Embed Size (px)
Citation preview

Pflanzenfarbstoffe: Thomas Niedenthal
Hinweis Bei dieser Datei handelt es sich um eine Wissenschaftliche Hausarbeit (1. Staatsexamensarbeit), die am Fachbereich Chemie der Philipps-Universität Marburg angefertigt wurde. Weitere Wissenschaftliche Hausarbeiten können auf der Seite http://www.chids.de/veranstaltungen/wiss_hausarbeit.html eingesehen und heruntergeladen werden. Zudem stehen auf der Seite www.chids.de weitere Versuche, Lernzirkel und Experimentalvortäge bereit.
Dr. Ph. Reiß, im Juli 2007

Erste Staatsprüfung für das Lehramt an Gymnasien
Wissenschaftliche Hausarbeit im Fach Chemie
„Pflanzenfarbstoffe“
vorgelegt von
Thomas Niedenthal
Hersfelder Str. 18
36151 Burghaun
Gutachter: Dr. Philipp Reiß
Mai 2007

Inhaltsverzeichnis
1. Einleitung 1
2. Fachwissenschaftliche Aspekte 3 2.1. Farbigkeit - was ist das? 3
2.2. Die Lichtreaktion der Photosynthese 9
2.3. Chlorophyll 15 2.3.1. Varianten - Molekülstruktur 15
2.3.2. Extraktion von Blattgrün 19
Versuch 1: Kaltextraktion von Blattgrün 19
Versuch 2: Heißextraktion von Blattrün 21
Vergleich der Extraktionsmethoden 22
2.3.3. Versuche zum Nachweis charakteristischer
Strukturelemente
23
Versuch 3: Darstellung der Phaeophytine 23
Versuch 4: Nachweis des Magnesium-Ions 25
Versuch 5: Verseifung der Chlorophylle 27
2.3.4. Versuch zum Nachweis der Sauerstoffproduktion 30
Versuch 6: Sauerstoffnachweis mit Indigo 30
2.4. Carotinoide 34 2.4.1. Varianten - Molekülstruktur 34
2.4.2. Extraktion des bekanntesten Carotinoids: β-Carotin 38
Versuch 7: Extraktion von β-Carotin aus klein-
geschnittenen Möhren mit Aceton
39
Versuch 8: Extraktion von β-Carotin aus klein-
geschnittenen Möhren mit n-Heptan
40
Versuch 9: Extraktion von β-Carotin aus
geraspelten Möhren mit Aceton
41
Versuch 10: Extraktion von β-Carotin aus
getrockneten Möhren mit Soxhlet-
Apparatur
42
Vergleich der verschiedenen Methoden 44

2.4.3. Nachweis des Strukturelementes Doppelbindung 45
Versuch 11: Elektrophile Addition von Brom an
β-Carotin
45
2.4.4. Schutzfunktion des β-Carotins für das Chlorophyll 49
Versuch 12: β-Carotin als Radikalfänger 49
Versuch 13: Desaktivierung von Singulett-
Sauerstoff
54
2.5. Chromatographie von Blattfarbstoffen 60 2.5.1. Erläuterung der unterschiedlichen Methoden 60
2.5.2. Praktische Umsetzung 64
Versuch 14: Chromatographie nach Tswett 64
Versuch 15: Chromatographie mit Tafelkreide 66
Versuch 16: Herstellung von DC-Karten 68
Versuch 17: Dünnschichtchromatographie 69
Versuch 18: Präparative Dünnschichtchromato-
graphie und photometrische
Untersuchung
71
3. Methodisch-didaktische Umsetzung 87
3.1. Ziele des Chemieunterrichtes 87
3.2. Didaktische Vorüberlegungen zur Behandlung
des Themas „Pflanzenfarbstoffe“ im Unterricht
88
3.3. Grobplanung der Unterrichtseinheit 90
4. Schlusswort 94
5. Literatur 95
6. Abbildungsverzeichnis 97
7. Anlagen 101 Anlage 1: R- und S-Sätze 102
Anlage 2: Versuchsvorschrift „Kaltextraktion von
Blattfarbstoff“
105
Anlage 3: Versuchsvorschrift „Heißextraktion von
Blattfarbstoff“
106
Anlage 4: Versuchsvorschrift „Darstellung der Phaeophytine“ 107

Anlage 5: Versuchsvorschrift „Nachweis des Magnesium-
Ions“
108
Anlage 6: Versuchsvorschrift „Verseifung der Chlorophylle“ 109
Anlage 7: Versuchsvorschrift „Nachweis der Sauerstoff-
produktion“
110
Anlage 8: Versuchsvorschrift „Extraktion von β-Carotin aus
kleingeschnittenen Möhren mit Aceton“
111
Anlage 9: Versuchsvorschrift „Extraktion von β-Carotin aus
kleingeschnittenen Möhren mit n-Heptan“
112
Anlage 10: Versuchsvorschrift „Extraktion von β-Carotin aus
geraspelten Möhren mit Aceton“
113
Anlage 11: Versuchsvorschrift „Extraktion von β-Carotin aus
getrockneten Möhren mit Soxhlet-Apparatur“
114
Anlage 12: Versuchsvorschrift „Elektrophile Addition von
Brom an β-Carotin“
115
Anlage 13: Versuchsvorschrift „β-Carotin als Radikalfänger“ 116
Anlage 14: Versuchsvorschrift „Desaktivierung von
Singulett-Sauerstoff“
117
Anlage 15: Versuchsvorschrift „Chromatographie nach
Tswett“
119
Anlage 16: Versuchsvorschrift „Chromatographie mit
Tafelkreide“
121
Anlage 17: Versuchsvorschrift „Herstellung von DC-Karten“ 122
Anlage 18: Versuchsvorschrift „Dünnschichtchromato-
graphie“
123
Anlage 19: Versuchsvorschrift „Präparative Dünnschicht-
chromatographie und photometrische
Untersuchung“
124

1. Einleitung
Das subjektive Empfinden eines Menschen wird ganz entscheidend
mitbestimmt durch die unterschiedlichsten Farbeindrücke. Eine besondere
Rolle spielt hier die Farbenvielfalt in der Natur. Pflanzen synthetisieren
viele unterschiedliche Farbstoffe.
Ich möchte einige Beispiele anführen: Tomate – Lycopin (rot), Curcuma-
Wurzel – Curcumin (gelb), Kornblume – Cyanidin (blau), Karotte – Carotin
(orange), alle grünen Pflanzenteile – Chlorophyll.
Im Rahmen einer wissenschaftlichen Hausarbeit ist es nicht möglich,
Pflanzenfarbstoffe umfassend zu behandeln. Deshalb beschränke ich mich
in meiner Arbeit auf Pflanzenfarbstoffe, die in grünen Blättern enthalten
sind: die Chlorophylle und die Carotinoide.
Zunächst werde ich darstellen, warum ein Stoff farbig ist und in welcher
Beziehung Stoff, Licht und resultierende Farbe stehen. Dies werde ich in
Kapitel 2.1. erläutern.
In den Kapiteln 2.3., 2.4. und 2.5. gehe ich auf Chlorophylle, Carotinoide
und Chromatographie ein. Zu Beginn eines jeden Kapitels stelle ich die
wesentlichen wissenschaftlichen Erkenntnisse dar. Im Anschluss daran
folgen die entsprechenden Versuche. Bei der Auswahl der Versuche habe
ich darauf geachtet, dass diese von Schülern – überwiegend selbständig –
durchgeführt werden können. Dies impliziert die schnelle und einfache
Durchführbarkeit der Versuche sowie die Verwendung von möglichst
gefahrstoffunbedenklichen Chemikalien.
Ich habe mich auf schulrelevante Versuche beschränkt, weil ich im
folgenden Kapitel 3. die Behandlung des Themas „Pflanzenfarbstoffe“ in
der Schule darstellen möchte.
Zunächst müssen didaktische Vorüberlegungen bzgl. Lehrplan,
Lerngruppengröße und Lernvoraussetzungen der Schüler angestellt werden.
Es müssen eindeutige Lernziele formuliert werden. Sie müssen dem
Leistungsvermögen der Schüler angepasst sein. Dies hat die didaktische
Reduktion des komplexen Themengebietes zu Folge.
1

Im letzten Teil des Kapitels geht es um die methodisch-didaktische
Umsetzung. Ich werde Mittel und Wege aufzeigen, wie dieses doch recht
komplexe Stoffgebiet Schülern nahe gebracht werden kann.
2

2. Fachwissenschaftliche Aspekte
2.1. Farbigkeit – was ist das?
Der Farbeindruck eines Stoffes entsteht dadurch, dass dieser Licht aus dem
sichtbaren Bereich des Spektrums (λ=380-780 nm) selektiv zu absorbiert,
den der Rest reflektiert oder durchlässt.
Der sichtbare Bereich zwischen 380 und 780 nm ist jedoch nur ein kleiner
Teil der elektromagnetischen Strahlung, wie folgende Abbildung zeigt:
Wellenlänge in cm
10-10 10-8 10-6 10-4 10-2 1 100
Gamma-
strahlen
Röntgen-
strahlen
Infrarot-
strahlen
UV
Mikrowellen
Radio-
wellen
400 500 600 700 800 Wellenlänge in nm (sichtbarer Bereich)
Abb. 1: Spektrum elektromagnetischer Wellen
Trifft Licht aus dem Bereich des sichtbaren Spektrums auf einen
Gegenstand, so kann das komplette Licht absorbiert, reflektiert bzw.
durchgelassen werden. Wird das komplette Licht absorbiert, so erscheint
der Gegenstand schwarz. Wird das komplette Licht reflektiert bzw.
durchgelassen, so erscheint der Gegenstand farblos. Wird Licht nur zum
Teil absorbiert, so ergibt die Summe der Spektralfarben, abzüglich der
3

absorbierten Spektralfarben, den Farbeindruck. Man spricht auch von der
Komplementärfarbe.[23]
Spektralfarben
violett indigo blau blau-grün
grün gelb-grün
gelb orange rot
gelb-grün
gelb orange rot purpur violett indigo blau blau-grün
nach Absorption einer Spektralfarbe wahrgenommene Komplementärfarbe
Abb. 2: Übersicht über Spektralfarben und ihre zugehörige Komplementärfarbe
Farbstofftheorie
Schon im 19. Jahrhundert vermuteten Wissenschaftler, dass die
unterschiedlichen Farbeindrücke durch verschiedenartige Moleküle bewirkt
werden. Es muss Moleküle geben, die kurzwelliges, energiereiches Licht
aufnehmen können und wieder andere, die langwelliges, energiearmes Licht
absorbieren können. Man sah einen Zusammenhang zwischen der Farbigkeit
eines Stoffes und der Struktur seiner Moleküle.
Bereits im Jahre 1868 gelang den deutschen Chemikern Carl Gräbe und
Carl Liebermann die Entfärbung bekannter Farbstoffe durch Reduktion. Sie
formulierten die folgenden Annahme, konnten sie aber noch nicht beweisen.
„Diejenigen Farbstoffe, zu welchen sich Wasserstoff hinzuaddirt, müssen
entweder Elemente mit unvollständig gesättigten Valenzen besitzen, oder es
sind in ihnen irgend welche Atome in einer innigeren Lagerung, als zu
ihrem Zusammenhange im Molecül nothwendig ist, enthalten.“[18]
Heute würde man diesen Sachverhalt mit dem Begriff
„Mehrfachbindungen“ beschreiben.
Bereits acht Jahre später, also 1876, veröffentlichte der deutsche Chemiker
Otto Nikolaus Witt die „Chromophortheorie“. Er ging davon aus, dass die
Farbstoffnatur bedingt ist durch die gleichzeitige Anwesenheit einer
4

farbstoffgebenden und einer salzbildenden Gruppe in einem Molekül. Die
farbstoffgebende Gruppe bezeichnete er als „Chromophor“. Ein Molekül,
das ein Chromophor besitzt, dem aber noch die salzbildende Gruppe fehlt,
nannte er „Chromogen“. Als Beispiel führte er Stickstoffdioxid, das
Chromophor des Nitroanilins und des Nitrophenols, an, während
Nitrobenzol das Chromogen für diese Moleküle darstellt. Erst im Jahre 1888
– also 12 Jahre später – nannte er die salzbildenden Gruppen
„Auxochrome“. Diese Bezeichnung ist heute noch gebräuchlich.[23]
Weil sich die praktischen Möglichkeiten zur Strukturaufklärung von
Farbstoffen ständig verbesserten, führte die Chromophortheorie in ihrer
Anwendung zu Schwierigkeiten. So ließen sich z.B. die Farbigkeit der
Triphenylmethanfarbstoffe oder auch die der Xanthenfarbstoffe nicht
ausreichend mit der Chromophortheorie deuten. Rudolf Nietzki
veröffentlichte deshalb um 1890 seine „Chinonhypothese“. Er hatte erkannt,
dass sowohl die von Witt betrachteten Farbstoffe, als auch die
Triphenylmethan- und Xanthenfarbstoffe mit mindestens einer chinoiden
Teilstruktur formulierbar sind.
Zu Beginn des 20. Jahrhunderts war es schließlich möglich, die Masse eines
Teilchens, das die Energie sichtbaren Lichtes in Form von
Eigenschwingungen aufgenommen hatte, zu berechnen. Man schloss
daraus, dass Elektronen am Farbeindruck eines Stoffes beteiligt sein
müssen. Im Jahr 1908 teilte J. Stark die Valenzelektronen in gesättigte,
ungesättigte und gelockerte Valenzelektronen ein. Die Anwendung des
klassischen Valenzbegriffes bei den Farbstoffen gelang jedoch nicht. Mit
der zuvor von Friedrich Karl Johannes Thiele entwickelten
„Partialvalenzhypothese“ versuchte H. Kaufmann das Problem zu lösen. Er
formulierte hierzu einen Valenzausgleich außerhalb des Systems, bzw. um
das System herum:
„Die benzoiden und chinoiden Formeln der auxochromhaltigen Nitrokörper
stellen zwei Gegensätze dar, zwischen welchen alle möglichen Uebergänge
bestehen. Die dazwischen liegenden Zustände lassen sich mit Hülfe von
Partialvalenzen ausdrücken. Die Nitrophenolsalze z.B. erhalten demnach
etwa die Formel:
5

NO2
NaO
Abb. 3: Partialvalenz eines Nitrophenolsalzes
wobei die punktirte Linie einen Ausgleich freier Valenz zwischen
Auxochrom und Chromophor darstellt.“[18]
Was jedoch zur damaligen Zeit niemand erkannte oder wagte zu
formulieren, war der Valenzausgleich innerhalb des Systems. Hinzu kam
großer Widerstand gegen die Vermutung, auch organische Moleküle
könnten ionisch sein. Diese Annahme verstärkte sich erst nach dem Ersten
Weltkrieg.
Weiter geht H. Kaufmann auf den Begriff „Auxochrom“ zurück. Aufgrund
einiger gegenläufiger Effekte der Chromophore nannte er diese
„Antiauxochrome“.
Auch K. Gebhard ging im Jahr 1911 davon aus, dass sich Farbstoffe durch
weiträumig verbrückte Partialvalenzen auszeichnen. Er formulierte die
Partialvalenzen innerhalb des Moleküls. An diese Theorie knüpfte auch das
Polymethinkonzept von W. König an. Auch er war der Auffassung, dass
nicht eine Partialvalenzbrücke um das System herum sondern ein
Valenzausgleich innerhalb des Systems die Farbigkeit eines organischen
Moleküls verursacht.
Etwa zur gleichen Zeit wie W. König arbeiteten R. Witzinger und
W. Dilthey an einer Theorie, welche direkt an die Chromophortherie von
O. Witt und R. Nietzky anknüpfte. Die beiden Wissenschaftler betrachteten
die koordinative Sättigung an jedem einzelnen Atom innerhalb des
Moleküls. Atome, in denen sie den eigentlichen Chromophor des Systems
sahen, markierten sie mit einem Punkt („Bonner Punkt“). Hieraus leitet sich
auch der Begriff „Bonner Farbstofftheorie“ ab. Beispielsweise sollte das
zentrale Carbeniumion der Triphenylmethanfarbstoffe deren eigentlicher
6

Chromophor sein. Bei dieser Theorie wurde aber immer noch verkannt, dass
das komplette Elektronensystem eines Moleküls für die Farbe maßgeblich
ist.
Etwa um 1930 wurde die „Mesomerielehre“ herangezogen, um die
Abhängigkeit von Struktur und Farbe zu klären. Sie war zuvor von
deutschen, englischen und amerikanischen Wissenschaftlern aufgestellt
worden. Mit Hilfe der Mesomerielehre konnte bewiesen werden, dass nicht
einzelne Atome oder Atomgruppen der Chromophor eines Farbstoffes sind
sondern das komplette π-Elektronensystem. Der Farbeindruck ist abhängig
von der Anzahl der Elektronen und deren Verschiebbarkeit. Hat ein System
z.B. eine große Anzahl von π-Elektronen, die leicht beweglich sind – es sind
also in der Theorie viele mesomere Grenzstrukturen formulierbar – so ist die
zur Anregung benötigte Energie gering und die erscheinende Farbe ist
dunkel. Umgekehrt ist die Farbe hell, wenn viel Energie benötigt wird, um
wenige, schwer bewegliche π-Elektronen anzuregen. Die freien
Elektronenpaare eventuell vorhandener Heteroatome spielen beim
Farbeindruck nur eine untergeordnete Rolle.[18]
Grundsätzlich benötigt ein organischer Farbstoff ein Kohlenstoffgerüst mit
konjugierten Doppelbindungen, an welchem sich geeignete Substituenten
befinden. Diese Substituenten werden nach ihren mesomeren Eigenschaften
unterschieden. In diesem Zusammenhang wurden auch die bereits geprägten
Begriffe Auxochrom, Chromophor und Chromogen (O. Witt) sowie der
Begriff Antiauxochrom (H. Kaufmann) neu definiert. Als Auxochrome
werden seither Substituenten bezeichnet, welche Elektronendonatoren sind,
also einen +M-Effekt ausüben. Analog sind Antiauxochrome als
Elektronenakzeptoren definiert. Sie üben einen –M-Effekt aus. Der
Chromophor ist der Teil des Moleküls, der die leicht anregbaren
π-Elektronen enthält. Die Gesamtheit aus Chromophor, Auxochrom und
Antiauxochrom wird als Chromogen bezeichnet.
Heute zieht man die MO-Theorie zur Deutung der Farbigkeit heran.
Licht ist in der Lage, Elektronen anzuregen. Diese gehen dabei vom highest
occupied molecul orbital (HOMO) in das lowest unoccupied molecul orbital
(LUMO), den ersten angeregten Zustand, über. Beim Zurückfallen in den
Grundzustand wird die aufgenommene Energie meist als Wärme frei.[11]
7

HOMO
LUMO
∆ E
Grundzustand 1. angeregterZustand
Abb. 4: Übergang von HOMO nach LUMO am Beispiel des Butadiens
Die Energiedifferenz zwischen HOMO und LUMO, ∆E, entspricht dabei
genau der Wellenlänge λ des absorbierten Lichtes. Dabei gilt:
∆E = h • ν = λhc λ =
Ehc∆
mit: ∆E: Energiedifferenz zwischen HOMO und LUMO in kJ/mol
h: Plancksche Konstante, h = 6,626 • 10-34 J • s
ν: Frequenz in s-1
c: Lichtgeschwindigkeit, c = 2,998 • 108 m • s-1
λ: Wellenlänge in nm
Man erkennt, dass nach obiger Gleichung die Absorptionswellenlänge
umgekehrt proportional zum Abstand zwischen Grundzustand und
angeregtem Zustand ist. Somit wird die Absorption um so langwelliger, je
kleiner ∆E ist.[23]
8

2.2. Die Lichtreaktion der Photosynthese
Unter Photosynthese versteht man die Fähigkeit grüner Pflanzen und
Bakterien, mit Hilfe von Lichtenergie und anorganischen Vorstufen
organische Verbindungen zu synthetisieren. Man denkt dabei sofort an die
Kohlenstoffassimilation, bei der aus dem in der Luft vorkommenden
Kohlenstoffdioxid Kohlenhydrate gebildet werden. Die Lichtenergie wird
genutzt, um Ammoniumstickstoff aus aufgenommenem Nitrat
(Nitratassimilation) zu bilden sowie Sulfat zu Sulfid zu reduzieren
(Sulfatassimilation). Auch der Kohlenstoff im Kohlenstoffdioxid und der
Stickstoff im Nitrat werden bei der Photosynthese reduziert. Die zur
Reduktion benötigten Elektronen entstammen bei grünen Pflanzen dem
Wasser. Bei der Lichtreaktion (Teilreaktion der Photosynthese) werden aus
dem Photosynthesepigment Chlorophyll nach der Absorption von Licht
Elektronen freigesetzt und über Elektronentransportketten auf Ferredoxin
übertragen. Das dadurch reduzierte Ferredoxin dient nun als
Elektronendonator bei der Schwefel- und Stickstoffassimilation. Es ist auch
in der Lage, oxidierte Purinnucleotide (NADP+) unter Bildung der
Reduktionsmittel NADPH + H+ zu reduzieren. Die Lichtreaktion läuft an
den Thylakoidmembranen, welche sich im Stroma der Chloroplasten
befinden, ab. Außerdem wird bei der Lichtreaktion auch ATP gebildet.
Dieses und das Reduktionsmittel NADPH werden zur
Kohlenstoffassimilation genutzt. Die Synthese von Kohlenhydraten aus
Kohlenstoffdioxid wird auch als „Dunkelreaktion“ bezeichnet. Der Name
Dunkelreaktion rührt daher, dass diese Reaktion nicht direkt lichtabhängig
ist. Sie würde auch bei entsprechender Verfügbarkeit von ATP und NADPH
im Dunkeln ablaufen. Jährlich werden etwa 275 Milliarden Tonnen
Kohlenstoffdioxid durch Pflanzen gebunden. Die dazu benötige Menge an
Lichtenergie liegt bei 1025 J pro Jahr.[20][21]
Photosynthesepigmente – Chlorophylle und Carotinoide
Der Prozess der Photosynthese beginnt mit der Absorption von Photonen.
Diese Absorption erfolgt durch Photosynthesepigmente, die dabei in einen
angeregten Zustand übergehen. Hierbei sind die Chlorophylle von zentraler
9
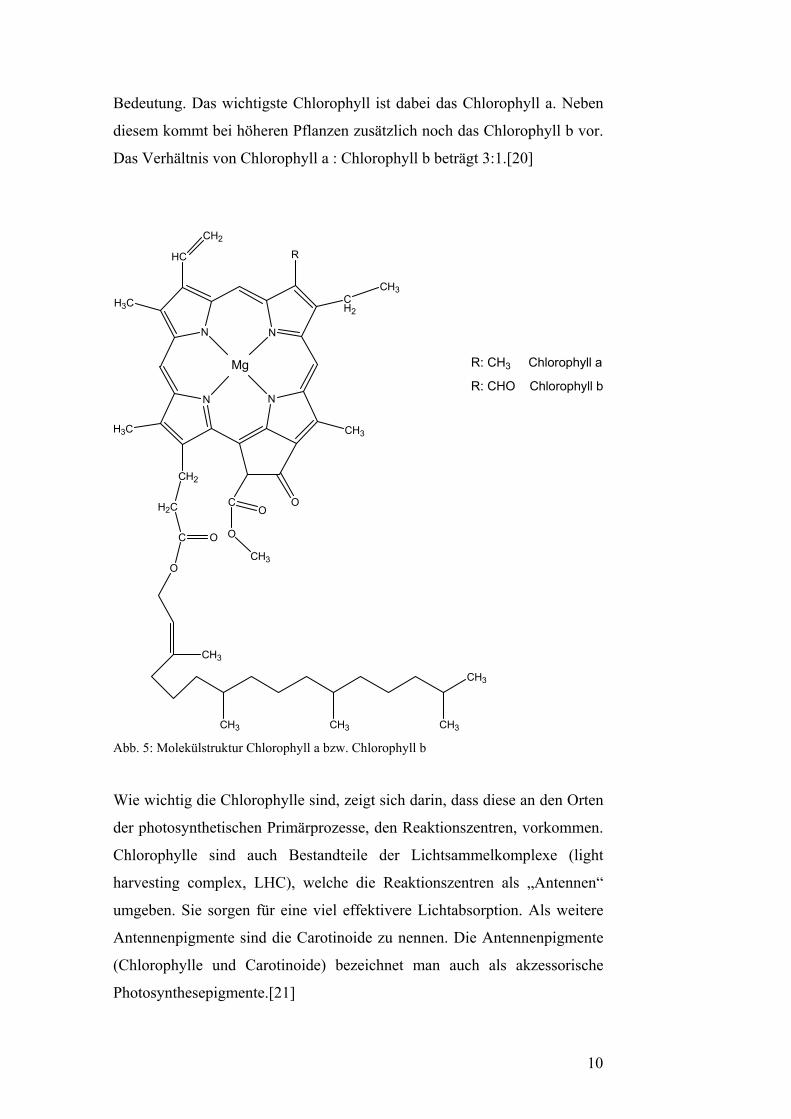
Bedeutung. Das wichtigste Chlorophyll ist dabei das Chlorophyll a. Neben
diesem kommt bei höheren Pflanzen zusätzlich noch das Chlorophyll b vor.
Das Verhältnis von Chlorophyll a : Chlorophyll b beträgt 3:1.[20]
N
N N
N
O
H3C
HC
CH2
R
CH2
CH3
CH3
C
O
O
CH3
CH2
H2C
C
O
CH3
CH3 CH3
CH3
CH3
O
H3C
Mg R: CH3 Chlorophyll a
R: CHO Chlorophyll b
Abb. 5: Molekülstruktur Chlorophyll a bzw. Chlorophyll b
Wie wichtig die Chlorophylle sind, zeigt sich darin, dass diese an den Orten
der photosynthetischen Primärprozesse, den Reaktionszentren, vorkommen.
Chlorophylle sind auch Bestandteile der Lichtsammelkomplexe (light
harvesting complex, LHC), welche die Reaktionszentren als „Antennen“
umgeben. Sie sorgen für eine viel effektivere Lichtabsorption. Als weitere
Antennenpigmente sind die Carotinoide zu nennen. Die Antennenpigmente
(Chlorophylle und Carotinoide) bezeichnet man auch als akzessorische
Photosynthesepigmente.[21]
10

