Embed Size (px)
Citation preview

Z Rheumatol 2006 · 65:32–43
DOI 10.1007/s00393-006-0039-2
Online publiziert: 8. Februar 2006
© Springer Medizin Verlag 2006
B. Manger1, · E. Mengel2 · R. Schaefer3 · C. Haase4 · J. Seidel5 · H. Michels6
1 Universität Erlangen-Nürnberg, Medizinische Klinik III, Erlangen2 Universitäts-Kinderklinik, Mainz3 Universitätsklinikum, Medizinische Klinik und Poliklinik D, Münster4 Klinikum der Friedrich-Schiller Universität Jena, Klinik für Kinder- und Jugendmedizin,
Jena5 SRH - Waldklinikum Gera, Klinik für Kinder- und Jugendmedizin, Gera6 Rheumaklinik für Kinder und Jugendliche, Garmisch-Partenkirchen
M. Gaucher, M. Fabry und Mukopolysaccharidose Typ IWie kann der Rheumatologe diese Patienten erkennen?
Einleitung
Lysosomale Speicherkrankheiten sind
seltene erbliche Stoffwechselerkran-
kungen, bei denen der Mangel eines En-
zyms zur lysosomalen Speicherung des
nun nicht mehr abbaubaren Substrats
führt. Zu diesen Erkrankungen zählen
die beiden Sphingolipidosen M. Gau-
cher und M. Fabry, sowie eine Reihe von
Mukopolysaccharidosen sowie Oligosac-
charidosen.
Die erste Beschreibung einer Patientin
mit M. Gaucher findet sich in der 1882 er-
schienenen Dissertation des Dermatolo-
gen Philippe Charles Ernest Gaucher [13].
1898 beschrieb der Dermatologe Jona-
than Fabry erstmals das später nach ihm
benannte Krankheitsbild einer „purpura
haemorrhagica nodularis“ [12]. Erst Jahr-
zehnte später wurde jeweils die Zusam-
mensetzung des Speichermaterials aufge-
klärt und die bei M. Gaucher bzw. M. Fa-
bry gespeicherten Lipide Glukozerebrosid
[1] und Globotriaosylzeramid [31] wurden
isoliert und charakterisiert. In den 60er
Jahren des 20. Jahrhunderts konnte der
jeweils zugrunde liegende Defekt der ly-
sosomalen Enzyme, die durch das GBA-
Gen auf Chromosom 1q21 bzw. das α-Ga-
laktosidase A-Gen auf dem X-Chromo-
som kodiert werden, nachgewiesen wer-
den [4, 5]. Anschließend begann die Su-
che nach kausalen Behandlungsmöglich-
keiten, die schließlich zur Entwicklung
von Enzymersatztherapien führte.
Auch die verschiedenen Mukopolysac-
charidosen sind meist nach ihrem Erst-
beschreiber benannt, wobei sich das von
dem österreichischen Arzt Meinhard von
Pfaundler und seiner deutschen Assisten-
tin Gertrud Hurler 1919 erstmals beschrie-
bene Krankheitsbild des M. Hurler [17]
und die 1962 von einem amerikanischen
Augenarzt beschriebene milde oder Er-
wachsenenform, der M. Scheie [28], als
verschiedene Ausprägungsgrade der Mu-
kopolysaccharidose Typ I (MPS I) erwie-
sen und so heute als MPS I – Hurler und
MPS I – Scheie bezeichnet werden. Bei-
de Formen werden durch einen Defekt
des Iduronidase-Gens auf Chromosom
4p16.3 verursacht. Auch für MPS I konnte
inzwischen eine Enzymersatztherapie ent-
wickelt werden.
Präparate für eine Enzymersatzthera-
pie sind in Deutschland seit 1994 für den
M. Gaucher und seit 2001 für den M. Fa-
bry zugelassen, seit Juni 2003 auch für die
MPS I. Durch diese über lange Jahre er-
reichten Fortschritte in der Pathogenese-
und Therapieforschung sind einige lyso-
somale Speichererkrankungen damit heu-
te therapierbar geworden.
Die bisherige klinische Erfahrung
zeigt, dass durch eine frühzeitige The-
rapie sonst irreversible Komplikationen
verhindert werden können und daher
eine rechtzeitige Diagnosestellung an-
zustreben ist. Die Schlüsselsymptome
zu erkennen und an diese sehr seltenen
Erkrankungen zu denken, ist dabei oh-
ne Zweifel eine Herausforderung. Nicht
selten suchen Patienten mit lysosoma-
len Speichererkrankungen initial wegen
Symptomen von Seiten des muskulos-
kelettalen Systems einen Arzt auf. Da-
her sollte insbesondere der Rheumato-
loge (Internist oder Pädiater) die Symp-
tome dieser Erkrankungen kennen und
einordnen können.
Mit diesem Ziel diskutierte im Rah-
men eines Roundtablegesprächs eine
Gruppe von Rheumatologen die Schlüs-
selsymptome dieser Erkrankungen im in-
terdisziplinären Austausch mit Ärzten, die
in spezialisierten Zentren Patienten mit
M. Gaucher, M. Fabry und MPS I behan-
deln. Über die Ergebnisse dieser Diskussi-
on wird im Folgenden berichtet.
32 | Zeitschrift für Rheumatologie 1 · 2006
Übersichten


Morbus Gaucher
Kasuistik
Bei Frau E. fiel im Alter von 17 Jahren
erstmals eine Schwellung und Überwär-
mung des linken Sprunggelenks auf. BSG
40/80 mm n.W., Antistreptolysintiter
1200 E. Nach einer 4-wöchigen Behand-
lung mit Naproxen war die Patientin be-
schwerdefrei. Im Alter von 20 Jahren trat
nun eine Schwellung des rechten Knie-
gelenks auf und es wurde erstmals eine
leichte Vergrößerung der Milz festgestellt.
Herzecho unauffällig. Antistreptolysinti-
ter nun 1600 E. BSG 45/80 mm n.W. Un-
ter der Verdachtsdiagnose rheumatisches
Fieber wurde die Patientin mit Predniso-
lon 2 mg/kg/die und Naproxen sowie Pe-
nicillin behandelt. Zwei Jahre später kam
es zu einer erneuten Schwellung des rech-
ten Kniegelenks, woraufhin ein erneuter
Behandlungszyklus mit Prednisolon und
Naproxen erfolgte. Die Behandlung mit
Penicillin wurde fortgeführt.
Im Alter von 23 Jahren wurde die Pati-
entin erstmals schwanger. Im 6. Schwan-
gerschaftsmonat wurde bei akuten,
schwersten Hüftschmerzen eine linkssei-
tige Hüftkopfnekrose diagnostiziert. Ein
Zusammenhang zu den Steroidbehand-
lungen wurde vermutet. Die Hüftkopfne-
krose wurde konservativ behandelt. Per
Sectio gebar Frau E. eine gesunde Toch-
ter. In der Folgezeit hatte die Patientin an-
haltende Schmerzen und nahm kontinu-
ierlich NSAR.
Im Alter von 29 Jahren stürzte die Pa-
tientin beim Eislaufen. Zu diesem Zeit-
punkt fiel erstmals eine Panzytopenie
(Leukozyten 2,5/nl; Hb 10,1 g/dl; Throm-
bozyten 82/nl) sowie eine nun deutliche
Milzvergrößerung (bipolar 20,5 cm) auf.
Im Knochenmarksausstrich wurden jetzt
Speicherzellen bzw. Gaucherzellen gefun-
den. Angesichts der typischen Befund-
konstellation von Splenomegalie, Kno-
chenbefall und Panzytopenie wurde dar-
aufhin die zur Sicherung der Diagnose
notwendige Enzymanalytik veranlasst.
Die Enzymaktivität der β-Glukozerebro-
sidase war auf 4 verringert, so dass nun
nach 12 Jahren Krankheitsverlauf die Di-
agnose Morbus Gaucher biochemisch ge-
sichert wurde. Die Chitotriosidaseaktivi-
tät im Plasma war mit 24 340 nmol/ml/
h massiv erhöht. Die Patientin wies kei-
ne neurologischen Symptome auf (nicht-
neuronopathische Verlaufsform).
Pathophysiologie
Der Morbus Gaucher, eine autosomal-re-
zessiv übertragene Erbkrankheit, ist die
häufigste lysosomale Speichererkrankung
mit einer Häufigkeit von etwa 1:50000–
60000 [21, 26]. Pathobiochemisch liegt
der Erkrankung ein Mangel des lysoso-
malen Enzyms β-Glukozerebrosidase zu
Grunde. Dies führt zur Speicherung von
Glukozerebrosiden in Gewebsmakropha-
gen [6], die dann als Gaucherzellen be-
zeichnet werden. In den letzten Jahren
wurde deutlich, dass pathologisch akti-
vierte Makrophagen, die zahlreiche bioak-
tive Proteine sezernieren, eine Rolle in der
Pathogenese der Erkrankung spielen [16,
29] und auch immunologische Verände-
rungen (Stimulation des B-Zell-Systems,
Immunoglobulinopathien, Störungen der
Granulozytenfunktion, Autoantikörper-
bildung) erklären.
Vor allem die Erhöhung des lysosoma-
len Enzyms Chitotriosidase, die mit der
Speicherbeladung des Körpers und der
Schwere der Erkrankung korreliert und
zur Therapiesteuerung eingesetzt werden
kann, ist Gegenstand zahlreicher Untersu-
chungen der letzten Jahre [16].
