Embed Size (px)
Citation preview

420 J. SCHO~MULLE~ und S. VIERK6TTER:
adsorbiert an einer, wie vorher dimensionierten, mit PetroNther getrankten S~ule yon Alu- miniumoxyd (BROCKMAN~). Mit etwa 50 ml Petrol/~ther wird anteilig fettfrei gewaschen und danaeh der Farbstoff mit einer LSsung yon ~thanol und Eisessig (5 ~- 1) eluiert.
Das Eluat dampft man mit einem FSn zur Troekne and nimmt den l~iickstand mi~ wenigen Tropfen Chloroform auf. Diese LSsung wird chromatographiert.
Unter Anwendung der bei dem Nachweis yon Annatto und Carotin (Abschn. III) beschriebenen Drehteehnik l&g~ man mit dem Elutionsmittel Benzol das dem Curcumin anhaftende Fet t aus dem Startpunkt seitlieh heraustreten.
Sodann wird mit dem Elutionsmittel I I I bis zur vollen SteighShe (10 era) ehromatographiert. Die Farbzone erreicht~ die gleiehe SteighShe, wie die mit aufzutragende Vergleichssubstanz. Die Tfipfelreaktion mit SbCla-LSsung ergib~ eine kri~ftige I%otf~rbung.
Zusammen/assung
Es werden E insa tzm6gl ichke i ten der Df innsch ieh tehromatograph ie Ifir die lebens- mi t t e l ana ly t i s che Prax i s aufgezeigt . Neben dem Naehweis fet t l6sl ieher , syn the t i seher Farbs tof fe werden Arbe i t svorschr i f t en fiir den Naehweis yon A n n a t t o nnd Carot in in Margar ine und Kgse, sowie yon Pap r ika - und Cureumafarbs tof f in W u r s t mi tgete i l t . Ube r den Naehweis yon Safran in Baekwaren wird an anderer Stelle ber ichte t .
Phosphate und organische Phosphorverbindungen in Lebensmitte ln
X I . Mittei lung
Pr~iparative Darstellung yon Inosittriphosphat aus enzymatischen Phytins~iurehydrolysaten
Von
J. SCHORM()LLER und S. VIERKOTTER*
MitteiZung aus dem Institut /iir Lebensmittelchemie und Lebensmitteltechnologie der Technischen Universit~it Berlin**
Mit 2 Textabbildungen
(Eingegangen am 25. Oktober 1961)
In vorhergehenden Arbe i ten haCten wir uns zun~chst mi t der pap ie rchromato- graphischen Trennung yon Inos i tphosphors~urees te rn besch~ft igt 1 nnd dann die pri~parative Isol ierung solcher Es t e r aus sauren sowie enzymat i schen H y d r o l y s a t e n des ~Iexa-Phosphors i iurees ters in Angrif f genommen ~. Es war uns seinerzei t n icht gelungen, im Bereich der Tri- und Te t r aphospha t e nach saurer Hydro ly se des H e x a p h o s p h a t e s eine rest lose Trennung dieser be iden t I y d r o i y s e n p r o d u k t e durch- zuffihren. I n de r vor l iegenden Arbe i t haben wi t diese Versuche wieder aufgenommen. W i r ber ich ten hier fiber ein Verfahren zur pri~parat iven Gewinnung yon re inem I n o s i t t r i p h o s p h a t aus enzymat i schen Phyt ins i~urehydro lysa ten mi~ Hilfe der Sgulen- chromatographie . Gleichzeit ig gelang es uns, du tch verbesser~e Ex t r ak t ionsbed in - gungen bei der Gewinnung eines Phy ta se -Trockenprgp~ra t e s aus WeizenMeie die
* Auszug aus der Diplomarbeit S. VIERKSTTEI~,: Untersuchungen zur Trennung enzymatischer Phytins~urehydrolysate. Techn. Univ. Berlin 1961.
