Embed Size (px)
Citation preview

Synthesen, EPR- und ENDOR-Untersuchungen
phenyl- und cycloalkylsubstituierter RadikaliJnen
aromatischer Kohlenwasserstoffe
Inaugural-Dissertation
zur Erlangung der Doktorwürde
des Fachbereichs Chemie
der Freien Universität Berlin
vorgelegt von
Kornelia Grein
aus Frankfurt am Main

Gutachter Professor Dr. H. Kurreck
Professor Dr. w. Broser
Tag d~r mündlichen Prlifung: 4. 12. 1981

Die Ergebnisse der vorliegenden Dissertation wurden t';ilweise
bereits publiziert:
K. Grein, B. Kirste, H. Kurreck
Chem. Ber. ill' 254 (1981)


INHALTSVERZEICHNIS
1.
2.
3.
3. 1 .
Einleitung
Problemstellung
Allgemeiner Teil
Grundlagen der magnetischen Resonanzmethod~n
EPR, ENDOR, TRIPLE
3.1 .1. Grundlagen der EPR
3.1.2. Grundlagen der ENDOR-Methode
3.1.3. EPR und ENDOR an deuterierten Verbindungen
3.1.4. TRIPLE-Resonanz
3. 2. Molekül-Orbital-Modelle
3.2.1. HMO-Modell
3.2.2. Verfeinerte MO-Modelle
3.2.3. Ladungsabhängigkeit der Kopplungskonstanten
3.2.4. HFS durch B-Protonen
3.2.5. "Long-Range"-Hyperfeinwechselwirkungen
3.2.6. Phenylhyperkonjugation
4.
4 • 1 •
4 .2.
4. 3.
Spezieller Teil
Namen und Struktur der untersuchten Verbindunge.:
Synthesen
NMR-Spektren
Seite
7
9
1 0
1 0
10
15
18
19
21
22
23
26
27
28
32
35
35
37
41
4. 3. 1. NMR-Spektren der phenylsubstituierten Verbindun·;en 41
4.3.2. NMR-Spektren der 9-Cycloalkylanthracene 44
4.4. Massenspektren 46
4.5. EPR- und ENDOR-Spektren 48
4.5.1. EPR- und ENDOR-Spektren der phenylsubstituierte~
Radikalanionen 48
4.5.2. EPR- und ENDOR-Spektren der phenylsubstituierten 65
Radikalkationen 4.5.3. EPR- und ENDOR-Spektren der Radikalanionen der
9-Cycloalkylanthracene 77

Seite
4.6. Diskussion 84
4.6.1. Diskussion der Meßergebnisse der phenylsub-
stituierten Verbindungen 84
4.6.2. Diskussion der Meßergebnisse der 9-Cycloalkyl-
4. 7.
5.
5. 1.
5. 2.
5.3.
6.
7.
anthracene
Untersuchung weiterer Radikalkationen
Experimenteller Teil
Messungen
Radikalerzeugung
Darstellung der Verbindungen
Zusammenfassung
Literaturverzeichnis
90
93
10 1
101
101
102
106
108

7
Die Strukturaufklärung organischer Moleküle durch ~oderne
spektroskopische Methoden stellt nicht nur eine Vereinfachung
gegenüber den klassischen Methoden, sondern gleichzeitig auch
eine Informationserweiterung dar. Neben der IR- und Massen
spektroskopie sind vor allem die magnetischen Resonan?methoden
zu zunehmender Bedeutung gelangt. Während die NMR-Spektrosko
pie an diamagnetischen Molekülen durchgeführt wird, dient die
Elektronenspinresonanz (EPR) zur Untersuchung paramagnetischer
Verbindungen. Die beiden letztgenannten Methoden liefE,rn viel
fältige Informationen über die untersuchten Verbindungen, die
weit über eine Konstitutionssicherung hinausreichen. So ist es
beispielsweise möglich, Aussagen über Struktur und Stabilität
verschiedener Konformerseiner Verbindung zu treffen. Durch An
wendung der EPR-Technik gelingt es, die Spindichteverteilung
eines Moleküls zu ermitteln, und so die Möglichkeit zur Bewer
tung von Modellrechnungen zu eröffnen.
Die wichtigsten, aus EPR-Messungen an Dublettsystemen er
hältlichen Meßgrößen sind der g-Faktor und die Hyperfeinwech
selwirkungsparameter (HFS-Kopplungskonstanten) . Die Untersu
chung zentrenreicher Moleküle mit niedriger Symmetrie führt
zu EPR-Spektren, die keine eindeutige Interpretation zulas
sen, da durch die Vielzahl der Linien Uberlagerungen auftre
ten und so keine vollständige Auflösung erreicht wird. Die
ENDOR-Methode (Elektron-Kern-Doppelresonanz) stellt bE,son
ders in solchen Fälleneine wichtige Ergänzung dar. Durch An
wendung dieser Doppelresonanzmethoden, bei der zusätzlich zum
Mikrowellenfeld eine Bestrahlung der Probe mit einem Radio
frequenzfeld erfolgt, wird ein Auflösungsgewinn erzielt. Aus
den ENDOR-Spektren, deren Linienzahl gegenüber EPR-Spektren
meist drastisch vermindert ist, lassen sich die Kopplungskon
stanten direkt ablesen. Um die Zuordnung der Kopplungskonstan
ten zu den Molekülpositionen treffen zu können, ist unter an
derem die Ermittlung ihrer Vorzeichen von Bedeutung. Während
unter bestimmten Voraussetzungen eine absolute Vorzeichenbe
sti~~ung mit der NMR möglich ist, stellt die TRIPLE-Technik
(Elektron-Kern-Kern-Dreifachresonanz) eine allgemein anwend
bare Methode zur Besti~~ung der relativen Vorzeichen der Kopp
lungskonstanten dar. Eine weitere wichtige Hilfe zum Treffen

- 8
der Zuordnung ist die spezifische Deuterierung einzelner Molekül
positionen, da in einem ENDOR-Spektrum die Protonen- und Deute
rium-Linien gut unterscheidbar sind.
Da die meisten organischen Moleküle diamagnetisch sind, müssen
sie zu EPR- und ENDOR-Untersuchungen in ihre Radikale übergeführt
werden. Eine Voraussetzung zur Durchführung der Messungen ist die
ausreichende Lebensdauer der paramagnetischen Spezies. Geeignete
Systeme sind aromatische Verbindungen, da die entsprechenden Ra
dikale meist die erforderliche Stabilität besitzen.
An solchen Systemen sind viele Modellrechnungen durchgeführt
worden. Für planare, aromatische n-systeme zeigen die aufgrund
einfacher MO-Modelle getroffenen Voraussetzungen oft zufrieden
stellende Übereinstimmung mit experimentell ermittelten Werten.
Bei der Behandlung von verdrillten, aromatischen System treten
jedoch signifikante Widersprüche zu experimentellen Daten auf.
Sowohl hinsichtlich der g-Faktoren /1,2/ als auch der Hyper
feinkopplungskonstanten treten Anomalien im Vergleich zu pla
naren n-Systemen auf. So wird di.e konventionelle Reihenfolge
der Hyperfeinkopplungskonstanten der Phenylprotonen \a~aral> laH th l»iaH t I nicht eingehalten /3-5/. Vielmehr nimmt der · or o me a Betrag der meta-Protonenkopplungskonstante drastisch zu und
kann gleich dem der ortho- und para-Protonenkopplungskonstan
ten bzw. sogar größer werden als jener. Diese Abweichungen konn
ten durch Berücksichtigung der n-a-Wechselwirkung zum Teil er
klärt werden. Da bisher nur an einer kleinen Zahl von Verbin
dungen dieser als Phenylhyperkonjugation bezeichnete Effekt
überprüft werden konnte, schien die Synthese und Untersuchung
weiterer verdrillter, aromatischer Kohlenwasserstoffe sinnvoll.
Eine weitere Gruppe interessanter Verbindungen stellen die
alkyl- und cycloalkylsubstituierten n-Systeme dar. Die EPR
und ENDOR-Untersuchungen ihrer Radikale geben Informationen
über die Geometrie der Verbindungen und gestatten das Studium
der Spintransfermechanismen in die durch mehrere a-Bindungen
vom n-System getrennten Zentren. Um die Abhängigkelt des
Spintransfers von der räumlichen Anordnung zu untersuchen,
sind Systeme mit eingeschränkter Beweglichkeit der Alkylreste,
wie sie beispielsweise bei Ringsystemen vorliegen, besonders
geeignet.

- 9 -
Aufgabe der vorliegenden Arbeit ist es, einerseits geeigne
te Modellsubstanzen zum weiteren Studium der Phenylhyperkonjuga
tion zu synthetisieren, Methoden zur Erzeugung paramagnetischer
Radikalionen zu testen und die Radikale durch EPR-, ENDOR- und
TRIPLE-Messungen zu untersuchen. Andererseits sollen die Aus
wirkungen der sterischen Anordnung von cycloalkylsubstituier
ten n-Systemen auf den Spintransfer zu den Alkylprotonen durch
Synthese entsprechender Radikale und Messungen mit den genann
ten magnetischen Resonanzmethoden behandelt werden.
Zum Studium der Phenylhyperkonjugation sollen solche phenyl
substituierten Verbindungen dargestellt werden, die aufgrund
der sterischen Wechselwirkungen der Substituenten mit anderen
Molekülteilen eine starke Verdrillung der Phenylringe erwarten
lassen. So sollen in die ortho-Positionen der Phenylsubstituen
ten des 9,10-Diphenylanthracens Methylgruppen eingeführt wer
den, da sie die Wechselwirkungen zu den peri-ständigen H-Atomen
verstärken und den Verdrillungswinkel vergrößern sollten. Es
sollen spezifische Deuterierungen in den Fällen durchgeführt
werden, in denen sie zur Zuordnung erforderlich sind.
Zur Untersuchung sterischer Effekte bei gesättigten Ring
systemen sollen verschiedene 9-Cycloalkylanthracene syntheti
siert werden. Die neutralen Moleküle beider Gruppen sollen in
ihre Radikalionen übergeführt werden, und durch EPR-, ENDOR
und TRIPLE-Messungen untersucht werden. Nach Ermittlung von
Größe und Vorzeichen der Kopplungskonstanten, deren Zuordnung
zu den Molekülpositionen und der Verifizierung der getroffenen
Zuordnung durch Simulation der EPR-Spektren soll ein Vergleich
der experimentell ermittelten Daten mit Werten aus quantenme
chanischen Modellrechnungen erfolgen.

- 10 -
3. Allgemeiner Teil
3.1. Grundlagen der magnetischen Resonanzmethoden EPR, ENDOR, TRIPLI
In den folgenden Abschnitten sollen die Grundlagen der EPR- und
ENDaR-Spektroskopie sowie das TRIPLE-Experiment beschrieben werden.
Die Behandlung der genannten Methoden soll unter Beschränkung auf
die für die vorliegende Arbeit relevanten Aspekte erfolgen.
Zum Nachweis und zur Untersuchung von paramagnetischen Ver
bindungen eignet sich hervorragend die paramagnetische Elektro
nenresonanz (EPR) als die empfindlichste Methode. Das ungepaar
te Elektron, das in einem solchen System auftritt, wird durch
Masse, Ladung und Drehimpuls beschrieben. Für organische Mole
küle kann letzterer dem Eigendrehimpuls, dem Spin S, gleichge
setzt werden. In einem äußeren, statischen Magnetfeld B kann
das über die Beziehung (1) mit dem ElektronenspinS verknüpf
te magnetische Dipolmoment ~s keine beliebigen Orientierungen
einnehmen.
~s
g -g ~B S g-Faktor des freien Elektrons
~B Bohrsches Magneton
( 1 )
Die Komponente von bezüglich der Feldachse kann nur zwei
Werte annehmen, die durch die Spinquantenzahlen bzw.
m =+l bestimmt sind, was einer parallelen bzw. antiparallelen s 2 Orientierung der Spins in Bezug zur Richtung des Magnetfeldes B
entspricht.
Die im feldfreien Raum vorliegende Entartung der Energiezu
stände des Elektrons wird durch Einwirkung des ~agnetfeldes B
aufgehoben (Zeeman-Effekt). Es erfolgt eine Aufspaltung in zwei
Energieterme mit der Zusatzenergie
E (2)

- 11 -
Ein Ubergang zwischen den Zeeman-Niveaus entsprechend einer
Spinumkehr des Elektrons wird durch Einstrahlen eines zweiten,
senkrecht zu B rotierenden Magnetfeldes B1 induziert, wenn die
Resonanzbedingung
h V ( 3)
erfüllt ist. Ftir ein freies Elektron entspricht dieser Ubergang
einer einzigen Linie. In einem paramagnetischen organischen Mo
lekül erfährt jedoch das Elektron Wechselwirkungen mit inneren
Magnetfeldern, die durch die magnetischen Momente der Atomker
ne und eventuell vorhandener weiterer ungepaarter Elektronen
hervorgerufen werden. Das führt zu einer Aufspaltung der Zeeman
-Niveaus, und im EPR-Experiment findet man ein Spektrum, das
durch Uberlagerung einer Vielzahl von Linien entsteht.
Aus der Lage des Zentrumsdes Signals kann der g-Faktor des
Radikals ermittelt werden, der eine ftir jedes Radikal charak
teristische Proportionalitätskonstante zwischen der Resonanz
frequenz v und dem Magnetfeld B darstellt. Die Abweichung vom
g-Faktor des freien Elektrons (ge=2.0023) ist durch nicht voll
ständig zu vernachlässigende Spin-Bahn-Wechselwirkungen zu er
klären /6,7/. Bei den Radikalionen aromatischer Kohlenwasser
stoffe beträgt die g-Faktor-Verschiebung ßg im allgerneinen we
niger als 5·10-4 • Ftir planare, aromatische Kohlenwasserstoff
radikale hat Stone einen linearen Zusammenhang zwischen der
g-Faktorverschiebung ßg und dem HUckel-Energiekoeffizienten
x0
des einfach besetzten Orbitals (s. Kap. 4) postuliert /8/.
(4)
Abweichungen von dieser Geraden werden bei der g-Faktor-Be
stirnrnung nichtebenerRadikale gefunden. Die Untersuchungen sol
cher aromatischer n-Systeme, die verdrillte Phenylsubstituenten
tragen, zeigten eine erhebliche, negative Abweichung von der
Stone'schen Geraden /1/.

- 12
Die quantenmechanische Beschreibung der EPR bedient sich
des Spin-Harnilton-Operators, eines Energie-Operators, der die
Wechselwirkungen der Elektronen- und Kernspins untereinander
sowie mit dem äußeren Magnetfeld beschreibt. Durch Anwendung
des Spin-Harnilton-Operators auf die Spinfunktionen lassen
sich die Energieeigenwerte erhalten.
Für ein Dublett-Radikal in isotroper, flüssiger Lösung mit
einem Elektron und einem Kern (S = 1/2, I = 1/2) lautet der in
der Hochfeldnäherung (B » a, mit B II z) vereinfachte Spin-Hamil
ton-Operator:
H g iJ.B B - gk j.J.k B i + a h §z I ( 5) z
mit g iJ.B B s Elektron-Zeeman-Term ~
gk ;.t.k B I = Kern-Zeernan-Term A z
a h s I isotrope Hyperfeinwechselwirkunq z z
Die Eigenfunktionen dieses Operators sind einfache Produkte
der Spinfunktionen lms' > • Damit lauten die Energieeigenwer-
te unter Vernachlässigung des Kern-Zeeman-Terms:
E ( 6)
Durch die magnetische Wechselwirkung zwischen dem ungepaar
ten Elektron und einem Atomkern des untersuchten Moleküls mit
Kernspin I wird eine Aufspaltung der Energieniveaus in 2I + 1
Subniveaus hervorgerufen. Die daraus resultierende Aufspaltung
des EPR-Signals eines organischen Radikals wird als Hyperfein
struktur bezeichnet. Die Kern-Elektron-Wechselwirkungen be
stehen aus einem anisotropen und einem isotropen Anteil. Der
anisotrope Anteil ist in isotroper, flüssiger Lösung aufgrund
der Ausrnittelung der Dipol-Dipol-Wechselwirkung durch die Mo
lekülbewegung im allgemeinen zu vernachlässigen. Der isotrope
Anteil der HFS ist von der Spindichteam Ort des Kerns iflrk)i 2
und vorn Kern-g-Faktor gk abhängig. Er wird beschrieben durch
den Kopplungsparameter a (in MHz).
a = 2;.t.o 3h
( 7)

- 13 -
Unter Berücksichtigung der EPR-Auswahlregeln !Ams=! 1, ßmr=O)
ergibt sich für die Resonanzfeldstärken eine symmetrische Anord
nung der Resonanzsignale am B0
, dem Zentrum der Resonanz.
B = B 0
a
Für mehrere Kerne gilt analog:
B B - 1: 0 j
(8)
( 9)
Die Gesamtaufspaltung A eines EPR-Spektrums, die dem Abstand
der beiden äußersten Hyperfeinlinien entspricht, wird durch Glei
chung 10 beschrieben:
A r 2 I. I a ·I j J J
( 1 0)
Für die Gesamtaufspaltung eines Kohlenwasserstoffradikals
ergibt sich damit:
A ( 11 )
Die Summation erfolgt über die u Kohlenstoffzentren, die
die wechselwirkenden Protonen tragen.
Durch Wechselwirkung mit n äquivalenten Kernen werden 2ni+1
Linien erhalten. Bei mehreren Sätzen äquivalenter Kerne wächst
die Linienzahl multiplikativ.
Für eine im Magnetfeld B befindliche paramagnetische Probe
ist im thermischen Gleichgewicht eine ungleiche Besetzung der
Zeeman-Niveaus zu erwarten. Gemäß der Boltzmann-Verteilung
exp (- ( 12) n
tritt ein Populationsüberschuß des energetisch tieferliegen
den Niveaus auf. Durch Bestrahlung der paramagnetischen Probe
mit der Resonanzfrequenz v werden sowohl Ubergänge vom unteren
zum oberen Niveau als auch in umgekehrter Richtung induziert,
deren Ubergangswahrscheinlichkeiten gleich sind. Durch gleich-

- 14 -
zeitige Absorption wie Emission sollte nach kurzer Zeit eine
Gleichbesetzung der Niveaus erfolgen. Ein Absorptionssignal
kann nur deswegen registriert werden, weil durch strahlungs
lose Energieübertragung mit der Umgebung, sogenannter Spin
-Gitter-Relaxation, der Populationsausgleich verhindert wird.
Eine ungleiche Besetzung der Energieniveaus ruft, makroskopisch
betrachtet, eine Längsmagnetisierung der Probe parallel zu B
hervor, die durch Mikrowellenabsorption eine Änderung erfährt.
Die Zeit, die das System zur Wiederherstellung der Boltzmann
-Verteilung benötigt, wird als longitudinale Relaxationszeit T 1 bezeichnet. Neben dieser Längsmagnetisierung wird auch eine
Quermagnetisierung senkrecht zu B beobachtet, die dadurch her
vorgerufen wird, daß bei Bestrahlung der Probe mit dem Magnet
feld B1 die Spins in gleicher Phasenlage präzessieren. Die Zeit,
die zum Abbau der Quermagnetisierung notwendig ist, wird als
transversale Relaxationszeit T2 bezeichnet.
Die Linienbreite eines EPR-Signals wird durch die Relaxations
zeit T2
bestimmt. Alle Faktoren, die die Phasenbeziehungen der
Spins beeinflussen, tragen damit zur Linienbreite bei. Durch
die Bewegung benachbarter paramagnetischer Teilchen wird das
Magnetfeld am Ort des Elektrons moduliert, was, wie auch die
Inhomogenität des Magnetfeldes, zur Unschärfe der Resonanz
führt. Die diese Prozesse beschreibende Relaxationszeit, die
Spin-Spin-Relaxationszeit T2 ', ist Teil der transversalen
Relaxationszeit T2
. Die durch Spin-Gitter-Relaxation bewirk-
ten Spinumkehrprozesse rufen ebenfalls eine Änderung der Quer
magnetisierung hervor. Damit trägt auch die longitudinale Re
laxationszeit T1
zur Linienbreite bei, die durch folgende Be
ziehung gegeben ist:
1 T,
2 + T'
2
2
Tz { 13)
Um bei einem EPR-Experiment schmale Linien zu erhalten,
müssen die Bedingungen so gewählt werden, daß keine extrem
kurzen Relaxationszeiten Ti und Tz' auftreten. sowohl für
T 1 wie für Tz' führen hohe Radikalkonzentrationen ( 10-4M!
zu einer Verkürzung. Weiterhin ist die Temperatur bzw. die
Viskosität des Lösungsmittels für beide Relaxationszeiten

- 15 -
von großer Bedeutung. Da jedoch die Veränderung der Temperatur
und damit der Viskosität des Lösungsmittels zu einer gegensätz
lichen Beeinflussung von T1
bzw. T 2 ' führt, muß die optimale
Meßtemperatur experimentell bestimmt werden.
Beim ENDOR-Experiment (ENDOR = ~lektron ~uclear DOuble
~esonance) wird eine, sich in einem statischen Magnetfeld
befindliche, paramagnetische Probe resonanzhaft sowohl mit
einem Mikrowellenfeld als auch mit einem Radiofrequenzfeld
bestrahlt. Die Bestrahlung durch das RF-Feld bewirkt die
partielle Entsättigung eines EPR-Ubergangs, da durch die zu
sätzlichen NMR-Ubergänge neue Relaxationswege eröffnet wer
den und führt somit zu einer Intensitätszunahme des EPR
-Signals. Im ENDOR-Spektrum wird die EPR-Signalintensitäts
änderung in Abhängigkeit von der Frequenz des RF-Feldes re
gistriert. Das ENDOR-Experiment kann am einfachen Beispiel
eines Dublettradikals mit einem Elektron und einem Kern
(8=112, I~112) erläutert werden. Durch Anwendung des Spin
-Rarnilton-Operators (Gleichung 5) auf die Spinfunktionen
! ms, mi > werden die Energieeigenwerte erhalten (in Fre
quenzeinheiten) .
E (ms, m1 Jih ms- vk m1 + a ms m1 mit g ~B B I h
vk gk ~k B I h
( 14)
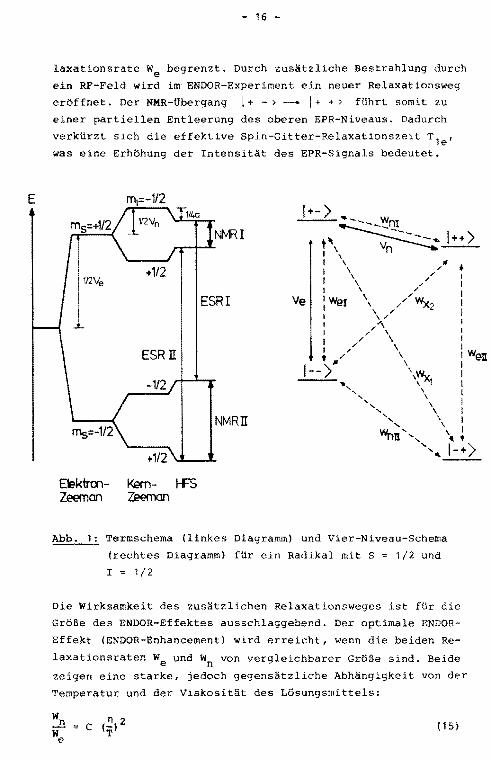
Die graphische Darstellung der Energieniveaus kann in einem
Termschema bzw. in einem übersichtlicheren Vier-Niveau-Schema
erfolgen (Abb. 1). Im letzteren sind außerdem die möglichen
Relaxationswege, charakterisiert durch die Relaxationsraten W,
eingezeichnet. Für Protonen sind die Kreuzrelaxationsprozesse
wx 1 ' wx2 meist zu vernachlässigen. Betrachtet man den Fall ei
ner sättigenden Einstrahlung des EPR-Ubergangs I, die zu einer
Gleichbesetzung der Niveaus I+ - > und i - - > führt, ist die
EPR-Linienintensität von der strahlungsfreien Spin-Gitter-Re-
E
- 16 -
laxationsrate We begrenzt. Durch zusätzliche Bestrahlung durch
ein RF-Feld wird im ENDOR-Experiment ein neuer Relaxationsweg
eröffnet. Der NMR-tlbergang l + - > - I+ + > fUhrt somit zu
einer partiellen Entleerung des oberen EPR-Niveaus. Dadurch
verkürzt sich die effektive Spin-Gitter-Relaxationszeit Tle'
was eine Erhöhung der Intensität des EPR-Signals bedeutet.
j 1f2Ve
!
E~ktrooZeeman
Kern- f-FS Zeeman
NMRI
ESRI
NMRli
1+-> --- ""----wni
t' ~ . .!:'----.... I•+> \ V~------
1 \ I \ " I \ /" ~ I \ .,/ I I \ ;" I
Ve I Wer , / Wx2 I I ' " I \; I
//\ I / \ I
I "/ \ I t / ' 'Wen
1-->" \ 'fx I ---.. , \ 1 I
', \ I ', \ I
' \ I ', \ I
' \ I Wnu',, \ t
........ , 1-+ >
Abb. 1; Termschema (linkes Diagramm) und Vier-Niveau-Schema
(rechtes Diagramm) für ein Radikal mit S = 1/2 und
I 1/2
Die Wirksamkeit des zusätzlichen Relaxationsweges ist für die
Größe des ENDOR-Effektes ausschlaggebend. Der optimale ENDOR
Effekt (ENDOR-Enhancement) wird erreicht, wenn die beiden Re
laxationsraten We und Wn von vergleichbarer Größe sind. Beide
zeigen eine starke, jedoch gegensätzliche Abhängigkeit von der
Temperatur und der Viskosität des Lösungsmittels:
( 15)

- 17 -
Durch die Wahl eines geeigneten Lösungsmittels und günstiger
Meßtemperatur kann eine Optimierung (Wn :::<We) erfolgen.
Mit den Auswahlregeln für EPR (~ms = :1, Ami = 0) und denen
für NMR {Ams = 0, Ami = !1) erhält man je zwei Ubergänge. Da
das ENDOR-Experiment einen Nachweis eines NMR-Uberganges, re
gistriert durch EPR-Signalintensitätsänderung, darstellt, sind
dessen Auswahlregeln hierbei bestimmend. Damit ergibt sich die
ENDOR-Resonanzbedingung (a in Frequenzeinheiten)
vENDOR ( 1 6)
Die Linienlagen im ENDOR-Spektrum sind abhängig von dem
Größenverhältnis zwischen der freien Kernfrequenz vk und dem
Kopplungsparameter a. Für vk < }a/21 sind die ENDOR-Linien sym
metrisch zu a/2 angeordnet; der Abstand zwischen den Linien
beträgt 2 vk. Im Falle für vk> ja/21, wie er im allgemeinen
für Protonen zutreffend ist, befinden sich die Linien mit dem
Abstand a symmetrisch um vk angeordnet.
vk < I a/2j ~ a/2 * a/2 ~ vk * a/2
Die Protonen- bzw. Heterokernkopplungen zugehörigen Linien
gruppieren sich in den meisten Fällen um verschiedene Zentren
und sind somit unterscheidbar.
Im ENDOR-Spektrum eines Dublett-Radikals werden für jeden
Satz äquivalenter Kerne zwei Linien erhalten, da alle NMR
-Ubergänge innerhalb eines ms-Zustandes frequenzentartet
sind. Die Zahl der ENDOR-Linien wächst bei mehreren Sätzen
äquivalenter Kerne additiv. Da die Zahl der EPR-Linien je
doch multiplikativ ansteigt, ist die Liniendichte eines
ENDOR-Spektrums gegenüber der eines EPR-Spektrums wesentlich
vermindert. Der dadurch hervorgerufene Auflösungsgewinn und
die Möglichkeit einer exakten Bestimmung der Kopplungskon
stanten bedeuten die wichtigsten Vorteile der ENDOR-Methode
gegenüber der EPR. Die Nachteile sind eine gegenüber der
EPR-Methode verminderte Empfindlichkeit auf 1-10%, das Feh
len der Information über die Zahl der äquivalenten Kerne und
der erhebliche apparative Aufwand.

