Embed Size (px)
Citation preview

302. Bd. 196919
S ynthe se
G. Zinner und R. Stoffel
des unsubstituierten 3,5-Dioxo-1,2,4-oxadiaeolidins 34. Mitt. iiber Hydroxylamin-Derivate’)
Aus dem Institut fi ir Pharmazeutische Chemie der Technischen Universitat Braunschweig
(Eingegangen am 15. November 1968)
Durch Addition von Hydroxylamin an Alkoxycarbonylisocyanate erhiilt man 1-Hydroxy- 3 - a l k o x y c a r b o n y l h , die im alkalischen Milieu zum unsubstituierten 3,6-Dioxo- 1,2,4-oxadiazolidin cyclisieren. Diesea liiBt sich in 2- und 4-Stellung methylieren und mit
Xanthydrol xenthylieren.
Synthesis of 3,5-Dioxo-1,2,4-oxadiazoIidine Part 34. Hydroxylamine Derivatives
By addition of hydroxylamine to alkoxJrcarbonyliso~y~ates, l-hydroxy-3-alkoxy~rbonyl m a s were received which cyclisize in alkali to unsubstituted 3.5-dioxo-1.2.4-oxadiazolidine.
This can be methylated in 2- and 4-position and reacts with xanthydrol.
uber Synthesen auf dem Gebiet der 3,5-Dioxo-1,2,4-oxadiazolidine haben wir bereits mehrmala berichbeta). Nachstehend wird die Darstellung des unsubstituier- ten Stammkorpem mitgeteilt. Sie gelang durch Alkalibehandlung von in 3-Stellung alkoxycarbonylierten Derivaten (5) des Hydroxyharnstoffs, die man durch Addition von Hydroxylamin an Alkoxycarbonylisocyanate (3) erhiilt.
Fiir die Darstellung von 3 empfahl sich die Methode von Spezkab und Smith3), niimlich Behandlung der betreffenden Siiureamide, hier also der Carbamidsaure- ester 1 mit Oxalylchlorid. Wir beobachteten jedoch die Bildung beachtlicher Mengen der betreffenden N,N’-Bis(acy1)-oxamide 2, die sich wesentlich einschriinken lie& wenn man den bei der Reaktion entstehenden Chlorwasserstoff nicht nur durch spiiteres Erhitzen, sondern unter zusiitzlichem Durchleiten von getrocknetem Stick- stoff entfernt.
3,5-Dioxo-1,2,4-oxadiazolidin (4) schmilzt bei 103-105 ’ ; sein IR-Spektrum zeigt die in allen bisher dargestellten Derivaten charakteristisch bei rerhiiltnismiiflig kleinen Wellenlingen auftretenden zwei Carbonyl-Banden (5,50 und 5,74 p ) .
1) 33. Mitt.: a. Z i n w und E. Diierkop, Arch. Pharmaz. 30I, 776 (1968). 2) a) a. Zinner, Arch. Pharmaz. 294,765 (1961); 296,420 (1963); b) U. Zinner und R.-0. We6er,
8 ) A . J . Spezkdeund L. R. Smith, J. org. Chemistry, a ) 27, 3743 (1962); 28,1805 (1963); b) 30, Arch. Pharmaz. 298,580 (1965).
4306 (1966); c) Org. Syntheses 46, 16 (1966).