** Die Arbeit wurde durch den Verband der Chemischen Industrie gef6rdert. Hierffir danken wir auch an dieser Stelle.
1 SC~O~i~LL~R, J., u. G. Wi~DIG: Diese Z. 105, 397 (1957). SCKO~iiLL~R, J., u. G. BR~SSAV: Diese Z. 113, 387, 484 (1960).

Phosphate und organische Phosphorverbindungen in Lebensmitteln. XI 421
Ausbeute u m 20~o zu steigern. I m Laufe dieser Arbei t und bei sp~teren Mit te i lungen wenden wir ffir die einzelnen Inosi tphosphorsi iuren in Anlehnung an DESJO~]~t~T u n d P~T~K ~ folgende Bezeichnungen an :
Inos i tmonophosph~t IP~ Inos i t t e t raphospha t I P t Inos i td iphosphat IP~ Inos i tpen taphospha t IP~ Inos i t t r iphosphat IP~ Inos i thexaphosphat IP~
Die yon uns bisher benu tz t en Abkfirzungen, vor ahem ffir Mono- u n d Diphosphat (IMP u n d IDP) sind aueh ffir Inosinphosphate im Gebraueh, so dab eine ~mderung unserer Nomenk la tu r zweekm~Big erseheint, u m Migverstgndnisse zu vermeiden.
Eigene Versuche I. Darstellung des Enzym-Troc~enpriiparates
300 g frische Weizenkleie wurden in Portionen yon je 100 g mit 500 ml Wasser angerfihrt und bei 20000 U/rain homogenisiert. Die breiartigen Suspensionen wurden vereinigt und 2~/~--3 Std zur Extraktion des Enzyms langsam gerfihrt. Die ttomogenisierung ermSglicht eine ~vesentlich bessere Extraktion des Enzyms als das bisher gefibte Zerquetschen der Kleie in der Kugelmfihle. Naeh beendeter Extraktion wurde die Suspension fiber eine Nutsehe (ohne eingelegtes Filter) abgesaugt. Nach dem Zentrifugieren des Filtrates (10 rain bei 3000 U/rain) und nach 33°/oiger S£ttigung der zentrifugierten LSsung wurde der entstehende Niederschlag abzentrifugiert und verworfen. Die weitere Behandlung des Ansatzes geschah nach frfiher ~ mitgeteilten Angaben. Nach dem Trocknen der letzten Acetonf~llung im Vakuumexsiccator fiber CaC12 wurden 3,3 g Enzymtrockenpr~parat erhalten, dessert Aktivit~t dem eines frfiher beschriebenen Phytase- pr~parates entsprach 3 (vgl. Abb. 1 dieser Arbeit). Ersatz der frfiher gefibten Extraktion in der Kugelmiihle durch die ()ben erw~hnte Homogenisierung steigerte die Ausbeute an Ferment- trockenpr~parat um rund 20O/o .
IL Enzymatische Hydrolyse des Na-Phytates
1. U m ffir die im pr/~parutiven MM3stab vorgesehenen Bebrtitungs~ns/~tze Anha l t spunk te fiber die notwendige t tydrolyseduuer u n d das gfinstigste Verhi~ltnis von Substrat- zu Enzymkonzen t r a t i on zu erhalten, wurde in einer Reihe yon Vor- versuehen die Konzen t r a t i on an Subs t ra t -P kons t an t gehalten, die an E n z y m jedoch abgestuft .
a) 2,6554 g Na-Phytat wurden in Wasser gelSst, mit Essigs~ure auf pE 5,2520 ° c (= 5,5537 ° c) gebraeht und im Megkolben auf 100 ml aufgeffillt.
