Embed Size (px)
Citation preview

Research Collection
Doctoral Thesis
Oxydativer Abbau reduzierender Zucker und Diels'scherAnhydro-hexosazone
Author(s): Kreis, Konrad
Publication Date: 1953
Permanent Link: https://doi.org/10.3929/ethz-a-000090330
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 2256
OxydativerAbbau reduzierender Zucker und
Z)/e/s scher Anhydro-hexosazone
VON DER
EIDGENÖSSISCHEN TECHNISCHEN HOCHSCHULE IN ZÜRICH
ZUR ERLANGUNG DER WÜRDE EINES
DOKTORS DER TECHNISCHEN WISSENSCHAFTEN
GENEHMIGTE -
PROMOTIONSARBEIT
VORGELEGT VON
Konrad Kreis
dipl. Ing.-Chem. ETH
von Ermatingen (TG)
Referent : Herr Prof. Dr. L. Ruzicka
Korreferent: Herr P.-D. Dr. E. Hardegger
v J
Zürich 1953
Dissertationsdruckerei Leemann AG

Leer - Vide - Empty

MEINEN LIEBEN ELTERN
IN DANKBARKEIT GEWIDMET

Leer - Vide - Empty

Meinen verehrten Lehrern
Herrn Prof. Dr. L. Ruzicka
und
Herrn P.-D. Dr. E. Hardegger
möchte ich für das fördernde Interesse, das sie dieser
Arbeit stets entgegenbrachten, und die vielen wert¬
vollen Ratschläge und Anregungen, die sie mir erteilten,
aufrichtig danken.

Leer - Vide - Empty

Inhaltsverzeichnis
Theoretischer Teil 9
Einleitung 9
Oxydativer Abbau reduzierender Zucker in alkalischer Lösung mit
molekularem Sauerstoff 13
Reaktionsmechanismus der Oxydation reduzierender Zucker in alka¬
lischer Lösung mit molekularem Sauerstoff 14
Abbau von Hexosen, Hexose-Derivaten und Disacchariden 19
Abbau von Pentosen und Vitamin C 21
Abbau von Hexuronsäuren und von 3,6-Anhydro-D-glucose 23
Die Konstitution des Diels'aohen „Anhydro-D-glucose-phenylosazons" 25
Versuche zur Herstellung von Anhydro-hexosazonen einseitig zweifach
substituierter Hydrazine 27
Abbau von Diels'aohen Anhydro-hexose-phenylosazonen 32
Anhydro-benzimidazole 35
Experimenteller Teil 37
Zusammenfassung 63
7

Leer - Vide - Empty

Theoretischer Teil
Einleitung
Die Konstitutionsermittlung organischer Verbindungen stützt
sich auf die Kenntnis zuverlässiger Abbaumethoden.
Auf dem Gebiete der Zucker bestehen zum Zwecke der Konsti-
tutions- und Konfigurationsbestimmung heute drei prinzipiell ver¬
schiedene Arten (a, b, c) des Abbaus, die sich gegenseitig aufs beste
ergänzen.Den älteren, klassischen Abbaumethoden (a) der Zuckerchemie
(Wohl, Weermann, Ruff) ist gemeinsam, dass sie von der Aldehyd¬bzw. Lactol-Gruppe der Aldosen, bzw. von der Carboxyl-Gruppeder Aldonsäuren aus, die schrittweise Verkürzung der Kohlenstoff-
ket'te des Zuckermoleküls, jeweils um ein C-Atom, erlauben. Die
analytische wie präparative Durchführung dieser Methoden ist
umständlich, die Isolierung der Abbauprodukte, deren Ausbeuten
meist zwischen 15 und 30 % der Theorie hegen, gestaltet sich schwie¬
rig.Der Abbau (b) nach Haworth ist ausschliesslich analytisch von
Bedeutung und in der praktischen Durchführung ebenfalls lang¬
wierig. Er erlaubt — neben beschränkten Aussagen über die Konfi¬
guration von Monosacchariden — die Bestimmung der Grösse der
Lactol-Ringe und in höheren Sacchariden auch der Verknüpfungs¬stellen der Monosaccharid-Bausteine. Die Kohlenhydrat-Molekülewerden beim Abbau nach Haworth an den äther- und acetalartigen
Verknüpfungsstellen gespalten und die C-Ketten dort aufgesprengt.Der oxydative Abbau (c) der Kohlenhydrate mit Perjodsäure
und mit Bleitetraacetat ist neueren Datums. Er vermittelt sowohl
die Grösse der Lactol-Ringe und die Lage der Monosaccharid-
9

Verknüpfungsstellen wie auch die nach Haworth (b) nicht erfass¬
baren Konfigurationen der äther- und acetalartig gebundenen C-
Atome der Kohlenhydrat-Molekeln. Bei der Oxydation mit Perjod-säure und mit Bleitetraacetat bleiben gerade jene Atomverbände
im Kohlenhydrat-Gerüst erhalten, welche nach Haworth (b) abgebautwerden. Die Ausbeute an Spaltprodukten ist nahezu quantitativ.Die Methoden sind ebensogut analytisch wie präparativ geeignetund einfach in der Durchführung.
In neuerer Zeit sind weitere Methoden (a) zum stufenweisen
Abbau reduzierender Zucker veröffentlicht worden.
Nach F. Weygand und A. Bergmann1) werden die abzubauenden
Aldosen in N-Glykoside umgewandelt. Aus den N-Glykosiden wer¬
den mittels der Amadori-Umlagerung Aryl-isoglykosamine gewon¬
nen und letztere durch eine katalytische Oxydation mit Platin und
Sauerstoff in ammoniakalischer Lösung zu den um ein C-Atom
niedrigeren Aldonsäuren abgebaut. Auf diese Weise wird zum Bei¬
spiel aus Glucose Arabonsäure in einer Ausbeute von ca. 50 % der
Theorie erhalten.
HO—OH
IHC—OH
HO¬
CH. -4 V-NH-CH
0
HC—OH
HC O
CH„ -N=CH
IHC—OH
CH,OH.
COOH
CH.OH
CH _^~~X—NH—CH,Ic=o
CH„OH
Ein weiterer Abbau führt nach F. Weygand und R. Löwenfeld*)von den Aldose-oximen, mittels Fluor-2,4-dinitro-benzol, in einer
1) B. 80, 261 (1947).
2) B. 83, 559 (1950).
*) Konfiguration unbestimmt.
10

Stufe zu den um ein C-Atom verkürzten Aldosen. Die Ausbeuten
beim Abbau von Hexosen liegen bei ca. 50 % und beim Abbau von
Pentosen bei ca. 10% der Theorie.
HC=NOH HC=N—O—/ >—N02
NO,
HCN
+
CHO
OH
NO,
CH2OH
Der zurzeit bezüglich Ausbeute — Aldohexosen werden durch¬
schnittlich mit einer Ausbeute von 60 % zu den Aldopentosen abge¬baut — wohl beste Abbau wurde im Jahre 1952 von D. L. Mac-
Donald und H. 0. L. Fischer3) veröffentlicht. Nach dieser Methode
werden aus den leicht zugänglichen, acetylierten Aldosemercaptalendurch Oxydation mit Perphthalsäure, unter Abspaltung von Essig¬
säure, die ungesättigten 1,1-Disulfone hergestellt. Letztere werden
mittels Hydrazin gespalten und das entstandene Hydrazon der um
ein C-Atom verkürzten Aldose in den freien Zucker übergeführt.
8—C,
IHC—S
HC—OAc
IAcO—CH
IR
+I
CHO
IHC—OH
IHO—CH
IR
C2H6
S02-C2H5IO—fc>02 • O2H5
- CH
IAcO—CH
IR
CH2-(S02-C2H5)2+
HC=NNH2
HO—CH
IR
4-
CHO
IHO—CH
IR
Nach diesen zum Teil recht komplizierten Methoden zeichnet
sich der von 0. Spengler und A. Pfannenstiel*) entwickelte oxyda-
3) Am. Soc. 74, 2087 (1952).
4) Z. Wirtschaftsgruppe Zuckerind. 85, 546 (1935); DRP. 618164, 620248.
11

tive Abbau reduzierender Zucker in alkalisch-wässrigen Lösungenmit molekularem Sauerstoff durch grosse Einfachheit aus. In Aus¬
beuten von 30—80% der Theorie werden aus Aldosen die nächst
niedrigeren Aldonsäuren erhalten.
In Fortsetzung der Untersuchungen von H. El Khadem5) ver¬
suchte ich den noch wenig bekannten oxydativen Abbau nach
Spengler und Pfannenstiel durch Anwendung auf verschiedene
Zucker und Zucker-Derivate weiter auszubauen. In der vorliegendenArbeit werden, nach einem kurzen Hinweis auf die Literatur und
einem Überblick auf die bisher entwickelten Anschauungen vom
Mechanismus des Spengler 'sehen Abbaus, die eigenen Versuche zum
Abbau verschiedener Monosaccharide, Monosaccharid-Derivate,
Disaccharide und Uronsäuren, sowie die Isolierung und Charakteri¬
sierung der Oxydationsprodukte besprochen.Im Zusammenhang mit eigenen Arbeiten über den oxydativen
Zuckerabbau nach Spengler und Pfannenstiel stehen Versuche über
die Konstitutionsermittlung der Diels'schen Anhydro-hexosazone,die mit salpetriger Säure zu 2,5-Anhydro-pentonsäuren abgebautwerden konnten.
Die zu Vergleichszwecken benötigte 2,5-Anhydro-D-arabon-säure lässt sich erwartungsgemäss aus 3,6-Anhydro-glucose durch
Oxydation mit Sauerstoff in alkalischer Lösung bereiten.
Weitere Versuche zur Umwandlung des Diels'schen „Anhydro-
glucose-phenylosazons" in ein Anhydro-osazon des Methylphenyl-bzw. Benzylphenylhydrazins führte zu einem gemischt substituier¬
ten Anhydro-osazon des Phenyl- und Benzylphenyl-hydrazins, des¬
sen Konstitution nicht vollständig aufgeklärt werden konnte.
Es wurde versucht, die durch Oxydation von ZH'efe-Anhydro-osazonen erhaltenen Anhydro-pentonsäuren in die Benzimidazole
überzuführen, um deren Konstitution imVergleich mit den analogen,von Link und Moore6) erstmals beschriebenen und auf anderem
Wege hergestellten Anhydro-benzimidazolen zu bestimmen.
Die Isolierung der Anhydro-pentonsäuren aus den Oxydations¬produkten der Diels'schen Anhydro-osazone bildet einen weiteren
5) Vgl. Diss. ETH (1950).
6) C. F. Huebner, R. Lohmar, R. J. Dimler, S. Moore und K. P. Link,J. Biol. Chem. 159, 503 (1945).
12

Beweis für die Richtigkeit der von E. Hardegger und Mitarbeitern7)entwickelten Anschauungen über die Konstitution der Diels 'sehen
Anhydro-hexosazone.
Oxydativer Abbau reduzierender Zucker in alkalischer Lösung mit
molekularem Sauerstoff
Da schon in der Dissertation von H. El Khadem5) ein Überblick
über die Literatur gegeben wurde, erübrigt sich hier deren ausführ¬
liche Besprechung.Die Vorgeschichte zum oxydativen Abbau nach Spengler und
Pfannenstieli) verzeichnet eine Anzahl wenig erfolgreicher Oxyda¬tionsversuche: Framm8) beobachtete, dass durch längeres Einleiten
von Luft in wässrig-alkalische Zuckerlösung ein Abbau der Zucker¬
moleküle zu Carbonsäuren mit verschiedener Kettenlänge statt¬
findet. E. Bucher und J. Meisenheimer9) isolierten bei der analog
durchgeführten Oxydation von Fructose in schlechter Ausbeute
Ameisensäure, Glykolsäure und Erythronsäure; in den nicht kristal¬
lisierten Oxydationsprodukten wurden mehrere Pentonsäuren ver¬
mutet. Nach Spoehr10), der neben 65% Ameisensäure nur ganz
wenig Arabonsäure aus der Oxydation von Glucose gewinnen konnte,
isolierte auch Glattfeld11) Arabonsäure als Phenylhydrazid in nur
10-proz. Ausbeute. J. U. Nef12) fand im Verlauf seiner weitschweifi¬
gen Arbeiten über die Oxydation reduzierender Zucker in alkalischer
Lösung eine noch bedeutend grössere Anzahl von sauren Oxy¬
dationsprodukten.Alle diese älteren Abbauversuche sind weder präparativ noch
analytisch von Bedeutung. Erst als im Jahre 1935 0. Spengler und
A. Pfannenstiel*) zur Oxydation von verdünnten Zuckerlösungen
7) Vgl. E. Hardegger und E.Schreier, Helv. 35, 232, 623 (1952); H.EI
Khadem, E. Schreier, O. Stöhr und E. Hardegger, Helv. 35, 993 (1952).
8) F. Framm, Arch. Physiol. 46, 587 (1896).
9) B. 39, 4217 (1906).
10) Am. 50, 135 (1913).
u) J. U. Nef, 0. F. Hedenburg und J. W. E. Glattfeld, Am. Soc. 39, 1638
(1917).
12) A. 403, 204 (1914).
13

unter milden Bedingungen molekularen Sauerstoff verwendeten und
durch Messung der Sauerstoff-Aufnahme eine zu weitgehende Oxy¬dation verhinderten, gelang es, die gegenüber dem zur Oxydationverwendeten Zucker um ein C-Atom verkürzten Aldonsäuren in
Ausbeuten bis zu 75% zu isolieren. Später fand Isbell13), dass die
Geschwindigkeit des Abbaus und die Höhe der Ausbeute an Aldon¬
säuren stark von der Art des verwendeten Zuckers abhängt. Nach
Isbell konnten aus D-Galactose nur 40 % der Theorie D-Lyxonsäure— in Form des Phenylhydrazids — gewonnen werden, während aus
D-Glucose über 85 % D-Arabonsäure — als Kaliumsalz isoliert —
erhalten wurden.
Reaktionsmechanismus der Oxydation reduzierender Zucker
in alkalischer Lösung mit molekularem Sauerstoff
Die Einwirkung von Alkalien in verdünnten wässrigen Lösungenauf reduzierende Zucker wurde von Lobry de Bruyn und Alberda van
Ekensteinu) schon im Jahre 1895 näher untersucht. Sie fanden, dass
z. B. Glucose teilweise in Fructose und die epimere Mannose umge¬
lagert wird. Neben dieser Isomerisierung treten in geringerem MasseNebenreaktionen auf, wie z. B. die Bildung von verschiedenen
Saccharinsäuren15) und Fructosanen16). Weiter beeinflussen zum
Teil auch die Kationen den Gang der Umlagerungen. Bleihydro¬xyd14) und Calciumhydroxyd17) verhindern bei Zimmertemperaturdie Bildung von Fructose neben Mannose aus Glucose, während mit
Natriumhydroxyd17) aus Glucose nur Fructose gebildet wird.
Konzentrierteres Alkali bewirkt neben dieser Isomerisierung auch
Spaltungen der Zuckermoleküle in Teilstücke mit verschiedener
Kohlenstoffzahl. Je nachdem die Spaltung beim Kohlenstoff-Atom
1, 2 oder 3 eintritt, werden Formaldehyd und Aldopentosen, Glykol-aldehyd und Aldotetrosen oder Dioxyaceton und Glycerinaldehyd
13) H. S. Isbell, J. Res. Natl. Bur. Stand. 29, 227 (1942).
14) C. A. Lobry de Bruyn und W. Alberda van Ekenstein, R. 14, 203 (1895) ;
15, 92 (1896); 16, 257, 262, 272, 282 (1897); 18, 147 (1899).
15) Vgl. H. Kiliani, B. 15, 701, 2953 (1882); B. 18, 631, 2517 (1885).
16) Vgl. L. Sattler und F. W. Zerban, Ind. Eng. Chem. 37, 1133 (1945).
17) A. Kusin, B. 69, 1041 (1936).
14

gebildet18). Durch Isomerisierung und teilweise Rekombination der
Spaltstücke wird die Vielzahl der entstehenden Produkte noch weiter
erhöht. Neben Milchsäure, in einer Ausbeute bis zu 60 %19), konnten
Ameisensäure, Essigsäure, Dioxybuttersäure, Glycerinaldehyd,
Dioxyaceton, Methylglyoxal etc. isoliert werden. Weiter entstehen
durch Polymerisation in kleinen Mengen Harze und „Polysaccha¬ride" unbekannter Konstitution12).
CHO
IHC—OH ^
-*•
IR
R
Zur Erklärung der nach ihnen benannten Umlagerungen nahmen
Lobry de Bruyn und Alberda van Ekensteinu) die intermediäre Bil¬
dung eines 1,2-Endiols an, das durch die zeitweilige Aufhebung der
Asymmetrie am C-Atom 2 die Entstehung der epimeren Aldosen
und die Umwandlung von Aldosen in Ketosen erklärt.
Beweise für die Anwesenheit einer Doppelbindung im Zucker¬
molekül erbückte man18) in der Tatsache, dass reduzierende Zucker
in alkalischer Lösung grosse Mengen Jod verbrauchen und weiter
leicht durch Sauerstoff zwischen Ct und C2 gespalten werden.
Die Endiol-Theorie wird durch Untersuchungen von Tropperund De Witt Stetten20), wie auch von Sowden und Schaffer21),
gestützt. Die beiden Forschergruppen fanden, dass durchEinwirkungvon Alkali auf reduzierende Zucker in schwerem Wasser bei 25°
18) Vgl. Zusammenfassungen der Einwirkung von Alkali auf red. Zucker:
W. L. Evans, Chem. Rev. 31, 537 (1942); W. W. PigmanunAR. M.Goepp jr.,
Chemistry of the Carbohydrates, 71 (1948).
19) Vgl. W. L. Evans, Chem. Rev. 6, 281 (1929).
20) Y. J. TropperundJ. R. De Witt Stetten, J. Biol. Chem. 189, 191 (1951).
21) J. G. Sowden und R. Schaffer, Am. Soc. 74, 499, 505 (1952).
15
HC—OH
IIC—OH
IR
CH.OH
Ic=o
CHO
IHO—CH
IR

