Embed Size (px)
Citation preview

Optische Beobachtung von oberflachengebundenen
und frei beweglichen Nanopartikeln
Von der
Fakultat fur Naturwissenschaften
der Universitat Duisburg-Essen
(Campus Duisburg)
zur Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften
genehmigte Dissertation
von
Christiane Finder
aus Berlin
Duisburg 2005

Berichterstatter: Prof. Dr. Christian Mayer
Prof. Dr. Karl Molt
Tag der mundlichen Prufung: 06. April 2005

Danksagung
Mein besonderer Dank gilt Herrn Prof. Dr. C. Mayer fur die Uberlassung des
interessanten Themas sowie fur seine fortwahrende Diskussionsbereitschaft wahrend
der Erstellung dieser Arbeit. Herrn Prof. Dr. K. Molt danke ich herzlich fur die
Ubernahme des Korreferates.
Ich danke Herrn Dr. R. H. G. Muller fur die große Hilfs- und Diskussionsbe-
reitschaft bezuglich der digitalen Bildverarbeitung in den letzten Jahren. Fur die Hilfe
bei technischen Fragestellungen und der technischen Umsetzung bedanke ich mich bei
Herrn U. Bachorski und den Mitarbeitern der Elektrowerkstatt. Herrn M.
Zahres danke ich fur die vielen praktischen Tipps bezuglich der Mikroskopie.
Allen Mitarbeiterinnen und Mitarbeitern des EU-Projektes CHANIL danke ich fur
die gute Kooperation, insbesondere Dr. K. Pfeiffer und H. Schulz fur den stetigen
Informations- und Ideenaustausch. Besonderer Dank geht an die EU fur die finanzielle
Unterstutzung.
Den Mitarbeiterinnen und Mitarbeitern des Fachgebietes Physikalische Chemie danke
ich fur die freundliche Arbeitsatmosphare. Besonders gilt mein Dank Michael Wohl-
gemuth fur die vielen anregenden Diskussionen. Alina Bauer danke ich fur ihre
Hilfsbereitschaft in den letzten Wochen. Ilka Broekmann und Natascha Emme-
richs gilt ein ganz besonderer Dank fur die tolle Zeit in unserem Buro.
Meinen Freunden Frank und Corinna danke ich herzlichst fur die aufopfernde Durch-
sicht des Manuskripts.
Das großte Dankeschon widme ich meinen Eltern. Ihr habt mich auch uber die große
Entfernung wahrend der ganzen Zeit konsequent unterstutzt und an mich geglaubt.

Das Schonste, was wir entdecken konnen,
ist das Geheimnisvolle.
Albert Einstein

Inhaltsverzeichnis
1 Einleitung 1
2 Optische Methoden 3
2.1 Fluoreszenzmikroskopie . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2.1.1 Physikalische und chemische Betrachtung der Fluoreszenz . . . . 6
2.1.2 Auflicht-Fluoreszenzmikroskopie . . . . . . . . . . . . . . . . . . 10
2.2 Dunkelfeldmikroskopie . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
2.2.1 Bildentstehung im Mikroskop und das Auflosungsvermogen . . . 20
2.3 Digitale Bildverarbeitung . . . . . . . . . . . . . . . . . . . . . . . . . . 26
2.3.1 Bildgewinnung mittels einer CCD-Kamera . . . . . . . . . . . . 27
2.3.2 Digitalisierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
2.3.3 Bildverarbeitung und -analyse . . . . . . . . . . . . . . . . . . . 29
3 Oberflachengebundene Nanopartikel 31
3.1 Nanoimprintlithographie und deren Anwendung . . . . . . . . . . . . . 32
3.2 Herkommliche Methoden zur Qualitatssicherung . . . . . . . . . . . . . 37
3.2.1 Rasterelektronenmikroskopie (SEM) . . . . . . . . . . . . . . . . 37
3.2.2 Rasterkraftmikroskopie (AFM) . . . . . . . . . . . . . . . . . . 41
4 Brownsche Bewegung von frei beweglichen Nanopartikeln 45
4.1 Theoretische Grundlagen zur Brownschen Bewegung . . . . . . . . . . . 46
4.2 Experimentelle Beobachtung der Brownschen Bewegung . . . . . . . . . 52
i

ii INHALTSVERZEICHNIS
5 Experimenteller Teil 57
5.1 Mikroskopie und digitale Bildverarbeitung . . . . . . . . . . . . . . . . 57
5.1.1 Auflicht-Fluoreszenzmikroskopie . . . . . . . . . . . . . . . . . . 57
5.1.2 Durchlicht-Dunkelfeldmikroskopie . . . . . . . . . . . . . . . . . 59
5.1.3 Digitale Bildverarbeitung . . . . . . . . . . . . . . . . . . . . . . 59
5.2 Oberflachengebundene Nanopartikel . . . . . . . . . . . . . . . . . . . . 60
5.2.1 Fluoreszenzspektroskopische Untersuchungen . . . . . . . . . . . 60
5.2.2 Stabilitatsbestimmung des Farbstoffes . . . . . . . . . . . . . . . 60
5.2.3 Bestimmung und Charakterisierung von Defekten auf den Pra-
gungen und den Stempeln . . . . . . . . . . . . . . . . . . . . . 61
5.2.4 Qualitatssicherung wahrend einer Serie von Nanopragungen . . 64
5.2.4.1 Stempel und Polymer . . . . . . . . . . . . . . . . . . 64
5.2.4.2 Prageprozess und Qualitatssicherung mittels Fluores-
zenzmikroskopie . . . . . . . . . . . . . . . . . . . . . 65
5.3 Großenbestimmung von frei beweglichen Nanopartikeln . . . . . . . . . 69
5.3.1 Polystyrolstandards . . . . . . . . . . . . . . . . . . . . . . . . . 69
5.3.1.1 Polystyrolstandards in der Durchlicht-Dunkelfeldmikros-
kopie . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
5.3.1.2 Polystyrolstandards in der Auflicht-Fluoreszenzmikros-
kopie . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
5.3.2 Probenvorbereitung . . . . . . . . . . . . . . . . . . . . . . . . . 70
5.3.2.1 Partikelkonzentrationen in der Dunkelfeldmikroskopie . 71
5.3.2.2 Partikelkonzentrationen in der Fluoreszenzmikroskopie 71
5.3.3 Messung und Datenauswertung . . . . . . . . . . . . . . . . . . 71
5.3.3.1 Großenbestimmung mittels der Dunkelfeldmikroskopie 72
5.3.3.2 Großenbestimmung mittels der Fluoreszenzmikroskopie 73
6 Ergebnisse und Diskussion 75
6.1 Oberflachengebundene Nanopartikel . . . . . . . . . . . . . . . . . . . . 75
6.1.1 Charakterisierung des Fluoreszenzmarkers . . . . . . . . . . . . 76

INHALTSVERZEICHNIS iii
6.1.1.1 Fluoreszenzspektroskopische Untersuchungen des Farb-
stoffes . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
6.1.1.2 Bestimmung der minimalen Farbstoffkonzentration . . 80
6.1.1.3 Bestimmung der Stabilitat des Farbstoffes . . . . . . . 83
6.1.2 Qualitatssicherung in der Nanoimprintlithographie . . . . . . . . 88
6.1.2.1 Charakterisierung der auftretenden Defekte in der Pra-
gung . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
6.1.2.2 Detektion von Defekten auf dem Stempel und auf der
Pragung . . . . . . . . . . . . . . . . . . . . . . . . . . 98
6.1.2.3 Bestimmung der Qualitat einer Nanopragung . . . . . 103
6.1.2.4 Qualitatssicherung an einem Stempel . . . . . . . . . . 108
6.2 Frei bewegliche Nanopartikel . . . . . . . . . . . . . . . . . . . . . . . . 120
6.2.1 Bildverarbeitung und -analyse zur Großenbestimmung von frei
beweglichen Nanopartikeln . . . . . . . . . . . . . . . . . . . . . 122
6.2.2 Großenbestimmung mittels der Dunkelfeldmikroskopie . . . . . 126
6.2.3 Großenbestimmung mittels der Fluoreszenzmikroskopie . . . . . 130
6.2.4 Vergleich der mikroskopischen Methoden . . . . . . . . . . . . . 132
7 Zusammenfassung 137
A Bildverarbeitungsprogramme 139
A.1 Mathematica Programm . . . . . . . . . . . . . . . . . . . . . . . . . . 139
A.2 Makro zur Bildverarbeitung und -analyse . . . . . . . . . . . . . . . . . 140
A.3 Excel Makro zur Spurenanalyse von frei beweglichen Nanopartikeln . . 146
A.4 Excel Makro zur Erstellung der Histogramme der Großenverteilungen . 154
Literaturverzeichnis 161

iv INHALTSVERZEICHNIS

Kapitel 1
Einleitung
Die Nanotechnologie wird als die Schlusseltechnologie des 21. Jahrhunderts bezeichnet.
Der Begriff Nano kommt aus dem Griechischen (nanos) und bedeutet Zwerg. Die Na-
notechnologie bewegt sich in einem Großenbereich unter 100 nm. Als Erfinder dieser
Technologie gilt der amerikanische Physiker Robert Feynman, der 1959 auf einem Tref-
fen der Amerikanischen Physikalischen Gesellschaft die visionare Rede mit dem Titel
”There’s plenty of room at the bottom“ halt [1]. Das Wort Nanotechnologie pragt 1974
der Japaner Norio Taniguchi. Seine Definition umfasst alle Verfahrenstechniken, die zu
Dimensionen unter 100 nm fuhren oder mit einer Genauigkeit im Nanometerbereich
arbeiten [2].
Die Nanotechnologie beschaftigt sich mit der Entwicklung von Materialien im Na-
nometerbereich und der Erforschung der Eigenschaften dieser Materialien, um diese
Aspekte in technische Entwicklungen umzusetzen. Dieses interdisziplinare Forschungs-
gebiet vereint Wissenschaftler unterschiedlichster Fachrichtungen, wie Chemiker, Phy-
siker, Materialwissenschaftler, Elektrotechniker, Biologen und Mediziner, um nur einige
zu nennen. Daraus leitet sich auch die Vielzahl der Anwendungsmoglichkeiten ab, die
in Nanochemie, Nanomaterialien, Nanoelektronik, Nanooptik oder Nanoanalytik un-
terteilt werden konnen [2].
Diese Arbeit befasst sich mit der Nanoanalytik, das heißt der Untersuchung von Eigen-
1

2 1. Einleitung
schaften von Materialien und Bauelementen im Nanometerbereich. Die Grundlage der
hier vorgestellten Analysemethoden bildet die optische Mikroskopie in Verbindung mit
der digitalen Bildverarbeitung. Es ist zwischen zwei Analysemethoden zu unterschei-
den: Der Beobachtung von oberflachengebundenen Nanopartikeln und der Beobachtung
von frei beweglichen Nanopartikeln.
Die Analysemethode zur Beobachtung und Untersuchung von oberflachengebundenen
Nanopartikeln kann u. a. im Bereich der Qualitatssicherung in der Nanoimprintlitho-
graphie angewendet werden (vgl. Kapitel 6.1.2). Hierbei handelt es sich um eine neue
Methode zur Herstellung nanostrukturierter Oberflachen, z. B. in der Halbleiterindus-
trie (siehe Kapitel 3). Mittels der Fluoreszenzmikroskopie und der digitalen Bildverar-
beitung konnen Defekte in den Pragungen charakterisiert und die Einheitlichkeit der
Pragungen bestimmt werden. Ferner ist eine quantitative Qualitatskontrolle an dem
Pragestempel moglich.
Die Analysemethode zur Beobachtung frei beweglicher Nanopartikel dient u. a. zur
Großenbestimmung von in Wasser suspendierten Nanopartikeln (Kapitel 6.2). In Ab-
hangigkeit von den Eigenschaften der Nanopartikel konnen verschiedene mikroskopische
Methoden eingesetzt werden. Die Auswertung basiert auf der von Einstein und Smo-
luchowksi entwickelten Gleichung zur Beschreibung der Brownschen Bewegung sowie
den darauf folgenden mikroskopischen Untersuchungen verschiedener Wissenschaftler
Anfang des 20. Jahrhunderts [3, 4, 5, 6, 7].
Die Entwicklung dieser Analysemethoden hat das Ziel, im Vergleich zu den herkomm-
lich angewendeten Methoden kosteneffizientere Moglichkeiten aufzuzeigen, die ferner
durch eine hohe Benutzerfreundlichkeit und eine sehr gute Reproduzierbarkeit charak-
terisiert sind.

Kapitel 2
Optische Methoden
Obwohl die Geschichte der Naturwissenschaft in engem Zusammenhang mit der Ge-
schichte der Mikroskopie steht, gilt das Mikroskop erst seit Beginn des 20. Jahrhunderts
als Symbol der Wissenschaft. Heute wird das Mikroskop als Forschungsinstrument in
einer großen Anzahl von unterschiedlichen Anwendungsgebieten genutzt, wie im Be-
reich der Biologie und der Medizin, der Nahrungsmittelindustrie, der Textiltechnik, im
Bereich der Mineralogie und der Geologie oder der Elektroindustrie, zum Beispiel zur
Untersuchung von Werkstoffeigenschaften [8, 9].
Die Erfindung des Mikroskops steht in engem Zusammenhang mit der Erfindung des
Fernrohres Anfang des 17. Jahrhunderts. Als Erfinder werden sowohl Hans und Zacha-
rias Jansen aus Holland als auch Johannes Kepler, der das astronomische Fernrohr ent-
wickelt hat, genannt. Auch Galileo Galilei nutzt das Fernrohr durch einen veranderten
Abstand zwischen Objektiv und Okularlinse als Vergroßerungsglas fur mikroskopische
Beobachtungen. Der Begriff Mikroskop stammt von den Mitgliedern der Academia dei
Lincei (Luchsaugige) in Rom, zu denen auch Galilei gehort. Schon in der ersten Half-
te des 17. Jahrhunderts wird das einfache, aus nur einer Linse bestehende Mikroskop
entwickelt. Der Niederlander Anthony van Leuwenhoek, der erste Mikroskopiker von
Weltruf, baut um 1665 zum ersten Mal ein Mikroskop mit einem Abbildungsmaßstab
von 200 : 1. Zu seinen Entdeckungen gehoren u. a. die Faserstruktur der Augenlin-
se, die Querstreifung der Muskelfaser sowie die ersten Beobachtungen von Bakterien.
3

4 2. Optische Methoden
Aus dieser Zeit stammt auch die Entdeckung des Aufbaus der Zelle durch den eng-
lischen Physiker und Naturforscher Robert Hooke, der den Begriff Zelle pragt. 1669
stellt Isaac Newton die Emanationstheorie auf. Er postuliert, dass das Licht eine Teil-
chenstrahlung ist, der er zur Erklarung der Lichterscheinung bestimmte Eigenschaften
zuschreiben kann. Im Jahre 1677 stellt Christian Huygens die Undulationstheorie auf,
die die Lichtausbreitung als Wellenbewegung beschreibt. Diese Theorie wird erst 1802
von Thomas Young durch Interferenzversuche bestatigt. Im 18. Jahrhundert stagniert
die Entwicklung der Mikroskope, da die storenden Farbfehler der Linsen nicht beseitigt
werden konnen. Erst zu Beginn des 19. Jahrhunderts kommt es durch J. von Fraun-
hofer zu einer neuen Ara der technischen Optik. Er entdeckt bei der Zerlegung des
Sonnenlichts im Spektrum mehrere schwarze Linien, wohingegen das Licht von ganz
bestimmten Farbtonen im Spektrum fehlt. Aufgrund der Lage und der Verteilung der
Linien konnen diese den chemischen Elementen im außeren Teil der Sonne zugeord-
net werden. Auf seinen Arbeiten basiert die Berechnung von Okularen und Objektiven
[8, 9].
Ende des 19. Jahrhunderts gelingt es, optisches Glas zu schmelzen. Zuvor entwickelt E.
Abbe seine Theorie zur Bildentstehung im Mikroskop, die die Berechnung der Wirkung
eines abbildenden optischen Systems ermoglicht. Auf der Basis dieser Berechnungen
stellt O. Schott in Jena 1879 die ersten optischen Glaser her. Seit 1886 vertreibt C.
Zeiss auf den Berechnungen von Abbe basierende apochromatische Objektive, die im
Vergleich zu den Achromaten fur drei anstelle von zwei Farben des Spektrums korri-
giert sind. Damit tritt zunachst ein Abschluss in der Entwicklung der Mikroskopoptik
ein. Man hat mit den zur Verfugung stehenden Mitteln die theoretisch mogliche Auflo-
sungsgrenze erreicht. Eine Steigerung der Auflosungsgrenze ist nur durch Verwendung
kurzerer Wellenlangen moglich [9, 10].

2.1 Fluoreszenzmikroskopie 5
2.1 Fluoreszenzmikroskopie
Zur Erhohung der Auflosung bei der mikroskopischen Abbildung empfiehlt E. Abbe
1878 die Anwendung von ultraviolettem Licht. 1904 entwickelt A. Kohler die Ultra-
violettmikroskopie. Durch die Nutzung einer Lichtquelle mit einer Wellenlange von
260 nm kann das Doppelte des bisherigen Auflosungsvermogens erreicht werden. Aus
der unbedeutenden Ultraviolettmikroskopie leitet sich die Fluoreszenzmikroskopie ab.
Die ersten handelsublichen, mit Kohlebogenlampen ausgestatteten Fluoreszenzmikros-
kope bieten C. Reichert und O. Heimstadt 1911 in Wien und 1913 C. Zeiss und H.
Lehmann an. Die Untersuchungen beschranken sich auf pflanzliche Zellen und Gewe-
be, die Primarfluoreszenz aufweisen. In der 1929 von P. Ellinger eingefuhrten Vital-
mikroskopie wird das Praparat zum ersten Mal mit einem Fluorochrom behandelt.
1933 entwickelt M. Haitinger die Sekundarfluoreszenz, in der das zu untersuchende
Objekt mit einem Fluorochrom eingefarbt wird. Bei Bestrahlung mit UV-Licht emit-
tiert dieses Fluoreszenzlicht. Der als Fluorochromierung bezeichnete Vorgang macht die
Fluoreszenzmikroskopie auch fur die Zoologie interessant. Basierend auf dem Fluoro-
chrom Fluoresceinisothiocyanat (FITC), das N. H. Kaplan und A. H. Coon zusammen
entwickeln, wird 1941 die Immunofluoreszenz-Mikroskopie, eine spezielle Methode zur
Fluorochromierung von Antikorpern, eingefuhrt, die heute zu den Standardmethoden
gehort. Weitere Einflusse auf die Fluoreszenzmikroskopie haben E. M. Brumberg und
J. S. Ploem. Brumberg rustet das Fluoreszenzmikroskop mit Interferenzfiltern, so ge-
nannten dichromatischen Teilerspiegeln, aus. Ploem entwickelt die nach ihm benannte
epi-Beleuchtungseinrichtung (Ploemopak 1 und Ploemopak 2) mit austauschbaren di-
elektrischen Teilerspiegeln und Interferenzfiltern fur einfache sowie Mehrwellenlangen-
Fluoreszenzmikroskopie. 1960 werden die Kohlebogenlampen durch die heute ublichen
Quecksilber-Hochdrucklampen ersetzt [9, 11, 12, 13].

6 2. Optische Methoden
2.1.1 Physikalische und chemische Betrachtung der Fluores-
zenz
Die Fluoreszenz ist eine Art der moglichen Lichterscheinungen, die unter dem Oberbe-
griff Lumineszenz zusammengefasst werden. Die Lumineszenz beschreibt die Emission
von Licht im sichtbaren, im UV- und im IR-Spektralbereich basierend auf dem Elektro-
nenubergang von einem energetisch hoheren Zustand auf einen energetisch niedrigeren
Zustand. Die Art der Lumineszenz ist dabei von der Art der Anregung der Elektro-
nen eines Molekuls abhangig. Wird die zur Anregung notwendige Energie durch die
Absorption von Photonen, z. B. in Form von ultraviolettem oder blauem Licht, er-
halten, spricht man von Photolumineszenz. Diese wird unterteilt in Fluoreszenz und
Phosphoreszenz. Der Rahmen dieser Arbeit beschrankt sich auf die physikalischen und
chemischen Eigenschaften der Fluoreszenz. Weitere bekannte Arten der Lumineszenz,
die nicht behandelt werden, sind Chemo-, Bio-, Radio-, Mechano-, Thermo- oder Elekt-
rolumineszenz [13, 14].
Ein Molekul kann durch Absorption von Photonen von dem Singulett-Grundzustand
(S0) in eines der Schwingungsniveaus der angeregten Zustande (S1 oder S2) ubergehen.
Der Ubergang eines Elektrons in ein energetisch hoheres Orbital fuhrt zu einer ver-
anderten Ladungsverteilung in dem Molekul. Die Zeit der Absorption liegt innerhalb
von 10−15 Sekunden, wobei sich gemaß dem Franck-Condon-Prinzip die Kernabstande
nicht wesentlich andern. Die Ubergange zwischen dem S0-Zustand und den Singulett-
und Triplettzustanden sind schematisch in dem Jablonski-Diagramm in Abbildung 2.1
dargestellt. Die Ruckkehr des Elektrons in den S0-Zustand kann durch verschiedene
Desaktivierungsprozesse, z. B. uber Emissionsmechanismen oder strahlungslose Me-
chanismen, erfolgen, in denen die zusatzliche Energie an die Umgebung abgegeben
wird. Zu den Emissionsmechanismen zahlen die Fluoreszenz und die Phosphoreszenz,
wahrend die innere Umwandlung (engl.: internal conversion (IC)), die Schwingungsre-
laxation (engl.: vibrational relaxation (VR)) und der Interkombinationsubergang (engl.:
intersystem crossing (ISC)) zu den strahlungslosen Mechanismen gehoren [13, 15, 16].

2.1 Fluoreszenzmikroskopie 7
�������
��
�
�
��
��
��
�������� ���
��
���������
��
� �
� �
� � ������
�������
��
��
Abbildung 2.1: Jablonski Diagramm. VR = Schwingungsrelaxation; IC = Innere
Umwandlung; ISC = Interkombinationsubergang [15, 16].
Bei dem Interkombinationsubergang wechselt das Molekul von einem angeregten Singu-
lettzustand unter Spinumkehr in einen angeregten Triplettzustand. Unter Schwingungs-
relaxation fallt das Molekul auf das geringste Schwingungsniveau des T1-Zustands, von
dem aus die Ruckkehr in den Grundzustand unter Emission eines Photons moglich ist.
Die Ruckkehr von dem T1-Zustand in den Grundzustand wird Phosphoreszenz genannt.
Diese Art der Photolumineszenz soll hier jedoch nicht naher erlautert werden [13].
Bei der inneren Umwandlung erfolgt der strahlungslose Ubergang von einem hoheren
elektronischen Zustand in einen energetisch niedrigeren Zustand ohne Veranderung der
Spinmultiplizitat. Durch Kollision mit anderen Molekulen, z. B. den Losemittelmoleku-
len, wird bei der Schwingungsrelaxation die Schwingungsenergie in Warme umgewan-
delt. Dabei relaxiert das angeregte Molekul in ungefahr 10−11 Sekunden auf das kleinste
Schwingungsniveau des ersten angeregten Zustands bzw. auf das Schwingungsniveau,
das nach der Boltzmann-Verteilung dem thermischen Gleichgewicht entspricht. Die
Schwingungsrelaxation kann auch von dem S1(ν′ = 0)-Zustand in den Grundzustand

8 2. Optische Methoden
erfolgen. Dieser Ubergang ist jedoch aufgrund der relativ großen Energiedifferenz zwi-
schen dem S1- und dem S0-Zustand wesentlich langsamer. Die Geschwindigkeit dieses
Prozesses liegt in der Großenordnung der Geschwindigkeit der Fluoreszenz [13, 16, 17].
Das Phanomen der Fluoreszenz entsteht durch den Ubergang eines Molekuls von dem
angeregten Singulettzustand S1(ν′ = 0) unter Emission eines Photons in den S0-
Zustand. Die Lebensdauer des angeregten S1-Zustands und damit auch die Abklingzeit
der Fluoreszenz betragt ungefahr 10−9 bis 10−8 Sekunden. Nach dem Gesetz von Sto-
kes ist die Wellenlange der Emission aufgrund der geringeren Energie des emittierten
Photons langer als die der Anregung. Die Wellenlange der Emission und somit die
Quantenenergie des emittierten Photons hangt ausschließlich von dem Energieunter-
schied zwischen dem S1- und dem S0-Zustand und nicht von der Quantenenergie des
absorbierten Photons ab (Stokes-Fluoreszenz). Die Anti-Stokes-Fluoreszenz stellt eine
Ausnahme dar, in der die Energie der Emission hoher ist als die der Anregung. Das
Molekul ist bereits in einem hoheren Schwingungsenergieniveau, bevor es das Photon
absorbiert, oder die zusatzlich benotigte Energie wird durch thermische Energie zuge-
fuhrt. Die Emission von Photonen mit der gleichen Energie wie der der absorbierten
Photonen heißt Resonanz-Fluoreszenz. Diese wird jedoch nur in Festkorpern oder Ga-
sen, aber niemals in Flussigkeiten beobachtet [13].
Substanzen, die Fluoreszenzlicht ausstrahlen konnen, werden Fluorochrome oder Fluo-
rophore genannt. Fluorochrome sind alle Farbstoffe, die ein nicht fluoreszierendes Ob-
jekt fluoreszieren lassen, wahrend Fluorophore lediglich Atomgruppierungen organi-
scher Verbindungen sind, die die Fluoreszenz ermoglichen. Die Fluoreszenz wird in
zwei Erscheinungen unterteilt, der Primarfluoreszenz und der Sekundarfluoreszenz. Pri-
marfluoreszenz ist die Eigenschaft einiger Substanzen, von sich aus bei Anregung mit
kurzwelliger Strahlung Fluoreszenzlicht zu emittieren. Diese Art der Fluoreszenz wird
auch als Eigen- oder Autofluoreszenz bezeichnet. Bei der Autofluoreszenz handelt es
sich um eine ungewollte Fluoreszenzerscheinung, die z. B. bei mikroskopischen Objekti-
ven auftreten kann. Da die meisten zu untersuchenden Objekte keine Primarfluoreszenz
2.1 Fluoreszenzmikroskopie 9
aufweisen, werden Fluorochrome zum Farben bestimmter Objektstrukturen verwendet.
Dieser Farbeprozess wird Fluorochromierung genannt und die so auftretende Fluores-
zenz wird als Sekundarfluoreszenz bezeichnet [12].
� � � � � � � � � � � � �
�����
����
� �
� �
� � � � � � � � � � � � � � � �
� �
� � � �
� � � �
� � � ! � � � � � � � " � � � � � � �
���#��$������������
������������
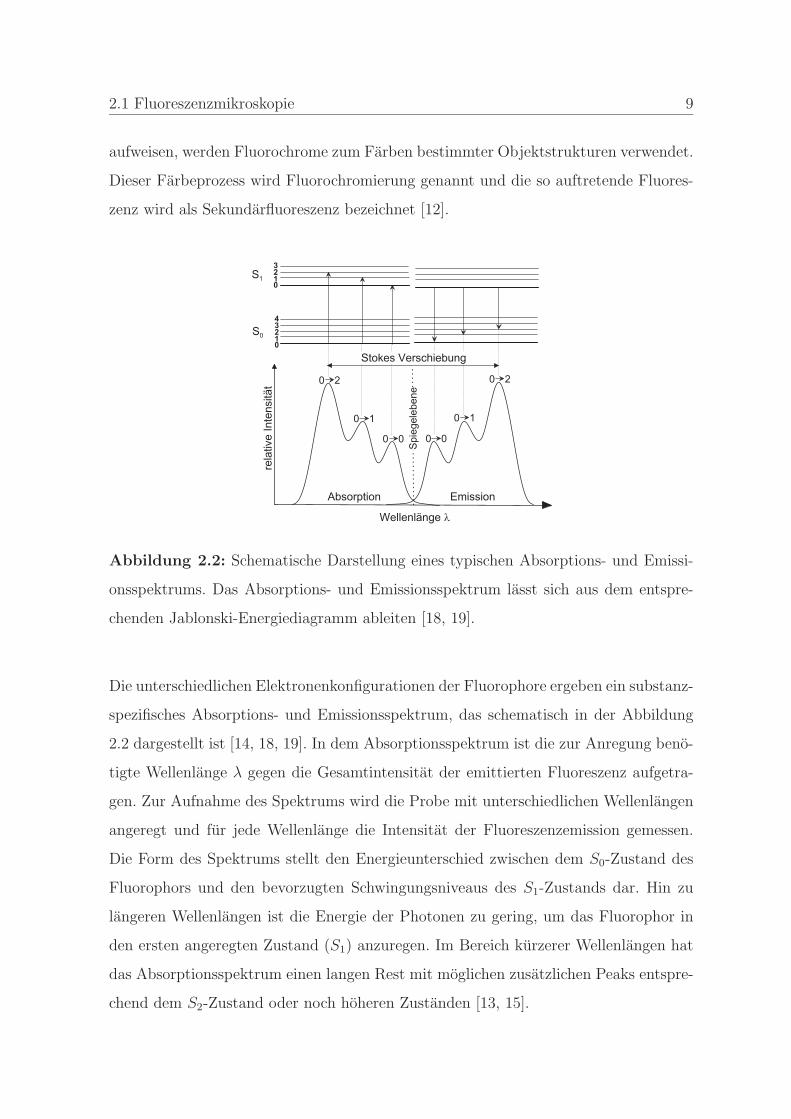
Abbildung 2.2: Schematische Darstellung eines typischen Absorptions- und Emissi-
onsspektrums. Das Absorptions- und Emissionsspektrum lasst sich aus dem entspre-
chenden Jablonski-Energiediagramm ableiten [18, 19].
Die unterschiedlichen Elektronenkonfigurationen der Fluorophore ergeben ein substanz-
spezifisches Absorptions- und Emissionsspektrum, das schematisch in der Abbildung
2.2 dargestellt ist [14, 18, 19]. In dem Absorptionsspektrum ist die zur Anregung beno-
tigte Wellenlange λ gegen die Gesamtintensitat der emittierten Fluoreszenz aufgetra-
gen. Zur Aufnahme des Spektrums wird die Probe mit unterschiedlichen Wellenlangen
angeregt und fur jede Wellenlange die Intensitat der Fluoreszenzemission gemessen.
Die Form des Spektrums stellt den Energieunterschied zwischen dem S0-Zustand des
Fluorophors und den bevorzugten Schwingungsniveaus des S1-Zustands dar. Hin zu
langeren Wellenlangen ist die Energie der Photonen zu gering, um das Fluorophor in
den ersten angeregten Zustand (S1) anzuregen. Im Bereich kurzerer Wellenlangen hat
das Absorptionsspektrum einen langen Rest mit moglichen zusatzlichen Peaks entspre-
chend dem S2-Zustand oder noch hoheren Zustanden [13, 15].

10 2. Optische Methoden
In dem Emissionsspektrum ist die relative Intensitat des emittierten Lichtes zu dessen
Wellenlange aufgetragen. Es zeigt die Fluoreszenzintensitatsverteilung bei der Anre-
gung mit einer bestimmten Wellenlange. Nach dem Stokesschen Gesetz sind die Wel-
lenlangen der Emission langer als die der Anregung. Der Hochstwert entspricht dem
Ubergang von dem geringsten Schwingungsniveau des S1-Zustands in das bevorzugte
Schwingungsniveau des S0-Zustands. Hin zu kurzeren Wellenlangen fallt das Spektrum
steil ab, da die Energie der emittierten Photonen nicht hoher sein kann als die Dif-
ferenz zwischen den geringsten Schwingungsniveaus von S1 und S0. Im langerwelligen
Bereich hat das Spektrum einen langen Rest, da die Fluoreszenz fast immer durch den
Ubergang des geringsten Schwingungsniveaus von S1 zu dem geringsten Schwingungs-
niveaus von S0 entsteht.
Beide Spektren entsprechen Wahrscheinlichkeitsverteilungen. Das Absorptionsspektrum
gibt die Wahrscheinlichkeit wieder, mit der ein Photon einer bestimmten Quanten-
energie absorbiert wird und Fluoreszenz erzeugt. Das Emissionsspektrum stellt die
Wahrscheinlichkeit dar, mit der ein Photon einer bestimmten Quantenenergie emit-
tiert wird. Der Spiegelbildcharakter der Spektren resultiert aus der Verknupfung der
Wahrscheinlichkeit, mit der ein Elektron in ein bestimmtes Schwingungsenergieniveau
des S0-Zustands zuruckfallt, mit der Wahrscheinlichkeit, mit der sich das Elektron
vor der Anregung in einer bestimmten Position des S0-Zustands aufgehalten hat. Die
Differenz zwischen den beiden Intensitatsmaxima wird Stokes Verschiebung genannt.
Dieser Unterschied in den Wellenlangen ist die Grundlage fur die Beobachtung von
Fluoreszenz mittels der Fluoreszenzmikroskopie [13, 15, 19].
2.1.2 Auflicht-Fluoreszenzmikroskopie
Es gibt zwei Arten der Fluoreszenzmikroskopie, die Durchlicht-(dia)- und die Auflicht-
(epi)-Fluoreszenzmikroskopie, die sich nur durch die Anordnung der Lichtquelle un-
terscheiden [14]. Bei der hier vorgestellten Methode handelt es sich um die Auflicht-
Fluoreszenzmikroskopie. In Abbildung 2.3 sind die Komponenten eines Auflicht-Fluores-
zenzmikroskops inklusive des Beleuchtungsstrahlengangs und des Abbildungsstrahlen-

2.1 Fluoreszenzmikroskopie 11
gangs dargestellt. Die Anordnung der Komponenten entspricht der von A. Kohler ent-
wickelten Beleuchtungsanordnung, bei der ein optisch erzeugtes homogenes Lichtfeld
entscheidend ist.
�
%
& '
(
) * � � � � # � � � �
� %
� &
� '
� +
� )
� (
� *
�
Abbildung 2.3: Schematische Darstellung eines Auflicht-Fluoreszenzmikroskops nach
dem Kohlerschen Beleuchtungsverfahren [14, 13].
1 Universal-Mikroskop; 2 Objekttisch mit Probentrager; 3 Objektivrevolver mit
Objektiv; 4 Lampenhaus; 5 Fluoreszenzilluminator mit Filterblock; 6 Trinoku-
lar; 7 Anschlussmoglichkeit fur weitere Beleuchtung; 8 Hilfsspiegel; 9 Quecksilber-
Hochdrucklampe; 10 Kollektorlinse; 11 Hilfslinsen; 12 Leuchtfeldblende; 13 Erreger-
filter; 14 Sperrfilter; 15 Dichromatischer Teilerspiegel; 16 Objektivlinse; 17 Probe; 18
Sehfeldblende mit Zwischenbildebene; 19 Okularlinse; 20 Auge oder Kamera; grun:
Abbildungsstrahlengang; blau: Beleuchtungsstrahlengang.
Das von der Lichtquelle ausgesandte kurzwellige Licht wird zunachst von einer Kollek-
torlinse (10) auf die Leuchtfeldblende (12) gelenkt, die die Große des Beleuchtungsfeldes

12 2. Optische Methoden
bestimmt. Dadurch entspricht das Beobachtungsfeld dem Beleuchtungsfeld, so dass die
Belichtung außerhalb dieses Bereichs liegender Teile durch Streulicht verhindert und
der Kontrast verbessert wird. Von der Leuchtfeldblende wird das Licht auf das Erre-
gerfilter (13) gelenkt. Es trifft dann auf einen dichromatischen Teilerspiegel (15), der
das Licht zu dem Objektiv (16) lenkt. Das Objektiv dient hierbei zusatzlich als Kon-
densor, der die Anregungsstrahlung im Objektfeld zentriert, die zum Teil vom Objekt
absorbiert und als langerwelliges Fluoreszenzlicht emittiert wird. Das emittierte Licht
wird im Objektiv gesammelt und wieder auf den dichromatischen Teilerspiegel gelei-
tet. Da in der Fluoreszenzmikroskopie die Probe selbst als neue Lichtquelle fungiert, ist
nur die Apertur des Objektivs fur die Kohlersche Beleuchtung von Bedeutung, so dass
die maximale Auflosung von dem verwendeten Objektiv abhangig ist. Der dichromati-
sche Teilerspiegel lasst das Fluoreszenzlicht durch, das anschließend auf ein Sperrfilter
(14) trifft. Dieses absorbiert gegebenenfalls das restliche vom Objekt reflektierte An-
regungslicht, so dass ausschließlich das emittierte Licht zum Okular (19) geleitet wird.
Auf diese Weise ergibt sich ein farbiges Bild auf dunklem Untergrund, wobei die Farbe
von der Art der Fluoreszenz abhangig ist [12, 14, 20].
Im Vergleich zu der Durchlicht-Fluoreszenzmikroskopie besteht durch die gleichzeitige
Nutzung des Objektivs als Kondensor bei der Auflicht-Fluoreszenzmikroskopie der Vor-
teil nur einer optischen Achse. Das erleichtert die Handhabung beim Fokussieren des
Lichtes auf die Probe und verhindert ein Ausbleichen der Probe durch Streulicht. Der
Einsatz hochaperturiger Immersionsobjektive fuhrt zu helleren Fluoreszenzbildern als
bei der Durchlicht-Methode. Außerdem lasst sich die Auflicht-Fluoreszenzmikroskopie
mit anderen Methoden, wie zum Beispiel mit der Phasenkontrast-, Dunkelfeld- oder
Hellfeld-Mikroskopie, zu Simultan- oder Alternativverfahren kombinieren. Bei der Ver-
wendung von Auflicht ist es ferner moglich, auch lichtundurchlassige Proben, wie zum
Beispiel Si-Wafer, zu untersuchen. Die Nachteile der Auflicht-Fluoreszenzmikroskopie
beschranken sich auf die hoheren Kosten, z. B. fur die notwendige Beleuchtungseinrich-
tung, die zu der kostenintensiveren Mikroskopieausstattung zahlt, und fur die Objektive
mit einer hohen numerischen Apertur und einem geringen Arbeitsabstand [13, 14, 20].
2.1 Fluoreszenzmikroskopie 13
Wie aus der Abbildung 2.3 hervorgeht, zahlen die Lichtquelle, die verschiedenen Filter
im Strahlengang und die verwendeten Objektive zu den wichtigsten Komponenten der
Auflicht-Fluoreszenzmikroskopie. Im Nachfolgenden wird daher kurz auf die charakte-
ristischen Eigenschaften einiger dieser Komponenten eingegangen.
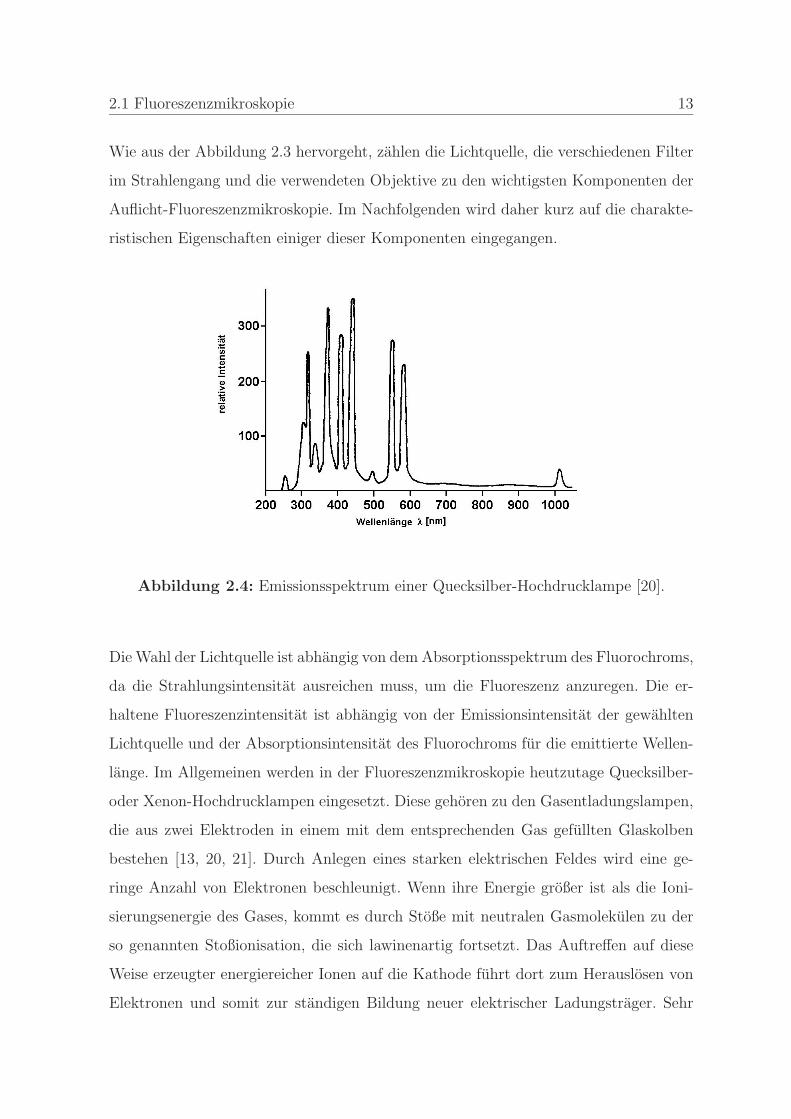
Abbildung 2.4: Emissionsspektrum einer Quecksilber-Hochdrucklampe [20].
Die Wahl der Lichtquelle ist abhangig von dem Absorptionsspektrum des Fluorochroms,
da die Strahlungsintensitat ausreichen muss, um die Fluoreszenz anzuregen. Die er-
haltene Fluoreszenzintensitat ist abhangig von der Emissionsintensitat der gewahlten
Lichtquelle und der Absorptionsintensitat des Fluorochroms fur die emittierte Wellen-
lange. Im Allgemeinen werden in der Fluoreszenzmikroskopie heutzutage Quecksilber-
oder Xenon-Hochdrucklampen eingesetzt. Diese gehoren zu den Gasentladungslampen,
die aus zwei Elektroden in einem mit dem entsprechenden Gas gefullten Glaskolben
bestehen [13, 20, 21]. Durch Anlegen eines starken elektrischen Feldes wird eine ge-
ringe Anzahl von Elektronen beschleunigt. Wenn ihre Energie großer ist als die Ioni-
sierungsenergie des Gases, kommt es durch Stoße mit neutralen Gasmolekulen zu der
so genannten Stoßionisation, die sich lawinenartig fortsetzt. Das Auftreffen auf diese
Weise erzeugter energiereicher Ionen auf die Kathode fuhrt dort zum Herauslosen von
Elektronen und somit zur standigen Bildung neuer elektrischer Ladungstrager. Sehr

14 2. Optische Methoden
große Entladungsstrome bewirken eine so starke Erhitzung der Elektroden, dass die
Elektronen aus der Kathode durch Gluhemission herausgelost werden, woraus die Bo-
genentladung resultiert [22, 23].
Das Emissionsspektrum einer herkommlichen Quecksilber-Hochdrucklampe ist in der
Abbildung 2.4 wiedergegeben. Es besteht aus einem Grundkontinuum, das im ultravio-
letten, sichtbaren und infraroten Spektralbereich etwa die gleiche Starke aufweist. Die
charakteristischen Spektrallinien sind in der Tabelle 2.1 wiedergegeben.
Tabelle 2.1: Die charakteristischen Spektrallinien einer Quecksilber-Hochdrucklampe
mit ihren relativen Intensitaten bezogen auf die Linie bei 546,1 nm [13].
Wellenlange [nm] rel. Intensitat [%] Wellenlange [nm] rel. Intensitat [%]
365,0 ultraviolett 0,74 435,8 blau 1,40
365,5 ultraviolett 0,17 546,1 grun 1,00
366,3 ultraviolett 0,12 577,0 gelb 0,14
404,7 violett 0,74 579,0 gelb 0,15
Aufgrund der großen Strahlungsintensitat im ultravioletten, violetten sowie blauen
Spektralbereich werden Quecksilberlampen als Universallampe fur die Fluoreszenzmi-
kroskopie eingesetzt, wahrend Xenonlampen nur bei Blau- oder Grunanregung verwen-
det werden [13, 14, 20].
Neben den spektralen Eigenschaften des Fluorochroms und der gewahlten Lichtquelle
ist die Verwendung der passenden Filter zur Trennung bestimmter Wellenlangen des
Lichtes fur die Gute der Fluoreszenzmikroskopie entscheidend. Wie aus der Abbildung
2.5 hervorgeht, umfasst weißes Licht alle Spektralfarben. Diese konnen mit Hilfe ei-
nes Prismas sichtbar gemacht und entsprechend ihrer Wellenlangen (in Nanometer)
unterteilt werden.

2.1 Fluoreszenzmikroskopie 15
� � �
� � , �� � # �� � � � #
- � � . � � �/ � " � �
� � # � � �� � � �
$ � � � � � �
� � � � " � � � # �
���
��,�
��#�
$������
� � " � � � # � � � � � " � � � # �
� � � � # $ � � � � � � � � 0 � # � � �
��#���
% � � & � � ' � � + � � ( � � ) � �
����
Abbildung 2.5: Dispersion des Lichtes [14].
Der Farbbereich erstreckt sich von violett uber blau, grun, gelb, orange bis rot. Der
ultraviolette Bereich liegt kurzwellig vom violetten Bereich und ist genau wie der in-
frarote Bereich, der langwellig vom roten Bereich liegt, fur das menschliche Auge nicht
sichtbar [14].
In der Fluoreszenzmikroskopie wird eine Vielzahl von unterschiedlichen Filtern ein-
gesetzt, die durch ihre Durchlassigkeitskurve (Abbildung 2.6) charakterisiert werden.
Die Durchlassigkeit τ ist definiert als das Verhaltnis der Lichtintensitat It, nachdem
das Licht das Filter passiert hat, zu der Intensitat I des einfallenden Lichtes:
τ =It
I. (2.1)
Fur das Absorptionsvermogen D eines Filters, das auch als optische Dichte bezeichnet
wird, gilt:
D = log1
τ= − log τ. (2.2)
In der Abbildung 2.6 sind die Durchlassigkeitskurven eines Erregerfilters und eines
Sperrfilters wiedergegeben. Die gepunktete Kurve reprasentiert das Emissionsspektrum
des Fluoreszenzlichtes, das im Bereich der Durchlassigkeitskurve des Sperrfilters liegen
muss. Im Idealfall sollten die Kurven des Erreger- und des Sperrfilters nicht uberlappen,
da eine Uberlappung aus folgenden Grunden in der Praxis zu Problemen fuhren kann:

16 2. Optische Methoden
1��"�������!�
����
� ��������
� � � � � � � � � � � � � � 2 � 3� � �
� � � � � � � 0 � � � � �
� � � � � 0 � � � � �
� � � � � � � � � � � � � � � � � �
Abbildung 2.6: Durchlassigkeitskurve eines Erreger- und Sperrfilters [12].
- Viele fluoreszierende Substanzen haben die Emissionsbanden in dem Spektralbe-
reich, der zum Teil mit dem wirksamsten Anregungsbereich uberlappt.
- Das Emissionszentrum vieler Fluorochrome liegt sehr nah an dem Anregungsma-
ximum [12].
Aus der Form der Durchlassigkeitskurve ergeben sich die Bezeichnungen der eingesetz-
ten Filter wie Kurzpass, Langpass, Breitband oder Schmalband. Um bei einem Filter ei-
ne selektive Durchlassigkeit von einem bestimmten Wellenlangenbereich zu gewahrleis-
ten, stehen verschiedene optische Instrumente als Bausteine zu Verfugung: Farbglaser,
Gelantine- oder Flussigkeitsfilter, Interferenzfilter oder Teilerspiegel oder verschiedene
Prismen [13].
Farbglaser, deren Farbe durch den Zusatz von Ionen mit einer homogenen Verteilung
im Glas zustande kommt, eignen sich gut als Erregerfilter. Sie lassen bevorzugt Licht in
einem bestimmten, sehr schmalen Wellenlangenbereich durch. Hingegen werden Farb-
glaser, deren Farbe durch submikroskopische Kristalle von Sulfiden und Seleniden ent-
steht, aufgrund der hohen Durchlassigkeit im langwelligen Bereich und einer geringen
Durchlassigkeit im kurzwelligen Bereich als Sperrfilter eingesetzt [13, 20].

