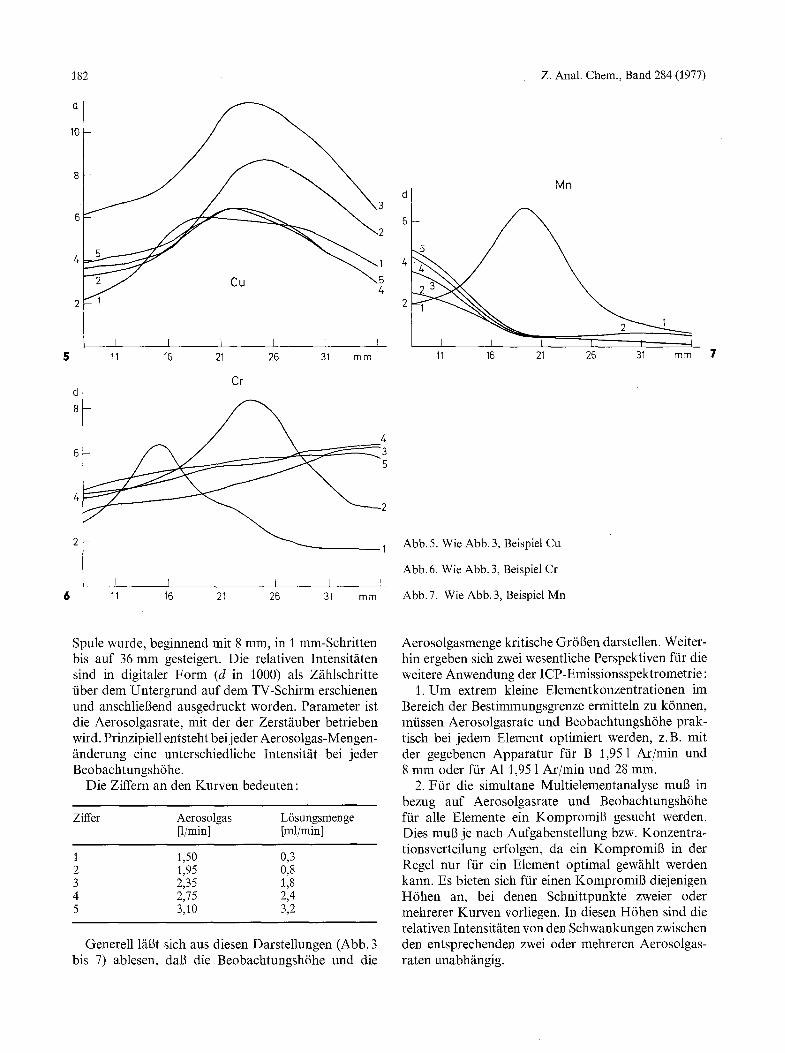
Abb. 5. Wie Abb. 3, Beispiel Cu
Abb. 6. Wie Abb. 3, Beispiel Cr
Abb. 7. Wie Abb. 3, Beispiel Mn
Spule wurde, beginnend mit 8 mm, in 1 mm-Schritten bis auf 36 mm gesteigert. Die relativen Intensit~iten sind in digitaler Form (d in 1000) als Zfihlschritte fiber dem Untergrund auf dem TV-Schirm erschienen und anschlie6end ausgedruckt worden. Parameter ist die Aerosolgasrate, mit der der Zerstfiuber betrieben wird. Prinzipiell entsteht bei j eder Aerosolgas-Mengen- ~inderung eine unterschiedliche Intensitfit bei jeder Beobachtungsh6he.
