Embed Size (px)
Citation preview

Neue Detektionsmethoden für den
Einsatz in der Hochdurchsatz-
Katalysatorforschung basierend auf dem
photoakustischen Effekt
Dissertation
zur Erlangung des Doktorgrades
der Naturwissenschaften
vorgelegt der Fakultät für Chemie
der Ruhr-Universität Bochum
von
Thorsten Johann
geboren in Mannheim
2002

Hab keine Angst vor Perfektion – du erreichst sie nie. Salvador Dali

Meiner Mutter, die all dies erst ermöglicht hat

Danksagung
Mein großer Dank gilt Herrn Prof. Dr. F. Schüth für die Bereitstellung des interessanten Themas. Die überaus kompetente Betreuung und die Möglichkeit eigene Ideen frei zu entwickeln und zu verwirklichen haben einen großen Anteil am Gelingen dieser Arbeit. Mein weiterer Dank gilt der Feinmechanischen Werkstatt I und II am Max-Planck-Institut für Kohlenforschung für alle Reaktorbauten und die Konstruktion der analytischen Arrays. Besonderer Dank gebührt hier Herrn H.-W. Schmidt und Herrn Weiler als Leiter der Werkstätten und allen Mitarbeitern insbesondere Herrn Sebastian Plankert. Für die Hilfe bei der Entwicklung der elektrischen und elektronischen Aufbauten bedanke ich mich bei Herrn Ihländer von der elektronischen Werkstatt am Max-Planck-Institut für Kohlenforschung. Für die Hilfe bei der Charakterisierung der Proben bedanke ich mich ganz herzlich bei den analytischen Abteilungen des Max-Planck Instituts, insbesondere bei Herrn Spliethoff für TEM- und EDX-Messungen, sowie Frau Dr. C. Weidenthaler für ihre Hilfe bei XRD-Fragen. Bei der hte AG möchte ich mich insbesondere bei Herrn Dr. Armin Brenner, Dr. Stephan Schunk und Dr. Torsten Zech für die fruchtbaren Diskussionen während der zahlreichen Besprechungen bedanken. Weiterhin gilt mein Dank den dortigen Werkstätten, hier insbesondere Herrn Heiko Hoffmann und Herrn Oliver Koechel. Bei Herrn Manfred Schwickardi möchte ich mich für die gute, kooperative Zusammenarbeit bei der Entwicklung der Parallelsynthese bedanken. Für zahlreiche fruchtbare Diskussionen über physikalische Hintergründe der Photoakustik danke ich Herrn Dr. Frank Marlow. Herrn Oliver Busch und Herrn Dr. W. Schmidt danke ich für das Korrekturlesen der Arbeit. Besonders bedanken möchte ich mich bei Herrn Oliver Busch und Frau Catharina Klanner für spannende abendliche Diskussionen im In- und Ausland. Meiner Familie und Yvette Schollmeier danke ich für die Geduld während der Promotion. Abschließend möchte ich mich bei meinen Kollegen am Max-Planck Institut für die phantastische Arbeitsatmosphäre, die ich sicherlich so schnell nicht wieder finden werde, bedanken: Dr. Patrik Ågren, Pablo Arnal, Holger Althues, José Bellosta von Colbe, Dr. Werner Brijoux, Oliver Busch, Dr. Patrick Bussian, Dr. Gerald Chaplais, Dr. Wenting Dong, Dr. Nathalie Dufau, Dr. David Farruseng, Manuela Germann, Dr. Christian Hoffmann, Yasemin Ilhan, Dr. Michael Janicke, Dr. Martin Kalwei, Dr. Stefan Kaskel, Harry Kestenbaum, Christoph Kiener, Catharina Klanner, Dr. Freddy Kleitz, Thomas Kruppa, Mark Kirchhoff, Dr. Anhui Lu, Dr. Armin Lange de Oliveira, Dr. Rabi Panda, Dr. Sohyun Park, Alberto Joaristi, Justus Loerke, Piotr Krawiec, Dr. Yachun Mao, Dr. Frank Marlow, Mamatha Mayanna, Inga Ritzkopf, Dr. Jürgen Sauer, Klaus Schlichte, Dr. Wolfgang Schmidt, Yvette Schollmeier, Manfred Schwickardi, Dr. Dan Shantz, Dr. Meike Thieme, Dr. Akira Taguchi, Dr. Stuart Thomson, Sascha Vukojevic, Dr. Yanquin Wang, Helga Wasilewski, Dr. Claudia Weidenthaler, Özlem Weiß, Dr. Annette Wingen, Dr. Anke Wolf, Ulrich Wüstefeld, Dr. Chia-min Yang.

Inhaltsverzeichnis
1 Einleitung ............................................................................................................................... 1
2 Motivation und Zielsetzung.................................................................................................. 3 2.1 Aktuelle Situation der Katalysatorforschung ................................................................... 3 2.2 Der Hochdurchsatz-Ansatz .............................................................................................. 4 2.3 Zielsetzung ....................................................................................................................... 7
3 Stand der Forschung............................................................................................................. 9 3.1 Kombinatorische Methoden im Rahmen der Katalysatorforschung ................................ 9
3.1.1 Hochdurchsatz-Synthese ......................................................................................... 10 3.1.2 Hochdurchsatz-Reaktorkonzepte ............................................................................ 13 3.1.3 Hochdurchsatz-Analyse .......................................................................................... 15 3.1.4 Datenmanagement ................................................................................................... 17
3.2 Photoakustischer und photothermischer Effekt.............................................................. 19 3.3 Kohlenmonoxid-Oxidation............................................................................................. 21 3.4 Oxidative Dehydrierung kurzkettiger Alkane zu Alkenen............................................. 24
4 Experimentelles ................................................................................................................... 27 4.1 Synthese ......................................................................................................................... 27 4.2 Katalyse.......................................................................................................................... 28
4.2.1 Kohlenmonoxid-Oxidation...................................................................................... 29 4.2.2 Oxidative Dehydrierung von Ethan......................................................................... 34
4.3 Charakterisierungsmethoden.......................................................................................... 39 4.3.1 Röntgenpulverdiffraktometrie (XRD)..................................................................... 39 4.3.2 Stickstoffsorption .................................................................................................... 40 4.3.3 Transmissionselektronenmikroskopie (TEM)......................................................... 41 4.3.4 Energiedispersive Analyse von Röntgenstrahlen (EDX) ........................................ 42
5 Entwicklung und Prinzip der Photoakustischen Spektroskopie..................................... 44 5.1 Einführung...................................................................................................................... 44 5.2 Geschichte ...................................................................................................................... 45 5.3 Prinzip der Photoakustik ................................................................................................ 47
5.3.1 Strahlungsquellen .................................................................................................... 47 5.3.2 Licht-Materie-Interaktion........................................................................................ 52 5.3.3 Photoakustische Effekte in gasförmigen Medien.................................................... 55
5.3.3.1 Grundlegende Gleichungen.............................................................................. 56 5.3.3.2 Zelldesign eines photoakustischen Aufbaus .................................................... 57
5.3.4 Photoakustische Effekte in nichtgasförmigen Medien............................................ 61
6 Entwicklung der photoakustischen Analytiken................................................................ 63 6.1 Mikrofontest ................................................................................................................... 63 6.2 Lasersystem.................................................................................................................... 68 6.3 Freifeld-Aufbau.............................................................................................................. 70
6.3.1 Modellsystem .......................................................................................................... 71 6.3.2 Aufbau..................................................................................................................... 71 6.3.3 Automatisierung ...................................................................................................... 73 6.3.4 Parallelisierung........................................................................................................ 75 6.3.5 Evaluierung der Analytik ........................................................................................ 77 6.3.6 Zusammenfassung und Diskussion ......................................................................... 79
6.4 Resonanter Aufbau......................................................................................................... 80

6.4.1 Modellsystem .......................................................................................................... 80 6.4.2 Resonatorentwicklung............................................................................................. 81
6.4.2.1 Erste photoakustische Messungen.................................................................... 81 6.4.2.2 Resonanzrohr-Konzept..................................................................................... 85 6.4.2.3 Optimierung der Resonanzröhre ...................................................................... 88
6.4.3 Parallelisierung........................................................................................................ 92 6.4.4 Automatisierung ...................................................................................................... 95 6.4.5 Evaluierung der Analytik ........................................................................................ 99 6.4.6 Zusammenfassung und Diskussion ....................................................................... 101
7 Weiterentwicklung der photoakustischen Analytik....................................................... 102 7.1 Motivation .................................................................................................................... 102 7.2 Experimente mit kleineren Flüssen .............................................................................. 103 7.3 Experimente mit geringerer Laserleistung ................................................................... 110 7.4 Zusammenfassung und Diskussion .............................................................................. 111
8 Parallele Feststoffsynthese................................................................................................ 113 8.1 Materialien mit hoher Oberfläche ................................................................................ 113 8.2 Aktivkohlen als Exotemplate ....................................................................................... 114 8.3 Automatisierung und Parallelisierung der Präparation ................................................ 115 8.4 Selektion der Katalysatorzusammensetzung................................................................ 117 8.5 Materialien und Charakterisierung............................................................................... 118 8.6 Gesamtprozess und Diskussion.................................................................................... 124
9 Katalytische Tests.............................................................................................................. 125 9.1 Kohlenmonoxid-Oxidation........................................................................................... 125 9.2 Oxidative Dehydrierung von Ethan.............................................................................. 127
10 Zusammenfassung und Ausblick ................................................................................... 130
11 Anhang ............................................................................................................................. 135

1 Einleitung
Nichts auf der Welt ist so kraftvoll wie eine Idee, deren Zeit gekommen ist.
Victor Hugo
In den letzten Jahren fand in der Pharmaindustrie eine rasante Entwicklung durch den Einsatz
kombinatorischer Methoden statt. Durch diese neuen Ansätze wurde ein gesamter
Industriezweig revolutioniert. Herkömmliche traditionell sequenzielle Forschung wurde durch
kombinatorisch parallele Forschung ergänzt. Teststände und Syntheserouten wurden
zusätzlich automatisiert. Heute werden in fast menschenleeren Labors tausende Proben
innerhalb kürzester Zeit hergestellt, getestet und ausgewertet. Dies geschieht 24 Stunden am
Tag, 7 Tage pro Woche, 365 Tage im Jahr.
Es stellt sich die Frage, was geschieht, wenn wir diese Techniken auch auf anderen Gebieten
der Chemie einsetzen? Zum Beispiel in der heterogenen Katalyse?
Die gleiche Entwicklung?
In der Chemie ist die Suche nach aktiven Feststoffkatalysatoren nach wie vor häufig eine
„Trial and Error“ Prozedur. Obwohl in der theoretischen Chemie große Fortschritte erzielt
wurden, ist es noch nicht möglich, ab-initio einen aktiven Feststoffkatalysator für den Einsatz
in der heterogenen Katalyse theoretisch vorherzusagen. Aller Wahrscheinlichkeit nach wird
dies auch in nächster Zukunft nicht möglich sein. Um Informationen zu gewinnen und tiefere
Einblicke in die Wirkungsweise der Katalysatoren zu erhalten, kommt es darauf an,
wirkungsvolle Materialien zu finden, welche uns bisher Unbekanntes zeigen können. Dies
geschieht meist nicht durch logisches Vorgehen und den Einsatz chemischen Wissens. Oft
erweist sich eine zufällige, völlig inaktiv geglaubte Mischung als erstaunlich aktiv. Dies sind
die Augenblicke, in denen ein neuer Katalysator gefunden wird, der auf eine Weise arbeitet,
die der Wissenschaft vorher noch nicht bekannt war. Hier können wir Neues lernen und sind
vielleicht einen Schritt weiter im Verständnis der Funktion eines Katalysators.
1

1 Einleitung
Ein schnelleres Testen erlaubt uns eine breit angelegte Suche, um auch solche Substanzen,
welche durch rein wissenschaftliches Vorgehen wahrscheinlich ausgeschlossen würden, zu
testen. Wenn die Zeit aufgrund hoher Testgeschwindigkeit nur noch eine untergeordnete
Rolle spielt, können wir uns ein solches „unlogisches“ Vorgehen erlauben. Vielleicht können
wir dann Unerwartetes finden und unseren Wissensstand vergrößern. Möglicherweise wird
dann in Zukunft irgendwann einmal ein zielgerichtetes Suchen auf einem Niveau möglich,
welches ein schnelles Testen überflüssig macht.
Dies ist im Augenblick und in absehbarer Zukunft noch als Utopie zu bezeichnen. Noch
können auf diesem Weg keine Materialien verlässlich gefunden werden.
Schon in den letzten Jahren fand die Hochdurchsatz-Testung zunehmende Akzeptanz als
geeignete Strategie, um die Entdeckung von neuen Feststoffkatalysatoren zu beschleunigen.
Auch in den nächsten Jahren wird bei der Suche nach Katalysatoren verstärkt auf ein
schnelles Testen wert gelegt werden. Obwohl bereits von interessanten Entwicklungen zur
Produktanalyse berichtet wurde, besteht unter anderem noch dringender Bedarf an schnellen
und parallelen Analysetechniken1,2,3,4. Des weiteren stellt die Integration automatisierter
Synthesen von Katalysatoren in den Gesamtprozess ein Problem dar. Dies ist verständlich, da
es sich bei den meisten Präparationstechniken um einen Technologietransfer aus der
pharmazeutischen Forschung handelt, welche vor allem mit Flüssigkeiten arbeitet. Bei der
Suche nach Feststoffkatalysatoren und generell bei der Feststoffsynthese sind jedoch meist
mehrere Aggregatzustände mit einer großen Vielfalt physikalischer Parameter involviert. Die
meisten automatisch arbeitenden Roboter sind hierfür nicht geeignet und können nur eine
kleine Bandbreite von Substanzen mit speziellen physikalischen Eigenschaften verarbeiten.
Um diese Hindernisse zu umgehen, gilt es, neue Wege zu finden, um bestehende Techniken
in automatisierte Synthesen zu integrieren oder neue Synthesen zu entwickeln, welche mit
bestehenden Techniken durchgeführt werden können. Ebenso müssen auf den technischen
Gebieten Entwicklungen erfolgen, mit deren Hilfe Feststoffsynthesen problemlos
durchgeführt werden können.
Gegenstand der vorliegenden Arbeit ist es, Beiträge zur Lösung dieser Probleme zu leisten.
Sowohl schnellere analytische Methoden als auch geeignete Präparationstechniken werden
entwickelt, beschrieben und ihr Nutzen in Bezug auf einen potenziellen Einsatz in der
Hochdurchsatz-Forschung erläutert.
2

2 Motivation und Zielsetzung
Auf viele der durch Forschung und Technik erzeugten Probleme gibt es keine
andere Antwort als durch neue Forschung und bessere Technik.
Hubert Markl
2.1 Aktuelle Situation der Katalysatorforschung
Feststoffkatalysatoren werden in 80-90 % aller im industriellen Maßstab durchgeführten
katalytischen Prozesse benötigt und haben damit eine Schlüsselstellung in Bezug auf
Wirtschaftlichkeit und Umweltfreundlichkeit in der chemischen Industrie. Die Umsätze der
Katalysatorindustrie liegen jährlich zur Zeit bei etwa 8-10 Milliarden Dollar. Ein Vergleich
von Katalysatorkosten und dem Wert der mit den Katalysatoren dargestellten Materialien
zeigt, dass der Wert des Katalysators meist mehrere Größenordnungen unter der mit ihm
erzielten Wertschöpfung liegt5,6.
Man kann geteilter Meinung sein und Bedenken gegenüber dem „sinnlosen Testen“ von
„irrwitzig zusammengesetzten“ Materialien aufgrund gesteigerter Testkapazität haben.
Tatsächlich sollte der Hochdurchsatz-Ansatz nicht als losgelöstes eigenständiges Vorgehen
gesehen werden. Die Aufklärung von Reaktionsmechanismen, die Nutzung von Erkenntnissen
aus erhaltenen Daten und weitere systematische Planung sind wichtige Bestandteile der
gesamten Katalysatorentwicklung. Eine wirkungsvolle Symbiose von gesteigerter
Testkapazität, Verständnis der Funktion eines Materials und hieraus resultierendem
strategischem Vorgehen ist das Gesamtbild, in dessen Zusammenhang die Hochdurchsatz-
Forschung gesehen werden sollte.
Es liegt auf der Hand, dass schon ein einziger neu aufgefundener Katalysator, welcher nach
einem neuen Wirkungsmechanismus arbeitet, die mechanistischen Studien weit voranbringt.
Diese Materialien lassen sich schwer mit bisher bekanntem Wissen vorhersagen. Hier zahlt
sich eine hohe Testkapazität aus und ein scheinbar „überflüssiger, sinnloser Test“ bringt
vielleicht wertvolle Ergebnisse hervor. Es ist klar, dass wir keinesfalls zur Zeit schon all das
3

2 Motivation und Zielsetzung
Wissen besitzen, um genau zu verstehen, wie eine katalysierte Reaktion an einer Grenzschicht
letztendlich abläuft.
Es liegt nun bei den Forschern auf dem Gebiet der Hochdurchsatz-Testung zu zeigen, dass ein
schnelles, paralleles Testen nicht nur die Generierung einer immensen Datenflut zur Folge
hat, sondern diese Daten auch wirkungsvoll für die Untersuchung und das Verständnis der
Arbeitsweise eines Katalysators verwendet werden können. Eventuell werden hierdurch sogar
Voraussagen ermöglicht. Letztgenanntes stellt den Ansatz für eine erfolgreiche Modellierung
von Feststoffsystemen für die Katalyse dar. Wirkungsvolle, den aktiven Katalysator
beschreibende Parameter, welche auf der Basis der katalytischen Tests gefunden werden
könnten, würden dann in den Vordergrund treten. Diese Parameter sind zur Zeit trotz
intensiver Bemühungen und unterschiedlicher Ansätze erst in Ansätzen bekannt und es bleibt
abzuwarten, ob diese Ansätze mittels der Hochdurchsatz-Methoden erweitert werden können.
2.2 Der Hochdurchsatz-Ansatz
Der Gesamtprozess der Hochdurchsatz-Katalysatorforschung besteht aus mehreren,
miteinander eng verbundenen Teilbereichen.
Automatisierungstechnologien
SyntheseTestung
Datenmanagement
Abbildung 1 Schematische Darstellung des Hochdurchsatz-Prozesses. Alle einzelnen Bereiche sind für
den Gesamtprozess wichtig und im besten Falle automatisierbar.
Erst wenn alle Teile, die in Abbildung 1 dargestellt sind, richtig und mit gleicher
Geschwindigkeit zusammenarbeiten, ist ein effizienter Gesamtprozess möglich. Die
zeitlimitierenden Faktoren sind von Fall zu Fall unterschiedlich. Generell ist es nicht einfach
4

2.2 Der Hochdurchsatz-Ansatz
möglich, die Forschung zu beschleunigen, indem man größere eventuell gebündelte oder
parallele Reaktoren konstruiert. Die Synthese der benötigten Materialien ist ein ebenso nicht
zu vernachlässigender Punkt. Da die Präparation der meisten Materialien nach wie vor
manuelle Arbeit bedeutet, ist dies ein nicht zu unterschätzender Engpass und Ansatzstelle
vieler Entwicklungsanstrengungen. Zur Zeit ist es aufgrund ungenügender Methoden
Feststoffe automatisiert zu verarbeiten nicht oder nur mit sehr großem Kostenaufwand
möglich, diesen Prozess durch eine hochentwickelte Robotik erledigen zu lassen. Es müssen
sowohl neue automatisierbare Syntheseroutinen als auch eine entsprechend problemorientierte
Technologie entwickelt werden, um diese Arbeit zukünftig sowohl parallel als auch
automatisiert durchzuführen.
Die Datenauswertunng, die dritte Komponente des Hochdurchsatz-Prozesses, könnte
vielleicht einmal zum wichtigsten Punkt im Gesamtprozess werden. Hier liegt die
Schlüsselstelle der Hochdurchsatz-Forschung. Es wird versucht, durch geschickte
Verknüpfungen in den immensen Datenmengen Parallelen zu finden, welche Materialien sich
im speziellen Fall für eine Reaktion als Katalysator eignen. Hierüber könnten vielleicht noch
unbekannte physikalische oder präparative Faktoren isoliert und unschätzbar wertvolle
Informationen für die theoretische Betrachtung der Katalyse gewonnen werden. Die
Entwicklung auf diesem Gebiet steckt allerdings noch in der Anfangsphase. Hier ist zukünftig
die interdisziplinäre Zusammenarbeit von Forschern vieler Fachbereiche wie zum Beispiel der
Chemie, der Informatik und der Mathematik notwendig, um eine effektive Lösung zu
entwickeln.
Der Entwicklungsprozess eines industriell nutzbaren Katalysators kann im Rahmen der
Hochdurchsatz-Forschung in mehrere separate Stufen eingeteilt werden:
Phase I
Diese Stufe wird auch als „Primary Screening“ bezeichnet und umschreibt ein Vorgehen, bei
welchem mit sehr hohem Probendurchsatz gearbeitet wird. Aufgrund der hohen Testrate ist
man gezwungen, mit einem Minimum an Daten zu arbeiten. Der durch den hohen
Probendurchsatz bedingte geringe Informationsgehalt der Einzelmessung muss ausreichend
Erkenntnis liefern, um einen erfolgreichen Kandidaten eindeutig zu identifizieren. Den
Informationsgehalt der Einzelmessung zu erhöhen und gleichzeitig die Testrate konstant zu
halten oder zu vergrößern ist hierbei einer der Schwerpunkte der Entwicklungsanstrengungen.
Um in späteren Untersuchungsstadien die Anzahl der negativ getesteten Kandidaten gering zu
5

2 Motivation und Zielsetzung
halten, gilt es, schon in Phase I, soweit möglich, das Material unter später realen
Einsatzbedingungen zu testen. Hier besteht noch großer Entwicklungsbedarf, da die
bisherigen Testverfahren meist weit entfernt von diesen Reaktionsbedingungen arbeiten
müssen. Häufig ist man zur Zeit noch darauf angewiesen, die Reaktionsbedingungen an die
im Rahmen der eingesetzten Analytik möglichen Parameter anzupassen. In Phase I werden
lediglich die wenigen, Erfolg versprechenden Materialien analytisch exakt untersucht, was
aufgrund des hohen Durchsatzes von bis zu einigen Tausend Proben am Tag für alle nicht
möglich wäre.
Phase II
Die als „Secondary Screening“ bezeichnete Phase II soll die auf der vorherigen Stufe positiv
getesteten Katalysatoren detaillierter untersuchen. Charakteristisch für diese Stufe ist eine
größere Katalysatormenge, welche unter Bedingungen getestet werden kann, die schon fast
den technischen Bedingungen entsprechen. In dieser Phase wird meist ein genaueres
Analyseverfahren herangezogen und die Materialien längere Zeit getestet, um ihr
Aktivierungs- und Desaktivierungsverhalten zu ergründen. Da der Zeitbedarf pro getestetem
Material auf dieser Stufe erheblich ansteigt, zeigen sich gerade hier die Vorteile des parallelen
Arbeitens. Parallelreaktoren konnten in dieser Phase bisher erfolgreich eingesetzt und die
Testzeit entsprechend dem Parallelisierungsfaktor reduziert werden. Gewöhnlich liegt der
Parallelisierungsgrad auf dieser Stufe bei bis zu 100. Aufgrund der größeren Substanzmenge,
welche bei bis zu 100 mg liegen kann, ist es auf dieser Stufe möglich, detaillierte
Informationen über Morphologie und weitere Beschaffenheiten des Katalysators zu erhalten.
Um auf dieser Stufe den Katalysator zu optimieren, werden kleine systematische Variationen
der Zusammensetzung durchgeführt und hierdurch der Kandidat für spätere Einsätze in
Großanlagen ermittelt.
Auch wenn die Unterteilung in zwei Phasen sinnvoll ist, existieren auch Methoden welche
nicht eindeutig einer dieser beiden Klassen zugeordnet werden können und schon mit
geringen Substanzmengen erstaunliche Ergebnisse liefern. Eine genaue Beschreibung der
einzelnen Methoden wird in Kapitel 3.1 gegeben.
6

2.3 Zielsetzung
2.3 Zielsetzung
Im Rahmen dieser Arbeit sollten neue analytische Techniken für die Hochdurchsatz-
Katalysatorforschung entwickelt werden. Ziel sollte es nicht nur sein, die Katalysatortestung
zu beschleunigen, sondern auch eine Methode zu etablieren, mit deren Hilfe on-line Echtzeit-
Messungen der Produktzusammensetzungen der katalytischen Umsätze möglich sind. Eines
der wichtigsten Ziele war hierbei die Vereinigung eines parallelen Reaktorkonzeptes mit der
Möglichkeit on-line Analysen durchzuführen. Der Vorteil einer solchen Methode ist, dass sie
längere Testzeiten durch eine parallele Arbeitsweise ermöglicht. Hierbei besteht die
Möglichkeit parallel Informationen über Anfahrverhalten der Katalysatoren zu erhalten, was
einen tieferen Einblick in deren Wirkungsmechanismen erlaubt. Gleichzeitig ist die
Vergleichbarkeit der einzelnen Materialien durch die lückenlose, parallele Datenakkumulation
gewährleistet.
Bei kurzen Testzeiten, welche aufgrund des hohen Durchsatzes notwendig sind, werden
eventuell Desaktivierungsprozesse nicht mehr gemessen. Durch eine parallele
Analysemethode können die Testzeiten länger gewählt werden, wodurch auf einer frühen
Stufe der Testung bereits Materialien ausgeschlossen werden können, welche durch
Desaktivierung nach kurzer Zeit inaktiv geworden sind.
Ferner ist es möglich erst später aktive Komponenten zu finden, welche sonst ausgeschlossen
werden. Dies ist wichtig, da manche Materialien, bevor sie katalytisch aktiv werden, erst eine
Formierungsphase durchlaufen und somit erst nach längeren Testzeiten gefunden werden
können.
Eine viel versprechende Methode, welche diese Problemstellung lösen könnte, ist die
Photoakustik. Sie kann die Vorteile sowohl des schnellen als auch des parallelen Testens
vereinen. Als optisches Verfahren ist sie in der Lage viele Analysen parallel durchzuführen
und würde daher eine simultane Datenakkumulation ermöglichen.
Weiteres Ziel dieser Arbeit war es, diese Analytik in einem realen Testprogramm einzusetzen
und neue aktive Materialien in diesen Tests zu finden. Die entwickelte Analysenmethode
sollte hierdurch evaluiert werden und ihre Brauchbarkeit unter realen Einsatzbedingungen
zeigen. Hierfür musste eine Beschleunigung der gesamten Testroutine entwickelt werden, da
mit Hilfe der schnellen Analytik auch eine schnellere Präparation der Testmaterialien nötig
wurde. Während des gesamten Entwicklungsprozesses wurde also immer im Hinblick auf
einen späteren Einsatz in einer automatisierten Hochdurchsatz-Anlage gearbeitet. Daher
7

2 Motivation und Zielsetzung
wurde eine ebenfalls automatisierbare Präparationsroutine zur Entwicklung der
photoakustischen Detektion etabliert.
8

3 Stand der Forschung
Wissen heißt wissen, wo es geschrieben steht.
Albert Einstein
3.1 Kombinatorische Methoden im Rahmen der Katalysatorforschung
Seit Ende der 80er Jahre zeichnet sich vor allem in der pharmazeutischen Wirkstoffforschung
ein Trend ab, der allgemein als „Kombinatorische Chemie“ bezeichnet wird. Durch den
Druck, neue Wirkstoffe in immer kürzerer Zeit finden zu müssen, um den ökonomischen
Anforderungen des Marktes standzuhalten, war die pharmazeutische Forschung gezwungen,
von althergebrachten Strategien abzuweichen. Dies bewirkte einen Paradigmenwechsel im
gesamten forschungsstrategischen Denken. Die lange Zeit von ca. 15 Jahren bis zur
Marktreife eines Wirkstoffes erhöhte den innovativen Druck auf einem Gebiet, dessen größte
Erfolge hauptsächlich auf zufälligen Entdeckungen beruhen.
Viele pharmazeutischen Wirk-Substanzen wurden und werden noch heute nicht durch ein
rationales Design entdeckt. Die theoretische Chemie ist noch immer weit von einem
zuverlässigen Modell entfernt, um aktive Substanzen maßschneidern zu können. Um den
großen Parameterräumen Herr zu werden, versuchte man, durch eine Steigerung der Anzahl
der durchgeführten Experimente einen Vorteil zu erlangen. Dies resultierte in einer
Parallelisierung der Experimente in miniaturisierter Ausführung und einer weitgehenden
Automatisierung der Routineprozeduren. Heute beginnen auch Forscher auf anderen
Gebieten, wie zum Beispiel der Materialforschung, sich dieser Ansätze zu bedienen. Bisher
konnte hierbei weitgehend eine Entwicklung parallel zu der in der kombinatorischen Chemie
beobachtet werden.
Die Verwendung des Begriffes „Kombinatorik“ ist zur Zeit noch etwas verwirrend. Auf dem
Gebiet der Materialforschung wurde er von Xiang et al. 1995 eingeführt. Es wurden
unterschiedliche Ausgangsmaterialien, sogenannte Bausteine verwendet, welche durch
unterschiedliche Kombination zu neuen Materialien führten7. Diese Arbeitsweise entspricht
der klassischen Definition des Begriffes „kombinatorisch“, der für die Verknüpfung aus
9

3 Stand der Forschung
mehreren definierten Fragmenten benutzt wird, welche unterschiedlich kombiniert werden.
Diese grundlegenden Arbeiten gaben der „Kombinatorischen Materialforschung“ ihren
Namen. Heute wird dieser Begriff teilweise noch für ein Vorgehen benutzt, welches richtiger
als „Hochdurchsatz-“ oder „Parallel-Verfahren“ bezeichnet werden sollte. Er beschreibt hier
lediglich die Diversität, also die Verschiedenheit der Substanzen einer Probensammlung.
Solche auch Bibliotheken genannte Probenkollektionen von unterschiedlichen Materialien
können auch durch klassisch kombinatorische Methoden generiert werden, es ist jedoch keine
notwendige Bedingung.
Eines der Gebiete, in denen die kombinatorische Materialforschung bisher bereits große
Erfolge erzielen konnte, ist die Katalysatorforschung. Im folgenden sollen daher nun vor
allem Entwicklungen der kombinatorischen Materialforschung im Rahmen der
Hochdurchsatz-Katalysatorforschung beschrieben werden.
Man unterscheidet heutzutage mehrere Stufen der Entwicklung eines für katalytische
Anwendungen aktiven Materials. In Kapitel 2.2 wurden diese Phasen bereits erläutert. In
frühen Entwicklungsstadien werden bei sehr hohen Parallelisierungsgraden wertvolle
Informationen über die Materialien erhalten. Häufig kommen hier Materialbibliotheken von
mehreren Tausend Einzelsubstanzen zum Einsatz, welche zum Beispiel auf Wafern
aufgebracht werden. Die Analysemethoden sind meist optischer Natur, da bisher nur bei
diesen Methoden eine ausreichende Parallelisierung möglich ist. In späteren Stadien arbeitet
man meist mit größeren Katalysatormengen im Milligramm- bis Grammbereich und führt die
Experimente mit wesentlich geringeren Parallelisierungsgraden durch.
Im späteren Verlauf wird in der vorliegenden Arbeit die „Kombinatorische
Materialforschung“ in der heterogenen Katalyse logisch in die Bereiche Synthese,
Reaktionsführung, Analyse und Datenmanagement getrennt.
Soweit möglich, wird der Begriff „Hochdurchsatz“ verwendet, um jede Verwechslung mit der
klassischen „Kombinatorik“ auszuschließen.
3.1.1 Hochdurchsatz-Synthese
Bei den Hochdurchsatz-Synthesen muss man zwischen Molekül- und Feststoffsynthese
unterscheiden. Die Entwicklungen in der Hochdurchsatz-Synthese sind hierbei häufig nicht
universell einsetzbar, sondern auf einen dieser beiden Synthesebereiche beschränkt. Im
folgenden werden vor allem Entwicklungen auf dem Gebiet der Feststoffsynthesen
beschrieben.
10

3.1 Kombinatorische Methoden im Rahmen der Katalysatorforschung
11
1970 stellte Hanak durch die physikalische Technik des Co-Sputterns die wohl erste
kombinatorische Materialbibliothek her8. Kombinationen von bis zu drei unterschiedlichen
Substanzen wurden auf ihre Eignung als Supraleiter geprüft. Schon damals wurde bei binären
Systemen eine um den Faktor 30 erhöhte Effizienz im Vergleich zur klassischen
sequenziellen Vorgehensweise erzielt. Das Potenzial dieser Methode wurde jedoch noch nicht
erkannt und war aufgrund mangelnder zusätzlicher technischer Komponenten, wie zum
Beispiel Automatisierungsmethoden, noch unzureichend, da ein sinnvoller Gesamtprozess
noch nicht zu etablieren war.
Die Entwicklung der Split/Pool-Methode 1988 von Furka im Rahmen der Molekülsynthesen
wird als Durchbruch der kombinatorischen Vorgehensweise betrachtet9,10. Die vielseitige, voll
kombinatorische Methode ermöglichte es, eine Vielzahl von unterschiedlichen Substanzen in
kurzer Zeit herzustellen. Zuerst war diese Methode auf die Synthese von Molekülen
beschränkt. Im Laufe der Jahre wurde diese Methode jedoch sukzessive entwickelt und
erweitert, so dass, wie später berichtet wird, diese Methode heutzutage auch für die Synthese
von Feststoffen herangezogen werden kann.
Die Entwicklung der Split/Pool-Methodik wurde in Ansätzen bereits 1985 von Houghton
beschrieben, der eine Modifizierung und Parallelisierung der Merrifield-Festphasensynthese
von Peptiden durch Split/Pool-Ansätze11 vorstellte. Da hierbei große Produktmengen anfallen,
gestaltet sich deren spätere Identifizierung teilweise problematisch. Nicolaou et al.12 und
Mornan et al.13 beschrieben 1995 beide eine auf Radiofrequenzmarkern basierende Methode,
um die bei der Synthese anfallenden Produkte zu identifizieren. Alternativ zu diesem
Verfahren existiert auch eine Reihe weiterer paralleler Techniken, wie zum Beispiel die
Maskentechnik von Fodor et al.14 oder von Geysen et al. die Multi-Pin Methode15. Bei diesen
Methoden wird das Material mit einem räumlichen Parameter versehen, welcher ein späteres
Auffinden bzw. eine Analyse der Einzelsubstanz innerhalb der Bibliothek erlaubt und eine
Markierung der Produkte im Vorfeld überflüssig macht.
2002 wurde erstmals der Einsatz der echt kombinatorischen Split/Pool-Synthesen im Rahmen
der Feststoffsynthese beschrieben16. Seit kurzem wird diese Synthesetechnik daher auch
industriell von der hte AG in Heidelberg als Standardprozedur zur
Feststoffkatalysatorsynthese eingesetzt. Der Transfer dieser für Moleküle geeigneten
Methodik in die Materialsynthese ermöglicht es, Materialien nicht nur durch physikalische,
sondern auch chemische Methoden sowohl in Fest- als auch Flüssigphase zu synthetisieren.
Man interessiert sich hierbei erst nach erfolgreich abgeschlossenem Test für die eigentliche
Zusammensetzung der Materialien, welche in Form kleiner Kügelchen komplett automatisiert

3 Stand der Forschung
als Voll- oder auch Schalenkatalysator synthetisiert werden können. Durch die drastische
Reduktion der Zahl der einzelnen Syntheseschritte bei dieser Methode, ist eine hohe
Effizienzsteigerung möglich. Meist werden jedoch bei der Feststoffsynthese, wie es sich
mittlerweile auch in der Pharmaforschung durchgesetzt hat, mit Parallelverfahren räumlich
getrennte und dadurch identifizierte Materialien hergestellt.
Eine eindrucksvolle Synthese wurde 1997 von Sun et al. durchgeführt17. Auf der Suche nach
neuen Phosphoren auf der Basis von Metalloxiden wurde hierbei eine Synthese mit Hilfe
eines Tintenstrahldruckers durchgeführt, wobei die einzelnen Druckköpfe mit den
unterschiedlichen Precursorlösungen gefüllt waren.
SyntheseplanungSyntheseplanung ParalleleSynthese
ParalleleSynthese
Langsam,niedriger Durchsatz
Räumlich adressierbar, mittlerer bis hoher
Durchsatz
Split/PoolSehr hoher Durchsatz
Langsam,niedriger Durchsatz
Räumlich adressierbar, mittlerer bis hoher
Durchsatz
Split/PoolSehr hoher Durchsatz
TraditionelleSynthese
TraditionelleSynthese
Split/PoolSynthese
Split/PoolSynthese
Langsam,bis sehr schnell,
sehr genau
Schnell,relativ genau
Sehr schnell,Entschlüsselungsstrategien
notwendig
Langsam,bis sehr schnell,
sehr genau
Schnell,relativ genau
Sehr schnell,Entschlüsselungsstrategien
notwendig
Präparative Methoden Screening-Methoden
Abbildung 2 Vergleich zwischen traditionellen, parallelen und Split/Pool-Verfahren18.
Ein Vergleich der unterschiedlichen Synthesen führt zu der in Abbildung 2 gezeigten
sinnvollen Unterteilung. Für die leistungsstärkste Variante unter den unterschiedlichen
Methoden, die Split/Pool-Synthese, existieren zur Zeit noch keine Publikationen zu
erfolgreich dargestellten Materialien. Die generierten Bibliotheken weisen hier keine räumlich
getrennten einzelnen Verbindungen auf. Man benötigt demnach spezielle
Entschlüsselungstechniken, welche meist auf einer nachträglichen spektroskopischen Analyse
beruhen.
Die räumliche Trennung bei parallel synthetisierten Materialien ermöglicht, da die Produkte
hier leicht zugeordnet werden können, im Vergleich zu Split/Pool-Synthesen eine einfache
Zuordnung der Produkte. Weiter ist, abhängig vom Syntheseaufwand, mit parallelen
Methoden noch immer ein mittlerer bis hoher Durchsatz zu erreichen. Reddington et al.
12

3.1 Kombinatorische Methoden im Rahmen der Katalysatorforschung
13
verwendete zum Beispiel die oben erwähnte Tintenstrahl-Variante, um Pt-Ru-Os-Ir-
Bibliotheken für die elektrochemische Methanol-Reformierung zu synthetisieren2. Es wurden
Bibliotheken von 645 verschiedenen Einzelmaterialien generiert, indem verschiedene
Metallsalzlösungen verwendet wurden.
Holzwarth et al. stellten erstmals eine Hochdurchsatz Synthese vor, die auf Sol-Gel-Chemie
basiert4. In den Vertiefungen einer Schieferplatte wurden über sukzessive Pipettierschritte der
einzelnen Sole amorphe mikroporöse Mischoxide hergestellt. Cong et al. führten eine
ähnliche Synthese auf modifizierten Quarzwafern durch und kamen auf eine
Bibliothekengröße von 144 Einzelmaterialien, bestehend aus Einzelelementen
unterschiedlicher Mo/V/Nb/O-Verhältnisse19.
Hoffmann et al. verwendeten einen modifizierten Pipettierroboter, der in der Lage war, Co-
und Auffällungsreaktionen durchzuführen20. Die Modifizierung bestand in speziell hierfür
konstruierten Schüttel- und Filtermodulen, wodurch Suspensionen und teilweise Feststoffe
zuverlässig gehandhabt werden konnten. Es wurde gezeigt, dass die Reproduzierbarkeit dieses
Systems der konventionellen manuellen Synthese mindestens ebenbürtig, wenn nicht sogar
überlegen ist. Durch diese Technik, die auch von Simons et al. angewandt wird, können
Materialien in größerem Maßstab im Schüttgut-Format erhalten werden, wodurch ein Einsatz
in Reaktoren, die nahe an technischen Bedingungen arbeiten, ermöglicht wird21.
Rodemerck et al. beschreiben eine Incipient-Wetness-Methode, wobei Schüttgut-Bibliotheken
durch die Benetzung von vorgelegten Trägermaterialien mit Hilfe eines Pipettierroboters
generiert werden22,23.
Auch zeolithische Materialien wurden durch Hochdurchsatz-Synthesen hergestellt24,25. Die
Hydrothermalsynthese wird in parallelen Autoklavenblöcken durchgeführt. Es werden sowohl
Schüttgut, als auch auf Wafer aufgebrachte Materialien auf diese Weise erhalten.
3.1.2 Hochdurchsatz-Reaktorkonzepte
Viele katalytischen Reaktionen laufen nicht über einen einzigen Elementarschritt ab. Häufig
sind mehrere, teilweise komplexe Reaktionsschritte miteinander gekoppelt. Um in solchen
Systemen zuverlässige Aussagen über die Güte eines Katalysators im späteren Einsatz zu
erhalten, muss ein Test nahe an technischen Bedingungen durchgeführt werden. Nur in einem
solchen System kann ein Katalysator Formierungsschritte und Induktionsperioden
durchlaufen, welche sich auch im späteren Einsatz abspielen. Hier befindet sich eine
potenzielle Engstelle im Gesamtprozess der Hochdurchsatz-Techniken. Die Zeit, die ein

3 Stand der Forschung
Katalysator braucht, um sein Aktivitätsmaximum zu erreichen und eventuell wieder zu
desaktivieren, kann nicht verkürzt werden, will man aussagekräftige Daten erhalten. Aus
diesem Grund ist die Konstruktion geeigneter Reaktorsysteme, in denen parallel aber auch
nahe an den späteren Einsatzbedingungen getestet werden kann, von großer Wichtigkeit. Man
kommt aus zeitlichen Gründen nicht an einer parallelen Testung vorbei, sequenzielles
Vorgehen ist hier ausgeschlossen.
Es existieren unterschiedliche Ansätze bei der Konstruktion eines Parallelreaktors. Einige
Gruppen verwenden lediglich „parallel“ nebeneinander angeordnete Reaktoren, welche noch
zusätzlich individuelle Gasversorgungen besitzen. Dies ist bis zu einem kleinen
Parallelisierungsgrad vielleicht sinnvoll, steigert jedoch den apparativen Aufwand bei höheren
Parallelisierungsgraden in nicht mehr akzeptable Größenordnungen. Eine schon
weiterentwickelte Stufe ist eine gemeinsame Gasversorgung der individuellen Reaktoren,
welche von Gruppen wie Blackstone et al.26 und Perez-Ramirez et al.27 beschrieben wird.
Eine Reihe von Reaktoren wird auch von den Forschern des ACA Berlin beschrieben. Es
werden 15-fach Festbett-Reaktoren22, 256-fach Monolithreaktoren23 und 64-fach
Keramikreaktoren verwendet28. Gemeinsam ist diesen ein einziger Reaktorkörper, welcher
durch ein einziges Heizsystem auf Reaktionstemperatur gehalten wird. Von Zech et al. wird
ein 35-fach mikrostrukturierter Reaktor beschrieben29. Weitergehende Entwicklungen von
Zech et al. beschäftigen sich mit der Miniaturisierung der Mikroreaktoren, was einen höheren
Parallelisierungsgrad ermöglichen soll und in Richtung des „catalysis on a chip“-Konzeptes
führt29.
Um die Einsatzfähigkeit der Reaktoren zu erhöhen, beschäftigen sich einige Gruppen mit
neuen Konzepten, die sowohl höheren Druck, höhere Temperatur, als auch agressivere
Medien als Testparameter erlauben. Die Entwicklung geeigneter Systeme für die Drei-
Phasen-Katalyse stellt eine weitere Herausforderung dar. Desrosiers et al. berichten über zwei
verschiedene 96-fach 3-Phasen-Hochdruckreaktoren, welche diskontinuierlich betrieben
werden. Die Analytik wird bei diesen Systemen off-line durch Standardsysteme wie
Gaschromatographie oder Kernspinresonanz durchgeführt30. Weitere Systeme, bestehend aus
gekoppelten Autoklaven, werden von Lukas et al. eingesetzt31.
Von unterschiedlichen Firmen werden bereits kommerzielle Lösungen für die Hochdurchsatz-
Reaktorproblematik angeboten. So bietet Parr Instrument Company drei unterschiedliche
Größen von teils gekoppelten Autoklaven an32. Argonaut Technologies vertreibt ebenfalls ein
kommerziell erhältliches System basierend auf dem Konzept gekoppelter Autoklaven33.
Johnson Matthey berichtet über ein bis zu 96-fach parallelisierbares System zur Durchführung
14

3.1 Kombinatorische Methoden im Rahmen der Katalysatorforschung
15
von Oxidationen in Flüssigphase21,34,35. Dieses System kann im Bereich von 0-200°C und von
0 bis 40 bar arbeiten.
Eine Reihe von unterschiedlichen Reaktoren wird von Hoffmann et al. beschrieben. Für die
Untersuchung der Kohlenmonoxid-Oxidation wurde ein 16-fach Messingreaktor entwickelt,
welcher aus einem Reaktorkörper mit gemeinsamer Gaszuführung besteht20. Ein
weiterentwickelter 49-fach Edelstahlreaktor, ebenfalls mit gemeinsamer Gaszuführung, wurde
über ein Multiportventil an zwei sequenziell geschaltete Gaschromatographen angeschlossen.
Mittels dieses Aufbaus wurde die Methanoxidation untersucht36. Für Hochdruckanwendungen
bei der Methanolsynthese wird ein diesem Reaktor ähnliches Konzept verwendet, welches
zusätzlich Drücken bis 65 bar standhalten kann37. Für die 3-Phasen-Hydrierung von
Crotonaldehyd werden zwei spezielle Rührreaktoren beschrieben. Die Analyse der
Reaktionsmischung wird hierbei off-line im Anschluss an die Reaktion durchgeführt38.
3.1.3 Hochdurchsatz-Analyse
Es kann prinzipiell zwischen parallelen und sequenziellen Analysetechniken unterschieden
werden. Die parallelen Methoden sind meist eleganter und wesentlich schneller, wobei sie
weniger Information liefern und die Produkte teilweise im voraus bekannt sein müssen.
Optischen Methoden eignen sich aus praktikablen Gründen am besten für eine parallele
Analyse. 1996 stellten Moates et al.3 eine auf Infrarot-Thermographie basierende Methode zur
Untersuchung von Reaktionen vor, bei denen eine exotherme oder endotherme Wärmetönung
auftrat. Eine Infrarot-Kamera wurde über einer Katalysatorbibliothek bestehend aus 16
verschiedenen Materialien platziert und die Wasserstoff-Oxidation untersucht, indem die
Temperaturunterschiede während der Reaktion mit Hilfe des Kamerabildes ausgewertet
wurden. Diese Methode wurde im Nachhinein von Holzwarth et al.4 verfeinert, so dass
Temperaturunterschiede von 0,1 °C noch zuverlässig detektiert werden konnten. Die Grenzen
dieses Verfahrens liegen in der Notwendigkeit einer Wärmetönung während der Reaktion,
weder können thermoneutrale Reaktionen verfolgt, noch eine Aussage über die während der
Reaktion erhaltenen Selektivitäten angestellt werden.
Weitere optische Methoden, die in der parallelen Testung angewendet werden, sind die
Fluoreszenzmessungen. Reddington et al. beschreiben für die Elektrooxidation von Methanol
einen pH- abhängigen Fluoreszenzfarbstoff, welcher auf während der Reaktion entstehende
Protonen reagiert2. Mit Hilfe einer CCD-Kamera kann so über die Intensität der Fluoreszenz