2.1 Fluoreszenzmikroskopie 17
Interferenzfilter bestehen aus zwei semitransparenten Spiegeloberflachen, die durch eine
dunne Schicht eines Dielektrikums getrennt sind. Die Interferenz des einfallenden Lich-
tes kommt durch mehrfache Reflexion an den Spiegeloberflachen zustande. Das fuhrt
zu der Durchlassigkeit von Licht der gewunschten Wellenlange und dem ganzzahligen
Vielfachen der Wellenlange erster Ordnung, wahrend andere Wellenlangen reflektiert
werden. Die Dicke und der Brechungsindex der dielektrischen Schicht, die Anzahl der
Schichten sowie ihre Anordnung bestimmen die Eigenschaften des Durchlassigkeitsbe-
reiches und die Menge an Hintergrunddurchlassigkeit. Die Bandbreite und der maxi-
male Wert der Durchlassigkeit hangen großtenteils von den optischen Eigenschaften
der reflektierenden Schichten ab. In der Praxis werden Interferenzfilter typischerweise
als dichromatischer Teilerspiegel eingesetzt [13, 20].
Der dichromatische Teilerspiegel, der auch als Reflexions-Kurzpassfilter, Farbteiler oder
Reflektor bezeichnet wird, spielt eine Schlusselrolle in der Auflicht-Fluoreszenzmikros-
kopie. Die Eigenschaften dieses Interferenzfilters werden gewohnlich fur Auflicht bei
45◦ angegeben. Die Filter sind in den Filterblocken so eingesetzt, dass sie im Anre-
gungslicht als Spiegel dienen und somit das Licht unter 45◦-Einfall in Richtung des
Praparates lenken, wahrend sie bei Beobachtung der Fluoreszenz als Durchlassfilter
verwendet werden [14, 20, 24].
Kurzpassfilter sind Interferenzfilter mit einer steilen Kante in einem bestimmten Spek-
tralbereich, wobei die Sperrung im langwelligen Licht erfolgt, d. h., sie weisen eine
Durchlassigkeit im kurzwelligen Bereich auf. Sie werden als Erregerfilter benutzt. Lang-
passfilter haben ebenfalls eine steile Kante in einem bestimmten Spektralbereich, aller-
dings erfolgt die Sperrung im kurzwelligen Bereich. Sie haben somit eine hohe Durchlas-
sigkeit im langwelligen Bereich und werden bevorzugt als Sperrfilter eingesetzt [13, 20].
Die Kombination aus Kurzpass- und Langpassfilter ergibt einen Bandpassfilter, wie
in Abbildung 2.7 dargestellt. Die Verteilung der Durchlassigkeit dieses Filters ent-
spricht einer Gaußschen Glockenkurve mit der hochsten Durchlassigkeit τmax bei einer
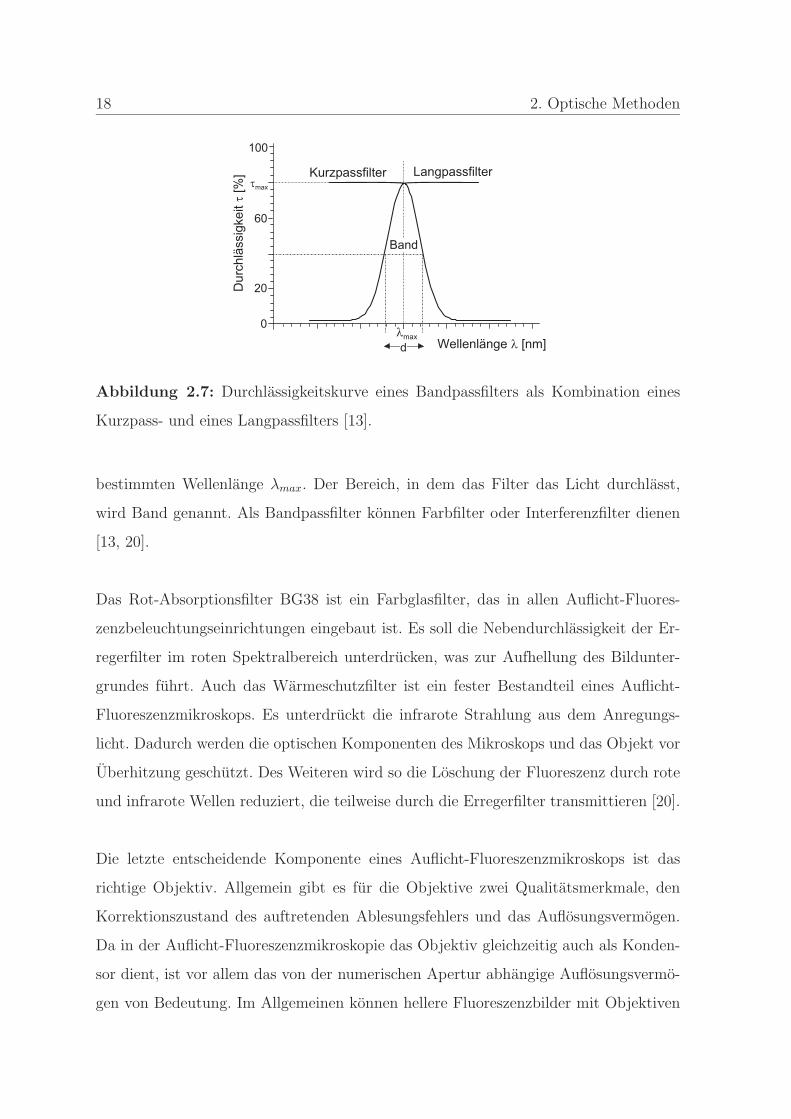
18 2. Optische Methoden
� # 4
5 � � � � � � � � � � � � � � 2 � 3
�
�
+ �
� � �
1��"�������!�
���� �26
3
� # 4
/ # � � � # � � 0 � � � � �7 � � � � # � � 0 � � � � �
8 # � 5
Abbildung 2.7: Durchlassigkeitskurve eines Bandpassfilters als Kombination eines
Kurzpass- und eines Langpassfilters [13].
bestimmten Wellenlange λmax. Der Bereich, in dem das Filter das Licht durchlasst,
wird Band genannt. Als Bandpassfilter konnen Farbfilter oder Interferenzfilter dienen
[13, 20].
Das Rot-Absorptionsfilter BG38 ist ein Farbglasfilter, das in allen Auflicht-Fluores-
zenzbeleuchtungseinrichtungen eingebaut ist. Es soll die Nebendurchlassigkeit der Er-
regerfilter im roten Spektralbereich unterdrucken, was zur Aufhellung des Bildunter-
grundes fuhrt. Auch das Warmeschutzfilter ist ein fester Bestandteil eines Auflicht-
Fluoreszenzmikroskops. Es unterdruckt die infrarote Strahlung aus dem Anregungs-
licht. Dadurch werden die optischen Komponenten des Mikroskops und das Objekt vor
Uberhitzung geschutzt. Des Weiteren wird so die Loschung der Fluoreszenz durch rote
und infrarote Wellen reduziert, die teilweise durch die Erregerfilter transmittieren [20].
Die letzte entscheidende Komponente eines Auflicht-Fluoreszenzmikroskops ist das
richtige Objektiv. Allgemein gibt es fur die Objektive zwei Qualitatsmerkmale, den
Korrektionszustand des auftretenden Ablesungsfehlers und das Auflosungsvermogen.
Da in der Auflicht-Fluoreszenzmikroskopie das Objektiv gleichzeitig auch als Konden-
sor dient, ist vor allem das von der numerischen Apertur abhangige Auflosungsvermo-
gen von Bedeutung. Im Allgemeinen konnen hellere Fluoreszenzbilder mit Objektiven

2.2 Dunkelfeldmikroskopie 19
hoherer numerischer Apertur erreicht werden, durch die viel Anregungslicht auf dem
Praparat konzentriert wird. In der Auflicht-Fluoreszenzmikroskopie sind nicht nur die
Lichtstarke und die Anregungsstarke, sondern auch die Helligkeit der Objektfluoreszenz
und dadurch die Helligkeit des Fluoreszenzbildes proportional zur vierten Potenz der
Objektivapertur, wodurch auch schwachere Vergroßerungen hellere Bilder ergeben. Ein
weiteres wichtiges Kriterium der in der Auflicht-Fluoreszenzmikroskopie eingesetzten
Objektive ist, dass sie frei von Autofluoreszenz sind [14, 20].
2.2 Dunkelfeldmikroskopie
Die Methode der Dunkelfeldmikroskopie ist bereits seit Beginn des 19. Jahrhunderts
bekannt. 1837 macht J. B. Reade auf sie aufmerksam. Er spricht von einer Hintergrund-
beleuchtung. Im Jahre 1850 baut F. H. Wenham den ersten konzentrischen paraboli-
schen Dunkelfeldkondensor. Zu Beginn des 20. Jahrhunderts entwickeln Siedentopf und
Zsigmondy die Spaltultramikroskopie, die einen Spezialfall der Dunkelfeldmikroskopie
darstellt. Hierbei wird das Beobachtungslicht senkrecht zur Beobachtungsrichtung auf
die Probe eingestrahlt. Siedentopf konstruiert 1910 bei Zeiss in Jena den heute bekann-
ten Kardioidkondensor [25].
Die Dunkelfeldmikroskopie gehort zu den Methoden der Kontrastmikroskopie, die ei-
ne Vergroßerung des Unterschiedes in der Lichtintensitat zwischen dem Objekt und
seiner Umgebung ermoglichen. Die Differenz zwischen den Lichtintensitaten kleiner,
dicht beieinander liegender Objektstrukturen, die gerade noch von einem Lichtempfan-
ger, wie zum Beispiel dem Auge oder einer CCD-Kamera, wahrnehmbar ist, wird als
Unterschiedsschwelle bezeichnet. Die Differenz der Lichtintensitaten I oberhalb dieses
Schwellenwertes ist der Kontrast K, der durch die Gleichung 2.3 beschrieben werden
kann:
K =IUmfeld − IObjekt
IUmfeld + IObjekt
. (2.3)

20 2. Optische Methoden
� �
�
� � � �
� � �
1 � " ! � � # �9 � : � ! � � � � � � �9 � : � ! �
/ � " � � ; � � � � �
Abbildung 2.8: Strahlengang eines Kardioidkondensors [12].
Charakteristisch fur die Dunkelfeldmikroskopie sind ein dunkles Umfeld und helle Ob-
jekte. Im Idealfall ist daher die Intensitat des Umfeldes Null oder nahe Null, wodurch
sich ein Wert von −1 fur den Kontrast ergibt. Dieses charakteristische Dunkelfeldbild
entsteht nicht durch Einstrahlung von direktem Licht auf die Probe, sondern durch
indirektes Licht, das durch Brechung, Beugung oder Reflexion an dem Objekt in das
Objektiv gelangt. Hierfur werden spezielle Dunkelfeldkondensoren verwendet. Zu die-
sen gehoren in der Durchlicht-Dunkelfeldmikroskopie der Paraboloidkondensor und der
Kardioidkondensor, die sich in der Art der Strahlenfuhrung unterscheiden. Die großere
Bedeutung hat der Kardioidkondensor, dessen Strahlengang in Abbildung 2.8 darge-
stellt ist [12, 21]. Er besteht aus zwei Glasspiegeln, von denen der erste eine spharische
Oberflache besitzt und der zweite eine Kardioide. Dadurch tritt das vom Konden-
sor kommende Licht in Form eines Kegelmantels mit einer inneren und einer außeren
Begrenzung hervor, woraus eine innere und eine außere Kondensorapertur resultiert
[12, 26].
2.2.1 Bildentstehung im Mikroskop und das Auflosungsver-
mogen
Airy berucksichtigt als Ausgangsbasis fur die Bildentstehung im Mikroskop nicht mehr
nur die geometrische Optik, sondern vielmehr die Wellentheorie des Lichtes, so dass

2.2 Dunkelfeldmikroskopie 21
das mikroskopische Bild als das Ergebnis von Interferenzvorgangen zu betrachten ist.
Dafur zerlegt er das Objekt in seine einzelnen Punkte und untersucht deren Abbil-
dung. Das Gesamtbild ergibt sich durch Zusammensetzen der einzelnen Bilder unter
Berucksichtigung der Koharenzverhaltnisse. Die Airysche Betrachtungsweise setzt die
Unterscheidung der Objekte in Selbstleuchter und Nichtselbstleuchter voraus [21, 27].
�
< � = �
= �
= �
� �
�
9 � 9 � < � 9 !
� � =# �
� �
Abbildung 2.9: Das Airysche Beugungsscheibchen. a) Die Entstehung des Beugungs-
scheibchens eines selbst leuchtenden Punktes nach Airy. b) Das Bild des Beugungs-
scheibchens und dessen Intensitatsverteilung [21]. Σ Elementarwelle, Σ′ Wellenfront,
Oe Objektebene, Ob Objektiv, Ze Zwischenbildebene, Ok Okular, I Intensitat.
Das Bild eines selbst leuchtenden Punktes erscheint als eine flachenhaft ausgebreitete
Lichtverteilung, die durch Lichtbeugung und Interferenzvorgange entsteht, dem Beu-
gungsscheibchen (siehe Abbildung 2.9).
Die von einem selbst leuchtenden Punkt ausgehende Wellenfront Σ sollte idealerweise
bei einem aberrationsfreien Objektiv als entsprechende Kugelflache Σ′ im Bildraum
erscheinen. Nach dem Huygensschen Prinzip kann jeder Punkt dieser Wellenfront das

22 2. Optische Methoden
Ausgangszentrum einer neuen Elementarwelle sein. In der Zwischenbildebene Ze kon-
nen die Elementarwellen aufgrund ihrer Koharenz interferieren. A′0 ist das Bild des Mit-
telpunktes der ursprunglichen Wellenfront Σ. Die Lichtamplitude in diesem Punkt ist
die Summe der Amplituden aller dort ankommenden Elementarwellen. Die entsprechen-
de Lichtintensitat ist proportional zum Quadrat der Amplitude. Die Elementarwellen,
die in jedem anderen beliebigen Punkt A′1 in der Zwischenbildebene zusammenkommen,
haben unterschiedliche Phasen. Zum Beispiel kommt es bei entgegengesetzten Phasen,
wie es in der Abbildung 2.9 dargestellt ist, zur Ausloschung. Das Beugungsscheibchen
ergibt sich aus den Betragen der Intensitaten aller Punkte in der Zwischenbildebene.
Die von den einzelnen Punkten eines Selbstleuchters ausgesandten Elementarwellen
sind untereinander nicht koharent, so dass es nicht zur Interferenz kommt. Folglich
ergibt sich das Gesamtbild eines Selbstleuchters nach der Airyschen Betrachtungsweise
aus der Summation der einzelnen Beugungsfiguren. Die Intensitat entspricht der Sum-
me der Amplitudenquadrate [21, 27].
Selbstleuchter kommen in der Mikroskopie eher selten vor. Die meisten mikroskopischen
Objekte sind Nichtselbstleuchter, die zur Sichtbarmachung im Mikroskop mit einer
fremden Lichtquelle bestrahlt werden mussen. Die Bildentstehung bei Nichtselbstleuch-
tern ist wesentlich komplizierter als bei den Selbstleuchtern. Zur Vereinfachung wird
angenommen, dass ein nicht selbst leuchtendes Objekt aus einer sehr großen Entfernung
von einer punktformigen Lichtquelle beleuchtet wird, so dass die von der Lichtquelle
ausgehende Elementarwelle senkrecht auf das Objekt trifft. Somit sind alle von dem
Objekt ausgehenden Elementarwellen untereinander koharent. In der Abbildung 2.10
ist die Entstehung des Bildes eines nicht selbst leuchtenden Objektes dargestellt. Die
in der Zwischenbildebene ankommenden Elementarwellen erzeugen aufgrund der Ko-
harenz fur jeden Punkt die gleiche Amplitudenverteilung. Somit erzeugt jeder Punkt
des Objektes ein Beugungsscheibchen, welche in der Zwischenbildebene miteinander
interferieren. Die Intensitat des daraus resultierenden Beugungsscheibchens ist dem
Quadrat der Summe aller Amplituden proportional [27].

2.2 Dunkelfeldmikroskopie 23
< �
9 �
9 �
/ � " � � ; � � � � �
� � � � � 5 �� � � � � � � � � �
4
>
� � �
�
� � > � " � �8 � � � � � � � � " � � � � " � � �
� � �
Abbildung 2.10: Strahlengang und Bildentstehung bei einem Nichtselbstleuchter [27].
Oe Objektebene, Ob Objektiv, Ze Zwischenbildebene, R1, R2 Amplituden, I Inten-
sitat.
Im Gegensatz zu Airy geht E. Abbe in seiner Theorie zur Bildentstehung im Mikros-
kop davon aus, dass das Bild durch die Beugung des Lichtes an dem Objekt bestimmt
wird. Seine Betrachtungsweise bezieht sich nur auf nicht selbst leuchtende Objekte,
da die Beleuchtung mittels einer punktformigen Lichtquelle vorausgesetzt wird. Die
von der Lichtquelle ausgehenden Elementarwellen werden an dem Objekt gebeugt und
im Objektiv gebundelt, so dass es in der hinteren Brennebene zur Interferenz kommt.
Das entstehende Beugungsbild wird primares Interferenzbild genannt. Dieses ist ent-
scheidend fur das in der Zwischenbildebene entstehende sekundare Interferenzbild, das
durch Interferenz der von dem primaren Interferenzbild kommenden Elementarwellen
entsteht. Das sekundare Interferenzbild entspricht nur bei vollstandiger Beteiligung al-
ler Elementarwellen des primaren Interferenzbildes absolut dem Objekt [21, 26, 27].

24 2. Optische Methoden
�
� � � �? ? �
�
� �� ? % � � � % �
# � � �
? � ? � � �
Abbildung 2.11: Die Auflosung eines Objektes bei unterschiedlichen Offnungswinkeln
2σ eines Objektivs [26]. a) Eintritt des Haupt- und des ersten Nebenmaxima bei kleinem
Offnungswinkel. b) Eintritt weiterer Nebenmaxima bei großerem Offnungswinkel.
Nach der Theorie von Abbe ist zur Auflosung eines Objektes die Interferenz von minde-
stens zwei Beugungsmaxima in der Zwischenbildebene erforderlich, wie es in Abbildung
2.11 a) dargestellt ist. Diese ist gegeben, wenn der halbe Offnungswinkel des Objektivs
σ betragt. Mit zunehmendem Offnungswinkel 2σ erhoht sich die Zahl der in das Ob-
jektiv eintretenden Maxima (Abbildung 2.11 b)), wodurch die Scharfe der Abbildung
erhoht wird. Das Auflosungsvermogen eines Mikroskops definiert einen Grenzwert, der
den minimalen Abstand d zwischen zwei Objekteinzelheiten angibt, bei dem diese ge-
rade noch zu unterscheiden sind. Definitionsgemaß gelten zwei Punkte immer dann
als eindeutig aufgelost, wenn zwischen den Hauptmaxima der beiden Beugungsfiguren
ein vollstandiges Minimum auftritt. Da die Auflosung eines Bildes zusatzlich von der
Wahrnehmung des Betrachters, wie z. B. durch das menschliche Auge oder durch eine
Kamera, abhangig ist, wird von einem so genannten kritischen Bereich der Auflosung
gesprochen, also einem Grenzbereich, in dem die Deutlichkeit der Auflosung langsam
abnimmt [27].
Nach den Theorien von Airy und Abbe lasst sich die theoretische Auflosungsgren-
ze, unterhalb derer zwei Objektpunkte nicht mehr klar voneinander getrennt werden
konnen, wie folgt berechnen:
dmin =λ
AObj
. (2.4)
Der Grenzwert fur die noch auflosbare Gitterkonstante bzw. den noch auflosbaren Ab-
stand zweier Objektpunkte dmin ist demnach von der Wellenlange des Lichtes λ und

2.2 Dunkelfeldmikroskopie 25
von der Apertur des Objektivs AObj abhangig. Die numerische Apertur A eines opti-
schen Systems entspricht dem Produkt aus der Brechzahl n des zwischen dem Objekt
und dem Objektiv befindlichen Mediums und dem Sinus des halben Offnungswinkels
σ im Objektraum [21]:
A = n · sin σ . (2.5)
Nach der Abbeschen Theorie stellt der oben genannte Grenzwert den Fall dar, dass bei
der Betrachtung eines Gitters genau zwei Hauptmaxima (0. und 1. Ordnung) an der
Bildentstehung beteiligt sind. Bei der Betrachtung zweier Objektpunkte eines Nicht-
selbstleuchters nach Airy existiert zwischen den beiden Hauptmaxima ein eindeutiges
Minimum. Aus der Praxis ist bekannt, dass selbst bei Uberlagerung zweier Beugungs-
scheibchen noch eine eindeutige Auflosung moglich ist, sofern fur den Abstand zwischen
den Objektpunkten gilt:
dmin =3
4· λ
AObj
. (2.6)
Bei den bisherigen Betrachtungen des Auflosungsvermogens wird von einer punktfor-
migen Lichtquelle ausgegangen, deren Lichtwellen senkrecht auf das Objekt treffen. Der
Einfallswinkel α betragt somit Null. Das Auflosungsvermogen kann durch eine schiefe
Beleuchtung, wie sie z. B. in der Dunkelfeldmikroskopie verwendet wird, gesteigert wer-
den (s. Abbildung 2.12). Entsprechend der Darstellung in der Abbildung 2.12 a) ist der
Einfallswinkel des Lichtes dann großer Null. Die vom Licht ausgehenden Elementarwel-
len treffen mit einem Gangunterschied δ auf das Objekt, der von der Gitterkonstanten
d und von dem Sinus des Einfallswinkels abhangig ist:
δ = d · sin α . (2.7)
Die von den einzelnen Objektpunkten erzeugten Beugungsscheibchen interferieren in
der Zwischenbildebene. Bei der Summation der einzelnen Bilder der Amplituden muss
der Phasenwinkel φ berucksichtigt werden. In der Abbildung 2.12 b) betragt der Gang-
unterschied δ zwischen den Amplituden eine halbe Wellenlange. Fur den Grenzwert
der Auflosung bei schiefer Beleuchtung ergibt sich damit:
dmin =λ
2 · AObj
. (2.8)

26 2. Optische Methoden
5�
�
� � � � � 5 � � �� � � � � � � � � � � �
� � �
�# � � �
Abbildung 2.12: Die Bildentstehung bei einem Nichtselbstleuchter mit schiefer Be-
leuchtung [26]. a) Der Gangunterschied δ der Lichtwellen bei schiefer Beleuchtung. b)
Die Verteilung der Beleuchtungsintensitat im Bild eines Nichtselbstleuchters bei schie-
fer Beleuchtung.
Somit ist die Auflosungsgrenze bei schiefer Beleuchtung wesentlich kleiner als bei gera-
der Beleuchtung. Die Dunkelfeldmikroskopie unterscheidet sich von der schiefen Hell-
feldbeleuchtung dadurch, dass bei ihr der Einfallswinkel des beleuchtenden Lichtes
großer ist als der halbe Offnungswinkel des Objektivs. Dadurch kann das nullte Haupt-
maximum nicht in das Objektiv eintreten.
2.3 Digitale Bildverarbeitung
Bis zum Anfang des 20. Jahrhunderts ist es Wissenschaftlern vorbehalten, ihre Er-
gebnisse verbal oder grafisch zu dokumentieren. Anfang des 20. Jahrhunderts nutzen
immer mehr Wissenschaftler die Fotografie, um ihre Entdeckungen festzuhalten. Diese
Moglichkeit bringt der Mikroskopie einen enormen Aufschwung, vor allem bei der Un-
tersuchung der Brownschen Bewegung von suspendierten Teilchen [28]. Einen erneuten
Aufschwung bekommt die Mikroskopie gegen Ende des 20. Jahrhunderts mit der Ein-
fuhrung der digitalen Bildverarbeitung. Besonders in der Kolloidchemie wie auch bei
der Qualitatssicherung stellen mikroskopische Methoden verbunden mit der digitalen
Bildverarbeitung interessante Untersuchungsmoglichkeiten dar.

2.3 Digitale Bildverarbeitung 27
� � � � 8 � �
8 � � 5 � � � � " � � � @ � # 0 � ! ?� � � � � � � � �
� � � � � � � � � � " � � � @ � # 0 � ! ! # � � �
# � # � � � � � � 8 � � 5$ � � A � ! � � � ! � �
� � � " � � � � � � # � �� � � # � � � � � � � 8 � � 5$ � � � � 1 ? � � � �
� � � � � � " � � � �# � � � � � � � � � �1 # � � � 0 � � # �
8 � � 5 � � - � � � � � �
1 � � � � # � � � � � � � � � 8 � � 5 $ � � # � � � � � � � � � � � 5 � ? # � # � > � �
' '
� )
� )
� )
� )
� � ' � � '
� � ' � � '
�
�
�
�
�
�
�
� � � � � � �
�
�
�
�
�
�
�� � � � �
+ �
+ �
+ �
+ �
& '
& '
& '
& '
& '
& '
& '
& '
% �
% � % �
% �
% �� ' � ' � �
� � � � � � �
Abbildung 2.13: Die Umwandlung eines analogen Bildes in ein digitalisiertes Bild.
Die klassische Bildverarbeitung setzt sich, wie in der Abbildung 2.13 dargestellt, aus
vier verschiedenen Operationen zusammen: Der Bildgewinnung, der Digitalisierung, der
Bildverarbeitung und der Bildanalyse.
2.3.1 Bildgewinnung mittels einer CCD-Kamera
Die CCD-Kamera besitzt einen Chip, der aus einer zweidimensionalen Matrix von licht-
empfindlichen Halbleiterelementen konstanter Große besteht, den so genannten Charge
Coupled Device (CCD) (aus dem Englischen sinngemaß ubersetzt: ladungsgekoppelter
Halbleiter). Ein CCD-Chip ist unterteilt in einen Belichtungsbereich und einen lichtun-
empfindlichen Speicherbereich. Die von dem Bild kommenden Photonen bewirken bei
dem Auftreffen auf die Halbleiterelemente die Freisetzung von Elektronen, so dass elek-
trische Ladung erzeugt wird. Diese wird in der Akkumulationsphase gesammelt. Nach
einer bestimmten Zeit erfolgt die Auslesephase, in der die Ladung punktweise und an-

28 2. Optische Methoden
schließend zeilenweise ausgelesen und in elektrische Spannung umgewandelt wird. Diese
kann schließlich auf ein Speichermedium ubertragen werden.
Es gibt zwei Moglichkeiten des Auslesens der Zeilenelemente: Das Halbbild- und das
Vollbildverfahren. Das Halbbildverfahren wird in der Fernsehtechnik nach der europai-
schen CCIR-Norm (Comite Consultatif International des Radiocommunications) ver-
wendet. Dabei werden zunachst alle ungeraden Zeilen ausgelesen und danach alle ge-
raden Zeilen. Die beiden Halbbilder werden im 50 Hz Takt gesendet. Das Vollbild wird
aus den beiden Halbbildern im Wechsel zusammengesetzt und somit praktisch mit 25
Hz dargestellt. Zur Messung von bewegten Objekten, zum Beispiel in der Qualitats-
sicherung, kann dieses Verfahren zu einer Verschiebung der beiden Halbbilder fuhren.
Dieser so genannte Kammeffekt ergibt ein verzerrtes Bild, das zu Messfehlern beitragen
kann. Daher wird bei bewegten Objekten das Vollbildverfahren angewendet. In diesem
Verfahren werden alle Zeilen des Bildes ihrer Reihenfolge nach ausgelesen und mit 25
Hz ubertragen [29, 30].
2.3.2 Digitalisierung
Der Prozess der Digitalisierung, das heißt die Umwandlung eines analogen Bildes in
ein fur den Rechner verarbeitbares Datenformat, setzt sich aus zwei Schritten zusam-
men: Der Rasterung und der Quantisierung. Bei dem Prozess der Rasterung wird ein
zweidimensionales Gitter mit einer bestimmten Gitterkonstanten uber das analoge Bild
gelegt. Die Große der einzelnen Gitterelemente bestimmt die Gute der Auflosung. Ein
solches Gitterelement wird als Pixel (engl.: picture element) bezeichnet. Seine Große
ist dabei durch die Große der Halbleiterelemente des CCD-Chips gegeben.
Bei der Quantisierung werden die gemessenen Beleuchtungsstarken auf eine begrenzte
Anzahl diskreter Werte abgebildet. Typischerweise erfolgt die Zuordnung der Grau-
werte mit 8 Bit, wodurch 256 Grauwerte resultieren. Die mittlere Beleuchtungsstarke
eines Bildpunktes wird dann dem Grauwert gleichgesetzt, von dem er am geringsten
abweicht.

2.3 Digitale Bildverarbeitung 29
Die Digitalisierung eines analogen Bildes fuhrt damit zu einer zweidimensionalen Bild-
matrix B mit m bzw. n Bildpunkten p in x- bzw. y-Richtung:
B(x, y) =
⎛⎜⎜⎜⎜⎜⎜⎝
p(1, 1) p(1, 2) · · · p(1, n)
p(2, 1) p(2, 2) · · · p(2, n)...
......
...
p(m, 1) p(m, 2) · · · p(m,n)
⎞⎟⎟⎟⎟⎟⎟⎠
. (2.9)
Die Bildverarbeitungskarte (engl.: frame grabber) ist ein wesentlicher Bestandteil eines
Bildverarbeitungssystems. Sie dient zunachst zur Umwandlung des analogen Bildes in
ein digitales Bild (A/D-Wandler). Die digitalen Bilddaten werden in dem Bildspeicher
der Bildverarbeitungskarte gespeichert. Von dort konnen sie in dem Hauptspeicher des
Computers abgelegt werden. Des Weiteren erfolgt mittels der Bildverarbeitungskarte
auch die Ruckwandlung eines digitalen Bildes in ein analoges Bild (D/A-Wandler), um
die Daten z. B. auf einem Monitor darzustellen [31].
2.3.3 Bildverarbeitung und -analyse
Die Bildverarbeitung dient zur Verbesserung der Bildqualitat. Mittels verschiedener
Operationen konnen Unscharfen oder Storungen in einem Bild entfernt oder Merkmale
extrahiert werden. Zu den bildverarbeitenden Operationen gehoren u. a. Subtrakti-
onsverfahren, Weichzeichnen und verschiedene Filteroperationen. Bei den Filtern ist
zwischen Tiefpass- und Hochpassfiltern zu unterscheiden. Im Rahmen dieser Arbeit
wird ausschließlich die Tiefpassfilterung genutzt, bei der die hohen Frequenzanteile ei-
nes Bildes durch die Uberlagerung einer entsprechenden Matrixoperation abgeschwacht
oder uberlagert werden (vgl. Kapitel 6.2). Das Herausfiltern starker Details, wie z. B.
das Abflachen der Kanten eines Objektes, wird als Glattung der Grauwerte bezeichnet.
Die Folge ist eine gewisse Unscharfe des Bildes, die jedoch keine Auswirkung auf die
anschließende Bildanalyse hat [32].
Bei der Bildanalyse steht das Erkennen und Auswerten von Merkmalen des gesam-
ten Bildes im Vordergrund. Dies umfasst z. B. die Bestimmung geometrischer Formen

30 2. Optische Methoden
und deren Koordinaten im Bild sowie die Auswertung der Grauwertverteilung. Die
Grauwertverteilung kann in einem Grauwerthistogramm dargestellt werden, in dem
die Haufigkeit der Grauwerte eines Bildes gegen die Grauwerte aufgetragen wird. Die
Bestimmung der Koordinaten bestimmter Objekte kann z. B. zur Verfolgung der Be-
wegung dieser Objekte herangezogen werden. Hierzu werden die in zeitlich aufeinan-
der folgenden Bildern gefundenen Objekte zugeordnet und deren Verschiebungsvekto-
ren bestimmt. Das Verfolgen der Partikelspuren wird allgemein in der Literatur als
Particle Tracking bezeichnet. Die Objektverfolgung kann einer Vielzahl von Anwen-
dungsbereichen dienen, z. B. die Untersuchung von technischen Stromungsvorgangen
in Automobilen und Flugzeugen, der Bewegung von Dummys in Crash-Tests der Au-
tomobilindustrie oder zur Analyse der Brownschen Bewegung in der Kolloidchemie
[31, 32].

Kapitel 3
Oberflachengebundene
Nanopartikel
Der Begriff Nanotechnologie beschreibt einen sehr weiten Bereich in der heutigen For-
schung, der die klassischen Naturwissenschaften bis hin zu den technologischen Wis-
senschaften umfasst. Entsprechend groß ist der Bereich moglicher Anwendungen. Ein
wichtiger Anwendungsbereich fur nanostrukturierte Oberflachen liegt in der Fertigung
von Leiterbahnen oder Bauelementen auf Si-Wafern fur die Halbleiterindustrie.
Zu einer der wichtigsten Methoden in der CMOS-Technologie (complementary metal-
oxide-semiconductor) ist in den letzten Jahren die optische Lithographie geworden.
Hierbei wird mit monochromatischem Licht unter zur Hilfenahme einer Maske ein
Muster auf einen mit einem Fotolack beschichteten Si-Wafer ubertragen. Durch die
Nutzung immer kurzerer Wellenlangen sowie der Optimierung der Oberflachen, der
verwendeten Polymere und der notwendigen Masken konnen immer kleinere Struktu-
ren erzeugt werden. Dennoch stoßt die optische Lithographie an ihre physikalischen
Grenzen. Aufgrund der enormen Kosten fur die notwendige hoch entwickelte Ausstat-
tung und unter dem Aspekt, dass diese Methode nach derzeitigem Kenntnisstand nur
zur Herstellung von Strukturen bis zu 100 nm geeignet zu sein scheint, wird im Rah-
men der fortschreitenden Miniaturisierung gegenwartig nach Alternativen gesucht. Zu
der nachsten Generation lithographischer Methoden zahlen die extreme Ultraviolett
31

32 3. Oberflachengebundene Nanopartikel
� � � � � � # � � � � ? � # 0 � � �
� � � > � � � " � � " � �
� � � � � � � � � B � � 9 � � 5 � � � C � �
1 � � " ! � �
� � � � � � � � � � � � D � E � D � � � � 5 � �
� ! , � � � � � � � 5 � � � � 0 � � � � �5 � � � � � � � � � �
F � � � � � � � � � � � � � �
Abbildung 3.1: Der Prozess der Nanoimprintlithographie (NIL).
(EUV)-, die Rontgen (X-Ray)-, die Elektronenstrahl (EB)- sowie die Ionenprojektions-
lithographie (IPL) [33].
Es gibt jedoch auch eine große Anzahl an Moglichkeiten, Nanostrukturen durch eine
Art Kopierprozess, ahnlich der konventionellen Lithographie, zu erzeugen. Eine dieser
Methoden ist die Nanoimprintlithographie, die im Folgenden vorgestellt wird.
3.1 Nanoimprintlithographie und deren Anwendung
Die Nanoimprintlithographie (NIL), die auch unter dem englischen Begriff Hot Embos-
sing (Heißpragung) in der Literatur zu finden ist, wird 1995 von Chou et. al. eingefuhrt
[34, 35]. Das Prinzip ist in Abbildung 3.1 schematisch dargestellt. Ein Stempel mit
einer strukturierten Oberflache und ein Substrat, auf dem eine dunne Polymerschicht
aufgebracht ist, sind parallel ubereinander angeordnet. Das Substrat wird auf eine Tem-
peratur erhitzt, die uber der Glasubergangstemperatur Tg des zu formenden Polymers
liegt. Der Stempel wird unter Druck auf das Substrat gepresst, so dass das Polymer die
Hohlraume des Stempels ausfullt. Nach einer Abkuhlphase wird der Stempel von dem
Substrat getrennt. Die Polymerreste werden anschließend in einem Atzprozess, wie bei-
spielsweise der Reaktiven-Ionen-Atzung (engl.: Reactive Ion Etching (RIE)), entfernt.
Die Auflosung, die hierbei erreicht werden kann, wird u. a. von der Struktur und der

3.1 Nanoimprintlithographie und deren Anwendung 33
Qualitat des verwendeten Stempels bestimmt. Zurzeit werden drei verschiedene Mate-
rialien zur Herstellung der Stempel verwendet: Siliziumdioxid (SiO2), Polymere und
Nickel (Ni). Bei der Herstellung der SiO2-Stempel wird ein Polymerfilm auf einem Si-
Wafer aufgebracht. Mittels hochauflosender Elektronenstrahllithographie (EBL) wird
das entworfene Muster in die Polymerschicht geschrieben. Anschließend werden entwe-
der Chrom oder nanometergroße Wolframpartikel auf die Probe gebracht. Durch den
anschließenden Atzprozess wird das Relief in die SiO2-Schicht ubertragen. Auf diese
Weise ist es moglich, Strukturen unter 20 nm zu erzeugen [36]. Die Nutzung von Po-
lymerstempeln soll den Herstellungsprozess vereinfachen und die anfallenden Kosten
reduzieren. Hierzu wird in einem NIL-Prozess die Kopie eines konventionellen SiO2-
oder Ni-Stempels in einem thermisch stabilen Polymer erzeugt. Das verwendete Po-
lymer verhalt sich bei der Pragung thermoplastisch, durch Erhohung der Temperatur
setzt nachtraglich eine Vernetzung des Polymers ein. Daraus resultiert schließlich ein
duroplastischer Stempel, der wiederum als Vorlage in einem weiteren Prageprozess ver-
wendet werden kann. Somit konnen in kurzer Zeit zwei Kopien, Positiv und Negativ, des
Hauptstempels erzeugt werden. Die geringere Lebensdauer der Polymerstempel wird
durch die niedrigen Herstellungskosten und den schnellen Herstellungsprozess kompen-
siert [37]. Durch Verwendung eines elektronenstrahlempfindlichen Polymersystems ist
eine direkte Musterubertragung mittels EBL moglich. Das Polymer basiert auf einem
Epoxydharz. Die Vernetzung findet durch eine mittels Elektronenstrahlen injizierte sau-
rekatalysierte kationische Polymerisation statt [38]. Die Herstellung von Ni-Stempeln
ist sehr kostenintensiv und aufwandig. Dennoch finden sie vor allem in der Industrie
Anwendung, da sie gegenuber den Si-Stempeln flexibler und weniger zerbrechlich sind.
Ein wesentliches Problem, das in der Nanoimprintlithographie auftritt, ist die Adhasion
des gepragten Polymers an der Stempeloberflache, was zu Defekten in der gepragten
Oberflache fuhren kann. Das Adhasionsverhalten des Polymers ist besonders stark in
Bereichen mit einer dichten Struktur zu beobachten. In diesen Bereichen ist die Flache
zwischen Polymer und Stempel und somit auch deren Wechselwirkungen großer als zwi-
schen Polymer und Substrat. Durch Verwendung einer Antihaftbeschichtung auf der

34 3. Oberflachengebundene Nanopartikel
Stempeloberflache kann die Adhasionskraft zwischen Stempel und Polymer herabge-
setzt werden. Am bekanntesten sind Antihaftbeschichtungen aus Polytetrafluoroethen
(PTFE). Jaszweski et. al. beschreiben 1997 eine Methode zur Auftragung einer Anti-
haftbeschichtung mit PTFE aus einem CH4/H2- und einem CHF3-Plasma auf einem
mikrostrukturierten Ni-Stempel. Bei der Pragung von Polycarbonat (PC) und Polyme-
thylmethacrylat (PMMA) wird eine Diffusion der Fluoratome von der PTFE-Schicht
in das gepragte Material beobachtet. Der Heißprageprozess kann uber 50 mal wieder-
holt werden [39]. Scheer et. al. wenden 1999 eine Antihaftbeschichtung aus PTFE auf
einer nanostrukturierten Stempeloberflache an, um sowohl thermoplastische als auch
duroplastische Polymere mittels NIL zu pragen. Beide Polymere konnen unter opti-
malen Prozessbedingungen gepragt werden. Durch die Antihaftbeschichtung konnen
außerdem die Pragetemperatur und die Pragezeit herabgesetzt werden [40]. Eine neue
Methode zum Auftragen einer Antihaftbeschichtung wird von Montelius et. al. 2002
vorgestellt. Hierbei wird eine Monoschicht aus Tridecafluoro-(1,1,2,2)-tetrahydrooktyl-
trichlorsilan (F13 − TCS) in der Gasphase auf die Oberflache eines Si/SiO2-Stempels
aufgetragen. Chlorsilan reagiert spontan mit der SiO2-Oberflache unter Eliminierung
von HCl, so dass die Antihaftschicht kovalent an die Stempeloberflache gebunden ist.
Es zeigt sich eine deutliche Verbesserung der Pragungen in einem thermoplastischen
Polymer. Diese Methode eignet sich jedoch nicht bei der Verwendung von Ni- oder
Polymerstempeln [41, 42].
Bei der Einfuhrung der NIL verwenden Chou et. al. PMMA zur Pragung, das auch
heute noch haufig eingesetzt wird [34, 35]. In dieser noch jungen Technologie entwickeln
die meisten Arbeitsgruppen jedoch ihre eigenen Polymere, die fur ihre speziellen An-
wendungsbereiche entsprechend angepasst sind. Aus der großen Anzahl der moglichen
Polymere werden daher in dieser Arbeit nur drei Polymere vorgestellt, die im Rahmen
des EU-Projektes CHANIL (Chances for a Nanoimprint Lithography based fabrication)
von den Partnern aus der Industrie entwickelt werden.
Mr − I 8000 (Microresist Technology (MRT), Berlin) ist ein thermoplastisches Po-

3.1 Nanoimprintlithographie und deren Anwendung 35
lymer, das aus einem Methacrylat gewonnen wird. Die Glasubergangstemperatur von
Tg = 107 ◦C entspricht nahezu der von PMMA (Tg = 105 ◦C). Durch Einbringen
verschiedener aromatischer Gruppen ist ein gezieltes Einstellen der Glasubergangstem-
peratur moglich. Außerdem kann dadurch die Widerstandsfahigkeit bei der Plasmaat-
zung, die uber der von PMMA liegt, weiter erhoht werden. Das Polymer weist eine
sehr hohe Genauigkeit bei der Musterubertragung auf, so dass es sich gut fur einfache
Prageprozesse eignet, in denen auch auf eine Antihaftbeschichtung verzichtet werden
kann [43, 44].
Mr − I 9000 (MRT, Berlin) ist ein duroplastisches Polymer, das ebenfalls aus einem
Methacrylat gewonnen wird. Aufgrund von Allyl-Resten ist es zur Kreuzpolymerisation
fahig. Die Allylendgruppen sind resonanzstabilisiert, so dass die Kreuzpolymerisation
hohere Temperaturen erfordert (> 120 ◦C). Das Vorpolymer hat eine Glasubergangs-
temperatur von Tg = 63 ◦C. Das Polymer zeigt eine gute Widerstandsfahigkeit bei der
Plasmaatzung. Das Pragen dieses Materials erfordert eine Antihaftbeschichtung, zeigt
unter diesen Bedingungen aber sehr gute Pragequalitaten. Es eignet sich besonders gut
fur die Herstellung von kompletten Polymerstempeln [37, 43, 44].
Mr − I 6000 (MRT, Berlin) ist ein Elektronenstrahl empfindliches Polymer, das aus
einem Epoxydharz synthetisiert wird. Die Glasubergangstemperatur des Vorpolymers
liegt bei Tg = 65 ◦C. Die Vernetzungsreaktion, eine saurekatalysierte kationische Poly-
merisation, wird durch den Elektronenstrahl initiiert. Das Hauptanwendungsgebiet ist
die Herstellung von Polymer-Si-Stempeln, die durch direkte Musterubertragung mit-
tels Elektronenstrahl erzeugt werden [38].
Zur Musterubertragung der Struktur der Polymerschicht in die darunter liegende Schicht
gibt es zwei Moglichkeiten: Das Atzverfahren oder der Lift-Off-Prozess. Das Atzverfah-
ren kann unterteilt werden in nass- und trockenchemisches Atzen. In dem nassche-
mischen Atzverfahren wird ein oxidatives Losemittel eingesetzt. Zu den bekanntesten
basischen Losemitteln, die in der Halbleiterindustrie zum Atzen von Si-Wafern einge-

36 3. Oberflachengebundene Nanopartikel
setzt werden, zahlt eine KOH-Losung. In diesem anisotropen Prozess ist die Atzrate
in der <111>-Kristallebene wesentlich niedriger als in der <100>-Ebene [45]. In dem
trockenchemischen Atzverfahren werden haufig per- bzw. hochfluorierte Kohlenstoffe,
wie z. B. CF4, CHF3 oder C2F6, als reaktive Atzgase eingesetzt. Es existiert eine Viel-
zahl an unterschiedlichen Trockenatzmethoden, daher soll im Folgenden nur die sehr
haufig angewendete Methode des reaktiven Ionenatzens (RIE) erlautert werden. Das
zu atzende Substrat wird in einem Parallel-Platten-Reaktor, der mit einem Plasma
gefullt ist, direkt auf der Kathode platziert. Durch Gasentladung werden in dem Plas-
ma sowohl reaktive Ionen als auch neutrale Radikale erzeugt. Die erzeugten Kationen
werden mit hoher Energie zu dem Substrat hin beschleunigt und reagieren mit diesem.
Durch eine verstarkte Atzung in vertikaler Richtung gewahrleistet RIE die geforderte
Anisotropie sowie ein hohes Aspektverhaltnis, also das Verhaltnis der Tiefe der geatz-
ten Struktur zu ihrer horizontalen Ausdehnung [46].
In dem Lift-Off-Prozess, der schematisch in Abbildung 3.2 dargestellt ist, wird auf
die strukturierte Polymeroberflache eine Metallschicht aufgedampft. Mit einem Lose-
mittel, z. B. Aceton, oder einem Ablacker konnen anschließend die gewunschten Poly-
merschichten mit der Metallschicht entfernt werden [47].
# � � � " �
Abbildung 3.2: Der Lift-Off-Prozess [47]. a) Nanostrukturierte Polymeroberflache
nach NIL. b) Beschichtung mit einem Metall. c) Struktur nach Entfernen der Polymer-
schicht.
Die Anwendungsgebiete fur die mit der Nanoimprintlithographie erzeugten nanome-
tergroßen Strukturen sind sehr vielfaltig. Die ursprungliche Idee ist die Herstellung
von Speichermedien. Schon 1997 gelingt es Chou et. al., eine Nano-Compact Disk mit
400 Gbit/in2 Speicherdichte herzustellen [48]. Derzeit konnen bereits nanostrukturierte
Flachen mit einer Dichte von > 500 Gbit/in2 erzeugt werden [49]. Weitere Anwendungs-

3.2 Herkommliche Methoden zur Qualitatssicherung 37
moglichkeiten aus dem Bereich der elektronischen Bausteine bestehen in der Herstellung
von Quantenpunktkontakten [50], Feldeffekttransistoren [51], dreidimensionalen Struk-
turen, wie z. B. T-Gates [52], neuen ballistischen Bausteinen (TBJ − Devices) [53],
optischen Bausteinen [54] oder nanoskalierten GaAs-Photodetektoren [55]. Ein weiteres
Anwendungsgebiet ist der biotechnologische Bereich, z. B. die Entwicklung von Nano-
biosensoren [56] oder nanofluiden Tunneln mit einer sehr hohen Dichte zur Isolierung
von Biomolekulen [57].
3.2 Herkommliche Methoden zur Qualitatssicherung
Der große Bereich der Anwendungsmoglichkeiten macht die Nanoimprintlithographie
(NIL) zu einer interessanten Methode fur die Industrie im Hinblick auf die geforderte
Miniaturisierung von Strukturen. Ein wesentlicher Aspekt fur die Anwendung im indu-
striellen Bereich ist bei den bisherigen Forschungsaktivitaten weitestgehend vernach-
lassigt worden, namlich die Qualitatssicherung der Stempel und Substrate. In diesem
Kapitel werden die derzeit ublicherweise verwendeten Methoden zur Untersuchung na-
nostrukturierter Oberflachen in der NIL-Technologie erlautert: Die Rasterelektronenmi-
kroskopie (engl: Scanning Electron Microscopy (SEM)) und die Rasterkraftmikroskopie
(engl: Atomic Force Microscopy (AFM)).
3.2.1 Rasterelektronenmikroskopie (SEM)
Die Rasterelektronenmikroskopie nutzt eine Elektronenstrahlsonde zum Abtasten von
Oberflachen. Die Abbildung der Oberflache kommt durch die Wechselwirkung der Elek-
tronen mit der Materie des Objektes zustande. Zur Elektronenstrahlerzeugung dient
eine W - oder LaB6-Kathode. Durch thermische Emission findet der Elektronenaustritt
aus der Kathode in das Vakuum in Richtung der Anode statt. Ein charakteristischer
Kennwert der Kathode ist der Durchmesser des so genannten Crossover, der den klein-
sten Strahlenquerschnitt zwischen der Kathode und der Anode beschreibt. Der Durch-
messer des Crossover wird durch mehrere magnetische Elektronenlinsen verkleinert, so
dass der auf der Oberflache des Objektes vorliegende Durchmesser der Elektronensonde

38 3. Oberflachengebundene Nanopartikel
schließlich 50−200 A betragt. Mit Hilfe des Rastergenerators wird das Objekt zeilenfor-
mig durch die Elektronensonde abgetastet. Dabei werden zwei Paare von Ablenkspulen
verwendet (je ein Paar fur die Ablenkung in x- und y-Richtung), die transversale ma-
gnetische Felder erzeugen. Der mit einer Verkleinerung der abgetasteten Objektflache
verbundene Vergroßerungseffekt wird durch die Abschwachung des Spulenstroms er-
reicht. Die Fokussierung erfolgt durch die Veranderung des Objektivlinsenstroms. Zur
Fokussierung wird eine moglichst hohe Vergroßerung gewahlt. Die durch die Wechsel-
wirkung zwischen dem Elektronenstrahl und dem Objekt emittierten oder ruckgestreu-
ten Elektronen werden von den Detektoren aufgenommen. Diese Signale werden durch
den Videoverstarker intensiviert und dienen schließlich zur Helligkeitsmodulation der
Bildrohre [58, 59].
Die Entstehung des SEM-Bildes beruht auf der Wechselwirkung zwischen den Primar-
elektronen (PE) aus der Elektronenstrahlsonde und der Materie des Objektes, wobei
es zur Emission von Sekundarelektronen (SE) und zur Ruckstreuung von Elektronen
(RE) kommt. Bei einer unelastischen Streuung der PE tritt haufig ein Energieverlust
kleiner 50 eV auf, wodurch langsame SE mit Energien kleiner 50 eV erzeugt werden.
Diese SE, die zur Gruppe 1 gehoren, treten aus einer sehr dunnen Oberflachenschicht
mit einer Tiefe von 10 − 100 A aus. Sie bestimmen die Hochauflosung des SEM, da
ihr Austrittsgebiet von der Austrittstiefe und dem Durchmesser des Primarstrahls be-
stimmt wird. Die RE entstehen durch elastische Streuung des Primarstrahls an den
Atomkernen des Objektes. Die Elektronen werden an den Atomkernen in solch großen
Winkeln abgelenkt, dass sie die Probe wieder verlassen konnen. Das Energiespektrum
dieser Elektronen liegt in einem Bereich zwischen der Energie der PE bis hinab zu
50 eV. Die RE sind stark von der Kernladungszahl Z abhangig, wodurch sie einen
Vergleich der Zusammensetzungen der verschiedenen Phasen ermoglichen. Beim Ver-
lassen der Oberflache kann es auch durch die RE zu einem Austritt von SE aus der
Oberflachenschicht kommen (Gruppe 2). Der Anteil dieser Gruppe an der Intensitat
des SE-Signals ist proportional zu dem Ruckstreukoeffizienten, der mit Z und dem
Einfallswinkel des Primarstrahls ansteigt. Eine weitere Gruppe an SE (Gruppe 3) wird

3.2 Herkommliche Methoden zur Qualitatssicherung 39
durch das Auftreffen der RE auf die Wande der Probenkammer erzeugt. Nur der An-
teil der Gruppe 1 an dem Gesamtsignal (25− 50 %) enthalt die Informationen mit der
hohen raumlichen Auflosung, die maßgeblich durch den Durchmesser des Elektronen-
strahls bestimmt werden. Der restliche Anteil, der durch die Gruppe 2 und 3 bestimmt
ist, enthalt Informationen, die von den RE geliefert werden, das heißt, die in tieferen
Schichten der Probe entstehen. Dieser Anteil stellt ein Hintergrundsignal dar, das zu
einer Abschwachung des Kontrastes und im gewissen Umfang zu einer Abschwachung
der Auflosung fuhrt [58, 59].
Wegen der zwei unterschiedlichen Arten von detektierten Elektronen (SE und RE)
erfolgt im SEM eine Unterscheidung zwischen der Abbildung mit SE und der Abbil-
dung mit RE. Der wichtigste Kontrasteffekt bei der Abbildung mit SE ist der Kontrast
durch Flachenneigung, auch Reliefkontrast genannt. Dieser ist abhangig von der Aus-
beute an SE, die der pro Strahlelektron freigesetzten Anzahl der SE entspricht, und
von dem Ruckstreukoeffizienten, der wiederum abhangig von dem Einfallswinkel des
Primarelektronenstrahls auf die Probenoberflache ist. Der Kontrast der Abschattung
entsteht durch die Anordnung des Detektors im Probenraum und dessen Geometrie.
Die von dem Detektor wegzeigenden Flachen erscheinen dunkel, weil nur ein Teil der
erzeugten SE von dem Detektor aufgenommen werden. Diese beiden Kontrasteffek-
te fuhren zu dem topographischen Kontrast in einem Bild mit SE, das einen dreidi-
mensionalen Eindruck der Oberflachentopographie vermittelt. Kanten und vorstehende
Objektstrukturen fuhren dazu, dass die Diffusionswolke starker angeschnitten wird, wo-
durch die Ausbeute an RE erhoht wird. Dadurch erhoht sich auch der Anteil der SE
der Gruppe 2 und 3, die an dem SE-Signal beteiligt sind. Der so genannte Kanteneffekt
bewirkt folglich eine Verschlechterung der Auflosung der Kanten. Auch bei der Abbil-
dung mit RE ist der Reliefkontrast entscheidend. Er ist eine Folge der Ausblendung
der RE durch Erhebungen, die einen Schatten bilden, und durch die Abhangigkeit der
Ausbeute der RE von dem Neigungswinkel der Oberflache. Durch eine entsprechende
Anordnung des RE-Detektors im Probenraum und die Neigung der Oberflache hin zu
dem Detektor kann die Anzahl der detektierten RE erhoht werden. In dem Bild erge-

40 3. Oberflachengebundene Nanopartikel
ben sich scharfe Schatten, so dass bei kleineren Vergroßerungen ein besserer Eindruck
der Oberflachentopographie als in einem SE-Bild entsteht. Bei einer ebenen Oberfla-
che ist kein Reliefkontrast vorhanden. In diesem Fall dominiert der Materialkontrast.
Dieser ergibt sich durch die Abhangigkeit des Ruckstreukoeffizienten von Z. Der Ma-
terialkontrast ist sehr empfindlich, so dass verschiedene Phasen mit unterschiedlichen
Mittelwerten der Kernladungszahl getrennt werden konnen. Die Auflosungsgrenze der
SE-Bilder ist durch den Durchmesser des abtastenden Elektronenstrahls begrenzt. Nur
der Anteil der SE der Gruppe 1 bildet die Auflosung, da die Austrittsorte der SE der
Gruppen 2 und 3 sowie der RE eine sehr hohe Verteilung aufweisen und daher ein sehr
schlechtes Auflosungsvermogen zeigen [59].
Die Untersuchung mittels SEM kann zu Veranderungen der Probe durch den Elek-
tronenbeschuss fuhren. Zur Schadigung der Probe kommt es vor allem durch Kon-
tamination, Objekterwarmung, Strahlenschaden oder Aufladungserscheinungen. Diese
konnen durch entsprechende Einstellungen des SEM und durch spezielle Probenvorbe-
reitungen verringert oder sogar ausgeschlossen werden. Vor der Messung im SEM muss
die Probe grundlich gereinigt werden, um die Bildung der Kontaminationsschicht ge-
ring zu halten. Ferner werden die Objekte haufig mit einer elektrisch leitenden Schicht
uberzogen, um z. B. Aufladungserscheinung zu vermeiden. Das dazu verwendete Mate-
rial muss eine gute elektrische Leitfahigkeit, eine gute Adhasion sowie eine chemische
Stabilitat zeigen. Zusatzlich muss die Bildung eines gleichmaßigen Films mit geringer,
einheitlicher Schichtdicke, der keine strukturellen Eigenschaften aufweist, gewahrleistet
sein, um die Topographie der Probe nicht zu verandern. Am haufigsten werden dafur
Edelmetalle wie Gold und Platin oder Legierungen, wie z. B. AuPd, verwendet, die
durch Hochvakuumbedampfung oder Kathodenzerstaubung aufgetragen werden kon-
nen [58].
SEM-Kontrolle in der Halbleiterindustrie kann wichtige Informationen hinsichtlich Pro-
zessfehler, wie z. B. Polymer- und Atzruckstande, oder kritischer Dimensionen bringen.
Eine industrielle Kontrolle schließt aber jegliche Art der Probenvorbereitung aus. Die

3.2 Herkommliche Methoden zur Qualitatssicherung 41
Probenvorbereitung wurde die Eigenschaft des Wafers verandern, so dass dieser an-
schließend nicht in den Produktionsprozess zuruckgebracht werden konnte. Zusatzlich
muss ein hoher zeitlicher Aufwand bei der Untersuchung eines kompletten Wafers be-
rucksichtigt werden.
3.2.2 Rasterkraftmikroskopie (AFM)
Die Rasterkraftmikroskopie, die 1986 von Binnig et. al. eingefuhrt wird, gehort zusam-
men mit der Rastertunnelmikroskopie (STM) zu der Gruppe der Rastersondenmikro-
skopie. Der Vorteil der AFM gegenuber der STM besteht darin, dass auch elektrisch
nicht leitende Materialien untersucht werden konnen [60].
Der Aufbau eines Rasterkraftmikroskops ist schematisch in Abbildung 3.3 dargestellt.
Eine sehr feine Spitze tastet die Oberflache der Probe in einem Rasterverfahren zeilen-
formig ab. Dabei werden die Anziehungs- und Abstoßungskrafte zwischen der verwen-
deten Spitze und der Oberflache der Probe gemessen. Die AFM bietet nicht nur die
Moglichkeit, die Oberflachentopographie abzubilden, sondern es konnen auch elektro-
statische und magnetische Krafte, Reibungskrafte und Elastizitaten bestimmt werden.
Im Hinblick auf die Anwendung zur Qualitatskontrolle wird hier jedoch nur auf die
Abbildung der Oberflachentopographie eingegangen [61].
� � � � �
� # � � � � � $ � �� � � � �
/ # � � �
� � � � � � �� � � � � � � � � � ?� � � � � 5 � � 5 �
7 � � � � � � � � � � � � � �� � � � � � � � � � 51 # � � � $ � � # � � � � � � � �
Abbildung 3.3: Schematische Darstellung der Rasterkraftmikroskopie (AFM).