Klinische Symptome und Prognose
Prinzipiell werden nicht-neuropathische
(85–95 der Fälle) von neuropathischen
Verlaufsformen (5–15 der Fälle) unter-
schieden, wobei die Übergänge zwischen
den Typen fließend sind und die Zuord-
nung zu einem bestimmten Typ nicht im-
mer klar ist [33].
Das klinische Bild der nicht-neuropa-
thischen Verlaufsform des Morbus Gau-
cher wird durch Splenomegalie, Hepato-
Abb. 1 8 Hüftkopfnekrose links bei Morbus Gaucher. (Freundli-cherweise zur Verfügung gestellt von Prof. Schumacher, Kinderra-diologie Universitätskinderklinik Mainz)
Abb. 2 9 Gibbusbildung bei Osteopenie und Lun-genbeteiligung (ver-mehrte interstitielle Zeichnung) bei Morbus Gaucher. (Freundlicher-weise zur Verfügung ge-stellt von Prof. Schuma-cher, Kinderradiologie Universitätskinderklinik Mainz)
34 | Zeitschrift für Rheumatologie 1 · 2006
Übersichten

megalie, Knochenbeteiligung und Zyto-
penie geprägt. Manifestationsalter (vom
Kleinkindesalter bis in die 8. Lebensde-
kade) sowie Progression der Erkrankung
sind ausgesprochen variabel. Allgemein
gilt, je früher die ersten Symptome der Er-
krankung beobachtet werden, desto kom-
plikationsreicher ist der natürliche Ver-
lauf der Erkrankung. Allerdings können
auch bei Erstmanifestationen im Erwach-
senenalter innerhalb weniger Jahre schwe-
re Knochenkomplikationen auftreten.
Hämatologische Symptome des Mor-
bus Gaucher sind die im Kindesalter meist
im Vordergrund stehende Anämie, im Er-
wachsenenalter fast immer eine Throm-
bopenie und in der Regel erst bei fortge-
schrittener Erkrankung eine Leukope-
nie. Die bei allen symptomatischen Pati-
enten in unterschiedlichem Ausmaß be-
obachtete Splenomegalie ist häufig das
zur Diagnose führende Erstsymptom. Fi-
brotische Areale und Regionen extrame-
dullärer Blutbildung können rundherdar-
tig imponieren. Milzinfarkte können unter
dem Bild eines akuten Abdomens ablau-
fen, aber auch asymptomatisch sein. Auch
die Leber ist bei den meisten Patienten
vergrößert. Sie nimmt aber fast nur dann
überdimensionale Ausmaße an, wenn der
Patient splenektomiert wurde. Ein Drittel
der Patienten weist bei Diagnosestellung
Erhöhungen der Lebertransaminasen auf.
Einschränkungen der Leberfunktion, Le-
berfibrose und Leberversagen treten gele-
gentlich im fortgeschrittenen Krankheits-
verlauf auf und werden im Zeitalter der
Enzymersatztherapie nicht mehr beob-
achtet.
Die Skelettbeteiligung, die die Lebens-
qualität der Gaucher-Patienten beson-
ders stark einschränkt, wird durch Kno-
chenmarkinfiltration und fehlerhaftes
Knochen-Remodelling verursacht. Fol-
ge sind Osteopenie, chronische Kno-
chenschmerzen, pathologische Frak-
turen, Osteonekrosen und avaskuläre In-
farzierungen (. Abb. 1 und 2). Akuter,
schwerster Knochenschmerz mit Fieber
und deutlicher Einschränkung des Allge-
meinzustandes charakterisieren den aku-
ten Knocheninfarkt. Prädeliktionsstellen
der Osteonekrosen sind der Hüftkopf,
Lendenwirbelkörper und proximaler Hu-
merus [27].
Zusammenfassung · Summary
Z Rheumatolo 2006 · 65:32–43
DOI 10.1007/s00393-006-0039-2
© Springer Medizin Verlag 2006
B. Manger · E. Mengel · R. Schaefer · C. Haase · J. Seidel · H. Michels
M. Gaucher, M. Fabry und Mukopolysaccharidose Typ I
Zusammenfassung
Die lysosomalen Speichererkrankungen
Morbus Gaucher, Morbus Fabry und
MPS I zählen zu den seltenen erblichen
Stoffwechselerkrankungen, die durch ei-
ne Enzymersatztherapie heute behandel-
bar sind. Damit durch eine frühzeitige The-
rapie sonst irreversible Komplikationen
verhindern werden können, ist eine recht-
zeitige Diagnosestellung wichtig. Weil Pa-
tienten mit diesen Speichererkrankungen
wegen Symptomen von Seiten des mus-
kuloskelettalen Systems nicht selten einen
Rheumatologen aufsuchen können, soll-
te dieser die Symptome dieser seltenen Er-
krankungen erkennen und einordnen kön-
nen. Anhand von Kasuistiken zu M. Gau-
cher, M. Fabry und MPS I (hier M. Scheie)
werden die Schlüsselsymptome diskutiert,
die der Rheumatologe (Internist oder Päd-
iater) für die differenzialdiagnostische Ab-
klärung dieser Patienten kennen sollte. Zu-
sätzlich werden – neben einer kurzen Ein-
führung in die Pathophysiologie – Hinwei-
se zur Prognose und Therapie gegeben.
Schlüsselwörter
Lysosomale Speichererkrankungen ·
M. Gaucher · M. Fabry · Mukopolysaccha-
ridose Typ I
Gaucher disease, Fabry disease and mucopolysaccharidosis type I – How can the rheumatologist recognise these patients?
Summary
The lysosomal storage diseases Gaucher
disease, Fabry disease and MPS I are rare
inheritable metabolic disorders that are
now treatable with enzyme replacement
therapy. In order to avoid irreversible com-
plications, an early diagnosis and initiation
of therapy is important. Due to the muscu-
loskeletal symptoms associated with these
storage diseases, patients are likely to visit
a rheumatologist, who should, therefore,
be able to recognise and diagnose the-
se rare diseases. On the basis of the causal
factors behind Gaucher disease, Fabry di-
sease und MPS I (here Scheie syndrome),
key symptoms that the rheumatologist (in-
ternist or paediatrician) should be famili-
ar with for the differential diagnosis of the-
se patients will be discussed. In addition, a
short introduction to the pathophysiology
and data on the prognosis and therapy for
these diseases will be presented.
Keywords
Lysosomal storage diseases · Gaucher
disease · Fabry disease · Mucopolysaccha-
ridosis type I
35Zeitschrift für Rheumatologie 1 · 2006 |
Therapie
Bevor die Enzymersatztherapie zur Verfü-
gung stand, war die Behandlung des Mor-
bus Gaucher rein symptomatisch. Bei aus-
geprägter Splenomegalie mit transfusions-
pflichtiger Zytopenie oder mechanischen
Problemen war oft eine Splenektomie un-
umgänglich. Akute Knochenkrisen be-
durften der Behandlung mit potenten An-
algetika und strikter Bettruhe. Orthopä-
dische Operationen, wie Knie- und Hüft-
gelenksersatz sowie Stabilisierungen der
Wirbelsäule, wurden im Verlauf nötig.
Inzwischen können die Organmani-
festationen der nicht-neuronopathischen
und der chronisch-neuronopathischen
Verlaufsform des Morbus Gaucher mit
der Enzymersatztherapie (Imiglucerase)
erfolgreich behandelt bzw. vorbeugend
vermieden werden [3]. 1991 wurden be-
reits die ersten Patienten in Deutschland
behandelt. Im Allgemeinen berichten die
Patienten zunächst über eine beeindru-
ckende Verbesserung des Allgemeinzu-
standes und der Lebensqualität. Im wei-
teren Verlauf innerhalb des ersten Thera-
piejahres kommt es zur Reduktion der Or-
gangröße und Blutbildverbesserungen. Bei
den meisten Patienten sind nach 24 Mona-
ten Behandlung Zytopenien und Hepato-
megalie nicht mehr nachweisbar, während
die Milz häufig auch nach einigen Thera-
piejahren leicht vergrößert bleibt. Das Auf-
treten akuter Knochenschmerzen kann bei
den meisten Patienten bereits im ersten
Jahr zuverlässig verhindert werden. Die
Knochenmarkinfiltration und die Kno-
chenmineralisation sprechen langsamer
auf die Therapie an; der maximale The-
rapieeffekt wird gelegentlich erst nach 4–
5 Jahren beobachtet [25].
Die Manifestation des Morbus Gau-
cher im Kindesalter ist zumeist mit einem
schweren Krankheitsverlauf und vor
allem mit Knochenkomplikationen und
Wachstumsretardierung vergesellschaf-
tet. Ein früher Beginn der Enzymersatz-
therapie sowie eine optimale Dosierung
verhindern Komplikationen, führen zu
einer nahezu unbehinderten, kindlichen
Entwicklung und unbeeinträchtigten Le-
bensqualität [2, 14, 18].
Seit Jahren werden niedrigdosierte ver-
sus hochdosierte Dosisregime auf dem
Hintergrund der enormen Kosten disku-
tiert. In Deutschland hat sich sehr erfolg-
reich eine dem Schweregrad der Erkran-
kung angepasste, individuelle Dosierung
durchgesetzt [23].
