Embed Size (px)
Citation preview

1920 A . Klemer, B. Brandt, U. Hofmeister und E. R. Riiter
Liebigs Ann. Chem. 1983,1920- 1929
Synthese einiger Chlordesoxyglycoside und ihr Einsatz zur Darstellung von Desoxy- und Aminodesoxy-Zuckern
Almuth Klemer *, Burkhard Brandt, Ulrich Hofmeister und Ernst Ruiner Riiter
Organisch-Chemisches Institut der Universitat Miinster, Orltans-Ring 23, D-4400 Miinster
Eingegangen am 25. April 1983
Die isolierten sekundaren Hydroxygruppen der 2- und 3-0-Benzoyl-4,6-0-benzylidenhexo- pyranoside 2 - 5 reagieren mit Dichlormethylen(dimethyl)ammoniumchlorid (I) in Pyridin stereo- spezifisch zu den inversen Chlordesoxypyranosiden 6 - 9. Methyl-a-o-lyxofuranosid (11 b) ergibt Methyl-3,5-dichlor-3,5-didesoxy-2-O-dimethylcarbamoyl-a-~-arabinofuranosid (13). Methyl-6- chlor-6-desoxy-4-O-dimethylcarbamoyl-a-o-galactosid wird zu Methyl-a-D-fucosid reduziert, 9 in drei Schritten zu Benzyl-B-o-abequosid (16). Aus 6 wird das 3-Amino-3-desoxyglucosid 17 und iiber die 3-Azido-6-brom-6-desoxy-Verbindung 18 das 3,6-Diamino-3,6-didesoxyglucosid 20 ge- wonnen.
Synthesis of Chlorodeoxyglycosides and their Use in Preparation of Deoxy and Aminodeoxy Sugars
Isolated secondary hydroxy groups of 2- or 3-0-benzoyl-4,6-0-benzylidene hexopyranosides 2 - 5 react with dichloromethylene(dimethyl)ammonium chloride (1) in pyridine stereospecifically to yield the inverse chlorodeoxy pyranosides 6 - 9. Methyl a-o-lyxopyranoside (11 b) yields methyl 3,5-dichloro-3,5-didesoxy-2-~-dimethylcarbamoyl-a-~-arabinofuranoside (13). Methyl 6-chloro- 6-deoxy-4-O-dimethylcarbamoyl-a-o-galactopyranoside is reduced to give methyl a-o-fucoside, 9 in three steps to give benzyl 0-D-abequoside (16). 6 is converted into the 3-amino-3-deoxy glucoside 17 and oia the 3-azido-3-deoxy compound 18 into the 3,6diamino-3,6-dideoxy-a-~-gluco- pyranoside 20.
In friiheren Arbeiten berichteten wir bereits iiber regio- und stereoselektive Synthe- sen von Chlordesoxy-Zuckern mittels des hochreaktiven Dichlormethylen(dimethy1)- ammoniumchlorids *) (1). Einfache Durchfiihrung, gute Ausbeuten und vielseitige An- wendungsmoglichkeiten kennzeichnen das Verfahren.
a) Ungeschiitzte Methylhexopyranoside ergeben mit 1 in Acetonitril und Zusatz von Natriumazid (zur Herabsetzung der Reaktivitat von 1) 6-Chlor-6-desoxy-4-0-dimethyl- carbamoylglycoside I ) .
b) Kohlenhydrate mit freien vicinalen Hydroxygruppen reagieren in Pyridin ohne Anwesenheit von Natriumazid zu intermediaren cyclischen Dimethyliminiumsalzen, die stereospezifisch durch Riickseitenangriff des Chlorid-Ions in Chlor-desoxy-cr-t)-(di- methylcarbamoy1)glycoside iibergehen".
*) Kauflich zu erwerben bei Fa. EGA Chemie als ,,Dichlormethylenimmoniumchlorid".
0 Verlag Chemie GmbH, D-6940 Weinheim, 1983 0170-2041/83/1112-1920$ 02.50/0

Synthese von Chlordesoxy-, Desoxy- und Aminodesoxyglycosiden 1921
c) Kohlenhydrate mit isolierten Hydroxygruppen werden mit 1 zu den entsprechen- den Chlordesoxy-Produkten substituiert, sekundare reagieren stereospezifisch unter Platz~echsel~).
Arbeiten von Stick und Copeland4), die dieses Verfahren aufgriffen, veranlassen uns, uber unsere zwischenzeitlich erzielten weiteren Ergebnisse zu berichten. Daruber hinaus zeigen wir an Hand einiger ausgewahlter Chlordesoxy-Zucker einfache Zugange zu Aminodesoxy- und Didesoxy-Zuckern, die auf Grund ihrer Vorkommen in mikrobio- logischen Wirkstoffeii von besonderem Interesse sind.
Synthese von Chlordesoxy-Zuckern 1) Nachdem sich gezeigt hatte, daB die 5-Hydroxygruppe des 1,2-O-Isopropyliden-c-
~-glucofurano-6,3-~actons~~ oder die des entsprechenden 3,6-Anhydroglucofuranose- Derivates’) mit 1 in hohen Ausbeuten zum zugehorenden 5-Chlor-5-desoxy-~-ido- konfigurierten Produkt reagierte, erprobten wir Substitutionsreaktionen an sekunda- ren Hydroxygruppen von Hexopyranosen benachbart zu axialen oder aquatorialen, grol3eren Schutzgruppen. Es kamen vier bekannte und einfach zugangliche 4,6-0- Benzyliden-benzoyl-hexopyranoside zum Einsatz (siehe Tab. 3).
Tab. 1. Darstellung von Chlordesoxy-Zuckern mit Dichlormethylen(dimethyl)ammoniumchlorid (1)
Edukt Reaktionsbe- ding u n g e n Produkt Vo Ausb.
