Embed Size (px)
Citation preview

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
Teil
IV
PhänomenologischeBegründung derThermodynamik
33
33.1 Entwicklung der Thermodynamik . . . . . . . . . . . . . . . . . . . . . .109833.2 Was ist Thermodynamik? . . . . . . . . . . . . . . . . . . . . . . . . . .110433.3 Temperatur, Zustandsgrößen und Zustandsänderungen . . . . . . . . .110733.4 Arbeit und Wärme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .111433.5 Die idealen Gasgesetze . . . . . . . . . . . . . . . . . . . . . . . . . . .111633.6 Der erste Hauptsatz . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .111833.7 Der zweite Hauptsatz: 1. Teil . . . . . . . . . . . . . . . . . . . . . . . .112433.8 Der zweite Hauptsatz: 2. Teil . . . . . . . . . . . . . . . . . . . . . . . .1134
Aufgaben . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1139Lösungen zu den Aufgaben . . . . . . . . . . . . . . . . . . . . . . . . .1141Ausführliche Lösungen zu den Aufgaben . . . . . . . . . . . . . . . . .1143Literatur . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1149
Wie verhalten sichphysikalische Systeme aussehr vielen Teilchen?
Wie können Temperaturund Wärme physikalischbeschrieben werden?
Wie arbeitenWärmekraftmaschinen?
Welche physikalischenVorgänge können spontanablaufen, welche nicht?
M. Bartelmann et al., Theoretische Physik, DOI 10.1007/978-3-642-54618-1_33, © Springer-Verlag Berlin Heidelberg 2014

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1098 33 Phänomenologische Begründung der Thermodynamik
Dieses Kapitel führt die wesentlichen Konzepte der Thermodynamikund ihre Axiome auf eine Weise ein, die keinen Bezug zur mikro-skopischen Natur der Materie nimmt. Es folgt damit zum einender historischen Entwicklung, welche die Thermodynamik ausge-hend von den Begriffen „warm“ und „kalt“ über das Bedürfnis,Wärmekraftmaschinen zu verstehen, bis hin zur Formulierung ih-rer sogenannten Hauptsätze genommen hat. Zum anderen zeigt es,wie und warum die Thermodynamik axiomatisch aufgebaut werdenkann, ohne eine präzise Vorstellung vom Aufbau der Materie zu ha-ben.
Zentral in diesem Kapitel sind die drei Hauptsätze, die aus his-torisch-konventionellen Gründen mit null beginnend nummeriertwerden und die als Axiome der Thermodynamik gelten können. Dernullte Hauptsatz definiert den physikalischen Temperaturbegriff, dererste Hauptsatz legt fest, wie verschiedene Formen von Energie in-einander umgewandelt werden können, und der zweite Hauptsatzklärt anhand des Begriffs der Entropie, welche dieser Umwandlun-gen überhaupt physikalisch möglich sind.
Zur Formulierung der Hauptsätze werden weitere Begriffe benö-tigt, die im Lauf des Kapitels eingeführt werden. Besonders wichtigsind die Begriffe der Zustands- und Prozessgrößen. Neben der For-mulierung der Hauptsätze ist es das wesentliche Anliegen diesesKapitels zu begründen, warum die Thermodynamik als allgemeineTheorie von den Energieumwandlungen makroskopischer Systemeso fundamental ist, dass sie die Revolutionen der Relativitätstheo-rien und der Quantentheorie weitgehend unverändert überstehenkonnte, gerade weil sie von jeder detaillierten mikroskopischen In-formation absieht.
Oft wird in diesem und in den folgenden Kapiteln allgemein undabstrakt von „physikalischen Systemen“ die Rede sein. Unter ei-nem „System“ verstehen wir hier einen wohldefinierten Ausschnittder Welt, der durch einen geschlossenen Rand von seiner Umge-bung abgegrenzt werden kann, aber nicht notwendigerweise davonisoliert ist. Wir unterscheiden mikroskopische und makroskopischeSysteme nach der Anzahl ihrer Freiheitsgrade. Bei welcher Anzahlvon Freiheitsgraden ein System als makroskopisch gelten kann, wirdspäter durch den Begriff der Stoffmenge und durch die Avogadro-Zahl einen präzisen Sinn bekommen.
Die Hauptsätze der Thermodynamik werden in diesem Kapitel vorallem anhand der idealen Gasgesetze und verschiedener Kreispro-zesse erläutert und vertieft. Das Kapitel schließt mit einer Diskussi-on der Entropiezunahme bei irreversiblen Prozessen, insbesonderebei Wärmeleitung und Mischungsvorgängen, anhand derer dasKonzept des reversiblen Ersatzprozesses eingeführt wird.
Die fünf Kapitel von Teil IV über Thermodynamik und statisti-sche Physik bilden fünf Durchgänge durch ähnliche Themen. JederDurchgang vertieft die vorige Diskussion und fügt neue Konzeptehinzu. Auf Kap. 33, in dem die Thermodynamik phänomenologischeingeführt wird, folgt ihre statistische Begründung (Kap. 34). Ins-besondere die Entropie wird dann auf eine Abzählung möglicherZustände zurückgeführt. In Kap. 35 werden die bis dahin entwickel-ten Konzepte erweitert und auf eine Reihe verschiedener Systemeangewandt, die von idealen zu realen Gasen und Phasenübergän-gen reichen. In Kap. 36 wird der Begriff der Ensembles geschärft
und mit dem Konzept der Zustandssummen versehen, von denengezeigt wird, dass in ihnen alle Information thermodynamischerSysteme im Gleichgewicht enthalten ist. Das abschließende Kap. 37stellt schließlich dar, wie die bis dahin eingeführten Konzepte aufquantenphysikalische Systeme erweitert werden können.
33.1 Entwicklungder Thermodynamik
Warm und kalt
Die physikalische Wärmelehre hat sich mit der Industrialisie-rung wesentlich aus dem Bedürfnis heraus entwickelt, die Funk-tion und das Verhalten von Wärmekraftmaschinen zu verstehen.Wärmekraftmaschinen sind periodisch arbeitende Maschinen,die in jedem Zyklus eine gewisse Wärmemenge aufnehmen,eine geringere Wärmemenge abgeben und dabei Arbeit ver-richten. Abbildung 33.1 zeigt als Beispiel eine Niederdruck-dampfmaschine. Gerade zu Beginn des Maschinenzeitaltershaben Ingenieure erheblich dazu beigetragen, die wesentlichenKonzepte der Wärmelehre zu klären und ihre Begriffe zu schär-fen. Das Verständnis der grundlegenden thermodynamischenGrößen Temperatur und Wärme ging mit dem technischen Fort-schritt Hand in Hand.
Entscheidende Schritte auf dem Weg zu einer Physik der Wärmewaren die Entwicklung von Methoden zur Messung der Tem-peratur, die Entdeckung der Gasgesetze, die Vorstellung vonWärme als einer Größe mit Substanzcharakter und die Erfor-schung des Zusammenhangs zwischen Wärme und Arbeit.
Die Schärfung der Begriffe „warm“ und „kalt“ zu einer Tem-peraturskala wurde von der Entwicklung erster sogenannterThermoskope und Thermometer begleitet, die zunächst dembloßen Nachweis einer Erwärmung dienten, aber zunehmendgenaue Messungen erlaubten.
Bereits im ägyptischen Alexandria hatte man die Tatsache ge-kannt und genutzt, dass sich Luft bei Wärmezufuhr ausdehnt.Der wesentliche Fortschritt bei der Messung von Temperaturenwurde durch den Übergang zum geschlossenen Flüssigkeits-thermometer möglich. Er wurde wohl zunächst bei den Un-tersuchungen in der florentinischen Akademie vollzogen undwird Ferdinand II., Herzog von Toskana, zugeschrieben. Abge-schlossen hat ihn später um 1700 der Danziger Physiker DanielFahrenheit (1686–1736).
Fahrenheits Temperaturskala
Um negative Temperaturen möglichst zu vermeiden, leg-te Fahrenheit den Nullpunkt seiner Temperaturskala aufdie niedrigste Temperatur fest, die er im strengen Win-ter 1708/09 in Danzig feststellen konnte: Sie lag bei

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.1 Entwicklung der Thermodynamik 1099
Teil
IV
Abb. 33.1 Stehende Einzylinderdampfmaschine der Silbermine „Alte Elisa-beth“ in Freiberg (Sachsen). Die Maschine wurde 1848 nach dem Watt’schenPatent gebaut und hatte eine Leistung von 12 PS
�17;8 ıC. Den Gefrierpunkt des Wassers legte er bei32 ıF fest. Für die Umrechung von Celsius- in Fahren-heit-Grade ergibt sich daraus die Formel
.Fahrenheit/ D 32
17;8.Celsius/ C 32 � 9
5.Celsius/ C 32 :
J
Ausdehnungsgesetze von Gasen
Für das Studium der Ausdehnungsgesetze von Gasen wurdees wichtig, mit Luftpumpen den Gasdruck innerhalb weiterGrenzen ändern zu können. Den Zusammenhang zwischen demDruck P eines Gases und seinem Volumen V untersuchte derirische Naturforscher Robert Boyle (1627–1692) um die Jahre1660/1661. Richard Townley (1629–1707) wiederholte BoylesVersuche und erkannte das Gesetz, das wir heute in der FormPV D const schreiben. Boyle veröffentlichte es im Jahre 1669als Townley’sches Gesetz, und der französische Physiker EdmeMariotte (vermutlich 1620–1684) gab es 1676 ebenfalls an.
Die einfachen Volumenverhältnisse, in denen Gase zu Ver-bindungen zusammentreten, wurden im Jahre 1805 von dememinenten deutschen Naturforscher Alexander von Humboldt(1769–1859) und dem französischen Physiker und ChemikerJoseph Gay-Lussac (1778–1850) anhand der Elektrolyse vonWasser in Sauerstoff und Wasserstoff ermittelt. In allgemeiner
Form wurden sie 1808 von Gay-Lussac und dem englischenNaturforscher und Lehrer John Dalton (1766–1844) ausgespro-chen.
Das Gesetz über die Abhängigkeit von Volumen und Druckeines Gases von seiner Temperatur wurde 1833 ebenfalls vonGay-Lussac entdeckt. Er fand, dass Druck und Volumen genü-gend verdünnter Gase bei sonst gleichbleibenden Bedingungenlinear mit der Temperatur zunehmen, unabhängig davon, welcheGase er betrachtete. Ein wesentlicher Aspekt des gleicharti-gen Verhaltens vieler verschiedener Gase war aber schon indem Gesetz der einfachen ganzzahligen Volumenverhältnissebei Verbindungen von Gasen erkannt worden, also ebenfallsschon 1808. Der italienische Naturwissenschaftler Amedeo Avo-gadro (1776–1856) deutete es 1811, indem er postulierte, dassbei gegebenem Druck und gegebener Temperatur in einem be-stimmten Volumen eines Gases jeweils dieselbe Anzahl vonMolekülen enthalten ist.
Wärmestoff und Wärmekapazität
Lange herrschte die Meinung vor, Wärme sei eine Substanz(Caloricum genannt), die von einem Körper auf einen anderenübergehen könne und dabei der Menge nach unverändert blei-be. Von einer Substanz ähnlich elementaren Charakters, demsogenannten Phlogiston, wurde angenommen, dass es Körperndurch Erwärmung zugeführt werde und bei Verbrennung ent-weiche. Diese Meinung hat ihren Ursprung in der Erfahrung,dass Stoffe verschiedene Wärmekapazitäten haben: Ihre Tem-peratur erhöht sich bei der Zufuhr gleicher Wärmemengen umverschiedene Beträge.
Kalorimeter wurden schon ab der Mitte des 18. Jahrhunderts be-nutzt. Bereits 1760 wurden die Schmelzwärme des Eises und dieVerdampfungswärme des Wassers von dem Chemiker JosephBlack (1728–1799) recht genau kalorimetrisch gemessen. Blackwar der Lehrer von James Watt (1736–1819). Ihm verdankenwir viele quantitative Begriffe der physikalischen Wärmeleh-re, wie z. B. Wärmemenge, spezifische Wärme, latente Wärmesowie Schmelz- und Siedetemperatur. Insbesondere erkannteBlack, dass sich beim Erwärmen von Eis die Temperatur solange nicht ändert, bis das Eis vollständig geschmolzen ist. Erschloss daraus, dass Temperatur und Wärme im Sinn einer In-tensität und einer Quantität unterschieden werden müssen.
Kalorimeter
Kalorimeter vergleichen die aufgenommene Wärme mitbekannten Wärmemengen. Beispielsweise dient im Eis-kalorimeter die Schmelzwärme von Wassereis als Ver-gleichsgröße. Anhand der Wärmemenge, die notwendigist, um die Temperatur einer bestimmten Menge Was-sers um einen bestimmten Betrag zu erhöhen, wurde die

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1100 33 Phänomenologische Begründung der Thermodynamik
Kalorie (cal) als Einheit der Wärmemengen festgelegt.Sie wurde schließlich 1948 offiziell abgeschafft, langenachdem man erkannt hatte, dass Wärme eine Form derEnergie ist und daher in Joule gemessen werden kann.Dennoch wird sie heute noch an vielen Stellen verwen-det. J
Mit seinen Untersuchungen widerlegte Black die zu seiner Zeitverbreitete Meinung, dass die spezifische Wärme für Körper mitgleichem Volumen proportional zu ihrer Masse und somit Dich-te sein sollte. Black meinte allerdings, dass seine Messungenim Widerspruch zu einer Theorie der Wärme stünden, die Wär-me durch Bewegung zu erklären suchte. Tatsächlich lieferte diekinetische Theorie erst 100 Jahre später mithilfe des Gleichver-teilungssatzes (Abschn. 36.2) eine Antwort auf diese zunächstoffengebliebene Frage, wie die Wärmekapazität von Gasen mitder kinetischen Theorie der Wärme in Einklang zu bringen wä-re.
Die Vorstellung, Wärme sei eine unzerstörbare Substanz,die zwischen Körpern fließen könne, herrschte bis ins 19.Jahrhundert vor.
Wärme und Arbeit
Der uns heute wohlbekannte Zusammenhang zwischen Wär-me und Arbeit wurde nur langsam aufgedeckt. Ausgehend vonder Dampfkugel des griechischen Mathematikers und Inge-nieurs Heron, der vermutlich im 1. Jahrhundert in Alexandrialebte, führte der Weg über die atmosphärische Maschine desdeutschen Juristen, Physikers und Erfinders Otto von Gueri-cke (1602–1686) und die Pulvermaschine des niederländischenAstronomen, Mathematikers und Physikers Christiaan Huygens(1629–1695) zur Dampfmaschine.
Der als Offizier, Politiker, Physiker und Erfinder tätige Ame-rikaner Benjamin Thompson (Graf von Rumford, 1753–1814)stellte um 1800 fest, dass ein Pferd in einer Stunde ungefähr450 Kilokalorien (kcal) Wärme erzeugen kann. Offenbar hatThompson seinem Pferd weniger zugemutet als der schottischeErfinder James Watt (1736–1819), der seinem Pferd eine grö-ßere Stärke zumaß: Mit einer Pferdestärke (1 PS D 735;5 W)kommt man in der Stunde auf 630 kcal. Thompson hatte sichzunächst die schwierige Aufgabe gestellt, das Gewicht derWärmesubstanz zu bestimmen. In seiner Darlegung findet sichbereits der vorsichtige Hinweis, dass sich sein negatives Resul-tat leicht verstehen ließe, wenn die Wärme nicht als Substanz,sondern als Folge innerer Bewegungen angesehen würde.
Der wundeste Punkt der Substanztheorie der Wärme war je-doch ihr Unvermögen, auf einfache Weise die Wärmeerzeugungdurch Reibung zu erklären. Eine Erklärung ging davon aus, dass
ein Körper bei Reibung seine Wärmekapazität verringert. Miteiner Untersuchung der beim Ausbohren von Kanonenrohrenauftretenden Wärmeerscheinungen versuchte Thompson, derWärmestofftheorie den Todesstoß zu versetzen. Aber obwohlThompsons experimentelle Ergebnisse durchaus auf Anerken-nung stießen, wurden sie noch zu Anfang des 19. Jahrhundertsauf der Grundlage der Vorstellung von einer Wärmesubstanz zudeuten versucht.
Die theoretischen Grundlagen der Thermodynamik wurdenschließlich in nur drei Jahrzehnten zwischen 1822 und 1851gelegt. Diese Entwicklung begann mit dem französischen Ma-thematiker und Physiker Jean Baptiste Joseph Fourier (1768–1830), der 1822 in seinem Buch Analytische Theorie der Wärmedie Wärmeleitung mathematisch behandelte (Fourier 1823 TS1 ,1955). Ziel seiner Untersuchungen war es, die an verschiedenenPunkten eines Körpers von Thermometern angezeigten Tem-peraturen und deren zeitliche Änderung zu berechnen. Um diedabei auftretenden Gleichungen zu lösen, schuf er die Fourier-Transformation (Abschn. 13.1).
Fouriers Untersuchungen kommt für den Fortschritt sowohl derMathematik als auch der Physik eine grundlegende Bedeutungzu. In Einklang mit der vorherrschenden Meinung vertrat jedochauch er die Ansicht, dass Wärme ein unveränderlicher Stoffsei. Dieser Wärmestoff sollte eine gewichtslose oder zumindestsehr leichte, unsichtbare und unzerstörbare Substanz sein – einwahrhaftig sehr eigenartiger Stoff, der an den Äther erinnert,der als Trägersubstanz elektromagnetischer Wellen angenom-men wurde. Würde der Wärmestoff einem Körper zugefügt,dann erhöhte sich seine Temperatur und sein Zustand ändertesich. Neben diese Ansicht trat allmählich eine andere Vorstel-lung, der zufolge die Temperatur eines Körpers ein Ausdruckder Bewegung seiner kleinsten Bestandteile ist.
Wärme und Arbeit in Kreisprozessen
Der entscheidende Fortschritt gelang dem französischen Inge-nieur und Physiker Nicolas Léonard Sadi Carnot (1796–1832;Abb. 33.2) mit seiner nur 45-seitigen Abhandlung Betrachtun-gen über die bewegende Kraft des Feuers und die zur Entwi-ckelung dieser Kraft geeigneten Maschinen von 1824 (Carnot1995). Außer dieser Arbeit hat Carnot nichts veröffentlicht.Acht Jahre nach der Veröffentlichung seines Büchleins erkrank-te er an Scharlach und starb während einer Cholera-Epidemie inParis im Alter von nur 36 Jahren.
Carnot zufolge wird Arbeit verrichtet, wenn der hypothetischeWärmestoff von höherer zu tieferer Temperatur herabsinkt. ImIdealfall ist die dergestalt vom Wärmestoff gelieferte Arbeitproportional zur Temperaturdifferenz geteilt durch die Endtem-peratur. Carnots These von der Unzerstörbarkeit der Wärme, inwelcher Gestalt auch immer diese auftreten möge, ist zwar nichtkorrekt, doch seine weiteren Aussagen sind richtig und weisenbereits den Weg zum zweiten Hauptsatz der Thermodynamik.In Carnots Nachlass findet sich auch die These bekräftigt, dass
TS1 Zu diesem Zitat fehlt die Quellenangabe im Abschnitt Literatur. Bitte ergänzen Sie diese.

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.1 Entwicklung der Thermodynamik 1101
Teil
IV
Abb. 33.2 Nicolas Léonard Sadi Carnot (1796–1832)
die Temperatur eines Körpers ein Ausdruck innerer Bewegungs-energie sei. Ohne Begründung gibt er dort auch an, dass dieWärmemenge einer Kilokalorie gleichwertig zu einer Arbeitvon 3628;5 J sei, und schlägt vor, wie diese Gleichwertigkeitzu messen wäre.
Carnots Arbeit wurde 1834 von dem französischen Physi-ker und Ingenieur Benoît Paul Émile Clapeyron (1799–1864)aufgenommen. Clapeyron brachte Carnots Aussagen in einemathematische Form, und zwar schon in diejenige, in der heu-te noch der Carnot’sche Kreisprozess in der Physik beschriebenwird. Der Carnot’sche Kreisprozess ist befreit vom Wärmestoffund dessen Unzerstörbarkeit. An die Stelle des Wärmestoffs tratdie Energie. Jedem Körper, beispielsweise einem Dampf odereinem Gas, wird unter definierten Bedingungen eine ganz be-stimmte innere Energie zugeschrieben. Diese wird somit alseine eindeutige Eigenschaft des Zustands eines Körpers ange-sehen. Sie kann sich infolge einer Erwärmung oder Abkühlungdes Körpers ändern oder indem an dem Körper Arbeit verrichtetwird.
Energieumwandlungen und der erste Hauptsatz
Diese Überlegungen nahmen vorweg, was heute im erstenHauptsatz der Thermodynamik formuliert wird. Dieser Haupt-
satz kann als eine Definition der Wärme angesehen werden. Ersetzt die Änderung der inneren Energie eines Körpers und dievom oder am Körper verrichtete Arbeit einer Wärmeänderunggleich. Damit wurde das entscheidende Problem der Wärme-stofftheorie aufgegeben, das in der Annahme bestanden hatte,man könne die Menge an Wärmestoff angeben, die in einemKörper enthalten sei. Indem der erste Hauptsatz ausdrücklichnur übertragene Wärmemengen angibt und sie durch verrich-tete Arbeit und Änderungen der inneren Energie quantifiziert,verzichtet er auf absolute Angaben einer Wärmemenge, die demKörper innewohnen könnte. Insbesondere erlaubt er damit auch,dass die insgesamt aufgenommene oder abgegebene Wärme-menge beim Übergang von einem Zustand eines Körpers ineinen anderen davon abhängen kann, wie der eine in den an-deren Zustand übergeht.
Die Unterscheidung von Wärme und Arbeit werden wir inAbschn. 34.1 und 36.2 genauer betrachten, wenn wir die ther-modynamischen Größen physikalischer Systeme mit der Physikihrer mikroskopischen Freiheitsgrade in Beziehung setzen.
Innere Energie
An die Stelle des Wärmestoffs trat um die Mitte des19. Jahrhunderts die innere Energie. Sie wird als eine Grö-ße aufgefasst, die den thermischen Zustand eines Körperskennzeichnet. Die innere Energie kann durch zu- oderabgeführte Wärme verändert werden oder dadurch, dassArbeit am oder vom Körper verrichtet wird.
Dieser entscheidende konzeptionelle Schritt führte zu einemder drei Axiome, die der heutigen Thermodynamik zugrun-de liegen. Als kennzeichnend für den thermischen Zustandeines Körpers wurde nun nicht mehr sein Gehalt an einemschwer fasslichen Wärmestoff angesehen, sondern seine innereEnergie, die später als Ausdruck des Bewegungszustands sei-ner mikroskopischen Bestandteile gedeutet wurde. Die innereEnergie wird als eine Größe angesehen, die durch den makro-skopisch charakterisierbaren Zustand eines Körpers eindeutigvorgegeben ist. Wenn ein Körper von einem Zustand in einenanderen übergeht, kann seine innere Energie zu Beginn und amEnde dieses Vorgangs lediglich von diesen Zuständen abhängen,nicht aber davon, auf welche Weise der Körper aus dem einemin den anderen Zustand überführt wurde.
Im Gegensatz dazu erweist sich die Wärmemenge als abhängigvom Weg, auf dem der Übergang in einen betrachteten Zustanderfolgt. Damit wird es ebenso unmöglich, sinnvoll davon zusprechen, der Körper enthalte eine bestimmte Menge Wärme,wie es unsinnig wäre zu sagen, im Körper stecke eine bestimm-te Arbeit. Wärme und Arbeit sind demnach keine Eigenschafteneines thermischen Zustands mehr. Stattdessen charakterisierensie eine Form des Energieaustauschs zwischen zwei Körpern.Damit werden Kreisprozesse gedanklich erst möglich, in denensich ein Körper auf einem Weg von seinem Ausgangszustand

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1102 33 Phänomenologische Begründung der Thermodynamik
Abb. 33.3 Rudolf Clausius (1822–1888), © akg-images/bildwissedition
entfernt und auf einem anderen dorthin zurückkehrt. Die vomKörper aufgenommene oder abgegebene Wärme und die vonoder an ihm verrichtete Arbeit werden sich in der Regel aufbeiden Wegen unterscheiden. Damit wird einsehbar, wie inperiodisch ablaufenden Prozessen Wärme in mechanische Ar-beit umgewandelt werden kann, wie also Wärmekraftmaschinenphysikalisch gedeutet und verstanden werden können.
In den folgenden Jahrzehnten setzte sich die Erkenntnis durch,dass übertragene Wärmemengen und verrichtete Arbeit Formenvon Energie seien. Die Äquivalenz von chemischer Energie,Wärme und Arbeit waren dem Koblenzer NaturwissenschaftlerFriedrich Mohr (1806–1879) schon seit 1837 und dem Chemi-ker Justus von Liebig (1803–1873) seit 1842 bekannt. Die Fol-gerungen des deutschen Physikers Rudolf Clausius (1822–1888;Abb. 33.3) über die spezifischen Wärmen oder den Tempera-turverlauf des Druckes gesättigten Dampfes stellten wichtigeSchritte bei der Entwicklung der Thermodynamik dar.
Entropie und der zweite Hauptsatz
Clausius musste bei seinen Untersuchungen feststellen, dassder erste Hauptsatz zur Erklärung der thermodynamischen Er-scheinungen nicht ausreicht und dass noch ein weiteres Axiomformuliert werden muss. Zwar erklärt der erste Hauptsatz, wiedie Änderung der Wärme mit der Änderung der inneren Ener-gie sowie mit der Arbeit in Beziehung steht, wenn ein Körpervon einem Zustand in einen anderen übergeht. Er erklärt abernicht, welche von allen denkbaren Zustandsänderungen über-haupt möglich sind.
Dementsprechend diente Clausius die Tatsache als Ausgangs-punkt seiner Überlegungen, dass Wärme nicht von sich aus von
einem kälteren zu einem wärmeren Körper übergeht. Zunächststellte er fest, dass sich in einem auf umkehrbare, aber sonstbeliebige Weise durchlaufenen Kreisprozess alle Wärmeände-rungen geteilt durch die Temperatur, bei der sie stattfinden, zunull summieren. Diese Größe, deren Änderung als eine auf dieTemperatur bezogene Wärmeänderung darstellbar ist, muss da-her eine Zustandsgröße des Körpers sein, denn sie kann nichtdavon abhängen, wie der Körper in seinen jeweiligen Zustandgelangt ist. Für diese neue Zustandsgröße führte Clausius ab1865 den neu geschaffenen Begriff der Entropie ein. Sie istvon großem praktischem Nutzen, weil sie bestimmt, welche Zu-standsänderungen überhaupt möglich sind. Daher begegnet manihr heute auch im Alltag der Ingenieure.
Entropie
Die Entropie bestimmt, welche von allen denkbaren Zu-standsänderungen makroskopischer Systeme überhauptmöglich sind.
Wir werden in Abschn. 34.3 sehen, dass die Entropie auf dieAnzahl der Möglichkeiten zurückgeführt werden kann, durchdie ein makroskopisches System mikroskopisch realisiert wer-den kann.
Kinetische Theorie: Innere Energieals Bewegung
Nach der Entdeckung des Erhaltungssatzes der Energie, denwir heute als ersten Hauptsatz der Thermodynamik bezeichnen,rückte die kinetische Theorie der Materie in den Vordergrund.Ihr Durchbruch begann mit den Arbeiten des britischen Phy-sikers James Joule (1818–1889), des deutschen Physikers undChemikers August Karl Krönig (1822–1879) und insbesonderedes bereits genannten Rudolf Clausius. Neben seinen bahnbre-chenden Arbeiten zur makroskopischen Thermodynamik, der erden Entropiebegriff schenkte, legte Clausius auch den Grund-stein für die mikroskopische Theorie der Wärme. In seiner 1857in den Annalen der Physik erschienenen Arbeit Über die Art derBewegung, welche wir Wärme nennen (Clausius 1857) leitete erab, wie der Gasdruck von der mittleren kinetischen Energie
hEkini D m
2
˝v 2˛
(33.1)
der Translationsbewegung der Moleküle abhängt. Die Energie,die in möglichen Schwingungen und Rotationen der Moleküleenthalten sein kann, erschien in diesem Zusammenhang nochnicht.
Clausius führte auch den Begriff der mittleren freien Weglängefür Moleküle in Gasen ein.