eine Einlagerung von ca. 1 Mol Deuterium pro Mol Zucker statt¬
findet. Ferner fanden Sowden und Schaffer eine geringe Einlagerungvon Deuterium an C3 der Zuckermolekel, was leicht durch eine
Wanderung der Endiol-Gruppe erklärt werden kann.
Neben Lobry de Bruyn und Alberda van Ekenstein wurde die
Wanderung der Endiol-Doppelbindung vor allem von Nef12) zur
Erklärung der Vielzahl der gebildeten Spaltprodukte bei der Ein¬
wirkung von Alkali auf reduzierende Zucker herangezogen.Da ein 2,3-Endiol von einem 1,2-Endiol energetisch kaum sehr
verschieden sein wird, ist es nicht recht erklärlich, warum die beiden
Endiole nicht stets in ähnlichen Mengen nebeneinander vorkommen
sollen. Das 2,3-Endiol lässt sich jedoch, wie die Einlagerungsversuchemit Deuterium und der beinahe quantitative Spengler'sehe Abbau
von Glucose zeigen, nur in kleinen Mengen nachweisen. Weiter
lassen sich mit der Endiol-Theorie weder die irreversiblen Neben¬
reaktionen der Lobry de Bruyn'sehen Umlagerung noch der Einfluss
der Kationen22) richtig erklären. Gerade die Einwirkung von Katio¬
nen auf den Gang der Lobry de Bruyn''sehen Umlagerung lässt ver¬
muten, dass letztere in Wirklichkeit viel komplizierter — möglicher¬weise über intermediäre Metall-Komplexe — verläuft.
Fredenhagen und Bonhoeffer23) wie auch Goto 2i) fanden im Gegen¬satz zu den Arbeitsgruppen um Tropper und Sowden, dass bei alkali¬
katalysierten Umlagerungen reduzierender Zucker in schwerem
Wasser eine Einlagerung von Deuterium in die Zuckermolekel erst
bei 45° eintritt. Auf Grund dieser Ergebnisse können die schon bei
tieferer Temperatur vor sich gehenden alkalischen Umlagerungenreduzierender Zucker nicht mehr nach der alten Endiol-Theorie
erklärt werden. Fredenhagen schlug deshalb folgenden Mechanismus
vor:
HC= 0 HO—CH
I + IHC—OH 0=CH
O
H/ \|HO—C CH
I IHC ß—OH
|\0/H_
CH.OH
2 Ic=o
22) Vgl. Seite 14.
23) H. Fredenhagen und K. F. Bonhoeffer, Z. physikal. Chem. A181, 392
(1938).
24) K. Goto, Chem. Soc. Japan, 63, 217 (1942).
16

Die alkalische Umlagerung von Aldosen in Ketosen kann auch
ohne dieses dimere Zwischenprodukt von Fredenhagen erklärt wer¬
den, wenn man in Analogie zur Benzilsäure-Umlagerung eine intra¬
molekulare Wanderung eines Wasserstoff-Anions annimmt26).
CHO
IHC—OH
IR
C=0C | ^(H)C—Ö :
I (")R
H
HC—Ö : (-)
C=0
IR
C=0
IR
Weder mit dieser noch mit der Formulierung von Fredenhagenlässt sich der grosse Jodverbrauch reduzierender Zucker in alkali¬
schen Lösungen erklären.
Nachdem der Mechanismus der alkalischen Umlagerungen redu¬
zierender Zucker noch kaum aufgeklärt werden konnte, ist es nicht
weiter verwunderlich, dass auch der Mechanismus der Oxydationreduzierender Zucker in alkalischer Lösung mit molekularem Sauer¬
stoff ebenfalls nicht genau bekannt ist.
Spengler und Pfannenstiel*) formulierten wie Nef12) den oxyda-tiven Abbau reduzierender Zucker als Aufspaltung des 1,2-Endiolsin Aldonsäure und Ameisensäure, was nach modernen Anschauun¬
gen sehr unwahrscheinlich ist. Für die Formulierung von Evans26),wonach die oxydative Aufspaltung in a-Stellung zur Endiol-Doppel-
bindung erfolgen soll, fehlt jeder Beweis.
Bei der alkalischen Oxydation von L-Sorbose mit Sauerstoff
wurde sowohl die erwartete L-Xylonsäure wie auch die 2-Keto-L-
gulonsäure isoliert27). Die alkalische Oxydation lässt sich demnach
über folgende Zwischenstufen formulieren:
CHO
IHC—OH
HC—OH
IIC—OH
CHO
ICO
COOH
ICO
HCOOH
+
-> COOH
Ein ähnlicher Mechanismus kann auch bei der alkalischen Oxy¬dation von Vitamin C, resp. Dehydro-L-ascorbinsäure, mittels Sauer¬
stoff zu L-Threonsäure und Oxalsäure angenommen werden.
25) G. R. Noller, Chemistry of Organic Compounds, 354 (1951).
2«) Vgl. W. L. Evans, Chem. Rev. 31, 537 (1942).
2') Vgl. O. Dalmer und K. Heyns, USP. 2190377 (1939).
17

Bei den bisherigen Untersuchungen über den alkalischen Oxy¬dationsverlauf basieren die theoretischen Folgerungen immer auf
den isolierten Endprodukten, da Zwischenstufen nie gefasst werden
konnten. Exakte Messungen der Reaktionsgeschwindigkeiten wur¬
den bis zu den Untersuchungen von Bamford und Collins28) im Jahre
1950 nie durchgeführt. Diese Autoren versuchten am Beispiel der
D-Glucose die Reaktionsgeschwindigkeit der Oxydation bei einer
bestimmten Temperatur in Abhängigkeit von Alkali- und Zucker-
Konzentration sowie vom Sauerstoffdruck zu messen. Sie fanden,
dass in sehr verdünnten Glucose-Lösungen (ca. 0,05 Mol/1) und bei
Sauerstoffdruck über 400 mm Hg — in Bestätigung der Resultate
von Spengler und Pfannenstiel — fast in quantitativer Ausbeute
Arabonsäure und Ameisensäure gebildet wird; dass dabei ferner die
Reaktionsgeschwindigkeit nur von der Konzentration des Zuckers
und dem Sauerstoffdruck abhängt, dessen Einfluss sich mit stei¬
gendem Druck bis ca. 1000 mm Hg langsam einem Maximum nähert.
Durch Zugabe von Peroxyd und oberflächenvergrössernden Stoffen,
wie pulverisiertem Glas oder Bariumsulfat, konnte die Reaktionsge¬
schwindigkeit des oxydativen Abbaus von Glucose zu Arabonsäure
nicht geändert werden. Die Autoren schlössen daraus auf das Vorlie¬
gen einer Ionenreaktion. AufGrund der reaktionskinetischen Berech¬
nungen wurden zwei voneinander verschiedene ionenartige Zwischen¬
stufen als geschwindigkeitsbestimmende Faktoren postuliert. Der
Oxydationsverlauf wird von Bamford u. Collins wie folgt formuliert:
CHOr /°) iHC (-)
/OHHC )
-
1! ' 4~V II —)C—OH C=-0 ]
1 L 1 J
CH2OH
HCOOH
+
COOH <-
CH„OH
CHO
(-)Oa—C—OHI
B
\
c=o
CH,OH
Glucose (=) —A'
Saocharinsäuren
8) C. H. Bamford und J. R. Collins, Proc. R. Soc. A 204, 62, 85 (1950).
18

Im vorstehenden Reaktionsschema bewirkt ein Hydroxylion amaktivierten C-Atom 2 der Aldohexose eine Protonenabspaltung, was
zur Ausbildung eines durch Mesomerie stabilisierten Anions A führt.
Durch die Einwirkung von Sauerstoff wird daraus das Peroxyd-ion B
gebildet, das weiter in Ameisensäure und eine Pentonsäure zerfällt.
Die Bildung des Peroxyd-ions B verhindert den Übergang des Ions A
in ein doppelt negativ geladenes Anion A', das den Ausgangspunktfür die Umlagerungen zu den Saccharinsäuren bildet. Das Reak¬
tionsschema erklärt, warum bei hohem Sauerstoffdruck, resp. gros¬
sem Peroxydgehalt, die Oxydation fast quantitativ zu den Penton¬
säuren führt.
Die Interpretationen von Bamford und Collins sind nur bei klei¬
nen Zucker- und Alkali-Konzentrationen gültig, was ihren Wert
für eine präparative Auswertung beeinträchtigt. Ferner lässt sich
damit das unterschiedliche Verhalten der einzelnen Zucker und der
Einfluss der Kationen ebenso wenig wie nach den alten Theorien
erklären.
Abbau von Hexosen, Hexose-Derivaten und Disacchariden29)
Aus früheren Arbeiten ist bekannt30), dass die besten Ausbeuten
an Aldonsäuren in stark verdünnten wässrigen Lösungen von ca.
0,01 bis 0,05 Mol Zucker pro Liter und ca. 0,5 Mol Alkali pro Liter
erzielt werden können. Für das präparative Arbeiten sind die grossen
Flüssigkeitsmengen unpraktisch. Ich versuchte daher bei der Oxy¬dation von D-Glucose (I), unter besonders intensiver Durchmischungder alkalischen Zuckerlösung mit Sauerstoff mittels eines Vibrators,
die Konzentration von Alkali und Zucker zu steigern. Im Verlauf
einer grösseren Anzahl von Versuchen, die im experimentellen Teil
nicht beschrieben sind, gelang es schliesslich, bei einer Konzentra¬
tion von 0,6 Mol Glucose pro Liter und einem 30-proz. Alkali-
Überschuss, in weniger als der Hälfte der üblichen Oxydationszeitnoch eine Ausbeute von 73 % an D-Arabonsäure (IV) — als Kalium¬
salz isoliert — zu erzielen. Mit ß-Pentaacetyl-D-glucose (II) und
D-Glucosamin-hydrochlorid (III) erfolgte der Abbau zu D-Arabon¬
säure (IV) fast ebenso gut wie bei D-Glucose.
29) Vgl. E. Hardegger, K. Kreis und H. El Khadem, Helv. 35, 618 (1952).
30) Vgl. C. H. BamfordimdB. J. Collins, Proc. R. Soc. A204, 62, 85 (1950).
19

Zur Charakterisierung von D-Arabonsäure (IV) wurden das
.Kaliumsalz V13), das Brucinsalz VI31), das Phenylhydrazid VII32)und das noch unbekannte S-Benzylthiuroniumsalz VIII hergestellt.
Im Gegensatz zum gut gehenden Abbau der D-Glucose konnte
aus D-Galactose (IX) nur in 43-proz. Ausbeute D-Lyxonsäure (X)als Phenylhydrazid XI isoliert werden13).
Der einzige Hinweis, dass auch Disaccharide mit Alkali und Sauer¬
stoff abgebaut werden können, findet sich in der Patentschrift von
Spengler und Pfannenstiel*), doch scheint die Isolierung von kristal¬
lisierten Abbauprodukten aus Maltose (XII) nicht gelungen zu sein.
Ich habe ausser Maltose (XII) auch Cellobiose (XV) und Lactose
(XIX) abgebaut und erwartungsgemäss drei Hexosido-pentonsäu-ren in Form ihrer kristallisierten Brucinsalze isoliert, nämlich die
3-[a-D-Glucosido]-D-arabonsäure (XIII), die 3-[/3-D-Glucosido] -D-
arabonsäure (XVI) und die 3-[/?-D-Galactosido]-D-arabonsäure
(XX). Die Kristallisation des Brucinsalzes XIV aus den Oxydations¬
produkten der Maltose (XII) war schwierig durchzuführen; sie
erfolgte langsam, auch wenn das Präparat bereits in reiner Form
vorlag und angeimpft wurde.
c)HOAcO—(
AcO—
}H
—OAcCHO
|—NH3C1c}HO
__
—OAc
—
HC 61
—
CH2OH CH2OAc CH2OH CH2OH
I /-" II _^—"" III 1 IX
+ ir-"" k +
H(
H
(
DOOH
h
?OOH
K-Salz : V
1
H
(
ICOOH
h
300H
Brucinsalz VI
2H2OH
PhenylhydrazidS-Benzylthiuronium-Salz
VII
VIII
(3H2OH
Phenyl¬
hydrazid
IV -X XI
x) Vgl. J. W. E. Glattfeld, Am. 50, 135 (1910).
2) Vgl. H. Ohle und O. Behrend, B. 60, 1166 (1927).
20

Aus den gereinigten Brucinsalzen der Disaccharidsäuren XVI
und XX Hessen sich die bereits bekannten, kristallisierten Calcium-
salze XVIII und XXII gewinnen33)34).
CH2OH CH2OH CH2OH
/l-0\ l/Uo\! _ J-% t/kH-CH2°HOH OH OH
XII XIII
Brueinsalz: XIV
CHaOH CH2OH CH2OH
/'—°\ /~°\ /'~°\ /oh"CH2°H
H0l\(^|/K°XI\^|/0H~*
HO^PY^'^COOHOH OH OH
XV XVI
Brueinsalz: XVII
Calciumsalz : XVIII
CH2OH CH2OH CH2OH.
HOJ—Ox /\—Ox HO ,1—0\ /UrCH2OH'<- >IX0XI<?i,>U - <?i>°XsiCOOH1H OH OH
XIX XX
Brueinsalz: XXI
Calciumsalz: XXII
Abbau von Pentosen und Vitamin C35)
Durch Oxydation mit Alkali und Sauerstoff wurden D-Ribose
(XXIII) und L-Arabinose (XXVI) zu D-Erythronsäure36)37), bzw.
L-Erythronsäure38) abgebaut. Aus D-Xylose (XXIX) gewann man
33) Vgl. P. A. Levene und M. L. Wolfrom, J. Biol. Chem. 77, 671 (1928).
34) Vgl. P. A. Levene und O. Wintersteiner, J. Biol. Chem. 75, 315 (1927).
35) Vgl. E. Hardegger, K. Kreis und H. El Khadem, Helv. 34, 2343 (1951).
36) Vgl. R. C. Hockett und B. S. Millmann, Am. Soe. 63, 2587 (1941).
37) Vgl. E. L. Jackson und C. S. Hudson, Am. Soe. 60, 989 (1938).
38) Vgl. N. K. Bichtmyer und C. S. Hudson, Am. Soe. 64, 1609 (1942).
21
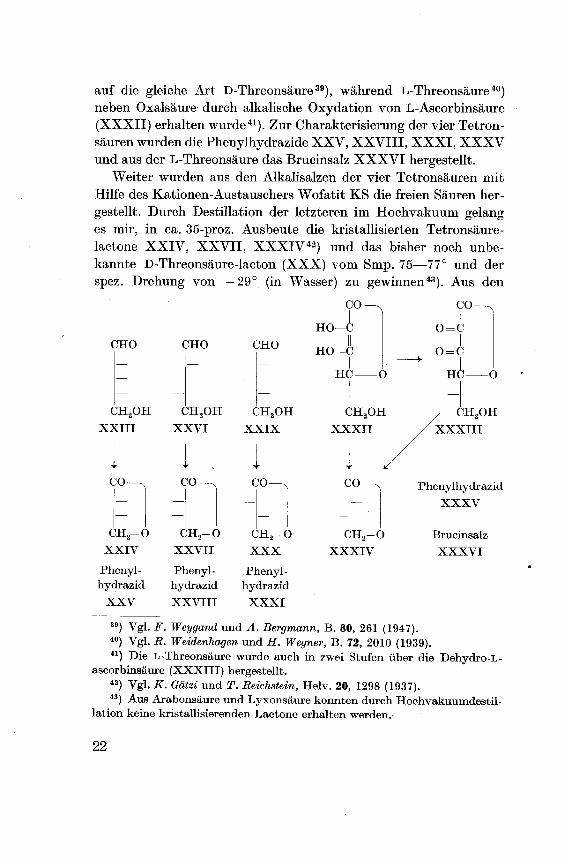
auf die gleiche Art D-Threonsäure39), während L-Threonsäure40)neben Oxalsäure durch alkalische Oxydation von L-Ascorbinsäure
(XXXII) erhalten wurde41). Zur Charakterisierung der vier Tetron¬
säuren wurden die Phenylhydrazide XXV, XXVIII, XXXI, XXXV
und aus der L-Threonsäure das Brucinsalz XXXVI hergestellt.Weiter wurden aus den Alkalisalzen der vier Tetronsäuren mit
Hilfe des Kationen-Austauschers Wofatit KS die freien Säuren her¬
gestellt. Durch Destillation der letzteren im Hochvakuum gelanges mir, in ca. 35-proz. Ausbeute die kristallisierten Tetronsäure-
lactone XXIV, XXVII, XXXIV42) und das bisher noch unbe¬
kannte D-Threonsäure-lacton (XXX) vom Smp. 75—77° und der
spez. Drehung von —29° (in Wasser) zu gewinnen43). Aus den
CO—. CO—.
CHO
XXIII
4-
co—
XXIV
Phenyl -
hydrazid
XXV
CHO
CH2OH
XXVI
CO-
CH2-0
XXVII
Phenyl-
hydrazid
XXVIII
CHO
CH2OH
XXIX
4-
co—-.
CH2-0
XXX
Phenyl -
hydrazid
XXXI
HO—C
IIHO-C
IHC- -O
0 = C
Io=c
IHC O
CH2OH
XXXII
+
co-
XXXIV
CH2OH
XXXIII
Phenylhydrazid
XXXV
Brucinsalz
XXXVI
39) Vgl. F. Weygand und A. Bergmann, B. 80, 261 (1947).40) Vgl. R. Weidenhagen und H. Wegner, B. 72, 2010 (1939).41) Die L-Threonsäure wurde auch in zwei Stufen über die Dehydro-L-
ascorbinsäure (XXXIII) hergestellt.42) Vgl. K. Gätzi und T. Reichstein, Helv. 20, 1298 (1937).43) Aus Arabonsäure und Lyxonsäure konnten durch Hochvakuumdestil¬
lation keine kristallisierenden Lactone erhalten werden.-
22