- 18 -
3.1.3. EPR und ENDOR an deuterierten Verbindungen
Da Protonen und Deuteronen unterschiedliche magnetische
Eigenschaften aufweisen (s. Tabelle 1), sind charakteristi
sche Unterschiede in den EPR- und ENDOR-Spektren zu erwar
ten, wenn Protonen selektiv durch Deuteronen ersetzt werden.
Tabelle 1: Eigenschaften von Protonen und Deuteronen
natürliche Häufigkeit (%)
Kernspin Ik
Kern-g-Faktor gk
99.986
0.5
5.5854
2H=D
0.014
1.0
0.8574
Das EPR Spektrum einer deuterierten Verbindung ist auf
grunddes Kernspins (I=1) linienreicher als das Spektrum der
entsprechenden protonenhaltigen Verbindung, gemäß der 21+1
Orientierungsmöglichkeiten des Spins. Die Deuteriumkopplungs
parameter aD sind um das Verhältnis der Kern-g-Faktoren
gk(H)/gk(D) = 6.51 gegenüber den entsprechenden Protonen
kopplungsparametern aH vermindert. Dadurch wird eine Verrin
gerung der Gesamtaufspaltung hervorgerufen, die neben der
Erhöhung der Linienzahl im allgemeinen zu einer Verminderung
der Auflösung durch Deuterierung führt. Einen Auflösungsge
winn durch H/D-Austausch erhält man im EPR-Experiment nur
dann, wenn dieser an einer Position kleiner Spindichte durch
geführt wurde, so daß die Deuteriumkopplungskonstante klein
ist gegen die Linienbreite.
Bei einem Deuterium-ENDOR-Spektrum werden Linien erhalten,
die sich symmetrisch mit dem Abstand laD/21 um die freie Deu
teronenfrequenz vD anordnen, die um den Faktor 6.51 kleiner
ist als vH (v 0 ~ 2.2 MHz, VH ~ 14 MHz). Im ENDOR-Spektrum
sind Deuteronen- und Protonenlinien sehr gut unterscheidbar,
da sie in unterschiedlichen Frequenzbereichen registriert
werden.

- 19 -
Durch das ENDOR-Experiment gelingt eine genaue Bestimmung
der Kopplungskonstanten, nicht jedoch die Bestimmung ihrer
Vorzeichen. Durch die Anwendung der TRIPLE-Technik, die eine
Elektron-Kern-Kern-Dreifachresonanzmethode darstellt, sind
zusätzliche Informationen zu gewinnen.
Beim general-TRIPLE-Experiment wird, während wie im ENDOR
-Experiment ein RF-Feld durch den Bereich der NMR-Resonanz
frequenzen gesweept wird, ein zweiter NMR-Ubergang sättigend
eingestrahlt. Dadurch werden weitere Relaxationsraten indu
ziert, die die Intensität der ENDOR-Linien beeinflussen. Aus
den Intensitätsänderungen können eindeutige Rückschlüsse auf
die relativen Vorzeichen der Kopplungskonstanten gezogen werden.
Für den einfachsten Fall eines Monoradikals mit zwei nicht
äquivalenten Kernen mit dem Spin r 1 = r 2 = 1/2 werden acht
Energieniveaus erhalten, die durch Spinrelaxationswege ver
bunden sind. Dieses System kann durch ein elektrisches Netz
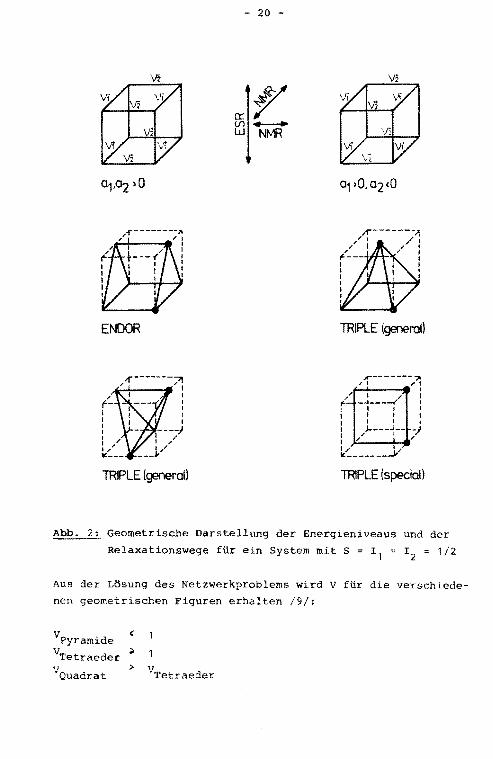
werk /9/ simuliert werden (s. Abb. 2). Die acht Energieniveaus
sind dabei an den Ecken eines Würfels angeordnet, die verti
kalen Würfelkanten entsprechen den Elektronspinrelaxationswe
gen, die horizontalen den Kernspinrelaxationswegen (Abb. 2).
Durch Sättigung eines NMR-Ubergangs wird der entsprechende
Relaxationsweg kurzgeschlossen, was sich durch Zusammenziehen
der entsprechenden Würfelkante geometrisch darstellen läßt.
Für das ENDOR-Experiment erhält man als geometrische Figur
ein Prisma, unabhängig davon, ob a 1 und a 2 gleiches oder ent
gegengesetztes Vorzeichen besitzen. Das general-TRIPLE-Experi
ment führt entweder zu einer Pyramide oder zu einem Tetraeder
in Abhängigkeit von den relativen Vorzeichen der Kopplungs
konstanten und von der Wahl der "Pump"frequenz.
Das special-TRIPLE-Experiment /10/, bei dem gleichzeitig
sowohl der hoch- wie der tieffrequente NMR-Ubergang eines
Kernes eingestrahlt wird, ergibt ein Quadrat.
Da sich der TRIPLE-Effekt in der Intensität der ENDOR-Li
nien manifestiert, wird er durch das Verhältnis V der TRIPLE
-Signalintensität zu der ENDOR-Signalintensität beschrieben.
ENDOR
,-r-------:;'1 l l
I I I l
----} I / I ,
___ 1.,""
TRIPLE !general)
- 20 -
n::lr VI--W NMR
TRIPLE (generutl
r-------.,. /I /I
I 1:_ _J ___ ~.... : I I I I I l I I I
l , }---+-;I
1/ t., "-- _____ _J,
TRIPLE (special)
Abb. 2: Geometrische Darstellung der Energieniveaus und der
Relaxationswege für ein System mit S = r1
Aus der Lösung des Netzwerkproblems wird V für die verschiede
nen geometrischen Figuren erhalten /9/:
V Pyramide
VTetraeder ~
V Quadrat > V Tetraeder

- 21 -
Die Verringerung der TRIPLE- gegenüber der ENDOR-Linien
intensität erfolgt also bei einem Pyramiden-Experiment, beim
Tetraeder-Experiment wird das TRIPLE-Signal dagegen vergrößert.
Damit ergeben sich für die Vorzeichenbestimmung folgendeRegeln:
Wird der Tieffrequenz-NMR-Ubergang v; eines Kerns gepumpt, er
hält man, wenn beide Kopplungskonstanten gleiches Vorzeichen
besitzen, für den Tieffrequenz-NMR-Ubergang v 1 des anderen
Kerns ein Pyramiden-Experiment, also eine Verringerung der +
Signalintensität. Der Hochfrequenz-NMR-Uhergang v 1 dieses
Kerns führt dagegen zu einem Tetraeder, also einer Vergröße
rung der Linie. Analog dazu wird bei der sättigenden Einstrah
lung eines Hochfrequenz-NMR-Uhergangs v; das Hochfrequenz-NMR
-Signal des anderen Kerns verkleiner't, dessen Tieffrequenz-
-NMR-Signal aber vergrößert, wenn die Vorzeichen der Kopp-
lungskonstanten gleich sind.
Haben die Kopplungskonstanten ungleiches Vorzeichen, ver
halten sich die Linienintensitäten entgegengesetzt der obigen
Beschreibung. Für ein special-TRIPLE-Experiment wird stets
eine Intensitätserhöhung der Signale erhalten.
3.2. Molekül-Orbital-Modelle
Durch die in Kapitel 3.1. beschriebenen Methoden können
Größe und Vorzeichen der Kopplungskonstanten a bestimmt wer
den. Diese Kopplungskonstante, hervorgerufen durch Wechsel
wirkung zwischen Kern- und Elektronenspin, ist ein Maß für
die Spindichte an dem betreffenden Atom. Die Kenntnis von
a gestattet Rückschlüsse auf die Elektronenspinverteilung
innerhalb des betrachteten Radikals unter Zugrundelegung ge
eigneter Beziehungen zwischen ai und der Spindichte . Damit
ist eine Oberprüfung theoretisch gefundener Spindichtever
teilungen möglich.
Eine Reihe von Verfahren zur Berechung der Spinverteilung
in rr-Radikalen ist bekannt. In diesem Kapitel sollen einige
MO-Modelle qualitativ beschrieben werden, soweit sie im Zu
sammenhang mit dieser Arbeit von Bedeutung sind.

- 22 -
3.2.1. HMO-Modell
Das einfachste Modell zur Beschreibung von n-Systemen ist
das Hückel-Modell /11/, das von der Unabhängigkeit der n-Elek
tronen von den anderen Elektronen des Moleküls und der n-Elek
tronen untereinander ausgeht. Die Molekülwellenfunktion 'i wird
durch einen LCAO-Ansatz (~inear ~ombination of ~tomic Qrbitals)
näherungsweise beschrieben.
( 1 7)
Damit werden aus 2m Atomorbitalen .p 2m Molekülorbitale 'i
gebildet, wobei die Koeffizienten ci~ so gewählt werden, daß
sich eine minimale Energie des Moleküls ergibt.
Durch Lösen der Schrödinger-Gleichung
E ' ( 18)
werden die Energiewerte
( 19)
erhalten, wobei a., ein Coulombintegral eines AOs (j) ~, und ß,
ein Resonanzintegral, für alle C-Atome konstant sind; xi ist
der Hückel'sche Energiekoeffizient. Die Spindichte ist durch
das Quadrat der Eigenfunktion (genauer ''*l des einfach be
setzten Orbitals 'j gegeben. Aufgrund der Normierung von '
und der Orthonorrnierung von (j) ergibt sich
(20)
wobei die Quadrate der AC-Koeffizienten den n-Spinpopulationen
an den Zentren ~ entsprechen.
Demnach sollten nur positive Spinpopulationen erhalten
werden.
( 21 )

- 23 -
3.2.2. Verfeinerte MO-Modelle
Schon für planare Kohlenwasserstoffe zeigt das HMO-Modell
bereits einige Schwächen:
1. Der Ausschluß der Wechselwirkung zwischen n- und o-Orbi
talen steht im Gegensatz zu der experimentell nachgewie
senen ungepaarten Spindichte am Ort der Protonen.
2. Es werden nur positive Spinpopulationen vorhergesagt;
experimentell werden aber auch negative Spinpopulatio
nen ermittelt und zwar an solchen Positionen, für die
das Quadrat der AO-Koeffizienten nach HMO-Rechnungen
nahezu Null oder Null ist.
Zur Lösung der Widersprüche wird das Auftreten von Polari
sationsmechanismen angenommen.
Am Ort der a-Protonen, sie sind direkt an ein zum n-System
gehöriges C-Atom gebunden, wird durch n-o-Spinpolarisation ein
endlicher Wert ungepaarter Spindichte erzeugt. Hält sich eines
der bindenden o-Elektronen nahe dem Kohlenstoffatom, das andere
nahe dem Proton auf, so sind zwei Spinanordnungen möglich (Abb.
3). Nach der Hund'schen Regel wird der Zustand höherer Multi
plizität (I) gegenüber dem niedriger Multiplizität (II) ener
getisch bevorzugt. Damit wird am Ort des Protons eine Spindich
te PH proportional zur Spindichte oc am Nachbarkohlenstoffatom Tt
I n
~bb. 3: Schematische Darstellung der n-o-Spinpolarisation

- 24 -
mit entgegengesetztem Vorzeichen induziert. Der daraus resul
tierende Protonenkopplungsparameter aH ist somit ebenfalls die
ser Spindichte o~ proportional. Dieser Zusammenhang wird in der
empirisch gefundenen McConnell-Beziehung
(22)
dargestellt. Damit ist es möglich, aus den experimentell er
haltenen Kopplungskonstanten aH auf die Spindichte des Nach
barkohlenstoffatoms zurückzuschließen. Die theoretisch nach
verschiedenen Methoden ermittelten Werte von Q variieren von
-2.3 bis -2.8 mT, das negative Vorzeichen von Q entspricht
dem entgegengesetzten Vorzeichen von aH gegenüber P~. Das Auftreten negativer Spindichten läßt sich durch n-n
-Spinpolarisation analog zur n-o-Spinpolarisation erklären.
In Abbildung 4 wird ein Ausschnitt eines n-Systems gezeigt,
dessen ungepaartes Elektron das MO ~j besetzt. Die Aufent
haltswahrscheinlichkeit dieses Elektrons sei groß am C-Atom 1
und verschwindend klein am C-Atom 2. Auch hier tritt eine Be
vorzugung des Spinzustandes höherer Multiplizität (I) auf.
Am Zentrum hoher n-Spindichte wird diese durch positive Bei
träge erhöht, am Zentrum verschwindender Spindichte erfolgt
eine Erniedrigung durch negative Beiträge, was zu negativen
Spindichten führen kann.
I II
Abb.~ Schematische Darstellung der n-n-Spinpolarisation

- 25 -
Nach verfeinerten Rechenverfahren, z.B. dem McLachlan-Ver
fahren /12/, das der n-n-Spinpolarisation Rechnung trägt, wer
den für weitgehend planare n-Systeme Spinpopulationen erhalten,
die in guter Ubereinstimmung mit experi~entellen Ergebnissen
sind. Für die nach dem McLachlan-Verfahren berechnete Spin
dichte ~ ergibt sich nach Gleichung 23 ein Wert, der sich von 2 dem nach dem HMO-Modell erhaltenen Wert (cj~) um den Beitrag
durch Polarisation unterscheidet.
p~ 11
2 11 c. ~V JV
(23)
Während die beiden genannten Verfahren nur die Wechselwir
kungen der pz-Orbitale betrachten, werden bei den all-valence
-electron-Verfahren auch die anderen Atomorbitale mit der Be
schränkung auf die Valenzschale berücksichtigt. Als Beispiele
seien die Verfahren CNDO (complete neglect of differential
overlap) und INDO (intermediate neglect of differential over
lap) erwähnt /13/. Sie stellen semiempirische Verfahren dar,
wobei durch die teilweise Berücksichtigung von Uberlappungen
und interelektronischen Wechselwirkungen das INDO-Verfahren
oft zu befriedigenden Ubereinstimmungen mit experimentellen
Ergebnissen führt. Die Durchführung von "ab initio"-Rechnun
gen gelingt nur an kleinen Modellverbindungen, bei polyzentri
schen Molekülen ist sie bisher noch nicht möglich.
3.2.3. Ladungsabhängigkeit der Kopplungskonstanten
Nach dem HMO-Modell sollten sich für Radikalanionen und Radi
kalkationen alternierender Systeme gleiche Spinpopulationen erge
ben, da für diese Systeme das "pairing theorem" gilt. Die einfach
besetzten HMOs alternierender Kohlenwasserstoffe lauten folgender
maßen.
Kation lji m
f.i
c miJ, q,IJ. (24)
'i'm+ 1 [ c ;t (25)
f.i m+11J. f.i
Anion

- 26 -
wobei c ! cm+ 1" ist. Damit sind die Quadrate der Koeffizien-2 m~.t ..
ten cju für Kation und Anion gleich. Bei nicht alternierenden
Systemen werden für Radikalanionen bzw. Radikalkationen unter
schiedliche Spindichteverteilungen erwartet und auch experimen
tell bestätigt, da hier das "pairing theorem" keine Gültigkeit
hat.
Auch verfeinerte MO-Modelle wie z.B. das McLachlan Verfah
ren liefern gleiche Spindichteverteilungen für Radikalanionen
und -kationen alternierender Systeme. Experimentell wurden aber
auch bei solchen Systemen Änderungen der Kopplungskonstanten
beim Ubergang vom Radikalanion zum -kation gefunden. Um diese
Ladungsabhängigkeit der Kopplungskonstanten zu berücksichtigen,
schlugen verschiedene Autoren eine M6difizierung der McConnell
-Beziehung vor. Zum einen wurde von Colpa und Bolton /14/ eine
Beziehung zwischen dem Kopplungsparameter a und der Spinpopu
lation p entwickelt, die eine Oberschußladung s berücksichtigt:
H a. ~
(26)
Zum anderen wurde von Giacometti, Nordio und Pavan /15/ un
ter Einführung eines Korrekturterms durch Störungsrechnung eine
ähnliche Beziehung erhalten:
(27)
Die in Gleichung 26 und Gleichung 27 auftauchenden Parameter
Q und K bzw. Q1 und o2 müssen empirisch bestimmt werden. Da in
beide Gleichungen unbekannte Größen (E bzw. c) eingehen, können
diese Beziehungen nicht zur Ermittlung der Spinpopulation be
nutzt werden.
3.2.4. HFS durch ß-Protonen
Der freie s-Anteil der Elektronen am Ort der a-Protonen
kommt, wie oben beschrieben, durch rr-o-Spinpolarisation zu
stande. Die am Ort der ß-Protonen nachgewiesene Spindichte
kann nicht durch Spinpolarisation alleine hervorgerufen wer
den. Deren Beitrag reicht nicht aus, um die großen ß-Proto-
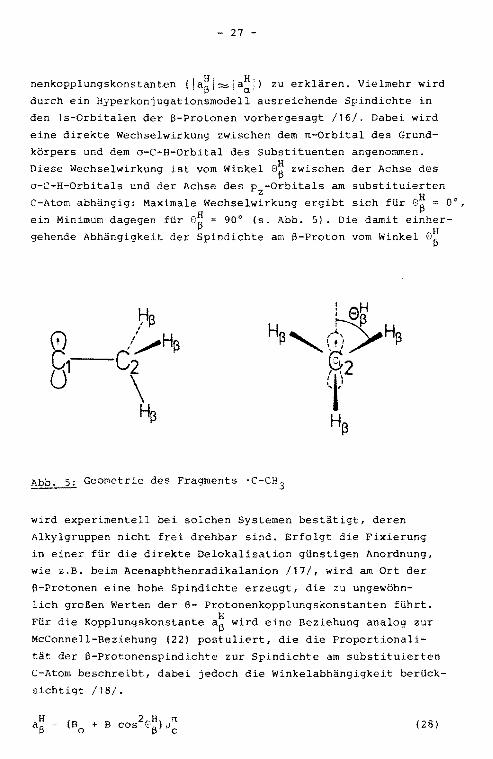
- 27 -
nenkopplungskonstanten (ja~l~la~!) zu erklären. Vielmehr wird
durch ein Hyperkonjugationsmodell ausreichende Spindichte in
den 1s-Orbitalen der 8-Protonen vorhergesagt /16/. Dabei wird
eine direkte Wechselwirkung zwischen dem n-Orbital des Grund
körpers und dem a-C-H-Orbital des Substituenten angenommen.
Diese Wechselwirkung ist vorn Winkel 8~ zwischen der Achse des
o-C-H-Orbitals und der Achse des pz-Orbitals arn substituierten
C-Atorn abhängig: Maximale Wechselwirkung ergibt sich für 8~ = 0°,
ein Minimum dagegen für G~ = 90° (s. Abb. 5). Die damit einher
gehende Abhängigkeit der Spindichte arn 8-Proton vorn Winkel 8~
Geometrie des Fragments ·C-CH3
wird experimentell bei solchen Systemen bestätigt, deren
Alkylgruppen nicht frei drehbar sind. Erfolgt die Fixierung
in einer für die direkte Delokalisation günstigen Anordnung,
wie z.B. beim Acenaphthenradikalanion /17/, wird am Ort der
ß-Protonen eine hohe Spindichte erzeugt, die zu ungewöhn-
lich großen Werten der ß- Protonenkopplungskonstanten führt.
Für die Kopplungskonstante a~ wird eine Beziehung analog zur
McConnell-Beziehung (22) postuliert, die die Proportionali
tät der ß-Protonenspindichte zur Spindichte am substituierten
C-Atom beschreibt, dabei jedoch die Winkelabhängigkeit berück
sichtigt /18/.
(28)

- 28 -
Die Kopplungskonstante und dle Spindichte des ß-Protons wei
sen dasselbe Vorzeichen auf wie die Spinpopulation am a-C-Atom.
Die positive Konstante B ergibt sich aus experimentellen Daten
zu 5.0 - 6.0 mT. Die experimentelle Bestimmung von B0
/19/ führt
zu kleinen Werten von 0.3 - 0.4 mT, so daß B0
gegenüber B im
allgemeinen zu vernachlässigen ist. Damit vereinfacht sich Glei
chung 28 zu
(29)
Für frei rotierende Methylgruppen, wie sie in flüssiger Lö
sung auftreten, ist der Mittelwert der Winkelfunktion zu bil
den. Damit lautet Gleichung 25
(30)
H Die experimentelle Ermittlung der Kopplungskonstanten aß
gestattet also nach Gleichung 28 bzw. Gleichung 29 und Glei
chung 30 einen Rückschluß auf die Spinverteilung des Systems
zu ziehen.
3.2.5. "Long-Range" - Hyperfeinwechselwirkunge~
Auch durch Kerne, die durch drei oder mehr o-Bindungen vom
n-C-Atom getrennt sind, also y-, 5- und E-Protonen, wird e:
ne Hyperfeinwechselwirkung beobachtet. Auch hier gilt es, den
wirksamsten der zwei grundsät,zlich möglichen Spintransferme
chanismen, die Spinpolarisation bzw. die Spindelokalisation,
zu ermitteln. Zahlreiche Arbeiten beschäftigen sich mit dem
Spintransfer zu y-Protonen. Als Beispiele sollen hier die
Untersuchungen an Radikalen von Semidionen und Semichinanen
/20/, an Nitroxid-Radikalen /21,22/ und an Alkylradikalen
/23, 24/ erwähnt werden. Obereinstimmend wird in den oben
genannten Publikationen eine starke Abhängigkeit der y-Pro
tonenkopplungskonstante von der sterischen Anordnung des
Systems festgestellt. Den y-Protonen, die durch eipe "Zick
-Zack"-Kette von o-B1ndungen mit dem spintragenden n:-Orbital
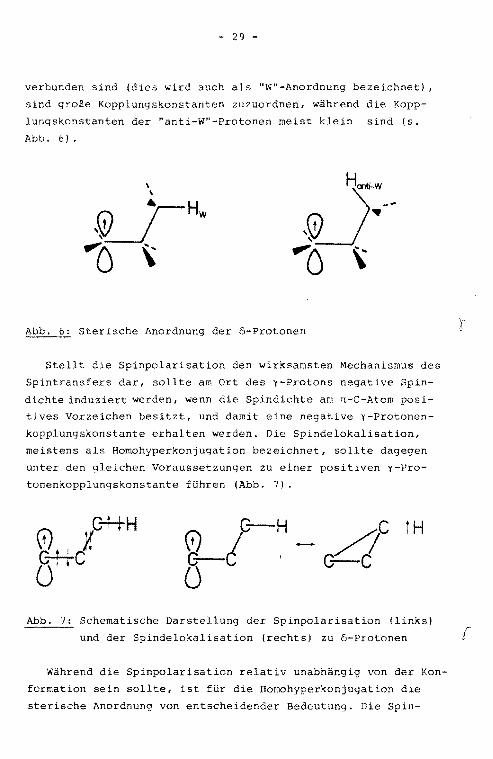
- 29 -
verbunden sind (dies wird auch als "W"-Anordnung bezeichnet) ,
sind große Kopplungskonstanten zuzuordnen, während die Kopp
lungskonstanten der "anti-W"-Protonen meist klein sind (s.
Abb. 6).
Sterische Anordnung der 5-Protonen
Stellt die Spinpolarisation den wirksamsten Mechanismus des
Spintransfers dar, sollte am Ort des y-Protons negative Spin
dichte induziert werden, wenn die Spindichte am n-C-Atom posi
tives Vorzeichen besitzt, und damit eine negative y-Protonen
kopplungskonstante erhalten werden. Die Spindelokalisation,
meistens als Homohyperkonjugation bezeichnet, sollte dagegen
unter den gleichen Voraussetzungen zu einer positiven y-Pro
tonenkopplungskonstante führen (Abb. 7).
r.\ C-H-H \T/ X csrrc
\t) 1c-H ~ /)C G-e , r?--c 0
!H
. 7: Schematische Darstellung der Spinpolarisation (links)
und der Spindelokalisation (rechts) zu 5-Protonen
Während die Spinpolarisation relativ unabhängig von der Kon
formation sein sollte, ist für die Homohyperkonjugation die
sterische Anordnung von entscheidender Bedeutung. Die Spin-
r

- 30 -
dichte der ~-Protonen, die sich bezüglich des -Orbitals in der
"W"-Anordnung befinden, wird deshalb durch einen großen, positi
ven Beitrag durch Homohyperkonjugation erhöht werden. Diese di
rekte Spindelokalisation wird für die "Anti-W"-y-Protonen zu
vernachlässigen sein.
Für einige Radikale, insbesondere für n-Propyl- bzw. n-Butyl
radikale, wurde die Spinverteilung nach verschiedenen Methoden
berechnet. Dabei liefern Rechnungen nach dem INDO-Verfahren
/25/ und auch mit der VB-Methode /26/ sowohl positive als auch
negative Werte für die y-Protonenkopplungskonstanten in Abhän
gigkeit von der geometrischen Anordnung. Bei beiden Verfahren
werden die Werte für aHY unter Variation der Winkel 9c bzw. eH ß y ermittelt (s. Abb. 8).
Abb. 8: Geometrie des Fragments ·C-CH2 -cH 3
Bei einer ekliptischen
des pz-Orbitals (6~ 0")
Anordnung der Methylgruppe bezüglich
wird für 9H 180° entsprechend der y
großer, positiver Wert für aH erhalY
"W"-Anordnung
ten; für eH y
gibt sich für
ein maximal
o•, was einer "anti-W"-Anordnung entspricht, er
aH ein negatives Vorzeichen. Die durch Anwendung y
des "ab inltid'-Verfahrens /27, 28/ erhaltenen Kopplungskonstan-
ten unterscheiden sich teilweise schon im Vorzeichen von den
Wen:en aus den anderen Modellrechnungen. Bei den genannten
Verfahren wird der Spintransfer als eine Kombination der bei
den oben aufgeführten Mechanismen aufgefaßt. Eine Entscheidung,
welches Rechenverfahren die beste Obereinstimmung mit den ex-

- 31 -
perimentellen Werten liefert, konnte nicht getroffen werden,
da nur in wenigen Fällen eine Vorzeichenbestimmung der Kopp
lungskonstanten durchgeführt wurde.
Einige Arbeiten behandeln auch den Spintransfer zu 6-Pro
tonen. Dazu wurden beispielsweise EPR-Untersuchungen an Radi
kalen bizyklischer Semidione /29/ und an Radikalen verschie
dener Ameisensäurealkylester /30/ sowie NMR-contact-shift-Mes
sungen an einer Reihe von Aminradikalen /31/ durchgeführt.
Auch hier wird analog zu den y-Protonen eine hohe Spindichte
am 6-Proton ermittelt, wenn sich dieses in einer koplanaren
"Zick-Zack"-Anordnung bezüglich des spintragenden -Orbitals
des a-C-Atoms befindet. Diese Konformation begünstigt wiederum
einen Konjugationsmechanismus und sollte also zu hohen, posi
tiven Spindichten am Ort der betreffenden Protonen führen. Die
Spinpolarisation über vier O-Bindungen führt ebenfalls zu ei
ner positiven Spindichte am Ort der 6-Protonen. Die Effizienz
dieses Spintransfermechanismus wird aber bei zunehmender Ent
fernung vom n-System abnehmen und deshalb nur kleine Beiträge
zur Spindichte liefern. Für 6-Protonen, die nicht in der "Zick
-Zack"-Konformation vorliegen, sollten deswegen kleine, posi
tive Kopplungskonstanten erhalten werden. Die Durchführung
von Modellrechnungen (INDO, EH-SCF) führte in Einstimmung
mit diesen Uberlegungen zu großen, positiven Spindichten
für 6-Protonen in der "Zick-Zack"-Anordnung, für die ande-
ren 6-Protonen wurden wesentlich kleinere Spindichten er
halten.

