Embed Size (px)
DESCRIPTION
Das technisch-wissenschaftliche Journal TechnoPharm richtet sich an Experten und Entscheider, die in Pharmaunternehmen und Zulieferbetrieben für Planung, Installation, Betrieb und Wartung von Produktionsanlagen und nicht zuletzt für F&E zuständig sind.
Citation preview

APV – Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e.V.
23890 ISSN 2191- 8341
www.ecv.de
Fermentation intensivierenGezielte Sauerstoffanreicherung erhöht die Ausbeute von Biotech-Prozessen
Fabrikumbau bei laufender HerstellungSicherheit in der Produktion hat oberste Priorität
Reinigung und Sauberkeit in der ProduktionSerie Teil 2: Technische Verunreinigungen bestimmen
Prozessorientierte WiegesystemePraktische Hilfestellungen für Planer und Anwender
04 ∙ 2014

Abgelegt auf: Vorstufe:GK:ECV:Satz:TechnoPharm:Archiv:TP_2013-04:Anzeigensatz-keine-Druck-PDFs:fette-TP-2013-04-216x303.indd Zuletzt gesichert: 10.07.14 (01:38:37 Uhr)
TH
E N
EW
FE
SE
RIE
S NEW FE SERIES+ Design –
Simplicity as a key to efficiency+ 360° accessibility+ Guaranteed easy and safe operation
www.fette-compacting.com

Biotech 2.O(Six Bags statt Six Packs)
Sehr geehrter Leserdes TechnoPharm Journals,
die erste kommerzielle Nutzung derrekombinanten Biotechnologie liegtbereits über 30 Jahre zurück. 1983wurde rekombinantes Insulin, vonGenentech entwickelt, von der FDAzur Vermarkung durch Eli Lilly zuge-lassen. Warum wird die Biotechnolo-gie trotz dieses gereiften Alters im-mer noch als Zukunftstechnologieangesehen? Die Antwort ist einfach:alle stakeholder dieser Querschnitts-technologie haben dazu beigetragen,dass relevante Technologiebausteineweiterentwickelt, zum Teil neu erfun-den wurden und sich jetzt zu einemStand der Technik mit völlig neuenPerspektiven synergistisch integrie-ren.
Der erste Fermenter zur Herstel-lung von rekombinanten Proteinenaus tierischen Zellkulturen im10000 l Maßstab war ein umgebauterE.coli Fermenter, das erste Gerät zurgroßtechnischen Ultra- und Diafil-tration war der Konzentrierung vonOrangensaft entliehen, die Fermenta-tionstiter lagen bei 0,1 g pro Literund die ersten Anlagenbetreiber wa-ren keine Biotechnologen, sondernChemiker bzw. Mikrobiologen, dieder Naturstoffchemie entliehen wa-ren. Lange Zeit waren die „Six Packs“Anlagen zur Herstellung von rekom-binanten Proteinen aus tierischerZellkultur im 6 × 10000 l Maßstabdas Maß aller Dinge. Welche wesent-lichen Elemente haben zu einem Pa-radigmenwechsel bei der Entwick-lung und Herstellung rekombinanterProteine beigetragen?
. Die Fermentations-Titer als Maßder Produktivität erhöhten sichvon 0,1 g/l auf bis zu 5 – 10 g/l, eineSteigerung um den Faktor von50 – 100.
. Das Produkt-Portfolio fokussiertsich auf monoklonale Antikörperaus tierischen Zellkulturen; derenSpezifität und Wirksamkeit wur-den erheblich gesteigert, auchdurch Derivatisierungen („armedantibodies“), was zur Reduzierungder notwendigen Dosis und damitzur Reduktion der zu produzie-renden Mengen führt.
. Die Fokussierung auf monoklonaleAntikörper erlaubt die Etablierungeiner Plattformtechnologie mitentsprechenden Vorteilen für Ent-wicklung und Herstellung.
. Die fragmentierte Zulieferindustriewurde konsolidiert und ist heute inder Lage „one stop shop“ Lösungenfür die Plattformtechnologie an-zubieten.
. Single-use Anwendungen ersetzenmehr und mehr Edelstahllösungenund führen zu einem Paradigmen-wechsel im Anlagenbau.
. Biosimilars wurden Realität underhöhen in positiver Weise denInnovationsdruck für die biophar-mazeutische Industrie.
. Der Studiengang Biotechnologieist an vielen Universitäten undHochschulen in Forschung undLehre etabliert und erweist sich alsInnovationsbeschleuniger.
Die Integration all dieser Entwick-lungen wird zur Ablösung der klassi-schen Six Packs mit 6 × 10000 l Re-aktorkapazität führen. In vielen Fäl-len werden modulare Produktions-
anlagen mit 6 × 2000 l single-use Re-aktoren genügen, um die entspre-chenden Märkte zu beliefern. DerNachteil des „economy of scales“wird durch geringere Investitions-kosten und -risiken, geringere Anla-genkomplexität und durch einenschnelleren Marktzugang kompen-siert.
Diese Fakten können in einemSatz aggregiert werden: „Six Bagsstatt Six Packs“. In diesem Sinne:Auf die nächsten 30 Jahre Biotech-nologie!
Ihr Hermann Allgaier
Dr. Hermann AllgaierSterile, Respiratory & SpecialtyOperations, TGOTeva Pharmaceuticals
Editorial
Dr. Hermann Allgaier

