Embed Size (px)
Citation preview

Homogene Katalyse der starken und schwachen
Wechselwirkungen:
Der Hafnium-Effekt bei Constrained Geometry
Katalysatoren und
neuartige Komplexe mit hyperverzweigten Liganden
Dissertation
zur Erlangung des Doktorgrades Dr. rer. nat.
der Fakultät für Naturwissenschaften der Universität Ulm
vorgelegt von
Sandra Meinhard
aus Ulm
2010

Amtierender Dekan: Prof. Dr. A. Groß
Erstgutachter: Prof. Dr. Dr. h. c. B. Rieger
Zweitgutachter: PD Dr. U. Ziener
Tag der mündlichen Prüfung: 26.10.2010

I Constrained Geometry Katalysatoren in Homo- und
Copolymerisationen: Der Hafnium-Effekt

Inhaltsverzeichnis
Abkürzungen .............................................................................................................................. - 3 -
I Constrained Geometry Katalysatoren in Homo- und Copolymerisationen: Der Hafnium-
Effekt .......................................................................................................................................... - 5 -
I.1 Grundlagen ................................................................................................................. - 5 -
I.1.1 Einleitung ............................................................................................................... - 5 -
I.1.2 Meilensteine in der Geschichte der Kunststoffe3 ................................................... - 8 -
I.1.2.1 Kunststoffe in der Informationstechnik .......................................................... - 8 -
I.1.2.2 Kunststoffe als Werkstoffe ............................................................................. - 9 -
I.1.2.3 Kunststoffe als Klebstoffe ............................................................................ - 10 -
I.1.2.4 Chemiefasern ................................................................................................ - 11 -
I.1.3 Homogene Katalyse ............................................................................................. - 12 -
I.1.3.1 Metallocene .................................................................................................. - 13 -
I.1.3.2 „Metallocene“ – Cyclopentadienylverbindungen der 4. Nebengruppe ........ - 13 -
I.1.3.3 Constrained Geometry Katalysatoren .......................................................... - 14 -
I.1.3.4 Zirkonium- und Hafnium-Metallocene im Vergleich .................................. - 18 -
I.1.3.5 Hafnium ........................................................................................................ - 21 -
I.1.4 Polymerisation – Vom Monomer zum Makromolekül ........................................ - 24 -
I.1.4.1 Aktivierung ................................................................................................... - 25 -
I.1.4.2 Mechanismus ................................................................................................ - 29 -
I.1.4.3 Abbruchreaktionen ....................................................................................... - 30 -
I.2 Aufgabenstellung ..................................................................................................... - 32 -
I.3 Synthese der Katalysatoren ...................................................................................... - 33 -
I.3.1 Überblick .............................................................................................................. - 33 -
I.3.2 Kristallstrukturen .................................................................................................. - 36 -
I.4 Polymerisationen ...................................................................................................... - 39 -
I.4.1 Experimenteller Teil ............................................................................................. - 39 -
I.4.1.1 Arbeitsmaterialien ........................................................................................ - 39 -
I.4.1.2 Polymerisationsapparatur ............................................................................. - 39 -
I.4.1.3 Polymercharakterisierung ............................................................................. - 41 -
I.4.1.4 Durchführung der Polymerisationen ............................................................ - 41 -
I.4.1.5 Parameterbestimmung .................................................................................. - 42 -
I.4.2 Polymerisation von Ethen .................................................................................... - 47 -

Inhaltsverzeichnis
- 2 -
I.4.3 Polymerisation von Ethen mit Propen .................................................................. - 70 -
I.4.4 Polymerisation von Ethen mit Norbornen ............................................................ - 85 -
I.4.5 Polymerisation von Ethen mit 1,3-Cyclohexadien ............................................. - 102 -
I.4.6 Fehlerbetrachtung ............................................................................................... - 116 -
I.5 Zusammenfassung .................................................................................................. - 124 -
I.6 Summary ................................................................................................................ - 133 -
I.7 Experimenteller Teil ............................................................................................... - 142 -
I.7.1 Arbeitstechniken und –materialien ..................................................................... - 142 -
I.7.2 Ligandensynthese ............................................................................................... - 143 -
I.7.2.1 1-(Chlorodimethylsilyl)-2, 3, 4, 5-tetramethyl-cyclopentadien ................. - 143 -
I.7.2.2 (N-tButylamino)(dimethyl)(2, 3, 4, 5-tetramethylcyclo-pentadienyl)silan - 144 -
I.7.3 Komplexsynthese ............................................................................................... - 145 -
I.7.3.1 Dilithium(N-tButylamido)(dimethyl)(2, 3, 4, 5-tetramethyl-
cyclopentadienyl)silan ................................................................................................ - 145 -
I.7.3.2 Tetrachlorobis(tetrahydrofuran)zirkonium(IV), ZrCl4(thf)2 ...................... - 146 -
I.7.3.3 Tetrachlorobis(tetrahydrofuran)hafnium(IV), HfCl4(thf)2 ......................... - 146 -
I.7.3.4 (N-tbutylamido)dimethyl(tetramethyl-�5-cyclopentadienyl)silan-
zirkonium(IV)dichlorid .............................................................................................. - 147 -
I.7.3.5 (N-tbutylamido)dimethyl(tetramethyl-�5-cyclopentadienyl)silan-
hafnium(IV)dichlorid ................................................................................................. - 148 -
I.7.3.6 Kristallographische Daten .......................................................................... - 149 -
I.7.4 Voraktivierung der Komplexe ............................................................................ - 152 -
I.8 Literatur .................................................................................................................. - 153 -

Abkürzungen
- 3 -
ABKÜRZUNGEN
1* (N-tbutylamido)dimethyl(tetramethyl-�5-cyclopentadienyl)silanzirkonium(IV)-
dichlorid
1 Katalysatorsystem bestehend aus 1*, TiBA und [Ph3P]+[B(C6F5)4-]
2* (N-tbutylamido)dimethyl(tetramethyl-�5-cyclopentadienyl)silanhafnium(IV)-
dichlorid
2 Katalysatorsystem bestehend aus 2*, TiBA und [Ph3P]+[B(C6F5)4-]
1,3-CHD 1,3-Cyclohexadien
abs. absolut
n-BuLi n-Butyl-Lithium tBu tert-Butyl
CGC constrained geometry catalyst = Katalysator mit eingeschränkter Geometrie
CHD Cyclohexadien
Cl Chlor(id)
Cp Cyclopentadien(yl)
Cp* Pentamethylcyclopentadien(yl)
Cp� Tetramethylcyclopentadien(yl)
CpA- Cyclopentadienylamido-
Cp�A Tetramethylcyclopentadienylamido-
D Polydispersität
DSC Differential Scanning Calorimetry
Flu Fluorenyl
Hf Hafnium
HfCp�A (N-tbutylamido)dimethyl(tetramethyl-�5-cyclopentadienyl)silanhafnium(IV)-
dichlorid
HPLC High Performance Liquid Chromatography
HT-GPC High Temperature Gel Permeations Chromatography
Ind Indenyl
Kat. Katalysator
LCB Langkettenverzweigungen (Long Chain Branches)
M Metall

Abkürzungen
- 4 -
MAO Methylalum(in)oxan
Me Methyl
Mn Zahlenmittel des Molekulargewichts
Mp Peakmaximum
Mw Gewichtsmittel des Molekulargewichts
NMR Nuclear Magnetic Resonance
n.v. nicht vorhanden
PCHD Poly(1,3-cyclohexadien)
PCHE Polycyclohexylen
PDI Poly Dispersity Index (Polydispersität)
PE Polyeth(yl)en
PP Polyprop(yl)en
PPP Polyparaphenylen
RT Raumtemperatur
Sdp. Siedepunkt
Smp. Schmelzpunkt
Temp. Temperatur
Tg Glasübergangstemperatur
THF Tetrahydrofuran
Ti Titan
TiBA Triisobutylaluminium
TiCp�A (N-tbutylamido)dimethyl(tetramethyl-�5-cyclopentadienyl)silantitan(IV)dichlorid
TMA Trimethylaluminium
Tritylborat Trityltetrakis(pentafluorophenyl)borat [Ph3P]+[B(C6F5)4-]
Zr Zirkonium
ZrCp�A (N-tbutylamido)dimethyl(tetramethyl-�5-cyclopentadienyl)silanzirkonium(IV)-
dichlorid

I.1 Grundlagen
- 5 -
I CONSTRAINED GEOMETRY KATALYSATOREN
IN HOMO- UND COPOLYMERISATIONEN:
DER HAFNIUM-EFFEKT
I.1 Grundlagen
I.1.1 Einleitung
Kunststoffe sind in unserem Leben allgegenwärtig. So oder ähnlich beginnen die meisten
Diplom- und Doktorarbeiten, die sich mit dem Thema Kunststoffe im Allgemeinen oder
Polyolefine im Speziellen befassen. Die Bedeutung und Wichtigkeit für das alltägliche Leben
wird durch die Angabe der Jahrestonnagen und anderer, wenig greifbarer Zahlen vor Augen
geführt. Doch wie kann und soll man sich eine Zahl von beispielsweise 180 Millionen Tonnen
Thermoplaste1 vorstellen? Ich möchte Sie an dieser Stelle auf ein Gedankenexperiment
mitnehmen. Sind Kunststoffe aus unserem heutigen Leben noch wegzudenken? Betrachten wir
einmal die ersten 15 Minuten am Tag einer Durchschnittsfamilie ohne Plastik!
Familie Schmidt hat schlecht geschlafen. Die Nacht war ungewohnt und unbequem, denn auf die
moderne Kaltschaummatratze mussten sie leider verzichten, ebenso auf die klimakontrollierte
Vier-Jahreszeiten-Steppdecke. Neben dem Bett steht nicht der Funkwecker mit LCD-Anzeige
sondern ein robustes Modell aus Metall. Aber heute sorgte sowieso die Sonne fürs Aufwachen:
der Fensterladen schirmt das Licht eben nicht so gut ab wie der Außenrollo. Weiter geht’s in die
Küche zum Kaffeekochen: der kommt heute nicht fertig gemahlen aus dem vakuumversiegelten
Aromapack sondern in ganzen Bohnen aus dem Jutesack. Auch auf die geliebte Kaffeemaschine
muss Herr Schmidt verzichten. In Omas altem Porzellanfilter wird selbst aufgebrüht. Das
Badezimmer wirkt spärlich eingerichtet. Der Duschvorhang fehlt, ebenso die Waschmaschine,
und die Toilette sieht äußerst merkwürdig aus: Holzsitz und Metallspülkasten ersetzen den
Kunststoff. In der Dusche gibt es keinen Wellness-Duschkopf mehr. Das Wasser kommt
vielmehr aus der leicht angerosteten Brause von oben. Die geliebten Shampoo- und Duschgel-
Flaschen sind weg – nachdem aber sowieso wichtige Inhaltsstoffe fehlen würden, greifen die
Schmidts heute zu Kernseife und Zitronensaft mit Honig. Während Herr Schmidt vorher schon
Schwierigkeiten bei der Rasur mit dem Messer hatte, so trifft es jetzt die Damenwelt genauso
hart. Die Haare werden heute an der Luft trocknen müssen. Haarspray, -schaum oder -gel bleiben

I.1 Grundlagen
- 6 -
ebenfalls nur ein Wunsch, selbstverständlich trifft das auch für alle Kosmetika zu – bestenfalls
gibt es ein Körperpuder. Zähneputzen gestaltet sich schwieriger. Eine Rosshaarbürste oder ein
Schwämmchen muss die Zahnbürste, wie wir sie kennen, ersetzen, von einer elektrischen
Zahnputzstation ganz zu schweigen. Dennoch geht es gut gelaunt ans Frühstück, denn hier gibt
es nach wie vor Brötchen, Butter, Marmelade, nur dass die Milch nicht aus dem Tetrapack
kommt. Die Tischplatte ist nicht kratzfest versiegelt und die Stuhlpolster sind auch nicht mehr
Schmutz abweisend. Das Vesperbrot kommt nicht in die Tupperware®dose sondern ins
Butterbrotpapier und die Wasserflasche ist etwas schwerer – Glas statt PET. Leider hat es das
Wetter mit den Schmidts heute nicht gut gemeint: es regnet. Das ist besonders ärgerlich, denn es
gibt keine Wasser abweisende Gore-Tex®-Jacke, keine atmungsaktiven aber wasserdichten
Schuhe und der Regenschirm aus Baumwolle nützt auch nicht viel. Wenigstens muss niemand
auf die Aktentasche aus Leder verzichten – allerdings gehen die Kinder heute ohne den leichten
Rucksack mit stabilisierendem Rückenpolster in die Schule. Das Notebook existiert ebenfalls
nicht, das Auto steht still und der Bus hat sich stark verändert: Holzbänke statt der bequemen
Schalensitze, und auch sonst ist vieles unbequemer geworden – einschließlich des Fahrgefühls,
denn die Reifen bieten noch einen ganz anderen Fahr„komfort“.
Ich denke, bereits jetzt ist deutlich geworden, dass wir im Kunststoffzeitalter leben und dass
Polymere die Werkstoffe des 20. und 21. Jahrhunderts sind. Als Chemiefasern, Klebstoffe,
Werkstoffe und in der Informationstechnik sind Kunststoffe unersetzlich geworden. Klar, es ist
schwer zu sagen, wie sich andere Materialien wie z. B. Metalle oder natürliche Rohstoffe wie
Cellulose ohne die Erfindung der künstlichen Polymere weiterentwickelt hätten. Doch
Kunststoffe begleiten uns ein ganzes Leben. Sie bereichern und erleichtern unser Dasein in so
vielen Bereichen und auf vielfältigste Art und Weise. Stellen Sie sich doch mal selbst die Frage:
Wollen Sie noch ohne Kunststoffe auskommen?

I.1 Grundlagen
- 7 -
Abb. 1: Kunststoffe im Alltag2 - ein kleiner Auszug.
Die Allgegenwart der Kunststoffe könnte leicht zu der Annahme verleiten, dass die
Innovationspotentiale dieser Werkstoffklasse nahezu erschöpft seien. Doch weit gefehlt!
Bescherte uns die erste Kunststoffrevolution den großen Siegeszug – angefangen vom
Nylonstrumpf über das Einweggeschirr bis zum Fernsehgehäuse –, dann hat die zweite
Revolution gerade erst begonnen.3

I.1 Grundlagen
- 8 -
I.1.2 Meilensteine in der Geschichte der Kunststoffe3
Im folgenden Kapitel wird die junge aber große Geschichte der Kunststoffe auf dem Weg zur
zweiten Revolution umrissen. Dabei wird zwischen den Bereichen Informationstechnik,
Werkstoffe, Klebstoffe und Chemiefasern unterschieden.
I.1.2.1 Kunststoffe in der Informationstechnik
1869 Celluloid-Film: Im Jahre 1869 durchfeuchtete J. W. Hyatt - der amerikanische Erfinder,
Drucker und Vater der Kunststoffe schlechthin - gewöhnliches Papier mit Salpetersäure und
verknetete die entstandene Nitrocellulose mit Campher. Er erhielt dadurch einen hornartigen
Stoff, der sich sehr gut formen ließ: Celluloid. Eigentlich suchte Hyatt nach einem
widerstandsfähigen Material für Buchdruckwalzen, bald sollte sich herausstellen, dass aus
diesem Material wesentlich kostbarere Dinge wie Perlen, Billardkugeln, Schmucksachen und
Spielzeuge gefertigt werden konnten. Als G. Eastman, Chef des Kodak-Konzerns 1884 den
fotografischen Film auf Celluloidbasis zum Patent anmeldete, erlebte die Cellulose-Industrie
einen sensationellen Aufschwung.
1928 – 1934 Tonband: Die erste Idee, den Magnetismus zum Aufzeichnen von Tönen zu
verwenden, hatte 1898 der Physiker V. Poulsen. Dem anfangs gebräuchlichen Stahlband war
aber nur ein kurzes Leben vergönnt. Bei der thermischen Zersetzung von Eisencarbonyl entsteht
ein äußerst feines Metallpulver, das sich hervorragend in einer Kunststoff-Folie, die als
Trägermaterial diente, verteilen ließ. 1934 wurden die ersten 50.000 Meter Tonband ausgeliefert.
1953 Makrolon-CD: H. Schnell fand, dass ein Kondensationsprodukt aus Phenol und Aceton
mit Hilfe von Phosgen in Polykohlensäureester überführt werden konnte. Fünf Jahre später
gelang es schließlich, den Polycarbonaten die noch fehlenden Eigenschaften eines gut
verarbeitbaren Kunststoffs zu verleihen und das Produkt unter dem Namen Makrolon auf den
Markt zu bringen – das Material für Compact Discs.
1973 Flüssigkristall-Bildschirm: Zu Beginn der 1960er Jahre machte sich bei der Radio
Corporation of America (RCA) eine Forschergruppe Gedanken über mögliche Anwendungen
von Flüssigkristallen. Heute macht der Flachbildschirm der Braun’schen Röhre nicht nur in
Notebook-PCs ernsthaft Konkurrenz.

I.1 Grundlagen
- 9 -
I.1.2.2 Kunststoffe als Werkstoffe
1909 – 1929 Synthetischer Kautschuk: Als die Preise für Naturkautschuk 1906 aufgrund der
dramatisch gestiegenen Nachfrage mit bis zu 28 Mark pro Kilogramm dem Wochenlohn eines
Industriearbeiters entsprachen, schrieb ein deutsches Chemieunternehmen einen Preis in Höhe
von 20.000 Mark für denjenigen Chemiker aus, dem es innerhalb von drei Jahren gelingen
würde, ein Verfahrung zur Herstellung von Kautschuk oder eines Ersatzes zu entwickeln.
F. Hofmann synthetisierte und polymerisierte Isopren, das unabhängige Prüfer als „veritablen
Kautschuk“ charakterisierten.
1912 PVC: Im Jahre 1912 hatte F. Klatte ein patentreifes Verfahren entwickelt, das die Synthese
des Vinylchlorids, der Vorstufe des PVC, aus Acetylen und Salzsäure in Gegenwart von Salzen
des Quecksilbers zum Gegenstand hatte. Polyvinylchlorid begegnen wir heute wegen seiner
herausragenden Eigenschaften fast überall – nicht nur in Form von Fußbodenbelag.
1933 Polyethylen: Die Geburtsstunde des Polyethylens war 1933, als R. O. Gibson und E. W.
Fawcett wissen wollte, wie sich Ethen unter Überdruck verhält. Sie heizten das Gas in einer
stählernen Bombe auf 170 °C bei einem Druck von 1900 Atmosphären und erhielten ein
wachsartiges Produkt. 1939 waren die Vorbereitungen für ein großtechnisches Verfahren
abgeschlossen und die Produktion begann. Nach dem 2. Weltkrieg suchte K. Ziegler nach
geeigneten Katalysatoren, um die Polymerisation auch im Niederdruckbereich durchführen zu
können. Durch die Verwendung eines Aluminium/Zirkonium-Katalysator erhielt er ein
Polyethylen, das dem Hochdruck-Polyethylen hinsichtlich der Elastizität und anderer chemischer
Eigenschaften deutlich überlegen war. PE kommt heute als umweltfreundlicher und wieder
verwertbarer Werkstoff in nahezu allen Bereichen des täglichen Lebens – von der
Frischhaltefolie bis zur Mülltonne – vor.
1937 – 1941 Polyurethan: Die Experimente von O. Bayer mit Isocyanaten führten 1937 zu einer
Patentschrift, in der von einem „neuartigen und wertvollen hochmolekularen Produkt“ die Rede
war. Im zweiten Weltkrieg wurden auf der Suche nach besseren Kautschuksorten die
Eigenschaften elastischer Polyurethan-Gießmassen untersucht. Einige der Gießmassen waren mit
Blasen durchsetzt, denen Bayer auf die Schliche kommen wollte. Als er die
Kohlendioxidabspaltung, die zur Blasenbildung geführt hatte, absichtlich herbeiführte, fand er

I.1 Grundlagen
- 10 -
den Polyurethan-Schaumstoff, der aus unserem heutigen Leben beim besten Willen nicht mehr
wegzudenken ist.
1951 Styropor: Seit 1949 arbeitete F. Stastny an einem Verfahren zur Verschäumung des
Polystyrols. Zwei Jahre später hatte er das richtige Treibmittel und die beste Mischung gefunden.
Zu 98 % aus Luft bestehend wird Styropor (Styrol + porös) heute vor allem in der
Verpackungstechnik, Isolationstechnik oder im Metallguss eingesetzt.
I.1.2.3 Kunststoffe als Klebstoffe
Klebstoffe benutzten die Menschen bereits in vorchristlicher Zeit. Die Sumerer gewannen 3000
v. Chr. einen Leim aus tierischen Häuten, 1500 v. Chr. verwendeten die Ägypter tierische Leime
für Furnierarbeiten. Die erste Leimfabrik nahm 1690 in Holland ihren Betrieb auf, während
F. Sichel 1889 den ersten gebrauchsfertigen Pflanzenleim vorstellte.
1909 Phenolharze: Das Zeitalter der Klebstoffe auf der Basis synthetischer Rohstoffe begann
1909 als L. H. Baekeland ein Verfahren zur Phenolharz-Härtung zum Patent anmeldete. Noch im
selben Jahr wurden die Flügel der Fokker erstmals mit Phenolharzen geklebt.
1958 – 1960 Cyanacrylat-Klebstoffe: Die so genannten Sofort-, Blitz- oder Sekundenkleber
basierten auf Cyanacrylat und kamen 1958 erstmals in den USA auf den Markt. Der
Einkomponenten-Klebstoff zeichnet sich durch rasche Aushärtung auf nahezu allen Oberflächen
unterschiedlichster Beschaffenheit aus. So werden Cyanacrylat-Klebstoffe nicht nur von
Hobbybastlern geschätzt, sondern kommen auch in technologisch anspruchsvollen Gebieten wie
der Elektronik-Industrie zum Verkleben von Chips oder sogar der Medizintechnik zum
Verbinden von feinsten Nervenfasern zum Einsatz.
1981 Lösemittelfreie Klebstoffe: Lösemittelfreie Klebstoffe basieren auf feinsten
Polymerpartikeln, die im „natürlichsten“ aller Lösungsmittel, Wasser, verteilt sind. Sie sind
umweltfreundlich, geruchsfrei und sehr gut für die Anwendung auf Holz, Papier oder Textilien
geeignet.

I.1 Grundlagen
- 11 -
I.1.2.4 Chemiefasern
18924 Viskose: Ausgehend von Cellulose synthetisierten C. F. Cross, E. J. Bevan und C. Beadle
mit Hilfe von Lauge und Schwefelkohlenstoff eine sirupähnliche Flüssigkeit. Diese pressten sie
durch feine Gold- oder Platindüsen in ein Säurebad und erhielten so feine glänzende Fäden, die
sich leicht verspinnen ließen. Auch heute ist Viskose nach wie vor die wichtigste Chemiefaser
auf der Basis von Cellulose – weltweit werden jährlich rund 3 Mio. Tonnen für die
Textilindustrie produziert.
1935 Nylon: W. H. Carothers schuf 1935 das Superpolyamid 6.6 aus Hexamethylendiamin und
Adipinsäure. Nylon, ist so fein wie Seide aber fester als Baumwolle. Als am 15. Mai 1940
erstmals Nylonstrümpfe auf den amerikanischen Markt kamen, wurden allein in New York City
innerhalb weniger Stunden vier Millionen Paare verkauft – damit avancierte Carothers zum
Favoriten der Damenwelt.
1937 Elasthan: Die elastische Chemiefaser auf der Basis von Polyurethan ist verglichen mit
Gummi reißfester, leichter und haltbarer. Dennoch hielt das Elasthan erst rund 25 Jahre später
Einzug in die Textilbranche – vor allem im Bereich der Sportbekleidung.
1980 Mikrofasern: Die Mikrofaser verbindet den Vorteil der heutigen Polyesterfäden mit den
Eigenschaften äußerst feiner Fäden. Mikrofasern sind hundertmal feiner als Menschenhaar,
10.000 Meter wiegen noch nicht mal ein Gramm. Ihre problemlose Kombinierbarkeit mit Seide,
Wolle, Baumwolle oder Viskose hat dazu geführt, dass über 75 % dieser Materialien heute für
modische Bekleidung eingesetzt werden.
Auch aus der Historie wird deutlich: Ohne geeignete Werkstoffe wären nicht nur die meisten
Errungenschaften der Technik auf der Strecke geblieben. Vielmehr müssten wir auch im
täglichen Leben auf nahezu sämtliche lieb gewordenen Gewohnheiten – von der Mobilität bis
zur Schönheitspflege und vom Informationsbedarf bis hin zum Freizeitvergnügen – wohl oder
übel verzichten. So heißt es in einer aktuellen Studie des US-Handelsministeriums: „Die
Erforschung neuer Werkstoffe gehört zu den innovativsten und wichtigsten
Schlüsseltechnologien des 21. Jahrhunderts überhaupt.“3

I.1 Grundlagen
- 12 -
I.1.3 Homogene Katalyse
Katalyse (griech.: katalysis = Auflösung, Umsturz) ist die Bezeichnung für die Beeinflussung der
Geschwindigkeit einer chemischen Reaktion durch die Gegenwart eines Stoffes (des
Katalysators), der die Reaktion scheinbar unverändert übersteht. Es wird zwischen homogener
und heterogener Katalyse unterschieden. Bei der homogenen Katalyse gehört der Katalysator der
gleichen Phase an wie das Reaktionssystem (z. B. flüssiger Katalysator in flüssiger
Reaktionsmischung gelöst); bei der heterogenen Katalyse liegt der Katalysator im Allgemeinen
als Feststoff vor, d.h. die Reaktanden (flüssig oder gasförmig) und der Katalysator sind einander
berührende, jedoch verschiedene Phasen.5
Die heterogene Katalyse ist in der Industrie nach wie vor dominierend. Die entscheidenden
Argumente sind hierbei die thermische Stabilität und die einfache Rückgewinnung der
heterogenen Katalysatoren. Gleichwohl bietet die homogene Variante einzigartige
Möglichkeiten. Der größte Vorteil liegt in dem eindeutig definierten molekularen Aufbau und
der hohen Strukturvariabilität der eingesetzten Verbindungen. Dieses Prinzip ermöglicht die
Entwicklung maßgeschneiderter Katalysatoren. Oftmals kann die homogene Spezies Schritt für
Schritt auf eine bestimmte Problemlösung hin optimiert werden.6 Für die Herstellung von
Polyolefinen stellt die Gruppe Metallocene solch maßgeschneiderte Werkzeuge dar.7
Neben den Metallocenen gibt es eine Vielzahl weiterer Katalysatorgruppen, die sich ebenfalls
sehr gut für die Olefinpolymerisation eignen.8 Diese sind allerdings nicht Inhalt dieser Arbeit.

I.1 Grundlagen
- 13 -
I.1.3.1 Metallocene
Namensgeber der Metallocene ist das Ferrocen (siehe Abb. 2). Der Name leitet sich vom
lateinischen „ferrum“ für Eisen und „cene“ in Analogie zum englischen „benzene“ ab. Es
handelt sich um so genannte Sandwich-Verbindungen, bei denen ein Metallatom zwischen zwei
aromatische Cyclopentadienyl-Ringe eingebettet ist. Nach den IUPAC-Regeln gilt dieser Name
streng genommen nur für Bis(�5-cyclopentadienyl)-Metallkomplexe, in denen die Ringe parallel
angeordnet sind. Für bahnbrechende Studien an Metallocenen und anderen Cyclopentadienyl-
Verbindungen teilten sich E. O. Fischer und G. Wilkinson 1973 den Chemie-Nobelpreis.5
Abb. 2: Ferrocen.
I.1.3.2 „Metallocene“ – Cyclopentadienylverbindungen der 4. Nebengruppe
Wilkinson stellte im Jahre 1953 mit Cp2TiCl2 das erste „Metallocen“ der Gruppe 4 her.9 Bereits
Mitte der neunziger Jahre waren weit über 1000 Strukturvariationen von gewinkelten
Metallocenen der allgemeinen Formel Cp2MCl2 bekannt.7
Abb. 3: Titanocendichlorid.
Ansatzpunkte zur Modifikation der Grundstruktur bieten das Metallatom, die Halogenatome und
die �-Liganden. Bei letzteren sind es besonders die Zahl und die Stellung der Substituenten, die
in weiten Grenzen variiert werden können. Zur Klasse der π-Liganden, die sich vom
Cyclopentadien ableiten, gehören auch Inden und Fluoren. Aus strukturchemischer Sicht lassen
sich diese Verbindungen im Wesentlichen in die beiden großen Bereiche der unverbrückten und
verbrückten Metallocene einteilen.

I.1 Grundlagen
- 14 -
Die verbrückten Metallocene, auch „ansa-Metallocenea“ genannt, wurden erstmals von
Brintzinger Anfang der achtziger Jahre beschrieben (siehe Abb. 4).10,11 Die Brücke schränkt die
Beweglichkeit der �-Liganden stark ein und fixiert die Geometrie um das Zentralatom.7 Durch
die Verwendung solcher Komplexe konnte mit homogenen Systemen erstmals isotaktisches
Polypropylen hergestellt werden12,13 – die Geburtsstunde der stereospezifischen Metallocen-
katalysatoren.14 Ein weiterer Meilenstein markiert das von Ewen und Razavi 1988 vorgestellte
ansa-Metallocen (siehe Abb. 4), mit dem sich mit hoher Aktivität und Stereoselektivität
syndiotaktisches PP herstellen lässt.15
Abb. 4: Ansa-Metallocene nach Brintzinger (links) sowie Ewen und Razavi (rechts).
I.1.3.3 Constrained Geometry Katalysatoren
Eine Weiterentwicklung der im vorangehenden Kapitel beschriebenen Metallocene sind die so
genannten „constrained geometry catalysts“ – kurz CGC. Es handelt sich um Halbsandwich-
Verbindungen, bei denen – verglichen mit den klassischen Metallocenen – ein Ringfragment
durch einen Amido-Liganden ersetzt wird. Der Stickstoff wiederum ist über eine Silizium-
Brücke mit dem verbliebenen Ring verbunden.
Abb. 5: Der constrained geometry Katalysator.
a lat. ansa = Henkel

I.1 Grundlagen
- 15 -
Der zugehörige Ligand wurde 1990 erstmals von Bercaw16 und Okuda17 vorgestellt, während die
Entwicklung der Katalysatoren durch die Industrie, vor allem Dow18 und Exxon19,
vorangetrieben wurde. Der Ausdruck „constrained geometry complex“ wurde ursprünglich von
Stevens et al. geprägt. Er stand für Komplexe, in denen ein �-gebundener Ligand (z. B. Cp oder
ein Derivat) mit einem der anderen Liganden am selben Metallzentrum in einer Art und Weise
verbunden ist, so dass der Winkel an diesem Metall zwischen des Centroiden des �-Systems und
dem zusätzlichen Liganden kleiner ist, als in vergleichbaren unverbrückten Komplexen.18 Diese
Definition schließt allerdings auch viele andere ansa-Komplexe mit �5-�1-Koordination mit ein.
Mittlerweile wurde der Begriff auch auf Komplexe mit sehr langen Brücken ausgeweitet, obwohl
durch die Flexibilität in der langen Brücke praktisch keine Spannung mehr vorhanden ist.20
Eine Besonderheit der CGCs liegt – wie der Name schon vermuten lässt – in der
außergewöhnlichen Geometrie. Abb. 6 zeigt die Koordinationssphären eines klassischen
verbrückten Metallocens und eines CGCs. Der Winkel, der von den beiden entscheidenden
Liganden aufgespannt wird (rot gekennzeichnet), liegt bei den CGCs 20 – 30° unter dem der
bekannten ethyl-verbrückten Verbindungen. Dadurch öffnet sich der Raum auf der anderen Seite
des aktiven Zentrums und ermöglicht so den Angriff auch sterisch anspruchsvoller Monomere.
Abb. 6: Koordinationssphären im Vergleich: Klassischer Brintzinger10-Komplex vs. CGC.
Neben der besonderen Sterik zeigen die CGCs zudem außergewöhnliche elektronische
Eigenschaften. Ungewöhnlich ist dabei bereits die Stabilität dieser Komplexe. Ein Ansatz zur
Diskussion von Struktur- und Bindungsverhältnissen in Übergangsmetallorganylen stellt die
18-Valenzelektronen-Regel dar. Sie geht auf die Arbeiten von Sidgwick21 aus dem Jahr 1927
zurück und besagt, dass stabile Übergansmetallkomplexe dann vorliegen, wenn die Summe der
Metall(d)-Elektronen und der von den Liganden zur Bindung beigesteuerten Elektronen 18
beträgt. Der CGC verfügt allerdings nur über 12 Elektronen. Unter der Berücksichtigung, dass
der Stickstoff ein freies Elektronenpaar trägt, welches er unter Umständen dem Metall zur
Verfügung stellen kann, resultieren maximal 14 Elektronen. Damit ist das Metallzentrum stark

I.1 Grundlagen
- 16 -
Lewis-sauer und besitzt eine große Affinität gegenüber Elektronen – auch aus elektronisch
anspruchsvollen Monomeren.
Ausgehend vom Original-CGC sind eine Vielzahl eng verwandter Komplexe synthetisiert
worden.22 Das Ligandensystem kann durch Veränderung des Cyclopentadienyl-Fragments, der
Amido-Gruppe, der ansa-Brücke oder durch eine Kombination dieser Möglichkeiten modifiziert
werden (siehe Abb. 7). Es sollte beachtet werden, dass ein unsymmetrisches Substitutionsmuster
am Cp-Liganden zu chiralen Komplexen führt.
Abb. 7: Modifikation von Constrained Geometry Komplexen.
Bei der Abwandlung des Cp-Fragments sind neben den gewöhnlichen Cp-, Cp�-, Indenyl- und
Fluorenyl- Fragmenten auch Alkyl- und Aryl-substituierte Cp-Ringe wie C5H3R (R = Me, tBu,
Benzyl) oder auch kompliziertere Substitutionsmuster am Ring mit sterisch anspruchsvolleren
Resten untersucht worden.20 Alt et al. synthetisierten beispielsweise eine Reihe Alkyl- und
�-Alkenyl-substituierter, Amido-funktionalisierter Cp- und Indenyl-Liganden, die zu den
entsprechenden Ti-, Zr- und Hf-CGCs umgesetzt und bei der Polymerisation von Ethen
eingesetzt wurden.23 Variation des Amido-Fragments kann sowohl die Sterik als auch die
Elektronik des Übergangsmetalls entscheidend beeinflussen. Einfache Aryl- und Alkyl-
Abänderungen scheinen allgegenwärtig, daneben sind auch chirale Reste oder zusätzliche
Donatoren (vgl. Abb. 15) gebräuchlich. Der Stickstoff kann überdies durch Phosphor ersetzt
werden, ebenso wurde der R2N--Ligand durch die isoelektrischen RO- oder RS- ausgetauscht.20
Stevens et al. schreiben die ungewöhnlichen Eigenschaften der CGCs der durch die Brücke
verursachten Spannung zu.18 Den Veränderungen der Brücke gilt daher besondere
Aufmerksamkeit. Die am häufigsten angewandte Verknüpfung ist nach wie vor die originale
SiMe2-Gruppe. Austausch einer Methyl-Gruppe am Silizium führt zu einem chiralen Si-Atom.
Neben Silizium sind einige wenige weitere Elemente für die kurze, einatomige Brücke wie
Kohlenstoff (H2C=C oder RHC), Bor (R2NB) und Phosphor (tBuP) beschrieben worden.
Desgleichen gilt für verlängerte Brücken von zwei bis drei Atomen.20

I.1 Grundlagen
- 17 -
Ursprünglich wurde der CpA-Ligand von Bercaw für die Synthese von Sc(III)-Komplexen
genutzt.16 Kurz danach berichtete Okuda17 von Titan-Verbindungen und Dow Chemical Co.18
sowie Exxon Chemical Co.19 patentierten die außergewöhnlichen Eigenschaften der GruppeIV-
Kongeneren. Neben den Übergangsmetallen der Gruppen drei und vier gibt es zahlreiche
Beispiele für CGCs anderer Elemente, speziell aus den frühen Nebengruppen. Komplexe des
Vanadium (seltener Nb. Ta), Chrom, Molybdän und Wolfram sind verbreitet, sogar Rhenium-
und Eisen-Spezies sind bekannt. CpA-Verbindungen der Gruppe-III-Elemente Al, Ga, In oder
der Gruppe-V-Vertreter P, As, Sb wurden beschrieben, haben aber mit den eigentlichen CGCs
aufgrund fehlender Orbitale für eine �5-Koordination mit dem Cp-Fragment nicht mehr viel zu
tun.20

I.1 Grundlagen
- 18 -
I.1.3.4 Zirkonium- und Hafnium-Metallocene im Vergleich
Wesentlicher Inhalt dieser Arbeit ist der Vergleich der Polymerisationseigenschaften des
Zirkonium- und Hafnium-Cp�A-Komplexes. Das folgende Kapitel handelt von bisherigen
Ergebnissen zu Merkmalen isostruktureller Zr- und Hf-Metallocene.
Es gibt keine Hafnium-Mineralien. Die Ionenradien von Zr4+ und Hf4+ sind aufgrund der
Lanthanoiden-Kontraktion fast gleich (Zr4+: 72 pm, Hf4+: 71 pm)24. (Die Angaben der
Ionenradien sind nicht einheitlich. Sowohl Riedel24 als auch CRC25 berichten von 72 pm für Zr4+
und 71 pm für Hf4+. Kaminsky dagegen gibt in seiner Veröffentlichung „Comparison of
zirconocene and hafnocene catalysts …“ Zr4+ mit 74 pm und Hf4+ mit 75 pm an26, was bedeuten
würde, dass Hafnium größer als Zirkonium ist.). Hafnium ist in Zirkoniummineralien enthalten,
in denen es Zirkonium diadoch vertritt. Dies ist auch die Ursache dafür, dass Hafnium erst 134
Jahre nach dem Zirkonium entdeckt worden ist.24 Bedingt durch die große Ähnlichkeit von
Zirkonium und Hafnium ist die Hafnium-Gewinnung aus Zirkoniumerzen außerordentlich
langwierig und kostspielig. Heute wird Hafnium nur als Nebenprodukt bei der Herstellung von
so genanntem Reactor-grade-Zirkonium für die Nuklearindustrie gewonnen.5
Als einfachstes Modell für den Vergleich von Zirkonium- und Hafniummetallocenen sind die
ursprünglichen Cp2MCl2-Komplexe (vgl. I.1.3.1) hervorragend geeignet. Nachfolgend werden
drei Beispiele aus der Literatur aufgegriffen. Rausch und Chien untersuchten deren Verhalten in
Ethenpolymerisationen mit MAO-Aktivierung.27
Abb. 8: Zirkonocen- und Hafnocendichlorid.
In ihrer Studie stellten sie fest, dass die Aktivität der Hafnium-Spezies nur 2/3 der Aktivität der
Zr-Analoga beträgt. Diesen Unterschied schrieben sie der größeren Anzahl aktiver Zentren im
Zr-Katalysator zu (durch radioaktive Markierung bestimmte 80 % aktive Stellen bei Zr, 52 % für
Hf). Der Hf-Katalysator zeigte eine geringere Abhängigkeit i) des Molekulargewichts von der
Polymerisationstemperatur und ii) der Aktivität von der Übergangsmetallkonzentration. Bei
70°C waren die Molekulargewichte der Hafnium-Polymere höher, bei 25°C galt das Gegenteil.
Dieselben Katalysatorsysteme hat die Veröffentlichung von Koivumäki zur
Copolymerisation von Ethen mit 1-Oktadeken zum Thema.28 Beim Vergleich der erhaltenen

I.1 Grundlagen
- 19 -
Polymere fällt auf, dass die Hafnium-Systeme größere Reaktivität gegenüber 1-Oktadeken
zeigen. Um einen Anteil des Comonomers von 6 mol-% im Produkt zu erreichen, benötigten die
Zirkonium-Katalysatoren eine dreifach höhere Comonomerkonzentration. Die Hafnium-Spezies
führten zu höheren Molekulargewichten. Obwohl reaktiver gegenüber 1-Oktadeken, zeigte
Cp2HfCl2 nur ein Zehntel der Aktivität seines Zr-Verwandten. Während die Zr-Aktivität mit
steigender Ethenkonzentration fortwährend stieg, blieb sie im Hafnium-Fall nahezu konstant.
Bochmann untersuchte die entsprechenden Cp2MMe2-Systeme (M = Ti, Zr, Hf) unter
Aktivierung mittels [CPh3]+[B(C6F5)4]
- bzw. [PhNHEt2]+[B(C6F5)4]
-.29 Deren Aktivität bei der
Polymerisation von Ethen steigt in der Reihe Ti << Hf < Zr an, die Aktivität der Zr- und Hf-
Komplexe ist vergleichbar mit denen der Dichloro-Spezies mit MAO als Cokatalysator. Die
Temperaturabhängigkeit der Aktivität spiegelt ebenfalls die Ergebnisse zu den Cp2MCl2/MAO-
Systeme von Rausch und Chien27 wieder.
Ähnliche Erkenntnisse brachte der Vergleich Ethyl-verbrückter Bis-Indenylmetallocene (siehe
Abb. 9). Für die Copolymerisation von Ethen mit Propen stellten Starck et al. fest, dass die
Experimente mit Et(Ind)2Hf und MAO zwar die niedrigeren Katalysatoraktivitäten ergeben,
jedoch die Polymere mit den deutlich höheren Molmassen. Auch bei der Synthese von Ethen-
1-Okten-Copolymeren wurden mit der Hafnium-Spezies das größere Molekulargewicht sowie
die höheren Comonomer-Einbauraten erzielt.30
Abb. 9: Et(Ind)2Zr und Et(Ind)2Hf.
Zusammenfassend lässt sich über die Metallocene der vierten Nebengruppe feststellen, dass der
Austausch des Metalls keinerlei Verbesserungen im Hinblick auf die technische Verwendbarkeit
brachte. Den Zirkonium-Systemen kommt die größte Bedeutung zu, da die Kombination ihrer
Eigenschaften sie für die Anwendung prädestiniert. Die meisten Titanocene sind unter den
technischen Polymerisationsbedingungen thermisch labil und somit nur wenig aktiv.
Strukturanaloge Hafniumderivate geben zwar Polymere mit deutlich höheren Molmassen, doch
betragen ihre Aktivitäten maximal ein Zehntel der entsprechenden Zirkonocene7, und sie sind
deutlich teurer (siehe oben).

I.1 Grundlagen
- 20 -
Durch die Sonderstellung der Constrained-Geometry-Katalysatoren stellt sich die Frage nach den
Vorzügen von Hafnium oder Zirkonium von neuem. Erste Hinweise auf unterschiedliche Trends
zu den klassischen Metallocenen liefert die theoretische Chemie. Ziegler et al. betrachteten die
Ethen Insertion in die M-CH3-Bindung des unsubstituierten CGC [(SiH2-C5H4-NH)MCH3]+ in
einer DFT-Studie.31
Abb. 10: Constrained Geometry Katalysator für DFT-Studien.
Studien der ursprünglichen kationischen Gruppe-4-Metallocene mit zwei Cp-Liganden deuten
darauf hin, dass die katalytische Aktivität bei der Olefin-Polymerisation von Titan nach
Zirkonium zunimmt. Dieser Trend kann mit der Beobachtung erklärt werden, dass die aktive
Stelle im Falle von Titan durch die kürzere M-Cp-Distanz beengter ist. Für Katalysatoren vom
CGC-Typ sollte die sterische Hinderung nicht so entscheidend sein, und Experimente lassen
vermuten, dass die jeweilige Titan-Verbindung am aktivsten ist. Im Vergleich der Reaktions-
Exothermien des Insertionsprozesses, stellten Ziegler et al. fest, dass sie für Ti(IV)-, Zr(IV)- und
Hf(IV)-basierte Katalysatoren ziemlich ähnlich sind. Auf der anderen Seite nehmen die
Insertionsbarrieren der Gruppe-IV-basierten CGCs in der Reihe Ti < Zr � Hf zu, die kalkulierten
Werte liegen bei 3,8 kcal/mol für Ti, 5,1 kcal/mol für Zr und 5,8 kcal/mol für Hf. Die Ergebnisse
der DFT-Studie lassen vermuten, dass die Insertion beim Halbsandwich-Komplex sterisch
ungehindert abläuft, wohingegen sie bei den Bis-Cp-.Metallocen „Voll“-Sandwiches signifikant
stärker gehindert ist. Die Unterschiede zwischen den Zirkonium- und Hafniumspezies werden als
gering eingeschätzt im Hinblick auf die Struktur des Katalysators, die �-Komplexe, die
Übergangszustände, die Propyl-Produkte und die relativen Energien.31
Demgegenüber konnten in dieser Arbeit signifikante Differenzen der Zr- und HfCp�A-Komplexe
hinsichtlich des Polymerisationsverhaltens festgestellt werden.

I.1 Grundlagen
- 21 -
I.1.3.5 Hafnium
Das Interesse an Hafnium-Katalysatoren allgemein lässt sich anhand der Treffer-Zahlen einer
Literatur-Recherche mit SciFinder Scholar ablesen.a Während für „catalyst titanium“ über
46.000 Treffer und für „catalyst zirconium“ knapp 19.000 Einträge erzielt werden, so sind es für
„catalyst hafnium“b gerade noch 2.200. Für „constrained geometry hafnium“ gibt es nur noch 7
Treffer. Die ersten Hinweise auf den HfCp�A-Komplex (η5:η1-C5Me4SiMe2NtBu)Hf(IV)Cl2
finden sich in den Patenten von Canich32 und von Stevens et al.18 (siehe Abb. 11).
Abb. 11: Der HfCp�A-Komplex: (ηηηη5:ηηηη1-C5Me4SiMe2NtBu)Hf(IV)Cl2.
Stevens beschreibt erst in Beispiel 107 von 107 die Synthese von 0,077 g (!) des Komplexes.
Weiterhin heißt es in einem folgenden Polymerisationsexperiment „a small amount of
polyethylene was recovered“. Canich zeigt darüber hinaus die Synthese weiterer Hf-Komplexe
(u. a. anderer Rest am Stickstoff, siehe Abb. 12) auf, jedoch ohne zugehörige Analytik.
Abb. 12: HfCpA-Komplexe nach Canich32.
a Recherche vom 06.09.2007 b Die Suche nach „Hf“ empfiehlt sich nicht, da hierbei auch viele Dokumente, die „THF“ enthalten, zu den Treffern
zählen.

I.1 Grundlagen
- 22 -
Die durchgeführten Polymerisationen dagegen sind erfolgreicher und somit auch detaillierter
angeführt. So wurde beispielsweise in einem zehnminütigen Experiment bei der Polymerisation
von Ethen eine Aktivität von 103.400 kg/(mol×h) erzielt.32 Die Synthese von HfCp�A
einschließlich ausführlicher Analytik wird erstmals von Okuda et al. veröffentlicht.33 Der
Niederschlag wird als mikrokristallin beschrieben, doch die Abweichung der Elementaranalyse
ist deutlich. Eine Röntgen-Strukturanalyse wurde nicht veröffentlicht. Alt et al.34 berichten über
den Hafnium-Katalysator mit unsubstituiertem Cp-Ring [C4H5-SiMe2-NtBu]HfCl2. Die Aktivität
bei der Polymerisation von Ethen ist mit 100 kg/(mol×h) allerdings deutlich geringer als die der
analogen Zirkonium- und Titankomplexe (1100 bzw. 4500 kg/(mol×h)). Dias et al. beschreiben
die Röntgenstruktur eines Hafnium-Komplexes mit Fluorenyl-Ligand und einer um eine
Methyleneinheit verlängerten Brücke (siehe Abb. 13).35
Abb. 13: Hafnium-amido-fluorenyl-Komplex nach Dias et al.35
Pakkanen et al. synthetisierten einen Hafnium-CGC durch Reaktion von Hf(NMe2)4 mit einem
auf einer Oberfläche angeknüpften CpA-Liganden (siehe Abb. 14).36 Der immobilisierte
Katalysator zeigte allerdings nur geringe Aktivität (ca. 11 kg/(mol×h)) bei der Polymerisation
von Ethen bei 80 °C mit MAO als Aktivator. Die entsprechenden Zirkonium-Systeme sind um
den Faktor 20 aktiver.37
Abb. 14: Immobilisierter Constrained-Geometry Hafnium Komplex nach Pakkanen et al.36

I.1 Grundlagen
- 23 -
Okuda et al. isolierten Zirkonium- und Hafnium-Komplexe mit einem 2-methoxyethyl-amido-
funktionalisierten Cp�-Liganden.38,39 Der dreizähnige Ligand wurde entwickelt, um die starke
Elekrophilie des Metall-Zentrums durch die Anwesenheit eines zusätzlichen Zwei-Elektronen-
Donators abzuschwächen. Neben der Dichloro-Spezies (siehe Abb. 15) sind Mono- und Di-
Alkyl-Spezies zugänglich und bemerkenswert stabil.39 Die Polymerisation von Ethen und
1-Hexen mit den entsprechenden Katalysatoren führte jedoch zu schwankenden Ergebnissen und
vergleichsweise geringen Aktivitäten.40
Abb. 15: Hafnium-Komplex mit dreizähnigem CpA-Liganden nach Okuda et al.38

I.1 Grundlagen
- 24 -
I.1.4 Polymerisation – Vom Monomer zum Makromolekül
Polymer ist im eigentlichen Sinne des Wortes die Bezeichnung für das "Mehrfache" (griech.:
poly) eines Teilchens (griech.: meros).5 Fachsprachlich wurde ursprünglich – und wird auch
noch heute vielfach – mit Polymer ein aus vielen diskreten Grundbausteinen aufgebautes
Molekül bezeichnet, der Begriff Polymer also synonym mit „Makromolekül“ und
„Polymermolekül“ verwendet. Nach einer Definition der IUPAC sollte aber unter Polymer eine
Substanz verstanden werden, die sich aus einem Kollektiv chemisch einheitlich aufgebauter, sich
in der Regel aber hinsichtlich Polymerisationsgrad, Molmasse und Kettenlänge unterscheidender
Makromoleküle (Polymermoleküle) zusammensetzt („makromolekulare Stoffe“).5
Polymere oder makromolekulare Stoffe werden aufgebaut, indem kleine Moleküle (Monomere)
durch geeignete chemische Reaktionen zu großen Molekülen aneinandergereiht werden. Diese
Polymerisation kann ausgelöst werden durch Einwirkung von Initiatoren, Wärme, ionisierender
Strahlung oder Licht. Bei der Insertionspolymerisation (oder Koordinations-Insertion) wird die
Polymerisation durch Ziegler-Natta-Katalysatoren, in jüngerer Zeit verstärkt auch durch
Metallocene, initiiert.
Abb. 16: Wichtige Vertreter der Metallocene – Katalysatoren für die Insertionspolymerisation.
I.1 Grundlagen
- 25 -
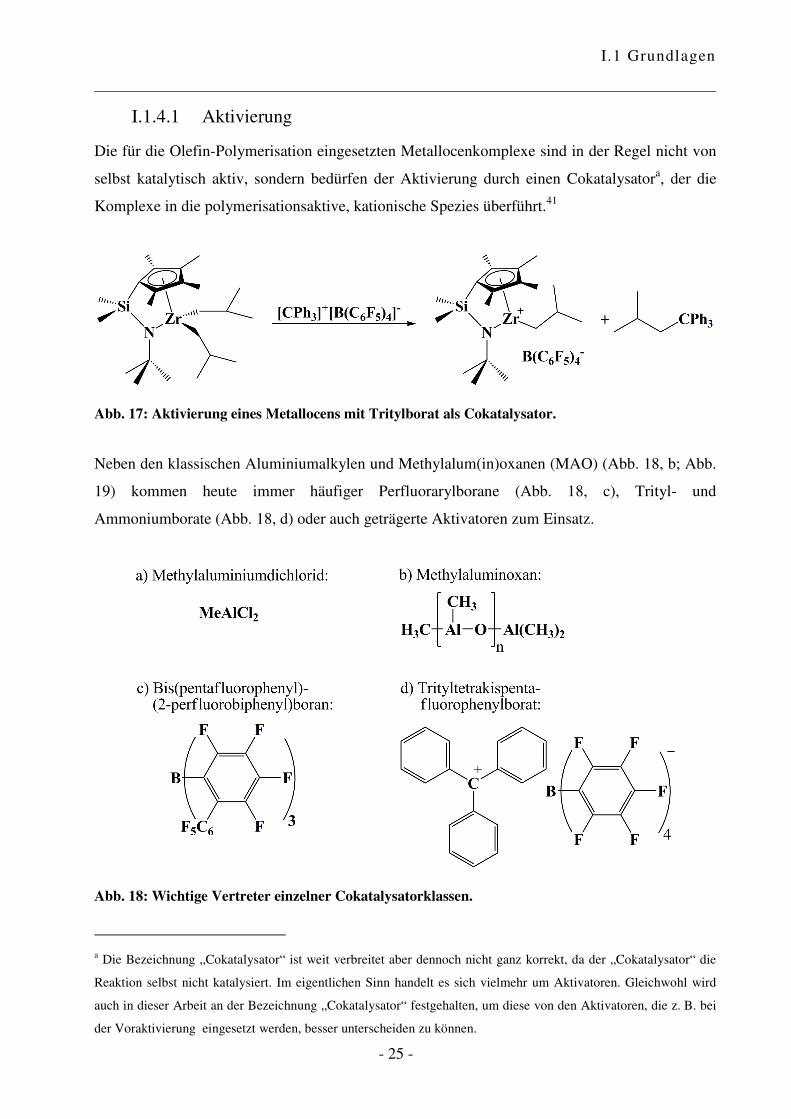
I.1.4.1 Aktivierung
Die für die Olefin-Polymerisation eingesetzten Metallocenkomplexe sind in der Regel nicht von
selbst katalytisch aktiv, sondern bedürfen der Aktivierung durch einen Cokatalysatora, der die
Komplexe in die polymerisationsaktive, kationische Spezies überführt.41
Abb. 17: Aktivierung eines Metallocens mit Tritylborat als Cokatalysator.
Neben den klassischen Aluminiumalkylen und Methylalum(in)oxanen (MAO) (Abb. 18, b; Abb.
19) kommen heute immer häufiger Perfluorarylborane (Abb. 18, c), Trityl- und
Ammoniumborate (Abb. 18, d) oder auch geträgerte Aktivatoren zum Einsatz.
Abb. 18: Wichtige Vertreter einzelner Cokatalysatorklassen.
a Die Bezeichnung „Cokatalysator“ ist weit verbreitet aber dennoch nicht ganz korrekt, da der „Cokatalysator“ die
Reaktion selbst nicht katalysiert. Im eigentlichen Sinn handelt es sich vielmehr um Aktivatoren. Gleichwohl wird
auch in dieser Arbeit an der Bezeichnung „Cokatalysator“ festgehalten, um diese von den Aktivatoren, die z. B. bei
der Voraktivierung eingesetzt werden, besser unterscheiden zu können.

I.1 Grundlagen
- 26 -
Aluminiumalkyle wie Trialkylaluminium oder Alkylaluminiumchloride sind wichtige
Komponenten in der klassischen heterogenen Ziegler-Natta-Katalyse. Ebenso wurde in der
frühen Literatur über eine Vielzahl homogener Katalysatoren basierend auf Aluminiumalkylen
als Cokatalysatoren berichtet. Jedoch waren deren Möglichkeiten beschränkt: Sie sind nicht in
der Lage, Propen oder höhere �-Olefine zu polymerisieren. Zahlreiche Versuche zur
Verbesserung der Katalysatorsysteme wurden unternommen. Erst der Zufall half Sinn und
Kaminsky diese Herausforderung zu meistern. Sie beobachteten eine überraschend hohe
Aktivität nach Zusatz von Wasser zu dem halogenfreien, polymerisations-inaktiven
Cp2ZrMe2/AlMe3-System – die Geburtsstunde des MAO.42,43 Der Zusatz von Wasser stellte
einen Widerspruch zu gängigen Theorien dar. Wasser ist das Katalysatorgift schlechthin.
Metallocene hydrolysieren heftig und schnell zu sehr stabilen Hafnium- bzw. Zirkoniumoxid-
hydroxiden und den entsprechenden Liganden. Allerdings beschrieben Reichert und Meyer
bereits 1973, dass geringe Mengen Wasser im System Cp2TiCl2/AlEtCl2 einen stark
geschwindigkeitserhöhenden Einfluss ausüben.44 Letztendlich brachte eine – wie sich später
herausstellen sollte – unsachgemäß verschlossene Trimethylaluminium-Flasche, die zu
unerklärlich hohen Aktivitäten führte, den Stein ins Rollen. Schnell zeigte sich, dass partiell
hydrolysiertes Trimethylaluminium als Cokatalysator in vielen Fällen dem reinen TMA
vorzuziehen ist.
Heute wird MAO bewusst durch kontrollierte Hydrolyse von Trimethylaluminium unter
Entwicklung von Methan hergestellt. Trotz intensiver Bemühung von akademischer wie auch
industrieller Seite ist die exakte Zusammensetzung und Struktur des MAO bis heute weder völlig
geklärt noch verstanden. Bekannt ist, dass es sich um komplex gebaute Oligomere handelt, die
aus fünf bis zwanzig –O-Al(Me)-Einheiten aufgebaut sind. Zusätzlich kann überschüssiges
Trimethylaluminium koordiniert sein. Als Strukturen werden lineare Ketten, Ringe und Cluster
vorgeschlagen.45
Abb. 19: Darstellung und wichtigste Strukturen des MAO.45

I.1 Grundlagen
- 27 -
Trotz seiner einzigartigen Effektivität als Cokatalysator bleibt MAO und die
Aktivierungssequenz eine „black box“. Abhängig vom eingesetzten Salz-Hydrat, das in der
Synthese als H2O-Quelle dient, und den exakten Synthesebedingungen können die MAO-
aktivierten Metallocene sehr stark in ihrer Aktivitäta variieren. Die Wirkung des MAO beruht
zum einen auf einer Methylierung des eingesetzten Metallocendichlorids, zum anderen besitzt es
eine lewis-acide Funktion: Es bewirkt die Abstraktion eines Methylanions unter Bildung der
eigentlichen polymerisationsaktiven Spezies. Das dabei entstandene [MAO-Me]--Anion
koordiniert vermutlich nicht oder nur schwach an das Metallocen-Kation, so dass es die
Koordination des Olefins nicht behindert. Noch wenig geklärt ist die Ausbildung einer
stabilisierenden Umgebung für das hochaktive Kation, eventuell in der Art eines Wirt-Gast- oder
Kronen-Alumoxan-Komplexes. Ein Vergleich mit Enzymen, in denen das kleine aktive Zentrum
durch eine große organische Hülle geschützt wird, bietet sich ebenfalls an.41
Abb. 20: Vorschlag zum Mechanismus der Aktivierung eines Metallocens durch MAO am Beispiel
von Zirkonocendichlorid.46
Zur erfolgreichen Aktivierung mit MAO bedarf es eines großen Überschusses gegenüber dem
Metallocen (Al : Metall � 1000 : 1), der mit Vorliegen eines ungünstigen Gleichgewichts
gedeutet wird.41 Zudem fungiert MAO als „scavanger“, der Verunreinigungen oder eventuell
vorhandene Wasserreste abfangen kann. Wie bereits kurz angesprochen, liegen in
Methylaluminoxan zwei Arten von Trimethylaluminium vor: freies und assoziiertes TMA. Hohe
Überschüsse an Aluminium können allerdings Abbruchreaktionen begünstigen (siehe I.1.4.3).
Die Aktivierung mittels Trityltetrakis(pentafluorophenyl)borat kommt ohne den hohen
Cokatalysator-Anteil aus (Borat : Katalysator � 6 : 1). Das Trityl-Kation Ph3C+ ist ein starkes
Hydrid- und Alkylid-Abstraktions-Reagenz. In Kombination mit Tetrakispentafluorophenylborat
a Mit dem Begriff „Aktivität“ wird in der Literatur nicht einheitlich umgegangen. So sind verschiedene Einheiten
wie kg/(mol×h) oder kg/(mol×h×bar) gebräuchlich.

I.1 Grundlagen
- 28 -
[B(C6F5)4]-, einem nicht oder nur schwach koordinierenden Anion, wurde [Ph3P]+[B(C6F5)4]
- als
effektiver Cokatalysator für die Aktivierung von Metallocenen entwickelt. Die entstehenden
Katalysatoren sind hoch aktiv für die Olefinpolymerisation.45
Metallocene, die als Dichloro-Spezies vorliegen, müssen um durch Tritylborat aktiviert werden
zu können, zunächst in die Dialkyl-Spezies überführt werden. Dies kann zum Beispiel durch
Umsetzung mit Triisobutylaluminium (TiBA) geschehen und wird später (Kapitel I.7.4) genauer
erklärt. Die Reaktion einer solchen alkylierten Spezies – in Abb. 21 gezeigt am Beispiel von
[(�5-C5Me4)SiMe2(N-tBu)]ZrMe2 – mit [Ph3P]+[B(C6F5)4]- in Toluol führt zu einem Aren-
Komplex, der die schwache Koordinationsfähigkeit des Borat-Anions widerspiegelt.45
Abb. 21: Aktivierung des CGC-Katalysators mit Tritylborat.
Rieger et al. hatten gezeigt, dass die Aktivierung mittels Trityltetrakis(pentafluorophenyl)borat
im Vergleich zur Aktivierung mit MAO bei Hafnocenen zu höheren Aktivitäten führt.47 Erst
kürzlich wurde beschrieben, dass die Frage der Aktivität in erster Linie eine Frage der
Aktivierung ist.48 Dies hatten früherer Arbeiten zu den CGCs bereits angedeutet.49 Daher wurde
bei den in dieser Arbeit durchgeführten Polymerisationen ausschließlich die Aktivierung mit
Tritylborat angewandt.
I.1 Grundlagen
- 29 -
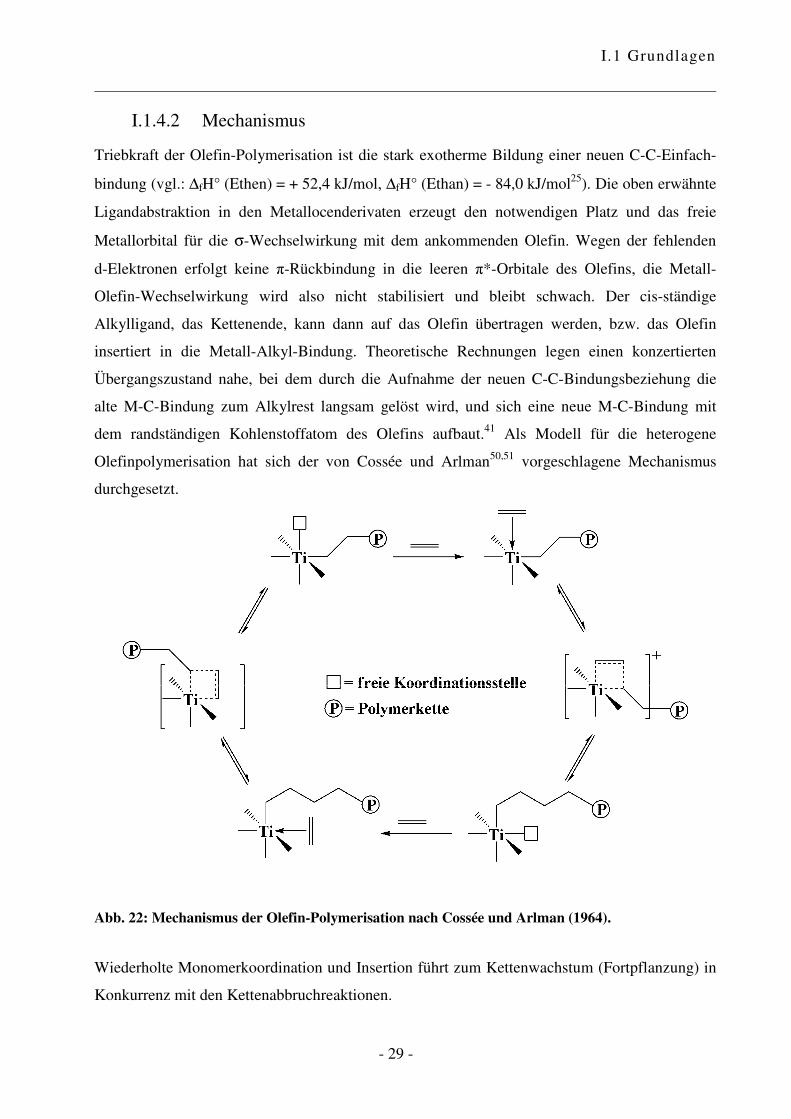
I.1.4.2 Mechanismus
Triebkraft der Olefin-Polymerisation ist die stark exotherme Bildung einer neuen C-C-Einfach-
bindung (vgl.: ∆fH° (Ethen) = + 52,4 kJ/mol, ∆fH° (Ethan) = - 84,0 kJ/mol25). Die oben erwähnte
Ligandabstraktion in den Metallocenderivaten erzeugt den notwendigen Platz und das freie
Metallorbital für die σ-Wechselwirkung mit dem ankommenden Olefin. Wegen der fehlenden
d-Elektronen erfolgt keine �-Rückbindung in die leeren �*-Orbitale des Olefins, die Metall-
Olefin-Wechselwirkung wird also nicht stabilisiert und bleibt schwach. Der cis-ständige
Alkylligand, das Kettenende, kann dann auf das Olefin übertragen werden, bzw. das Olefin
insertiert in die Metall-Alkyl-Bindung. Theoretische Rechnungen legen einen konzertierten
Übergangszustand nahe, bei dem durch die Aufnahme der neuen C-C-Bindungsbeziehung die
alte M-C-Bindung zum Alkylrest langsam gelöst wird, und sich eine neue M-C-Bindung mit
dem randständigen Kohlenstoffatom des Olefins aufbaut.41 Als Modell für die heterogene
Olefinpolymerisation hat sich der von Cossée und Arlman50,51 vorgeschlagene Mechanismus
durchgesetzt.
Abb. 22: Mechanismus der Olefin-Polymerisation nach Cossée und Arlman (1964).
Wiederholte Monomerkoordination und Insertion führt zum Kettenwachstum (Fortpflanzung) in
Konkurrenz mit den Kettenabbruchreaktionen.

I.1 Grundlagen
- 30 -
I.1.4.3 Abbruchreaktionen
Polymerisationen im industriellen Maßstab machen die Kontrolle des Molekulargewichts
unumgänglich. Meistens dient hierbei Wasserstoff als chemische Schere. Katalysator wie auch
Polymerrest werden hydriert und das Kettenwachstum somit gestoppt.
Abb. 23: Abbruch durch Hydrierung.
Doch auch ohne äußeres Zutun können Abbruchreaktionen nicht ganz verhindert werden. Die
wichtigste Kettenabbruchreaktion ist die �-Hydrideliminierung (Abb. 25 a), bei der ein Hydrid
der Polymerkette auf das Metallatom übertragen wird. Das Katalysatorzentrum wird hierbei
nicht deaktiviert. Die entstandenen Polymere haben olefinischen Endgruppen und können unter
Umständen als Comonomer wieder in die Kette eingebaut werden. Durch geschickte Wahl der
Liganden (sterisch anspruchvoll) kann diese Art des Abbruchs unterdrückt werden, ebenso ist sie
nach Insertion bestimmter sterisch anspruchsvoller Monomere (wie Norbornen) nicht möglich.52
Abb. 24: Insertion von Norbornena.
a Nach der Norbornen-Insertion befindet sich das �-H-Atom des Norbornens in einer endo-Position. Die
Grundvoraussetzung für den Kettenabbruch durch �-Hydrid-Eliminierung ist jedoch die Coplanarität der Metall-C-
Bindung und des �-H-Atoms. Durch die endo-Position kann der typische viergliedrige Übergangszustand nicht
gebildet werden. Auch die zweite Möglichkeit, die �-Hydrid-Eliminierung unter Teilnahme des Brücken-H-Atoms,
ist aufgrund der Bredt’schen Regel nicht möglich.
I.1 Grundlagen
- 31 -
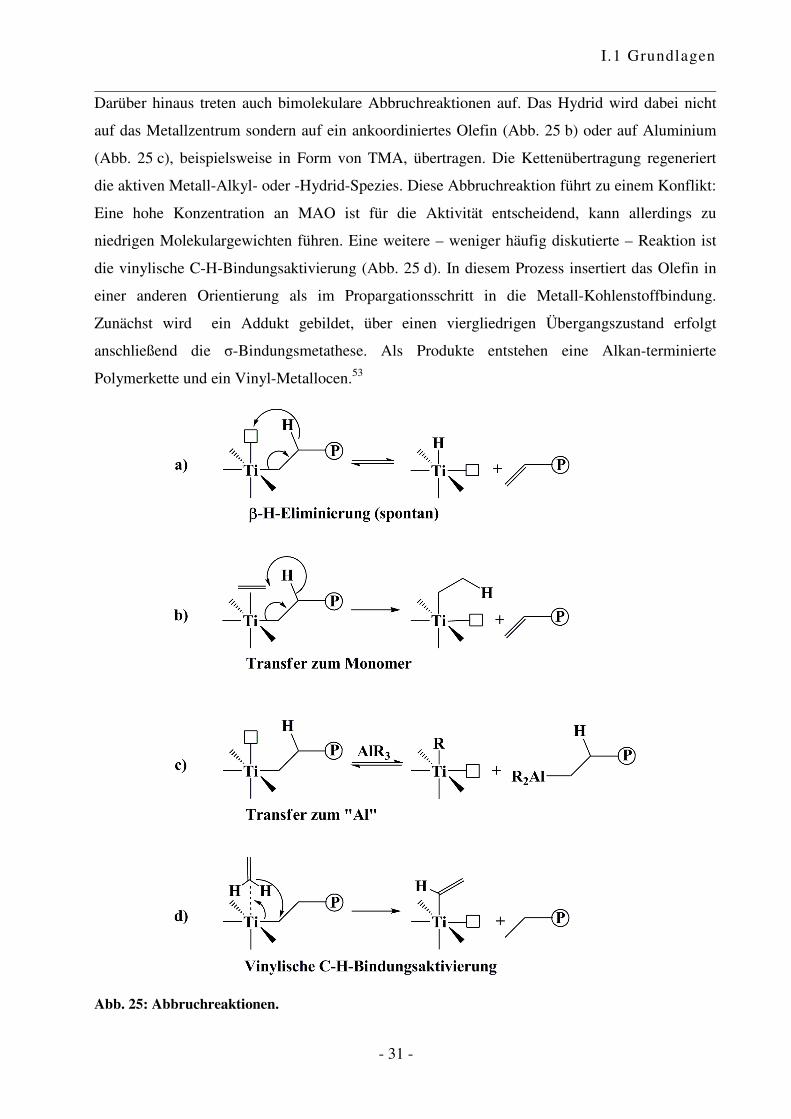
Darüber hinaus treten auch bimolekulare Abbruchreaktionen auf. Das Hydrid wird dabei nicht
auf das Metallzentrum sondern auf ein ankoordiniertes Olefin (Abb. 25 b) oder auf Aluminium
(Abb. 25 c), beispielsweise in Form von TMA, übertragen. Die Kettenübertragung regeneriert
die aktiven Metall-Alkyl- oder -Hydrid-Spezies. Diese Abbruchreaktion führt zu einem Konflikt:
Eine hohe Konzentration an MAO ist für die Aktivität entscheidend, kann allerdings zu
niedrigen Molekulargewichten führen. Eine weitere – weniger häufig diskutierte – Reaktion ist
die vinylische C-H-Bindungsaktivierung (Abb. 25 d). In diesem Prozess insertiert das Olefin in
einer anderen Orientierung als im Propargationsschritt in die Metall-Kohlenstoffbindung.
Zunächst wird ein Addukt gebildet, über einen viergliedrigen Übergangszustand erfolgt
anschließend die �-Bindungsmetathese. Als Produkte entstehen eine Alkan-terminierte
Polymerkette und ein Vinyl-Metallocen.53
Abb. 25: Abbruchreaktionen.

I.2 Aufgabenstellung
- 32 -
I.2 Aufgabenstellung
Ziel dieser Arbeit war das Erforschen der katalytischen Eigenschaften zweier isostruktureller
Zirkonium- bzw. Hafnium-Constrained-Geometry-Komplexe (siehe Abb. 26). Besonderes
Augenmerk galt der Frage, ob der Hafnium-Effekt auch für Halbsandwichverbindungen wie die
CGCs Gültigkeit besitzt. Die Darstellung und Analytik der Komplexe ist in Kapitel I.3
beschrieben.
Abb. 26: In dieser Arbeit untersuchte Constrained-Geometry-Komplexe.
Nach der erfolgreichen Synthese wurden zunächst die Polymerisationsparameter anhand
zahlreicher Experimente mit Ethen festgelegt. Besonderes Augenmerk galt jedoch den
Copolymerisationen. Hierfür wurden neben Propen Norbornen als sterisch anspruchsvolles und
1,3-Cyclohexadien als elektronisch anspruchsvolles Monomer gewählt (siehe Abb. 27). Die
Ergebnisse der Polymerisationsexperimente befinden sich in den Kapiteln I.4.2 bis I.4.5. Es
schließt sich eine ausführliche Fehlerbetrachtung in Kapitel I.4.6 an.
Abb. 27: Eingesetze Monomere.
Von großem Interesse war der Vergleich zwischen Zirkonium- und Hafnium, nicht nur bezüglich
der Komplexe sondern vor allem hinsichtlich eventueller Unterschiede der synthetisierten
Kunststoffe. Die Polymeranalytik spielte hierbei eine sehr wichtige Rolle. Die ausführliche
Diskussion der Ergebnisse erfolgt in den Kapiteln I.4.2 bis I.4.5.