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.1 Entwicklung der Thermodynamik 1103
Teil
IV
Abb. 33.4 James Clerk Maxwell (1831–1879), © nickolae – Fotolia.com
Statistische Begründung der Thermodynamik
Der schottische Physiker James Clerk Maxwell (1831–1879;Abb. 33.4) untersuchte, wie sich eine große Zahl von Teilchenverhält, die nach den Gesetzen der klassischen Mechanik wieKugeln aneinanderstoßen. In seiner Arbeit On the dynamicaltheory of gases. – Part I. On the motions and collisions ofperfectly elastic spheres (Maxwell 1860) gab er die Ableitungder nach ihm benannten Verteilungsfunktion der Teilchenge-schwindigkeiten an. Auch das bekannte Äquipartitions- oderGleichverteilungsgesetz, nach dem auf jeden Freiheitsgrad derGaspartikel die gleiche Energie entfällt, wurde von Maxwellaufgestellt.
Der nächste wesentliche Schritt auf dem Weg zur mikrosko-pischen Begründung der Thermodynamik war die statistischeAuffassung der Entropie durch den österreichischen Physikerund Philosophen Ludwig Boltzmann (1844–1906; Abb. 33.5),dargelegt in seiner Arbeit Über die mechanische Bedeutung deszweiten Hauptsatzes der Wärmetheorie (Boltzmann 1866). Die-se entstand aus dem Bedürfnis, auch den zweiten Hauptsatzder Thermodynamik mithilfe der kinetischen Theorie deutenzu können. Es zeigte sich bald, dass ausschließlich mecha-nische Prinzipien zur Deutung des zweiten Hauptsatzes nichtausreichen, insbesonders bei irreversiblen Prozessen. In seinerArbeit Über die Beziehung zwischen dem zweiten Hauptsatzeder mechanischen Wärmetheorie und der Wahrscheinlichkeits-rechnung etc. (Boltzmann 1877) gelang Boltzmann eine statis-tische Deutung der Entropie. Seiner fundamentalen Erkenntniszufolge ist die Entropie eines Gases von Partikeln in einembestimmten makroskopischen Zustand proportional zum Loga-rithmus der Anzahl der Mikrozustände, in denen die Teilchenden gegebenen Makrozustand verwirklichen können. Die uni-
Abb. 33.5 Ludwig Boltzmann (1844–1906), © akg/Science Photo Library
verselle Boltzmann-Konstante
kB � 1;3806 � 10�23 J K�1 (33.2)
tritt dabei als Proportionalitätskonstante auf.
Die statistische Behandlung von thermodynamischen Vorgän-gen im Rahmen der klassischen Physik erreichte zu Beginn des20. Jahrhunderts ihren Höhepunkt mit den Untersuchungen desamerikanischen Physikers Josiah Willard Gibbs (1839–1903).Im Zentrum seiner Überlegungen stand die Verteilungsfunk-tion im Phasenraum eines mechanischen Systems mit einersehr großen Anzahl von Freiheitsgraden. Verschiedenen Vertei-lungsfunktionen entsprechen dabei verschiedene physikalischeSituationen. Die wichtigste Rolle spielte dabei das sogenannteGibbs’sche kanonische Ensemble, für das sich die Verteilungs-funktion im Phasenraum als proportional zum sogenanntenBoltzmann-Faktor
e�H=kBT (33.3)
erweist, in dem die Hamilton-Funktion H mit der thermischenEnergie kBT verglichen wird, die der absoluten Temperatur Tentspricht. Eine solche Verteilung beschreibt das Verhalten ei-nes physikalischen Systems im thermischen Gleichgewicht mitseiner Umgebung.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1104 33 Phänomenologische Begründung der Thermodynamik
Abb. 33.6 Josiah Willard Gibbs (1839–1903), © akg/Science Photo Library
Mikroskopische Begründung der Thermodynamik
Die kinetische Theorie der Wärme ermöglichte eine mi-kroskopische Begründung der thermodynamischen Ge-setze.
Wie sich später herausstellte, genügen die Erscheinungen derMikrophysik nicht den Gleichungen der klassischen Mechanik,sondern den Gesetzen der Quantenmechanik. Die Herleitungstreng gültiger Gesetze der makroskopischen Physik aus Wahr-scheinlichkeitsaussagen über Elementarereignisse der Mikro-physik gelang nicht nur aus der klassischen Mechanik heraus,sondern auch ausgehend von der Quantenmechanik.
Gleichartige Teilchen sind in der Quantenmechanik nicht unter-scheidbar (Abschn. 31.1). Dies hat insbesondere für Fermionen,z. B. Elektronen oder Protonen, weitreichende Folgen: Auf-grund des Pauli-Prinzips können zwei identische Fermionennicht im gleichen Mikrozustand sein. Auch für identische Bo-sonen, z. B. Photonen, sind nicht alle denkbaren Mikrozuständeerlaubt. Diese Einschränkungen beeinflussen die Abzählungvon Elementarereignissen und damit die Gesetze der makros-kopischen Physik. Identische Fermionen folgen der Fermi-Dirac-Verteilung, während identische Bosonen der Bose-Ein-stein-Verteilung folgen. Obwohl aufgrund dieser Erkenntnissedie Abzählverfahren angepasst werden mussten, die auf Fer-
mionen bzw. Bosonen angewendet werden müssen, blieben dieGrundlagen der Thermodynamik auch für Quantensysteme gül-tig.
Wie wir im abschließenden Kap. 37 von Teil IV sehen wer-den, kann der Übergang von der klassischen zur quantalenBeschreibung mikroskopischer Vorgänge in der Thermodyna-mik dadurch bewerkstelligt werden, dass der Phasenraum derklassischen Mechanik durch einen geeigneten quantenmechani-schen Zustandsraum und dass Wahrscheinlichkeitsverteilungendurch Dichteoperatoren ersetzt werden. Davon abgesehen, blei-ben die fundamentalen Konzepte der Thermodynamik und sogarihre formalen Gleichungen unverändert.
33.2 Was ist Thermodynamik?
Verzicht auf mikroskopische Information
Wir werden in diesem Buch nacheinander zwei Wege zurThermodynamik beschreiten. Zunächst werden wir die Ther-modynamik makroskopisch und phänomenologisch einführen,d. h., wir lernen in diesem Kapitel die Thermodynamik alsdenjenigen Teil der Physik kennen, der sich mit den Umwand-lungen der Energie in solchen physikalischen Systemen befasst,die aus sehr vielen Einzelteilen zusammengesetzt sind.
Dieser erste Teil dient zum einen dazu, die Grundlagen derThermodynamik aus fundamentalen Beobachtungen heraus zuentwickeln und ihre Begriffe zu klären. Zum Zweiten hat erdie Aufgabe anzudeuten, wie weit thermodynamische Begriffebei der Beschreibung physikalischer Phänomene tragen kön-nen, ohne dass darauf zurückgegriffen werden müsste, wie ihreKonzepte in der Statistik des Verhaltens von vielen Teilchen be-gründet liegen. Zum Dritten schließlich soll dieser erste Teilbereits eine Vorstellung davon erwecken, dass und warum dieThermodynamik als der vielleicht universellste durch Erfahrunggesicherte Teil der Physik angesehen werden kann. Die Haupt-sätze der Thermodynamik überspannen die gesamte Physik,gerade weil sie auf eine Begründung durch spezielle Modell-vorstellungen verzichten.
Die Thermodynamik handelt von den Möglichkeiten, Energieumzuwandeln. Dabei geht es nicht um den Energieaustauschzwischen Systemen, die aus nur wenigen Komponenten zusam-mengesetzt sind, denn der kann durch die Quantenmechanik, dieklassische Mechanik oder andere Theorien der Physik präzisebeschrieben werden. Vielmehr geht es in der Thermodynamikdarum, solche Gesetze zu finden, nach denen Energieumwand-lungen in physikalischen Systemen verlaufen, die aus sehrvielen Komponenten bestehen, möglichst weitgehend unabhän-gig davon, wie diese physikalischen Systeme aufgebaut undzusammengesetzt sein mögen.
Wie ist so etwas überhaupt möglich? Wie könnten wir hof-fen, die Umwandlung verschiedener Energieformen davon zuabstrahieren, wie und zwischen welchen Arten physikalischer

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.2 Was ist Thermodynamik? 1105
Teil
IV
Systeme sie umgewandelt werden? Wie könnten wir hoffen, all-gemeine Gesetze zur Beschreibung von Energieumwandlungenaufzustellen, ohne Bezug darauf nehmen zu müssen, wie dieEnergie im Einzelnen aufgenommen, abgegeben und transpor-tiert werden mag?
Es ist die bestaunenswerte, fundamentale Erkenntnis der phä-nomenologischen Thermodynamik, dass dies durchaus möglichist. Energieumwandlungen physikalischer Systeme lassen sichtatsächlich durch eine kleine Menge grundlegender Aussagenso weitgehend beschreiben, dass präzise Vorhersagen über daszu erwartende Verhalten einer Vielzahl konkreter Systeme ge-troffen werden können.
Darauf beruhen zugleich die Stärke und die Schwierigkeit derThermodynamik: In ihrer phänomenologischen Form wurde sieaus dem Bemühen heraus entwickelt, die Funktionsweise derDampfmaschinen zu verstehen und in physikalische Begriffe zufassen. In dieser Form stammt sie aus dem 18. und 19. Jahrhun-dert, wie wir einleitend beschrieben haben. Dennoch hat sie dieRevolutionen der Physik unbeschadet und in ihren Grundlagenunverändert überstanden, die im 20. Jahrhundert überall sonst inder Physik kaum einen Stein auf dem anderen gelassen haben.Die spezielle Relativitätstheorie ebenso wie die Quantenmecha-nik haben die Konzepte der Thermodynamik nicht in ihremKern berührt. Stattdessen haben sich Konzepte, die letztlich ausder Thermodynamik stammen, sehr fruchtbar auf weit entfern-te Gebiete der Physik ausgewirkt. Ein bezwingendes Beispieldafür ist die Hawking-Strahlung Schwarzer Löcher. In diesemSinne darf die Thermodynamik als die universellste, robustesteTheorie der Physik gelten.
Diese weitreichende Wirkung kann die Thermodynamik gera-de deswegen entfalten, weil sie sich nicht um die konkretenphysikalischen Vorgänge kümmert, die zum Energieaustauschzwischen elementaren Bestandteilen physikalischer Systemeführen und gehören. Thermodynamik beschreibt physikalischeSysteme nicht auf der mikroskopischen Ebene einzelner oderweniger Freiheitsgrade.
Gültigkeitsbereich der Thermodynamik
Die Thermodynamik beschreibt den Energieaustauschzwischen physikalischen Systemen mit vielen Freiheits-graden und ihrer Umgebung. Sie sieht dabei bewusst vonden mikroskopischen Vorgängen der Energieübertragungab.
Zum Beispiel wird durch die Hamilton’schen Gleichungen derklassischen Mechanik beschrieben, wie die als harte, struk-turlose Kugeln aufgefassten Teilchen eines idealen Gases an-einanderstoßen und dabei Impuls aufeinander übertragen (Ab-schn. 7.1). Die Thermodynamik interessiert sich nicht für dieDetails dieser mechanischen Abläufe. Wie Atome oder andereQuantensysteme aufgrund ihrer Wechselwirkung mit elektro-magnetischen Feldern Energie aufnehmen und abgeben, wiedadurch quantenmechanische Zustände an- oder abgeregt wer-den, geht die Thermodynamik nichts an (vgl. dazu Kap. 29).
Die Thermodynamik nimmt lediglich an, dass es solche Gesetzegibt, die den Zuständen der kleinsten Bestandteile physikali-scher Systeme eine Energie zuzuordnen erlauben.
Makroskopische Systeme, Stoffmenge
Die Thermodynamik kann deswegen von der mikroskopischenPhysik absehen, die sich zwischen den elementaren Bestand-teilen physikalischer Systeme abspielt, weil sie sich mit denEigenschaften solcher Systeme befasst, die aus sehr vielen ele-mentaren Bestandteilen zusammengesetzt sind. Wie wir sehenwerden, bedingt diese sehr große Zahl mikroskopischer Be-standteile, dass Fluktuationen um Mittelwerte vernachlässigbarklein werden. Die Thermodynamik ist in diesem Sinne eineTheorie makroskopischer Systeme. Die Begriffe mikro- undmakroskopisch sind in diesem Zusammenhang absichtlich lo-se bestimmt. Sie bedeuten in unserem Sprachgebrauch, dassmikroskopische Systeme aus höchstens wenigen Teilchen beste-hen und wenige Freiheitsgrade haben, während makroskopischeSysteme sehr viele Teilchen enthalten und sehr viele Freiheits-grade haben. Was „wenig“ oder „viel“ in diesem Zusammen-hang bedeuten, wird anhand der Avogadro-Zahl ersichtlich. Siequantifiziert den Begriff der Stoffmenge n.
Mit der Stoffmenge ist die Anzahl der Teilchen oder „Moleküle“eines Stoffs gemeint, die in einem betrachteten physikalischenSystem enthalten sind. Die Stoffmenge wurde 1971 als physi-kalische Grundgröße eingeführt und bekam die Einheit „Mol“(mol) zugeordnet.
Avogadrozahl und Stoffmenge
Die Stoffmenge bezeichnet die Anzahl der Moleküle oderTeilchen eines physikalischen Systems. Sie wird in Molangegeben. Ein Mol eines Stoffes enthält ebenso vieleTeilchen, wie Atome in 12 g des Kohlenstoffisotops 12Centhalten sind.
Dies entspricht gerade der Avogadro-Zahl mit dem unge-heuer großen Wert
NA D 6;02214129 .27/ � 1023 Teilchen
mol: (33.4)
Entsprechend beträgt die Stoffmenge eines Systems aus NTeilchen
n D N
NA: (33.5)
Die Avogadro-Zahl wird besonders in der älteren Literaturgelegentlich auch als Loschmidt-Zahl bezeichnet.
Teilchenzahlen von der Größenordnung der Avogadro-Zahlkennzeichnen demnach die Stoffmengen makroskopischer Sys-teme. Solche Systeme, die etwa 1 mol Teilchen umfassen, kom-men auf Massen von einigen bis zu einigen Hundert Gramm.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1106 33 Phänomenologische Begründung der Thermodynamik
Makroskopische Stoffmengen
Wie viele Teilchen enthält 1 m3 Luft, und welcher Stoff-menge entspricht das?
Unter Normalbedingungen hat Luft eine Dichte von1;293 kg m�3. In ausreichender Näherung besteht ihr Vo-lumen zu 78 % aus Stickstoff, zu 21 % aus Sauerstoff undzu 1 % aus Argon. Die Massen eines StickstoffmolekülsN2, eines Sauerstoffmoleküls O2 und eines ArgonatomsAr betragen mN2 D 28 u, mO2 D 32 u und mAr D 40 uin der atomaren Masseneinheit 1 u D 1;6605 � 10�24 g.Die mittlere Masse eines „Luftteilchens“ ist daher Nm D.0;78 � 28 C 0;21 � 32 C 0;01 � 40/ u D 28;96 u D4;81 � 10�23 g. 1 m3 Luft muss sich daher aus 1;293 �103=4;81 � 10�23 D 2;69 � 1025 Teilchen zusammenset-zen, das sind n D 44;7 mol. J
Wie diese Teilchen im Einzelnen, also auf mikroskopischerEbene, miteinander wechselwirken, kümmert die Thermodyna-mik nicht. Sie abstrahiert gerade davon, wie Energie zwischenden mikroskopischen Freiheitsgraden ausgetauscht wird. Sienimmt lediglich an, dass zwischen ihnen Energie ausgetauschtwerden kann. Durch die extrem große Zahl von Freiheitsgradenin makroskopischen Systemen spielen Fluktuationen im Ener-gieaustausch kaum mehr eine Rolle. Zwar tragen sehr viele Teil-chen dazu bei, aber alle aufgrund derselben mikroskopischenphysikalischen Gesetze. Dadurch kommt ein makroskopischerEnergieaustausch zustande, der von seiner mikrophysikalischenRealisierung nichts mehr weiß.
Über das Verhalten einzelner mikroskopischer Freiheitsgrademacht die phänomenologische Thermodynamik daher ganz be-wusst keine Aussagen. Zu keinem Zeitpunkt können wir denOrt, die Größe oder die Geschwindigkeit eines individuellenGasteilchens angeben, das Teil eines makroskopischen Systemsist. An die Stelle einer physikalischen Beschreibung aller Frei-heitsgrade anhand mikroskopischer Zustandsgrößen tritt in derThermodynamik die Beschreibung großer Gesamtheiten vonMolekülen durch makroskopische Zustandsgrößen.
Während die mikroskopischen Zustandsgrößen klassischer oderquantaler Vielteilchensysteme durch die Menge der Orte undImpulse oder durch den (reinen oder gemischten) Zustandsvek-tor aller Teilchen gegeben wären, kennzeichnen makroskopi-sche Zustandsgrößen messbare Eigenschaften des Gesamtsys-tems.
Makroskopische Zustandsgrößen
Die Thermodynamik verzichtet bewusst darauf, allemikroskopischen Bestandteile eines makroskopischenSystems durch die Angabe ihrer Zustandsgrößen exaktzu beschreiben. Stattdessen beschränkt sie sich darauf,makroskopische Systeme durch Zustandsgrößen zubeschreiben, die makroskopisch als Eigenschaften großer
Gesamtheiten vieler Teilchen mit vielen Freiheitsgradenmessbar sind.
Beispiele für makroskopische Zustandsgrößen sind der Druck,die Temperatur, das Volumen oder die Teilchenzahl eines ma-kroskopischen Systems. Da es sich dabei um makroskopischdefinierte Größen handelt, lassen sich Druck oder Temperaturfür ein einzelnes Molekül gar nicht angeben, ebenso wenig wiemögliche andere makroskopische Zustandsgrößen.
Axiome der Thermodynamik
Die Thermodynamik kann demzufolge als die physikalischeTheorie von den Gesetzmäßigkeiten der makroskopischen Ener-gieumwandlungen aufgefasst werden. Sie kommt mit drei Axio-men aus, die als die Hauptsätze der Thermodynamik bezeichnetwerden. Das erste Axiom definiert den Begriff der Temperatur.Es wird oft als nullter Hauptsatz bezeichnet. Das zweite Axi-om, erster Hauptsatz genannt, bestimmt den Zusammenhangzwischen den Änderungen verschiedener Formen der Energieeines physikalischen Systems. Das dritte Axiom, der zweiteHauptsatz, legt fest, welche Arten der Energieumwandlung phy-sikalischer Systeme zulässig sind.
Axiome oder Hauptsätze der Thermodynamik
Die Thermodynamik baut auf drei Axiomen auf, Haupt-sätze genannt. Sie definieren den Begriff der Temperatur,die Umwandlung verschiedener Energieformen ineinan-der und die physikalisch erlaubten Energieumwandlun-gen.
Am Ende dieses Abschnitts sei noch eine etwas weiter gehen-de Bemerkung erlaubt. In der Thermodynamik erweist sich wiefast überall in der theoretischen Physik, dass es entscheidend fürihren Erfolg ist, dass physikalische Systeme hierarchisch geglie-dert werden können. Skalen können so eingeführt werden, dassverschiedene Eigenschaften physikalischer Systeme klar von-einander getrennt werden können.
Mikro- und Makrophysik sind nicht nur durch Faktoren zweioder drei in den relevanten Größen voneinander verschieden,sondern durch Faktoren, deren Größenordnung im Bereich derThermodynamik durch die Avogadro-Zahl gekennzeichnet wer-den. Im Bereich der Quantenmechanik legt das Planck’scheWirkungsquantum h eine Skala fest, die in makroskopischenSystemen ebenfalls um viele Größenordnungen überschrittenwird.
Die Existenz absoluter Skalen, die damit zusammenhängen-de Isolierbarkeit physikalischer Systeme und die wiederumdadurch begründete Möglichkeit, auf einer Ebene einer Skalen-hierarchie von den jeweils anderen Ebenen zu abstrahieren, sind

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.3 Temperatur, Zustandsgrößen und Zustandsänderungen 1107
Teil
IV
Vertiefung: Messung und Einheiten des Druckes
Da der Druck als diejenige Kraft messbar ist, die ein Kör-per einer Volumenänderung pro Fläche entgegensetzt, ist dieSI-Einheit des Druckes ein Newton pro Quadratmeter, abge-kürzt Pascal mit dem Einheitenzeichen Pa:
1 Pa D 1N
m2D 1
kg
m s2:
Die Einheit Pascal ist nach dem eminenten französischenMathematiker, Physiker und Philosophen Blaise Pascal(1623–1662) benannt. Ein Druck von 1 Pa ist gegenüber all-täglichen Drücken recht klein. Deshalb wird oft das Bar (bar,vom griechischen barýs = schwer) als Einheit verwendet,
1 bar D 105 Pa D 105 N
m2D 10
N
cm2;
das mit einem Vorfaktor 106 aus dem cgs-System abgeleitetist (1 bar D 1 Mdyn cm�2). In technischen Anwendungenwird zudem häufig die Einheit Atmosphäre (at) verwendet,die durch die Gewichtskraft einer Masse von 1 kg definiertist, die auf einer Fläche von 1 cm2 lastet. Aus dieser Defi-nition ergibt sich die Umrechnung zwischen den Einheiten
Atmosphäre, Bar und Pascal:
1 at D 9;81N
kg� 1
kg
cm2D 9;81
N
cm2
D 0;981 bar D 9;81 � 104 Pa :
Die Physikalische Atmosphäre (atm) dagegen ist als derjeni-ge Druck definiert, den eine Quecksilbersäule von 760 mmHöhe bei einer Temperatur von 0 ıC auf dem Erdbodenausübt. Mithilfe der Dichte des Quecksilbers von �Hg D13;59 g cm�3 bei dieser Temperatur ergibt sich
1 atm D 760 mm � 9;81N
kg� 13;59
g
cm3
D 10;13N
cm2D 1;013 bar :
Dieser Wert definiert den Normaldruck der Erdatmosphäream Erdboden. Der Druck einer 1 mm hohen Quecksilbersäu-le wird nach dem italienischen Physiker und MathematikerEvangelista Torricelli (1608–1647) auch als Torricelli (Torr)bezeichnet.
entscheidende Vorbedingungen dafür, dass wir überhaupt theo-retische Physik betreiben können.
Skalenhierarchie
Beispiele für solche Skalenhierarchien lassen sich über-all in der Physik in großer Zahl finden. KontinuierlicheMedien wie etwa Flüssigkeiten sind ein Beispiel für eineräumliche Skalenhierarchie. Als Flüssigkeiten oder Flui-de werden solche physikalische Systeme bezeichnet, indenen die mittlere freie Weglänge der mikroskopischenTeilchen sehr klein gegenüber der Skala ist, auf der räum-liche Veränderungen des Fluids betrachtet werden, diewiederum sehr klein gegenüber den Abmessungen desGesamtsystems ist. J
33.3 Temperatur, Zustandsgrößenund Zustandsänderungen
Temperatur
Was bedeutet es, wenn wir sagen, „draußen ist es kalt“? Was be-deutet es, wenn wir sagen, „der Tee ist warm“? Wir bezeichnen
damit Situationen, in denen uns unser körperliches Empfindenanzeigt, dass physikalische Systeme in unserer Umgebung, sei-en es unsere Umwelt oder ein Tee, einen fühlbaren Unterschiedzu unserem Körper aufweisen. Diesen Unterschied bezeichnenwir mit „wärmer“ oder „kälter“. Die damit benannte Qualitätphysikalischer Systeme nennen wir „ihre Temperatur“. UnserEmpfinden erlaubt nur eine grobe Bestimmung der Temperatur:„wärmer“, „kälter“ und „ungefähr gleich warm“, vielleicht nochergänzt durch Steigerungen wie „heiß“ oder „eiskalt“.
Alltägliche Erfahrung zeigt weiterhin, dass sich die Tempera-turen wärmerer Körper und kälterer Körper aneinander anglei-chen, wenn sie in Kontakt miteinander gebracht werden. Derheiße Tee kühlt sich ab, bis er ungefähr gleich warm wie seineUmgebung ist. Die Milch aus dem Kühlschrank erwärmt sich,wenn sie dem Kühlschrank entnommen wird. Nach einer gewis-sen Zeit, die vom jeweils betrachteten physikalischen Systemabhängt, kann unser Empfinden keinen Temperaturunterschiedzwischen dem Tee und der Umgebung bzw. zwischen der Milchund ihrer Umgebung mehr feststellen.
Erfahrungen dieser Art werden im physikalischen Begriff derTemperatur genauer gefasst. Wenn zwei physikalische Syste-me verschiedener Temperatur miteinander in Kontakt gebrachtwerden, gleichen sich ihre Temperaturen einander an. Thermi-sches Gleichgewicht und damit ein Ende der Angleichung trittdann ein, wenn die Temperaturen gleich sind. Diese Aussagewird als nullter Hauptsatz zum ersten Axiom der Thermodyna-mik.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1108 33 Phänomenologische Begründung der Thermodynamik
Nullter Hauptsatz der Thermodynamik
Makroskopischen physikalischen Systemen wird eineTemperatur zugeordnet. Als thermisches Gleichgewichtzwischen zwei Systemen wird der Zustand der beidenSysteme bezeichnet, in dem sich ihre Temperaturen an-einander angeglichen haben.
Mathematisch betrachtet, errichtet der Begriff der Temperatureine Äquivalenzrelation auf der Menge aller makroskopischenphysikalischen Systeme. Es gibt Systeme mit niedrigeren undhöheren Temperaturen. Solche Systeme, deren Temperaturengleich sind, befinden sich zueinander im thermischen Gleich-gewicht.
Physikalischen Begriffen müssen Messvorschriften zugeordnetwerden, um sie präzise zu definieren und quantifizierbar werdenzu lassen. Die Definition der Temperatur, die der nullte Haupt-satz gibt, nimmt eine solche Messvorschrift für die Temperaturbereits vorweg.
Temperaturen werden durch Thermometer gemessen. Als Ther-mometer eignet sich jedes physikalische System, das seineTemperatur in eine andere, leicht messbare physikalische Grö-ße übersetzt. Das kann die Volumenausdehnung dieses Systemssein, die einfach an einem Längenmaßstab abgelesen werdenkann. Es kann der elektrische Widerstand sein, der bei Metallenmit der Temperatur zu-, bei Halbleitern mit der Temperatur ab-nimmt. Sobald sich thermisches Gleichgewicht zwischen demThermometer und dem System eingestellt hat, mit dem esin Kontakt gebracht wurde, muss das Thermometer die Tem-peratur dieses Systems angenommen haben. Aufgrund seinerKonstruktion bildet es diese Temperatur auf eine andere, z. B.geometrische oder elektrische Eigenschaft ab, die geeignetenMessmethoden zugänglich ist.
Wir werden in Abschn. 33.5 sehen, dass es in der Physik eineabsolute Temperaturskala gibt, deren Grade in Kelvin (K) ange-geben werden.
Temperaturmessung
Vielleicht erscheint dieses Beispiel trivial, aber es ist dochnützlich, sich die empirische Bedeutung des Tempera-turbegriffs zu verdeutlichen. Definitionsgemäß tritt ther-misches Gleichgewicht zwischen zwei Systemen dannein, wenn die Temperaturen der beiden Systeme gleichsind. Exakt diese Definition legen wir jeder Temperatur-messung zugrunde: Wenn wir wissen wollen, wie warmoder kalt es heute draußen ist, bringen wir ein Sytem,eben ein Thermometer, in thermischen Kontakt mit ei-nem anderen System, nämlich in diesem Fall mit derLuft vor dem Haus. Sobald sich der Thermometerstandnicht mehr ändert, hat sich thermisches Gleichgewicht
eingestellt. Die Temperatur des Thermometers ist danndefinitionsgemäß gleich der Temperatur der Außenluft.Die thermische Ausdehnung der Thermometerflüssigkeit,die etwa durch ein Röhrchen auf eine Dimension redu-ziert wird, übersetzt die Temperatur in eine Länge, dienun leicht geometrisch ablesbar ist. J
Zustandsgrößen und Prozessgrößen
Anhand der Temperatur können wir nun den Begriff der Zu-standsgröße präzisieren. Dazu betrachten wir eine sehr kleine,als differenziell gedachte Änderung dT der Temperatur, die sichaus differenziellen Änderungen zweier unabhängiger Variablerx und y ergeben möge. Diese beiden Variablen sollen makrosko-pisch messbare Eigenschaften des Systems sein, wie z. B. Druckund Volumen.
Die Änderung der Temperatur aufgrund der Änderungen dx unddy in den beiden Variablen x und y beträgt
dT D Xdx C Ydy ; (33.6)
wobei X und Y die partiellen Ableitungen
X D @T
@xund Y D @T
@y(33.7)
sind. Die zweiten Ableitungen sind (unter genügend allgemei-nen Bedingungen, um die wir uns hier nicht kümmern wollen)vertauschbar:
@X
@yD @2T
@x@yD @2T
@y@xD @Y
@x: (33.8)
Dieser Zusammenhang zwischen den partiellen Ableitungenvon X und Y ist notwendig und hinreichend dafür, dass dT DXdx C Ydy ein vollständiges Differenzial ist. Dies ist äquivalentzur Wegunabhängigkeit von Linienintegralen in der x-y-Ebene,
ZZ
Z0
.Xdx C Ydy/ DZZ
Z0
dT D T.Z/ � T.Z0/ ; (33.9)
die von einem Anfangspunkt oder -zustand Z0 zu einem End-zustand Z in der x-y-Ebene führen. Daraus folgt sofort, dassfür jeden geschlossenen Weg in der x-y-Ebene, d. h. für jedenbeliebigen Kreisprozess, das geschlossene Wegintegral ver-schwindet: I
.Xdx C Ydy/ D 0 : (33.10)

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.3 Temperatur, Zustandsgrößen und Zustandsänderungen 1109
Teil
IV
Frage 1Vergegenwärtigen Sie sich nochmals, vielleicht anhand der Dis-kussion in Abschn. 1.6, welcher Zusammenhang zwischen derWegunabhängigkeit des Integrals über eine Funktion und derRotation dieser Funktion besteht.
Aufgrund von Beziehungen wie (33.10) definiert ein vollstän-diges Differenzial eine Zustandsgröße des Systems: SolcheGrößen hängen nur noch von dem (makroskopischen) Zustanddes Systems ab, aber nicht davon, wie das System in diesen Zu-stand gelangt ist. Bei festgehaltenem Ausgangszustand Z0 und(willkürlicher) Festlegung seiner Temperatur T.Z0/ hat jederthermodynamische Zustand Z eine wohlbestimmte TemperaturT.Z/. Mehr über vollständige und unvollständige Differenzia-le wird im Kasten „Vertiefung: Vollständige und unvollständigeDifferenziale“ besprochen.
Man unterscheidet zwischen extensiven und intensiven Zu-standsgrößen. Eine Zustandsgröße heißt extensiv, wenn sie derStoffmenge des betrachteten Systems proportional ist, und in-tensiv, wenn sie von ihr unabhängig ist. Es ist eine wichtigeempirische Tatsache, dass die thermodynamischen Größen inguter Näherung entweder extensiv oder intensiv sind. So sindVolumen, innere Energie und Entropie extensiv, während Druckund Temperatur intensiv sind.
Zustandsgrößen
Zustandsgrößen kennzeichnen den Zustand makroskopi-scher physikalischer Systeme auf eine Weise, die un-abhängig davon ist, wie das System in seinen Zustandgelangt ist. Mathematisch entsprechen ZustandsgrößenFunktionen der makroskopischen Freiheitsgrade, von de-nen vollständige Differenziale gebildet werden können,weil sie nicht von dem Weg abhängen dürfen, über denein System in seinen Zustand gelangt ist.
Intensive Zustandsgrößen hängen nicht von der Stoffmen-ge des Systems ab, während extensive Zustandsgrößenproportional zur Stoffmenge sind.
Vielleicht ist hier ein Vergleich mit konservativen Kräften inder klassischen Mechanik angebracht. Für konservative Kräfteexistiert ein Potenzial, dessen (positiver oder negativer) Gradi-ent die Kraft ist. Die Arbeit, die verrichtet werden muss, wennein Körper in einem konservativen Kraftfeld bewegt wird, istunabhängig vom konkreten Weg, den der Körper nimmt.
Bei der Wahl derjenigen Zustandsgrößen, mit denen wir einphysikalisches System beschreiben wollen, besteht eine ge-wisse Freiheit. Wir nennen einen kleinstmöglichen Satz vonZustandsgrößen, der zur kompletten Beschreibung der thermo-dynamischen Zustände eines Systems notwendig ist, vollstän-dig. Die Zustandsgrößen eines vollständigen Satzes sind unab-hängig, während alle weiteren Zustandsgrößen abhängig sind.
Die abhängigen Größen sind dann Funktionen der unabhängi-gen Größen. Die Anzahl der unabhängigen Zustandsgrößen istgleich der Anzahl makroskopischer Freiheitsgrade des thermo-dynamischen Systems. Die unabhängigen Zustandsgrößen sindKoordinaten des Zustandsraumes.
Zustandsgrößen eines idealen Gases
Ideale Gase sind beispielsweise vollständig durch ihreTemperatur T , ihren Druck P, ihr Volumen V und ihreTeilchenzahl N gekennzeichnet. Diese Größen sind abernicht alle unabhängig, weil der Druck eines idealen Gasesdurch die Kombination von Teilchenzahl, Volumen undTemperatur bestimmt ist. Deswegen können beispielswei-se die Temperatur, das Volumen und die Teilchenzahl alsein vollständiger Satz unabhängiger Zustandsgrößen ge-wählt werden. J
Im Unterschied zu einer infinitesimalen Temperaturänderungdefinieren eine infinitesimale Wärmezufuhr oder eine infini-tesimale abgegebene Arbeit keine vollständigen Differenziale,da die Wärmezufuhr oder die abgegebene Arbeit nicht zu ver-schwinden brauchen, wenn das System in einem Kreisprozessauf einem geschlossenen Weg durch den Zustandsraum geführtwird. Die am Körper verrichtete Arbeit verändert die potenzi-elle Energie des Körpers, die ihrerseits nicht vom Weg abhängtund daher einer Zustandsgröße vergleichbar ist. Die verrichteteArbeit wird in der Regel davon abhängen, auf welchem Weg sieeine Zustandsänderung herbeiführt. Im Unterschied zu den Zu-standsgrößen nennt man Wärme und Arbeit Prozessgrößen undbezeichnet sie in der Literatur oft mit ıQ und ıW . Alternativwerden auch die Schreibweisen μ Q oder d0Q statt ıQ verwen-det.
Prozessgrößen
Im Unterschied zu den Zustandsgrößen kennzeichnenProzessgrößen nicht den Zustand eines makroskopischenphysikalischen Systems, sondern den Weg, auf dem dasSystem in seinen Zustand gelangt ist. Aus mathematischerSicht haben Prozessgrößen keine vollständigen Differen-ziale.
Homogene Flüssigkeiten, Gase oder Dämpfe sind die einfachs-ten thermodynamischen Systeme. Deren thermodynamischeZustände sind durch die mechanischen Zustandsgrößen DruckP und Volumen V sowie durch die thermische Zustandsgröße Tund die Teilchenzahl N charakterisiert. Der Druck ist im All-gemeinen eine Funktion von T , V und N, womit die Zahl derunabhängigen Variablen des Systems von vier auf drei reduziertwird. Die funktionale Abhängigkeit
f .P; T; V; N/ D 0 ; (33.11)