Abgelegt auf: F:\GK\ECV\Satz\Pharmind\PI_2013-07\Anzeigensatz-keine-Druck-PDFs\letzner-pi-2013-07-216x303.indd Zuletzt gesichert: 11.07.13 (12:53:19 Uhr)
Enthärtung im „sanitary design“
L Komplett in EdelstahlL Heisswasser- Sanitisierung
Umkehrosmose / Elektrodeionisation
L Letzner-Kohlensäure-PatentL Wassereinsparung bis zu 50 %
Mehrstufen Druckkolonnendestillation
L Anti-Rouging KonzeptL Vollisolierung L Leistungsregelung
LetzTOC / Mehrkanal online TOC Messung
L gemäß Ph.Eur. USP. JP16 complianceL Automatischer Systemseignungstest (SST)
Letzner Pharmawasseraufbereitung GmbH · Robert-Koch-Str. 1 · 42499 HückeswagenTel. +49(0)2192/83883 · Telefax +49(0)2192/921733 · www.letzner.de

TERMINE 178
FOKUS BIOTECHNOLOGIE
Kaltenegger, Johann 180Intensivierung aerober Fermentationendurch gezielte SauerstoffanreicherungSichere Beherrschung der Prozess-Parameter alsVoraussetzung
MASCHINEN- UND ANLAGENBAU
Enderlein, Gita 186Umbau während laufender Produktion – na und!Von der Notwendigkeit effizient und strukturiertzu agieren, um höchste Sicherheit zu gewähr-leisten
König-Birk, Juliane 192Serie: Sauberkeit und Reinigung in derProduktionTeil 2: Technische Verunreinigungen bestimmen
REINRAUM
Erens, Stefan 198Reinraum-Qualifizierungsmessungen imSterilbereich – Teil 2Regulatorische Ableitung und risikobasierteFestlegung
PROZESS- UND VERFAHRENSTECHNIK
Welser, Rita 207Media Fills
PROZESS- UND VERFAHRENSTECHNIK
Steinmetz, Konrad 210Prozessorientierten Wiegesystemengehört die ZukunftPraktische Hilfestellungen zur systematischenLösungskonzeption
Lettau, Ulrich; Quick, Andreas 214Prozessdaten: Komplexität wirdtransparent und beherrschbar
SPEKTRUM 224
MESSE
Hans Herrmann Letzner 226Lounges 2014 – Eine Retrospektive
PRODUKTE 231
IMPRESSUM 232
Inhaltsverzeichnis
TechnoPharm 4, Nr. 4, 177 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 177Inhaltsverzeichnis
BeiratsgremiumKonstantin Clevermann, Dortmund · Prof. Dr. Jürgen Hannemann, Biberach · Dr. Udo Janske, Darmstadt · Prof. Dr. Gerd Kutz,Detmold · Heinz Kudernatsch, Nürnberg · Hans Ulrich Petereit, Darmstadt · Dr. Elke Sternberger-Rützel, Freiberg a.N. · Dr. MikeSchäfers, Eschweiler · Prof. Dr. Hartwig Steckel, Kiel · Dr. Frank Stieneker, Hofheim · Roland Szymoniak, Frankfurt am Main · Dr.Jochen Thies, Warendorf · Dipl. Ing. Frank Wilde, Basel · Prof. Dr. Ing. Dominik Rabus, Forchtenberg · Dipl. Ing. Frank Lehmann,Allschwil (Schweiz)
APVnewsNachrichten und Mitteilungen von der Arbeitsgemeinschaft fürPharmazeutische Verfahrenstechnik e.V.(Ausgabe 04/14, nach S. 232)

APVAPV – Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik, Kurfürstenstr. 59, 55118, Mainz, Tel. + 49 (0) 6131-9769-0,Fax +49 (0) 6131-9769-69, e-mail: [email protected]
21./22.10.2014 ENNIGERLOH Praktikum / Feuchtgranulieren
COG O-RING-AKADEMIEC. Otto Gehrckens GmbH + Co. KG, Gehrstücken 9, 25421 Pinneberg, Tel + 49 (0) 4101 5002 – 0, Fax +49 (0) 4101 5002-83,e-mail: [email protected]
18.09.2014 PINNEBERG Sicheres Abdichten mit O-Ringen
CONCEPT HEIDELBERGCONCEPT HEIDELBERG GmbH, Rischerstr. 8, 69123 Heidelberg, Tel. + 49 (0) 6221-84440, Fax +49 (0) 6221-844434,e-mail: [email protected]
09.–11.09.2014 MARBURG Lyophilisation kompakt – Technologie der Lyophilisation zum Anfassen
11.09.2014 BASEL (CH) GDP in der Schweiz – Besonderheiten im Arzneimittelvertrieb
16.09.2014 HEIDELBERG Reinigungsvalidierung kompakt
23./24.09.2014 MANNHEIM GMP-/GDP-Anforderungen an Lager und Transport
FORUM INSTITUTFORUM Institut für Management GmbH, Postfach 10 50 60, 69040 Heidelberg, Tel + 49 (0) 6221-500 500,Fax +49 (0) 6221-500 505, e-mail: [email protected]
16.09.2014 BONN MR and Decentralised Procedure – Your Regulatory Strategy
30.09.2014 KÖLN Arzneimittelfälschungsschutz – Kennzeichnung, Verpackung und Fälschungsschutz von Arzneimitteln
PCS GMBHPCS GmbH, Goldschmiedeweg 1a, 32051 Herford, Tel. + 49 (0)5221 69418-0, Fax +49 (0)5221 69418-29,e-mail: [email protected]
17.09.2014 HAMBURG Gute Lagerhaltungs-Praxis
23.10.2014 MANNHEIM Reinigungsvalidierung
PDA EUROPEParenteral Drug Association, Adalbertstraße 9, 16548 Glienicke/Nordbahn
16./17.09.2014 BRUSSELS (BE) Pharmaceutical Freeze Drying Technology
18./19.09.2014 BRUSSELS (BE) Development of a Freeze Drying Process
PTS TRAINING SERVICEPTS Training Service, Postfach 4308, 59737 Arnsberg, Tel.: + 49 (0) 2932-51477, Fax +49 (0) 2932-51674, e-mail: [email protected]
16.09.2014 WIEN (AT) GDP Good Distribution Practice
18.09.2014 ISERLOHN Herstellung und gesicherte Lagerung von Betäubungsmitteln
22./23.09.2014 MÜLLHEIM/BASEL(CH)
Risikobasierte Herstellung hochwirksamer Substanzen bei F. Hoffmann-La Roche AG vor Ort erleben
23.–25.09.2014 SCHEER Experte für Herstellung
23./24.09.2014 WIESBADEN Computervalidierung Modul 1 / Grundlagen, Regeln, GAMP 5
07.10.2014 OLTEN (CH) GDP Gute Vertriebspraxis
07.10.2014 FULDA GMP Inspektion in der Pharmatechnik
08./09.10.2014 FULDA Qualifizierung von Feststofftechnologien
23.10.2014 DARMSTADT Leitung der Herstellung und Produktions Abweichungen kompakt
TTC – TECHNOLOGY TRAINING CENTERTechnology Training Center (TTC), Werner-Glatt-Straße 1, 79589 Binzen, Tel. + 49 (0) 7621 664-535,e-mail: [email protected]
23.–25.09.2014 WEIMAR Continuous Particle processing
07.–09.10.2014 PRATTELN (CH) Tabletten Coating
21.–23.10.2014 PRATTELN (CH) Pan Coating
Termine
TechnoPharm 4, Nr. 4, 178 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)178 Termine