erheblichen Destillationsrückständen, die aus Estoliden zu be¬
stehen scheinen, lassen sich durch erneute Behandlung mit Lauge,Wofatit KS und Destillation im Hochvakuum weitere Mengen der
Lactone gewinnen.Die Herstellung des D,L-Erythronsäure-lactons vom Smp. 92°
erfolgte durch Umkristallisieren gleicher Mengen D- und L-Lacton
XXIV und XXVII. Das Erythronsäure-lacton ist ein Racemat, da
die Mischprobe mit D-Lacton XXIV eine Schmelzpunktserniedri¬
gung von 6° gab.Ob das noch unbekannte D,L-Threonsäure-lacton vom Smp.
48—50° ebenfalls ein Racemat darstellt, konnte auf Grund von
Mischproben mit D-Lacton XXX nicht sicher entschieden werden.
Die Threonsäure-lactone sind im Gegensatz zu den Erythron-säure-lactonen sehr hygroskopisch; sie zerfhessen an der Luft. In
kristallisierter Form sind sie nur bei strengstem Feuchtigkeits-Aus-schluss, also z. B. im Hochvakuum eingeschmolzen, unbeschränkt
haltbar.
Abbau von Hexuronsäuren und von 3,6-Anhydro-D-glucose44)
Überraschenderweise gelingt es, die gegen Alkali recht empfind¬lichen Hexuronsäuren, nämlich die D-Mannuronsäure (XXXVII),die D-Galacturonsäure (XXXVIII) und D-Glucuron (XXXIX) zur
D-Arabo-trioxyglutarsäure (XL) abzubauen. Die TrioxyglutarsäureXL wurde in einer Ausbeute bis zu 40 % der Theorie als Brucinsalz
XLI, bzw. Dikaliumsalz XLII, als Dibenzimidazol XLIII und
Pikrat XLIX isoliert.
Zu Vergleichszwecken wurden aus den Diamiden45) der Ribo-
trioxy-glutarsäure (XLV) und der Xylo-trioxyglutarsäure (XLVIII)ebenfalls die Dibenzimidazole, XLVI und XLIX, sowie deren
Pikrate XLVII und L hergestellt.Die nach Linie und ilioore46) bereiteten Dibenzimidazole wie
**) Vgl. E. Hardegger, K. Kreis und D. Spitz, Helv. 35, 958 (1952).
45) Hergestellt von D. Spitz; vgl. Diss. ETH (1952).
46) B. Lohmar, B. J. Dimler, S. Moore und K. P. Link, J. Biol. Chem. 143,
551 (1942).
23

auch deren Pikrate sind zur Charakterisierung der verschiedenen
Trioxyglutarsäuren hervorragend geeignet47).Anhand der gut charakterisierten Trioxyglutarsäuren ist es
leicht möglich, im Vergleich mit den entsprechenden, Zucker- bzw.
Schleimsäuren48), die Konfiguration der abgebauten Hexuronsäuren
CHO CHOCHO
COOH
XXXVII
COOH
XXXVIII
O—CH
CO
XXXIX
COOH
COOH
XL
Brucinsalz : XLI
Kaliumsalz: XLII
C17H1603N4: XLIII
D-Arabo-trioxyglutar-säure-dibenzimidazol
CONH,
CONH2
XLV
CONH,
CONH2
XLVIII
4-
XLVI XLIX
Ribo- Xylo-
trioxyglutarsäure-dibenzimidazol
4-
Pikrat
XLVII
Pikrat
L
Pikrat: XLIV
CHO
CH
O—CH2
LI
COOH
I-CH
O—CH2
LH
Brucinsalz: LUI
47) Vgl. Zusammenfassung über Benzimidazolert/. B. Wright, Chem. Rev.
48, 397 (19S1); N. K. Richtmyer, Advances in Carbohydrate Chemistry, Vol.
6, 175 (1951).
48) Die Zucker- bzw. Schleimsäuren werden aus Hexuronsäuren durch
Oxydation mit Salpetersäure hergestellt.
24

zu bestimmen. So sind von den 16 stereoisomeren Aldohexuron-
säuren 14 auf diese Weise identifizierbar. Nur zwischen D- und L-
Alluronsäure kann nicht unterschieden werden.
Als Vergleichspräparat für die als Abbauprodukte der Diels'-
schen Anhydro-hexosazone bedeutungsvollen Anhydro-penton-säuren wurde 3,6-Anhydro-D-glucose (LI) mit Alkali und Sauerstoff
zur 2,5-Anhydro-D-arabonsäure (LH) abgebaut und letztere als
Brucinsalz LUI vom Smp. 126—127° und der spez. Drehung von
— 34,1° (in Wasser) isoliert. Im Gegensatz zu den Penton-, Hexon-,
Zucker-, Schleim- und Trioxyglutarsäuren gelang es nach der von
Link und Moore*®) beschriebenen Methode nicht, aus der 2,5-Anhy¬dro-D-arabonsäure ein kristallisiertes Benzimidazol herzustellen.
DieKonstitution des Di«îs'schen„Anhydro-D-glucose-phenylosazons"
Wie Diels und Mitarbeiter49) fanden, lassen sich die Phenylosa-
zone der Hexosen, Pentosen und Disaccharide leicht durch Behan¬
deln mit Ameisensäure in der Kälte oder durch Kochen in Metha¬
nol mit einer Spur Schwefelsäure unter Abspaltung von 1 Mol
Wasser in gut kristallisierende Monoanhydro-Derivate50) über¬
führen, die sich im Aussehen kaum von den Phenylosazonenunterscheiden.
Da die Konstitution der Phenylosazone51) bis heute nicht ein¬
deutig abgeklärt werden konnte — es sind theoretisch über 100
tautomere Formen möglich —, ist es nicht weiter verwunderlich,
dass bis in die neueste Zeit Unklarheiten über den Bau der Diels-
schen Anhydro-osazone bestanden. Neben den vielen möglichenIsomeren und Tautomeren ist der Umstand, dass die Anhydro-
osazone zum Teil sehr stabile Hydrate52) bilden, für die Auswertung
der experimentellen Ergebnisse sehr erschwerend.
49) O. Diels und R.Meyer, A. 519, 157 (1935); 0. Diels, B.Meyer und
0. Onnen, A. 525, 94 (1938).50 ) Nur Maltose bildet ein Dianhydro-phenylosazon.
51) Vgl. Zusammenfassung von E. O. V. Percival, Advances in Carbo¬
hydrate Chemistry, Vol. 3, 23 (1948).
62) Vgl. E. Hardegger und E. Schreier, Helv. 35, 623 (1952).
25

Diels (1. c.) glaubte, nach Verwerfung einer carbocyclischen
Desoxy-inosose-osazon-Struktur, auf Grund experimenteller Ergeb¬nisse das aus D-Fructose-phenylosazon gewonnene Anhydro-osazonals 3,6-Anhydro-D-fructose-phenylosazon (LIV) formulieren zu kön¬
nen. Später „bewies" Diels aber, nach missglückten Versuchen zur
Wasserabspaltung aus N-alkylierten Hexose-phenylosazonen und
neuer Interpretation früherer Ergebnisse, eine Pyrazolstruktur LVfür das aus D-Fructose-phenylosazon gewonnene Anhydro-osazon.
Etwas später schrieben Percival und Mitarbeiter53), hauptsäch¬lich auf Grund negativer Versuchsergebnisse, im Gegensatz zu Diels,
dem „Mono-anhydro-fructose-phenylosazon" die bicyclische Form
LVI zu.
In unserem Laboratorium wurden von E. Hardegger, H. El Kha-
dem, E. Schreier und G. Stöhr Untersuchungen über die Diels 'sehen
Anhydro-hexosazone durchgeführt54). Als Ergebnis dieser Arbeiten,
HC=NNH-C6H6 N=CH
C=N-NH-C6H5 C6H5—N C-NHNH• C6H5 f—C—NH
-CH
O—CH2
LIV
//
LV
HC N—C6H5I
O—CH2
LVI
HC=NNH C6H5 N CH1 1
(
H(-1
H C6H5 C6H5--N C=NNHC6H5
()H2—C) CH2—0
LVII LVIII
HO
HO
0 NH
CH2.
53) Vgl. Literaturzusammenstellung bei E. Hardegger und E. Schreier,Helv. 35, 232 (1952).
") E. Hardegger und E. Schreier, Helv. 35, 232, 623 (1952) ; H. El Khadem,E. Schreier, G. Stöhr und E. Hardegger, Helv. 35, 993 (1952).
26

die bereits andernorts55) ausführlich diskutiert wurden, folgt, dass
dem Diels 'sehen „Anhydro-glucose-phenylosazon" die Konstitution
LVII des 3,6-Anhydro-D-psicose-penylosazons zukommen dürfte.
Für die aus andern Hexose- und Disaccharid-phenylosazonen56)nach Diels hergestellten Mono-anhydro-osazone wurden analogeStrukturen postuliert.
Die experimentellen Befunde von E. Hardegger und Mitarbeitern
widerlegen eindeutig die von Percival für das Diels'sehe „Anhy¬
dro-glucose-phenylosazon" aufgestellte Konstitutionsformel LVI.
Gegen die Pyrazolformel LV von Diels und die von Hardegger und
Mitarbeitern zeitweilig in Betracht gezogene spirocyclische Formel
LVIII sprechen viele, aber vorläufig nur indirekte Beweise.
Um in direkter Beweisführung die Konstitution des 3,6-Anhydro-
D-psicose-phenylosazons (LVII) für das Diels'sche „Anhydro-glu¬
cose-phenylosazon" festzulegen, wurden die folgenden Umsetzungen
vorgesehen:
a) Herstellung eines Mono-anhydro-hexose-osazons des einseitigzweifach substituierten Methylphenyl- bzw. Benzylphenylhydra-zins nach zwei Methoden, nämlich:
1. durch Abspaltung von Wasser nach Diels aus diesen Osazonen
und
2. durch Einführung zweier Benzylgruppen in ein nach Diels her¬
gestelltes Anhydro-isopropyliden-phenylosazon.
b) Abbau der Diels'schen Anhydro-hexosazone durch Abspaltungder Osazonreste zu Spaltstücken, wie z. B. 3,6-Anhydro-D-psicoson,
3,6-Anhydro-2-keto-D-allonsäure oder 2,5-Anhydro-D-ribonsäureaus Diefe-Anhydro-glucose-osazon.
Versuche zur Herstellung von Anhydro-hexoseosazonen
einseitig zweifach substituierter Hydrazine
Nach Angaben von Diels57) ist es nicht möglich, aus Alkylphenyl-hexosazonen Anhydro-alkyl-phenylosazone herzustellen. Diels und
Meyer (1. c.) konnten beim Behandeln von Fructose-methylphenyl-
55) Vgl. E. Schreier, Diss. ETH (1953) ; Dissertation von G. Stöhr erscheint
demnächst.
56) Vgl. S. Bayne, Soc. 1952, 4998.
") O. Diels und R. Meyer, A. 519, 157 (1935).
27

und Äthylphenyl-osazon, mit Wenig Schwefelsäure in Methanol,
immer nur kristallisiertes unverändertes Ausgangsmaterial isolieren.
Diels folgerte aus dem Nichteintreten der Wasserabspaltung bei den
Hexosazonen mit zweifach substituierten Hydrazonresten, dass an
der Wasserabspaltung aus Fructose-phenylosazon eines der beiden
am Stickstoff haftenden Wasserstoff-Atome beteiligt sei. Die Formu¬
lierung des aus Fructose-phenylosazon hergestellten Anhydro-osa-zons als Pyrazolon-Derivat ist als Ausdruck dieser Interpretationzu deuten.
Meine eigenen Wasserabspaltungsversuche mit Fructose-methyl-
phenylosazon bestätigten die Diels 'sehen Ergebnisse nicht. Ich fand,dass bei längerdauerndem Einwirken von Schwefelsäure in Methanol
das Methylphenylosazon verändert wird und kein kristallisiertes
Ausgangsmaterialmehr isoliert werden kann. Die Reaktionsproduktekonnten nicht identifiziert werden, da sie auch nach mehrmaligem
Chromatographieren nicht kristallisierten. Die Deutung der Was¬
serabspaltungsversuche von Diels und Meyer erscheint demnach
fragwürdig.Versuche zur Wasserabspaltung nach Diels aus dem Fructose-
osazon des Benzylphenylhydrazins stiessen auf unerwartete Schwie¬
rigkeiten in der Bereitung des Ausgangsmaterials.BenzylphenylosazonevonHexosenwiez.B. LXI sind unbekannt.
Ein von Neuberg58) aus D-Fructose und Benzylphenylhydrazin her¬
gestelltes Osazon vom Smp. 190° konnte später von 0frier59) als
ein gemischtes Osazon, nämlich D-Fructose-benzylphenyl-phenyl-osazon (LXII) identifiziert werden, das der Autor ebenfalls aus Glu-
cose-phenylosazon durch Behandeln mit Benzylphenylhydrazingewinnen konnte.
Aus einer grösseren Anzahl von Versuchen, die ich mit D-Glucose
(I), D-Fructose (LIX) und D-Glucoson (LX) durchführte, konnte
nach Behandeln mit frisch gereinigtem Benzylphenylhydrazin in
wässrig-alkoholischer Essigsäure ein Osazon LXII vom Smp. 194
bis 195° in einer Ausbeute von ca. 16% isoliert werden. Das vom
hartnäckig anhaftenden Kristallwasser befreite Präparat erwies
58) O. Neuberg, B. 35, 959 (1902).
59) R. Ofner, M. 25, 615, 1153 (1904); B. 37, 2624 (1904).
28

sich als identisch mit dem von Neuberg und Ofner hergestellten
Benzylphenyl-phenylosazon.Über die Haftstelle der Benzylphenyl- bzw. Phenylhydrazon-
Gruppe im Ofner 'sehen Präparat ist nichts Sicheres bekannt. Ofner60konnte das gemischte Osazon auch aus D-Glucose-benzylphenyl-hydrazon durch Behandeln mit Phenylhydrazin herstellen. In Ana¬
logie zu den sich widersprechenden Resultaten der Untersuchungenvon Votocek61) an Methylphenyl-phenylosazonen zur Bestimmungder Haftstellen der Methylphenyl- bzw. Phenylhydrazon-Gruppenist es nicht möglich, aus der Lage des Benzylphenylrestes im Glucose-
hydrazon sichere Schlüsse auf dessen Lage im Osazon zu ziehen.
Da für die vorstehend beschriebenen Versuche reines Benzyl-
phenylhydrazin verwendet wurde, erschien zunächst die Herkunft
des gemischten Glucose-osazons des Phenyl- und Benzylphenyl-
hydrazins rätselhaft. Eine Erklärung fand sich dann in der Beobach¬
tung von Ofner62), wonach infolge der leichten Zersetzlichkeit des
Benzylphenylhydrazins stets Phenylhydrazin in der Reaktions¬
lösung vorhanden ist. Die Zersetzung von Benzylphenylhydrazinführt nach Ofner, neben Phenylhydrazin, zum Benzylphenylhydra-zon des Benzaldehyds vom Smp. 111°, das nach längerem Stehen
auch aus den Mutterlaugen des Benzylphenyl-phenylosazons LXII
isoliert werden konnte.
Durch Abspaltung von Wasser nach Diels gelang es leicht, das
D-Pructose-benzylphenyl-phenylosazon (LXII) in ein Anhydro-osa-zon LXIII vom Smp. 205° überzuführen, das in Analogie zu früheren
Arbeiten von E. Hardegger und Mitarbeitern57) über das Diels'sche
,,Anhydro-glucose-phenylosazon" als 3,6-Anhydro-D-psicose-benzyl-
phenyl-phenylosazon (LXIII) bezeichnet wurde.
Das Anhydro-osazon LXIII konnte durch Behandeln mit Aceton
und wenig Schwefelsäure in ein Isopropyliden-Derivat LXIV vom
Smp. 132—133° übergeführt werden. Über die Lage des Isopro-
pyliden-Restes, der willkürlich zwischen C4 und C5 angenommen
wurde, können keine Aussagen gemacht werden.
oo) R. Ofner, M. 25, 1153 (1904).
61) E. Votocek und R. Vondràcek, B. 37, 3848 (1904).
62) R. Ofner, M. 25, 593 (1904).
29

Bei der Behandlung von 3,6-Anhydro-D-glucose (LI) mit Benzyl-
phenylhydrazin konnte nur das Benzylphenylhydrazon LXV vom
Smp. 156—157° isoliert werden.
CHO
ÇeH5
C=NN-CH2-C6H5
CbHs
CH2OH
I \
CH2OH
LXI
CHO
ICO
LX
CHO HC=NN-CH2-C6H5
CßHs
CH r CH
CH2OH O—CH2
LI
O—CH2
LXV
CH2OH / HC=NN • CH2 • C6H5
CO
CH2OH
LIX LXII
ÇsH5
C=NNH-CJL
HC-
HC=NN-CH2-C6H5
C=NNH-C6H5
HC N
CH2—O
LXIII
HC-OxI
HC-O--
ÎCH2-
LXIV
>Ipd
O
Ein zweiter Weg zur Herstellung von Anhydro-hexosazonen ein¬
seitig zweifach substituierter Hydrazine ergibt sich durch die Ein¬
führung von Methyl- oder Benzyl-Gruppen in den Osazon-Rest der
Diels 'sehen Anhydro-hexose-phenylosazone.
Percival63) konnte aus D-Glucose-phenylosazon durch Behandeln
mit Dimethylsulfat und Alkali ein öliges Tetramethyl-Derivat her¬
stellen, das er als 3,4,5-Trimethyl-D-glucose-methylphenyl-phenyl-osazon identifizierte. Die Herstellung eines total methylierten Tetra-
methyl-glucose-methylphenylosazons gelang ihm nicht. Analoge
Methylierungsversuehe mit Anhydro-osazonen sind nicht bekannt
geworden.
6S) Vgl. L. L. Engel, Am. Soc. 57, 2419 (1935) ; E. E. Percival und E. O. V.
Percival, Soc. 1935, 1398; 1937, 1320.
30