Methyl-2-0-benzoyl-4,6-0- benzyliden-a-o-gluco- pyranosid6) (2)
Methyl-3-O-benzoyl-4,6-0- benzyliden-a-o-glucopy- ranosid7) (3)
Methyl-2-O-benzoyI-4,6-0- benzyliden-a-o-allopy- ranosid*) (4)
Benzyl-2-O-benzoyl-4,S-O- benzyliden-P-o-galacto- pyranosid9.10) (5)
80 - 1 10°C, 2.5 h
80- 100°C, 2.5 h
80- 100°C, 5h
80°C, l h
Methyl-2-O-benzoyl-4,6-0- 90 benzyliden-3-chlor-3-des- oxy-a-D-allopyranosid (6) Methyl-3-O-benzoyl-4,6-0- 90 benzyliden-2-chlor-2-des- oxy-a-o-mannopyranosid (7) Methyl-2-O-benzoyl-4,6-0- 90 benzyliden-3-chlor-3-des- oxy-a-o-glucopyranosid (8) Benzyl-2-O-benzoyl-4,6-0- 80 benzyliden-3-chlor-3-desoxy- b-o-gulopyranosid (9)
Am gunstigsten verliefen auch diese Umsetzungen in wasserfreiem Pyridin. Abspal- tungen oder Wanderungen von Schutzgruppen wurden niemals beobachtet. Wichtig ist es, die Umsetzungen bei hoherer Temperatur und rnit mehrfachem UberschuB an 1 zu starten. Alle Edukte bildeten unter diesen Bedingungen die zugehorenden, bisher unbe- kannten inversen Chlordesoxy-Zucker in hohen Ausbeuten (siehe Tab. 1). Nebenpro- dukte sind geringe Mengen dimerer Carbonate rnit gleicher Konfiguration wie das Edukt. Sie brauchen in der Regel fur weiterfuhrende Synthesen nicht abgetrennt zu werden. Diese Nebenreaktion wird durch tiefere Temperaturen und nur geringen Re- agenzuberschuI3 stark begunstigt. Z. B. entstand aus 2 bei 60°C und 1.5fach molarer
Liebigs Ann. Chern. 1983
1922 A . Klemer, B. Brandt, U. Hofmeister und E. R. Riiter
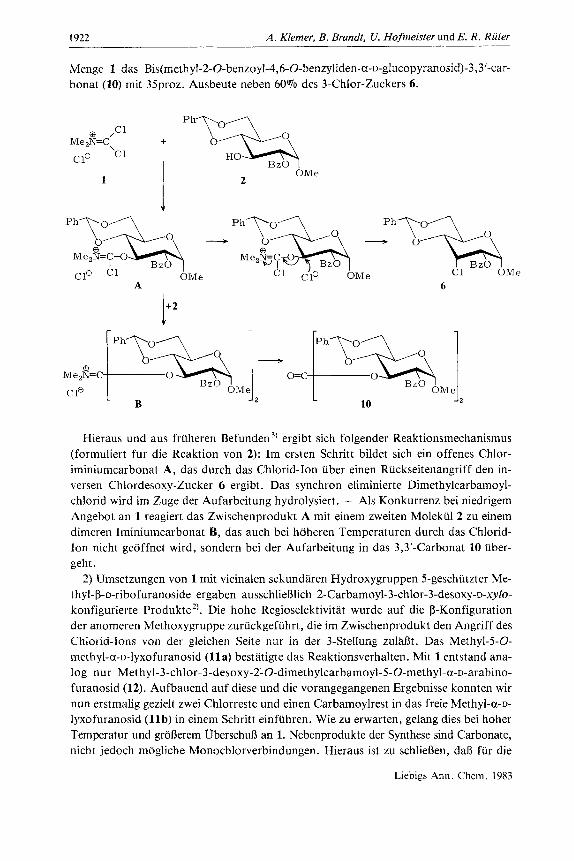
Menge 1 das Bis(methyl-2-O-benzoyl-4,6-O-benzyliden-~-~-glucopyranosid)-3,3~-~ar- bonat (10) mit 35proz. Ausbeute neben 60% des 3-Chlor-Zuckers 6.
Hieraus und aus fruheren Befunden 3, ergibt sich folgender Reaktionsmechanismus (formuliert fur die Reaktion von 2): Im ersten Schritt bildet sich ein offenes Chlor- iminiumcarbonat A, das durch das Chlorid-Ion uber einen Ruckseitenangriff den in- versen Chlordesoxy-Zucker 6 ergibt. Das synchron eliminierte Dimethylcarbamoyl- chlorid wird im Zuge der Aufarbeitung hydrolysiert. - Als Konkurrenz bei niedrigem Angebot an 1 reagiert das Zwischenprodukt A mit einem zweiten Molekul 2 zu einem dimeren Iminiumcarbonat B, das auch bei hoheren Temperaturen durch das Chlorid- Ion nicht geoffnet wird, sondern bei der Aufarbeitung in das 3,3‘-Carbonat 10 uber- geht .