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1110 33 Phänomenologische Begründung der Thermodynamik
Vertiefung: Vollständige und unvollständige Differenziale
Eine Funktion F.x; y/, die hier der Einfachheit halber nur vonzwei Veränderlichen abhängen soll, hat das vollständige Dif-ferenzial
dF D @F.x; y/
@xdx C @F.x; y/
@ydy DW A.x; y/dx C B.x; y/dy ;
wobei wir die beiden Funktionen A.x; y/ und B.x; y/ zur Ab-kürzung der partiellen Ableitungen eingeführt haben.
Nehmen wir umgekehrt zwei beliebige Funktionen C.x; y/und D.x; y/, dann ist die Größe
ıG.x; y/ WD C.x; y/dx C D.x; y/dy
im Allgemeinen nicht das vollständige Differenzial einerFunktion G.x; y/. Die beiden Koeffizientenfunktionen C undD können dann nicht als partielle Ableitungen einer Funk-tion G.x; y/ nach den beiden Variablen x und y dargestelltwerden.
Eine notwendige und hinreichende Bedingung dafür, dass eseine Funktion G gibt, deren Ableitungen die Koeffizienten-funktionen C und D sind, lässt sich leicht finden. Nehmenwir zunächst an, es gäbe eine solche Funktion, dann wärendie Funktionen C und D die Komponentenfunktionen desGradienten von G:
�C.x; y/D.x; y/
�D rG.x; y/ :
Da die Rotation eines Gradienten identisch verschwindet,folgt daraus die notwendige Bedingung
@C.x; y/
@yD @D.x; y/
@x
dafür, dass ıG D dG ein vollständiges Differenzial ist.Wenn die Funktionen auf einem einfach zusammenhängen-den Gebiet definiert sind, zeigt der Stokes’sche Satz, dassdiese Bedingung auch hinreichend ist. Ein Differenzial istdemnach genau dann vollständig, wenn die Rotation desVektorfeldes verschwindet, dessen Komponenten die Ko-effizientenfunktionen des Differenzials sind. Angelehnt andie Bezeichnungsweise von Differenzialformen werden voll-ständige Differenziale auch exakt genannt.
Als Beispiel setzen wir
C.x; y/ D a ; D.x; y/ D bx
y
mit beliebigen Konstanten a und b. Da
0 D @C.x; y/
@y¤ @D.x; y/
@xD b
y
ist, kann ıG D Cdx C Ddy kein vollständiges Differenzialsein. Für infinitesimale dx und dy ist die Größe ıG zwar in-finitesimal klein, kann aber nicht als infinitesimale Differenzzwischen infinitesimal benachbarten Werten einer FunktionG aufgefasst werden. Daher heißt sie unvollständiges Diffe-renzial.
Eine Funktion ˛.x; y/ heißt integrierender Faktor, wenn˛.x; y/ ŒC.x; y/dx C D.x; y/dy� ein vollständiges Differenzi-al ist. Nach dem oben Gesagten ist dies genau dann der Fall,wenn die Rotation des Vektors mit den Komponenten ˛Cund ˛D verschwindet, woraus die Bedingung
C
˛
@˛
@y� D
˛
@˛
@xD @D.x; y/
@x� @C.x; y/
@y
an ˛ folgt.
Frage 2Leiten Sie diese Bedingung selbst her. Finden Sie einen inte-grierenden Faktor so, dass ıG.x; y/ aus dem vorigen Beispielein vollständiges Differenzial wird.
In zwei Dimensionen besitzt jedes Differenzial ! D Xdx CYdy mit beliebigen Koeffizientenfunktionen X.x; y/ undY.x; y/ einen integrierenden Faktor ˛. Dies ist leicht einzuse-hen, weil die dafür notwendige und hinreichende Bedingungr �.˛!/ D 0 in zwei Dimensionen genau eine Gleichung, inmehr als zwei Dimensionen aber mehr Gleichungen für denintegrierenden Faktor liefert. In drei oder mehr Dimensionenist der integrierende Faktor daher überbestimmt und existiertnicht mehr notwendigerweise. Ein Beispiel dafür finden Siein Aufgabe 33.9.
Selbst wenn ıG unvollständig ist, kann das Integral
2Z
1
ıG
von einem Anfangszustand .x1; y1/ zu einem Endzustand.x2; y2/ dennoch als Summe über infinitesimale Größen ver-standen und berechnet werden, nur wird es in der Regeldavon abhängen, wie wir vom Anfangs- zum Endzustandgelangen. Im Gegensatz dazu wird ein Integral über ein voll-ständiges Differenzial dF nicht vom Weg abhängen, weil
2Z
1
dF D F.x2; y2/ � F.x1; y1/
nur vom Anfangs- und Endpunkt abhängen kann. Dadurchwird der Zusammenhang zwischen vollständigen Differen-zialen und Funktionen hergestellt, die als Gradient einesPotenzials dargestellt werden können, wie wir sie im Zu-sammenhang mit konservativen Kraftfeldern in Abschn. 1.6besprochen hatten.

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.3 Temperatur, Zustandsgrößen und Zustandsänderungen 1111
Teil
IV
33.1 Mathematischer Hintergrund: Homogene Funktionen
Funktionen f W Rn � U ! R, für die
f .�x/ D �kf .x/
mit � 2 R gilt, heißen homogen vom Grad k in x. Dievorangegangene Definition extensiver und intensiver Zu-standsgrößen lässt sich mathematisch dadurch ausdrücken,dass extensive Zustandsgrößen homogene Funktionen vomGrad 1 in der Stoffmenge sind, während intensive Zustands-größen homogene Funktionen vom Grad 0 sind. Auf dieBedeutung homogener Funktionen insbesondere in der klas-sischen Mechanik und der Quantenmechanik wurde schon inAbschn. 3.7 und 30.2 hingewiesen.
Für homogene Funktionen vom Grad k gilt der Euler’scheSatz für homogene Funktionen,
x � r f .x/ D kf .x/ ;
wobei r f .x/ der Gradient von f .x/ nach dem Argument xist. Dieser Satz ist leicht einzusehen, indem man die voran-gegangene Gleichung nach � ableitet,
x � r f .�x/ D k�k�1f .�x/ ;
und dann � D 1 setzt. Eingeschränkt auf Funktionen einerVariablen bedeutet der Euler’sche Satz insbesondere, dassfür die Ableitung jeder Potenzfunktion f .x/ D xp gilt:
f 0.x/ D pf .x/
x:
Wir werden später sehen, dass die Entropie S eine extensiveZustandsgröße ist, die als Funktion der inneren Energie U,des Volumens V und der Teilchenzahl N geschrieben werdenkann. Also gilt für die Entropie
S.�U; �V; �N/ D �S.U; V; N/ ;
und ihre Ableitungen nach U, V und N müssen aufgrund desEuler’schen Satzes durch
U@S
@UC V
@S
@VC N
@S
@ND S
mit der Entropie selbst verbunden sein.
Druck P Volumen V
Gleichgewichts-zustand
Temperatur T
Zustandsflache
Abb. 33.7 Zustandsfläche in einem Zustandsraum, der hier durch Druck, Vo-lumen und Temperatur aufgespannt wird. Die Zustandsfläche wird durch dieZustandsgleichung f .P; V; T/ D 0 festgelegt
die den Zusammenhang zwischen Druck, Volumen, Temperaturund Teilchenzahl bestimmt, wird als (thermische) Zustandsglei-chung bezeichnet.
Die Funktion f in (33.11) spezifiziert das betrachtete System.Stellt man den Zustand eines solchen Systems bei festgehalte-ner Teilchenzahl N durch einen Punkt in dem dreidimensionalen
Zustandsraum dar, der durch die Größen P, V und T aufge-spannt wird, dann definiert die Zustandsgleichung eine Flächein diesem Raum. Eine solche Zustandsfläche ist in Abb. 33.7schematisch dargestellt. Jeder Punkt dieser Fläche stellt einenGleichgewichtszustand des Systems dar. Gleichgewichtszustän-de sind solche, die sich zeitlich nicht oder nur so langsamändern, dass ihre Änderung in der betrachteten Situation uner-heblich ist. In der Thermodynamik versteht man unter einemZustand, wenn es nicht ausdrücklich anders angegeben ist, im-mer einen Gleichgewichtszustand.
Zustandsänderungen
Einen Übergang von einem thermodynamischen Zustand ineinen anderen bezeichnet man als thermodynamische Zustands-änderung. Wenn der Ausgangszustand Z1 ein Gleichgewichts-zustand ist, kann eine Zustandsänderung nur durch eine Än-derung der äußeren Bedingungen hervorgerufen werden, denendas System unterworfen ist.
Die Änderung jedes Gleichgewichtszustands eines thermody-namischen Systems kommt durch Wechselwirkungen mit derUmgebung des Systems zustande. Beispielsweise kann die Um-gebung an einem thermodynamischen System Arbeit verrichten,oder das System selbst kann umgekehrt an der Umgebung Ar-beit verrichten. Zwischen einem System und seiner Umgebungkann ein Wärme- oder sogar ein Stoffaustausch stattfinden.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1112 33 Phänomenologische Begründung der Thermodynamik
Stoffe Arbeit
Warme
System
Umgebung
Abb. 33.8 Ein makroskopisches physikalisches System kann auf verschiedeneWeisen mit seiner Umgebung in Kontakt treten
Eine sehr große Umgebung im thermischen Gleichgewicht, de-ren Temperatur sich bei Entnahme einer endlichen Wärmemen-ge praktisch nicht ändert, nennt man Wärmebad, Wärmereser-voir oder einfach Reservoir. Die erwähnten Austauschprozessezwischen System und Umgebung werden in Abb. 33.8 schema-tisch gezeigt.
Wärmereservoire
Das Konzept eines Wärmereservoirs verweist auf denBegriff der Skalenhierarchie, von dem in Abschn. 33.2die Rede war. Natürlich existiert kein unendlich großesWärmereservoir, aus dem sich ein kleineres thermodyna-misches System unbegrenzt bedienen kann. Entscheidendfür die Argumentation ist aber nur, dass das Wärme-reservoir sehr viel mehr innere Energie enthält als dasbetrachtete thermodynamische System. Für eine FlascheBier, die gekühlt werden soll, kann ein Teich in besterNäherung als Wärmereservoir gelten. J
Offene, geschlossene und abgeschlossene Systeme
Ein System heißt
offen, wenn alle gezeigten Austauschprozesse erlaubtsind;geschlossen, wenn es keinen Stoffaustausch gibt;anergisch, wenn es keinen Arbeitsaustausch gibt;(adiabatisch) isoliert, wenn kein Wärmeaustauschmöglich ist;thermisch isoliert, wenn weder Arbeits- noch Wärme-austausch möglich sind;abgeschlossen, wenn weder Energie- noch Stoffaus-tausch möglich sind.
Das Adjektiv „adiabatisch“ leitet sich von den griechi-schen Worten a für „nicht“ und diabaínein für „hindurch-gehen“ ab.
Abgeschlossene Systeme gibt es in der Realität nicht, da sieeine unendlich gute Isolierung hinsichtlich aller möglichenAustauschvorgänge mit der Umgebung voraussetzen. Hierzugehört auch eine perfekte Wärmeisolierung des Systems. Einnäherungweise abgeschlossenes System stellt z. B. eine Ther-moskanne dar. Geschlossene Systeme können Energie, z. B.in Form von Wärme und Arbeit, über die Systemgrenze aus-tauschen. Die Systemgrenze ist allerdings für Stoffaustauschundurchlässig. Bei offenen Systemen können sowohl Teilchenals auch Energie über die Systemgrenze treten. Ein Beispiel ei-nes offenen Systems ist eine Gasflasche mit geöffnetem Ventiloder eine Tasse Kaffee.
Sprudelflasche
Eine Sprudelflasche befinde sich mit geschlossenem Ver-schluss im Kühlschrank. Die Flasche und ihr Verschlussdefinieren ein gegenüber dem Kühlschrank geschlossenesSystem, weil sie den Austausch von Sprudel mit der Um-gebung verhindern. Wärmeaustausch ist aber möglich;dazu wurde die Flasche ja in den Kühlschrank gestellt.Wird die Sprudelflasche aus dem Kühlschrank genom-men und auf den Küchentisch gestellt, beginnt Wärmeaus der Küche in die Flasche zu fließen. Wird zusätzlichder Verschluss geöffnet, verrichtet das freigesetzte Koh-lendioxid Arbeit an der Umgebungsluft. Sprudelwasserkann jetzt in die Umgebung gelangen; das System ist nunoffen. J
Eine Zustandsänderung Z1 ! Z2 heißt quasistatisch, wenn dieäußeren Bedingungen sich so langsam ändern, dass das Sys-tem in jedem Augenblick näherungsweise im Gleichgewicht ist.Während einer quasistatischen Zustandsänderung verlässt dasSystem seine Zustandsfläche nicht und wandert entlang einerKurve über die Zustandsfläche, die durch die Prozessführungbestimmt ist.
Eine Zustandsänderung ist reversibel, wenn einer zeitlichenUmkehr der Änderung der äußeren Parameter eine zeitlicheUmkehr der Folge von Zuständen entspricht. In einem solchenFall kann der Ausgangszustand des Systems ohne bleibendeVeränderungen in der Umgebung wiederhergestellt werden.
Quasistatische Zustandsänderungen
Quasistatische Zustandsänderungen sind ein weiteresBeispiel für eine Skalenhierarchie, wie sie in Ab-schn. 33.2 erwähnt wurde. Gelegentlich werden quasista-tische Zustandsänderungen als „unendlich langsam“ be-schrieben, was dann leicht den Eindruck erweckt, solcheZustandsänderungen seien von vornherein völlig unrea-listisch. Gemeint ist damit aber, dass die Zustandsgrößen

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.3 Temperatur, Zustandsgrößen und Zustandsänderungen 1113
Teil
IV
quasistatischeExpansion:Arbeitsabgabe,Warmeverlust
quasistatischeKompression:Arbeitsaufnahme,Warmegewinn
Abb. 33.9 Beispiele für reversible Zustandsänderungen eines Gases, entwederdurch quasistatische Expansion (oben) oder quasistatische Kompression (unten)
des Systems während des Prozesses wesentlich langsa-mer geändert werden, als diejenigen Prozesse ablaufen,durch welche die Freiheitsgrade des Systems wechselwir-ken und ein neues Gleichgewicht herstellen können. Obeine Zustandsänderung als langsam oder schnell angese-hen werden kann, ist daher immer eine Frage nach demVerhältnis zwischen den Zeitskalen der daran beteiligtenProzesse. J
Jeder reversible Prozess ist quasistatisch, aber nicht jeder qua-sistatische Prozess ist reversibel. Ein Prozess Z1 ! Z2 heißtdagegen irreversibel, wenn bei der umgekehrten Prozessfüh-rung Z2 ! Z1 in der Umgebung Veränderungen zurückbleiben.Bei einem solchen Kreisprozess ändert sich dann die Umgebungdes thermodynamischen Systems. Alle wirklichen, in der Naturrealisierten Prozesse laufen mehr oder weniger irreversibel ab.Bei der reversiblen Gasexpansion in Abb. 33.9 expandiert dasGas sehr langsam und verrichtet dabei Arbeit. Beim Umkehr-prozess wird diese Arbeit eingesetzt, um das Gas quasistatischin seinen Ausgangszustand zu verdichten. Abbildung 33.10zeigt dagegen eine irreversible Gasexpansion. Nach dem Her-ausziehen der Trennwand strömt ein Teil des Gases von derlinken in die rechte Hälfte des Behälters. Dabei wird keine Ar-beit an der Umgebung verrichtet. Um das Gas danach wiederin die linke Seite zu verdichten, muss die Umgebung Arbeit amGas verrichten.
schnelles Herausziehenund Hineinschiebender Trennwand
Nichtgleichgewichts-zustand geht insGleichgewicht
Abb. 33.10 Beispiel für eine irreversible Zustandsänderung. In einem Kolbenwird schnell eine Trennwand verschoben. Das Gas dehnt sich plötzlich aus undgeht dann in einen neuen Gleichgewichtszustand über
Zustandsänderungen
Eine Zustandsänderung eines Systems heißt quasistatisch,wenn sie so langsam verläuft, dass das System wäh-rend der gesamten Zustandsänderung näherungsweise imGleichgewicht bleibt. Reversibel sind solche Prozesse, diedas System auch in umgekehrter Richtung durchlaufenkann, wenn die äußeren Parameter so verändert werden,dass sie am Ende des umgekehrten Verlaufs auch wiederihre ursprünglichen Werte annehmen. Reversible Prozessesind quasistatisch, quasistatische Prozesse aber nicht un-bedingt reversibel.
Zustandsänderungen eines Luftballons
Ein Beispiel für eine Zustandsänderung, die zwar quasi-statisch, aber irreversibel verläuft, ist das sehr langsameEntweichen der Luft aus einem Ballon mit einer leichtporösen Hülle. Während die Luft langsam ausströmt,manchmal über Tage, bleibt dem Ballon stets ausreichendZeit, sich kontinuierlich auf ein verändertes mechani-sches Gleichgewicht einzustellen. J

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1114 33 Phänomenologische Begründung der Thermodynamik
Eine reversible Zustandsänderung eines einfachen Systems,das durch Druck, Volumen und Temperatur vollständig ge-kennzeichnet ist, definiert eine Kurve auf der Zustandsfläche,weil das System während reversibler Zustandsänderungen dieZustandsfläche nicht verlassen darf. Die Projektionen solcherKurven aus der Zustandsfläche auf die P-V-Ebene tragen jenach der Art der Zustandsänderung besondere Namen, z. B. Iso-thermen bei festgehalter Temperatur, Adiabaten bei adiabatischabgeschlossenen Systemen, Isochoren bei konstantem Volumenund Isobaren bei konstantem Druck.
Frage 3Betrachten Sie möglichst viele physikalische Vorgänge in Ih-rem Alltag: die Mischung von Milch mit Tee oder Kaffee, dasAufpumpen eines Fahrradreifens, die Abkühlung, die Sie imSchwimmbad erleben und viele andere mehr. Überlegen Siesich bei jedem dieser Vorgänge: Verläuft er irreversibel oderin ausreichender Näherung reversibel? Könnte man denselbenVorgang so gestalten, dass er reversibel ablaufen könnte, undwie könnte das gegebenenfalls geschehen?
33.4 Arbeit und Wärme
Arbeit
Betrachten wir nun eine Substanz, die wir im allgemeinen Sinnals Fluidum bezeichnen, womit sowohl eine Flüssigkeit alsauch ein Gas gemeint sein kann. Sie befinde sich in einemzylindrischen Gefäß mit einem beweglichen Kolben der Quer-schnittsfläche A. Aufgrund ihres Druckes P übt die Flüssigkeitdie Kraft PA auf den Kolben aus. Wird der Kolben um den in-finitesimalen Weg dh verschoben, verrichtet die Flüssigkeit dieinfinitesimale Arbeit
ıW D PAdh D PdV : (33.12)
Diese Gleichung gilt nicht nur für die hier gewählte zylindrischeBegrenzung des Volumens. Für beliebig geformte Voluminaund für beliebige Formänderungen der Begrenzungsfläche mussman nur alle gegebenenfalls infinitesimal kleinen Volumenän-derungen längs der Oberfläche aufsummieren. Für negatives dV,also bei einer Kompression, wird gegen den Druck Arbeit ander Flüssigkeit verrichtet, sodass ıW negativ wird. Die infi-nitesimale Arbeit ıW kann wie die Wärme kein vollständigesDifferenzial sein, weil sie im Allgemeinen davon abhängt, ent-lang welchen Weges im Zustandsraum die Arbeit am Systemverrichtet wird.
Beispielsweise werden in der Regel verschiedene Arbeitsbeträ-ge notwendig sein, um ein Gas im einen Fall zunächst ohneWärmeaustausch zu komprimieren und danach wieder auf dieAusgangstemperatur abzukühlen oder im anderen Fall bei kon-stanter Temperatur um denselben Faktor zu komprimieren.
Volumen V
DruckP
P = P1
P = P2
Expansion
Kompression
Abb. 33.11 Indikatordiagramm der Dampfmaschine
Das bereits von James Watt für Dampfmaschinen eingeführ-te, sogenannte Indikatordiagramm in Abb. 33.11 zeigt einenKreisprozess im P-V-Diagramm. Längs der oberen Isobaren mitdem Druck P1 ist der Dampfzylinder an einen Hochdruckkes-sel angeschlossen, längs der unteren mit dem niedrigeren DruckP2 < P1 an einen Niederdruckkessel. Der absteigende bzw.aufsteigende Kurvenast bedeutet Expansion bzw. Kompression.Der Flächeninhalt misst den Betrag des geschlossenen Integrals
IP dV D
IıW ; (33.13)
der ja gerade deswegen nicht verschwinden muss, weil ıW keinvollständiges Differenzial ist.
Wärme und Wärmekapazitäten
Da die Dampfmaschine nach einem Umlauf wieder im Aus-gangspunkt des Indikatordiagramms ankommen und daher un-verändert aus dem Umlauf hervorgehen muss, kann sie diesenArbeitsbetrag nur auf Kosten der zugeführten Wärme verrich-ten, sodass bei einem geschlossenen Umlauf
IıQ D
IıW (33.14)
gelten muss. Um Wärme zu quantifizieren, wurde eine eigeneEinheit eingeführt, die Kalorie.
Kalorie
Die Wärmemenge, die 1 g Wasser bei Atmosphärendruckvon 14;5 auf 15;5 ıC erwärmt, heißt eine Kalorie.

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.4 Arbeit und Wärme 1115
Teil
IV
Die Wärme, die ein Körper pro Kilogramm seiner Masse beieiner Temperaturänderung �T aufnehmen oder abgeben kann,heißt spezifische Wärme oder spezifische Wärmekapazität undwird mit c bezeichnet.
Wenn die Wärme bei konstantem Volumen zugeführt wird, be-wirkt sie eine Temperatur- und Druckerhöhung der Flüssigkeit,ohne dass durch den Körper eine Druck-Volumen-Arbeit PdVverrichtet werden könnte. Wird die Wärme dagegen bei kon-stantem Druck zugeführt, wird sie zum Teil dafür eingesetzt, dieFlüssigkeit gegen die Umgebung expandieren zu lassen. Des-halb wird sich die Temperatur bei gegebener Wärmezufuhr beikonstantem Druck weniger verändern als bei konstantem Volu-men.
Daher müssen die spezifischen Wärmekapazitäten cV bei kon-stantem Volumen und cP > cV bei konstantem Druck unter-schieden werden. Für Gase ist der Unterschied zwischen denbeiden spezifischen Wärmekapazitäten wesentlich.
Für eine Flüssigkeit oder ein Gas der Masse m gelten demnachdie Gleichungen
�QV D m cV �T oder �QP D m cP�T (33.15)
für die bei konstantem Volumen bzw. konstantem Druck aufge-nommenen Wärmemengen. Daraus und aus der oben gegebenenDefinition der Kalorie folgt beispielsweise für Wasser bei 15 ıCmit �T D 1 K die spezifische Wärme bei konstantem (Atmo-sphären-)Druck (siehe hierzu (33.24)):
cP D 1kcal
K kgD 426;9
kpm
K kgD 4185
J
K kg: (33.16)
Die Wahl einer geeigneten Temperaturskala wird in Ab-schn. 33.5 nachgeholt.
Anstelle der spezifischen Wärmekapazitäten pro Masse benutztman häufig auch die Wärmekapazitäten pro Stoffmenge, die so-genannten Molwärmen oder molaren Wärmen, cmol
V und cmolP .
Eine Stoffmenge n einer Flüssigkeit oder eines Gases nimmtbei einer Temperaturänderung �T die Wärmemengen
�QV D n cmolV �T oder �QP D n cmol
P �T (33.17)
auf oder gibt diese ab, je nachdem, ob dabei das Volumen oderder Druck konstant gehalten wird.
Wärmekapazität
Die Wärmemenge, die ein Körper pro Masse oder proStoffmenge bei einer Temperaturänderung �T aufnehmenoder abgeben kann, ist seine spezifische Wärmekapazitätc oder seine molare Wärmekapazität cmol. Wärmekapazi-täten bei konstantem Druck sind größer als bei konstantemVolumen,
CP > CV ; (33.18)
weil bei konstantem Druck ein Teil der aufgenommenenWärme als Druck-Volumen-Arbeit gegen die Umgebungverrichtet werden muss.
Die Wärmekapazität ist ein Beispiel für eine sogenannte Ant-wort- oder Responsefunktion eines thermodynamischen Sys-tems, die anzeigt, wie das System auf eine Änderung äußererZustandsparameter reagiert. Weitere Beispiele für Response-funktionen werden in Abschn. 35.1 besprochen, darunter derAusdehnungskoeffizient und die Kompressibilität.
Mischungstemperatur
Damit können wir sofort angeben, welche Temperatursich einstellen wird, wenn zwei gemeinsam isolierte Sys-teme in thermischen Kontakt gebracht werden. Wenn kei-nerlei mechanische Arbeit ausgeübt wird, muss �Q1 D��Q2 gelten, woraus
�Q1 C �Q2 D 0 D m1
TZ
T1
cV1.T0/dT 0
C m2
TZ
T2
cV2 .T 0/dT 0
(33.19)
folgt, wenn cV die spezifische Wärme pro Masse beikonstantem Volumen ist. Wenn zudem noch cV von derTemperatur zumindest in genügender Näherung unabhän-gig ist, erhalten wir daraus
m1cV1.T � T1/ C m2cV2.T � T2/ D 0 : (33.20)
Dies liefert die Mischungstemperatur
T D m1cV1 T1 C m2cV2 T2
m1cV1 C m2cV2
; (33.21)
die bei gleichen Wärmekapazitäten allein durch das Mas-senverhältnis bestimmt wird:
T D m1T1 C m2T2
m1 C m2: (33.22)
J
Molare Wärmekapazitäten für einige Gase bei 20 ıC sind inTabelle 33.1 angegeben. Beachten Sie, dass diese Wärmekapa-zitäten für ein-, zwei- und mehratomige Moleküle jeweils etwagleich groß sind.
Bei allen Reibungsvorgängen wird Arbeit in Wärme oder innereEnergie umgewandelt. Dies wurde schon früh von James Pres-cott Joule (1818–1889) und Julius Robert Mayer (1814–1878)erkannt. Dabei erweist sich die aufgewendete Arbeit als propor-tional zur erzeugten Wärme,
ıW D �ıQ ; (33.23)

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1116 33 Phänomenologische Begründung der Thermodynamik
Tab. 33.1 Molare Wärmekapazitäten einiger Gase bei 20 ıC
Stoff molare Masse cmolP cmol
V
[g mol�1] [J mol�1 K�1] [J mol�1 K�1]He 4,003 20,94 12,85Ar 39,94 20,89 12,69H2 2,02 28,83 20,45Luft 29 29,14 20,78CH4 16,04 35,59 27,21CO2 44,01 36,84 28,48
mit einem von den Versuchsbedingungen unabhängigen Propor-tionalitätsfaktor
� D 426;9kpm
kcalD 4;185
J
cal: (33.24)
Wärme wird dadurch auch anhand ihrer Einheit als Energieformidentifiziert, die in Joule angegeben wird.
33.5 Die idealen Gasgesetze
Experimentell wurde schon früh in der Geschichte der Ther-modynamik gefunden, dass sich alle untersuchten Gase sehrähnlich verhalten, sofern sie nur hinreichend verdünnt sind.Diese interessante und wichtige Feststellung wird im Kon-zept des idealen Gases zum Prinzip erhoben: Als ideal wirdhier zunächst ein Gas angesehen, dessen thermodynamische Ei-genschaften nicht oder nur vernachlässigbar wenig von seinerZusammensetzung abhängen. Wir werden in Abschn. 35.3 einegenaue mikroskopische Begründung für diese Idealisierung an-geben. Ein reales Gas kommt dem Verhalten des idealen Gasesumso näher, je schwerer es bei Normaldruck zu verflüssigen ist,je tiefer also sein Siedepunkt ist.
Sieht man im Sinne dieser Idealisierung von der Zusammenset-zung des Gases ab, bleiben als makroskopische Zustandsgrößeneines idealen Gases nur der Druck P, das Volumen V, dieTemperatur T und die Zahl N der Moleküle verfügbar. Ge-setzmäßige Zusammenhänge zwischen diesen Zustandsgrößenwurden schon früh entdeckt und untersucht.
Die Gesetze von Boyle-Mariotte und Gay-Lussac
Die Zustandsgleichung des idealen Gases ist bei konstanterTemperatur T und Teilchenzahl N durch das Boyle-Mariot-te’sche Gesetz gegeben:
PV D const : (33.25)
Führen wir statt des Volumens V die Dichte � ein, gilt ebensogut
P
�D const ; (33.26)
wiederum bei konstanter Temperatur.
Während das Gesetz von Boyle-Mariotte feststellt, dass derDruck eines Gases bei konstanter Temperatur indirekt propor-tional zu seinem Volumen ist, fasst das Gesetz von Gay-Lussacdie beiden experimentellen Befunde zusammen, dass sowohlder Druck bei konstantem Volumen als auch das Volumen beikonstantem Druck jeweils linear von der Temperatur abhängen:
P
TD const ;
V
TD const : (33.27)
Zudem stellt sich heraus, dass Druck oder Volumen bei sonstgleichen Bedingungen proportional zur Stoffmenge n sind.
Gesetze von Boyle-Mariotte und von Gay-Lussac
Das Gesetz von Boyle-Mariotte besagt, dass das Produktaus Druck und Volumen eines idealen Gases bei konstan-ter Temperatur konstant ist:
PV D const bei T D const : (33.28)
Das Gesetz von Gay-Lussac stellt fest, dass Druck bzw.Volumen eines idealen Gases proportional zur Temperatursind, wenn das Volumen bzw. der Druck konstant gehaltenwerden:
P / T bei V D const ;
V / T bei P D const :(33.29)
Achtung Bei der Formulierung des Gesetzes von Gay-Lussac mag man sich die Frage stellen, wie es experimentellüberhaupt aufgestellt werden kann, ohne in Zirkelschlüsse zugeraten. Gasthermometer scheiden offensichtlich zur Tempera-turmessung aus, weil das Verhalten von Gasen unter Tempera-turänderungen ja gerade der Gegenstand der Untersuchung ist.Bei anderen Thermometern muss man zumindest sicher sein,dass sie linear auf Temperaturänderungen reagieren. Veröffent-lichungen von Joseph Gay-Lussac legen die Vermutung nahe,dass er bereits Quecksilberthermometer verwendet und derenLinearität im verwendeten Temperaturbereich sorgfältig abge-wogen hat. J
Fasst man diese experimentellen Befunde in einer Gleichungzusammen, lautet sie
PV D nR .T � T0/ ; (33.30)
worin die allgemeine Gaskonstante R als Proportionalitätsfaktorauftritt und der Nullpunkt T0 der Temperaturskala noch mög-lichst geschickt gewählt werden muss.
Achtung Beachten Sie, dass die Gaskonstante R letztlich dieSteigung der Temperaturskala bestimmt und deswegen von derWahl der Temperaturskala abhängen muss. J
Die ursprüngliche Temperaturskala des schwedischen Ma-thematikers und Geodäten Anders Celsius (1701–1744)verwendete als Fixpunkte die Temperaturen des Gefrier- und

