Embed Size (px)
Citation preview

Ruhr-Universität Bochum
PD Dr. med. Moritz Meins
Dienstort: Ärztliche Partnerschaft Wagner Stibbe
Göttingen
Mutationen im MFN2-Gen als Ursache der hereditären peripheren Polyneuropa-
thie Charcot-Marie-Tooth Typ 2
Kumulative
Inaugural-Dissertation
zur
Erlangung des Doktorgrades der Medizin
einer
Hohen Medizinischen Fakultät
der Ruhr-Universität Bochum
vorgelegt von
Kathrin Engelfried
aus Esslingen (am Neckar)
2007

Dekan: Prof. Dr. med. G. Muhr
Referent: PD Dr. med. M. Meins
Korreferent: Prof. Dr. med. M. Vorgerd
Tag der mündlichen Prüfung: 17. 06. 2008

Für meine Familie

Inhaltsverzeichnis -I-
Inhaltsverzeichnis
Abkürzungsverzeichnis................................................................................... II
Tabellenverzeichnis ........................................................................................ IV
Abbildungsverzeichnis................................................................................... IV
1 Einleitung................................................................................................... 1
1.1 Charcot-Marie-Tooth Neuropathien (CMT) ...................................... 1
1.1.1 Elektrophysiologische und histopathologische Einteilung............. 2
1.1.2 Molekulargenetik der CMT............................................................ 4
1.2 Physiologie und Pathophysiologie der Mitochondrien .................. 6
1.2.1 Das MFN2-Gen............................................................................. 7
1.2.2 Aufbau, Lokalisation und Funktion des MFN2-Proteins ................ 8
2 Zielsetzung der Arbeit ............................................................................ 10
3 Methode und Ergebnisse ....................................................................... 11
3.1 Mutationsanalyse des MFN2-Gens................................................. 11
3.2 Ergebnisse ....................................................................................... 11
4 Diskussion............................................................................................... 14
5 Literaturverzeichnis................................................................................ 17
6 Danksagung ............................................................................................ 24
7 Lebenslauf ............................................................................................... 25
8 Veröffentlichung ..................................................................................... 27

Abkürzungsverzeichnis -II-
Abkürzungsverzeichnis
AS Aminosäure
ATP Adenosintriphosphat
bp Basenpaare
C- Carboxy-
Cc coiled-coil
CMT Charcot-Marie-Tooth Neuropathie
CMT1 Charcot-Marie-Tooth Neuropathie Typ 1
CMT2 Charcot-Marie-Tooth Neuropathie Typ 2
Da Dalton
del Deletion
DNA desoxyribonucleic acid, Desoxyribonukleinsäure
EGR2 Early-Growth-Response-2-Gen
et al. und andere
F Frau
fzo fuzzy onions
GARS Glycyl-tRNA-Synthetase-Gen
GDAP1 Ganglioside-Induced-Differentiation-Associated-Protein-Gen
GJB1 Gap-Junction-Protein-Beta1-Gen
GTPase Guanosintriphosphatase
HR1 heptad repeat regions 1
HR2 heptad repeat regions 2
HMSN Hereditäre Motorisch-Sensorische Neuropathie
HMSN V Hereditäre Motorisch-Sensorische Neuropathie Typ V
HMSN VI Hereditäre Motorisch-Sensorische Neuropathie Typ VI
HSPB1 Heat-Shock-27kDa-Protein-1-Gen
ins Insertion
k Kilo
kb Kilobasenpaare
KIF1B Kinesin-Family-Member-1B-Gen
LMNA Lamin-A/C-Gen
LITAF Lipopolysaccharide-Induced-TNF-Factor-Gen
M Mann
MFN1 Mitofusin 1-Gen
MFN1 Mitofusin 1-Protein
MFN2 Mitofusin 2-Gen

Abkürzungsverzeichnis -III-
MFN2 Mitofusin 2-Protein
mNLG motorische Nervenleitgeschwindigkeit
MPZ Myelin-Protein-Zero-Gen
mtDNA mitochondriale DNA
NEFL Neurofilament-Light-Polypeptide-Gen
NLG Nervenleitgeschwindigkeit
m/s Meter pro Sekunde
N- Amino-
OPA1 Optic-Atroph-1-Gen
PCR polymerase chain reaction, Polymerasekettenreaktion
p kurzer Arm eines Chromosoms; pico (10-12)
PMP22 Peripheral-Myelin-Protein-22-Gen
q langer Arm eines Chromosoms
RAB7 Ras-Associated-Protein-7-Gen
SSCP single strand conformation polymorphism
SNPs single nucleotide polymorphisms
TM Transmembran
Nukleobasen/Nukleotide:
A Adenin/Adenosin
C Cytosin/Cytidin
G Guanin/Guanidin
T Thymin/Thymidin
U Uracil/Uridin
P Purin
Y Pyrimidin
N beliebige(s) Nukleobase/Nukleotid

Abkürzungsverzeichnis -IV-
Aminosäuren:
A Ala Alanin M Met Methionin
C Cys Cystein N Asn Asparagin
D Asp Asparaginsäure P Pro Prolin
E Glu Glutaminsäure Q Gln Glutamin
F Phe Phenylalanin R Arg Arginin
G Gly Glycin S Ser Serin
H His Histidin T Thr Threonin
I Ile Isoleucin V Val Valin
K Lys Lysin W Trp Tryptophan
L Leu Leucin Y Tyr Tyrosin
Tabellen- und Abbildungsverzeichnis
Tabellenverzeichnis
Tab. 1: ................................................................................................................ 4
Tab. 2: ................................................................................................................ 5
Tab. 3: .............................................................................................................. 13
Tab. 4: .............................................................................................................. 13
Abbildungsverzeichnis
Abb. 1 ................................................................................................................. 1
Abb. 2 ................................................................................................................. 2
Abb. 3 ................................................................................................................. 7
Abb. 4 ................................................................................................................. 8
Abb. 5 ............................................................................................................... 12
Einleitung 1
1 Einleitung
1.1 Charcot-Marie-Tooth Neuropathien (CMT)
Unter Charcot-Marie-Tooth Neuropathien (CMT), auch bekannt unter der Be-
zeichnung Hereditäre Motorisch-Sensorische Neuropathien (HMSN), versteht
man eine Gruppe von genetisch heterogenen Erkrankungen des peripheren
Nervensystems. Die CMT zählen mit einer Prävalenz von etwa 1/2500 zu den
häufigsten vererbbaren Erkrankungen des Nervensystems [49].
Die Erkrankungen, die nach ihren Entdeckern Jean-Martin Charcot, Pierre Ma-
rie und Howard Tooth benannt sind, wurden erstmals 1886 dokumentiert. Die
Hauptsymptome sind progressive symmetrisch ausgeprägte Paresen mit dista-
lem Beginn, welche hauptsächlich die Beine, besonders die kleinen Fußmus-
keln und die peronealen Muskeln betreffen. Die Muskeleigenreflexe der Beine
fehlen oft bereits im frühen Stadium der Erkrankung. Charakteristisch sind
Steppergang und Storchenbeine sowie Hohlfüße (Pes cavus) mit Krallenzehen.
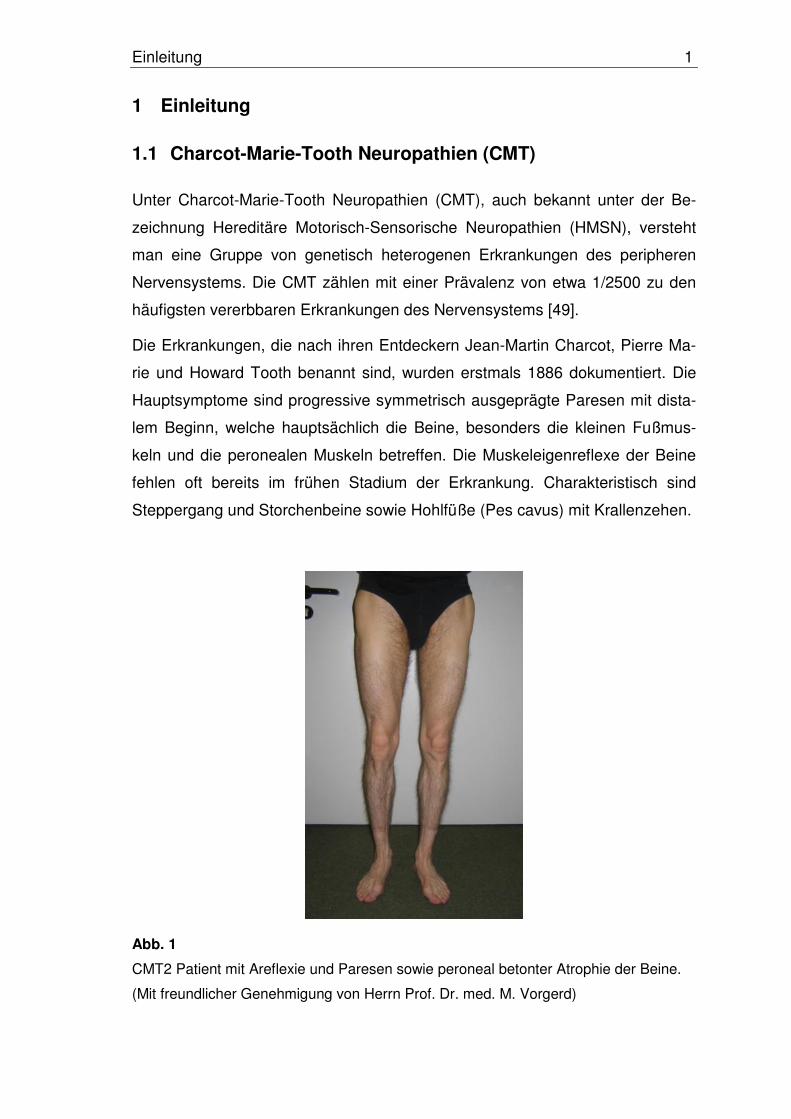
Abb. 1
CMT2 Patient mit Areflexie und Paresen sowie peroneal betonter Atrophie der Beine.
(Mit freundlicher Genehmigung von Herrn Prof. Dr. med. M. Vorgerd)
Einleitung 2
Die Ursache der Paresen liegt in einer durch Neuropathie bedingten Unterver-
sorgung der Muskulatur mit Nervenimpulsen. Trotz Bewegung tritt ein Abbau
der Muskulatur ein. Dieser Vorgang wird als neurogene Muskelatrophie be-
zeichnet. Es kommt zunehmend zum Verlust der motorischen und sensorischen
Fähigkeiten.
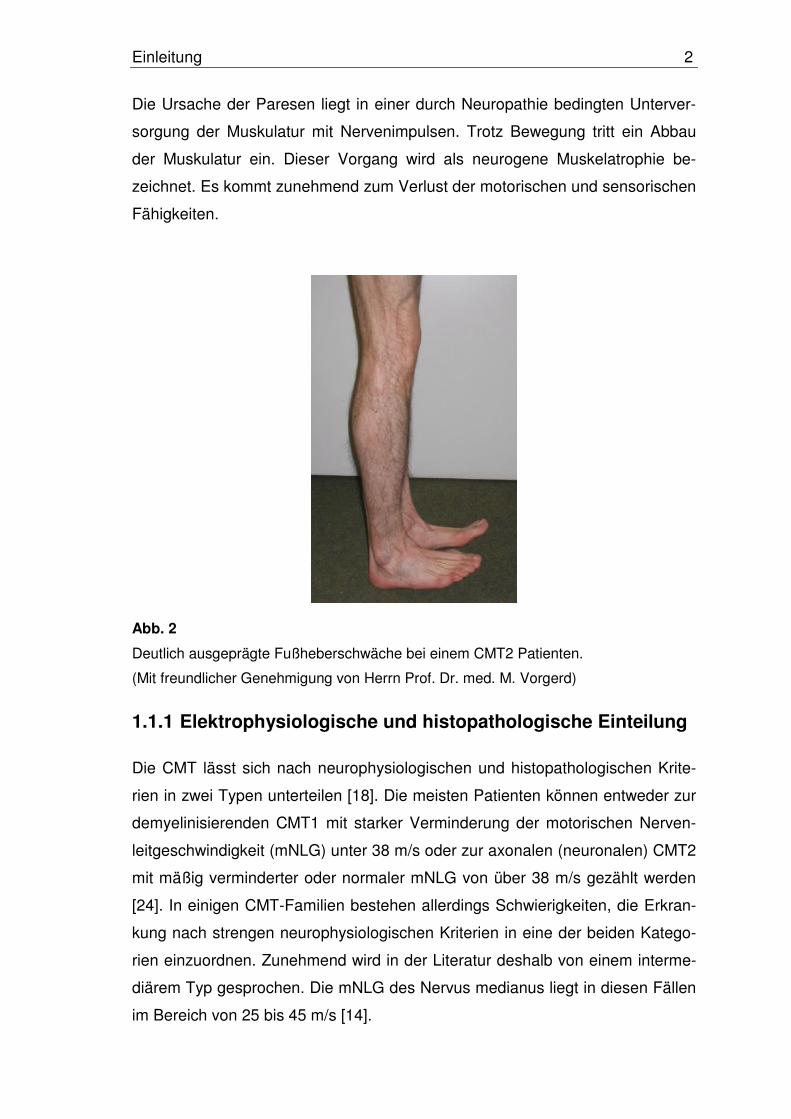
Abb. 2
Deutlich ausgeprägte Fußheberschwäche bei einem CMT2 Patienten.
(Mit freundlicher Genehmigung von Herrn Prof. Dr. med. M. Vorgerd)
1.1.1 Elektrophysiologische und histopathologische Einteilung
Die CMT lässt sich nach neurophysiologischen und histopathologischen Krite-
rien in zwei Typen unterteilen [18]. Die meisten Patienten können entweder zur
demyelinisierenden CMT1 mit starker Verminderung der motorischen Nerven-
leitgeschwindigkeit (mNLG) unter 38 m/s oder zur axonalen (neuronalen) CMT2
mit mäßig verminderter oder normaler mNLG von über 38 m/s gezählt werden
[24]. In einigen CMT-Familien bestehen allerdings Schwierigkeiten, die Erkran-
kung nach strengen neurophysiologischen Kriterien in eine der beiden Katego-
rien einzuordnen. Zunehmend wird in der Literatur deshalb von einem interme-
diärem Typ gesprochen. Die mNLG des Nervus medianus liegt in diesen Fällen
im Bereich von 25 bis 45 m/s [14].