b) Die iiir den Ansa~z benStigte Menge an Enzym-Troekenpr~parat wurde im N5rser mit wenig Puffer [0,2 m-Acetatpuffer p~ 5,2520° c (= 5,5537o c)] angeteigt, dutch Rfihren in mehr Puffer gelSst, nach 1 stfindigem Stehen filtriert und mit Puffer auf 100 nil aufgefiillt. Gegen Mikroorganismenbefa]l wurde ein kleiner Kristall Thymol zu- gesetzt. Beide LOsungen wurden im Thermostaten 1 Std auf 37 ° C vorgew~rmt, ebenso das Be- brfitungsgef~iB im Brutschrank. Den vereinigten L5sungen Will.- den sofort und dann in weiteren Abst~nden bei 37 ° C Proben ent- nommen, die naeh Sistierung
Tabelle 1. Versuchsdaten der orientierenden Ansiitze
Versuch 3
Ester-P-Konzentration (#g/ml) . Enzym-Konzentration (#g/ml) . Verh~ltnis Substrat-P zu Enzym-
I~onzentration . . . . . . . Puffer . . . . . . . . . . . .
Temperatur
Yersuch Versuch 1 2
2460 2455 2455 2500 1250 625
1:1 1:0,5 1:0,25 0,1m-Acetat)uffer
p~5,537 ° C 37°C
I DESJO]~ERT, A., u. F. PETEK: C. •. Acad. Sei. (Paris) 241, 1343 (1955); Bull. Soc. chim. biol. (Paris) 38, 871 (1956).
SC~O~iiLLER, J., 11. G. B~ssAv: Zit. S. 420, Anm. 2; daselbst S. 494. SCHOnMi~LLER, J., 11. G. B~SSAU: Zit. S. 420, Anm. 2; daselbst Abb. 3, S. 496.
422 J. SCHO~i~LL~ und S. VIERKOTTER:
der Fermentaktivi~gt mit Trichloressigsaure sowie nach Neutralisation des Filtrates gegen p-Nitrophenol wie friiher beschrieben auf ihren P-Gehalt untersuch$ wurden.
Die Phosphorbestimmung erfolgte auch hier nach der in unserem Institu~ fiblichen Methode yon LowRY und LOPEZ ~.
Das verwendete Na-Phytat (Fa. Ciba AG, Wehr/Baden) enthielt 0,39% anorganischen P und 18,85% Gesamt-P. Daraus erreehnet sich ein Ester-P-Gehult yon 18,46%.
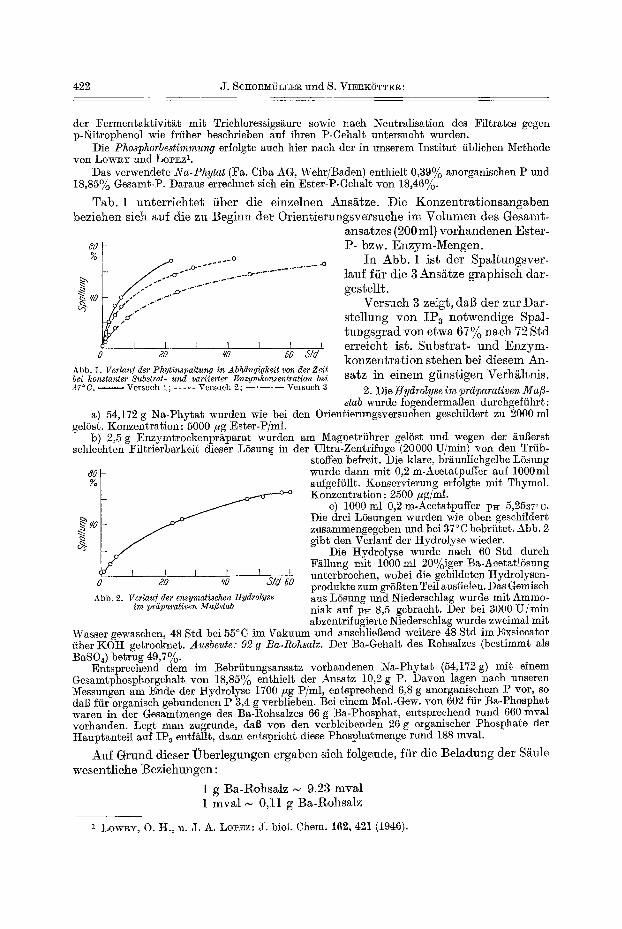
Tab. 1 unter r ich te t fiber die einzelnen Ans/~tze. Die Konzen t ra t ionsangaben beziehen sieh auf die zu Beginn der Orientierungsversuche ira Volumen des Gesamt-
ansatzes (200 ml) vorhandenen Ester-
~E I I I 1 I I ) o 20 ~o 50 J/d
Abb. 1. Verlauf der Phytinspaltung in Abh~ngigkeit yon der Zeit bei konstanter Substrat- and variierter Enzymkonzentration bei 37 ° C. Versuch i; . . . . . ¥ersuch 2; . . . . . Versuch 3
P- bzw. Enzym-Mengen. I n Abb. 1 ist der Spal tungsver-
lauf ffir die 3 Ans/~tze graphisch dar- gestellt.
