Embed Size (px)
Citation preview

Mehrfachbindungen zwischen Ubergangsmetallen und ,,nackten" Hauptgruppenelementen: Brucken zwischen der anorganischen Festkorperchemie und der Organometallchemie
Von Wolfgang A. Herrmann*
In memoriam Wilhelm KIemm
Die Chemie der Metall-Metall-Mehrfachbindung hat in den vergangenen beiden Jahrzehn- ten eine stiirmische Entwicklung erlebt, wobei insbesondere die Organometallchemie groBe Erfolge zu verzeichnen hatte. Die Entwicklung zielgerichteter Synthesen neuer Ubergangs- metallkomplexe mit Einfach-, Doppel-, Dreifach- und Vierfachbindungen zwischen den Metallatomen sowie die Untersuchung von deren Reaktivitat sind derzeit prominente Ar- beitsgebiete. Die Hoffnung auf Verallgemeinerungs- und Ordnungsprinzipien ist eine Triebfeder dieser Aktivitaten, doch darf auch das weitergesteckte Ziel, Metall-Metall-Mehr- fachbindungssysteme als Katalysatoren zu nutzen, nicht aus dem Auge verloren werden. Alle Aspekte, unter denen man bisher Metall-Metall-Mehrfachbindungen untersucht und beurteilt hat, interessieren auch bei der Verbindungsklasse, die bisher ein Schattendasein gefuhrt hat : Komplexverbindungen mit Mehrfachbindungen zwischen lfbergangsmetallato- men und substituentenfreien (,,nackten") Hauptgruppenelementatomen. Urspriinglich Zu- fallsentdeckungen, lassen die wenigen derzeit namhaft zu machenden Beispiele ausbaufa- hige Synthesekonzepte erkennen. Wenn, was zu erwarten ist, weitere Strukturtypen dieser Verbindungsklasse bekannt werden, diirfte dieser Zweig der prsparativen Organometallche- mie beispielsweise bei der Komplexstabilisierung und der Aktivierung kleiner Molekule groBe Bedeutung erlangen. Um die Thematik umfassend zu beleuchten, wird die Komplex- chemie nackter Kohlenstoff-, Stickstoff- und Sauerstoffliganden hier nur soweit beriicksich- tigt, wie dadurch praparative und strukturchemische Beziehungen zu den Komplexverbin- dungen ihrer hoheren Homologen sichtbar werden.
1. Einleitung
Monumental und reich ausgestattet priisentiert sich das Gebaude der Organornetallchemie nach 40jahriger Arbeit. Die Metallgeriiste sind haufig uppig verziert und mitunter kaum mehr erkennbar",21. Ausgeschopft ist die Reichhal- tigkeit an Strukturen noch lange nicht, denn erst in junge- rer Zeit wurden die Bauplane bekannt, die ausbaufahige Konzepte sowie Verwandtschaftsbeziehungen zu anderen Teilbereichen der Chemie erkennen l a ~ s e n [ ~ , ~ ~ . Offenbart die Vielfalt kohlenstoffhaltiger Ligandsysteme den nahtlo- sen Ubergang zur Organischen Chemie, so ist die Geriist- architektur polynuclearer Clustermolekule den Struktu- ren anorganischer Festkorper nicht fremd und wie dort durch Synthesebedingungen und Atomradienverhaltnisse miteinander verknupfter Elemente sowie deren Elektro- nenhaushalt gepragt. Der anorganischen Festkdrperche- mie nicht unahnlich, umfaBt die bunte Substanzpalette der Organometallchemie den weiten Bereich von kleinen Ele- mentensembles uber mittelgroRe Cluster bis zu metallarti- gen Multiatomverbanden, wie sie uns etwa die Gold- und Palladiumchemie in der kubisch-dichtest gepackten ,,fie- senkugel" A U ~ ~ [ P ( C ~ H ~ ) ~ ] , ~ C I ~ - Vorstufe einer neuen Goldmodifikation mit Aul3-Kuboktaedem! - bzw. in ei- nem wasserloslichen Pd570f ,o-Giganten (240 & 13 Ligan- den) vorfiihren[s-81.
[*I Prof. Dr. W. A. Herrmann Anorganisch-chemisches Institut der Technischen UniversitBt Milnchen LichtenbergstraBe 4, D-8046 Garching
Polyatomare Clusterstrukturen diirfen nicht aus dem Auge verloren werden, wenn im vorliegenden Aufsatz Ubergangsmetalle und substituentenfreie Hauptgruppen- elemente vorwiegend unter dem Aspekt der zwischen ih- nen vorliegenden Mehrfachbindungen besprochen werden sollen. Buchhalterische Prizision ist hier nicht angestrebt, vielmehr sollen Zusammenhange zwischen Genese und Struktur solcher Komplexverbindungen einerseits sowie zwischen niedrig- und hochkoordinierten nackten Haupt- gruppenelementen andererseits sichtbar gemacht werden. Eine Beschrankung auf die Kohlenstoff-, Stickstoff- und Sauerstoffgruppen des Periodensystems erscheint zur Ver- meidung ausufemder Aufziihlungen angeraten. Obwohl generalisierende Aussagen noch verfriiht sind, sol1 der Versuch gemacht werden, fur Mehrfachbindungen zwi- schen lfbergangsmetallen und substituentenfreien Haupt- gruppenelementen typische Reaktivitiitsmuster zu benen- nen.
2. Substituentenfreie Elemente der 4. Hauptgruppe als Briickenliganden
2.1. Kafigstrukturen
Wurden noch vor etlichen Jahren Komplexverbindun- gen mit ,,interstitiellen" Kohlenstoffatomen als Laborato- riumskuriositaten betrachtet, so sind Cluster dieses Typs heute gezielt synthetisierbar, wenngleich die Reaktivitaten der zwischen mehrere ijbergangsmetallatome eingebette-
Angew. Chern. 98 (1986) S7-77 Q VCH Verlagsgesellschafr mbH. 0-6940 Weinheim. 1986 W44-8249/86/0101-0057 0 02.50/0 57

ten Kohlenstoffatome noch immer Ratsel aufgeben und noch langst nicht klassifizierbar sindl'-'']. Aliphatische Kohlenwasserstoffe und fast jede beliebige kohlenstoffhal- tige Ligandvorstufe eignen sich zur Darstellung von Carbi- doclustern mit interstitiellen C-Atomen. Die Lehrbuch- beispiele 1-4 (Abb. 1) sind nicht zuletzt um des asthe- tischen Genusses willen wiedergegeben.
1
Abb. 1. Nackte Kohlenstoffatome in isolierbaren, diskreten Ubergangsme- tallclustern rnit fiinf-, sechs- und achtatomigen Metdllpolyederstrukturen (C- Atome sind rchwarz. Metallatome schraffiert und 0-Atome weiB dargestellt). Mi1 Ausnahme des Fe5C-Clusters 1, der ein exponiertes Kohlenstoffatom aufweist, reprasentieren diese Komplexe Metallverbindungen mit interstitiel- len (,,interstitial", ..encapsulated") Kohlenstoffliganden. ~ [FesC(CO)Is] I : Das fiinffach koordinierte Kohlenstoffatom befindet sich 15 pm unterhalb der Grundflsche der quadratischen Fel-Pyramide; die FeC-Abstlnde betra- gen 189 (basal) und 196 pm (apical). - [RubC(C0)17] 2: Ein oktaedrischer, rnit dem strukturanalogen Anion [Fe6C(C0),,l2- isoelektronischer 86e-Kom- plex. 16 der 17 Carbonylliganden sind durch Striche dargestellt. ~
[RhoC(CO),,]2- 3: Das Kohlenstoffatom befindet sich im Zentrum eines tri- gonalen Rh,-F'rismas (sechs terminale, neun kantenverbriickende CO-Ligan- den; 90e-Cluster). - [Co,C(CO),,l2- 4 : Ein interstitieller Carhidokomplex mit quadratisch-antiprismatischer Geriiststruktur (Co-C 195-220 pm).
Die Fe,C- und Ru,C-Kafigstrukturen 1 bzw. 2 repra- sentieren die Konstitutionstypen E (tetragonal-pyramidal) bzw. F (oktaedrisch) in Schema 1, bei denen keine Mehr- fachbindungen zwischen dem Molekulzentrum E (hier ein Kohlenstoffatom) und den Ubergangsmetallatomen M auftreten konnen. Trotz unterschiedlicher Kafigarchitektur tragen die nackten C-Atome stets vier Elektronen zum La- dungshaushalt der Clustermolekule bei.
M*=E=M* MLE-M M, M,E=M*
linear linear trigonal-planar
A B C
tetraedrisch tetragonal- oktaedrisch pyramidal
D E F
Schema I . Koordinationsmoglichkeiten fiir substituentenfreie Elemente der 4. Hauptgruppe.
Es fallt auf, da13 die Konstitutionsmoglichkeiten A und B (linear) sowie C (trigonal-planar) in der Organometall-
chemie des Kohlenstoffs fehlen"], obwohl sich gerade diese Strukturtypen fur die hoheren Homologen als stabil erweisen (siehe Abschnitte 2.2-2.4). Als exemplarisch fur te- traedrische Koordination (Typ D) mogen die Organo- quecksilber-Verbindungen C(HgR)4 (R = Alkyl, Aryl) an- gesehen werdenl
Einen substituentenfreien Zinnliganden fanden F. G. A. Stone et al. erstmals im Jahre 1966 im Komplex
einem Cluster, der sich nach R. HofSmann rnit der Isolo- bal-Anal~gie[~] als ,,anorganisches Spiropentan" beschrei- ben la&, und in dem das Zinnatom verzerrt tetraedrisch koordiniert ist (Abb. 2; Schema 1, Typ D). Urn vierbindige
d Abb. 2. MaOstabsgetreue Strukturskizze des rnit Spiropentan isolobalen, an- ntihernd D2d-symmetrischen Fe4Sn-Clusters 5. Die Zinn-Eisen-AbstBnde lie- gen bei 254pm. die FeSnFe-Winkel betragen 69 und 133". Man nimmt schwache Wechselwirkungen zwischen den 5d,*- sowie 5d.,-Orbitalen des vierbindigen Zinnatoms und den 3d.~_,~-Orbitalen der Eisenatome an.
Silicium- und Germaniumliganden gruppieren sich Carbo- nylcobalt-Fragmente in den strukturchemisch abgesicher- ten Komplexen 6a und 9 (Abb. 3; Schema 1, Typ D)[ls-2z1,
G?p0 I ,Si,
(co),cg/j~o(co~ (co)3co,~,co(co), co' (CO), (CO),
6a , E=SI M- colco). 7 6b. E-Ge: M- ColCOl. 6c , E-Go. M- MnCO),
8a. E=Sn 8b. E=Pb
9
Analoge Strukturverhaltnisse darf man wohl auch fur die interessante FeCo,Si-Verbindung 7 annehmen, die Malisch et al. durch Metallierung des Silylliganden in der Vorstufe [($-C,Hs)Fe(CO)2(SiH,)1 rnit Octacarbonyldicobalt in die Hande gefallen war[2o1, sowie fur die von Schrnid et al. aus elementarem Zinn und Blei bei deren Einwirkung auf Oc- tacarbonyldicobalt erhaltenen Komplexe 8a bzw. 8b'''. I9l.
[*I Eine mogliche Ausnahme reprssentiert der strukturchemisch bisher nicht charakterisierte Porphyrinkomplex [(p-C)(Fe(TPP)J,KTPP=Tetraphenyl- porphyrin): vgl. D. Mansuy, J.-P. Lecomte, J.-C. Chottard, J.-F. Bartoli, Inorg. Chem. 20 (1981) 3119.
58 Angew. Chem. 98 (1986) 57-77
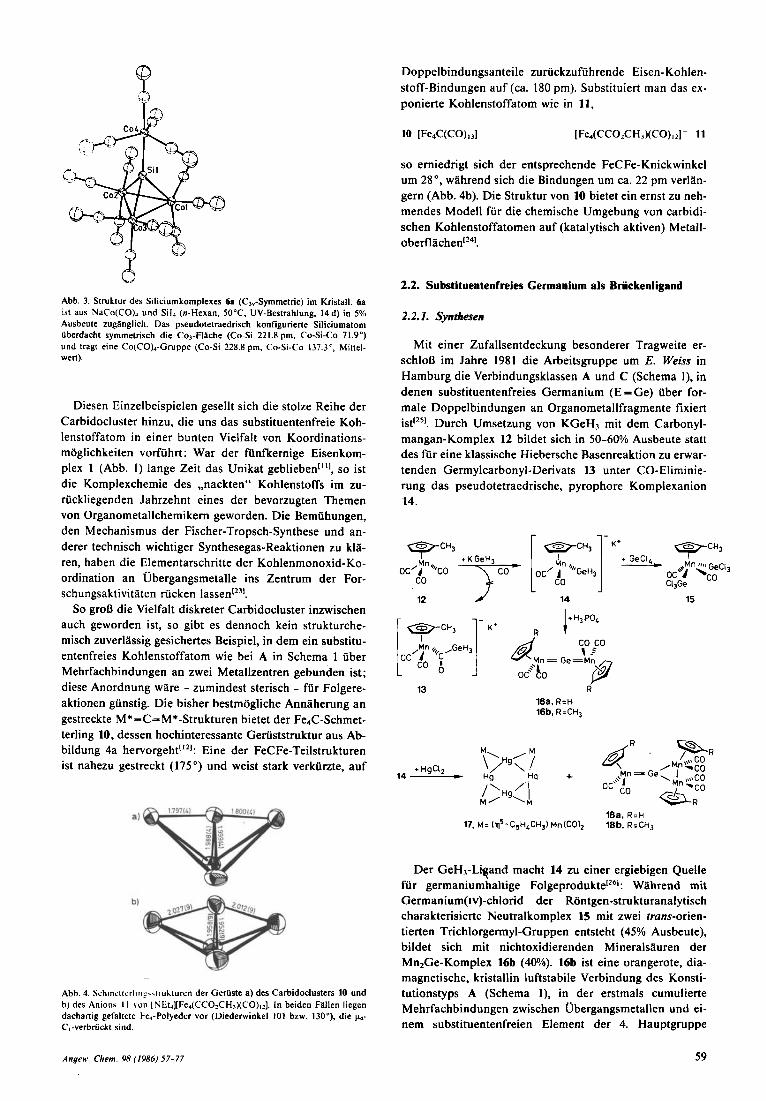
Abb. 3. Struktur des Siliciumkomplexes 6. (C,,-Symmetrie) im Kristall. 6. ist aus N ~ C O ( C O ) ~ und Sil, (n-Hexan, 50°C, UV-Bestrahlung, 14 d) in 5% Ausbeute zugilnglich. Das pseudotetraedrisch konfigurierte Siliciumatom iiberdacht symmetrisch die Co,-Flilche (Co-Si 22 1.8 pm. Co-Si-Co 71.9") und tragt eine Co(CO)4-Cruppe (Co-Si 228.8 pm, Co-Si-Co 137.3'. Millel- wert).
Diesen Einzelbeispielen gesellt sich die stolze Reihe der Carbidocluster hinzu, die uns das substituentenfreie Koh- lenstoffatom in einer bunten Vielfalt von Koordinations- moglichkeiten vorfuhrt: War der funfkernige Eisenkom- plex 1 (Abb. 1) lange Zeit das Unikat geblieben'l'], so ist die Komplexchemie des ,,nackten" Kohlenstoffs im zu- riickliegenden Jahrzehnt eines der bevorzugten Themen von Organometallchemikern geworden. Die Bemuhungen, den Mechanismus der Fischer-Tropsch-Synthese und an- derer technisch wichtiger Synthesegas-Reaktionen zu kla- ren, haben die Elementarschritte der Kohlenmonoxid-Ko- ordination an Ubergangsmetalle ins Zentrum der For- schungsaktivitiiten riicken lassen[231.
So grof3 die Vielfalt diskreter Carbidocluster inzwischen auch geworden ist, so gibt es dennoch kein strukturche- misch zuverliissig gesichertes Beispiel, in dem ein substitu- entenfreies Kohlenstoffatom wie bei A in Schema 1 uber Mehrfachbindungen an zwei Metallzentren gebunden ist; diese Anordnung ware - zumindest sterisch - fur Folgere- aktionen giinstig. Die bisher bestmogliche Anniiherung an gestreckte M*=C=M*-Strukturen bietet der Fe,C-Schmet- terling 10, dessen hochinteressante Geriiststruktur aus Ab- bildung 4a hervorgeht"21: Eine der FeCFe-Teilstrukturen ist nahezu gestreckt (175") und weist stark verkurzte, auf
Abb. 4. Sc.hrneiir.rliiiF..iiuhturen der Geriiste a) des Carbidoclusters 10 und b) des Anion\ I I \on INEtJ][Fe.(CC02CH~)(C0)111. In beiden Fallen liegen dachartig gefaltete Fe,-Polyeder vor (Diederwinkel 101 bzw. 130"), die p,- CI-verbriickt sind.
Doppelbindungsanteile zuriickzufiihrende Eisen-Kohlen- stoff-Bindungen auf (ca. 180 pm). Substituiert man das ex- ponierte Kohlenstoffatom wie in 11,
so erniedrigt sich der entsprechende FeCFe-Knickwinkel um 28", wahrend sich die Bindungen um ca. 22 pm verlan- gern (Abb. 4b). Die Struktur von 10 bietet ein ernst zu neh- mendes Modell fur die chemische Umgebung von carbidi- schen Kohlenstoffatomen auf (katalytisch aktiven) Metall- ~befflachen["~.
2.2. Substituentenfreies Germanium als Briickeoligand
2.2.1. Synthesen
Mit einer Zufallsentdeckung besonderer Tragweite er- schloR im Jahre 1981 die Arbeitsgruppe um E. Weiss in Hamburg die Verbindungsklassen A und C (Schema I), in denen substituentenfreies Germanium (E = Ge) uber for- male Doppelbindungen an Organometallfragmente fixiert i ~ t ' ~ ' ~ . Durch Umsetzung von KGeH3 mit dem Carbonyl- mangan-Komplex 12 bildet sich in 50-60% Ausbeute statt des fur eine klassische Hiebersche Basenreaktion zu erwar- tenden Germylcarbonyl-Derivats 13 unter CO-Eliminie- rung das pseudotetraedrische, pyrophore Komplexanion 14.