I.3 Synthese der Katalysatoren
- 33 -
I.3 Synthese der Katalysatoren
I.3.1 Überblick
Über die letzten Jahre haben sich viele verschiedene Routen für die Synthese der CGCs
entwickelt. Dabei kann zwischen zwei generellen Ansätzen unterschieden werden: i) die
Synthese der Ligand-Vorstufe und anschließende Komplexierung mit einem Übergangsmetall
und ii) die Einführung der Brücke durch Reaktion in der Ligandensphäre eines
Übergangsmetallkomplexes. Ersterer wurde weit ausführlicher untersucht und zahlreiche
Varianten wurden entwickelt. Die Erfolg versprechendsten Wege führen über die Dimetallierung
der Liganden-Vorstufe gefolgt von einer Salz-Eliminierungs-Reaktion beziehungsweise die
direkte Reaktion der Liganden-Vorstufe mit Übergangsmetallamiden unter Amin-
Eliminierung.20
Bedingt durch die geringe Symmetrie des CpA-Liganden gibt es zwei Routen für die Synthese
des Ligandensystems. Die erste, häufiger verwendete, startet mit dem Cyclopentadienyl-
Fragment. Zunächst wird mittels Salzeliminierung die Silylbrücke eingeführt. Im Anschluss wird
die Amingruppe an dieses Zwischenprodukt ankondensiert.54 Die zweite Möglichkeit dreht die
Reihenfolge der Reaktionsschritte um. Zuerst wird Dichlordimethylsilan mit dem Amin
verknüpft. Dieses Fragment kann anschließend in quantitativer Reaktion mit dem
Cyclopentadienyl-Salz verbunden werden.55
Abb. 28: Synthesewege zum CpA-Ligand.
In Vorarbeiten wurden beide Strategien nachvollzogen und miteinander verglichen.49 Hierbei
konnte festgestellt werden, dass Syntheseweg 1 für die Darstellung des Cp�-Liganden zu
bevorzugen ist. Die Deprotonierung von Cp� mit BuLi, die nachfolgende Umsetzung mit
Dichlordimethylsilan und die Kondensationsreaktion mit dem Amin verlaufen alle quantitativ,
die Gesamtausbeute beträgt 89 %. Die Reinheit der Zwischenprodukte ist ausreichend, so dass
auf eine destillative Reinigung verzichtet werden kann. Die Vorteile des zweiten Synthesewegs

I.3 Synthese der Katalysatoren
- 34 -
wie z. B. das späte Einbringen des wertvollen Cp�-Fragments und die einfache Herstellung des
Silylamins in sehr guter Reinheit sind dennoch nicht zu vernachlässigen. Er ist daher sehr gut für
die Modifikation des Liganden (z. B. Fluorenyl statt Cp�) geeignet.
Wie bereits am Anfang des Kapitels erwähnt, kann die Synthese der entsprechenden CpA-
Komplexe auf verschiedene Arten erfolgen. In der klassischen Salzmetathese-Route wird
Zirkonium- oder Hafniumtetrachlorid mit dem Dilithiumsalz des Liganden umgesetzt (siehe
Abb. 29). Eine andere Metallierungs-Strategie nutzt den Vorteil der Acidität des protonierten
CpA-Liganden: anstelle der Tetrachloride kommen Tetrakis(dialkylamide) zum Einsatz. Petersen
zeigte, dass diese Eliminierungs-Route bei der Umsetzung einer 1 : 1 Reaktionsmischung des
protonierten Liganden mit Zr(NMe2)4 den CpA-Zr(NMe2)2-Komplex in 75 %-iger Ausbeute
liefert.56 Die Bis(dialkylamid)-Komplexe können mit Ammoniumchlorid oder einem
Überschuss Me3SiCl in die Dichloro-Spezies überführt werden, obgleich auch Amin-Addukte
gefunden worden sind.54 Darüber hinaus sind ausgehend vom vorgefertigten Liganden weitere
Synthesewege wie die Toluol-Eliminierung (Umsetzung des Liganden mit Titantetrabenzyl), die
Amin-assistierte HCl-Eliminierung (Reaktion des Liganden mit TiCl4 in Anwesenheit von
Triethylamin) und weitere Spezialfälle wie die Me3SiCl-Eliminierung oder die kombinierte LiCl
und Me3SiCl-Eliminierung bekannt.20
Abb. 29: Komplexsynthese nach der Salz-Eliminierungs-Route.
Die Synthese der Katalysatoren durch Reaktion in der Ligandensphäre des Übergangsmetalls ist
weniger ausführlich erforscht und in der Anwendbarkeit stark beschränkt. So führt die Reaktion
von [{(�5-C5H4)(SiMe2Cl)}TiCl3] mit NH2R und NEt3 (oder Li[NHR]) zum korrespondierenden
Komplex (siehe Abb. 30). Auf ähnliche Art und Weise sind auch Zirkonium- und
Hafniumkomplexe wie [{(�5-C5H4)(SiMe2Cl)}M(�5-1,3-tBuC5H3)Cl] zugänglich. Allerdings
wurde bisher weder die Synthese von Indenyl- noch von Fluorenyl-Analoga beschrieben –
möglicherweise aufgrund von Schwierigkeiten. Erst kürzlich wurde die Synthese von
Tetramethylcyclopentadienyl-Komplexen durch Reaktion von [{(�5-C5Me4)(SiMeClX)}TiCl3]

I.3 Synthese der Katalysatoren
- 35 -
(X = H, Cl) mit zwei Äquivalenten LiNHtBu zugänglich. Diese Variante könnte für die einfache
Modifikation des Amido-Fragments von Interesse sein.20
Abb. 30: CGC-Komplexsynthese durch Reaktion in der Ligandensphäre.
In dieser Arbeit wurden die Komplexe in Anlehnung an die Synthesen von Petersen56 und
Okuda33 ausgehend von ZrCl4(thf)2 bzw. HfCl4(thf)2 und dem Dilithiumsalz des Liganden
dargestellt (siehe Abb. 29).
Die Darstellung von Tetrachlorobis(tetrahydrofuran)zirkonium(IV) oder –hafnium(IV) erfolgte
nach der Methode von Manzer.57 Diese Tetrahydrofuran-Komplexe kommen häufig bei der
Synthese organometallischer Komplexe der frühen Übergangsmetalle zur Anwendung. Während
die wasserfreien Metallhalogene oft zu Disproportionierungsreaktionen neigen und die
Nitrilderivate MXn(NCR)y aufgrund der Reaktivität der Nitrile mit vielen anderen Reagenzien
wenig geeignet sind, stellen die Tetrahydrofuran-Komplexe eine nützliche Alternative für die
Synthese einer Vielzahl von metallorganischen Komplexen dar.57
Während [(C5Me4)SiMe2(N-tBu)]ZrCl2 in verschiedenen Quellen erwähnt wird18, 33, 56, 58, 59, gab
es bisher nur wenige Hinweise auf die Existenz des analogen Hafniumkomplexes.18, 19, 33 Die
Kristallstruktur des Hafnium-Komplexes [(C5Me4)SiMe2(N-tBu)]HfCl2 ist seit der Diplomarbeit
von Gröner49 bekannt.
I.3 Synthese der Katalysatoren
- 36 -
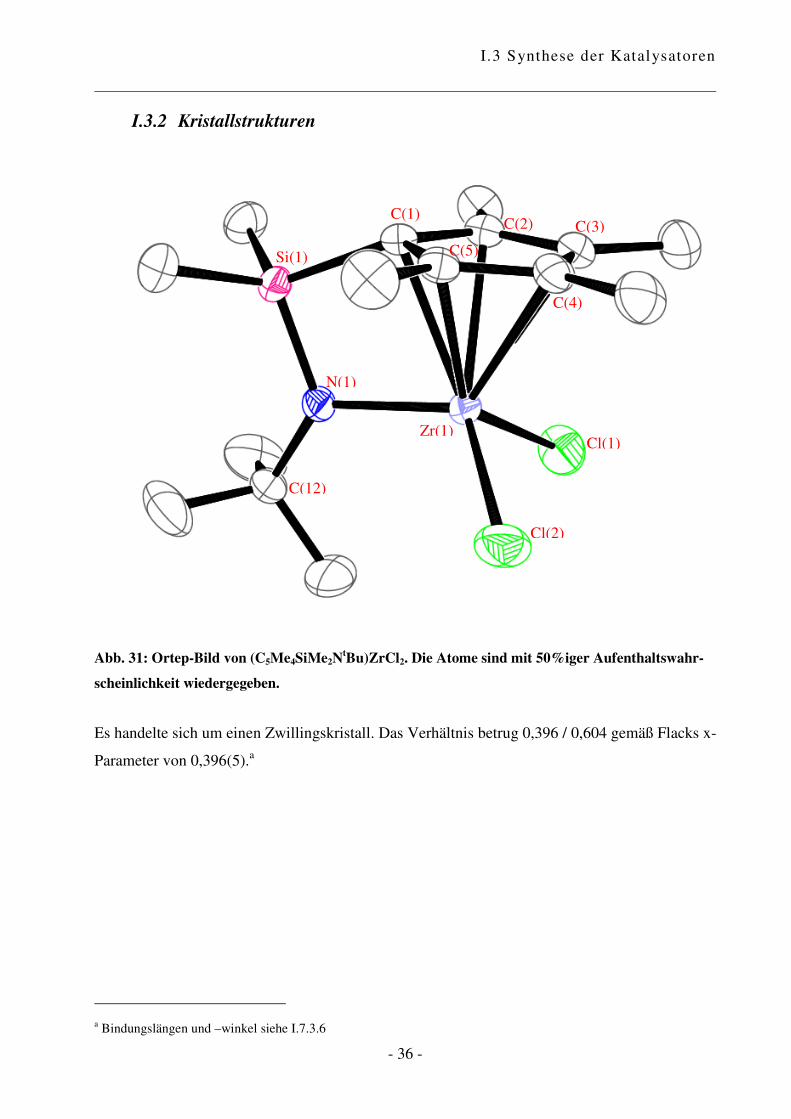
I.3.2 Kristallstrukturen
Abb. 31: Ortep-Bild von (C5Me4SiMe2NtBu)ZrCl2. Die Atome sind mit 50%iger Aufenthaltswahr-
scheinlichkeit wiedergegeben.
Es handelte sich um einen Zwillingskristall. Das Verhältnis betrug 0,396 / 0,604 gemäß Flacks x-
Parameter von 0,396(5).a
a Bindungslängen und –winkel siehe I.7.3.6
Zr(1) Cl(1)
Cl(2)
N(1)
C(1) C(2) C(3)
C(4)
C(5) Si(1)
C(12)
I.3 Synthese der Katalysatoren
- 37 -
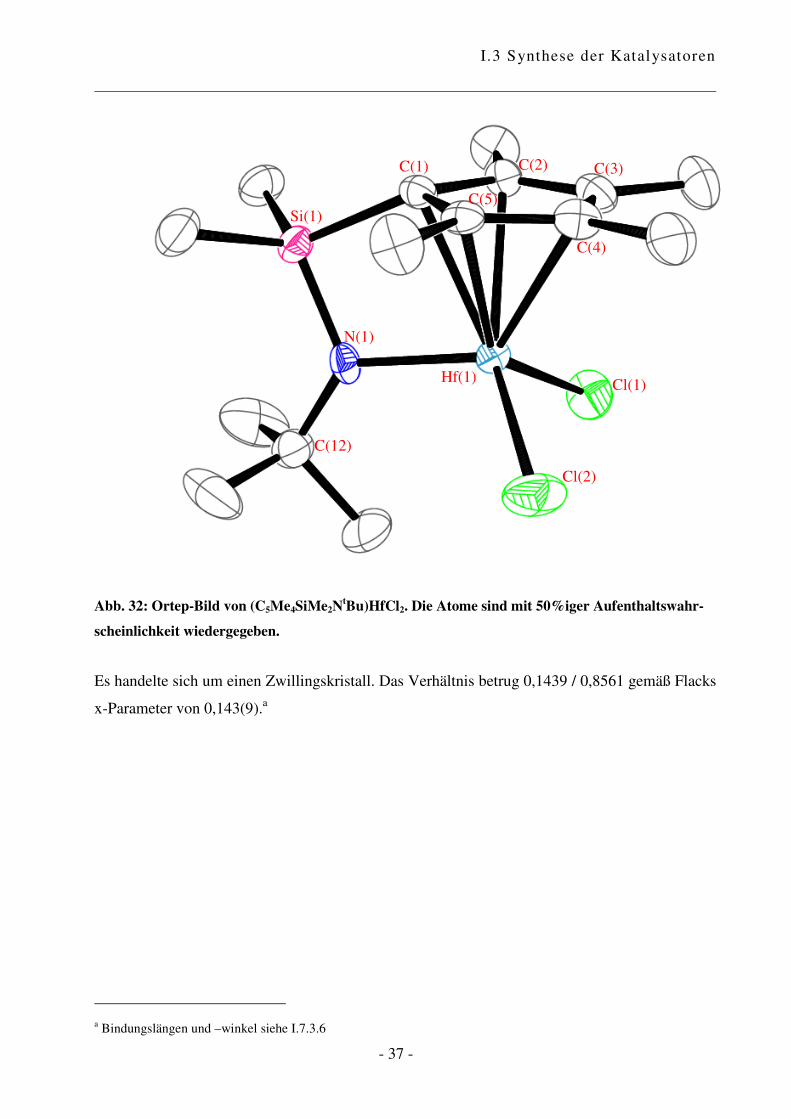
Abb. 32: Ortep-Bild von (C5Me4SiMe2NtBu)HfCl2. Die Atome sind mit 50%iger Aufenthaltswahr-
scheinlichkeit wiedergegeben.
Es handelte sich um einen Zwillingskristall. Das Verhältnis betrug 0,1439 / 0,8561 gemäß Flacks
x-Parameter von 0,143(9).a
a Bindungslängen und –winkel siehe I.7.3.6
C(1) C(2)
Cl(1)
C(3)
Hf(1)
C(4)
C(5)
C(12)
N(1)
Cl(2)
Si(1)

I.3 Synthese der Katalysatoren
- 38 -
Der CpA-Komplex weist eine dreibeinige Pianostuhl-Konfiguration mit pseudotetrahedraler
Anordnung der vier Liganden auf. Es liegt CS-Symmetrie vor.
Im Festkörper ist das Metallatom nicht zentrosymmetrisch an den Fünfring gebunden, was die
unterschiedlichen Abstände des Metalls zu den Ring-Kohlenstoffatomen von 2,399(2) bis
2,539(3) Å für Zr zeigen. Diese verzerrten Bindungslängen, bei denen das Brücken-C-Atom dem
Metall am nächsten ist, ist eine Konsequenz der Molekülorbitalstruktur des Cp-Anions.
Die Zirkonium-Stickstoff-Bindungslänge von 2,056(2) Å ist im typischen Bereich für einen
Amido-Ligand, der zu einem d0 Zirkonium-Zentrum mit einer starken �-Bindung gebunden ist:
Cp*Zr(NHCMe3)3 2,00(1), 2,02(1); MeZr[NHSi(CMe3)3)]3 2,039(7); [Zr(�5:�1-
C5H4CH2CH2CH2NMe)Cl(CH2Ph)]2 1,988(2)58, 2,034 (C13H8-SiMe2-NtBu)ZrCl2.
34
Das Stickstoff-Atom ist trigonal planar; die Summe der Bindungswinkel Si(1)–N(1)–C(12),
C(12)–N(1)–Zr/Hf(1) und Si(1)–N(1)–Zr/Hf(1) ist 360°. Das N-Atom ist sp2-hybridisiert, das
freie Elektronenpaar befindet sich in einem pz-Orbital und kann mit einem Hybridorbital des
Zentralmetalls in Wechselwirkung treten. Die Metall-N-Bindungslänge und die Hybridisierung
am Stickstoffatom deuten darauf hin, dass der Amidosubstituent in diesem Komplex als Vier-
Elektronen-Ligand wirkt, und die ansa-Halbsandwichverbindung folglich als 14 Elektronen-
Komplex vorliegt.
Beim Vergleich des Zirkonium- mit dem analogen Hafnium-Komplex zeigt sich, dass die
Bindungen, die vom Zentralmetall ausgehen, in der Hafnium-Spezies grundsätzlich kürzer sind.
Das Hafnium-Atom ist stärker an den Cyclopentadienyl-Ring und an das Stickstoffatom
gebunden, wodurch sich der Winkel centroid–Zr/Hf(1)–N(1) von 101,98° für Zirkonium auf
103,17° vergrößert. Als Konsequenz daraus rücken die beiden Chlor-Atome näher zusammen,
was durch eine Verkleinerung des Winkels Cl(1)–Zr/Hf(1)–Cl(2) deutlich wird. Die Winkel
zwischen centroid–Zr/Hf(1) und den beiden Chlor-Atomen sind im Hafnium-Komplex größer als
in der entsprechenden Zirkonium-Verbindung.
Es bleibt die Frage, welche Konsequenzen sich aus diesen Strukturen ergeben. Die Unterschiede
in den Bindungslängen und –winkeln sind nicht gewaltig, aber durch aus in einem Maß, das
entscheidenden Einfluss auf das Verhalten haben kann. Die Struktur des Hafnium-Komplexes
bietet etwas mehr Platz für ankoordinierende Moleküle, was sich in einer größeren Toleranz
gegenüber anspruchsvollen Monomeren zeigen könnte. Gleichzeitig bedeutet das aber, dass es
auch für Abbruchreaktionen wie �-Hydrid-Eliminierung mehr Raum gibt. Dies wiederum könnte
durch die stärkere Hafnium-Kohlenstoff-Bindung kompensiert werden.60 Die Verkleinerung des
Chlor-Metall-Chlor-Winkels vom Zirkonium zum Hafnium kann sich in einer höheren Turn-
Over-Zahl zeigen, wobei diese vermutlich mehr von der Elektronik abhängig ist.

I.4 Polymerisationen
- 39 -
I.4 Polymerisationen
I.4.1 Experimenteller Teil
I.4.1.1 Arbeitsmaterialien
Das für die Polymerisationen verwendete Toluol stammt von Fa. Merck (p.a.-Qualität) und
wurde vor Gebrauch 16 h über Lithiumaluminiumhydrid gerührt, anschließend 5 h unter
Rückfluss gekocht, abdestilliert und entgast. Das Toluol zum Reinigen des Autoklaven wurde
über Aluminiumoxid (90 aktiv neutral von Merck) gereinigt und anschließend entgast.
Triisobutylaluminium (TiBA) 1,1 M in Toluol stammt von Acros Organics.
Trityltetrakispentafluorophenylborat wurde im Fortgeschrittenen-Praktikum für Anorganische
Chemie nach der Vorschrift von Lambert et al.61 synthetisiert.
Ethen (Reinheit 2.7) und Propen (Reinheit 2.6) wurden über Molekularsieb (4Å) getrocknet.
Restsauerstoff wurde durch einen handelsüblichen Kupfer(I)-Katalysator entfernt.
1,3-Cyclohexadien wurde über Aluminiumoxid (90 aktiv neutral von Merck) getrocknet und
frisch destilliert. Norbornen (Merck) wurde als Feststoff in ein Schlenkrohr eingewogen,
sekuriert und anschließend in Toluol gelöst.
Alle Polymerisationen wurden unter Argon (Reinheit 5.0) durchgeführt. Dabei wurden Standard-
Schlenktechniken angewendet. Der Autoklav wurde nach jeder Polymerisation mechanisch
gesäubert und mit einem Gemisch aus 300 mL trockenem Toluol und 5 mL TiBA 1 h bei 80 °C
gereinigt.
I.4.1.2 Polymerisationsapparatur
Die Polymerisationsversuche wurden in einem 1L-Büchi-Stahlautoklaven (Abb. 34)
durchgeführt. Druck und Temperatur wurden während den Polymerisationen konstant gehalten
und mittels eines Druckreglers (P-602; ±50 mbar) und eines Thermostaten (Julabo-Thermostat;
ATX-Serie) kontrolliert. Die gereinigten Monomergase wurden kontinuierlich in den Autoklaven
eingeleitet; die Steuerung der zugefügten Gasmenge erfolgte durch einen Durchflussregler
(Bronkhorst flow-meter F-201C).
Der Start der Polymerisation erfolgte in situ durch die Zugabe der Cokatalysator-Lösung mittels
einer HPLC-Pumpe (Waters M 600).

I.4 Polymerisationen
- 40 -
Abb. 33: Blockschaltbild der Polymerisationsanlage.
Abb. 34: Foto des Druckreaktors.
HPLC-
Pumpe
Legende: Vorrat Cx = Ethen- bzw. Propengasflasche Rein. Cx = Reinigungstürme für Ethen bzw. Propen M/S Control = Master/Slave Kontrolle durchgezogene Linie = Massenfluss (z. B. Gas, Aktivatorlösung) gestrichelte Linie = Datenfluss Zwischen M/S Control sowie Thermostat besteht eine zusätzliche Verbindung mit dem Daten aufzeichnenden Computer.
Thermostat
Rein. C2
Rein. C3
Fluss- messer
Fluss- messer
M/S Control Druck-
messer
Zugabe-Hahn
HPLC- Pumpe
Vorrat C3
Vorrat C2

I.4 Polymerisationen
- 41 -
I.4.1.3 Polymercharakterisierung
Die NMR-Messungen wurden bei einer Temperatur von 373 K an einem Bruker AMX-500
Spektrometer durchgeführt. Als Lösungsmittel wurde Brombenzol-d5 (Deutero GmbH)
verwendet. Für die 13C-NMR-Spektren der Polymere wurden standardmäßig 4000 scans
aufgenommen.
Molekulargewichte und Molekulargewichtsverteilungen wurden durch Hochtemperatur-
Gelpermeationschromatographie (HT-GPC) bestimmt. Dazu wurde ein Gerät der Firma Waters
(Alliance GPC 2000; 145°C; 1,2,4-Trichlorbenzol, Fluss: 0,5 mL/min) mit einem
Differentialrefraktometer (RI) als Detektor verwendet. Die Auswertung erfolgte relativ gegen
einen Polystyrol-Standard.
Glasübergangstemperaturen und Schmelzpunkte wurden kalorimetrisch an einem Perkin-Elmer
DSC-7 gemessen. Von -50 °C bis 180 °C wurde mit einer Heizrate von 10 °C/min aufgeheizt,
anschließend 0,1 Minuten bei 180 °C gehalten und in Schritten von 10 °C/min wieder auf -50 °C
abgekühlt. Nachdem die Temperatur für 0,3 Minuten bei -50 °C gehalten worden war, wurde
erneut mit 10 °C/min auf 180 °C geheizt. Es wurden die Werte des zweiten Aufheizvorgangs
ausgewertet.
I.4.1.4 Durchführung der Polymerisationen
Der Katalysator wurde durch Lösen in Toluol/TiBA (Al : Zr/Hf = 100 : 1) und 60 min Rühren
bei 60 °C voraktiviert (siehe I.7.4).
Abb. 35: Die Katalysatorsysteme 1 und 2.
Die Polymerisationen wurden in 300 mL Toluol durchgeführt. Zum Entfernen von Restwasser
und anderen Verunreinigungen wurde 1 mL Triisobutylaluminium (1,1 mmol/mL in Toluol)

I.4 Polymerisationen
- 42 -
zugesetzt. Nach kurzem Rühren wurde eine Lösung des voraktivierten Katalysators zugegeben.
Die Reaktionslösung wurde auf Polymerisationstemperatur erwärmt, anschließend wurde Ethen
bis zum gewählten Druck aufgepresst. Zum Starten der Polymerisation wurde die entsprechende
Menge Tritylborat-Lösung mittels einer HPLC-Pumpe zugegeben. Der Verlauf der
Polymerisation wurde online über den Gasfluss kontrolliert. Der Abbruch der Polymerisationen
erfolgte durch Schließen des Gaseinlassventils und vorsichtiges Dekomprimieren des Reaktors.
Die Polymerisationslösung wurde in einem Liter leicht saurem Methanol gefällt. Das Polymer
wurde mit Methanol gewaschen und im Vakuumtrockenschrank bei 60 °C bis zur
Gewichtskonstanz getrocknet.
Für die Copolymerisationen mit 1,3-Cyclohexadien und Norbornen wurde die Toluol-Menge auf
290 mL reduziert. Die Zugabe des Comonomers erfolgte nach dem TiBA, vor dem Katalysator.
Beim Ausfällen der 1,3-Cyclohexadien-Copolymere wurde 2,6-Di-tert-butyl-4-methylphenol als
Stabilisator zugesetzt.
I.4.1.5 Parameterbestimmung
Das Ergebnis von Polymerisationsreaktionen wird von unterschiedlichsten Faktoren beeinflusst,
die Auswirkungen auf die Katalysatoraktivität und die Polymereigenschaften haben können. Es
ist sinnvoll, eine Vorauswahl an Parametern zu treffen, um möglichst schnell einen Eindruck
vom Leistungsvermögen des Katalysators zu bekommen. Im folgenden Kapitel wird das
Konzept dieser Auswahl erläutert.
Bei der Festlegung von Einflussgrößen ist es das oberste Ziel, gleich bleibende
Reaktionsbedingungen zu erreichen. Nur so kann es gelingen, reproduzierbare und vergleichbare
Ergebnisse sowie einheitliche Produkte zu bekommen. Ganz entscheidend ist hierfür die schnelle
Einstellung des Gleichgewichts zwischen Gas- und flüssiger Phase, damit die Konzentration des
Monomers in der Reaktionslösung konstant ist.a Der zweite wesentliche Faktor ist die
Temperatur. Die Parameter müssen so gewählt werden, dass die freiwerdende Reaktionswärme
schnellstmöglich abgeführt werden kann. Unter Anwendung der RGT-Regel als erste Näherung,
führt eine Erhöhung der Temperatur um 10 K zu einer Verdoppelung bis Vervierfachung der
Reaktionsgeschwindigkeit.5 Folglich können selbst kleine Temperaturschwankungen große
a Die Konzentration des Monomers in Lösung hängt entscheidend von der Konzentration des gebildeten Polymers
ab. In dieser Arbeit wurde das Semi-Batch-Verfahren angewendet. Anders als bei der Batch-Methode werden hier
zwar Edukte nachgegeben, jedoch keine Produkte abgeführt, wie es in der kontinuierlichen Methode zusätzlich der
Fall ist.

I.4 Polymerisationen
- 43 -
Auswirkungen auf das Reaktionsergebnis haben und eine vergleichende Beurteilung der
Katalysatoren erschweren.
Die ersten Parameter werden durch die Wahl der Polymerisationsanlage (siehe I.4.1.2) fest-
gelegt. Dadurch sind bereits die Reaktorgröße und das –oberflächenmateriala sowie die
Geometrie des Rührers und Strömungsbrechers bestimmt. Die Reaktorgröße gibt die
Ansatzgröße und damit z. B. Lösungsmittelvolumen oder Scavangermenge vor. Zur
bestmöglichen Homogenisierung der Reaktionslösung und zur schnelleren Einstellung des
Gleichgewichts wird für die Geschwindigkeit des Rührers die maximal empfohlene
Umdrehungszahl gewählt. Die Leistung des Thermostaten gab den Temperaturbereich von 30 bis
70 °C vor. Zudem bestehen aus Sicherheitsgründen Begrenzungen für den Druck (hier maximal
6 bar).6263
Als Lösungsmittel wurde Toluol verwendet. Es lässt sich sehr gut mittels Standardmethoden
trocknenb und hat einen höheren Siedepunkt als gängige aliphatische Kohlenwasserstoffe (Sdp.
Toluol = 112 °C, Sdp. Hexan = 69 °C, Sdp. Heptan = 98 °C)64, was höhere Reaktions-
temperaturen erlaubt. Die verwendeten Monomergase, Katalysator, Aktivator sowie die
polymeren Produkte sind in Toluol in einem weiten Temperaturbereich sehr gut löslich. Der
Aktivator kann zudem als toluolische Lösung käuflich erworben werden. Beim Scavanger wurde
auf Triisobutylaluminium zurückgegriffen, da dieses bereits als Aktivator dient. Es sind auch
andere Aluminiumalkylverbindungen wie MAO gebräuchlich, diese würden eine neue
Komponente darstellen und Raum für unerwünschte Nebenreaktionen eröffnen (siehe auch
Abbruchreaktionen Kapitel I.1.4.3).
In Vorarbeiten ist es gelungen, erste Ergebnisse in Hinblick auf den geeigneten Druck sowie den
Cokatalysator zu erlangen.49 Innerhalb dieser Arbeit wurde das optimale Katalysator-
Cokatalysator-Verhältnis nochmals untersucht. Wie in Diagramm 1 ersichtlich ist, steigt die
Aktivität mit Zunahme der Cokatalysator-Menge zunächst stark an. Bei einem Verhältnis von
6:1 wird die optimale Konzentration an katalytisch aktiven Zentren erreicht. Diese Beobachtung
entspricht dem bekannten Verhalten.65 Für alle folgenden Experimente wurde daher ein
Verhältnis Cokatalysator zu Katalysator von 6:1 verwendet.
a Reaktionen an aktiven Oberflächen können zu unerwünschten Nebenprodukten führen und einen Einfluss auf den
Verlauf sowie das Ergebnis der Polymerisationsreaktion haben. Übersichtsartikel zu heterogenen Katalysatoren
siehe Lit. 62 und 63. b Es wurden grundsätzlich Restfeuchtigkeits-Werte von ca. 5 ppm erreicht.
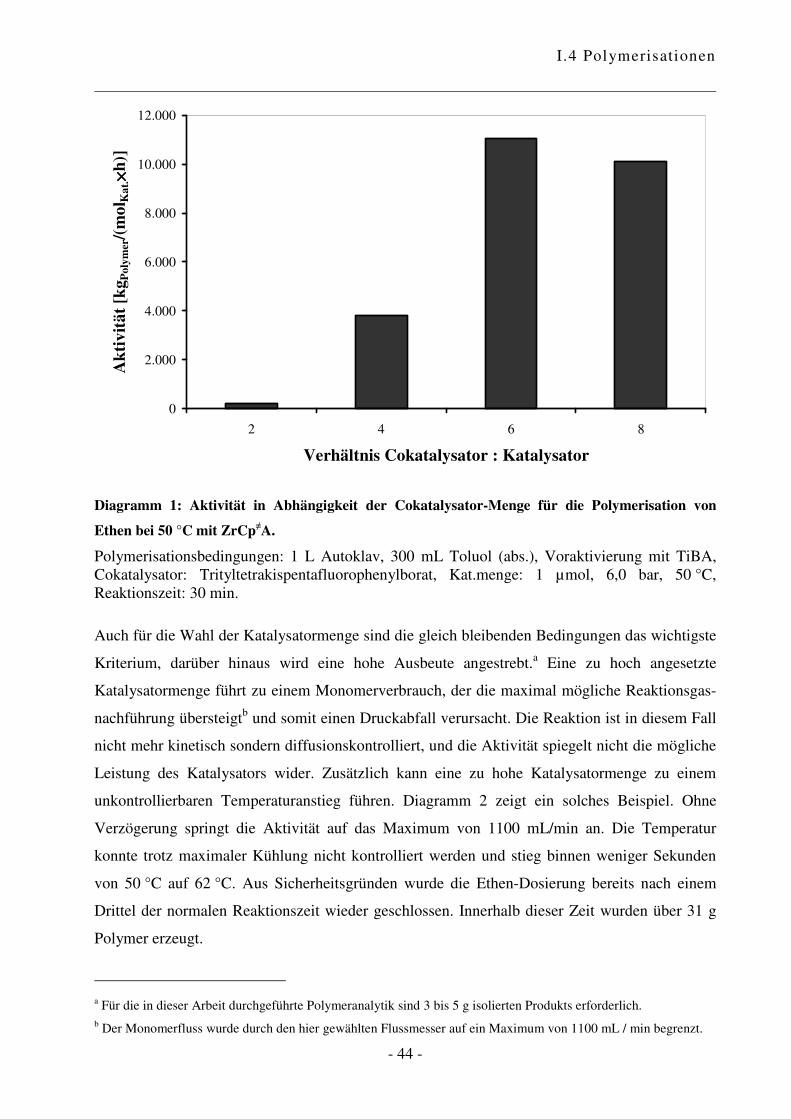
I.4 Polymerisationen
- 44 -
0
2.000
4.000
6.000
8.000
10.000
12.000
2 4 6 8
Verhältnis Cokatalysator : Katalysator
Akt
ivit
ät [
kgP
olym
er/(
mol
Kat
.×× ××h)
]
Diagramm 1: Aktivität in Abhängigkeit der Cokatalysator-Menge für die Polymerisation von
Ethen bei 50 °C mit ZrCp�A.
Polymerisationsbedingungen: 1 L Autoklav, 300 mL Toluol (abs.), Voraktivierung mit TiBA, Cokatalysator: Trityltetrakispentafluorophenylborat, Kat.menge: 1 µmol, 6,0 bar, 50 °C, Reaktionszeit: 30 min.
Auch für die Wahl der Katalysatormenge sind die gleich bleibenden Bedingungen das wichtigste
Kriterium, darüber hinaus wird eine hohe Ausbeute angestrebt.a Eine zu hoch angesetzte
Katalysatormenge führt zu einem Monomerverbrauch, der die maximal mögliche Reaktionsgas-
nachführung übersteigtb und somit einen Druckabfall verursacht. Die Reaktion ist in diesem Fall
nicht mehr kinetisch sondern diffusionskontrolliert, und die Aktivität spiegelt nicht die mögliche
Leistung des Katalysators wider. Zusätzlich kann eine zu hohe Katalysatormenge zu einem
unkontrollierbaren Temperaturanstieg führen. Diagramm 2 zeigt ein solches Beispiel. Ohne
Verzögerung springt die Aktivität auf das Maximum von 1100 mL/min an. Die Temperatur
konnte trotz maximaler Kühlung nicht kontrolliert werden und stieg binnen weniger Sekunden
von 50 °C auf 62 °C. Aus Sicherheitsgründen wurde die Ethen-Dosierung bereits nach einem
Drittel der normalen Reaktionszeit wieder geschlossen. Innerhalb dieser Zeit wurden über 31 g
Polymer erzeugt.
a Für die in dieser Arbeit durchgeführte Polymeranalytik sind 3 bis 5 g isolierten Produkts erforderlich. b Der Monomerfluss wurde durch den hier gewählten Flussmesser auf ein Maximum von 1100 mL / min begrenzt.
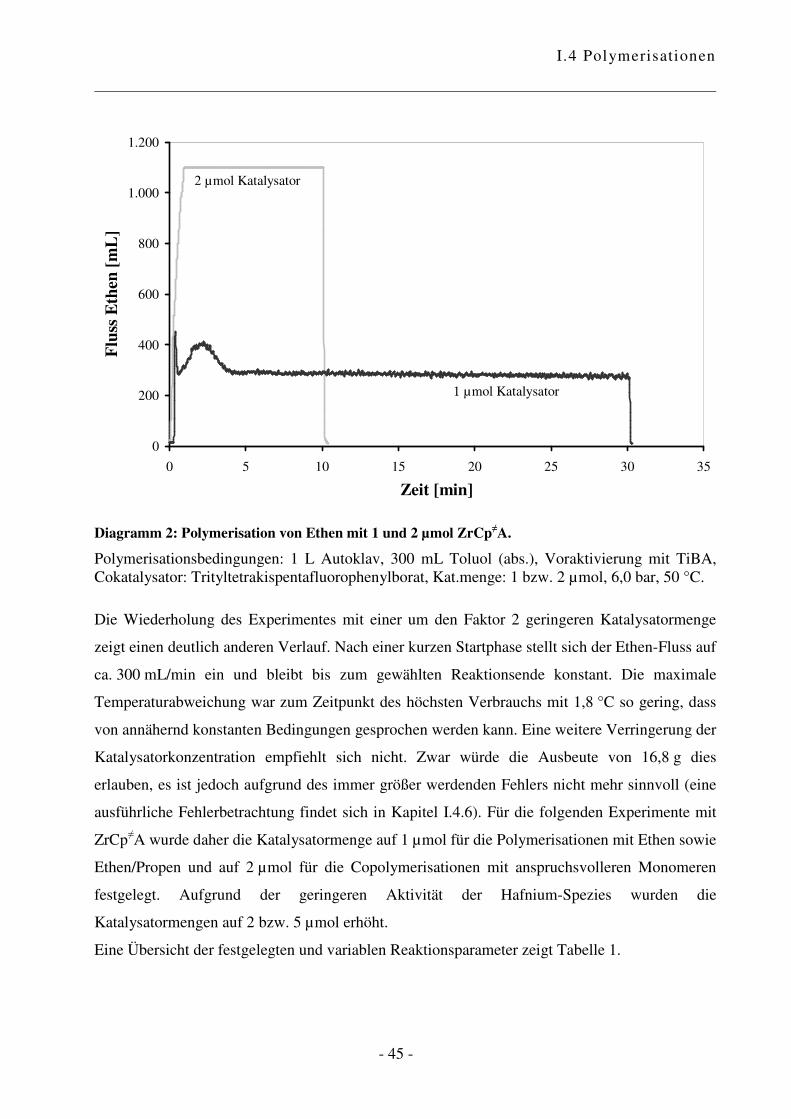
I.4 Polymerisationen
- 45 -
0
200
400
600
800
1.000
1.200
0 5 10 15 20 25 30 35
Zeit [min]
Flu
ss E
then
[m
L]
2 µmol Katalysator
1 µmol Katalysator
Diagramm 2: Polymerisation von Ethen mit 1 und 2 µmol ZrCp�A.
Polymerisationsbedingungen: 1 L Autoklav, 300 mL Toluol (abs.), Voraktivierung mit TiBA, Cokatalysator: Trityltetrakispentafluorophenylborat, Kat.menge: 1 bzw. 2 µmol, 6,0 bar, 50 °C.
Die Wiederholung des Experimentes mit einer um den Faktor 2 geringeren Katalysatormenge
zeigt einen deutlich anderen Verlauf. Nach einer kurzen Startphase stellt sich der Ethen-Fluss auf
ca. 300 mL/min ein und bleibt bis zum gewählten Reaktionsende konstant. Die maximale
Temperaturabweichung war zum Zeitpunkt des höchsten Verbrauchs mit 1,8 °C so gering, dass
von annähernd konstanten Bedingungen gesprochen werden kann. Eine weitere Verringerung der
Katalysatorkonzentration empfiehlt sich nicht. Zwar würde die Ausbeute von 16,8 g dies
erlauben, es ist jedoch aufgrund des immer größer werdenden Fehlers nicht mehr sinnvoll (eine
ausführliche Fehlerbetrachtung findet sich in Kapitel I.4.6). Für die folgenden Experimente mit
ZrCp�A wurde daher die Katalysatormenge auf 1 µmol für die Polymerisationen mit Ethen sowie
Ethen/Propen und auf 2 µmol für die Copolymerisationen mit anspruchsvolleren Monomeren
festgelegt. Aufgrund der geringeren Aktivität der Hafnium-Spezies wurden die
Katalysatormengen auf 2 bzw. 5 µmol erhöht.
Eine Übersicht der festgelegten und variablen Reaktionsparameter zeigt Tabelle 1.
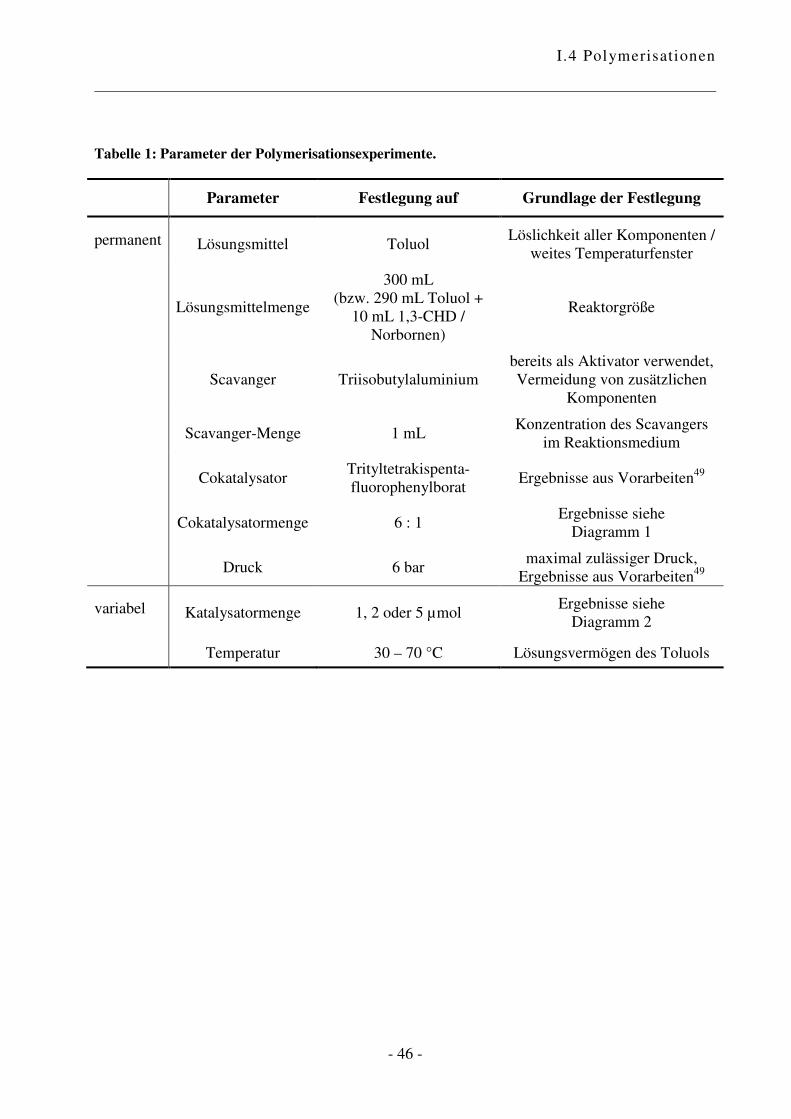
I.4 Polymerisationen
- 46 -
Tabelle 1: Parameter der Polymerisationsexperimente.
Parameter Festlegung auf Grundlage der Festlegung
permanent Lösungsmittel Toluol Löslichkeit aller Komponenten /
weites Temperaturfenster
Lösungsmittelmenge
300 mL (bzw. 290 mL Toluol +
10 mL 1,3-CHD / Norbornen)
Reaktorgröße
Scavanger Triisobutylaluminium bereits als Aktivator verwendet, Vermeidung von zusätzlichen
Komponenten
Scavanger-Menge 1 mL Konzentration des Scavangers
im Reaktionsmedium
Cokatalysator Trityltetrakispenta-fluorophenylborat
Ergebnisse aus Vorarbeiten49
Cokatalysatormenge 6 : 1 Ergebnisse siehe
Diagramm 1
Druck 6 bar maximal zulässiger Druck,
Ergebnisse aus Vorarbeiten49
variabel Katalysatormenge 1, 2 oder 5 µmol Ergebnisse siehe
Diagramm 2
Temperatur 30 – 70 °C Lösungsvermögen des Toluols

I.4 Polymerisationen
- 47 -
I.4.2 Polymerisation von Ethen
Abb. 36: Synthese von Polyethen.
Um Aufschlüsse über das Polymerisationsverhalten der CGCs zu erhalten, wurde in den ersten
Experimenten Ethen homopolymerisiert. Ethen als das kleinste Olefin ist sterisch am wenigsten
anspruchsvoll und wird in der Regel am schnellsten polymerisiert. Daher ist es ideal geeignet,
um die Leistungsfähigkeit der Katalysatoren auszutesten. Sind die optimalen Bedingungen für
die Polymerisation von Ethen gefunden, können diese auch auf die folgenden
Copolymerisationen übertragen werden, ohne dass befürchtet werden muss, dass die Reaktion
unkontrollierbar schnell voranschreitet.
Das erste Kriterium für die Beurteilung eines Katalysators ist die Aktivität. Sie wird bestimmt
über die Masse des erzeugten Polymers in Bezug auf Polymerisationszeit und eingesetzte
Katalysatormenge (siehe Gleichung 1).a
eit[h]Reaktionsz[mol]Stoffmenge
ge[kg]Polymermen Aktivität
rKatalysato ×=
Gleichung 1: Berechnung der Aktivität.
Bei den hier durchgeführten Experimenten zeigten die Katalysatoren mit 6×103 – 40×103
kg/(mol×h) sehr hohe Aktivitätenb, wobei die von 1 um den Faktor 5-7 größer als die von 2 ist.
Dieses Verhalten der Gruppe-4-Metallocene wurde bereits in Kapitel I.1.3.4 diskutiert und
a Mit dem Begriff „Aktivität“ wird in der Literatur nicht einheitlich umgegangen. So sind verschiedene Einheiten
wie kgPolymer/(molKatalysator×h), kgPolymer/(molKatalysator×h×bar) oder kgPolymer/(gMetall×h) gebräuchlich. Ein Maß für die
Effizienz eines Katalysators ist neben der Aktivität auch die Wechselzahl (engl. Turnover Number, TON). Sie gibt
die Anzahl an Formelumsätzen an, die ein bestimmter Katalysator beschleunigen kann, ehe er inaktiv wird. Die
Turnover Frequency (TOF) ist die Zahl an Umsätzen pro Zeiteinheit. Durch diese verschiedenen Definitionen ist ein
Vergleich von Katalysatoren ohne aufwändige Umrechnung sehr schwierig wenn nicht sogar unmöglich, da oft
nicht alle Daten, die dazu relevant wären, veröffentlicht werden. Zur Aktivität siehe auch: Kapitel I.1.4.1. b In ihrem Aufsatz über die Olefinpolymerisation stufen Britovsek, Gibson und Wass eine Aktivität über
1.000 kg/(mol×h×bar) als sehr hoch ein.8
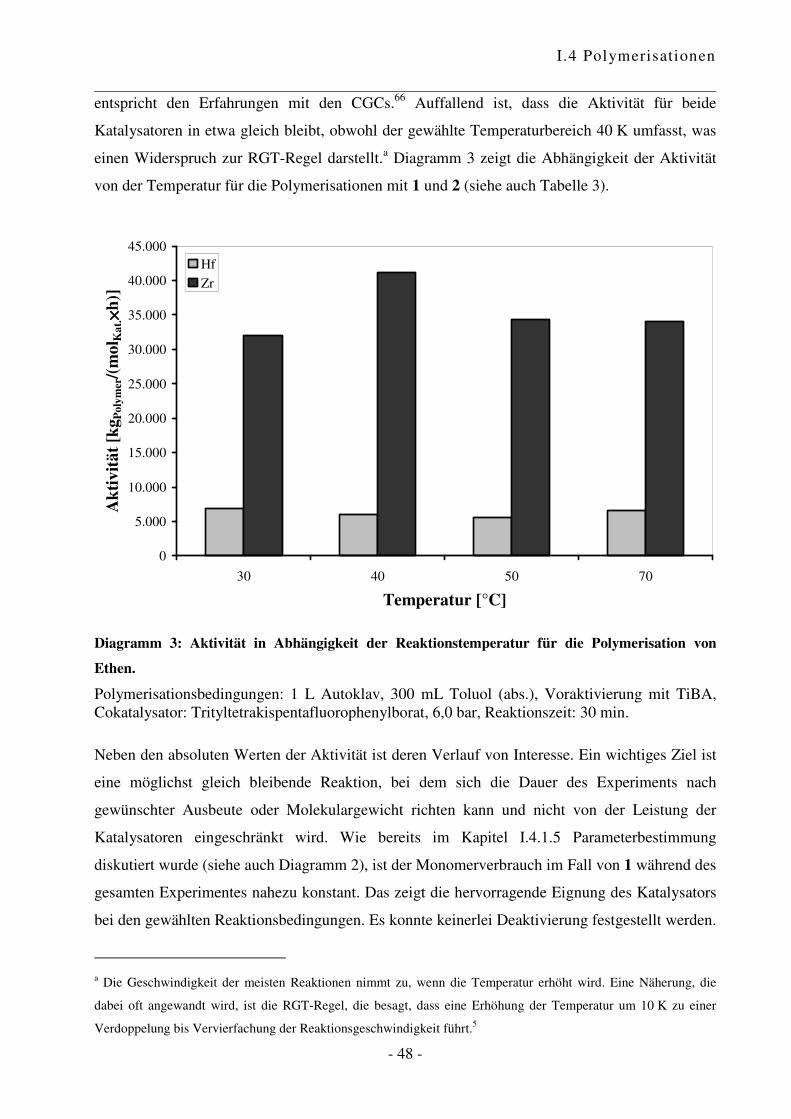
I.4 Polymerisationen
- 48 -
entspricht den Erfahrungen mit den CGCs.66 Auffallend ist, dass die Aktivität für beide
Katalysatoren in etwa gleich bleibt, obwohl der gewählte Temperaturbereich 40 K umfasst, was
einen Widerspruch zur RGT-Regel darstellt.a Diagramm 3 zeigt die Abhängigkeit der Aktivität
von der Temperatur für die Polymerisationen mit 1 und 2 (siehe auch Tabelle 3).
0
5.000
10.000
15.000
20.000
25.000
30.000
35.000
40.000
45.000
30 40 50 70
Temperatur [°C]
Akt
ivit
ät [
kgP
olym
er/(
mol
Kat
.×× ××h)
]
HfZr
Diagramm 3: Aktivität in Abhängigkeit der Reaktionstemperatur für die Polymerisation von
Ethen.
Polymerisationsbedingungen: 1 L Autoklav, 300 mL Toluol (abs.), Voraktivierung mit TiBA, Cokatalysator: Trityltetrakispentafluorophenylborat, 6,0 bar, Reaktionszeit: 30 min.
Neben den absoluten Werten der Aktivität ist deren Verlauf von Interesse. Ein wichtiges Ziel ist
eine möglichst gleich bleibende Reaktion, bei dem sich die Dauer des Experiments nach
gewünschter Ausbeute oder Molekulargewicht richten kann und nicht von der Leistung der
Katalysatoren eingeschränkt wird. Wie bereits im Kapitel I.4.1.5 Parameterbestimmung
diskutiert wurde (siehe auch Diagramm 2), ist der Monomerverbrauch im Fall von 1 während des
gesamten Experimentes nahezu konstant. Das zeigt die hervorragende Eignung des Katalysators
bei den gewählten Reaktionsbedingungen. Es konnte keinerlei Deaktivierung festgestellt werden.
a Die Geschwindigkeit der meisten Reaktionen nimmt zu, wenn die Temperatur erhöht wird. Eine Näherung, die
dabei oft angewandt wird, ist die RGT-Regel, die besagt, dass eine Erhöhung der Temperatur um 10 K zu einer
Verdoppelung bis Vervierfachung der Reaktionsgeschwindigkeit führt.5
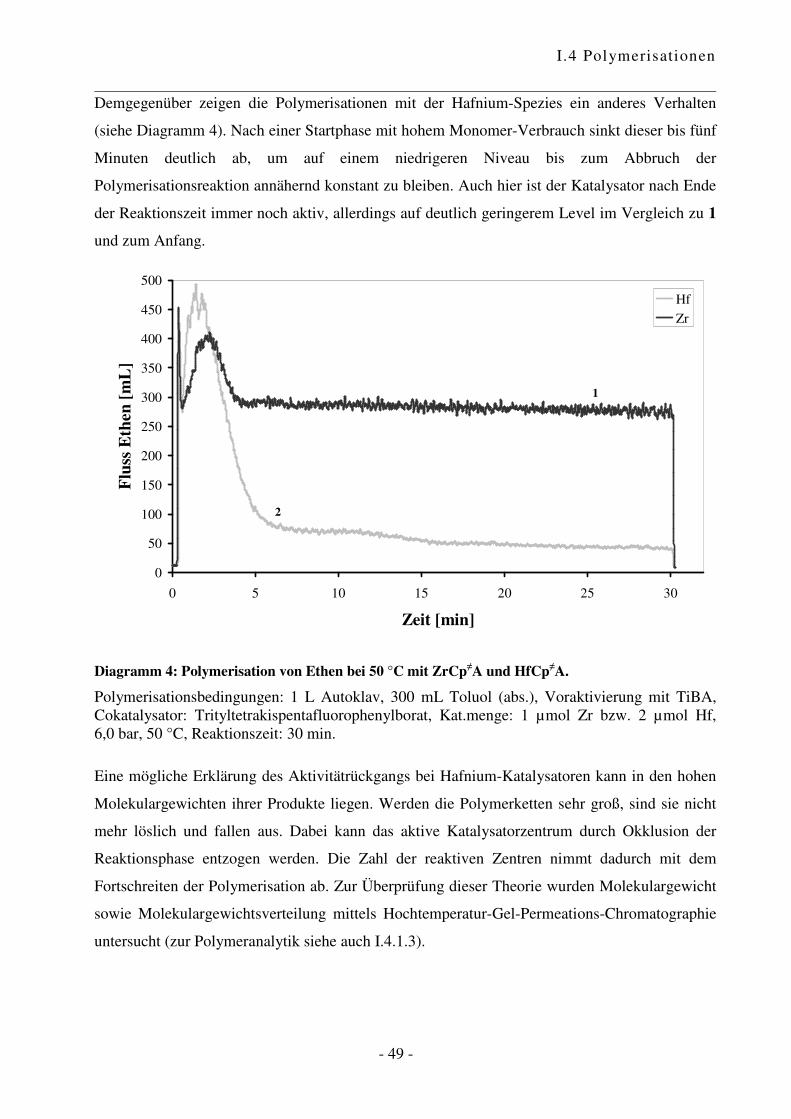
I.4 Polymerisationen
- 49 -
Demgegenüber zeigen die Polymerisationen mit der Hafnium-Spezies ein anderes Verhalten
(siehe Diagramm 4). Nach einer Startphase mit hohem Monomer-Verbrauch sinkt dieser bis fünf
Minuten deutlich ab, um auf einem niedrigeren Niveau bis zum Abbruch der
Polymerisationsreaktion annähernd konstant zu bleiben. Auch hier ist der Katalysator nach Ende
der Reaktionszeit immer noch aktiv, allerdings auf deutlich geringerem Level im Vergleich zu 1
und zum Anfang.
0
50
100
150
200
250
300
350
400
450
500
0 5 10 15 20 25 30
Zeit [min]
Flu
ss E
then
[m
L]
HfZr
1
2
Diagramm 4: Polymerisation von Ethen bei 50 °C mit ZrCp�A und HfCp�A.
Polymerisationsbedingungen: 1 L Autoklav, 300 mL Toluol (abs.), Voraktivierung mit TiBA, Cokatalysator: Trityltetrakispentafluorophenylborat, Kat.menge: 1 µmol Zr bzw. 2 µmol Hf, 6,0 bar, 50 °C, Reaktionszeit: 30 min.
Eine mögliche Erklärung des Aktivitätrückgangs bei Hafnium-Katalysatoren kann in den hohen
Molekulargewichten ihrer Produkte liegen. Werden die Polymerketten sehr groß, sind sie nicht
mehr löslich und fallen aus. Dabei kann das aktive Katalysatorzentrum durch Okklusion der
Reaktionsphase entzogen werden. Die Zahl der reaktiven Zentren nimmt dadurch mit dem
Fortschreiten der Polymerisation ab. Zur Überprüfung dieser Theorie wurden Molekulargewicht
sowie Molekulargewichtsverteilung mittels Hochtemperatur-Gel-Permeations-Chromatographie
untersucht (zur Polymeranalytik siehe auch I.4.1.3).
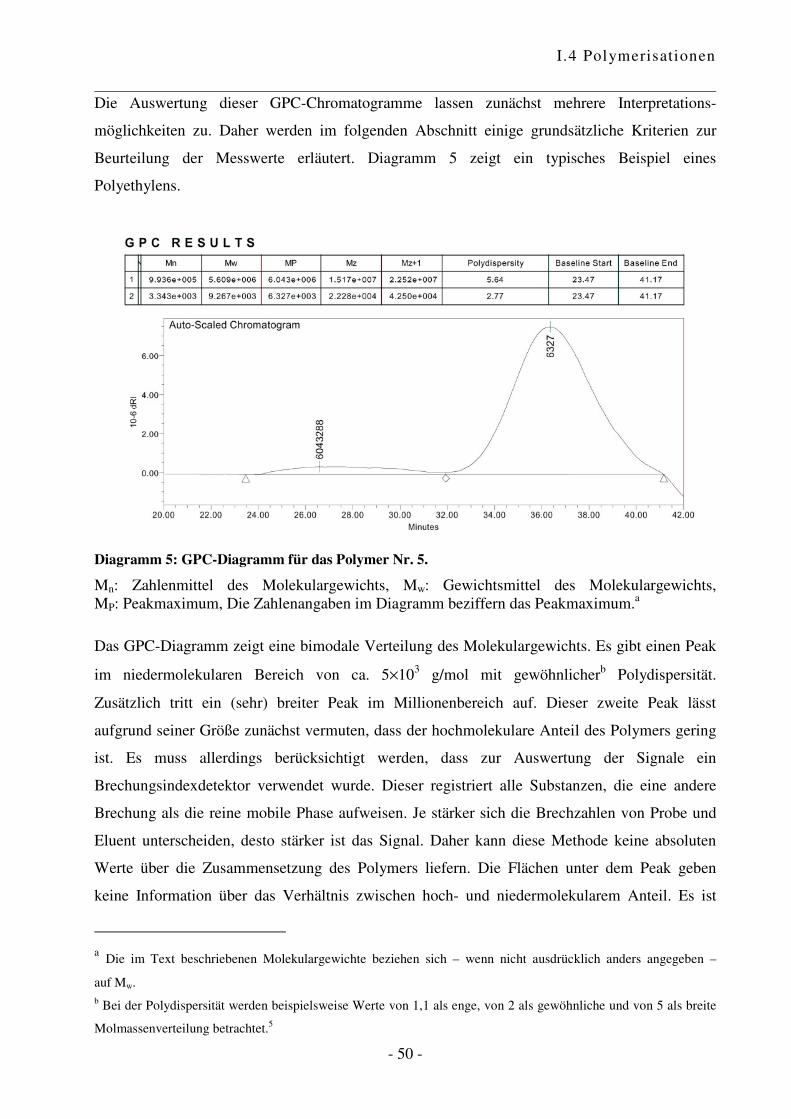
I.4 Polymerisationen
- 50 -
Die Auswertung dieser GPC-Chromatogramme lassen zunächst mehrere Interpretations-
möglichkeiten zu. Daher werden im folgenden Abschnitt einige grundsätzliche Kriterien zur
Beurteilung der Messwerte erläutert. Diagramm 5 zeigt ein typisches Beispiel eines
Polyethylens.
Diagramm 5: GPC-Diagramm für das Polymer Nr. 5.
Mn: Zahlenmittel des Molekulargewichts, Mw: Gewichtsmittel des Molekulargewichts, MP: Peakmaximum, Die Zahlenangaben im Diagramm beziffern das Peakmaximum.a
Das GPC-Diagramm zeigt eine bimodale Verteilung des Molekulargewichts. Es gibt einen Peak
im niedermolekularen Bereich von ca. 5×103 g/mol mit gewöhnlicherb Polydispersität.
Zusätzlich tritt ein (sehr) breiter Peak im Millionenbereich auf. Dieser zweite Peak lässt
aufgrund seiner Größe zunächst vermuten, dass der hochmolekulare Anteil des Polymers gering
ist. Es muss allerdings berücksichtigt werden, dass zur Auswertung der Signale ein
Brechungsindexdetektor verwendet wurde. Dieser registriert alle Substanzen, die eine andere
Brechung als die reine mobile Phase aufweisen. Je stärker sich die Brechzahlen von Probe und
Eluent unterscheiden, desto stärker ist das Signal. Daher kann diese Methode keine absoluten
Werte über die Zusammensetzung des Polymers liefern. Die Flächen unter dem Peak geben
keine Information über das Verhältnis zwischen hoch- und niedermolekularem Anteil. Es ist
a Die im Text beschriebenen Molekulargewichte beziehen sich – wenn nicht ausdrücklich anders angegeben –
auf Mw. b Bei der Polydispersität werden beispielsweise Werte von 1,1 als enge, von 2 als gewöhnliche und von 5 als breite
Molmassenverteilung betrachtet.5

I.4 Polymerisationen
- 51 -
denkbar, dass beispielsweise olefinische Endgruppen einen stärkeren Einfluss auf den
Brechungsindex haben als die CH2-Gruppen innerhalb der Polymerkette. Durch
Abbruchreaktionen wie �-H-Eliminimerung oder Transfer zum Monomer entstehen Oligomere
mit einer Doppelbindung, die den niedermolekularen Anteil darstellen (siehe auch Kapitel
I.1.4.3). Prozentual gesehen ist der Doppelbindungsgehalt der Oligomere pro eingebautem
Monomer deutlich höher als der der hochmolekularen Polymerkette. Das würde bedeuten, dass
die niedermolekularen Anteile ein höheres Signal des Detektors bewirken als die langkettigen
Polymere mit geringerem Doppelbindungsanteil.
Um das genaue Verhältnis zwischen hoch- und niedermolekularem Polymeranteil bestimmen zu
können, muss das Polymer fraktioniert werden. Üblicherweise werden bei Fraktionierungen die
Polymere mehrfach mit heißen Lösungsmitteln versetzt (oder soxhletiert), um niedermolekulare
Anteile zu extrahieren.67 Es wurde durch GPC-Messungen festgestellt, dass die hoch- bzw.
ultrahoch-molekularen Polymere unter diesen Bedingungen depolymerisieren. Diagramm 6 und
Diagramm 7 zeigen dieselbe Polymerprobe.
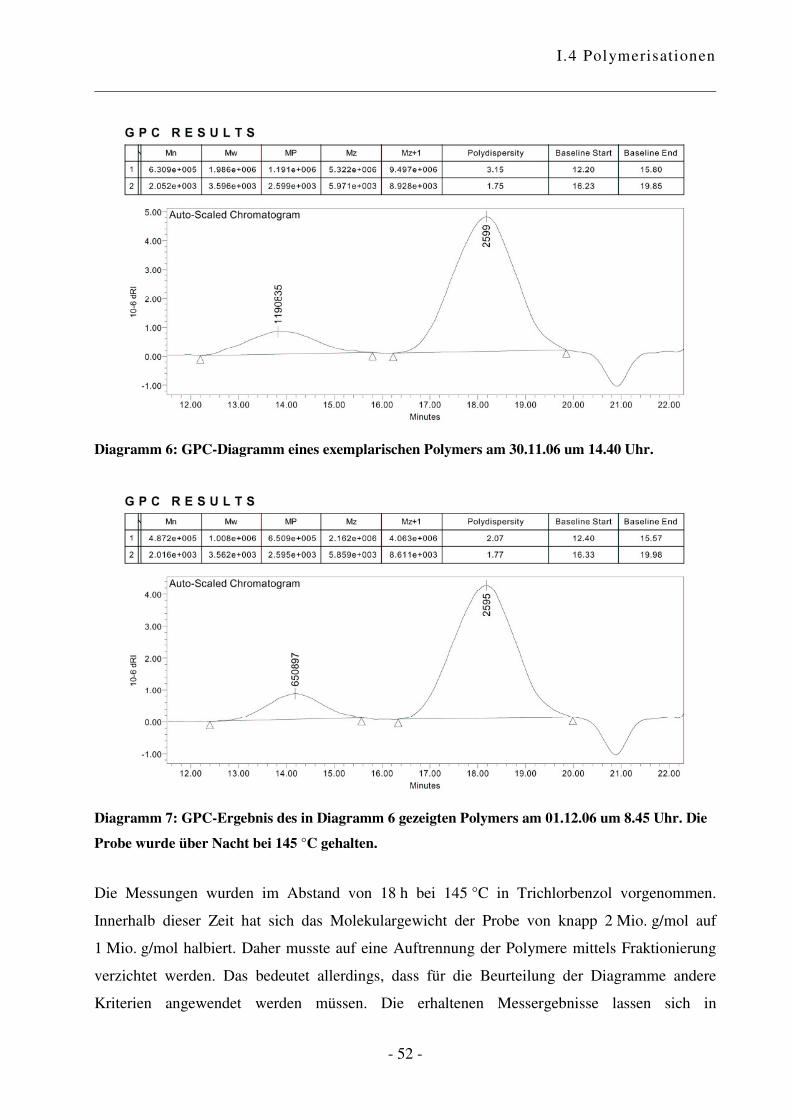
I.4 Polymerisationen
- 52 -
Diagramm 6: GPC-Diagramm eines exemplarischen Polymers am 30.11.06 um 14.40 Uhr.
Diagramm 7: GPC-Ergebnis des in Diagramm 6 gezeigten Polymers am 01.12.06 um 8.45 Uhr. Die
Probe wurde über Nacht bei 145 °C gehalten.
Die Messungen wurden im Abstand von 18 h bei 145 °C in Trichlorbenzol vorgenommen.
Innerhalb dieser Zeit hat sich das Molekulargewicht der Probe von knapp 2 Mio. g/mol auf
1 Mio. g/mol halbiert. Daher musste auf eine Auftrennung der Polymere mittels Fraktionierung
verzichtet werden. Das bedeutet allerdings, dass für die Beurteilung der Diagramme andere
Kriterien angewendet werden müssen. Die erhaltenen Messergebnisse lassen sich in
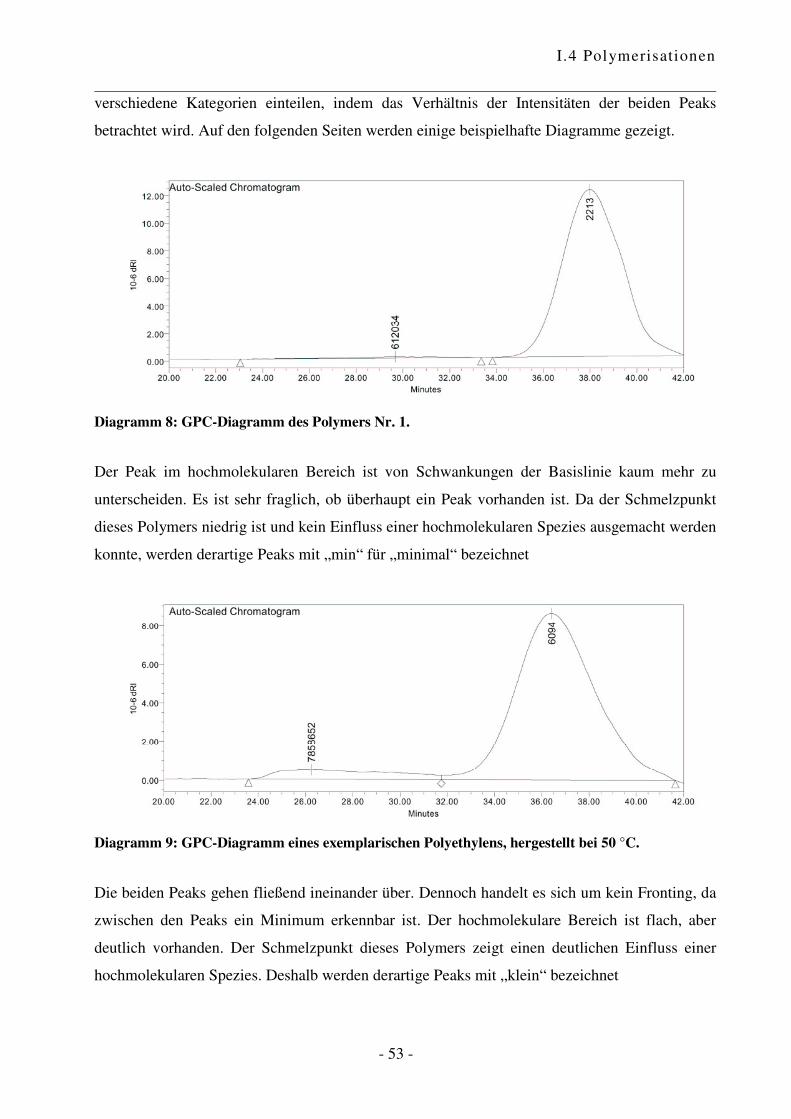
I.4 Polymerisationen
- 53 -
verschiedene Kategorien einteilen, indem das Verhältnis der Intensitäten der beiden Peaks
betrachtet wird. Auf den folgenden Seiten werden einige beispielhafte Diagramme gezeigt.
Diagramm 8: GPC-Diagramm des Polymers Nr. 1.
Der Peak im hochmolekularen Bereich ist von Schwankungen der Basislinie kaum mehr zu
unterscheiden. Es ist sehr fraglich, ob überhaupt ein Peak vorhanden ist. Da der Schmelzpunkt
dieses Polymers niedrig ist und kein Einfluss einer hochmolekularen Spezies ausgemacht werden
konnte, werden derartige Peaks mit „min“ für „minimal“ bezeichnet
Diagramm 9: GPC-Diagramm eines exemplarischen Polyethylens, hergestellt bei 50 °C.
Die beiden Peaks gehen fließend ineinander über. Dennoch handelt es sich um kein Fronting, da
zwischen den Peaks ein Minimum erkennbar ist. Der hochmolekulare Bereich ist flach, aber
deutlich vorhanden. Der Schmelzpunkt dieses Polymers zeigt einen deutlichen Einfluss einer
hochmolekularen Spezies. Deshalb werden derartige Peaks mit „klein“ bezeichnet
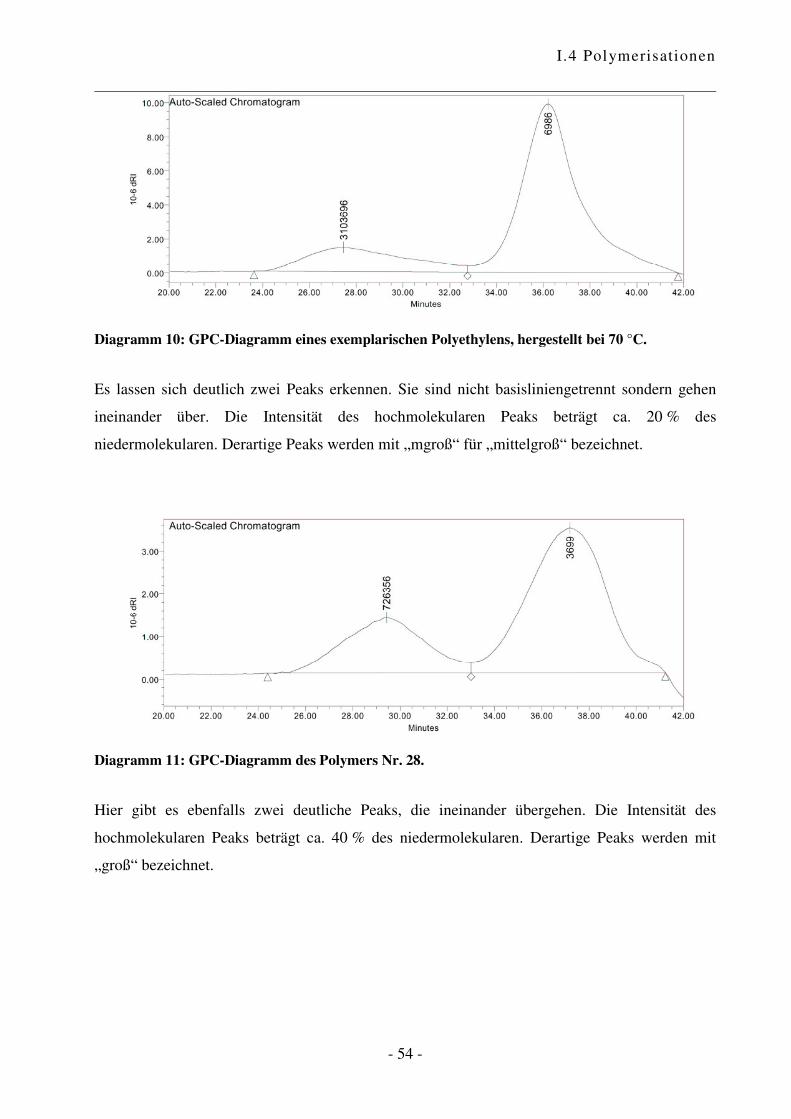
I.4 Polymerisationen
- 54 -
Diagramm 10: GPC-Diagramm eines exemplarischen Polyethylens, hergestellt bei 70 °C.
Es lassen sich deutlich zwei Peaks erkennen. Sie sind nicht basisliniengetrennt sondern gehen
ineinander über. Die Intensität des hochmolekularen Peaks beträgt ca. 20 % des
niedermolekularen. Derartige Peaks werden mit „mgroß“ für „mittelgroß“ bezeichnet.
Diagramm 11: GPC-Diagramm des Polymers Nr. 28.
Hier gibt es ebenfalls zwei deutliche Peaks, die ineinander übergehen. Die Intensität des
hochmolekularen Peaks beträgt ca. 40 % des niedermolekularen. Derartige Peaks werden mit
„groß“ bezeichnet.
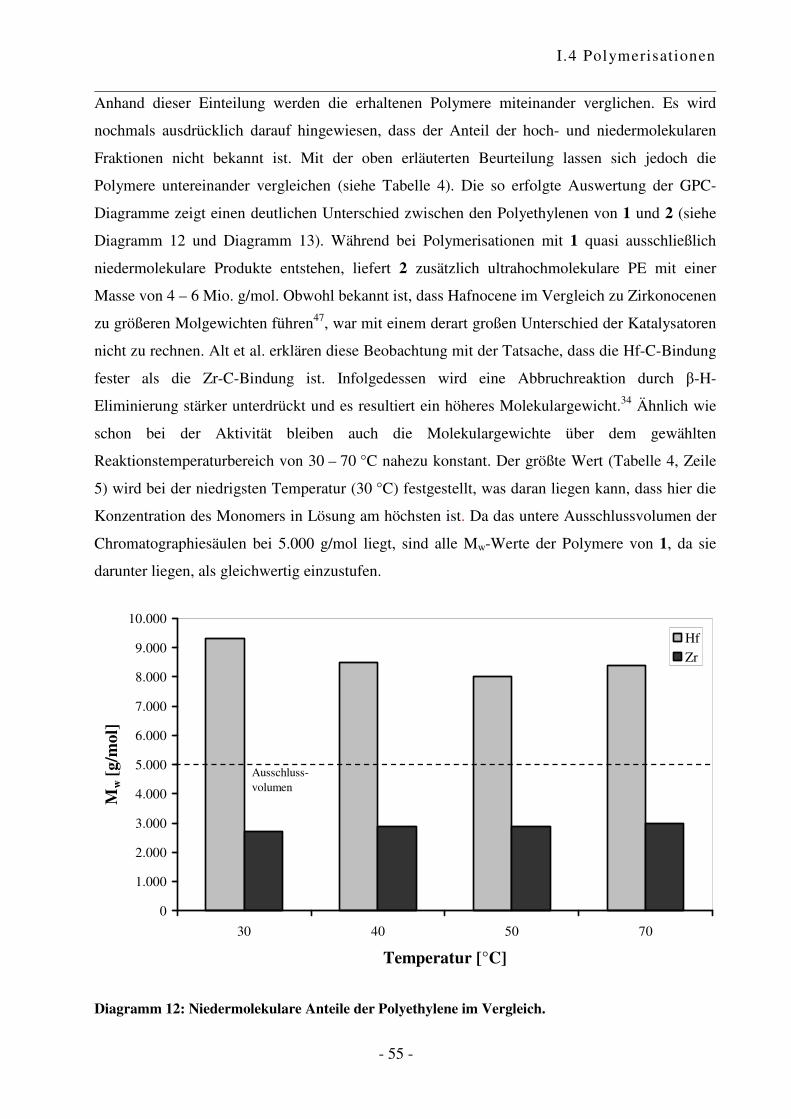
I.4 Polymerisationen
- 55 -
Anhand dieser Einteilung werden die erhaltenen Polymere miteinander verglichen. Es wird
nochmals ausdrücklich darauf hingewiesen, dass der Anteil der hoch- und niedermolekularen
Fraktionen nicht bekannt ist. Mit der oben erläuterten Beurteilung lassen sich jedoch die
Polymere untereinander vergleichen (siehe Tabelle 4). Die so erfolgte Auswertung der GPC-
Diagramme zeigt einen deutlichen Unterschied zwischen den Polyethylenen von 1 und 2 (siehe
Diagramm 12 und Diagramm 13). Während bei Polymerisationen mit 1 quasi ausschließlich
niedermolekulare Produkte entstehen, liefert 2 zusätzlich ultrahochmolekulare PE mit einer
Masse von 4 – 6 Mio. g/mol. Obwohl bekannt ist, dass Hafnocene im Vergleich zu Zirkonocenen
zu größeren Molgewichten führen47, war mit einem derart großen Unterschied der Katalysatoren
nicht zu rechnen. Alt et al. erklären diese Beobachtung mit der Tatsache, dass die Hf-C-Bindung
fester als die Zr-C-Bindung ist. Infolgedessen wird eine Abbruchreaktion durch �-H-
Eliminierung stärker unterdrückt und es resultiert ein höheres Molekulargewicht.34 Ähnlich wie
schon bei der Aktivität bleiben auch die Molekulargewichte über dem gewählten
Reaktionstemperaturbereich von 30 – 70 °C nahezu konstant. Der größte Wert (Tabelle 4, Zeile
5) wird bei der niedrigsten Temperatur (30 °C) festgestellt, was daran liegen kann, dass hier die
Konzentration des Monomers in Lösung am höchsten ist. Da das untere Ausschlussvolumen der
Chromatographiesäulen bei 5.000 g/mol liegt, sind alle Mw-Werte der Polymere von 1, da sie
darunter liegen, als gleichwertig einzustufen.
0
1.000
2.000
3.000
4.000
5.000
6.000
7.000
8.000
9.000
10.000
30 40 50 70
Temperatur [°C]
Mw [
g/m
ol]
HfZr
Ausschluss-volumen
Diagramm 12: Niedermolekulare Anteile der Polyethylene im Vergleich.
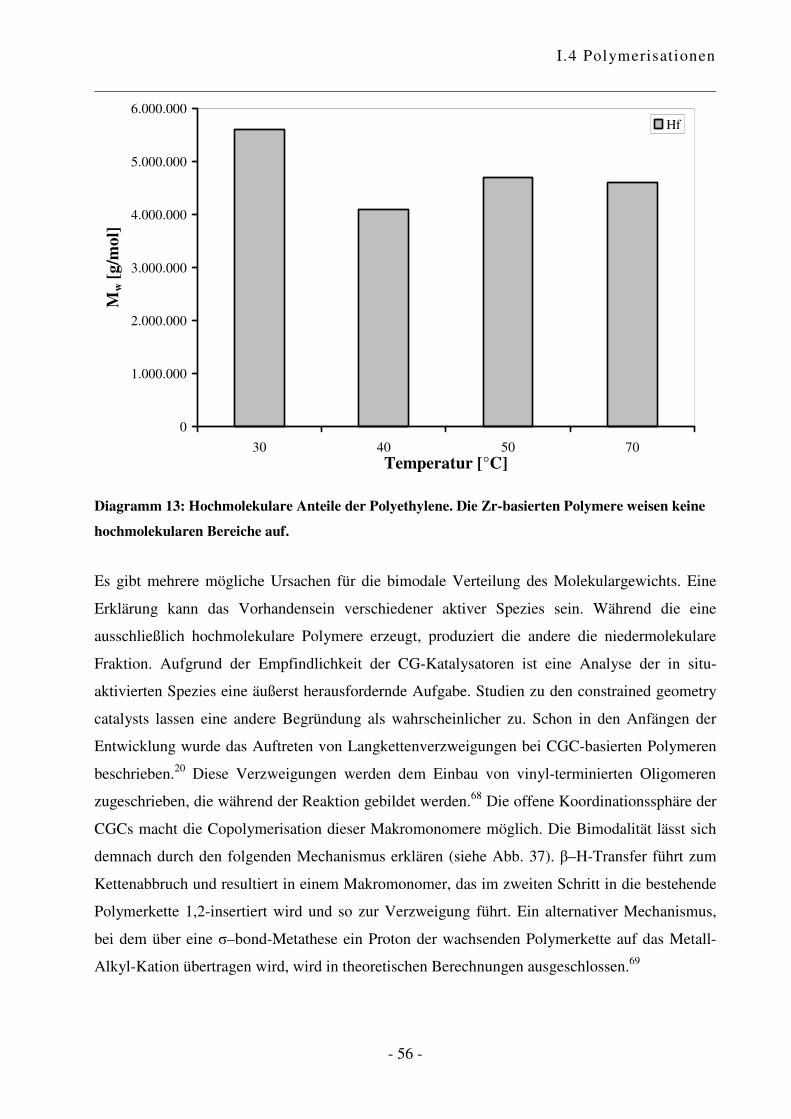
I.4 Polymerisationen
- 56 -
0
1.000.000
2.000.000
3.000.000
4.000.000
5.000.000
6.000.000
30 40 50 70Temperatur [°C]
Mw [
g/m
ol]
Hf
Diagramm 13: Hochmolekulare Anteile der Polyethylene. Die Zr-basierten Polymere weisen keine
hochmolekularen Bereiche auf.
Es gibt mehrere mögliche Ursachen für die bimodale Verteilung des Molekulargewichts. Eine
Erklärung kann das Vorhandensein verschiedener aktiver Spezies sein. Während die eine
ausschließlich hochmolekulare Polymere erzeugt, produziert die andere die niedermolekulare
Fraktion. Aufgrund der Empfindlichkeit der CG-Katalysatoren ist eine Analyse der in situ-
aktivierten Spezies eine äußerst herausfordernde Aufgabe. Studien zu den constrained geometry
catalysts lassen eine andere Begründung als wahrscheinlicher zu. Schon in den Anfängen der
Entwicklung wurde das Auftreten von Langkettenverzweigungen bei CGC-basierten Polymeren
beschrieben.20 Diese Verzweigungen werden dem Einbau von vinyl-terminierten Oligomeren
zugeschrieben, die während der Reaktion gebildet werden.68 Die offene Koordinationssphäre der
CGCs macht die Copolymerisation dieser Makromonomere möglich. Die Bimodalität lässt sich
demnach durch den folgenden Mechanismus erklären (siehe Abb. 37). �–H-Transfer führt zum
Kettenabbruch und resultiert in einem Makromonomer, das im zweiten Schritt in die bestehende
Polymerkette 1,2-insertiert wird und so zur Verzweigung führt. Ein alternativer Mechanismus,
bei dem über eine �–bond-Metathese ein Proton der wachsenden Polymerkette auf das Metall-
Alkyl-Kation übertragen wird, wird in theoretischen Berechnungen ausgeschlossen.69