Die Ziffern an den Kurven bedeuten'
Ziffer Aerosolgas L6sungsmenge [1/min] [ml/min]
1 1,50 0,3 2 1,95 0,8 3 2,35 1,8 4 2,75 2,4 5 3,10 3,2
Generell lfi6t sich aus diesen Darstellungen (Abb. 3 bis 7) ablesen, da6 die Beobachtungsh6he und die
Aerosolgasmenge kritische Gr613en darstellen. Weiter- hin ergeben sich zwei wesentliche Perspektiven ffir die weitere Anwendung der ICP-Emissionsspektrometrie:
1. Um extrem kleine Elementkonzentrationen im Bereich der Bestimmungsgrenze ermitteln zu k6nnen, mfissen Aerosolgasrate und Beobachtungsh6he prak- tisch bei jedem Element optimiert werden, z.B. mit der gegebenen Apparatur ffir B 1,951 Ar/min und 8 mm oder ffir A1 1,95 1 Ar/min und 28 mm.
2. F fir die simultane Multielementanalyse muB in bezug auf Aerosolgasrate und Beobachtungsh6he ffir alle Elemente ein Kompromil3 gesucht werden. Dies mug je nach Aufgabenstellung bzw. Konzentra- tionsverteilung erfolgen, da ein Kompromil3 in der Regel nur ftir ein Element optimal gewfihlt werden kann. Es bieten sich ftir einen Kompromil3 diejenigen H6hen an, bei denen Schnittpunkte zweier oder mehrerer Kurven vorliegen. In diesen H6hen sind die relativen Intensitfiten von den Schwankungen zwischen den entsprechenden zwei oder mehreren Aerosolgas- raten unabhfingig.
K. Ohls et al. : ICP-Emissionsspektrometrie zur Multielementanalyse 183
Water 10 mm
I i I I J ~ 1 10 20 30 mm 0 1 2 3
ml / min
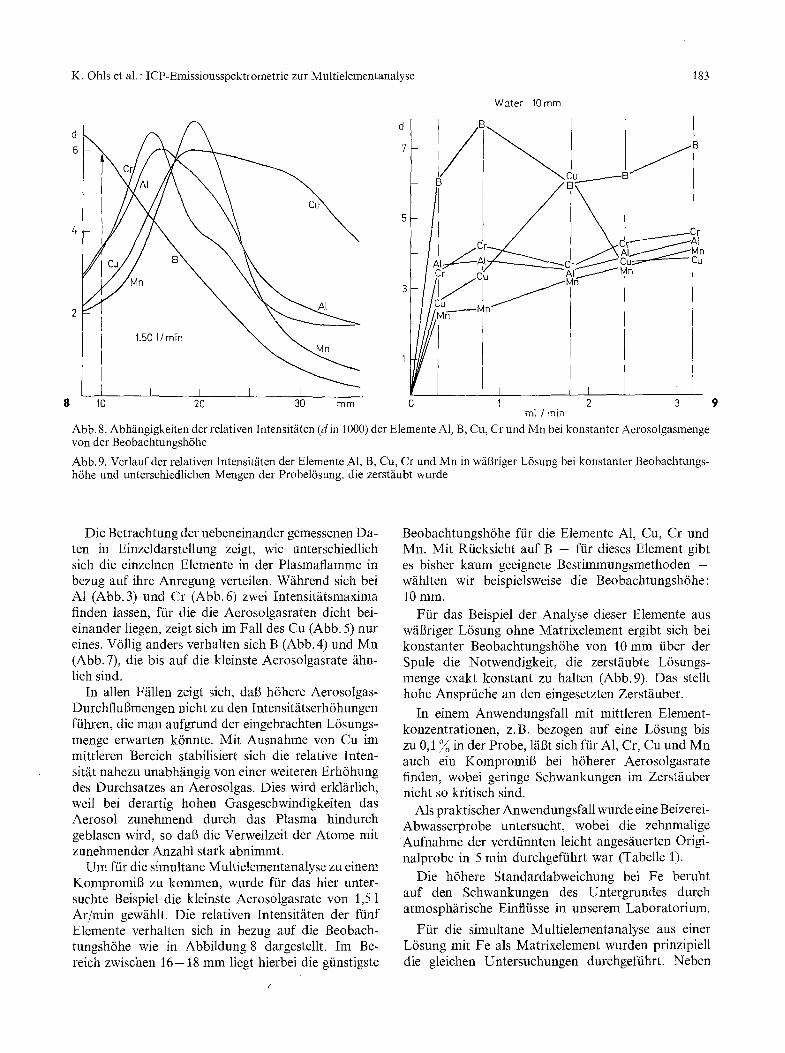
Abb. 8. Abhfingigkeiten der relativen Intensitfiten (d in 1000) der glemente A1, B, Cu, Cr und Mn bei konstanter Aerosolgasmenge vonder Beobachtungsh6he
Abb. 9. Verlauf der relativen Intensit/iten der Elemente A1, B, Cu, Cr und Mn in w/il3riger L6sung bei konstanter Beobachtungs- h6he und unterschiedlichen Mengen der Probel6sung, die zerstfiubt wurde
Die Betrachtung der nebeneinander gemessenen Da- ten in Einzeldarstellung zeigt, wie unterschiedlich sich die einzelnen Elemente in der Plasmaflamme in bezug auf ihre Anregung verteilen. W/ihrend sich bei A1 (Abb. 3) und Cr (Abb. 6) zwei Intensit~tsmaxima finden lassen, ftir die die Aerosolgasraten dicht bei- einander liegen, zeigt sich im Fall des Cu (Abb. 5) nur eines. V6llig anders verhalten sich B (Abb. 4) und Mn (Abb. 7), die bis auf die kleinste Aerosolgasrate fihn- lich sind.