- 32 -
3.2.6. Phenylhyperkonjugation
Bei phenylsubstituierten aromatischen n-Systemen, die eine
starke Verdrillung des Substituenten aufgrund sterischer Hin
derung durch das peri-ständige H-Atom erwarten lassen, werden
neben der erwähnten Abweichung von der Stone'schen Geraden
(vgl. Kap. 3.1.1.) für die meta-Protonenkopplungskonstanten
unerwartet große Werte erhalten. Beide Anomalien sind durch
Anwendung einfacher MO-Modelle nicht erklärbar.
Für ein verdrilltes n-System, bei dem sich der Phenylsub
stituent an einem Zentrum positiver Spindichte des Grundkör
pers befindet und um den Winkel 0 gegenüber der Ebene des
Grundkörpers verdreht ist (Abb. 9), erfolgt eine Delokalisa
tion von positiver Spindichte in den Phenylring. An den ortho-
Abb. 9: Geometrie eines phenylsubstituierten Aromaten
und para-Kohlenstoffatomen sollten so relativ große, positive
Spinpopulationen erhalten werden, die aufgrund der n-o-Spil'
polarisation zu großen ortho- und para-Protonenkopplungskon
stanten mit negativem Vorzeichen führen sollten. Nach ~!cLachlan
werden für die meta-Kohlenstoffatome kleine, negative Spinpopu
lationen erwartet, d.h. für die meta-Protonenkopplungskonstante
wird ein kleiner, positiver Wert vorhergesagt. Die verminderte

- 33
Delokalisation bei Zunahme des Verdrillungswinkels soll zu ei
ner Verkleinerung der Beträge aller Phenylprotonenkopplungs
konstanten annähernd proportional zu cos 2e führen. Die kon
ventionelle Reihenfolge- la~aral > la~rthol >> ia~etalwird de~nach nicht geändert. Experimentell werden aber bei
nicht ebenen n-Systemen große, positive Werte für die Kopp
lungskonstanten der meta-Protonen gefunden. Bei starker Ver
drillung des Phenylsubstituenten kann der Betrag von aH t H H me a
gleich dem von a th und a werden oder er kann sogar or o para größer werden als jener/3, 4/.
Eine zusätzliche n-o-Delokalisation wird nach dem Phenyl
hyperkonjugationsmodell berücksichtigt, das in Anlehnung an
das Methylhyperkonjugationsmodell entwickelt wurde /32/. Nach
diesem Modell werden durch Linearkombination der Orbitale des II II
o-Systems des Phenylsubstituenten (C (2px , 2py , ) ,
H (1s 11) ) - pseudo-p -Orbitale solcher Symmetrie konstruiert, z
daß eine Konjugation mit dem n-System des Grundkörpers möglich
wird. Diese direkte Wechselwirkung (=Hyperkonjugation) ermög
licht eine direkte Delokalisation von positiver Spindichte aus
dem n-System des Grundkörpers in das 1s-Orbital des meta-Pro
tons. Mit zunehmender Verdrillung des Substituenten wird eine
Zunahme der meta-Protonenkopplungskonstanten proportional zu
0 vorhergesagt /4, 33/. Die Proportionalitätskonstante k
sollte eine Ladungsabhängigkeit in der Weise zeigen, daß eine
Vergrößerung beim Ubergang vom Radikalanion zum Radikalkation
auftritt. Die para-Protonenkopplungskonstante sollte aus Sym
metriegründen von der n-o-Delokalisation unbeeinflußt blei
ben, ebenso wird für die ortho-Protonenkopplungskonstante kei
ne Änderung erwartet. Für die Beträge dieser beiden Kopplungen
werden Abnahmen proportional zu cos 20 bei Zunahme von 0 vor
hergesagt. Die Berechnung der meta-Protonenkopplungskonstan
ten liefert Werte, die im Vergleich zu den experimentellen
Werten wesentlich zu groß sind. Dies deutet auf eine Oberbe
wertung der n-o-Delokalisation in diesem Modell hin.
Ähnliche Ergebnisse wie das Phenylhyperkonjugationsmodell
werden durch INDO-all-Valenzelektronenrechnungen erhalten
/33, 34/, die an Molekülfragmenten durchgeführt wurden. An
schaulich können die für verschiedene Winkel 0 berechneten
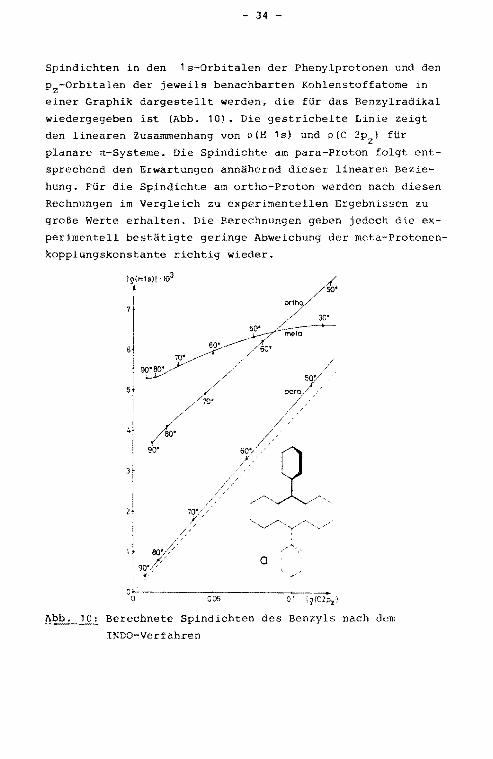
- 34 -
Spindichten in den 1s-Orbitalen der Phenylprotonen und den
p2-0rbitalen der jeweils benachbarten Kohlenstoffatome in
einer Graphik dargestellt werden, die für das Benzylradikal
wiedergegeben ist (Abb. 10). Die gestrichelte Linie zeigt
den linearen Zus~menhang von p(H 1s) und p(C 2p2
) für
planare n-Systeme. Die Spindichte am para-Proton folgt ent
sprechend den Erwartungen annähernd dieser linearen Bezie
hung. Für die Spindichte am ortho-Proton werden nach diesen
Rechnungen im Vergleich zu experimentellen Ergebnissen zu
große Werte erhalten. Die Berechnungen geben jedoch die ex
perimentell bestätigte geringe Abweichung der meta-Protonen
kopplungskonstante richtig wieder.
005
Abb. 10: Berechnete Spindichten des Benzyls nach dem
INDO-Verfahren

- 3S
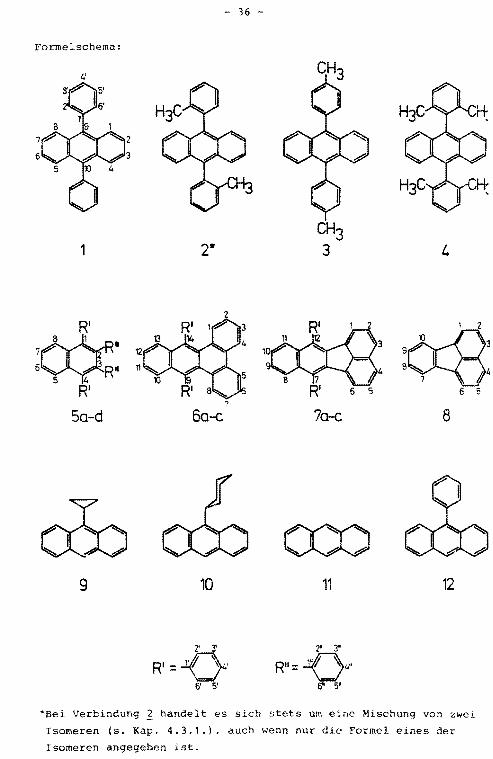
4.1. Namen und Struktur der untersuchten Verbindungen
Rationale Namen der untersuchten Verbindungen
9,10-Diphenylanthracen
2 9,10-Bis(2-methylphenyl)anthracen
3 9,10-Bis(4-methylphenyl)anthracen
4 9,10-Bis(2,6-dimethylphenyl)anthracen
1,2,3,4-Tetraphenylnaphthalin
Sb 2,3-Diphenyl-1,4-bis([Dslphenyl)naphthalin
Sc 1,4-Diphenyl-2,3-bis([Dslphenyl)naphthalin
Sd 1,4-Bis([3,S-D2 Jphenyl)-2,3-bis([DslPhenyl)naphthalin
6a 9,14-Diphenylbenzo[b]triphenylen
6b 9,14-Bis([Dslphenyl)benzo[b)triphenylen
6c 9,14-Diphenyl-[10,11,12,13-D4 Jbenzo[b]triphenylen
7a 7,12-Diphenylbenzo[k]fluoranthen
7b 7,12-Bis([Dslphenyl)benzo[k]fluoranthen
7,12-Diphenyl-[8,9,10,11-D4 ]-benzo[k]fluoranthen
8 Fluoranthen
9 9-Cyclopropylanthracen
10 9-Cyclohexylanthracen
11 Anthracen
12 9-Phenylanthracen
Deuterierte Positionen der Verbindungen 5 - 7
Verbindung
Sa
Sb
Sc
Sd
6a
6b
6c
7a
7b
7c
R'
2'-6'
3., 5.
2'-6'
2'-6'
R''
2 1 '-6 f I
2 O O -6 I O
10-13
8-11
- 36 -
Formelschema:
4'
1 2*
2 1 2 R'
~' 'QQr Q:g' 12 1 9 •
s Rn 4 e J ~
5 4 R• R' s 5 6 5
5a-d 6a-c 7a-c 8
9 10 11 12
*Bei Verbindung 2 handelt es sich stets um eine Mischung von zwei
Isomeren (s. Kap. 4.3.1.), auch wenn nur die Formel eines der
Isomeren angegeben ist.
37 -
4.2. Synthesen
Die Darstellung der substituierten, aromatischen Systeme
wurde auf zwei grundsätzlich verschiedenen Wegen vorgeno~~en:
1. Die Einführung der Substituenten in Position 9 bzw. 9
und 10 des Anthracengrundkörpers erfolgte durch Umset
zung der entsprechenden Carbonylverbindung, also Anthron
bzw. Anthrachinon, mit einer metallorganischen Komponente.
2. Bei der Synthese der übrigen Verbindungen wurde das
anellierte System erst im letzten Reaktionsschritt ge
bildet.
Zur Darstellung der disubstituierten Anthracene 1 bis 4
wurde also Anthrachinon mit dem entsprechenden Aryllithium
umgesetzt. Anschließend wurden die nach Hydrolyse entstande
nen Biscarbinole mit Jodwasserstoff in Eisessig zu den Koh
lenwasserstoffen reduziert /35/. Die Verbindungen l bis l• ebenso wie lf, wurden in der dieser Arbeit vorangegangenen
Diplomarbeit synthetisiert.
Hl
1. Syntheseweg (Variante zur Darstellung disubstituierter Anthracene)
Die Darstellung der Verbindungen ~ bis 2 erfolgte auf dem
zweiten der oben angeführten Reaktionswege. Die Verbindungen
5a - ~ wurden durch Umsetzung des entsprechenden Tetraphenyl
cyclopentadienons mit Dehydrobenzol, erhalten durch thermische
Zersetzung von Diphenyliodonium-o-carboxylat in Triglyme syn
thetisiert /36/. Die teildeuterierten Tetraphenylcyclopenta
dienone wurden durch Kondensation von Benzil mit Dibenzylketon
in Triethylenglykol unter Basenzusatz erhalten /37, 38/. Da
bei wurde perdeuteriertes Dibenzylketon (5b), perdeuteriertes
Benzil (Sc) bzw. perdeuteriertes Benzil und teildeuteriertes
Dibenzylketon (5d) verwendet. Die perdeuterierten Ausgangs
verbindungen wurden in früheren Arbeiten in der Arbeitsgruppe
- 38
0
2. Syntheseweg

- 39 -
H. Kurreck, von K. Hinr ichs und U. Mennenga, dargestellt. Zur Synthese
von [D 4 ]Dibenzylketon wurde p-Toluidin selektiv deuteriert /39/,
aus dem entstandenen 4-Amino-[3,5-D 2 ]toluol wurde durch Diazotie
rung und anschließende Reduktion mit hypophosphoriger Säure
[3,5-D2 ]Toluol erhalten /40/. Dieses wurde zum Benzylbromid
bromiert, mit Natriumcyanid zum Benzylcyanid umgesetzt und
daraus durch Hydrolyse [3,5-D2 ]Phenylessigsäure gewonnen.
Diese wurde mit Calciumhydroxid in ihr Calciumsalz überge-
führt, aus dem durch Destillation das [D4 JDibenzylketon er
halten wurde /41/.
Die Verbindungen ~ und 7a, ~wurden analog zu d
durch Umsetzung von Dehydrobenzol mit 1,3-Diphenyl-2H-cyclopenta
[l]phenanthren-2-on (Phencyclon) bzw. 7,9-Diphenyl-SH-cyclopenta
[a]acenaphthylen-8-on (Acenaphthencyclon) dargestellt, die wie
derum durch Kondensation von Dibenzylketon mit 9,10-Phenanthren
chinon bzw. 1,2-Acenaphthenchinon erhalten wurden /37/. Zur
Synthese von 6b und wurden mit perdeuteriertem Dibenzylketon
hergestellte Cyclone eingesetzt. Die Darstellung von und 7c
erfolgte mit Dehydrobenzol aus diazotierter, perdeuterierter
Anthranilsäure /36/. Zur Deuterierung der Anthranilsäure wur-
de ihr Kaliumsalz einer Austauschreaktion durch unter Zu-
satzeines Pt-Katalysators unterworfen /42/.
Die 9-Cycloalkylanthracene 2 und lQ wurden durch Umsetzung
von Anthron mit der Cycloalkyl-Grignard-Verbindung hergestellt.
Nach anschließender Hydrolyse mit verdünnter Salzsäure wurde
direkt der Kohlenwasserstoff erhalten /43/.
~ ~
H H
R-Mg-Br .. od. R-Li
1. Syntheseweg (Variante zur Darstellung rr,onosubstituierter Anthracene)
Im Falle von Verbindung lQ entstand ein Gemisch aus 95%
Cyclohexylanthracen und 5% Anthracen, das vor der EPR- und
ENDOR-Untersuchung getrennt werden mußte. Eine Synthese von
9-Cyclobutyl- und 9-Cyclopentylanthracen ist auf diesem Wege

40 -
nicht möglich. Eine analoge Umsetzung von Cyclobutyl- bzw.
Cyclopentylmagnesiumbromid mit Anthron führte zur Reduktion
von Anthron zum Anthracen. Ebenso scheiterten die Versuche
zur Darstellung der 9,10-Dicycloalkylanthracene. Bei dem Ver
such, Anthrachinon mit dem entsprechenden Cycloalkyllithium
analog zur Darstellung der 9,10-Diarylanthracene umzusetzen,
wurde nahezu quantitativ unumgesetztes Anthrachinon erhalten.
Auch der Ersatz der Lithiumalkyle durch Grignard-Verbindungen
führte nicht zum gewünschten Produkt. Auch hier wurde der
größte Teil des Anthrachinons nicht umgesetzt. Da vermutet
wurde, daß die schlechte Löslichkeit von Anthrachinon we
sentlich zu der beobachteten Reaktionsträgheit beiträgt,
wurde versucht, die Verwendung von Anthrachinon zu umgehen.
So wurde zum einen versucht, analog zur Darstellung von
9,10-Dimethylanthracen /44/, Anthron in das 10-Cycloalkyl
anthron zu überführen, das anschließend durch übliche
Grignard-Reaktion zum 9,10-Dicycloalkylanthracen umgesetzt
werden sollte. Hierbei entstand schon im ersten Reaktions
schritt ein Produktgemisch, aus dem das 10-Cycloalkylanthron
nicht isoliert werden konnte. Zum anderen sollte Oktahydro
anthrachinon, hergestellt durch Addition von Benzoehinan und
Butadien, durch Grignard-Reaktion in das Biscarbinol überge
führt werden, aus dem durch Dehydratisierung und Dehydrierung
das entsprechende 9,10-Dicycloalkylanthracen gebildet werden
sollte. Auch bei diesem Reaktionsweg fand keine Umsetzung der
Carbonylverbindung zum Biscarbinol statt.
Die Darstellung des 9-Phenylanthracens (U0 erfolgte durch
Umsetzung von Anthron mit Phenyllithium. Auch hier wurde der
Kohlenwasserstoff direkt nach der Hydrolyse gebildet.

- 41 -
4.3.1. NMR-Spektren der phenylsubstituierten Verbindungen
Mit Hilfe der NMR gelang es, bei einigen der dargestellten
Verbindungen den Verdrillungswinkel zu bestimmen, bei einer
anderen Verbindung konnte der Nachweis für das Vorliegen von
zwei Isomeren geführt werden. 13c-NMR-Untersuchungen am 9,10-Diphenylanthracen hatten
ergeben /4/, daß die Elektronendichteverteilung im Anthracen
grundgerüst durch die Einführung von Phenylsubstituenten keine
Änderung erfährt. Das 1H-NMR-Spektrum des 9,10-Diphenylanthracens
zeigt im Bereich der aromatischen Protonen neben dem Phenylpro
tonenmultiplett ein AA'XX'-System /45/, das durch die Protonen
in Position 1 und 2 des Anthracengrundkörpers hervorgerufen
wird. Im Vergleich zum unsubstituierten Anthracen zeigen die-
se Protonen eine Hochfeldverschiebung, deren Ursache in der
Anisotropie des Ringstromes der Phenylringe liegt. Unter die-
ser Voraussetzung wurde unter Verwendung von Ringstromdaten
und Ergebnissen von Röntgenstrukturanalysen die Abhängigkeit
des Verdrillungswinkels 0 von der relativen chemischen Ver
schiebung 6 des Protons im Bezug zum unsubstituierten Grund
körper dargestellt /4/, die in Abb. 11 wiedergegeben ist. Die-
se Beziehung ist ebenso auf andere kondensierte aromatische
Systeme, zum Beispiel Naphthalin, anwendbar.
6 ppm
•0.!1
8
-0.5
Abb. 11: Abhängigkeit der relativen chemischen Verschiebung des
Protons 1 vom Verdrillungswinkel
* Aufnahme der NMR-Spektren: K. Roth, M. Brauer, G. Dreke
- 42 -
1 Die 'H-NMR-Spektren der Verbindungen 1 - 7 weisen im Be-
reich der aromatischen Protonen jeweils ein AA'XX'-System auf.
Eine Ausnahme stellt Verbindung ~ mit zwei solchen Systemen
dar. Durch die Bestimmung von vA v (H-1) aus diesen Spektren
kann in einigen Fällen der Verdrillungswinkel der Phenylringe
ermittelt werden. Der Verdrillungswinkel bei Verbindung 5 be
zieht sich auf die Phenylringe in 1,4-Position. Bei den Ver
bindungen ~ und i werden durch die ortho-ständigen Methylgrup
pen zusätzliche Einflüsse auf Proton 1 wirksam, was zur Ver
stärkung der Hochfeldverschiebung führt und somit keine Aussa
ge über den Verdrillungswinkel zuläßt. Die relativen chemi
schen Verschiebungen der Verbindungen 1 4 und die daraus
resultierenden Verdrillungswinkel sind in Tabelle 4 zusammen
gefaSt.
3
5a
Relative chemische Verschiebungen der Protonen an
Position 1 und Verdrillungswinkel
o(ppm)
0.24
0.23
0.18
8
Das 1H-NMR-Spektrum der Verbindung ~ besteht neben zweier
Singuletts bei 1.52 bzw. 1.94 ppm, hervorgerufen durch inäqui
valente Methylgruppen, aus zwei AA'XX'-Systemen und weiteren,
nicht analysierten Aromatenrnultipletts (Abb. 12a) . Dies deutet
darauf hin, daß bei der Synthese des Di-o-tolylanthracens zwei
verschiedene Stereoisomere gebildet werden:
+
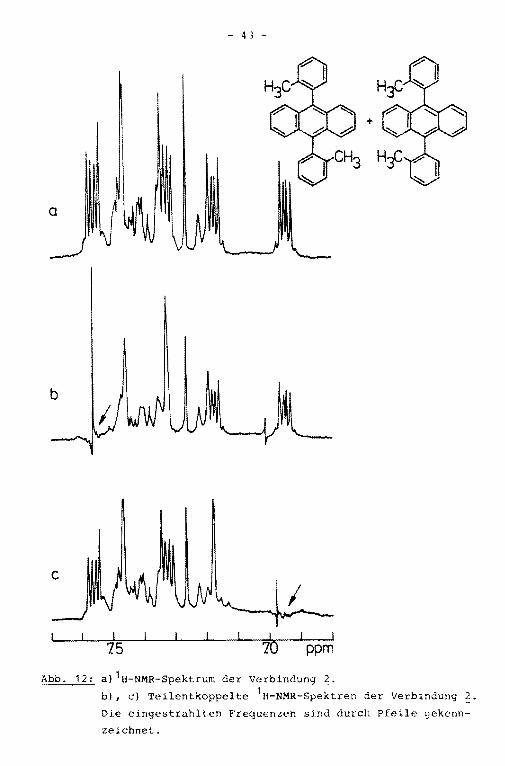
- 43 -
a
Abb. 12: a) 1H-NMR-Spektrum der Verbindung 2.
b), c) Teilentkoppelte 1H-NMR-Spektren der Verbindung~Die eingestrahlten Frequenzen sind durch Pfeile gekenn
zeichnet.

- 44 -
Aus den Integralen der entsprechenden Signale ist zu ent
nehmen, daß beide Isomere in annähernd gleichen Mengen gebil
det werden. Zum Nachweis, daß es sich um zwei verschiedene
Moleküle handelt, wurde die Doppelresonanztechnik angewendet.
Durch Einstrahlung von v (H-1) der einen Spezies wird ein
partiell entkoppeltes NMR-Spektrum erhalten (Abb. 12b), das
bei v (H-2) derselben Spezies nur noch einen einzelnen Peak
anstelle der durch Spin-Spin-Wechselwirkung hervorgerufenen
Liniengruppe zeigt. Das AA'XX'-System, der zweiten Spezies
bleibt dabei erhalten. Analog dazu vereinfacht sich bei Ein
strahlung von v (H-2) der zweiten Spezies der AA'-Teil, wäh
rend beide Liniengruppen der ersten Spezies unverändert
bleiben (Abb. 12c).
4.3.2. NMR-Spektren der 9-Cycloalkylanthracene
Die NMR-Spektren der Verbindungen 9 und (Abb. 13) lassen
sich jeweils in zwei Bereiche gliedern: in den Bereich der
Protonen am Aromaten und in den der Alkylprotonen. Beim
9-Cyclopropylanthracen werden im Aromatenbereich vier Si
gnale erhalten. Die Protonen in Position 1 und 8 bzw. 4 und 5 des Anthracengrundkörpers rufen die Dubletts bei
8.56 bzw. 7.80 ppm hervor, wobei die Verschiebung der Pro
tonen in Position 1 und 8 nach tieferem Feld durch die
Anisotropie des Cyclopropylringes hervorgerufen wird. Das
Proton in Position 10 führt zu einem Singulett bei 8.17 ppm,
das Multiplett bei 7.30 ppm ist den Protonen in den Posi
tionen 2, 3, 6 und 7 zuzuordnen. Das Intensitätsverhältnis
der einzelnen Signale stimmt mit der getroffenen Zuordnung
überein. Der Bereich der Alkylprotonen besteht aus drei Si
gnalen: einem Signal, entstanden aus der Oberlagerung zweier
Tripletts bei 2.37 ppm, das durch das ß-Proton hervorgerufen
wird und zwei Signalen höherer Multiplizität bei 1.34 b~w.
0.72 ppm, die von den beiden Paaren inäquivalenter y-Proto
nen verursacht werden. Das Intensitätsverhältnis beträqt
entsprechend dieser Zuordnung 1:2:2.
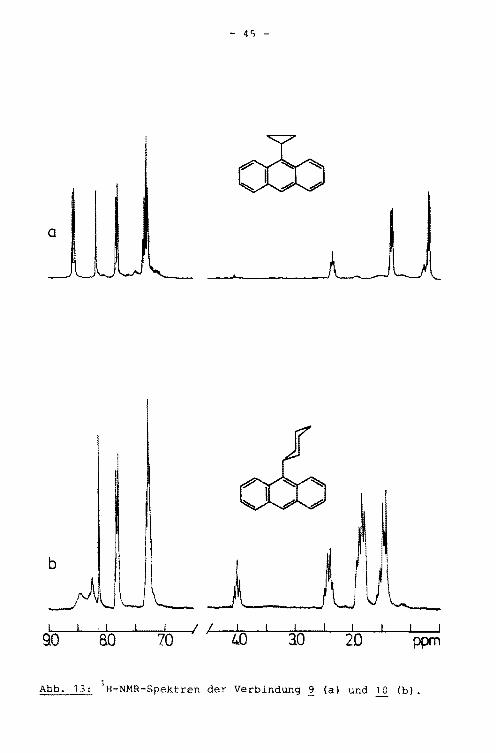
- 45 -
a
b UL . I I I t. I I I 80 70
j ' 40 30 20 ppm 90
Abb. 13: 1H-NMR-Spektren der Verbindung 2 (a) und 10 (b).