Seit 2003 ist auch Miglustat zur Behand-
lung des Morbus Gaucher zugelassen. Mig-
lustat ist ein Inhibitor der Glukosylzera-
mid-Synthase und reduziert die Synthese
von Glykosphingolipiden. Aufgrund seiner
geringeren Effektivität und seines ungüns-
tigeren Nebenwirkungsprofils [8] wurde
das Medikament nur für den Einsatz bei
leicht bis mittelschwer betroffenen Pati-
enten, für die eine Enzymsubstitutionsthe-
rapie nicht in Frage kommt, zugelassen.
Morbus Fabry
Kasuistik
Im Alter von 26 Jahren stellte sich erstmals
eine junge Frau vor, die seit ihrem zehn-
ten Lebensjahr unter Schmerzen in Hän-
den und Füßen, anfangs häufig mit Fieber
einhergehend, und körperlicher Schwäche
gelitten hatte.
Die Eltern hatten mit ihrem Kind meh-
rere Pädiater konsultiert. Ein Kinderarzt
vermutete Wachstumsschmerzen, ein
zweiter diagnostizierte einen Morbus Still,
da zum Zeitpunkt der Untersuchung eine
diskret beschleunigte Blutsenkung auffäl-
lig gewesen war. Therapeutisch erhielt das
Mädchen Metamizol, Indomethacin und
schließlich Glukokortikoide ohne durch-
greifenden Erfolg. Schließlich mussten
Opiate eingesetzt werden, um zumindest
eine symptomatische Linderung zu erzie-
len. Noch einmal suchten die Eltern mit
ihrem Kind einen weiteren Pädiater auf,
der einen Schmerzmittelmissbrauch ver-
mutete und jugendpsychiatrische Betreu-
ung empfahl.
Inzwischen war das Mädchen 16 Jah-
re alt und man wandte sich nun an einen
Internisten, der neben den Schmerzen
Hauteffloreszenzen (. Abb. 3), eine Pro-
teinurie und erneut eine leicht beschleu-
nigte Senkung fand. Diese Konstellati-
on wurde als Vaskulitis interpretiert, und
die Patientin erhielt erneut Glukokortiko-
ide, die sie über die nächsten 5 Jahre ein-
nahm.
Mit 21 Jahren kam es neben den be-
kannten brennenden Schmerzen in Hän-
den und Füßen zu schmerzhaften Be-
schwerden in beiden Hüften. Der hin-
zugezogene Orthopäde veranlasste eine
MRT der Hüftgelenke, und es fanden sich
Hüftkopfnekrosen beidseits, die schließ-
lich mit Endoprothesen versorgt wurden.
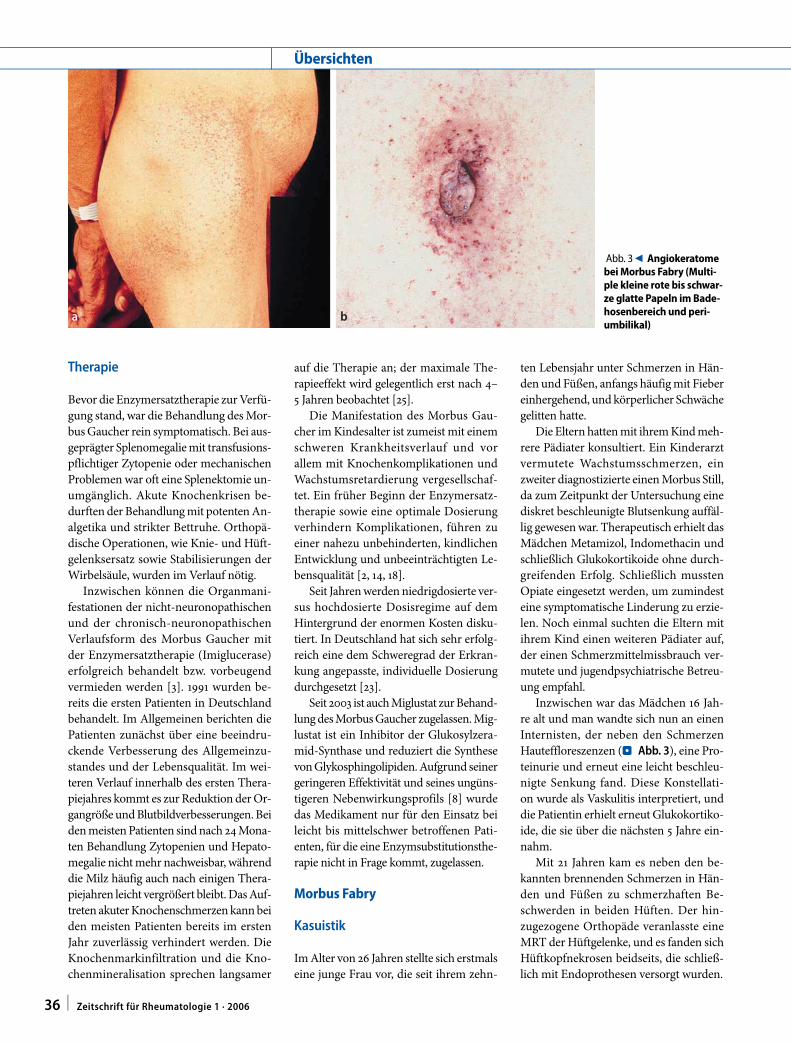
Abb. 3 9 Angiokeratome bei Morbus Fabry (Multi-ple kleine rote bis schwar-ze glatte Papeln im Bade-hosenbereich und peri-umbilikal)
36 | Zeitschrift für Rheumatologie 1 · 2006
Übersichten
Während dieser Zeit wurde die Stero-
idtherapie, wenn auch in relativ niedriger
Dosis (5 mg jeden zweiten Tag), unbeirrt
fortgesetzt, und als die Patientin 26 Jah-
re alt war, kam es zu einer zunehmenden
Sehverschlechterung. Man vermutete eine
Katarakt im Rahmen der langjährigen Ste-
roidbehandlung und strengte eine opthal-
mologische Untersuchung an. Tatsächlich
fand sich bei der Spaltlampen-Untersu-
chung des Auges eine Linsentrübung, dar-
über hinaus fielen jedoch auch Verände-
rungen der Cornea im Sinne einer Cornea
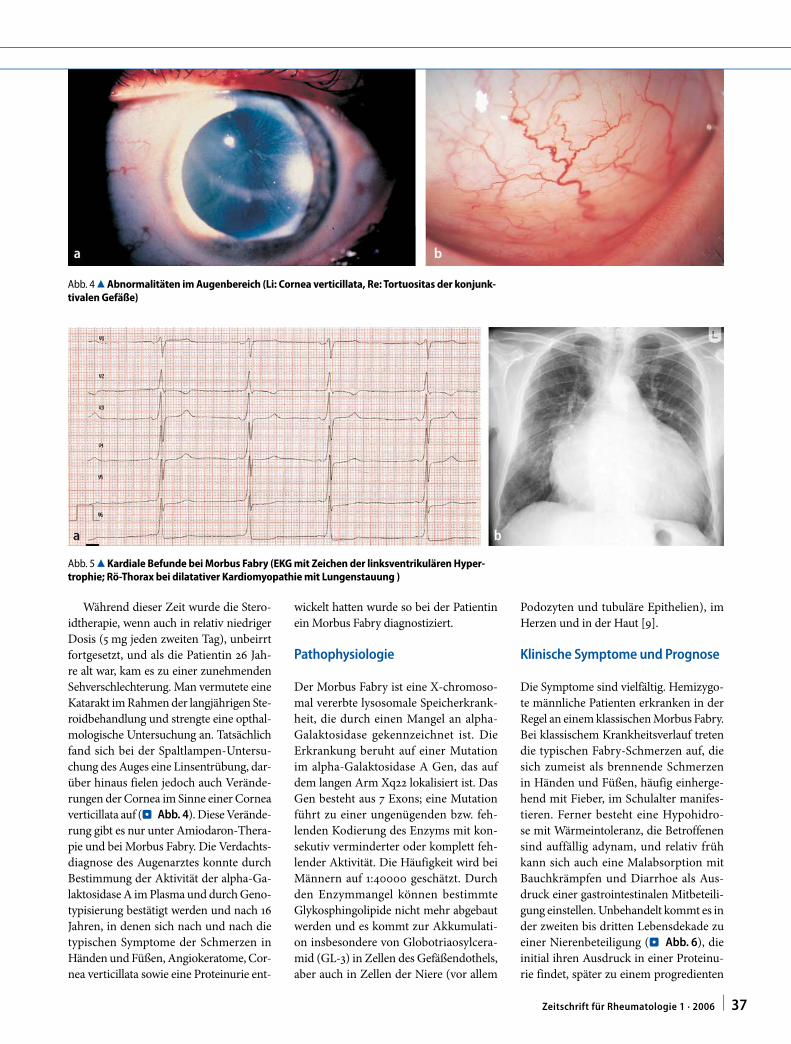
verticillata auf (. Abb. 4). Diese Verände-
rung gibt es nur unter Amiodaron-Thera-
pie und bei Morbus Fabry. Die Verdachts-
diagnose des Augenarztes konnte durch
Bestimmung der Aktivität der alpha-Ga-
laktosidase A im Plasma und durch Geno-
typisierung bestätigt werden und nach 16
Jahren, in denen sich nach und nach die
typischen Symptome der Schmerzen in
Händen und Füßen, Angiokeratome, Cor-
nea verticillata sowie eine Proteinurie ent-
wickelt hatten wurde so bei der Patientin
ein Morbus Fabry diagnostiziert.