I.4 Polymerisationen
- 57 -
Abb. 37: Mechanismus für die Bildung von Langkettenverzweigungen in Ethen-Homo-
polymerisationen70.
Welche Auswirkungen sich durch die Langkettenverzweigungen auf die Polymereigenschaften
ergeben und wie diese erkannt und bestimmt werden können, wird im folgenden Abschnitt näher
erläutert. Metallocen-Polymere leiden aufgrund ihrer hohen Viskositäten oftmals unter
schlechter Verarbeitbarkeit. Das Einbringen von Langkettenverzweigungen kann dies
verbessern. Yan, Wang und Zhu haben den Effekt der Langkettenverzweigungen auf die
rheologischen Eigenschaften Metallocen-basierter Polymere untersucht.71 Sie fanden einen
starken Einfluss der LCBs auf die rheologischen Eigenschaften, während der Effekt auf die
mechanischen Eigenschaften, wie Schmelzenthalpie und Dichte, eher gering waren. Die langen
Verzweigungen verstärken die Verschlaufungen und reduzieren das hydrodynamische Volumen
– zwei Faktoren, die die Rheologie in entgegengesetzter Weise beeinflussen. Durch LCBs wird
die stationäre Viskosität erhöht, der Schmelzfluss erniedrigt und die Strukturviskosität
verbessert. Ebenso zeigt sich ein Effekt auf die viskoelastischen Eigenschaften. Speichermodul,
Entspannungsmodul und Strangaufweitung werden erhöht, was zu einer Steigerung der
Elastizität des Polymers führt. Zusammenfassend lässt sich sagen, dass lineare Polyethylene, die
Langkettenverzweigungen aufweisen, gegenüber herkömmlichen PE stark verbesserte
Eigenschaften zeigen. Ihre Verarbeitbarkeit ist – dank der Verzweigungen – ähnlich den
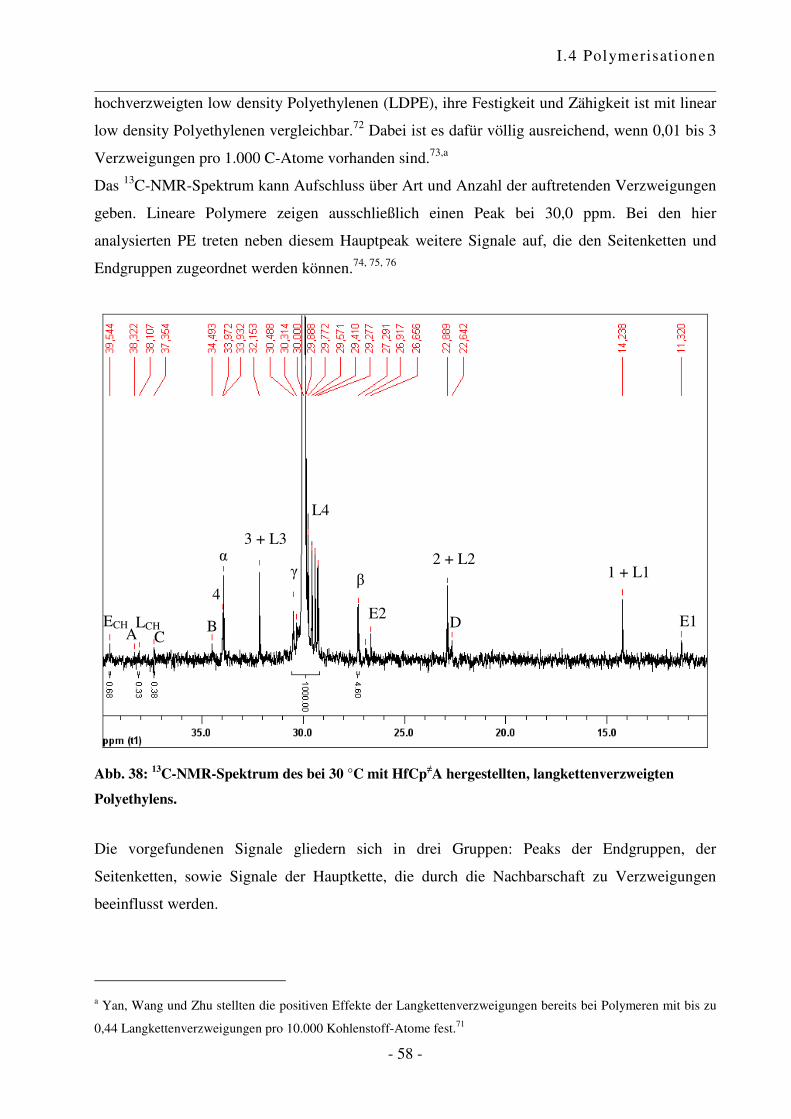
I.4 Polymerisationen
- 58 -
hochverzweigten low density Polyethylenen (LDPE), ihre Festigkeit und Zähigkeit ist mit linear
low density Polyethylenen vergleichbar.72 Dabei ist es dafür völlig ausreichend, wenn 0,01 bis 3
Verzweigungen pro 1.000 C-Atome vorhanden sind.73,a
Das 13C-NMR-Spektrum kann Aufschluss über Art und Anzahl der auftretenden Verzweigungen
geben. Lineare Polymere zeigen ausschließlich einen Peak bei 30,0 ppm. Bei den hier
analysierten PE treten neben diesem Hauptpeak weitere Signale auf, die den Seitenketten und
Endgruppen zugeordnet werden können.74, 75, 76
Abb. 38: 13C-NMR-Spektrum des bei 30 °C mit HfCp�A hergestellten, langkettenverzweigten
Polyethylens.
Die vorgefundenen Signale gliedern sich in drei Gruppen: Peaks der Endgruppen, der
Seitenketten, sowie Signale der Hauptkette, die durch die Nachbarschaft zu Verzweigungen
beeinflusst werden.
a Yan, Wang und Zhu stellten die positiven Effekte der Langkettenverzweigungen bereits bei Polymeren mit bis zu
0,44 Langkettenverzweigungen pro 10.000 Kohlenstoff-Atome fest.71
2 + L2 3 + L3
1 + L1 4
L4
ECH E1 E2 LCH
�
�
B A C D

I.4 Polymerisationen
- 59 -
Sind die Molekulargewichte niedrig, können die Endgruppen (siehe Abb. 39) erkannt werden.
Die Kohlenstoffatome Nr. 5 und 6 erscheinen bei 139,1 bzw. 114,4 ppm. Das Signal des zur
vinylischen Endgruppe benachbarten C-Atoms (Nr. 4) liegt bei 34,0 ppm.
Abb. 39: Endgruppen eines Polyethylens.
Die Kohlenstoffatome 1, 2 und 3 sowie die Enden der Langkettenverzweigungen L1, L2 und L3
(siehe Abb. 40) sind äquivalent und im NMR nicht voneinander zu unterscheiden. L4 ist bereits
auf der Schulter des Hauptpeaks bei 29,6 ppm zu finden. Neben den Langkettenverzweigungen
(Hexyl und länger) können hier ebenfalls Ethyl-Verzweigungen erkannt werden. Die Signale der
Ethyl-Ketten liegen bei 11,3 ppm für E1 und 26,6 ppm für E2.
Abb. 40: Ethyl- und Langkettenverzweigungen bei Polyethylenen.
Die tertiären Kohlenstoffatome LCH und ECH, von denen die Verzweigung ausgeht, sind
intensitätsschwach und befinden sich Bereich von 37 bis 40 ppm. Die Ursache des
vergleichsweise kleinen Signals liegt zum einen im verringerten Kern-Overhauser-Effekt für
CH-Gruppen. Des Weiteren stehen einem einzelnen tertiären C-Atom jeweils drei �-, �- und -
C-Atome gegenüber: links und rechts der Verzweigung in der Hauptkette sowie in der
Seitenkette. Das Signal der A-CH2-Gruppen befindet sich bereits im Bereich des Hauptpeaks bei
29,3 – 30,5 ppm.

I.4 Polymerisationen
- 60 -
Eine Besonderheit tritt auf, wenn zwei Verzweigungen sehr nahe beieinander liegen (siehe Abb.
41). Die A-CH2-Gruppe der Hauptkette, die sich zwischen zwei Verzweigungen befindet,
erscheint tieffeldverschoben bei 38,3 ppm und damit im Bereich der Signale der tertiären
C-Atome, wo auch der Peak der Verzweigungspunkte C liegt. Die B-CH2-Gruppen treten in der
Nähe der �-Gruppen bei 34,5 ppm auf, die D-CH2-Gruppen in Nachbarschaft des (2+L2)-Peaks
bei 22,6 ppm.
Abb. 41: Einbau zweier Verzweigungen direkt hintereinander.
Tabelle 2: Zuordnung der NMR-Signale nach Literatur Liu74, Axelson75 und Meinhard76.
Signal [ppm] Cxa Zuordnung Abstand Signal [ppm] Cx Zuordnung
11,3 P E1 32,2 S 3 + L3
14,2 P 1 + L1 33,9 S � + �’
22,6 S D 34,0 S 4
22,9 S 2 + L2 34,5 S B
26,7 S E2 37,4 T LCH
26,9 n.b. n.b. 38,1 T LCH
27,3 S � 38,3 S A
29,3 – 30,3 S A 39,5 T ECH
29,6 S L4 114,4 S 6
30,5 S 139,1 T 5
n.b.: nicht bestimmt
a P steht für ein primäres, S für ein sekundäres und T für ein tertiäres C-Atom

I.4 Polymerisationen
- 61 -
Die Auswertung der NMR-Spektren zeigt deutlich, dass mit HfCp�A verzweigte Polymere
entstanden sind. Es treten Signale von Ethylverzweigungen auf, was bedeutet, dass der
Katalysator neben der Polymerisationsreaktion in der Lage ist, Ethen zu 1-Buten zu dimerisieren.
Signale von Butyl-Verzweigungen, deren charakteristischer Peak bei 23,4 ppm zu finden ist77,
liegen hier nicht vor, jedoch die von längeren Seitenketten. Bemerkenswert ist das Auftreten der
Peaks für den Sonderfall von zwei Verzweigungen direkt hintereinander. Dies zeigt erneut, dass
der CG-Katalysator sehr gut in der Lage ist, sterisch anspruchsvolle Monomere zu
polymerisieren.a Das Auftreten der Signale der vinylischen Endgruppen kann nur von
niedermolekularen Anteilen herrühren, ihr prozentualer Anteil in den ultrahochmolekularen
Polymeren (Molgewichte von 6 Mio. g/mol bedeuten über 200.000 Ethylen-Einheiten) ist viel zu
gering.b
Eine quantitative Beurteilung der NMR-Spektren, die es erlaubt, eine Aussage über die Anzahl
der Verzweigungen zu machen, ist komplex. Axelson, Levy und Mandelkern haben die
Schwierigkeiten, die bei der Integration der Signale auftreten, ausführlich erörtert.75 Selbst bei
sorgfältig durchgeführten Langzeitexperimenten ist der Fehler unter anderem aufgrund des
Kern-Overhauser-Effekts sehr groß. Für eine grobe Abschätzung der Anzahl der Verzweigungen
eignen sich die ausgeprägten CH2-Gruppen-Peaks am besten. Dazu wird die Signalgruppe der
Hauptkette von 29,2 bis 30,6 ppm auf 1000 gesetzt. Das Integral der �-CH2-Gruppen spiegelt
dann die Zahl der Verzweigungen pro 1000 C-Atome wider. Da bei Langkettenverzweigungen
pro Verzweigung drei, bei Ethyl-Verzweigungen zwei �-Atome auftreten, wird das bestimmte
Integral für eine grobe Näherung durch 3 dividiert (Methode 1). Eine weitere Abschätzung der
Verzweigungszahlen erfolgt, indem die Integrale der Signale der CH-Gruppen (ECH, LCH und C)
addiert werden (Methode 2). Anhand dieser Methoden ergeben sich für die hier hergestellten
Polymere Verzweigungszahlen von ca. einer Verzweigung pro 1000 C-Atome (siehe
Diagramm 14 und Tabelle 5).
a Der Einbau zweier Verzweigungen direkt nacheinander tritt mit größter Wahrscheinlichkeit am Reaktionsende auf,
wenn überschüssiges Ethen vor dem Öffnen des Reaktors abgelassen wird. Durch das Entfernen von Ethen steigt die
Konzentration der Makromonomere an, bis letztlich ausschließlich Oligoethylene als Monomere zur Verfügung
stehen. Dieser Zeitpunkt direkt vor dem Ausschlauchen der Reaktionslösung in das Abbruchreagenz
Methanol/Salzsäure wird zwar so kurz wie möglich gehalten, kann jedoch nicht vermieden werden. b Bei der Bestimmung der Verzweigungen anhand des NMR-Spektrums birgt die schlechte Löslichkeit der
ultrahochmolekularen Polymere eine Fehlerquelle. Trotz sorgfältiger Probenpräparation besteht die Möglichkeit,
dass während der Messung das Polymer nicht komplett gelöst vorlag. Die niedermolekularen Anteile sind sehr viel
besser löslich, daher ist nicht auszuschließen, dass das Spektrum einen erhöhten Anteil des niedermolekularen
Polymerbereichs zeigt.
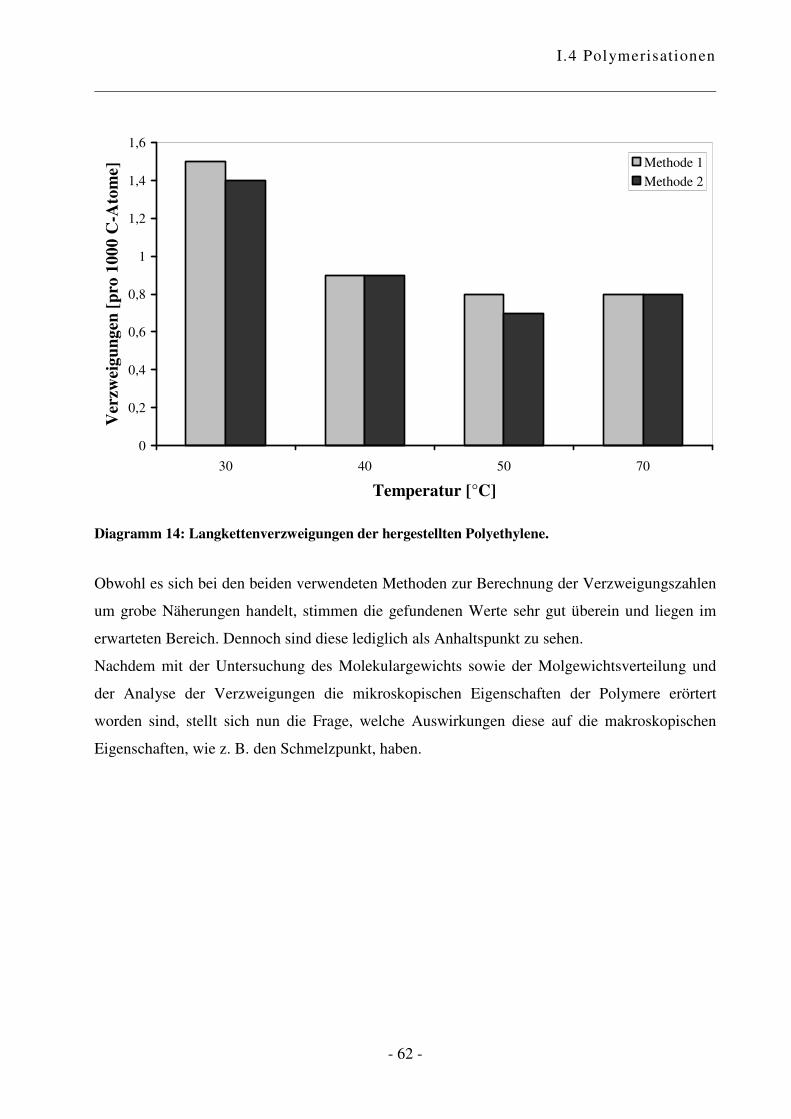
I.4 Polymerisationen
- 62 -
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
30 40 50 70
Temperatur [°C]
Ver
zwei
gung
en [
pro
1000
C-A
tom
e] Methode 1Methode 2
Diagramm 14: Langkettenverzweigungen der hergestellten Polyethylene.
Obwohl es sich bei den beiden verwendeten Methoden zur Berechnung der Verzweigungszahlen
um grobe Näherungen handelt, stimmen die gefundenen Werte sehr gut überein und liegen im
erwarteten Bereich. Dennoch sind diese lediglich als Anhaltspunkt zu sehen.
Nachdem mit der Untersuchung des Molekulargewichts sowie der Molgewichtsverteilung und
der Analyse der Verzweigungen die mikroskopischen Eigenschaften der Polymere erörtert
worden sind, stellt sich nun die Frage, welche Auswirkungen diese auf die makroskopischen
Eigenschaften, wie z. B. den Schmelzpunkt, haben.

I.4 Polymerisationen
- 63 -
Wie schon in den vorangehenden Abschnitten erwähnt, gibt es eine Vielzahl von Faktoren, die
das Schmelzverhalten von Makromolekülen beeinflussen. Im Vergleich zu niedermolekularen,
kristallinen Substanzen existiert bei Polymeren kein scharfer Schmelzpunkt, sondern ein
Schmelzbereich, der von der Probenvorgeschichte und der Aufheizrate abhängig ist.78 Deshalb
wurde bei der Aufnahme der DSC-Diagramme eine festgelegte Methode mit zwei
Aufheizvorgängen angewendet, wobei nur der zweite Aufheizvorgang zur Auswertung
herangezogen wird (vergleiche I.4.1.3). Entscheidenden Einfluss auf die Schmelztemperatur von
Polymeren haben das Molekulargewicht, die Einheitlichkeit der Kettenlängen (Polydispersität),
Verzweigungen sowie die Kristallinität. Kristallisation setzt kristallisierbare Ketten voraus. Nicht
kristallisierbar sind vernetzte oder verzweigte Polymere und statistische Copolymere. Ist der
Anteil der Verzweigungen oder der Comonomereinheiten nur gering, wird die Kristallinität zwar
verringert, jedoch nicht vollständig unterdrückt.78 Die Kettenlänge wirkt sich ebenfalls auf die
Kristallinität aus. Je kürzer die Makromoleküle, desto höher ist der prozentuale Anteil an
Kettenenden, die Fehlstellen im Kristall darstellen, meist nicht in das Kristallgitter eingebaut
werden und damit eine Erniedrigung der Schmelztemperatur bewirken. Breite Molekular-
gewichtsverteilungen führen zu uneinheitlichen Schmelzpunkten und resultieren in großen
Schmelzbereichen, polydisperse Polymere können mehrere Schmelzpeaks aufweisen. Die
Schmelztemperaturen von Polymerhomologen nehmen mit steigender Molmasse zu und
erreichen schließlich einen Endwert, da der Einfluss der Endgruppen mit steigender Molmasse
immer geringer wird.79
Die DSC-Schmelzkurven der hergestellten Polymere sind zunächst ungewöhnlich, wobei auch
Alt et al. außergewöhnliche DSC-Kurven mit CGCs feststellen konnten.80 Anstelle eines
scharfen Peaks tritt ein weiter Schmelzbereich. Für den Hafnium-Katalysator zeigen die Kurven
einen lang gezogenen Peak während mit dem Zirkonium-Katalysator ein größerer Bereich mit
drei Maxima erhalten wird. Beispielhaft zeigen dies Diagramm 15 und Diagramm 16. Diagramm
17 stellt den jeweils höchsten Schmelzpeak in Abhängigkeit der Reaktionstemperatur dar.
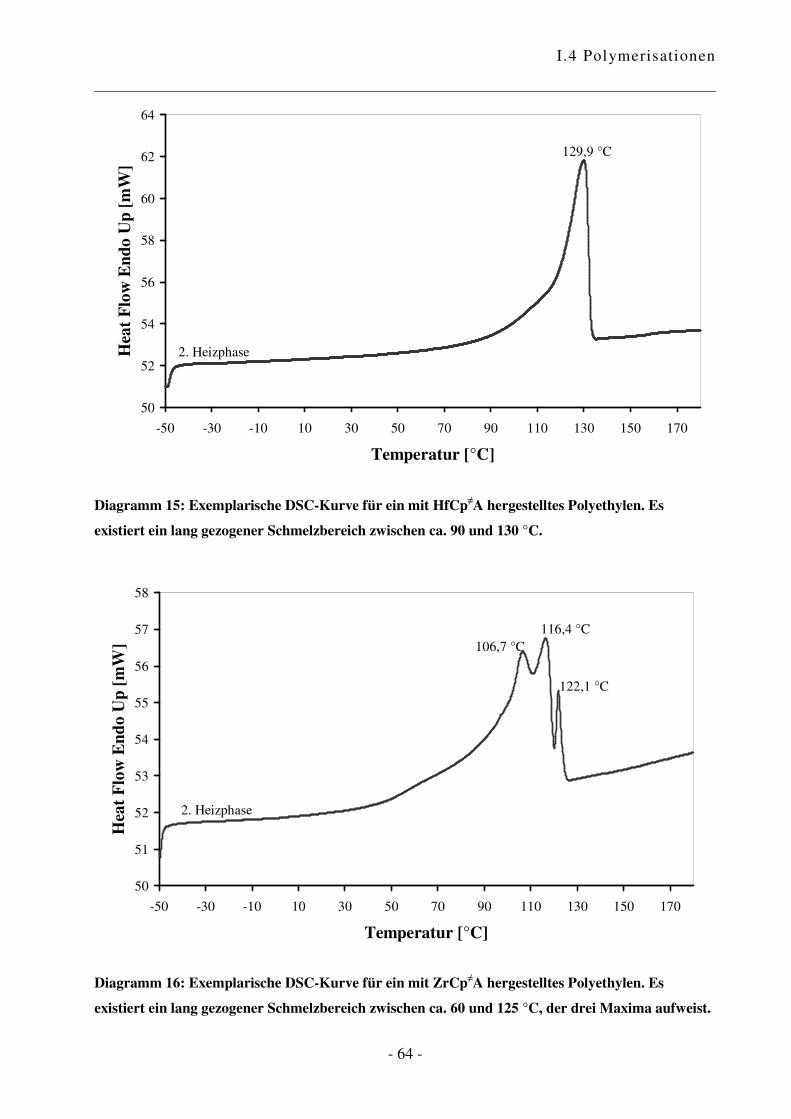
I.4 Polymerisationen
- 64 -
50
52
54
56
58
60
62
64
-50 -30 -10 10 30 50 70 90 110 130 150 170
Temperatur [°C]
Hea
t F
low
End
o U
p [m
W]
2. Heizphase
129,9 °C
Diagramm 15: Exemplarische DSC-Kurve für ein mit HfCp�A hergestelltes Polyethylen. Es
existiert ein lang gezogener Schmelzbereich zwischen ca. 90 und 130 °C.
50
51
52
53
54
55
56
57
58
-50 -30 -10 10 30 50 70 90 110 130 150 170
Temperatur [°C]
Hea
t F
low
End
o U
p [m
W]
2. Heizphase
106,7 °C116,4 °C
122,1 °C
Diagramm 16: Exemplarische DSC-Kurve für ein mit ZrCp�A hergestelltes Polyethylen. Es
existiert ein lang gezogener Schmelzbereich zwischen ca. 60 und 125 °C, der drei Maxima aufweist.
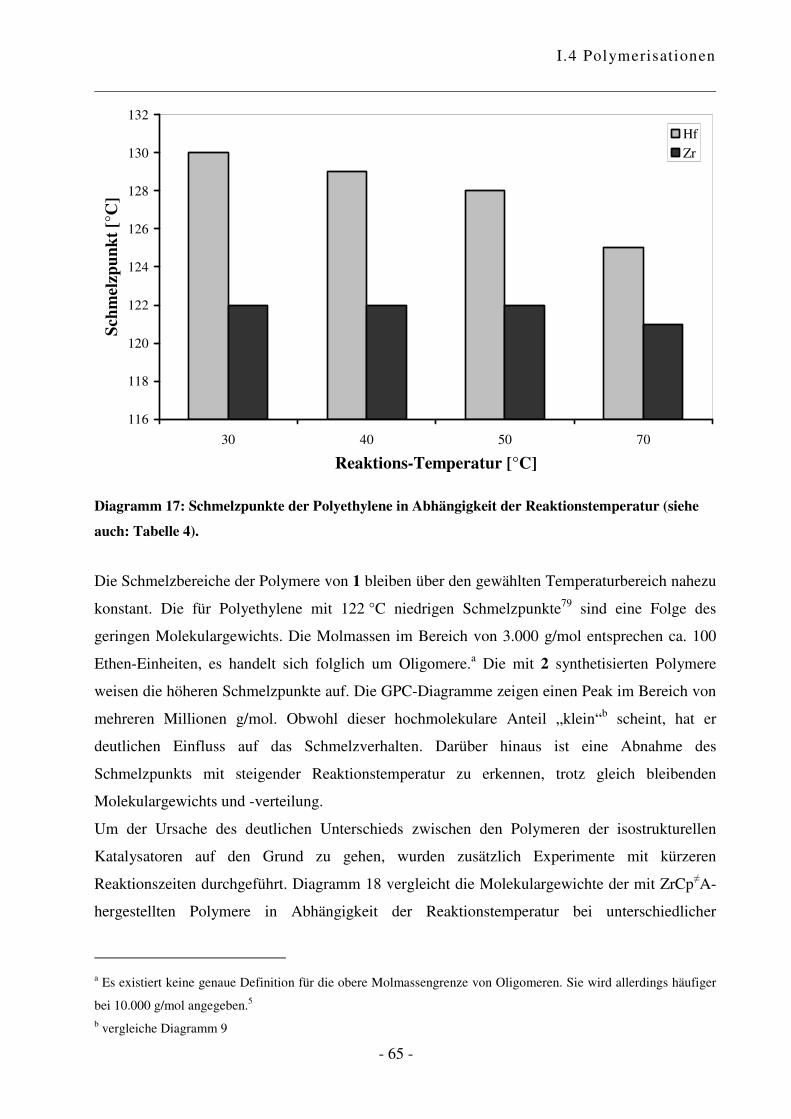
I.4 Polymerisationen
- 65 -
116
118
120
122
124
126
128
130
132
30 40 50 70
Reaktions-Temperatur [°C]
Schm
elzp
unkt
[°C
]HfZr
Diagramm 17: Schmelzpunkte der Polyethylene in Abhängigkeit der Reaktionstemperatur (siehe
auch: Tabelle 4).
Die Schmelzbereiche der Polymere von 1 bleiben über den gewählten Temperaturbereich nahezu
konstant. Die für Polyethylene mit 122 °C niedrigen Schmelzpunkte79 sind eine Folge des
geringen Molekulargewichts. Die Molmassen im Bereich von 3.000 g/mol entsprechen ca. 100
Ethen-Einheiten, es handelt sich folglich um Oligomere.a Die mit 2 synthetisierten Polymere
weisen die höheren Schmelzpunkte auf. Die GPC-Diagramme zeigen einen Peak im Bereich von
mehreren Millionen g/mol. Obwohl dieser hochmolekulare Anteil „klein“b scheint, hat er
deutlichen Einfluss auf das Schmelzverhalten. Darüber hinaus ist eine Abnahme des
Schmelzpunkts mit steigender Reaktionstemperatur zu erkennen, trotz gleich bleibenden
Molekulargewichts und -verteilung.
Um der Ursache des deutlichen Unterschieds zwischen den Polymeren der isostrukturellen
Katalysatoren auf den Grund zu gehen, wurden zusätzlich Experimente mit kürzeren
Reaktionszeiten durchgeführt. Diagramm 18 vergleicht die Molekulargewichte der mit ZrCp�A-
hergestellten Polymere in Abhängigkeit der Reaktionstemperatur bei unterschiedlicher
a Es existiert keine genaue Definition für die obere Molmassengrenze von Oligomeren. Sie wird allerdings häufiger
bei 10.000 g/mol angegeben.5 b vergleiche Diagramm 9
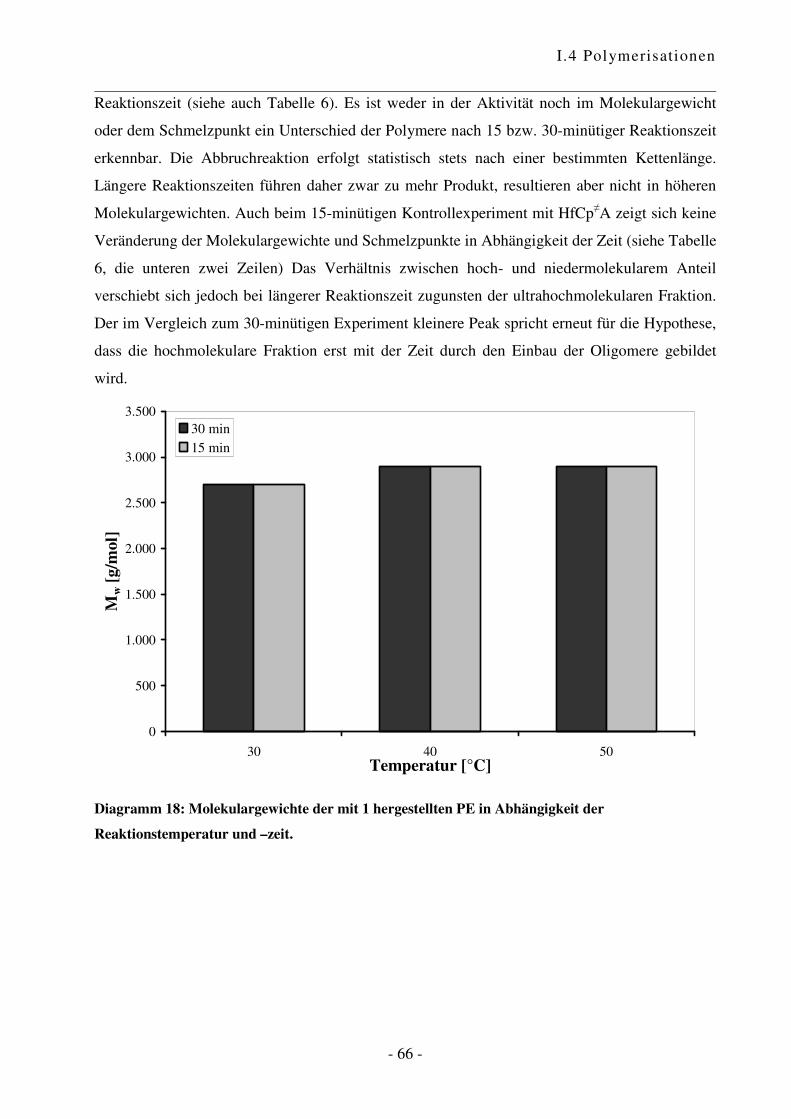
I.4 Polymerisationen
- 66 -
Reaktionszeit (siehe auch Tabelle 6). Es ist weder in der Aktivität noch im Molekulargewicht
oder dem Schmelzpunkt ein Unterschied der Polymere nach 15 bzw. 30-minütiger Reaktionszeit
erkennbar. Die Abbruchreaktion erfolgt statistisch stets nach einer bestimmten Kettenlänge.
Längere Reaktionszeiten führen daher zwar zu mehr Produkt, resultieren aber nicht in höheren
Molekulargewichten. Auch beim 15-minütigen Kontrollexperiment mit HfCp�A zeigt sich keine
Veränderung der Molekulargewichte und Schmelzpunkte in Abhängigkeit der Zeit (siehe Tabelle
6, die unteren zwei Zeilen) Das Verhältnis zwischen hoch- und niedermolekularem Anteil
verschiebt sich jedoch bei längerer Reaktionszeit zugunsten der ultrahochmolekularen Fraktion.
Der im Vergleich zum 30-minütigen Experiment kleinere Peak spricht erneut für die Hypothese,
dass die hochmolekulare Fraktion erst mit der Zeit durch den Einbau der Oligomere gebildet
wird.
0
500
1.000
1.500
2.000
2.500
3.000
3.500
30 40 50Temperatur [°C]
Mw [
g/m
ol]
30 min15 min
Diagramm 18: Molekulargewichte der mit 1 hergestellten PE in Abhängigkeit der
Reaktionstemperatur und –zeit.

I.4 Polymerisationen
- 67 -
Zusammenfassung
Das Katalysatorsystem 1 produziert mit sehr hoher Aktivität niedermolekulare Polyethylene mit
niedrigen Schmelzpunkten. Die Reaktionszeit sowie die –temperatur zeigten dabei keinen
Einfluss auf die Kettenlänge der Produkte. Katalysatorsystem 2 polymerisiert ebenfalls mit sehr
hoher Aktivität Ethen, allerdings um den Faktor 5-7 geringer als bei 1. Während der Verlauf der
Polymerisation bei 1 über die gesamte Reaktionszeit konstant ist, geht die Aktivität bei 2 zurück.
Dieser Aktivitätsrückgang lässt sich durch die hohen Molekulargewichte der Produkte erklären:
Wird das Molekulargewicht zu groß, fallen die Polymere aus und entziehen dem Gleichgewicht
aktive Zentren durch Okklusion. Die mit 2 erhaltenen PE zeigen eine bimodale Molekular-
gewichtsverteilung mit einem nieder- und einem ultrahochmolekularen Anteil. Der Einfluss der
Reaktionstemperatur auf das Molekulargewicht ist gering. Eine weitere Besonderheit der
Produkte von 2 sind die Langkettenverzweigungen, die durch den Einbau der niedermolekularen
Oligomere entstehen. Die so erhaltenen ultrahochmolekularen Polyethylene weisen ungefähr
eine Langkettenverzweigung pro 1000 C-Atome auf, wodurch sich die Polymereigenschaften
hinsichtlich der Verarbeitbarkeit verbessern. Bemerkenswert bei der Bildung der LCBs mit 2 ist
das Auftreten des Sonderfalls der Insertion zweier Langkettenverzweigungen direkt
nacheinander. Das spricht für die außergewöhnliche Geometrie des Katalysators. Neben
Langkettenverzweigungen konnten zudem Ethyl-Verzweigungen erkannt werden, die darauf
schließen lassen, dass der HfCp�A in der Lage ist, Ethen zu 1-Buten zu dimerisieren.
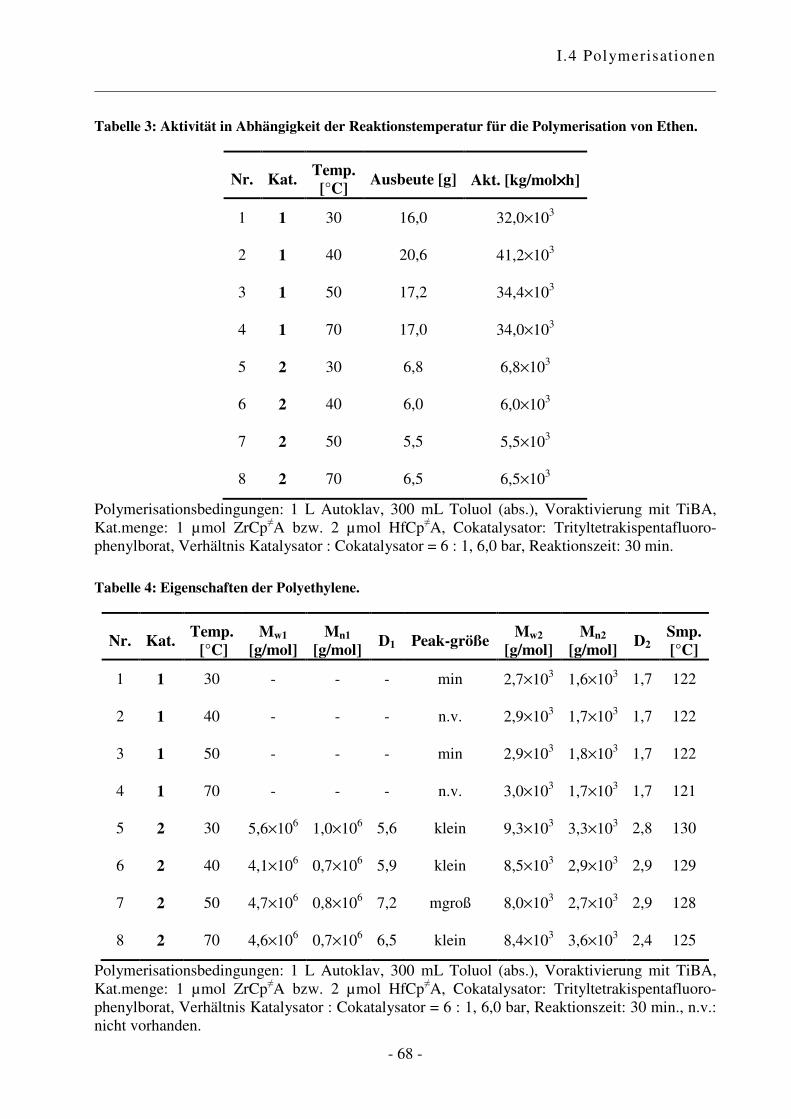
I.4 Polymerisationen
- 68 -
Tabelle 3: Aktivität in Abhängigkeit der Reaktionstemperatur für die Polymerisation von Ethen.
Nr. Kat. Temp. [°C] Ausbeute [g] Akt. [kg/mol××××h]
1 1 30 16,0 32,0×103
2 1 40 20,6 41,2×103
3 1 50 17,2 34,4×103
4 1 70 17,0 34,0×103
5 2 30 6,8 6,8×103
6 2 40 6,0 6,0×103
7 2 50 5,5 5,5×103
8 2 70 6,5 6,5×103
Polymerisationsbedingungen: 1 L Autoklav, 300 mL Toluol (abs.), Voraktivierung mit TiBA, Kat.menge: 1 µmol ZrCp�A bzw. 2 µmol HfCp�A, Cokatalysator: Trityltetrakispentafluoro-phenylborat, Verhältnis Katalysator : Cokatalysator = 6 : 1, 6,0 bar, Reaktionszeit: 30 min.
Tabelle 4: Eigenschaften der Polyethylene.
Nr. Kat. Temp. [°C]
Mw1
[g/mol] Mn1
[g/mol] D1 Peak-größe Mw2
[g/mol] Mn2
[g/mol] D2 Smp. [°C]
1 1 30 - - - min 2,7×103 1,6×103 1,7 122
2 1 40 - - - n.v. 2,9×103 1,7×103 1,7 122
3 1 50 - - - min 2,9×103 1,8×103 1,7 122
4 1 70 - - - n.v. 3,0×103 1,7×103 1,7 121
5 2 30 5,6×106 1,0×106 5,6 klein 9,3×103 3,3×103 2,8 130
6 2 40 4,1×106 0,7×106 5,9 klein 8,5×103 2,9×103 2,9 129
7 2 50 4,7×106 0,8×106 7,2 mgroß 8,0×103 2,7×103 2,9 128
8 2 70 4,6×106 0,7×106 6,5 klein 8,4×103 3,6×103 2,4 125
Polymerisationsbedingungen: 1 L Autoklav, 300 mL Toluol (abs.), Voraktivierung mit TiBA, Kat.menge: 1 µmol ZrCp�A bzw. 2 µmol HfCp�A, Cokatalysator: Trityltetrakispentafluoro-phenylborat, Verhältnis Katalysator : Cokatalysator = 6 : 1, 6,0 bar, Reaktionszeit: 30 min., n.v.: nicht vorhanden.
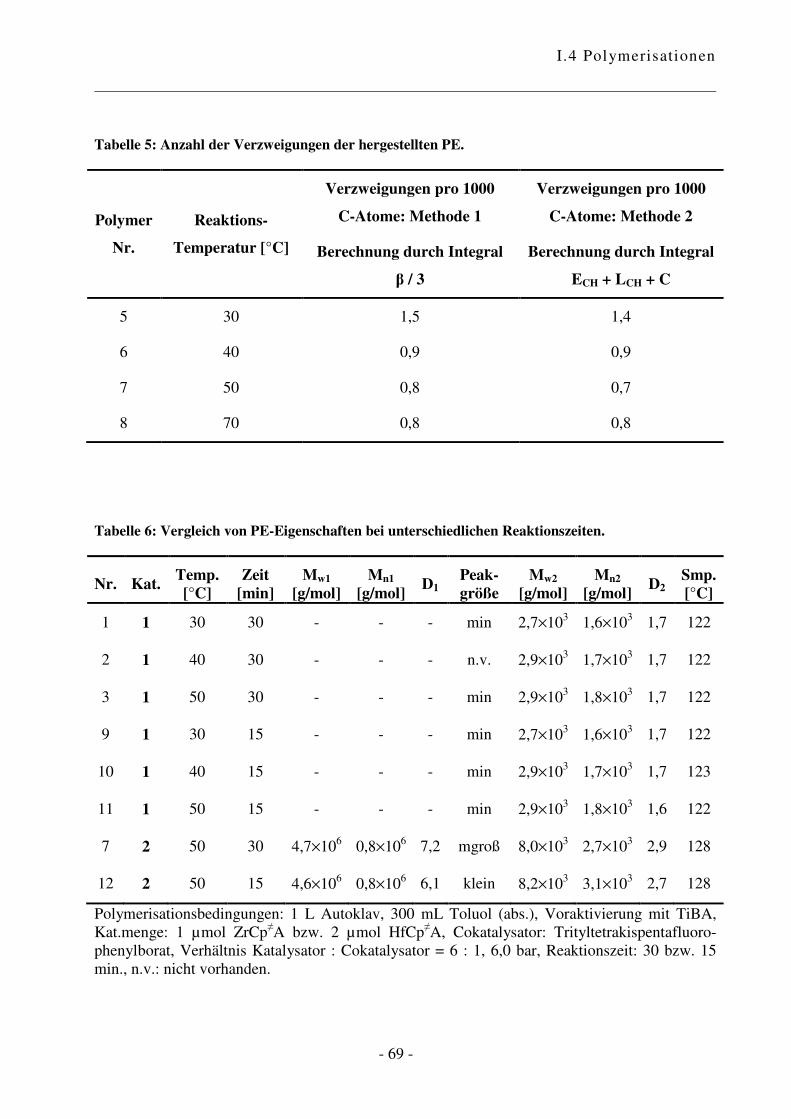
I.4 Polymerisationen
- 69 -
Tabelle 5: Anzahl der Verzweigungen der hergestellten PE.
Polymer
Nr.
Reaktions-
Temperatur [°C]
Verzweigungen pro 1000
C-Atome: Methode 1
Berechnung durch Integral
� / 3
Verzweigungen pro 1000
C-Atome: Methode 2
Berechnung durch Integral
ECH + LCH + C
5 30 1,5 1,4
6 40 0,9 0,9
7 50 0,8 0,7
8 70 0,8 0,8
Tabelle 6: Vergleich von PE-Eigenschaften bei unterschiedlichen Reaktionszeiten.
Nr. Kat. Temp. [°C]
Zeit [min]
Mw1
[g/mol] Mn1
[g/mol] D1
Peak-größe
Mw2
[g/mol] Mn2
[g/mol] D2
Smp. [°C]
1 1 30 30 - - - min 2,7×103 1,6×103 1,7 122
2 1 40 30 - - - n.v. 2,9×103 1,7×103 1,7 122
3 1 50 30 - - - min 2,9×103 1,8×103 1,7 122
9 1 30 15 - - - min 2,7×103 1,6×103 1,7 122
10 1 40 15 - - - min 2,9×103 1,7×103 1,7 123
11 1 50 15 - - - min 2,9×103 1,8×103 1,6 122
7 2 50 30 4,7×106 0,8×106 7,2 mgroß 8,0×103 2,7×103 2,9 128
12 2 50 15 4,6×106 0,8×106 6,1 klein 8,2×103 3,1×103 2,7 128
Polymerisationsbedingungen: 1 L Autoklav, 300 mL Toluol (abs.), Voraktivierung mit TiBA, Kat.menge: 1 µmol ZrCp�A bzw. 2 µmol HfCp�A, Cokatalysator: Trityltetrakispentafluoro-phenylborat, Verhältnis Katalysator : Cokatalysator = 6 : 1, 6,0 bar, Reaktionszeit: 30 bzw. 15 min., n.v.: nicht vorhanden.

I.4 Polymerisationen
- 70 -
I.4.3 Polymerisation von Ethen mit Propen
Abb. 42: Synthese von Ethen-Propen-Copolymeren.
Polyolefine, die durch Copolymerisation von Ethen mit Propen entstehen, weisen gegenüber
reinen Polyethylenen eine größere Anzahl an Verzweigungen auf. Dadurch sinkt die
Kristallinität der Polymere, was zu einer niedrigeren Dichte führt. Polyolefine zeichnen sich
durch ihre hohe Chemikalienbeständigkeit, eine sehr geringe Wasserdampfdurchlässigkeit und
physiologische Unbedenklichkeit aus. Sie finden daher häufig als Verpackungsmittel in der
Lebensmittelindustrie sowie als Lager- und Transportgefäße Anwendung.5
Bei der Durchführung von Copolymerisationen gewinnen im Vergleich zu den
Homopolymerisationen zusätzliche Reaktionsparameter an Bedeutung: die beiden wichtigsten
sind die Sättigung der Reaktionslösung und die Nachführung der Monomere. Zur Sättigung
können Reingase oder ein – beliebig einstellbares – Monomerengemisch verwendet werden. Bei
den hier durchgeführten Experimenten wurde erstere Variante mit Ethen allein gewählt, da die
Aktivität der Katalysatoren beim Aufpressen eines Gemisches oder ausschließlich Propen bei
den gewählten Drücken und Temperaturen sehr niedrig ist.a Im Verlauf der Polymerisation
wurde mit Hilfe der Master-Slave-Steuerung ein Gemisch von Propen und Ethen im Verhältnis
10:1 nachgegeben. Dieser Faktor trägt den stark unterschiedlichen Löslichkeiten der beiden
verwendeten Monomere Rechnungb und wurde angewandt, um den Propen-Gehalt in der Lösung
nach Reaktionsbeginn schnell zu erhöhen. Mit dem Start nimmt durch die Nachführung von zehn
Mal mehr Propen als Ethen und durch den Verdrängungsprozess die Propenkonzentration in
Lösung zu. Da immer mehr Propen zur Verfügung steht, erhöht sich die Wahrscheinlichkeit für
den Propen-Einbau, was Auswirkungen auf die Aktivität und die Polymereigenschaften hat. Ziel
dieses Versuchsaufbaus war es, die Toleranz der beiden Katalysatoren gegenüber Propen zu
testen. Das Hauptaugenmerk gilt dem Vergleich zwischen Zr und Hf und der Frage, wie sich die
a Entsprechende Versuche führten zu keinen messbaren Ausbeuten. Gelegentlich wurden geringe Mengen einer sehr
niedermolekularen Fraktion erhalten. b Zur Sättigung der toluolischen Reaktionslösung werden in dem hier beschriebenen System (1 L Autoklav, 300 mL
Toluol, Druck 6,0 bar)bei 70 °C 1,3 L Ethen beziehungsweise 14 L Propen benötigt.

I.4 Polymerisationen
- 71 -
sterischen und elektronischen Unterschiede zwischen Hafnium- und Zirkonium-Komplex bei
höheren Monomeren auswirken.
Als erstes Kriterium für den Vergleich der beiden Katalysatoren wird wiederum die Aktivität
betrachtet. Wie schon bei der Ethen-Homopolymerisation liegt diese für beide Katalysatoren auf
einem sehr hohen Level, ist jedoch niedriger als zuvor. Da die Aktivität bei 2 weniger stark
zurückgeht, beträgt der Unterschied zwischen 1 und 2 hier nur noch ungefähr den Faktor vier bis
sechs (siehe Diagramm 19 und Tabelle 9) – bei den Ethen-Homopolymerisationen war dieser
fünf bis sieben (siehe I.4.2). Das lässt auf eine größere Toleranz des Hafnium-Katalysators
gegenüber Propen schließen. Unterschiede zeigen sich im Verhalten gegenüber verschiedenen
Reaktionstemperaturen. Während die Aktivität für 1 über den gewählten Reaktionstemperatur-
bereich annähernd konstant bleibt, gibt es bei 2 eine deutliche Steigerung von 2.000 auf
5.100 kg/(mol×h) mit zunehmender Temperatur – bei den Ethen-Polymerisationen zeigten noch
beide Katalysatoren konstante Werte, obwohl eine Zunahme der Aktivität der gängigen Theorie
entspräche.a
Die geringen Aktivitäten für die Polymerisation von Ethen/Propen bei Aufpressen eines
Monomeren-Gemischs sowie bei der Polymerisation von Propen sind erstaunlich. Die CG-
Katalysatoren sind dafür bekannt, dass sie sterisch anspruchsvolle Monomere polymerisieren
können81, offenbaren hier jedoch Schwächen. Shiomura et al. hatten ähnliche Schwierigkeiten
bei der Propen-Polymerisation. Sie erhielten bei 20 °C und einem Druck von 2,94 bar mit dem
Fluorenyl-Zirkonium-Katalysator ebenfalls nur sehr geringe Mengen (50 mg) eines öligen
Materials bzw. weniger als 30 mg wachsartige Materialien, die nicht charakterisiert werden
konnten.82 83
a Aus der Arrhenius-Gleichung ��
���
� −
×= RT
EA
eAk geht hervor, dass die Geschwindigkeitskonstante k mit
zunehmender Temperatur größer wird. Je größer die Temperatur, desto größer wird demnach auch die
Reaktionsgeschwindigkeit v gemäß [A]kv ⋅= .83 Als Konsequenz daraus sollten sich für die hier durchgeführten
Polymerisationsreaktionen bei höherer Reaktionstemperatur größere Ausbeuten und gesteigerte Aktivitäten ergeben.
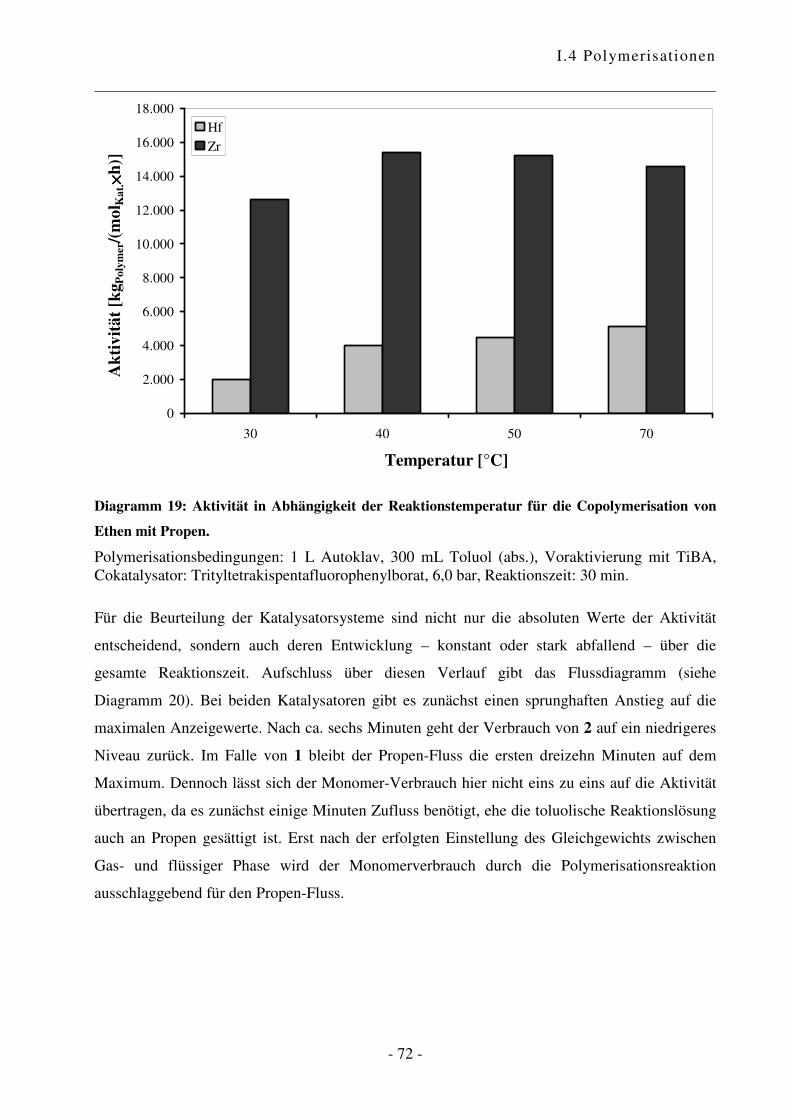
I.4 Polymerisationen
- 72 -
0
2.000
4.000
6.000
8.000
10.000
12.000
14.000
16.000
18.000
30 40 50 70
Temperatur [°C]
Akt
ivit
ät [
kgP
olym
er/(
mol
Kat
.×× ××h)
]HfZr
Diagramm 19: Aktivität in Abhängigkeit der Reaktionstemperatur für die Copolymerisation von
Ethen mit Propen.
Polymerisationsbedingungen: 1 L Autoklav, 300 mL Toluol (abs.), Voraktivierung mit TiBA, Cokatalysator: Trityltetrakispentafluorophenylborat, 6,0 bar, Reaktionszeit: 30 min.
Für die Beurteilung der Katalysatorsysteme sind nicht nur die absoluten Werte der Aktivität
entscheidend, sondern auch deren Entwicklung – konstant oder stark abfallend – über die
gesamte Reaktionszeit. Aufschluss über diesen Verlauf gibt das Flussdiagramm (siehe
Diagramm 20). Bei beiden Katalysatoren gibt es zunächst einen sprunghaften Anstieg auf die
maximalen Anzeigewerte. Nach ca. sechs Minuten geht der Verbrauch von 2 auf ein niedrigeres
Niveau zurück. Im Falle von 1 bleibt der Propen-Fluss die ersten dreizehn Minuten auf dem
Maximum. Dennoch lässt sich der Monomer-Verbrauch hier nicht eins zu eins auf die Aktivität
übertragen, da es zunächst einige Minuten Zufluss benötigt, ehe die toluolische Reaktionslösung
auch an Propen gesättigt ist. Erst nach der erfolgten Einstellung des Gleichgewichts zwischen
Gas- und flüssiger Phase wird der Monomerverbrauch durch die Polymerisationsreaktion
ausschlaggebend für den Propen-Fluss.
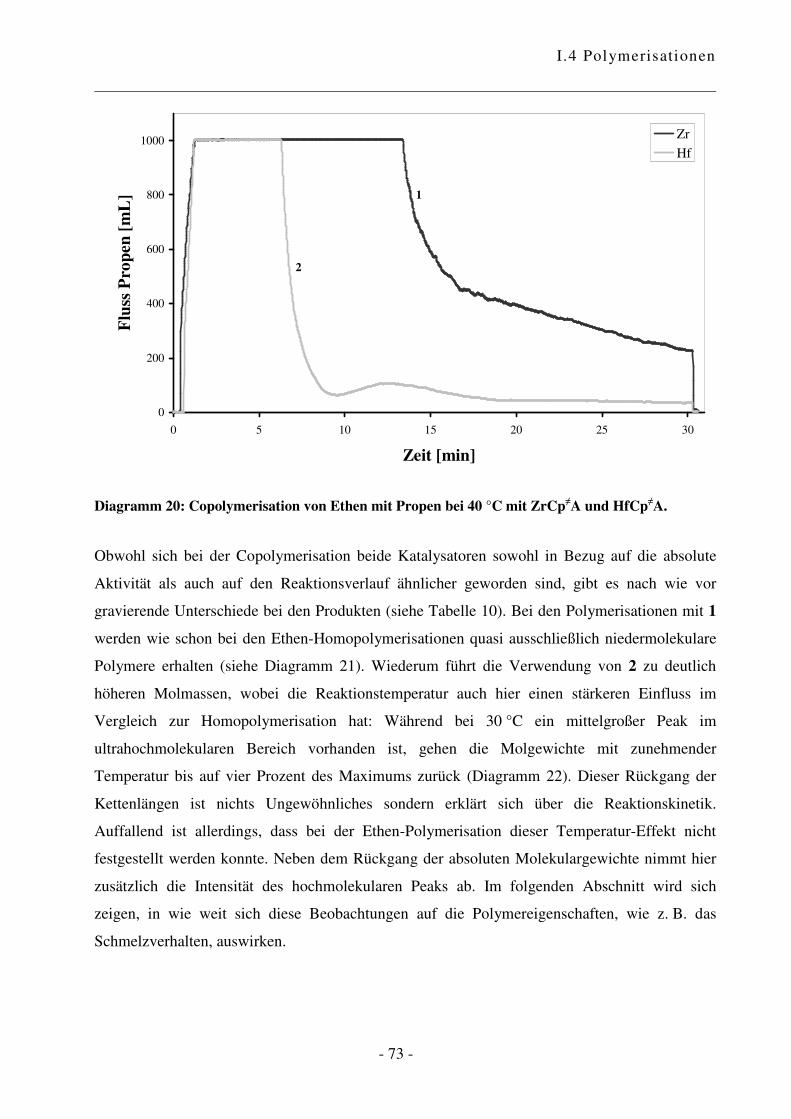
I.4 Polymerisationen
- 73 -
0
200
400
600
800
1000
0 5 10 15 20 25 30
Zeit [min]
Flu
ss P
rope
n [m
L]
ZrHf
1
2
Diagramm 20: Copolymerisation von Ethen mit Propen bei 40 °C mit ZrCp�A und HfCp�A.
Obwohl sich bei der Copolymerisation beide Katalysatoren sowohl in Bezug auf die absolute
Aktivität als auch auf den Reaktionsverlauf ähnlicher geworden sind, gibt es nach wie vor
gravierende Unterschiede bei den Produkten (siehe Tabelle 10). Bei den Polymerisationen mit 1
werden wie schon bei den Ethen-Homopolymerisationen quasi ausschließlich niedermolekulare
Polymere erhalten (siehe Diagramm 21). Wiederum führt die Verwendung von 2 zu deutlich
höheren Molmassen, wobei die Reaktionstemperatur auch hier einen stärkeren Einfluss im
Vergleich zur Homopolymerisation hat: Während bei 30 °C ein mittelgroßer Peak im
ultrahochmolekularen Bereich vorhanden ist, gehen die Molgewichte mit zunehmender
Temperatur bis auf vier Prozent des Maximums zurück (Diagramm 22). Dieser Rückgang der
Kettenlängen ist nichts Ungewöhnliches sondern erklärt sich über die Reaktionskinetik.
Auffallend ist allerdings, dass bei der Ethen-Polymerisation dieser Temperatur-Effekt nicht
festgestellt werden konnte. Neben dem Rückgang der absoluten Molekulargewichte nimmt hier
zusätzlich die Intensität des hochmolekularen Peaks ab. Im folgenden Abschnitt wird sich
zeigen, in wie weit sich diese Beobachtungen auf die Polymereigenschaften, wie z. B. das
Schmelzverhalten, auswirken.
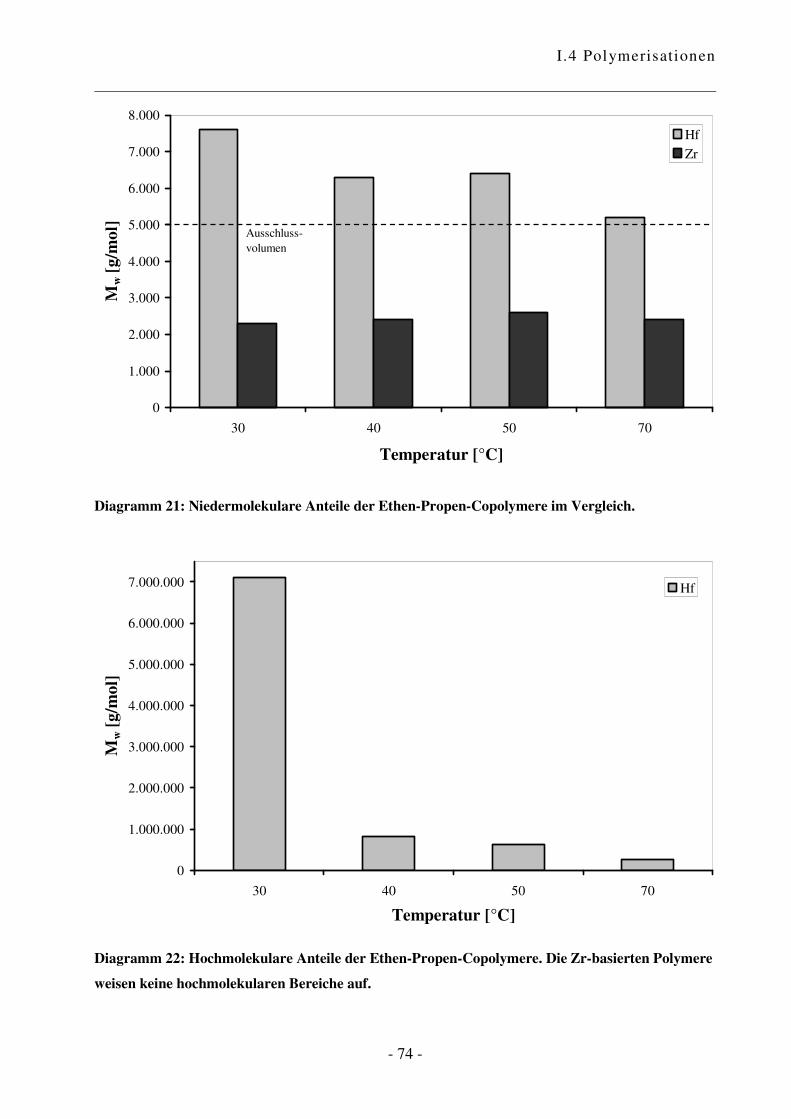
I.4 Polymerisationen
- 74 -
0
1.000
2.000
3.000
4.000
5.000
6.000
7.000
8.000
30 40 50 70
Temperatur [°C]
Mw [
g/m
ol]
HfZr
Ausschluss-volumen
Diagramm 21: Niedermolekulare Anteile der Ethen-Propen-Copolymere im Vergleich.
0
1.000.000
2.000.000
3.000.000
4.000.000
5.000.000
6.000.000
7.000.000
30 40 50 70
Temperatur [°C]
Mw [
g/m
ol]
Hf
Diagramm 22: Hochmolekulare Anteile der Ethen-Propen-Copolymere. Die Zr-basierten Polymere
weisen keine hochmolekularen Bereiche auf.
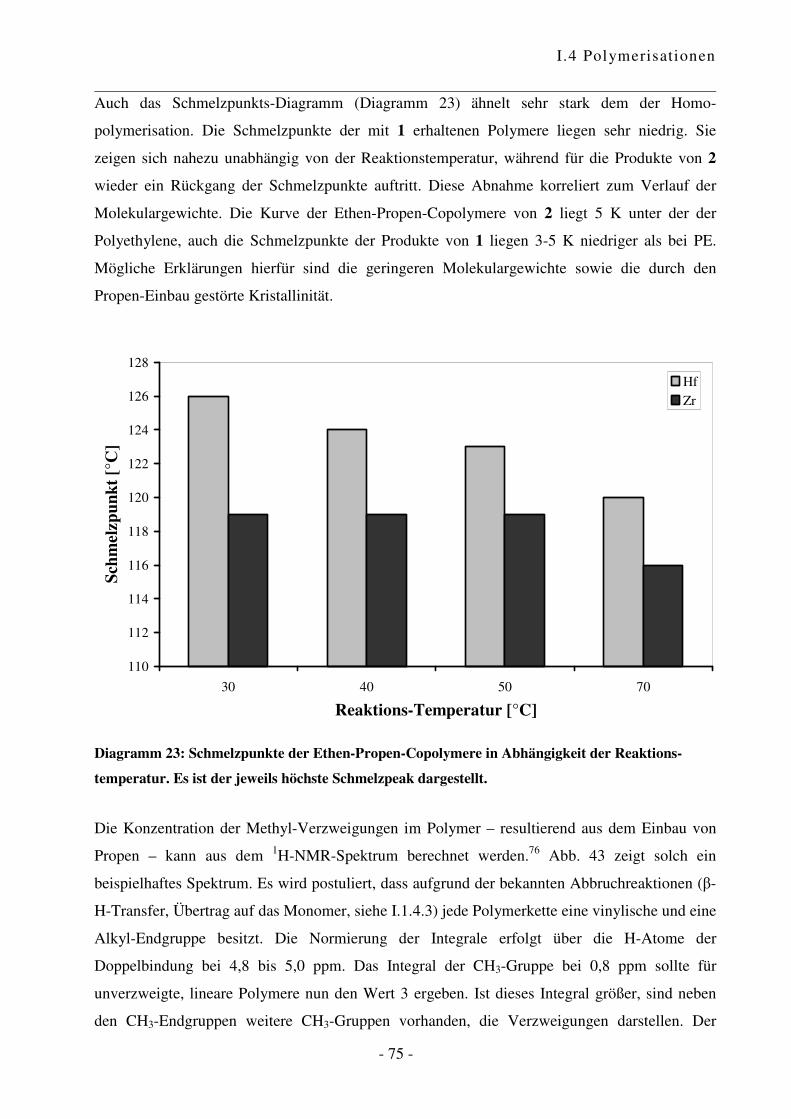
I.4 Polymerisationen
- 75 -
Auch das Schmelzpunkts-Diagramm (Diagramm 23) ähnelt sehr stark dem der Homo-
polymerisation. Die Schmelzpunkte der mit 1 erhaltenen Polymere liegen sehr niedrig. Sie
zeigen sich nahezu unabhängig von der Reaktionstemperatur, während für die Produkte von 2
wieder ein Rückgang der Schmelzpunkte auftritt. Diese Abnahme korreliert zum Verlauf der
Molekulargewichte. Die Kurve der Ethen-Propen-Copolymere von 2 liegt 5 K unter der der
Polyethylene, auch die Schmelzpunkte der Produkte von 1 liegen 3-5 K niedriger als bei PE.
Mögliche Erklärungen hierfür sind die geringeren Molekulargewichte sowie die durch den
Propen-Einbau gestörte Kristallinität.
110
112
114
116
118
120
122
124
126
128
30 40 50 70
Reaktions-Temperatur [°C]
Schm
elzp
unkt
[°C
]
HfZr
Diagramm 23: Schmelzpunkte der Ethen-Propen-Copolymere in Abhängigkeit der Reaktions-
temperatur. Es ist der jeweils höchste Schmelzpeak dargestellt.
Die Konzentration der Methyl-Verzweigungen im Polymer – resultierend aus dem Einbau von
Propen – kann aus dem 1H-NMR-Spektrum berechnet werden.76 Abb. 43 zeigt solch ein
beispielhaftes Spektrum. Es wird postuliert, dass aufgrund der bekannten Abbruchreaktionen (�-
H-Transfer, Übertrag auf das Monomer, siehe I.1.4.3) jede Polymerkette eine vinylische und eine
Alkyl-Endgruppe besitzt. Die Normierung der Integrale erfolgt über die H-Atome der
Doppelbindung bei 4,8 bis 5,0 ppm. Das Integral der CH3-Gruppe bei 0,8 ppm sollte für
unverzweigte, lineare Polymere nun den Wert 3 ergeben. Ist dieses Integral größer, sind neben
den CH3-Endgruppen weitere CH3-Gruppen vorhanden, die Verzweigungen darstellen. Der
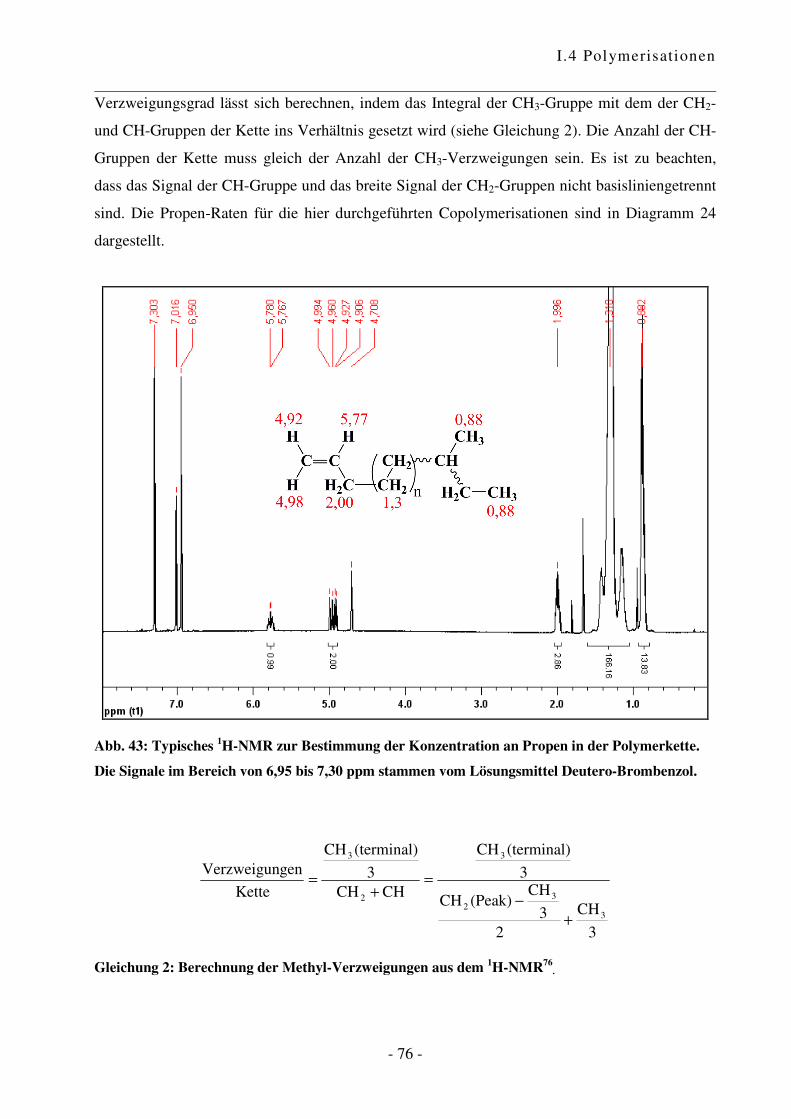
I.4 Polymerisationen
- 76 -
Verzweigungsgrad lässt sich berechnen, indem das Integral der CH3-Gruppe mit dem der CH2-
und CH-Gruppen der Kette ins Verhältnis gesetzt wird (siehe Gleichung 2). Die Anzahl der CH-
Gruppen der Kette muss gleich der Anzahl der CH3-Verzweigungen sein. Es ist zu beachten,
dass das Signal der CH-Gruppe und das breite Signal der CH2-Gruppen nicht basisliniengetrennt
sind. Die Propen-Raten für die hier durchgeführten Copolymerisationen sind in Diagramm 24
dargestellt.
Abb. 43: Typisches 1H-NMR zur Bestimmung der Konzentration an Propen in der Polymerkette.
Die Signale im Bereich von 6,95 bis 7,30 ppm stammen vom Lösungsmittel Deutero-Brombenzol.
3
CH
23
CH(Peak)CH
3
(terminal)CH
CHCH3
(terminal)CH
Kette
genVerzweigun
3
32
3
2
3
+−
=+
=
Gleichung 2: Berechnung der Methyl-Verzweigungen aus dem 1H-NMR76.
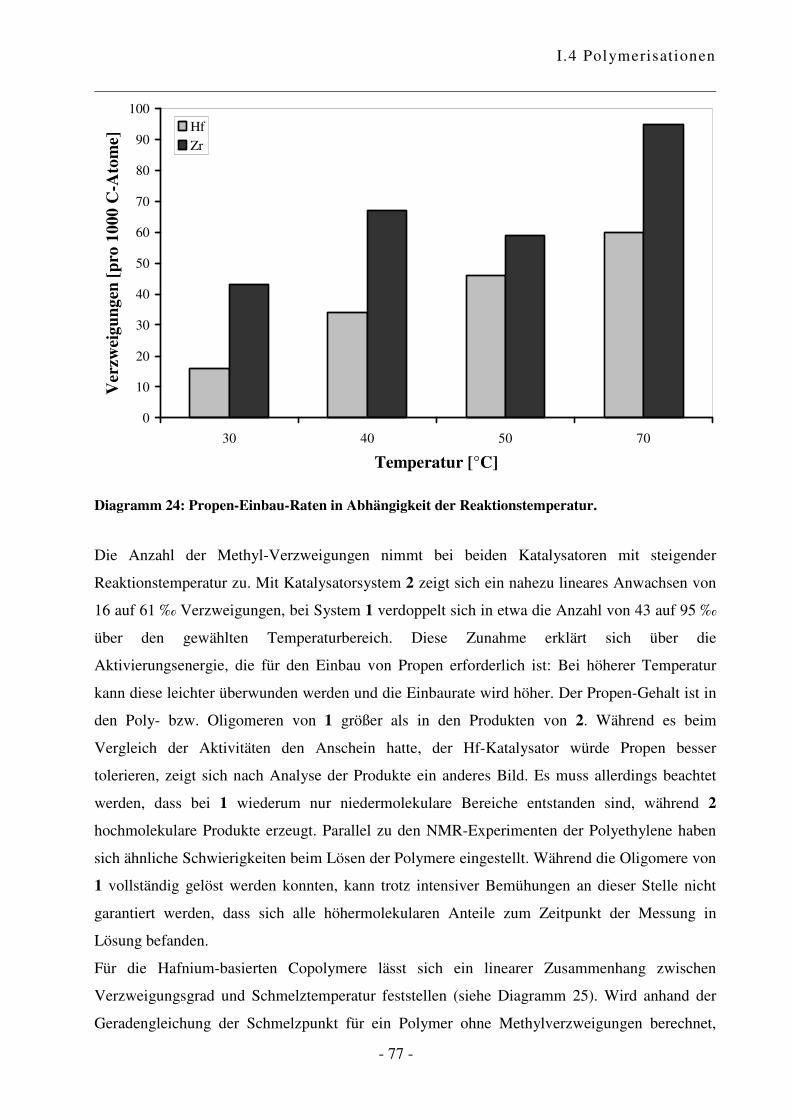
I.4 Polymerisationen
- 77 -
0
10
20
30
40
50
60
70
80
90
100
30 40 50 70
Temperatur [°C]
Ver
zwei
gung
en [
pro
1000
C-A
tom
e]HfZr
Diagramm 24: Propen-Einbau-Raten in Abhängigkeit der Reaktionstemperatur.
Die Anzahl der Methyl-Verzweigungen nimmt bei beiden Katalysatoren mit steigender
Reaktionstemperatur zu. Mit Katalysatorsystem 2 zeigt sich ein nahezu lineares Anwachsen von
16 auf 61 ‰ Verzweigungen, bei System 1 verdoppelt sich in etwa die Anzahl von 43 auf 95 ‰
über den gewählten Temperaturbereich. Diese Zunahme erklärt sich über die
Aktivierungsenergie, die für den Einbau von Propen erforderlich ist: Bei höherer Temperatur
kann diese leichter überwunden werden und die Einbaurate wird höher. Der Propen-Gehalt ist in
den Poly- bzw. Oligomeren von 1 größer als in den Produkten von 2. Während es beim
Vergleich der Aktivitäten den Anschein hatte, der Hf-Katalysator würde Propen besser
tolerieren, zeigt sich nach Analyse der Produkte ein anderes Bild. Es muss allerdings beachtet
werden, dass bei 1 wiederum nur niedermolekulare Bereiche entstanden sind, während 2
hochmolekulare Produkte erzeugt. Parallel zu den NMR-Experimenten der Polyethylene haben
sich ähnliche Schwierigkeiten beim Lösen der Polymere eingestellt. Während die Oligomere von
1 vollständig gelöst werden konnten, kann trotz intensiver Bemühungen an dieser Stelle nicht
garantiert werden, dass sich alle höhermolekularen Anteile zum Zeitpunkt der Messung in
Lösung befanden.
Für die Hafnium-basierten Copolymere lässt sich ein linearer Zusammenhang zwischen
Verzweigungsgrad und Schmelztemperatur feststellen (siehe Diagramm 25). Wird anhand der
Geradengleichung der Schmelzpunkt für ein Polymer ohne Methylverzweigungen berechnet,
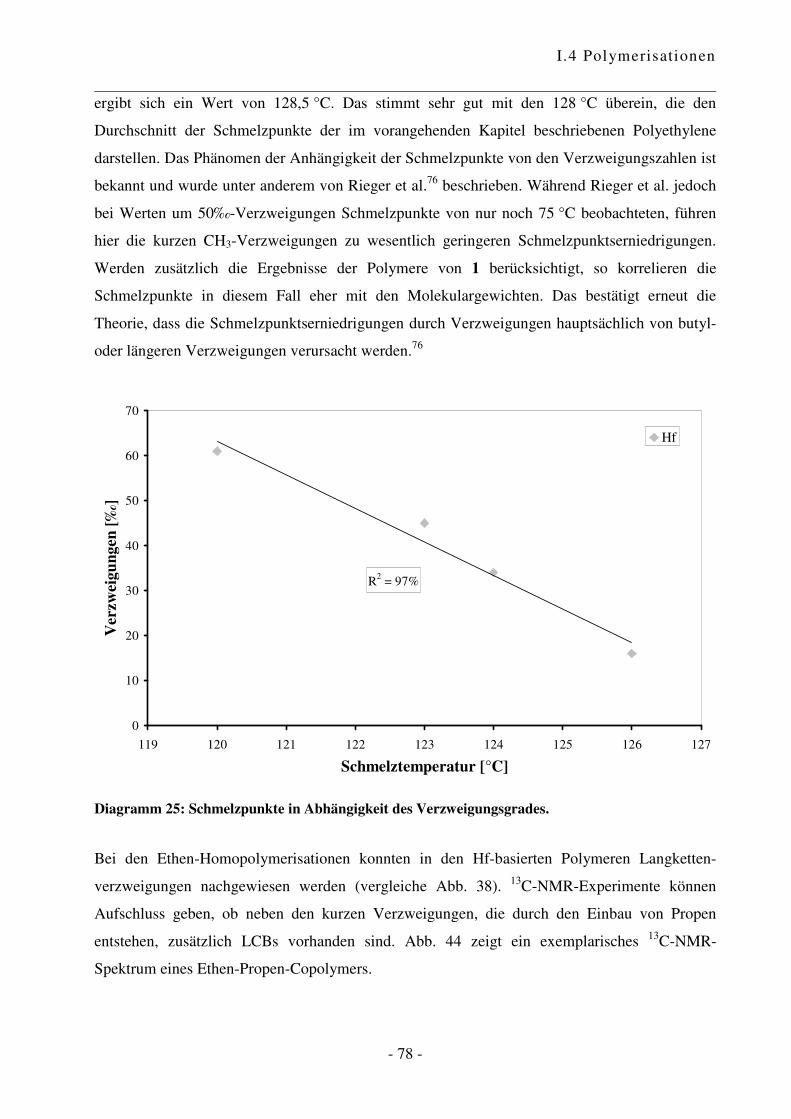
I.4 Polymerisationen
- 78 -
ergibt sich ein Wert von 128,5 °C. Das stimmt sehr gut mit den 128 °C überein, die den
Durchschnitt der Schmelzpunkte der im vorangehenden Kapitel beschriebenen Polyethylene
darstellen. Das Phänomen der Anhängigkeit der Schmelzpunkte von den Verzweigungszahlen ist
bekannt und wurde unter anderem von Rieger et al.76 beschrieben. Während Rieger et al. jedoch
bei Werten um 50‰-Verzweigungen Schmelzpunkte von nur noch 75 °C beobachteten, führen
hier die kurzen CH3-Verzweigungen zu wesentlich geringeren Schmelzpunktserniedrigungen.
Werden zusätzlich die Ergebnisse der Polymere von 1 berücksichtigt, so korrelieren die
Schmelzpunkte in diesem Fall eher mit den Molekulargewichten. Das bestätigt erneut die
Theorie, dass die Schmelzpunktserniedrigungen durch Verzweigungen hauptsächlich von butyl-
oder längeren Verzweigungen verursacht werden.76
R2 = 97%
0
10
20
30
40
50
60
70
119 120 121 122 123 124 125 126 127
Schmelztemperatur [°C]
Ver
zwei
gung
en [
‰]
Hf
Diagramm 25: Schmelzpunkte in Abhängigkeit des Verzweigungsgrades.
Bei den Ethen-Homopolymerisationen konnten in den Hf-basierten Polymeren Langketten-
verzweigungen nachgewiesen werden (vergleiche Abb. 38). 13C-NMR-Experimente können
Aufschluss geben, ob neben den kurzen Verzweigungen, die durch den Einbau von Propen
entstehen, zusätzlich LCBs vorhanden sind. Abb. 44 zeigt ein exemplarisches 13C-NMR-
Spektrum eines Ethen-Propen-Copolymers.
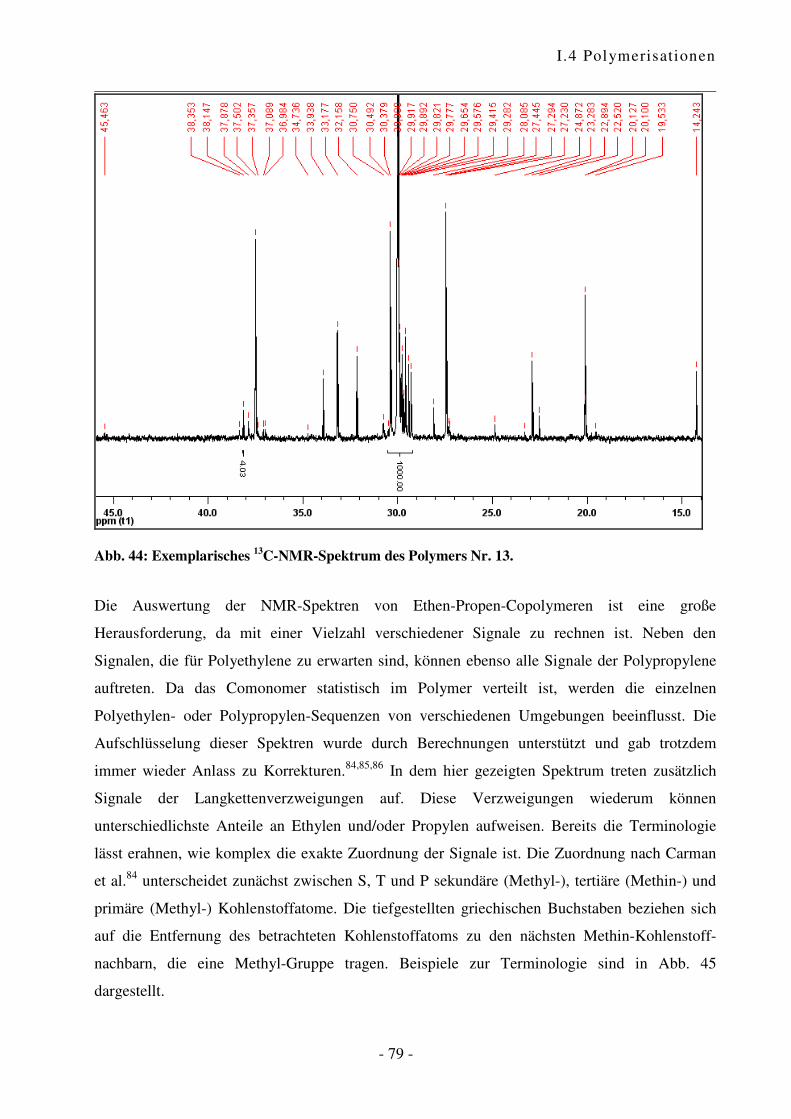
I.4 Polymerisationen
- 79 -
Abb. 44: Exemplarisches 13C-NMR-Spektrum des Polymers Nr. 13.
Die Auswertung der NMR-Spektren von Ethen-Propen-Copolymeren ist eine große
Herausforderung, da mit einer Vielzahl verschiedener Signale zu rechnen ist. Neben den
Signalen, die für Polyethylene zu erwarten sind, können ebenso alle Signale der Polypropylene
auftreten. Da das Comonomer statistisch im Polymer verteilt ist, werden die einzelnen
Polyethylen- oder Polypropylen-Sequenzen von verschiedenen Umgebungen beeinflusst. Die
Aufschlüsselung dieser Spektren wurde durch Berechnungen unterstützt und gab trotzdem
immer wieder Anlass zu Korrekturen.84,85,86 In dem hier gezeigten Spektrum treten zusätzlich
Signale der Langkettenverzweigungen auf. Diese Verzweigungen wiederum können
unterschiedlichste Anteile an Ethylen und/oder Propylen aufweisen. Bereits die Terminologie
lässt erahnen, wie komplex die exakte Zuordnung der Signale ist. Die Zuordnung nach Carman
et al.84 unterscheidet zunächst zwischen S, T und P sekundäre (Methyl-), tertiäre (Methin-) und
primäre (Methyl-) Kohlenstoffatome. Die tiefgestellten griechischen Buchstaben beziehen sich
auf die Entfernung des betrachteten Kohlenstoffatoms zu den nächsten Methin-Kohlenstoff-
nachbarn, die eine Methyl-Gruppe tragen. Beispiele zur Terminologie sind in Abb. 45
dargestellt.