Einleitung 3
Dem neurophysiologischen Befund entsprechend findet sich bei der CMT1 als
histologisches Korrelat eine Demyelinisierung der Markscheide. Durch wieder-
holte De- und Remyelinisierung der Nerven bilden sich histopathologisch ein
nachweisbarer segmentaler Markscheidenzerfall und sogenannte Zwiebelscha-
lenformationen aus [55].
Bei Patienten mit CMT2 findet sich histologisch primär weder Markscheidenzer-
fall noch Zwiebelschalenformation. Morphologisch dominiert der Verlust von
großen myelinisierten Axonen. Es lassen sich Zeichen der Regeneration mit
einer großen Anzahl an kleinen dünnen myelinisierten Axonen finden [55]. Die
NLG ist meist normal oder nur geringfügig erniedrigt. Die Amplitude der motori-
schen und sensorischen Aktionspotentiale ist jedoch erniedrigt [24].
Manche Familien mit CMT-Neuropathien zeigen zusätzlich zu den bereits ge-
nannten Hauptsymptomen noch eine große Variation begleitender Symptome.
Auch der Erbgang der CMT ist nicht einheitlich: In vielen Familien ist eine auto-
somal–dominante Vererbung der CMT zu erkennen. Es gibt jedoch auch Fami-
lien mit autosomal-rezessiver oder geschlechtsgebundener (X-chromosomaler)
Vererbung. Deshalb wurde neben der Einteilung in die Hauptformen CMT1 und
CMT2 noch eine Abgrenzung weiterer Unterformen vorgenommen [35].
Weiterhin ist auch eine Subklassifizierung der CMT1 und der CMT2 erfolgt. Die
Unterscheidung dieser Subtypen ist in den meisten Fällen darin begründet,
dass Mutationen in unterschiedlichen Genen mit unterschiedlicher chromoso-
maler Lokalisation zur Ausprägung einer CMT1 bzw. CMT2 führen. Die Zuord-
nung eines Patienten oder einer Familie zu einem bestimmten Subtyp ist einer-
seits durch den Nachweis der ursächlichen Mutation in einem bestimmten CMT-
Krankheitsgen, durch Familienuntersuchungen (Kopplungsanalysen) oder durch
zusätzliche, für einen bestimmten CMT-Subtyp spezifische Symptome möglich.
Auf folgender Internetseite kann eine ständig aktualisierte Mutationsdatenbank
der CMT1- und CMT2-Erkrankungen mit begleitenden Symptomen eingesehen
werden: http://www.molgen.ua.ac.be/CMTMutations.
Unter anderem werden Symptome wie spastische Paresen, Optikusatrophie,
Neuropathien der Hirnnerven, Glaukom und Neutropenie beobachtet [64]. Bei-
spielsweise wird die CMT2 mit Optikusatrophie als HMSN VI subklassifiziert [8]
und die CMT2 mit Pyramidenbahnzeichen ist auch als HMSN V bekannt [25].
Einleitung 4
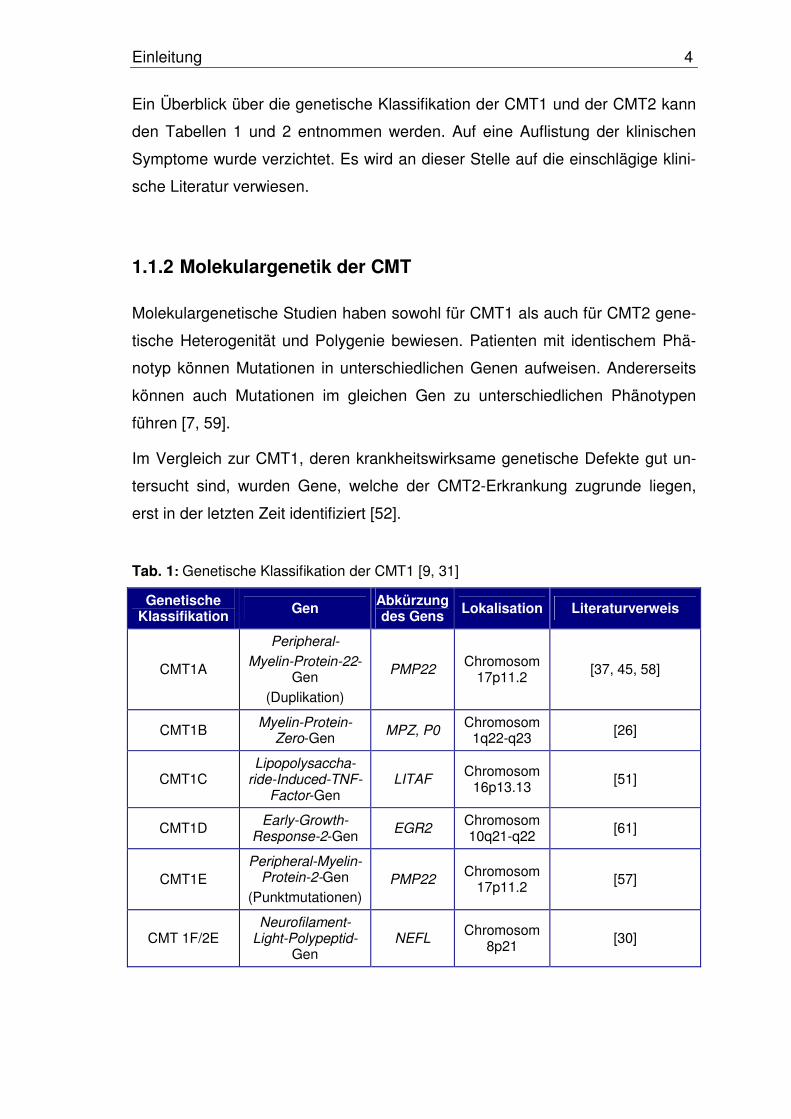
Ein Überblick über die genetische Klassifikation der CMT1 und der CMT2 kann
den Tabellen 1 und 2 entnommen werden. Auf eine Auflistung der klinischen
Symptome wurde verzichtet. Es wird an dieser Stelle auf die einschlägige klini-
sche Literatur verwiesen.
1.1.2 Molekulargenetik der CMT
Molekulargenetische Studien haben sowohl für CMT1 als auch für CMT2 gene-
tische Heterogenität und Polygenie bewiesen. Patienten mit identischem Phä-
notyp können Mutationen in unterschiedlichen Genen aufweisen. Andererseits
können auch Mutationen im gleichen Gen zu unterschiedlichen Phänotypen
führen [7, 59].
Im Vergleich zur CMT1, deren krankheitswirksame genetische Defekte gut un-
tersucht sind, wurden Gene, welche der CMT2-Erkrankung zugrunde liegen,
erst in der letzten Zeit identifiziert [52].
Tab. 1: Genetische Klassifikation der CMT1 [9, 31]
Genetische Klassifikation Gen Abkürzung
des Gens Lokalisation Literaturverweis
CMT1A
Peripheral-
Myelin-Protein-22-Gen
(Duplikation)
PMP22 Chromosom
17p11.2 [37, 45, 58]
CMT1B Myelin-Protein-
Zero-Gen MPZ, P0
Chromosom 1q22-q23
[26]
CMT1C Lipopolysaccha-
ride-Induced-TNF-Factor-Gen
LITAF Chromosom
16p13.13 [51]
CMT1D Early-Growth-
Response-2-Gen EGR2
Chromosom 10q21-q22
[61]
CMT1E Peripheral-Myelin-
Protein-2-Gen
(Punktmutationen)PMP22
Chromosom 17p11.2
[57]
CMT 1F/2E Neurofilament-
Light-Polypeptid-Gen
NEFL Chromosom
8p21 [30]
Einleitung 5
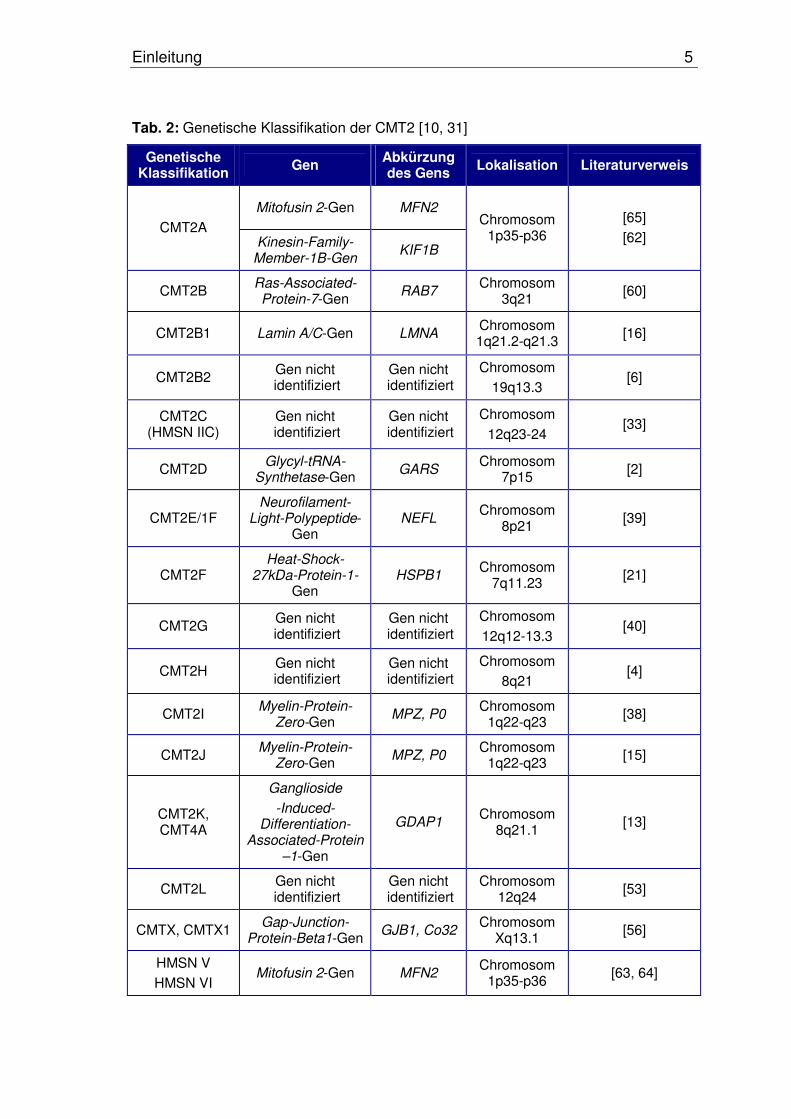
Tab. 2: Genetische Klassifikation der CMT2 [10, 31]
Genetische Klassifikation Gen Abkürzung
des Gens Lokalisation Literaturverweis
Mitofusin 2-Gen MFN2
CMT2A Kinesin-Family-
Member-1B-Gen KIF1B
Chromosom 1p35-p36
[65]
[62]
CMT2B Ras-Associated-Protein-7-Gen
RAB7 Chromosom
3q21 [60]
CMT2B1 Lamin A/C-Gen LMNA Chromosom 1q21.2-q21.3
[16]
CMT2B2 Gen nicht
identifiziert Gen nicht
identifiziertChromosom
19q13.3 [6]
CMT2C (HMSN IIC)
Gen nicht identifiziert
Gen nicht identifiziert
Chromosom
12q23-24 [33]
CMT2D Glycyl-tRNA-
Synthetase-Gen GARS
Chromosom 7p15
[2]
CMT2E/1F Neurofilament-
Light-Polypeptide-Gen
NEFL Chromosom
8p21 [39]
CMT2F Heat-Shock-
27kDa-Protein-1-Gen
HSPB1 Chromosom
7q11.23 [21]
CMT2G Gen nicht
identifiziertGen nicht
identifiziertChromosom
12q12-13.3 [40]
CMT2H Gen nicht
identifiziertGen nicht
identifiziertChromosom
8q21 [4]
CMT2I Myelin-Protein-
Zero-Gen MPZ, P0
Chromosom 1q22-q23
[38]
CMT2J Myelin-Protein-
Zero-Gen MPZ, P0
Chromosom 1q22-q23
[15]
CMT2K, CMT4A
Ganglioside
-Induced- Differentiation-
Associated-Protein –1-Gen
GDAP1 Chromosom
8q21.1 [13]
CMT2L Gen nicht
identifiziertGen nicht
identifiziertChromosom
12q24 [53]
CMTX, CMTX1 Gap-Junction-
Protein-Beta1-GenGJB1, Co32
Chromosom Xq13.1
[56]
HMSN V
HMSN VI Mitofusin 2-Gen MFN2
Chromosom 1p35-p36
[63, 64]