2) Umsetzungen von 1 mit vicinalen sekundaren Hydroxygruppen 5-geschutzter Me- thyl-P-o-ribofuranoside ergaben ausschliefilich 2-Carbamoyl-3-chlor-3-desoxy-~-xylo- konfigurierte Produkte”. Die hohe Regioselektivitat wurde auf die p-Konfiguration der anomeren Methoxygruppe zuriickgefiihrt, die im Zwischenprodukt den Angriff des Chlorid-Ions von der gleichen Seite nur in der 3-Stellung zulaRt. Das Methyl-5-O- methyl-a-o-lyxofuranosid (11 a) bestatigte das Reaktionsverhalten. Mit 1 entstand ana- log nur Methyl-3-chlor-3-desoxy-2-O-dimethylcarbamoyl-5-O-methyl-a-~-arabino- furanosid (12). Aufbauend auf diese und die vorangegangenen Ergebnisse konnten wir nun erstmalig gezielt zwei Chlorreste und einen Carbamoylrest in das freie Methyl-a-o- Iyxofuranosid ( l lb ) in einem Schritt einfuhren. Wie zu erwarten, gelang dies bei hoher Temperatur und grdnerem Uberschufi an 1. Nebenprodukte der Synthese sind Carbonate, nicht jedoch mogliche Monochlorverbindungen. Hieraus ist zu schliefien, dal3 fur die
Liebigs Ann. Chem. 1983
Synthese von Chlordesoxy-, Desoxy- und Aminodesoxyglycosiden 1923
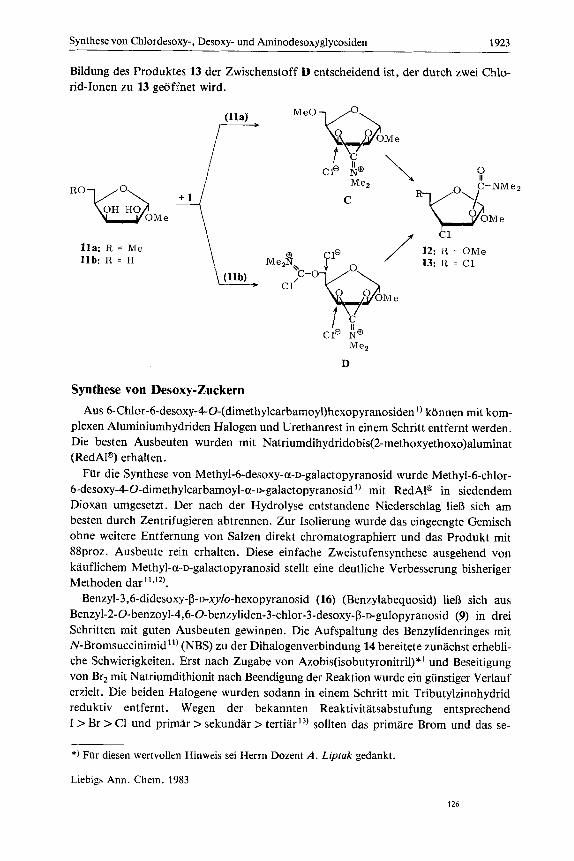
Bildung des Produktes 13 der Zwischenstoff D entscheidend ist, der durch zwei Chlo- rid-Ionen zu 13 geoffnet wird.
C
12: R = OMe 13: R = C1
1la: R = Me l l b : R = H
CI0 NQ
D Me2
Synthese von Desoxy-Zuckern Aus 6-Chlor-6-desoxy-4-O-(dimethylcarbamoyl)hexopyranosiden konnen mit kom-
plexen Aluminiumhydriden Halogen und Urethanrest in einem Schritt entfernt werden. Die besten Ausbeuten wurden rnit Natriumdihydridobis(2-methoxyethoxo)aluminat (RedAP) erhalten.
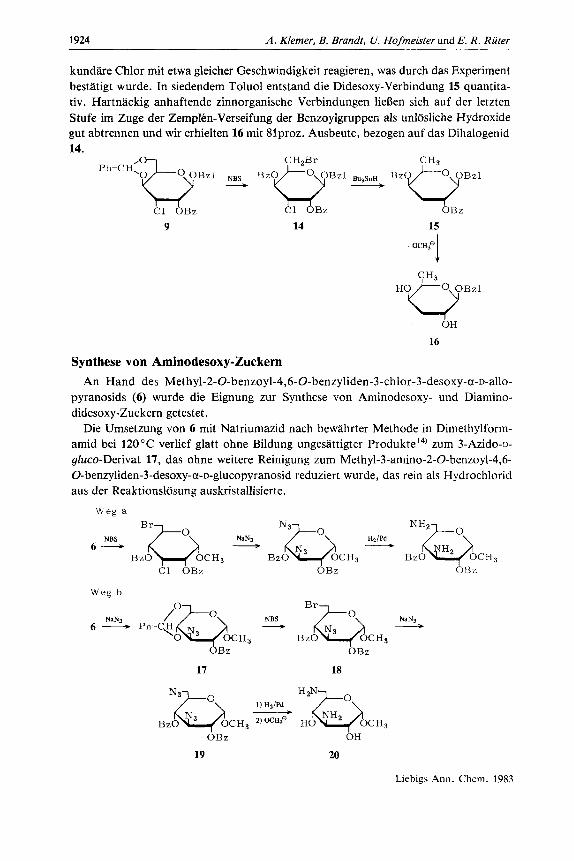
Fur die Synthese von Methyl-6-desoxy-a-~-galactopyranosid wurde Methyl-6-chlor- 6-desoxy-4-O-dimeth~~lcarbamoyl-a-~-galactopyranosid rnit RedAl@ in siedendem Dioxan umgesetzt. Der nach der Hydrolyse entstandene Niederschlag lie13 sich am besten durch Zentrifugieren abtrennen. Zur Isolierung wurde das eingeengte Gemisch ohne weitere Entfernung von Salzen direkt chromatographiert und das Produkt mit 88proz. Ausbeute rein erhalten. Diese einfache Zweistufensynthese ausgehend von kauflichem Methyl-a-D-galactopyranosid stellt eine deutliche Verbesserung bisheriger Methoden dar"s12). Benzyl-3,6-didesoxy-(3-~-xylo-hexopyranosid (16) (Benzylabequosid) lieR sich aus
Benzyl-2-O-benzoyl-4,6-O-benzyliden-3-chlor-3-desoxy-(3-~-gulopyranosid (9) in drei Schritten rnit guten Ausbeuten gewinnen. Die Aufspaltung des Benzylidenringes mit N-Bromsuccinimid '') (NBS) zu der Dihalogenverbindung 14 bereitete zunachst erhebli- che Schwierigkeiten. Erst nach Zugabe von Azobis(isobutyronitril)*) und Beseitigung von Br, rnit Natriumdithionit nach Beendigung der Reaktion wurde ein gunstiger Verlauf erzielt. Die beiden Halogene wurden sodann in einem Schritt mit Tributylzinnhydrid reduktiv entfernt. Wegen der bekannten Reaktivitatsabstufung entsprechend I > Br > C1 und primiir > sekundar > tertiar13) sollten das primare Brom und das se-
*) Fur diesen wertvollen Hinweis sei Herrn Dozent A. Liptak gedankt.