Das „stabile" Isopropyliden-Derivat des Diels 'sehen „Anhydro-glucose-phenylosazons" weist in der Formulierung nach E. Hard-
egger und E. Schreier nur zwei aktive Wasserstoffatome im Osazon-
Rest auf. Die Substitution dieser beiden beweglichen Wasserstoff¬
atome durch Benzyl-Gruppen wurde auf folgenden Wegen versucht,die nur zum Teil im experimentellen Teil beschrieben sind:
a) Kochen des Isopropyliden-Derivates mit Benzylchlorid, zum
Teil unter Zusatz von wenig Pyridin, bzw. Natriumjodid.b) Umsetzen des Isopropyliden-Derivates mit überschüssigem
Methyl-magnesiumbromid und nachfolgende Behandlung mit Ben¬
zylchlorid.
c) Behandeln des Isopropyliden-Derivates mit Benzyljodid, bzw.
Benzylbromid unter Zusatz von Silberoxyd.In den Versuchen a) und b) wurde das Isopropyliden-Derivat
LXVI nicht verändert; es konnte aus den Reaktionslösungen wieder
kristallisiert zurückgewonnen werden.
Bei der Behandlung c) mit Benzyljodid, bzw. Benzylbromid und
Silberoxyd wurde kein Ausgangsmaterial mehr isoliert. Die Reak¬
tionsprodukte, welche als rote Öle vorlagen, wurden mehrmals an
Aluminiumoxyd chromatographiert. Trotz scheinbarer Einheitlich¬
keit kristallisierten die Öle auch nach längerem Stehen nicht64).Die Verbrennungswerte von C, H und N einiger Chromatogramm-Fraktionen konnten nicht interpretiert werden.
Analoge Versuche mit Benzylbromid und Silberoxyd bei Glyoxal-phenylosazon blieben erfolgslos, da Glyoxal-osazon durch Silberoxydsofort zu 1, 2-Bis-phenylazo-äthylen65) oxydiert wird.
Die Resultate der Versuche c) lassen nicht entscheiden, ob Ben¬
zyl-Gruppen in das Isopropyliden-Derivat eingeführt werden konn¬
ten oder ob das Anhydro-osazon ähnlich wie Glyoxal-osazon oxydiertwurde.
Alle vorstehend beschriebenen Versuche wurden auch mit dem
von Diels66) erstmals hergestellten „labilen" Isopropyliden-Derivatdes „Anhydro-glucose-phenylosazons" vorgenommen.
64) Destillationsversuche im Hochvakuum lassen auf hartnäckig anhaf¬
tende Verunreinigungen schließen.
65) Vgl. H. v. Pechmann, B. 21, 2751 (1888); A. 262, 265 (1891); R. Stollé,B. 59, 1742 (1926).
66) 0. Diels und R. Meyer, A. 519, 157 (1935).
31
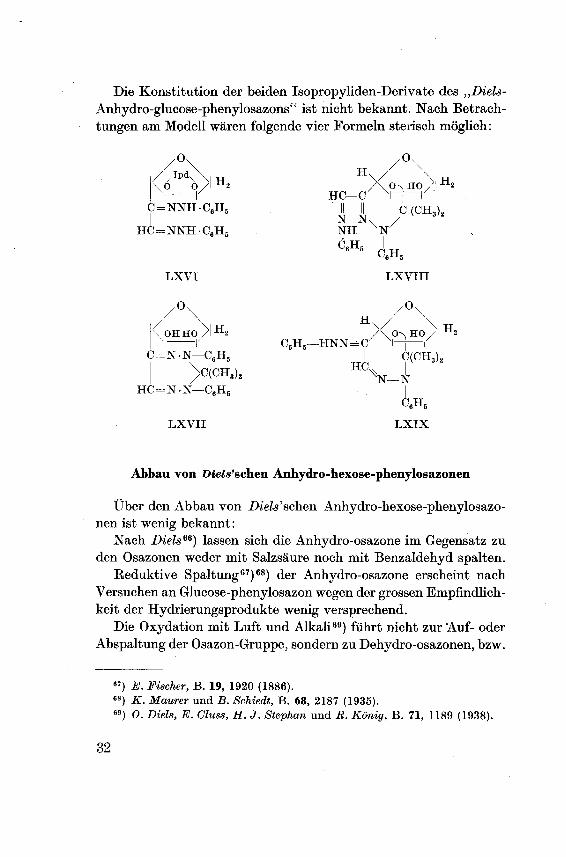
Die Konstitution der beiden Isopropyliden-Derivate des „Diels-
Anhydro-glucose-phenylosazons" ist nicht bekannt. Nach Betrach¬
tungen am Modell wären folgende vier Formeln sterisch möglich:
xIpd,
»I H2
LXVI
/°\OH HO,
H,
C=N-N—C6H5
f )>C(CH3)2HC=N-N—C6H5
H
HC—C
°\\
HO.IH,
NH XN
C (CH3)2
C«H5CeH5
LXVIII
/CK
H
/\O^ HO
C6H5—HNN=C N!~—I
IH,
HCC(CH3)2
\.
N—N
LXVII LXIX
Abbau von Diels'schen Anhydro-hexose-phenylosazonen
Über den Abbau von Diels'schen Anhydro-hexose-phenylosazo-nen ist wenig bekannt:
Nach Diels66) lassen sich die Anhydro-osazone im Gegensatz zu
den Osazonen weder mit Salzsäure noch mit Benzaldehyd spalten.Reduktive Spaltung67)68) der Anhydro-osazone erscheint nach
Versuchen an Glucose-phenylosazon wegen der grossen Empfindlich¬keit der Hydrierungsprodukte wenig versprechend.
Die Oxydation mit Luft und Alkali69) führt nicht zur Auf- oder
Abspaltung der Osazon-Gruppe, sondern zu Dehydro-osazonen, bzw.
6') E. Fischer, B. 19, 1920 (1886).
68) K. Maurer und B. Schiedt, B. 68, 2187 (1935).
69) 0. Diels, E. Cluss, H. J. Stephan und R. König, B. 71, 1189 (1938).
32

Anhydro-dehydro-osazonen, deren Konstitution nicht sicher bekannt
ist und die zur Konstitutionsermittlung der Diels 'sehen Anhydro-osazone kaum geeignet erscheinen.
Auch die Spaltung mit Alkali erscheint wenig erfolgversprechend.Nach Diels und Mitarbeitern70) führt die Einwirkung von Alkali bei
Osazonen zu einer Aldolspaltung zwischen C2 und C3 und gibt, neben
Glyoxal-osazon, bzw. 1,2-Bis-phenyl-azo-äthylen, ein nicht identifi-
zieïbares Gemisch von vermutlich mehreren Zuckern, bzw. Saccha¬
rinsäuren.
Die Einwirkung von Hydroxylamin-hydrochlorid in heissem Al¬
kohol führt nach Diels unter Zerstörung des C-Gerüstes zu Deri¬
vaten der Pyrazols71).Da bekannt ist, dass Hydrazone von salpetriger Säure oxydativ
gespalten werden72), schien es aussichtsreich, eine analoge Spaltungder Osazone, bzw. Anhydro-osazone zu versuchen. Gemäss der von
E. Hardegger und Mitarbeitern54) angenommenen Konstitution-
formel LVII der Diels 'sehen Anhydro-hexosazone waren als Spalt¬
produkte nach Einwirkung von salpetriger Säure das 3,6-Anhydro-oson, die 3,6-Anhydro-2-keto-säure, bzw. unter Verlust des C-Atoms
1 die 2,5-Anhydro-pentose, bzw. 2,5-Anhydro-pentonsäure, zu er¬
warten.
Die Behandlung von 3,6-Anhydro-D-psicose-phenylosazon (LVII)mit Natriumnitrit in Eisessig führte nach Aufarbeitung der dunkel¬
roten Oxydationsprodukte zu einer hellbraunen wässrigen Lösung,die sauer reagierte und gegenüber I'ehling'scher Lösung starkes
Reduktionsvermögen zeigte. Die sauren Anteile konnten in Form
eines gut kristallisierenden, unscharf bei 260—265° schmelzenden
Brucinsalzes LXXV mit der spezifischen Drehung von —41° (in
Wasser) in einer Ausbeute von 9,5 % der Theorie gewonnen werden.
Aus dem 3,6-Anhydro-D-galactose-phenylosazon (LXX) wurde
in gleicher Weise in einer Ausbeute von 39 % der Theorie ein gutkristallisierendes Brucinsalz LXXII mit dem unscharfen Zerset-
70) 0. Diels, B. Meyer und 0. Onnen, A. 525, 94 (1936).
71) 0. Diels, B.Meyer und O. Onnen, A. 525, 94 (1936); vgl. O. Stöhr,
Diss. ETH (1953).
72) E. Bamberger und W. Bemsel, B. 36, 57, 347, 359 (1903); M. Busatvnd
H. Kunder, B. 49, 317 (1916).
33
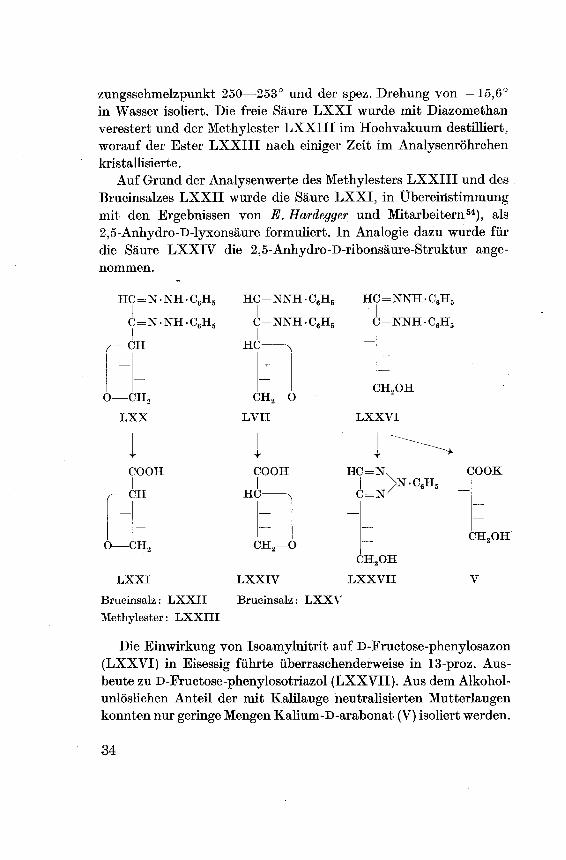
zungsschmelzpunkt 250—253° und der spez. Drehung von —15,6°
in Wasser isoliert. Die freie Säure LXXI wurde mit Diazomethan
verestert und der Methylester LXXIII im Hochvakuum destilliert,
worauf der Ester LXXIII nach einiger Zeit im Analysenröhrchenkristallisierte.
Auf Grund der Analysenwerte des Methylesters LXXIII und des
Brucinsalzes LXXII wurde die Säure LXXI, in Übereinstimmungmit den Ergebnissen von E. Hardegger und Mitarbeitern54), als
2,5-Anhydro-D-lyxonsäure formuliert. In Analogie dazu wurde für
die Säure LXXIV die 2,5-Anhydro-D-ribonsäure-Struktur ange¬
nommen.
C=N-NH-C6H5
CH
O—CH2
LXX
HO¬
CH,,—O
LVII LXXVI
4-
COOH
ICH
4-
COOH
IHO n
4-
HC=NN
C=N/
COOK
CH2OHO CH2
CH2OH
LXXI LXXIV LXXVII V
Brueinsalz: LXXII Bruoinsalz: LXXV
Methylester: LXXIII
Die Einwirkung von Isoamylnitrit auf D-Fructose-phenylosazon
(LXXVI) in Eisessig führte überraschenderweise in 13-proz. Aus¬
beute zu D-Fructose-phenylosotriazol (LXXVII). Aus dem Alkohol¬
unlöslichen Anteil der mit Kalilauge neutralisierten Mutterlaugenkonnten nur geringe Mengen Kalium-D-arabonat (V) isoliert werden.
34

Anhydro-benzimidazole
Zur Konstitutionsaufklärung der vorstehend beschriebenen 2,5-
Anhydro-pentonsäuren sind die von Link und Moore'73) aufgefun¬denen Anhydro-benzimidazole wichtig geworden.
Wie Hashins und Hudson1*) im Jahre 1939 fanden, lassen sich die
Aldonsäuren durch Kondensation mit o-Phenylendiamin in die
Benzimidazole überführen. Nach der von Link und Moore11") ausge¬
arbeiteten Vorschrift wurde es möglich, die Benzimidazole der ver¬
schiedenen Aldonsäuren in guter Ausbeute zu gewinnen.Die Benzimidazole sind amphoter. Sie bilden mit Säuren Salze, von
denen sich die Pikrate für die Charakterisierung der Benzimidazole
als wichtig erwiesen. In starken Laugen sind die Benzimidazole
löslich. Mit Kupfer- und Silberionen geben sie schwer lösliche Salze.
Zur Charakterisierung der Aldon-, Zucker-, Schleim- und Trioxy-
glutarsäuren sind die Benzimidazole und deren Pikrate dank ihrer
guten Kristallisationsfähigkeit und des scharfen Schmelzpunktessehr geeignet47).
Nach Link und Moore15) wird aus den Pentonsäure-benzimi-
dazolen beim Erwärmen auf 180° in Gegenwart von Salzsäure und
Zinkchlorid 1 Mol Wasser abgespalten. Die Anhydro-benzimidazolewurden in der Folge als Derivate der 2,5-Anhydro-pentonsäurenformuliert. Die Oxydation der Anhydro-pentonsäure-benzimidazolemit Perjodsäure führte nicht zu den erwarteten Dialdehyden, son¬
dern unter Bildung von 2 Mol Ameisensäure und 1 Mol Formaldehydzur 2-Benzimidazol-carbonsäure. Ein analoger abnormaler Verlauf
der Oxydation mit Perjodsäure wurde auch an andern Verbindun¬
gen, die aktivierte Wasserstoff-Atome enthalten, beobachtet76). Den
Beweis der Lage des Anhydroringes bei Anhydro-benzimidazolenerbrachten Link und Huebner11) durch Synthese des 2,5-Anhydro-
73) 8. Moore und K. P. Link, J. Org. Chem. 5, 637 (1940); G. F. Huebner,
R. Lohmar, R. J. Dirnler, S. Moore und K. P. Link, J. Biol. Chem. 159, 503
(1945).
74) W. F. Hashins und C. S. Hudson, Am. Soc. 61, 1266 (1939).
75) S. Moore und K. P. Link, J. Biol. Chem. 133, 293 (1940).
76) Vgl. -R. C. Hockett, M. H. Nickerson und W. H. Reeder, Am. Soc. 66,
472 (1944).
77) C. F. Huebner und K. P. Link, J. Biol. Chem. 186, 387 (1950).
35

L-xylonsäure-benzimidazols (LXXXI) aus 1,4-Anhydro-D-sorbit
(LXXVIII), der durch selektive Oxydation mit Perjodsäure in die
2,5-Anhydro-L-xylose (LXXIX) und weiter in die 2,5-Anhydro-L-
xylonsäure (LXXX) übergeführt wurde. Das Benzimidazol LXXXI
der Anhydro-L-xylonsäure erwies sich nach Schmelzpunkt und spez.
Drehung als Antipode des aus D-Xylonsäure-benzimidazol herge¬stellten Anhydro-Derivates.
CH2OH
_
CHO
r—CH r~CH
O—CH2 O—CH2
LXXVIII LXXIX
Ich habe versucht, die von mir aus 3,6-Anhydro-D-galactose-
phenylosazon (LXX) hergestellte 2,5 - Anhydro - D - lyxonsäure
(LXXI)78), deren Konstitution noch unbewiesen ist, sowie die aus
3,6-Anhydro-D-glucose (LI) gewonnene 2,5-Anhydro-D-arabon-säure (LH)79) in die Anhydro-benzimidazole umzuwandeln. Die
Aufarbeitung der schlecht kristallisierenden Anhydro-benzimidazolewar schwierig. Die geringen Substanzmengen erlaubten keine sichere
Identifizierung der Derivate.
Nach den in unserm Laboratorium durchgeführten Untersu¬
chungen54) über Diels'sche Anhydro-osazone und Anhydro-osotria-zole liegt die Voraussage nahe, dass aus den Benzimidazolen der
Bibon- und Arabonsäure ein 2,5-Anhydro-ribonsäure-benzimidazolund aus Lyxonsäure-benzimidazol das 2.5-Anhydro-lyxonsäure-ben-zimidazol entstehen sollte.
Das von mir hergestellte, vielleicht nicht ganz reine Benzimidazol
der 2,5-Anhydro-lyxonsäure (LXXII) schmolz bei 216—217°; im
Gegensatz zu dem von Link und Moore75) aus D-Lyxonsäure her¬
gestellten, bei 200—204° schmelzenden Anhydro-benzimidazol. Der
Vergleich ihrer Pikrate konnte aus Mangel an Substanz nicht durch¬
geführt werden.
'8) Vgl. Seite 33 und 34.
"») Vgl. Seite 25.
NH
COOH
ICH
C
-CH
O—CH2
LXXX
O—CH2
LXXXI
36