- 46 -
Das NMR-Spektrum des 9-Cyclohexylanthracens zeigt im Bereich
der aromatischen Protonen drei Signale. Die breiten Signale im
Tieffeldbereich sind auf Verunreinigungen der Probe zurückzu
führen. Neben dem Singulett von Proton 10 und einem Multiplett
der Protonen 2, 3, 6 und 7 bei 7.29 ppm, tritt bei 7.82 ppm
ein Signal auf, das durch Uberlagerung der beiden Dubletts
der Protonen 1 und 8 bzw. 4 und 5 entstanden ist. Aufgrund
der fehlenden Anisotropie des Cyclohexylringes sind die che
mischen Verschiebungen dieser Protonen gleich. Weiterhin zeigt
das Spektrum vier Signale der Alkylprotonen: das Signal des
B-Protons bei 4.01 ppm, das Signal der beiden s-Protonen bei
2.44 ppm und die Signale der y- und o-Protonen bei 1.88 bzw.
1.47 ppm. Das Verhältnis der Peakintensitäten (1:2:4:4) ist
im Einklang mit der 1-lolekülstruktur.
In den Massenspektren der Verbindungen ~ - Z zeichnen sich
die Molpeaks M+ und M++ durch hohe Intensität aus, welche ihre
Ursache in der Stabilität der entsprechenden aromatischen
Ionen hat. Die hohe Intensität der Molpeaks ist Voraussetzung
für die Ermittlung des Deuterierungsgrades, da er anband des
M+-Peaks bestimmt wird. Dazu ist jedoch das Auftreten von
Fragmentierungsreaktionen, insbesondere von Wasserstoffab
spaltungen auszuschließen. Bei der experimentellen Durchfüh
rung wird deshalb die Elektronenenergie, die normalerweise
70 eV beträgt, solange gesenkt, bis keine Fragmentierungen
mehr beobachtet werden. Diese bleiben im allgemeinen bei
Elektronenenergien unter 30 eV aus. Treten in Massenspektren
deuterierter Verbindungen bei verminderter Elektronenenergie
(M-1)+-Peaks auf, werden diese nicht durch Dehydrierungen
hervorgerufen, sondern sie stellen die Molpeaks solcher Mo
leküle dar, die ein D-Atom weniger tragen. Bei der Auswer
tung der Massenspektren ist zu berücksichtigen, daß auch
Moleküle mit einem oder mehr 13c-Atomen auftreten können.
Es muß deshalb eine Korrektur der ausgemessenen Signalin
tensitäten um den Beitragdurch die 13c-Atorne erfolgen. Da
*Aufnahme der Massenspektren: G. Holzmann, M. Franke, B. Merten, u. Ostwald

- 47 -
die natürliche Häufigkeit des Kohlenstoffisotops 1
1.12% be
trägt, ergibt sich für die Intensität des (M+1)•-Peaks n·1.12%
des M•-Peaks, wobei n die Zahl der C-Atome darstellt. Für das
Tetraphenylnaphthalin (c 34 H24 ) beträgt deshalb die Intensität
des (M+1)•-Peaks 3a.1% des ?-lolpeaks. Bei solch großen Molekü
len ist auch das Vorkommen von Molekülen mit zwei 13c-Atomen
nicht zu vernachlässigen. Die Intensität des (M+2)•-Peaks be
trägt 1.12% des (M-1)+-Peaks, dies entspricht im obigen
Beispiel 7% des Molpeaks.
Zur Berechnung des Deuterierungsgrades wurde die Summe der
korrigierten Peakintensitäten gleich 100 gesetzt, der Anteil
jedes Peaks entspricht dann dem Prozentsatz der Moleküle mit
dem entsprechenden Molekulargewicht. Der Deuterierungsgrad
nach Benz /46/ wird ermittelt, indem man die Summe aller ge
fundenen D-Atome durch die Anzahl der möglichen D-Atome di
vidiert. Die Deuteriumverteilung und der Deuterierungsgrad
der D-haltigen Verbindungen sind in Tabelle 5 zusammengestellt.
Verbindung
Sc
Sd
6b
6c
7b
Deuteriumverteilung und Deuterierungsgrad der
Verbindungen ~ - 2 Deuteriumverteilung
2.2% Da, 16.4% D9
, a1 .4% D10
6.3% D9' 93.7% D10
3.4% D 11' 15.7% D12' 39.7% D13' 41 • 2% D14 2.5% D7, 10.3% Da, 29.7% D
9, 57.5% D10
9.9% D2' 4a.a% D3 , 41.3% D4 36.1% D
9, 63.9% DlO
9.a% D2
, 50.9% D3
, 39.3% D4
Deuterierungsgrad
93.9%
99.4%
94.2%
94.2%
a2.a%
96.4%
a2.4%

48 -
4.5. EPR- und ENDOR-Spektren
4.5.1. EPR- und ENDOR-Spektren der phenylsubstituierten
Radikalanionen
Für die EPR- und ENDOR-Untersuchungen wurden die Verbin
dungen 1 - 2 mit Kalium zu den entsprechenden Radikalanionen
reduziert. Als Lösungsmittel wurde Dimethoxyethan (DME) ver
wendet.
Die Ermittlung der Protonenkopplungskonstanten erfolgte mit
Hilfe der ENDOR-Spektren. Die relativen Vorzeichen wurden durch
TRIPLE-Messungen ermittelt. Die Zuordnung der Kopplungskonstan
ten zu den Molekülpositionen erfolgte aufgrund der ermittelten
Vorzeichen, in einigen Fällen durch Untersuchung teildeuterier
ter Spezies und durch Analogschlüsse zu bekannten Systemen.
Die EPR-Spektren wurden unter Zugrundelegung der erhaltenen
Kopplungskonstanten simuliert. Durch Vergleich der experimen
tell ermittelten mit den simulierten EPR-Spektren konnten die
getroffenen Zuordnungen verifiziert \oJerden.
Obereinstimmend zeigen die ENDOR-Spektren der Verbindungen
l·e bis ±" 9 jeweils zwei Linienpaare, die relativ großen Kopp
lungskonstanten <laH I"' 7.2- 7.7 MHz bzw. 4.0 4.3 MHz) zu
gehören. In Analogie zu Untersuchungen an spezifisch deuterier
ten Systemen /4/ können sie den Grundkörperprotonen in den Po
sitionen 1, 4, 5, 8 bzw. 2, 3, 6, 7 zugeordnet werden. Die Pro
tonenkopplungskonstanten des Anthracengrundkörpers haben nega
tives Vorzeichen (vgl. Lit. /4/), die Vorzeichen der Arylpro
tonenkopplungskonstanten, ihr Betrag ist kleiner als 0.9 MHz,
werden relativ zu denen des Grundkörpers bestimmt.
Die EPR- und ENDOR-Spektren sowie die Zuordnung der Kopp
lungskonstanten des Radikalanions von 9,10-Diphenylanthracen
(1. 9 ) sind literaturbekannt /4/.
- Im ENDOR-Spektrum des Radikalanions ~-e sind vier der auf
grund der Molekülsyrn~etrie zu erwartenden sieben Linienpaare
aufgelöst (Abb. 14). Die beiden größten Kopplungskonstanten
werden dem Anthracengrundkörper zugeordnet, die beiden klei
neren den Tolylsubstituenten. Durch das TRIPLE-Experiment
konnte ein Auflösungsgewinn erzielt werden. Es zeigte sich,
daß im ENDOR-Spektrum Linien verschiedener Protonen .innerhalb
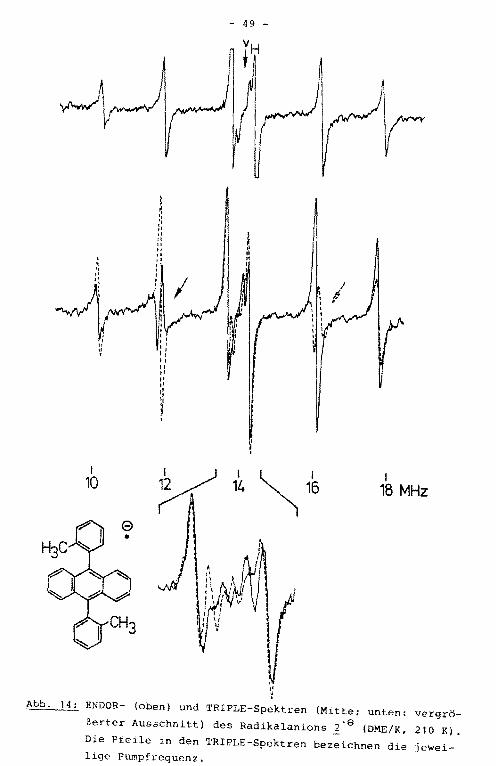
I 10
I
" \1
'
~ '• ., :· ,, " " .. " ,,
- 49 -
I 14
! ,, ,. I I
\
' ,. 1
I 16
I 18 MHz
Abb. 14: ENDOR- (oben) und TRIPLE-Spektren (Mitte; unten: vergrö
ßerter Ausschnitt) des Radikalanions ~·a (DME/K, 210 K).
Die Pfeile in den TRIPLE-Spektren bezeichnen die ieweilige Pumpfrequenz.
- so -
0.2mT .. B
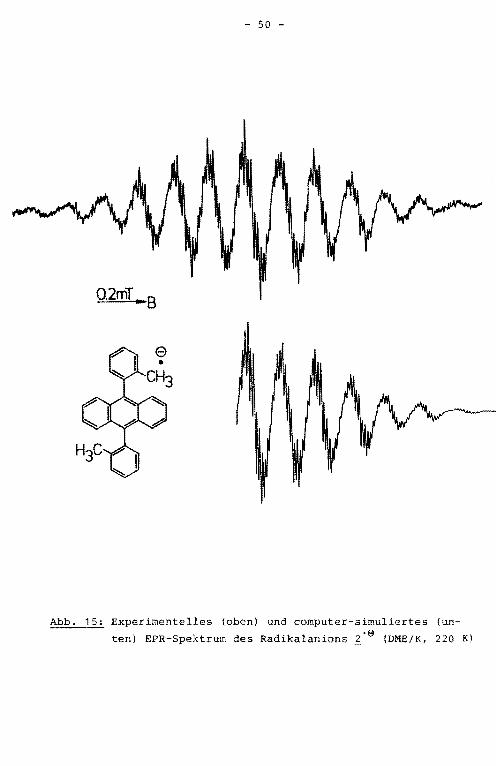
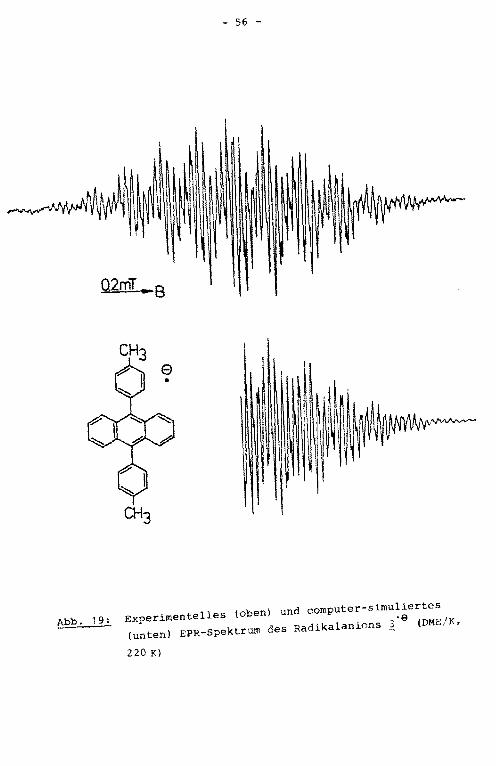
Abb. 15: Experimentelles (oben) und computer-simuliertes (un
ten) EPR-Spektrum des Radikalanions ~·e (DME/K, 220 K)

- 51 -
der Linienbreite von110kHz zusammenfallen. Aus dem unter hoch
auflösenden Bedingungen geschriebenen TRIPLE-Spektrum konnte
aus der Änderung der Signalform des Linienpaares mit laHI ~ 0.6 MHz geschlossen werden, daß es sich um eine Oberlagerung
der Signale zu drei Kopplungskonstanten mit unterschiedlichen
Vorzeichen handelt. Die kleinste dieser Kopplungskonstanten
ist positiv, die beiden anderen, dem Betrag nach gleich gro-
ßen Kopplungen, haben entgegengesetzte Vorzeichen. Weiterhin
ließ sich die kleinste Kopplungskonstante mit 80 kHz entneh
men. Da die Protonenkopplungskonstanten des Anthracengrund
körpers negatives Vorzeichen besitzen, ist das Vorzeichen der
Kopplungskonstanten der ortho- und para-Protonen der Arylreste
ebenfalls negativ, das der meta-Protonen und der Methylproto
nen in ortho-Position jedoch positiv. Unter dieser Vorausset
zung ergibt sich folgende Interpretation, die durch Simulation
des hochaufgelösten EPR-Spektrums verifiziert wurde (Abb. 15).
Die kleinste (positive) Kopplungskonstante muß aufgrund der
Multiplizität den Ortho-Methylprotonen zugeordnet werden. Die
verbleibenden beiden positiven Kopplungskonstanten sind den
inäquivalenten meta-Protonen zuzuordnen, wobei deren Zuordnung
zu den Positionen 3' und 5' nicht möglich ist. Die Zuordnung
der beiden negativen Kopplungskonstanten zur ortho- bzw. para-•6 -Position erfolgte in Analogie zu den Ergebnissen von l , in-
dem die dem Betrag nach größere Kopplungskonstante als den
ortho-Protonen zugehörig bestimmt wird. Die Richtigkeit die
ser Interpretation wird auch durch die Ergebnisse aus der Mes
sung von 4' 6 bestätigt (s.u.). - ·6
Das ENDOR-Spektrum der Verbindung ! zeigt vier statt der
hier erwarteten fünf Linienpaare (Abb. 16). Mit Hilfe des
TRIPLE-Experiments ließ sich zeigen, daß die kleinsten Kopp
lungskonstanten unterschiedliche Vorzeichen besitzen und so
mit im TRIPLE-Spektrum unterscheidbar werden, die dazugehöri
gen ENDOR-Linien fallen jedoch innerhalb der Linienbreite von
100 kHz zusammen. Die negative dieser beiden Kopplungskonstan
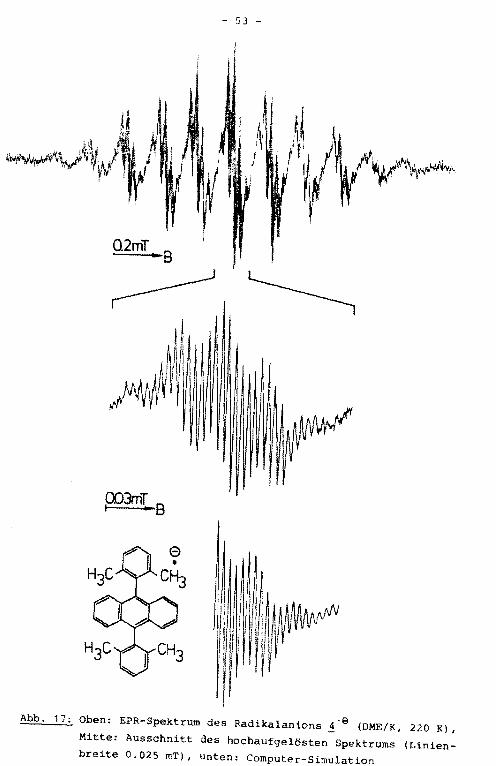
ten wird den para-Protonen zugeordnet. Durch Simulation des
hochaufgelösten EPR-Spektrums (Abb. 17) ließ sich die Multipli
zität der hfs-Kopplungskonstanten bestimmen, die eine eindeuti
ge Zuordnung der beiden positiven Kopplungskonstanten zu den
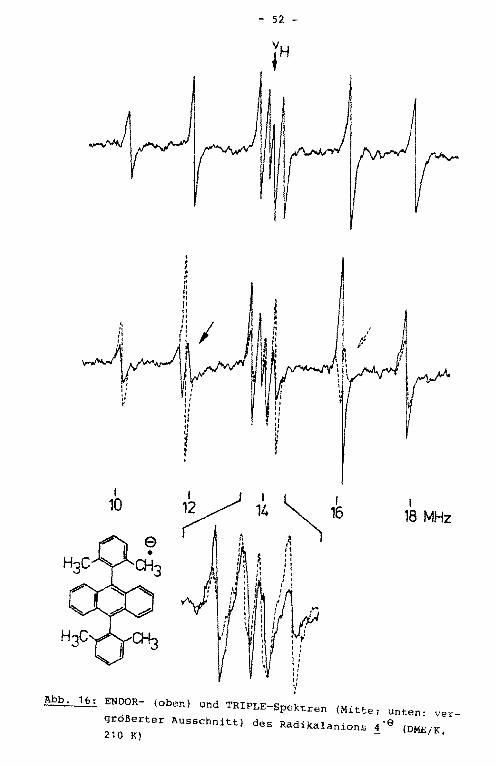
! 10
• ~ .. II
" ., l• ,, " ··; " , ' I' I
I I I'
'· ,, " II .. •• ,, ,. i I . I
~ 1\ I' I
! '
- 52 -
j ,, .. ., I ,, a '• r: :
I 14 \
11 '' <I ,.
I I , ' . ' ' I I I
I 16
I 18 MHz
Abb. 16: ENDOR- (oben) und TRIPLE-Spektren (Mitte; unten: ver
größerter Ausschnitt) des Radikalanions 4" 9 (DME/K, 210 K)
- 53 -
Abb. 17: Oben: EPR-Spektrum des Radikalanions ~·e (DME/K, 220 Kl,
Mitte: Ausschnitt des hochaufgelösten Spektrums (Linien
breite 0.025 mT), unten: Computer-Simulation

- 54 -
Molekülpositionen ermöglichte. Die größere ist demnach den meta
-Protonen, die kleinere den Ortho-Methylprotonen zugehörig. Die
beiden größten, negativen Kopplungen werden wiederum von den
Grundkörperprotonen hervorgerufen.
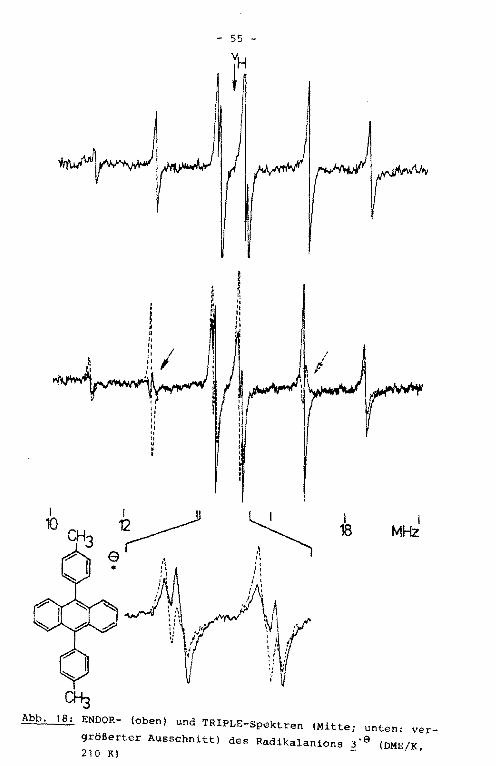
Aus dem ENDOR-Spektrum der Verbindung 3'9 werden vier der
aufgrund der Molekülsymmetrie erwarteten fünf Linienpaare er
halten (Abb. 18). Aus dem unter hochauflösenden Bedingungen
geschriebenen TRIPLE-Spektrum ist die fünfte Kopplungskonstan
te erhältlich. Die äußeren Linien der ENDOR- und TRIPLE-Spek
tren sind den Kopplungskonstanten des Grundkörpers, die Signa
le in der Mitte des Spektrums denen der Substituenten zugehö
rig. Die größte der Tolylprotonenkopplungskonstante hat nega
tives Vorzeichen und wird somit den ortho-Protonen zugeordnet.
Die Zuordnung der positiven Kopplungskonstanten in der Weise,
daß die größere der beiden den meta-Protonen, die kleinere den
Methylprotonen zugerechnet wird, kann durch Simulation des
EPR-Spektrums aufgrund der unterschiedlichen Multiplizität
bewiesen werden (Abb. 19). Die Kopplungskonstanten der Ver
bindungen 1'9 bis i·e einschließlich ihrer Zuordnung sind
in Tabelle 8, Kapitel 4.5.2., zusammengestellt.
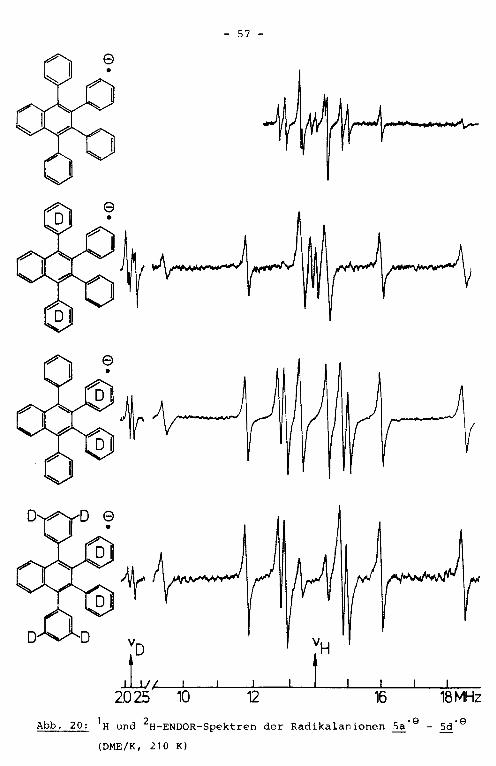
Für das Radikalanion sa' 9 sind aus Symmetriegründen acht
Sätze von Protonenkopplungskonstanten zu erwarten, sofern nicht
aufgrund einer gehinderten Rotation der Phenylreste zusätzliche
Inäquivalenzen der jeweiligen ortho- bzw. meta-Positionen auf
treten. Das hochaufgelöste ENDOR-Spektrum dieser Verbindung
zeigt sieben Linienpaare (Abb. 20). Um die Zuordnung der er
mittelten Kopplungskonstanten zu den verschiedenen Phenylre
sten bzw. zum Naphthalingrundkörper treffen zu können, wurden
die teildeuterierten Verbindungen 5b' 9 und sc' 9 untersucht.
In ihren ENDOR-Spektren fehlen die Protonenlinien der deu
terierten Positionen. Erwartungsgemäß erhält man jeweils
fünf Linienpaare von 1H-ENDOR-Linien. Damit lassen sich alle
acht Kopplungskonstanten ermitteln. Die relativen Vorzeichen
wurden durch ein TRIPLE-Experiment bestimmt (Abb. 21). Die
Änderung der Signalform des Linienpaarec mit; a · ~ 0.8 MHz
läßt sich mit einer Uberlagerung der Linien zu drei Kopplungs
konstanten mit unterschiedlichen Vorzeichen erklären. Diese
Interpretation stimmt mit den Ergebnissen aus den ENDOR-Messun
gen der teildeuterierten Verbindungen Sb•e- Sd•e überein.
I
y e
i ,, '• ,, h ,, '• f,
~ ..
I t-'
'• ,, : j j.
: I r '" '' ~ ; " ·: .
I 18
I MHz
Abb. 18: ENDOR- (oben) und TRIPLE-Spektren (Mitte; unten: ver
größerter Ausschnitt) des Radikalanions d' 9 (DME/K, 210 K)
- 56 -
02mT -B
CH3
Abb. 19: Experimentelles (oben) und computer-simuliertes
(unten) EPR-Spektrum des Radikalanions l·e (DME/K,
220 Kl
- 57 -
VD VH
L~, ~~~ _.____.__t ~~ ~~ 20 25 10 12 16 18 MHz
1 2 . . ·9 ·9 Abb. 20: H und H-ENDOR-Spektren der Rad1kalan1onen Sa - Sd
(DME/K, 210 K)

- 58 -
I \_I 14 ~
4
I 16
I I 18M Hz
Abb. 21: TRIPLE-Spektrum (unten vergrößerter Ausschnitt) des
Radikalanions 5a·e (DME/K, 210 K)
Durch selektive Deuterierung in den meta-Positionen der
Phenylsubstituenten in 1,4-Stellung konnte bestätigt werden,
daß die positive Kopplungskonstante den meta-Protonen zuzu
ordnen ist. Durch Simulation des EPR-Spektrums der Verbindung
5d·e ließ sich aufgrund der unterschiedlichen Multiplizität
die Zuordnung der Kopplungskonstanten mit negativen Vorzei
chen zu den ortho- und para-Positionen dieser Phenylreste
treffen. Ein Analogschluß kann für die Phenylreste in 2,3-
-Stellung gezogen werden (s. Tab. 6). Bei den Verbindungen
Sb·e- ·e konnten 2H-ENDOR-Linien nachgewiesen werden, de
ren vollständige Auflösung jedoch nicht gelang.
Aus dem ENDOR-Spektrum der Verbindung 6a" 9 lassen sich
sieben Kopplungskonstanten entnehmen (Abb. 22). Da die bei
den Linien in der Mitte des Spektrums durch Uberlagerung
der Signale von je zwei Kopplungskonstanten mit entgegenge
setzten Vorzeichen zustandekomrnen, sind alle aufgrund der
Molekülsymmetrie erwarteten neun Kopplungskonstanten durch
das TRIPLE-Experiment auflösbar (Abb. 23). Um d1e Zuordnung