Einleitung 6
Das mutierte Gen Mitofusin 2 (MFN2), welches in dieser Arbeit untersucht wur-
de, wird für die Entstehung der CMT2A verantwortlich gemacht [5, 65]. Zhao et
al. beschrieb eine Mutation im Kinesin-Family-Member-1B-Gen (KIF1B) in einer
großen CMT2A Familie [62]. Jedoch wurden bis jetzt keine weiteren Mutationen
im KIF1B-Gen entdeckt, welche in Zusammenhang mit CMT2A Patienten ste-
hen. 2004 gelang es Züchner et al. erstmalig, in sieben großen CMT2A Famili-
en mittels Kopplungsanalysen zahlreiche Mutationen im MFN2-Gen zu lokalisie-
ren [65]. Zwischenzeitlich wurden ebenfalls von anderen Arbeitsgruppen Muta-
tionen im MFN2-Gen, die mit CMT2A assoziiert sind, beschrieben [19, 32, 36,
63, 64].
1.2 Physiologie und Pathophysiologie der Mitochondrien
In der Mitte des 19. Jahrhunderts entdeckte Flemming Mitochondrien als an-
färbbare Granula im Zytoplasma. Sie nehmen eine zentrale Position im zellulä-
ren Metabolismus und in energiegewinnenden Prozessen ein. Diese Energie
benötigt die Zelle zum Wachstum, zur Differenzierung und zur Biogenese von
Organellen. Sie wird durch den Zitratzyklus, die oxidative Phosphorylierung und
schließlich durch die ATP-Synthese geliefert. Mitochondrien sind auch an weite-
ren Prozessen wie Fettsäureabbau, Hämbiosynthese und der Biosynthese von
Eisen-Schwefel-Komplexen beteiligt [46].
Mitochondrien formen dynamische, netzwerkartige Strukturen, die durch ständi-
ge Teilungs- und Fusionsprozesse aufrecht erhalten werden [43]. Dabei können
diese Organellen ihre Anzahl, Form und Proteinkomposition auf unterschied-
lichste metabolische Differenzierungsstadien einstellen [48].
In Hepatozyten oder Fibroblasten finden sich längliche Mitochondrien mit einer
Länge von rund 4 µm. In einigen Zelltypen wie z.B. Muskelzellen mit hohem
Energiebedarf nehmen die Mitochondrien über 30% des Zellvolumens ein. Sie
bilden Netzwerke aus und ordnen sich entlang der energieverbrauchenden
Strukturen an [3]. Die Ausbildung umfangreicher mitochondrialer Netzwerke
innerhalb der Zelle stellt einen wichtigen Beitrag zur Sicherstellung der Energie-
versorgung in den Zellkompartimenten dar [50]. Untersuchungen an Synapsen
haben gezeigt, dass es in diesen Strukturen zu einer bemerkenswert großen

Einleitung 7
Ansammlung an Mitochondrien kommt [44]. Ist die Ausbildung von Netzwerken
gestört und der Transport entlang des Axons über eine relativ große Distanz
nicht möglich, so kann die Funktion des peripheren Nervs nicht aufrecht erhal-
ten werden. Die Folge ist axonale Degeneration.
Die Prävalenz neuronaler Erkrankungen, welche mit Mutationen in mitochondri-
alen Genen assoziiert sind, zeigt die große Bedeutung der Mitochondrien für die
Neuronen. So weisen beispielsweise mitochondriale Enzephalomyopathien,
welche durch Mutationen der mitochondrialen DNA (mtDNA) verursacht sind,
oft charakteristische neurologische Symptome auf [17, 54]. Mutationen im Pa-
raplegin-Gen, das eine ATPase in der inneren Mitochondrienmembran kodiert,
verursachen eine Form der Hereditären Spastischen Paraplegie (HSP). Tierex-
perimente an Mäusen zeigen abnormale Mitochondrienstrukturen bevor es zu
einer axonalen Degeneration kommt [22].
Auch bei CMT2-Patienten ist die mitochondriale Funktion gestört. Nur wenige
Informationen über die Histopathologie der Mitochondrien in CMT2-Patienten
sind bis jetzt bekannt. Ebenso unklar ist, welche mitochondrialen Dysfunktionen
genau vorliegen [11]. Weitere Studien werden nötig sein, um neue Erkenntnisse
über mitochondriale und zelluläre Effekte zu erhalten und zu einem besseren
Verständnis der neuronalen Biologie zu gelangen.
1.2.1 Das MFN2-Gen
Das MFN2-Gen kodiert das Protein Mitofusin 2 (MFN2) und ist auf Chromosom
1p.36.2 lokalisiert [5]. Der offene Leserahmen umspannt Exon 3 bis 19.
Abb. 3
Schematische Darstellung des MFN2-Gens, bestehend aus 19 Exons. Der offene Le-serahmen umspannt Exon 3 bis Exon 19. Funktionelle Domänen des Gens: GTPase-Region, Cc-Region (coiled coil), TM-Region (Transmembranfragment). (modifiziert nach Züchner et al. 2004)

Einleitung 8
In Hefen, sowie in der Fruchtfliege Drosophila melanogaster, wird die mito-
chondriale Fusion durch die nukleär kodierte GTPase fuzzy onions (fzo) kontrol-
liert [12, 23, 27]. Im Menschen und in Mäusen existieren zwei homologe Protei-
ne des Fzo-Proteins, Mitofusin 1 (MFN1) und MFN2. Sie sind beide essentiell
für die embryonale Entwicklung und für die mitochondriale Fusion [12]. MFN2
ist eines der Gene, welches direkt an der Aufrechterhaltung des mitochondria-
len Netzwerks beteiligt ist. Mutationen im Ganglioside-Induced-Differentiation-
Associated-Protein-1-Gen (GDAP1 Gen) führen bei Überexpression des Prote-
ins ebenfalls zu eine Störung von Fusionsprozessen. Eine Fragmentierung von
Mitochondrien ist die Folge [41, 42]. Autosomal-rezessiv vererbte Mutationen im
GDAP1-Gen wurden in Patienten mit CMT4A gefunden.
1.2.2 Aufbau, Lokalisation und Funktion des MFN2-Proteins
Das MFN2-Protein ist ein Transmembranprotein, welches in der Außenmemb-
ran der Mitochondrien verankert ist. Der Aufbau des Proteins gliedert sich in
eine GTPase-Domäne, welche die Energie für mitochondriale Fusionsprozesse
sicherstellt, ein Transmembranfragment (TM), welches das Protein in der Mito-
chondrienwand verankert und zwei angrenzende coiled-coil (Cc) Strukturen
(HR1 (heptad repeat regions 1) und HR2 (heptad repeat regions 1)), die wichtig
für die Verknüpfung unterschiedlicher Mitochondrien sind.
Abb. 4
Schematische Darstellung des MFN2-Proteins in der mitochondrialen Außenmembran (AM). Die GTPase stellt Energie für Fusionsprozesse bereit. HR1 und HR2 Regionen erfüllen eben-falls wichtige Aufgaben bei der Verknüpfung von Mitochondrien.

Einleitung 9
Die Cc-Strukturen bilden antiparallele Dimere, welche Mitochondrien miteinan-
der verbinden, die bereit sind zu fusionieren [29, 34]. Eine intakte Funktion der
GTPase ist unverzichtbar für die Funktion des Mitofusins [20, 23, 34].

Zielsetzung der Arbeit 10
2 Zielsetzung der Arbeit
Ziel der vorliegenden Arbeit war es, Patienten mit der klinischen Diagnose
CMT2 mittels molekulargenetischer Methoden auf Mutationen im MFN2-Gen zu
untersuchen. Es sollte ermittelt werden, ob im untersuchten Patientenkollektiv
eine Assoziation zwischen Mutationen in diesem Gen und der CMT2-
Erkrankung existiert. Weiterhin war zu fragen, ob es sich bei einer gegebenen
Assoziation sogar um eine der Hauptursachen für die CMT2-Erkrankung han-
delt. Diese Studie sollte somit einen Beitrag zur Klärung der Krankheitsursache
liefern.
Die molekulare Diagnostik stellt ein bedeutendes Instrumentarium zur Früher-
kennung von erblichen Erkrankungen dar. Zu Beginn dieser Studie lagen nur
begrenzte Kenntnisse über häufig betroffene Gene der CMT2-Erkrankung vor.
Daher sollte diese Studie einen wichtigen Beitrag zur Klärung dieser Fragestel-
lung liefern, und kann mit den Ergebnissen entscheidende Empfehlungen für
die molekulare CMT2-Diagnostik geben.

Methode und Ergebnisse 11
3 Methode und Ergebnisse
3.1 Mutationsanalyse des MFN2-Gens
Für die molekulargenetischen Analysen standen 73 Proben von CMT2-
Patienten zur Verfügung.
Für die Mutationsanalyse wurde Polymerasekettenreaktion (PCR), single strand
conformation polymorphism (SSCP) und Sequenzierung genutzt. Die gesunden
Kontrollproben wurden mit Hilfe geeigneter Restriktionsenzyme geschnitten, um
zu beweisen, dass sie die Mutation der entsprechenden Patientenprobe nicht
tragen.
Zur SSCP-Analyse wurden die denaturierten Patientenproben auf zwei jeweils
in der Zusammensetzung unterschiedliche Polyacrylamidgele (PAA) aufgetra-
gen und mittels Elektrophorese aufgetrennt. Auffällige Proben, welche sich im
Laufverhalten von den anderen unterschieden, wurden erneut amplifiziert und
sequenziert. Die genaue Aufschlüsselung der Basensequenz durch die Se-
quenzierung gab Hinweise auf das Vorliegen einer Mutation in der entspre-
chenden Probe.
Um auszuschließen, dass der jeweilige Basenaustausch einen seltenen Poly-
morphismus darstellt, wurden pro Mutation mindestens 200 gesunde Kontroll-
chromosomen durch Restriktionsenzyme geschnitten, auf ein Agarosegel auf-
getragen und elektrophoretisch getrennt.
3.2 Ergebnisse
Die Mutationsanalyse des MFN2-Gens zeigte insgesamt sechs Mutationen,
welche in der Literatur zuvor noch nicht beschrieben wurden.
Diese Mutationen ließen sich in sechs unterschiedlichen Patienten nachweisen.
Jeder der sechs Patienten zeigte jeweils eine Mutation in einem Exon. Insge-
samt konnten in fünf unterschiedlichen der 19 Exons Mutationen nachgewiesen
werden.