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.5 Die idealen Gasgesetze 1117
Teil
IV
des Siedepunktes von Wasser bei Normaldruck, d. h. bei ei-nem Luftdruck von 1 atm D 1;013 bar. Der Bereich zwischendiesen Fixpunkten, gemessen mit einem Quecksilberthermo-meter, wird in 100 „Grad“ genannte gleich lange Abschnitteeingeteilt, was auch zu der historischen Bezeichnung des „hun-dertteiligen Thermometers“ geführt hat. Bei Normaldruck liegtder Gefrierpunkt des Wassers bei 0 ıC, sein Siedepunkt bei100 ıC. (Kurioserweise hatte Celsius zunächst den Gefrierpunktdes Wassers bei 100ıC und den Siedepunkt bei 0ıC festgelegt,was später sinnvollerweise vertauscht wurde.)
Die absolute Temperaturskala
Experimente mit vielen verschiedenen hinreichend verdünntenGasen zeigen, dass im Grenzfall eines idealen Gases das Ver-hältnis
.PV/100 ıC
.PV/0 ıCD 1;3661 (33.31)
beträgt. Mithilfe dieses Messwertes folgt aus dem experimen-tellen Befund (33.30)
1;3661 D 100 ıC � T0
0 ıC � T0; (33.32)
woraus
T0 D ��
100
0;3661
�ıC D �273;15 ıC (33.33)
folgt. Verschieben wir die Temperaturskala so, dass ihr Null-punkt bei der Temperatur T0 D �273;15 ıC liegt, behalten aberdie Celsius’sche Teilung in 100ıC zwischen dem Gefrier- unddem Siedepunkt des Wassers bei, erhalten wir die Temperaturin Kelvin:
TŒK� D TŒ ıC� C 273;15 : (33.34)
Verwenden wir diese absolute Temperaturskala in (33.30), ver-einfacht sich das experimentell gefundene Gesetz (33.30) zurZustandsgleichung eines idealen Gases.
Zustandsgleichung eines idealen Gases
Die Zustandsgleichung eines idealen Gases lautet
PV D nRT ; (33.35)
wenn die Temperatur in Kelvin angegeben wird. Darintritt die allgemeine Gaskonstante R als Proportionalitäts-faktor auf. Bei T D 0 K verschwindet formal das ProduktPV aus Druck und Volumen. Das ist eine unrealistischeFolge der Annahme, dass das Gas ideal sei. Jedes rea-le Gas verflüssigt sich bei einer geringen, aber endlichenTemperatur.
Die Zustandsgleichung für ideale Gase (33.35) enthält die Stoff-menge n der im Volumen V enthaltenen Mole.
International wurde 1967 die Größe
T D 273;16 K (33.36)
als Tripelpunkt des Wassers fixiert. Das ist derjenige Punkt imZustandsraum, bei dem Wasser zugleich in festem, flüssigemund gasförmigem Aggregatzustand vorliegt.
Absolute Temperatur
Es gibt eine absolute Temperaturskala. Ihr Nullpunkt bei�273;15 ıC kann dadurch definiert werden, dass dortder Druck eines idealen Gases verschwindet. Absolu-te Temperaturen werden in Kelvin (K) angegeben. DieTemperaturskala wird dadurch festgelegt, dass dem Tri-pelpunkt des Wassers die Temperatur 273;16 K D 0;01ıCzugewiesen wird.
Um Temperaturen zu unterscheiden, die auf verschiedenen Ska-len gemessen werden, unterscheiden wir von jetzt an, wo esnötig wird, die absolute Temperatur T von der Temperatur # ,die in Grad Celsius gemessen wird.
Gaskonstante und Boltzmann-Konstante
Bei einer Temperatur von # D 0 ıC und Normaldruck von1 atm D 1;013 bar nimmt ein Mol eines idealen Gases – d. h.einer Gasmenge, deren Masse der Molekülmasse in Grammentspricht – ein Volumen von 22;4141 l ein. Aus diesen empiri-schen Zahlenwerten erhalten wir die universelle Gaskonstante
R D 1;013 bar � 22;4141 dm3 mol�1
273;15 K
D 8;3145J
mol K(33.37)
oder, ausgedrückt in Kalorien:
R D 1;986cal
mol K: (33.38)
Ideale Gasgleichung mit Teilchenzahl
Setzen wir anstelle der Stoffmenge n das Verhältnis n DN=NA ein und definieren die Konstante kB D R=NA, soerhalten wir für die Zustandsgleichung idealer Gase dieweitere Fassung
PV D NkBT : (33.39)

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1118 33 Phänomenologische Begründung der Thermodynamik
Die hier neu eingeführte Konstante
kB D R
NAD 1;3806488 .13/ � 10�23 J K�1 (33.40)
ist die universelle Boltzmann-Konstante, die schon in der Ein-führung unter (33.2) erwähnt wurde. Sie stellt den Zusammen-hang zwischen der Temperatur- und der Energieskala her.
Achtung Die Boltzmann-Konstante ist insofern eine unnö-tige Naturkonstante, als sie lediglich zwischen den willkürlichgewählten Einheiten der absoluten Temperatur und der Ener-gie umzurechnen ermöglicht. Umgekehrt könnte man den Wertder Boltzmann-Konstante exakt festlegen und damit das Kelvinneu definieren. So schlägt das Consultative Committee for Units(CCU) des Bureau International des Poids et Mesures im Ent-wurf einer Broschüre zur Neudefinition der SI-Einheiten vor,die Boltzmann-Konstante auf exakt 1;38065 � 10�23 J K�1 fest-zusetzen und damit zugleich das Kelvin neu zu betimmen. J
Häufig ist es nützlich, die Boltzmann-Konstante nicht in J K�1,sondern in eV K�1 anzugeben. Dann beträgt sie
kB D 8;6173 � 10�5 eV K�1 : (33.41)
Frage 4Welcher Temperatur entspricht die Ionisationsenergie einesWasserstoffatoms von 13;6 eV?
33.6 Der erste Hauptsatz
Nachdem die Stofftheorie der Wärme als unhaltbar erkannt wor-den war, trat an die Stelle des Wärmestoffs oder Caloricums dieinnere Energie, die sich infolge von Wärme- und Arbeitsaus-tausch mit der Umgebung ändern kann.
Arbeit, Wärme und innere Energie
Bei einer beliebigen thermodynamischen Zustandsänderungseien ıQ die vom System aufgenommene Wärme und ıW dievom System verrichtete Arbeit. Der erste Hauptsatz der Ther-modynamik stellt nun axiomatisch eine Beziehung zwischendiesen Größen und der inneren Energie her.
Erster Hauptsatz der Thermodynamik
Jedem thermodynamischen System wird die innere Ener-gie U als eine Zustandsgröße zugeordnet. Die innereEnergie wächst mit der zugeführten Wärme und nimmtum die vom System an seiner Umwelt verrichtete Arbeit
ab. Für ein geschlossenes System beträgt die infinitesima-le Änderung der inneren Energie
dU D ıQ � ıW ; (33.42)
wenn es die infinitesimale Wärme ıQ aufnimmt und dieinfinitesimale Arbeit ıW an der Umgebung verrichtet. Dieinnere Energie ist nur bis auf eine additive Konstante be-stimmt.
Im Gegensatz zu den Prozessgrößen Wärme und Arbeit ist dieinnere Energie eine Zustandsgröße und hat daher ein vollständi-ges Differenzial dU. Für jeden Kreisprozess gilt daher
IdU D 0 : (33.43)
Für ein abgeschlossenes System, das an seiner Umwelt keineArbeit verrichten kann und das auch mit ihr keine Wärme aus-tauscht, gilt wegen ıW D 0 und ıQ D 0 sogar dU D 0. Unterdieser Voraussetzung ist also die innere Energie U konstant; sieist dann eine Erhaltungsgröße. Das gilt natürlich auch, wenninnerhalb eines abgeschlossenen Systems irreversible Prozessestattfinden sollten, die das System aus einem Nichtgleichge-wichts- in einen Gleichgewichtszustand überführen.
Bei der Zustandsänderung eines adiabatischen Systems, daszwar Arbeit an der Umgebung verrichten kann, aber keine Wär-me mit ihr austauscht, hat auch die Arbeit ein vollständigesDifferenzial, weil dann wegen ıQ D 0 aufgrund des erstenHauptsatzes dU D �ıW gelten muss. In guter Näherung ver-läuft ein Prozess dann adiabatisch, wenn er sehr viel schnellerabläuft, als ein Wärmeaustausch mit der Umwelt erfolgen kann.Ein Beispiel für adiabatisch verlaufende Schwingungen sinddie Schallwellen, die im Kasten „Vertiefung: Schallwellen undSchallgeschwindigkeit“ in Abschn. 35.3 näher besprochen wer-den.
Eine andere, aber vergleichbare Vereinfachung ergibt sich füranergische Systeme, die keine Arbeit an ihrer Umgebung ver-richten, aber mit ihr Wärme austauschen können. Dann istıW D 0, daher dU D ıQ, sodass in einem solchen Fall auchdas Differenzial der Wärme vollständig ist.
Für den allgemeinen Fall eines geschlossenen Systems betrach-ten wir noch einen beliebig verlaufenden Kreisprozess. Da sichdie innere Energie als Zustandsgröße nach jedem vollständigenUmlauf eines Kreisprozesses nicht geändert haben darf, aus-gedrückt durch (33.43), muss in einem Kreisprozess die demSystem insgesamt zugeführte Wärme gleich der dabei vom Sys-tem verrichteten Arbeit sein:
IıQ D
IıW : (33.44)
Dies präzisiert die Aussage, die wir noch ohne die Kenntnisder inneren Energie unter (33.14) getroffen hatten: Aus einem

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.6 Der erste Hauptsatz 1119
Teil
IV
Kreisprozess geht ein System unverändert hervor, d. h., seineinnere Energie muss am Ende des Kreisprozesses auf ihren An-fangswert zurückkehren. Über die Einzelbeträge von Wärmeund Arbeit während des Kreisprozesses sagt diese Gleichheitjedoch nichts aus.
Eine andere häufig benutzte Formulierung des ersten Haupt-satzes ist: Es lässt sich kein Perpetuum mobile erster Artkonstruieren. Ein Perpetuum mobile erster Art ist eine gedach-te, periodisch arbeitende Maschine, die während eines UmlaufsEnergie abgibt und an seinem Ende wieder in ihren Ausgangs-zustand zurückkehrt. Diese Version des ersten Hauptsatzesfolgt einfach aus (33.43), denn ein Umlauf entspricht einemKreisprozess, und Energie kann dabei weder entstehen nochvernichtet werden.
Innere Energie und Wärmekapazitäteneines idealen Gases
Wir untersuchen nun die innere Energie am Beispiel eines idea-len Gases. Als unabhängige Zustandgrößen wählen wir beifestgehaltener Teilchenzahl N das Volumen V und die Tempe-ratur T , sodass das vollständige Differenzial dU in der Form
dU D�
@U
@V
�
TdV C
�@U
@T
�
V
dT (33.45)
geschrieben werden kann. Die eingeklammerten partiellen Ab-leitungen mit Subskript bedeuten, dass während der Ableitun-gen diejenige Größe konstant gehalten werden soll, die imSubskript steht. Diese zunächst vielleicht etwas eigenartig wir-kende Notation wird in Abschn. 35.1 genauer erläutert.
Mit dem idealen Gas im Volumen V werde nun der Gay-Lussac-Versuch durchgeführt: Wie in Abb. 33.12 gezeigt, wird das Gasin einem adiabatisch isolierten System nach dem schnellen Her-ausziehen einer Trennwand von V auf V C�V expandiert. VomSystem wird weder Arbeit verrichtet, noch wird ihm Wärme zu-geführt, sodass
�U D 0 (33.46)
gelten muss. Nach dieser irreversiblen Expansion wird �T D 0gemessen, d. h. sie verläuft zumindest im Rahmen der Messge-nauigkeit isotherm! Dies gilt für beliebige Volumenänderungen�V. Aufgrund dieses empirischen Befunds impliziert (33.45),dass für ein ideales Gas die partielle Ableitung der inneren Ener-gie nach dem Volumen bei konstanter Temperatur verschwindenmuss, sodass die innere Energie eines idealen Gases nur von sei-ner Temperatur abhängen kann:
U D U.T/ C U0 : (33.47)
Wir berufen uns hier allein auf die gegebene empirische Be-gründung, bleiben die theoretische Begründung von (33.47)schuldig und reichen sie später in Abschn. 35.3 nach.
V ΔV
V + ΔV
Abb. 33.12 Der Versuch von Gay-Lussac
Innere Energie eines idealen Gases
Bei fester Teilchenzahl hängt die innere Energie einesidealen Gases allein von seiner Temperatur, aber nicht vonseinem Volumen ab.
Mithilfe des Resultats (33.47) wenden wir uns der Beziehungzwischen den Molwärmen cmol
P bei konstantem Druck und cmolV
bei konstantem Volumen zu. Dazu betrachten wir zwei Zu-standsänderungen: Die eine soll bei konstantem V stattfinden,also das System aus dem Zustand .V; T/ in den infinitesimal be-nachbarten Zustand .V; T C dT/ überführen. Dabei ändert sichwegen des konstant gehaltenen Volumens die innere Energie umden Betrag
dU D ıQjVDconst D ncmolV dT : (33.48)
Die andere Änderung soll bei konstantem Druck P stattfindenund den Zustand .V; T/ in den Zustand .V C dV; T C dT/ über-führen. Dabei muss sich die innere Energie um den Betrag
dU D ıQjPDconst � PdV D ncmolP dT � PdV (33.49)
ändern. Nun verwenden wir das ideale Gasgesetz (33.39), umdie Volumenänderung dV bei konstantem Druck mit der Tem-peraturänderung dT in Beziehung zu setzen:
PdV D nRdT : (33.50)
Damit ergibt sich aus (33.49)
dU D n�cmol
P � R�
dT (33.51)

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1120 33 Phänomenologische Begründung der Thermodynamik
Vertiefung: Einige Eigenschaften idealer Gase
Aus dem empirisch gefundenen Gesetz (33.39) können wirbereits einige wichtige Ergebnisse für ideale Gase ableiten.Wir setzen hier voraus, dass die Teilchenzahl N festgehaltenbleibt.
Zunächst hängt die Arbeit, die ein ideales Gas durch eineVolumenänderung verrichtet, durch
�W DZ
P.V/dV D NkB
ZT.V/dV
V
davon ab, wie sich die Temperatur während einer Volu-menänderung verhält. Bleibt die Temperatur etwa dadurchkonstant, dass das Gas während der Ausdehnung mit einemWärmebad der Temperatur T verbunden bleibt, verrichtet dasGas bei einem solchen isothermen Übergang vom VolumenV1 zum Volumen V2 > V1 die Arbeit
�W D NkBT
V2Z
V1
dV
VD NkBT ln
V2
V1:
Dehnt sich das Gas aber isobar gegen einen konstanten äu-ßeren Druck aus, muss es die Arbeit
�W D P
V2Z
V1
dV D P.V2 � V1/
verrichten.
Betrachten wir eine kleine Volumenänderung dV, für die einideales Gas die Arbeit ıW D PdV aufbringen muss. Bei
konstantem äußeren Druck muss aufgrund der Zustandsglei-chung (33.39)
0 D dP D NkB
�dT
V� TdV
V2
�D NkBT
V
�dT
T� dV
V
�
gelten, also muss die relative Temperatur- gerade gleich derrelativen Volumenänderung sein:
dT
TD dV
V:
Da die innere Energie eines idealen Gases nur von der Tem-peratur abhängt, muss sie bei einer isothermen Expansionkonstant bleiben. Nach dem ersten Hauptsatz muss dann dievom System aufgenommene Wärme restlos als Arbeit abge-geben werden:
ıQ D ıW :
Die Arbeit �W aufgrund einer isothermen Volumenände-rung lässt sich aber leicht berechnen, wie wir bereits zuBeginn dieses Kastens gesehen hatten. Damit es isothermvom Volumen V1 auf das Volumen V2 expandieren kann,muss einem idealen Gas also die Wärmemenge
ıQ D NkBT lnV2
V1
zugeführt werden.
bei konstantem Druck. Bei beiden Prozessen seien T und dTgleich. Da die innere Energie eines idealen Gases nur von derTemperatur abhängt, muss dann auch die Änderung dU derinneren Energie in beiden Fällen gleich sein. Daher folgt aus(33.48) und (33.49)
cmolP � cmol
V D R (33.52)
für die Differenz der molaren Wärmekapazitäten eines idealenGases. Die experimentellen Werte in Tabelle 33.2 liegen in derTat nicht weit weg vom Wert R D 8;3145 J mol�1 K�1 der uni-versellen Gaskonstante.
Wärmekapazitäten des idealen Gases
Die Differenz der molaren Wärmekapazitäten cmolP und
cmolV eines idealen Gases ist gleich der idealen Gaskon-
stanten R.
Tab. 33.2 Differenz zwischen den molaren Wärmekapazitäten cmolP und cmol
Veiniger realer Gase. Im Grenzfall eines idealen Gases beträgt die Differenz R D8;3145 J mol�1 K�1
Stoff CP � CV in J mol�1 K�1
He 8;09Ar 8;25H2 8;38Luft 8;36CH4 8;38CO2 8;35
Eine detaillierte Behandlung der Wärmekapazitäten, andererResponsefunktionen und der Zusammenhänge zwischen ihnenfolgt in Abschn. 35.1. Die dort abgeleiteten, allgemeinen Er-gebnisse werden in Abschn. 35.3 auf das ideale Gas angewandt.Dadurch wird die hier bestimmte Differenz der Wärmekapa-zitäten eines idealen Gases in einen größeren Zusammenhanggestellt.
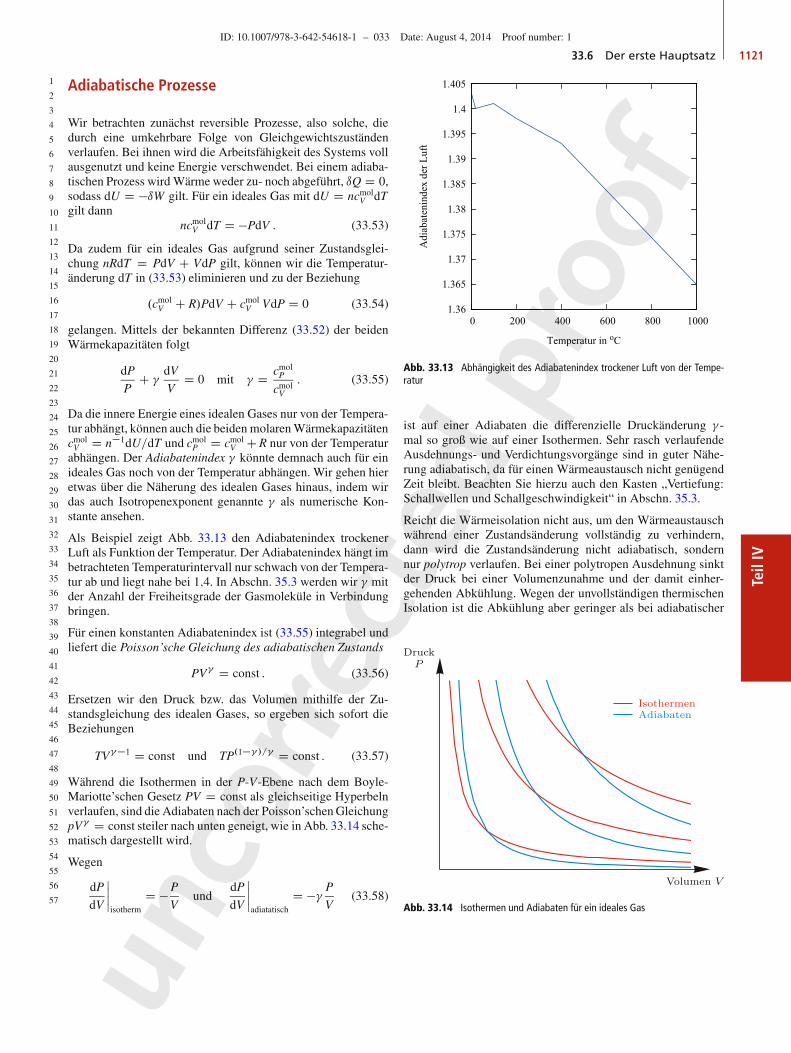
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.6 Der erste Hauptsatz 1121
Teil
IV
Adiabatische Prozesse
Wir betrachten zunächst reversible Prozesse, also solche, diedurch eine umkehrbare Folge von Gleichgewichtszuständenverlaufen. Bei ihnen wird die Arbeitsfähigkeit des Systems vollausgenutzt und keine Energie verschwendet. Bei einem adiaba-tischen Prozess wird Wärme weder zu- noch abgeführt, ıQ D 0,sodass dU D �ıW gilt. Für ein ideales Gas mit dU D ncmol
V dTgilt dann
ncmolV dT D �PdV : (33.53)
Da zudem für ein ideales Gas aufgrund seiner Zustandsglei-chung nRdT D PdV C VdP gilt, können wir die Temperatur-änderung dT in (33.53) eliminieren und zu der Beziehung
.cmolV C R/PdV C cmol
V VdP D 0 (33.54)
gelangen. Mittels der bekannten Differenz (33.52) der beidenWärmekapazitäten folgt
dP
PC �
dV
VD 0 mit � D cmol
P
cmolV
: (33.55)
Da die innere Energie eines idealen Gases nur von der Tempera-tur abhängt, können auch die beiden molaren Wärmekapazitätencmol
V D n�1dU=dT und cmolP D cmol
V C R nur von der Temperaturabhängen. Der Adiabatenindex � könnte demnach auch für einideales Gas noch von der Temperatur abhängen. Wir gehen hieretwas über die Näherung des idealen Gases hinaus, indem wirdas auch Isotropenexponent genannte � als numerische Kon-stante ansehen.
Als Beispiel zeigt Abb. 33.13 den Adiabatenindex trockenerLuft als Funktion der Temperatur. Der Adiabatenindex hängt imbetrachteten Temperaturintervall nur schwach von der Tempera-tur ab und liegt nahe bei 1;4. In Abschn. 35.3 werden wir � mitder Anzahl der Freiheitsgrade der Gasmoleküle in Verbindungbringen.
Für einen konstanten Adiabatenindex ist (33.55) integrabel undliefert die Poisson’sche Gleichung des adiabatischen Zustands
PV� D const : (33.56)
Ersetzen wir den Druck bzw. das Volumen mithilfe der Zu-standsgleichung des idealen Gases, so ergeben sich sofort dieBeziehungen
TV��1 D const und TP.1��/=� D const : (33.57)
Während die Isothermen in der P-V-Ebene nach dem Boyle-Mariotte’schen Gesetz PV D const als gleichseitige Hyperbelnverlaufen, sind die Adiabaten nach der Poisson’schen GleichungpV� D const steiler nach unten geneigt, wie in Abb. 33.14 sche-matisch dargestellt wird.
Wegen
dP
dV
ˇˇisotherm
D � P
Vund
dP
dV
ˇˇadiatatisch
D ��P
V(33.58)
1.36
1.365
1.37
1.375
1.38
1.385
1.39
1.395
1.4
1.405
0 200 400 600 800 1000
Adia
bat
enin
dex
der
Luft
Temperatur in oC
Abb. 33.13 Abhängigkeit des Adiabatenindex trockener Luft von der Tempe-ratur
ist auf einer Adiabaten die differenzielle Druckänderung � -mal so groß wie auf einer Isothermen. Sehr rasch verlaufendeAusdehnungs- und Verdichtungsvorgänge sind in guter Nähe-rung adiabatisch, da für einen Wärmeaustausch nicht genügendZeit bleibt. Beachten Sie hierzu auch den Kasten „Vertiefung:Schallwellen und Schallgeschwindigkeit“ in Abschn. 35.3.
Reicht die Wärmeisolation nicht aus, um den Wärmeaustauschwährend einer Zustandsänderung vollständig zu verhindern,dann wird die Zustandsänderung nicht adiabatisch, sondernnur polytrop verlaufen. Bei einer polytropen Ausdehnung sinktder Druck bei einer Volumenzunahme und der damit einher-gehenden Abkühlung. Wegen der unvollständigen thermischenIsolation ist die Abkühlung aber geringer als bei adiabatischer
Volumen V
DruckP
IsothermenAdiabaten
Abb. 33.14 Isothermen und Adiabaten für ein ideales Gas