Chlorophylle absorbieren Licht im Bereich von 400 – 480 nm (blau) und
550 – 700 nm (gelb bis rot). Im Bereich zwischen 480 nm und 550 nm ist
nur eine sehr geringe Lichtabsorption durch Chlorophyll feststellbar. Da
dies der Bereich des grünen Lichtes ist, erscheinen chlorophyllhaltige
Pflanzenteile grün. Man spricht man auch von der „Grünlücke“. Diese
Lücke wird teilweise durch das Photosynthesepigment β-Carotin, ein
Carotinoid geschlossen, das genau in diesem Bereich ein lokales
Absorptionsmaximum besitzt.[5]
Wichtig für die Lichtabsorption der Photosynthesepigmente ist das
Vorhandensein von delokalisierten π-Elektronen. Im Falle der Chlorophylle
liegen diese Elektronen in einem Ringsystem (Porphyrinring) vor. Dieses
ermöglicht es ihnen nicht nur zu oszillieren sondern zusätzlich noch im
Ringsystem zu zirkulieren. Durch geringe Energien ist es möglich, die π-
Elektronen aus dem Grundzustand (S0) auf höhere Energieniveaus
anzuheben. Hierbei geht das Molekül in einen angeregten Zustand über.
Von zentraler Bedeutung sind der erste Singulettzustand (S1), welcher einer
Rotabsorption entspricht, und der zweite Singulettzustand (S2), der einer
Blauabsorption entspricht. Beiden Zuständen ist gemeinsam, dass zwei
ungepaarte Elektronen mit antiparallelem Spin vorliegen. Beim Prozess der
Anregung hat also keine Spinumkehr stattgefunden. Ein weiterer wichtiger
angeregter Zustand ist der erste Triplettzustand (T1). Dieser kann nur aus
dem ersten Singulettzustand heraus erreicht werden, da dessen Lebensdauer
mit etwa 15 • 10-6 s lang genug für eine Spinumkehr ist. Im Triplettzustand
liegen folglich zwei ungepaarte Elektronen mit parallelem Spin vor. Der
zweite Singulettzustand ist mit 10-12 s zu kurzlebig für eine Spinumkehr.
Der Triplettzustand des Chlorophylls ist für die Photosynthese nicht
bedeutsam. Beim Übergang in den Grundzustand kann die Energie nur in
Form von Wärme und Phosphoreszenz abgegeben werden. Allerdings ist ein
Chlorophyllmolekül im Triplettzustand in der Lage, Sauerstoff zu einem
Singulett-Sauerstoff anzuregen. Dieser ist chemisch sehr reaktiv und wirkt
gewebeschädigend. Das β-Carotin ist in der Lage, die Energie vom
Singulett-Sauerstoff zu übernehmen, ohne dabei selbst zerstört zu werden.
Man spricht auch von „quenchen“. Dadurch wird eine Schädigung des
Gewebes vermieden. [20][21]
11
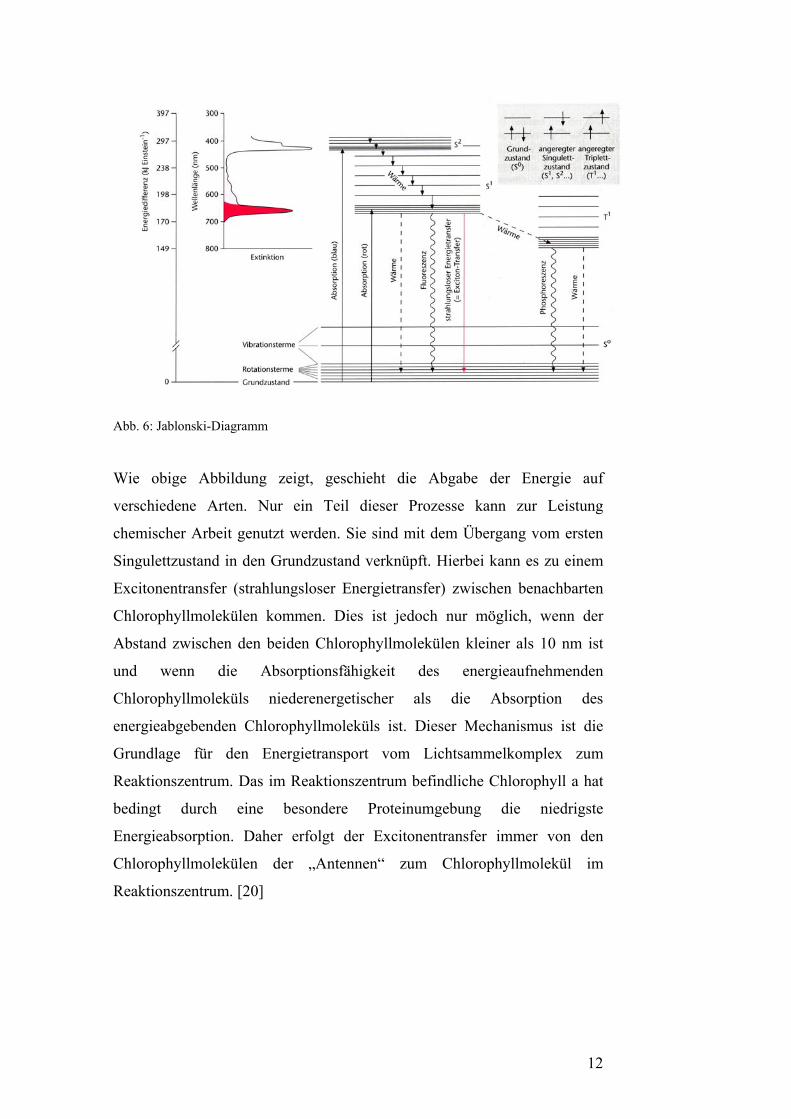
Abb. 6: Jablonski-Diagramm
Wie obige Abbildung zeigt, geschieht die Abgabe der Energie auf
verschiedene Arten. Nur ein Teil dieser Prozesse kann zur Leistung
chemischer Arbeit genutzt werden. Sie sind mit dem Übergang vom ersten
Singulettzustand in den Grundzustand verknüpft. Hierbei kann es zu einem
Excitonentransfer (strahlungsloser Energietransfer) zwischen benachbarten
Chlorophyllmolekülen kommen. Dies ist jedoch nur möglich, wenn der
Abstand zwischen den beiden Chlorophyllmolekülen kleiner als 10 nm ist
und wenn die Absorptionsfähigkeit des energieaufnehmenden
Chlorophyllmoleküls niederenergetischer als die Absorption des
energieabgebenden Chlorophyllmoleküls ist. Dieser Mechanismus ist die
Grundlage für den Energietransport vom Lichtsammelkomplex zum
Reaktionszentrum. Das im Reaktionszentrum befindliche Chlorophyll a hat
bedingt durch eine besondere Proteinumgebung die niedrigste
Energieabsorption. Daher erfolgt der Excitonentransfer immer von den
Chlorophyllmolekülen der „Antennen“ zum Chlorophyllmolekül im
Reaktionszentrum. [20]
12

Antenne Reaktions- Antenne zentrum
Licht Licht
Abb. 7: Schematische Darstellung des Excitonentransfers von den Antennen zum
Reaktionszentrum
Nur in seltenen Fällen wird das Chlorophyllmolekül im Reaktionszentrum
direkt durch ein Photon angeregt. Da das Chlorophyllmolekül die
aufgenommene Energie an kein Molekül mit niedrigerer Energieabsorption
weiterleiten kann, gibt es unter Bildung eines positiv geladenen Radikals ein
Elektron ab. Unter Aufnahme eines Elektrons aus der Wasserspaltung (s.u.)
geht das Chlorophyllmolekül wieder in den Grundzustand zurück. Bei
optimalen Bedingungen läuft dieser Prozess etwa 100 – 200 mal pro
Sekunde ab.
Chl a Chl a→∆E +• + e-
Der Prozess der Ladungstrennung ist der entscheidende Schritt bei der
Photosynthese. Die kurzlebige Anregungsenergie der Photonen ist in ein
elektrisches Potential umgesetzt worden, welches wesentlich langlebiger ist.
Dieses Potential kann nun in chemische Arbeit umgewandelt werden. [20]
Beschreibung des Elektronentransportes
Bereits 1937 stellte R. Hill bei Untersuchungen von belichteten
Blattextrakten fest, dass diese in Gegenwart von künstlichen
Elektronenakzeptoren (A), wie z.B. Fe3+, Disauerstoff entwickeln. Bei
dieser nach ihm benannten „Hill-Reaktion“ wird als Elektronendonator
13

ausschließlich Wasser benötigt. Kohlenstoffdioxid ist an dieser Reaktion
nicht beteiligt.
2 H2O + 4 A 4 A →Licht - + 4 H+ + O2
Daraus folgt, dass der bei der Photosynthese entwickelte Sauerstoff aus dem
Wasser stammt und nicht aus dem Kohlenstoffdioxid. Die Reduktion von
Kohlenstoffdioxid zu Kohlenhydraten ist also ein Prozess, den man von der
Lichtreaktion trennen muss, die Dunkelreaktion (s.o.).
Untersuchungen haben gezeigt, dass der natürliche Elektronenakzeptor der
„Hill-Reaktion“ NADP+ ist. [20]
N
H
C
H
H
H
NH2
O
2 H2O + 2
N
C
H
H
H
NH2
O
+ 2 H+ + O2
H H
2
NADP+ NADPH
Es werden zwei in Serie geschaltete Lichtreaktionen benötigt, um NADP+
mit den Elektronen des Wassers zu reduzieren. Diese Reaktionen laufen in
den Photosystemen I und II ab, welche nach der Reihenfolge ihrer
Entdeckung nummeriert wurden. Tatsächlich beginnt die Photosynthese mit
der Anregung eines Chlorophyll-Moleküls im Photosystem II. Die
aufgenommene Energie wird von dort über Elektronentransportketten auf
den Cytochrom-b6/f-Komplex übertragen. Über andere
Elektronentransportketten gelangen die Elektronen schließlich zum
Chlorophyllmolekül im Reaktionszentrum vom Photosystem I. [21]
Die Chlorophyllmoleküle der Reaktionszentren der jeweiligen
Photosysteme unterscheiden sich in ihrem Absorptionsverhalten. Das
Chlorophyllmolekül im Reaktionszentrum des Photosystems II absorbiert
maximal bei 680 nm. Man bezeichnet es deshalb auch als P680. Da das
14

Chlorophyllmolekül im Reaktionszentrum des Photosystems I bei 700 nm
maximal absorbiert, wird es analog P700 genannt. [21]
2.3. Chlorophyll
2.3.1. Varianten - Molekülstruktur
Der Begriff „Chlorophyll“ leitet sich aus dem griechischen ab und bedeutet
„grünes Blatt“.
Erst zu Beginn des 20. Jahrhunderts begannen die Forschungen zur
Strukturaufklärung des Chlorophylls. Der deutsche Chemiker Richard
Willstätter stellte zu dieser Zeit erste Forschungen zu Photosynthese und
Chlorophyll sowie dem strukturell ähnlichen Häm an. [8]
Im Jahr 1940 konnte der Chemiker Hans Fischer schließlich die Struktur des
Chlorophylls aufklären. Hierzu zerlegte er ein Chlorophyllmolekül in
kleinere definierte Bruchstücke. Deren Struktur bewies er durch ihre
Synthese. [8]
Zwanzig Jahre später konnten die US-amerikanischen Chemiker
Woodward, Strell und Treibs die von Fischer entwickelte Chlorophyllformel
durch zwei Totalsynthesen beweisen. [8]
Einteilung
Grundbaustein der Chlorophylle ist das Pyrrol.
N
H
Abb. 8: Molekülstruktur Pyrrol
Sind diese Pyrrole über Methinbrücken zu einem Ring geschlossen, so
spricht man von einem Porphyrin. [21]
15

N
NH N
HN
Abb. 9: Molekülstruktur Porphyrin
Chlorophyll enthält zusätzlich noch ein Magnesium-Ion als Zentralatom.
Jedes Stickstoffatom der vier Pyrrolringe ist kovalent an das Magnesium
gebunden. Es handelt sich um vier gleichwertige, nicht unterscheidbare
Bindungen. Deshalb bezeichnet man den Porphyrinring als vierzähnigen
Chelatliganden. Außerdem besitzt Chlorophyll noch einen
charakteristischen isozyklischen Fünferring sowie verschiedene
Substituenten an den Pyrrolringen. [7][22]
16
N
N N
N
O
H3C
HC
CH2
R1
CH2
CH3
CH3
CH2
H2C
C
CH3
CH3 CH3
CH3
CH3
O
H3C
Mg
7
A B
CD
17
O
Phytol
E
R2R3
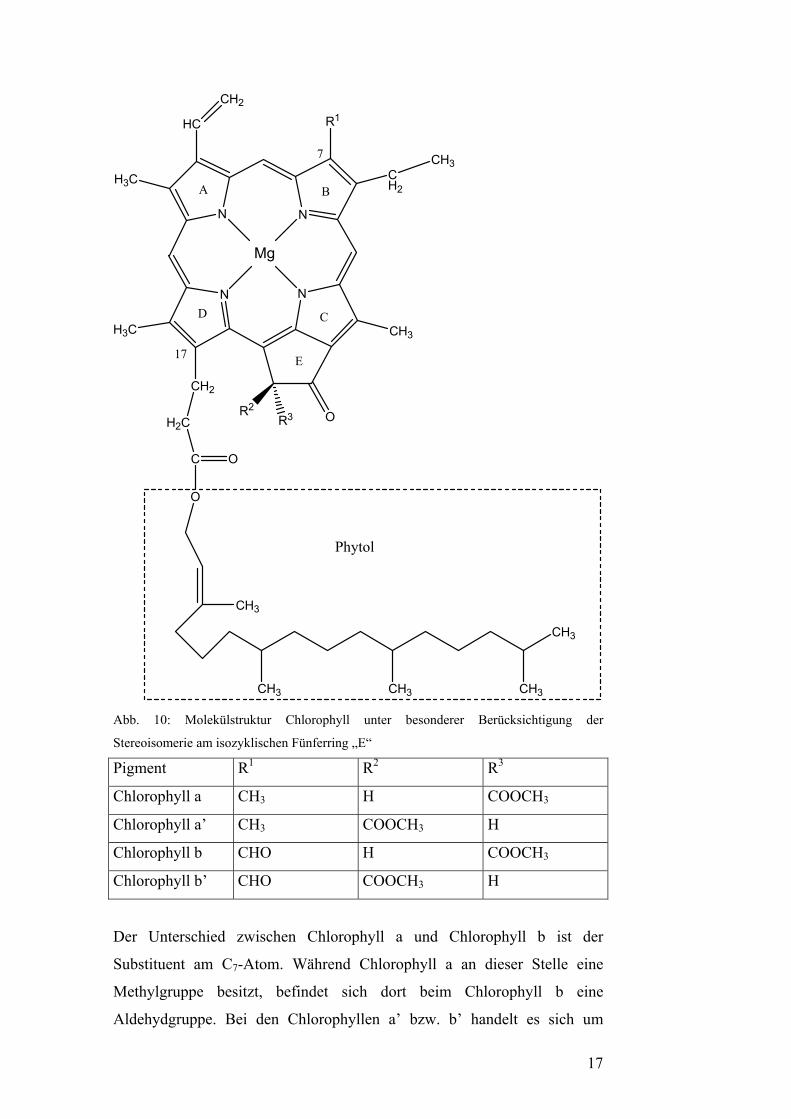
Abb. 10: Molekülstruktur Chlorophyll unter besonderer Berücksichtigung der
Stereoisomerie am isozyklischen Fünferring „E“
Pigment R1 R2 R3
Chlorophyll a CH3 H COOCH3
Chlorophyll a’ CH3 COOCH3 H
Chlorophyll b CHO H COOCH3
Chlorophyll b’ CHO COOCH3 H
Der Unterschied zwischen Chlorophyll a und Chlorophyll b ist der
Substituent am C7-Atom. Während Chlorophyll a an dieser Stelle eine
Methylgruppe besitzt, befindet sich dort beim Chlorophyll b eine
Aldehydgruppe. Bei den Chlorophyllen a’ bzw. b’ handelt es sich um
17

Stereoisomere der Chlorophylle a bzw. b. Die jeweiligen Isomere
unterscheiden sich funktionell nicht, jedoch besitzen sie unterschiedliche
polare Eigenschaften und kommen nur in geringer Konzentration in
Pflanzenzellen vor. (vgl. Versuch: Präparative Dünnschicht-
chromatographie). [7]
Allen Chlorophyllen gemeinsam ist der Propionylrest am C17-Atom. Mit
diesem ist im Fall der Chlorophylle a und b der langkettige Alkohol Phytol
verestert. Dieser lipophile Alkohol dient der Verankerung der
Chlorophyllmoleküle im lipophilen Innenbereich der Chlorophyll-
bindeproteine. Diese sitzen in den Antennen oder in den Reaktionszentren.
Strukturell handelt es sich bei Phytol um einen C20-Körper, ein Diterpen
(vgl. Carotinoide). [20] [5]
Ein Chlorophyllmolekül ohne Phytolrest wird als Chlorophyllid bezeichnet
(vgl. Versuch: Verseifung der Chlorophylle). [24]
Wird aus den Chlorophyllmolekülen das Zentralatom entfernt, so erhält man
die Stoffgruppe der Phaeophytine (vgl. Versuch: Darstellung der
Phaeophytine). Die Phaephytine sind auch Bestandteil der
Elektronentransportkette im Photosystem II. Dort übernehmen sie
Elektronen von angeregten P680 Molekülen und leiten diese über
Plastochinon zum Cytochrom-b6/f-Komplex. [21]
Vorkommen und Funktion der Chlorophylle
Wie schon im Kapitel „Lichtreaktion der Photosynthese“ beschrieben
handelt es sich bei den Chlorophyllen um Photosynthesepigmente der
grünen Pflanzen. Sie absorbieren Photonen und gehen dabei in einen
angeregten Zustand über, den Beginn des Photosyntheseprozesses.[21]
Neben grünen Pflanzen betreiben aber auch andere Organismen
Photosynthese. So verfügen z.B. Cyanobakterien über Chlorophyll a.
Rotalgen (Rhodophyta) enthalten zusätzlich noch Chlorophyll c, welches
sich strukturell nur durch einen anderen Rest am C17-Atom vom
Chlorophyll a unterscheidet. Grünalgen (Chlorophyta) enthalten
Chlorophyll a und b. Auch einzellige Flagellaten (Geißeltierchen) der
Gattung Euglena besitzen Chlorophyll a und b. Kommen diese in Pfützen
18
und Gräben in großen Mengen vor, so verfärbt sich das Wasser grün.
[20][25]
2.3.2. Extraktion von Blattgrün
Um Blattgrün untersuchen zu können, muss dieses zuerst aus dem
pflanzlichen Gewebe extrahiert werden.
Verschiedene Methoden der Extraktion werde ich in diesem Abschnitt
beschreiben.
Versuch 1: Kaltextraktion von Blattgrün [5][24]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Aceton F, Xi 11-36-66-67 9-16-26
Methanol T, F 23/24/25-
39/23/24/25
7-16-36/37-45
Calciumcarbonat - - -
Seesand - - -
Geräte:
Mörser mit Pistill, Trichter, Faltenfilter, Becherglas, Messpipette 20 mL,
Pipettierhilfe
Durchführung:
Im Abzug werden etwa 10 g frische grüne Blätter in einem Mörser mit einer
Spatelspitze Calciumcarbonat, etwas feinem Seesand sowie 1 mL Aceton
versetzt. Alternativ kann man auch tiefgekühlten Spinat („ohne Sahne“)
verwenden. Dieser sollte jedoch vor seiner Verwendung mittels Faltenfilter
oder Toilettenpapier ausgepresst werden.
Das Gemenge im Mörser wird nun mit einem Pistill zu einem feinen Brei
zerrieben, mit 20 mL Aceton versetzt und 20 Minuten stehen gelassen.
Danach wird durch einen Faltenfilter abfiltriert und zweimal mit 5 mL
19

Aceton nachgespült. Das Filtrat wird zur weiteren Verwendung
lichtgeschützt in einem verschließbaren Gefäß aufbewahrt.
Analog kann man auch mit Methanol extrahieren. Die Art der
Folgeversuche entscheidet über das zu verwendende Extraktionsmittel (vgl.
2.5.2. Versuch: Präparative Dünnschichtchromatographie)
Beobachtung:
Bereits beim Abfiltrieren kann man nebenbei einen chromatographischen
Effekt beobachten. Der Blattextrakt wandert aufgrund kapillarer Kräfte am
Filterpapier nach oben. Mehrere gelbe und grünen Banden sind zu erkennen.
In der Vorlage sammelt sich eine intensiv grüne Lösung.
Abb. 11: Chromatographischer Effekt auf Filterpapier
Auswertung:
Beim Mörsern werden die Pflanzenzellen zerstört. Blatteigene Säuren
können nun Chlorophyll verseifen (vgl. Versuch: Verseifung der
Chlorophylle). Um dies zu verhindern, wird beim Mörsern eine Spatelspitze
Calciumcarbonat zugesetzt, das die blatteigenen Säuren neutralisiert.
CaCO3(s) + H2O(l) Ca2+(aq) + HCO3
-(aq) + OH-
(aq)
H3O+(aq) + OH-
(aq) 2 H2O(l)
Die Blattfarbstoffe lösen sich im polaren Lösungsmittel Aceton bzw.
Methanol.
20
Entsorgung:
Der Faltenfilter wird getrocknet und in der Feststofftonne entsorgt.
Versuch 2: Heißextraktion von Blattgrün [5]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Aceton F, Xi 11-36-66-67 9-16-26
Methanol T, F 23/24/25-
39/23/24/25
7-16-36/37-45
Geräte:
Reagenzglas, Reagenzglasklammer, Wasserbad, Siedesteinchen, Becherglas
Durchführung:
In ein Reagenzglas werden 2-3 Siedesteinchen und 2 gerollte Blätter (z.B.
Ficus benjamina, Hedera helix, Tilia chordata) gegeben. Anschließend wird
im Abzug mit Aceton überschichtet und im Wasserbad mehrmals kurz
aufgekocht. Wenn das Lösungsmittel eine intensiv grüne Farbe
angenommen hat, überführt man dieses in ein Becherglas. Die Blätter im
Reagenzglas werden ein weiteres Mal mit Aceton überschichtet und erneut
mehrmals kurz aufgekocht. Die Lösung wird in das Becherglas
dazugegossen. Zum Aufkonzentrieren wird bis zur Trockne eingedampft
und in wenig Aceton wieder aufgenommen.
Entsprechend lässt sich statt Aceton auch Methanol verwenden.
Beobachtung:
Bereits nach kurzer Zeit färbt sich das Lösungsmittel grün. Bei den Blättern
kann nach Beendigung des Versuches eine Entfärbung festgestellt werden.
21

Auswertung:
Blattfarbstoffe lösen sich im polaren Lösungsmittel Aceton bzw. Methanol.
Bedingt durch die Tatsache, dass ganze Blätter verwendet wurden -
Pflanzenzellen wurden nicht zerstört – kann keine Verseifung stattfinden.
Deshalb kann auf Calciumcarbonat verzichtet werden.
Entsorgung:
Die Blätter werden in der Feststofftonne entsorgt.
Vergleich: Extraktionsmethoden - Lösungsmittel
Sowohl Aceton als auch Methanol sind in der Lage, Blattfarbstoffe aus
pflanzlichem Gewebe zu extrahieren. Untersucht man jedoch die
verschiedenen Extrakte chromatographisch, dann zeigen sich Unterschiede.
Bei der Verwendung von Aceton macht es keinen Unterschied, ob kalt oder
heiß extrahiert wurde. Im Chromatogramm können maximal zwei
Xanthophylle, β-Carotin sowie Chlorophyll a und b aufgetrennt werden.
Setzt man bei der Chromatographie jedoch einen Heißextrakt mit Methanol
ein, dann werden zusätzlich sichtbar Chlorophyll a’ und b’ sowie ein drittes
Xanthophyll (vgl. Versuche zur Chromatographie).
Für die meisten Versuche mit Blattgrün ist ein Acetonextrakt völlig
ausreichend.
22

2.3.3. Versuche zum Nachweis von charakteristischen Strukturelementen
Versuch 3: Darstellung der Phaeophytine [24]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Salzsäure
2 mol/L
- - -
Geräte:
Reagenzglas, Stopfen, Pasteurpipette
Durchführung:
Ein Reagenzglas wird etwa 2 cm hoch mit der Blattextraktlösung befüllt.
Anschließend werden einige Tropfen Salzsäure, 2 mol/L, zugesetzt, das
Reagenzglas mit einem Stopfen verschlossen und geschüttelt.
Beobachtung:
Die Lösung im Reagenzglas verfärbt sich braun.
Abb. 12: linkes Reagenzglas: reiner Blattextrakt, rechtes Reagenzglas Blattextrakt mit
Salzsäure (Phaeophytin)
23

Auswertung:
Wie bereits im Kapitel 2.3.1. beschrieben, befindet sich im Zentrum eines
jeden Chlorophyllmoleküls ein Magnesium-Ion als Zentralatom. Der
Porphyrinring ist der zugehörige Ligand des Komplexes. Jedes der vier
Stickstoffatome im Porphyrinring trägt mit einem Elektronenpaar zur
Komplexbildung bei. Durch milde Säurebehandlung wird der Komplex
zerstört. Zwei Protonen ersetzen das zentrale Magnesium-Ion. Ein
Chlorophyllmolekül ohne Magnesium-Ion als Zentralatom wird als
Phaeophytin bezeichnet.
Chlorophyll(solv) + 2 H3O+(aq) Phaeophytin(solv) + 2 H2O(l) + Mg2+
(aq)
Durch das Herauslösen des Magnesium-Ions verliert das Chlorophyll seine
typischen Absorptionseigenschaften. Die Lösung färbt sich braun.
Entsorgung:
Die Lösung kann neutral im Behälter für organische Lösungsmittel entsorgt
werden.
24
Versuch 4: Nachweis des Magnesium-Ions [22]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Salzsäure
4 mol/L
Xi 36/37/38 26
Titangelb-Lösung
(gesättigt)
- - -
Natronlauge
2 mol/L
C 35 26-36/37/39-45
Magnesiumchlorid - - -
Spinat - - -
Geräte:
Erlenmeyerkolben 100 mL, Spatel, Glasstab, Bunsenbrenner, Dreifuß mit
Drahtnetz, Messpipette 10 mL, Pipettierhilfe, Faltenfilter, Trichter
Durchführung:
Etwa 10 g Spinat werden mit 10 mL Salzsäure, 4 mol/L, im Abzug
aufgekocht. Anschließend wird die farblose Lösung abgefiltert. Das Filtrat
wird mit Natronlauge, 2 mol/L, neutralisiert und mit entionisiertem Wasser
auf 100 mL aufgefüllt.
In einem zweiten Erlenmeyerkolben wird eine Spatelspitze
Magnesiumchlorid in etwa 100 mL entionisiertem Wasser gelöst. Diese
Magnesiumchloridlösung dient als Vergleichslösung.
Beide Lösungen werden mit Natronlauge, 2 mol/L, alkalisch gemacht und
mit drei Tropfen Titangelb-Lösung versetzt.
Beobachtung:
Durch Zusatz von Natronlauge kommt es in beiden Fällen zu einer leichten
Trübung der Lösung.
In beiden Lösungen fällt nach Zusatz der Titangelb-Lösung ein roter
Feststoff aus.
25

Abb. 13 und 14: Magnesiumchlorid-Lösung und Extrakt aus Spinat vor und nach Zugabe
von Titangelb-Lösung. Im jeweils linken Erlenmeyerkolben befindet sich die
Magnesiumchlorid-Lösung, im jeweils rechten Erlenmeyerkolben der Extrakt aus Spinat.
Auswertung:
Die beobachtete Trübung wird durch entstehendes Magnesiumhydroxid
hervorgerufen. Dieses ist weiß und schwerlöslich.
Mg2+(aq) + 2 OH-
(aq) Mg(OH)2(s)
Die Magnesium-Ionen werden bei Zusatz einer Titangelb-Lösung im
alkalischen Milieu als roter Komplex gefällt. Es handelt sich um eine
charakteristische Nachweisreaktion für Magnesium-Ionen. Allerdings ist die
genaue Struktur des entstehenden Komplexes nicht bekannt.
Da man in beiden Fällen zur gleichen Beobachtung kommt, ist gezeigt, dass
durch Säureeinwirkung Magnesium-Ionen aus dem Chlorophyll gelöst
werden.
Entsorgung:
Die Lösungen können neutral im Behälter für organische Lösungsmittel
entsorgt werden.
26
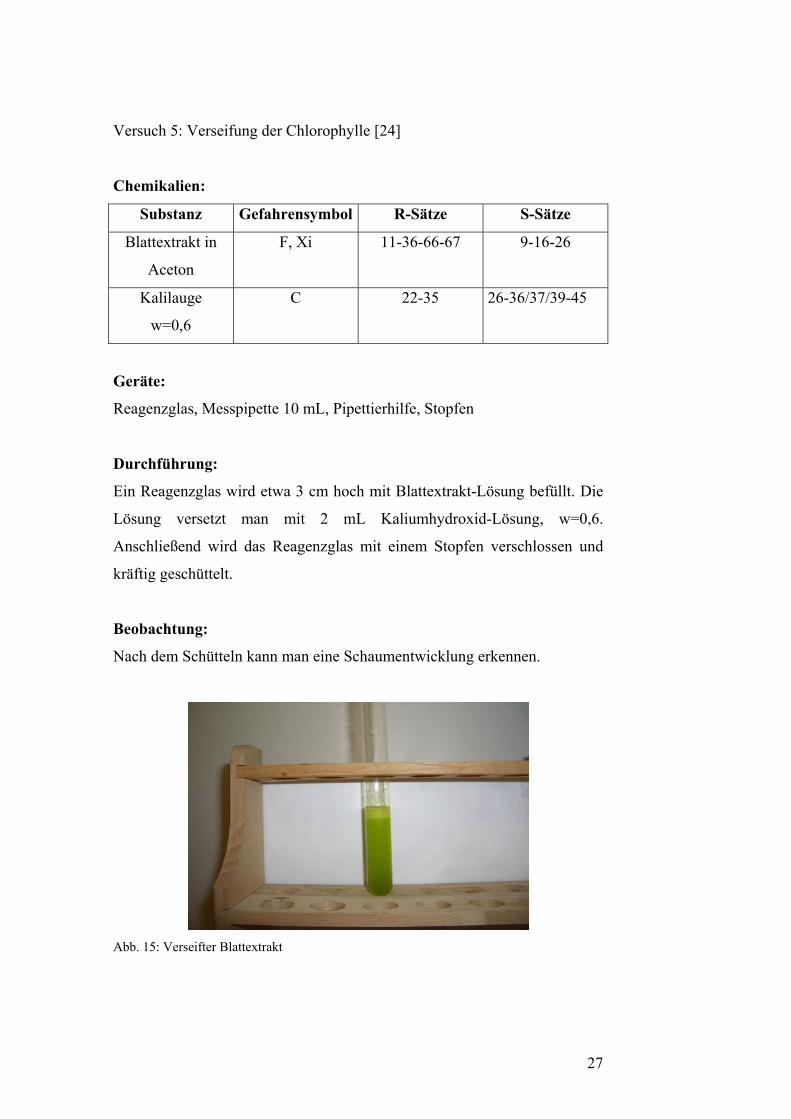
Versuch 5: Verseifung der Chlorophylle [24]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Kalilauge
w=0,6
C 22-35 26-36/37/39-45
Geräte:
Reagenzglas, Messpipette 10 mL, Pipettierhilfe, Stopfen
Durchführung:
Ein Reagenzglas wird etwa 3 cm hoch mit Blattextrakt-Lösung befüllt. Die
Lösung versetzt man mit 2 mL Kaliumhydroxid-Lösung, w=0,6.
Anschließend wird das Reagenzglas mit einem Stopfen verschlossen und
kräftig geschüttelt.
Beobachtung:
Nach dem Schütteln kann man eine Schaumentwicklung erkennen.
Abb. 15: Verseifter Blattextrakt
27