3 Stand der Forschung
ein ortsaufgelöstes pH-Wert-Bild erstellt werden und Rückschlüsse über die Aktivität der
Katalysatoren angestellt werden.
Eine Methode, welche auf der Verwendung von UV-Laserlicht beruht, ist die von Senkan et
al. eingeführte resonanzverstärkte Multiphotonenionisierung (REMPI)39,40. Kennt man die
genauen Absorptionsfrequenzen eines Produktmoleküls, ist es mit dieser Methode möglich,
selektiv ein Molekül zu ionisieren und den durch diese Spezies entstehenden Ionenstrom zu
messen. Mit dieser Methode wurde die Dehydrierung von Cyclohexan zu Benzol untersucht.
Hierbei wurde ein räumlich adressierbares Mikroelektrodenarray verwendet, wobei die
Elektroden direkt dem Produktgasstrom ausgesetzt waren. Da die exakten
Absorptionsfrequenzen für diese Methode nur mit hohem experimentellem Aufwand
zugänglich sind, wurde lediglich die Konzentration von Benzol verfolgt. Eine weitere
laserbasierte Detektionsmethode ist die sogenannte „Photothermal Deflection“ (PTD) welche
auf der Änderung des Brechungsindexes eines gasförmigen Mediums bei Änderung der
Temperatur beruht. Cong et al. benutzten zur Detektion von Ethen in einer Ethanumgebung
dieses Verfahren, um die oxidative Dehydrierung von Ethan zu Ethen zu untersuchen19. Es
wurde ein Infrarot-Anregungslaser verwendet, dessen Strahlung von Ethen absorbiert wurde
und die Ablenkung eines zweiten Laserstrahls gemessen. Über den Betrag der Ablenkung
konnten Rückschlüsse auf die Konzentration von Ethen gezogen werden. Notwendig
geworden war diese Entwicklung, da diese beiden Substanzen nicht durch die
Fragmentierungsmuster im Massenspektrum auseinander gehalten werden konnten. Hiervon
abgesehen ist die Massenspektrometrie jedoch eine ausgezeichnete Methode, da sie
hinlänglich schnell und sehr vielseitig einsetzbar ist. Konstruktionsbedingt wird diese
Methode jedoch sequenziell eingesetzt. Viele Gruppen, die sich mit Hochdurchsatz-
Techniken beschäftigen, haben diese Technik in abgewandelter Form bereits angewandt. Die
Symyx-Gruppe hat ein Massenspektrometrieverfahren in Form zweier koaxial verlaufender
Kapillaren entwickelt41,1. Durch den Mantel im äußeren Teil eines Kapillarsystems wird ein
Eduktgasstrom dem Katalysator zugeführt, während durch die innere Kapillare ein Teil des
Produktgasstromes abgesaugt und massenspektrometrisch untersucht wird. Über eine X/Y-
Positionierung können die Einzelsubstanzen der gesamten Bibliothek sequenziell angesteuert
werden. Dieses Verfahren ist in der Lage, innerhalb einer Minute einen Katalysator zu
vermessen. Es wird bei diesem Verfahren jeweils nur der gemessene Katalysator von der
Rückseite mit einem CO2-Laser geheizt. Orschel et al. umgehen dies, indem die gesamte
Bibliothek geheizt und über eine Maske der entsprechende Katalysator, welcher gerade
vermessen wird, von der Umgebung abgeschirmt wird42.
16

3.1 Kombinatorische Methoden im Rahmen der Katalysatorforschung
17
All die bisher erwähnten massenspektrometrischen Verfahren arbeiten während der
Untersuchung weit entfernt von technischen Bedingungen und können daher nur für
anfängliche Studien dienen. Es existieren zur Zeit eine Reihe weiterer ortsaufgelöster
massenspektrometrischer Aufbauten, welche die Reaktionsräume voneinander abschirmen
und somit einzelne Produktgasströme erzeugen. Die einzelnen Bereiche werden hierbei meist
kontinuierlich geheizt und mit Eduktgas durchströmt, um eine Analyse nahe an technischen
Bedingungen zu gewährleisten. Senkan et al. beschreiben eine Untersuchung der
Dehydrierung von Cyclohexan zu Benzol auf diese Weise, wobei ein stationäres
Massenspektrometer verwendet wird. Ein Reaktor, welcher aus gestapelten Trägerplatten
besteht, auf welchen das Katalysatormaterial aufgebracht ist, wird zur Analyse derart
positioniert, dass die Probennahme nur zwischen zwei Platten erfolgen kann43. Auf diese
Weise werden bis zu 80 unterschiedliche Materialien sequenziell untersucht. Weitere
Systeme, basierend auf Monolithen, einer positionierbaren Probennahmekapillare und
Rohrbündelreaktoren, in denen die Katalysatoren sequenziell durchströmt werden, sind von
Rodemerck et al. beschrieben22,23.
Von Hoffmann et al. wird eine Analyse basierend auf Gaschromatographie eingesetzt. Die
Reaktionen werden in speziell entwickelten Parallelreaktoren durchgeführt, welche durch
Mulitportventile jeweils nur den Produktstrom eines Katalysators auf die Probenschleife des
Gaschromatographen leiten20,36. Zur Beschleunigung werden zwei auf unterschiedlichen
Temperaturen arbeitende Gaschromatographen sequenziell geschaltet. Es ist hierdurch
möglich, die Testzeit pro Kanal auf unter 4 Minuten zu senken, da Heiz- und Kühlzeiten der
Gaschromatographen wegfallen.
Busch et al. beschreiben eine Methode basierend auf der Farbänderung eines auf Filterpapier
aufgebrachten organischen Farbstoffs, welcher auf NOx anspricht. Sie untersuchten hiermit
die NOx-Zersetzung in einem 49-fach Parallelreaktor, indem ein einziges imprägniertes
Filterpapier dem Gasstrom aller Kanäle ausgesetzt wird, und können hiermit ortsspezifische
Aussagen über die NOx-Konzentration treffen44.
3.1.4 Datenmanagement
Um die Hochdurchsatz-Forschung sinnvoll einsetzen zu können, darf die Datenverarbeitung
nicht vernachlässigt werden. Die aus einem Experiment gewonnenen Ergebnisse liefern nicht
nur Aufschluss über den durchgeführten Test, sondern sollten auch in einem größeren
Zusammenhang mit anderen Versuchen gesehen werden, um Trends innerhalb der

3 Stand der Forschung
Parameterräume festzustellen. Dies wurde mittlerweile auch von den auf diesem Gebiet
tätigen Firmen und vielen Forschern erkannt. Die Methoden, die hierfür zur Zeit noch zur
Verfügung stehen, sind jedoch noch im Entwicklungsstadium5. Steht man vor dem Problem
ein Mehrkomponentensystem systematisch zu testen, erkennt man schnell, dass dies selbst mit
Hochdurchsatz-Methoden kaum zu bewältigen ist. Wie von Symyx berichtet wurde, entstand
bei einer industriell durchgeführten Materialsuche innerhalb von zwei Jahren ein
Datenvolumen von mehreren Terabyte. Diese enorme Datenmenge stellt schon allein ihrer
Größe wegen höchste Ansprüche an eine Datenbank. Durch die multidimensionalen
Parameterräume ist es außerdem unmöglich, ohne Hilfe eines Computers und eines
leistungsfähigen Algorithmus die Daten zu sichten und sinnvolle Beziehungen zwischen
einzelnen Positionen herzustellen. Ziel der Anstrengungen ist es, Querverbindungen zwischen
den einzelnen Materialien zu finden und hierdurch die Anzahl der nötigen Experimente zu
verringern. Theoretisch wäre ein relationales Datenbanksystem hierzu in der Lage,
erschwerend kommt jedoch hinzu, dass für eine Evaluierung der Daten noch keine Erfolg
versprechenden Algorithmen zur Verfügung stehen.
Von einer Voraussage, welche eine Testung des gesamten Parameterraumes überflüssig
machen würde, ist man zur Zeit noch weit entfernt. Neuronale Netzwerke und andere
theoretische Konzepte gelten als viel versprechende Ansätze für diese Problemstellung, zur
Zeit ist jedoch noch keine vollständig entwickelte Methode bekannt. Einer dieser Ansätze
wird von Wolf et al.45 angewandt. Die Forscher nutzen bei der Entwicklung eines
Katalysators für die oxidative Dehydrierung von Propan einen genetischen evolutionären
Ansatz, welcher aus den besten Materialien einer Bibliothek gezielt über Punkt- oder
Kreuzmutationen neue Generationen von Katalysatoren generiert. Durch dieses Vorgehen
wurden in den ersten Generationen Verbesserungen der Aktivität erzielt, welche im Mittel
beträchtlich anstieg. Leider stagnierte diese Steigerung in späteren Generationen.
Das „Data Mining“, wie die Suche nach Querverbindungen auch genannt wird, gilt als eine
der anspruchsvollsten Aufgaben auf dem Gebiet der Datenverarbeitung in der Hochdurchsatz-
Forschung. Eventuell werden hierdurch in zukünftigen Bibliotheken noch vor den Tests
Prognosen über die Aktivität möglich.
18

3.2 Photoakustischer und photothermischer Effekt
19
3.2 Photoakustischer und photothermischer Effekt
In diesem Abschnitt werden nur kurz neuere Anwendungen des photoakustischen bzw.
photothermischen Effektes diskutiert. Eine genauere Behandlung, welche sowohl historische
Entwicklung als auch theoretische Betrachtungen insbesondere der Photoakustischen
Spektroskopie umfasst, ist im Abschnitt „Entwicklung und Prinzip der Photoakustischen
Spektroskopie“ zu finden.
Der photoakustische Effekt wird heutzutage in vielen Gebieten der Wissenschaft angewendet.
Nach anfänglichen Startschwierigkeiten im vorletzten Jahrhundert erfuhr er eine Renaissance,
nachdem technische Neuerungen wie das Mikrofon oder der Laser zur Verfügung standen.
Die Anwendungsgebiete umfassen mittlerweile vor allem Spurengasanalyse, zerstörungsfreie
Materialuntersuchungen, zahlreiche biologische und umweltrelevante Fragestellungen und
viele weitere. Die wohl bekannteste Anwendung ist die photoakustische Spektroskopie.
Die photoakustische Spektroskopie im Bereich der Gasanalytik wird intensiv von vielen
Gruppen weltweit betrieben. Sigrist et al. berichten über ein entwickeltes Hochdruck CO2-
Lasersystem, mit welchem durch die Druckverbreiterung der einzelnen Emissionslinien
kontinuierlich im Wellenlängenbereich um 11 µm spektroskopiert werden kann. Der Laser
wird bei 11,5 bar betrieben und es können Gase wie Kohlendioxid, Ethen, Ammoniak,
Methanol und Ozon bis in den unteren ppm Bereich detektiert werden46,47,48. Schmohl et al.
beschreiben eine Ammoniak-Detektion durch den Einsatz von Halbleiterlasern49. Von Sigrist
et al. existieren Untersuchungen zu Lasern, welche durch Frequenzmischung im spektral sehr
interessanten Bereich um 3000 cm-1 (3,3 µm) arbeiten. Über die Schwingungen der
Kohlenwasserstoffe können hiermit Konzentrationen bis in den ppm Bereich detektiert
werden50. Von den Forschern wird auch ein mobiles System zur Untersuchung von
umweltrelevanten Spurengasen beschrieben, welches in Feldstudien eingesetzt wird51.
Weitere Lasersysteme werden von Hess angewendet. Er beschreibt den Einsatz eines
Festkörperlasers, welcher gepulst zur Detektion von Methan in Kombination mit einem OPO
(Optischer Parametrischer Oszillator) eingesetzt wird. Der OPO ist hierbei als optisches
Element notwendig um die gewünschte Frequenz zu erhalten. Um die Detektionsgrenze in
den unteren ppb Bereich zu senken, wird ein auf zwei Kammern beruhendes differentielles
Messsystem eingesetzt52. Auch andere Diodenlasersysteme werden zur Untersuchung von
Spurengasen bis hinab in den ppb Bereich verwendet53. Von Beenen et al. werden

3 Stand der Forschung
Untersuchungen mit Lasern durchgeführt, bei denen der Laser zuerst in eine optische Faser
eingekoppelt und so direkt in die Detektionszelle geleitet wird54.
Der wohl am häufigste eingesetzte Laser ist der CO2-Laser, welcher von Gruppen wie Li et al.
und Yamulki et al. zur Untersuchung von umweltrelevanten Gasen wie den Halogenalkanen
und Alkanen sowie Stickoxiden eingesetzt wird55,56. Markusev et al. beschreiben einen CO2-
Laser Aufbau für die Untersuchung von Multiphoton-Prozessen57. Simultane Untersuchungen
der Konzentration von Methan und Ethylen mittels eines Diodenlasers werden von Boschetti
et al. beschrieben, wobei isolierte Banden des jeweiligen Gases im Bereich von 5900 und
6250 cm-1 (1,7 bzw. 1,6 µm) verglichen werden58. Sigrist et al. betrachten Multikomponenten-
Systeme theoretisch über den Zusammenhang von Phase und Intensität des Signals und
vergleichen dies mit real gemessenen Signalen59.
Auch dynamische Prozesse können mittels schneller Druckaufnehmer untersucht werden.
Chattopadhyay et al. beschreiben die erfolgreiche Detektion einer Zwischenstufen der
Photocyclisierung von Triphenylamin mit Hilfe eines auf 2 Hz gepulsten Stickstoff-Lasers
und können auf diese Weise die Existenz einer langlebigen Zwischenstufe der Reaktion
untermauern60. Resonante Phänomene beim Energietransfer in Gasen wurden schon 1992 von
Hammerich et al. mittels zeitaufgelöster Photoakustik untersucht61. Selbst angeregte Zustände
können durch photoakustische Methoden spektroskopiert werden62.
Eine Reihe weiterer Untersuchungen behandeln das Gebiet der zerstörungsfreien
Materialanalyse. Sigrist beschreibt die photoakustische Untersuchung der Adhäsion von
Beschichtungen, indem ein lasergenerierter akustischer Puls eine Oberflächenschicht in
Schwingungen versetzte, welche durch einen piezogesteuerten Displacement-Sensor
detektiert werden63. Hoshimiya et al. untersuchen die inneren Oberflächen von
Röhrensystemen, indem die Verstärkung der als Resonator benutzten Probenröhre gemessen
und über die Verstärkergüte die Defekte auf der Innenseite quantitativ erfasst werden64.
Verschiedene Umwandlungsprozesse von Alumophosphaten werden von koreanischen
Forschern untersucht. Man findet eine unterschiedliche Stärke des photoakustischen Signals
von unterschiedlichen Alumophosphaten bei unterschiedlichen Temperaturen, was eine
Phasenumwandlung zwischen diesen Modifikationen erkennen lässt65.
Eine Reihe mikroskopischer Anwendung der Photoakustik werden von Thomas in einem
interessanten Übersichtsartikel beschrieben, der sich vorrangig mit der zweidimensionalen
Auflösung und Darstellung von Oberflächen beschäftigt. Er stellt fest, dass die Wellenlänge
bei diesen Anwendungen nicht wie im Fall der optischen Mikroskopie die Auflösung
determiniert sondern die Gesetze des Nahfelds66.
20

3.3 Kohlenmonoxid-Oxidation
Jensen beschreibt auf dem Gebiet der Miniaturisierung von photoakustischen
Detektionseinheiten auch für die vorliegende Arbeit relevante Ergebnisse. Er führt eine
photoakustische Detektion in einem Mikrokanal durch und beobachtete eine sich ändernde
Propankonzentration67. Kim et al. verfolgen das während der Wassergas-Shift-Reaktion an
Eisenoxid entstehende Kohlendioxid mit photoakustischen Methoden68.
Weitere interessante Arbeiten behandeln die Kopplung von Fluoreszenz und Photoakustik,
wobei die Forscher zwei sich ergänzende Messtechniken kombinieren. Während die
Photoakustik die strahlungslosen Übergänge detektiert, werden durch Fluoreszenzmessungen
gerade die Übergänge unter Aussendung von Photonen erfasst69. Im Rahmen biologischer
Studien existieren eine Reihe Untersuchungen der durch Pflanzen abgegebenen Gase70,71.
Umweltrelevante Studien in der Atmosphäre über Städten, wie z.B. Mexico City, bedienen
sich ebenfalls der Photoakustik72. Yang et al führen Studien an Rind- und Schweinefleisch
durch, wobei die Photoakustik als Charakterisierungsmethode benutzt wird und das Fleisch in
einer Zelle für Feststoffuntersuchungen mit einem Laser periodisch angeregt wird73. Es
können hiermit Veränderungen im Fleisch anhand des Signals gemessen werden. Eine
interessante Studie betrachtet sogar die Ethenkonzentration im menschlichen Atem nach
einem Besuch der Testperson in einem Solarium mit definierter UV-Bestrahlung74.
Bemerkenswert ist hierbei die Nachweisgrenze von Ethen, welche bei 6 ppt liegt.
3.3 Kohlenmonoxid-Oxidation
Die Kohlenmonoxid-Oxidation spielt eine wichtige Rolle bei technischen Prozessen. Auch in
vielen Prozessen des Alltags ist sie von großer Relevanz. Da Kohlenmonoxid bei fast allen
Verbrennungen meist als unerwünschtes Nebenprodukt auftritt, ist dessen Entfernung aus
dem Abgasstrom eine wichtige Aufgabe. Die hier wahrscheinlich wichtigste Anwendung ist
die Abgasreinigung durch Totaloxidation zu Kohlendioxid, welche heutzutage in jedem
Kraftfahrzeug durch den allgemein so bezeichneten „Katalysator“ bewerkstelligt wird. Die
Bundesrepublik hatte im Jahre 2000 einen Kohlenmonoxid-Ausstoß von 4,8·106 t, 60%
hiervon entfallen auf den Straßenverkehr. Aufgrund der hohen Toxizität ist man bemüht, den
Kohlenmonoxid-Ausstoß so weit wie möglich noch unter den im Vergleich zu Vorjahren
schon beachtlich niedrigen Wert zu reduzieren. Dies ist neben Kraftstoffeinsparungen nur
durch verbesserte Katalysatoren möglich, welche schon bei tiefen Temperaturen eine
Totaloxidation ermöglichen.
21

3 Stand der Forschung
Zur Zeit verwendet man für die Oxidation von Kohlenmonoxid bei der Autoabgasreinigung
Mischkatalysatoren aus Platin, Palladium und Rhodium, welche auf mit γ-Al2O3 beschichtete
Monolithen aufgebracht werden. Diese Systeme müssen hierbei nicht nur Kohlenmonoxid
oxidieren, sondern gleichzeitig auch andere Reaktionen katalysieren, wie zum Beispiel die
Reduktion von toxischen Stickoxiden zu Stickstoff oder die Totaloxidation von
unverbranntem Kraftstoff. Der effektive Arbeitsbereich dieser Systeme liegt häufig über 300
°C. Für Anwendungen, welche unter dieser Temperatur liegen und im folgenden als
Tieftemperaturanwendungen bezeichnet werden, sind daher andere Systeme notwendig.
Weitere hierfür denkbarere Einsatzgebiete wären zum Beispiel in Gasmasken, CO2-Lasern,
Gassensoren oder Brennstoffzellen zu sehen.
Das Hopcalit-System, das älteste Kohlenmonoxid-Oxidationskatalysatorsystem, besteht aus
gemischten Mangan-Kupfer-Oxiden und wird noch heute in Gasmasken eingesetzt75. Es ist
jedoch nur bedingt stabil gegen Feuchtigkeit und erst über Raumtemperatur wirklich
ausreichend aktiv, was die Einsatzmöglichkeiten drastisch reduziert. Es existieren eine Reihe
von weiteren Verbindungen welche in der Kohlenmonoxid-Oxidation eingesetzt wurden, wie
zum Beispiel Perowskite oder einige Silber-Mangan-Oxide76.
In den letzten Jahren werden neben den Übergangsmetallen verstärkt geträgerte
Edelmetallsysteme untersucht. Hier zeigen Systeme wie Pd/SnO2 oder Pt/SnO2 erstaunliche
Aktivitäten, die teilweise schon unter 100 °C zu hohen Umsätzen führen77,78.
Erklärungsansätze für die Aktivität dieser Materialien sind wahrscheinlich in der
Legierungsbildung zu suchen oder auf synergetische Effekte bei Adsorptionsprozessen
zurückzuführen78,79,80,81.
Unter der großen Zahl von bekannten Katalysatoren, welche in der Kohlenmonoxid-
Oxidation aktiv sind, stechen vor allem die von Haruta beschriebenen Goldkatalysatoren
heraus. Es handelt sich hierbei um Gold, welches auf unterschiedlichen Trägern, wie z.B.
Titanoxid, Kobaltoxid oder Eisenoxid, aufgebracht wird. Sie überraschen mit Aktivitäten bei
–76 °C bei einer Raumgeschwindigkeit von 20 000 ml h-1 g-1Katalysator 82. Weshalb diese
Systeme so überaus aktiv sind, konnte bisher noch nicht zufrieden stellend geklärt werden. Es
existieren mehrere Modelle zur Erklärung. Wie von Schubert et al. beschrieben, sind es
wahrscheinlich zwei unterschiedliche Reaktionsmechanismen, welche vor allem von der
Trägersubstanz determiniert werden. Aus kinetischen Untersuchungen kann man eine Klasse
von „inaktiven“ Trägermaterialien, zu denen auch Siliciumoxid und Aluminiumoxid gehören,
finden und eine zweite „aktive“ Klasse, welche Träger wie Titanoxid, Eisenoxid oder auch
Kobaltoxid beinhaltet. Generell kann bei aktiven Trägern ein aktiverer Katalysator erhalten
22

3.3 Kohlenmonoxid-Oxidation
werden, was wahrscheinlich auf ein Mitwirken des Trägers bei der Adsorption des Sauerstoffs
hinweist. Bei den inaktiven Trägern liegt dieser dann lediglich an der Goldoberfläche
adsorbiert vor83.
Zu der Synthese der Goldkatalysatoren und der daraus resultierenden Aktivität sind viele sich
teilweise widersprechende Untersuchungen in der Literatur zu finden, auf welche hier nicht
genauer eingegangen werden kann. Generell Übereinstimmung herrscht lediglich darin, dass
eine reduktive Vorbehandlung der Katalysatoren sich negativ auf die Aktivität auswirkt84,85.
Weiterhin kann auch behauptet werden, dass nicht nur die elektronischen Veränderungen
innerhalb der Goldpartikel, welche durch die hohe Dispersion und die hierdurch kleinen
Partikel zustande kommt, für das Verhalten verantwortlich gemacht werden können. Vielmehr
dürften auch Metall-Träger-Wechselwirkungen eine Rolle spielen85,86. Zu diesem Punkt und
dem genauen Mechanismus dieser Reaktion existieren eine Reihe von unterschiedlichen
Untersuchungen. Einig ist man sich lediglich darin, dass der Sauerstoff nicht aus der
Gasphase sondern als adsorbierte Spezies reagiert. Die Kohlenmonoxid-Adsorptionsplätze
wurden anhand von Infrarot-Experimenten untersucht. Es zeigte sich, dass sowohl auf dem
Träger, dem Goldpartikel als auch an der Grenzfläche Träger/Goldpartikel Kohlenmonoxid
adsorbiert werden kann87,88,89. Die aktive Metallspezies wird ebenfalls kontrovers diskutiert.
Die meisten Gruppen betrachten metallisches Gold als verantwortlich, wobei es XPS-
Untersuchungen gibt, welche oxidisches Gold für die Aktivität verantwortlich zeigen90.
12
3
a
b
Abbildung 3 Mögliche stark vereinfachte Reaktionswege bei der Kohlenmonoxid-Oxidation an
Goldkatalysatoren83.
In Abbildung 3 sind grob die wichtigsten Reaktionswege, welche in der Literatur diskutiert
werden, graphisch dargestellt. Es können für jeden der möglichen Wege experimentelle
Belege gefunden werden. Die einfache direkte Adsorption von Sauerstoff an den
Goldpartikeln (Weg 1), bei der beide Reaktionspartner adsorbiert vorliegen müssen, gibt dem
Träger lediglich eine stabilisierende Funktion, die die hohe Dispersion des Goldes
23

3 Stand der Forschung
aufrechterhält91,92. Andere Vorschläge legen eine Adsorption am Träger oder an der
Grenzfläche zwischen Gold und Träger nahe84,93. Eine Adsorption auf dem Trägermaterial
wird durch ESR-Messungen gestützt94. Als weitere Variation wird die Dissoziation des
Sauerstoffes (Weg 2a) diskutiert. Bei der direkten Reaktion würden intermediär
carbonathaltige Spezies gebildet werden (Weg 2b)95. Dieser Weg wird jedoch von vielen
Gruppen abgelehnt83,85. Auch ein Weg über Gerüstsauerstoff in der Art eines Mars-van-
Krevelen Mechanismus oder über einen Spill-over auf die Goldoberfläche wäre denkbar (Weg
3)84,93.
Zusammenfassend bleibt zu sagen, dass die Kohlenmonoxid-Oxidation, obwohl eine
eigentlich sehr einfache Reaktion, dennoch zumindest im Fall des Goldes noch nicht
vollständig verstanden ist.
Die Kohlenmonoxid-Oxidation ist eine in der chemischen Literatur häufig beschriebene
Reaktion. Es existieren neben den Goldsystemen eine große Zahl an weiteren untersuchten
Systemen. Durch ihren relativ einfachen Ablauf eignet sie sich hervorragend für die
Untersuchung elementarer Prozesse im Ablauf katalytischer Reaktionen. Ihre Kinetik ist an
vielen Katalysatoren ein schönes Beispiel für einen Langmuir-Hinshelwood-Mechanismus96.
Der einfache Ablauf dieser Reaktion und das definierte einzige Produkt Kohlendioxid
prädestinierte diese Reaktion aufgrund der nicht zu erwartenden Nebenprodukte für die später
beschriebenen Experimente, die im Rahmen dieser Arbeit durchgeführt wurden.
3.4 Oxidative Dehydrierung kurzkettiger Alkane zu Alkenen
Olefine sind die wichtigsten Bausteine der modernen petrochemischen Industrie.
Hauptsächlich wird der Bedarf an diesen Grundstoffen heute durch Steam-Cracking und
Fluidized Catalytic Cracking (FCC) gedeckt. Diese Verfahren sind ausgereift und optimiert,
können den Bedarf aufgrund der Nebenprodukte jedoch nicht beliebig decken. Da die
Nachfrage an diesen Stoffen zunehmend steigt, müssen daher neue Verfahren entwickelt
werden, um gezielt die gewünschten Grundbausteine wie Ethylen für Polyethylen und vor
allem Propylen für Polypropylen zu decken97.
Versuche, den steigenden Bedarf über eine Dehydrierung von Alkanen zu decken, scheitern
an thermodynamischen Grenzen und der hohen Tendenz der Kohlenstoffabscheidung, was zu
kurzen Lebenszeiten der Katalysatorsysteme führt98. Eine Möglichkeit, diese Probleme zu
überwinden, ist die Zugabe von Sauerstoff zur Reaktionsmischung. Hierdurch wird der
24

3.4 Oxidative Dehydrierung kurzkettiger Alkane zu Alkenen
25
Wasserstoff aus dem Gleichgewicht entfernt und eventuell auftretender Kohlenstoff beseitigt.
Die Ökonomie des Verfahrens wird hierdurch beträchtlich gesteigert. Obwohl sich weltweit
viele Arbeitsgruppen intensiv mit der oxidativen Dehydrierung (ODH) beschäftigen, gelang
es bislang noch nicht, einen wirtschaftlichen Prozess für eine industrielle Applikation zu
entwickeln. Das Hauptproblem liegt hierbei zur Zeit bei den geringen Olefin-Selektivitäten,
die typischerweise eine Ausbeute von weniger als 30% bewirken. Im folgenden soll kurz ein
Überblick über die unterschiedlichen Ansätze und Katalysatorsysteme gegeben werden.
Redox-Katalysatoren
Die meisten Arbeiten beschäftigen sich mit Oxiden der Übergangsmetalle. Diese
redoxaktiven Katalysatoren bewirken häufig eine Reaktion, welche einer Mars-van-Krevelen-
Kinetik gehorcht. Das Oxidmaterial wird in einem initiierenden Schritt von dem
Kohlenwasserstoff reduziert und in einem Folgeschritt durch Sauerstoff aus der Gasphase
reoxidiert. Cavani et al. und Kung et al. haben ausführliche Übersichtsartikel über die ODH
kurzkettiger Alkane geschrieben98,99. Es herrscht Übereinstimmung, dass die Spaltung der C-
H-Bindung der geschwindigkeitsbestimmende Schritt ist. Dies wird aus den unterschiedlichen
Reaktionsgeschwindigkeiten der unterschiedlichen Kohlenwasserstoffe geschlossen, welche
mit der Stabilität dieser Bindung korreliert werden kann. Abgesehen von Ethan und Ethen
reagieren Alkene generell schneller als Alkane, was es erschwert, einen geeigneten
Katalysator speziell für Propan/Propen zu finden100. Es werden daher große Anstrengungen
unternommen, um eine geeignete Prozessführung zu finden, in der das im ersten Schritt
gebildete Alken in den Folgeschritten nicht mit Sauerstoff in Berührung kommt. Dies wird bei
zyklischer Reaktionsführung zum einen mit getrennten Reaktions- und Reoxidationsschritten
versucht101, zum anderen durch eine Trennung in zwei unabhängige katalytisch durchgeführte
Prozesse. In einem ersten Schritt wird hierbei durch einen guten Dehydrierungskatalysator das
Alken gebildet und in einem zweiten der Wasserstoff durch einen selektiven
Oxidationskatalysator entfernt102.
Edelmetall-Katalysatoren
Edelmetalle sind als gute Verbrennungskatalysatoren bekannt. Bei oxidativen
Dehydrierungen werden mit Platin beschichtete Monolithreaktoren verwendet. Die
Kontaktzeiten sind hierbei extrem kurz und liegen im Bereich einer Millisekunde103. Es wird
ein Mechanismus diskutiert, der zunächst über die Totaloxidation der Alkane abläuft. Durch
die bei dieser Reaktion frei werdende Wärmeenergie wird dann ein thermisches Cracken bei

3 Stand der Forschung
genügend geringen Sauerstoffkonzentrationen bewirkt und eine oxidative Dehydrierung
eingeleitet104,105.
Redoxinaktive Katalysatoren
Auf dem Gebiet der redoxinaktiven Katalysatoren existieren eine Reihe von Untersuchungen
zur oxidativen Dehydrierung von Ethan. Verbreitet werden hierfür Alkalimetalloxide und
Seltenerdoxide eingesetzt100. Zur oxidativen Dehydrierung von Propan existieren weit
weniger Arbeiten. Bemerkenswert ist die teilweise hohe Ausbeute an Ethen bei der ODH von
Propan106. Dies ist auf eine Spaltung des Kohlenstoffgerüsts zum einen durch
Gasphasenreaktionen oder Reaktionen mit lockerem Gerüstsauerstoff zurückzuführen.
Inwieweit der Propanumsatz katalytisch bedingt wird, bleibt unklar. Einige Gruppen wie
Sinev et al.107 und Dahl et al.108 favorisieren die radikalische Reaktion in der Gasphase,
welche durch eine Oberflächenreaktion initiiert wird und über Radikal-Oberflächen-
Reaktionsschritte verläuft. Andere Gruppen wie auch Bujevskaja et al. erklären ihre
Ergebnisse ausschließlich über katalytische Beiträge und schließen einen Gasphasenanteil
aus106,109,110.
Unkatalysierte Reaktion
Es existieren auch Arbeiten zur nicht-katalysierten oxidativen Dehydrierung kurzkettiger
Alkane, welche häufig als Pyrolyse oder Oxycracking bezeichnet wird111,112. Der
Mechanismus dieser Reaktion wird allgemein als radikalische Kettenreaktion, ähnlich dem
thermischen Cracken beschrieben. Xu et al.113 und Lemonidou et al.114 vergleichen die
katalysierte mit der nicht-katalysierten Reaktion und finden, dass ohne Katalysator die besten
Umsätze erzielt werden.
Generell bleibt bei allen Katalysatortypen anzumerken, dass oberhalb von 600 °C eine
Reaktion, welche ausschließlich katalytisch ohne homogene Gasphasenschritte abläuft,
äußerst unwahrscheinlich erscheint.
26

4 Experimentelles
Es ist besser, sich von Erfahrungen als von Zielen leiten zu lassen.
Martin Walser
4.1 Synthese
Eine genaue Beschreibung des gesamten Prozesses und der dargestellten Materialien befindet
sich im Kapitel „Parallele Feststoffsynthese“. Die Synthese beruht im Groben auf einer
Imprägnierung von Aktivkohle durch unterschiedliche Metallsalzlösungen. Das
Lösungsvolumen sollte hierbei komplett von der Kohle aufgenommen werden können. Die so
mit Metallsalzen beladene Kohle wird im Anschluss an die Imprägnierung verbrannt, wobei
die Metalle in oxidischer Form zurückbleiben. Die hierdurch erhaltenen Materialien weisen
eine sehr hohe Oberfläche auf und sind daher für katalytische Anwendungen von großem
Interesse. Um einen hohen Probendurchsatz zu ermöglichen, wurden die Synthesen teilweise
automatisiert mittels eines Pipettierroboters (Gilson XL 232) durchgeführt.
Abbildung 4 Pipetierroboter Gilson XL 232 (rechts) mit Präparations-Aufbau für die Synthese der
Metalloxide (links). Links unten ist die Präparationsplatte für die gleichzeitige Synthese
von 77 Metalloxiden zu sehen. Links oben befinden sich die Vorratslösungen.
27

4 Experimentelles
28
Die Präparationsgefäße wurden zu Anfang der Synthese mit einem definierten Volumen
Aktivkohle befüllt. Die Imprägnierung wurde von einem kommerziellen Pipettierroboter
durchgeführt. Der Gesamtaufbau ist in Abbildung 4 gezeigt. Bei den in dieser Arbeit
durchgeführten Synthesen entsprach die Imprägnierung den Bedingungen der Incipient-
Wetness-Methode, was bedeutet, dass gerade so viel Precursorlösung transferriert wurde, wie
der Feststoff aufnehmen konnte. Die Zusammensetzung der Precursorlösung wurde durch
einen zufälligen Algorithmus bestimmt, der zuerst entschied, ob ein Precursor enthalten ist
oder nicht, und im Anschluss hieran den relativen Anteil ermittelte, den der gewählte
Precursor zur gesamten Zusammensetzung beisteuert. Die Precursorlösungen wurden erst in
eine Mischzone pipettiert (linke Seite Mitte in Abbildung 4) und nach einer Durchmischung
durch Einblasen von Luft in die eigentlichen Reaktionsgefäße transferiert. Fluka 05120 und
Darco® KB-B, -100 von Aldrich wurden als Aktivkohlen verwendet. Als Metallverbindungen
dienten Hydrate der Nitratsalze von Fluka. Alle Tränklösungen waren gesättigt. Die
imprägnierten Proben wurden im Anschluss hieran kalziniert, wobei eine Abdeckung aus
Quarzwolle verwendet wurde, um ein Aufwirbeln der Oxidpartikel während der
Kohlenstoffverbrennung zu verhindern und somit eine Kreuzkontamination auszuschließen.
Die getränkten Kohlen wurden langsam innerhalb drei Stunden auf 500 °C erhitzt. Nach zwei
weiteren Stunden bei 500 °C, was zu vollständiger Verbrennung der Kohle führte, kühlten die
Substanzen auf Raumtemperatur ab. Um die Temperaturrampen des abschließenden
Verbrennungsprozesses möglichst variabel zu gestalten und somit eine große Bandbreite an
Syntheseparametern zu gewährleisten, wurden Quarzgefäße verwendet und ein
Glimmermaterial, welches Temperaturen von über 1000 °C dauerhaft standhalten konnte, als
Konstruktionsmaterial der Syntheseplatte gewählt.
4.2 Katalyse
In diesem Abschnitt werden nur die apparativen Reaktoraufbauten und Reaktionsparameter
besprochen. Die photoakustische Analytik ist in Kapitel 6 genauer beschrieben. Als
Modellreaktionen wurden die Kohlenmonoxid-Oxidation und die oxidative Dehydrierung von
Ethan herangezogen. Für beide Reaktionen wurden Hochdurchsatz-Teststände entwickelt und
die im Rahmen dieser Arbeit dargestellten Materialien in diesen Testständen auf ihre Aktivität
untersucht. Für ausgewählte Katalysatoren wurden Tests im größeren Maßstab in

4.2 Katalyse
konventionellen Einrohrreaktoren durchgeführt, um die neu entwickelten Analysesysteme zu
evaluieren.
4.2.1 Kohlenmonoxid-Oxidation
Apparativer Aufbau
Für die Testung der Katalysatorbibliotheken für die Kohlenmonoxid-Oxidation wurde ein im
Max-Planck-Institut für Kohlenforschung konstruierter 16-fach Parallelreaktor eingesetzt20.
a) b)
102
15
60
15
187,2
37
95
18
R3,25
Abbildung 5 a) Schematische Zeichnung der Seitenansicht des 16-fach Parallelreaktors und b) Foto
der Seitenansicht. Es sind deutlich die Bohrungen (vier) für die Katalysatoraufnahmen
zu erkennen.
Das Reaktorkonzept stammte in erster Linie von Dr. C. Hoffmann. Der Reaktorgrundkörper
ist in Abbildung 5 gezeigt und beschrieben. Er besteht im wesentlichen aus einem durch fünf
individuelle Heizpatronen beheizbaren Messingblock in den die 16 Bohrungen für die
Katalysatoraufnahme eingebracht sind und einem durch vier Heizpatronen ebenfalls
beheizbaren Deckel. Die Heizpatronen haben eine Gesamtleistung von 1350 W. Die
Temperaturregelung erfolgte über ein in die Mitte des Reaktorblockes eingebrachtes
Thermoelement und einen Regler der Firma Eurotherm (Typ 2216e). Der Regler wurde durch
eine auf LabVIEW basierende Routine automatisiert betrieben, konnte jedoch auch manuell,
sowohl direkt am Gerät als auch vom Steuerrechner aus, eingestellt werden.
29

4 Experimentelles
30
a) b)
40 60 80 100 120 140 160 180 200 220
40
60
80
100
120
140
160
180
200
220
Tem
pera
tur i
st [°
C]
Temperatur soll [°C]
Abbildung 6 a) Blick in den 16-fach Parallelreaktor von oben, man erkennt die Lage der Heizpatronen
(vertikale Linien) zwischen den Katalysatorpatronen und b) Temperaturverteilung des
Reaktors mit Abweichungen von der Solltemperatur.
Die zu vermessenden Materialien wurden in Edelstahlpatronen abgefüllt und einzeln in den
Messingblock eingesetzt. Das Konstruktionsmaterial Messing gewährleistete durch die gute
Wärmeleitfähigkeit eine homogene Temperaturverteilung. Die Abweichungen von der
Solltemperatur betrugt hierbei wie in Abbildung 6 b) zu sehen ist maximal ein Grad, wobei
die Abweichung der einzelnen Positionen relativ zueinander ebenfalls ein Grad betrug.
�����������������������������������������������
Parallelreaktor
CO
Luft
������������������������������
��������������
�������������� ����������
����������������������
����������������������������
����������� ����
���
������
Gasversorgung PC: Steuerung und Auswertung
CO2
����������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������
����������������������������������������������������������������������������������������� PA Analytik
400
Abbildung 7 Schematischer Aufbau der Testanlage für die Kohlenmonoxid-Oxidation für den
parallelen Betrieb.

4.2 Katalyse
Die Gasversorgung und der Aufbau der Testanlage sind schematisch in Abbildung 7 gezeigt.
Die Gaszuleitung erfolgte über Bronkhorst F-201C Massendurchflussregler, welche durch
einen Steuerrechner und eine hierfür entwickelte, auf LabVIEW basierende, Software
programmiert und automatisiert betrieben werden konnten. Um eine größere Flexibilität des
Systems zu gewährleisten, war auch eine manuelle Einstellung am Steuerrecher abweichend
von der Programmierung möglich.
Um homogene Flussbedingungen zu gewährleisten, wurden in den Böden der zur
Katalysatoraufnahme bestimmten Edelstahlpatronen poröse Fritten mit einer Porenweite von
4 µm oder 10 µm verwendet. Für Katalysatormaterialien, welche über die Aktivkohle-Route
hergestellt wurden, erwies sich eine Porenweite von 10 µm als besser geeignet, da kleinere
Porenweiten teilweise zur Verstopfung des Kanals führten. Eine gemeinsame Gaszuleitung
versorgte alle 16 Katalysatorschüttungen mit dem gleichen Eduktgas, wobei durch eine
Diffusorplatte die Separation der einzelnen Kanäle sichergestellt wurde. Diese Platte bestand
im wesentlichen aus einer Scheibe, welche mit 16 kleinen Bohrungen ausgestattet war. Die
Lineargeschwindigkeit des Gasstroms wird innerhalb der Bohrungen derart erhöht, dass eine
Rückdiffusion verhindert wird. Durch die gleichmäßige Verteilung auf die 16 Positionen hat
die Lage der einzelnen Positionen relativ zum Gaseinlass keinen Einfluss auf den gemessenen
Umsatz. Die Bohrungen der Diffusorplatte wurden von einem anfänglichen Durchmesser von
2 mm auf 0,5 mm verringert, was eine 16-fache Steigerung der Flussgeschwindigkeit
bewirkte. Durch diese Punkte wurde letztendlich eine homogene Flussverteilung
sichergestellt, da der gesamte Druckabfall hauptsächlich vor und nach der
Katalysatorschüttung stattfand.
F lachd ichtu ng
a
b c
O -R ing
O -R ingR and g äng igkeit
K reu zko ntam in ationD iffu sorp latte
Abbildung 8 Mögliche Undichtigkeiten und Dichtkonzept: O-Ring-Dichtung a ist vor allem gegen
Undichtigkeiten nach außen, Dichtung c gegen Randgängigkeiten und Dichtung b eine
Zusatzdichtung gegen Kreuzkontaminationen.
31

4 Experimentelles
32
Den komplizierten Dichtungsverhältnissen welche schematisch in Abbildung 8 gezeigt sind,
wurde mit einer Dreifach-Dichtung Rechnung getragen. Es wurden 16 einzelne Flach-
Dichtungen gegen die Randgängigkeit (Dichtung c) und O-Ring-Dichtungen für
Undichtigkeiten nach außen (Dichtung a) und eine eventuelle Kreuzkontamination der
unterschiedlichen Positionen im Reaktorkörper (Dichtung b) verwendet. Anfängliche
Probleme bei der Dichtigkeit konnten durch Experimente mit unterschiedlichen
Dichtungsdicken und verschiedenen Dichtungsmaterialien relativ rasch behoben werden.
Getestet wurden Dichtungen aus Silikon, Perbunan und Viton. Es stellte sich heraus, dass
Perbunan eine zu geringe Temperaturstabilität aufwies und oberhalb von 150°C innerhalb
kurzer Zeit die Formstabilität verlor, wobei die Dichtungen stark ausgasten. Dichtungen aus
Silikon oder Viton zeigten sich in ihrer Temperaturstabilität im Bereich bis 210 °C als
ausreichend, mussten jedoch beide nach ca. 15 Katalysatorchargen regelmäßig gewechselt
werden. Eine Dicke der Flach-Dichtungen von 2 mm erwies sich in mehreren Testläufen als
optimal geeignet. Die Leitungen, welche das Produktgas führten, bestanden ausnahmslos aus
Edelstahl. Für alle Verbindungen wurde ein Klemm-Schneidring-Dichtsystem (Swagelok)
verwendet.
Der Einrohr-Festbett-Reaktor für die Evaluierung der Analytik bestand im wesentlichen aus
einem vertikal angeordneten Rohrreaktorsystem, bestehend aus drei konzentrisch
angeordneten Röhren. Das innere Quarzrohr, welches einen Durchmesser von 4 mm hatte,
enthielt die Katalysatorschüttung und war am unteren Ende mit einer Quarzfritte
abgeschlossen. Das mittlere Rohr bestand aus Duranglas und war mit einer Heizwicklung
versehen, welche das System auf bis zu 300 °C heizen konnte. Umgeben war das gesamte
System von einer Edelstahlwandung, welche auf Bedarf mit einem Kühlmedium durchströmt
und somit auf bis zu –100 °C gekühlt werden konnte. Dieses Kühlsystem wurde im Rahmen
dieser Arbeit nicht verwendet, wurde jedoch von Frau Dr. A. Wolf erfolgreich zur
Untersuchung von hochaktiven Kohlenmonoxid-Oxidationskatalysatoren basierend auf
Goldsystemen eingesetzt20. Die Steuerung der Temperaturregelung und der Bronkhorst F-
201C Massendurchflussregler erfolgte ausschließlich manuell.
Die Konzentration an Kohlendioxid wurde mittels nicht dispersiver Infrarot-Spektroskopie
ermittelt. Es wurde ein Gerät der Firma Hartmann & Braun, Typ URAS 3E, verwendet. Das
Messverfahren basiert auf den unterschiedlichen Infrarotabsorptionen von Kohlenmonoxid
und Kohlendioxid im Bereich von 2,5 bis 12 µm. Durch diese Unterschiede wird über

4.2 Katalyse
Temperatur- bzw. Druckschwankungen zwischen einer Mess- und einer Vergleichskammer
eine Membran unterschiedlich gewölbt. Dieser Vorgang wird gemessen und angezeigt.
a) b)
Luft
���������������
���������������
����������
����������������
����������
CO URAS-Analytik
��������������������������������������������������������������������������������������������������������������������������������������������
�������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������
������������������������������������������������������������������������������������������������������������������������������
Temperatursteuerung
Flusssteuerung
Reaktor
Abbildung 9 a) Schematischer Aufbau des Einrohr-Festbett-Reaktor Teststandes und b) schematische
Zeichnung des Reaktors.
In Abbildung 9 sind der Aufbau der Einrohr-Festbett-Reaktor-Anlage und der dreiwandige
Reaktor schematisch dargestellt.
Reaktionsbedingungen
Im Parallelreaktor wurden je 40 mg Katalysator als Pulver direkt aus der Synthese ungesiebt
eingewogen. Als Reaktionsgas wurde ein Prüfgas von Messer-Griesheim mit einer
Zusammensetzung von 10 % Kohlenmonoxid in Stickstoff mit synthetischer Luft auf eine
Konzentration von 3 bzw. 1 % Kohlenmonoxid absolut verdünnt. Ein Gesamtgasstrom von
200 ml·min-1, woraus sich ein Fluss von 12,5 ml·min-1 pro Katalysatorschüttung ergab,
bedingte eine Raumgeschwindigkeit von 18750 ml·h-1g-1Katalysator. Üblich ist normalerweise
33

4 Experimentelles
34
eine Angabe der Raumgeschwindigkeit in GHSV (Gas Hourly Space Velocity), welche sich
aus
( )
⋅=
hZeitrvolumenKatalysatoGasvolumenGHSV 1
ergibt115. Diese Norm war jedoch aufgrund der unterschiedlichen Dichten der in dieser Arbeit
eingesetzten Materialien unpraktisch. Es wurde daher eine auf die Masse bezogene
Kenngröße verwendet. Ein Vergleich der Umsatzkurven gestaltet sich aufgrund der
unterschiedlichen Formen der Messkurven als äußerst schwierig. Es wurde daher der T1/2
Wert herangezogen, bei dem 50 % des vorhandenen Kohlenmonoxids oxidiert waren. Für die
katalytischen Messungen wurde die Temperatur schrittweise mit 1 °C pro Minute von
Raumtemperatur auf 180 °C erhöht. Im Anschluss lies man das System langsam wieder auf
Raumtemperatur abkühlen um das gesamte Zünd-/Lösch-Verhalten zu beobachten.
Für Vergleichstests wurden die Temperatur/Umsatz-Kurven ausgewählter Katalysatoren
zusätzlich in oben beschriebenem Einrohr-Festbett-Reaktor (Abbildung 9) vermessen. Die
Katalysatormaterialien wurden, um genauere Daten zu erhalten, vor ihrem Einsatz gepresst
und gesiebt, wobei eine Sieb-Fraktion zwischen 250 und 500 µm eingesetzt wurde. Dieses
Material wurde im Anschluss in beiden Systemen, parallel mit photoakustischer Analytik und
individuell mit angeschlossener URAS Analytik erneut vermessen. Aus experimentellen
Gründen wurde jeweils eine Konzentration von 1 % Kohlenmonoxid (Raumgeschwindigkeit
von 21600 ml·h-1g-1Katalysator) eingestellt, was 50 mg Probe im Parallel- und im Einrohrreaktor
entsprach.
4.2.2 Oxidative Dehydrierung von Ethan
Apparativer Aufbau
Für die oxidative Dehydrierung von Ethan wurde im frühen Teststadium ein 16-fach
Parallelreaktor verwendet. Der Reaktorgrundkörper wurde von der hte AG in Heidelberg
erworben.