12 J 14 15
13 d 16a, R = H 16b. R=CHJ
Der GeH3-L$and macht 14 zu einer ergiebigen Quelle fur germaniumhaltige Folgeprodukte1261: Wahrend mit Germanium(1v)-chlorid der Rontgen-strukturanalytisch charakterisierte Neutralkomplex 15 mit zwei rrans-orien- tierten Trichlorgermyl-Gruppen entsteht (45% Ausbeute), bildet sich mit nichtoxidierenden Mineralsawen der Mn,Ge-Komplex 16b (40%). 16b ist eine orangerote, dia- magnetische, kristallin luftstabile Verbindung des Konsti- tutionstyps A (Schema l), in der erstmals cumulierte Mehrfachbindungen zwischen Ubergangsmetallen und ei- nem substituentenfreien Element der 4. Hauptgruppe
Anyew. Chem. 98 (1986) 57-77 59

nachweisbar waren. LaBt man dagegen Quecksilber(i1)- chlorid auf das Komplexanion 14 einwirken, so entsteht neben dem ebenen (!) sternfirrmigen Mn,H&-Komplex 17 (55%) in 1% Au~beute[”~ der Mn,Ge-Komplex 18b (Typ C), der als auf Umwegen gebildetes Oxidationsprodukt von 14 aufgefaI3t werden kann. Die Bildungsweise dieser damals in der Germaniumchemie spektakularen Komplex- verbindungen 16b und 18b ist noch immer ungewil3. Un- strittig ist, daB die Ge-H-Bindungen in 14 ausgesprochen substitutionslabil sind, was den Einsatz von GeH, als er- giebigere Quelle fur ,,nackte“ Cermaniumliganden nahe- legte, zumal oxidative Additionen der Organogermane R,GeH,-, (n = 0-3) in der Ubergangsmetallchemie wohl- dokumentiert sind12x1. So gelang uns die Synthese der Mn,Ge-Verbindung 18a durch Umsetzung des solvenssta- bilisierten Organometallfragments
I(rlS-CsHs)Mn(CO),(thfll 19
mit German in Tetrahydrofuran (THF‘) bei Raumtempera- Diese Methode liefert erheblich bessere Ausbeuten
(40-60%) und umgeht die Darstellung von KGeH, aus GeH4 und Kalium. Es sei bereits an dieser Stelle darauf hingewiesen, da13 sich binare Elementhydride EH, ganz allgemein bestens zur Einfiihrung der nackten Hauptgrup- penelemente E in Ubergangsmetallkomplexe eignen.
Insbesondere die franzosische Arbeitsgruppe um Corriu hat in zahlreichen Publikationen gezeigt, da13 sich Organo- silane glatt an koordinativ ungesattigte Organometallfrag- mente unter Spaltung der Si-H-Bindung addieren und da- bei Hydrido(sily1)-Komplexe ergebenIgnl. SiH, verhalt sich nicht prinzipiell anders, wenn es rnit dem 16e-Fragment
reagiert und dabei den Mn2-Komplex
liefert, der nach einer Rantgen-Strukturanalyse erwar- tungsgemBR ein gewinkeltes MnSiMn-Skelett rnit zwei manganstandigen Hydridoliganden aufwei~tl~’~. Versuche zur thermolytischen H2-Abspaltung rnit dem Ziel der Syn- these des leider immer noch unbekannten Siliciumkomple- xes
sind bisher fehlgeschlagen; dies diirfte auf die vergleichs- weise hohe Stabilitgt der SiH-Bindungen im Komplex 21 zuriickzufiihren sein. Solange keine Gegenbeweise vorlie- gen, ist als Erklarung fur die metallzentrierte Bildung sub- stituentenfreier Germaniumliganden die schematische Schrittfolge (a) als Arbeitshypothese plausibel.
Rontgen-Strukturanalysen haben gezeigt, da13 die Mn,Ge-Verbindungen 18a und 18b (Typ C) im Gegensatz
zu den einfacher gebauten Mn,Ge-Derivaten 16a und 16b (Typ A) sterisch iiberladen ~ i n d ~ ~ ~ - ” ~ . So war zu erwarten, daB ein sperrigerer Ligand in der Vorstufe 19 zur selekti- ven Bildung von Komplexen rnit lineurem MnGeMn-Ge- riist fuhren wurde. Einen Beweis hierfur liefert die Syn- these der Verbindung 16c durch Urnsetzung von [($- CsMe,)Mn(CO)2(thf)] 20 rnit German in Gegenwart von Schwefelsaure[2‘.’’’. Wahrend die Anlagerung eines weite- ren 16e-Fragments (z. B. [(q’-C5HS)Mn(CO),]) an eine der beiden MnGe-Mehrfachbindungen der Cyclopentadienyl- Verbindung 16a moglich ist und glatt zum Mn3Ge-System 18a fiihrt1321, bleibt diese Additionsreaktion im Falle des Pentamethylcyclopentadienyl-Derivats 16c aus.
f l M n = G e = M n . .
20
c rCH3
16 C
Schon wegen ihrer Grolje drangen Germaniumatome nach einer Aufweitung ihrer Koordinationssphare und be- vorzugen unter Preisgabe von Mehrfachbindungen einen Einbau in Clusterstrukturen. So stellte unlangst die neu- seelandische Arbeitsgruppe um Muckuy und Nicholson die strukturchemisch faszinierenden CoCe-Cluster 23 (Abb. 5)
Abb. 5. E n Germaniumatom als Spirozentrum. blrul lur \on 23 im Kristall. 23 ist rnit [ ( ~ & I ) ( F ~ ~ ( C O ) , ~ ~ ] 5 (Abb. 2) isovalenzelektronisch und hat zw6lf terminale und zwei kantenverbriickende Carbonylliganden. Die beiden ge- schlossenen Co,Ge-Teilstrukturen sind zueinander nahezu orthogonal (Die- derwinkel 96”). Die Vdlenzwinkel um das Spirozentrum betragen 66 und 135 ’. Das C2-symmetrische Molekiil hat die gleiche Geriistgeometrie wie das Komplexion [ ( ~ A S ) ( ( ~ ~ ’ - C ~ H ~ ) ~ C O ~ ( C O ) ~ ] ~ ~ + 53 (Abb. 24).
und 24 (Abb. 6) her - zwei treffliche Beispiele fur tetra- edrisch bzw. tetragonal-pyramidal strukturierte M,Ge- bzw. M&e-Systeme (Konstitutionstypen D und E,
Schema 1). lhre Synthese gelingt durch Einwirkung von German bzw. Digerman auf Octacarbonyldicobalt[2’. 221.
Bereits im Jahre 1977 hatten Schrnid et al. uber den auch aus 23 durch thermolytische CO- Abspaltung zuganglichen Co4Ge-Komplex 6b berichtet1’8”1. Interessanterweise las- sen sich auch Alkylgermane wie CH,GeH, mit Carbonyl- metallkomplexen zu substituentenfreien Germaniumligan-
Angew. Chem. 98 (1986) 57-77

den abbauen. In keinem der bisher untersuchten Falle blie- ben die GeGe-Bindungen intakt, wenn man Polygerman- Gemische mit Organometallkomplexen umsetzte12',221. In den Hohlraum eines oktaedrischen M,-Ensembles lassen sich Germaniumatome aufgrund ihrer GroDe irn Gegen- satz zu Kohlenstoffatomen (Abschnitt 2.1) nicht einbauen.
Abb. 6. Struhlur des Co,Ge2-Clusten 24 im Kristall. Das oktaedrische closo- CoJGe2-Geriist tragt elf CO-Liganden an den vier Cobaltatomen. Stimtliche CoCe-Abstande liegen im Bereich zwischen 239 und 243 pm. 24 entsteht bei thermischer Belastung der isolierbaren Vorltiuferspezies der Zusammenset- zung [Ge:Co,(CO):o] (Struktur unbekannt, vermutlich ein Ge-Spirocyclus).
2.2.2. Strukturen
Es uberrascht nicht, daD von den Germaniumliganden in den polynuclearen Co,Ge,-Komplexen 23 und 24 keine Mehrfachbindungen ausgehen, da derartige Cluster durch Aggregation kleinerer, hochreaktiver Molekule, z. B. sol- cher mit Mehrfachbindungen, entstehen. Erst wenn deren kinetische Stabilisierung moglich ist, konnen Mehrfach- bindungssysteme, wie sie unzweifelhaft in den Mangan- komplexen 16a-c und 18a,b vorliegen, isoliert werden. Al- lein die extrem kurzen MnCe-Abstande in 16c (Abb. 7)
Abb. 7. Struktur der Mn:Ge-Verbindung 16c (Typ A. Schema I ) im Kristall. Die Struktur zeichnet sich durch ein praktisch lineares Mn-Ge=Mn-System (Mn-Ge 218(2) pm, Mn-Ge-Mn 179(1)") und anticlinale Konformation aus (vgl. Abb. 10).
sind iiberzeugend genug fur cumulierte Mehrfachbindun- gen des Strukturtyps A. Die Mn=Ge-Bindungen sind ca. 218 pm (Tabelle 1 ) lang, womit sie dem Wert der uber- bruckten MnMn-Dreifachbindung (217.0( 1) pm) im ver- wandten Komplex ((qS-CSMe5)2Mn2(p-CO)3] sehr nahe-
k ~ m m e n ~ ~ ~ . " ] ; dies ist verstandlich, da die Kovalenzra- dien von Germanium und Mangan, zurnindest in [(q5-C5Rs)Mn(C0)2]-Derivaten, nahezu gleich ~ ind"~ ' .
Abb. 8. Struktur der Mn,Ge-Verbindung 18b in1 hi ihldl l (trigonat-planar konfiguriertes Gemst, Typ C, Schema 1). Die exocyclische Mn*-Ge-Bindung (225.0( 1) pm) ist formal eine Doppelbindung, wahrend die endocyclischen Mn-Ge-Bindungen (236.0(1) und 236.4(1) pm) als Einfachbindungen aufzu- fassen sind. Die CHXiruppen sind weggelassen.
Authentische Einfachbindungen zwischen Germanium und Mangan sind mindestens 25 pm langer, wobei als Re- ferenzverbindungen die Komplexe [(CO)5Mn(GeH3)] (249 pm, Elektronenbeugung in der Gasphase), [(CO),Mn(GePh,)] (260 pm) sowie [(p-GeMe2)(Mn2(C0),)] (248-252 pm) herangezogen werden konnen'36-3M1. Im Zwi- schenbereich um 225 pm sind die formalen Doppelbindun- gen der trigonal-planaren Mn3Ge-Kornplexe 18 (Struktur von 18b siehe Abb. 8) angesiedelt (Typ C), deren Reaktivi- tat in Abschnitt 2.2.4 diskutiert wird.
2.2.3. Bindungsverhiltnisse
Aus Elektronenbilanzgriinden (Edelgasregel) klassifi- zierte man den linearen Mn2Ge-Komplex 16b in der Origi- nalpublikation als ,,metallorganisches Allen" mit sp-hybri- disiertem Germanium als Z e n t r a l a t ~ m [ ~ ~ ' , Diese Betrach- tungsweise ist mittlerweile durch Fenske-Hall-Rechnungen und aufgrund schwingungsspektroskopischer und struktur- chemischer lndizien dahingehend revidiert worden, daD jetzt Dreifachbindungsanteile diskutiert ~ e r d e n ' ~ ~ ] . Eine Allenstruktur wiirde fur das MnGeMn-Geriist eine Rota- tionsbarriere von rund 70 kcal/mol bedeuten, wohingegen bereits aus den IR-Spektren das Vorliegen eines Rotame- rengemisches hervorgeht. Zur detaillierten MO-Betrach- tung dieses Problems sei auf die Originalliteratur verwie- sen1391, doch sei hier unter Verwendung der Orbitalskizzen von Abbildung 9 zusammenfassend festgehalten, daB die metallzentrierten Orbitale 2a" und 3a' energetisch sehr Bhnlich sind und daB das n-Elektronensystem nahezu Zy- lindersymmetrie hat. Analoge Bindungsverhaltnisse liegen in den isovalenzelektronischen Schwefel- und Selenkom- plexen der allgemeinen Formel [(~-E)((~5-C5R5)Cr(C0)2)21 (E=S, Se; R = H , CH3) vor (vgl. Abschnitt 4). Vom Mn2Ge-Komplex 16c treten im Kristall beide anticlinale Konformere 16A und 16B auf, wahrend die ebenfalls denkbare antiperiplanare Konformation 16C nicht beob-
Angew. Chum. 98 (1986) 57-77 61

Tabelle 1. Strukturtyp und Bindungslangen in ausgewshlten Komplexen mit Bindungen zwiscben ubergangsmetallen und ,,nackten" Hauptgruppenelementen [a].
G I H J
K
J
R
R R
P
248
226.8(4)* (P-W)
270(2)
242.q8)
245.Y( I )
166 158
186 I92
210 ( P - M n ) 218 (P-Fe)
279.9( 1) 281.4( 1)
234'5) (As-Cr) 235.3 234.7. (As-Mn)
207. I ( I ) 207.6( I ) 2 I7.2( I) 203.4(2) 212.8(1) 218.1(1)
220.6( I ) 221. I ( I ) 229.8(2) 232.3( I )
Te-Mn I(~Te)l(tlS-C,Me,)Mn(CO)2tzl~ Q 245.9(2) Te-Mn [(lr,-Te)l(rlS-CsHs)Mn(CO)~t,l 65 T 246-251 Tee-V [(lr-~ee)l(d~~e)v(Co)31~1 59c Q 251.4(3)
252.2(3)
[a] Alle Komplexverbindungen sind im Text mit Literaturverweisen erwshnt. Die Bezeichnung der Strukturtypen bezieht sich auf die Schemata I , 3 und 4. Die (formalen) Bindungsordnungen sind nach der Edelgasregcl klassifiziert. - Abkilrzungen: dppe = Bis(diphenylphosphino)ethan, np, =Tris(diphenylphosphino- ethyl)amin, p, = I , I , I -Tris(diphenylphosphinomethyl)ethan, HB(pz), = Hydridotris(3,5-dimethylpyrazolyl)borat.
62 Angew. Chem. 98 (1986) 57-77

oc co
anti-Konformation
gauche-Konformation
Abb. Y. .)cheiii.itischc U;lr,it.llung der wichtigsten n-Orbital-Wechselwirkun- gen in linearen Komplexen vom Typ A (Schema 1) der allgemeinen Formel [(r-E)((rl'-C,H,)M(CO)LJL] (hier: E=Ge. M=Mn) in der anti- (oben) und gouehe-Konformation (unten). Dargestellt sind die p,-Orbitale des monoato- maren Brilckenliganden. Dieselbe Betrachtungsweise ist auf isovalenzelek- tronische Komplexe des Typs R (Schema 4) anwendbar, z. 9. auf 57a, b (Ah- schnitt 4.1).
achtet wird (siehe Abb. 10). 16b liegt im Kristall nur als antiperiplanares Konformer vor.
16A l6B 16C
Ahh. 10. AntIchl1.de ( 16A und 16H j w a l e .miperiplanare Konfonnationen (16C) der M=E=M-Komplexe vom Typ A (Schema 1) in Newman-Projek- tion.
2.2.4. Reaktivitit
Mehrfachbindungssysteme konnen sich erst dann durch Reaktivitat ausweisen, wenn sie exponiert und dem Angriff von Reaktionspartnern zugiinglich sind. Gerade in Orga- nometallkomplexen rnit ihren zahlreichen, oft sehr spem- gen Hilfsliganden ist dies nicht immer der Fall, doch hat man rnit den hier in Rede stehenden Mn,Ge-Verbindun- gen Gliick: So iibertrilgt Diazomethan bereits zwischen - 20 und 0°C unter N,-Entwicklung seine Methylen- gruppe auf die sehr reaktive MnGe-Doppelbindung von Ma, wobei in 85-95% Ausbeute das Metallaspiran 25 ent- steht ; die MnGe-Einfachbindungen bleiben erwartungsge- ma0 unverilndert[*''I. Wie aus 'H-NMR-Spektren bei ver- schiedenen Temperaturen (CD2Cl,) hervorgeht, hat 25 bei Raumtemperatur eine ,,fluktuierende" Struktur: Unterhalb - 50°C erscheinen drei gut aufgelbte CSHS-Signale, von denen zwei bei Temperaturerhohung breiter werden ; bei ca. 20°C enthalt das Spektrum nur noch zwei Linien fur C5Hs-Gruppen und ein sehr breites CH,-Signal. Dieser Be- fund kann sowohl rnit einer entgegengesetzten Rotation der beiden dreigliedrigen Ringe am Spirozentrum Ge als auch rnit einer cis/rruns-Isomerisierung innerhalb des Zweikernfragmentes [(q5-CsHs)2Mn2(CO)4] gedeutet wer- den.
Chemisch ist interessant, da0 25 oberhalb ca. 50°C in Losung langsam und beim Schmelzen ( I 12°C) sofort unter quantitativer Freisetzung von Ethylen zerfilllt, wobei die
Vorstufe 18a zuriickgebildet wird. In Gegenwart von H2 er- gibt die Thermolyse zusatzlich Methan als Spaltprodukt. Die Methylen-Eliminierung klassifiziert 25 in Einklang rnit den NMR-Daten und im Gegensatz zur vergleichbaren Tellur- verbindung [(p-TeCH2){(qs-CsMes)Mn(CO)2}z] (siehe Ab-
schnitt 4.3.1) als thermolabiles Metallaspiran, in dem erst- mals die Methylenverbriickung eines substituentenfreien Hauptgruppenelements und eines Ubergangsmetalls realisiert ist. Das mit der Methylengruppe isolobale Fe(CO),-Fragmenti3I laDt sich an die Doppelbindungen von 16b addieren, wobei es aber zu konsekutiven Metathe- sereaktionen kommt, an deren Ende die spirocyclischen Komplexe 26 (8%) und 27 (21%) stehen; nach Rontgen- Strukturanalysen gehen in 26 und 27 vom Germaniuma- tom keine Mehrfachbindungen mehr aus; es ist jetzt Spiro- zentrurn (Abb. 1 I ) , das nur iiber Einfachbindungen rnit Ei- sen- und Manganatomen verknupft ist (siehe Tabelle 1). Bereits hingewiesen wurde auf die Addition des rnit CH2- und Fe(CO),-Fragmenten isolobalen (qs-CsH5)Mn(C0)2- Systems an den linearen Mn2Ge-Komplex 16a, wobei das trigonal-planare Mn3Ge-Derivat 18a ent~teh t '~~ ' .