Auswertung:
Der Phytolrest des Chlorophylls ist über eine Esterbindung mit dem
Propionylrest am C17-Atom des Porphyrinringes verbunden. Durch Zusatz
von Säuren oder Laugen, kann die Esterbindung gespalten werden. Im
vorliegenden Versuch handelt es sich um eine basische Esterspaltung. Man
spricht auch von einer Verseifung.
Mechanistisch lässt sich die Verseifung folgendermaßen darstellen: [11]
Zu besseren Übersicht ist der Phytolrest mit C20H39 abgekürzt, das
Ringgerüst mit R.
Im ersten Schritt greift ein Hydroxid-Ion nucleophil am positiv polarisierten
Carbonylkohlenstoff der Esterbindung an. Dabei wird das bindende
π-Elektronenpaar der Carbonylgruppe zum Carbonylsauerstoff verschoben.
RC
O
OC20H39
HO
RC
O
OC20H39
OH
Im zweiten Schritt greift ein Elektronenpaar des formal negativ geladenen
Sauerstoffatoms nucleophil an einem Wasserstoffatom eines
Wassermoleküls an. Dabei entsteht ein Diol und ein Hydroxid-Ion.
RC
O
OC20H39
OH
O
H
H
RC
OH
OC20H39
OH
HO+
Unter Abspaltung eines Protons kommt es schließlich zur Spaltung der
Estergruppe. Dabei entstehen als Produkte eine Carbonsäure und ein
Alkoholat-Ion.
28

RC
OH
OC20H39
O
H
- H+
RC
O
OH
+ O C20H39
In einem letzen irreversiblen Schritt greift das Alkoholat nucleophil am
Proton der Carbonsäuregruppe an. Es entstehen dabei Alkohol und
Carboxylat-Ion (Verseifung). Das Carboxylat-Ion ist gut
mesomeriestabilisiert und besitzt als Gegenion ein Kaliumion aus der
Kaliumhydroxid-Lösung.
RC
O
OH
+ O C20H39
RC
O
O
+ HO C20H39
+ K+
K+
Bezogen auf ein Chlophyllmolekül handelt es sich beim Carboxylat-Ion um
das so genannte Chlorophyllid und beim Alkohol um Phytol.[24]
Durch Schaumbildung kann die Seifenentstehung gezeigt werden.
Entsorgung:
Die Lösung kann neutral im Behälter für organische Lösungsmittel entsorgt
werden.
29
2.3.4. Versuch zum Nachweis der Sauerstoffproduktion mit Indigo
Vorbemerkung: Gewinnung des Farbstoffes Indigo [14]
Bei „Indigo“ handelt es sich um einen aus der in Indien beheimateten
Pflanze Indigofera tinctoria gewonnenen Farbstoff. In dieser Pflanze liegt
das farblose Glucosid Indican vor. Es wird gewonnen und zu Indigo
verarbeitet. Durch enzymatische Hydrolyse wird das Glucosid zunächst in
Glycose und Indoxyl gespalten. Durch Luftsauerstoff werden dann zwei
Indoxyl-Moleküle zu einem Indigo-Molekül oxidiert. Dieses Indigo-
Molekül ist blau und wasserunlöslich. Um es in eine wasserlösliche Form zu
bringen, wird es mit Natriumdithionit in alkalischer Lösung reduziert.
Heute wird Indigo nach dem von Adolf von Baeyer entwickeltem Verfahren
aus ortho-Nitrobenzaldehyd und Aceton hergestellt.
Verwendung findet der Farbstoff in der Textilindustrie (Küpenfärbung mit
Indigo).
Versuch 6: Nachweis der Sauerstoffproduktion [3]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Indigopulver - - -
Natriumdithionit Xn 7-22-31 7/8-26-28-43
Natronlauge
w=0,1
C 35 26-37/39-45
Ethanol F 11 7-16
Wasserpest - - -
Geräte:
Mörser mit Pistill, Bunsenbrenner, Dreifuß mit Drahtnetz, Thermometer
Schraubdeckelgläser 100 mL,
30

Durchführung:
In einem Mörser werden 0,9 g Indigo mit etwas Ethanol und 10 mL
Natronlauge, w=0,1, verrieben. Anschließend überführt man die Suspension
in 300 mL Wasser, das auf 70 °C vorgewärmt wurde. Danach rührt man so
viel Natriumdithionit hinzu, bis die Lösung gerade gelb-grün wird.
In die Schraubdeckelgläser gibt man je einen Spross Wasserpest.
Anschließend befüllt man die beiden Gläser bis zum oberen Rand mit der
Lösung. Eventuell auftretende Blaufärbung wird unter Einrühren von
Natriumdithionit beseitigt. Danach werden die Gläser verschlossen
Das erste Schraubdeckelglas wird vor eine Lichtquelle gestellt, das andere
lichtgeschützt aufbewahrt.
Beobachtung:
Nach etwa 90 Minuten hat die hell stehende Lösung eine blaue Farbe
angenommen. Die Farbe der dunkel gelagerten Lösung ist unverändert.
Abb. 16 und 17: Dunkel gelagerte und belichtete Wasserpestpflanzen
Auswertung:
Indigo ist ein Redox-Indikator. In der oxidierten Form handelt es sich um
den Farbstoff selbst. Dieser ist blau und wasserunlöslich. Die reduzierte
Form wird Leukoindigo genannt. Sie ist wasserlöslich und gelb-grün.
31
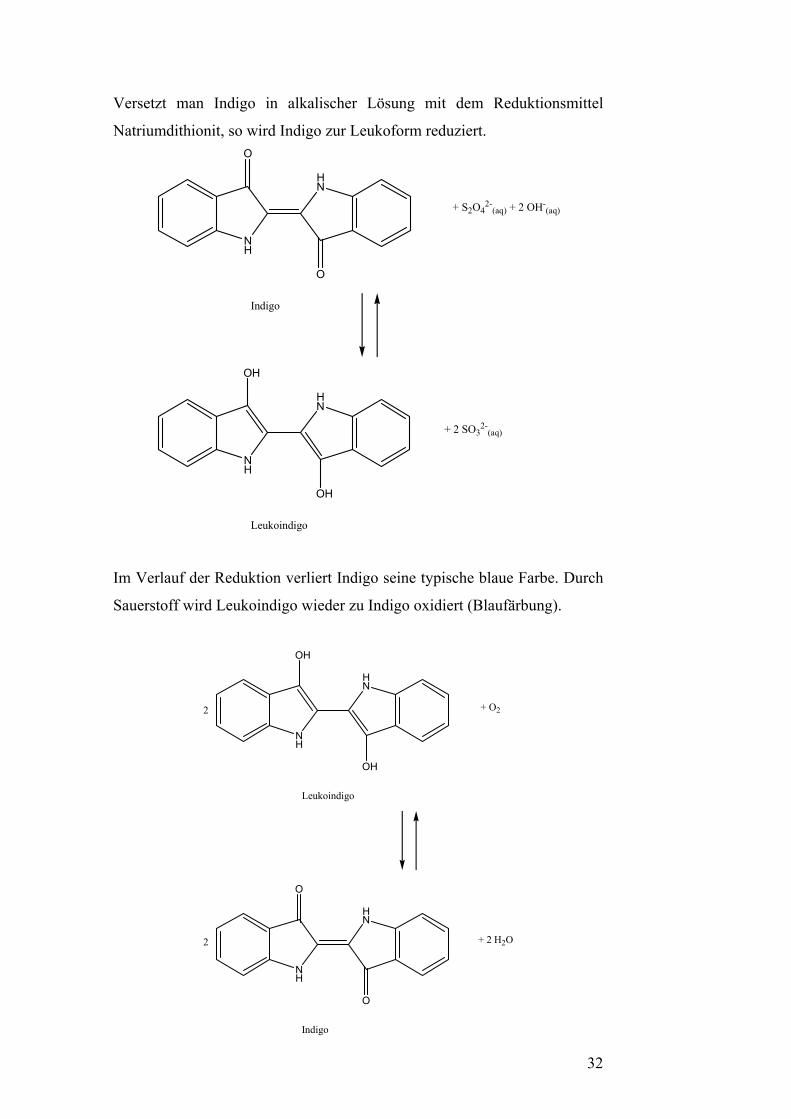
Versetzt man Indigo in alkalischer Lösung mit dem Reduktionsmittel
Natriumdithionit, so wird Indigo zur Leukoform reduziert.
NH
O
HN
O
NH
OH
HN
OH
+ S2O42-
(aq) + 2 OH-(aq)
+ 2 SO32-
(aq)
Indigo
Leukoindigo
Im Verlauf der Reduktion verliert Indigo seine typische blaue Farbe. Durch
Sauerstoff wird Leukoindigo wieder zu Indigo oxidiert (Blaufärbung).
NH
OH
HN
OH
+ O2
Leukoindigo
NH
O
HN
O
Indigo
2
2 + 2 H2O
32

Im belichteten Gefäß färbt sich Leukoindigo blau. Da das Gefäß
geschlossen war, kann das Oxidationsmittel Sauerstoff nicht aus der Luft
stammen. Es muss also von der Pflanze selbst produziert worden sein.
Im unbelichteten Gefäß bleibt die Blaufärbung aus. Folglich wurde kein
Sauerstoff produziert.
Durch diesen Versuch kann gezeigt werden, dass im Verlauf der
Photosynthese Sauerstoff gebildet wird und die Photosynthese lichtabhängig
ist.
Entsorgung:
Die Lösungen werden neutral im Behälter für organische Lösungsmittel
entsorgt.
33

2.4. Carotinoide
2.4.1. Varianten - Molekülstruktur
Carotinoide sind Pflanzenfarbstoffe, die im gesamten Pflanzenreich
vorkommen.
Bereits im Jahr 1837 bezeichnete Berzelius die gelben, alkohollöslichen
Pigmente des Herbstlaubes als „Xanthophylle“.[2]
Fremy und Stokes konnten 1860 und 1884 als erste nachweisen, dass diese
Farbstoffe auch in grünen Blättern vorkommen.[2]
Erst im Jahr 1911 gelang es dem russischen Botaniker und Biochemiker
Mikail Tswett die gelben Pigmente chromatographisch aufzutrennen.
Aufgrund ihrer unterschiedlichen Löslichkeit in organischen Lösungsmitteln
unterteilte er die Pigmente in die Gruppe der „Xanthophylle“ und in die
Gruppe der „Carotine“. Als Überbegriff für beide Gruppen wählte er den
Begriff „Carotinoide“.[2]
In den zwanziger Jahren des letzten Jahrhunderts gelang es schließlich
verschiedenen Wissenschaftlern (u.a. Karrer, Kuhn, Heilbron und
Zechmeister), die Strukturen verschiedener Carotinoide aufzuklären.
Bis zum heutigen Tag wird die Einteilung der Carotinoide in Carotine und
Xanthophylle nach Tswett beibehalten. Heute versteht man unter Carotinen
reine Kohlenwasserstoffe, unter Xanthophyllen mit Sauerstoff oxidierte
Kohlenwasserstoffe. [5]
Einteilung
Die Carotinoide gehören zur Stoffklasse der Isoprenoiden. Dieser
Oberbegriff leitet sich vom Grundbaustein aller Isoprenoide, dem Isopren,
ab.
Abb. 18: Molekülstruktur Isopren
34

Während der Biosynthese der Carotinoide werden zwei Isopreneinheiten zu
einem Monoterpen (C10) zusammengelagert. Vier dieser Monoterpene
ergeben schließlich das Grundgerüst der Carotinoide, ein Tetraterpen
(8 Isoprene, C40).[21]
Dabei handelt es sich um langgestreckte Moleküle mit
40 Kohlenstoffatomen in einer Reihe mit konjugierten Doppelbindungen.
Die Carotinoide sind gelb bis rot gefärbt. Die Farbe rührt von den
chromophoren Eigenschaften der linear konjugierten Doppelbindungen her.
An diesen Doppelbindungen sind cis-trans-Isomere denkbar. Zumeist liegen
die Carotinoide in der all-trans-Form vor.[5]
Man unterscheidet bei Carotinoiden zwischen Primär- und
Sekundärcarotinoiden.
Primärcarotinoide sind die Carotinoide, die direkt an den
Photosyntheseprozessen beteiligt sind. Sie zeichnen sich durch je einen
Iononring an den Enden der Kohlenstoffkette aus und sie haben drei lokale
Maxima im sichtbaren Spektrum. Alle diese Maxima liegen unter 480 nm.
Häufig vorkommende Primärcarotinoide sind β-Carotin, ein Vertreter der
Carotine, sowie Lutein, Violaxanthin und Neoxanthin als Vertreter der
Xanthophylle.[5]
35

LuteinHO
OH
ViolaxanthinHO
OH
O
O
NeoxanthinHO
O
HO
OH
β−Carotin
Abb. 19 – 22: Häufig vorkommende Primärcarotinoide
Sekundärcarotinoide sind nicht direkt an der Photosynthe beteiligt. Sie
werden nur von Pflanzen gebildet, die unter Stress stehen, z.B. Dauerlicht
oder Sickstoffmangel. [5]
Strukturell unterscheiden sich die Sekundär- von den Primärcarotinoiden
durch das Fehlen eines oder auch beider Iononringe. Diese können durch
einen substituierten Pentanring ersetzt sein. Im sichtbaren Bereich des
Spektrums haben die Sekundärcarotinoide nur ein lokales Maximum. [5]
Vorkommen und Funktion der Carotinoide
Vorkommen und Funktion von Farbstoffen sind in der Natur eng
miteinander verknüpft.
In Blättern dienen Carotinoide als akzessorische Photosynthesepigmente.
Außerdem sind sie am Aufbau des Photosystems II beteiligt und schützen
vor Photooxidation. [5]
Als Lockstoff für Insekten kommen Carotinoide in Blüten vor,
z.B. Violaxanthin in Veilchen (Viola). Auch in Früchten können
36

Carotinoide als Lockstoff vorkommen, z.B. Lycopin in Tomaten (Solanum
lycopersicum). [5]
Aber auch in anderen Organen von Pflanzen können Carotinoide
vorkommen. So lagert beispielsweise die Möhre (Daucus carota) β-Carotin
in der rübenförmigen Wurzel ein. [20]
Alle diese plasmochromen (membrangebundenen) Farbstoffe werden in
Plastiden (Chromoplasten, Chloroplasten) angehäuft. [17]
Außerdem kommen Carotinoide in Schalen von Crustaceaen, im Fleisch
von Fischen und in Federn oder im Eigelb von Vögeln vor. Da aber nur
Pflanzen und einige Bakterien und Pilze in der Lage sind, Carotinoide zu
synthetisieren, müssen diese Tiere die Carotinoide mit der Nahrung
aufgenommen haben.
37

2.4.2. Extraktion des bekanntesten Carotinoides: β-Carotin
Die Gewinnung der Primärcarotinoide β-Carotin, Lutein, Neoxanthin und
Violaxanthin aus Blattgrün erfolgt mittels einer präparativen
Dünnschichtchromatographie von grünem Blattextrakt. Die Banden werden
von der DC-Alufolie abgekratzt und in Aceton gelöst. Nachdem das
Kieselgel abzentrifugiert ist, erhält man hochreine Lösungen o.g.
Primärcarotinoide in Aceton. Das Verfahren wird in Abschnitt 2.5.2.
beschrieben.
Behandelt man in der Schule Primärcarotinoide, so wird man sich
insbesondere bei Versuchen hauptsächlich auf β-Carotin beschränken, da es
entweder bereits in der Sammlung vorhanden ist, oder sich relativ einfach
und schnell in großen Mengen extrahieren lässt. Typische Eigenschaften
oben genannter Carotinoide, wie z.B. Farbe, Existenz von Doppelbindungen
und die Funktionen in der Pflanze lassen sich am Beispiel des β-Carotins als
Stellvertreter der Carotinoide gut veranschaulichen.
38

Versuch 7: Extraktion von β-Carotin mit Aceton aus kleingeschnittenen
Möhren [9]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Aceton F, Xi 11-36-66-67 9-16-26
Möhre - - -
Geräte:
Messer, 2 Erlenmeyerkolben 250 mL
Durchführung:
Eine frische Möhre wird gewaschen und geschält. Etwa die Hälfte der
Möhre wird in kleine Scheiben geschnitten und in den Erlenmeyerkolben
gegeben. Anschließend werden die Möhrenscheiben im Abzug mit Aceton
überschichtet. Der Erlenmeyerkolben wird nun etwa 2 Minuten geschwenkt.
Danach wird die Lösung in den zweiten Erlenmeyerkolben dekantiert.
Beobachtung:
Bereits nach etwa 20 Sekunden hat sich das Aceton gelb gefärbt. Nach dem
Dekantieren kann man beobachten, dass die Möhrenscheiben ausgebleicht
sind.
Auswertung:
Aceton ist in der Lage, β-Carotin aus dem Stroma der Chromoplasten
herauszulösen. [17]
Entsorgung:
Die Möhrenscheiben können getrocknet als Feststoffabfall entsorgt werden.
Die Lösung von β-Carotin in Aceton kann für weitere Versuche aufbewahrt
werden. Falls diese nicht mehr benötigt wird, entsorgt man sie im Behälter
für organische Lösungsmittel.
39

Versuch 8: Extraktion von β-Carotin mit n-Heptan aus kleingeschnittenen
Möhren [9]
Bei diesem Versuch soll geprüft werden, ob sich n-Heptan besser als Aceton
eignet, β-Carotin zu extrahieren.
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
n-Heptan F, Xn, N 11-38-50/53-65-
67
9-16-29-33-60-
61-62
Möhre - - -
Geräte, Durchführung, Beobachtung, Entsorgung vgl. Versuch: Extraktion
von β-Carotin mit Aceton aus kleingeschnittenen Möhren.
Auswertung:
n-Heptan ist ähnlich wie Aceton gut geeignet, β-Carotin aus dem Stroma der
Chloroplasten herauszulösen. Für weitergehende Versuche mit β-Carotin-
Extrakten kommt es entscheidend auf das Lösungsmittel n-Heptan oder
Aceton an. Genau dies muss bei der Wahl des Extraktionsmittels
berücksichtigt werden. [17]
40

Versuch 9: Extraktion von β-Carotin mit Aceton aus geraspelten Möhren
Es soll geprüft werden, ob die Verwendung geraspelter Möhren zu einer
höheren β-Carotin-Ausbeute führt.
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Aceton F, Xi 11-36-66-67 9-16-26
Möhre - - -
Geräte:
Messer, Küchenreibe, 2 Erlenmeyerkolben 250 mL
Vorbereitung:
Eine frische Möhre wird gewaschen und geschält. Etwa die Hälfte der
Möhre wird auf einer Küchenreibe kleingeraspelt und in den
Erlenmeyerkolben gegeben.
Durchführung, Beobachtung, Entsorgung vgl. Versuch: Extraktion von
β-Carotin mit Aceton aus kleingeschnittenen Möhren.
Auswertung:
Es lässt sich feststellen, dass die Ausbeute an β-Carotin nicht erhöht ist. Die
Gelbfärbung des Lösungsmittels ist vergleichbar (siehe Versuch: Extraktion
von β-Carotin mit Aceton aus kleingeschnittenen Möhren und Versuch:
Extraktion von β-Carotin mit n-Heptan aus kleingeschnittenen Möhren).
41

Versuch 10: Heißextraktion von β-Carotin mit Petrolether in einer Soxhlet-
Apparatur [24]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Petrolether
(60/95)
F, Xn, N 11-38-48/20-
51/53-62-65-67
9-16-33-36/27-
60-62
Möhre - - -
Geräte:
Messer, Petrischale, Trockenschrank, Soxhlet-Apparatur, Rotations-
verdampfer
Abb. 23: Soxhlet-Apparatur
Durchführung:
Eine frische Möhre wird gewaschen, geschält und in kleine Stücke
geschnitten. Diese werden in eine Petrischale gegeben und im
Trockenschrank bei 80 °C getrocknet. Nach etwa einer halben Stunde wird
das β-Carotin in einer Soxhlet-Apparatur im Abzug mit etwa 400 mL
42

Petrolether extrahiert. Der Versuch wird beendet, wenn der in den
Rundkolben zurückfließende Petrolether farblos ist (etwa nach
60 Minuten). Je nach Weiterverwendungszweck kann die Lösung am
Rotationsverdampfer noch eingeengt werden.
Beobachtung:
Mit zunehmender Versuchsdauer kann man eine Intensivierung der
Gelbfärbung des Petrolethers im Rundkolben beobachten.
Nach Versuchsende sind die Möhrenstücke in der Papphülse ausgebleicht.
Auswertung:
Wie die Lösungsmittel Aceton und n-Heptan ist auch Petrolether in der
Lage, β-Carotin aus der Möhre herauszulösen. Nach Einengen am
Rotationsverdampfer erhält man eine sehr konzentrierte Lösung.
Entsorgung:
Die Möhrenstückchen werden getrocknet als Feststoffabfall entsorgt.
Die Lösung von β-Carotin in Petrolether kann für weitere Versuche
aufbewahrt werden. Falls man sie nicht mehr benötigt, wird sie im Behälter
für organische Lösungsmittel entsorgt.
43

Vergleich der verschiedenen Extraktionsmethoden
Alle oben beschriebenen Extraktionsverfahren eignen sich zur Extraktion
von β-Carotin aus Möhren. Die Konzentration der verschiedenen Lösungen
reicht bei allen Versuchen aus, um weitere Experimente mit β-Carotin
durchzuführen.
Abschließend lässt sich feststellen, dass die einfache Extraktionsmethode
Möhrenscheiben mit Lösungsmittel zu überschichten, zu einem guten,
brauchbaren Ergebnis führt. Die Aspekte Zeit und Nutzen harmonieren sehr
gut. Gerade in der Schule ist es wichtig, dass Versuche schnell
durchzuführen sind und mit hoher Wahrscheinlichkeit funktionieren. Dies
ist hier der Fall. Die Extraktion aus Möhrenraspeln und die Heißextraktion
liefern vergleichbare Ergebnisse, der Zeitaufwand ist jedoch viel zu hoch.
Für die Heißextraktion spricht allerdings die Tatsache, dass man bei einer
Durchführung eine relativ große Menge an konzentrierter Lösung erhält.
Diese kann dunkel und kühl über einen längeren Zeitraum gelagert werden,
so dass man bei Bedarf schnell darauf zurückgreifen kann.
44

2.4.3. Nachweis des Strukturelementes Doppelbindung:
Elektrophile Addition
Eine wichtige Eigenschaft der Carotinoide ist deren Polaritiät. Auf diesen
Sachverhalt werde ich im Abschnitt 2.5. Chromatographie von
Blattfarbstoffen näher eingehen.
Bezüglich ihrer Struktur handelt es sich bei den Carotinoiden um mehrfach
ungesättigte Kohlenwasserstoffe. Dies möchte ich mit dem folgenden
Versuch zeigen.
Versuch 11: Elektrophile Addition von Brom an β-Carotin [15]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Brom in Phthal-
säurediethylester
Xn - -
β-Carotin in Aceton F, Xi 11-36-66-67 9-16-26
gesättigte
Natriumthiosulfatlösung
- - -
Geräte:
Demo-Reagenzglas mit Septum, Einwegspritze mit Kanüle
Durchführung:
Im Abzug wird ein Demo-Reagenzglas etwa 2 cm hoch mit einer Lösung
von β-Carotin in Aceton befüllt. Dieses Reagenzglas wird mit einem
Septum verschlossen. Mit einer Einwegspitze wird nun 1 mL einer Lösung
von Brom in Phtalsäurediethylester durch das Septum in das Reagenzglas
eingebracht.
Beobachtung:
Die Braunfärbung von Brom verschwindet sofort. Setzt man weiter
Bromlösung zu, wird diese zwar immer noch entfärbt, jedoch verliert dann
auch die β-Carotin-Lösung langsam ihre charakteristische gelbe Farbe.
45
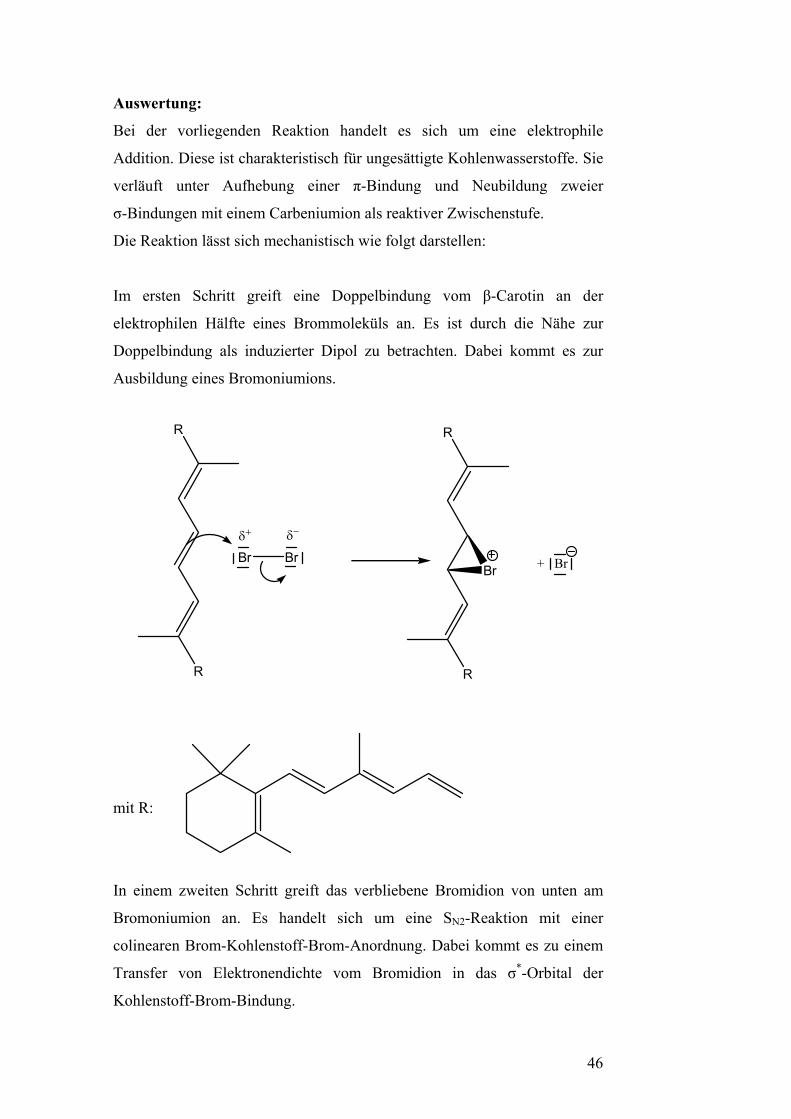
Auswertung:
Bei der vorliegenden Reaktion handelt es sich um eine elektrophile
Addition. Diese ist charakteristisch für ungesättigte Kohlenwasserstoffe. Sie
verläuft unter Aufhebung einer π-Bindung und Neubildung zweier
σ-Bindungen mit einem Carbeniumion als reaktiver Zwischenstufe.
Die Reaktion lässt sich mechanistisch wie folgt darstellen:
Im ersten Schritt greift eine Doppelbindung vom β-Carotin an der
elektrophilen Hälfte eines Brommoleküls an. Es ist durch die Nähe zur
Doppelbindung als induzierter Dipol zu betrachten. Dabei kommt es zur
Ausbildung eines Bromoniumions.
R
R
Br Brδ+ δ−
R
R
Br + Br
mit R:
In einem zweiten Schritt greift das verbliebene Bromidion von unten am
Bromoniumion an. Es handelt sich um eine SN2-Reaktion mit einer
colinearen Brom-Kohlenstoff-Brom-Anordnung. Dabei kommt es zu einem
Transfer von Elektronendichte vom Bromidion in das σ*-Orbital der
Kohlenstoff-Brom-Bindung.
46
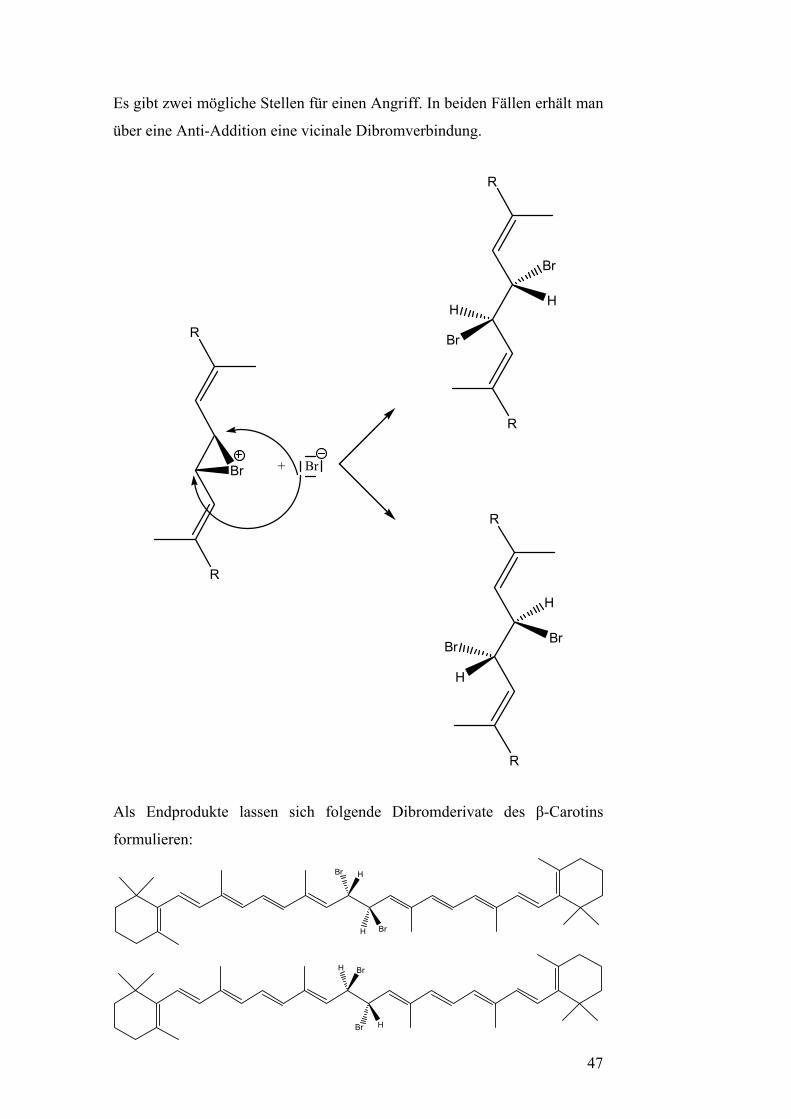
Es gibt zwei mögliche Stellen für einen Angriff. In beiden Fällen erhält man
über eine Anti-Addition eine vicinale Dibromverbindung.
R
R
Br + Br
R
R
H
Br
Br
H
R
R
Br
H
H
Br
Als Endprodukte lassen sich folgende Dibromderivate des β-Carotins
formulieren:
Br
H
H
Br
H
Br
Br
H
47