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1122 33 Phänomenologische Begründung der Thermodynamik
Ausdehnung. Infolgedessen fällt eine solche Polytrope genann-te Kurve im P-V-Diagramm weniger steil ab als eine Adiabate.Ihre Gleichung ist
PV˛ D const (33.59)
mit dem Polytropenindex ˛ � � . Bis hierher gilt die Betrach-tung polytroper Zustandsänderungen für allgemeine thermody-namische Systeme.
Wenden wir nun das Ergebnis (33.59) auf ein ideales Gas an,folgen daraus mithilfe der Zustandsgleichung unmittelbar dieBeziehungen
TV˛�1 D const und TP.1�˛/=˛ D const ; (33.60)
aus denen wir die Zusammenhänge
dT
TD .1 � ˛/
dV
Vund
dT
TD ˛ � 1
˛
dP
P(33.61)
zwischen den relativen Änderungen der Temperatur, des Vo-lumens und des Druckes bei polytropen Zustandsänderungenschließen können.
Frage 5Vergewissern Sie sich, dass für ein ideales Gas (33.60) aus(33.59) folgt und dass sich daraus (33.61) ergibt.
Bei einer adiabatischen oder einer allgemeineren polytropenExpansion wird das Reservoir der inneren Energie des idealenGases in Anspruch genommen. Zur Berechnung der dabei ver-richteten Arbeit schreiben wir
�W DV2Z
V1
P dV D nR
V2Z
V1
T
VdV D � nR
˛ � 1
T2Z
T1
dT
D � nR
˛ � 1.T2 � T1/ ;
(33.62)
wobei zunächst die Zustandsgleichung des idealen Gases unddann (33.61) verwendet wurde. Bei einer Expansion (Entspan-nung) des Gases ist T2 < T1. Wie erwartet, verrichtet das Gasdann Arbeit an seiner Umgebung.
Abkühlung einer Gaspatrone
Ein vielleicht aus dem alltäglichen Leben bekanntes Bei-spiel dafür ist die Abkühlung einer Gaspatrone, wie siebeispielsweise zur Herstellung von Sprudelwasser ver-wendet werden. Wird die Gaspatrone geöffnet, strömtdas enthaltene Gas aus, verdrängt einen Teil der Umge-bungsluft, muss dafür Arbeit aufwenden und kühlt sichdeswegen so weit ab, dass das Kondenswasser auf ihrerOberfläche zu Reif gefrieren kann. J
Die Enthalpie als Zustandsgröße
Oft ist es in der Thermodynamik nützlich, Zustandsgrößen sozu wählen, dass sie der Situation bestmöglich angepasst sind,die gerade beschrieben werden soll. In Abschn. 35.1 werdenwir ausführlich die sogenannten thermodynamischen Potenzia-le besprechen und dabei zeigen, wie solche Zustandsgrößensystematisch konstruiert werden können. Im Rahmen der phä-nomenologischen Thermodynamik, die wir in diesem Kapitelbehandeln, stellen wir neben der inneren Energie nur die für dieTechnik wichtige Zustandsgröße Enthalpie H vor, die durch dieSumme
H D U C PV (33.63)
definiert ist. Der Begriff leitet sich ab von den altgriechischenWörtern en für „in“ und thalpéin für „erwärmen“, „erhitzen“;das Formelzeichen H spielt auf die englische Bezeichnung heatcontent an.
Mithilfe des vollständigen Differenzials dU D ıQ � PdV derinneren Energie, das uns der erste Hauptsatz vorgibt, folgt fürdie infinitesimale Änderung der Enthalpie
dH D ıQ C VdP : (33.64)
Bei konstantem Druck, dP D 0, ist demnach dH die dem Sys-tem von außen zugeführte Wärme, dH D ıQ bei P D const.Daraus folgt für die molare Wärmekapazität cmol
P bei konstan-tem Druck
cmolP D 1
n
�@H
@T
�
P
; (33.65)
während für die molare Wärmekapazität cmolV bei konstantem
Volumen
cmolV D 1
n
�@U
@T
�
V
(33.66)
gilt.
Um die Bedeutung der Enthalpie in der Technik aufzuklären,stellen wir zunächst fest, dass alle Wärmekraftmaschinen nur inperiodischer Folge mithilfe eines strömenden ArbeitsmediumsArbeit verrichten können. Ein Schwungrad sorgt bei solchenMaschinen durch seine Trägheit für einen gleichmäßigen, pe-riodischen Arbeitsablauf. Zur Beschreibung solcher Situationenhat man den Begriff der technischen Arbeit geschaffen, der an-hand von Abb. 33.15 erläutert werden soll.
Im ersten Zeitabschnitt einer Arbeitsperiode strömt ein Arbeits-medium mit konstantem Druck P1 in den Zylinder ein. Es musssich dadurch Platz schaffen, dass es den Kolben bis zur Stel-lung 1 verschiebt, wodurch das Volumen V1 entsteht. Dabeiverrichtet das Medium über den Kolben die Verdrängungsar-beit P1V1. Im zweiten Zeitabschnitt der Arbeitsperiode wirddas Zuflussventil geschlossen. Das Arbeitsmedium dehnt sichweiter aus und verschiebt den Kolben bis zur Stellung 2 mitVolumen V2. Dabei sinkt der Druck des Arbeitsmediums vonP1 auf P2. Die über den Kolben verrichtete Verdrängungsarbeit
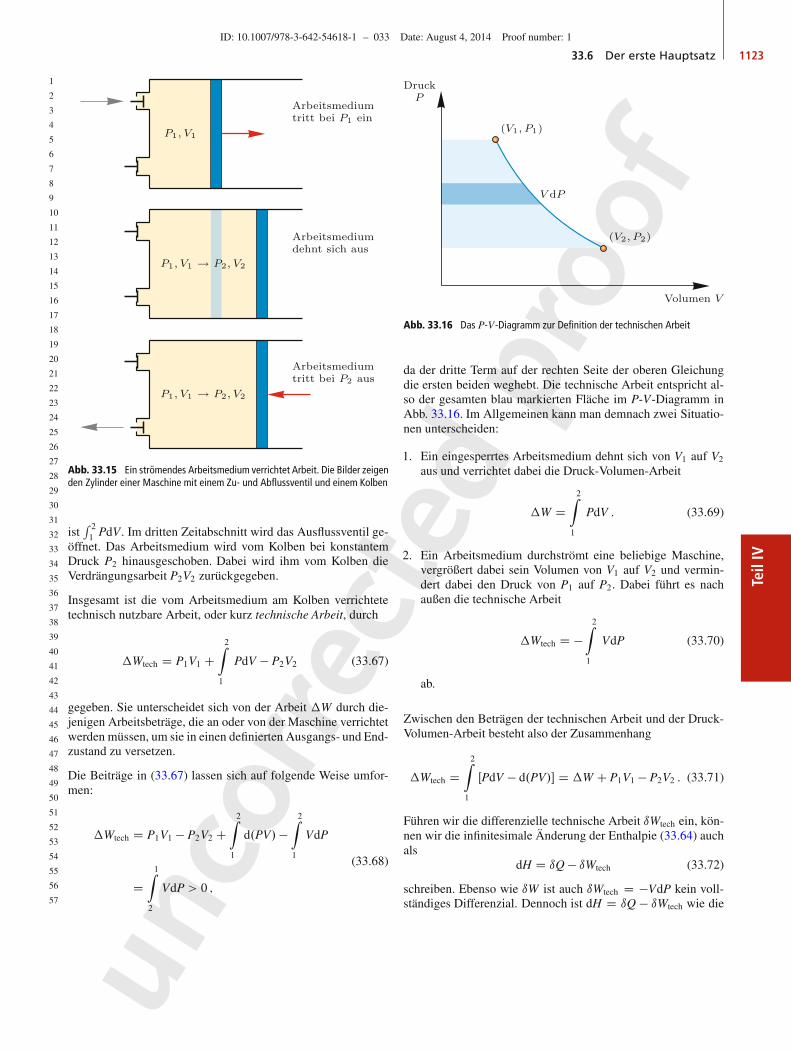
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.6 Der erste Hauptsatz 1123
Teil
IV
P1, V1
Arbeitsmediumtritt bei P1 ein
P1, V1 → P2, V2
Arbeitsmediumdehnt sich aus
P1, V1 → P2, V2
Arbeitsmediumtritt bei P2 aus
Abb. 33.15 Ein strömendes Arbeitsmedium verrichtet Arbeit. Die Bilder zeigenden Zylinder einer Maschine mit einem Zu- und Abflussventil und einem Kolben
istR 2
1 PdV. Im dritten Zeitabschnitt wird das Ausflussventil ge-öffnet. Das Arbeitsmedium wird vom Kolben bei konstantemDruck P2 hinausgeschoben. Dabei wird ihm vom Kolben dieVerdrängungsarbeit P2V2 zurückgegeben.
Insgesamt ist die vom Arbeitsmedium am Kolben verrichtetetechnisch nutzbare Arbeit, oder kurz technische Arbeit, durch
�Wtech D P1V1 C2Z
1
PdV � P2V2 (33.67)
gegeben. Sie unterscheidet sich von der Arbeit �W durch die-jenigen Arbeitsbeträge, die an oder von der Maschine verrichtetwerden müssen, um sie in einen definierten Ausgangs- und End-zustand zu versetzen.
Die Beiträge in (33.67) lassen sich auf folgende Weise umfor-men:
�Wtech D P1V1 � P2V2 C2Z
1
d.PV/ �2Z
1
VdP
D1Z
2
VdP > 0 ;
(33.68)
(V1, P1)
(V2, P2)
V dP
Volumen V
DruckP
Abb. 33.16 Das P-V-Diagramm zur Definition der technischen Arbeit
da der dritte Term auf der rechten Seite der oberen Gleichungdie ersten beiden weghebt. Die technische Arbeit entspricht al-so der gesamten blau markierten Fläche im P-V-Diagramm inAbb. 33.16. Im Allgemeinen kann man demnach zwei Situatio-nen unterscheiden:
1. Ein eingesperrtes Arbeitsmedium dehnt sich von V1 auf V2
aus und verrichtet dabei die Druck-Volumen-Arbeit
�W D2Z
1
PdV : (33.69)
2. Ein Arbeitsmedium durchströmt eine beliebige Maschine,vergrößert dabei sein Volumen von V1 auf V2 und vermin-dert dabei den Druck von P1 auf P2. Dabei führt es nachaußen die technische Arbeit
�Wtech D �2Z
1
VdP (33.70)
ab.
Zwischen den Beträgen der technischen Arbeit und der Druck-Volumen-Arbeit besteht also der Zusammenhang
�Wtech D2Z
1
ŒPdV � d.PV/� D �W C P1V1 � P2V2 : (33.71)
Führen wir die differenzielle technische Arbeit ıWtech ein, kön-nen wir die infinitesimale Änderung der Enthalpie (33.64) auchals
dH D ıQ � ıWtech (33.72)
schreiben. Ebenso wie ıW ist auch ıWtech D �VdP kein voll-ständiges Differenzial. Dennoch ist dH D ıQ � ıWtech wie die

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1124 33 Phänomenologische Begründung der Thermodynamik
innere Energie ein vollständiges Differenzial und definiert (bisauf eine additive Konstante) die Zustandsgröße Enthalpie.
Innere Energie und Enthalpie
Während Änderungen der inneren Energie durch die Dif-ferenz aus der zugeführten Wärme und der Druck-Volu-men-Arbeit zustande kommen, gibt eine Enthalpieände-rung die Differenz aus zugeführter Wärme und technischnutzbarer Arbeit an.
Aufgrund der Beziehung (33.72) hängen infinitesimale Beträgeder Druck-Volumen-Arbeit und der technischen Arbeit durch
ıWtech D ıW � d.PV/ (33.73)
zusammen. Wir werden in Abschn. 35.1 sehen, dass dies typischfür Größen ist, die sich durch sogenannte Legendre-Transfor-mationen auseinander ergeben.
33.7 Der zweite Hauptsatz: 1. Teil
Der erste Hauptsatz formuliert, wie verschiedene Formen derEnergie ineinander umgewandelt werden können. Er beschreibt,wie die innere Energie eines physikalischen Systems entwe-der durch Wärmezu- oder -abfuhr verändert werden kann oderdurch Arbeit, die durch das System an der Umwelt oder am Sys-tem durch die Umwelt verrichtet wird. Aber er beschreibt nicht,welche Zustandsänderungen thermodynamischer Systeme über-haupt möglich sind.
Wir haben oben zwischen reversiblen und irreversiblen Vor-gängen unterschieden und dabei reversible Vorgänge als solchegekennzeichnet, die langsam genug ablaufen, dass sie als eineFolge von Gleichgewichtszuständen beschrieben werden kön-nen. Natürliche Vorgänge sind nie völlig reversibel, könneneinem reversiblen Ablauf aber unter Umständen beliebig nahekommen.
Natürliche, fast reversible Vorgänge
Ein Beispiel für einen natürlichen, fast reversiblen Vor-gang ist ein Luftballon, der sich in der Sonne ausdehntund im Schatten wieder zusammenzieht. Während der Er-wärmung passt sich sein Volumen so an, dass der Druckin seinem Inneren dem Außendruck gleich wird. DieseZustandsänderung verläuft in sehr guter Näherung umge-kehrt ab, während sich der Ballon wieder abkühlt. J
Reversible Vorgänge stellen idealisierte Grenzfälle von realis-tischen Prozessen dar. Schon früh während der Entwicklungder Thermodynamik wurde daher die Notwendigkeit erkannt,das Ausmaß der Irreversibilität bei physikalischen Vorgängen
zu messen. Dies wurde durch die Einführung einer neuen Zu-standsgröße möglich, die Rudolf Clausius mit dem aus demgriechischen entlehnten Kunstwort Entropie bezeichnete.
Im Jahre 1834 brachte der französische Physiker und IngenieurBenoît Paul Clapeyron die bahnbrechende Arbeit von Sadi Car-not in eine mathematische Form, nämlich bereits in diejenigeForm, in welcher der Carnot’sche Kreisprozess heute noch inder Physik beschrieben wird. Dieser Kreisprozess führt auf-grund makroskopischer Konzepte an den Begriff der Entropieheran und erklärt dabei die Wirkungsweise einer Wärmekraft-maschine. Wir werden in Abschn. 34.3 sehen, wie die Entropiedurch die statistische Physik mikroskopisch begründet wird.
Carnot’scher Kreisprozess:Wirkungsgrad mit idealem Gas
Stellen wir uns ein warmes Reservoir mit der Temperatur T1
und ein kaltes Reservoir mit der Temperatur T2 < T1 vor. Zwi-schen beiden Reservoiren wollen wir eine Wärmekraftmaschinebetreiben. Das Arbeitsmedium sei ein ideales Gas, das wir unsdurch ein hinreichend verdünntes reales Gas verwirklicht den-ken können.
In Abschn. 35.6 werden wir Kreisprozesse noch einmal auf ei-ne abstraktere Weise behandeln und dabei sehen, dass wir unsvon der Beschränkung auf ein konkretes Arbeitsmedium voll-kommen lösen können. In einem Carnot-Prozess wechseln sichnun die folgenden vier Teilprozesse periodisch ab, die im P-V-Diagramm in Abb. 33.17 dargestellt sind.
1. Das Gas steht in Kontakt mit dem warmen Reservoir undexpandiert isotherm bei der Temperatur T1 von einem Volu-men V1 zu einem Volumen V2 > V1. Die Zustandsänderungdes Gases wird durch die Zustandsgleichung
PV D nRT D const (33.74)
des idealen Gases beschrieben und im P-V-Diagramm durchein Hyperbelstück dargestellt. Bei dieser isothermen Expan-sion verrichtet das Gas die Arbeit �W12, die noch bestimmtwerden muss.
2. Im zweiten Teilprozess isolieren wir das Gas von demwarmen Reservoir und von seiner Umgebung. Bei abneh-mendem Druck dehnt es sich adiabatisch weiter aus. Wiewir in Abschn. 33.6 gesehen hatten, verläuft eine Adiabateim P-V-Diagramm steiler als eine Isotherme. Sie wird durch(33.56) beschrieben,
PV� D const ; (33.75)
worin der Adiabatenindex � > 1 auftritt. Bei dieser adia-batischen Expansion verringert sich die Temperatur wegender an der Umgebung verrichteten Arbeit. Wir führen dieExpansion bis zu demjenigen Volumen V3 durch, bei dem
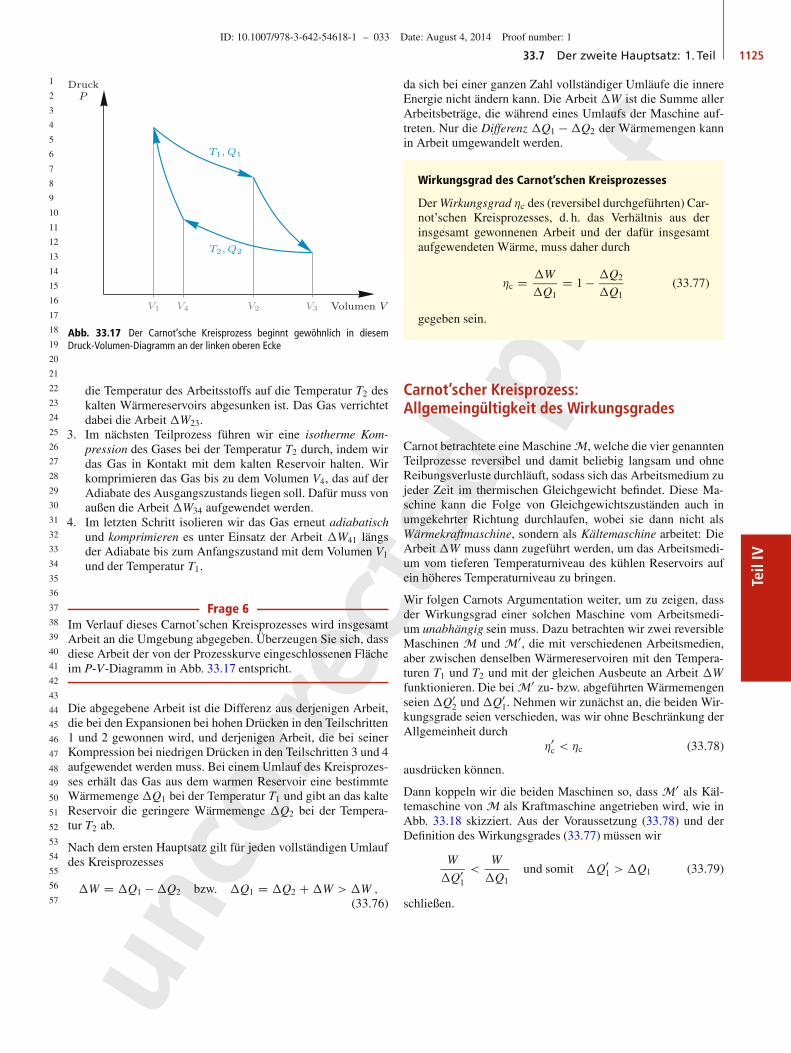
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.7 Der zweite Hauptsatz: 1. Teil 1125
Teil
IV
Volumen V
DruckP
V4 V3V2V1
T2, Q2
T1, Q1
Abb. 33.17 Der Carnot’sche Kreisprozess beginnt gewöhnlich in diesemDruck-Volumen-Diagramm an der linken oberen Ecke
die Temperatur des Arbeitsstoffs auf die Temperatur T2 deskalten Wärmereservoirs abgesunken ist. Das Gas verrichtetdabei die Arbeit �W23.
3. Im nächsten Teilprozess führen wir eine isotherme Kom-pression des Gases bei der Temperatur T2 durch, indem wirdas Gas in Kontakt mit dem kalten Reservoir halten. Wirkomprimieren das Gas bis zu dem Volumen V4, das auf derAdiabate des Ausgangszustands liegen soll. Dafür muss vonaußen die Arbeit �W34 aufgewendet werden.
4. Im letzten Schritt isolieren wir das Gas erneut adiabatischund komprimieren es unter Einsatz der Arbeit �W41 längsder Adiabate bis zum Anfangszustand mit dem Volumen V1
und der Temperatur T1.
Frage 6Im Verlauf dieses Carnot’schen Kreisprozesses wird insgesamtArbeit an die Umgebung abgegeben. Überzeugen Sie sich, dassdiese Arbeit der von der Prozesskurve eingeschlossenen Flächeim P-V-Diagramm in Abb. 33.17 entspricht.
Die abgegebene Arbeit ist die Differenz aus derjenigen Arbeit,die bei den Expansionen bei hohen Drücken in den Teilschritten1 und 2 gewonnen wird, und derjenigen Arbeit, die bei seinerKompression bei niedrigen Drücken in den Teilschritten 3 und 4aufgewendet werden muss. Bei einem Umlauf des Kreisprozes-ses erhält das Gas aus dem warmen Reservoir eine bestimmteWärmemenge �Q1 bei der Temperatur T1 und gibt an das kalteReservoir die geringere Wärmemenge �Q2 bei der Tempera-tur T2 ab.
Nach dem ersten Hauptsatz gilt für jeden vollständigen Umlaufdes Kreisprozesses
�W D �Q1 � �Q2 bzw. �Q1 D �Q2 C �W > �W ;(33.76)
da sich bei einer ganzen Zahl vollständiger Umläufe die innereEnergie nicht ändern kann. Die Arbeit �W ist die Summe allerArbeitsbeträge, die während eines Umlaufs der Maschine auf-treten. Nur die Differenz �Q1 � �Q2 der Wärmemengen kannin Arbeit umgewandelt werden.
Wirkungsgrad des Carnot’schen Kreisprozesses
Der Wirkungsgrad �c des (reversibel durchgeführten) Car-not’schen Kreisprozesses, d. h. das Verhältnis aus derinsgesamt gewonnenen Arbeit und der dafür insgesamtaufgewendeten Wärme, muss daher durch
�c D �W
�Q1D 1 � �Q2
�Q1(33.77)
gegeben sein.
Carnot’scher Kreisprozess:Allgemeingültigkeit des Wirkungsgrades
Carnot betrachtete eine MaschineM, welche die vier genanntenTeilprozesse reversibel und damit beliebig langsam und ohneReibungsverluste durchläuft, sodass sich das Arbeitsmedium zujeder Zeit im thermischen Gleichgewicht befindet. Diese Ma-schine kann die Folge von Gleichgewichtszuständen auch inumgekehrter Richtung durchlaufen, wobei sie dann nicht alsWärmekraftmaschine, sondern als Kältemaschine arbeitet: DieArbeit �W muss dann zugeführt werden, um das Arbeitsmedi-um vom tieferen Temperaturniveau des kühlen Reservoirs aufein höheres Temperaturniveau zu bringen.
Wir folgen Carnots Argumentation weiter, um zu zeigen, dassder Wirkungsgrad einer solchen Maschine vom Arbeitsmedi-um unabhängig sein muss. Dazu betrachten wir zwei reversibleMaschinen M und M0, die mit verschiedenen Arbeitsmedien,aber zwischen denselben Wärmereservoiren mit den Tempera-turen T1 und T2 und mit der gleichen Ausbeute an Arbeit �Wfunktionieren. Die beiM0 zu- bzw. abgeführten Wärmemengenseien �Q0
2 und �Q0
1. Nehmen wir zunächst an, die beiden Wir-kungsgrade seien verschieden, was wir ohne Beschränkung derAllgemeinheit durch
�0
c < �c (33.78)
ausdrücken können.
Dann koppeln wir die beiden Maschinen so, dass M0 als Käl-temaschine von M als Kraftmaschine angetrieben wird, wie inAbb. 33.18 skizziert. Aus der Voraussetzung (33.78) und derDefinition des Wirkungsgrades (33.77) müssen wir
W
�Q0
1
<W
�Q1und somit �Q0
1 > �Q1 (33.79)
schließen.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1126 33 Phänomenologische Begründung der Thermodynamik
M M′
T1 > T2
T2
W
Q2 Q′2
Q′1Q1
Abb. 33.18 Eine Wärmekraftmaschine M wird mit einer Kältemaschine M0
gekoppelt
Dem heißeren Reservoir muss also in diesem Fall durch dieKältemaschine M0 mehr Wärme zugeführt werden, als ihm dieKraftmaschine M zuvor entzogen hat. Durch die Kopplung derMaschinen muss demnach insgesamt die Wärme �Q0
1 � �Q1
aus dem kälteren Reservoir entnommen und dem wärmerenReservoir zugeführt werden. Der Gesamteffekt der beiden ge-koppelten Maschinen wäre also, dass die Wärmemenge �Q0
1 ��Q1 vom tieferen Temperaturniveau T2 zum höheren Tempe-raturniveau T1 übergeben würde, ohne dass im Ganzen Arbeitverrichtet wurde und ohne dass eine andere Änderung inM oderM0 zurückgeblieben ist.
Dieses Ergebnis widerspricht der Erfahrung, die durch dasClausius’sche Postulat ausgedrückt wird.
Clausius’sches Postulat
Wärme kann nicht von selbst von einem niederen zu ei-nem höheren Temperaturniveau übergehen.
Der Widerspruch zwischen dem aus der Erfahrung abgeleitetenClausius’schen Postulat und dem Ergebnis unserer Überlegun-gen zu den gekoppelten Maschinen wird behoben, indem wirdie Voraussetzung verschiedener Wirkungsgrade aufgeben: Bei-de Wirkungsgrade müssen demnach gleich groß sein,
�0
c D �c ; (33.80)
damit die Kopplung zwischen einer idealen Wärmekraft- undeiner idealen Kältemaschine nicht in Konflikt mit der Erfahrunggerät.
Demzufolge müssen alle reversiblen Maschinen, die nur beizwei Temperaturen T1 und T2 Wärme mit der Umgebung
austauschen, denselben Wirkungsgrad haben. Dann kann derWirkungsgrad aber nur von diesen beiden Temperaturen abhän-gen. Wegen (33.77) muss entsprechend
�Q1
�Q2D f .T1; T2/ (33.81)
gelten, wobei f eine universelle, vom Arbeitsmedium und vonder Konstruktion der Wärmemaschine unabhängige, dimensi-onslose Funktion sein muss.
Carnot’scher Kreisprozess:Wärme und absolute Temperatur
Diese Funktion f .T1; T2/ lässt sich nun weiter spezifizieren.Dazu führen wir neben den beiden Temperaturniveaus T1 undT2 < T1 ein weiteres Wärmereservoir mit einer beliebig gedach-ten Referenztemperatur T0 ein, die nicht notwendig zwischenden beiden Temperaturen T1 und T2 liegen muss. Diese Tem-peratur T0 kann z. B. als Bezugspunkt einer Temperaturskalaangesehen werden. Ohne vorerst genauer festlegen zu müssen,was 1ı genau sei, können wir z. B. sagen, dass T0 D 1ı seinmöge.
Die beiden Temperaturniveaus T1 und T2 werden nun durchdie beiden reversiblen Carnot-Maschinen M1 und M2 mit demReferenzniveau T0 verbunden. Die Maschine M1 werde alsWärmekraftmaschine betrieben, die Maschine M2 als Kälte-maschine. Dabei sei die Maschine M2 so konstruiert, dass sieaus dem Referenzreservoir bei T0 gerade dieselbe Wärmemen-ge �Q0 aufnimmt, welche die Maschine M1 dorthin abgibt.
Da beide Maschinen als ideale Carnot-Maschinen reversibelarbeiten, entsprechen sie gekoppelt einer einfachen Carnot-Ma-schine M, die direkt zwischen den beiden Niveaus T1 und T2
als Wärmekraftmaschine betrieben wird. Daher tritt das Refe-renzreservoir bei der Wärmebilanz nicht in Erscheinung: Dieeinfache Maschine M zwischen den Niveaus T1 und T2 arbeitetmit denselben Wärmemengen wie die zusammengesetzte Ma-schine, die aus den beiden Maschinen M1 und M2 besteht, diezwischen den Niveaus T1 und T0 bzw. T0 und T2 arbeiten. Neben(33.81) gelten dann die Gleichungen
�Q1
�Q0D f .T1; T0/ und
�Q0
�Q2D f .T0; T2/ : (33.82)
Durch Multiplikation dieser beiden Gleichungen und Vergleichmit (33.81) entsteht
f .T1; T2/ D f .T1; T0/ f .T0; T2/ : (33.83)
Setzt man hier T1 D T2 und berücksichtigt, dass dann �Q1 D�Q2 sein muss, weil auf derselben Isotherme expandiert undkomprimiert wird, muss die linke Seite f .T1; T1/ D 1 sein. Die

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.7 Der zweite Hauptsatz: 1. Teil 1127
Teil
IV
Vertauschung der Argumente in f muss also gemäß (33.83) zumKehrwert führen:
f .T0; T1/ D 1
f .T1; T0/: (33.84)
Wenden wir diese Einsicht auf (33.83) an, folgt
f .T1; T2/ D f .T1; T0/
f .T2; T0/: (33.85)
Da die Zwischentemperatur T0 beliebig gewählt wurde und alsBezugspunkt einer Temperaturskala dienen kann, kürzen wirf .T; T0/ D Nf .T/ ab. Zusammen mit (33.84) können wir danndas Verhältnis der Wärmemengen (33.82) in der Form
�Q1
�Q2D
Nf .T1/
Nf .T2/(33.86)
schreiben. Die Funktion Nf .T/ muss zudem streng monotonwachsen, damit die aufgenommenen oder abgegebenen Wär-men mit der Temperatur anwachsen.
Es liegt nun nahe, diese streng monotone Funktion der Tempera-tur, Nf .T/, der Einfachheit halber als den Zahlenwert einer neuenTemperaturskala zu verwenden und sie, versehen mit einer ge-eigneten Einheit, mit der Temperatur selbst zu identifizieren.Als Einheit liegt die Referenztemperatur T0 nahe. Wir definierendementsprechend T D Nf .T/T0 als die neue, absolute Tempera-turskala, die uns die Diskussion von Kreisprozessen nahelegt.Aufgrund von (33.86) würde dann die Wärmemenge, die einphysikalisches System aus einem Reservoir der Temperatur Tentnehmen oder daran abgeben kann, proportional zur Tempe-ratur selbst werden:
�Q1
�Q2D T1
T2: (33.87)
Für ein ideales Gas stellt es sich als durchaus sinnvoll heraus,die Funktion Nf .T/T0 mit der Temperatur T zu identifizieren. Wirhaben in Abschn. 33.6 gesehen, dass die Arbeit, die unter iso-thermen, adiabatischen oder polytropen Bedingungen von oderan einem idealen Gas verrichtet wird, proportional zur absolu-ten Temperatur ist. Unter isothermen Bedingungen, unter denensich die innere Energie des idealen Gases nicht verändert, mussdann aufgrund des ersten Hauptsatzes auch die Wärme propor-tional zur Temperatur sein.
Unter allgemeinen Zustandsänderungen müsste dann auch dieinnere Energie proportional zur Temperatur sein. Für das idea-le Gas würde dies wegen (33.48) und (33.51) bedeuten, dassseine Wärmekapazitäten bei konstantem Volumen ebenso wiebei konstantem Druck unabhängig von der Temperatur seinmüssten. Wir werden später eine überzeugende theoretische Be-gründung dafür finden.
Aufgrund dieser Überlegungen identifizieren wir abschließenddie Funktion Nf .T/T0 mit einer neuen Temperaturskala T und er-warten aufgrund der Verhältnisse beim idealen Gas, dass dieseTemperatur mit der absoluten Temperatur gleichgesetzt werdenkann, die bereits in Abschn. 33.5 eingeführt wurde.
Wirkungsgrad des Carnot’schen Kreisprozesses
Aus (33.77) schließen wir auf die Gleichung
�c D 1 � T2
T1(33.88)
für den Wirkungsgrad der Carnot-Maschine.
Er kann nur dann gleich eins werden, wenn das kalte Reservoirdie Temperatur T2 D 0 K oder #2 D �273;15 ıC hat. Brin-gen wir (33.87) in die Form �Q1=T1 D �Q2=T2, sehen wir,dass bei einem Carnot’schen Kreisprozess gleich große Beträgeder Größe �Q=T aufgenommen und freigesetzt werden. Daraufkommen wir in Abschn. 33.7 zurück.
Wirkungsgrad einer Dampfmaschine
Der Wirkungsgrad �c ist der maximale mögliche theore-tische Wirkungsgrad. Er kann nur dann erreicht werden,wenn alle möglichen Teilprozesse reversibel ablaufen.In der Praxis ist der Wirkungsgrad � stets (viel) kleinerals �c. Als Beispiel betrachten wir eine Dampfmaschine,deren heißes Reservoir 100 ıC und deren kaltes Reser-voir 20 ıC hat. Ihr Wirkungsgrad kann maximal �c D1 � 293=373 D 0;214 sein. Eine Verbesserung ist durcheine Druckerhöhung im heißen Behälter möglich, weildann die Siedetemperatur des Wassers höher liegt. Bei ei-nem Druck von 10 bar beträgt sie etwa TSiede D 180 ıC.Bei dieser Temperatur des heißen Reservoirs steigt derWirkungsgrad auf �c D 0;35. Moderne Verbrennungsmo-toren erreichen Wirkungsgrade zwischen 0;35 und 0;5. J
Kreisprozesse mit idealem Gas:Absolute Temperaturskala und Entropie
Wir betrachten nun den Carnot’schen Kreisprozess im Detail amkonkreten Beispiel eines idealen Gases.
Carnot’scher Kreisprozess mit idealem Gas
Da die innere Energie eines idealen Gases nur von derTemperatur abhängt, ist sie während eines isothermenProzesses konstant. Wie wir zum Abschluss des Kastens„Vertiefung: Einige Eigenschaften idealer Gase“ in Ab-schn. 33.6 gezeigt haben, wird dann die Wärmemenge
�Q D �W DV2Z
V1
PdV D nRT lnV2
V1(33.89)
aufgenommen.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1128 33 Phänomenologische Begründung der Thermodynamik
Volumen V
DruckP
T2
T1
S1 = S4
S2 = S3
Q12
Q34
1
2
3
4
Abb. 33.19 Der Carnot’sche Kreisprozess im Detail. Die beiden rotenPfeile sollen andeuten, dass im oberen Teil Wärme in die Maschine fließt,im unteren wieder heraus. Die von den Kurvenstücken umschlossene Flä-che ist ein Maß für die verrichtete Arbeit
Für den in Abb. 33.19 gezeigten Kreisprozess betragendemnach die zu- und abgeführten Wärmen
�Q12 D nRT1 lnV2
V1und
�Q34 D nRT2 lnV3
V4:
(33.90)
Da die Zustandspaare 2 und 3 mit den Volumina V2 undV3 bzw. 4 und 1 mit den Volumina V4 und V1 jeweils aufAdiabaten liegen, gilt zudem aufgrund von (33.57)
T1V��11 D T2V��1
4 und T1V��12 D T2V��1
3 :(33.91)
Daraus ergeben sich die Verhältnisse
V2
V1D V3
V4(33.92)
der Volumina. Setzen wir diese Volumenverhältnisse indie beiden Gleichungen (33.90) ein und dividieren dieerste durch die zweite Gleichung, kürzen sich die Loga-rithmen heraus, sodass wir
�Q12
�Q34D T1
T2(33.93)
erhalten. Dies entspricht gerade der Beziehung (33.87)zwischen den aufgenommenen und abgegebenen Wär-men und den Temperaturen, bei denen sie aufgenommenoder abgegeben werden. Dieses Ergebnis bekräftigt noch-mals, dass die Definition der absoluten Temperatur, diewir aus dem Carnot’schen Kreisprozess abgeleitet haben,mit derjenigen übereinstimmt, die wir aus den gemesse-nen Eigenschaften des idealen Gases abgeleitet hatten.
J
Nun gehen wir zu einer allgemeineren reversiblen Zustands-änderung des idealen Gases über. Der erste Hauptsatz und dieZustandsgleichung führen auf
ıQrev D dU C PdV D nCVdT C nRTdV
V: (33.94)
Da wir bereits festgestellt haben, dass die innere Energie desidealen Gases nicht vom Volumen abhängt, aber proportionalzur Temperatur ist, können wir die Wärmekapazität CV bei kon-stantem Volumen als unabhängig sowohl von der Temperaturals auch vom Volumen ansehen. Indem wir (33.94) durch dieTemperatur T dividieren, erhalten wir
ıQrev
TD X.T/dT C Y.V/dV : (33.95)
Im Gegensatz zu ıQrev ist dies ein vollständiges Differenzial,da X unabhängig von V und Y unabhängig von T sind. Wiejedes vollständige Differenzial definiert ıQrev=T eine Zustands-größe, die wir durch Integration des Differenzials von einemAusgangszustand Z0 zu einem Endzustand Z gewinnen:
S.Z/ � S.Z0/ DZZ
Z0
ıQrev
T�
ZZ
Z0
dS : (33.96)
Wenn der Prozess reversibel geführt wird, ist das Wegintegraldavon unabhängig, auf welchem Weg wir vom Anfangs- zumEndzustand gelangen. Bis auf eine Integrationskonstante kannS demnach nur von den unabhängigen Zustandsgrößen abhän-gen, die den Zustand Z definieren. Die neue Zustandsgröße S,die hier anhand der Eigenschaften eines idealen Gases definiertwurde, bezeichnen wir mit Rudolf Clausius als Entropie. Diesesans Griechische angelehnte Kunstwort bedeutet „Verwandelbar-keit“, „Umlauf“ oder „Wendung“.
Entropie eines idealen Gases
Insbesondere für ein ideales Gas mit festgehaltener Teil-chenzahl N besteht aufgrund von (33.94) der Zusammen-hang
S.T; V/ D nCV lnT
T0C nR ln
V
V0C S0 (33.97)
zwischen den unabhängigen Zustandsgrößen .T; V/ undder Entropie S, wobei S0 D S.T0; V0/ abgekürzt wurde.
Im Allgemeinen, namentlich wenn das Arbeitsmedium keinideales Gas ist, muss bei der Integration von (33.96) in Betrachtgezogen werden, dass die Wärmekapazität CV von der Tempe-ratur abhängen kann.
Bei adiabatischen und reversiblen Zustandsänderungen bleibtdie Entropie konstant, denn dafür gilt gerade ıQrev D 0. Insolchen Fällen muss eine mögliche Temperaturänderung gerade