Abgelegt auf: Vorstufe:GK:ECV:Satz:TechnoPharm:TP_2014-04:Anzeigensatz-keine-Druck-PDFs:phast-TP-2014-03_216x303.indd Zuletzt gesichert: 15.04.14 (12:38:48 Uhr)
We support you with our new services:
• NMR structural analysis• aerosol characterization• particle characterization (e. g. SEM, EDX, Microflow Digital Imaging)• biopharmaceutical mass spectrometry (e. g. peptid mass fingerprinting)• reference standard characterization and qualification
COMPETENCE JOINS QUALITY
APVAPV – Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik, Kurfürstenstr. 59, 55118, Mainz, Tel. + 49 (0) 6131-9769-0,Fax +49 (0) 6131-9769-69, e-mail: [email protected]
21./22.10.2014 ENNIGERLOH Praktikum / Feuchtgranulieren
COG O-RING-AKADEMIEC. Otto Gehrckens GmbH + Co. KG, Gehrstücken 9, 25421 Pinneberg, Tel + 49 (0) 4101 5002 – 0, Fax +49 (0) 4101 5002-83,e-mail: [email protected]
18.09.2014 PINNEBERG Sicheres Abdichten mit O-Ringen
CONCEPT HEIDELBERGCONCEPT HEIDELBERG GmbH, Rischerstr. 8, 69123 Heidelberg, Tel. + 49 (0) 6221-84440, Fax +49 (0) 6221-844434,e-mail: [email protected]
09.–11.09.2014 MARBURG Lyophilisation kompakt – Technologie der Lyophilisation zum Anfassen
11.09.2014 BASEL (CH) GDP in der Schweiz – Besonderheiten im Arzneimittelvertrieb
16.09.2014 HEIDELBERG Reinigungsvalidierung kompakt
23./24.09.2014 MANNHEIM GMP-/GDP-Anforderungen an Lager und Transport
FORUM INSTITUTFORUM Institut für Management GmbH, Postfach 10 50 60, 69040 Heidelberg, Tel + 49 (0) 6221-500 500,Fax +49 (0) 6221-500 505, e-mail: [email protected]
16.09.2014 BONN MR and Decentralised Procedure – Your Regulatory Strategy
30.09.2014 KÖLN Arzneimittelfälschungsschutz – Kennzeichnung, Verpackung und Fälschungsschutz von Arzneimitteln
PCS GMBHPCS GmbH, Goldschmiedeweg 1a, 32051 Herford, Tel. + 49 (0)5221 69418-0, Fax +49 (0)5221 69418-29,e-mail: [email protected]
17.09.2014 HAMBURG Gute Lagerhaltungs-Praxis
23.10.2014 MANNHEIM Reinigungsvalidierung
PDA EUROPEParenteral Drug Association, Adalbertstraße 9, 16548 Glienicke/Nordbahn
16./17.09.2014 BRUSSELS (BE) Pharmaceutical Freeze Drying Technology
18./19.09.2014 BRUSSELS (BE) Development of a Freeze Drying Process
PTS TRAINING SERVICEPTS Training Service, Postfach 4308, 59737 Arnsberg, Tel.: + 49 (0) 2932-51477, Fax +49 (0) 2932-51674, e-mail: [email protected]
16.09.2014 WIEN (AT) GDP Good Distribution Practice
18.09.2014 ISERLOHN Herstellung und gesicherte Lagerung von Betäubungsmitteln
22./23.09.2014 MÜLLHEIM/BASEL(CH)
Risikobasierte Herstellung hochwirksamer Substanzen bei F. Hoffmann-La Roche AG vor Ort erleben
23.–25.09.2014 SCHEER Experte für Herstellung
23./24.09.2014 WIESBADEN Computervalidierung Modul 1 / Grundlagen, Regeln, GAMP 5
07.10.2014 OLTEN (CH) GDP Gute Vertriebspraxis
07.10.2014 FULDA GMP Inspektion in der Pharmatechnik
08./09.10.2014 FULDA Qualifizierung von Feststofftechnologien
23.10.2014 DARMSTADT Leitung der Herstellung und Produktions Abweichungen kompakt
TTC – TECHNOLOGY TRAINING CENTERTechnology Training Center (TTC), Werner-Glatt-Straße 1, 79589 Binzen, Tel. + 49 (0) 7621 664-535,e-mail: [email protected]
23.–25.09.2014 WEIMAR Continuous Particle processing
07.–09.10.2014 PRATTELN (CH) Tabletten Coating
21.–23.10.2014 PRATTELN (CH) Pan Coating
Termine
TechnoPharm 4, Nr. 4, 178 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)178 Termine