Bedingt durch die Addition von Brom wird der Chromophor des β-Carotins
zerstört. Die Anzahl der verschiebbaren Elektronen im konjugierten
π-Elektronensystem und die Größe des π-Elektronensystems selbst werden
reduziert. Außerdem sind die Elektronen nicht mehr so leicht verschiebbar.
Deshalb ändert sich der Farbeindruck. Damit wird auch erklärbar, warum
das β-Carotin seine charakteristische Farbe verliert.
Entsorgung:
Bromhaltige Lösungen werden mit Natriumthiosulfatlösung versetzt und in
der Tonne für organische Lösungsmittel entsorgt.
2 Na+(aq) + S2O3
2-(aq) + 4 Br2(solv) + 15 H2O 2 Na+
(aq) + 2 SO42-
(aq) + 8 Br-
(aq) + 10 H3O+(aq)
48
2.4.4. Schutzfunktion des β-Carotins für das Chlorophyll
Carotinoide schließen – wie schon beschrieben – die Grünlücke. Sie sind am
Aufbau des Photosystems II beteiligt. Außerdem haben Carotinoide noch
eine weitere wesentliche Aufgabe: sie schützen die Photosysteme vor
Photooxidation durch freie Radikale und Singulett-Sauerstoff.
Diese Schutzfunktion möchte ich in den folgenden Versuchen
veranschaulichen.
Versuch 12: β-Carotin als Radikalfänger [9]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Tetraiodethen - - -
n-Heptan F, Xn, N 11-38-50/53-65-
67
9-16-29-33-60-
61-62
β-Carotin in n-
Heptan
F, Xn, N 11-38-50/53-65-
67
9-16-29-33-60-
61-62
Geräte:
Diaprojektor, Hebebühne, kleine Küvetten, Becherglas 50 mL, Messpipette
20 mL, Einwegspritzen mit Kanülen, Analysenwaage, Magnetrührer mit
Rührfisch
Durchführung:
Es werden in 20 mL n-Heptan 0,11 g Tetraiodethen gelöst. Hierzu wird die
Lösung im Wasserbad auf 45 °C erhitzt und mittels Magnetrührer gerührt.
Sobald die Lösung eine blass-violette Farbe hat, wird das Erhitzen beendet.
Mittels Einwegspritze werden zwei Küvetten mit Tetraiodethen-Lösung
befüllt. In eine der Küvetten werden zusätzlich 2-3 Tropfen einer Lösung
von β-Carotin in n-Heptan gegeben.
49

Beide Küvetten werden anschließend auf einer Hebebühne vor die Linse
eines eingeschalteten Diaprojektors gestellt.
Beobachtung:
Schon nach etwa 20 Sekunden kann man in der Küvette ohne β-Carotin eine
tiefviolette Färbung der Lösung erkennen. Bei der anderen Küvette ist keine
Veränderung zu beobachten. Erst nach etwa 8 Minuten färbt sich die
Lösung mit β-Carotin ebenfalls tief violett.
Abb. 24 und 25: Belichtete Küvetten, links nach 20 Sekunden, rechts nach 8 Minuten.
Jeweils in der rechten Küvette die mit β-Carotin versetzte Probe.
Auswertung:[11]
Bei der hier vorliegenden Reaktion handelt es sich um eine
Radikalkettenreaktion. Im ersten Schritt kommt es durch Lichteinwirkung
zu einer homolytischen Bindungsspaltung der Kohlenstoff-Iod-Bindung im
Tetraiodethenmolekül.
Kettenstart – Bildung von Radikalen:
I
I I
I
h ν
I
I
I
+ I
50

Das beim Kettenstart entstandene Iodradikal kann im nun folgenden Schritt
ein Tetraiodethenmolekül angreifen.
Kettenreaktion:
I
I I
I
+ I
I
I
I
+ I2
Bei diesem Schritt kommt es erneut zu einer homolytischen
Bindungsspaltung der Kohlenstoff-Iod-Bindung im Tetraiodethenmolekül.
Bei der Rekombination von zwei Iodradikalen kommt es zur Bildung von
elementarem Iod. Dieses verursacht die violette Farbe der Lösung in der
Küvette ohne β-Carotin.
In der Küvette mit β-Carotin entsteht in einem ersten Schritt ebenfalls ein
Iodradikal. Das besondere ist nun, dass β-Carotin in der Lage ist, das
Iodradikal durch radikalische Addition anzulagern. Dabei greift das
Iodradikal die Doppelbindung in der Mitte des β-Carotins an. Diese weist
die geringste Elektronendichte auf. Alle anderen Doppelbindungen weisen
eine höhere Elektronendichte bedingt durch +I-Effekte benachbarter
Methylgruppen auf.
51
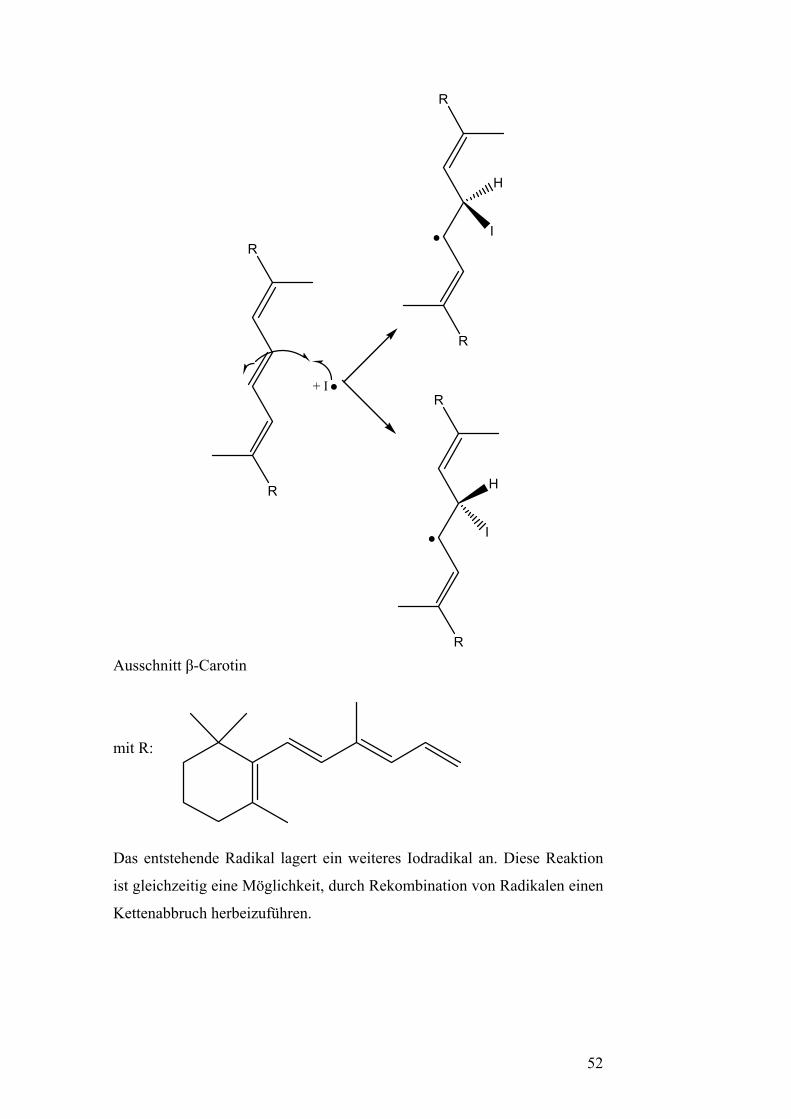
R
R
+ I
R
R
H
I
R
R
H
I
Ausschnitt β-Carotin
mit R:
Das entstehende Radikal lagert ein weiteres Iodradikal an. Diese Reaktion
ist gleichzeitig eine Möglichkeit, durch Rekombination von Radikalen einen
Kettenabbruch herbeizuführen.
52
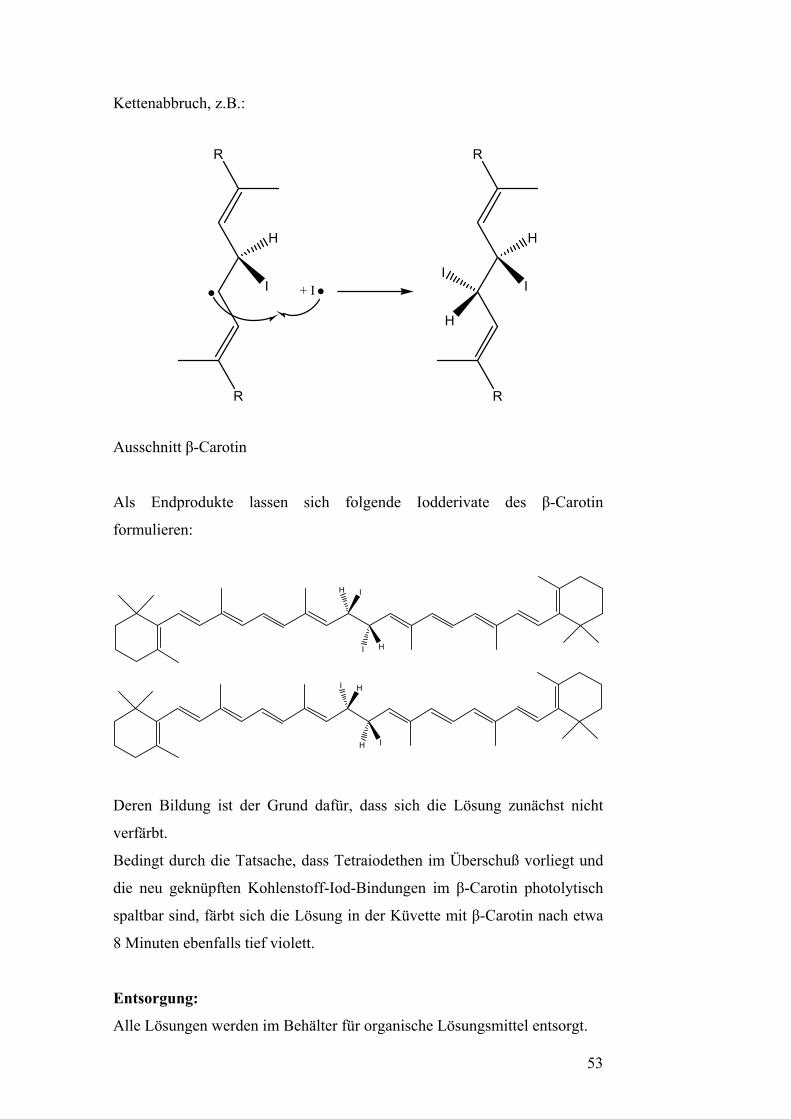
Kettenabbruch, z.B.:
R
R
H
I + I
R
R
H
II
H
Ausschnitt β-Carotin
Als Endprodukte lassen sich folgende Iodderivate des β-Carotin
formulieren:
H
I
I
H
I
H
H
I
Deren Bildung ist der Grund dafür, dass sich die Lösung zunächst nicht
verfärbt.
Bedingt durch die Tatsache, dass Tetraiodethen im Überschuß vorliegt und
die neu geknüpften Kohlenstoff-Iod-Bindungen im β-Carotin photolytisch
spaltbar sind, färbt sich die Lösung in der Küvette mit β-Carotin nach etwa
8 Minuten ebenfalls tief violett.
Entsorgung:
Alle Lösungen werden im Behälter für organische Lösungsmittel entsorgt.
53
Versuch 13: Desaktivierung von Singulett-Sauerstoff durch β-Carotin,
auch als „Quenchen“ bezeichnet [6][21]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
β-Carotin in Aceton F, Xi 11-36-66-67 9-16-26
Mangandioxid Xn 20/22 25
Salzsäure, konz. C 34-37 26-45
Wasserstoffperoxid-
Lösung, w = 0,3
C 34 3-26-36/37/39-
45
Natronlauge,
2 mol/L
C 35 26-36/37/39-45
gesättigte
Natriumthiosulfatlösung
- - -
Geräte:
Becherglas, Pasteurpipetten, Reagenzglas mit durchbohrtem Gummistopfen,
Gärröhrchen
Durchführung:
Vorbereitung:
Vor der eigentlichen Versuchsdurchführung muss zunächst eine alkalische
Wasserstoffperoxid-Lösung angesetzt werden. Hierzu mischt man 2 molare
Natronlauge mit 30 %iger Wasserstoffperoxid-Lösung im Verhältnis 5:1.
1. Schritt: Erzeugung von Singulett-Sauerstoff
Hierzu befüllt man ein Reagenzglas 1 cm hoch mit Mangandioxid. Dieses
wird im Abzug mit konzentrierter Salzsäure überschichtet. Das Reagenzglas
wird mit einem durchbohrten Gummistopfen, in dem ein Gärröhrchen
steckt, verschlossen. Das Gärröhrchen wird mit der zuvor angesetzten
alkalischen Wasserstoffperoxid-Lösung befüllt.
54

2. Schritt: Desaktivierung von Singulett-Sauerstoff.
Die sich im Gärröhrchen befindliche alkalische Wasserstoffperoxid-Lösung
wird mit einem Tropfen β-Carotin-Lösung versetzt.
Beobachtung:
Zu 1.: Gasblasen, die durch die alkalische Wasserstoffperoxid-Lösung
strömen, sind von einem hellroten Leuchten umgeben.
Zu 2.: Es ist kein Leuchten mehr zu beobachten.
Auswertung:
Begriffsklärung Singulett-Sauerstoff:
Dem MO-Schema für das Disauerstoff-Molekül im Grundzustand kann man
entnehmen, dass die beiden am schwächsten gebundenen Elektronen je ein
antibindendes π*-Orbital besetzen.[6]
2s 2s
σ
σ
σ
σ2s
*2s
2px
*2px
2px2py 2pz 2pz2px 2py
π π
π π
2py 2pz
*2py*2pz
O-Atom O2-Molekül O-Atom
Energie
Abb. 26: Schematisches Energieniveaudiagramm für Disauerstoff im Grundzustand
55

Diese beiden Elektronen weisen parallele Spins auf. Dieser Triplett-Zustand
wird mit 3Σg- bezeichnet und ist der tiefste elektronische Zustand des
molekularen Sauerstoffs. Durch Energiezufuhr kann der Triplett-Sauerstoff
angeregt werden. Es ergeben sich zwei energiereichere Zustände, die jedoch
nicht den gleichen Betrag an Energie aufweisen. Im energiereicheren der
beiden Zustände (1 Σ +g) verbleibt jedes Elektron in seinem Orbital, jedoch
weisen die Elektronen antiparallelen Spin auf. Die Lebensdauer dieses
Singulett-Sauerstoffs beträgt 10-9 Sekunden. Unter Energieabgabe von 63,1
kJ/mol geht der Singulett-Sauerstoff 1 Σ +g zum Singulett-Sauerstoff 1 ∆
∆
g
(Lebensdauer 10-4 Sekunden) über. Nun besetzen beide Elektronen ein
Orbital (antiparalleler Spin), das zweite Orbital ist elektronenleer. Unter
weiterer Energieabgabe ( 94,5 kJ/mol) geht der Singulett-Sauerstoff 1g
zum Triplett-Sauerstoff (3 Σ -g) über. [6]
π π*2py
*2pzLebensdauer
10-9 s
10-4 s
8
1O2 (1 g+)Σ
1O2 (1 g)
Σ3O2 (3 g-)
∆
Abb. 27: Elektronenanordnungen im Grundzustand und den beiden angeregten Zuständen
eines Disauerstoff-Moleküls
Darstellung Singulett-Sauerstoff [6]
Versetzt man Mangandioxid mit konzentrierter Salzsäure, so entstehen
Chlorgas, Wasser und Manganchlorid.
MnO2(s) + 4 H3O+(aq) + 4 Cl-
(aq) Mn2+(aq) + 2 Cl-
(aq) + Cl2(g) + 6 H2O(l)
Strömt dieses Chlorgas durch eine alkalische Wasserstoffperoxid-Lösung,
so entstehen zuerst Hypochlorit-Ionen. Diese reagieren im weiteren Verlauf
56

mit Hydroperoxid zu Chlorperoxid-Ionen. Die Chlorperoxid-Ionen
wiederum zerfallen in Chlorid-Ionen und Singulett-Sauerstoff.
Cl2(g) Cl2(aq)
Cl2(aq) + 2 Na+(aq) + 2 OH-
(aq) ClO-
(aq) + Cl-(aq) + 2 Na+
(aq) + H2O
H2O2(aq) + OH-(aq) HO2
-(aq) + H2O
HO2-(aq) + ClO-
(aq) ClOO-(aq) + OH-
(aq)
ClOO-(aq) Cl-
(aq) + 1O2(aq)
Der Zerfall der Chlorperoxid-Ionen lässt sich mit folgenden Lewis-Formeln
darstellen:
O O Cl O O Cl+
Man erkennt, dass im Sauerstoffmolekül keine ungepaarten Elektronen
vorliegen.
Da Singulett-Sauerstoff 1O2 (↑↓ ) (1 Σ +g) instabil ist, zerfällt dieser zuerst in
Singulett-Sauerstoff 1O2 (↑↓ ) (1 ∆ g) und schließlich in Triplett-Sauerstoff 3O2 ( ) (↑↑ 3 Σ -
g).
1O2 (1Σg
+) 1O2 (1∆g) 1O2 1O2 (1∆g) 3O2 3O2 (3Σg
-)
Der Zerfall von Singulett-Sauerstoff 1O2 verläuft unter Aussendung von
Licht. Während des ersten Schrittes ist dieses Licht nicht sichtbar, da es im
infraroten Bereich liegt ( λ ≈ 1900 nm). Während des zweiten Schrittes ist
dieses Licht sichtbar (gelb-orange 633 nm und hellrot 703 nm).
Die Aussendung von Licht lässt sich wie folgt erklären:
Zwei angeregte Sauerstoffmoleküle vom Typ 1 ∆ g-O2 stoßen zusammen und
gehen unter Austausch von je einem Elektron in den Grundzustand 3 Σ -g
57

über. Der Elektronenaustausch erfolg so, dass jedes Sauerstoffmolekül
wieder zwei Elektronen mit parallelem Spin besitzt.[19]
1O2 (↑↓ ) + 1O2 ( ) ↑↓ 3O2 ( ) + ↑↑ 3O2 (↓↓ )
Desaktivierung von Singulett-Sauerstoff durch Carotinoide
Carotinoide können Singulett-Sauerstoff nach dem „Dexter-Mechanismus“
desaktivieren. Die Energieübertragung erfolgt über einen doppelten
Elektronenaustausch. Der Elektronendonator (Singulett-Sauerstoff)
überträgt das energetisch angeregte Elektron auf den Elektronenakzeptor
(Carotinoid). Der Elektronenakzeptor wiederum gibt ein nicht angeregtes
Elektron an den Elektronendonator zurück. Beide Schritte erfolgen mehr
oder weniger konzertiert. Notwendigerweise müssen sich bei diesem
Mechanismus die Orbitale von Elektronendonator und Elektronenakzeptor
überlappen. Der Mechanismus wirkt nur im Nahbereich von Molekülen bis
etwa 10 Å. Während der Reaktion gehen sowohl der Sauerstoff als auch das
Carotinoid vom Singulett- in den Triplettzustand über. [10]
1O2 + 1Car 3O2 + 3Car
Dieser Schritt wird auch als „Quenchen“ bezeichnet, d.h. Carotinoide
übernehmen die Energie des Singulett-Sauerstoffs ohne dabei selbst zerstört
zu werden.[21]
In einem letzten Schritt geht das Carotinoid durch ein Intersystem Crossing
in den Singulettzustand zurück. [7]
3Car 1Car
Entstehung und Desaktivierung von Singulett-Sauerstoff in grünen Blättern
Innerhalb der Lebenszeit der angeregten Chlorophyll-Moleküle ist es
möglich, dass vor der photosynthetischen Ladungstrennung ein gewisser
Teil der Anregungszustände durch Intersystem Crossing in Triplettzustände
58

übergeht. Diese liegen energetisch höher als der Triplettzustand des
Sauerstoffs und können deshalb durch Energieübertragung den Sauerstoff
(3O2) in den elektronisch angeregten Singlett-Sauerstoff (1O2) überführen.
Dieser Singulett-Sauerstoff geht mit fast allen organischen Molekülen
Oxidationsreaktionen ein. Dies führt somit zur oxidativen Zerstörung von
Proteinen, Pigmenten insbesondere auch von Chlorophyll. Aus diesem
Grund ist es wichtig, Singulett-Sauerstoff schnellstmöglich unschädlich zu
machen. Dies ist die Aufgabe der Carotinoide im Blattgrün. Ohne
Carotinoide würden alle grünen Pflanzenteile durch Photooxidation
zerstört. [7]
Entsorgung:
Nachdem alle Lösungen mit Natriumthiosulfat-Lösung versetzt wurden,
können diese neutral im Behälter für Schwermetallabfall entsorgt werden.
59

2.5. Chromatographie
2.5.1. Erläuterung der unterschiedlichen Methoden
Der Begriff „Chromatographie“ leitet sich aus dem Griechischem ab und
bedeutet so viel wie „Farbenschreiben“. Die Entwicklung der
Chromatographie geht auf den russischen Botaniker und Biochemiker
Mikail Tswett zurück, der 1906 die Lösung eines grünen Blattextraktes
durch eine mit Calciumcarbonat gefüllte Säule leitete. Dabei fand eine
Auftrennung des Extraktes statt. Er nannte das Präparat „Chromatogramm“
und die Methode „Chromatographie“. [16]
In der heutigen Zeit versteht man unter Chromatographie physikalische
Methoden, bei denen eine Auftrennung eines Stoffes durch Verteilung
zwischen einer ruhenden und einer beweglichen Phase erfolgt. Die ruhende
Phase wird auch als „stationäre Phase“ bezeichnet, die bewegliche Phase als
„mobile Phase“. Während des Verfahrens strömt die mobile Phase an der
stationären vorbei. Beide Phasen dürfen nicht miteinander mischbar sein.
Aus den unterschiedlichen physikalisch-chemischen Vorgängen, welche bei
einer Chromatographie ablaufen, ergibt sich eine Grobeinteilung in zwei
Hauptgruppen.[16]
Bei der Verteilungschromatographie verteilen sich die zu trennenden Stoffe
auf einem polaren Flüssigkeitsfilm eines Festkörpers als stationärer Phase
und einer Flüssigkeit oder einem Gas als mobiler Phase.[16]
Verteilen sich die zu trennenden Stoffe auf einen Festkörper mit
physikalischen Oberflächenkräften (stationäre Phase) und eine Flüssigkeit
oder ein Gas (mobile Phase), so spricht man von
Adsorptionschromatographie. [16]
Eine wesentlich exaktere Einteilung der verschiedenen
Chromatographieverfahren erzielt man, wenn man diese nach der
jeweiligen, angewandten Ausführungstechnik untergliedert. Folgende Arten
der Chromatographie sind heute gebräuchlich: Papierchromatographie (PC),
Dünnschichtchromatographie (DC), Säulenchromatographie (SC),
Gaschromatographie (GC). [6]
60

Papierchromatographie
Die einfachste Art der Chromatographie ist die Papierchromatographie. Als
stationäre Phase dient hier spezielles Chromatographiepapier. Ist dieses
nicht verfügbar, so kann man - insbesondere in der Schule - auch auf
Filterpapier zurückgreifen. Das zu trennende Stoffgemisch löst sich im
polaren Wasserfilm („Cellulose-Wasser-Komplex“) des Papiers. Als mobile
Phase dient ein Fließmittel, meist ein organisches Lösungsmittel oder
Lösungsmittelgemisch. Somit kann man die Papierchromatographie als
Verteilungschromatographie auffassen. Hier ist es besonders wichtig, dass
die mobile Phase nur schwer bzw. nicht mit Wasser mischbar ist.[6]
Zur Durchführung einer Papierchromatographie wird das Gemisch auf die
stationäre Phase aufgetragen. Die darüber strömende mobile Phase löst das
Gemisch – oder auch nur Teile davon – aus der stationären Phase heraus
und transportiert es vom Ort der Auftragung (Start) weg. Dabei gilt, je
besser sich ein Stoff in der mobilen Phase löst, desto weiter wird er von ihr
fortbewegt. Umgekehrt gilt, je schlechter sich ein Stoff in der mobilen Phase
löst, desto näher verbleibt er am Start.
Aus dem Quotienten aus der Entfernung des Stoffes vom Start und der
Entfernung der Fließmittelfront vom Start ergibt sich der Retentionsfaktor
oder auch Rf-Wert genannt.[6]
Entfernung des Stoffes vom Start
Entfernung der Fließmittelfront vom StartRf =
Dünnschichtchromatographie
Eine weit verbreitetes Verfahren der Chromatographie ist die
Dünnschichtchromatographie. Als stationäre Phase dient eine maximal
1 mm dünne Trennschicht, die auf eine Trägerplatte aufgebracht wird.
Mögliche Materialien der Trägerplatte sind Glas, Kunststoff oder
Aluminium. Das Trennschichtmaterial entscheidet über die physikalisch-
chemischen Vorgänge, die eine Trennung bewirken:
61