- 59 -
Tabelle 6: Experimentelle und berechnete Kopplungskonstanten
des Radikalanions 5a" 9 (MHz)
Positionen H b) H c) a !! ber - exp
51 8 -9.05 -9.31
6, 7 -4.07 -2.17
2 I 1 6 ,a) -1.69 -1.58
3 > 1 S'a) +0.80 +0.51 4. al -2.09 -2.22
2 I I 1 6 I' a) -0.68 -0.64
3 I t I 5 "a) +0.15 +0.25 4' ,a) -0.83 -0.99
a) Diese Positionsangaben gelten für zwei äquivalente Phenyl
reste, siehe Formelschema.
b) DME/K, 210 K; ENDOR-Daten, experimenteller Fehler : 10 kHz.
c) HMO/McLachlan-Rechnung mit A = 1.2, Q -64.5 MHzund Ver-
drillungswinkeln 58° und 8''
der Kopplungskonstanten zu den Phenylresten und zu den Posi
tionen 10-13 treffen zu können, wurden die ENDOR-Spektren
der in den betreffenden Molekülpositionen deuterierten Ver
bindungen 6b.e und 6c" 9 herangezogen. Dort fehlen die ent
sprechende~1H-ENDOR-Linien bzw. sie sind im Falle unvoll
ständiger Deuterierung von stark verminderter Intensität.
Im Falle von 6c" 9 konnte die Zuordnung zu den Positionen
10-13 durch das 2H-ENDOR-Spektrum bestätigt werden. Dieses
besteht aus zwei Linienpaaren, die sich symmetrisch um die
freie Deuteronenfrequenz von 2.2 MHz anordnen. Die 2H-Kopp
lungskonstanten sind um den Faktor 6.51 gegenüber den jewei
ligen 1H-Kopplungskonstanten vermindert. Im Falle von 6b·e
werden nur zwei breite 2H-ENDOR-Linien erhalten. Die z~den drei Deuteriumkopplungskonstanten (jaDI = 150- 320kHz) zu
gehörigen Signale sind nicht auflösbar. Die weitere Zuord-
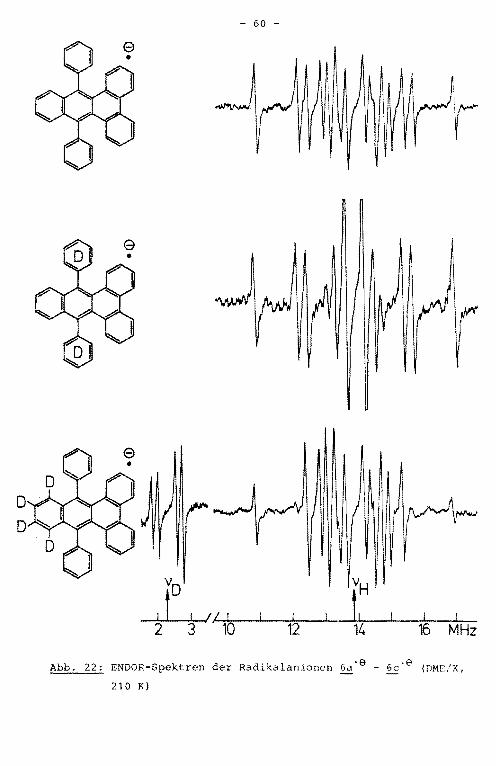
D D
- 60
VD VH
lt I /1-/~t~~--~~--~~t~~--~~~~~~~ 2 3 '' 10 12 14 16 MHz
Abb. 22: ENDOR-Spektren der Radikalanionen 6a·e - .e (DME/K,
210 K)
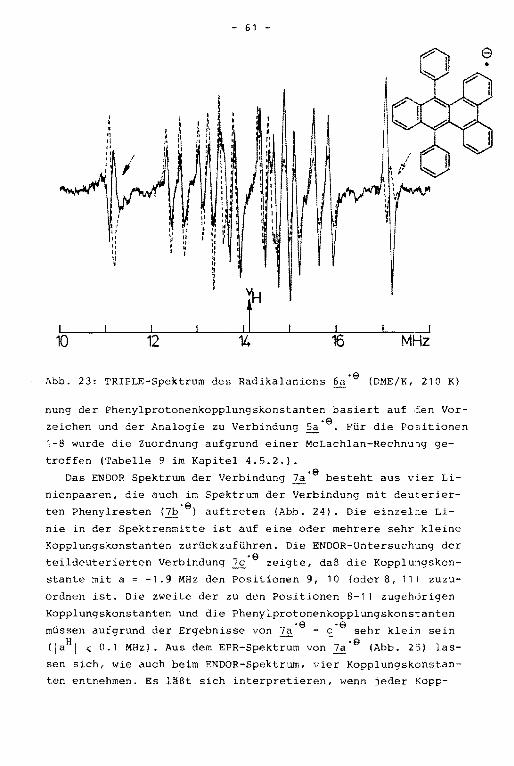
10
' • ' ,, ,,
12
- 61 -
.I 14 16 MHz
Abb. 23: TRIPLE-Spektrum des Radikalanions 6a·e (DME/K, 210 K)
nung der Phenylprotonenkopplungskonstanten basiert auf ~en Vor
zeichen und der Analogie zu Verbindung ·e. Für die Positionen
1-8 wurde die Zuordnung aufgrund einer McLachlan-Rechnu"lg ge
troffen (Tabelle 9 im Kapitel 4.5.2.).
Das ENDOR Spektrum der Verbindung ·e besteht aus vier Li
nienpaaren, die auch im Spektrum der Verbindung mit deucerier
ten Phenylresten (7b. 9 ) auftreten (Abb. 24). Die einzeL:e Li
nie in der Spektrenmitte ist auf eine oder mehrere sehr kleine
Kopplungskonstanten zurückzuführen. Die ENDOR-Untersuch"J.ng der
teildeuterierten Verbindung ·e zeigte, daß die Koppl u:1gskon
stante mit a = -1.9 MHzden Positionen 9, 10 (oder8, 11) zuzu
ordnen ist. Die zweite der zu den Positionen 8-11 zugehörigen
Kopplungskonstanten und die Phenylprotonenkopplungskons~anten
müssen aufgrundder Ergebnisse von 7a·e- c" 9 sehr klein sein H - ~ ·9
(ja I < 0.1 MHz). Aus dem EPR-Spektrum von 7a (Abb. 25) las-
sen sich, wie auch beim ENDOR-Spektrum, vier Kopplungskonstan
ten entnehmen. Es läßt sich interpretieren, wenn jeder Kopp-
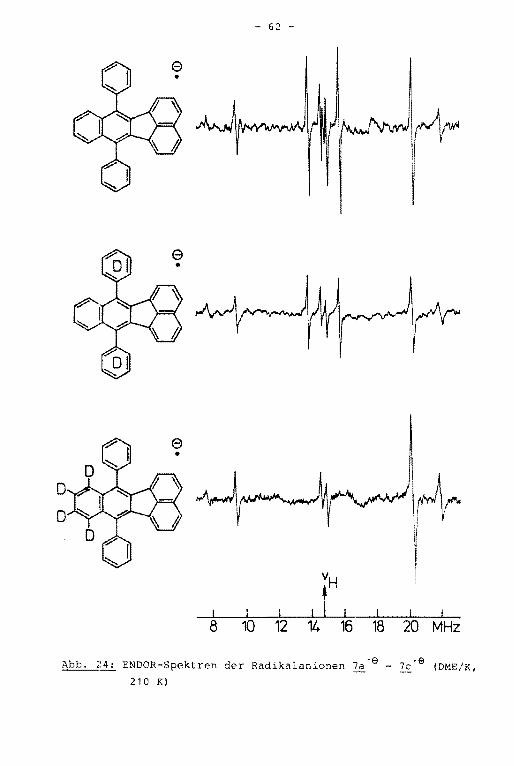
D
D
8 10
- 62 -
12 14 16
=.:.._::..::.::.. ENDOR-Spektren der Radikalanionen
210 K)
I I I
18 20 MHz
·e ·e (DME/K,
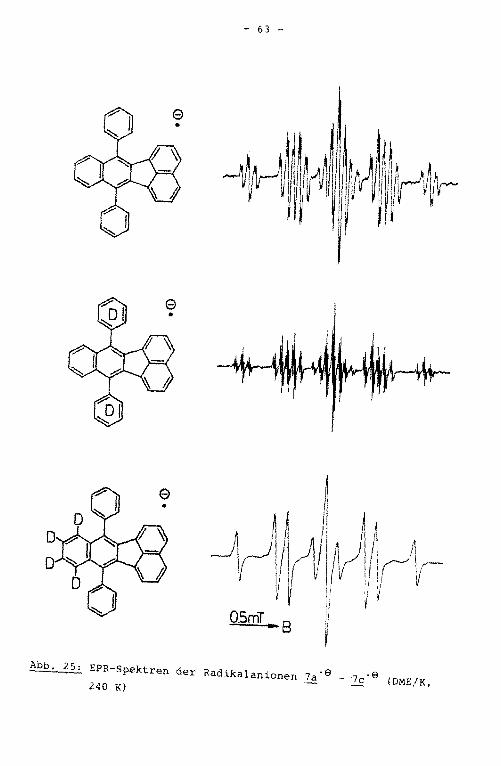
- 63 -
0.5mT B f
Abb. 25: EPR-Spektren der Radikalanionen 7a' 8 - ·e (DME/K, 240 K)

- 64 -
lungskonstanten zwei Protonen zugeordnet werden. Das EPR-Spek
trum von 7b·e unterscheidet sich davon lediglich durch eine
etwas verbesserte Auflösung, die durch den H/D-Austausch an
den Phenylringen verursacht wird. Das EPR-Spektrum von 7c" 8
besteht aus neun Komponenten, die durch die zwei Sätze zu je
zwei Protonen mit den beiden größten Kopplungskonstanten her
vorgerufen werden. Die Linien der kleineren Protonenkopplungs
konstanten wie auch der etwa gleich großen Deuteronenkopplung
sind nicht auflösbar.
Die Zuordnung der Kopplungskonstanten erfolgte, soweit sie
nicht durch Untersuchung der teildeuterierten Spezies gelang,
unter Berücksichtigung der durch TRIPLE-Messung ermittelten
Vorze.ichen aufgrund einer .HcLachlan-Rechnung sowie durch Ver
gleich mit dem Fluoranthenradikalanion 8"8
/47/ (Tabelle 7).
Tabel~e 7: Kopplungskonstanten der Radikalanionen 7a·e und
8 ·e (MHz)
7a ·e 8·e
Positionen H b) aH c) Positionen b) a - exp - ber exp
1 , 6 -10.71 -10.04 1 , 6 -10.99
2, 5 -+0.33 + 1 . 71 2, 5 -0.42
3' 4 -14.18 15. 1 8 3, 4 -14.65
8, 1 1 .; ! o .1 I +0. 1 7 7, 1 0 -0.24
9, 1 0 -1.88 -0.60 8, 9 -3.41
2' - 6 ,a) ,;; I 0. 1 : "i 0. 3 i
a-c) Vgl. Fußnoten zu Tabelle 6. Für die Rechnung wurde ein
ilerdrillungswinkel von 8• 40° angeno=en.
Die EPR- und ENDOR-Spektren des vorangegangenen Abschnitts
wurden vorwiegend von B. Kirste aufgenommen, der auch die Si
mulationen der entsprechenden EPR-Spektren und die McLachlan
-Rechnungen durchführte. Freundlicherweise wurde von Herrn
M. Plalo das Programm ISOSIH zur Spektrensimulation und von
Herrn 1'. Rakowski das Prograr.1m zur ~lci.achlan-Rechnung zur
Verfügung gestellt.

65 -
4.5.2. EPR- und ENDOR-Spektren der phenylsubstituierten
Radikalkationen
Die Darstellung der Radikalkationen der Verbindungen 1 4
und 6a ~ erfolgte nach einer für Kohlenwasserstoffe neuen
Methode. Die Oxidation w~rde mit Benzoylperoxid in einem Ge
misch aus Trifluoressigsäure und Toluol durchgeführt. Der Me
chanismus der dabei ablaufenden Reaktion ist ungeklärt. ~ach
dieser Methode ließen sich keine Radikalkationen der Vertin
dungen 2 und ~ erzeugen. Deren Darstellung nach konvertio
nellen Methoden, z.B. H2so4 oder CHC1 3/cH 3No 2 , wurde bisrer
nicht durchgeführt. Die oxidative Behandlung von - c lie-
ferte zwar paramagnetische Spezies, deren Struktur jedoct nicht
mit der eines monomeren Kations vereinbar ist (s. Kap. 4.7.).
Die EPR- und ENDOR-Untersuchung der Radikalkationen wtrde
analog zu der der Radikalanionen durchgeführt. Auch hier konn
te durch Simulation der experimentellen EPR-Spektren die Rich
tigkeit der aufgrund der ENDOR- und TRIPLE-Messungen erfolgten
Zuordnung der Kopplungskonstanten zu den Molekülpositonerc be
stätigt werden.
In Analogie zu den Radikalanionen sind in den ENDOR-Spektren
der Radikalkationen von 1 4 die Linien der Grundk6rperproto-
nenkopplungskonstanten von denen der Substituenten zu unt.er
scheiden. Die Kopplungskonstanten der Protonen des Anthracen
gerüsts betragen 7-8 MHz bzw. 3.3-3.5 MHz, ihr Vorzeichen ist
negativ. Der Betrag der Arylprotonenkopplungskonstanten !.st
kleiner als 2 MHz, ihr Vorzeichen wird relativ zu den Vorzei
chen der Kopplungskonstanten des Grundkörpers bestimmt.
EPR- und auch ENDOR-Untersuchungen von .!_·@ in H2so4 sind be
kannt /4, 33, 48/. Die Messungen von .!_·!& in Toluol/Trifluor
essigsäure liefern Spektren, die sich durch schmale Linien
und ein sehr gutes Signal-Rausch-Verhältnis auszeichnen
(Abb. 26). Die daraus ermittelten Kopplungskonstanten st:.rn
rnen mit den Literaturwerten überein. '@
Aus dem ENDOR-Spektrum der Verbindung l werden secho;
Kopplungskonstanten erhalten (Abb. 27). Die Vorzeichenbe-
stimmung durch das TRIPLE-Experirnent ergibt für die größte
der Arylprotonenkopplungskonstanten ein positives Vorzei··
chen. Sie wird den meta-Protonen zugeordnet. Eine Inäqui-
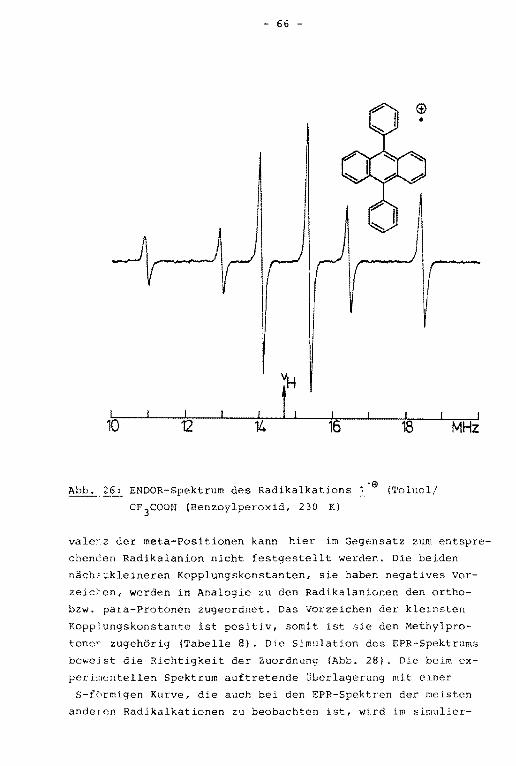
- 66 -
10 12 14 16 18
Abb .. 26: ENDOR-Spektrum des Radikalkations 1·e (Toluol/
CF 3COOH (Benzoylperoxid, 230 Kl
MHz
valerz der meta-Positionen kann hier im Gegensatz zum entspre
chencen Radikalanion nicht festgestellt werden. Die beiden
näch~tkleineren Kopplungskonstanten, sie haben negatives Vor
zeic>en, werden in Analogie zu den Radikalanionen den ortho
bzw. para-Protonen zugeordnet. Das Vorzeichen der kleinsten
Kopplungskonstante ist positiv, somit ist sie den Methylpro
toner zugehörig !Tabelle 8). Die Simulation des EPR-Spektrums
beweist die Richtigkeit der Zuordnung (Abb. 28). Die beim ex
perimentellen Spektrum auftretende 1Jberlagerung mit einer
S-förmigen Kurve, die auch bei den EPR-Spektren der meisten
anderen Radikalkationen zu beobachten ist, wird im simulier-
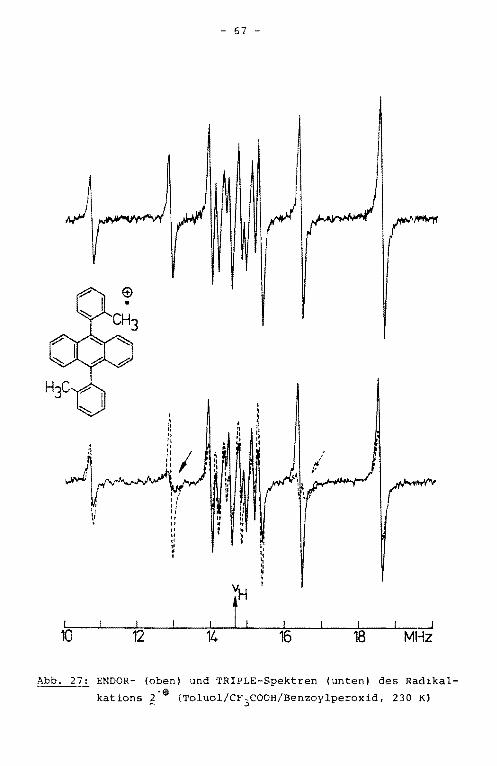
- 67 -
I I
I > •' • •' ,, ., •' •'
~ I•
I .. .. II • I
' ,. '• ,. ~ " •i
" ,, " • " ~ ,, ,,
' ~ ,. ' •' I
~ I
!, t
I 1 I I _.J
12 14 16 18 MHZ 10
Abb. 27: ENDOR- (oben) und TRIPLE-Spektren (unten) des Radikal
kations 2·~ (Toluol/CF3COOH/Benzoylperoxid, 230 K)
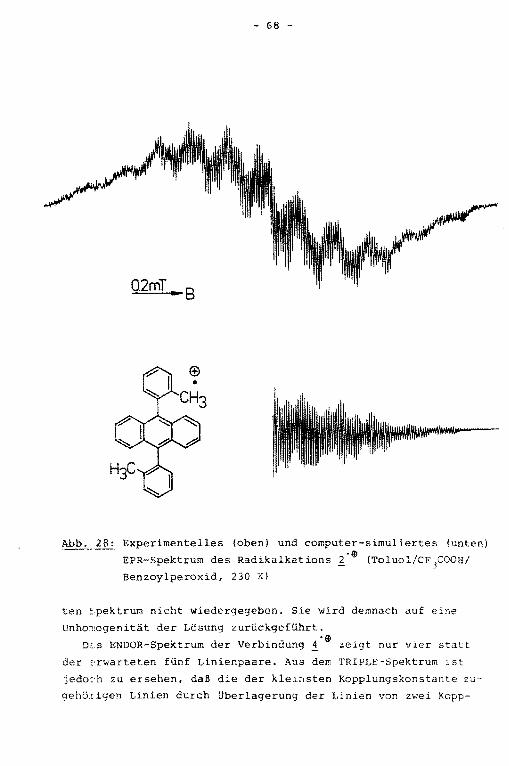
- 68 -
Q2mT B
Abb._28: Experimentelles (oben) und computer-simuliertes (unten) ·@
EPR-Spektrum des Radikalkations 2 (Toluol/CF 3COOH/
Benzoylperoxid, 230 K)
ten ~,pektrum nicht wiedergegeben. Sie wird demnach auf eine
Unho~ogenitlt der Lösung zurückgeführt. ·~ Des ENDOR-Spektrum der Verbindung ! zeigt nur vier statt
der erwarteten fünf Linienpaare. Aus de~ TRIPLE-Spektrum lSt
jedo2h zu ersehen, daß die der kleinsten Kopplungskonstante zu
geh6Iigen Linien durch Oberlagerung der Linien von zwei Kopp-
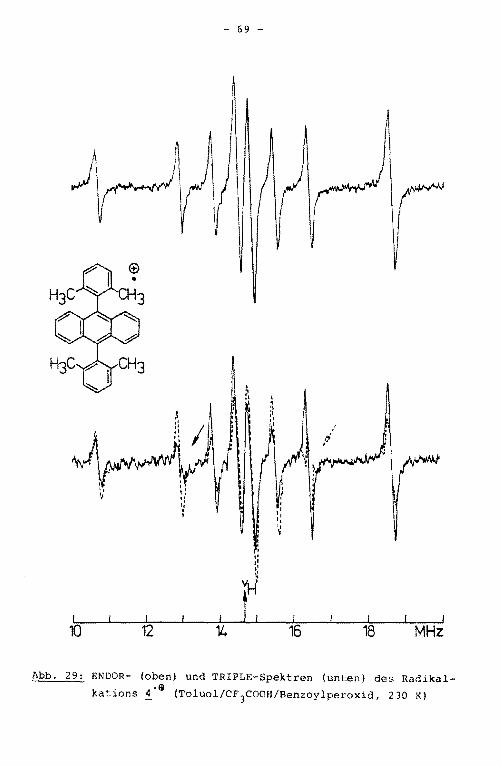
- 69 -
' ., :·
.. ,. •' ~
YHI
I I I t I I I I 1k0--L-~Q~~~~~~~1~6--~~18.-~M"HT-z
Abb. 29: ENDOR- (oben) und TRIPLE-Spektren (unten) des Radikal
kations 4·$ (Toluol/CF3COOH/Benzoylperoxid, 230 K)
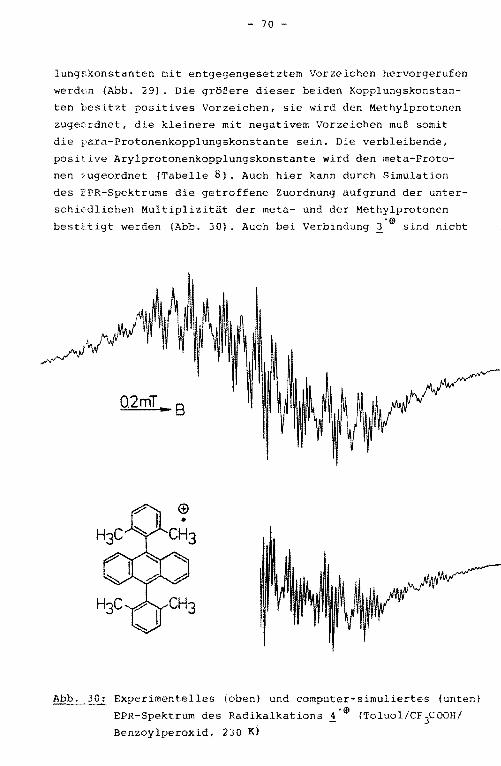
- 70
1 ungE:konstanten mit entgegengesetztem Vorzeichen hervorgerufen
werdEn (Abb. 29). Die größere dieser beiden Kopplungskonstan
ten besitzt positives Vorzeichen, sie wird den Methylprotonen
zugeordnet, die kleinere mit negativem Vorzeichen muß somit
die para-Protonenk.opplungskonstante sein. Die verbleibende,
positive Arylprotonenkopplungskonstante wird den meta-Proto
nen 2ugeordnet (Tabelle 8). Auch hier kann durch Simulation
des E:PR-Spektrums die getroffene Zuordnung aufgrund der unter
schiE·dlichen Mul tiplizität der meta- und der Methylprotonen
besU.tigt werden (Abb. 301. Auch bei Verbindung 2"@ sind nicht
0.2mT 8
Experimentelles (oben) und computer-simuliertes (unten)
EPR-Spektrum des Radikalkations 4•$ (Toluol/CF3COOH/
Benzoylperoxid, 230 Ki
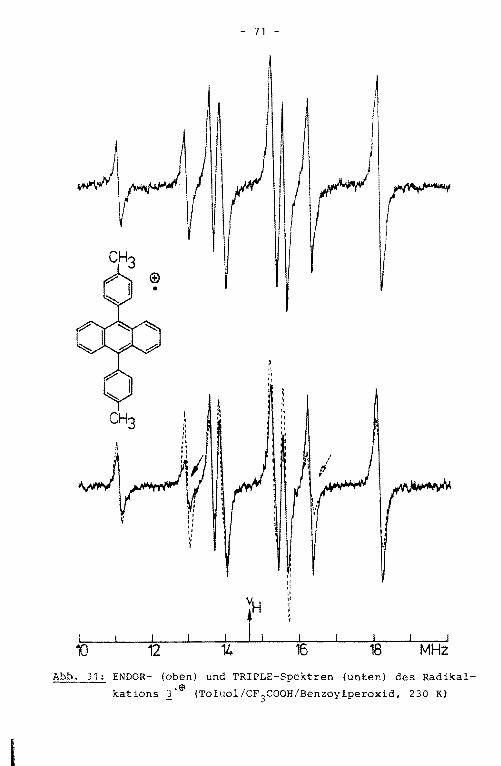
l
- 71 -
10 12
' ,, ,, ,, ,, ,, " : I
f
16 18 MHz
Abb. 31: ENDOR- (oben) und TRIPLE-Spektren (unten) des Radikal
kations 3.$ (Toluol/CF3COOH/Benzoylperoxid, 230 K)
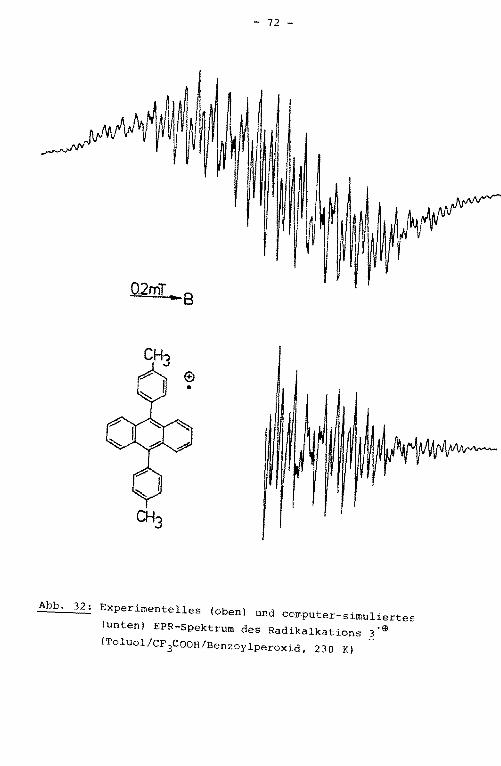
- 72 -
02mT -B
Abb. 32: Experimentelles (oben} und computer-simuliertes
(unten) EPR-Spektrum des Radikalkations d·~ (Toluol/CF3COOH/Benzoylperoxid, 230 K)