42 3. Oberflachengebundene Nanopartikel
Zum Abtasten der Probe bestehen zwei Moglichkeiten: Entweder wird die Probe uber
eine fixierte Spitze bewegt oder die Spitze wird uber die fixierte Probe bewegt. Die
letztere Methode hat den Vorteil, dass die Bewegung der geringeren Masse eine hohere
Genauigkeit und eine schnellere Abtastgeschwindigkeit bewirkt. Die Spitze ist unter
einem federnden Ausleger, dem so genannten Cantilever, angebracht. Im Allgemeinen
werden die Cantilever mit integrierter Spitze aus Si3N4 oder Si hergestellt. Die Feder-
konstante eines Cantilevers liegt in einer Großenordnung von 0, 1 bis 0, 01 N/m. Die
Ruckseite ist mit Gold, Platin oder Chrom beschichtet, damit der Laserstrahl reflektiert
werden kann. Der Laserstrahl wird auf den Cantilever fokussiert, der das Licht uber
einen Spiegel zu einer Viersegment-Photodiode reflektiert. Die Steuerung der Spitze
erfolgt mittels eines piezokeramischen Antriebs. Hierbei werden meistens Rohrenscan-
ner verwendet, die aus einer Innenelektrode und einer in vier Segmente aufgeteilten
Außenelektrode bestehen. Durch das Anlegen unterschiedlicher Spannungen zwischen
der Innen- und den vier Außenelektroden konnen die einzelnen Rohren des Scanners
gestreckt oder gestaucht werden, wodurch die Bewegung in x- und y-Richtung ge-
steuert wird. Die erzielte Genauigkeit betragt den Bruchteil eines Atomdurchmessers.
Zwischen der Spitze und der Probe kommt es zu Anziehungskraften, z. B. Kapillar-,
Adhasions- und Van-der-Waals-Kraften, und zu Abstoßungskraften, z. B. durch Ion-
Ion-Wechselwirkungen oder Pauli-Kraften. Diese Wechselwirkungen resultieren in einer
Verbiegung des Cantilevers. Die Starke der Auslenkung wird mittels der Veranderung
des Lichtsignals in der Photodiode gemessen. Ein elektronischer Regelkreis in Verbin-
dung mit einem Piezo fur die z-Richtung sorgt dafur, dass die ursprungliche Hohe
schnell wieder erreicht wird. Damit ist eine Zuordnung der Auslenkung in z-Richtung
zu jedem Rasterpunkt in der x, y-Ebene moglich. Mittels der digitalen Bildverarbeitung
wird so ein dreidimensionales Bild der Topographie der Oberflache erhalten. Zur Ver-
meidung von Unregelmaßigkeiten und zur Kalibrierung wird vor dem Abtasten einer
Probe eine Kraft-Distanz-Kurve aufgenommen [61, 62].
In der Rasterkraftmikroskopie gibt es zwei Betriebsarten: Den Kontaktmodus und den
Nicht-Kontaktmodus. Zur Bestimmung der Oberflachentopographie wird haufig der

3.2 Herkommliche Methoden zur Qualitatssicherung 43
Kontaktmodus verwendet. In diesem beruhrt die Spitze direkt die Oberflache der Pro-
be. Hierbei konnen zwei Betriebsmodi verwendet werden: Der constant hight-Modus
und der constant force-Modus. Im ersten wird der Ruckkopplungsmechanismus ausge-
schaltet und der Cantilever wird auf einer konstanten Hohe gehalten. Die Ablenkung
des Cantilevers wird gemessen. Im zweiten Modus ist die Steigung des Cantilevers mit
dem Ruckkopplungsmechanismus abgestimmt. Dadurch wird die Kraft und folglich das
Durchbiegen des Cantilevers konstant gehalten. Im Nicht-Kontaktmodus gibt es keinen
direkten Kontakt zwischen der Spitze und der Probe. Der Cantilever wird hierbei in
eine harmonische Schwingung mit seiner Resonanzfrequenz versetzt. Die anziehenden
Van-der-Waals-Krafte fuhren zu einer Verschiebung des Abstandes zwischen der Spitze
und der Probe und damit zu einer Veranderung der Resonanz. Der Vorteil dieser Me-
thode ist, dass eine Deformation von empfindlichen Proben, die durch die Beruhrung
mit der Spitze im Kontaktmodus entstehen kann, vermieden wird [61].

44 3. Oberflachengebundene Nanopartikel

Kapitel 4
Brownsche Bewegung von frei
beweglichen Nanopartikeln
Die als Brownsche Bewegung bekannte Zickzackbewegung in Wasser suspendierter Teil-
chen wird erstmals im Jahr 1827 von dem englischen Botaniker Robert Brown entdeckt.
Er erkennt bei der Untersuchung wassriger Suspensionen, dass diese Bewegungserschei-
nung von den Teilchen selbst stammt und weder auf lebendige Organismen, noch auf
Stromungen oder Verdampfungen zuruckzufuhren ist [63].
Bei der Brownschen Bewegung beschreibt ein suspendiertes Teilchen eine sehr unre-
gelmaßige Bahn, die aus kurzen, geradlinigen Abschnitten zusammengesetzt ist. In
Abbildung 4.1 ist die Bewegung eines suspendierten Teilchens schematisch dargestellt.
Viele Wissenschaftler versuchen in den darauf folgenden Jahren, diese Bewegung sowohl
experimentell als auch theoretisch zu klaren. Erst die Arbeiten von Albert Einstein aus
den Jahren 1905 und 1906 machen die Brownsche Bewegung durch physikalisch messba-
re Großen zuganglich. Er sieht in der Bewegung suspendierter Teilchen eine Folge der
Warmebewegung der Molekule. Basierend auf der molekularkinetischen Warmetheorie
entwickelt er die folgende Gleichung:
NA =R · T · t
3 · π · η · r · ∆2. (4.1)
45

46 4. Brownsche Bewegung von frei beweglichen Nanopartikeln
>
4
>
4
Abbildung 4.1: Die Brownsche Bewegung eines suspendierten Teilchens. Die Ver-
schiebung eines Teilchens ∆ in einem bestimmten Zeitintervall t kann auf eine beliebige
Achse projiziert werden.
In dieser ist R die allgemeine Gaskonstante, T die Temperatur in Kelvin, NA die
Avogadro-Konstante, η die Viskositat der umgebenden Flussigkeit und ∆2 das zu er-
wartende Verschiebungsquadrat.
Im Folgenden wird zunachst auf die theoretische Untersuchung der Brownschen Bewe-
gung eingegangen, wobei die Arbeiten von Einstein im Vordergrund dieser Betrachtung
stehen. Anschließend wird ein kurzer Uberblick uber die experimentellen Arbeiten auf
diesem Gebiet seit Anfang des 20. Jahrhunderts gegeben.
4.1 Theoretische Grundlagen zur Brownschen Be-
wegung
Die Theorie zur Brownschen Bewegung haben Albert Einstein und Marian von Smo-
luchowski unabhangig voneinander entwickelt [3, 4, 5, 6]. In den heutigen Lehrbuchern
wird bei der Beschreibung der Brownschen Bewegung jedoch uberwiegend auf den theo-
retischen Arbeiten Einsteins aufgebaut [64, 65]. Einstein weist bei seinen theoretischen

4.1 Theoretische Grundlagen zur Brownschen Bewegung 47
Betrachtungen aus dem Jahre 1905 zuerst darauf hin [3]:”Es ist moglich, daß die hier
zu behandelnden Bewegungen mit der sogenannten”Brownschen Molekularbewegung“
identisch sind; die mir erreichbaren Angaben uber letztere sind jedoch so ungenau, daß
ich mir hieruber kein Urteil bilden konnte.“ Heutzutage ist der Zusammenhang allge-
mein bekannt.
" � "
"
G
5 " B 5 4 � H � �
G � E � �
# �
� �
Abbildung 4.2: Die Bewegung der Teilchen in einem Zylinder. a) Der Teilchenfluss
in Richtung eines Konzentrationsgradienten [66]. b) Die lineare Verschiebung eines
Teilchens in einem bestimmten Zeitintervall t [64].
Einsteins Betrachtungen basieren auf der Definition des Diffusionskoeffizienten mittels
des ersten Fickschen Gesetzes (Gleichung 4.2) und auf dem Stokesschen Gesetz der
laminaren Stromung um eine Kugel (Gleichung 4.13). Zur Beschreibung der unregel-
maßigen Bewegung der Teilchen wird ein Zylinder betrachtet, der durch eine Quer-
schnittsflache A in zwei gleich große Zylinderabschnitte mit den Konzentrationen c1
und c2 geteilt ist (Abbildung 4.2). Aufgrund unterschiedlicher Konzentrationen in den
beiden Abschnitten (c1 > c2) liegt ein Konzentrationsgefalle in Richtung c2 vor. Das
erste Ficksche Gesetz beschreibt den Materiefluss J in einem solchen Zylinder, d. h., es
gibt an, wie viele Teilchen dN in einem bestimmten Zeitintervall dt die Querschnitts-

48 4. Brownsche Bewegung von frei beweglichen Nanopartikeln
flache A passieren:
J =dN
A · dt= −D · dc
dx⇔ dN
dt= −D · A · dc
dx. (4.2)
D ist der Diffusionskoeffizient, und der Quotient dcdx
beschreibt den Konzentrationsgra-
dienten, der senkrecht zu der Flache A besteht. Fur die Berechnung des Diffusionskoef-
fizienten zeigt Einstein zwei Moglichkeiten. Im ersten Fall wird die lineare Verschiebung
der Teilchen in beide Richtungen in einem bestimmten Zeitintervall dt betrachtet. Die
Zahl der Teilchen, die die Querschnittsflache A in der Zeit t von rechts nach links pas-
sieren, entspricht der Zahl der Teilchen, die die entgegengesetzte Bewegung vollfuhren:
N1 =1
2· A · c1 · ∆
tund N2 =
1
2· A · c2 · ∆
t. (4.3)
Die Anzahl der Teilchen, die die Flache insgesamt in einem bestimmten Zeitintervall
passieren, ist damit gegeben durch:
dN
dt=
1
2· A · ∆(c1 − c2)
t. (4.4)
Fur den Konzentrationsgradienten gilt dann:
− dc
dx=
c1 − c2
∆. (4.5)
Durch Einsetzen von Gleichung 4.4 und Gleichung 4.5 in das erste Ficksche Gesetz
(Gleichung 4.2) ergibt sich die Beziehung zwischen dem Diffusionskoeffizienten und
dem Verschiebungsquadrat, die als das Diffusionsgesetz von Einstein und Smoluchowski
bekannt ist:
D =∆2
2 · t . (4.6)
Im zweiten Fall geht Einstein zur Beschreibung des Diffusionskoeffizienten von einer
kinetischen Betrachtung aus. Er nimmt an, dass die Teilchen die Querschnittsflache A
mit einer gleichformigen Geschwindigkeit v passieren:
dN
dt= A · c · v . (4.7)
Im stationaren Zustand kann die Geschwindigkeit v als Quotient aus der konstant
treibenden Kraft FTeilchen des Teilchens und dem Widerstandsfaktor � beschrieben
werden. Einsetzen in Gleichung 4.7 ergibt dann:
dN
dt=
A · c · FTeilchen
� . (4.8)

4.1 Theoretische Grundlagen zur Brownschen Bewegung 49
"
" � I � 5 "
� 4 �
� 4
4
4 � I � 5 4
Abbildung 4.3: Darstellung der treibenden Kraft, die durch ein Konzentrationsgefalle
ausgeubt wird [64].
Zur Beschreibung der treibenden Kraft, die ein Mol Teilchen durch ein Konzentrati-
onsgefalle erfahrt, wird ebenfalls ein Zylinder mit der Grundflache A herangezogen,
der in Abbildung 4.3 dargestellt ist. Betrachtet werden zunachst zwei Flachen mit der
Konzentration c auf der Hohe x und der Konzentration c + dc auf der Hohe x + dx.
Auf diese Flache wirkt ein kinetischer Druck p, der allgemein als p = R · T · c definiert
werden kann, wobei R die allgemeine Gaskonstante und T die Temperatur in Kelvin ist.
Die zugehorige Kraft entspricht dem Produkt aus der Flache und dem auf diese Flache
wirkenden Druck. Damit ergeben sich folgende Krafte fur die jeweiligen Flachen:
Fx1 = A · R · T · c ,
Fx2 = A · R · T · (c + dc) .
Die Differenz der entgegengesetzt wirkenden Krafte entspricht der Kraft, die auf die
Materie zwischen den beiden Flachen wirkt:
Fx = −A · R · T · dc . (4.9)
Die Anzahl der Teilchen zwischen den beiden Flachen ist gegeben mit A · c ·dx. Daraus
resultiert, dass auf ein Mol Teilchen folgende Kraft wirkt:
Fx = −R · Tc
· dc
dx= −R · T · d ln c
dx. (4.10)

50 4. Brownsche Bewegung von frei beweglichen Nanopartikeln
Mittels der Avogadro-Konstante lasst sich die auf ein Teilchen wirkende Kraft beschrei-
ben:
FTeilchen = − R · TNA · c · dc
dx. (4.11)
Das Einsetzen der Gleichungen 4.8 und 4.11 in das erste Ficksche Gesetz (Gleichung
4.2) fuhrt zur folgenden Gleichung fur den Diffusionskoeffizienten:
D =R · TNA · � . (4.12)
Die Abhangigkeit des Widerstandsfaktors � von dem Radius r und der Viskositat η
des die Kugel umgebenden Mediums wird von Stokes folgendermaßen beschrieben:
� = 6 · π · r · η . (4.13)
Diese Gleichung fuhrt schließlich zu dem Diffusionsgesetz von Stokes und Einstein:
D =R · T
NA · 6 · π · η · r . (4.14)
Das Diffusionsgesetz von Einstein und Smoluchowski (Gleichung 4.6) eingesetzt in das
Diffusionsgesetz von Stokes und Einstein (Gleichung 4.14) ergibt schließlich die Glei-
chung zur Beschreibung der Brownschen Bewegung (Gleichung 4.1). Da es sich bei der
Brownschen Bewegung um eine Zufallsbewegung handelt (vgl. Abbildung 4.1), kann
bei einer genugend großen Anzahl von Einzelmessungen das Verschiebungsquadrat ∆2
durch das mittlere Verschiebungsquadrat ∆2 in Gleichung 4.1 ersetzt werden.
Das Diffusionsgesetz von Einstein und Smoluchowski wird 1908 von P. Langevin trotz
eines anderen Ausgangspunktes bestatigt [67]. Bei der Bestimmung der Avogadro-
Konstante im Jahre 1914 erkennt I. Nordlund den Einfluss der Nahe der Wande auf die
Bewegung der Partikel [68]. Dieser Einfluss lasst sich mittels der Newtonschen Flus-
sigkeit erklaren. In dem Modell wird die Schicht einer Flussigkeit mit einer konstanten
Geschwindigkeit v gegen eine zweite Schicht verschoben, so dass eine laminare Stro-
mung zu beobachten ist (Abbildung 4.4). Die Fließgeschwindigkeit jeder Schicht hangt
linear von der außeren Kraft F ab, die die Flussigkeitsschichten bewegt. Die an der

4.1 Theoretische Grundlagen zur Brownschen Bewegung 51
$
�
5 4
5 $
4
Abbildung 4.4: Schematische Darstellung der Laminarbewegung der Newtonschen
Flussigkeit zur Definition der Viskositat [65].
Wand anhaftende Schicht ist stationar, wahrend die Bewegung der darauf folgenden
Schichten zunimmt. Die außere Kraft entspricht dem Betrag nach der Reibungskraft
FRW , die von der Viskositat η der Flussigkeit abhangt:
FRW = η · A · dv
dx, (4.15)
wobei A die Oberflache der Schicht ist. Der Geschwindigkeitsgradient dvdx
tendiert mit
zunehmendem Abstand x von der Wand gegen null, so dass auch die durch die Wand
hervorgerufene Reibungskraft gegen null geht. Folglich verlangsamt die Wechselwirkung
zwischen der Wand und dem Teilchen die Brownsche Bewegung nur in der Nahe der
Wand [64].
Dieser Einfluss wird von H. A. Lorentz durch das Verhaltnis von dem Widerstand
fur die Bewegung einer Kugel in der Nahe der Wand gegenuber dem Widerstand fur
die Bewegung einer Kugel in einem unbegrenzten Medium, wie folgt, berechnet:
FR
FR + FRW
=1
1 + 9r16a
, (4.16)
wobei r der Radius der Kugel und a der Abstand des Kugelzentrums zur Wand ist [69].
I. Nordlund fuhrt in Anlehnung an die Formel von Lorentz eine Korrekturformel ein
[68]. In dieser berucksichtigt er, dass einerseits der Abstand des Kugelzentrums von
der Wand bei der Bewegung des Teilchens standig variiert und dass andererseits in
der Mikroskopie zwei Wande, namlich Objekttrager und Deckglas, die Bewegung des

52 4. Brownsche Bewegung von frei beweglichen Nanopartikeln
Teilchens beeinflussen. Er berechnet den durchschnittlichen Widerstand auf eine Kugel
in einem begrenzten Medium F , indem er annimmt, dass die Wahrscheinlichkeit fur
jeden Wert des Abstandes zwischen Kugelzentrum und Wand , r bis d − r, gleich ist:
F∞ =F
d − 2r
∫ d−r
d
da
(1 + 9r16a
)(1 + 9r16(d−a)
). (4.17)
F∞ in Gleichung 4.17 ist die Kraft, die in einem unbegrenzten Medium auf ein Teil-
chen wirkt, d entspricht dem Abstand zwischen Deckglas und Objekttrager, also der
Schichtdicke der Probe, r ist der Radius des Teilchens und a ist der Abstand des Teil-
chenzentrums von der Wand. Der Term 1 + 9r16a
entspricht dem Term aus der Formel
nach Lorentz (Gleichung 4.16) und berucksichtigt die Anderung des Widerstandes auf
das Teilchen aufgrund der Wechselwirkung zur nahen Wand, wahrend der zweite Term
1 + 9r16(d−a)
die Anderung des Widerstandes aufgrund der Wechselwirkung mit der an-
deren Wand einbezieht. Durch Integration ergibt sich fur den Reibungswiderstand:
F∞ = F · x(4.18)
F =F∞x
mit dem Korrekturfaktor x, der wie folgt definiert ist:
x = 1 − 9r(16d + 9r)
16(d − 2r)(8d + 9r)ln
16d − 7r
25r. (4.19)
Da η ∼ F ist, kann entsprechend die scheinbare Viskositat η aus der Viskositat fur ein
unbegrenztes Medium berechnet werden:
η =η∞x
. (4.20)
4.2 Experimentelle Beobachtung der Brownschen
Bewegung
Die Brownsche Bewegung beschaftigt bereits vor den theoretischen Arbeiten von A.
Einstein und M. von Smoluchowski etliche Wissenschaftler. Schon 1900 untersucht F.

4.2 Experimentelle Beobachtung der Brownschen Bewegung 53
Exner die Abhangigkeit der Brownschen Bewegung von der Teilchengroße und der Tem-
peratur an Gummiguttsuspensionen durch die Bestimmung der Positionen der Teilchen
mittels des Abbeschen Zeichenapparates auf berußten Glasplatten [70].
1903 entwickeln R. Zigmondy und H. Siedentopf das Ultramikroskop, einen Vorgan-
ger des heutigen Dunkelfeldmikroskops. Sie untersuchen submikroskopische Teilchen
und entwickeln eine Methode zu deren Großenbestimmung. Der Ansatz basiert auf der
Berechnung der durchschnittlichen Masse an metallischem Gold in einem Kubikmilli-
meter Glas. Die Methode ist jedoch aufgrund zahlreicher Vereinfachungen und vieler
Fehlerquellen sehr ungenau [71].
T. Svedberg entdeckt 1906 folgende Abhangigkeit des Verschiebungsquadrates von der
Zeit und der Viskositat des Mediums: ∆2 = const · tη. Ohne Kenntnis der Arbeiten von
Einstein entspricht diese Beziehung der Gleichung von Einstein (vgl. Gl. 4.1) unter der
Annahme, dass die Temperatur und der Radius der Teilchen konstant sind [7, 28].
Nach Veroffentlichung der molekularkinetischen Theorie der Brownschen Bewegung
von A. Einstein unternimmt M. Seddig im Jahre 1907 als Erster eine quantitative
Untersuchung auf diesem Gebiet. Er nimmt die Verschiebung einer wassrigen Zinn-
obersuspension auf einer Fotoplatte mit einem Zeitabstand von 0, 1 Sekunden auf und
vermisst anschließend die Positionen. Seine Ergebnisse sind aufgrund eines Tempera-
turfehlers wahrend des Experiments alle großer als die berechneten [72].
Im darauf folgenden Jahr bestimmt M. V. Henri die mittlere Verschiebung von Kau-
tschukpartikeln in gleich großen Zeitintervallen. Mittels eines Kinematographen kann
er die genauen Positionen der Partikel aufzeichnen. Seine experimentellen Ergebnisse
sind insgesamt viel großer als die nach der Theorie von Einstein berechneten Werte [73].
Noch im selben Jahr konnen M. J. Perrin und sein Schuler M. Chaudesaigues die
Gultigkeit der Theorie von Einstein experimentell bestatigen. Chaudesaigues unter-

54 4. Brownsche Bewegung von frei beweglichen Nanopartikeln
sucht eine Gummiguttsuspension direkt unter dem Mikroskop und markiert mittels
eines Zeichenapparates die Positionen der Teilchen in Zeitintervallen von 30 Sekun-
den. Er verwendet zwei unterschiedliche Teilchengroßen und variiert die Viskositat des
umgebenden Mediums. Seine Ergebnisse zeigen den Einfluss des Radius, der Tempe-
ratur und der Viskositat auf die Brownsche Bewegung entsprechend den theoretischen
Arbeiten Einsteins. Ferner ermittelt er die Avogadro-Konstante mit einem Wert von
NA = 6, 4 · 1023 mol−1 [74, 75].
1909 setzt T. Svedberg zusammen mit Innouye seine Arbeiten auf dem Gebiet der
Brownschen Bewegung fort. Mittels des Ultramikroskops und eines extra fur diese
Arbeiten konstruierten Zeichenapparates bestimmen sie die Verschiebung von Gold-
hydrosolen in genau definierten Zeitintervallen. Fur einige ihrer hergestellten Sole mit
kleinem Radius stimmen die ermittelten Verschiebungen mit den nach Einsteins Theo-
rie berechneten Verschiebungen uberein [28].
Eine wichtige Verbesserung der Ergebnisse erzielte I. Nordlund 1914 mit der Einfuh-
rung der Korrekturformel zur Berucksichtigung des Einflusses der Wande auf die Kugel
(siehe Gl. 4.17) [68].
Auch in den darauf folgenden Jahren beschaftigen sich viele Wissenschaftler mit die-
ser Thematik. Mit der Entwicklung der dynamischen Lichtstreuung, der ebenfalls die
Brownsche Bewegung zugrunde liegt, tritt das Interesse an den mikroskopischen Me-
thoden jedoch in den Hintergrund.
Im Jahre 1988 stellt H. Kruglak eine moderne Version der Camera Obscura von Per-
rin vor. Hierzu wird eine TV-Kamera auf einem Mikroskop angebracht, die die Be-
obachtung der Brownschen Bewegung von Latexpartikeln mit einem Durchmesser von
0, 93− 1, 305 µm auf einem Monitor ermoglicht. In definierten Zeitintervallen wird die
Position eines Teilchens auf einer auf dem Monitor haftenden Folie markiert. Somit
konnen die Verschiebungswerte eines Teilchens bestimmt werden, aus denen anschlie-

4.2 Experimentelle Beobachtung der Brownschen Bewegung 55
ßend deren Standardabweichung sowie die Avogadro-Konstante berechnet werden [76].
W. Schaertl und H. Sillescu nutzen 1992 zur Untersuchung der dynamischen Eigen-
schaften von Kolloidpartikeln die computergestutzte digitale Bildverarbeitung. Auf
einem Mikroskop, das sowohl mit Dunkelfeldbeleuchtung als auch mit Fluoreszenz-
beleuchtung ausgestattet ist, wird zur Aufzeichnung der Bewegung der Partikel eine
hoch lichtempfindliche Videokamera angebracht. Die Auswertung der Bewegungen er-
folgt mittels eines Computers. Um die Wechselwirkung zwischen Kolloidpartikeln und
Glaswand moglichst gering zu halten, werden die Glaser mit Octadecyltrichlorsilan
beschichtet. Durch Hinzufugen von so genannten Spacer-Partikeln wird eine genau
definierte Schichtdicke erzielt. Das Ziel dieser Arbeit ist die Bestimmung des Diffu-
sionskoeffizienten in Abhangigkeit des Verhaltnisses der Partikelgroße zur Schichtdicke
und die Bestimmung der Abhangigkeit der Diffusion von der Partikelkonzentration [77].
Der Schwerpunkt der weiteren Arbeiten auf diesem Gebiet Ende des 20. Jahrhun-
derts liegt in der Entwicklung verschiedener Algorithmen zum Particle Tracking (vgl.
Kapitel 2.3.3) [77, 78, 79]. Diese Algorithmen werden vor allem zur Berechnung der
Diffusionskoeffizienten von kolloidalen Partikeln in einem Radienbereich von 300− 750
nm herangezogen. Ein weiterer wichtiger Aspekt ist die Verbesserung der Statistik.
Wahrend zu Beginn nur die Beobachtung der Verschiebungen eines Partikels moglich
ist, besteht nun die Moglichkeit, gleichzeitig mehr als 100 Partikel zu beobachten [80].
Bei dem Algorithmus, der in dem Kapitel 6.2.1 beschrieben wird, steht die Bestimmung
der Partikelgroßen und deren Verteilung von einer großen Anzahl einzelner kolloidaler
Nanopartikel im Vordergrund [81, 82, 83].
Die Beobachtung der Brownschen Bewegung mittels digitaler Bildverarbeitung dient
mittlerweile nicht mehr nur der Bestimmung des Diffusionskoeffizienten oder der Avoga-
dro-Konstante, vielmehr besteht auch ein wesentliches Interesse in der Untersuchung
der Dynamik in biochemischen Systemen. Beispielsweise untersucht Forstner im Jahre
2003 raumliche Inhomogenitaten in Membranen monomolekularer Lipidschichten. Zur

56 4. Brownsche Bewegung von frei beweglichen Nanopartikeln
Bestimmung der Spuren von fluoreszierenden Nanopartikeln in diesen Schichten und
des Diffusionskoeffizienten wird der Particle Tracking-Algorithmus nach Crocker et al.
herangezogen [84].

Kapitel 5
Experimenteller Teil
5.1 Mikroskopie und digitale Bildverarbeitung
Der in dieser Arbeit verwendete experimentelle Aufbau ist bereits in der Abbildung
2.13 in dem Kapitel 2.3 schematisch dargestellt. Dieser Aufbau ist prinzipiell unabhan-
gig von dem verwendeten Mikroskop. Wahrend zur Beobachtung von oberflachenge-
bundenen Nanopartikeln ausschließlich die Auflicht-Fluoreszenzmikroskopie verwendet
wird, kann die Großenbestimmung von frei beweglichen Nanopartikeln sowohl mittels
der Auflicht-Fluoreszenz- als auch der Durchlicht-Dunkelfeldmikroskopie erfolgen. Die
Ausstattung und die technischen Daten der entsprechenden Mikroskope werden im
Folgenden erlautert.
5.1.1 Auflicht-Fluoreszenzmikroskopie
Zur Untersuchung fluoreszierender Objekte wird das Universalmikroskop ORTHO-
PLAN (Leitz, Wetzlar, Deutschland) mit den aufgefuhrten Objektiven (Leitz) verwen-
det:
100x Objektiv NA: 0, 90 Planachromat (Leitz)
50x Objektiv NA: 0, 85 Planachromat (Leitz)
40x Objektiv NA: 0, 65 Planachromat (Lomo)
32x Objektiv NA: 0, 60 Planachromat (Leitz)
20x Objektiv NA: 0, 40 Planachromat (Leitz)
57

58 5. Experimenteller Teil
Das Mikroskop ist zusatzlich mit der Auflicht-Fluoreszenzbeleuchtung LRF 4/22 (Leica,
Wetzlar, Deutschland) ausgestattet, die die folgenden Komponenten enthalt:
Anregungsfilter 450 − 490 nm
Dichromatischer Teilerspiegel 510 nm
Langpassfilter 515 nm
Quecksilber-Hochdrucklampe HBO 103W/2 Osram, Munchen, Deutschland
Zum Anbringen der CCD-Kamera auf dem Trinokular stehen zwei Adapter, so genann-
te C-Mount (1x und 0, 5x) der Firma Leica, Wetzlar, Deutschland, zur Verfugung.
Der Kreuztisch des Mikroskops, der in der Abbildung 5.1 wiedergegeben ist, ist mit zwei
bipolaren Schrittmotoren (TANDON, Typ: KP4M2-203, Conrad, Hirschau, Deutsch-
land) fur die Bewegung in x- und y-Richtung nachgerustet, die uber eine Schrittmotor-
steuerkarte SMC1500 (Conrad, Hirschau, Deutschland) computergesteuert angetrieben
werden konnen.
Abbildung 5.1: Der Kreuztisch des Auflicht-Fluoreszenzmikroskops von der Unter-
seite. Zwei Schrittmotoren, die uber eine Schrittmotorsteuerung angetrieben werden,
ermoglichen die computergesteuerte Bewegung des Tisches in x- und y-Richtung.

5.1 Mikroskopie und digitale Bildverarbeitung 59
5.1.2 Durchlicht-Dunkelfeldmikroskopie
Die Ausstattung des Universalmikroskops BIOLAR (PZO, Polen) besteht aus folgenden
Komponenten:
10x Objektiv NA: 0, 24 Achromat PZO
40x Objektiv NA: 0, 75 Wasserimmersion Achromat LOMO
Dunkelfeldkondensor NA: 1, 2 − 1, 4 Olimmersion Typ: Kardioid PZO
Halogenlampe 24V/250W Osram
C-Mount 1x PZO
In den Strahlengang der Beleuchtung wird zur Filterung des infraroten Lichtanteils eine
mit Wasser gefullte Kuvette eingebaut, um eine Erwarmung der Probe zu vermeiden
[81, 82].
5.1.3 Digitale Bildverarbeitung
Zur Bildaufnahme und Bildanalyse werden folgende Komponenten verwendet:
CCD-Kamera: KP-F1 Hitachi, Japan
effektive Pixel: 782(H) x 582(V)
Pixelgroße 8, 3 × 8, 3 µm
1/25 sek/Bild
Bildverarbeitungskarte: Occulus F/64-DSP Coreco, Kanada
Bildverarbeitungssoftware: PicColor� FIBUS, Dusseldorf,
Deutschland
Personalcomputer: Pentium III, 950 MHz
Intel 440BX, 512 MByte
40 GByte Festplattenspeicher

60 5. Experimenteller Teil
5.2 Oberflachengebundene Nanopartikel
5.2.1 Fluoreszenzspektroskopische Untersuchungen
Die UV-Spektren werden mit einem Spektrophotometer Lambda 40 der Firma Perkin-
Elmer, Massachusetts, USA aufgenommen.
Das Lumineszenzspektrometer LS50B und die zugehorige Auswertungssoftware FL-
WinLAB (Version 1.10) der Firma Perkin-Elmer, Massachusetts, USA dienen zur Auf-
nahme der Fluoreszenzspektren.
Zur Entwicklung eines fluoreszierenden Polymers werden die Polymere mr − I 8000
und mr − I 9000 der Firma Microresist Technology (MRT), Berlin, Deutschland von
MRT mit den drei Farbstoffen Fluorescein, Anthracen und einem Perylidenderivat mar-
kiert. Fur die Messungen der UV- und der Fluoreszenzspektren werden die Polymere
in Dichlormethan mit einer Konzentration von c = 1 g/l gelost.
5.2.2 Stabilitatsbestimmung des Farbstoffes
Die polymeren Feststoffe werden gemorsert und fein verteilt auf einen Objekttrager ge-
geben. Dieser wird unter dem Fluoreszenzmikroskop fur mehr als 80 Minuten belichtet.
Wahrend der Belichtung werden Sequenzen mit je 218 Bildern mit unterschiedlichen
Zeitabstanden ∆t aufgenommen. Wahrend der gesamten Belichtungszeit werden drei
Aufnahmeblocke in Abstanden von 30 Minuten getatigt. Ein Aufnahmeblock setzt sich
wiederum aus drei Aufnahmeeinheiten zusammen, die wie folgt gegliedert sind.
Nr. Einheit ∆t Anzahl Bilder
1. Sequenz 0, 04 sek. 218
Pause 120 sek. 0
2. Sequenz 0, 08 sek. 218
Pause 120 sek. 0
3. Sequenz 0, 2 sek. 218
Pause 120 sek. 0

5.2 Oberflachengebundene Nanopartikel 61
Der Helligkeitsverstarker der Kamera ist manuell eingestellt, so dass ein fester Wert fur
alle Messungen gesetzt ist. Der gewahlte Bildausschnitt betragt 60, 87 µm × 60, 87 µm.
Zur punktuellen Bestimmung der Grauwerte in jedem Bild werden vier verschiedene
Positionen festgesetzt und deren Grauwerte bestimmt.
Punkt x-Koordinate [Pixel] y-Koordinate [Pixel]
a 280 200
b 400 260
c 370 300
d 370 322
Die Bildverarbeitungssoftware gibt die Grauwerte automatisch aus. Die Daten werden
dann mit den zugehorigen Zeiten in eine Excel-Tabelle ubertragen und grafisch ausge-
wertet.
Zur flachenhaften Bestimmung der Grauwerte wird ein Grauwerthistogramm von der
Bilddiagonalen des zehnten Bildes der ersten Aufnahmeeinheit (mit t = 0, 04 Sekun-
den) eines jeden Blockes gewahlt. Zur Erstellung der Grauwerthistogramme steht ein
in der Bildverarbeitungssoftware implementiertes Programm zur Verfugung, das die
Grauwerte der Diagonalen in einer Datei abspeichert. Die Datei kann in Excel impor-
tiert und entsprechend bearbeitet werden. Die Diagramme werden mit Excel oder mit
Origin erstellt.
5.2.3 Bestimmung und Charakterisierung von Defekten auf
den Pragungen und den Stempeln
Die untersuchten Nanopragungen werden von der Arbeitsgruppe Mikrostrukturtechnik
von Frau Prof. Dr.-Ing. H.-C. Scheer des Fachbereichs Elektro- und Informationstech-
nik der Bergischen Universitat Wuppertal, Deutschland hergestellt. Hierbei handelt es
sich um Si-Wafer, die mittels Spin-Coating mit dem mit einem Perylidenfarbstoff mar-
kierten Polymer mr − I 8000 der Firma MRT beschichtet sind. Die entsprechenden

62 5. Experimenteller Teil
Parameter fur die Nanoimprintlithographie, die Antihaftbeschichtung sowie die Her-
stellung des Stempels sind der Literatur zu entnehmen [40, 41, 85].
Der Stempel besteht aus poly-Si/SiO2 auf einem Si-Wafer. Die Struktur wird von
der Firma VTT Electronics, Finnland mittels UV-Lithographie und Trockenatzung
hergestellt. Die Struktur des kompletten Stempels setzt sich aus vier 20 × 20 mm2
großen, quadratischen Feldern zusammen. In der Abbildung 5.2 ist die Struktur eines
solchen quadratischen Feldes schematisch dargestellt.
JJJ
J JJ JJ J
� �
��
8
8 8 8 8
�
1
1
�
�
Abbildung 5.2: Schematische Darstellung der Strukturen auf der Stempeloberflache.
A: 100× 100 µm2 große Flachen; B: 50× 50 µm2 große Flachen; C: Y-Baugruppen; D:
5 µm breite Linien und Abstande; E: 400 nm breite Linien und Abstande; F: Bander
bestehend aus positiven und negativen Punkten und Linien.
Es besteht aus vier 5× 5 mm2 großen Bereichen mit Linien und Abstanden einer Breite
von 400 nm (E) bzw. von 5 µm (D) sowie mit mikrometergroßen Y-Baugruppen (C).
Zwischen diesen vier Bereichen befinden sich die so genannten Bander mit positiven
und negativen Linien und Punkten (F). Die vier Bereiche sind von einem Rand um-
geben, der sich aus 3 × 3 mm2 großen Quadraten zusammensetzt (A und B). Diese
sind wiederum in Felder einer Große von 300× 300 µm2 unterteilt, in deren Mitte sich

5.2 Oberflachengebundene Nanopartikel 63
erhohte 100×100 µm2 (A) bzw. 50×50 µm2 große, quadratische Flachen (B) befinden
[85, 86].
Die Untersuchungen mittels der Fluoreszenzmikroskopie werden mit den folgenden Ob-
jektiven und den entsprechenden Großen der Bildausschnitte durchgefuhrt:
Objektiv Bildausschnitt
100x/0, 90 130 × 90 µm2
50x/0, 85 265 × 180 µm2
32x/0, 60 415 × 275 µm2
In der nachfolgenden Tabelle 5.1 sind die in Kapitel 6.1.2 dargestellten fluoreszenzmi-
kroskopischen Aufnahmen mit den verwendeten Objektiven aufgelistet.
Tabelle 5.1: Verwendete Objektive fur die einzelnen fluoreszenzmikroskopischen Auf-
nahmen und die entsprechenden Felder auf dem Stempel.
Bildnr. Felder Objektiv Bildnr. Felder Objektiv
6.12 E 50x 6.17 a) E 50x
6.13 D 50x b) E 50x
6.14 a) C 50x c) E 50x
b) C 50x d) E 50x
6.15 a) A 32x 6.18 a) F 100x
b) A 50x b) F 100x
c) A 100x 6.19 a) D 50x
6.16 a) A 32x b) D 50x
b) A 50x c) D 50x
Der Helligkeitsverstarker der Kamera ist fixiert. Die Auswertung der fluoreszenzmikro-
skopischen Aufnahmen erfolgt, wie in Kapitel 5.2.2 beschrieben, mit der Bildverarbei-
tungssoftware PicColor und den darin implementierten Programmen.

64 5. Experimenteller Teil
Die Profilometriemessungen werden mit einem Profilometer Veeco Dektak 3ST der
Firma Veeco Tucson Inc., Arizona, USA gemessen. Die SEM-Bilder sind mit einem
Rasterelektronenmikroskop Topcon SM300 der Firma ISS Group Services Ltd., Man-
chester, USA aufgenommen [87].
5.2.4 Qualitatssicherung wahrend einer Serie von Nanopra-
gungen
5.2.4.1 Stempel und Polymer
Fur die Untersuchung der Qualitat der Pragestempel in einer Serie von Nanopragungen
werden drei verschiedene Stempel verwendet:
1. Der Stempel besteht aus 1“ Si/SiO2-Wafer (1“ = 1 Zoll). Er wird mittels Elek-
tronenstrahllithographie, einem Lift-Off-Prozess mit Chrom und der reaktiven
Ionenatzung hergestellt.
2. Der Stempel besteht aus dem Polymer mr − I 6000 auf einem 1“ Wafer.
3. Der Si-Stempel wird durch UV-Lithographie und reaktive Ionenatzung erzeugt.
Die Herstellungsmethoden und Nachbehandlungen sowie die Eigenschaften der Stem-
pel sind in Kapitel 3.1 beschrieben [83].
Die Stempel 1 und 2 besitzen dieselbe strukturierte Oberflache, die in Abbildung 5.3
schematisch dargestellt ist. Die Oberflache des Stempels entspricht einer 3 × 3 An-
ordnung der Felder, die in einem Abstand von 4 mm voneinander entfernt liegen. Die
strukturierten Felder 2, 4, 6, 8 (Even) sind 200×200 µm2 groß. Sie haben einen Bereich
mit einzelnen Punkten in einer Großenordnung von 20 − 100 nm und drei Bereiche
mit 100 − 200 nm breiten Linien in Abstanden von 300 nm. Die strukturierten Felder
1, 3, 5, 7, 9 (Odd) haben eine Flache von 100× 100 µm2. Bei den Strukturen handelt es
sich um 100 − 200 nm breite Linien mit einem Abstand von 200 nm und Punkten in
einer Großenordnung von 2 µm, 1 µm und 200 nm.