4.2 Katalyse
Abbildung 10 Edelstahlreaktor der Firma hte AG.
Die Konstruktion beruht auf einer Weiterentwicklung eines ursprünglich an der Universität
Frankfurt entworfenen und schon bei der Kohlenmonoxid-Oxidation eingesetzten
Messingreaktors. In Abbildung 10 ist der geschlossene Reaktor (links) bzw. eine Aufnahme
von oben, welche die Bohrungen für die Katalysatoraufnahmen zeigt (rechts), abgebildet.
Die Bohrungen für die Katalysatorpatronen waren in einer 4x4 Matrix angeordnet. Da
Edelstahl ein schlechter Wärmeleiter ist, wurde über insgesamt 24 Heizpatronen, welche
parallel zu den Katalysatorbohrungen von unten in den Reaktorkörper eingebracht waren, eine
ausreichende Wärmezufuhr gewährleistet.
200 250 300 350 400 450 500150
200
250
300
350
400
450
500
550
Tem
pera
tur i
st [°
C]
Temperatur soll [°C]
Abbildung 11 Temperaturverteilung und Dichtungen des hte Reaktors. Der Reaktor kommt mit
lediglich 2 Dichtungen aus. Im Vergleich hierzu wurden für den Messingreaktor 33
Einzeldichtungen verwendet.
Wie aus Abbildung 11 (linke Seite) hervorgeht, ist auch bei verwendetem Edelstahlreaktor
eine homogene Temperaturverteilung gegeben.
35

4 Experimentelles
36
Es wurde ein spezielles Dichtverfahren eingesetzt, welches mit nur zwei Dichtungen (rechte
Seite Abbildung 11) eine Dichtigkeit bis zu 60 bar und 600 °C gewährleistete. Eine erste
Flachring-Dichtung verhinderte das Ausströmen des Gases aus dem Reaktor, während eine
weitere Flach-Dichtung mit 16 Löchern für die einzelnen Katalysatorpatronen sowohl
Rändgängigkeit verhinderte als auch die Katalysatorpositionen voneinander isolierte. Die
Dichtungen bestanden aus Graphit, was den Einsatz stark oxidierender Reaktionsbedingungen
in Verbindung mit hohen Drücken und Temperaturen einschränkte. Da die einzelnen Bauteile
über Schneidringe mit dem Graphit in Verbindung standen, musste die Dichtung spätestens
nach 10 Einsätzen erneuert werden. Bei richtiger Anwendung und Verschrauben des Deckels
mit einem Drehmomentschlüssel war dieses Dichtverfahren sehr zuverlässig und einfach
anzuwenden. Die Schrauben bestanden aus verzinktem Stahl, welcher zusätzlich vor jedem
Einsatz mit einem silberhaltigen Hochtemperatur-Schmiermittel eingefettet wurde, um ein
Diffusionsschweissen bei hohen Temperaturen zu verhindern. Aufgrund von
Sublimationseffekten musste der Einsatz von cadmierten Schrauben vermieden werden.
Durch die Zinkbeschichtung bildete sich ein zusätzlicher Film aus Zinkoxid, welcher die
Leichtgängigkeit beim Öffnen des Systems sicherstellte. Da die Verzinkung nach 5 Einsätzen
vollständig absublimiert war, mussten die Schrauben regelmäßig ausgewechselt werden. Wie
schon beim 16-fach Messingreaktor, stellte auch hier eine Diffusorplatte eine homogene
Flussverteilung sicher, wobei ebenfalls zusätzlich die Katalysatorpatronen mit Edelstahlfritten
der Porenweite 10 µm verschlossen waren.
Die Temperaturregelung des Reaktorsystems erfolgte über ein in die Mitte des
Reaktorblockes eingebrachtes Thermoelement und einen Regler der Firma Eurotherm (Typ
2216e). Der Regler wurde durch eine auf LabVIEW basierende Routine automatisiert
betrieben, konnte jedoch auch manuell, sowohl direkt am Gerät als auch vom Steuerrechner
aus, eingestellt werden.

4.2 Katalyse
Parallelreaktor
C2H4
Luft
��������������
�������������
������������� ���������
����������
����������������������������
���������� ��������
���
������
��������������������������������������
Gasversorgung PC: Steuerung und Auswertung
C2H6
��������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������
��������������������������������������������������������������������������������������� PA Analytik
400
Abbildung 12 Schematischer Aufbau der Anlage zur parallelen Untersuchung der oxidativen
Dehydrierung von Ethan.
Die Gasversorgung und der Aufbau der Testanlage sind schematisch in Abbildung 12 gezeigt.
Die Gaszuleitung erfolgte über Bronkhorst F-201C Massendurchflussregler, welche durch
einen Rechner und eine ebenfalls auf LabVIEW basierende Software automatisiert betrieben
und programmiert werden konnten. Die Leitungen, welche das Produktgas führten, bestanden
ausnahmslos aus Edelstahl. Für alle Verbindungen wurde ein von der hte AG entwickeltes
System bestehend aus gasdichten Einschraubmodulen verwendet.
Der Einrohr-Festbett-Reaktor für die Evaluierung der Analytik bestand im wesentlichen aus
einem vertikalen Rohr aus Quarzglas mit einem Innendurchmesser von 10 mm. Das
Reaktionsrohr befand sich in einem Ofen, welcher eine Temperatur von 950°C erreichen
konnte.
37

4 Experimentelles
38
C2H4
Luft
���������������
���������������
����������
����������������
����������
Flussteuerung
C2H6
400
���������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������
GC1 GC2
������������������������������������������������������������������������������ ������������������
Temperatursteuerung
Reaktor
Abbildung 13 Schematischer Aufbau des Einrohr-Festbett-Reaktors. Die Analyse erfolgte über zwei
sequenziell geschaltete Gaschromatographen, deren Betriebstemperaturen 90 °C (GC1)
und 235 °C (GC2) betrugen.
Ein schematischer Aufbau der Anlage ist in Abbildung 13 gezeigt. Auf Höhe der
Katalysatorschüttung, welche durch eine Quarzfritte gehalten wurde, befand sich ein
Thermoelement, wodurch eine relativ exakte Temperatureinstellung in der Reaktionszone
ermöglicht wurde. Die Regelung erfolgte über einen Thermoregler der Firma Eurotherm (818
P), welcher manuell eingestellt wurde. Zusätzlich war ein Thermoelement im Quarzrohr in
der Katalysatorschüttung angebracht, um die Temperatur im Katalysatormaterial zu messen.
Die Gaszuleitung erfolgte über manuell geregelte Bronkhorst F-201C
Massendurchflussregler. Die Analyse erfolgte über zwei in Serie geschaltete
Gaschromatographen (HP 6940).
Reaktionsbedingungen
Für die oxidative Dehydrierung wurden aus analytischen Gründen nur 8 Kanäle benutzt. Die
Katalysatoren wurden direkt aus der Synthese ungesiebt eingewogen. Es wurden jeweils 50
mg Katalysator verwendet. Bei einem Gesamtfluss von 300 ml·min-1 (2 % Ethan in Luft), was
einem konstanten Fluss von 37.5 ml·min-1 pro Katalysatorschüttung entspricht, wurden die
Materialien in Temperaturschritten von 50 °C im Bereich von 300 bis 550 °C getestet, wobei
die jeweiligen Temperaturen für 30 min gehalten wurden. Die Flussbedingungen entsprachen
einer Raumgeschwindigkeit von 45000 ml·h-1g-1Katalysator.

4.3 Charakterisierungsmethoden
4.3 Charakterisierungsmethoden
39
Für Vergleichstests wurden die Temperatur/Umsatz-Kurven ausgewählter Katalysatoren
zusätzlich in oben beschriebenem Einrohr-Festbett-Reaktor vermessen. Die ausgesuchten
Materialien wurden zum Vergleich erneut in großem Ansatz dargestellt und in beiden
Systemen, parallel mit photoakustischer Analytik und individuell mit in Serie geschalteten
Gaschromatographen (HP 6940) vermessen, wobei 100 mg Katalysatormaterial für den
Einrohrreaktor verwendet wurden.
Zur Charakterisierung von sowohl kristallinen als auch amorphen Festkörpern existieren eine
Reihe von unterschiedlichen Methoden. Hier werden die zur Untersuchung von exemplarisch
ausgewählten gemischten Metalloxiden verwendeten Charakterisierungsmethoden
beschrieben.
4.3.1 Röntgenpulverdiffraktometrie (XRD)
Zeigt ein Feststoff eine regelmäßig periodische Fernordnung der Atome, so werden
Röntgenstrahlen an den Elektronendichtezentren, den Streuzentren der Substanz, gebeugt und
erzeugen durch Interferenz ein Beugungsbild. Die Beugung an den Ebenen des Kristalls
gehorcht der bekannten Bragg Gleichung:
Θ⋅⋅=⋅ sin2 dn λ
n steht hierbei für eine ganze natürliche Zahl, λ für die Wellenlänge der Röntgenlichtes, d für
den Netzebenenabstand und Θ für den Einfallswinkel des Röntgenstrahles.
Das hieraus resultierende Diffraktogramm kann zur Bestimmung der Zusammensetzung und
darüber hinaus zur Phasenidentifikation herangezogen werden. Dies erfolgt meist durch
Vergleich mit Diffraktogrammen aus Datenbanken. Neben Informationen über
Zusammensetzung und Modifikation können zusätzlich aus der Form der Reflexe auch
Anhaltspunkte über die Größe der kristallinen Bereiche erhalten werden, woraus auf
Partikelgrößen geschlossen werden kann.

4 Experimentelles
40
Grundlage für diese Berechnungen bildet die Scherrer-Gleichung:
Θ⋅⋅
=cosL
k λβ
β ist hierbei die integrale Breite des Reflexes, k eine von der Geometrie des Partikels
abhängige Konstante (wird meist, so auch hier, auf 0,89 gesetzt), λ die Wellenlänge der
Röntgenstrahlung und Θ der Winkel des einfallenden Röntgenstrahles. L ist der gesuchte
Kristallitdurchmesser.
Eine Reflexverbreiterung kann auch durch Präparationsfehler und gerätespezifische Parameter
bewirkt werden. Um diesen Faktoren Rechnung zu tragen, sollten die Werte vor einer
Messung durch Testmessungen im entsprechenden Winkelbereich unter Verwendung eines
hochkristallinen Standards ermittelt werden. Fehler, welche aus intrinsischen
Stoffeigenschaften resultieren, wie z.B. Dehnungen und Spannungen, können mit Hilfe einer
Entfaltung ermittelt werden. Dies gelingt aufgrund der lorentzförmigen Reflexform, welche
durch die Kristallitgröße bewirkt wird. Im Vergleich hierzu liefern die anderen Effekte ein
gaussförmiges Reflexprofil, welches herausgerechnet werden kann.
Die Pulverdiffraktogramme wurden mit Diffraktometern des Typs STADI P der Firma Stoe
aufgenommen. Die Messungen wurden sowohl in Debye-Scherrer-Geometrie in Transmission
als auch, um Störungen durch Fluoreszenz der enthaltenen Atomspezies zu vermeiden, in
Reflexion in Bragg-Brentano-Geometrie unter Verwendung von CuKα-Strahlung der
Wellenlängen 1,54051 Å und 1,5443 Å durchgeführt. In Reflexion wurden die Proben im
Winkelbereich von 15 bis 80° (2Θ) mit 0,04 Schritten und 100 Sekunden pro Punkt
vermessen.
4.3.2 Stickstoffsorption
Eine Methode, die Oberfläche eines Feststoffes zu bestimmen, stellt die Messung des
adsorbierten Gasvolumens bei einer definierten Temperatur dar. Dieses auch Sorption
genannte Verfahren wurde im Rahmen dieser Arbeit ausschließlich mit Stickstoff als
Adsorptiv durchgeführt. Es handelt sich bei der Sorption von Stickstoff um eine reine
Physisorption, welche bei einer Temperatur von 77 K, der Temperatur von flüssigem
Stickstoff, durchgeführt wurde. Aus einer Auftragung des adsorbierten Volumens gegen den

4.3 Charakterisierungsmethoden
41
Druck p/p0 (p0 = Gleichgewichtsdampfdruck von Stickstoff bei 77 K) ergibt sich somit eine
Isotherme. Die gesamte Messung bestand aus einem aufsteigenden Ast, der
Adsorptionsmessung, sowie einem absteigenden Ast, der Desorptionsmessung. Aus der Form
der Messkurven kann auf unterschiedliche Porenbeschaffenheit, wie Mikro- oder Mesoporen,
geschlossen werden. Unter Annahme einer monodispersen Partikelverteilung kann bei kleinen
Partikeln auch das Zwischenkornvolumen und die Partikelgröße berechnet werden. Die
Berechnung der Oberfläche erfolgte nach dem Verfahren von Brunauer, Emmet und Teller
(BET-Verfahren)116. Die dem Verfahren zugrunde liegenden Gleichungen sind:
( )( )
CnppC
Cnppnpp
momoad
0
0
0 /11/1
/ −+=
−
mit
AmomoBET NSnS =
Mit bekanntem Smo (durchschnittlicher Flächenbedarf eines Stickstoffmoleküles bei 77 K
16,2·10-20 m2) und der Avogadrozahl NA kann hieraus SBET, die spezifische Fläche des
Substrates, berechnet werden. p steht hier für den gemessenen Dampfdruck und p0 für den
Sättigungsdampfdruck von Stickstoff bei 77 K. nmo steht für die adsorbierte Stoffmenge in der
Monoschicht, nad für die gesamte adsorbierte Stoffmenge. C ist eine dimensionslose
Kennzahl.
Da diese Methode die Multischicht-Adsorption berücksichtigt, ist sie sowohl für meso- und
makroporöse Materialien als auch für unporöse Substanzen geeignet.
Alle Messungen wurden mit einem ASAP 2010 C Gerät der Firma Micromeritics
durchgeführt. Die Proben wurden zuvor bei 200 °C vier Stunden im Vakuum aktiviert und bei
77 K mit statisch-volumetrischem Verfahren vermessen.
4.3.3 Transmissionselektronenmikroskopie (TEM)
Bei dieser Untersuchungsmethode wird ein Elektronenstrahl im Hochvakuum auf die Probe
fokussiert und produziert durch unterschiedliche Schwächungen oder Beugungen innerhalb
des Substrates ein Abbild von diesem. Die Proben dürfen wegen des Intensitätsverlustes eine

4 Experimentelles
42
Dicke von wenigen 100 nm nicht überschreiten. Da Elektronen eine weit geringere
Wellenlänge (Faktor 105 kleiner) als herkömmliches Licht besitzen, ist die physikalisch
mögliche Maximalvergrößerung bei diesem Verfahren weit höher als bei herkömmlicher
Mikroskopie. Die Auflösung kann bis zu 2 Å betragen und wird vorrangig durch
unterschiedliche Beschleunigungsspannungen der Elektronen beeinflusst. Mit dieser Technik
ist es möglich, bei Hochauflösung (HRTEM) bis in atomare Bereiche vorzudringen und selbst
einzelne Netzebenen in kristallinen Domänen sichtbar zu machen. Man erhält hierbei sowohl
Informationen über die Morphologie der Gesamtprobe als auch über die Beschaffenheit
einzelner Domänen oder Partikel. Problematisch ist hierbei wie bei allen mikroskopischen
Verfahren die Repräsentationsgüte der Einzelmessung. Da von kleinen mikroskopischen
Ausschnitten auf eine makroskopische Probe geschlossen wird, sind mehrere Messungen über
den gesamten Probenbereich notwendig, um repräsentative verlässliche Daten zu erhalten.
Die in dieser Arbeit gezeigten TEM Aufnahmen wurden von Herrn B. Spliethoff an einem
Hitachi HF 2000 Elektronenmikroskop mit kaltem Feldemitter (CFE) aufgenommen. Die
maximale Beschleunigungsspannung betrug 200 kV.
4.3.4 Energiedispersive Analyse von Röntgenstrahlen (EDX)
Bei der Interaktion von Elektronenstrahlen mit Materie wird immer ein gewisser Betrag an
Röntgenstrahlung durch inneratomare Relaxationseffekte aus den Schalen der Elektronenhülle
frei. Diese Strahlung ist elementspezifisch und tritt als positiver Nebeneffekt auch bei
mikroskopischen Verfahren, wie z.B. der Transmissionselektronenmikroskopie, auf, welche
mit Elektronenstrahlen arbeiten. Die Informationen kommen aus einem Bereich, der unter der
Oberfläche nur wenige Mikrometer in die Tiefe reicht. Detektiert werden können meist nur
Elemente oberhalb der Ordnungszahl 11, da die leichten Elemente nur sehr wenig
Röntgenstrahlung freisetzen. Es sind jedoch auch Geräte erhältlich welche mit
Berylliumfenstern ausgerüstet sind und bis hinab zu Kohlenstoff mit einer Ordnungszahl von
6 messen können. Die Quantifizierung bei kleinen Ordnungszahlen ist jedoch relativ ungenau
und erreicht auch bei höheren Ordnungszahlen eher einen semiquantitativen Status. Elemente
höherer Ordnungszahl können bis hinab zu einem Gewichtsprozent noch zuverlässig
qualitativ detektiert werden.
Bei ortsaufgelösten Bestimmungen ist dieses Verfahren hervorragend geeignet, um
Variationen der ungefähren Zusammensetzung auf kleinstem Raum, z.B. innerhalb eines
einzigen Partikels, zu analysieren.

4.3 Charakterisierungsmethoden
43
Die im Rahmen dieser Arbeit durchgeführten EDX Messungen wurden in obigem
Transmissionselektronenmikroskop von Herrn B. Spliethoff mit einem Gerät der Firma Noran
durchgeführt. Zur Detektion wurde ein Si(Li)-Detektor benutzt. Die maximale Ortsauflösung
betrug 2 nm.

5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
Theorie und Praxis sind eins wie Leib und Seele, und wie
Seele und Leib liegen sie großenteils miteinander im Streit.
Marie Freifrau von Ebner-Eschenbach
5.1 Einführung
Seit Jahrzehnten haben sich spektroskopische Methoden in Chemie und Physik als
unverzichtbar zur Lösung analytischer Probleme erwiesen und sind heutzutage aus den
modernen Naturwissenschaften nicht mehr wegzudenken. Üblicherweise werden diese
Methoden in zwei große Gruppen unterteilt. Die erste Gruppe macht sich die Analyse der
durch eine Probe hindurchgedrungenen Photonen zunutzte. Diese transmittierten Photonen
werden in Absorptionsmessungen unter anderem in IR- oder UV/Vis-Geräten herangezogen
und liefern direkte Informationen über den Teil des Lichtes, welcher nicht direkt mit der zu
untersuchenden Materie wechselwirkt. Die zweite Gruppe analysiert physikalische
Eigenschaften der Photonen, welche von der zu untersuchenden Probe gestreut oder reflektiert
wurden. Es werden also direkte Auskünfte über Art und Größe der Wechselwirkung zwischen
Photon und Materie erhalten.
Strahlungslose Übergängedurch Stossprozesse
Absorption
Abbildung 14 Schematische Darstellung eines photoakustischen Prozesses. Nach der Absorption kann
die Energie entweder durch die Umkehrung des Absorptionsprozesses unter Aussendung
eines Photons relaxieren oder durch strahlungslose Prozesse in translatorische Energie
umgewandelt werden.
44

5.2 Geschichte
Die Photoakustik geht einen alternativen Weg, indem direkt die Energieabsorption der Probe
als Funktion des einfallenden Lichtstrahls gemessen wird (Abbildung 14). Dies gelingt bei
gasförmigen Proben, falls die in Form von Licht aufgenommene Energie durch interne
strahlungslose Umwandlungsprozesse (internal conversion oder IC) teilweise in
translatorische Energie umgewandelt wird. Diese von der ursprünglichen aufgenommenen
Form unterschiedliche Energie kann durch Zuhilfenahme geeigneter Detektoren analysiert
werden. Die Photoakustik nutzt hierfür gewöhnliche Mikrofone und fängt die durch die
absorbierenden Materialien generierten thermisch erzeugten Druckwellen aus den
Übergängen auf. Da die Intensität lediglich von der Zahl der absorbierenden Zentren und der
Stärke des einfallenden Lichtes abhängt, kann man hieraus Rückschlüsse über die
Zusammensetzung der zu analysierenden Proben ziehen. Es wird quasi eine
Konzentrationsmessung ermöglicht.
5.2 Geschichte
Der photoakustische Effekt wurde zuerst im Jahre 1880 von Alexander Graham Bell
beschrieben117. Er berichtete der „American Association for the Advancement of Science”
von der zufälligen Entdeckung des photoakustischen Effektes in Feststoffen.
Abbildung 15 Alexander Graham Bell bei dem Versuch ein Phototelefon zu entwickeln. Gezeigt ist die
Empfängereinheit bei der er den photoakustischen Effekt erstmals bemerkte118.
Er entdeckte diesen Effekt bei den Arbeiten an einem Photophon, welches Töne in Form von
Licht übertragen sollte. Der apparative Aufbau, den er zum Empfang benutzt ist in Abbildung
15 gezeigt. Er fand, dass unterschiedliche Materialien, wenn sie einem unterbrochenen
45

5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
46
Sonnenstrahl ausgesetzt waren, einen hörbaren Ton produzierten. Weiter konnte er einen
Zusammenhang zwischen der Intensität des Tones und sowohl Farbe als auch Form der
Materialien finden. Laut seinen Beobachtungen waren die Töne von porösen, schwarzen
Materialien am lautesten119. Sowohl Wilhelm Conrad Roentgen als auch John Tyndall
begannen, nachdem sie von diesen Ergebnissen gehört hatten, ebenfalls mit Experimenten auf
diesem Gebiet. In einer Reihe von Experimenten wurden Feststoffe, Flüssigkeiten und Gase
untersucht120,121. Wie man fand, wurde die Frequenz des generierten Tones lediglich von der
Modulation des einfallenden Lichtes bestimmt. Da zu dieser Zeit die Gasgesetzte schon gut
untersucht waren, konnte man sich den Zusammenhang zwischen Energieabsorption und
nachfolgender Ausdehnung des Mediums leicht über Volumenausdehnung und Druckanstieg
erklären. Bei Flüssigkeiten und Feststoffe bereitete die Interpretation zu dieser Zeit noch
Schwierigkeiten.
Als Detektor verwendete man ausschließlich das menschliche Ohr. Dies und die Tatsache,
dass dem Effekt keine praktische Bedeutung beigemessen wurde, trug dazu bei, dass er als
Kuriosität abgetan wurde und die Photoakustik in Vergessenheit geriet. Ungefähr 50 Jahre
lang wurden keine weiteren Experimente auf diesem Gebiet durchgeführt, bis zur Entdeckung
des Mikrofons. Da nun auch die Reproduzierbarkeit der Messergebnisse sichergestellt war,
wurden erste Experimente durchgeführt und 1938 der photoakustische Effekt von Viengerow
aus Leningrad erstmals zur Konzentrationsbestimmung von Einzelgasen in Gasmischungen
über die Infrarot-Absorption von Gasen herangezogen. Als Lichtquelle benutzte er die
Schwarzkörperstrahlung eines Nernst-Stifts. Mit seinen Aufbauten war er in der Lage,
Kohlendioxid in Stickstoff bis zu einer Konzentration von 0,2 Volumenprozenten zu
detektieren und zu quantifizieren122. 1946 wurde ein von Luft weiterentwickeltes, differenziell
arbeitendes Luftanalysegerät, welches drei Jahre zuvor von ihm entworfen worden war,
kommerziell erhältlich. Mit diesem Aufbau konnte nun Kohlendioxid bis zu einer
Konzentration von einigen ppm gemessen werden123. Ab 1948 erkannte man, dass diese
Technologie durch genaue Phasenanalysen des Signals erfolgversprechend zur Untersuchung
der Lebensdauer von Energiezuständen in Molekülen eingesetzt werden konnte124. Dies blieb
nach der Erfindung der Spektrophotometer bis Anfang der 70er Jahre auch die
Hauptanwendung der Photoakustischen Spektroskopie (PAS)125. Als dann Laser Einzug in die
Wissenschaft hielten, konnten Experimente nicht nur an gasförmigen Proben, sondern
aufgrund der starken Strahlungsquellen auch an Flüssigkeiten und Feststoffen durchgeführt
werden.

5.3 Prinzip der Photoakustik
Es bleibt festzuhalten, dass, obwohl das Prinzip schon seit über 120 Jahren bekannt ist, die
Photoakustik seither wenig Verbreitung gefunden hat.
5.3 Prinzip der Photoakustik
Im folgenden sollen in aller Kürze die notwendigen Grundlagen geschildert werden, um die
Photoakustik als Messmethode zu verstehen. Eine tiefere Behandlung der theoretischen
Grundsätze überschreitet den Rahmen dieser Arbeit.
5.3.1 Strahlungsquellen
Das photoakustische Signal wird durch Energieabsorption der Moleküle in einer
Gasmischung hervorgerufen. Die Energie wird durch Photonen in das System eingebracht.
Quellen wie Elektronen- oder Ionenstrahlen, welche theoretisch auch verwendet werden
können, sollen hier außer Acht bleiben. Im folgenden werden die möglichen Photonenquellen
und ihre Vor- und Nachteile beschrieben. Die Quellen sind hierbei in die zwei wichtigsten
Gruppen unterteilt, die inkohärenten Glühquellen oder Lichtbögen und die kohärenten
Laserquellen. Im Rahmen dieser Arbeit werden lediglich Charakteristika dieser Systeme
beschrieben. Die teilweise komplexen theoretischen Beschreibungen sollen hier nicht
diskutiert werden.
Inkohärente Strahlungsquellen
Die wichtigsten Vertreter dieser Klasse von Strahlern sind die schon erwähnten glühenden
Strahler und Leuchtbogenlampen. Die Energie, die von einer inkohärenten Glühquelle
ausgeht, kann in erster Näherung durch die Gleichungen für einen schwarzen Strahler
beschrieben werden. Nach Stefan-Boltzmann ist die gesamte Strahlungsintensität bei einem
schwarzen Strahler proportional zur vierten Potenz der Temperatur. Die Strahlungsverteilung
wird durch die bekannte Planck´sche Strahlungsformel wiedergegeben.
47
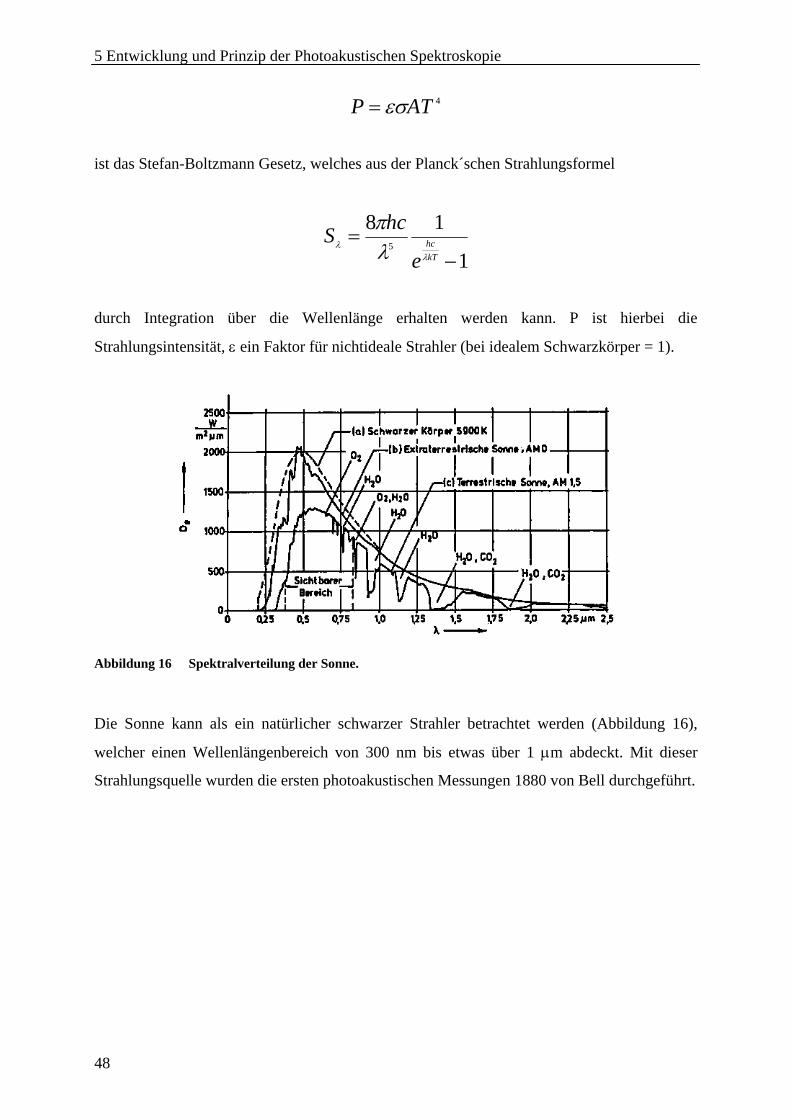
5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
48
4ATP εσ=
ist das Stefan-Boltzmann Gesetz, welches aus der Planck´schen Strahlungsformel
1
185
−=
kThc
e
hcSλ
λ λπ
durch Integration über die Wellenlänge erhalten werden kann. P ist hierbei die
Strahlungsintensität, ε ein Faktor für nichtideale Strahler (bei idealem Schwarzkörper = 1).
Abbildung 16 Spektralverteilung der Sonne.
Die Sonne kann als ein natürlicher schwarzer Strahler betrachtet werden (Abbildung 16),
welcher einen Wellenlängenbereich von 300 nm bis etwas über 1 µm abdeckt. Mit dieser
Strahlungsquelle wurden die ersten photoakustischen Messungen 1880 von Bell durchgeführt.
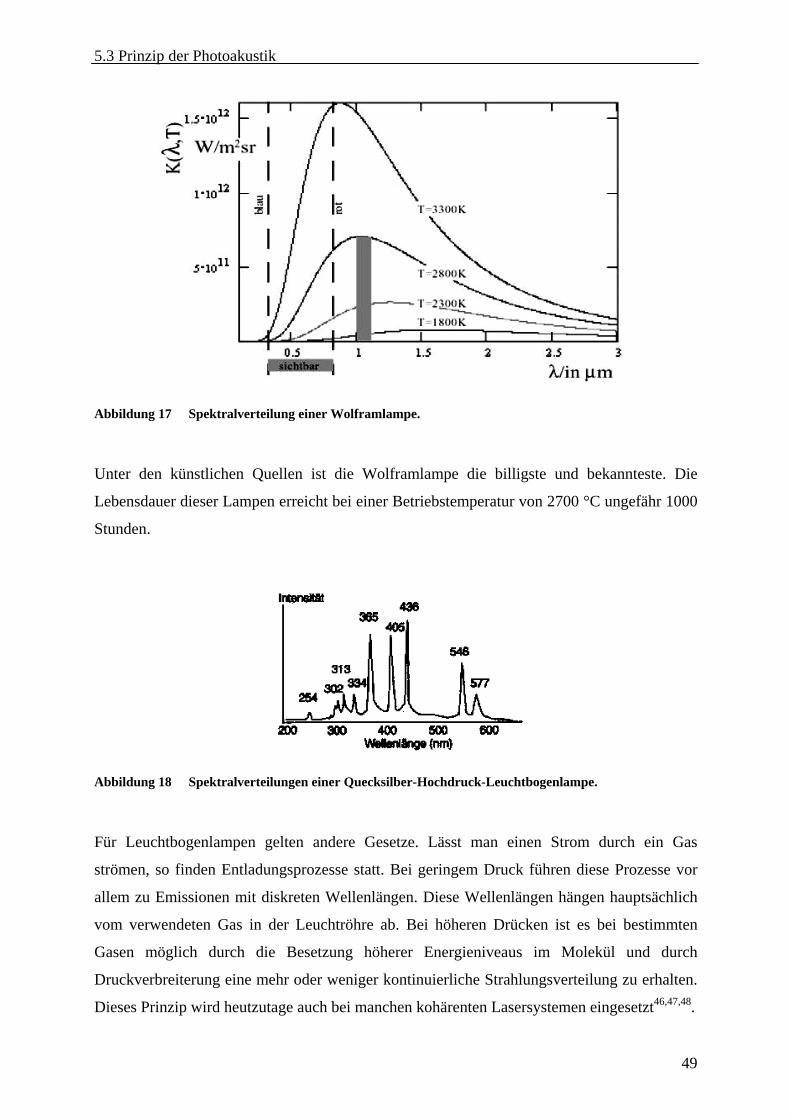
5.3 Prinzip der Photoakustik
Abbildung 17 Spektralverteilung einer Wolframlampe.
Unter den künstlichen Quellen ist die Wolframlampe die billigste und bekannteste. Die
Lebensdauer dieser Lampen erreicht bei einer Betriebstemperatur von 2700 °C ungefähr 1000
Stunden.
Abbildung 18 Spektralverteilungen einer Quecksilber-Hochdruck-Leuchtbogenlampe.
Für Leuchtbogenlampen gelten andere Gesetze. Lässt man einen Strom durch ein Gas
strömen, so finden Entladungsprozesse statt. Bei geringem Druck führen diese Prozesse vor
allem zu Emissionen mit diskreten Wellenlängen. Diese Wellenlängen hängen hauptsächlich
vom verwendeten Gas in der Leuchtröhre ab. Bei höheren Drücken ist es bei bestimmten
Gasen möglich durch die Besetzung höherer Energieniveaus im Molekül und durch
Druckverbreiterung eine mehr oder weniger kontinuierliche Strahlungsverteilung zu erhalten.
Dieses Prinzip wird heutzutage auch bei manchen kohärenten Lasersystemen eingesetzt46,47,48.
49

5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
50
Für den Infrarot-Bereich ist es schwieriger geeignete Strahlungsquellen zu finden. Eine der
einfachsten und weit verbreiteten Quellen ist der Globar, der, wenn er mit Thoriumdioxid
beschichtet ist, bis zu Wellenlängen von über 30 µm verwendet werden kann. Der Nernst
Stift, eine Mischung von Thorium-, Yttrium- und Zirconiumoxid, ist ebenfalls ein geeigneter
Infrarot-Strahler, der vor allem im nahen Infrarot verwendet werden kann und bis zu
Wellenlängen von 14 µm wesentlich intensiver ist als ein herkömmlicher Globar. Es bleibt
festzuhalten, dass die inkohärenten Infrarot-Quellen generell wesentlich schwächer sind als
die UV/Vis-Quellen und mehr Aufwand betrieben werden muss, um im Infraroten
photoakustische Spektroskopie (PAS) zu betreiben.
Als Nachteil der inkohärenten Quellen ist ihre unzureichende Intensität zu betrachten. Da ihre
Leistung über einen weiten spektralen Bereich verteilt ist, steht bei einer bestimmten
Wellenlänge nur ein Bruchteil der Gesamtintensität zur Verfügung. Weiter können sie aus
obigem Grund meist nur nach einer geeigneten optischen Filterung für spektroskopische
Messungen verwendet werden, da ein Einstrahlen mit einer breitbandigen Infrarot-Strahlung
ein selektives Messen hinfällig machen würde.
Bei einer Messung mit hoher Auflösung ist also erstens ein großer Aufwand zu betreiben um
die gewünschte Wellenlänge zu erhalten, und zweitens steht diese dann nur noch bei sehr
geringer Strahlstärke zur Verfügung. Dem entgegen steht die einfache Handhabung der
Strahlquellen. Generell liegt der finanzielle Aufwand weit unter dem kohärenter Quellen, so
dass für Anwendungen, bei denen nur geringe Strahlstärken benötigt werden, dieses System
durchaus seine Berechtigung findet. Weiterhin existieren für fast alle Bereiche des Spektrums
Strahlquellen, so dass durch Modifikation des Filteraufbaus schnell auf eine neue
Wellenlänge justiert werden kann.
Kohärente Strahlungsquellen
Als kohärente Strahler werden fast ausschließlich die mittlerweile weit verbreiteten, 1960
entwickelten LASER-Systeme eingesetzt. Sie bestehen im wesentlichen aus einem optischen
Resonator, welcher bei bestimmten Bedingungen durch Rückkopplungseffekte eine
Verstärkung des Lichtes bewirkt. Ein gewöhnlicher 20 mW Diodenlaser kann im Gegensatz
zu den üblichen Strahlquellen, welche eine spektrale Leistungsdichte von bis zu
10 mW·mm-2nm-1 aufweisen können, eine Leistungsdichte von bis zu 1010 mW·mm-2nm-1
haben. Dies wird durch die extrem schmalbandige Emission und die räumliche Fokussierung
bewirkt, welche üblicherweise mit der Laseremission verbunden sind.

5.3 Prinzip der Photoakustik
Der Laservorgang tritt ein, wenn eine Inversion der Zustandsbesetzung zweier energetisch
verschiedener Zustände besteht. Ein höheres Niveau ist also stärker besetzt als das niedrigere.
Platziert man dieses aktive Medium in einem geeigneten Resonator, kann nun ein Photon
kaskadenartig eine stimulierte Emission verursachen. Der Resonator besteht hierbei lediglich
aus zwei planparallelen Spiegeln, welche die Photonen stetig reflektieren und nur einen Teil
der Leistung auskoppeln, um den Laserprozess aufrecht zu erhalten. Die durch diesen
Vorgang generierte Strahlung hat eine hohe optische Reinheit und ausgezeichnete räumliche
Kohärenz. Die Besetzungs-Inversion kann unterschiedlichste Ursachen haben, z.B. optisches
Pumpen oder Anregungen durch Stöße mit freien Elektronen aus Gasentladungen.
Der schwerwiegendste Nachteil der Laser ist, dass sie lediglich in der Lage sind Strahlung
einer oder weniger diskreter Wellenlängen zu liefern. Die häufigsten Laser dieser Klasse,
welche im Sichtbaren oder UV-Bereich eingesetzt werden, sind der Argon-Ionen-, der Rubin-,
der Nd:YAG- und der Helium-Neon-Laser. Im Infraroten Bereich sind CO- und CO2-Laser
die häufigsten Systeme.
Obwohl mittlerweile für fast jede Wellenlänge Laser existieren, sind die hohen Kosten, die
mit den meisten Wellenlängen einhergehen, ein nicht zu vernachlässigender Faktor.
Theoretisch ist jede Wellenlänge realisierbar, doch die „günstigen“ Lasersysteme, wie z.B.
der CO2-Gaslaser, finden aus Kostengründen den häufigsten Einsatz. Von Vorteil ist auch die
hohe Strahlintensität, die mehrere 100 W betragen kann. Er wird daher häufig auch für viele
technische Einsätze, wie z.B. Schweißarbeiten, verwendet. Leider ist der
Wellenlängenbereich dieses Systems auf ein Fenster von 5-7 µm für CO- und 9–11 µm für
CO2–Laser beschränkt. Lasersysteme wie der Farbstofflaser oder ein Verfahren, welches eine
geeignete Wellenlänge über Frequenzmischung generiert, sind aufgrund des komplexen
Aufbaus und der Unhandlichkeit im Betrieb dieser Systeme meist unterlegen.
Eine Alternative bei Messaufbauten, welche weniger sensitiv sein sollen und daher mit
geringeren Strahlintensitäten auskommen, sind einige Diodenlaser. Diese werden heute in
Massenproduktion hergestellt und sind daher relativ günstig. Diese Laser haben allerdings
Leistungen, die sich deutlich unterhalb von 1 W bewegen. Ein besonderes Augenmerk kommt
hier dem für den kompletten Infrarotbereich erhältlichen Bleisalzlaser zu. Dieses System kann
nach einmaliger Anschaffung durch Wechseln der Diode leicht auf eine andere Wellenlänge
eingestellt werden. Die Problematik besteht lediglich in der Stabilisierung der Wellenlänge,
welche durch eine exakte Temperaturregelung bei tiefen Temperaturen (-270 bis –140 °C)
erfolgen muss. Eine einzige Diode ist so in der Lage einen Wellenlängenbereich von bis zu
100 reziproken Zentimetern abzudecken.
51

5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
52
Eine genaue Beschreibung des im Rahmen dieser Arbeit verwendeten CO2-Lasers findet sich
in Abschnitt 6.2.
5.3.2 Licht-Materie-Interaktion
Der photoakustische Effekt beruht auf der Absorption von Licht durch Materie. Dieser
Prozess kann logisch in zwei Teilschritte eingeteilt werden. Die Absorption, die determiniert,
welche Spezies angeregt wird, und die darauf folgenden weit komplexeren
Relaxationsprozesse.
Die Energie kann in einem einzigen Schritt oder innerhalb des Moleküls auf unterschiedlichen
Wegen schrittweise abgegeben werden. Teilweise können so detaillierte Informationen über
die energetische Struktur der absorbierenden Spezies erhalten werden.
Absorption und Relaxation
Wird ein Photon von einem Gasmolekül absorbiert, so versetzt es dieses in einen angeregten
Zustand. Die Energie kann in Form von Rotations-, Schwingungs- oder auch elektronischer
Energie im Molekül gespeichert werden. Es gibt nun mehrere Wege wie die Energie wieder
abgegeben werden kann.
1. Reemission des Photons und Rückkehr in den Ausgangszustand.
Folge: Strahlungsemission
2. Eine chemische Reaktion wird eingeleitet, wodurch Bindungen umgeordnet werden.
Folge: Photochemie
3. Die Energie wird durch Stossprozesse auf weitere Gasmolekül der selben oder einer
anderen Spezies übertragen.
Folge: Energietransfer
Ist die ursprüngliche Anregung elektronischer Natur, spielen alle diese drei Wege eine
gleichberechtigte Rolle, was den Gesamtprozess verkompliziert aber eine interessante
Möglichkeit darstellt, physikalische Daten über das entsprechende System zu erhalten. Ist die
Anregung dagegen auf Schwingungsfreiheitsgrade beschränkt, so ist die Lebensdauer der

5.3 Prinzip der Photoakustik
energetisch angeregten Zustände lang genug, um eine Relaxation über Stossprozesse zu
ermöglichen, und die Energie zu schwach, um photochemische Prozesse einzuleiten.
Während der Stöße wird die Energie portionsweise in kleinen Schritten abgegeben, wobei sie
bei Gasen größtenteils in translatorische Energie übergeht. Dies führt letztendlich zu einer
Aufheizung der Probe. Natürlich sind während dieser Vorgänge jederzeit auch
Strahlungsprozesse von Photonen geringeren Energieinhaltes möglich. Bei eintretender
Emission von Strahlung ist zusätzlich zu photoakustischen bzw. photothermischen
Messungen auch eine Analyse der laserinduzierten Fluoreszenz (LIF) möglich.
Für photoakustische bzw. photothermische Prozesse ist lediglich der Teil der Energie wichtig,
der in Form thermischer Energie bzw. Bewegung übrig bleibt. Die hierdurch bewirkte
Aufheizung hat makroskopisch einen lokalen Druckanstieg zur Folge, der sich gleichmäßig
im Messmedium ausbreitet. Der Unterschied der photoakustischen und der photothermischen
Analyse liegt in der Art, wie diese Druckschwankungen detektiert werden.
Detektor
a
b
c
Detektionslaser
Messlaser
Chopper
Laser
Laser
Laser
Abbildung 19 Vergleich von a) herkömmlicher Absorptions-Spektroskopie, b) photothermischer
Analyse und c) photoakustischer Analyse.
Die Photoakustik misst direkt den Druck über spezielle Mikrofonaufbauten, während
photothermische Analysen die Änderung physikalischer Parameter durch den Druck bzw. den
Temperaturanstieg messen. Wie in Abbildung 19 gezeigt, geschieht dies z.B. durch einen
Detektionslaser, welcher auf den sich ändernden Brechungsindex der Probe anspricht. Die
53

5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
54
Ablenkung des Laserstrahles können gemessen und hieraus Rückschlüsse auf den
Brechungsindex bzw. die thermische Energie gezogen werden.
Wird die Anregung des Systems nun periodisch durchgeführt, so entsteht in der Messzelle
eine periodische Druckschwankung, was makroskopisch einem Ton entspricht.
Moleküle
Laserstrahl
Relaxation
Abbildung 20 Schematische Darstellung des photoakustischen Effekts in Gasen. Wichtig für eine
effektive Modulation ist eine vollständige Relaxation vor dem nächsten Laserpuls.
Wie aus Abbildung 20 ersichtlich, ist die Erzeugung eines photoakustischen Signals auf eine
wiederkehrende energetische Anregung zurückzuführen, die großteils in Bewegungsenergie
umgewandelt wird. Die Anregungsrate ist hierbei langsam im Vergleich zur Relaxation des
Systems. Der Anregungsprozess wird meist durch mechanische Chopper oder direkt durch
modulierte Lasersysteme periodisch durchgeführt.
Weitere Relaxationsprozesse
Wie schon im vorherigen Abschnitt angedeutet, ist in einem photoakustischen Experiment
weit mehr Information enthalten als nur die über die Zahl der Absorptionszentren zugängliche
Konzentration der spektroskopierten Spezies.

5.3 Prinzip der Photoakustik
Photoakustische Analyse
ChemischeReaktion
Transport-koeff.
Schall-geschw.
Wärme-kapazität
Gas-konstante
EnergieTransfer
Virial-koeff.
Abbildung 21 Einige, abgesehen von einfachen Konzentrationsmessungen, durch Photoakustik
zugängliche Prozesse und physikalische Größen.
Aus einer genauen, eventuell zeitlich aufgelösten Signalanalyse, lassen sich Informationen
über Gaskonstante, kinetische Größen und weitere dynamische Prozesse erhalten, wie in
Abbildung 21 gezeigt ist. Vor allem die Kombination mit weiteren Spektroskopiearten, wie
z.B. LIF-Messungen, ist eine sinnvolle Ergänzung. Hierdurch können Relaxationswege der
Energie verfolgt werden, die der Photoakustik verborgen bleiben.
In obiger Abbildung sind lediglich die direkt zugänglichen Informationen aus
photothermischen bzw. photoakustischen Messungen abgebildet. Bei gekoppelten
Relaxationsprozessen wie z.B. ein Energietransfer in Freiheitsgrade der Translation mit
anschließender Emission eines Photons, welches mittels anderer Spektroskopiearten detektiert
wird, lassen sich direkte Einblicke in die Energieschemata der Moleküle erhalten. Weiter sind
z.B. über eine genaue Phasenanalyse sowohl Spinumkehrprozesse als auch andere
elektronische Vorgänge detektierbar. Eine umfassende Behandlung dieser Phänomene würde
den Rahmen dieser kurzen Abhandlung sprengen.
5.3.3 Photoakustische Effekte in gasförmigen Medien
Eine umfassende Herleitung der dieses Phänomen beschreibenden Gleichungen ist im
Rahmen dieser Arbeit nicht möglich, dennoch sollen einige der fundamentalen empirischen
und theoretischen Gleichungen der Photoakustik kurz behandelt werden. Im folgenden wird
auf die unterschiedlichen photoakustischen Aufbauten eingegangen und einige Probleme
sowie Lösungen beim Bau eines photoakustischen Messsystems beschrieben.
55

5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
56
5.3.3.1 Grundlegende Gleichungen126
Eine der grundlegenden Gleichungen, welche die Anregung von Schallwellen durch
Absorption von Energie durch Gasmoleküle beschreibt, lautet im Fall eines einfachen
Systems mit zwei energetischen Zuständen:
( ) ( )[ ]( )2
212210
10
0
22
222 πγω
ωττδτ
ω−−
−−
−
+++= tiS
v
eBIBI
BIC
kENp
p ist hierbei der entstehende Druck, Cv die Wärmekapazität bei konstantem Volumen, N die
Zahl der Teilchen pro Volumeneinheit, E der relative Wert des oberen Energieniveaus, τ
dessen Lebensdauer und τS die Lebensdauer welche durch Relaxationsprozesse durch
Kollisionen bedingt wird. B ist eine Konstante die den Einsteinkoeffizient für stimulierte
Emission enthält, I0 die Intensität des einfallenden Lichtes und der Rest beinhaltet den
zeitlichen Verlauf, welcher durch eine Exponentialfunktion wiedergegeben werden kann126.
Diese Gleichung lässt sich für zwei wichtige Grenzfälle folgendermaßen vereinfachen. Ist die
Laserleistung gering und entsprechend 2BI0 << τ-1, lautet die Gleichung:
( )( )2
2122
0
22
12 πγω
τωδ
ττ
ω+−
+
≅=− ti
Sv
eBI
CkENqp
q ist das vom Mikrofon aufgenommene Signal und ist äquivalent zu –p. Für die Photoakustik
lassen sich hieraus folgende wichtige Punkte ableiten. Die Intensität des Messsignals steigt
linear mit zunehmender Laserintensität. Weiter steigt die Intensität auch proportional zu N2,
was den Einfluss der Gasdichte verdeutlicht und da die Lebensdauer, verursacht durch
Stosseffekte, mit steigender Temperatur abnimmt und folglich τS2 kleiner wird, steigt die
Intensität auch mit steigender Temperatur.