Methode und Ergebnisse 12
Abb. 5
Struktur des MFN2-Proteins. Es sind funktionelle Domänen und publizierte Mutationen abgebildet. Die durch vorliegende Studie entdeckten Mutationen sind unterstrichen. Mit HMSN V und HMSN VI assoziierte Mutationen sind mit * und + gekennzeichnet [19].
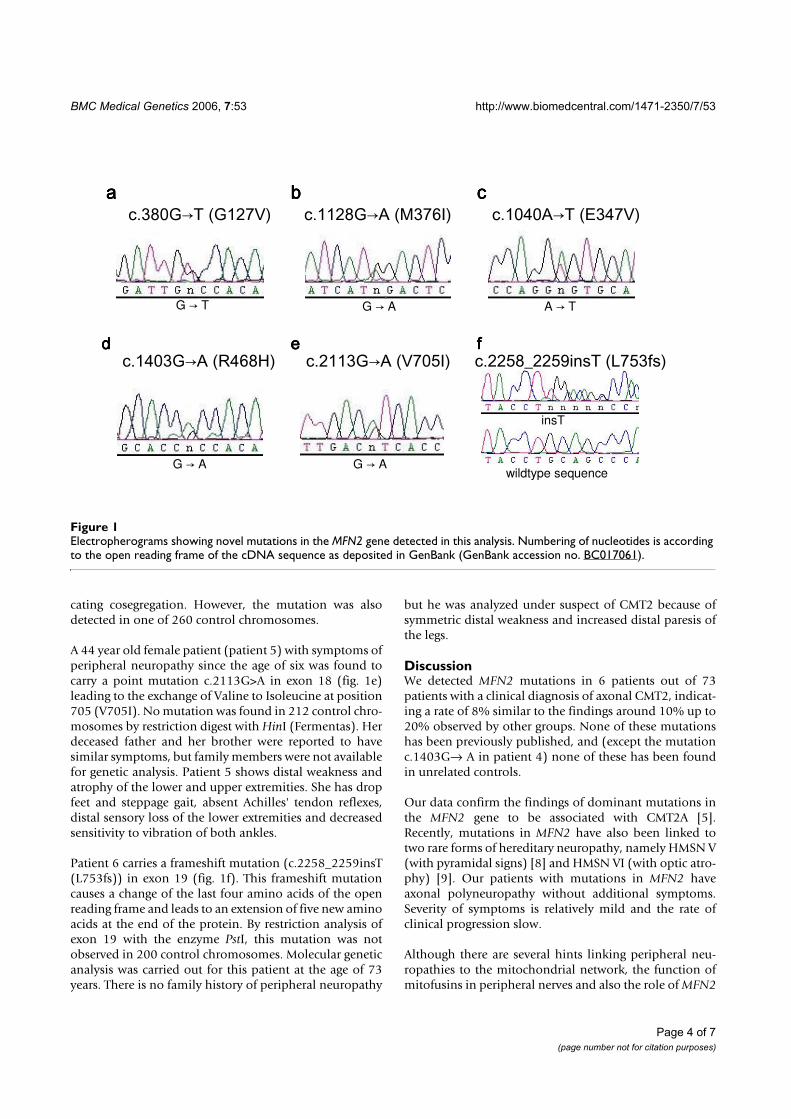
Eine Mutation in Exon 5 (c.380G>T (G127V)) wurde in der Patientenprobe einer
58 Jahre alten Frau gefunden. Ich entdeckte zwei weitere Mutationen in Exon
11 (c.1128G>A (M376I) und c.1040A>T (E347V)) bei einem 34- bzw. bei einem
32-jährigen Mann. Eine Mutation wurde bei einer 27 Jahre alten Frau in Exon
14 lokalisiert (c.1403G>A (R468H)). Auch in Exon 18 bzw. in Exon 19 konnte
bei einer 44 Jahre alten Patientin bzw. bei einem 64 Jahre altem Patienten je-
weils eine Mutation aufgespürt werden (c.2113G>A (V705I) und
c.2258_2259insT (L753fs)). Der Phänotyp aller Patienten ist in Tabelle 3 und 4
den entsprechenden Mutationen zugeordnet.
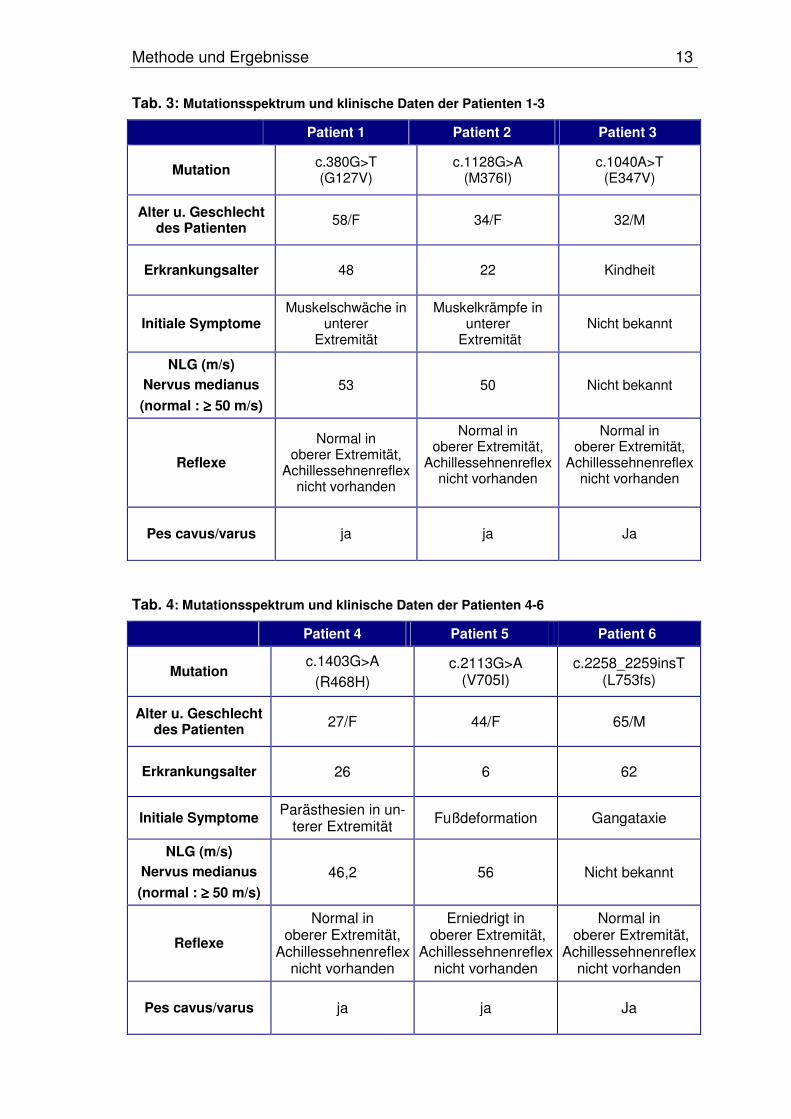
Methode und Ergebnisse 13
Tab. 3: Mutationsspektrum und klinische Daten der Patienten 1-3
Patient 1 Patient 2 Patient 3
Mutation c.380G>T (G127V)
c.1128G>A (M376I)
c.1040A>T (E347V)
Alter u. Geschlecht des Patienten 58/F 34/F 32/M
Erkrankungsalter 48 22 Kindheit
Initiale Symptome Muskelschwäche in
unterer Extremität
Muskelkrämpfe in unterer
Extremität Nicht bekannt
NLG (m/s)
Nervus medianus
(normal : ≥≥≥≥ 50 m/s)
53 50 Nicht bekannt
Reflexe
Normal in oberer Extremität,
Achillessehnenreflex nicht vorhanden
Normal in oberer Extremität,
Achillessehnenreflexnicht vorhanden
Normal in oberer Extremität,
Achillessehnenreflexnicht vorhanden
Pes cavus/varus ja ja Ja
Tab. 4: Mutationsspektrum und klinische Daten der Patienten 4-6
Patient 4 Patient 5 Patient 6
Mutation c.1403G>A
(R468H)c.2113G>A
(V705I)c.2258_2259insT
(L753fs)
Alter u. Geschlecht des Patienten 27/F 44/F 65/M
Erkrankungsalter 26 6 62
Initiale Symptome Parästhesien in un-terer Extremität Fußdeformation Gangataxie
NLG (m/s)
Nervus medianus
(normal : ≥≥≥≥ 50 m/s)
46,2 56 Nicht bekannt
Reflexe
Normal in oberer Extremität,
Achillessehnenreflex nicht vorhanden
Erniedrigt in oberer Extremität,
Achillessehnenreflex nicht vorhanden
Normal in oberer Extremität,
Achillessehnenreflex nicht vorhanden
Pes cavus/varus ja ja Ja

Diskussion 14
4 Diskussion
In der vorliegenden Studie konnten bei einem CMT2-Kollektiv von 73 Patienten
in sechs Fällen Mutationen nachgewiesen werden. Keine der Mutationen wurde
zuvor in der Literatur beschrieben und bis auf eine Mutation ließ sich keine der
weiteren Mutationen in anonymen Kontrollen nachweisen [32, 36, 63-65].
Somit beträgt die Mutationsrate dieses Kollektivs 8 %. Die Rate ist mit den De-
tektionsraten von 10-20 % anderer Arbeitsgruppen vergleichbar. Meine Daten
unterstützen die These, dass Mutationen im MFN2-Gen eine große Rolle bei
der Pathogenese von Charcot-Marie-Tooth Neuropathien vom Typ 2 einneh-
men.
Obwohl Patienten mit HMSN V und HMSN VI ebenfalls Mutationen im MFN2-
Gen aufweisen, ließen sich in Patienten dieser Studie keine zusätzlichen Sym-
ptome wie Pyramidenbahnzeichen und Optikusatrophie nachweisen [64]. Die
Patientensymptome in der vorliegenden Studie können als mäßig ausgeprägt
beschrieben werden und die Progression verläuft relativ langsam. Bei zwei Drit-
tel der Patienten manifestierte sich die Erkrankung erst relativ spät (>10 Jahre
alt, late onset). Im Vordergrund stehen Fußdeformationen, Muskelschwäche
und Sensitivitätsverlust in der unteren Extremität. Nur ein Patient zeigte eine
Abschwächung der Reflexe der oberen Extremität (c.2113G>A (V705I)). Keiner
der Patienten ist bis jetzt rollstuhlabhängig. Die mNLG weist in allen Fällen auf
eine axonale Polyneuropathie hin.
In der Literatur wird zunehmend der Zusammenhang von MFN2-Mutationen
und peripheren Polyneuropathien hergestellt [65]. Die Physiologie der mito-
chondrialen Netzwerke im peripherem Nerv, die Rolle des Proteins MFN2 und
letztendlich die Auswirkungen von Mutationen im MFN2-Gen sind allerdings nur
teilweise bekannt.
Mitochondrien sind aktive Organellen, welche ständigen Teilungs- und Fusions-
prozessen unterworfen sind [43]. Sie bilden in Zellen netzwerkartige Strukturen
aus, um die Energieversorgung auch über große Distanzen aufrecht zu erhal-
ten. In der Fruchtfliege Drosophila melanogaster, wie auch in Hefen, wird die
mitochondriale Fusion von der GTPase fzo kontrolliert [12, 23, 27]. In Menschen
und Mäusen kontrollieren, homolog zu fzo, MFN1 und MFN2 die Fusionspro-
zesse der Mitochondrien. Es wurde nachgewiesen, dass ein Fehlen von MFN1

Diskussion 15
oder MFN2 zu fehlerhafter Embryonalentwicklung und vollständiger Fragmen-
tierung von mitochondrialen Netzwerken führt [12]. Eine Fusionsaktivität konnte
nicht mehr beobachtet werden. Die mitochondriale Morphologie ist ebenso ent-
scheidend für das Zellwachstum, das mitochondriale Membranpotential und die
Zellatmung.
MFN2 ist in der äußeren Membran der Mitochondrien verankert. Der N-
Terminus sowie der C-Terminus ragen in das Zytosol hinein. Der N-Terminus
besteht aus einer GTPase Domäne und wird über eine Cc-Domäne mit dem
TM-Fragment, welches das Protein in der Membran befestigt, verbunden. Dem
TM-Fragment ist auf der C-terminalen Seite eine zweite Cc-Region angeschlos-
sen. Eine funktionierende GTPase-Domäne ist unverzichtbar für die Funktion
des Proteins [20, 23, 47]. Ebenso wichtig ist die Funktion der Cc-Regionen.
Über diese werden Verbindungen mit benachbarten Mitochondrien hergestellt,
um Fusionsprozesse durchführen zu können [28].
Ein weiteres Gen ist ebenfalls bei Fusionsprozessen der Mitochondrien betei-
ligt. Optic-Atrophy-1- (OPA1)- Mutationen sind eine der häufigsten Ursachen für
autosomal-dominant vererbte Optikusatrophien [1]. Kürzlich konnte auch eine
Beteiligung von MFN2-Mutationen an der Entstehung von Optikusatrophien
nachgewiesen werden [64].
GDAP1-Mutationen führen zu einer Überexpression des Proteins und dadurch
zu Störungen mitochondrialer Fusionsprozesse. Autosomal-rezessive Mutatio-
nen lassen sich bei CMT4A-Patienten finden [41, 42].
Zum Zeitpunkt der Veröffentlichung sind 22 verschiedene Mutationen im MFN2-
Gen bekannt, welche eine CMT2 oder eine HMSN V und HMSN VI auslösen
[32, 36, 63-65]. Bis heute ist noch nicht geklärt, wie es zu der recht unterschied-
lichen Genotyp-Phänotyp-Relation kommt [65]. Weitere Untersuchungen zur
Dynamik von Mitochondrien werden nötig sein, um zu verstehen, warum Muta-
tionen in unterschiedlichen Genen zu gleichen oder unterschiedlichen Phänoty-
pen führen.
Obwohl andere Arbeitsgruppen Mutationen in besonderen hot spot Regionen
des MFN2-Gens beschreiben (aa94, aa236-251, aa273-284, aa357-364), wur-
de in diesem Kollektiv keine der bisher entdecken Mutationen wieder gefunden.
Dies könnte darauf hindeuten, dass die unterschiedlichen untersuchten Patien-