Liebigs Ann. Chem. 1983
126
1924 A . Klemer, B. Brandt, U. Hofmeister und E. R. Riiter
kundare Chlor mit etwa gleicher Geschwindigkeit reagieren, was durch das Experiment bestatigt wurde. In siedendem Toluol entstand die Didesoxy-Verbindung 15 quantita- tiv. Hartnackig anhaftende zinnorganische Verbindungen lieBen sich auf der letzten Stufe im Zuge der Zemplen-Verseifung der Benzoylgruppen als unlosliche Hydroxide gut abtrennen und wir erhielten 16 mit 8lproz. Ausbeute, bezogen auf das Dihalogenid 14.
p h - c H x O b ,O Bzl NBS BzOQ CHzBr Bzl BuaSnH BzO@Bzl
------?) __i)
C1 OBz OB z
9 14 15
1 - OCH3O
HoeBz1 OH
16
Synthese von Aminodesoxy-Zuckern An Hand des Methyl-2-O-benzoyl-4,6-O-benzyliden-3-chlor-3-desoxy-~-~-allo-
pyranosids (6) wurde die Eignung zur Synthese von Aminodesoxy- und Diamino- didesoxy-Zuckern getestet.
Die Umsetzung von 6 mit Natriumazid nach bewahrter Methode in Dimethylform- amid bei 120 "C verlief glatt ohne Bildung ungesattigter P r o d ~ k t e ' ~ ) zum 3-Azido-D- gluco-Derivat 17, das ohne weitere Reinigung zum Methyl-3-amino-2-O-benzoyl-4,6- O-benzyliden-3-desoxy-a-~-glucopyranosid reduziert wurde, das rein als Hydrochlorid aus der Reaktionslosung auskristallisierte.
Weg a
6 - NBS '0 __t NaN3 No - H d P d NH@ocH3
BzO OCH3 BZO OCH3 B z 0 C l OBz OBZ 6BZ
W e g b
6-Ph-&Q NaN3 % 5 OCH, B z 0 OCH,
B Z OBZ
17 18
OBz OH
19 20
Liebigs Ann. Chem. 1983

Synthese von Chlordesoxy-, Desoxy- und Aminodesoxyglycosiden 1925
Zur Darstellung von Methyl-3,6-diamino-3,6-didesoxy-a-o-glucopyranosid (20) wur- den 2 Synthesewege untersucht. Weg a), der ausgehend von 6 in nur 3 Schritten zum angestrebten Produkt fiihren konnte, erwies sich als unvorteilhaft. Nach Aufspaltung des Benzylidenrings init N-Bromsuccinimid verlief die weitere Umsetzung rnit Natrium- azid unvollstandig und unter Bildung mehrerer Nebenprodukte, deren Abtrennung nur durch aufwendige urid verlustreiche Chromatographie moglich war.
Sehr giinstig verlief jedoch die Synthese nach Weg b). Das 3-Azido-2-O-benzoyl-4,6- 0-benzyliden-3-deso:tyglucosid 17 liefl sich nach bewahrter Methode in hohen Ausbeu- ten in die 3-Azido-6~-brom-3,6-didesoxy-Verbindung 18 iiberfiihren. Nach chromato- graphischer Reinigung konnten die weiteren Schritte zu 20 wie oben ohne Zwischeniso- lierung der Diazidoverbindung 19 mit einer Ausbeute von 90% bezogen auf 18, vollzo- gen werden.
Negativ verliefen (alle Versuche, das Halogen in a-Chlor-a-desoxy-(dimethylcarb- amoy1)pyranosiden oder -furanosiden durch die Azidgruppe zu substituieren. Z. B. la- gen Methyl-3-chlor-3,6-didesoxy-2-0-dimethylcarbamoyl-cc-o-altropyranosid und 12 in Dimethylformamid/Natriumazid bei 100 "C noch nach 3 Tagen nahezu unverandert vor. Auch unter Pha.sentransferbedingungen z. B. rnit Aliquat oder Tributyl(hexa- decyl)phosphoniumbromidt6) trat keine Substitution ein. D a im Gegensatz dazu 6 glatt reagierte, mussen die: Fehlschlage auf den benachbarten trans-standigen Urethanrest zuriickzufiihren sein. Er schirmt offenbar das Molekiil derart ab, da13 ein Ruckseiten- angriff durch die Azidgruppe ausgeschlossen ist .
Wir danken der Deulschen Forschungsgemeinschaft und dem Verband der Chemischen Indu- srrie fur die finanzielle Unterstutzung dieser Arbeit.
Experimenteller Teil Allgemeine Methodem: Schmelzpunkte (unkorrigiert): Thermopan-Heiztischmikroskop der Fa. Reichert. - Optische
Drehungen: Polarimeteir Perkin-Elmer 241; 10-cm-Kiivetten, Natrium-D-Linie, 20°C. - 'H- NMR-Spektren: Gerat Ieol JNM-PMX 60 (60 MHz) und Gerat Varian WM-300 (300 MHz); 5-mm-Rohrchen, 35 "C, Tetramethylsilan als innerer Standard. - Massenspektren: Gerat SM-1 und CH-1 der Fa. Varian MAT; ElektronenstoR (70 eV). - Dunnschichtchromatographie (DC): Polygram-Fertigfolien, 0.25 mm Kieselgel mit Fluoreszenzindikator UV 254 der Fa. Macherey, Nagel & Co.; Anfarbung durch Bespruhen mit konz. Schwefelsaure und Erhitzen auf 120°C. - Saulenchromatographie (SC): Kieselgel60 (KorngroBe 0.063 - 0.200 mm) der Fa. Merck; Lauf- mittel A: Diisopropylether/Aceton (9/1), Laufmittel B: Toluol/Aceton (19/1), Laufmittel C: ToluoVAceton (9/1), Laufmittel D: Essigester/Methanol (411).