5.3 Prinzip der Photoakustik
Bei hohen Laserleistungen und dementsprechend 2BI0 >> τ-1 lässt sich die Gleichung
umformen zu:
( )2
0
22 1 πγωδτ
ω+−−≅ ti
S
v
eBIC
kENq
Es wird hier klar, dass sich die Intensität eines Signals nicht beliebig durch eine Steigerung
der Laserintensität erhöhen lässt. Bei zu hohen Leistungen wird lediglich eine 1/I0
Abhängigkeit gefunden. Dies lässt sich anschaulich so verdeutlichen, dass man nicht stärker
pumpen kann als es das obere Energieniveau „aufnehmen kann“. Das obere Niveau muss in
der Lage sein, die Energie in dem Maß abzugeben, wie es sie aufnimmt. Tritt die 1/I0
Abhängigkeit auf, nennt man dies Sättigungseffekt.
Experimentell stellt man oft eine Abhängigkeit des Messsignals der folgenden Form fest.
XNlq ≈
Wobei X eine Konstante ist, die alle Einflüsse beinhaltet. Dies gilt jedoch nur für kleine
Konzentrationen des Messgases und nur angenähert. Die Intensität ist hier linear von der
Zelllänge l abhängig, weshalb häufig sogenannte Multipass Zellen verwendet werden. Weiter
steigt die Intensität linear mit der Konzentration des Gases N, was auch im Rahmen dieser
Arbeit innerhalb der Fehlergrenzen festgestellt werden konnte.
5.3.3.2 Zelldesign eines photoakustischen Aufbaus
Es existiert eine Vielzahl an unterschiedlichen Zellenkonzepten in der Gas-Photoakustik,
deren einzige Gemeinsamkeit darin besteht, dass ein Mikrofon zur Detektion benutzt wird.
Welcher Aufbau am besten für ein Experiment geeignet ist, hängt von der Problemstellung ab.
57

5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
58
b
a
c
bekannt
unbekannt
Laser
d
e
Abbildung 22 Einige Zellkonzepte in der photoakustischen Spektroskopie. a) gewöhnliche
nichtresonante oder resonante Zelle, b) Multipass-Zelle, c) Zelle mit zusätzlichen
akustischen Filtern, d) differentielle Zelle zur Untergrundsubtraktion, e) Zelle im Innern
eines Laserresonators.
Die typischerweise verwendeten Aufbauten sind in Abbildung 22 gezeigt. Meist werden
einfache Zellen wie in a gezeigt verwendet. Modifikationen hiervon sind in b und c gezeigt.
Die Verbesserung liegt zum einen in einer Verlängerung des Absorptionspfads (b) oder in
einer besseren akustischen Filterung (c). Das in d gezeigte Verfahren wird häufig zur
Untergrundsubtraktion eingesetzt, da alle konstruktionsbedingten Störungen und zusätzliche
störende Gaseinflüsse herausgerechnet werden können. Auch das Verfahren e wird zur
Messung eingesetzt, ist jedoch weit weniger verbreitet als die anderen Methoden.
Ein idealer Aufbau sollte das gewünschte Messsignal verstärken, ohne eventuelle
Störgeräusche zu erzeugen. Mögliche auftretende Störgeräusche können mehrere Ursachen
haben.
Akustische Störgeräusche
Wird der Strahl durch Fenstermaterial absorbiert, so treten Störgeräusche auf, die die gleiche
Frequenz haben wie das Messsignal. Auch durch an den Wänden gestreutes Licht können
Störungen im System auftreten. Letzteres trägt allerdings weit weniger zur Störung bei als die
Fensterabsorptionen.
Durch ein differenzielles Design, wie es in Abbildung 22 d zu sehen ist, lassen sich durch
Zellgeometrie und –aufbau auftretende Störungen leicht eliminieren.

5.3 Prinzip der Photoakustik
Weitere Störgeräusche entstehen aus den den Messaufbau umgebenden Schallquellen, wie
z.B. dem Chopper, der in vielen Aufbauten als modulierendes Element eingesetzt wird. Durch
eine geeignete Isolierung der Messzelle lassen sich diese Störeinflüsse leicht beheben.
Sollen wie auch in dieser Arbeit Messungen an strömenden Gasen durchgeführt werden, so
treten weitere Störgeräusche auf, welche sich nur schwer durch konstruktive Maßnahmen
unterdrücken lassen. Abhilfe schafft hier, das Gas im Knotenpunkt der stehenden Welle in
den Resonator zu injizieren. So wird eine Störung der resonanten Welle weitestgehend
vermieden.
Elektronische Störgeräusche
Alle im System vorhandenen elektronischen Bauteile, wie z.B. der Verstärker, produzieren
ein Störsignal. Eine durch diese Ursachen entstehende Störung lässt sich nur durch den
Einbau eines besseren und meist auch teureren Bauteils mit entsprechend niedrigen
Störwerten lösen. Man sollte generell beachten, dass Kabel so kurz wie möglich gewählt
werden sollten. Vor allem vor der Verstärkung des Messsignals ist dies häufig eine
Störungsursache, da eine vor der Verstärkung eingetretene Störung entsprechend verstärkt
wird und somit wesentlich zur Gesamtstörung beiträgt. Nach einer geeigneten Vorverstärkung
räumlich nahe am eigentlichen Mikrofon ist das Messsignal weniger anfällig für in die
Leitungen eingekoppelte Störungen.
Mikrofongeräusche
Dieser Störfaktor kann wie andere elektronische Einflüsse nur durch ein entsprechend gut
konstruiertes Mikrofon vermieden werden. Die unterschiedlichen Mikrofontypen sind hier in
ihrer Störanfälligkeit stark unterschiedlich. Generell bleibt anzumerken, dass sich
Elektretmikrofone in der Photoakustik bewährt haben und durch ihre Sensitivität auch
großteils verwendet werden.
Brownsche Bewegung
Dieser Punkt soll nur der Vollständigkeit halber erwähnt werden, da er in der
Spurengasanalyse eine Rolle spielen kann. Dieser Effekt kann durch keinen apparativen
Aufbau gemindert werden, besitzt aber in den meisten Fällen auch keinen großen Einfluss auf
die Güte einer Messung. In der Literatur wird jedoch beschrieben, dass die reine thermische
Fluktuation eine resonante Mode in einem Messaufbau anregen kann126.
59

5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
60
Generell kann man bei der Arbeit mit photoakustische Zellen zwei unterschiedliche
Arbeitsweisen unterscheiden. Zum einen den nichtresonanten Betrieb, bei welchem lediglich
ein Füllkörper für das zu spektroskopierende Gas zur Verfügung gestellt wird und zum
zweiten den resonanten Betrieb. Hierbei wird eine geometrische Eigenschaft der Zelle zur
Verstärkung des Messsignals genutzt, indem die Modulation der Anregung des Signals
periodisch resonante Moden der Zelle anregt.
Nichtresonante Zellen
Auch wenn die Anwendung dieser Arbeitsweise aufgrund mangelnder Verstärkung
unvorteilhaft erscheint, ist sie unter bestimmten Voraussetzungen manchmal vorteilhaft.
Mikrofon
Abbildung 23 Schematische Zeichnung einer nichtresonanten Zelle. Die Druckpulse können an allen
seitlichen Stellen der Zelle mit gleicher Intensität abgegriffen werden.
Der Leistungsgrad einer Zelle im nichtresonanten Betrieb (Abbildung 23) lässt sich durch die
sogenannte Zellkonstante F ausdrücken, welche das Vermögen der Zelle widerspiegelt,
absorbierte Strahlungsenergie in akustische Energie umzuwandeln. Diese Zellkonstante wird
meist experimentell über Messungen mit einem bekannten Gas ermittelt. Die Intensität des
akustischen Signals nimmt nach außen mit der bekannten 1/r2 Abhängigkeit ab.
Mikrofon
Resonante Zellen
Abbildung 24 Schematische Zeichnung einer resonanten Zelle. Die Signalstärke hängt stark von der
Position des Mikrofons an der Zellenwand ab.

5.3 Prinzip der Photoakustik
Bei dieser Arbeitsweise (schematisch in Abbildung 24) wird die Zellkonstante nur im
resonanten Betrieb um den sogenannten Gütefaktor Q erweitert, welcher größer 1 ist.
antnichtresonresonant QFF =
Q spiegelt das Verhältnis der in der stehenden Welle gespeicherten akustischen Energie zu
den Energieverlusten, bewirkt durch inelastische Interaktion mit den Zellwänden oder
Übertragung der Energie in andere Freiheitsgrade, wieder.
longitudinal radial azimuthal
Abbildung 25 Resonante Moden einer Zylinderzelle. Neben der longitudinalen Resonanz existieren
noch die radiale und azimuthale Resonanz. Auch alle Kopplungen dieser Schwingungen
sind möglich.
Die resonanten Moden einer Zylinderzelle (schematisch dargestellt in Abbildung 25) lassen
sich durch die bekannten Bessel-Funktionen berechnen, auf welche hier nicht weiter
eingegangen wird. Es existieren drei verschiedene Arten isolierter resonanter Moden, welche
auch als Koppelschwingung auftreten können.
5.3.4 Photoakustische Effekte in nichtgasförmigen Medien
Da die photoakustischen Vorgänge in Feststoffen und Flüssigkeiten ein zu komplexes Thema
darstellen, um umfassend behandelt zu werden, erfolgt an dieser Stelle eine kurze Einführung.
Die photoakustischen Prozesse in Flüssigkeiten oder Feststoffen waren lange Zeit umstritten.
Es existierte eine Vielzahl von Erklärungsversuchen. Direkt nach der Entdeckung des
photoakustischen Effektes ging Bell davon aus, dass aus Poren der Probe die Luft während
der Einstrahlung herausgepresst wird, wodurch eine Druckerhöhung in der Probenkammer
61

5 Entwicklung und Prinzip der Photoakustischen Spektroskopie
62
bewirkt wird119. Rayleigh glaubte an den Transfer der Energie in Freiheitsgrade der
Schwingung des Feststoffes127, während Mercadier128 und Preece129 der Meinung waren, dass
der Feststoff die Energie lediglich an das ihn umgebende Gas abgibt.
Mikrofon
Fenster
Probe
Piezokristall
Probe
Akustische Anbindung
Abbildung 26 Photoakustische Zellen für Feststoffe. Neben der links gezeigten Gas-Mikrofon Kopplung
ist auch die Messung der Phononanregung über die Kopplung an einen Piezokristall
möglich, wie rechts gezeigt.
Heute wissen wir, dass beide Effekte eine Rolle spielen. Durch Messaufbauten wie in
Abbildung 26 wurde mit einem an den Feststoff gekoppelten Gas-Mikrofon festgestellt, dass
die Energie hauptsächlich an das umgebende Gas abgegeben wird. Durch piezoelektrische
Messungen fand man aber auch einen zwar geringen aber vorhandenen Übertrag auf andere
Freiheitsgrade des Feststoffes über eine Anregung von Gitterschwingungen.
Eine umfassende Behandlung des photoakustischen Effektes bei Feststoffen wird in der
Rosencwaig-Gersho-Theorie gegeben126.

6 Entwicklung der photoakustischen Analytiken
Daß all unsere Erkenntnis mit der Erfahrung anfange, daran ist gar kein Zweifel.
Immanuel Kant
6.1 Mikrofontest
In der Photoakustik werden eine Reihe unterschiedlicher Mikrofone eingesetzt. Meist sind die
Mikrofone speziell auf die entsprechenden Problemstellungen zugeschnitten.
Mikrofon
Gasauslaß
SpiegelLaserstrahl
Abbildung 27 Ursprüngliche Idee einer photoakustischen Analyseeinheit. Der Laserstrahl regt parallel
alle Kanäle an, wobei er durch Spiegel umgelenkt wird. Die Intensität und Abstände der
Signalursprünge sollte über die Laufzeit und bekannte Distanz aus dem Interferenzsignal
berechnet werden.
Das in Abbildung 27 gezeigte Konzept wurde zu Beginn des Projektes erstellt und zeigt einen
schematischen Aufbau eines zweidimensionalen Detektionsarrays. Da ein solcher Aufbau
nicht nur die Intensität eines Signals sondern auch den Zeitversatz sehr genau erfassen muss,
wurden mit einer Reihe kommerzieller Mikrofone Abstandsmessungen über die Laufzeit
63

6 Entwicklung der photoakustischen Analytiken
einzelner Schallpulse durchgeführt, um die Eignung der Mikrofone diesbezüglich zu
überprüfen.
Abstandsmessungen
In einer ersten Serie von Experimenten wurden mehrere Mikrofone unterschiedlichen Bautyps
getestet. Ziel war es ein Mikrofon zu finden, welches zum einen eine hohe Empfindlichkeit
aufwies und zum zweiten noch genügend schnell war, um die einzelnen Signale aufzulösen.
Dies ist problematisch, da diese Eigenschaften aus konstruktionstechnischen Gründen nicht
gleichzeitig in einem Mikrofon verwirklicht werden können130. Da für spätere Anwendungen
nicht ausgeschlossen werden konnte, dass mehrere Mikrofone (bis zu einem pro Kanal) in
einem Analytikaufbau untergebracht werden müssen, spielte auch die Kostenfrage eine
entscheidende Rolle.
Mikrofone können im großen und ganzen in vier unterschiedliche Bauklassen eingeteilt
werden:
Dynamische Mikrofone
Dieser Mikrofontyp wird auch Tauchspulen-Mikrofon genannt und erzeugt das Signal, indem
ein Metallkern in einer Spule durch die Schallwellen hin und her bewegt wird.
Charakteristisch für diesen Mikrofontyp ist das Unvermögen hochfrequente Signale
aufzulösen, da der Kern ein hohes Trägheitsmoment besitzt.
Kondensatormikrofone
Die Kondensatormikrofone nutzen eine metallisierte Membran als eine Elektrode eines
polarisierbaren Kondensators. Für die Messung wird eine Spannungsversorgung benötigt,
welche den Kondensator vorpolarisiert. Die Schallwellen ändern durch Druck auf die
Membran die Kapazität dieses Bauteils. Diese Änderung der Kapazität kann mit der
Schallwelle korreliert werden.
Elektretmikrofone
Elektrete sind Kunststoffe in denen eine Vorspannung quasi eingefroren wurde. Somit können
zusätzliche Spannungsversorgungen wie im Fall des Kondensatormikrofons umgangen
werden. Ansonsten sind diese Typen jedoch prinzipgleich, wobei sie wesentlich kleiner
64

6.1 Mikrofontest
gebaut werden können. Nachteilig ist das sehr schwache Messsignal, welches vorverstärkt
werden muss.
Kristallkapselmikrofone
Bei diesem Mikrofontyp wird ein Ende eines Piezokristalls mit einer Membran versehen. Das
andere Ende wird fixiert. Schallwellen üben nun über die Membran einen Druck auf den
Kristall aus, wodurch eine Spannung erzeugt wird. Hieraus resultiert das Signal.
MD33 MD60 CC20K C252 CC30
Frequenzbereich 4-8 kHz 5-15 kHz 1-10 kHz 3- 15 kHz 3-10 kHz
Empfindlichkeit 0.25 V · bar-1 · kHz-1 0.25 V · bar-1 · kHz-1 -70 dB -60 dB -60 dB
Typ Dynamisch Dynamisch Kristall Kristall Kristall
Tabelle 1 In der Abstandsmessung getestete Mikrofone.
Die in Tabelle 1 aufgelisteten Mikrofone wurden zuerst auf ihren möglichen Einsatz
überprüft. Da sowohl Kondensator- als auch Elektretmikrofone eine Spannungsversorgung für
Messung bzw. Verstärkung benötigen, wurden sie bei den Voruntersuchungen nicht mit
einbezogen.
Für den Testaufbau wurde ein Wassertropfen, welcher nach einer Fallstrecke von 10 cm auf
eine Metallplatte fiel zur Erzeugung einer Schallwelle gewählt. Eine Peristaltikpumpe regelte
kontinuierlich die Tropfenrate, wobei ein Oszilloskop jeweils 256 solcher Ereignisse mittelte.
OszilloskopTrigger Mikrofon
Mikrofon zur Abstandsmessung
Abbildung 28 Testaufbau zur Abstandsmessung. Das Tropfenereignis wurde sowohl über ein Mikrofon
zur Triggerung des Oszilloskops verwendet, als auch zur Abstandsmessung über ein
zweites Mikrofon.
65
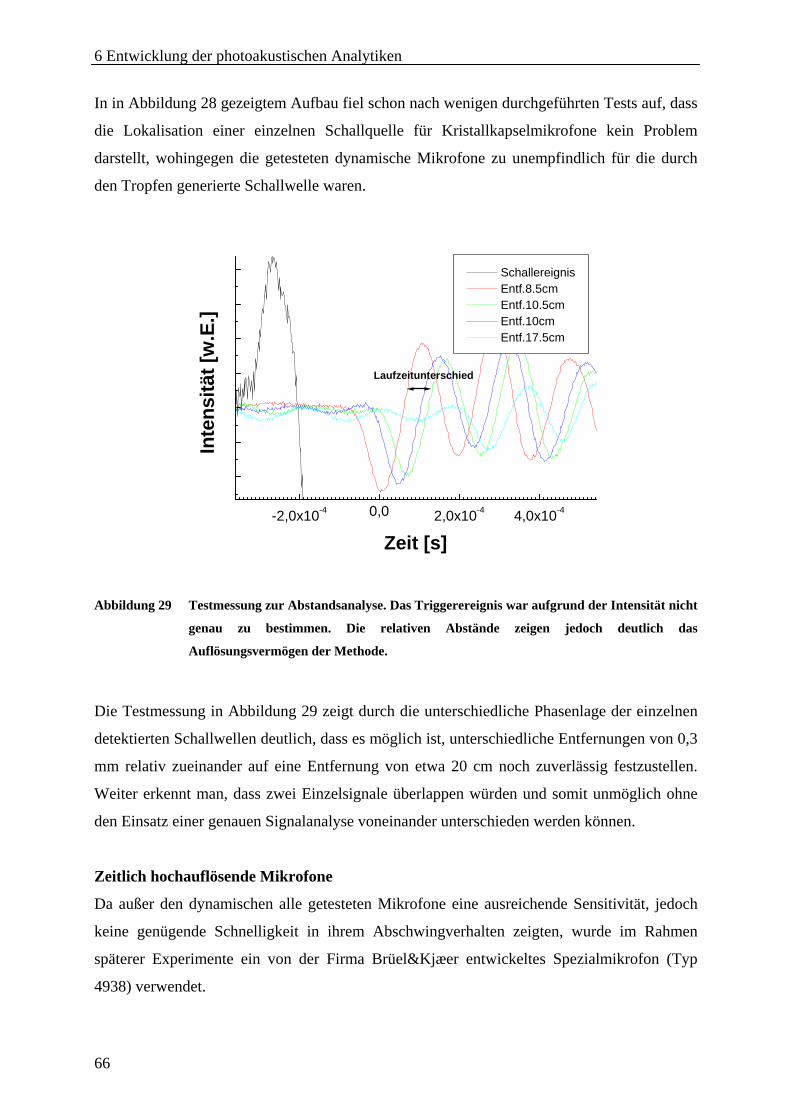
6 Entwicklung der photoakustischen Analytiken
In in Abbildung 28 gezeigtem Aufbau fiel schon nach wenigen durchgeführten Tests auf, dass
die Lokalisation einer einzelnen Schallquelle für Kristallkapselmikrofone kein Problem
darstellt, wohingegen die getesteten dynamische Mikrofone zu unempfindlich für die durch
den Tropfen generierte Schallwelle waren.
-2,0x10-4 0,0 2,0x10-4 4,0x10-4
Laufzeitunterschied
Schallereignis Entf.8.5cm Entf.10.5cm Entf.10cm Entf.17.5cm
Inte
nsitä
t [w
.E.]
Zeit [s]
Abbildung 29 Testmessung zur Abstandsanalyse. Das Triggerereignis war aufgrund der Intensität nicht
genau zu bestimmen. Die relativen Abstände zeigen jedoch deutlich das
Auflösungsvermögen der Methode.
Die Testmessung in Abbildung 29 zeigt durch die unterschiedliche Phasenlage der einzelnen
detektierten Schallwellen deutlich, dass es möglich ist, unterschiedliche Entfernungen von 0,3
mm relativ zueinander auf eine Entfernung von etwa 20 cm noch zuverlässig festzustellen.
Weiter erkennt man, dass zwei Einzelsignale überlappen würden und somit unmöglich ohne
den Einsatz einer genauen Signalanalyse voneinander unterschieden werden können.
Zeitlich hochauflösende Mikrofone
Da außer den dynamischen alle getesteten Mikrofone eine ausreichende Sensitivität, jedoch
keine genügende Schnelligkeit in ihrem Abschwingverhalten zeigten, wurde im Rahmen
späterer Experimente ein von der Firma Brüel&Kjæer entwickeltes Spezialmikrofon (Typ
4938) verwendet.
66

6.1 Mikrofontest
Abbildung 30 Spezialmikrofon der Firma Brüel&Kjæer
Es handelte sich hierbei um das in Abbildung 30 gezeigte Kondensatormikrofon, welches im
Bereich der Explosionsanalytik für zeitliche Analysen eingesetzt wird. Es besitzt eine ¼ Zoll
Membran, welche innerhalb weniger 10 Mikrosekunden wieder in den Ausgangszustand
zurück schwingt. Die Kosten, welche im Fall des verwendeten Mikrofons bei 5000 € inclusive
Vorverstärker liegen, erlaubten jedoch keinen Einsatz in Aufbauten, welche mit mehreren
Mikrofonen arbeiteten.
Ergebnis
Aus den Voruntersuchungen ging hervor, dass die Sensitivität im betrachteten Fall eher eine
untergeordnete Rolle spielen würde, da im Rahmen der Hochdurchsatz-Analyse eher im
Bereich von mehreren Prozenten Detektionsgas gearbeitet wird. Für die Spurengasanalytik
gestaltet sich dies problematischer, da hier hohe Sensitivitäten gefragt sind. Dennoch fiel die
Wahl der Mikrofone für die späteren Aufbauten auf keine der Typen, welche zu Beginn
getestet wurden, da andere Faktoren immer klarer in den Vordergrund traten und die Wahl
entscheidend beeinflussten. Als wichtigste Faktoren für die Durchführbarkeit der Experimente
standen im vorliegenden Fall erstens ein sehr schnelles Mikrofon und zweitens sehr kleine
Mikrofone im Vordergrund, um den Aufbau möglichst kompakt zu gestalten. Beides konnte
weder durch Kristallkapsel- noch durch dynamische Mikrofone bewerkstelligt werden. Die
Entscheidung fiel daher im Fall der entwickelten Freifeld-Analytik auf das oben erwähnte
Kondensatormikrofon von Brüel&Kjær und im Fall der resonanten Analytik auf sehr kleine
Mikrofone des Elektret-Typs, welche in bezug auf Empfindlichkeit die gleichen
Testeigenschaften wie die oben untersuchten Kristallkapselmikrofone hatten und für unter 1 €
zu erhalten waren.
67

6 Entwicklung der photoakustischen Analytiken
6.2 Lasersystem
Zu Beginn der Arbeiten wurde ein Lasersystem gesucht, welches sowohl einfache
zuverlässige Bedienbarkeit als auch hohe Leistung miteinander kombinieren sollte. Die
Emission sollte im Bereich der Molekülschwingungsbanden liegen, da die Anregungsenergie
nicht ausreichen sollte eine elektronische Anregung zu bewirken, um photochemische
Reaktionen zu vermeiden. Wie im Kapitel 5.3.1 beschrieben, erfüllt das CO2-Lasersystem alle
obigen Kriterien und besitzt zusätzlich den Vorteil, dass es schon für unter 10000 € erhältlich
ist.
Der im Rahmen dieser Arbeit verwendete Laser war ein 25 W CO2-Laser, der in Einzelteilen
von der Firma DEOS erworben wurde. Die Anregung erfolgte über einen Radiofrequenz-
Generator. Die Modulation des Lasers erfolgte über eine TTL-Steuerung des Radiofrequenz-
Generators, der Frequenzen bis zu 25 kHz noch zuverlässig einhalten konnte. TTL
(Transistor-Transistor-Logik) bezeichnet ein Signal, welches entweder 5 oder 0 Volt beträgt
und somit den logischen Bedeutungen EIN oder AUS entspricht. Die Justage der Pulsdauer
wurde ebenfalls über dieses Modul durch eine Variation der TTL-Signalverhältnisse
gewährleistet. Eine Leistungsregelung erfolgte durch laserinterne Überlagerung des TTL-
Signals am Radiofrequenz-Generator mit einer 25 kHz Frequenz unterschiedlichen
Puls/Pause-Verhältnisses. Das TTL-Signal zur Steuerung des Lasers wurde direkt von dem im
System befindlichen Signalgenerator der Firma RISys Typ 9514 übernommen, um nicht auf
die ungenaue Zeittaktung des Rechners angewiesen zu sein, welcher auch gleichzeitig die
anderen Steuerprozesse und Auswertungen durchführen musste. Die Ansteuerung des
Signalgenerators erfolgte über eine RS 232 Schnittstelle und die entsprechenden ASCII Codes
unter LabVIEW. Es war hierdurch während der Messungen möglich, auch automatisiert den
Laser zu programmieren und bei Bedarf abzuschalten.
68

6.2 Lasersystem
69
Abbildung 31 Abbildung des 25 W CO2-Lasersystems (links) und des steuernden Signalgenerators
(RISys GmbH Typ 9514).
Die Kühlung des Systems, welches in Abbildung 31 gezeigt ist, erfolgte durch sieben
Ventilatoren, welche auf Laserkopf und Radiofrequenz-Generator verteilt wurden. Nachteilig
an diesem Aufbau war die hohe Lautstärke der Lüftung, die bei späteren Experimenten über
eine geeignete Schallisolierung ausgeblendet werden musste. Der Vorteil des Systems bestand
in einer gesteigerten Mobilität, die es erlaubte, den Laser auch in Räumlichkeiten ohne
geeignete Kühlwasserversorgung über einen längeren Zeitraum zu betreiben.
Die Justage des Lasers erfolgte über ein in den Aufbau integriertes Justage-Shuttermodul. In
diesem Modul wurde ein sichtbarer Diodenlaser kolinear in den Strahlengang eingekoppelt
und somit eine genaue Justierung des ansonsten unsichtbaren CO2-Laserstrahles ermöglicht.
Zusätzlich konnte der Strahlengang manuell verschlossen werden.
Da der verwendete Laser eine Strahlaufweitung von 7,3 mrad besaß, stand für die Detektion
lediglich eine optische Pfadlänge von ca. einem Meter zur Verfügung. Da der Shutter hiervon
die ersten 25 cm benötigte und die letzten 25 cm schon eine beträchtliche Aufweitung zeigten,
wurden nur die mittleren 50 cm benutzt. Auf eine Fokussierung des Strahls wurde im Rahmen
dieser Arbeit verzichtet.
Der CO2-Laserprozess
Ein kommerzieller CO2-Laser arbeitet mit einer Gasmischung von 70 % Helium, 20 %
Stickstoff und lediglich 10 % Kohlendioxid. Das Helium ist für den Abtransport der während
des Betriebs in der Gasmischung entstehenden Wärme verantwortlich, während die
eigentliche Anregung des Kohlendioxid-Moleküls durch zuvor angeregten Stickstoff bewirkt

6 Entwicklung der photoakustischen Analytiken
wird. Da während der Laserprozesse auch Zerfallserscheinungen des Kohlendioxids auftreten,
sind der Lasermischung Gase wie Sauerstoff oder Kohlenmonoxid zugesetzt, die eine
Rückreaktion ermöglichen sollen.
(001) 2349 cm-1
CO2-Deformations-Schwingung
CO2-Asymmetrische-Streckschwingung
(010) 667 cm-1
(020)
N2-Schwingung
Stossübertragung
Anregung
Laserprozess9,4 µm
(010) 1388 cm-1
CO2-Symmetrische-Streckschwingung
Laserprozess10,4 µm
Abbildung 32 Schema der Laserprozesse eines CO2-Lasers. Die eigentliche Anregung erfolgt über
Stickstoff-Moleküle, welche die Energie bei Stossprozessen auf das Kohlendioxid
übertragen.
Wie man in Abbildung 32 sieht, findet der eigentliche Laserübergang vom Niveau 001 der
asymmetrischen Streckschwingung in das 1. angeregte Schwingungsniveau der
symmetrischen Streckschwingung oder das 2. Schwingungsniveau der Deformations-
Schwingung statt. Diese Niveaus sind bei Raumtemperatur zu weniger als einem Prozent
besetzt.
6.3 Freifeld-Aufbau
Im Rahmen der vorliegenden Arbeit sollen die beiden entwickelten analytischen Aufbauten
getrennt voneinander behandelt werden. Die Unterteilung erscheint sinnvoll, da die
Entwicklungen sowohl unterschiedliche Zielmoleküle hatten als auch unterschiedliche
Detektionstechniken nutzten.
70

6.3 Freifeld-Aufbau
6.3.1 Modellsystem
Zur Detektion von Molekülen mit großem Absorptionskoeffizient wurde exemplarisch die
oxidative Dehydrierung von Ethan zu Ethen gewählt.
a) b)
Abbildung 33 Infrarot Spektren131 von a) Ethan und b) Ethen. Man erkennt deutlich, dass im Bild a)
keine signifikante Absorptionsbande im Bereich der Laserstrahlung liegt, während bei b)
eine hohe Absorption zu erkennen ist.
Wie man in Abbildung 33 sieht, besitzt Ethen im Bereich der CO2-Laseremission bei 10,6 µm
eine hohe Absorption und liefert daher bei einer Anregung einen deutlichen Druckpuls, der
ohne zusätzliche Verstärkung aus dem Hintergrundrauschen herausgefiltert werden können
sollte. Weiterhin erkennt man aus dem Vergleich von Ethan- und Ethen-Spektrum, dass Ethan
keine Absorption in diesem Bereich zeigt. Ein Ethan/Luft-Hintergrund sollte also die
störungsfreie Detektion von Ethen nicht beeinträchtigen.
6.3.2 Aufbau
Der apparative Aufbau wurde bezüglich der Reaktoren bereits in Kapitel 4.2.2 behandelt. Hier
soll nun die Analytik im Vordergrund stehen.
71

6 Entwicklung der photoakustischen Analytiken
Abbildung 34 Photoakustische Freifeld-Analytik. Die Minima im Diagramm entsprechen den einzelnen
Kanälen. Die gezeigte Messung wurde nach einer 256-fach Mittelung erhalten.
Da es bei der Detektion eines einzelnen Druckpulses auf die zeitliche Auflösung des Signals
ankommt, wurde mit einem speziell für diese Anforderung entwickelten Mikrofon der Firma
Brüel&Kjæer (Typ 4938) gearbeitet. Das Mikrofonsignal wurde, wie aus Abbildung 34
hervorgeht, zuerst von einem Oszilloskop (Tektronix TDS 380) aufgenommen, welches eine
Mittelung von bis zu 256 Messungen durchführen konnte und hierdurch das Signal/Rausch-
Verhältnis erheblich verbesserte. Das gemittelte Signal wurde von einem Computer aus dem
Oszilloskop ausgelesen und von einer Auswertungssoftware basierend auf LabVIEW auf
vorhandene Druckpulse untersucht. Dies wurde durch eine Programmroutine durchgeführt,
welche zuerst die im Signal vorhandenen Extremstellen ermittelte, was durch rechnerinterne
Ableitung des kompletten Signalverlaufs bewerkstelligt wurde. Hierauf wurde in einem
Bereich um die entsprechende Extremstelle das Signal integriert, wobei ein symmetrischer
Peak vorausgesetzt wurde. Es konnten hierbei manuell sowohl Schwellwerte für Extrema als
auch Werte für die Integrationsbreite gewählt werden. Im Routinebetrieb wurden einmal
gefundene Peakpositionen automatisch als konstante Integrationsgrenzen auf der Zeitachse
übernommen, wodurch die Detektion von extrem niedrigen Werten unterhalb des zuvor
eingestellten Schwellwertes möglich wurde und selbst eine Messung der Intensität, welche
dem 0 % Umsatz entsprach, möglich war. Dies war notwendig, da bei geringen Signalstärken
aufgrund der hohen Intensität der Hintergrundgeräusche keine Druckpulse automatisiert
gefunden werden konnten.
72

6.3 Freifeld-Aufbau
Im Routinebetrieb wurde vor jeder neuen Messung das System mit einer Prüfgasmischung,
welche einen Umsatz von 100 % simulierte, auf den entsprechenden Umsatzwert kalibriert,
wobei gleichzeitig der Abstand der Druckpulsursprünge durch die Laufzeit der Signale
ermittelt wurde. Im Anschluss hieran wurde das Signal an den entsprechenden Stellen der
Pulse auf der Zeitachse bei einer Mischung integriert, welche einen Umsatz von 0 %
simulierte. Diese Kalibrierroutine ermöglichte es, unabhängig von eventuell zwischenzeitlich
aufgetretenen Dejustagen zu arbeiten.
6.3.3 Automatisierung
Abgesehen von der zur Reaktorführung notwendigen Automatisierung waren für den
automatisierten Routinebetrieb noch weitere Programmierschritte notwendig, um einen
reibungslosen Ablauf von Analytik und Prozessführung zu gewährleisten.
Gasfluss Temperatur
Eurotherm
RS-485 EI-bisynch
3 BronkhorstMFC
analog 0 - 5 V
Zeit t3 Gasfluss Vi(t)1 Temperatur T(t)Laserkontrolle (t)Kalibrierung (t) Temperatur
Umsatz zu EthenGasfluss Ethan, Luft,Ethen
�������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������
������������������
Protokoll-Logfile
��������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������
TektronixTDS 380
RS-232
Mikrofon&
Verstärker
Brüel&KjaerType 4938
Laser
DEOS LC-25
25 Watt
Puls GeneratorQuantum
9514 Series
Labview-Modul
Input
PC (Win NT, TCP-IP)
�������������
����������������������
��������������
����������
������������������������������������������������������������
Output
Abbildung 35 Schematische Zeichnung der elektronischen Verknüpfung und des Datenflusses bei der
photoakustischen Freifeld-Analytik. Die Reaktionskontrolle und die Analytik sind, wie
aus der Abbildung zu erkennen ist, voneinander unabhängige Routinen.
Computer sind bei der Durchführung mancher Prozesse nicht in der Lage, ein genaues,
zeitlich konstantes Signal zu liefern. Dies liegt an der schwankenden Rechenleistung, welche
73

6 Entwicklung der photoakustischen Analytiken
normalerweise von einem einzigen Prozessor aufgebracht werden muss. Für alle zeitlich exakt
zu steuernden Prozesse wurde daher die Zeitbasis des Signalgenerators, welcher sich im
System befand und im Normalfall lediglich die Lasersteuerung übernahm, verwendet.
Wie aus dem in Abbildung 35 gezeigten Schema hervorgeht, konnte die Anlage über ein
Protokoll, welches in einem kommerziellen Tabellenkalkulationsprogramm erstellt werden
konnte, gesteuert werden. Im vorliegenden Fall wurden nach Ablauf von einer Minute neue
Steuerwerte eingelesen und zu den entsprechenden Komponenten wie Massendurchflussregler
und Temperatursteuerung übertragen. Das Oszilloskop wurde über eine RS 232 Schnittstelle
mit den entsprechenden ASCII Codes angesteuert und ausgelesen. Hierbei war aufgrund des
langsamen Datentransfers zwischen Oszilloskop und Rechner lediglich alle zwei Sekunden
eine Messung möglich.
Abbildung 36 Benutzeroberfläche der Freifeld-Analytik.
Das Mikrofon und der Vorverstärker arbeiteten unabhängig und versorgten das Oszilloskop
kontinuierlich mit Daten. Das Oszilloskop führte ebenfalls kontinuierlich die Mittelungen
durch und gab lediglich auf Anweisung ein aktuelles Bild des Monitors an den Rechner
weiter. Die Signalauswertung erfolgte wie oben beschrieben, wobei die Werte entweder als
echte Messwerte oder als korrigierte Umsatzwerte gespeichert werden konnten. Im
Messprotokoll standen nach erfolgreicher Messung Daten über Flüsse, Temperatur und
gemessene Konzentrationen aller Kanäle in zuvor festgelegter Form in Abständen von zwei
Sekunden zur Verfügung.
Die Benutzeroberfläche, welche in Abbildung 36 gezeigt ist, ermöglichte es alle Einstellungen
und Änderungen manuell durchzuführen. Für den Routinebetrieb konnten jedoch auch
automatisiert zu definierten Zeitpunkten, welche in Form von Laufzeiten in das
Steuerprogramm eingegeben wurden, festgelegte Prozesse, wie z.B. Laser an/aus,
74

6.3 Freifeld-Aufbau
Signalgeneratoreinstellungen oder Kalibrierroutinen, gestartet und automatisch durchlaufen
werden.
6.3.4 Parallelisierung
Im vorliegenden Fall waren einer Parallelisierung nur durch zwei Faktoren Grenzen gesetzt.
Zum einen durch die Entfernung vom Signalursprung zum Mikrofon, welche aufgrund der
1/r2 Abhängigkeit der Signalintensität bei zu großen Distanzen zu nicht mehr messbaren
Signalen führte, und zum zweiten durch ein zu dichtes Aufeinanderfolgen der einzelnen
Kanäle, was eine Überlappung der einzelnen Signale zur Folge hatte. Letzter Punkt stellte
hohe Anforderungen an das Mikrofon, da ein möglichst kleinen Abstand angestrebt war. In
Kapitel 6.1 wird auf die unterschiedlichen Mikrofontypen eingegangen sowie auf die
notwendigen Charakteristika eines Mikrofons, mit dessen Hilfe die Signale aufgelöst werden
können.
Puls
Signal(minimal)
Distanz
Laufzeitunterschied durch Gaswolke
Abbildung 37 Berechnung der Laufzeit des akustischen Signals. Die Zeit setzt sich aus der Entfernung
zwischen den einzelnen Kanälen und der Ausdehnung der Gaswolke zusammen. Das
Signal wird idealisiert als eine vollständige Schwingung betrachtet. Die Dämpfung kann
im realen Experiment wesentlich mehr Zyklen in Anspruch nehmen.
Durch einfache Berechnungen, welche in Abbildung 37 verdeutlicht sind, lassen sich sehr
schnell die minimalen Abstände zwischen den Kanälen abschätzen. Setzt man eine
Schallgeschwindigkeit von rund 340 m·s-1 voraus und arbeitet bei einer eine Pulslänge von ca.
50 µs, so ergibt sich als minimaler Abstand bei einer kompletten Schwingung, welche in
erster Näherung doppelt so lang sein sollte wie die Anregungsphase (Pulsdauer), eine
75
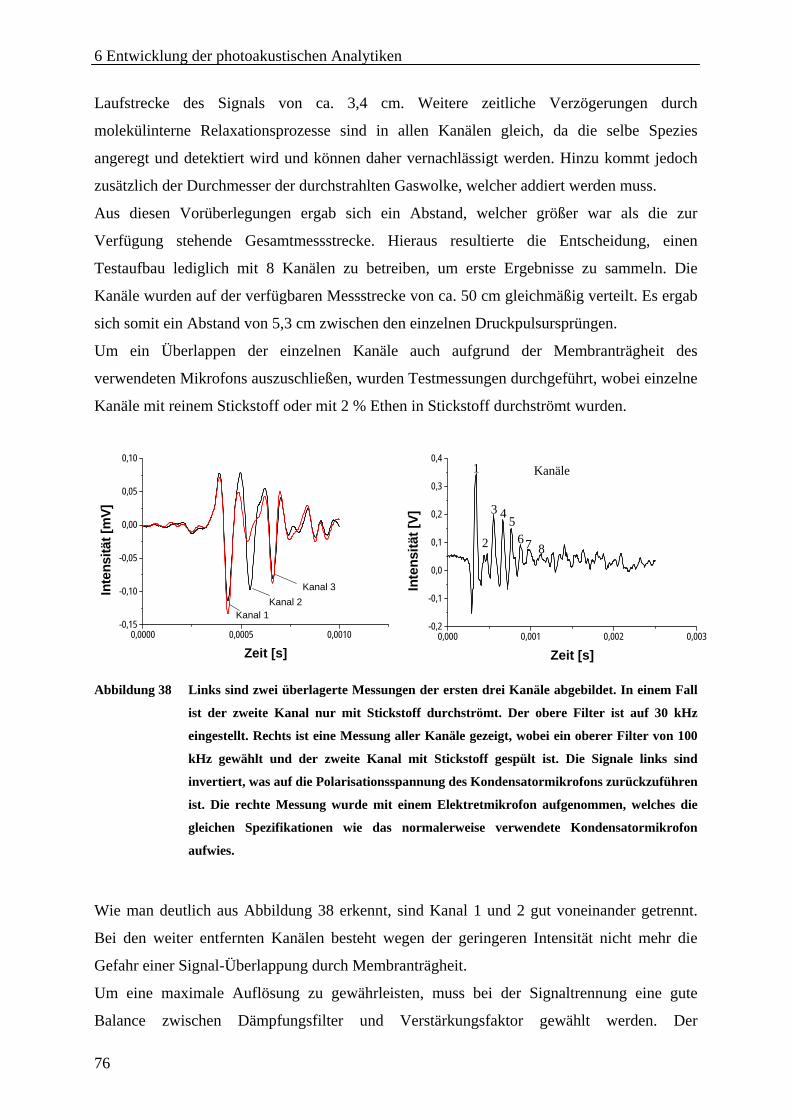
6 Entwicklung der photoakustischen Analytiken
Laufstrecke des Signals von ca. 3,4 cm. Weitere zeitliche Verzögerungen durch
molekülinterne Relaxationsprozesse sind in allen Kanälen gleich, da die selbe Spezies
angeregt und detektiert wird und können daher vernachlässigt werden. Hinzu kommt jedoch
zusätzlich der Durchmesser der durchstrahlten Gaswolke, welcher addiert werden muss.
Aus diesen Vorüberlegungen ergab sich ein Abstand, welcher größer war als die zur
Verfügung stehende Gesamtmessstrecke. Hieraus resultierte die Entscheidung, einen
Testaufbau lediglich mit 8 Kanälen zu betreiben, um erste Ergebnisse zu sammeln. Die
Kanäle wurden auf der verfügbaren Messstrecke von ca. 50 cm gleichmäßig verteilt. Es ergab
sich somit ein Abstand von 5,3 cm zwischen den einzelnen Druckpulsursprüngen.
Um ein Überlappen der einzelnen Kanäle auch aufgrund der Membranträgheit des
verwendeten Mikrofons auszuschließen, wurden Testmessungen durchgeführt, wobei einzelne
Kanäle mit reinem Stickstoff oder mit 2 % Ethen in Stickstoff durchströmt wurden.
0,0000 0,0005 0,0010-0,15
-0,10
-0,05
0,00
0,05
0,10
Kanal 3Kanal 2
Kanal 1
Inte
nsitä
t [m
V]
Zeit [s] 0,000 0,001 0,002 0,003
-0,2
-0,1
0,0
0,1
0,2
0,3
0,4
Inte
nsitä
t [V]
Zeit [s]
Kanäle1
2
5
8
3
6
4
7
Abbildung 38 Links sind zwei überlagerte Messungen der ersten drei Kanäle abgebildet. In einem Fall
ist der zweite Kanal nur mit Stickstoff durchströmt. Der obere Filter ist auf 30 kHz
eingestellt. Rechts ist eine Messung aller Kanäle gezeigt, wobei ein oberer Filter von 100
kHz gewählt und der zweite Kanal mit Stickstoff gespült ist. Die Signale links sind
invertiert, was auf die Polarisationsspannung des Kondensatormikrofons zurückzuführen
ist. Die rechte Messung wurde mit einem Elektretmikrofon aufgenommen, welches die
gleichen Spezifikationen wie das normalerweise verwendete Kondensatormikrofon
aufwies.
Wie man deutlich aus Abbildung 38 erkennt, sind Kanal 1 und 2 gut voneinander getrennt.
Bei den weiter entfernten Kanälen besteht wegen der geringeren Intensität nicht mehr die
Gefahr einer Signal-Überlappung durch Membranträgheit.
Um eine maximale Auflösung zu gewährleisten, muss bei der Signaltrennung eine gute
Balance zwischen Dämpfungsfilter und Verstärkungsfaktor gewählt werden. Der
76

6.3 Freifeld-Aufbau
Dämpfungsfilter sorgt für eine Verstärkung lediglich innerhalb des interessanten
Frequenzbereiches, während außerhalb die Störsignale gedämpft werden. Diese elektronische
Dämpfung sollte sinnvollerweise innerhalb der maximalen Arbeitsfrequenzen des Mikrofons
liegen. Für den entwickelten Messaufbau stellte sich ein unterer Filter von 20 Hz und ein
oberer Filter von 30 bzw. 100 kHz als am besten geeignet heraus. Die hohe obere Grenze
welche auch noch vom Mikrofon erfasst werden konnte, bedingte die schnelle messbare
Abschwingrate. Bei anderen Mikrofonen ist ein oberer Filter in dieser Höhe wegen
mangelnder Auflösung physikalisch nicht sinnvoll. Niederfrequente Störungen von außen
konnten mit der unteren 20 Hz Grenze erfolgreich gedämpft werden.
6.3.5 Evaluierung der Analytik
Um die Analytik zu evaluieren, wurden Experimente hinsichtlich der Repetitionfrequenz und
der Laserpulslänge durchgeführt, wodurch Effekte auf die Signalgüte experimentell ermittelt
werden sollten.
0,0 2,0x10-4 4,0x10-4 6,0x10-4 8,0x10-4 1,0x10-3 1,2x10-3 1,4x10-3-4
-2
0
2
Zeit [s]
Inte
nsitä
t [V
]
50 Mikrosekunden 25 Mikrosekunden
Abbildung 39 Messung des Signalverlaufs bei unterschiedlichen Anregungspulslängen.
Erwartungsgemäß sind die Druckpulse länger, wenn die Laserpulsdauer steigt.
Erwartungsgemäß wächst, wie in Abbildung 39 zu sehen ist, die Druckpulslänge bei längeren
Laserpulszeiten. Bei unterschiedlichen Wiederholfrequenzen konnte im unteren
Frequenzbereich bis ca. 500 Hz kein Einfluss auf die Signalstärke gefunden werden.
77

6 Entwicklung der photoakustischen Analytiken
Für spätere Messungen wurde eine Pulslänge von 35 µs verwendet, was bei 10 bzw. 100 Hz
einer durchschnittlichen Strahlintensität von 10 bzw. 100 Milliwatt entsprach und somit noch
genügend Energie für die Messung bereitstellte. Durch die kurzen Pulse war eine
ausreichende Signalseparation ebenfalls gewährleistet.
Da Messungen im Bereich von 0 bis 2 % Ethen durchgeführt werden sollten, musste noch die
Linearität der gesamten Messprozedur, welche nicht nur Signalaufnahme, sondern auch
Integrationsroutine umfasste, experimentell bestätigt werden. Zu diesem Zweck wurden
Konzentrationsrampen gefahren und bei den entsprechenden Konzentrationen mehrere
Messungen durchgeführt, um die Stabilität und Driftanfälligkeit empirisch zu ermitteln.
0,0 0,5 1,0 1,5 2,0-0,5
0,0
0,5
1,0
1,5
2,0
2,5
Expe
rimen
telle
r Wer
t [%
]
Theoretischer Wert [%]
0,0 0,5 1,0 1,5 2,0-0,5
0,0
0,5
1,0
1,5
2,0
2,5
Expe
rimen
telle
r Wer
t [%
]
Theoretischer Wert [%]
Abbildung 40 Bestimmung der Linearität im Messbereich. Links ist der erste und rechts der sechste
Messkanal gezeigt. Wie deutlich zu sehen ist, ist im Rahmen der Messfehler eine lineare
Korrelation zwischen Konzentration und Signalstärke gegeben.
Wie deutlich aus den Messungen in Abbildung 40 hervorgeht, war im gesamten Bereich von 0
bis 2 % Ethen eine Linearität im Rahmen der Messfehler gewährleistet. Die Fehler
errechneten sich aus den Standardabweichungen von 30 Messungen innerhalb einer Minute
bei gleicher Konzentration.
Eine neue Analytik sollte immer an einem schon erprobten System gemessen werden. Daher
wurden abschließend im Rahmen dieser Arbeit an ausgewählten Katalysatoren
Vergleichsmessungen durchgeführt. Es wurden Daten aus parallel akustischen Messungen mit
Werten verglichen, welche in einem Einrohr Reaktor mit Gaschromatographie-Analytik
erhalten wurden.
78