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.7 Der zweite Hauptsatz: 1. Teil 1129
Teil
IV
durch eine entsprechende Volumenänderung kompensiert wer-den. Für ein ideales Gas ergibt (33.97) dann
CV lnT
T0D �R ln
V
V0: (33.98)
Wenn sich ein ideales Gas adiabatisch ausdehnen soll, musses sich daher entsprechend abkühlen. Bei konstantem Volumenoder bei konstanter Temperatur steigt die Entropie logarith-misch mit der Temperatur oder dem Volumen an.
Frage 7Rechnen Sie explizit nach, dass bei einer adiabatischen Expan-sion eines idealen Gases, für die wir TV��1 D const gezeigthaben, die Entropie unverändert bleibt.
Achtung Wir betonen nochmals, dass wir bei der Herleitungvon (33.97) vorausgesetzt haben, dass die Wärmekapazität beikonstantem Volumen temperaturunabhängig sei. In den meis-ten nichtidealen Systemen verringert sich die Wärmekapazitäteines Körpers bei Erniedrigung der Temperatur, was sich beson-ders bei tiefen Temperaturen bemerkbar macht. Eine genauereTheorie der Wärmekapazitäten bei tiefen Temperaturen mussQuanteneffekte berücksichtigen, die in Abschn. 37.7 bespro-chen werden. J
Spezielle reversible Kreisprozesse
Bei Kreisprozessen kann das Arbeitsmedium entweder in einemgeschlossenem System zirkulieren (geschlossener Kreispro-zess) oder ein offenes System durchströmen. Bei einem offenenProzess wird das Arbeitsmedium nach einem oder mehrerenTeilprozessen ausgestoßen. Dieses „Ausstoßen“ können wir alsWärmeabgabe an die Umgebung betrachten, die dann die Rol-le eines Wärmereservoirs übernimmt. Für den kontinuierlichenBetrieb muss im Kreisprozess genauso viel Stoff aufgenommenwerden wie abgegeben wurde.
Im P-V-Diagramm rechts- oder im Uhrzeigersinn laufende Pro-zesse beschreiben Wärmekraftmaschinen, während links- odergegen den Uhrzeigersinn laufende Prozesse Kältemaschinenoder Wärmepumpen beschreiben. Bei einer Wärmepumpe wirdunter Arbeitszufuhr Wärme von einem niedrigeren zu einemhöheren Temperaturniveau gepumpt. Die auf dem hohen Tem-peraturniveau anfallende Wärme kann z. B. zum Heizen genutztwerden (Wärmepumpenheizung). Bei der Kältemaschine wirddie Abkühlung des Kältemittels benutzt, um ein anderes Fluidabzukühlen. Für den Wärmekraftprozess ist die abgegebene Ar-beit der Nutzen und die zugeführte Wärme der Aufwand. Füreine Kältemaschine ist die zugeführte Arbeit der Aufwand unddie dem kalten Reservoir entzogene Wärme der Nutzen. Bei derWärmepumpe ist die dem wärmeren Reservoir zugeführte Wär-me der Nutzen.
Bei der nun folgenden Diskussion verschiedener spezieller Rea-lisierungen von Kreisprozessen nehmen wir ein ideales Gas alsArbeitsmedium an.
Den Carnot’schen Kreisprozess haben wir wegen seiner his-torischen und konzeptionellen Bedeutung bereits im Detailbesprochen, sodass wir hier nur noch einmal seine wichtigstenEigenschaften zusammenfassen. Der Carnot’sche Kreisprozessist als theoretischer Vergleichsprozess von großer Bedeutungund gibt Aufschluss über die Güte anderer Prozesse, die beidenselben Temperaturen der jeweiligen heißen und kalten Wär-mereservoire ablaufen. In seinen vier Teilschritten, die im P-V-Diagramm in Abb. 33.19 dargestellt wurden, treten die folgen-den Arbeitsbeträge auf:
1 ! 2: isotherme Expansion,�W12 D �Q12 D nRT1 ln V2=V1,2 ! 3: adiabatische Expansion,�W23 D �nCV.T2 � T1/,3 ! 4: isotherme Kompression,�W34 D �Q34 D nRT2 ln V4=V3,4 ! 1: adiabatische Kompression,�W41 D �nCV.T1 � T2/.
Wir hatten unter (33.91) und (33.92) gesehen, dass aus demAdiabatengesetz PV� D const die Beziehung V4=V3 D V1=V2
zwischen den Volumina folgt, sodass die vom Medium verrich-tete Arbeit gleich
�W D �W12 C �W23 C �W34 C �W41
D nR.T1 � T2/ lnV2
V1
(33.99)
ist. Daraus ergibt sich direkt der schon aus (33.88) bekannteWirkungsgrad:
�c D �W
�Q12D 1 � T2
T1: (33.100)
Der hier beschriebene rechtslaufende Prozess beschreibt eineideale Wärmekraftmaschine, während der linkslaufende Pro-zess eine ideale Wärmepumpe bzw. eine ideale Kältemaschinedarstellt. Für die Kältemaschine ist die Leistungszahl durch
"K D �Q43
�WD T2
T1 � T2(33.101)
definiert, wobei für den linkslaufenden Prozess �Q43 D��Q34 ist und �W das Vorzeichen umkehrt. Für die Wärme-pumpe ist
"W D �Q21
�WD T1
T1 � T2: (33.102)
Joule’scher oder Brayton’scher Kreisprozess Der Joule’scheoder Brayton’sche Kreisprozess (benannt nach dem amerika-nischen Maschinenbauingenieur George Brayton, 1830–1892;
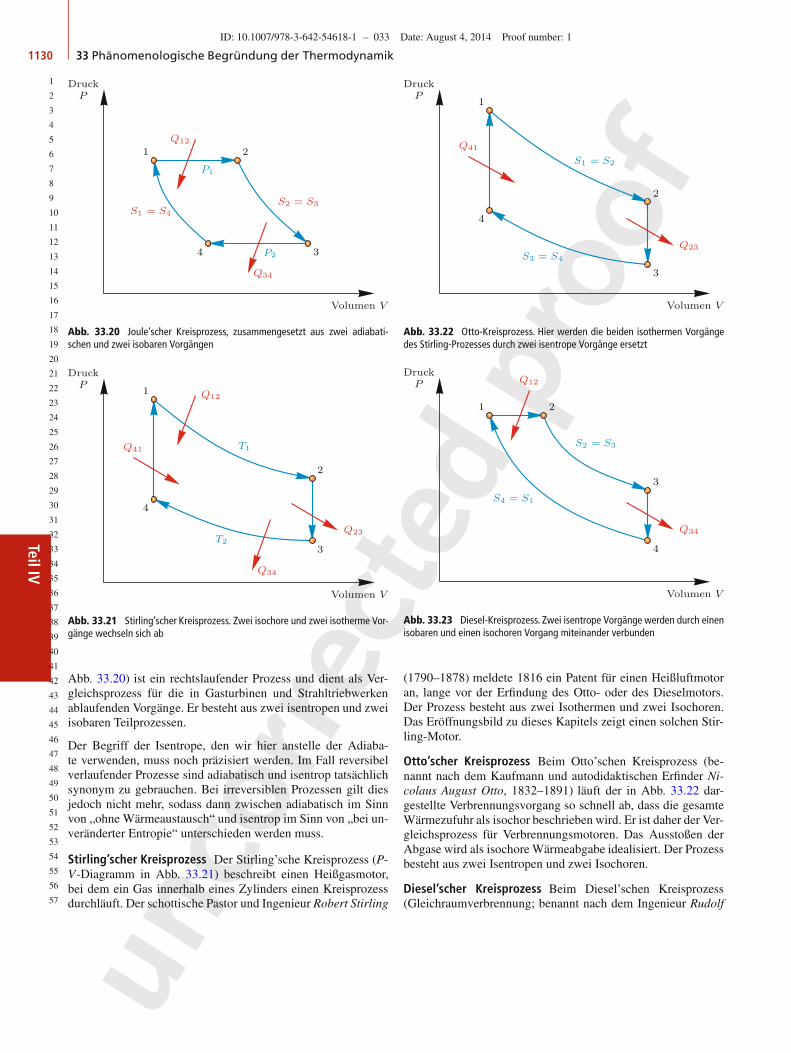
TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1130 33 Phänomenologische Begründung der Thermodynamik
Volumen V
DruckP
P1
P2
S1 = S4S2 = S3
Q12
Q34
1 2
34
Abb. 33.20 Joule’scher Kreisprozess, zusammengesetzt aus zwei adiabati-schen und zwei isobaren Vorgängen
Volumen V
DruckP
T2
T1
Q12
Q34
Q41
Q23
1
2
3
4
Abb. 33.21 Stirling’scher Kreisprozess. Zwei isochore und zwei isotherme Vor-gänge wechseln sich ab
Abb. 33.20) ist ein rechtslaufender Prozess und dient als Ver-gleichsprozess für die in Gasturbinen und Strahltriebwerkenablaufenden Vorgänge. Er besteht aus zwei isentropen und zweiisobaren Teilprozessen.
Der Begriff der Isentrope, den wir hier anstelle der Adiaba-te verwenden, muss noch präzisiert werden. Im Fall reversibelverlaufender Prozesse sind adiabatisch und isentrop tatsächlichsynonym zu gebrauchen. Bei irreversiblen Prozessen gilt diesjedoch nicht mehr, sodass dann zwischen adiabatisch im Sinnvon „ohne Wärmeaustausch“ und isentrop im Sinn von „bei un-veränderter Entropie“ unterschieden werden muss.
Stirling’scher Kreisprozess Der Stirling’sche Kreisprozess (P-V-Diagramm in Abb. 33.21) beschreibt einen Heißgasmotor,bei dem ein Gas innerhalb eines Zylinders einen Kreisprozessdurchläuft. Der schottische Pastor und Ingenieur Robert Stirling
Volumen V
DruckP
S3 = S4
S1 = S2
Q41
Q23
1
2
3
4
Abb. 33.22 Otto-Kreisprozess. Hier werden die beiden isothermen Vorgängedes Stirling-Prozesses durch zwei isentrope Vorgänge ersetzt
Volumen V
DruckP
S2 = S3
S4 = S1
Q12
Q34
1 2
3
4
Abb. 33.23 Diesel-Kreisprozess. Zwei isentrope Vorgänge werden durch einenisobaren und einen isochoren Vorgang miteinander verbunden
(1790–1878) meldete 1816 ein Patent für einen Heißluftmotoran, lange vor der Erfindung des Otto- oder des Dieselmotors.Der Prozess besteht aus zwei Isothermen und zwei Isochoren.Das Eröffnungsbild zu dieses Kapitels zeigt einen solchen Stir-ling-Motor.
Otto’scher Kreisprozess Beim Otto’schen Kreisprozess (be-nannt nach dem Kaufmann und autodidaktischen Erfinder Ni-colaus August Otto, 1832–1891) läuft der in Abb. 33.22 dar-gestellte Verbrennungsvorgang so schnell ab, dass die gesamteWärmezufuhr als isochor beschrieben wird. Er ist daher der Ver-gleichsprozess für Verbrennungsmotoren. Das Ausstoßen derAbgase wird als isochore Wärmeabgabe idealisiert. Der Prozessbesteht aus zwei Isentropen und zwei Isochoren.
Diesel’scher Kreisprozess Beim Diesel’schen Kreisprozess(Gleichraumverbrennung; benannt nach dem Ingenieur Rudolf
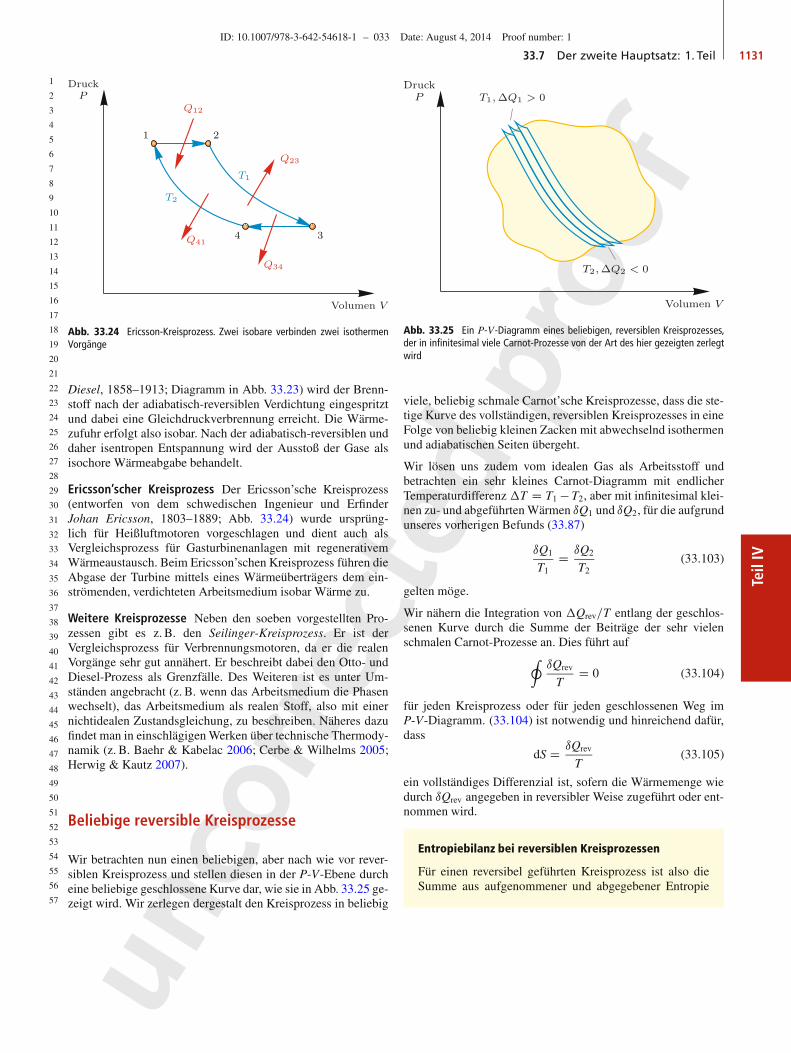
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.7 Der zweite Hauptsatz: 1. Teil 1131
Teil
IV
Volumen V
DruckP
T1
T2
Q12
Q34
Q41
Q23
1 2
34
Abb. 33.24 Ericsson-Kreisprozess. Zwei isobare verbinden zwei isothermenVorgänge
Diesel, 1858–1913; Diagramm in Abb. 33.23) wird der Brenn-stoff nach der adiabatisch-reversiblen Verdichtung eingespritztund dabei eine Gleichdruckverbrennung erreicht. Die Wärme-zufuhr erfolgt also isobar. Nach der adiabatisch-reversiblen unddaher isentropen Entspannung wird der Ausstoß der Gase alsisochore Wärmeabgabe behandelt.
Ericsson’scher Kreisprozess Der Ericsson’sche Kreisprozess(entworfen von dem schwedischen Ingenieur und ErfinderJohan Ericsson, 1803–1889; Abb. 33.24) wurde ursprüng-lich für Heißluftmotoren vorgeschlagen und dient auch alsVergleichsprozess für Gasturbinenanlagen mit regenerativemWärmeaustausch. Beim Ericsson’schen Kreisprozess führen dieAbgase der Turbine mittels eines Wärmeüberträgers dem ein-strömenden, verdichteten Arbeitsmedium isobar Wärme zu.
Weitere Kreisprozesse Neben den soeben vorgestellten Pro-zessen gibt es z. B. den Seilinger-Kreisprozess. Er ist derVergleichsprozess für Verbrennungsmotoren, da er die realenVorgänge sehr gut annähert. Er beschreibt dabei den Otto- undDiesel-Prozess als Grenzfälle. Des Weiteren ist es unter Um-ständen angebracht (z. B. wenn das Arbeitsmedium die Phasenwechselt), das Arbeitsmedium als realen Stoff, also mit einernichtidealen Zustandsgleichung, zu beschreiben. Näheres dazufindet man in einschlägigen Werken über technische Thermody-namik (z. B. Baehr & Kabelac 2006; Cerbe & Wilhelms 2005;Herwig & Kautz 2007).
Beliebige reversible Kreisprozesse
Wir betrachten nun einen beliebigen, aber nach wie vor rever-siblen Kreisprozess und stellen diesen in der P-V-Ebene durcheine beliebige geschlossene Kurve dar, wie sie in Abb. 33.25 ge-zeigt wird. Wir zerlegen dergestalt den Kreisprozess in beliebig
Volumen V
DruckP T1, ΔQ1 > 0
T2, ΔQ2 < 0
Abb. 33.25 Ein P-V-Diagramm eines beliebigen, reversiblen Kreisprozesses,der in infinitesimal viele Carnot-Prozesse von der Art des hier gezeigten zerlegtwird
viele, beliebig schmale Carnot’sche Kreisprozesse, dass die ste-tige Kurve des vollständigen, reversiblen Kreisprozesses in eineFolge von beliebig kleinen Zacken mit abwechselnd isothermenund adiabatischen Seiten übergeht.
Wir lösen uns zudem vom idealen Gas als Arbeitsstoff undbetrachten ein sehr kleines Carnot-Diagramm mit endlicherTemperaturdifferenz �T D T1 � T2, aber mit infinitesimal klei-nen zu- und abgeführten Wärmen ıQ1 und ıQ2, für die aufgrundunseres vorherigen Befunds (33.87)
ıQ1
T1D ıQ2
T2(33.103)
gelten möge.
Wir nähern die Integration von �Qrev=T entlang der geschlos-senen Kurve durch die Summe der Beiträge der sehr vielenschmalen Carnot-Prozesse an. Dies führt auf
IıQrev
TD 0 (33.104)
für jeden Kreisprozess oder für jeden geschlossenen Weg imP-V-Diagramm. (33.104) ist notwendig und hinreichend dafür,dass
dS D ıQrev
T(33.105)
ein vollständiges Differenzial ist, sofern die Wärmemenge wiedurch ıQrev angegeben in reversibler Weise zugeführt oder ent-nommen wird.
Entropiebilanz bei reversiblen Kreisprozessen
Für einen reversibel geführten Kreisprozess ist also dieSumme aus aufgenommener und abgegebener Entropie

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1132 33 Phänomenologische Begründung der Thermodynamik
Übersicht: Prozessgrößen und Wirkungsgrade einiger Kreisprozesse
Joule’scher Kreisprozess Wechsel von isobaren und adia-batischen Teilprozessen (Abb. 33.20):
1 ! 2 W isobare Wärmezufuhr,�Q12 D nCP.T2 � T1/ ; �W12 D nR.T2 � T1/,2 ! 3 W adiabatische Expansion,�W23 D �nCV.T3 � T2/,3 ! 4 W isobare Wärmeabfuhr,�Q34 D nCP.T4 � T3/ ; �W34 D nR.T4 � T3/,4 ! 1 W adiabatische Kompression,�W41 D �nCV.T1 � T4/,Volumenarbeit:�W D nCP.T1 � T2 C T3 � T4/,Wirkungsgrad:� D �W=�Q12 D 1 � .T3 � T4/=.T2 � T1/.
Stirling’scher Kreisprozess Wechsel von isothermen undisochoren Teilprozessen (Abb. 33.21):
1 ! 2 W isotherme Expansion,�W12 D �Q12 D nRT1 ln V2=V1,2 ! 3 W isochore Abkühlung,�Q23 D nCV.T2 � T1/,3 ! 4 W isotherme Kompression,�W34 D �Q34 D nRT2 ln V4=V3 D nRT2 ln V1=V2,4 ! 1 W isochore Erwärmung,�Q41 D nCV.T1 � T2/,Volumenarbeit:�W D nR.T1 � T2/ ln.V2=V1/,Wirkungsgrad:� D �W=�Q12 D 1 � T2=T1.
Otto-Kreisprozess Wechsel von adiabatischen und isocho-ren Teilprozessen (Abb. 33.22):
1 ! 2 W adiabatische Expansion,�W12 D �nCV.T2 � T1/,2 ! 3 W isochore Druckminderung,�Q23 D nCV.T3 � T2/,3 ! 4 W adiabatische Kompression,�W34 D �nCV.T4 � T3/,4 ! 1 W isochore Druckerhöhung,�Q41 D nCV.T1 � T4/,Volumenarbeit:�W D nCV.T1 � T2 C T3 � T4/,
Wirkungsgrad:� D �W=�Q41 D 1 � .T2 � T3/=.T1 � T4/.
Diesel-Kreisprozess Zwei adiabatische Teilprozesse imWechsel mit einem isobaren und einem isochoren Prozess(Abb. 33.23):
1 ! 2 W isobare Expansion,�Q12 D nCP.T2 � T1/ ; �W12 D nR.T2 � T1/,2 ! 3 W adiabatische Expansion,�W23 D �nCV.T3 � T2/,3 ! 4 W isochore Druckminderung,�Q34 D nCV.T4 � T3/,4 ! 1 W adiabatische Kompression,�W41 D �nCV.T1 � T4/,Volumenarbeit:�W D nCP.T2 � T1/ C nCV.T4 � T3/,Wirkungsgrad:� D �W=�Q12 D 1 � .T3 � T4/=Œ�.T2 � T1/�.
Ericsson-Kreisprozess Wechsel von isothermen und isoba-ren Teilprozessen (Abb. 33.24):
1 ! 2 W isobare Expansion,�Q12 D nCP.T1 � T2/ ; �W12 D nR.T1 � T2/,2 ! 3 W isotherme Expansion,�W23 D �Q23 D nRT1 ln V3=V2 D nRT1 ln P1=P2,3 ! 4 W isobare Kompression,�Q34 D nCP.T2 � T1/ ; �W34 D nR.T2 � T1/,4 ! 1 W isotherme Kompression,�W41 D �Q41 D nRT2 ln V1=V4 D nRT2 ln P2=P1,Volumenarbeit:�W D nR.T1 � T2/ ln.P1=P2/,Wirkungsgrad:� D �W=�Q23 D 1 � T2=T1.
Frage 8Verwenden Sie die für isotherme bzw. adiabatische Zu-standsänderungen gültigen Beziehungen PV D const undTV��1 D const, um die Beziehung T3=T2 D T4=T1 her-zuleiten. Vereinfachen Sie damit die Wirkungsgrade desJoule’schen und des Otto-Kreisprozesses.
immer gleich null:
IdS D 0 : (33.106)
Der Faktor 1=T in dS D ıQrev=T erweist sich damit für belie-bige Arbeitsmedien als ein sogenannter integrierender Faktor.Im Kasten „Vertiefung: Vollständige und unvollständige Diffe-renziale“ in Abschn. 33.3 wird die Existenz eines integrierendenFaktors etwas näher beleuchtet und eine Bestimmungsgleichung

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.7 Der zweite Hauptsatz: 1. Teil 1133
Teil
IV
für ihn angegeben. Für ! D ıQrev ist der integrierende Faktorgerade die inverse ideale Gastemperatur.
Die Zustandsgröße S, die Entropie, gibt Auskunft über die maxi-mal aus einem System gewinnbare mechanische Arbeit: GemäßıQ2 D T2ıS ist die wieder an das kältere Reservoir abzugeben-de Wärme umso kleiner, je kleiner ıS ist.
Arbeit und Entropie
Bei festgehaltener aufgenommener Wärmemenge �Q1 istdaher der Anteil von �Q1, der in Arbeit umgewandeltwerden kann, umso größer, je kleiner der Betrag j�Sj derEntropie ist, der während eines Umlaufs zunächst von derWärmekraftmaschine aufgenommen und dann wieder ab-gegeben wird.
Da die Entropie als Zustandsgröße unabhängig von dem Wegsein muss, über den ein System in einen Zustand gelangt ist,muss es erlaubt sein, Entropieunterschiede entlang beliebigerreversibler Zustandsänderungen zu berechnen.
Entropieunterschied
Man berechnet den Unterschied der Entropien zweier be-liebiger Zustände Z1 und Z2 durch
S.Z2/ � S.Z1/ DZ2Z
Z1
ıQrev
T: (33.107)
Die auszuführende Integration hat im Allgemeinen nichts da-mit zu tun, wie das System in Wirklichkeit von Z1 nach Z2
gelangt. Die wirklichen Prozesse sind immer mehr oder weni-ger irreversibel. Unsere Vorschrift (33.107) setzt dagegen einenhinzugedachten und beliebigen Ersatzprozess voraus, der auf re-versible Weise von Z1 nach Z2 gelangt. Wie dieser Ersatzprozessausgeführt wird, muss gleichgültig sein, da dS ein vollständi-ges Differenzial ist. Die Entropiedifferenz kann nicht von demWeg abhängen, der von Z1 nach Z2 führt! Entropieänderungenkönnen daher unabhängig vom wahren Prozessverlauf anhandgeschickt gewählter reversibler Ersatzprozesse bestimmt wer-den.
Achtung Für ein abgeschlossenes System ist die innere Ener-gie konstant, sodass ıW D ıQ gilt. Falls keine Wärme zu- oderabgeführt wird, braucht die Entropie aber trotz ıQ D 0 nichtkonstant zu sein, da irreversible Wechselwirkungen zwischenTeilen des Systems möglich sind. J
So nimmt beim Versuch von Gay-Lussac, der in Abb. 33.12dargestellt ist, die Entropie eines idealen Gases zu, obwohldas System weder Arbeit noch Wärme mit der Umgebung aus-tauscht. Da die Überströmung von V1 nach V2 > V1 adiabatisch
geschieht, ist ıQ D 0, sodass für den wirklichen Verlauf
Z2Z
Z1
ıQ
TD 0 (33.108)
gelten muss – unabhängig davon, wie der Integrand währenddes schnellen, turbulenten Übergangs variiert! Führen wir dage-gen einen reversiblen Ersatzprozess längs einer Isotherme, dennAnfangs- und Endzustände haben beim Überströmen dieselbeTemperatur, dann nimmt gemäß (33.97) die Entropie proportio-nal zum Logarithmus der relativen Volumenvergrößerung beimÜberströmen zu:
�S D nR lnV2
V1: (33.109)
Entropie in abgeschlossenen Systemen
Die Entropie eines abgeschlossenen Systems ist nur fürreversibel geführte Prozesse konstant.
Besteht ein inhomogenes System aus mehreren homogenen Teil-systemen, dann ist für jedes Teilsystem
dSi D ıQrev;i
Ti(33.110)
ein vollständiges Differenzial, wenn Ti die absolute Tempera-tur des i-ten Teilsystems und ıQrev;i die Wärme ist, die das i-teTeilsystem mit der Umgebung oder den anderen Teilsystemenauf reversible Weise austauscht. Die Summe der Differenziale
dS DX
i
dSi DX
i
ıQrev;i
Ti(33.111)
ist ebenfalls wieder ein vollständiges Differenzial.
Frage 9Warum brauchen die Wärmeübergänge zwischen den homoge-nen Teilsystemen des Systems nicht berücksichtigt zu werden?Argumentieren Sie zunächst selbst und lesen Sie dann den fol-genden Absatz.
Hier ist es wichtig einzusehen, dass die Wärmeübergänge zwi-schen den homogenen Teilsystemen des Systems nicht berück-sichtigt werden müssen. Da diese Übergänge reversibel seinsollen, müssen sie zwischen Teilsystemen gleicher Tempera-tur stattfinden, denn jede Wärmeleitung wäre irreversibel. Dieswird in Abschn. 33.8 näher besprochen. Findet aber ein rever-sibler Wärmeaustausch zwischen den Teilsystemen i und j statt,dann trägt dieser bis auf ein Vorzeichen auf gleiche Weise zu�Qrev;i und �Qrev;j in (33.111) bei. Da Ti D Tj gelten muss,kompensieren sich die Terme in der Summe (33.111). Somitbrauchen wir in ıQrev;i nur die dem i-ten Teilsystem von derUmgebung zugeführte Wärme zu berücksichtigen.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1134 33 Phänomenologische Begründung der Thermodynamik
Haben alle Teilsysteme die gleiche Temperatur, dann verein-facht sich (33.111) wieder zu
dS D 1
T
X
i
ıQrev;i D ıQrev
T: (33.112)
Eine wichtige Eigenschaft der Entropie ist offensichtlich: DieEntropie eines homogenen, im thermischen Gleichgewicht be-findlichen Systems wächst mit seiner Stoffmenge, denn dieWärmemenge �Q, die beim Übergang des Systems von ei-nem beliebigen Ausgangszustand in den betrachteten Zustandin jedem Teilprozess verbraucht wird, ist proportional zu seinerStoffmenge. Deshalb ist die Entropie eines Systems gleich derSumme der Entropien der Teilsysteme.
Extensivität
Die Entropie ist eine extensive Zustandsgröße. Wie imKasten „Mathematischer Hintergrund 33.1: HomogeneFunktionen“ erläutert wird, ist sie deswegen eine homo-gene Funktion vom Grad 1 in der Stoffmenge.
Bevor wir zur Diskussion irreversibler Prozesse übergehen,fassen wir hier das wichtigste Ergebnis dieses Abschnitts zu-sammen.
Der zweite Hauptsatz für reversible Prozesse
Für reversibel geführte Prozesse ist das Differenzial
dS D ıQrev
T(33.113)
vollständig. Es definiert eine Zustandsgröße S, die Entro-pie. Bei einem vollständigen Umlauf eines reversibelgeführten Kreisprozesses ändert sich die Entropie nicht:
IdS D
IıQrev
TD 0 : (33.114)
33.8 Der zweite Hauptsatz: 2. Teil
Wir kommen nun zu einer weiteren zentralen und entscheidendwichtigen Funktion der Entropie. Zwar lässt sich die Entropiewie die Energie nicht vernichten. Im Gegensatz zur Energiekönnen wir aber Entropie durch irreversible Prozesse erzeugen.Die Zunahme der Entropie für abgeschlossene Systeme ist derGegenstand des zweiten Teiles des zweiten Hauptsatzes, den wirnun diskutieren. Die bei irreversiblen Prozessen erzeugte Entro-pie ist ein Maß für die Dissipation der Energie, d. h. für derenUmwandlung in innere, thermische Energie.
Irreversible Prozesse
Wir nehmen jetzt an, dass die Kraftmaschine M in Abb. 33.18nicht umkehrbar, also irreversibel arbeite. Dann können wirwieder wie vorher beweisen, dass � > �0 unmöglich ist, weildann wieder Wärme vom kälteren ins wärmere Reservoir trans-portiert werden könnte. Da der Prozess nun jedoch nicht mehrumkehrbar ist, sind die Wirkungsgrade aber auch nicht mehrgleich. Stattdessen gilt
� < �0 : (33.115)
Der reversible Carnot’sche Kreisprozess hat einen größerenWirkungsgrad als irgendein zwischen gleichen Temperaturenbei gleicher Leistung arbeitender irreversibler Carnot-Prozess.Letzterer verlangt mehr Wärme bei gleicher Arbeitsleistung,�Q1 > �Q0
1. Wir folgern aus
T2
T1D �Q0
2
�Q0
1
D 1 � �0 < 1 � � D �Q2
�Q1(33.116)
die Ungleichung �Q1=T1 < �Q2=T2, wobei �Q1 die vom hei-ßen Reservoir der Temperatur T1 aufgenommene Wärme und�Q2 die an das kalte Reservoir der Temperatur T2 abgeführteWärme ist, wie sie in Abb. 33.18 skizziert sind. Für ein sehrschmales Diagramm eines Carnot-Prozesses erhalten wir des-halb
ıQ1
T1<
ıQ2
T2: (33.117)
Auf dieselbe Weise wie bei der Diskussion der reversibelarbeitenden Kraftmaschine ergibt sich für einen teilweise irre-versiblen Kreisprozess, wenn wir die abgegebene Wärmen ıQ2
negativ rechnen: IıQ
T< 0 : (33.118)
Wir zerlegen den Kreisprozess in zwei Abschnitte Z0 ! Z undZ ! Z0 und nehmen an, dass der zweite Abschnitt reversibelgeführt wird. Dann ergibt sich
0 >
ZZ
Z0
ıQ
TC
Z0Z
Z
ıQrev
TD
ZZ
Z0
ıQ
TC S.Z0/ � S.Z/ (33.119)
oder
S.Z/ � S.Z0/ >
ZZ
Z0
ıQ
T: (33.120)
Diese Ungleichung gilt nun auch für ein beliebig zusammen-gesetztes System. Bei irreversiblen Prozessen Z1 ! Z2 inadiabatisch abgeschlossenen Systemen verschwindet das Inte-gral, und entsprechend wächst die Entropie an:
S.Z/ > S.Z0/ : (33.121)