I.4 Polymerisationen
- 80 -
Abb. 45: Nomenklatur der Methyl-Kohlenstoffatome in Ethen-Propen-Copolymeren.
Darüber hinaus wird für die Zuordnung der Langkettenverzweigungen die selbe Nomenklatur
wie in Kapitel I.4.2 (Tabelle 2, Abb. 40 und Abb. 41) verwendet. Zur besseren Unterscheidung
sind die Sequenzen der Langkettenverzweigungen kursiv gedruckt. In Tabelle 7 sind die LCBs
der Ethen-Propen-Copolymere aufgeführt, in der nachfolgenden Tabelle 8 sind die wichtigsten
Bereiche aufgeschlüsselt. Analog zu den Polyethylenen ergeben sich hier Verzweigungszahlen
von ca. einer Verzweigung pro 1000 C-Atome. Obwohl 1 nur niedermolekulare Polymere
erzeugt, konnten auch hier in den NMR-Spektren die Signale in den für LCBs charakteristischen
Bereichen gefunden werden.
Tabelle 7: Anzahl der Langketten-Verzweigungen der hergestellten Ethen-Propen-Copolymere.
Polymer Nr. Kat. Temp. [°C] Verzweigungen pro 1000 C-Atome
Berechnung durch Integral � / 3
13 1 30 0,9
14 1 40 1,3
15 1 50 1,3
16 1 70 2,3
17 2 30 1,2
18 2 40 0,9
19 2 50 1,1
20 2 70 1,7
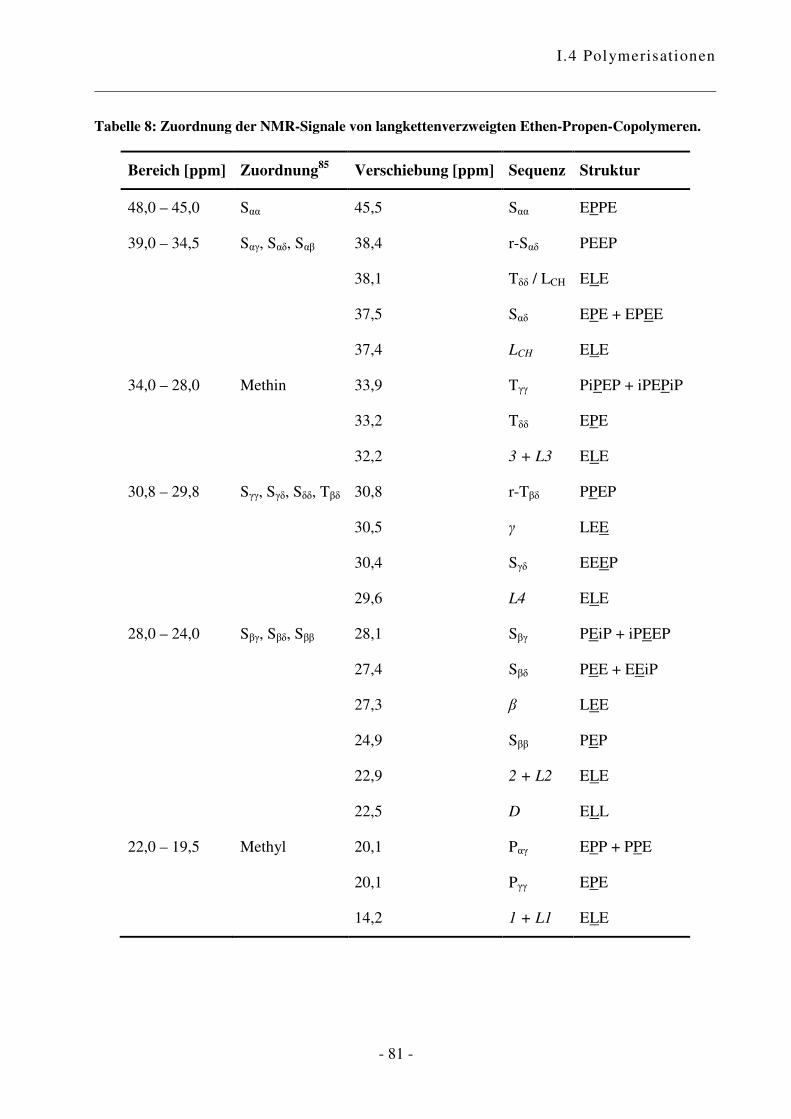
I.4 Polymerisationen
- 81 -
Tabelle 8: Zuordnung der NMR-Signale von langkettenverzweigten Ethen-Propen-Copolymeren.
Bereich [ppm] Zuordnung85 Verschiebung [ppm] Sequenz Struktur
48,0 – 45,0 S�� 45,5 S�� EPPE
39,0 – 34,5 S�, S�A, S�� 38,4 r-S�A PEEP
38,1 TAA / LCH ELE
37,5 S�A EPE + EPEE
37,4 LCH ELE
34,0 – 28,0 Methin 33,9 T PiPEP + iPEPiP
33,2 TAA EPE
32,2 3 + L3 ELE
30,8 – 29,8 S, SA, SAA, T�A 30,8 r-T�A PPEP
30,5 � LEE
30,4 SA EEEP
29,6 L4 ELE
28,0 – 24,0 S�, S�A, S�� 28,1 S� PEiP + iPEEP
27,4 S�A PEE + EEiP
27,3 � LEE
24,9 S�� PEP
22,9 2 + L2 ELE
22,5 D ELL
22,0 – 19,5 Methyl 20,1 P� EPP + PPE
20,1 P EPE
14,2 1 + L1 ELE

I.4 Polymerisationen
- 82 -
Zusammenfassung
Katalysator 1 produziert mit sehr hoher Aktivität langkettenverzweigte, niedermolekulare Ethen-
Propen-Copolymere mit niedrigen Schmelzpunkten. Die Reaktionstemperatur hat dabei keinen
Einfluss auf die Kettenlänge. Katalysator 2 polymerisiert ebenfalls mit sehr hoher Aktivität
Ethen mit Propen. Die erhaltenen Polymere weisen ca. eine Langkettenverzweigung pro 1000 C-
Atome auf und zeigen eine bimodale Molekulargewichtsverteilung mit einem ultrahoch- bis
hochmolekularen Anteil. Das Molekulargewicht fällt mit steigender Reaktionstemperatur. Der
Propen-Anteil in den mit 1 und 2 hergestellten Copolymeren steigt mit zunehmender
Reaktionstemperatur an, die Einbauraten liegen zwischen 15 und 95 Methyl-Verzweigungen pro
1000 C-Atome. Katalysator 1 erreicht dabei jeweils etwas höhere Einbauraten als 2, allerdings
handelt es sich bei 1 lediglich um Oligomere. Die Schmelzpunkte der hochmolekularen
Copolymere nehmen mit zunehmender Propen-Konzentration ab, es existiert ein linearer
Zusammenhang.
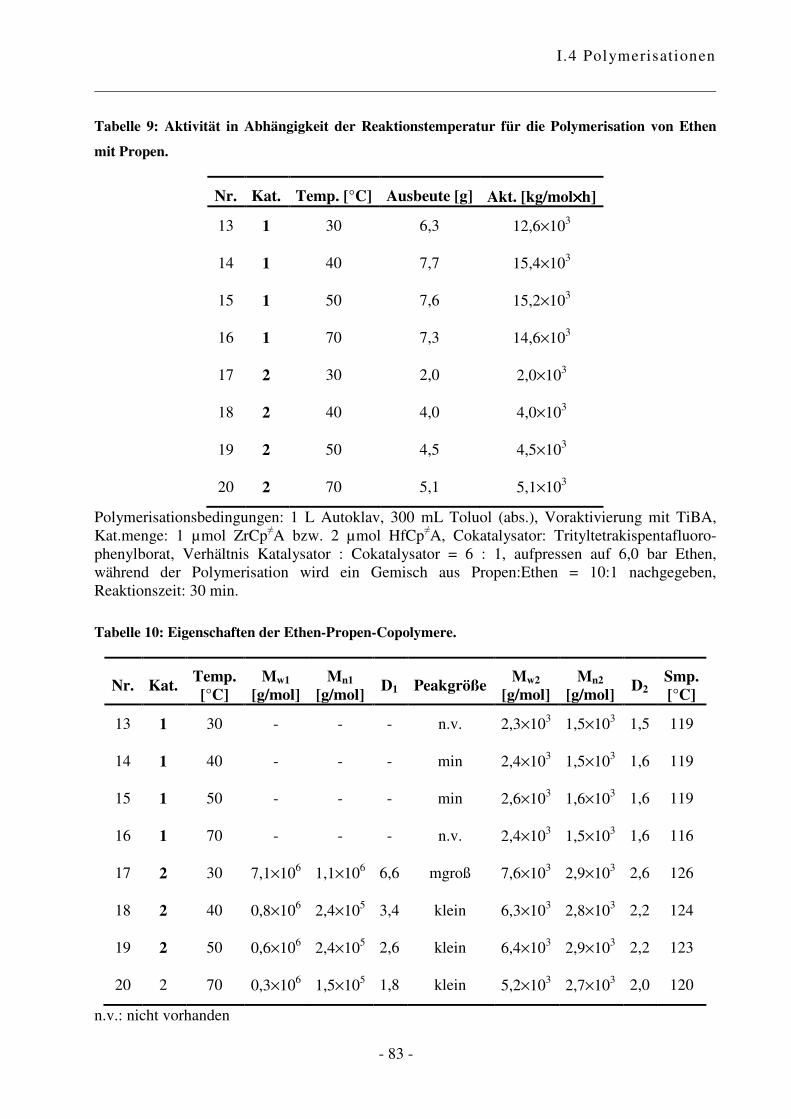
I.4 Polymerisationen
- 83 -
Tabelle 9: Aktivität in Abhängigkeit der Reaktionstemperatur für die Polymerisation von Ethen
mit Propen.
Nr. Kat. Temp. [°C] Ausbeute [g] Akt. [kg/mol××××h]
13 1 30 6,3 12,6×103
14 1 40 7,7 15,4×103
15 1 50 7,6 15,2×103
16 1 70 7,3 14,6×103
17 2 30 2,0 2,0×103
18 2 40 4,0 4,0×103
19 2 50 4,5 4,5×103
20 2 70 5,1 5,1×103
Polymerisationsbedingungen: 1 L Autoklav, 300 mL Toluol (abs.), Voraktivierung mit TiBA, Kat.menge: 1 µmol ZrCp�A bzw. 2 µmol HfCp�A, Cokatalysator: Trityltetrakispentafluoro-phenylborat, Verhältnis Katalysator : Cokatalysator = 6 : 1, aufpressen auf 6,0 bar Ethen, während der Polymerisation wird ein Gemisch aus Propen:Ethen = 10:1 nachgegeben, Reaktionszeit: 30 min.
Tabelle 10: Eigenschaften der Ethen-Propen-Copolymere.
Nr. Kat. Temp. [°C]
Mw1
[g/mol] Mn1
[g/mol] D1 Peakgröße Mw2
[g/mol] Mn2
[g/mol] D2
Smp. [°C]
13 1 30 - - - n.v. 2,3×103 1,5×103 1,5 119
14 1 40 - - - min 2,4×103 1,5×103 1,6 119
15 1 50 - - - min 2,6×103 1,6×103 1,6 119
16 1 70 - - - n.v. 2,4×103 1,5×103 1,6 116
17 2 30 7,1×106 1,1×106 6,6 mgroß 7,6×103 2,9×103 2,6 126
18 2 40 0,8×106 2,4×105 3,4 klein 6,3×103 2,8×103 2,2 124
19 2 50 0,6×106 2,4×105 2,6 klein 6,4×103 2,9×103 2,2 123
20 2 70 0,3×106 1,5×105 1,8 klein 5,2×103 2,7×103 2,0 120
n.v.: nicht vorhanden
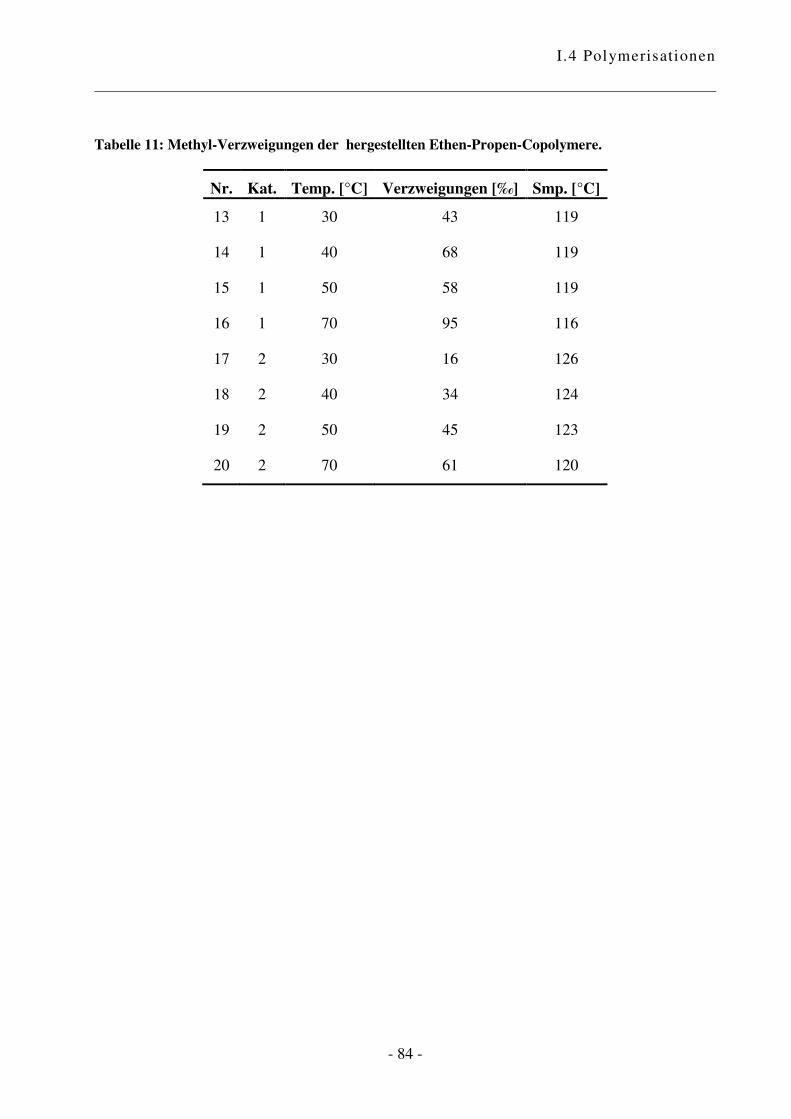
I.4 Polymerisationen
- 84 -
Tabelle 11: Methyl-Verzweigungen der hergestellten Ethen-Propen-Copolymere.
Nr. Kat. Temp. [°C] Verzweigungen [‰] Smp. [°C]
13 1 30 43 119
14 1 40 68 119
15 1 50 58 119
16 1 70 95 116
17 2 30 16 126
18 2 40 34 124
19 2 50 45 123
20 2 70 61 120

I.4 Polymerisationen
- 85 -
I.4.4 Polymerisation von Ethen mit Norbornen
Bei der Polymerisation von Ethen mit cyclischen Olefinen wie Norbornen oder Cyclopenten
entstehen Cycloolefincopolymere – die so genannten COC (siehe Abb. 46).87 Ethen-Norbornen-
Copolymere zeichnen sich insbesondere durch Eigenschaften wie hohe Transparenz, hohe
Brechungsindices, niedrige Dichten, hohe Glastemperaturen, gute Festigkeit und Steifigkeit,
Hydrolysestabilität und geringe Wasseraufnahme aus.7 Dadurch eignet sich diese Materialklasse
besonders für optische Anwendungen wie z. B. Compact Discs, Linsen, optische Fasern und
Filme.88 Die erste kommerzielle Anlage zur Herstellung von COCs wurde im Jahr 2000 in
Oberhausen in Betrieb genommen. Unter dem Handelsnamen Topas® vertreibt die Ticona GmbH
ein Ethen-Norbornen-Copolymer, das aufgrund der Umweltverträglichkeit die hohen
Anforderungen der Medizintechnologie und Lebensmittelindustrie erfüllt.89
Abb. 46: Cycloolefincopolymer aus Ethen und Norbornen.
Metallocen- und CGC-katalysierte Ethen-Norbornen-Copolymerisationen wurden ausführlich
von Ruchatz und Fink diskutiert. In ihren Studien betrachteten sie verschiedene Titan- und
Zirkonium-Katalysatoren.52,90,91,92 Intensive Forschung an Metallocen-basierten COCs erfolgte
auch in der Gruppe um Prof. Kaminsky.88,93,94,95,96 Cyclopentadienylamido-Titan-Systeme und
davon abgeleitete Verbindungen waren Gegenstand einer Publikation von McKnight und
Waymouth97 sowie der Dissertation von Tran.98 Die Verwendung eines Hafnium-CGCs für die
Ethen-Norbornen-Copolymerisation wurde Harrington und Crowther99 sowie in der
Patentliteratur100 beschrieben. Das folgende Kapitel gilt der Untersuchung des Verhaltens und
insbesondere dem Vergleich von ZrCp�A und HfCp�A bei der Copolymerisation von Ethen mit
Norbornen.
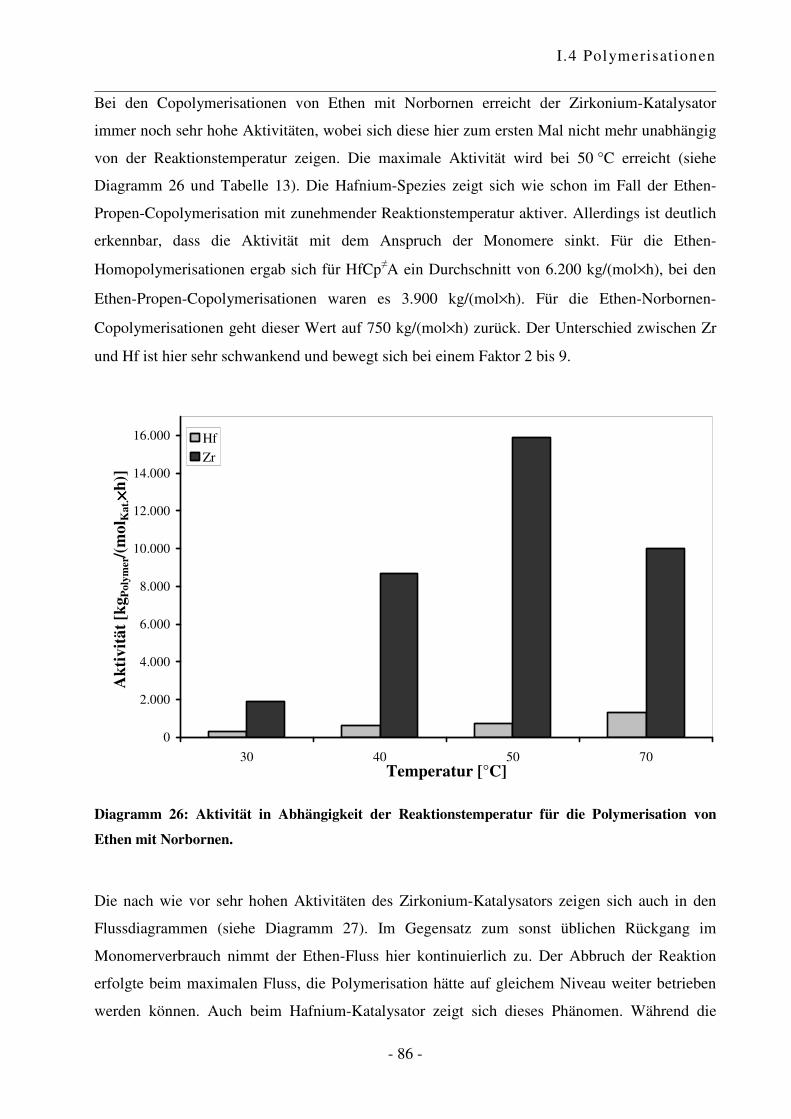
I.4 Polymerisationen
- 86 -
Bei den Copolymerisationen von Ethen mit Norbornen erreicht der Zirkonium-Katalysator
immer noch sehr hohe Aktivitäten, wobei sich diese hier zum ersten Mal nicht mehr unabhängig
von der Reaktionstemperatur zeigen. Die maximale Aktivität wird bei 50 °C erreicht (siehe
Diagramm 26 und Tabelle 13). Die Hafnium-Spezies zeigt sich wie schon im Fall der Ethen-
Propen-Copolymerisation mit zunehmender Reaktionstemperatur aktiver. Allerdings ist deutlich
erkennbar, dass die Aktivität mit dem Anspruch der Monomere sinkt. Für die Ethen-
Homopolymerisationen ergab sich für HfCp�A ein Durchschnitt von 6.200 kg/(mol×h), bei den
Ethen-Propen-Copolymerisationen waren es 3.900 kg/(mol×h). Für die Ethen-Norbornen-
Copolymerisationen geht dieser Wert auf 750 kg/(mol×h) zurück. Der Unterschied zwischen Zr
und Hf ist hier sehr schwankend und bewegt sich bei einem Faktor 2 bis 9.
0
2.000
4.000
6.000
8.000
10.000
12.000
14.000
16.000
30 40 50 70Temperatur [°C]
Akt
ivit
ät [
kgP
olym
er/(
mol
Kat
.×× ××h)
]
HfZr
Diagramm 26: Aktivität in Abhängigkeit der Reaktionstemperatur für die Polymerisation von
Ethen mit Norbornen.
Die nach wie vor sehr hohen Aktivitäten des Zirkonium-Katalysators zeigen sich auch in den
Flussdiagrammen (siehe Diagramm 27). Im Gegensatz zum sonst üblichen Rückgang im
Monomerverbrauch nimmt der Ethen-Fluss hier kontinuierlich zu. Der Abbruch der Reaktion
erfolgte beim maximalen Fluss, die Polymerisation hätte auf gleichem Niveau weiter betrieben
werden können. Auch beim Hafnium-Katalysator zeigt sich dieses Phänomen. Während die
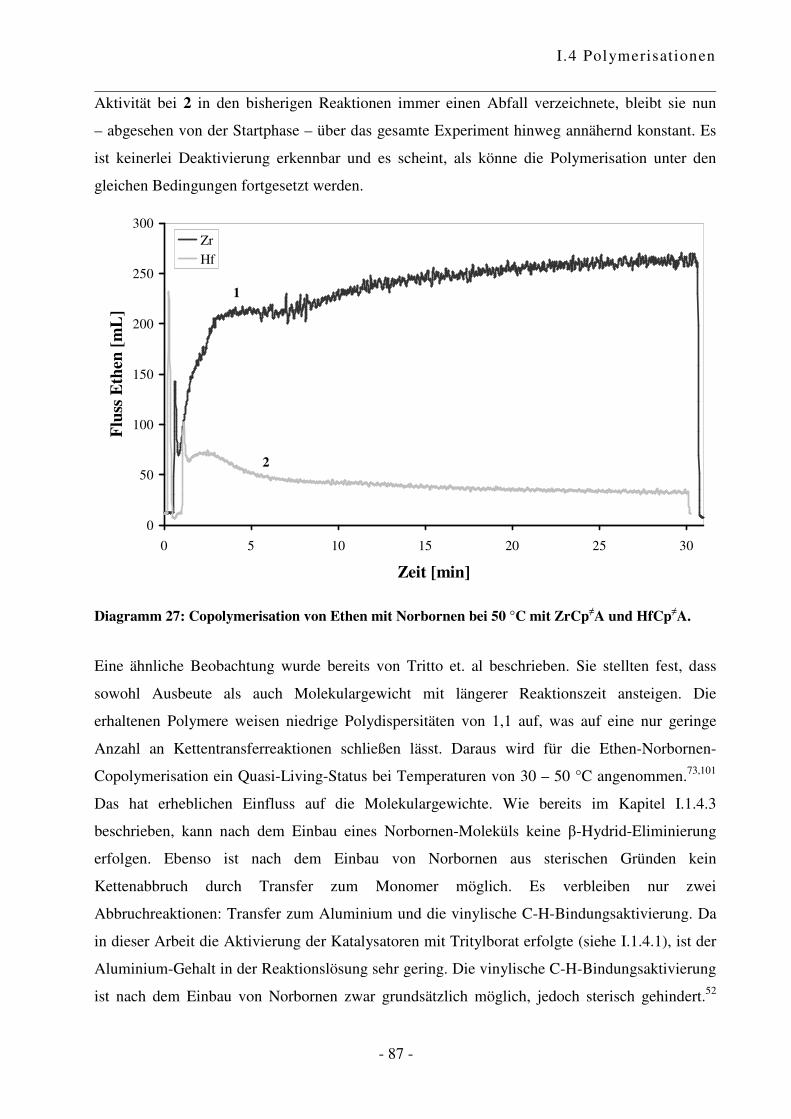
I.4 Polymerisationen
- 87 -
Aktivität bei 2 in den bisherigen Reaktionen immer einen Abfall verzeichnete, bleibt sie nun
– abgesehen von der Startphase – über das gesamte Experiment hinweg annähernd konstant. Es
ist keinerlei Deaktivierung erkennbar und es scheint, als könne die Polymerisation unter den
gleichen Bedingungen fortgesetzt werden.
0
50
100
150
200
250
300
0 5 10 15 20 25 30
Zeit [min]
Flu
ss E
then
[m
L]
ZrHf
1
2
Diagramm 27: Copolymerisation von Ethen mit Norbornen bei 50 °C mit ZrCp�A und HfCp�A.
Eine ähnliche Beobachtung wurde bereits von Tritto et. al beschrieben. Sie stellten fest, dass
sowohl Ausbeute als auch Molekulargewicht mit längerer Reaktionszeit ansteigen. Die
erhaltenen Polymere weisen niedrige Polydispersitäten von 1,1 auf, was auf eine nur geringe
Anzahl an Kettentransferreaktionen schließen lässt. Daraus wird für die Ethen-Norbornen-
Copolymerisation ein Quasi-Living-Status bei Temperaturen von 30 – 50 °C angenommen.73,101
Das hat erheblichen Einfluss auf die Molekulargewichte. Wie bereits im Kapitel I.1.4.3
beschrieben, kann nach dem Einbau eines Norbornen-Moleküls keine �-Hydrid-Eliminierung
erfolgen. Ebenso ist nach dem Einbau von Norbornen aus sterischen Gründen kein
Kettenabbruch durch Transfer zum Monomer möglich. Es verbleiben nur zwei
Abbruchreaktionen: Transfer zum Aluminium und die vinylische C-H-Bindungsaktivierung. Da
in dieser Arbeit die Aktivierung der Katalysatoren mit Tritylborat erfolgte (siehe I.1.4.1), ist der
Aluminium-Gehalt in der Reaktionslösung sehr gering. Die vinylische C-H-Bindungsaktivierung
ist nach dem Einbau von Norbornen zwar grundsätzlich möglich, jedoch sterisch gehindert.52
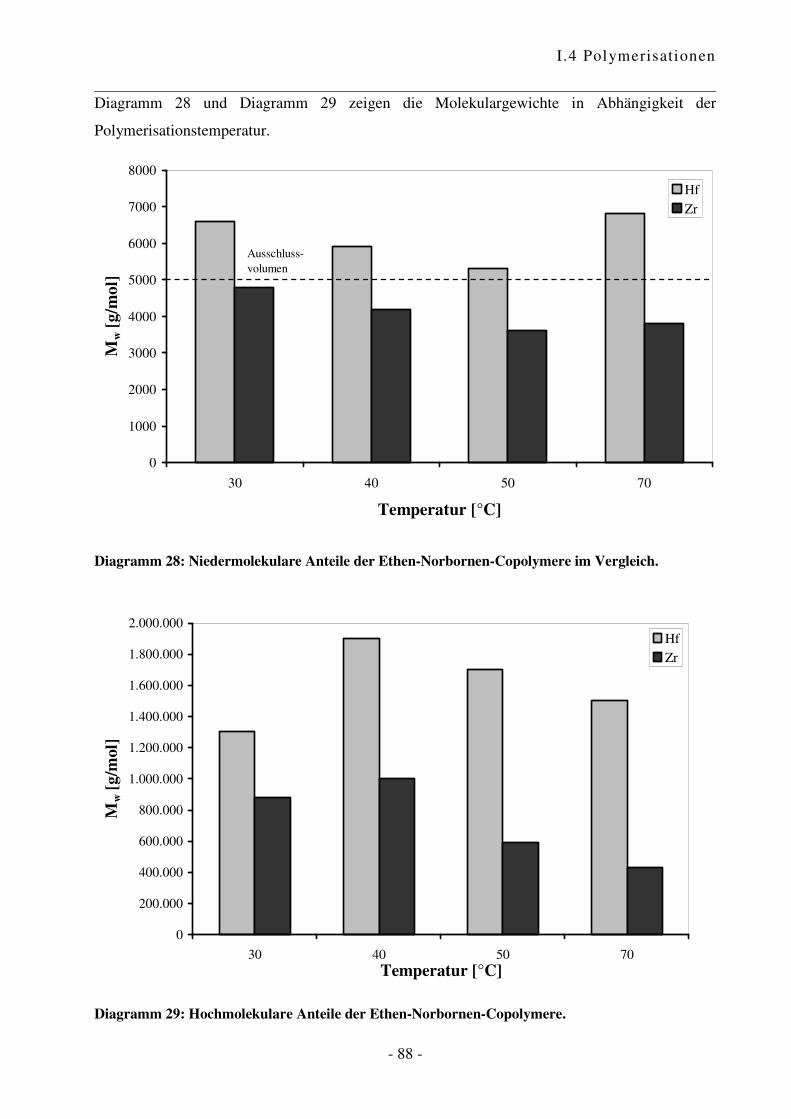
I.4 Polymerisationen
- 88 -
Diagramm 28 und Diagramm 29 zeigen die Molekulargewichte in Abhängigkeit der
Polymerisationstemperatur.
0
1000
2000
3000
4000
5000
6000
7000
8000
30 40 50 70
Temperatur [°C]
Mw [
g/m
ol]
HfZr
Ausschluss-volumen
Diagramm 28: Niedermolekulare Anteile der Ethen-Norbornen-Copolymere im Vergleich.
0
200.000
400.000
600.000
800.000
1.000.000
1.200.000
1.400.000
1.600.000
1.800.000
2.000.000
30 40 50 70Temperatur [°C]
Mw [
g/m
ol]
HfZr
Diagramm 29: Hochmolekulare Anteile der Ethen-Norbornen-Copolymere.

I.4 Polymerisationen
- 89 -
Die oben beschriebenen Besonderheiten bei der Ethen-Norbornen-Copolymerisation führen zum
ersten Mal dazu, dass auch mit ZrCp�A hochmolekulare Polymeranteile erhalten werden.
Dennoch sind – verglichen mit der Hafnium-Spezies – sowohl Molmasse als auch die GPC-
Peakgröße geringer. Molekulargewicht und Norbornen-Gehalt im Copolymer sind eng
miteinander verknüpft. Der Einbau eines Norbornen-Moleküls führt im Vergleich zum Einbau
eines Ethen-Moleküls zu einer größeren Zunahme an Molekulargewicht, erfordert aber die
Überwindung einer größeren Aktivierungsenergie. Ruchatz und Fink fanden (mit einer
Ausnahme) höhere Molmassen bei größerem Comonomergehalt im Polymer.52 Die Einbaurate
an Norbornen lässt sich aus dem 13C-NMR berechnen.88,91,97,98,102 Die Zuordnung der Signale
wird dadurch erschwert, dass im Polymer pro eingebautem Norbornen-Molekül zwei chirale
Zentren entstehen. Studien haben gezeigt, dass Norbornen in Metallocen-katalysierten
Copolymerisationen ausschließlich in 2,3-exo-cis-Konfiguration insertiert (siehe Abb. 47).102
Abb. 47: Bezeichnung der Kohlenstoffatome in einem Ethen-Norbornen-Copolymer. C1 bis C7
kennzeichnen die Kohlenstoffatome des Norbornens, C�, C�, C� und C� die der Ethen-Sequenzen.
Sind im Copolymer ausschließlich isolierte Norbornen-Einheiten vorhanden, erscheinen im 13C-NMR nur vier Signale, wobei das Signal der Kohlenstoffatome C5 und C6 kaum von denen
der CH2-Hauptkette (C�, C�, C und CA) zu unterscheiden sind. Abb. 48 zeigt ein exemplarisches
NMR-Spektrum für ein mit ZrCp�A hergestelltes Ethen-Norbornen-Copolymer. Bei 30,0 ppm
tritt das Signal der für Polyethylene typischen CH2-Hauptkette auf. Die Signale bei 14,2, 22,9,
32,2 und 33,9 ppm stellen alkylische Endgruppen dar (vergleiche Abb. 38). Das Vorhandensein
isolierter Norbornen-Einheiten zeigt sich anhand der Signale bei 33,2 ppm für C7, bei 41,7 ppm
für C1 und C4 sowie bei 47,2 ppm für C2 und C3.
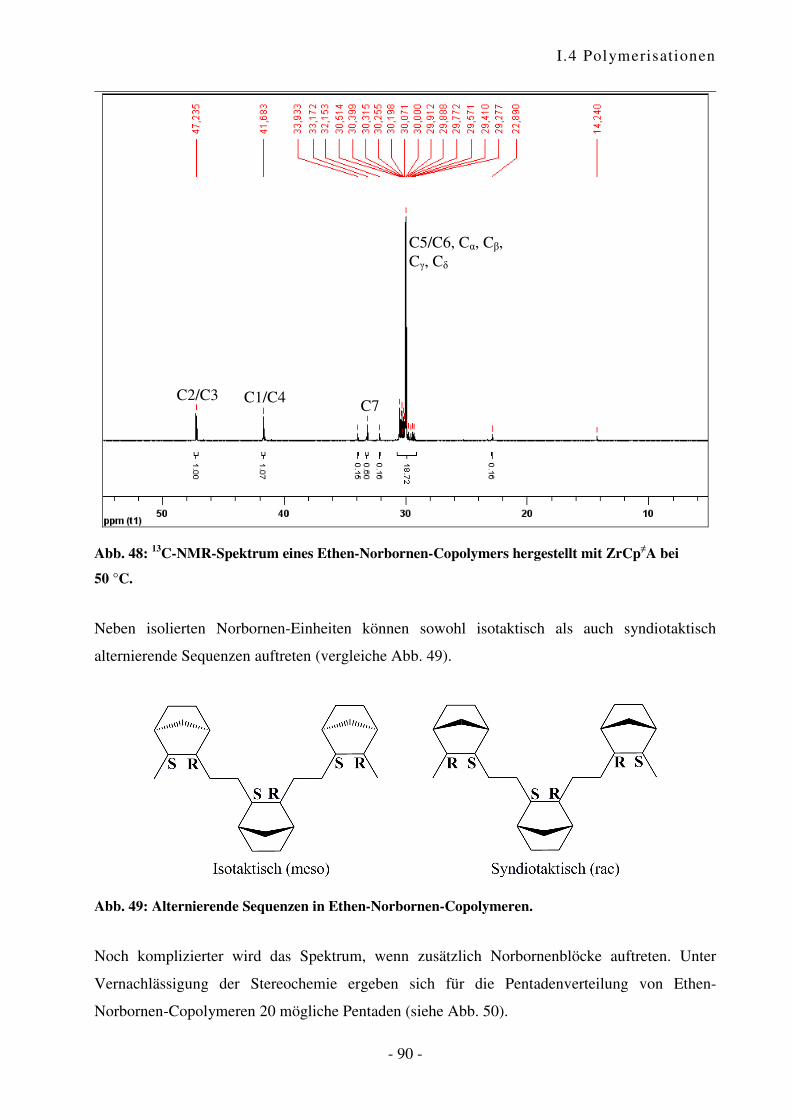
I.4 Polymerisationen
- 90 -
Abb. 48: 13C-NMR-Spektrum eines Ethen-Norbornen-Copolymers hergestellt mit ZrCp�A bei
50 °C.
Neben isolierten Norbornen-Einheiten können sowohl isotaktisch als auch syndiotaktisch
alternierende Sequenzen auftreten (vergleiche Abb. 49).
Abb. 49: Alternierende Sequenzen in Ethen-Norbornen-Copolymeren.
Noch komplizierter wird das Spektrum, wenn zusätzlich Norbornenblöcke auftreten. Unter
Vernachlässigung der Stereochemie ergeben sich für die Pentadenverteilung von Ethen-
Norbornen-Copolymeren 20 mögliche Pentaden (siehe Abb. 50).
C2/C3 C1/C4 C7
C5/C6, C�, C�, C, CA
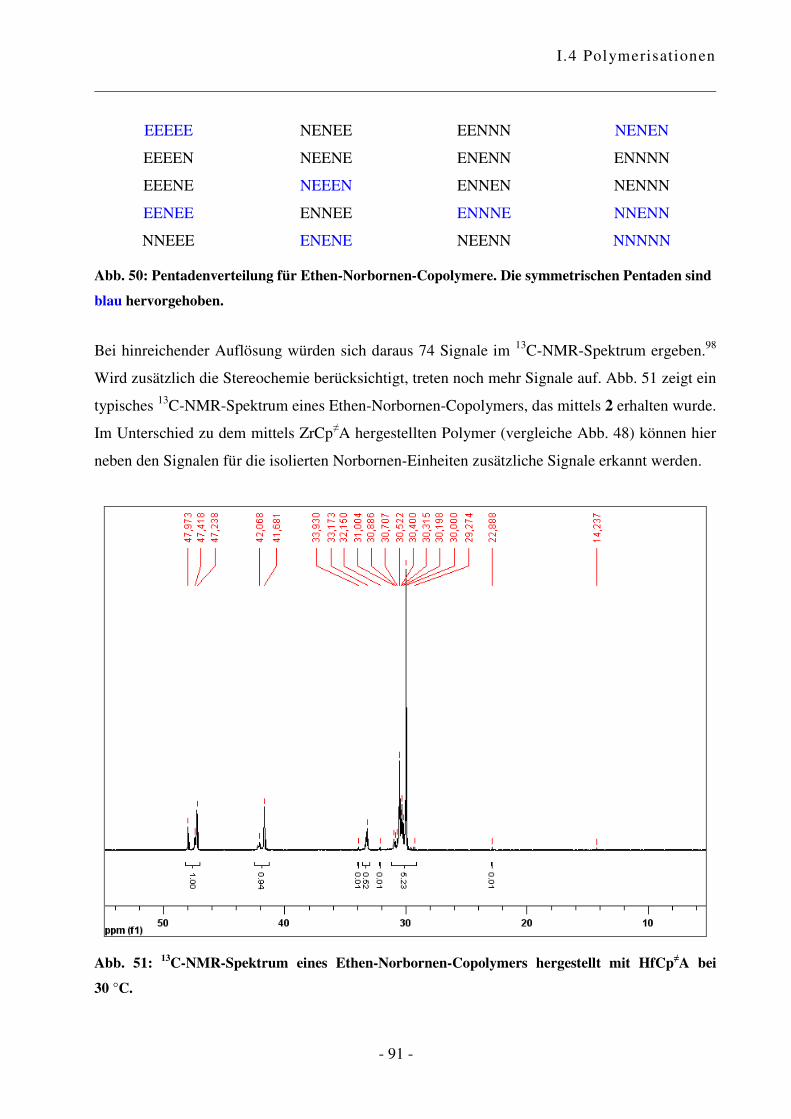
I.4 Polymerisationen
- 91 -
EEEEE NENEE EENNN NENEN
EEEEN NEENE ENENN ENNNN
EEENE NEEEN ENNEN NENNN
EENEE ENNEE ENNNE NNENN
NNEEE ENENE NEENN NNNNN
Abb. 50: Pentadenverteilung für Ethen-Norbornen-Copolymere. Die symmetrischen Pentaden sind
blau hervorgehoben.
Bei hinreichender Auflösung würden sich daraus 74 Signale im 13C-NMR-Spektrum ergeben.98
Wird zusätzlich die Stereochemie berücksichtigt, treten noch mehr Signale auf. Abb. 51 zeigt ein
typisches 13C-NMR-Spektrum eines Ethen-Norbornen-Copolymers, das mittels 2 erhalten wurde.
Im Unterschied zu dem mittels ZrCp�A hergestellten Polymer (vergleiche Abb. 48) können hier
neben den Signalen für die isolierten Norbornen-Einheiten zusätzliche Signale erkannt werden.
Abb. 51: 13C-NMR-Spektrum eines Ethen-Norbornen-Copolymers hergestellt mit HfCp�A bei
30 °C.
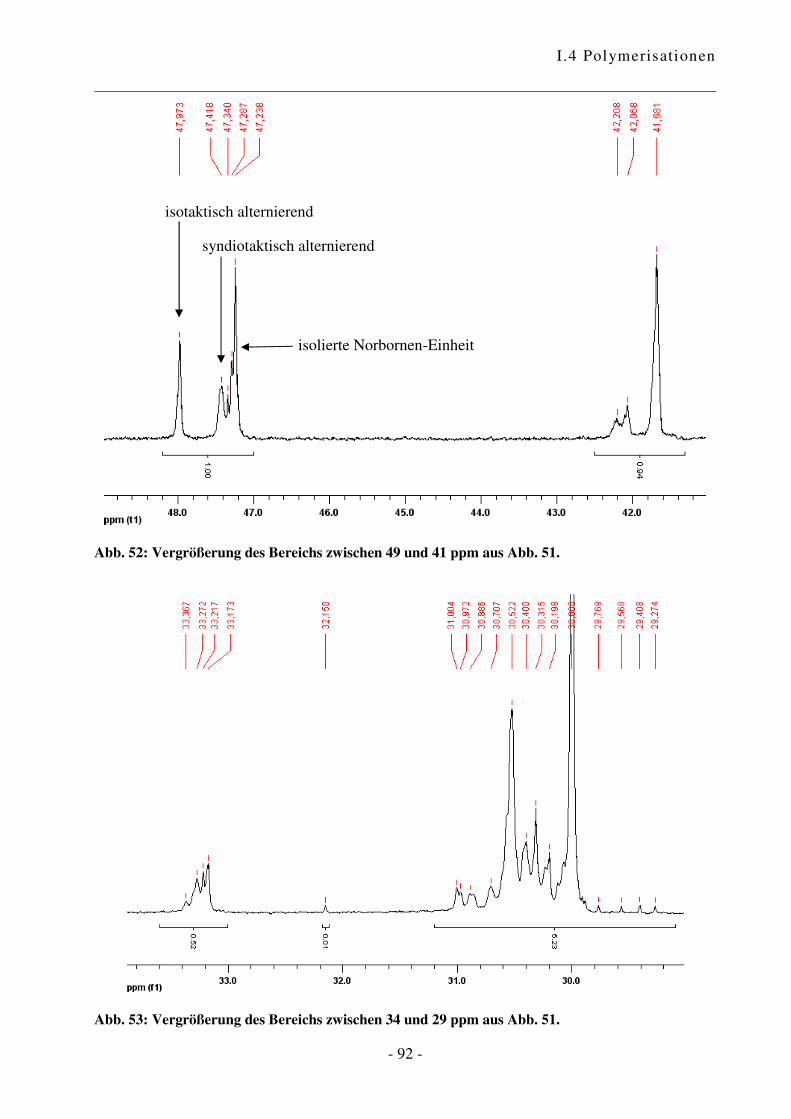
I.4 Polymerisationen
- 92 -
Abb. 52: Vergrößerung des Bereichs zwischen 49 und 41 ppm aus Abb. 51.
Abb. 53: Vergrößerung des Bereichs zwischen 34 und 29 ppm aus Abb. 51.
isotaktisch alternierend
syndiotaktisch alternierend
isolierte Norbornen-Einheit

I.4 Polymerisationen
- 93 -
Die Zuordnung der Signale ist in Tabelle 16 erklärt. Der Hafnium-Katalysator ist in der Lage,
neben isolierten Norbornen-Molekülen auch alternierende Norbornen-Ethen-Sequenzen
(vergleiche Abb. 49) in das Copolymer einzubauen. Hierin zeigt sich erneut ein deutlicher
Unterschied zwischen Zirkonium- und Hafnium-CGC. Das Vorhandensein sowohl isolierter
Norbornen-Einheiten als auch alternierender Ethen-Norbornen-Sequenzen lässt zudem auf die
Bildung eines Block-Copolymers (PE – Ethen-Norbornen-Copolymer – PE) schließen. In den
Bereichen, die für die Insertion zweier oder mehrerer Norbornen-Einheiten direkt nacheinander
(sog. Dyaden und Triaden) charakteristisch sind, konnten keine Signale gefunden werden. Die
Tendenz zur Bildung alternierender Ethen-Norbornen-Sequenzen wurde auch für andere CGC-
Systeme beschrieben.97,99 Es stellt sich die Frage, ob es – wie bei den Ethen-Homopolymerisa-
tionen und Ethen-Propen-Copolymerisationen – zur Bildung von Langkettenverzweigungen
kommt. Tritto, Mühlhaupt et al. haben über langkettenverzweigte Ethen-Norbornen-Copolymere
durch CGC-Katalyse berichtet.73 Als aktive Spezies wurde das System TiCp�A mit MAO-
Cokatalysator verwendet (vergleiche Abb. 54).
Abb. 54: Das von Tritto, Mühlhaupt et al. verwendete Katalysatorsystem TiCp�A mit
Methylaluminoxan.73
Sie bestätigten das Vorhandensein von Langkettenverzweigungen durch Größenausschluss-
chromatographie und rheologische Untersuchungen. Ein Nachweis durch NMR-Spektroskopie
erfolgte nicht. Es wurde jedoch zu Bedenken gegeben, dass es noch nicht geklärt sei, ob die
Verschiebung der Signale, die für LCBs in Polyethylenen charakteristisch sind, bei Ethen-
Norbornen-Copolymeren ebenso Gültigkeit besitzen.73 In den in dieser Arbeit erhaltenen NMR-
Spektren konnten keine Signale gefunden werden, die auf die Bildung von Langketten-
verzweigungen schließen lassen würden. Nichtsdestotrotz liegt die Vermutung nahe, dass
derartige Verzweigungen entstanden sind.

I.4 Polymerisationen
- 94 -
Die Bestimmung des Norbornen-Gehalts im Copolymer kann über die Integration der
charakteristischen Bereiche des 13C-NMR-Spektrums erfolgen. Die Signalbereiche werden dabei
häufig sehr weit gefasst (ca. 4 bis 10 ppm).98 Um Fehler durch die hier zu sehenden Polymer-
Endgruppen zu vermeiden, wurden die Integrations-Grenzen enger gesetzt (vergleiche Tabelle
12). Die Anteile an Norbornen im Copolymer können über Gleichung 3 berechnet werden73,98,102
und sind in Diagramm 30 sowie in Tabelle 15 dargestellt.
Tabelle 12: Integrationsgrenzen für die Bestimmung der Norbornen-Gehalte.
� 13C [ppm] Bereich Zuordnung Integrationsgrenzen
für ZrCp�A
Integrationsgrenzen
für HfCp�A
56,0 – 44,8 A C2, C3 47,4 – 47,1 48,2 – 47,0
44,8 – 36,8 B C1, C4 41,85 – 41,55 42,5 – 41,3
36,8 – 32,8 C C7 33,3 – 33,1 33,6 – 33,0
32,8 – 27,8 D C5, C6, C�,
C�, C, CA 30,7 – 29,1 31,2 – 29,1
I(D)2,5I(C)I(B)I(A)
)]C,C,C,I(CC6)[I(C5,21
I(C7)]C4)I(C1,C3)[I(C2,51
X
A��
N⋅
++=
+
++=
Gleichung 3: Berechnung des Norbornen-Anteils in Ethen-Norbornen-Copolymeren. XN bezeichnet
den Molenbruch an Norbornen, I steht für Integral.
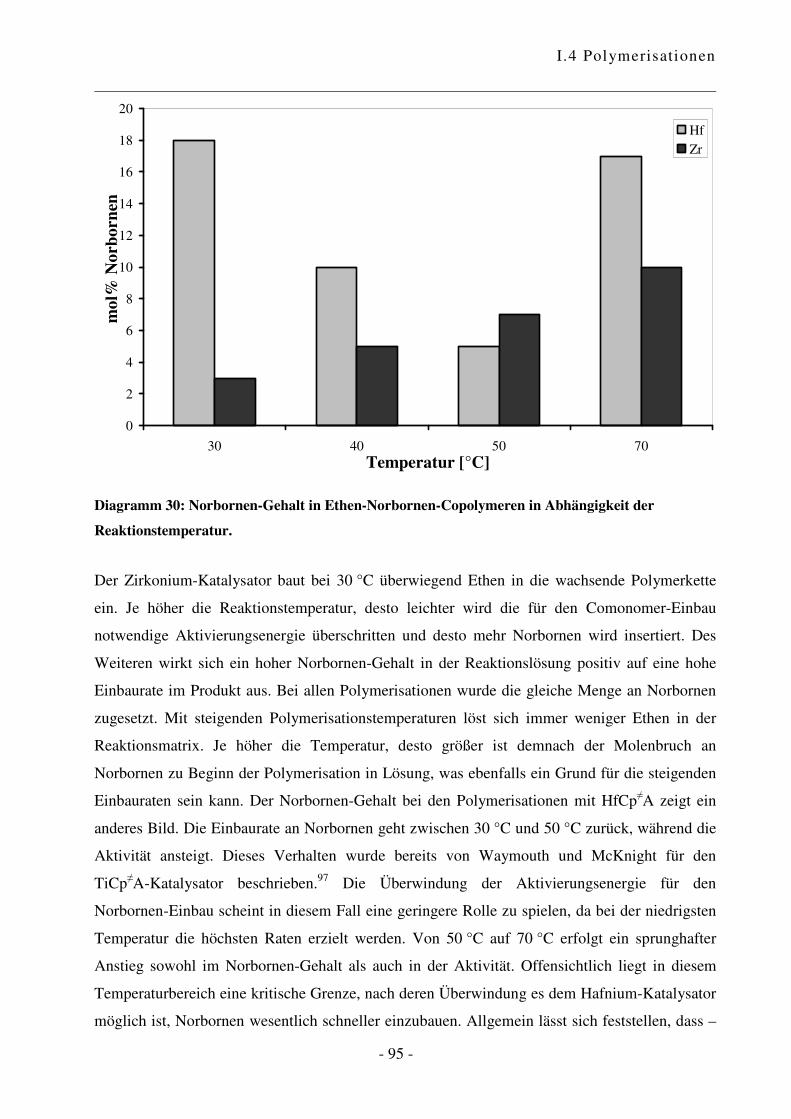
I.4 Polymerisationen
- 95 -
0
2
4
6
8
10
12
14
16
18
20
30 40 50 70Temperatur [°C]
mol
% N
orbo
rnen
HfZr
Diagramm 30: Norbornen-Gehalt in Ethen-Norbornen-Copolymeren in Abhängigkeit der
Reaktionstemperatur.
Der Zirkonium-Katalysator baut bei 30 °C überwiegend Ethen in die wachsende Polymerkette
ein. Je höher die Reaktionstemperatur, desto leichter wird die für den Comonomer-Einbau
notwendige Aktivierungsenergie überschritten und desto mehr Norbornen wird insertiert. Des
Weiteren wirkt sich ein hoher Norbornen-Gehalt in der Reaktionslösung positiv auf eine hohe
Einbaurate im Produkt aus. Bei allen Polymerisationen wurde die gleiche Menge an Norbornen
zugesetzt. Mit steigenden Polymerisationstemperaturen löst sich immer weniger Ethen in der
Reaktionsmatrix. Je höher die Temperatur, desto größer ist demnach der Molenbruch an
Norbornen zu Beginn der Polymerisation in Lösung, was ebenfalls ein Grund für die steigenden
Einbauraten sein kann. Der Norbornen-Gehalt bei den Polymerisationen mit HfCp�A zeigt ein
anderes Bild. Die Einbaurate an Norbornen geht zwischen 30 °C und 50 °C zurück, während die
Aktivität ansteigt. Dieses Verhalten wurde bereits von Waymouth und McKnight für den
TiCp�A-Katalysator beschrieben.97 Die Überwindung der Aktivierungsenergie für den
Norbornen-Einbau scheint in diesem Fall eine geringere Rolle zu spielen, da bei der niedrigsten
Temperatur die höchsten Raten erzielt werden. Von 50 °C auf 70 °C erfolgt ein sprunghafter
Anstieg sowohl im Norbornen-Gehalt als auch in der Aktivität. Offensichtlich liegt in diesem
Temperaturbereich eine kritische Grenze, nach deren Überwindung es dem Hafnium-Katalysator
möglich ist, Norbornen wesentlich schneller einzubauen. Allgemein lässt sich feststellen, dass –
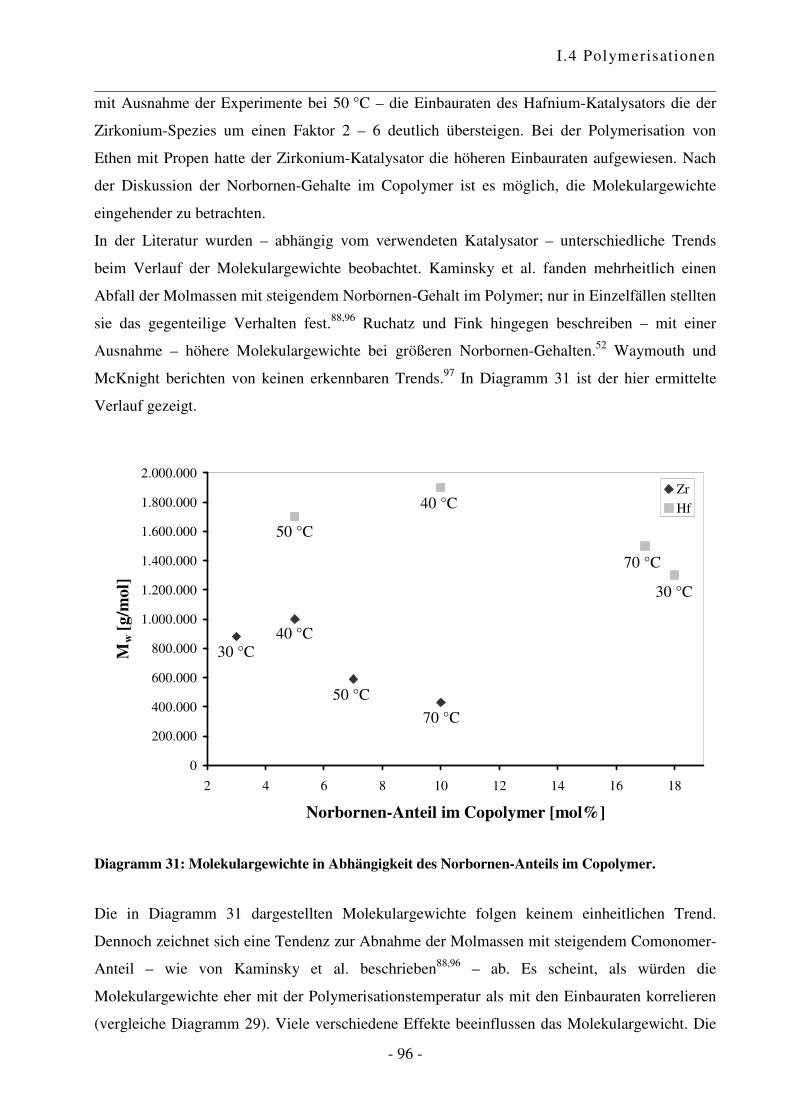
I.4 Polymerisationen
- 96 -
mit Ausnahme der Experimente bei 50 °C – die Einbauraten des Hafnium-Katalysators die der
Zirkonium-Spezies um einen Faktor 2 – 6 deutlich übersteigen. Bei der Polymerisation von
Ethen mit Propen hatte der Zirkonium-Katalysator die höheren Einbauraten aufgewiesen. Nach
der Diskussion der Norbornen-Gehalte im Copolymer ist es möglich, die Molekulargewichte
eingehender zu betrachten.
In der Literatur wurden – abhängig vom verwendeten Katalysator – unterschiedliche Trends
beim Verlauf der Molekulargewichte beobachtet. Kaminsky et al. fanden mehrheitlich einen
Abfall der Molmassen mit steigendem Norbornen-Gehalt im Polymer; nur in Einzelfällen stellten
sie das gegenteilige Verhalten fest.88,96 Ruchatz und Fink hingegen beschreiben – mit einer
Ausnahme – höhere Molekulargewichte bei größeren Norbornen-Gehalten.52 Waymouth und
McKnight berichten von keinen erkennbaren Trends.97 In Diagramm 31 ist der hier ermittelte
Verlauf gezeigt.
0
200.000
400.000
600.000
800.000
1.000.000
1.200.000
1.400.000
1.600.000
1.800.000
2.000.000
2 4 6 8 10 12 14 16 18
Norbornen-Anteil im Copolymer [mol%]
Mw [
g/m
ol]
ZrHf
Diagramm 31: Molekulargewichte in Abhängigkeit des Norbornen-Anteils im Copolymer.
Die in Diagramm 31 dargestellten Molekulargewichte folgen keinem einheitlichen Trend.
Dennoch zeichnet sich eine Tendenz zur Abnahme der Molmassen mit steigendem Comonomer-
Anteil – wie von Kaminsky et al. beschrieben88,96 – ab. Es scheint, als würden die
Molekulargewichte eher mit der Polymerisationstemperatur als mit den Einbauraten korrelieren
(vergleiche Diagramm 29). Viele verschiedene Effekte beeinflussen das Molekulargewicht. Die
30 °C
30 °C 40 °C
40 °C
50 °C
50 °C
70 °C
70 °C
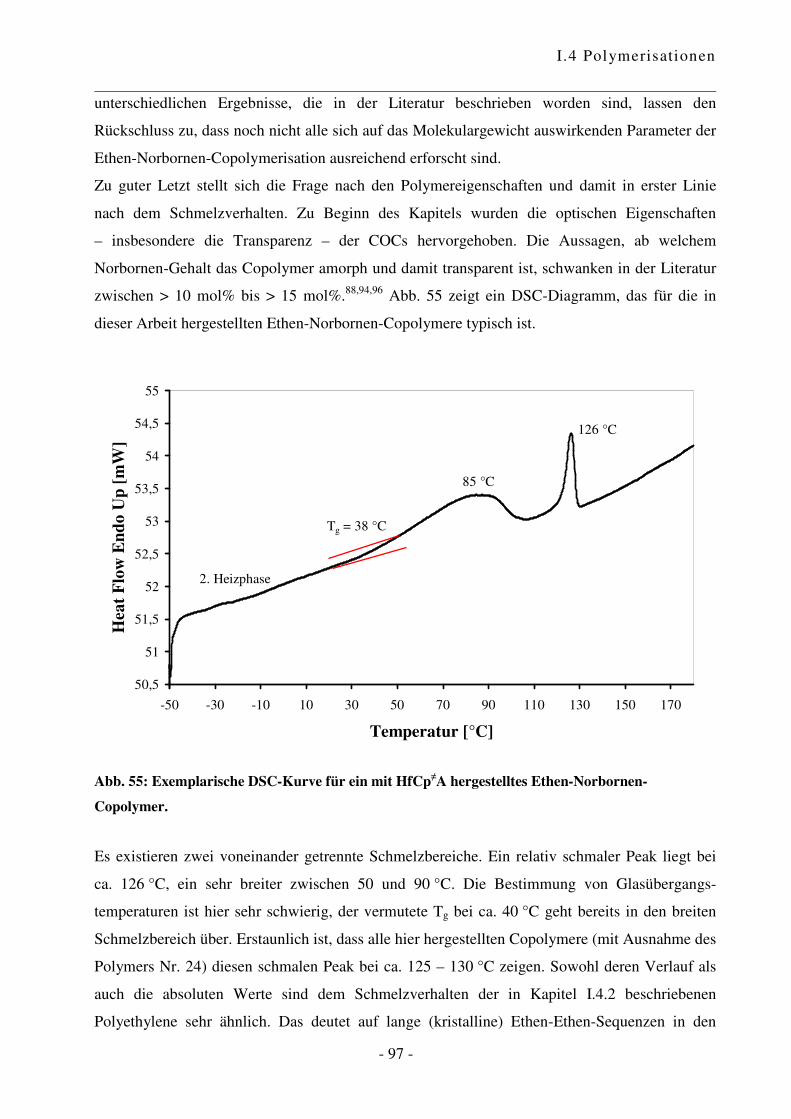
I.4 Polymerisationen
- 97 -
unterschiedlichen Ergebnisse, die in der Literatur beschrieben worden sind, lassen den
Rückschluss zu, dass noch nicht alle sich auf das Molekulargewicht auswirkenden Parameter der
Ethen-Norbornen-Copolymerisation ausreichend erforscht sind.
Zu guter Letzt stellt sich die Frage nach den Polymereigenschaften und damit in erster Linie
nach dem Schmelzverhalten. Zu Beginn des Kapitels wurden die optischen Eigenschaften
– insbesondere die Transparenz – der COCs hervorgehoben. Die Aussagen, ab welchem
Norbornen-Gehalt das Copolymer amorph und damit transparent ist, schwanken in der Literatur
zwischen > 10 mol% bis > 15 mol%.88,94,96 Abb. 55 zeigt ein DSC-Diagramm, das für die in
dieser Arbeit hergestellten Ethen-Norbornen-Copolymere typisch ist.
50,5
51
51,5
52
52,5
53
53,5
54
54,5
55
-50 -30 -10 10 30 50 70 90 110 130 150 170
Temperatur [°C]
Hea
t F
low
End
o U
p [m
W]
2. Heizphase
85 °C
126 °C
Tg = 38 °C
Abb. 55: Exemplarische DSC-Kurve für ein mit HfCp�A hergestelltes Ethen-Norbornen-
Copolymer.
Es existieren zwei voneinander getrennte Schmelzbereiche. Ein relativ schmaler Peak liegt bei
ca. 126 °C, ein sehr breiter zwischen 50 und 90 °C. Die Bestimmung von Glasübergangs-
temperaturen ist hier sehr schwierig, der vermutete Tg bei ca. 40 °C geht bereits in den breiten
Schmelzbereich über. Erstaunlich ist, dass alle hier hergestellten Copolymere (mit Ausnahme des
Polymers Nr. 24) diesen schmalen Peak bei ca. 125 – 130 °C zeigen. Sowohl deren Verlauf als
auch die absoluten Werte sind dem Schmelzverhalten der in Kapitel I.4.2 beschriebenen
Polyethylene sehr ähnlich. Das deutet auf lange (kristalline) Ethen-Ethen-Sequenzen in den
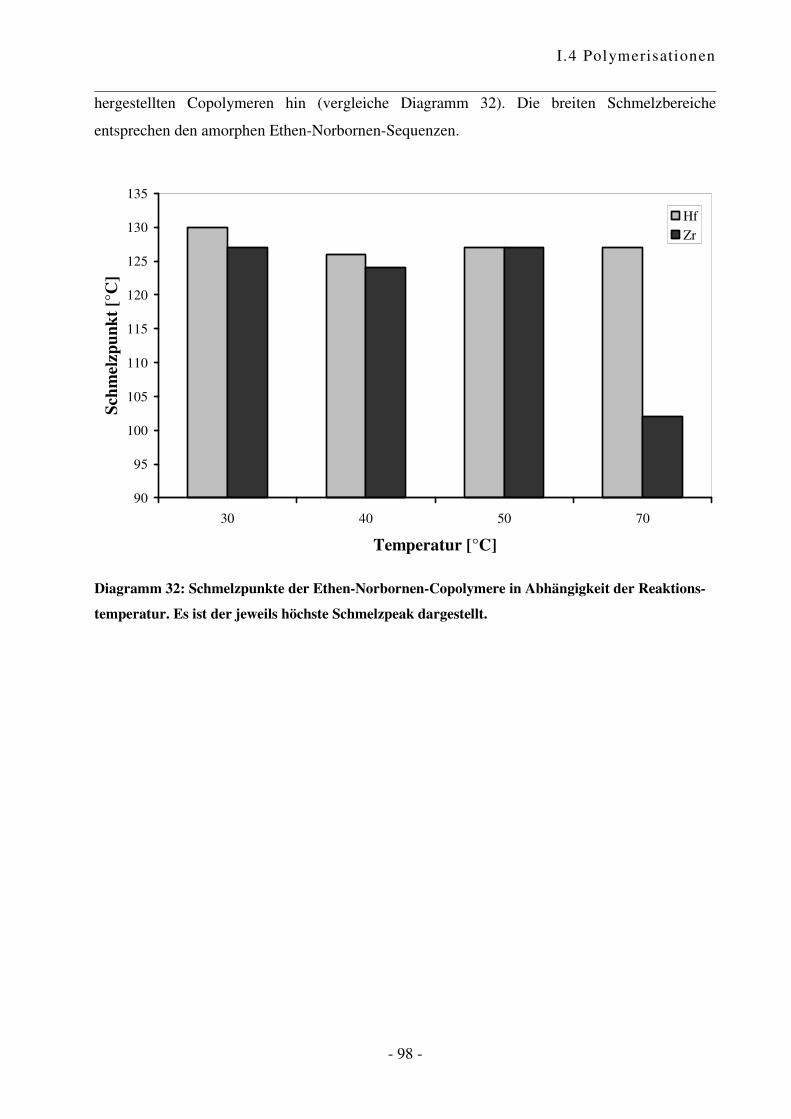
I.4 Polymerisationen
- 98 -
hergestellten Copolymeren hin (vergleiche Diagramm 32). Die breiten Schmelzbereiche
entsprechen den amorphen Ethen-Norbornen-Sequenzen.
90
95
100
105
110
115
120
125
130
135
30 40 50 70
Temperatur [°C]
Schm
elzp
unkt
[°C
]
HfZr
Diagramm 32: Schmelzpunkte der Ethen-Norbornen-Copolymere in Abhängigkeit der Reaktions-
temperatur. Es ist der jeweils höchste Schmelzpeak dargestellt.