6.3 Freifeld-Aufbau
300 350 400 450 500 550-0,02
-0,01
0,00
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
Konz
entra
tion
Ethe
n [%
abs
.]
Temperatur [°C]
Parallele photoakustische Messung Gaschromatographische Messung
Abbildung 41 Vergleich der parallel photoakustischen Messung und der gaschromatographischen
Messung im Einrohr-Reaktor. Die Gaschromatographie-Messungen liegen, wenn man die
Fehlergrenzen betrachtet, genau im Bereich des photoakustischen Verfahrens. Des
getestete Material hatte die Zusammensetzung La12Ba2Pb9Th7Mn27Ni24Cu19Ox.
Wie man in Abbildung 41 deutlich sieht, sind die Werte, die mit dem Gaschromatographen
ermittelt wurden, innerhalb der Messungenauigkeit mit denen des photoakustischen Aufbaus
identisch.
6.3.6 Zusammenfassung und Diskussion
Der photoakustische Freifeld-Aufbau stellt für bekannte Reaktionen mit Produkten, welche
durch Lasersysteme anregbare selektive Banden hoher Absorptionswahrscheinlichkeit
aufweisen, eine gute Methode dar, um eine echtzeit-parallele Messung durchzuführen. Da
jede Spülzeit bzw. eine umständliche Gasführung vermieden wird, werden die Messungen
quasi „direkt am Katalysator“ durchgeführt. Berücksichtigt man die Analysenzeit und die
Spülzeit der Abgasleitung, kommt man somit auf eine zeitliche Verzögerung, welche
unterhalb von 10 s liegt. Da die Messungen parallel durchgeführt werden, ergeben sich echte
Vergleichsmöglichkeiten der einzelnen Katalysatoren untereinander. Die Geschwindigkeit der
Messung liegt zur Zeit aufgrund der nicht optimierten Datenverarbeitung noch im Bereich
weniger Sekunden. Ein zeitlimitierender Faktor ist hierbei der langsame Datentransfer von
Oszilloskop zum Computer, was durch eine rechnerinterne Oszilloskopkarte verbessert
werden könnte. Es würde hierdurch eine Steigerung der Geschwindigkeit erreicht, welche
lediglich durch die Busgeschwindigkeit des Rechners limitiert wäre. Weiter begrenzt die
79

6 Entwicklung der photoakustischen Analytiken
langsame Mittelung die Geschwindigkeit, welche ebenfalls durch rechnerinterne digitale
Signalverarbeitung gelöst werden könnte. Limitierende Grenze wäre damit letztendlich die
Repetitionsfrequenz des Lasers.
Nachteilig ist die hohe Störanfälligkeit des Systems. Selbst zu hohe Leistung der
Abzugsanlage konnte im vorliegenden Fall die Ergebnisse beeinträchtigen bzw. im Extremfall
eine Messung unmöglich machen. Dies bleibt bei einer zukünftigen Anwendung in Form
geeigneter Abschirmungen zu berücksichtigen.
Theoretisch wäre bei der entwickelten Methode auch ein zweidimensionaler Aufbau denkbar,
indem mit zwei Mikrofonen für die unterschiedlichen Raumrichtungen eine
mehrdimensionale Analyse durchgeführt würde. In diesem Fall müsste allerdings eine
kugelsymmetrische Reflektionscharakteristik im gesamten System gewährleistet sein, um eine
Kugelsymmetrie der Signale auch bei Überlagerung mit eventuell vorhandenen Echos zu
garantieren, was eine Auswertung der unterschiedlichen räumlichen Laufzeiten ermöglichen
würde.
Eine Optimierung des vorhandenen Systems müsste sich auch durch Verbesserung der
akustischen Güte des Analysenraumes bewerkstelligen lassen. Bei der vorliegenden Arbeit
stellte die Beherrschung bzw. Vermeidung der im System intern reflektierten Druckpulse
eines der Hauptprobleme dar. In den Beispielmessungen sind diese störenden Echos immer
sichtbar, konnten jedoch reproduziert werden und gingen somit nicht in die Interpretation der
Ergebnisse mit ein.
6.4 Resonanter Aufbau
Im Gegensatz zum Freifeld-Aufbau war die Entwicklung des parallelen Resonanz-Aufbaus
eine weitaus anspruchsvollere Aufgabe. Es galt hierbei nicht nur, einen für die Detektion von
Molekülen mit kleinem Absorptionskoeffizienten geeigneten Resonator zu entwickeln,
sondern diesen auch noch den geometrischen Gegebenheiten des Systems möglichst
platzsparend anzupassen, wobei die Resonanzverstärkung weitgehend erhalten bleiben sollte.
6.4.1 Modellsystem
Bei der Detektion von Molekülen mit kleinen Absorptionskoeffizienten wurde als
Modellsystem die Oxidation von Kohlenmonoxid zu Kohlendioxid gewählt. Kohlendioxid
80

6.4 Resonanter Aufbau
besitzt im Bereich der CO2-Laserstrahlung eine sehr schwache Absorption. Dies liegt daran,
dass der Laservorgang zwischen angeregten Energieniveaus stattfindet und diese Niveaus bei
Raumtemperatur nur schwach zu weniger als einem Prozent besetzt sind. Die Anregung durch
den Laserstrahl erfolgt bei Kohlendioxid aus diesen thermisch nur schwach besetzten
Schwingungsniveaus. Die direkte Laseranregung des Kohlendioxids aus diesem Niveaus ist,
da es sich um eine heiße Bande handelt, also weitaus weniger günstig. Im Abschnitt 6.2 ist
dieser Vorgang genauer erklärt und bildlich aufgezeigt.
Betrachtet man das Infrarot-Spektrum von Kohlenmonoxid, so stellt man keine Absorption im
Bereich der verwendeten Laserstrahlung fest. Eine Messung von Kohlendioxid in einem
Kohlenmonoxid/Luft-Untergrund sollte also bei entsprechend empfindlicher Messzelle
möglich sein, wie auch häufig in der Literatur beschrieben wird (Abschnitt 3.2).
6.4.2 Resonatorentwicklung
Der Hauptteil der Entwicklungsarbeit bestand in der Konstruktion eines parallel einsetzbaren
Resonators. In der Literatur gibt es eine Vielzahl unterschiedlicher, teilweise mit exzellenten
Eigenschaften ausgestatteten Resonatorkonstruktionen. Diese sind jedoch meist im Hinblick
auf maximale Sensitivität für den Einsatz in der Spurengasanalyse gebaut und nur selten klein
genug, um parallel benutzt zu werden. Es galt daher, einen an unsere Anforderungen
angepassten eigenen Resonator zu entwickeln. Dieser Resonator sollte folgenden Kriterien
genügen:
1. Genügend hohe Sensitivität
2. Parallelisierbarkeit
3. Permanent mit Messgas durchströmbar
4. Kompakte Bauweise
6.4.2.1 Erste photoakustische Messungen
Da nach ersten Messversuchen klar war, dass Kohlendioxid nicht ohne Resonanzverstärkung
detektiert werden konnte, wurden in einer provisorischen Zelle, welche unterschiedliche
Kantenlängen hatte, erste Testmessungen durchgeführt.
81

6 Entwicklung der photoakustischen Analytiken
Verstärker +Mikrofon
LaserMesszelle
Resonanzröhre
Oszilloskop
TTL-Generator
Strahlfalle
-2,0x10-4-1,0x10-4 0,0 1,0x10-4 2,0x10-4 3,0x10-4 4,0x10-4
-2
-1
0
1
2
3
4 TTL Leermessung CO2
Inte
nsitä
t [V]
Zeit [s]
Abbildung 42 Aufbau der ersten photoakustischen Experimente (links) und Messung von Kohlendioxid
in der mit Trockeneis teilgefüllten Zelle im Vergleich zur leeren Zelle (rechts).
Für die Detektion wurde ein Elektretmikrofon mit kompakter Vorverstärkerstufe eingesetzt.
Abbildung 43 Schaltbild des modifizierten Mikrofonbausatzes 302155-66 (Conrad).
Der Verstärker, dessen Schaltplan in Abbildung 43 gezeigt ist, basierte auf einer modifizierten
Schaltung eines Bausatzes (Conrad Best.Nr. 302155-66).
Es wurden mit Hilfe dieses Aufbaus Messungen bei unterschiedlichen Modulationsfrequenzen
durchgeführt. Der Innenraum der Zelle konnte hierbei verändert werden. Die in Abbildung 42
rechts gezeigte Messung wurde außerhalb jeder Resonanz aufgenommen die durch die
Zellgeometrie hätte entstehen können, was durch Variation der Zelldimension experimentell
bestätigt werden konnte. Der Laser wurde im Gegenteil auf einer Resonanzfrequenz des
Schlauches, welcher Mikrofon und Zelle verband moduliert. Auf diese Weise sollte die Zelle
lediglich eine lokal begrenzte Gasatmosphäre sicherstellen und nicht die Signalintensität
82

6.4 Resonanter Aufbau
während der Messung beeinflussen. Die unterschiedlichen Atmosphären während der
Messung wurden durch eine leere Zelle bzw. Trockeneis, welches sich am Zellenboden
befand, bewirkt. Das wichtigste Ergebnis aus diesem Experiment war der Nachweis der
Machbarkeit einer photoakustischen Detektion mit den von uns gewählten Mikrofon- und
Verstärker-Komponenten ohne zusätzliche Resonanzverstärkung durch das das Messgas
beinhaltende Bauteil.
Im nächsten Entwicklungsstadium sollte nun sowohl eine kontrolliert einstellbare
Konzentration an Kohlendioxid möglich sein als auch eine Messung in einem strömenden
Medium, um sich den realen Einsatzbedingungen anzunähern.
Laser
Oszilloskop
Signalgenerator
Gas
Strahlfalle
Resonanzröhre(PE)
Laser
Oszilloskop
Signalgenerator
Gas
StrahlfalleResonanzröhre
(Stahl)
PE-Schlauch
Mikrofon (in PE-Röhre)
Mikrofon
Abbildung 44 Testaufbauten der Experimente für kontrollierte Konzentrationsmessungen. Links ist
eine Anregung im Freifeld mit nachfolgender resonanter Signalabgreifung gezeigt.
Rechts wird das Signal direkt im Innern des Resonators erzeugt.
In Anlehnung an die in Abbildung 42 gezeigten ersten Experimente wurde nun ein
Messverfahren ohne Absorptionsvorgang im Innern des Resonators angestrebt. Es wurde
hierfür eine Freifeld-Methodik mit der schon im vorherigen Experiment erfolgreichen
resonanten Signalabgreifung kombiniert. Dieser Aufbau ist in Abbildung 44 links gezeigt.
Leider erwies sich dieses Konzept nicht als erfolgreich. Dass in den in Abbildung 42
gezeigten Experimenten diese Methode erfolgreich war, liegt wahrscheinlich an den höheren
Kohlendioxidkonzentrationen, welche sich in der Messzelle im Aufbau in Abbildung 42
einstellen konnten. Im Gegensatz hierzu wurde nun bei Konzentrationen unter 10 %
Kohlendioxid gemessen, was den späteren realen Einsatzbedingungen entsprach. Daraufhin
83

6 Entwicklung der photoakustischen Analytiken
wurde eine Resonanzröhre konstruiert, in deren Innern Messgas und Laserstrahl
zusammentrafen, wodurch ein periodisches, durch die Röhrengeometrie resonant verstärktes
Signal erzeugt werden sollte. Diese Weiterentwicklung des Messaufbaus ist schematisch in
Abbildung 44 rechts dargestellt. Das Signal wird hier direkt im Innern des Resonators erzeugt.
Abbildung 45 Bild des ersten parallelen Aufbaus zur Messung unterschiedlicher Kohlendioxid
Konzentrationen.
Um erste Anhaltspunkte über eine mögliche Parallelisierung zu erhalten, wurde dieser
Aufbau, wie in Abbildung 45 durch ein Foto gezeigt, bereits in diesem frühen Stadium
vierfach parallel angefertigt. Der in den Abbildungen erwähnte PE- oder Silikon-Schlauch
erwies sich experimentell als notwendig, was wahrscheinlich in Dämpfungseffekten des
Schlauchmaterial begründet liegt. Zu Anfang konnte bei einer direkten Anbindung des
Mikrofons an die Resonanzröhre nur ein stark instabiles Signal beobachtet werden. Diese
dämpfende Verbindung wurde daher in allen folgenden Aufbauten eingesetzt.
0,0 5,0x10-5 1,0x10-4 1,5x10-4 2,0x10-4 2,5x10-4 3,0x10-4
-0,4
-0,3
-0,2
-0,1
0,0
0,1
0,2
0,3
0,4 36000 PPM 32000 PPM 28000 PPM 24000 PPM 20000 PPM 16000 PPM 12000 PPM 8000 PPM 4000 PPM 0 PPM
Inte
nsitä
t [V]
Zeit [s]
0 10000 20000 30000 40000
-0,45
-0,40
-0,35
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
Inte
nsitä
t [V]
Konzentration [PPM]
Abbildung 46 Oszilloskopsignal der Konzentrationsmessung von Kohlendioxid (links) und Korrelation
von Konzentration und Signalintensität (rechts). Die Konzentrationen wurden jeweils
schrittweise erhöht und bei Erreichen des Maximalwerts von 36000 ppm wieder
schrittweise auf 0 erniedrigt. Hieraus ergeben sich die beiden Äste der Korrelation.
84

6.4 Resonanter Aufbau
Die in Abbildung 46 links gezeigte Messung zeigt das Oszilloskopsignal bei
unterschiedlichen Kohlendioxidkonzentrationen. Rechts sind die gemessenen Signalstärken
gegen die Kohlendioxidkonzentration aufgetragen, wobei eine lineare Korrelation zwischen
gemessener Signalstärke und Konzentration an Kohlendioxid zu erkennen ist. Da die
Messungen an strömenden Medien durchgeführt wurden, benötigt eine Anbindung der
Einzelröhre an den Reaktorausgang zur on-line-Messung lediglich eine gasdichten
Verbindung.
Es war mit diesem Aufbau möglich, bis hinab zu Konzentrationen von 4000 ppm zuverlässig
zu arbeiten. Die Akkumulationszeit der Daten betrug weniger als 10 Sekunden pro Messung,
was in der langsamen Mittelung des Oszilloskops begründet lag. Da die Messungen resonant
durchgeführt wurden, ist die Geschwindigkeit der Signalabgreifung lediglich ein technisches
Problem, welches durch die Wiederholfrequenz von mehreren 1000 Anregungen pro Sekunde
determiniert wird. Für eine zuverlässige Messung sollten physikalisch einige dieser
Anregungszyklen ausreichend sein, was eine theoretische Messgeschwindigkeit von weit
unter einer Sekunde ermöglichen würde.
Da im Rahmen dieser Arbeit keine höheren Konzentrationen gemessen werden sollten,
wurden oberhalb von 10 % Kohlendioxid keine Testmessungen durchgeführt. Daher liegen
über diesen Bereich keine Informationen vor. Es ist anzunehmen, dass die Gesetzmäßigkeiten
für kleine Konzentrationen bei höheren Konzentrationen nicht mehr gelten.
Während der bisher beschriebenen Messungen wurde bereits klar, dass eine Parallelisierung
mit einer genauen Justage der Resonanzfrequenz und des Laserstrahles verbunden sein würde,
da es nicht ohne weiteres möglich war, Messungen mit dem in Abbildung 45 gezeigten
Aufbau an mehreren Kanälen gleichzeitig durchzuführen. Die Parallelisierung wird gesondert
in Kapitel 6.4.3 beschrieben. Weiter wurde deutlich, dass schon im Stadium früher
Experimente eine Automatisierung der Datenakkumulation angestrebt werden sollte, da mit
den Messungen ein hoher manueller Auswertungsaufwand einherging.
6.4.2.2 Resonanzrohr-Konzept
Die folgenden Messungen wurden bereits mit einem vorläufig automatisierten System
durchgeführt, auf welches hier nicht weiter eingegangen werden soll. Eine Beschreibung des
letztendlich in den Routinemessungen verwendeten Aufbaus nebst Software erfolgt in Kapitel
6.4.4.
85

6 Entwicklung der photoakustischen Analytiken
Abbildung 47 Verstellbare Resonanzröhre.
Für weitere Messungen wurde die in Abbildung 47 gezeigte, verstellbare Resonanzröhre
konstruiert, mit deren Hilfe automatisierte Messungen auf einem Kanal durchgeführt werden
konnten. Durch die Verstellbarkeit sollten genauere Erkenntnisse über die in der Röhre
ablaufenden Resonanzphänomene erhalten werden. Dieser Aufbau arbeitete bereits mittels
einer Lock-In-Verstärkung. Dieses Verstärkungskonzept erwies sich während der Messungen
als überaus sensitiv und unanfällig gegenüber Störungen, so dass es für weitere Aufbauten
standardmäßig eingesetzt wurde.
Lock-In-Verstärkerprinzip
Die Entscheidung für eine Lock-In-Verstärkung fiel aufgrund der guten Filtereigenschaften
dieser Systeme. Prinzipiell filtert ein solcher Verstärker über eine Multiplikation des
Messsignals mit einem Referenzsignal.
90°
Signal-In
Reference
Output
Abbildung 48 Lock-In-Prinzip. Links ist das Signal, welches gegen die Referenzphase um 90 °
verschoben ist, rechts die in-Phase-Situation. Nur wenn die Phase übereinstimmt, ist eine
optimale Verstärkung gegeben. Falls das Signal nicht in der Frequenz übereinstimmt, ist
keine Verstärkung möglich. Moderne Verstärker arbeiten zusätzlich mit einem rein
integrativen Verfahren, welches jede Welle mit übereinstimmender Frequenz verstärkt
(unabhängig von der Phasenlage).
Diese in Abbildung 48 verdeutlichte Verstärkungsvariante eignet sich demnach hervorragend
für den Einsatz bei periodischen Signalen, welche noch aus starkem Rauschen herausgefiltert
werden können. Da man durch den den Laser modulierenden Signalgenerator bereits ein,
86
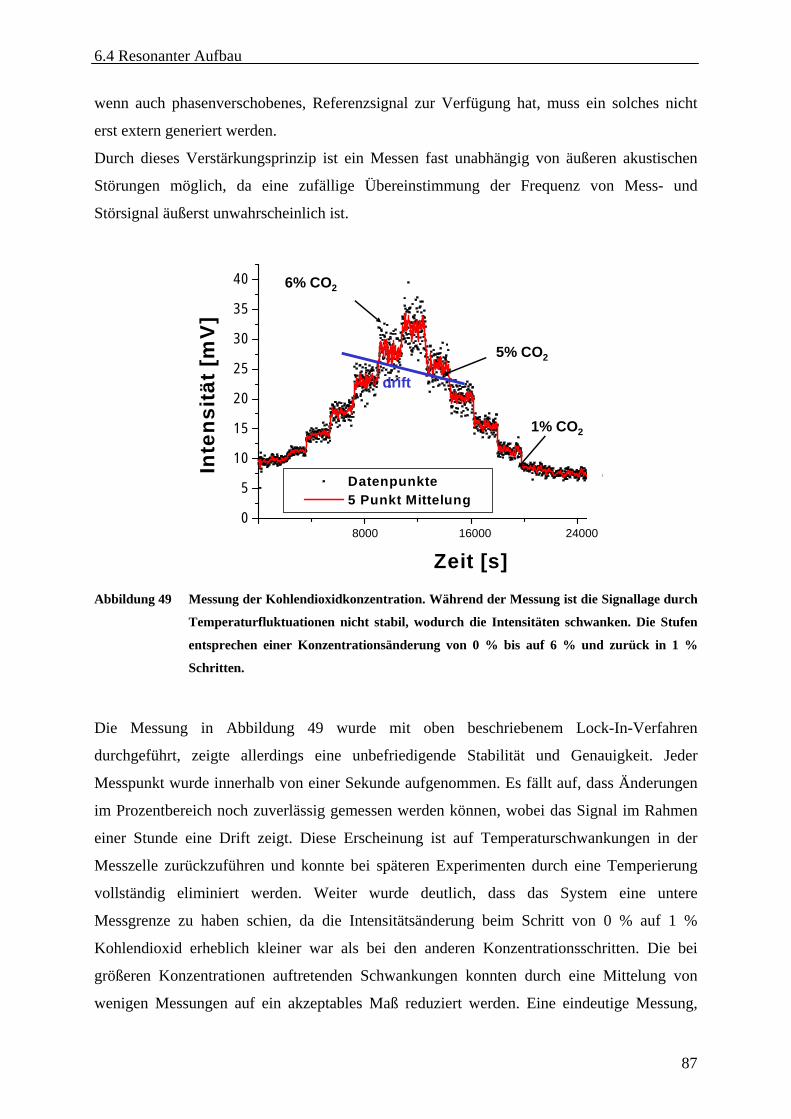
6.4 Resonanter Aufbau
wenn auch phasenverschobenes, Referenzsignal zur Verfügung hat, muss ein solches nicht
erst extern generiert werden.
Durch dieses Verstärkungsprinzip ist ein Messen fast unabhängig von äußeren akustischen
Störungen möglich, da eine zufällige Übereinstimmung der Frequenz von Mess- und
Störsignal äußerst unwahrscheinlich ist.
0
5
10
15
20
25
30
35
40
Datenpunkte 5 Punkt Mittelung
Inte
nsitä
t [m
V]
Zeit [s]
6% CO2
1% CO2
5% CO2
drift
24000160008000
Abbildung 49 Messung der Kohlendioxidkonzentration. Während der Messung ist die Signallage durch
Temperaturfluktuationen nicht stabil, wodurch die Intensitäten schwanken. Die Stufen
entsprechen einer Konzentrationsänderung von 0 % bis auf 6 % und zurück in 1 %
Schritten.
Die Messung in Abbildung 49 wurde mit oben beschriebenem Lock-In-Verfahren
durchgeführt, zeigte allerdings eine unbefriedigende Stabilität und Genauigkeit. Jeder
Messpunkt wurde innerhalb von einer Sekunde aufgenommen. Es fällt auf, dass Änderungen
im Prozentbereich noch zuverlässig gemessen werden können, wobei das Signal im Rahmen
einer Stunde eine Drift zeigt. Diese Erscheinung ist auf Temperaturschwankungen in der
Messzelle zurückzuführen und konnte bei späteren Experimenten durch eine Temperierung
vollständig eliminiert werden. Weiter wurde deutlich, dass das System eine untere
Messgrenze zu haben schien, da die Intensitätsänderung beim Schritt von 0 % auf 1 %
Kohlendioxid erheblich kleiner war als bei den anderen Konzentrationsschritten. Die bei
größeren Konzentrationen auftretenden Schwankungen konnten durch eine Mittelung von
wenigen Messungen auf ein akzeptables Maß reduziert werden. Eine eindeutige Messung,
87

6 Entwicklung der photoakustischen Analytiken
welche Aussagen über ± 0,5 % anstellen konnte, war somit innerhalb von 5 Sekunden
möglich.
6.4.2.3 Optimierung der Resonanzröhre
Zur Optimierung der Messung wurden eine Reihe von Änderungen an der Resonatorröhre
durchgeführt, von denen im folgenden lediglich die die Intensität steigernden bzw. das Signal
stabilisierenden Modifizierungen aufgezeigt werden sollen.
Um die in Abbildung 49 erkennbare Streuung der Messwerte und Instabilitäten zu beheben,
wurde eine Reihe von Veränderungen durchgeführt, welche die Detektionszone stabilisieren
bzw. das Signal am Mikrofon verstärken sollten.
50
100
150
200
250
300
350
400
129630
ResonanzMikrofonkennlinie
Inte
nsiä
t[w
.E.]
Frequenz [kHz]
Abbildung 50 Mikrofonkennlinie und Resonanzverstärkung der Resonanzröhre. Die Röhrenresonanz
wurde durch die direkte Einkopplung eines Signals entsprechender Frequenz in die
Detektionszone ermittelt. Bewerkstelligt wurde dies durch einen Referenzton, welcher
durch einen Schallleiter direkt in die Mitte der Detektionszone geleitet wurde. Da diese
auch der Ursprung der Druckpulse ist, wurde dieser experimentelle Aufbau als genügend
genaue Simulation eines Anregungsprozesses gewählt. Es ist oberhalb 6 kHz ein günstiger
Arbeitsbereich mit relativ kleinen Schwankungen zu erkennen.
Aus den in Abbildung 50 gezeigten bekannten Resonanzfrequenzen des Aufbaus konnte ein
optimaler Arbeitsbereich ermittelt werden, welcher sich aus der Filtereigenschaft der
Resonanzröhre und dem Arbeitsfenster des Mikrofons zusammensetzte. Es war hieraus zu
erkennen, dass eine Anregungsfrequenz von ca. 6 kHz die beste resultierende Signalintensität
haben sollte. Aus der hierfür errechneten Wellenlänge der Longitudinalresonanz von 5,8 cm
88

6.4 Resonanter Aufbau
ergab sich eine neue Resonatorkonstruktion, welche für optimale Resonanzbedingungen den
in Abbildung 51 gezeigten Aufbau besitzen sollte.
Mikrofon
PE-Schlauch
Gasfluss
Querrohr
Abbildung 51 Rechts abgebildet ist die optimale Resonanzbedingung in einem zylindrischen
Musterrohr. Wie ersichtlich, wurde die Verstärkung durch eine longitudinale Mode
bewirkt. Das Mikrofon sollte hierbei den gleichen Abstand zum Röhrenboden haben wie
der Laserstrahl zum oberen Ende (oder ein ganzzahliges Vielfaches). Der Zwischenraum
sollte ein ganzzahliges Vielfaches hiervon sein. Zu beachten ist die Verschiebung der
stehenden Druckwelle relativ zur stehenden Welle der Molekülgeschwindigkeiten von
90 °. Links zu sehen ist die neue Resonanzröhre, welche zusätzlich mit einem Querrohr
zur Stabilisierung des Messsignals ausgestattet ist.
Wie aus Abbildung 51 (rechts) folgte, fand die Resonanz in einer longitudinalen Mode statt.
Die longitudinale Resonanzverstärkung war aus der Anregungsposition theoretisch von
vornherein zu erwarten. Ebenfalls neu im optimierten Aufbau war die ungewöhnliche Position
des Mikrofons seitlich am Resonator. Eine maximale Amplitude sollte wie aus Abbildung 51
(rechts) ersichtlich auch am Ende der Röhre erwartet werden. Experimentell war jedoch an
der seitlichen Position eine deutliche Stabilisierung zu bemerken. Dies könnte auf elastische
Effekte des schon anfänglich verwendeten Schlauchmaterials (PE oder Silikon)
zurückzuführen sein, welches an der seitlichen Position der Druckwelle nicht nachgeben
konnte. Neu war auch das in Abbildung 51 links gezeigte Querrohr, welches die
Detektionszone umgab. Wie sich zeigte, war dieses Rohr für eine starke Verstärkung des
Signals verantwortlich. Dies ist verständlich, da die Detektionszone ein nicht abgeschirmter
offener Bereich ist und durch das Rohr eine gewisse Gasführung bewirkt wird. Es konnte
89

6 Entwicklung der photoakustischen Analytiken
gezeigt werden, dass die Länge des Rohrs ab einer gewissen kritischen Länge von etwa einem
Zentimeter bei weiterer Verlängerung keinen weiteren signifikanten verstärkenden Einfluss
auf das Messsignal hatte. Die verstärkende Wirkung dürfte also lediglich in einer
Stabilisierung der Flussverhältnisse innerhalb der Detektionszone begründet liegen, wodurch
Konzentrationsschwankungen im Anregungsbereich während der Messung vermieden
wurden. Im Anregungsbereich ist laut empirischer Gesetze der Photoakustik ein linearer
Anstieg des Messsignals in Abhängigkeit von der Länge des Absorptionspfads zu erwarten
(siehe Kapitel 5.3). Dies bestätigte sich hier jedoch nicht, was zeigt, dass das in unseren
Aufbauten verwendete Querrohr nicht als Absorptionspfad betrachtet werden kann, sondern
wie bereits erwähnt, eher der Gasführung dient.
0 5 0 0 1 0 0 0 1 5 0 0 2 0 0 0 2 5 0 02
4
6
8
1 0
1 2
1 4
1 6
Ko
nze
ntr
ati
on
[w
.E.]
Z e it [s ]
0% CO2
0,3% CO2
0,6% CO2
Abbildung 52 Messung der Konzentration unter 1 % Kohlendioxid. Die Stufen entsprechen einer
Konzentrationsänderung von 0 % bis auf 0,6 % und zurück in 0,1 % Schritten. Unter 0,3
% ist der Überlagernde Einfluss einer Störwelle zu erkennen.
Mit diesen optimierten Resonatoren war eine erhebliche Steigerung der Messgenauigkeit zu
verzeichnen. Diese ging soweit, dass der Bereich unter einem Prozent Kohlendioxid genauer
untersucht werden konnte, wobei ein Verlauf entsprechend der in Abbildung 52 gezeigten
Messkurve gefunden wurde. Man erkennt deutlich, dass das Messsignal zuerst mit steigender
Kohlendioxidkonzentration in der Intensität sinkt. Nach Überschreiten eines kritischen Werts,
welcher bei 0,3 % Kohlendioxid liegt, steigt es weiter linear bis auf 3 % Kohlendioxid absolut
an (in dieser Messung nicht mehr gezeigt).
Dieses Verhalten deutet auf eine Überlagerung mit einer konstanten Störwelle hin, was auch
anhand der Phasenanalyse erkannt werden konnte. Da durch diesen Effekt eine eindeutige
Messung der Konzentration in diesem Bereich nicht möglich war, wurden Anstrengungen
unternommen, um diesen Effekt auszuschalten. Dies gelang, indem der Lock-In-Verstärker
90
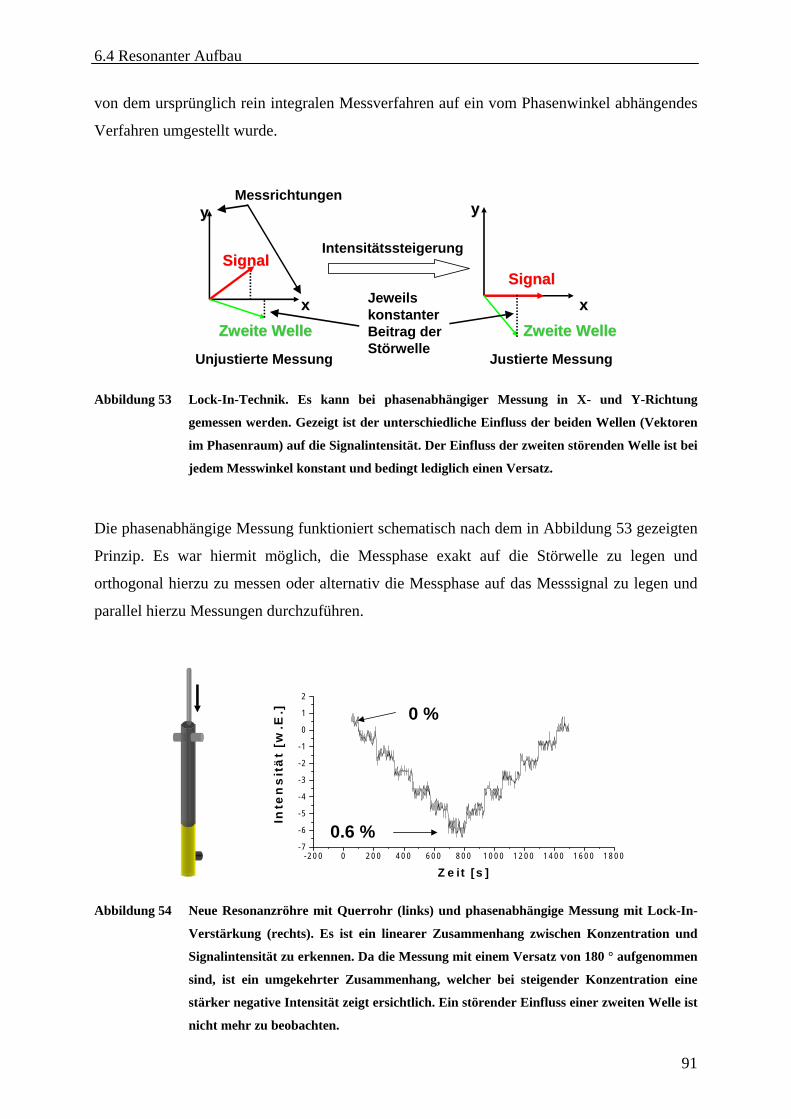
6.4 Resonanter Aufbau
von dem ursprünglich rein integralen Messverfahren auf ein vom Phasenwinkel abhängendes
Verfahren umgestellt wurde.
xx
yy
Zweite WelleZweite Wellex x
yy
Zweite WelleZweite Welle
Unjustierte Messung Justierte Messung
Intensitätssteigerung
Messrichtungen
Jeweils konstanter Beitrag der Störwelle
SignalSignalSignalSignal
Abbildung 53 Lock-In-Technik. Es kann bei phasenabhängiger Messung in X- und Y-Richtung
gemessen werden. Gezeigt ist der unterschiedliche Einfluss der beiden Wellen (Vektoren
im Phasenraum) auf die Signalintensität. Der Einfluss der zweiten störenden Welle ist bei
jedem Messwinkel konstant und bedingt lediglich einen Versatz.
Die phasenabhängige Messung funktioniert schematisch nach dem in Abbildung 53 gezeigten
Prinzip. Es war hiermit möglich, die Messphase exakt auf die Störwelle zu legen und
orthogonal hierzu zu messen oder alternativ die Messphase auf das Messsignal zu legen und
parallel hierzu Messungen durchzuführen.
-2 0 0 0 2 0 0 4 0 0 6 0 0 8 0 0 1 0 0 0 1 2 0 0 1 4 0 0 1 6 0 0 1 8 0 0-7
-6
-5
-4
-3
-2
-1
0
1
2
Inte
ns
itä
t [w
.E.]
Z e it [s ]
0 %
0.6 %
Abbildung 54 Neue Resonanzröhre mit Querrohr (links) und phasenabhängige Messung mit Lock-In-
Verstärkung (rechts). Es ist ein linearer Zusammenhang zwischen Konzentration und
Signalintensität zu erkennen. Da die Messung mit einem Versatz von 180 ° aufgenommen
sind, ist ein umgekehrter Zusammenhang, welcher bei steigender Konzentration eine
stärker negative Intensität zeigt ersichtlich. Ein störender Einfluss einer zweiten Welle ist
nicht mehr zu beobachten.
91

6 Entwicklung der photoakustischen Analytiken
Bei genaueren Untersuchungen wurde gefunden, dass der störende Anteil der Gesamtwelle
von der Konzentration des Kohlendioxid unabhängig war. Da dieser Anteil wie erwähnt, bei
Variationen der Konzentration konstant blieb, kann angenommen werden, dass die Störwelle
durch Absorptions- bzw. Streueffekte des Laserstrahls von der Resonanzröhre herrührt.
Alternativ könnte die Störung durch Absorption von Spurengasen bewirkt werden, welche
ebenfalls im Bereich der CO2-Laserstrahlung Absorption zeigen oder durch resonanten
Energieübertrag durch Kohlendioxid angeregt werden. Wäre ein Beitrag einer resonanten
Übertragung der Energie auf ein Schwingungsniveau des Stickstoffmoleküls in der Gasphase
verantwortlich, sollte nicht erwartet werden, dass der Beitrag von der
Kohlendioxidkonzentration unabhängig ist. Solche Effekte können, wenn die
Relaxationsprozesse der Fremdgase eine relativ zum Kohlendioxid unterschiedliche Zeit in
Anspruch nehmen, eine Welle mit einer verschobenen Phase bewirken61. Da die Justage des
Lasers einen großen Einfluss auf die Störwelle hatte und bei schlechter Justage die Störwelle
intensiver war, wurde die Störung somit mit großer Wahrscheinlichkeit durch ein Signal von
der Resonanzröhre hervorgerufen.
Mit Hilfe der Lock-In-Messtechnik war es möglich, bei weiteren Messungen ein fast
störungsfreies Messsignal, wie auch das in Abbildung 54 gezeigte, mit einer linearen
Korrelation zwischen Intensität und Konzentration zu erhalten. Die Dauer einer Messung
reduzierte sich aufgrund der stark gestiegenen Signalstabilität, die eine Mittelung unnötig
machte, auf unter eine Sekunde.
6.4.3 Parallelisierung
Da die Messgeschwindigkeit mit der oben beschriebenen optimierten Resonanzröhre und
entsprechender Verstärkungstechnik genügend schnell war, wurde anstelle des geplanten
parallelen Vorgehens eine sequenzielle Methodik eingesetzt. Der wesentlich geringere
apparative Aufwand lag auf der Hand, da mit einem einzigen Lock-In-Verstärker gearbeitet
werden konnte und keine multiple Signalverarbeitung vom Rechner zu bewältigen war.
92

6.4 Resonanter Aufbau
Abbildung 55 Parallele Röhren
Abbildung 55 zeigt die Anordnung verstellbarer Resonanzröhren. Es ist zu erkennen, dass
zwischen den einzelnen Kanälen, will man die verwendbare maximal nutzbare Länge des
unfokussierten Strahlengangs von 50 cm nicht überschreiten, nur noch ein Raum von ca. 3
mm blieb. Dies bedeutet, dass einmal eingebaute Röhren nur noch bedingt nachträglich
eingestellt werden konnten und der anfänglichen Justage große Aufmerksamkeit geschenkt
werden musste.
Jede Röhre wurde mit einem eigenen Mikrofon ausgestattet, wovon jedes einen eigenen
Vorverstärker besaß. Für die Verstärker wurde ein Multiplexer-Verstärker-Modul entwickelt.
Die einzelnen Verstärker waren nach dem in Abbildung 43 gezeigten Muster des vorher
verwendeten Einzelverstärkers gebaut und wurden von einer gemeinsamen 9 V-Quelle
betrieben. Die Eingänge eines 16-fach Multiplexers (PCLD-788 von Spectra) waren mit den
jeweiligen Verstärkern verbunden. Der Multiplexer wurde automatisch rechnergesteuert
betrieben. Der Ausgang der Multiplexerkarte war mit einer BNC-Buchse verbunden, an
welche der Lock-In-Verstärker angeschlossen werden konnte. Dieser Aufbau ermöglichte ein
sequenzielles Messen, wobei jeweils nur ein einziges Mikrofon auf den Lock-In-Verstärker
geschaltet wurde.
Mit diesem parallelen Aufbau wurden Testmessungen an einem Standardkatalysator (2 % Pt
auf Al2O3) durchgeführt.
93

6 Entwicklung der photoakustischen Analytiken
140 160 180 200
0
20
40
60
80
100
120 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Umsa
tz [%
]
Temperatur [°C]
Al2O3
Abbildung 56 Paralleler Katalysatortest. Verwendet wurde der früher beschriebene Messingreaktor.
Eingesetzt wurde eine Gasmischung von 3 % CO in Luft bei einer Raumgeschwindigkeit
von 18750 ml·h-1g-1Katalysator. Die Nummern sind die einzelnen Katalysatorpositionen.
Position 16 ist mit reinem Al2O3 gefüllt, weshalb kein Umsatz erwartet werden sollte. Alle
anderen Positionen sind mit 2% Pt auf Al2O3 gefüllt. Die Detektion von Kohlendioxid in
Kanal 16 zeigt ein Übersprechen der benachbarten Kanäle (Cross Talk).
Die Testbedingungen entsprachen den im experimentellen Teil beschriebenen. Um ein
Übersprechen der Kanäle zu überprüfen, wurde eine Position abweichend von allen anderen
mit reinem Al2O3 befüllt. Das Ergebnis in Abbildung 56 zeigt einen 100-prozentigen Umsatz
auf fast allen Kanälen, was bei Edelmetallkatalysatoren auch zu erwarten war. Der mit
Aluminiumoxid befüllte Kanal zeigte allerdings einen Umsatz von ca. 70 %, was eindeutig
auf einen Einfluss durch andere Messkanäle zurückgeführt werden konnte. In weiteren
Testmessungen wurde nach einigen Modifizierungen des Reaktors, welcher teilweise zu den
Messfehlern beitrug, ein weiteres Übersprechen im Reaktorraum ausgeschlossen. Ein
Restanteil an vorgetäuschter Aktivität konnte nur auf einen konstruktionsbedingten Fehler
innerhalb der Analytik zurückgeführt werden. Um dies zu überprüfen, wurden die einzelnen
Kanäle mit ZnSe-Fenstern ausgestattet, was eine Diffusion von Gas aus der Nachbarröhre in
den Messkanal verhindern und Schallwellen blockieren sollte.
94

6.4 Resonanter Aufbau
110 120 130 140 150 160 170
-100
102030405060708090
100
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Umsa
tz [%
]
Temperatur [°C]
ÜbersprechenÜbersprechen
Abbildung 57 Paralleler Katalysatortest. Kanal 14 und 15 sind mit ZnSe-Fenstern abgeschirmt.
Auffällig ist besonders der vorgetäuschte Umsatz in Kanal 5 und 7 im Bereich eines
hohen Umsatzes in Kanal 6. Dieses Übersprechen wird vor allem bei Umsätzen über 50
% in den Nachbarkanälen gefunden, falls diese nicht wie Kanal 14 durch ZnSe-Fenster
abgeschirmt sind.
In Abbildung 57 ist eine Vergleichsmessung gezeigt. Kanal 6 und 14 sind mit dem
Testkatalysator befüllt, wobei die Resonatoren 14 und 13 mit einem ZnSe-Fenster bestückt
sind. Es ist deutlich zu erkennen, dass Kanal 6 bei Aktivität des Katalysators die beiden
benachbarten Kanäle 5 und 7 beeinflusst und in diesen eine Aktivität vortäuscht. Im
Gegensatz hierzu zeigen die Nachbarkanäle von Kanal 14 keinen erkennbaren
Aktivitätsanstieg. Da die Geometrie der Messanordnung linear ist, die Geometrie des
Reaktorkörpers jedoch einer 4x4 Matrix entspricht, kann jede Störung innerhalb des
Reaktorkörpers von vornherein ausgeschlossen werden.
Aufgrund dieser Ergebnisse folgte eine Umrüstung der Resonanzröhren, welche einseitig mit
einem ZnSe-Fenster ausgestattet wurden und somit unabhängig voneinander arbeiten konnten.
6.4.4 Automatisierung
Aufgrund der hohen Datenmengen wurde für die photoakustische Analytik eine
automatisierte Auswertungsroutine entwickelt. Da die einzelnen Temperaturprogramme
95

6 Entwicklung der photoakustischen Analytiken
teilweise bis zu zwölf Stunden dauerten, war eine komplette Automatisierung des gesamten
Systems eine logische Folge.
Laser
Resonanzröhren
Multiplexer
Lock-In Verstärker
Puls Generator
Abbildung 58 Verknüpfung der einzelnen elektronischen Bauteile im routinemäßig betriebenen
automatisierten Aufbau zur parallelen Untersuchung der Kohlenmonoxid-Oxidation.
Das Schema in Abbildung 58 zeigt die elektronische Verknüpfung der einzelnen Bestandteile.
Dieses System wurde für die Routinemessungen eingesetzt und 24 Stunden am Tag
kontinuierlich betrieben. Die Zeitbasis für das System war der Signalgenerator, welcher
sowohl das Signal für die Lasermodulation bereitstellte, als auch das Referenzsignal für die
Lock-In-Verstärkung generierte. Da es sich um ein vom Computer unabhängiges System
handelte, konnte eine hohe zeitliche Konstanz sichergestellt werden. Das Signal der
Mikrofone ging in die Vorverstärker der Multiplexer-Einheit. Von hier aus wurde jeweils ein
Kanal auf den Lock-In-Verstärker gelegt, welcher das Signal an den Computer weiterleitete.
96

6.4 Resonanter Aufbau
���������
Gasfluss Temperatur
Eurotherm
RS-485 EI-bisynch
3 BronkhorstMFC
analog 0 - 5 V
Zeit t3 Gasfluss Vi(t)1 Temperatur T(t)Laserkontrolle (t)Kalibrierung (t) Temperatur
Umsatz zu CO2
Gasfluss CO,N2,CO2
������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������
������������������������
Protokoll-Logfile
������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������������
Lock-InPerkin Elmer 5105
RS-232
Multiplexer
Spectra PCLD-788
Mikrofone&
Verstärker
Conrad Nr. 1929 10-66
Laser
DEOS LC-25
25 Watt
Puls Generator
9514 Series
Labview-Modul
Input
PC (Win NT, TCP-IP)
���������������������
������������
����������������������
����������������������������
��������������������
�������������
��������������������������������������
Output
��������������
Abbildung 59 Schematische Zeichnung der elektronischen Verknüpfung und des Datenflusses bei der
photoakustischen Resonanz-Analytik. Die Reaktionskontrolle und die Analytik sind, wie
aus der Abbildung zu erkennen ist, voneinander unabhängige Routinen. Die einzige
Verknüpfung zwischen den Bauteilen besteht in der Doppelfunktion des Puls Generators
zur Steuerung des Lasers und des Lock-In-Verstärkers.
Abbildung 59 verdeutlicht den Datenfluss. Es ist erkenntlich, dass sich die Prozessführung
analog zur Freifeld-Analytik gestaltete. Die Steuerung der für die Analyse verantwortlichen
Bauteile erfolgte durch eine unter LabVIEW entwickelte Routine. Da der Lock-In-Verstärker
nicht für die Aufnahme von mehreren sequenziellen, unterschiedlichen Signalen geeignet war,
musste experimentell ein Wert ermittelt werden, innerhalb dessen alle aufgenommenen Daten
des Vorgängerkanals gelöscht und die Messung lediglich mit Daten des aktuellen Kanals
durchgeführt wurde. Dies geschah über eine Kompromisslösung bezüglich der Zeitkonstante.
Es mussten für eine Messung genügend Signalzyklen bereitgestellt werden, um ein stabiles
Signal zu erhalten, jedoch musste mindestens der zehnfache Wert als Pause eingehalten
werden, um eine Einstellung auf den nächsten Kanal zu ermöglichen. Durch eine
softwareseitige Synchronisation von Steuerbefehl an die Lock-In-Einheit und Schaltprozess
auf den nächsten Kanal wurde dieses anfänglich auftretende Problem behoben. Als
Integrationskonstante τ wurde ein Wert von 30 Millisekunden gewählt. Der eigentliche
Schaltprozess erfolgte über vier Digitalleitungen, welche mittels einer beliebigen
Kombination der Bitwerte 1 oder 0 die 16 Kanäle freischalten konnten. In diesem Rahmen
wurde mit der Firma Advantech ein für LabVIEW Anwendungen verbesserter Treiber
97