Intensivierung aeroberFermentationen durch gezielteSauerstoffanreicherungSichere Beherrschung der Prozess-Parameter als Voraussetzung
Johann Kaltenegger . Linde AG – Geschäftsbereich Linde Gas, Unterschleißheim
Korrespondenz: Johann Kaltenegger, Linde AG – Geschäftsbereich Linde Gas, Carl-von-Linde-Str. 25 – 85716 Unterschleißheim;e-mail: [email protected]
Einleitung
Die Anreicherung der Prozessluftdurch Zusatz von reinem Sauerstoff(O2) kann bei vielen biotechnologi-schen Anwendungen Qualität, Effi-zienz und Produktivität steigern.Diese positiven Effekte lassen sichvor allem für aerobe Fermentationennutzen, wobei der Sauerstoff-Einsatzin jedem Fall auch sicherheitsrele-vanten Kriterien anzupassen ist undeine entsprechende Hardware vo-raussetzt.
Aerobe Fermentationen (Abb. 1)durch Mikroorganismen kommen inzahlreichen Bereichen der pharma-zeutischen und industriellen Bio-technologie zum Einsatz. Ein Grunddafür: Aus nachwachsenden undleicht verfügbaren Rohstoffen iso-lierte Edukte bilden ideale Ausgangs-stoffe für mitunter recht komplexaufgebaute Produkte, welche auf reinchemischem Wege nicht ohne wei-teres darstellbar wären.
Anwendungen finden sich in zahl-reichen biotechnischen Prozessenwie bei der Produktion von pharma-zeutischen Proteinen und tech-nischen Enzymen. Vorwiegend aufder Basis von Kohlehydraten alsNährstoff spielen aerobe Fermenta-tionen mittlerweile auch bei der Her-
stellung einer ganzen Reihe von Mas-senprodukten eine zentrale Rolle.Neben großvolumigen Bausteinenfür Arzneimittel wie Antibiotika zäh-len hierzu beispielsweise organischeSäuren wie Zitronensäure, Futtermit-teladditive wie die Aminosäure Lysin,aber auch Biopolymere. TierischeZellen kommen vorzugsweise zumEinsatz, um komplexe Proteine wiemonoklonale Antikörper zu pro-duzieren.
Das eingesetzte Zellmaterial –Bakterienstämme wie Escherichiacoli oder speziell gezüchtete tierischeZellkulturen oder auch spezielle Pilz-kulturen – findet in Fermentations-
reaktoren ideale Bedin-gungen: Die Behälter zu-meist aus Edelstahl sindmit einer wässrigenNährstofflösung befüllt.Sie können sowohl be-heizt als auch gekühltwerden. Außerdem lässtsich der Inhalt differen-ziert begasen und rüh-ren. Bei 30 bis 40 GradCelsius vermehren sichdie meisten der gegen-wärtig kommerziell ge-nutzten Zellen beson-ders rasch (Abb. 2).
Neben zuckerhalti-gem Nährstoff benötigt das Zell-material zur Erzeugung der ge-wünschten Produkte vor allem auchSauerstoff (O2). Denn viele Fermenta-tionen in der pharmazeutischen und
Fokus BioTech
TechnoPharm 4, Nr. 4, 180–184 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)180 Kaltenegger . Intensivierung aerober Fermentationen
Abb. 1 – Anwendungen aerober Fermentationen durchMikroorganismen finden sich in zahlreichen biotechnischenProzessen wie bei der Produktion von pharmazeutischenProteinen und technischen Enzymen (Quelle: Linde).
Abb. 2 – Produktionslinie mit Erntebehälter ineiner Insulinproduktion (Quelle: Linde).