In allen Ffillen zeigt sich, dab h6here Aerosolgas- Durchflui3mengen nicht zu den Intensit/itserh6hungen ftihren, die man aufgrund der eingebrachten L6sungs- menge erwarten k6nnte. Mit Ausnahme von Cu im mittleren Bereich stabilisiert sich die relative Inten- sit/it nahezu unabh/ingig yon einer weiteren Erh6hung des Durchsatzes an Aerosolgas. Dies wird erklgrlich, weil bei derartig hohen Gasgeschwindigkeiten das Aerosol zunehmend durch das Plasma hindurch geblasen wird, so dab die Verweilzeit der Atome mit zunehmender Anzahl stark abnimmt.
Um ffir die simultane Multielementanalyse zu einem Kompromi6 zu kommen, wurde ftir das hier unter- suchte Beispiel die kleinste Aerosolgasrate von 1,5 1 Ar/min gewfihlt. Die relativen Intensit/iten der ftinf Elemente verhalten sich in bezug auf die Beobach- tungsh6he wie in Abbildung 8 dargestellt. Im Be- reich zwischen t 6 - 1 8 mm liegt hierbei die giinstigste
Beobachtungsh6he ffir die Elemente A1, Cu, Cr und Mn. Mit Rficksicht auf B - ffir dieses Element gibt es bisher kaum geeignete Bestimmungsmethoden - wtihlten wir beispielsweise die Beobachtungsh6he: 10 mm.
F fir das Beispiel der Analyse dieser Elemente aus w/il3riger L6sung ohne Matrixelement ergibt sich bei konstanter Beobachtungsh6he von 10 mm fiber der Spule die Notwendigkeit, die zerstfiubte L6sungs- menge exakt konstant zu haken (Abb. 9). Das stellt hohe Ansprfiche an den eingesetzten Zerstfiuber.
In einem Anwendungsfall mit mittleren Element- konzentrationen, z.B. bezogen auf eine L6sung bis zu 0,1 ~ in der Probe, 1/iBt sich ffir A1, Cr, Cu und Mn auch ein Kompromi6 bei h6herer Aerosolgasrate finden, wobei geringe Schwankungen im Zerst~iuber nicht so kritisch sind.
Als praktischer Anwendungsfall wurde eine Beizerei- Abwasserprobe untersucht, wobei die zehnmalige Aufnahme der verdfinnten leicht anges~iuerten Origi- nalprobe in 5 min durchgeffihrt war (Tabelle 1).
Die h~Shere Standardabweichung bei Fe beruht auf den Schwankungen des Untergrundes durch atmosph~irische Einftiisse in unserem Laboratorium.