6 Entwicklung der photoakustischen Analytiken
entwickelt, da anfänglich eine korrekte Steuerung der Digitalkanäle der Messkarte (PCL 812
PG von Spectra) nicht möglich war. Die Datenauswertung wurde direkt von der Lock-In-
Einheit übernommen, die über eine RS 232 Schnittstelle dem Computer den aktuellen
Messwert übermittelte. Der Rechner übernahm hierauf die Umrechnung in Umsatzprozente.
Alle weiteren Befehle an die Lock-In-Einheit erfolgten ebenfalls über diese Schnittstelle.
Um den Einfluss eventueller Dejustagen zwischen den einzelnen Messungen auszuschließen,
wurde die Anlage vor jeder Messung erneut kalibriert. Diese Routine nahm etwa zehn
Minuten in Anspruch und konnte automatisiert durchlaufen werden. Die Resonatoren wurden
hierbei mit den Gasmischungen für 0 und 100 % Umsatz gespült und die Intensitäten als Basis
für die spätere Umrechnung in Umsatzprozente gespeichert.
Die Programmierung für den automatisierten Betrieb erfolgte über ein Protokoll, welches in
einem kommerziellen Tabellenkalkulationsprogramm erstellt werden konnte. Nach Ablauf
von einer Minute wurden jeweils neue Steuerwerte eingelesen und zu den entsprechenden
Komponenten wie Massendurchflussregler und Temperatursteuerung übertragen. Die
Schaltprozesse zwischen den einzelnen Kanälen erfolgten im Routinebetrieb alle drei
Sekunden. Eine Messung innerhalb einer Sekunde war ebenfalls möglich, jedoch verbunden
mit einer ungenaueren Datenerfassung. Der Wert für die Messrate wurde manuell am Rechner
festgelegt und konnte nicht automatisiert eingegeben bzw. verändert werden.
Abbildung 60 Benutzeroberfläche der Resonanz-Analytik.
Die Benutzeroberfläche des fertigen Steuerprogramms ist in Abbildung 60 gezeigt. Die
Messwerte konnten entweder als echte gemessene Werte in Volt oder als korrigierte
98
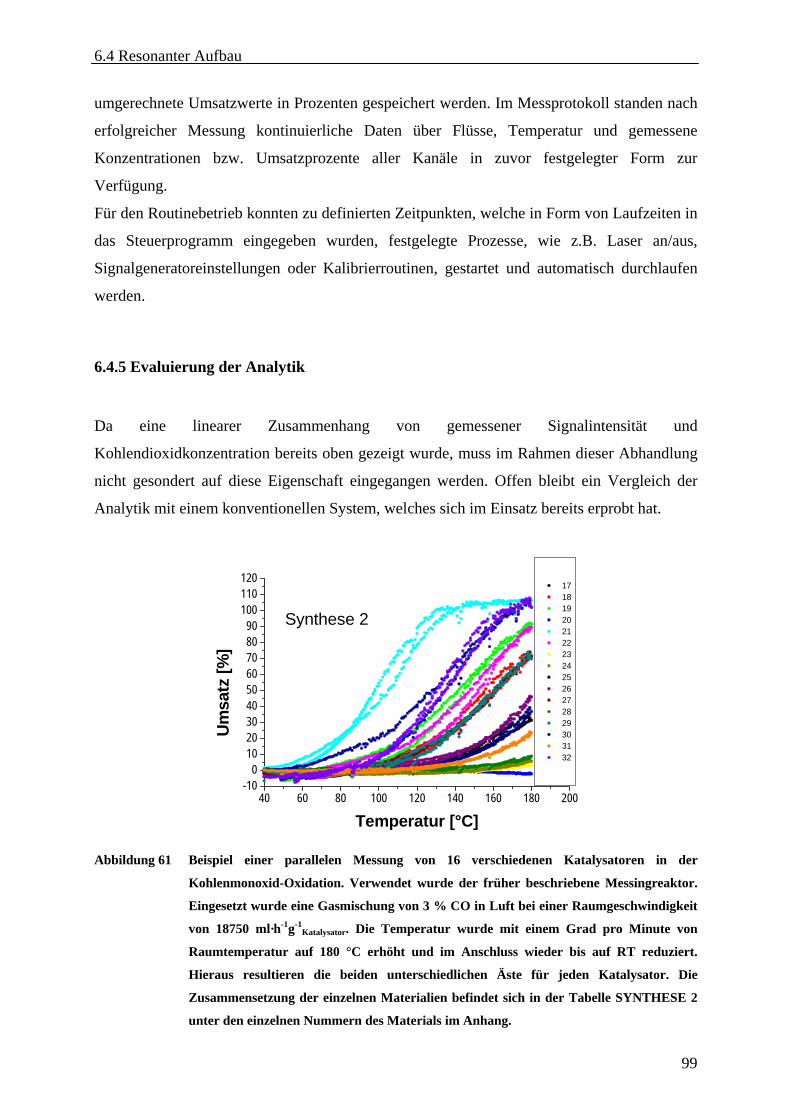
6.4 Resonanter Aufbau
umgerechnete Umsatzwerte in Prozenten gespeichert werden. Im Messprotokoll standen nach
erfolgreicher Messung kontinuierliche Daten über Flüsse, Temperatur und gemessene
Konzentrationen bzw. Umsatzprozente aller Kanäle in zuvor festgelegter Form zur
Verfügung.
Für den Routinebetrieb konnten zu definierten Zeitpunkten, welche in Form von Laufzeiten in
das Steuerprogramm eingegeben wurden, festgelegte Prozesse, wie z.B. Laser an/aus,
Signalgeneratoreinstellungen oder Kalibrierroutinen, gestartet und automatisch durchlaufen
werden.
6.4.5 Evaluierung der Analytik
Da eine linearer Zusammenhang von gemessener Signalintensität und
Kohlendioxidkonzentration bereits oben gezeigt wurde, muss im Rahmen dieser Abhandlung
nicht gesondert auf diese Eigenschaft eingegangen werden. Offen bleibt ein Vergleich der
Analytik mit einem konventionellen System, welches sich im Einsatz bereits erprobt hat.
40 60 80 100 120 140 160 180 200-10
0102030405060708090
100110120
Synthese 2
17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32
Umsa
tz [%
]
Temperatur [°C]
Abbildung 61 Beispiel einer parallelen Messung von 16 verschiedenen Katalysatoren in der
Kohlenmonoxid-Oxidation. Verwendet wurde der früher beschriebene Messingreaktor.
Eingesetzt wurde eine Gasmischung von 3 % CO in Luft bei einer Raumgeschwindigkeit
von 18750 ml·h-1g-1Katalysator. Die Temperatur wurde mit einem Grad pro Minute von
Raumtemperatur auf 180 °C erhöht und im Anschluss wieder bis auf RT reduziert.
Hieraus resultieren die beiden unterschiedlichen Äste für jeden Katalysator. Die
Zusammensetzung der einzelnen Materialien befindet sich in der Tabelle SYNTHESE 2
unter den einzelnen Nummern des Materials im Anhang.
99

6 Entwicklung der photoakustischen Analytiken
Abbildung 61 zeigt einen parallelen Test, wobei einer der Katalysatoren durch seine im
Vergleich zu den anderen herausragende Aktivität auffällt. Ausgewählte Katalysatoren,
welche einen guten Umsatz in der Kohlenmonoxid-Oxidation zeigten, wie auch dieser
Katalysator mit der Nummer 21, wurden zusätzlich in einem Einrohr-Reaktor mit einer
URAS-Analytik vermessen. Auf diese Weise sollten die Ergebnisse der photoakustischen
Messung überprüft werden, um einen Vergleich der photoakustischen Analytik mit
kommerziell erhältlichen Systemen zu ermöglichen.
40 60 80 100 120 140 160 180 200-20
0
20
40
60
80
100
120
Temperatur [°C]
Umsa
tz [%
]
URAS Parallel
Abbildung 62 Vergleich der parallel photoakustischen Messung und der URAS-Messung im Einrohr-
Reaktor. Die URAS-Messungen liegen exakt zwischen den einzelnen Ästen der parallelen
Messung. Temperaturunterschiede von ± 5 °C können vernachlässigt werden.
Wie aus Abbildung 62 ersichtlich ist, liegt die Umsatzkurve, welche mit der konventionellen
URAS-Messung erhalten wurde, direkt zwischen den Zünd- und Lösch-Kurven der
photoakustischen Messung. Die Abweichung von maximal 5 °C kann vernachlässigt werden
und ist teils auf die transiente Messung bei der Parallelanalyse zurückzuführen. Die
Unterschiede in den beiden Ästen aus der photoakustischen Messung sind auf das
Wärmemanagement des Reaktorgrundkörpers und der hieraus verfälschten
Temperaturmessung zurückzuführen. Dies liegt an der Lage des Temperaturfühlers, welcher
in der Mitte des Reaktors liegt und die lokale Temperatur der einzelnen Position, welche
hiervon bei der transienten Messung abweichen kann, nicht erfasst.
Abschließend bleibt noch anzumerken, dass vor jeder Messung eine Justage der Phasenlage
des Messsignals notwendig war, was für alle Kanäle zusammen etwa zehn Minuten in
Anspruch nahm. Mit dieser Justage wurde sichergestellt, dass leichte Verschiebungen der
100

6.4 Resonanter Aufbau
Phase sich aufgrund der geringen Steigung des Signalverlaufs an Extremstellen nicht oder nur
in geringem Maße auf die gemessene Gesamtintensität des Signals auswirkten.
6.4.6 Zusammenfassung und Diskussion
Die photoakustische Detektion mit Resonanzverstärkung stellt bei Reaktionen mit bekannten
Produkten eine gute und schnelle Alternative zu bereits verwendeten Methoden dar. Der
parallele Aufbau gestattet ein extrem schnelles Messen, welches zwar im vorliegenden Fall
sequenziell durchgeführt wurde, jedoch auch parallel denkbar wäre. Anfängliche Bedenken,
dass die einzelnen Kanäle sich beeinflussen könnten, eine Absorption des Laserstrahls in einer
Resonanzröhre also eine Abschwächung des Signals in der folgenden Röhre zur Folge haben
könnte, bestätigten sich nicht oder konnten zumindest im verwendeten Aufbau nicht detektiert
werden. Da die Signalanregung parallel erfolgte, ist die Information an jedem Mikrofon zu
jeder Zeit abgreifbar und somit echtzeit-parallel verfügbar, nötig hierfür ist lediglich ein
multipler Lock-In-Verstärker.
Der Aufbau, welcher für jeden Kanal ein eigenes Mikrofon vorsieht, mag im ersten
Augenblick als Nachteil erscheinen. Da die Kosten pro Mikrofon jedoch unter einem Euro
liegen, ist dieser Punkt zumindest in finanzieller Hinsicht kein Nachteil. Bezüglich der
Schnelligkeit ist der resonante Aufbau in der Lage, Informationen über alle Katalysatoren
selbst im sequenziellen Modus innerhalb einer Minute zu erhalten. Diese Geschwindigkeit
lässt einen Vergleich der einzelnen Positionen noch in akzeptabler Weise zu.
Die Konstruktion macht den resonanten Aufbau völlig unempfindlich gegenüber Störungen
wie Luftströmung oder Umgebungsgeräusche, so dass dieser Aufbau hervorragend für den
Einsatz in jedweder Umgebung geeignet ist. Diese macht ihn auch für einen industriellen
Einsatz geeignet. Weiterhin ist er sehr leicht und zuverlässig zu bedienen und benötigt kein
Aufsichtspersonal.
Der einzige manuelle Eingriff in den Gesamtprozess, sieht man von dem Befüllen der
Katalysatorpatronen ab, ist die manuelle Phasenjustage zu Beginn einer Messung. Diese kann
jedoch, wenn kleine Schwankungen im unteren Prozentbereich vernachlässigt werden können,
entfallen.
101

7 Weiterentwicklung der photoakustischen Analytik
Wir sind alle ständig Teil eines Experimentes, nur manche merken das nicht ... Aber
die, die die Experimente machen, sind ebenfalls wieder Teil eines Experimentes ...
Wolfgang Reus
7.1 Motivation
Die bisher vorgestellten Systeme haben bereits überaus vielseitige Einsatzmöglichkeiten.
Durch eine kompakte Konstruktion und die hohe Messgeschwindigkeit ist eine Verwendung
dieser Analytik in vielen Einsatzbereichen denkbar.
Für einige Problemstellungen in Systemen mit höherem Parallelisierungsgrad kann es jedoch
wünschenswert sein, mit geringeren Flüssen zu arbeiten. Die bisher durchgeführten
Messungen bewegten sich im Bereich von mehreren 10 ml·min-1 pro Kanal. Es wurden daher
Untersuchungen zur Flussverringerung, sowohl im Freifeld als auch in Resonatoren,
durchgeführt. In diesem Zusammenhang wurde auch die minimal notwendige Messzeit
untersucht, da ein geringerer Fluss unausweichlich mit einer längeren Spülzeit der
Resonatoren zusammenhängt. Für eine erfolgreiche on-line-Testung sollte diese Spülzeit so
kurz wie möglich sein, um jederzeit Informationen über den gerade untersuchten Kanal
erhalten zu können.
Um das Potenzial dieser Methode weiter auszuschöpfen, wurden außerdem Untersuchungen
über die minimal notwendige Laserleistung unternommen. Sollte eine Weiterentwicklung des
Systems in bezug auf geringere Laserleistung möglich sein, würde dieser Optimierungsschritt
den Einsatz neuer Lasersysteme in anderen Wellenlängenbereichen erlauben. Insbesondere
der Bleisalzlaser, welcher kontinuierlich im gesamten Infrarotbereich zwischen 2 bis 20 µm
arbeiten kann, wäre ein sehr interessantes, universell einsetzbares Lasersystem. Hierdurch
würde der Nachweis vieler interessanter Moleküle möglich, welche in diesem Bereich eine
selektive Absorptionsbande besitzen.
102

7.2 Experimente mit kleineren Flüssen
7.2 Experimente mit kleineren Flüssen
103
0 1 2 3 4 5 6 7 8 9 10 12 13 14 1511
CO-Laser
CO2-Laser
Bleisalz-Laser
Wellenlänge [µm]
Abbildung 63 Zugängliche spektrale Einsatzbereiche der Bleisalzlaser und der gitterabstimmbaren
CO- und CO2-Lasersysteme.
Theoretisch wäre für einen breiteren Einsatz auch die Anwendung eines gitterabstimmbaren
CO-oder CO2-Lasers, deren spektrale Bereiche in Abbildung 63 gezeigt sind, denkbar. Diese
Lasertypen besitzen Emissionsstärken von bis zu 1 W pro Emissionslinie. Aufgrund der
Einschränkung auf den Wellenlängenbereich zwischen 5 bis 7 bzw. 9 bis 11 µm sind sie
jedoch nur für spezielle Moleküle, welche in diesen Bereichen absorbieren, nützlich. Für
Messverfahren, die mit einer Strahlintensität von 1 mW arbeiten können, wäre ein Einsatz der
Bleisalzlaser möglich und hiermit der gesamte spektrale Bereich als Arbeitsfeld offen. Dies
würde auch die C-N-Dreifachbindung und die C-O-Doppelbindung als Detektionsziel
erlauben. Diese Bindungen kommen in vielen für die Industrie wichtigen Zwischenprodukten
vor. Moleküle mit diesen Gruppen sind daher mögliche zukünftige Analyten.
Apparativer Aufbau
Um Untersuchungen mit beliebig kleinen Flüssen durchführen zu können, wurde ein
Gasversorgungssystem zusammen mit der hte AG in Heidelberg konstruiert und am Max-
Planck-Institut für Kohlenforschung in den vorhandenen Photoakustikaufbau integriert. Es
war zu erwarten, dass die Detektion mit geringen Signalstärken einhergehen würde, weshalb
die oxidative Dehydrierung von Ethan als Testreaktion eingesetzt wurde. Es sollte durch die
starke Absorptionsbande des Ethen im Bereich der Laserstrahlung ein starkes Signal erhalten
werden.

7 Weiterentwicklung der photoakustischen Analytik
104
Luft �����������
����������� ��������
���������������������������������
������
���������
C2H6
����������������������������������
�����
�����������������������
������������������������
�������������
�������������������Messzelle
������������������������
�����������������������������������������������������������������������������C2H4
Split-Ventil
Reservoir
Reservoir
Split-Ventil
Druckluft-Ventil
Abbildung 64 Schematische Zeichnung der Gasversorgung. Die Druckreservoire stellen sicher, dass die
Massenflussregler nicht durch Druckschwankungen im System ungenau arbeiten. Über
die beiden Split-Ventile wird der eigentliche Nachdruck eingeregelt und der Fluss in der
Messzone justiert.
Anfängliche Stabilisierungsprobleme kleiner Gasströme mit Aufbauten ohne
Nachdruckregelung der Massendurchflussregler hatten starke Schwankungen des Flusses am
Ausgang des druckluftgesteuerten Ventils zur Folge und führten daher zu starken
Schwankungen der Signalintensität. Eine Version, in der diese Probleme durch ein Reservoir
gelöst wurden, welches einen konstanten Nachdruck der Massendurchflussregler sicherstellte,
ist in Abbildung 64 gezeigt. Mit dieser Gasversorgung wurden alle nachfolgenden
Experimente durchgeführt.
In der für die Versuche verwendeten Gasversorgung wurden die Gasmischungen über jeweils
zwei manuell geregelte Massendurchflussregler der Firma Brooks (HFC-202) eingestellt.
Über ein Nadelventil konnte der jeweilige Nachdruck der Regler an einem durchflossenen
Druckgefäß, welches die Gasmischung enthielt, geregelt werden. Der Nachdruck war für den
Fluss verantwortlich, welcher sich am Analysatorausgang einstellte. Die Gasmischung wurde
über Glaskapillaren mit verschiedenen Durchmessern in die Analysenzone eingedüst.
Experimentell stellte sich ein Durchmesser von 100 µm als optimal heraus, weshalb alle hier
beschriebenen Experimente mit diesen Kapillaren durchgeführt wurden. Um die Zeit zu
bestimmen, die das System zum Spülen benötigte, konnten zwei unterschiedliche
Mischkonzentrationen separat eingestellt werden. Ein druckluftgesteuertes Ventil konnte
zwischen diesen Mischungen in sehr kurzen Zeiten manuell oder automatisch in
programmierbaren Zeitabständen hin und her schalten.

7.2 Experimente mit kleineren Flüssen
105
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,00,0
0,5
1,0
1,5
2,0
2,5
3,0
Druck [bar]
Flu
ss [m
l/min
]
Abbildung 65 Nachdruck/Fluss-Relationen der Messkanäle. Mit Druck ist der über die beiden Split-
Ventile eingestellte Nachdruck gemeint. Der Fluss bezieht sich auf die Analysenzone und
wurde mittels eines kalibrierten Massenflussmessers bestimmt. Der zu erwartende lineare
Zusammenhang, welcher im Bereich unterhalb 1 ml/min nicht gefunden wird, ist
wahrscheinlich auf Ungenauigkeiten des Massenflussmessers zurückzuführen.
Der Fluss, welcher in der Analysenzone herrschte, konnte über den Nachdruck der
Massendurchflussregler, welcher am Reservoir abgelesen wurde, ermittelt werden. Die
Korrelation dieser Werte ist in Abbildung 65 gezeigt.
Die Steuerung der Analyseneinheit und Auswertung der Messdaten erfolgte über eine
Kombination der bei der Untersuchung der Freifeld- und Resonanz-Analytik entwickelten
Hard- und Software. Anfängliche Versuche, die Experimente mit Hilfe des Lock-In-
Verstärkers durchzuführen, scheiterten an der mangelndem Periodizität des Messignales. Die
Daten wurden daraufhin mit dem schon im Freifeld-Aufbau verwendeten Oszilloskop
aufgezeichnet und direkt über eine RS 232 Schnittstelle ausgelesen und analysiert. Die
Analyseroutine entsprach der des Freifeld-Aufbaus und bediente sich der Integration der
ermittelten Intensität der Schallpulse. Über eine Kalibrierung konnte somit der prozentuale
Anteil des Messgases ermittelt werden. Für anfängliche Experimente wurden die Messsignale
jedoch manuell ausgewertet.

7 Weiterentwicklung der photoakustischen Analytik
106
Abbildung 66 Vollständige Messzelle im Einsatz (links) und einzelne Module mit unterschiedlichem
Zellvolumen mit Mikrofoneinsatz (mitte und rechst). Die linken Module sind für Freifeld-
Messungen, die rechten mit an den Analysenkanal angekoppeltem Mikrofon.
Da die Freifeld-Experimente erfolglos verliefen, wurde eine eigens für diese Problemstellung
geeignete Messplattform mit einzelnen Zellenmodulen entwickelt. Die Plattform, welche über
einen Federmechanismus gehalten wurde, ist in Abbildung 66 gezeigt. Sie konnte durch
mehrere Justageschrauben in ihrer Höhe und Neigung einfach verändert werden. An diese
Zelle wurde die Glaskapillare des druckluftgesteuerten Ventils angeschlossen. Die einzelnen
Messzellen (Module, Abbildung 66 Mitte) waren beidseitig offen, so dass das Gas schnell und
ungehindert ausströmen konnte und durch die Messzelle lediglich eine Gasführung bewirkt
wurde. Diese Führung bewirkte eine Stabilisierung der Gasmischung innerhalb der Messzone
und ermöglichte die Detektion.
Abbildung 67 Schematische Zeichnung des Messzellentisches (links) und der Messmodule (rechts). Das
Zentralloch mit 2 mm Durchmesser in der Mitte der Module (rechts) war der eigentliche
Messkanal und diente zur Gasführung bzw. zur Stabilisierung der Detektionszone. Es
wurden für die Messungen Module mit einer Messkanallänge von 2, 4, 6 und 8 mm
gefertigt.
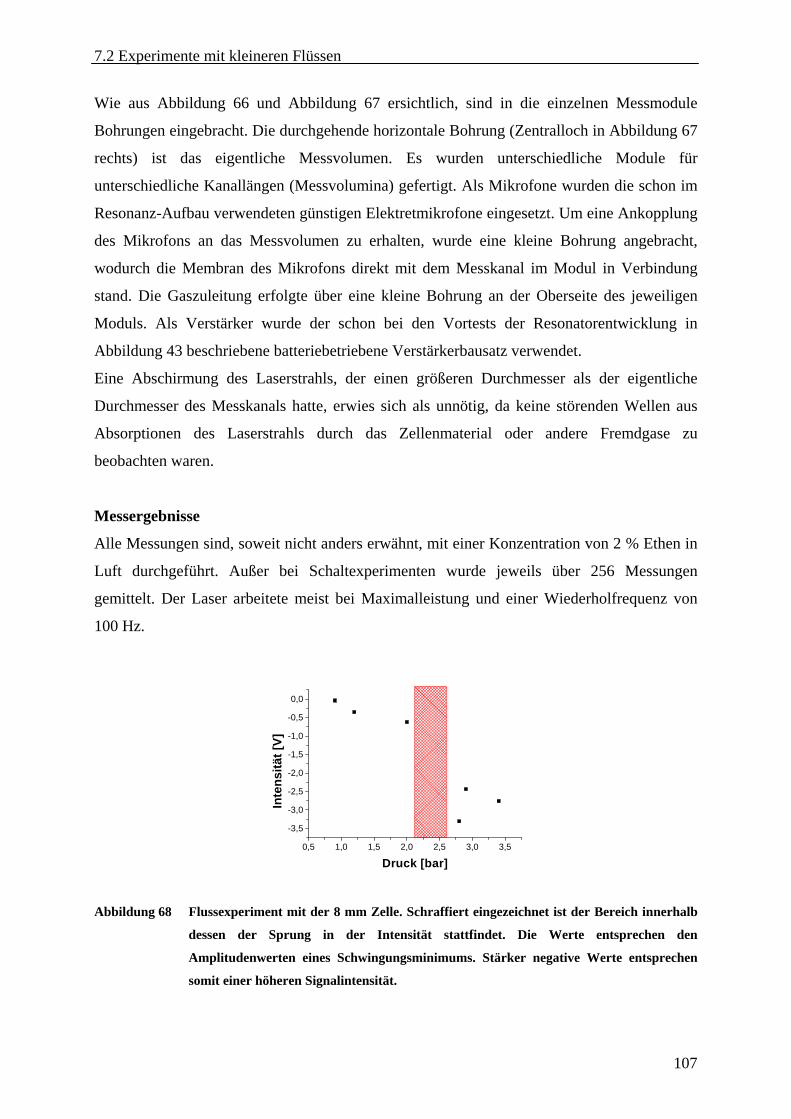
7.2 Experimente mit kleineren Flüssen
107
Wie aus Abbildung 66 und Abbildung 67 ersichtlich, sind in die einzelnen Messmodule
Bohrungen eingebracht. Die durchgehende horizontale Bohrung (Zentralloch in Abbildung 67
rechts) ist das eigentliche Messvolumen. Es wurden unterschiedliche Module für
unterschiedliche Kanallängen (Messvolumina) gefertigt. Als Mikrofone wurden die schon im
Resonanz-Aufbau verwendeten günstigen Elektretmikrofone eingesetzt. Um eine Ankopplung
des Mikrofons an das Messvolumen zu erhalten, wurde eine kleine Bohrung angebracht,
wodurch die Membran des Mikrofons direkt mit dem Messkanal im Modul in Verbindung
stand. Die Gaszuleitung erfolgte über eine kleine Bohrung an der Oberseite des jeweiligen
Moduls. Als Verstärker wurde der schon bei den Vortests der Resonatorentwicklung in
Abbildung 43 beschriebene batteriebetriebene Verstärkerbausatz verwendet.
Eine Abschirmung des Laserstrahls, der einen größeren Durchmesser als der eigentliche
Durchmesser des Messkanals hatte, erwies sich als unnötig, da keine störenden Wellen aus
Absorptionen des Laserstrahls durch das Zellenmaterial oder andere Fremdgase zu
beobachten waren.
Messergebnisse
Alle Messungen sind, soweit nicht anders erwähnt, mit einer Konzentration von 2 % Ethen in
Luft durchgeführt. Außer bei Schaltexperimenten wurde jeweils über 256 Messungen
gemittelt. Der Laser arbeitete meist bei Maximalleistung und einer Wiederholfrequenz von
100 Hz.
0,5 1,0 1,5 2,0 2,5 3,0 3,5
-3,5
-3,0
-2,5
-2,0
-1,5
-1,0
-0,5
0,0
Inte
nsitä
t [V]
Druck [bar]
Abbildung 68 Flussexperiment mit der 8 mm Zelle. Schraffiert eingezeichnet ist der Bereich innerhalb
dessen der Sprung in der Intensität stattfindet. Die Werte entsprechen den
Amplitudenwerten eines Schwingungsminimums. Stärker negative Werte entsprechen
somit einer höheren Signalintensität.

7 Weiterentwicklung der photoakustischen Analytik
108
Bei der Untersuchung der Flussabhängigkeit wurde bei Verwendung der 8 mm Zelle der
Druck im Reservoir um mehrere Bar reduziert, was einer Flussverringerung von 1,8 ml·min-1
auf 0,25 ml·min-1 entsprach. Die Signalstärke reduzierte sich, wie aus Abbildung 68 zu
erkennen ist, augenblicklich innerhalb eines schmalen Fensters auf einen Bruchteil des
ursprünglichen Wertes. Da die Signale manuell ausgewertet wurden und die dargestellten
Amplitudenwerte aus einem Schwingungsminimum entnommen sind, sinken die Werte bei
steigender Intensität auf stärker negative Werte. Oberhalb dieses Druckbereichs konnte ein
stabiles Signal beobachtet werden und es fanden nur geringe Änderungen der Signalstärke bei
weiteren Druckänderungen statt. Unterhalb dieses Druckbereichs waren Signalschwankungen
die Regel und nur eine geringe Signalstärke vorhanden. Starke Intensitätseinbussen konnten
auch bei verkürztem Messkanal gefunden werden. Da die Intensität bei den längeren Zellen
zu groß für eine saubere Verstärkung war, musste das Signal durch zusätzliche Filz- oder
Teflonbandbeschichtungen am Mikrofon gedämpft werden. Eine genaue Ermittlung des die
Intensität reduzierenden Faktors war aufgrund dieser unterschiedlichen Dämpfungen und der
daraus resultierenden mangelnden Vergleichbarkeit der unterschiedlichen Zellen nicht
möglich. Die starke Zunahme der Signalintensität relativ zur Nullmarke oberhalb von 2,25 bar
(1 ml·min-1) könnte durch stabile Flussverhältnisse in der Detektionszone oberhalb dieses
Druckwertes bewirkt werden. Diese Interpretation stützt auch die schwache Änderung
oberhalb dieses Schwellenwertes und die starken Schwankungen bei kleinen Drucken.
Messungen mit den 2 und 4 mm langen Modulen ergaben ebenfalls Instabilitäten des Signals
bei kleinen Ethenkonzentrationen.
0 200 400 600 800 1000 1200-10
0102030405060708090
100110120
50% soll
Inte
nsitä
t [%
]
Zeit [s] 0 50 100 150 200 250
-20
0
20
40
60
80
100
120
Inte
nsitä
t [%
]
Ze it [s]
100%
50%
20%
0 21000 20001500500 500
Abbildung 69 Stäbilitätskontrolle bei kürzeren Zellen im Vergleich zu längeren. Die Konzentration
wurde von 2 % Ethen auf 0 % reduziert um untere Detektionsgrenzen zu erkennen. Im
Fall der 8 mm Zelle (rechts) kann noch zuverlässig bei 0,4 % Ethen abs. gemessen
werden. Die kürzere 4 mm Zelle (links) ist schon bei einem Prozent hierzu nicht mehr in
der Lage.

7.2 Experimente mit kleineren Flüssen
109
Wie aus Abbildung 69 ersichtlich, können Konzentrationen, die unterhalb von 1 % Ethen
liegen, zuverlässig nur noch mit den längeren Zellen gemessen werden. Die Instabilität des
Messsignals könnte mit der mangelnden Ausbildung einer stabilen Messzone
zusammenhängen. Dies ist auch aufgrund der beobachteten Einflüsse der Abzugsanlage zu
vermuten. Bei mangelnder Abschirmung und starker Lüftung konnte im Freifeld ein stark
destabilisierender Einfluss auf die Ausbildung einer stabilen Messzone gefunden werden.
Dieser Effekt könnte sich auch bei zu kurzem Messkanal bemerkbar machen. Aus den obigen
Gründen wurden alle weiteren Messungen direkt mit der 8 mm Zelle durchgeführt.
Zur Untersuchungen der minimal notwendigen Messzeit wurde abwechselnd der Gasstrom in
der Detektionszone von 2 % Ethan auf 2 % Ethen und umgekehrt gewechselt. Das
resultierende Signal am Oszilloskop wurde mittels manueller Zeitnahme verfolgt. Die Zeit,
innerhalb der das Messsignal des Ethen-Kanals auf Null abfiel (entspricht dem 2 % Ethan
Kanal), wurde jeweils festgehalten. Aufgrund des langsamen Datentransfers zwischen
Oszilloskop und Computer ist ein automatisierter Betrieb der aufgebauten Anlage wenig
sinnvoll. Die Messungen wurden daher ausschließlich mit manueller Zeitnahme durchgeführt.
0 1 2 3 4 5 6 7 81,31,41,51,61,71,81,92,02,12,22,32,42,52,6
Däm
pfun
gsze
it [s
]
Kanallänge [mm]
0 2 4 6 8 10 12 14 16
1,4
1,6
1,8
2,0
2,2
2,4
2,6
2,8
3,0
3,2
3,4
Däm
pfun
gsze
it [s
]
Mittelungsfaktor
Abbildung 70 Abhängigkeit der Schaltzeit vom Mittelungsfaktor (rechts) und der Länge der Messzelle
(links). Unter Dämpfungszeit ist der Zeitraum zu verstehen, bei dem auf dem Oszilloskop
keine Welle mehr zu erkennen war. Die Zeitmessung erfolgte mittels einer Stoppuhr, was
die großen Fehlergrenzen erklärt.
Die Schaltzeit nimmt, wie aus Abbildung 70 zu erkennen ist, etwas mit der Kanallänge zu,
man erkennt jedoch deutlich, dass die Länge des Messkanals keinen signifikanten Einfluss
hat. Dies ist bei einem Fluss von 1,5 ml·min-1 auch nicht zu erwarten, da das Zellvolumen im

7 Weiterentwicklung der photoakustischen Analytik
110
Fall der 8 mm Zelle nur etwa 25 mm3 beträgt und daher innerhalb einer Sekunde einmal
komplett ausgetauscht werden sollte.
Höhere Mittelungen, welche die Genauigkeit der Einzelmessung stark erhöhen, schlagen sich
hingegen bei oszilloskopinterner Mittelung in stark verlängerten Messintervallen nieder. Im
verwendeten Aufbau war daher die minimale Schaltzeit von der Mittelung des Oszilloskops
abhängig. Bei einer Signalstabilität welche einer Genauigkeit von ± 0,2 % Ethen entsprach lag
dieser Wert bei ca. 3 Sekunden, da eine 16-fach Mittelung benötigt wurde.
Theoretisch ist die Schaltzeit bei schnellerer rechnerinterner Mittelung durch die
Geschwindigkeit des Gasaustauschs bestimmt und sollte im Bereich einer Sekunde liegen.
7.3 Experimente mit geringerer Laserleistung
In oben beschriebenem Aufbau wurde der schon für die anderen Experimente verwendete
CO2-Laserstrahl durch einen Filter um den Faktor 1000 gedämpft. Der IR-ND-Filter hatte
einen Durchmesser von 25 mm, eine Dicke von 1 mm und war in die Öffnung des Abzuges,
in den der Strahl eingekoppelt wurde, eingebaut.
Abbildung 71 Faktor 1000 Filter (links) und Laserleistungsmessgerät (rechts) für hohe
Strahlintensitäten (> 1 W).
Eine exakte Messung dieser geringen Laserleistung war aufgrund mangelnder Detektoren
nicht mehr möglich, weshalb lediglich die ungedämpfte Laserleistung mit dem in Abbildung
71 rechts gezeigten für hohe Laserleistungen geeigneten Messgerät (Powerwizard 250 der
Firma Synrad) gemessen wurde. Es wurde hierauf eine Dämpfung um exakt den Faktor 1000
durch den in Abbildung 71 links abgebildeten Filter angenommen. Die Messbedingungen
entsprachen den Bedingungen für Maximalintensität bei den Flussmessungen. Im einzelnen
waren dies ein Fluß von ca. 1,5 ml·min-1 und eine maximale Laserleistung, was ca. 30 Watt
ungedämpft entsprach.

7.4 Zusammenfassung und Diskussion
Bei den Messungen musste am Oszilloskop der höchste Mittelungswert von 256 Messungen
eingestellt werden, um ein stabiles Messsignal zu erhalten. Selbst unter diesen Bedingungen
gelang es nicht, eine Messgenauigkeit zu erhalten, wie sie bei den oben beschriebenen
Schaltexperimenten möglich war.
-0 ,0 0 0 2 0 ,0 0 0 0 0 ,0 0 0 2 0 ,0 0 0 4 0 ,0 0 0 6
-0 ,0 1 6-0 ,0 1 4-0 ,0 1 2-0 ,0 1 0-0 ,0 0 8-0 ,0 0 6-0 ,0 0 4-0 ,0 0 20 ,0 0 00 ,0 0 20 ,0 0 40 ,0 0 60 ,0 0 80 ,0 1 0
Z e it [s ]
Inte
ns
itä
t [V
]
2 % E th a n 2 % E th e n
0,0 0,5 1,0 1,5 2,00,006
0,008
0,010
0,012
0,014
Konzentration Ethen [%]
Inte
nsitä
t [V]
Abbildung 72 Zwei unterschiedliche Konzentrationsmessungen. Links ist eine Vergleichsmessung von 2
% Ethen bzw. 2 % Ethan in einem Stickstoffuntergrund gezeigt. Rechts sind die
Amplitudenwerte mehrerer Konzentrationsmessungen gegen die Konzentrationen
aufgetragen. Es findet sich in diesem Bereich ein proportionaler Zusammenhang.
Die Ergebnisse der Messungen von 0 bis 2 % Ethen in Luft im Vergleich sind in Abbildung
72 gezeigt. Zwischen den beiden Konzentrationen ist ein deutlicher Unterschied des
Signalverlaufs zu erkennen. Die Stabilität des Signals war jedoch nicht ausreichend hoch, um
automatisierte Messungen bei kleineren Konzentrationen unter Verwendung der
Integrationsroutine durchzuführen.
7.4 Zusammenfassung und Diskussion
Es ist möglich, die photoakustischen Aufbauten, wie sie im Rahmen dieser Arbeit entwickelt
wurden, bis hinab zu Flüssen von 1,5 ml·min-1 pro Kanal zuverlässig zu betreiben. Die
Messungen wurden ohne Resonanzverstärkung durchgeführt, womit
Konzentrationsunterschiede bis 0,1 % Ethen absolut detektiert werden können. Die
Schaltzeiten des Systems erlauben Messung innerhalb von 2 Sekunden. Die Limitierung stellt
der Datentransfer von Oszilloskop zu Computer und die Mittelungsgeschwindigkeit des
Oszilloskops dar. Bei geringeren Laserleistungen ist das System nur noch in der Lage bei 256-
facher Mittelung verlässliche Ergebnisse zu liefern.
111

7 Weiterentwicklung der photoakustischen Analytik
112
Die optimale Lösung wäre ein System mit möglichst hoher Genauigkeit und schneller
Schaltzeit. Die störenden Faktoren bei oben beschriebenen Experimenten konnten im Rahmen
dieser Arbeit nicht minimiert werden. Im folgenden werden einige leicht durchführbare
Verbesserungen diskutiert, welche bei einer Anwendung die aufgetretenen Mängel des
Systems beheben sollten.
Die Experimente wurden ohne jede akustische Isolation durchgeführt, was einen hohen Pegel
an Hintergrundstörgeräuschen bedingte. Der Laser wurde, wie oben beschrieben, durch sieben
voneinander unabhängige Lüfter gekühlt, welche einen hohen Geräuschpegel bei den
Messungen verursachten. Diesen störenden Nebeneffekt könnte man bei einer erneuten
Konstruktion des Systems durch geeignete Isolationsmaterialien leicht beheben.
Die Mittelungen wurden von einem Oszilloskop durchgeführt, das nicht in der Lage war, mit
einer befriedigenden Geschwindigkeit zu arbeiten. Dieses Problem könnte durch eine
kommerziell erhältliche schnelle rechnerinterne Messkarte umgangen werden, wobei eine
maximal mögliche Datenakkumulation durch die Wiederholfrequenz des Lasers determiniert
ist. Durch eine rechnerinterne Mittelung sollten dann auch Messungen mit Laserleistungen im
unteren Milliwattbereich möglich sein, wie aus den schon durchgeführten Experimenten bei
hohen Mittelungsfaktoren ersichtlich ist. Die Genauigkeit der Messmethode würde durch eine
derartige Optimierung um ein Vielfaches gesteigert werden und sollte somit allen
Anforderungen genügen.
Weitere Verbesserungen könnten auch durch resonanten Betrieb des Systems erzielt werden.
Dies würde zu einer zusätzlichen Verstärkung führen und auch die Detektion von anderen
Molekülen mit kleineren Absorptionskoeffizienten erlauben. Die Resonanzen eines
Messkanals, wie er in den hier vorgestellten Experimenten verwendet wurde, würden jedoch
im Bereich von etwa 170 kHz liegen, setzt man eine radiale Resonanz und daher festgelegte
Wellenlänge im Bereich von 2 mm voraus. Dies würde den Einsatz normaler Mikrofone
ausschließen und die Verwendung eines Ultraschall-Transducers erfordern.

113
8 Parallele Feststoffsynthese
Lass den Computer für dich arbeiten, aber werde nie Sklave der technischen Systeme.
Sergio Pininfarina
8.1 Materialien mit hoher Oberfläche
Materialien mit hoher Oberfläche sind eine interessante Stoffklasse für die heterogene
Katalyse. Da die meisten Reaktionen an der Oberfläche eines Feststoffes oder durch
Diffusionsprozesse im Feststoff ablaufen, ist es wichtig, ein schon aktives Material über die
Oberflächenbeschaffenheit weiter optimieren zu können. Es existieren eine Reihe von
Methoden, um hohe Oberflächen auf präparativem Weg zu erhalten. Dies sind z.B.
Fällungsreaktionen oder über Sol-Gel-Chemie gesteuerte Routen. Eine weitere Methode,
durch welche zum Beispiel auch die Zeolithe dargestellt werden, ist die sogenannte
templatgesteuerte Synthese.
Endotemplat
Kalzinierung
Kalzinierung
Exotemplat
Abbildung 73 Schematischer Vergleich der Endo- und Exotemplat-Synthese.
Wie in Abbildung 73 zu sehen ist, kann man hierbei zwischen Endotemplaten und
Exotemplaten unterscheiden. Die Endotemplate sind meist Moleküle oder Teilchen, welche
die Vorstufen des zu synthetisierenden Materials über strukturgebende Effekte um sich herum
anlagern. Sie werden vor der eigentlichen Synthese in die Lösung der Vorstufen des Materials
eingebracht und im Anschluss hieran durch Prozesse wie z.B. kontrollierten Abbrand entfernt.

8 Parallele Feststoffsynthese
114
Durch diesen Schritt entstehen an den Stellen, die zuvor das Templat einnahm, Hohlräume,
welche später die hohen inneren Oberflächen verursachen. Das Material wird hierbei zu
einem hochporösen Feststoff.
Im Fall der Exotemplate wird der Reaktionsraum selbst schon während der Synthese durch
den strukturgebenden restriktiven Effekt des Exoskeletts beschränkt. Es kann sich daher nur
ein kleiner Partikel ausbilden. Das Exotemplat, welches während der Synthese wie ein
Schwamm das eigentliche Material umschließt, muss im Anschluss hieran durch geeignete
Methoden möglichst „schonend“ entfernt werden. Dies kann durch die gleichen Methoden
wie bei Endotemplaten geschehen. Nach der Entfernung des Exoskeletts bleibt das
gewünschte Material in Form von Partikeln, welche aufgrund ihres kleinen Durchmessers
eine hohe äußere Oberfläche besitzen, zurück.
Der Syntheseweg der Materialien mit hoher Oberfläche wurde am Max-Planck-Institut für
Kohlenforschung von M. Schwickardi entwickelt. Die Parallelisierung und anschließende
Automatisierung erfolgte in Zusammenarbeit mit O. Busch.
8.2 Aktivkohlen als Exotemplate
Wie festgestellt wurde, eignen sich Aktivkohlen aufgrund ihrer großen inneren Oberfläche
hervorragend als Exotemplate für die Synthese von Metalloxiden mit hoher Oberfläche132,133.
Die BET-Oberfläche der Kohle sollte hierbei höher als 1000 m2·g-1 sein, um ausreichende
Saugfähigkeit zu gewährleisten. Eine genaue Beschreibung der begünstigenden Parameter der
aktivkohlebasierten Synthese, welche generell von mehreren Faktoren abhängt, wurde von
Schwickardi et al. publiziert132.
Temperatur [°C]
20
40
60
80
100TG [%]
100 200 300 400 500 600 700 800 900 10000
Aktivkohle
Cr(NO3)3 trockenMg(NO3)2
Cr(NO3)3
Cu(NO3)2
Abbildung 74 TG-Messungen unterschiedlich beladener Aktivkohlen. Beladene Aktivkohlen sind
deutlich früher vollständig verbrannt.

8.3 Automatisierung und Parallelisierung der Präparation
8.3 Automatisierung und Parallelisierung der Präparation
115
Wie aus Abbildung 74 deutlich wird, verbrennt die Kohle bei Beladung mit Metallnitraten
schon bei niedrigen Temperaturen vollständig. Dies begünstigt die Synthese von Materialien
mit hohen Oberflächen, da Sinterprozesse weitgehend vermieden werden. Es ist anzunehmen,
dass sich in der Kohle redoxkatalysierte Verbrennungsprozesse abspielen. Diese Prozesse
laufen bei manchen Metallen, wie z.B. Cr oder Cu, so heftig ab, dass von einem kontrollierten
Abbrand nicht mehr gesprochen werden kann. Durch solche stark beschleunigte
Verbrennungen besteht die Möglichkeit einer lokalen Temperaturerhöhung, was zum einen
ein Segregieren bestimmter Komponenten bewirken kann und zum anderen die Oberfläche
durch Sinterprozesse erniedrigt. Diesen Prozessen muss daher besondere Aufmerksamkeit
geschenkt werden, will man eine Synthese auf diesem Weg in größerem Maßstab
durchführen.
Auffällig ist, dass in Kohlen die Synthese von bestimmten Modifikationen einiger Materialien
möglich ist, die sich ohne die unterstützende Funktion des Exotemplats erst bei wesentlich
höheren Temperaturen ausbilden. So konnten bei tiefen Temperaturen zahlreiche Spinelle und
Perowskite mit hohen Oberflächen dargestellt werden132.
Die Automatisierung der durch Aktivkohle gestützten Präparation bot sich aufgrund des
Synthesewegs, der abgesehen von der Kohle selbst ausschließlich die Handhabung von
Flüssigkeiten beinhaltete geradezu an. Der einzige Schritt, bei dem Feststoffe gehandhabt
werden mussten, war die Abfüllung der Aktivkohlen. Alle weiteren Syntheseschritte konnten
durch einen kommerziellen Pipettierroboter durchgeführt werden.
Abbildung 75 Quarzgefäße für die parallele Synthese (links) und definierte Standard-Abfüllvolumina
(rechts).

8 Parallele Feststoffsynthese
116
Um das Abwiegen des Feststoffs zu umgehen, wurden mehrere standardisierte Maßvolumina
gefertigt. Das Abfüllen definierter Volumen ist manuell innerhalb kurzer Zeit zu
bewerkstelligen und erspart zeitraubende Wägeschritte. Die Handhabung des Feststoffs stellte
somit keinen zeitlimitierenden Schritt mehr im Gesamtprozess dar. Die in Abbildung 75 zu
sehenden Standard-Abfüllvolumina zeigten im Einsatz bei 250 mg Gesamteinwaage eine
Genauigkeit von ± 10 mg was für die Synthese ein akzeptabler Wert war. Wie festgestellt
wurde, hatte ein durch diesen Abfüllschritt bewirkter kleiner Überschuss an Kohle keine
Veränderung in der Beschaffenheit des dargestellten Feststoffes zur Folge. Um ein schnelles
Einfüllen zu gewährleisten, war die Öffnung des Füllvolumens etwas kleiner gewählt als der
Durchmesser des Reaktionsgefäßes. Der manuelle Aufwand pro Probe lag mit dieser Prozedur
unter zehn Sekunden.
Bei der Parallelisierung der Reaktion wurde durch die unterschiedliche Zusammensetzung
und die hieraus resultierende unterschiedliche Geschwindigkeit der Verbrennungsreaktion
eine räumliche Abtrennung der Reaktionsgefäße notwendig. Alternativ könnte der Zufluss an
Sauerstoff gedrosselt werden, um ein Zünden der Mischung zu vermeiden.
Abbildung 76 Präparationsplatte mit 77 Positionen (links) und Abdeckung bestehend aus Quarzwolle
(rechts).
Die in Abbildung 76 gezeigte apparative Lösung hatte zum einen den Vorteil, dass die
Reaktionsgefäße während der Verbrennung räumlich voneinander getrennt waren, als auch
einen positiven Drosseleffekt der Luftzufuhr. Dies wurde durch die poröse Quarzwolle über
den Gefäßen ermöglicht, welche eine kontinuierliche Luftzufuhr gewährleistete, verwirbelte
Partikel aber aufhielt und somit eine Kreuzkontamination der einzelnen Materialien
verhinderte. Während der Verbrennung wurde über der Probe ein schützendes Kohlendioxid-
Polster erzeugt, was eine zusätzliche Diffusionsstrecke für den Sauerstoff bedeutete. Die
Verbrennung wurde hierdurch etwas gebremst und erfolgte somit kontrollierter. In

8.4 Selektion der Katalysatorzusammensetzung
8.4 Selektion der Katalysatorzusammensetzung
117
Vergleichsexperimenten konnte bei einzelnen Materialien durch den Einsatz dieser
Diffusionsbarriere die Oberfläche um bis zu 50 % gesteigert werden.
Über die Entwicklung des Katalysatordesigns wurde bereits in Kapitel 3.1 ausführlich
berichtet. Da zur Zeit noch kein geeigneter Algorithmus existiert, der es vermag,
erfolgversprechende Materialien einer Bibliothek von unterschiedlichen Katalysatoren zu
ermitteln, wurde ein zufälliges Vorgehen gewählt. Die Auswahl übernahm ein auf LabVIEW
basierendes Computerprogramm. Die Selektion der Startbibliotheken lief über zwei
gekoppelte Zufallsroutinen, welche die Zusammensetzungen ermittelten.
Zufällige Auswahl von Bestandteilen aus 10
möglichen
Zufällige Auswahl der relativen
Zusammensetzung
Verifikation derPräparationsparameter
des Roboters
Nein
DisketteJa
Abbildung 77 Schematische Darstellung der Katalysatorselektion (links) und Benutzeroberfläche des
Präparationsprogramms (rechts).
Der Ablauf dieser Routine und ein Bild der Benutzeroberfläche des entwickelten Programms
sind in Abbildung 77 gezeigt. In einem ersten Schritt wurde aus bis zu zehn möglichen
Precursorlösungen ermittelt, welche unterschiedlichen Metallkomponenten im fertigen
Material enthalten sein sollten. Für dieses Zahlenarray bestehend aus 1 für enthalten oder 0
für nicht enthalten wurde dann für die im Material enthaltenen Komponenten (entsprechend
der Zahl 1) in einem zweiten Schritt ein zufälliges relatives Verhältnis ermittelt. Dieses
Verhältnis wurde basierend auf den bekannten Konzentrationen der Precursorlösungen
automatisch in Volumina für den Pipettierroboter umgerechnet. In einem letzten Schritt wurde
die Durchführbarkeit dieser Synthese getestet. Unsinnige Zusammensetzungen, welche aus

8 Parallele Feststoffsynthese
118
technischen Gründen nicht durch den Roboter darzustellen waren (zu geringes
Pipettiervolumen), wurden in dieser Phase automatisch aussortiert. Abschließend wurden die
Daten auf ein für den Roboter verständliches Format übersetzt und automatisch in Form der
entsprechenden Syntheseanweisungen auf eine Diskette gespeichert.
8.5 Materialien und Charakterisierung
Aufgrund der hohen Zahl synthetisierter Materialien war nur die Analyse einiger
ausgewählter Vertreter der entsprechenden Substanzklassen möglich. Es kann angenommen
werden, dass diese Substanzen aufgrund gleicher Syntheseparameter und ähnlichen
Inhaltsstoffen charakteristisch und repräsentativ für alle dargestellten Materialien sind.
Stellvertretend sollen nun zwei in der Kohlenmonoxid-Oxidation erfolgreich getestete
Materialien diskutiert werden. Sowohl das Material der relativen atomaren
Metallzusammensetzung Fe:Cu:Ni:Mn von 17:30:25:27 (Material 1) als auch das Material
Cu:Ni:Mn von 35:33:32 (Material 2) zeigten bezüglich ihrer Fähigkeit, Kohlenmonoxid bei
tiefen Temperaturen zu oxidieren, gute Eigenschaften. Der T1/2-Wert lag bei Material 1 bei
102 °C (± 5 °C), bei Material 2 bei 115 °C (± 5 °C).
2Theta10.0 20.0 30.0 40.0 50.0 60.0 70.0
Material 1
Material 2
Inte
nsitä
t [w
.E.]
2Theta20.0 30.0 40.0 50.0 60.0 70.0
Inte
nsitä
t [w
.E.] Material 2
Abbildung 78 Vergleich der Transmissionsaufnahmen von Material 1 im Vergleich zu Material 2
(links). Das Überlagerte Diffraktogramm eines Kupferoxides, lässt den Schluss zu, dass
bei Material 2 ein höherer Anteil kristallines CuO vorhanden ist. Die Abbildung rechts
zeigt eine Überlagerung mit einem Cu1Ni0,5Mn1,5O4 Spinell. Die restlichen Reflexe können
sowohl diesem als auch weiteren ähnlich zusammengesetzten Spezies mit gleichem
Reflexmuster zugeordnet werden. Eine genaue Ermittlung der Zusammensetzung ist
nicht möglich.