Kopplungskonstanten (MHz) der Radikalanionen 1 -9_ 4 ·9a) und der
Radikalkationen 1·~ 4 ·~ b)
Positionen 1 ·9 d) 1·~ z·e 2 ·EI! 3 ·9 3"~ 4 ·9 4·~
I 4, 5' 8 -7.29 -7.43 -7.56 -7.83 -7.41 -7.04 -7.70 -7.88
21 31 6, 7 -4.08 -3.43 -4.20 -3.50 -4.11 -3.32 -4.24 -3.47
2. I
6 ,c) -0.86 -1 . 2 7 +O.o8';l +0.27;) -0.84 -1.44 +0. 1 Oe) +0.38 8)
-0.63 -0.98
3., 5'c) +0.65 +1. 27 +0.56, +1. 34 +0.67 + 1. 34 +0.64 + 1. 64 +0.63
4 ,c) -0.65 -1.27 -0.32 -0.52 +0.59e) +1. -0.21 -0.32
a) DME/K1 210 K.- b) Toluol/CF3COOH/Benzoylperoxid, 230 K. a) 1 b) experimenteller
Fehler! 10kHz. - c) Diese Positionsangaben gelten für die beiden äquivalenten
Phenylreste. d) Werte aus Lit. /4/. - e) Kopplungskonstanten der Methylprotonen.
-...J w

- 74 -
alle Linien im ENDOR-Spektrum aufgelöst. Wiederum ergibt sich
aus dem TRIPLE-Experirnent, daß im ENDOR-Spektrum die Linien
der zwei kleinsten Kopplungskonstanten mit entgegengesetztem
Vorzeichen innerhalb der Linienbreite von 130 kHz zusammenfal
len (Abb. 31). Die dem Betrage nach größten Kopplungskonstan
ten werden dem Anthracengrundkörper, die größte der Arylproto
nenkopplungskonstanten den Methylprotonen, die nächstkleinere,
die ein negatives Vorzeichen besitzt, den Protonen in den
ortho-Positionen und die kleinste mit positivem Vorzeichen
den rneta-Protonen zugeordnet (Tabelle 8). Die Simulation des
EPR-Spektrums zeigt wieder die Richtigkeit der Interpretation
von ENDOR- und TRIPLE-Spektrum (Abb. 32). Das simulierte Spek
trum stellt zwar keine exakte Wiedergabe des experimentellen
Spektrums dar, ein Austausch der meta- und der Methylprotonen
kopplungskonstanten führt jedoch zu einer wesentlich schlech
teren Ubereinstimrnung.
Für Verbindung ~·al werden aus dem ENDOR-Spektrum acht Kopp
lungskonstanten erhalten (Abb. 33). Die beiden kleinen Linien
bei 14.3 bzw. 15.0 MHz sind auf Verunreinigungen der Probe zu
rückzuführen, denn sie sind in den Spektren der teildeuterier
ten Spezies 6b·al und 6c·~ nicht zu sehen und sie zeigen keinen
TRIPLE-Effekt (Abb. 34). Die Zuordnung der Kopplungskonstanten
zu den Molekülpositionen erfolgte durch Untersuchung der teil-•@ •IJ)
deuterierten Verbindungen 6b und 6c , durch Vorzeichenbe-
sti~~ung und durch Vergleich mit Modellrechnungen. Durch Deu
terierung der Phenylringe können die Kopplungskonstanten des
Grundkörpers von denen der Substituenten unterschieden werden.
Im ENDOR-Spektrum der Verbindung 6b.@ fehlen die entsprechen
den Protonenlinien, bzw. sie sind im Falle der unvollständigen
Deuterierung einer Position von geringer Intensität, ebenso
wie bei den entsprechenden Radikalanionen. Das Deuterium
-ENDOR-Spektrum ist unaufgelöst, es zeigt ein Linienpaar, des
sen Abstand von 0.26 MHz mit einem Mittelwert der berechneten
Phenyl-D-Kopplungskonstanten übereinstirnmt. Aus dem ENDOR-Spek
trum der Verbindung 6c·al ist zu ersehen, daß die beiden größten
Kopplungskonstanten, sie haben negatives 'Jorzeichen, den Pro
tonen in Position 10 - 13 des Grundkörpers zuzuordnen sind.
Wie schon beim korrespondierenden Anion sind hier aufgrund
0
0
- 75 -
•$ •$ Abb. 33: ENDOR-Spektren der Radikalkationen 6a - 6c
(Toluol/CF3COOH/Benzoylperoxid, 230 K)
'• ,, • • I I
•
76 -
·~ Abb. 34: TRIPLE-Spektrum des Radikalkations 6a (Toluol/CF 3COOH/
Benzoylperoxid, 230 K)
der nicht vollständigen Deuterierung die entsprechenden 1H-ENDOR
-Linien noch sichtbar, ihre Intensität ist jedoch erheblich ver
mindert. Die Eindeutigkeit der Zuordnung wird durch das 2H-ENDOR
-Spektrurn bewiesen. Es besteht aus zwei Linienpaaren, aus deren
Abstand die zugehörigen Deuteriumkopplungskonstanten ermittel
bar sind (a~ = 1.12 MHz, a~ = 0.48 MHzl, die um den Faktor 6.51
gegenüber den Protonenkopplungskonstanten verkleinert sind. Die
Zuordnung zu den Positionen 1 - 8 erfolgte durch Vergleich der
experimentell ermittelten mit berechneten Werten (Tabelle 9).

- 77 -
Experimentelle und berechnete Kopplungskonstanten ·9 •Eil
der Radikalionen 6a und 6a
H ·9 b) aH ·Eil c) H d) Positionen a exp (6a ) (6a ) a exp ber
1 1 8 +0. +0.2 +0.40
2' 7 -2.92e) -2.04el -1.2 5
3, 6 -1,19e) 1. 04 e) -0.97
4, 5 -0.57el -o.o8el +0.09
10, 13 -6.03 -7.39 -7.29
11 , 12 -3.51 -3.16 -1 . 99
2', 6,a) 1. 6 7 -1.84 -1.66
3' , s,a) +0.98 +1. 39 +0.55 4 ,a) -2.09 -1 • 84 -2.08
a), b) Vgl. Fußnoten zu Tabelle 6. - c) Toluol/CF3COOH/Benzoyl
peroxid, 230K; ENOOR-Oaten, experimenteller Fehler + 10 kHz. -
d) HMO-McLachlan-Rechnung mit>. = 1.2, Q -64.5 MH~ und Ver
drillungswinkel 0' = 55°. - e) Die Zuordnung zu den Positionen
ist experimentell nicht abgesichert.
9-Cycloalkylanthracene
Von beiden Cycloalkylanthracenen ließen sich nur Radikal
anionen darstellen. Die Behandlung mit Benzoylperoxid im Tri
fluoressigsäure/Toluol-Gemisch führte kurzzeitig zu schwach
blauen Lösungen, deren Färbung möglicherweise durch entspre
chende Radikalkationen hervorgerufen wurde. Die Lebensdauer
dieser farbigen Verbindungen war jedoch auch bei tiefen Tem
peraturen und bei Ausschluß von Sauerstoff durch Probenberei
tung im Hochvakuum zu kurz, um ~lessungen durchzuführen. Die
Radikalanionen wurden auch hier durch Reduktion mit Kalium
in DME erzeugt.
Das ENDOR-Spektrum des 9-Cyclopropylanthracenradikalanions ·6 2 zeigt sieben Linienpaare (Abb. 35). Durch das TRIPLE-Experi-
ment wird ein Auflösungsgewinn erreicht: Es zeigt sich, daß die
beiden kleinsten Kopplungskonstanten unterschiedliche Vorzei-
- 78 -
6 8 10 12 14 16 18 20 22 MHz
---'-'""--- ENDOR- (oben) und TRIPLE-Spektren \unten) des Radikal
anions ~·e (DME/K, 200 K)
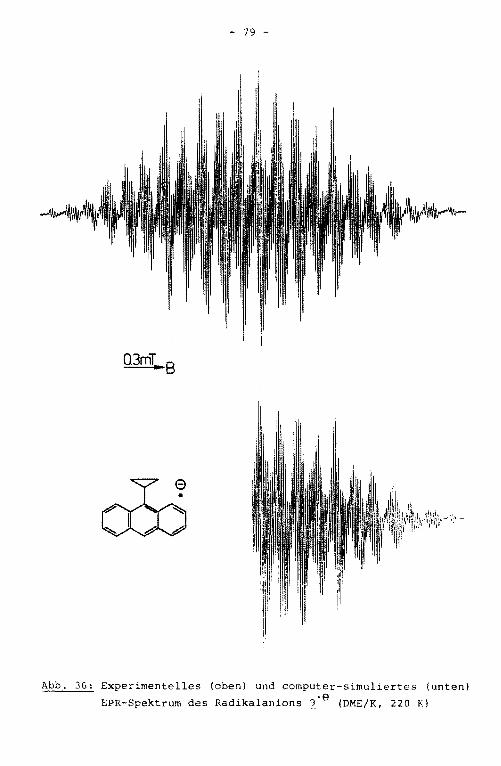
- 79
03mT B
~ y ~~ ax)
Abb. 36: Experimentelles (oben) und computer-simuliertes (unten)
EPR-Spektrum des Radikalanions 2·e (DME/K, 220 K)

- 80 -
chen besitzen, ihre Linien fallen im ENDOR-Spektrurn innerhalb
der Linienbreite von 120 kHz zusammen. Somit werden in Uberein
stimmung mit der Molekülsymmetrie acht Kopplungskonstanten er
halten. Deren Zuordnung zu den Molekülpositionen erfolgt für
den Anthracengrundkörper in Analogie zu bekannten, in 9-Stel
lung substituierten Anthracenen, beispielsweise dem 9-Methyl
und 9-Ethylanthracen /49, 50/, zum anderen basiert sie auf
der Vorzeichenbestimmung (Tabelle 10l. Die größte der gemesse
nen Kopplungskonstanten besitzt ein positives Vorzeichen, sie
wird deshalb dem ß-Proton zugeordnet, die beiden kleinsten
Kopplungskonstanten sind den beiden Sitzen inäquivalenter
y-Protonen zugehörig. Soweit stimmt die getroffene Zuordnung
mit der in der Literatur /43/ angegebenen überein, wobei je
doch die Autoren keine Inlquivalenz der y-Protonen feststel-
Positionen
1 , 8
2, 7
3, 6
4, 5
1 0
B y'
y''
Experimentelle und berechnete Kopplungskonstanten
des Radikalanions 9' 6 (MHz)
H a) H b) a exp a ber
-8.00 -6.80
-3.46 -2.13
-4.86 -1.04
-7. 10 -7.71
-15.04 -19.92
+18.60 +24.30
+0.45c) +0.51
-0.49c) -0.55
a)DME/K, 200 K; experimenteller Fehler : 10 kHz. -
b)Die INDO-Rechnungen wurden unter Verwendung folgender
Bindungnlängen und Bindungswinkel durchgeführt:
(C m··C :1.400Ä,c -c 1 .h:1.600A aro . arom. arom. a 1p • call'ph -c 1 .ph : 1.520 A: c -H: 1.080 l , . a 1 . o 2 arom. Caliph. -H: 1.080 A; Sp : 120°, L::,.: C-C-C: 60°, H-C-H: 130°)
clDie Zt;ordnung dieser Werte zu den Molekülpositionen ist
experimentell nicht gesichert