der Spezialchemie-Industrie verlau-fen aerob. Das Molekül ist essenziellfür die Atmung und intensiviertStoffwechselprozesse. Eine idealeVersorgung mit Zucker und Sauer-stoff spiegelt sich entsprechend di-rekt im Ertrag wider. Häufig sorgtein Rührwerk für eine gute Durch-mischung von Nährstofflösung undSauerstoff im Fermenter. Wachsendie Zellen, steigt der Sauerstoff-Be-darf. Entsprechend muss die Rührer-geschwindigkeit erhöht werden. Dasführt zu einer stärkeren Dispergie-rung der eingebrachten Luftblasenund damit zu einer Erhöhung desSauerstoffeintrags.
Erhöhung desSauerstoff-Gehaltes
Der für die aerobe Fermentation be-nötigte Sauerstoff wird klassisch be-reitgestellt, indem Umgebungsluftdem Fermenter zugeführt und darineffektiv verteilt wird. Der O2-Gehaltvon Luft ist mit knapp 21 Vol.-Pro-zent jedoch vielfach zu gering, umüber alle Phasen des Fermentations-vorganges eine genügend hohe Kon-zentration gelöster und damit für dieeingesetzten Zellen verwertbarerSauerstoff-Moleküle bereitzustellen.Das gilt insbesondere, wenn hoheWachstumsraten, hohe Zelldichtensowie hochviskose oder sehr hoch-wertige Produkte verlangt werden.
Die möglichen Gründe für einederartige Limitation sind vielfältig:So können die charakteristischen Ei-genschaften der jeweiligen Fermen-terbrühe – wie z.B. ein besondershoher Bedarf an Sauerstoff oder stei-gende Viskosität des Produktes – zuSauerstoff-Mangel in der wässrigenPhase beitragen. Darüber hinaus sindEngpässe bei der Bereitstellung derLuft oder apparativ bedingte Limita-tionen beim Fermenterbetrieb, wiez.B. eine begrenzte Rührerdrehzahl,besonders in der großtechnischenProduktion nicht selten.
Eine naheliegende und zuneh-mend angewandte Maßnahme zurErhöhung des Gehaltes an gelöstemSauerstoff in der Brühe ist die Erhö-
hung des O2-Partialdruckes in derBrutluft. Unter Normalbedingungenliegt dieser wenig über 0,21 bar. Erlässt sich jedoch durch Zugabe vontechnischem Sauerstoff flexibel erhö-hen – und damit den Erfordernissender jeweiligen Fermentationsphaseanpassen. Durch diese Verfahrens-modifikation lässt sich die Effizienzdes Fermentationsprozesses häufigdeutlich steigern. Im Extremfall kanndie Prozessintensivierung bis hin zurausschließlichen Anwendung vonreinem Sauerstoff reichen.
Vorteile von technischemSauerstoff für mikrobielleProzesse und Zellkulturen
Die Anreicherung der Brutluft mitreinem Sauerstoff kann sowohl dieQualität als auch die Effizienz aer-ober Fermentationsprozesse signifi-kant steigern. Bei mikrobiellen Pro-zessen bedeutet das: Sie laufenschneller ab. Raum und Zeit werdenoptimal ausgenutzt. Hohe Zelldich-ten lassen sich so realisieren. Da ins-gesamt eine geringere Gasmenge imFermenter benötigt wird, reduzierensich Energiebedarf und Abgasstrom.Zusätzlich wird die Sauerstoffversor-gung bei niedrigerer Begasungsrateverbessert. Und die Gelöstsauer-stoff-Konzentration (Dissolved Oxy-gene, DO) ist einfach und sicher steu-erbar. Zellkulturprozesse profitierendarüber hinaus durch eine Vermin-derung der Schädigung von Zellen.Zudem gewährleistet die gleichblei-bend gesicherte Gasqualität stabilereProzesse.
Diese qualitativen Verbesserun-gen steigern unmittelbar die Wirt-schaftlichkeit [1]. Denn den größtenKostenanteil bei einem Fermentati-onsprozess stellen die Rohmateria-lien wie Glukose etc. dar. Die Kostenfür Sauerstoff sind im Verhältnisdazu niedrig. Und schon eine geringeAnreicherung mit Sauerstoff kanneine deutliche Produktivitätssteige-rung bewirken. Versuche im Laborhaben bereits gezeigt, dass aerobeFermentationen von mehr Sauerstoffdeutlich profitieren.
Um die Vorteile für Biotech-Pro-zesse im Produktionsmaßstab zu un-termauern, hat Linde neben Wirt-schaftlichkeitsberechnungen auchComputersimulationen durchgeführt,letztere in enger Zusammenarbeit mitden im Anlagenbau tätigen Kollegenvon Linde Engineering Dresden. DieBasis bildete das Bakterium Escheri-chia coli in einem 50 Kubikmeter gro-ßen Fermenter – ein gängiges Systemin der Biotechnologie zum Beispielzur Insulinherstellung. Mithilfe der Si-mulationsmodelle konnte genau un-tersucht werden, wie viel Biomassesich bei normaler Luftbegasung undbei Sauerstoffanreicherung über dieZeit bildet.
Die Ergebnisse sprechen für sich:Wird der O2-Gehalt auf 30 Prozenterhöht, haben sich durch die damiterzielbare hohe Wachstumsrate be-reits nach 15 Stunden 50 Gramm Bio-masse pro Liter gebildet. Zum Ver-gleich: Bei Zugabe reiner Umgebungs-luft in den Fermenter, sind nach 60Stunden erst 30 Gramm Biomassepro Liter entstanden. Zudem lassensich durch mehr Sauerstoff im Reak-tionsgefäß auch höhere Zelldichten,also mehr Mikroorganismen pro Vo-lumen, kultivieren. Bei kürzerer Reak-tionsdauer sinkt ferner die aufgrunddes Energiestoffwechsels der Mi-kroorganismen benötigte Subs-tratmenge merklich ab. Eine bessereSubstratausnutzung hinsichtlich dergewünschten Produkte ist die Folge.Durch den zusätzlichen Sauerstoffwird die Apparatur zum „Hochleis-tungsfermenter“. Der Biotech-Prozessläuft besser ab.
Ein weiterer Vorteil: Es bildet sichoft weniger Schaum im Bioreaktor.Dank des höheren Sauerstoffanteilsmuss insgesamt weniger Gas durchdie Lösung strömen. Auf so genanntechemische Entschäumer, die denBiotech-Prozess stören können, lässtsich gegebenenfalls verzichten. Aberdas schnelle Wachstum der Mikroor-ganismen birgt auch Risiken. Wennder Zellstoffwechsel auf Hochtourenläuft, wird deutlich mehr Wärmeproduziert. Und das macht leistungs-fähigere Kühlsysteme erforderlich.
TechnoPharm 4, Nr. 4, 180–184 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 181Kaltenegger . Intensivierung aerober Fermentationen