Pathophysiologie
Der Morbus Fabry ist eine X-chromoso-
mal vererbte lysosomale Speicherkrank-
heit, die durch einen Mangel an alpha-
Galaktosidase gekennzeichnet ist. Die
Erkrankung beruht auf einer Mutation
im alpha-Galaktosidase A Gen, das auf
dem langen Arm Xq22 lokalisiert ist. Das
Gen besteht aus 7 Exons; eine Mutation
führt zu einer ungenügenden bzw. feh-
lenden Kodierung des Enzyms mit kon-
sekutiv verminderter oder komplett feh-
lender Aktivität. Die Häufigkeit wird bei
Männern auf 1:40000 geschätzt. Durch
den Enzymmangel können bestimmte
Glykosphingolipide nicht mehr abgebaut
werden und es kommt zur Akkumulati-
on insbesondere von Globotriaosylcera-
mid (GL-3) in Zellen des Gefäßendothels,
aber auch in Zellen der Niere (vor allem
Podozyten und tubuläre Epithelien), im
Herzen und in der Haut [9].
Klinische Symptome und Prognose
Die Symptome sind vielfältig. Hemizygo-
te männliche Patienten erkranken in der
Regel an einem klassischen Morbus Fabry.
Bei klassischem Krankheitsverlauf treten
die typischen Fabry-Schmerzen auf, die
sich zumeist als brennende Schmerzen
in Händen und Füßen, häufig einherge-
hend mit Fieber, im Schulalter manifes-
tieren. Ferner besteht eine Hypohidro-
se mit Wärmeintoleranz, die Betroffenen
sind auffällig adynam, und relativ früh
kann sich auch eine Malabsorption mit
Bauchkrämpfen und Diarrhoe als Aus-
druck einer gastrointestinalen Mitbeteili-
gung einstellen. Unbehandelt kommt es in
der zweiten bis dritten Lebensdekade zu
einer Nierenbeteiligung (. Abb. 6), die
initial ihren Ausdruck in einer Proteinu-
rie findet, später zu einem progredienten
Abb. 4 8 Abnormalitäten im Augenbereich (Li: Cornea verticillata, Re: Tortuositas der konjunk-tivalen Gefäße)
Abb. 5 8 Kardiale Befunde bei Morbus Fabry (EKG mit Zeichen der linksventrikulären Hyper-trophie; Rö-Thorax bei dilatativer Kardiomyopathie mit Lungenstauung )
37Zeitschrift für Rheumatologie 1 · 2006 |
Nierenversagen führt und schließlich eine
Nierenersatztherapie notwendig macht.
Im Rahmen der kardialen Manifestati-
on findet sich typischerweise eine hy-
pertrophe Kardiomyopathie (. Abb. 5),
neurologisch kann es schon früh (zwei-
te und dritte Lebensdekade) zu transito-
rischen Ischämien, Apoplexen oder auch
zu einem Hörsturz kommen. Für die Di-
agnosefindung wichtige Zeichen sind die
Angiokeratome der Haut (Badehosenbe-
reich) und die Cornea verticillata [9].
Frauen sind von der Fabry-Krank-
heit häufiger betroffen als früher ange-
nommen, dafür in der Regel aber weniger
schwer erkrankt als Männer. Dies kann
sich bei den heterozygoten Patientinnen
als milde Form der klassischen Fabry-Er-
krankung manifestieren oder es treten
auch so genannte Organ-Varianten (z.B.
kardiale oder renale Varianten) auf. Hier-
bei fehlen die klassischen Symptome völ-
lig, es besteht keine Gefäßbeteiligung, und
es findet sich lediglich eine progrediente
linksventrikuläre Hypertrophie oder eine
geringgradige Proteinurie. In solchen Fäl-
len kann die Erkrankung meist nur durch
Biopsie des betroffenen Organs oder
durch eine positive Familienanamnese di-
agnostiziert werden [20]. Zahlreiche he-
terozygote Frauen entwickeln zeitlebens
überhaupt keine Krankheitszeichen (rei-
ne Konduktorinnen).
Unbehandelt ist die Prognose beson-
ders für die männlichen Fabry-Patienten
ungünstig. Die Mehrzahl der Betroffenen
wird zwischen dem 30. und 40. Lebens-
jahr dialysepflichtig, und das Überleben
an der Dialyse ist deutlich reduziert, da
bei dialysepflichtigen Fabry-Patienten ei-
ne gesteigerte kardio- und cerebrovasku-
läre Morbidität und Mortalität besteht.
Die Nierenbeteiligung bei heterozygoten
Frauen verläuft grundsätzlich günstiger
als bei den hemizygoten männlichen Pa-
tienten; so kann über lange Strecken ei-
ne Proteinurie bestehen, ohne dass es zu
einem Abfall der glomerulären Filtrati-
onsrate kommt. In vielen Fällen stellt sich
eine chronische Niereninsuffizienz erst im
höheren Lebensalter (ab der 6. Lebensde-
kade) ein. Zur terminalen Niereninsuffizi-
enz kommt es nur bei einem kleinen Pro-
zentsatz der betroffenen Frauen [9].
Therapie
Eine Enzymersatztherapie mit gentech-
nisch hergestellter alpha-Galaktosida-
se (Agalsidase alfa oder Agalsidase beta)
sollte bei hemizygoten Männern mit Auf-
treten der ersten typischen Symptome, in
der Regel Akroparästhesien, begonnen
werden. Bei heterozygoten Frauen ist es
häufig das Ausmaß der Nieren- bzw. der
Herzbeteiligung, an der die Indikation zur
Enzymersatztherapie festgemacht wird.
Bei männlichen Patienten wird die Thera-
pie in der Regel so frühzeitig wie möglich
(in der Regel zwischen dem 10. und 20.
Lebensjahr) begonnen, während die re-
nale bzw. kardiale Beteiligung bei hetero-
zygoten Frauen häufig erst zwischen der 4.
und 6. Lebensdekade ein therapeutisches
Eingreifen erforderlich macht. Heterozy-
gote Frauen mit Organ-Varianten (kardi-
ale oder renale Variante) nehmen in der
Regel einen unvorhersehbaren Verlauf, so
dass die Entscheidung zur Enzymersatz-
therapie sehr individuell getroffen werden
muss [10].
Die Enzymersatztherapie erfolgt in-
travenös, wobei für Agalsidase alfa eine
Dosierung von 0,2 mg/kg alle 2 Wochen
empfohlen wird. Für Agalsidase beta be-
trägt die Dosierung 1 mg/kg alle 2 Wo-
chen; unter dieser Dosis kommt es zu
einem völligen Verschwinden von GL-3
aus der Zirkulation, nach 6 Monaten sind
im Gefäßendothel keine GL-3 Ablage-
rungen mehr nachweisbar [11]. GL-3 Ab-
lagerungen in Kardiomyozyten oder Po-
dozyten der Niere benötigen, je nach Aus-
maß der GL-3 Speicherung, 12–36 Monate
um sich weitgehend zurückzubilden [32].
Auch die kardialen GL-3 Ablagerungen
bilden sich unter Enzymersatztherapie
zurück, und es kommt dadurch zu einer
echokardiographisch nachweisbaren Ver-
besserung der linksventrikulären Pump-
funktion [34].
Entscheidend für den Erfolg der Enzy-
mersatztherapie ist zum einen ein mög-
lichst frühzeitiger Therapiebeginn, noch
bevor strukturelle Organveränderungen
(interstitielle Fibrose) eingetreten sind,
zum anderen eine ausreichend hoch do-
sierte Enzymsubstitution.
Mukopolysaccharidose Typ I (MPS I – Scheie/MPS I – Hurler)
Kasuistik
Im Folgenden werden drei Geschwister
vorgestellt, bei denen die Diagnose MPS I
– Typ Scheie gestellt wurde.