- 81 -
len konnten. Weiterhin konnte die Richtigkeit dieser Zuordnung
durch Simulation des hochaufgelösten EPR-Spektrums bestätigt
werden (Abb. 36) . Die weitergehende Zuordnung der y-Protonen
erfolgt mit Hilfe von INDO-Rechnungen, die von W. Broser durch
geführt wurden /51/. Diese erhalten für die exo-ständigen
y-Protonen, sie werden nun als y' bezeichnet, eine Kopplungs
konstante mit positivem Vorzeichen, für die endo-ständigen
Protonen (y' ') dagegen eine negative Kopplungskonstante (s.
Abb. 39 im Kapitel 4.6.2.).
Aus dem ENDOR-Spektrum des Radikalanions des 9-Cyclohexyl-
anthracens ' 8 ) sind neun Kopplungskonstanten erhältlich
(Abb. 37). Aus dem TRIPLE-Spektrum ist ersichtlich, daß die
ENDOR-Linien zu der zweitkleinsten Kopplungskonstante durch
Uberlagerung der Linien zu zwei Kopplungskonstanten mit ent
gegengesetztem Vorzeichen zustandekommen. Die Zuordnung der
Grundkörperprotonenkopplungskonstanten erfolgt analog zu Ver
bindung 2· 8 . Dabei wird jedoch eine Irräquivalenz der zum
Cyclohexylring peri-ständigen Protonen festgestellt. Die
größte der positiven Kopplungskonstanten wird wiederum dem
8-Proton zugeordnet. Die Zuordnung der übrigen Kopplungskon
stanten basiert auf INDO-Rechnungen /51/: danach werden die
positiven Kopplungskonstanten den 6-Protonen in axialer (6a)
bzw. in äquatorialer (6e) Anordnung zugerechnet, die negati
ve Kopplungskonstante wird den vier y-Protonen zugeordnet
(Tabelle 1 1). Eine eindeutige Bestätigung der getroffenen
Zuordnung kann in diesem Fall nicht durch Simulation des
EPR-Spektrums erfolgen, da dieses durch die Vielzahl der
Kopplungen keine gute Auflösung zeigt. Trotz der guten Uber
einstimmung des simulierten mit dem experimentellen Spektrum
kann die Zuordnung zu den y- und 6-Protonen nicht als gesi
chert angesehen werden (Abb. 38). Eine eindeutige experimen-
telle Zuordnung würde die spezifische Deuterierung der ent
sprechenäen Positionen des Cyclohexylringes erfordern.
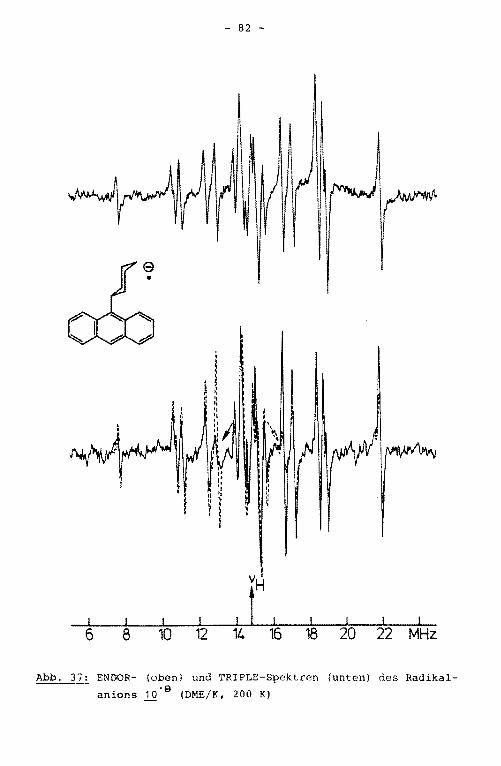
6 8 10
l . l I 'I 1 ., . '•
12
II ., :• ,. " I
' I' ,. I' I
II •r II :r ! ~
l
- 82 -
I
}, I I I I
14 16 18 20 22 MHz
ENDOR- (oben) und TRIPLE-Spektren (unten) des Radikal
anions 10" 9 (DME/K, 200 K)
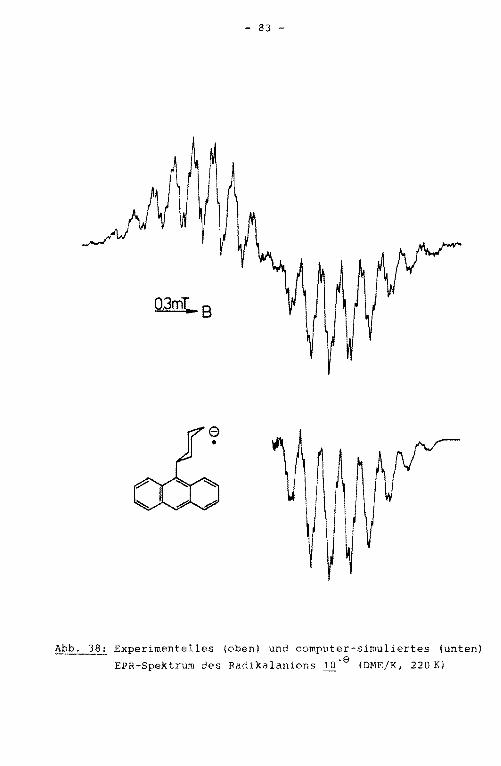
- 83
0.3mT. B
~~~~- Experimentelles (oben) und computer-simuliertes (unten)
EPR-Spektrum des Radikalanions 10' 9 (DME/K, 220K)

- 84 -
Tabelle 11: Experimentelle und berechnete Kopplungskonstanten
des Radikalanions 10" 8 (MHz)
H a) H b) Positionen a a ber exp
-8.08 -7.20
2, 7 -3.60 -1.86
31 6 -4.76 -1.27
41 5 -7.38 -7.51
8 -8.25 -7.54
10 -14.19 -19.11
ß +1. 61 + 1. 28
Ya -0. 68°) -1.30
Ye -0.68°) -1.27
6 +0.34°) +0.44 a
+0.89° 1 6 +1. 53
e
a)' c) Siehe Fußnoten Tabelle 10
b) Die INDO-Rechnungen wurden unter Verwendung von Standard
bindungslängen und Standardbindungswinkeln durchgeführt:
(C -c : 1.4oo Ä, c -c 1 . h: 1.540 Ä arom. arom. arom. a ~p . c
1. h -c
1. h: 1.540 A; c -H: 1.080 Ä, a ~p . a ~p . o
2 arom. 3 C 1 . h -H: 1.080 A; Sp 120", Sp : 109,5")
a ~P .
4.6. Diskussion
4.6.1. Diskussion der Meßergebnisse der phenylsubstituierten
Verbindungen
Im folgenden soll diskutiert werden, welche Mechanismen für
den Spintransfer in die verdrillten Phenylsubstituenten der
untersuchten Radikalionen verantwortlich sind. Dabei ist die
Abhängigkeit deJ.· Ringprotonenkopplungskonstanten vom Verdril
lungswinkel 0 zwischen den Ebenen des Grundge~üsts und der
Phenylsubstituenten von besonderem Interesse. Reine n-MO-Model
le lassen die Reihenfolge der Phenylprotonenkopplungskonstan-H . ' H I . H I ten - Ia I > Ia· th 1 » ja t -erwarten, wobei alle Kopp-para or o me a
lungen mit zunehmendem Winkel 0 abnehmen. Unter Berlicksichti-

- 85 -
gung der n-o-Delokalisation wird jedoch diese Winkelabhängigkeit
nur für die ortho- und para-Protonenkopplungskonstanten erwar
tet, dagegen wird für die meta-Protonenkopplungskonstante eine
Vergrößerung bei Zunahme von e bzw. eine Unabhängigkeit vom
Verdrillungswinkel erwartet (vgl. Kapitel 3.2.6.).
In der Reihe Diphenylanthracen lll , Di-o-tolylanthracen (~)
und Dixylylanthracen Ii) ist durch sukzessive Einführung der
Methylgruppen in die ortho-Positionen der Phenylringe eine Zu
nahme des Verdrillungswinkels zu erwarten. Die Verdrillungs
winkel für die neutralen Moleküle Diphenylanthracen lll und
Di-p-tolylanthracen lll sind nach den NMR-Messungen ungefähr
gleich (62° bzw. 60°). Aus den EPR- und ENDOR-Meßergebnissen ·9 ·9 der Radikalanionen 1 - i (Tabelle 8) ist zu ersehen, daß
die Beträge der Kopplungskonstanten des Grundkörpers in der ·9 ·9 ·9 ·e Reihenfolge - 1 , l , ~ , i - stetig zunehmen. Dieser
Befund entspricht einer Zunahme des Verdrillungswinkels in
gleicher Richtung, die bewirkt, daß vermindert n-Spindichte
aus dem Grundkörper in die Substituenten abfließt. Die für
4' 9 ermittelten Werte entsprechen fast denen des unsubsti
~uierten Anthracen-Radikalanions (a~ = -7.71 MHz, a~ = -4.26
MHz, a~ 14.97 MHz; K/DME, 240K). Hieraus ist zu schließen,
daß die Xylylreste nahezu senkrecht zum Grundgerüst stehen.
Ubereinstimmend mit der Änderung der Grundkörperprotonenkopp
lungskonstanten findet man in der genannten Reihenfolge eine
starke Abnahme der Beträge der para-Protonenkopplungskonstan
ten, wobei Verbindung 3' 9 unter der Annahme von laH I= H - para
jap-CH3
j berücksichtigt wird. Die meta-Protonenkopplungskon-
stanten haben in der untersuchten Reihe nahezu gleiche Werte;
sie zeigen sich als unabhängig vom Verdrillungswinkel e. Wäh
rend die Zunahme des Verdrillungswinkels von 1' 9 nach 2 ' 9 und
4' 9 durch die verstärkte Wechselwirkung der o~tho-Met~ylgruppen mit den peri-ständigen H-Atomen verursacht wird, ist sie
für }.9 durch induktive Effekte der Methylgruppen erklärbar.
Der +I-Effekt der Methylgruppe in para-Position führt zu ei
ner erhöhten Elektronendichte im Phenylring, die bei negati
ver Ladung des Systems zu einer Vermindung der Elektronende
lokalisation unter Vergrößerung des Verdrillungswinkels zwi
schen Grundkörper und Substituent im Vergleich zum neutralen
Molekül führt.

- 86 -
Die Meßergebnisse der Radikalkationen bestätigen ebenfalls
die Erwartungen der Zunahme des Verdrillungswinkels in der
Reihe - l·m, ~·$, 4•$ -. Damit übereinstimmend wird in dieser
Reihe wiederum eine Vergrößerung der Beträge der Kopplungskon
stanten der Grundkörperprotonen beobachtet, während gleichzei
tig die Beträge der para-Protonenkopplungskonstanten erheblich
abnehmen. Beim Vergleich der Meßwerte der Radikalkationen fal
len die dem Betrag nach kleinen Werte der Grundkörperprotonen
kopplungskonstanten für 3•$ auf, die auf einen verminderten -b ·E& h "b .e Verdrillungswinkel gegenü er 1 und auc gegenu er l deu-
ten, was auch durch die betragsmäßig sehr große ortho-Proto
nenkopplungskonstante bestätigt wird. Auch dieser Befund läßt
sich durch den positiven induktiven Effekt der para-ständigen
Methylgruppe erklären. Die positive Ladung des Systems einer
seits und die durch die Methylgruppe erhöhte Elektronendichte
des Phenylringes andererseits führen zur Erhöhung der Deloka
lisation unter Verminderung des Verdrillungswinkels. Die Be
trachtung der meta-Kopplungen zeigt, daß diese im Vergleich
zu den übrigen Phenylprotonenkopplungen nur geringe Verände
rungen bei Zunahme des Verdrillungswinkels zeigen. Dieser Be
fund, unterstützt durch die Konstanz der meta-Protonenkopp
lungskonstanten der Radikalanionen, ist durch eine Kompensa
tion der bei zunehmender Verdrillung verminderten n-Spindich
tedelokalisation durch gleichzeitig verstärkte n-o-Delokali
sation zu erklären. Dieses Ergebnis steht in Obereinstimmung
mit INDO-Rechnungen, die annähernd gleiche Werte der meta
-Kopplungenbei Variation von e fordern (vgl. Kapitel 3.2.6.).
Beim Ubergang vom Radikalanion zum Radikalkation werden
Änderungen aller Kopplungskonstanten gefunden. Während für
die Beträge der Kopplungskonstanten des Grundkörpers sowohl
Vergrößerungen als auch Verkleinerungen auftreten, werdenfür
alle Arylprotonenkopplungskonstanten beim Radikalkation dem
Betrag nach größere Werte erhalten als beim entsprechenden
Radikalanion. Die Anwendung der die Ladungsabhängigkeit der
Kopplungskonstanten berücksichtigenden Beziehungen (26) bzw.
(27) erschien nicht sinnvoll, da sie auf Spindichten aus
MO-Verfahren basieren, und so in jedem Fall für die Phenyl
protonenkopplungen falsche Werte liefern. Weiterhin lassen

- 87 -
sie nur Veränderungen der Beträge der Kopplungen im Sinne von
Vergrößerungen zu, was den experimentellen Befunden widerspricht.
Die Betrachtung der meta-Protonenkopplungskonstanten zeigt
eine starke Vergrößerung beim Ubergang vom Radikalanion zum
Radikalkation, die durch Berechnungen aufgrund des Hyperkon
jugationsmodell auch vorhergesagt wird (vgl. Kap. 3.2.6.).
Die Meßergebnisse zeigen aber auch eine, jedoch wesentlich
schwächere Zunahme der Beträge der beiden übrigen Phenylpro
tonenkopplungen. Dieser Befund deutet auf eine Zunahme der
n-o-Delokalisation beim Ubergang vom Anion zum Kation hin.
Betrachtet man nämlich den Betrag des Verhältnisses der meta
zur para-Protonenkopplungskonstanten, so ergibt sich beia~
und i für das Kation ein wesentlich größerer Wert von lamet~~ als beim Anion. Offenbar ist für den Spintransfermecha- para
nismus nicht nur der Verdrillungswinkel sondern ebenfalls
die Ladung des Radikals von Bedeutung.
Eine sehr starke Vergrößerung der Methylkopplungen ist bei •!Jj •!Jj
den Verbindungen ~ bis i im Vergleich zu den entsprechen-
den Radikalanionen festzustellen. Dieser in der Literatur
vielfach beobachtete Effekt wurde anhand des Methylhyperkon
jugationsmodells erklärt /49, 52/. Der Spintransfer zu den
ß-Protonen einer Methylgruppe wird im wesentlichen durch
Hyperkonjugation hervorgerufen, welche durch zunehmende Po
sitivierung des substituierten a-C-Atoms begünstigt wird und
damit zu einer Vergrößerung der Aufspaltung durch die Methyl
protonen führt.
Bemerkenswert ist noch das Ergebnis, daß die Kopplungskon
stanten der ortho-ständigen Methylgruppen der Radikalionen
von 2 und 4 sehr klein sind. Während die Kopplungskonstanten
der Methylprotonen in para-Stellung bei 1" 8 erwartungsgemäß
einen vergleichbaren Betrag wie die der entsprechenden para
-Protonen bei 1" 8 zeigen, sind die Kopplungskonstanten der - ·e ·lll
Methylgruppen von ~ und auch ~ dem Betrag nach wesent-
lich kleiner als die des ortho-Protons und auch kleiner als
die des para-Protons. Möglicherweise ist hierfür eine direk
te Wechselwirkung der ortho-ständigen Methylgruppen mit dem
n-System des Grundkörpers durch den Raum verantwortlich.

- 88 -
Für das Radikalanion des 1,2,3,4-Tetraphenylnaphthalins (5a)
findet man für die Phenylprotonenkopplungskonstanten sowohl der
Phenylringe in 1,4-Position wie auch in 2,3-Position die kon
ventionelle Reihenfolge Ja~aral > Ja~rthol > la~etal . Demnach liefert hier der Phenylhyperkonjugationsmechanismus keinen do
minierenden Betrag. Die Begründung dafür ist in verminderten
Verdrillungswinkeln der Phenylringe im Vergleich zu den Verbin
dungen 1 bis ! zu suchen. Eine genauere Betrachtung der Kopp
lungskonstanten zeigt, daß der Betrag des Verhältnisses von
meta- zu para-Kopplung für die Phenylreste in 1,4-Position
etwa doppelt so groß ist wie für die 2,3-Position (0.38 bzw.
0.18), was auf einen merklichen Anteil der rt-o-Delokalisation
im ersteren Fall deutet. Die Wechselwirkung mit dem perl-stän
digen H-Atom führt demnach zu einer stärkeren Verdrillung als
die mit einem benachbarten Phenylring. Dies konnte auch an
phenylsubstituierten Phenalenylen beobachtet werden /53/. Aus
den EPR- und ENDaR-Untersuchungen von 1-Phenyl-, 2-Phenyl- und
1,2-Diphenylphenalenyl ergab sich jeweils für den Phenylring , meta in Position 1 ein wesentlich größerer Wert von ,._a ____ l als
para für den Phenylring in Position 2, was auf einen v~rstä kten
Einfluß der hyperkonjugativen Spindelokalisation und einen
größeren Verdrillungswinkel des Phenylringes 1 hindeutet.
Um die Verdrillungswinkel im Tetraphenylnaphthalinradikal
anion abschätzen zu können, wurden HMO/McLachlan-Rechnungen
für verschiedene Winkel durchgeführt. Die beste Ubereinstim
mung mit dem Experiment ergab sich dabei für Verdrillungswin
kel der Phenylreste von etwa 60° (1,4-Position) bzw. 50°.
(2,3-Position; s. Tabelle 6). Die Verdrillungswinkel der
Phenylringe in Position waren mit der NMR zu 55° bestimmt
worden, die berechneten Werte sind demnach möglicherweise
etwas zu groß, ihre Größenordnung scheint jedoch vernünftig.
Beide Methoden erhalten für das Tetraphenylnaphthalin Werte,
die wesentlich größer als die für die monosubstituierten Ver
bindungen 1- bzw. 2-Phenylnaphthalin abgeschätzten sind
(20-30°) /54, 55/.
Die Untersuchung des Radikalanions von ergab ebenso
wie bei 5a· 8 für die meta-Protonenkopplungskonstanten die
dem Betrage nach kleinsten Werte der Phenylprotonenkopp-

- 89 -
lungskonstanten (Tabelle9).Auch hier leistet offenbar die Phenyl
hyperkonjugation einen ungeordneten Beitrag zum Spintransfer, was
wiederum auf nicht ausreichend große Verdrillungswinkel zurück
zuführen ist. Betrachtet man wieder das Verhältnis von meta- zu
para-Kopplung, so ist eine Vergrößerung seines Betrages beim
Ubergang vorn Anion zum Kation festzustellen {0.26 bzw. 0.76).
Dieser Befund zeigt, ebenso wie bei den Verbindungenluna !, daß die positive Ladung beim Radikalkation zu einer Verstär-
kung des hyperkonjugativen Spintransfers führen kann. Zur Ab
schätzung des Verdrillungswinkels wurde eine HMO/McLachlan
-Rechnung durchgeführt. Diese liefert für Radikalanionen und
Radikalkationen gleiche Spinpopulationen und damit werden nach
der McConnell-Beziehung auch gleiche Kopplungskonstanten erhal
ten. Die beste Ubereinstimmung der berechneten mit den experi
mentellen Werten wird dabei für einen Verdrillungswinkel von
55° erhalten. Nach den die Ladungsabhängigkeit der Kopplungs
konstanten berücksichtigenden Beziehungen (26) und (27) wer-
den beim Ubergang vom Anion zum Kation ausschließlich Ver
größerungen der Beträge der Kopplungskonstanten erhalten.
Folglich war keine Obereinstimmung der experimentellen mit
den nach den genannten Beziehungen berechneten Werten zu er
warten. Deshalb wurde auf diese Berechnung verzichtet.
Im Falle des Radikalanions ·e war die Frage von Inter
esse, ob es eher als ein Naphthalin- oder als Fluoranthen
derivat zu beschreiben wäre. Sowohl das Experiment als auch
die HMO-McLachlan-Rechnung beantworten diese Frage eindeutig
im letzteren Sinne (Tabelle 7). Hieraus folgt allerdings,
daß die n-Spinpopulation in den Positionen 7 und 12 und da
mit auch die Protonenkopplungen der Phenylreste sehr klein
sind. Eine Aussage über den Spindichtetransfer in die Phenyl
ringe ist dementsprechend hier nicht möglich.

90 -
4.6.2 Diskussion der Meßergebnisse der Cycloalkylanthracene
Durch Analyse der experimentell ermittelten Kopplungskonstan
ten der Verbindungen 2' 9 und lQ'9 sind Informationen sowohl
Über die Molekülstruktur als auch über die Spintransfermecha
nismen zu gewinnen. Dabei stellen Modellrechnungen eine wert
volle Hilfe dar.
Aus einem Vergleich der ß-Protonenkopplungskonstanten bei
der Verbindungen ergibt sich, daß sich die geometrische Anord
nung der ß-Protonen im Bezug zum n-System des Grundkörpers
drastisch unterscheiden muß, denn a~ 12. 9 ) ist bei vergleich
barer Spindichte im Grundkörper um mehr als den Faktor 10
größer als a~ ( ' 9 ) • Für beide Verbindungen kann man an
nehmen, daß durch Wechselwirkungen des Substituenten mit
den perl-ständigen H-Atomen keine freie Drehbarkelt der Sub
stituenten gegeben ist. Vielmehr werden sie durch Drehung
um die Ca - c 6 - Bindung Schwingungen um ihre Gleichgew~chts
konformation ausführen, die zur Variation des Winkelse~ füh
ren (vgl. Kapitel 3.2.4.). Der Winkel0~ zwischen der Achse
des cr-C-H-Orbitals und der des pz-Orbitals des u-C-Atoms
gibt über das Maß der hyperkonjugativen Wechselwirkung Aus
kunft. Der Beitrag der n-cr-Delokalisation zur Spindichte er
reicht für 0~ = 0° seinen maximalen Wert, für e~ = 90° ist
er minimal. Die energieärmsten Konformationen beider Mole
küle wurden durch Berechnungen nach dem MINDO-Verfahren be
stimmt /51/. Dieses stellt ein modifiziertes INDO-Verfahren
zur Ermittlung der Energien verschiedener Konformere dar.
Beim 9-Cyclopropylanthracen wurde die energieärmste Konfor
mation für den Winkel 9~ = 0° bestimmt. Für diesen Winkel
ist eine maximale n-o-Delokalisation möglich, die zu hoher
Spindichte am Ort des ß-Protons führt und somit auch den ho
hen Wert der ß-Protonenkopplungskonstante erklärt. Diese
energieärmste Konformation zeigt CS-Symmetrie, wobei die
Spiegelebene senkrecht zur Ebene des Anthracens steht und
die C-Atome 9, 10 und c6
in dieser Ebene liegen {Abb. 39).
Ubereinstimmend damit werden zwei Paare jeweils äquivalen
ter l-Protonen gefunden und auch die Protonen in den Posi
tionen 1 und 8 des Anthracens sind identisch. Ganz anders
sind dagegen die Verhältnisse beim 9-Cyclohexylanthracen.
I
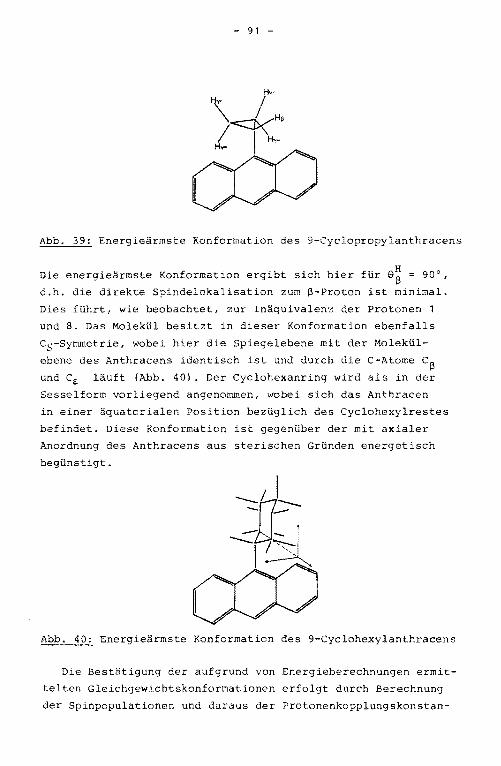
- 91 -
Abb. 39: Energieärmste Konformation des 9-Cyclopropylanthracens
Die energieärmste Konformation ergibt sich hier für 9~ = 90°,
d.h. die direkte Spindelokalisation zum ß-Proton ist minimal.
Dies führt, wie beobachtet, zur Inäquivalenz der Protonen 1
und 8. Das Molekül besitzt in dieser Konformation ebenfalls
Cs-Symrnetrie, wobei hier die Spiegelebene mit der Molekül
ebene des Anthracens identisch ist und durch die C-Atorne Cß
und CE läuft (Abb. 40). Der Cyclohexanring wird als in der
Sesselform vorliegend angenommen, wobei sich das Anthracen
in einer äquatorialen Position bezüglich des Cyclohexylrestes
befindet. Diese Konformation ist gegenüber der mit axialer
Anordnung des Anthracens aus sterischen Gründen energetisch
begünstigt.
Abb. 40: Energieärmste Konformation des 9-Cyclohexylanthracens
Die Bestätigung der aufgrund von Energieberechnungen ermit
telten Gleichgewichtskonformationen erfolgt durch Berechnung
der Spinpopulationen und daraus der Protonenkopplungskonstan-

- 92 -
tennachder INDO-Methode /51/. Die beste Obereinstimmung mit
den experimentellen Werten ergibt sich dabei für die beschrie
benen energieärmsten Konformationen. Bei Verbindung 2' 9 wird
erwartungsgemäß ein sehr großer Wert für die ß-Protonenkopp
lungskonstante erhalten, der bei stärkerer Berücksichtigung
der Schwingungen um die Gleichgewichtslage noch bessere Ober
einstimmung mit dem experimentellen Wert zeigen würde. Für die
y-Protonenkopplungskonstanten wird sowohl ein positiver als
auch ein negativer Wert berechnet. Eine positive Kopplungskon
stante, sie wird für die exo-ständigen Protonen erhalten, be
deutet aber, daß der dominierende Beitrag zum Spintransferme
chanismus zu den entsprechenden Protonen die n-o-Delokalisa
tion durch den Raum darstellt. Die dazu notwendige sterische
Anordnung zum n-System ist für die exo-ständigen Protonen of
fenbar gegeben. Sie liegen nahezu in der koplanaren "W"-Anord
nung vor, die einen hyperkonjugativen Spintransfermechanismus
begünstigt {vgl. Kapitel 3.2.5.). Für die endo-st&ndigen y-Pro
tonen, für sie trifft eine "anti-W"-Anordnung zu, wird überein
stimmend mit diesereinfachen Betrachtungweise auch nach der
INDO-Rechnung eine negative Kopplungskonstante erhalten, die
auf die Dominanz der Spinpolarisation beim Spintransfer zurück
zuführen ist. Aus der INDO-Rechnung ergibt sich für Verbindung
lQ·e entsprechend der Molekülsymmetrie ein kleiner, positiver
Wert für die ß-Protonenkopplungskonstante. Für die beiden y-Pro
tonenkopplungskonstanten werden fast identische, negative Wer
te erhalten, die auf verschwindende Beiträge der n-o-Spindelo
kalisation und Dominanz der Spinpolarisation deuten. Auch hier
läßt sich somit die einfache "Zick-Zack"-Regel bestätigen, da
hier alle y-Protonen in einer "anti-W"-Konformation vorliegen.
Ebenso gültig erweist sich diese Regel für die o-Protonen. Das
o-Proton in äquatorialer Position befindet sich am Ende einer
"Zick-Zack"-Kette bezüglich des spintragenden p2-0rbitals am
C-Atom 9, also einer Anordnung, die zu einer Begünstigung der
konjugativen Spindelokalisation führen soll. Obereinstimmend
damit liefert die INDO-Rechnung fLir die oe-Protonen relativ
große, positive Kopplungskonstanten. Für die -Protonen wer
den dagegen geringere, ebenfalls positive Kopplungskonstanten
erhalten, wobei zu berücksichtigen ist, daß bei einem durch

- 93
vier o-Bindungen vom n-System getrennten Proton ebenfalls die
Spinpolarisation zu einer positiven, wenn auch geringen Spin
population führt.
4.7. Untersuchung weiterer Radikalkationen
Die Anwendung der in Kapitel 4.5.2. beschriebenen Methode
zur Darstellung von Radikalkationen lieferte in einigen Fäl:!.en
radikalisehe Spezies, deren Struktur nicht geklärt werden konn
te. So werden bei der Oxidation von
7,10-Diphenylbenzo[k]fluoranthen (7a- ~)
Anthracen l..!.ll
9-Phenylanthracen (~)
Radikale erhalten, die eindeutig nicht die monomeren Radikal
kationen der entsprechenden Verbindungen darstellen.
Bei Verbindung 7a handelt es sich um einen nicht-altcernie
renden Kohlenwasserstoff, für den aufgrunddes Fehlens der
"pairing"-Eigenschaften die Spindichteverteilungen im Radikal
anion und Radikalkation verschieden sein sollen. Aus diesem
Grunde schien es interessant, das entsprechende RadikaJ.kation
zu untersuchen. Das ENDOR-Spektrum des Radikalkations, ent-
standen aus Verbindung zeigt acht Linienpaare (Abb. 41).
Im ENDOR-Spektrum der entsprechenden teildeuterierten Spezies
aus Verbindung 7b fehlen die Linien, die den beiden kleinsten
Kopplungskonstanten zugehören. Die Vorzeichenbestimmung die
ser beiden Kopplungskonstanten ist schvlierig, da die zugehö
rigen Linien keinen eindeutigen TRIPLE-Effekt zeigen (s. Abb.
42}. Während die größere der beiden Kopplungskonstanten posi
tiv zu sein scheint, ist über das Vorzeichen der kleineren
Kopplung keine Aussage möglich. Aus dem EPR-Spektrum dieses
Radikals, das aus einer mäßig strukturierten Einhüllenden be
steht, kann die Gesamtaufspaltung A zu ca. 16.6 G bestimmt
werden. Im 1H-ENDOR-Spektrum des Radikalkations aus Verbin-
dung zeigen zwei Linienpaare erheblich verminderte Inten-
sität, damit kann die Zuordnung der zugehörigen Kopplungskon
stanten zu den Positionen 8-11 der Ausgangsverbindung ge
troffen werden. Im Deuterium-ENDOR-Spektrum sind die ent-
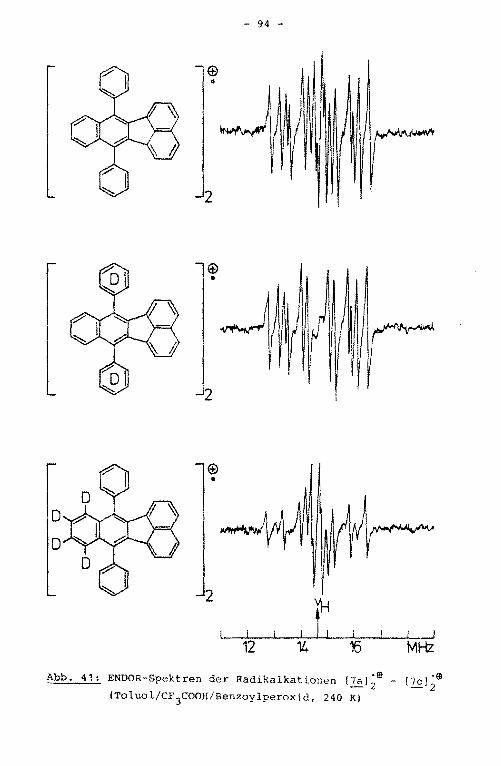
0
0
2
@ •
2
@ •
2
- 94
YH ,t, 12 16
Abb. 41: ENDOR-Spektren der Radikalkationen [7a];e
(Toluol/CF3COOH/Benzoylperoxid, 240 K)
I I
MHz

- 95 -
16
Abb. 42: TRIPLE-Spektrum des Radikalkations 7a
CF3
COOH/Benzoylperoxid, 240 K)
I I MHz
(Toluol/
sprechenden Linien nicht aufgelöst. Die Interpretation der be
schriebenen Spektren führt zu der Annahme, daß die untersuchte
Spezies ein unsymmetrisches, dimeres Radikalkation darstellt.
Diese These wird durch folgende Uberlegungen gestützt:
1. Mit dem aus den ENDOR-Spektren erhaltenen Kopplungskon
stanten müßte sich für das monomere Radikalkation 7b·~ eine Gesamtaufspaltung A~9.14 G ergeben, unter der An
nahme eines Dirneren wird dagegen ein Wert von ca. 15 G
erhalten, der in der Größe des experimentell ermittel
ten Wertes liegt.
2. Die aus der HMO-McLachlan-Rechnung erhaltenen Kopplungs
konstanten stimmen sehr schlecht mit den experimentellen
Werten überein, während bei den Radikalanionen eine gute
Ubereinstimrnung festgestellt wurde (vgl. Tabelle 7). Zum
Beispiel werden von der Rechnung auch positive Grundkör
perprotonenkopplungskonstanten vorhergesagt, das Experi
ment liefert jedoch für alle Kopplungskonstanten der
Grundkörperprotonen negatives Vorzeichen.
3. Da sich aus der Untersuchung des Radikals aus 7b ergibt,
daß die Phenylkopplungen zum Teil äquivalent sind und

- 96 -
deswegen nur zwei ENDOR-Linienpaare hervorrufen, sollte
für ein monomeres Radikalkation ein ENDOR-Spektrum aus
sieben Linienpaaren resultieren. Die experimentell ge
fundenen acht Linienpaare sind durch Inäquivalenzen im Be
reich der Protonen 1-6 erklärbar. Im Monomeren, und auch