26 6 27
Abb. I I . Struktur der Additions/Metathese-Produkte der Umsetzungen von Fe2(COb rnit dem Mn2Ge-Komplex 16b: Links: [(g.,-Ge)lFe2(CO)u)21 26: rechts: [(g.,-Ge)~[FeZ(CO)u][Fe(CO)~~[(~5-C,H,Me)Mn(CO)~]J~ 27. Abstande siehe Tabelle 1 .
2.3. Substituentenfreies Zinn als Briickenligand
Substituentenfreies Zinn liegt nicht nur in den prototy- pischen M4Sn-Spirocyclen mit verzerrt tetraedrischer M4- Geriistgeometrie und Zinn-Metall-Einfachbindungen vor (siehe Abschnitt 2.1). Neuerdings gibt es auch trigonal-pla- nar konfigurierte Systeme (Konstitutionstyp C, Schema 1). So wurden die beiden isovalenzelektronischen M3Sn- Komplexe 28 und 29 auf vollig verschiedenen Wegen er-
Angew. Chem. 9X (1986) 57-77 63
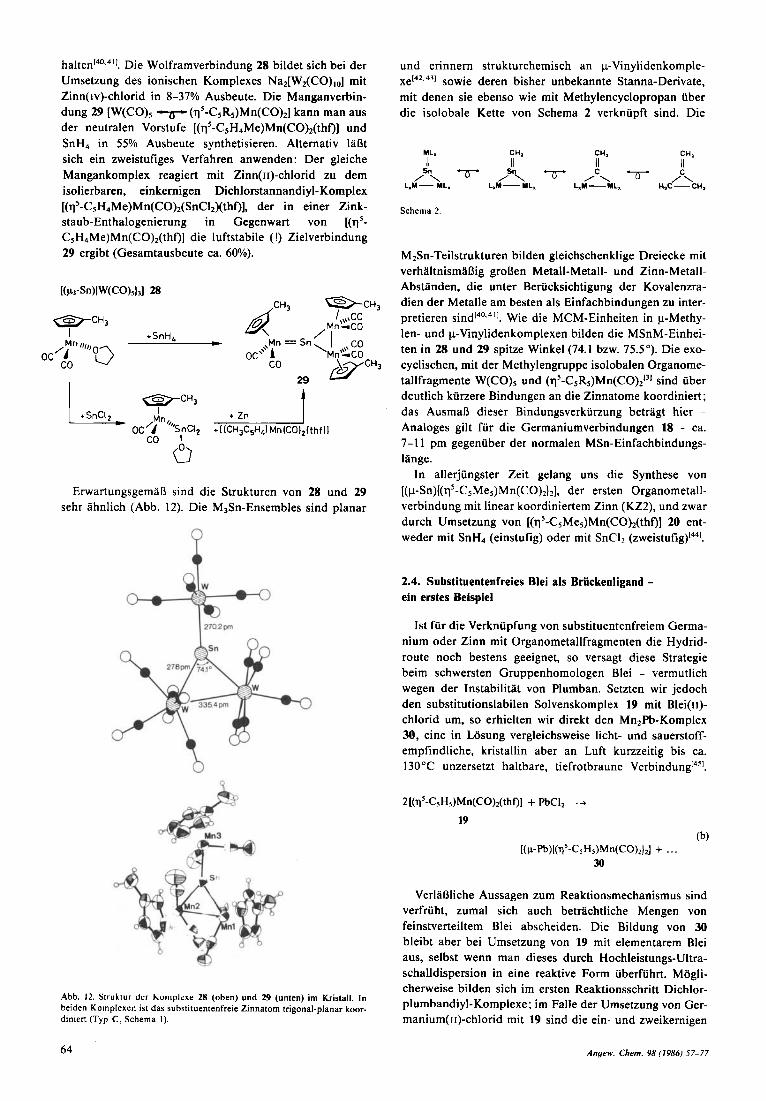
halten140.4'1. Die Wolframverbindung 28 bildet sich bei der Umsetzung des ionischen Komplexes Na2[W,(CO),,] mit Zinn(iv)-chlorid in 8-37% Ausbeute. Die Manganverbin- dung 29 [W(CO), '6 (qS-C5RS)Mn(CO),] kann man aus der neutralen Vorstufe [(q5-CsH4Me)Mn(CO),(thf)l und SnH, in 55% Ausbeute synthetisieren. Alternativ 1aDt sich ein zweistufiges Verfahren anwenden: Der gleiche Mangankomplex reagiert rnit Zinn(1r)-chlorid zu dem isolierbaren, einkernigen Dichlorstannandiyl-Komplex [(qs-CsH4Me)Mn(CO)2(SnC12)(thf, der in einer Zink- staub-Enthalogenierung in Gegenwart von [($- CSH4Me)Mn(CO)Z(thf)] die luftstabile (!) Zielverbindung 29 ergibt (Gesamtausbeute ca. 60%).
u
ErwartungsgemaO sind die Strukturen von 28 und 29 sehr ahnlich (Abb. 12). Die M3Sn-Ensembles sind planar
Q b
Abb. 12. Struktui dci homplrw 28 (oben) und 29 (unten) im Kristall. In beiden Komplexen ist dab substituentenfreie Zinnatom trigonal-planar koor- diniert (Typ C, Schema 1).
und erinnern strukturchemisch an p-Vinylidenkomple- xe142.431 sowie deren bisher unbekannte Stanna-Derivate, rnit denen sie ebenso wie rnit Methylencyclopropan uber die isolobale Kette von Schema 2 verknupft sind. Die
Schema 2
M2Sn-Teilstrukturen bilden gleichschenklige Dreiecke rnit verhaltnismtil3ig groDen Metall-Metall- und Zinn-Metall- Abstanden, die unter Beriicksichtigung der Kovalenzra- dien der Metalle am besten als Einfachbindungen zu inter- pretieren ~ i n d [ ~ ' ? ~ ' ~ . Wie die MCM-Einheiten in p-Methy- len- und p-Vinylidenkomplexen bilden die MSnM-Einhei- ten in 28 und 29 spitze Winkel (74.1 bzw. 75.5"). Die exo- cyclischen, mit der Methylengruppe isolobalen Organome- tallfragmente W(CO), und (q5-CsRs)Mn(C0)2131 sind uber deutlich kurzere Bindungen an die Zinnatome koordiniert ; das AusmaD dieser Bindungsverkurzung betragt hier -
Analoges gilt fur die Germaniumverbindungen 18 - ca. 7-1 1 pm gegenuber der normalen MSn-Einfachbindungs- Iange.
In allerjungster Zeit gelang uns die Synthese von [(p-Sn){(q'-CSMes)Mn(CO)d,I, der ersten Organometall- verbindung rnit linear koordiniertem Zinn (KZ2), und zwar durch Umsetzung von [(qS-CsMe5)Mn(CO),(thf)] 20 ent- weder mit SnH, (einstufig) oder rnit SnCI, (zweistufig)[441.
2.4. Substituentenfreies Blei als Briickenligand - ein erstes Beispiel
1st fur die Verknupfung von substituentenfreiem Germa- nium oder Zinn rnit Organornetallfragmenten die Hydrid- route noch bestens geeignet, so versagt diese Strategie beim schwersten Gruppenhomologen Blei - vermutlich wegen der Instabilitat von Plumban. Setzten wir jedoch den substitutionslabilen Solvenskomplex 19 rnit Blei(i1)- chlorid um, so erhielten wir direkt den Mn2Pb-Komplex 30, eine in Losung vergleichsweise licht- und sauerstoff- empfindliche, kristallin aber an Luft kurzzeitig bis ca. 130°C unzersetzt haltbare, tiefrotbraune Verbind~ng[~''.
VerlaDliche Aussagen zum Reaktionsmechanismus sind verfriiht, zumal sich auch betrlchtliche Mengen von feinstverteiltem Blei abscheiden. Die Bildung von 30 bleibt aber bei Umsetzung von 19 mit elementarem Blei aus, selbst wenn man dieses durch Hochleistungs-Ultra- schalldispersion in eine reaktive Form uberfuhrt. Mogli- cherweise bilden sich im ersten Reaktionsschritt Dichlor- plumbandiyl-Komplexe; im Falle der Umsetzung von Ger- manium(1r)-chlorid rnit 19 sind die ein- und zweikernigen
64 Angew. Chem. 98 (1986) 57-77

Dichlorgermandiyl-Komplexe [(q'-CsH5)Mn(C0)2(GeC12)] (als Solvensaddukt) bzw. [(p-GeCIz)((qS-CsHS)Mn(C0)2)2] (solvensfrei) in Substanz faBba~[~ ' .~ '~.
In der Komplexverbindung 30 liegt erstmals ein substi- tuentenfreies Bleiatom vor, das uber Mehrfachbindungen an Ubergangsmetalle fixiert ist. Das MnPbMn-Geriist ist praktisch linear (177.2( I)'), so daR keine Kontakte zwi- schen den Manganatomen bestehen und das Briickenele- ment Blei die Koordinationszahl 2 hat (Abb. 13).
Abb. 13. Struktur \on 3U im Criaiall. Ausgewihlte Abstande [pm] und Win- kel ["]: Mn-Pb 245.9(1), Mn-CI l85(2), Mn-C2 181(1), C1-01 111(2), C2-02 113(2); Mn-Pb-Mn' 177.2(1), Mn-CI-01 l74.1(1), Mn-C2-02 174(1); Tor- sionswinkel C2-Mn-Mn8-CI - 1 15.2(5).
Nach den fur die analogen Germaniumkomplexe 16a und 16c (Mn-Ge 2 18-220 pm) entwickelten Bindungsvor- stellungen sollten vom bruckenstandigen Bleiatom glei- chermaBen cumulierte Doppelbindungen rnit Dreifachbin- dungsanteilen ausgehen. Die MnPb-Abstande (245.9( 1) pm) rechtfertigen diese Anschauung. Die Llnge von MnPb-Einfachbindungen kann durch Extrapolation be- kannter Strukturdaten einigermaBen zuverlassig zu etwa 265-270 pm abgeschatzt werdenr4']; MnPb-Doppelbindun- gen sollten um mindestens 20 pm kiirzer und damit urn etwa 20 pm langer als MnGe-Doppelbindungen sein. Da- mit setzt sich in der Bleichemie jener Trend fort, den Hurt- ner et al. und wir fur Germanium- und Zinnkomplexe be- obachtet hatten (siehe Tabelle I ) .
2.5. Existieren Carbinkomplexe M* d - M und ihre ,,schweren Briider" M*=E-M?
Stehen schon bei der gestreckten Konstitution A (Schema 1) Dreifachbindungsanteile in Rede, so erhebt sich zurecht die Frage nach der Verifizierbarkeit einer un- symmetrischen Elementverbriickung von (unterschiedli- chen) Ubergangsmetallen im Sinne des ebenfalls linearen Konstitutionstyps B. Bisher Papier geblieben, weckt diese Verbindungsklasse nach den Erfahrungen rnit Fischers C a r b i n k ~ m p l e x e n ~ ~ " ~ berechtigte Hoffnungen: 1st M* ein 15e- und M ein 17e-Organometallfragment, so la& sich die Isolobal-Beziehung
HCEC-H - M*=C-H M*=C-M M*=E-M (4 B
herstellen. Ob die ,,schweren Bruder" des Kohlenstoffs - Silicium, Germanium. Zinn und Blei - carbinanaloge Komplexe des Strukturtyps B rnit colinearen Einfach- und Dreifachbindungen dulden, ist ungewiB, doch sehen wir wenigstens fur die Kohlenstoffkornplexe M*=C-M eine reelle Chance: Sollte es beispielsweise nicht m6glich sein,
Fischer-Carbine L,M*=C-X mit rnetallhaltigen le-Ligan- den zu verknupfen, sei es durch ,,Salzmetathese" (z. B. rnit Na[Mn(CO),]; X= Halogen, SiR3 etc.) oder durch oxida- tive Addition (X = H; z. B. an [(qS-C5RS)Ir(CO)]-Fragmen- te; CH-Aktivierung)? Hier sind wieder einmal Phantasie und Geschick der Metallorganiker aufgerufen, um diese Liicke auf der schon recht bunten Palette wichtiger Konsti- tutionstypen zu schlieBen!
3. Substituentenfreie Elemente der 5. Hauptgruppe als Briickenliganden
3.1. Nitridoliganden
Die komplette Stickstoffgruppe prlsentiert sich substitu- entenfrei und metallgebunden in zahlreichen Strukturva- rietlten, deren prominenteste in Schema 3 zusammengetra-
MEE-M M A E L M M E E
terminal linear symmetrlsch linear unsymmetrisch
G H 1
m I
pyramidal Irigonal-planar trigonal-olanar
J K L M m
Schema 3. Koordin~lionrmoglichkeilen tur wh\liluenrenlrcir Llcmcnie der 5. Hauptgruppe.
gen sind. Wlhrend der nackte Kohlenstoffligand vorzugs- weise eingekapselt in polynuclearen Metallpolyedern an- zutreffen ist, findet man bei seinem Nachbarn Stickstoff nicht selten ein-, zwei- und dreikernige Komplexe rnit Mehrfachbindungen zwischen dem Nitridoliganden und ~bergangsmetallen[sO~sll, Wie Srruhle und Dehnicke in ei- nem lesenswerten Aufsatz gezeigt habenIs2l, verfiigen ins- besondere anorganische Azide (z. B. Chlor- und Iodazid), Stickstofftrichlorid und gelegentlich auch Hydrazin sowie Ammoniak und Kaliumamid iiber ein hervorragendes Syn- thesepotential zur Einfuhrung des substituentenfreien Stickstoffliganden in ~bergangsmetallkomplexe.
Als Schulbeispiele fur ein- und zweikernige Nitridokom- plexe mit terminalem Nitridoliganden (C) bzw. rnit sym-
a) TN
"e- CI
31
Br I I
32a 32b
Abb. 14. Nackte N ,-Llgdnden in con- und rweikernlgen Komplexverbindun- gen (oben) mit vereinfachten MO-Bindungsschemata (unten). a) Struktur von [OsNC15]'- 31 (0s-N 161.4(13), 0s-CI,. 260.5, Os-CIi, 236.4(4) pm (Mittel- wen)): b) Ausschnitt aus der Kolumnarstmktur yon (ReNCI,), 32. (Re-N 158(4) und 248(4) pm, Re-CI 227 pm (Mittelwert)); c) Struktur von ITa2NBrlo]'- 3Zb (Ta-N 185.9, Ta-Br,. 273.8(6), Ta-Br., 251.4(3) pm (Mittel- wene)).
Anyew. Chem. 98 (1986) 57-77 65

metrisch (H) oder unsymmetrisch (I) angeordneten Stick- stoffbriicken mogen die in Abbildung 14 gezeigten Verbin- dungen genugen: Eine typische, extrem kurze Metall- Stickstoff-Dreifachbindung (16 1.4 pm) findet sich im pseu- dooktaedrischen Komplexanion [ O S N C I ~ ] ~ - 3lrS3’, das zu- gleich ein treffliches Beispiel fur den extremen trans-Ef- fekt eines endstandigen Nitridoliganden bietet (Abb. 14a); die Bindung zwischen dem Metallatom und dem zur MEN-Bindung trans-standigen Liganden ist bis zu 30 pm liinger als die zwischen dem Metallatom und dem gleichen cis-standigen Liganden! Haufig wirkt sich der trans-Effekt so stark aus, da13 die gegeniiberliegende Position unbesetzt bleibt. Ein Musterbeispiel hierfur ist das in Liisungen von [MoNCI,]’- in Methylenchlorid nachweisbare Dissoziati- onsgleichgewicht (d)l5,].
[MoNClSl2- ---1 [MoNCIJ + C1- (4
Andere strukturchemisch wohldokumentierte Beispiele fur terminale Metall-Stickstoff-Dreifachbindungen sind die tetragonal-pyramidalen Komplexanionen [MNCI,]- (M = Mo, W, Re, Ru, 0s); hier liegen die Metall-Stickstoff- Abstande zwischen 166 pm (Mo) und 157 pm (Ru)[”]. Dreifachgebundene Nitridoliganden sind neben den iso- elektronischen Carbingruppen (CR) die starksten n-Dona- toren in der Komplexchemie. Wie die schematische Dar- stellung in Abbildung 14a unten zeigt, la13t sich die M=N- Bindung als Uberlagerung einer o-Bindung und zweier entarteter x-Bindungen verstehen, wobei das freie Elektro- nenpaar dem Nitridoliganden nur schwach basische Ei- genschaften verleiht.
Unter bestimmten Voraussetzungen koordiniert das freie Elektronenpaar des Nitridoliganden ein weiteres Metall- komplexfragment; dabei entsteht eine lineare N-ver- briickte Anordnung, wie sie etwa in der Kolumnarstruktur von Tetrachloro(nitrido)rhenium 32a vorliegt (Abb. 14b)[’’]. Es resultiert dann eine stark asymmetrische Briik- kenkonstitution mit sehr unterschiedlichen Bindungslan- gen (158 und 248 pm); die kurze Bindung ist wie eine ter- minale MEN-Bindung als Dreifachbindung zu interpretie- ren, wahrend die lange einer a-Bindung entspricht, bei der das besetzte sp-Hybridorbital des Stickstoffatoms mit ei- nem unbesetzten d2sp3-Hybridorbital (Abb. 14b unten)
33 Ahh. 15. Ilindungrvt.rhdlitiisau dcr Ir,l\i-htr~nplexes 33, des bisher einzigen Beispiels fur trigonal-planar konfigurierte N,-Briicken ( J in Schema 3). - Ein T-formig koordiniertes Stickstoffatom fand man im Komplex [Mo3N(0)(CO),(~’-CsHs),l; siehe N. D. Feasey. S. A. R. Knox, A. C. Orpen, J. Chem. SOC. Chem. Commun. 1982. IS.
uberlappt. Bestehen die Voraussetzungen fur den Aufbau zweier entarteter d,-p,-d,-Dreizentren-z-Molekiilorbitale, die jeweils mit einem Elektronenpaar besetzt sind (Abb. 14c unten), so kommt es wie etwa im Fall des Komplex- anions [TazNBrlol3- 32b zur Bildung einer symmetrischen
Nitridobriicke (Ta-N 186 pm[’61), wobei das Stickstoffatom iiber colineare Doppelbindungen an die beiden Metall- atome fixiert ist.