Beide Eltern sind gesund und deut-
scher Abstammung. Nach unauffälliger
Schwangerschaft und Geburt verlief die
frühkindliche Entwicklung des ersten
Sohnes altersgerecht bei normaler Intel-
ligenz und ohne Dysmorphiezeichen. Im
Alter von drei Jahren erfolgte wegen an-
haltend beklagter Schmerzen in den Hän-
den erstmals die Vorstellung in einer kin-
derrheumatologischen Sprechstunde. Es
zeigten sich beginnende palmare Ver-
härtungen im Sehnenbereich mit Beuge-
kontrakturen. Die Schmerzen und Bewe-
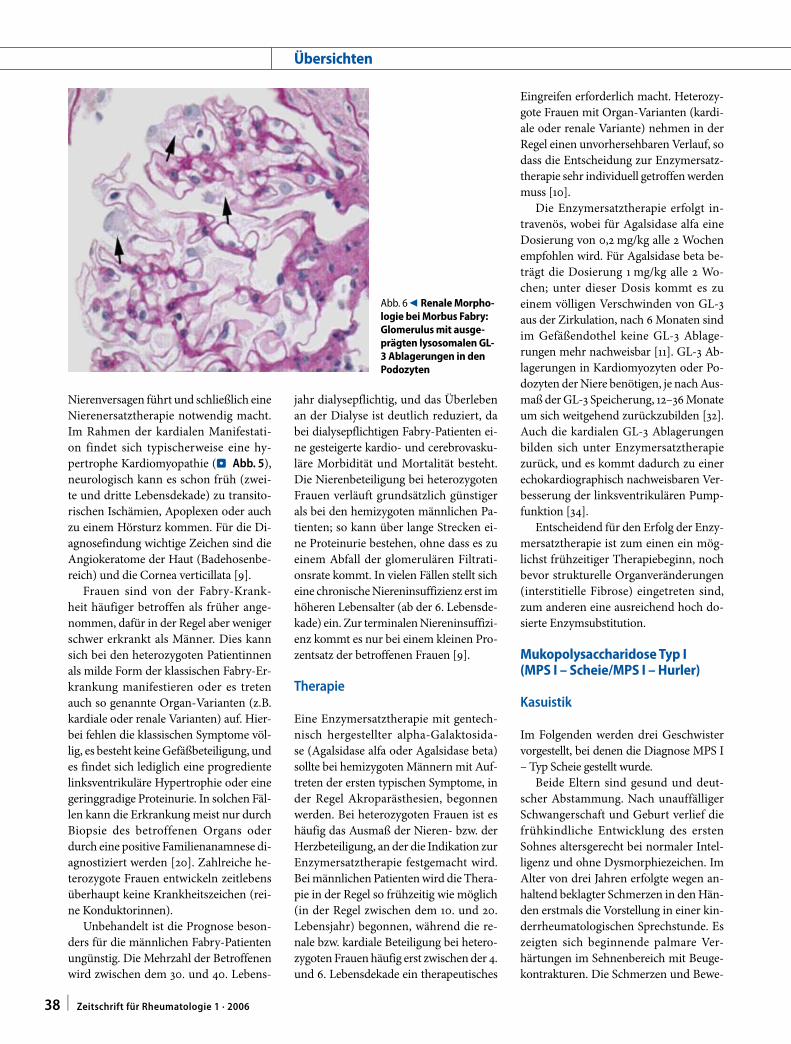
Abb. 6 9 Renale Morpho-logie bei Morbus Fabry: Glomerulus mit ausge-prägten lysosomalen GL-3 Ablagerungen in den Podozyten
38 | Zeitschrift für Rheumatologie 1 · 2006
Übersichten

gungseinschränkungen nahmen im Ver-
lauf zu, ohne dass periphere Zeichen eines
entzündlichen Geschehens oder Hinwei-
se auf eine Autoimmunerkrankung fest-
zustellen waren. Klinisch traten wei-
ter zunehmend die Kontrakturen im Be-
reich der Fingerend- und Fingergrund-
gelenke in den Vordergrund. Es war kein
vollständiger Faustschluss mehr möglich
(. Abb. 7), während Ellenbogen und
Kniegelenke ohne Bewegungseinschrän-
kung blieben.
Im Alter von 5 Jahren zeigte sich eine
progrediente beidseitig symmetrisch auf-
tretende Achillessehnenverkürzung mit
folgender Spitzfußstellung (. Abb. 8).
Aus den Fußdeformitäten resultierten
dauerhafte Schmerzen. Erneut ergab sich
bei der Diagnostik kein Anhaltspunkt für
ein entzündliches oder autoimmunolo-
gisches Geschehen. Therapeutische Ver-
suche mit nichtsteroidalen Antirheumati-
ka erbrachten keine Besserung.
Im Rahmen der durchgeführten Dia-
gnostik fiel bei dem nunmehr neunjäh-
rigen Patienten eine beidseitige Cornea-
trübung, sowie eine mittelgradige Mitral-
und leichtgradige Trikuspidalinsuffizienz
bei sonst normalen parenchymatösen Or-
ganen, und unauffälligen Befunden im ce-
rebralen MRT auf. Eine Hepatosplenome-
galie wurde nicht beobachtet.
Aufgrund dieser Befundkonstellation
sowie wegen des zunehmend schlechteren
klinischen Befundes wurde die Ausschei-
dung der Glykosaminoglykane im 24-
Stunden-Sammelurin gemessen. Diese
zeigte sich mit 10 mg/mmol Kreatinin bei
einem Normwert von 7,5 mg/mmol Kre-
atinin grenzwertig erhöht. Die daraufhin
durchgeführte enzymatische Untersu-
chung der Alpha-Iduronidase in den Leu-
kozyten und Fibroblasten ergab eine deut-
lich verminderte Aktivität des Enzyms mit
ca. 1 Restaktivität. Hierauf konnten zwei
Mutationen im Alpha-Iduronidase-Gen
bei compound Heterozygotie Q 70 X und
R 89 W nachgewiesen werden. Die Mu-
tation Q 70 X führt im Falle einer Ho-
mozygotie zu einer MPS I – Typ Hurler.
Die „mildere“ Mutation R 89 W ist bis-
her nicht beschrieben. Beide Mutationen
zusammen rufen den familiären Scheie-
Phänotyp hervor.
Beide jüngeren Brüder waren zunächst
in klinisch gutem Allgemeinzustand und
ohne klinische Symptome. Bei beiden er-
gab sich jedoch eine erhöhte Ausschei-
dung von Glykosaminoglykanen im
Urin und eine verminderte Enzymaktivi-
tät. Auch gelang ein positiver Mutations-
nachweis, so dass die Diagnose MPS I –
Scheie für alle Kinder der Familie nach-
gewiesen werden konnte. Beide jüngeren
Brüder zeigten inzwischen Fingerkon-
trakturen und eine beginnende Spitzfuß-
stellung, röntgenologisch ist eine Verbrei-
terung der Metakarpalknochen zu beob-
achten. Derzeit werden die Patienten mit
einer wöchentlichen Enzymersatztherapie
behandelt.
Pathophysiologie und Genetik
Bei der Mukopolysaccharidose Typ I
(MPS I) handelt es sich um eine autoso-
mal-rezessiv vererbte Speichererkran-
kung, bei der ein gestörter Abbau der lyso-
somalen Glykosaminoglykane Dermatan-
und Heparansulfat vorliegt. Ursächlich
hierbei sind Mutationen im α-L-Iduroni-
dase-Gen, die zu einer verminderten Ak-
tivität des Enzyms führen. Durch den En-
zymmangel werden die Glykosamino-
glykane nicht abgebaut und in den Lyso-
somen abgelagert. Über noch weitgehend
ungeklärte Pathomechanismen kommt es
zu Fehlfunktionen auf Zell-, Gewebe- und
Organebene. Entsprechend der Gewebe-
verteilung der Glykosaminoglykane sind
insbesondere Gehirn, Bindegewebe, Kor-
tikalis, Epiphysenfugen, Herz, Leber und
Nieren betroffen.
Die geschätzte Inzidenz einer leich-
teren Verlaufsform der MPS I ohne ZNS-
Abb. 7 7 Kontrakturen im Bereich der Fingerge-lenke, kein Faustschluss
mehr möglich (MPS I)
Abb. 8 8 Spitzfußstellung mit bleibender Fußdeformität (MPS I)
Abb. 9 8 Craniofaziale Dysmorphiezeichen („Wasserspeiergesicht“) bei MPS I – Hurler; ausladendes, vorgewölbtes Abdomen, Tat-zenhände, plumpe Füße, geringe Hepato-megalie
39Zeitschrift für Rheumatologie 1 · 2006 |

Beteiligung liegt bei ungefähr 1:500000
[22].
Klinische Symptome und Prognose
Wie bei den meisten Speicherkrankungen
ist die klinische Bandbreite der MPS I Er-
krankung sehr breit. Dabei können vom
klassischen Morbus Hurler über die In-
termediärform Scheie/Hurler zur milde-
ren Form des Morbus Scheie verschiedene
Verlaufsformen vorliegen.
Beim Vollbild des Morbus Hurler tre-
ten, beginnend in den ersten Lebens-
jahren, die typischen craniofazialen Dys-
morphiezeichen mit flacher breiter Na-
senwurzel, geschwollenen Lippen und
insgesamt vergröberten Gesichtszügen
auf („Wasserspeiergesicht“, . Abb. 9).
Zunehmend entwickelt sich daneben
eine psychomotorische Retardierung, au-
ßerdem eine zum Teil rasch progrediente
viszerale Organbeteiligung sowie Herz-
klappeninsuffizienzen und Kardiomyo-
pathien mit Funktionsstörungen [7]. Ei-
ne Hepatosplenomegalie gehört ebenfalls
zum klassischen Vollbild der MPS I. Ty-
pischerweise treten in Folge von Binde-
gewebsschwächen Leisten- und Nabel-
hernien auf, und es werden progrediente
Gelenkfehlstellungen mit Beugekontrak-
turen sowie tatzenartige Verformungen
der Hände und plumpe Füße diagnosti-
ziert.
Scheie-Patienten haben im Gegen-
satz zu Hurler-Patienten ein normales
Äußeres ohne vergröberte Gesichtszü-
ge und können eine normale Körpergrö-
ße erreichen. Kennzeichnend sind Symp-
tome am Bewegungsapparat, sowie Aor-
tenklappenbeteiligung und Hornhauttrü-
bung. Die Gelenkkontrakturen sind häu-
fig an der Hand am stärksten ausgeprägt
(Tatzenhand, . Abb. 10) und können in
der Verbindung mit einem Karpaltunnel-
Syndrom zu einem gravierenden Funkti-
onsverlust führen. Eine obstruktive Atem-
wegserkrankung kann bereits in der Ju-
gend eine Tracheostomie notwendig ma-
chen [15].
Von den Symptomen des Bewegungs-
apparates müssen neurologische Funkti-
onseinschränkungen aufgrund einer En-
ge des kraniozervikalen Überganges mit
konsekutiver Myelopathie abgegrenzt
werden, welche gelegentlich auch bei Pa-
tienten mit attenuierter Verlaufsform be-
obachtet wird [22, 15].
