Embed Size (px)
Citation preview

Maus Herpesvirus 68 (MHV-68) Infektion – Modell für die Epstein-Barr Virus-
Infektion des Menschen
Von der Medizinischen Fakultät der Rheinisch-Westfälischen Technischen Hochschule Aachen
zur Erlangung des akademischen Grades eines Doktors der Medizin genehmigte Dissertation
vorgelegt von
Matthias Engler
aus
Frankfurt am Main
Berichter: Herr Universitätsprofessor Dr. med. Klaus Ritter Herr Privatdozent Dr. med. Martin Georg Häusler Tag der mündlichen Prüfung: 4. November 2008 Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.

Für Helena und Ronja
In Gedenken an Steffi

I
Inhaltsverzeichnis
1 Einleitung
Maus Herpesvirus 68 (MHV-68) Infektion – Modell für die EBV-
Infektion des Menschen
1
Das Maus Herpesvirus 68 2
Zuordnung des Virus und dessen Struktur 2
Infektionsverlauf bei Mäusen 3
Immunreaktion auf MHV-68 Infektionen 4
Die latente Persistenz von MHV-68 5
Bekannte Modelle einer ZNS-Infektion mit MHV-68 5
Ziel dieser Arbeit 6
2 Material
2.1 Tiere und Organe 7
2.2 Gewebeschnitte 7
2.3 Zelllinien 7
2.4 Bakterien 7
2.5 Vektoren 8
2.6 Oligonukleotide 9
2.7 Antikörper 10
2.7.1 Primärantikörper 10
2.7.2 Sekundärantikörper 10
2.7.3 Tertiärantikörper 10
2.8 Virus 10
2.9 Spezielle Testsysteme 10
2.10 Chemikalien 11
2.11 Enzyme 11
2.12 Geräte 12
2.13 Zellkulturmedien 12
2.13.1 Medium für BHK-21 Zellen 12

II
2.13.2 Medium für primäre Mauslymphozyten 12
2.13.3 Medium für etablierte MHV-68 positive Lymphozytenkultur 13
2.14 Verwendete Lösungen und Puffer 13
2.15 Materialien zur Zellkultur 14
3 Methoden
3.1 Organentnahme und Aufbewahrung 14
3.2 Kultivierung von BHK-21 Zellen 14
3.3 Isolieren von Lymphozyten aus MHV-68-infizierten Mäusen 14
3.4 Archivieren adhärenter Zellen 15
3.5 Archivieren von Zellen aus Suspensionskultur 15
3.6 Isolieren von DNA 16
3.6.1 Gewebe 16
3.6.2 Blut und Zellsuspensionen 16
3.7 Isolieren von RNA 16
3.8 Spaltung mit DNase 16
3.9 C-DNA Synthese 17
3.10 Photometrische Bestimmung der DNA-Konzentration 18
3.11 Polymerase-Kettenreaktion (PCR) 18
3.11.1 Konventionelle PCR 18
3.11.2 Bedingungen für spezifische PCR-Reaktionen 19
3.11.3 Agarosegelelektrophorese 20
3.12 Real-Time-PCR 21
3.12.1 Nachweis der Amplifikate mittels SYBR Green I 22
3.12.2 Bedingungen für die Real-Time-PCR 22
3.13 Indirekter Immunfluoreszenztest (IFT) 24
3.13.1 Immunfluoreszenztest für Zellen 24
3.13.2 Immunfluoreszenztest an Gewebeschnitten 25
3.14 Vermehrung von MHV-68 26
3.15 Virustitration 26
3.16 Anzucht von MHV-68 aus Gewebeproben 27
3.17 Herstellung von Größenstandards für die Real-Time-PCR 27
3.17.1 Plasmidgenerierung und Transformation 28

III
3.17.2 Selektion der transformierten Bakterien 28
3.17.3 DNA-Minipräparation 28
3.17.4 Spaltung mit Restriktionsendonucleasen 28
3.17.5 DNA-Maxipräparation 29
3.18 Auswertung der Daten 29
4 Ergebnisse
4.1 Etablierung neuer erforderlicher Methoden 30
4.1.1 Quantifizierung der infektiösen Virionen mittels Virustitration 30
4.1.2 Qualitativer Nachweis viraler RNA und quantitativer Nachweis viraler
DNA
30
4.1.2.1 RNA-Nachweismethoden 30
4.1.2.2 Etablierung einer quantitativen Real-Time PCR zum Nachweis viraler
DNA
30
4.1.3 Methode zum Nachweis von Virusantigen im Gewebe 32
4.1.4 Etablierung einer durch MHV-68-Infektion immortalisierten B-Zelllinie 34
4.2 Morbidität und Mortalität waren vom Alter bei Infektion abhängig 35
4.3 Nachweis von MHV-68 DNA in verschiedenen Organen 37
4.4 Transkriptionsaktivität und Infektiosität von MHV-68 38
4.4.1 Nachweis auf RNA-Ebene 38
4.4.2 Direkter Nachweis von Virionen mittels des zytopathischen Effekts 40
4.5 Verlauf der Virusmengen 40
4.6 Nachweis von Virusantigen in peripheren Geweben 41
4.7 ZNS-Infektion nach nasaler Inokulation 43
4.7.1 Nachweis von Virus-DNA im ZNS 43
4.7.2 Nachweis von Virusantigen im ZNS 44
4.7.3 Vergleich virologischer und immunhistochemischer Befunde 46
5 Diskussion
5.1 Die peripheren Organe im Verlauf der Infektion 49
5.2 Natürliche Infektion des ZNS 50

IV
5.3 Zeitlicher Verlauf von Infektion und Immunabwehr 51
5.4 Ort der Infektion und Replikation 52
5.5 Ausblick 53
6 Zusammenfassung 54
7 Literaturverzeichnis 55
A Anhang 59
Abkürzungsverzeichnis 59
Abbildungsverzeichnis 60
Danksagung 62
Erklärung zur Datenaufbewahrung 63
Lebenslauf 64

1
1. Einleitung
Maus Herpesvirus 68 Infektion als Modell für die Epstein-Barr-Virus-
Infektion des Menschen
Das Epstein-Barr Virus (EBV), ein Gammaherpesvirus, verursacht das Krankheitsbild
der infektiösen Mononukleose. Das Virus persistiert lebenslang latent in B-Zellen und
kann, insbesondere bei immunkompromittierten Patienten, zu lymphoproliferativen
Erkrankungen führen. Wichtige Komplikationen der akuten oder reaktivierten EBV-
Infektion sind Meningoenzephalitis, Periventrikulitis, Hydrozephalus und
Temporallappenepilepsien. Bis zu 20 Prozent entzündlicher Schäden des ZNS bei
Kindern könnten durch das EBV verursacht sein (Häusler et al. 2002). Es gab bisher
kein Tiermodell für die EBV-Infektion. Mit der Entdeckung des murinen Herpesvirus
68 (MHV-68) (Blaskovic et al. 1980) stand dann ein tierisches Gammaherpesvirus
zur Verfügung, dessen klinische Manifestation der EBV-Infektion des Menschen sehr
ähnlich ist.
MHV-68 wurde, wie im Folgenden beschrieben, den Gammaherpesviren zugeordnet.
Der Krankheitsverlauf ähnelt der durch EBV verursachten Mononukleose. Die
entscheidende Frage war, ob das Virus auch neuropathologische Potenz besitzt.
Sowohl EBV als auch MHV-68 führen zu transienten, lytischen Infektionen von
Epithelzellen, wobei EBV Zellen des Oropharynx befällt, während MHV-68
vorwiegend in Alveolarepithelzellen repliziert wird. Gleichzeitiges Ziel sind B-
Lymphozyten, in denen eine latente Infektion etabliert wird. Beide Wirte reagieren mit
Proliferation von T- und B-Zellen. Diese polyklonale Lymphozytenaktivierung führt
dabei zu einer Vermehrung der mit Virus infizierten B-Zellen. Die Virusinfektion
induziert eine polyklonale B-Zellaktivierung, die mit der Produktion von Antikörpern
vielfältiger Spezifität verbunden ist.
Die latent infizierten Lymphozyten bleiben lebenslang in den Wirtsorganismen und
können bei Immunsuppression zu einer Reaktivierung der Infektion oder zu
Malignomen führen, wie z.B. Burkitt-Lymphom oder Nasopharynxkarzinom bei EBV,
verschiedene Lymphome bei MHV-68.
Da nur Menschen und andere Primaten Wirte des EBV sind, waren Untersuchungen
zur Pathogenität auf klinische Fälle oder in-vitro-Untersuchungen beschränkt. So

2
konnten viele Fragen zum Infektionsverlauf nicht beantwortet werden. Die große
Ähnlichkeit in Genomaufbau und Pathogenese von EBV und MHV-68 machen es
möglich, die MHV-68-Infektion der Maus als Modell für die EBV-Infektion des
Menschen mit Schwerpunkt auf neurologischen Komplikationen zu entwickeln.
Das Maus Herpesvirus 68
Zuordnung des Virus und dessen Struktur
Das murine Herpesvirus 68 wurde 1980 aus freilebenden Rötelmäusen (Myodes
glareolus) nach Passage in Hirngewebe neugeborener Mäuse isoliert (Blaskovic et
al. 1980).
Der Neurotropismus und die elektronenmikroskopisch dargestellte Virusstruktur
deuteten auf ein alpha-Herpesvirus hin. Nach der Genomanalyse wurde das Virus
den Gammaherpesviren zugeordnet (Efstathiou et al. 1990).
MHV-68 und verwandte (Gamma)herpesviren sind unter wildlebenden Mäusen weit
verbreitet (Nash et al. 2001;Blasdell et al. 2003), ähnlich wie EBV beim Menschen.
MHV-68 kann in vitro in verschiedenen Epithel- oder Fibroblastzelllinien
zytopathische Effekte verursachen. Gut geeignet für die Vermehrung des Virus sind
BHK-21 Zellen.
In bestimmten Zelllinien kann auch eine latente Infektion etabliert werden, z.B. in
NS0, einer Mausmyelomzelllinie (Sunil-Chandra et al. 1993).
Das Genom von MHV-68 besteht aus einer doppelsträngigen DNA von 118 kbp
Länge. Es enthält 73 offene Leserahmen, von denen die meisten Homologien zu
anderen Gammaherpesviren aufweisen.
Es gibt einige virusspezifische Abschnitte. Dazu gehören die Gene M 1-4 und 8
Abschnitte, die für virale tRNA kodieren (Nash et al. 2001, Bowden et al. 1997).
Die in dieser Arbeit untersuchten Gene sind mit ihren Funktion und den
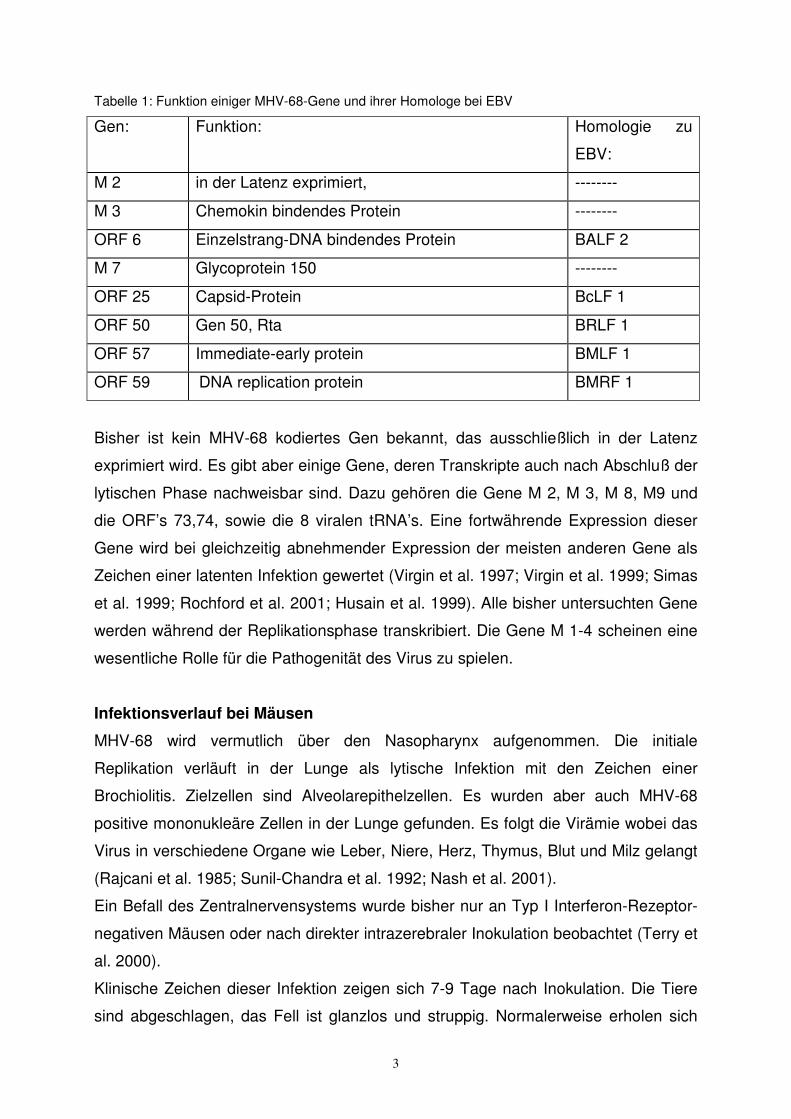
dazugehörigen homologen EBV-Genen in Tabelle 1 zusammengefasst.
3
Tabelle 1: Funktion einiger MHV-68-Gene und ihrer Homologe bei EBV
Gen:
Funktion: Homologie zu
EBV:
M 2 in der Latenz exprimiert, --------
M 3 Chemokin bindendes Protein --------
ORF 6 Einzelstrang-DNA bindendes Protein BALF 2
M 7 Glycoprotein 150 --------
ORF 25 Capsid-Protein BcLF 1
ORF 50 Gen 50, Rta BRLF 1
ORF 57 Immediate-early protein BMLF 1
ORF 59 DNA replication protein BMRF 1
Bisher ist kein MHV-68 kodiertes Gen bekannt, das ausschließlich in der Latenz
exprimiert wird. Es gibt aber einige Gene, deren Transkripte auch nach Abschluß der
lytischen Phase nachweisbar sind. Dazu gehören die Gene M 2, M 3, M 8, M9 und
die ORF’s 73,74, sowie die 8 viralen tRNA’s. Eine fortwährende Expression dieser
Gene wird bei gleichzeitig abnehmender Expression der meisten anderen Gene als
Zeichen einer latenten Infektion gewertet (Virgin et al. 1997; Virgin et al. 1999; Simas
et al. 1999; Rochford et al. 2001; Husain et al. 1999). Alle bisher untersuchten Gene
werden während der Replikationsphase transkribiert. Die Gene M 1-4 scheinen eine
wesentliche Rolle für die Pathogenität des Virus zu spielen.
Infektionsverlauf bei Mäusen
MHV-68 wird vermutlich über den Nasopharynx aufgenommen. Die initiale
Replikation verläuft in der Lunge als lytische Infektion mit den Zeichen einer
Brochiolitis. Zielzellen sind Alveolarepithelzellen. Es wurden aber auch MHV-68
positive mononukleäre Zellen in der Lunge gefunden. Es folgt die Virämie wobei das
Virus in verschiedene Organe wie Leber, Niere, Herz, Thymus, Blut und Milz gelangt
(Rajcani et al. 1985; Sunil-Chandra et al. 1992; Nash et al. 2001).
Ein Befall des Zentralnervensystems wurde bisher nur an Typ I Interferon-Rezeptor-
negativen Mäusen oder nach direkter intrazerebraler Inokulation beobachtet (Terry et
al. 2000).
Klinische Zeichen dieser Infektion zeigen sich 7-9 Tage nach Inokulation. Die Tiere
sind abgeschlagen, das Fell ist glanzlos und struppig. Normalerweise erholen sich

4
die Tiere nach 2-3 Tagen. Das Versterben der Tiere ist selten und hängt von der
initialen Virusdosis ab (Sunil-Chandra et al. 1992).
Nach oraler oder intragastraler Inokulation übersteht MHV-68 auch eine
Magenpassage und führt zu einer lytischen Infektion von Darmepithelzellen. Auch
hier folgt eine latente Infektion von B-Lymphozyten. In der Lunge lassen sich
Virionen nicht nachweisen. Untersuchungen an wild lebenden Mäusen zeigen die
Lunge als Hauptort der Viruspersistenz. Der Respirationstrakt scheint der wichtigste
Eintrittsort für das Virus zu sein (Blasdell et al. 2003; Peacock et al. 2000).
Immunreaktion auf MHV-68 Infektionen
Interferon α und β besitzen eine antivirale Wirkung und haben damit eine wichtige
Funktion bei der Abwehr und Überwindung von akuten und chronischen
Virusinfektionen. Interferonrezeptor-negative Mäuse zeigen eine schwere Beteiligung
der Lunge und eine rasche Ausbreitung des MHV-68. Die Infektion mit sonst nicht
tödlichen Dosen (105 pfu) führt bei diesen Tieren in 80-90 Prozent zum Tod (Dutia et
al. 1999c, Nash et al. 2001).
CD 4+ T-Zellen stimulieren die spezifische B-Zellantwort. Dabei beschleunigen sie
auch die Vermehrung der MHV-68 positiven B-Zellen. Die Produktion spezifischer
Antikörper hängt ebenso von den MHV-68-positiven B-Zellen ab. Interessanterweise
entstehen durch polyklonale B-Zellstimulation wesentlich mehr Antikörper, die kein
erregerspezifisches Epitop aufweisen, als solche, die sich direkt gegen MHV-68
richten.
Wenige Tage nach der Infektion werden in Milz und Lymphknoten IgM-Antikörper
gebildet, um nach etwa einer Woche von IgG abgelöst zu werden, vor allem IgG2 a/b.
12 Wochen nach Infektion ist in diesen Organen kaum noch Immunglobulinbildung
nachzuweisen. Diese Aufgabe übernehmen B-Zellen im Knochenmark zunehmend
ab der 2. Woche p.i. Auch hier überwiegen IgG2 a und b. (Usherwood et al. 1996b;
Sangster et al. 2000).
CD 8+ T-Zellen sind entscheidend für die Überwindung der MHV-68-Infektion.
Normalerweise kommt es nach 4-6 Wochen zur Ausheilung der akuten Infektion
durch die Wirkung von zytotoxischen T-Zellen und spezifischer neutralisierender
Antikörper. Fehlen CD 8+ Zellen, resultiert eine verstärkte Beteiligung der Lunge und
eine beschleunigte Ausbreitung des Virus. Auch rezidivierende Infektionen in der
Lunge wurden beobachtet. CD 8+ Zellen sind nicht in der Lage, eine latente Infektion

5
zu verhindern, scheinen aber die Zahl infizierter B-Zellen zu kontrollieren (Stevenson
et al. 1998;1999; Sangster et al. 2000; Etisham et al. 1993).
Die latente Persistenz von MHV-68
Bei infizierten Tieren können noch Monate bis Jahre nach der Infektion Virusantigene
bzw. virale DNA in dendritischen Zellen, Makrophagen, Alveolarepithelzellen und B-
Lymphozyten nachgewiesen werden, diese Zellen sind latent infiziert (Nash et al.
2001; Sunil-Chandra et al. 1992; Steward et al. 1998; Usherwood et al. 1996a; Flaño
et al. 2000).
Vereinzelt wurde bis zu 60 Tage nach Infektion Virus-DNA in Leber, Niere,
Knochenmark und Peyer’schen Plaques gefunden. In den Lungen infizierter Tiere
kann noch nach 12 Monaten Virus-DNA nachgewiesen werden, die betroffenen
Zellen sind Alveolarepithelzellen (Flaño et al. 2003; Steward et al. 1998).
Das Virus gelangt von der Lunge über die Lymphbahn in mediastinale Lymphknoten,
wo der Kontakt zu den B-Zellen hergestellt wird. Die Infektion der B-Zellen führt initial
zu einer raschen Zunahme dieser Zellen, um sich dann dauerhaft auf einem Niveau
von ca. 1 auf 5*105 Milzzellen einzupendeln.
Das Virusgenom liegt in den dauerhaft infizierten Zellen episomal vor (Steward et al.
1998).
Interessanterweise ist es bislang nicht gelungen, in vitro eine MHV-68 positive B-
Zelllinie zu etablieren. Es gelingt zwar, die Zellen in vitro mit Virionen zu infizieren,
eine Zirkularisierung der DNA jedoch bleibt aus, ebenso eine Immortalisierung der
Zellen. Je nach Kulturbedingungen sterben die Zellen nach 2-6 Wochen ab (Dutia et
al. 1999).
Bekannte Modelle einer ZNS-Infektion mit MHV-68
Die intrazerebrale Inokulation des Virus, aber auch die Infektion
immunkompromittierter Tiere, die mit sehr hoher Viruskonzentration im Blut
einhergeht, kann zu einer Infektion des ZNS mit replikativer Virusvermehrung führen.
Zeichen der Infektion sind in den Meningen und perivaskulär zu finden. Dies deutet
auf eine hämatogene Ausbreitung hin.
Neben den Meningen und Ependymzellen sind Pyramidenzellen des Hippocampus,
Oligodendrozyten und Bergmannsche Gliazellen betroffen (Terry et al. 2000). Diese
Form der Infektion endet in der Regel letal.