8.5 Materialien und Charakterisierung
Der in Abbildung 78 gezeigte XRD-Vergleich lässt erkennen, dass zumindest die kristallinen
Domänen in beiden Fällen gleich zusammengesetzt sind. Aufgrund der Vielzahl von fast
kontinuierlichen Mischphasen lässt sich jedoch keine Aussage über die exakte
Phasenzusammensetzung anstellen. Eine Vergleich mit Datanbankeinträgen (PDFZ) von
Cu1Ni0,5Mn1,5O4 zeigt, dass die Reflexe der Probe mit dem Muster des
Literaturdiffraktogramms übereinstimmen, was eine Spinellstruktur nahelegt. Es sind jedoch
unterschiedlichste Zusammensetzungen der kristallinen Domänen möglich, welche stark von
dem gezeigten Beispiel abweichen können. Da zudem das Vorliegen amorpher Bereiche bzw.
von Kristalliten unter 2 nm möglich ist, könnte ein Teil der Probe röntgenamorph sein. Da
mehr als 20 mögliche Zusammensetzungen allein aus der PDFZ-Datenbank erhalten wurden,
wurden keine weiteren Anstrengungen unternommen, über Röntgendiffraktogramme die
exakte Phasenzusammensetzung zu ermitteln. Aus einer Reflexverbreiterung konnte jedoch
erfolgreich ein Wert von ca. 15 nm für die kristallinen Domänen gefunden werden, was sich
mit Untersuchungen aus TEM-Experimenten deckt.
0,0 0,2 0,4 0,6 0,8 1,0
0
20
40
60
80
100
120
140
Ads.
Vol
umen
cm
3 /g
Relativer Druck p/p0
58 m2/g
17,4% Fe
25,2% Ni
27,4% Mn
29,9% Cu
0,0 0,2 0,4 0,6 0,8 1,00
20
40
60
80
100
Ads.
Vol
umen
cm
3 /g
Relativer Druck p/p0
53 m2/g
32,9% Ni
32,1% Mn
35,0% Cu
Abbildung 79 BET-Messungen von Material 1 und 2 im Vergleich.
Die BET-Messungen in Abbildung 79 zeigen in beiden Fällen eine Oberfläche im Bereich
von 60 m2·g-1. Dies ist für Oxide welche Kupfer enthalten ein erstaunlich hoher Wert, da diese
Substanzen stark zur Segregation und Versinterung neigen. Der geringe, wenn überhaupt
relevante Unterschied von 5 m2·g-1 könnte zwar für die unterschiedliche Aktivität
verantwortlich gemacht werden, dies erscheint aber nicht sehr überzeugend. Aus den
Messungen geht klar hervor, dass es sich nicht um poröse Materialien handelt, da sonst eine
Hysterese anderer Form erhalten werden müsste. Man erkennt deutlich, dass die Probe in
Form kleiner Partikel vorliegt.
119

8 Parallele Feststoffsynthese
120
Die einzige Methode, welche Klarheit über die Proben verschafft und die Erfolglosigkeit
anderer Messmethoden erklärt, ist die Transmissionselektronenmikroskopie.
Abbildung 80 TEM-Aufnahmen von Material 1 (links) und 2 (rechts) bei 30000-facher Vergrößerung
im Vergleich.
Bei einer 30000-fachen Vergrößerung (Abbildung 80) sehen die beiden Materialien noch
relativ ähnlich aus. Bei genauerem Betrachten fällt jedoch schon hier auf, dass Material 2 eine
etwas gröbere Körnung zu haben scheint und die Homogenität der Probe etwas schlechter ist.
Dies wird auch an anderen Stellen gefunden.
Abbildung 81 TEM-Aufnahmen von Material 1 (links) und 2 (rechts) bei 500000-facher Vergrößerung
im Vergleich. Man erkennt deutlich die kristallinen Bereiche.
Eine Betrachtung bei 500000-facher Vergrößerung zeigt deutlich, dass das Material in beiden
Fällen hoch kristallin ist, wobei die einzelnen Domänen zwischen 2 und 20 nm Durchmesser

8.5 Materialien und Charakterisierung
haben, was mit der Bestimmung über Reflexverbreiterungen aus XRD-Untersuchungen
übereinstimmt.
Gleichbedeutend mit diesem Ergebnis ist, dass es nur mit ortsaufgelösten Messungen möglich
ist, eine genaue Aussage über die Beschaffenheit der Materialien anzustellen, da Methoden,
die das Bulk-Material analysieren, hier versagen.
Abbildung 82 Ortsaufgelöste EDX Messungen an Material 1.
a b c d e f g h i j
Mn [%] 24,91 31,32 31,29 11,43 22,22 24,48 22,19 12,12 28,12 32,43
Fe [% ] 27,61 22,01 19,38 28,61 43,80 38,87 43,68 30,03 29,02 25,16
Ni [%] 21,79 21,13 24,45 12,60 14,31 15,20 12,34 49,36 22,85 18,07
Cu [%] 22,42 18,45 16,25 47,36 15,68 19,49 19,20 8,48 16,19 20,58
Tabelle 2 Positionen und Zusammensetzungen aus Abbildung 82.
Bei Untersuchungen von Material 1 fällt, wie aus Abbildung 82 und Tabelle 2 zu erkennen ist,
auf, dass immer alle Elemente in signifikanten prozentualen Mengen gefunden werden.
121
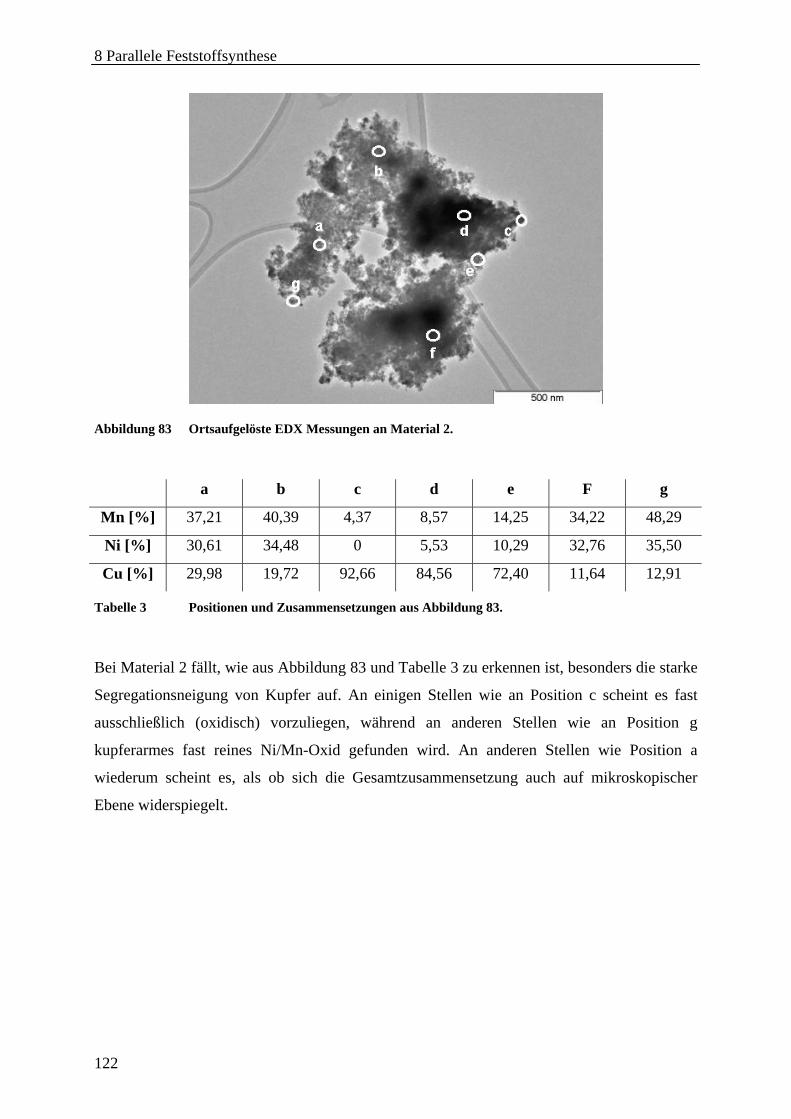
8 Parallele Feststoffsynthese
122
Abbildung 83 Ortsaufgelöste EDX Messungen an Material 2.
a b c d e F g
Mn [%] 37,21 40,39 4,37 8,57 14,25 34,22 48,29
Ni [%] 30,61 34,48 0 5,53 10,29 32,76 35,50
Cu [%] 29,98 19,72 92,66 84,56 72,40 11,64 12,91
Tabelle 3 Positionen und Zusammensetzungen aus Abbildung 83.
Bei Material 2 fällt, wie aus Abbildung 83 und Tabelle 3 zu erkennen ist, besonders die starke
Segregationsneigung von Kupfer auf. An einigen Stellen wie an Position c scheint es fast
ausschließlich (oxidisch) vorzuliegen, während an anderen Stellen wie an Position g
kupferarmes fast reines Ni/Mn-Oxid gefunden wird. An anderen Stellen wie Position a
wiederum scheint es, als ob sich die Gesamtzusammensetzung auch auf mikroskopischer
Ebene widerspiegelt.

8.5 Materialien und Charakterisierung
a b c
Mn [%] 0 49,8 13,0
Ni [%] 2,5 42,5 11,4
Cu [%] 86,9 0 70,1
Abbildung 84 Ortsaufgelöste EDX und Zusammensetzungen bei Material 2.
Ein Extrembeispiel ist in Abbildung 84 gezeigt. Innerhalb einer mikroskopischen Distanz von
nur 50 nm wechselt die Zusammensetzung des Materials 2 von reinem Kupferoxid auf reines
Ni/Mn-Oxid. In der Mitte kann eine Art Übergangszone gefunden werden in der die einzelnen
Materialien aufgrund der Befunde aus den XRD-Messungen wahrscheinlich als Mischkristall
und nicht einzeln nebeneinander vorliegen.
Wenn man die Ergebnisse aus den TEM-Aufnahmen interpretiert, so legt die aus TG-
Messungen gefundene frühe Zersetzung von Kupfernitrat nahe, dass zumindest bei Material 2
eine überwiegende Abscheidung von reinem Kupferoxid stattfindet, bevor es zur Zersetzung
der Nitrate der anderen Metalle kommt. Da die Segregation bei Material 1 nicht oder nur in
sehr geringem Umfang gefunden wird, die thermische Behandlung beider Proben jedoch
identisch war, kann die Vermutung angestellt werden, dass Eisen die Ursache hierfür sein
könnte. Da Eisennitrat schon bei Zimmertemperatur in Kontakt mit Kohle nitrose Gase
entwickelt und die Zersetzung schon bei sehr niedrigen Temperaturen beginnt, bildet sich
eventuell ein wie auch immer geartetes „Gerüst“ aus Eisenoxiden oder gemischten
Kupfer/Eisen, Oxiden/Nitraten, welches eine Stützfunktion übernimmt und eine Segregation
von reinem Kupferoxid verhindert, wodurch der hohe Dispersionsgrad von Cu
aufrechterhalten wird, bis sich die anderen Nitrate zersetzen und ein Mischkristall gebildet
werden kann.
123

8 Parallele Feststoffsynthese
124
8.6 Gesamtprozess und Diskussion
Die parallele auf Aktivkohlen basierende Präparation von Metalloxiden mit hoher Oberfläche
kann als eine weitere Methode der Hochdurchsatz-Synthesen betrachtet werden. Der
entwickelte Prozess ist in der Lage, innerhalb eines Tages 77 Proben zu generieren, wobei der
manuelle Aufwand bei etwa 15 Minuten liegt. Ein solches Verfahren kann einen 16-fachen
Parallelreaktor und selbst höher parallelisierte Systeme leicht mit der nötigen Probenmenge
versorgen.
Da die Syntheseroute über die Nitrate der Metalle läuft und von den meisten Metallen die
Nitrate leicht zu handhaben sind, stellt die Synthese keinerlei Gefahr dar und kann
unbeaufsichtigt in einem Abzug durchgeführt werden. Die Metallnitrate sind zudem in
Lösung stabil und günstig als Feststoffe zu erwerben, was es in unserem Fall ermöglichte, den
Roboter mit Vorratsgefäßen für mehrere 100 Synthesen auszustatten und vielfach
hintereinander ohne Wechsel der Vorratslösungen zu betreiben. Die Heizraten können in ihrer
Geschwindigkeit in einem weiten Fenster variiert und bis zu Maximaltemperaturen von 1000
°C gewählt werden, was auch die Synthese von Hochtemperaturmodifikationen erlaubt. Es
steht hiermit ein weiter Parameterraum zur Verfügung.
Die zufällige Katalysatorselektion stellt eine gute Alternative für unbekannte Systeme dar, bei
denen ein systematisches Vorgehen noch zu früh wäre. Es können hiermit in einigen ersten,
zufällig gewählten Bibliotheken Treffer in weiteren systematischen Tests optimiert werden,
wobei das Wissen des Chemikers erst bei der Optimierung gefragt ist. Natürlich würden
hierdurch gewonnene Erkenntnisse dann die zufällige Selektion ablösen.
Da die besten gefundenen Materialien in der Kohlenmonoxid-Oxidation Cu und Mn
enthalten, ist eine Verwandtschaft zum bereits bekannten Hopcalit-System wahrscheinlich.
Bemerkenswert ist jedoch, dass dieses Material im Rahmen dieser Arbeit innerhalb einer
Woche zufällig gefunden wurde, was das Potenzial dieser kombinierten Präparations- und
Analysentechnik zeigt.

9 Katalytische Tests
Immer, wenn wir etwas Neues anfangen, wollen wir es besser machen, als das Vergangene.
Nur, um dann festzustellen, es das nächste Mal wieder besser zu machen.
Damaris Wieser
Im folgenden Abschnitt werden nun die wichtigsten Ergebnisse der katalytischen Messungen
gezeigt, die im Rahmen dieser Arbeit durchgeführt wurden. Es handelt sich bei den
vorgestellten Katalysatoren ausschließlich um Materialien, welche nach der im Abschnitt
„Parallele Feststoffsynthese“ beschriebenen Syntheseanleitung automatisiert hergestellt
wurden. Die Messungen wurden mit den photoakustischen Aufbauten durchgeführt, deren
Entwicklung im Abschnitt „Entwicklung der photoakustischen Analytiken“ beschrieben
wurde.
9.1 Kohlenmonoxid-Oxidation
20 40 60 80 100 120 140 160 180 200-20
0
20
40
60
80
100
120
Synthese 1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Umsa
tz [%
]
Temperatur [°C]
20 40 60 80 100 120 140 160 180 200-20
0
20
40
60
80
100
120 Synthese 1 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32
Umsa
tz [%
]
Temperatur [°C]
125

9 Katalytische Tests
40 60 80 100 120 140 160 180 200-20
0
20
40
60
80
100
120
Synthese 2 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Umsa
tz [%
]
Temperatur [°C]
40 60 80 100 120 140 160 180 200-20
0
20
40
60
80
100
120
Synthese 2 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48
Umsa
tz [%
]
Temperatur [°C]
60 80 100 120 140 160 180-20
0
20
40
60
80
100Synthese 3 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Umsa
tz [%
]
Temperatur [°C] 60 80 100 120 140 160 180
-20
0
20
40
60
80
100
Synthese 3
17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32
Umsa
tz [%
]
Temperatur [°C] Abbildung 85 Einige photoakustische Messung mit dem resonanten Aufbau. Die obigen Messungen
zeigen einen Ausschnitt aus den getesteten Materialien und wurden innerhalb von 3
Tagen aufgenommen. Die genauen Zusammensetzungen der Materialien finden sich im
Anhang.
Einige der parallel durchgeführten Kohlenmonoxid-Oxidationsmessungen sind in Abbildung
85 aufgelistet.
Die Hysterese, welche bei fast allen Messungen deutlich zu sehen ist, ist auf das
Wärmemanagement des Reaktorgrundkörpers zurückzuführen. Da die Temperatur im Inneren
des Reaktorblockes gemessen wird, ist beim Aufwärmen eine zu hohe und beim Abkühlen ein
zu niedrige Temperatur aufgrund der langsameren Übertragung der Wärme auf die
Katalysatoren und umgekehrt zu finden. Weiterhin erfasst die Temperaturmessung lediglich
einen Mittelwert des gesamten Reaktors und nicht den exakten Wert in der
Katalysatorschüttung.
126
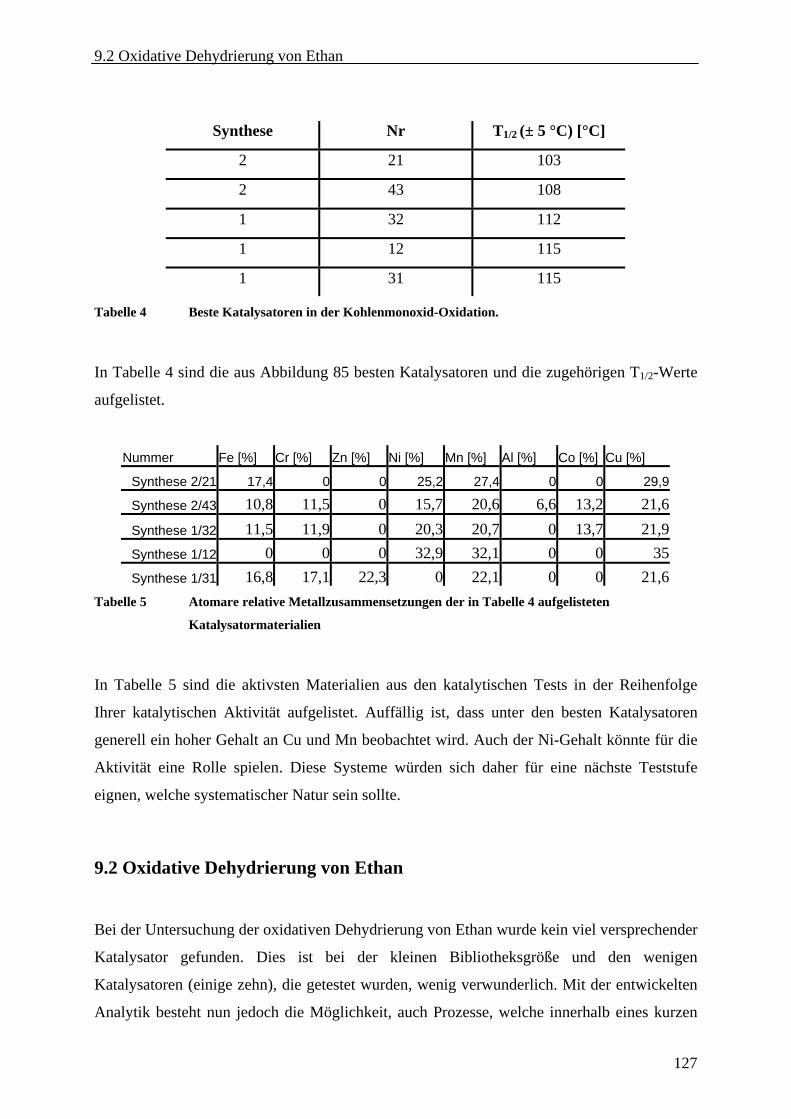
9.2 Oxidative Dehydrierung von Ethan
Synthese Nr T1/2 (± 5 °C) [°C]
2 21 103
2 43 108
1 32 112
1 12 115
1 31 115
Tabelle 4 Beste Katalysatoren in der Kohlenmonoxid-Oxidation.
In Tabelle 4 sind die aus Abbildung 85 besten Katalysatoren und die zugehörigen T1/2-Werte
aufgelistet.
Nummer Fe [%] Cr [%] Zn [%] Ni [%] Mn [%] Al [%] Co [%] Cu [%]
Synthese 2/21 17,4 0 0 25,2 27,4 0 0 29,9
Synthese 2/43 10,8 11,5 0 15,7 20,6 6,6 13,2 21,6 Synthese 1/32 11,5 11,9 0 20,3 20,7 0 13,7 21,9 Synthese 1/12 0 0 0 32,9 32,1 0 0 35 Synthese 1/31 16,8 17,1 22,3 0 22,1 0 0 21,6
Tabelle 5 Atomare relative Metallzusammensetzungen der in Tabelle 4 aufgelisteten
Katalysatormaterialien
In Tabelle 5 sind die aktivsten Materialien aus den katalytischen Tests in der Reihenfolge
Ihrer katalytischen Aktivität aufgelistet. Auffällig ist, dass unter den besten Katalysatoren
generell ein hoher Gehalt an Cu und Mn beobachtet wird. Auch der Ni-Gehalt könnte für die
Aktivität eine Rolle spielen. Diese Systeme würden sich daher für eine nächste Teststufe
eignen, welche systematischer Natur sein sollte.
9.2 Oxidative Dehydrierung von Ethan
Bei der Untersuchung der oxidativen Dehydrierung von Ethan wurde kein viel versprechender
Katalysator gefunden. Dies ist bei der kleinen Bibliotheksgröße und den wenigen
Katalysatoren (einige zehn), die getestet wurden, wenig verwunderlich. Mit der entwickelten
Analytik besteht nun jedoch die Möglichkeit, auch Prozesse, welche innerhalb eines kurzen
127

9 Katalytische Tests
Zeitfensters ablaufen, zuverlässig zu verfolgen. Es wurden während der durchgeführten
Messungen in manchen Fällen zu Beginn einer angefahrenen Temperatur, direkt nach der
Temperaturerhöhung, ein deutlich gesteigerter Umsatz gefunden. Dieser brach nach kurzer
Zeit zusammen und konnte bei erneuter Messung nicht wieder gefunden werden, was einen
nichtreversiblen stöchiometrischen Prozess vermuten lässt oder einen katalytischen Prozess
mit extrem kurzer Katalysatorstandzeit. Es kann vermutet werden, dass der Umsatz auf durch
die Synthese bewirkte Eigenschaften des Katalysators zurückzuführen ist. Denkbar wäre hier
ein Einfluss von Cu-(I) welches durch Teilreduktion beim Erhitzen mit der reduzierend
wirkenden Kohle entstanden sein könnte. Diese Ergebnisse müssen jedoch noch genauer
untersucht werden, was nicht das Ziel dieser Arbeit war.
200 250 300 350 400 450 500-1
0
1
2
3
4
Umsa
tz [%
]
Temperatur [°C]
Abbildung 86 Photoakustische Messung mit dem Freifeldaufbau. Das getestete Material hatte die
Zusammensetzung La12Ba2Pb9Th7Mn27Ni24Cu19Ox.
Die Messung in Abbildung 86 zeigt einen Katalysator, welcher bei 550 °C einen Umsatz zu
Ethen von maximal drei Prozent in Bezug auf das eingesetzte Ethan zeigt. Dieser Katalysator
wurde auch in gaschromatographischen Experimenten zur Evaluierung des Systems
herangezogen und bereits in Abschnitt 6.3 erwähnt. Nun sollen noch weitere Ergebnisse der
Gaschromatographie-Untersuchungen, welche die Reaktion charakterisieren, gezeigt werden.
Auch bei gaschromatographischen Analysen ist ein Ansteigen der Ethenkonzentration bei
hohen Temperaturen zu finden, wie bereits in Abschnitt 6.3.5 in Abbildung 41 beschrieben
wurde.
128
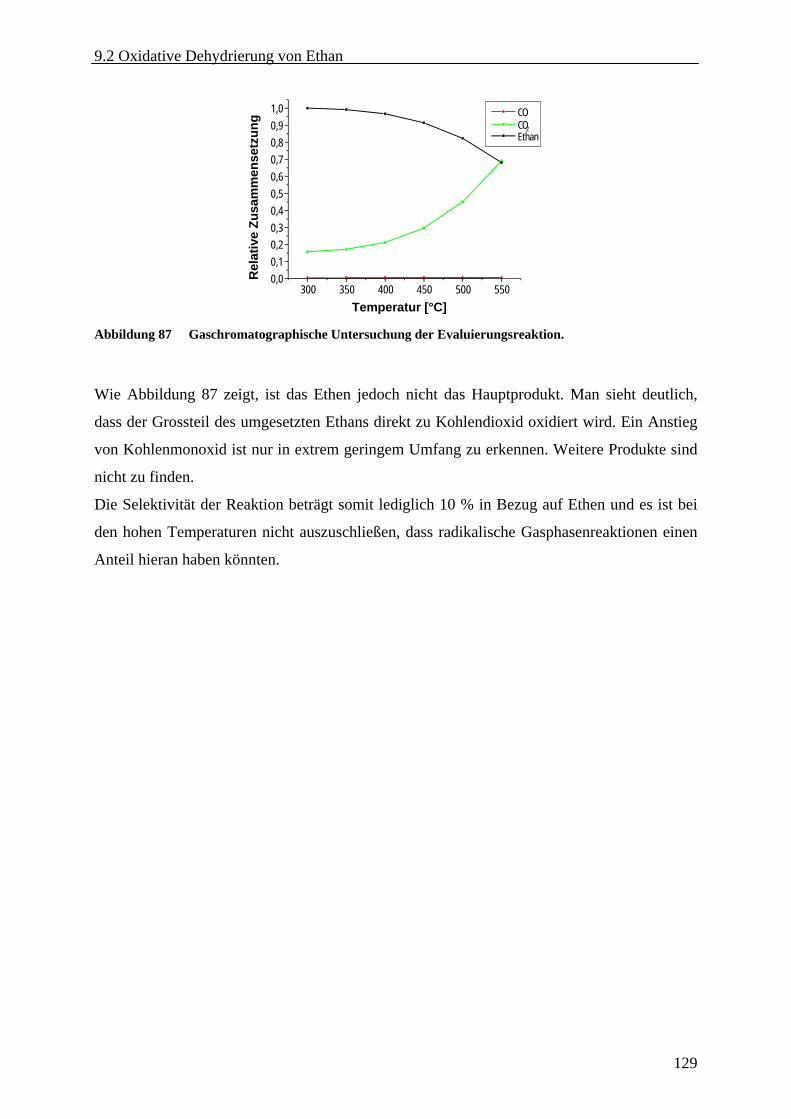
9.2 Oxidative Dehydrierung von Ethan
300 350 400 450 500 5500,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
Temperatur [°C]
Rel
ativ
e Zu
sam
men
setz
ung CO
CO2
Ethan
Abbildung 87 Gaschromatographische Untersuchung der Evaluierungsreaktion.
Wie Abbildung 87 zeigt, ist das Ethen jedoch nicht das Hauptprodukt. Man sieht deutlich,
dass der Grossteil des umgesetzten Ethans direkt zu Kohlendioxid oxidiert wird. Ein Anstieg
von Kohlenmonoxid ist nur in extrem geringem Umfang zu erkennen. Weitere Produkte sind
nicht zu finden.
Die Selektivität der Reaktion beträgt somit lediglich 10 % in Bezug auf Ethen und es ist bei
den hohen Temperaturen nicht auszuschließen, dass radikalische Gasphasenreaktionen einen
Anteil hieran haben könnten.
129

10 Zusammenfassung und Ausblick
Alle großen Taten und alle großen Gedanken haben in ihren Anfängen etwas
Lächerliches. Die bedeutenden Werke werden oft an einer Straßenecke oder
in der Windfangtür eines Restaurants geboren.
Albert Camus
Die unterschiedlichen Bereiche, welche im Rahmen dieser Arbeit behandelt wurden, zeigen
die vielfältigen, teilweise interdisziplinären Schnittstellen im Gebiet der Hochdurchsatz-
Katalysatorforschung. Ziel aller Forscher, welche auf diesen Gebieten tätig sind, ist es, den
Gesamtprozess der Entwicklung eines neuen aktiven Materials zu beschleunigen. Die
Engstellen des Testprozesses, welche hierbei auftreten, sind von Fall zu Fall verschieden und
nicht selten nur durch Zusammenarbeit von Forschern unterschiedlicher Fachrichtungen
lösbar. Generell liegen sie in einem der in früheren Abschnitten genannten grundlegenden
Teilbereiche, aus denen sich die Hochdurchsatz-Katalysatorforschung zusammensetzt. Im
einzelnen sind dies die Synthese, der eigentliche Test und die Verarbeitung der resultierenden
Daten nebst Auswertungsroutine. Eine Automatisierung dieser Abläufe umschließt den
Gesamtprozess und sollte daher immer im Auge behalten werden, um nach erfolgreich
entwickelter manueller Routine den Prozess noch weiter optimieren zu können.
Der Schwerpunkt dieser Arbeit lag in der Entwicklung neuer analytischer Methoden im
Bereich der Hochdurchsatz-Katalysatorforschung. Diese sollten in der Lage sein, erstmals
sowohl parallel als auch hinreichend schnell Messungen on-line durchzuführen. Hiermit sollte
nach erfolgtem Test ein realistischer Vergleich der einzelnen Katalysatoren auf den
unterschiedlichen Positionen eines Reaktors möglich sein. Tendenzen sollten schon während
des eigentlichen Tests erkannt und analysiert werden können. Dies war aufgrund der bisher
eingesetzten sequenziellen Methoden noch nicht standardmäßig möglich. Die am häufigsten
verwendeten Analysesysteme, wie z.B. Gaschromatographie-Aufbauten, konnten lediglich
eine Testrate von bis zu einem Katalysator pro Minute erreichen, was einen Vergleich bei
hohen Parallelisierungsgraden nur begrenzt zuließ. Auch die weit verbreiteten
massenspektrometrischen Methoden liegen bei der für einen Katalysator benötigten Zeit in
diesem Bereich.
130

Im Rahmen dieser Arbeit wurden zwei unterschiedliche Analysesysteme entwickelt, welche
für Moleküle mit unterschiedlich starker Absorption der eingesetzten Anregungsstrahlung
geeignet sind. Hierbei stellt die Freifeld-Analytik ein sehr elegantes Verfahren dar, falls das
detektierte Molekül einen hohen Absorptionskoeffizienten aufweist und der generierte
Druckpuls ohne Resonanzverstärkung ausgewertet werden kann. Ein Laser generiert hierbei
in unterschiedlichen Gasmischungen in der Analysenzone jeweils der Konzentration der
Zielmoleküle entsprechend intensive Druckpulse. Diese Pulse durchlaufen zeitlich versetzt
den Raum. Die Detektion basiert auf der zeitlichen Auswertung der Druckpulskurve, welche
durch den Laser generiert wird. Die einzelnen Pulse können durch ihre Laufzeit in räumliche
Distanzen umgerechnet werden, was eine Zuordnung zu den einzelnen Messkanälen erlaubt.
Über die Intensität des Signals kann die Konzentration der Zielmoleküle ermittelt werden. Die
Geschwindigkeit der Messung aller Kanäle wird bei geeigneter Datenverarbeitung in den
Bereich einer einzigen Sekunde zurückgedrängt. Die Resonanzanalytik stellt im Gegensatz
hierzu eine eher langsame Methode dar, welche immerhin noch eine Einzelmessung innerhalb
einer Sekunde durchführen kann und daher im sequenziellen Betrieb eine Gesamtmesszeit
entsprechend dem Parallelisierungsfaktor benötigt. Sollte dieser Punkt sich störend auswirken,
kann sie durch den Einsatz multipler Verstärker leicht in eine echte parallele Messmethode
umgewandelt werden. Dies erfordert jedoch weitere Investitionen. Die Detektion basiert
hierbei auf reiner Lautstärkemessung des Signals innerhalb der Resonanzröhre, in der die
durch den Laser generierten Druckpulse resonant zu einer stehenden Welle verstärkt werden.
Der Nachteil des sequenziellen Betriebs wird durch eine hervorragende Signalstabilität und
eine Unempfindlichkeit gegen äußere Störfaktoren wie Geräuschkulisse oder starken Luftzug
aufgewogen. Es wird hierdurch eine Detektion von Molekülen mit sehr schwachen
Absorptionen mit einer hohen Zuverlässigkeit möglich.
Nach erfolgreicher Etablierung dieser schnellen und parallelen analytischen Verfahren wurde
der Gesamtprozess der Katalysatorentwicklung weiter optimiert, indem eine ausreichend
schnelle Präparation der Testmaterialien entwickelt wurde. Dies war notwendig, da keine
Methode in der Lage war, eine ausreichende Menge unterschiedlicher Substanzen für den
stark beschleunigten Test/Analyse-Ablauf zu liefern. Hierzu wurde eine Synthese, welche für
die Darstellung von Vollkatalysatoren aus Oxidmaterialien mit besonders hohen Oberflächen
entwickelt worden war, parallelisiert und automatisiert. Die Katalysatorselektion wurde durch
ein Computerprogramm zufällig getroffen. Dieser Ansatz eignet sich für die Startphase einer
Katalysatortestung, wenn noch kein Ansatzpunkt für ein strategisches Vorgehen existiert oder
131

10 Zusammenfassung und Ausblick
neue Wirkmechanismen gefunden werden sollen. Im weiteren Verlauf sollte dieser Ansatz
durch strategische Methoden erweitert oder abgelöst werden.
Das Zusammenspiel von neuer Analytik und schneller Synthese ermöglichte aufgrund der
hohen Testraten die Akkumulation großer Datenmengen. Aktuell gibt es Anstrengungen von
Forschern sowohl im Max-Planck-Institut für Kohlenforschung als auch in Frankreich am
CNRS in Lyon, mit Hilfe dieser Daten zuverlässige Algorithmen zu entwickeln, welche ein
weiteres strategisches Vorgehen aufgrund bestimmter Deskriptoren erlauben.
Die im ersten Teil der Arbeit entwickelten Systeme ergänzen und erweitern die aktuell
existierenden analytischen Methoden um eine echtzeit-parallele Methode, welche on-line
eingesetzt werden kann. Mit den photoakustischen Techniken wurde eine gute Alternative zu
existierenden analytischen Verfahren geschaffen, welche bei bestimmten Anforderungen den
bisherigen Methoden vor allem in der Testrate weit überlegen ist. Die Informationstiefe
verlangt jedoch eine genaue Kenntnis der untersuchten Reaktion. Ein Einsatz ist hierdurch vor
allem im Bereich der Optimierung der Katalysatoren von Interesse, bei denen es weniger auf
die genaue Untersuchung des Produktspektrums als auf die Testgeschwindigkeit ankommt. In
diesen Fällen hat man mit der entwickelten Methode eine extrem zuverlässige und den Zweck
erfüllende genügend schnelle Analytik in Händen. Es bleibt noch anzumerken, dass der
Einsatz dieser Technik nicht für Untersuchungen mit unbekanntem Produktspektrum geeignet
ist, weshalb sie idealerweise durch die Gaschromatographie ergänzt wird und diese nach
anfänglichen Studien ablösen kann.
Die Wahl der geeigneten Analytik wird durch das Zielmolekül determiniert. Moleküle mit
kleinen Absorptionskoeffizienten sind mit der resonanten Analytik-Methode quantitativ zu
verfolgen. Moleküle mit großem Absorptionskoeffizienten können sowohl mit Freifeld-
Analytik als auch durch Resonanzverstärkung gemessen werden. Weiterhin hat man einen
Kompromiss zwischen Schnelligkeit und Genauigkeit einzugehen. Prinzipiell spricht nichts
gegen eine parallele Nutzung des Resonanz-Aufbaus außer den im Vergleich zu anderen
Aufbauten hohen Kosten.
Die Ergebnisse aus den weitergehenden Experimenten, welche im zweiten Teil dieser Arbeit
durchgeführt wurden, zeigen, dass die Analytik sehr vielseitig an unterschiedliche
Problemstellungen angepasst werden kann. Das Potenzial der photoakustischen Detektion im
Rahmen der Hochdurchsatz-Forschung ist mit den beiden hier entwickelten Methoden bei
weitem noch nicht ausgeschöpft und weitere Entwicklungen erscheinen daher durchaus
lohnend. Interessant ist, dass für eine Messung ein Produktstrom von lediglich einem
Milliliter pro Minute genügt. Dies erlaubt den Einsatz in Systemen, welche hochgradig
132

parallelisiert und hierdurch notwendigerweise miniaturisiert sind. Ein Einsatz dieses
Verfahrens in Phase I einer Testung erscheint hierdurch überaus sinnvoll.
Ferner ist eine Reduzierung der für die Analyse benötigten Laserleistung äußerst interessant.
Hierdurch werden weitere spektrale Bereiche zugänglich. Wie gezeigt werden konnte, kann
die Messung noch zuverlässig im Bereich weniger Milliwatt durchgeführt werden, was vor
allem den Bleisalzlaser für zukünftige Anwendungen überaus interessant erscheinen lässt. Es
würden mit diesem Lasersystem keine Limitierungen durch unzugängliche spektrale Bereiche
mehr bestehen, da praktisch der gesamte Infrarot-Bereich durch dieses Lasersystem abgedeckt
werden kann. Es wäre hiermit jedes gasförmige Molekül, welches eine selektive Bande im
Infraroten besitzt, potenziell detektierbar. Dies ist im entwickelten System noch nicht
möglich, kann aber leicht durch den Austausch der Laserquelle bewerkstelligt werden.
Aktuell ist durch den CO2-Laser lediglich der Bereich um 940 cm-1 (10,6 µm) abgedeckt.
Zwar zeigen viele Moleküle in diesem Bereich eine Absorption, jedoch ist hier die
Wahrscheinlichkeit eine selektive Bande zu finden sehr gering.
Zur Zeit wird eine Weiterentwicklung dieses Analysesystems bei der hte AG in Heidelberg
auf ihren industriellen Routineeinsatz getestet. Im Rahmen der Hochschulforschung ist das
System in den Routinebetrieb übernommen worden und wird bei der Kohlendioxid-Detektion
eingesetzt.
Nach der Entwicklung der Echtzeit-Analytik wurde im dritten Teil dieser Arbeit das Problem
der Katalysatorpräparation behandelt. Mit den in unserer Gruppe eingesetzten kohlebasierten
Synthesen gelang es, einen parallelen automatisierten Gesamtprozess zu entwickeln, welcher
Materialien mit im Vergleich zu unkontrolliertem Abbrand sehr hohen Oberflächen lieferte.
Vor allem in der Kohlenmonoxid-Oxidation zeigten sich diese Materialien in den
katalytischen Tests als äußerst interessante Stoffklasse und es konnten in kürzester Zeit
hinreichend aktive Materialien für weitere systematische Studien gefunden werden. Die
Synthese des einzelnen Materials nahm hierbei noch immer einen Tag in Anspruch, was an
der langsamen Abkühlrate von 500 °C auf Raumtemperatur lag, der hohe
Parallelisierungsgrad von 77 Materialien pro Synthese erlaubte es dennoch, den Teststand am
Rande der maximalen Kapazität zu betreiben. Da das Temperaturprogramm für einen
katalytischen Test für einen Zeitraum von ca. sieben Stunden erstellt wurde, lag der Durchsatz
bei drei Katalysatorchargen pro Tag, was einer Anzahl von 48 Materialien entsprach. Würde
der Bedarf nach schnellerer Testung bestehen, sollte ein neuer Reaktor eingesetzt werden, da
sowohl Analytik als auch Präparation noch nicht vollständig ausgelastet sind. Man erkennt
hier sehr gut, dass in der Hochdurchsatz-Katalysatorforschung ein gelöstes Problem bzw. ein
133

10 Zusammenfassung und Ausblick
schnellerer Prozess lediglich neue Engstellen in anderen Bereichen des Gesamtprozesses
aufzeigt.
Durch diese Testroutine gelang es letztendlich, innerhalb nur weniger Tage im Bereich der
Kohlenmonoxid-Oxidation Materialien zu finden, welche den meisten geträgerten Edelmetall-
Katalysatoren in ihrer Aktivität bei tiefen Temperaturen weit überlegen sind.
Abschließend kann festgestellt werden, dass eine erfolgreiche neue Testroutine entwickelt
wurde, welche sowohl viele Materialien herstellen als auch die katalytische Untersuchung
dieser Materialien bewältigen kann. Photoakustischen Methoden wurden erfolgreich in der
Hochdurchsatzforschung parallel und sequenziell zur on-line Untersuchung eingesetzt und mit
diesen Techniken innerhalb kürzester Zeit neue aktive Materialien in der Kohlenmonoxid-
Oxidation gefunden.
134

11 Anhang
A Synthetisierte Materialien Die Angaben in den folgenden Tabellen beziehen sich auf das relative Metallverhältnis.
Durch die Synthese liegen die dargestellten Materialien oxidisch vor, was die
Zusammensetzung verändert. Da eine Angabe dieser Werte nicht möglich ist, sind hier
lediglich die für die Synthese eingestellten Metallverhältnisse aufgelistet.
Synthese 1
Nummer Fe [%] Cr [%] Zn [%] Ni [%] Mn [%] Al [%] Co [%] Cu [%]
1 15,6 0 23,1 0 0 11,4 20,4 29,6 2 13,8 14,9 18,5 0 17,8 0 14,8 20,2 3 0 13,3 17,9 0 21,3 8,3 15,2 23,9 4 17,6 21,4 28,9 0 0 0 0 32,1 5 100 0 0 0 0 0 0 0 6 17,9 18,7 0 25,2 27,5 10,7 0 0 7 14,9 14,4 0 19,7 0 9,8 15 26,2 8 0 0 32,4 0 32,6 13,3 21,7 0 9 20,1 19,7 29,6 30,6 0 0 0 0
10 26,1 0 0 0 55,8 18,1 0 0 11 100 0 0 0 0 0 0 0 12 0 0 0 32,9 32,1 0 0 35 13 16,9 0 0 25 29,4 10 18,8 0 14 0 0 27,5 0 32,2 0 0 40,3 15 15,1 16 18,1 17,6 0 8,9 0 24,3 16 0 0 0 0 47,3 22,2 30,5 0 17 0 13,4 19,3 0 21 8,7 14,4 23,3 18 25,5 25,6 33,2 0 0 15,7 0 0 19 0 17,6 31,3 28,9 0 0 22,2 0 20 37,1 0 0 36,6 0 26,2 0 0 21 0 0 17,9 22,2 25,7 9,3 0 25 22 30,4 0 0 0 69,6 0 0 0 23 12,9 10,8 0 18 17,1 10,2 12,4 18,7 24 0 0 39,2 47,3 0 13,5 0 0 25 24,4 26,9 0 37,1 0 11,6 0 0
135

11 Anhang
26 15,6 0 18,5 0 23,4 0 16 26,5 27 15,6 13,6 19,2 0 24,2 8,8 18,6 0 28 0 0 30,8 0 0 12,7 20,8 35,7 29 15,1 15,8 0 23 0 0 17,3 28,8 30 37,1 0 0 0 0 19,3 43,6 0 31 16,8 17,1 22,3 0 22,1 0 0 21,6 32 11,5 11,9 0 20,3 20,7 0 13,7 21,9
Synthese 2
Nummer Fe [%] Cr [%] Zn [%] Ni [%] Mn [%] Al [%] Co [%] Cu [%]
1 13,9 14,7 0 19,9 25,4 8,8 17,3 0 2 8,8 12,9 16,6 16,9 18,3 7,6 0 18,8 3 14 14,7 0 21 0 8,2 15,9 26,3 4 16,8 19,1 0 24,9 0 9,4 0 29,8 5 0 23,6 0 0 37,3 17,5 21,7 0 6 17,4 18,6 0 23,9 29,8 10,3 0 0 7 0 15,9 22,2 22,1 0 11,2 0 28,6 8 0 15,9 0 0 28 0 19,8 36,2 9 0 0 31,5 31,9 0 0 0 36,6
10 0 38,9 0 0 0 28,9 32,2 0 11 0 0 0 45,1 0 0 25,9 29 12 0 20,6 32,5 0 36,5 10,4 0 0 13 19,5 0 23,6 25,5 31,3 0 0 0 14 19 0 0 36 0 19,9 25,1 0 15 0 51,2 48,8 0 0 0 0 0 16 22,4 22,4 0 29,8 0 0 25,4 0 17 18,4 17,8 0 0 30,4 11,9 21,5 0 18 14,9 13,6 18,9 0 24,6 11,6 16,5 0 19 0 0 17,3 17,8 22,9 6,9 13,1 21,9 20 36,3 0 31,6 32,1 0 0 0 0 21 17,4 0 0 25,2 27,4 0 0 29,9 22 10,5 0 16,1 15,9 18,3 6,4 12,8 19,9 23 26,4 27,4 0 0 0 16,2 30 0 24 0 0 0 0 0 19,2 28,9 51,9 25 0 21,4 22,9 0 0 0 18 37,7 26 15,8 19,4 21 0 0 0 17,2 26,5 27 0 15,8 27,1 0 25,9 11,3 19,9 0 28 0 0 42,5 0 0 13,2 44,3 0 29 12,5 0 18,5 21,6 22,9 7,6 16,9 0
136
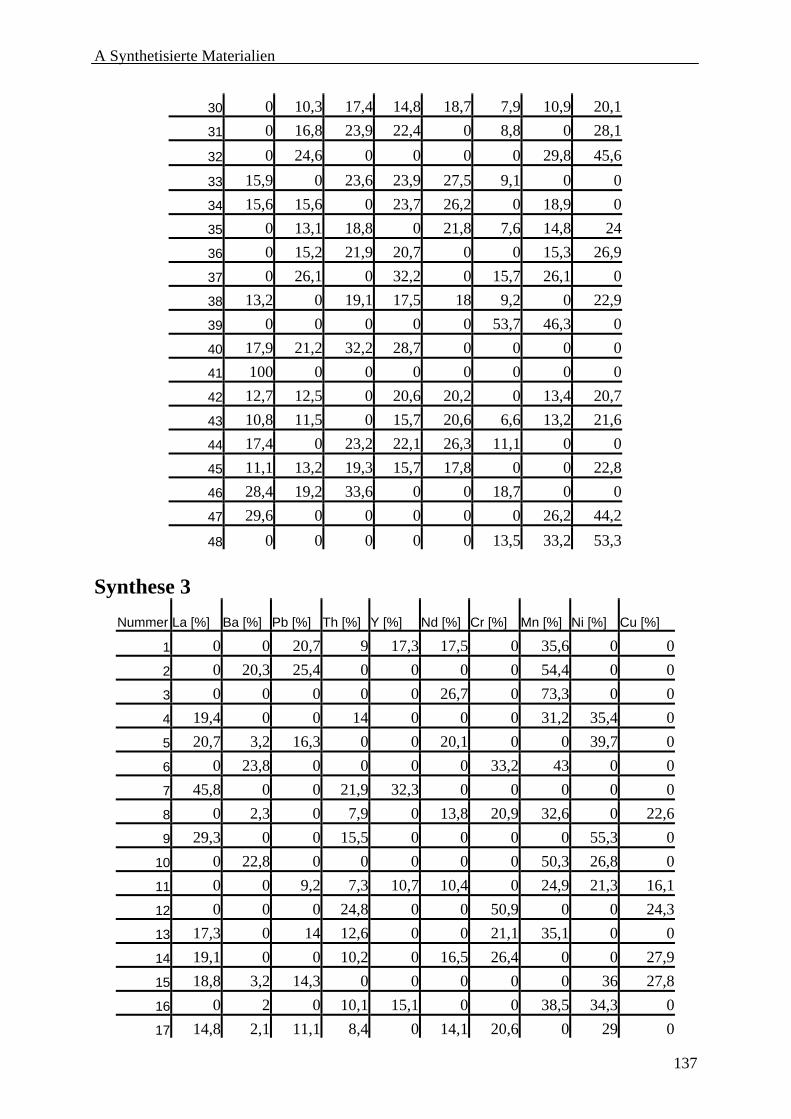
A Synthetisierte Materialien
30 0 10,3 17,4 14,8 18,7 7,9 10,9 20,1 31 0 16,8 23,9 22,4 0 8,8 0 28,1 32 0 24,6 0 0 0 0 29,8 45,6 33 15,9 0 23,6 23,9 27,5 9,1 0 0 34 15,6 15,6 0 23,7 26,2 0 18,9 0 35 0 13,1 18,8 0 21,8 7,6 14,8 24 36 0 15,2 21,9 20,7 0 0 15,3 26,9
0 26,1 0 32,2 0 15,7 26,1 0 38 13,2 0 19,1 17,5 18 9,2 0 22,9 39 0 0 0 0 0 53,7 46,3 0 40 17,9 21,2 32,2 28,7 0 0 0 0 41 100 0 0 0 0 0 0 0 42 12,7 12,5 0 20,6 20,2 0 13,4 20,7 43 10,8 11,5 0 15,7 20,6 6,6 13,2 21,6 44 17,4 0 23,2 22,1 26,3 11,1 0 0 45 11,1 13,2 19,3 15,7 17,8 0 0 22,8 46 28,4 19,2 33,6 0 0 18,7 0 0 47 29,6 0 0 0 0 0 26,2 44,2 48 0 0 0 0 0 13,5 33,2 53,3
37
Synthese 3
Nummer La [%] Ba [%] Pb [%] Th [%] Y [%] Nd [%] Cr [%] Mn [%] Ni [%] Cu [%]
1 0 0 20,7 9 17,3 17,5 0 35,6 0 0 2 0 20,3 25,4 0 0 0 0 54,4 0 0 3 0 0 0 0 0 26,7 0 73,3 0 0 4 19,4 0 0 14 0 0 0 31,2 35,4 0 5 20,7 3,2 16,3 0 0 20,1 0 0 39,7 0 6 0 23,8 0 0 0 0 33,2 43 0 0 7 45,8 0 0 21,9 32,3 0 0 0 0 0 8 0 2,3 0 7,9 0 13,8 20,9 32,6 0 22,6 9 29,3 0 0 15,5 0 0 0 0 55,3 0
10 0 22,8 0 0 0 0 0 50,3 26,8 0 11 0 0 9,2 7,3 10,7 10,4 0 24,9 21,3 16,1 12 0 0 0 24,8 0 0 50,9 0 0 24,3 13 17,3 0 14 12,6 0 0 21,1 35,1 0 0 14 19,1 0 0 10,2 0 16,5 26,4 0 0 27,9 15 18,8 3,2 14,3 0 0 0 0 0 36 27,8 16 0 2 0 10,1 15,1 0 0 38,5 34,3 0 17 14,8 2,1 11,1 8,4 0 14,1 20,6 0 29 0
137
11 Anhang
18 18 1,9 0 0 0 17,3 0 0 35,9 27 19 29,5 9,6 0 0 0 0 0 0 60,9 0 20 16,7 3 0 8,2 12,3 16,2 22,2 0 0 21,4 21 0 0 7,7 0 8,7 9,7 14,4 23,6 20,5 15,4 22 0 2,3 0 8,4 0 14,2 22 0 30,1 22,9 23 0 2,2 8,8 6,8 9,3 10,8 0 24,3 21,6 16,2 24 12,2 1,8 9,2 7,2 0 0 0 27,4 23,7 18,5 25 0 6 17,8 0 0 0 0 0 41,2 34,9 26 0 0 17,3 10,2 0 17,3 23,3 0 31,9 0 27 16,4 0 10,5 0 0 0 20,9 27,6 0 24,7 28 0 0 21,7 0 0 0 23,3 31,6 0 23,3 29 12,8 0 0 0 0 0 16,7 27,3 24,5 18,7 30 19,9 3,1 0 11,3 0 0 26,2 39,5 0 0 31 0 11,4 0 26,7 0 0 61,8 0 0 0 32 0 0 0 14,5 15,9 0 0 0 37,5 32,1
138