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.8 Der zweite Hauptsatz: 2. Teil 1135
Teil
IV
Entropie bei irreversiblen Prozessen
Läuft in einem abgeschlossenen System ein Prozess adia-batisch ab, d. h. ohne Wärmeaustausch mit der Umge-bung, so erhöht sich im irreversiblen Fall die Entropie undbleibt im reversiblen Fall erhalten.
Immer dann, wenn Energie dissipiert wird, wird zugleich Entro-pie erzeugt. Die Begründung dafür haben wir in Abschn. 33.7gefunden: Je mehr Entropie bei einem Kreisprozess transpor-tiert wird, umso geringer wird der Betrag der Arbeit, die durchdie entsprechende Maschine verrichtet werden kann. Da alleProzesse, die in der Natur von selbst ablaufen, mit Energiedissi-pation einhergehen, folgt daraus, dass die Entropie die Richtungangibt, in der Prozesse ablaufen, wenn wir ein System sichselbst überlassen.
Angelehnt daran hat der Mathematiker und theoretische Physi-ker Arnold Sommerfeld (1868–1951) die Natur mit einer Firmaverglichen: Dem ersten Hauptsatz kommt dabei die Bedeutungder Buchhaltungsabteilung zu, während der zweite Hauptsatzden Vorstand bildet.
Entropiezunahme bei Wärmeleitung
Betrachten wir nun ein abgeschlossenes System, das aus zweizunächst getrennten Behältern mit den Volumina V1 und V2
besteht. Beide Behälter mögen homogene Stoffe im thermi-schen Gleichgewicht bei den Temperaturen T1 und T2 > T1
enthalten. Wir bringen nun die Behälter in thermischen Kon-takt miteinander. Aufgrund der Wärmeleitung vom heißen zumkalten Behälter gehen die beiden Stoffmengen ohne Änderungihres Volumens in einen neuen Gleichgewichtszustand über.
Zur Berechnung der Entropieänderung wird der tatsächliche,irreversibel verlaufende Vorgang in dem abgeschlossenen Sys-tem, bei dem ja weder Wärme noch Arbeit mit der Umgebungausgetauscht werden, durch einen reversiblen Prozess im ge-schlossenen System ersetzt.
Dies ist ein Beispiel dafür, wie die Entropieänderung als Än-derung einer Zustandsgröße anhand eines reversiblen Ersatz-prozesses berechnet werden kann. Nur der Anfangs- und derEndzustand eines Systems sind für die Entropieänderung maß-geblich, aber nicht, wie das System vom Anfangs- in denEndzustand gelangt.
Wir bezeichnen den Wert der Entropie des Gesamtsystems zuBeginn des Wärmeübergangs mit Si und den Wert am Ende desProzesses mit Sf. Nach dem Clausius’schen Postulat, das dieAlltagserfahrung zum Prinzip erhebt, wird die Wärme vom wär-meren Reservoir auf das kältere übergehen, also von T2 auf T1.
Frage 10
Wie wäre ein reversibler Übergang der Wärme realisierbar?Überlegen Sie zunächst selbst und lesen Sie dann den folgen-den Absatz.
Reversibel ist dies nur möglich, wenn der heiße Behälter durchthermischen Kontakt mit einer Folge von Wärmereservoirenmit abnehmender Temperatur und der kalte Behälter umgekehrtdurch eine Folge von Wärmereservoiren mit steigender Tempe-ratur thermisch aneinander angeglichen werden, bis sie beidedie Mischungstemperatur erreichen.
Betrachten wir den ersten Schritt dieser Folge. Der heiße Be-hälter mit der Ausgangstemperatur T2 wird mit einem Wärme-reservoir der Temperatur T2 � dT , der kalte Behälter mit derAusgangstemperatur T1 dagegen mit einem Wärmereservoir derTemperatur T1 C dT in Kontakt gebracht. Der Temperaturunter-schied dT muss so klein gewählt werden, dass der Vorgang alsreversibel betrachtet werden kann.
Dabei soll der heißere Stoff die Wärme ıQrev abgeben und derkältere Stoff die Wärme ıQrev aufnehmen. Dies entspricht ei-nem reversiblen Übergang der Wärme ıQrev vom heißeren zumkälteren Stoff. Damit haben wir den Beginn des irreversiblenProzesses eines direkten Wärmeausgleichs durch den erstenSchritt eines reversiblen Prozesses ersetzt. Die Entropieände-rung bei diesem Schritt ist jetzt berechenbar:
dS D dS1 C dS2 D ıQrev
T1� ıQrev
T2
D ıQrev
T1T2.T2 � T1/ > 0 :
(33.122)
Enthalten beide Behälter ideale Gase, dann können wir Sf � Si
explizit berechnen. Da sich die innere Energie des Gesamt-systems bei der Wärmeleitung nicht ändert, stellt sich eineEndtemperatur ein, die sich aus den anteiligen Stoffmengen n1
und n2 in den beiden Behältern anhand von
Tf D n1
nT1 C n2
nT2 ; n D n1 C n2 (33.123)
berechnen lässt. Der Einfachheit halber nehmen wir in (33.123)an, dass die Wärmekapazitäten der beteiligten Gase gleichseien, und beziehen uns auf die Gleichung für die Mischung-stemperatur, die im Beispielkasten „Mischungstemperatur“ inAbschn. 33.4 hergeleitet wurde. Wegen (33.97) gilt dann
Sf � Si DfZ
i
dS1 CfZ
i
dS2
D n1CV lnTf
T1C n2CV ln
Tf
T2:
(33.124)

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1136 33 Phänomenologische Begründung der Thermodynamik
Der natürliche Logarithmus ist eine konkave Funktion, wasabschließend in Abschn. 33.8 noch vertieft wird. Damit ist Fol-gendes gemeint: Konkav heißen Funktionen f W R � I ! R,deren Graphen in jedem Intervall Œx1; x2� R nicht unter dergeraden Verbindung von .x1; f .x1// und .x2; f .x2// liegen,
f .tx1 C .1 � t/x2/ tf .x1/ C .1 � t/f .x2/ ; (33.125)
wobei der Parameter t zwischen x1 und x2 interpoliert, 0 �t � 1. Wenn eine Funktion f auf dem Intervall I � R zwei-mal differenzierbar ist, so ist sie dort genau dann konkav, wennf 00 � 0 ist. Da die zweite Ableitung des natürlichen Logarith-mus ln g.x/ D �x�2 für alle x 2 RC negativ ist, ist er sogarstrikt konkav. Dann gilt statt des -Zeichens ein >-Zeichen in(33.125), sodass für alle x; y > 0 und alle t 2 .0; 1/
ln .tX C .1 � t/Y/ t ln X C .1 � t/ ln Y (33.126)
gilt. Das Gleichheitszeichen in (33.125) gilt dann nur für t D 0oder t D 1.
Teilen wir die linke Gleichung in (33.123) zunächst durch T1,setzen X D 1, Y D T2=T1 sowie t D n1=n und entsprechend1�t D n2=n, wiederholen diese Schritte nach Vertauschung von1 und 2 und wenden jeweils (33.126) an, ergeben sich sofort dieUngleichungen
lnTf
T1 n2
nln
T2
T1und ln
Tf
T2 n1
nln
T1
T2; (33.127)
wobei das Gleichheitszeichen links bei n1 D 0 und rechts bein2 D 0 gilt. Eingesetzt in (33.124) folgt dann wiederum Sf Si.Die beiden Entropien sind nur dann gleich groß, falls T1 D T2
ist.
Mischung von Gasen
Schließlich kommen wir noch zur Mischung von Gasen. Ge-geben seien Stoffmengen n1 eines Gases im Volumen V1 undn2 eines anderen Gases im Volumen V2, beide mit demselbenDruck P und derselben Temperatur T . Im Ausgangszustand sol-len die Gase durch eine Wand getrennt sein. Dann entfernenwir die Trennwand, sodass jedem der beiden Gase das VolumenV1 C V2 zur Verfügung steht. Der reversible Ersatzprozess läuftnach Max Planck (1858–1947) wie folgt ab:
Die beiden Gase mögen der Einfachheit halber jeweils das glei-che Volumen V einnehmen. Die in Abb. 33.26 gezeichnetenZylinder lassen sich ineinander verschieben. Die gestrichelt ge-zeichneten Böden jedes der beiden Zylinder seien durchlässigfür die Gasart im jeweils anderen Zylinder, sodass Mischungnur im hellgrün unterlegten Volumen eintritt. Durch Auseinan-derziehen der Zylinder kann wieder vollständige Entmischungerreicht werden, und dies zeigt, dass der Ersatzprozess für dieMischung reversibel ist. Am Ende des Vorgangs nimmt das
Gas 1 Gas 2
Abb. 33.26 Zur Mischung von Gasen
Stoffgemisch gerade das Volumen V ein, das jedes Gas ein-zeln zu Beginn ausfüllte. Da bei dem Mischvorgang selbst keineWärme mit der Umgebung ausgetauscht wird, ändert sich dieEntropie dabei nicht.
Anders als beim Planck’schen Ersatzprozess ändert sich beimtatsächlichen Mischvorgang das Volumen des Gesamtsystems,und damit ist eine Entropieänderung verbunden. Für ideale Gasemuss man die Entropiezunahme bei einer Mischung daher be-rechnen, indem man diese in zwei Schritte zerlegt. Zuerst wirddas Gas 1 vom Volumen V1 reversibel und isotherm auf das Vo-lumen V1CV2 gebracht und analog das Gas 2 von V2 auf V1CV2.Die gesamte dabei auftretende Entropieänderung ist positiv:
Sf � Si D �S1 C �S2
D n1R lnV1 C V2
V1C n2R ln
V1 C V2
V2> 0 :
(33.128)
Danach nimmt man eine reversible Mischung auf diePlanck’sche Art vor, wobei das Endvolumen wie im irreversi-blen Fall V1 C V2 ist. Hier tritt keine Entropieänderung auf.Setzen wir wie oben angekündigt voraus, dass die Tempera-tur und der Druck beider Gase rechts und links der Trennwandgleich sind, so ergibt sich die Vereinfachung
Sf � Si D R
�n1 ln
n1 C n2
n1C n2 ln
n1 C n2
n2
�: (33.129)
Mischt man nun zwei gleiche Gase, dann erhält man das soge-nannte Gibbs’sche Paradoxon, benannt nach seinem EntdeckerJosiah Willard Gibbs (1839–1903): Entfernt man die Trenn-wand zwischen zwei gleichen Gasen, so ist makroskopischnichts geschehen, also müsste �S D 0 gelten. Jedoch ergibtsich nach (33.129) eine positive Mischentropie. Das Gibbs’scheParadoxon wird uns in Abschn. 35.3 noch einmal begegnen.
Diese Entropiezunahme ist nach der klassischen Vorstellungauch korrekt. Jedem Atom würde eine Nummer verliehen, undman könnte sich vorstellen, Atome mit gerader und ungeraderNummer zu mischen. Nach den heutigen quantenphysikalischenVorstellungen sind Atome bzw. Moleküle, die aus den glei-chen Elementarteilchen bestehen, allerdings ununterscheidbar,da sie quantenmechanisch durch dieselben Wellenfunktionenbeschrieben werden, weshalb auch keine Entropiezunahme beiMischung gleicher Stoffe beobachtet werden kann. Aus diesemGrund tritt das Paradoxon in der modernen (quantentheoreti-schen) Physik nicht mehr auf. In der Quantenstatistik gibt es

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
33.8 Der zweite Hauptsatz: 2. Teil 1137
Teil
IV
z. B. keinen stetigen Übergang zwischen zwei Gasen, von de-nen das eine aus der Stoffmenge n1 Cn2 Helium und das anderezu den Stoffmengen n1 aus Helium und n2 aus Wasserstoff be-steht.
Abschließend fügen wir noch einige allgemeinere Bemerkun-gen an. Wir haben gesehen, dass bei irreversiblen Prozessen dieGesamtentropie eines Systems immer zunimmt. Das heißt, esgilt für jeden irreversiblen Prozess
S >
TZ
0
ıQ
T: (33.130)
Zum Beispiel kann der Temperaturausgleich zwischen zweiKörpern reversibel (wie im Carnot’schen Kreisprozess) oder ir-reversibel erfolgen. Im letzteren Fall ist der Entropiezuwachsgrößer. Bis zu welchem Wert kann aber nun die Entropieanwachsen, bevor ihre Zunahme zum Stillstand kommt? DieEntropie wächst jeweils bis zu ihrem Maximalwert, der demGleichgewichtszustand des Systems entspricht. Früher oderspäter kommen alle veränderlichen Systeme ins Gleichge-wicht – in den Zustand maximaler Entropie. William Thomson(1824–1907), der 1. Baron Kelvin, schloss daraus, dass „derWelt der Wärmetod droht“ – womit nicht ein Zustand maxima-ler Temperatur gemeint ist.
Das wesentliche Ergebnis der bisherigen Diskussion in diesemAbschnitt können wir nun zusammenfassen.
Der zweite Hauptsatz für irreversible Prozesse
Bei irreversibel verlaufenden Prozessen nimmt die Entro-pie zu. Da die Entropie als Zustandsgröße nicht davonabhängt, wie ein System von einem Gleichgewichtszu-stand in einen anderen gelangt, kann die Entropiezunahmeanhand eines reversiblen Ersatzprozesses berechnet wer-den.
Konkavität der Entropie
Wir haben in (33.97) gesehen, dass die Entropie eines idealenGases vom Logarithmus der Temperatur und des Volumens ab-hängt. Da der Logarithmus eine konkave Funktion ist, wie in
Abschn. 33.8 gezeigt wurde, ist die Entropie eines idealen Gaseszumindest konkav bezüglich der Temperatur und des Volumens.
Aus dem bisher Gesagten folgt aber allgemeiner, dass die Entro-pie immer eine konkave Funktion bezüglich der Zustandsgrößensein muss, die als ihre Argumente auftreten. Davon wollen wiruns noch überzeugen.
Seien zwei Teilsysteme aus derselben Substanz gegeben, diedurch ihre inneren Energien U1;2, Volumina V1;2 und Teil-chenzahlen N1;2 gekennzeichnet sind. Wir fassen .U; V; N/ derEinfachheit halber zu einer abstrakten Zustandsgröße X zusam-men,
X1 D .U1; V1; N1/ ; X2 D .U2; V2; N2/ ; (33.131)
und bezeichnen die Entropien der beiden Teilsysteme mit S1 DS.X1/ und S2 D S.X2/.
Bringen wir beide Teilsysteme in Kontakt, stellt sich ein neuesGleichgewicht ein. Das Gesamtsystem trennen wir adiabatischso in Teilsysteme auf, dass deren Teilchenzahlen wieder durchN1 und N2 gegeben sind. Ihre Entropien seien S0
1 und S0
2, ih-re Gesamtentropie sei S D S0
1 C S0
2. Diese beiden Teilsystemebringen wir mittels eines adiabatischen Prozesses wieder in ih-re Anfangszustände zurück. Dann gilt aufgrund des zweitenHauptsatzes
S.X1/ C S.X2/ � S0
1 C S0
2 D S.X1 C X2/ : (33.132)
Als extensive Zustandsgröße ist die Entropie zudem homogenvom Grad 1, weshalb aus (33.132) direkt
S
�X1 C X2
2
� 1
2ŒS.X1/ C S.X2/� (33.133)
folgt. Sei nun t D j=2n mit j D 0; 1; : : : ; 2n. Für n D 0 undn D 1 belegen die bisherigen Ergebnisse bereits, dass
S .tX1 C .1 � t/X2/ tS.X1/ C .1 � t/S.X2/ (33.134)
gilt. Wenn nun (33.134) für ein beliebiges n und alle j D0; 1; : : : 2n vorausgesetzt werden kann, überzeugt man sichleicht davon, dass (33.134) dann auch für n C 1 undj D 0; 1; : : : 2nC1 gilt. Aufgrund vollständiger Induktion gilt(33.134) folglich für alle n. Da S in den Zustandsgrößen ste-tig sein muss, ergibt sich daraus die Gültigkeit von (33.134) füralle t 2 Œ0; 1�. Also ist die Entropie eine konkave Funktion.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1138 33 Phänomenologische Begründung der Thermodynamik
Vertiefung: Zur Bestimmung der absoluten Entropie
Bei der Temperatur T gilt aufgrund der Definition der Entro-pie
�S DTZ
0
ıQrev
T 0D ST � S0 :
Nun gibt es in der Thermodynamik noch einen dritten Haupt-satz, auch als Nernst’sches Theorem bezeichnet, auf den wirin Abschn. 34.3 zurückkommen werden. Nach diesem Satzverschwindet die Entropie am absoluten Nullpunkt, S0 D 0.Aufgrund dessen können wir den Entropieunterschied �S re-lativ zum absoluten Nullpunkt mit der Entropie S und ebensomit ST identifizieren und
S DTZ
0
ıQrev
T 0
schreiben. Findet die Erwärmung von T D 0 nach T bei kon-stantem Druck statt, dann ist
S D n
TZ
0
CP.T 0/dT 0
T 0
:
Auf dem Weg vom absoluten Nullpunkt bis zur Tempera-tur T geschieht jedoch einiges mit einem Stoff, und dabeiändert sich seine Wärmekapazität. Sehen wir uns dies fürTetrachlorkohlenstoff CCl4 näher an. Er liegt bei einer an-genommenen Zimmertemperatur von 298;1 K als eine Flüs-sigkeit vor, die gerade zu verdampfen beginnt.
Bei tiefen Temperaturen sind die Wärmekapazitäten reinerkristalliner Stoffe proportional zur dritten Potenz der Tem-peratur,
CP D b T3 für T . T0 D 10 K ;
wie in Abschn. 37.7 gezeigt wird. Die Proportionalitäts-konstante hat für CCl4 den Wert 3;14 � 10�3J mol�1 K4.Bei Tf D 225;4 K ändert sich der Aufbau des Kristall-gitters des bei diesen Temperaturen gefrorenen Stoffes; eserfolgt ein Übergang von einer festen Phase in eine ande-re. Dafür wird eine Wärmemenge von �Qf D 4524 J mol�1
verbraucht. Bei Ts D 250;2 K schmilzt der Kristall. DieSchmelzwärme, die für die Umwandlung des Kristalls in ei-ne Flüssigkeit benötigt wird, beträgt �Qs D 2416 J mol�1.Für die Verdampfung bei Td D 298;1K werden �Qd D32:407 J mol�1 aufgebraucht. Daraus ergibt sich für dieEntropie (in J mol�1 K�1)
STd D b
T0Z
0
T2 dT CTfZ
T0
CpdT
TC �Qf
Tf
CTsZ
Tf
CPdT
TC �Qs
TsC
TdZ
Ts
CPdT
TC �Qd
Td:
Bei Td D 298;1 K hat CCl4 einen Dampfdruck von14:819 Pa. Um den Dampf auf den Normaldruck von101325 Pa zu bringen, muss man ihn komprimieren. Dabeiverringert sich die Entropie um
�S D R lnV101325
V14819:
Die Integranden CP=T sind entsprechend den experimen-tellen Werten von CP Funktionen der Temperatur. Die In-tegrationen werden numerisch ausgeführt. Die einzelnenEntropieanteile pro Mol und ihre Summe sind in der folgen-den Tabelle zusammengestellt.
Insgesamt treten drei Phasenübergänge auf, nämlich bei derUmordnung des Kristallgitters, beim Schmelzen und beimVerdampfen. In allen drei Fällen musste Wärme zugeführtwerden. Solche Wärmebeträge, die während Phasenüber-gängen auftreten können (nicht müssen!), heißen latenteWärmen. Beachten Sie, dass die latenten Wärmen bei denPhasenübergängen wesentlich zur Gesamtbilanz beitragen.
Entropiebeitrag J mol�1 K�1
ST0 D bR T0
0 T2 dT 1;05STf � ST0 (numerisch) 151;89�Sf (Phasenübergang) 20;05STs � STf (numerisch) 12;89�Ss (Schmelzen) 9;67STd � STs (numerisch) 22;81�Sd (Verdampfung) 108;57�S (Kompression) �15;4Entropie insgesamt 311;22
Ähnlich verfährt man mit anderen Stoffen. Die Entropien ei-niger Feststoffe, Flüssigkeiten und Gase bei 298 K sind inder nächsten Tabelle zusammengestellt.
Aggregatzustand Stoff EntropieJ mol�1 K�1
Feststoffe Graphit, C 5;7Diamant, C 2;4Iod, I2 116;1
Flüssigkeiten Benzol, C6H6 173;3Wasser, H2O 69;9Quecksilber, Hg 76;0
Gase Methan, CH4 186;1Kohlendioxid, CO2 213;6Wasserstoff, H2 130;6Helium, He 126;0
Wie wir sehen werden, lässt sich die Entropie eines Körpersauch theoretisch berechnen. In der Luft unseres Zimmers, ineinem Stück Kreide – in jedem Körper steckt eine bestimmteEntropie.

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
Aufgaben 1139
Teil
IV
Aufgaben
Die Aufgaben gliedern sich in drei Kategorien: Anhand der Verständnisfragen können Sie prüfen, ob Sie die Begriffe und zentralenAussagen verstanden haben, mit den Rechenaufgaben üben Sie Ihre technischen Fertigkeiten und die Beweisaufgaben geben IhnenGelegenheit, zu lernen, wie man Beweise findet und führt.
� leichte Aufgaben mit wenigen Rechenschritten�� mittelschwere Aufgaben, die etwas Denkarbeit und unter Umständen die Kombination verschiedener Konzepte erfordern��� anspruchsvolle Aufgaben, die fortgeschrittene Konzepte (unter Umständen auch aus späteren Kapiteln) oder eigene mathe-
matische Modellbildung benötigen
Lösungshinweise am Ende des Kapitels helfen Ihnen, falls Sie bei einer Aufgabe partout nicht weiterkommen. Dort finden Sieauch die Lösungen – betrügen Sie sich aber nicht selbst und schlagen Sie erst nach, wenn Sie selber zu einer Lösung gekommensind. Ausführliche Lösungswege, Beweise und Abbildungen finden Sie auf der Website zum Buch.
Viel Spaß und Erfolg bei den Aufgaben!
33.1 � Adiabaten bei vorgegebener innerer EnergieDie innere Energie eines Systems sei durch U D aP2V mit ei-ner positiven Konstanten a gegeben. Finden Sie die Adiabatendieses Systems in der P-V-Ebene.
33.2 � Gleichgewicht unter verschiedenen Bedin-gungen Ein Hohlzylinder sei durch einen Kolben in zweiKammern geteilt. Die Wände des Hohlzylinders seien vollkom-men isolierend. Im Ausgangszustand sei der Kolben zunächstunbeweglich arretiert und ebenfalls vollkommen isolierend. DieKammern 1 und 2 haben anfänglich die Volumina V1 und V2 undenthalten N1 bzw. N2 Moleküle desselben idealen Gases (z. B.Helium). Im Ausgangszustand seien die Drücke in den Kam-mern 1 und 2 gleich P1 und P2.
(a) Welcher neue Gleichgewichtszustand stellt sich ein, wennder Kolben wärmedurchlässig (diathermisch) wird?
(b) Welcher neue Gleichgewichtszustand stellt sich an Stelle desZustands aus Teilaufgabe a ein, wenn sich der diathermischeKolben auch noch frei bewegen kann?
(c) Ändert sich der Gleichgewichtszustand aus Teilaufgabe b,wenn der diathermische, bewegliche Kolben für die Gasmo-leküle durchlässig wird?
33.3 �� Mechanische Arbeit durch Rühren Ein Gasbefinde sich in einem Kolben mit dem Volumen V unter einemDruck P. Für adiabatische Zustandsänderungen gilt die Bezie-hung
PV5=3 D const : (33.135)
In den Kolben ist ein Propeller eingebaut. Lässt man den Propel-ler mit der Umlauffrequenz f D 240 Umdrehungen pro Sekunderotieren, so ist aufgrund der Viskosität des Gases ein Drehmo-ment von M D 104 dyn cm erforderlich. Zugleich misst man beikonstantem Volumen und thermischer Isolierung des Gases eine
Druckerhöhung von
dP
dtD 2
3
Mf
V
dyn
cm2s: (33.136)
(Das Drehmoment M, multipliziert mit der Umlauffrequenz f ,ergibt die Leistung, die am Propeller erbracht werden muss. Ge-teilt durch das Volumen entsteht eine Energiedichte, die demGas pro Zeiteinheit zugeführt wird. Dies entspricht einer Druck-erhöhung pro Zeiteinheit.)
Mithilfe eines isochoren Prozesses eines derartigen Typs be-stimme man die innere Energie U D U.P; V/ für einenbeliebigen Gleichgewichtszustand mit dem Volumen V unddem Druck P relativ zu einem willkürlich gewählten Refe-renzzustand .P0; V0/. Wählen Sie U0 als innere Energie desReferenzzustands.
33.4 ��� Zustandsänderung eines Paramagneten EineStoffmenge n einer paramagnetischen Substanz genüge der ther-mischen Zustandsgleichung
M D na
TB0 ; (33.137)
wobei M die Magnetisierung in Reaktion auf das äußere ma-gnetische Feld B0 und a > 0 eine positive Konstante seien. Dieinnere Energie U der Substanz sei eine reine Funktion der Tem-peratur, U D U.T/. Für tiefe Temperaturen gelte
U.T/ D nbT4 ; (33.138)
wobei wieder b > 0 eine positive Konstante sei.
(a) Geben Sie die Form der Isothermen im jB0j�jMj-Diagramman.
(b) Bestimmen Sie die molaren Wärmekapazitäten(1) bei konstantem äußerem Feld B0 und(2) bei konstanter Magnetisierung M.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1140 33 Phänomenologische Begründung der Thermodynamik
Begründen Sie den Unterschied, falls Sie einen finden.(c) Magnetisieren Sie die Substanz bei der Temperatur T0 iso-
therm von M1 nach M2 > M1 und entmagnetisieren Sie danndie Substanz adiabatisch von M2 zurück nach M1. WelcheTemperatur stellt sich am Ende ein? Welche Arbeitsbeträgeund welche Wärmemengen werden auf den beiden Wegenmit der Umgebung ausgetauscht?
33.5 �� Kreisprozesse als Wärmepumpen
(a) Betrachten Sie eine Carnot- und eine Stirling-Maschine alsWärmepumpen und berechnen Sie explizit deren Leistungs-zahlen.
(b) Bestimmen Sie explizit die technische Arbeit, die beideWärmepumpen verrichten.
Lösungshinweis: Begründen Sie zunächst, warum die Leis-tungszahl einer Wärmepumpe durch (33.102) plausibel definiertist. Bestimmen Sie dann alle anfallenden Wärmemengen undArbeitsbeträge. Gehen Sie davon aus, dass das Arbeitsmediumals ideales Gas beschrieben werden kann.
33.6 � Entropie eines geheizten Zimmers Stellen Siesich ein gewöhnliches Zimmer vor, in dem die Raumluft durchein ideales Gas beschrieben werden kann. Wenn das Zimmergeheizt wird, nehmen dann die innere Energie und die Entropieder Raumluft zu, ab, oder bleiben sie gleich?
Lösungshinweis: Überlegen Sie zunächst, welche Zustands-größen der Raumluft im Zimmer konstant bleiben, während esgeheizt wird.
33.7 � Entropieänderung bei einem irreversiblenProzess In einem Behälter mit wärmeundurchlässigen Wändenund dem Gesamtvolumen V1 ist durch einen Schieber ein Teil-volumen V0 < V1 abgetrennt, in dem sich ein Mol eines idealen,einatomigen Gases der Temperatur T0 befindet. Die Zustands-gleichung lautet PV D RT , wobei wie üblich P der Druckund R die Gaskonstante sind. Von der inneren Energie U wer-den wir in Abschn. 35.3 sehen, dass sie mit der Temperaturdurch U D 3RT=2 zusammenhängt. Der Schieber wird kräf-tefrei herausgezogen, und das Gas expandiert auf das VolumenV1. Nachdem sich ein neues Gleichgewicht eingestellt hat, wirddas Gas mit dem Kolben quasistatisch auf das ursprüngliche Vo-lumen V0 komprimiert.
(a) Wie groß ist die Entropieänderung bei diesem Prozess?(b) Berechnen Sie die Endtemperatur Tf des Gases und die
Kompressionsarbeit W , ausgedrückt durch .U0; V0; T0/.
33.8 ��� Entropie von Gasmischungen
(a) In einem Gefäß mit dem Volumen V befinde sich einGemisch aus zwei Isotopen eines idealen Gases mit denTeilchenzahlen N1 und N2 im thermischen Gleichgewichtmit einem Wärmebad der Temperatur T . Berechnen Siedie Entropie S D S.U; V; N; N1; N2/ des Gemischs, wobeiN D N1 C N2 und U die gesamte innere Energie seien.
(b) Ein adiabatisch isolierter Behälter sei durch einen wärme-undurchlässigen, kräftefrei beweglichen Schieber in zweigleich große Kammern geteilt. Nach dem Herausziehen desSchiebers sind die Kammern durch eine starke, wärme-durchlässige Membran getrennt, die nur für das Isotop 1durchlässig ist.Wir bezeichnen mit hochgestellten, eingeklammerten Indi-zes die Kammer, mit tiefgestellten Indizes die Teilchensorte;N.2/
1 ist also beispielsweise die Anzahl der Teilchen der Sor-te 1 in der Kammer 2.Berechnen Sie die Teilchenzahlen N.1/
1 und N.2/1 des Isotops
1 in den beiden Kammern, ferner die Temperatur T und dieDrücke P.1/ und P.2/ in den beiden Kammern im endgülti-gen Gleichgewichtszustand. Der Ausgangszustand sei durchN.1/
1 D 0;5 NA, N.1/2 D 0;75 NA, V.1/ D 5 dm3 D V.2/,
T.1/ D 300 K, N.2/1 D NA, N.2/
2 D 0;5 NA und T.2/ D 250 Kgegeben.
Lösungshinweis: Die Entropie eines idealen Gases ist
S.U; V; N/ D N
N0S.U0; V0; N0/
C kBN ln
"V
V0
�U
U0
�3=2 �N0
N
�5=2#
:
(33.139)
Verwenden Sie an geeigneter Stelle, dass sich die Teilchen-zahlen in den beiden Kammern so einstellen müssen, dass dieEntropie unter den gegebenen Voraussetzungen maximiert wird.Bedenken Sie dabei, dass auch die Gesamtzahl N der Teilchenvon der Anzahl N1 abhängt.
33.9 �� Vollständige und unvollständige Differenzia-le
(a) Stellen Sie fest, ob die Differenziale(1)
xdy � ydx
x2 C y2; .x; y/ 2 R2 n .0; 0/ (33.140)
(2)
.y � x2/dx C .x C y2/dy ; .x; y/ 2 R2 (33.141)
(3).2y2 � 3x/dx � 4xydy ; .x; y/ 2 R2 (33.142)
vollständig oder geschlossen sind. Der Begriff des geschlos-senen Differenzials wird in den Hinweisen zur Aufgabenäher erläutert.
(b) Integrieren Sie die vollständigen Differenziale aus Teilauf-gabe a.
(c) Wie lautet das Differenzial aus Teilaufgabe a1 in Polarkoor-dinaten?
(d) Berechnen Sie die Kurvenintegrale über das Differenzial ausTeilaufgabe a1 längs folgender Wege:(1) entlang eines Kreises um den Mittelpunkt .0; 0/ mit dem
Radius a;