Experimenteller Teil1)
Abbau reduzierender Monosaccharide, Disaccharide und
Uronsäuren mit Sauerstoff in alkalischer Lösung
D-Arabonsäure (IV) aus D-Glucose (I)
Eine Lösung von 1,8 g ( = 10 Millimol) D-Glucose (I) in 150 ccm
Wasser wurde mit 3,5 g (= 11,1 Millimol) kristallisiertem Barium-
hydroxyd versetzt und unter Sauerstoff vier Tage bei Zimmer¬
temperatur geschüttelt, wobei 260 ccm Sauerstoff (ber. 250 ccm 02)verbraucht wurden. Das überschüssige Bariumhydroxyd wurde als
Karbonat gefällt, durch Celit abfiltriert und das gelbliche Filtrat
anschliessend durch eine Säule mit 30 ccm Wofatit KS laufen gelas¬sen. Die freien Säuren wurden zusammen mit 100 ccm Waschwasser
zur Entfernung der Ameisensäure vorsichtig auf dem Wasserbad
im Wasserstrahlvakuum vollständig eingedampft. Der gelbe, zäh¬
flüssige Rückstand wurde in wenig warmem Wasser aufgenommenund mit verdünnter Kalilauge neutralisiert. Nach mehrmaligemUmfallen aus Wasser-Äthanol konnten 1,42 g Kalium-D-arabonat
(V) erhalten werden, was einer Ausbeute von 69 % der Theorie
entspricht.36 g ( = 200 Millimol) D-Glucose (I) wurden in 100 ccm Wasser
gelöst und 30 g ( = 534 Millimol) Kaliumhydroxyd, gelöst in 250 ccm
Wasser, im Verlauf von 5 Stunden zugetropft, wobei die alkalische
Zuckerlösung mittels eines Vibromischers intensiv mit Sauerstoff
durchwirbelt wurde. Die Lösung erwärmte sich nur leicht und ver-
) Alle Schmelzpunkte sind korrigiert.
37

färbte sich gegen das Ende der Sauerstoffaufnahme hellbräunlich2).Die Oxydation wurde nach 48 Stunden abgebrochen, da der genaue
Endpunkt der Sauerstoffaufnahme nicht beobachtet werden konnte.
Aus der Hälfte der Oxydationslösung wurden, wie oben beschrieben,
15,8 g rohes Kalium-D-arabonat (V) isoliert, was einer Ausbeute von
73 % der Theorie entspricht.
D-Arabonsäure (IV) aus jß-Pentaacetyl-D-glucose (II)
3,9 g ( = 10 Millimol) ß-Pentaacetyl-D-glucose (II) wurden zu
einer eiskalten Lösung von 7 g (= 125 Millimol) Kaliumhydroxydin 150 ccm Wasser gegeben und die Mischung fünf Tage bei Zimmer¬
temperatur unter Sauerstoff geschüttelt, wobei 300 ccm Sauerstoff
(ber. 250 ccm 02) aufgenommen wurden. Die gelbliche Lösung wurde
durch 140 ccm Wofatit KS filtriert und aus Filtrat und Wasch-
wasser die Essigsäure durch mehrmaliges Eindampfen im Wasser¬
strahlvakuum abgetrieben. Der gelbliche Rückstand wurde in 10
ccm Wasser gelöst und nach Zugabe von 0,7 g Kaliumcarbonat die
heiße Lösung mit 40 ccm Methanol versetzt, worauf beim Erkalten
der Lösung 1,4 g (=37% der Theorie) Kalium-D-arabonat (V)auskristallisierten.
D-Arabonsäure (IV) aus D-Glucosamin-hydrochlorid (III)
1,5 g (= 7 Millimol) D-Glucosamin-hydrochlorid (III) und 3 g
( = 9 Millimol) krist. Bariumhydroxyd wurden acht Tage bei Zimmer¬
temperatur unter Sauerstoff geschüttelt. Der Verbrauch an Sauer¬
stoff betrug 260 ccm (ber. 175 ccm 02). Die Lösung wurde auf
bekannte Weise, unter Verwendung von 40 ccm Wofatit KS, auf¬
gearbeitet. Die Ausbeute an Kalium-D-arabonat (V) betrug 1,0 g
( = 70,5 % der Theorie).Kalium-D-arabonat (V). Das Kaliumarabonat wurde dreimal aus
Wasser-Methanol umkristallisiert und das Analysen-Präparat 24
Stunden bei 60° im Hochvakuum getrocknet.
2) Konzentrierte Zuckerlösungen verfärben sich bei der Oxydation mit
Alkali und Sauerstoff unter Wärmeentwicklung schnell dunkelbraun. Die
Oxydationsprodukte sind schwierig aufzuarbeiten und die Ausbeute an
Aldonsäuren ist gering.
38

4,046 mg Subst. gaben 4,385 mg C02 und 1,679 mg H20
C5H906K Ber. C 29,40 H 4,44%Gef. C 29,57 H 4,64%
[«Id = - 4,8° (c = 0,5 in Wasser)
Brucinsalz VI der D-Arabonsäure. 1 g D-Arabonsäure (IY) wurde
in 5 com Wasser gelöst und 2,7 g Brucin in 10 com heissem Methanol
zugegeben, worauf nach einiger Zeit das Brucinsalz kristallisierte.
Das überschüssige Brucin wurde durch mehrmaliges Ausschütteln
der wässrigen Lösung mit Chloroform entfernt. Das aus Wasser-
Methanol umkristallisierte Präparat vom Smp. 158° wurde zur
Analyse 48 Stunden bei 20° im Hochvakuum getrocknet.
3,654 mg Subst. gaben 7,709 mg C02 und 2,020 mg H20
C28H36O10N2,l,5H2O Ber. C 57,23 H 6,34%Gef. C 57,57 H 6,19%
[a]D = - 23° (c = 1,1 in Wasser)
D-Arabonsäure-phenylhydrazid (VII). 1 g Arabonsäure wurde in
5 ccm Wasser gelöst, 0,7 ccm Phenylhydrazin zugegeben und die
Mischung einige Minuten auf dem Wasserbad erwärmt. Das Phenyl-
hydrazid VII fiel beim Erkalten aus. Das aus Methanol-Essigesterumkristallisierte Präparat vom Smp. 208—209° (u. Zers.) wurde zur
Analyse 24 Stunden bei 50° im Hochvakuum getrocknet.
3,638 mg Subst. gaben 6,862 mg C02 und 2,074 mg H20
CnH1605N2 Ber. C 51,55 H 6,29%Gef. C 51,49 H 6,38%
[a]D = _13° (e = 0,5 in Methanol)
S-Benzyl-thiuronium-T>-arabonat (VIII). 0,6 g Barium-D-ara-
bonat wurden in 5 ccm Methanol suspendiert und 0,52 g Benzyl-thiuronium-sulfat, gelöst in 10 ccm heissem Methanol, zugegeben.
Ausgefallenes Bariumsulfat wurde abfiltriert und das Benzylthiu-ronium-arabonat (VIII) mit Aceton gefällt. Das aus Methanol-
Aceton umkristallisierte Analysenpräparat vom Smp. 144—145°
wurde 24 Stunden bei 40° im Hochvakuum getrocknet.
3,632 mg Subst. gaben 6,249 mg C02 und 1,977 mg H20
C13H20O6N2S Ber. C 46,98 H 6,07%Gef. C 46,95 H 6,10%
[<x]D = + 3,6° (c = 0,5 in Methanol)
39

D-Lyxonsäure (X) aus D-Galactose (IX)
9 g ( = 50 Millimol) D-Galactose (IX) wurden in 200 ccm Wasser
und 18 g ( = 57 Millimol) krist. Bariumhydroxyd in 300 ccm Wasser
gelöst. Die vereinigten Lösungen wurden vier Tage bei Zimmer¬
temperatur unter Sauerstoff geschüttelt, wobei 1370 ccm Sauerstoff
(ber. 1250 ccm 02) verbraucht wurden. Nach Sättigung mit C02wurde die Mischung durch Celit und anschliessend durch 120 ccm
Wofatit KS filtriert. Das mit 250 ccm Waschwasser verdünnte Fil¬
trat wurde im Wasserstrahlvakuum zur Trockene eingedampft.D-Lyxonsäure-phenylhydrazid (XI). Der Eindampfrückstand
wurde in 10 ccm Wasser gelöst, nach Zugabe von 5 ccm Phenyl¬
hydrazin zwanzig Minuten auf dem Wasserbad erwärmt und erneut
im Vakuum zur Trockene verdampft. Das aus Methanol-Essigesterumkristallisierte Lyxonsäure-phenylhydrazid (XI) (5,5 g = 43 %der Theorie) vom Smp. 164° (u. Zers.) wurde zur Analyse 48 Stun¬
den bei 20° im Hochvakuum getrocknet.
3,859 mg Subst. gaben 7,278 mg C02 und 2,198 mg H20
CuH1605N2 Ber. C 51,55 H 6,29%Gef. C 51,47 H 6,37%
[a]D = - 10° (c = 1,3 in Wasser)
3-[a-D-Glucosido]-D-arabonsäure (XIII) aus Maltose (XII)
Eine Suspension von 20 g ( = 63 Millimol) krist. Bariumhydroxydin 150 ccm Wasser wurden unter Sauerstoff von einem Vibrator mit
Gasbläschen durchwirbelt. Im Verlauf von 2—3 Stunden wurde
eine Lösung von 18 g (= 50 Millimol) Maltose (XII) in 200 ccm
Wasser zur alkalischen Suspension des Bariumhydroxyds zugetropft.Die Oxydation der Maltose (XII) erfolgte in der stets von Sauerstoff¬
bläschen durchsetzten Mischung unter leichter Erwärmung. Nach
22 Stunden waren 1250 ccm Sauerstoff (ber. 1250 ccm 02) ver¬
braucht. Die weitere Oxydation wurde abgebrochen, die Mischungmit C02 gesättigt und nacheinander durch Celit und 120 ccm Wofa¬
tit KS filtriert. Das Filtrat wurde mit 240 ccm Waschwasser verei¬
nigt und unter fortwährend mittels einer Kapillare zugesetztemMethanol auf dem Wasserbad im Wasserstrahlvakuum vorsichtig
40

zur Trockne eingedampft3). Die rohe 3-[a-D-Glucosido]-D-arabon-säure (XIII) von ca. 17g Gewicht wurde als gelbliche, sehr zäh¬
flüssige Masse erhalten.
Brucinsalz XIV. Die rohe Säure XIII von ca. 17 g wurde in
30 ccm heissem Wasser gelöst und mit einer heissen Lösung von
24 g Brucin in 50 ccm Methanol versetzt. Im Verlauf von 20 Minuten
wurde das Methanol auf dem Wasserbad abgedampft, die Mischungauf 20° gekühlt und zur Entfernung des überschüssigen Brucins
mehrmals mit Chloroform ausgeschüttelt. Die im Vakuum zu einem
Sirup eingeengte Lösung wurde vorsichtig mit Methanol bis zur
beginnenden Trübung versetzt und angeimpft4). Das Brucinsalz
XIV kristallisierte sehr langsam im Verlauf mehrerer Tage in Warzen
aus. Das mit Alkohol-Methanol (1:1) gewaschene Derivat wog 7,2 g.Aus den Mutterlaugen liessen sich nochmals 3,2 g Brucinsalz XIV
gewinnen, was zusammen einer Ausbeute von 28 % der Theorie
entspricht. Das wiederholt aus Wasser, bzw. Wasser-Äthanol umkri¬
stallisierte Präparat vom Smp. 152—154° (u. Zers.) wurde zur Ana¬
lyse 48 Stunden im Hochvakuum bei Zimmertemperatur getrocknet.
3,810 mg Subst. gaben 7,717 mg C02 und 2,181 mg H20C34H46015N2,1H20 Ber. C 55,13 H 6,53%
Gef. C 55,27 H 6,41%[a]D = + 50° (c = 0,5 in Wasser)
3-[/?-D-Glucosido]-D-arabonsaure (XVI) aus Cellobiose (XV)
9,4 g (= 26 Millimol) Cellobiose (XV) wurden in 100 ccm Wasser
gelöst und zu 11g (=32 Millimol) krist. Bariumhydroxyd in 150
ccm Wasser zugetropft, wobei nach der oben beschriebenen Weise
mit Sauerstoff oxydiert wurde. Nach 20 Stunden waren 800 ccm
Sauerstoff (ber. 670 ccm 02) aufgenommen worden. Die Aufarbei¬
tung der 3-[jS-D-Glucosido]-D-arabonsäure (XVI) erfolgte ebenfalls
auf die bekannte Weise. Die in Form einer gelblich, zähflüssigenMasse anfallende Säure XVI wog 8,3 g.
3) Bei unvorsichtigem Arbeiten wird die a-Glucosidbindung schnell durch
die bei der Oxydation gebildete Ameisensäure hydrolysiert.4) Durch längeres Zerreiben unter Äther wurden aus gefälltem Brucinsalz
XIV Impfkristalle erhalten.
41

Brucinsalz XVII. Aus 8,3 g roher Säure XVI wurden 7,6 gBrucinsalz XVII erhalten, was einer Ausbeute von 37 % der Theorie
entspricht. Das aus Wasser-Methanol mehrmals umkristallisierte
Präparat vom Snap. 149—150° (u. Zers.) wurde zur Analyse 48
Stunden bei Zimmertemperatur im Hochvakuum getrocknet.
3,796 mg Subst. gaben 6,987 mg C02 und 2,365 mg H20
C34H46015N2,4H20 Ber. C 50,14 H 6,85%Gef. C 50,23 H 6,97%
[a]D = - 6,2° (c = 1,1 in Wasser)
Calciumsalz XVIII. 2,4 g Brucinsalz XVII vom Smp. 149—150°
wurden in 300 ccm Wasser gelöst und nach Zugabe von 0,2 g Cal-
ciumhydroxyd unter Umschütteln leicht erwärmt. Das ausgefalleneBrucin wurde durch mehrmaliges Ausschütteln der wässrigen Lösungmit Chloroform entfernt. Aus der im Wasserstrahlvakuum eingeeng¬ten Lösung kristallisierte das Calcium-3-[j8-D-glucosido]-D-arabonat(XVIII), nach Zugabe von Äthanol, im Verlauf einiger Stunden.
Das aus Wasser-Äthanol mehrmals umkristallisierte Analysenprä¬parat wurde 48 Stunden bei Zimmertemperatur im Hochvakuum
getrocknet.
3,894 mg Subst. gaben 5,286 mg C02 und 1,946 mg H20
C22H40O28Ca . Ber. C 37,08 H 5,66%Gef. C 36,92 H 5,60%
[*1d = + 15>5° (c = 0,9 in Wasser)
3-[/?-D-GaIactosido]-D-arabonsäure (XX) aus Lactose (XIX)
Die Oxydation von 18 g (= 50 Millimol) Lactose (XIX), gelöstin 350 ccm Wasser, erfolgte nach Zugabe von 20 g ( = 63 Millimol)krist. Bariumhydroxyd, unter Aufnahme von ca. 1600 ccm Sauer¬
stoff (ber. 1250 ccm 02), im Verlaufe von 20 Stunden. Die Aufarbei¬
tung der Oxydationsprodukte erfolgte wie bei der Oxydation der
Maltose (XII). Die rohe, gelbliche Säure XX wog 16,5 g.
Brucinsalz XXI. Aus 8,2 g roher Säure XX wurden 10,1g
Brucin-3-[j8-D-galactosido]-D-arabonat (XXI) vom Smp. 144 bis
145° (u. Zers.) erhalten, was einer Ausbeute von 26% der Theorie
entspricht. Das mehrmals aus Wasser-Äthanol umkristalHsierte
Präparat wurde zur Analyse 48 Stunden bei Zimmertemperatur imHochvakuum getrocknet.
42

3,702 mg Subst. gaben 7,184 mg C02 und 2,199 mg H20
C34H46015N2,3H20 Ber. C 52,61 H 6,72%Gef. C 52,96 H 6,63%
[a]D=-3,4° (c = 1,2 in Wasser)
Calciumsalz XXII. 2,7 g Brucinsalz XXI vom Smp. 144—145°
(u. Zers.) führten auf die bekannte Weise zu 0,71 g Calcium-3-[,8-D-
galactosido]-D-arabonat (XXII). Das aus Wasser-Methanol mehr¬
mals umkristallisierte Derivat wurde zur Analyse 48 Stunden bei
Zimmertemperatur im Hochvakuum getrocknet.
3,760 mg Subst. gaben 4,604 mg C02 und 2,027 mg H20
C22H40O23Ca,4H2O Ber. C 33,67 H 6,16%Gef. C 33,42 H 6,03%
[a]D = + 32° (c = 0,4 in Wasser)
D-Erythronsäure-lacton (XXIV) ans D-Ribose (XXIII) 5)
4,5 g ( = 30 Millimol) D-Ribose (XXIII) und 4,5 g ( = 80 Millimol)
Kaliumhydroxyd wurden in je 100 ccm Wasser gelöst und die bei
0° vereinigten Lösungen in einem Hydrierkolben unter Sauerstoff
geschüttelt. Nach ötägigem Schütteln bei Zimmertemperatur waren
735 ccm Sauerstoff (ber. 720 ccm 02) aufgenommen worden. Die
leicht gelbliche Lösung wurde durch 100 ccm Wofatit KS filtriert
und zusammen mit 200 ccm Waschwasser im Vakuum auf dem
Wasserbad zur Trockene eingedampft. Der in Methanol aufge¬nommene hellgelbe, zähflüssige Rückstand wurde in Kugelröhrchenin Portionen von ca. je 1 g vorsichtig bei 140—160° im Hochvakuum
destilliert. Die Destillate wogen je 0,6—0,7 g und kristallisierten im
Verlauf einiger Stunden. Nach dem Umkristallisieren aus Essigesterund Sublimation im Hochvakuum bei 90° schmolz das analysen¬reine Präparat bei 101—102°.
3,794 mg Subst. gaben 5,638 mg C02 und 1,762 mg H20
C4H604 Ber. C 40,68 H 5,12%Gef. C 40,55 H 5,20%
[«Id = ~ 71° (c = °'8 in Wasser)
5) Mitbearbeitet von N. Nikoloff.
43