F fir die simultane Multielementanalyse aus einer L6sung mit Fe als Matrixelement wurden prinzipiell die gleichen Untersuchungen durchgefiihrt. Neben
184 Z. Anal. Chem., Band 284 (1977)
F e d
1.5 I/min ~ 1.95 I/rain 12
- Cu 10
8
6
4
2
1 0 11 16 21 26 31 mm 11 16 21 26 31 mm
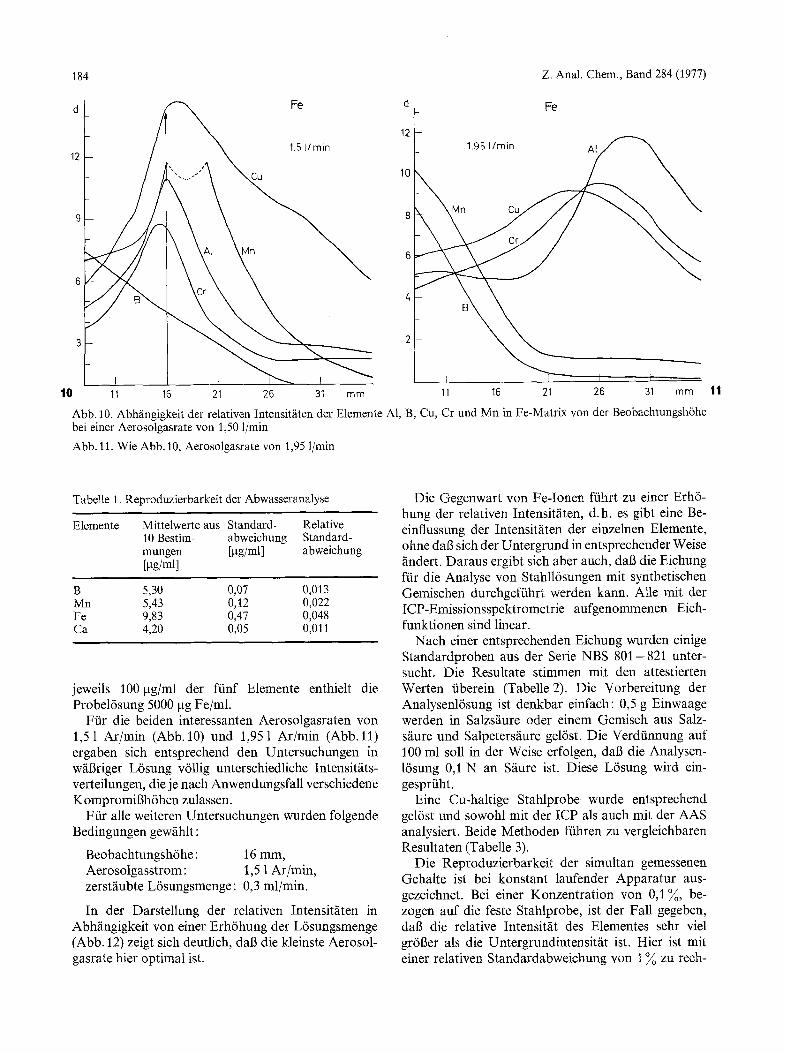
Abb. 10. Abh/ingigkeit der relativen lntensit~iten der Elemente A1, B, Cu, Cr und Mn in Fe-Matrix von der Beobachtungsh6he bei einer Aerosolgasrate yon 1,50 1/min
Abb. 11. Wie Abb. 10, Aerosolgasrate von 1,95 l/rain
tl
Tabelle 1. Reproduzierbarkeit der Abwasseranalyse
Elemente Mittelwerte aus Standard- 10 Bestim- abweichung mungen [lag/ml] [gg/ml]
Relative Standard- abweichung
B 5,30 0,07 0,013 Mn 5,43 0,12 0,022 Fe 9,83 0,47 0,048 Ca 4,20 0,05 0,011
jeweils 100 gg/ml der ftinf Elemente enthielt die Probel6sung 5000 gg Fe/ml.