6
Ziel dieser Arbeit
Die bekannten Mausinfektionsmodelle sollen so abgewandelt werden, dass die
Primoinfektion stets auch zu einem Befall des ZNS führt. Zum Erreichen dieses Ziels
waren neben den infektionskinetischen Untersuchungen an der Maus auch
geeignete virusdiagnostische und molekularbiologische Methoden zu etablieren. Im
Einzelnen waren folgende Aufgaben zu lösen:
1. Anzucht des Virus, Vermehrung und Bestimmen des Infektionstiters
2. Bestimmen der geeigneten Infektionsmethode und des optimalen
Infektionszeitpunktes
3. Quantifizierung von Viren im ZNS und in ausgewählten Organen
4. Etablieren einer Real-Time-PCR zum quantitativen Virusnachweis
5. Nachweis von Transkriptionsaktivität im Gewebe
6. Nachweis viraler m-RNA mittels Nukleinsäureamplifikation
7. Nachweis viraler Antigene in Gewebeschnitten
8. Etablieren einer permanent wachsenden MHV-68-positiven Zelllinie

7
2 Material
Für die Versuche wurden die im Folgenden aufgeführten Materialien verwendet.
Allgemein gebräuchliche Laborchemikalien werden nicht im Einzelnen genannt.
2.1 Tiere und Organe
Verwendet wurden BALB/c (Acy1s, Alada, Carb, Cas1a, Cd5b, Cd72b, Cd8ab, Cd8b1b,
Ce2a, Es1b, Esda, pi1a, Gusa, H6pdb, Hbab, Hbbd, Hc1, Idh1a, Mod1a, Mup1a, Pepca,
Pgm1a, Thy1b, Trfb)
Mäuse von Taconic, Dänemark. Es wurden jeweils Muttertiere mit einem kompletten
Wurf verwendet. Die Tierversuche wurden genehmigt von der Bezirksregierung Köln
unter der Nummer: 50.203.2-AC 2,43/02.
Die Organe wurden entsprechend der Fragestellung den Tieren entnommen
(detaillierte Angaben zur Versuchsplanung finden sich im Abschnitt 3.1).
2.2 Gewebeschnitte
Die fixierten und paraffinierten Gewebeschnitte wurden freundlicherweise von Frau
Evelyn Alberg, Institut für Neuropathologie des Universitätsklinikums Aachen (ehem.
Direktor: Prof. Dr. Schröder) zur Verfügung gestellt.
2.3 Zelllinien
Die Zelllinie BHK-21 wurde 1961 aus Nierengewebe neugeborener Hamster etabliert
(MacPherson et al., Virology 16: 147-151 (1962). Die Zellen sind immortal und
wachsen adhärent. Sie eignen sich für die Vermehrung von MHV-68. Die Zelllinie
stammt aus der Deutschen Sammlung von Mikroorganismen und Zellkulturen
(DSMZ) in Braunschweig.
2.4 Bakterien
Es wurde der Laborstamm E.coli One Shot™ TOP 10 (Invitrogen) vom Genotyp F-
mrcA ∆(mrr-hsdRMS-mcrBC) Φ80lacZ∆M15 ∆lacX74recA1 deoR araD139 D(ara-
leu)7697 galU galK rpsL (StrR) endA1 nupG verwendet.
Diese Bakterien wurden zur Vermehrung der Gen-50 Amplifikate (Plasmide)
verwendet.

8
2.5 Vektoren
Der Vektor pcDNA 3.1/V5-His TOPO (Invitrogen™) liegt linear mit zwei freien dT’
Enden vor, so dass PCR-Produkte direkt einkloniert werden können. Er trägt ein
Ampicillinresistenzgen zur Selektion transformierter Bakterien.
Der Vektor ist in Abb. I dargestellt.
Abb. I: pGlow-TOPO Vektor (Invitrogen™)
9
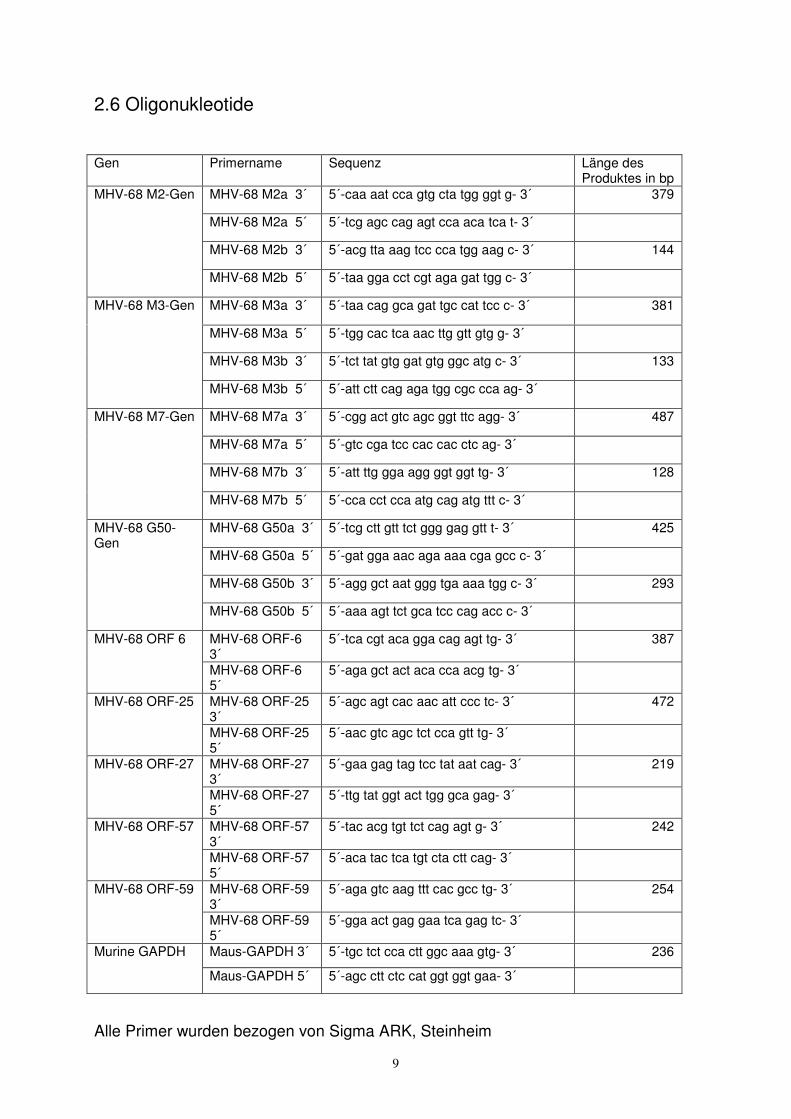
2.6 Oligonukleotide
Alle Primer wurden bezogen von Sigma ARK, Steinheim
Gen Primername
Sequenz Länge des Produktes in bp
MHV-68 M2a 3´ 5´-caa aat cca gtg cta tgg ggt g- 3´ 379
MHV-68 M2a 5´ 5´-tcg agc cag agt cca aca tca t- 3´
MHV-68 M2b 3´ 5´-acg tta aag tcc cca tgg aag c- 3´ 144
MHV-68 M2-Gen
MHV-68 M2b 5´ 5´-taa gga cct cgt aga gat tgg c- 3´
MHV-68 M3a 3´ 5´-taa cag gca gat tgc cat tcc c- 3´ 381
MHV-68 M3a 5´ 5´-tgg cac tca aac ttg gtt gtg g- 3´
MHV-68 M3b 3´ 5´-tct tat gtg gat gtg ggc atg c- 3´ 133
MHV-68 M3-Gen
MHV-68 M3b 5´ 5´-att ctt cag aga tgg cgc cca ag- 3´
MHV-68 M7a 3´ 5´-cgg act gtc agc ggt ttc agg- 3´ 487
MHV-68 M7a 5´ 5´-gtc cga tcc cac cac ctc ag- 3´
MHV-68 M7b 3´ 5´-att ttg gga agg ggt ggt tg- 3´ 128
MHV-68 M7-Gen
MHV-68 M7b 5´ 5´-cca cct cca atg cag atg ttt c- 3´
MHV-68 G50a 3´ 5´-tcg ctt gtt tct ggg gag gtt t- 3´ 425
MHV-68 G50a 5´ 5´-gat gga aac aga aaa cga gcc c- 3´
MHV-68 G50b 3´ 5´-agg gct aat ggg tga aaa tgg c- 3´ 293
MHV-68 G50-Gen
MHV-68 G50b 5´ 5´-aaa agt tct gca tcc cag acc c- 3´
MHV-68 ORF-6 3´
5´-tca cgt aca gga cag agt tg- 3´ 387 MHV-68 ORF 6
MHV-68 ORF-6 5´
5´-aga gct act aca cca acg tg- 3´
MHV-68 ORF-25 3´
5´-agc agt cac aac att ccc tc- 3´ 472 MHV-68 ORF-25
MHV-68 ORF-25 5´
5´-aac gtc agc tct cca gtt tg- 3´
MHV-68 ORF-27 3´
5´-gaa gag tag tcc tat aat cag- 3´ 219 MHV-68 ORF-27
MHV-68 ORF-27 5´
5´-ttg tat ggt act tgg gca gag- 3´
MHV-68 ORF-57 3´
5´-tac acg tgt tct cag agt g- 3´ 242 MHV-68 ORF-57
MHV-68 ORF-57 5´
5´-aca tac tca tgt cta ctt cag- 3´
MHV-68 ORF-59 3´
5´-aga gtc aag ttt cac gcc tg- 3´ 254 MHV-68 ORF-59
MHV-68 ORF-59 5´
5´-gga act gag gaa tca gag tc- 3´
Maus-GAPDH 3´ 5´-tgc tct cca ctt ggc aaa gtg- 3´ 236 Murine GAPDH
Maus-GAPDH 5´ 5´-agc ctt ctc cat ggt ggt gaa- 3´

10
2.7 Antikörper
2.7.1 Primärantikörper:
Die Antikörper gegen MHV-68 wurden nach Infektion der Mäuse zu
unterschiedlichen Zeitpunkten aus dem Blut gewonnen.
2.7.2 Sekundärantikörper:
Kaninchen anti- Maus-Immunglobuline FITC-konjugiert (DAKO, Dänemark)
oder Ziege anti-Maus-Immunglobuline nicht konjugiert (DAKO, Dänemark)
2.7.3 Tertiärantikörper:
Kaninchen anti-Ziege-Immunglobuline FITC-konjugiert (DAKO, Dänemark)
2.8 Virus
MHV-68 wurde uns freundlicherweise zur Verfügung gestellt von Herr Dr. M.
Messerle, Abteilung für Virologie, Max von Pettenkofer Institut, LMU-München. Die
Viren wurden in BHK-21 Zellen vermehrt und bei -70 °C gelagert.
2.9 Spezielle Testsysteme
Desoxynucleoside Triphosphate Set Roche, Mannheim
EasyPrep® Plasmid Mini Prep Kit Biozym, Oldendorf
LightCycler FastStart DNA Master SYBR Green I Roche, Mannheim
HiSPEED Plasmid Purification Kit Qiagen, Hilden
Omniscript® RT Kit Qiagen, Hilden
QIAamp® DNA-Blood Mini-Kit Qiagen, Hilden
QIAamp® DNA Mini-Kit Qiagen, Hilden
RedTaq™ ReadyMix™ PCRReactionMix Sigma, Steinheim
Rneasy® Mini-Kit Qiagen, Hilden
Taq-Polymerase Kit Qiagen, Hilden

11
2.10 Chemikalien
Ampicillin Roche, Mannheim
Bovines Serumalbumin (BSA) PAA, Österreich
Desoxynucleosid Triphosphate Set Roche, Mannheim
Dextransulfat 500 Na-Salz Serva, Heidelberg
DNA-Molecular Weight Marker XIV (100 bp) Roche, Mannheim
Eindeckmedium BAG, Lich
Ethidiumbromid (10 mg/ml) Sigma, Steinheim
Evansblue 2 % Euroimmun, Lübeck
Fetales Kälberserum (FKS) Biochrom KG, Berlin
L-Glutamin (Stammlsg. 200 mM) Sigma, Taufkirchen
Lipopolysaccharide (E-coli O55:B5) Sigma, Steinheim
Milchpulver Sigma, Steinheim
Minimum Essential Medium Eagle Sigma, Taufkirchen
Nicht essentielle Aminosäuren 100* (NEA) Biochrom KG, Berlin
Penicillin (200 mM)-Streptomycin (1 mg/ml)-Lösung Sigma, Taufkirchen
Primer p(DT) 15 for c-DNA synthesis Roche, Mannheim
PufferR+mitBSA
MBI Fermentas
RNAlater (Konservierungsstoff für RNA-haltige
Proben)
Ambion, England
RPMI-1640 Medium Sigma, Taufkirchen
Trypsin Sigma, Steinheim
2.11 Enzyme
DNase I (10 U/µl) Roche, Mannheim
Hind III (10 U/µl) MBI Fermentas, St.Leon-
Rot
Protease Qiagen, Hilden
Protector RNase Inhibitor (40 U/µl) Roche, Mannheim
Proteinase K Qiagen, Hilden

12
Xho I (10 U/µl) MBI Fermentas, St.Leon-
Rot
2.12 Geräte
Centrifuge 5403 (Kühlzentrifuge) Eppendorf, Hamburg
Eppendorf centrifuge 5414c (für1,5/2ml
Reaktionsgefäße)
Eppendorf, Hamburg
Fluoreszenzmikroskop Zeiss, Jena
Gene Quant II (RNA/DNA-Calculator) Pharmacia Biotech
Labofuge GL (für Zellsusp.) Kendro, Langenselbold
Lichtmikroskop Zeiss, Jena
Lightcycler Roche, Mannheim
Retsch Mixer Mill 300 (MM 300) Retsch, Haan
Thermocycler UNO II Biometra, Göttingen
Thermomixer 5436 Eppendorf, Hamburg
2.13 Zellkulturmedien
2.13.1 Medium für BHK-21 Zellen
Minimum Essential Medium Eagle
5 % FKS (1 % für Virustitrationsversuche) (v/v)
2 % Penicillin-Streptomycin (v/w)
0,2 % NEA (v/w)
2.13.2 Medium für primäre Mauslymphozyten
RPMI-1640
20 % FKS (v/v)
2 % Penicillin-Streptomycin (v/w)
2 mM L-Glutamin
12,5 µg/ml Lipopolysaccharide (E-coli O55:B5)
20 µg/ml Dextransulfat
0,2 % NEA (v/w)

13
2.13.3 Medium für etablierte MHV-68 positive Lymphozytenkultur
RPMI-1640
10 % FKS (v/v)
2 % L-Glutamin (v/w)
2 % Streptomycin-Penicillin (v/w)
0,2 % NEA (v/w)
2.14 Verwendete Lösungen und Puffer
1x PBS (pH 7,4)
NaCl 140 mM
KCl 3 mM
Na2HPO4 10 mM
K2HPO4 2 mM
0,5x TBE
Tris-Puffer 45 mM
Essigsäure 45 mM
EDTA (pH 8,0) 1 mM
LB-Medium (pH 7,0)
NaCl 1,0 % (w/v)
Pepton 1,0 % (w/v)
Hefeextrakt 0,5 % (w/v)
SOC-Medium
Bactotrypton 2,0 %
Hefeextrakt 0,5 %
NaCl 10 mM
KCl 25 mM
MgCl2 10 mM
MgSO4 10 mM
Glucose 20 mM

14
2.15 Materialien für Zellkulturen
Cellstar Gewebekulturflaschen (50 ml/ 25 cm²) Greiner Bio One, Frickenhausen
Falcon Multiwell 24 Well Becton Dickinson Europe
Chamber-Slide System Lab-Tek II Nalge Nunc int. Corp., USA
3 Methoden
3.1 Organentnahme und -aufbewahrung
Die Mäuse wurden bei Krankheitszeichen oder Tod, ansonsten an den Tagen 7, 14
oder 35 nach Virusinokulation getötet. Dies geschah mit einer Überdosis Isofluran.
Es folgte eine Herzpunktion zur Blutgewinnung. Anschließend wurden die Organe
unter sterilen Bedingungen entnommen. Für die virologischen Untersuchungen
wurden die Proben bei -70°C archiviert. Für die histolopathologische Aufarbeitung
wurden die Proben in 4 %iger Paraformaldehyd-Lösung fixiert. Folgende Organe und
Gewebe wurden entnommen:
Milz, Leber, Niere, Herz, Lunge, Psoasmuskel, Thymus, Großhirn, Kleinhirn
3.2 Kultivierung von BHK-21 Zellen
BHK-21 Zellen wurden in 25 cm² Zellkulturflaschen bei 37 °C in
wasserdampfgesättigter Atmosphäre mit 5 % CO2 kultiviert.
Die Zellen wurden nach jeweils 7 Tagen subkultiviert: nach Trypsinierung lösten sich
die Zellen vom Boden der Kulturflaschen ab. Sie wurden in 10 ml frischem
Kulturmedium (v/v) resuspendiert. Diese Suspension wurde mit Medium auf 100 ml
aufgefüllt, je 10 ml wurden auf neue Kulturflaschen verteilt.
3.3 Isolieren von Lymphozyten aus MHV-68 infizierten Mäusen
Milzgewebe MHV-68-positiver Mäuse wurde unmittelbar nach Entnahme durch
Zellfilter (Greiner Bioone) gedrückt. Das entstandene Zellhomogenisat wurde mittels
Ficoll-Dichtegradientenzentrifugation aufgetrennt. Sichtbare lymphozytenhaltige
Banden wurden isoliert und die Zellen dreimal mit eiskaltem PBS gewaschen.