Phenylhydrazid XXV. 0,3 g D-Erythronsäure-lacton (XXIV)wurden in 5 ccm Methanol mit 0,3 ccm Phenylhydrazin 10 Minuten
am Rückfluss gekocht. Das Methanol wurde im Wasserstrahl¬
vakuum entfernt und der Rückstand aus Methanol-Äther umkri¬
stallisiert. Zur Analyse wurde das bei 129—130° schmelzende
Phenylhydrazid XXV 48 Stunden bei 70° im Hochvakuum getrock¬net.
3,659 mg Subst. gaben 7,098 mg C02 und 2,068 mg H20
C10H14O4H2 Ber. C 53,09 H 6,24%Gef. C 52,93 H 6,24%
[a]D = + 19° (o = 0,3 in Wasser)
L-Erythronsäure-lacton (XXVII) aus L-Arabinose (XXXVI)6)
3,6 g ( = 24 Millimol) L-Arabinose (XXXVI) und 4 g (= 71 Milli-
mol) Kaliumhydroxyd wurden in je 120 ccm Wasser gelöst. Die
gekühlten Lösungen wurden zusammengegeben und bei Zimmer¬
temperatur unter Sauerstoff geschüttelt. Die Oxydation erfolgtesehr langsam7), sie wurde abgebrochen, als nach 14 Tagen 280 ccm
Sauerstoff (ber. 580 ccm 02) aufgenommen worden waren. Die durch
100 ccm Wofatit KS filtrierte Lösung wurde zusammen mit ca.
250 ccm Waschwasser im Vakuum bei 80° zur Trockene eingedampftund der gelbliche, glasige Rückstand dreimal mit je 10 ccm heissem
Methanol extrahiert. Die filtrierten, vom Methanol befreiten Aus¬
züge wurden im Kugelrohr bei 140—150° im Hochvakuum destilliert.
Das kristallisierende Lacton XXVII wog 0,9 g (= 33 % der Theorie)und schmolz bei 100—102°. Durch Umkristallisieren des Lactons
XXVII aus Essigester stieg der Smp. auf 103—104°. Das Analysen¬
präparat wurde im Hochvakuum bei 90° sublimiert.
3,820 mg Subst. gaben 5,687 mg C02 und 1,761 mg H20
C4H604 Ber. 0 40,68 H 5,12%Gef. C 40,63 H 5,16%
[a]D = + 72° (c = 0,5 in Wasser)
6) Mitbearbeitet von P. Geistlich.
') Die Gründe für den langsamen Verlauf der Oxydation sind unbekannt;andere Ansätze mit L-Arabinose zeigten normale Sauerstoffaufnähme.
44

Phenylhydrazid XXVIII. Das analog wie oben hergestellte Prä¬
parat hatte einen Smp. von 130—131° und wurde zur Analyse48 Stunden bei 70° im Hochvakuum getrocknet.
3,692 mg Subst. gaben 7,203 mg C02 und 2,036 mg H20
C10H14O4N2 Ber. C 53,09 H 6,24%Gef. C 53,25 H 6,17%
Wd = - 17° (c = 1 in Wasser)
D,L-Ery'thronsäure-lacton. Je 10 mg D-Lacton XXIV und L-
Lacton XXVII wurden in wenig heissem Essigester gelöst. Das
D,L-Erythronsäure-lacton kristallisierte beim Abkühlen der Lösung.Nach Umkristallisieren aus Essigester schmolz das Laeton bei 91 bis
92°. Das Analysenpräparat wurde 48 Stunden bei Zimmertempera¬tur im Hochvakuum getrocknet.
3,770 mg Subst. gaben 5,593 mg C02 und 1,717 mg H20
C4H604 Ber. C 40,68 H 5,12%Gef. C 40,49 H 5,13%
[a]D = 0° (o = 1 in Wasser)
Die Mischung von D,L-Lacton mit wenig D-Erythronsäure-lacton schmolz bei 86°.
D-Threonsäure-lacton (XXX) aus D-Xylose (XXIX)
Eine wie bei der Erythronsäure-Herstellung bereitete und
gekühlte Lösung von 4,5 g ( = 30 Millimol) D-Xylose (XXIX) und
4 g ( = 71 Millimol) Kaliumhydroxyd in 260 com Wasser hatte nach
9tägigem Schütteln 740 cem Sauerstoff (ber. 720 cem 02) aufgenom¬men. Die Aufarbeitung zu D-Threonsäure-lacton (XXX) erfolgtein gleicher Weise wie bei den Erythronsäure-lactonen. Das destil¬
lierte Laeton XXX (1,2 g = 34% der Theorie) kristallisierte nach
einigem Stehen und schmolz bei 70—73°. Das aus Essigester-Ätherumkristallisierte Präparat vom Smp. 75—77° wurde zur Analyse24 Stunden bei Zimmertemperatur im Hochvakuum getrocknet.
3,765 mg Subst. gaben 5,590 mg C02 und 1,732 mg H20
C4H604 Ber. C 40,68 H 5,12%Gef. C 40,54 H 5,14%
[a]D = - 29° (c = 0,8 in Wasser)
45

Das Lacton XXX ist hygroskopisch und mußte in Präparaten-röhrchen, die im Hochvakuum zugeschmolzen wurden, aufbewahrt
werden.
PMnylhydrazid XXXI. Das Phenylhydrazid XXXI der D-
Threonsäure wurde wie die analogen Präparate der Erythronsäure
hergestellt und aufgearbeitet. Das aus Methanol-Äther umkristalli¬
sierte Derivat wurde zur Analyse 24 Stunden bei 60° im Hoch¬
vakuum getrocknet.
3,778 mg Subst. gaben 7,337 rag C02 und 2,100 mg H20
C10HuO4N2 Ber. C 53,09 H 6,24%Gef. C 52,99 H 6,22%
Md —
— 30° (c = 0,5 in Wasser)
L-Threonsäure-lacton (XXXIV) aus L-Ascorbinsäure (XXXII)
Zu einer Lösung von 8,8 g (= 50 Millimol) Ascorbinsäure XXXII
in 100 ccm Wasser wurde unter Sauerstoff eine Lösung von 14 g
( = 250 Millimol) Kaliumhydroxyd in 100 ccm Wasser im Verlauf
von 4 Stunden zugetropft. Vom Beginn des Zutropfens der Kali¬
lauge an wurde die Mischung mit Hilfe eines Vibrators intensiv
mit Sauerstoff durchwirbelt. Die Oxydation erfolgte unter leichter
Erwärmung der Mischung und kam nach 15 Stunden, unter Auf¬
nahme von 1250 ccm Sauerstoff (ber. 1200 ccm 02), zu Ende. Nach
Filtration durch 300 ccm Wofatit KS wurde die saure Lösung mit
ca. 10 g in wenig Wasser gelöstem Calciumacetat versetzt, vom aus¬
geschiedenen Calciumoxalat (ca. 6,3 g) abfiltriert, nochmals durch
100 ccm Wofatit KS laufen gelassen und zusammen mit dem Wasch¬
wasser bei ca. 80° im Wasserstrahlvakuum zur Trockene einge¬dampft. Das im Kugelrohr bei 140—150° im Hochvakuum destil¬
lierte Lacton XXXIV kristallisierte nach einigen Stunden. Nach
mehrmaligem Umkristallisieren aus trockenem Essigester-Ätherstieg der Smp. von 72—74° auf 74—76°. Das Analysenpräparatwurde 24 Stunden bei 20° im Hochvakuum getrocknet.
3,530 mg Subst. gaben 5,272 mg C02 und 1,588 mg H20C4H604 Ber. C 40,68 H 5,12%
Gef. C 40,76 H 5,03%Md = + 30° (c = 0,9 in Wasser)
Das Lacton XXXIV erwies sich ebenfalls als sehr hygroskopisch.
46

L-Threonsäure-lacton (XXXIV) aus Dehydro-L-ascorbinsäure
(XXXIII)
1,76 g ( = 10 Millimol) L-Ascorbinsäure (XXXII) wurden in
30 ccm Wasser 4 Tage unter Sauerstoff geschüttelt. Der Sauerstoff¬
verbrauch betrug 115 ccm. Die wässrige Lösung der Dehydro-ascorbinsäure XXXIII wurde eingefroren, mit 2 g ( = 35 Millimol)
Kaliumhydroxyd, gelöst in 10 ccm kaltem Wasser, versetzt und
erneut unter Sauerstoff bei Zimmertemperatur geschüttelt. Nach
24 Stunden waren abermals 165 ccm Sauerstoff verbraucht worden.
Die Aufarbeitung erfolgte, wie vorstehend beschrieben, unter Zugabevon 2 g Calciumacetat, Behandln ng mit Wofatit KS und Destilla¬
tion im Hochvakuum. Das kristallisierte Destillat erwies sich nach
Schmelzpunkt, Mischprobe und spez. Drehung als L-Threonsäure-
lacton (XXXIV).
Phenylhydrazid XXXV. Das aus Methanol-Äther umkristalli¬
sierte und 48 Stunden bei 70° im Hochvakuum getrocknete Ana¬
lysenpräparat schmolz bei 160—]61°.
3,700 mg Subst. gaben 7,210 mg C02 und 2,052 mg H20
Ci0H14O4N2 Ber. C 53,09 H 6,24%Gef. C 53,18 H 6,21%
Md = + 29° (c = 0,3 in Wasser)
Brucinsalz XXXVI. 1,5 g rohe, nicht destillierte L-Threonsäure
wurden in 20 ccm heissem Methanol gelöst, 4,7 g Brucin zugegebenund die Lösung 15 Minuten auf dem Wasserbad erwärmt. Nach dem
Abkühlen kristallisierte das Brucinsalz XXXVI der L-Threonsäure
im Verlauf einiger Stunden aus. Es wurde in Wasser gelöst, mit
Chloroform mehrmals ausgeschüttelt und aus der eingeengten wäss-
rigen Lösung durch Zugabe von Aceton kristallisiert. Das Analysen¬
präparat vom Smp. 211—212° (u. Zers.) wurde 12 Stunden bei
70° im Hochvakuum getrocknet.
3,784 mg Subst. gaben 8,452 mg C02 und 2,160 mg H20
C27H3409N2 Ber. C 61,12 H 6,46%Gef. C 60,95 H 6,39%
[<x]d = ~ 27° (° = !>3 in Wasser)
47

D, L-Threonsäure-lacton
Je 1 g analysenreines D- und L-Threonsäure-lacton (XXX und
XXXIV) wurden im Hochvakuum zusammengeschmolzen und hier¬
auf in wenig heissem Essigester gelöst. Beim Erkalten kristallisierte
das bei 48—50° schmelzende D,L-Lacton aus. (Falls die übersättigte
Lösung nicht kristallisiert, kann mit einer Spur D-Lacton angeimpft
werden.) Das sehr hygroskopische Präparat wurde zur Analyse über
Phosphorpentoxyd getrocknet und im Schweinchen eingewogen.
4,250 mg Subst. gaben 6,213 mg COa und 1,979 mg H20
C4H604 Ber. C 40,68 H 5,12%
Gef. C 39,89 H 5,21%
C4H604,1/8H20 Ber. C 39,90 H 5,21%
[a]D = 0° (c = 1 in Wasser)
D-Arabo-trioxyglutarsäure (XL) aus D-Mannuronsäure (XXXVII)8)
Die auf 0° gekühlte Lösung von 5,9 g ( = 11 Millimol) mannuron-
saurem Barium in 60 ccm Wasser wurde zu einer Lösung von 10 g
(=31 Millimol) krist. Bariumhydroxyd in 120 ccm eiskaltem Was¬
ser gegeben. Die Oxydation erfolgte bei Zimmertemperatur durch
reinen Sauerstoff unter intensiver Durchwirbelung mittels eines
Vibrators. Nach 20 Stunden waren 350 ccm Sauerstoff (ber. 273 ccm
02) verbraucht worden; das überschüssige Barium wurde durch
Einleiten von Kohlendioxyd gefällt. Die Lösung wurde durch Celit
und anschliessend durch 80 ccm Wofatit KS nitriert. Das fast farb¬
lose Filtrat wurde zusammen mit dem Waschwasser im Vakuum
auf dem Wasserbad zur Trockene eingedampft. Der bräunliche
Rückstand wog 2,5 g.
Dibrwinsalz XLI. Der Rückstand von 2,5 g wurde in 40 ccm
Wasser aufgenommen, mit einer Lösung von 12 g Brucin in 20 ccm
heissem Methanol versetzt und anschliessend das Methanol auf dem
Wasserbad abgetrieben. Nach Erkalten wurde das überschüssige
8) Die durch Hydrolyse von Alginsäure nach H. A. Spoehr, Arch. Biochem.
14, 153 (1947) hergestellte Mannuronsäure lag als rohes Brucinsalz vor. Das
aus dem Brucinsalz bereitete mannuronsäure Barium wurde in ungereinigtemZustand für die Oxydation verwendet.
48

Brucin mehrmals mit Chloroform ausgeschüttelt und die wässrige
Lösung im Vakuum eingedampft. Das Brucinsalz XLI der Arabo-
trioxyglutarsäure kristallisierte aus wenig Wasser im Verlaufe eini¬
ger Tage in Warzen (3,8 g = 39% der Theorie). Das aus Wasser
umkristallisierte Analysenpräparat vom Smp. 152—154° (u. Zers.)wurde 24 Stunden bei 75° im Hochvakuum getrocknet und im
Schweinchen eingewogen.
3,744 mg Subst. gaben 8,656 mg C02 und 2,064 mg H20
C51H60O15N4 Ber. C 63,22 H 6,24%Gef. C 63,09 H 6,17%
[<x]D = - 17,5° (c = 0,8 in Wasser)
Die nicht kristallisierenden Anteile aus der Herstellung des
Brucinsalzes XLI wurden auf freie Säure (0,8 g) verarbeitet, die
nach der unten gegebenen Vorschrift 130 mg D-Arabo-trioxyglutar-säure-dibenzimidazol (XLIII) vom Smp. 227—229° (u. Zers.) lie¬
ferte.
D-Arabo-trioxyglutarsäure (XL) aus D-Galacturonsäure (XXXVIII)
Dibenzimidazol XLIII. Es wurde nach der bekannten Methode
1,13 g (= 5,8Millimol) D-Galacturonsäure (XXXVIII) in 40 ccm
Wasser gelöst und mittels Sauerstoff in Gegenwart von 3 g ( = 29
Millimol) krist. Bariumhydroxyd oxydiert. Nach 48 Stunden waren
180 ccm Sauerstoff (ber. 145 ccm 02) verbraucht worden. Nach der
Behandlung mit C02 und 20 ccm Wofatit KS wurde die rohe, freie
Säure von ca. 1 g Gewicht in das Dibenzimidazol XLIII übergeführt.Das einmal aus Methanol-Wasser umkristallisierte Derivat wog
0,54 g und schmolz bei 226—228° (u. Zers.).
D-Arabo-trioxyglutarsäure (XL) aus D-Glucuron (XXXIX)
Dikaliumsalz XLII. Nach der üblichen Vorschrift wurden 1 g
(=6,6 Millimol) Glucuron (XXXIX) und 1,1g (= 20 Millimol)
Kaliumhydroxyd in 40 ccm Wasser gelöst und mit Sauerstoff oxy¬
diert. Nach 28 Stunden waren 150 ccm Sauerstoff verbraucht wor¬
den (ber. 140 ccm 02). Nach der Filtration durch 40 ccm Wofatit
49

KS und Abtreiben der Ameisensäure wurde die freie Arabo-trioxy-
glutarsäure XL in 10 com Wasser gelöst und mit 0,8 g Kaliumcar-
bonat versetzt. Der im Vakuum auf die Hälfte eingeengten Lösungwurde bis zur beginnenden Trübung Alkohol zugesetzt, worauf das
Dikaliumsalz XLII in Nadeln (0,17 g = 12% der Theorie) auskri¬
stallisierte. Das mehrmals aus Wasser-Äthanol umkristallisierte
Analysenpräparat wurde 24 Stunden bei 50° im Hochvakuum
getrocknet.
3,930 mg Subst. gaben 3,335 mg C02 und 0,843 mg H20
C5H607K Ber. C 23,43 H 2,36%Gef. C 23,15 H 2,40%
[a]D = - 5,5° (c = 1,2 in Wasser)
Aus den nichtkristallisierenden Mutterlaugen des Dikalium-
salzes XLII konnten noch 0,17 g Dibenzimidazol XLIII vom Smp.227—229° (u. Zers.) gewonnen werden.
D-Arabo-trioxyglutarsäure-dibenzimidazol (XLIII)
0,5 g ( = 2,8 Millimol) D-Arabo-trioxyglutarsäure-diamid9), 0,8 g
( = 7,4 MiUimol) o-Phenylendiamin, 1,1 ccm konz. Salzsäure, 0,6 ccm
konz. Phosphorsäure und 2,5 ccm Diäthylenglykol wurden unter
Umschütteln zusammengegeben und bei 100° gehalten, bis eine
homogene Lösung entstand. Die Lösung wurde zwei Stunden auf
135° ±5° im Ölbad erwärmt, wobei Wasser abgespalten wurde und
die Reaktionsmischung sich dunkel färbte. Noch heiss wurde mit
ca. 5 ccm Wasser verdünnt und nach Zugabe von wenig Aktivkohle
durch Celit filtriert. Dem noch warmen Filtrat wurde bis zur alkali¬
schen Reaktion tropfenweise konz. Ammoniak zugegeben, worauf
das Dibenzimidazol XLIII als brauner Niederschlag ausfiel. Das
abfiltrierte, mit Wasser, Aceton und Äther gewaschene Benzimida-
zol XLIII wurde in 50 ccm Wasser suspendiert, in der Wärme durch
Zugabe von verdünnter Salzsäure gelöst und nach dem Entfärben
mit Norit wieder mit Ammoniak gefällt. Das getrocknete Di¬
benzimidazol wog 0,75 g. Das mehrmals aus Wasser-Methanol
umkristallisierte, bei 232—234° (u. Zers.) schmelzende Analysen¬
präparat wurde 24 Stunden bei 70° im Hochvakuum getrocknet.
9) Hergestellt von D. Spitz; vgl. Diss. ETH (1951).
50