Ffir die beiden interessanten Aerosolgasraten yon 1,51 Ar/min (Abb. 10) und 1,951 Ar/min (Abb. 11) ergaben sich entsprechend den Untersuchungen in w/i6riger L6sung v611ig unterschiedliche Intensitfits- verteilungen, die je nach Anwendungsfall verschiedene Kompromigh6hen zulassen.
Ftir alle weiteren Untersuchungen wurden folgende Bedingungen gewfihlt:
Beobachtungsh6he: 16 mm, Aerosolgasstrom: 1,5 1 Ar/min, zerst~iubte L6sungsmenge: 0,3 ml/min.
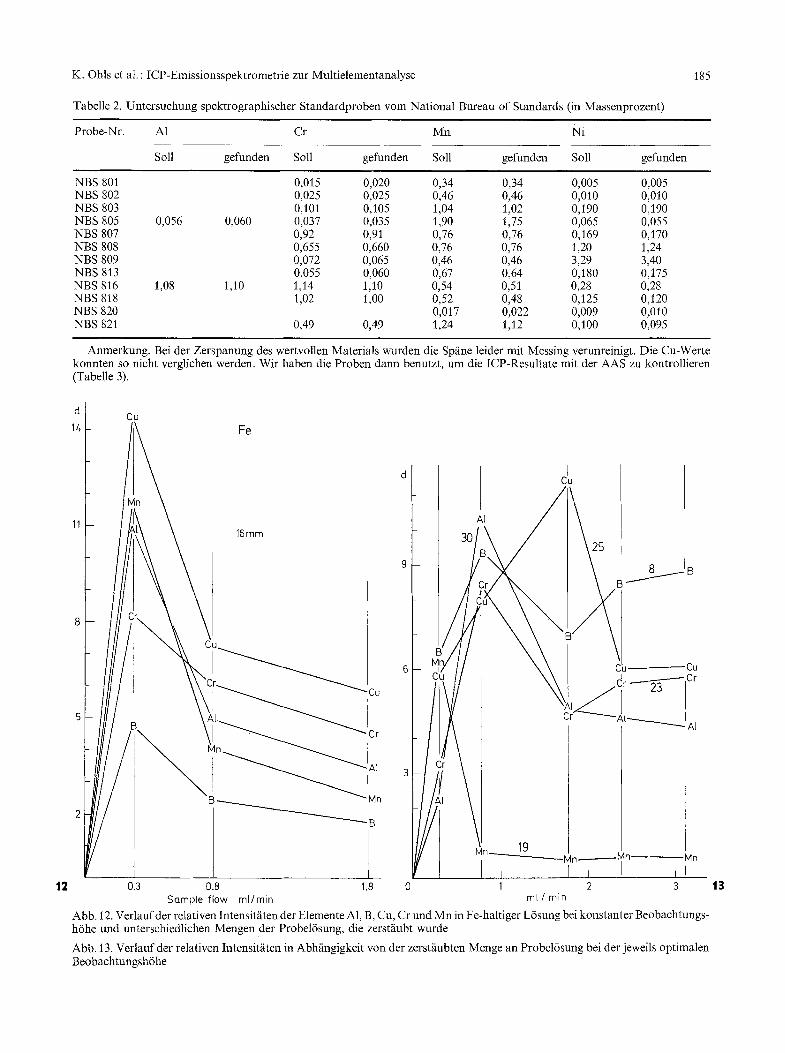
In der Darstellung der relativen Intensitfiten in Abh~ingigkeit yon einer Erh6hung der L6sungsmenge (Abb. 12) zeigt sich deutlich, dab die kleinste Aerosol- gasrate hier optimal ist.