Allgemeine Arbeitsoorschriyt fur die Darstellung der Chlordesoxy-Zucker 6 - 9, 12 und 13: Ei- ne Losung des Ausgangszuckers (Tab. 1) in wasserfreiem Pyridin wird unter Feuchtigkeitsaus- schluR auf die Reaktionstemperatur gebracht und anschlieRend unter Ruhren im N2-Gegenstrom mit Dichlormethylen(dirnethy1)ammoniumchlorid (1) versetzt. Nach wenigen Minuten farbt sich die Reaktionslosung tieibraun. - Aufarbeitung fur die Produkte 6, 7, 8, 12 und 13: Die Reak- tionslosung wird bei 40°C i. Vak. zur Trockne eingedampft und restliches Pyridin durch 2maliges Eindampfen mit Toluol unter gleichen Bedingungen entfernt. Man versetzt den Ruckstand mit Ether/Wasser (1 : 1) und schuttelt aus. Die waRrige Phase wird noch 2mal mit je 100 ml Ether ex- trahiert. Man trocknet die vereinigten Etherphasen, klart gegebenenfalls rnit Aktivkohle und
Liebigs Ann. Chem. 1983
126*

1926 A. Klemer, 3. Brandt, (1. Hofmeister und E. R. Riiter
dampft zur Trockne ein. Das Rohprodukt, welches in der Regel meist zu Weiterverarbeitung di- rekt eingesetzt werden kann, 1aBt sich durch Umkristallisation oder durch Saulenchromatogra- phie reinigen.
Methyl-2-O-benzoyl-4,6-O-benzyliden-3-chlor-3-desoxy-a-~-ullopyranosid (6): Ansatz: 500 mg (1.29 mmol) 2, 530 mg (3.5 mmol) 1. Reaktionsbedingungen: llO°C, 30 min; dann 80°C, 2 h in 100 ml Pyridin. Reinigung des Rohproduktes durch Saulenchromatographie rnit Laufmittel A. Ausb. 470 mg (go%), Schmp. 58 -63 "C (nach 3 Monaten kristallin), [a]? = + 77.4 ( c = 1.0 in Chloroform). - 'H-NMR (CDCI,): 6 = 3.5 (s; 3H, OCH,), 3.58 - 3.92 und 4.28 - 4.42 (m; 4H,
5 Hz, J2,, = 2 Hz; 1 H, 2-H), 7.3-8.2 (m; 10H, Phenyl-H). - MS (70 eV): m/e = 4041406 (1.2/0.4%, M'), 36/337 (19/4.6%), 2791281 (1.5/0.8%), 105 (100%).
C2~H2,CIO6 (404.8) Ber. C 62.30 H 5.23 Gef. C 63.20 H 5.22
4-H, 5-H, 6-H, 6'-H), 5.22 (d, Ji.2 = 5 Hz; l H , I-H), 5.64 (s; 1 H, Ph-CH), 5.70 (dd, Jz,1 1
Bis(methyl-2-O-benzoyl-4,6-O-benzyliden-a-~-glucopyrunosid)-3,3'-carbonat ( 10): Ansatz: 500 rng (1.29 mmol) 2, 350 mg (2.1 mmol) 1. Reaktionsbedingungen: 60"C, 6 h in 100 ml wasser- freiem Pyridin. Reinigung des Rohprodukts durch Saulenchromatographie mit Laufmittel A. Ausb. 320 mg (61%) 6; 190 mg (35%) 10 mit Schmp. 2O8-21O0C, [a]z,0 = + 152.6 (c = 1.0 in Chloroform). - 'H-NMR (CDCI,): 6 = 3.58-4.38 (m; je 2 x 1 H, 4-H, 5-H, 6-H, 6'-H), 5.02
Benzyliden-H), 5.57 (t; 2 x l H , 3-H), 7.10- 8.0 (m; 2 x 5H, Phenyl-H). - MS (70 eV): m/e =
797/798/799/800 (11/35/16/4%, M'), 6451646 (1.5/0.8%), 579 (0.6%), 4291430 (15/3.3%), 105 (200%).
C&,201, (798.8) Ber. C 64.66 H 5.30 Gef. C 64.38 H 5.18
(dd, J2,i = 3.8 Hz, J2 ,3 10 Hz; ~ x I H , 2-H), 5.20 (d; 2 ~ 1 H , 1-H), 5.38 ( s ; 2 ~ 1 H ,
Methyl-3-0-benzoyl-4,6-O-benzyliden-2-chlor-2-desoxy-a-~-mannopyrunosid (7): Ansatz: 500mg (1.29 mmol) 3,530mg (3.5 mmol) 1. Reaktionsbedingungen: 11O"C, 30min; dann 80°C 2 h in 100 ml Pyridin. Reinigung durch Saulenchromatographie mit Laufmittel A. Ausb. 470 mg (90%) Sirup [a]? = -29.5 (c = 1.0 in Chloroform). - 'H-NMR (CDCI,): 6 = 3.40 (s; 3H, OCH3),3.45-4.40(m;4H,4-H,5-H,6-H,6'-H),4.6(dd,J2,, = 4 H z , J 2 , , = 1.8Hz; lH,2-H) , 5.52 (dd, J2 , , = 4 Hz, J3,4 = 9.8 Hz; l H , 3-H), 7.2- 8.14 (m; 10H, Phenyl-H). - MS (70 eV): m/e = 403/404/405/406 (7/9.8/4.8/3.4%, M'), 373 (1.4%), 257 (5%), 255 (20%), 149 (15.4%), 105 (100%).