3,648 mg Subst. gaben 8,394 mg C02 und 1,641 mg H20
C17H1603N4 Ber. C 62,95 H 4,97%Gef. C 62,79 H 5,03%
[«Id = - 47° (c = °'3 in Methanol)
Pikrat XLIV. Die aus einer erkalteten Lösung von 60 mg D-
Arabo-trioxyglutarsäure-dibenzimidazol (XLIII) und 75 mg Pikrin¬
säure in 5 ccm Alkohol-Wasser (1/1) ausgeschiedenen feinen, gelbenNädelchen vom Smp. 228—230° (u. Zers.) wurden einige Male aus
Methanol umkristallisiert und zur Analyse 48 Stunden bei 60° im
Hochvakuum getrocknet.
3,762 mg Subst. gaben 6,164 mg C03 und 0,910 mg H20
C29H22O17N10 Ber. C 44,51 H 2,83%Gef. C 44,71 H 2,71%
[«Id = - 14° (° = °>2 in Methanol)
Ribo-trioxyglutarsäure-dibenzimidazol (XLVI)
Nach der oben gegebenen Vorschrift wurden aus 0,55 g Ribo-
trioxyglutarsäure-diamid (XLV)9) 0,74 g Dibenzimidazol XLVI vom
Smp. 258—260° (u. Zers.) gewonnen. Das Analysenpräparat wurde
48 Stunden bei 60° im Hochvakuum getrocknet.
3,630 mg Subst. gaben 8,402 mg C02 und 1,579 mg H20
C17H1603N4 Ber. C 62,95 H 4,97%Gef. C 63,17 H 4,88%
[a]D = 0°")
Pikrat XLVII. Das 48 Stunden bei 60° im Hochvakuum getrock¬nete Analysenpräparat schmolz bei 225—227° (u. Zers.).
3,690 mg Subst. gaben 8,402 mg C02 und 0,943 mg H30
C29H22O17NI0 Ber. C 44,51 H 2,83%Gef. C 44,55 H 2,86%
[a]D = 0o")
Xylo-trioxyglutarsäure-dibenzimidazol (XLIX)
Nach bekannter Vorschrift wurden aus 0,16 g Xylo-trioxyglutar-
säure-diamid9) (XLVIII) 0,165 g Dibenzimidazol XLIX vom Smp.230—231° (u. Zers.) gewonnen. Das Präparat wurde zur Analyse48 Stunden bei 60° im Hochvakuum getrocknet.
10) Optisch inaktiv.
51

4,230 mg Subst. gaben 9,772 mg C02 und 1,865 mg H20
C17H1603N4 Ber. C 62,95 H 4,97%Gef. C 63,04 H 4,93%
[*]d = 0°10)
Pikrat L. Das in feinen gelben Nädelchen vom Smp. 220—222°
(u. Zers.) kristallisierende Präparat wurde zur Analyse 24 Stunden
bei 70° im Hochvakuum getrocknet.
3,750 mg Subst. gaben 6,150 mg C02 und 0,985 mg H20
CmHjjO17N10 Ber. 0 44,51 H 2,83%Gef. 0 44,76 H 2,94%
[a]D = 0°10)
2,5-Anhydro-D-arabonsäure (LH) aus 3,6-Anhydro-D-gIucose (LI)11)
Brucinsalz LUI. lg ( = 6,2 Millimol) Anhydro-D-glucose LI
wurde in 40 com Wasser gelöst und zu 1,2 g (= 20 Millimol) Kalium¬
hydroxyd in 50 ccm Wasser gegeben, um bei Zimmertemperatur in
Sauerstoffatmosphäre und unter Verwendung eines Vibromischers
oxydiert zu werden. Nach 2 Tagen wurde die Oxydation abgebro¬
chen, nachdem ca. 170 ccm Sauerstoff (ber. 150 ccm 02) verbraucht
worden waren. Die Lösung wurde durch 30 ccm Wofatit KS filtriert
und das Filtrat zusammen mit 70 ccm Waschwasser aufdem Wasser¬
bad im Vakuum zur Trockene eingedampft. Der gelbliche Rück¬
stand (0,8 g) wurde in 10 ccm Wasser gelöst und mit 2,8 g Brucin
in 10 ccm heissem Methanol versetzt; das Methanol auf dem Wasser¬
bad abgetrieben und die erkaltete Lösung zur Entfernung des über¬
schüssigen Brucins mehrmals mit Chloroform ausgeschüttelt. Aus
der stark eingeengten wässrigen Lösung kristallisierte das Brucin¬
salz LUI im Verlauf mehrerer Tage in Warzen aus (1,2 g = 34%der Theorie).
Das Analysenpräparat vom Smp. 126—127° wurde mehrmals aus
Wasser und zuletzt aus Alkohol und Wasser (5/1) umkristallisiert
und 48 Stunden bei 60° im Hochvakuum getrocknet.
3,820 mg Subst. gaben 8,100 mg C02 und 2,193 mg H20
C28H3109N2,2H20 Ber. C 58,11 H 6,61%Gef. C 57,87 H 6,43%
[«]D = - 34,1° (c = 0,4 in Wasser)
n) Die 3,6-Anhydro-D-glucose wurde nach H. Ohle, L. von Vargha und
H. Erlbach, B. 61, 1211 (1928) hergestellt.
52

Herstellung und Abbau von Hexcisazonen und Diels'schen Anhydro-hexose- osazonen
D-Fructose-benzylphenyl-phenylosazon (LXII) aus D-Fructose
(LIX)12)
Zu einer Lösung von 6,2 g ( == 34 Millimol) D-Fructose (LIX) in
40 com 50-proz. Essigsäure wurde eine Lösung von 21 g ( = 120 Milli¬
mol) a-Benzylphenylhydrazin13) in 55 ccm 80-proz. Essigsäure gege¬
ben. Die klare gelbe Lösung wurde wenige Minuten auf dem Wasser¬
bad erwärmt, wobei sie sich unter Rotfärbung trübte. Unter häufi¬
gem Umschütteln wurde 20 Minuten bei Zimmertemperatur stehen
gelassen und dann mit 200 ccm Wasser versetzt. Nach 24 Stunden
wurde die wässrige Schicht vom ausgeschiedenen schwarzen, zäh¬
flüssigen Ol abgegossen, aus demi — nach einmaligem Umfallen aus
Methanol-Wasser und Lösen in wenig Methanol — im Verlauf eini¬
ger Tage feine gelbe Nädelchen kristallisierten. Die Isolierung der
geringen Mengen Osazon LXII war schwierig, da sich die sehr zäh¬
flüssigen Nebenprodukte nur schwer abfiltrieren und mit Alkohol,
bzw. Äther auswaschen liessen. Nach einmaligem Umkristallisieren
aus Aceton wog das Osazon LXII 1,8 g (= 16% der Theorie). Zur
Analyse wurde das Präparat vom Smp. 194—195° (u. Zers.) mehr¬
mals aus Aceton umkristallisiert und 24 Stunden bei 70° im Hoch¬
vakuum getrocknet.
3,797 mg Subst. gaben 9,222 mg C02 und 2,225 mg H202,776 mg Subst. gaben 0,315 ccm N2 (24°, 730 mm)
C26H2804:N4,1/4H20 Ber. C 66,28 H 6,34 N 12,37%Gef. C 66,28 H 6,56 N 12,53%
Md = - 30° (c = 0,2 in Methanol)
Nach mehrwöchigem Stehen konnten aus den Mutterlaugenweitere gelbliche, grob nadeiförmige Kristalle — vermutlich Benzal-
12) Entsprechende Ansätze wurden auch mit D-Glucose (I) durchgeführt,wobei das gleiche Osazon LXII in schlechterer Ausbeute isoliert werden
konnte.
13) Das verwendete Benzylphenylhydrazin wurde nach der Vorschrift
von R. Ofner, M. 25, 593 (1904) frisch hergestellt und im Hochvakuum bei
170—172° destilliert.
53

benzylphenylhydrazon14) — (ca. 1 g) isoliert werden, die nach Um¬
kristallisieren aus Aceton bei 107—108° schmolzen. Zur Analysewurde das Präparat 24 Stunden bei Zimmertemperatur im Hoch¬
vakuum getrocknet.
3,746 mg Subst. gaben 11,427 mg C02 und 2,247 mg H20
C20H18N2 Ber. C 83,75 H 6,35%Gef. C 83,25 H 6,71%
Versuch zur Herstellung von D-Fructose-benzylphenylosazon (LXI)aus D-Glucoson (LX)15)
1,2 g (= 6,7 Millimol) D-Glucoson (LX), gelöst in 30 com 50-proz.
Essigsäure, wurde mit 3 g ( = 17 Millimol) frisch bereitetem Benzyl-
phenylhydrazin in 50 ccm Alkohol versetzt. Nach kurzem Erwär¬
men auf dem Wasserbad und Zugabe von 200 ccm Wasser bildete
sich ein dunkler, zähflüssiger Niederschlag, von dem nach 2tägigemStehen die überstehende wässrige Schicht abgegossen wurde. Das
zähflüssige Öl wurde in 20 ccm Methanol aufgenommen, konnte
aber auch nach längerem Stehen weder aus Methanol noch Aceton
zum Kristallisieren gebracht werden.
3,6-Anhydro-D-fructose-benzylphenyIhydrazon (LXV) aus
3,6-Anhydro-D-glucose (LI) n)
0,7 g ( = 4,2 Millimol) 3,6-Anhydro-D-glucose (LI), gelöst in 10
ccm 50-proz. Essigsäure, wurden mit 3,0 g ( = 17 Millimol) Benzyl-
phenylhydrazin, gelöst in 20 ccm 50-proz. Essigsäure, versetzt und
unter zeitweiligem Umschütteln 1 Stunde stehen gelassen. Die
Lösung färbte sich schnell rot, und bald bildete sich ein dunkler,
öliger Niederschlag. Nach Zugabe von 50 ccm Wasser erfolgte die
weitere Aufarbeitung wie im vorstehend beschriebenen Ansatz zur
Herstellung von Fructose-benzylphenyl-phenylosazon. Es konnten
14) Nach R. Ojner, M. 25, 593 (1904), konnte aus älterem Benzylphenyl-
hydrazin neben Phenylhydrazin das bei 111D schmelzende Benzal-benzyl-
phenylhydrazon isoliert werden.
15) Das verwendete D-Glucoson wurde nach der Vorschrift von R. Weiden¬
hagen, Z. Wirtschaftsgruppe Zuckerind. 87, 711 (1937), aus D-Glucose her¬
gestellt.
54

aber nur geringe Mengen farbloses Benzylphenylhydrazon LXV
vom Smp. 156—157° isoliert werden. Zur Analyse wurde das mehr¬
mals aus Methanol, bzw. Alkohol umkristallisierte Präparat 24 Stun¬
den bei 60° im Hochvakuum getrocknet.
3,666 mg Subst. gaben 8,961 mg C02 und 2,155 mg H20
C19H2204N2 Ber. C 66,65 H 6,48%Gef. C 66,71 H 6,58%
[«Id = - 18>4° (c = °>25 in Methanol)
Auch aus zwei weiteren Ansätzen mit gleichen Mengen Anhydro-
L>-glucose LI und Benzylphenylhydrazin konnte trotz längeremErwärmen (1/2, resp. l1/2 Stunden) auf dem Wasserbad kein Osazon
isoliert werden.
3,6-Anhydro-D-psicose-benzylphenyl-phenylosazon (LXIII)16) aus
D-Fructose-benzylphenyl-phenylosazon (LXII)
0,54 g ( = 1 Millimol) Benzylphenyl-phenylosazon LXII aus D-
Fructose wurden in 15 ccm heissem Methanol suspendiert, mit 0,04
ccm 20-proz. Schwefelsäure versetzt und 9 Stunden am Rückfluss
gekocht, worauf die gegen das Ende rotbraun gewordene Reaktions-
lösung mit ca. 1 g fein pulverisiertem Bariumcarbonat kurz auf¬
gekocht wurde. Nach Filtration durch wenig Celit wurde das neu¬
trale Filtrat auf dem Wasserbad im Wasserstrahlvakuum zur Trok-
kene eingedampft. Der braune Rückstand wurde in 10 ccm heissem
Alkohol aufgenommen, woraus nach dem Abkühlen das Anhydro-osazon LXIII (0,11 g) in hellgelben Nädelchen kristallisierte. Das
Analysenpräparat schmolz nach mehrfachem Umkristallisieren aus
Alkohol, bzw. Aceton bei 205—206° (u. Zers.). Die Mischprobe mit
reinem Ausgangsmaterial ergab eine deutliche Depression. Zur Ana¬
lyse wurde 48 Stunden bei 70° im Hochvakuum getrocknet.
gaben 9,437 mg C02 und 2,046 mg H20
gaben 0,405 ccm N2 (24°, 721 mm)
Ber. C 69,75 H 6,09 N 13,02%Gef. C 69,45 H 6,17 N 12,97%
(c = 0,15 in Methanol)
16) InAnalogie zum 3,6-Anhydro-I)-psieose-phenylosazonals 3,6-Anhydro-D-psicose-benzylphenyl-phenylosazon formuliert.
3,708 mg Subst.
3,404 mg Subst.
c25H26o3isr4
[a]D =-210°
55

3,6-Anhydro-4,5-isopropyliden-D-psicose-benzylphenyl-
phenylosazon (LXIV)17)
150 mg Anhydro-benzylphenyl-phenylosazon LXIII, gelöst in
10 ccm wasserfreiem Aceton, wurden mit einem Tropfen konz.
Schwefelsäure versetzt und über Nacht stehen gelassen. Zur Ent¬
fernung der Schwefelsäure wurde die rotgefärbte Lösung mit ca.
2 g pulverisiertem Bariumcarbonat 2 Stunden geschüttelt und an¬
schliessend durch Celit filtriert. Nach dem Einengen der neutralen
Lösung kristallisierte das Aceton-Derivat LXIV in gelben Nadeln
(35 mg) aus. Das Präparat, das nach dreimaligem Umkristallisieren
aus Aceton bei 132—133° schmolz, wurde zur Analyse 48 Stunden
bei Zimmertemperatur im Hochvakuum getrocknet.
3,689 mg Subst. gaben 9,616 mg C()2 und 2,202 mg H203,456 mg Subst. gaben 0,577 ccm N2 (22°, 719 mm)
C28H30O3N4 Ber. C 71,47 H 6,43 N 11,91%Gef. C 71,14 H 6,68 N 11,94%
[a]D = - 70,6° (c = 0,2 in Methanol)
Isopropyliden-Derivate18) aus 3,6-Anhydro-D-psicose-phenylosazon
(LVII)
Labile Modifikation: 24,5 g ( = 72 Millimol) des nach Diels (1. c.)
hergestellten 3,6-Anhydro-D-psicose-phenylosazons (LVII) wurden
in 700 ccm wasserfreiem Aceton gelöst, mit 3,9 ccm konz. Schwefel¬
säure versetzt und über Nacht stehen gelassen. Zur Entfernung der
Schwefelsäure wurde die dunkelrot verfärbte Lösung 24 Stunden
mit 140 g fein pulverisiertem Bariumcarbonat geschüttelt und der
Niederschlag durch Celit abfiltriert. Aus dem neutralen Filtrat, das
am Wasserstrahlvakuum auf ca. 100 ccm eingeengt worden war, kri¬
stallisierte nach Animpfen das unreine labile Aceton-Derivat in
roten Warzen (8,1 g = 29,5 % der Theorie) im Verlauf einiger Stun¬
den aus. Zur Reinigung wurde das Isopropyliden-Derivat in 250
ccm heissem Benzol gelöst und durch eine Säule mit 100 g Alumi¬
niumoxyd der Aktivität III filtriert. Nach dem Abkühlen der
benzolischen Lösung kristallisierte das 3,6-Anhydro-isopropyliden-D-psicose-phenylosazon in feinen gelben Nadeln, deren Schmelz-
17) Unbewiesene Konstitution, vgl. dazu Seite 29.
18) Konstitution unbekannt, vgl. dazu Seite 32.
56

punkt nach mehrmaligem Umkristallisieren aus Methanol-Benzol,bzw. Methanol von 159—161° auf 167—168° (u. Zers.) stieg. Das
Analysenpräparat wurde 24 Stunden bei 70° im Hochvakuum
getrocknet.
3,754 mg Subst. gaben 9,120 mg C02 und 2,152 mg H203,339 mg Subst. gaben 0,443 mg com N2 (24°, 728 mm)
C21H2403N4 Ber. C 66,30 H 6,36 N 14,72%Gef. C 66,30 H 6,42 N 14,61%
Md = - 80° (c = 0,12 in Methanol)
Stabile Modifikation. 6,0 g ( = 15,9 Millimol) labiles 3,6-Anhydro-
isopropyliden-D-psicose-phenylosazon18) aus obigem Ansatz wurde
in 250 com Acetonitril 12 Stunden am Rückfluss gekocht, wobei
sich die anfänglich gelbe Lösung nach hellrot verfärbte. Das Aceto¬
nitril wurde auf dem Wasserbad im Vakuum vollständig abgedampftund der Rückstand in ca. 100 ccm heissem Methanol aufgenommen,woraus nach dem Abkühlen, im Verlauf einiger Stunden, das stabile
Isopropyliden-Derivat18) in orangen Nadeln kristallisierte. Nach
Umkristallisieren aus Methanol, bzw. Aceton wurde das 3,6-Anhydro-
4,5-isopropyliden-D-psicose-phenylosazon vom Smp. 219—220° (u.
Zers.) zur Analyse 24 Stunden bei 70° im Hochvakuum getrocknet.
3,730 mg Subst. gaben 9,049 mg C02 und 2,123 mg H20
2,948 mg Subst. gaben 0,389 com N2 (23° 731 mm)
C21H2403N4 Ber. C 66,30 H 6,36 N 14,73%Gef. C 66,21 H 6,37 N 14,63%
[<x]D = - 175° (c = 0,1 in Methanol)
Versuch zur N-Benzylierung vom 3,6-Anhydro-4,5-isopropyliden-
D-psicose-phenylosazon
2,5 g ( = 6,6 Millimol) stabiles Anhydro-isopropyliden-D-psicose-
phenylosazon19) wurden in 10 ccm wasserfreiem Benzol gelöst und
25 g (= 115 Millimol) Benzyljodid zugegeben. Im Verlauf von 3
Stunden wurden 14 g ( = 60,5 Millimol) frisch bereitetes und getrock¬netes Silberoxyd in Portionen von ca. ] g zugesetzt und mit Hilfe
eines Vibromischers in der Lösung fein verteilt. Die gegen das Ende
der Silberoxyd-Zugabe leicht erwärmte Reaktionsmischung wurde
19) Gleiche Ansätze wurden mit labilem Isopropyliden-Derivat durch¬
geführt und dabei die gleichen Ergebnisse erzielt.
57