I.4 Polymerisationen
- 99 -
Zusammenfassung
Katalysator 1 produziert mit sehr hoher Aktivität statistische Ethen-Norbornen-Copolymere, die
ausschließlich isolierte Norbornen-Einheiten aufweisen. Katalysator 2 polymerisiert ebenfalls
mit hoher Aktivität Ethen mit Norbornen. Die erhaltenen Polymere weisen neben isolierten
Norbornen-Molekülen alternierende Ethen-Norbornen-Sequenzen auf. Die Copolymere beider
Katalysatoren zeigen eine bimodale Molekulargewichtsverteilung mit einem ultrahoch- bis
hochmolekularen Anteil, wobei mit 2 die deutlich höheren Molmassen erreicht werden. Der
Norbornen-Anteil in den Copolymeren steigt bei 1 mit zunehmender Reaktionstemperatur an,
der Trend bei 2 ist uneinheitlich. Die Einbauraten liegen zwischen 3 und 18 mol%. Katalysator 2
erreicht dabei (mit einer Ausnahme) höhere Einbauraten als 1.
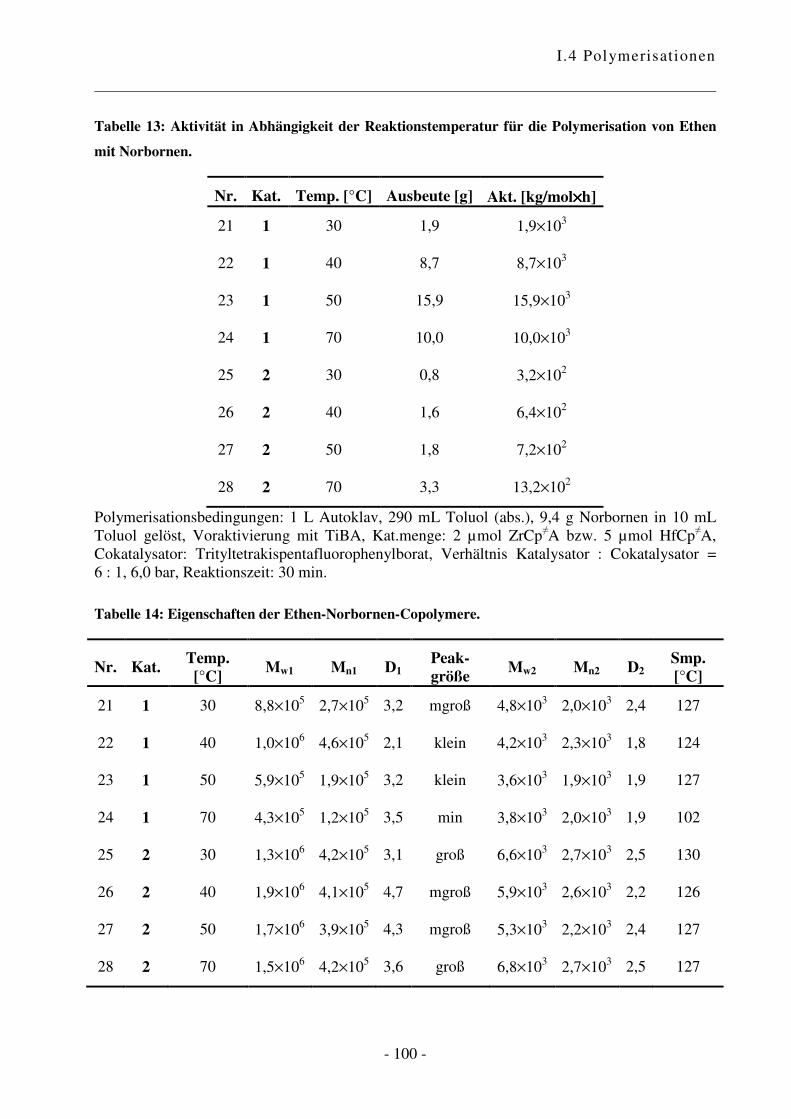
I.4 Polymerisationen
- 100 -
Tabelle 13: Aktivität in Abhängigkeit der Reaktionstemperatur für die Polymerisation von Ethen
mit Norbornen.
Nr. Kat. Temp. [°C] Ausbeute [g] Akt. [kg/mol××××h]
21 1 30 1,9 1,9×103
22 1 40 8,7 8,7×103
23 1 50 15,9 15,9×103
24 1 70 10,0 10,0×103
25 2 30 0,8 3,2×102
26 2 40 1,6 6,4×102
27 2 50 1,8 7,2×102
28 2 70 3,3 13,2×102
Polymerisationsbedingungen: 1 L Autoklav, 290 mL Toluol (abs.), 9,4 g Norbornen in 10 mL Toluol gelöst, Voraktivierung mit TiBA, Kat.menge: 2 µmol ZrCp�A bzw. 5 µmol HfCp�A, Cokatalysator: Trityltetrakispentafluorophenylborat, Verhältnis Katalysator : Cokatalysator = 6 : 1, 6,0 bar, Reaktionszeit: 30 min.
Tabelle 14: Eigenschaften der Ethen-Norbornen-Copolymere.
Nr. Kat. Temp. [°C]
Mw1 Mn1 D1 Peak-größe
Mw2 Mn2 D2 Smp. [°C]
21 1 30 8,8×105 2,7×105 3,2 mgroß 4,8×103 2,0×103 2,4 127
22 1 40 1,0×106 4,6×105 2,1 klein 4,2×103 2,3×103 1,8 124
23 1 50 5,9×105 1,9×105 3,2 klein 3,6×103 1,9×103 1,9 127
24 1 70 4,3×105 1,2×105 3,5 min 3,8×103 2,0×103 1,9 102
25 2 30 1,3×106 4,2×105 3,1 groß 6,6×103 2,7×103 2,5 130
26 2 40 1,9×106 4,1×105 4,7 mgroß 5,9×103 2,6×103 2,2 126
27 2 50 1,7×106 3,9×105 4,3 mgroß 5,3×103 2,2×103 2,4 127
28 2 70 1,5×106 4,2×105 3,6 groß 6,8×103 2,7×103 2,5 127
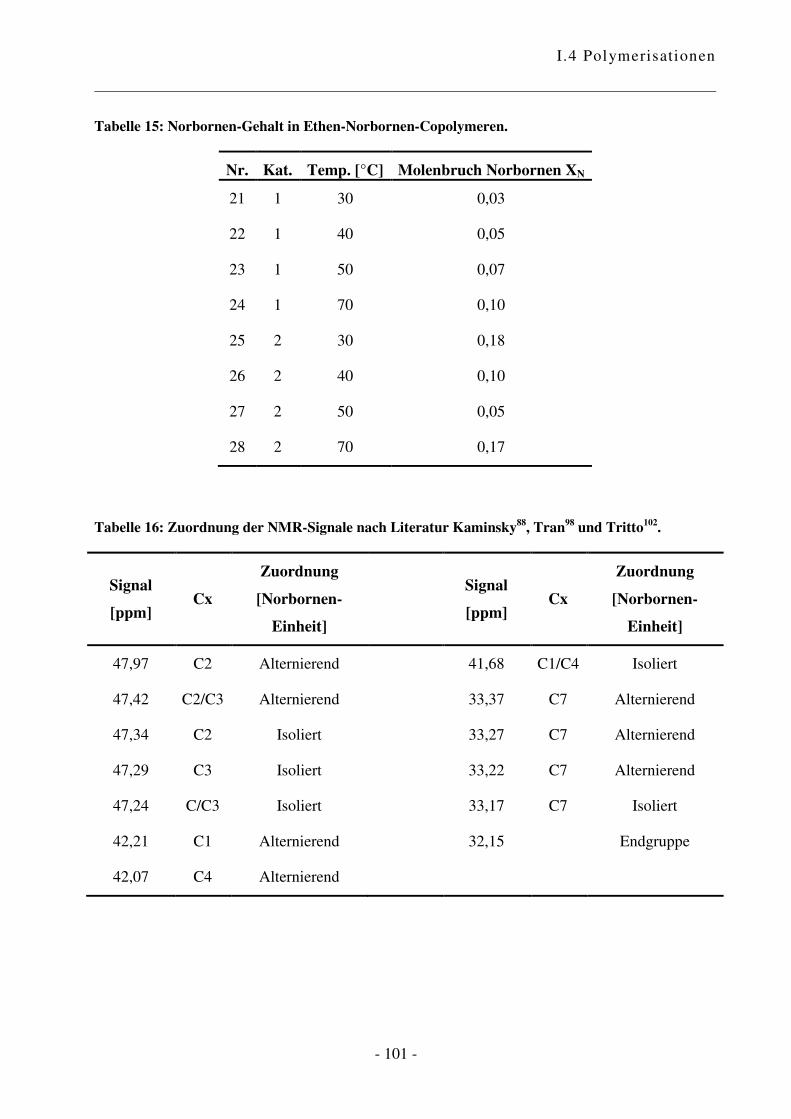
I.4 Polymerisationen
- 101 -
Tabelle 15: Norbornen-Gehalt in Ethen-Norbornen-Copolymeren.
Nr. Kat. Temp. [°C] Molenbruch Norbornen XN
21 1 30 0,03
22 1 40 0,05
23 1 50 0,07
24 1 70 0,10
25 2 30 0,18
26 2 40 0,10
27 2 50 0,05
28 2 70 0,17
Tabelle 16: Zuordnung der NMR-Signale nach Literatur Kaminsky88, Tran98 und Tritto102.
Signal
[ppm] Cx
Zuordnung
[Norbornen-
Einheit]
Abstand
Signal
[ppm] Cx
Zuordnung
[Norbornen-
Einheit]
47,97 C2 Alternierend 41,68 C1/C4 Isoliert
47,42 C2/C3 Alternierend 33,37 C7 Alternierend
47,34 C2 Isoliert 33,27 C7 Alternierend
47,29 C3 Isoliert 33,22 C7 Alternierend
47,24 C/C3 Isoliert 33,17 C7 Isoliert
42,21 C1 Alternierend 32,15 Endgruppe
42,07 C4 Alternierend

I.4 Polymerisationen
- 102 -
I.4.5 Polymerisation von Ethen mit 1,3-Cyclohexadien
In den vorherigen Kapiteln wurden sterische Effekte bei der Polymerisation durch die
Constrained Geometry Katalysatoren untersucht, indem Propen bzw. Norbornen als Comonomer
eingesetzt worden sind. Der folgende Abschnitt gilt der Copolymerisation von Ethen mit
1,3-Cyclohexadien, das als Monomer sowohl sterisch als auch elektronisch anspruchsvoll ist.
Abb. 56: Polymerisation von Ethen mit 1,3-Cyclohexadien. Das 1,3-CHD-Molekül kann sowohl 1,2-
als auch 1,4-insertiert werden.
Besonders das konjugierte Doppelbindungssystem stellt eine Herausforderung für Katalysatoren
dar. Wird ein Molekül unter Spaltung einer konjugierten Doppelbindung in die Polymerkette
eingebaut, so wird dabei weniger Energie frei als bei vergleichbaren Mono-Enen (siehe auch
Kapitel I.1.4.2). Die dafür verantwortliche Delokalisationsenergie lässt sich durch den Vergleich
der Hydrierungswärmen von Cyclohexen und 1,3-Cyclohexadien abschätzen (siehe Abb. 57).
Abb. 57: Hydrierungswärmen von Cyclohexen und Cyclohexadien103
Bei der Hydrierung der nichtkonjugierten Doppelbindung des Cyclohexens werden 119 kJ/mol
freigesetzt, während es für eine konjugierte Bindung des 1,3-Cyclohexadiens nur 111 kJ/mol
sind. Die Hydrierwärme von Ethen zu Ethan beträgt demgegenüber BH° = - 136,9 kJ/mol. Dieser

I.4 Polymerisationen
- 103 -
Vergleich macht deutlich, dass es sich bei 1,3-Cyclohexadien um ein elektronisch wie auch
sterisch anspruchsvolles Monomer handelt.
Schwierigkeiten bei der Polymerisation von konjugierten cyclischen Diolefinen sind bekannt.104
Viele verschiedene Systeme wurden auf ihre Eignung für die 1,3-CHD-Homopolymerisation
getestet. In der Literatur sind radikalische105, kationische106,107 und anionische
Polymerisationen106,108 sowie Ziegler-Katalysen106,107,108,109 beschrieben worden. Diese
Synthesen brachten allerdings nur niedermolekulare Poly(1,3-cyclohexadien)e hervor, ehe Natori
von der kontrollierten lebenden anionischen Polymerisation von 1,3-CHD mit n-Butyllithium
und Tetramethylethylendiamin berichtete.110,111 Auf Basis dieses neuen Systems gelang es,
weitere 1,3-CHD-Homopolyere112,113, Star-Shaped Polymere113,114, Star-Block Polymere114 und
Block Co-, Ter- und Quaterpolymere mit Stryrol, Isopren und Butadien herzustellen.115,116,117
Polycyclohexadien ist aufgrund seiner thermischen, chemischen und mechanischen
Eigenschaften ein interessantes Polydien.116 Die meisten PCHDs sind jedoch unlöslich und
weisen Schmelztemperaturen oberhalb von 300 °C auf. Da der Schmelzpunkt oberhalb des
Zersetzungspunkts liegt, können diese Polymere nicht in Schmelzen (z. B. Extrusion, Spritzguss)
verarbeitet werden.118 Von Anfang an galt daher das Interesse der post-polymeren Reaktionen.
PCHD ist ein einzigartiger Precursor für nachfolgende chemische Modifizierungen.113
Abb. 58: Post-polymere Weiterverarbeitung von PCHD.
Bereits in der frühen Literatur wurde über die Aromatisierung von PCHD zu Polyparaphenylen
berichtet.106,107,108,109 Aufgrund des konjugierten �-Elektronen-Systems kann PPP vom Isolator
zum elektrischen Leiter werden, wenn es mit einem geeigneten Dotierungsreagenz versetzt
wird.119 Darüber hinaus sind weitere interessante Eigenschaften wie Elektrolumineszenz
beschrieben worden.120 Durch Hydrierung von Polycyclohexadien entsteht Polycyclohexylen.
PCHE weist eine Glasübergangtemperatur von 231 °C – der höchste je für Kohlenwasserstoff-
Polymere festgestellte Wert –, eine geringe spezifische Dichte, hohe Temperaturbeständigkeit
und einen hohen Elastizitätsmodul auf.117,121

I.4 Polymerisationen
- 104 -
Eine Idee zur Verbesserung der Verarbeitbarkeit der Polycyclohexadiene in Lösung und in
Schmelze liegt in Copolymerisationsreaktionen. Die meisten konventionellen Katalysator-
systeme werden jedoch durch die Zugabe konjugierter 1,3-Dienen wie z. B. Butadien zur
Polymerisationsmatrix vergiftet.118 Die ersten katalytischen (Homo-)Polymerisationen von
1,3-CHD wurden mit Nickel-Komplexen beschrieben.118,122 Naga setzt zum ersten Mal den
Constrained Geometry Katalysator TiCp�A für Copolymerisationen von Ethen mit
verschiedenen Cycloolefinen und Cyclodiolefinen – jedoch nicht 1,3-CDH – ein.123 Eine
ausführliche Untersuchung der 1,3-CHD-Homo- und -Copolymerisation mit Ethen findet sich
bei Mühlhaupt et. al. Als Katalysatoren verwendeten sie Nickeldiiminkomplexe sowie
verschiedenartig substituierte Titan-Halbsandwichverbindungen wie TiCp�A mit MAO als
Aktivator.118 Wie schon bei den Homopolymeren bieten die so hergestellten Copolymere
interessante Möglichkeiten zur weiteren Umsetzung (siehe Abb. 59).
Abb. 59: Möglichkeiten zur Modifizierung von Ethen-1,3-Cyclohexadien-Copolymeren.
Im folgenden Abschnitt wird nun zum ersten Mal das Verhalten von Zirkonium- und Hafnium-
CGCs bei der Copolymerisation von Ethen mit 1,3-CHD untersucht.
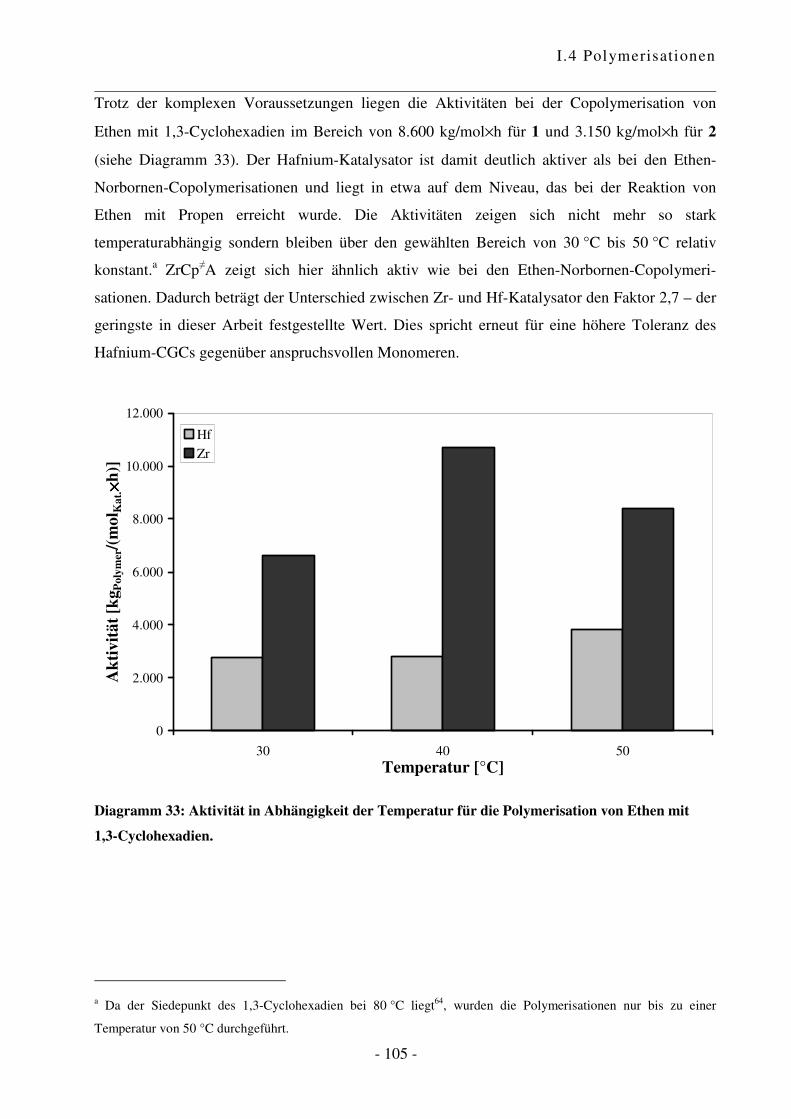
I.4 Polymerisationen
- 105 -
Trotz der komplexen Voraussetzungen liegen die Aktivitäten bei der Copolymerisation von
Ethen mit 1,3-Cyclohexadien im Bereich von 8.600 kg/mol×h für 1 und 3.150 kg/mol×h für 2
(siehe Diagramm 33). Der Hafnium-Katalysator ist damit deutlich aktiver als bei den Ethen-
Norbornen-Copolymerisationen und liegt in etwa auf dem Niveau, das bei der Reaktion von
Ethen mit Propen erreicht wurde. Die Aktivitäten zeigen sich nicht mehr so stark
temperaturabhängig sondern bleiben über den gewählten Bereich von 30 °C bis 50 °C relativ
konstant.a ZrCp�A zeigt sich hier ähnlich aktiv wie bei den Ethen-Norbornen-Copolymeri-
sationen. Dadurch beträgt der Unterschied zwischen Zr- und Hf-Katalysator den Faktor 2,7 – der
geringste in dieser Arbeit festgestellte Wert. Dies spricht erneut für eine höhere Toleranz des
Hafnium-CGCs gegenüber anspruchsvollen Monomeren.
0
2.000
4.000
6.000
8.000
10.000
12.000
30 40 50Temperatur [°C]
Akt
ivit
ät [
kgP
olym
er/(
mol
Kat
.×× ××h)
]
HfZr
Diagramm 33: Aktivität in Abhängigkeit der Temperatur für die Polymerisation von Ethen mit
1,3-Cyclohexadien.
a Da der Siedepunkt des 1,3-Cyclohexadien bei 80 °C liegt64, wurden die Polymerisationen nur bis zu einer
Temperatur von 50 °C durchgeführt.
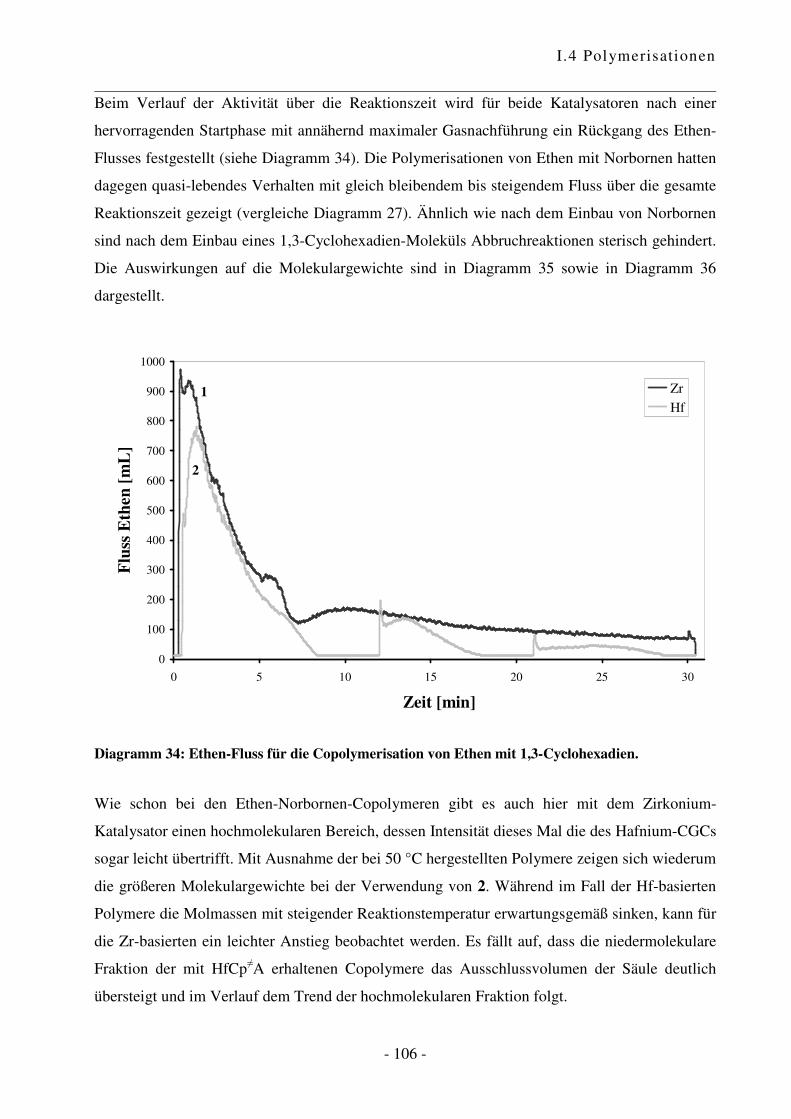
I.4 Polymerisationen
- 106 -
Beim Verlauf der Aktivität über die Reaktionszeit wird für beide Katalysatoren nach einer
hervorragenden Startphase mit annähernd maximaler Gasnachführung ein Rückgang des Ethen-
Flusses festgestellt (siehe Diagramm 34). Die Polymerisationen von Ethen mit Norbornen hatten
dagegen quasi-lebendes Verhalten mit gleich bleibendem bis steigendem Fluss über die gesamte
Reaktionszeit gezeigt (vergleiche Diagramm 27). Ähnlich wie nach dem Einbau von Norbornen
sind nach dem Einbau eines 1,3-Cyclohexadien-Moleküls Abbruchreaktionen sterisch gehindert.
Die Auswirkungen auf die Molekulargewichte sind in Diagramm 35 sowie in Diagramm 36
dargestellt.
0
100
200
300
400
500
600
700
800
900
1000
0 5 10 15 20 25 30
Zeit [min]
Flu
ss E
then
[m
L]
ZrHf
1
2
Diagramm 34: Ethen-Fluss für die Copolymerisation von Ethen mit 1,3-Cyclohexadien.
Wie schon bei den Ethen-Norbornen-Copolymeren gibt es auch hier mit dem Zirkonium-
Katalysator einen hochmolekularen Bereich, dessen Intensität dieses Mal die des Hafnium-CGCs
sogar leicht übertrifft. Mit Ausnahme der bei 50 °C hergestellten Polymere zeigen sich wiederum
die größeren Molekulargewichte bei der Verwendung von 2. Während im Fall der Hf-basierten
Polymere die Molmassen mit steigender Reaktionstemperatur erwartungsgemäß sinken, kann für
die Zr-basierten ein leichter Anstieg beobachtet werden. Es fällt auf, dass die niedermolekulare
Fraktion der mit HfCp�A erhaltenen Copolymere das Ausschlussvolumen der Säule deutlich
übersteigt und im Verlauf dem Trend der hochmolekularen Fraktion folgt.
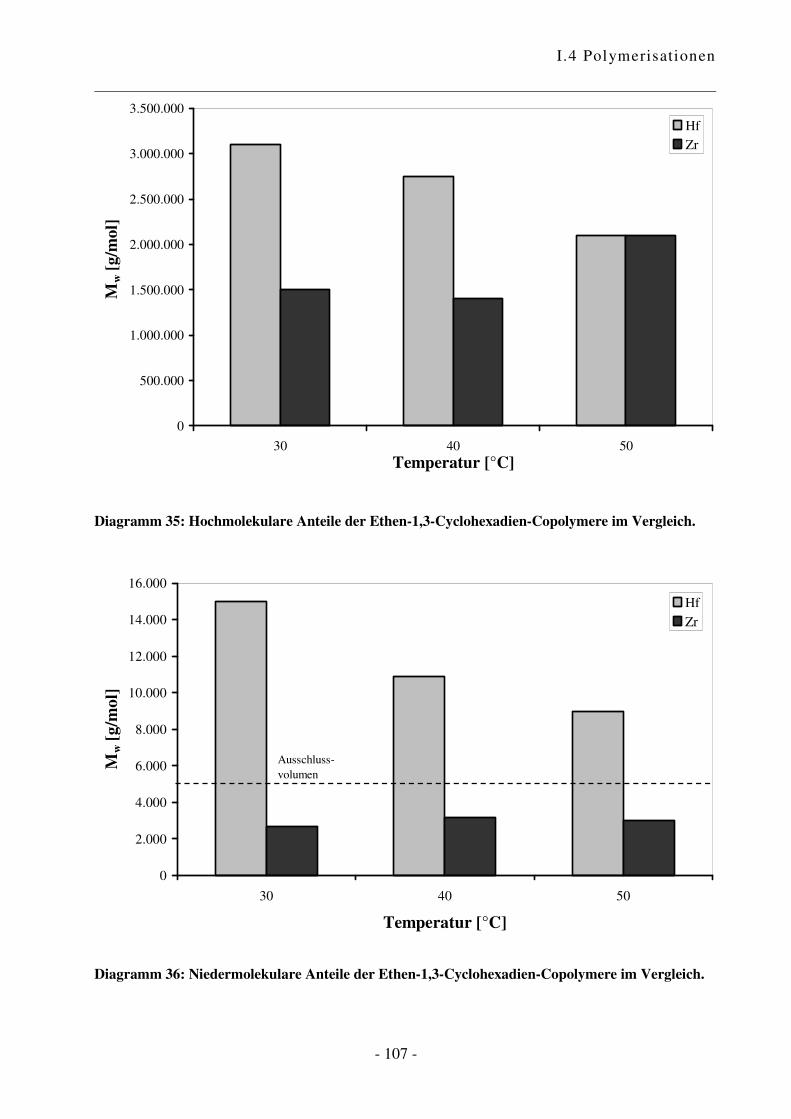
I.4 Polymerisationen
- 107 -
0
500.000
1.000.000
1.500.000
2.000.000
2.500.000
3.000.000
3.500.000
30 40 50Temperatur [°C]
Mw [
g/m
ol]
HfZr
Diagramm 35: Hochmolekulare Anteile der Ethen-1,3-Cyclohexadien-Copolymere im Vergleich.
0
2.000
4.000
6.000
8.000
10.000
12.000
14.000
16.000
30 40 50
Temperatur [°C]
Mw [
g/m
ol]
HfZr
Ausschluss-volumen
Diagramm 36: Niedermolekulare Anteile der Ethen-1,3-Cyclohexadien-Copolymere im Vergleich.

I.4 Polymerisationen
- 108 -
Analog zu den vorherigen Kapiteln stellt sich an diesem Punkt der Diskussion die Frage nach
den Einbauraten. 1,3-Cyclohexadien kann über 1,2- oder 1,4-Insertion in die wachsende
Polymerkette eingebaut werden. Für die beiden Einbauwege ergeben sich unterschiedliche
Verhältnisse an olefinischen zu allylischen Protonen (siehe Abb. 60). Durch Vergleich der
Integrale im 1H-NMR-Spektrum kann das Verhältnis an 1,2- zu 1,4-insertierten Cyclohexadien-
Einheiten berechnet werden.111,118 Der CHD-Gehalt in Ethen-1,3-Cyclohexadien-Copolymeren
ergibt sich – unabhängig von der Einbauart – aus dem Verhältnis der olefinischen Protonen zu
den CH2-Protonen der Polymer-Hauptkette.118
Abb. 60: 1,2- und 1,4-Insertion von 1,3-Cyclohexadien. Bei der 1,2-Insertion beträgt das Verhältnis
HA zu HB 2:3, bei der 1,4-Insertion 2:2.
Bei allen in dieser Arbeit hergestellten Polymeren können im 1H-NMR-Spektrum Endgruppen
erkannt werden (vergleiche Abb. 43 im Kapitel der Ethen-Propen-Copolymerisation). Dies
erschwert die Bestimmung des Comonomergehalts deutlich, da alle für die Berechnung
geeigneten Signale von Signalen der Endgruppen überlagert werden. Zur Verdeutlichung wird
auf der folgenden Seite ein NMR-Spektrum eines bei 50 °C mit 2 hergestellten Polyethylens mit
einem Spektrum eines ebenfalls bei 50 °C mit 2 hergestellten Ethen-1,3-Cyclohexadien-
Copolymer verglichen (Abb. 61 und Abb. 62). Die Zuordnung der Signale ist in Tabelle 19
angegeben.
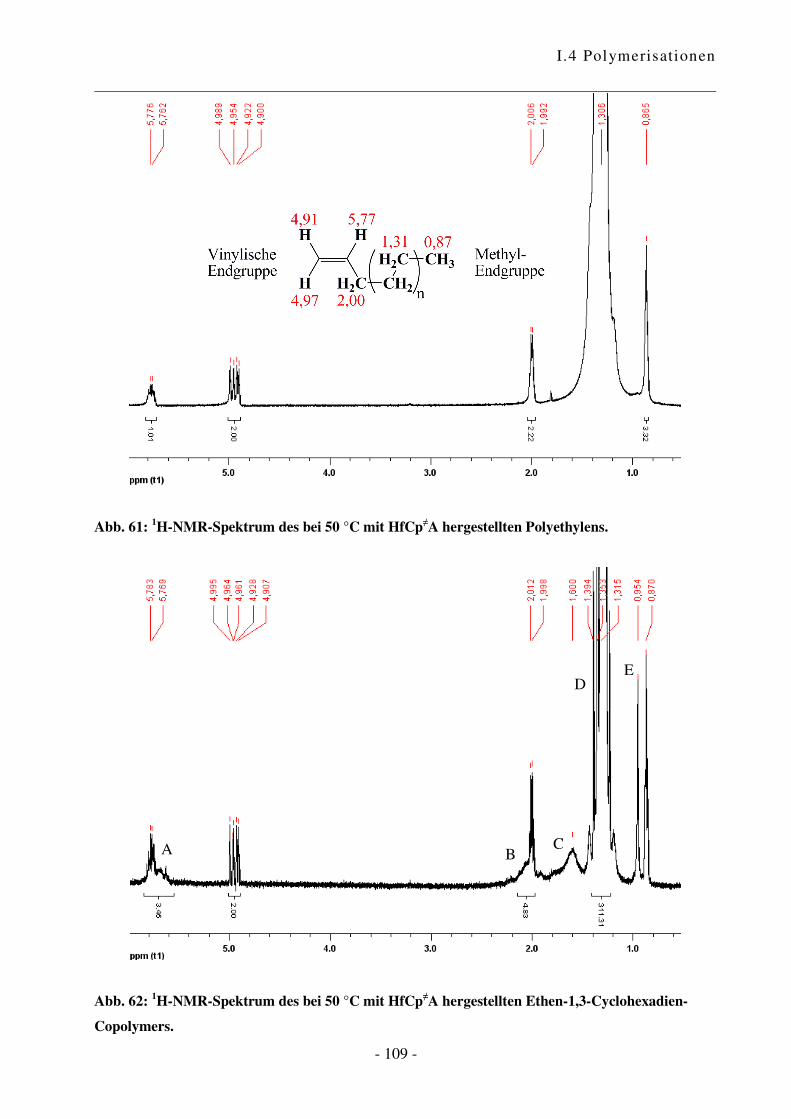
I.4 Polymerisationen
- 109 -
Abb. 61: 1H-NMR-Spektrum des bei 50 °C mit HfCp�A hergestellten Polyethylens.
Abb. 62: 1H-NMR-Spektrum des bei 50 °C mit HfCp�A hergestellten Ethen-1,3-Cyclohexadien-
Copolymers.
B C
E D
A
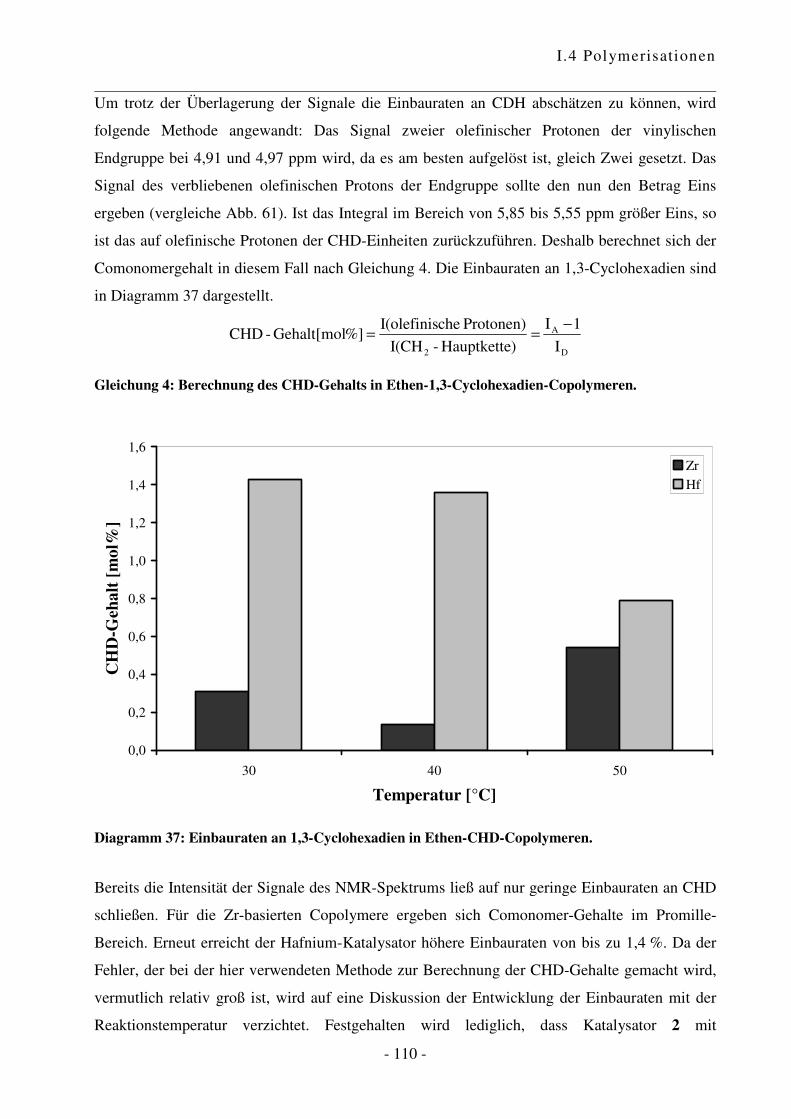
I.4 Polymerisationen
- 110 -
Um trotz der Überlagerung der Signale die Einbauraten an CDH abschätzen zu können, wird
folgende Methode angewandt: Das Signal zweier olefinischer Protonen der vinylischen
Endgruppe bei 4,91 und 4,97 ppm wird, da es am besten aufgelöst ist, gleich Zwei gesetzt. Das
Signal des verbliebenen olefinischen Protons der Endgruppe sollte den nun den Betrag Eins
ergeben (vergleiche Abb. 61). Ist das Integral im Bereich von 5,85 bis 5,55 ppm größer Eins, so
ist das auf olefinische Protonen der CHD-Einheiten zurückzuführen. Deshalb berechnet sich der
Comonomergehalt in diesem Fall nach Gleichung 4. Die Einbauraten an 1,3-Cyclohexadien sind
in Diagramm 37 dargestellt.
D
A
2 I
1I
)Hauptkette-I(CHProtonen) cheI(olefinis
%]Gehalt[mol-CHD−
==
Gleichung 4: Berechnung des CHD-Gehalts in Ethen-1,3-Cyclohexadien-Copolymeren.
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
30 40 50
Temperatur [°C]
CH
D-G
ehal
t [m
ol%
]
ZrHf
Diagramm 37: Einbauraten an 1,3-Cyclohexadien in Ethen-CHD-Copolymeren.
Bereits die Intensität der Signale des NMR-Spektrums ließ auf nur geringe Einbauraten an CHD
schließen. Für die Zr-basierten Copolymere ergeben sich Comonomer-Gehalte im Promille-
Bereich. Erneut erreicht der Hafnium-Katalysator höhere Einbauraten von bis zu 1,4 %. Da der
Fehler, der bei der hier verwendeten Methode zur Berechnung der CHD-Gehalte gemacht wird,
vermutlich relativ groß ist, wird auf eine Diskussion der Entwicklung der Einbauraten mit der
Reaktionstemperatur verzichtet. Festgehalten wird lediglich, dass Katalysator 2 mit

I.4 Polymerisationen
- 111 -
durchschnittlich 1,2 % Comonomer-Einheiten vier Mal mehr CHD einbaut als Katalysator 1, der
durchschnittliche Einbauraten von 0,3 % erzielt. Die breiten Signale im 1H-NMR-Spektrum
lassen zudem eine Aufschlüsselung zwischen 1,2- und 1,4-Insertion nicht zu. Mühlhaupt et al.
hatten bei ihrer Untersuchung des TiCp�A-Katalysators ausschließlich 1,4-insertierte CHD-
Einheiten festgestellt. Sie zeigten zudem, dass CHD-Gehalte in der Polymermatrix von über
85 mol% notwendig sind, um Einbauraten > 3 mol% im Polymer zu erzielen.118 Unter diesem
Gesichtspunkt sind die mit HfCp�A erzielten Werte durchaus beachtlich.
Die Molekulargewichte der Zr-basierten Copolymere erscheinen unter dem Aspekt der
Einbauraten außergewöhnlich. Während bei der Homopolymerisation von Ethen nur
niedermolekulare Anteile erhalten wurden, reicht ein geringer – mit 0,3 % beinahe als
sporadischer anzusehender – Einbau an 1,3-Cyclohexadien aus, um das Molekulargewicht auf
Werte von durchschnittlich 1.700.000 g/mol zu steigern. Bei der Copolymerisation von Ethen
mit Norbornen war es ein Comonomer-Gehalt von durchschnittlich 6,3 mol%, der Molekular-
gewichte von 730.000 g/mol ermöglichte. Die außergewöhnliche Molmassensteigerung bei den
Zr-basierten Copolymeren durch geringe Einbauraten an 1,3-CHD eröffnen völlig neue
Perspektiven. Während Wasserstoff in der Industrie zur Begrenzung der Molekulargewichte
eingesetzt wird, könnte mit dem hier festgestellten Effekt das Beimischen eines Comonomers zur
Steigerung der Kettenlänge eingesetzt werden.
Mühlhaupt et al. berichteten, dass die Molmassen der Copolymere mit geringeren
Konzentrationen an CHD in der Reaktionsmatrix steigen. Die höchsten zahlengemittelten
Molekulargewichte erreichten Werte von Mn = 240.000 g/mol.118 Diese Werte konnten in den
hier gezeigten Beispielen vor allem mit 2 weit übertroffen werden (vergleiche Tabelle 18).
Der letzte Diskussionspunkt gilt auch in diesem Kapitel den Polymereigenschaften. Ethen-1,3-
Cyclohexadien-Copolymere zählen wie die Ethen-Norbornen-Copolymere zur viel
versprechenden Materialklasse der COCs. Für Polymere mit alicyclischen Strukturen in der
Hauptkette ist eine dramatische Verbesserung der thermischen und chemischen Stabilität sowie
mechanischen Stärke vorausgesagt worden.111 Mit steigenden Einbauraten an CHD sind
abnehmende Schmelzpunkte zu erwarten.118 Da die hier vorgefundenen Comonomer-Gehalte
niedrig sind, zeigen sich nur geringe Auswirkungen auf die Schmelztemperaturen (vergleiche
Diagramm 38). Die Schmelzbereiche der mit 2 hergestellten Copolymere liegen mit
durchschnittlich 126 °C ungefähr 3 °C unterhalb der Kurven, die für Homo-PE erhalten worden
sind. Das kann durchaus als ein Effekt der durch den CHD-Einbau gestörten Kristallinität
angesehen werden. Bei den Zr-basierten Polymeren zeigt sich ein anderes Bild. Dadurch, dass im
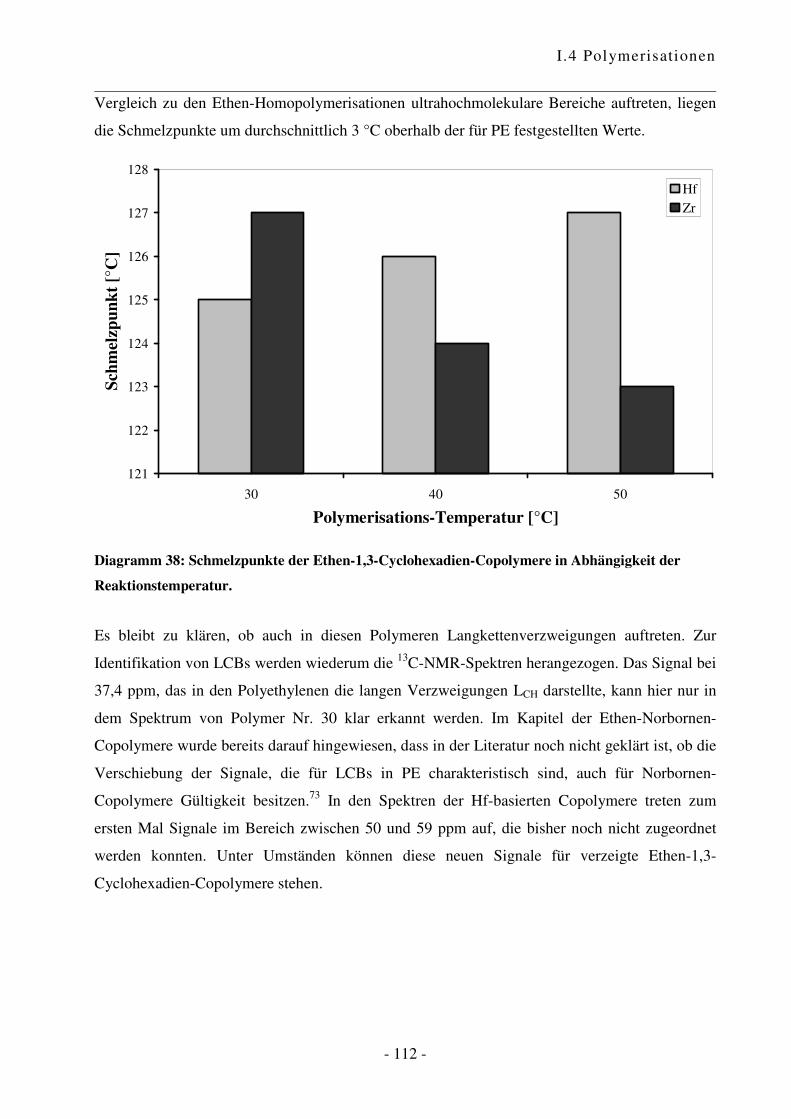
I.4 Polymerisationen
- 112 -
Vergleich zu den Ethen-Homopolymerisationen ultrahochmolekulare Bereiche auftreten, liegen
die Schmelzpunkte um durchschnittlich 3 °C oberhalb der für PE festgestellten Werte.
121
122
123
124
125
126
127
128
30 40 50
Polymerisations-Temperatur [°C]
Schm
elzp
unkt
[°C
]
HfZr
Diagramm 38: Schmelzpunkte der Ethen-1,3-Cyclohexadien-Copolymere in Abhängigkeit der
Reaktionstemperatur.
Es bleibt zu klären, ob auch in diesen Polymeren Langkettenverzweigungen auftreten. Zur
Identifikation von LCBs werden wiederum die 13C-NMR-Spektren herangezogen. Das Signal bei
37,4 ppm, das in den Polyethylenen die langen Verzweigungen LCH darstellte, kann hier nur in
dem Spektrum von Polymer Nr. 30 klar erkannt werden. Im Kapitel der Ethen-Norbornen-
Copolymere wurde bereits darauf hingewiesen, dass in der Literatur noch nicht geklärt ist, ob die
Verschiebung der Signale, die für LCBs in PE charakteristisch sind, auch für Norbornen-
Copolymere Gültigkeit besitzen.73 In den Spektren der Hf-basierten Copolymere treten zum
ersten Mal Signale im Bereich zwischen 50 und 59 ppm auf, die bisher noch nicht zugeordnet
werden konnten. Unter Umständen können diese neuen Signale für verzeigte Ethen-1,3-
Cyclohexadien-Copolymere stehen.

I.4 Polymerisationen
- 113 -
Zusammenfassung
Die Katalysatoren 1 und 2 produzieren mit sehr hoher Aktivität Ethen-1,3-Cyclohexadien-
Copolymere. Alle erhaltenen Polymere zeigen eine bimodale Molekulargewichtsverteilung mit
einem ultrahochmolekularen Anteil, wobei das Molekulargewicht mit steigender
Reaktionstemperatur – bis auf eine Ausnahme – zurückgeht. Die erzielten Molmassen sind bei
den Hafnium-basierten Copolymeren – mit einer Ausnahme – um den Faktor Zwei größer als die
der Zr-basierten. 1,3-Cyclohexadien scheint dabei ein geeigneter Molmassenregulator hin zu
höheren Molekulargewichten für die Polymerisation mit 1 zu sein. Die Einbauraten an 1,3-CHD
liegen für 1 im Promille-Bereich, bei 2 werden bis zu 1,4 mol% Comonomer-Anteil erreicht. Die
Auswirkungen auf die Schmelzkurven der Polymere sind aufgrund der geringen Gehalte an 1,3-
Cyclohexadien gering. Da Vergleichswerte in der Literatur fehlen konnte noch nicht geklärt
werden, ob die vorliegenden Copolymere Langkettenverzweigungen aufweisen.
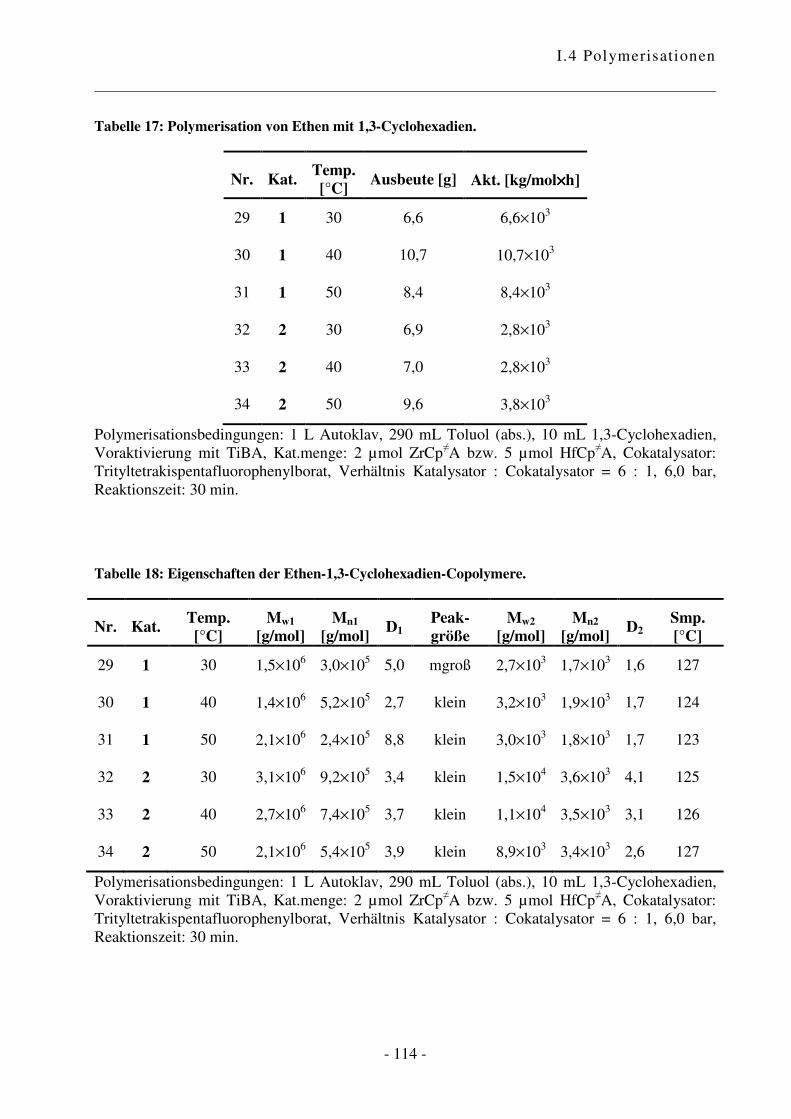
I.4 Polymerisationen
- 114 -
Tabelle 17: Polymerisation von Ethen mit 1,3-Cyclohexadien.
Nr. Kat. Temp. [°C] Ausbeute [g] Akt. [kg/mol××××h]
29 1 30 6,6 6,6×103
30 1 40 10,7 10,7×103
31 1 50 8,4 8,4×103
32 2 30 6,9 2,8×103
33 2 40 7,0 2,8×103
34 2 50 9,6 3,8×103
Polymerisationsbedingungen: 1 L Autoklav, 290 mL Toluol (abs.), 10 mL 1,3-Cyclohexadien, Voraktivierung mit TiBA, Kat.menge: 2 µmol ZrCp�A bzw. 5 µmol HfCp�A, Cokatalysator: Trityltetrakispentafluorophenylborat, Verhältnis Katalysator : Cokatalysator = 6 : 1, 6,0 bar, Reaktionszeit: 30 min.
Tabelle 18: Eigenschaften der Ethen-1,3-Cyclohexadien-Copolymere.
Nr. Kat. Temp. [°C]
Mw1 [g/mol]
Mn1 [g/mol]
D1 Peak-größe
Mw2 [g/mol]
Mn2 [g/mol]
D2 Smp. [°C]
29 1 30 1,5×106 3,0×105 5,0 mgroß 2,7×103 1,7×103 1,6 127
30 1 40 1,4×106 5,2×105 2,7 klein 3,2×103 1,9×103 1,7 124
31 1 50 2,1×106 2,4×105 8,8 klein 3,0×103 1,8×103 1,7 123
32 2 30 3,1×106 9,2×105 3,4 klein 1,5×104 3,6×103 4,1 125
33 2 40 2,7×106 7,4×105 3,7 klein 1,1×104 3,5×103 3,1 126
34 2 50 2,1×106 5,4×105 3,9 klein 8,9×103 3,4×103 2,6 127
Polymerisationsbedingungen: 1 L Autoklav, 290 mL Toluol (abs.), 10 mL 1,3-Cyclohexadien, Voraktivierung mit TiBA, Kat.menge: 2 µmol ZrCp�A bzw. 5 µmol HfCp�A, Cokatalysator: Trityltetrakispentafluorophenylborat, Verhältnis Katalysator : Cokatalysator = 6 : 1, 6,0 bar, Reaktionszeit: 30 min.

I.4 Polymerisationen
- 115 -
Tabelle 19: Zuordnung der Signale im 1H-NMR-Spektrum für Ethen-1,3-CHD-Copolymere.
Signal-
bereich
Verschiebung118
[ppm] Zuordnung Bemerkung
A 5,59 olefinische Protonen,
HA
hier überlagert von olefinischen Protonen
der vinylischen Endgruppe
B 2,02 allylische Protonen,
HB
hier überlagert von allylischen Protonen
der vinylischen Endgruppe
C 1,65 CH2-Ring überlappt partiell bereits mit Bereich D118
D 1,31 CH2-Hauptkette
E 0,91 Methyl-Endgruppe nicht für die Bestimmung des
Comonomer-Anteils geeignet
Tabelle 20: Cyclohexadien-Gehalte in Ethen-1,3-CHD-Copolymeren.
Nr. Kat. Temp. [°C] CHD-Gehalt [mol%]
29 1 30 0,3
30 1 40 0,1
31 1 50 0,5
32 2 30 1,4
33 2 40 1,4
34 2 50 0,8

I.4 Polymerisationen
- 116 -
I.4.6 Fehlerbetrachtung
Schon bei der Herstellung der empfindlichen Katalysatorlösungen kommt es zu den so
genannten zufälligen Fehlern bei der Einwaage und beim Abmessen von Flüssigkeiten. Kleine
Schwankungen in der Konzentration der Katalysatorlösung können aufgrund der geringen
verwendeten Mengen großen Einfluss auf die Polymerisationsergebnisse haben. Im folgenden
Kapitel wird eine Fehlerbetrachtung zur tatsächlich eingesetzten Katalysatormenge durchgeführt.
Hierbei gehen nur die Geräte- und Ablesefehler ein – Fehler aufgrund von Einwirken von z. B.
Luft können nicht abgeschätzt werden, wirken sich allerdings gravierend auf die Ergebnisse aus.a
Die Herstellung einer Katalysatorlösung wird folgendermaßen durchgeführt: Die gewünschte
Katalysatormenge wird in der Glovebox eingewogen (Fehler 1). Die entsprechenden Mengen an
TiBA und Toluol werden berechnet. TiBA wird mit einer Spritze abgemessen und in einen
Schlenkkolben gegeben (Fehler 2). Toluol wird ebenfalls mit einer Spritze abgemessen und
zugefügt (Fehler 3). Jetzt wird das komplette Gemisch zum abgewogenen Katalysator zugegeben
(hier wird von keinem Fehler ausgegangen, vorherige Fehler beim Abmessen bleiben bestehen).
Die fertige Katalysatorlösung wird kurz vor der Polymerisation entnommen (Fehler 4).
Es wird eine exemplarische Berechnung des Fehlers bei der Herstellung von Katalysator-
lösungen durchgeführt. Im vorliegenden Fall wurden folgende Mengen verwendet:
Fehler 1: Einwaage(Komplex), Sartorius Waage: 6,1 mg ± 0,5 mg
Fehler 2: Menge(TiBA), 2 mL Spritze: 1,1 mL ± 0,1 mL
Fehler 3: Menge(Toluol), 20 mL Spritze: 11,1 mL ± 1 mL
Fehler 4: Entnahme der Katalysator-Lösung, 5 mL Spritze: 5 mL ± 0,2 mL
Für die Katalysatormenge gilt:
[mL]EntnahmeTiBA[mL])](Toluol[mLM[g/mol]
10[mg]EinwaageVcn Kat.lösung
3Komplex
Kat. ⋅+⋅
⋅=⋅=
−
Gleichung 5: Berechnung der Katalysatormenge.
a Das äußerst reaktive Katalysatorsystem zersetzt sich sofort bei Kontakt mit Wasser z. B. aus der Luft. Jedes Öffnen
des Schlenkrohres – auch im Argon-Gegenstrom – stellt ein unvermeidbares Risiko dar. Daher wurde jeden Tag
eine frische Katalysatorlösung verwendet.

I.4 Polymerisationen
- 117 -
Der Fehler setzt sich demnach zusammen aus:
Entnahmen
TiBAn
Toluoln
Einwaagen
Fehler∂
∂+
∂
∂+
∂
∂+
∂
∂=
Gleichung 6: Fehler bei der Berechnung der Katalysatormenge.
Für das hier vorgestellte Verfahren ergibt sich daher:
TiBA)(TolMWaage
BEntn.TiBA)(TolMEntn.Waage
BTiBA
TiBA)(TolM
Entn.WaageBTol
TiBA)(TolMEntn.
BWaage
Entnahmen
TiBAn
Toluoln
Einwaagen
Fehler
2
2
+⋅⋅+
+⋅
⋅⋅+
+⋅
⋅⋅+
+⋅⋅=
∂
∂+
∂
∂+
∂
∂+
∂
∂=
1,1mL)(11,1mL498,87gmol6,1mg0,2mL
1,1mL)(11,1mL498,87g
mol5mL6,1mg0,1mL
1,1mL)(11,1mL498,87gmol5mL6,1mg1mL
1,1mL)(11,1mL498,87gmol5mL0,5mg
2
2
+⋅
⋅⋅+
+⋅
⋅⋅⋅+
+⋅
⋅⋅⋅+
+⋅
⋅⋅=
= 0,082 Cmol + 0,41 Cmol +0,041 Cmol +0,2 Cmol
= ± 0,734 Cmol
Das entspricht bei Entnahme von 5 Cmol Katalysator einem Fehler von 15 %. Für eine
errechnete Katalysatoraktivität von 3000 kg/(mol×h) würde das bedeuten, dass der wahre Wert –
nur unter Berücksichtigung des Fehlers in der Katalysatormenge - zwischen 2616 und 3516
kg/(mol×h) liegt.a
Zur Überprüfung der Reproduzierbarkeit wurde die Polymerisation von Ethen mit HfCp�A bei
50 °C als Standard-Experiment gewählt und insgesamt acht Mal durchgeführt. Die Ergebnisse
sind in Tabelle 21 zusammengefasst.
a Zusätzlich entsteht ein Fehler durch ungenaues Abmessen der Zeit, durch eventuelle unvollständige Entnahme des
Produkts aus dem Autoklaven und durch die ungenaue Auswaage des entstandenen Polymers.

I.4 Polymerisationen
- 118 -
Tabelle 21: Polymerisation von Ethen mit HfCp�A bei 50 °C
Nr. Kat. Ausbeute
[g] Akt. [kg/mol××××h] Mw1
[g/mol] Mn1
[g/mol] D1 Mw2
[g/mol] Smp. [°C]
7 2 5,5 5,5×103 4,70×106 8,10×105 5,9 8,0×103 128
35 2 9,3 9,3×103 5,50×106 6,75×105 8,2 7,7×103 129
36 2 5,1 5,1×103 6,38×106 7,50×105 8,5 10,25×103 128
37 2 3,1 3,1×103 10,01×106 12,75×105 7,9 9,6×103 129
38 2 4,8 4,8×103 8,07×106 10,50×105 7,7 9,5×103 130
39 2 4,5 4,5×103 7,52×106 18,60×105 4,0 7,9×103 131
40 2 3,8 3,8×103 4,15×106 7,50×105 5,6 7,3×103 128
41 2 5,4 5,4×103 4,65×106 6,60×105 7,0 8,6×103 129
Die Mittelwerte berechnen sich nach:
�=
=n
1iix
n1
x
Gleichung 7: Berechnung des Mittelwertes.124
Die Streuung der Werte ist ein Maß für die Genauigkeit der Mittelwertangabe. Die Genauigkeit
wird am korrektesten durch die Standardabweichung � angegeben. Sie bestimmt die
durchschnittliche zufällige Abweichung vom Mittelwert und wird häufig als „mittlerer
(quadratischer) Fehler der Einzelmessung“ bezeichnet:
1n
)x(x�
n
1i
2i
−
−
=�
=
Gleichung 8: Berechnung der Standardabweichung.124
xi: Einzelwerte, x : Mittelwert, n: Anzahl der Wiederholungsdurchführungen, n-1: Freiheitsgrad. Im Zähler des Bruches steht die Summe der Abweichungsquadrate der Einzelwerte zum Mittelwert.

I.4 Polymerisationen
- 119 -
Daraus ergeben sich für die Messwerte in Tabelle 21 folgende Ergebnisse:
Tabelle 22: Mittelwerte und Standardabweichungen für die Ergebnisse aus Tabelle 21.
Akt.
[kg/mol××××h] Mw1
[g/mol] Mn1
[g/mol]
Mittelwert 5,19×103 6,373×106 9,80×105
Standardabweichung 1,85×103 2,032×106 4,13×105
Standardabweichung/Mittelwert 36 % 32 % 42 %
Im Falle der Aktivität beträgt die Standardabweichung 36 % des Mittelwertes. Da dieser Wert
etwas zu hoch erscheint, wird ein Test auf Ausreißer durchgeführt. Mit Hilfe des Q-Tests wird
überprüft, ob sich Ausreißer unter den Messwerten befinden, die dann von der Rechnung
ausgeschlossen werden. Der Eliminierungsquotienten Q wird mit folgenden Gleichungen
ermittelt:
maxmin
1minmin
xx
xxQ
−
−=
+ maxmin
1-maxmax
xx
xxQ
−
−=
Gleichung 9: Berechung der Eliminierungsquotienten beim Q-Test.
Der Zahlenwert ist mit Tabellenwerten zu vergleichen, die für eine bestimmte
Wahrscheinlichkeit das Entscheidungskriterium des Einzelwertes zur Mittelwertbildung
entscheiden. Sofern der zu errechnende Quotient Q gleich oder größer als der Vergleichswert ist,
wird der Einzelmesswert eliminiert.
Tabelle 23: Literaturwerte des Eliminierungskoeffizienten für n Messungen125
n Eliminierungsquotient
Q 90 %
Eliminierungsquotient
Q 96 %
Eliminierungsquotient
Q 99 %
7 0,51 0,59 0,68
8 0,47 0,54 0,63

I.4 Polymerisationen
- 120 -
Für die Aktivität des Experiments Nr. 35 von 9.300 kg/(mol×h) ergibt sich ein Q-Wert von:
61,0300.9100.3
500.5300.9
xx
xxQ
maxmin
1-maxmax≈
−
−=
−
−=
Gleichung 10: Q-Test für Experiment Nr. 35.
Das Ergebnis bedeutet, dass es sich bei dem vorliegenden Wert mit 96%iger Wahrscheinlichkeit
um einen Ausreißer handelt. Nach Ausschluss dieses Wertes wird mit den verbliebenen 7
Messwerten für den jeweils höchsten und niedrigsten erneut ein Q-Test durchgeführt. In beiden
Fällen handelt es sich um keinen Ausreißer (siehe Gleichung 11 und Gleichung 12).
Q-Test für den Wert aus Experiment Nr. 7 von 5.500 kg/(mol×h):
04,0500.5100.3
400.5500.5
xx
xxQ
maxmin
1-maxmax≈
−
−=
−
−=
Gleichung 11: Q-Test für Experiment Nr. 7.
Q-Test für den Wert aus Experiment Nr. 37 von 3.100 kg/(mol×h):
29,0500.5100.3
800.3100.3
xx
xxQ
maxmin
1minmin≈
−
−=
−
−=
+
Gleichung 12: Q-Test für Experiment Nr. 37.
Mit den bestätigten 7 Werten werden Mittelwerte und Standardabweichungen erneut berechnet.
Tabelle 24: Ausreißer-bereinigte Mittelwerte und Standardabweichungen für die Ergebnisse aus
Tabelle 21.
bereinigte Aktivität
[kg/mol××××h] Aktivität über alle Werte
[kg/mol××××h]
Mittelwert 4.600 5.190
Standardabweichung 880 1.850
Standardabweichung/Mittelwert 19 % 36 %
Nach Ausschluss des Ausreißers beträgt die Standardabweichung für die Aktivität nur noch 19 %
des Mittelwerts. Dieser Wert korreliert sehr gut mit dem vorhergesagten Fehler von 15 % und
bestätigt die ordnungsgemäße Durchführung der Experimente.

I.4 Polymerisationen
- 121 -
Die Standardabweichungen für die Molekulargewichte mit 33 % für Mw und 42 % für Mn sind
höher als erwartet. Es muss berücksichtigt werden, dass mehrere Faktoren dafür verantwortlich
sind. Die Streuung in der Aktivität hat Auswirkungen auf die Molmassen, hinzu kommt ein
Fehler durch die verwendete Analysenmethode. Um die Ungenauigkeit bei der Bestimmung des
Molekulargewichts durch GPC-Messungen besser abschätzen zu können, wurde die Probe Nr. 7
insgesamt acht Mal vorbereitet und vermessen. Die Ergebnisse sind in Tabelle 25 dargestellt. Die
berechneten Mittelwerte und Standardabweichungen werden in Tabelle 26 gezeigt.
Tabelle 25: Ergebnisse der GPC-Messungen für Polymer Nr. 7.
Mw [g/mol] Mn [g/mol] Mp [g/mol] PDI
7a 5,93×106 12,65×105 3,52×106 4,7
7b 4,91×106 8,35×105 3,13×106 5,9
7c 4,81×106 8,00×105 3,45×106 6,0
7d 4,68×106 7,75×105 2,43×106 6,0
7e 4,33×106 8,20×105 2,58×106 5,3
7f 3,90×106 5,40×105 1,54×106 7,2
7g 4,66×106 7,30×105 2,23×106 6,4
7h 4,51×106 7,35×105 2,73×106 6,1
Tabelle 26: Mittelwerte und Standardabweichungen für die GPC-Messungen aus Tabelle 25.
Mw [g/mol] Mn [g/mol] Mp [g/mol] PDI
Mittelwert 4,670×106 8,08×105 2,68×106 5,9
Standardabweichung 5,66×105 1,92×105 6,21×105 0,7
Standardabweichung/Mittelwert 12 % 24 % 23 % 12 %

I.4 Polymerisationen
- 122 -
Auch hier wird mit dem größten und kleinsten Wert ein Q-Test auf Ausreißer durchgeführt.
Q-Test für den Wert aus Experiment Nr. 7a, Tabelle 25, von 5,93×106 g/mol:
50,01093,51090,3
1091,41093,5
xx
xxQ
66
66
maxmin
1-maxmax≈
⋅−⋅
⋅−⋅=
−
−=
Gleichung 13: Q-Test für Messwert 7a.
Es handelt sich mit 90%iger Wahrscheinlichkeit um einen Ausreißer. Der Wert wird als
Ausreißer ausgeschlossen und mit den verbliebenen 7 Werten auf weitere Ausreißer getestet. In
beiden Fällen handelt es sich um keine Ausreißer (siehe Gleichung 14 und Gleichung 15)
Q-Test für den nun höchsten Wert aus Experiment Nr. 7b von 4,91×106 g/mol:
10,01091,41090,3
1081,41091,4
xx
xxQ
66
66
maxmin
1-maxmax≈
⋅−⋅
⋅−⋅=
−
−=
Gleichung 14: Q-Test für Messwert 7b.
Q-Test für den kleinsten Wert aus Experiment Nr. 7h von 3,90×106 g/mol:
43,01091,41090,3
1033,41090,3
xx
xxQ
66
66
maxmin
1minmin≈
⋅−⋅
⋅−⋅=
−
−=
+
Gleichung 15: Q-Test für Messwert 7h.
Mit den verbliebenen 7 Werten werden die bereinigten Mittelwert und Standardabweichungen
berechnet.
Tabelle 27: Ausreißer-bereinigte Mittelwerte und Standardabweichungen für die GPC-Messungen
aus Tabelle 25.
Mw bereinigt
[g/mol]
Mn bereinigt
[g/mol]
Mp bereinigt
[g/mol]
PDI
bereinigt
Mittelwert 4,51×106 7,51×105 2,580×106 6,1
Standardabweichung 3,31×105 0,93×105 5,74×105 0,6
Standardabweichung/Mittelwert 7 % 12 % 22 % 10 %

I.4 Polymerisationen
- 123 -
Die Fehler für die Molekulargewichte liegen mit ca. 10 % im Bereich des Erwarteten. Der größte
Fehler entsteht vermutlich bei der manuellen Auswertung der Kurven, daher ist es besonders
wichtig, dass diese immer von der gleichen Person vorgenommen wird. Auffallend ist, dass
ausgerechnet Mp, das automatisch von der Bearbeitungssoftware bestimmt wird, die höchste
Ungenauigkeit aufweist. Trotz sorgfältiger Auswertung und Bearbeitung der Proben lassen sich
Ungenauigkeiten, die beispielsweise durch Temperatur-, Druck- oder Flussschwankungen im
Chromatographen verursacht werden, nicht verhindern.

I.5 Zusammenfassung
- 124 -
I.5 Zusammenfassung
Seit der ersten Veröffentlichung im Jahre 1990 gilt den Übergangsmetallkomplexen mit
verbrückten Cyclopentadienylamido-Liganden großes Interesse sowohl auf akademischer Seite
als auch in der Industrie. Speziell die so genannten „Constrained Geometry“ Katalysatoren der
vierten Nebengruppe werden äußerst erfolgreich in Homo- und Copolymerisationen eingesetzt.
Die Polymere, die dabei entstehen, weisen einzigartige Mikrostrukturen und dadurch bedingt
außergewöhnliche Materialeigenschaften auf.20
Abb. 63: Die „Constrained Geometry“ Katalysatoren (ηηηη5:ηηηη1-C5Me4SiMe2NtBu)Zr(IV)Cl2 und
(ηηηη5:ηηηη1-C5Me4SiMe2NtBu)Hf(IV)Cl2 aktiviert mittels Triisobutylaluminium und Trityltetrakis-
(pentafluorophenyl)borat.
Die meisten Veröffentlichungen im Bereich der Metallocene befassen sich mit Titan- und
Zirkoniumkomplexen während die Hafnium-Analoga ein Schattendasein fristen, da sie häufig
die geringeren Aktivitäten aufweisen, oftmals schwerer zugänglich und deutlich teurer sind. Im
aktuellsten Übersichtsartikel zu den CGCs ist im gesamten Polymerisationsteil keine einzige
Reaktion durch eine Hafnium-Verbindung beschrieben.20 Ziel dieser Arbeit war die
Untersuchung der Unterschiede der in Abb. 63 dargestellten isostrukturellen CGCs
(η5:η1-C5Me4SiMe2NtBu)Zr(IV)Cl2 und (η5:η1-C5Me4SiMe2N
tBu)Hf(IV)Cl2. Ausgangspunkt für
das Interesse an der Hf-Spezies ist der bei den klassischen Metallocenen im Jahre 2002 von Prof.
Rieger et al. erstmals beschriebene „erste Hafnium-Effekt“.47 Werden die Gruppe-4-Metallocene
mittels Trityltetrakis(pentafluorophenyl)borat aktiviert, zeigen die erhaltenen Polymere deutlich
höhere Molekulargewichte, wenn das Zentralmetall Hafnium an Stelle von Zirkonium ist. In
dieser Arbeit soll geklärt werden, ob dieser erste Hafnium-Effekt auch für die Klasse der
Constrained Geometry Katalysatoren Gültigkeit besitzt.