Die Gegenwart yon Fe-Ionen fiihrt zu einer Erh6- hung der relativen Intensitfiten, d.h. es gibt eine Be- einflussung der Intensit~iten der einzelnen Elemente, ohne dal3 sich der Untergrund in entsprechender Weise /indert. Daraus ergibt sich aber auch, dab die Eichung ffir die Analyse von Stahll6sungen mit synthetischen Gemischen durchgeftihrt werden kann. Alte mit der ICP-Emissionsspektrometrie aufgenommenen Eich- funktionen sind linear.
Nach einer entsprechenden Eichung wurden einige Standardproben aus der Serie NBS 801-821 unter- sucht. Die Resultate stimmen mit den attestierten Werten iiberein (Tabelle 2). Die Vorbereitung der Analysenl6sung ist denkbar einfach: 0,5 g Einwaage werden in Salzs~iure oder einem Gemisch aus Salz- s/iure und Salpeters/iure gel6st. Die Verdiinnung auf 100 ml soll in der Weise erfolgen, dab die Analysen- [6sung 0,1 N an S~ure ist. Diese L6sung wird ein- gesprfiht.
Eine Cu-haltige Stahlprobe wurde entsprechend gel6st und sowohl mit der ICP als auch mit der AAS analysiert. Beide Methoden fiihren zu vergleichbaren Resultaten (Tabelle 3).
Die Reproduzierbarkeit der simultan gemessenen Gehalte ist bei konstant laufender Apparatur aus- gezeichnet. Bei einer Konzentration yon 0,1%, be- zogen auf die feste Stahlprobe, ist der Fall gegeben, dab die relative Intensit/it des Elementes sehr viel gr66er als die Untergrundintensit/it ist. Hier ist mit einer relativen Standardabweichung yon 1% zu rech-
K. Ohls et al. : ICP-Emissionsspektrometrie zur Multielementanalyse
Tabelle 2. Untersuchung spektrographischer Standardproben vom National Bureau of Standards (in Massenprozent)
185
Probe-Nr. A1 Cr Mn Ni
Soll gefunden Soll gefunden Soll gefunden Soll gefunden
NBS 801 0,015 0,020 0,34 0,34 0,005 0,005 NBS 802 0,025 0,025 0,46 0,46 0,010 0,010 NBS 803 0,101 0,105 1,04 1,02 0,190 0,190 NBS 805 0,056 0,060 0,037 0,035 1,90 1,75 0,065 0,055 NBS 807 0,92 0,91 0,76 0,76 0,169 0,170 NBS 808 0,655 0,660 0,76 0,76 1,20 1,24 NBS 809 0,072 0,065 0,46 0,46 3,29 3,40 NBS 813 0,055 0,060 0,67 0,64 0,180 0,175 NBS 816 1,08 1,10 1,14 1,10 0,54 0,51 0,28 0,28 NBS 818 1,02 1,00 0,52 0,48 0,125 0,120 NBS 820 0,017 0,022 0,009 0,010 NBS 821 0,49 0,49 1,24 1,12 0,100 0,095
1 2
Anmerkung. Bei der Zerspanung des wertvollen Materials wurden die Sp~ine leider mit Messing verunreinigt. Die Cu-Werte konnten so nicht verglichen werden. Wir haben die Proben dann benutzt, um die ICP-Resultate mit der AAS zu kontrollieren (Tabelle 3).