Trennschichtmaterialien physikalisch-chemische Vorgänge
Aluminiumoxid (Al2O3) Adsorption
Kieselgel (SiO2) Adsorption, daneben auch
Verteilung, Ionenaustausch und
Siebwirkung möglich
Cellulose meist Verteilung, aber auch
Adsorption möglich.
Polyamide strukturbedingte Siebwirkung
Zur Durchführung einer Dünnschichtchromatographie wird das zu
trennende Substanzgemisch meist in Form eines Punktes auf der DC-Karte
aufgetragen. Diese wird nun senkrecht in eine mit Fließmittel befüllte DC-
Kammer getaucht. Durch kapillare Vorgänge steigt das Fließmittel nach
oben und führt die Gemischbestandteile unterschiedlich weit mit.[6]
Säulenchromatographie
Bei der Säulenchromatographie gibt es zwei Arten, die zur Anwendung
kommen: die klassische Säulenchromatographie und die Hochdruck-
Flüssigkeitschromatographie (HPLC, high pressure liquid chromatography).
Bei der klassischen Säulenchromatographie wird eine Glassäule mit einem
Durchmesser von mindestens 1 cm mit der stationären Phase gefüllt.
Meistens verwendet man Aluminiumoxid oder Kieselgel mit einem
Teilchendurchmesser von 100 bis 200 µm. Anschließend gibt man von oben
die zu trennende, gelöste Substanz in die Säule. Nun lässt man so lange die
flüssige mobile Phase durch die Säule fließen, bis die einzelnen Substanzen
des Gemisches die Säule getrennt verlassen haben (Elution). Die Trennung
wird hierbei durch die unterschiedlichen Wechselwirkungen der Substanzen
mit mobiler und stationärer Phase erzielt. Die Verweildauer der einzelnen
Phasen in der Säule wird als Retentionszeit bezeichnet. [6]
Das Prinzip der Hochdruck-Flüssigkeitschromatographie ist analog.
Allerdings besteht hier das Chromatographierohr meist aus Edelstahl mit
einem Durchmesser von 2-6 mm. Der Teilchendurchmesser der stationären
Phase (Aluminiumoxid oder Kieselgel) ist mit maximal 50 µm entsprechend
62

kleiner. Um eine höhere Durchflussgeschwindigkeit der mobilen Phase zu
erzielen, wird diese mit hohem Druck durch die Säule gepresst. [6]
Gaschromatographie
Ebenso wie bei der Säulenchromatographie befindet sich die stationäre
Phase in einer Säule. Hier wird zwischen gepackten Säulen und
Kapillarsäulen unterschieden. Gepackte Säulen haben einen Durchmesser
von 2-6 mm und eine Länge von einem halben bis etwa 20 Metern. Als
Adsorbens werden auch hier Aluminiumoxid oder Kieselgel eingesetzt.
Kapillarsäulen haben einen Durchmesser von 0,2-0,6 mm und eine Länge
von bis zu 3000 m. Als Adsorptionsmittel dient in diesem Fall eine flüssige
stationäre Phase auf der Innenseite der Kapillaren (Dünnfilmkapillare).
Wird die Kapillare mit dem Adsorbens gefüllt, so handelt es sich um eine
Dünnschichtkapillare. In allen Fällen ist die mobile Phase ein Gas
(Wasserstoff, Helium, Argon, usw.). [16]
Zur Trennung wird das Substanzgemisch in einen vorgeheizten Block
eingespritzt. Dies gewährleistet eine unmittelbare Verdampfung des
Gemisches. Zu beachten ist hierbei, dass sich die Bestandteile des
Gemisches beim Verdampfen nicht zersetzen. Das gasförmige Gemisch
wird nun mit dem Trägergas durch die Säule gedrückt. Wie schon bei der
Säulenchromatographie wird auch hier die Trennung durch unterschiedliche
Wechselwirkungen der Substanzen mit der mobilen bzw. stationären Phase
erzielt. Am Ende der Säule befindet sich bei der Gaschromatographie ein
Detektor. Dieser Detektor misst Zeit und Volumen, die charakteristischen
Kenngrößen bei der Gaschromatographie. [16]
63
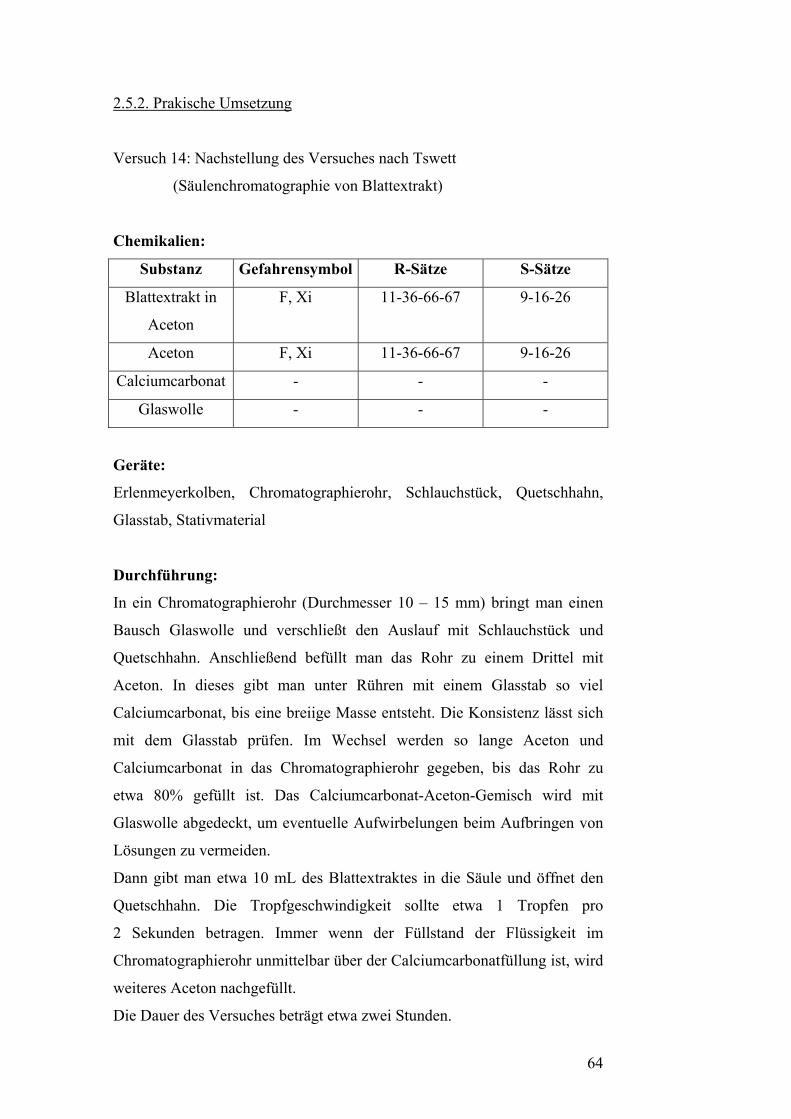
2.5.2. Prakische Umsetzung
Versuch 14: Nachstellung des Versuches nach Tswett
(Säulenchromatographie von Blattextrakt)
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Aceton F, Xi 11-36-66-67 9-16-26
Calciumcarbonat - - -
Glaswolle - - -
Geräte:
Erlenmeyerkolben, Chromatographierohr, Schlauchstück, Quetschhahn,
Glasstab, Stativmaterial
Durchführung:
In ein Chromatographierohr (Durchmesser 10 – 15 mm) bringt man einen
Bausch Glaswolle und verschließt den Auslauf mit Schlauchstück und
Quetschhahn. Anschließend befüllt man das Rohr zu einem Drittel mit
Aceton. In dieses gibt man unter Rühren mit einem Glasstab so viel
Calciumcarbonat, bis eine breiige Masse entsteht. Die Konsistenz lässt sich
mit dem Glasstab prüfen. Im Wechsel werden so lange Aceton und
Calciumcarbonat in das Chromatographierohr gegeben, bis das Rohr zu
etwa 80% gefüllt ist. Das Calciumcarbonat-Aceton-Gemisch wird mit
Glaswolle abgedeckt, um eventuelle Aufwirbelungen beim Aufbringen von
Lösungen zu vermeiden.
Dann gibt man etwa 10 mL des Blattextraktes in die Säule und öffnet den
Quetschhahn. Die Tropfgeschwindigkeit sollte etwa 1 Tropfen pro
2 Sekunden betragen. Immer wenn der Füllstand der Flüssigkeit im
Chromatographierohr unmittelbar über der Calciumcarbonatfüllung ist, wird
weiteres Aceton nachgefüllt.
Die Dauer des Versuches beträgt etwa zwei Stunden.
64

Beobachtung:
Im oberen Bereich des Calciumcarbonat-Aceton-Gemisches sind zwei
dünne gelb-braune Banden zu erkennen. An diese schließt sich eine grüne
Bande an, die sich fast bis nach unten erstreckt.
Abb. 28: Ausschnitt aus dem Chromatographierohr
Auswertung:
Bei den beiden gelb-braunen Banden handelt es sich um zwei
unterschiedliche Xanthophylle. Diese polaren Carotinoide werden von
relativ unpolaren Fließmittel Aceton nicht sehr weit mitgeführt. Das im
Vergleich zu den Xantophyllen unpolarere Chlorophyll wird wesentlich
weiter mitgeführt.
Die Chromatographie nach Tswett stellt eine sehr einfache Form der
Chromatographie dar. Es kann gezeigt werden, dass in einem Blattextrakt
außer dem grünen Farbstoff zwei weitere Farbstoffe enthalten sind.
Entsorgung:
Die organischen Lösungen können neutral im Behälter für organische
Lösungsmittel entsorgt werden. Das Füllmaterial des
Chromatographierohres wird getrocknet und in der Feststofftonne entsorgt.
65

Versuch 15: Chromatographie mit Tafelkreide
Mit sehr viel weniger Aufwand kann der Versuch nach Tswett mit
Tafelkreide durchgeführt werden. Die mobile und die stationäre Phase sind
identisch mit denen des vorangegangenen Versuches. Tafelkreide hat bereits
die Form einer Säule. Ein Glasrohr wird nicht benötigt.
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Tafelkreide - - -
Geräte:
Weithalserlenmeyerkolben 250 mL
Durchführung:
Ein komplettes Stück Tafelkreide wird im Trockenschrank 15 Minuten bei 110 °C getrockenet. Anschließend wird ein Weithalserlenmeyerkolben etwa 3 mm hoch mit der
Blattextrakt-Lösung befüllt und die Tafelkreide senkrecht freistehend in den
Erlenmeyerkolben gestellt. Nach ca. 5 Minuten kann die Tafelkreide aus
dem Erlenmeyerkolben entnommen werden.
Beobachtung:
Im unteren Bereich der Kreide ist eine dunkelbraune Bande zu erkennen.
Darüber befindet sich eine weitgestreckte und unscharfe hellgrüne Bande.
Daran schließt sich eine scharfe dunkelgrüne Bande an. Über dieser Bande
befindet sich eine dicke gelbe Bande.
Abb. 29: Tafelkreide nach erfolgter Chromatographie
66

Auswertung:
Mit diesem Versuch lässt sich zeigen, dass in einem Blattextrakt mindestens
vier Farbstoffe enthalten ist. Wie auch im vorigen Versuch sind die
Xanthophylle nicht sehr weit vom Fließmittel mitgenommen worden
(braune Bande). Allerdings ist hier nur ein Xanthophyllgemisch zu erkennen
und nicht zwei aufgetrennte Xanthophylle wie im vorangegangenen
Versuch. Es gelingt sogar, zwei verschiedene grüne Blattfarbstoffe
aufzutrennen: Chlorophyll a (dunkelgrün) und b (hellgrün).
Die Chlorophylle a und b sind durch den Phytol-Rest weit weniger polar als
die Xanthophylle. Sie werden deshalb weiter vom Fließmittel transportiert.
Die beiden Chlorophylle unterscheiden sich nur im Rest am C7-Atom
(Chlorophyll a: Methylgruppe, Chlorophyll b: Aldehydgruppe).
Chlorophyll a mit der im Vergleich zu Chlorophyll b unpolareren
Methylgruppe wird noch etwas weiter vom Fließmittel mitgenommen. Bei
der abschließenden dicken gelben Bande handelt es sich um β-Carotin,
welches bei der vorangegangenen Chromatographie nicht zu erkennen war.
Da dieses vollkommen unpolar ist, steigt es am weitesten mit nach oben.
Entsorgung:
Die Tafelkreide kann in der Feststofftonne entsorgt werden.
67
Bei den beiden durchgeführten Methoden konnte gezeigt werden, dass in
grünem Blattextrakt mehrere Farbstoffe enthalten sind. Allerdings ist der
Trenneffekt nicht sehr gut. Eine wesentlich bessere Trennwirkung wird
durch die Dünnschichtchromatographie erzielt.
Versuch 16: Herstellung von DC-Karten [24]
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Kieselgel - - -
Calciumhydroxid Xi 41 22-24-26-39
Ascorbinsäure
5 • 10-3 mol/L
- - -
Calciumchlorid Xi 36 22-24
Geräte:
Becherglas, Glasplatte 20 x 20 cm, Objektträger, Exsikkator, KPG-Rührer
komplett, Stativmaterial, Glasstab, Auftragegerät für Kieselgel (Degessa,
Heidelberg)
Durchführung:
In einem Becherglas werden 20 g Kieselgel, 12 mg Calciumhydroxid und
44 mL 5 • 10-3 molare Ascorbinsäurelösung eine Stunde lang gerührt. Die
breiige Masse wird anschließend mit einem Streichgerät auf die Glasplatte
aufgebracht. Zusätzlich werden mehrere Objektträger beschichtet. Dies
geschieht durch „Ausrollen“ mit einem Glasstab. Die hergestellten Platten
werden nun eine Stunde im Trockenschrank bei 100 °C getrocknet. Nach
3-5 Tagen Lagerung in einem Exsikkator können die Platten verwendet
werden.
68
Versuch 17: Dünnschichtchromatographie von Blattextrakt [5]
Vorbemerkung:
Die Versuchsergebnisse der Dünnschichtchromatographien mit selbst
hergestellten DC-Karten waren unzureichend. Es erfolgte nur eine äußert
undeutliche Auftrennung. Deshalb werde ich bei den folgenden Versuchen
auf DC-Karten aus industrieller Herstellung zurückgreifen.
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Petrolether
(40-60 °C)
F, Xn, N 11-38-48/20-
51/53-62-65-67
9-16-33-36/37-
60-62
Isopropanol F 11 7-16
Geräte:
DC-Kammer, DC-Mikrokarte (5 x 10 cm, Kieselgel auf Aluminiumfolie),
Fön, Glaskapillaren, Messpipette 10 mL, Pipettierthilfe
Durchführung:
Zu Beginn wird die DC-Kammer in einem Abzug mit dem Fließmittel
befüllt und verschlossen. Das Fließmittel besteht aus 10 mL Petrolether,
1 mL Isopropanol und einem Tropfen entionisiertem Wasser.
Der Blattextrakt wird als dünner Strich 1 cm vom unteren Rand und 1 cm
von den seitlichen Rändern der DC-Karte mit einer Glaskapillare
aufgetragen. Anschließend wird die DC-Karte trocken gefönt und erneut
Blattextrakt aufgetragen. Dieser Vorgang wird 8-10 mal wiederholt.
Anschließend wird die Karte in die DC-Kammer gestellt. Wenn sich die
Fließmittelfront etwa 1 cm unterhalb des oberen Randes der DC-Karte
befindet, wird die Chromatographie beendet und die Fließmittelfront
markiert.
69

Beobachtung:
Nach etwa 15 Minuten ist dieses Ziel erreicht. Im unteren Bereich der DC-
Karte ist eine breite gelb-braune Bande zu erkennen. Darauf folgen zwei
dünne, grüne Banden. Unterhalb der Fließmittelfront ist eine scharfe, gelbe
Bande zu erkennen.
Auswertung:
Wie schon im Versuch mit Tafelkreide werden die Xanthophylle nicht sehr
weit vom Fließmittel mitgeführt und auch nicht aufgetrennt. Man erkennt
aber eine deutliche Auftrennung von Chlorophyll b und darüber
Chlorophyll a. Deutlich weiter oben folgt die Bande des β-Carotins.
Messwerte:
Entfernung Start und Fließmittelfront: 8 cm
Stoff Entfernung
Stoff und Start
Rf-Wert
β-Carotin 6,5 cm 0,81
Chlorophyll a 3,4 cm 0,43
Chlorophyll b 3,1 cm 0,39
Xanthophylle 1,7 cm 0,21
Entsorgung:
Das Fließmittel wird im Behälter für organische Lösungsmittel entsorgt. Die
DC-Karte wird in der Feststofftonne entsorgt.
70
Versuch 18: Präparative Dünnschichtchromatographie mit photometrischer
Untersuchung [5]
Will man die einzelnen Banden einer Dünnschichtchromatographie näher
untersuchen, so bietet sich die Durchführung einer präparativen
Dünnschichtchromatographie an.
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Methanol
T, F 23/24/25-
39/23/24/25
7-16-36/37-45
Petrolether
(40-60 °C)
F, Xn, N 11-38-48/20-
51/53-62-65-67
9-16-33-36/37-
60-62
Isopropanol F 11 7-16
Aceton F, Xi 11-36-66-67 9-16-26
Dünnschichtchromatographie:
Geräte:
DC-Kammer, DC-Karte (20 x 20 cm, Kieselgel auf Aluminiumfolie), Fön,
Glaskapillaren, Messpipette 10 mL, Pipettierthilfe
Vorbemerkung:
Die Dünnschichtchromatographie von Blattfarbstoffen habe ich mehrmals
durchgeführt. Dabei habe ich festgestellt, dass die Auftrennung in die
Inhaltsstoffe wesentlich besser gelingt, wenn man das Blattgrün mit
Methanol extrahiert hat. Offensichtlich werden die einzelnen Blattfarbstoffe
besser herausgelöst. Die größeren Anteile der einzelnen Stoffe im Extrakt
spiegeln sich dann auch in einem verbesserten Chromatographieergebnis
wieder.
71

Durchführung:
Zu Beginn wird die DC-Kammer in einem Abzug mit dem Fließmittel
befüllt und verschlossen. Das Fließmittel setzt sich aus 20 mL Petrolether,
2 mL Isopropanol und zwei Tropfen entionisiertem Wasser zusammen.
Das Auftragen des Blattextraktes erfolgt wie bereits beim Versuch
„Dünnschichtchromatographie“ beschrieben.
Beobachtung:
Nach etwa 50 Minuten ist eine ausreichende Auftrennung der einzelnen
Stoffe erreicht. Man erkennt insgesamt acht Banden. Im unteren Bereich der
Karte werden drei gelb-braune Banden sichtbar. Daran schließen sich vier
grüne Banden an. In Nähe der Fließmittelfront befindet sich eine gelbe
Bande.
Abb. 30: Ergebnis der Dünnschichtchromatographie eines Blattextraktes in Methanol
72
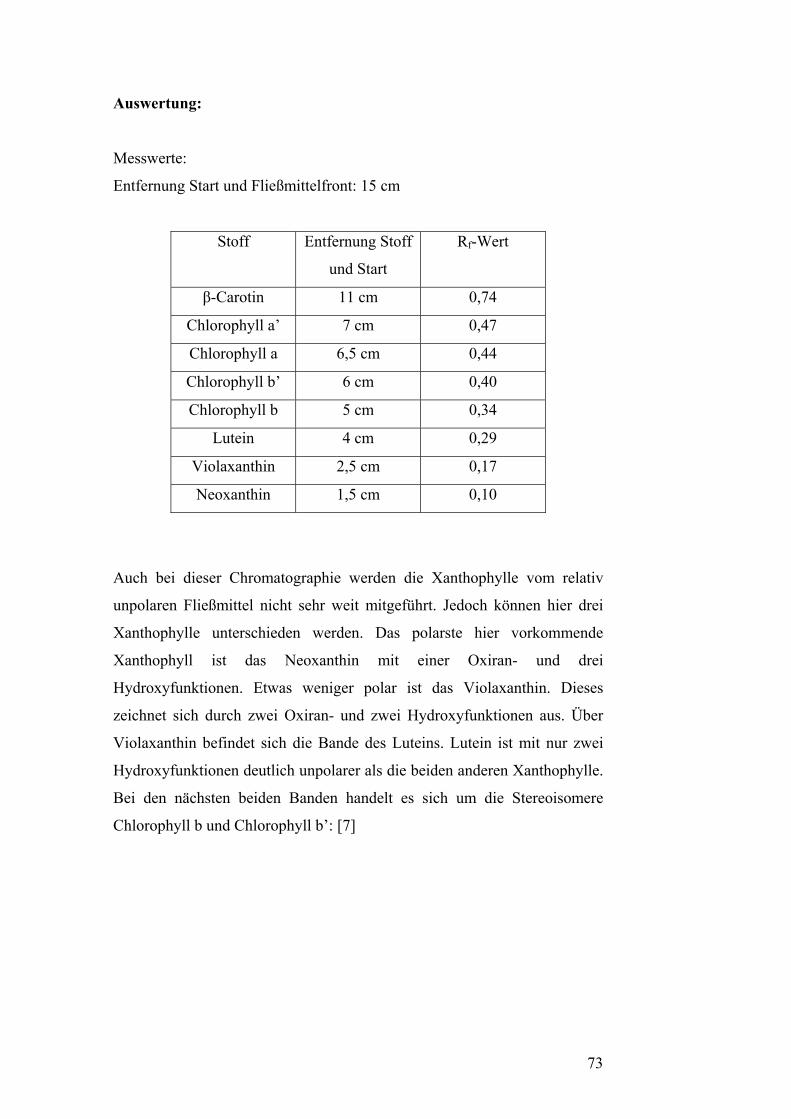
Auswertung:
Messwerte:
Entfernung Start und Fließmittelfront: 15 cm
Stoff Entfernung Stoff
und Start
Rf-Wert
β-Carotin 11 cm 0,74
Chlorophyll a’ 7 cm 0,47
Chlorophyll a 6,5 cm 0,44
Chlorophyll b’ 6 cm 0,40
Chlorophyll b 5 cm 0,34
Lutein 4 cm 0,29
Violaxanthin 2,5 cm 0,17
Neoxanthin 1,5 cm 0,10
Auch bei dieser Chromatographie werden die Xanthophylle vom relativ
unpolaren Fließmittel nicht sehr weit mitgeführt. Jedoch können hier drei
Xanthophylle unterschieden werden. Das polarste hier vorkommende
Xanthophyll ist das Neoxanthin mit einer Oxiran- und drei
Hydroxyfunktionen. Etwas weniger polar ist das Violaxanthin. Dieses
zeichnet sich durch zwei Oxiran- und zwei Hydroxyfunktionen aus. Über
Violaxanthin befindet sich die Bande des Luteins. Lutein ist mit nur zwei
Hydroxyfunktionen deutlich unpolarer als die beiden anderen Xanthophylle.
Bei den nächsten beiden Banden handelt es sich um die Stereoisomere
Chlorophyll b und Chlorophyll b’: [7]
73
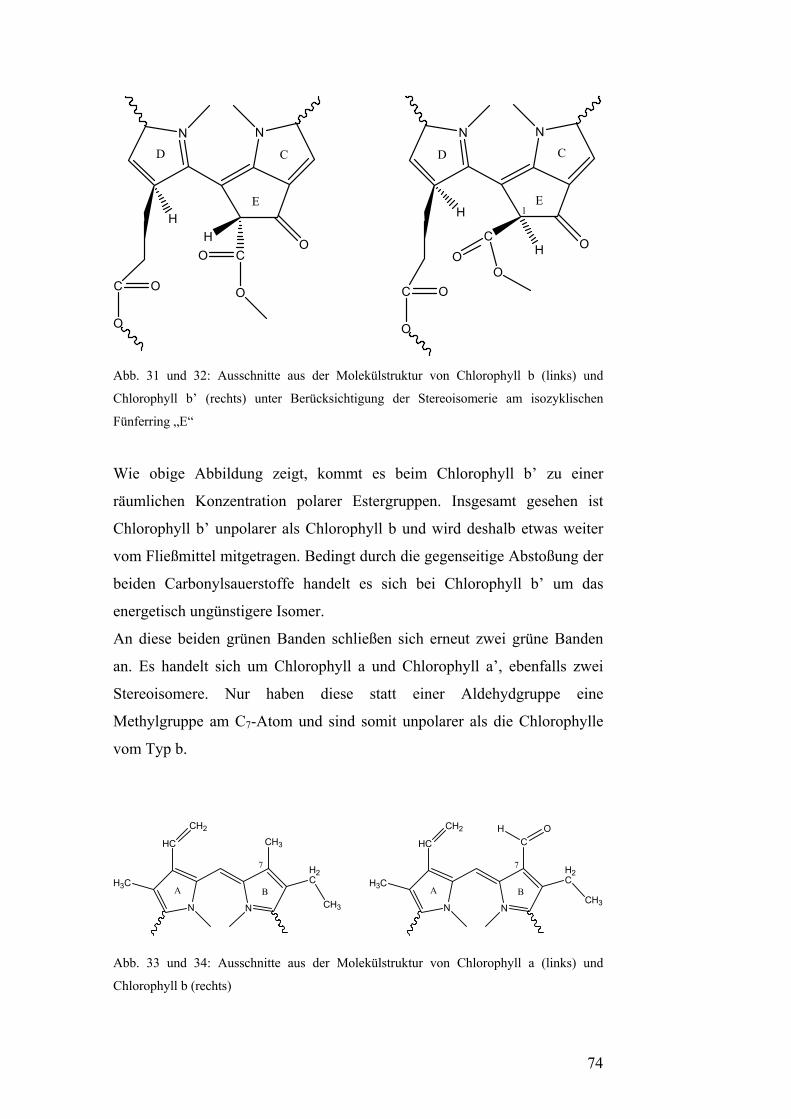
N N
O
N N
OHC
CH
H H
C CO
O OO
O O
O O
D D
E E
CC
1
Abb. 31 und 32: Ausschnitte aus der Molekülstruktur von Chlorophyll b (links) und
Chlorophyll b’ (rechts) unter Berücksichtigung der Stereoisomerie am isozyklischen
Fünferring „E“
Wie obige Abbildung zeigt, kommt es beim Chlorophyll b’ zu einer
räumlichen Konzentration polarer Estergruppen. Insgesamt gesehen ist
Chlorophyll b’ unpolarer als Chlorophyll b und wird deshalb etwas weiter
vom Fließmittel mitgetragen. Bedingt durch die gegenseitige Abstoßung der
beiden Carbonylsauerstoffe handelt es sich bei Chlorophyll b’ um das
energetisch ungünstigere Isomer.
An diese beiden grünen Banden schließen sich erneut zwei grüne Banden
an. Es handelt sich um Chlorophyll a und Chlorophyll a’, ebenfalls zwei
Stereoisomere. Nur haben diese statt einer Aldehydgruppe eine
Methylgruppe am C7-Atom und sind somit unpolarer als die Chlorophylle
vom Typ b.
N N
H3C
HC
CH2
CH3
H2C
7
A B
N N
H3C
HC
CH2
C
H2C
7
A B
OH
CH3CH3
Abb. 33 und 34: Ausschnitte aus der Molekülstruktur von Chlorophyll a (links) und
Chlorophyll b (rechts)
74