5.2 Oberflachengebundene Nanopartikel 65
�
%
+
'
&
(
)
*
Abbildung 5.3: Schematische Darstellung der Anordnung der strukturierten Felder
der Stempel 1 und 2. Die Felder 1, 3, 5, 7, 9 sind 100×100 µm2 groß, die Felder 2, 4, 6, 8
haben eine Große von 200 × 200 µm2.
Der Stempel 3 entspricht dem in Kapitel 5.2.3 beschriebenen und in der Abbildung
5.2 schematisch dargestellten Stempel. Alle drei Stempel sind mit einer Antihaftbe-
schichtung aus Tridecafluoro-(1,1,2,2)-tetrahydrooktyl-trichlorsilan (F13 − TCS) ver-
sehen [41, 42]. Zum Pragen wird das fluoreszenzmarkierte Polymer mr − I 8000 der
Firma MRT verwendet.
5.2.4.2 Prageprozess und Qualitatssicherung mittels Fluoreszenzmikrosko-
pie
Die Nanopragungen werden in einem 2, 5“ Nanoimprinter der Firma Obducat, Malmo,
Schweden erzeugt. Diese Serienpragungen sind eine Kooperation mit dem Institut der
Materialwissenschaften von Frau Prof. C. M. Sotomayor Torres der Bergischen Uni-
versitat Wuppertal, Deutschland, dem Institut fur Festkorperphysik von Herrn Prof.
L. Montelius der Universiat Lund, Schweden und dem Institut fur Physikalische Che-
mie von Herrn Prof. C. Mayer der Universitat Duisburg-Essen, Deutschland, die am
Standort der Universitat Lund durchgefuhrt werden. Die Prozessparameter der jeweili-
gen Pragungen sind den nachfolgenden Tabellen zu entnehmen. Der Druck betragt bei
allen Pragungen 50 bar [88].
66 5. Experimenteller Teil
Tabelle 5.2: Parameter zur Nanopragung einer Serie mit einem 1“ Si/SiO2 Nano-
stempel (Stempel 1).
Nr. Aufheiz- Abkuhl- Temp. Nr. Aufheiz- Abkuhl- Temp.
rate [min.] rate [min.] [◦C] rate [min.] rate [min.] [◦C]
1 4, 03 3, 34 180 11 3, 06 4, 20 180
2 3, 14 3, 14 180 12 3, 07 4, 21 180
3 3, 23 3, 24 180 13 3, 08 4, 28 180
4 3, 02 3, 56 180 14 3, 34 3, 25 180
5 3, 02 4, 56 180 15 3, 30 4, 15 190
6 2, 52 4, 10 180 16 3, 30 4, 14 190
7 2, 50 4, 09 180 17 3, 37 3, 97 190
8 2, 50 4, 10 180 18 4, 04 5, 57 210
9 2, 44 4, 15 180 19 4, 10 5, 49 210
10a 3, 55 6, 25 180 20 3, 53 6, 44 210
alaterale Verschiebung des Stempels wahrend der Pragung
Tabelle 5.3: Pragebedingungen bei einer seriellen Nanopragung mit einem 1“ Stempel
bestehend aus dem Polymer mr − I 6000 (Stempel 2).
Nr. Aufheizrate [min.] Abkuhlrate [min.] Temperatur [◦C]
1 3, 27 3, 50 180
2 3, 05 3, 56 180
3 2, 56 4, 09 180
4 2, 54 4, 38 180
5 2, 58 4, 25 180
6 2, 50 4, 29 180
7 2, 50 4, 50 180
8 2, 44 4, 55 180
9 3, 11 3, 48 180
10 2, 57 5, 07 180
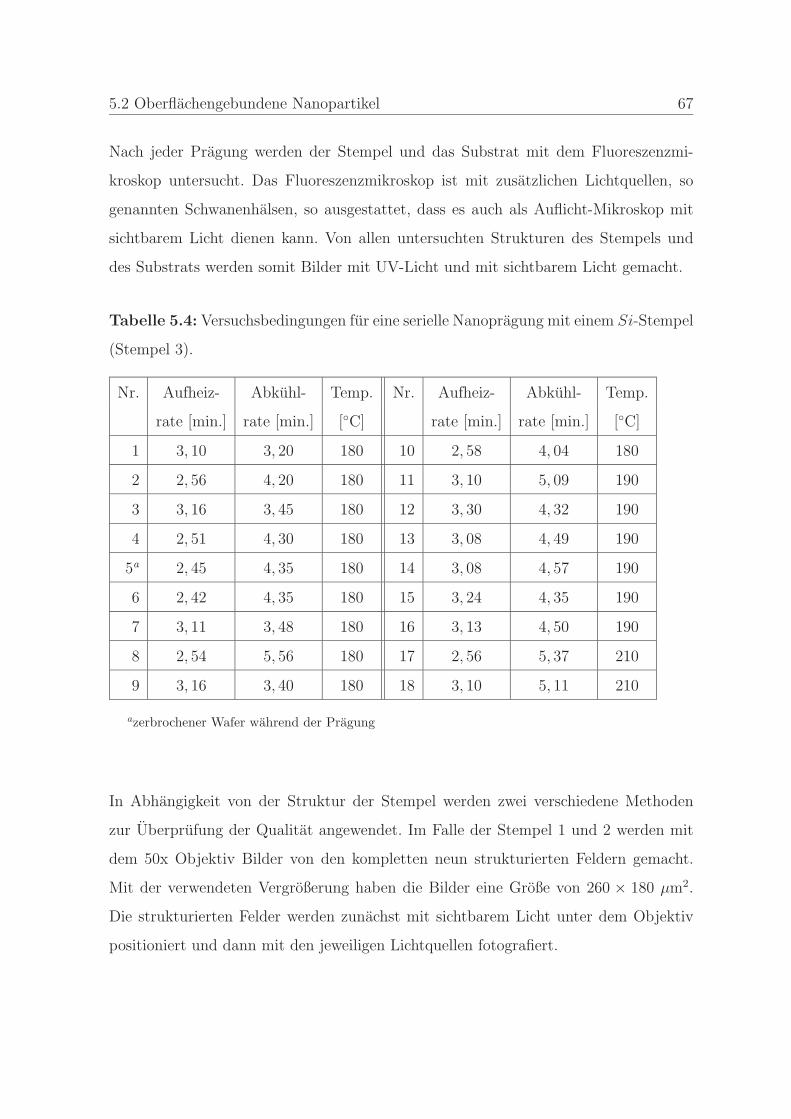
5.2 Oberflachengebundene Nanopartikel 67
Nach jeder Pragung werden der Stempel und das Substrat mit dem Fluoreszenzmi-
kroskop untersucht. Das Fluoreszenzmikroskop ist mit zusatzlichen Lichtquellen, so
genannten Schwanenhalsen, so ausgestattet, dass es auch als Auflicht-Mikroskop mit
sichtbarem Licht dienen kann. Von allen untersuchten Strukturen des Stempels und
des Substrats werden somit Bilder mit UV-Licht und mit sichtbarem Licht gemacht.
Tabelle 5.4: Versuchsbedingungen fur eine serielle Nanopragung mit einem Si-Stempel
(Stempel 3).
Nr. Aufheiz- Abkuhl- Temp. Nr. Aufheiz- Abkuhl- Temp.
rate [min.] rate [min.] [◦C] rate [min.] rate [min.] [◦C]
1 3, 10 3, 20 180 10 2, 58 4, 04 180
2 2, 56 4, 20 180 11 3, 10 5, 09 190
3 3, 16 3, 45 180 12 3, 30 4, 32 190
4 2, 51 4, 30 180 13 3, 08 4, 49 190
5a 2, 45 4, 35 180 14 3, 08 4, 57 190
6 2, 42 4, 35 180 15 3, 24 4, 35 190
7 3, 11 3, 48 180 16 3, 13 4, 50 190
8 2, 54 5, 56 180 17 2, 56 5, 37 210
9 3, 16 3, 40 180 18 3, 10 5, 11 210
azerbrochener Wafer wahrend der Pragung
In Abhangigkeit von der Struktur der Stempel werden zwei verschiedene Methoden
zur Uberprufung der Qualitat angewendet. Im Falle der Stempel 1 und 2 werden mit
dem 50x Objektiv Bilder von den kompletten neun strukturierten Feldern gemacht.
Mit der verwendeten Vergroßerung haben die Bilder eine Große von 260 × 180 µm2.
Die strukturierten Felder werden zunachst mit sichtbarem Licht unter dem Objektiv
positioniert und dann mit den jeweiligen Lichtquellen fotografiert.

68 5. Experimenteller Teil
Zur Untersuchung der Qualitat des Stempels 3 wird eine Sequenz von Bildern entlang
einer Achse in einem Halbautomatikverfahren aufgenommen. In dem Halbautomatik-
verfahren wird der Mikroskoptisch mittels der Schrittmotoren computergesteuert um
einen bestimmten, festgelegten Abstand in der x-Richtung verschoben. Zur Untersu-
chung wird das 20x Objektiv verwendet, so dass der Bildausschnitt eine Große von
560 × 405 µm2 hat. Der Mikroskoptisch wird somit nach jeder Aufnahme jeweils um
560 µm entlang der x-Richtung verschoben. Von jeder Position wird sowohl ein Bild
mit UV-Beleuchtung als auch ein Bild mit sichtbarem Licht aufgenommen. Die Kamera
wird uber ein Makro der Bildverarbeitungssoftware so gesteuert, dass das Auslosen der
Kamera uber die Computertastatur erfolgt, sobald der Mikroskoptisch die gewunschte
Position erreicht hat. Die Bilder werden als Sequenz mit fortlaufender Nummer gespei-
chert.
Sowohl in dem manuellen Verfahren, das fur die Stempel 1 und 2 angewendet wird,
als auch in dem Halbautomatikverfahren, das fur die Untersuchung des Stempels 3
genutzt wird, wird zur Aufnahme der Bilder mit UV-Beleuchtung der Kontrast mittels
der Bildverarbeitungssoftware auf 150 und die Helligkeit auf 220 gesetzt. Zur Bear-
beitung und Auswertung der digitalen Bilder wird ein Auswertungsprogramm mit der
Software Mathematica (Version 4.1, c©1988 − 2000 Wolfram Research Inc., Illinois,
USA) geschrieben, das im Anhang A.1 angefuhrt wird.

5.3 Großenbestimmung von frei beweglichen Nanopartikeln 69
5.3 Großenbestimmung von frei beweglichen Nano-
partikeln
5.3.1 Polystyrolstandards
5.3.1.1 Polystyrolstandards in der Durchlicht-Dunkelfeldmikroskopie
Zur Uberprufung der Großenbestimmung von Nanopartikeln werden die in der Ta-
belle 5.5 aufgefuhrten Polystyrolpartikelstandards (Nanosphere� Size Standard, Duke
Scientific Corporation, Palo Alto, Californien, USA) eingesetzt.
Tabelle 5.5: Ubersicht der verwendeten Polystyrolstandards zur Großenbestimmung
mittels der Durchlicht-Dunkelfeldmikroskopie.
Name 3100A 3240A 3400A
Nennradius 50 nm 120 nm 200 nm
zertifizierter Radiusa 51 nm ± 1, 5 nm 120 nm ± 3 nm 202 nm ± 2 nm
Standardabweichunga 1, 5 nm 1, 65 nm 2, 95 nm
hydrodynamischer
Radiusb
50 − 52, 5 nm 119, 5 − 122, 5 nm 207, 5 − 217, 5 nm
aLt. Herstellerangaben mittels Transmissionselektronenmikroskopie (TEM) bestimmt.bLt. Herstellerangaben mittels Photonenkorrelationsspektroskopie (PCS) ermittelt.
Die Standards werden als wassrige Suspensionen mit einer Dichte von 1, 05 g/cm3 und
einem Feststoffanteil von 1 Gew.% geliefert.
5.3.1.2 Polystyrolstandards in der Auflicht-Fluoreszenzmikroskopie
Die Großenbestimmung von Nanopartikeln mittels der Fluoreszenzmikroskopie erfolgt
an zwei fluoreszierenden Partikelstandards:
a) Fluoreszierender Polystyrolstandard G200 (Nanosphere� Size Standard, Duke
Scientific Corporation, Palo Alto, Californien, USA): Nennradius 100 nm, Stan-

70 5. Experimenteller Teil
dardabweichung 1 nm, Absorptionsmaximum bei 468 nm, Emissionsmaximum bei
508 nm, Dichte der wassrigen Suspension 1, 05 g/cm3, 1 Gew.% Feststoffanteil.
b) Nanospharen aus PMMA, die mit Cumarin 6 markiert sind. Diese werden von
Zentel et. al. hergestellt [89]. Zentel et. al. bestimmen einen mittleren Radius von
165 nm aus SEM-Bildern. Das Absorptionsmaximum des Farbstoffes liegt bei 458
nm und das Emissionsmaximum bei 505 nm.
5.3.2 Probenvorbereitung
Die Messungen wassriger Suspensionen mit mikroskopischen Methoden erfordern Ob-
jekttrager und Deckglaser mit hydrophoben Oberflachen. Zur Beschichtung der Glaser
werden diese zunachst mit konzentrierter Schwefelsaure gereinigt, dann nacheinander
mit Wasser und Ethanol gespult und schließlich getrocknet. Restliche Staubpartikel
konnen mit Druckluft entfernt werden. In einer Glovebox werden die gereinigten Gla-
ser unter Stickstoffatmosphare mit Octadecytrichlorsilan (Aldrich Chemical Company,
Missouri, USA) beschichtet und uber Nacht getrocknet [77]. Uberschussige Silanreste
auf der Oberflache werden mit n-Hexan entfernt.
Ein Tropfen eines verdunnten Polystyrolstandards wird auf einen Objekttrager gegeben
und unter einem Deckglas gleichmaßig verteilt. Um Stromungen in der Probe und das
Austrocknen der Probe zu verhindern, wird der Spalt zwischen dem Objekttrager und
dem Deckglas mit flussigem Wachs verschlossen [81, 82].
Zur Verdunnung der Polystyrolstandards werden im Allgemeinen 0, 03 ml der gekauften
Suspensionen mit 100 ml Wasser versetzt. Die Berechnung der nachfolgend angegebe-
nen Partikelkonzentrationen basiert auf den Angaben des Herstellers, dass die original
Suspensionen einen Feststoffanteil von 1 Gew.% und eine Dichte von 1, 05 g/cm3 haben.

5.3 Großenbestimmung von frei beweglichen Nanopartikeln 71
5.3.2.1 Partikelkonzentrationen in der Dunkelfeldmikroskopie
Zur Bestimmung unimodaler Großenverteilungen werden folgende Partikelkonzentra-
tionen in Abhangigkeit von dem Partikelradius eingesetzt:
c 51nm = 3, 06 · 1012 Teilchen/l
c100nm = 2, 17 · 1011 Teilchen/l
c120nm = 4, 78 · 1011 Teilchen/l
c202nm = 1, 04 · 1011 Teilchen/l
Die Messung einer bimodalen Großenverteilung erfolgt an einer Mischung aus zwei
Partikelstandards (120 nm und 202 nm) mit folgenden Konzentrationen:
c120nm = 2, 41 · 1011 Teilchen/l
c202nm = 3, 91 · 1011 Teilchen/l
5.3.2.2 Partikelkonzentrationen in der Fluoreszenzmikroskopie
Zur Großenbestimmung von Nanopartikeln mittels der Fluoreszenzmikroskopie werden
die nachstehenden Partikelkonzentrationen eingesetzt:
c100nm = 7, 24 · 109 Teilchen/l
c165nm � 1010 Teilchen/l
5.3.3 Messung und Datenauswertung
Alle Messungen zur Partikelgroßenbestimmung werden unabhangig von der mikrosko-
pischen Methode mit folgenden Parametern durchgefuhrt.
Bildgroße 240 × 240 Pixel
Aufnahmerate 25 Bilder/sek.
Zeit t 0, 08 sek.
Anzahl Bilder pro Sequenz 218
Anzahl Sequenzen pro Messung 30

72 5. Experimenteller Teil
Der Umrechnungsfaktor von Pixel in Mikrometer wird mittels einer geeichten Objekt-
mikrometerskala (Leitz) bestimmt, die eine Skalierung von 200 Teilen auf 2 mm hat.
Die ermittelten Umrechnungsfaktoren fur die jeweiligen Mikroskope mit den entspre-
chenden Objektiven sind nachfolgend aufgefuhrt.
Dunkelfeldmikroskop 40x Objektiv 0, 2402 µm/Pixel
Fluoreszenzmikroskop 50x Objektiv 0, 4167 µm/Pixel
5.3.3.1 Großenbestimmung mittels der Dunkelfeldmikroskopie
Entsprechend dem Umrechnungsfaktor betragt die in der Dunkelfeldmikroskopie ver-
wendete Bildgroße 57, 7× 57, 7 µm2. Fur alle Aufnahmen werden die Standardeinstel-
lungen der CCD-Kamera gewahlt:
Kontrast : 0
Helligkeit : 127 .
Die Parameter der einzelnen Messungen sind der Tabelle 5.6 zu entnehmen. Die Aus-
wertung der Sequenzen erfolgt ohne die SUB128-Prozedur (siehe Kapitel 6.2.1).
Tabelle 5.6: Messparameter zur Großenbestimmung von Nanopartikeln mittels der
Dunkelfeldmikroskopie.
Poylstyrolstandard Temperatur [◦C] Schichtdicke [µm] Schwellenwert [Pixel]
r = 51 nm 22, 5 4 125
r = 100 nm 20, 8 3 200
r = 120 nm 22, 5 5 145
r = 202 nm 22, 7 5 180
r1/r2 = 120/202 nm 20, 5 5 200

5.3 Großenbestimmung von frei beweglichen Nanopartikeln 73
5.3.3.2 Großenbestimmung mittels der Fluoreszenzmikroskopie
Die Bildgroße bei Verwendung der Fluoreszenzmikroskopie betragt 100 × 100 µm2.
In der Tabelle 5.7 sind die notwendigen Parameter angegeben. Die Auswertung der
Sequenzen erfolgt mit dem SUB128-Verfahren.
Tabelle 5.7: Messparameter zur Großenbestimmung von Nanopartikeln mittels der
Fluoreszenzmikroskopie.
Poylstyrolstandard r = 100 nm r = 165 nm
Aufnahme: Kontrast 100 0
Helligkeit 200 127
Auswertung: Temperatur [◦C] 21, 8 20, 7
Schichtdicke [µm] 4 6
Grauschwellenwert 135 135

74 5. Experimenteller Teil

Kapitel 6
Ergebnisse und Diskussion
6.1 Oberflachengebundene Nanopartikel
Die Nanotechnologie hat in den letzten Jahren weltweit in allen Bereichen der Wis-
senschaft und Technik einen starken Entwicklungsprozess erlebt. Besonders im Bereich
der Halbleitertechnologie sind die ublichen Methoden zur Darstellung von Nanostruk-
turen, wie z. B. die optische Lithographie, bedingt durch die Wellenlange des Lichtes
an ihre Auflosungsgrenzen gelangt. Die zunehmende Miniaturisierung von Strukturen
erfordert somit die Entwicklung neuer Methoden, zu denen auch die Nanoimprintlitho-
graphie zahlt. Die Forschungsaktivitaten in der Nanoimprintlithographie sind vielfaltig.
Sie reichen von der Entwicklung spezieller Polymere uber die Optimierung des Prage-
prozesses bzw. der Kombination verschiedener lithographischer Methoden bis hin zur
Entwicklung neuer Pragemaschinen. Der Aspekt der Qualitatssicherung wird jedoch
weitestgehend vernachlassigt. Im Allgemeinen wird die Rasterelektronenmikroskopie
(SEM) oder die Rasterkraftmikroskopie (AFM) zur Darstellung der nanostrukturier-
ten Oberflachen herangezogen. Diese Methoden erfordern jedoch zum Teil eine Pro-
benvorbereitung der zu untersuchenden Probe oder die Messung benotigt den direkten
Kontakt zur Probe. Bei einer industriellen Anwendung wird dadurch ein Zuruckbringen
des Wafers in den Produktionsprozess ausgeschlossen (s. Kapitel 3.1).
Die herkommliche optische Mikroskopie wird zur Inspektion der nanostrukturierten
75

76 6. Ergebnisse und Diskussion
Oberflachen nicht eingesetzt, da ihre Auflosungsgrenze aufgrund der Wellenlange des
Lichtes nicht ausreicht, um Strukturen unter 500 nm abzubilden. Aus dem Bereich
der Mikrobiologie ist jedoch bekannt, das mittels der Fluoreszenzmikroskopie noch ein-
zelne fluoreszenzmarkierte Molekule aufgelost werden konnen [12]. Aus diesem Grund
scheint sie auch zur Qualitatssicherung an den gepragten Nanostrukturen und den
verwendeten Stempeln geeignet. Ihr Vorteil gegenuber den ublicherweise verwendeten
Methoden besteht darin, dass es sich im Vergleich zu SEM und AFM um eine kosten-
gunstige Methode zur Detektion von Nanostrukturen handelt. Die Untersuchung der
Wafer und Stempel erfolgt dabei ohne direkten Kontakt zur Oberflache. Fur die in-
dustrielle Anwendung ware somit ein Zuruckbringen des untersuchten Wafers in den
Produktionsprozess gewahrleistet. Weitere Anforderungen, die an eine solche Methode
gestellt werden, sind eine einfache und unkomplizierte Handhabung sowie eine schnelle
und reproduzierbare Auswertung.
In diesem Kapitel wird eine auf der Fluoreszenzmikroskopie basierende Methode zur
Qualitatssicherung in der Nanoimprintlithographie vorgestellt. Der Anwendungsbereich
ist vielfaltig. Er reicht von der Detektion der verschiedenen Pragedefekte auf dem Wa-
fer uber die Kontrolle der Pragetiefe des Polymers und deren Einheitlichkeit bis hin
zur automatischen Kontrolle eines Pragestempels wahrend einer Serie von Pragungen.
6.1.1 Charakterisierung des Fluoreszenzmarkers
Die in der Nanoimprintlithographie herkommlich verwendeten Polymere, wie z. B. Po-
lymethylmethacrylat (PMMA), weisen keine Primarfluoreszenz im sichtbaren Bereich
auf. Daher erfordert die Anwendung der Fluoreszenzmikroskopie zur Qualitatssiche-
rung die Entwicklung eines mit einem Fluoreszenzfarbstoff markierten Polymers. An
den Farbstoff werden verschiedene Anforderungen gestellt:
- Er sollte die Eigenschaften des Polymers bei der Pragung nicht wesentlich veran-
dern.
- Er sollte kovalent an das Polymer gebunden sein, um eine gleichmaßige Verteilung

6.1 Oberflachengebundene Nanopartikel 77
des Farbstoffes zu gewahrleisten.
- Die Anregung der Fluoreszenz muss wegen des verwendeten Filtersystems im
Bereich von 450 − 490 nm, die Emission uber 510 nm liegen.
- Die Intensitat der Fluoreszenz muss uber einen langeren Zeitraum stabil sein, um
die Uberprufung eines kompletten Wafers zu ermoglichen.
Die Entwicklung des mit einem Fluoreszenzfarbstoff markierten Polymers erfolgt im
Rahmen des EU-Projektes CHANIL in Kooperation mit der Firma Micro Resist Tech-
nology (MRT), die fur die Entwicklung und Optimierung der Synthese des Polymers
verantwortlich ist.
6.1.1.1 Fluoreszenzspektroskopische Untersuchungen des Farbstoffes
Die von MRT herkommlich fur die Nanoimprintlithographie entwickelten Polymere
mr-I 8000 und mr-I 9000 basieren auf einem Methacrylat, das mit unterschiedlichen
aromatischen oder cyclischen, aliphatischen Resten versehen werden kann (vgl. Kapitel
3.1). Beide Polymere zeigen keine oder nur eine außerst schwache Eigenfluoreszenz und
sind somit in der reinen Form nicht fur die fluoreszenzmikroskopischen Untersuchungen
geeignet. Damit diese Polymere eine Sekundarfluoreszenz aufweisen, mussen sie mit
einem Fluoreszenzfarbstoff markiert werden. Als Fluoreszenzmarker fur diese Polymere
stehen Fluoreszenzfarbstoffe, die auf Fluorescein, Anthracen und Peryliden basieren,
zur Diskussion. Die Strukturformeln der Ausgangsfarbstoffe sind in der Abbildung 6.1
dargestellt. Die Eigenschaften der mit den drei Farbstoffen markierten Polymere werden
zunachst in zwei Kategorien unterteilt:
a) Die Position der Spektrallinien im Spektrum: Die Linie der Absorption sollte in
dem UV-Bereich liegen, der leicht mit einer Standardlichtquelle, wie z. B. einer
Quecksilber-Hochdrucklampe, zuganglich ist. Die Emission sollte hierbei sichtbar
sein und nicht durch das Polymer absorbiert werden.

78 6. Ergebnisse und Diskussion
b) Die Intensitat der Emission: Die Intensitat der Emission sollte so hoch wie moglich
sein, um eine moglichst empfindliche Detektion zu gewahrleisten. Das schließt eine
minimale Loschung durch das Polymer mit ein.
� �
� K %
� 9 9K
K
�
� � L � # � � # � � � " � � � � � 5 � �" > " � � � " � ? # � � � � # � � � " � � � � � � � �
# �
� �
" � �
5 �
C �C�
99K 9
� 9 9 K
Abbildung 6.1: Ubersicht uber das verwendete Polymer und die zur Diskussion ste-
henden Farbstoffe. a) Methacrylat als Basis des Polymers, b) auf Fluorescein basie-
render Farbstoff, c) auf Anthracen basierender Farbstoff, d) auf Peryliden basierender
Farbstoff.
Die Ergebnisse des Vorversuchs sind nach obigen Aspekten qualitativ in der Tabelle
6.1 gegenubergestellt.
Tabelle 6.1: Qualitative Einteilung der spektralen Eigenschaften der Polymere, die
mit Fluorescein, Anthracen und Peryliden markiert sind.
Farbstoff Position der Spektrallinien Intensitat der Emission
Fluorescein ++ +
Anthracen ++ −−Peryliden ++ ++
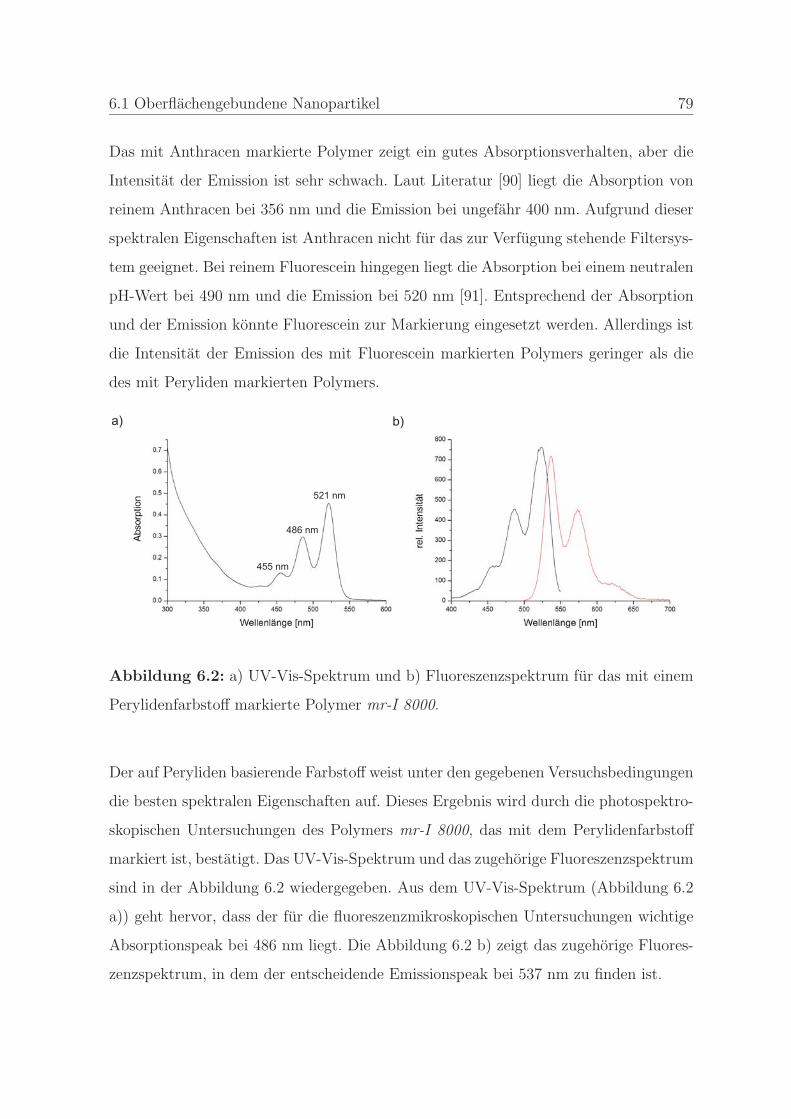
6.1 Oberflachengebundene Nanopartikel 79
Das mit Anthracen markierte Polymer zeigt ein gutes Absorptionsverhalten, aber die
Intensitat der Emission ist sehr schwach. Laut Literatur [90] liegt die Absorption von
reinem Anthracen bei 356 nm und die Emission bei ungefahr 400 nm. Aufgrund dieser
spektralen Eigenschaften ist Anthracen nicht fur das zur Verfugung stehende Filtersys-
tem geeignet. Bei reinem Fluorescein hingegen liegt die Absorption bei einem neutralen
pH-Wert bei 490 nm und die Emission bei 520 nm [91]. Entsprechend der Absorption
und der Emission konnte Fluorescein zur Markierung eingesetzt werden. Allerdings ist
die Intensitat der Emission des mit Fluorescein markierten Polymers geringer als die
des mit Peryliden markierten Polymers.
a) b)
455 nm
486 nm
521 nm
Abbildung 6.2: a) UV-Vis-Spektrum und b) Fluoreszenzspektrum fur das mit einem
Perylidenfarbstoff markierte Polymer mr-I 8000.
Der auf Peryliden basierende Farbstoff weist unter den gegebenen Versuchsbedingungen
die besten spektralen Eigenschaften auf. Dieses Ergebnis wird durch die photospektro-
skopischen Untersuchungen des Polymers mr-I 8000, das mit dem Perylidenfarbstoff
markiert ist, bestatigt. Das UV-Vis-Spektrum und das zugehorige Fluoreszenzspektrum
sind in der Abbildung 6.2 wiedergegeben. Aus dem UV-Vis-Spektrum (Abbildung 6.2
a)) geht hervor, dass der fur die fluoreszenzmikroskopischen Untersuchungen wichtige
Absorptionspeak bei 486 nm liegt. Die Abbildung 6.2 b) zeigt das zugehorige Fluores-
zenzspektrum, in dem der entscheidende Emissionspeak bei 537 nm zu finden ist.
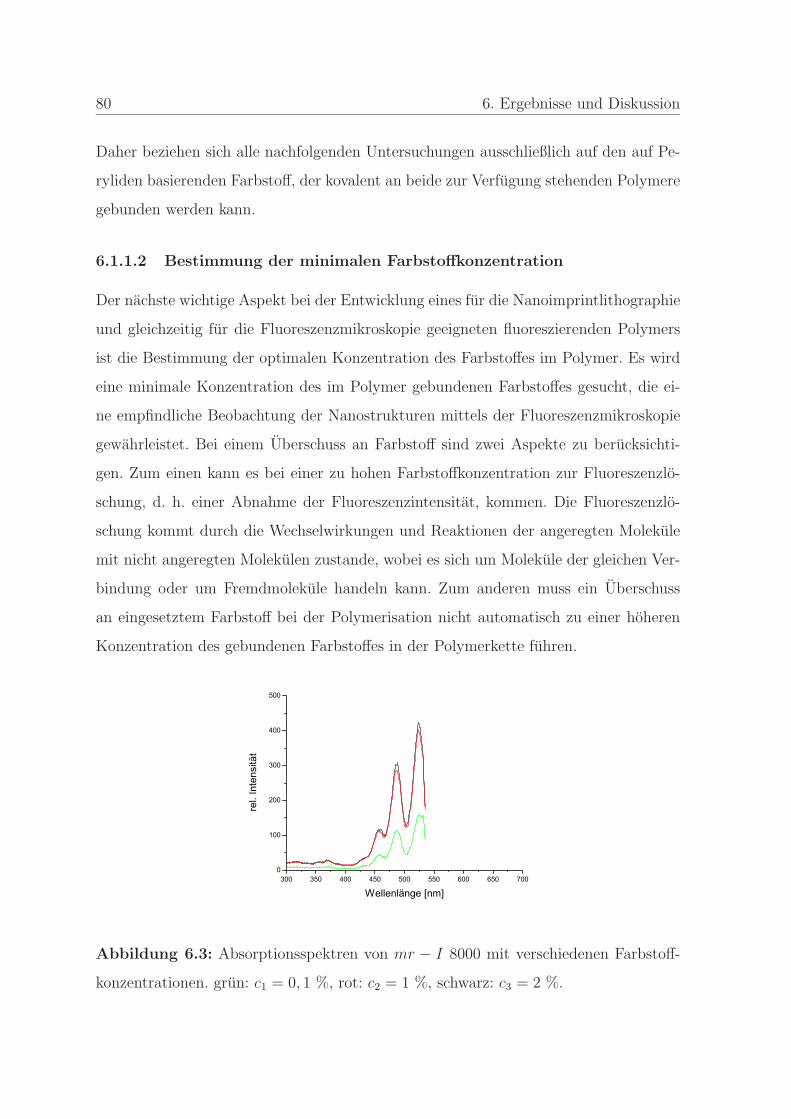
80 6. Ergebnisse und Diskussion
Daher beziehen sich alle nachfolgenden Untersuchungen ausschließlich auf den auf Pe-
ryliden basierenden Farbstoff, der kovalent an beide zur Verfugung stehenden Polymere
gebunden werden kann.
6.1.1.2 Bestimmung der minimalen Farbstoffkonzentration
Der nachste wichtige Aspekt bei der Entwicklung eines fur die Nanoimprintlithographie
und gleichzeitig fur die Fluoreszenzmikroskopie geeigneten fluoreszierenden Polymers
ist die Bestimmung der optimalen Konzentration des Farbstoffes im Polymer. Es wird
eine minimale Konzentration des im Polymer gebundenen Farbstoffes gesucht, die ei-
ne empfindliche Beobachtung der Nanostrukturen mittels der Fluoreszenzmikroskopie
gewahrleistet. Bei einem Uberschuss an Farbstoff sind zwei Aspekte zu berucksichti-
gen. Zum einen kann es bei einer zu hohen Farbstoffkonzentration zur Fluoreszenzlo-
schung, d. h. einer Abnahme der Fluoreszenzintensitat, kommen. Die Fluoreszenzlo-
schung kommt durch die Wechselwirkungen und Reaktionen der angeregten Molekule
mit nicht angeregten Molekulen zustande, wobei es sich um Molekule der gleichen Ver-
bindung oder um Fremdmolekule handeln kann. Zum anderen muss ein Uberschuss
an eingesetztem Farbstoff bei der Polymerisation nicht automatisch zu einer hoheren
Konzentration des gebundenen Farbstoffes in der Polymerkette fuhren.
Abbildung 6.3: Absorptionsspektren von mr − I 8000 mit verschiedenen Farbstoff-
konzentrationen. grun: c1 = 0, 1 %, rot: c2 = 1 %, schwarz: c3 = 2 %.
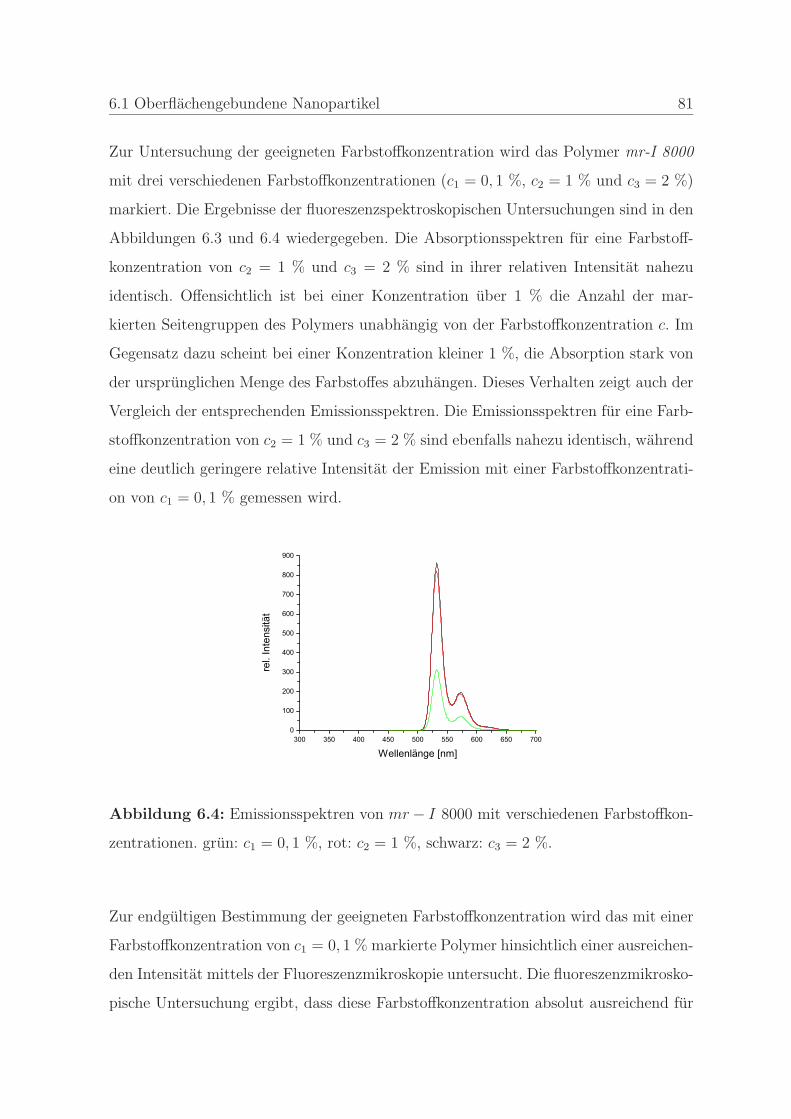
6.1 Oberflachengebundene Nanopartikel 81
Zur Untersuchung der geeigneten Farbstoffkonzentration wird das Polymer mr-I 8000
mit drei verschiedenen Farbstoffkonzentrationen (c1 = 0, 1 %, c2 = 1 % und c3 = 2 %)
markiert. Die Ergebnisse der fluoreszenzspektroskopischen Untersuchungen sind in den
Abbildungen 6.3 und 6.4 wiedergegeben. Die Absorptionsspektren fur eine Farbstoff-
konzentration von c2 = 1 % und c3 = 2 % sind in ihrer relativen Intensitat nahezu
identisch. Offensichtlich ist bei einer Konzentration uber 1 % die Anzahl der mar-
kierten Seitengruppen des Polymers unabhangig von der Farbstoffkonzentration c. Im
Gegensatz dazu scheint bei einer Konzentration kleiner 1 %, die Absorption stark von
der ursprunglichen Menge des Farbstoffes abzuhangen. Dieses Verhalten zeigt auch der
Vergleich der entsprechenden Emissionsspektren. Die Emissionsspektren fur eine Farb-
stoffkonzentration von c2 = 1 % und c3 = 2 % sind ebenfalls nahezu identisch, wahrend
eine deutlich geringere relative Intensitat der Emission mit einer Farbstoffkonzentrati-
on von c1 = 0, 1 % gemessen wird.
Abbildung 6.4: Emissionsspektren von mr − I 8000 mit verschiedenen Farbstoffkon-
zentrationen. grun: c1 = 0, 1 %, rot: c2 = 1 %, schwarz: c3 = 2 %.
Zur endgultigen Bestimmung der geeigneten Farbstoffkonzentration wird das mit einer
Farbstoffkonzentration von c1 = 0, 1 % markierte Polymer hinsichtlich einer ausreichen-
den Intensitat mittels der Fluoreszenzmikroskopie untersucht. Die fluoreszenzmikrosko-
pische Untersuchung ergibt, dass diese Farbstoffkonzentration absolut ausreichend fur
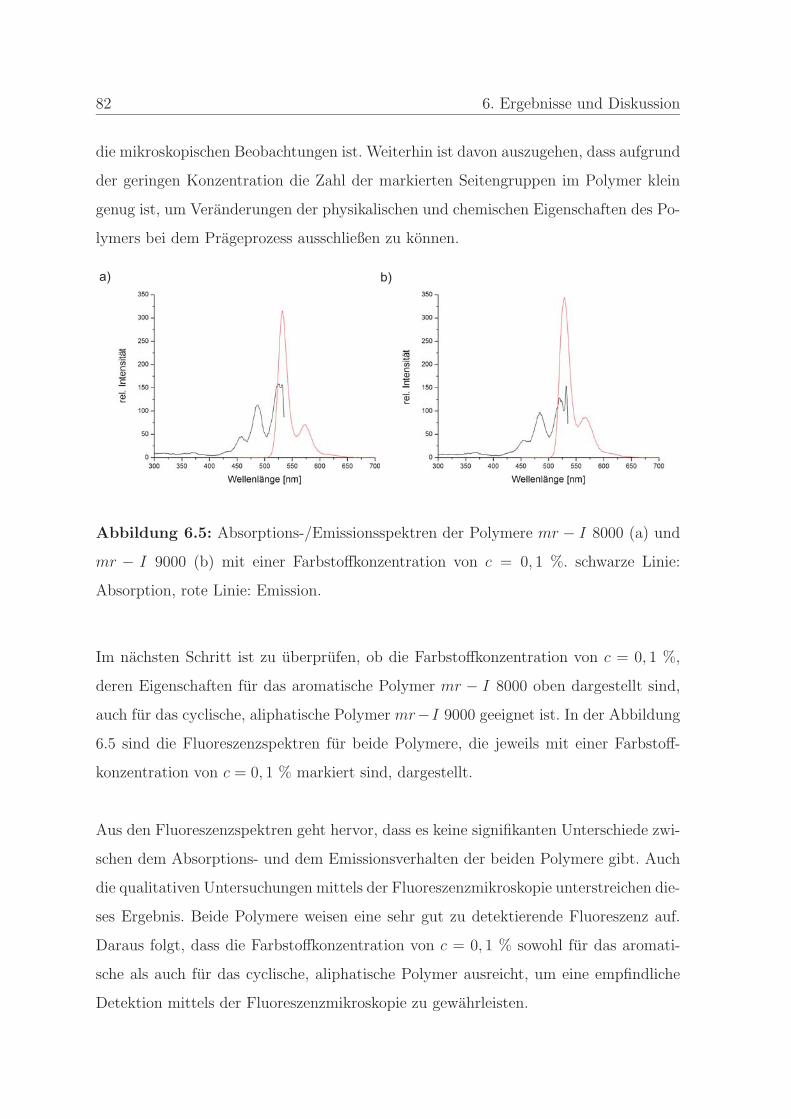
82 6. Ergebnisse und Diskussion
die mikroskopischen Beobachtungen ist. Weiterhin ist davon auszugehen, dass aufgrund
der geringen Konzentration die Zahl der markierten Seitengruppen im Polymer klein
genug ist, um Veranderungen der physikalischen und chemischen Eigenschaften des Po-
lymers bei dem Prageprozess ausschließen zu konnen.
a) b)
Abbildung 6.5: Absorptions-/Emissionsspektren der Polymere mr − I 8000 (a) und
mr − I 9000 (b) mit einer Farbstoffkonzentration von c = 0, 1 %. schwarze Linie:
Absorption, rote Linie: Emission.
Im nachsten Schritt ist zu uberprufen, ob die Farbstoffkonzentration von c = 0, 1 %,
deren Eigenschaften fur das aromatische Polymer mr − I 8000 oben dargestellt sind,
auch fur das cyclische, aliphatische Polymer mr−I 9000 geeignet ist. In der Abbildung
6.5 sind die Fluoreszenzspektren fur beide Polymere, die jeweils mit einer Farbstoff-
konzentration von c = 0, 1 % markiert sind, dargestellt.
Aus den Fluoreszenzspektren geht hervor, dass es keine signifikanten Unterschiede zwi-
schen dem Absorptions- und dem Emissionsverhalten der beiden Polymere gibt. Auch
die qualitativen Untersuchungen mittels der Fluoreszenzmikroskopie unterstreichen die-
ses Ergebnis. Beide Polymere weisen eine sehr gut zu detektierende Fluoreszenz auf.
Daraus folgt, dass die Farbstoffkonzentration von c = 0, 1 % sowohl fur das aromati-
sche als auch fur das cyclische, aliphatische Polymer ausreicht, um eine empfindliche
Detektion mittels der Fluoreszenzmikroskopie zu gewahrleisten.
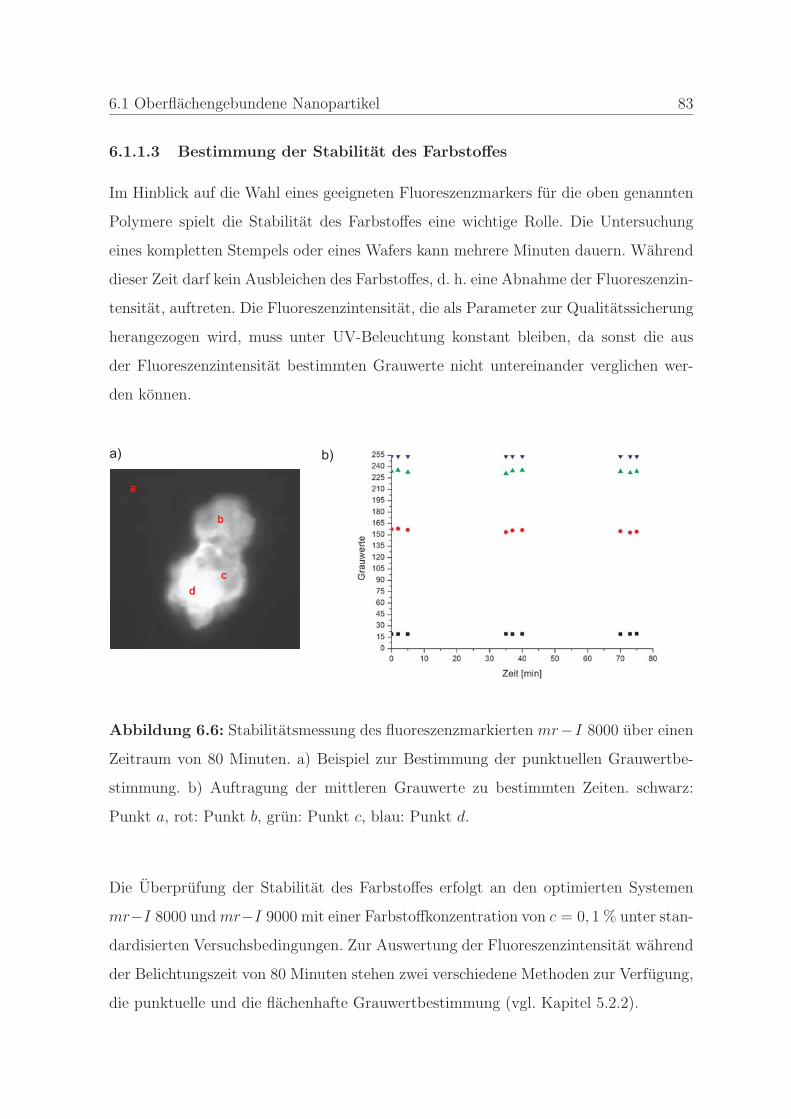
6.1 Oberflachengebundene Nanopartikel 83
6.1.1.3 Bestimmung der Stabilitat des Farbstoffes
Im Hinblick auf die Wahl eines geeigneten Fluoreszenzmarkers fur die oben genannten
Polymere spielt die Stabilitat des Farbstoffes eine wichtige Rolle. Die Untersuchung
eines kompletten Stempels oder eines Wafers kann mehrere Minuten dauern. Wahrend
dieser Zeit darf kein Ausbleichen des Farbstoffes, d. h. eine Abnahme der Fluoreszenzin-
tensitat, auftreten. Die Fluoreszenzintensitat, die als Parameter zur Qualitatssicherung
herangezogen wird, muss unter UV-Beleuchtung konstant bleiben, da sonst die aus
der Fluoreszenzintensitat bestimmten Grauwerte nicht untereinander verglichen wer-
den konnen.
� � � �
Abbildung 6.6: Stabilitatsmessung des fluoreszenzmarkierten mr−I 8000 uber einen
Zeitraum von 80 Minuten. a) Beispiel zur Bestimmung der punktuellen Grauwertbe-
stimmung. b) Auftragung der mittleren Grauwerte zu bestimmten Zeiten. schwarz:
Punkt a, rot: Punkt b, grun: Punkt c, blau: Punkt d.
Die Uberprufung der Stabilitat des Farbstoffes erfolgt an den optimierten Systemen
mr−I 8000 und mr−I 9000 mit einer Farbstoffkonzentration von c = 0, 1 % unter stan-
dardisierten Versuchsbedingungen. Zur Auswertung der Fluoreszenzintensitat wahrend
der Belichtungszeit von 80 Minuten stehen zwei verschiedene Methoden zur Verfugung,
die punktuelle und die flachenhafte Grauwertbestimmung (vgl. Kapitel 5.2.2).
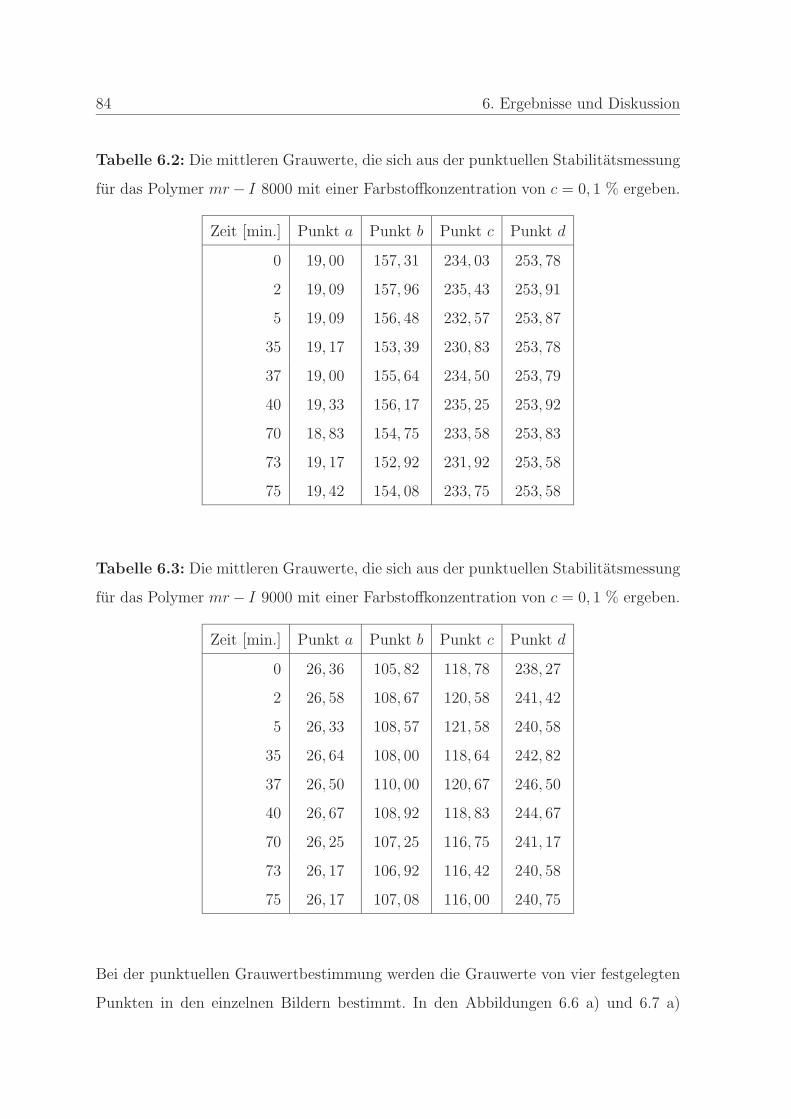
84 6. Ergebnisse und Diskussion
Tabelle 6.2: Die mittleren Grauwerte, die sich aus der punktuellen Stabilitatsmessung
fur das Polymer mr − I 8000 mit einer Farbstoffkonzentration von c = 0, 1 % ergeben.
Zeit [min.] Punkt a Punkt b Punkt c Punkt d
0 19, 00 157, 31 234, 03 253, 78
2 19, 09 157, 96 235, 43 253, 91
5 19, 09 156, 48 232, 57 253, 87
35 19, 17 153, 39 230, 83 253, 78
37 19, 00 155, 64 234, 50 253, 79
40 19, 33 156, 17 235, 25 253, 92
70 18, 83 154, 75 233, 58 253, 83
73 19, 17 152, 92 231, 92 253, 58
75 19, 42 154, 08 233, 75 253, 58
Tabelle 6.3: Die mittleren Grauwerte, die sich aus der punktuellen Stabilitatsmessung
fur das Polymer mr − I 9000 mit einer Farbstoffkonzentration von c = 0, 1 % ergeben.
Zeit [min.] Punkt a Punkt b Punkt c Punkt d
0 26, 36 105, 82 118, 78 238, 27
2 26, 58 108, 67 120, 58 241, 42
5 26, 33 108, 57 121, 58 240, 58
35 26, 64 108, 00 118, 64 242, 82
37 26, 50 110, 00 120, 67 246, 50
40 26, 67 108, 92 118, 83 244, 67
70 26, 25 107, 25 116, 75 241, 17
73 26, 17 106, 92 116, 42 240, 58
75 26, 17 107, 08 116, 00 240, 75
Bei der punktuellen Grauwertbestimmung werden die Grauwerte von vier festgelegten
Punkten in den einzelnen Bildern bestimmt. In den Abbildungen 6.6 a) und 6.7 a)
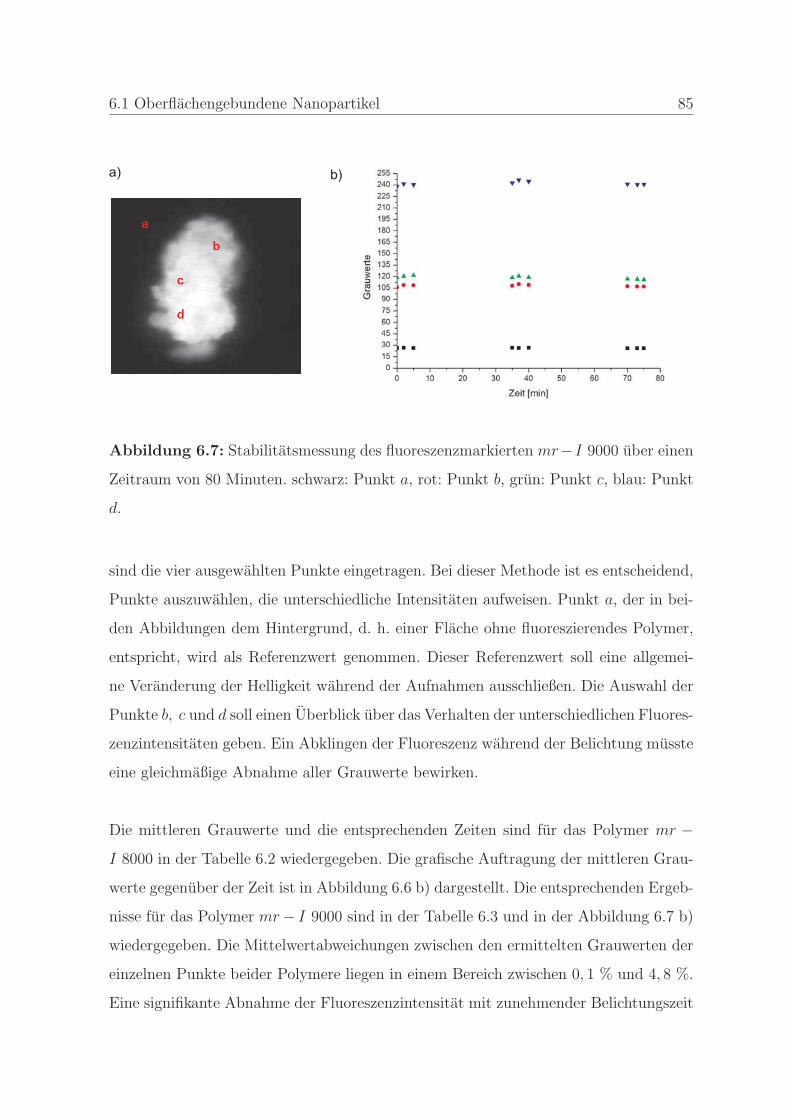
6.1 Oberflachengebundene Nanopartikel 85
� � � �
Abbildung 6.7: Stabilitatsmessung des fluoreszenzmarkierten mr−I 9000 uber einen
Zeitraum von 80 Minuten. schwarz: Punkt a, rot: Punkt b, grun: Punkt c, blau: Punkt
d.
sind die vier ausgewahlten Punkte eingetragen. Bei dieser Methode ist es entscheidend,
Punkte auszuwahlen, die unterschiedliche Intensitaten aufweisen. Punkt a, der in bei-
den Abbildungen dem Hintergrund, d. h. einer Flache ohne fluoreszierendes Polymer,
entspricht, wird als Referenzwert genommen. Dieser Referenzwert soll eine allgemei-
ne Veranderung der Helligkeit wahrend der Aufnahmen ausschließen. Die Auswahl der
Punkte b, c und d soll einen Uberblick uber das Verhalten der unterschiedlichen Fluores-
zenzintensitaten geben. Ein Abklingen der Fluoreszenz wahrend der Belichtung musste
eine gleichmaßige Abnahme aller Grauwerte bewirken.
Die mittleren Grauwerte und die entsprechenden Zeiten sind fur das Polymer mr −I 8000 in der Tabelle 6.2 wiedergegeben. Die grafische Auftragung der mittleren Grau-
werte gegenuber der Zeit ist in Abbildung 6.6 b) dargestellt. Die entsprechenden Ergeb-
nisse fur das Polymer mr − I 9000 sind in der Tabelle 6.3 und in der Abbildung 6.7 b)
wiedergegeben. Die Mittelwertabweichungen zwischen den ermittelten Grauwerten der
einzelnen Punkte beider Polymere liegen in einem Bereich zwischen 0, 1 % und 4, 8 %.
Eine signifikante Abnahme der Fluoreszenzintensitat mit zunehmender Belichtungszeit