I.5 Zusammenfassung
- 125 -
Anhand von Homo- und Copolymerisationsexperimenten wurde analysiert, inwieweit die beiden
Katalysatoren zu unterschiedlichen Mikrostrukturen in den erzeugten Polymeren führen. Um die
vorgefundenen Effekte beurteilen zu können und kleinste strukturelle Unterschiede festzustellen,
wurden zunächst die Kristallstrukturen der Komplex-Vorstufen miteinander verglichen.
Abb. 64: Pov-Ray gerenderte, durch Röntgenbeugung bestimmte Kristallstruktur von
(ηηηη5:ηηηη1-C5Me4SiMe2NtBu)Hf(IV)Cl2. Die Atome sind mit 50% Aufenthaltswahrscheinlichkeit
wiedergegeben. Die Wasserstoffatome werden zur besseren Übersichtlichkeit nicht dargestellt.
Atome: Chlor, Hafnium, Kohlenstoff, Silizium, Stickstoff.
Es zeigt sich, dass die Bindungen, die vom Zentralmetall ausgehen, in der Hafnium-Spezies
grundsätzlich kürzer sind. Das Hafnium-Atom ist stärker an den Cyclopentadienyl-Ring und an
das Stickstoffatom gebunden, wodurch sich der Winkel centroid–Metall–N von 101,98° für
Zirkonium auf 103,17° vergrößert. Als Konsequenz daraus rücken die beiden Chlor-Atome näher
zusammen, was durch eine Verkleinerung des Winkels Cl(1)–Metall–Cl(2) deutlich wird. Die
Winkel zwischen centroid–Metall und den beiden Chlor-Atomen sind im Hafnium-Komplex
größer als in der entsprechenden Zirkonium-Verbindung. Die Geometrie des Hafnium-
Komplexes bietet somit bei Polymerisationsreaktionen etwas mehr Platz für ankoordinierende
Moleküle. Gleichzeitig gibt es jedoch auch für Abbruchreaktionen wie �-Hydrid-Eliminierung
mehr Raum.
Hf Cl(1) N
Si
Cl(2)

I.5 Zusammenfassung
- 126 -
DFT-Rechnungen ließen darauf schließen, dass die Unterschiede zwischen Zirkon- und
Hafniumspezies gering sind in Hinblick auf die Struktur des Katalysators, die �-Komplexe, die
Übergangsstrukturen, die Propyl-Produkte und die relativen Energien.31 Demgegenüber konnten
in allen hier durchgeführten Homo- und Copolymerisationsexperimenten deutliche
Abweichungen beim Polymerisationsverhalten der beiden Katalysator-Systeme festgestellt
werden.
Bei der Diskussion der Ergebnisse von Polymerisationen, ist es notwendig, sich Gedanken über
mögliche Fehlerquellen zu machen und sich damit die Aussagefähigkeit der Messwerte vor
Augen zu führen. Um die Qualität der durchgeführten Experimente zu überprüfen, wurde
deshalb in Kapitel I.4.6 eine ausführliche Fehlerbetrachtung durchgeführt. Unter
Berücksichtigung der unvermeidbaren und definitiv vorhandenen Abweichungen vom „wahren“
Messwert, wurden auch die Ergebnisdiskussionen in dieser Arbeit geführt.
Bevor Katalysatoren detailliert untersucht werden können, ist es notwendig, Grenzen
festzulegen, innerhalb derer die verschiedenen Einflussgrößen variiert werden. Zahlreiche
Parameter nehmen Einfluss auf den Verlauf von Polymerisationsreaktionen. In Kapitel I.4.1.5
wurden umfangreiche Untersuchungen mit Ethen durchgeführt, um beispielsweise Druck,
Temperatur, Katalysatormenge oder Cokatalysator festzulegen. Um einen möglichst
umfassenden Überblick über die Katalysatoren zu erhalten, wurden in Sterik und Elektronik
unterschiedlich anspruchsvolle Comonomere eingesetzt (siehe Abb. 65).
Abb. 65: Eingesetzte (Co-)Monomere.
Bereits bei den ersten Homo-Polymerisationsexperimenten mit dem „einfachsten“ Monomer
Ethen werden die außergewöhnlichen Eigenschaften der CGCs deutlich. Beide Katalysator-
systeme zeigen sehr hohe Aktivitäten von bis zu 41.200 kgPolymer/molKatalysator×h, wobei 1 um den
Faktor 5-7 aktiver als 2 ist. Während 1 ausschließlich niedermolekulare Polyethylene mit
Molmassen von ca. 3.000 g/mol erzeugt, entstehen durch 2 Produkte, die eine bimodale
Molekulargewichtsverteilung mit einem ultrahochmolekularen Bereich aufweisen. Die
Reaktionstemperaturen von 30 – 70 °C haben dabei nur einen geringen Einfluss auf die
Molmasse (vgl. Diagramm 39). Eine Besonderheit, die bereits für andere CGCs beschrieben

I.5 Zusammenfassung
- 127 -
worden ist, sind die Langkettenverzweigungen, die bei den mittels Hafnium-Katalysator
hergestellten Polymeren auftreten. Es wird angenommen, dass diese Langkettenverzweigungen
durch den Einbau von vinyl-terminierten Oligomeren entstehen, die während der Reaktion
gebildet werden.68 Die offene Koordinationssphäre der CGCs macht die Copolymerisation der
Makromonomere möglich. Durch den Mechanismus zur Bildung der LCBs erklärt sich auch die
bimodale Molekulargewichtsverteilung der Produkte (vgl. Abb. 66, Kapitel I.4.2).
Abb. 66: Mechanismus für die Bildung von Langkettenverzweigungen in Ethen-Homo-
polymerisationen.70
Die Polymerisationsergebnisse für die Ethen-Propen-Copolymerisationen zeigen ein ähnlich
erstaunliches Bild. Wiederum entstehen mit 1 ausschließlich niedermolekulare Kunststoffe,
wohingegen 2 zu langkettenverzweigten Produkten mit bimodaler Molekulargewichtsverteilung
führt. Während die Molmassen bei den Ethen-Homopolymerisationen nur geringfügig von der
Reaktionstemperatur beeinflusst werden, ist hier ein deutlicher Rückgang mit steigenden
Temperaturen erkennbar (vergleiche Diagramm 39).
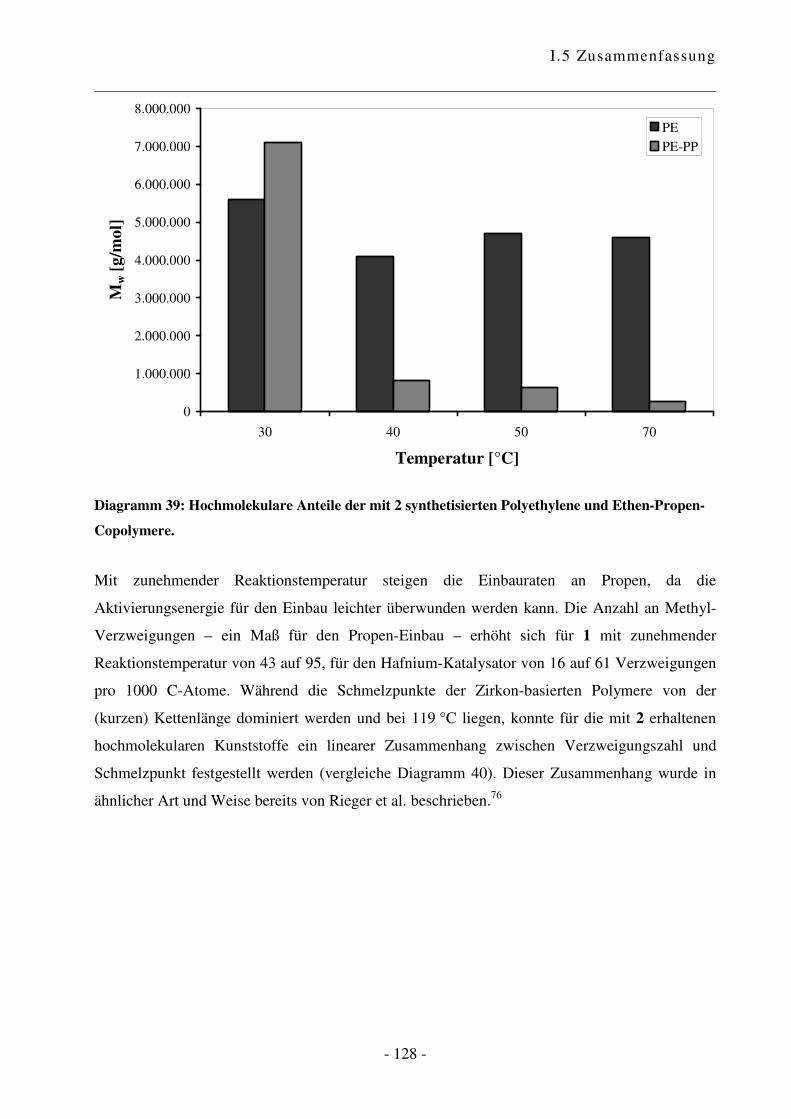
I.5 Zusammenfassung
- 128 -
0
1.000.000
2.000.000
3.000.000
4.000.000
5.000.000
6.000.000
7.000.000
8.000.000
30 40 50 70
Temperatur [°C]
Mw [
g/m
ol]
PEPE-PP
Diagramm 39: Hochmolekulare Anteile der mit 2 synthetisierten Polyethylene und Ethen-Propen-
Copolymere.
Mit zunehmender Reaktionstemperatur steigen die Einbauraten an Propen, da die
Aktivierungsenergie für den Einbau leichter überwunden werden kann. Die Anzahl an Methyl-
Verzweigungen – ein Maß für den Propen-Einbau – erhöht sich für 1 mit zunehmender
Reaktionstemperatur von 43 auf 95, für den Hafnium-Katalysator von 16 auf 61 Verzweigungen
pro 1000 C-Atome. Während die Schmelzpunkte der Zirkon-basierten Polymere von der
(kurzen) Kettenlänge dominiert werden und bei 119 °C liegen, konnte für die mit 2 erhaltenen
hochmolekularen Kunststoffe ein linearer Zusammenhang zwischen Verzweigungszahl und
Schmelzpunkt festgestellt werden (vergleiche Diagramm 40). Dieser Zusammenhang wurde in
ähnlicher Art und Weise bereits von Rieger et al. beschrieben.76
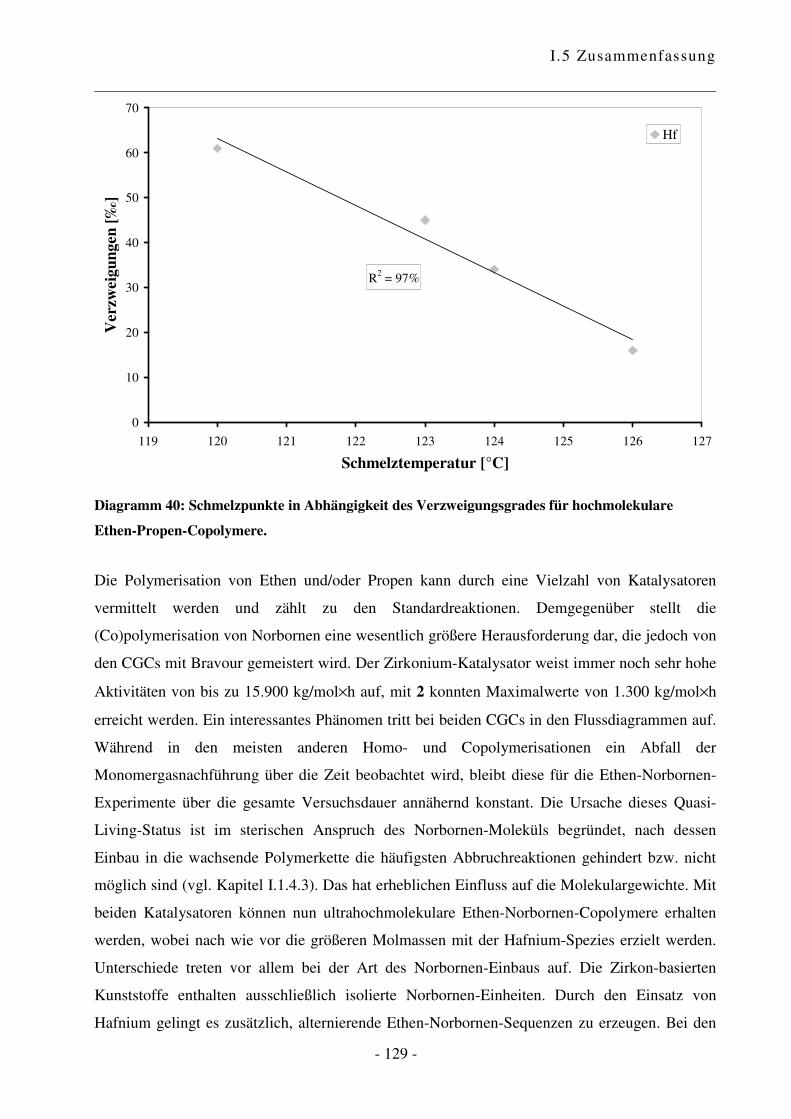
I.5 Zusammenfassung
- 129 -
R2 = 97%
0
10
20
30
40
50
60
70
119 120 121 122 123 124 125 126 127
Schmelztemperatur [°C]
Ver
zwei
gung
en [
‰]
Hf
Diagramm 40: Schmelzpunkte in Abhängigkeit des Verzweigungsgrades für hochmolekulare
Ethen-Propen-Copolymere.
Die Polymerisation von Ethen und/oder Propen kann durch eine Vielzahl von Katalysatoren
vermittelt werden und zählt zu den Standardreaktionen. Demgegenüber stellt die
(Co)polymerisation von Norbornen eine wesentlich größere Herausforderung dar, die jedoch von
den CGCs mit Bravour gemeistert wird. Der Zirkonium-Katalysator weist immer noch sehr hohe
Aktivitäten von bis zu 15.900 kg/mol×h auf, mit 2 konnten Maximalwerte von 1.300 kg/mol×h
erreicht werden. Ein interessantes Phänomen tritt bei beiden CGCs in den Flussdiagrammen auf.
Während in den meisten anderen Homo- und Copolymerisationen ein Abfall der
Monomergasnachführung über die Zeit beobachtet wird, bleibt diese für die Ethen-Norbornen-
Experimente über die gesamte Versuchsdauer annähernd konstant. Die Ursache dieses Quasi-
Living-Status ist im sterischen Anspruch des Norbornen-Moleküls begründet, nach dessen
Einbau in die wachsende Polymerkette die häufigsten Abbruchreaktionen gehindert bzw. nicht
möglich sind (vgl. Kapitel I.1.4.3). Das hat erheblichen Einfluss auf die Molekulargewichte. Mit
beiden Katalysatoren können nun ultrahochmolekulare Ethen-Norbornen-Copolymere erhalten
werden, wobei nach wie vor die größeren Molmassen mit der Hafnium-Spezies erzielt werden.
Unterschiede treten vor allem bei der Art des Norbornen-Einbaus auf. Die Zirkon-basierten
Kunststoffe enthalten ausschließlich isolierte Norbornen-Einheiten. Durch den Einsatz von
Hafnium gelingt es zusätzlich, alternierende Ethen-Norbornen-Sequenzen zu erzeugen. Bei den
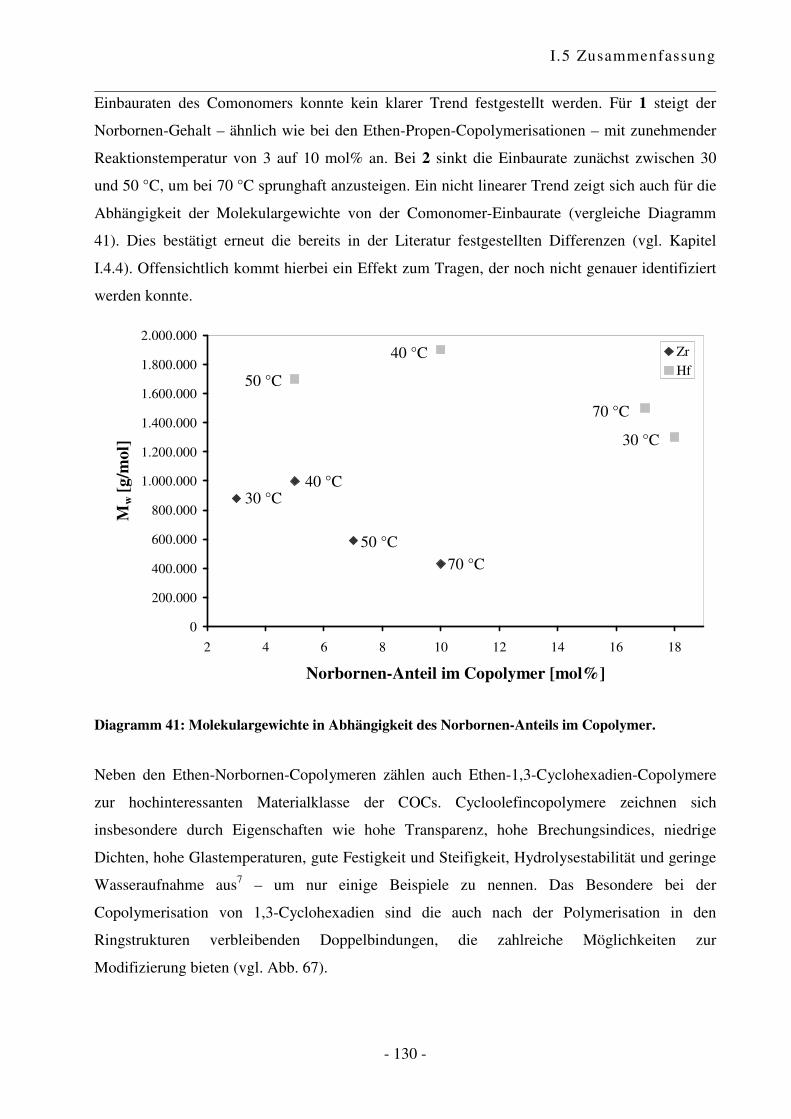
I.5 Zusammenfassung
- 130 -
Einbauraten des Comonomers konnte kein klarer Trend festgestellt werden. Für 1 steigt der
Norbornen-Gehalt – ähnlich wie bei den Ethen-Propen-Copolymerisationen – mit zunehmender
Reaktionstemperatur von 3 auf 10 mol% an. Bei 2 sinkt die Einbaurate zunächst zwischen 30
und 50 °C, um bei 70 °C sprunghaft anzusteigen. Ein nicht linearer Trend zeigt sich auch für die
Abhängigkeit der Molekulargewichte von der Comonomer-Einbaurate (vergleiche Diagramm
41). Dies bestätigt erneut die bereits in der Literatur festgestellten Differenzen (vgl. Kapitel
I.4.4). Offensichtlich kommt hierbei ein Effekt zum Tragen, der noch nicht genauer identifiziert
werden konnte.
0
200.000
400.000
600.000
800.000
1.000.000
1.200.000
1.400.000
1.600.000
1.800.000
2.000.000
2 4 6 8 10 12 14 16 18
Norbornen-Anteil im Copolymer [mol%]
Mw [
g/m
ol]
ZrHf
Diagramm 41: Molekulargewichte in Abhängigkeit des Norbornen-Anteils im Copolymer.
Neben den Ethen-Norbornen-Copolymeren zählen auch Ethen-1,3-Cyclohexadien-Copolymere
zur hochinteressanten Materialklasse der COCs. Cycloolefincopolymere zeichnen sich
insbesondere durch Eigenschaften wie hohe Transparenz, hohe Brechungsindices, niedrige
Dichten, hohe Glastemperaturen, gute Festigkeit und Steifigkeit, Hydrolysestabilität und geringe
Wasseraufnahme aus7 – um nur einige Beispiele zu nennen. Das Besondere bei der
Copolymerisation von 1,3-Cyclohexadien sind die auch nach der Polymerisation in den
Ringstrukturen verbleibenden Doppelbindungen, die zahlreiche Möglichkeiten zur
Modifizierung bieten (vgl. Abb. 67).
30 °C
30 °C 40 °C
40 °C
50 °C
50 °C
70 °C
70 °C

I.5 Zusammenfassung
- 131 -
Abb. 67: Möglichkeiten zur weiteren Umsetzung von Ethen-1,3-Cyclohexadien-Copolymeren.
Bei den hier durchgeführten Experimenten zeigen beide Katalysatoren sehr hohe Aktivitäten und
ultrahochmolekulare Produkte. Obwohl die Einbauraten an 1,3-CHD für 1 im Promille-Bereich
liegen, hat der Einbau entscheidende Auswirkung auf die Kettenlänge. Ein Einsatz von 1,3-CHD
als Molmassenregulator hin zu höheren Molekulargewichten ist für die Polymerisationen mit
dem Zirkonium CGC durchaus denkbar. Wiederum setzt sich der Trend der höheren Molmassen
für die mit dem Hafnium-Katalysator erzeugten Polymere durch. Auch bei den Comonomer-
Einbauraten erreicht 2 mit bis zu 1,4 mol% die deutlich höheren Werte.
Zusammenfassend lässt sich sagen, dass in der vorliegenden Arbeit ein Zirkonium und ein
Hafnium Constrained Geometry Komplex synthetisiert, umfangreich untersucht und in
zahlreichen Polymerisationsreaktionen getestet worden sind. Es stellte sich heraus, dass der so
genannte „erste Hafnium-Effekt“ auch für die Klasse der CGCs Gültigkeit besitzt. Trotz der
Ähnlichkeit der Kristallstrukturen zeigen die Katalysatoren ein grundlegend verschiedenes
Polymerisationsverhalten. Die Unterschiede in der Metall-C-Bindungsstärke scheinen gering,
führen jedoch dazu, dass sich das Verhältnis der Kettenabbruchreaktionen zu den
Kettenwachstumsreaktionen so verändert, dass der Hafnium-Katalysator ultrahochmolekulare

I.5 Zusammenfassung
- 132 -
Polymere und der Zirkonium-Katalysator Oligomere erzeugt. Erst durch den Einsatz sterisch
anspruchsvoller Monomere, nach deren Einbau Abbruchreaktionen gehindert sind, erreicht auch
1 hochmolekulare Produkte. Bei den Einbauraten stellt man fest, dass 2 mit zunehmendem
Conomomer-Anspruch eine größere Variabilität gegenüber 1 zeigt. Einbau von
Makromonomeren – auch direkt nacheinander, langkettenverzweigte Copolymere, alternierende
Ethen-Norbornen-Sequenzen, kontrollierte Insertionspolymerisation von Monomeren, die
standardmäßig anionisch polymerisiert werden (müssen): All das ist mit dem Hafnium-CGC
möglich. In dieser Arbeit hat sich erneut gezeigt, dass Hafnium-Katalysatoren zu Unrecht
vernachlässigt werden. Es bleibt spannend, wann die zahlreichen positiven Ergebnisse der letzten
Jahre in Forschung und Industrie Gehör und Anwendung finden.

I.6 Summary
- 133 -
I.6 Summary
Since 1990, transition metal complexes with bridged cyclopentadienylamido-ligands have
aroused great interest in academia and industry. Especially, the so-called “constrained geometry
catalysts” of the fourth subgroup are successfully applied in homo- and copolymerization
reactions. The corresponding polymers possess uniform microstructures and extraordinary
material properties.20
Fig. 1: “Constrained geometry catalysts” (ηηηη5:ηηηη1-C5Me4SiMe2NtBu)Zr(IV)Cl2 and
(ηηηη5:ηηηη1-C5Me4SiMe2NtBu)Hf(IV)Cl2 activated by triisobutylaluminium and trityltetrakis(penta-
fluorophenyl)borate.
Most publications on metallocenes consider exclusively titanium and zirconium complexes
whereas the hafnium analogues are of minor interest, since they often show lower activities, they
are more difficult to synthesize and more expensive. Within the polymerization part of the latest
review on CGCs not one single hafnium-mediated reaction is described.20 This work’s principal
goal was the investigation of the two isostructural CGCs (η5:η1-C5Me4SiMe2NtBu)Zr(IV)Cl2 and
(η5:η1-C5Me4SiMe2NtBu)Hf(IV)Cl2 (see Fig. 1). Starting point for the interest in hafnium
species is the so-called “first hafnium effect” that was initially found for classical metallocenes
by Rieger et al.47 On activating group-4-metallocenes by trityltetrakis(pentafluorophenyl)borate,
the obtained polymers show considerably higher molecular weights if the central metal is
hafnium instead of zirconium. Aim of this work is to investigate, whether this first hafnium
effect is valid for the class of CGCs.

I.6 Summary
- 134 -
By means of homo- and copolymerization experiments it has been analyzed to what extend the
two catalysts lead to different microstructures in the resulting polymers. In order to evaluate the
discovered effects and to find out smallest structural differences, first of all the crystal structures
of the complex precursors have been compared.
Fig. 2: By Means of X-ray diffraction defined Crystal structure of (ηηηη5:ηηηη1-C5Me4SiMe2NtBu)-
Hf(IV)Cl2. The atoms are drawn as 50% ellipsoids. Hydrogen atoms are omitted for clarity. Atoms:
chlorine, hafnium, carbon, silica, nitrogen.
This reveals that bonds, coming from the metal are in principle shorter for the hafnium species.
The hafnium atom is bonded stronger to the cyclopentadienyl ring as well as the nitrogen atom,
resulting in an enlargement of the angle centroid–metal–N of 101.98° for zirconium to 103.17°.
As a consequence, the both chlorine atoms approach, shown by a decrease of the angle Cl(1)–
metal–Cl(2). The angles between centroid-metal and the chlorine atoms are larger in the hafnium
complex than in the corresponding zirconium compound. Therefore, the geometry of the
hafnium complex provides more open space for coordinating monomers in polymerization
reactions as well as for termination reactions, like �-hydride-elimination.
Hf Cl(1) N
Si
Cl(2)

I.6 Summary
- 135 -
DFT-calculations had suggested that the differences between zirconium and hafnium species are
minor regarding to catalyst structure, �-complexes, transition states, propyl-products and relative
energies.31 In contrast, striking differences between the polymerization performances of both
catalysts could be discovered in all of the homo- and copolymerization experiments that have
been carried out.
Preliminary to a detailed discussion of polymerization results, it is necessary to think about
potential nonconformities and to consider the informational value of the results. In order to prove
the experiments’ quality, a detailed error discussion is enclosed in chapter I.4.6. Under
consideration of these inevitable but definitively existent discrepancies, the results in this work
have been disputed.
Before the catalysts are studied in detail, it is necessary to define the boundary, in which the
influencing values are varied. The course of polymerization reactions is influenced by numerous
parameters. Chapter I.4.1.5 deals with substantial investigations on the polymerization of
ethylene to predefine the reaction parameters like pressure, temperature, catalyst amount or
cocatalyst. In order to gain a preferably broad overview about the two catalysts, monomers and
comonomers with different sterical and electronical requirements were applied (see Fig. 3).
Fig. 3: Comonomers used in this work.
Already, the first homopolymerization reactions of the “simplest” monomer ethylene reveal the
extraordinary properties of CGCs. Both catalyst systems show very high activities up to
41.200 kgpolymer/molcatalyst×h with 1 exceeding 2 by a factor of 5-7. However, 1 produces only
lower molecular weight polyethylene with molar masses of about 3.000 g/mol, whereas 2 leads
to products, showing a bimolal molecular weight distribution including an ultra-high molecular
weight region. There is a lesser influence of the reaction temperatures (30 – 70 °C) on molecular
masses (see Fig. 5). A noteworthy ability that has already been reported for other CGCs, is the
formation of long chain branches that appear in polymers synthesized by 2. It is assumed that
these LCBs evolve from vinyl-terminated oligomers that are built during the polymerization
reaction.68 The CGCs’ open coordination sphere enables copolymerization of macromonomers.

I.6 Summary
- 136 -
Also, the bimodal molecular weight distribution is explained by the mechanism of long chain
branch formation (see Fig. 4, chapter I.4.2).
Fig. 4: Mechanism for LCB-formation in ethylene homopolymerization.70
In the same way the results of the ethylene-propylene-copolymerization experiments are
astonishing. Again, while 1 produces only low molecular weight waxes, 2 leads to long chain
branched products with bimodal molecular weight distribution. Molar masses of polyethylenes
had scarcely been influenced by reaction temperatures whereas for polyethylene-co-propylenes a
distinct decrease is observed (see Graph 1).
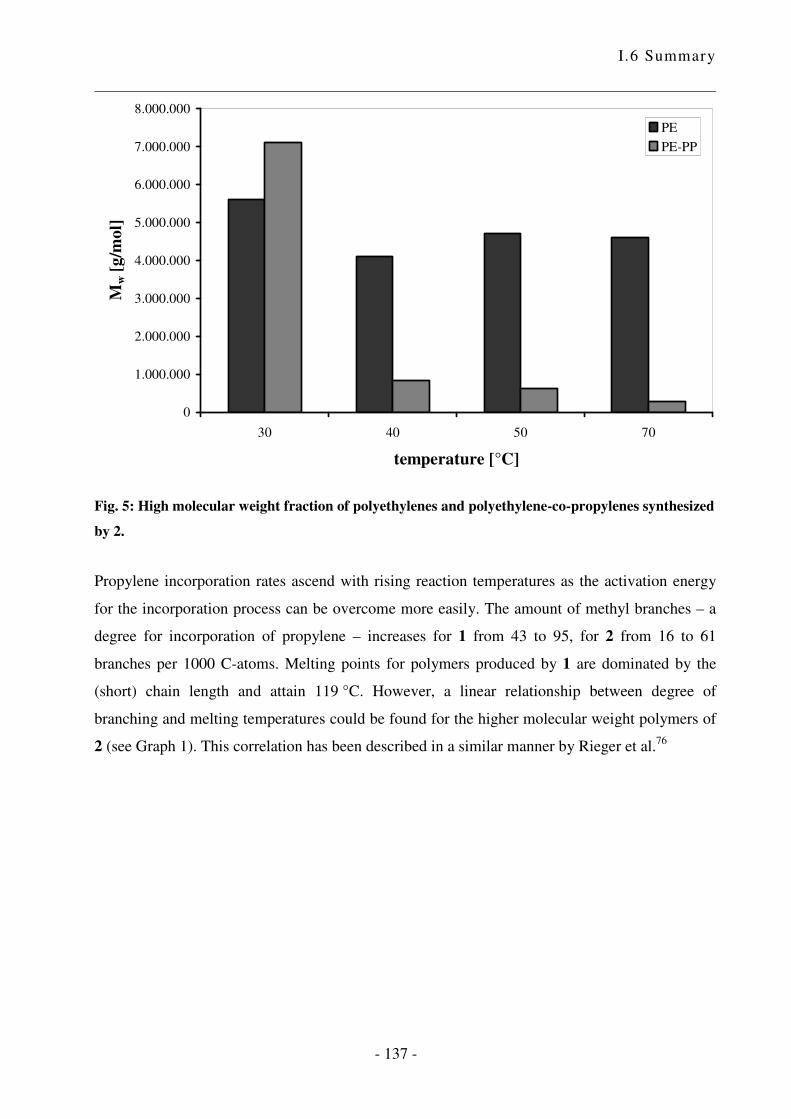
I.6 Summary
- 137 -
0
1.000.000
2.000.000
3.000.000
4.000.000
5.000.000
6.000.000
7.000.000
8.000.000
30 40 50 70
temperature [°C]
Mw [
g/m
ol]
PEPE-PP
Fig. 5: High molecular weight fraction of polyethylenes and polyethylene-co-propylenes synthesized
by 2.
Propylene incorporation rates ascend with rising reaction temperatures as the activation energy
for the incorporation process can be overcome more easily. The amount of methyl branches – a
degree for incorporation of propylene – increases for 1 from 43 to 95, for 2 from 16 to 61
branches per 1000 C-atoms. Melting points for polymers produced by 1 are dominated by the
(short) chain length and attain 119 °C. However, a linear relationship between degree of
branching and melting temperatures could be found for the higher molecular weight polymers of
2 (see Graph 1). This correlation has been described in a similar manner by Rieger et al.76
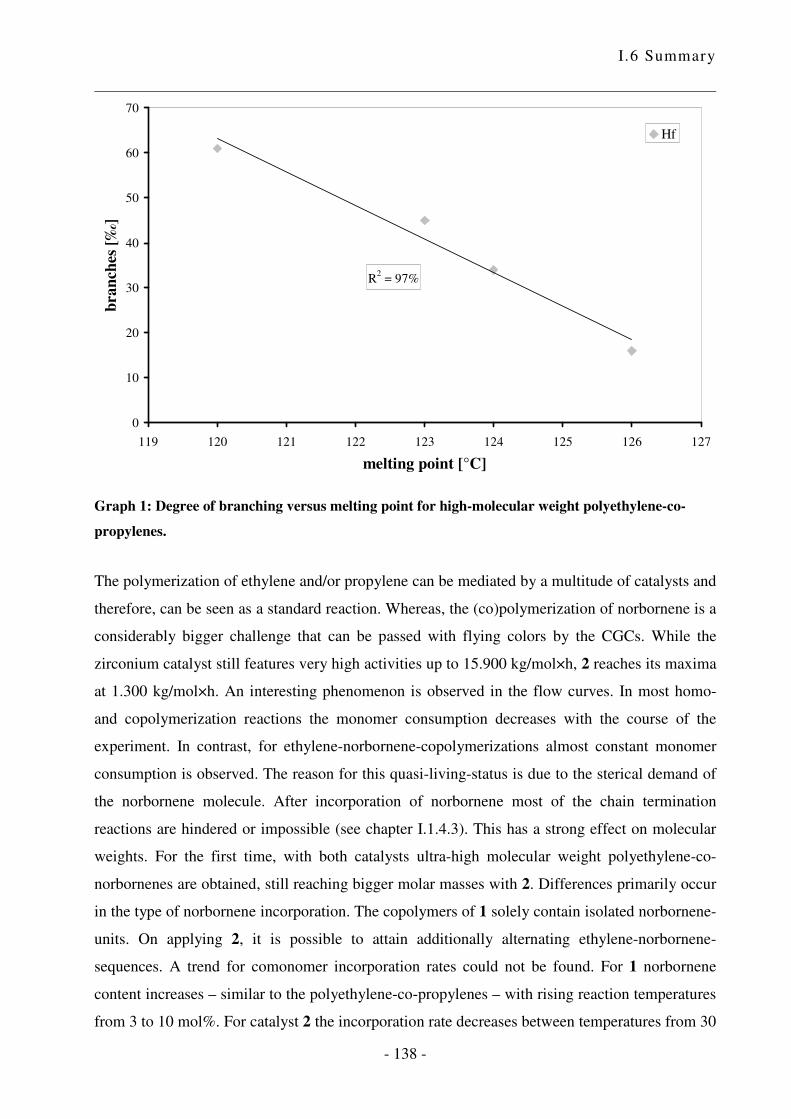
I.6 Summary
- 138 -
R2 = 97%
0
10
20
30
40
50
60
70
119 120 121 122 123 124 125 126 127
melting point [°C]
bran
ches
[‰
]
Hf
Graph 1: Degree of branching versus melting point for high-molecular weight polyethylene-co-
propylenes.
The polymerization of ethylene and/or propylene can be mediated by a multitude of catalysts and
therefore, can be seen as a standard reaction. Whereas, the (co)polymerization of norbornene is a
considerably bigger challenge that can be passed with flying colors by the CGCs. While the
zirconium catalyst still features very high activities up to 15.900 kg/mol×h, 2 reaches its maxima
at 1.300 kg/mol×h. An interesting phenomenon is observed in the flow curves. In most homo-
and copolymerization reactions the monomer consumption decreases with the course of the
experiment. In contrast, for ethylene-norbornene-copolymerizations almost constant monomer
consumption is observed. The reason for this quasi-living-status is due to the sterical demand of
the norbornene molecule. After incorporation of norbornene most of the chain termination
reactions are hindered or impossible (see chapter I.1.4.3). This has a strong effect on molecular
weights. For the first time, with both catalysts ultra-high molecular weight polyethylene-co-
norbornenes are obtained, still reaching bigger molar masses with 2. Differences primarily occur
in the type of norbornene incorporation. The copolymers of 1 solely contain isolated norbornene-
units. On applying 2, it is possible to attain additionally alternating ethylene-norbornene-
sequences. A trend for comonomer incorporation rates could not be found. For 1 norbornene
content increases – similar to the polyethylene-co-propylenes – with rising reaction temperatures
from 3 to 10 mol%. For catalyst 2 the incorporation rate decreases between temperatures from 30
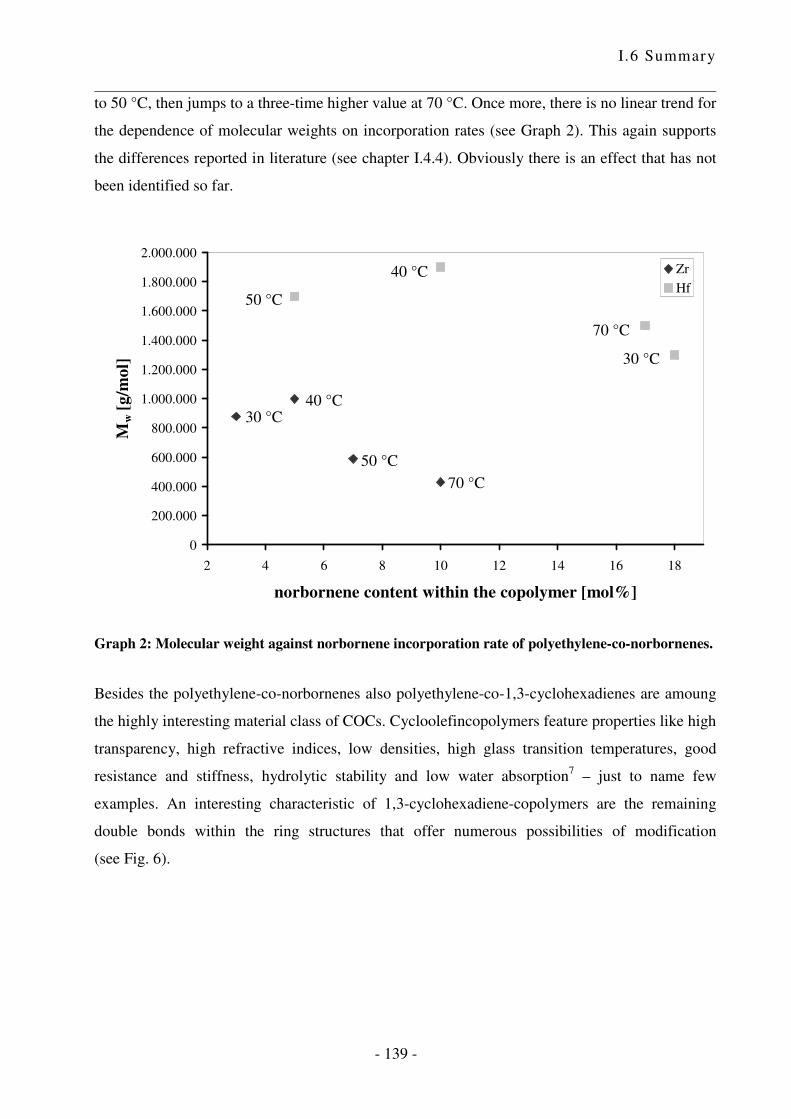
I.6 Summary
- 139 -
to 50 °C, then jumps to a three-time higher value at 70 °C. Once more, there is no linear trend for
the dependence of molecular weights on incorporation rates (see Graph 2). This again supports
the differences reported in literature (see chapter I.4.4). Obviously there is an effect that has not
been identified so far.
0
200.000
400.000
600.000
800.000
1.000.000
1.200.000
1.400.000
1.600.000
1.800.000
2.000.000
2 4 6 8 10 12 14 16 18
norbornene content within the copolymer [mol%]
Mw [
g/m
ol]
ZrHf
Graph 2: Molecular weight against norbornene incorporation rate of polyethylene-co-norbornenes.
Besides the polyethylene-co-norbornenes also polyethylene-co-1,3-cyclohexadienes are amoung
the highly interesting material class of COCs. Cycloolefincopolymers feature properties like high
transparency, high refractive indices, low densities, high glass transition temperatures, good
resistance and stiffness, hydrolytic stability and low water absorption7 – just to name few
examples. An interesting characteristic of 1,3-cyclohexadiene-copolymers are the remaining
double bonds within the ring structures that offer numerous possibilities of modification
(see Fig. 6).
30 °C
30 °C 40 °C
40 °C
50 °C
50 °C
70 °C
70 °C
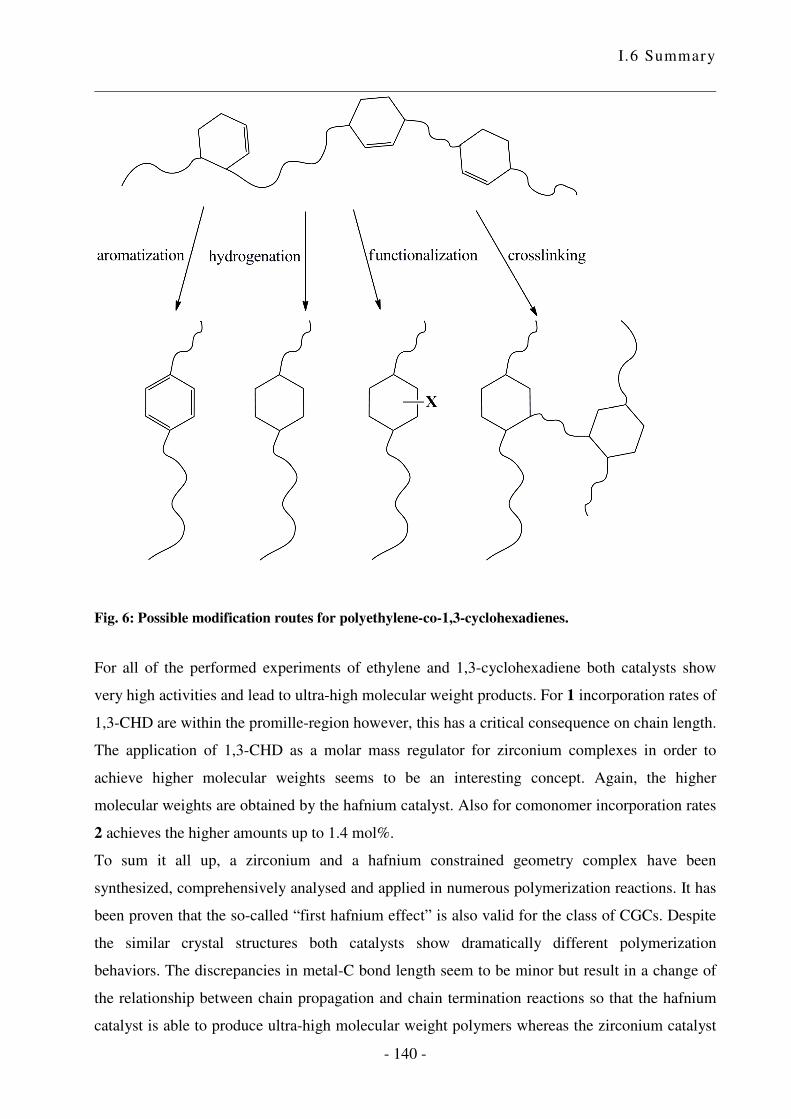
I.6 Summary
- 140 -
Fig. 6: Possible modification routes for polyethylene-co-1,3-cyclohexadienes.
For all of the performed experiments of ethylene and 1,3-cyclohexadiene both catalysts show
very high activities and lead to ultra-high molecular weight products. For 1 incorporation rates of
1,3-CHD are within the promille-region however, this has a critical consequence on chain length.
The application of 1,3-CHD as a molar mass regulator for zirconium complexes in order to
achieve higher molecular weights seems to be an interesting concept. Again, the higher
molecular weights are obtained by the hafnium catalyst. Also for comonomer incorporation rates
2 achieves the higher amounts up to 1.4 mol%.
To sum it all up, a zirconium and a hafnium constrained geometry complex have been
synthesized, comprehensively analysed and applied in numerous polymerization reactions. It has
been proven that the so-called “first hafnium effect” is also valid for the class of CGCs. Despite
the similar crystal structures both catalysts show dramatically different polymerization
behaviors. The discrepancies in metal-C bond length seem to be minor but result in a change of
the relationship between chain propagation and chain termination reactions so that the hafnium
catalyst is able to produce ultra-high molecular weight polymers whereas the zirconium catalyst

I.6 Summary
- 141 -
only leads to oligomers. Only on applying sterically demanding monomers, after the
incorporation of which chain termination reactions are hindered, higher molecular weight
products can also be achieved with 1. For the comonomer incorporation rates 2 shows greater
variability than 1 with increasing comonomer demand. Incorporation of macromonomers – also
consecutively – alternating ethylene-norbornene-sequences, controlled insertion polymerization
reaction of monomers that are usually polymerized in an anionically way: all these things are
possible for the hafnium-CGC. Within this work it has been shown that there is no reason why
hafnium catalysts should be neglected. It remains exciting when these numerous positive results
of the last years will be considered and applied in science and industry.

I.7 Experimenteller Teil
- 142 -
I.7 Experimenteller Teil
I.7.1 Arbeitstechniken und –materialien
Alle experimentellen Arbeiten wurden unter Anwendung von Standard-Schlenktechniken
durchgeführt. Die Komplexsynthesen erfolgten in einer Glovebox der Fa. Braun, welche mit
Argon 4.8 betrieben wurde.
Die verwendeten Lösungsmittel wurden wie folgt getrocknet: THF wurde mehrere Tage über
Lithiumaluminiumhydrid gerührt und vor Gebrauch destilliert. Pentan wurde über
Aluminiumoxid gereinigt. Toluol (p.a.) wurde mittels Lithiumaluminiumhydrid getrocknet. Das
Toluol, welches in der Glovebox verwendet wurde, wurde zusätzlich über Natrium/Benzophenon
getrocknet. Durch dreimaliges Einfrieren, Evakuieren und Auftauen wurde Sauerstoff entfernt.
Anschließend wurde das Toluol mittels Vakuum-Transfer in ein Schlenkgefäß überführt. Benzol-
d6 wurde ebenfalls über Natrium/Benzophenon getrocknet und dieser zusätzlichen Reinigung
unterzogen. Das in der Glovebox verwendete Pentan wurde analog zum Toluol über
Natrium/Benzophenon getrocknet und über Einfrieren/Auftauen und Vakuumtransfer entgast.
Folgende Chemikalien wurden bei der Fa. Merck erworben und wie erhalten eingesetzt: n-
Butyllithium, Lithiumaluminiumhydrid, Dichlordimethylsilan, tert-Butylamin. Zirkonium- und
Hafniumtetrachlorid stammen von der Fa. ABCR. Tetramethylcyclopentadien wurde nach der
Methode von Beagley et al. selbst hergestellt. 49,126

I.7 Experimenteller Teil
- 143 -
I.7.2 Ligandensynthese
I.7.2.1 1-(Chlorodimethylsilyl)-2, 3, 4, 5-tetramethyl-cyclopentadien
Ansatz:
1, 2, 3, 4-Tetramethylcyclopentadien: 52 mmol 6,4 g
Tetrahydrofuran: 280 mL
n-Butyllithium, 15%ig in Hexan 60 mmol 38 mL
Dichlordimethylsilan: 160 mmol 21,0 g
Pentan 140 mL
Durchführung:
1, 2, 3, 4-Tetramethylcyclopentadien (6,4 g, 52 mmol) wird in 200 mL THF gelöst. Bei -78 °C
wird n-Butyllithium in Hexan langsam zugetropft und anschließend drei Tage bei RT gerührt.
Die milchig-trübe Suspension wird bei -78°C zu einer Lösung von Dichlordimethylsilan (21,0 g,
160 mmol) in 80 mL THF zugegeben und zwei Tage bei RT weiter gerührt.
Das Lösungsmittel wird im Vakuum entfernt. Der Rückstand wird in 100 mL Pentan wieder
aufgenommen. Es entsteht eine hellgelbe Lösung mit weißem, mikrokristallinem Niederschlag,
der abfiltriert wird. Der Rückstand wird zwei Mal mit je 20 mL Pentan gewaschen. Nach
Einengen des Filtrats im Vakuum resultiert eine gelbe Flüssigkeit (10,4 g; 48 mmol; 93 %).
NMR: 1H-NMR (400 MHz, CDCl3):
δ [ppm]: 0,25 (s, 6 H, 2 Si-CH3); 1,83 (s, 6 H, 2 CH3); 1,99 (s, 6 H, 2 CH3); 3,09 (br s,
1H, Cp-H). 13C-NMR (100 MHz, CDCl3):
δ[ppm]: 0,6; 11,2; 14,2; 56,4; 131,4; 137.8.

I.7 Experimenteller Teil
- 144 -
I.7.2.2 (N-tButylamino)(dimethyl)(2, 3, 4, 5-tetramethylcyclo-
pentadienyl)silan
Ansatz:
1-(Chlorodimethylsilyl)-2, 3, 4, 5-tetramethyl-
cyclopentadien:
48 mmol 10,4 g
Tetrahydrofuran: 190 mL tButylamin: 146 mmol 11,0 g
Pentan: 130 mL
Durchführung:
1-(Chlorodimethylsilyl)-2, 3, 4, 5-tetramethylcyclopentadien (48 mmol; 10,4 g) wird in 150 mL
Tetrahydrofuran gelöst. Unter Eiskühlung wird eine Lösung von tButylamin (146 mmol; 11,0 g)
in 40 mL Tetrahydrofuran zugetropft. Die Reaktion wird bei RT vier Tage gerührt, es bildet sich
dabei ein weißer Niederschlag.
Das Lösungsmittel wird im Vakuum entfernt. Der Rückstand wird in 60 mL Pentan wieder
aufgenommen und die Lösung mittels Filterkanüle vom Niederschlag abgetrennt. Der
Niederschlag wird mit insgesamt 70 mL Pentan in drei Schritten gewaschen. Nach Einengen des
Filtrats am Vakuum resultiert eine gelb-grüne, klare Flüssigkeit (11,9 g; 47 mmol; 98 %).
NMR: 1H-NMR (400 MHz, CDCl3):
δ[ppm]: 0,03 (s, 6 H, 2 Si-CH3); 0,47 (br s, 1 H, N-H); 1,15 (s, 9 H, t-Butyl); 1,81 (s, 6 H,
2 CH3); 1,98 (s, 6 H, 2 CH3); 2,84 (br s, 1H, Cp-H). 13C-NMR (100 MHz, CDCl3):
δ[ppm]: 0,9; 11,1; 14,7; 33,7; 49,3; 57,0; 133,3; 135,3.

I.7 Experimenteller Teil
- 145 -
I.7.3 Komplexsynthese
I.7.3.1 Dilithium(N-tButylamido)(dimethyl)(2, 3, 4, 5-tetramethyl-
cyclopentadienyl)silan
Ansatz:
(N-tButylamino)(dimethyl)(2, 3, 4, 5-tetramethyl-
cyclopentadienylsilan:
47 mmol 11,9 g
Diethylether: 250 mL
n-Butyllithium, 15%ig in Hexan: 95 mmol 60 mL
Pentan: 60 mL
Durchführung:
(N-tButylamino)(dimethyl)(2, 3, 4, 5-tetramethylcyclopentadienyl)silan (11,9 g; 47 mmol) wird
in 250 mL Diethylether gelöst. Bei –78°C wird n-Butyllithium (60 mL, 95 mmol) innerhalb von
1 h zugegeben und über Nacht bei RT weiter gerührt.
In der gelben Lösung ist weißer Niederschlag ausgefallen. Das Lösungsmittel wird abgezogen
und der Rückstand in 60 mL Pentan wieder aufgenommen. Die Lösung wird vom Niederschlag
abgetrennt und der Niederschlag am Vakuum getrocknet. Es resultiert ein schwach gelbes Pulver
(11,9 g; 45 mmol; 95 %).
NMR: 1H-NMR (400 MHz, THF-d8):
δ[ppm]: 0,02 (s, 6 H, 2 Si-CH3); 0,87 (s, 9 H, t-Butyl); 1,63 (s, 6 H, 2 CH3); 1,80 (s, 6 H,
2 CH3). 13C-NMR (100 MHz, THF-d8):
δ[ppm]: 7,1; 11,8; 15,0; 34,6; 102,8; 112,3; 117,1.
Das quartäre C-Atom der tButyl-Gruppe konnte nicht bestimmt werden.

I.7 Experimenteller Teil
- 146 -
I.7.3.2 Tetrachlorobis(tetrahydrofuran)zirkonium(IV), ZrCl4(thf)2
Zirkonium(IV)chlorid ist ein weißer, sublimierender Feststoff, der aus Zickzack-Ketten
aufgebaut ist, in denen kantenverknüpfte ZrCl6-Oktaeder vorliegen. Er hydrolysiert zu dem
beständigen Oxidchlorid ZrOCl2×8 H2O.24
Ansatz:
ZrCl4: 15 mmol 3,5 g
Dichlormethan: 50 mL
Tetrahydrofuran: 60 mmol 4,3 g; 5 mL
Durchführung:
Zirkoniumchlorid (3,5 g, 15 mmol) wird in 50 mL Dichlormethan suspendiert. Unter Eiskühlung
wird Tetrahydrofuran (60 mmol; 5 mL) zugetropft. Nach beendeter Zugabe wird für weitere 20
min bei Raumtemperatur gerührt. Das Lösungsmittel wird im Vakuum abgezogen. Es resultiert
ein weißes Pulver (5,6 g; 14,9 mmol; 99 %).
I.7.3.3 Tetrachlorobis(tetrahydrofuran)hafnium(IV), HfCl4(thf)2
Ansatz:
HfCl4: 14 mmol 4,6 g
Dichlormethan: 30 mL
Tetrahydrofuran: 60 mmol 4,3 g; 4,9 mL
Durchführung:
Hafniumchlorid (4,6 g; 14 mmol) wird in 30 mL Dichlormethan suspendiert. Unter Eiskühlung
wird eine Lösung von 10 mL Tetrahydrofuran in 20 mL Dichlormethan zugetropft. Nach
beendeter Zugabe wird 1 h bei Raumtemperatur weitergerührt. Nach Abziehen des
Lösungsmittels im Vakuum verbleibt Tetrachlorobis(tetrahydrofuran)-hafnium(IV) als weißes
Pulver in fast quantitativer Ausbeute.

I.7 Experimenteller Teil
- 147 -
I.7.3.4 (N-tbutylamido)dimethyl(tetramethyl-�5-cyclopentadienyl)silan-
zirkonium(IV)dichlorid
Ansatz:
Ligand-Dilithiumsalz (siehe I.7.3.1 ): 4 mmol 1,06 g
ZrCl4(thf)2: 4 mmol 1,52 g
Toluol: 30 mL
Pentan: 7 mL
Durchführung:
Der Ligand (1,06 g; 4 mmol) wird in 15 mL Toluol suspendiert. Die Ligand-Suspension wird auf
-30 °C gekühlt. Unter Rühren wird ZrCl4(thf)2 (1,52 g, 4 mmol) portionsweise zugegeben und
fünf Tage bei Raumtemperatur weiter gerührt.
Die Lösung wird vom sehr feinen, weißen Niederschlag abfiltriert. Das Filtrat wird am Vakuum
eingeengt. Der gelbliche, kristalline Rückstand wird in 8 mL Toluol wieder aufgenommen und
erneut filtriert. Das Filtrat wird sechs Tage bei -30°C gelagert. Nach Entfernen der Mutterlauge
werden die erhaltenen Kristalle mit 3 mL Pentan gewaschen. Anschließend wird erneut aus 7 mL
Toluol umkristallisiert. Die nun farblosen Kristalle werden noch zwei Mal mit je 2 mL Pentan
gewaschen. (η5:η1-C5Me4SiMe2NtBu)Zr(IV)Cl2 kristallisiert als farblose Plättchen (0,68 g,
41 %).
CHN-Analyse: C15H27Cl2NSiZr
Berechnet: C: 43,77 H: 6,61 N: 3,40
Gefunden: C: 43,79 H: 6,58 N: 3,31
NMR: 1H-NMR (400 MHz, Benzol-d6): δ[ppm]: 0,56 (s, 6 H, 2 Si-CH3); 1,41 (s, 9 H, t-Butyl);
2,10 (s, 6 H, 2 CH3); 2,20 (s, 6 H, 2 CH3). 13C-NMR (100 MHz, Benzol-d6): δ[ppm]: 6,0; 11,9; 14,8; 33,1; 56,6; 101,2; 131,5;
134,2.

I.7 Experimenteller Teil
- 148 -
I.7.3.5 (N-tbutylamido)dimethyl(tetramethyl-�5-cyclopentadienyl)silan-
hafnium(IV)dichlorid
Ansatz:
Ligand-Dilithiumsalz (siehe I.7.3.1): 4 mmol 1,0 g
HfCl4(thf)2: 4 mmol 1,9 g
Toluol (sublimiert): 20 mL
Durchführung:
Der Ligand (1,0 g; 4 mmol) wird in 20 mL Toluol suspendiert. Die Ligand-Suspension wird auf
-30 °C gekühlt. Unter Rühren wird HfCl4(thf)2 portionsweise zugegeben. Es wird vier Tage bei
Raumtemperatur gerührt.
Die Lösung wird vom sehr feinen, weißen Niederschlag abfiltriert. Das Filtrat wird bei -30°C
gelagert. (η5:η1-C5Me4SiMe2NtBu)Hf(IV)Cl2 kristallisiert als farblose Plättchen.
CHN-Analyse: C15H27Cl2HfNSi
Berechnet: C: 36,11 H: 5,46 N: 2,81
Gefunden: C: 36,15 H: 5,48 N: 2,79
NMR: 1H-NMR (400 MHz, Benzol-d6):
δ[ppm]: 0,56 (s, 6 H, 2 Si-CH3); 1,41 (s, 9 H, t-Butyl); 2,10 (s, 6 H, 2 CH3); 2,20 (s, 6 H,
2 CH3). 13C-NMR (100 MHz, Benzol-d6):
δ[ppm]: 6,3; 11,6; 14,4; 33,9; 55,3; 100,8; 130,1; 131,8.

I.7 Experimenteller Teil
- 149 -
I.7.3.6 Kristallographische Daten
Kristalldaten:
Zr Hf
Summenformel C15H27Cl2NSiZr C15H27Cl2HfNSi
Molekulargewicht [g/mol] 411,59 498,86
Kristallgröße [mm] 0,35 x 0,32 x 0,12 0,26 x 0,24 x 0,20
Symmetrie der Einheitszelle Orthorhombisch Orthorhombisch
Raumgruppe P212121 P212121
Zellenlänge (a) [Å] 8,3500(17) 8,3341(7)
Zellenlänge (b) [Å] 13,680(3) 13,6819(12)
Zellenlänge (c) [Å] 16,770(3) 16,801(2)
α [°]α [°]α [°]α [°] 90 90
β [°]β [°]β [°]β [°] 90 90
γ [°]γ [°]γ [°]γ [°] 90 90
Zellvolumen [ų] 1915,6(7) 1915,6(3)
Z 4 4
Berechnete Dichte [mg/m³] 1,427 1,730
Absorptionskoeffizient [mm-1] 0,907 5,779
F(000) 848 976
Datenaufnahme:
Zr Hf
Wellenlänge [Å] λ = 0,71073 λ = 0,71073
T [K] 223 223
2ΘΘΘΘ Bereich [°] 2,43 – 26,01 2,42 − 25,93
hkl-Bereich -10 <= h <=10,
-16 <= k <= 16,
-20 <= l <= 20
-10 <= h <=10,
-16 <= k <= 16,
-20 <= l <= 20
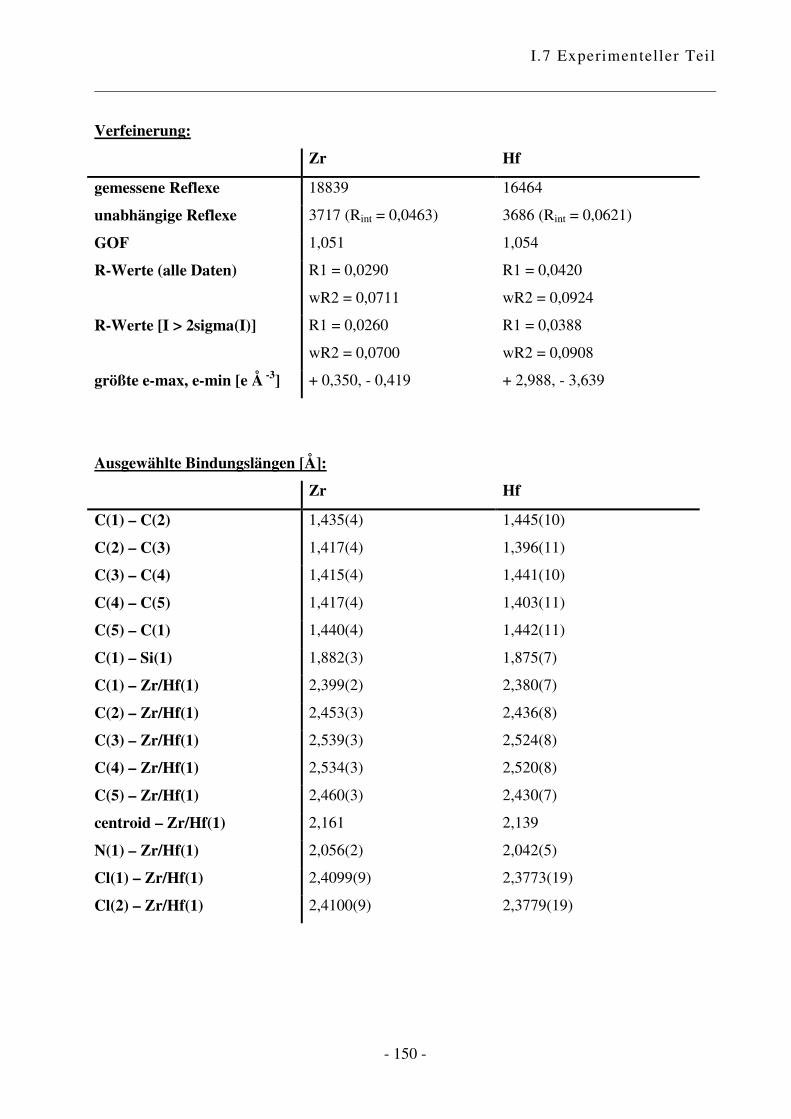
I.7 Experimenteller Teil
- 150 -
Verfeinerung:
Zr Hf
gemessene Reflexe 18839 16464
unabhängige Reflexe 3717 (Rint = 0,0463) 3686 (Rint = 0,0621)
GOF 1,051 1,054
R-Werte (alle Daten) R1 = 0,0290
wR2 = 0,0711
R1 = 0,0420
wR2 = 0,0924
R-Werte [I > 2sigma(I)] R1 = 0,0260
wR2 = 0,0700
R1 = 0,0388
wR2 = 0,0908
größte e-max, e-min [e Å -3] + 0,350, - 0,419 + 2,988, - 3,639
Ausgewählte Bindungslängen [Å]:
Zr Hf
C(1) – C(2) 1,435(4) 1,445(10)
C(2) – C(3) 1,417(4) 1,396(11)
C(3) – C(4) 1,415(4) 1,441(10)
C(4) – C(5) 1,417(4) 1,403(11)
C(5) – C(1) 1,440(4) 1,442(11)
C(1) – Si(1) 1,882(3) 1,875(7)
C(1) – Zr/Hf(1) 2,399(2) 2,380(7)
C(2) – Zr/Hf(1) 2,453(3) 2,436(8)
C(3) – Zr/Hf(1) 2,539(3) 2,524(8)
C(4) – Zr/Hf(1) 2,534(3) 2,520(8)
C(5) – Zr/Hf(1) 2,460(3) 2,430(7)
centroid – Zr/Hf(1) 2,161 2,139
N(1) – Zr/Hf(1) 2,056(2) 2,042(5)
Cl(1) – Zr/Hf(1) 2,4099(9) 2,3773(19)
Cl(2) – Zr/Hf(1) 2,4100(9) 2,3779(19)
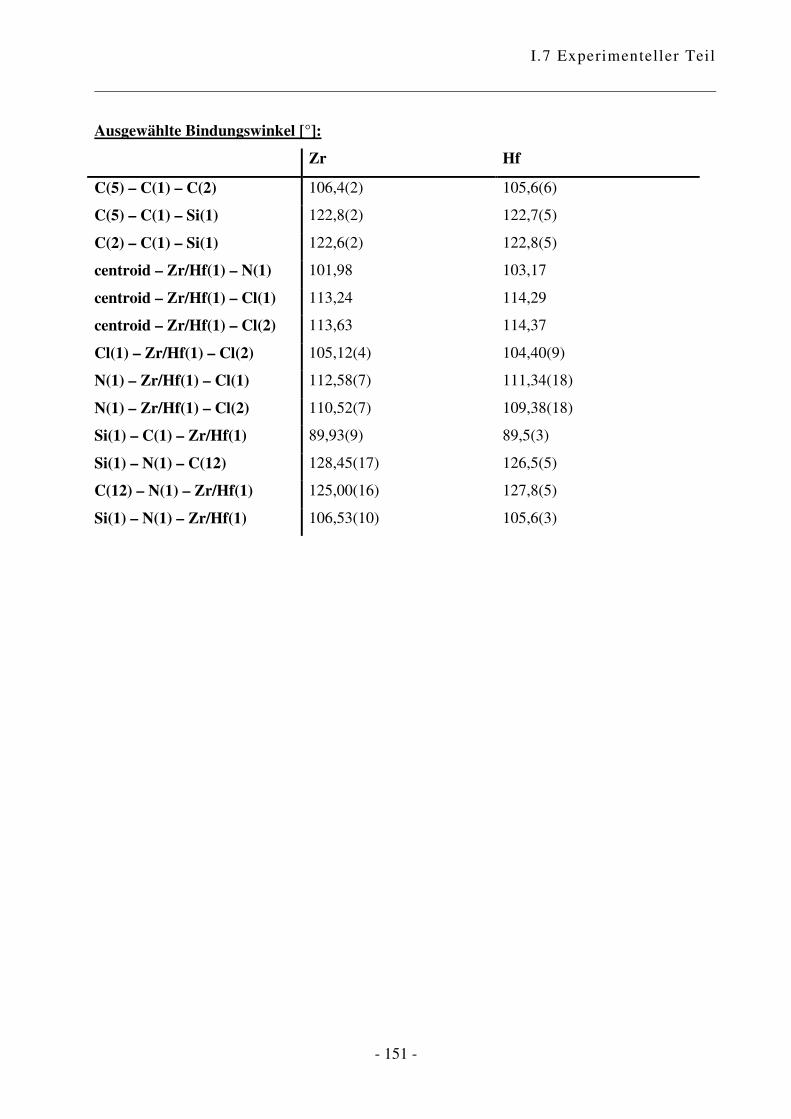
I.7 Experimenteller Teil
- 151 -
Ausgewählte Bindungswinkel [°]:
Zr Hf
C(5) – C(1) – C(2) 106,4(2) 105,6(6)
C(5) – C(1) – Si(1) 122,8(2) 122,7(5)
C(2) – C(1) – Si(1) 122,6(2) 122,8(5)
centroid – Zr/Hf(1) – N(1) 101,98 103,17
centroid – Zr/Hf(1) – Cl(1) 113,24 114,29
centroid – Zr/Hf(1) – Cl(2) 113,63 114,37
Cl(1) – Zr/Hf(1) – Cl(2) 105,12(4) 104,40(9)
N(1) – Zr/Hf(1) – Cl(1) 112,58(7) 111,34(18)
N(1) – Zr/Hf(1) – Cl(2) 110,52(7) 109,38(18)
Si(1) – C(1) – Zr/Hf(1) 89,93(9) 89,5(3)
Si(1) – N(1) – C(12) 128,45(17) 126,5(5)
C(12) – N(1) – Zr/Hf(1) 125,00(16) 127,8(5)
Si(1) – N(1) – Zr/Hf(1) 106,53(10) 105,6(3)

I.7 Experimenteller Teil
- 152 -
I.7.4 Voraktivierung der Komplexe
Wie bereits im Kapitel I.1.4.1 beschrieben, werden die synthetisierten Komplexe vor der
Polymerisationsreaktion durch Tritylborat voraktiviert. Als Prä-Aktivator dient hierbei
Triisobutylaluminium im Verhältnis Al : Metall = 100 : 1.
Ansatz:
HfCp�A-Komplex: 22,5 Cmol 11,2 mg
TiBA, 1,1 M in Toluol: 2,2 mmol 2,0 mL
Toluol: 9,3 mL
Durchführung:
Der Komplex wird in der Glovebox in ein Schlenkrohr vorgelegt. In einem zweiten Schlenkrohr
werden TiBA und Toluol gemischt. Dabei wird die TiBA-Menge gemäß der Komplex-Einwaage
berechnet. Die Toluol-Menge wird so gewählt, dass die entstehende Katalysatorlösung eine
Konzentration von 2 Cmol/mL hat. Das vorherige Mischen ist zwingend notwendig. Bei direkter
Zugabe von Toluol könnte ein Teil des Katalysators durch Restwasser im Toluol hydrolysiert
und in das polymerisationsinaktive Hydroxid überführt werden. Zugabe von unverdünntem
TiBA könnte zu einer heftigen Reaktion führen (zweifache Alkylierung und anschließende
Abspaltung einer Alkyl-Kette, wodurch die aktive Spezies bereits außerhalb des Autoklaven
gebildet wird) und damit unerwünschte Nebenprodukte erzeugen. Durch das Mischen wird das
TiBA verdünnt und Restwasser im Toluol vom Aluminiumalkyl abgefangen. Die Toluol-TiBA-
Lösung wird zum Komplex zugegeben und 1 h bei 60 °C gerührt.

I.8 Literatur
- 153 -
I.8 Literatur
1 Trendbericht Makromolekulare Chemie 2006 in Nachrichten aus der Chemie, 55, März 2007, 306 - 310.
www.gdch.de 2 http://www.bransoneurope.net/it/53456697100c2cc02/index.html 3 Froböse, R.; Jopp, K., Fußball, Fashion, Flachbildschirme. 1 ed.; WILEY-VCH Verlag GmbH & Co. KGaA:
Weinheim, 2006; p 309. 4 Cross, C. F.; Bevan, E. J.; Beadle, C., 1892 (Viscose Syndicate) British Patent 8 700 (1892/1893) 5 Römpp-Lexikon online: http://www.roempp.com/prod/ 6 Herrmann, W. A.; Cornils, B., Angew. Chem. 1997, 109, 1074 - 1095. 7 Aulbach, M.; Küber, F., Chemie in unserer Zeit 1994, 28, (4), 197 - 208. 8 Übersichtsartikel: Britovsek, G. J.; Gibson, V. C.; Wass, D. F., Angew. Chem. 1999, 111, 448 - 468. 9 Wilkinson, G.; Pauson, P. L.; Birmingham, J. M.; Cotton, F. A., J. Am. Chem. Soc. 1953, 75, (4), 1011 - 1012. 10 Wild, F. R. W. P.; Zsolnai, L.; Huttner, G.; Brintzinger, H. H., J. Organomet. Chem. 1982, 232, (3), 233 - 247. 11 Wild, F. R. W. P.; Wasiucionek, M.; Huttner, G.; Brintzinger, H. H., J. Organomet. Chem. 1985, 288, (1), 63 - 67. 12 Ewen, J. A., J. Am. Chem. Soc. 1984, 106, 6355 - 6364. 13 Kaminsky, W.; Külper, K.; Brintzinger, H. H.; Wild, F. R. W. P., Angew. Chem. 1985, 97, 507 - 508. 14 Übersichtsartikel: Brintzinger, H.-H.; Fischer, D.; Mülhaupt, R.; Rieger, B.; Waymouth, R., Angew. Chem. 1995,
107, 1255 - 1283. 15 Ewen, J. A.; Jones, R. L.; Razavi, A.; Ferrara, J. D., J. Am. Chem. Soc. 1988, 110, 6255 - 6256. 16 Shapiro, P. J.; Bunel, E.; Schaefer, W. P.; Bercaw, J. E., Organometallics 1990, 9, 867 - 869. 17 Okuda, J., Chem. Ber. 1990, 123, 1649 - 1651. 18 Stevens, J. C.; Timmers, F. J.; Wilson, D. R.; Schmidt, G. F.; Nickias, P. N.; Rosen, R. K.; Knight, G. W.; Lai, S.-
Y. EP 0 416 815 A2, 1990. 19 Canich, J. A. M.; Turner, H. WO 92/12162, 1992. 20 Braunschweig, H.; Breitling, F. M., Coordination Chemistry Reviews 2006, 250, 2691 - 2720. 21 Sidgwick, N. V., The Electronic Theory of Valency. Oxford Univ Press: 1927; p 310. 22 Übersichtsartikel: Cano, J.; Kunz, K., J. Organomet. Chem. 2007, 692, 4411 - 4423. 23 Alt, H. G.; Reb, A.; Milius, W.; Weis, A., J. Organomet. Chem. 2001, 628, 169 - 182. 24 Riedel, E., Anorganische Chemie. 2 ed.; de Gruyter: Berlin, New York, 1990; p 849. 25 Lide, D. R., Handbook of Chemistry and Physics. 86 ed.; CRC Press: 2005. 26 Heiland, K.; Kaminsky, W., Makromol. Chem. 1992, 193, 601 - 610. 27 Mallin, D. T.; Rausch, M. D.; Chien, J. C. W., Polymer Bulletin 1988, 20, 421 - 425. 28 Koivumäki, J., Polym. Bull. 1995, 34, 413 - 418. 29 Bochmann, M.; Lancaster, S. J., J. Organomet. Chem. 1992, 434, C1- C5. 30 Starck, P.; Lehtinen, C.; Löfgren, B., Die Angewandte Makromolekulare Chemie 1997, 249, 115 - 135. 31 Fan, L.; Harrison, D.; Woo, T. K.; Ziegler, T., Organometallics 1995, 14, 2018 - 2026. 32 Canich, J. A. M. Olefin Polymerization Catalysts. 0 420 436 A1, 1990. 33 Okuda, J.; Amor, F.; Plooy, K. E. du; Eberle, T.; Hultzsch, K. C.; Spaniol, T. P., Polyhedron 1998, 17, (7), 1073 -
1080.

I.8 Literatur
- 154 -
34 Alt, H. G.; Föttinger, K.; Milius, W., J. Organomet. Chem. 1999, 572, 21 - 30. 35 Dias, H. V. R.; Wang, Z.; Bott, S. G., J. Organomet. Chem. 1996, 508, 91 - 99. 36 Juvaste, H.; Pakkanen, T. T.; Iiskola, E. I., J. Organomet. Chem. 2000, 606, 169 - 175. 37 Juvaste, H.; Pakkanen, T. T., Organometallics 2000, 19, 1729 - 1733. 38 Amor, F.; Plooy, K. E. du; Spaniol, T. P.; Okuda, J., J. Organomet. Chem. 1998, 558, 139 - 146. 39 Amor, F.; Spaniol, T. P.; Okuda, J., Organometallics 1997, 16, 4765 - 4767. 40 Amor, F.; Butt, A.; Plooy, K. E. du; Spaniol, T. P.; Okuda, J., Organometallics 1998, 17, 5836 - 5849. 41 Janiak, C.; Klapötke, T. M.; Meyer, H.-J., Moderne Anorganische Chemie. 2 ed.; Walter de Gruyter GmbH &
Co.: Berlin, 2003; p 801. 42 Sinn, H.; Kaminsky, W.; Vollmer, H.-J.; Woldt, R., Angew. Chem. 1980, 92, (5), 396 - 402. 43 Sinn, H.; Kaminsky, W., Adv. Organomet. Chem. 1980, 18, 99 - 149. 44 Reichert, K. H.; Meyer, K. R., Die Makromolekulare Chemie 1973, 169, 163 - 176. 45 Chen, E. Y.-X.; Marks, T. J., Chem. Rev. 2000, 100, 1391 - 1434. 46 Rieger, B. Vorlesungsskript zur Vorlesung „Homogene Katalyse: Polymerisationskatalyse“ Uni Ulm, 2003. 47 Rieger, B.; Troll, C.; Preuschen, J., Macromolecules 2002, 35, 5742 - 5743. 48 Diesner, T.; Troll, C.; Rieger, B., Topics in Organometallic Chemistry 2009, 26 (Metal Catalysts in Olefin
Polymerization), 47-63. 49 Gröner, S. Neue Hafnium(IV)-Katalysatoren für die Ethen/Dien-Copolymerisation. Universität Ulm, Ulm, 2004. 50 Cossée, P., J. Catal. 1964, 3, 80 - 88. 51 Arlman, E. J.; Cossée, P., J. Catal. 1964, 3, 99 - 104. 52 Ruchatz, D.; Fink, G., Macromolecules 1998, 31, 4684 - 4686. 53 Woo, T. K.; Fan, L.; Ziegler, T., Organometallics 1994, 13, 2252 - 2261. 54 Shapiro, P. J.; D.Cotter, W.; Schaefer, W. P.; Labinger, J. A.; Bercaw, J. E., J. Am. Chem. Soc. 1994, 116, 4623 -
4640. 55 McKnight, A. L.; Waymouth, R. M., Chem. Rev. 1998, 98, 2587 - 2598. 56 Carpenetti, D. W.; Kloppenburg, L.; Kupec, J. T.; Petersen, J. L., Organomet. 1996, 15, 1572 - 1581. 57 Manzer, L. E., Inorg. Synth. 1982, 21, 135 - 137. 58 Okuda, J.; Schattenmann, F. J.; Wocadlo, S.; Massa, W., Organometallics 1995, 14, 789 - 795. 59 Kloppenburg, L.; Dissertation, Universität Münster, 1997. 60 King, W. A.; Bella, S. Di; Lanza, G.; Khan, K.; Duncalf, D. J.; Cloke, F. G. N.; Fragala, I. L.; Marks, T. J., J. Am.
Chem. Soc. 1996, 118, 627 - 635. 61 Lambert, J. B.; Lin, L.; Keinan, S., Org. Biomol. Chem. 2003, 1, 2559 - 2565. 62 Hattori, H., Chem. Rev. 1995, 95, 537 - 558. 63 Mizuno, N.; Misono, M., Chem. Rev. 1998, 98, 199 - 217. 64 Becker, H. G.; Berger, W.; Domschke, G.; Fanghänel, E.; Fischer, M.; Gentz, F.; Gewald, K.; Gluch, R.; Mayer,
R.; Müller, K.; Pavel, D.; Schmidt, H.; Schollberg, K.; Schwetlick, K.; Seiler, E.; Zeppenfeld, G., Organikum. 20
ed.; Wiley-VCH: Weinheim, 1999; p 793. 65 Langjährige, unveröffentlichte Erfahrungswerte der Arbeitsgruppe um Prof. Rieger. 66 Alt, H. G.; Reb, A.; Milius, W.; Weis, A., J. Organomet. Chem. 2001, 628, 169 - 182.