44*
Archiv der Z i n n e r und Stoffel Pharmazie
-~ 692
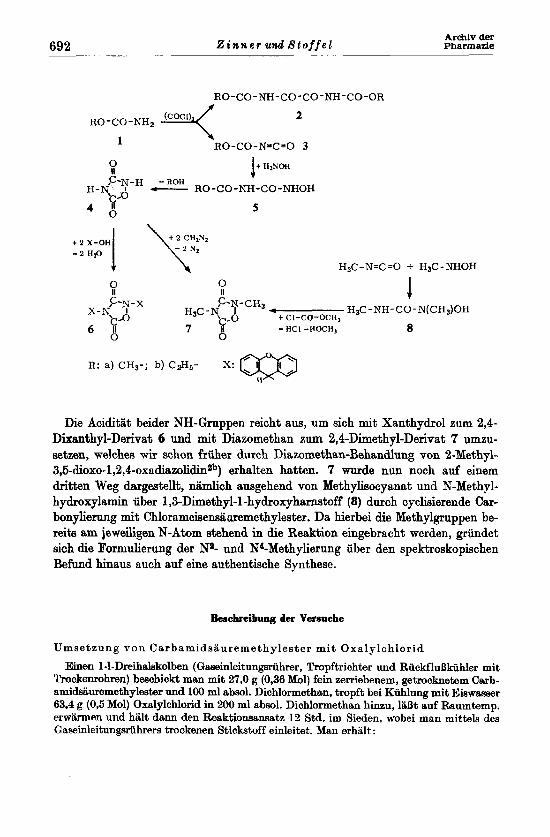
RO-CO-NH-CO-CO-NH-CO-OR 2
RO-CO-N=C=O 3 it €izNOH
1
- ROH B c- RO-CO-NH-CO-NHOH
5 0
H&-N=C=O + HsC-NHOH
R: a) CH3-; b) ‘28,- X:
Die Aciditlit beider NH-Gruppen reicht aw, M sich mit Xanthydrol zum 2,4- Dixanthyl-Derivat 6 und mit Diazomethan zum 2,4-Dimethyl-Derivat 7 umzu- SetZen, welches wir schon friiher durch Diazomethm-Behandlung von 2-Methyl- 3,6-dioxo-1,-2,4-oxadia~olidin~~) erhalten hatten. 7 wurde nun noch auf einem dritten Weg dargestellt, nlimlich awgehend von Methylisocyanat und N-Methyl- hydroxylamin iiber 1,3-Dirnethpl-l-hydroxyharstoff (8) durch cyclisierende Car- bonylierung mit Chlorameisensii uremethylester. Da hierbei die Methylgruppen be- reits am jeweiligen N-Atom stehend in die Reaktion eingebracht werden, griindet sich die Formulierung der N2- und WMethylierung uber den spektroskopischen Befund hinaus such auf eine authentische Synthese.
Beschreibung der Versuche
Umsetzung von Carbamidsiiuremethylester mit Oxalylchlorid Einen 1-1-Dreiialskolben (Gaaeinleitungsriihrer, Tropftrichter und RiickfluBkiihler mit
Trockenrohren) beschickt man mit 27,O g (0,36 Mol) fein zerriebenem, getrocknetem Carb- amidsiiuremethylester und 100 ml absol. Dichlormetbn, tropft bei Kiihlung mit Eiswasser 63,4 g (0,5 Mol) Oxalylchlorid in 200 ml absol. Dichlormethan hinzu, liBt auf Raumtemp. erwsrmen und hiilt dam den Raaktionsansatz 12 SM. im Sieden, wobei man mittels des Gaseinleitungsriihrers trockenen Stickstoff einleitet. Man erhiilt :

302. Bd. 1969/9 S ynthese des u d s t i t u i e r t e n 3,5-Di0x0-1.2,4-oxndiaw1icEins 693
~~ _ _ _ _
a) als festes Produkt wechselnde Mengen von S,S'-Bis(methoxycarbony1)-oxa- mid (24, welches aus Waaser umkristallisiert ab 215O sintert und bei 220' schmilzt'); Y (c=o) 1785/cm, 1760/cm und 1710/cm.
C ~ H B N ~ O ~ (2043) Ber.: C 35,30 H 3.95 N 13,72 Gef.: C 35,21 H 3,89 N 13,77
b) als Fliissigkeit Met ho x y carbon y li so c y ana t (3 a), Sap. 35-45 26-29" (iiberein- stimmend mit der Lit.8b); ng0 1,4062; Ausbeute nach zweimaliger frakt. Destillation 17,l g (47% d. Th.).