Radiologisch zeigen sich thorakal spa-
tenförmig verbreiterte Rippen („Ruder-
blattrippen“) und, als weitere Hinweise
auf eine Speichererkrankung, abgerunde-
te Skapulae und plump verbreiterte Me-
takarpalknochen, Grund- und Mittelpha-
langen. Gemeinsam mit der Hüftdysplasie
werden diese Veränderungen unter dem
Begriff der Dysostosis multiplex subsu-
miert. Bei der Intermediärform Scheie/
Hurler treten Symptome später und zu-
nächst in geringerem Maße auf, können
jedoch ungleich schneller progredient
und in Schüben verlaufen [22].
Therapie
Bei der schweren Form der Mukopolysac-
charidose Typ I, dem Morbus Hurler, ist
eine frühzeitige Diagnosestellung vor dem
3. Lebensjahr anzustreben, da in diesem
Alter noch eine Knochenmarkstransplan-
tation als Therapieoption zur Verfügung
steht [24, 19, 30]. Derzeitige Therapieop-
timierungsstudien überprüfen, ob eine
Kombinationstherapie von Enzymersatz-
therapie und Knochenmarkstransplanta-
tion zu noch besseren Therapieergebnis-
sen führt.
Für alle anderen Patienten mit MPS I,
die nicht der klassischen Hurler-Verlaufs-
form entsprechen, ist die Enzymersatzthe-
rapie mit Laronidase der einzig mögliche
kausale Therapieansatz.
Im Jahr 2004 wurden die Ergebnisse ei-
ner internationalen, placebo-kontrollier-
ten Phase III Studie mit rekombinanter,
menschlicher α-L-Iduronidase (Laroni-
dase) publiziert. Demnach verbessert die
Enzymersatztherapie bei Patienten mit
MPS I signifikant sowohl die forcierte Vi-
talkapazität als auch die Gehstrecke im
6-Minuten-Gehtest. Ebenso wurde eine
Reduzierung der Glykosaminoglykan-
Ausscheidung und der Lebervergröße-
rung beobachtet. Eine Verbesserung des
Schlafapnoe-Indexes und der Schulterfle-
xion wurde bei den stark betroffenen Pati-
enten gesehen [35]. Frühzeitige Diagnose-
stellung, bevor irreversible Schäden einge-
treten sind, ist entscheidend für den The-
rapieerfolg.
Irreversible Endorganschäden bedür-
fen gegebenenfalls der chirurgischen In-
tervention (kraniozervikale Dekompres-
sion, Herzklappenersatz, Hornhauttrans-
planation, etc.) bzw. der supportiven The-
rapie (Krankengymnastik, ACE-Hemmer,
etc.).
Diskussion
Durch die modernen Möglichkeiten der
Enzymersatztherapie oder Stammzell-
transplantation sind lysosomale Speicher-
krankheiten behandelbar geworden. Um
durch einen möglichst frühen Therapie-
beginn Organschäden zu vermeiden, ist
eine rechtzeitige Diagnosestellung dieser
seltenen hereditären Krankheitsbilder es-
sentiell. Da bei allen drei beschriebenen
Syndromen muskuloskelettale Beschwer-
den den Krankheitsverlauf prägen kön-
nen, sollten insbesondere rheumatolo-
gisch tätige Internisten, Orthopäden und
Pädiater die Symptome kennen und zur
Früherkennung beitragen:
Die pathophysiologische Grundlage
für die Symptome von Seiten des Bewe-
gungsapparates bei Morbus Gaucher ist
die progrediente Knochenmarksinfiltra-
tion und -verdrängung durch lipidspei-
chernde Gewebsmakrophagen (Gaucher-
zellen). Als Konsequenz dieser Infiltration
kommt es zur Aktivierung entzündlicher
Mechanismen und des Gerinnungssys-
tems. Da sich die Knochenmarksinfiltra-
tion zentrifugal ausbreitet, sind die proxi-
Abb. 10 9 Tatzenhände mit Beugekontrakturen bei MPS I
40 | Zeitschrift für Rheumatologie 1 · 2006
Übersichten
malen Enden von Röhrenknochen meist
stärker betroffen als die distalen, die Epi-
physen werden typischerweise ausgespart.
Klinisch äußert sich dies als Knochen-
schmerzen, Osteopenie, fokale Osteoly-
sen und -sklerosen sowie pathologische
Frakturen und Osteonekrosen. Hauptprä-
dilektionsstellen für letztere sind Femur-
und Humeruskopf sowie Lendenwirbel-
körper. Bei gelenknahen Prozessen kön-
nen stark schmerzhafte Schwellungen mit
Fieber und Allgemeinsymptomatik plötz-
lich aus völliger Beschwerdefreiheit her-
aus auftreten. Bei unauffälligen Befun-
den auf konventionellen Röntgenaufnah-
men können Knocheninfarkte und Kno-
chenmarksinfiltration gut mittels MRT
oder Knochenmarksszintigraphie mittels 99mTc-Schwefelkolloid dargestellt werden.
Differentialdiagnostisch kann problema-
tisch sein, dass Erkrankungen mit Mani-
festation am Knochen, wie z.B. multiples
Myelom, Arthritiden oder idiopathische
Osteonekrosen, sowohl Differentialdiag-
nose als auch Komplikation des Morbus
Gaucher sein können.
Durch die Ablagerung von Glykosphin-
golipiden in Gefäßendothelien, glatten
Muskelzellen und vielen anderen Gewe-
ben, wie Niere, Nerven, Herz, Augen ist die
Symptomatik des Morbus Fabry vielfältig
und die rechtzeitige Diagnosestellung an-
spruchsvoll. Frühsymptome im Kindesal-
ter sind Akroparästhesien und Schmer-
zen an Händen und Füßen, ohne dass
bei der rheumatologischen und neurolo-
gischen Untersuchung pathologische Be-
funde fassbar sind. Wegweisend kann ei-
ne begleitende Hypohidrose und Wärme-
oder Kälteunverträglichkeit sein, was zur
Fehldiagnose „Raynaud-Syndrom“ führen
kann. Im Erwachsenenalter treten Symp-
tome von Seiten der betroffenen Organ-
systeme in den Vordergrund, wie Protein-
urie und zunehmende Niereninsuffizienz,
Kardiomyopathie, kardio- und zerebro-
vaskuläre Ereignisse sowie Haut- und Au-
genbeteiligung. Aufgrund der möglichen
Symptomenkonstellationen stellt sich hier
für den Rheumatologen insbesondere die
Differentialdiagnose zu den systemischen
Autoimmunopathien.
Bei der MPS I führt der behinderte
Abbau von Glukosaminoglykanen zu ei-
ner abnormen Knorpelentwicklung und
zur Störung der enchondralen und in-
tramembranösen Ossifikation. Dies äu-
ßert sich bei der klassischen Form, der
MPS I – Hurler, in craniofazialen Dys-
morphien, Minderwuchs, Dysostosis
multiplex und Hüftdysplasien. Bei der
MPS I – Scheie und bei Intermediärfor-
men können jedoch auch Symptome von
Seiten des Bewegungsapparates im Vor-
dergrund stehen, ohne dass bei der Un-
tersuchung morphologische Auffällig-
keiten festzustellen sind. Es treten Be-
wegungseinschränkungen und Kontrak-
turen im Bereich der Finger mit Verdi-
ckungen von Palmaraponeurose und von
Sehnen mit Symptomen des schnellenden
Fingers auf. Mukopolysaccharidosen sind
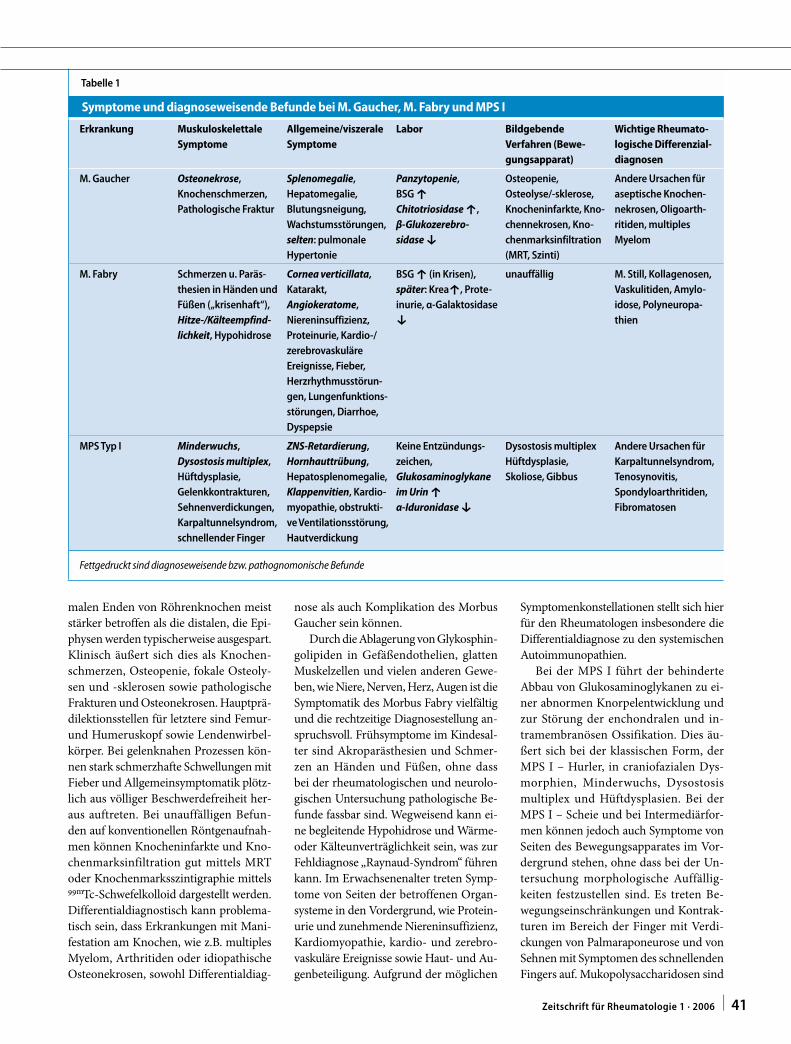
Tabelle 1