15
Die Lymphozyten wurden in Konzentrationen von 101 bis 106 Zellen/ml in
Spezialmedium (2.13.2) resuspendiert und in Vertiefungen von 24-Well-Platten
gegeben. Die Inkubation erfolgte bei 37 °C in wasserdampfgesättigter Atmosphäre
mit 5 % CO2. Die Zellen wurden mit 106 Viren/ml inkubiert.
Nachdem die Zellen eine stabile Teilungsquote erreicht hatten, wurden sie in 50 ml
Kulturflaschen umgesetzt. Die Vermehrung verlief weiter kontinuierlich.
3.4 Archivieren adhärenter Zellen
BHK-21-Zellen einer gut bewachsenen Zellkulturflasche wurden mittels Trypsinierung
vom Flaschenboden gelöst und in 2 ml Kulturmedium resuspendiert. Die
Zellsuspension wurde zu je 1 ml in Zentrifugengefäße gegeben und die Zellen
5 Minuten bei 1.600 rpm pelletiert (LabofugeGL, Kendro). Nach dem Dekantieren des
Überstands wurde jedes Zellsedimentpellet in 1 ml Lösung A resuspendiert. Nach
Zugabe von 1 ml Lösung B wurden die Lösungen durch Auf- und Abpipettieren
gemischt. Die Mischung wurde in Kryoröhrchen überführt und in einer Polystyrol-Box
bei -70 °C eingefroren. Nach 24 Stunden wurden die Röhrchen in flüssigen Stickstoff
(-196 °C) überführt.
Lösung A (für 10 Kryoröhrchen)
Medium ohne Zusätze 7,5 ml
FKS 2,5 ml
Lösung B (für 10 Kryoröhrchen)
Medium ohne Zusätze 5,5 ml
FKS 2,5 ml
DMSO 2,0 ml
3.5 Archivieren von Zellen aus Suspensionskultur
In Suspension wachsende Zellen wurden in 5 ml Einheiten in Zentrifugenröhrchen
überführt und weiterhin wurde entsprechend dem Einfrierprotokoll für adhärente
Zellen verfahren.

16
3.6 Isolieren von DNA
3.6.1 Gewebe:
Für das Isolieren von DNA aus solidem Gewebe wurden die Proben zunächst
homogenisiert. In ein 2 ml-Reaktionsgefäß wurde zu 25 mg Gewebe (Milz/ Thymus
10 mg) 180 µl Puffer sowie eine Stahlkugel mit 3 mm Durchmesser gegeben. Der
Ansatz wurde zweimal für 20 sec bei 30 Hz in der Zellmühle MM 300 (Firma
Retsch®) homogenisiert. Nach Zugabe von 20 µl Proteinase K (QIAmp DNA Mini-Kit)
wurde der Ansatz für 1 Stunde bei 56° C in einem Thermoblock schüttelnd inkubiert.
Die weitere Aufarbeitung erfolgte nach Herstellerangaben für das QIAmp DNA Mini-
Kit. Die DNA wurde in 200 µl Puffer AE eluiert und bei -20 °C gelagert.
3.6.2 Blut und Zellsuspensionen:
Für die Isolierung von DNA aus Blut oder Zellsuspensionen wurde das QIAmp DNA-
Blood Mini-Kit verwendet. Die Aufarbeitung erfolgte nach Herstellerangaben.
3.7 Isolieren von RNA
Gewebeproben wurden in 10 mg-Einheiten mit Hilfe einer 3 mm Stahlkugel und 350-
600 µl RLT-Puffer (RNeasy Kit, Qiagen) in der Zellmühle MM 300 2x für 1 Minute bei
20 Hz homogenisiert.
Die so entstandene Zellsuspension wurde nach Herstellerangaben mit dem RNeasy-
Kit weiterbehandelt. Die RNA wurde mit 30 µl RNase-freiem Wasser eluiert und bei
-70 °C gelagert.
3.8 Spaltung mit DNase
Nach Isolieren der RNA (3.7) kann eine Verunreinigung mit genomischer DNA nicht
völlig ausgeschlossen werden. Deshalb wurde die extrahierte RNA mit DNase I
behandelt. Dieses Enzym ist eine Doppelstrang-spezifische Endonuklease aus
Rinderpankreas.
Der Standard-Reaktionsansatz wurde eine Stunde bei 37 °C inkubiert. Die
Inaktivierung des Enzyms erfolgte über 10 Minuten bei 95 °C.
Standard-Reaktionsansatz:

17
RNA (50 ng – 2 µg) x µl
RNase Inhibitor (40 U/µl, Roche) 0,5 µl
Puffer für DNase I (10 x) 4,0 µl
DNase I, RNase-frei (10 U/µl, Roche) 2,0 µl
RNase-freies Aqua dest. ad 40,0 µl
Puffer für DNase I (10 x):
Tris HCl (pH 7,5) 100 mM
3.9 C-DNA Synthese
Da RNA nicht direkt mittels PCR amplifiziert werden kann, muss sie zunächst in
cDNA umgeschrieben werden. Dieses geschieht mit Hilfe einer RNA-abhängigen
DNA-Polymerase - der Reversen Transkriptase (RT). Als Primer wurde ein Oligo(dT)-
Primer eingesetzt, der an den poly(A)-Schwanz am 3’-Ende der mRNA bindet. Das
Enzym bindet an die freie 3’-OH-Gruppe des Primers und synthetisiert einen zur
RNA-Matrize komplementären cDNA-Strang.
Die Synthese einzelsträngiger cDNA erfolgte mit dem OmniscriptTM RT-Kit von
Qiagen nach Herstellerangaben. Es wurden RNA-Mengen zwischen 50 ng und 2 µg
als Matrize eingesetzt.
Die isolierte DNA-freie RNA wurde auf Eis aufgetaut und 5 Minuten bei 65 °C
denaturiert (Thermomixer 5436, Eppendorf). Nachdem eine geeignete Menge des
Standard-Reaktionsansatzes zugesetzt worden war, erfolgte die Transkription
1 Stunde bei 37 °C. Anschließend wurde der Reaktionsansatz für 5 Minuten auf
95 °C (Thermomixer 5436, Eppendorf) erhitzt, um die Reaktion zu stoppen, dann
sofort auf Eis gekühlt.
Standard-Reaktionsansatz:
RNA 20 µl
10 x RT-Puffer 4,0 µl
dNTP-Mix (5 mM) 4,0 µl
Oligo(dT)-Primer (10 µM, Roche) 4,0 µl
RNase Inhibitor (10 U/µl, Roche) 0,5 µl
Omniscript RT (4 U/µl) 2,0 µl
RNase-freies Aqua bidest. 15,5 µl

18
3.10 Photometrische Bestimmung der DNA-Konzentration
Das Vorhandensein von Nukleinsäuren kann photometrisch bei 260 nm im
Spektralphotometer GeneQuant II (Pharmacia Biotech) gemessen werden. Eine
Extinktion E260 = 1 entspricht einer Konzentration von 50 µg DNA pro ml Lösung.
Die DNA-Konzentration berechnet sich nach der Formel: DNA (µg/ml) = OD x
spezifische Verdünnung x 50.
3.11 Polymerase-Kettenreaktion (PCR)
Die Methode der polymerase chain reaction (PCR) (SAIKI et al., 1988) wurde im
Jahre 1987 von Kary B. Mullis entwickelt. Die Methode macht es möglich,
Nukleotidsequenzen in vitro millionenfach zu vervielfältigen. Dadurch werden auch
sehr geringe Mengen an DNA leicht nachweisbar.
Bei der PCR werden zwei Oligonukleotide (Primer) eingesetzt, die homolog zu den
beiden Enden der Zielsequenz sind. Die doppelsträngige DNA wird zunächst
denaturiert. In der Annealingphase hybridisieren die Primer an die einzelsträngige
Matrizen-DNA. Ein hitzestabiles Enzym, die Taq-Polymerase, synthetisiert während
der Elongation vom 3’-Ende der Primer ausgehend einen neuen DNA-Doppelstrang.
Durch die Wahl eines gegenläufig orientierten Oligonukleotid-Primerpaares kann
gezielt die DNA-Sequenz zwischen den beiden Primern vervielfältigt werden.
Aufgrund einer zyklischen Wiederholung der einzelnen Reaktionsschritte -
Denaturierung, Annealing und Elongation - wird die Matrizen-DNA exponentiell
amplifiziert.
3.11.1 Konventionelle PCR
Die PCR-Reaktionen wurden in einem Thermocycler mit Heizdeckel (Thermocycler
UNO II, Biometra) unter Hot-Start-Bedingungen durchgeführt.
Der Reaktionsansatz wurde mit Readymix™ hergestellt, der alle für die Amplifikation
nötigen Reagenzien (Nukleotide, Taq-Polymerase, MgCl, PCR-Puffer) in
Standardkonzentrationen enthält.

19
Standardreaktionsansatz:
DNA bzw. cDNA 5 µl
Readymix 25 µl
Primer 3’ (10 µM) 2 µl
Primer 5’ (10 µM) 2 µl
Aqua bidest 16 µl
Zur Erhöhung der Sensitivität wurde für ausgewählte Gene eine nested-PCR
durchgeführt, d.h. ein Teil des PCR-Produktes wird erneut in eine PCR eingesetzt.
Hierbei werden Primer verwendet, die innerhalb des bereits amplifizierten DNA-
Fragmentes liegen.
Standardreaktionsansatz:
PCR-Produkt 2 µl
Readymix 25 µl
Primer 3’ (10 µM) 2 µl
Primer 5’ (10 µM) 2 µl
Aqua bidest 19 µl
3.11.2 Bedingungen für spezifische PCR-Reaktionen
Die Amplifikation der MHV-68-Gene G 50, M 2, M 3 und M 7 wurde unter folgenden
PCR-Bedingungen durchgeführt:
Äußere PCR: Nested PCR:
94 °C 2 min 94 °C 2 min
94 °C 1 min 94 °C 1 min
60 °C 1 min x 25 63 °C 1 min x 35
72 °C 1 min 72 °C 1 min
72 °C 5 min 72 °C 5 min
4 °C ∞ 4 °C ∞
Die Amplifikation der Gene ORF 6, ORF 25, ORF 27, ORF 57 und ORF 59 wurde
unter folgenden PCR-Bedingungen durchgeführt:

20
PCR:
94 °C 2 min
94 °C 1 min
55 °C 1 min x 30-40
72 °C 1 min
72 °C 5 min
4 °C ∞
Die Amplifikation des Murine-GAPDH Gens wurde unter folgenden PCR-
Bedingungen durchgeführt:
PCR:
3.11.3 Agarosegel-Elektrophorese
Für den Nachweis von DNA-Amplifikaten nach PCR ist die Agarosegel-
Elektrophorese geeignet. Die negativ geladenen DNA-Amplifikate wandern im
elektrischen Feld und werden nach ihrer Größe aufgetrennt.
Die als Träger verwendete Agarose ist ein Polysaccharid aus Rotalgen. Es wird
durch Aufkochen in Puffer gelöst und polymerisiert beim Abkühlen. Durch Variation
der Agarosekonzentration können lineare DNA-Fragmente der Größe 0,1 bis 60 kb
getrennt und dargestellt werden [GASSEN & SCHRIMPF, 1999].
DNA-Fragmente ≤ 1 kb werden dabei auf 1,5 %ige TBE-Agarosegele aufgetragen.
Zur Herstellung der Gele wird das Agarose-Puffer-Gemisch solange in einer
Mikrowelle erwärmt, bis die Agarose gelöst ist. Nach Abkühlen auf 65 °C wird den
Agarosegelen Ethidiumbromid zugegeben. Dieses interkaliert in DNA und
94 °C 2 min
94 °C 1 min
54 °C 45 sec x 45
72 °C 1 min
72 °C 7 min
4 °C ∞

21
fluoresziert bei Anregung mit Lichtwellen von 302 nm, so dass eine optische
Darstellung der Banden möglich ist.
Parallel zu den Proben wird ein Größenmarker mit DNA-Fragmenten definierter
Länge aufgetragen. Dies erlaubt die Bestimmung der Größe der zu untersuchenden
Proben. Die Elektrophorese erfolgt in 0,5 x TBE-Puffer und die DNA wird bei 5 V/cm
Elektrodenabstand aufgetrennt. Die Dokumentation erfolgt durch die „BioDoc
Analyze Camera” (Biometra) und digitale Speicherung.
TBE-Puffer (0,5 x):
Tris 45 mM
Borsäure 45 mM
EDTA 1 mM
TBE-Gel, 1,5 %ig (500 ml):
Agarose 7,5 g
TBE-Puffer (0,5 x) 500 ml � erwärmen, auf 65 °C abkühlen
Ethidiumbromid (10 mg/ml) 5 µl
Farbmarker:
Ficoll Polymer 10 % (w/v)
Bromphenolblau 0,25 % (w/v)
Xylemcyanol 0,25 % (w/v)
EDTA 0,25 M
Größenmarker:
DNA-Längenstandards XIV, 100-Basenpaarleiter: 0-2500 bp (Roche)
3.12 Real-Time-PCR
Die LightCycler-Technologie kombiniert „rapid cycle-PCR” und „Real-Time-PCR”
miteinander. Verwendet werden hierzu Glaskapillaren mit nur 20 µl
Reaktionsvolumen, dadurch wird das Verhältnis des Flüssigkeitsvolumens zur
Oberfläche und damit die Temperaturübertragung optimiert. Schnelle

22
Temperaturveränderungen im Reaktionsgefäß sind somit möglich, so daß ein PCR-
Zyklus weniger als 30 Sekunden dauert (rapid cycle-PCR). Die Analyse der PCR-
Produkte erfolgt während der Amplifikation mittels Fluoreszenz (real-time-PCR). Ein
zusätzlicher Analysenschritt entfällt und die Gefahr von Kontaminationen wird
minimiert. Im Anschluß an die PCR wird der Schmelzpunkt des Amplikons ermittelt.
Für jedes Amplifikationsprodukt besteht eine charakteristische Schmelztemperatur.
Diese ist abhängig von den gewählten PCR-Bedingungen, den eingesetzten Primern
und der Amplifikationsmethode.
Die Detektion der amplifizierten PCR-Produkte erfolgte mittels SYBR Green I
Farbstoff (s.u.).
3.12.1 Nachweis der Amplifikate mittels SYBR Green I
Für den Nachweis von PCR-Amplifikaten mittels SYBR Green wurde das
LightCycler-FastStart DNA Master SYBR Green I-Kit (Roche) verwendet. Der
Farbstoff SYBR Green I interkaliert mit doppelsträngiger DNA. Durch Anregung mit
UV-Licht sendet SYBR Green I Lichtquanten aus, die bei 530 nm gemessen werden.
Die Detektion von PCR-Produkten erfolgt in der Elongationsphase, in der
doppelsträngige DNA entsteht. Die Messung erfolgt beim Übergang der lag-Phase in
die log-Phase. Da SYBR Green I nur beim Interkalieren angeregt werden kann, lässt
sich der Schmelzpunkt der Produkte bestimmen, indem in einem abschließenden
Schritt die Produkte erhitzt werden, bis bei erreichtem Schmelzpunkt die Fluoreszenz
plötzlich stark abnimmt (Denaturierung der doppelsträngigen DNA).
3.12.2 Bedingungen für die Real-Time PCR
Für die Amplifikation von Gen-50 wurden Primer verwendet, die auch in der
konventionellen PCR zum Einsatz kamen. Zusätzlich zu den Proben wurde ein
Größenstandard mitgeführt, d.h. eine bekannte Menge spezifischer DNA, die
ermöglicht, durch Vergleich der Fluoreszenzintensitäten die Menge der eingesetzten
DNA zu errechnen.
Als Größenstandard diente ein Vektor mit einklonierter Gen-50 Gensequenz. Hiervon
wurden verschiedene Verdünnungen eingesetzt (102 bis 106 Genomäquivalente/µl).
Auch hier wurde eine Nested-PCR durchgeführt. Dabei wurden in einem ersten
Schritt mittels konventioneller PCR DNA-Amplifikate der Proben gewonnen, die in
einem zweiten Schritt als Proben in eine Real-Time PCR eingesetzt wurden.

23
Gen-50 PCR:
Ansatz:
DNA 5 µl (bzw. 2 µl Standardverdünnung)
MgCl (2,5 mM) 2,5 µl
PCR-Puffer 10* 5 µl
dNTP’s (2,5 mM) 5 µl
Primer 3’ (10 pmol/µl) 2 µl
Primer 5’ 2 µl
Taq-Polymerase (5 U/µl) 0,5 µl
Aqua bidest 28 µl (bzw. 31 µl)
PCR-Bedingungen:
94 °C 2 min
94 °C 1 min
60 °C 1 min x 15
72 °C 1 min
72 °C 5 min
4 °C ∞
Real-Time PCR von Gen-50:
Ansatz:
PCR-Produkt (äußere PCR) 2 µl
MgCl (25 mM) 2,5 µl
Primer 3’ 1 µl
Primer 5’ 1 µl
Fast Start Reaktionsmix 2 µl
Der Fast Start Reaktionsmix enthält PCR-Puffer, Taq-Polymerase, MgCl (10 mM)
und dNTP’s.

24
Realtime-PCR-Bedingungen:
Denaturierung 10 min 95°C
Amplifikation 0 s 95 °C
10 s 63 °C x 45
5 s 72 °C
Schmelzkurve ab 83 °C
Kühlung 1min 40 °C
3.13 Indirekter Immunfluoreszenztest (IFT)
Der indirekte Immunfluoreszenztest dient dem Nachweis von Virusantigenen in
Zellen bzw. Geweben. Dabei werden Antikörper gegen virale Epitope mit der Probe
inkubiert. Es resultiert eine Antigen-Antikörperreaktion, die durch
fluoreszenzmarkierte Anti-Antikörper nachweisbar wird. Durch Anregung mit UV-
Licht, z.B. bei 519 nm werden spezifische Virusantigene unter dem
Fluoreszenzmikroskop lokalisiert. Da die Methoden für Zellen und Gewebe sich recht
stark unterscheiden, werden sie getrennt dargestellt.
3.13.1 Immunfluoreszenztest für Zellen
a) Adhärente Zellen
Zu einem Monolayer ausgewachsene BHK-21-Zellen wurden mit Trypsin abgelöst
(siehe 3.2) und in 35 ml Medium (5 % FKS v/v) resuspendiert. Je 500 µl der
Zellsuspension wurde in Chamber-Slide-Objektträger gegeben.
MHV-68 negative BHK-Zellen wurden bis zum vollständigen Bewuchs inkubiert
(37 °C, 90 % Luftfeuchtigkeit, 5 % CO2).
Für MHV-68 positive Zellkulturen wurden die Kammern 24 h nach Befüllung mit 104
pfu (siehe 3.15) Virus beimpft und weitere 2 Tage inkubiert.
Anschließend wurde das Medium entfernt und die Kammern von den Objektträgern
gelöst.
Um die Zellen auf den Objektträgern zu fixieren wurden diese für 10 Minuten in
eiskaltes Aceton gegeben. Die fixierten Zellen wurden bis zur Versuchsdurchführung
bei -20 °C gelagert.

25
b) Vorbereitung der Suspensionszellen
Aus den Kulturflaschen wurde 1 ml Zellsuspension entnommen und bei 1500 rpm
(Eppendorf 5415c) 10 min zentrifugiert. Das entstandene Zellpellet wurde in 100 µl
PBS resuspendiert. Je 10 µl dieser Suspension wurden in ein Auftragsfeld eines 10-
Feld-Objektträgers pipettiert. Nach 30-minütiger Trocknung bei 37°C erfolgte die
Fixierung mittels eiskaltem Aceton für 10 Minuten.
Durchführung des Immunfluoreszenztestes:
Die Zellen auf den Feldern wurden mit 10 µl MHV-68-Antiserum (Verdünnung 1:200
in PBS v/v) überschichtet und 30 min bei 37 °C in einer feuchten Kammer inkubiert.
Anschließend wurden die Objektträger 3x5 min in PBS gewaschen, um nicht
gebundene Antikörper zu entfernen.
Die einzelnen Felder wurden dann mit 10 µl Sekundärantikörper (Kaninchen anti-
Maus IgG, FITC-gekoppelt 1:30 in PBS V/V) überschichtet und 30 min bei 37 °C in
einer feuchten Kammer inkubiert.
Danach wurde wiederum 3x5 min in PBS gewaschen, wobei beim 3. Waschschritt 50
µl Evans Blue zur Gegenfärbung zugegeben wurden.
Die fertig bearbeiteten Präparate wurden mit einem Tropfen „antifading“
Eindeckmedium und einem Deckglas eingedeckt und direkt am
Fluoreszenzmikroskop ausgewertet oder bei -20 °C gelagert.
3.13.2 Immunfluoreszenztest an Gewebeschnitten
Zunächst mussten die Schnitte entparaffiniert und gewässert werden. Hierzu wurden
die Objektträger in Küvetten 20 min in Xylol, anschließend in absteigender Reihe je
7 min in 100 %igem, 96 %igem und 70 %igem Ethylalkohol und abschließend für
2 min in Aqua bidest. inkubiert.
Durch die Fixierung und Paraffinierung der Schnitte leiden die Antigene. Zur
Verbesserung der Antigenität wurden die Proben in Zitratpuffer 20 min gekocht und
langsam wieder auf Raumtemperatur gebracht.
Vor der weiteren Behandlung wurden die Schnitte 3*5 min in PBS gewaschen.
Um unspezifische Antigenbindungsstellen zu blockieren wurden die Gewebe noch 30
min in 100 ml PBS (supplementiert mit 1 g BSA und 0,2 g Milchpulver) inkubiert
(geblockt) und anschließend 3x5 min in PBS gewaschen.