Versueh 3 zeigt, dab der zur Dar- stellung yon I P 3 notwendige Spal- tungsgrad yon etwa 67~o nach 72 Std erreicht ist. Substra t - a n d Enzym- konzen t ra t ion stehen bei diesem An- satz in einem gfinstigen Verh/~ltnis.
2. Die Hydrolyse im pri~parativen Marl- 8tab wurde Iogendermal3en durchgeffihrt:
a) 54,172 g I~a-Phytat wurden wie bei den Orientierungsversuehen geschildert zu 2000 ml gelSs$. Konzentration: 5000 #g Ester-P/ml.
b) 2,5 g Enzymtrockenpr/~parat wurden am Magnetriihrer gelSst und wegen der ~uBers~ schleehten Ffltrierbarkeit dieser LSsung in der Ultra-Zentrifuge (20000 U/rain) yon den Trfib-
stoffen befreit. Die klare, br~iunlichgelbe LSsung
I I I 1 1 1 zo ~o J/d 6o
Abb. 2. Ve~'lauf de~' enzymatischen Hyd~'olyse im prdiparativen Ma~stab
wurde dann mit 0,2 m-Ace~atpuffer auf 1000ml aufgeffillt. Konservierung erfolgte mit Thymol. Konzentration: 2500 gg/ml.
c) 1000 ml 0,2 m-Aceta~puffer pr~ 5,2537 ° c. Die drei LSsungen wurden wie oben geschildert zusammengegeben and bei 37 °C bebriitet. Abb. 2 gibt den Verlauf der I-Iydrolyse wieder.
Die Hydrolyse wurde nach 60 Std durch F~llung mit I000 ml 20°/oiger Ba-AeetatlSsung unterbrochen, wobei die gebildeten I-Iydro]ysen- produkte zum grSBten Tefl uusfielen. Das Gemisch aus L5sung und Iqiederschlag wurde mit Ammo- niak auf plz 8,5 gebracht. Der bei 3000 U/rain abzentrifugierte Niedersehlag wurde zweimal mit
Wasser gewaschen, 48 Std bei 55°C im Vakuum und anschliel]end weitere 48 Std im Exsiccator (iber KOH ge~rocknet. Ausbeute: 92 g Ba.Rohsalz. Der B~-Gehalt des ltohsalzes (bestimmt als BaS04) betrug 49,7%.
Entsprechend dem im Bebrfitungsansatz vorhandenen Na-Phytat (54,172 g) mit einem Gesamtphosphorgehalt yon 18,85% enthielt der Ansatz 10,2 g P. D~von lagen naeh unseren Messungen am Ende der Hydrolyse 1700 #g P/ml, entspreehend 6,8 g anorganischem P vor, so dab fiir organisch gebundenen P 3,4 g verblieben. Bei einem Mol.-Gew. yon 602 flit Ba-Phosphat waren in der Gesamtmenge des Ba-Rohsalzes 66 g Ba-Phosphat, entspreehend fund 660 mval vorh~nden. Leg~ man zugrunde, d~l~ yon den verbleibenden 26 g organischer Phosphate tier Hauptanteil auf IP 3 entf~ll~, dann entsprieht diese Phosphatmenge fund 188 reval.