Bei den hoheren Homologen hlufiger anzutreffen, kennt die trigonal-planare Konstitution J in der Komplexchemie des nackten Stickstoffs nur das Einzelbeispiel des von Grq- 5 t h et al. untersuchten Iridiumkomplexes 33Is7]; die Ir3N- Baugruppe hat drei gleich lange IrN-Bindungen (192 pm). Die Bindungsverhaltnisse zeigt schematisch Abbildung 15 : Neben drei a-Bindungen gibt es drei p,-d,-Uberlappungen zwischen dem besetzten N(p,)-Orbital und je einem dxL- Orbital der drei Ubergangsmetallatome.
3.2. Cluster mit interstitiellen Atomen der Stickstoffgruppe
Um hier die Beziehung zur strukturchemisch faszinie- renden Klasse der Carbidocluster (Abschnitt 2.1) henu- stellen, sei a n Cluster mit interstitiellen Stickstoffatomen erinnert, deren erstes Beispiel der Mailander Gruppe um Martinengo bei der Umsetzung von K2[Co6(CO),s] mit Ni- trosyltetrafluoroborat in die Hande gefallen war[? Nach Abbildung 17a laBt sich das Geriist von
(auch ein Komplex mit Rh statt Co ist bekannt) als trigo- nales Prisma mit angenaherter D,,-Symmetrie beschrei- ben, in dessen Hohlraum sich der nackte StickskoMigand befindet; die strukturelle Analogie zu den isoelektroni- schen Carbidoclustern [M6C(C0),,l2- (M =Co, Rh; siehe 3 in Abb. 1) ist offenkundig. Die sechs Cobalt-Stickstoff- Abstande sind nahezu gleich (um 194 pm), womit dem in- terstitiellen Stickstoffatom ein Kovalenzradius von 67 pm zuzuordnen ist. Eine ahnliche Synthese - Desoxygenierung einer Nitrosylgruppe - fuhrte GludfeZter et al. ausgehend von [N(PPh3)2][Fe(CO)3NO] und Fe3(C0)1z zum Fe,N-Clu- ster
dessen vierkerniges, naherungsweise C2,-symmetrisches Anion (Abb. 16) eine Fe4-Schmetterlingsstruktur aufweist.
Abb. 16. Links: Struktur des Anions von [N(PPhl)ll[Fe,N(CO)121 35 im Kri- stall. Rechts: Geriiststruktur mit Bindungslangen. Der N,-Ligand ist expo- niert und daher reaktiv; zum Beispiel ist das Anion mit HJPOJ protonierbar, wobei die Neutralkomplexe [HFe,N(CO),,] und [Fe3(p3-NH)(CO),,] entste- hen.
Wie die Rontgen-Strukturanalyse ergab, ist der nackte Stickstoffligand exponiert und - wie seine leichte Proto- nierbarkeit zeigt - offenbar recht elektronenreich. Wie im
66 Angew. C‘hrm. 911 (19841 57-77

isosteren Fe,C-Cluster 10 liegt eine praktisch lineare (!) Fe3-N-Fe4-Atomsequenz (179.0") mit auffallend kurzen Bindungen vor (ca. 177 pm); die gewinkelte Fel-N-Fe2- Teilstruktur hat deutlich langere Fe-N-Bindungen (ca. 190 pm).
Mit dem Hinweis auf das spektakulare Rh,P-Ion
[Rh,P(CO),,J'- 36
gelingt an dieser Stelle der Briickenschlag zur Organome- tallchemie des substituentenfreien Phosphors. Als Quelle fur das nackte Phosphoratom, das im Innern eines quadra- tischen, auf einer Seite uberdachten Rh8-Antiprismas ein- gekapselt ist (Abb. 17b) konnte uberraschend Triphenyl- phosphan genutzt werden: 36 entsteht bei der Umsetzung
34 36 37
38 39
Abb. 17. Cluster mit iiiterrtitiellen tlernenten drr Stickstoffgruppe. a) Das Ion [Co,N(CO)J 34 hat ein trigonal-prismatisches Geriist; in der analogen Rhodiumverbindung sind fur das zentrale N-Atom IsN-NMR-spektrosko- pisch sechs magnetisch ilquivalente Metallatome nachweisbar. Das N-Atom hat die Koordinationszahl (KZ) 6. - b) Eingekapselt in ein iiberdachtes qua- dratisches Antiprisma findet sich der P,-Ligand im Ion IRhpP(CO),,12- 36 (KZ9 fur Phosphor). - c) Soil ein P-Atom in einen kleineren M.-Kifig, so mu0 dieser mindestens an einer Seite gedffnet sein, wie dies im Ion [Co6pCCO)161- 37 (KZ 6 fur Phosphor) der Fall ist. - d) und e) Zunehmender Platzbedarf bei den schwereren Homologen Amen und Antimon bedingt die noch geriumigeren Hdhlen in den Mlo- und M,,-KLfigstrukturen der Ionen IR~,,AS(CO)~~~'- 38 (KZ 10 fur Arsen) bzw. [Rh12Sb(C0)27]3- 39 (KZ 12 fur Antimon).
von Ph3P mit [Rh(CO)2(acac)] (acac = Acetylacetonat) un- ter einem CO-Druck von 300 bar'601. Lehrreich erweist sich hier die Frage nach dem Raumbedarf eines nackten Atoms der Stickstoffgruppe. Abbildung 17 zeigt die kleinstmogli- chen closo-Cluster mit interstitiellen N-, P-, As- und Sb- Atomen. Genugt dem Stickstoffatom noch ein geschlosse- nes Mb-GefiSt (hier: trigonales Pr ima, Abb. 17a), so erfor- dert Phosphor schon einen M,-Kiifig (Abb. 17b) oder zu- mindest eine offene korbartige M6-Struktur wie wir sie im Komplex
in Abbildung 17c erkennenl6']. Als iiberzeugende Beweis- stiicke fur den stark zunehmenden Platzbedarf der hoheren Homologen mogen die uberdacht quadratisch-antiprisma- tische pi,,-Arsen- 38 und die ikosaedrische p,2-Antimon- verbindung 39 aus der stolzen Phalanx der Rhodiumkom- plexe akzeptiert werden (Abb. 17d bzw. 37e)[62,633.
38 [ Rhl"As(CO)Z$ - [Rh12Sb(C0)2i]3 39
Der Kovalenzradius von Antimon ist immerhin doppelt so groR wie der von Stickstoff (N 70, P 110, As 120, Sb 140, Bi I50 pm). Eingekapseltes Bismut kennt man bislang nicht, doch ist dieses Element sein eigener Baumeister bei Clusterionen wie Bii+ (trigonale Bipyramide, Dlh), Bi;+ (quadratisches Antiprisma, D4) oder Bi:+ (dreifach fla- chenuberbriicktes trigonales Prisma, Cjh = D3,,). Fur die verwandten Elemente Antimon, Arsen, Phosphor, Blei, Zinn und Germanium haben insbesondere die Gruppen um uon Schnering und Corhett eine faszinierende Vielfalt von Homopolyionen in Zintl-Phasen praparativ erschlossen und strukturchemisch abgesichert (2. B. P:r, Ge;-/Ge;-,
ment-Cluster konnen ihrerseits kleinere Atome einkapseln, wofur beispielhaft an das basische Blei(ri)-perchlorat [Pb60(OH),(CI04),- H20] erinnert sei, in dem das 0-Atom in einem Pb,-Tetraeder eingeschlossen i~t[~'I; auch das ba- sische Beryllium(i1)-acetat [Be40(O2CCH&.] verdient hier erwahnt zu werden~"I.
sn",-, pbz- und As3- 11 ) 164-69) . Solche Hauptgruppenele-
3.3. Substituentenfreie ein- und zweiatomige Briickenliganden mit Phosphor, Amen und Antimon
Den trigonal-planaren Koordinationstyp K (Schema 3) mit dreifach metalliertem Phosphor im Zentrum verifizier- ten erstmals Hutfner et al. an den Komplexen 40 und 41[72.731. Die Synthesestrategie beruht auf der Enthalo- genierung von Halogenphosphandiyl-(=PX) oder Phos- phortrihalogenid-Liganden (PX,). Die heteronucleare Mn2Fe2-Verbindung 40 wird durch Umsetzung von [ (I~'-C,H,)M~(CO)~(PB~,)~ mit dem notorischen Halogen- fanger Fe2(C0)9 e r h a l t e r ~ ~ ~ ~ ] . Die beiden Phosphoratome in 40 liegen mit ihren jeweils drei benachbarten Metallato- men in einer Ebene (maximale Abweichung 5 pm), wobei der Interplanarwinkel von der Orthogonalitat nur wenig abweicht (94.1") (Abb. 18 oben). Das leere p-Orbital der nackten Phosphorliganden wird durch Ruckbindung aus Mn-Donororbitalen aufgefullt; die Mangan-Phosphor- Bindung ist mit 210 pm noch kiirzer als die ebenfalls durch Mangan-d,-Phosphor-p,-Wechselwirkung verkurzten Bin- dungen in einfachen Phosphandiyl(Ph0sphiniden)-Kom- plexen des Mangans.
Der Cr2-Komplex [(p-PBr){Cr(CO)S}2] reagiert mit dem Komplexanion [(qs-C,HS)W(CO),]- unter Salzmetathese zum p3-Phosphor-Komplex 41, einem schonen Beispiel fur den Strukturtyp K in Schema 3. Die Details der Struktur von 41 sind gut mit einer Interpretation vertraglich, nach der die exocyclische Phosphor-Chrom-Bindung einen er- heblich hoheren n-Bindungsanteil hat als die endocycli- sche (Abb. 18 unten).
Als vie1 bestauntes Kuriosum der Organometallchemie des Phosphors sei hier auch auf den P2-Komplex
Aiigew. Chrm. 98 (1986) 57-77 67
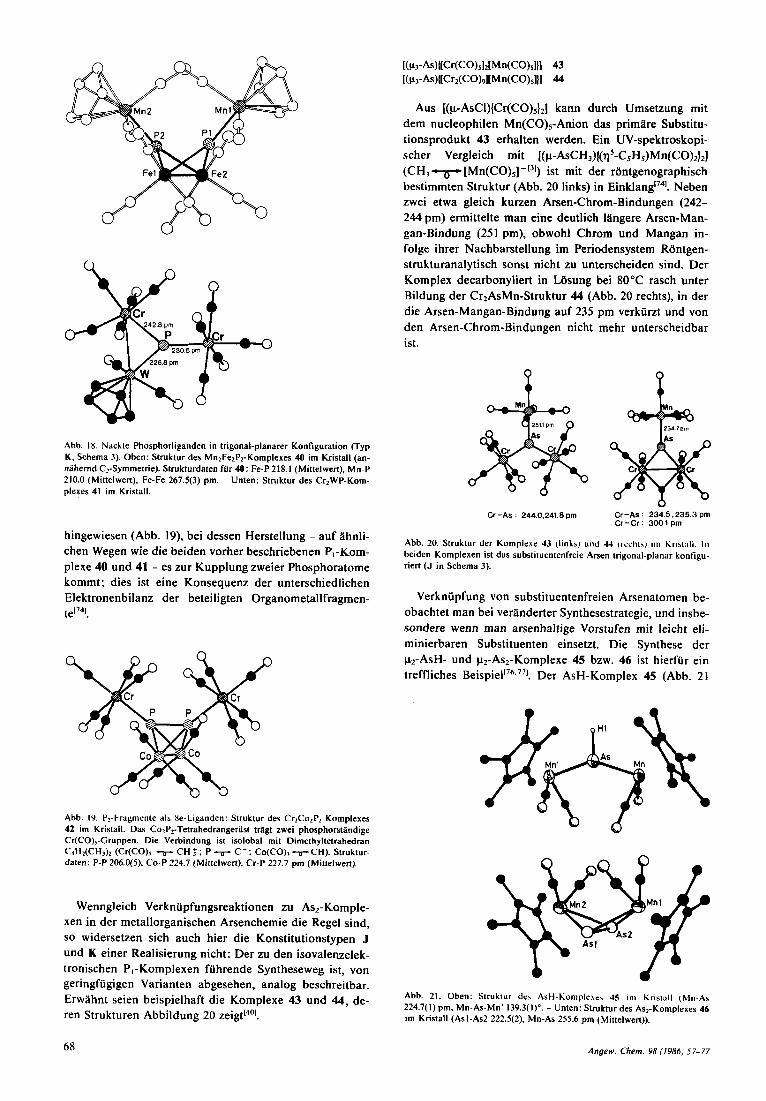
c\ n
Abb. 18. Nackte Phosphorliganden in trigonal-planarer Konfiguration (Typ K. Schema 3). Oben: Struktur des MnlFelPz-Komplexes 40 im Kristall (an- niihernd C2-Symmetrie). Strukturdaten fur 40: Fe-P 218.1 (Mittelwert), Mn-P 210.0 (Mittelwert), Fe-Fe 267.5(3) pm. - Unten: Struktur des CrzWP-Kom- plexes 41 im Kristdll.
hingewiesen (Abb. 19), bei dessen Herstellung - auf ahnli- chen Wegen wie die beiden vorher beschriebenen PI-Kom- plexe 40 und 41 - es zur Kupplung zweier Phosphoratome kommt : dies ist eine Konsequenz der unterschiedlichen Elektronenbilanz der beteiligten Organometallfragmen- te174'.
Abb. IY . P:-tragmente als Xe-Liganden: Struktur des Cr2Co,P2-Komplexes 42 im Kristall. Das CozP,-Tetrahedrangerust tritgt zwei phosphorstandige Cr(CO)5-Gruppen. D ie Verbindung ist isolobal mi1 Dimethyltetrahedran CAH~(CH,)~ (Cr(CO), -p C H I ; P T C- ; CO(CO)ITCH). Struktur- daten: P-P 206.0(5). Co-P 224.7 (Mittelwert), Cr-P 227.7 pm (Mittelwert).
Wenngleich Verkniipfungsreaktionen zu As,-Komple- xen in der metallorganischen Arsenchemie die Regel sind, SO widersetzen sich auch hier die Konstitutionstypen J und K einer Realisierung nicht: Der zu den isovalenzelek- tronischen P,-Komplexen fiihrende Syntheseweg ist, von geringfiigigen Varianten abgesehen, analog beschreitbar. Eraahnt seien beispielhaft die Komplexe 43 und 44, de- ren Strukturen Abbildung 20 zeigt1401.
Aus [(p-AsCI)(Cr(CO),},] kann durch Umsetzung mit dem nucleophilen Mn(C0)5-Anion das primare Substitu- tionsprodukt 43 erhalten werden. Ein UV-spektroskopi- scher Vergleich mit [(~-ASCH,){(~~-C,H,)M~(CO),)~] ( C H l ~ [Mn(CO),]-~'l) ist mit der rhtgenographisch bestimmten Struktur (Abb. 20 links) in Einklang1741. Neben zwei etwa gleich kurzen Arsen-Chrom-Bindungen (242- 244 pm) ermittelte man eine deutlich lgngere Arsen-Man- gan-Bindung (251 pm), obwohl Chrom und Mangan in- folge ihrer Nachbarstellung im Periodensystem Rontgen- strukturanalytisch sonst nicht zu unterscheiden sind. Der Komplex decarbonyliert in Losung bei 80°C rasch unter Bildung der Cr2AsMn-Struktur 44 (Abb. 20 rechts), in der die Arsen-Mangan-Bindung auf 235 pm verkiirzt und von den Arsen-Chrom-Bindungen nicht mehr unterscheidbar ist.
Cr-As: 244.0.241.8pm Cr-As: 2345.235.3pm Cr - Cr : 300.1 pm
Abb. 20. Struktur der Komplexe 43 (Ilnhrj urid U (iccIii3i in hri,tall. In beiden Komplexen ist das substituentenfreie Arsen rrigonal-planar konfigu- riert (J in Schema 3).
Verkniipfung von substituentenfreien Arsenatomen be- obachtet man bei verlnderter Synthesestrategie, und insbe- sondere wenn man arsenhaltige Vorstufen mit leicht eli- minierbaren Substituenten einsetzt. Die Synthese der p2-AsH- und p2-As2-Komplexe 45 bzw. 46 ist hierfiir ein treffliches B e i ~ p i e I ' ~ ~ . ~ ~ ' . Der ASH-Komplex 45 (Abb. 21
Abb. 21. Oben: Struktur deb AaH-honipleie\ 45 1111 Krlsi,tII (hln-As 224.7(1) pm, Mn-As-Mn' 139.3(1)". - Unten: Struktur des As2-Komplexes 46 im Kristall (Asl-As2 222.5(2), Mn-As 255.6 pm (Mittelwert)).