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
Aufgaben 1141
Teil
IV
(2) entlang einer Kurve, welche die Punkte A-B-C-D-A ab-wechselnd durch gerade radiale Linien oder Kreisbögenverbindet, wobei A D .r1; 0/, B D .r2; 0/, C D.r2 cos '; r2 sin '/ und D D .r1 sin '; r1 cos '/ mit r2 >r1 seien.
Lösungshinweis: In einer Bezeichnungsweise, die aus derTheorie der Differenzialformen entlehnt ist, wird ein Diffe-renzial als geschlossen bezeichnet, wenn die Rotation seinerKoeffizientenfunktionen überall auf dem Definitionsbereich desDifferenzials existiert und verschwindet. Sei beispielsweise
!x.x; y/dx C !y.x; y/dy (33.143)
ein Differenzial in zwei Dimensionen. Dann ist die Rotation derKoeffizientenfunktionen durch
@!y.x; y/
@x� @!x.x; y/
@y(33.144)
bestimmt. Um zu sehen, ob ein zweidimensionales Differenzialgeschlossen ist, gilt es also zu prüfen, ob dieser Ausdruck ver-schwindet.
Wir erinnern daran, dass ein Differenzial ! genau dann voll-ständig (oder exakt) ist, wenn es eine Funktion ˛ gibt, derenDifferenzial ! ist, ! D d˛. Jedes vollständige Differenzialmuss auch geschlossen sein. Umgekehrt kann ein Differenzial,das nicht geschlossen ist, auch nicht vollständig sein.
33.10 �� Bestimmung eines integrierenden FaktorsZeigen Sie, dass das Differenzial
! D x1dx2 C dx3 (33.145)
keinen integrierenden Faktor besitzt.
Lösungshinweis: Eine Differenzial ! hat einen integrieren-den Faktor f genau dann, wenn d.f !/ D 0 ist. Dafür mussdie Rotation der Koeffizientenfunktionen des Differenzials f !verschwinden, woraus sich notwendige und hinreichende Be-dingungen dafür ergeben, dass ! einen integrierenden Faktorhat.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1142 33 Phänomenologische Begründung der Thermodynamik
Lösungen zu den Aufgaben
33.4
(a) Die Isothermen im jB0j � jMj-Diagramm sind Ursprungsge-raden mit der Steigung naT�1.
(b) Die Wärmekapazitäten sind(1)
CB0 D 4bT3 C aV
T2B2
0 (33.146)
bei konstantem äußeren Magnetfeld B0 und(2)
CM D 4bT3 (33.147)
bei konstanter Magnetisierung.
33.5 Die Formel
"W D T1
T1 � T2(33.148)
erweist sich im Fall der Carnot-Maschine als richtig, währendsie bei der Stirling-Maschine größer ist. Die gesamte technischeArbeit pro Umlauf ist in diesen Fällen gerade die negative Ar-beit.

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
Ausführliche Lösungen zu den Aufgaben 1143
Teil
IV
Ausführliche Lösungen zu den Aufgaben
33.1 Die Adiabaten sind durch ıQ D 0 gekennzeichnet, auf-grund des ersten Hauptsatzes also durch dU D �ıW . Mit demangegebenen Ausdruck für U erhalten wir daraus zunächst
a�2PVdP C P2dV
� D �PdV : (33.149)
Trennung der Variablen P und V ergibt
2adP
1 C aPD �dV
V; (33.150)
woraus durch Integration
2 ln .1 C aP/ D � ln V C const (33.151)
oder
.1 C aP/2 D V0
V, P D 1
a
rV0
V� 1
!
(33.152)
folgt. Der zweite Ausdruck ist die gewünschte Form der Adia-baten im P-V-Diagramm.
33.2
(a) Wenn der Kolben diathermisch wird, gleichen sich die Tem-peraturen der beiden Kammern einander an, aber nicht dieDrücke. Da die Teilvolumina V1 und V2 ebenso wie die Teil-chenzahlen N1 und N2 unverändert bleiben müssen, mussnach dem Temperaturausgleich
P0
1V1
N1D P0
2V2
N2) P0
1
P0
2
D V2N1
V1N2(33.153)
gelten. In beiden Teilvolumina stellt sich die Temperatur aufden Wert
T 0
1 D P0
1V1
N1kBD P0
2V2
N2kBD T 0
2 (33.154)
ein.(b) Wenn sich nun der Kolben zusätzlich noch frei bewegen
kann, gleichen sich auch die Drücke einander an. Die Teil-volumina müssen sich entsprechend einstellen, woraus
1 D V00
2 N1
V00
1 N2) V00
1
V00
2
D N1
N2(33.155)
folgt.(c) Wenn zusätzlich Teilchenaustausch zugelassen wird, ändert
sich der Gleichgewichtszustand nicht mehr, weil beide Teil-volumina dieselbe Teilchensorte enthalten.
33.3 Wir geben eine Lösung dieser Aufgabe in zwei Schrittenan, die beide adiabatisch verlaufen sollen. Zunächst entfernenwir uns bei ausgeschaltetem Propeller mittels reiner Druck-Vo-lumen-Arbeit vom Ausgangszustand Z0 D .P0; V0/ zu einemZustand Z0 D .P0; V/, der bereits das Volumen des Endzustandshaben soll. Vom Zustand Z0 aus begeben wir uns dann mittelseines isochoren Prozesses weiter zum Endzustand Z D .P; V/,wozu der Propeller in Betrieb genommen wird.
Da der Übergang von .P0; V0/ nach .P0; V/ adiabatisch war, istıQ D 0, daher dU D �ıW D �PdV. Daraus und mithilfeder angebeben adiabatischen Beziehung zwischen Druck undVolumen erhalten wir sofort für den ersten Schritt
U D U0 � P0
VZ
V0
�V0
NV�5=3
d NV
D U0 C 3
2P0V0
�V0
NV�2=3
ˇˇˇ
V
V0
D U0 C 3
2P0V0
"�V0
V
�2=3
� 1
#
:
(33.156)
Während der isochoren Zustandsänderung ist dV D 0, sodassArbeit allein dadurch verrichtet wird, dass der Propeller im Me-dium rührt. Die dabei am Medium verrichtete und daher negativzu zählende Arbeit erhalten wir zunächst als Produkt aus derKraft F und der Weglänge ds,
dW D �Fds D �Frd' D �Md' ; (33.157)
indem wir das Drehmoment M D Fr identifizieren. Die Arbeitergibt sich daher aus
dW D �Md'
dtdt D �Mf dt : (33.158)
Mithilfe dieser Rührarbeit soll der Druck von P0 auf P erhöhtwerden. Da die isochore Druckerhöhung pro Zeiteinheit ge-messen wurde, können wir daraus bestimmen, wie lang derPropeller dazu rühren muss, denn dazu muss
P � P0 DZ
dP
dtdt D 2
3
Mf
V�t (33.159)
gelten. Daraus erhalten wir zunächst das Zeitintervall
�t D 3
2
V
Mf.P � P0/ : (33.160)
Während dieses Zeitintervalls wird vom Medium insgesamt dieArbeit
W D �Mf �t D �3
2V.P � P0/ (33.161)
verrichtet.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1144 33 Phänomenologische Begründung der Thermodynamik
Aufgrund des adiabatischen Zusammenhangs zwischen Druckund Volumen können wir noch den Druck P0 durch das Volumenausdrücken:
P0 D�
V0
V
�5=3
P0 : (33.162)
Insgesamt folgt so aus beiden Schritten der einfache Ausdruck
U D U0 C 3
2P0V0
"�V0
V
�2=3
� 1
#
C 3
2V
"
P ��
V0
V
�5=3
P0
#
D U0 C 3
2.PV � P0V0/ (33.163)
für die innere Energie.
33.4
(a) Die Isothermen sind einfach zu finden: Ist T D const, sindjMj und jB0j direkt zueinander proportional, d. h., die Iso-thermen sind Ursprungsgeraden im jB0j � jMj-Diagrammmit der Steigung naT�1.
(b) Die molaren Wärmekapazitäten bei konstanter Zustandsgrö-ße X sind durch
CX D 1
n
�ıQ
dT
�
XDconst(33.164)
definiert.(1) Von dem äußeren Magnetfeld B0 wird für eine Änderung
dM der Magnetisierung die Arbeit
ıW D �VB0dM (33.165)
verrichtet, wobei sich die Änderung der MagnetisierungdM auf
dM D � na
T2B0dT (33.166)
reduziert, denn dB0 D 0 (siehe hierzu die Diskussionin 15). Da ıW als Arbeit am System eingeführt wurde,müssen wir �ıW als Arbeit des Systems in den ers-ten Hauptsatz einsetzen. Aus dem ersten Hauptsatz folgtdann
ıQ D dU C ıW D 4nbT3dT C naV
T2B2
0dT ; (33.167)
woraus sofort die molare Wärmekapazität bei konstan-tem äußeren Magnetfeld
CB0 D 4bT3 C aV
T2B2
0 (33.168)
abgelesen werden kann.(2) Bei konstanter Magnetisierung wird keine Arbeit ver-
richtet. Dann ist aufgrund des ersten Hauptsatzes
ıQ D dU D 4nbT3dT (33.169)
und daherCM D 4bT3 : (33.170)
Der Unterschied rührt daher, dass im Fall eines konstan-ten äußeren Magnetfeldes die Magnetisierung durch eineTemperaturänderung verändert wird, sodass dem Sys-tem für eine gegebene Temperaturänderung eine größereWärmemenge zugeführt werden muss.
(c) Bei einer isothermen Magnetisierung ändert sich die inne-re Energie nicht, da sie allein von der Temperatur abhängt,dT D 0 D dU. Aus dem ersten Hauptsatz folgt dann
ıQ D ıW D �VB0dM D �VT0
naM � dM : (33.171)
Integration ergibt
�Q12 D �W12 D �VT0
2na
�M2
2 � M21
�< 0 : (33.172)
Bei der adiabatischen Entmagnetisierung ist definitionsge-mäß ıQ D 0, daher dU D �ıW oder
4nbT3dT D VT
naM � dM : (33.173)
Daraus folgt für den Temperaturverlauf während der Entma-gnetisierung
T2dT D V
4n2abM � dM (33.174)
und durch Integration für die Endtemperatur
Tend D�
T30 C 3VT
8n2ab
�M2
1 � M22
��1=3
: (33.175)
Dabei fällt keine Wärme an. Vom System wird dabei die Ar-beit
�W21 D ��U21 D �nb�T4
end � T40
�(33.176)
verrichtet.
33.5
(a) Beachten Sie zunächst, dass beide Prozesse linksläufig be-trieben werden müssen, damit sie als Wärmepumpen funk-tionieren. Wir beginnen jeweils bei Schritt 4 und durchlaufendie Kreisprozesse in der Folge 4 ! 3 ! 2 ! 1 ! 4.Der Gewinn, den man aus dem Betrieb der Wärmepumpeziehen möchte, ist die an das warme Reservoir übergebe-ne Wärmemenge Q21 (weitere Wärmebeträge können zwarauftreten, heben sich aber hier heraus). Dafür muss die Ar-beit aufgewendet werden, die insgesamt in allen Schrittender Kreisprozesse anfällt. Zwar kehren sich die Vorzeichender Arbeitsbeträge gegenüber dem Betrieb des Kreisprozes-ses als Wärmekraftmaschine um, aber dies geschieht auchbei der Wärmemenge �Q21 D ��Q12, die jetzt abgegeben

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
Ausführliche Lösungen zu den Aufgaben 1145
Teil
IV
statt aufgenommen wird. Wir können also im Fall des Car-not-Prozesses
"W D �Q12
�WD �W12
�W12 C �W34(33.177)
schreiben, denn die Arbeitsbeträge �W23 und �W41 hebensich heraus. Daraus folgt zunächst
"W D T1 ln V2=V1
T1 ln V2=V1 C T2 ln V4=V3: (33.178)
Berücksichtigt man, dass die Volumina wegen der adiabati-schen Zustandsänderungen 3 ! 4 und 4 ! 1 durch
V3
V2D V4
V1) V3
V4D V2
V1(33.179)
verbunden sein müssen, folgt sofort
"W D T1
T1 � T2: (33.180)
Beim Stirling-Prozess treten die zusätzlichen Wärmebeträge�Q14 und �Q32 auf, von denen �Q14 ebenfalls an das war-me Reservoir übergeben wird. Insgesamt wird demnach dieWärmemenge
�Q D �Q14 C �Q21 D nCV.T1 � T2/ C nRT1 lnV2
V1(33.181)
an das warme Reservoir übertragen. Die gesamte Arbeit, diewährend eines Umlaufs aufgewendet werden muss, bestehtaus den zwei Beiträgen �W43 und �W21 und beträgt insge-samt
�W D �W43 C �W21 D nR.T1 � T2/ lnV2
V1: (33.182)
Die Leistungszahl ist also
"W D T1
T1 � T2C CV
R ln.V2=V1/; (33.183)
größer als die Leistungszahl der als Wärmepumpe eingesetz-ten Carnot-Maschine.
(b) Die technische Arbeit, die bei einer Zustandsänderung 1 !2 verrichtet werden muss, ist durch
�Wtech;12 D2Z
1
VdP D2Z
1
d.PV/ �2Z
1
PdV
D .PV/2 � .PV/1 � �W12
D nR.T2 � T1/ � �W12
(33.184)
gegeben und mit der Arbeit verbunden. Bei einem isother-men Prozess bleibt das Produkt aus Druck und Volumenunverändert, sodass einfach
�Wtech;12 D ��W12 (33.185)
gilt. Bei einem isochoren Prozess ist dagegen �W12 D 0,weshalb
�Wtech;12 D nR.T2 � T1/ (33.186)
ist. Bei einem adiabatischen Prozess ist
�W12 D2Z
1
PdV D ��U12 D �nCV.T2 � T1/ ; (33.187)
die gesamte technische Arbeit demnach
�Wtech;12 D nR.T2 � T1/ C nCV.T2 � T1/
D nCP.T2 � T1/ :(33.188)
Aus diesen Ergebnissen folgt für den Carnot-Prozess, derbekanntlich aus zwei isothermen und zwei adiabatischen Zu-standsänderungen zusammengesetzt ist:
�Wtech D ��W C nCP.T2 � T1/ C nCP.T1 � T2/
D ��W : (33.189)
Für den Stirling-Prozess lautet die Rechnung
�Wtech D ��W12 � �W34 C nR.T2 � T1/ C nR.T1 � T2/
D ��W12 � �W34 (33.190)
und führt damit zu demselben Ergebnis.
33.6 Ein gewöhnliches Zimmer hat ein konstantes Volumen.Die Raumluft steht zudem in Verbindung mit der Außenluft,sodass sich auch ihr Druck nicht ändern wird. Wenn daher Pund V beide konstant sind, muss eine Temperaturerhöhung auf-grund der Zustandsgleichung des idealen Gases bedeuten, dassdie Stoffmenge der Raumluft in demselben Maß abnimmt, wiedie (absolute) Temperatur im Zimmer zunimmt.
Da die innere Energie eines idealen Gases sowohl zur Tempera-tur als auch zur Stoffmenge proportional ist, die Stoffmenge beider Heizung aber in demselben Maß abnehmen muss, in demdie Temperatur zunimmt, muss die innere Energie dabei kon-stant bleiben.
Die Entropie dagegen ist als extensive Größe zwar zur Stoff-menge proportional, aber sie steigt lediglich logarithmisch mitder Temperatur an. Da die Entropie konkav in der Temperaturist, überwiegt die Abnahme aufgrund der verringerten Stoff-menge die Zunahme aufgrund der erhöhten Temperatur: DieEntropie der Raumluft nimmt daher durch die Heizung ab.
33.7
(a) Die Entropieänderung ergibt sich allein aus der Vergröße-rung des Volumens, da das Gas mit der Umgebung keineWärme oder Arbeit austauschen kann. Daher ist
�S D R lnV1
V0: (33.191)
Die Temperatur kann sich dabei nicht ändern, da die innereEnergie unverändert bleibt.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1146 33 Phänomenologische Begründung der Thermodynamik
(b) Für die quasistatische (und zudem adiabatische) Kompressi-on gilt aufgrund des ersten Hauptsatzes
dU D �PdV ) 3R
2dT D �RT
VdV : (33.192)
Variablentrennung und Integration ergibt sofort
�Tf
T0
�3=2
D V1
V0) Tf D T0
�V1
V0
�2=3
: (33.193)
Die Kompressionsarbeit ist gleich der Änderung der innerenEnergie, daher
W D Uf � U0 D 3R
2.Tf � T0/ (33.194)
D 3R
2T0
�V1
V0
�2=3
� U0 D U0
"�V1
V0
�2=3
� 1
#
:
33.8
(a) Nach Vorgabe ist die Entropie des i-ten Gases durch
Si.Ui; V; Ni/ D Ni
N0S.U0; V0; N0/ (33.195)
C kBNi ln
"V
V0
�Ui
U0
�3=2 �N0
Ni
�5=2#
gegeben. Die Gesamtentropie S ist die Summe der beidenEntropien S1 und S2, die aber hier noch nicht durch diegesamte innere Energie U D U1 C U2 ausgedrückt sind,sondern durch U1 und U2 getrennt. Die inneren Energien U1
und U2 folgen daraus, dass die innere Energie eine extensiveGröße ist, sodass im vorliegenden Fall
Ui D Ni
NU (33.196)
gelten muss. Die Gesamtentropie des Gemischs aus beidenIsotopen ist daher
S.U; V; N; N1; N2/
D N
N0S.U0; V0; N0/
C kBN1 ln
"V
V0
�N1
N
U
U0
�3=2�N0
N1
�5=2#
C kBN2 ln
"V
V0
�N2
N
U
U0
�3=2�N0
N2
�5=2#
:
(33.197)
Eine längere, aber nicht schwierige Umformung führtschließlich auf die Form
S.U; V; N; N1; N2/ D N
N0S.U0; V0; N0/ (33.198)
C NkB ln
"V
V0
�U
U0
�3=2#
� kB
2X
iD1
Ni lnNi
N� 5
2kBN ln
N
N0
des Ergebnisses.(b) Wir bezeichnen Größen im Anfangszustand mit einem Quer-
strich; NT.1/ ist also die Anfangstemperatur in Kammer 1.Zunächst stellen wir fest, dass sich die Temperaturen der bei-den Kammern im Gleichgewichtszustand angeglichen habenmüssen, da die Trennwand diathermisch wird und dahereinen Temperaturausgleich zulässt. Weiterhin wird die ge-samte innere Energie erhalten bleiben, da das Gesamtsystemaus beiden Kammern adiabatisch isoliert ist und beim Über-gang ins Gleichgewicht keine Arbeit verrichtet wird. Darauserhalten wir zunächst die Temperatur T im Gleichgewicht,
T D NT.1/ NN.1/ C NT.2/ NN.2/
NN.1/ C NN.2/; (33.199)
da die Wärmekapazitäten der beiden Gasisotope als gleichvorausgesetzt werden können.Als Nächstes bestimmen wir die Teilchenzahlen N.k/
i imGleichgewicht. Da die Membran nur für die Teilchen derSorte 1 durchlässig wird, müssen die Teilchenzahlen derSorte 2 unverändert bleiben:
N.1/2 D NN.1/
2 ; N.2/2 D NN.2/
2 : (33.200)
Die Teilchenzahlen der Sorte 1 in beiden Kammern folgt ausder Forderung, dass die Gesamtentropie gegenüber Verände-rungen der Teilchenzahlen N.1/
1 und N.2/1 maximiert werden
muss. Da die gesamte Anzahl der Teilchen der Sorte 1 un-verändert bleiben muss, gilt
N.1/1 C N.2/
1 D NN.1/1 C NN.2/
1 D NN1 : (33.201)
Es genügt also, die Gesamtentropie nach einer der beidenTeilchenzahlen N.k/
1 abzuleiten, z. B. nach N.1/1 . Aus
@�S.1/ C S.2/
�
@N.1/1
D 0 (33.202)
folgt damit sofort
@S.1/
@N.1/1
D @S.2/
@N.2/1
(33.203)
als Gleichgewichtsbedingung, wobei für jede der beidenEntropien S.k/ der Ausdruck eingesetzt werden muss, den

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
Ausführliche Lösungen zu den Aufgaben 1147
Teil
IV
wir in Teilaufgabe a abgeleitet hatten. Eine etwas langwieri-ge, aber nicht schwierige Rechnung führt zunächst auf
�N.2/
1
5=2
U.1/
N.1/
!3=2
D�
N.1/1
5=2
U.2/
N.2/
!3=2
:
(33.204)Die Verhältnisse U=N auf beiden Seiten der Gleichung füh-ren auf dieselbe Konstante, weil die innere Energie eineextensive Größe ist. Daher fallen die Verhältnisse U=N aufbeiden Seiten der letzten Gleichung heraus, und wir erhaltenals einfache Gleichgewichtsbedingung
N.1/1 D N.2/
1 : (33.205)
Damit können wir schließlich noch die Drücke in beidenKammern berechnen, denn nach der idealen Gasgleichungist
P.k/ D N.k/
V.k/kBT D N.k/
1 C NN.k/2
V.k/kBT : (33.206)
Mit den angegebenen Zahlen kommen wir schließlich aufdie Gleichgewichtstemperatur
T D 1;25 � 300 C 1;5 � 250
2;75K D 272;73 K ; (33.207)
die Teilchenzahlen
N.1/1 D 0;75 NA D N.2/
1 (33.208)
und die Drücke
P.1/ D R1;5 mol � 272;73 K
5;0 � 10�3 m3D 6;8 � 105 N
m2;
P.2/ D R1;25 mol � 272;73 K
5;0 � 10�3 m3D 5;7 � 105 N
m2:
(33.209)
33.9
(a) Dem Hinweis folgend prüfen wir zunächst, ob die beidengegebenen Differenzialformen geschlossen sind:(1)
@
@x
x
x2 C y2D 1
x2 C y2� 2x2
.x2 C y2/2;
@
@y
�y
x2 C y2D 2y2
.x2 C y2/2� 1
x2 C y2:
(33.210)
Da die Differenz dieser beiden Ausdrücke verschwindet,ist das Differenzial geschlossen.
(2)@
@x.x C y2/ D 1 ;
@
@y.y � x2/ D 1 ; (33.211)
sodass auch dieses Differenzial geschlossen ist.
(3)
@
@x.�4xy/ D �4y ;
@
@y.2y2 � 3x/ D 4y ; (33.212)
wodurch sich dieses Differenzial als nicht geschlossenerweist.
Nun prüfen wir, welche der geschlossenen Differenzialevollständig sind; das dritte kommt von vornherein nichtmehr in Betracht. Für das erste und das zweite suchen wirFunktionen ˛.x; y/ in zwei Dimensionen so, dass die gege-benen Differenziale identisch zu
d˛ D @˛
@xdx C @˛
@ydy (33.213)
sind. Für das Differenzial aus Teilaufgabe a1 lässt sich zwarlokal eine Funktion angeben, deren Differenzial Teilaufgabea1 ergibt, nämlich arctan y
x , aber die ist am Ursprung nichtdefiniert. Das Differenzial ist daher zwar geschlossen, abernicht vollständig. Dagegen gilt für das Differenzial aus Teil-aufgabe a2
.y � x2/dx C .x C y2/dy D d�
y3
3C xy � x3
3
�; (33.214)
wodurch es sich als exakt herausstellt.(b) Das Integral des einzigen exakten Differenzials aus Teil-
aufgabe a, des Differenzials aus Teilaufgabe a2, ist leichtausgeführt:Z
.y � x2/dx C .x C y2/dy� D xy � x3
3C y3
3C const ;
(33.215)daR
xdy D Rydx ist.
(c) Mit x D r cos ' und y D r sin ' ist natürlich x2 C y2 D r2
und
xdy D r cos ' .sin 'dr C r cos 'd'/ und
ydx D r sin ' .cos 'dr � r sin 'd'/ :(33.216)
Die Differenz dieser beiden Gleichungen ergibt
xdy � ydx D r2�sin2 ' C cos2 '
�d' D r2d' ; (33.217)
sodass das Differenzial aus Teilaufgabe a1 schlicht gleichd' ist.
(d) (1) Mit dem vorigen Ergebnis ist das Integral über den Kreisbesonders leicht auszuführen:
Z
Kreis
xdy � ydx
x2 C y2D
2 Z
0
d' D 2 : (33.218)
(2) Da das Differenzial aus Teilaufgabe a1 gleich d' ist,tragen die radial verlaufenden Kurvenanteile A-B undC-D nicht zu seinem Integral bei. Die verbleibendenStücke B-C und D-A sind Kreisbögen um den Ursprungmit jeweils demselben Öffnungswinkel, aber entgegen-gesetzter Orientierung. Daher muss in diesem Fall dasKurvenintegral verschwinden.

TeilIV
ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1148 33 Phänomenologische Begründung der Thermodynamik
33.10 Im gegebenen Fall kann f von den drei Variablen.x1; x2; x3/ abhängen. Wir müssen prüfen, ob die Rotation von
f .x1; x2; x3/
0
@0x1
1
1
A (33.219)
verschwindet. Daraus ergeben sich für die Funktion f unmittel-bar die Bedingungen
x1@f
@x1D f ;
@f
@x2� x1
@f
@x3D 0 ;
@f
@x1D 0 ; (33.220)
von denen sich die erste und die dritte offenbar gegenseitig aus-schließen. Daher kann ! keinen integrierenden Faktor haben.

ID: 10.1007/978-3-642-54618-1 – 033 Date: August 4, 2014 Proof number: 1
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
Literatur 1149
Teil
IV
Literatur
Baehr, H.D., Kabelac, S.: Thermodynamik, Grundlagen undtechnische Anwendungen. Springer, TS2 (2006)
Boltzmann, L.: Über die Mechanische Bedeutung des ZweitenHauptsatzes der Wärmetheorie. Sitzungsberichte der kaiser-lichen Akademie der Wissenschaften LIII, 195–220 (1866)
Boltzmann, L: Über die Beziehung zwischen dem zweitenHauptsatze der mechanischen Wärmetheorie und der Wahr-scheinlichkeitsrechnung, respective den Sätzen über dasWärmegleichgewicht. Sitzungsberichte der kaiserlichen Aka-demie der Wissenschaften LXXVI, 373–435 (1877)
Carnot, S.: Betrachtungen über die bewegende Kraft des Feuersund die zur Entwickelung dieser Kraft geeigneten Maschi-nen. Ed. Wilhelm Ostwald. Harri Deutsch, TS2 (1995)
Cerbe, G., Wilhelms, G.: Technische Thermodynamik. Theo-retische Grundlagen und praktische Anwendungen. HanserFachbuchverlag, TS2 (2005)
Clausius, R.: Über die Art der Bewegung, welche wir Wärmenennen. Ann. Phys. 176, 353–380 (1857)
Fourier, J.B.J.: Théorie analytique de la chaleur. Koebner, TS2
(1883) TS3
Fourier, J.B.J.: The analytical theory of heat. Ed. A. Freeman.Dover, TS2 (1955)
Herwig, D., Kautz, C.H.: Technische Thermodynamik. PearsonStudium, TS2 (2007)
Maxwell, J. C.: On the dynamical theory of gases. – Part I. Onthe motions and collisions of perfectly elastic spheres. Philos.Mag. 19, 19–32 (1860)
TS2 Bitte geben Sie den Verlagsort an.
TS3 Verweis im Text nicht aufgefunden. Bitte angeben.



![Husserl Studies Volume 13 Issue 1 1996 [Doi 10.1007%2Fbf00117141] J. N. Mohanty -- Kant and Husserl](https://img.pdfslide.org/doc/110x75/55cf98d9550346d0339a0a08/husserl-studies-volume-13-issue-1-1996-doi-1010072fbf00117141-j-n-mohanty.jpg)

