Auf Grund dieser l~berlegungen ergaben sieh folgende, fiir die Beladusg der S/~nle wesentliche Beziehungen :
1 g Ba-Rohsalz ~ 9,23 mva l 1 royal ~ 0,11 g Ba-Rohsalz
I Low~¥, O. H., u. g. A. Lop,z: J. biol. Chem. 162, 421 (1946).

Phosphate und organische Phosphorverbindungen in Lebensmitteln. XI 423
I I I . Sdulenchromatographische Trennung des Phytinsdurehydrolysates
Da wir uns weitgehend ffiihere Erfahrungen unseres Arbeitskreises 1 zunutze machen konnten, geniigten wenige Vorversuche, um zu folgender Arbeitsvorschrift zu gelangen, die sich fiir die Isolierung yon 1P 3 eignete:
8,25 g im M5rser fein zerriebenes Ba-Rohsalz wurden mit wenig Wasser zu einer breiartigen Suspension verarbeitet und naeh und naeh mit 120 ml Amberlite IR 120 (H+-Form, 20--50 mesh; Kapazit~t 2,3 mval/ml) unter dauerndem Riihren versetzt. Naeh wenigen Minuten war bis auf eine geringe Triibung weitgehende ~berffihrung in die freien Sauren erfolgt. Zur restlosen Ent- fernung der Ba++-Ionen wurden L5sung und ttarz auf eine mit demselben Harz beschickte S~ule gegeben, die L5sung bei einer Tropfgeschwindigkeit yon 1 ml/min abgezogen und das Harz mit Wasser bis zur neutralen Reaktion des Eluates nachgewaschen. Die Menge des in der S~ule vorbereiteten Harzes war auf eine Kapazit~t yon 1 ml Harz/1 mwl Ba ++ berechnet, dies ent- sprach im vorliegenden Falle bei einem Ba-Gehalt des Rohsalzes yon 49,7% etwa 60 ml Harz, so dab fiir eine vollst~ndige ~Yberfiihrung des Rohsalzes in die ~reien S~uren insgesamt 180 ml Harz notwendig waren.
Die hier verwendete Harzmenge lag welt fiber der, wie sie der Kapazit~t des Harzes entsprach. Wir machten jedoch die Beobachtung, dab bei der Anwendung ldeinerer Mengen der Austausch um vieles langsamer vor sich geht. Offenbar ist im letztgenannten Fall die Bertihrungsfl~che zwischen Harz und Suspension zu gering und damit die Bfldung der ffeien S~uren verzSgert. Die freigesetzten S~uren besitzen auBerdem noch zus~tzlich ein LSsungsverm6gen fiir die Ba-Inosit- Phosphate.
Die nach dem oben beschriebenen Arbeitsgang erhaltene LSsung der freien S~uren wurde mit 2 n-NaOH gegen p-Nitrophenol neutralisiert und im Vakuum (17 mm Hg) bei einer Wasser- bad~emperatur yon 45 o C auI ein Volumen yon 5--10 ml eingeengt.