Umsetzung von Carbamids~ure&thy les t e r mit Oxalylchlorid Nach der gleichen Vorschrift erhiilt man am 35,7 g (0,36 Mol) CarbamiUWthyIesbr
und 400 ml Dichlormethan mit 63,5 g (0,5 Mol) Oxalylchlorid in 100 ml Dichlormethan
a) nachEinengenderReaktiom1osungi.Vak. wechselnddengen N,N'-Bis(6thoxy- carbony1)-oxamid (2b), Schmp. 169-172' (Wasser) (Lit.5) 170,5' (Athanol)); qC+) 1790/cm, 1755/cm und 1705/cm.
C8H,,N,0, (232,O) Ber.: C 41,38 H 5,21 N 12,07 Gef.: C 40,77 H 5,24 N 1131
b) nach zweimaliger frakt. Destillation 20,6 g (50% d. Th.) Bthoxycarbonyliso- cyanat (3b), Sdp.,, 20-22' (Lit. 6, Sdp.,8, 115-116'); n$o 1,4070.
3-Methoxycarbonyl-1 -hydroxyharnstoff (5a) Unter Riihren und AusschluB von Feuchtigkeit tropft man bei 10" zu 1,65 g (0,05 Mol)
Hydroxylamin(-Bese) in 50 ml a b d . Dioxan langsam 6,l g (0,05 Mol) 39 in 30 ml ebsol. Dioxan, riihrt noch 30 Min. bei Raumtemp. und weitere 30 Mm. auf dem Wasserbad. Dazu~ bringt man i. Vak. zur Trockne Md kristallisiert den Ruckatand aus Athanol. Schmp. 148-150°, bleuviolette Farbreaktion mit Eisen(II1)-chloridliisung; Y ( ~ - ~ ) 1725/cm und 1665/cm; Ausbeute 6,6 g (98% d. Th.).
C&N,O4 (1%,1) Ber.: C 26,86 H 4,47 N 20,89 Gef.: C 27,09 H 4,41 N 20,85
3-~thoxycarbonyl-l-hydroxyhernstoff (5b) In gleicher Weise erhiilt man e m liquimol. Uengen Hydroxylamin(-Baae) und 3b in
95proz. Ausbeute farblose Kristalle vom Schmp. 146-147' (1 T. Athanol und 9 T. Chloro- form), blauviolette Farbreaktion mit Eisen(1II)-chloridliung; Y ( ~ , ~ ) 1715/cm und 1665 /cm.
c&NzOd (14821) Ber.: C 32,44 H 5,44 N 18,91 Gef.: C 32,44 H 5,59 N 18,85
4 ) H . Najer und P . Mabille, C . R. h6bd. SBances -4oad. Sci. 242, 2737 (1966) geben Schmp.
5, Th. Bornwater, Recueil Trav. chim. Pays-Bas 31, 122 (1912). 6) 0. Diela und B. Wolf, Ber. dtsch. chem. Ges. 39, 688 (1906).
270-271' an.

69 4 Z i n n e r and B t o f f e l Archiv der Pharmazie
3,5 - D i o xo - 1,2,4 - o xadi a z oli di n (4) 0,06 Mol511 oder 5b werden mit 40 ml Wasser angeschwemmt, zuniichst mit 20 ml 3 n
KOH versetzt und bei 40" mit einem Glasstab bis zur Lijsung geriihrt. Dam erwarmt man so lange auf 70°, bis die Farbreaktion einer neutralieierten Probe mit Eisen(III)-chlorid- h u n g negativ ausfiillt ; es ist darauf zu achten, daD die Reaktionslosung selbst alkalisch bleibt, was einen weiteren Zusatz von insgeaamt etwe 16 m13 n KOH erfordert. Nach dem Erkalten wird rnit verd. HC1 angesiiuert, i. Vak. zur Trockne gebracht, der Ruckstand 2mal mit 10 ml absol. Aceton extrahiert. Die acetonische Lasung engt man so weit ein, &I3 die Substanz gerade noch in LGsung bleibt, aber mitgelostes KCl bereits ausfallt. Man filtriert, versetzt dasFiltrat mit etwas Chloroform und Petrolather und l i D t in der mite auskristallisieren. Schmp. 103-105° ; qC=,,) 1820/cm und 1740/cm; Ausbeute 4,O g (78% d. Th.).