I.8 Literatur
- 155 -
67 Übersicht: Francuskiewicz, F., Polymer Fractionation.; Springer Verlag Berlin and Heidelberg GmbH & Co. KG:
1994; p 225. 68 Stevens, J. C., Stud. Surf. Catal. 1996, 101, 11 - 20. 69 Woo, T. K.; Margl, P. M.; Ziegler, T., Organometallics. 1997, 16, 3454 - 3468. 70 Li, L.; Metz, M. V.; Li, H.; Chen, M.-C.; Marks, T. J.; Liable-Sands, L.; Rheingold, A. L., J. Am. Chem. Soc.
2002, 124, 12725 - 12741. 71 Yan, D.; Wang, W.-J.; Zhu, S., Polymer 1999, 40, 1737 - 1744. 72 Lai, S.-Y.; Wilson, J. R.; Knight, G. W.; Stevens, J. C.; Chum, P.-W. S., US 5 272 236, 1993. 73 Thorshaug, K.; Mendichi, R.; Boggioni, L.; Tritto, I.; Trinkle, S.; Friedrich, C.; Mülhaupt, R., Macromolecules
2002, 35, 2903 - 2911. 74 Liu, W.; III, D. G. Ray; Rinaldi, P. L., Macromolecules 1999, 32, 3817 - 3819. 75 Axelson, D. E.; Levy, G. C.; Mandelkern, L., Macromolecules 1979, 12, 41 - 52. 76 Meinhard, D.; Wegner, M.; Kipiani, G.; Hearley, A.; Reuter, P.; Fischer, S.; Marti, O.; Rieger, B., J. Am. Chem.
Soc. 2007, 129, 9182 - 9191. 77 Galland, G. B.; Silva, L. P. Da; Dias, M. L.; Crossetti, G. L.; Ziglio, C. M.; Filgueiras, C. A. L., J. Polym. Sci.
Part A: Polym. Chem. 2004, 42, 2171 - 2178. 78 Tieke, B., Makromolekulare Chemie. 2 ed.; WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim: Weinheim,
2005; p 368. 79 Elias, H.-G., Makromoleküle Band 2: Physikalische Strukturen und Eigenschaften. 6 ed.; Wiley-VCH: Weinheim,
2001; p 673. 80 Alt, H. G.; Föttinger, K.; Milius, W., J. Organomet. Chem. 1999, 572, 21 - 30. 81 Brant, P.; Canich, J. A. M. WO 93/12151, 1993. 82 Shiomura, T.; Asanuma, T.; Inoue, N., Macromol. Rapid Commun. 1996, 17, 9 - 14. 83 Atkins, P. W., Physikalische Chemie. 2 ed.; VCH Verlagsgesellschaft mbH: Weinheim, 1996; p 1106. 84 Carman, C. J.; Harrington, R. A.; Wilkes, C. E., Macromolecules 1977, 10, (3), 536 - 544. 85 Cheng, H. N., Macromolecules 1984, 17, 1950 - 1955. 86 Wang, W.-J.; Zhu, S., Macromolecules 2000, 33, 1157 - 1162. 87 Übersichtsartikel: Cherdron, H.; Brekner, M.-J.; Osan, F., Angew. Makromol. Chem. 1994, 223, 121 - 133. 88 Kaminsky, W.; Beulich, I.; Arndt-Rosenau, M., Macromol. Symp. 2001, 173, 211 - 225. 89 www.ticona.com 90 Ruchatz, D.; Fink, G., Macromolecules 1998, 31, 4669 - 4673. 91 Ruchatz, D.; Fink, G., Macromolecules 1998, 31, 4674 - 4680. 92 Ruchatz, D.; Fink, G., Macromolecules 1998, 31, 4681 - 4683. 93 Kaminsky, W.; Noll, A., Polym. Bull. 1993, 31, 175 - 182. 94 Kaminsky, W.; Engehausen, R.; Kopf, J., Angew. Chem. 1995, 107, 2469 - 2471. 95 Arndt, M.; Engehausen, R.; Kaminsky, W.; Zoumis, K., J. Mol. Catal. A: Chem. 1995, 101, 171 - 178. 96 Kaminsky, W.; Tran, P.-D.; Werner, R., Macromol. Symp. 2004, 213, 101 - 108. 97 McKnight, A. L.; Waymouth, R. M., Macromolecules 1999, 32, 2816 - 2825. 98 Tran, P.-D.; Dissertation, Universität Hamburg, 2003. 99 Harrington, B. A.; Crowther, D. J., J. Mol. Catal. A: Chem. 1998, 128, 79 - 84.

I.8 Literatur
- 156 -
100 Harrington, B. A. WO 96/40806, 1996. 101 Tritto, I.; Boggioni, L.; Sacchi, M. C.; Locatelli, P.; Ferro, D. R., Macromol. Symp. 2004, 213, 109 - 121. 102 Tritto, I.; Marestin, C.; Boggioni, L.; Zetta, L.; Provasoli, A.; Ferro, D. R., Macromolecules 2000, 33, 8931 -
8944. 103 Streitwieser, A.; Heathcock, C. H., Organische Chemie. ed.; Verlag Chemie: Weinheim, 1980; p 1489. 104 Po, R.; Santi, R.; Cardaci, M. A., J. Polym. Sci. Part A: Polym. Chem. 2000, 38, 3004 - 3009. 105 Stücklen, H.; Thayer, H.; Willis, P., J. Am. Chem. Soc. 1940, 62, 1717 - 1719. 106 Lefebre, G.; Dawans, F., J. Polym. Sci.: Part A 1964, 2, 3277 - 3295. 107 Frey, D. A.; Hasegawa, M.; Marvel, C. S., J. Polym. Sci.: Part A 1963, 1, 2057 - 2065. 108 Cassidy, P. E.; Marvel, C. S.; Ray, S., J. Polym. Sci. Part A: Polym. Chem. 1965, 3, 1553 - 1565. 109 Marvel, C. S.; Hartzell, G. E., J. Am. Chem. Soc. 1959, 81, 448 - 452. 110 Natori, I., Macromolecules 1997, 30, 3696 - 3697. 111 Natori, I.; Inoue, S., Macromolecules 1998, 31, 4687 - 4694. 112 Hong, K.; Mays, J. W., Macromolecules 2001, 34, 782 - 786. 113 Williamson, D. T.; Elman, J. F.; Madison, P. H.; Pasquale, A.J.; Long, T. E., Macromolecules 2001, 34, 2108 -
2114. 114 Hong, K.; Wan, Y.; Mays, J. W., Macromolecules 2001, 34, 2482 - 2487. 115 Hong, K.; Mays, J.W., Macromolecules 2001, 34, 3540 - 3547. 116 Tsoukatos, T.; Avgeropoulos, A.; Hadjichristidis, N.; Hong, K.; Mays, J. W., Macromolecules 2002, 35, 7928 -
7935. 117 Natori, I.; Imaizumi, K., Macromol. Symp. 2000, 157, 143 - 150. 118 Heiser, D. E.; Okuda, J.; Gambarotta, S.; Mülhaupt, R., Macromol. Chem. Phys. 2005, 206, 195 - 202. 119 Übersichtsartikel zu PPP: Kovacic, P.; Jones, M. B., Chem. Rev. 1987, 87, 357 - 379. 120 Gin, D. L.; Conticello, V.t P.; Grubbs, R. H., J. Am. Chem. Soc. 1994, 116, 10507 - 10519. 121 Natori, I.; Imaizumi, K.; Yamagishi, H.; Kazunori, M., J. Polym. Sci. Part B: Polym. Phys. 1998, 36, 1657 -
1668. 122 Po, R.; Santi, R.; Cardaci, M. A., J. Polym. Sci. Part A: Polym. Chem. 2000, 38, 3004 - 3009. 123 Naga, N., J. Polym. Sci. Part A: Polym. Chem. 2005, 43, 1285 - 1291. 124 Kuchling, H., Taschenbuch der Physik. 16. ed.; Fachbuchverlag Leipzig im Carl Hanser Verlag: 1999, p 708. 125 Skript zum Praktikum “Quantitative Analyse” Universität Ulm, Abteilung Analytische Chemie und
Umweltchemie, Dr. U. Reuter. 126 Beagley, P.; Davies, P.; Adams, H.; White, C., Can. J. Chem. 2001, 79, 731 - 741.

II Auf dem Weg zu chiralen, hyperverzweigten
Polyglycerolen als Liganden für enantioselektive
Katalysen

Inhaltsverzeichnis
- 157 -
Abkürzungen .......................................................................................................................... - 159 -�
II� Auf dem Weg zu chiralen, hyperverzweigten Polyglycerolen als Liganden für
enantioselektive Katalysen ..................................................................................................... - 160 -�
II.1� Einleitung ............................................................................................................... - 160 -�
II.1.1� Chirale Moleküle .............................................................................................. - 160 -�
II.1.2� Dendritische Polymere ..................................................................................... - 162 -�
II.2� Grundlagen ............................................................................................................. - 164 -�
II.2.1� Synthese hyperverzweigter Polymere .............................................................. - 164 -�
II.2.2� Nebenreaktionen ............................................................................................... - 167 -�
II.2.3� Aufgabenstellung ............................................................................................. - 168 -�
II.3� Ergebnisdiskussion ................................................................................................. - 169 -�
II.3.1� Auswahl geeigneter Starter-Moleküle .............................................................. - 169 -�
II.3.1.1� Hydroxyphenylphosphane als Starter ..................................................... - 172 -�
II.3.1.2� Diphenylphosphin als Starter ................................................................. - 175 -�
II.3.2� Auf dem Weg zu neuen Komplexen ................................................................ - 177 -�
II.3.3� Charakterisierung und Synthese hyperverzweigter Polyglycidole .................. - 185 -�
II.3.3.1� Polymeranalytik ..................................................................................... - 185 -�
II.3.3.2� Einfluss verschiedener Reaktionsparameter ........................................... - 190 -�
II.3.3.3� Kontrollierte Monomerzugabe zur Steuerung der Produkte .................. - 191 -�
II.3.3.4� Aufarbeitung der Roh-Polymere ............................................................ - 193 -�
II.4� Zusammenfassung .................................................................................................. - 196 -�
II.5� Summary ................................................................................................................ - 198 -�
II.6� Experimenteller Teil ............................................................................................... - 200 -�
II.6.1� Arbeitstechniken und –materialien ................................................................... - 200 -�
II.6.2� Polymercharakterisierung ................................................................................. - 200 -�
II.6.3� Synthese hyperverzweigter Polyglycidole ....................................................... - 201 -�
II.6.3.1� Polymerisation von Glycidol mit Phenol als Starter .............................. - 201 -�
II.6.3.2� Polymerisation von Glycidol mit Natriumphenolat als Starter .............. - 202 -�
II.6.3.3� Polymerisation von Glycidol mit Triphenylphosphin als Starter ........... - 203 -�
II.6.3.4� Polymerisation von Glycidol mit Diphenylphosphin als Starter ............ - 203 -�
II.6.4� Synthese von Para-hydroxyphenyl-phosphanen .............................................. - 204 -�
II.6.4.1� Synthese von Diphenyl-[p-methoxy-phenyl]-phosphan ........................ - 204 -�

Inhaltsverzeichnis
- 158 -
II.6.4.2� Synthese von Diphenyl-[p-hydroxy-phenyl]-phosphan-hydrobromid ... - 205 -�
II.6.4.3� Synthese von Diphenyl-[p-hydroxy-phenyl]-phosphan ......................... - 206 -�
II.6.4.4� Synthese von Tris-[p-methoxy-phenyl]-phosphan ................................. - 207 -�
II.6.4.5� Synthese von Tris-[p-hydroxy-phenyl]-phosphan-hydrobromid ........... - 208 -�
II.6.4.6� Synthese von Tris-[p-hydroxy-phenyl]-phosphan ................................. - 209 -�
II.6.5� Synthese der Phosphan-Komplexe ................................................................... - 210 -�
II.6.5.1� Synthese von Bis[Tris-(p-hydroxyphenyl)phosphin)]Palladium(II)chlorid
………………………………………………………………………….- 210 -�
II.7� Literatur .................................................................................................................. - 211 -�

Abkürzungen
- 159 -
ABKÜRZUNGEN
DB Verzweigungsgrad (degree of branching)
COD 1,5-Cyclooktadien
GPC Gel-Permeations-Chromatographie
HOAc Essigsäure
IR Infrarot
MALDI-TOF Matrix assisted laser desorption ionization time of flight
Mw Gewichtsmittel des Molekulargewichts
NaOH Natriumhydroxid
PD Polydispersität
Ph Phenyl
pKa negativ dekadischer Logarithmus des Ka-Werts
(Säuredissoziationskonstante)
pKb negativ dekadischer Logarithmus des Kb-Werts
(Basendissoziationskonstante)
PPh3 Triphenylphosphan / Triphenylphosphin
ROMBP ring-opening multibranching polymerization (Ring-Öffnungs-Multi-
Verzweigungs-Polymerisation)
RT Raumtemperatur
THF Tetrahydrofuran
TMNO Trimethyl-N-Oxid
VE Valenzelektronen

II.1 Einleitung
- 160 -
II AUF DEM WEG ZU CHIRALEN,
HYPERVERZWEIGTEN POLYGLYCEROLEN ALS
LIGANDEN FÜR ENANTIOSELEKTIVE KATALYSEN
II.1 Einleitung
II.1.1 Chirale Moleküle
“We can expect that they [monophosphanes] will play a role of increasing importance in many
aspects of organometallic catalysis. We hope that this review will encourage practitioners of
asymmetric catalysis to consider the potential of chiral monodentate phosphines and to
investigate this area which has been quite neglected till now.”1
Chirale Moleküle spielen in der Natur eine große Rolle. Die steigende Nachfrage nach
enantiomerenreinen Pharmazeutika, Agrochemikalien, Geruchsstoffen und anderen
Feinchemikalien hat das Feld der asymmetrischen Katalyse vorangetrieben.2 Die Aktualität
dieses Themas zeigt sich auch im Nobelpreis des Jahres 2001 in Chemie, mit dem S. Knowles
und R. Noyori „für ihre Arbeiten über chiral katalysierende Hydrierungsreaktionen“ sowie
K. B. Sharpless „für seine Arbeiten über chiral katalysierende Oxidationsreaktionen“
ausgezeichnet wurden.3
Für enantioselektive Synthesen wird ein chiraler Katalysator benötigt, der z. B. ein prochirales
Olefin bevorzugt von einer Seite koordiniert. Die Erkennung und Einstellung der
Vorzugskonformation geschieht meistens durch chirale Liganden, die zwischen Ligand und
Substrat ein „chirales Loch“ in der Koordinationssphäre des Metalls schaffen.4 Dazu werden
häufig Phosphan-Liganden eingesetzt.a Der Ort, von dem die chirale Information ausgeht, kann
sehr unterschiedlich sein. In Abb. 1 sind einige Beispiele wichtiger Phosphan-Liganden gezeigt.
Beim Molekül Dipamp geht die Chiralität vom Phosphor aus, bei Chiraphos oder Norphos ist das
chirale C-Atom direkt an den Phosphor gebunden, bei Diop befindet sich die Information weiter
entfernt in der Kohlenstoff-Brücke und bei Binap ist die Chiralität durch Einfrieren der
Konformation des Moleküls verankert.
a Phosphane sind aufgrund ihrer �- und �-Donator- sowie ihrer �-Akzeptor-Eigenschaften besonders für die
Komplexierung später Übergangsmetalle geeignet.
II.1 Einleitung
- 161 -
Abb. 1: Chirale Phosphanliganden.2,4
In dieser Arbeit sollen neue chirale Liganden synthetisiert werden, die sich von den klaren
Strukturen ihrer Vorgänger unterscheiden. Diese neuen Liganden sollen – nach dem Vorbild der
Proteine – eine chirale „Wolke“ um das katalytisch aktive Metallzentrum bilden (vgl. Abb. 2).
Der Aufbau dieser chiralen Umgebung soll durch hochverzweigte Polymere geschehen.
Abb. 2: Metallkomplex, bei dem das Zentralmetall wie in einer chiralen Wolke eingebettet ist.

II.1 Einleitung
- 162 -
II.1.2 Dendritische Polymere
Hochverzweigte oder dendritische Polymere werden in die zwei großen Klassen der Dendrimere
und der hyperverzweigten Polymere unterteilt. Dendrimere (von griech.: dendron = Baum) sind
monodisperse, perfekt verzweigte Makromoleküle, die schrittweise aufgebaut werden (vgl. Abb.
3). Durch Verknüpfung von jeweils zwei oder mehr Monomeren mit dem bereits gebundenen
Monomer wächst mit jedem Schritt die Zahl der Endgruppen exponentiell an, so dass am Ende
eine kugelförmige Struktur entsteht.5
Abb. 3: Schematische Darstellung eines Dendrimers.
Dendrimere faszinierten die Forscher zunächst aufgrund ihrer ästhetischen Schönheit und hohen
Symmetrie.6 Ihre typische Architektur wirkt sich auf die physikalischen Eigenschaften aus.7
Diese werden in erster Linie bestimmt durch die globulare Struktur und die sehr hohe Zahl an
funktionellen Gruppen, die beispielsweise zu der für Makromoleküle sehr guten Löslichkeit
führen. Eine weitere Besonderheit zeigt sich bei der Viskosität. An Stelle eines rapiden Anstiegs
der intrinsischen Viskosität mit zunehmendem Molekulargewicht – wie für die meisten Polymere
beobachtet – erreicht die Viskosität ein Maximum, welches bereits bei relativ geringer
Generationenzahl erreicht wird. Ein ganz großer Nachteil der perfekten Makromoleküle ist
jedoch die aufwändige, mehrstufige Synthese, die in jeder Zwischenstufe Aufreinigungsschritte
erfordert.8 Gerade aber in Spezialanwendungen wie z. B. der Diagnostik, Pharmazeutik,
Gentechnik, Sensorik und Katalysatortechnik, in denen nur geringste Materialmengen bereits
essentiell für die Wirkung sind, haben diese Moleküle ihr Potenzial gezeigt und sind daher eher
als Wirkstoffe und weniger als klassische Polymere zu bezeichnen.9

II.1 Einleitung
- 163 -
Hyperverzweigte Polymere sind den Dendrimeren sehr ähnlich. Es handelt sich ebenfalls um
hoch verzweigte jedoch „fehlerhafte“ Moleküle mit unregelmäßigen Strukturen (vgl. Abb. 4).
Ihre einzigartigen Eigenschaften sind wiederum auf die kugelförmige Struktur sowie die große
Anzahl an terminalen Endgruppen zurückzuführen. Im Gegensatz zu den Dendrimeren zeigen sie
jedoch auch wesentliche Faktoren von konventionellen Polymeren wie zum Beispiel Molekular-
gewichtsverteilungen oder Isomere.10 Aufgrund Ihrer Ähnlichkeit werden hyperverzweigte
Polymere als wirtschaftlicher Ersatz für Dendrimere diskutiert. Ihr großer Vorteil liegt in der
bedeutend einfacheren Synthese, die bereits als Einstufen-Reaktion gelingen kann.11,12
Abb. 4: Schematischer Vergleich eines perfekten Dendrimers (links) und eines hyperverzweigten
Polymers (rechts) mit zentralem Kern und funktionellen Randgruppen.

II.2 Grundlagen
- 164 -
II.2 Grundlagen
II.2.1 Synthese hyperverzweigter Polymere
Bei der Synthese hyperverzweigter Polymere wird häufig auf ein altbekanntes Konzept
zurückgegriffen. Bereits im Jahre 1952 beschrieb Flory in der Theorie die Synthese von
verzweigten Polymeren durch Polykondensation von ABx-Molekülen.13 Nach Flory entstehen
hoch verzweigte Polymere, wenn bei der Polymerisation von ABx-Monomeren mit x>1 die
funktionelle Gruppe A ausschließlich mit der funktionellen Gruppe B reagiert (vgl. Abb. 5).
Weitere wichtige Voraussetzungen für eine erfolgreiche Synthese sind die Abwesenheit von
Neben- und intramolekularen Zyklisierungsreaktionen sowie die identische Aktivität der
B-Funktionalitäten.14
Abb. 5: Hyperverzweigtes Polymer, aufgebaut aus einem AB2-Monomer.
Ein äußerst interessantes Molekül, das für den Aufbau eines hyperverzweigten Polymers
geeignet ist, ist 2,3-Epoxy-1-propanol – auch Glycidol oder Glycerol genannt (vgl. Abb. 6).
Glycidol ist ein chirales latentes AB2-Monomer. Die Funktionalität A entspricht der positiv
polarisierten und damit elektrophilen CH2-Gruppe des Rings, die beiden nukleophilen
Sauerstoff-Atome stellen die B-Funktionalität dar.
Abb. 6: Glycidol – ein latentes AB2-Monomer.

II.2 Grundlagen
- 165 -
Damit aus Glycidol ein echtes AB2-Monomer wird, muss die zweite Sauerstoff-Funktionalität
zugänglich werden, indem der Ring geöffnet wird. Treibende Kraft für diese Reaktion ist die
Reduktion der Ringspannung. Dies kann auf unterschiedliche Arten geschehen. Bei der
kationischen Ring-Öffnungs-Polymerisation von Glycidol greift ein Elektrophil am Sauerstoff
des Oxiran-Rings an (vgl. Abb. 7). Als Initiatoren können sowohl Brönstedt-Säuren (wie z. B.
Salzsäure) als auch Lewis-Säuren (z. B. Bortrifluorid15, Aluminiumtrichlorid) verwendet
werden.16,17 Es entsteht dabei ein sp2-hybridisiertes planares Carbokation, das achiral ist. Die
wertvolle chirale Information des Monomers geht folglich bei der kationischen Polymerisation
verloren.
Abb. 7: Kationische Ringöffnung von Glycidol.18
Eine wesentlich elegantere Methode zur Ringöffnung, bei der die chirale Information erhalten
bleibt, ist die anionische Ring-Öffnungs-Polymerisation. Ein Nukleophil greift regioselektiv am
sterisch weniger gehinderten, positiv polarisierten C-Atom der CH2-Gruppe des Oxirans an (vgl.
Abb. 8).
Abb. 8: Anionische Ringöffnung von Glycidol.
Diese Reaktion ist eine SN2-Substitution mit einem Alkoxid als Abgangsgruppe. Das Sauerstoff-
Atom des Rings stellt wiederum ein Nukleophil dar und kann entweder ein weiteres Monomer
II.2 Grundlagen
- 166 -
angreifen oder in einer intra- oder intermolekularen Reaktion ein H-Atom einer anderen
Alkohol-Funktionalität abstrahieren. Dadurch, dass bei diesem Mechanismus das chirale C-Atom
erhalten bleibt, ist der Aufbau von chiralen, hyperverzweigten Polymeren möglich (vgl.
Abb. 9).19
Abb. 9: Chirales, hyperverzweigtes Polyglycidol.

II.2 Grundlagen
- 167 -
II.2.2 Nebenreaktionen
Anionische Polymerisationen sind äußerst empfindliche Systeme. Damit die Initiator-Moleküle
nicht zu unerwünschten Reaktionen führen, müssen zahlreiche Nebenreaktionen berücksichtigt
werden. Zur Vermeidung spontaner Abbruchreaktionen, ist es notwendig, protische
Lösungsmittel (wie z. B. Wasser), Sauerstoff und Kohlendioxid von der Reaktionsmatrix
fernzuhalten.20 Darüber hinaus gibt es weitere Aspekte, die für die Versuchsplanung wichtig
sind. Die Ring-Öffnungs-Polymerisation von Glycidol verläuft stark exotherm. Wird die
Reaktionswärme nicht schnell genug abgeführt, steigt die Reaktionsgeschwindigkeit mit
zunehmender Temperatur weiter an und die Reaktion verläuft unkontrolliert, sie „geht durch“.
Bei hinreichend hoher Temperatur der Reaktionslösung kann die Kondensation zweier Glycidol-
Moleküle unter Wasserabspaltung erfolgen (vgl. Abb. 10). Dies hat nicht nur den Verlust von
Monomer und das Erzeugen niedermolekularer Nebenprodukte zur Folge. Das freigesetzte
Wasser-Molekül reagiert mit wachsenden Polymerketten und führt somit zum Kettenabbruch.
Aus diesem Grund müssen derartige Nebenreaktionen zwingend verhindert werden.
Abb. 10: Kondensation zweier Glycidol-Moleküle unter Abspaltung von Wasser.
Eine andere unerwünschte Reaktion ist die Autopolymerisation von Glycidol. Bereits geringe
Mengen einer Base sind ausreichend, um ein hochverzweigtes Polymer aufzubauen (vgl.
Abb. 11). Durch Autopolymerisation entstandene hyperverzweigte Makromoleküle weisen
keinen funktionellen Kern auf und können damit nicht in beabsichtigter Weise als Liganden
dienen.
Abb. 11: Autopolymerisation von Glycidol.

II.2 Grundlagen
- 168 -
II.2.3 Aufgabenstellung
Erinnern wir uns an die Vision in der Einleitung vom Metall-Atom, eingebettet in einer chiralen
Wolke zurück. Diese chirale Umgebung soll durch hyperverzweigtes, chirales Polyglycidol
aufgebaut werden (vgl. Abb. 12). Damit die Vision Realität werden kann, muss eine geeignete
Kern-Gruppe gefunden werden, die zum einen an das Metall-Zentrum koordinieren kann, zum
anderen die anionische Ring-Öffnungs-Polymerisation von Glycidol ermöglicht.
Abb. 12: Metallkomplex, dessen Liganden aus chiralem Polyglycidol bestehen.
Ziel dieser Arbeit ist die Synthese eines hyperverzweigten Polyglycidols mit funktionellem Kern
sowie dessen Umsetzung zu einem neuartigen Komplex.

II.3 Ergebnisdiskussion
- 169 -
II.3 Ergebnisdiskussion
II.3.1 Auswahl geeigneter Starter-Moleküle
Für die Herstellung von Polymeren mit funktionalisiertem Kern über anionische Ring-Öffnungs-
Multi-Verzweigungs-Polymerisation (ROMBP) ergibt sich die Schwierigkeit, dass der
verwendete Initiator ein sehr starkes Nukleophil und gleichzeitig eine schwache Base sein muss,
gleichwohl auch keine starke Säure sein darf. Während zahlreiche nicht-nukleophile Basen wie
z. B. bizyklische Amidine oder sperrige Lithiumdialkylamide bekannt sind, ist die Suche nach
einem nicht-basischen Nukleophil eine wesentlich größere Herausforderung.
Bei allen Diskussionen um starke oder schwache Säuren und Basen darf nicht vergessen werden,
dass sich die gebräuchlichen pKa- und pKb-Werte in aller Regel auf wässrige Systeme beziehen.
Für nichtwässrige Systeme – wie die Polymerisation von Glycidol – können die Tabellenwerke
lediglich Hinweise geben, absolute Voraussagen sind jedoch nicht möglich. Um einen Eindruck
davon zu erhalten, wie stark basisch ein Molekül sein muss, damit es zur Deprotonierung und
anschließenden Autopolymerisation von Glycidol kommt, werden zunächst Säurestärken
verschiedener Alkohole betrachtet (vgl. Abb. 13).
Abb. 13: Säurestärke von Alkoholen im Vergleich.18
Die pKa-Werte für die meisten aliphatischen Alkohole liegen zwischen 14 und 16. Nach der
Gleichung pKa + pKb = 14 wird (in wässrigen Systemen!) eine Base mit einem pKb-Wert kleiner
Null benötigt, damit die OH-Gruppe des Glycidols deprotoniert wird. Moleküle, die diese
Voraussetzung erfüllen, zählen zu den sehr starken Basen wie beispielsweise HO- oder H2N-.
Nach dieser Rechnung ist die Autopolymerisation durch basische Nukleophile daher als
grundsätzlich möglich aber nicht sehr wahrscheinlich einzustufen.

II.3 Ergebnisdiskussion
- 170 -
Eine Stoffklasse, die sowohl für ihre Nukleophilie als auch für ihre Komplexbildung bekannt ist,
sind die Amine (vgl. Abb. 14). Das freie Elektronenpaar am Stickstoff erlaubt den Angriff an der
elektrophilen CH2-Gruppe des Glycidol-Rings. Setzt man hierfür tertiäre Amine wie
beispielsweise Triethylamin ein, so entsteht ein quartäres Ammoniumion. Quartäre
Ammoniumionen können allerdings nicht mehr als komplexbildende Liganden fungieren. Aus
diesem Grund werden bevorzugt sekundäre Amine wie Diethyl- oder Diisopropylamin
eingesetzt.
Abb. 14: Verschiedene Amine als mögliche Starter für die Polymerisation von Glycidol.
Die Eignung der sekundären Amine als funktioneller Kern bleibt fraglich. In Abb. 15 ist die
Reaktion von Diethylamin mit Glycidol gezeigt. Nach dem Initiierungsschritt entsteht ein
quartäres Ammoniumion, welches ein H+-Ion sowohl in intra- als auch intermolekularen
Reaktionen an ein negativ geladenes Sauerstoffion abgeben kann. Diese Protonenabgabe führt
allerdings – unabhängig davon, zu welchem Zeitpunkt der Polymerisationsreaktion sie geschieht
– zur Absättigung einer wachsenden Kette und damit zur klassischen Abbruchreaktion In dem
Moment, in dem das freie Elektronenpaar des Stickstoffs wieder zur Verfügung steht (vgl. blau
gezeichnete Strukturformel in Abb. 15), kann theoretisch eine weitere Anlagerung an Glycidol
erfolgen. Das auf diese Art entstandene quartäre Ammoniumion kommt wiederum als
komplexbildender Ligand nicht mehr in Frage. Damit Amine funktionelle Kerne für
hyperverzweigte Polymere darstellen können, müsste idealerweise das Proton bis zum geplanten
Ende der Polymerisationsreaktion schützend am Stickstoff verbleiben und erst durch die
Aufarbeitung entfernt werden. Experimentelle Untersuchungen werden zeigen müssen, ob dies
so möglich ist.
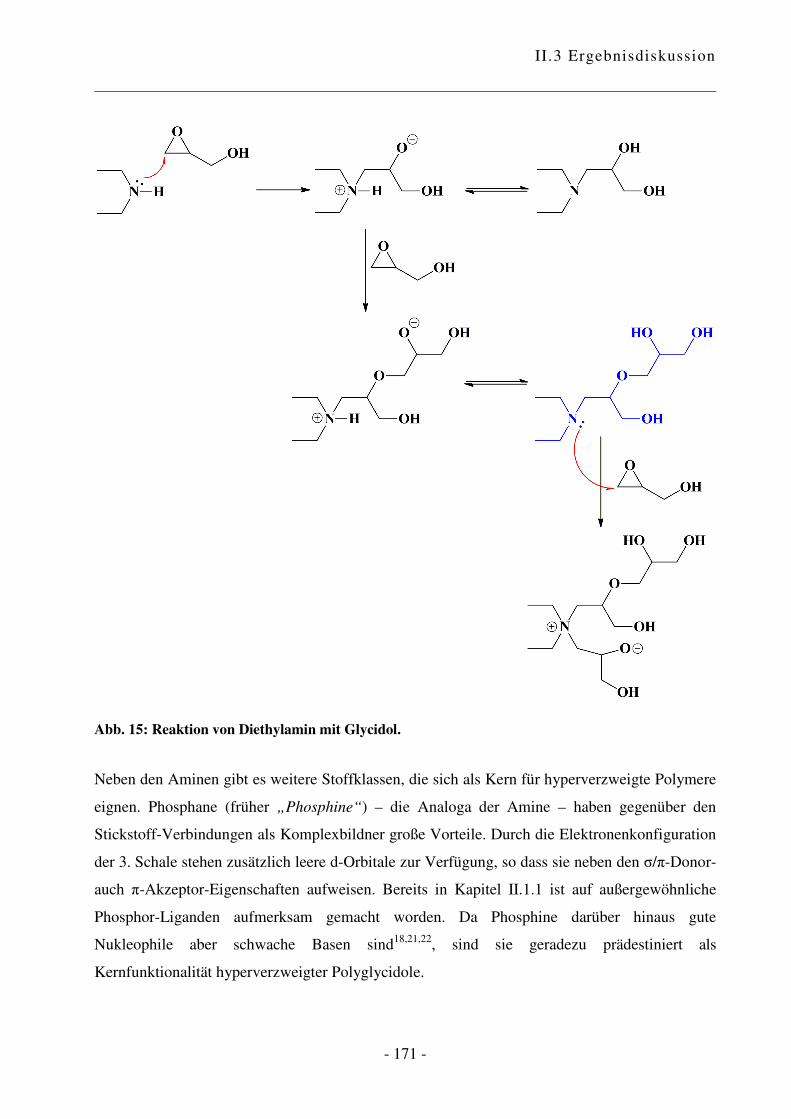
II.3 Ergebnisdiskussion
- 171 -
Abb. 15: Reaktion von Diethylamin mit Glycidol.
Neben den Aminen gibt es weitere Stoffklassen, die sich als Kern für hyperverzweigte Polymere
eignen. Phosphane (früher „Phosphine“) – die Analoga der Amine – haben gegenüber den
Stickstoff-Verbindungen als Komplexbildner große Vorteile. Durch die Elektronenkonfiguration
der 3. Schale stehen zusätzlich leere d-Orbitale zur Verfügung, so dass sie neben den �/�-Donor-
auch �-Akzeptor-Eigenschaften aufweisen. Bereits in Kapitel II.1.1 ist auf außergewöhnliche
Phosphor-Liganden aufmerksam gemacht worden. Da Phosphine darüber hinaus gute
Nukleophile aber schwache Basen sind18,21,22, sind sie geradezu prädestiniert als
Kernfunktionalität hyperverzweigter Polyglycidole.

II.3 Ergebnisdiskussion
- 172 -
II.3.1.1 Hydroxyphenylphosphane als Starter
Im Jahr 1996 beschrieben Fell et al. die Synthese von ethoxyliertem Tris-(p-hydroxyphenyl)-
phosphan als Komplexligand für den Rhodiumcarbonyl-Katalysator (vgl. Abb. 16).23 Dieses
Molekül war Vorbild für einen möglichen Initiator der Polymerisation von Glycidol.
Abb. 16: Synthese eines ethoxylierten Phosphan-Liganden nach Fell et al.23
Damit Tris-(p-hydroxyphenyl)phosphan ein möglicher Kandidat für ein Precursormolekül wird,
müssen zunächst offene Fragestellungen beantwortet werden. Nach dem Vorbild von Fell et al.
soll die Polymerkette von der OH-Funktionalität gestartet werden. Das bedeutet, dass
vorangehend geklärt werden muss, ob Phenol oder Phenolat geeignete Initiatoren sind (vgl.
Abb. 17).
Abb. 17: Phenol (links) und Natriumphenolat (rechts).
In verschiedenen Polymerisationsreaktionen konnte nachgewiesen werden, dass das neutrale
Phenol allein zu keiner Polymerisation von Glycidol führt, während Natriumphenolat als
möglicher Initiator verwendet werden kann. (vgl. Synthesen II.6.3.1 und II.6.3.2). Im nächsten
Schritt ist es notwendig herauszufinden, ob das Phosphor-Atom selbst die Polymerisation
initiieren kann. Hierfür wurde die Polymerisation von Glycidol durch unsubstituiertes
Triphenylphosphin untersucht (vgl. Synthese II.6.3.3). Es hat sich gezeigt, dass PPh3 ebenfalls
ein geeignetes Starter-Molekül ist. Daraus ergeben sich neue Herausforderungen aber auch neue
Chancen. Tris-(p-hydroxyphenyl)phosphan bzw. dessen Salze können sowohl vom Sauerstoff-
als auch von Phosphor-Atom ausgehend die Polymerisation initiieren. Durch Anlagerung eines

II.3 Ergebnisdiskussion
- 173 -
Glycidol-Moleküls an das Phosphor-Atom steht dieses für spätere Komplexbildungsreaktionen
nicht mehr zur Verfügung. Somit muss das Phosphor-Atom an dem Angriff auf Glycidol
gehindert werden. Am besten lassen sich Phosphane schützen, indem ein stabiler Komplex
gebildet wird. Ein möglicher Lösungsansatz ist demnach bereits im ersten Schritt den Komplex
zu synthetisieren und davon ausgehend die hyperverzweigte Umgebung aufzubauen.
Grundsätzlich stellt sich die Frage, ob die Polymerkette zwingend von drei Seiten (drei
substituierte Phenylringe) ausgehend wachsen muss oder soll, um die später gewünschte chirale
Umgebung zu erzeugen, oder ob mono- oder disubstituierte Phenylphosphane ebenso
interessante Initiatoren darstellen (vgl. Abb. 18).
Abb. 18: Phosphine als mögliche Starter für die Polymerisation von Glycidol.
Als Darstellungsmethode für (Para-hydroxy-phenyl)phosphane ist die Entmethylierung der
(Methoxyphenyl)phosphane sehr gut geeignet.24,25,26 Zunächst werden die Mono-, Di- oder Tri-
p-methoxy-Derivate über eine Grignard-Reaktion ausgehend von Bromanisol und der
entsprechenden Chlorphenylphosphan-Verbindung hergestellt (vgl. Abb. 19). Durch Brom- oder
Iodwasserstoffsäure reagieren die Methoxy- zu den entsprechenden Hydroxyverbindungen.
Gleichzeitig wird der Phosphor protoniert und es bilden sich quartäre Hydrobromid- bzw.
Hydroiodidsalze aus. Diese Verbindungen sind sehr stabil und können gut aufbewahrt werden,
da das quartäre Phosphor-Atom vor Oxidation geschützt ist. Für die Umwandlung der Phosphor-
Salze in die gewünschten Produkte gibt es verschiedene Möglichkeiten wie beispielsweise das
Lösen in verdünnter Natronlauge und die anschließende Neutralisation.
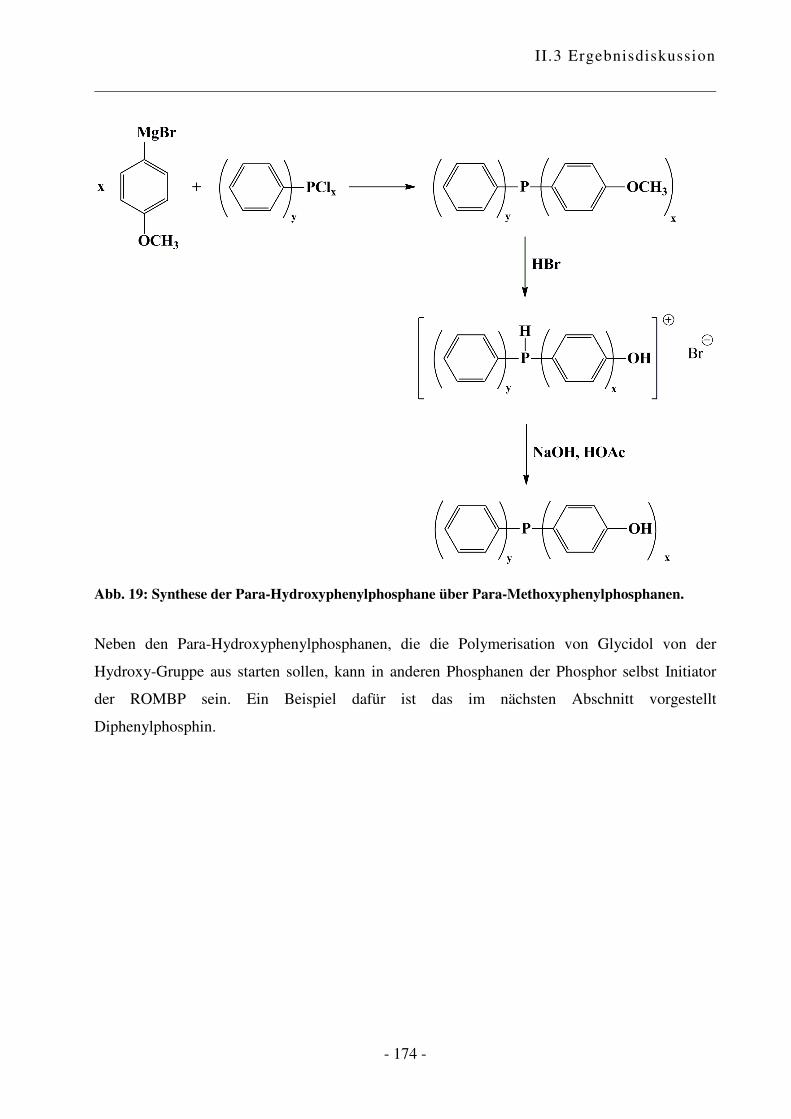
II.3 Ergebnisdiskussion
- 174 -
Abb. 19: Synthese der Para-Hydroxyphenylphosphane über Para-Methoxyphenylphosphanen.
Neben den Para-Hydroxyphenylphosphanen, die die Polymerisation von Glycidol von der
Hydroxy-Gruppe aus starten sollen, kann in anderen Phosphanen der Phosphor selbst Initiator
der ROMBP sein. Ein Beispiel dafür ist das im nächsten Abschnitt vorgestellt
Diphenylphosphin.
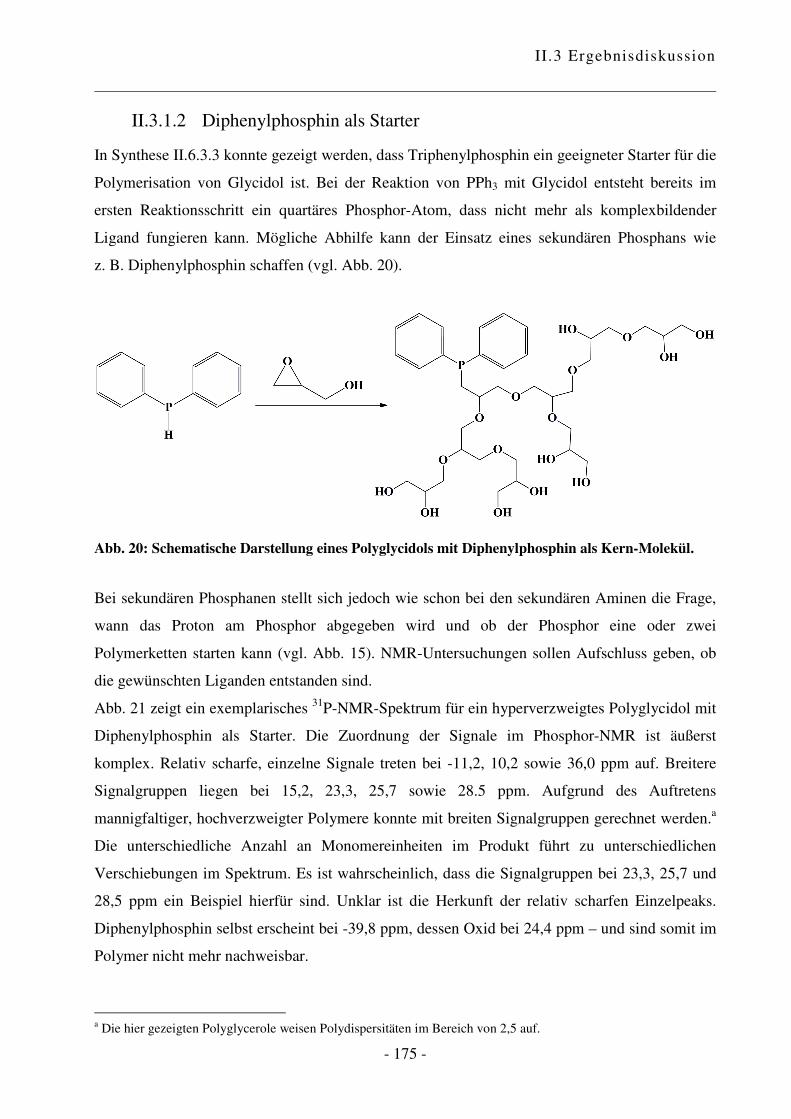
II.3 Ergebnisdiskussion
- 175 -
II.3.1.2 Diphenylphosphin als Starter
In Synthese II.6.3.3 konnte gezeigt werden, dass Triphenylphosphin ein geeigneter Starter für die
Polymerisation von Glycidol ist. Bei der Reaktion von PPh3 mit Glycidol entsteht bereits im
ersten Reaktionsschritt ein quartäres Phosphor-Atom, dass nicht mehr als komplexbildender
Ligand fungieren kann. Mögliche Abhilfe kann der Einsatz eines sekundären Phosphans wie
z. B. Diphenylphosphin schaffen (vgl. Abb. 20).
Abb. 20: Schematische Darstellung eines Polyglycidols mit Diphenylphosphin als Kern-Molekül.
Bei sekundären Phosphanen stellt sich jedoch wie schon bei den sekundären Aminen die Frage,
wann das Proton am Phosphor abgegeben wird und ob der Phosphor eine oder zwei
Polymerketten starten kann (vgl. Abb. 15). NMR-Untersuchungen sollen Aufschluss geben, ob
die gewünschten Liganden entstanden sind.
Abb. 21 zeigt ein exemplarisches 31P-NMR-Spektrum für ein hyperverzweigtes Polyglycidol mit
Diphenylphosphin als Starter. Die Zuordnung der Signale im Phosphor-NMR ist äußerst
komplex. Relativ scharfe, einzelne Signale treten bei -11,2, 10,2 sowie 36,0 ppm auf. Breitere
Signalgruppen liegen bei 15,2, 23,3, 25,7 sowie 28.5 ppm. Aufgrund des Auftretens
mannigfaltiger, hochverzweigter Polymere konnte mit breiten Signalgruppen gerechnet werden.a
Die unterschiedliche Anzahl an Monomereinheiten im Produkt führt zu unterschiedlichen
Verschiebungen im Spektrum. Es ist wahrscheinlich, dass die Signalgruppen bei 23,3, 25,7 und
28,5 ppm ein Beispiel hierfür sind. Unklar ist die Herkunft der relativ scharfen Einzelpeaks.
Diphenylphosphin selbst erscheint bei -39,8 ppm, dessen Oxid bei 24,4 ppm – und sind somit im
Polymer nicht mehr nachweisbar.
a Die hier gezeigten Polyglycerole weisen Polydispersitäten im Bereich von 2,5 auf.
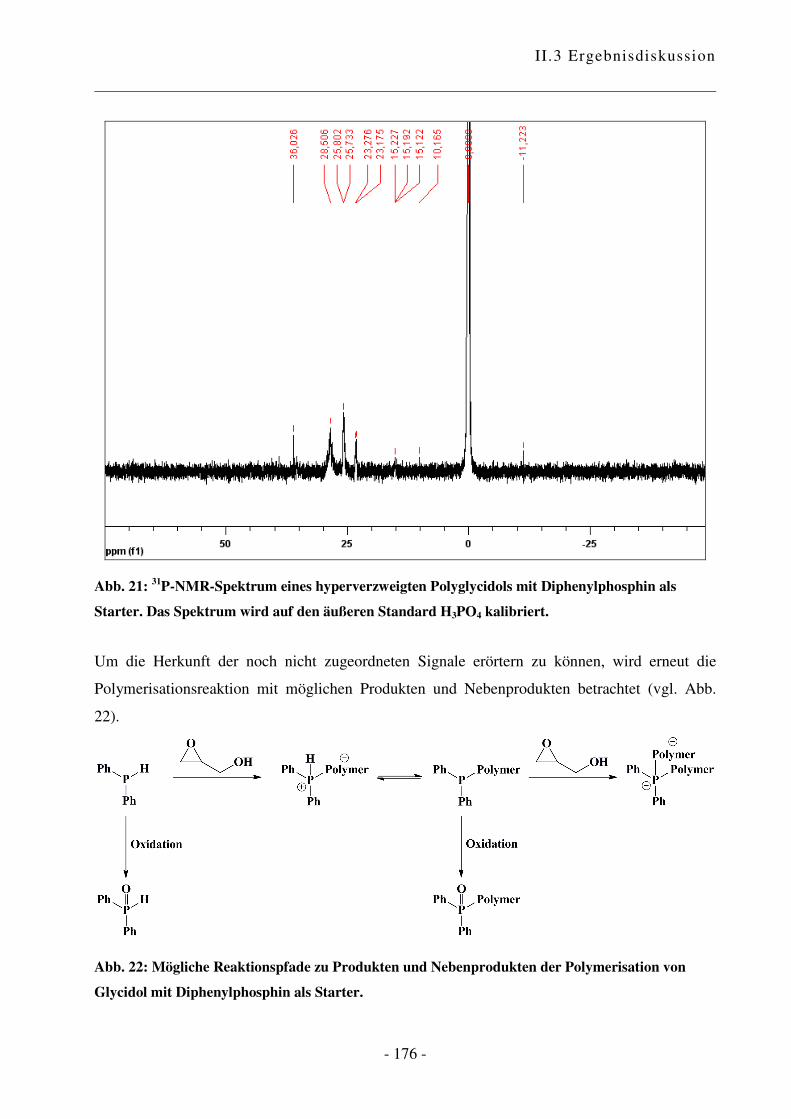
II.3 Ergebnisdiskussion
- 176 -
Abb. 21: 31P-NMR-Spektrum eines hyperverzweigten Polyglycidols mit Diphenylphosphin als
Starter. Das Spektrum wird auf den äußeren Standard H3PO4 kalibriert.
Um die Herkunft der noch nicht zugeordneten Signale erörtern zu können, wird erneut die
Polymerisationsreaktion mit möglichen Produkten und Nebenprodukten betrachtet (vgl. Abb.
22).
Abb. 22: Mögliche Reaktionspfade zu Produkten und Nebenprodukten der Polymerisation von
Glycidol mit Diphenylphosphin als Starter.

II.3 Ergebnisdiskussion
- 177 -
Neben der gewünschten Ph2P-Polymer-Spezies, die selbst schon zu einer Reihe an Signalen im
Spektrum führen wird, muss mit verschiedenen quartären Phosphorsalzen sowie mit
Phosphanoxiden gerechnet werden. Für Phosphor-NMR-Spektren gilt die allgemeine Tendenz,
dass mit zunehmender Bindigkeit des Phosphors die NMR-Signale zu hohem Feld verschoben
werden.27 Da sowohl die Oxidation des Phosphors als auch die Bildung eines quartären Salzes
die Bindigkeit erhöhen und damit zu ähnlichen Verschiebungen führen können, ist es noch nicht
gelungen, die Struktur des Phosphans mit hyperverzweigtem Glycidol-Substituent aus dem NMR
zweifelsfrei zu klären. Ein Beweis dafür, dass das gewünschte Molekül entstanden ist, wäre eine
erfolgreiche Komplexierung des Liganden.
II.3.2 Auf dem Weg zu neuen Komplexen
Zur Synthese der neuartigen Komplexe werden zwei verschiedene Strategien verfolgt (vgl.
Abb. 23). Der erste Ansatz besteht in der Bildung eines „herkömmlichen“ Phosphankomplexes,
der mit Substituenten ausgestattet ist, die die Polymerisation von Glycidol initiieren können. Die
zweite Darstellungsroute verfolgt den Aufbau der neuen Komplexe durch den Einsatz
hyperverzweigter Polyglycidole mit funktionalisiertem Kern.
Abb. 23: Schematische Darstellung der beiden Strategien zur Synthese neuartiger Komplexe.
Die in Kapitel II.3.1.1 beschriebenen Para-hydroxyphenylphosphane eignen sich hervorragend
für die erste Synthesestrategie. Für den Aufbau des Phosphan-Komplexes wird mit 1,5-Cyclo-
oktadienpalladium(II)chlorid ein gängiger Palladium-Precursor eingesetzt.28 Das
Reaktionsschema ist in Abb. 24 gezeigt.
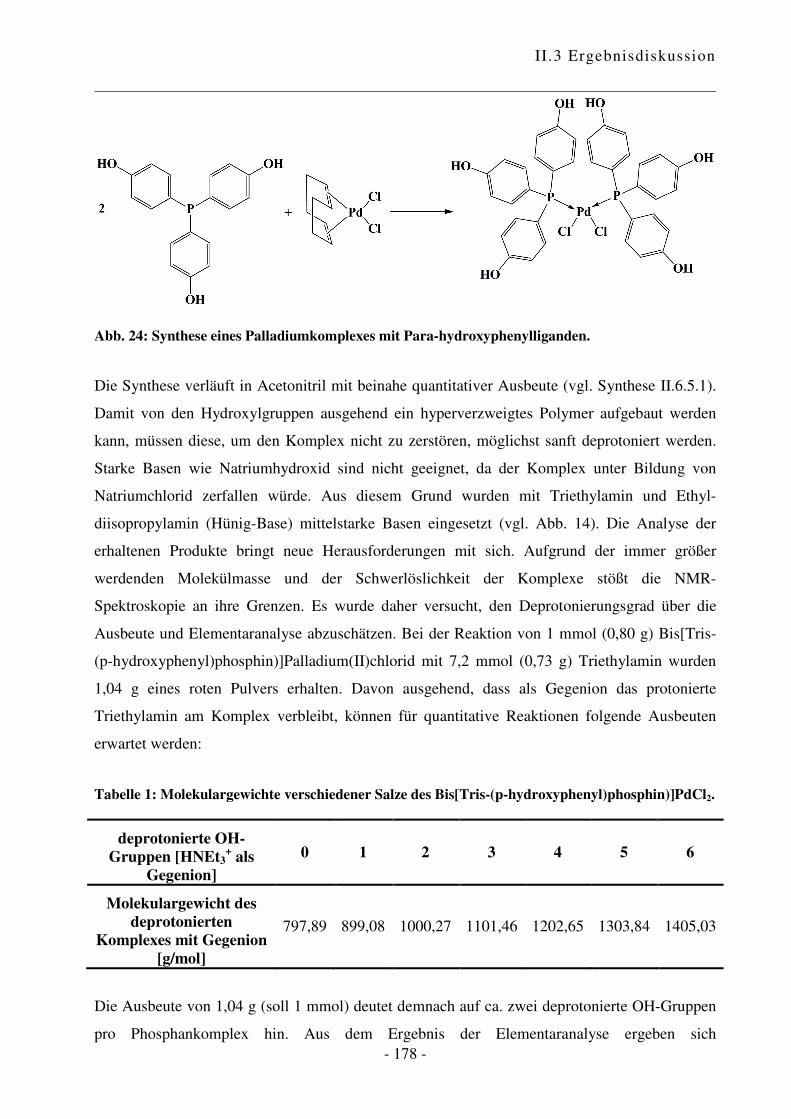
II.3 Ergebnisdiskussion
- 178 -
Abb. 24: Synthese eines Palladiumkomplexes mit Para-hydroxyphenylliganden.
Die Synthese verläuft in Acetonitril mit beinahe quantitativer Ausbeute (vgl. Synthese II.6.5.1).
Damit von den Hydroxylgruppen ausgehend ein hyperverzweigtes Polymer aufgebaut werden
kann, müssen diese, um den Komplex nicht zu zerstören, möglichst sanft deprotoniert werden.
Starke Basen wie Natriumhydroxid sind nicht geeignet, da der Komplex unter Bildung von
Natriumchlorid zerfallen würde. Aus diesem Grund wurden mit Triethylamin und Ethyl-
diisopropylamin (Hünig-Base) mittelstarke Basen eingesetzt (vgl. Abb. 14). Die Analyse der
erhaltenen Produkte bringt neue Herausforderungen mit sich. Aufgrund der immer größer
werdenden Molekülmasse und der Schwerlöslichkeit der Komplexe stößt die NMR-
Spektroskopie an ihre Grenzen. Es wurde daher versucht, den Deprotonierungsgrad über die
Ausbeute und Elementaranalyse abzuschätzen. Bei der Reaktion von 1 mmol (0,80 g) Bis[Tris-
(p-hydroxyphenyl)phosphin)]Palladium(II)chlorid mit 7,2 mmol (0,73 g) Triethylamin wurden
1,04 g eines roten Pulvers erhalten. Davon ausgehend, dass als Gegenion das protonierte
Triethylamin am Komplex verbleibt, können für quantitative Reaktionen folgende Ausbeuten
erwartet werden:
Tabelle 1: Molekulargewichte verschiedener Salze des Bis[Tris-(p-hydroxyphenyl)phosphin)]PdCl2.
deprotonierte OH-Gruppen [HNEt3
+ als Gegenion]
0 1 2 3 4 5 6
Molekulargewicht des deprotonierten
Komplexes mit Gegenion [g/mol]
797,89 899,08 1000,27 1101,46 1202,65 1303,84 1405,03
Die Ausbeute von 1,04 g (soll 1 mmol) deutet demnach auf ca. zwei deprotonierte OH-Gruppen
pro Phosphankomplex hin. Aus dem Ergebnis der Elementaranalyse ergeben sich
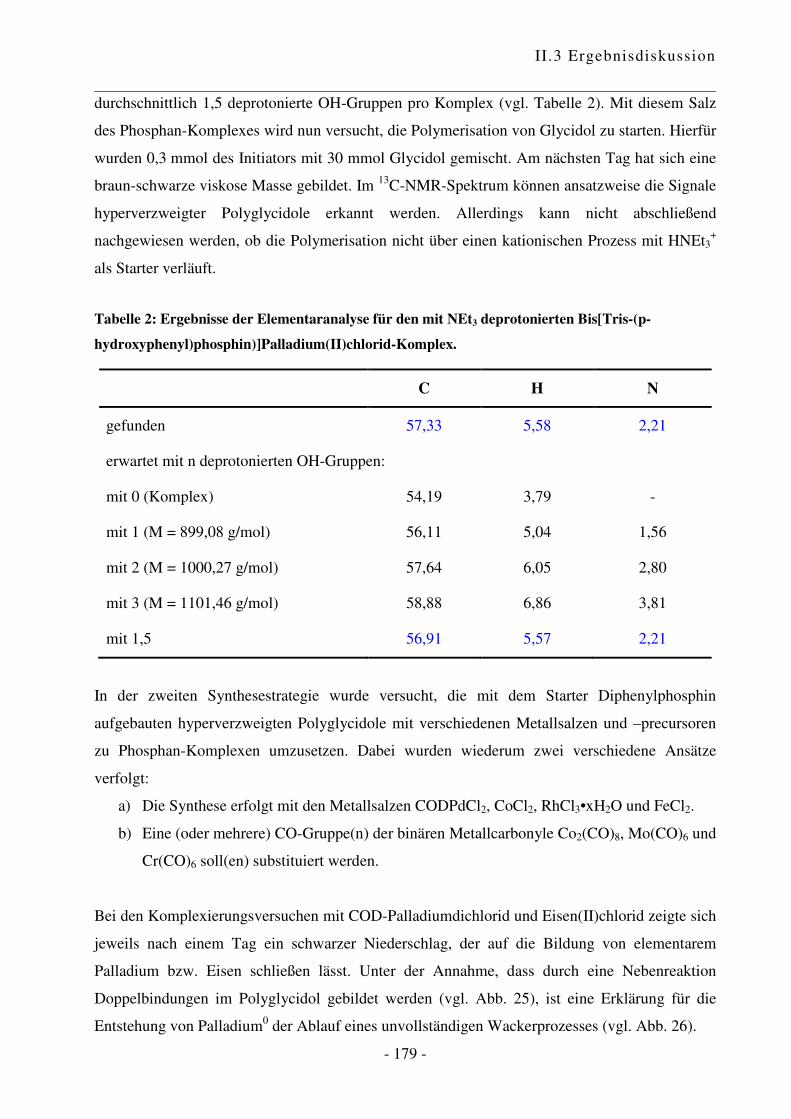
II.3 Ergebnisdiskussion
- 179 -
durchschnittlich 1,5 deprotonierte OH-Gruppen pro Komplex (vgl. Tabelle 2). Mit diesem Salz
des Phosphan-Komplexes wird nun versucht, die Polymerisation von Glycidol zu starten. Hierfür
wurden 0,3 mmol des Initiators mit 30 mmol Glycidol gemischt. Am nächsten Tag hat sich eine
braun-schwarze viskose Masse gebildet. Im 13C-NMR-Spektrum können ansatzweise die Signale
hyperverzweigter Polyglycidole erkannt werden. Allerdings kann nicht abschließend
nachgewiesen werden, ob die Polymerisation nicht über einen kationischen Prozess mit HNEt3+
als Starter verläuft.
Tabelle 2: Ergebnisse der Elementaranalyse für den mit NEt3 deprotonierten Bis[Tris-(p-
hydroxyphenyl)phosphin)]Palladium(II)chlorid-Komplex.
C H N
gefunden 57,33 5,58 2,21
erwartet mit n deprotonierten OH-Gruppen:
mit 0 (Komplex) 54,19 3,79 -
mit 1 (M = 899,08 g/mol) 56,11 5,04 1,56
mit 2 (M = 1000,27 g/mol) 57,64 6,05 2,80
mit 3 (M = 1101,46 g/mol) 58,88 6,86 3,81
mit 1,5 56,91 5,57 2,21
In der zweiten Synthesestrategie wurde versucht, die mit dem Starter Diphenylphosphin
aufgebauten hyperverzweigten Polyglycidole mit verschiedenen Metallsalzen und –precursoren
zu Phosphan-Komplexen umzusetzen. Dabei wurden wiederum zwei verschiedene Ansätze
verfolgt:
a) Die Synthese erfolgt mit den Metallsalzen CODPdCl2, CoCl2, RhCl3•xH2O und FeCl2.
b) Eine (oder mehrere) CO-Gruppe(n) der binären Metallcarbonyle Co2(CO)8, Mo(CO)6 und
Cr(CO)6 soll(en) substituiert werden.
Bei den Komplexierungsversuchen mit COD-Palladiumdichlorid und Eisen(II)chlorid zeigte sich
jeweils nach einem Tag ein schwarzer Niederschlag, der auf die Bildung von elementarem
Palladium bzw. Eisen schließen lässt. Unter der Annahme, dass durch eine Nebenreaktion
Doppelbindungen im Polyglycidol gebildet werden (vgl. Abb. 25), ist eine Erklärung für die
Entstehung von Palladium0 der Ablauf eines unvollständigen Wackerprozesses (vgl. Abb. 26).
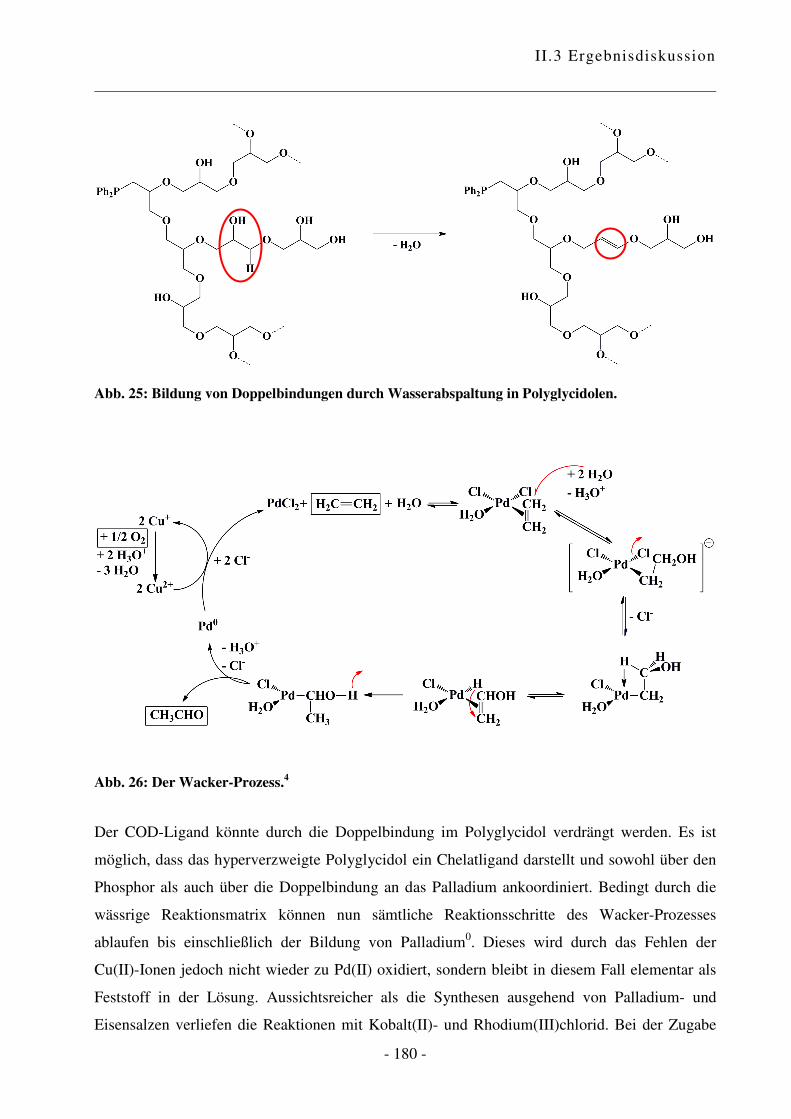
II.3 Ergebnisdiskussion
- 180 -
Abb. 25: Bildung von Doppelbindungen durch Wasserabspaltung in Polyglycidolen.
Abb. 26: Der Wacker-Prozess.4
Der COD-Ligand könnte durch die Doppelbindung im Polyglycidol verdrängt werden. Es ist
möglich, dass das hyperverzweigte Polyglycidol ein Chelatligand darstellt und sowohl über den
Phosphor als auch über die Doppelbindung an das Palladium ankoordiniert. Bedingt durch die
wässrige Reaktionsmatrix können nun sämtliche Reaktionsschritte des Wacker-Prozesses
ablaufen bis einschließlich der Bildung von Palladium0. Dieses wird durch das Fehlen der
Cu(II)-Ionen jedoch nicht wieder zu Pd(II) oxidiert, sondern bleibt in diesem Fall elementar als
Feststoff in der Lösung. Aussichtsreicher als die Synthesen ausgehend von Palladium- und
Eisensalzen verliefen die Reaktionen mit Kobalt(II)- und Rhodium(III)chlorid. Bei der Zugabe

II.3 Ergebnisdiskussion
- 181 -
von Kobaltchlorid zum gelösten hyperverzweigten Phosphan-Liganden verfärbte sich die Lösung
zunächst nach blau, nach einem Tag Reaktionszeit war die Lösung gelb gefärbt. Im Fall von
Rhodiumchlorid verfärbte sich die anfangs dunkelrote Lösung nach gelbbraun. Leider ist es trotz
des Einsatzes verschiedener Analysemethoden nicht gelungen, die erfolgreiche Komplexbildung
zweifelsfrei zu bestätigen. In beiden Fällen zeigen die 13C-NMR-Spektren nahezu identische
Signale wie die Edukte. In den Phosphor-Spektren erscheint jeweils ein einzelnes zusätzliches
Signal (bei -72,5 ppm im Fall von Kobalt, bei -68,4 ppm im Fall von Rhodium), das jedoch nicht
eindeutig zugeordnet werden kann. Auch bei den Massenspektren erhält man unklare Ergebnisse.
In beiden Spektren treten neben den bekannten Peaks der Liganden einige intensitätsschwache
neue Signale auf. Diese Signale können allerdings keiner der gewünschten Komplexspezies
zugeordnet werden.
Da die erfolgreiche Synthese neuartiger Komplexe in erster Linie an der schwierigen Analyse
der erhaltenen Produkte gehindert wird, wurde die zweite Synthesestrategie über binäre
Metallcarbonyle entwickelt. Für die Untersuchungen an Metallcarbonylen und Derivaten ist vor
allem die Infrarot-Spektroskopie hervorragend geeignet. Grundlage für diese Analyse ist die
Tatsache, dass sich die C-O-Streckschwingung in erster Näherung gut isoliert von anderen
Schwingungen des Moleküls betrachten lässt. Die Lage der CO-Valenzschwingung wird durch
die Metall-C-Bindung beeinflusst. Generell gilt, dass eine Zunahme der M-C-Bindungsordnung
eine Abnahme der C-O-Bindungsordnung nach sich zieht. Je schwächer die CO-Bindung, desto
weniger Energie ist zur Anregung der entsprechenden Schwingung notwendig.4 Die Lage und
Anzahl der Schwingungen der binären Metallcarbonyle ändert sich, sobald ein CO-Ligand gegen
einen anderen Liganden ausgetauscht wird. Die IR-Spektroskopie kann somit eine sehr
aussagekräftige Methode zur Überwachung des Reaktionsfortschritts sein.
Die Carbonylsubstitution an Komplexen mit 18 Valenzelektronen (wie z. B. Co2(CO)8, Mo(CO)6
und Cr(CO)6) verläuft nach einem dissoziativen Mechanismus, d.h. über eine Zwischenstufe
niedrigerer Koordinationszahl.29,30
Abb. 27: Mechanismus der Carbonylsubstitution bei 18 VE-Komplexen.31

II.3 Ergebnisdiskussion
- 182 -
Durch thermische Anregung (teilweise bereits bei Raumtemperatur) wird in einer relativ
langsamen Reaktion vom Hexacarbonylkomplex eine CO-Gruppe abgespalten. Es entsteht ein
ungesättigter 16 VE-Komplex an den sich rasch ein neuer Ligand anlagert, damit wieder eine
stabile Verbindung mit 18 Valenzelektronen gebildet wird. Eine mögliche Reaktionsgleichung
für die Synthese eines Komplexes mit hyperverzweigtem Ligand ist in Abb. 28 gezeigt.
Abb. 28: Synthese eines neuartigen Komplexes mit hyperverzweigtem Polyglycidol-Ligand.
Neben der thermisch induzierten Abspaltung einer Carbonyl-Gruppe gibt es die Möglichkeit, den
Ligandenaustausch durch Zusätze wie z. B. Pyridin-N-oxid oder Trimethyl-N-Oxid (TMNO) zu
beschleunigen. In den ersten kinetischen Studien über den Sauerstoff-Atom-Transfer auf
Metallcarbonyle wurde die Reaktion von M(CO)6 (M = Cr, Mo, W) mit PPh3 in der Gegenwart
von TMNO beschrieben (vgl. Abb. 29). Verglichen mit den Reaktionsgeschwindigkeiten der
thermischen Substitution verläuft der Ligandenaustausch unter Zusatz von TMNO extrem
schnell.
Abb. 29: Mechanismus des TMNO-assistierten Ligandenaustauschs binärer Metallcarbonyle.30
Die schnelle Substitution ist auf die effiziente Umsetzung eines CO-Liganden zurückzuführen.
Das nukleophile TMNO greift das Kohlenstoffatom einer Carbonylgruppe an und oxidiert diese
zu Kohlendioxid. CO2 ist ein schlechter Ligand und gleichzeitig eine gute Abgangsgruppe. Es
entsteht die äußerst reaktive, koordinativ ungesättigte Zwischenstufe M(CO)5, die sehr schnell
den zur Verfügung stehenden Liganden annektiert.30 Der in dieser Arbeit verwendete
hyperverzweigte Polyglycidol-Ligand mit Diphenylphosphin-Kern ist dem PPh3 ähnlich und
könnte daher einen ähnlichen Reaktionsverlauf zeigen. Für die hier durchgeführten Synthesen

II.3 Ergebnisdiskussion
- 183 -
wurden beide vorgestellten Wege zum Ligandenaustausch nachvollzogen. Eine Übersicht über
ausgewählte Synthesen gibt Tabelle 3.
Tabelle 3: Ausgewählte Synthesen von Metallcarbonylen mit neuartigen Phosphan-Liganden.
Nr. Metallcarbonyl Lösungsmittel Zusätze Temp., Zeit Farbe des Produkts
01 Co2(CO)8
orange-braun MeOH - RT, 18 h lila-violett
02 Mo(CO)6
farblos MeOH -
1.) RT, 24 h 2.) Rückfluss,
2 h
1.) unverändert 2.) dunkelbraun
03 Mo(CO)6
farblos (in Lösung mit TMNO gelblich)
Acetonitril TMNO 1.) RT, 24 h
2.) Rückfluss, 2,5 h
1.) unverändert (gelblich)
2.) gelb-orange
04 Mo(CO)6
farblos (in Lösung mit TMNO gelblich)
MeOH + Acetonitril
TMNO RT, 72 h etwas intensivere
Gelbfärbung
05 Cr(CO)6
farblos MeOH TMNO RT, 24 h grün
Ausgehend von Kobaltchlorid deutete zunächst eine Farbänderung der Lösung von orange-braun
auf violett auf eine erfolgreiche Synthese hin. Löst man Co2(CO)8 ohne weitere Zusätze in
Methanol stellt sich jedoch eine ähnliche Farbänderung ein. Das führt zu der Vermutung, dass
sich das Kobalt im hyperverzweigten Polyglycidol quasi gelöst hat. Auch bei den anderen
Synthesen gab der Reaktionsverlauf wenig Aufschlüsse. Weder in den NMR noch in den
Massenspektren konnten Hinweise auf neue Komplexe gefunden werden. Idee hinter der
Synthese ausgehend von Carbonylkomplexen war die zusätzliche Möglichkeit zur Analytik
mittels IR-Spektroskopie. Doch auch diese Methode stößt hier an ihre Grenzen. Die zahlreichen
OH-Gruppen des hyperverzweigten Polyglycidols führen zu einer so starken Absorption, dass
die ansonsten deutlich hervortretenden Carbonyl-Banden oftmals mit nur 97,5% Transmission
nur sehr schwach erscheinen und beinahe im Rauschen untergehen. Dennoch war es möglich, für
alle durchgeführten Synthesen neue IR-Banden im Produkt zu erkennen (vgl. Tabelle 4). In der
Literatur wurden für Carbonylkomplexe der Summenformel LM(CO)5 mit L = Phosphanligand
(wie z. B. PPh3, P(OMe)3 oder PCl3) Absorptionsbanden im Bereich zwischen 2150 und
1700 cm-1 beschrieben.4,29,32 Für alle hergestellten Verbindungen konnten Signale in diesem
Bereich identifiziert werden. Dennoch kann dies nicht als eindeutiges Indiz für die erfolgreiche
Synthese gewertet werden, da sich beispielsweise auch Lösungsmittelkomplexe wie L3Mo(CO)3
mit L = Acetonitril gebildet haben könnten.

II.3 Ergebnisdiskussion
- 184 -
Tabelle 4: Gemessene IR-Absorptionsbanden für binäre Metallcarbonyle und neu synthetisierte
Komplexe.
Substanz IR-Absorptionsbanden
v~ [cm-1]
Co2(CO)8 (fest) 2031, 1850
Co2(CO)8 in MeOH 2042
Komplex Nr. 1 1993, 1890
Mo(CO)6 fest 2002, 1970
Mo(CO)2 in MeOH 2122 (sehr breit)
Komplex Nr. 2 2071, 2020, 1976, 1946
Komplex Nr. 3 2063, 2023, 1930, 1879, 1780
Komplex Nr. 4 2066, 1979, 1934
Cr(CO)6 (Literaturwert29) 2000
Komplex Nr. 5 2068
Es gibt etliche Hinweise auf die erfolgreiche Synthese der neuartigen Komplexe, letztendlich
bewiesen werden konnte sie jedoch nicht. Um neue Verbindungen hinreichend charakterisieren
zu können, sind üblicherweise Methoden wie NMR-Spektroskopie, Elementaranalyse,
Röntgenstrukturanalyse und Bestimmung des Schmelzpunkts erforderlich. Diese
Analyseverfahren stoßen bei den hyperverzweigten Liganden an ihre Grenzen. Im Gegensatz zu
den perfekt verzweigten, monodispersen Dendrimeren zählen die hochverzweigten Moleküle zu
den Polymeren und sind damit auch durch die für Polymere zugänglichen Analysemethoden
eingeschränkt. Im folgenden Kapitel wird die Charakterisierung der Polyglycidole erläutert.
Darüber hinaus werden Möglichkeiten zur Beeinflussung der Synthese vorgestellt, die
zukünftige Komplexbildungsversuche erleichtern könnten.