Das vollkommen unpolare β-Carotin wird am weitesten durch das
Fließmittel nach oben geführt.
Aufnahme der Absorptionsspektren[5]:
Geräte:
Spatel, Zentrifugengläser, Photometer (Shimadzu MPS 2000), Küvetten
Durchführung:
Zur Aufnahme der Absorptionsspektren werden die Banden einzeln mit
einem Spatel abgekratzt und in jeweils ein Zentrifugenglas überführt. Das
Kieselgel wird nun mit 3 mL Aceton überschichtet und die Farbstoffe
eluiert. Anschließend wird zentrifugiert, der Überstand kann für die
Spektroskopie verwendet werden.
Alle Absorptionsspektren werden im Bereich von 760-360 nm
aufgenommen. Zusätzlich zu den isolierten Banden wird auch ein Spektrum
des Blattextraktes aufgenommen.
75
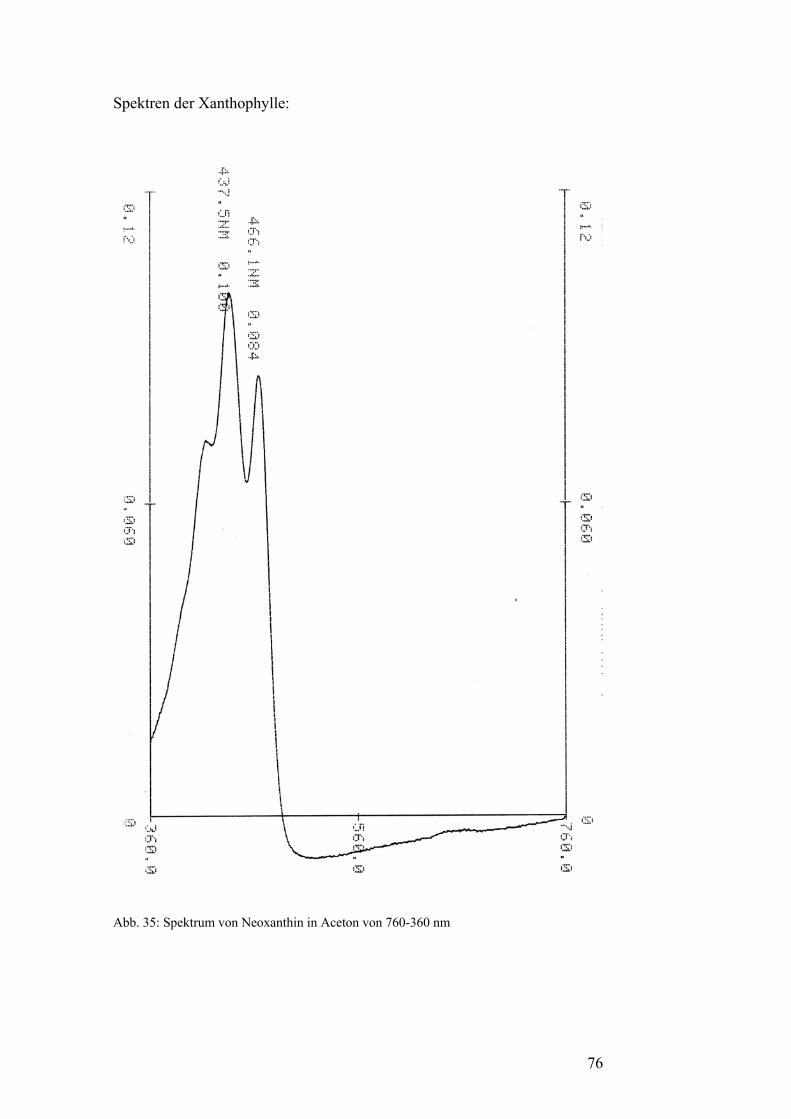
Spektren der Xanthophylle:
Abb. 35: Spektrum von Neoxanthin in Aceton von 760-360 nm
76
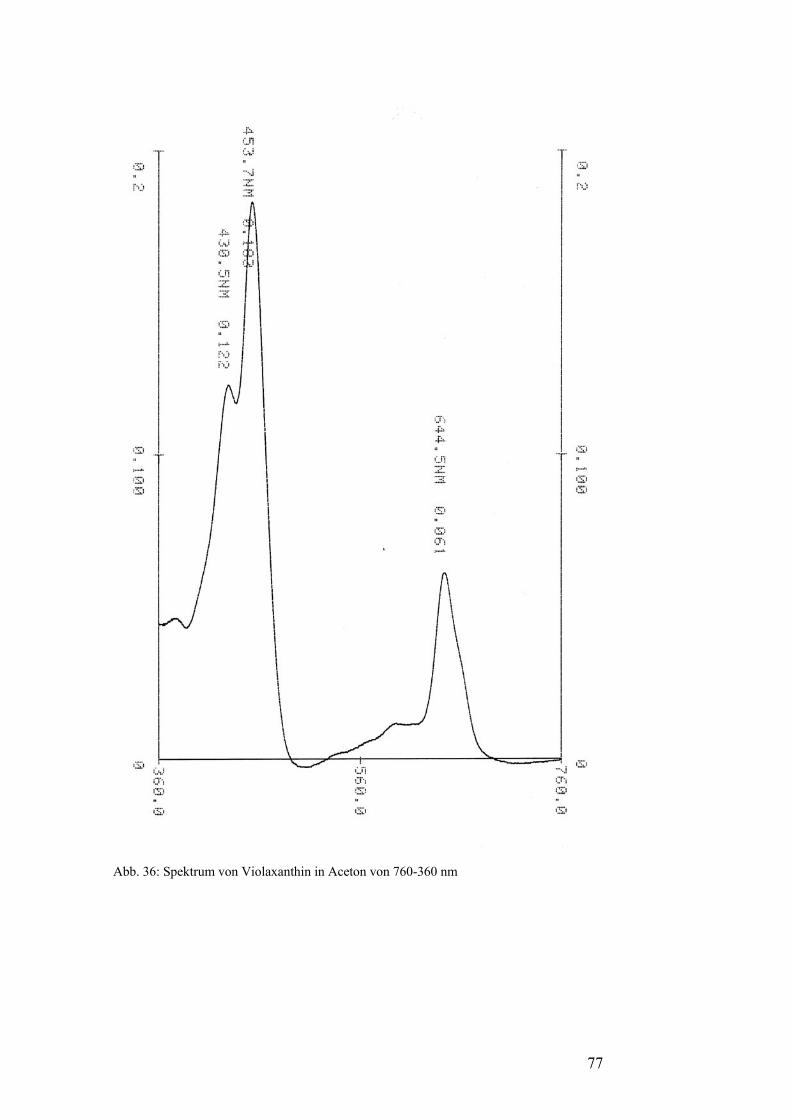
Abb. 36: Spektrum von Violaxanthin in Aceton von 760-360 nm
77
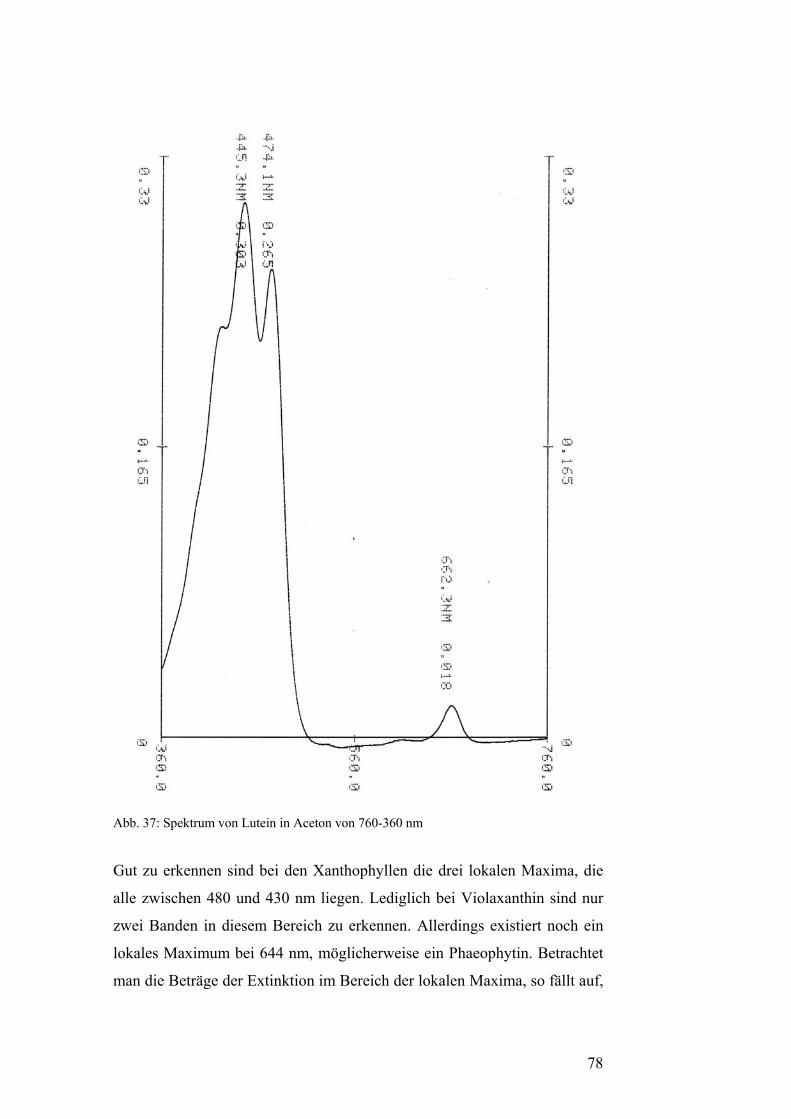
Abb. 37: Spektrum von Lutein in Aceton von 760-360 nm
Gut zu erkennen sind bei den Xanthophyllen die drei lokalen Maxima, die
alle zwischen 480 und 430 nm liegen. Lediglich bei Violaxanthin sind nur
zwei Banden in diesem Bereich zu erkennen. Allerdings existiert noch ein
lokales Maximum bei 644 nm, möglicherweise ein Phaeophytin. Betrachtet
man die Beträge der Extinktion im Bereich der lokalen Maxima, so fällt auf,
78
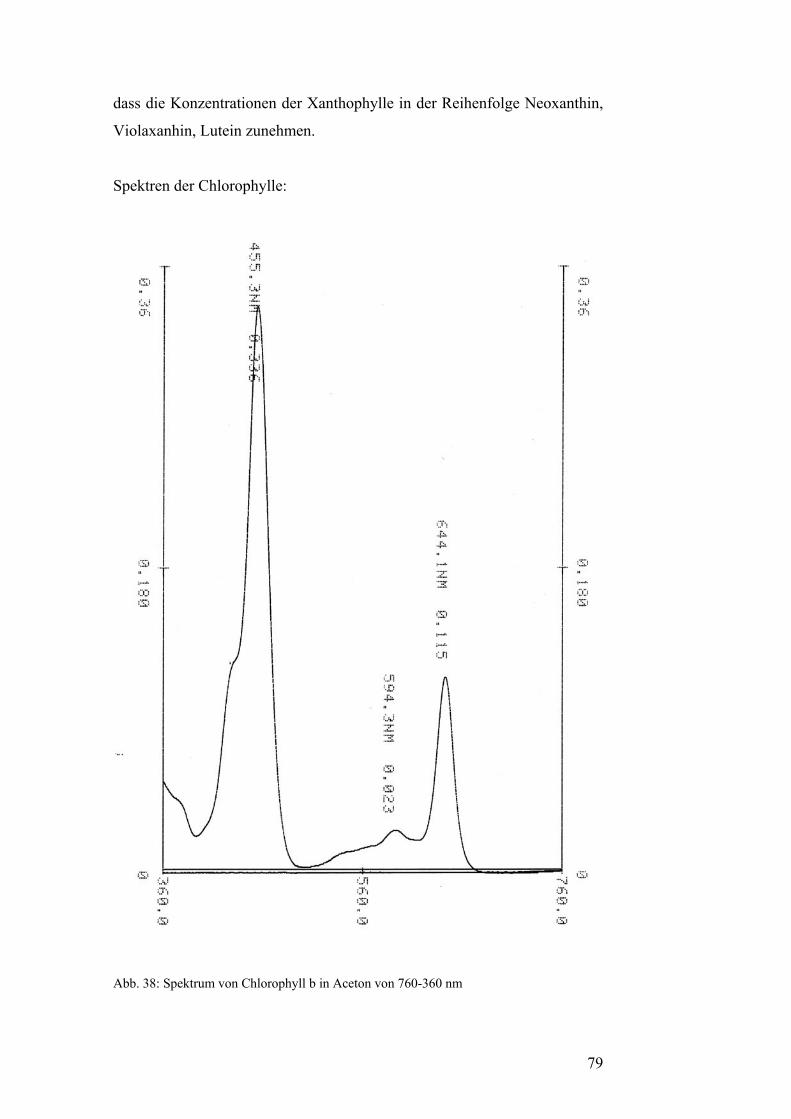
dass die Konzentrationen der Xanthophylle in der Reihenfolge Neoxanthin,
Violaxanhin, Lutein zunehmen.
Spektren der Chlorophylle:
Abb. 38: Spektrum von Chlorophyll b in Aceton von 760-360 nm
79
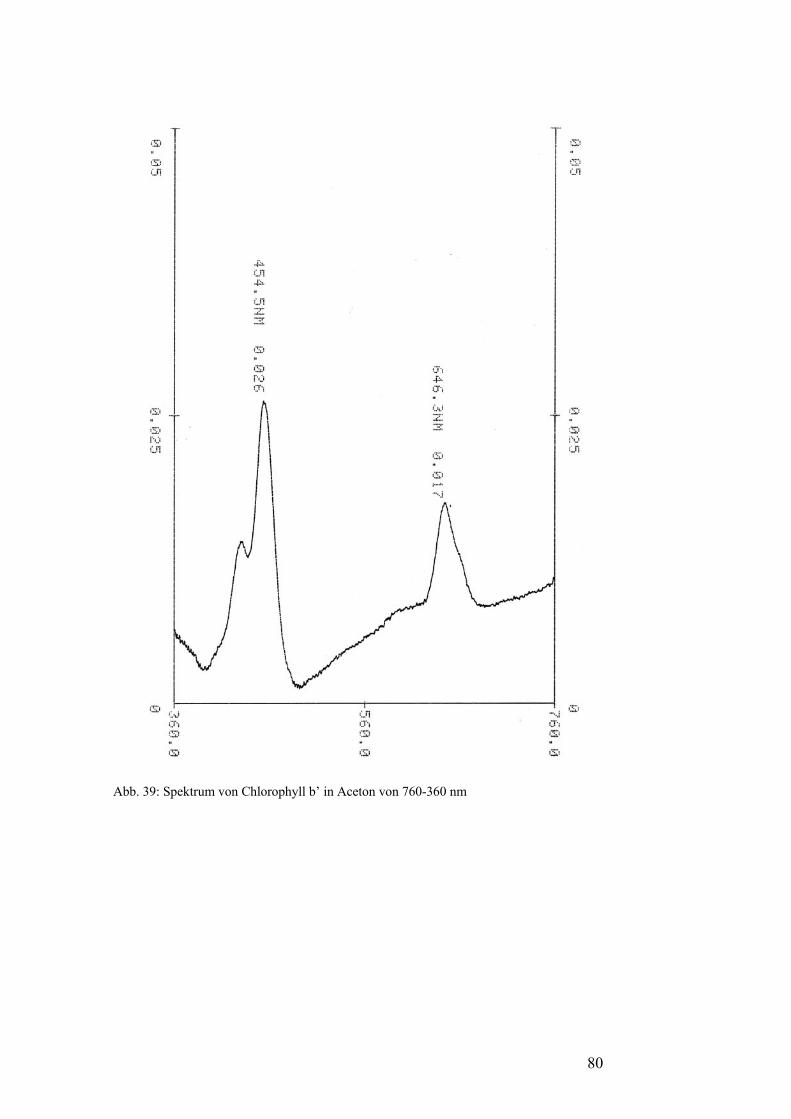
Abb. 39: Spektrum von Chlorophyll b’ in Aceton von 760-360 nm
80
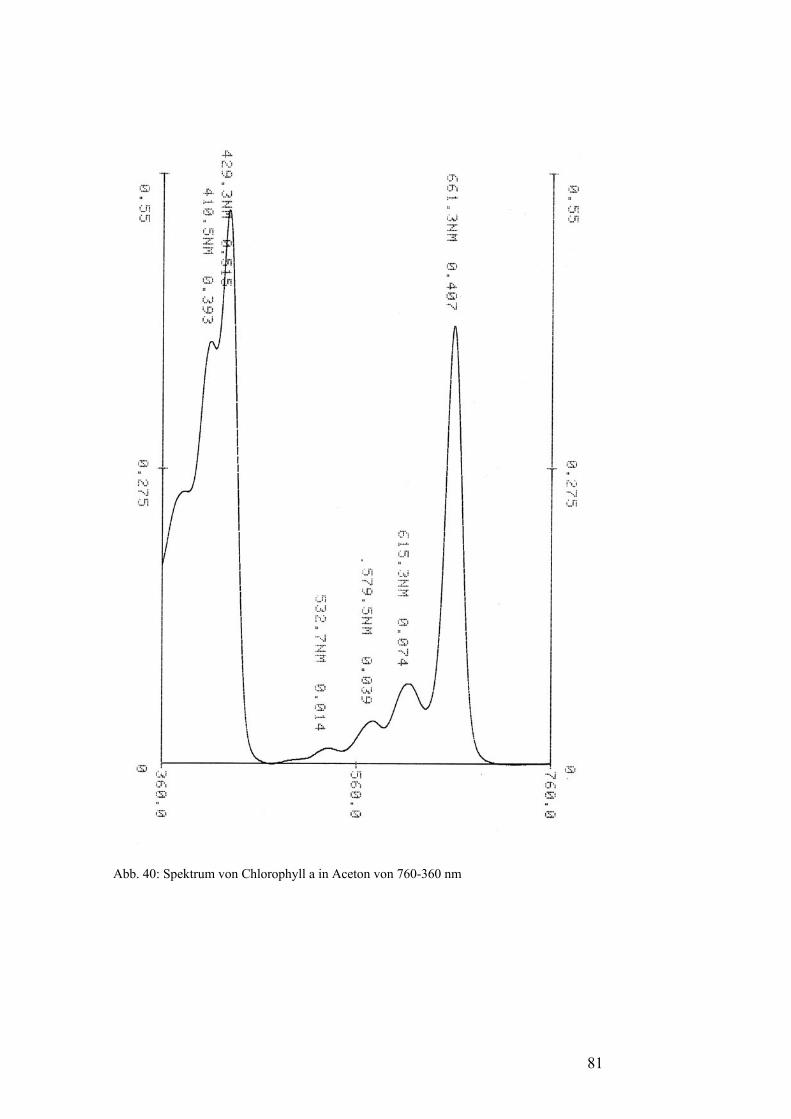
Abb. 40: Spektrum von Chlorophyll a in Aceton von 760-360 nm
81
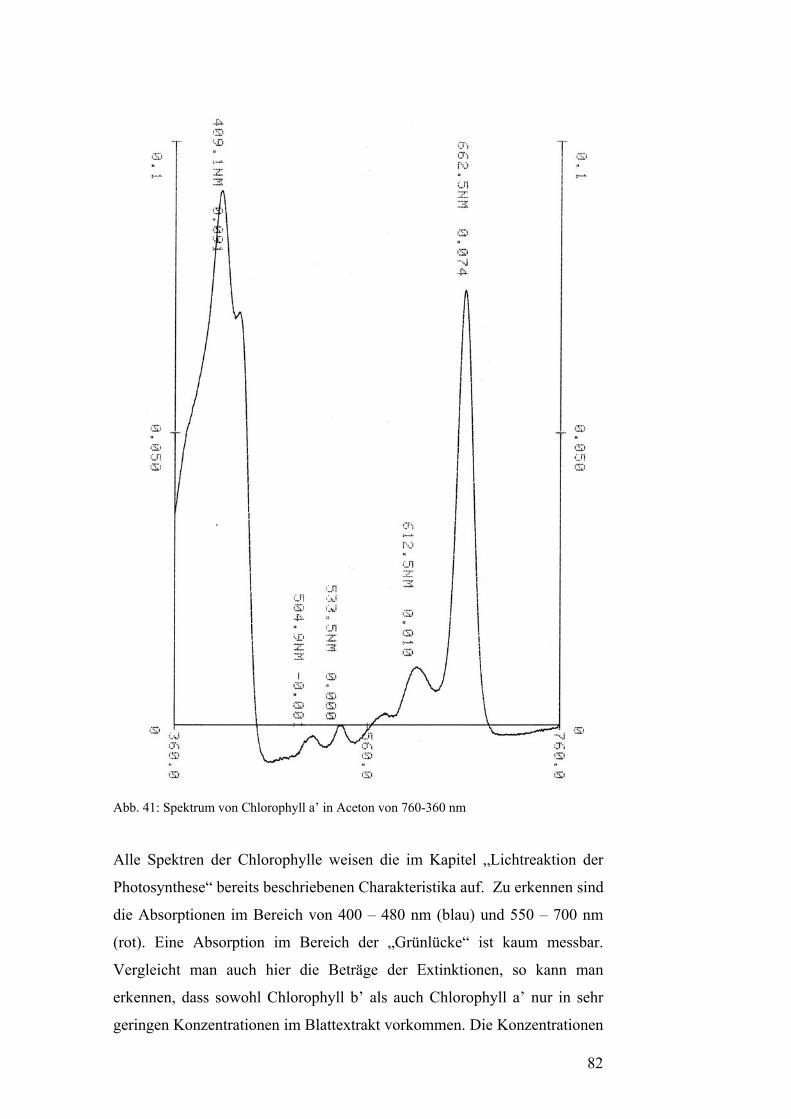
Abb. 41: Spektrum von Chlorophyll a’ in Aceton von 760-360 nm
Alle Spektren der Chlorophylle weisen die im Kapitel „Lichtreaktion der
Photosynthese“ bereits beschriebenen Charakteristika auf. Zu erkennen sind
die Absorptionen im Bereich von 400 – 480 nm (blau) und 550 – 700 nm
(rot). Eine Absorption im Bereich der „Grünlücke“ ist kaum messbar.
Vergleicht man auch hier die Beträge der Extinktionen, so kann man
erkennen, dass sowohl Chlorophyll b’ als auch Chlorophyll a’ nur in sehr
geringen Konzentrationen im Blattextrakt vorkommen. Die Konzentrationen
82

der Chlorophylle a und b ist um ein Vielfaches höher. Begründen lässt sich
dies dadurch, dass es sich bei diesen Chlorophyllen um die energetisch
günstigeren Isomere (d.h. die mit geringerem Energieinhalt) handelt. Diese
werden in der Natur üblicherweise bevorzugt synthetisiert. Vergleicht man
die maximalen Extinktionen von Chlorophyll a und b miteinander, so
erkennt man, dass Chlorophyll a in höherer Konzentration vorkommt als
Chlorophyll b.
83
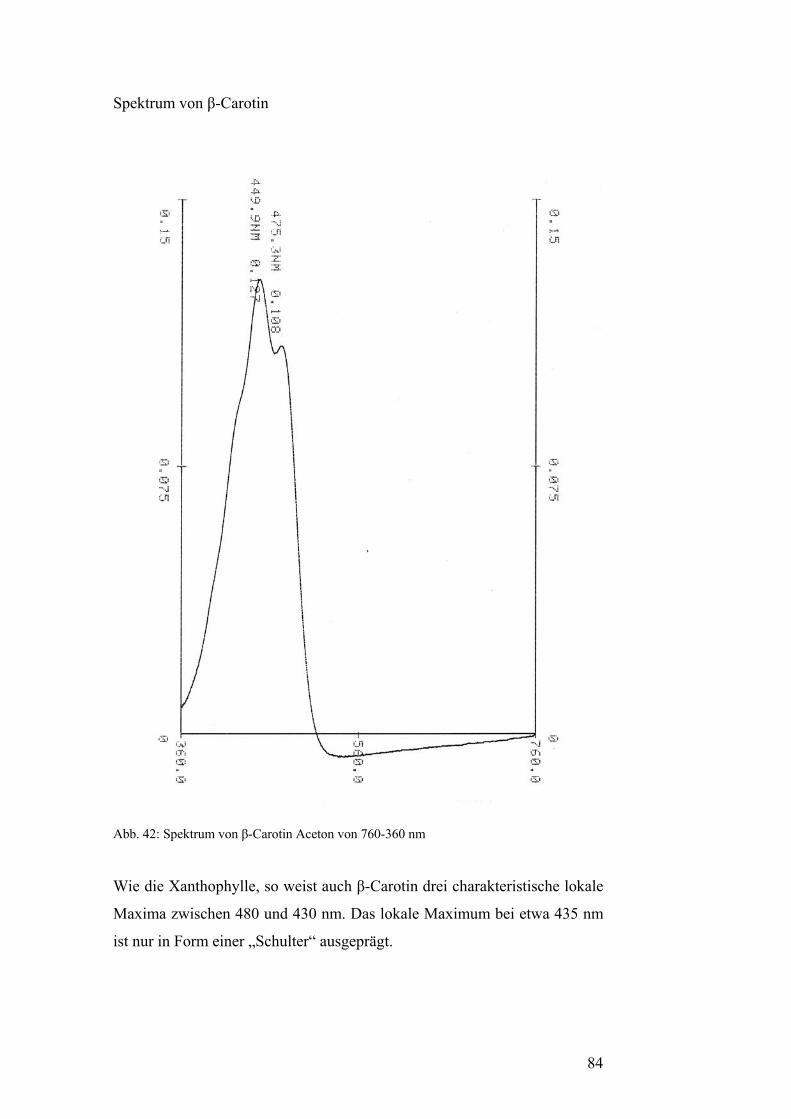
Spektrum von β-Carotin
Abb. 42: Spektrum von β-Carotin Aceton von 760-360 nm
Wie die Xanthophylle, so weist auch β-Carotin drei charakteristische lokale
Maxima zwischen 480 und 430 nm. Das lokale Maximum bei etwa 435 nm
ist nur in Form einer „Schulter“ ausgeprägt.
84
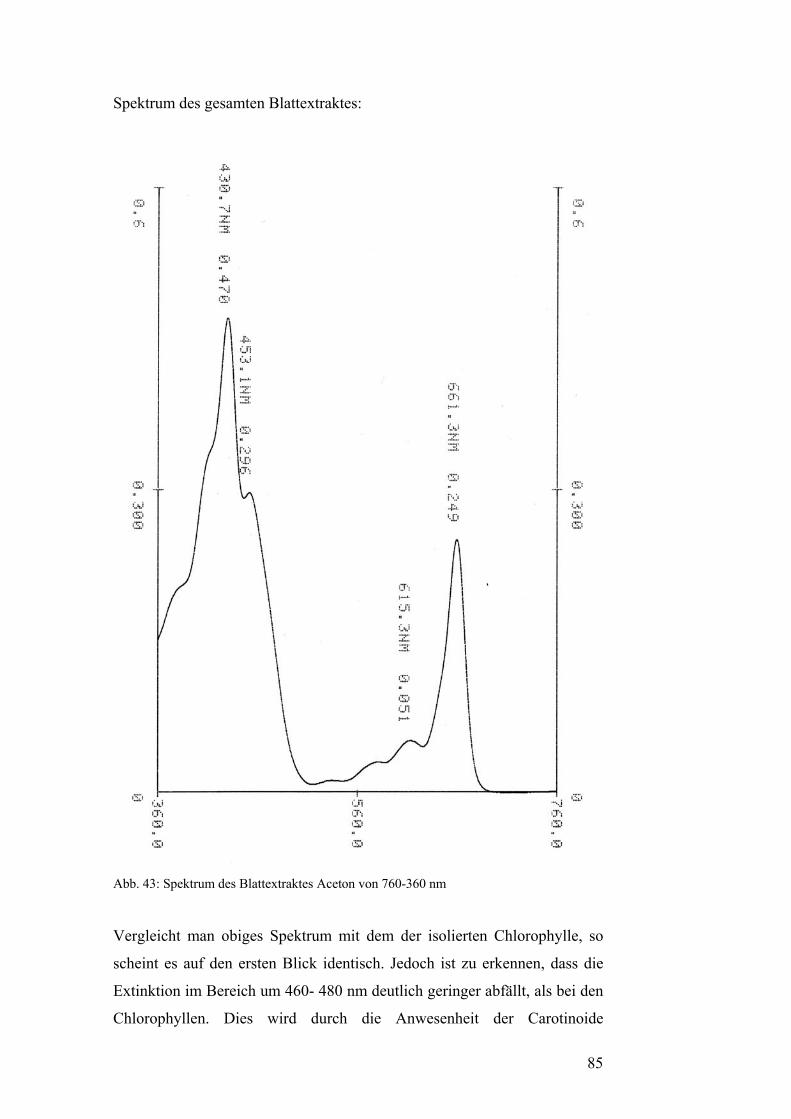
Spektrum des gesamten Blattextraktes:
Abb. 43: Spektrum des Blattextraktes Aceton von 760-360 nm
Vergleicht man obiges Spektrum mit dem der isolierten Chlorophylle, so
scheint es auf den ersten Blick identisch. Jedoch ist zu erkennen, dass die
Extinktion im Bereich um 460- 480 nm deutlich geringer abfällt, als bei den
Chlorophyllen. Dies wird durch die Anwesenheit der Carotinoide
85

verursacht. Wie auch in Pflanzen unterstützen sie die Lichtabsorption in
diesem Bereich und helfen die „Grünlücke“ etwas zu schließen.
Entsorgung:
Das Fließmittel sowie die Lösungen der Blattfarbstoffe werden im Behälter
für organische Lösungsmittel entsorgt, die DC-Karte in der Feststofftonne.
86

3. Methodisch-didaktische Umsetzung
3.1. Aufgaben und Ziele des Chemieunterrichtes[12]
Chemieunterricht soll Schüler befähigen, zukünftige Lebenssituationen zu
bewältigen sowie demokratische Entscheidungsprozesse kritisch zu
betrachten. Dazu gehört insbesondere eine zeitgemäße
naturwissenschaftliche Grundausbildung. Nur dadurch wird es möglich,
fortschreitende Technisierung, gesellschaftliche und wirtschaftliche
Situationen und Forderungen zu verstehen und zu beurteilen.
Den Schülern wird im Chemieunterricht das Verständnis im Umgang mit
Stoffen vermittelt. Sie werden befähigt, Vorgänge in der Natur zu verstehen,
ihren Ablauf vorauszusagen und zu beeinflussen.
Ein weiterer Schwerpunkt des Chemieunterrichtes ist die Vermittlung
wissenschaftlicher Arbeitsmethoden. Deshalb haben Experimente im
Chemieunterricht einen hohen Stellenwert. Anregungen zum selbständigen
und forschenden Lernen können durch Schülerexperimente oder
experimentelle Hausaufgaben gegeben werden. Durch selbsttätiges Lernen
wird die immer wieder von der Gesellschaft geforderte Methodenkompetenz
verbessert.
Ein wesentliches Ziel des Chemieunterrichtes muss es auch sein, Schüler zu
befähigen, Erlerntes in Alltagssituationen anzuwenden.
Zusätzlich zu den oben genannten Zielen sollen die Schüler in der Oberstufe
lernen, sachkompetent und verantwortungsbewusst zu entscheiden und zu
handeln.
Schließlich sollen Schüler u.a. auch durch den Chemieunterricht
schrittweise zu Studierfähigkeit und der allgemeinen Hochschulreife
herangeführt werden.
87