Zur Trennung der Phosphate wurde eine Anionen-Austauschersgule (3,6 ×22 era) benutzt, die mit 200 ml Dowex 2X8 (20--50 mesh; Kapazit~t 1,4 mval/ml) gefiillt war. 50% des Harzes waren vorher mit n-Na-AeetatlSsung vollst~ndig in die Acetatform iibergefiihrt und dann mit der anderen, in der C1--Form vorliegenden Halite im Becherglas gut gemiseht worden. Die Kapazit~t der Saule betrug 280 mval. Die teflweise t)-berfiihrung des Austauschers in die Aeetatform ist notwendig, um eine quantitative Fixierung der Hydrolysenprodukte, insbesondere der niederen Ester, an der S£ule zu erreichen. Die ttydroxydform ist weniger geeignet, da der Austauseher dureh Laugen leicht gesch~digt wird 1. Auf die so vorbereitete S~ule wurde die eingeengte LSsung des tIydrolysates gegeben; die S~ule wurde mit 200--250 ml Wasser bei langsamer Tropfgeschwin- digkeit (0,5 ml/min) nachgewaschen. Die vo]lst~ndige Fixierung der aufgegebenen Phosphate wurde wie iiblich mit dLem Phosphat-Test nach HA~S und IS~WOOD ~ naehgeprfift. Anorga- nisches und organisches Phosphat lassen sich nach FLv, cKs.~sws.r~ u. Mitarb2 unterscheiden.
Nach den Befunden yon POSTERNAK 4 sowie yon DESJOBERT u n d PETEK 5 sind nach etwa 66%iger enzymatischer Spal tung des Inosi thexaphosphates n u r noch IP3, IP2, I P 1 u n d P 0 ~ - - im Ansatz vorhanden. I P 1 geht wegen der leichtenLSs]ichkeit des Ba-Salzes n icht in den Ba-Niederschlag des Rohsalzes. Dieser Tatsaehe sowie dem Ziel einer Isol ierung des I P 3 Reehnung t ragend genfigte es in unserem Fall, un te r weitgehender Schonung des I P 3 die anderen noeh vorhandenen X o m p o n e n t e n (IP 2 und PO~-- ) zu~'~ammen zu eluieren. Wie wir sehon fr/iher fanden, bestehen zwischen der Anz~hl der Phospha tgruppen im Inosi tmoleki i l u n d den R r W e r t e n einerseits, den ftir d:ie Eluierung des jeweiligen Phosphatrestes notwendigen HC1- Konzen t ra t ionen andererseits Beziehungen der Art, dal] die l e t z tgenann ten Konzen- t r a t ionen proport ional dem Quadra t der Phosphorgruppen-Anzahl im Inosi tmolekti l zunehmen 6.
En t spreehend dieser Beziehung: 0,02 norma l × a 2 (a = Anzuhl der Phosphat- gruppen) wurden POI/-- , f~r das diese Beziehung zw~r nieht gilt, das aber bereRs mi t
SCJ~O~tt~L~, J., u. G. BRasses: Zit. S. 420, Anm. 2. I-lANES, C. S., u. ~". A. ISttERWOOD: Nature (Loud.) 164, 1107 (1949). FLEeK~NST~IN, A., E. GER~ACH U. J. J~NK~: Natm'wiss. 40, 462 (1953). POSTS, R~K, S., u. T~. POST~R~K: Helv. chim. Acta 15, 1165 (1928). D]ssJo~]~RT, A., u. F. P ~ K : Zit. S. 421, Anm. 1. SCJ~OR~Ln~R, J., u. G. BRESSAV: Zit. S. 420, Anm. 2; daselbst S. 390.

424 J. SCtIOI~I~IULLER und S. VIERKOTTER:
0,03 n-tiC1 eluiert wird, und I P 2 mit der fiir das Diphosphat wirksamen Konzen- t ra t ion yon 0,08 n-tiC1 eluiert. Zu Beginn der Elut ion findet zun/~ehst ein Austausch der lockerer fixierten Aceta t - Ionen start. Die Elut ion yon P O ~ - - und IP~ wurde auch hier mit dem oben erw/~hnten Phosphat -Tes t kontrolliert. I m vorliegenden Ansatz gen/igten etwa 1500 ml 0,08 n-He1 zur vollst/~ndigen Elution. Diese Menge ist na- t/irlich yon Fall zu Fall weehselnd und abhgngig yore Anteil des in Aeeta t form vor- liegende n Harzes, da auch das Acetat yon der Saule entfernt werden muB.