Symptome und diagnoseweisende Befunde bei M. Gaucher, M. Fabry und MPS I
Erkrankung Muskuloskelettale
Symptome
Allgemeine/viszerale
Symptome
Labor Bildgebende
Verfahren (Bewe-
gungsapparat)
Wichtige Rheumato-
logische Differenzial-
diagnosen
M. Gaucher Osteonekrose,
Knochenschmerzen,
Pathologische Fraktur
Splenomegalie,
Hepatomegalie,
Blutungsneigung,
Wachstumsstörungen,
selten: pulmonale
Hypertonie
Panzytopenie,
BSG è
Chitotriosidase è,
β-Glukozerebro-
sidase é
Osteopenie,
Osteolyse/-sklerose,
Knocheninfarkte, Kno-
chennekrosen, Kno-
chenmarksinfiltration
(MRT, Szinti)
Andere Ursachen für
aseptische Knochen-
nekrosen, Oligoarth-
ritiden, multiples
Myelom
M. Fabry Schmerzen u. Paräs-
thesien in Händen und
Füßen („krisenhaft“),
Hitze-/Kälteempfind-
lichkeit, Hypohidrose
Cornea verticillata,
Katarakt,
Angiokeratome,
Niereninsuffizienz,
Proteinurie, Kardio-/
zerebrovaskuläre
Ereignisse, Fieber,
Herzrhythmusstörun-
gen, Lungenfunktions-
störungen, Diarrhoe,
Dyspepsie
BSG è (in Krisen),
später: Kreaè, Prote-
inurie, α-Galaktosidase
é
unauffällig M. Still, Kollagenosen,
Vaskulitiden, Amylo-
idose, Polyneuropa-
thien
MPS Typ I Minderwuchs,
Dysostosis multiplex,
Hüftdysplasie,
Gelenkkontrakturen,
Sehnenverdickungen,
Karpaltunnelsyndrom,
schnellender Finger
ZNS-Retardierung,
Hornhauttrübung,
Hepatosplenomegalie,
Klappenvitien, Kardio-
myopathie, obstrukti-
ve Ventilationsstörung,
Hautverdickung
Keine Entzündungs-
zeichen,
Glukosaminoglykane
im Urin è
α-Iduronidase é
Dysostosis multiplex
Hüftdysplasie,
Skoliose, Gibbus
Andere Ursachen für
Karpaltunnelsyndrom,
Tenosynovitis,
Spondyloarthritiden,
Fibromatosen
Fettgedruckt sind diagnoseweisende bzw. pathognomonische Befunde
41Zeitschrift für Rheumatologie 1 · 2006 |

die häufigsten Ursachen für ein Karpal-
tunnelsyndrom im Kindesalter. Eine Ver-
dickung der Achillessehne kann die diffe-
renzialdiagnostische Abgrenzung zu einer
Spondyloarthritis erforderlich machen,
typischerweise sind die Bindegewebsver-
änderungen bei MPS Typ I jedoch wenig
schmerzhaft, klinische und serologische
Entzündungszeichen fehlen.
Die wichtigsten Symptome und dia-
gnoseweisenden Befunde sind in . Ta-
belle 1 zusammengefasst.
Zusammenfassung
Zusammenfassend bleibt festzuhalten,
dass für Patienten mit Morbus Gaucher,
Morbus Fabry und Morbus Scheie mitt-
lerweile Therapien zur Verfügung ste-
hen, die ihnen bei rechtzeitigem Behand-
lungsbeginn erlauben, ein nahezu norma-
les und beschwerdefreies Leben zu führen.
Organ- und Gelenkschäden können ver-
mieden oder zumindest abgemildert wer-
den. Voraussetzung für einen optima-
len Effekt der Enzymersatztherapie ist
die rechtzeitige Identifizierung der Pati-
enten, das heißt die Diagnosestellung vor
der Manifestation bleibender Schäden, sei
es am Bewegungsapparat oder im Bereich
der inneren Organe.
Der Krankheitsbeginn liegt überwie-
gend im Kindes- oder Jugendalter, wenn
auch die Prävalenz in dieser Altersgrup-
pe entsprechend dem Anteil von Kindern
und Jugendlichen in unserer Bevölkerung
nur etwa 20 der Gesamtpatientenzahl
ausmacht. Deshalb sind für die frühzeitige
Diagnosestellung zunächst die Fachärzte
für Kinder- und Jugendmedizin, wegen
der Manifestationen am Bewegungsappa-
rat vor allem die Kinder- und Jugendrheu-
matologen gefordert.
Bei der Seltenheit dieser Erkrankungen
und der anfänglichen Ähnlichkeit mit ver-
schiedenen entzündlichen und auch nich-
tentzündlichen rheumatischen Krank-
heiten handelt es sich um eine schwierige,
aber bei Beachtung der klinischen Beson-
derheiten lösbare Aufgabe. Wichtig ist es,
die lysosomalen Speicherkrankheiten dif-
ferentialdiagnostisch überhaupt zu erwä-
gen. Dies sollte insbesondere immer dann
geschehen, wenn durch die bekannten
rheumatischen Erkrankungen nicht plau-
sibel erklärte Symptomenkonstellationen
auftreten, etwa wenn bei einem Kind mit
Fingerkontrakturen bei fehlenden humo-
ralen Entzündungszeichen ein Karpaltun-
nelsyndrom und schließlich eine Horn-
hauttrübung beobachtet werden (→ Mor-
bus Scheie) oder wenn eine aseptische
Knochennekrose von einer Splenomega-
lie begleitet ist (→ Morbus Gaucher).
Sollte die Diagnose im Kindes- bzw. Ju-
gendalter noch nicht gestellt worden sein,
ist der internistische Rheumatologe gefor-
dert. Nach Erreichen des Erwachsenenal-
ters sind in der Regel verschiedene Symp-
tome deutlicher geworden, andere Manifes-
tationen, insbesondere im Bereich der inne-
ren Organe oder der Augen, eventuell erst
hinzugekommen. Bei der im Erwachsenen-
alter viel größeren Zahl von Patienten mit
entzündlich-rheumatischen Erkrankungen
wie der rheumatoiden Arthritis und großer
Variabilität auch bei diesen Erkrankungen
selbst bleibt die Identifizierung der seltenen
Scheie-, Fabry- oder Gaucher-Patienten eine
schwierige, anspruchsvolle, aber auch dring-
liche Herausforderung. Auch hier gilt es, be-
sondere Befundkonstellationen zu regis-
trieren, insbesondere, wenn es sich um Pa-
tienten handelt, deren Erkrankungsbeginn
im Kindesalter liegt. Wir sind es den betrof-
fenen Menschen und ihren Familien schul-
dig, dass sie in den Genuss der den Krank-
heitsverlauf entscheidend verbessernden
Therapie kommen, so von schweren kör-
perlichen Leiden verschont bleiben, sozi-
al integriert einer normalen Arbeit nachge-
hen können und nicht frühberentet werden
müssen.
Diese Publikation beruht auf den Er-
gebnissen eines Roundtablegesprächs, das
am 22. 9. 2004 in Frankfurt stattfand und
an dem neben den Autoren teilnahmen:
M. Backhaus/Berlin, K. Bandilla/Wiesba-
den, H.-I. Huppertz/Bremen, H.L. Kell-
ner/München, E. Reinhold-Keller/Ham-
burg, E. Weißbarth-Riedel/Hamburg.
Das Roundtablegespräch wurde durch
einen unrestricted educational grant der
Firma Genzyme GmbH unterstützt.
Korrespondierender AutorProf. Dr. B. Manger
Medizinische Klinik III,Universität Erlangen-Nürnberg,Krankenhausstraße 12, 91054 ErlangenE-Mail: [email protected]
Literatur
1. Aghion H (1934) La maladie de Gaucher dans
l’enfance: forme cardiorenale. Paris: Faculte de
Medecine de Paris
2. Allison JW, James CA, Arnold GL et al (1998) Re-
conversion of bone marrow in Gaucher disease
treated with enzyme therapy documented by MR.