86 6. Ergebnisse und Diskussion
ist jedoch in den Diagrammen nicht zu erkennen.
� � � � �
� � � � � � � � � � � � � � � � � � � � � �
� � � � � � � � � � � � � � � � � � � � � � � � �
� � � � � � � � � ! � � � � � � � " � � # � � � � � � � �
� � � �
�����
Abbildung 6.8: Schematische Darstellung der Stabilitatsmessung nach der Methode
der flachenhaften Grauwertbestimmung.
Um dieses Ergebnis zu verifizieren, werden die wahrend der Belichtung aufgenommenen
Bilder zusatzlich mit der zweiten Methode, der flachenhaften Grauwertbestimmung,
ausgewertet. Bei der flachenhaften Grauwertbestimmung wird ein Grauwerthistogramm
entlang der Bilddiagonalen, wie es schematisch in Abbildung 6.8 dargestellt ist, erstellt.
Zur Uberprufung der Fluoreszenzstabilitat werden die Grauwerthistogramme eines Bil-
des einer Sequenz bei t1 mit dem entsprechenden Bild einer Sequenz bei t2 usw. ver-
glichen. In der Abbildung 6.9 sind die Grauwerthistogramme eines Bildes bei drei ver-
schiedenen Zeiten in einem Diagramm dargestellt. Der Vergleich der Abbildungen 6.9
a), b) und c) zeigt, dass die Auswertung unabhangig von der Wahl des Bildes ist. Alle
drei Diagramme zeigen nahezu identisches Verhalten. Ferner ist aus den Diagrammen
zu entnehmen, dass auch mit dieser Methode kein signifikanter Unterschied, d. h. keine
Abnahme der Fluoreszenzintensitat, nach mehr als 80 Minuten registriert werden kann.
Die Stabilitat des cyclischen, aliphatischen Polymers mr−I 9000 wird ebenfalls mittels
der flachenhaften Grauwertbestimmung gepruft. Aus der Abbildung 6.10 geht hervor,
dass dieses Polymer ein identisches Verhalten zu dem aromatischen Polymer aufweist.
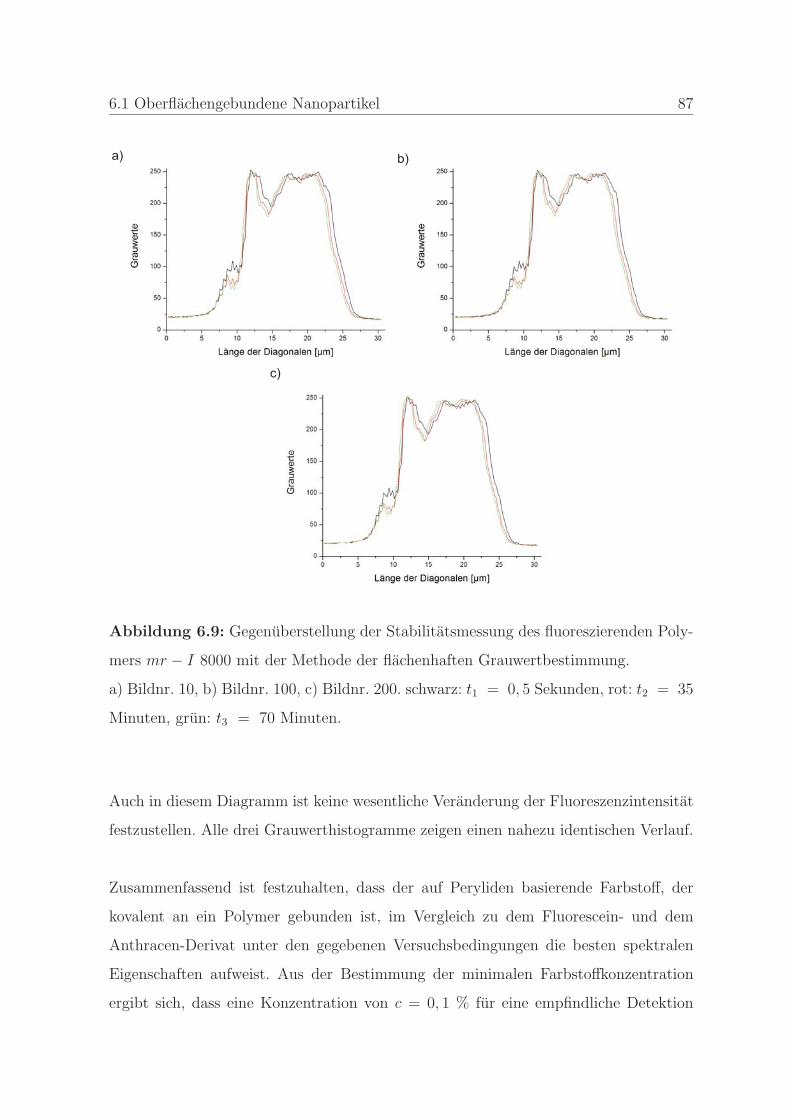
6.1 Oberflachengebundene Nanopartikel 87
a) b)
c)
Abbildung 6.9: Gegenuberstellung der Stabilitatsmessung des fluoreszierenden Poly-
mers mr − I 8000 mit der Methode der flachenhaften Grauwertbestimmung.
a) Bildnr. 10, b) Bildnr. 100, c) Bildnr. 200. schwarz: t1 = 0, 5 Sekunden, rot: t2 = 35
Minuten, grun: t3 = 70 Minuten.
Auch in diesem Diagramm ist keine wesentliche Veranderung der Fluoreszenzintensitat
festzustellen. Alle drei Grauwerthistogramme zeigen einen nahezu identischen Verlauf.
Zusammenfassend ist festzuhalten, dass der auf Peryliden basierende Farbstoff, der
kovalent an ein Polymer gebunden ist, im Vergleich zu dem Fluorescein- und dem
Anthracen-Derivat unter den gegebenen Versuchsbedingungen die besten spektralen
Eigenschaften aufweist. Aus der Bestimmung der minimalen Farbstoffkonzentration
ergibt sich, dass eine Konzentration von c = 0, 1 % fur eine empfindliche Detektion

88 6. Ergebnisse und Diskussion
Abbildung 6.10: Stabilitatsmessung des fluoreszierenden Polymers mr − I 9000 mit
der Methode der flachenhaften Grauwertbestimmung. schwarz: t1 = 0, 5 Sekunden,
rot: t2 = 35 Minuten, grun: t3 = 70 Minuten.
mittels der Fluoreszenzmikroskopie vollkommen ausreichend ist. Ferner ist die rela-
tive Fluoreszenzintensitat dabei unabhangig von dem verwendeten Polymer, so dass
sowohl das thermoplastische Polymer mr − I 8000 als auch das duroplastische Poly-
mer mr − I 9000, markiert mit der minimalen Farbstoffkonzentration, zur fluoreszenz-
mikroskopischen Untersuchung herangezogen werden konnen. Aufgrund der geringen
Konzentration ist davon auszugehen, dass sich die Prageeigenschaften des Polymers im
Vergleich zu den unmarkierten Polymeren nicht verandern. Wie die Stabilitatsmessun-
gen zeigen, weisen beide markierten Polymere eine sehr hohe Fluoreszenzstabilitat bei
einer Belichtung unter UV-Licht von uber einer Stunde auf. Somit eignen sich beide
markierten Polymere optimal fur den Einsatz zur Qualitatssicherung in der Nanoim-
printlithographie.
6.1.2 Qualitatssicherung in der Nanoimprintlithographie
Die Entwicklung der fluoreszierenden Polymere zur Anwendung in der Nanoimprint-
lithographie ermoglicht die Detektion von nanostrukturierten Oberflachen mittels der
Fluoreszenzmikroskopie unterhalb der Auflosungsgrenze eines herkommlichen Auflicht-

6.1 Oberflachengebundene Nanopartikel 89
Mikroskops. Nachfolgend werden die verschiedenen Anwendungsmoglichkeiten fur die
Qualitatssicherung in der Nanoimprintlithographie aufgezeigt. Zunachst werden die
auftretenden Defekte charakterisiert, um einen Uberblick uber ihre verschiedenen Arten
zu geben. Ein Vergleich der fluoreszenzmikroskopischen Bilder und deren Auswertun-
gen mit den Ergebnissen der in der Nanoimprintlithographie ublichen Methoden, wie
der Rasterelektronenmikroskopie (SEM) oder der Profilometrie, wird die Moglichkei-
ten hervorheben, die sich durch die Fluoreszenzmikroskopie ergeben. Ferner wird eine
halbautomatische Kontrolle eines Pragestempels wahrend einer Serie von Pragungen
vorgestellt.
6.1.2.1 Charakterisierung der auftretenden Defekte in der Pragung
In der Nanoimprintlithographie wird die Struktur eines Stempels in die Polymerschicht
des Substrates ubertragen, wobei ein direkter Kontakt zwischen Stempel und Substrat
besteht. Das Prinzip der Nanoimprintlithographie wird in Kapitel 3.1 naher beschrie-
ben. Eine gute Kopie der Stempelstruktur in der Polymerschicht erfordert eine gute
Anpassung des Polymers an die Struktur durch einen ausreichenden Polymerfluss. Folg-
lich hangt die Qualitat einer Pragung in der Nanoimprintlithographie von einer Viel-
zahl verschiedener Faktoren ab, die den Polymerfluss beeinflussen. Zu den wesentlichen
Parametern gehoren Pragetemperatur und -druck sowie die Pragezeit. Weitere Fakto-
ren, die die Qualitat einer Pragung bestimmen, sind die Dicke der auf dem Substrat
aufgebrachten Polymerschicht und die Art der Strukturen des Stempels [43, 86, 92, 93].
Die Pragetemperatur, die hoher sein muss als die Glasubergangstemperatur des verwen-
deten Polymers, und der angewendete Druck bestimmen die Viskositat des Polymers
und damit dessen Fließeigenschaft. Aufgrund der Viskositat des Polymers wird eine be-
stimmte Zeit benotigt, damit das Polymer die Kavitaten des Stempels ausfullen kann
[85, 86, 92]. Die Art der Stempelstrukturen wird in negativ oder positiv strukturierte
Oberflachen unterteilt. Laut Literatur hat ein positiver Stempel erhohte Strukturen auf
einer großen vertieften Flache, hingegen wird bei vertieften Strukturen in einer großen
erhohten Flache von einem negativen Stempel gesprochen [94]. Ein weiterer wichtiger

90 6. Ergebnisse und Diskussion
Aspekt hinsichtlich der Strukturen des Stempels ist die Große der Strukturen, ihre
Form und die Umgebung eines strukturierten Feldes. Diese Faktoren bestimmen die
notwendige Dicke der Polymerschicht, die auf dem Substrat mittels Spin-Coating auf-
getragen wird [93].
A
D
E
C
F
B
Abbildung 6.11: Schematische Abbildung des Pragestempels mit den zugehorigen
fluoreszenzmikroskopischen Abbildungen der Pragungen der jeweiligen Bereiche.
A: 100× 100 µm2 große Flachen; B: 50× 50 µm2 große Flachen; C: Y-Baugruppen; D:
5 µm breite Linien und Abstande; E: 400 nm breite Linien und Abstande; F: Bander
bestehend aus positiven und negativen Punkten und Linien.
Der Stempel, der zur Charakterisierung der auftretenden Defekte und zur Bestimmung
der Qualitat der Pragung (vgl. Kapitel 6.1.2.3) verwendet wird, ist schematisch in der
Abbildung 6.11 wiedergegeben. Die Benennung der Felder entspricht der Abbildung 5.2
aus Kapitel 5.2.3. Die fluoreszenzmikroskopischen Aufnahmen der Pragungen der ein-
zelnen Felder sollen einen Uberblick uber die jeweiligen Strukturen liefern. Der Stempel
hat Strukturen unterschiedlicher Formen, wie die Y-Baugruppen (C), Linien (D und E),

6.1 Oberflachengebundene Nanopartikel 91
Punkte (F) und große erhohte Flachen (A und B). Sehr charakteristisch sind ferner die
unterschiedlichen Großen der Strukturen, die im Bereich von 400 nm bis 100 µm liegen
sowie die positiven und negativen Strukturen im Feld F. Diese verschiedenen Struktu-
ren und Strukturgroßen dienen der praktischen Untersuchung der Fließeigenschaften
der Polymere, da sie die Grenzbereiche der Nanoimprintlithographie abdecken.
a) b)
Abbildung 6.12: Beispiel fur einen Klebedefekt im Bereich der 400 nm breiten Linien
und Abstande. a) Fluoreszenzmikroskopische Abbildung; b) SEM-Bild
Zu den aus der Literatur bekannten Defekten, die in der Nanoimprintlithographie auf-
treten, gehoren die Klebedefekte und die Fließdefekte. Die Klebedefekte, wie beispiel-
haft in Abbildung 6.12 dargestellt, konnen durch die Adhasion des Polymers an der
Stempeloberflache entstehen. Durch den direkten Kontakt der Stempeloberflache mit
dem Polymer kann es beim Pragen zum Anhaften des Polymers an der Stempeloberfla-
che kommen. Beim Abnehmen des Stempels reißt der Polymerfilm von dem Substrat
ab und haftet an dem Stempel. Besonders haufig treten diese Defekte in Bereichen
kleiner Strukturen mit einer hohen Dichte auf. Mit einer Antihaftbeschichtung soll die
Adhasion des Polymers an der Stempeloberflache verringert bzw. vermieden werden
[41, 42]. Im nachfolgenden Kapitel 6.1.2.2 wird besonders auf diese Art der Defekte
und ihre Auswirkungen eingegangen.
Der Klebedefekt in der Abbildung 6.12 a) wird am Rande des Feldes E detektiert.
Aus der fluoreszenzmikroskopischen Aufnahme ist zu erkennen, dass zwei benachbarte

92 6. Ergebnisse und Diskussion
400 nm breite Linien am Ende abgerissen sind. Einige Linien weiter wiederholt sich ein
solcher Defekt bei einer anderen 400 nm breiten Linie. In dem SEM-Bild 6.12 b) ist der
Klebedefekt mit wesentlich hoherer Vergroßerung dargestellt. Die abgerissenen Enden
der zwei 400 nm breiten Linien sind deutlich zu sehen. Dennoch ergibt der Vergleich
der beiden Bilder, dass selbst mit einer geringen Vergroßerung (50x Objektiv) dieser
Klebedefekt im Fluoreszenzmikroskop detektiert und mit ausreichender Auflosung dar-
gestellt werden kann.
Die so genannten Fließdefekte treten in einer großen Zahl von Variationen auf. Hier-
zu sind unter anderem Defekte, die auf einer konvexen Verkrummung des Polymers
beruhen, und labyrinthformige Strukturen zu zahlen [86, 87]. Charakteristisch fur die
Fließdefekte sind unvollstandig ausgefullte Strukturen in der Pragung, die aus einem
unzureichenden Materialtransport resultieren. Dieser kann durch eine zu geringe Pra-
getemperatur, einen zu geringen Druck oder eine zu kurze Pragezeit entstehen. Die
Fließdefekte treten vor allem bei großen Flachen auf. Ein Beispiel fur einen Fließde-
fekt ist in der Abbildung 6.13 wiedergegeben. Wie dem fluoreszenzmikroskopischen
Bild (6.13 a)) zu entnehmen ist, handelt es sich um das Feld D mit 5 µm breiten Li-
nien und Abstanden, das von einer großen unstrukturierten Flache umgeben ist. Der
Fließdefekt kommt dadurch zustande, dass die Fließeigenschaften des Polymers nicht
ausreichen, um die an das Feld D angrenzende vertiefte Flache des Stempels vollstandig
auszufullen. Dem fluoreszenzmikroskopischen Bild sind zwei SEM-Aufnahmen (6.13 b)
und c)) gegenubergestellt. Das SEM-Bild 6.13 b) hat eine ahnliche Auflosung wie das
fluoreszenzmikroskopische Bild. Die Fließdefekte am Rand des periodisch strukturier-
ten Feldes sind zwar zu erkennen, werden jedoch durch die fluoreszenzmikroskopische
Aufnahme ebenso gut aufgelost. Das zweite SEM-Bild 6.13 c) zeigt den fur SEM-
Bilder charakteristischen 3D-Effekt. Durch den veranderten Betrachtungswinkel und
eine hohere Auflosung konnen deutlich drei Schichtdicken in der Polymerschicht er-
kannt werden. Die erste Schicht (1) entspricht der anfanglichen Polymerschicht, die
mittels Spin-Coating gleichmaßig auf das Substrat aufgetragen wird. Durch den Pra-
geprozess findet eine Verschiebung dieser ursprunglich gleichmaßigen Polymerschicht

6.1 Oberflachengebundene Nanopartikel 93
statt: Zum einen wird das Polymer durch die erhohten Stempelstrukturen wahrend des
Pragens verdrangt, d. h., es gibt eine Abnahme der Polymerschicht in diesen Berei-
chen, und zum anderen findet gleichzeitig eine Anreicherung des verdrangten Polymers
in den Hohlraumen des Stempels statt, so dass in diesen Bereichen eine Zunahme der
Polymerschicht beobachtet werden kann. Die Schicht (2) in der Abbildung 6.13 c), die
niedriger ist als die Schichten (1) und (3), entspricht dem Bereich, in dem das Polymer
durch die Stempelstruktur verdrangt wird. Die hochste Schichtdicke ist bei der Schicht
(3) zu erkennen, da hier das Polymer die Hohlraume des Stempels gleichmaßig ausfullt.
a) b)
c)
1
2
3
2
3 1
Abbildung 6.13: a) Fluoreszenz-, b) SEM-Bild (von oben) und c) SEM-Bild (3D-
Ansicht) eines Fließdefektes in der Nanoimprintlithographie.
Die Bildung der drei unterschiedlichen Schichtdicken durch Fließdefekte ist bereits aus
der Literatur bekannt [93]. Bisher werden diese mittels SEM oder AFM detektiert. Diese
drei verschiedenen Schichten konnen jedoch auch dem fluoreszenzmikroskopischen Bild
entnommen werden. Die dreidimensionale Ansicht des SEM-Bildes 6.13 c) kann zwar in

94 6. Ergebnisse und Diskussion
dem fluoreszenzmikroskopischen Bild 6.13 a) nicht wiedergegeben werden, da die ver-
wendete mikroskopische Methode eine Auflichtmethode ist und somit nur Bilder von
oben aufgenommen werden konnen. Dennoch ist es moglich, die drei Polymerschichten
anhand der unterschiedlichen Grauwerte zu unterscheiden. Die dunklen, fast schwar-
zen Bereiche im Fluoreszenzbild haben Grauwerte von 20 bis 40. Mit zunehmender
Helligkeit der Grautone, die mit einer Zunahme der Fluoreszenzintensitat verbunden
ist, steigen die Grauwerte. Der hochste erreichbare Grauwert betragt 255 und ent-
spricht der Farbe Weiß. In der Abbildung 6.13 a) sind die drei verschiedenen Schichten,
die in dem SEM-Bild detektiert werden, markiert. Die Schicht (3) ist gekennzeichnet
durch die hellsten Grautone und damit verbunden sehr großen Grauwerten, so dass eine
große Menge an fluoreszierendem Polymer vorliegt. Dies ist der Bereich, in dem durch
das Ausfullen der Stempelhohlraume eine Anhaufung des Polymers vorliegt. Hingegen
weist die Schicht (2) dunkle Grautone auf, d. h., es liegen kleine Grauwerte vor. Das
korrespondiert mit einer geringeren Menge an fluoreszierendem Polymer, die in dem
Bereich der erhohten Stempelstrukturen zu finden ist. Die anfangliche Polymerschicht,
die vor der Pragung auf dem Substrat vorliegt, ist gekennzeichnet durch einen mittleren
Grauwert und entspricht einem mittleren Grauton. Die Schicht (1) weist einen solchen
mittleren Grauton auf. Die Beurteilung von Schichtdicken mittels der Grauwertbestim-
mung wird im Kapitel 6.1.2.3 eingehender besprochen.
a) b)
Abbildung 6.14: Vergleich der Pragungen bei unterschiedlichen Pragezeiten. a) Pra-
gezeit t = 10 Minuten, b) Pragezeit t = 1 Minute

6.1 Oberflachengebundene Nanopartikel 95
Ein weiteres Beispiel fur einen Fließdefekt zeigt das fluoreszenzmikroskopische Bild
6.14 b), dem eine saubere Pragung der gleichen Struktur gegenuber gestellt ist. Beide
Bilder zeigen einen Ausschnitt aus dem Feld C mit den Y-Baugruppen des Prage-
stempels. Wahrend im linken Bild a) die Strukturen der Y-Baugruppe vollstandig und
gleichmaßig ausgefullt sind, sind die Stege der Y-Baugruppe in dem rechten Bild b)
nur unvollstandig gefullt. Dies ist an den unterschiedlichen Grauwerten innerhalb der
Stege zu erkennen. Zwischen diesen beiden Pragungen wird die Haltezeit variiert. Bei
der linken Pragung (Abbildung 6.14 a)) betragt die Haltezeit 10 Minuten, wahrend bei
der rechten Pragung (b) der Stempel nur eine Minute in das Polymer gepresst wird.
Offensichtlich reicht die kurzere Zeit fur das viskose Polymer, das durch die erhohten
Stempelstrukturen verdrangt wird, nicht aus, um die Kavitaten des Stempels komplett
und gleichmaßig auszufullen.
a) b)
c)
Abbildung 6.15: Fließdefekte am Rande einer großen unstrukturierten Flache.
a, b) Der gepragte Bereich rund um das 100 × 100 µm Rechteck. c) Vergroßerung der
labyrinthformigen Struktur aus b).

96 6. Ergebnisse und Diskussion
Eine weitere Art der Fließdefekte, die haufig und insbesondere an den Randern von
großen unstrukturierten Flachen auftritt, ist eine fortschreitende Polymerfront. Zu den
großen unstrukturierten Flachen sind bei dem verwendeten Stempel (Abbildung 6.11)
die 100 × 100 µm2 Rechtecke der mit A gekennzeichneten Felder zu zahlen. Ein Bei-
spiel fur eine solche Polymerfront ist in der Abbildung 6.15 wiedergeben. Die fluores-
zenzmikroskopischen Bilder 6.15 a) und b) zeigen den gepragten Bereich rund um ein
100 × 100 µm2 Rechteck. Das Rechteck ist nahezu kreisformig von der Polymerfront
umgeben. Aufgrund der helleren Grauwerte, auch im Vergleich zu der den Kreis umge-
benden Flache, kann hier von einer hoheren Schichtdicke ausgegangen werden. In der
Literatur ist diese Art von Fließdefekt hinlanglich bekannt. Es wird von einer blumen-
formigen Struktur gesprochen [86, 95, 96]. Diese blumenformige Struktur ist in dem
Bild a) zu erkennen. Sie entsteht durch eine seitliche Verschiebung des verdrangten
Polymers ausgehend von den Kanten der erhohten Strukturen des Stempels in dessen
Hohlraume. Die AFM- und SEM-Untersuchungen haben ergeben, dass sich das ver-
drangte Polymer vertikal an der Wand der erhohten Struktur des Stempels bewegt.
Der Polymerfluss wird dabei von der vertikalen Verschiebung des Polymers entlang der
Stempelwand und der seitlichen Verschiebung des unter der erhohten Stempelstruktur
verdrangten Polymers beeinflusst, so dass die entstehende Polymerfront in den SEM-
Aufnahmen als eine sich ausbreitende Welle erscheint [86, 96].
Am Rand dieser blumenformigen Polymerfront sowie am Rand der vertieften Flache
des Stempels sind labyrinthartige Strukturen zu erkennen. Eine Vergroßerung dieser
Strukturen ist in der Abbildung 6.15 c) dargestellt. Auch dieser Defekt ist haufig zu be-
obachten, insbesondere bei den großen unstrukturierten Flachen. Diese labyrinthartigen
Strukturen treten oftmals in Kombination mit der zuvor beschriebenen Polymerfront
auf. Es wird vermutet, dass diese Strukturen ebenfalls auf einer lateralen Stoßwelle mit
abnehmender Starke beruhen, die durch die Kombination der vertikalen Verschiebung
des Polymers entlang der erhohten Stempelstrukturen und der lateralen Verschiebung
des Polymers aufgrund der Verdrangung durch die erhohten Stempelstrukturen zustan-
de kommt. Die Detektion dieser Strukturen war bisher nur mit SEM oder AFM moglich

6.1 Oberflachengebundene Nanopartikel 97
[86, 92, 96].
a) b)
Abbildung 6.16: Konvexe Verkrummung des Polymers in der Mitte eines unstruk-
turierten 100 × 100 µm2 großen Feldes. a) Saubere Pragung ohne Fließdefekt (32x
Objektiv). b) Deutliche Polymeranhaufung in der Mitte des Feldes (50x Objektiv).
Wie bereits erwahnt, treten vor allem in Bereichen großer unstrukturierter Flachen eine
Vielzahl von unterschiedlichen Fließdefekten auf. In der Abbildung 6.16 ist ein Defekt
dargestellt, der auf einer konvexen Verkrummung des Polymers beruht. Im Vergleich
dazu ist im linken Bild a) eine saubere Pragung ohne Defekte gezeigt. Diese Art eines
Fließdefektes wird haufig in Zusammenhang mit den auf der Polymerfront beruhenden
Fließdefekten beobachtet. Trotz der unterschiedlichen Vergroßerung der Aufnahmen ist
der Unterschied deutlich zu erkennen. In dem rechten Bild (6.16 b)) ist in der Mitte
der 100 × 100 µm2 großen Flache deutlich eine Zunahme der Grauwerte zu beobach-
ten. Daraus ist zu schließen, dass der Anteil an fluoreszierendem Polymer zur Mitte
hin zunimmt. Die Entstehung solcher Verkrummungen ist noch nicht geklart. Es wird
vermutet, dass es sich um Relaxationseffekte handelt, da die Verkrummung mit zu-
nehmenden Alter der Proben steigt [86, 92]. Bei den hier vorgestellten Beispielen sind
die Pragungen jedoch nur einen Tag alt. Diese Verkrummung konnte auch durch die
Adhasion zur Stempeloberflache oder durch Kapillarkrafte entstehen.
Die Variationen der Defekte, die in der Nanoimprintlithographie entstehen konnen,
sind vielfaltig. Bisher konnte die Entstehung der Defekte in keinen direkten Zusammen-

98 6. Ergebnisse und Diskussion
hang mit den Pragebedingungen gebracht werden [86]. Die hier vorgestellten Defekte
zeigen, dass die Fluoreszenzmikroskopie die Detektion samtlicher bislang bekannter De-
fekte ermoglicht. Mittels der Fluoreszenzmikroskopie konnen sowohl die unterschied-
lichsten Klebedefekte als auch die Fließdefekte deutlich detektiert werden. Der Vorteil
gegenuber der Rasterelektronenmikroskopie besteht darin, dass die Proben durch die
Untersuchung mit der Fluoreszenzmikroskopie nicht zerstort werden. Die Fluoreszenz-
mikroskopie erfordert keine Probenvorbereitung und hat wahrend der Messung keinen
Kontakt zur Probenoberflache. Die Untersuchung mit der Rasterelektronenmikrosko-
pie erfordert hingegen eine Beschichtung der Proben mit einer Goldschicht. Aufgrund
dieser Beschichtung sind weitere Untersuchungen mit anderen Methoden, wie z. B. der
Fluoreszenzmikroskopie, ausgeschlossen.
6.1.2.2 Detektion von Defekten auf dem Stempel und auf der Pragung
Im vorangegangenen Kapitel 6.1.2.1 werden die in der Nanoimprintlithographie be-
kannten Defekte in Bezug auf die Pragung charakterisiert. Hinsichtlich einer Quali-
tatssicherung ist daher die Auswirkung dieser Defekte auf den Stempel zu uberprufen.
Eine besondere Bedeutung kommt hierbei den Klebedefekten zu. Diese Defekte kom-
men vor allem in Bereichen sehr feiner Strukturen mit einer hohen Dichte vor. In diesen
Bereichen sind die Wechselwirkungen zwischen der Oberflache des Stempels und dem
Polymer wesentlich großer als die Wechselwirkungen zwischen Substrat und Polymer.
Dabei ist sehr haufig zu beobachten, dass die von dem Substrat abgerissenen Polymer-
strukturen auf dem Stempel haften.
Zwei Beispiele fur die Auswirkung von Klebedefekten auf dem Stempel, die in der
Pragung detektiert werden konnen, sind in der Abbildung 6.17 dargestellt. In beiden
Fallen handelt es sich um Defekte im Bereich der 400 nm breiten Linien und Abstan-
de, der dem Feld E in der Abbildung 6.11 entspricht. Bei dem vorliegenden Stempel
hat dieses Feld die feinsten Linien und Abstande und dadurch die hochste strukturelle
Dichte. Folglich reprasentiert es einen fur die Klebedefekte kritischen Bereich in der
Nanoimprintlithographie. Bei beiden Pragungen (Abbildung 6.17 a) und c)) sind große

6.1 Oberflachengebundene Nanopartikel 99
a) b)
c) d)
Abbildung 6.17: Klebedefekt im Bereich der 400 nm breiten Linien und Abstande
auf der Pragung (a, c) und dem Stempel (b, d).
Bereiche der Polymerstruktur von dem Substrat abgerissen. In den Bildern a) und c)
entspricht das den dunklen Bereichen, also den Bereichen mit den kleineren Grauwer-
ten. Die Defekte werden annahernd spiegelbildlich auf der entsprechenden Stelle der
Stempeloberflache detektiert (Abbildung 6.17 b) und d)). Da der Stempel von sich
aus nicht fluoreszieren sollte, sind bei einem sauberen Stempel nur kleine Grauwerte,
entsprechend dem Hintergrund, zu erwarten. Grauwerte, die uber diesem Hintergrund-
grauwert liegen, sind den Polymerresten zu zuordnen. Das Bild d) stellt einen besonders
stark mit Polymer verunreinigten Stempel dar, da hier große Bereiche des Polymerfilms
auf dem Stempel haften.
Die Felder F des in der Abbildung 6.11 dargestellten Stempels reprasentieren eben-
falls Bereiche mit einer hohen Strukturdichte und kleinen Strukturen. Von besonderem
Interesse in diesem Bereich ist die Nachbarschaft von positiven und negativen Stempel-

100 6. Ergebnisse und Diskussion
a) b)
Abbildung 6.18: Fluoreszenzmikroskopische Aufnahmen von einem Klebedefekt im
Bereich der Bander a) auf der Pragung und b) auf dem Stempel.
strukturen. Positiv und negativ strukturierte Oberflachen haben ein unterschiedliches
Prageverhalten. Aufgrund des Materialtransportes begunstigt eine anfanglich hohe Po-
lymerschicht die Pragung von positiven Stempeln. Da durch die kleinen erhohten Struk-
turen eines positiven Stempels nur wenig Polymer verschoben wird, wird zum Auffullen
der großen vertieften Hohlraume ausreichend Polymer benotigt. Bei der Pragung eines
negativen Stempels beeinflusst eine zu hohe Menge an Polymer die Pragung eher ne-
gativ, da die Hohlraume des Stempels klein sind. Somit ist eher eine anfanglich dunne
Polymerschicht von Vorteil. Bei der Pragung von negativen und positiven Strukturen,
wie im vorliegenden Fall, hangt die gewahlte Schichtdicke von dem Muster des Stem-
pels ab [93].
In der Abbildung 6.18 ist ein Beispiel fur einen Klebedefekt im Feld F dargestellt.
In der Pragung (Abbildung 6.18 a)) ist zu erkennen, dass diese so genannten Bander
aus Struktureinheiten unterschiedlicher Großen und Formen bestehen. Viele der nega-
tiven Punkte sind von dem Substrat abgerissen. Ebenso ist ein großer Teil der 400 nm
breiten Linien und Abstande im oberen Bereich der Abbildung zerstort. Im Bereich der
positiven Punkte sind Fehlstellen zu sehen, die jedoch eher auf Fließdefekte, die deut-
lich am Rand des Feldes auftreten, zuruckzufuhren sind. Die großen unstrukturierten
Hohlraume des Stempels sind unzureichend aufgefullt. Auf dem Stempel (Abbildung

6.1 Oberflachengebundene Nanopartikel 101
6.18 b)) wird ein großer Teil der Polymerreste detektiert, die in der Pragung im Bereich
der negativen Punkte und der 400 nm breiten Linien fehlen. Wie in dem vorangegan-
genen Beispiel zeigt sich wieder ein annahernd spiegelbildliches Verhalten der Pragung
und des Stempels.
a) b)
c)
Abbildung 6.19: Gegenuberstellung von einem Klebedefekt im Bereich der 5 µm
breiten Linien und Abstande a) der Pragung und b) des Stempels. c) Optimale Pragung
im Bereich der 5 µm breiten Linien und Abstande.
Die bisherigen Beispiele der Klebedefekte bezogen sich auf die bekannten kritischen
Bereiche eines Stempels in der Nanoimprintlithographie. Dass es bei einer nicht opti-
malen Wahl der Prozessparameter auch zu schlechten Pragungen und entsprechenden
Defekten in eigentlich unkritischen Bereichen kommen kann, soll die Abbildung 6.19
demonstrieren. Sie zeigt das Feld D des Stempels mit 5 µm breiten Linien und Abstan-
den. Die Pragung (Abbildung 6.19 a)) zeigt eindeutig, dass sich die Struktur wahrend
des Pragens nicht vollstandig ausgebildet hat. Die Strukturen der Hohlraume sind nur

102 6. Ergebnisse und Diskussion
unzureichend ausgefullt. Zum Vergleich ist in der Abbildung 6.19 c) die Struktur einer
guten Pragung aus diesem Bereich gezeigt. Die Hohlraume des Stempels sind vollstan-
dig und gleichmaßig aufgefullt, wahrend das Polymer der anfanglichen Schicht durch die
erhohten Strukturen verdrangt wird. Dieses Muster ist in Bild 6.19 a) nur in der rechten
oberen Ecke schwach zu erkennen. Die Abbildung 6.19 b) zeigt die entsprechende Stelle
auf dem Stempel nach der nicht optimalen Pragung. Auf der Stempeloberflache konnen
eindeutig feine Faden detektiert werden, die in der Pragestruktur fehlen. Wieder zeigt
sich ein fast spiegelbildliches Verhalten von Pragung und Stempel.
Die Qualitatssicherung an den Stempeln ist ein Gebiet, das bisher weitestgehend ver-
nachlassigt wird. Die dargestellten Beispiele verdeutlichen die Notwendigkeit einer Qua-
litatssicherung in der Nanoimprintlithographie. Bisher unbekannt ist jedoch die Aus-
wirkung des an der Stempeloberflache anhaftenden Polymers bei einer Serie von Pra-
gungen. Es ist nicht auszuschließen, dass die Polymerreste auf dem Stempel bei einer
darauf folgenden Pragung in den Prageprozess mit einfließen und somit keine storenden
Effekte verursachen. Andererseits ist es auch moglich, dass durch diese Polymerreste
weitere Klebedefekte begunstigt werden. Daher sollten Stempeloberflachen, die anhaf-
tende Polymerreste aufweisen, gereinigt werden. Ferner wird auch deutlich, dass die
Fluoreszenzmikroskopie nicht nur eine geeignete Methode zur Untersuchung der Pra-
gungen darstellt, sondern auch zur Qualitatssicherung des Stempels verwendet werden
kann. Ein besonderer Vorteil im Hinblick auf die Stempel ist darin zu sehen, dass auch
in diesem Fall kein Kontakt zur Oberflache erforderlich ist und somit Defekte auf der
Stempeloberflache durch die Methode der Qualitatssicherung ausgeschlossen werden
konnen. Hinzu kommt, dass das Material, aus dem der Stempel besteht, keinen Ein-
fluss auf die Qualitatssicherung mittels der Fluoreszenzmikroskopie hat. Treten mecha-
nische Fehler in der Stempelstruktur auf, sind diese mittels der Fluoreszenzmikroskopie
nur anhand der Pragung zu erkennen. In solchen Fallen ware eine Untersuchung des
Stempels mit SEM oder AFM notwendig.