Ansehliegend wurde mit 800--900 ml 0,2 n-HC1 das IP3 elniert, das Eluat mit 2 n-NaOH gegen p-Nitrophenol neutralisiert und das IPs mit 20 ml 40°/oiger Ba-AcetatlSsung gef/illt. Die fiberstehende LSsung wurde dekantiert, der Niederschlag abzentrifugiert, mehrmals am ttomo- genisator mit Wasser bis zur negativen Cl--l~eaktion gewasehen und im Exsiecator fiber KOH getroeknet. Ausbeute: 0,8 g Barium-Inosittriphosphat (bezogen auf das Ba-Rohsalz).
Danaeh liegen rund 10% des Ba-Rohsalzes als Ba-IPs vor. Beriicksichtigt man jedoch den hohen Gehalt an anorganischem Phospha t im Rohsalz (fund 71~o), so betr/~gt der Anteil des B a - I P 3 an der Gesamtmenge des organischen Phosphates etwa ein Drittel. W/ihrend wir frtiher 1 naeh der Methode POSTEENAKs 2 Ausbea ten an I P a yon 6% (bezogen auf eingesetztes Na-Phy ta t ) erreichten, erlaubt die besehriebene s/~ulenehromatographisehe Trennung eine Ausbeutesteigerung auf 16%. AuBerdem fallen bier, wie die papierehromatographische Pr/ifung zeigte, Produkte hohen Reinheitsgrades an. Auf dem Rundf i l te rchromatogramm 3 war I P a als scharf abge- grenzter Fleck neben geringen Verunreinigungen lokalisierbar. I m Vergleieh mit Modellsnbstanzen aus friiheren Untersuchungen nahm die yon uns isolierte Substanz den ihr zukommenden, zwisehen IP~ und I P 4 liegenden Platz ein.
IV. Reinigung und Analyse des Inosittriphosphates
Um geringe Mengen der papierchromatographiseh nachgewiesenen Verunreinigungen zn entfernen, wurde das Ba-Salz des Triphosphates in wenig Wasser suspendiert, mit Amberlite IR120
(It+-Form) in die freie S/~ure fibergeffihrt, dutch ein
Tabelle 2. Analyse des Inosittriphos- phates (Ba-SMz)
I c ~2 moI-C/P
Ber. 8,71 11,23 6,00:3,00 Gel. 8,69 11,14 6,00:2,96
Blaubandfilter filtriert, mit lriseh bereiteter Barytlauge bei p~ 6--7 wieder ausgef/~llt, je 10 rain bei 3000 und dann bei 15000 U/rain zentrifugiert und im Vakuum getrocknet.
Zur Analyse wurde die Substanz fiber KOH im Va- kuum naehgetroeknet. Aus den ermittelten Werten ffir Kohlenstoff ~ und Phosphor 5 wurde das molare C/P-Ver- h/~ltnis erreehnet.
V. Zur Strulctur des Inosittriphosphates
Uber die S t ruktur des IP 3 ist wenig bekannt geworden. CouRToIs und MAsso~ 6 oxydierten I P 3 mit I-Iypobromit, t tNOa--CrO 3 und Perjodsaure-Sehwefelsaure und schlossen aus den hier auf t re tenden Oxydat ionsprodukten, dab yon den drei zur Diskussion stehenden Strukturen (1-2-3 ; 1-2-4 and 1-3-5 fiir die freien Ott-Gruppen) nur die letzte in Frage komme, da nach ihren Untersuehungen zwei freie, neben-
1 Dissertation G. W/i~DIG: Untersuehungen zur papierchromatographisehen Trennung und Identifizierung der Inositphosphors~iuren. Teehn. Univ. Berlin 1958 (D 83).