26
Die Gewebeschnitte wurden mit 200 µl Primärantikörper (Mausserum 1:200 in PBS)
überschichtet und 30 min bei 37 °C in einer feuchten Kammer inkubiert.
Anschließend wurde 3x5 min in PBS gewaschen.
Weiterhin wurden die Schnitte mit 200 µl Sekundärantikörper (Ziege anti-Maus IgG
1:100 in PBS) überschichtet und 30 min bei 37 °C in einer feuchten Kammer
inkubiert, und wie bereits beschrieben gewaschen.
Die letzte Inkubation erfolgte mit 200 µl Tertiärantikörper (Kaninchen anti-Ziege IgG
gekoppelt mit FITC 1:30 in PBS) wiederum bei 37 °C in der feuchten Kammer.
Gewaschen wurde mit folgender Modifikation:
Beim letzten Waschschritt erfolgte die Zugabe von 50 µl Evansblue zum
Gegenfärben. Anschließend wurde eingedeckt und unter dem Fluoreszenzmikroskop
ausgewertet. Die Dokumentation erfolgte durch Photographie. Die Lagerung der
Objektträger erfolgte bei -20 °C.
3.14 Vermehrung von MHV-68
BHK-21 Zellen eignen sich für die Vermehrung von MHV-68. Die Viren durchlaufen in
den Zellen einen lytischen Zyklus.
Es wurden die Zellen einer gut bewachsenen Kulturflasche mit 2 ml Trypsin abgelöst,
in 50 ml Medium (BHK-21 Standardmedium) aufgenommen und auf 5 neue
Kulturflaschen verteilt. Nach 24 h Kultivierung bzw. bei Erreichen von 50-60 %
Konfluenz wurden die Zellen mit 100 µl Virussuspension (entspricht ca. 106 pfu)
infiziert.
Nach 3-4 Tagen wurden die Überstände gepoolt, bei 2000 rpm für 10 min in der
Labofuge GL (Kendro) zentrifugiert. Der Überstand wurde dekantiert, sterilfiltriert und
in 1 ml Aliquots bei -70 °C eingefroren. Die Pellets wurden verworfen. Der
Virusnachweis erfolgte mittels G-50 PCR oder durch Nachweis eines zytopathischen
Effektes.
3.15 Virustitration
Die Quantifizierung zytopathogener Viren erfolgt über den zytopathischen Effekt.
Dabei wird eine virushaltige Suspension in einer logarithmischen Reihe verdünnt. Mit
den Verdünnungen werden im Mehrfachansatz Zellkulturen infiziert. Die Menge an
infektiösem Material wird in plaque forming units (pfu) angegeben.

27
(Kärber G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche.
Arch. Exp. Pathol. Pharmakol. 162, 480-483 (1931).
Zur Quantifizierung der MHV-68 Virionen wurden BHK-21 Zellen in zwei 24-well-
Platten 24 h kultiviert, bis die Böden zu ca. 60 % bewachsen waren. Das
Kulturmedium wurde verworfen und die Zellen wurden mit Verdünnungen der
Virussuspension (1:10-3 bis 1:10-12) in BHK-Medium (1 % FKS) 10 Tage inkubiert. Die
Virusverdünnung wurde im Vierfach-Ansatz getestet. Die Kontrollen enthielten
virusfreies Medium.
Die Zellen wurden täglich unter dem Mikroskop kontrolliert und die Ergebnisse
protokolliert. Am 10. Tag nach Infektion wurde dokumentiert, welche Vertiefungen
noch bewachsen waren und welche nicht.
Die Verdünnungsstufen, bei denen eine teilweise oder vollständige Lyse vorlag,
wurden in die Kaerber-Formel (lgID50=L1,0+L(Int)x(S-0,5)) eingesetzt. Errechnet
wurde die Zahl an plaque forming units in der Stammlösung.
3.16 Anzucht von MHV-68 aus Gewebeproben
Infektiöse Viruspartikel im Gewebe wurden mittels Virustitration nachgewiesen. Den
Gewebeproben wurde 10 mg Material entnommen und in 1 ml BHK-Medium (2.13.1;
1 % FKS) zweimal für 20 Sekunden bei 30 Hz in der Zellmühle MM 300
homogenisiert. Die Zellreste wurden 10 min bei 1700 rpm in der Zentrifuge 5414c
abzentrifugiert.
Der Überstand wurde gewonnen und Verdünnungen von 1:10 bis 1: 10.000 (v/v)
hergestellt. Weiter wurde wie bei der Virustitration (3.15) verfahren. Ein Absterben
der mit den Verdünnungen inkubierten BHK-21 Zellen wurde als positiver Befund
gewertet.
3.17 Herstellung von Größenstandards für die Real-Time-PCR
Für die Quantifizierung von DNA mittels Real-Time PCR wird eine definierte Menge
an spezifischer DNA benötigt, die als Standard dient und es möglich macht, durch
Vergleich der Fluoreszenzintensität die Menge an gesuchter DNA in der Probe zu
berechnen. Dazu sind Vektoren geeignet, die den spezifischen Genabschnitt
enthalten. Es resultieren genau quantifizierbare DNA-Einheiten. Sie lassen sich in

28
beliebiger Menge in Bakterien vermehren und können für die Quantifizierung
spezifischer PCR-Produkte verwendet werden.
3.17.1 Plasmidgenerierung und Transformation
Als spezifische DNA wurde der G-50 Abschnitt zwischen den G-50 a Primern
gewählt. Dieser DNA-Abschnitt wurde mittels konventioneller PCR amplifiziert.
Zur Verminderung von Verunreinigungen wurde das Produkt in einem Agarosegel
getrennt, die spezifische Bande wurde ausgeschnitten. Mit dem Gel-Extraction-Kit
(Qiagen) wurde die DNA aus dem Gel extrahiert. Die aufgereinigten DNA-Fragmente
wurden in pcDNA 3.1 Vektoren kloniert. Die Vektoren liegen linear vor, haben zwei
freie Thymin-Enden, so daß sich die PCR-generierte DNA mit ihren zwei freien
Adenin-Enden problemlos einfügen läßt. Es entsteht ein zirkulärer Vektor.
Dieser wurde mittels Hitzeschocktransformation in kompetente E. coli (One Shot™
TOP 10) eingebracht.
3.17.2 Selektion der transformierten Bakterien
Die Bakterien wurden in SOC-Medium aufgenommen und für 1 h bei 37 °C unter
Schütteln inkubiert. Anschließend wurden die Bakterien auf LB-Agar mit
Ampicillingehalt ausplattiert und über Nacht bei 37 °C bebrütet. Die Vektoren
enthalten ein Ampicillin-Resistenzgen, so dass nur Klone wachsen, die den Vektor
enthalten.
Die Kulturen wurden mit einer Öse aufgenommen und einzeln in jeweils 2 ml LB-
Medium mit Ampicillin suspendiert und nochmals über Nacht bebrütet. Danach
wurden sie direkt weiterverarbeitet oder bei 4 °C gelagert.
3.17.3 DNA-Minipräparation
Die Plasmid-Mini-Präparation erfolgte mit 1-2 ml der über Nacht gewachsenen
Bakterienkulturen mit dem EasyPrep Plasmid Mini Prep Kit nach Herstellerangaben.
3.17.4 Spaltung mit Restriktionsendonucleasen
Der Erfolg einer Klonierung wurde durch Spaltung mit Restriktionsendonukleasen
überprüft. Bei der anschließenden Gelelektrophorese konnten die spezifischen G-50
Genabschnitte dargestellt werden.

29
Benutzt wurden die Restriktionsenzyme Hind III und Xho I. Gemeinsam mit den
anderen unten angegebenen Reagenzien wurde für 1 Stunde bei 37 °C inkubiert und
die Lösung anschließend sofort auf Eis gegeben.
Plasmidlösung 10 µl
Puffer R+ 10* 2 µl
Xho I (10 U/µl) 0,4 µl
Hind III (10 U/µl) 0,4 µl
Aqua bidest. 7,2 µl
Die Produkte wurden über ein TBE-Gel geschickt, bei Banden in der spezifischen
Länge konnte man davon ausgehen, daß diese Vektoren den gewünschten
Genabschnitt enthielten.
3.17.5 DNA-Maxipräparation:
Die DNA-Maxipräparation wurde aus 200 ml einer über Nacht gewachsenen
Bakteriensuspension durchgeführt. Die Isolierung der Plasmide aus dieser
Bakteriensuspension erfolgte mit dem HIGHSPEED Plasmid Purification Kit nach
Herstellerangaben. Es folgte eine Fällung mit 2,5 Volumenanteil eiskaltem absoluten
Ethanol und 0,1 Volumenanteil 3 M Natriumacetat für 20 min bei 4 °C und 15000 rpm
(Eppendorf-Centrifuge 5403). Das DNA-Pellet wurde mit 70 %igem Ethanol für 10
min bei 4 °C und 15000 rpm gewaschen, vollständig getrocknet und in 100 µl Aqua
bidest. resuspendiert.
3.18 Auswertung der Daten
Aufgrund der Datenstruktur erfolgte eine rein deskriptive Auswertung der Daten. Für
die Diagramme wurde, soweit nicht anders beschrieben, jeweils der Median der
genannten Population ermittelt.

30
4 Ergebnisse
4.1 Etablierung neuer erforderlicher Methoden
4.1.1 Quantifizierung der infektiösen Virionen mittels Virustitration
Die Infektion der Mäuse erfordert definierte Mengen infektiöser Viruspartikel, die
mittels Titration auf geeigneten Zellkulturen ermittelt wurden. Da die BHK-21 Zellen
unter gewöhnlichen Kulturbedingungen nicht die erforderliche Zeit vital blieben,
wurde der FKS-Gehalt so lange variiert, bis ein stabiler Bewuchs der Kulturplatten
über 10 Tage gewährleistet war. Als optimal stellte sich eine FKS-Konzentration von
1% heraus. Nach 5-6 Tagen konnten keine Veränderungen des Bewuchses mehr
festgestellt werden, 10 Tage der Kultivierung waren also ausreichend, um alle
Veränderungen sicher zu erfassen. Mit dieser Methode wurde ein Virusgehalt von
107 pfu/ ml Stammlösung ermittelt. Für eine Infektion der Tiere mit etwa 105 pfu
wurden 10 µl Stammlösung verwendet.
4.1.2 Qualitativer Nachweis viraler RNA und quantitativer Nachweis viraler DNA
4.1.2.1 RNA-Nachweismethoden
Der Nachweis viraler Transkriptionsaktivität im Gewebe gelang mittels Isolierung von
mRNA. Die erfolgreiche Isolierung wurde durch Nachweis von Maus-GAPDH belegt,
einem Housekeeping-Gen ähnlich der humanen GAPDH. Die aus den verschiedenen
Organen gewonnene RNA wurde dann auf verschiedene virale Gene untersucht, die
jeweils entweder dem lytischen Zyklus oder der Latenzperiode zugeordnet werden
(siehe 3.11.1 und 1.2.6). Die nötigen Nukleinsäureamplifikationstechniken wurden
nach institutseigenen Standardprotokollen etabliert und mit isolierter Virus-DNA
getestet.
4.1.2.2 Etablierung einer quantitativen Real-Time PCR zum Nachweis viraler Gen-
50-DNA
Um die Virusmenge im Gewebe zu bestimmen, wurde ein neuer Weg beschritten.
Bisher wurde die Virusmenge unter Ausnutzung des zytopathischen Effektes,
halbquantitativer konventioneller PCR oder anderer Methoden bestimmt. Für dieses
Projekt wurde eine Real-Time-PCR etabliert. Dabei wurden definierte Mengen von
Gen-50 DNA mitgeführt, die eine Quantifizierung der eingesetzten Menge an viraler
31
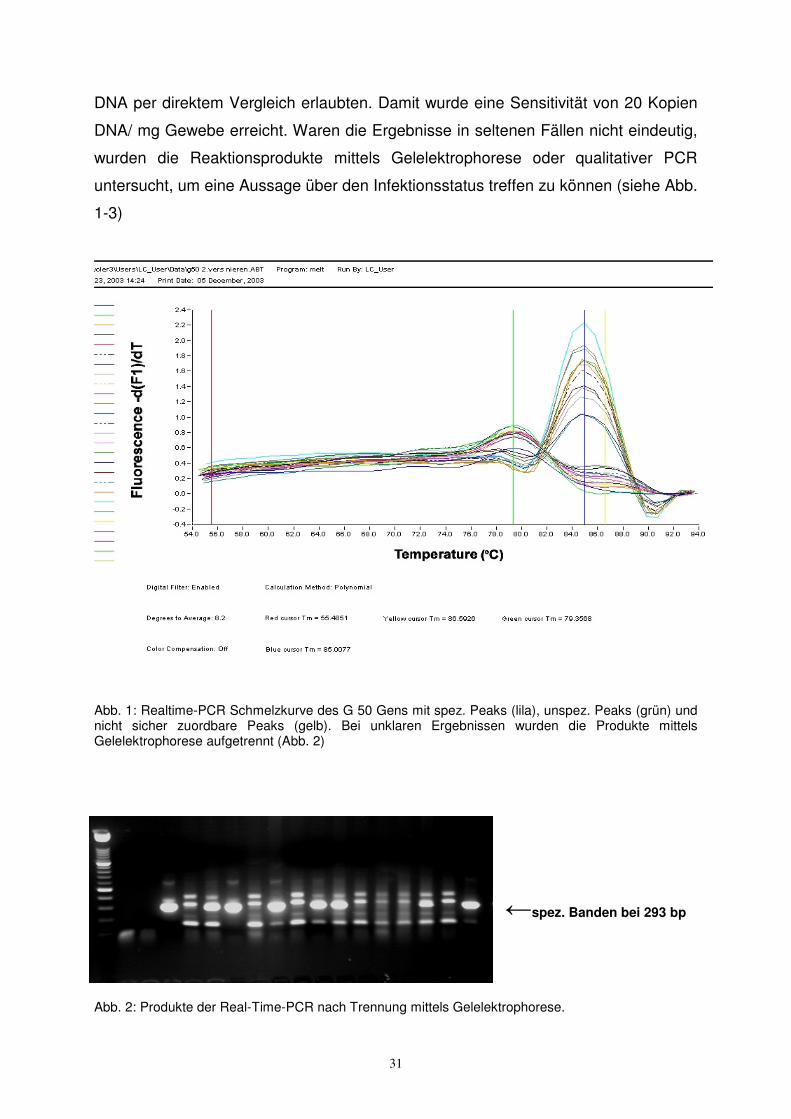
DNA per direktem Vergleich erlaubten. Damit wurde eine Sensitivität von 20 Kopien
DNA/ mg Gewebe erreicht. Waren die Ergebnisse in seltenen Fällen nicht eindeutig,
wurden die Reaktionsprodukte mittels Gelelektrophorese oder qualitativer PCR
untersucht, um eine Aussage über den Infektionsstatus treffen zu können (siehe Abb.
1-3)
Abb. 1: Realtime-PCR Schmelzkurve des G 50 Gens mit spez. Peaks (lila), unspez. Peaks (grün) und nicht sicher zuordbare Peaks (gelb). Bei unklaren Ergebnissen wurden die Produkte mittels Gelelektrophorese aufgetrennt (Abb. 2)
←spez. Banden bei 293 bp
Abb. 2: Produkte der Real-Time-PCR nach Trennung mittels Gelelektrophorese.

32
← spez. Banden bei 133 bp
Abb. 3: M3 nested PCR mit spezifischen Banden bei 133 bp (Pfeil).
4.1.3 Methode zum Nachweis von Virusantigen im Gewebe
Eine weitere Möglichkeit für den Virusnachweis und eine sichere Lokalisation der
viralen Antigene bot die indirekte Immunfluoreszenz. Dafür wurden entparaffinierte
Gewebeschnitte mit dem Serum MHV-68-Antikörper-positiver Mäuse inkubiert. Die in
diesem Serum befindlichen spezifischen Antikörper banden an Virusantigen und
wurden in einem zweiten Schritt nachgewiesen. Die Ergebnisse der
Fluoreszenzuntersuchungen waren aber noch nicht zufriedenstellend. Offenbar
waren die viralen Antigene noch nicht ausreichend freigelegt. Daher schlossen sich
an die Entparaffinierung noch weitere Schritte zur Demaskierung der antigenen
Strukturen an. Dazu wurden die Gewebeschnitte 20 Minuten in Zitratpuffer gekocht
und nach dem Abkühlen in PBS gewaschen. Anschließend wurden die
Gewebeproben noch 30 Minuten in einer Lösung aus PBS, Milchpulver und bovinem
Serumalbumin inkubiert.
Der Nachweis MHV-68-spezifischer Antikörper in den Maussera gelang an
viruspositiven BHK-21-Zellen. Verwendung von anti-Maus-IgG als
Sekundärantikörper führte zu intensiver zytoplasmatischer und membranständiger
Fluoreszenz an den virusinfizierten Zellen. Verdünnung des Serums führte
erwartungsgemäß zu einer Signalabschwächung, bei einer Verdünnung von 1:2048
war Fluoreszenz nicht mehr nachweisbar. Entsprechend wurde eine
Arbeitsverdünnung von 1:200 gewählt (Bilder 4/5).
Eine weitere Verbesserung der Ergebnisse brachte die Einführung eines
Zwischenschrittes in Form eines nicht konjugierten Sekundärantikörpers, so dass
erst in einem dritten Schritt fluoreszierende Antikörper eingesetzt wurden. In Leber,
Lunge, Herz, Großhirn, Niere und Kleinhirn fand sich spezifische Färbung

33
ausschließlich in den Schnitten infizierter Tiere, deshalb kann man die Ergebnisse
der Immunfluoreszenztests als verwertbar ansehen. Probleme bereiteten Milzen und
Thymi, hier traten auch bei den Kontrolltieren unspezifische Signale auf. Daher
wurden die entsprechenden Organschnitte nicht ausgewertet.
Abb. 4: BHK 21 Zellen inkubiert mit MHV-68-positivem Serum 1:1024
Abb. 5: BHK 21 inkubiert mit negativem Mausserum 1:50