Diskussion 16
tenkollektive verschiedener Arbeitsgruppen ethnische Unterschiede aufweisen.
Möglich wäre auch, dass zu wenig Patienten für diese Studie zur Verfügung
standen und somit auch weniger Mutationen gefunden werden konnten.
Fünf von sechs Mutationen in unserem Kollektiv sind missense-Mutationen.
Eine Mutation befindet sich direkt in der für die GTPase kodierenden Region
(G127V). Zwei Mutationen sind zwischen der GTPase-Region (E347V) und der
ersten Cc-Region (M376I) lokalisiert. Eine Mutation befindet sich ebenfalls nahe
der ersten Cc-Region (R468H). Zwei weitere Mutationen betreffen den Bereich
oberhalb der TM-Region (V705I) und der zweiten Cc-Region (L753fs) (siehe
Abb.5).
Die Mutation E347V liegt in einer hoch konservierten Region des Gens, welche
möglicherweise unterstützende Funktionen beim Fusionieren von Mitochondrien
einnimmt und somit essentiell für die Funktion von MFN2 zu sein scheint [28].
In Deletionskonstrukten für MFN2 wurde gezeigt, dass GTPase-abhängige In-
teraktionen zwischen dem N-Terminus und der sich am C-Terminus des Prote-
ins befindenden Cc-Region stattfinden. Diese Interaktionen dienen ebenfalls der
Fusion von Mitochondrien. Es wird auch vermutet, dass der C-Terminus wichti-
ge Regionen enthält, welche das Targeting zwischen Protein und Mitochond-
rienmembran sicherstellen [28].
Die Frameshiftmutation L753fs befindet sich am Ende der zweiten Cc-Region
am C-terminalem Ende des Proteins. Das Protein unterscheidet sich nur in vier
Aminosäuren und einer Substitution von 5 weiteren Aminosäuren vom ur-
sprünglichem Protein. Weitere Studien von Promotor-Regionen werden nötig
sein, um zu klären, ob diese Region bei der Ausbildung von CMT2 beteiligt ist.
Zu Beginn dieser Studie waren nur wenige Daten über CMT2-Erkrankungen
und Mutationen im MFN2-Gen bekannt. Diese Studie konnte eine Assoziation
zwischen MFN2-Mutationen und CMT2-Neuropathien nachweisen. Somit liefert
sie neben anderen Veröffentlichungen einen wichtigen Beitrag zur Erforschung
von hereditären Neuropathien und deren Ursachen. Die Mutationsrate von 8 %
in diesem Patientenkollektiv weist darauf hin, dass MFN2-Mutationen zu den
häufigsten Ursachen der CMT2 zählen. Eine molekulargenetische Diagnostik
des MFN2-Gens sollte deshalb in die Laborroutine zur Testung aufgenommen
werden.

Literaturverzeichnis 17
5 Literaturverzeichnis
[1] Alexander, C., Votruba, M., Pesch, U.E., Thiselton, D.L., Mayer, S., Moo-re, A., Rodriguez, M., Kellner, U., Leo-Kottler, B., Auburger, G., Bhatta-charya, S.S. and Wissinger, B. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26, 211-215.
[2] Antonellis, A., Ellsworth, R.E., Sambuughin, N., Puls, I., Abel, A., Lee-Lin, S.Q., Jordanova, A., Kremensky, I., Christodoulou, K., Middleton, L.T., Sivakumar, K., Ionasescu, V., Funalot, B., Vance, J.M., Goldfarb, L.G., Fischbeck, K.H. and Green, E.D. (2003). Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72, 1293-1299.
[3] Bakeeva, L.E., Chentsov Yu, S. and Skulachev, V.P. (1978). Mitochon-drial framework (reticulum mitochondriale) in rat diaphragm muscle. Bio-chim Biophys Acta 501, 349-369.
[4] Barhoumi, C., Amouri, R., Ben Hamida, C., Ben Hamida, M., Machghoul, S., Gueddiche, M. and Hentati, F. (2001). Linkage of a new locus for au-tosomal recessive axonal form of Charcot-Marie-Tooth disease to chro-mosome 8q21.3. Neuromuscul Disord 11, 27-34.
[5] Ben Othmane, K., Middleton, L.T., Loprest, L.J., Wilkinson, K.M., Len-non, F., Rozear, M.P., Stajich, J.M., Gaskell, P.C., Roses, A.D., Pericak-Vance, M.A. and et al. (1993). Localization of a gene (CMT2A) for auto-somal dominant Charcot-Marie-Tooth disease type 2 to chromosome 1p and evidence of genetic heterogeneity. Genomics 17, 370-375.
[6] Berghoff, C., Berghoff, M., Leal, A., Morera, B., Barrantes, R., Reis, A., Neundorfer, B., Rautenstrauss, B., Del Valle, G. and Heuss, D. (2004). Clinical and electrophysiological characteristics of autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2B) that maps to chromo-some 19q13.3. Neuromuscul Disord 14, 301-306.
[7] Boerkoel, C.F., Takashima, H., Garcia, C.A., Olney, R.K., Johnson, J., Berry, K., Russo, P., Kennedy, S., Teebi, A.S., Scavina, M., Williams, L.L., Mancias, P., Butler, I.J., Krajewski, K., Shy, M. and Lupski, J.R. (2002). Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann Neurol 51, 190-201.
[8] Chalmers, R.M., Bird, A.C. and Harding, A.E. (1996). Autosomal domi-nant optic atrophy with asymptomatic peripheral neuropathy. J Neurol Neurosurg Psychiatry 60, 195-196.
[9] Charcot-Marie-Tooth Neuropathy Type 1. (Zugriff vom 01.05.2007). http://www.ncbi.nlm.nih.gov/books/bv.fcgi?indexed=google&rid=gene.chapter.cmt1

Literaturverzeichnis 18
[10] Charcot-Marie-Tooth Neuropathy Type 2. (Zugriff vom 01.05.2007). http://www.ncbi.nlm.nih.gov/books/bv.fcgi?indexed=google&rid=gene.chapter.cmt2
[11] Chen, H. and Chan, D.C. (2006). Critical dependence of neurons on mi-tochondrial dynamics. Curr Opin Cell Biol 18, 453-459.
[12] Chen, H., Detmer, S.A., Ewald, A.J., Griffin, E.E., Fraser, S.E. and Chan, D.C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochon-drial fusion and are essential for embryonic development. J Cell Biol 160, 189-200.
[13] Cuesta, A., Pedrola, L., Sevilla, T., Garcia-Planells, J., Chumillas, M.J., Mayordomo, F., LeGuern, E., Marin, I., Vilchez, J.J. and Palau, F. (2002). The gene encoding ganglioside-induced differentiation-associated pro-tein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet 30, 22-25.
[14] Davis, C.J., Bradley, W.G. and Madrid, R. (1978). The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve bi-opsy studies. I. Clinical, genetic and electrophysiological findings and classification. J Genet Hum 26, 311-349.
[15] De Jonghe, P., Timmerman, V., Ceuterick, C., Nelis, E., De Vriendt, E., Lofgren, A., Vercruyssen, A., Verellen, C., Van Maldergem, L., Martin, J.J. and Van Broeckhoven, C. (1999). The Thr124Met mutation in the pe-ripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain 122 (Pt 2), 281-290.
[16] De Sandre-Giovannoli, A., Chaouch, M., Kozlov, S., Vallat, J.M., Tazir, M., Kassouri, N., Szepetowski, P., Hammadouche, T., Vandenberghe, A., Stewart, C.L., Grid, D. and Levy, N. (2002). Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 70, 726-736.
[17] Dimauro, S. and Davidzon, G. (2005). Mitochondrial DNA and disease.Ann Med 37, 222-232.
[18] Dyck, P.J. and Lambert, E.H. (1968). Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neu-rol 18, 603-618.
[19] Engelfried, K., Vorgerd, M., Hagedorn, M., Haas, G., Gilles, J., Epplen, J.T. and Meins, M. (2006). Charcot-Marie-Tooth neuropathy type 2A: no-vel mutations in the mitofusin 2 gene (MFN2). BMC Med Genet 7, 53.
[20] Eura, Y., Ishihara, N., Yokota, S. and Mihara, K. (2003). Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem (Tokyo) 134, 333-344.

Literaturverzeichnis 19
[21] Evgrafov, O.V., Mersiyanova, I., Irobi, J., Van Den Bosch, L., Dierick, I., Leung, C.L., Schagina, O., Verpoorten, N., Van Impe, K., Fedotov, V., Dadali, E., Auer-Grumbach, M., Windpassinger, C., Wagner, K., Mitrovic, Z., Hilton-Jones, D., Talbot, K., Martin, J.J., Vasserman, N., Tverskaya, S., Polyakov, A., Liem, R.K., Gettemans, J., Robberecht, W., De Jonghe, P. and Timmerman, V. (2004). Mutant small heat-shock protein 27 cau-ses axonal Charcot-Marie-Tooth disease and distal hereditary motor neu-ropathy. Nat Genet 36, 602-606.
[22] Ferreirinha, F., Quattrini, A., Pirozzi, M., Valsecchi, V., Dina, G., Broccoli, V., Auricchio, A., Piemonte, F., Tozzi, G., Gaeta, L., Casari, G., Ballabio, A. and Rugarli, E.I. (2004). Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest 113, 231-242.
[23] Hales, K.G. and Fuller, M.T. (1997). Developmentally regulated mito-chondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 90, 121-129.
[24] Harding, A.E. and Thomas, P.K. (1980). The clinical features of heredi-tary motor and sensory neuropathy types I and II. Brain 103, 259-280.
[25] Harding, A.E. and Thomas, P.K. (1984). Peroneal muscular atrophy with pyramidal features. J Neurol Neurosurg Psychiatry 47, 168-172.
[26] Hayasaka, K., Himoro, M., Sato, W., Takada, G., Uyemura, K., Shimizu, N., Bird, T.D., Conneally, P.M. and Chance, P.F. (1993). Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat Genet 5, 31-34.
[27] Hermann, G.J. and Shaw, J.M. (1998). Mitochondrial dynamics in yeast.Annu Rev Cell Dev Biol 14, 265-303.
[28] Honda, S., Aihara, T., Hontani, M., Okubo, K. and Hirose, S. (2005). Mu-tational analysis of action of mitochondrial fusion factor mitofusin-2. J Cell Sci 118, 3153-3161.
[29] Honda, S. and Hirose, S. (2003). Stage-specific enhanced expression of mitochondrial fusion and fission factors during spermatogenesis in rat testis. Biochem Biophys Res Commun 311, 424-432.
[30] Houlden, H. and Reilly, M.M. (2006). Molecular genetics of autosomal-dominant demyelinating Charcot-Marie-Tooth disease. Neuromolecular Med 8, 43-62.
[31] Inherited Peripheral Neuropathy Mutation Database. (Zugriff vom 01.05.2007). http://www.molgen.ua.ac.be/CMTMutations
[32] Kijima, K., Numakura, C., Izumino, H., Umetsu, K., Nezu, A., Shiiki, T., Ogawa, M., Ishizaki, Y., Kitamura, T., Shozawa, Y. and Hayasaka, K.

Literaturverzeichnis 20
(2005). Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet 116, 23-27.
[33] Klein, C.J., Cunningham, J.M., Atkinson, E.J., Schaid, D.J., Hebbring, S.J., Anderson, S.A., Klein, D.M., Dyck, P.J., Litchy, W.J., Thibodeau, S.N. and Dyck, P.J. (2003). The gene for HMSN2C maps to 12q23-24: a region of neuromuscular disorders. Neurology 60, 1151-1156.
[34] Koshiba, T., Detmer, S.A., Kaiser, J.T., Chen, H., McCaffery, J.M. and Chan, D.C. (2004). Structural basis of mitochondrial tethering by mito-fusin complexes. Science 305, 858-862.
[35] Kuhlenbäumer, G., Young, P., Hunermund, G., Ringelstein, B. and Stog-bauer, F. (2002). Clinical features and molecular genetics of hereditary peripheral neuropathies. J Neurol 249, 1629-1650.
[36] Lawson, V.H., Graham, B.V. and Flanigan, K.M. (2005). Clinical and e-lectrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology 65, 197-204.
[37] Lupski, J.R., de Oca-Luna, R.M., Slaugenhaupt, S., Pentao, L., Guzzetta, V., Trask, B.J., Saucedo-Cardenas, O., Barker, D.F., Killian, J.M., Gar-cia, C.A., Chakravarti, A. and Patel, P.I. (1991). DNA duplication associ-ated with Charcot-Marie-Tooth disease type 1A. Cell 66, 219-232.
[38] Marrosu, M.G., Vaccargiu, S., Marrosu, G., Vannelli, A., Cianchetti, C. and Muntoni, F. (1998). Charcot-Marie-Tooth disease type 2 associated with mutation of the myelin protein zero gene. Neurology 50, 1397-1401.
[39] Mersiyanova, I.V., Perepelov, A.V., Polyakov, A.V., Sitnikov, V.F., Dadali, E.L., Oparin, R.B., Petrin, A.N. and Evgrafov, O.V. (2000). A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a muta-tion in the neurofilament-light gene. Am J Hum Genet 67, 37-46.
[40] Nelis, E., Berciano, J., Verpoorten, N., Coen, K., Dierick, I., Van Gerwen, V., Combarros, O., De Jonghe, P. and Timmerman, V. (2004). Auto-somal dominant axonal Charcot-Marie-Tooth disease type 2 (CMT2G) maps to chromosome 12q12-q13.3. J Med Genet 41, 193-197.
[41] Nelis, E., Erdem, S., Van Den Bergh, P.Y., Belpaire-Dethiou, M.C., Ceu-terick, C., Van Gerwen, V., Cuesta, A., Pedrola, L., Palau, F., Gabreels-Festen, A.A., Verellen, C., Tan, E., Demirci, M., Van Broeckhoven, C., De Jonghe, P., Topaloglu, H. and Timmerman, V. (2002). Mutations in GDAP1: autosomal recessive CMT with demyelination and axonopathy.Neurology 59, 1865-1872.
[42] Niemann, A., Ruegg, M., La Padula, V., Schenone, A. and Suter, U. (2005). Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol 170, 1067-1078.