68 Angew. Chem. 98 (1986) 57-77
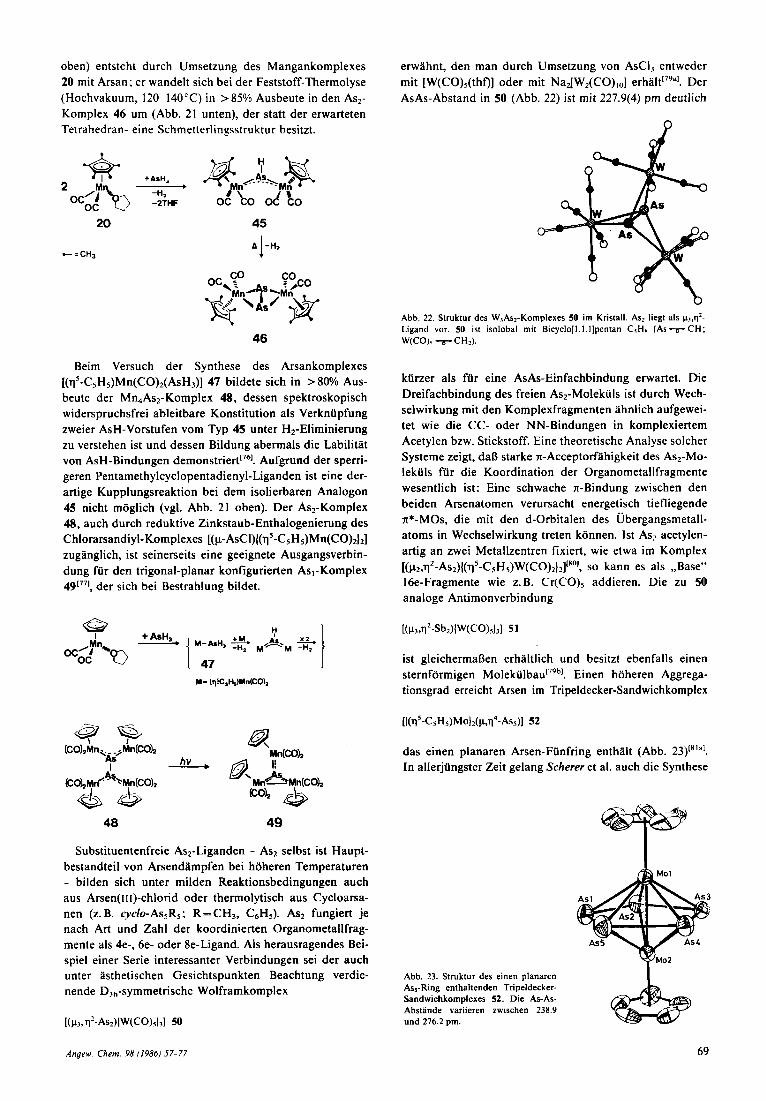
oben) entsteht durch Umsetzung des Mangankomplexes 20 mit Arsan; er wandelt sich bei der Feststoff-Thermolyse (Hochvakuum, 120-140°C) in >85% Ausbeute in den As2- Komplex 46 um (Abb. 21 unten), der statt der erwarteten Tetrahedran- eine Schmetterlingsstruktur besitzt.
20
c : CH,
46
Beim Versuch der Synthese des Arsankomplexes [(q5-C5H,)Mn(CO),(AsH3)] 47 bildete sich in > 80% Aus- beute der Mn,As2-Komplex 48, dessen spektroskopisch widerspruchsfrei ableitbare Konstitution als Verknupfung zweier ASH-Vorstufen vom Typ 45 unter H2-Eliminiemng zu verstehen ist und dessen Bildung abermals die Labilitat von AsH-Bindungen d e m ~ n s t r i e r t ~ ~ ~ ] . Aufgrund der sperri- geren Pentamethylcyclopentadienyl-Liganden ist eine der- artige Kupplungsreaktion bei dem isolierbaren Analogon 45 nicht moglich (vgl. Abb. 21 oben). Der As2-Komplex 48, auch durch reduktive Zinkstaub-Enthalogenierung des Chlorarsandiyl-Komplexes [ (~ -ASCI){ (~ ' -C ,H, )M~(C~)~}~] zuganglich, ist seinerseits eine geeignete Ausgangsverbin- dung fur den trigonal-planar konfigurierten As,-Komplex 49[771, der sich bei Bestrahlung bildet.
48 49
Substituentenfreie As2-Liganden - As2 selbst ist Haupt- bestandteil von Arsendampfen bei hoheren Temperaturen - bilden sich unter milden Reaktionsbedingungen auch aus Arsen(iii)-chlorid oder thermolytisch aus Cycloarsa- nen (z.B. cyc/o-AsSR5; R=CH3, C6H5). As2 fungiert j e nach Art und Zahl der koordinierten Organometallfrag- mente als 4e-, 6e- oder 8e-Ligand. Als herausragendes Bei- spiel einer Serie interessanter Verbindungen sei der auch unter asthetischen Gesichtspunkten Beachtung verdie- nende D,,-symmetrische Wolframkomplex
erwahnt, den man durch Umsetzung von AsCI, entweder rnit [W(CO),(thf)] oder rnit Na,[W,(CO),,] erhalt[7y"1. Der AsAs-Abstand in 50 (Abb. 22) ist rnit 227.9(4) pm deutlich
Abb. 22. Struktur des W,As2-Komplexes 50 im Kristall. As2 liegt als pl,q2- Ligand vor. 50 ist isolobal mit Bicyclo[l.l.l]pentan CcHx (As -a- CH; W(C0)- -C'HZ).
kurzer als fur eine AsAs-Einfachbindung erwartet. Die Dreifachbindung des freien As,-Molekuls ist durch Wech- selwirkung mit den Komplexfragmenten ahnlich aufgewei- tet wie die CC- oder NN-Bindungen in komplexiertem Acetylen bzw. Stickstoff. Eine theoretische Analyse solcher Systeme zeigt, daB starke n-Acceptorfahigkeit des As2-Mo- lekuls fur die Koordination der Organometallfragmente wesentlich ist: Eine schwache n-Bindung zwischen den beiden Arsenatomen verursacht energetisch tiefliegende n*-MOs, die rnit den d-Orbitalen des Ubergangsmetall- atoms in Wechselwirkung treten konnen. 1st Asz acetylen- artig an zwei Metallzentren fixiert, wie etwa im Komplex [(p2,qz-A~2){(qS-C5HS)W(CO)2]2]~Hnl, so kann es als ,,Base*' 16e-Fragmente wie z.B. Cr(CO), addieren. Die zu 50 analoge Antimonverbindung
ist gleichermaBen erhaltlich und besitzt ebenfalls einen sternformigen M o l e k i i l b a ~ [ ~ ~ ~ ~ . Einen hoheren Aggrega- tionsgrad erreicht Arsen im Tripeldecker-Sandwichkomplex
das einen planaren Arsen-Funfring enthalt (Abb. 23)[""]. In allerjungster Zeit gelang Scherer et al. auch die Synthese
Abb. 23. Struktur des einen planaren Ass-Ring enthaltenden Tripeldecker- Sandwichkomplexes 52. Die As-As- Abstande variieren zwischen 238.9 und 276.2 pm.
Anges. Chem. Y8 (19861 57-77 69

eines analogen Komplexes mit cyclo-P, als planarer Ein- heit im Zwischendeckl" '1.
Den Konstitutionstyp M von Schema 3 findet man in metallorganischen Arsonium-lonen (spirocyclische Struk- tur = ,,halboffenes" Tetraeder!), wofur das Co,As-Kom- plexion 53 ein Beispiel gibt (Abb. 24)["'.
Q
Abb. 24 Struktur dcs Co,As-Komplexions 53 im Kristall. Co-As 227 (Mittel- wert), Co-Co 260, C o . . .Co 414 pm.
3.4. Substituentenfreie Bismutliganden
Auch Bismut als grobtes und schwerstes Element der 5. Hauptgruppe findet sich als substituentenfreier Ligand in Ubergangsmetallkomplexen. Wenngleich bisher kein Bei- spiel einer Metall-Bismut-Mehrfachbindung (Strukturty- pen G-K in Schema 3) bekannt ist, so haben M3Bi-Geriist- fragmente ihre Stabilitat mehrfach unter Beweis gestellt (Konstitutionstyp L). Zuganglich entweder aus metalli- schem Bismut, Bismut(ir1)-chlorid oder Natriumbismu- tat(v), enthalten die Cobalt- und Eisenkomplexe 54 bzw. 55 pyramidal konfigurierte Bi-Briicken (Abb. 25)[1Rn.751.
Auch der einzige bisher bekannte Bi2-Komplex 56 weist wie die As- und Sb-Analoga 50 bzw. 51179'.bl keine Metall- Hauptgruppenelement-Mehrfachbindung auf, enthalt da- fur aber erhebliche Mehrfachbindungsanteile zwischen den beiden Bismutatomen (281.8(3) pm)179C1. Bedeutung konnten losliche Bi-Cluster fur die Aufklarung des Mecha- nismus der industriell wichtigen Ammonoxidation von Propen (,,SOHIO-ProzeB") gewinnen, die hochaktive Bis- mut/Wolframoxid-Katalysatoren erfordert.
4. Substituentenfreie Elemente der 6. Hauptgruppe als Bruckenliganden
Von Sauerstoff und seinen Homologen kennt man eine ausgedehnte Komplexchemie. Die Anknupfung an Uber- gangsmetallzentren fuhrt zu den Konstitutionstypen 0 - T (Schema 4), wenn man mehrkernige Clusterverbindungen auber Betracht IiiBt. Substituentenfreier Sauerstoff bevor- zugt als Komplexligand die terminale (0 ) oder die sym- metrisch verbriickende Koordination (P). Verfugt das Me- tallatom iiber leere d-Orbitale, so werden - vor allem bei den hoheren Homologen von Sauerstoff - Komplexe mit Mehrfachbindungen (d,-p,-Wechselwirkungen) gebildet (Q-T). Doch auch bei Sauerstoff kennt man lineare und trigonal-planare Strukturen; sie sind z. B. typisch fur die Ionen [Ru20C110]4--1n31 und [Si2O7I6- (sowie Coesit)lsol bzw. [O(HgCI),]+ und Metalldioxide vom Rutiltyp. Die hochste Koordinationszahl erreicht ein Chalkogenatom im doppelt uberdachten quadratischen Antiprisma (KZ 10) des Clu- sterions [ Rh loS(CO)22]2 -Ix4].
M*=E
lerminal gewnke l l gewmkelt
0 P a M*
M* 4'7-
l inear pyramidal triganal-planar
R S T
Schema 4. Koordinationsmoglichkci[sn lur auhbuiuentenfrele tlemente der 6. Hauptgruppe.
4.1. Gewinkelte und lineare Koordination
@-
a Abh. 25. Wuhcurcn dcr pyramidal gebduten p,-Bi-Komplexe 54 (oben) und 55 (unten) im Kristall. 54 hat keine Co-Co-Bindung: 55 hat drei Fe-Fee- Bindungen. Durch die geringeren MM-Abstande im Komplex 55 ist don die M,Ri-Geornetrie stdrker pyramidalisiert (Fe-Bi-Fe 62.6", Mittelwert) als bei 54 (Co-Bi-Co 107", Mittelwert). Den lridiumkomplex [(p3-Bi){lr,(CO).,l], der 55 Lhnelt, beschrieben Schrnid et al. [ 19bI.
Beginnen wir mit den Uberraschungen, die uns substitu- entenfreie Schwefel-, Selen- und Tellurliganden in den letzten Jahren beschert haben. Komplexe mit gewinkelten S,-Briicken (Typ P) sind heutzutage Legion, und sie wer- den hier nicht weiter behandelt. Die Synthese und Struk- turaufklgrung der Cr,S-Verbindung 57a (Typ R ; Abb. 26) durch Legrdins et al. im Jahre 1979 war ein Meilenstein der Organometallchemie~xsl. Sie erhielten 57a durch Um-
setzung von Trithiazylchlorid, N3S3C13, mit dem Komplex- ion [(T~~-C=,H~)C~(CO)~]-. Heute existieren fur die Syn- these derartiger Systeme mit cumulierten Dreifachbin- dungen die unterschiedlichsten Strategien; sie sind in
70 Angew. Chern. 98 (1986) 57-77
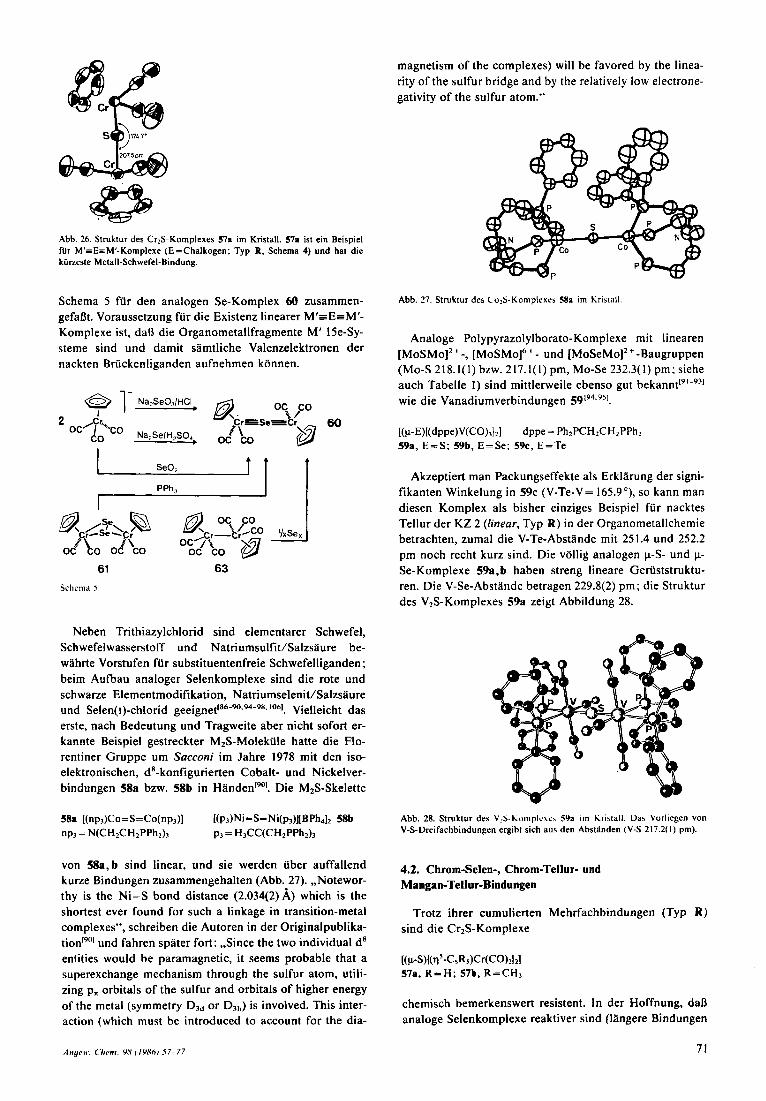
Abh. 26. Struktur des Cr:S-Komplexes 57r im Kristall. 57r ist ein Beispiel fur M'=E=M'-Komplexe (E=Chalkogen: Typ R, Schema 4) und hat die kiirzeste Metall-Schwefel-Bindung.
Schema 5 fur den analogen Se-Komplex 60 zusammen- gefaBt. Voraussetzung fur die Existenz h e a r e r M'=E=M'- Komplexe ist, daB die Organometallfragmente M' I Se-Sy- steme sind und damit samtliche Valenzelektronen der nackten Briickenliganden aufnehmen konnen.
63
Neben Trithiazylchlorid sind elementarer Schwefel, Schwefelwasserstoff und Natriumsulfit/Salzsaure be- wahrte Vorstufen fur substituentenfreie Schwefelliganden; beim Aufbau analoger Selenkomplexe sind die rote und schwarze Elementmodifikation, Natriumselenit/Salzsaure und Selen(1)-chlorid geeignet186-w.94-98. . V ielleicht das erste, nach Bedeutung und Tragweite aber nicht sofort er- kannte Beispiel gestreckter M,S-Molekiile hatte die Flo- rentiner Cruppe um Sacconi im Jahre 1978 mit den iso- elektronischen, d*-konfigurierten Cobalt- und Nickelver- bindungen 58a bzw. 58b in Handeniwl. Die M2S-Skelette
von 58a,b sind linear, und sie werden iiber auffallend kurze Bindungen zusammengehalten (Abb. 27). ,,Notewor- thy is the Ni-S bond distance (2.034(2) A) which is the shortest ever found for such a linkage in transition-metal complexes", schreiben die Autoren in der Originalpublika- t i ~ n [ ~ " ~ und fahren spater fort: ,,Since the two individual d8 entities would be paramagnetic, it seems probable that a superexchange mechanism through the sulfur atom, utili- zing pn orbitals of the sulfur and orbitals of higher energy of the metal (symmetry D3d or D3,,) is involved. This inter- action (which must be introduced to account for the dia-
magnetism of the complexes) will be favored by the linea- rity of the sulfur bridge and by the relatively low electrone- gativity of the sulfur atom.''
Ahh. 27. Struktur des Co2S-Kornplrxr, M a im Krislall
Analoge Polypyrazolylborato-Komplexe mit linearen [MoSMoJ2+-, [MoSMo16+- und [M~SeMo]~+-Baugruppen (Mo-S 218.1(1) bzw. 217.1(1) pm, Mo-Se 232.3(1) pm; siehe auch Tabelle 1) sind mittlerweile ebenso gut bekannti9'-93J wie die Vanadiumverbindungen 59[94.951.
Akzeptiert man Packungseffekte als Erklarung der signi- fikanten Winkelung in 59c (V-Te-V= 165.9"), so kann man diesen Komplex als bisher einziges Beispiel fur nacktes Tellur der KZ 2 (linear, Typ R) in der Organometallchemie betrachten, zumal die V-Te-Abstlnde mit 251.4 und 252.2 pm noch recht kurz sind. Die vollig analogen p-S- und p- Se-Komplexe 59a,b haben streng lineare Ceriiststruktu- ren. Die V-Se-Abstande betragen 229.8(2) pm; die Struktur des V,S-Komplexes 59a zeigt Abbildung 28.
Abb. 28. Struktur des V,S-homplu\u.\ 5% im liri\tdll . 1)ds Vorliegen von V-S-Dreifachhindungen ergiht sich aus den Ahstanden (V-S 217.2( I ) pm).