34
4.1.4 Etablierung einer durch MHV-68-Infektion immortalisierten B-Zelllinie
Eine wichtige Aufgabe bestand darin, eine immortale, MHV-68-positive Zelllinie zu
schaffen. Hierzu wurden Lymphozyten aus Milzen von infizierten Tieren isoliert. Nach
der Kultivierung wurden die Zellen mit MHV-68 superinfiziert. Innerhalb von 24
Stunden kam es zur Clusterbildung. Im weiteren Verlauf proliferierten die Zellen im
Cluster rasch.
Innerhalb von zwei Wochen ließ die Teilungsaktivität nach und die überwiegende
Zahl der Zellen starb ab. Wenige Zellen blieben danach über einen Zeitraum von vier
Wochen vital und wurden weiter kultiviert. Nach dieser Zeit setzte bei drei Kulturen
ohne weitere Behandlung erneut Clusterbildung und Proliferation ein. Die
Wachstumsgeschwindigkeit der Zellen war so groß, daß sie versuchsweise in einem
RPMI Lymphozytenmedium, supplementiert mit 10 % FKS kultiviert wurden. Diese
Veränderung der Bedingungen wurde gut toleriert.
Die Zellkulturen wurden erfolgreich auf DNA von Maus-GAPDH (ein
mausspezifisches housekeeping-Protein) untersucht, um zu belegen, daß es sich
um Mauszellen handelt (Abb. 6). Weiterhin wurde nachgewiesen, daß die Zellen
MHV-68 DNA (G 50 nested-PCR) enthielten. Im Überstand konnte in geringerer
Menge ebenfalls Virus-DNA nachgewiesen werden (Abb. 7).
Mittels IFT konnten auch Virusantigene an bzw. in den Zellen dargestellt werden
(Abb. 8/9). Nach etwa 50 erfolgreichen Passagen wurden von allen drei Klonen
Zellen eingefroren, die restlichen Zellen werden inzwischen seit Jahren erfolgreich
weiterkultiviert.
Abb. 6: Nachweis von muriner GAPDH (236 bp) Abb. 7: Nachweis von G 50 Gen (293 bp) in B- Zelllinie

35
Abb. 8: MHV-68 positive Lymphozytenkultur inkubiert mit PBS
Abb. 9: MHV-68 positive Lymphozytenkultur inkubiert mit
MHV-68-Antikörper 1:64
4.2 Morbidität und Mortalität waren vom Alter bei Infektion abhängig
Grundlage dieser Arbeit sind im Wesentlichen drei Versuchsreihen mit insgesamt 95
Tieren. Davon wurden 39 Tiere neonatal (maximal 24 h nach Geburt) infiziert, 19
Tiere 4-6 Tage nach der Geburt und 17 Tiere 18-22 Tage nach der Geburt. 20 Tiere
wurden nicht infiziert und als Kontrolltiere separiert gehalten. Die Tiere wurden eine,
drei oder fünf Wochen nach Infektion getötet und präpariert. Alle Tiere wurden
quantitativ auf Virus-DNA untersucht. Folgende Organe wurden bei allen Mäusen
untersucht: Leber, Lunge, Milz, Niere, Thymus und Muskel. Die anderen Organe
(Großhirn, Kleinhirn, Herz) wurden nur bei den Tieren der 3. Versuchsreihe
untersucht. Blut konnte aus methodischen Gründen nicht bei allen Tieren
entnommen werden.
36
Von den infizierten Tieren (n=75) waren 51 MHV-68-positiv; dies entspricht einer
Infektionsrate von 73 %. Gefressene Tiere wurden hier nicht berücksichtigt. Sowohl
Morbidität als auch Mortalität wurde nur bei den neonatal infizierten Tieren
beobachtet. Fünf dieser Tiere wurden tot aufgefunden, acht sind von ihren Müttern
gefressen worden und zwei mussten wegen deutlicher neurologischer Ausfälle
vorzeitig entnommen werden.
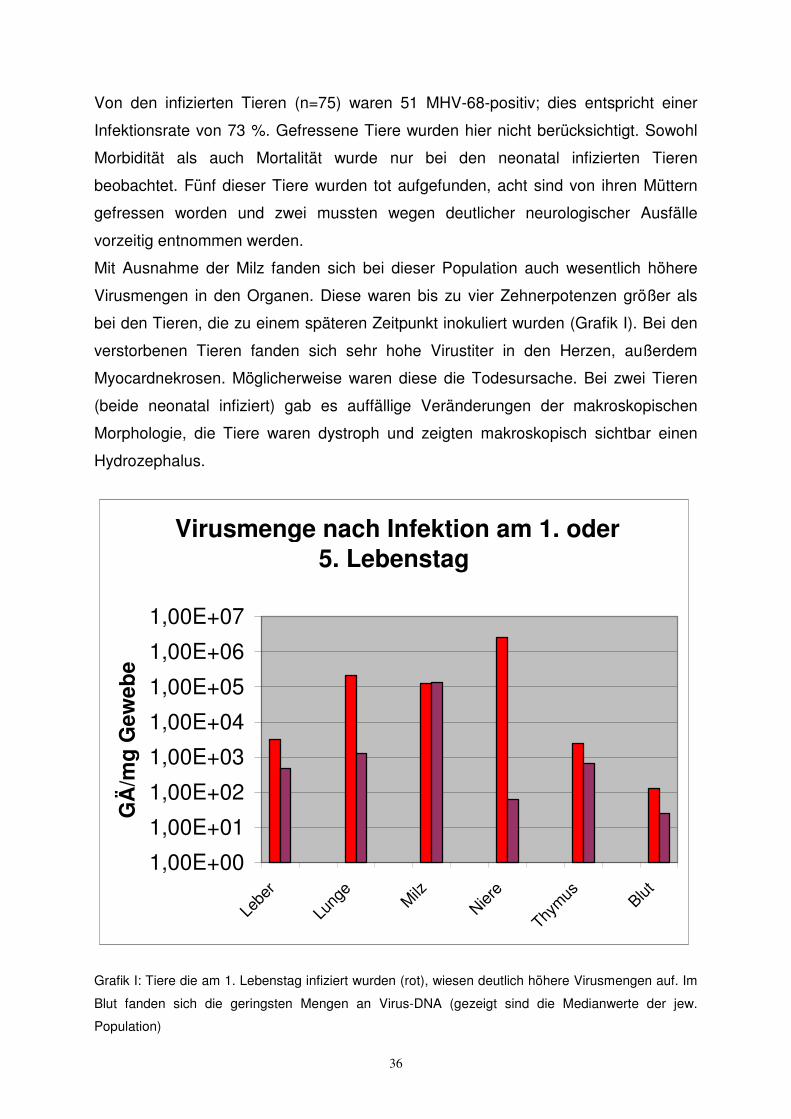
Mit Ausnahme der Milz fanden sich bei dieser Population auch wesentlich höhere
Virusmengen in den Organen. Diese waren bis zu vier Zehnerpotenzen größer als
bei den Tieren, die zu einem späteren Zeitpunkt inokuliert wurden (Grafik I). Bei den
verstorbenen Tieren fanden sich sehr hohe Virustiter in den Herzen, außerdem
Myocardnekrosen. Möglicherweise waren diese die Todesursache. Bei zwei Tieren
(beide neonatal infiziert) gab es auffällige Veränderungen der makroskopischen
Morphologie, die Tiere waren dystroph und zeigten makroskopisch sichtbar einen
Hydrozephalus.
Virusmenge nach Infektion am 1. oder 5. Lebenstag
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1,00E+04
1,00E+05
1,00E+06
1,00E+07
Lebe
r
Lung
eM
ilzNier
e
Thym
us Blut
GÄ
/mg
Gew
ebe
Grafik I: Tiere die am 1. Lebenstag infiziert wurden (rot), wiesen deutlich höhere Virusmengen auf. Im
Blut fanden sich die geringsten Mengen an Virus-DNA (gezeigt sind die Medianwerte der jew.
Population)
37
4.3 Nachweis von MHV-68 DNA in verschiedenen Organen
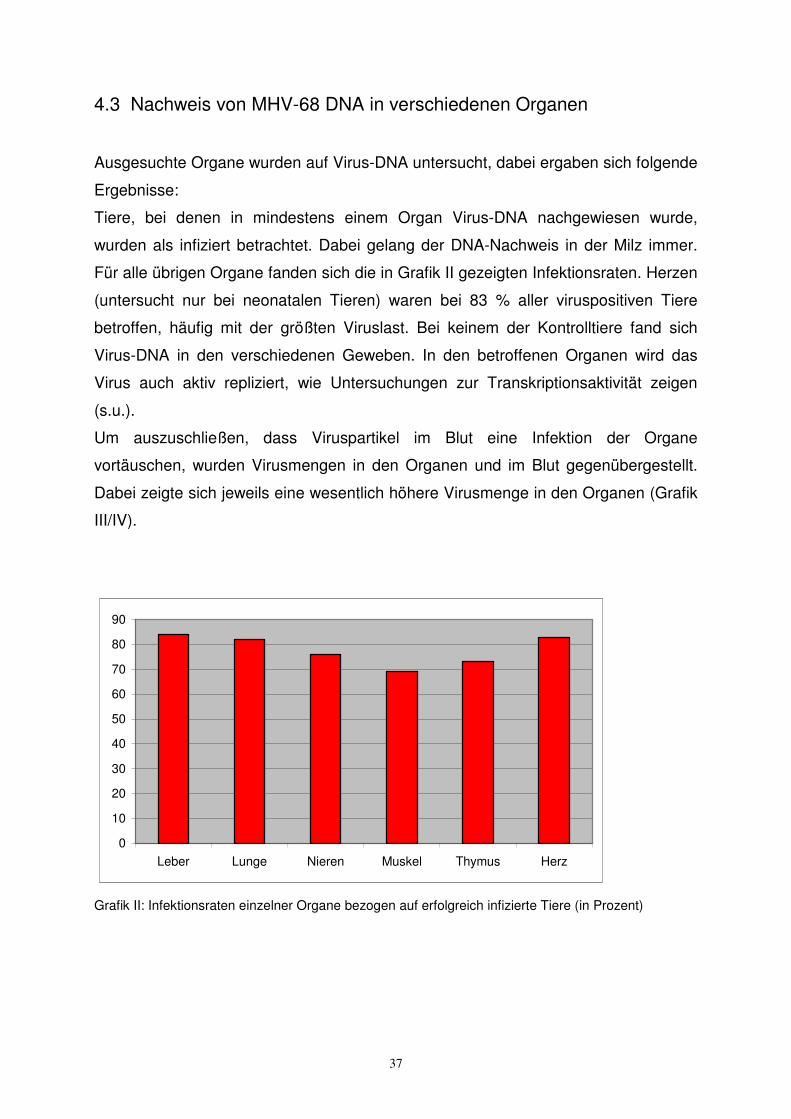
Ausgesuchte Organe wurden auf Virus-DNA untersucht, dabei ergaben sich folgende
Ergebnisse:
Tiere, bei denen in mindestens einem Organ Virus-DNA nachgewiesen wurde,
wurden als infiziert betrachtet. Dabei gelang der DNA-Nachweis in der Milz immer.
Für alle übrigen Organe fanden sich die in Grafik II gezeigten Infektionsraten. Herzen
(untersucht nur bei neonatalen Tieren) waren bei 83 % aller viruspositiven Tiere
betroffen, häufig mit der größten Viruslast. Bei keinem der Kontrolltiere fand sich
Virus-DNA in den verschiedenen Geweben. In den betroffenen Organen wird das
Virus auch aktiv repliziert, wie Untersuchungen zur Transkriptionsaktivität zeigen
(s.u.).
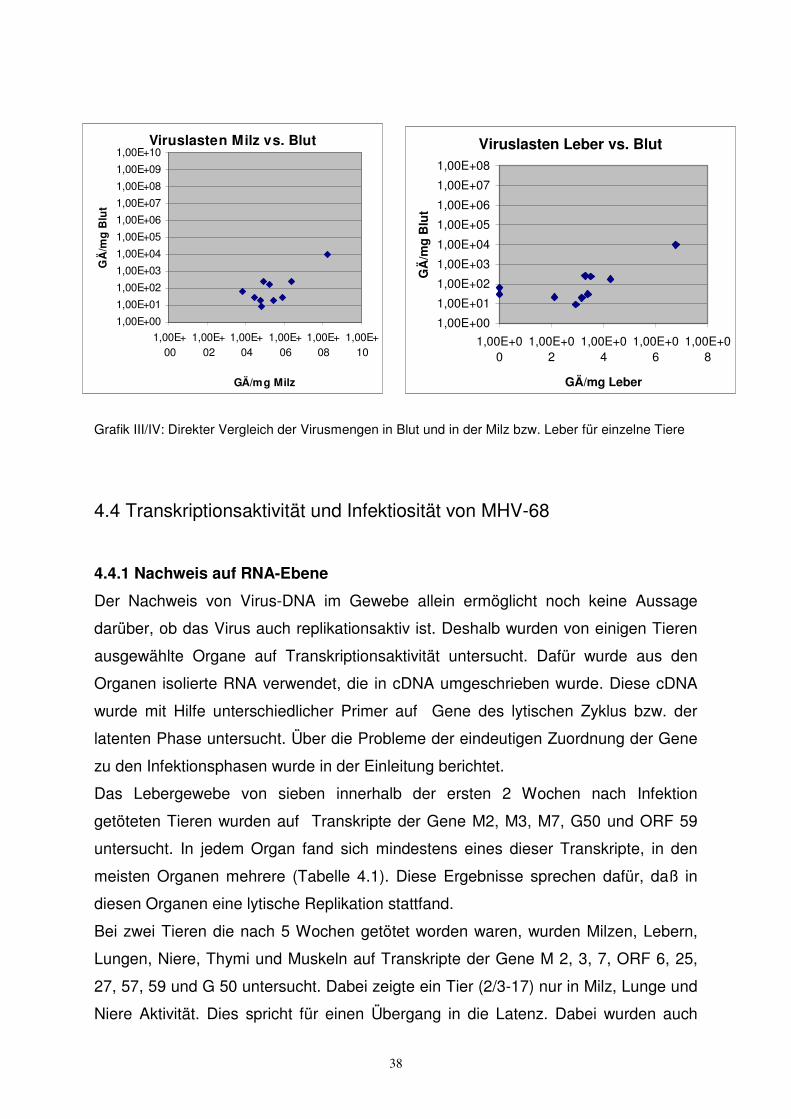
Um auszuschließen, dass Viruspartikel im Blut eine Infektion der Organe
vortäuschen, wurden Virusmengen in den Organen und im Blut gegenübergestellt.
Dabei zeigte sich jeweils eine wesentlich höhere Virusmenge in den Organen (Grafik
III/IV).
0
10
20
30
40
50
60
70
80
90
Leber Lunge Nieren Muskel Thymus Herz
Grafik II: Infektionsraten einzelner Organe bezogen auf erfolgreich infizierte Tiere (in Prozent)
38
Viruslasten Leber vs. Blut
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1,00E+04
1,00E+05
1,00E+06
1,00E+07
1,00E+08
1,00E+00
1,00E+02
1,00E+04
1,00E+06
1,00E+08
GÄ/mg Leber
GÄ
/mg
Blu
t
Grafik III/IV: Direkter Vergleich der Virusmengen in Blut und in der Milz bzw. Leber für einzelne Tiere
4.4 Transkriptionsaktivität und Infektiosität von MHV-68
4.4.1 Nachweis auf RNA-Ebene
Der Nachweis von Virus-DNA im Gewebe allein ermöglicht noch keine Aussage
darüber, ob das Virus auch replikationsaktiv ist. Deshalb wurden von einigen Tieren
ausgewählte Organe auf Transkriptionsaktivität untersucht. Dafür wurde aus den
Organen isolierte RNA verwendet, die in cDNA umgeschrieben wurde. Diese cDNA
wurde mit Hilfe unterschiedlicher Primer auf Gene des lytischen Zyklus bzw. der
latenten Phase untersucht. Über die Probleme der eindeutigen Zuordnung der Gene
zu den Infektionsphasen wurde in der Einleitung berichtet.
Das Lebergewebe von sieben innerhalb der ersten 2 Wochen nach Infektion
getöteten Tieren wurden auf Transkripte der Gene M2, M3, M7, G50 und ORF 59
untersucht. In jedem Organ fand sich mindestens eines dieser Transkripte, in den
meisten Organen mehrere (Tabelle 4.1). Diese Ergebnisse sprechen dafür, daß in
diesen Organen eine lytische Replikation stattfand.
Bei zwei Tieren die nach 5 Wochen getötet worden waren, wurden Milzen, Lebern,
Lungen, Niere, Thymi und Muskeln auf Transkripte der Gene M 2, 3, 7, ORF 6, 25,
27, 57, 59 und G 50 untersucht. Dabei zeigte ein Tier (2/3-17) nur in Milz, Lunge und
Niere Aktivität. Dies spricht für einen Übergang in die Latenz. Dabei wurden auch
Viruslasten Milz vs. Blut
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1,00E+04
1,00E+05
1,00E+06
1,00E+07
1,00E+08
1,00E+09
1,00E+10
1,00E+00
1,00E+02
1,00E+04
1,00E+06
1,00E+08
1,00E+10
GÄ/mg Milz
GÄ
/mg
Blu
t

39
Gene exprimiert, die eher dem lytischen Zyklus zugeordnet werden (M7, G50). Das
andere Tier (2/3-18) zeigte in allen Organen ein breites Spektrum an Aktivität. Es
bestand keine Präferenz für bestimmte Gene. Dieses Tier war zum Zeitpunkt der
Organentnahme deutlich dystroph und hatte histologisch einen Hydrozephalus (Abb.
10). Diese ausgeprägte ubiquitäre Virusaktivität könnte Zeichen einer persistierenden
aktiven Infektion sein.
Tabelle 4.1 Expression viraler Gene in verschiedenen Geweben
Primer Tier/Organ
G 50 M 2 M 3 M 7 ORF 6
ORF 25
ORF 27
ORF 57
ORF 59
2/3-17 Milz + + + - - - - - - Leber - - - - - - - - - Lunge + - - - - - - - - Niere - - - + - - - - - Thymus - - - - - - - - - Muskel - - - - - - - - - 2/3-18 Milz + + + + + + + + + Leber + - - - - - - + + Lunge - - - - - - - - - Niere + + + + + + + + + Thymus + + + + + + - - + Muskel - - - - - - - - - Leber: 3/1-2 - - - - + 3/1-3 + + + + + 3/2-4 + + + + + 3/4-6 - + + + + 3/1-8 - - - + + 3/1-10 + + + + + 3/5-18 + + + + +
Abb. 10: Maus 2/3-18 im Vergleich mit gleich alten Kontrolltieren
40
4.4.2 Direkter Nachweis von Virionen mittels des zytopathischen Effekts
Für den Nachweis infektiöser Virionen wurden verschiedene Verdünnungsstufen
homogenisierter Organe (Leber, Lunge, Milz) auf BHK-21 Zellen inkubiert. Bei 2
Tieren (2/2-10, 3/7-24), die 24 und 32 Tage nach Infektion getötet wurden ließ sich
kein Virus rekultivieren. Bei 2 Tieren (3/2-4 und 3/2-8), die 9 Tage nach Infektion
getötet wurden, war dies möglich. Bei diesen Tieren wurden aus Leber- und
Milzgewebe sowie aus dem Großhirn infektiöse Virionen rekultiviert.
Dies stimmt mit dem bekannten Modell überein, nach dem die Infektion nach 10-14
Tagen ein Maximum erreicht. Hier kann Virus isoliert werden. Danach geht MHV-68
in die Latenz über und MHV-68 ist nicht mehr direkt anzuzüchten.
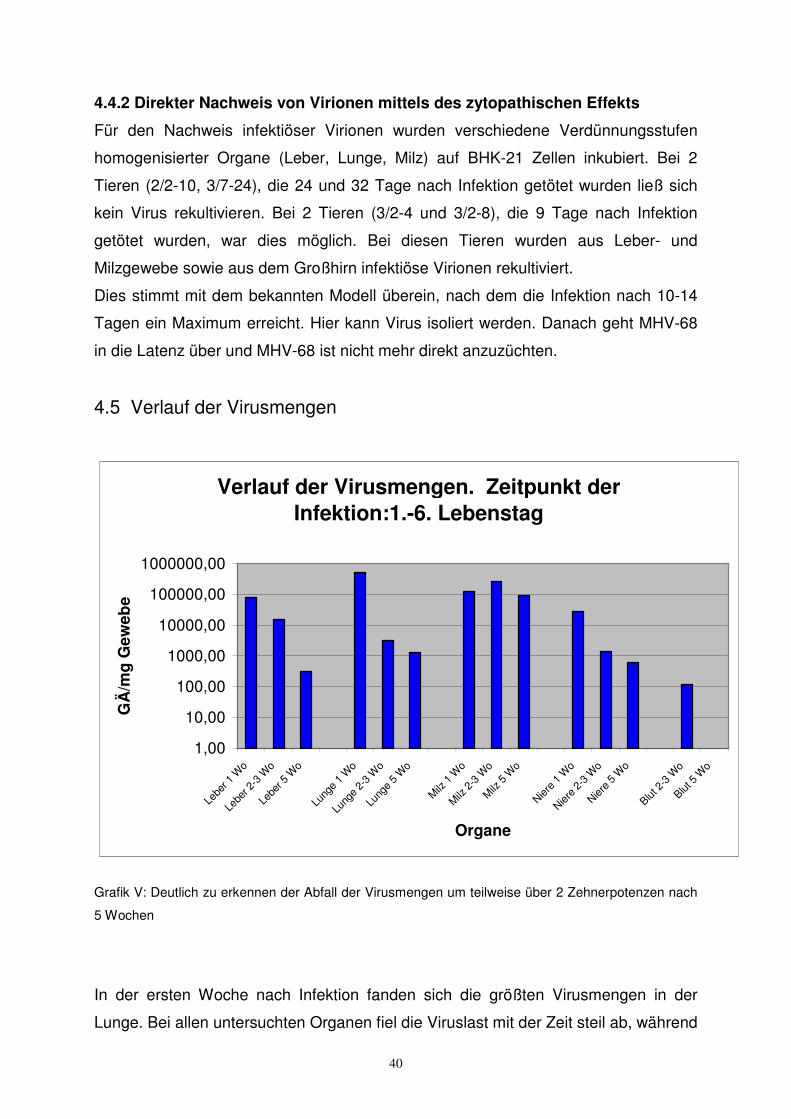
4.5 Verlauf der Virusmengen
Verlauf der Virusmengen. Zeitpunkt der Infektion:1.-6. Lebenstag
1,00
10,00
100,00
1000,00
10000,00
100000,00
1000000,00
Lebe
r 1 W
o
Lebe
r 2-3
Wo
Lebe
r 5 W
o
Lung
e 1
Wo
Lung
e 2-
3 W
o
Lung
e 5
Wo
Milz
1 W
o
Milz
2-3
Wo
Milz
5 W
o
Niere
1 W
o
Niere
2-3
Wo
Niere
5 W
o
Blut 2
-3 W
o
Blut 5
Wo
Organe
GÄ
/mg
Gew
ebe
Grafik V: Deutlich zu erkennen der Abfall der Virusmengen um teilweise über 2 Zehnerpotenzen nach
5 Wochen
In der ersten Woche nach Infektion fanden sich die größten Virusmengen in der
Lunge. Bei allen untersuchten Organen fiel die Viruslast mit der Zeit steil ab, während