Literaturverzeichnis 21
[43] Nunnari, J., Marshall, W.F., Straight, A., Murray, A., Sedat, J.W. and Walter, P. (1997). Mitochondrial transmission during mating in Sac-charomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol Biol Cell 8, 1233-1242.
[44] Palay, S.L. (1956). Synapses in the central nervous system. J Biophys Biochem Cytol 2, 193-202.
[45] Raeymaekers, P., Timmerman, V., Nelis, E., De Jonghe, P., Hoogendijk, J.E., Baas, F., Barker, D.F., Martin, J.J., De Visser, M., Bolhuis, P.A. and et al. (1991). Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group. Neuromuscul Disord 1, 93-97.
[46] Reichert, A.S. and Neupert, W. (2004). Mitochondriomics or what makes us breathe. Trends Genet 20, 555-562.
[47] Santel, A. and Fuller, M.T. (2001). Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114, 867-874.
[48] Shaw, J.M. and Nunnari, J. (2002). Mitochondrial dynamics and division in budding yeast. Trends Cell Biol 12, 178-184.
[49] Skre, H. (1974). Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet 6, 98-118.
[50] Skulachev, V.P. (2001). Mitochondrial filaments and clusters as intracel-lular power-transmitting cables. Trends Biochem Sci 26, 23-29.
[51] Street, V.A., Bennett, C.L., Goldy, J.D., Shirk, A.J., Kleopa, K.A., Tempel, B.L., Lipe, H.P., Scherer, S.S., Bird, T.D. and Chance, P.F. (2003). Muta-tion of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C. Neurology 60, 22-26.
[52] Suter, U. and Scherer, S.S. (2003). Disease mechanisms in inherited neuropathies. Nat Rev Neurosci 4, 714-726.
[53] Tang, B.S., Luo, W., Xia, K., Xiao, J.F., Jiang, H., Shen, L., Tang, J.G., Zhao, G.H., Cai, F., Pan, Q., Dai, H.P., Yang, Q.D., Xia, J.H. and Evgra-fov, O.V. (2004). A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2L) maps to chromosome 12q24. Hum Genet 114, 527-533.
[54] Taylor, R.W. and Turnbull, D.M. (2005). Mitochondrial DNA mutations in human disease. Nat Rev Genet 6, 389-402.
[55] Thomas, P.K., King, R.H., Small, J.R. and Robertson, A.M. (1996). The pathology of charcot-marie-tooth disease and related disorders. Neuro-pathol Appl Neurobiol 22, 269-284.

Literaturverzeichnis 22
[56] Timmerman, V., De Jonghe, P., Spoelders, P., Simokovic, S., Lofgren, A., Nelis, E., Vance, J., Martin, J.J. and Van Broeckhoven, C. (1996). Linkage and mutation analysis of Charcot-Marie-Tooth neuropathy type 2 families with chromosomes 1p35-p36 and Xq13. Neurology 46, 1311-1318.
[57] Valentijn, L.J., Baas, F., Wolterman, R.A., Hoogendijk, J.E., van den Bosch, N.H., Zorn, I., Gabreels-Festen, A.W., de Visser, M. and Bolhuis, P.A. (1992). Identical point mutations of PMP-22 in Trembler-J mouse and Charcot-Marie-Tooth disease type 1A. Nat Genet 2, 288-291.
[58] Valentijn, L.J., Bolhuis, P.A., Zorn, I., Hoogendijk, J.E., van den Bosch, N., Hensels, G.W., Stanton, V.P., Jr., Housman, D.E., Fischbeck, K.H., Ross, D.A. and et al. (1992). The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A. Nat Genet 1, 166-170.
[59] Vance, J.M. (2000). The many faces of Charcot-Marie-Tooth disease.Arch Neurol 57, 638-640.
[60] Verhoeven, K., De Jonghe, P., Coen, K., Verpoorten, N., Auer-Grumbach, M., Kwon, J.M., FitzPatrick, D., Schmedding, E., De Vriendt, E., Jacobs, A., Van Gerwen, V., Wagner, K., Hartung, H.P. and Timmer-man, V. (2003). Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet 72, 722-727.
[61] Warner, L.E., Mancias, P., Butler, I.J., McDonald, C.M., Keppen, L., Koob, K.G. and Lupski, J.R. (1998). Mutations in the early growth re-sponse 2 (EGR2) gene are associated with hereditary myelinopathies.Nat Genet 18, 382-384.
[62] Zhao, C., Takita, J., Tanaka, Y., Setou, M., Nakagawa, T., Takeda, S., Yang, H.W., Terada, S., Nakata, T., Takei, Y., Saito, M., Tsuji, S., Haya-shi, Y. and Hirokawa, N. (2001). Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell 105, 587-597.
[63] Zhu, D., Kennerson, M.L., Walizada, G., Züchner, S., Vance, J.M. and Nicholson, G.A. (2005). Charcot-Marie-Tooth with pyramidal signs is ge-netically heterogeneous: families with and without MFN2 mutations. Neu-rology 65, 496-497.
[64] Züchner, S., De Jonghe, P., Jordanova, A., Claeys, K.G., Guergueltche-va, V., Cherninkova, S., Hamilton, S.R., Van Stavern, G., Krajewski, K.M., Stajich, J., Tournev, I., Verhoeven, K., Langerhorst, C.T., de Vis-ser, M., Baas, F., Bird, T., Timmerman, V., Shy, M. and Vance, J.M. (2006). Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol 59, 276-281.

Literaturverzeichnis 23
[65] Züchner, S., Mersiyanova, I.V., Muglia, M., Bissar-Tadmouri, N., Ro-chelle, J., Dadali, E.L., Zappia, M., Nelis, E., Patitucci, A., Senderek, J., Parman, Y., Evgrafov, O., Jonghe, P.D., Takahashi, Y., Tsuji, S., Peri-cak-Vance, M.A., Quattrone, A., Battaloglu, E., Polyakov, A.V., Timmerman, V., Schröder, J.M. and Vance, J.M. (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuro-pathy type 2A. Nat Genet 36, 449-451.

Danksagung
6 Danksagung
Mein Dank gilt vielen Leuten, die mich auf verschiedenste Art und Weise unter-
stütz haben:
Herrn PD Dr. med. Moritz Meins und Herrn Prof. Dr. med. Jörg T. Epplen für
eine hervorragende Betreuung,
den Mitarbeitern der Abteilung für Humangenetik Bochum für meine erlernten
technischen Fähigkeiten und dafür, dass sie immer ein offenes Ohr für mich
hatten,
Herrn Prof. Dr. med. Matthias Vorgerd,
Stefan Woeste,
Alexandra Engelfried.
Vielen Dank!

Lebenslauf
Lebenslauf
PERSÖNLICHE DATEN:
Vorname: Kathrin
Nachname: Engelfried
Geburtstag: 27. Juli 1978
Geburtsort: Esslingen (am Neckar)
AUSBILDUNG/ STUDIUM/ FORTBILDUNG:
1989 – 1998 Friedrich-Harkort-Gymnasium Herdecke, Abitur
1999 Ausbildung zur Rettungssanitäterin, DRK Lan-
desschule Münster
1999 – 2001 Ausbildung zur Rettungsassistentin, Westfalen-
Schulen Dortmund; Feuerwehr Hattingen
2001 – 2003 Ruhr-Universität Bochum, Vorklinik
September 2003 Physikum
seit 2003 Ruhr-Universität Bochum, klinischer Abschnitt
Juni 2004 Beginn einer experimentellen, prospektiven
Doktorarbeit in der Abteilung für Humangene-
tik, Ruhr-Universität Bochum
Oktober 2004 – Februar 2005 Wahlseminare Schmerztherapie, Wahlseminar
Akupunktur, Wahlseminar EKG-Analyse,
Wahlseminar Anästhesie, Wahlseminar Na-
turheilkunde an der Ruhr-Universität Bochum
März 2006 Wahlseminar Echokardiographie und Herzge-
räusche, Gernsbach
August 2006 Wahlseminar Asthma und Allergie, Davos
(Schweiz)

Lebenslauf
August 2006 – August 2007 Praktisches Jahr im Allgemeinen Krankenhaus
in Hagen, Wahlfach: Anästhesie
voraussichtlich Mai 2008 Zweiter Abschnitt der Ärztlichen Prüfung
FAMULATUREN:
Februar 2004 – März 2004 Marienhospital Witten, Famulatur in der Kinder-
klinik
März 2004 – April 2004 Berufsgenossenschaftliche Kliniken Berg-
mannsheil, Famulatur in der Abteilung für Anäs-
thesie, Intensivmedizin und Schmerztherapie
August 2004 – September 2004 Ruhr-Universität Bochum, Famulatur in der Ab-
teilung für Humangenetik
September 2004 Dr. med Holz & Dr. med Bocksch, Famulatur in
einer Praxis für Innere Medizin (Schwerpunkt
Gastroenterologie)
Juli 2005 – September 2005 University of Southern California, Los Angeles,
USA, Famulatur in der Abteilung für Pulmono-
logie und Intensivmedizin des Los Angeles
County Hospital
Oktober 2005 Uniklinik St. Josef Hospital Bochum, Famulatur
in der Abteilung für Neurologie
April 2006 Marienhospital Witten, Famulatur in der Abtei-
lung für Anästhesie und Intensivmedizin
Juni 2006 University Hospital Galway, Irland, Famulatur in
der interdisziplinären Notaufnahme (Accident &
Emergency)

BioMed Central
Page 1 of 7
(page number not for citation purposes)
BMC Medical Genetics
Open AccessResearch article
Charcot-Marie-Tooth neuropathy type 2A: novel mutations in the mitofusin 2 gene (MFN2)Kathrin Engelfried1, Matthias Vorgerd2, Michaela Hagedorn1, Gerhard Haas3,Jürgen Gilles4, Jörg T Epplen1 and Moritz Meins*1
Address: 1Department of Human Genetics, Ruhr-University Bochum, Germany, 2Department of Neurology, Neuromuscular Center Ruhrgebiet, Ruhr-University Bochum, Germany, 3Neurology, Evangelische Stiftung Tannenhof, Remscheid, Germany and 4Neurology, St.-Marien-Hospital, Lünen, Germany
Email: Kathrin Engelfried - [email protected]; Matthias Vorgerd - [email protected];Michaela Hagedorn - [email protected]; Gerhard Haas - [email protected]; Jürgen Gilles - [email protected]; Jörg T Epplen - [email protected]; Moritz Meins* - [email protected]
* Corresponding author
Abstract
Background: Charcot-Marie-Tooth neuropathies are a group of genetically heterogeneous
diseases of the peripheral nervous system. Mutations in the MFN2 gene have been reported as the
primary cause of Charcot-Marie-Tooth disease type 2A.
Methods: Patients with the clinical diagnosis of Charcot-Marie-Tooth type 2 were screened using
single strand conformation polymorphism (SSCP). All DNA samples showing band shifts in the
SSCP analysis were amplified from genomic DNA and cycle sequenced.
Results: We analyzed a total of 73 unrelated patients with a clinical diagnosis of CMT 2. Overall,
novel mutations were detected in 6 patients. c.380G>T (G127V), c.1128G>A (M376I), c.1040A>T
(E347V), c.1403G>A (R468H), c.2113G>A (V705I), and c.2258_2259insT (L753fs).
Conclusion: We confirmed a significant role of mutations in MFN2 in the pathogenesis of
Charcot-Marie-Tooth disease type 2.
BackgroundCharcot-Marie-Tooth neuropathies (CMT), also named ashereditary motor and sensory neuropathies (HMSN) are agroup of genetically heterogeneous diseases of the periph-eral nervous system. CMT has been classified into twomajor subgroups. A severe reduction of nerve conductionvelocities (NCV) with NCV < 38 m/s is found in the demy-elinating CMT type 1. The axonal type (CMT type 2) ischaracterized by preferential degeneration of the axonshowing amplitude reductions in nerve conduction stud-ies but only mildly reduced NCV. Both CMT1 and CMT2
have been recognized to be genetically heterogeneous.Several genetic loci have been defined for both clinicaltypes. CMT2A was located to chromosome 1p35–p36 [1-3]. Although a mutation in KIF1B was published in a largeCMT2A family, no further mutations were detected inother families with CMT2A [4,5]. Züchner et al. identifiedthe MFN2 gene by means of linkage studies and reportedmutations in MFN2 in seven large families with linkage tothe CMT2A locus [5]. Recently, other groups confirmedthe MFN2 gene as the primary cause of CMT2A [6-8]. Wescreened 73 unrelated patients with the clinical diagnosis
Published: 08 June 2006
BMC Medical Genetics 2006, 7:53 doi:10.1186/1471-2350-7-53
Received: 03 April 2006Accepted: 08 June 2006
This article is available from: http://www.biomedcentral.com/1471-2350/7/53
© 2006 Engelfried et al; licensee BioMed Central Ltd.This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