B Abbildungsverzeichnis
B Abbildungsverzeichnis Abbildung 1 Schematische Darstellung des Hochdurchsatz-Prozesses. Alle einzelnen
Bereiche sind für den Gesamtprozess wichtig und im besten Falle automatisierbar. ....................................................................................................4
Abbildung 2 Vergleich zwischen traditionellen, parallelen und Split/Pool-Verfahren. ..........12 Abbildung 3 Mögliche stark vereinfachte Reaktionswege bei der Kohlenmonoxid-
Oxidation an Goldkatalysatoren83.......................................................................23 Abbildung 4 Pipetierroboter Gilson XL 232 (rechts) mit Präparations-Aufbau für die
Synthese der Metalloxide (links). Links unten ist die Präparationsplatte für die gleichzeitige Synthese von 77 Metalloxiden zu sehen. Links oben befinden sich die Vorratslösungen. .....................................................................27
Abbildung 5 a) Schematische Zeichnung der Seitenansicht des 16-fach Parallelreaktors und b) Foto der Seitenansicht. Es sind deutlich die Bohrungen (vier) für die Katalysatoraufnahmen zu erkennen. ...................................................................29
Abbildung 6 a) Blick in den 16-fach Parallelreaktor von oben, man erkennt die Lage der Heizpatronen (vertikale Linien) zwischen den Katalysatorpatronen und b) Temperaturverteilung des Reaktors mit Abweichungen von der Solltemperatur. ....................................................................................................30
Abbildung 7 Schematischer Aufbau der Testanlage für die Kohlenmonoxid-Oxidation für den parallelen Betrieb. ........................................................................................30
Abbildung 8 Mögliche Undichtigkeiten und Dichtkonzept: O-Ring-Dichtung a ist vor allem gegen Undichtigkeiten nach außen, Dichtung c gegen Randgängigkeiten und Dichtung b eine Zusatzdichtung gegen Kreuzkontaminationen.........................................................................................31
Abbildung 9 a) Schematischer Aufbau des Einrohr-Festbett-Reaktor Teststandes und b) schematische Zeichnung des Reaktors. ...............................................................33
Abbildung 10 Edelstahlreaktor der Firma hte AG.....................................................................35 Abbildung 11 Temperaturverteilung und Dichtungen des hte Reaktors. Der Reaktor kommt
mit lediglich 2 Dichtungen aus. Im Vergleich hierzu wurden für den Messingreaktor 33 Einzeldichtungen verwendet. ................................................35
Abbildung 12 Schematischer Aufbau der Anlage zur parallelen Untersuchung der oxidativen Dehydrierung von Ethan....................................................................37
Abbildung 13 Schematischer Aufbau des Einrohr-Festbett-Reaktors. Die Analyse erfolgte über zwei sequenziell geschaltete Gaschromatographen, deren Betriebstemperaturen 90 °C (GC1) und 235 °C (GC2) betrugen. ......................38
Abbildung 14 Schematische Darstellung eines photoakustischen Prozesses. Nach der Absorption kann die Energie entweder durch die Umkehrung des Absorptionsprozesses unter Aussendung eines Photons relaxieren oder durch strahlungslose Prozesse in translatorische Energie umgewandelt werden. ................................................................................................................44
Abbildung 15 Alexander Graham Bell bei dem Versuch ein Phototelefon zu entwickeln. Gezeigt ist die Empfängereinheit bei der er den photoakustischen Effekt erstmals bemerkte. ...............................................................................................45
Abbildung 16 Spektralverteilung der Sonne...............................................................................48 Abbildung 17 Spektralverteilung einer Wolframlampe..............................................................49 Abbildung 18 Spektralverteilungen einer Quecksilber-Hochdruck-Leuchtbogenlampe............49 Abbildung 19 Vergleich von a) herkömmlicher Absorptions-Spektroskopie, b)
photothermischer Analyse und c) photoakustischer Analyse. .............................53
139

11 Anhang
Abbildung 20 Schematische Darstellung des photoakustischen Effekts in Gasen. Wichtig für eine effektive Modulation ist eine vollständige Relaxation vor dem nächsten Laserpuls. .............................................................................................54
Abbildung 21 Einige, abgesehen von einfachen Konzentrationsmessungen, durch Photoakustik zugängliche Prozesse und physikalische Größen. .........................55
Abbildung 22 Einige Zellkonzepte in der photoakustischen Spektroskopie. a) gewöhnliche nichtresonante oder resonante Zelle, b) Multipass-Zelle, c) Zelle mit zusätzlichen akustischen Filtern, d) differentielle Zelle zur Untergrundsubtraktion, e) Zelle im Innern eines Laserresonators. ....................58
Abbildung 23 Schematische Zeichnung einer nichtresonanten Zelle. Die Druckpulse können an allen seitlichen Stellen der Zelle mit gleicher Intensität abgegriffen werden. .............................................................................................60
Abbildung 24 Schematische Zeichnung einer resonanten Zelle. Die Signalstärke hängt stark von der Position des Mikrofons an der Zellenwand ab. .............................60
Abbildung 25 Resonante Moden einer Zylinderzelle. Neben der longitudinalen Resonanz existieren noch die radiale und azimuthale Resonanz. Auch alle Kopplungen dieser Schwingungen sind möglich......................................................................61
Abbildung 26 Photoakustische Zellen für Feststoffe. Neben der links gezeigten Gas-Mikrofon Kopplung ist auch die Messung der Phononanregung über die Kopplung an einen Piezokristall möglich, wie rechts gezeigt. ............................62
Abbildung 27 Ursprüngliche Idee einer photoakustischen Analyseeinheit. Der Laserstrahl regt parallel alle Kanäle an, wobei er durch Spiegel umgelenkt wird. Die Intensität und Abstände der Signalursprünge sollte über die Laufzeit und bekannte Distanz aus dem Interferenzsignal berechnet werden. ........................63
Abbildung 28 Testaufbau zur Abstandsmessung. Das Tropfenereignis wurde sowohl über ein Mikrofon zur Triggerung des Oszilloskops verwendet, als auch zur Abstandsmessung über ein zweites Mikrofon. .....................................................65
Abbildung 29 Testmessung zur Abstandsanalyse. Das Triggerereignis war aufgrund der Intensität nicht genau zu bestimmen. Die relativen Abstände zeigen jedoch deutlich das Auflösungsvermögen der Methode. .................................................66
Abbildung 30 Spezialmikrofon der Firma Brüel&Kjæer ...........................................................67 Abbildung 31 Abbildung des 25 W CO2-Lasersystems (links) und des steuernden
Signalgenerators (RISys GmbH Typ 9514). ........................................................69 Abbildung 32 Schema der Laserprozesse eines CO2-Lasers. Die eigentliche Anregung
erfolgt über Stickstoff-Moleküle, welche die Energie bei Stossprozessen auf das Kohlendioxid übertragen. .............................................................................70
Abbildung 33 Infrarot Spektren von a) Ethan und b) Ethen. Man erkennt deutlich, dass im Bild a) keine signifikante Absorptionsbande im Bereich der Laserstrahlung liegt, während bei b) eine hohe Absorption zu erkennen ist................................71
Abbildung 34 Photoakustische Freifeld-Analytik. Die Minima im Diagramm entsprechen den einzelnen Kanälen. Die gezeigte Messung wurde nach einer 256-fach Mittelung erhalten. ..............................................................................................72
Abbildung 35 Schematische Zeichnung der elektronischen Verknüpfung und des Datenflusses bei der photoakustischen Freifeld-Analytik. Die Reaktionskontrolle und die Analytik sind, wie aus der Abbildung zu erkennen ist, voneinander unabhängige Routinen. .............................................73
Abbildung 36 Benutzeroberfläche der Freifeld-Analytik. ..........................................................74 Abbildung 37 Berechnung der Laufzeit des akustischen Signals. Die Zeit setzt sich aus der
Entfernung zwischen den einzelnen Kanälen und der Ausdehnung der Gaswolke zusammen. Das Signal wird idealisiert als eine vollständige
140

B Abbildungsverzeichnis
Schwingung betrachtet. Die Dämpfung kann im realen Experiment wesentlich mehr Zyklen in Anspruch nehmen......................................................75
Abbildung 38 Links sind zwei überlagerte Messungen der ersten drei Kanäle abgebildet. In einem Fall ist der zweite Kanal nur mit Stickstoff durchströmt. Der obere Filter ist auf 30 kHz eingestellt. Rechts ist eine Messung aller Kanäle gezeigt, wobei ein oberer Filter von 100 kHz gewählt und der zweite Kanal mit Stickstoff gespült ist. Die Signale links sind invertiert, was auf die Polarisationsspannung des Kondensatormikrofons zurückzuführen ist. Die rechte Messung wurde mit einem Elektretmikrofon aufgenommen, welches die gleichen Spezifikationen wie das normalerweise verwendete Kondensatormikrofon aufwies. ............................................................................76
Abbildung 39 Messung des Signalverlaufs bei unterschiedlichen Anregungspulslängen. Erwartungsgemäß sind die Druckpulse länger, wenn die Laserpulsdauer steigt.....................................................................................................................77
Abbildung 40 Bestimmung der Linearität im Messbereich. Links ist der erste und rechts der sechste Messkanal gezeigt. Wie deutlich zu sehen ist, ist im Rahmen der Messfehler eine lineare Korrelation zwischen Konzentration und Signalstärke gegeben. ..........................................................................................78
Abbildung 41 Vergleich der parallel photoakustischen Messung und der gaschromatographischen Messung im Einrohr-Reaktor. Die Gaschromatographie-Messungen liegen, wenn man die Fehlergrenzen betrachtet, genau im Bereich des photoakustischen Verfahrens. Des getestete Material hatte die Zusammensetzung La12Ba2Pb9Th7Mn27Ni24Cu19Ox. ..........................................................................79
Abbildung 42 Aufbau der ersten photoakustischen Experimente (links) und Messung von Kohlendioxid in der mit Trockeneis teilgefüllten Zelle im Vergleich zur leeren Zelle (rechts).............................................................................................82
Abbildung 43 Schaltbild des modifizierten Mikrofonbausatzes 302155-66 (Conrad). ..............82 Abbildung 44 Testaufbauten der Experimente für kontrollierte Konzentrationsmessungen.
Links ist eine Anregung im Freifeld mit nachfolgender resonanter Signalabgreifung gezeigt. Rechts wird das Signal direkt im Innern des Resonators erzeugt. .............................................................................................83
Abbildung 45 Bild des ersten parallelen Aufbaus zur Messung unterschiedlicher Kohlendioxid Konzentrationen. ...........................................................................84
Abbildung 46 Oszilloskopsignal der Konzentrationsmessung von Kohlendioxid (links) und Korrelation von Konzentration und Signalintensität (rechts). Die Konzentrationen wurden jeweils schrittweise erhöht und bei Erreichen des Maximalwerts von 36000 ppm wieder schrittweise auf 0 erniedrigt. Hieraus ergeben sich die beiden Äste der Korrelation. ....................................................84
Abbildung 47 Verstellbare Resonanzröhre. ...............................................................................86 Abbildung 48 Lock-In-Prinzip. Links ist das Signal, welches gegen die Referenzphase um
90 ° verschoben ist, rechts die in-Phase-Situation. Nur wenn die Phase übereinstimmt, ist eine optimale Verstärkung gegeben. Falls das Signal nicht in der Frequenz übereinstimmt, ist keine Verstärkung möglich. Moderne Verstärker arbeiten zusätzlich mit einem rein integrativen Verfahren, welches jede Welle mit übereinstimmender Frequenz verstärkt (unabhängig von der Phasenlage).......................................................................86
Abbildung 49 Messung der Kohlendioxidkonzentration. Während der Messung ist die Signallage durch Temperaturfluktuationen nicht stabil, wodurch die Intensitäten schwanken. Die Stufen entsprechen einer Konzentrationsänderung von 0 % bis auf 6 % und zurück in 1 % Schritten.......87
141

11 Anhang
Abbildung 50 Mikrofonkennlinie und Resonanzverstärkung der Resonanzröhre. Die Röhrenresonanz wurde durch die direkte Einkopplung eines Signals entsprechender Frequenz in die Detektionszone ermittelt. Bewerkstelligt wurde dies durch einen Referenzton, welcher durch einen Schallleiter direkt in die Mitte der Detektionszone geleitet wurde. Da diese auch der Ursprung der Druckpulse ist, wurde dieser experimentelle Aufbau als genügend genaue Simulation eines Anregungsprozesses gewählt. Es ist oberhalb 6 kHz ein günstiger Arbeitsbereich mit relativ kleinen Schwankungen zu erkennen...............................................................................................................88
Abbildung 51 Rechts abgebildet ist die optimale Resonanzbedingung in einem zylindrischen Musterrohr. Wie ersichtlich, wurde die Verstärkung durch eine longitudinale Mode bewirkt. Das Mikrofon sollte hierbei den gleichen Abstand zum Röhrenboden haben wie der Laserstrahl zum oberen Ende (oder ein ganzzahliges Vielfaches). Der Zwischenraum sollte ein ganzzahliges Vielfaches hiervon sein. Zu beachten ist die Verschiebung der stehenden Druckwelle relativ zur stehenden Welle der Molekülgeschwindigkeiten von 90 °. Links zu sehen ist die neue Resonanzröhre, welche zusätzlich mit einem Querrohr zur Stabilisierung des Messsignals ausgestattet ist. .........................................................................89
Abbildung 52 Messung der Konzentration unter 1 % Kohlendioxid. Die Stufen entsprechen einer Konzentrationsänderung von 0 % bis auf 0,6 % und zurück in 0,1 % Schritten. Unter 0,3 % ist der Überlagernde Einfluss einer Störwelle zu erkennen. .........................................................................................90
Abbildung 53 Lock-In-Technik. Es kann bei phasenabhängiger Messung in X- und Y-Richtung gemessen werden. Gezeigt ist der unterschiedliche Einfluss der beiden Wellen (Vektoren im Phasenraum) auf die Signalintensität. Der Einfluss der zweiten störenden Welle ist bei jedem Messwinkel konstant und bedingt lediglich einen Versatz............................................................................91
Abbildung 54 Neue Resonanzröhre mit Querrohr (links) und phasenabhängige Messung mit Lock-In-Verstärkung (rechts). Es ist ein linearer Zusammenhang zwischen Konzentration und Signalintensität zu erkennen. Da die Messung mit einem Versatz von 180 ° aufgenommen sind, ist ein umgekehrter Zusammenhang, welcher bei steigender Konzentration eine stärker negative Intensität zeigt ersichtlich. Ein störender Einfluss einer zweiten Welle ist nicht mehr zu beobachten. ...................................................................................91
Abbildung 55 Parallele Röhren..................................................................................................93 Abbildung 56 Paralleler Katalysatortest. Verwendet wurde der früher beschriebene
Messingreaktor. Eingesetzt wurde eine Gasmischung von 3 % CO in Luft bei einer Raumgeschwindigkeit von 18750 ml·h-1g-1
Katalysator. Die Nummern sind die einzelnen Katalysatorpositionen. Position 16 ist mit reinem Al2O3 gefüllt, weshalb kein Umsatz erwartet werden sollte. Alle anderen Positionen sind mit 2% Pt auf Al2O3 gefüllt. Die Detektion von Kohlendioxid in Kanal 16 zeigt ein Übersprechen der benachbarten Kanäle (Cross Talk). ........................................................................................................94
Abbildung 57 Paralleler Katalysatortest. Kanal 14 und 15 sind mit ZnSe-Fenstern abgeschirmt. Auffällig ist besonders der vorgetäuschte Umsatz in Kanal 5 und 7 im Bereich eines hohen Umsatzes in Kanal 6. Dieses Übersprechen wird vor allem bei Umsätzen über 50 % in den Nachbarkanälen gefunden, falls diese nicht wie Kanal 14 durch ZnSe-Fenster abgeschirmt sind. ...............95
142

B Abbildungsverzeichnis
Abbildung 58 Verknüpfung der einzelnen elektronischen Bauteile im routinemäßig betriebenen automatisierten Aufbau zur parallelen Untersuchung der Kohlenmonoxid-Oxidation...................................................................................96
Abbildung 59 Schematische Zeichnung der elektronischen Verknüpfung und des Datenflusses bei der photoakustischen Resonanz-Analytik. Die Reaktionskontrolle und die Analytik sind, wie aus der Abbildung zu erkennen ist, voneinander unabhängige Routinen. Die einzige Verknüpfung zwischen den Bauteilen besteht in der Doppelfunktion des Puls Generators zur Steuerung des Lasers und des Lock-In-Verstärkers. .....................................97
Abbildung 60 Benutzeroberfläche der Resonanz-Analytik.........................................................98 Abbildung 61 Beispiel einer parallelen Messung von 16 verschiedenen Katalysatoren in
der Kohlenmonoxid-Oxidation. Verwendet wurde der früher beschriebene Messingreaktor. Eingesetzt wurde eine Gasmischung von 3 % CO in Luft bei einer Raumgeschwindigkeit von 18750 ml·h-1g-1
Katalysator. Die Temperatur wurde mit einem Grad pro Minute von Raumtemperatur auf 180 °C erhöht und im Anschluss wieder bis auf RT reduziert. Hieraus resultieren die beiden unterschiedlichen Äste für jeden Katalysator. Die Zusammensetzung der einzelnen Materialien befindet sich in der Tabelle SYNTHESE 2 unter den einzelnen Nummern des Materials im Anhang. ............................................99
Abbildung 62 Vergleich der parallel photoakustischen Messung und der URAS-Messung im Einrohr-Reaktor. Die URAS-Messungen liegen exakt zwischen den einzelnen Ästen der parallelen Messung. Temperaturunterschiede von ± 5 °C können vernachlässigt werden. ....................................................................100
Abbildung 63 Zugängliche spektrale Einsatzbereiche der Bleisalzlaser und der gitterabstimmbaren CO- und CO2-Lasersysteme. .............................................103
Abbildung 64 Schematische Zeichnung der Gasversorgung. Die Druckreservoire stellen sicher, dass die Massenflussregler nicht durch Druckschwankungen im System ungenau arbeiten. Über die beiden Split-Ventile wird der eigentliche Nachdruck eingeregelt und der Fluss in der Messzone justiert. .......................104
Abbildung 65 Nachdruck/Fluss-Relationen der Messkanäle. Mit Druck ist der über die beiden Split-Ventile eingestellte Nachdruck gemeint. Der Fluss bezieht sich auf die Analysenzone und wurde mittels eines kalibrierten Massenflussmessers bestimmt. Der zu erwartende lineare Zusammenhang, welcher im Bereich unterhalb 1 ml/min nicht gefunden wird, ist wahrscheinlich auf Ungenauigkeiten des Massenflussmessers zurückzuführen...................................................................................................105
Abbildung 66 Vollständige Messzelle im Einsatz (links) und einzelne Module mit unterschiedlichem Zellvolumen mit Mikrofoneinsatz (mitte und rechst). Die linken Module sind für Freifeld-Messungen, die rechten mit an den Analysenkanal angekoppeltem Mikrofon...........................................................106
Abbildung 67 Schematische Zeichnung des Messzellentisches (links) und der Messmodule (rechts). Das Zentralloch mit 2 mm Durchmesser in der Mitte der Module (rechts) war der eigentliche Messkanal und diente zur Gasführung bzw. zur Stabilisierung der Detektionszone. Es wurden für die Messungen Module mit einer Messkanallänge von 2, 4, 6 und 8 mm gefertigt.................................106
Abbildung 68 Flussexperiment mit der 8 mm Zelle. Schraffiert eingezeichnet ist der Bereich innerhalb dessen der Sprung in der Intensität stattfindet. Die Werte entsprechen den Amplitudenwerten eines Schwingungsminimums. Stärker negative Werte entsprechen somit einer höheren Signalintensität....................107
Abbildung 69 Stäbilitätskontrolle bei kürzeren Zellen im Vergleich zu längeren. Die Konzentration wurde von 2 % Ethen auf 0 % reduziert um untere
143

11 Anhang
Detektionsgrenzen zu erkennen. Im Fall der 8 mm Zelle (rechts) kann noch zuverlässig bei 0,4 % Ethen abs. gemessen werden. Die kürzere 4 mm Zelle (links) ist schon bei einem Prozent hierzu nicht mehr in der Lage. ..................108
Abbildung 70 Abhängigkeit der Schaltzeit vom Mittelungsfaktor (rechts) und der Länge der Messzelle (links). Unter Dämpfungszeit ist der Zeitraum zu verstehen, bei dem auf dem Oszilloskop keine Welle mehr zu erkennen war. Die Zeitmessung erfolgte mittels einer Stoppuhr, was die großen Fehlergrenzen erklärt. ...............................................................................................................109
Abbildung 71 Faktor 1000 Filter (links) und Laserleistungsmessgerät (rechts) für hohe Strahlintensitäten (> 1 W). ................................................................................110
Abbildung 72 Zwei unterschiedliche Konzentrationsmessungen. Links ist eine Vergleichsmessung von 2 % Ethen bzw. 2 % Ethan in einem Stickstoffuntergrund gezeigt. Rechts sind die Amplitudenwerte mehrerer Konzentrationsmessungen gegen die Konzentrationen aufgetragen. Es findet sich in diesem Bereich ein proportionaler Zusammenhang. .............................111
Abbildung 73 Schematischer Vergleich der Endo- und Exotemplat-Synthese.........................113 Abbildung 74 TG-Messungen unterschiedlich beladener Aktivkohlen. Beladene
Aktivkohlen sind deutlich früher vollständig verbrannt. ...................................114 Abbildung 75 Quarzgefäße für die parallele Synthese (links) und definierte Standard-
Abfüllvolumina (rechts). ....................................................................................115 Abbildung 76 Präparationsplatte mit 77 Positionen (links) und Abdeckung bestehend aus
Quarzwolle (rechts). ..........................................................................................116 Abbildung 77 Schematische Darstellung der Katalysatorselektion (links) und
Benutzeroberfläche des Präparationsprogramms (rechts). ..............................117 Abbildung 78 Vergleich der Transmissionsaufnahmen von Material 1 im Vergleich zu
Material 2 (links). Das Überlagerte Diffraktogramm eines Kupferoxides, lässt den Schluss zu, dass bei Material 2 ein höherer Anteil kristallines CuO vorhanden ist. Die Abbildung rechts zeigt eine Überlagerung mit einem Cu1Ni0,5Mn1,5O4 Spinell. Die restlichen Reflexe können sowohl diesem als auch weiteren ähnlich zusammengesetzten Spezies mit gleichem Reflexmuster zugeordnet werden. Eine genaue Ermittlung der Zusammensetzung ist nicht möglich. .................................................................118
Abbildung 79 BET-Messungen von Material 1 und 2 im Vergleich. .......................................119 Abbildung 80 TEM-Aufnahmen von Material 1 (links) und 2 (rechts) bei 30000-facher
Vergrößerung im Vergleich. ..............................................................................120 Abbildung 81 TEM-Aufnahmen von Material 1 (links) und 2 (rechts) bei 500000-facher
Vergrößerung im Vergleich. Man erkennt deutlich die kristallinen Bereiche. .120 Abbildung 82 Ortsaufgelöste EDX Messungen an Material 1. ................................................121 Abbildung 83 Ortsaufgelöste EDX Messungen an Material 2. ................................................122 Abbildung 84 Ortsaufgelöste EDX und Zusammensetzungen bei Material 2. .........................123 Abbildung 85 Einige photoakustische Messung mit dem resonanten Aufbau. Die obigen
Messungen zeigen einen Ausschnitt aus den getesteten Materialien und wurden innerhalb von 3 Tagen aufgenommen. Die genauen Zusammensetzungen der Materialien finden sich im Anhang. ..........................126
Abbildung 86 Photoakustische Messung mit dem Freifeldaufbau. Das getestete Material hatte die Zusammensetzung La12Ba2Pb9Th7Mn27Ni24Cu19Ox. ...........................128
Abbildung 87 Gaschromatographische Untersuchung der Evaluierungsreaktion. .................129
144

Literaturverzeichnis 1 P. Cong, R.D. Doolen, Q. Fan, D.M. Giaquinta, S. Guan, E.W. McFarland, D.M.
Poojary, K. Self, H.W. Turner, W.H. Weinberg, Angew. Chem. 1999, 111, 507-512;
Angew. Chem. Int. Ed. Engl. 1999, 38, 484.
2 E. Reddington, A. Sapienza, B. Gurau, R. Viswanathan, S. Sarangapani, E.S. Smotkin,
T.E. Mallouk, Science, 1998, 280, 1735.
3 F.C. Moates, M. Somani, J. Annamalai, J.T. Richardson, D. Luss, R.C. Willson Ind.
Eng. Chem. Res. 1996, 35, 4801.
4 A. Holzwarth, H.W. Schmidt, W.F. Maier, Angew. Chem. 1998, 110, 2788-2792;
Angew. Chem. Int. Ed. Engl. 1998, 37, 2644.
5 S. Senkan, Angew. Chem. 2001, 113, 322.
6 J.M. Newsam, F. Schüth, Biotechnology and Bioengineering 1998/1999, 61, 203.
7 X.D. Xiang, X. Sun, G. Briceno, Y. Lou, K.A. Wang, H. Chang, W.G.W. Freedman,
S.W. Chen, P.G. Schultz, Science 1995, 268, 1738
8 J.J. Hanak, J. Mat. Sci. 1970, 5, 964.
9 Á. Furka, F. Sebestyén, M. Asgedom, G. Dibó, 14th Int. Congr. Biochem. Prague,
Czechoslovakia, 1988, 5, 47.
10 Á. Furka, F. Sebestyén, M. Asgedom, G. Dibó, Int. J. Peptide Protein Res. 1991, 37,
487.
11 R.A. Houghten, Proc. Natl. Acad. Sci. USA 1985, 82, 5131.
12 K.C. Nicolaou, X.Y. Xiao, Z. Parandoosh, A. Senyei, M.P. Nova, Angew. Chem. 1995,
107, 2476.
13 E.J. Moran, S. Sarshar, J.F. Cargill, M.M. Shahbaz, A. Lio, A.M.M. Mjalli, R.W.
Armstrong, J. Am. Chem. Soc. 1995, 117, 10787.
14 S.P.A. Fodor, R.J. Leighton, M.C. Pirrung, L. Stryer, A.T. Lu, D. Solas, Science 1991,
251, 767.
15 H.M. Geysen, R.H. Meloen, S.J. Barteling, Proc. Natl. Acad. Sci. USA 1984, 81, 3998.
16 J. Klein, O. Laus, J.M. Newsam, S.A. Schunk, A. Straßer, A. Sundermann, U. Vietze,
T. Zech, Tagungsband XXXV. Jahestreffen Deutscher Katalytiker, 44.
17 X.D. Sun, K.A. Wang, Y. Yoo, W.G.W. Freedman, C. Gao, X.D. Xiang, P.G. Schultz,
Adv. Mater. 1997, 9, 1046.
145

18 B. Jandeleit, D.J. Schaefer, T.S. Powers, H.W. Turner, W.H. Weinberg, Angew. Chem.
Int. Ed. 1999, 38, 2494.
19 P. Cong, A. Dehestani, R.D. Doolen, D.M. Giaquinta, S. Guan, V. Markov, D.M.
Poojary, K. Self, H.W. Turner, W.H. Weinberg, Proc. Nat. Acad. Sci. 1999, 96,
11077.
20 C. Hoffmann, A. Wolf, F. Schüth, Angew. Chem. Int. Ed. 1999, 38, 2800.
21 K.E. Simons, Topics in Catalysis 2000, 13, 201.
22 U. Rodemerck, P. Ignaszewski, M. Lucas, P. Claus, Chem. Ing. Tech. 1999, 71, 873.
23 U. Rodemerck, P. Ignaszewski, M. Lucas, P. Claus, M. Baerns, Topics in Catalysis
2000, 13, 249.
24 D.E. Akporiaye, I.M. Dahl, A. Karlsson, R. Wendelbo, Angew. Chem. Int. Ed. 1998,
37, 609.
25 J. Klein, C.W. Lehmann, H.W. Schmidt, W.F. Maier, Angew. Chem. Int. Ed. 1998, 38,
3369.
26 C.M. Blackstone, J.J. Lerou, Het Ingenieursblad 1986, 55, 289.
27 J. Perez-Ramierez, R.J. Berger, G. Mul, F. Kapteijn, J.A. Moulijn, Catal. Today 2000,
60, 93.
28 A. Richter, M. Langpape, S. Kolf, G. Grubert, R. Eckelt, J. Radnik, A. Schneider,
M.M. Pohl, R. Fricke, Appl. Catal. B-Env. 2002, 36, 261.
29 P. Claus, D. Hönicke, T. Zech, Catal. Today 2001, 67, 319.
30 P. Desrosiers, A. Guram, A. Hagemeyer, B. Jandeleit, D.M. Poorjary, H. Turner, H.
Weinberg, Catal. Today 2001, 67, 397.
31 M. Lucas, P. Claus, Chem. Ing. Tech. 2001, 73, 252.
32 www.parrinst.de
33 www.argotech.com
34 K. Griffin, S. Bennett, P. Johnston, Abstr. Pap. Am. Chem. Apr. 1 2001, 184-IEC Part
1, 221.
35 http://www.chemicals.matthey.com/services.asp?id=2
36 C. Hoffmann, H.W. Schmidt, F. Schüth, J. Catal. 2001, 198, 348.
37 C. Kiener, M. Kurtz, H. Wilmer, C. Hoffmann, H.W. Schmidt, J.D. Grunwaldt, M.
Muhler and F. Schüth, J. Catal. special issue, in press.
38 S. Thomson, C. Hoffmann, S. Ruthe, H.W. Schmidt, F. Schüth, Appl. Catal. A-
General 2001, 220, 253.
39 S.M. Senkan, S. Ozturk, Angew. Chem. Int. Ed. 1999, 38, 791.
146

40 S.M. Senkan, Nature 1998, 394, 350.
41 W.H. Weinberg, E.W. McFarland, P. Cong, S. Guan, WO 98/15969, 1996.
42 M. Orschel, J. Klein, H.W. Schmidt, W.F. Maier, Angew. Chem. Int. Ed. 1999, 38,
2791.
43 S. Senkan, K. Krantz, S. Ozturk, V. Zengin, I. Onal, Angew. Chem. Int. Ed. 1999, 38,
2794.
44 O.M. Busch, C. Hoffmann, T.R.F. Johann, H.W. Schmidt, W. Strehlau, F. Schüth, J.
Am. Chem. Soc. 2002, 124, 13527.
45 D. Wolf, O.V. Buyevskaja, M. Baerns, Appl. Catal. A: General, 2000, 200, 63.
46 M.W. Sigrist, Infr. Phys. & Tech. 1995, 36, 415.
47 P. Repond, M.W. Sigrist, J. Quant. Electr. 1996, 32, 1549.
48 P. Repond, M.W. Sigrist, Appl. Opt. 1996, 35, 4065.
49 A. Schmohl, A. Miklos, P. Hess, Appl. Optics, 2002, 41, 1815.
50 A. Bohren, M.W. Sigrist, Infr. Phys. & Tech. B, 1997, 38, 423.
51 P.L. Meyer, M.W. Sigrist, Rev. Sci. Instrum. 1990, 61, 1779.
52 G.C. Liang, H.H. Liu, A.H. Kung, A. Mohacsi, A. Miklos, P. Hess, J. Phys. Chem. A,
2000, 104, 10179.
53 V.A. Kapitanov, Y.N. Ponomarev, K. Song, H.K. Cha, J. Lee, Appl. Phys. B, 2001, 73,
745.
54 A. Beenen, R. Niessner, Appl. Spec. 1999, 53, 1040.
55 C.P. Li, J. Davis, Appl. Optics, 1979, 18, 3541.
56 S. Yamulki, S.C. Jarvis, J. Geophys. Res. 1999, 104, 5463.
57 D.D. Markusev, M. Terzic, P. Vujkovic Cvijin, J. Jovanovic-Kurepa, Appl. Photon.
Tech. G.A. Lampropoulos et al. (Ed.), Plenum Press, New York, 1995, 241.
58 A. Boschetti, D. Bassi, E. Jacob, S. Iannotta, L. Ricci, M. Scotoni, Appl. Phys. B,
2002, 74, 273.
59 M.A. Moeckli, C. Hilbes, M.W. Sigrist, Appl. Phys. B, 1998, 67, 449.
60 N. Chattopadhyay, C. Serpa, P. Purkayastha, L.G. Arnaut, S.J. Formosinho, Phys.
Chem. Chem. Phys. 2001, 3, 70.
61 M. Hammerich, A. Ólafsson, J. Henningsen, Chem. Phys. 1992, 163, 173.
62 J Lalevee, X. Allonas, J.P. Fourassier, Abstr. Pap. Amer. Chem. Soc. 2001, 222, 516.
63 M.W. Sigrist, Opt. Eng. 1995, 34, 1916.
64 T. Hoshimiya, K.y. Ishikawa, S. Kikuchi, Anal. Sci. 2001, 17, s469.
65 M.H. Ryoo, H. Chon, J. Chem. Soc. Faraday Trans. 1997, 93, 3259.
147

66 R.L. Thomas, Anal. Sci. 2001, 17, s1 und Zitate.
67 S.L. Firebaugh, K.F. Jensen, M.A. Schmidt, Journal of Microelectromechanical
Systems, 2001, 10, 232.
68 S.J. Kim, I.S. Byun, H.Y. Han, H.L. Ju, S.H. Lee, J.G. Choi, Appl. Catal. A: General,
2002, 234, 35.
69 C.K. Williamson, G.N. Coleman, Instr. Sci. & Techn. 1994, 22, 1.
70 F. Kühnemann, W. Urban, Physikalische Blätter, 2000, 56, 43.
71 H. Dahnke, J. Kahl, G. Schüler, W. Boland, W. Urban, F. Kühnemann, Appl. Phys. B,
2000, 70, 275.
72 V. Altuzar, M. Pacheco, S.A. Tomas, J.L. Arriaga, O. Zelaya-Angel, F. Sanchez-
Sinencio, Anal. Sciences, 2001, 17, 541.
73 H. Yang, J. Irudayaraj, Lebensmittel-Wissenschaft und Teechnologie-Food Science
and Technology, 2001, 34, 402.
74 F.J.M. Harren, R. Berkelmans, K. Kuiper, S. te Lintel Hekkert, P. Scheepers, R.
Dekhuijzen, P. Hollander, D.H. Parker, Appl. Phys. Lett. 1999, 24, 1761.
75 N.W. Cant, N.J. Ossipoff, Catal. Today 1997, 36, 125.
76 S. Imamura, H. Sawada, K. Uemura, S. Ishida, J. Catal 1988, 109, 198.
77 J.E. Drawdy, G.B. Hoflund, S.D. Gardner, E. Yngvadottier, D.R. Schryer, Surface and
Interface Analysis 1990, 16, 369.
78 K. Grass, H.G. Lintz, J. Catal 1997, 172, 446.
79 S.D. Gardner, G.B. Hoflund, M.R. Davidson, D.R. Schryer J. Catal 1989, 115, 132.
80 G.C. Bond, L.R. Molloy, M.L. Fuller, J. Chem. Soc. Chem. Commun. 1975, 796.
81 M. Sheintuch, J. Schmidt, Y. Lechthman, Appl. Catal. 1989, 49, 55.
82 M. Haruta, Catal. Today 1997, 36, 153.
83 M.M. Schubert, S. Hackenberg, A.C. van Veen, M. Muhler, V. Plzak, R.J. Behm, J.
Catal. 2001, 197, 113.
84 A.K. Tripathi, V.S. Kamble, N.M. Gupta, J. Catal. 1999, 187, 332.
85 D. Horváth, L. Toth, L. Guczi, Catal. Lett. 2000, 67, 117.
86 M.A. Bollinger, M.A. Vannice, Appl. Catal. B, 1996, 8, 417.
87 S. Minicó, S. Sciré, C. Crisafulli, A.M. Visco, S. Galvagno, Catal. Lett. 1997, 47, 273.
88 F. Boccuzzi, E. Guglielminotti, F. Pinna, G. Strukul, Surf. Sci. 1997, 377-379, 728.
89 F. Boccuzzi, A. Chiorino, S. Tsubota, M. Haruta, J. Phys. Chem. 1996, 100, 3625.
90 E.D. Park, J.S. Lee, J. Catal. 1999, 186, 1.
91 M. Mavrikakis, P. Stoltze, J.K. Nørskov, Catal. Lett. 2000, 64, 101.
148

92 M. Valden, S. Pak, X. Lai, D.W. Goodman, Catal. Lett. 1998, 56, 7.
93 J.D. Grunwaldt, A. Baiker, J. Phys. Chem. B 1999, 103, 1002.
94 A.I. Kozlov, A.P. Kozlova, H. Liu, Y. Iwasawa, Appl. Catal. A: General, 1999, 182, 9.
95 F. Bocuzzi, A. Chiorino, S. Tsubota, M. Haruta, Catal. Lett. 1994, 29, 225.
96 T. Engel, G. Ertl, “Oxidation of Carbon Monoxide” in: D.A. King, D.P. Woodruff
(Eds.), “The Chemical Physics of Solid Surfaces and Heterogeneous Catalysis”, Band
4, Kapitel 3, Amsterdam 1982.
97 S. Zehnder, Hydrocarb. Process. 1998, 77(2), 59.
98 F. Cavani, F. Trifido, Catal.Today 1995, 24, 307.
99 H.H. Kung, Adv.Catal. 1994, 1.
100 M.M. Bhasin, J.H. McCain, B.V. Vora, T. Imai,P.R. Pujado, Appl. Catal. A-Gen.
2001, 221, 397.
101 R.K. Grasselli, D.L. Stern, J.G. Tsikoyiannis, Appl. Catal. A-Gen. 1999, 189, 9.
102 R.K. Grasselli, D.L. Stern, J.G. Tsikoyiannis Appl. Catal. A-Gen. 1999, 189, 1.
103 L.D. Schmidt, Stud. Surf. Sci. Catal. 2000, 130A(International Congress on Catalysis,
2000, Pt. A), 61.
104 L.D. Schmidt, J. Siddall, M. Bearden, Aiche J. 2000, 46, 1492.
105 A. Beretta, L. Piovesan, P. Forzatti, J. Catal. 1999, 184, 455.
106 W.D. Zhang, X.P. Zhou, D.L. Tang, H.L. Wan, K. Tsai, Catal. Lett. 1994, 23, 103.
107 M.Y. Sinev, L.Y. Margolis, V.Y. Bychkov, V.N. Korchak, Stud. Surf. Sci. Catal.
1997, 110, 327.
108 I.M. Dahl, K. Grande, K.J. Jens, E. Rytter, A. Slagtern, Appl. Cat. 1991, 77, 163.
109 O.V. Buyevskaya, M. Baerns, Catal. Today 1998, 42, 315.
110 Q.J. Ge, B. Zhaorigetu, C.Y. Yu, W.Z. Li, H.Y. Xu, Catal. Lett. 2000, 68,59.
111 V.R. Choudhary, V.H. Rane, A.M. Rajput, AIChE J. 1998, 44, 2293.
112 E. Ranzi, T. Faravelli, P. Gaffuri, E. Garavaglia, A. Goldaniga, Ind. Eng. Chem.Res.
1997, 36, 3336.
113 M. Xu, J.H. Lunsford, React.Kinet.Catal.Lett. 1996, 57, 3.
114 A.A. Lemonidou, A.E. Stambouli, Appl.Catal. A, 1998, 171, 325.
115 Ullmann´s Encyclopedia of Industrial Chemistry, A5, Catalysis and Catalysts, 1986,
357.
116 S. Brunauer, P.H. Emmet, E. Teller, J. Am. Chem. Soc. 1938, 60, 309.
117 A.G. Bell, Am. J. Sci. 1880, 20, 305.
118 http://www.bell-labs.com/org/physicalsciences/timeline/span2.html#
149

119 A.G. Bell, Philos. Mag. 1881, 11, 510.
120 J. Tyndall, Proc. R. Soc. Lond. 1881, 31, 307.
121 W.C. Roentgen, Philos. Mag. 1881, 11, 308.
122 M.L. Veingerof, Dokl. Akad. Nauk SSSR 1938, 19, 687. Aus A. Rosenzwaig,
Photoacoustics and Photoacoustic Spectroscopy, Chemical Analysis Vol. 57, Wiley,
New York.
123 K.F. Luft, Z. Tech. Phys. 1943, 24, 97.
124 G. Gorelik, Dokl. Akad. Nauk SSSR 1946, 54, 779. Aus A. Rosenzwaig,
Photoacoustics and Photoacoustic Spectroscopy, Chemical Analysis Vol. 57, Wiley,
New York.
125 P.V. Slobodskaya, Izv. Akad. Nauk SSSR, Ser. Fiz. 1948, 12, 656. Aus A. Rosenzwaig,
Photoacoustics and Photoacoustic Spectroscopy, Chemical Analysis Vol. 57, Wiley,
New York.
126 A. Rosenzwaig, Photoacoustics and Photoacoustic Spectroscopy, Chemical Analysis
Vol. 57, Wiley, New York.
127 Rayleigh, Nature (Lond.), 1881, 23, 274.
128 M.E. Mercadier, C. R. Hebd. Serv. Acad. Sci. 1881, 92, 409. Aus A. Rosenzwaig,
Photoacoustics and Photoacoustic Spectroscopy, Chemical Analysis Vol. 57, Wiley,
New York.
129 W.H. Preece, Proc. R. Soc. Lond. 1881, 31, 506.
130 I.G. Calasso, M.W. Sigrist, Rev. Sci. Instr. 1999, 70, 4569.
131 Aus http://spectra.galactic.com/
132 M. Schwickardi, T. Johann, W. Schmidt, F. Schüth, Chem. Mater. 2002, 14, 3913.
133 M. Ozawa, M. Kimura, J. Mater. Sci. Lett. 1990, 9, 446.
150

Dipl.- Chem. Thorsten Johann
Lebenslauf PERSÖNLICHE DATEN: Name : Johann Vorname : Thorsten Robert Florian geb. am : 26.08.1973 Geburtsort : Mannheim SCHULISCHER WERDEGANG: Grundschule : 1980-1984 - Gustav-Wiederkehr-Schule, MA-Sandhofen Gymnasium : 1984-1993 - Peter-Petersen-Gymnasium, MA-Schönau Abitur : 10.05.1993 - Peter-Petersen-Gymnasium, MA-Schönau AKADEMISCHER UND WEITERER WERDEGANG: Oktober 1993 Immatrikulation an der Ruprecht Karls Universität in Heidelberg (Chemie als Diplomstudiengang). Vordiplom im Dezember 1995 (Proctor&Gamble Preis für herausragende Leistungen). Im August 1996 Aufnahme in die „Studienstiftung des deutschen Volkes“ als Stipendiat. Von Oktober 1996 bis April 1997 Chemiestudium an der University of Bristol (Erasmus Stipendium). Von April 1997 bis Juni 1998 Abschluß des Studiums und mündliche Prüfungen. Diplomarbeit „Untersuchungen der CVD von GaN“ von August 1998 bis April 1999 an der Ruhr-Universität in Bochum im Arbeitskreis von Prof. R.A. Fischer, betreut von Prof. J. Wolfrum aus Heidelberg. 19. April 1999 Verleihung des akademischen Grades „Diplom–Chemiker“ durch die Ruprecht Karls Universität in Heidelberg. April 1999 bis November 1999 Forschungsaufenthalt an der Ruhr-Universität in Bochum bei Prof. R.A. Fischer. Dezember 1999 bis Dezember 2002, Promotion über „Neue Analytische Methoden bei der high-throughput Katalysator Forschung“ am Max-Planck-Institut für Kohlenforschung in Mülheim an der Ruhr bei Prof. F. Schüth. Seit Januar 2003 als Chemiker bei der BASF AG in Ludwigshafen angestellt.


![X-Ray diffraction XRD Röntgenbeugung 2T [°] Method-XRD... · (Bsp. C-C Bindung: 1.54 Å; 1.54 x 10-8 cm) Elementarzelle mit a = 1000 pm, Größe des Kristalls: 0.1 - 0.5 mm](https://img.pdfslide.org/doc/110x75/5b95dfb609d3f27f5b8d328d/x-ray-diffraction-xrd-roentgenbeugung-2t-method-xrd-bsp-c-c-bindung.jpg)


