41
sie in der Milz stabil blieb. Das bestätigt die Bedeutung der Milz für die latente
Infektion. Die anderen untersuchten Organe waren regelmäßig mit betroffen, über die
Kontamination mit Viren im Blut hinaus. Dies war besonders deutlich bei den sehr
früh infizierten (1.-6. Lebenstag) Tieren.
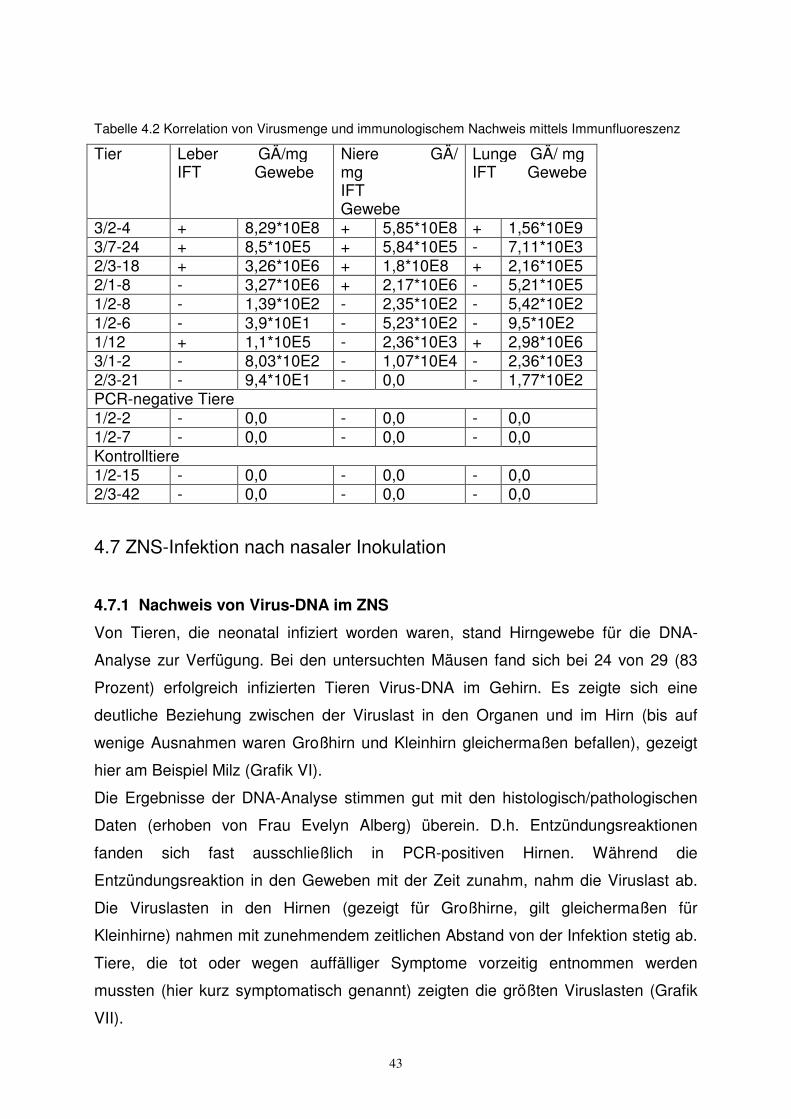
4.6 Nachweis von Virusantigen in peripheren Geweben
Nach entsprechender Vorbehandlung (siehe 3.13) wurden die Organschnitte der
Tiere mit den verschiedenen Antikörpern inkubiert. Es zeigte sich, daß die
Nachweisgrenze für Virusantigene bei dieser Methode recht hoch liegt, d.h. nur bei
Tieren mit hohen Virusmengen gelang der Nachweis von Fluoreszenz, wie Tabelle
4.2 zeigt. Insgesamt konnte ab etwa 105 GÄ/ mg Gewebe Antigen nachgewiesen
werden. Besonders eindrucksvolle Ergebnisse wurden bei Leber, Lunge und Hirn
erzielt.
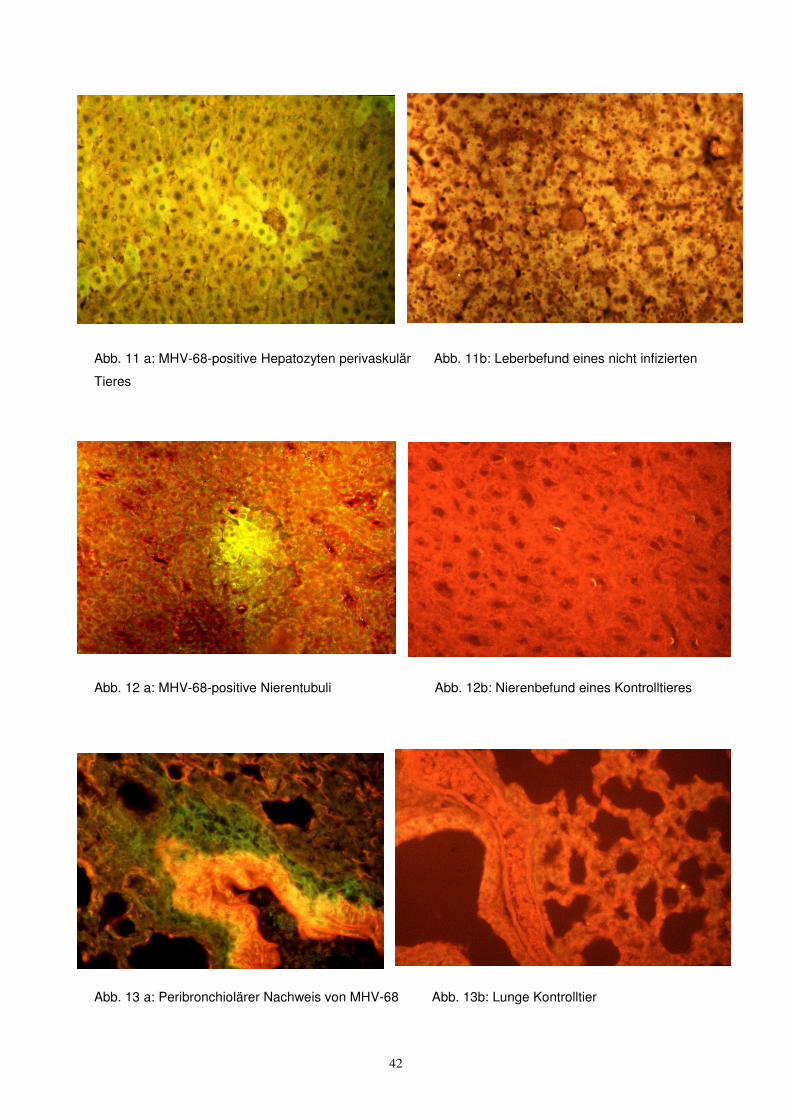
Bei den Lebern wurden generell Hepatozyten angefärbt, und zwar einzeln oder in
Gruppen über den ganzen Schnitt verteilt, allerdings mit Betonung der perivaskulären
Zonen (Abb. 11). Insgesamt fanden sich 4 positive Leberschnitte in 9 PCR-positiven
Versuchstieren, keine in den Kontrollen, allerdings 1 pos. Leber in einem PCR-
negativen Versuchstier. Die Nieren boten eher unscharf begrenzte Zonen der
Fluoreszenz, teilweise schienen Tubuluszellen betroffen, teilweise das Tubuluslumen
(Abb. 12). Hier waren 4 von 9 Organen positiv, 3 davon übereinstimmend mit dem
Leberbefund. In den Kontrollen und negativen Versuchstieren fand sich nichts.
Anders als erwartet zeigte sich Fluoreszenz in den Lungen vorwiegend
peribronchiolär und interstitiell (Abb. 13), nicht in Alveolarzellen. Insgesamt waren 3
von 9 untersuchten Organen positiv, alle parallel zum Leberbefund, keine der
Kontrollen.
Herzen waren 2 positiv, hier fand sich zusätzlich ein positiver Befund bei einem PCR-
negativen Tier (dieses Tier zeigte auch den pos. Leberbefund).
42
Abb. 11 a: MHV-68-positive Hepatozyten perivaskulär Abb. 11b: Leberbefund eines nicht infizierten
Tieres
Abb. 12 a: MHV-68-positive Nierentubuli Abb. 12b: Nierenbefund eines Kontrolltieres
Abb. 13 a: Peribronchiolärer Nachweis von MHV-68 Abb. 13b: Lunge Kontrolltier
43
Tabelle 4.2 Korrelation von Virusmenge und immunologischem Nachweis mittels Immunfluoreszenz
Tier Leber GÄ/mg IFT Gewebe
Niere GÄ/ mg IFT Gewebe
Lunge GÄ/ mg IFT Gewebe
3/2-4 + 8,29*10E8 + 5,85*10E8 + 1,56*10E9 3/7-24 + 8,5*10E5 + 5,84*10E5 - 7,11*10E3 2/3-18 + 3,26*10E6 + 1,8*10E8 + 2,16*10E5 2/1-8 - 3,27*10E6 + 2,17*10E6 - 5,21*10E5 1/2-8 - 1,39*10E2 - 2,35*10E2 - 5,42*10E2 1/2-6 - 3,9*10E1 - 5,23*10E2 - 9,5*10E2 1/12 + 1,1*10E5 - 2,36*10E3 + 2,98*10E6 3/1-2 - 8,03*10E2 - 1,07*10E4 - 2,36*10E3 2/3-21 - 9,4*10E1 - 0,0 - 1,77*10E2 PCR-negative Tiere 1/2-2 - 0,0 - 0,0 - 0,0 1/2-7 - 0,0 - 0,0 - 0,0 Kontrolltiere 1/2-15 - 0,0 - 0,0 - 0,0 2/3-42 - 0,0 - 0,0 - 0,0
4.7 ZNS-Infektion nach nasaler Inokulation
4.7.1 Nachweis von Virus-DNA im ZNS
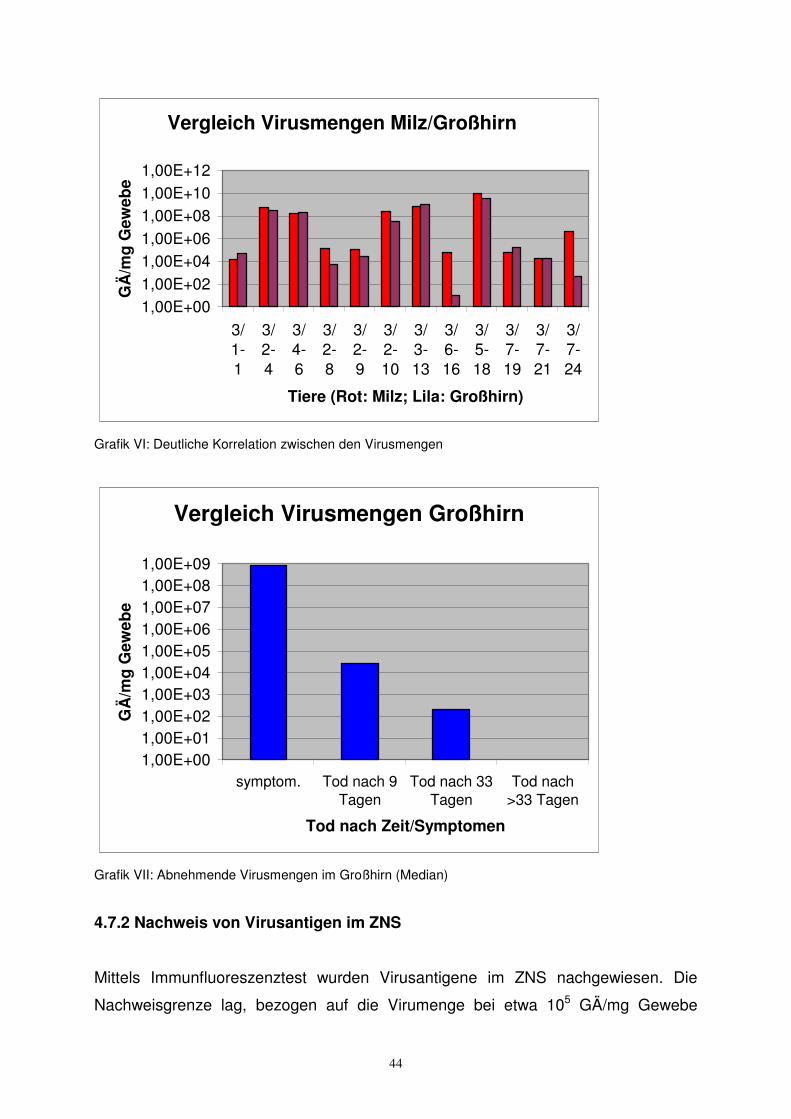
Von Tieren, die neonatal infiziert worden waren, stand Hirngewebe für die DNA-
Analyse zur Verfügung. Bei den untersuchten Mäusen fand sich bei 24 von 29 (83
Prozent) erfolgreich infizierten Tieren Virus-DNA im Gehirn. Es zeigte sich eine
deutliche Beziehung zwischen der Viruslast in den Organen und im Hirn (bis auf
wenige Ausnahmen waren Großhirn und Kleinhirn gleichermaßen befallen), gezeigt
hier am Beispiel Milz (Grafik VI).
Die Ergebnisse der DNA-Analyse stimmen gut mit den histologisch/pathologischen
Daten (erhoben von Frau Evelyn Alberg) überein. D.h. Entzündungsreaktionen
fanden sich fast ausschließlich in PCR-positiven Hirnen. Während die
Entzündungsreaktion in den Geweben mit der Zeit zunahm, nahm die Viruslast ab.
Die Viruslasten in den Hirnen (gezeigt für Großhirne, gilt gleichermaßen für
Kleinhirne) nahmen mit zunehmendem zeitlichen Abstand von der Infektion stetig ab.
Tiere, die tot oder wegen auffälliger Symptome vorzeitig entnommen werden
mussten (hier kurz symptomatisch genannt) zeigten die größten Viruslasten (Grafik
VII).
44
Vergleich Virusmengen Milz/Großhirn
1,00E+00
1,00E+02
1,00E+04
1,00E+06
1,00E+08
1,00E+10
1,00E+12
3/1-1
3/2-4
3/4-6
3/2-8
3/2-9
3/2-10
3/3-13
3/6-16
3/5-18
3/7-19
3/7-21
3/7-24
Tiere (Rot: Milz; Lila: Großhirn)
GÄ
/mg
Gew
ebe
Grafik VI: Deutliche Korrelation zwischen den Virusmengen
Vergleich Virusmengen Großhirn
1,00E+001,00E+011,00E+021,00E+031,00E+041,00E+051,00E+061,00E+071,00E+081,00E+09
symptom. Tod nach 9Tagen
Tod nach 33Tagen
Tod nach>33 Tagen
Tod nach Zeit/Symptomen
GÄ
/mg
Gew
ebe
Grafik VII: Abnehmende Virusmengen im Großhirn (Median)
4.7.2 Nachweis von Virusantigen im ZNS
Mittels Immunfluoreszenztest wurden Virusantigene im ZNS nachgewiesen. Die
Nachweisgrenze lag, bezogen auf die Virumenge bei etwa 105 GÄ/mg Gewebe