II.3 Ergebnisdiskussion
- 185 -
II.3.3 Charakterisierung und Synthese hyperverzweigter Polyglycidole
II.3.3.1 Polymeranalytik
Eine wichtige Methode zur Analyse der hyperverzweigten Polymere stellt die NMR-
Spektroskopie dar. Damit die Signale im Spektrum zugeordnet werden können, ist ein Überblick
über die potentiell auftretenden Gruppen notwendig. Abb. 30 zeigt ein Polyglycerol und dessen
wichtigste Struktureinheiten. Der nukleophile Starter bildet den Kern (engl.: core) des
Makromoleküls. Es treten perfekt dendritische (D), lineare (L) und terminale (T) Einheiten auf.
Dadurch, dass Glycidol ein unsymmetrisches Molekül ist, gibt es zweierlei lineare Einheiten.
Falls das Kettenwachstum mit der sekundären Hydroxylgruppe voranschreitet, entsteht eine
lineare 1,3-Einheit (L13). Setzt sich das Polymer am Ende der primären Hydroxylgruppe fort,
wird eine lineare 1,4-Einheit (L14) gebildet. Reagieren beide Hydroxylgruppen weiter, resultiert
eine verzweigte, dendritische Einheit (D). Am Ende der wachsenden Kette erscheinen die
terminalen Einheiten (T) mit zwei Hydroxyendgruppen.
Abb. 30: Schematische Darstellung eines Polyglycerols unter Kennzeichnung wichtiger
Struktureinheiten.
Die charakteristischen Verschiebungen der Kohlenstoffatome der verschiedenen Struktur-
einheiten sind von Sunder, Hanselmann, Frey und Mülhaupt beschrieben worden33 und in
Abb. 31 dargestellt.
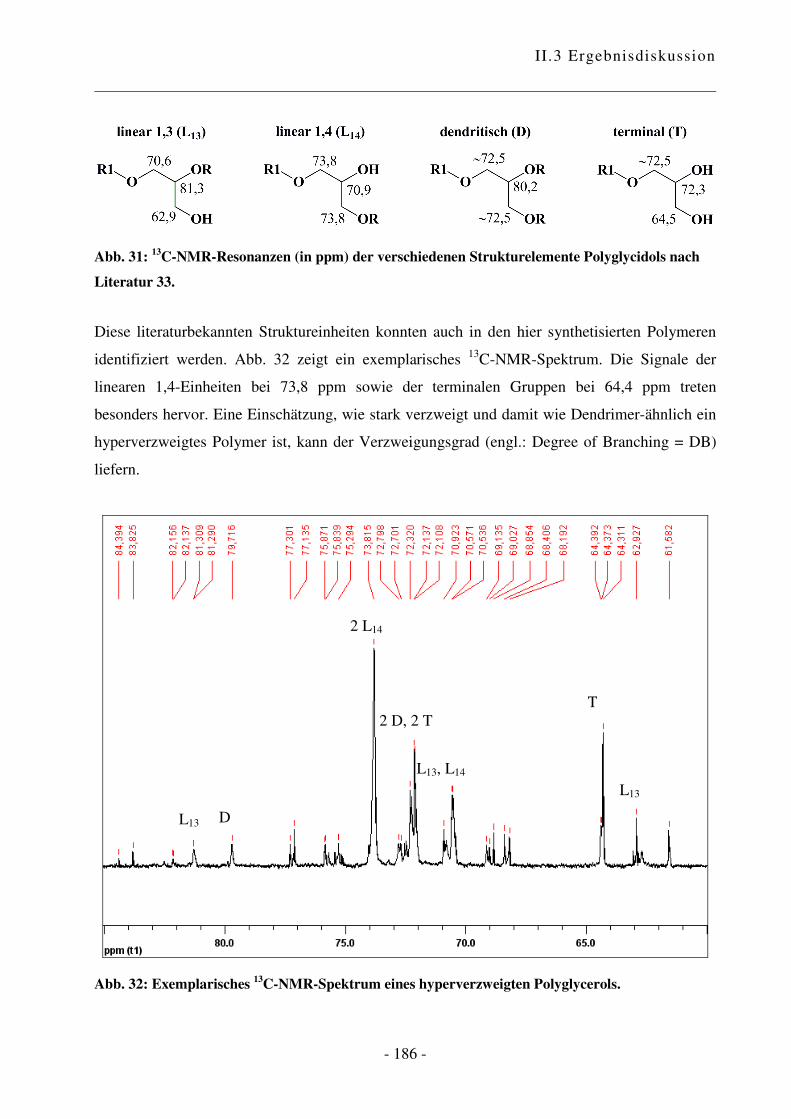
II.3 Ergebnisdiskussion
- 186 -
Abb. 31: 13C-NMR-Resonanzen (in ppm) der verschiedenen Strukturelemente Polyglycidols nach
Literatur 33.
Diese literaturbekannten Struktureinheiten konnten auch in den hier synthetisierten Polymeren
identifiziert werden. Abb. 32 zeigt ein exemplarisches 13C-NMR-Spektrum. Die Signale der
linearen 1,4-Einheiten bei 73,8 ppm sowie der terminalen Gruppen bei 64,4 ppm treten
besonders hervor. Eine Einschätzung, wie stark verzweigt und damit wie Dendrimer-ähnlich ein
hyperverzweigtes Polymer ist, kann der Verzweigungsgrad (engl.: Degree of Branching = DB)
liefern.
Abb. 32: Exemplarisches 13C-NMR-Spektrum eines hyperverzweigten Polyglycerols.
L13
L13, L14
D
2 L14
2 D, 2 T T
L13

II.3 Ergebnisdiskussion
- 187 -
Der Verzweigungsgrad stellt das Verhältnis aus dendritischen Einheiten zu allen vorhandenen
Struktureinheiten dar. Für perfekte Dendrimere, in denen ausschließlich dendritische Einheiten
auftreten, beträgt er Eins, für vollständig lineare Moleküle ist er Null. Theoretische
Betrachtungen haben gezeigt, dass mittels statistischer Polymerisation von AB2-Monomeren –
der so genannten Ein-Topf-Synthese – unabhängig vom Kernmolekül maximal ein DB von 0,5
erreicht werden kann.34 Durch das Verdünnungsprinzip und die „slow addition technique“ ist
theoretisch ein DB von 0,67 möglich.35
Aus dem in Abb. 32 gezeigten NMR-Spektrum lässt sich über die in Gleichung 1 gezeigte
Formel der Verzweigungsgrad der synthetisierten Polymere berechnen:
1413 LL2D
2DDB
++=
Gleichung 1: Berechnung des Verzweigungsgrads hyperverzweigter Polyglycidole.34
Eine beispielhafte Bestimmung des DB für drei exemplarische Polymere ist in Tabelle 5 gezeigt.
Über das Verhältnis der Integrale ergeben sich für die hier gezeigten Produkte Verzweigungs-
grade von 33% für Polymer 1 sowie 32% für Polymer 2 und 3. Dieses Ergebnis bestätigt die
erfolgreiche Synthese der hyperverzweigten Polyglycidole, lässt allerdings Raum für
Verbesserungen in Hinblick auf den Reaktionsverlauf.
Tabelle 5: Berechnung des Verzweigungsgrads dreier exemplarischer Polyglycerole.
Struktureinheit Verschiebung [ppm] Relative Intensität
Polymer 1 Polymer 2 Polymer 3
L13 81,0 – 82,0 1,00 1,00 1,00
D 79,5 – 80,5 1,08 1,31 1,17
2 L14 73,5 – 74,5 9,79 12,27 13,10
2 D, 2 T 71,5 – 73,1 9,01 11,33 10,71
L13, L14 69,8 – 71,3 5,22 6,87 6,12
T 64,0 – 65,0 3,41 4,44 4,85
L13 62,0 – 63,5 1,77 2,18 2,04
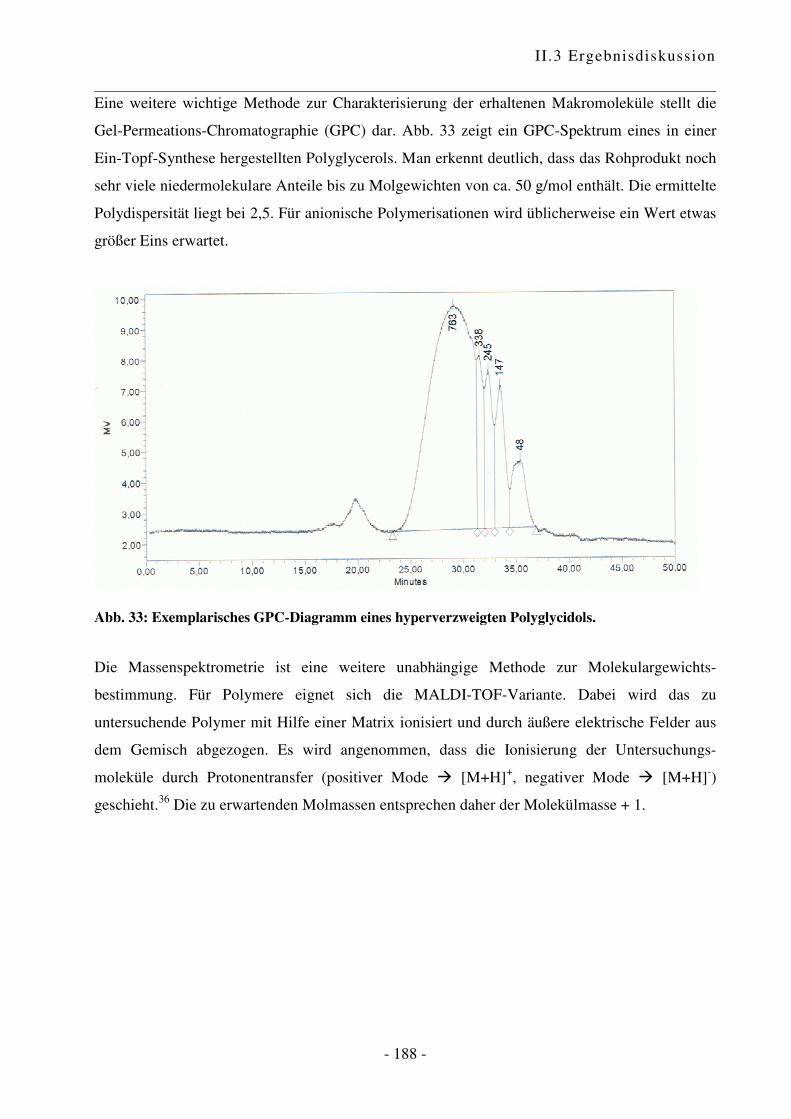
II.3 Ergebnisdiskussion
- 188 -
Eine weitere wichtige Methode zur Charakterisierung der erhaltenen Makromoleküle stellt die
Gel-Permeations-Chromatographie (GPC) dar. Abb. 33 zeigt ein GPC-Spektrum eines in einer
Ein-Topf-Synthese hergestellten Polyglycerols. Man erkennt deutlich, dass das Rohprodukt noch
sehr viele niedermolekulare Anteile bis zu Molgewichten von ca. 50 g/mol enthält. Die ermittelte
Polydispersität liegt bei 2,5. Für anionische Polymerisationen wird üblicherweise ein Wert etwas
größer Eins erwartet.
Abb. 33: Exemplarisches GPC-Diagramm eines hyperverzweigten Polyglycidols.
Die Massenspektrometrie ist eine weitere unabhängige Methode zur Molekulargewichts-
bestimmung. Für Polymere eignet sich die MALDI-TOF-Variante. Dabei wird das zu
untersuchende Polymer mit Hilfe einer Matrix ionisiert und durch äußere elektrische Felder aus
dem Gemisch abgezogen. Es wird angenommen, dass die Ionisierung der Untersuchungs-
moleküle durch Protonentransfer (positiver Mode � [M+H]+, negativer Mode � [M+H]-)
geschieht.36 Die zu erwartenden Molmassen entsprechen daher der Molekülmasse + 1.
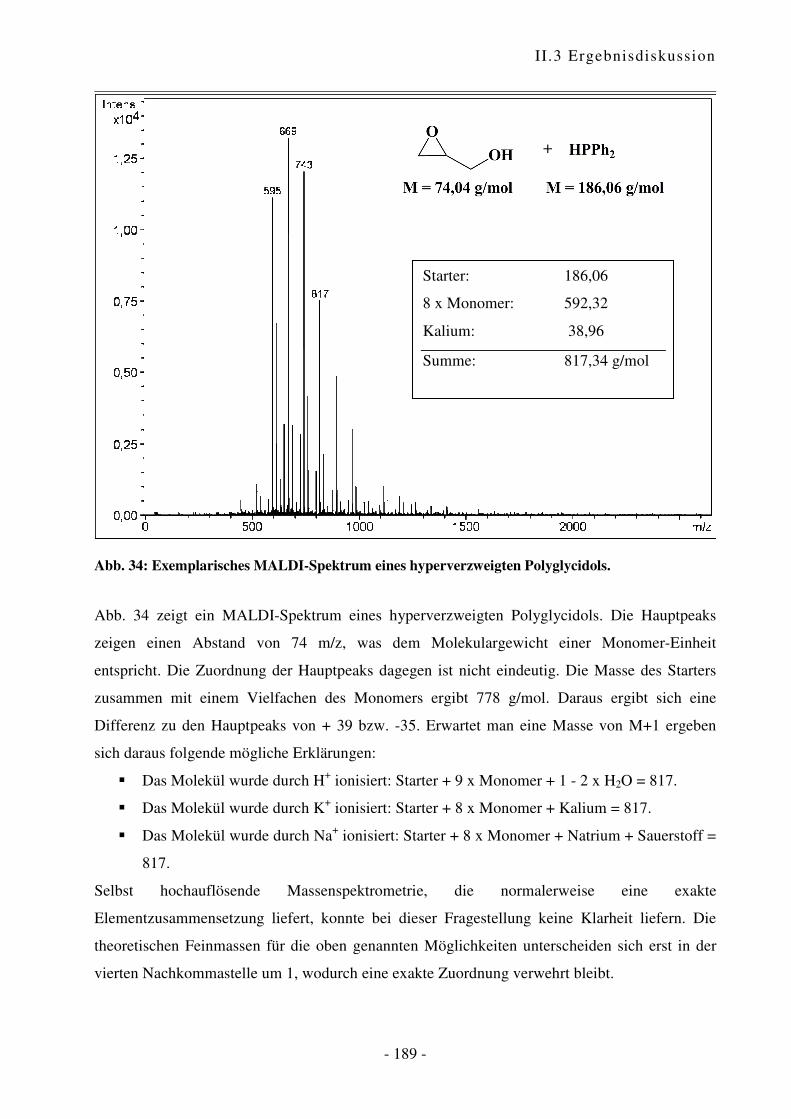
II.3 Ergebnisdiskussion
- 189 -
Abb. 34: Exemplarisches MALDI-Spektrum eines hyperverzweigten Polyglycidols.
Abb. 34 zeigt ein MALDI-Spektrum eines hyperverzweigten Polyglycidols. Die Hauptpeaks
zeigen einen Abstand von 74 m/z, was dem Molekulargewicht einer Monomer-Einheit
entspricht. Die Zuordnung der Hauptpeaks dagegen ist nicht eindeutig. Die Masse des Starters
zusammen mit einem Vielfachen des Monomers ergibt 778 g/mol. Daraus ergibt sich eine
Differenz zu den Hauptpeaks von + 39 bzw. -35. Erwartet man eine Masse von M+1 ergeben
sich daraus folgende mögliche Erklärungen:
� Das Molekül wurde durch H+ ionisiert: Starter + 9 x Monomer + 1 - 2 x H2O = 817.
� Das Molekül wurde durch K+ ionisiert: Starter + 8 x Monomer + Kalium = 817.
� Das Molekül wurde durch Na+ ionisiert: Starter + 8 x Monomer + Natrium + Sauerstoff =
817.
Selbst hochauflösende Massenspektrometrie, die normalerweise eine exakte
Elementzusammensetzung liefert, konnte bei dieser Fragestellung keine Klarheit liefern. Die
theoretischen Feinmassen für die oben genannten Möglichkeiten unterscheiden sich erst in der
vierten Nachkommastelle um 1, wodurch eine exakte Zuordnung verwehrt bleibt.
Starter: 186,06
8 x Monomer: 592,32
Kalium: 38,96
Summe: 817,34 g/mol

II.3 Ergebnisdiskussion
- 190 -
II.3.3.2 Einfluss verschiedener Reaktionsparameter
Nachdem im vorherigen Kapitel geeignete Startermoleküle für die Polymerisation von Glycidol
ausgewählt und die geeigneten Methoden zur Polymercharakterisierung vorgestellt worden sind,
wird nun der Einfluss verschiedener Parameter auf den Reaktionsverlauf und damit die
entstehenden Produkte untersucht. Wie bereits in Kapitel II.2.2 beschrieben, reagiert das System
äußerst sensibel auf Wärmeeintrag. Aus diesem Grund wurden Polymerisationen bei
verschiedenen Temperaturen zwischen -20 und +50 °C durchgeführt (vgl. Tabelle 6).
Tabelle 6: Polymerisation von Glycidol bei verschiedenen Reaktionstemperaturen.
Temperatur [°C]
Initiator
PPh3
Initiator
HPPh2
-20 nach 24 h: schwach gelbe,
wenig viskose Lösung
0 nach 24 h: schwach gelbe,
niedrig-viskose Lösung nach 24 h: farblose und wenig viskose Lösung
5 nach 13 Tagen: Reaktionsmatrix ist hellgelb und hochviskos geworden
10 nach 10 Tagen: Reaktionsmatrix ist
schwach gelb und hochviskos geworden
RT nach 24 h: gelbe, viskose Lösung nach 24 h: orange,
hochviskose Lösung
35 Reaktionsmatrix ist bereits nach 1 h
hochviskos und orange-braun gefärbt
Reaktionsmatrix ist bereits nach 30 min hochviskos und
dunkelbraun gefärbt
50
heftige Reaktion unter Rauchentwicklung und Braunfärbung, Reaktionsmatrix ist bereits nach 1 h
hochviskos
Die Synthesen haben gezeigt, dass bereits geringe Temperaturunterschiede deutliche
Auswirkungen auf den Reaktionsverlauf und damit die entstehenden Produkte haben. Bei
Temperaturen oberhalb Raumtemperatur ist die Polymerisation quasi unkontrollierbar.
Besonders die Abführung der bei der exothermen Ringöffnung freigesetzten Wärme ist wichtig.
Aus diesem Grund wird für nachfolgende Forschungsvorhaben ein doppelwandiges Glasgefäß
empfohlen, das eine schnelle Temperatursteuerung mittels Umlaufkühler erlaubt.

II.3 Ergebnisdiskussion
- 191 -
II.3.3.3 Kontrollierte Monomerzugabe zur Steuerung der Produkte
In den bisher aufgezeigten Reaktionen wurden Initiator und Monomer bereits zu Beginn der
Reaktion vollständig miteinander gemischt. Die Reaktion verläuft im Anschluss völlig „sich
selbst überlassen“ und kann – außer über die Temperatur – von außen nicht mehr beeinflusst
werden. In der Literatur sind so genannte „core dilution“ oder „slow addition techniques“
beschrieben worden, die die Herstellung von Polymeren mit geringerer Polydispersität
erlauben.37,38 Frey et al. konnten in Computer-Simulationen zeigen, dass die Polydispersität
hyperverzweigter Polymere ausgehend von ABm-Monomeren kontrolliert werden kann. Für
AB2-Monomere wie Glycidol wurde die hypothetische Formel
fPD
11+=
mit f = Funktionalität des Kerns beschrieben.37 Das bedeutet, dass mit steigender Anzahl
potentieller Startergruppen am Kernmolekül die zu erwartende Polydispersität des hyper-
verzweigten Polymers abnimmt. Polyfunktionale Startermoleküle sind demnach besonders
empfehlenswert. Neben dem positiven Einfluss auf die Einheitlichkeit der Makromoleküle kann
über die Technik der langsamen Monomerzugabe auch der Verzweigungsgrad in positiver Art
beeinflusst werden.37
Die Methode langsamen Monomerzugabe wurde in verschiedenen Experimenten untersucht.
Hierfür wurde der Starter zunächst mit einer geringen Menge Glycidol gemischt und in
kontrollierten Zeitabständen weiteres Monomer hinzugegeben. Der direkte Vergleich zu in Ein-
Topf-Synthesen hergestellten Polymeren zeigt, dass die Molekulargewichte bei langsamer
Monomerzugabe höher liegen. Eine Änderung der Polydispersität konnte nicht beobachtet
werden (vgl. Tabelle 7). Der aus den NMR-Spektren bestimmte Verzweigungsgrad liegt wie zu
erwarten war für die Ein-Topf-Synthese deutlich niedriger als für die kontinuierliche
Monomerzugabe.

II.3 Ergebnisdiskussion
- 192 -
Tabelle 7: Vergleich der Ein-Topf-Synthese mit der Methode der langsamen Monomerzugabe bei
Polyglycidolen.
Polymer 4 Polymer 5
Initiator HPPh2, 2 mmol HPPh2, 2 mmol
Glycidol 5 mL (75 mmol) insg. 5 mL (75 mmol)
Temperatur RT RT
Art der Monomerzugabe alles auf einmal zu Beginn der
Reaktion
3 mL zu Beginn der Reaktion,
anschließend alle 30 min
0,2 mL nachgegeben
Molekulargewicht Mw über
den gesamten Bereich, PD 460 g/mol, 2,2 580 g/mol, 2,2
Molekulargewicht Mw des
höchsten Signals, PD 690 g/mol, 1,2 880 g/mol, 1,2
Verzweigungsgrad
(aus NMR) 20% 27%
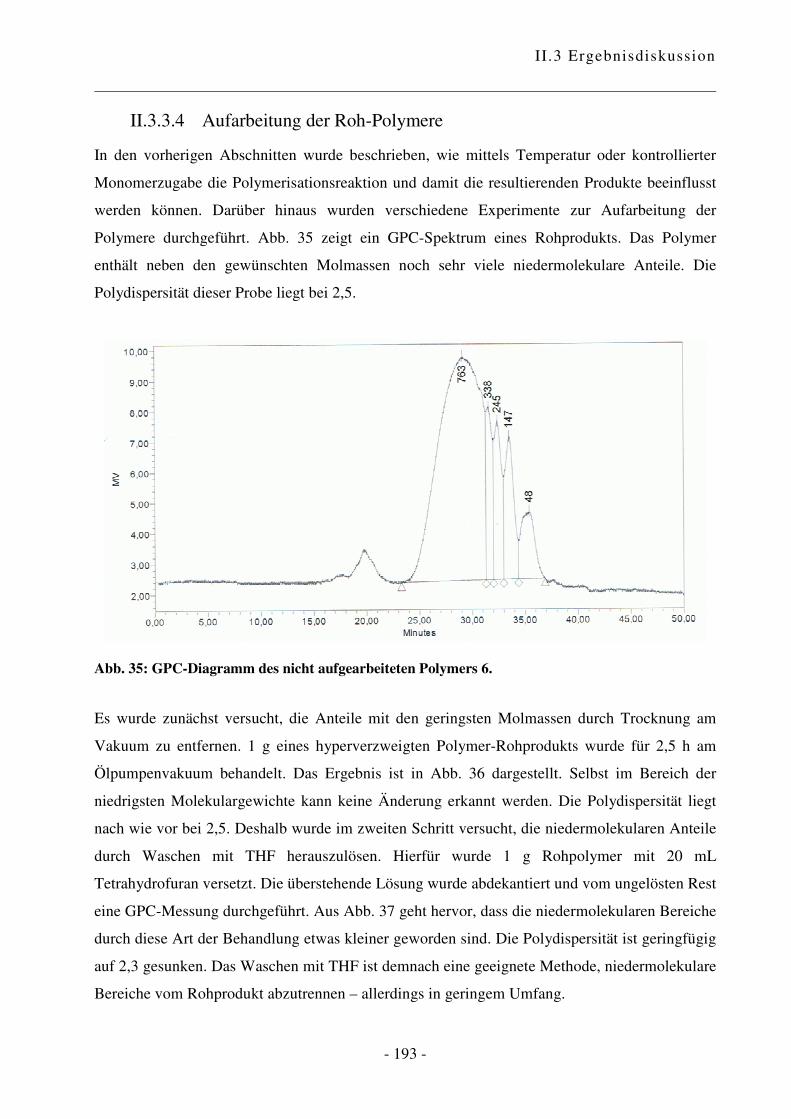
II.3 Ergebnisdiskussion
- 193 -
II.3.3.4 Aufarbeitung der Roh-Polymere
In den vorherigen Abschnitten wurde beschrieben, wie mittels Temperatur oder kontrollierter
Monomerzugabe die Polymerisationsreaktion und damit die resultierenden Produkte beeinflusst
werden können. Darüber hinaus wurden verschiedene Experimente zur Aufarbeitung der
Polymere durchgeführt. Abb. 35 zeigt ein GPC-Spektrum eines Rohprodukts. Das Polymer
enthält neben den gewünschten Molmassen noch sehr viele niedermolekulare Anteile. Die
Polydispersität dieser Probe liegt bei 2,5.
Abb. 35: GPC-Diagramm des nicht aufgearbeiteten Polymers 6.
Es wurde zunächst versucht, die Anteile mit den geringsten Molmassen durch Trocknung am
Vakuum zu entfernen. 1 g eines hyperverzweigten Polymer-Rohprodukts wurde für 2,5 h am
Ölpumpenvakuum behandelt. Das Ergebnis ist in Abb. 36 dargestellt. Selbst im Bereich der
niedrigsten Molekulargewichte kann keine Änderung erkannt werden. Die Polydispersität liegt
nach wie vor bei 2,5. Deshalb wurde im zweiten Schritt versucht, die niedermolekularen Anteile
durch Waschen mit THF herauszulösen. Hierfür wurde 1 g Rohpolymer mit 20 mL
Tetrahydrofuran versetzt. Die überstehende Lösung wurde abdekantiert und vom ungelösten Rest
eine GPC-Messung durchgeführt. Aus Abb. 37 geht hervor, dass die niedermolekularen Bereiche
durch diese Art der Behandlung etwas kleiner geworden sind. Die Polydispersität ist geringfügig
auf 2,3 gesunken. Das Waschen mit THF ist demnach eine geeignete Methode, niedermolekulare
Bereiche vom Rohprodukt abzutrennen – allerdings in geringem Umfang.
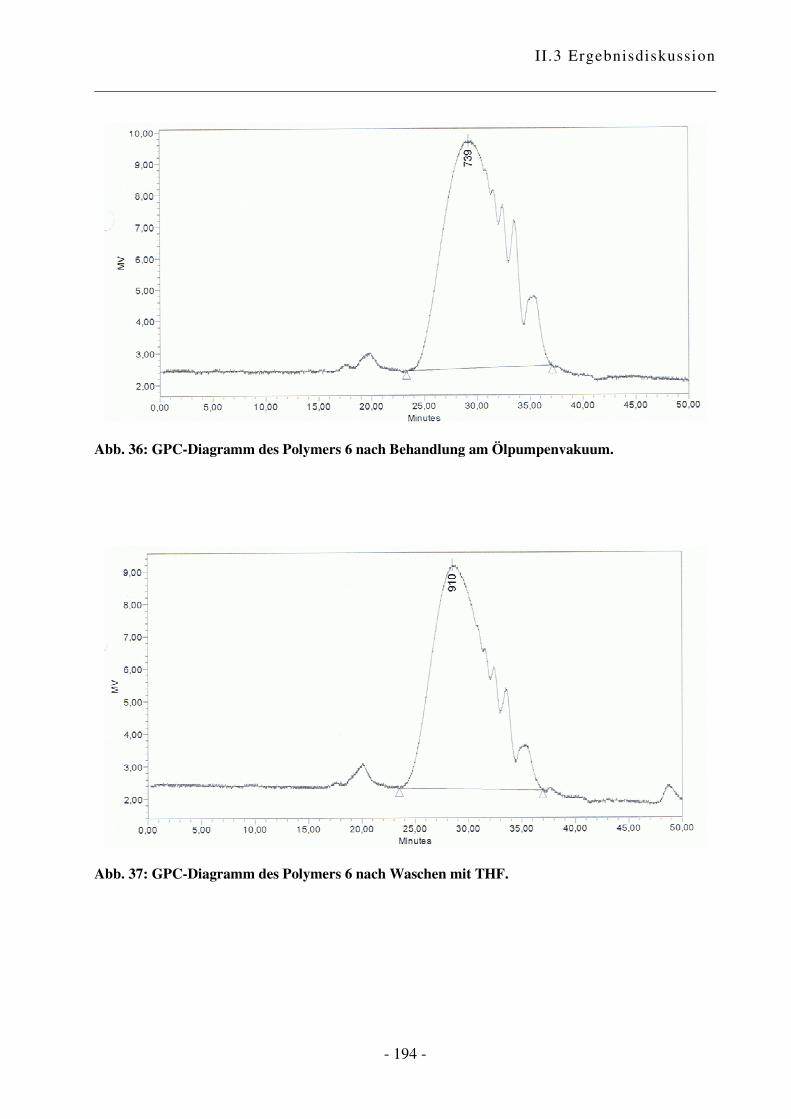
II.3 Ergebnisdiskussion
- 194 -
Abb. 36: GPC-Diagramm des Polymers 6 nach Behandlung am Ölpumpenvakuum.
Abb. 37: GPC-Diagramm des Polymers 6 nach Waschen mit THF.
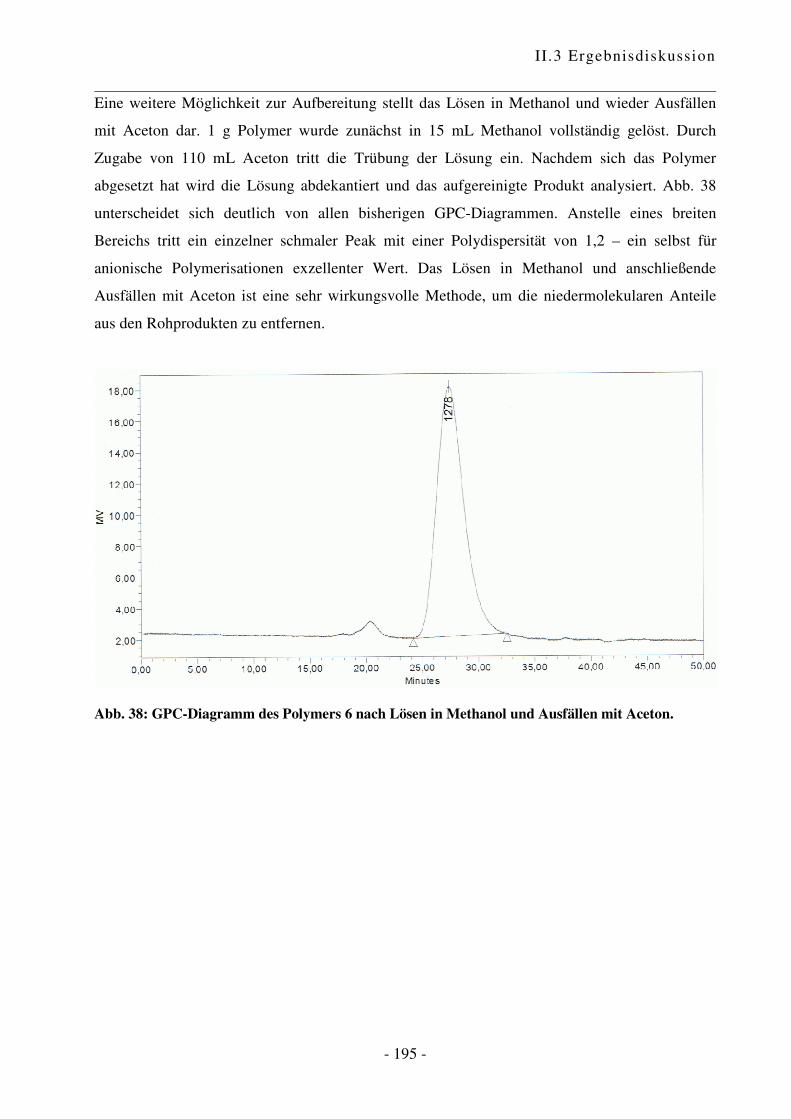
II.3 Ergebnisdiskussion
- 195 -
Eine weitere Möglichkeit zur Aufbereitung stellt das Lösen in Methanol und wieder Ausfällen
mit Aceton dar. 1 g Polymer wurde zunächst in 15 mL Methanol vollständig gelöst. Durch
Zugabe von 110 mL Aceton tritt die Trübung der Lösung ein. Nachdem sich das Polymer
abgesetzt hat wird die Lösung abdekantiert und das aufgereinigte Produkt analysiert. Abb. 38
unterscheidet sich deutlich von allen bisherigen GPC-Diagrammen. Anstelle eines breiten
Bereichs tritt ein einzelner schmaler Peak mit einer Polydispersität von 1,2 – ein selbst für
anionische Polymerisationen exzellenter Wert. Das Lösen in Methanol und anschließende
Ausfällen mit Aceton ist eine sehr wirkungsvolle Methode, um die niedermolekularen Anteile
aus den Rohprodukten zu entfernen.
Abb. 38: GPC-Diagramm des Polymers 6 nach Lösen in Methanol und Ausfällen mit Aceton.

II.4 Zusammenfassung
- 196 -
II.4 Zusammenfassung
Chirale, hyperverzweigte Polyglycidole sind höchst interessante Makromoleküle, die über die
anionische Ring-Öffnungs-Multi-Verzweigungs-Polymerisation (ROMBP) in einer Ein-Topf-
Synthese zugänglich sind.19 Durch die Wahl eines geeigneten Startermoleküls ist es möglich, die
Polymere mit einem funktionellen Kern auszustatten, der Raum für Folgereaktionen bietet. Ziel
dieser Arbeit war die Synthese neuartiger Komplexe mit hyperverzweigten Polyglycidolen als
Liganden (vgl. Abb. 39).
Abb. 39: Schematische Darstellung eines neuartigen Metallkomplexes, dessen Liganden aus
chiralen, hyperverzweigten Polymeren bestehen.
Damit die anionische Aufbaureaktion der Polymere gelingen kann, muss eine Vielzahl an
Nebenreaktionen beachtet werden. Es konnte gezeigt werden, dass Phosphane wie
Diphenylphosphin aufgrund ihrer guten Nukleophilie aber schwachen Basizität als Initiatoren für
die Polyglycidol-Synthese hervorragend geeignet sind. Dabei wurde der Einfluss verschiedener
Reaktionsparameter wie Temperatur oder die Art der Monomerzugabe auf die erhaltenen
Produkte untersucht. Es ist gelungen, Produktmerkmale wie Molekulargewicht oder
Verzweigungsgrad durch die gezielte Einstellung der Einflussgrößen zu steuern. Bei der
Aufarbeitung der Rohpolymere konnte gezeigt werden, dass mit der Wahl der geeigneten
Methode Polydispersitäten von unter 1,2 erreicht werden können.
Zum Aufbau der neuartigen Komplexe wurden zwei unterschiedliche Strategien verfolgt (vgl.
Abb. 40). Der erste Ansatz besteht in der Bildung eines „herkömmlichen“ Phosphankomplexes,

II.4 Zusammenfassung
- 197 -
der mit Substituenten ausgestattet ist, die die Polymerisation von Glycidol initiieren können. Die
zweite Darstellungsroute verfolgt den Aufbau der neuen Komplexe durch den Einsatz
hyperverzweigter Polyglycidole mit funktionalisiertem Kern.
Abb. 40: Strategien zur Synthese neuartiger Komplexe.
In der ersten Synthesestrategie wurde der Komplex Bis[Tris-(p-hydroxyphenyl)phosphin)]-
Palladium(II)chlorid hergestellt, deprotoniert und als Starter für die Glycidol-Polymerisation
verwendet. Es entstand dabei eine hochviskose Masse, die auf die gelungene ROMBP schließen
lässt. Für den zweiten Darstellungsweg wurde ein hyperverzweigtes Polyglycidol mit Phosphan-
Kern in zahlreichen Komplexierungsreaktionen mit diversen Metallsalzen und Metall-
Carbonylprecursoren umgesetzt. Es gibt etliche Hinweise auf die erfolgreiche Synthese der
neuartigen Komplexe, letztendlich bewiesen werden konnte sie jedoch nicht. Um neue
Verbindungen hinreichend charakterisieren zu können, sind üblicherweise Methoden wie NMR-
Spektroskopie, Elementaranalyse, Röntgenstrukturanalyse und Bestimmung des Schmelzpunkts
erforderlich. Diese Analyseverfahren stoßen bei den hyperverzweigten Liganden an ihre
Grenzen. Im Gegensatz zu den perfekt verzweigten, monodispersen Dendrimeren zählen die
hochverzweigten Moleküle zu den Polymeren und sind damit auch auf die für Polymere
zugänglichen Analysemethoden beschränkt.
Nichtsdestotrotz hat die Idee der neuartigen Komplexe mit chiralen, hyperverzweigten
Polyglycidolliganden nichts von ihrer Brisanz verloren. Mit der Verbesserung der bestehenden
und der Entwicklung neuer Analyseverfahren werden in absehbarer Zeit geeignete Methoden zur
Verfügung stehen, die die Weiterentwicklung dieses Themas beschleunigen werden.

II.5 Summary
- 198 -
II.5 Summary
Chiral hyperbranched polyglycidols are highly interesting macromolecules that can be produced
in a one-pot synthesis by ring-opening multi-branching polymerization of glycidol (ROMBP).33
The initiator molecule will be present as functional core within the product, which offers the
possibility of further reactions. Aim of this work was the synthesis of newly complexes bearing
hyperbranched polyglycerols as ligands (see Fig. 1).
Fig. 1: Newly metal complex, bearing ligands out of chiral, hyperbranched polymers.
In order to succeed in the anionic polymerization it is necessary to consider numerous side
reactions. It has been shown that phosphanes (e.g. diphenylphosphane) are, due to their good
nucleophilicity but weak basicity, excellent initiators for the synthesis of polyglycerols. The
influence of several reaction parameters like temperature or kind of monomer addition has been
studied in detail. Therefore, it has been possible to adjust product properties like molecular
weight or degree of branching by varying the appropriate determining factors. Reprocessing
methods for the crude polymers have been found so that polydispersities smaller than 1.2 are
enabled.
For the synthesis of the new complexes two different strategies have been pursued (see Fig. 2).
The first route consists in the formation of a “conventional” phosphane complex containing
substituents that can initiate the polymerization of glycidol. The second strategy is the building
of the new complexes by application of polyglycidols with functionalized core.

II.5 Summary
- 199 -
Fig. 2: Strategies to synthesize new complexes.
Within the first strategy the complex bis[tris-(p-hydroxyphenyl)phosphane)]palladium(II)-
chloride has been synthesized, deprotonated and used as initiator for the polymerization of
glycidol. A highly viscous product has been obtained that suggests the successful ROMBP. For
the second synthesis route a hyperbranched polyglycerol including a phosphane core has been
brought to reaction with several metal salts and metal carbonyl precursors. There are many hints,
that the syntheses have been successful, but there is no final evidence. In order to characterize
new molecules adequately, methods like NMR-spectroscopy, elemental analysis, X-ray
diffraction analysis and determination of the melting point are necessary. All these methods
reach their limits for the hyperbranched ligands. In contrast to the perfectly branched,
monodispers dendrimers hyperbranched molecules are among the class of polymers and
therefore limited to analytics suitable for polymers.
Nevertheless, the idea of new complexes with chiral, hyperbranched polyglycidol ligands is still
a hot topic. With the improvement of the established methods as well as the development of new
analytics there will be the chance to advance within this subject.

II.6 Experimenteller Teil
- 200 -
II.6 Experimenteller Teil
II.6.1 Arbeitstechniken und –materialien
Alle experimentellen Arbeiten wurden unter Anwendung von Standard-Schlenktechniken unter
Argonatmosphäre durchgeführt.
Glycidol (96%, GC) wurde von sigmaaldrich bezogen, vor Gebrauch destilliert und unter bei
5 °C unter Argon aufbewahrt. Lösungsmittel wie Diethylether und Tetrahydrofuran wurden vor
Gebrauch durch Destillation über Natrium absolutiert. Alle Lösungsmittel für die Darstellung der
Phosphane wurden vor Gebrauch gründlich entgast.
Phenol (p.a.), Triphenylphosphin (zur Synthese), Chlordiphenylphosphin, Magnesium,
Ammoniumchlorid, Methanol (>99,9%), HBr (47%ig), THF, Essigsäure, Natriumhydroxid, PCl3
(>99%), Natrium, Dichlormethan, Ethanol und Acetonitril stammen von der Firma Merck,
Diphenylphosphin (99%) von ABCR, p-Bromanisol (99%) von sigmaaldrich und wurden ohne
weitere Aufreinigung eingesetzt. CODPdCl2 wurde im anorganischen Fortgeschrittenen-
praktikum nach der Vorschrift von Brauer28 synthetisiert.
II.6.2 Polymercharakterisierung
Die NMR-Messungen wurden bei einer Temperatur von 298 K an einem Bruker Avance 400
Spektrometer durchgeführt. Als Referenz der chemischen Verschiebung diente Tetramethylsilan
sowie bei 31P-NMR-Messungen 85%ige Phosphorsäure (H3PO4).
Molekulargewichte und Molekulargewichtsverteilungen wurden durch Gelpermeations-
chromatographie (GPC) bestimmt. Als Säulen kamen PSS Suprema Säulen (Polymer Standard
Service, Mainz, Deutschland) zum Einsatz. Das Gerät der Firma Waters bestand aus einem
Autosampler 717, Controller 600, Brechnungsindexdetektor sowie einem Entgaser. Die
Messungen wurden bei Raumtemperatur mit Wasser (Lichrosolv, Merck) bei einer
Flussgeschwindigkeit von 1 mL/min durchgeführt. Die Auswertung erfolgte gegen PSS PEG
Standards.
Die MALDI-Spektren wurden an einem Bruker Daltronics Reflex III mit Dithranol als Matrix
aufgenommen.

II.6 Experimenteller Teil
- 201 -
II.6.3 Synthese hyperverzweigter Polyglycidole
II.6.3.1 Polymerisation von Glycidol mit Phenol als Starter
Ansatz:
Glycidol (frisch destilliert): 30 mmol 2 mL
Phenol: 1 mmol 94 mg
Durchführung:
Phenol (1 mmol, 94 mg) wird in einem Schlenkrohr vorgelegt. Anschließend wird Glycidol
(2 mL, 30 mmol) auf einmal hinzugegeben und bei RT gerührt. Das Voranschreiten der Reaktion
wird durch Entnahme von NMR-Proben nach 5 h sowie nach einem Tag kontrolliert. In den
NMR-Spektren können ausschließlich die nicht umgesetzten Edukte erkannt werden.
NMR: 1H-NMR (400 MHz, MeOD):
δ [ppm]: Phenol: 6,78 (m, 2 H, meta-H); 7,16 (m, 3 H, ortho-H, para-H).
Glycidol: 2,63 (m, 1 H, CH2-Ring); 2,76 (m, 1 H, CH2-Ring); 3,01 (m,
1 H, CH-Ring); 3,48 (m, 1 H, CH2-OH); 3,79 (m, 1 H, CH2-OH). 13C-NMR (100 MHz, MeOD):
δ[ppm]: Phenol: 116,2 (s, 2 C, C2/C6); 120,5 (s, 1 C, C4); 130,4 (s, 2 C, C3/C5),
158,3 (s, 1 C, C1).
Glycidol: 44,9 (s, 1 C, CH2-Ring); 53,4 (s, 1 C, CH-Ring); 63,5 (s, 1 C,
CH2-OH).

II.6 Experimenteller Teil
- 202 -
II.6.3.2 Polymerisation von Glycidol mit Natriumphenolat als Starter
Ansatz:
Glycidol (frisch destilliert): 30 mmol 2 mL
Natriumphenolat: 0,8 mmol 95 mg
Durchführung:
Natriumphenolat (0,8 mmol, 95 mg) wird in einem Schlenkrohr vorgelegt. Anschließend wird
Glycidol (2 mL, 30 mmol) auf einmal hinzugegeben und bei RT gerührt. Das Voranschreiten der
Reaktion wird durch Entnahme von NMR-Proben nach 5 h sowie nach einem Tag kontrolliert.
Am nächsten Tag ist die Lösung bereits hochviskos und deutet somit auf eine erfolgreiche
Polymerisation hin. Ein Indiz dafür, dass die Polymerisation vom Sauerstoff des Phenolat-Ions
ausgegangen ist, ist die Verschiebung der Kerne im NMR im Vergleich zu reinem Phenol.
NMR: 1H-NMR (400 MHz, MeOD):
δ[ppm]: Phenol-Polymer: 6,93 (m, 2 H, meta-H); 7,27 (m, 3 H, ortho-H, para-H).
13C-NMR (100 MHz, MeOD):
δ[ppm]: Phenol-Polymer: 115,6 (s, 2 C, C2/C6); 121,8 (s, 1 C, C4); 130,4 (s, 2 C,
C3/C5), 160,3 (s, 1 C, C1).

II.6 Experimenteller Teil
- 203 -
II.6.3.3 Polymerisation von Glycidol mit Triphenylphosphin als Starter
Ansatz:
Glycidol (frisch destilliert): 30 mmol 2 mL
Triphenylphosphin: 1 mmol 262 mg
Durchführung:
Triphenylphosphin (1 mmol, 262 mg) wird in einem Schlenkrohr vorgelegt und 0,5 mL Glycidol
zugegeben. Nach 15, 45 und 105 min werden jeweils weitere 0,5 mL Glycidol zugefügt.
Zunächst entsteht eine milchig-weiße Suspension. Nach der letzten Zugabe wird die Suspension
heller, 20 min später ist die Lösung klar und schwach gelb gefärbt. Weiterrühren der
Reaktionslösung bei RT. Nach einem Tag ist die Lösung ist fest geworden.
II.6.3.4 Polymerisation von Glycidol mit Diphenylphosphin als Starter
Ansatz:
Glycidol (frisch destilliert): 30 mmol 2 mL
Diphenylphosphin: 1 mmol 190 mg
0,17 mL
Durchführung:
Glycidol (30 mmol, 2 mL) wird in ein Schlenkrohr vorgelegt und Diphenylphosphin (1 mmol,
190 mg, 0,17 mL) zugegeben. Es wird bei RT gerührt über Nacht. Nach einem Tag ist die
Lösung ist fest geworden und hat sich orange gefärbt.

II.6 Experimenteller Teil
- 204 -
II.6.4 Synthese von Para-hydroxyphenyl-phosphanen
II.6.4.1 Synthese von Diphenyl-[p-methoxy-phenyl]-phosphan
Ansatz:
p-Bromanisol: 73 mmol 13,67 g
Chlordiphenylphosphin: 100 mmol 2,43 g
Magnesium-Späne: 73 mmol 16,12 g,
13,4 mL
Diethylether: 200 mL
Ammoniumchlorid-Lösung (entgast)
Methanol (entgast)
Durchführung:
Zu 2,4 g Mg-Späne in 160 mL Diethylether wird ein Gemisch von 40 mL Ether mit
p-Bromanisol (73 mmol, 13,67 g) bei RT zugetropft. Nach Anspringen des Grignards wird für
4 h unter Rückfluss gekocht und abschließend bei RT über Nacht gerührt. Zur abgetrennten
Grignard-Lösung wird Chlordiphenylphosphin (100 mmol, 2,43 g) in 40 mL Ether langsam bei
0 °C zugetropft und bei RT über Nacht gerührt. Die Reaktionsmischung wird durch Zugabe von
entgaster Ammoniumchlorid-Lösung hydrolysiert. Die etherische Phase wird abgetrennt und
über Natriumsulfat getrocknet. Einengen am Vakuum führt zunächst zu einem gelben Öl
(Rohausbeute 17,88 g). Umkristallisation aus 5 mL Methanol ergibt einen weißen kristallinen
Feststoff.
NMR: 13C-NMR (100 MHz, CDCl3):
δ [ppm]: 160,4 (s, C4); 137,8 (d, 1J = 10.3 Hz, C5); 135,6 (d, 2J = 21.4 Hz, C2);
133,4 (d, 2J = 19.1 Hz, C6); 128,4 (d, 3J = 9.3 Hz, C7); 128,4 (s, C8);
128,4 (d, 1J = 6.7 Hz, C1); 114,2 (d, 3J = 8.1 Hz, C3); 55,1 (s, O-CH3).
31P-NMR (CDCl3): δ [ppm]: - 6,8.
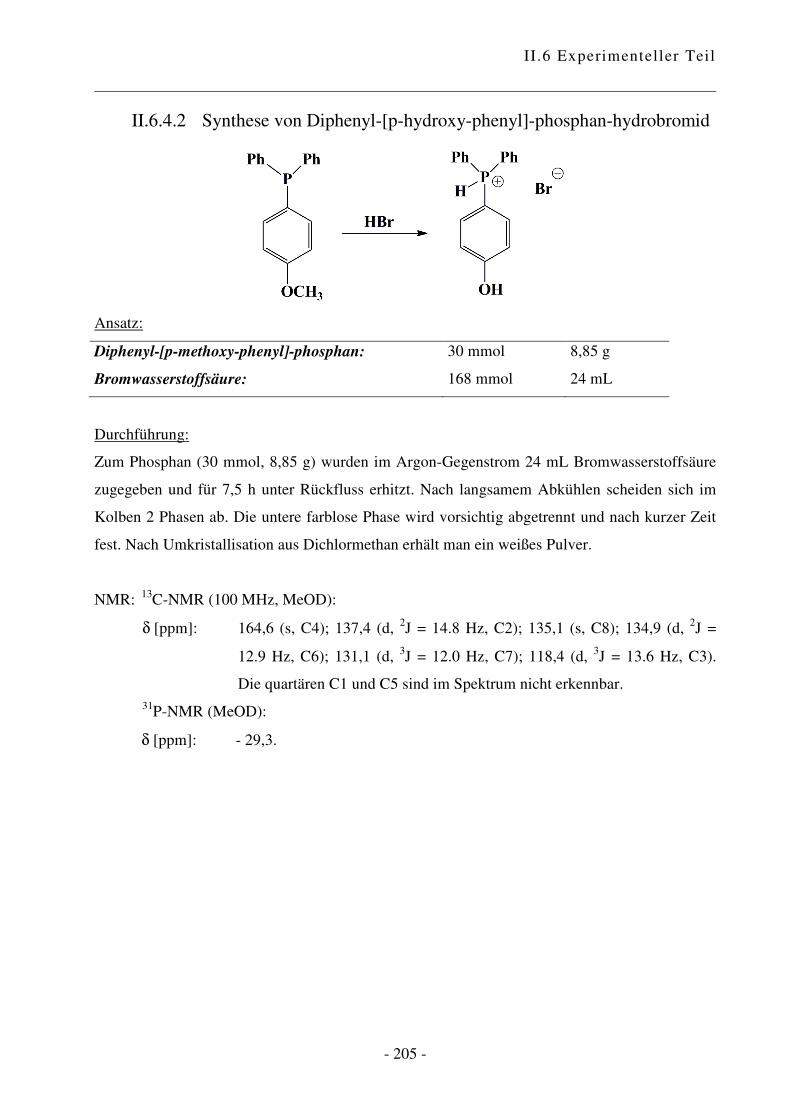
II.6 Experimenteller Teil
- 205 -
II.6.4.2 Synthese von Diphenyl-[p-hydroxy-phenyl]-phosphan-hydrobromid
Ansatz:
Diphenyl-[p-methoxy-phenyl]-phosphan: 30 mmol 8,85 g
Bromwasserstoffsäure: 168 mmol 24 mL
Durchführung:
Zum Phosphan (30 mmol, 8,85 g) wurden im Argon-Gegenstrom 24 mL Bromwasserstoffsäure
zugegeben und für 7,5 h unter Rückfluss erhitzt. Nach langsamem Abkühlen scheiden sich im
Kolben 2 Phasen ab. Die untere farblose Phase wird vorsichtig abgetrennt und nach kurzer Zeit
fest. Nach Umkristallisation aus Dichlormethan erhält man ein weißes Pulver.
NMR: 13C-NMR (100 MHz, MeOD):
δ [ppm]: 164,6 (s, C4); 137,4 (d, 2J = 14.8 Hz, C2); 135,1 (s, C8); 134,9 (d, 2J =
12.9 Hz, C6); 131,1 (d, 3J = 12.0 Hz, C7); 118,4 (d, 3J = 13.6 Hz, C3).
Die quartären C1 und C5 sind im Spektrum nicht erkennbar. 31P-NMR (MeOD):
δ [ppm]: - 29,3.

II.6 Experimenteller Teil
- 206 -
II.6.4.3 Synthese von Diphenyl-[p-hydroxy-phenyl]-phosphan
Ansatz:
Diphenyl-[p-methoxy-phenyl]-phosphan-
hydrobromid:
21,3 mmol 7,66 g
Natrium: 21,3 mmol 0,49 g
Ethanol (entgast): 50 mL
Dichlormethan (entgast): 45 mL
Durchführung:
Zunächst wird Natrium (0,49 g) in 50 mL Ethanol unter Bildung von Natriumethanolat gelöst.
Die Natriumethanolat-Lösung wird zum Phosphan gegeben und über Nacht bei RT gerührt. Nach
Einengen am Vakuum entsteht ein Öl, das in 45 mL Dichlormethan wieder aufgenommen wird.
Die Lösung wird vom NaBr abfiltriert und am Vakuum zu einem fast farblosen Pulver eingeengt.
NMR: 13C-NMR (100 MHz, MeOD):
δ [ppm]: 159,8 (s, C4); 139,5 (d, 1J = 10.5 Hz, C5); 136,8 (d, 2J = 21.8 Hz, C2);
134,3 (d, 2J = 19.2 Hz, C6); 129,5 (s, C8); 129,4 (d, 3J = 7.0 Hz, C7);
116,8 (d, 3J = 8.2 Hz, C3). 31P-NMR (MeOD):
δ [ppm]: - 36,8.
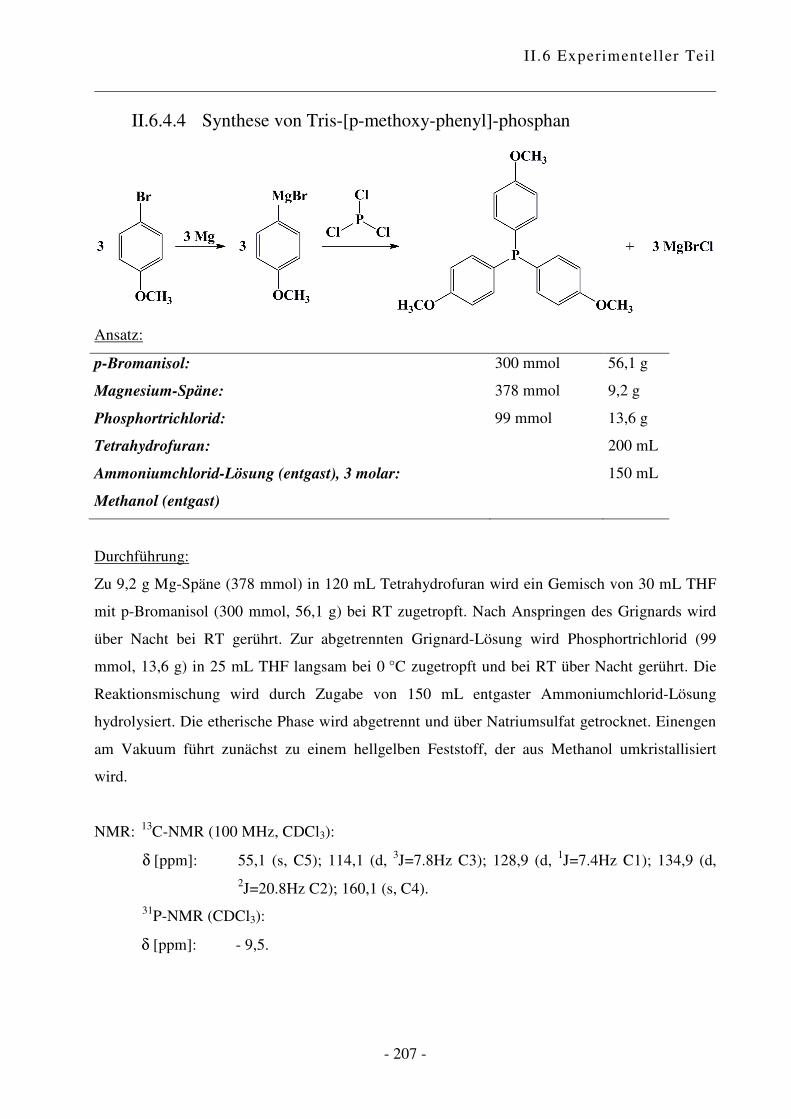
II.6 Experimenteller Teil
- 207 -
II.6.4.4 Synthese von Tris-[p-methoxy-phenyl]-phosphan
Ansatz:
p-Bromanisol: 300 mmol 56,1 g
Magnesium-Späne: 378 mmol 9,2 g
Phosphortrichlorid: 99 mmol 13,6 g
Tetrahydrofuran: 200 mL
Ammoniumchlorid-Lösung (entgast), 3 molar: 150 mL
Methanol (entgast)
Durchführung:
Zu 9,2 g Mg-Späne (378 mmol) in 120 mL Tetrahydrofuran wird ein Gemisch von 30 mL THF
mit p-Bromanisol (300 mmol, 56,1 g) bei RT zugetropft. Nach Anspringen des Grignards wird
über Nacht bei RT gerührt. Zur abgetrennten Grignard-Lösung wird Phosphortrichlorid (99
mmol, 13,6 g) in 25 mL THF langsam bei 0 °C zugetropft und bei RT über Nacht gerührt. Die
Reaktionsmischung wird durch Zugabe von 150 mL entgaster Ammoniumchlorid-Lösung
hydrolysiert. Die etherische Phase wird abgetrennt und über Natriumsulfat getrocknet. Einengen
am Vakuum führt zunächst zu einem hellgelben Feststoff, der aus Methanol umkristallisiert
wird.
NMR: 13C-NMR (100 MHz, CDCl3):
δ [ppm]: 55,1 (s, C5); 114,1 (d, 3J=7.8Hz C3); 128,9 (d, 1J=7.4Hz C1); 134,9 (d,
2J=20.8Hz C2); 160,1 (s, C4). 31P-NMR (CDCl3):
δ [ppm]: - 9,5.

II.6 Experimenteller Teil
- 208 -
II.6.4.5 Synthese von Tris-[p-hydroxy-phenyl]-phosphan-hydrobromid
Ansatz:
Tris-[p-methoxy-phenyl]-phosphan: 49 mmol 17,4 g
Bromwasserstoffsäure: 70 mL
Durchführung:
Zum Phosphan (49 mmol, 17,4 g) wurden im Argon-Gegenstrom 70 mL Bromwasserstoffsäure
zugegeben und für 5,5 h unter Rückfluss erhitzt. Nach langsamem Abkühlen hat sich im Kolben
ein orange-farbener Niederschlag gebildet, der abgetrennt und getrocknet wird. Der Niederschlag
wird in Methanol gelöst, vom nicht umgesetzten Edukt abfiltriert und am Vakuum bis zum
Erhalt eines farblosen Pulvers eingeengt.
NMR: 13C-NMR (100 MHz, MeOD):
δ [ppm]: 106,8 (d, 1J=96.7Hz C1); 118,6 (d, 2J=14.7Hz C2); 137,1 (d, 3J=13.1Hz
C3); 165,2 (d, 4J=2.9Hz C4). 31P-NMR (MeOD):
δ [ppm]: - 28,4.
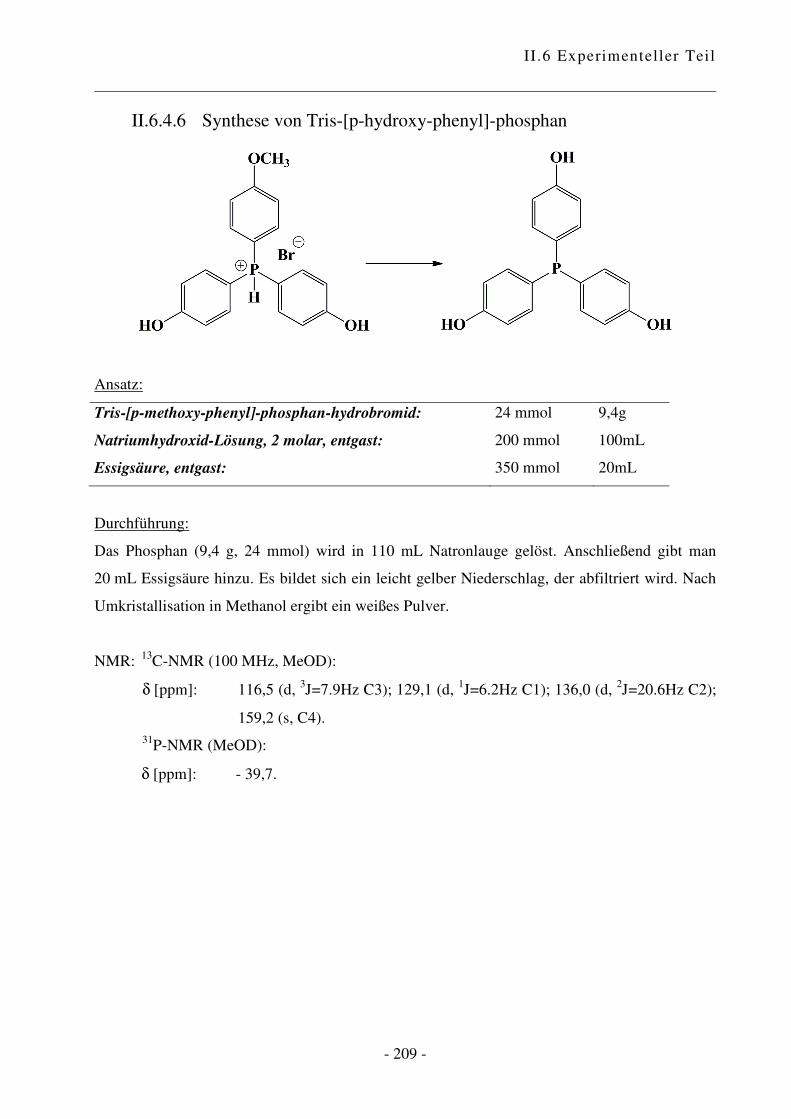
II.6 Experimenteller Teil
- 209 -
II.6.4.6 Synthese von Tris-[p-hydroxy-phenyl]-phosphan
Ansatz:
Tris-[p-methoxy-phenyl]-phosphan-hydrobromid: 24 mmol 9,4g
Natriumhydroxid-Lösung, 2 molar, entgast: 200 mmol 100mL
Essigsäure, entgast: 350 mmol 20mL
Durchführung:
Das Phosphan (9,4 g, 24 mmol) wird in 110 mL Natronlauge gelöst. Anschließend gibt man
20 mL Essigsäure hinzu. Es bildet sich ein leicht gelber Niederschlag, der abfiltriert wird. Nach
Umkristallisation in Methanol ergibt ein weißes Pulver.
NMR: 13C-NMR (100 MHz, MeOD):
δ [ppm]: 116,5 (d, 3J=7.9Hz C3); 129,1 (d, 1J=6.2Hz C1); 136,0 (d, 2J=20.6Hz C2);
159,2 (s, C4). 31P-NMR (MeOD):
δ [ppm]: - 39,7.

II.6 Experimenteller Teil
- 210 -
II.6.5 Synthese der Phosphan-Komplexe
II.6.5.1 Synthese von Bis[Tris-(p-hydroxyphenyl)phosphin)]Palladium(II)-
chlorid
Ansatz:
CODPdCl2: 6 mmol 1,7 g
Tris-[p-hydroxy-phenyl]-phosphan: 15 mmol 4,5 g
Acetonitril, entgast: 430 mL
Durchführung:
Tris-(p-hydroxyphenyl)phosphin (15 mmol, 4,5 g) wurde in 180mL Acetonitril gelöst und zu
einer Lösung von CODPdCl2 (6 mmol, 1,7 g) in 250mL Acetonitril bei 0 °C zugetropft. Nach
beendeter Zugabe wurde 30 min im Eisbad und anschließend 1,5 h bei RT gerührt. Anschließend
wurde die erhaltene Suspension filtriert und der gelbe Niederschlag am Vakuum getrocknet
NMR: 13C-NMR (100 MHz, MeOD):
δ [ppm]: 116,2 (d, 3J=14.4Hz C3); 137,8 (d, 2J=12.0Hz C2); 159.2 (s, C4).
Elementaranalyse: erwartet: C: 54,19 H: 3,79
gefunden: C: 54,02 H: 4,14 N: 0,90
(Im Produkt befanden sich Reste von Acetonitril.)

II.7 Literatur
- 211 -
II.7 Literatur
1 Lagasse, F.; Kagan, H. B., Chem. Pharm. Bull. 2000, 48, (3), 315 - 324. 2 Tang, W.; Zhang, Xumu, Chem. Rev. 2003, 103, 3029 - 3069. 3 http://nobelprize.org, Pressemitteilung: Der Nobelpreis in Chemie 2001. 4 Janiak, C.; Klapötke, T. M.; Meyer, H.-J., Moderne Anorganische Chemie. 2 ed.; Walter de Gruyter GmbH & Co.:
Berlin, 2003; p 801. 5 Römpp-Lexikon online: http://www.roempp.com/prod/ 6 Übersichtsartikel: Voit, B. I., Acta Polymer. 1995, 46, 87 - 99. 7 Übersichtsartikel: Bosman, A. W.; Janssen, H. M.; Meijer, E. W., Chem. Rev. 1999, 99, 1665 - 1688. 8 Übersichtsartikel: Fréchet, J. M. J.; Hawker, C. J.; Gitsov, I.; Leon, J. W., Pure Appl. Chem. 1996, A33, (10), 1399
- 1425. 9 Voit, B., Chemie in Dresden 2004, 94 - 99. 10 Kim, Y. H., J. Polym. Sci. Part A: Polym. Chem. 1998, 36, 1685 - 1698. 11 Uhrich, K. E.; Hawker, C. J.; Fréchet, J. M. J.; Turner, S. R., Macromolecules 1992, 25, 4583 - 4587. 12 Lapienis, G.; Penczek, S., Biomacromolecules 2005, 6, 752 - 762. 13 Flory, P. J., J. Am. Chem. Soc. 1952, 74, 2718 - 2723. 14 Voit, B., J. Polym. Sci. Part A: Polym. Chem. 2000, 38, 2505 - 2525. 15 Royappa, A. T.; Vogt, M. L.; Sharma, V., J. Appl. Polym. Sci. 2004, 91, 1344 - 1351. 16 Tokar, R.; Kubisa, P.; Penczek, S.; Dworak, A., Macromolecules 1994, 27, 320 - 322. 17 Dworak, A.; Walach, W.; Trzebicka, B., Macromol. Chem. Phys. 1995, 196, 1963 - 1970. 18 Streitwieser, A.; Heathcock, C. H., Organische Chemie. ed.; Verlag Chemie: Weinheim, 1980; p 1489. 19 Sunder, A.; Mülhaupt, R.; Haag, R.; Frey, H., Macromolecules 2000, 33, 253 - 254. 20 Tieke, B., Makromolekulare Chemie. 2 ed.; WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim: Weinheim,
2005; p 368. 21 Henderson, W. A.; Streuli, C. A., J. Am. Chem. Soc. 1960, 82, 5791 - 5794. 22 Henderson, W. A.; Buckler, S. A., J. Am. Chem. Soc. 1960, 82, 5794 - 5800. 23 Jin, Z.; Yan, Y.; Zuo, H.; Fell, B., J. prakt. Chem. 1996, 338, 124 - 128. 24 Neunhoeffer, O.; Lamza, L., Chem. Ber. 1961, 94, 2514 - 2518. 25 Neunhoeffer, O.; Lamza, L., Chem. Ber. 1961, 94, 2519 - 2521. 26 Senear, A. E.; Valient, W.; Wirth, J., J. Org. Chem. 1960, 25, 2001 - 2006. 27 Berger, S.; Braun, S.; Kalinowski, H.-O., NMR-Spektroskopie von Nichtmetallen. Bd. 3. 31P-NMR-Spektroskopie.
ed.; Georg Thieme Verlag: Stuttgart, 1993; p 205. 28 Brauer, G.; Handbuch der Präparativen Anorganischen Chemie. 3. Band, Ferdinand Enke Verlag: Stuttgart,1981. 29 Elschenbroich, Ch.; Salzer, A., Organometallchemie. 3 ed.; Teubner Studienbücher Chemie: Stuttgart, 1993; p
562. 30 Übersichtsartikel: Basolo, F., Polyhedron 1990, 9, (13), 1503 - 1535. 31 Darensbourg, D. J.; Brown, T. L., Inorg. Chem. 1968, 7, (8), 1679 - 1680. 32 Magee, T. A.; Matthews, C. N.; Wang, T. S.; Wotiz, J. H., J. Am. Chem. Soc. 1961, 83, 3200 - 3203. 33 Sunder, A.; Hanselmann, R.; Frey, H.; Mülhaupt, R., Macromolecules 1999, 32, 4240 - 4246.

II.7 Literatur
- 212 -
34 Hölter, D.; Burgath, A.; Frey, H., Acta Polymer. 1997, 48, 30 - 35. 35 Hölter, D.; Frey, H., Acta Polymer. 1997, 48, 298 - 309. 36 Hesse, M.; Meier, H.; Zeeh, B., Spektroskopische Methoden in der organischen Chemie. Thieme: Stuttgart; New
York, 2002; 6. überarb. Aufl., p 437. 37 Hanselmann, R.; D.Hölter; Frey, H., Macromolecules 1998, 31, 3790 - 3801. 38 Bharathi, P.; Moore, J. S., Macromolecules 2000, 33, 3212 - 3218.

Danksagung
Danksagung
Mein großer Dank gilt Prof. Rieger für die Aufnahme in seine Abteilung und die Möglichkeit,
gleich zwei herausfordernde Themen zu bearbeiten. Das in mich gesetzte Vertrauen und die
Freiräume, die Themen zu entwickeln, haben mich sehr motiviert. Gespräche mit Ihnen haben
mir stets gezeigt, wie großartig Forschung ist, und wie wir Chemiker dazu beitragen, die Welt
ein Stück zu verbessern. Danke für die Geduld und Unterstützung in den letzten zwei für mich
persönlich nicht einfachen Jahren.
Ich bedanke mich ebenso herzlich bei PD Dr. Ulrich Ziener für die Erstellung des zweiten
Gutachtens.
Ganz herzlicher Dank gilt allen meinen Kollegen. Das Arbeitsklima in der Abteilung war – und
das wird mir jeder bestätigen – phänomenal. Eigentlich könnte ich alle Mitglieder der Abteilung
hier aufzählen – ich schätze Euch sehr. Besondere Erwähnung verdienen meine Laborkollegen
Tobi und Phips. Tobi hat mich quasi „großgezogen“, und Phips hat mir unglaublich viel Spaß
und Freude im Labor gebracht. Von Knut habe ich das Handwerk zum Polymerisieren gelernt,
wir beide hielten am Ende die Fahnenstange der Metallocene oben. Die Diskussionen mit
Marcus – vor allem nach dem Umzug der Abteilung – habe ich immer sehr genossen, egal ob’s
um Polymere, Bewerbungen oder Kochrezepte ging. Vielen Dank meinen Kochknechten und –
mägden Uwe, Markus, Lena, Shirin, Rantje und Johannes für die geleistete Arbeit und den Spaß,
den wir gemeinsam hatten. Dank gilt auch allen Technikern in und außerhalb der Abteilung, die
mich durch Analytik-Messungen oder Anlagenbau unterstützt haben, v.a. Siggi und Petra.
Hervorheben möchte ich Elvira, deren Motivation, Fleiß und Zeitmanagement mir immer ein
Vorbild waren.
Während meiner Zeit an der Uni hab ich zwei unglaubliche Sekretärinnen kennen gelernt: Fe
und Sigrid. Fe hat ein Selbstverständnis anderen zu helfen, wie ich es noch nie erlebt habe, und
findet das alles dann „ganz normal“. Wenn es nur mehr Menschen wie Dich gäbe! Sigrid hat
immer ein offenes Ohr für die großen und kleinen Sorgen vor allem kleiner Leute. Egal ob es um
Fragen zur Verwaltung geht oder um persönliche Probleme, bei ihr ist man immer gut
aufgehoben!
Meine Familie hat mich zu dem Menschen gemacht, der ich heute bin. Ich danke Euch für die
Unterstützung und den Beistand. Dadurch, dass ich Euch zeigen musste, das Chemie nicht
immer nur schlecht ist, sondern ganz wesentlich in allen Bereichen des Lebens mitwirkt, habe
ich viel hinterfragt und viel gewonnen. Vielleicht könnt Ihr ja irgendwann mal mit Stolz ein von
mir entwickeltes Waschmittel oder Klebeband benutzen.

Danksagung
Für eine wissenschaftliche Arbeit ist es unglaublich bereichernd, wenn nicht sogar zwingend
erforderlich, Diskussionspartner zu haben, denen man auch mal die berühmten „blöden“ Fragen
stellen kann, die ehrliche und offene Kritik üben, die aber auch loben und staunen können. Und
wenn man dann noch jemanden hat, der mit einem durch alle Höhen und Tiefen geht und diese
sogar noch versteht, weil er sie selbst auch schon erlebt hat, dann ist das einfach ein ganz großes
Glück. Lieber Dieter, Du hast mich schon mein ganzes Studium begleitet und großen Anteil
daran, dass aus mir eine Chemikerin mit Leib und Seele geworden ist. Ich liebe Dich – ganz
herzlichen Dank für alles!
Zum Schluss möchte ich mich bei all denen bedanken, die bei der Entstehung dieser Arbeit in
irgendeiner Weise beteiligt waren – sei es als Diskussionspartner, als Ratgeber, als Techniker,
als moralische Stütze oder einfach mal als willkommene Ablenkung. Ihr alle habt mitgeholfen,
dass die drei Jahre Doktorarbeit eine unvergessliche Zeit für mich waren!
Das Erstellen meiner Dissertationsschrift hat am Ende weitaus länger gedauert, als gedacht,
gehofft und als es hätte sein sollen. Ich danke allen, die den Glauben an mich nicht verloren
haben und mir durch diese schwere Zeit geholfen haben!

Eidesstattliche Erklärung
Hiermit erkläre ich, dass ich die vorliegende Arbeit von mir selbstständig und
nur unter Verwendung der angeführten Quellen und Hilfsmittel angefertigt habe.
Dresden, den
(Sandra Meinhard)
Die vorliegende Arbeit wurde im Zeitraum zwischen November 2004 und
November 2007 unter der Betreuung von Prof. Dr. Dr. h. c. B. Rieger,
Anorganische Chemie II, Universität Ulm, durchgeführt.





















![Enantioselektive Katalyse, IX. Neue optisch reine 3,4-Bis ...anorganik.uni-tuebingen.de/aknagel/VEROEFFEN/19931260505_ftp.pdf · U. Nagel, T. Krink Enantioselektive Katalyse, IX"]](https://img.pdfslide.org/doc/110x75/5e1385baf543157800699fef/enantioselektive-katalyse-ix-neue-optisch-reine-34-bis-u-nagel-t-krink.jpg)