6.1 Oberflachengebundene Nanopartikel 103
6.1.2.3 Bestimmung der Qualitat einer Nanopragung
Die Qualitat einer Nanopragung wird nicht nur durch eine genaue Kopie der Stempel-
struktur bestimmt, sondern auch durch eine gleichmaßige Pragetiefe der Strukturen.
Wie in dem Kapitel 6.1.2.1 bereits gezeigt wird, konnen unterschiedliche Dicken der
Polymerschicht mittels der Fluoreszenzmikroskopie detektiert werden (vgl. Abbildung
6.13). Im Allgemeinen wird die Profilometrie zur Bestimmung von Schichtdicken ver-
wendet. Durch diese Methode kann der Querschnitt einer Pragung direkt bestimmt
werden. Bei dieser Methode ist der direkte Kontakt zu der Polymeroberflache nicht
zu vermeiden, da die Probe mit einer feinen Spitze abgetastet wird. Nachfolgend sind
die Ergebnisse der Messungen mit der Profilometrie den fluoreszenzmikroskopischen
Aufnahmen und Auswertungen gegenubergestellt. Die Profilometriemessungen werden
von der Arbeitsgruppe von Frau Prof. Dr.-Ing. H.-C. Scheer durchgefuhrt.
Bei der Charakterisierung der Defekte ist gezeigt worden, dass die Grenzen der Na-
noimprintlithographie eher in der Abbildung großer unstrukturierter Flachen als in der
Abbildung kleiner strukturierter Flachen liegen. Bei der Pragung von großen erhohten
Flachen des Stempels muss das verdrangte Polymer in die benachbarten Hohlraume
transportiert werden. Da das Verschieben des Polymers, das unter der erhohten Stem-
pelstruktur zunachst komprimiert wird, entsprechend viel Zeit benotigt, mussen die
Prozessparameter Pragezeit, Temperatur und Druck optimal eingestellt werden. Bei
einer zu kurzen Pragezeit oder zu niedrigem Druck und Temperatur kann der Polymer-
transport nicht vollstandig abgeschlossen werden, so dass Polymerreste unter der er-
hohten Stempelstruktur verbleiben. Dieses kann beim Abheben des Stempels zu einem
Defekt fuhren, der als Verkrummung bezeichnet wird [95]. Dieser ist zu der Gruppe der
Fließdefekte zu zahlen (vgl. Abbildung 6.16). Aufgrund dieser Problematik erscheint es
sinnvoll, die Gleichmaßigkeit und somit auch die Qualitat einer Pragung in diesem in
der Nanoimprintlithographie kritischen Bereich zu prufen. Bei dem verwendeten Stem-
pel, der in der Abbildung 6.11 dargestellt ist, ist dieser Bereich in den Feldern A zu
finden. Hierbei handelt es sich um 3 × 3 mm2 große Felder mit 100 × 100 µm2 Qua-
draten, die bereits im Zusammenhang mit den Fließdefekten (vgl. Abbildungen 6.15
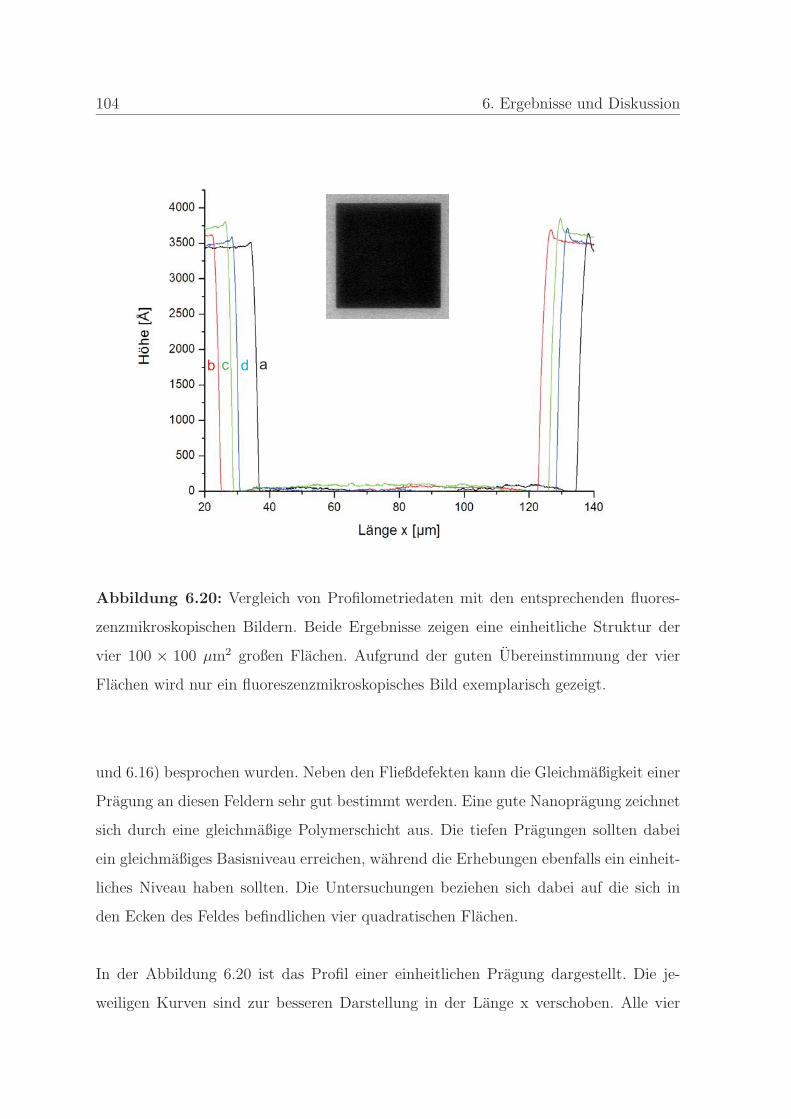
104 6. Ergebnisse und Diskussion
b c d a
Abbildung 6.20: Vergleich von Profilometriedaten mit den entsprechenden fluores-
zenzmikroskopischen Bildern. Beide Ergebnisse zeigen eine einheitliche Struktur der
vier 100 × 100 µm2 großen Flachen. Aufgrund der guten Ubereinstimmung der vier
Flachen wird nur ein fluoreszenzmikroskopisches Bild exemplarisch gezeigt.
und 6.16) besprochen wurden. Neben den Fließdefekten kann die Gleichmaßigkeit einer
Pragung an diesen Feldern sehr gut bestimmt werden. Eine gute Nanopragung zeichnet
sich durch eine gleichmaßige Polymerschicht aus. Die tiefen Pragungen sollten dabei
ein gleichmaßiges Basisniveau erreichen, wahrend die Erhebungen ebenfalls ein einheit-
liches Niveau haben sollten. Die Untersuchungen beziehen sich dabei auf die sich in
den Ecken des Feldes befindlichen vier quadratischen Flachen.
In der Abbildung 6.20 ist das Profil einer einheitlichen Pragung dargestellt. Die je-
weiligen Kurven sind zur besseren Darstellung in der Lange x verschoben. Alle vier
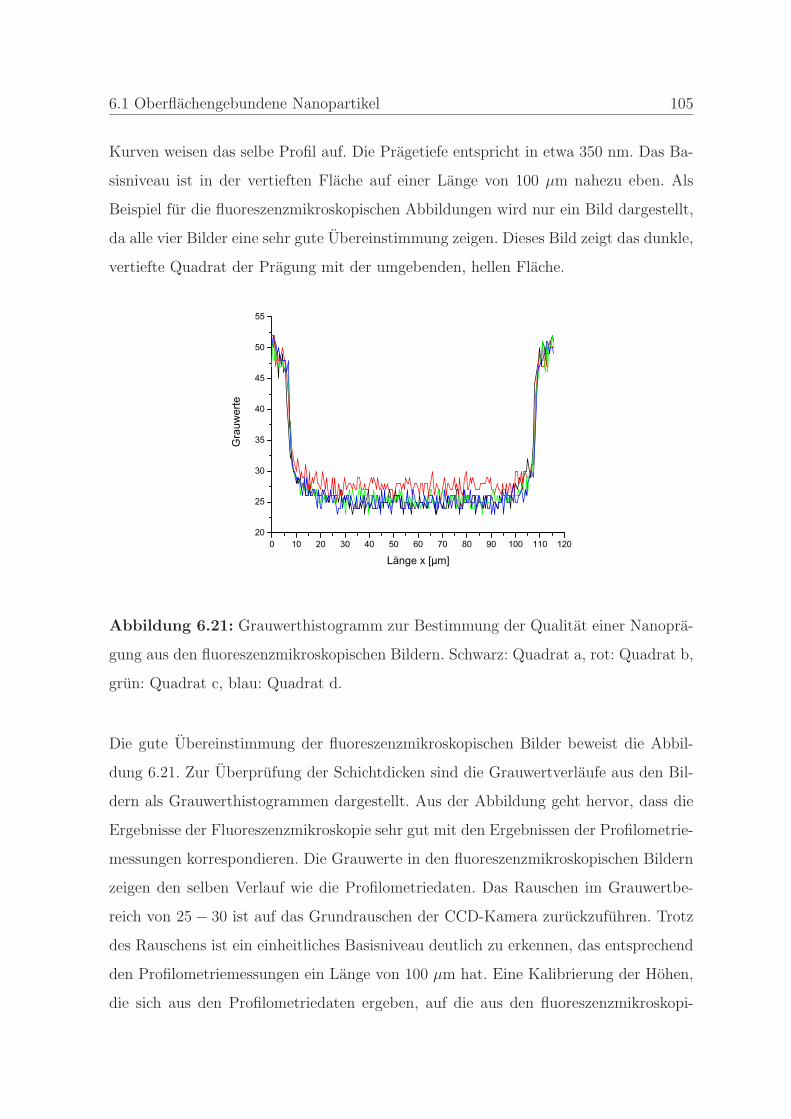
6.1 Oberflachengebundene Nanopartikel 105
Kurven weisen das selbe Profil auf. Die Pragetiefe entspricht in etwa 350 nm. Das Ba-
sisniveau ist in der vertieften Flache auf einer Lange von 100 µm nahezu eben. Als
Beispiel fur die fluoreszenzmikroskopischen Abbildungen wird nur ein Bild dargestellt,
da alle vier Bilder eine sehr gute Ubereinstimmung zeigen. Dieses Bild zeigt das dunkle,
vertiefte Quadrat der Pragung mit der umgebenden, hellen Flache.
Abbildung 6.21: Grauwerthistogramm zur Bestimmung der Qualitat einer Nanopra-
gung aus den fluoreszenzmikroskopischen Bildern. Schwarz: Quadrat a, rot: Quadrat b,
grun: Quadrat c, blau: Quadrat d.
Die gute Ubereinstimmung der fluoreszenzmikroskopischen Bilder beweist die Abbil-
dung 6.21. Zur Uberprufung der Schichtdicken sind die Grauwertverlaufe aus den Bil-
dern als Grauwerthistogrammen dargestellt. Aus der Abbildung geht hervor, dass die
Ergebnisse der Fluoreszenzmikroskopie sehr gut mit den Ergebnissen der Profilometrie-
messungen korrespondieren. Die Grauwerte in den fluoreszenzmikroskopischen Bildern
zeigen den selben Verlauf wie die Profilometriedaten. Das Rauschen im Grauwertbe-
reich von 25 − 30 ist auf das Grundrauschen der CCD-Kamera zuruckzufuhren. Trotz
des Rauschens ist ein einheitliches Basisniveau deutlich zu erkennen, das entsprechend
den Profilometriemessungen ein Lange von 100 µm hat. Eine Kalibrierung der Hohen,
die sich aus den Profilometriedaten ergeben, auf die aus den fluoreszenzmikroskopi-
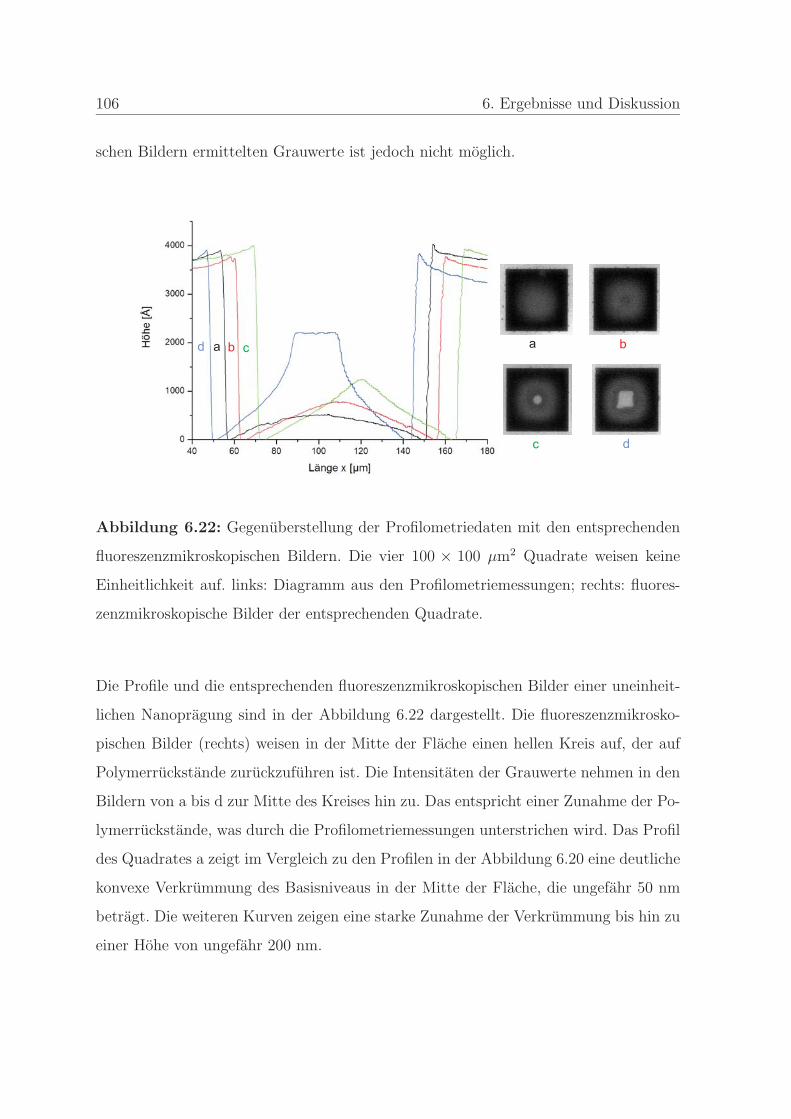
106 6. Ergebnisse und Diskussion
schen Bildern ermittelten Grauwerte ist jedoch nicht moglich.
a b
c d
d ca b
Abbildung 6.22: Gegenuberstellung der Profilometriedaten mit den entsprechenden
fluoreszenzmikroskopischen Bildern. Die vier 100 × 100 µm2 Quadrate weisen keine
Einheitlichkeit auf. links: Diagramm aus den Profilometriemessungen; rechts: fluores-
zenzmikroskopische Bilder der entsprechenden Quadrate.
Die Profile und die entsprechenden fluoreszenzmikroskopischen Bilder einer uneinheit-
lichen Nanopragung sind in der Abbildung 6.22 dargestellt. Die fluoreszenzmikrosko-
pischen Bilder (rechts) weisen in der Mitte der Flache einen hellen Kreis auf, der auf
Polymerruckstande zuruckzufuhren ist. Die Intensitaten der Grauwerte nehmen in den
Bildern von a bis d zur Mitte des Kreises hin zu. Das entspricht einer Zunahme der Po-
lymerruckstande, was durch die Profilometriemessungen unterstrichen wird. Das Profil
des Quadrates a zeigt im Vergleich zu den Profilen in der Abbildung 6.20 eine deutliche
konvexe Verkrummung des Basisniveaus in der Mitte der Flache, die ungefahr 50 nm
betragt. Die weiteren Kurven zeigen eine starke Zunahme der Verkrummung bis hin zu
einer Hohe von ungefahr 200 nm.
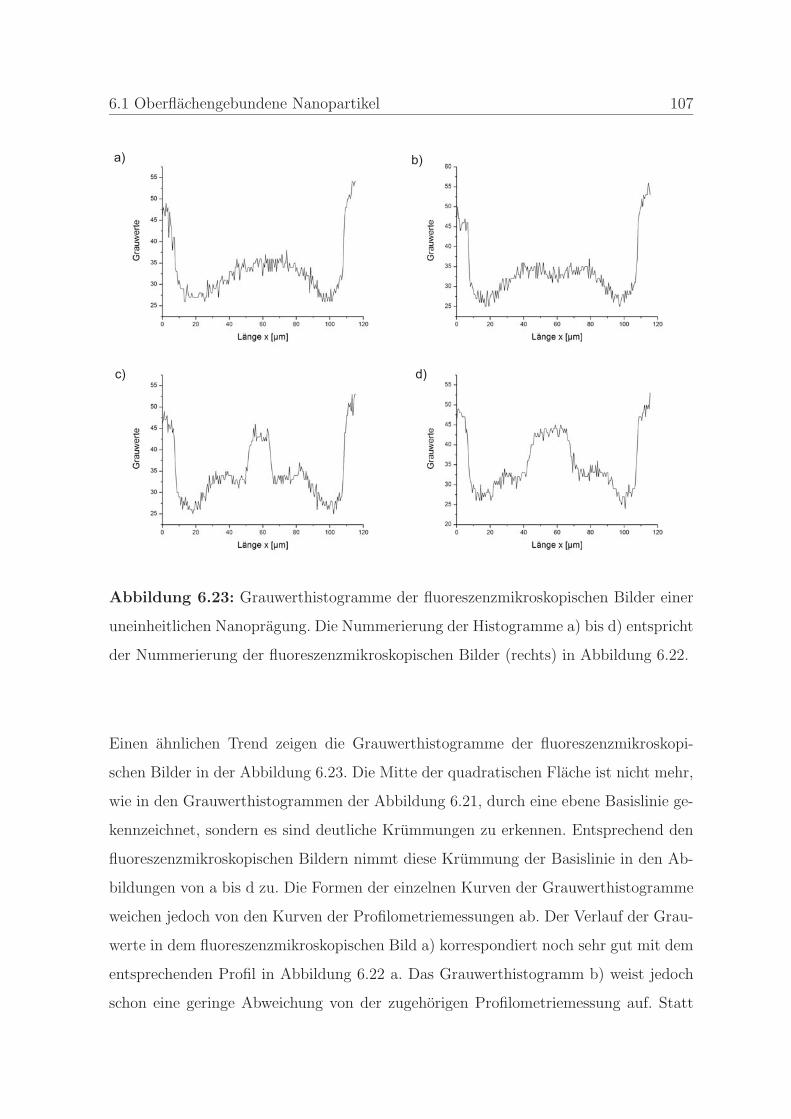
6.1 Oberflachengebundene Nanopartikel 107
a) b)
c) d)
Abbildung 6.23: Grauwerthistogramme der fluoreszenzmikroskopischen Bilder einer
uneinheitlichen Nanopragung. Die Nummerierung der Histogramme a) bis d) entspricht
der Nummerierung der fluoreszenzmikroskopischen Bilder (rechts) in Abbildung 6.22.
Einen ahnlichen Trend zeigen die Grauwerthistogramme der fluoreszenzmikroskopi-
schen Bilder in der Abbildung 6.23. Die Mitte der quadratischen Flache ist nicht mehr,
wie in den Grauwerthistogrammen der Abbildung 6.21, durch eine ebene Basislinie ge-
kennzeichnet, sondern es sind deutliche Krummungen zu erkennen. Entsprechend den
fluoreszenzmikroskopischen Bildern nimmt diese Krummung der Basislinie in den Ab-
bildungen von a bis d zu. Die Formen der einzelnen Kurven der Grauwerthistogramme
weichen jedoch von den Kurven der Profilometriemessungen ab. Der Verlauf der Grau-
werte in dem fluoreszenzmikroskopischen Bild a) korrespondiert noch sehr gut mit dem
entsprechenden Profil in Abbildung 6.22 a. Das Grauwerthistogramm b) weist jedoch
schon eine geringe Abweichung von der zugehorigen Profilometriemessung auf. Statt

108 6. Ergebnisse und Diskussion
einer Zunahme der konvexen Verkrummung ist in der Mitte eine leichte Abnahme der
Grauwerte zu beobachten. Entsprechend ist dies in dem fluoreszenzmikroskopischen
Bild 6.22 b als dunkler Punkt in der Mitte zu erkennen. In den darauf folgenden Grau-
werthistogrammen c) und d) nehmen die Grauwerte, wie erwartet, in der Mitte der
Flache zu, aber sie weisen anstelle eines kontinuierlichen Anstiegs, wie in den Profilo-
metriemessungen, eher einen sprunghaften Anstieg auf. Der Sprung in den Grauwerten
ist auch in den zugehorigen fluoreszenzmikroskopischen Bildern zu sehen. In diesen
Bildern (6.22 c und d) liegt um die hellste Flache in der Mitte erkennbar ein dunkler
Ring. Da eine dreidimensionale Abbildung mittels der Fluoreszenzmikroskopie nicht
moglich ist, handelt es sich hierbei vermutlich um einen Abbildungsfehler, der sich aus
der berg- bzw. vulkanformigen Struktur des Polymerruckstandes ableiten lasst.
Die Fluoreszenzmikroskopie erweist sich als gute Alternative fur die Untersuchung der
Qualitat einer Pragung. Der Vergleich mit den Profilometriemessungen zeigt, dass bei-
de Messmethoden dieselben Trends aufzeigen. Eine direkte Bestimmung der Dicke der
Polymerschicht ist mittels der Fluoreszenzmikroskopie zwar nicht moglich. Im Hinblick
auf die Qualitatssicherung an Nanopragungen uberwiegen jedoch die Vorteile gegen-
uber der Profilometrie. Hierzu zahlen die bereits angesprochene kontaktfreie Detektion
der Oberflache und eine sehr schnelle Auswertung. Die beobachteten Trends konnen
qualitativ direkt aus den fluoreszenzmikroskopischen Aufnahmen abgeleitet werden.
Die entsprechenden Grauwerthistogramme unterstreichen diese quantitativ.
6.1.2.4 Qualitatssicherung an einem Stempel
In den vorangegangen Kapiteln werden mittels der Fluoreszenzmikroskopie die in der
Nanoimprintlithographie auftretenden Defekte dargestellt und charakterisiert. Die Fluo-
reszenzmikroskopie ist eine geeignete Alternative zur Qualitatssicherung an Nanopra-
gungen zu den bisher ublicherweise verwendeten Methoden wie SEM, AFM oder der
Profilometrie. Nachfolgend wird nun betrachtet, wie sich die durch adhasive Polymer-
ruckstande auftretenden Defekte auf der Stempeloberflache in einer Serie von Pragun-
gen verhalten. Durch den direkten Kontakt zwischen der Stempeloberflache und dem

6.1 Oberflachengebundene Nanopartikel 109
Substrat sind grundsatzlich drei Hypothesen zur Entstehung und Fortpflanzung der
Defekte wahrend der Pragung moglich [97]:
1. In einem perfekten Prageprozess entstehen keine Defekte und somit kann auch
keine Fortpflanzung der Defekte stattfinden.
2. Wahrend des Pragezyklus entstehen Defekte, die sich nicht in den darauf folgen-
den Pragungen fortsetzen. Die willkurliche Entstehung der Defekte kann zu einer
nachfolgenden Pragung ohne Defekte fuhren. Es wird auch von einem Selbstrei-
nigungsprozess gesprochen.
3. Die entstehenden Defekte setzen sich in den nachfolgenden Pragungen fort. Das
Ergebnis ware ein extremer Verlust der Genauigkeit der Musterubertragung.
Im Hinblick auf einen industriellen Produktionsprozess, d. h. einer Serie von Pragungen,
wird ein halbautomatisches Verfahren, das auf der Fluoreszenzmikroskopie basiert, zur
Qualitatssicherung an einem Stempel vorgestellt. Serien von Nanopragungen werden
mit drei verschiedenen Stempeln durchgefuhrt, die nach jeder Pragung auf Defektstel-
len untersucht werden (vgl. Kapitel 5.2.4.2). Im Vordergrund dieser Untersuchung steht
dabei nicht, wie in den vorangegangenen Kapiteln, die Charakterisierung der Defekte,
sondern vielmehr deren Quantifizierung.
Die Qualitat eines Stempels wird anhand der Menge der auf der Stempeloberflache
anhaftenden Polymerruckstande charakterisiert. In der Abbildung 6.24 sind zwei ge-
gensatzliche Zustande einer Stempeloberflache dargestellt. Die Bilder zeigen eines der
100×100 µm2 großen Felder (Odd-Struktur) des ersten Stempels (vgl. Kapitel 5.2.4.2).
Den Fluoreszenzbildern sind mit sichtbarem Licht aufgenommene Bilder gegenuberge-
stellt. Auf einer sauberen Stempeloberflache (Abbildung 6.24 a)) werden keine fluo-
reszierende Objekte detektiert, so dass das entsprechende Fluoreszenzbild einheitlich
dunkel ist. Eine stark verunreinigte Stempeloberflache (Abbildung 6.24 b)) hingegen
weist eine Anhaufung von Polymerruckstanden in Form von fluoreszierenden Objekten
auf, die im Fluoreszenzbild als helle Bildpunkte erscheinen.

110 6. Ergebnisse und Diskussion
a) b)
c) d)
10
0µ
m
Abbildung 6.24: Fluoreszenzbilder zweier verschiedener Stempelzustande. Die Bilder
a) und b) sind die Fluoreszenzbilder, die dazu korrespondierenden Bilder c) und d)
werden mit sichtbarem Licht aufgenommen. Auf dem linken Bild a) konnen keine Po-
lymerruckstande detektiert werden, wahrend im rechten Bild b) eine starke Adhasion
des Polymers auf der Stempeloberflache beobachtet werden kann. In den Bildern c)
und d) konnen dieselben Effekte auch mit sichtbarem Licht beobachtet werden, wobei
der Kontrast wesentlich geringer ist.
In den mit sichtbarem Licht aufgenommenen Bildern wird ebenfalls die Zunahme des
Polymers auf der Stempeloberflache beobachtet. Die Abbildung 6.24 d) weist im Ver-
gleich zu der Abbildung 6.24 c) einen starkeren Kontrast der Strukturen innerhalb
des 100 × 100 µm2 großen Feldes auf. Der Unterschied in diesen zwei Bildern ist je-
doch wesentlich undeutlicher als bei den entsprechenden Fluoreszenzaufnahmen. Nur
bei genauer Betrachtung ist zu erkennen, dass neben dem starkeren Kontrast noch an
einer weiteren Stelle (roter Kreis) auf der Stempeloberflache Polymerruckstande zu fin-

6.1 Oberflachengebundene Nanopartikel 111
den sind. Ein weiterer Nachteil der Auflicht-Mikroskopie mit sichtbarem Licht besteht
darin, dass es aufgrund der Wellenlange des sichtbaren Lichtes nicht moglich ist, kleine-
re Strukturen aufzulosen. Daher werden ausschließlich die fluoreszenzmikroskopischen
Aufnahmen der Stempeloberflachen fur die nachfolgende Auswertung herangezogen.
a) b)
c)
Länge [Pixel]
Länge [Pixel]
Länge [Pixel]
Bre
ite[P
ixel]
Bre
ite[P
ixel]
Bre
ite[P
ixel]H
öhe
[Pix
el]
Höhe
[Pix
el]
Höhe
[Pix
el]
Abbildung 6.25: Bildverarbeitungsprozesse zur Bestimmung der Qualitat eines Stem-
pels in einer Serienpragung. a) Originalbild als 3D-Grafik. b) Korrektur der Verzerrung
des Bildes. c) Weichzeichnen der Grafik.
Zur Beurteilung der Qualitat eines Stempels wird das Auswertungsprogramm, das im
Anhang A wiedergeben ist, verwendet. Hierbei werden die Bilder in Form von digi-
talisierten Daten eingelesen und bearbeitet. Das Programm besteht im Wesentlichen
aus drei Prozessen zur Bildverarbeitung: Der Korrektur der Verzerrung des Original-
bildes, dem Weichzeichnen und einer finalen Anpassung des Bildausschnittes an den
entscheidenden Grauwertbereich. In der Abbildung 6.25 sind die Auswirkungen der
bildverarbeitenden Prozesse zur Korrektur der Verzerrung des Originalbildes und des

112 6. Ergebnisse und Diskussion
Weichzeichnens grafisch dargestellt. Das originale 3D-Bild zeigt einen Abfall der Grau-
wertintensitaten in Richtung der Lange des Bildes. Dieser Intensitatsverlust liegt in
einem Bereich von ungefahr zehn Pixeln. Zur Korrektur dieses Abfalls wird fur jedes
Bild ein entsprechendes Kompensationsbild erstellt. Dafur wird zunachst eine lineare
Kompensationsgerade der funften Spalte eines Bildes berechnet. Das Produkt aus der
Anzahl der Spalten des Bildes und der Kompensationsgeraden ergibt das Kompensati-
onsbild. Aus der Subtraktion des Kompensationsbildes von dem Originalbild resultiert
ein neues Bild (Abbildung 6.25 b)), dessen Grundflache auf den Wert Null festgelegt
wird.
a) b)
0 10 20 30 40 50 60 70
10
20
30
40
50
Abbildung 6.26: Verbesserung des Signal-Rausch-Verhaltnisses durch die Bildverar-
beitungsprozedur des Weichzeichnens. a) Das Originalbild als 2D-Grafik. b) Das weich-
gezeichnete Bild als 2D-Grafik.
Zur Verbesserung des Signal-Rausch-Verhaltnisses, das aus den bei der Bildaufnah-
me gewahlten Kontrast- und Helligkeitseinstellungen resultiert, wird zur Bildverarbei-
tung das Weichzeichnen durchgefuhrt. Das Weichzeichnen kann mittels unterschied-
licher Methoden vollzogen werden. In dieser Arbeit wird der mittlere Grauwert von
zehn benachbarten Pixeln berechnet und eingesetzt. Das neu berechnete Bild hat so-
mit eine neue Große, die um ein Zehnfaches kleiner ist als die ursprungliche Bildgroße.
Die Pixelgroße sollte so gewahlt sein, dass einerseits das Signal-Rausch-Verhaltnis aus-
reichend verbessert wird, andererseits aber nicht zu viele Bildinformationen verloren
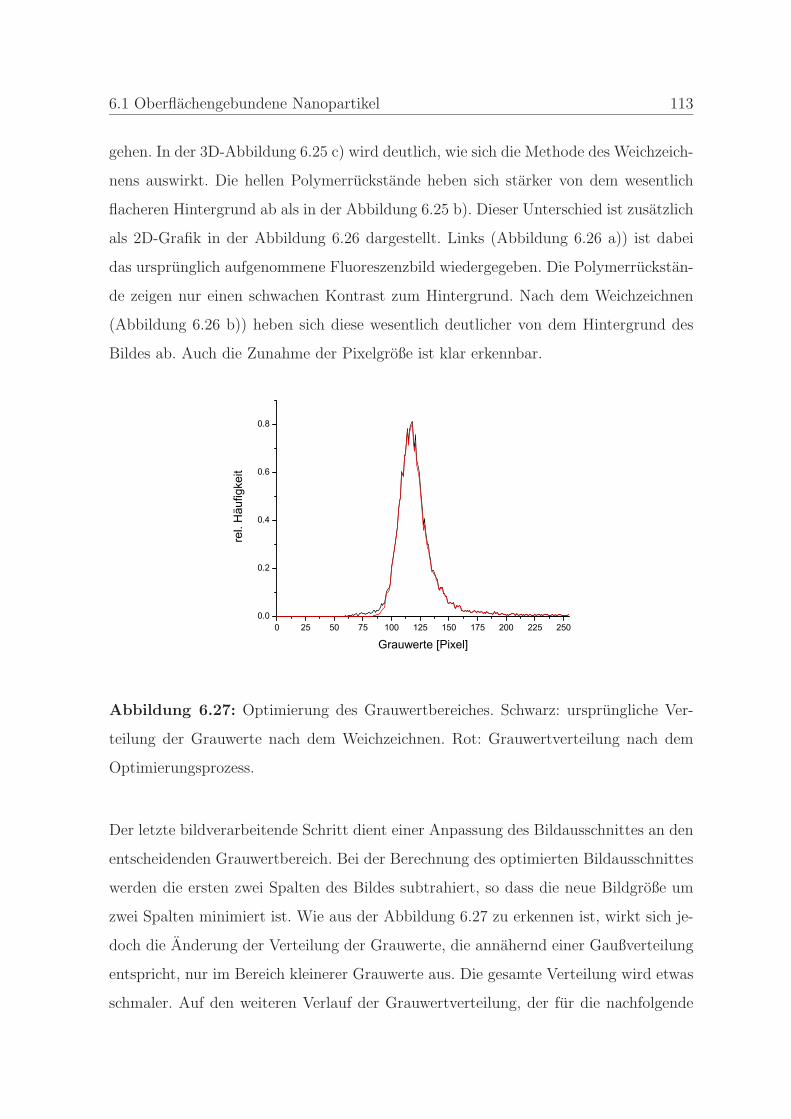
6.1 Oberflachengebundene Nanopartikel 113
gehen. In der 3D-Abbildung 6.25 c) wird deutlich, wie sich die Methode des Weichzeich-
nens auswirkt. Die hellen Polymerruckstande heben sich starker von dem wesentlich
flacheren Hintergrund ab als in der Abbildung 6.25 b). Dieser Unterschied ist zusatzlich
als 2D-Grafik in der Abbildung 6.26 dargestellt. Links (Abbildung 6.26 a)) ist dabei
das ursprunglich aufgenommene Fluoreszenzbild wiedergegeben. Die Polymerruckstan-
de zeigen nur einen schwachen Kontrast zum Hintergrund. Nach dem Weichzeichnen
(Abbildung 6.26 b)) heben sich diese wesentlich deutlicher von dem Hintergrund des
Bildes ab. Auch die Zunahme der Pixelgroße ist klar erkennbar.
Abbildung 6.27: Optimierung des Grauwertbereiches. Schwarz: ursprungliche Ver-
teilung der Grauwerte nach dem Weichzeichnen. Rot: Grauwertverteilung nach dem
Optimierungsprozess.
Der letzte bildverarbeitende Schritt dient einer Anpassung des Bildausschnittes an den
entscheidenden Grauwertbereich. Bei der Berechnung des optimierten Bildausschnittes
werden die ersten zwei Spalten des Bildes subtrahiert, so dass die neue Bildgroße um
zwei Spalten minimiert ist. Wie aus der Abbildung 6.27 zu erkennen ist, wirkt sich je-
doch die Anderung der Verteilung der Grauwerte, die annahernd einer Gaußverteilung
entspricht, nur im Bereich kleinerer Grauwerte aus. Die gesamte Verteilung wird etwas
schmaler. Auf den weiteren Verlauf der Grauwertverteilung, der fur die nachfolgende
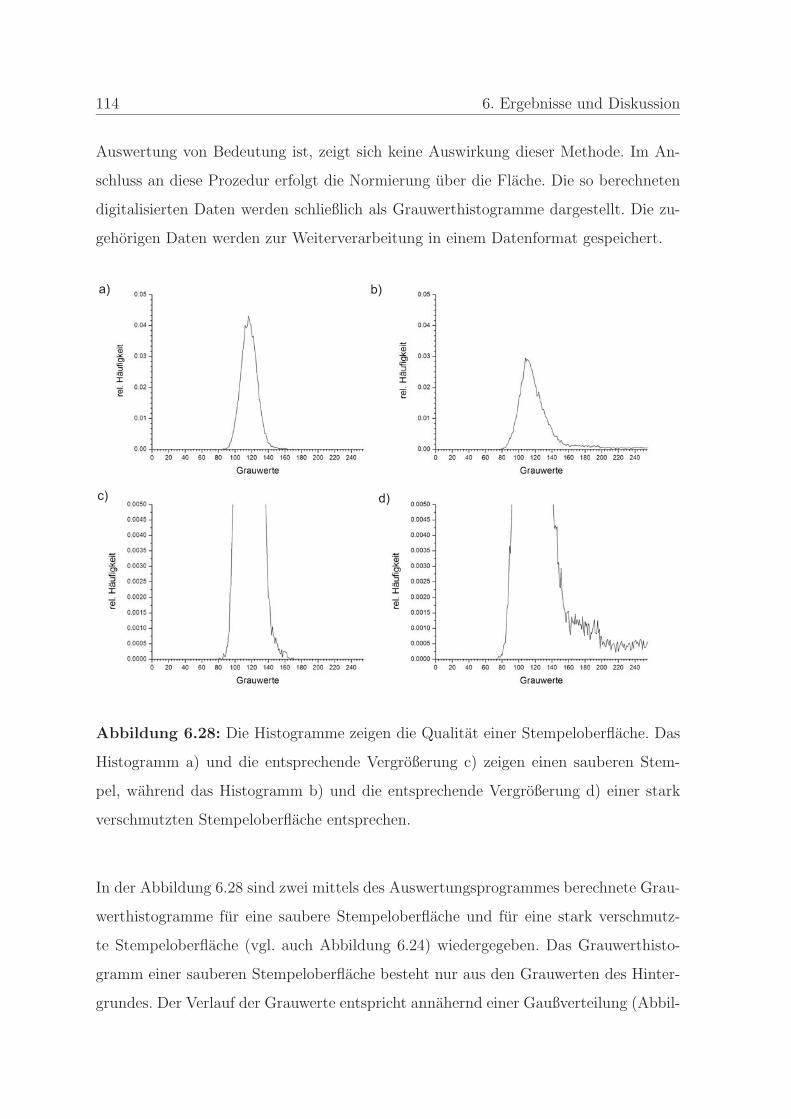
114 6. Ergebnisse und Diskussion
Auswertung von Bedeutung ist, zeigt sich keine Auswirkung dieser Methode. Im An-
schluss an diese Prozedur erfolgt die Normierung uber die Flache. Die so berechneten
digitalisierten Daten werden schließlich als Grauwerthistogramme dargestellt. Die zu-
gehorigen Daten werden zur Weiterverarbeitung in einem Datenformat gespeichert.
a) b)
c) d)
Abbildung 6.28: Die Histogramme zeigen die Qualitat einer Stempeloberflache. Das
Histogramm a) und die entsprechende Vergroßerung c) zeigen einen sauberen Stem-
pel, wahrend das Histogramm b) und die entsprechende Vergroßerung d) einer stark
verschmutzten Stempeloberflache entsprechen.
In der Abbildung 6.28 sind zwei mittels des Auswertungsprogrammes berechnete Grau-
werthistogramme fur eine saubere Stempeloberflache und fur eine stark verschmutz-
te Stempeloberflache (vgl. auch Abbildung 6.24) wiedergegeben. Das Grauwerthisto-
gramm einer sauberen Stempeloberflache besteht nur aus den Grauwerten des Hinter-
grundes. Der Verlauf der Grauwerte entspricht annahernd einer Gaußverteilung (Abbil-
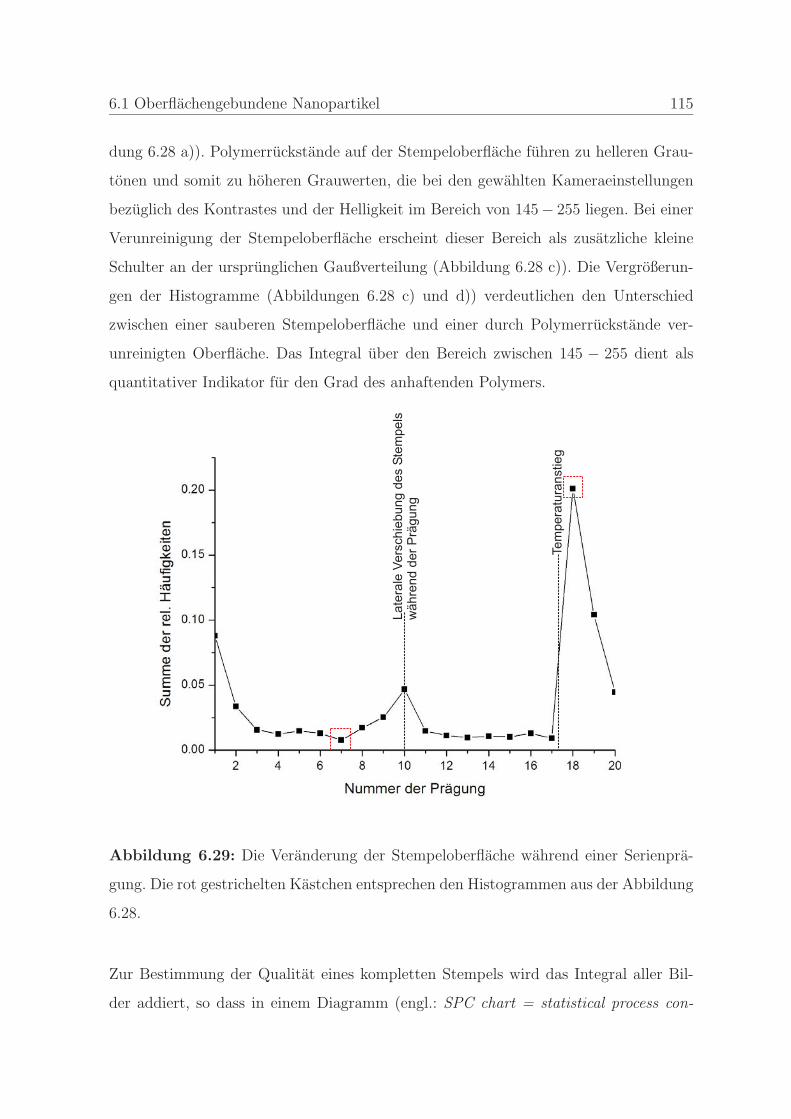
6.1 Oberflachengebundene Nanopartikel 115
dung 6.28 a)). Polymerruckstande auf der Stempeloberflache fuhren zu helleren Grau-
tonen und somit zu hoheren Grauwerten, die bei den gewahlten Kameraeinstellungen
bezuglich des Kontrastes und der Helligkeit im Bereich von 145− 255 liegen. Bei einer
Verunreinigung der Stempeloberflache erscheint dieser Bereich als zusatzliche kleine
Schulter an der ursprunglichen Gaußverteilung (Abbildung 6.28 c)). Die Vergroßerun-
gen der Histogramme (Abbildungen 6.28 c) und d)) verdeutlichen den Unterschied
zwischen einer sauberen Stempeloberflache und einer durch Polymerruckstande ver-
unreinigten Oberflache. Das Integral uber den Bereich zwischen 145 − 255 dient als
quantitativer Indikator fur den Grad des anhaftenden Polymers.L
ate
rale
Ve
rsch
ieb
un
gd
es
Ste
mp
els
wä
hre
nd
de
rP
räg
un
g
Te
mp
era
tura
nstie
g
Abbildung 6.29: Die Veranderung der Stempeloberflache wahrend einer Serienpra-
gung. Die rot gestrichelten Kastchen entsprechen den Histogrammen aus der Abbildung
6.28.
Zur Bestimmung der Qualitat eines kompletten Stempels wird das Integral aller Bil-
der addiert, so dass in einem Diagramm (engl.: SPC chart = statistical process con-

116 6. Ergebnisse und Diskussion
trol chart) die Veranderungen wahrend einer Serie von Pragungen dargestellt werden
konnen. Ein solches Diagramm ist in der Abbildung 6.29 fur den ersten Stempel wie-
dergegeben. Nach der ersten Pragung weist die Stempeloberflache eine relativ große
Menge an adhasiven Polymerruckstanden auf. Diese nehmen nach den nachsten bei-
den Pragungen aufgrund des Selbstreinigungsprozesses stark ab [97, 98]. Ab der dritten
Pragung zeigt die Stempeloberflache keine starke Verschmutzung. Die Summe der Inte-
grale liegt in einem Bereich von 0, 01−0, 025. Die im Diagramm erkennbare Abweichung
von diesem Zustand nach der 10. Pragung ergibt sich aus einem experimentellen Pro-
blem, das wahrend des Prageprozesses auftritt. Bei der 10. Pragung findet eine laterale
Verschiebung des Stempels statt, so dass das Polymer verstarkt in den Hohlraumen
des Stempels anhaftet. Die Stempeloberflachen der darauf folgenden Pragungen weisen
wieder eine sehr geringe Verschmutzung auf, deren Summen der Integrale ebenfalls in
einem Bereich von 0, 01− 0, 025 liegen. Eine starke Zunahme an anhaftenden Polymer
zeigt die Stempeloberflache nach der 18. Pragung. Bei dieser Pragung wird die Tempe-
ratur im Vergleich zu der vorherigen Pragung um 20 ◦C erhoht. Wieder kann bei den
darauf folgenden Untersuchungen eine Abnahme der Polymerruckstande entsprechend
des Selbstreinigungsprozesses beobachtet werden.
Wie bereits in den Kapiteln 6.1.2.1 und 6.1.2.2 erlautert wurde, ist die Anordnung
der Strukturen in einem Feld und die Art dieser Strukturen bei der Diskussion der
auftretenden Defekte relevant. Fur die Stempel 1 und 2 werden die Defekte der glei-
chen Felder einer Pragung addiert, wodurch ein quantitativer Vergleich zwischen den
Defekten der 100× 100 µm2 großen Felder und der 200× 200 µm2 großen Felder mog-
lich ist. Die kritischen Bereiche dieser Felder sind vor allem die Linien mit festgelegten
Abstanden zueinander, da bei diesen Strukturen eine sehr hohe Dichte vorliegt. Beide
Felder haben 200 nm und 100 nm breite Linien. Sie unterscheiden sich jedoch in den
entsprechenden Abstanden zwischen den Linien. Die Abstande zwischen den Linien
in dem 200 × 200 µm2 großen Feld sind 300 nm breit, wahrend die Abstande in dem
100 × 100 µm2 großen Feld nur 200 nm betragen. Folglich ist die Strukturdichte in
dem 100× 100 µm2 großen Feld hoher als in dem 200× 200 µm2 großen Feld, wodurch
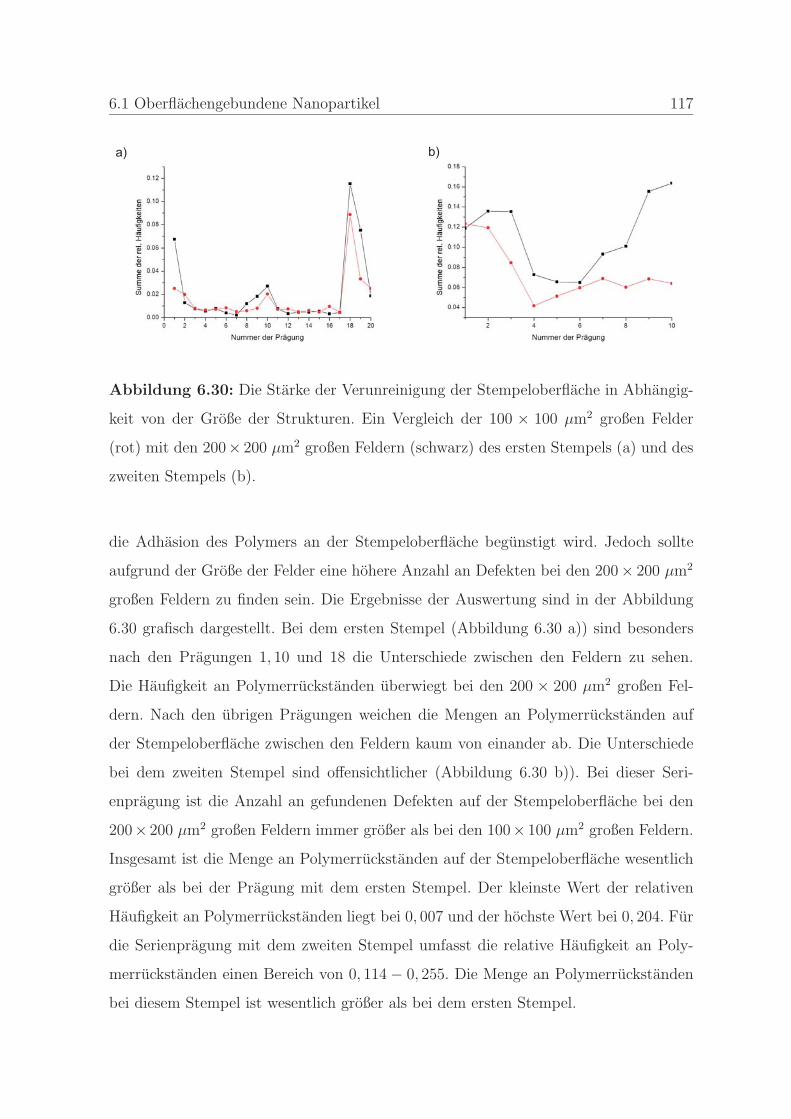
6.1 Oberflachengebundene Nanopartikel 117
a) b)
Abbildung 6.30: Die Starke der Verunreinigung der Stempeloberflache in Abhangig-
keit von der Große der Strukturen. Ein Vergleich der 100 × 100 µm2 großen Felder
(rot) mit den 200× 200 µm2 großen Feldern (schwarz) des ersten Stempels (a) und des
zweiten Stempels (b).
die Adhasion des Polymers an der Stempeloberflache begunstigt wird. Jedoch sollte
aufgrund der Große der Felder eine hohere Anzahl an Defekten bei den 200× 200 µm2
großen Feldern zu finden sein. Die Ergebnisse der Auswertung sind in der Abbildung
6.30 grafisch dargestellt. Bei dem ersten Stempel (Abbildung 6.30 a)) sind besonders
nach den Pragungen 1, 10 und 18 die Unterschiede zwischen den Feldern zu sehen.
Die Haufigkeit an Polymerruckstanden uberwiegt bei den 200 × 200 µm2 großen Fel-
dern. Nach den ubrigen Pragungen weichen die Mengen an Polymerruckstanden auf
der Stempeloberflache zwischen den Feldern kaum von einander ab. Die Unterschiede
bei dem zweiten Stempel sind offensichtlicher (Abbildung 6.30 b)). Bei dieser Seri-
enpragung ist die Anzahl an gefundenen Defekten auf der Stempeloberflache bei den
200× 200 µm2 großen Feldern immer großer als bei den 100× 100 µm2 großen Feldern.
Insgesamt ist die Menge an Polymerruckstanden auf der Stempeloberflache wesentlich
großer als bei der Pragung mit dem ersten Stempel. Der kleinste Wert der relativen
Haufigkeit an Polymerruckstanden liegt bei 0, 007 und der hochste Wert bei 0, 204. Fur
die Serienpragung mit dem zweiten Stempel umfasst die relative Haufigkeit an Poly-
merruckstanden einen Bereich von 0, 114 − 0, 255. Die Menge an Polymerruckstanden
bei diesem Stempel ist wesentlich großer als bei dem ersten Stempel.
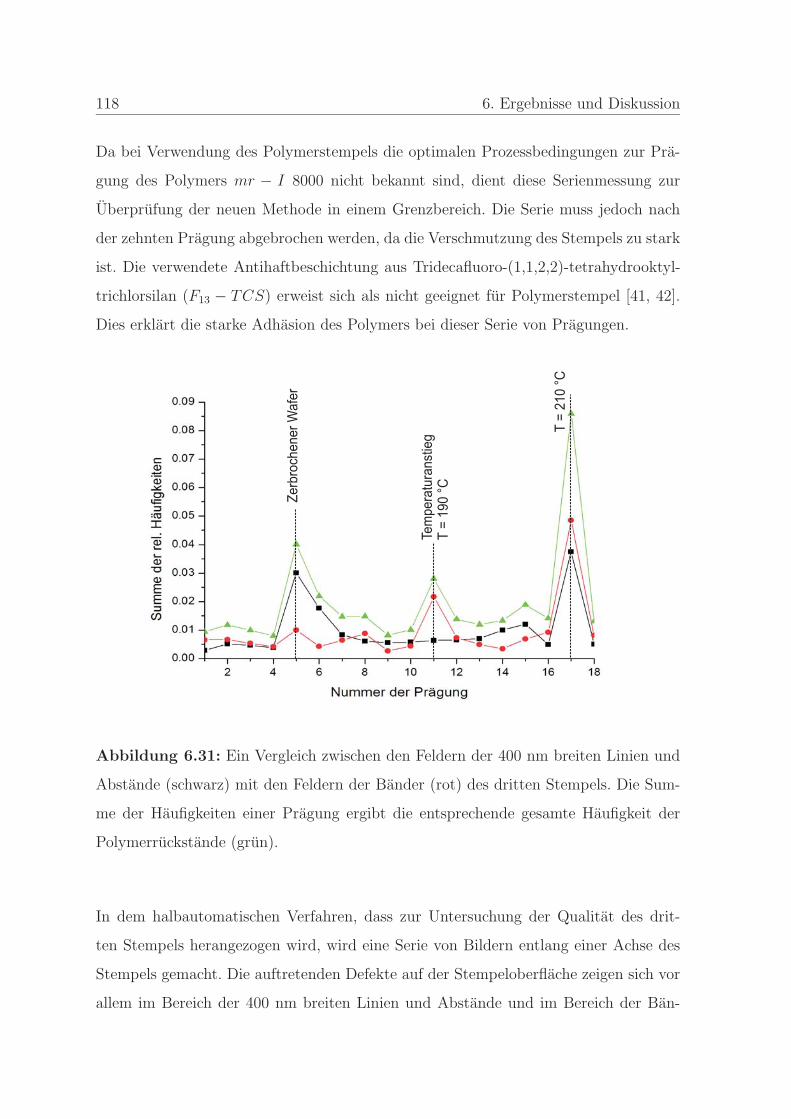
118 6. Ergebnisse und Diskussion
Da bei Verwendung des Polymerstempels die optimalen Prozessbedingungen zur Pra-
gung des Polymers mr − I 8000 nicht bekannt sind, dient diese Serienmessung zur
Uberprufung der neuen Methode in einem Grenzbereich. Die Serie muss jedoch nach
der zehnten Pragung abgebrochen werden, da die Verschmutzung des Stempels zu stark
ist. Die verwendete Antihaftbeschichtung aus Tridecafluoro-(1,1,2,2)-tetrahydrooktyl-
trichlorsilan (F13 − TCS) erweist sich als nicht geeignet fur Polymerstempel [41, 42].
Dies erklart die starke Adhasion des Polymers bei dieser Serie von Pragungen.
Tem
pe
ratu
ran
stie
gT
=1
90
°C
T=
21
0°C
Ze
rbro
che
ne
rW
afe
r
Abbildung 6.31: Ein Vergleich zwischen den Feldern der 400 nm breiten Linien und
Abstande (schwarz) mit den Feldern der Bander (rot) des dritten Stempels. Die Sum-
me der Haufigkeiten einer Pragung ergibt die entsprechende gesamte Haufigkeit der
Polymerruckstande (grun).
In dem halbautomatischen Verfahren, dass zur Untersuchung der Qualitat des drit-
ten Stempels herangezogen wird, wird eine Serie von Bildern entlang einer Achse des
Stempels gemacht. Die auftretenden Defekte auf der Stempeloberflache zeigen sich vor
allem im Bereich der 400 nm breiten Linien und Abstande und im Bereich der Ban-

6.1 Oberflachengebundene Nanopartikel 119
der. Der Verlauf der Polymerruckstande in diesen Bereichen ist in dem Diagramm 6.31
wiedergegeben. Die rote Linie entspricht dem Verlauf im Bereich der Bander, wahrend
die schwarze Linie die Summe an Polymerruckstanden im Bereich der 400 nm brei-
ten Linien und Abstande darstellt. Der grune Verlauf ist die Summe der Defekte in
beiden Bereichen. Ein direkter Zusammenhang zwischen der Struktur und den auftre-
tenden Defekten wird nicht gefunden. Dennoch zeigt auch dieses Diagramm eine gute
Ubereinstimmung mit den experimentellen Daten. Die Auswirkungen von Tempera-
turerhohungen und wahrend der Pragung auftretenden Schwierigkeiten spiegeln sich in
dem Diagramm wider. Wie schon bei der Untersuchung des ersten Stempels liegen auch
bei dieser Messung die relativen Haufigkeiten fur eine fast saubere Stempeloberflache
in einem Bereich von 0, 008 − 0, 086 (gruner Verlauf). Ein direkter Vergleich zwischen
dem ersten Stempel und dem dritten Stempel ist nicht moglich, da unterschiedlich
große Ausschnitte der Oberflache untersucht werden.
Zusammenfassend ist festzuhalten, dass die Fluoreszenzmikroskopie eine geeignete Al-
ternative zur Detektion von oberflachengebundenen Nanopartikeln ist. Bezuglich der
Qualitatssicherung in der Nanoimprintlithographie ist eine Charakterisierung der auf-
tretenden Defekte sowohl bei der Pragung als auch bei dem Stempel moglich. Dar-
uberhinaus lasst sich die Einheitlichkeit einer Pragung uber die Grauwertverteilun-
gen bestimmen. Die Ausstattung des Mikroskoptisches mit zwei Schrittmotoren zur
Bewegung in x- und y-Richtung ermoglicht die halbautomatische Untersuchung der
Pragestempel. Der Vorteil der Fluoreszenzmikroskopie im Vergleich zu den ublicher-
weise verwendeten Methoden liegt darin, dass alle Messungen ohne direkten Kontakt
zu der Probenoberflache erfolgen. Dies ermoglicht das Zuruckfuhren einer Probe in
den Produktionsprozess, z. B. im Hinblick auf eine industrielle Anwendung. Aufgrund
der kontaktfreien Messung ist auch eine Probenvorbereitung, z. B. eine zusatzliche Be-
schichtung der Oberflache, nicht notwendig. Dadurch ist eine schnelle Untersuchung
und eine einfache Handhabung gewahrleistet. Ein weiterer wesentlicher Aspekt, der
im Zusammenhang mit der Nanoimprintlithographie haufig diskutiert wird, sind die
anfallenden Kosten. Die vorgestellte Methode ist im Vergleich zu anderen Methoden

120 6. Ergebnisse und Diskussion
sehr kosteneffizient. Dies gilt auch fur die Methode zur Qualitatssicherung mittels der
Fluoreszenzmikroskopie in Verbindung mit der digitalen Bildverarbeitung.
6.2 Frei bewegliche Nanopartikel
Die Bestimmung der Partikelgroße und der Partikelgroßenverteilung hat in den letz-
ten Jahren einen bedeutenden industriellen Stellenwert eingenommen. Die Partikel-
große und deren Verteilung gehoren mit zu den wesentlichen Eigenschaften disperser
Systeme aus Latex, Pigmenten, keramischen Materialien oder Wirkstoffverbindungen.
Aufgrund der breiten Produktpalette reicht der Bereich der Partikelgroßen von klei-
ner 10 nm bis großer 100 µm. Entsprechend der Produktpalette disperser Systeme
gibt es auch eine Vielzahl von Methoden und Messinstrumenten basierend auf unter-
schiedlichen physikalischen Prinzipien, die zur Bestimmung der Partikelgroßen bzw.
der Partikelgroßenverteilung genutzt werden. Diese Methoden konnen in zwei Klassen
unterteilt werden:
- Zu der ersten Klasse zahlen diejenigen Methoden, die leicht zu bedienen sind und
deren Ausstattung weniger aufwandig sowie kostengunstig ist.
- Die zweite Klasse sollte ebenfalls einfach zu bedienen sein, erfordert aber eine
kompliziertere und kostenintensivere Ausstattung.
Zu den bekanntesten Methoden zahlen die Turbidimetrie, die dynamische und statische
Lichtstreuung, die Elektronenmikroskopie sowie die Disk- und die Ultrazentrifugation.
Einen Uberblick uber die Grundlagen der einzelnen Methoden gibt E. A. Collins [99].
Die Vor- und Nachteile der einzelnen Methoden sowie deren Entwicklung und Opti-
mierung in der Vergangenheit werden von T. Provder aufgezeigt [100]. Eine weltweite
Untersuchung von 17 Testlaboratorien innerhalb der Bayergruppe zeigt, zu welch un-
terschiedlichen Ergebnissen die Messungen der gleichen Partikelsuspensionen mit den
unterschiedlichen Methoden fuhren konnen [101]. Die Abweichungen der Zahlenmittel
liegen bei ±7 % und die Halbwertsbreiten der unimodalen Verteilungen sind bis zu
drei mal so groß, wahrend bimodale Verteilungen gar nicht aufgelost werden. Nur die

6.2 Frei bewegliche Nanopartikel 121
Elektronenmikroskopie und die Analytische Ultrazentrifuge weisen zuverlassige Bestim-
mungen unimodaler und bimodaler Verteilungen von Polystyrolpartikeln auf. Anhand
dieser Untersuchung wird die Empfindlichkeit der verschiedenen Methoden deutlich.
Diese Ergebnisse verdeutlichen, dass ein Bedarf an neuen, verbesserten Methoden zur
Bestimmung der Partikelgroßen besteht. Solche Methoden sollten folgende Anforderun-
gen erfullen:
- Benutzerfreundlichkeit
- Zuverlassigkeit bzw. Reproduzierbarkeit
- Schnelligkeit
- geringe Kosten
Die hier vorgestellte Methode basiert auf der optischen Verfolgung von Partikelspuren
zur Bestimmung des Partikelradius in Wasser suspendierter Nanopartikel. Dies ist eine
relativ alte Methode (vgl. Kapitel 4.2), die sich jedoch mit den modernen Techniken
der digitalen Bildverarbeitung und -analyse als eine neue vielversprechende Moglichkeit
zur schnellen Bestimmung von Partikelgroßen an geringen Mengen einer Suspension er-
weist. Ihr besonderer Vorteil im Vergleich zur dynamischen Lichtstreuung liegt in der
Fahigkeit, eine große Anzahl einzelner Partikel beobachten zu konnen, woraus sich ei-
ne hohe Zuverlassigkeit der resultierenden Großenverteilungen ergibt. Die Optimierung
der Methode erfolgt aufbauend auf den Arbeiten von M. Wohlgemuth [81, 82].
Im Folgenden werden zunachst die zur Auswertung herangezogenen Programme zur
Bildverarbeitung und Bildanalyse erlautert. Anschließend werden die Großenverteilun-
gen von verschiedenen Polystyrolstandards, die mit zwei unterschiedlichen mikroskopi-
schen Methoden gemessen werden, vorgestellt und diskutiert.