C2,H2,C1O6 (404.8) Ber. C 62.30 H 5.23 Gef. C 62.18 H 5.12
Methyl-2-O-benzoyl-4,6-O-benzyliden-3-chlor-3-desoxy-a-~-glucopyrunosid (8): Ansatz: 500 mg (1.29 mmol) 4, 530 mg (3.5 mmol) 1. Reaktionsbedingungen: 110°C 30 min; dann 80°C 5 h in 100 ml Pyridin. Reinigung durch Saulenchromatographie mit L,aufmitttel A. Ausb. 465 g (90%) Sirup, [a]? = +90.2 (c = 1 in Chloroform). - 'H-NMR (CDCI,): 6 = 3.38 (s; 3H, OCH3),3.5-3.98(m; 3H,5-H,6-H,6'-H),4.31(m;iH,4-H),4.52(dd; lH,3-H),5.06(d; l H , 1-H), 5.18 (m; l H , 2-H), 5.58 (s; l H , Phenyl-H), 7.15-8.25 (m; 10H, Phenyl-H). - MS (70 eV): m/e = 403/404/405/406 (7/12/2/3.3%, Mt) . 373 (lo%), 368 (0.3%), 3361338 ( O . l / O.O30ro), 255/157 (4.6/1.2Vo), 148/150 (90/16%), 149 (go%), 105 (100%).
CzlH2,CIO6 (404.8) Ber. C 62.30 H 5.23 Gef. C 62.30 H 5.19
Benzyl-2-O-benzoyl-4,6-O-benzylidend-chlor-3-desoxy-~-D-gu(runosid (9): Ansatz: 1.2 g (2.6 mmol) 5, 3.2 g (20 mmol) 1. Reaktionsbedingungen: 8O"C, 1 h in 250 ml Pyridin. Aufarbei- tung: Nach dern Einengen i. Vak. bei 40°C wird 3mal mit je 100 ml Toluol zur Entfernung des restlichen Pyridins eingedampft. Der Ruckstand wird mit 200 ml Chloroform aufgenommen und die schwere Losung ca. 3 - 4mal mit je 200 ml Wasser ausgeschiittelt. Die noch schwach gefarbte organische Phase wird mit Magnesiumsulfat getrocknet, das Losungsmittel verdampft, der Ruck- stand mit 200 ml Ether aufgenommen, mit Aktivkohle entfarbt und eingedampft. Der Ruckstand
Liebigs Ann. Chem. 1983

Synthese von Chlordesoxy-, Desoxy- und Aminodesoxyglycosiden 1927
wird rnit Laufmittel B chromatographiert. Ausb. 1.0 g (80%) 9 harter, weiRer Schaum, [ a l g = - 57 (c = 0.33 in Chloroform). - 'H-NMR [(CD3)2CO]: 6 = 4.81 (q,B, 6 A = 4.72, 6 , = 4.90, J = 12.5 Hz; 2H, Benz:yl-CH2), 4.75 (dd; IH, 3-H), 5.20 (d, .Tl,2 = 9 Hz; I H , 3-H), 5.47 (dd, J l , 2 = 9 Hz, J2,3 = 4 Hz; 1 H, 2-H), 5.72 (s; 1 H, Benzyliden-H), 7.1 - 8.1 (Aromaten-H). - MS (70 eV): m/e = 480/4.82 (0.19/0.06%, Mt ) , 479/481 (0.62/0.21%), 3891391 (1.54/0.50%), 3741376 (0.62/0.24%), 283/285 (12.59/4.43%), 105 (100%).
C,,H,,CIO, (480.9) Ber. C 67.43 H 5.24 Gef. C 67.67 H 5.55
Methyl-3-chlor-3-des~oxy-2-O-dimethylcarbamoyl-5-O-methy~-~-~arabinofuranosid (12): An- satz: 330 mg (1.85 mmol) l l a , 820 mg (5.05 mmol) 1. Reaktionsbedingungen: 80"C, 60 min in 15 ml Pyridin. Aufarbeitung: nach Aktivkohleklarung Rohausbeute ca. 9070, nach SC rnit Lauf- mittel C Ausb. 380 rng (77%) 12 mit [ a ] g = +52.2 (c = 0.81 in Chloroform). - 'H-NMR (CDC1,): 6 = 2.94 und 2.96 (2s, 6H, Urethan-CH,), 5.14 (dd, J2,3 = 3.8 Hz, J2, , = 0.9 Hz; 1 H, 2-H), 4.97 (s; I H , 1-HI. - MS (70 eV): m/e = 236/234 (5.7/2.3%, M ? - 31), 72 (100%).
CjoH,,CINO, (267.7) Ber. C 44.87 H 6.78 N 5.23 Gef. C 44.73 H 6.93 N 5.36
Methyl-3,5-dichlor-ji',5-didesoxy-2-O-dimethylcarbamoyl-a-~-arabinofuranosid (13): Ansatz: 230 mg (1.40 mmol) l l b , 1.2 g (7.39 mmol) 1. Reaktionsbedingungen: 81 "C, 15 min in 20 ml Py- ridin. Aufarbeitung: SC-Reinigung mit Laufmittel C. Ausb. 230 mg (61%) 13 mit [a]? = + 35.6 (c = 0.52 in Chloroform). - 'H-NMR (CDCI,): 6 = 2.92 und 2.95 (2s; 6H, Urethan-CH,), 5.01 (s; IH , 1-H), 5.16 (d, J2,, = 3 Hz; IH , 2-H). - MS (70 eV): m/e = 240/242/244 (815. l / l V o , M t ) , 72 (100%).