4.2. Chrom-Selen-, Chrom-Tellur- und Mangan-Tellur-Bindungen
Trotz ihrer cumulierten Mehrfachbindungen (Typ R) sind die Cr,S-Komplexe
chemisch bemerkenswert resistent. In der Hoffnung, daB analoge Selenkomplexe reaktiver sind (langere Bindungen
71
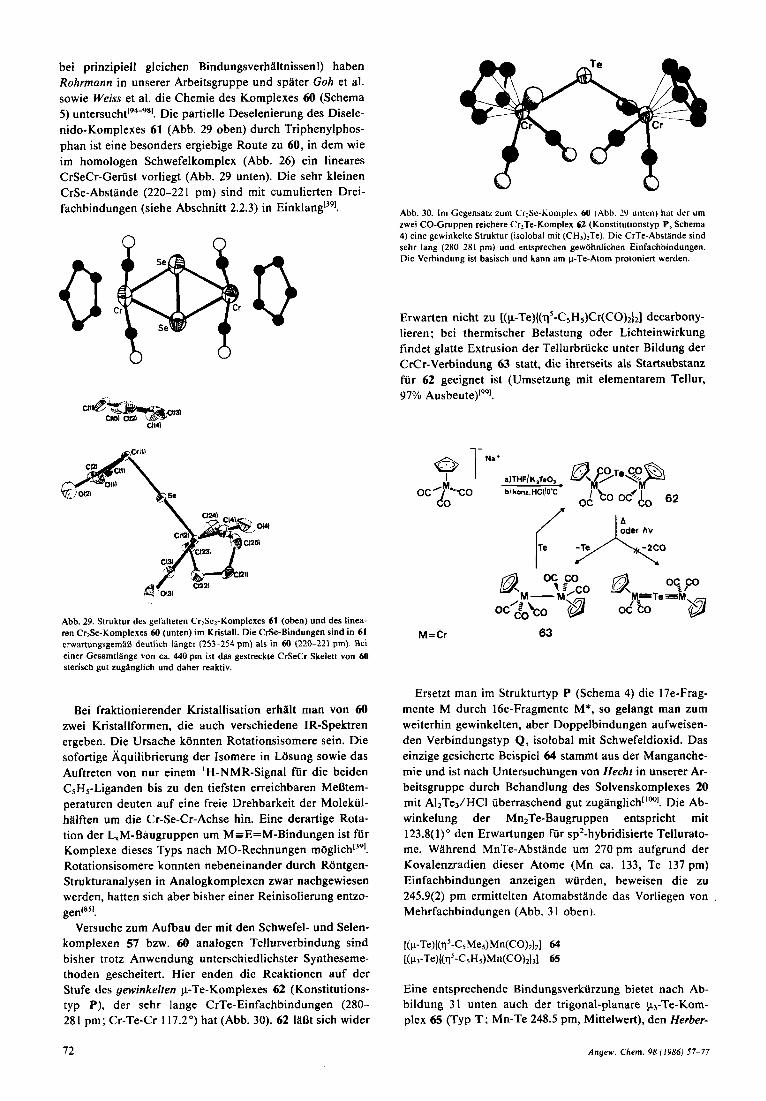
bei prinzipiell gleichen Bindungsverhaltnissen!) haben Rohrmann in unserer Arbeitsgruppe und spater Goh et al. sowie Weiss et al. die Chemie des Komplexes 60 (Schema 5) u n t e r s ~ c h t l ~ ~ - ~ ~ l . Die partielle Deselenierung des Disele- nido-Komplexes 61 (Abb. 29 oben) durch Triphenylphos- phan ist eine besonders ergiebige Route zu 60, in dem wie im homologen Schwefelkomplex (Abb. 26) ein lineares CrSeCr-Geriist vorliegt (Abb. 29 unten). Die sehr kleinen CrSe-Abstande (220-22 1 pm) sind mit cumulierten Drei- fachbindungen (siehe Abschnitt 2.2.3) in Einklang[391. Abb. 30. Im Cegensatz zum Cr:Sc-Komple,\ 6U (Abb. 3 unten) hat dcr um
zwei CO-Gruppen reichere Cr,Te-Komplex 62 (Konstitutionstyp P, Schema 4) eine gewinkelte Struktur (isolobal mit (CH,)>Te). Die CrTe-Abstande sind sehr lang (280-281 pm) und entsprechen gew6hnlichen Einfachbindungen. Die Vcrhindung ist basisch und kann am p-Te-Atom protoniert werden.
Erwarten nicht zu [(p-Te)((q5-C,HS)Cr(C0)2}2] decarbony- lieren; bei thermischer Belastung oder Lichteinwirkung findet glatte Extrusion der Tellurbriicke unter Bildung der CrCr-Verbindung 63 statt, die ihrerseits als Startsubstanz fur 62 geeignet ist (Umsetzung mit elementarem Tellur, 97% A u s b e ~ t e ) [ ~ ~ I .
00
n
Abb. 29. Struktur Jrr gefalteten Cr2SeZ-Komplexes 61 (oben) und des linea- ren CrzSe-Komplexes 60 (unten) im Kristall. Die CrSe-Bindungen sind in 61 crwartungsgernifl deutlich langer (253-254 pm) als in 60 (220-221 pm). Bei einer Gesamtlange von ca. 440 pm ist das gestreckte CrSeCr-Skelett von 60 sterisch gut zugtinglicb und daher reaktiv.
Bei fraktionierender Kristallisation erhalt man von 60 zwei Kristallformen, die auch verschiedene IR-Spektren ergeben. Die Ursache konnten Rotationsisomere sein. Die sofortige Aquilibrierung der Isomere in Losung sowie das Auftreten von nur einem ‘H-NMR-Signal fur die beiden C,H,-Liganden bis zu den tiefsten erreichbaren MeBtem- peraturen deuten auf eine freie Drehbarkeit der Molekiil- halften um die Cr-Se-Cr-Achse hin. Eine derartige Rota- tion der L,M-Baugruppen um M=E=M-Bindungen ist fur Komplexe dieses Typs nach MO-Rechnungen r n o g l i ~ h [ ~ ~ ] . Rotationsisomere konnten nebeneinander durch Rontgen- Strukturanalysen in Analogkomplexen zwar nachgewiesen werden, hatten sich aber bisher einer Reinisolierung entzo- gen[”s’.
Versuche zum Aufbau der mit den Schwefel- und Selen- komplexen 57 bzw. 60 analogen Tellurverbindung sind bisher trotz Anwendung unterschiedlichster Syntheseme- thoden gescheitert. Hier enden die Reaktionen auf der Stufe des gewinkelten p-Te-Komplexes 62 (Konstitutions- typ P), der sehr lange CrTe-Einfachbindungen (280- 281 pm; Cr-Te-Cr 117.2”) hat (Abb. 30). 62 laBt sich wider
M=Cr 63
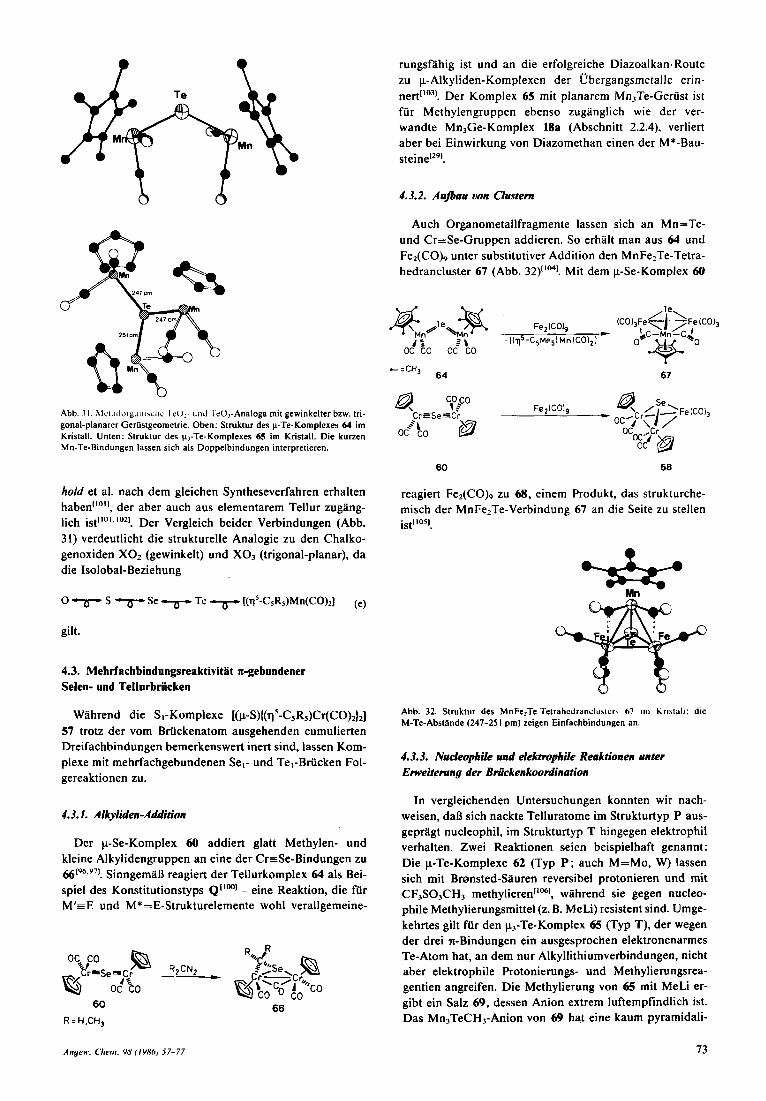
Ersetzt man im Strukturtyp P (Schema 4) die 17e-Frag- mente M durch 16e-Fragmente M*, so gelangt man zum weiterhin gewinkelten, aber Doppelbindungen aufweisen- den Verbindungstyp Q, isolobal mit Schwefeldioxid. Das einzige gesicherte Beispiel 64 stammt aus der Manganche- mie und ist nach Untersuchungen von Hecht in unserer Ar- beitsgruppe durch Behandlung des Solvenskomplexes 20 mit AI2Te3/HCI uberraschend gut zuganglich[‘m! Die Ab- winkelung der Mn2Te-Baugruppen entspricht mit I23.8( 1)” den Erwartungen fur sp2-hybridisierte Tellurato- me. Wahrend MnTe-Abstande um 270pm aufgrund der Kovalenzradien dieser Atome (Mn ca. 133, Te 137 pm) Einfachbindungen anzeigen wiirden, beweisen die zu 245.9(2) pm ermittelten Atomabstande das Vorliegen von Mehrfachbindungen (Abb. 31 oben).
Eine entsprechende Bindungsverkiirzung bietet nach Ab- bildung 3 1 unten auch der trigonal-planare p3-Te-Kom- plex 65 (Typ T ; Mn-Te 248.5 pm, Mittelwert), den Herber-
72 Anyew. Chem. 98 119861 57-77
t t Mn \
b b
Abb. 31. h l ~ ~ . i l l ~ i t ~ , i i i i ~ ~ l i ~ I t [J2- uiid IcO,-Analoga mit gewinkelter bzw. tri- gonal-planarer Gerustgeornetrie. Oben: Struktur des p-Te-Komplexes 64 irn Kristall. Unten: Struktur des p,-Te-Kornplexes 65 im Kristall. Die kurzen Mn-Te-Bindungen lassen sich als Doppelbindungen interpretieren.
hold et al. nach dem gleichen Syntheseverfahren erhalten habenl""l, der aber auch aus elementarem Tellur zugang-
3 1) verdeutlicht die strukturelle Analogie zu den Chalko- genoxiden X 0 2 (gewinkelt) und X 0 3 (trigonal-planar), da die Isolobal-Beziehung
lich iStllOI. 1021 . De r Vergleich beider Verbindungen (Abb.
gilt.
4.3. Mehrfachbindungsreaktivitat n-gebundener Selen- und Tellurbriicken
Wahrend die S1-Komplexe [(p-S)((qS-C5RS)Cr(C0)2)2] 57 trotz der vom Briickenatom ausgehenden cumulierten Dreifachbindungen bemerkenswert inert sind, lassen Kom- plexe mit mehrfachgebundenen Sel- und Te,-Briicken Fol- gereaktionen zu.
4.3.1. Alkyliden-Addition
Der p-Se-Komplex 60 addiert glatt Methylen- und kleine Alkylidengruppen a n eine der CrESe-Bindungen zu 66[y6.v71. SinngemaB reagiert der Tellurkomplex 64 als Bei- spiel des Konstitutionstyps Qltoo1 - eine Reaktion, die fur M'EE und M*=E-Strukturelemente wohl verallgemeine-
R = H.CH3
rungsfahig ist und an die erfolgreiche Diazoal kan-Route zu p-Alkyliden-Komplexen der Ubergangsmetalle erin- nertl'031. Der Komplex 65 mit planarem Mn3Te-Gerust ist fur Methylengruppen ebenso zuganglich wie der ver- wandte Mn3Ge-Komplex 18a (Abschnitt 2.2.4), verliert aber bei Einwirkung von Diazomethan einen der M*-Bau- ~ t e i n e l ~ ~ l .
4.3.2. Aujbau von Clustern
Auch Organometallfragmente lassen sich an Mn=Te- und Cr=Se-Gruppen addieren. So erhalt man aus 64 und Fe2(CO), unter substitutiver Addition den MnFe2Te-Tetra- hedrancluster 67 (Abb. 32)[Iw1. Mit dern p-Se-Komplex 60
c = CH3 64 67
60 68
reagiert Fe2(CO)9 zu 68, einem Produkt, das strukturche- misch der MnFe2Te-Verbindung 67 an die Seite zu stellen istiinsi * Mn
Abb. 32. Struktur des MnFe,Te-TetrahcdrancIuricr* 67 in1 hrirtdll; die M-Te-AbstBnde (247-25 1 prn) zeigen Einfachbindungen an.
4.3.3. Nucleophile und elektrophile Reaktionen unter Erweiterung der Bdckenkoordination
In vergleichenden Untersuchungen konnten wir nach- weisen, daB sich nackte Telluratome im Strukturtyp P aus- gepragt nucleophil, im Strukturtyp T hingegen elektrophil verhalten. Zwei Reaktionen seien beispielhaft genannt: Die p-Te-Komplexe 62 (Typ P; auch M=Mo, W) lassen sich mit Bronsted-Sauren reversibel protonieren und mit CF,S0,CH3 methylierenllM1, wahrend sie gegen nucleo- phile Methylierungsmittel (2. B. MeLi) resistent sind. Umge- kehrtes gilt fur den p,-Te-Komplex 65 (Typ T), der wegen der drei n-Bindungen ein ausgesprochen elektronenarmes Te-Atom hat, a n dem nur Alkyllithiumverbindungen, nicht aber elektrophile Protonierungs- und Methylierungsrea- gentien angreifen. Die Methylierung von 65 rnit MeLi er- gibt ein Salz 69, dessen Anion extrem luftempfindlich ist. Das Mn,TeCH,-Anion von 69 hat eine kaum pyramidali-
Anyew. (Aetn. 98 (1986) 57-77 73
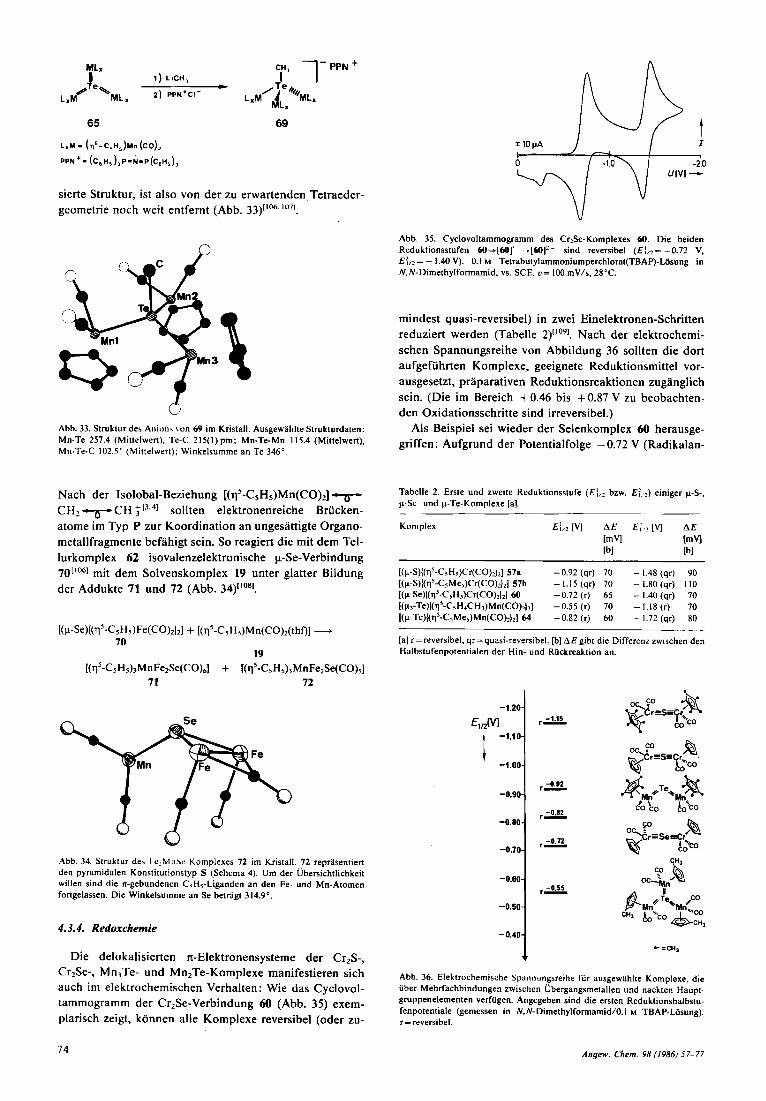
69
sierte Struktur, ist also von der zu erwartenden Tetraeder- geometrie noch weit entfernt (Abb. 33)"". 1')71.
n
Yn3 r3 Abb. 33. Struktur des Anion\ \on 6Y im Kristall. Ausgewahlte Strukturdaten: Mn-Te 257.4 (Mittelwert), Te-C 215(1) pm; Mn-Te-Mn 115.4 (Mittelwert), Mn-Te-C 102.5" (Mittelwert); Winkelsumme an Te 346".
Nach der Isolobal-Beziehung [(qs-C5H5)Mn(CO),] 7 CH2 '6 CH :[3.41 sollten elektronenreiche Brucken- atome irn Typ P zur Koordination an ungesattigte Organo- metallfragmente befahigt sein. So reagiert die mit dern Tel- lurkomplex 62 isovalenzelektronische p-Se-Verbindung 7OL'O6I mit dem Solvenskomplex 19 unter glatter Bildung der Addukte 71 und 72 (Abb. 34)"On1.
[(~-se)((r15-C5Hs)Fe(CO)2~21 + [(r15-C5H5)Mn(CO)z(thf)l d 70
19 [(q5-C5H5)3MnFe2Se(CO)6] + [(q5-C5H5)3MnFe2Se(C0)5]
71 72
W Abb. 34. Struktur des I e2Mti. \c homplexes 72 im Kristall. 72 reprasentiert den pyramidalen Konstitutionstyp S (Schema 4). Um der Ubersichtlichkeit willen sind die n-gebundenen C,Hs-Liganden a n den Fe- und Mn-Atomen fortgelassen. Die Winkelsumme an Se betragt 314.9".