45
(Tabelle 4.2). Es wurden Gewebeschnitte von zwei Kontrolltieren, von drei Tieren, bei
denen in keinem der untersuchten Gewebe Virus mittels PCR detektiert wurde, und
von acht infizierten Tieren untersucht. Hierbei wurden im ZNS 3 Regionen gesondert
betrachtet: der Cortex, der Hippocampus und das Kleinhirn. Kein Schnitt von
Kontrolltieren bzw. PCR-negativen Tieren zeigte eine spezifische Fluoreszenz.
Dagegen zeigten alle hier untersuchten PCR-positiven Tiere Leukozyteninfiltrate in
der Immunhistochemie. Bei vier dieser Tiere fanden sich auch positive Ergebnisse
bei den Immunfluoreszenztests:
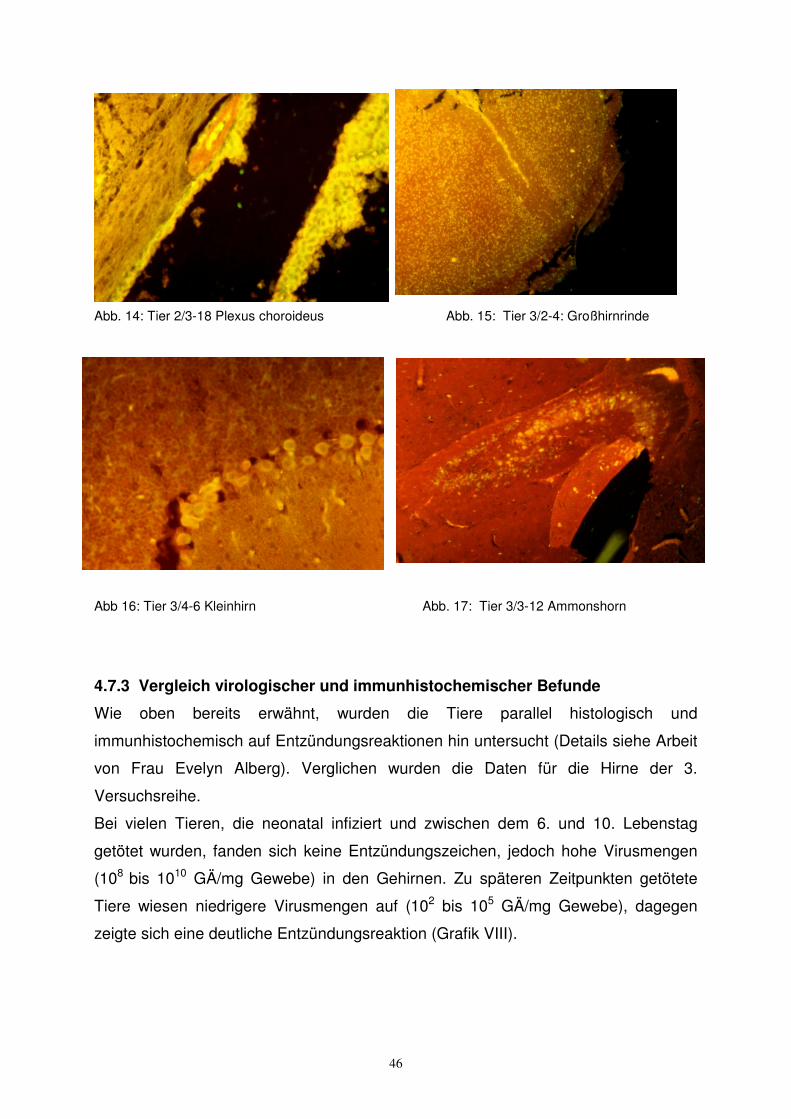
1. Tier 2/3-18: (neonatal infiziert, getötet nach 5 Wochen) Die Histologie zeigte
erweiterte Ventrikel mit periventrikulären Infiltraten. Diesem Befund
entsprechend fand sich im IFT Fluoreszenz an Ependymzellen und an
Zellen des Plexus Choroideus des 4. Ventrikels.
2. Tier 3/2-4: (neonatal infiziert, Tod nach 10 Tagen) Die Histologie zeigte eine
Meningitis und eine diffuse Enzephalitis. Der IFT zeigte diffuse
plasmatische Fluoreszenz von kortikalen und subkortikalen Zellen.
3. Tier 3/4-6: (neonatal infiziert, nach 16 Tagen wegen Ataxie vorzeitig entnommen)
Die Histologie zeigte eine diffuse Enzephalitis und Zerebellitis.
Fluoreszenz fand sich sowohl im Ammonshorn, als auch im Kleinhirn.
4. Tier 3/3-12: (neonatal infiziert, nach 10 Tagen tot aufgefunden) Die Histologie
zeigte eine diffuse Enzephalitis und ein Infiltrat im Temporallappen.
Die Immunfluoreszenz zeigte Fluoreszenz von Zellen des
Hippocampus und kortikaler Neurone.
46
Abb. 14: Tier 2/3-18 Plexus choroideus Abb. 15: Tier 3/2-4: Großhirnrinde
Abb 16: Tier 3/4-6 Kleinhirn Abb. 17: Tier 3/3-12 Ammonshorn
4.7.3 Vergleich virologischer und immunhistochemischer Befunde
Wie oben bereits erwähnt, wurden die Tiere parallel histologisch und
immunhistochemisch auf Entzündungsreaktionen hin untersucht (Details siehe Arbeit
von Frau Evelyn Alberg). Verglichen wurden die Daten für die Hirne der 3.
Versuchsreihe.
Bei vielen Tieren, die neonatal infiziert und zwischen dem 6. und 10. Lebenstag
getötet wurden, fanden sich keine Entzündungszeichen, jedoch hohe Virusmengen
(108 bis 1010 GÄ/mg Gewebe) in den Gehirnen. Zu späteren Zeitpunkten getötete
Tiere wiesen niedrigere Virusmengen auf (102 bis 105 GÄ/mg Gewebe), dagegen
zeigte sich eine deutliche Entzündungsreaktion (Grafik VIII).
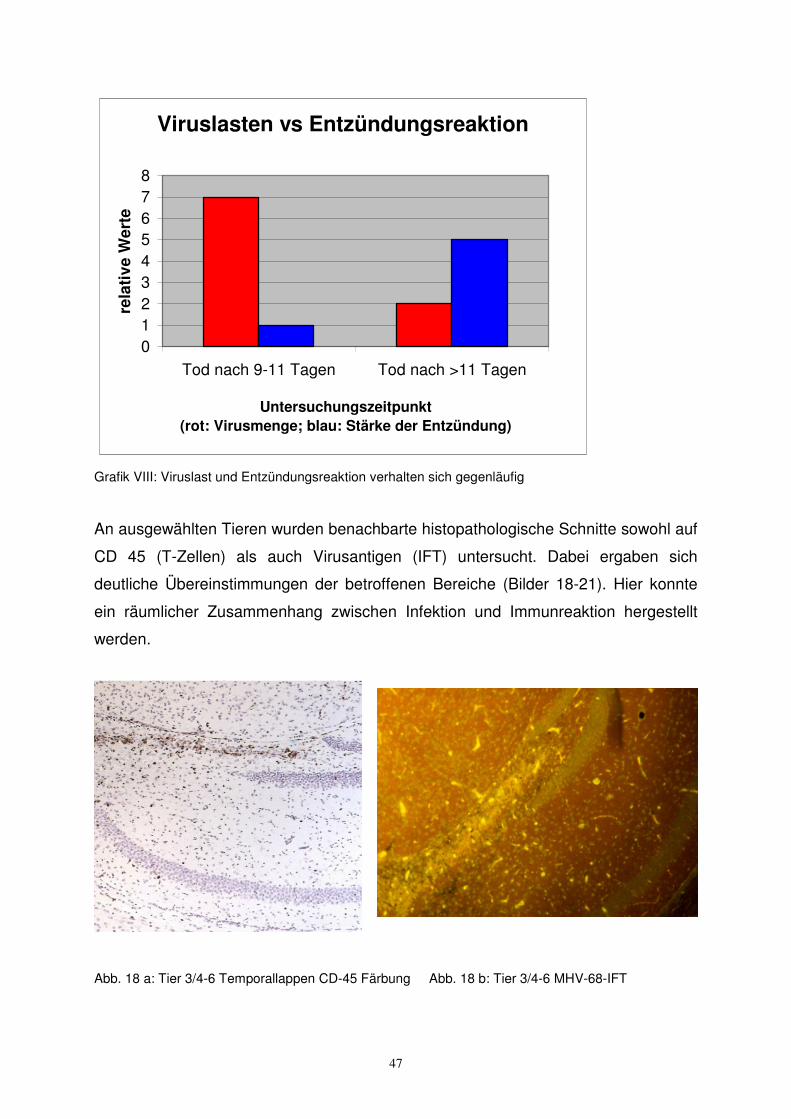
47
Viruslasten vs Entzündungsreaktion
012345678
Tod nach 9-11 Tagen Tod nach >11 Tagen
Untersuchungszeitpunkt (rot: Virusmenge; blau: Stärke der Entzündung)
rela
tive
Wer
te
Grafik VIII: Viruslast und Entzündungsreaktion verhalten sich gegenläufig
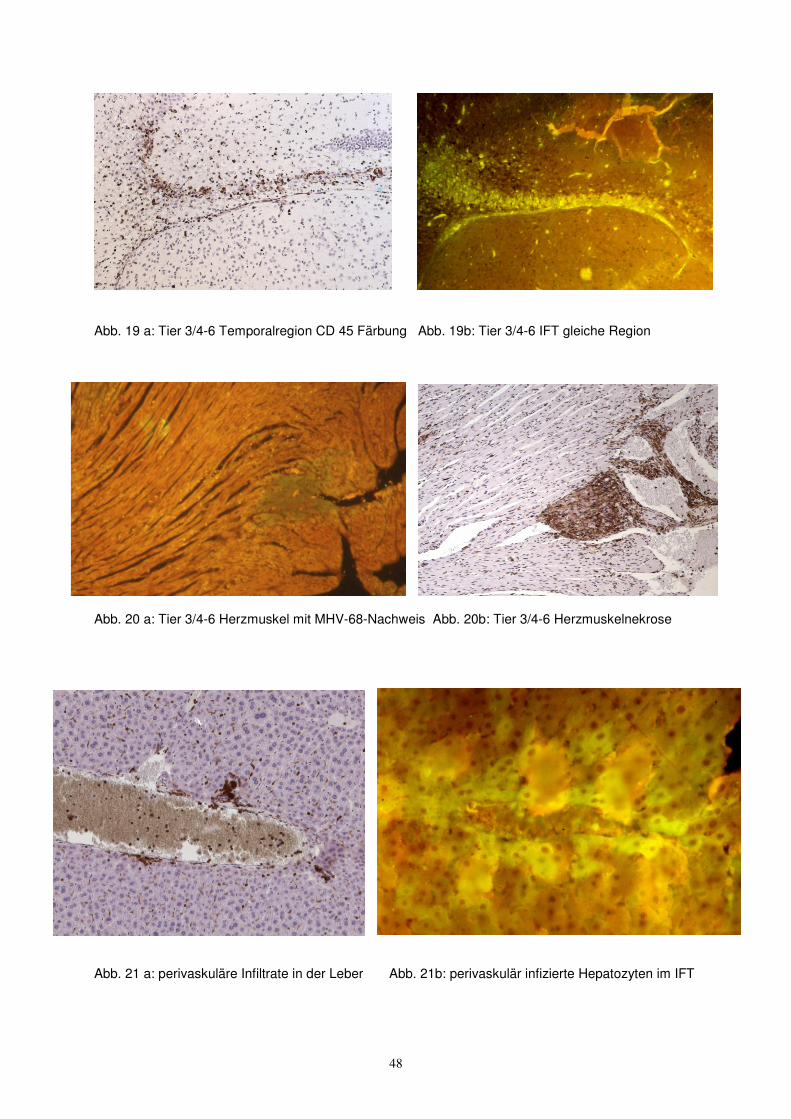
An ausgewählten Tieren wurden benachbarte histopathologische Schnitte sowohl auf
CD 45 (T-Zellen) als auch Virusantigen (IFT) untersucht. Dabei ergaben sich
deutliche Übereinstimmungen der betroffenen Bereiche (Bilder 18-21). Hier konnte
ein räumlicher Zusammenhang zwischen Infektion und Immunreaktion hergestellt
werden.
Abb. 18 a: Tier 3/4-6 Temporallappen CD-45 Färbung Abb. 18 b: Tier 3/4-6 MHV-68-IFT
48
Abb. 19 a: Tier 3/4-6 Temporalregion CD 45 Färbung Abb. 19b: Tier 3/4-6 IFT gleiche Region
Abb. 20 a: Tier 3/4-6 Herzmuskel mit MHV-68-Nachweis Abb. 20b: Tier 3/4-6 Herzmuskelnekrose
Abb. 21 a: perivaskuläre Infiltrate in der Leber Abb. 21b: perivaskulär infizierte Hepatozyten im IFT

49
5 Diskussion
Die MHV-68-Infektion der Maus gleicht im Ablauf und im klinischen Verlauf der EBV-
Infektion des Menschen. Ziel dieser Arbeit war es, das bestehende Maus-Modell zu
erweitern und den Ablauf der Infektion im ZNS darzustellen. Mit der Einführung einer
Real-Time-PCR für das G-50-Gen ist es erstmals möglich MHV-68-DNA quantitativ
im Gewebe nachzuweisen und quantitative Aussagen über den Verlauf der Infektion
in Organen und Geweben zu machen. Der Nachweis der viralen Gene M 2, 3, 7,
ORF 25, 57, 59 und des Gens 50 wurden etabliert; das macht den qualitativen
Nachweis für diese Gene sowohl auf DNA als auch auf RNA-Ebene möglich.
Der Nachweis viraler Antigene im Gewebe gelang mittels spezifischer Antikörper; so
konnte das Virus in den Organen lokalisiert werden. Das Vorkommen des Virus im
Gewebe wurde mit immunhistochemischen Daten zur Verteilung von
Entzündungszeichen verglichen. Mittels Zellkulturtechniken wurde infektiöses Virus
im Gewebe der infizierten Mäuse nachgewiesen.
Über die Atemwege gelangt MHV-68 normalerweise in den Organismus und führt zu
einer aktiven Infektion der Lunge, die etwa 10-14 Tage anhält. Es entwickelt sich
eine Virämie mit Befall verschiedener Organe, wie Leber, Niere und Herz. In B-
Lymphozyten entwickelt das Virus ein latentes Stadium. In den B-Lymphozyten ist
die Virus-DNA lebenslang nachweisbar, in anderen Organen sinkt die Virusmenge
nach 10-14 Tagen rasch bis unter die Nachweisgrenze ab.
5.1 Die peripheren Organe im Verlauf der Infektion
Der Virusnachweis in den peripheren Organen diente in erster Linie zur Erprobung
der neu entwickelten Methoden, dabei wurden die Daten, die an dem Bekannten
Modell erhoben wurden, teilweise bestätigt. Allerdings ergaben sich auch neue
Aspekte.
Die Virusreplikation findet offenbar nicht nur in der Lunge und dem lymphatischen
System sondern auch in anderen Organen in größerem Ausmaß statt. Vor allem ist
die Leber betroffen. Die Anwesenheit von Virionen im Lebergewebe unterstreicht
dies. Die Anfärbung von Hepatozyten im IFT zeigt, dass hier Parenchymzellen Ziel
der Virionen sind und nicht etwa Zellen des Immunsystems oder des Interstitiums.
Bislang waren lediglich MHV-68 Virionen mittels Zellkultur und ihre DNA mittels
nested PCR im Lebergewebe nachgewiesen worden (Blaskovic et al. 1984, Flaño et

50
al. 2003, Sunil-Chandra et al. 1992). Die von mir durchgeführte Quantifizierung der
viralen DNA und der Nachweis von Transkriptionsaktivität in der Leber sind neue
Erkenntnisse.
Die Nieren sind in der Regel mitbetroffen, bei einigen Tieren findet sich in ihnen
sogar die höchste Virusmenge. Die Frage, ob hier eine aktive Replikation des Virus
stattfindet, kann aufgrund der bisher erhobenen Daten nicht eindeutig beantwortet
werden. In Anbetracht der teilweise sehr hohen Viruslasten ist dies aber
wahrscheinlich. Möglicherweise werden die Virionen auch renal ausgeschieden (Abb.
12). Dafür spricht der Nachweis viraler Antigene in Tubulusepithelien (Rajcani et al.
1985).
Eine Beteiligung des Herzens im Sinne einer Myokarditis war bei den vorzeitig
verstorbenen Tieren unter Umständen die Todesursache. Die Untersuchten Mäuse
dieses Kollektivs hatten Viruslasten von etwa 109 GÄ/mg Gewebe im Herzgewebe,
was auch meist dem höchsten Wert bei dem entsprechenden Individuum entsprach.
Bei vier Tieren (3/4-6, 3/3-12, 3/3-13 und 3/5-18) wurde histologisch eine
Herzmuskelnekrose gesichert und bei Tier 3/4-6 gleichzeitig Virusantigen im Bereich
der Nekrose nachgewiesen. Bei einem anderen Versuchstier (3/1-1) wurde
Transkriptionsaktivität für Gene des lytischen Zyklus im Herzen nachgewiesen.
Andere Autoren hatten bisher nur den Nachweis von Virionen in der Herzmuskulatur
erbracht, genauere Daten lagen nicht vor (Blaskovic et al. 1984, Rajcani et al. 1985).
Die Lokalisation von Virusantigen in der Lunge weicht von den bisherigen
Erkenntnissen deutlich ab. Bei den hier untersuchten Tieren war virales Antigen
peribronchiolär nachweisbar, nicht in Alveolarepithelzellen, wie vermutet.
Möglicherweise war die Ursache auch hier die hohe Nachweisschwelle bei der
angewandten Methode. Es ist allerdings bekannt, daß MHV-68 bei Mäusen eine
Bronchiolitis verursacht (Nash et al. 2001).
5.2 Natürliche Infektion des ZNS
Eine Infektion neuronaler Zellen durch MHV-68 wurde bereits früh beschrieben. Im
Jahre 1980 wurde das Virus durch Hirnpassage an neugeborenen Mäusen isoliert
(Blaskovic et al. 1980). Allerdings existierte bislang kein Tiermodell für eine MHV-68
Infektion des ZNS auf natürlichem Wege.
MHV-68 DNA wurde im Hirn einer adulten C57BL/6J Maus 60 Tage nach Infektion
gefunden (Flaño et al. 2003). Ebenso gelang es MHV-68 in Neuronen des

51
Hippocampus, Bergmannschen Gliazellen, meningealen Zellen und ependymalen
Zellen zu identifizieren. Dies gelang jedoch nur nach intrathekaler Applikation der
Virionen, oder nach Infektion Interferonrezeptor-negativer Tiere (Terry et al. 2000).
Eine Infektion des ZNS nach intranasaler Inokulation von sieben Tage alten Wildtyp-
Mäusen gelang nicht. Im Rahmen unseres Projektes wurde der Versuch
unternommen, eine Infektion des ZNS mit MHV-68 an Wildtyp-Mäusen zu erreichen.
Diese Aufgabe wurde erfüllt. Die Untersuchungen belegen zweifelsfrei eine Infektion
des ZNS neugeborener Tiere. Die Infektion kam auf physiologischem Weg, über die
nasale Aufnahme der Virionen, zustande. Großhirn und Kleinhirn waren
gleichermaßen von der Infektion betroffen. Entsprechend konnten in einer anderen
Arbeitsgruppe Entzündungsreaktionen im Hirn mittels CD 45 Marker (T-Zellmarker)
nachgewiesen werden, hier auch vereinzelt bei älteren Tieren.
5.3 Zeitlicher Verlauf von Infektion und Immunabwehr
Das Virus etabliert sich sehr wahrscheinlich im Verlauf der Infektion früh im ZNS.
Bereits sieben Tage nach der Infektion ist virale DNA in verschiedenen Arealen des
ZNS nachweisbar. Eine immunhistochemisch darstellbare Abwehrreaktion war zu
diesem Zeitpunkt bei den meisten Tieren noch nicht vorhanden. Mit zunehmender
Immunreaktion nahm die Viruslast im ZNS ab.
Zeichen einer effektiven Virusclearance finden sich frühestens nach 10-14 Tagen.
Vorher ließen sich kaum Entzündungsreaktionen im ZNS nachweisen. Das späte
Einwandern von Lymphozyten ist einerseits auf das unreife Immunsystem
zurückzuführen, andererseits auf die Fähigkeit des Virus, durch das
chemokinbindende Protein M 3 die Chemotaxis zu behindern (Jensen et al. 2003).
Zwei von 95 Tieren (2,1 Prozent) blieben in der körperlichen Entwicklung sichtbar
zurück. Diese Tiere wiesen auch 5 Wochen nach Infektion noch in allen Organen
überdurchschnittlich hohe Viruslasten auf. Bei einem der Tiere (2/3-18) wurde eine
hohe Transkriptionsaktivität nachgewiesen. Diese Ergebnisse sprechen dafür, dass
hier ein Übergang in die Latenz nicht stattgefunden hat, sondern eine aktive,
persistierende Infektion besteht. Möglicherweise handelt es sich um eine anerge
Abwehrlage, da weder die Infektion ausreichend kontrolliert werden konnte, noch der
Organismus durch das Immunsystem letal geschädigt wurde. Dies ist vermutlich kein
seltenes Ereignis. Unter den neonatal infizierten Tieren (39) stellt diese Gruppe
immerhin 5,1 % der Mäuse.