Die aufwendigere apparative Aus-stattung verursacht aber nur zu Be-ginn Mehrkosten. Denn die Wirt-schaftlichkeitsberechnung zeigte,dass diese durch den schnellerenBiotech-Prozess, Einsparungen beiEnergie und Nährstoffmedium sowieeine geringere Abwasserproduktiondeutlich überkompensiert werden(Abb. 3).
AnlagentechnischeVoraussetzungen
Die Voraussetzung zur Erschließungder genannten Vorteile bildet einegeeignete Hardware zur Sauerstoff-Versorgung. Dabei ist zunächst zuentscheiden, wie der zusätzliche Sau-erstoff in den Fermenter gelangensoll. Theoretisch wäre es möglich,ihn als reines Gas direkt in den Fer-menter einzubringen. Allerdings sinddamit vermeidbare Risiken verbun-den, etwa in Hinblick auf lokal be-grenzt sehr hohe DO-Konzentratio-nen. Deshalb empfiehlt sich häufigeine Anreicherung der Zuluft vordem Eintritt in den Fermenter(Abb. 4).
Die Injektion des zusätzlichenSauerstoffs in das bestehende Versor-gungssystem erfolgt über OXY-MIX™, einen speziellen Gasmischer
(Abb. 5). Sein Düsensystem ist in ei-nem Lochkreis angeordnet und soausgelegt, dass das Gas entgegender Luftströmung in die Leitung ein-gebracht wird. Dadurch werden diebeiden Komponenten innerhalb kür-zester Zeit vollständig vermischt. Soist in der Luftleitung ohne wesentli-chen Druckverlust eine homogeneSauerstoff-Verteilung sichergestellt.D.h. Zonen mit zu hoher Sauerstoff-Konzentration werden zuverlässigvermieden. Damit gewährleistet derMischer auch eine hohe Betriebs-sicherheit.
Die sehr kompakt konstruierteKomponente kann über einenFlanschanschluss ohne großen tech-nischen oder finanziellen Aufwand ineine bestehende Luftleitung einge-baut werden, beispielsweise währendeines Routine-Stillstandes der Anla-
ge. Weil das System keine beweg-lichen Bauteile enthält, arbeitet esnach der Installation wartungsfrei.Konstruktion und Einbau des Sauer-stoff-Injektors realisiert Linde abge-stimmt auf Geometrie und Betriebs-bedingungen der Luftleitung. NebenEdelstahl (1.4571) sind auch anderefür Sauerstoff geeignete Werkstoffeverfügbar. Die individuelle Aus-legung basiert auf CFD-Simulationen(Computational Fluid Dynamics).
Die Mess- und Regel-strecke FLOWTRAIN®
ermöglicht die punkt-genaue Sauerstoff-Anrei-cherung per Touchpanel:Über eine speicherpro-grammierbare Steuerung(SPS) dosiert das Systemgasförmigen Sauerstoffexakt bis zur gewünsch-ten Konzentration in dieBrutluft. Dabei regeltFLOWTRAIN® den ge-nauen Grad der Sauer-stoff-Anreicherung in
Abhängigkeit vonder Brutluftmenge.
Im Gegensatz zumklassischen Verfah-ren ist jedoch nichtnur in der Brutluft-zuführung mit höhe-ren Sauerstoff-Gehal-ten als beim aus-schließlichen Betriebmit Luft zu rechnen,sondern unter Um-ständen auch im Fer-menterkopfraum so-wie in der Abgaslei-tung. Aufgrund diesererweiterten Rahmen-bedingungen ist dieSicherheit der Pro-zessführung nichtmehr automatischgegeben. Dies machtgrundsätzliche Über-legungen und beson-dere Maßnahmen zurRisikoeinschätzungsowie zur sicherenBetriebsweise unab-dingbar.
Parameter für einensicheren Sauerstoff-Einsatz
Um die Parameter für einen sicherenSauerstoff-Einsatz bei aeroben Fer-mentationen zuverlässig festlegenzu können, führte Linde eine umfas-sende, experimentell hinterlegte Stu-die durch [2]. Darin wurde das si-cherheitsrelevante Verhalten eines
Fokus BioTech
TechnoPharm 4, Nr. 4, 180–184 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)182 Kaltenegger . Intensivierung aerober Fermentationen
Abb. 3 – Vergleich – Sauerstoffanreicherung vs. Luftbegasung(Quelle: Linde).
Abb. 4 – Vereinfachtes Beispiel für die Implementierung einerSauerstoff-Versorgung in den Fermentationsprozess (Quelle: Linde,in Anlehnung an Heinzle, Develompent of Sustainable Biopro-cesses).
Abb. 5 – Die Injektion des zusätzlichen Sauerstoffs in dasbestehende Versorgungssystem erfolgt über einen speziellenGasmischer (Quelle: Linde).

Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2014-04\Anzeigensatz-keine-Druck-PDFs\sartorius-TP-2014-04-216x303.indd Zuletzt gesichert: 15.07.14 (01:34:54 Uhr)
Our new Flexsafe bags ensure an excellent and reproducible growth behavior
with the most sensitive production cell lines. The optimization of the resin
formulation, the complete control of our raw materials, the extrusion process
and the bag assembly guarantee a consistent lot-to-lot cell growth performance.
www.sartorius-stedim.com/flexsafe U S P – D S P – F + F
O N E F I L M F O R A L L
New Flexsafe Bag Family. New PE Film. New Benchmark.