4.3.4. Redoxchemie
Die delokalisierten n-Elektronensysteme der Cr,S-, Cr2Se-, Mn,Te- und Mn,Te-Komplexe manifestieren sich auch irn elektrochemischen Verhalten: Wie das Cyclovol- tammogramm der Cr,Se-Verbindung 60 (Abb. 35) exem- plarisch zeigt, konnen alle Komplexe reversibel (oder zu-
Abb. 35. Cyclovoltammogramm des Cr2Se-Komplexes 60. Die beiden Reduktionsstufen 60+[6Oj'--[60]'- sind reversibel (E1,*- -0.72 V, E:,2= - 1.40 V). 0.1 M Tetrabutylammoniumperchlorat(TBAP)-LBsung in N.N-Dimethylformamid, vs. SCE, u= 100 mV/s, 28°C.
mindest quasi-reversibel) in zwei Einelektronen-Schritten reduziert werden (Tabelle 2)['O9]. Nach der elektrocherni- schen Spannungsreihe von Abbildung 36 sollten die dort aufgefuhrten Kornplexe, geeignete Reduktionsmittel vor- ausgesetzt, praparativen Reduktionsreaktionen zuganglich sein. (Die irn Bereich +0.46 bis +0.87 V zu beobachten- den Oxidationsschritte sind irreversibel.)
Als Beispiel sei wieder der Selenkomplex 60 herausge- griffen: Aufgrund der Potentialfolge - 0.72 V (Radikalan-
Tabelle 2. Erste und zweite Reduktionssrufe p-Sc- und p-Te-Komplexe [a].
bzw. E f 2 ) einiger p-S-,
Komplex
~(~-S)I(~s-~sHs)C~CO)~I~I 57a -0.92 (qr) 70 - 1.48 (qr) 90 [(~-S)((llS-CsMe,)Cr(CO)~t~l 57b - 1.15 (qr) 70 - 1.80 (qr) I10 [(p-se)l(~'-C~H,)Cr(CO)~t~l 60 -0.72 (r) 65 - 1.40 (qr) 70 [(p7-Te)l(rls-CsH,CH,)Mn(CO)*t,l -0.55 (r) 70 - 1.18 (r) 70 [(p-Te)((rl'-CsMe,)Mn(CO)212] 64 -0.82 (r) 60 - 1.72 (qr) 80
[a] r=reversibel, qr=quasi-reversibel. [b] AE gibt die Differenz zwischen den Halbstufenpotentialen der Hin- und Ruckreaktion an.
-1.20
~ t n W l -1.10
-1.00 1
-0.90
-0.80
-0.10
-0.60
-0.50
- 0.40
CHn
-=CH3
Abb. 36. Elektrochemische Spannungsreihe f u r ausgewlhlte Komplexe. die iiber Mehrfachbindungen zwischen Ubergangsmetallen und nackten Haupt- gruppenelementen verfugen. Angegeben sind die ersten Reduktionshalbstu- fenpotentiale (gemessen in N.N-Dimethylformamid/O. I M TBAP-&sung). r = reversibel.
74 Angew. Chem. 98 (1986) 57-77

ion)/ - I .40 V (Dianion) war rnit Reduktionsmitteln, deren Halbstufenpotential im Zwischenbereich liegt, die Erzeu- gung des Radikalanions zu erwarten. Umsetzung mit Co- baltocen (E1,2[Cp,Co+/Cp2Co]= -0.77 V, N, N-Dimethyl- formamid[''"l) in Tetrahydrofuran/Diethylether ergibt bei - 30°C in sehr langsamet Reaktion die schwane, kristalli- ne, ionische Komplexverbindung
Abb. 37. Struktur des Komplexanions von 73 im Kristall (-60 "C). Das pla- nare (!) Cr,Se-Geriist reprasentiert eine neuartige Koordinationsart, die in Schema 4 noch nicht aufgefijhrt ist. Die salzartige Verbindung kristallisiert aus Tetrahydrofuran/Diethylether ( - 30 "C) mit einem Aquivalent THF.
Das Komplexanion dieser Verbindung hat ein planares (!) Cr,Se-Geriist rnit einem Cr2Se-Dreiring (Abb. 37). Die exo- und endocyclischen CrSe-Bindungen (227.9(0) bzw. 233.2(0) pm) weisen unterschiedlich hohe Mehrfachbin- dungsanteile auf; der CrCr-Abstand entspricht rnit 308.0 pm einer Einfachbindung. Wlhrend der lineare Cr2Se- Komplex 60 isoelektronisch rnit der Mn2Ce-Verbindung 16a ist und fur beide Systeme ganz ahnliche Bindungsver- hlltnisse diskutiert werden (vgl. Abschnitt 2.2.3), so ist das Cr3Se-Komplexanion von 73 isoelektronisch rnit der neu- tralen Mn,Ge-Verbindung 18a Schema 6). Das zentrale, nackte Selenatom tragt samtliche Valenzelektronen zur Bindung an die drei [(~l'-C~H~)Cr(CO)~]-Fragmente bei, die rnit CH- und CH :-Fragmenten isolobal sind. Fur das einfach geladene Komplexanion von 73 ergibt sich damit wie fur 18 wegen S e T C 2 - - C H - bzw. Ge 7 C eine Isolobal-Beziehung zu Methylency~lopropan~~~~~.
MnLx L,Mn=Ge=hlnL, L,Mn=Ge<
MnLx 16 a 18 a
Wie aus den 'H-NMR-Spektren ([D,]Aceton) hervor- geht, hat 73 eine ,,fluktuierende" Struktur: Bei - 80 "C er- scheinen drei gut aufgeloste C5H5-Signale, von denen zwei bei Temperaturerhohung breiter werden und sich ab ca. - 50 "C zu einem vereinigen. Dieser Befund kann mit ei- ner cis-trans-Isomerisierung im Zweikernfragment [(I$- C5HS)Cr(C0)2]2 erklart werden. Das Auftreten eines schar-
fen Signals fur die exocyclische [(q5-CSH5)Cr(C0)2]-Ein- heit zwischen - 80 und 25°C weist auf eine freie Drehbar- keit dieses Strukturelements um die CrSe-Bindung hin.
73 ist extrem sauerstoffempfindlich und gibt an Luft schlagartig, aber nicht quantitativ den Ausgangskomplex 60 zuriick. Der Komplex bleibt in Losung nur bei tiefen Temperaturen unversehrt und zerfallt bei Raumtemperatur rasch zum selenfreien, Rontgen-strukturanalytisch charak- terisierten Salz [(q5-C5H5)2Co]+[(q'-C5H5)Cr(C0)3]-i"2' und einem carbonylfreien [(C5H5)CrSe],-Cluster unbe- kannter Struktur. Aus der Zusammensetzung des diama- gnetischen (!) Komplexanions von 73 folgt, daB die Ein- elektronen-Reduktion von 60 Sekundarreaktionen nach sich ziehen kann, was im vorliegenden Fall zu Clustermo- lekiilen fuhrt. Mit dem Reduktionsmittel KO2 reagiert der Cr2Se-Komplex 60 in Gegenwart eines Kryptanden unter sofortiger Reduktion und Fragmentierung zum Rontgen- strukturanalytisch gesicherten Komplex [K(2.2.2-Kryp- tand)][(~f-C~H~)Cr(CO)~Se~]74[' l3].
5. Perspektiven
Substituentenfreie Elemente der Kohlenstoff-, Stick- stoff- und Sauerstoffgruppen haben wir strukturpragend als Zentralatome in Mehrfachbindungssystemen wie auch eingekapselt in den Hohlraumen von Ubergangsmetallclu- stern kennengelernt. Noch fehlt es der Substanzpalette an jener Reichhaltigkeit, die wir aus der nunmehr 20jahrigen Geschichte der Komplexe rnit Metall-Metall-Mehrfachbin- dungen kennen" 14', doch lassen die bisher angewendeten Synthesemethoden auf Ausbaufahigkeit des neu erschlos- senen Gebietes hoffen. Was die schweren, metallahnliche- ren Hauptgruppenelemente betrifft, so fallt hier die hohe ReaktivitLt der Elementmodifikationen sowie der binaren Hydride bei Umsetzungen mit koordinativ ungesattigten Organometallkomplexen auf. Die Anbindung an die Che- mie der Ubergangsmetalle erscheint nahtlos, denn sowohl die Atomradien als auch die elektronische Situation sind hier cum grano salis ahnlich. Strukturchemische Ver- wandtschaftsbeziehungen zur klassischen anorganischen Festktirperchemie sind offensichtlich, und es steht zu er- warten, daB bald auch Iso- und Heteropolyionen wie Se:+, Te:+, Teg-, Tei+ oder [Au2Te$- und [ A S , ~ T ~ ~ ] ~ - [ ' ~ . ~ . ' ' ' ~ ihren Weg in das priiparative Arsenal des Metallorganikers finden werden. Uber Mehrfachbin- dungen verkniipfte Elementensembles von fjbergangsme- tallen und substituentenfreien Hauptgruppenelementen versprechen eine reichhaltige Chemie, die jener der Me- tall-Metall-Mehrfachbindungssysteme nicht unahnlich sein sollte. Die Suche nach neuen Katalysatoren - schon immer eine Triebfeder der Organometallchemiker - durfte auch auf dem hier geschilderten Gebiet schon bald Erfolge zeitigen.
Dieser Aufsatz entspringt einer Vorlesung, die ich vor Stu- denten an der Technischen Universitat Miinchen gehalten habe und denen ich fur die vielen aufmerksamen Anmerkun- gen und klugen Fragen danke, die mich zu tieferem Nach- denken gebracht haben. Ebenso bin ich meinen Mitarbeitern dankbar. die im Laboratorium Teilbereiche der hier vorge- tragenen Thematik mit Geschick und ldeenreichtum sowie He$ und Ausdauer bearbeitet haben. So danke ich Basile Koumbouris (Arsenchemie). Heinz-Josef Kneuper (Germani- um-, Zinn- und Bleichemie). Jurgen Rohrmann (Schwefel-,
Angew. Chem. 98 (1986) 57-77 75

Selen- und Tellurchemie), Christian Hecht (Tellurchemie). Erdmuthe Voss (Siliciumchemie), R o y Horlein und Dr. Josef Weichmann (Germaniumchemie). Dr. Eberhardt Herdtweck aus unserem Laboratorium sowie den Arbeitsgruppen meiner Kollegen Manfred L. Ziegler (Universitat Heidelberg). Ivan Bernal (University of Houston) und Heinrich Noth (Universi- tat Miinchen) sei f u r ihre wertvollen Beitrage zur Struktur- chemie ebenso herzlich gedankt wie Herrn Kollegen Hans Bock (Universitat Frankfurt) fur intellektuelle Anregungen und praktische Beitrage zur Elektrochemie. Wie aus meinen Ausfuhrungen an vielen Stellen hervorgeht. verdankt das hier erstmals zusammenfassend dargestellte Forschungsge- biet vie1 den Arbeitsgruppen der Kollegen Herberhold (Uni- versitat Bayreuth), Huttner (Universitat Konstanz) und Weiss (Univemitat Hamburg), denen fur offenherzige Dis- kussionen meine Verbundenheit gilt. Freya und den Kindern danke ich f u r ihre Geduld.
Eingegangen am 14. Oktober 1985 [A 5631
[l] G. Wilkinson, F. G. A. Stone, E. W. Ahel (Hrsg.): Comprehensive Orga- nometallic Chemistry. Pergamon Press, Oxford 1982. Dds Monumental- werk besteht aus neun BBnden und gilt heute als Standardwerk der ele- mentorganischen Chemie; mit 8750 Seiten ist es die reichhaltigste In- formationsquelle.
[2] J. Buckingham (Hrsg.): Dictionary of Organometalltc Compounds. Chapman and Hall, London 1984. Das dreibBndige Werk referiert 14000 Organometallverbindungen.
131 Als leistungsfiihigstes Konzept fiir die Syntheseplanung hat sich die IsolobaCAnalogie enviesen: R. Hoffmann, Angew. Chem. 94 (1982) 725; Angew. Chem. Int . Ed. Enyl. 21 (1982) 711 (Nobel-Vortrag).
[4] Zusammenfassung iiber Anwendungen der Isolobal-Analogie in der metallorganischen Chemie: F. G . A. Stone, Angew. Chem. 96 (1984) 85; Angew. Chem. Int. Ed. Engl. 23 (1984) 85.
[5] Ubersichtsartikel: G . Schmid, Stnrcf. Bonding (Berlin) 62 (1985) 51. [6] Originalarbeiten: a) G . Schmid, R. F'feil, R Boese, F. Bandermann, S.
Meyer. G. H. M. Calis, J. W. A. van der Velden, Chem. Ber. 114 (1981) 3634: b) L. R. Wallenberg. J.-0. Bovin, G . Schmid, Surf: Sci. 156 (1985) 256; c) vgl. auch den Rh,,-Cluster [Rhss(PtBu,)12C120]: G. Schmid, U. Giebel, W. Huster, Inorg. Chim. Acta 85 (1984) 97.
171 Gold-Modilikation (Au&: G. Schmid et al., unver6ffentlicbt. Den ak- tuellen Hahepunkt strategisch wertvoller Clustersynthesen markiert der Komplex [ N i J I S e ~ ~ ( P P h ~ ) t ~ l : D. Fenske, J. Ohmer, J. Hachgenei, Angew. Chem. 97 (1985) 993; Angew. Chem. Int. Ed. Engl. 24 (1985) 993.
[8] M. N. Vargaftik, V. P. Zagorodnikov, P. Stolyarov, 1. 1. Moiseev, V. A. Likholobov, D. 1. Kochubey, L. Chuvilin, 1. Zaikovsky, K. 1. Zamaraev, G. 1. Timofeeva, J. Chem. Soc. Chem. Commun. 1985. 937.
191 Ubersichtsartikel (Cluster): a) P. Chini, J. Organomef. Chem. 200 (1980) 37; b) E. L. Muetterties, ibid. 2W (1980) 177; c) W. L. Gladfelter, G. L. Geoffroy, Adu. Organomel. Chem. 18 (1980) 207; d ) H. Vahren- kamp, ibid. 22 (1983) 169.
[lo] Ubersichtsartikel (Carbidocluster): P. Chini, G . Longoni. V. G. Albano, Adu. Organomet. Chem. 14 (1976) 285.
[Il l E. H. Braye, L. F. Dahl, W. HUbel, D. L. Wampler, J . Am. Chem. Soc. 84 (1962) 4633.
[It] J. S. Bradley, G . B. Ansell, M. E. Leonowicz, E. W. Hill, J. Am. Chem. Soc. 103 (1981) 4968.
[I31 a) J. S. Bradley, E. W. Hill, G . 8. Ansell, M. A. Modrick, Organometal- lies I (1982) 1634; b) J. S. Bradley, G. B. Ansell, E. W. Hill, J . Am. Chem. Soc. 701 (1979) 7417; c) M. R. Churchill. J. Wormald, J. Knight, M. J. Mays, ibid. 93 (1971) 3073.
1141 a) V. G. Albano, P. Chini, G . Ciani, M. Sansoni, D. Strumolo. 8. T. Heaton, S. Martinengo, J. Am. Chem. Soe. 98 (1976) 5027 ([CO~C(CO)~,]~- und [CoSC(CO),.]-): b) G. Ciani, C. DAlfonso, M. Freni, P. Remiti, A. Sironi, J. Chem. Soc. Chem. Commun. 1982. 339 ([Re7C(CO)Zl]2-); c) D. H. Fdrrar, P. F. Jackson, B. F. G. Johnson, J. Lewis. J. N. Nicholls, ibid. 1981, 415 ([RU~C(CO),~]); d ) D. F. Shriver, E. Holt, J. Organornet. Chem.. im Druck; e) J. H. Davis, M. A. Beno, J. M. Williams, J. Zimmie, M. Tachikawa, E. L. Muetterties, hoe. Natl. Acad. Sci. USA 78 (1981) 668.
[IS] Neuere Ubersichtsartikel: a) M. Tachikawa, E. L. Muetterties, h o g r . Inorg. Chem. 28 (1981) 203: b) J. S. Rradley, Adu. Organomef. Chem. 22 (1983) 1 .
1161 Beispiele: a) D. K. Breitinger, K. Geibel, W. Kress, R. Sendelbeck, J . Organomef. Chem. 191 (1980) 7; b) D. K. Breitinger, W. Kress, R. Sen- delbeck, K. Ishiwada, ibid. 243 (1983) 245.
1171 a) J. D. Cotton, J. Duckworth, S. A. R. Knox, P. F. Lindley, I . Paul, F. G. A. Stone, P. Woodward, Chem. Commun. 1966. 253: b) P. F. Lind- ley, P. Woodwdrd, J. Chem. Sor. A1967. 382; c) J. D. Cotton. S. A. R. Knox, 1. Paul, F. G. A. Stone, J. Chem. Sor. A 1967. 264.
1181 a) G. Schmid, G. Etzrodt, J . Organomef. Chem. 137 (1977) 367: b) C . Schmid, V. Blltzel, G. Etzrodt, ibid. 112 (1976) 345.
[I91 a) G . Etzrodt, R. Boese, G. Schmid, Chem. Bey. 112 (1979) 2574: b) W. Kruppd, D. BIaser, R. Boese, G . Schmid, 2. Naturjorsch. 8 3 7 (1982) 209.
[20] W. Malisch, H.-U. Wekel, I. Grob, F.-H. KBhler. Z. Nafurjorsch. 8 3 7 (1982) 601.
[21] R. F. Cerlach, K. M. Mackay, B. K. Nicholson, W. T. Robinson, J . Chem. Soc. Dalton Trans. 1981, 80.
[22] S. P. Foster, K. M. Mackay, B. K. Nicholson, Inorg. Chern. 24 (1985) 909.
I231 Metallorganische Aspekte der Fischer-Tropsch-Synthese: W. A. Herr- mann. Angew. Chem. 94 (1982) 118; Angew. Chem. Inf . Ed. Engl. 21 (1982) 117, zit. Lit.
[24] a) V. Ponec. Catal. Reu. Sci. Eng. 18 (1978) 1517; b) P. Bileon. J. N. Helle, W. M. H. Sdchtler, J. Carol. 58 (1978) 95.
1251 W. Gllde, E. Weiss, J . Oryanomef. Chem. 213 (1981) 451. [26] W. GBde, E. Weiss, Chem. Ber. 114 (1981) 2399: vgl. D. Melzer, E.
[27] W. Gllde, E. Weiss. Angew. Chem. 93 (1981) 796; Angew. Chem. Int. Ed.