nach 14 Stunden durch Celit filtriert und das dunkelrote Filtrat bis
zur beginnenden Trübung mit Petroläther (ca. 50 ccm) versetzt und
auf eine Säule von 100 g Aluminiumoxyd (Aktivität III) in Petrol-
äther-Benzol (2:1) gegeben. Nach einer anfänglich farblosen Frak¬
tion von Nebenprodukten, die verworfen wurde, konnte weiter mit
Benzol eine rotgefärbte Fraktion (1,4 g) eluiert werden, die erneut in
Benzol-Petroläther (1:2) aufgenommen und an 50 g Aluminium¬
oxyd (Aktivität III) chromatographiert wurde. Mit Benzol wurden
420 mg einer rötlichgelben, zähflüssigen Masse eluiert, die auch nach
längerem Stehen nicht kristallisierte. Zur weiteren Reinigung wurde
diese Fraktion nochmals an 10 g Aluminiumoxyd III chromato¬
graphiert und eine Mittelfraktion des nicht kristallisierenden Petrol-
äther-Benzol-Eluates zur Analyse 24 Stunden bei 80° im Hoch¬
vakuum getrocknet.
3,388 mg Subst. gaben 9,250 mg C02 und 1,985 mg H203,628 mg Subst. gaben 0,219 ocm N2 (22°, 732 mm)
Gef. C 74,51 H 6,56 N 6,73%
C35H3603N4 Ber. C 74,97 H 6,47 N 9,99%
C28H30O3N4 Ber. C 71,47 H 6,43 N 11.91%
Eine Mittelfraktion des Benzol-Äther-Eluates wurde zur Analyse24 Stunden bei 80° im Hochvakuum getrocknet.
3,556 mg Subst. gaben 9,613 mg CC)2 und 2,115 mg H203,688 mg Subst. gaben 0,219 ccm N2 (22°, 732 mm)
Gef. C 73,77 H 6,66 N 6,62%
Aus keiner Fraktion des Chromatogramms konnte ein kristalli¬
siertes Derivat isoliert werden. Eine weitere Reinigung mit Hilfe
der Kugelrohrdestillation im Hochvakuum misslang infolge der
Zersetzlichkeit der Reaktionsprodukte.
2,5-Anhydro-D-lyxonsäure (LXXI) aus 3,6-Anhydro-D-galactose-phenylosazon (LXX)
Zu einer Suspension von 4,0 g ( = 11 Mülimol) Anhydro-D-galac-
tose-phenylosazon (LXX) in 100 ccm Eisessig wurden unter häufi¬
gem Umschütteln bei Zimmertemperatur 6,0 g ( = 89 Mülimol)Natriumnitrit im Verlauf von 4 Stunden in kleinen Portionen zuge¬
geben, während das Anhydro-osazon unter steter Gasentwicklung
langsam in Lösung ging. Das anfänglich tief dunkelrote Reaktions-
58

gemisch hellte sich dabei bis gegen das Ende der Nitrit-Zugabeimmer mehr auf. Nach weiterem 3 stündigem Stehen wurde die
klare Lösung im Wasserstrahlvakuum, unter vorsichtigem Erwär¬
men, auf Sirupdicke eingeengt, 150 ccm Wasser zugegeben und das
ausgeschiedene, rötliche Öl mehrmals mit Äther ausgeschüttelt.Die wässrige Lösung wurde durch 120 ccm Wofatit KS nitriert und
das noch hellrote Mitrat zusammen mit ca. 250 ccm Waschwasser
im Vakuum bei Wasserbadtemperatur vollständig zur Trockene
eingedampft. Der bräunliche Rückstand von 880 mg reagiertesauer und reduzierte Fehling'sehe Lösung in der Wärme.
Brucinsalz LXXII. Der saure Oxydationsrückstand (880 mg)wurde in 10 ccm warmem Wasser aufgenommen und mit 2,8 g
Brucin in 20 ccm heissem Methanol versetzt, das Methanol nach
kurzem Erwärmen auf dem Wasserbad im Vakuum abgetriebenund überschüssiges Brucin nach dem Erkalten der wässrigen Lösungdurch mehrmaliges Ausschütteln mit Chloroform entfernt. Die wäss¬
rige Lösung wurde im Vakuum auf Sirupdicke eingeengt, worauf
das Brucinsalz LXXII nach einigen Stunden langsam auszukristalli-
sieren begann. Zusammen mit einer zweiten Kristallfraktion aus
den Mutterlaugen wog das rohe Brucinsalz LXXII 2,3 g (= 39%der Theorie). Das Brucinsalz, das unter Zersetzung unscharf bei
250—253° schmolz, wurde zur Analyse mehrmals aus Wasser umkri¬
stallisiert und 12 Stunden bei 90° im Hochvakuum getrocknet.
3,682 mg Subst. gaben 8,357 mg C02 und 2,141 mg H20
C28H3409N2 Ber. C 61,98 H 6,32%Gef. C 61,94 H 6,51%
[a]D = _ 15,6° (c = 0,4 in Wasser)
2,5-Anhydro-T>-lyxonsäure-meihylester (LXXIII). 120 mg aus
rohem Brucinsalz LXXII zurückgewonnene Säure LXXI wurden
in 10 ccm absolutem Methanol gelöst und mit Diazomethan-Lösungversetzt, bis keine nennenswerte Gasentwicklung mehr auftrat.
Nach 2 Stunden wurde das Methanol im Wasserstrahlvakuum abge¬trieben und das braune, zähflüssige Öl im Kugelrohr bei 120—170°
im Hochvakuum während einer Dauer von ca. 2 Stunden destilliert.
Die 70mgwiegendeMittelfraktion vomKp.140-170° wurde erneut im
Kugelrohr bei 130-170° im Hochvakuum destilliert und hievon die
farblose Mittelfraktion vom Kp. 150-160° in die Analyse gegeben.
59

3,740 mg Subst. gaben 6,036 mg 0O2 und 2,183 mg H20
C6H10O5 Ber. C 44,44 H 6,22%Gef. C 44,04 H 6,53%
Der im Analysenröhrchen eingeschmolzene, zähflüssige Ester
kristallisierte nach einigen Tagen.
2,5-Anhydro-T>-lyxonsäure-benzimidazol. Analog den früher be¬
schriebenen Methoden20) wurden 390 mg (= 2,6 Millimol) rohe
Anhydro-D-lyxonsäure LXXII — durch Zersetzung ihres Brucin-
salzes gewonnen — mit 310 mg ( = 2,9 Millimol) o-Phenylendiaminbei 135° umgesetzt. Aus dem schwarzen Reaktionsgemisch konnte
nach den üblichen Aufarbeitungsmethoden kein kristallisiertes
Benzimidazol erhalten werden. Das in Wasser gelöste Reaktions¬
produkt wurde im Vakuum zur Trockene eingedampft und der
dunkle Rückstand mehrmals mit heissem Alkohol extrahiert, wobei
durch stufenweises Einengen mitgelöstes Ammonchlorid von den
geringen Mengen (ca. 30 mg) unreinem Benzimidazol abgetrenntwerden konnten. Infolge Substanzmangels konnte das rohe Derivat
vom Smp. 216—217° nicht analysenrein erhalten werden.
2,5-Anhydro-D-ribonsäure (LXXIV) aus 3,6-Anhydro-D-psicose-
phenylosazon (LVII)
Analog der vorstehend beschriebenen Herstellung von Anhydro-
D-lyxonsäure (LXXI) wurden 2,1 g (=- 6 Millimol) 3,6-Anhydro-D-
psicose-phenylosazon (LVII) mit 3 g ( = 43 Millimol) Natriumnitrit
bei Zimmertemperatur im Verlaufe von 3 Stunden oxydiert. Die
Aufarbeitung erfolgte unter Verwendung von 70 ccm Wofatit KS
und führte zu 200 mg bräunlichem Rückstand, der ebenfalls sauer
reagierte und Fehling'sche Lösung in der Wärme reduzierte.
Brucinsalz LXXV. Der saure Rückstand von 200 mg Gewicht
wurde in wässriger Lösung mit 650 mg Brucin versetzt, kurz auf
dem Wasserbad erwärmt und nach dem Erkalten mehrmals mit
Chloroform ausgeschüttelt. Der Rückstand der im Vakuum einge¬
dampften wässrigen Lösung wog ca. 400 mg, woraus nach Aufneh¬
men in ganz wenig heissem Wasser das Brucinsalz LXXV im Ver¬
lauf einiger Stunden auskristallisierte. Zusammen mit weiteren
20) Vgl. Seite 50.
60

Kristallen aus der Mutterlauge wog das Derivat (LXXV) 310 mg
( = 9,5 % der Theorie). Nach mehrmaligem Umkristallisieren aus
Wasser-Alkohol schmolz das Brucinsalz LXXV unscharf unter Zer¬
setzung bei 260—265°. Das Analysenpräparat wurde 14 Stunden
bei 90° im Hochvakuum getrocknet.
3,598 mg Subst. gaben 8,090 mg C02 und 2,133 mg H20
C28H3409N2,1/4H20 Ber. C 61,46 H 6,36
Gef. C 61,36 H 6,63
[œ]D = - 41° (o = 0,2 in Wasser)
D-Fructose-phenylosotriazol (I^XXVII) aus D-Fructose-
phenylosazora (LXXVI)
Zu einer Suspension von 3,0 g ( = 8,3 Millimol) D-Fructose-
phenylosazon (LXXVI) in 40 ccm Eisessig wurden bei Zimmer¬
temperatur 5 ccm Isoamylnitrit21) gegeben, wobei sich die Reak¬
tionsmischung unter leichtem Erwärmen und Gasentwicklung rasch
dunkelrot verfärbte. Unter Eiskühlung wurden allmählich weitere
20 ccm Isoamylnitrit in Portionen von ca. 5 ccm zugegeben. Nach
8 Stunden wurde die klare, rote Lösung im Wasserstrahlvakuum
auf Sirupdicke eingeengt, mit 100 ccm Wasser versetzt und das
dabei ausgefallene dunkle, harzige Öl mit Äther dreimal ausge¬
schüttelt. Der braune Rückstand der im Wasserstrahlvakuum
sorgfältig zur Trockene eingedampften wässrigen Lösung wog 1,1g.Nachdem er in wenig heissem Wasser aufgenommen worden war,
kristallisierte sofort ein Teil des Oxydationsrückstandes aus. Im
ganzen konnten 0,29 g ( = 13 % der Theorie) kristallisiertes PräparatLXXVII isoliert werden, das nach fünfmaligem Umkristallisieren
aus Alkohol bei 195—196° schmolz. Zur Analyse wurde das Präparat24 Stunden bei 80° im Hochvakuum getrocknet.
gaben 7,770 mg C02 und 1,961 mg H20
gaben 0,495 ccm N2 (23°, 728 mm)Ber. C 54,32 H 5,69 N 15,84%Gef. C 54,09 H 5,59 N 15,95%
(e = 1,1 in Pyridin)
21) Aus einem früheren Oxydationsansatz von 3,0 g D-Fructosephenyl-osazon und 5 g Natriumnitrit in Eisessig bei Zimmertemperatur konnten
keine kristallisierenden Verbindungen isoliert werden.
3,920 mg Subst.
3,430 mg Subst.
C12H1504N3
'
[a]D =-82,6°
61

Kalïum-D-arabonat (V) aus D-Fructose-phenylosazon (LXXVI)
Die nicht kristallisierenden sauren Mutterlaugen aus dem vor¬
stehend beschriebenen Oxydationsansatz von D-Fructose-osazon
LXXVI wurden mit verdünnter Kalilauge vorsichtig neutralisiert,
im Wasserstrahlvakuum zur Trockene eingedampft und der braune
Rückstand mehrmals mit heissem Alkohol extrahiert. Aus dem in
Alkohol unlöslichen Rückstand (38 mg) kristallisierten — nach
Lösen in wenig Wasser und Versetzen mit Alkohol bis zur begin¬nenden Trübung — im Verlauf einiger Tage feine Nädelchen (ca.15 mg). Die Kristalle, vermutlich Kalium-D-arabonat (V), wurden
aus Wasser-Alkohol viermal umkristallisiert und zur Analyse 48
Stunden bei Zimmertemperatur im Hochvakuum getrocknet.
3,838 mg Subst. gaben 4,104 mg C02 und 1,527 mg H20
C5H906K Ber. C 29,40 H 4,44%Gef. C 29,18 H 4,45%
Die spez. Drehung konnte infolge Substanzmangels nicht be¬
stimmt werden.
62

Zusammenfassung
Unter den verschiedenen Methoden, nach welchen die Kohlen¬
stoffkette von Aldosen um ein C-Atom abgebaut werden kann, ist
die alkalische Oxydation mit molekularem Sauerstoffnach 0. Speng¬ler und A. Pfannenstiel nach Ausbeute und Einfachheit in der Aus¬
führung eine der besten. Der Abbau ist auch für Ketosen geeignet.Der Mechanismus dieses Abbaus ist infolge der komplizierten Umla-
gerungen, welche reduzierende Zucker in alkalischem Milieu erleiden,
bis heute nicht restlos abgeklärt.Meine eigenen Untersuchungen befassen sich mit dem Spengler-
schen Abbau von Hexosen, Hexose-Derivaten, Pentosen, Disaccha-
riden und Vitamin C. Die als Abbauprodukte anfallenden Aldon-
säuren konnten in Ausbeuten von 30—75% isoliert werden. Die
Säuren wurden in Form verschiedener, zum Teil erstmals beschrie¬
bener Derivate charakterisiert.
Für die durch oxydativen Abbau gewonnenen Tetronsäuren
wurde eine gute und einfache Methode zur Herstellung ihrer kristal¬
lisierten und bis anhin zum Teil noch unbekannten Lactone gefun¬den.
Im oxydativen Abbau von Hexuronsäuren zu Trioxyglutarsäu-ren wurde ein Weg gefunden, der die sichere Identifizierung von
14 der insgesamt 16 stereoisomeren Hexuronsäuren erlaubt. Die
erstmals hergestellten Dibenzimidazole und Dibenzimidazolpikrateder Trioxyglutarsäuren erwiesen sich als die zur Charakterisierung
geeignetsten Derivate.
Die Ergebnisse meiner Untersuchungen über den alkalischen
Abbau mit molekularem Sauerstoff sind in drei Publikationen
(Helv. 34, 2343 (1951); 35, 618, 958 (1952)) zusammengefasst.Im Zusammenhang mit der Konstitutionsaufklärung der Diels-
schen Anhydro-osazone wurde, ebenfalls nach der Spengler 'sehen
63

Methode, durch Abbau der 3,6-Anhydro-glucose die noch unbe¬
kannte 2,5-Anhydro-D-arabonsäure hergestellt.
2,5-Anhydro-D-ribonsäure und 2,5-Anhydro-D-lyxonsäure konn¬
ten aus den beiden Diels'sehen Anhydro-osazonen, dem 3,6-Anhydro
D-psicose-phenylosazon, bzw. 3,6-Anhydro-D-galactose-phenylosa-zon, durch oxydativen Abbau mittels salpetriger Säure hergestelltwerden. Die Brucinsalze der beiden Anhydro-pentonsäuren und der
Methylester der Anhydro-D-lyxonsäure sind kristallisiert.
Infolge Substanzmangels war es nicht möglich, die Benzimidazole
der Anhydro-säuren mit den von Link und Moore beschriebenen
Anhydro-pentonsäure-benzimidazolen zu vergleichen.In weiteren Versuchen, die ebenfalls im Zusammenhang mit der
Konstitutionsaufklärung der Diels 'sehen Anhydro-hexosazone ange¬
stellt wurden, sind Alkylierungen der Isopropyliden-Derivate des
Diels 'sehen „Anhydro-glucosazons" beschrieben. Die Herstellungvon Benzylphenyl-glucosazon, welche in Ergänzung dieser Arbeiten
versucht wurde, führte zum Ofner'sehen Benzylphenyl-phenyl-D-
glucosazon, aus dem ein bisher unbekanntes kristallisiertes An-
hydro-osazon und dessen Isopropyliden-Derivat gewonnen werden
konnten.
64

Lebenslauf
Am 18. Juni 1926 wurde ich als Sohn des Konrad Kreis von
Ermatingen, Kanton Thurgau, und der Maria, geborene Sauter,
in Ermatingen geboren. Nach Beendigung der dortigen Primär- und
Sekundärschulen besuchte ich die Kantonsschule in Frauenfeld, wo
ich im Sommer 1945 die Maturitätsprüfung des Typus C bestand.
Im Herbst des gleichen Jahres begann ich mit dem Studium der
Chemie an der Eidgenössischen Technischen Hochschule in Zürich
und erhielt im Winter 1949/50 das Diplom als Ingenieur-Chemiker.Seit April 1950 war ich im Laboratorium für organische Chemie
(Leitung Prof. Dr. L. Ruzicka) mit der Durchführung vorliegenderPromotionsarbeit beschäftigt.
April 1953
Konrad Kreis



