3.2. Didaktische Vorüberlegungen zur Unterrichtseinheit
„Pflanzenfarbstoffe“
Bevor man das Thema „Pflanzenfarbstoffe“ im Unterricht behandeln kann,
ist es notwendig, didaktische Vorüberlegungen anzustellen bezüglich des
Lehrplans, der Lerngruppengröße und der Lernvoraussetzungen, welche die
Schüler mitbringen.
Der Hessische Lehrplan Chemie, gymnasialer Bildungsgang G 8, sieht zwei
Zeitpunkte zur Behandlung des Themas „Farbstoffe“ vor und zwar in den
Leistungskursen der Jahrgangstufen 11G.2 und 12G.2. In beiden Fällen
handelt es sich um fakultative Unterrichtseinheiten. Der Lehrplan gibt die
Groblernziele vor. Bei der Durchführung der Unterrichtseinheit Farbstoffe
in der Jahrgangstufe 11G.2 sollen die inhaltlichen Schwerpunkte u.a. sein:
natürliche und synthetische Farbstoffe, Struktur und Lichtabsorption sowie
das Mesomerie-Modell.
In der Jahrgangstufe 12G.2 kann die Unterrichtseinheit Farbstoffe innerhalb
des Wahlbereiches Angewandte Chemie angeboten werden. Allgemein soll
„Angewandte Chemie“ zur Vertiefung und Ergänzung bereits erworbener
Lerninhalte beitragen. Speziell erarbeitet werden sollen in dieser
Unterrichtseinheit u.a.: der Zusammenhang zwischen Licht und Farbe,
Theorien der Farbigkeit, Farbstoffklassen und natürliche Farbstoffe.
Die Lerngruppengröße muss ebenfalls bei der Planung einer
Unterrichtseinheit berücksichtigt werden. Von ihr ist es unter anderem
abhängig, ob Schülerversuche durchgeführt werden können, oder ob alle
Versuche durch den Lehrer vorgeführt werden müssen. Die „Verordnung
über die Aufsicht der Schülerinnen und Schüler vom 20.12.2005“ des
hessischen Kultusministeriums legt folgende Rahmenbedingungen fest:
„In den Sekundarstufen I und II sollen in der Regel nicht mehr als
16 Schülerinnen und Schüler gleichzeitig experimentieren oder nicht mehr
als 8 Arbeitsgruppen gebildet werden. Wie viele Schülerinnen und Schüler
in einer einzelnen Gruppe arbeiten können, hängt ab
• von deren Erfahrungsstand,
• vom Gefährdungsgrad des durchzuführenden Experimentes,
88

• von den eingesetzten Geräten, Werkzeugen und Maschinen,
• von den Gefährlichkeitsmerkmalen der Gefahrstoffe, mit denen
umgegangen wird,
• von der Anlage und Größe des Raumes.
Auch in der Sekundarstufe II kann bei Schülerinnen und Schülern die
erforderliche Umsicht, die Erfahrungen im Experimentieren und die
jeweilige Sachkenntnis nicht ohne weiteres vorausgesetzt werden.“
Neben Lehrplanbezug und Lerngruppengröße sind die Lernvoraussetzungen
der Schüler von elementarer Bedeutung bei der Planung einer
Unterrichtseinheit. Die neu zu vermittelnden Inhalte müssen auf dem
aufbauen, was die Schüler bereits gelernt haben.
Es wird zeitlich nicht möglich sein, das Thema „Farbstoffe“ so umfassend,
wie im Lehrplan vorgesehen, im Unterricht zu behandeln.
Deshalb werden exemplarisch nur die Farbstoffe betrachtet, die in grünen
Blätter und Pflanzenteilen vorhanden sind. Dies reicht völlig aus, um die
geforderten Lernziele zu erreichen. Außerdem liegen diese
Pflanzenfarbstoffe im Erfahrungsbereich der Schüler und könnten deshalb
motivierend wirken.
Wie bereits beschrieben kann das Thema „Pflanzenfarbstoffe“ fakultativ in
den Jahrgangstufen 11G.2 und 12G.2 erarbeitet werden. Zu diesen
Zeitpunkten verfügen die Schüler bereits über fast alle Grundlagen, die zum
Verständnis der in meiner Examensarbeit ausgearbeiteten Versuche
notwendig sind. Bereits in der Jahrgangstufe 7G.1 erlernen Schüler
grundlegende Trennverfahren für Stoffgemische auf Grund
unterschiedlicher Polarität. Es werden auch schon einfache
chromatographische Trennverfahren durchgeführt. Der Großteil der
benötigten Grundlagen wird den Schülern in den Jahrgangstufen 10 und 11
vermittelt. Behandelt werden unter anderem die Mechanismen der
Verseifung, der elektrophilen Addition und der Radikalkettenreaktion.
Völlig neu werden für die Schüler die komplexen Molekülstrukturen der
Farbstoffe sein. Damit eng verbunden ist der Zusammenhang zwischen
Struktur und Farbe.
Da das Thema „Pflanzenfarbstoffe“ einen starken Bezug zur Biologie hat,
sind auch hier Voraussetzungen im Vorfeld zu überprüfen. In der
89

Jahrgangstufe 7G.3 wird in Biologie die Photosynthese erarbeitet. Hier
lernen die Schüler die Bedeutung von Licht für grüne Pflanzen auf
einfachstem Niveau.
3.3. Grobplanung der Unterrichtseinheit
Bei jeder Unterrichtseinheit ist der Einstieg wichtig. Den Schülern muss klar
werden, weshalb gerade dieses Thema für sie wichtig sein könnte.
Außerdem sollte der Einstieg möglichst an den Erfahrungsbereich der
Schüler angelehnt sein.
Ein möglicher Einstieg in die Unterrichtseinheit „Farbstoffe“ kann darin
bestehen, den Schülern verschiedene farbige Pflanzenteile vorzustellen, z.B.
Blätter, Blüten, Früchte. Ausgehend von den unterschiedlichen Farben kann
man dann auf die Fragestellung „Wie entstehen Farben?“ überleiten.
Exemplarisch kann das Problem am Farbstoff β-Carotin angegangen
werden. Seine charakteristische Farbe ist den Schülern von Karotten,
Softdrinks oder auch Schalentieren bekannt. Bei β-Carotin handelt es sich
um einen strukturell relativ einfach aufgebauten Farbstoff (keine
Verzweigungen, keine Heteroatome).
Zur Klärung der Kernfrage müssen zunächst experimentell Eigenschaften
und Strukturmerkmale des β-Carotins erarbeitet werden. Zuerst wird der
Stoff extrahiert. Das Trennverfahren „Extraktion“ ist den Schülern aus der
Jahrgangstufe 7G.1 bekannt. In der Jahrgangstufe 9G.1 haben sie erfahren,
dass es polare und unpolare Lösungsmittel gibt und sich „Gleiches in
Gleichem“ löst. Mit Hilfe der Extraktion lernen die Schüler eine erste
Eigenschaft von β-Carotin kennen: es ist vollkommen unpolar.
Durch polare Eigenschaften eines Moleküls allein lässt sich noch nicht seine
Farbigkeit erklären. Deshalb ist es wichtig, Strukturmerkmale des
β-Carotins aufzuzeigen. Das wichtigste Strukturmerkmal ist das System
konjugierter Doppelbindungen. Die in der Schule gebräuchlichste
Nachweisreaktion für Alkene ist die elektrophile Addition. Der Lehrplan
sieht vor, dass die Schüler diese Reaktion bereits in der Jahrgangstufe 10
zum Nachweis von Doppelbindungen kennen lernen. Im Leistungskurs der
Jahrgangstufe 11G.2 sollen der Reaktionstyp und der Mechanismus der
90

elektrophilen Addition erarbeitet werden, um Eigenschaften von
Kohlenstoff-Wasserstoff-Verbindungen zu deuten. Führt man eine
elektrophile Addition von Halogenen an β-Carotin durch, so können Schüler
erneut auf vorhandenes Wissen zurückgreifen und mit diesem ihre
Beobachtungen deuten. Die Schüler sollen erkennen, dass es sich bei
β-Carotin um einen polaren (langkettigen) und ungesättigten
(Doppelbindungen) Kohlenwasserstoff handelt.
Jetzt kann damit begonnen werden, einen Zusammenhang zwischen Licht,
Farbe und Struktur eines Stoffes herzustellen.
Die Betrachtung der mesomeren Eigenschaften des β-Carotins muss eine
zentrale Rolle spielen. In der Jahrgangstufe 11G.1 wurde der Begriff
Mesomerie bereits erarbeitet. Somit bringen die Schüler Kenntnisse mit
bezüglich konjugierter Doppelbindungen. Sie können bereist mesomere
Grenzstrukturen formulieren und deuten. Es dürfte deshalb den Schülern
nicht schwer fallen, das Mesomerie-Modell auf das β-Carotin zu übertragen.
Hiermit kann nun die Entstehung des Farbeindruckes gedeutet werden, der
entscheidend vom konjugierten π-Elektronensystem abhängig ist. Das
Prinzip lässt sich auch auf andere Pflanzenfarbstoffe übertragen.
Insgesamt lässt sich feststellen, dass mit der Untersuchung von β-Carotin
viele im Lehrplan geforderten Lernziele abgedeckt werden können.
Außerdem können die Schüler auf bereits erlerntes Fachwissen
zurückgreifen. Sie können es anwenden und somit auch gleichzeitig
wiederholen und vertiefen. Zur Vorbereitung auf einzelne
Unterrichtsstunden könnte man die Schüler auffordern, benötigtes
Fachwissen zu Hause aufzuarbeiten. Ständige Wiederholungen und
Vertiefungen von Wissen sind im Hinblick auf das Zentralabitur ein nicht zu
unterschätzender Lerneffekt.
Falls es die Zeit erlaubt, kann man innerhalb der Unterrichtseinheit
„Farbstoffe“ noch den Pflanzenfarbstoff Chlorophyll behandeln.
Zum Einstieg bietet sich die Photosynthese an. Sie ist den Schülern in
einfachster Form aus dem Biologieunterricht der Jahrgangstufe 7G.3
bekannt.
Auch hier ist es zunächst notwendig, diesen Blattfarbstoff aus dem
pflanzlichen Gewebe zu extrahieren, um anschließend Strukturelemente
91

praktisch aufzuzeigen und nachzuweisen. Sicherlich ist es in der Schule
nicht möglich, das Porphyringerüst durch Versuche nachzuweisen. Das
zentrale Magnesium-Ion und der lipophile Phytolrest können einfach und
schnell nachgewiesen werden. Hier können die Schüler erneut den Transfer
von Bekanntem zu (noch) Unbekannten leisten. Der Mechanismus der
alkalischen Esterhydrolyse ist den Schülern aus der Jahrgangstufe 11G.1
bekannt.
Nach Erarbeitung der Chlorophyllstruktur bietet es sich an – entsprechend
wie bei β-Carotin – die Farbe des Chlorophylls mit seiner Struktur in
Zusammenhang zu bringen (Mesomerie, konjugiertes π-Elektronensystem).
Zu diesem Zeitpunkt wird noch kein Schüler einen funktionellen
Zusammenhang zwischen β-Carotin und Chlorophyll vermuten. Um zu
zeigen, dass es einen solchen gibt, bietet sich die Chromatographie von
Blattgrün an. So erfahren Schüler, dass es sich bei Blattgrün keineswegs um
einen Reinstoff handelt, sondern um ein Gemisch von Pflanzenfarbstoffen,
u.a. Chlorophyll und β-Carotin. Jetzt werden sich sicherlich einige Schüler
fragen, weshalb in grünen Blättern β-Carotin enthalten ist und welche
Funktion es hat, obwohl doch Chlorophyll das Photosynthesepigment ist.
Um die Funktion des β-Carotins als Hilfspigment zu zeigen, können
Spektren von Chlorophyll und β-Carotin aufgenommen werden. Ein
Vergleich dieser Spektren zeigt dann, dass β-Carotin die Grünlücke schließt.
Verfügt die Schule nicht über ein Photometer oder ist es zeitlich nicht
möglich eigene Spektren aufzunehmen, so kann auf Spektren aus der
Literatur zurückgegriffen werden.
Schließlich kann noch die Schutzfunktion von β-Carotin für Chlorophyll
erarbeitet werden. Für das Verständnis der Radikalkettenreaktion verfügen
die Schüler bereits seit der Jahrgangstufe 10 über die notwendigen
Grundlagen. Die Erarbeitung der Desaktivierung von Singulett-Sauerstoff
muss allerdings auf einem niedrigeren Niveau - als in meiner Examensarbeit
beschrieben - erfolgen. Die MO-theoretischen Grundlagen können in der
Schule nicht erarbeitet werden. Es reicht völlig aus, den Schülern zu zeigen,
dass β-Carotin in der Lage ist, eine agressive, energiereiche Form des
Luftsauerstoffs unschädlich zu machen.
92

Anhand dieser Versuche können die Schüler den funktionellen
Zusammenhang zwischen Chlorophyll und β-Carotin herstellen.
In meinen Ausführungen zur Behandlung des komplexen Themas Farbstoffe
habe ich aufgezeigt, warum man exemplarisch vorgehen muss. Es ist
möglich, die meisten geforderten Lernziele durch die Behandlung eines
einzelnen Pflanzenfarbstoffs zu erreichen.
93

4. Schlusswort
Ziel dieser wissenschaftlichen Hausarbeit war es, das Thema
„Pflanzenfarbstoffe“ fachwissenschaftlich darzustellen und darüber hinaus
Möglichkeiten aufzuzeigen, wie das Thema im Unterricht umgesetzt werden
kann.
Bei der Erarbeitung von Unterrichtsinhalten im naturwissenschaftlichen
Unterricht sind geeignete Experimente unverzichtbar. Sie dienen der
Veranschaulichung fachwissenschaftlicher Aspekte und der
Erkenntnisgewinnung. Versuche wirken auf Schüler motivierend, bringen
Abwechslung in den Unterricht und fördern dadurch den Lernprozess.
Dabei ist Schülerexperimenten ein noch höherer Stellenwert einzuräumen
als den Lehrerversuchen. Genau dies habe ich bei der Auswahl meiner
Versuche berücksichtigt.
Durch selbsttätiges Experimentieren lernen Schüler grundlegende
wissenschaftliche Arbeitsmethoden kennen. Zum Gelingen eines
Experimentes trägt wesentlich sauberes und genaues Arbeiten bei. Die
Beobachtungsgabe wird geschult. Genaue Beobachtungen sind notwendige
Voraussetzung für eine Versuchsauswertung und die sich daran
anschließende Deutung.
Durch Schülerexperimente erhalten die Schüler Anregungen zum
selbständigen und forschenden Lernen. Durch diese Lernmethode bleiben
Lerninhalte nachhaltig im Gedächtnis der Schüler.
Durch das Zusammenwirken von praktischem und theoretischen Lernen
erlangen die Schüler eine verbesserte Methodenkompetenz. Gefördert
werden insbesondere die Teamkompetenz sowie die
Problemlösekompetenz. Genau dies wird von Politik, Wirtschaft und
Gesellschaft verstärkt gefordert. Durch Schülerexperimente kann auch das
Unterrichtsfach Chemie zur Entwicklung und Verbesserung der
Methodenkompetenz der Schüler beitragen.
94

5. Literatur [1] A. Batschauer, Vorlesung „Pflanzenphysiologie“, Fachbereich Biologie der Philipps-Universität Marburg, WS 2004/05 [2] H. Berger, „Funktion der Carotinoide bei der Photosynthese“, Marburg 1972 [3] F. Bukatsch, O. P. Krätz, G. Probeck und R. J. Schwanker, „So interessant ist Chemie“, Aulis Verlag Deubner & Co KG, 1997 [4] C. Buschmann und K. Grumbach, „Physiologie der Photosynthese“, Springer Verlag Heidelberg, New York, Tokyo 1985 [5] D. Dörnemann, Vorlesung und Arbeitsanleitung zum Kurs: „Sekundäre Pflanzeninhaltsstoffe“, Fachbereich Biologie der Philipps-Universität Marburg, 2003 [6] E. Gerstner, „Skriptum zum anorganisch-chemischen Praktikum für Lehramtskandidaten (Teil I und II)“, Marburg 1993 [7] D.-P. Häder, „Photosynthese“, Georg Thieme Verlag Stuttgart, New York 1999 [8] http://www.ch.tum.de/oc1/History/HansFischer.htm, letzter Zugriff am 11.04.07 [9] http://www.theochem.uni-duisburg.de/DC/material/carotin/carver.html, letzter Zugriff am 17.07.06 [10] C.-P. Jellen, Dissertation „Floureszenzspektroskopische Untersuchungen an Aggregaten von Porphyrinen und Carotinoporphyrinen“, Heinrich-Heine-Universität Düsseldorf, 2002 [11] U. Koert, Vorlesung „Organische Chemie für Studierende der Chemie“, Fachbereich Chemie der Philipps-Universität Marburg, WS 2003/04 [12] Lehrplan Chemie, Hessen, gymnasialer Bildungsgang, Jahrgangstufen 7G bis 12G [13] Lehrplan Biologie, Hessen, gymnasialer Bildungsgang, Jahrgangstufen 5G bis 12G [14] H. Meier, Ausarbeitung zum Experimentalvortrag: „Indigoide Farbstoffe“, Fachbereich Chemie der Philipps-Universität Marburg [15] T. Niedenthal, Ausarbeitung zum Experimentalvortrag: „Kohlenwasserstoffe“, Fachbereich Chemie der Philipps-Universität Marburg, 2006
95

[16] Noll, Ausarbeitung zum Experimentalvortrag: „Chromatographische Verfahren“, Fachbereich Chemie der Philipps-Universität Marburg, 1979 [17] W. Nultsch, „Mikroskopisch-Botanisches Praktikum“, Georg Thieme Verlag Stuttgart, New York 2001 [18] R. Peichert, „Die historische Entwicklung der Farbstofftheorie“, PdN-Ch. 8/38, 1989 [19] Riedel, „Anorganische Chemie“, Walter de Gruyter, Berlin 1999 [20] P. Sitte, E. W. Weiler, J. W. Kadereit, A. Bresinsky und C. Körner, „Strasburger, Lehrbuch der Botanik“, Spektrum Akademischer Verlag GmbH Heidelberg, Berlin, 2002 [21] Skript zum „Pflanzenphysiologischen Kurs“ für Diplomstudierende im Grundstudium und L3-Studierende im Hauptstudium, Fachbereich Biologie der Philipps-Universität Marburg, WS 2004/05 [22] Skript zum „Chemischen Praktikum für Studierende der Biologie und Humanbiologie“, Fachbereich Chemie der Philipps-Universität Marburg, 2004 [23] M. Schmidt, Ausarbeitung zum Experimentalvortrag: „Pflanzenfarbstoffe“, Fachbereich Chemie der Philipps-Universität Marburg, 1993 [24] W. Steuber und M. Schween, „Lehrerfortbildungskurs Farbstoffe und Lebensmittelfarben“, Fachbereich Chemie der Philipps-Universität Marbrug, 1987 [25] V. Storch und U. Welsch, „Kükenthal, Zoologisches Praktikum“, Spektrum Akademischer Verlag GmbH Heidelberg, Berlin, 2002
96

6. Abbildungsverzeichnis
Abb. 1: Spektrum elektromagnetischer Wellen
Abbildung (verändert) aus Riedel, „Anorganische
Chemie“, Walter de Gruyter,
Berlin 1999
3
Abb. 2: Übersicht über Spektralfarben und ihre zugehörige
Komplementärfarbe
Abbildung (verändert) aus Riedel, „Anorganische
Chemie“, Walter de Gruyter,
Berlin 1999
4
Abb. 3: Partialvalenz eines Nitrophenolsalzes
Abbildung aus R. Peichert, „Die historische Entwicklung
der Farbstofftheorie“,
PdN-Ch. 8/38, 1989
6
Abb. 4: Übergang von HOMO nach LUMO am Beispiel des
Butadiens
U. Koert, Vorlesung „Organische Chemie für Studierende
der Chemie“,
Fachbereich Chemie der Philipps-Universität Marburg,
WS 2003/04
8
Abb. 5: Molekülstruktur Chlorophyll a bzw. Chlorophyll b
Abbildung (verändert) aus D.-P. Häder, „Photosynthese“,
Georg Thieme Verlag Stuttgart, New York 1999
10
Abb. 6: Jablonski-Diagramm
Abbildung aus P. Sitte, E. W. Weiler, J. W. Kadereit,
A. Bresinsky und C. Körner, „Strasburger, Lehrbuch der
Botanik“, Spektrum Akademischer Verlag GmbH,
Heidelberg, Berlin, 2002
12
97

Abb. 7: Schematische Darstellung des Excitonentransfers von den
Antennen zum Reaktionszentrum
A. Batschauer, Vorlesung „Pflanzenphysiologie“,
Fachbereich Biologie der
Philipps-Universität Marburg, WS 2004/05
13
Abb. 8: Molekülstruktur Pyrrol
Abbildung aus P. Sitte, E. W. Weiler, J. W. Kadereit, A.
Bresinsky und C. Körner, „Strasburger, Lehrbuch der
Botanik“, Spektrum Akademischer Verlag GmbH,
Heidelberg, Berlin, 2002
15
Abb. 9: Molekülstruktur Porphyrin
Abbildung aus P. Sitte, E. W. Weiler, J. W. Kadereit, A.
Bresinsky und C. Körner, „Strasburger, Lehrbuch der
Botanik“, Spektrum Akademischer Verlag GmbH,
Heidelberg, Berlin, 2002
16
Abb. 10: Molekülstruktur Chlorophyll unter besonderer
Berücksichtigung der Stereoisomerie am isozyklischen
Fünferring „E“
Abbildung (verändert) aus D.-P. Häder, „Photosynthese“,
Georg Thieme Verlag Stuttgart, New York 1999
17
Abb. 11: Chromatographischer Effekt auf Filterpapier 20
Abb. 12: links: reiner Blattextrakt, rechts: Blattextrakt mit
Salzsäure (Phaeophytin)
23
Abb. 13
und 14:
Magnesiumchlorid-Lösung und Extrakt aus Spinat vor und
nach Zugabe von Titangelb-Lösung.
26
Abb. 15: Verseifter Blattextrakt 27
Abb. 16
und 17:
Dunkel gelagerte und belichtete Wasserpestpflanzen 31
Abb. 18: Molekülstruktur Isopren 34
Abb. 19 –
22:
Häufig vorkommende Primärcarotinoide
Abbildung aus D. Dörnemann, Arbeitsanleitung zum Kurs:
„Sekundäre Pflanzeninhaltsstoffe“,Fachbereich Biologie
der Philipps-Universität Marburg, 2003
36
98

Abb. 23: Soxhlet-Apparatur 42
Belichtete Küvetten 50
Abb. 26: Schematisches Energieniveaudiagramm für Disauerstoff
im Grundzustand
Abbildung aus E. Gerstner, „Skriptum zum anorganisch-
chemischen Praktikum für Lehramtskandidaten
(Teil I und II)“, Marburg 1993
55
Abb. 27: Elektronenanordnungen im Grundzustand und den beiden
angeregten Zuständen eines Disauerstoff-Moleküls
Abbildung aus E. Gerstner, „Skriptum zum anorganisch-
chemischen Praktikum für Lehramtskandidaten
(Teil I und II)“, Marburg 1993
56
Abb. 28: Ausschnitt aus dem Chromatographierohr 65
Abb. 29: Tafelkreide nach erfolgter Chromatographie 66
Abb. 30: Ergebnis der Dünnschichtchromatographie eines
Blattextraktes in Methanol
72
Abb. 31
und 32:
Ausschnitte aus der Molekülstruktur von Chlorophyll b
und Chlorophyll b’ unter Berücksichtigung der
Stereoisomerie am isozyklischen Fünferring „E“
Abbildung (verändert) aus D.-P. Häder, „Photosynthese“,
Georg Thieme Verlag Stuttgart, New York 1999
73
Abb. 33
und 34:
Ausschnitte aus der Molekülstruktur von Chlorophyll a
und Chlorophyll b
Abbildung (verändert) aus D.-P. Häder, „Photosynthese“,
Georg Thieme Verlag Stuttgart, New York 1999
74
Abb. 35: Spektrum von Neoxanthin in Aceton von 760-360 nm 76
Abb. 36: Spektrum von Violaxanthin in Aceton von 760-360 nm 77
Abb. 37: Spektrum von Lutein in Aceton von 760-360 nm 78
Abb. 38: Spektrum von Chlorophyll b in Aceton von 760-360 nm 79
Abb. 39: Spektrum von Chlorophyll b’ in Aceton von 760-360 nm 80
Abb. 40: Spektrum von Chlorophyll a in Aceton von 760-360 nm 81
Abb. 41: Spektrum von Chlorophyll a’ in Aceton von 760-360 nm 82
Abb. 24
und 25:
99

Abb. 42: Spektrum von β-Carotin in Aceton von 760-360 nm 84
Abb. 43: Spektrum des Blattextraktes in Aceton von 760-360 nm 85
100

7. Anlagen
101

Anlage 1
Alle in dieser Arbeit angegebenen R- und S-Sätze stammen vom
„Berufsgenossenschaftlichen Institut für Arbeitsschutz“
http://www.hvbg.de/d/bia/gestis/stoffdb/index.html
letzter Zugriff am 29.04.07
Bedeutung der R- und S-Sätze sowie der Gefahrensymbole der verwendeten
Chemikalien
R-Sätze:
R 7 Kann Brand verursachen
R 11 Leichtentzündlich
R 20/22 Gesundheitsschädlich beim Einatmen und Verschlucken
R 22 Giftig bei Berührung mit der Haut
R 23/24/25 Giftig beim Einatmen, Verschlucken und Berührung mit der
Haut
R 31 Entwickelt bei Berührung mit Säure giftige Gase
R 34 Verursacht Verätzungen
R 35 Verursacht schwere Verätzungen
R 36 Reizt die Augen
R 36/37/38 Reizt die Augen, Atmungsorgane und die Haut
R 37 Reizt die Atmungsorgane
R 38 Reizt die Haut
R 39/23/24/25 Giftig: ernste Gefahr irreversiblen Schadens durch
Einatmen, Berührung mit der Haut und durch Verschlucken
R 41 Gefahr ernster Augenschäden
R 48/20 Gesundheitsschädlich: Gefahr ernster Gesundheitsschäden
bei längerer Exposition durch Einatmen
R 50/53 Sehr giftig für Wasserorganismen, kann in Gewässern
längerfristig schädliche Wirkungen haben
R 51/53 Giftig für Wasserorganismen, kann in Gewässern
längerfristig schädliche Wirkungen haben
102

R 62 Kann möglicherweise die Fortpflanzungsfähigkeit
beeinträchtigen
R 65 Gesundheitsschädlich: kann beim Verschlucken
Lungenschäden verursachen.
R 66 Wiederholter Kontakt kann zu spröder oder rissiger Haut
führen
R 67 Dämpfe können Schläfrigkeit oder Benommenheit erzeugen
S-Sätze:
S 7 Behälter dicht geschlossen halten
S 7/8 Behälter trocken und dicht geschlossen halten
S 9 Behälter an einem gut gelüfteten Ort aufbewahren
S 16 Von Zündquellen fernhalten - Nicht rauchen
S 22 Staub nicht einatmen
S 24 Berührung mit der Haut vermeiden
S 25 Berührung mit den Augen vermeiden
S 26 Bei Berührung mit den Augen sofort gründlich mit Wasser
abspülen und Arzt konsultieren
S 28 Bei Berührung mit der Haut sofort abwaschen mit viel
Wasser und Seife abwaschen
S 29 Nicht in die Kanalisation gelangen lassen
S 33 Maßnahmen gegen elektrostatische Aufladungen treffen
S 36/37 Bei der Arbeit geeignete Schutzhandschuhe und
Schutzkleidung tragen
S 36/37/39 Bei der Arbeit geeignete Schutzhandschuhe, Schutzkleidung
und Schutzbrille/Gesichtsschutz tragen
S 37/39 Bei der Arbeit geeignete Schutzhandschuhe und
Schutzbrille/Gesichtsschutz tragen
S 39 Schutzbrille/Gesichtsschutz tragen
S 43 Zum Löschen Wasser oder Pulverlöschmittel verwenden
S 45 Bei Unfall oder Unwohlsein sofort Arzt zuziehen; wenn
möglich dieses Etikett vorzeigen
103