BMC Medical Genetics 2006, 7:53 http://www.biomedcentral.com/1471-2350/7/53
Page 2 of 7
(page number not for citation purposes)
of CMT2 and identified six new disease-causing muta-tions.
MethodsSubjects
We analyzed samples of 73 unrelated patients sent to ourdiagnostic laboratory with the request of genetic analysisfor Charcot-Marie-Tooth disease. Linkage analysis was notpossible in these individuals. Inclusion criteria for theMFN2 analysis were a clinical classification of CMT2,nerve conduction velocity (NCV) > 38 m/s, or histologicalfindings of axonal degeneration. EDTA blood sampleswere taken from patients after informed consent. DNAwas extracted from blood leukocytes by standard meth-ods. All control samples used to check the distribution ofsequence alterations mentioned in the results sectionswere taken from an anonymous collective of ethnicallymatched samples.
Mutation analysis of MFN2 gene
For mutation analysis in MFN2, primers were designed toamplify all exons including flanking intronic regions.Primer sequences are available on request. Exon 7 and 8,and exon 10 and 11, respectively, were each amplified inone amplicon including the intermediate intron. PCRswere performed in 96-well microtiter plates (ThermowellCostar Corning, NY) using a thermocycler (Biometra,Goettingen, Germany). Each well contained 50 ng DNA in10 µl reaction volume, GC buffer (Genecraft, Münster,Germany), 10 pMol of forward and reverse primer, 1 UTaq Polymerase (Genecraft, Münster, Germany), 2 mMolof each dNTP, and a MgCl2 concentration of 1 mM. ForSSCP analysis, 0.06 µl of [α32 P] dCTP (10 mCi/ml) wasincluded in the PCR. PCR conditions included initialdenaturation (2 min at 94°C), two initial cycles at 94°C(15 s) and 6°C and 3°C above the main annealing tem-perature (30 s) followed by 30 s at 72°C, and 28–32 cyclesof 94°C (15 s), annealing temperature (30 s) and finalelongation step at 72°C (30 s). Annealing temperaturewas 62°C except for exons 7/8, 10/11, 15, 16, 17, and 8(58°C). PCR products were digested with suitable restric-tion enzymes depending on the fragment size to optimizethe mutation detection rate by SSCP analysis. In the SSCPanalysis the denatured PCR products were separated bypolyacrylamide (PAA) gel electrophoresis using two dif-ferent conditions: 30% PAA (acrylamide/bisacrylamide:19/1) gel, 1xTBE, and either 10% glycerol or 5% glycerol/1 M urea. Electrophoresis was carried out at 55 W for 3–4h at 4°C. Gels were evaluated by autoradiography orexposure to a phosphoimager screen, using the corre-sponding software. Exon 13 was screened using SSCP andDHPLC. DNA samples showing band shifts in the SSCPanalysis were amplified from genomic DNA and cyclesequenced by standard protocols using the Megabace1000 (Amersham Bioscience, Freiburg, Germany). If pos-
sible, restriction analysis with a suitable restrictionenzyme was applied to confirm the mutation and to ana-lyse control samples. Enzymes were ordered from NewEngland Biolabs if not indicated otherwise.
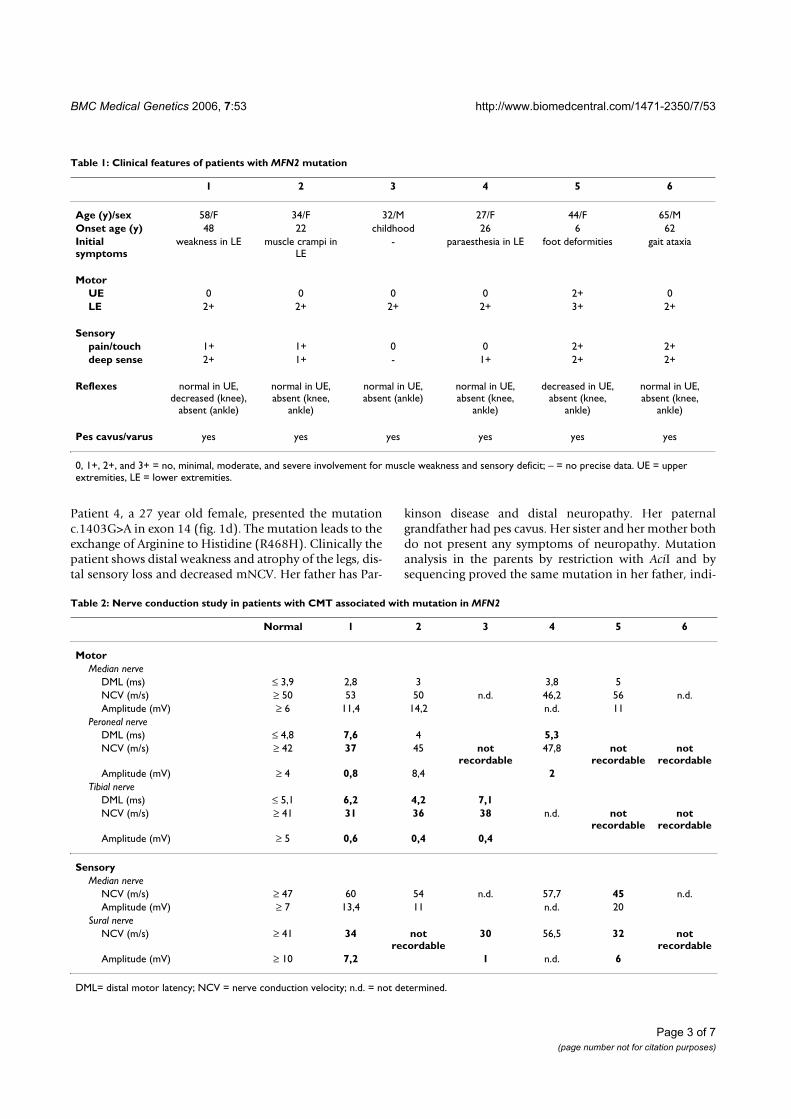
ResultsWe analyzed a total of 73 unrelated patients with a clinicaldiagnosis of CMT 2. Overall, novel mutations weredetected in 6 patients. The clinical features and the neuro-graphical data of these patients are summarized (tables 1and 2). Nerve biopsy has not been performed in any ofthese 6 patients.
The heterozygous point mutation c.380 G>T in exon 5 ofMFN2 (fig. 1a) was detected in a 58 year old femalepatient (patient 1), predicting the exchange of Glycine toValine at position 127 of the MFN2 protein (G127V). Themutation leads to a loss of a recognition site for the restric-tion enzyme Msc I, which was used to exclude this muta-tion in 97 control persons (194 control chromosomes).Clinically the patient shows a predominant axonal neu-ropathy with normal motor nerve conduction velocity(mNCV) in electrophysiological studies of the mediannerve. The family history is compatible with autosomal-dominant inheritance, but other family members werenot available for genetic analysis.
Patient 2, a 34 year old male, is heterozygous for the pointmutation c.1128G>A in exon 11, predicting the exchangeof Methionine to Isoleucine at position 376 (M376I) (fig.1b). This mutation is detectable by a loss of a NlaIIIrestriction site, and was excluded by restriction analysis in190 control chromosomes. The patient has distal sensoryloss in his legs, absent Achilles' tendon reflexes, pes cavusand severely reduced amplitudes of the compound motornerve action potentials of the peroneal and tibial nerves,but preserved motor nerve conduction velocities. Hismother, one brother and one sister reportedly have simi-lar symptoms of peripheral neuropathy but denied furtherneurophysiological testing or genetic analysis.
The point mutation c.1040A>T in exon 11, leading to theexchange of Glutamine to Valine at position 347 (E347V)of the protein, was detected in a 32 year old male (fig. 1c,patient 3). The mutation was not observed by SSCP anal-ysis in over 200 control chromosomes, detection byrestriction analysis was not possible. The parents of thepatient were reported to be healthy and unrelated, there isno history of peripheral neuropathies in the family. Thepatient has distal atrophy of his legs since childhood. Healso shows high-grade paresis of peroneus muscles inboth legs, severe pain in both legs and distal sensory loss.Electrophysiological studies indicate an axonal neuropa-thy with mildly decreased mNCV.
BMC Medical Genetics 2006, 7:53 http://www.biomedcentral.com/1471-2350/7/53
Page 3 of 7
(page number not for citation purposes)
Patient 4, a 27 year old female, presented the mutationc.1403G>A in exon 14 (fig. 1d). The mutation leads to theexchange of Arginine to Histidine (R468H). Clinically thepatient shows distal weakness and atrophy of the legs, dis-tal sensory loss and decreased mNCV. Her father has Par-
kinson disease and distal neuropathy. Her paternalgrandfather had pes cavus. Her sister and her mother bothdo not present any symptoms of neuropathy. Mutationanalysis in the parents by restriction with AciI and bysequencing proved the same mutation in her father, indi-
Table 2: Nerve conduction study in patients with CMT associated with mutation in MFN2
Normal 1 2 3 4 5 6
Motor
Median nerve
DML (ms) ≤ 3,9 2,8 3 3,8 5
NCV (m/s) ≥ 50 53 50 n.d. 46,2 56 n.d.
Amplitude (mV) ≥ 6 11,4 14,2 n.d. 11
Peroneal nerve
DML (ms) ≤ 4,8 7,6 4 5,3
NCV (m/s) ≥ 42 37 45 not recordable
47,8 not recordable
not recordable
Amplitude (mV) ≥ 4 0,8 8,4 2
Tibial nerve
DML (ms) ≤ 5,1 6,2 4,2 7,1
NCV (m/s) ≥ 41 31 36 38 n.d. not recordable
not recordable
Amplitude (mV) ≥ 5 0,6 0,4 0,4
Sensory
Median nerve
NCV (m/s) ≥ 47 60 54 n.d. 57,7 45 n.d.
Amplitude (mV) ≥ 7 13,4 11 n.d. 20
Sural nerve
NCV (m/s) ≥ 41 34 not recordable
30 56,5 32 not recordable
Amplitude (mV) ≥ 10 7,2 1 n.d. 6
DML= distal motor latency; NCV = nerve conduction velocity; n.d. = not determined.
Table 1: Clinical features of patients with MFN2 mutation
1 2 3 4 5 6
Age (y)/sex 58/F 34/F 32/M 27/F 44/F 65/M
Onset age (y) 48 22 childhood 26 6 62
Initial symptoms
weakness in LE muscle crampi in LE
- paraesthesia in LE foot deformities gait ataxia
Motor
UE 0 0 0 0 2+ 0
LE 2+ 2+ 2+ 2+ 3+ 2+
Sensory
pain/touch 1+ 1+ 0 0 2+ 2+
deep sense 2+ 1+ - 1+ 2+ 2+
Reflexes normal in UE, decreased (knee),
absent (ankle)
normal in UE, absent (knee,
ankle)
normal in UE, absent (ankle)
normal in UE, absent (knee,
ankle)
decreased in UE, absent (knee,
ankle)
normal in UE, absent (knee,
ankle)
Pes cavus/varus yes yes yes yes yes yes
0, 1+, 2+, and 3+ = no, minimal, moderate, and severe involvement for muscle weakness and sensory deficit; – = no precise data. UE = upper extremities, LE = lower extremities.
BMC Medical Genetics 2006, 7:53 http://www.biomedcentral.com/1471-2350/7/53
Page 4 of 7
(page number not for citation purposes)
cating cosegregation. However, the mutation was alsodetected in one of 260 control chromosomes.
A 44 year old female patient (patient 5) with symptoms ofperipheral neuropathy since the age of six was found tocarry a point mutation c.2113G>A in exon 18 (fig. 1e)leading to the exchange of Valine to Isoleucine at position705 (V705I). No mutation was found in 212 control chro-mosomes by restriction digest with HinI (Fermentas). Herdeceased father and her brother were reported to havesimilar symptoms, but family members were not availablefor genetic analysis. Patient 5 shows distal weakness andatrophy of the lower and upper extremities. She has dropfeet and steppage gait, absent Achilles' tendon reflexes,distal sensory loss of the lower extremities and decreasedsensitivity to vibration of both ankles.
Patient 6 carries a frameshift mutation (c.2258_2259insT(L753fs)) in exon 19 (fig. 1f). This frameshift mutationcauses a change of the last four amino acids of the openreading frame and leads to an extension of five new aminoacids at the end of the protein. By restriction analysis ofexon 19 with the enzyme PstI, this mutation was notobserved in 200 control chromosomes. Molecular geneticanalysis was carried out for this patient at the age of 73years. There is no family history of peripheral neuropathy
but he was analyzed under suspect of CMT2 because ofsymmetric distal weakness and increased distal paresis ofthe legs.
DiscussionWe detected MFN2 mutations in 6 patients out of 73patients with a clinical diagnosis of axonal CMT2, indicat-ing a rate of 8% similar to the findings around 10% up to20% observed by other groups. None of these mutationshas been previously published, and (except the mutationc.1403G→ A in patient 4) none of these has been foundin unrelated controls.
Our data confirm the findings of dominant mutations inthe MFN2 gene to be associated with CMT2A [5].Recently, mutations in MFN2 have also been linked totwo rare forms of hereditary neuropathy, namely HMSN V(with pyramidal signs) [8] and HMSN VI (with optic atro-phy) [9]. Our patients with mutations in MFN2 haveaxonal polyneuropathy without additional symptoms.Severity of symptoms is relatively mild and the rate ofclinical progression slow.
Although there are several hints linking peripheral neu-ropathies to the mitochondrial network, the function ofmitofusins in peripheral nerves and also the role of MFN2
Electropherograms showing novel mutations in the MFN2 gene detected in this analysisFigure 1Electropherograms showing novel mutations in the MFN2 gene detected in this analysis. Numbering of nucleotides is according to the open reading frame of the cDNA sequence as deposited in GenBank (GenBank accession no. BC017061).
������������� ����������������� ���������������� �������
��������� ������� ��������� � ������������� �������
G � T
G � A
A � TG � A
G � A
����
����
����
��������
����
wildtype sequence
insT