Pediatr Radiol 28:237–240
3. Barton NW, Brady RO, Dambrosia JM et al (1991)
Replacement therapy for inherited enzyme defi-
ciency – macrophage-targeted glucocerebrosida-
se for Gaucher’s disease. N Engl J Med 324:1464–
1470
4. Brady RO, Kanfer JN, Shapiro D (1965) Metabolism
of Glucocerebrosides. II. Evidence of an Enzymatic
Deficiency in Gaucher’s Disease. Biochem Biophys
Res Commun 18:221–225
5. Brady RO, Gal AE, Bradley RM et al (1967) Enzyma-
tic defect in Fabry’s disease. Ceramidetrihexosida-
se deficiency. N Engl J Med 276:1163–1167
6. Brady RO (1972) Lipidoses. Biochimie 54:723–733
7. Cleary MA, Wraith JE (1995) The presenting fea-
tures of mucopolysaccharidosis type IH (Hurler
syndrome). Acta Paediatr 84:337–339
8. Cox T, Lachmann R, Hollak C et al (2000) Novel oral
treatment of Gaucher’s disease with N-butyldeo-
xynojirimycin (OGT 918) to decrease substrate bio-
synthesis. Lancet 355:1481–1485
9. Desnick RJ, Ioannou Y, Eng CM (2001) Alpha-Galac-
tosidase A deficiency: Fabry disease. In: Scriver C,
Beaudet A, Sly W, Valle D (Hrsg) The Metabolic Ba-
sis of Inherited Disease. McGraw-Hill, New York,
pp 3733–3774
10. Desnick RJ, Brady R, Barranger J et al (2003) Fabry
disease, an under-recognized multisystemic disor-
der: expert recommendations for diagnosis, ma-
nagement, and enzyme replacement therapy. Ann
Intern Med 138:338–346
11. Eng CM, Guffon N, Wilcox WR et al (2001) Safe-
ty and efficacy of recombinant human alpha-ga-
lactosidase A–replacement therapy in Fabry’s di-
sease. N Engl J Med 345:9–16
12. Fabry J (1898) Ein Beitrag zur Kenntnis der purpura
haemorrhagica nodularis. Arch Dermatol Syph 43
13. Gaucher P (1882) De l’epithelioma primitif de la ra-
te, hypertrophie idiopathique de la rate sans leu-
cemie (doctoral thesis). Paris
14. Giraldo P, Pocovi M, Perez-Calvo J et al (2000) Re-
port of the Spanish Gaucher’s disease registry: cli-
nical and genetic characteristics. Haematologica
85:792–799
15. Hein LK, Hopwood JJ, Clements PR, Brooks DA
(2003) The alpha-L-iduronidase mutations R89Q
and R89W result in an attenuated mucopolysac-
charidosis type I clinical presentation. Biochim Bio-
phys Acta 1639:95–103
16. Hollak CE, van Weely S, van Oers MH, Aerts JM
(1994) Marked elevation of plasma chitotriosidase
activity. A novel hallmark of Gaucher disease. J Clin
Invest 93:1288–1292
17. Hurler G (1919) Über einen Typ multipler Abar-
tungen, vorwiegend am Skelettsystem. Z Kinder-
heilk 24:220
18. Kauli R, Zaizov R, Lazar L et al (2000) Delayed
growth and puberty in patients with Gaucher di-
sease type 1: natural history and effect of splenec-
tomy and/or enzyme replacement therapy. Isr
Med Assoc J 2:158–163
19. Krivit W (2004) Allogeneic stem cell transplantati-
on for the treatment of lysosomal and peroxisomal
metabolic diseases. Springer Semin Immunopa-
thol 26:119–132
42 | Zeitschrift für Rheumatologie 1 · 2006
Übersichten

20. MacDermot KD, Holmes A, Miners AH (2001) An-
derson-Fabry disease: clinical manifestations and
impact of disease in a cohort of 60 obligate carrier
females. J Med Genet 38:769–775
21. Meikle PJ, Hopwood JJ, Clague AE, Carey WF
(1999) Prevalence of lysosomal storage disorders.
Jama 281:249–254
22. Neufeld EF, Muenzer J (2001) The mucopolysac-
charidoses. In: Scriver CR, Beaudet AL, Valle D, Sly
W (Hrsg) The metabolic and molecular basis of in-
herited disease. McGraw Hill, New York, pp 3421–
3452
23. Niederau C, Rolfs A, vom Dahl S et al (2001) Dia-
gnose und Therapie des Morbus Gaucher. Aktuelle
Empfehlungen der deutschen Therapiezentren im
Jahr 2000. Med Klin 96:32–39
24. Peters C, Balthazor M, Shapiro EG et al (1996) Out-
come of unrelated donor bone marrow transplan-
tation in 40 children with Hurler syndrome. Blood
87:4894–4902
25. Poll LW, Maas M, Terk MR et al (2002) Response
of Gaucher bone disease to enzyme replacement
therapy. Br J Radiol 75 Suppl 1:A25–36
26. Poorthuis BJ, Wevers RA, Kleijer WJ et al (1999) The
frequency of lysosomal storage diseases in The Ne-
therlands. Hum Genet 105:151–156
27. Rosenthal DI, Barton NW, McKusick KA et al (1992)
Quantitative imaging of Gaucher disease. Radiolo-
gy 185: 841–845
28. Scheie H, Hambrick G, Barness L (1962) A newly re-
cognized forme fruste of Hurler’s disease (gargoy-
lism). Am J Ophthalm 53:753–769
29. Shoenfeld Y, Gallant LA, Shaklai M et al (1982)
Gaucher’s disease: a disease with chronic stimula-
tion of the immune system. Arch Pathol Lab Med
106:388–391
30. Staba SL, Escolar ML, Poe M et al (2004) Cord-
blood transplants from unrelated donors in pa-
tients with Hurler’s syndrome. N Engl J Med
350:1960–1969
31. Sweeley CC, Klionsky B (1963) Fabry’s Disease:
Classification as a Sphingolipidosis and Partial
Characterization of a Novel Glycolipid. J Biol Chem
238:3148–3150
32. Thurberg BL, Rennke H, Colvin RB et al (2002) Glo-
botriaosylceramide accumulation in the Fabry kid-
ney is cleared from multiple cell types after en-
zyme replacement therapy. Kidney Int 62:1933–
1946
33. Vellodi A, Bembi B, de Villemeur TB et al (2001) Ma-
nagement of neuronopathic Gaucher disease: a
European consensus. J Inherit Metab Dis 24:319–
327
34. Weidemann F, Breunig F, Beer M et al (2003) Im-
provement of cardiac function during enzyme re-
placement therapy in patients with Fabry disease:
a prospective strain rate imaging study. Circulation
108:1299–1301
35. Wraith JE, Clarke LA, Beck M et al (2004) Enzyme
replacement therapy for mucopolysaccharidosis I:
a randomized, double-blinded, placebo-control-
led, multinational study of recombinant human al-
pha-L-iduronidase (laronidase). J Pediatr 144:581–
588
Heide Koula-Jenik, Michael Miko,
Matthias Kraft, Ralf-Joachim Schulz
(Hrsg.)
Leitfaden ErnährungsmedizinMünchen: Elsevier, Urban & Fischer 2005, 896 S., (ISBN 3-437-56530-3), 49.95 EUR
Patienten stellen immer häufiger Fragen
zum Thema Ernährung, die gelegentlich
selbst Experten, wie Gastroenterologen
und Diabetologen, verunsichern können
– zum Beispiel: Ist die makrobiotische
Kostform empfehlenswert? Nicht zu jeder
Frage ist das passende Werk zur Hand.
Auch das Internet stellt keine Lösung dar,
weil die Quellen nicht immer seriös sind.
Ideal ist dagegen der „Leitfaden Ernäh-
rungsmedizin“, der sich am Curriculum
der Ernährungsmedizin der Bundesärz-
tekammer orientiert. Das fast 900 Seiten
umfassende Nachschlagewerk, erschienen
bei Elsevier (Urban & Fischer) gibt über fast
jeden Aspekt der Ernährung knapp und
wissenschaftlich fundiert Auskunft. Über
die Makrobiotik etwa ist auf eineinhalb
Seiten das Wichtigste zu erfahren: Ihre
Grundsätze, die Lebensmittelauswahl und
ihre ernährungsphysiologische Bewer-
tung; am Ende findet sich in einem Kasten
ein handlungsorientiertes Fazit. Bewertet
werden unter anderem auch die Brigitte-
Diät, Blutgruppendiät, die Mayr-Diät und
Krebsdiäten.
Auch Sonderdiäten werden vorgestellt,
etwa Diäten bei speziellen Systemerkran-
kungen, wie Rheumaerkrankungen und
Multiple Sklerose. Dies zeigt die Brauch-
barkeit des Leitfadens – denn wer würde
einen Patienten mit MS nicht gern darüber
informieren, dass gesättigte Fettsäuren
drastisch reduziert und Omega-3-Fettsäure
vermehrt aufgenommen werden sollten?
Nicht fehlen darf in einem ernährungs-
medizinischem Kompendium die enterale
Ernährung. In dem Kapitel „Klinische Er-
nährung“ fehlt allerdings der Hinweis, dass
die enterale Ernährung auch missbraucht
werden kann, etwa um Demenzkranke
zeitsparend zu ernähren – ein Aspekt, den
sogar der Gemeinsame Bundesausschuss
kritisiert hat.
Das umfangreiche Kapitel „Praxis der
Ernährungsmedizin“, in dem vor allem er-
nährungsbedingte Krankheiten behandelt
werden, versöhnt den Leser jedoch wieder.
Deutlich zeigt sich beim Thema Diabetes,
dass sich der Leitfaden auf dem neuesten
Erkenntnisstand befindet: Mit einem Aus-
rufezeichen markiert wurde etwa die Be-
merkung: „Die Einhaltung einer Diät oder
einer Mahlzeitenfrequenz/-verteilung gilt
als überholt.“ Auch wird mit dem Vorurteil
aufgeräumt, dass Fruktose für den Diabe-
tiker-Stoffwechsel besser sei als Haushalts-
zucker. Es sind Hinweise wie diese, die das
Nachschlagewerk auch für ernährungswis-
senschaftliche Laien attraktiv macht.
Kirsten Gaede (Berlin)
Buchbesprechungen
43Zeitschrift für Rheumatologie 1 · 2006 |