122 6. Ergebnisse und Diskussion
6.2.1 Bildverarbeitung und -analyse zur Großenbestimmung
von frei beweglichen Nanopartikeln
Die Bestimmung der Großenverteilung von frei beweglichen Nanopartikeln erfolgt uber
drei Makros, die in dem Anhang A angefuhrt sind. Das erste Makro (Anhang A.2)
enthalt dabei die wesentlichen Schritte der Bildverarbeitung und der Bildanalyse und
dient zur Bestimmung der Lage der Partikel in jedem Bild. Aus diesen Daten erfolgt
dann unter Anwendung des zweiten Makros (Anhang A.3) die Spurenanalyse, das so
genannte Particle Tracking. Die berechneten Großenverteilungen werden anschließend
mit Hilfe des dritten Makros (Anhang A.4) in Form von Balkenhistogrammen wieder-
gegeben, deren Daten in einer benachbarten Tabelle aufgefuhrt werden.
Das Makro A.2 besteht aus zwei Abschnitten. Der erste Abschnitt dient zur Bild-
verarbeitung, wahrend in dem zweiten Abschnitt die Bildanalyse erfolgt. Zur Bild-
verarbeitung werden zwei Methoden angewendet: Ein Subtraktionsverfahren, das als
SUB128-Verfahren bezeichnet wird, und ein Glattungsfilter, bei dem es sich um einen
5x5 Gaußfilter handelt.
Das SUB128-Verfahren dient zur Entfernung von Storungen in einem Bild. Hierbei
kann es sich um Defekte in oder auf der Glasoberflache handeln, wie z. B. Kratzer
oder anhaftende Partikel. Zur Entfernung dieser Storungen wird das erste Bild der
aufgenommenen Frequenz B(x, y)0 als Referenzbild von den darauf folgenden Bildern
B(x, y)z subtrahiert:
z′=n∑z′=0
B(x, y)z′ =z=n∑z=0
(B(x, y)z − B(x, y)0) (6.1)
mit n = Anzahl der Bilder einer Sequenz.
Zu dem neuen Bild B(x, y)z′ wird der mittlere Grauwert von 128 addiert:
z′=n∑z′=0
B(x, y)z′ =z′=n∑z′=0
B(x, y)z′ + 128, (6.2)
dadurch wird der Nullpegel des Bildes auf den mittleren Grauwert verlegt.

6.2 Frei bewegliche Nanopartikel 123
Im Anschluss an das SUB128-Verfahren wird jedes Bild mit einem 5x5 Gaußfilter ge-
glattet. Dieser Filter gehort zu den Tiefpassfiltern (vgl. Kapitel 2.3.3), die die hohen
Frequenzanteile eines Bildes durch eine Matrixoperation abschwachen oder uberlagern.
Dadurch findet eine Glattung der Grauwerte statt, die in einer gewissen Unscharfe
des Bildes resultieren. Auf die anschließende Bildanalyse hat diese Unscharfe keine
Auswirkung. Die uberarbeiteten Bilder werden als neue Sequenz abgespeichert [32]. In
der Abbildung 6.32 ist das Ergebnis der zwei bildverarbeitenden Schritte grafisch dar-
gestellt. Die Verschiebung des Nullpegels des Bildes auf den mittleren Grauwert von
128 ist an dem hellgrauen Hintergrund in dem neuen Bild deutlich zu erkennen. Die
dunklen Punkte in dem Bild entsprechen den Defekten, wahrend die hellen Objekte
die sich bewegenden Nanopartikel darstellen. Daruberhinaus zeigt sich der Effekt des
Gaußfilters in einer leichten Vergroßerung sowohl der Defekte als auch der Nanoparti-
kel. Der eigentliche Flachenschwerpunkt bleibt dabei jedoch unverandert.
_=
1. Bild der Sequenzn-tes Bild der Sequenz n-tes Bild der neuen Sequenz
Abbildung 6.32: Die Bildverarbeitung: Die Wirkung der SUB128-Prozedur und des
Gaußfilters.
Die so berechneten neuen Bildsequenzen werden anschließend automatisch abgespei-
chert. Uber die Anwendung der beiden bildverarbeitenden Prozeduren kann der Anwen-
der in Abhangigkeit von der verwendeten mikroskopischen Methode und der Qualitat
der Bilder entscheiden (siehe Kapitel 6.2.2 und 6.2.3).
Im zweiten Abschnitt des Makros erfolgt die Bildanalyse. Hierbei stehen das Erkennen

124 6. Ergebnisse und Diskussion
der Partikel und die Bestimmung der genauen Lage der Partikel im Bild im Vorder-
grund. Zur Merkmalserkennung wird ein Schwellenwert herangezogen. Die Bestimmung
des Schwellenwertes kann gegebenenfalls uber ein Grauwerthistogramm erfolgen. Als
Schwellenwert wird derjenige Grauwert gewahlt, der uber dem Minimum zwischen den
beiden Maxima liegt. Der Schwellenwert kann von dem Anwender in dem Makro vor-
gegeben werden. Zum Auffinden der Partikel wird das Bild ausgehend von der oberen
linken Ecke zeilenweise abgetastet, bis ein Grauwert gefunden wird, der großer bzw.
gleich dem Schwellenwert ist. Ist der Grauwert eines Bildpunktes hoher als die vorge-
gebene Grenze, dann wird der Rand des Objektes zur Uberprufung, ob es sich um ein
gultiges Objekt handelt, im Uhrzeigersinn abgetastet. Anschließend werden von einem
gultigen Objekt die x- und y-Koordinaten des Flachenschwerpunktes ermittelt und ta-
bellarisch in einer Datei gespeichert werden [82].
Der Algorithmus zum Particle Tracking ist im Anhang A.3 wieder gegeben. Er kann in
drei Hauptschritte unterteilt werden:
1. Zunachst werden die x- und y-Koordinaten der bei der Bildanalyse in dem ersten
Bild gefundenen Objekte in eine Spurenliste eingetragen.
2. Dann folgt das Einlesen der Koordinaten aller Objekte des darauf folgenden Bil-
des und der Vergleich dieser mit den im vorherigen Bild gefundenen. Die Zu-
ordnung der Partikelspuren erfolgt dabei uber die Berechnung der Lange der
Verschiebungsvektoren. Fur jedes Objekt des ersten Bildes werden die Langen
der Verschiebungsvektoren von allen in dem darauf folgenden Bild gefundenen
Objekten bestimmt. Der kleinste berechnete Vektor, d. h. der kleinste Abstand
zwischen zwei Objekten, wird als Zuordnungskriterium genommen. Die Werte
dieser beiden Objekte werden in der Spurenliste gespeichert.
3. Ist der Vektor zwischen zwei Objekten Null, dann erfolgt mittels des so genann-
ten Stillstandskriteriums die Uberprufung, in welchem Zeitraum keine Bewegung
des Objektes berechnet werden kann. Der Zeitraum, d. h. die Anzahl der aufein-
ander folgenden Bilder, kann von dem Anwender vorgegeben werden. Die Stan-

6.2 Frei bewegliche Nanopartikel 125
dardeinstellung betragt funf Bilder. Beim Uberschreiten dieses Zeitraums wird
das Objekt aus der Spurenliste geloscht und in eine so genannte Ausschlussliste
ubernommen.
Zur Vermeidung einer fehlerhaften Zuordnung der Partikelspuren werden anschließend
die folgenden Kriterien angewandt:
a) Uberprufung, ob zwei Objekte aus dem vorherigen Bild in dem darauf folgenden
die gleiche Position haben. Dies kann durch eine Kollision zweier Objekte her-
vorgerufen werden, die keine eindeutige Zuweisung der Partikelspuren erlaubt. In
diesem Fall werden die Spuren dieser Objekte in der Spurenliste beendet und die
Objekte in die Ausschlussliste aufgenommen.
b) Uberprufung, ob neue Objekte in dem darauf folgenden Bild gefunden wurden.
Neue Objekte konnen in einem Bild definiert werden, wenn ein weiteres Partikel
neu in den Bildausschnitt gelangt oder wenn sich zwei Partikel nach einer Kolli-
sion wieder einzeln bewegen. Zur Uberprufung, ob es sich um ein neues Objekt
oder um bereits bekannte Objekte handelt, wird die Ausschlussliste herangezo-
gen. Neue Objekte werden in die Spurenliste aufgenommen.
c) Uberprufung, ob ein zuvor definiertes Objekt in dem darauf folgenden Bild fehlt.
Die Ursache kann darin liegen, dass ein Partikel den Bildausschnitt verlassen hat.
In diesem Fall wird die Spur des Objektes in der Spurenliste beendet und in die
Ausschlussliste aufgenommen.
Anschließend werden fur die einzelnen Spuren die Verschiebungsquadrate in x- und
y-Richtung berechnet und in ein Tabellenblatt eingetragen.
In dem dritten Makro erfolgt schließlich die Berechnung der Radien der einzelnen Parti-
kel aus den zuvor bestimmten Verschiebungsquadraten (siehe Kapitel 4). Dazu werden
die Gleichungen 4.1, 4.19 und 4.20 herangezogen. Die hierfur notwendigen Variablen
konnen vor dem Start des Makros von dem Anwender in einem vorgefertigten Excel-
Tabellenblatt eingetragen werden, das auszugsweise in der Abbildung 6.33 dargestellt
ist. Aus den berechneten Radien wird dann die Großenverteilung der Partikel bestimmt,

126 6. Ergebnisse und Diskussion
Abbildung 6.33: Interaktive Abfrage der notwendigen Variablen.
die in Form eines Balkenhistogramms ausgegeben wird. Die zur Berechnung des Histo-
gramms notwendigen Parameter konnen ebenfalls in dem Excel-Tabellenblatt von dem
Anwender vorgegeben werden (siehe Abbildung 6.33).
6.2.2 Großenbestimmung mittels der Dunkelfeldmikroskopie
Zur Uberprufung der Methode werden vier Polystyrolstandards verschiedener Große
herangezogen. In der Abbildung 6.34 sind die Großenverteilungen dieser Standards in
Form von Balkenhistogrammen dargestellt. Jeder Balken kennzeichnet den prozentua-
len Anteil der gefundenen Nanopartikel in einem bestimmten Radienbereich. Durch-
schnittlich hat jedes Nanopartikel, das in dem Histogramm aufgenommen wird, mehr
als 50 aufeinander folgende Schritte absolviert. Dies entspricht einem Ensemble von
mehr als 100 unabhangigen Werten (∆x2 und ∆y2) fur eine eindimensionale quadrati-
sche Verschiebung. Die Standardabweichung der Partikelradien liegt typischerweise in
einem Bereich von 8, 5 % bis 13, 8 %. Die entsprechenden Partikelradien konnen mit
einer Fehlerwahrscheinlichkeit von weniger als 1 % Intervallen von 4 nm zugeordnet
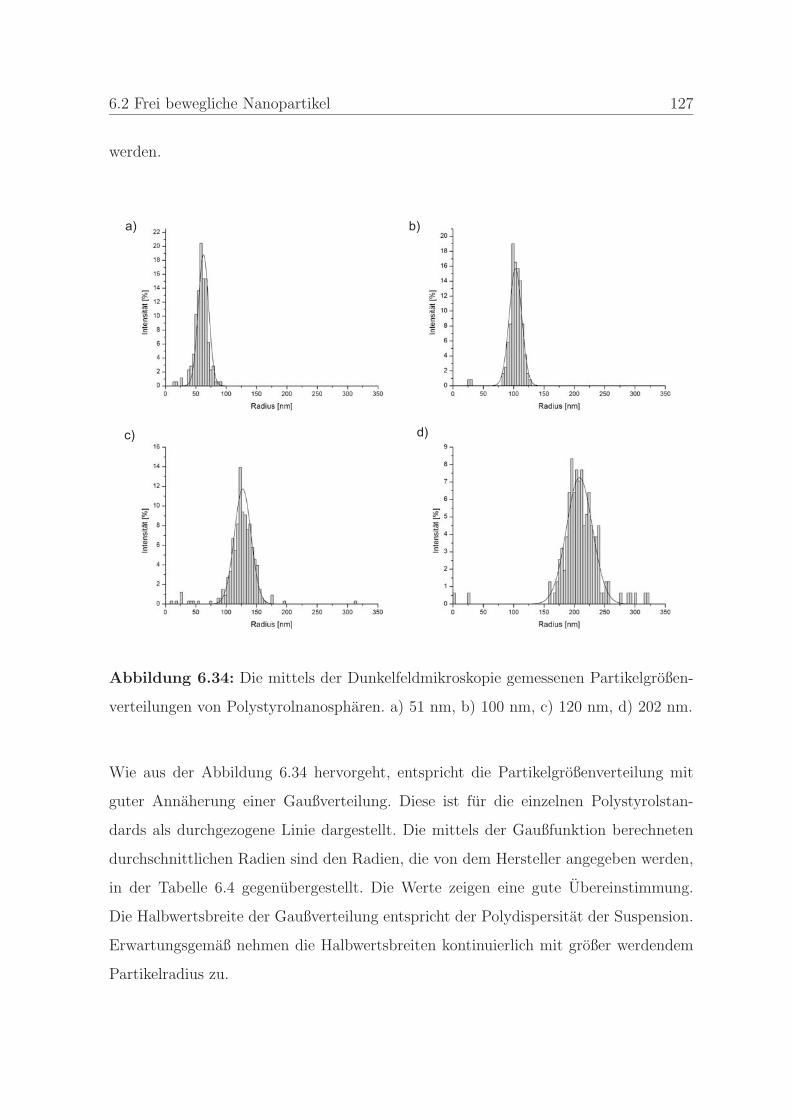
6.2 Frei bewegliche Nanopartikel 127
werden.
a) b)
c) d)
Abbildung 6.34: Die mittels der Dunkelfeldmikroskopie gemessenen Partikelgroßen-
verteilungen von Polystyrolnanospharen. a) 51 nm, b) 100 nm, c) 120 nm, d) 202 nm.
Wie aus der Abbildung 6.34 hervorgeht, entspricht die Partikelgroßenverteilung mit
guter Annaherung einer Gaußverteilung. Diese ist fur die einzelnen Polystyrolstan-
dards als durchgezogene Linie dargestellt. Die mittels der Gaußfunktion berechneten
durchschnittlichen Radien sind den Radien, die von dem Hersteller angegeben werden,
in der Tabelle 6.4 gegenubergestellt. Die Werte zeigen eine gute Ubereinstimmung.
Die Halbwertsbreite der Gaußverteilung entspricht der Polydispersitat der Suspension.
Erwartungsgemaß nehmen die Halbwertsbreiten kontinuierlich mit großer werdendem
Partikelradius zu.
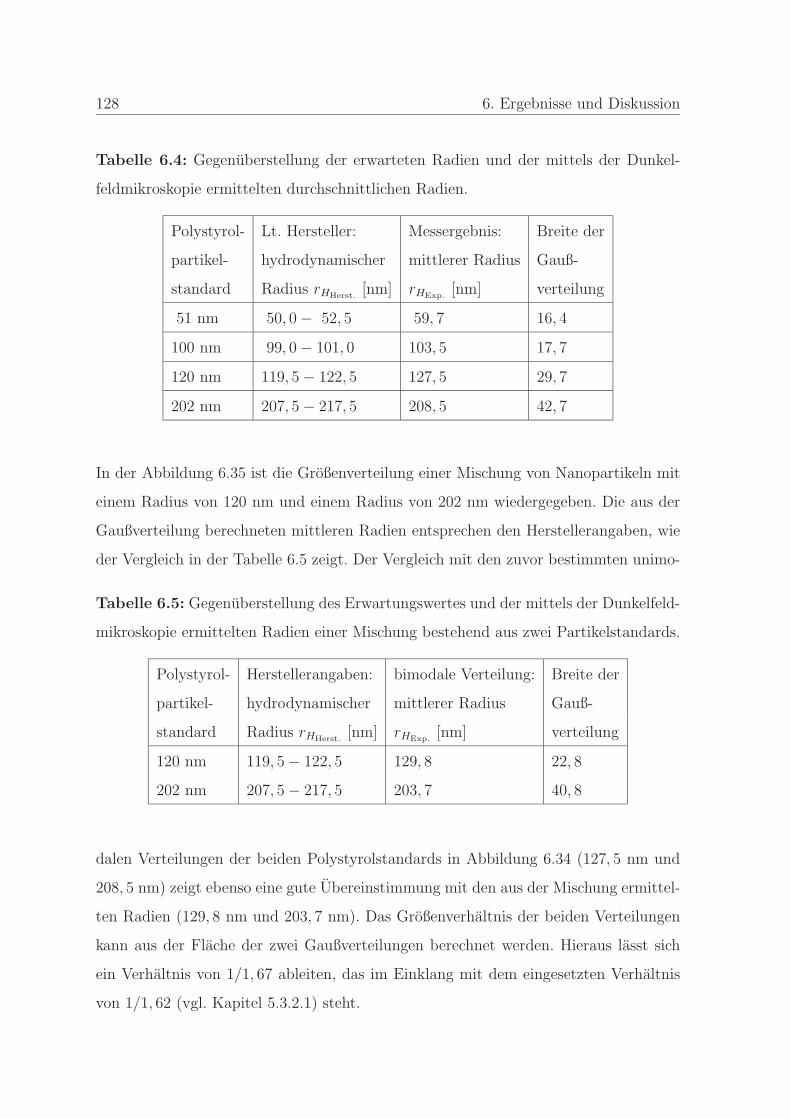
128 6. Ergebnisse und Diskussion
Tabelle 6.4: Gegenuberstellung der erwarteten Radien und der mittels der Dunkel-
feldmikroskopie ermittelten durchschnittlichen Radien.
Polystyrol- Lt. Hersteller: Messergebnis: Breite der
partikel- hydrodynamischer mittlerer Radius Gauß-
standard Radius rHHerst.[nm] rHExp.
[nm] verteilung
51 nm 50, 0 − 52, 5 59, 7 16, 4
100 nm 99, 0 − 101, 0 103, 5 17, 7
120 nm 119, 5 − 122, 5 127, 5 29, 7
202 nm 207, 5 − 217, 5 208, 5 42, 7
In der Abbildung 6.35 ist die Großenverteilung einer Mischung von Nanopartikeln mit
einem Radius von 120 nm und einem Radius von 202 nm wiedergegeben. Die aus der
Gaußverteilung berechneten mittleren Radien entsprechen den Herstellerangaben, wie
der Vergleich in der Tabelle 6.5 zeigt. Der Vergleich mit den zuvor bestimmten unimo-
Tabelle 6.5: Gegenuberstellung des Erwartungswertes und der mittels der Dunkelfeld-
mikroskopie ermittelten Radien einer Mischung bestehend aus zwei Partikelstandards.
Polystyrol- Herstellerangaben: bimodale Verteilung: Breite der
partikel- hydrodynamischer mittlerer Radius Gauß-
standard Radius rHHerst.[nm] rHExp.
[nm] verteilung
120 nm 119, 5 − 122, 5 129, 8 22, 8
202 nm 207, 5 − 217, 5 203, 7 40, 8
dalen Verteilungen der beiden Polystyrolstandards in Abbildung 6.34 (127, 5 nm und
208, 5 nm) zeigt ebenso eine gute Ubereinstimmung mit den aus der Mischung ermittel-
ten Radien (129, 8 nm und 203, 7 nm). Das Großenverhaltnis der beiden Verteilungen
kann aus der Flache der zwei Gaußverteilungen berechnet werden. Hieraus lasst sich
ein Verhaltnis von 1/1, 67 ableiten, das im Einklang mit dem eingesetzten Verhaltnis
von 1/1, 62 (vgl. Kapitel 5.3.2.1) steht.
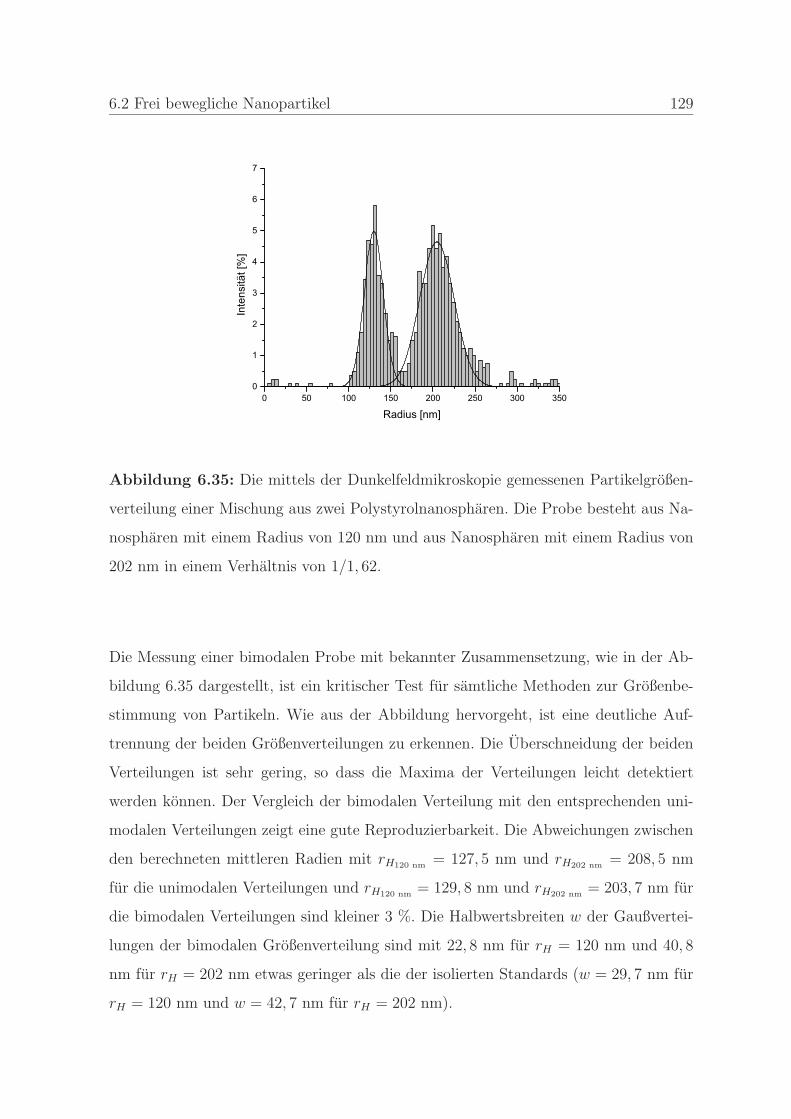
6.2 Frei bewegliche Nanopartikel 129
Abbildung 6.35: Die mittels der Dunkelfeldmikroskopie gemessenen Partikelgroßen-
verteilung einer Mischung aus zwei Polystyrolnanospharen. Die Probe besteht aus Na-
nospharen mit einem Radius von 120 nm und aus Nanospharen mit einem Radius von
202 nm in einem Verhaltnis von 1/1, 62.
Die Messung einer bimodalen Probe mit bekannter Zusammensetzung, wie in der Ab-
bildung 6.35 dargestellt, ist ein kritischer Test fur samtliche Methoden zur Großenbe-
stimmung von Partikeln. Wie aus der Abbildung hervorgeht, ist eine deutliche Auf-
trennung der beiden Großenverteilungen zu erkennen. Die Uberschneidung der beiden
Verteilungen ist sehr gering, so dass die Maxima der Verteilungen leicht detektiert
werden konnen. Der Vergleich der bimodalen Verteilung mit den entsprechenden uni-
modalen Verteilungen zeigt eine gute Reproduzierbarkeit. Die Abweichungen zwischen
den berechneten mittleren Radien mit rH120 nm = 127, 5 nm und rH202 nm = 208, 5 nm
fur die unimodalen Verteilungen und rH120 nm = 129, 8 nm und rH202 nm = 203, 7 nm fur
die bimodalen Verteilungen sind kleiner 3 %. Die Halbwertsbreiten w der Gaußvertei-
lungen der bimodalen Großenverteilung sind mit 22, 8 nm fur rH = 120 nm und 40, 8
nm fur rH = 202 nm etwas geringer als die der isolierten Standards (w = 29, 7 nm fur
rH = 120 nm und w = 42, 7 nm fur rH = 202 nm).

130 6. Ergebnisse und Diskussion
Die hier vorgestellte Methode zur Großenbestimmung von Nanopartikeln hat die Fa-
higkeit, in einer homogenen Suspension mehrere Komponenten verschiedener Großen
unabhangig voneinander zu beobachten und entsprechend auszuwerten. Die Anwesen-
heit kleinerer und großerer beweglicher Nanopartikel hat dabei scheinbar keinen bedeu-
tenden Einfluss auf die Detektion der einzelnen Partikel. Der Vergleich der berechneten
Flachen der Verteilungskurven und des eingesetzten Verhaltnisses beweist, dass die Me-
thode quantitative Daten der Großenverteilung ergibt.
Alle Messungen werden ohne das zur Bildverarbeitung genutzte SUB128-Verfahren
ausgewertet (s. Kapitel 6.2.1), da der Kontrast der Partikel hoch genug fur die Objekt-
erkennung ist. Es zeigt sich jedoch, dass das Auflosungsvermogen bei der Untersuchung
der Polystyrolstandards mittels der Dunkelfeldmikroskopie bei ungefahr 50 nm liegt.
Unterhalb dieser Grenze ist der Kontrast der Partikel sehr gering. Eine Auswertung
unter diesen Bedingungen erfordert eine nachtragliche Bildbearbeitung, d. h. die An-
wendung des SUB128-Verfahrens.
6.2.3 Großenbestimmung mittels der Fluoreszenzmikroskopie
Mittels der Fluoreszenzmikroskopie werden zwei Nanopartikel verschiedener Große un-
tersucht. Die Balkenhistogramme, die in Abbildung 6.36 dargestellt sind, werden unter
denselben Bedingungen berechnet wie bei der Dunkelfeldmikroskopie. Die daraus ab-
geleiteten Daten sind in der Tabelle 6.6 den bekannten Radien gegenubergestellt. Die
Abbildung 6.36 a) reprasentiert die Großenverteilung des fluoreszierenden Polystyrol-
standards, der laut Herstellerangaben einen Radius von 100 nm hat. Der berechnete
durchschnittliche Radius entspricht mit 100, 8 nm dem Erwartungswert und zeigt eine
gute Ubereinstimmung zu dem mittels der Dunkelfeldmikroskopie berechneten Wert
von 103, 5 nm. Die Standardabweichung betragt somit weniger als 1 %.
Die Großenverteilung der Nanopartikel aus PMMA, die mit Cumarin 6 markiert sind,
weist ein ahnliches Ergebnis auf. Der aus der Gaußverteilung ermittelte durchschnitt-
liche Radius betragt 169, 9 nm. Dieser steht im Einklang mit dem aus SEM-Bildern
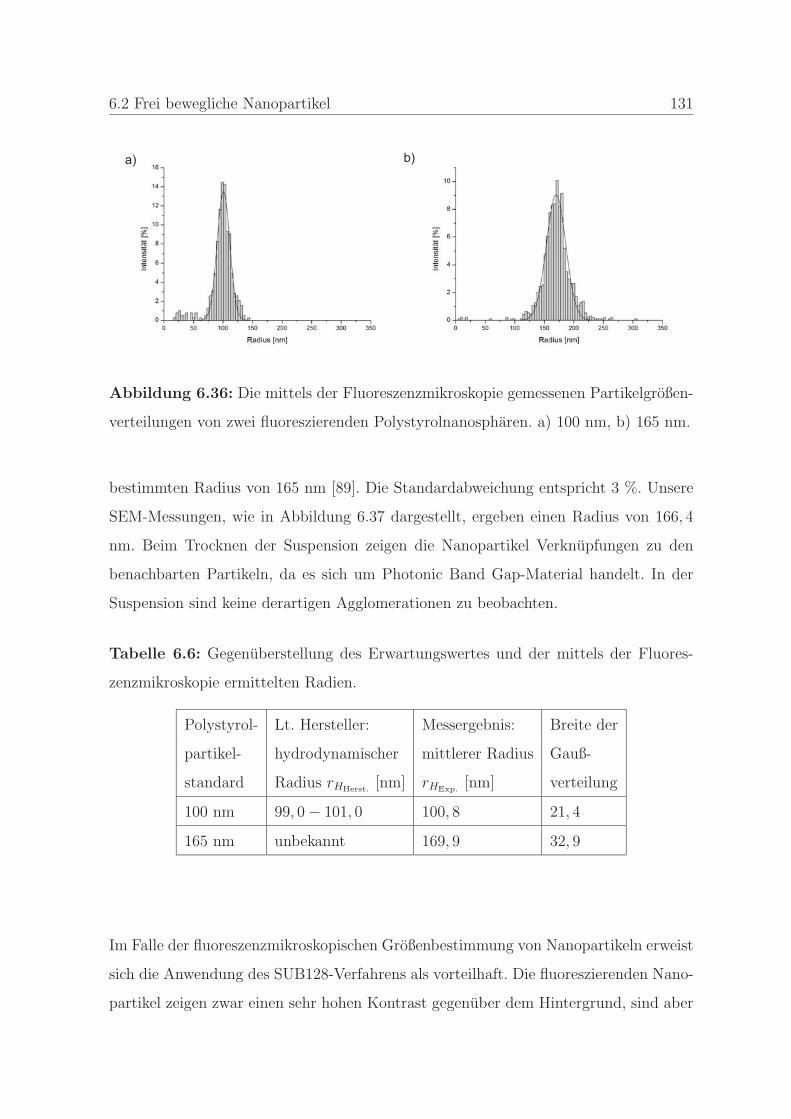
6.2 Frei bewegliche Nanopartikel 131
a) b)
Abbildung 6.36: Die mittels der Fluoreszenzmikroskopie gemessenen Partikelgroßen-
verteilungen von zwei fluoreszierenden Polystyrolnanospharen. a) 100 nm, b) 165 nm.
bestimmten Radius von 165 nm [89]. Die Standardabweichung entspricht 3 %. Unsere
SEM-Messungen, wie in Abbildung 6.37 dargestellt, ergeben einen Radius von 166, 4
nm. Beim Trocknen der Suspension zeigen die Nanopartikel Verknupfungen zu den
benachbarten Partikeln, da es sich um Photonic Band Gap-Material handelt. In der
Suspension sind keine derartigen Agglomerationen zu beobachten.
Tabelle 6.6: Gegenuberstellung des Erwartungswertes und der mittels der Fluores-
zenzmikroskopie ermittelten Radien.
Polystyrol- Lt. Hersteller: Messergebnis: Breite der
partikel- hydrodynamischer mittlerer Radius Gauß-
standard Radius rHHerst.[nm] rHExp.
[nm] verteilung
100 nm 99, 0 − 101, 0 100, 8 21, 4
165 nm unbekannt 169, 9 32, 9
Im Falle der fluoreszenzmikroskopischen Großenbestimmung von Nanopartikeln erweist
sich die Anwendung des SUB128-Verfahrens als vorteilhaft. Die fluoreszierenden Nano-
partikel zeigen zwar einen sehr hohen Kontrast gegenuber dem Hintergrund, sind aber

132 6. Ergebnisse und Diskussion
Abbildung 6.37: Der mittels SEM gemessene Radius von Nanospharen, die mit Cu-
marin 6 markiert sind.
aufgrund der verwendeten Optik sehr klein. Durch die Bildverarbeitung werden die Na-
nopartikel starker vom Hintergrund abgehoben, was die Objekterkennung vereinfacht.
6.2.4 Vergleich der mikroskopischen Methoden
Die Großenverteilungen der einzelnen Polystyrolstandards geben die Genauigkeit der
Methode wieder, die, wie am Beispiel des Polystyrolstandards mit einem Radius von
100 nm gezeigt, unabhangig von der verwendeten mikroskopischen Methode ist. Der
berechnete mittlere Radius stimmt generell mit den Angaben des Herstellers zu dem
hydrodynamischen Radius uberein. Die Abweichungen variieren zwischen 1 % (fur den
Standard mit einem Radius von 100 nm) und 14 % (fur den Standard mit einem Radius
von 51 nm).
Mit einzelnen Partikeln eines bekannten Radius r, die uber annahernd 50 Zeitintervalle
∆t beobachtet werden, ist die statistische Voraussetzung fur eine eindeutige Zuordnung
zu einem bestimmten Großenintervall n gegeben. Dabei ist das Großenintervall definiert
durch (rn−2 nm) < rn < (rn+2 nm). Erwartungsgemaß entsprechen alle Großenvertei-
lungen (vgl. Abbildung 6.34 und 6.36) in sehr guter Annaherung einer Gaußverteilung.

6.2 Frei bewegliche Nanopartikel 133
Die Halbwertsbreite hangt dabei offensichtlich von der durchschnittlichen Partikelgroße
ab, wie der Tabelle 6.4 zu entnehmen ist. Dieses Verhalten stimmt mit der erwarteten
Abweichung uberein, die durch den Herstellungsprozess dieser Partikel gegeben ist.
Die Qualitat einer Messung und des daraus resultierenden Histogramms hangt im We-
sentlichen von folgenden vier Parametern ab:
- der Schichtdicke d zwischen Objekttrager und Deckglas
- dem Zeitintervall ∆t zwischen zwei Bildern
- der Raumtemperatur T
- und dem gewahlten Grauschwellenwert
Die Bestimmung der Schichtdicke zwischen den beiden Glasoberflachen ist essenziell
fur die Berechnung der scheinbaren Viskositat nach Gleichung 4.20. In Bezug auf die
Beobachtung der Nanopartikel hat sie einen großen Einfluss auf die Qualitat der aufge-
nommenen Bilder. Je großer die Schichtdicke ist, desto haufiger findet eine Bewegung
der Partikel aus der Fokusebene in z-Richtung statt. Das erschwert die Beobachtung
der Bewegung der einzelnen Partikel uber einen langeren Zeitraum. Besonders haufig
tritt dieses Verhalten bei Partikeln mit einem Radius kleiner 100 nm auf. Die Messun-
gen zeigen, dass eine Schichtdicke in einem Bereich von 2 − 5 nm erforderlich ist, um
eine ausreichende Fokussierung zu gewahrleisten.
Ein weiterer wichtiger Parameter, der die Qualitat der Messungen beeinflusst, ist das
Zeitintervall ∆t zwischen zwei Bildern (vgl. Gleichung 4.1). Die Messungen haben er-
geben, dass ein Zeitintervall ∆t von 0, 08 Sekunden, d. h. 12, 5 Bilder pro Sekunde,
fur eine genaue Berechnung der Verschiebung in x- und y-Richtung erforderlich ist.
Messungen mit einem kleineren Zeitintervall von 0, 04 Sekunden (25 Bilder/Sekunde)
zeigen die Tendenz zu einer Verschiebung hin zu großeren Radien. Die Zeit zwischen
zwei Schritten eines Partikels ist nicht ausreichend, um die exakte Verschiebung des
Partikels zu registrieren. Diese Eigenschaft macht sich besonders bei Nanopartikeln be-
merkbar, die großer sind als 100 nm. Bei diesen Partikeln kann die Verschiebung in einer

134 6. Ergebnisse und Diskussion
Zeit von 0, 04 Sekunden in einem Bereich von nur einem Pixel liegen. Bei einer solch
kleinen Verschiebung werden die Partikel aufgrund des Stillstandskriteriums zwar nicht
aus der Auswertung genommen (vgl. Kapitel 6.2.1), dennoch konnen die Schwerpunkte
der Partikel nicht mehr exakt berechnet werden. Entsprechend Gleichung 4.1 werden
zu kleine Verschiebungswerte berechnet, wodurch sich zu große Radien ergeben. Mit
einem gewahlten Zeitintervall von 0, 08 Sekunden kann dieser Messfehler vermieden
werden.
Die in den Abbildungen 6.34, 6.35 und 6.36 dargestellten Ergebnisse zeigen, dass die
hier vorgestellte Methode unter Einbeziehung der Raumtemperatur zu den erwarteten
Ergebnissen fuhrt. Die Ergebnisse beweisen, dass ein Aufheizen der Probe wahrend
der Messung ausgeschlossen werden kann. Ein Aufheizen der Probe hatte eine Erho-
hung der Geschwindigkeit der Nanopartikel zur Folge, woraus eine Verschiebung der
Großenverteilung hin zu kleineren Radien resultieren wurde. Bei den Messungen mit
der Durchlicht-Dunkelfeldmikroskopie wird, wie in Kapitel 5.1.2 beschrieben, eine mit
Wasser gefullte Kuvette in den Strahlengang eingebracht, um das Aufheizen der Probe
durch den infraroten Lichtanteil zu vermeiden. Temperaturmessungen an verschiede-
nen Stellen am Mikroskop, wie z. B. an der Probe, auf dem Mikroskoptisch, in der
mit Wasser gefullten Kuvette sowie an verschiedenen Stellen im Messraum, zeigen eine
gute Ubereinstimmung. Bei den Messungen mit dem Fluoreszenzmikroskop ist es nicht
notwendig, einen Wasserfilter in dem Strahlengang vor der Probe anzubringen, da das
Mikroskop standardmaßig mit einem Warmeschutzfilter ausgestattet ist (vgl. 2.1.2).
Um ein Aufheizen der Probe ausschließen zu konnen, werden ebenfalls die Tempera-
turen an verschiedenen Stellen am Mikroskop gemessen. Die Messungen ergeben die
gleichen Ergebnisse wie bei Verwendung der Dunkelfeldmikroskopie. Die verschiedenen
Temperaturmessungen sowie die dargestellten Ergebnisse bestatigen die Verwendung
der Raumtemperatur zur Berechnung der Großenverteilungen mittels der Gleichung 4.1.
Ein fur die Auswertung besonders wichtiger Parameter ist der Grauschwellenwert, der
zur Objekterkennung vorgegeben werden muss (vgl. Kapitel 6.2.1). Der Grauwert ist

6.2 Frei bewegliche Nanopartikel 135
das einzige Kriterium, das fur die Unterscheidung zwischen Hintergrund und Objekt in
der Bildanalyse verantwortlich ist. Mittels des Grauwertes konnen folglich die Positio-
nen der Nanopartikel und deren Flachenschwerpunkte bestimmt werden. Der Grauwert
muss so gewahlt werden, dass er moglichst nah an dem Grauwert der Partikel liegt.
Dies fuhrt zu einer genaueren Bestimmung des Flachenschwerpunktes der Partikel und
somit zu einer genaueren Berechnung der Radien.
Zusammenfassend ist festzuhalten, dass diese Methode zur Bestimmung von Parti-
kelgroßen in einem Radienbereich von 50 nm bis 500 nm eine gute Alternative zu den
bisher verwendeten Methoden darstellt. Die hier vorgestellte Methode zeichnet sich
durch eine einfache Handhabung, sowohl bei der Probenvorbereitung als auch bei der
Messung, aus und ist sehr benutzerfreundlich. Die Ergebnisse, die eine hohe Uber-
einstimmung mit den Angaben des Herstellers aufweisen, unterstreichen die Zuver-
lassigkeit der Messungen. Der Vergleich zwischen der Dunkelfeldmikroskopie und der
Fluoreszenzmikroskopie zeigt, dass die Auswertung unabhangig von der verwendeten
mikroskopischen Methode ist, so dass auch die Anwendung anderer mikroskopischer
Methoden moglich ist.

136 6. Ergebnisse und Diskussion

Kapitel 7
Zusammenfassung
In dieser Arbeit werden zwei neuartige Analysemethoden aus dem Bereich der Nano-
technologie vorgestellt. Die Grundlage beider Methoden bildet die optische Mikroskopie
in Verbindung mit der digitalen Bildverarbeitung. Wahrend eine der Methode der Be-
obachtung und Analyse von oberflachengebundenen Nanopartikeln mit dem Ziel der
Qualitatssicherung in der Nanoimprintlithographie dient, handelt es sich bei der zwei-
ten um eine Methode zur Großenbestimmung von frei beweglichen Nanopartikeln.
Die Qualitatssicherung in der Nanoimprintlithographie erfolgt mittels der Fluoreszenz-
mikroskopie. Dazu werden die in der Nanoimprintlithographie herkommlich eingesetz-
ten Polymere mr − I 8000 und mr − I 9000 mit einem Derivat des Perylen-3,4,9,10-
tetracarboxlysauredianhydrids markiert, das kovalent an die Polymere gebunden ist.
Dieses Farbstoffderivat weist unter den gegebenen Versuchsbedingungen im Vergleich
zu Derivaten der Farbstoffe Anthracen oder Fluorescein die besten spektralen Eigen-
schaften auf. Fur eine empfindliche Detektion mittels der Fluoreszenzmikroskopie ist
eine Farbstoffkonzentration von 0, 1 % in dem Polymer ausreichend. Durch diese gerin-
ge Konzentration werden die Prageeigenschaften der Polymere nicht verandert. Beide
Polymere zeigen bei UV-Belichtung uber eine Stunde eine hohe Fluoreszenzstabilitat.
Die Untersuchungen der Pragungen und der Stempel zeigen, dass die Fluoreszenzmikro-
skopie eine schnelle und effiziente Charakterisierung der Defekte ermoglicht. Daruber
137

138 7. Zusammenfassung
hinaus lasst sich die Einheitlichkeit einer Pragung qualitativ bestimmen. Im Vergleich
zu den herkommlich verwendeten Methoden wie SEM, AFM oder der Profilometrie
erfordert die Fluoreszenzmikroskopie keine Probenvorbereitung. Die Analyse der Pro-
ben findet ohne Kontakt zur Probenoberflache statt, so dass im Hinblick auf einen
industriellen Prozess ein Zuruckfuhren der Probe in den Produktionskreislauf jederzeit
moglich ist. Unter diesem Aspekt erfolgt eine halbautomatische Qualitatskontrolle ei-
nes Stempels, deren Ergebnisse in einem SPC-Diagramm (statistische Prozesskontrolle)
quantitativ wiedergegeben werden.
Die Methode zur Großenbestimmung von frei beweglichen Nanopartikeln basiert auf
einem Particle Tracking-Algorithmus und ist unabhangig von der verwendeten Mikro-
skopiemethode. Gezeigt werden Großenverteilungen von Polystyrolpartikelstandards,
die sowohl mit der Dunkelfeldmikroskopie als auch mit der Fluoreszenzmikroskopie
bestimmt werden. Die vorgestellten unimodalen Großenverteilungen ergeben durch-
schnittliche Radien mit einer hohen Ubereinstimmung zu den Herstellerangaben. Die
Standardabweichungen liegen zwischen 1 % fur den 100 nm Standard und 15 % fur
den 51 nm Standard. Die bimodale Großenverteilung einer Mischung des 120 nm Stan-
dards und des 202 nm Standards bestatigt die hohe Zuverlassigkeit der Methode. Die
mittleren Radien zeigen eine gute Ubereinstimmung zu den Einzelmessungen. Ferner
kann das eingesetzte Verhaltnis der einzelnen Standards quantitativ bestatigt werden.
Beide Analysemethoden zeichnen sich aufgrund ihrer einfachen Handhabung durch
eine hohe Benutzerfreundlichkeit aus, da sie, wie im Falle der Qualitatssicherung in der
Nanoimprintlithographie, keine Probenvorbereitung erfordern oder, wie bei der Großen-
bestimmung der frei beweglichen Nanopartikel, durch eine schnelle Probenpraparation
charakterisiert sind. Die Ergebnisse unterstreichen die hohe Zuverlassigkeit und die
gute Reproduzierbarkeit beider Methoden. Auch im Hinblick auf die geringen entste-
henden Kosten stellen diese Methoden eine gute Alternative zu den bereits bekannten
Methoden dar.