d
14
11
Cu
Fe
16mm
dl Cu
I ~i ~ ~ c u
Cr
I B
j - - - - Cu
i ~ l AU
- - Mn
I pl 0.3 0.8 1.8 0 1 2 3
Sample f low m[ /min m [ / m i n
Abb. 12. Verlauf der relativen Intensitfiten der Elemente A1, B, Cu, Cr und Mn in Fe-haltiger L6sung bei konstanter Beobachtungs- h6he und unterschiedlichen Mengen der Probel6sung, die zerstfiubt wurde
Abb. 13. Verlauf der relativen Intensiditen in Abh~ingigkeit vonder zerstfiubten Menge an Probel6sung bei der jeweits optimalen Beobachtungsh6he
1 3

186
Tabelle 3, Vergleich der Cu-Werte ICP/AAS prozent)
(in Massen-
Probe CLIAA S CnICP
1 0,026 0,025 2 0,045 0,045 3 0,156 0,152 5 0,310 0,317 7 0,153 0,155 8 0,155 0,160 9 0,193 0,185
16 0,337 0,333 20 0,092 0,093
Tabelle 4. Grenzen der quantitativen Bestimmbarkeit (in Massenprozent) in Fe-haltigen LSsungen
Elemente Literatur [5] Literatur [6] Verfasser
A1 0,0004 0,0001 0,00001 B - 0,0005 0,000005 Cr 0,0002 0,00002 0,00001 Cu 0,00003 0,00001 0,00001 Mn 0,0003 0,00002 0,00001
nen, die in der Regel auch erreicht wird. F/Jr den Konzentrationsbereich von 0,01 -0,001 ~o steigt sr auf etwa 2 ~ an, sofern man sich etwa beim 50fachen Wert der Nachweisgrenze befindet. Im Bereich der Nachweisgrenze, wobei die gesamte Intensitat nicht sehr yon derjenigen des Untergrundes abweicht, wird die relative Standardabweichung > 10 ~o.
Wenn man an der Grenze der quantitativen Be- stimmbarkeit (maximal sr = 10 ~) arbeitet, so ist es erforderlich, die beiden kritischen Parameter - Aero- solgasrate und Beobachtungsh6he - ftir jedes Ele- ment optimal zu wfihlen. Bei der gegebenen Apparatur und den als Beispiel gewfihlten Elementen lfil3t sich aus der Darstellung der relativen Intensitfiten bei der optimalen Beobachtungsh6he in Abhangigkeit von der zerstfiubten L6sungsmenge (Abb. 13) die gtinstigste Aerosolgasrate ablesen:
A1 (30 mm): 1,95 1 Ar/min B (8 ram): 1,95 1 Ar/min Cu (25 turn): 2,35 1Ar/min Cr (23 ram): 1,95 1 Ar/min Mn (19 ram): 1,50 1Ar/min.
Auch hier lassen sich wieder drei Elemente bei einer einzigen Aerosolgasrate zusammenfassen. Die exakte Verfinderung der Beobachtungsh6he ist in allen Fallen erforderlich. Unter diesen Voraussetzungen ergeben sich als Bestimmungsgrenzen fiir A1, Cr, Cu und Mn jeweils 0,1 ~tg/ml und fiir B 0,05 ~tg/ml.
Der Vergleich mit Literaturangaben ist deshalb schwierig, weil man normalerweise nicht weil3, wie die
Z. Anal. Chem., Band 284 (1977)
Angaben ermittelt wurden. Unsere Werte stimmen etwa mit derartigen Literaturangaben tiberein (Ta- belle 4), wenn man sich nur auf die AnalysenlSsung bezieht.
Dies sagt zunachst nur soviel far die Praxis aus, dal3 die Methode relativ empfindlich ist. Nicht berficksich- tigt ist hier der in der Vorbereitung auftretende Verdfinnungsfaktor, der in der Regel >~ 100 betr/igt. Bezogen auf feste Stahlproben liegen dann die echten Bestimmungsgrenzen z.Z. im Bereich von 0,01 bis 0,001 ~. In dieser Gr613enordnung beginnen auch die bisher ver6ffentlichten Eichkurven far die Element- bestimmung aus Stfihlen [5].
5. Schluflbetrachtung
Die ICP-Emissionsspektrometrie ist auf dem Wege, eine wertvolle Ergfinzung far die Arbeitsmethodik in chemischen Laboratorien zu werden. Der Erfolg wird weit gr613er werden als derjenige der Atom- absorptionsspektrometrie, well einmal die simultane Analyse yon maximal 6 - 8 Elementen mSglich ist und zum anderen gerade die Elemente, z. B. B, Nb, Zr, Ce und Si, mit der ICP vorzfiglich bestimmbar sind, die mit der AAS grol3e Schwierigkeiten bereiten, sowie sich Elemente bestimmen lassen, z. B. S, P und F, die mit der AAS nicht direkt erfal3bar sind. Wenn man dann noch bedenkt, dab auch bereits Metalldampfe und Gase [6] erfolgreich analysiert werden, ist der weitere erfolgreiche Weg der ICP-Emissionsspektro- metrie nicht schwer vorherzusagen. In allen Fallen ergibt sich ein rationeller Effekt.