typischerweise als Nährstoff einge-setzten Zuckers mit einem natürlichvorkommenden Öl, das ebenfalls beiFermentationen Einsatz findet, ver-glichen. Besondere Beachtung fan-den dabei Ablagerungen, die sich ins-besondere während der Fermenta-tion aufbauen können und mit derenAuftreten auch in der Peripherie desFermenters zu rechnen ist. Sie kön-nen austrocknen, was vor allem beimehrere Tage andauernden Fermen-tationen nicht auszuschließen ist.Grundsätzlich haben solche Ablage-rungen das Potenzial sich zu entzün-den, was mit steigendem Sauerstoff-Partialdruck in der umgebenden At-mosphäre immer wahrscheinlicher
wird. Der in Abb. 6 beispielhaft ge-zeigte Versuchsaufbau wurde ge-wählt, um das Langzeitverhaltenvon trockenem Substrat zu unter-suchen.
Abb. 7 zeigt das Verhalten der inAbb. 6 gezeigten Probe bei deutlicherhöhtem Sauerstoff-Partialdruck so-wie bei einer Temperatur, die ober-halb der bei Fermentationen typi-scherweise auftretenden Temperatu-ren liegt und die über einen längerenZeitraum hinweg konstant gehaltenwurde. Die Bedingungen waren hiersowie bei weiteren Experimenten soeingestellt, dass die Ergebnisse alsfundierte Datengrundlage für weiter-gehende, individuell angepasste Si-cherheitsanalysen dienen können.
Zusammenfassend haben die Un-tersuchungen dieTendenz bestä-tigt, dass sich beierhöhtem Sauer-stoff-Partialdruckdie Wechselwir-kung von moleku-larem Sauerstoffmit organischenAblagerungen in-tensiviert. Diesführt vermehrtzur Bildung ther-misch instabilerSauerstoffverbin-dungen. Als be-sonders kritischgilt, wenn stark
ausgeprägte Ablagerungen (Schicht-dicke) über einen längeren Zeitraumhinweg bei hohem Sauerstoff-Par-tialdruck erhöhten Temperaturenausgesetzt sind. Dies kann unter Um-ständen zu einer schlagartigen Zer-setzungsreaktion führen, die mit ei-nem rapiden Druck- und Tempera-turanstieg einhergeht.
Die durchgeführten Untersuchun-gen haben ebenso ergeben, dass der(zusätzliche) Einsatz von reinemSauerstoff bei Fermentationen auchim technischen Maßstab sicher be-herrscht werden kann. Die Voraus-setzung hierfür bilden einige grund-legende Maßnahmen zur Anpassungdes ursprünglich luftbasierten Fer-
mentationsprozesses: Insbesonderedurch Überwachung – unter Um-ständen können auch kleinere kons-truktive Veränderungen notwendigsein – und nicht zuletzt kann auchdurch regelmäßige Inspektionen dieGefahr einer Selbstentzündung vonAblagerungen ausgeschlossen wer-den. Dieser vergleichsweise geringeAufwand steht dabei einer deutlichgesteigerten Fermentationsleistunggegenüber.
Fazit
Sowohl die Qualität und als auch dieEffizienz und Produktivität aeroberFermentationsprozesse lassen sichdurch die Anreicherung der Brutluftmit reinem Sauerstoff signifikantsteigern. Wirtschaftlichkeitsberech-nungen sowie Computersimulatio-nen von Linde belegen dabei Poten-ziale von bis zu 45 Prozent an Effi-zienzverbesserung. Eine Vorausset-zung zur Nutzung der aufgezeigtenVorteile bildet eine geeignete Hard-ware zur Sauerstoff-Versorgung un-ter Berücksichtigung der Sicherheits-standards. Bei Bedarf stellt Linde in-teressierten Kunden entsprechendesEquipment und Beratung zur Ver-fügung.
Interessante Möglichkeiten für einScale-up in den industriellen Maß-stab bietet das kürzlich eröffneteFraunhofer-Zentrum für Chemisch-Biotechnologische Prozesse CBP inLeuna. Für das Engineering der ver-fahrenstechnischen Einheiten sowiedie hierfür erforderliche Infrastruk-tur und benötigten Medien zeichnetedie Linde Engineering Dresden alsGeneralunternehmer verantwortlich.
Fachliteratur[1] Johanna Leisling, Erstellung eines Ver-
triebskonzepts zum Einsatz von tech-nischem Sauerstoff in der industriellenund pharmazeutischen Biotechnologie,Bachelor-Thesis, Hochschule für Ange-wandte Wissenschaften München, 2012
[2] Dr. Bernhard Schreiner, Einsatz von mitSauerstoff angereicherter Brutluft beiFermentationsprozessen, Linde-interneStudie, 2008
Fokus BioTech
TechnoPharm 4, Nr. 4, 180–184 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)184 Kaltenegger . Intensivierung aerober Fermentationen
PS 20070296 0,75l / 500bar Autoklav Druckaufnehmer: 100 barAuftraggeber: Bernhard Schreiner Starttemperatur: 20 °C Anfangsdruck: 1 barEinwaage: 80g Lagertemperatur: 60 °C Druck bei Lagertemp: 11 barProbengefäß: 200 ml Dewar, dickwandig, ausgeheizt max.Temperatur: 371 °C max. Druck: 74 bar
Glucosemonohydrat / Kieselgur (1:1) gelagert unter 10 bar Sauerstoffatmosphäre (Versuch 2)
0
50
100
150
200
250
300
350
400
0 12 24 36 48 60 72 84 96 108 120 132 144Zeit [h]
Tem
pera
tur [
°C]
0
10
20
30
40
50
60
70
80
Dru
ck [b
ar]
ProbeDruck
Ofen
Abb. 7 – Verhalten der in Abb. 6 gezeigten Probe (Quelle: Linde).
Abb. 6 – Versuchsaufbau zur Untersuchungdes Langzeitverhaltens von trockenem Subs-trat (Quelle: Linde).