in einem symmetrischen Dimeren, stellen sie drei Sätze
äquivalenter Protonen dar, im untersuchten Radikal wer
den vier verschiedene Protonensorten erhalten. Diese
Inäquivalenz bedingt jedoch, daß dem Radikalkation nicht
die in der Literatur für andere Kohlenwasserstoffradi
kalkationen häufig postulierte Struktur einer symmetri
schen Sandwichverbindung /56, 57, 58/ zukommt.
Bei der untersuchten Spezies handelt es sich deshalb vermut
lich nn ein dimeres Radikalkation, bei dem die Molekülteile
nicht spiegelbildlich zueinander angeordnet sind, sondern ge
genei:<ander etwas verdreht sind.
So·,.;ohl das monomere Radikalkation von Anthracen
(a1
= 18.22 MHz, a 2 = 8.63 MHz, a 3 = 3.87 MHz /57/; g
als a~ch dessen dimeres Radikalkation
2. 00257 /6/:
(a 1 = 9.11 MHz, a 2 = 3.98 MHz, a 3 = 1.99 MHz; g = 2.002562 /57/)
sind bekannt. Das durch Oxidation von Anthracen (l_l) mit Benzoyl
peroxid gebildete Radikal ist mit keiner der genannten Spezies
identisch, denn hinsichtlich der Kopplungskonstanten und des
g-Faktors wird keine Ubereinstimmung mit den obigen Werten ge
funde~. Aus den EPR- und ENDOR-Spektren (Abb. 43) des Radikals,
das a~ch durch Oxidation mit 2,3-Dichlor-5,6-dicyano-p-benzochinon
oder Azo-bis-isobutyronitril dargestellt werden kann, sind drei
Koppl~ngskonstanten und der g-Faktor ermittelbar:
(a 1 = 8.38 MHz (4H), a 2 = 1.79 MHz (2H), a 3 = 0.40 MHz; g = 2.00307'
Aufgr~nd der Abweichung des g-Faktors scheint es sich bei der
untersuchten Verbindung nicht um ein Kohlenwasserstoffradikal
zu handeln, vergleichbare g-Faktoren sind jedoch für sauerstoff
haltige radikalisehe Anthracenderivate gefunden worden /59, 60/.
So wicd für das Radikalkation des 9,10-Dihydroxyanthracens /59/
ein g-Faktor von 2.00309 ermittelt, die Kopplungskonstanten
dieses Radikals
(a 1 = 4.35 MHz, a 2 CH
3N0
2)
2.91 MHz, a 3 (OH)
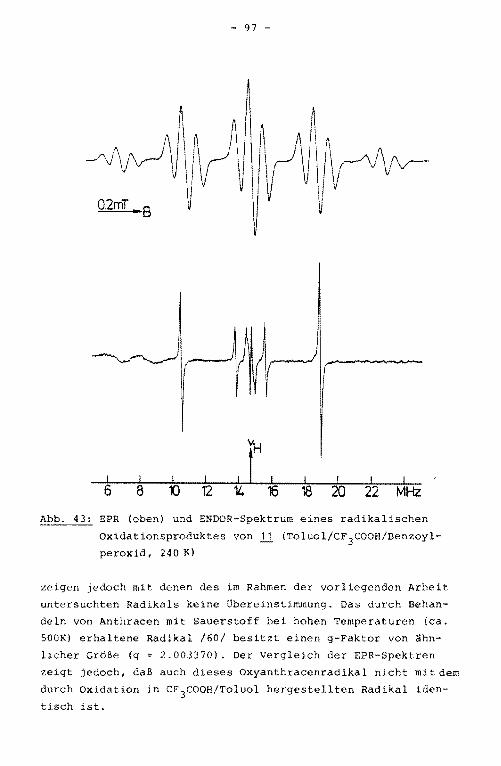
- 97 -
,I, I I
6 8 12 u. 16 18 20 22 MHz
Abb. 43: EPR (oben) und ENDOR-Spektrum eines radikalischen
Oxidationsproduktes von 11 (Toluol/CF3COOH/Benzoyl
peroxid, 240 K)
zeigen jedoch mit denen des im Rahmen der vorliegenden Arbeit
untersuchten Radikals keine Ubereinstimmung. Das durch Behan
deln von Anthracen mit Sauerstoff bei hohen Temperaturen (ca.
500K) erhaltene Radikal /60/ besitzt einen g-Faktor von ähn
licher Größe (g = 2.003370). Der Vergleich der EPR-Spektren
zeigt jedoch, daß auch dieses Oxyanthracenradikal nicht mitdem
durch Oxidation in CF 3COOH/Toluol hergestellten Radikal iden
tisch ist.
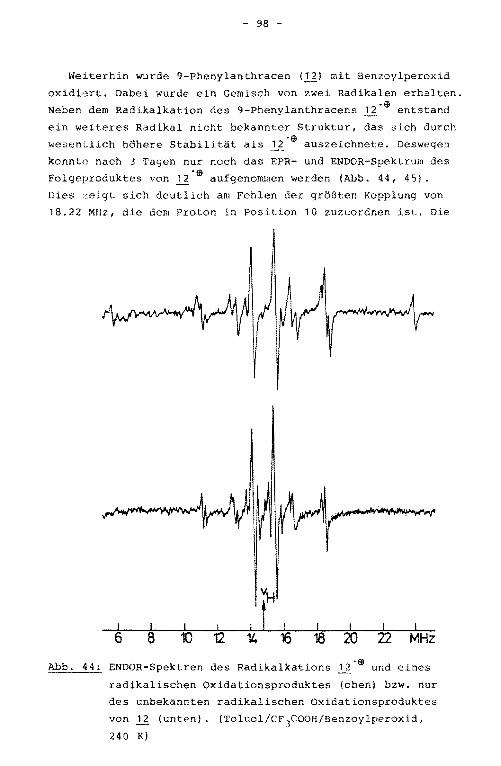
- 98 -
Weiterhin wurde 9-Phenylanthracen (j1) mit Benzoylperoxid
oxidiert. Dabei wurde ein Gemisch von zwei Radikalen erhalten.
Neben dem Radikalkation des 9-Phenylanthracens ·Efi entstand
ein weiteres Radikal nicht bekannter Struktur, das sich durch ·(&
wesen~lich höhere Stabilität als J1 auszeichnete. Deswegen
konnt•~ nach 3 Tagen nur noch das EPR- und ENDOR-Spektruro des 'Efi Folgeproduktes von J1 aufgenommen werden (Abb. 44, 45).
Dies zeigt sich deutlich am Fehlen der größten Kopplung von
18.22 MHz, die dem Proton in Position 10 zuzuordnen ist. Die
6 8 10 12 14 16 18 20 22 MHZ
Abb. 44: ENDOR-Spektren des Radikalkations 12·@ und eines
radikalischen Oxidationsproduktes (oben) bzw. nur
des unbekannten radikalischen Oxidationsproduktes
von J1 (unten). {Toluol/CF3COOH/Benzoylperoxid,
240 K)
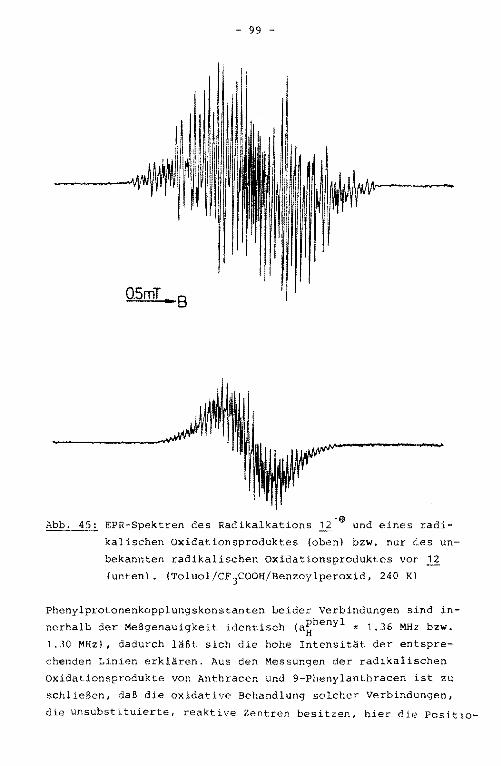
- 99 -
8
Abb. 45: EPR-Spektren des Radikalkations ~·~ und eines radi
kalischen Oxidationsproduktes (oben) bzw. nur ces un
bekannten radikalischen Oxidationsproduktes vor 12
(unten). (Toluol/CF 3COOH/Benzoylperoxid, 240 K)
Phenylprotonenkopplungskonstanten beider Verbindungen sind in
nerhalb der Meßgenauigkeit identisch (a~henyl = 1.36 MHz bzw.
1.30 MHz), dadurch läßt sich die hohe Intensität der entspre
chenden Linien erklären. Aus den Messungen der radikalischen
Oxidationsprodukte von Anthracen und 9-Phenylanthracen ist zu
schließen, daß die oxidative Behandlung solcher Verbindungen,
die unsubstituierte, reaktive Zentren besitzen, hier die Positio-

- 100 -
nen 10 bzw. 9 und 10, nicht zu Kohlenwasserstoffradikalkationen
führt. Werden die Radikalkationen solcher Verbindungen gebildet,
scheint ihre Lebensdauer sehr gering zu sein. Die Oxidation
führt aber zu anderen, wahrscheinlich sauerstoffhaltigen, sta
bilen Radikalen, deren Struktur ungeklärt ist. Um exakte Struk
turaussagen treffen zu können, ist es erforderlich, die genann
te Methode der Erzeugung von Radikalkationen an einer Reihe
weiterer, unsubstituierter, aromatischer Kohlenwasserstoffe
anzuwenden und die entstehenden radikaliseben Oxidationspro
dukte zu untersuchen.

- 101 -
5.1. Messungen
Die NMR-Spektren wurden in CDC1 3 mit dem Gerät Brukcr WH 270
aufgenommen.
Die Aufnahme der Massenspektren erfolgte mit dem Gerät CH 5-DF
Varian-MAT.
Die EPR-Messungen wurden mit den Elektronenresonanzspektro
metern AEG 12 X (125 kHz Feldmodulation) bzw. Bruker ER 220 D
(verwendete Feldmodulation 12.5 kHz) durchgeführt. Die Aufnah
me der ENDOR- und TRIPLE-Spektren erfolgte mit dem Elektronen
resonanzspektrometern AEG 20 XT tzw. Bruker ER 220 D mit in
diesem Laboratorium aufgebautem ENDOR-Zusatz /61/. Die Einstel
lung der Meßtemperatur erfolgte durch Regulierung eines Stick
stoffstromes (Temperiereinrichtungen der Firmen AEG und Bruker).
Die ENDOR-Spektren der Radikalanionen l·e, 2" 8 , .a sowie
aller Radikalkationen wurden mit einer digitalen Speicherein
heit (Nicolet 1174) kumuliert.
5.2. Radikalerzeugung
Die Radikalanionen wurden nach Standardverfahren /62/ durch
Reduktion der neutralen Verbindungen mit Kalium in Dimethoxyethan
(DME) erzeugt. Dazu wurde das Kalium durch mehrmalige Destilla
tion im Destillierarm des Probengefäßes gereinigt und als Spie
gel auf die Glaswand des dafür vorgesehenen Teils des Probenge
fäßes aufgebracht. Das gereinigte und über einer Natrium/Kalium
-Legierung getrocknete DME wurde in ein Vorratsbehälter des Pro
bengefäßes eindestilliert. Nach sorgfältigem Entgasen des Lö
sungsmittels durch wiederholtes Einfrieren und Auftauen an ei
nem Hochvakuumpumpstand wurde das Probengefäß abgeschmolzen.
Bringt man die Lösung nun in Kontakt mit dem Kaliumspiegel,
tritt die Reduktion zum Radikalanion ein. Änderungen der Ra
dikalkonzentrationen können durch Destillation des Lösungsmit
tels innerhalb des Probengefäßes erfolgen.
Zur Darstellung der Radikalkationen wurde die Substanz in
ein Meßröhrchen eingebracht und mit einem Gemisch aus ca.
0.3 ml Trifluoressigsäure und 3 ml Toluol versetzt. Nach Zu-

- 102 -
gabe von Benzoylperoxid entstanden innerhalb weniger Minuten bei
Raumtemperatur die Radikalkationen. Zur Durchführung der EPR
und ENDOR-Messungen wurden die Radikallösungen durch Perlen mit
Reinstickstoff von Sauerstoff befreit. Die Meßröhrchen wurden
mit Schliffstopfen verschlossen. Die Radikalkationen zeigten
bei Aufbewahrung bei -30°C eine Lebensdauer von mehreren Wochen.
5.3. Darstellung der Verbindungen
Die Beschreibung der Synthesen soll sich hier auf die in
der Literatur bisher nicht bekannten Verbindungen beschränken.
Zur Darstellung der übrigen Verbindungen sei auf die angege
bene Literatur verwiesen.
Die angegebenen Schmelzpunkte sind unkorrigiert, sie wur
den mit einem Schmelzpunktapparat nach Tottoli (Fa. Büchi)
bestimmt. Die Durchführung der Analysen erfolgte nach der
Methode von Pregl.
9,10-Bis(2,6-dimethylphenyl)anthracen (il
Durch langsames zutropfen unter Stickstoff von 15 g (0.081 mol)
2-Brom-1,3-dimethylbenzol in 50 ml wasserfreiem Ether zu 1.25 g
(0.18 mol) Lithium in wasserfreiem Ether wurde eine Xylyllithium
lösung erhalten. In diese wurden innerhalb 30 min 5.0 g (0.024
rnol) Anthrachinon eingetragen. Es entstand eine dickflüssige,
orange Mischung, die nach Verdünnen mit wasserfreiem Benzol
1 h unter Rückfluß erhitzt wurde. Nach vorsichtiger Hydrolyse
mit Eiswasser wurde ein Feststoff erhalten, aus dem nach Absau
gen und Trocknen durch Extraktion mit siedendem Essigsäureethyl
ester 7. 0 g ( 69.4% l 9, 1 0-Dihydr.;xy-9, 1 O-bis( 2, 6-dimethylphenyl)-
9,10-dihydroxyanthracen erhalten werden.
Das erhaltene Biscarbinol wurde ohne weitere Reinigung mit
8 ml 67 proz. HI-Lösung in 40 ml Eisessig versetzt und 15 min
unter Rückfluß erhitzt. Nach dem Abkühlen wurde der ausgefal
lene Kohlenwasserstoff abgesaugt, getrocknet und anschließend
unter Zusatz von Aktivkohle aus Benzol/Cyclohexan (1:1) umkri
stallisiert. Es wurden 5.9 g (92.1%) 1 erhalten, Schmp. 295-297"C.
c30H26 (386.5) Ber. c 93.22 H 6.78
Gef. C 93.30 H 6.79

- 103 -
1,2,3,4-Tetraphenylnaphthaline (~-Q)
Die verschieden deuterierten Tetraphenylnaphthaline Sb-d
wurden nach Literaturverfahren /36/ aus den entsprechenden
deuterierten Ausgangsverbindungen synthetisiert. Dazu wurden
0.5 g (0.0013 mol) des entsprechenden teildeuterierten Tetra
phenylcyclopentadienons in 1.5 g Triethylenglykoldimethylether
(Triglyme) gelöst und bei 180-185°C 1 g (0.0029 mol) Diphenyl
iodonium-o-carboxylat zugesetzt. Die nach Abkühlen durch Zu
satz vonEthanol ausgefallenen hellgelben Feststoffe wurden
aus Benzol/Ethanol umkristallisiert.
Ausb.: 0.30 g (52.2%), Schmp.: 198-199°C
Ausb.: 0.32 g (55.4%), Schmp.: 199-200°C
Ausb.: 0.31 g (54.0%), Schmp.: 197-198°C
Die Synthese der deuterierten Ausgangsverbindungen von Sb
und Sc ist bekannt /38/.
2,5-Bis([3, ]phenyl)-2,4-cyclopentadien-1-on
60 g (0.42 mol) 4-Aminotoluolhydrochlorid wurde in Portionen
zu 20 g in jeweils 40 ml D2o sechs Stunden erhitzt, wobei das
gebrauchte n2o der ersten Portionnach Abdestillation zur Aus
tauschreaktion der zweiten Portion benutzt wurde, anschließend
zu der der dritten Portion /39/. Dieser Vorgang wurde sechs
mal durchgeführt. Nach Zugabe von wässriger NaOH-Lsg. wurden
44.2 g (96.6%) 4-Amino-[3,5-D2]toluol erhalten. 32.7 g (0.3 mol)
4-Amino-[3,5-D2 ]toluol wurden in 110 ml konz. HCl und 40 ml H2o unter Eiskühlung vorsichtig mit 21.75 g (0.32 mol) NaN02 ver
setzt. Anschließend wurden unter weiterer Eiskühlung 468 ml
(4.5 mol) 50 proz. hypophosphoriger Säure zugegeben /40/. Nach
üblicher Aufarbeitung der organische Phase wurde diese destil
liert. Es wurden 15.6 g (55.0%) [3,5-D2 ]Toluol erhal-
ten. 15 g (0.16 mol) [D2 ]Toluol wurden unter üblichen Bedin
gungen zum [D2 ]Benzylbromid bromiert (Ausb.: 55%). Dieses
(15.2 g = 0.088 mol) wurde mit 6.4 g (0.13 mol) NaCN durch
Kochen in einem Gemisch aus 10 ml Wasser und 10 ml Ethanol
in das entsprechende Benzylcyanid übergeführt. Nach Abtren
nen der wässrigen Phase wurde durch Vakuumdestillation 7.45 g

- 104 -
(71.1%) [D2 ]Benzylcyanid erhalten. 5 g des [D 2 JBenzylcyanids wur
den in einem Gemisch aus 5 ml Wasser, 5 ml konz. H2so 4 und 5 ml Eis
essig erhitzt und nach dem Abkühlen kristallisierte die
Phenylessigsäure aus. Nach Umkristallisation aus Ligroin unter
Zusatz von wenig Ethanol wurden 5.2 g (90.5%) !D 2 ]Phenylessig
säure erhalten. Diese wurde in Ethanol gelöst und zu einer
Aufschlämmunq von 2 g (0. 029 mol) Ca (OH) 2 in 4 ml Wasser ge
geben. Der entstandene Niederschlag wurde abgesaugt, nach
dessen Trocknen bei 160°C verblieben 4.0 g (68.4%) glasar-
tiges Produkt. Aus diesem Ca-Salz wurde direkt durch Fest
stoffdestillation im Vakuum 1.6 g (58.0%) [D 4 JDibenzylketon
erhalten /41/. Die Kondensation zum [D 14 Jcyclopentadienon
wurde analog zu Verbindung Sb und ~ durch Erhitzen äquimola-
rer Mengen (0.002 mol) [D 4 ]Dibenzylketon mit [D 10 JBenzil in
Triethylenglykol unter Zusatz von 0.2 ml 40 proz. Benzyltri
methylammoniumhydroxidlösung erhalten /38/.
Ausb.: 0.72 g (90.9%) Schmp.: 217°C
9,10-Diphenylbenzo(b]triphenylene (~, ~)
7,12-Diphenylbenzo[k]fluoranthene (~, ~)
Die Kondensation zu den Kohlenwasserstoffen wurde analog
zu den Verbindungen~ durch Umsetzung von 1 g bzw. 0.5 g
Phencyclon und 1 g bzw. 0.5 g Acenaphthencyclon mit einer
entsprechenden Menge Diphenyliodonium-o-carboxylat in Triglyme
durchgeführt.
6a Ausb.: 0.72 g 164.6%), Schmp.: 282-283°C
c 34 H22 (430.6) Ber. C 94.85 H 5.15 Gef. C 94.72 H 5.03
6b Ausb.: 0.36 g (63.9%), Schmp.: 279-281°C
Ausb.: 0.71 g (62.9%), Schmp.: 271°C
c 32H20 (404.5) Ber. C 95.02 H 4.98 Gef.C94.72 H 5.03
7b Ausb.: 0.46 g (81.3%), Sch:rr.p.: 268-269°C

- 105 -
1,3-Bis([ ]phenyl)-2H-cyclopenta[l]phenanthren-2-on und
7,9-Bis([ phenyl)-8H-cyclopenta[a]acenaphthylen-8-on
0.52 g (0.025 mol) Phenanthrenchinonbzw. 0.6 g (0.003 mol)
Acenaphthenchinon wurden mit 0.55 g (0.0025 mol) bzw. 0.66 g
(0.003 mol) Dibenzylketon in Ethanol aufgeschlämmt. Nach Zu
gabe von ethanolischer KOH-Lsg. und anschließendem Erhitzen
fielen schwarze Kristalle aus, die abgesaugt und mit Ethanol
gewaschen wurden /37/.
[D 10 JPhencyclon :Ausb.: 0.77 g (78.2%), Schrnp.: 268-269°C
[D 10 JAcenaphthencyclon:Ausb.: 0.89 g (81.1%), Schmp.: 285-287°C
9,14-Diphenyl-[10,11,12,1 l-benzo(b]triphenylen (6c)
7,12-Diphenyl-(8,9,10,11 ]-benzo[k]fluoranthen (7c)
Die Deuterierung der Anthranilsäure wurde entsprechend der
Vorschrift von R.A. Russell und R.N. Warrener /42/ durchge
führt. Es konnte aus technischen Gründen nur eine Austausch
reaktion erfolgen, deshalb wird ein relativ niedriger Deu
terierungsgrad erhalten (0.3% D1
, 7.5% o2
, 41.0% o3
, 51.2%
D4
J 0.38 g (0.001 mol) Phencyclon bzw. 0.37 g (0.001 mol).
Acenaphthencyclon wurden in 5 ml siedendem Dioxan gelöst.
Dann wurden gleichzeitig Lösungen von 0.17 g (0.0012 mol)
[D 4 JAnthranilsäure und 0.2 ml Isopentylnitrit in Dioxan zu
getropft /36/. Nach Entfärbung der Lösung wurde das Lösungs
mittel abdestilliert. Aus dem öligen Rückstand kristalli
sierte durch Zugabe von Ethanol ein gelber Feststoff aus.
Nach Umkristallisieren aus Chloroform/Methanol wurde oe-reines Produkt erhalten.
6c Ausb.: 0.30 g (69.0%), Schmp.: 285°C
Ausb.: 0.25 g (61.9%), Schmp.: 270-271°C

- 106 -
6. Zusammenfassung
In dieser Arbeit wird der sterische Einfluß auf den Spin
transfer bei verdrillten phenylsubstituierten und cycloalkyl
substituierten aromatischen Kohlenwasserstoffradikalen be
schrieben. In einem allgerneinen Teil werden die theoretischen
Grundlagen der angewandten Untersuchungsmethoden erörtert und
einige, die Spinverteilungen behandelnde Modellverfahren vor
gestellt.
Es wurden die Synthesen von einer Reihe teils literaturbe
kannter, teils bisher nicht bekannter, phenylsubstituierter
Abkömmlinge des Anthracens, des Naphthalins, des Benzo[b]tri
phenylens und des Benzo[k]fluoranthens durchgeführt. In eini
gen Fällen wurden spezifische Deuterlerungen vorgenommen.
Mit Hilfe von ENDOR- und TRIPLE-Experirnenten wurden Größe
und relatives Vorzeichen der Hyperfeinkopplungskonstanten der
korrespondierenden Radikalanionen, bei einigen Verbindungen
auch der der Radikalkationen bestimmt. Die Kopplungskonstan
ten wurden den jeweiligen Molekülpositionen zugeordnet, die
getroffene Zuordnung wurde durch Simulation der experimen
tellen EPR-Spektren überprüft.
Es konnte gezeigt werden, daß für die phenylsubstituierten
Anthracene die konventionelle Reihenfolge der Phenylprotonen
kopplungskonstanten nicht eingehalten wird. Die Einführung
von Methylgruppen in die ortho-Positionen der Phenylringe
führte zu dem erwarteten Effekt der zunehmenden Verdrillung
der Substituenten, welche eine Zunahme des hyperkonjugativen
Spintransfermechanismus bewirkte. Neben dem sterischen Ein
fluß wurden auch Auswirkungen der Ladung auf das Ausmaß die
ser n-a-Delokalisation festgestellt. Weiterhin wurden bei
den Radikalen des 9,10-Di-p-tolylanthracens eine Ladungsab
hängigkeit des Verdrillungswinkels festgestellt. Für die übri
gen Verbindungen der Gruppe der phenylsubstituierten aromati
schen Kohlenwasserstoffe konnte kein dominierender Beitrag der
Phenylhyperkonjugation am Spintransfer festgestellt werden.
Dies wird auf verminderte Verdrillungswinkel im Vergleich zu
den erstgenannten Systemen zurückgeführt. Die für diese Syste
me durch Anwendung einer HMO-McLachlan-Rechnung ermittelten
Spindichteverteilungen zeigen gute Ubereinstimmungen mit den
experimentellen Werten.

- 107 -
Weiterhin wurden zwei 9-Cycloalkylanthracene dargestellt.
Auch von ihren Radikalanionen wurden mit Hilfe von EPR-, ENDOR
und TRIPLE-Messungen die Hyperfeinkopplungskonstanten einschließ
lich ihrer Vorzeichen bestimmt und deren Zuordnung zu den Mole
külpositionen getroffen. Wiederum wurden Simulationen der EPR
-Spektren durchgeführt.
Es konnte die Geometrie der beiden 9-Cycloalkylanthracene be
stimmt werden, dabei stellten INDO-Berechnungen dieser Systeme
eine wesentliche Hilfe dar. Es zeigte sich, daß sich die Anord
nungen der Cycloalkylsubstituenten bezüglich des Anthracengrund
körpers drastisch unterscheiden. Außerdem wurden Spindichten am
Ort der y- und auch der 6-Protonen nachgewiesen. Aus deren Größe
und Vorzeichen konnten im Einklang mit den erwähnten Modellrech
nungen Rückschlüsse auf den jeweils dominierenden Spintransfer
mechanismus gezogen werden.
Schließlich wurde eine neue Methode zur Erzeugung von Kohlen
wasserstoffradikalkationen gefunden. Diese scheint jedoch keine
universelle Anwendbarkeit zu besitzen. Nach den bisherigen Unter
suchungen ist sie vor allem zur Darstellung von Radikalkationen
substituierter Kohlenwasserstoffe geeignet. Trotz ihrer begrenz
ten Anwendbarkeit zeichnet sich diese Methode durch einige Vor
teile aus: Die Probenbereitung ist leicht durchzuführen, durch
die Verwendung von Toluol als Hauptbestandteil des Lösungsmittel
gemisches gelingt die Messung bei relativ niederigen Temperatu
ren, die erhaltenen Spektren zeigen meist ein sehr gutes Signal
-Rausch-Verhältnis und bei der Messung von deuterierten Verbin
dungen tritt kein H/D-Austausch ein.
Bei beiden untersuchten Substanzgruppen, den phenylsubstitu
ierten und den cycloalkylsubstituierten Verbindungen, zeigt sich,
daß der Spintransfer zu den Substituenten von deren sterischer
Anordnung bezüglich des spintragenden n-Systems wesentlich be
einflußt wird. Die Dominanz hyperkonjugativer Spintransferme
chanismen wird nur bei solchen Anordnungen der Substituenten
gefunden, die direkte Wechselwirkungen von a-Orbitalen der
Substituenten mit den n-Orbitalen des Grundkörpers zulassen.

- 108 -
7. Literaturverzeichnis
/1/ K. Möbius, M. Plato: z. Naturforsch. A 24, 1078 (1969)
/2/ R. Biehl, M. Plato, K. Möbius: Mol. Phys. 985 (1978)
/3/ R. Biehl, K.-P. Dinse, K. Möbius, M. Plato, H. Kurreck,
U. Mennenga: Tetrahedron 29, 363 (1973)
/4/ R. Biehl, K. Hinrichs, H. Kurreck, w. Lubitz, U. Mennenga,
K. Roth: J. Am. ehern. Soc. 99, 4278 (1977)
/5/ P. Devolder, G. Goudmand: ehern. Phys. }2 1 307 (1978)
/6/ B. G. Segal, M. Kaplan, G. K. Fraenkel: J. ehern. Phys.
4191 (1965)
/7/ K. Möbius: Z. Naturforsch. A 20, 1093 (1965)
/8/ A. J. Stone: Mol. Phys. ~, 509 (1963)
/9/ R. Biehl, M. Plato, K. Möbius: J. ehern. Phys. ~, 3515
( 197 5)
/10/ K.-P. Dinse, R. Biehl und K. Möbius: J. ehern. Phys. 21• 4335 (1974)
/11/ E. HUckel: Z. Phys. 70, 204 (1931); 72, 310 (1931); 76,
628 (1932); 86, 632 (1933)
/12/ A. D. McLachlan: Mol. Phys. l• 233 (1960)
/13/ J. A. Pople und D. L. Beveridge: "Approximate MO-Theory",
McGraw-Hill, New York (1970)
/14/ J. P. eolpa, J. R. Bolton: Mol. Phys. ~, 273 (1963)
/15/ G. Giacornetti, P. L. Nordio, M. V. Pavan: Theor. ehim.
Acta 1• 404 (1963)
/16/ R. S. Mulliken, C. A. Riehe, W. G. Brown: J. Am. Chem.
Soc . g, 41 ( 1 941 )
/17/ J. P. eolpa, E. de Boer: Phys. Lett. ~· 225 (1963); Mol.
Phys. z, 333 ( 1963/64)
/18/ H. e. Heller, H. M. Mceonnell: J. Chern. Phys. ~, 1535
( 1960)
/19/ A. Horsfield, J. R. Morton, B. H. Whiffen: Mol. Phys. !• 425 (1961)
/20/ G. A. Russell, J. J. McDonnell, P. R. Whittle, R. S. Givens,
R. G. Keske: J. Am. ehern. Soc. 1452 (1971)
/21/ Th. A. J. W. Wajer, A. Mackor, Th. J. de Boer: Rec. Trav.
Chim. 90, 568 ( 1971)
/22/ C. Morat, A. Rassat: Bull. Soc. Chirn. Fr. 1971, 891, 893

- 109 -
/23/ D. Griller, E. c. Horswill, K. U. Ingold: Mol. Phys 27,
1117 (1974)
/24/ T. Kawamura, M. Matsunaga, T. Yonezawa: J. Am. ehern. Soc .
.1_QQ, 92 ( 1978)
/25/ G. R. Underwood, v. L. Vogel, J.-A. Iorio: Mol. Phys. ~'
1093 (1973)
/26/ M. Barfield: J. Phys. Chem. 2!, 621 (1970)
/27/ Y. Ellinger, A. Rassat, R. Subra, G. Berthier: J. Am. Chem.
Soc. 2372 (1973)
/28/ H. B. Schlegel, F. W. King: J. Mol. Structure ~' 155 (1976)
/29/ G. A. Russell, G. W. Holland, K.-Y. Chang, R. G. Keske,
J. Mattox, e. s. c. Chung, K. Stanley, K. Schmitt, R.
Blankespoor, Y. Kosugi: J. Am. ehern. Soc. 96, 7237 (1974)
/30/ P. Smith, R. A. Kaba, L. M. Dorninguez, S. M. Denning: J.
Phys. ehern. !l• 162 (1977)
/31/ G. R. Underwood, H. S. Friedrnan: J. Am. ehern. Soc. 96,
4089 (1974)
/32/ M. Plato, K. Möbius: Z. Naturforsch. A 24, 1083
/33/ R. Biehl: Dissertation, Freie Universität Berlin 1974
/34/ J. A. Pople, D. L. Beveridge: J. ehern. Phys. i1• 4725 (1968)
/35/ A. Willemart: Bull. Soc. Chim. Fr. 2• 83 (1942)
/36/ L. F. Fieser: Organic Experiments, Raytheon Education
Company, Lexington 1968
/37/ W. Dilthey, F. Quint: J. Prakt. Chern. ~' 139 (1930)
/38/ K. Hinrichs, K. Kurreck, w. Niemeier: Tetrahedron 30,
315 ( 1974)
/39/ A. P. Best, Ch. L. Wilson: J. ehern. Soc. 1946, 239
/40/ N. Kornblum: Org. React. ~, 262 (1944)
/41/ H. Apitzsch: Ber. Dtsch. Chem. Ges. 37, 1428 (1904)
/42/ R. A. Russell, R. N. Warrener: J. Labelled eompd. Radio
pharrn. li• 239 (1978)
/43/ N. L. Bauld, J. D. McDermed, eh. E. Hudson, Y. S. Rim,
J. Zoeller, Jr., R. D. Gordon, J. S. Hyde: J. Am. Chern.
Soc. 2.1_, 6666 (1969)
/44/ W. E. Bachmann, J. M. ehemerda: J. Org. Chem. !r 583 (1939)
/45/ H. Günther: Angew. Chern. 84, 907 (1972)
/46/ W. Benz: Massenspektrometrie organischer Verbindungen,
Akademische Verlagsanstalt, Frankfurt am Main (1974)

Diese Arbeit wurde unter der Leitung von Herrn Professor
Dr. H. Kurreck im Institut für Organische Chemie der Freien
Universität Berlin angefertigt.
Herrn Professor Dr. H. Kurreck danke ich für die Uberlassung
des Themas und sein ständiges Interesse am Fortgang der Arbeit
sowie für viele anregende Diskussionen und wertvolle Ratschläge.
Ausdrücklich danken möchte ich auch Herrn Dr. B. Kirste für
seine Hilfe bei den Untersuchungen und seine engagierten Dis
kussionsbeiträge sowie Herrn Professor Dr. W. Broser für die
Durchführung der INDO-Rechnungen.
Schließlich danke ich allen namentlich nicht Genannten, die
zum Gelingen dieser Arbeit beigetragen haben.







![En route to multi-bridged metallocenes · PDF filePG Protecting Group . VII Ph Phenyl ... 5.4.7 2,2-Dimethyl-1,1,1-tris[1’-(tributylstannyl)ferrocenyl]propane (175 ... (1-methoxy-1-methylethyl)oxazoline](https://img.pdfslide.org/doc/110x75/5a972f777f8b9ad96f8d0ded/en-route-to-multi-bridged-metallocenes-protecting-group-vii-ph-phenyl-547.jpg)






![chemistry.mdma€¦ · Phenylmethacrylsäure auszogehen, bei diesem Verfalhren vie] reittere Veiclhter trenr;eade eutstehen. Phenyl methaeyylsäurengethyläther. 3C E 30 g Methylafkûhol](https://img.pdfslide.org/doc/110x75/605040e037f26c3922240c8d/phenylmethacrylsure-auszogehen-bei-diesem-verfalhren-vie-reittere-veiclhter.jpg)




![4.1 Allgemeine strukturelle Merkmale ... - uni-halle.de · SYNTHESE UND EIGENSCHAFTEN 41 Schema 4.2: Synthese der substituierten 4-[4-(4-n- Hexadecyloxy-phenyliminomethyl)benzoyloxymethyl]phenyl-4-(4-n-](https://img.pdfslide.org/doc/110x75/5ec203a564588249d76c85c3/41-allgemeine-strukturelle-merkmale-uni-hallede-synthese-und-eigenschaften.jpg)
![chemistry.mdma...Die Oxvdation $ Jahrg. 74 ist 3/194 Il N -LP- (2-Methoxy-phenyl) -äthyl]-pyridiniunj bromid (VI). Atis 2 g Bromid und 0.75 g Pyridin wie oben dargestellt. Das Produkt](https://img.pdfslide.org/doc/110x75/60dd9527f6bf256fb62c2936/-die-oxvdation-jahrg-74-ist-3194-il-n-lp-2-methoxy-phenyl-thyl-pyridiniunj.jpg)







![acid to give l-phenyl-imidazo[2,l-b]quinazoline-zfn.mpdl.mpg.de/data/Reihe_B/36/ZNB-1981-36b-0366.pdf · respectively. 2 a, b, d were cyclised with aceti to givc anhydrid imidazoimidazoline](https://img.pdfslide.org/doc/110x75/5e608de3b755c6406c271e13/acid-to-give-l-phenyl-imidazo2l-bquinazoline-zfnmpdlmpgdedatareiheb36znb-1981-36b-0366pdf.jpg)