C,H,N,O, (10271) Ber.: C 23,54 IE 1,98 N 27,45 &f.: C 23.54 R 2,29 N 27,54
2,4 - B i s ( x a n t h y 1 ) - 3,5 - di o x o - 1,2,4 - o s a d i a z 01 i di n (6) Man lost 0,125 g (1,25 mMol) 4 in 1 ml Methanol, versetzt mit 5,O g loproz. methanol.
Xanthydrollosung (2,5 mMol) und 2 ml Essigsaure, erwiirmt unter gelegentlichem Um- schiitteln 1 Std. auf 60', bringt i. Vak. zur Trockne und kristallisiert den Ruckstand 2mal ausibhanol. Schmp. 20%204"; Y ( ~ , ~ ) 181O/cm und 1740/cm; Ausbeute 0,5 g (86% d. Th.)
C,,HI,N,O, (462,5) Ber. : C 72,72 H 3,92 N 6,06 Gef.: C 72,21 H 4,29 K 6,03
2,4- Dime t h y 1 - 3,5 - di o xo - 1,2,4 - o x adi azol idi n (7) a) Man behandelt eine Losung von 0,3 g (3 mMol) 4 in 4,5 ml Methanol und 0,5 ml Was-
ser rnit einem tfberschuB einer Ltherischen Losung von Diazomethan, bringt i. Vak. zur Trockne und kristallisiert aus Athanol. Ausbeute 0,3 g (69% d. Th.).
b) 1,05 g (0,Ol Mol) 8 (nachetehend beschrieben) werden in 10 ml Athanol gelost und nach Zugabe von 10 ml einer lOproz. KOH unter Wasserkfilung und Umschutteln tropfen- weise mit zunitchst 1,0 g und dam noch so vie1 Chlorameisensiiuremethylester versetzt, bis die Lijsung einen pH-Wert von 7-8 zeigt und die Farbreaktion mit Eisen(II1)-chlorid- l 6 m g negativ awfallt. Man bringt i. Vak. zur Trockne, extrahiert den Ruckstand mit Chloroform und kristallisiert die dabei gewonnene Substanz aus Athanol. Ausbeute 0,9 g (62% d. Th.).
Schmp. 54-56' (LiL2b) 56"); ycs0) 1825/cm und 17151cm. CJ&N,O3 (130,1) Ber.: C 36,93 H 4,65 N 21,39
Gef.: C 37,37 H 4,81 N 21,25
1,3-Dimethyl- I-hydroxyharnstoff (8) gewann man in 84proz. Ausbeute aus Lquimolaren Mengen N-Methylhydroxylamin und Methylisocyanat in absol. Ather. Schmp. 83-85' (aus Chloroform mit Ather gefallt); V ( C = ~ ) 1660 /cm; blaue Farbreaktion mit Eisen(II1)-ohloridlosung.
C3H,N,O2 (lOa,l) Ber.: C 34,61 H 7,75 N 26,91 Gef.: C 34,40 H 7,72 N 27,26
Anschrift: Prof. Dr. G . Zinner, 33 Draunschweig, Beethovenstr. 55. [Ph 8741

![TPIP 藥師公會全聯會 · 2016. 2. 20. · Synthesis of 2-Substituted 3-0xo- 3H-naphtho[1', 2':4, 5]thiazolo[3, 2- b][1,2,4] triazines as Potential Anti- HIV Agents, J. Heterocyclic](https://img.pdfslide.org/doc/110x75/60f88489f5bbd1419e0390e2/tpip-eoefeoef-2016-2-20-synthesis-of-2-substituted-3-0xo-3h-naphtho1.jpg)