Anhang A
Bildverarbeitungsprogramme
A.1 Mathematica Programm
Remove["Global‘*"]
dateien=Import["d:\name.txt","Lines"];
anzahl=Length[dateien];
bild=Table[Import["d:/name/"<>dateien[[i]]],{i,1,anzahl}];
pixelgroesse=10;
bildn=Table[bild[[i,1,1]],{i,1,anzahl}];
a=Dimensions[bildn[[1]],2];
hohe=a[[1]];
breite=a[[2]];
kompensationsgerade=Table[Table[Fit[bildn[[i,5]] ,{1,x}, x],{x,1,breite}],
{i,1,anzahl}];
lkg=Length[kompensationsgerade];
b=Table[kompensationsgerade[[m,1]]=kompensationsgerade[[m,2]],{m,1,lkg}];
kompensationsbild=Table[Table[kompensationsgerade[[m]],{i,1,hohe}],
{m,1,lkg}];
bbildn=bildn;
Table[bbildn[[m]]=bildn[[m]]-kompensationsbild[[m]],{m, 1,lkg}];
abildn=Table[Flatten[bildn[[m]]],{m, 1,lkg}];
weichbild=bbildn;
c=Table[Dimensions[weichbild[[m]],2],{m,1,lkg}];
hoehe=c[[1,1]];
breite=c[[1,2]];
neuhoehe=Round[hoehe/pixelgroesse]-1;
neubreite=Round[breite/pixelgroesse]-1;
139

140 A. Bildverarbeitungsprogramme
hoeheoff=Table[i,{i,1,hoehe,pixelgroesse}];
breiteoff=Table[i,{i,1,breite,pixelgroesse}];
neubild=Table[Table[Sum[weichbild[[m,l,k]],
{k,breiteoff[[j]],breiteoff[[j]]+pixelgroesse},
{l,hoeheoff[[i]],hoeheoff[[i]]+pixelgroesse}]
/pixelgroesse^2,{i,1,neuhoehe},
{j,1,neubreite}],{m,1,lkg}];
kanteab=Table[neubild[[m,i]],{m,1,lkg},
{i,Round[pixelgroesse/6]+1,neuhoehe}];
d=Table[Dimensions[kanteab[[m]],2],{m,1,lkg}];
hoehe2=d[[1,1]];
breite2=d[[1,2]];
bildwerte=Table[Flatten[kanteab[[m]]],{m,1,lkg}];
histo=Table[Table[Count[Round[10*bildwerte[[m]]],i],
{i,-127,127}],{m,1,lkg}];
Flache=hoehe2*breite2;
histonorm=Table[histo[[m]]/Flache,{m,1,lkg}];
lhn=Length[histonorm[[1]]];
bildsum=Table[Apply[Plus,Table[histonorm[[m,i]],{m,1,lkg}]],{i,1,lhn}];
bildsum1=bildsum/lkg;
Export["d:\name.dat", Chop[N[bildsum1,5]]];
ListPlot[bildsum1,PlotJoined->True, PlotRange->{0,1}]
ListPlot[bildsum1,PlotJoined->True, PlotRange->{0,0.1}]
A.2 Makro zur Bildverarbeitung und -analyse
DEFINT Count
DEFINT Width
DEFINT Height
DEFINT Expnum
DEFINT Data_ID
DEFINT Numpic
DEFINT Tres
DEFINT Parnum
DEFINT Firstseq
transfer_active
enable_undo 0
get_roi
IVAR[40] := IVAR[10]

A.2 Makro zur Bildverarbeitung und -analyse 141
IVAR[41] := IVAR[11]
IVAR[42] := IVAR[12]
IVAR[43] := IVAR[13]
IVAR[44] := IVAR[14]
get_3d_image
IVAR[9] := 0
FOR n
IVAR[9]++
IF IVAR[9] = 1
load_pathname "d:\Pfad\name"
STRING[6] := "se"
IVAR[5] := 30
Width := 240
Height := 240
IVAR[4] := 218
save_pathname "d:\Pfad\subname"
ELSE
ENDIF
:
:
:
IF IVAR[9] = 8
load_pathname "d:\Pfad\name"
STRING[6] := "se"
IVAR[5] := 30
Width := 240
Height := 240
IVAR[4] := 218
save_pathname "d:\Pfad\subname"
ELSE
ENDIF
IVAR[7] := 0
WHILE IVAR[7] < IVAR[5]
IVAR[7]++
* Lade-Prozedur
roi_size Width Height 0 0
STRING[4] := CONCAT (STRING[6], LTOA(IVAR[7]), 4)
IVAR[1] := 0
FOR IVAR[4]

142 A. Bildverarbeitungsprogramme
STRING[1] := CONCAT (STRING[4], LTOA(IVAR[1]), 8)
load_filename STRING[1]
load_tiff
roi_to_d3d (IVAR[1])
IVAR[1]++
ENDFOR
Count := IVAR[4]
roi_size Width Height 0 0
select_active 0 1
d3d_to_roi 0 0
IVAR[2] := 0
select_active 0 2
select_second 0 1
FOR Count
d3d_to_roi 0 IVAR[2]
src_dst_op 5 128
const_op 5 100 4.0 0.0
gauss 5
roi_to_d3d (IVAR[2])
IVAR[2]++
ENDFOR
* Speicher-Prozedur
roi_size Width Height 0 0
IVAR[10] := 0
IF IVAR[10] = 1
save_compress (1)
ELSE
save_compress (0)
ENDIF
IVAR[3] := IVAR[4]
IVAR[1] := 0
FOR IVAR[3]
d3d_to_roi (0,IVAR[1])
STRING[1] := CONCAT (STRING[4], LTOA(IVAR[1]), 8)
save_filename STRING[1]
save_tiff(2)
IVAR[1]++
ENDFOR
save_compress (0)

A.2 Makro zur Bildverarbeitung und -analyse 143
ENDWHILE
ENDFOR
Expnum := 4
IVAR[9] := 0
FOR n
IVAR[9]++
IF IVAR[9] = 1
load_pathname "d:\Pfad\subname"
STRING[5] := "se0"
STRING[7] := "01"
STRING[6] := "se"
data_pathname "d:\Pfad\Dateiname"
Tres := 180
IVAR[5] := 30
Width := 240
Height := 240
IVAR[4] := 218
IVAR[2] := 0
WHILE IVAR[2] < IVAR[5]
IVAR[1] := 0
IVAR[2]++
IF IVAR[2] < 10
STRING[2] := CONCAT(STRING[5], LTOA(IVAR[2]), 4)
ELSE
STRING[2] := CONCAT(STRING[6], LTOA(IVAR[2]), 4)
ENDIF
STRING[3] := CONCAT(STRING[7], LTOA(IVAR[2]), 4)
data_filename STRING[3]
open_data_file 0 0 0 0
Data_ID := IVAR[10]
set_writef_id Data_ID
set_pix_area 2 1
set_append_obj 0
set_contour_jump 0
set_cutobj 0 0 0 0
roi_size Width Height 0 0
FOR IVAR[4]
Count := -1
STRING[4] := CONCAT (STRING[2], LTOA(IVAR[1]), 8)

144 A. Bildverarbeitungsprogramme
load_filename STRING[4]
load_tiff
set_append_obj 0
search_objects Tres 0
Parnum := IVAR[10]
writef_string "A"
writefln_long Parnum
FOR Parnum
Count++
object_data Count 1
writef_long FVAR[17]
writefln_long FVAR[18]
ENDFOR
writef_crlf
IVAR[1]++
ENDFOR
msg_interactive 0
close_data_file Data_ID
msg_interactive -1
ENDWHILE
ELSE
ENDIF
:
:
:
IVAR[9]++
IF IVAR[9] = 8
load_pathname "d:\Pfad\subname"
STRING[5] := "se0"
STRING[7] := "01"
STRING[6] := "se"
data_pathname "d:\Pfad\Dateiname"
Tres := 180
IVAR[5] := 30
Width := 240
Height := 240
IVAR[4] := 218
IVAR[2] := 0
WHILE IVAR[2] < IVAR[5]

A.2 Makro zur Bildverarbeitung und -analyse 145
IVAR[1] := 0
IVAR[2]++
IF IVAR[2] < 10
STRING[2] := CONCAT(STRING[5], LTOA(IVAR[2]), 4)
ELSE
STRING[2] := CONCAT(STRING[6], LTOA(IVAR[2]), 4)
ENDIF
STRING[3] := CONCAT(STRING[7], LTOA(IVAR[2]), 4)
data_filename STRING[3]
open_data_file 0 0 0 0
Data_ID := IVAR[10]
set_writef_id Data_ID
set_pix_area 2 1
set_append_obj 0
set_contour_jump 0
set_cutobj 0 0 0 0
roi_size Width Height 0 0
FOR IVAR[4]
Count := -1
STRING[4] := CONCAT (STRING[2], LTOA(IVAR[1]), 8)
load_filename STRING[4]
load_tiff
set_append_obj 0
search_objects Tres 0
Parnum := IVAR[10]
writef_string "A"
writefln_long Parnum
FOR Parnum
Count++
object_data Count 1
writef_long FVAR[17]
writefln_long FVAR[18]
ENDFOR
writef_crlf
IVAR[1]++
ENDFOR
msg_interactive 0
close_data_file Data_ID
msg_interactive -1

146 A. Bildverarbeitungsprogramme
ENDWHILE
ELSE
ENDIF
ENDFOR
enable_undo 1
A.3 Excel Makro zur Spurenanalyse von frei be-
weglichen Nanopartikeln
Sub A_Spurenanalyse()
’*** Teil 1: Spurerzeugung aus den einzelnen Bildpunktkoordinaten ***
’Variablen-Dimensionierung
Dim Spuren(1 To 65000, 0 To 300) As Single
Dim Distanz As Single
Dim DistVer(1 To 65000, 1 To 2) As Single
Dim AnzahlSpuren As Single
Dim NotizGes(1 To 65000, 0 To 1) As Integer
Dim DistCount(1 To 65000, 0 To 1) As Integer
Dim AnzahlSeq As Integer
Dim StepZeit As Single
Dim KaliWert As Single
Dim Temper As Single
Dim DisQu1 As Single
Dim DisQu2 As Single
Dim Zaehler As Single
Dim Nenner As Single
Dim DisQuFeld(1 To 65000, 1 To 3)
Dim Out(1 To 4000, 1 To 3) As Single
Dim StillStand As Integer
Dim BereichH(1 To 4000, 0 To 1) As Integer
Dim MinTeilAbstand As Single
Dim DistanzMin As Single
’Zuordnung der experimentellen Werte aus dem Excel-Tabellenblatt
AnzahlSeq = Worksheets("Tabelle1").Range("D3")
StepZeit = Worksheets("Tabelle1").Range("D4")

A.3 Excel Makro zur Spurenanalyse von frei beweglichen Nanopartikeln 147
KaliWert = Worksheets("Tabelle1").Range("D5")
KaliWert = KaliWert * 0.000001
Temper = Worksheets("Tabelle1").Range("D6")
Temper = Temper + 273.15
StillStand = Worksheets("Tabelle1").Range("D14")
Wahl1 = Worksheets("Tabelle1").Range("D17")
MinTeilAbstand = Worksheets("Tabelle1").Range("D18")
’1. Berechnung des Zahlers und des Nenners der
’Einstein-Smoluchowski-Gleichung
Zaehler = 8.31441 * Temper * StepZeit
Nenner = 3 * 3.14159265 * 0.0008905 * 6.02205 * (10 ^ 23)
’Offnen der Dat-Dateien im Excel-Format
For zIt = 1 To AnzahlSeq
Worksheets("Tabelle1").Range("E3").Value = zIt
If zIt = 1 Then
Workbooks.OpenText FileName:="D:\Pfad\name.Dat",
Origin:= xlWindows, StartRow:=1, DataType:=xlDelimited,
TextQualifier:= xlDoubleQuote, ConsecutiveDelimiter:=True,
Tab:=False, Semicolon:=False, Comma:=False, Space:=True,
Other:=False, FieldInfo:=Array(Array(1, 1), Array(2, 1),
Array(3, 1), Array(4, 1))
Range("A1:D65000").Select
Bereich1 = Selection
ActiveWorkbook.Close
End If
:
:
:
If zIt = 30 Then
Workbooks.OpenText FileName:="D:\Pfad\name.Dat",
Origin:= xlWindows, StartRow:=1, DataType:=xlDelimited,
TextQualifier:= xlDoubleQuote, ConsecutiveDelimiter:=True,
Tab:=False, Semicolon:=False, Comma:=False, Space:=True,
Other:=False, FieldInfo:=Array(Array(1, 1), Array(2, 1),
Array(3, 1), Array(4, 1))
Range("A1:D65000").Select
Bereich1 = Selection

148 A. Bildverarbeitungsprogramme
ActiveWorkbook.Close
End If
’Variablen-Wertstellungen
z0H2 = 0
For z0 = 1 To Bereich1(4, 2) Step 1
z0H = z0 + 4
z0H2 = z0H2 + 1
Spuren(z0H2, 0) = 0
Spuren(z0H2, 1) = 1
Spuren(z0H2, 2) = Bereich1(z0H, 2)
z0H2 = z0H2 + 1
Spuren(z0H2, 0) = 0
Spuren(z0H2, 1) = 1
Spuren(z0H2, 2) = Bereich1(z0H, 3)
Next z0
AnzahlSpuren = Bereich1(4, 2)
For z0 = 1 To 65000
DistVer(z0, 1) = 1000000
Next z0
sp1 = 2
sp2 = 3
’Vorfilterung von unbewegten Partikeln
z7 = 4
z5 = 1
z8 = 0
z6 = 0
If Wahl1 = 1 Then
For zFilt = 1 To StillStand
Do
z7 = z7 + 1
Loop Until Bereich1(z7, 1) = "A"
For z3 = 1 To AnzahlSpuren
z6 = z5 + 1
For z4 = 1 To Bereich1(z7, 2)
z4H = z4 + z7
Distanz = ((Spuren(z5, 2) - Bereich1(z4H, 2)) ^ 2 +
(Spuren(z6, 2) - Bereich1(z4H, 3)) ^ 2) ^ 0.5

A.3 Excel Makro zur Spurenanalyse von frei beweglichen Nanopartikeln 149
If Distanz = 0 Then
DistCount(z3, 1) = DistCount(z3, 1) + 1
End If
Next z4
If DistCount(z3, 1) = StillStand Then
Spuren(z5, 1) = 0
Spuren(z6, 1) = 0
z8 = z8 + 1
Out(z8, 1) = Spuren(z5, sp1)
z8 = z8 + 1
Out(z8, 1) = Spuren(z6, sp1)
End If
z5 = z5 + 2
Next z3
z5 = 1
Next zFilt
End If
For z3 = 1 To 65000
DistCount(z3, 1) = 0
Next z3
’Zuordnungsschleife der Spuren
z7 = 4
AnzahlSpuren = Bereich1(4, 2)
Do
z7 = z7 + 1
If Bereich1(z7, 1) = "A" Then
For z3 = 1 To 4000
BereichH(z3, 1) = 0
Next z3
For z3 = 1 To Bereich1(z7, 2)
z3H = z3 + z7
z8H = 1
For z4 = 1 To z8
z9H = z8H + 1
If Bereich1(z3H, 2) = Out(z8H, 1) And
Bereich1(z3H, 3) = Out(z9H, 1) Then
BereichH(z3, 1) = 1
End If

150 A. Bildverarbeitungsprogramme
z8H = z8H + 2
Next z4
Next z3
For z3 = 1 To Bereich1(z7, 2)
z3H = z3 + z7
For z4 = 1 To Bereich1(z7, 2)
z4H = z4 + z7
DistanzMin = ((Bereich1(z3H, 2) - Bereich1(z4H, 2)) ^ 2
+ (Bereich1(z3H, 3) - Bereich1(z4H, 3)) ^ 2) ^ 0.5
If DistanzMin < MinTeilAbstand And z3H <> z4H Then
BereichH(z3, 1) = 1
End If
Next z4
Next z3
z5 = 1
For z3 = 1 To AnzahlSpuren
z6 = z5 + 1
If Spuren(z6, 1) = 1 Then
For z4 = 1 To Bereich1(z7, 2)
z4H = z4 + z7
Distanz = ((Spuren(z5, sp1) - Bereich1(z4H, 2)) ^ 2
+ (Spuren(z6, sp1) - Bereich1(z4H, 3)) ^ 2) ^ 0.5
If Distanz < DistVer(z3, 1) And BereichH(z4, 1) = 0
Then
DistVer(z3, 1) = Distanz
DistVer(z3, 2) = z4
End If
Next z4
z4H = DistVer(z3, 2) + z7
If Spuren(z5, 1) = 1 Then
Spuren(z5, sp2) = Bereich1(z4H, 2)
Spuren(z6, sp2) = Bereich1(z4H, 3)
End If
If DistVer(z3, 1) = 0 Then
DistCount(z3, 1) = DistCount(z3, 1) + 1
Else
DistCount(z3, 1) = 0
End If
If DistCount(z3, 1) = StillStand Then

A.3 Excel Makro zur Spurenanalyse von frei beweglichen Nanopartikeln 151
Spuren(z5, sp2) = 0
Spuren(z6, sp2) = 0
Spuren(z5, 1) = 0
Spuren(z6, 1) = 0
z8 = z8 + 1
Out(z8, 1) = Spuren(z5, sp2)
z8 = z8 + 1
Out(z8, 1) = Spuren(z6, sp2)
End If
Else
DistVer(z3, 2) = 0
End If
z5 = z5 + 2
Next z3
z5 = 1
For z3 = 1 To AnzahlSpuren
z6 = z5 + 1
If DistVer(z3, 2) > 0 Then NotizGes(DistVer(z3, 2), 1) = 1
If Spuren(z6, 1) = 1 Then
For z4 = 1 To AnzahlSpuren
If DistVer(z3, 2) = DistVer(z4, 2) And z3 <> z4 Then
Spuren(z5, 1) = 0
Spuren(z6, 1) = 0
End If
Next z4
End If
If Spuren(z5, 1) = 1 Then
Spuren(z5, 0) = Spuren(z5, 0) + 1
Spuren(z6, 0) = Spuren(z6, 0) + 1
End If
z5 = z5 + 2
Next z3
z5 = 1
For z3 = 1 To AnzahlSpuren
z6 = z5 + 1
If Spuren(z5, 1) = 0 Then
Spuren(z5, sp2) = 0
Spuren(z6, sp2) = 0
End If

152 A. Bildverarbeitungsprogramme
z5 = z5 + 2
Next z3
For z3 = 1 To Bereich1(z7, 2)
If NotizGes(z3, 1) = 0 Then
OutH = 0
zDo1 = 1
zDo2 = 0
z3H = z3 + z7
While zDo2 < z8
zDo2 = zDo1 + 1
If Bereich1(z3H, 2) = Out(zDo1, 1) And
Bereich1(z3H, 3) = Out(zDo2, 1) Then
OutH = 1
End If
zDo1 = zDo1 + 2
Wend
If OutH = 0 Then
AnzahlSpuren = AnzahlSpuren + 1
PosY = AnzahlSpuren * 2
PosX = PosY - 1
z4 = z3 + z7
Spuren(PosX, sp2) = Bereich1(z4, 2)
Spuren(PosY, sp2) = Bereich1(z4, 3)
Spuren(PosX, sp1) = -1
Spuren(PosY, sp1) = -1
Spuren(PosX, 1) = 1
Spuren(PosY, 1) = 1
End If
End If
Next z3
sp1 = sp1 + 1
sp2 = sp1 + 1
z5 = 1
For z3 = 1 To 65000
DistVer(z3, 1) = 1000000
Next z3
For z3 = 1 To Bereich1(z7, 2)
NotizGes(z3, 1) = 0
Next z3

A.3 Excel Makro zur Spurenanalyse von frei beweglichen Nanopartikeln 153
End If
Loop Until z7 = 65000
If zIt = 5 Then
Worksheets("Tabelle2").Range("A1:IV400") = Spuren
End If
’ *** Teil 2: Berechnung des mittleren Verschiebungsquadrats ***
y5 = 1
For y1 = 1 To AnzahlSpuren
y6 = y5 + 1
If Spuren(y5, 0) > 0 Then
y2 = 1
Do
y2 = y2 + 1
Loop Until Spuren(y5, y2) > 0
yEnd = y2 + Spuren(y5, 0)
y2 = y2 + 1
For y3 = y2 To yEnd
y3H = y3 - 1
DisQu1 = DisQu1 + (KaliWert * (Spuren(y5, y3)
- Spuren(y5, y3H))) ^ 2
DisQu2 = DisQu2 + (KaliWert * (Spuren(y6, y3)
- Spuren(y6, y3H))) ^ 2
Next y3
If DisQu1 > 0 And DisQu2 > 0 Then
DisQuCounter = DisQuCounter + 1
DisQu1 = DisQu1 / Spuren(y5, 0)
DisQuFeld(DisQuCounter, 1) = DisQu1
DisQu1 = 0
DisQu2 = DisQu2 / Spuren(y6, 0)
DisQuFeld(DisQuCounter, 2) = DisQu2
DisQu2 = 0
DisQuFeld(DisQuCounter, 3) = Spuren(y5, 0)
End If
End If
y5 = y5 + 2
Next y1
For y1 = 1 To 65000
For y2 = 0 To 300

154 A. Bildverarbeitungsprogramme
Spuren(y1, y2) = 0
Next y2
Next y1
AnzahlSpurenSumme = AnzahlSpurenSumme + AnzahlSpuren
Next zIt
Worksheets("Tabelle1").Range("D22") = DisQuCounter
Worksheets("Tabelle1").Range("D21") = AnzahlSpurenSumme
Worksheets("Tabelle1").Range("N1:P65000") = DisQuFeld
End Sub
A.4 Excel Makro zur Erstellung der Histogramme
der Großenverteilungen
Sub B_Histogrammerstellung()
’ *** Teil 3: Berechnung der Radien***
Dim SumCurParNum1 As Single
Dim SumCurParNum2 As Single
Dim DisQu2(1 To 65000, 1 To 4) As Double
Dim Temp As Double
Dim TempK As Double
Dim Time As Double
Dim Eta As Double
Dim EtaConst As Double
Dim Controlling As Double
Dim Controlling2 As Double
Dim Density As Double
Dim ix As Double
Dim Distance As Double
Dim Histo(1 To 1000, 1 To 2) As Double
Dim MidNum As Double
Dim MidWei As Double
Dim Weight As Double
Dim WeightSum As Double
Dim Polydis As Double
Dim Wish1 As Single Dim

A.4 Excel Makro zur Erstellung der Histogramme der Großenverteilungen 155
Wish2 As Single
Dim Wish3 As Single
Dim Radii(1 To 150000, 1 To 2) As Double
Dim Radi1 As Double
Dim RadiX1 As Double
Dim RadiY1 As Double
Dim Delta2 As Double
Dim DeltaX2 As Double
Dim DeltaY2 As Double
Dim IntervalStart As Double
Dim IntervalEnd As Double
Dim IntervalSize As Double
Dim PolydisLinkeGrenze As Double
Dim PolydisRechteGrenze As Double
Dim HistoQuali As Double
Const R As Double = 8.31441
Const NA As Double = 6.02205 * (10 ^ 23)
Const Pi As Double = 3.141592654
’ Interaktives Einlesen benotigter Daten
Time = Worksheets("Tabelle1").Range("D4").Value
TempKorrektur = Worksheets("Tabelle1").Range("D16").Value
Temp = Worksheets("Tabelle1").Range("D6").Value
Temp = Temp + TempKorrektur
Density = Worksheets("Tabelle1").Range("D7").Value
Distance = Worksheets("Tabelle1").Range("D8").Value
SumCurParNum1 = Worksheets("Tabelle1").Range("D22").Value
’ Berechnung der Viskositat von Wasser bei gegebener Temperatur
Distance = Distance / 1000000
Eta = (10 ^ ((((1.3272 * (20 - Temp))
- (0.001053 * (Temp - 20) ^ 2))
/ (Temp + 105)) + 0.000867721)) * 0.001
EtaConst = Eta TempK = Temp + 273.15
’ Histogramm Berechnung
Wish1 = Worksheets("Tabelle1").Range("D15").Value
Wish2 = Worksheets("Tabelle1").Range("D10").Value

156 A. Bildverarbeitungsprogramme
’ Berechnung der Radien mit Schichtdicke, dx, dy und
Sicherheitsgrenze: DisQu -> Radii If Wish1 = 1 Then
Range("N1:P65000").Select
Bereich = Selection
Range("A1").Select
h = 0
CX = 0
CY = 0
Do
h = h + 1
CX = CX + 2
CY = CX - 1
DisQu2(h, 1) = Bereich(h, 1)
DisQu2(h, 2) = Bereich(h, 2)
DisQu2(h, 3) = Bereich(h, 3)
If DisQu2(h, 2) > 0 Then
DeltaY2 = DisQu2(h, 2)
Eta = EtaConst
For k = 1 To 10 Step 1
RadiY1 = (R * TempK * Time)
/ (3 * Pi * Eta * NA * DeltaY2)
Controlling = 16 * Distance / 7
If Controlling > RadiY1 Then
Controlling2 = ((9 * RadiY1 * ((16 * Distance)
+ (9 * RadiY1)) / (16 * (Distance - (2 * RadiY1))
* ((8 * Distance) + (9 * RadiY1))))
* (Log(((16 * Distance) - (7 * RadiY1)) / (25 * RadiY1))))
If Controlling2 < 1 Then
ix = 1 - Controlling2
Eta = EtaConst / ix
End If
End If
Next k
Radii(CX, 1) = RadiY1
Radii(CX, 2) = DisQu2(h, 3)
ElseIf DisQu2(h, 2) = 0 Then
Radii(CX, 1) = 0
Radii(CX, 2) = DisQu2(h, 3)
End If

A.4 Excel Makro zur Erstellung der Histogramme der Großenverteilungen 157
If DisQu2(h, 1) > 0 Then
DeltaX2 = DisQu2(h, 1)
Eta = EtaConst
For l = 1 To 10 Step 1
RadiX1 = (R * TempK * Time)
/ (3 * Pi * Eta * NA * DeltaX2)
Controlling = 16 * Distance / 7
If Controlling > RadiX1 Then
Controlling2 = ((9 * RadiX1 * ((16 * Distance)
+ (9 * RadiX1)) / (16 * (Distance - (2 * RadiX1))
* ((8 * Distance) + (9 * RadiX1))))
* (Log(((16 * Distance) - (7 * RadiX1)) / (25 * RadiX1))))
If Controlling2 < 1 Then
ix = 1 - Controlling2
Eta = EtaConst / ix
End If
End If
Next l
Radii(CY, 1) = RadiX1
Radii(CY, 2) = DisQu2(h, 3)
ElseIf DisQu2(h, 1) = 0 Then
Radii(CY, 1) = 0
Radii(CY, 2) = DisQu2(h, 3)
End If
Loop Until h = SumCurParNum1
SumCurParNum2 = SumCurParNum1 * 2
ElseIf Wish1 = 0 Then
Range("N1:P65000").Select
Bereich = Selection
Range("A1").Select
h = 0
Do
h = h + 1
DisQu2(h, 1) = Bereich(h, 1)
DisQu2(h, 2) = Bereich(h, 2)
DisQu2(h, 3) = Bereich(h, 3)
If DisQu2(h, 1) > 0 Or DisQu2(h, 2) > 0 Then
Delta2 = (DisQu2(h, 1) + DisQu2(h, 2)) / 2
Eta = EtaConst

158 A. Bildverarbeitungsprogramme
For l = 1 To 10 Step 1
Radi1 = (R * TempK * Time)
/ (3 * Pi * Eta * NA * Delta2)
Controlling = 16 * Distance / 7
If Controlling > Radi1 Then
Controlling2 = ((9 * Radi1 * ((16 * Distance)
+ (9 * Radi1)) / (16 * (Distance - (2 * Radi1))
* ((8 * Distance) + (9 * Radi1))))
* (Log(((16 * Distance) - (7 * Radi1)) / (25 * Radi1))))
If Controlling2 < 1 Then
ix = 1 - Controlling2
Eta = EtaConst / ix
End If
End If
Next l
Radii(h, 1) = Radi1
Radii(h, 2) = DisQu2(h, 3)
Else
Radii(h, 1) = 0
Radii(h, 2) = DisQu2(h, 3)
End If
Loop Until h = SumCurParNum1
SumCurParNum2 = SumCurParNum1 * 2
For z4 = SumCurParNum1 To SumCurParNum2 Step 1
z5 = z4 + 1
Radii(z5, 1) = 0
Radii(z5, 2) = 0
Next z4
End If
’ *** Teil 4: Histogrammerstellung***
’ Erstellung des Histogramms: Radii -> Histo
If Worksheets("Tabelle1").Range("E11") = "um" Then
IntervalStart = Worksheets("Tabelle1").Range("D11").Value * 0.000001
End If
If Worksheets("Tabelle1").Range("E11") = "nm" Then
IntervalStart = Worksheets("Tabelle1").Range("D11").Value
* 0.000000001
End If

A.4 Excel Makro zur Erstellung der Histogramme der Großenverteilungen 159
If Worksheets("Tabelle1").Range("E11") = "m" Then
IntervalStart = Worksheets("Tabelle1").Range("D11").Value
End If
If Worksheets("Tabelle1").Range("E11") = "mm" Then
IntervalStart = Worksheets("Tabelle1").Range("D11").Value * 0.001
End If
If Worksheets("Tabelle1").Range("E12") = "um" Then
IntervalEnd = Worksheets("Tabelle1").Range("D12").Value * 0.000001
End If
If Worksheets("Tabelle1").Range("E12") = "nm" Then
IntervalEnd = Worksheets("Tabelle1").Range("D12").Value
* 0.000000001
End If
If Worksheets("Tabelle1").Range("E12") = "m" Then
IntervalEnd = Worksheets("Tabelle1").Range("D12").Value
End If
If Worksheets("Tabelle1").Range("E12") = "mm" Then
IntervalEnd = Worksheets("Tabelle1").Range("D12").Value * 0.001
End If
IntervalSize = Worksheets("Tabelle1").Range("D13").Value
IntervalStep = (IntervalEnd - IntervalStart) / (IntervalSize - 1)
IntervalRight = IntervalStart + IntervalStep
IntervalLeft = IntervalStart
For z2 = 1 To IntervalSize Step 1
Histo(z2, 1) = (IntervalLeft + (IntervalStep / 2)) * 1000000000
For z8 = 1 To SumCurParNum2 Step 1
If Radii(z8, 1) >= IntervalLeft And Radii(z8, 1)
< IntervalRight And Radii(z8, 2) >= Wish2 Then
Histo(z2, 2) = Histo(z2, 2) + 1
TeilZaehl = TeilZaehl + 1
End If
Next z8
IntervalLeft = IntervalLeft + IntervalStep
IntervalRight = IntervalRight + IntervalStep
Next z2
’ Histogramm Ausgabe
For O = 1 To 1000 Step 1
Worksheets("Tabelle1").Range("Q1:Q1000").Cells(O) = Histo(O, 1)

160 A. Bildverarbeitungsprogramme
Worksheets("Tabelle1").Range("R1:R1000").Cells(O) = Histo(O, 2)
Next O
For z = 1 To 65000 Step 1
Radii(z, 1) = Radii(z, 1)
Next z
Beep
End Sub

Literaturverzeichnis
[1] R. Feynman, There’s plenty of Room at the Bottom, American Physical Society,
California Institute of Technology, 1956.
[2] Nanotruck: Reise in den Nanokosmos , Bundesministerium fur Bildung und For-
schung, 2004.
[3] A. Einstein, Uber die von der molekularkinetischen Theorie der Warme geforderte
Bewegung von in ruhenden Flussigkeiten suspendierten Teilchen, Annalen der
Physik , 17, 549 – 560, 1905.
[4] A. Einstein, Zur Theorie der Brownschen Bewegung, Annalen der Physik , 19,
371 – 381, 1906.
[5] M. von Smoluchowski, Zur kinetischen Theorie der Brownschen Molekularbewe-
gung und der Suspensionen, Annalen der Physik , 21, 756 – 780, 1906.
[6] A. Einstein, M. von Smoluchowski, Untersuchungen uber die Theorie der Brown-
schen Bewegung, Ostwalds Klassiker der exakten Wissenschaften, Bd. 199 und
207, Thun [u. a.], Frankfurt a. Main, 1997.
[7] T. Svedberg, Uber die Eigenbewegung der Teilchen in kolloidalen Losungen, Zeit-
schrift fur Elektrochemie, 12(47), 853 – 868, 1906.
[8] G. Goke, Streifzuge durch die Geschichte der Mikroskopie: 1. Die großen Erfolge
im 17. und 18. Jahrhundert, Mikrokosmos, 78(3), 76 – 81, 1989.
[9] H. Beyer, Historischer Ruckblick, Handbuch der Mikroskopie, 15 – 24, VEB Verlag
Technik, Berlin, 1977.
161

162 LITERATURVERZEICHNIS
[10] G. Goke, Streifzuge durch die Geschichte der Mikroskopie: 2. Der große Durch-
bruch im 19. Jahrhundert, Mikrokosmos, 78(4), 104 – 107, 1989.
[11] G. Goke, Streifzuge durch die Geschichte der Mikroskopie: 4. Die neuen Methoden
der Lichtmikorskopie im 20. Jahrhundert, Mikrokosmos, 78(8), 231 – 236, 1989.
[12] M. Pluta, Advanced Light Microscopy , Bd. 2, Elsevier, Amsterdam, 1993.
[13] F. W. D. Rost, Fluorescence microscopy , Bd. 1, Cambridge University Press,
Cambridge, 1992.
[14] J. S. Ploem, H. Tanke, Introduction to Fluorescence Microscopy , Bd. 10 von
Microscopy Handbooks, Oxford University Press, Oxford, 1987.
[15] J. R. Lakowicz, Principles of fluorescence spectrocospy , Plenum Press, New York,
1983.
[16] M. Klessinger, J. Michl, Lichtabsorption und Photochemie organischer Molekule,
Bd. 3, VCH, Weinheim, 1989.
[17] R. Winter, F. Noll, Methoden der Biophysikalischen Chemie, Teubner, Stuttgart,
1998.
[18] W. Schmidt, Optische Spektroskopie, VCH, Weinheim, 1994.
[19] B. Hermann, Fluorescence Microscopy , Bios Scientific Publishers, Oxford, 2.
Aufl., 1998.
[20] E. Becker, Fluoreszenzmikroskopie, Leitz, Wetzlar, 1983.
[21] G. Goke, Moderne Methoden der Lichtmikroskopie, Kosmos Verlag, Stuttgart,
1988.
[22] H. Vogel, Gerthsen Physik , Springer, Berlin, 18. Aufl., 1995.
[23] H. Lindner, Physik fur Ingenieure, Vieweg & Sohn, Braunschweig, 1991.

LITERATURVERZEICHNIS 163
[24] F. W. D. Rost, Quantitative fluorescence microscopy , Cambridge University
Press, Cambridge, 1991.
[25] G. Goke, Streifzuge durch die Geschichte der Mikroskopie: 3. Mikroskope fur
spezielle Aufgaben, Mikrokosmos, 78(5), 139 – 143, 1989.
[26] D. Gerlach, Das Lichtmikroskop, Thieme, Stuttgart, 1976.
[27] K. Michel, Die Grundzuge der Theorie des Mikroskops in elementarer Darstel-
lung , Wissenschaftliche Verlagsgesellschaft M.B.H., Stuttgart, 2. Aufl., 1964.
[28] T. Svedberg, Neuere Untersuchungen uber die Brownsche Bewegung, Jahrbuch
der Radioaktivitat und Elektronik , 10, 467 – 515, 1913.
[29] J. K. Henning Bassmann, Bildverarbeitung Ad Oculus, Springer, Berlin, 3. Aufl.,
1998.
[30] B. Jahne, Digitale Bildverarbeitung , Springer, Berlin, 4. Aufl., 1997.
[31] J. Klas, Digitale Bildverarbeitung , Moreno-Verlag, Buchloe, 1996.
[32] P. Haberacker, Digitale Bildverarbeitung , Carl Hanser Verlag, Munchen, 3. Aufl.,
1989.
[33] Technology Roadmap for Nanoelectronics, European Communities, Luxemburg,
2. Aufl., 2001.
[34] S. Y. Chou, P. R. Krauss, P. J. Renstrom, Imprint Sub-25 nm vias and trenches
in polymers, Appl. Phys. Lett., 67(21), 3114 – 3116, 1995.
[35] S. Y. Chou, P. R. Krauss, P. J. Renstrom, Nanoimprint lithography, J. Vac. Sci.
Technol. B , 14(6), 4129 – 4133, 1996.
[36] I. Maximov, E.-L. Sarwe, M. Beck, K. Deppert, M. Graczyk, M. H. Magnus-
son, L. Montelius, Fabrication of Si-based nanoimprint stamps with sub-20 nm
features, Microelectronic Engineering , 61 - 62, 449 – 454, 2002.

164 LITERATURVERZEICHNIS
[37] H. Schulz, D. Lyebyedyev, H.-C. Scheer, K. Pfeiffer, G. Bleidiessel, G. Grutzner,
J. Ahopelto, Master replication in to thermosetting polymers for nanoimprinting,
J. Vac. Sci. Technol. B , 18(6), 3582 – 3585, 2000.
[38] K. Pfeiffer, M. Fink, G. Ahrens, F. Reuther, J. Seekamp, S. Zankovych, C. M.
Sotomayor-Torres, I. Maximov, M. Beck, M. Graczyk, L. Montelius, H. Schulz, H.-
C. Scheer, F. Steingrueber, Polymer stamps for nanoimprinting, Microelectronic
Engineering , 61 - 62, 393 – 398, 2002.
[39] R. W. Jaszewski, H. Schift, P. Groning, G. Margaritondo, Properties of thin
anti-adhesive films used for the replication of microstructures in polymers, Mi-
croelectronic Engineering , 35, 381 – 384, 1997.
[40] H. Schulz, H.-C. Scheer, F. Osenberg, J. Engemann, Mask fabrication by nanoim-
print lithography using anti-sticking layers, 16th European Conference on Mask
Technology for integrated Circuits and Microcomponents, Bd. 3996, 244 – 249,
VDE Verlag, Berlin, 1999.
[41] M. Beck, M-Graczyk, I. Maximov, E.-L. Sarwe, T. G. I. Ling, M. Keil,
L.Montelius, Improving stamps for 10 nm level wafer scale nanoimprint litho-
graphy, Microelectronic Engineering , 61 - 62, 441 – 448, 2002.
[42] M. Beck, Development of Nanoimprint Lithography for Fabrication of Electroche-
mical Transducers, Doktorarbeit, Lund University, Lund, Schweden, 2003.
[43] H. Schulz, H.-C. Scheer, T. Hoffmann, C. M. Sotomayor-Torres, K. Pfeiffer,
G. Bleidiessel, G. Grutzner, J. Ahopelto, B. Heidari, New polymer materials
for nanoimprinting, J. Vac. Sci. Technol. B , 18(4), 1861 – 1865, 2000.
[44] K. Pfeiffer, M. Fink, G. Bleidiessel, G. Gruetzner, H. Schulz, H.-C. Scheer,
T. Hoffmann, C. M. Sotomayor-Torres, F. Gaboriau, C. Cardinaud, Novel li-
near and crosslinking polymers for nanoimprinting with high etch resistance,
Microelectronic Engineering , 53, 411 – 414, 2000.
[45] U. Hilleringmann, Mikrosystemtechnik auf Silizium, Teubner, Stuttgart, 1995.

LITERATURVERZEICHNIS 165
[46] M. Kohler, Etching in microsystem technology , Wiley-VCH, Weinheim, New
York, 1999.
[47] F. Volklein, T. Zetterer, Einfuhrung in die Mikrosystemtechnik , Vieweg, Braun-
schweig, Wiesbaden, 2000.
[48] P. R. Krauss, S. Y. Chou, Nano-compact disk with 400 Gbit/in2 storage density
fabricated using nanoimprint lithography and read with proximal probe, Appl.
Phys. Lett., 71(21), 3174 – 3176, 1997.
[49] F. Carcenac, C. Vieu, A. Lebib, Y. Chen, L. Manin-Ferlazzo, H. Launois, Fabri-
cation of high density nanostructures gratings (> 500 Gbit/in2) used as molds
for nanoimprint lithography, Microelectronic Engineering , 53, 163 – 166, 2000.
[50] I. Martini, D. Eisert, M. Kamp, L. Worschech, A. Forchel, Quantum point con-
tacts fabricated by nanoimprint lithography, Appl. Phys. Lett., 77(14), 2237 –
2239, 2000.
[51] L. Guo, P. R. Krauss, S. Y. Chou, Nanoscale silicon field effect transistors fabri-
cated using imprint lithography, Appl. Phys. Lett., 71(13), 1881 – 1883, 1997.
[52] D. S. Macintyre, Y. C. Chen, D. Lim, S. Thoms, Fabrication of T-gate structures
by nanoimprint lithography, J. Vac. Sci. Technol. B , 19(6), 2797 – 2800, 2001.
[53] I. Maximov, P. Carlberg, I. Shorubalko, W. Seifert, H. Q. Xu, L. Montelius,
L. Samuelson, Nanoimprint lithography for fabrication of three-terminal ballistic
junctions in InP/GaInAs, Nanotechnology , 13, 666 – 668, 2002.
[54] Z. Yu, W. Wu, L. Chen, S. Y. Chou, Fabrication of large area 100 nm pitch
gratings by spatial frequency doubling and nanoimprint lithography for subwa-
velength otpical applications, J. Vac. Sci. Technol. B , 19(6), 2816 – 2819, 2001.
[55] Z. Yu, J. Schablitsky, S. Y. Chou, Nanoscale GaAs metal-semiconductor-metal
photodetectors fabricated using nanoimprint lithography, Appl. Phys. Lett.,
74(16), 2381 – 2383, 1999.

166 LITERATURVERZEICHNIS
[56] L. Montelius, B. Heidari, M. Graczyk, I. Maximov, E.-L. Sarwe, T. G. I. Ling,
Nanoimprint- and UV-Lithography: Mix & Match process for fabrication of in-
terdigitated nanobiosensors, Microelectronic Engineering , 53, 521 – 524, 2000.
[57] H. Cao, Z. Yu, J. Whang, J. O. Tegenfeldt, R. H. Austin, E. Chen, W. Wu, S. Y.
Chou, Fabrication of 10 nm enclosed nanofluidic channels, Appl. Phys. Lett.,
81(1), 174 – 176, 2002.
[58] L. Reimer, G. Pfefferkorn, Raster-Elektronenmikroskopie, Springer Verlag, Berlin,
1977.
[59] E. Fuchs, H. Oppolzer, H. Rehme, Particle Beam Microanalysis , VCH, Weinheim,
1990.
[60] G. Binnig, C. F. Quate, C. Gerber, Atomic Force Microscope, Phys. Rev. Lett.,
56(9), 930 – 933, 1986.
[61] S. N. Magonov, M.-H. Whangbo, Surface Analysis with STM and AFM , VCH,
Weinheim, 1996.
[62] W. Heckl, O. Marti, Atomic Force Microscopy, Procedures in Scanning Probe
Microscopies, 85 – 148, John Wiley & Sons, Chichester, 1998.
[63] R. Brown, A brief account of microscopical observations made in the month on
June, July and August 1828 on the particles contained in the pollen of plants
and on the general existence of active molecules in organic and inorganic bodies,
Phil. Mag., 4, 161 – 173, 1828.
[64] E. A. Moelwyn-Hughes, Physikalische Chemie, Georg Thieme Verlag, Stuttgart,
1970.
[65] J. Eggert, Lehrbuch der Physikalischen Chemie, S. Hirzel Verlag, Stuttgart, 1968.
[66] P. W. Atkins, Physikalische Chemie, Georg Thieme Verlag, Stuttgart, 1990.
[67] M. P. Langevin, Sur la theorie du mouvement brownien, Comptes Rendus , 146,
530 – 533, 1908.

LITERATURVERZEICHNIS 167
[68] I. Nordlund, Eine neue Bestimmung der Avogadroschen Konstante aus der
Brownschen Bewegung kleiner, in Wasser suspendierter Quecksilberkugelchen,
Zeitschrift Physikalische Chemie, 87, 40 – 62, 1914.
[69] H. A. Lorentz, Ein allgemeiner Satz, die Bewegung einer reibenden Flussigkeit be-
treffend, nebst einigen Anwendungen desselben, Abhandlungen uber theoretische
Physik , 23 – 42, Teubner Verlag, Lepizig, Berlin, 1907.
[70] F. Exner, Notiz zu Browns Molekularbewegung, Annalen der Physik , 4(2), 843
– 847, 1900.
[71] H. Siedentopf, R. Zsigmondy, Uber Sichtbarmachung und Großenbestimmung
ultramikoskopischer Teilchen, mit besonderer Anwendung auf Goldrubinglaser,
Annalen der Physik , 4(10), 1 – 39, 1903.
[72] M. Seddig, Uber die Messung der Temperaturabhangigkeit der Brownschen Mo-
lekularbewegung, Physikalische Zeitschrift , 9, 465 – 468, 1908.
[73] M. V. Henri, Etude cinematographique des mouvements browniens, Comptes
Rendus , 146, 1024 – 1026, 1908.
[74] M. J. Perrin, La loi de Stokes et le mouvement brownien, Comptes Rendus , 147,
475 – 476, 1908.
[75] M. Chaudesaigues, Le mouvement brownien et la formule d’ Einstein, Comptes
Rendus , 147, 1044 – 1046, 1908.
[76] H. Kruglak, Brownian Movement and Avogadro’s Number, Journal of Chemical
Education, 65(8), 732 –734, 1988.
[77] W. Schaertl, H. Sillescu, Dynamics of Colloidal Hard Spheres in Thin Aqueous
Suspension Layers, Journal of colloid and interface science, 155, 313–318, 1993.
[78] Y. Grasselli, G. Bossis, Three-Dimensional Particle Tracking for the Characte-
rization of Micrometer-Size Colloidal Particles, Journal of colloid and interface
science, 170, 269–274, 1995.

168 LITERATURVERZEICHNIS
[79] J. C. Crocker, D. G. Grier, Methods of Digital Video Microscopy for Colloidal
Studies, Journal of colloid and interface science, 179, 298–310, 1995.
[80] P. Nakroshis, M. Amoroso, J. Legere, C. Smith, Measuring Boltzmann’s constant
using video microscopy of Brownian motion, American Journal of Physics, 71(6),
568 – 573, 2002.
[81] C. Finder, Beobachtung und Analyse von Brownschen Teilchenbewegungen mit
einem automatischen Bildauswertungssystem, Diplomarbeit, Gerhard-Mercator-
Universitat, Duisburg, Deutschland, 1999.
[82] M. Wohlgemuth, Diffusionsexperimente an Nanokapseldispersionen, Doktorar-
beit, Gerhard-Mercator-Universitat, Duisburg, Deutschland, 2002.
[83] C. Finder, M. Wohlgemuth, C. Mayer, Analysis of Particle Size Distribution by
Particle Tracking, Part. Part. Syst. Charact., 2004, im Druck.
[84] M. B. Forstner, D. S. Martin, A. M. Navar, J. A. Kas, Simultaneous Single-
Particle Tracking and Visualization of Domain Structure on Lipid Monolayers,
Langmuir , 19, 2003.
[85] N. Roos, T. Luxbacher, T. Glinsner, K. Pfeiffer, H. Schulz, H.-C. Scheer, Nanoi-
moprint Lithography with a Commercial 4 Inch Bond System for Hot Embossing,
Proceedings of the SPIE , 4343, 2001.
[86] H.-C. Scheer, H. Schulz, A contribution to the flow behaviour of thin polymer
films during hot embossing lithography, Microelectronic Engineering , 56, 311 –
332, 2001.
[87] C. Finder, C. Mayer, H. Schulz, H.-C. Scheer, M. Fink, K. Pfeiffer, Non-contact
measurements for inspection and imprint depth control on nanoimprint lithogra-
phy, Proc. SPIE , 4764, 218 – 223, 2002.
[88] C. Finder, M. Beck, J. Seekamp, K. Pfeiffer, P. Carlberg, I. Maximov, F. Reuther,
E.-L. Sarwe, S. Zankovich, J. Ahopelto, L. Montelius, C. Mayer, C. Sotomayor-

LITERATURVERZEICHNIS 169
Torres, Fluorescence microscopy for quality control in nanoimrpint lithography,
Microelectronic Engingeering , 67 -68, 623 – 628, 2003.
[89] M. Muller, R. Zentel, T. Maka, G. Romanov, C. Sotomayor-Torres, Dye contai-
ning polymer beads as photonic crystals, Chem. Mater., 12, 2508 – 2512, 2000.
[90] E. L. Wehry (Hg.), Modern Fluorescence Spectroscopy , Bd. 2, Heyden, New York,
1976.
[91] E. L. Wehry (Hg.), Modern Fluorescence Spectroscopy , Bd. 3, Heyden, New York,
1981.
[92] H.-C. Scheer, H. Schulz, D. Lyebyedyev, Strategies for Wafer-Scale hot Embossing
Lithography, Proc. SPIE , 4349, 86 – 89, 2001.
[93] H.-C. Scheer, H. Schulz, T. Hoffmann, C. M. Sotomayor-Torres, Problems of
the naoimprinting technique for nanometer scale pattern definition, J. Vac. Sci.
Technol. B , 16(6), 3917 – 3921, 1998.
[94] R. W. Jaszewski, H. Schift, J. Gobrecht, P. Smith, Hot embossing in polymers
as a direct way to pattern resist, Microelectronic Engineering , 41/42, 575 – 578,
1998.
[95] D. Lyebyedyev, H. Schulz, H.-C. Scheer, Process Development for Nanoimprint
Lithography, Jena, 2000.
[96] L. J. Heyderman, H. Schift, C. David, I. Gobrecht, T. Schweizer, Flow beha-
viour of thin polymer films used for hot embossing lithography, Microelectronic
Engineering , 54, 229 – 245, 2000.
[97] T. Bailey, B. Smith, B. J. Choi, M. Meissl, S. V. Sreenivasan, J. G. Ekerdt, C. G.
Wilson, Step and flash imprint lithography: Defect analysis, J. Vac. Sci. Technol.,
B 19(6), 2806 – 28190, 2001.

170 LITERATURVERZEICHNIS
[98] W. Wu, B. Cui, X.-Y. Sun, W. Zhang, L. Zhuang, L. Kong, S. Y. Chou, Large area
high density quantized mangetic disks fabricated using nanoimprint lithography,
J. Vac. Sci. Technol. B , 16(6), 2897 – 2904, 1998.
[99] E. A. Collins, Measurements of Particle Size and Particle Size Distribution, P. A.
Lovell (Hg.), Emulsion Polymerization and Emulsion Polymers , Kap. 12, 385 –
436, Wiley, Chichester, 1997.
[100] T. Provder, Challenges in particle size distribution measurements past, present
and for the 21st century, Progress in Organic Coatings, 32, 143 – 153, 1997.
[101] H. Lange, Comparative Test of Methods to Determine Particle Size and Particle
Size Distribution in the Submicron Range, Part. Part. Syst. Charact., 12, 148 –
157, 1995.

Lebenslauf
Personliche Daten
Name Christiane Finder
Geburtstag 12. 03. 1972
Geburtsort Berlin
Familienstand ledig
Schulbildung
08/1978–07/1983 Lauterbach-Grundschule in Berlin
08/1983–07/1991 Gymnasium Ernestinenschule zu Lubeck
Abschluss: Allgemeine Hochschulreife
Studium
10/1991–11/1999 Studium der Chemie
an der Gerhard-Mercator-Universitat Duisburg
Abschluss: Diplom-Chemikerin
01/2000 Promotion im Fachgebiet der Physikalische Chemie an der Gerhard-
Mercator-Universitat Duisburg
Thema: Optische Beobachtung von oberflachengebundenen und frei
beweglichen Nanopartikeln
10/2000–03/2003 Zusatzstudium der Wirtschaftswissenschaften
an der Fernunniversitat Hagen
Abschluss: Vordiplom

Beruflicher Werdegang
03/1996–12/1997 Studentische Hilfskraft im Fachbereich Maschinenbau an der Gerhard-
Mercator-Universitat Duisburg
02/1997–04/1997 Werkvertrag im Fachbereich Maschinenbau an der Gerhard-Mercator-
Universitat Duisburg
05/2000–12/2002 Task Leader fur den Standort Duisburg im EU-Projekt CHANIL
(”Chances for a Nanoimprint Lithography based fabrication“)
01/2003–04/2004 Wissenschaftliche Mitarbeiterin im Fachgebiet der Physikalische
Chemie an der Gerhard-Mercator-Universitat Duisburg
Publikationen
2002”Non-contact fluorescence measurements for inspection and imprint
depth control in nanoimprint lithography“
Ch. Finder, C. Mayer, H. Schulz, H.-C. Scheer, M. Fink, K. Pfeiffer
Proc. SPIE, 4764 (2002), 218 - 223
2003”Fluorescence microscopy for quality control in nanoimprint litho-
graphy“
Ch. Finder, M. Beck, J. Seekamp, K. Pfeiffer, P. Carlberg, I. Maxi-
mov, F. Reuther, E.-L. Sarwe, S. Zankovich, J. Ahopelto, L. Mon-
telius, C. Mayer, C.M. Sotomayor Torres
Microelectronic Engineering, 67 - 68 (2003), 623 - 628
2004”Analysis of Particle Size Distribution by Particle Tracking“
Ch. Finder, M. Wohlgemuth, C. Mayer
Part. Part. Syst. Charact., 21 (2004), 372 - 378
Duisburg, 27.09.2004

Selbstandigkeitserklarung
Hiermit erklare ich, die vorliegende Arbeit selbstandig ohne fremde Hilfe verfasst zu
haben und nur die angegebene Literatur und Hilfsmittel verwendet zu haben.
Christiane Finder
27. September 2004






![chr tab elektro vorspann - Buch.de - Bücher versandkostenfrei · SR-Speicher → 261 ... EN AW – 1199 [Al99,99] EN ... Oberfl ächenprüftechnik 759 Mess- und Prüftechnik Oberfl](https://img.pdfslide.org/doc/110x75/5e053f243f33f86e1164e096/chr-tab-elektro-vorspann-buchde-bcher-versandkostenfrei-sr-speicher-a-261.jpg)






