Bei der weiteren Anwendung werden noch einige praktische Schwierigkeiten zu bew~iltigen sein. Wir arbeiten z.Z. an der Vorbereitung und ICP-Analyse mit oxidischen Substanzen nach Schmelzaufschlul3. Hier spielt die L6slichkeit der zur Schmelze eingesetz- ten bzw. gebildeten Salze eine grol3e Rolle, da Unter- schreitungen des L6slichkeitsproduktes zur Kristalli- sation im Bereich der Capillare fiihren k6nnen. Das kann zum Einsatz anderer Aufschlul3techniken fiihren.
Weiterhin ist anzunehmen, dab sich die Stabilitfit der apparativen Systeme weiter erhShen wird, sowohl in der optischen Abbildung als auch durch den Ein- satz leistungsstabilisierter Generatoren. Dies kann die Arbeitsweise insofern verandern, als gegen einen internen Standard gemessen werden kann. Wir haben mit der Messung bei konstanter Zeit bisher die besseren Reproduzierbarkeiten.erreicht.
Das Hauptaugenmerk wird man in Zukunft auf die Entwicklung leistungsffihiger, stabiler Zerstfiubersy- sterne legen mfissen, denn das Einbringen der Analy- senl6sung ist wie im Fall der AAS der kritische Punkt. Die ICP-Emissionsspektrometrie hat bereits

K. Ohls et al. : ICP-Emissionsspektrometrie zur Multielementanalyse 187
durch die vorl iegenden Erfahrungen mit derart igen Zerst/iubersystemen und die Kenntnis der registrie- renden Emissionsspektrometr ie einen hohen Lei- s tungsstand erreicht. Dies wird sich dann bestfitigen, wenn die ICP-Spektrometr ie , simultan oder sequen- tiell, in vielen Labora tor ien eingesetzt wird.
Literatur
1. Barnes, R. M.: Mitt. auf dem ICP-Expertentreffen in Noordwijk, 9.-,11.6. 1976
2. Barnes, R. M., Schleicher, R.G. : Spectrochim. Acta 30B, 109-134 (1975)
3. Bernhard, A. E. : Applikationsbericht der Fa. Labtest, Los Angeles
4. Boumans, P. W. J. M.: Fresenius Z. Anal. Chem. 279, 1 - 16 (1976)
5. Butler, C. C., Kniseley, R. N., Fassel, V. A. : Anal. Chem. 47, 825- 829 (1975)
6. Dahlquist, R. L., Knoll, J. W., Hoyt, R. E.: 21. Cana- dian Spectroscopy Symposium, Ottawa 1974
7. Dickinson, G. W., Fassel, V. A. : Anal. Chem. 41, 1021- 1024 (1969)
8. Fassel, V. A., Kniseley, R. N. : Anal. Chem. 46, 1115A- 1120A (1974)
9. Greenfield, S., Jones, S. L., McGeachin, H. M., Smith, P. B. : Anal. Chim. Acta 74, 225-245 (1974)
10. Kranz, E.: Spectrochim. Acta 27B, 327-343 (1972) 11. Larson, G. F., Fassel, V. A., Scott, R. H., Kniseley, R. N. :
Anal. Chem. 47, 238-242 (1975) 12. Scott, R. H., Fassel, V. A., Kniseley, R. N., Nixon, D. E. :
Anal. Chem. 46, 75 -80 (1974) 13. Souilliart, J.-C., Robin, J.-P. : Analusis 1,427-433 (1972)





