BMC Medical Genetics 2006, 7:53 http://www.biomedcentral.com/1471-2350/7/53
Page 5 of 7
(page number not for citation purposes)
mutations in the pathogenesis of peripheral neuropathy isonly partly understood.
Mitochondria are active organelles forming a singledynamic network whose continuity is maintained by abalance of fission and fusion events [10]. In both yeastand drosophila, mitochondrial fusion is controlled by thenuclear-encoded mitochondrial transmembrane GTPasefuzzy onions (Fzo) [11-13]. In humans and mice MFN2and MFN1, two human homologues of Fzo exist and bothare essential for embryonic development and mitochon-drial fusion [13]. MFN2 is the second HMSN disease genedirectly involved in the maintenance of the mitochondrialnetwork. In contrast to mitofusins, overexpression of gan-glioside-induced differentiation associated protein 1(GDAP1) is interfering with mitochondrial fusion andinduces fragmentation of mitochondria, and recessivemutations in GDAP1 have been found in patients withCMT4A [14,15].
MFN2 is localized to the outer mitochondrial membrane.The protein contains two coiled-coil regions flanking atransmembrane segment and a GTPase domain. Func-tional coiled-coil regions are essential for tethering ofmitochondria before fusion [16-18]. An intact GTPasedomain is indispensable for the function of mitofusins[12,17,19]. Overall, more than 25 different mutations inMFN2 have been observed up to now in patients withCMT2A, but also HMSN V and HMSN VI. There is cur-
rently no explanation for the divergence of phenotypesassociated with mutations in MFN2 [5]. Many mutationsare predicted to affect the GTPase domain of the protein,but an equal proportion has been detected for the regionlinking the GTPase domain and the first coiled coil region(see fig. 2). The mutations described up to now seem tocluster in several hotspot regions (aa94, aa236–251,aa273–284, aa357–364), but we have not found anyrecurrent mutation.
Five out of six mutations described in this paper are mis-sense mutations, like the majority of mutations detectedin MFN2 up to now [5-8]. Of the five missense mutations,one (G127V) affects the GTPase domain, and two muta-tions change the region linking the GTPase domain andthe first coiled coil region (E347V and M376I). The twoother missense mutations (R468H and V705I) are pre-dicted to cause amino acid substitutions between thetransmembrane domain and the coiled coil regions (fig.2).
The missense mutation c.1403G→ A (R468H) is the firstdescribed in the region between the transmembranedomain and the C-terminal coiled coil region, and wasdetected in patient 4 and her symptomatic father, indicat-ing cosegregation of mutation and disease. The same sub-stitution was detected in one out of 130 anonymouscontrol samples given by blood donors of different agesassumed to be healthy. Since mild symptoms of CMT2
Structure of the MFN2 protein, showing functional domains and published mutationsFigure 2Structure of the MFN2 protein, showing functional domains and published mutations. Mutations described in this paper are underlined. Mutations associated with HMSN V and HMSN VI are marked with + and *, respectively.
GTPase C TM C
103 408261 433 614 646 724 752757
T105 M
G127V
H165D+
T206I*
I213TF223L
T236M
V244M
P251A R468H V705I L753fs
V69F
L76P
R94W*
R94Q
V273GR274Q
Q276R*
R280H
F284Y
E347VK357N
H361Y*
R364W*
M376I
R418X*
E424G
+ HMSN V (with pyramidal signs) (Zhu et al. 2005)
* HMSN VI (with optic atrophy) (Züchner et al.2006)

BMC Medical Genetics 2006, 7:53 http://www.biomedcentral.com/1471-2350/7/53
Page 6 of 7
(page number not for citation purposes)
may have been missed in a younger blood donor added tothe control collective, this does not exclude the substitionR468H as pathogenic, but it may also represent a raresequence variant. Molecular biological studies may benecessary to finally determine the nature of this substitu-tion.
Another novel mutation (c.1040A>T) found in patient 3in our series causes the change of the polar glutamine tothe non-polar valine at position 347. This amino acidexchange occurred in a highly conserved region of MFN2.This region is one of the indispensable segment of the pro-tein that might provides binding sites for the assembly[18].
Only one stop mutation, R418X, has been describedrecently in a patient with CMT6 [9]. Interestingly, theframeshift mutation c.2258_2259insT in our patient 6would result in a protein only differing by four aminoacids and an extension of further five new amino acids atthe C-terminus of the protein. This mutation may affectthe C-terminal coiled-coil domain at the end of the fzo-mitofusin domain. Analysis of MFN2 deletion constructsrevealed that GTPase-dependent interaction between theN-terminal and C-terminal tails of MFN2 through theircoiled-coil domains and a highly conserved domain in themost N-terminal region is essential for mitochondrialfusion [18]. On the other hand it has been suggested thatthe C-terminus of MFN2 contains determinants requiredfor targeting of the protein to the mitochondrial mem-brane [20].
Further studies of promoter domains in the MFN2 genewill help to clarify if these regions are involved in thedevelopment of CMT2.
ConclusionWe confirmed a significant role of mutations in MFN2 inthe pathogenesis of Charcot-Marie-Tooth disease type 2.
AbbreviationsCMT: Charcot-Marie-Tooth
CMT2A: Charcot-Marie-Tooth type 2A
GDAP1: ganglioside-induced differentiation associatedprotein 1
KIF1B: kinesin family member protein 1B
MFN2: mitofusin 2
mNCV: motor nerve conduction velocity
SSCP: single strand conformation polymorphism
Competing interestsThe author(s) declare that they have no competing inter-ests.
Authors' contributionsKE carried out the molecular genetic studies, KE and MMparticipated in the sequence alignment and drafted themanuscript. MH participated and assisted in moleculargenetic studies and the sequence alignment. MM con-ceived of the study, and participated in the design. JTE par-ticipated in its design and coordination and helped todraft the manuscript. MV, GH and JG made major contri-butions to the clinical characterisation of the patients. Allauthors read and approved the final manuscript.
AcknowledgementsWe thank the patients and their families for their cooperation
References1. Ben Othmane K, Middleton LT, Loprest LJ, Wilkinson KM, Lennon F,
Rozear MP, Stajich JM, Gaskell PC, Roses AD, Pericak-Vance MA, etal.: Localization of a gene (CMT2A) for autosomal dominantCharcot-Marie-Tooth disease type 2 to chromosome 1p andevidence of genetic heterogeneity. Genomics 1993,17(2):370-375.
2. Harding AE, Thomas PK: The clinical features of hereditarymotor and sensory neuropathy types I and II. Brain 1980,103(2):259-280.
3. Kuhlenbäumer G, Young P, Hunermund G, Ringelstein B, StogbauerF: Clinical features and molecular genetics of hereditaryperipheral neuropathies. J Neurol 2002, 249(12):1629-1650.
4. Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, YangHW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y,Hirokawa N: Charcot-Marie-Tooth disease type 2A caused bymutation in a microtubule motor KIF1Bbeta. Cell 2001,105(5):587-597.
5. Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J,Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgra-fov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, QuattroneA, Battaloglu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM:Mutations in the mitochondrial GTPase mitofusin 2 causeCharcot-Marie-Tooth neuropathy type 2A. Nat Genet 2004,36(5):449-451.
6. Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T,Ogawa M, Ishizaki Y, Kitamura T, Shozawa Y, Hayasaka K: Mitochon-drial GTPase mitofusin 2 mutation in Charcot-Marie-Toothneuropathy type 2A. Hum Genet 2005, 116(1-2):23-27.
7. Lawson VH, Graham BV, Flanigan KM: Clinical and electrophysio-logic features of CMT2A with mutations in the mitofusin 2gene. Neurology 2005, 65(2):197-204.
8. Zhu D, Kennerson ML, Walizada G, Züchner S, Vance JM, NicholsonGA: Charcot-Marie-Tooth with pyramidal signs is geneticallyheterogeneous: families with and without MFN2 mutations.Neurology 2005, 65(3):496-497.
9. Züchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V,Cherninkova S, Hamilton SR, Van Stavern G, Krajewski KM, Stajich J,Tournev I, Verhoeven K, Langerhorst CT, de Visser M, Baas F, BirdT, Timmerman V, Shy M, Vance JM: Axonal neuropathy with opticatrophy is caused by mutations in mitofusin 2. Ann Neurol2006, 59(2):276-281.
10. Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW, Walter P:Mitochondrial transmission during mating in Saccharomycescerevisiae is determined by mitochondrial fusion and fissionand the intramitochondrial segregation of mitochondrialDNA. Mol Biol Cell 1997, 8(7):1233-1242.
11. Hermann GJ, Shaw JM: Mitochondrial dynamics in yeast. AnnuRev Cell Dev Biol 1998, 14:265-303.
12. Hales KG, Fuller MT: Developmentally regulated mitochon-drial fusion mediated by a conserved, novel, predictedGTPase. Cell 1997, 90(1):121-129.

Publish with BioMed Central and every scientist can read your work free of charge
"BioMed Central will be the most significant development for
disseminating the results of biomedical research in our lifetime."
Sir Paul Nurse, Cancer Research UK
Your research papers will be:
available free of charge to the entire biomedical community
peer reviewed and published immediately upon acceptance
cited in PubMed and archived on PubMed Central
yours — you keep the copyright
Submit your manuscript here:
http://www.biomedcentral.com/info/publishing_adv.asp
BioMedcentral
BMC Medical Genetics 2006, 7:53 http://www.biomedcentral.com/1471-2350/7/53
Page 7 of 7
(page number not for citation purposes)
13. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC: Mito-fusins Mfn1 and Mfn2 coordinately regulate mitochondrialfusion and are essential for embryonic development. J Cell Biol2003, 160(2):189-200.
14. Nelis E, Erdem S, Van Den Bergh PY, Belpaire-Dethiou MC, CeuterickC, Van Gerwen V, Cuesta A, Pedrola L, Palau F, Gabreels-Festen AA,Verellen C, Tan E, Demirci M, Van Broeckhoven C, De Jonghe P,Topaloglu H, Timmerman V: Mutations in GDAP1: autosomalrecessive CMT with demyelination and axonopathy. Neurol-ogy 2002, 59(12):1865-1872.
15. Niemann A, Ruegg M, La Padula V, Schenone A, Suter U: Ganglio-side-induced differentiation associated protein 1 is a regula-tor of the mitochondrial network: new implications forCharcot-Marie-Tooth disease. J Cell Biol 2005,170(7):1067-1078.
16. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC:Structural basis of mitochondrial tethering by mitofusincomplexes. Science 2004, 305(5685):858-862.
17. Santel A, Fuller MT: Control of mitochondrial morphology by ahuman mitofusin. J Cell Sci 2001, 114(Pt 5):867-874.
18. Honda S, Aihara T, Hontani M, Okubo K, Hirose S: Mutationalanalysis of action of mitochondrial fusion factor mitofusin-2.J Cell Sci 2005, 118(Pt 14):3153-3161.
19. Eura Y, Ishihara N, Yokota S, Mihara K: Two mitofusin proteins,mammalian homologues of FZO, with distinct functions areboth required for mitochondrial fusion. J Biochem (Tokyo) 2003,134(3):333-344.
20. Rojo M, Legros F, Chateau D, Lombes A: Membrane topology andmitochondrial targeting of mitofusins, ubiquitous mamma-lian homologs of the transmembrane GTPase Fzo. J Cell Sci2002, 115(Pt 8):1663-1674.
Pre-publication historyThe pre-publication history for this paper can be accessedhere:
http://www.biomedcentral.com/1471-2350/7/53/prepub





