C,H,,C12N04 (272.1) Ber. C 39.72 H 5.56 N 5.15 Gef. C 40.14 H 5.73 N 5.25
Methyl-6-desoxy-a-irgalactopyranosid: Zu 4.9 g (17.2 mmol) Methyl-6-chlor-6-desoxy-4-0- dirnethylcarbarnoyl-a-o-galactopyranosid in 200 ml wasserfreiem Dioxan werden unter Sieden langsam 30 ml Natriurndihydridobis(2-methoxyethoxo)aluminat (RedAP) (70% in Toluol) zuge- tropft, und es wird 1 hi unter RuckfluR gekocht. Danach wird uberschussiges Reagenz rnit 200 ml Wasser zersetzt und die Suspension bei 1200 U h i n 10 min zentrifugiert. Die uberstehende triibe Losung wird dekantiert und i. Vak. eingeengt, der Ruckstand in 250 ml Methanol aufgenommen, filtriert und auf ca. 50 ml eingeengt. Die erneut ausgefallenen Salze werden durch SC mit Lauf- mittel D abgetrennt. Hierzu benutzt man eine weite Saule und ruhrt die Suspension in die oberen 5 cm der Kieselgelschicht ein, da die Tropfgeschwindigkeit sonst zu niedrig ist. Das Produkt kann aus EssigesterIPetrolether umkristallisiert werden. Ausb. 2.7 g (88%), Schrnp. 155 - 157 "C (Lit.17): Schmp. 157--158"C), [a]? = +I91 (c = 0.5 in Wasser) (Lit.17): [ a ] g = +196). Das Produkt stimmt mit einer nach Lit. 12) dargestellten authentischen Probe spektroskopisch vollstandig iiberein.
Benzyl-2.,4-di-O-benzoyl-6-brom-3-chlor-3,6-didesoxy-~-o-gulopyranosid (14): 1.92 g (4.0 mmol) 9 werden in 200 ml Tetrachlorkohlenstoff zum Sieden erhitzt. Man fiigt 3.0 g Bariumcarbonat und 785 mg (4.4 mmol) N-Bromsuccinimid hinzu und startet die Reaktion mit einer Spatelspitze Azo- bis(isobutyronitri1). Wenn die rotbraune Bromfarbe verschwunden ist (etwa nach 60 rnin), wird die Losung abfiltriert und der Ruckstand nochmals mit 30 ml Tetrachlorkohlenstoff gekocht. Die vereinigten Losungen wascht man zunachst 2mal mit 10proz. Natriumdithionit-Losung, dann 3mal mit 100 ml Wasser, trocknet mit Magnesiumsulfat und dampft i. Vak. zum Sirup ein. 14 wird saulenchromatographisch rnit Laufmittel B isoliert. Ausb. 1.04 g (46010) Sirup, sehr zer- setzlich. - MS (70 eV): m/e = 557/559/561 (0.5/0.5/0.2VoO, M + - l ) , 523/525 (0.13/0.14%), 467/469/471 (2.60/3.28/0.98%), 4151417 (0.34/0.39%), 401/403 (1.22/0.88%), 346/348/ 350 (3.38/4.31/1.13%1), 307 (32.10%), 105 (100%).
C2,HZ4BrClO6 (559.8) Ber. C 57.93 H 4.32 Gef. C 58.54 H 4.62
Liebigs Ann. Chem. 1983

1928 A. Klemer, B. Brandt, U. Hofmeister und E. R. Ruler
Benzyl-3,6-didesoxy-/3-~-xylo-hexopyranosid (Benzyl-b-o-abequosid, 16): 520 mg (0.93 mmol) 14 werden in 50 ml wasserfreiem Toluol gelost und unter FeuchtigkeitausschluR zum Sieden er- hitzt. Man gibt 1 ml Tributylzinnhydrid (dargestellt nach Lit. 18)) und eine Spatelspitze Azobis(is0- butyronitril) als Starter hinzu. Nach ca. 1 h ist die Umsetzung zu 15 quantitativ beendet, man ent- fernt das Losungsmittel i. Vak. Der Ruckstand, der noch zinnorganische Verbindungen enthalt, wird in 50 ml wasserfreiem Methanol gelost und mit 10 mi Natriummethylat-Losung (aus 300 mg Natrium) debenzoyliert. Nach 24 h bei Raumtemp. neutralisiert man mit Dowex 50-WX2 H', fil- lriert und engt i. Vak. ein. Nach SC mit Laufmittel D erhalt man 16 als farblosen Sirup. Ausb. 180 mg (81%) 16, nach zweimaliger Destillation bei 15O0C/O.2 Torr: [a];' = -89 (c = 0.6 in Chloroform), (Lit.19): [a]? = - 107 (c = 0.5 in Chloroform)). - 'H-NMR ([Ds]Pyridin): 6 =
1.43(d, J5,6 = 6.5Hz; 1H,6-H), 1.89(m,J2,, = 11 Hz,J3,, = 3 H z , J ~ , ~ = 14Hz; I H , 3-H,), 2.57 (m, JZ,, = 5 Hz, J3,4 = 3 Hz; 1 H, 3-H,), 3.73 (9; 1 H, 5-H), 3.84 (t?; 1 H, 4-H), 4.35 (m; 1 H, 2-H),4.64(d,Jl,, = 6Hz; lH,l-H),4.92(qA,,6, = 4.73,6, = 5 .11 , J= 11.5Hz;2H,Benzyl- CH,), 5.77 (breit; 2H, 2-OH, 4-OH), 8.1 -8.6 (Aromaten-H).
C,,H1804 (238.3) Ber. C 65.53 H 7.61 Gef. C 65.03 H 8.04
Me~hyl-3-amino-2-O-benzoyl-4,6-O-benzyliden-3-desoxy-ff-~-glucopyranosid: 500 mg (1.24 mmol) 6 in 200 ml wasserfreiem Dimethylformamid werden rnit 2.0 g (30 mmol) Natriumazid versetzt. Nach 4tagigem Riihren bei 120°C ist die Umsetzung zu 17 vollstandig. Es wird i . Vak. eingeengt und getrocknet. Ohne weitere Isolierung von 17 kann nun in 150 ml Methanol aufge- nommen werden und rnit Pd/C-Katalysator im H2-Strom hydriert werden. Nach 5 h ist die Um- setzung quantitativ. Der Katalysator wir abfiltriert und die Losung i. Vak. eingeengt. Das Gluco- pyranosid wird aus einer 50-ml-Etherlosung 3mal mit jeweils 30 ml Wasser ausgeschuttelt. Die vereinigten wafirigen Extrakte werden eingeengt und in 100 ml Ether gelost. Da das Amin nicht stabil ist, wird es sofort nach Lit. i9) rnit 10proz. ethanolischer HC1 in das Hydrochlorid uberge- fiihrt. Ausb. 315 mg (66V0, bez. auf 6) Hydrochlorid mit Schmp. 185°C (Zers.) (Lit.20): Schmp. 184.5-186"C), [a]$ = +64.1 (c = 1 in Chloroform) (Lit.2o): [a];' = +63.4 (in Chloro- form)).