52
5.4 Ort der Infektion und Replikation
Von besonderem Interesse war die Lokalisation des MHV-68 im ZNS. Zielorte
scheinen vor allem die Temporalregion, die Meningen, der Periventrikularraum und
perivaskuläre Zonen im Großhirn zu sein. Diese Regionen wurden sowohl mittels
Antigennachweis (IFT) direkt, als auch indirekt über Nachweis von
Entzündungsreaktionen (siehe Arbeit von Frau Evelyn Alberg, Institut für
Neuropathologie) ausgemacht.
Der Nachweis von MHV-68 in anderen Strukturen des ZNS – in 4 von 5 untersuchten
Rückenmarksproben und bei 2 von 2 untersuchten Augen fand sich Virus-DNA –
spricht für das Vorliegen von Virionen im Liquor. Ob in diesen Strukturen
möglicherweise auch eine Virusvermehrung erfolgt, oder ob die Gewebeproben
aufgrund ihres Liquorgehaltes positiv getestet wurden, kann nicht endgültig
beantwortet werden.
Die Häufigkeit der ZNS-Beteiligung scheint vor allem vom Alter der Tiere zum
Zeitpunkt der Infektion abzuhängen. Je jünger die Tiere sind, desto häufiger ist das
ZNS betroffen. Dabei spielen wahrscheinlich zwei Faktoren eine wichtige Rolle.
Einerseits fehlt den jungen Tieren noch eine effektive Immunabwehr, was die
Infektion wesentlich aggressiver verlaufen lässt, dafür spricht auch die hohe Letalität
in der entsprechenden Population. Die Folge sind wesentlich höhere Viruslasten im
Organismus, und diese wiederum erleichtern einen Übergang ins ZNS. Andererseits
ist die Blut-Hirnschranke wie auch das Immunsystem noch unausgereift, und damit
leichter zu überwinden.
Die Möglichkeit für das Virus über neuronale Strukturen in das ZNS einzudringen
erscheint gegenwärtig nicht sehr wahrscheinlich. Sowohl die Lokalisation der Viren
als auch der Entzündungsreaktionen (Hippocampus, Ependym, Meningen, corticale
weiße Substanz usw.) sprechen für eine Infiltration der Hirnstrukturen von den
Blutgefäßen aus.

53
5.5 Ausblick
Die Ergebnisse dieser Arbeit belegen eine ausgeprägte Beteiligung des ZNS nach
Infektion von Maus-Neonaten mit MHV-68. Es gibt Anhaltspunkte für die Lokalisation
der Viren im Gehirn. Damit besteht die Möglichkeit, die Aktivitäten des Virus im ZNS
genauer zu untersuchen. Die Reaktivierungsvorgänge des Virus können jetzt an der
neu etablierten MHV-68 positiven B-Zelllinie dargestellt werden.
Erkenntnisse über die Reaktivierung des Virus und sein Verhalten im Wirt helfen,
Strategien zur Entwicklung eines Impfschutzes gegen EBV zu finden.

54
6 Zusammenfassung
Eine schwerwiegende Komplikation der Epstein-Barr-Virus-Infektion im Kindesalter
ist der Befall des zentralen Nervensystems (ZNS), mit Folgen wie z.B. Epilepsien und
Enzephalitiden. Das gut etablierte Modell des dem EBV verwandten Maus-
Herpesvirus 68 (MHV-68), war bisher nicht für Infektionen des ZNS anwendbar, da
Infektionen auf natürlichem Wege nicht möglich waren.
Ziel dieser Arbeit war es, eine Strategie zu finden, die eine MHV-68-Infektion des
ZNS ermöglichte. Darüber hinaus war eine stabile, MHV-68-permissive Zelllinie zu
entwickeln, die für in-vitro-Untersuchungen geeignet ist. Folgende Ergebnisse
wurden erzielt:
- die Infektion des ZNS gelang nach intranasaler Applikation des MHV-68; der
Infektionserfolg wurde mittels quantitativem Nachweises viraler DNA im ZNS
und anderer Organe erbracht;
- der Nachweis viraler Antigene im ZNS und anderen Organen gelang mittels
Immunfluoreszenz;
- das Virus wurde aus Gewebe angezüchtet;
- die geeignete Infektiosdosis wurde ermittelt;
- das Virus wurde im ZNS lokalisiert;
- der Nachweis viraler m-RNA in Geweben als Ausdruck von
Replikationsaktivität war möglich;
- infektiöse Viruspartikel wurden in verschiedenen Geweben nachgewiesen;
- es ist gelungen, eine permanent wachsende, MHV-68-positive B-Zelllinie zu
etablieren.
Es ist gelungen, das bekannte MHV-68-Modell zu erweitern und für Infektionen des
ZNS zu nutzen. Der Infektionsweg und die bevorzugte Lokalisation der Viren im ZNS
wurden aufgespürt. Diese Ergebnisse können zusammen mit der permanent MHV-
68-positiven B-Zelllinie genutzt werden, die pathogenetische Wirkung des Virus auf
Zellen und Strukturen des ZNS weiter zu untersuchen.

55
7 Literatur
Blasdell K, Mc Cracken C, Morris A, Nash AA, Begon M, Bennet M & Steward
JP (2003): The wood mouse is a natural host for Murid herpesvirus 4. Journal of
Gen. Virol. 84: 111-113
Blaskovic D, Stancekova M, Svoboda J & Mistrikova J (1980): Isolation of five
strains of herpesviruses from two species of free living small rodents. Acta
virologica 24: 468
Blaskovic D, Stanekova D & Rajcani J (1984): Experimental pathogenesis of
murine herpesvirus in newborn mice. Acta Virologica 28: 225-231
Bowden RJ, Simas JP, Davis AJ & Efstathiou S (1997): Murine
gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during
latency. Journal of Gen. Virol. 78: 1675-1687
Dutia BM, Allen DJ, Dyson H & Nash AA (1999c): Type I interferons and IRF-1 play
a critical role in the control of a gammaherpesvirus infection. Virology 261: 173-
179
Dutia BM, Steward JP, Clayton RAE, Dyson H & Nash AA (1999a): Kinetic and
phenotypic changes in murine lymphocytes infected with murine
gammaherpesvirus-68 in vitro. Journal of Gen. Virol. 80: 2729-2736
Efstathiou S, Ho YM, Hall S, Styles CJ, Scott SD & Gompels VA (1990): Murine
herpesvirus 68 is genetically related to the γ-herpesviruses EBV + HVS. Journal of
Gen. Virology 71: 1365-1372
Ehtisham S, Sunil-Chandra NP & Nash AA (1993): Pathogenesis in mice deficient
in CD4+ and CD8+ T cells. Journal of Virology 67: 5247-5252
Flaño E, Husain SM, Sample JT, Woodland DL & Blackman MA (2000): Latent
murine γ-herpesvirus infection is established in activated B cells, dendritic cells
and macrophages. Journal of Immunology 165: 1074-1081

56
Flaño E, Kim IJ, Moore J, Woodland DL & Blackman MA (2003): Differential γ-
herpesvirus distribution in distinct anatomical locations and cell subsets during
persistent infection in mice. Journal of Immunology 170: 3828-3834
Häusler M, Ramaekers VT, Doenges M, Schweizer K, Ritter K & Schaade L
(2002): Neurological Complications of Acute and Persistent Epstein-Barr Virus
Infection in Paediatric Patients. Journal of Med. Virology 68: 253-263
Husain SM, Usherwood EJ, Dyson H, Coleclough C, Coppola MA, Woodland
DL, Blackman MA, Stewart JP & Sample JT (1999): Murine gammaherpesvirus
M2 gene is latency-associated and its protein a target for CD8+ T lymphocytes.
Proc. Natl. Acad. Sci. USA 96: 7508-7513
Jensen KK, Chen SC, Hipkin RW, Wiekowski MT, Schwarz MA, Chou CC, Simas
JP, Alcami A & Lira SA (2003): Disruption of CCL 21-induced chemotaxis in vitro
and in vivo by M 3, a chemokine-binding protein encoded by murine
gammaherpesvirus 68. Journal of Virology 77: 624-630
Macrae AI, Dutia BM, Milligan S, Allen DJ, Mistrikova J, Davison AJ, Nash AA &
Stewart JP (2001): Analysis of a novel strain of murine gammaherpesvirus
reveals a genomic locus important for acute pathogenesis. Journal of Virology 75:
5315-5327
Martinez-Guzman DA, Rickabough T, Wu TT, Brown H, Cole S, Song MJ, Tong L
& Sun R (2003): Transcription program of murine gammaherpesvirus 68. Journal
of Virology 77: 10488-10503
Nash AA, Dutia BM, Steward JP & Davison AJ (2001): Natural history of murine γ-
herpesvirus infection. Phil. Trans. R. Soc. Lond. B 356: 569-579
Neyts J & De Clercq E (1998): In vitro and in vivo inhibition of murine gamma
herpesvirus 68 replication by selected antiviral agents. Antimicrobial Agents and
Chemotherapie 42: 170-172

57
Peacock JW & Bost KL (2000): Infection of intestinal epithelial cells and
development of systemic disease following gastric instillation of murine
gammaherpesvirus -68. Journal of Gen. Virol. 81: 421-429
Rajcani J, Blaskovic D, Svoboda J, Ciampor F, Huckova D & Stanekova D
(1985): Pathogenesis of acute and persistent murine herpesvirus infection in mice.
Acta Virol. 29: 51-60
Rajcani J, Bustamante de Contreras LR & Svoboda J (1986): Corneal inoculation
of murine herpesvirus in mice: the absence of neural spread. Acta Virol. 31: 25-30
Rochford R, Lutzke ML, Alfinito RS, Clavo A & Cardin RD (2001): Kinetics of
murine gammaherpesvirus 68 gene expression following infection of murine cells
in culture and in mice. Journal of Virology 75: 4955-4963
Sangster MY, Topham DJ, D’Costa S, Cardin RD, Marion TN, Myers K & Doherty
PC (2000): Analysis of the virus-specific and nonspecific B cell response to a
persistent B-lymphotropic gammaherpesvirus. Journal of Immunology 164: 1820-
1828
Simas JP, Swann D, Bowden R & Efstathiou S (1999): Analysis of murine
gammaherpesvirus-68 transcription during lytic and latent infection. Journal of
Gen. Virol. 80: 75-82
Stevenson PG & Doherty PC (1998): Kinetic analysis of the specific host response
to a murine gammaherpesvirus. Journal of Virology 72: 943-949
Stevenson PG, Cardin RD, Christensen JP & Doherty PC (1999): Immunological
control of a murine gammaherpesvirus independent of CD8+ T cells. Journal of
Gen. Virol. 80: 477-483

58
Steward JP, Usherwood EJ, Ross A, Dyson H & Nash AA (1998): Lung epithelial
cells are a major site of murine gammaherpesvirus persistence. Journal of Exp.
Med. 187: 1941-1951
Sunil-Chandra NP, Arno J, Fazakerley J & Nash AA (1994): Lymphoproliferative
disease in mice infected with murine gammaherpesvirus 68. Am. Journal of
Pathology 145: 818-826
Sunil-Chandra NP, Efstathiou S & Nash AA (1993): Interactions of murine
gammaherpesvirus 68 with B and T cell lines. Virology 193: 825-833
Sunil-Chandra NP, Efstathiou S, Arno J & Nash AA (1992): Virological and
pathological features of mice infected with murine gammaherpesvirus 68. Journal
of Gen. Virol. 73: 2347-2356
Terry LA, Steward JP, Nash AA & Fazakerley JK (2000): Murine
gammaherpesvirus-68 infection of and persistence in the central nervous system.
Journal of Gen. Virol. 81: 2635-2643
Usherwood EJ, Ross AJ, Allen AJ & Nash AA (1996b): Murine
gammaherpesvirus-induced splenomegaly: a critical role for CD4+ T cells. Journal
of Gen. Virol. 77: 627-630
Usherwood EJ, Steward JP, Robertson K, Allen DJ & Nash AA (1996a): Absence
of splenic latency in murine gammaherpesvirus 68-infected B cell-deficient mice.
Journal of Gen. Virol. 77: 2819-2825
Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ &
Speck SH (1997): Complete sequence and genomic analysis of
gammaherpesvirus 68. Journal of Virology 71: 5894-5904
Virgin HW, Presti RM, Xi-Yang L, Liu C & Speck SH (1999): Three distinct regions
of the murine gammaherpesvirus 68 genome are transcriptionally active in latently
infected mice. Journal of Virology 73: 2321-2332

59
A Anhang
Abkürzungsverzeichnis
Abb. Abbildung
BHK-21 Baby Hamster Kidney
bp Basenpaare
BSA bovine serum albumine (Rinderserumalbumin)
CD cluster of differentiation
DMSO Dimethylsulfoxid
DNA desoxyribonucleic acid (Desoxyribonukleinsäure)
dNTPs Desoxynukleosidtriphosphate
EBV Epstein-Barr Virus
E. coli Escherichia coli
FITC Fluoresceinisothiocyanat
FKS Fetales Kälberserum
GÄ Genomäquivalent
GAPDH Glycerinaldehyd-3-Phosphat Dehydrogenase
h Stunde
IFT Immunfluoreszenztest
LB-Medium Luria Bertani-Medium
LMU Ludwig Maximilians Universität
MHV-68 murines Herpesvirus 68
min Minute
mRNA messenger RNA
n Stichprobengröße
NEA non essential aminoacid
PBS phosphate buffered saline
PCR polymerase chain reaction
RNA ribonucleic acid (Ribonukleinsäure)
rpm rotations per minute
RT Reverse Transkriptase
Taq Thermus aquaticus
TBE Tris-Borsäure-EDTA

60
Abbildungsverzeichnis
Abb. I pGlow-TOPO Vektor (der entsprechenden Gebrauchsanweisung der Firma Invitrogen entnommen)
8
Abb. 1 Real-Time-PCR Schmelzkurve des G 50 Gens 31
Abb. 2 Produkte der Real-Time-PCR nach Trennung mittels
Gelelektrophorese
31
Abb. 3 M 3 nested PCR mit spezifischen Banden bei 133 bp 32
Abb. 4 BHK-21 Zellen inkubiert mit MHV-68-positivem Serum 1:1024 33
Abb. 5 BHK-21 Zellen inkubiert mit negativem Mausserum 1:50 33
Abb. 6 Nachweis von muriner GAPDH (236 bp) 34
Abb. 7 Nachweis von G 50 Gen in B-Zellinie (293 bp) 34
Abb. 8 MHV-68 positive Lymphozytenkultur inkubiert mit PBS 35
Abb. 9 MHV-68 positive Lymphozytenkultur inkubiert mit MHV-68 Antikörper
1:64
35
Die Abbildungen 8 und 9 wurden freundlicherweise zur Verfügung
gestellt von Frau S. Kuropka; Doktorandin am Institut für
medizinische Mikrobiologie der RWTH-Aachen
Grafik I Virusmenge nach Infektion am 1. oder 5. Lebenstag 36
Grafik II Infektionsraten einzelner Organe bezogen auf erfolgreich infizierte
Tiere
37
Grafik III Direkter Vergleich der Virusmengen in Blut und Milz 38
Grafik IV Direkter Vergleich der Virusmengen in Blut und Leber 38
Abb. 10 Maus 2/3-18 im Vergleich mit gleichalten Kontrolltieren 39
Grafik V Verlauf der Virusmengen. Zeitpunkt der Infektion: 1.-6. Lebenstag 40
Abb. 11a MHV-68-positive Hepatozyten perivaskulär 42
Abb. 11b Leberbefund eines nicht infizierten Tieres 42
Abb. 12a MHV-68-positive Nierentubuli 42
Abb. 12b Nierenbefund eines Kontrolltieres 42
Abb. 13a Peribronchiolärer Nachweis von MHV-68 42
Abb. 13b Lunge eines Kontrolltieres 42
Grafik VI Vergleich Virusmengen Milz/ Großhirn 44
Grafik VII Vergleich Virusmengen Großhirn 44
Abb. 14 Tier 2/3-18 Plexus choroideus 46

61
Abb. 15 Tier 3/2-4 Großhinrinde 46
Abb. 16 Tier 3/4-6 Kleinhirn 46
Abb. 17 Tier 3/3-12 Ammonshorn 46
Grafik VIII Viruslasten vs Entzündungsreaktion 47
Abb. 18a Tier 3/4-6 Temporallappen CD-45 Färbung 47
Abb. 18b Tier 3/4-6 Temporallappen MHV-68-IFT 47
Abb. 19a Tier 3/4-6 Temporalregion CD-45 Färbung 48
Abb. 19b Tier 3/4-6 IFT gleiche Region 48
Abb. 20a Tier 3/4-6 Herzmuskel mit MHV-68 Nachweis 48
Abb. 20b Tier 3/4-6 Herzmuskelnekrose 48
Abb. 21a Perivaskuläre Infiltrate in der Leber 48
Abb. 21b Peivaskulär infizierte Hepatozyten im IFT 48

62
Danksagung
Mein besonderer Dank gilt Herrn Prof. Dr. K. Ritter für die Ermöglichung dieser
Arbeit, für wesentliche Anregungen sowohl bei der praktischen Durchführung, als
auch bei der Erstellung dieser Arbeit.
Herrn PD Dr. M. Häusler danke ich für die Aufnahme in das Projekt und dafür, dass
er mir jederzeit mit Rat und Tat zur Seite stand.
Frau Dr. S. Scheithauer und Herrn Dr. M. Kleines danke ich für die gute
Zusammenarbeit und Betreuung.
Ganz besonders danke ich Michele Peters und Kirsten Schellenberg, ohne deren
Hilfe ich den praktischen Teil der Arbeit sicher nicht beendet hätte. Außerdem
natürlich für die Kaffeepausen.
Weiterhin bedanke ich mich bei allen Doktoranden und Diplomanden am Institut bzw.
im MHV-Projekt für die gute Zusammenarbeit.
Evelyn danke ich für die Gewebeschnitte, ihre Gastfreundschaft und die schönsten
Feiern in Aachen.
Claudia danke ich dafür, dass sie mich beim Schreiben dieser Arbeit unterstützt hat,
und dafür, dass sie eine tolle Mutter ist.
Der größte Dank gilt meinen Eltern und meiner Schwester. Dafür, dass es sie gibt,
und dafür, dass ich das werden konnte, was ich heute bin.

63
Erklärung § 5 Abs. 1 zur Datenaufbewahrung
Hiermit erkläre ich, dass die dieser Dissertation zu Grunde liegenden Originaldaten
im Institut für Medizinische Mikrobiologie/ Virologie des Universitätsklinikums Aachen
hinterlegt sind.

64
Lebenslauf
Persönliche Daten Name: Matthias Engler Geboren am: 06.06.1975 in Frankfurt am Main Schulausbildung 1981 – 1985 Heiligenstockschule Hofheim am Taunus, Grundschule 1985 – 1991 Elisabethenschule Hofheim, staatlich anerkannte private Realschule 1991 – 1994 Main-Taunus-Schule Hofheim, Gymnasiale Oberstufe Abgeschlossen mit der allgemeinen Hochschulreife am 10.06.1994 Zivildienst/ Berufsausbildung Aug. 1994 – Okt. 1995 Zivildienst in Altenpflegeeinrichtung der Caritas in Hofheim Okt. 1995 – März 1996 Studium der Geschichte, Politologie und Philosophie an
der Johann Wolfgang Goethe-Universität in Frankfurt am Main
Okt. 1996 – Sep. 1999 Ausbildung zum Krankenpfleger an den Kliniken des Main-
Taunus-Kreises in Hofheim / Bad Soden Studium Okt. 1999 – Sep. 2004 Studium der Humanmedizin an der Rheinisch-
Westfälischen Technischen Hochschule in Aachen Okt. 2004 – März 2006 Fortsetzung des Studiums der Humanmedizin an der
Johann Wolfgang Goethe-Universität in Frankfurt am Main 03.05. 2006 Dritter Abschnitt der ärztlichen Prüfung Berufstätigkeit Seit 01.06.2006 Assistenzarzt an der Klinik für Kinder- und Jugendmedizin
des Klinikums Offenbach