S 60 Dieser Stoff und sein Behälter sind als gefährlicher Abfall zu
entsorgen
S 61 Freisetzung in die Umwelt vermeiden. Besondere
Anweisungen einholen, Sicherheitsdatenblatt zu Rate ziehen
S 62 Bei Verschlucken kein Erbrechen herbeiführen. Sofort
ärztlichen Rat einholen und Verpackung oder dieses Etikett
vorzeigen
Gefahrensymbole:
C Ätzend
F Leichtentzündlich
N Umweltgefährlich
T Giftig
Xi Reizend
Xn Gesundheitsschädlich
104
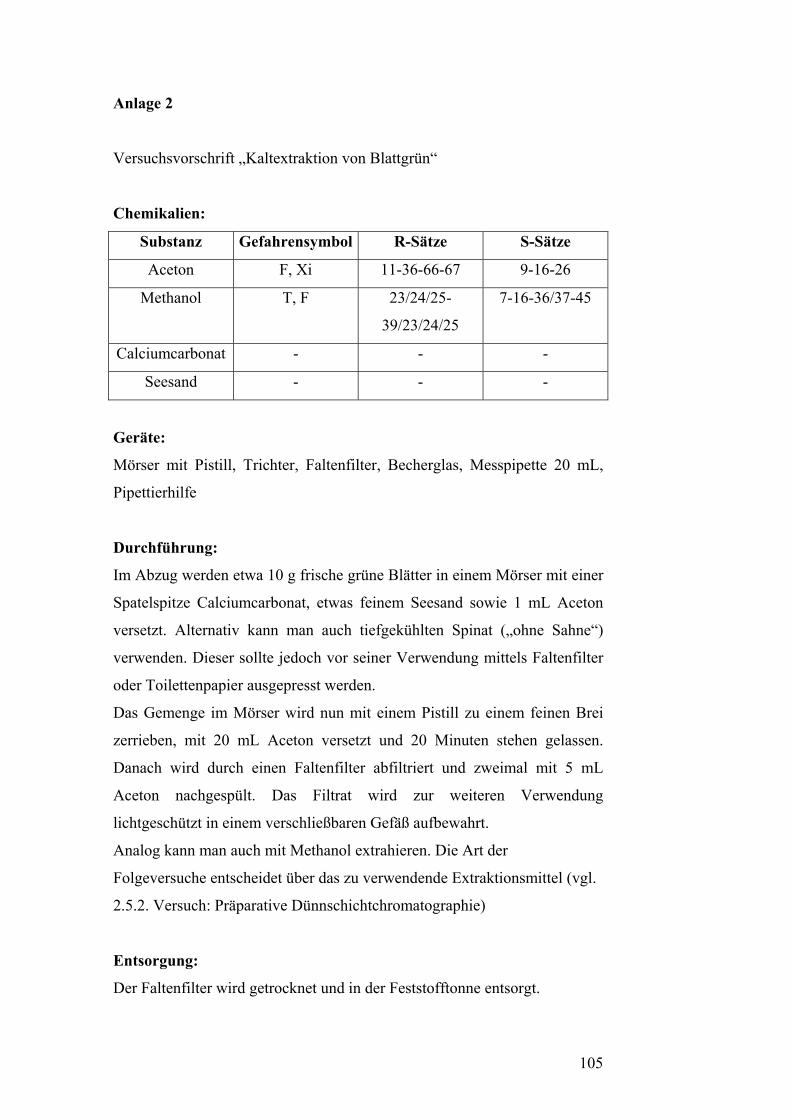
Anlage 2
Versuchsvorschrift „Kaltextraktion von Blattgrün“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Aceton F, Xi 11-36-66-67 9-16-26
Methanol T, F 23/24/25-
39/23/24/25
7-16-36/37-45
Calciumcarbonat - - -
Seesand - - -
Geräte:
Mörser mit Pistill, Trichter, Faltenfilter, Becherglas, Messpipette 20 mL,
Pipettierhilfe
Durchführung:
Im Abzug werden etwa 10 g frische grüne Blätter in einem Mörser mit einer
Spatelspitze Calciumcarbonat, etwas feinem Seesand sowie 1 mL Aceton
versetzt. Alternativ kann man auch tiefgekühlten Spinat („ohne Sahne“)
verwenden. Dieser sollte jedoch vor seiner Verwendung mittels Faltenfilter
oder Toilettenpapier ausgepresst werden.
Das Gemenge im Mörser wird nun mit einem Pistill zu einem feinen Brei
zerrieben, mit 20 mL Aceton versetzt und 20 Minuten stehen gelassen.
Danach wird durch einen Faltenfilter abfiltriert und zweimal mit 5 mL
Aceton nachgespült. Das Filtrat wird zur weiteren Verwendung
lichtgeschützt in einem verschließbaren Gefäß aufbewahrt.
Analog kann man auch mit Methanol extrahieren. Die Art der
Folgeversuche entscheidet über das zu verwendende Extraktionsmittel (vgl.
2.5.2. Versuch: Präparative Dünnschichtchromatographie)
Entsorgung:
Der Faltenfilter wird getrocknet und in der Feststofftonne entsorgt.
105
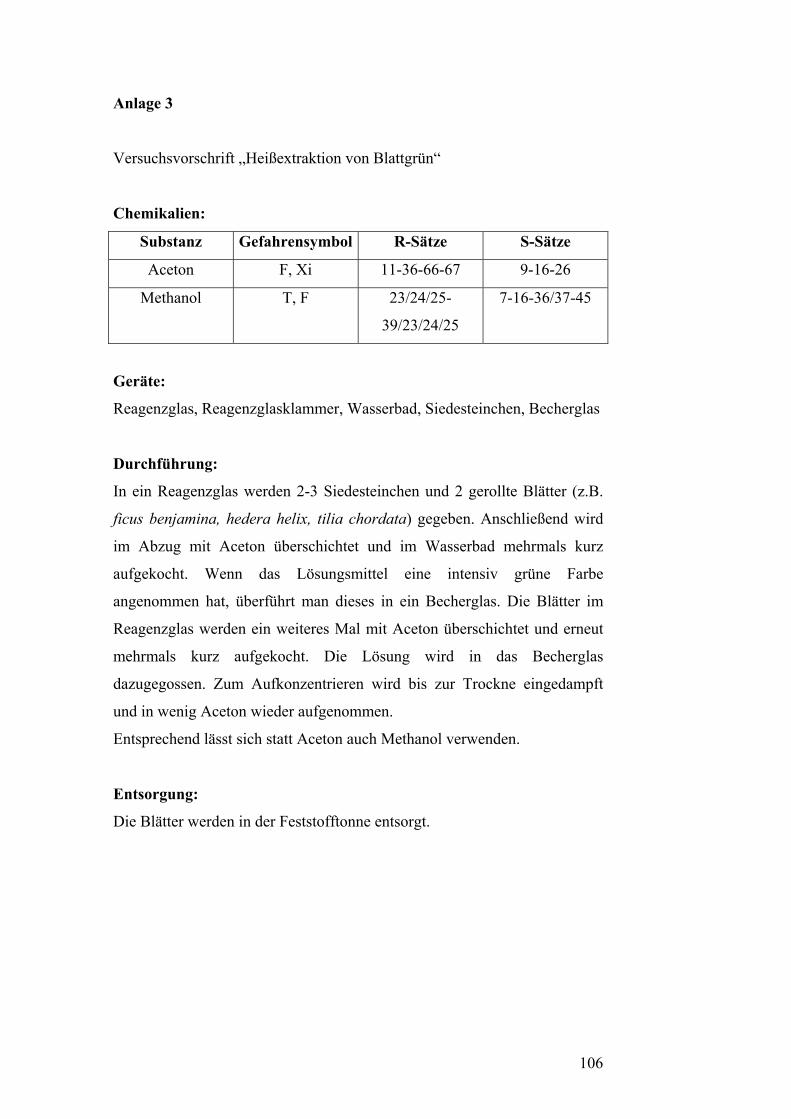
Anlage 3
Versuchsvorschrift „Heißextraktion von Blattgrün“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Aceton F, Xi 11-36-66-67 9-16-26
Methanol T, F 23/24/25-
39/23/24/25
7-16-36/37-45
Geräte:
Reagenzglas, Reagenzglasklammer, Wasserbad, Siedesteinchen, Becherglas
Durchführung:
In ein Reagenzglas werden 2-3 Siedesteinchen und 2 gerollte Blätter (z.B.
ficus benjamina, hedera helix, tilia chordata) gegeben. Anschließend wird
im Abzug mit Aceton überschichtet und im Wasserbad mehrmals kurz
aufgekocht. Wenn das Lösungsmittel eine intensiv grüne Farbe
angenommen hat, überführt man dieses in ein Becherglas. Die Blätter im
Reagenzglas werden ein weiteres Mal mit Aceton überschichtet und erneut
mehrmals kurz aufgekocht. Die Lösung wird in das Becherglas
dazugegossen. Zum Aufkonzentrieren wird bis zur Trockne eingedampft
und in wenig Aceton wieder aufgenommen.
Entsprechend lässt sich statt Aceton auch Methanol verwenden.
Entsorgung:
Die Blätter werden in der Feststofftonne entsorgt.
106
Anlage 4
Versuchsvorschrift „Darstellung der Phaeophytine“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Salzsäure
2 mol/L
- - -
Geräte:
Reagenzglas, Stopfen, Pasteurpipette
Durchführung:
Ein Reagenzglas wird etwa 2 cm hoch mit der Blattextraktlösung befüllt.
Anschließend werden einige Tropfen Salzsäure, 2 mol/L, zugesetzt, das
Reagenzglas mit einem Stopfen verschlossen und geschüttelt.
Entsorgung:
Die Lösung kann neutral im Behälter für organische Lösungsmittel entsorgt
werden.
107
Anlage 5
Versuchsvorschrift „Nachweis des Magnesium-Ions“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Salzsäure
4 mol/L
Xi 36/37/38 26
Titangelb-Lösung
(gesättigt)
- - -
Natronlauge
2 mol/L
C 35 26-36/37/39-45
Magnesiumchlorid - - -
Spinat - - -
Geräte:
Erlenmeyerkolben 100 mL, Spatel, Glasstab, Bunsenbrenner, Dreifuß mit
Drahtnetz, Messpipette 10 mL, Pipettierhilfe, Faltenfilter, Trichter
Durchführung:
Etwa 10 g Spinat werden mit 10 mL Salzsäure, 4 mol/L, im Abzug
aufgekocht. Anschließend wird die farblose Lösung abgefiltert. Das Filtrat
wird mit Natronlauge, 2 mol/L, neutralisiert und mit entionisiertem Wasser
auf 100 mL aufgefüllt.
In einem zweiten Erlenmeyerkolben wird eine Spatelspitze
Magnesiumchlorid in etwa 100 mL entionisiertem Wasser gelöst. Diese
Magnesiumchloridlösung dient als Vergleichslösung.
Beide Lösungen werden mit Natronlauge, 2 mol/L, alkalisch gemacht und
mit drei Tropfen Titangelb-Lösung versetzt.
Entsorgung:
Die Lösungen können neutral im Behälter für organische Lösungsmittel
entsorgt werden.
108
Anlage 6
Versuchsvorschrift „Verseifung der Chlorophylle“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Kalilauge
w=0,6
C 22-35 26-36/37/39-45
Geräte:
Reagenzglas, Messpipette 10 mL, Pipettierhilfe, Stopfen
Durchführung:
Ein Reagenzglas wird etwa 3 cm hoch mit Blattextrakt-Lösung befüllt. Die
Lösung versetzt man mit 2 mL Kaliumhydroxid-Lösung, w=0,6.
Anschließend wird das Reagenzglas mit einem Stopfen verschlossen und
kräftig geschüttelt.
Entsorgung:
Die Lösung kann neutral im Behälter für organische Lösungsmittel entsorgt
werden.
109
Anlage 7
Versuchsvorschrift „Nachweis der Sauerstoffproduktion“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Indigopulver - - -
Natriumdithionit Xn 7-22-31 7/8-26-28-43
Natronlauge
w=0,1
C 35 26-37/39-45
Ethanol F 11 7-16
Wasserpest - - -
Geräte:
Mörser mit Pistill, Bunsenbrenner, Dreifuß mit Drahtnetz, Thermometer
Schraubdeckelgläser 100 mL,
Durchführung:
In einem Mörser werden 0,9 g Indigo mit etwas Ethanol und 10 mL
Natronlauge, w=0,1, verrieben. Anschließend überführt man die Suspension
in 300 mL Wasser, das auf 70 °C vorgewärmt wurde. Danach rührt man so
viel Natriumdithionit hinzu, bis die Lösung gerade gelb-grün wird.
In die Schraubdeckelgläser gibt man je einen Spross Wasserpest.
Anschließend befüllt man die beiden Gläser bis zum oberen Rand mit der
Lösung. Eventuell auftretende Blaufärbung wird unter Einrühren von
Natriumdithionit beseitigt. Danach werden die Gläser verschlossen
Das erste Schraubdeckelglas wird vor eine Lichtquelle gestellt, das andere
lichtgeschützt aufbewahrt.
Entsorgung:
Die Lösungen werden neutral im Behälter für organische Lösungsmittel
entsorgt.
110
Anlage 8
Versuchsvorschrift „Extraktion von β-Carotin aus kleingeschnittenen
Möhren mit Aceton“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Aceton F, Xi 11-36-66-67 9-16-26
Möhre - - -
Geräte:
Messer, 2 Erlenmeyerkolben 250 mL
Durchführung:
Eine frische Möhre wird gewaschen und geschält. Etwa die Hälfte der
Möhre wird in kleine Scheiben geschnitten und in den Erlenmeyerkolben
gegeben. Anschließend werden die Möhrenscheiben im Abzug mit Aceton
überschichtet. Der Erlenmeyerkolben wird nun etwa 2 Minuten geschwenkt.
Danach wird die Lösung in den zweiten Erlenmeyerkolben dekantiert.
Entsorgung:
Die Möhrenscheiben können getrocknet als Feststoffabfall entsorgt werden.
Die Lösung von β-Carotin in Aceton kann für weitere Versuche aufbewahrt
werden. Falls diese nicht mehr benötigt wird, entsorgt man sie im Behälter
für organische Lösungsmittel.
111

Anlage 9
Versuchsvorschrift „Extraktion von β-Carotin aus kleingeschnittenen
Möhren mit n-Heptan“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
n-Heptan F, Xn, N 11-38-50/53-65-
67
9-16-29-33-60-
61-62
Möhre - - -
Geräte:
Messer, 2 Erlenmeyerkolben 250 mL
Durchführung:
Eine frische Möhre wird gewaschen und geschält. Etwa die Hälfte der
Möhre wird in kleine Scheiben geschnitten und in den Erlenmeyerkolben
gegeben. Anschließend werden die Möhrenscheiben im Abzug mit n-Heptan
überschichtet. Der Erlenmeyerkolben wird nun etwa 2 Minuten geschwenkt.
Danach wird die Lösung in den zweiten Erlenmeyerkolben dekantiert.
Entsorgung:
Die Möhrenscheiben können getrocknet als Feststoffabfall entsorgt werden.
Die Lösung von β-Carotin in n-Heptan kann für weitere Versuche
aufbewahrt werden. Falls diese nicht mehr benötigt wird, entsorgt man sie
im Behälter für organische Lösungsmittel.
112
Anlage 10
Versuchsvorschrift „Extraktion von β-Carotin mit Aceton aus geraspelten
Möhren“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Aceton F, Xi 11-36-66-67 9-16-26
Möhre - - -
Geräte:
Messer, Küchenreibe, 2 Erlenmeyerkolben 250 mL
Durchführung:
Eine frische Möhre wird gewaschen und geschält. Etwa die Hälfte der
Möhre wird auf einer Küchenreibe kleingeraspelt und in den
Erlenmeyerkolben gegeben. Anschließend wird mit Aceton im Abzug
überschichtet. Der Erlenmeyerkolben wird nun etwa 2 Minuten geschwenkt.
Danach wird die Lösung in den zweiten Erlenmeyerkolben dekantiert.
Entsorgung:
Die geraspelten Möhren können getrocknet als Feststoffabfall entsorgt
werden.
Die Lösung von β-Carotin in Aceton kann für weitere Versuche aufbewahrt
werden. Falls diese nicht mehr benötigt wird, entsorgt man sie im Behälter
für organische Lösungsmittel.
113

Anlage 11
Versuchsvorschrift „Heißextraktion von β-Carotin mit Petrolether in einer
Soxhlet-Apparatur“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Petrolether
(60/95)
F, Xn, N 11-38-48/20-
51/53-62-65-67
9-16-33-36/27-
60-62
Möhre - - -
Geräte:
Messer, Petrischale, Trockenschrank, Soxhlet-Apparatur, Rotations-
verdampfer
Durchführung:
Eine frische Möhre wird gewaschen, geschält und in kleine Stücke
geschnitten. Diese werden in eine Petrischale gegeben und im
Trockenschrank bei 80 °C getrocknet. Nach etwa einer halben Stunde wird
das β-Carotin in einer Soxhlet-Apparatur im Abzug mit etwa 400 mL
Petrolether extrahiert. Der Versuch wird beendet, wenn der in den
Rundkolben zurückfließende Petrolether farblos ist (etwa nach 60 Minuten).
Je nach Weiterverwendungszweck kann die Lösung am
Rotationsverdampfer noch eingeengt werden.
Entsorgung:
Die Möhrenstückchen werden getrocknet als Feststoffabfall entsorgt.
Die Lösung von β-Carotin in Petrolether kann für weitere Versuche
aufbewahrt werden. Falls man sie nicht mehr benötigt, wird sie im Behälter
für organische Lösungsmittel entsorgt.
114
Anlage 12
Versuchsvorschrift „Elektrophile Addition von Brom an β-Carotin“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Brom in Phthal-
säurediethylester
Xn - -
β-Carotin in Aceton F, Xi 11-36-66-67 9-16-26
gesättigte
Natriumthiosulfatlösung
- - -
Geräte:
Demo-Reagenzglas mit Septum, Einwegspritze mit Kanüle
Durchführung:
Im Abzug wird ein Demo-Reagenzglas etwa 2 cm hoch mit einer Lösung
von β-Carotin in Aceton befüllt. Dieses Reagenzglas wird mit einem
Septum verschlossen. Mit einer Einwegspitze wird nun 1 mL einer Lösung
von Brom in Phtalsäurediethylester durch das Septum in das Reagenzglas
eingebracht.
Entsorgung:
Bromhaltige Lösungen werden mit Natriumthiosulfatlösung versetzt und in
der Tonne für organische Lösungsmittel entsorgt.
115
Anlage 13
Versuchsvorschrift „β-Carotin als Radikalfänger“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Tetraiodethen - - -
n-Heptan F, Xn, N 11-38-50/53-65-
67
9-16-29-33-60-
61-62
β-Carotin in n-
Heptan
F, Xn, N 11-38-50/53-65-
67
9-16-29-33-60-
61-62
Geräte:
Diaprojektor, Hebebühne, kleine Küvetten, Becherglas 50 mL, Messpipette
20 mL, Einwegspritzen mit Kanülen, Analysenwaage, Magnetrührer mit
Rührfisch
Durchführung:
Es werden in 20 mL n-Heptan 0,11 g Tetraiodethen gelöst. Hierzu wird die
Lösung im Wasserbad auf 45 °C erhitzt und mittels Magnetrührer gerührt.
Sobald die Lösung eine blass-violette Farbe hat, wird das Erhitzen beendet.
Mittels Einwegspritze werden zwei Küvetten mit Tetraiodethen-Lösung
befüllt. In eine der Küvetten werden zusätzlich 2-3 Tropfen einer Lösung
von β-Carotin in n-Heptan gegeben.
Beide Küvetten werden anschließend auf einer Hebebühne vor die Linse
eines eingeschalteten Diaprojektors gestellt.
Entsorgung:
Alle Lösungen können im Behälter für organische Lösungsmittel entsorgt
werden.
116
Anlage 14
Versuchsvorschrift „Desaktivierung von Singulett-Sauerstoff durch
β-Carotin“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
β-Carotin in Aceton F, Xi 11-36-66-67 9-16-26
Mangandioxid Xn 20/22 25
Salzsäure, konz. C 34-37 26-45
Wasserstoffperoxid-
Lösung, w = 0,3
C 34 3-26-36/37/39-
45
Natronlauge,
2 mol/L
C 35 26-36/37/39-45
gesättigte
Natriumthiosulfatlösung
- - -
Geräte:
Becherglas, Pasteurpipetten, Reagenzglas mit durchbohrtem Gummistopfen,
Gärröhrchen
Durchführung:
Vorbereitung:
Vor der eigentlichen Versuchsdurchführung muss zunächst eine alkalische
Wasserstoffperoxid-Lösung angesetzt werden. Hierzu mischt man 2 molare
Natronlauge mit 30 %iger Wasserstoffperoxid-Lösung im Verhältnis 5:1.
1. Schritt: Erzeugung von Singulett-Sauerstoff
Hierzu befüllt man ein Reagenzglas 1 cm hoch mit Mangandioxid. Dieses
wird im Abzug mit konzentrierter Salzsäure überschichtet. Das Reagenzglas
wird mit einem durchbohrten Gummistopfen, in dem ein Gärröhrchen
steckt, verschlossen. Das Gärröhrchen wird mit der zuvor angesetzten
alkalischen Wasserstoffperoxid-Lösung befüllt.
117

2. Schritt: Desaktivierung von Singulett-Sauerstoff.
Die sich im Gärröhrchen befindliche alkalische Wasserstoffperoxid-Lösung
wird mit einem Tropfen β-Carotin-Lösung versetzt.
Entsorgung:
Nachdem alle Lösungen mit Natriumthiosulfat-Lösung versetzt wurden,
können diese neutral im Behälter für Schwermetallabfall entsorgt werden.
118
Anlage 15
Versuchsvorschrift „Nachstellung des Versuches nach Tswett“
(Säulenchromatographie von Blattextrakt)
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Aceton F, Xi 11-36-66-67 9-16-26
Calciumcarbonat - - -
Glaswolle - - -
Geräte:
Erlenmeyerkolben, Chromatographierohr, Schlauchstück, Quetschhahn,
Glasstab, Stativmaterial
Durchführung:
In ein Chromatographierohr (Durchmesser 10 – 15 mm) bringt man einen
Bausch Glaswolle und verschließt den Auslauf mit Schlauchstück und
Quetschhahn. Anschließend befüllt man das Rohr zu einem Drittel mit
Aceton. In dieses gibt man unter Rühren mit einem Glasstab so viel
Calciumcarbonat, bis eine breiige Masse entsteht. Die Konsistenz lässt sich
mit dem Glasstab prüfen. Im Wechsel werden so lange Aceton und
Calciumcarbonat in das Chromatographierohr gegeben, bis das Rohr zu
etwa 80% gefüllt ist. Das Calciumcarbonat-Aceton-Gemisch wird mit
Glaswolle abgedeckt, um eventuelle Aufwirbelungen beim Aufbringen von
Lösungen zu vermeiden.
Dann gibt man etwa 10 mL des Blattextraktes in die Säule und öffnet den
Quetschhahn. Die Tropfgeschwindigkeit sollte etwa 1 Tropfen pro
2 Sekunden betragen. Immer wenn der Füllstand der Flüssigkeit im
Chromatographierohr unmittelbar über der Calciumcarbonatfüllung ist, wird
weiteres Aceton nachgefüllt.
Die Dauer des Versuches beträgt etwa zwei Stunden.
119

Entsorgung:
Die organischen Lösungen können neutral im Behälter für organische
Lösungsmittel entsorgt werden. Das Füllmaterial des
Chromatographierohres wird getrocknet und in der Feststofftonne entsorgt.
120
Anlage 16
Versuchsvorschrift „Chromatographie mit Tafelkreide“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Tafelkreide - - -
Geräte:
Weithalserlenmeyerkolben 250 mL
Durchführung:
Ein komplettes Stück Tafelkreide wird im Trockenschrank 15 Minuten bei 110 °C getrockenet. Anschließend wird ein Weithalserlenmeyerkolben etwa 3 mm hoch mit der
Blattextrakt-Lösung befüllt und die Tafelkreide senkrecht freistehend in den
Erlenmeyerkolben gestellt. Nach ca. 5 Minuten kann die Tafelkreide aus
dem Erlenmeyerkolben entnommen werden.
Entsorgung:
Die Tafelkreide kann in der Feststofftonne entsorgt werden.
121
Anlage 17
Versuchsvorschrift „Herstellung von DC-Karten“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Kieselgel - - -
Calciumhydroxid Xi 41 22-24-26-39
Ascorbinsäure
5 • 10-3 mol/L
- - -
Calciumchlorid Xi 36 22-24
Geräte:
Becherglas, Glasplatte 20 x 20 cm, Objektträger, Exsikkator, KPG-Rührer
komplett, Stativmaterial, Glasstab, Auftragegerät für Kieselgel (Degessa,
Heidelberg)
Durchführung:
In einem Becherglas werden 20 g Kieselgel, 12 mg Calciumhydroxid und
44 mL 5 • 10-3 molare Ascorbinsäurelösung eine Stunde lang gerührt. Die
breiige Masse wird anschließend mit einem Streichgerät auf die Glasplatte
aufgebracht. Zusätzlich werden mehrere Objektträger beschichtet. Dies
geschieht durch „Ausrollen“ mit einem Glasstab. Die hergestellten Platten
werden nun eine Stunde im Trockenschrank bei 100 °C getrocknet. Nach
3-5 Tagen Lagerung in einem Exsikkator können die Platten verwendet
werden.
122
Anlage 18
Versuchsvorschrift „Dünnschichtchromatographie von Blattextrakt“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Aceton
F, Xi 11-36-66-67 9-16-26
Petrolether
(40-60 °C)
F, Xn, N 11-38-48/20-
51/53-62-65-67
9-16-33-36/37-
60-62
Isopropanol F 11 7-16
Geräte:
DC-Kammer, DC-Mikrokarte (5 x 10 cm, Kieselgel auf Aluminiumfolie),
Fön, Glaskapillaren, Messpipette 10 mL, Pipettierthilfe
Durchführung:
Zu Beginn wird die DC-Kammer in einem Abzug mit dem Fließmittel
befüllt und verschlossen. Das Fließmittel besteht aus 10 mL Petrolether,
1 mL Isopropanol und einem Tropfen entionisiertem Wasser.
Der Blattextrakt wird als dünner Strich 1 cm vom unteren Rand und 1 cm
von den seitlichen Rändern der DC-Karte mit einer Glaskapillare
aufgetragen. Anschließend wird die DC-Karte trocken gefönt und erneut
Blattextrakt aufgetragen. Dieser Vorgang wird 8-10 mal wiederholt.
Anschließend wird die Karte in die DC-Kammer gestellt. Wenn sich die
Fließmittelfront etwa 1 cm unterhalb des oberen Randes der DC-Karte
befindet, wird die Chromatographie beendet und die Fließmittelfront
markiert.
Entsorgung:
Das Fließmittel wird im Behälter für organische Lösungsmittel entsorgt. Die
DC-Karte wird in der Feststofftonne entsorgt.
123
Anlage 19
Versuchsvorschrift „Präparative Dünnschichtchromatographie mit
photometrischer Untersuchung“
Chemikalien:
Substanz Gefahrensymbol R-Sätze S-Sätze
Blattextrakt in
Methanol
T, F 23/24/25-
39/23/24/25
7-16-36/37-45
Petrolether
(40-60 °C)
F, Xn, N 11-38-48/20-
51/53-62-65-67
9-16-33-36/37-
60-62
Isopropanol F 11 7-16
Aceton F, Xi 11-36-66-67 9-16-26
Dünnschichtchromatographie
Geräte:
DC-Kammer, DC-Karte (20 x 20 cm, Kieselgel auf Aluminiumfolie), Fön,
Glaskapillaren, Messpipette 10 mL, Pipettierthilfe
Durchführung:
Zu Beginn wird die DC-Kammer in einem Abzug mit dem Fließmittel
befüllt und verschlossen. Das Fließmittel setzt sich aus 20 mL Petrolether,
2 mL Isopropanol und zwei Tropfen entionisiertem Wasser zusammen.
Der Blattextrakt wird als dünner Strich 1 cm vom unteren Rand und 1 cm
von den seitlichen Rändern der DC-Karte mit einer Glaskapillare
aufgetragen. Anschließend wird die DC-Karte trocken gefönt und erneut
Blattextrakt aufgetragen. Diese Vorgänge werden 8-10 mal wiederholt.
Anschließend wird die Karte in die DC-Kammer gestellt. Wenn sich die
Fließmittelfront etwa 1 cm unterhalb des oberen Randes der DC-Karte
befindet, wird die Chromatographie beendet und die Fließmittelfront
markiert.
124

Aufnahme der Absorptionsspektren
Geräte:
Spatel, Zentrifugengläser, Photometer, Küvetten
Durchführung:
Zur Aufnahme der Absorptionsspektren werden die Banden einzeln mit
einem Spatel abgekratzt und in jeweils ein Zentrifugenglas überführt. Das
Kieselgel wird nun mit 3 mL Aceton überschichtet und die Farbstoffe
eluiert. Anschließend wird zentrifugiert, der Überstand kann für die
Spektroskopie verwendet werden.
Alle Absorptionsspektren werden im Bereich von 760-360 nm
aufgenommen. Zusätzlich zu den isolierten Banden wird auch ein Spektrum
des Blattextraktes aufgenommen.
Entsorgung:
Das Fließmittel sowie die Lösungen der Blattfarbstoffe werden im Behälter
für organische Lösungsmittel entsorgt, die DC-Karte in der Feststofftonne.
125

Ich versichere hiermit, dass die vorliegende Arbeit selbständig verfasst,
keine anderen als die angegebenen Hilfsmittel verwendet und sämtliche
Stellen, die den benutzten Werken dem Wortlaut oder dem Sinne nach
entnommen sind, mit Quellenangaben kenntlich gemacht sind. Alle wörtlich
entnommenen Stellen sind als Zitate kenntlich gemacht.
Entsprechend gilt dies auch für alle Abbildungen.
Alle Datenträger, auf denen der Text und die Abbildungen der Arbeit
gespeichert wurden, befinden sich in meinem Besitz.
Burghaun, den 08.05.2007 __________________________














![FREUDE SCHENKEN AN WEIHNACHTEN · 2010-11-16 · [20] THOMAS SABO Uhr Classic 359 € [21] THOMAS SABO Uhr It Girl, Keramik 798 € [22] THOMAS SABO Ohrhänger 279 € [23] THOMAS](https://img.pdfslide.org/doc/110x75/5f54869d270bd47b5334c561/freude-schenken-an-weihnachten-2010-11-16-20-thomas-sabo-uhr-classic-359-a.jpg)