Methyl-3-azido-2,4-di-0-benzoyl-6-brom-3,6-didesoxy-a-~glucopyranosid (18): 500 mg (1.24 mmol) 6 werden wie bei der voranstehenden Darstellung beschrieben zur 3-Azido-Verbindung 17 umgesetzt. Das Rohprodukt wird mit einem 1.5fachen N-Bromsuccinimid-UberschuR und 1.2 g BaCO, in 100 ml CC14 2 h unter RiickfluR erhitzt. Der Niederschlag wird abfiltriert und rnit 300 ml l0proz. waRriger Natriumdithionit-Losung ausgeschuttelt. Nach der Phasentrennung wird die organische Phase im Rotationsverdampfer eingeengt. Der erhaltene Sirup wird saulen- chromatographisch mit Laufmittel A gereinigt. Ausb. 207 mg (42%) 18.
Methyl-3,6-diamino-3,6-didesoxy-a-~glucopyranosid (20): 200 mg 18 werden wie bei der Dar- stellung des Methyl-3-amino-2-O-benzoyl-4,6-O-benzyliden-3-desoxy-a-o-glucopyranosids be- schrieben, zur Diazido-Verbindung 20 umgesetzt und aufgearbeitet. Die Umsetzung betragt nach 5 d uber 90%. Das Rohpropdukt wird in wasserfreiem Methanol im H2-Strom mit Pd/C-Kata- lysator wahrend 7 h zur Diamino-Verbindung hydriert. Zur Debenzoylierung wird mit Natrium- methanolat-Losung versetzt und 20 nach ublicher Aufarbeitung kristallin erhalten. Ausb. 62 mg (80%) 20 mit Schmp. 162°C (Lit.,'): 162- 163"C), [a];' = + 153 (c = 0.9 in Wasser) (Lit.zi): [a]hO = + 152 (c = 0.93 in Wasser)).
A. Klemer, R. Lemmes und G. Nicolaus, Liebigs Ann. 1977, 177. 2, A . Klemer, R. Lemmes und K. Cirnander, Carbohydr. Res. 68, 383 (1979). 3, A. Klemer, (1. Hofmeister und R. Lemmes, Carbohydr. Res. 68, 391 (1979).
Liebigs Ann. Chem. 1983

Synthese von Chlordesoxy-, Desoxy- und Aminodesoxyglycosiden 1929
4) C. Copeland und R. V. Stick, Aust. J . Chem. 35, 2257 (1982).
6) F. A. Carey und K. 0. Hodgson, Carbohydr. Res. 12, 463 (1970). 7, J. G. Buchanan und J. P. Schwarz, Carbohydr. Res. 11, 383 (1969). *) H. Kdnig und H. Weidemann, Carbohydr. Res. 39, 374 (1975). sf S. A. Abbas und A. H. Hains, Carbohydr. Res. 39, 358 (1975).
lo) G. J. F. Chittenden und J. G. Buchanan, Carbohydr. Res. 11, 379 (1969). l i ) S. Hannessian und If. Plessas, J. Org. Chem. 34, 1035 (1969). 12) 0. T. Schmidt in Methods in Carbohydrate Chemistry (Edit. R. T. Whistler und M. L.
Wolfrom), Bd. I, S. 191, Academic Press, New York 1961. 13) H. Paulsen, H. Salzburger und H. Redlich, Chem. Ber. 108, 3589 (1976). 14) S. Hannessian und H. Plessas, J. Org. Chem. 34, 1053 (1969). 15) W. P. Reeves und M. L. Bahr, Synthesis 1976, 823. 16) J. Landini und F. Rolla, Synthesis 1976, 389. 17) Rodd’s Chemistry of Carbon Compounds (Edit. S. Coffey), 2. Aufl., Bd. I F, S. 519, Else-
Is) H. G. Kuivala, Synthesis 1970, 499. 19) J. Yoshimura, K. Sato, H. Haschimoto und K. Schimizu, Bull. Chem. SOC. Jpn. 50, 3305
*O) Cotton, Silk and Man Made Fibers Rersearch Assoc. ( E r f . R. D. Guthrie), Brit. Pat. 1007011
21) S. Inouye, Chem. Pharm. Bull. 14, 902 (1966).
R. Lemmes, Dissertation Univ. Miinster 1978.
vier, Amsterdam 1967.
(1977).
(13. Okt. 1965) [Chem. Abstr. 64, P 8290f (1966)l.
[104/83]
Liebigs Ann. Chem. 1983









![Problematik, Klinik und Beispiele der ... · PDF fileDMAA) [1,10]. Marine Einzeller sowie essbare Algen können Arsen in Form von Arseno-zuckern [8] und Arsenolipiden [9] 1000fach](https://img.pdfslide.org/doc/110x75/5a78995e7f8b9a7b698d5c0b/problematik-klinik-und-beispiele-der-110-marine-einzeller-sowie-essbare.jpg)






![Znr Datirnng einiger athenischer Archonten. I. Damasias. · 2011. 3. 7. · Znr Datirnng einiger athenischer Archonten. I. Damasias. Der Arohon Damasias, unter dem der arwv O'TEq>aviTl]](https://img.pdfslide.org/doc/110x75/60c3d7d8eb45ea3faa0a732f/znr-datirnng-einiger-athenischer-archonten-i-2011-3-7-znr-datirnng-einiger.jpg)