2 POST~R~AK, S., u. Tm POST~AK: Zit. S. 423, Anm. 4. 8 SCHOaMiiLL~, J., u. G. W/3RDm: Zit. S. 420, Anm. 1.
Die Kohlenstoffanalysen wurden dankenswerter Weise im Analytischen L~boratorium des Organischen Instituts der Techn. Univ. Berlin (Prof. F. BOttLMANN) durchgeffihrt.
Low~:f, O. It., u. J. A. LoPEz: Zit. S. 422, Anm. 1. 6 COU~ToIS, J., u. M. 3/IAsso~: Bull. Soc. chim. biol. (Paris) 32, 314; 326 (1950).

Phosphate und organisehe Phosphorverbindungen in Lebensmitteln. XI 425
einander liegende OH-Gruppen nicht vorkommen k6nnen. Danach wird dem I P 3 folgende Struktur zugesehrieben:
H OH \ / / c \
H203P--O--HC Ctt--0--PO3H2
HO--HC Ctt--OH \ /
H O--PO3H2
Ffir diese Ann~hme spricht der Befund, wonach die Priifung auf etwa vorliegende ~-G]ykol-Gruppierungen negativ verlief. Die weitere Annahme der Autoren, dal3 die Dephosphorylierung der Phytinsaure jeweils in meta-Stellung fortschreite, scheint fiir die ersten drei t tydrolysenstufen (Penta- bis Triphosphat) eine gewisse Berechti- gung zu haben. Fiir IP 1 haben jedoch neuere Untersuchungen ergeben, dab sowohl das yon ISEH~ 1 synthetisierte als auch das dutch enzymatische oder chemische t tydrolyse gewonnene Produkt ein Inosit-2-monophosphat ist 2. GI~A])o und BALLON 3 erhielten I P 3 nach der yon FoLc~ 4 angegebenen Methode durch a]kalische Hydrolyse yon Phosphoinositiden. Sie postulieren fiir das Triphosphat eine andere Struktur. Nach Perjodat-Oxydation, l~eduktion des entsprechenden Dia]dehydes und an- schlieBender Dephosphorylierung erhielten sic D-Iditol. Aus diesem Befund ergib]b sich zwingend die Struktu:~ des I P 3 als L-myo-Inosit-1,4,5(6)-triphosphat:
P P ]
Zusammen]assung Durch Ver£nderung der Extraktionsbedingungen bei der Gewinnung eines
Phy~ase-Trockenpriipara~es aus Weizenkleie konnte die Ausbeute betri~chtlich gesteigert werden. Mit I-Iil~e des Enzyms wurde Phytinsi~ure zu 66~o gespMten. Aus dem tIydrolysat wurde ein Bariuln-Rohsalz gewonnen und dieses durch Sgulen- chromatographic aufgetrennt. Das in der t tauptmenge anfallende Barium-Inosit- t r iphosphat wurde einer weiteren Reinigungsprozedur unterworfen und papier- ehromatographiseh sowie dureh Bestimmung des molaren C/P-Verhgltnisses identi- fiziert.
1 ISELIN, :B. ~.: J. Amer. Chem. Soc. 71, 3822 (1949). 2 BROWN, D. M., u. G. E. ItAnL: J. chem. Soc. 1959, 357. a GR~DO, C., u. C. E. BALLOU: J. biol. Chem. 236, 54 (1961).
FOLey, J. : J. biol. Chem. 177, 505 (1949).
![Phosphate Translocator of Isolated Guard-Cell Chloroplasts f ......["C]sorbitol as membrane-permeating and nonpermeating mark- ers and [32P]phosphate as tracer for phosphate. lhe affinities](https://img.pdfslide.org/doc/110x75/60796b9402c91a4d925da525/phosphate-translocator-of-isolated-guard-cell-chloroplasts-f-csorbitol.jpg)




