[28] P. Riviere, M. Riviere-Baudet, J. Satge in [I], Vol. 2. S. 399ff. I291 W. A. Hernnann, J. Weichmann, U. KListhardt, A. SchBfer, R. HBrlein,
C. Hecbt, E. Voss, R. Serrano. Angew. Chem. 95 (1983) 1019; Angew. Chem. Int . Ed. Engl. 22 (1983) 979; Angew. Chem. Suppl. 1983. 1543.
1301 Ubersichtsartikel: a) E. Colomer, R. J. P. Comu. Top. Curr. Cliem. 96 (1981) 79; b) R. J. Aylett, Ado. Inorg. Chem. Radiochern. 25 (1982) I .
1311 W. A. Herrmann, E. Voss, E. Cuggolz, M. L. Ziegler, J . Organornet. Chem. 284 (1985) 47.
(32) a) D. Melzer, E. Weiss, J. Organornet. Chern. 255 (1983) 335: b) ibid. 263 (1984) 67.
1331 J. D. Korp, 1. Bernal, R. Hbrlcin, R. Serrano. W. A. Herrmann. Chem. Ber. 118 (1985) 340.
134) W. A. Herrmann, R. Serrano. J. Weichmann, J. Oryanomet. Chem. 246 (1983) C 57.
[35] I. Bernal, J. D. Korp. W. A. Herrmann, R. Serrano, Chem. Ber. 117 (1984) 434.
1361 D. W. H. Rankin, A. Robertson. J . Organomet. Chem. 85 (1975) 225. [37] Yu. T. Struchkov, K. N. Anisimov, 0. P. Osipova, N. E. Kolobova. A.
[381 K. Triplett, M. D. Curtis. J. Am. Chem. Sor. 97 (1975) 5747. [39] N. M. Kostic. R F. Fenske, J. Organomet. Chem. 233 (1982) 337. [40] G. Huttner, U. Weber, B. Sigwarth, 0. Scheidsteger. H. Lang, L. Zsol-
[411 W. A. Herrmann, H.-J. Kneuper, E. Herdtweck, unveroffentlicht. [42] Zusammenfassung: M. 1. Bruce, A. G. Swincer, Adu. Organomef. Chem.
I431 Neuere Arbeit: W. A. Herrmann, C. Weber. M. L. Ziegler, 0. Serhadli,
1441 W. A. Herrmann. H.-J. Kneuper, unverBffentlicht. 1451 W. A. Herrmann, H.-J. Kneuper, E. Herdtweck, Angew. Chem. 97
[46] P. Jutzi, W. Steiner, Chem. Ber. 109 (1976) 3473. 1471 W. A. Herrmann, H.-J. Kneuper, E. Herdtweck, unverbffentlichte Er-
gebnisse (Synthese und Rbntgen-Strukturanalyse von [(pGeCIJ(q'- CsHs)Mn(C0)Zt2]: Mn-Ge 233.7(1), 236.7(1) pm; triklin).
[48] Vgl. beispielsweise die Diskussion in [99] iiher Kovalenzradien von Me- tallatomen und Bindungsordnungen unter Beriicksichtigung der ein- schliigigen Literatur: a) L. Pauling: Die Narur der chemisehen Bindung, 3. Aufl., Verlag Chemie, Weinheim 1976; b) A. F. Wells: Structural Inorganic Chemistry. 5. Aufl., Clarendon Press, Oxford 1985; siehe auch W. A. Herrmann, J. Weichmann, R. Serrano. K. Blechschmitt, H. Plisterer, M. L. Ziegler, Angew. Chem. Suppl. 1983. 363, FuUnote 32.
1491 a) Originalpublikation: E. 0. Fischer. G. Kreis. C. G . Kreiter, J. Mul- ler, G. Huttner, H. Lorenz, Angew. Chem. 85 (1973) 618; Angew. Chem. Int. Ed. Engl. ' I 2 (1973) 564; b) Zusammenfassungen: E. 0. Fischer, U. Schubert, 1. Organomet. Chem. 100 (1975) 59; E. 0. Fischer, Angew. Chem. 86 (1974) 651 (Nobel-Vortrag); Adu. Organomel. Chem. 14 (1976) I.
[SO] N. N. Greenwood, A. Edrnshaw: Chemistry of rhe Elements. Pergamon Press, Oxford 1984.
[5 I] F. A. Cotton, G . Wilkinson: Anorganische Chemie. 4. Aufl., Verlag Che- mie, Weinheim 1982.
1521 K. Dehnicke. J. Strahle, Angew. Chem. 93 (1981) 451; Angew. Chem. Int. Ed. Engl. 20 (1981) 413.
1531 D. Bright, J. A. Ibers, h r g . Chem. 8 (1969) 709. 1541 a) K. Dehnicke, W. Kolitsch, 2. Naturjbrsch. 8 3 2 (1977) 1485; b) W.
Kolitsch, K. Dehnicke, ibid. 8 2 5 (1970) 1080.
Weiss. ibid. I I 7 ( 1984) 2464.
Engl. 20 (1981) 803.
N. Nesmeyanov, Dokl. Akad. Nauk. SSSR I72 (1967) 15.
nai, J. Organomef. Chem. 282 (1985) 331, zit. Lit.
22 (1983) 60.
J. Organomet. Chem.. im Druck, zit. Lit.
(1985) 1060; Angew. Chem. Int. Ed. Engl. 24 (1985) 1062.
76 Angew. Chem. 98 (1986) 57-77

[SS] W. Liese. K. Dehnicke, 1. Walker, J. Strahle, Z. Naturforsch. 834 (1979)
IS61 K.-P. Frank. J. Strahle, J. Weidlein, Z. Natufursch. 835 (1980) 300. 1571 M. Ciechanowicz, W. P. Griffith, D. Pawson. A. C . Skapski, Chem.
Commun. 1971. 1176. (581 S. Martinengo, G. Ciani. A. Sironi, B. T. Heaton, 1. Mason, 1. Am.
Chem. SUC. 101 (1979) 7095. [59] D. E. Fjare. W. L. Gladfelter, J. Am. Chem. Soc. 103 (1981) 1572; vgl.
M. A. Collins. B. F. G. Johnson, J. Lewis, J. M. Mace, J. Moms, M. McPartlin, W. J. H. Nelson, J. Puga, P. R Raithby, J. Chem. SOC. Cliem. Commun. 1983. 689 (Ru4N- und Os,N-Cluster).
[60] I. L. Vidal. W. E. Walker, R. L. Pruett, R. C. Schoening, lnorg. Chem. 18 (1979) 129.
(61) P. Chini, G. Ciani, S. Martinengo, A. Sironi, L. Longhetti, B. T. Hea- ton, J. Chem. SOC. Chem. Commun. 1979. 188.
1621 J. Vidal, Inorg. Chem. 20 (1981) 243. 1631 J. Vidal, J. M. Troup, J . Organornet. Chem. 213 (1981) 351. [64] a) W. Schmettow, H. G. von Schnering. Angew. Chem. 89 (1977) 895;
Angew. Chem. Int. Ed. Engl. 16 (1977) 857; b) H. G. von Schnering, D. Fenske, W. Hoinle, M. Binnewies, K. Peters, Angew. Chem. 91 (1979) 755; Angew. Chem. Inf. Ed. Engl. 18 (1979) 679; c) eine lesenswerte Zusammenfassung iiber ,.Homonucleare Bindungen bei Hauptgrup- penelementen" bietet: H. G. von Schnering, Angew. Chem. 93 (1981) 44; Angew. Chem. Int. Ed. Engl. 20 (1981) 33.
[65] Zusammenfassung: H. SchAfer, B. Eisenmann, W. MOller, Angew. Chem. 85 (1973) 742: Angew. Chem. In,. Ed. Engl. I2 (1973) 694 und die don sowie die in [SO] Zitierte Literatur.
1661 a) P. A. Edwards, J. D. Corbett, Inorg. Chem. 16 (1977) 903; b) J. D. Corbett, P. A. Edwards, J. Am. Chem. Soc. 99 (1977) 3313; c) C. H. E. Belin, J. D. Corbett, A. Cisar. ibid. 99 (1977) 7163; d) S. C. Critchlow, J. D. Corbett, J . Chem. Soc. Chem. Commun. 1981. 236; e) R. C. Burns, J. D. Corbett, J . Am. Chem. SOC. 104 (1982) 2804; r) J. D. Corbett, D. G. Adolphson, D. J. Merriman, P. A. Edwards, F. J. Armatis, ibid. 97 (1975) 6267.
693.
1671 Ubersichtsartikel: J. D. Corbett, Progr. fnorg. Chem. 21 (1976) 129. 1681 R. W. Rudolph, W. L. Wilson, J. Am. Chem. Soc. 103 (1981) 2480, zit.
Lit.; F. Teixidor, M. L. Luetkens, R. W. Rudolph, ibid. 105 (1983) 149.
[69] a) 1. L. Lohr, fnorg. Chem. 20 (1981) 4229; b) R. C. Bums, R. J. Gilles- pie, J. A. Barnes, M. J. McGlinchey, ibid. 21 (1982) 799 (MO-Rechnun- gen); c) R. I. Cillespie, J. Passmore. Ado. Inorg. Chem. Radiochem. 17 (1975) 49.
[70] T. G. Spiro. D. H. Templeton, A. Zalkin, lnorg. Chem. 8 (1969) 856; vgl. auch die Struktur von [Pb40(Si(C,,H5)31n]: C. Gaffney, P. G. Harri- son, T. I. King, J. Chem. SOC. Chem. Commun. 1980. 1251.
[71] Siehe [SO], S. 136. (721 H. Lang, L. Zsolnai, G. Huttner, Angew. Chem. 95 (1983) 1016; Angew.
1731 Siehe 1401; vgl. auch [72]. 1741 H. Lang, L. Zsolnai, G. Huttner, Angew. Chem. 95 (1983) 1017; Angew.
Chem. Int. Ed. Engl. 22 (1983) 976; Angew. Chem. Suppl. 1983. 1463; vgl. auch: A. Vizi-Orosz, G. Palyi, L. Marko, R. Boese, G. Schmid, J. Organomef. Chem. 288 (1985) 179; C. F. Campana, A. Vizi-Orosz, G . Palyi, L. Marko, L. F. Dahl, fnorg. Chem. 18 (1979) 3054.
1751 a) K. H. Whitmire, C. B. Lagrone, M. R. Churchill, J. C. Fettinger, L. V. Biondi, Inorg. Chem. 23 (1984) 4227; b) M. R. Churchill, J. C. Fettin- ger, K. H. Whitmire, J . Organornet. Chem. 284 (1985) 13.
[76] W. A. Herrmann, B. Koumbouris, T. Zahn, M. L. Ziegler, Angew. Chem. 96 (1984) 802: Angew. Chem. Inf. Ed. Engl. 23 (1984) 812.
[77] W. A. Herrmann, B. Koumbouris, A. SchPfer, T. Zahn, M. L. Ziegler. Chem. Ber. 118 (1985) 2472.
[78] Originalarbeiten zu dieser Thematik: a) G. Huttner, J. Borm. L. Zsol- nai, J. Organomel. Chem. 263 (1984) C33, zit. Lit.; b) G. Huttner, H.-D. Miiller. A. Frank, H. Lorenz, Angew. Chem. 87 (1975) 714; Angew. Chem. Inf. Ed. Engl. 14 (1975) 705; c) B. Sigwarth, L. Zsolnai, 0. Scheidsteger. G. Huttner, J. Organomel. Chem. 235 (1982) 43, zit. Lit.
[79] a) B. Sigwarth, 1. Zsolnai, H. Berke. G. Huttner. J. Organomel. Chem. 226 (1982) CS; b) G. Huttner, U. Weber, B. Sigwarth, 0. Scheidsteger, Angew. Chem. 94 (1982) 210: Angew. Chem. Inf. Ed. Engl. 21 (1982) 41 1 ; c) G. Huttner, U. Weber, L. Zsolnai, Z. NarurjOsch. 837 (1982) 707.
[SO] G. Huttner, B. Sigwarth, 0. Scheidsteger. L. Zsolnai, 0. Orama, Orga- nometal1ic.s 4 (1985) 326; vgl. P. J. Sullivan, A. L. Rheingold, ibid. I (1982) 1547.
Chem. Int. Ed. Engl. 22 (1983) 976; Angew. Chem. Suppl. 1983. 1451.
[8l] a) A. L. Rheingold, M. J. Foley, P. J. Sullivan, J. Am. Chem. SOC. 104 (1982) 4727; b) 0. J. Scherer, J. Schwalb, G. Wolmershauser, W. Kaim, R CroO, Angew. Chem.. im Druck.
1821 C. F. Campana, L. F. Dahl, J. Organomer. Chem. 127 (1977) 209. [83] A. F. Wells: Sfrucfural Inorganic Chemistry. 4. Aufl., Clarendon Press,
1841 G. Ciani, L. Garlaschelli, A. Sironi. S. Martinengo, J. Chem. SOC.
[85] T. J. Greenhough, B. W. S. Kolthammer, P. Legzdins, J. Trotter, Inorg.
IS61 1. Y. Goh, T. W. Hambley, G. B. Robertson, J . Chem. SOC. Chem. Com-
(871 M. Herberhold, D. Reiner, B. Zimmer-Gasser, U. Schubert, Z. Nofur-
1881 W. Hieber, J. Gruber, Z. Anorg. Allg. Chem. 296 (1958) 91. 1891 M. Holler. A. Baitz, Chem. Ber. 109 (1976) 3147. 1901 C. Mealli, S. Midollini. L. Sacconi, lnorg. Chem. 17 (1978) 632. [9l] D. L. Lichtenberger, J. L. Hubbard, Inorg. Chem. 23 (1984) 2718. 1921 C. Potvin, J.-M. Manoli, J. M. Bregeault, Inorg. Chim. Acta 72 (1983)
103. 1931 S. Lincoln, S.-L. Soong, S. A. Koch, M. Sato, J. H. Enemark, Inorg.
Chem. 24 (1985) 1355. [94] J. Schiemann, P. Hilbener, E. Weiss, Angew. Chem. 95 (1983) 1021: An-
gew. Chem. Inf. Ed. Engl. 22 (1983) 980. [US] N. Albrecht. P. Hllbener, U. Behrens, E. Weiss, Chem. Ber. 118 (1985)
4059. [96] W. A. Herrmann, J. Rohrmann, A. SchBfer, J . Organomel. Chem. 265
(1984) C I . (971 W. A. Herrmann. J. Rohnndnn, H. Noth, C. K. Narula, I. Bernal, M.
Draux, J. Organomef. Chem. 284 (1985) 189; ibid. 289 (1985) C26. I981 L. Y. Goh, Ch. Wei, E. Sinn, 1. Chem. Soc. Chem. Commun. 1985.
462. 1991 W. A. Herrmann, J. Rohrmann, M. L. Ziegler, T. Zahn, J. Organomef.
Chem. 273 (1984) 221. [lo01 W. A. Herrmann, C. Hecht, M. L. Ziegler, B. Balbach, J. Chem. Soc.
Chem. Commun. 1984. 686. [loll M. Herberhold, D. Reiner, D. Neugebauer. Angew. Chem. 95 (1983) 46;
Angew. Chem. Int. Ed. Engl. 22 (1983) 59; Angew. Chem. Suppl. 1983. 20.
[I021 W. A. Herrmann, C. Bauer, J. Weichmann, J. Organomef. Chem. 243 (1983) C21.
[I031 Zusammenfassungen: a) W. A. Herrmann, Angew. Chem. 90 (1978) 855; Angew. Chem. f n t . Ed. Engl. 17 (1978) 800: b) J. Organomef. Chem. 250(1983) 319; c) Adu. Organomer. Chem. 2U( 1982) 159; d) Pure Appl. Chem. 54 (1982) 65; e) W. A. Herrmann in B. L. Shapiro (Hrsg.): Organometallic Compounds - Synfhesir. Structure. and Theory. Texas A & M University Press, College Station, TX, USA 1983, S. 383N.; f) J. E. Hahn, Progr. Inorg. Chem. 31 (1984) 205.
11041 W. A. Herrmann, C. Hecht, M. L. Ziegler, T. Zahn, J. Organornet. Chem. 273 (1984) 323.
[I051 W. A. Herrmann, J. Rohrmann, unveroffentlicht. [ I061 W. A. Hemnann, J. Rohrmann, C. Hecht. J. Organornet. Chrm. 290
[I071 W. A. Herrmann, C. Hecht, E. Herdtweck, unver8ffentlicht. [I081 W. A. Herrmann, J. Rohrmann, M. L. Ziegler, T. Zahn, J. Orgunomel.
[ I 0 9 1 H. Bock, A. Veltmann, unverBNentlicht; A. Veltmann, Diplomarbeit.
[I101 N. E. Murr, E. Laviron. Tetrahedron Leff. 1975. 875. [ I 1 I] W. A. Hemnann, J. Rohrmann, unveroffentlicht. - Das Radikalanion
von 60 wurde ESR-spektroskopisch charakterisiert (H. Bock et al., un- veraffentlicht).
11121 W. A. Herrmann, J. Rohrmann, M. L. Ziegler, G. Sergeson, unverof- fentlicht.
[ 1131 W. A. Herrmann, J. Rohrmann, unveroffentlicht. [ 1141 Monographie: F. A. Cotton, R. A. Walton: Multiple Bonds Between Me-
tal Atoms. Wiley, New York 1982; siehe auch den Aufsatz von M. H. Chisholm in diesem Heft: M. H. Chisholm, Angew. Chem. 98 (1986) 21; Angew. Chem. Inf. Ed. Engl. 25 (1986) Nr. I .
11151 a) R C. Haushalter, C. M. OConnor. J. P. Haushalter, A. M. Umarji, G. K. Shenoy, Angew. Chem. 96 (1984) 147; Angew. Chem. fnf. Ed. Engl. 23 (1984) 169; R. C. Haushalter, ibid. 97 (1985) 414 bzw. 24 (1985) 433; b) B. Eisenmann, H. Jordan, H. Schafer, ibid. 93 (1981) 21 I bzw. 20 (1981) 197.
Oxford 1975, S. 416f. und S. 664.
Chem. Commun. 1981. 563.
Chem. 18 (1979) 3543.
mun. 1983. 1458.
forsch. 8 3 5 (1980) 1281.
(1985) 53.
Chem. 295 (1985) 175.
Universitat Frankfurt 1984.
Angew. Chem. 98 11986) 57-77 77





