Abgelegt auf: Vorstufe:GK:ECV:Satz:TechnoPharm:TP_2014-04:Anzeigensatz-keine-Druck-PDFs:nurnberg-messe-TP-2014-04_216x303.indd Zuletzt gesichert: 05.05.14 (08:46:43 Uhr)
TP14_210x297_TechnoPharm_B.indd 1 30.04.14 07:25

Umbau während laufenderProduktion – na und!Von der Notwendigkeit effizient und strukturiert zu agieren, um höchste Sicherheitzu gewährleisten
Gita Enderlein . Chemgineering Technology GmbH & Co, KG, Ludwigshafen
Korrespondenz: Gita Enderlein, Heidelberge-mail: [email protected]; [email protected]
ZusammenfassungEin Investitionsprojekt lässt sich idealerweise nur als Neubau auf der grünen Wiese reali-sieren. Doch wann gibt es das schon? Stattdessen müssen immer häufiger Projekte imBestand während laufender Produktion ausgeführt werden. Das sind sicherlich keineoptimalen Voraussetzungen. Aber sie sind zu bewältigen.Das Eingreifen in die laufende Produktion verlangt eine intensive Planung und systema-tische Vorbereitung. Die wesentlichen Kriterien werden bereits in der ersten Planungs-phase festgelegt. Ein stimmiger Terminplan, ein strategisch ausgerichtetes Vorgehen, eindetaillierter Maßnahmenkatalog sowie klar und eindeutig zugewiesene Verantwortlich-keiten sind das Handwerkszeug für eine erfolgreiche Projektdurchführung.
Einleitung
Der Luxus, ein Projekt auf der grünenWiese neu planen und realisieren zukönnen, ist heute leider recht selten.Der Trend aktueller Projekte im glo-balen Pharmamarkt sieht üblicher-weise anders aus:
Die Implementierung weitererProduktionslinien, Optimierungs-,und Modernisierungsmaßnahmenoder die Erneuerung eines in dieJahre gekommenen Maschinenparksmüssen möglichst bei laufender Pro-duktion durchgeführt werden.
Ein gezieltes Vorproduzieren istaufgrund knapper Maschinen-, Per-sonal- und Lagerkapazitäten sowieder begrenzten Haltbarkeit der Zwi-schen- oder Endprodukte häufig nurbedingt möglich. Eine zeitlich be-grenzte Fremdvergabe der Produk-tion an externe Lohnhersteller istmeist auch nicht ohne weiteres
durchführbar. Die Firmen befindensich in der Zwangslage, den Produk-tionsausfall so kurz wie möglich zuhalten. Jede auftretende Produkti-onsbeeinträchtigung oder -unterbre-chung erzeugt Kosten, die zusätzlichzu den eigentlichen Projektkostenmit kalkuliert und in der Jahrespro-duktionsplanung frühzeitig berück-sichtigt werden müssen.
Längere Produktionsunterbre-chungen können empfindliche Kon-sequenzen haben, zum Beispiel einekostspielige Umlagerung von Lager-beständen. Die fatalen Folgen von Lie-ferschwierigkeiten können letztend-lich den Verlust von langjährigenKunden, die Regression der Markt-anteile, im schlimmsten Fall den Ver-lust einer Verkaufs-Vormachtstellungoder der Marktexklusivität bedeuten.Dies gilt es unter allen Umständen zuvermeiden. Viele Firmen scheuen sichdaher, diese sensiblen und schwieri-
gen Projekte anzugehen; die Notwen-digkeit wird kritisch hinterfragt.Letztendlich wird das Projekt wiederzur Seite gelegt und um ein weiteresJahr verschoben. Mit einem passen-den Rezept und entsprechendenMaßnahmen ist die Abwicklung sol-cher Projekte jedoch kein Hexenwerk.
Die Konzeptphase – dieerste und wichtigsteDiagnose
Das Projekt muss zunächst in einerFeasibility-Studie oder einer Kon-zeptphase auf seine Machbarkeitanalysiert werden. Die Studie hatfür diese Art von Projekt noch zu-sätzlich einen – für die weitere Pla-nung ganz entscheidenden – Outputzu liefern:. Aus welchen Schritten setzt sichdie Realisierung im Einzelnen zu-sammen?
Maschinen- und Anlagenbau
TechnoPharm 4, Nr. 4, 186–190 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)186 Enderlein – Umbau während laufender Produktion . na und!
AutorinGita Enderlein istChemie-Ingenieurin.Nach Ihrem Studiumwar sie unter ande-rem im Bereich Kli-ma- und Reinraum-technik bei der FirmaZander tätig. Es folg-ten u.a. Stationen beiUhde Pharma Con-sult, wo sie in Con-sulting- und Projekt-
geschäft in Pharma- und Kosmetikbereich tätig war.Nach elf Jahren bei Chemgineering wechselte sieAnfang 2014 zu AbbVie, wo sie als Projektleiterin fürPharma- und Bauprojekte eingesetzt ist. In ihrerFreizeit ist sie im Bergsport aktiv.

. Welche dieser Schritte sindzwangsläufig mit einem Produkti-onsstillstand verbunden und wa-rum?
. Welche Produktionsbereiche sindim Detail betroffen?
. Wann und wie lange ist ein Still-stand notwendig?
Das sind zunächst sehr einfach klin-gende Fragestellungen, die aber be-reits im Konzept detailliert evaluiertwerden müssen.
Dazu wird die Realisierungsphasedes Projektes in sinnvolle Bausequen-zen unterteilt. Eine Differenzierungund nachfolgende Klassifizierung derSequenzen bezugnehmend auf dieseFragestellungen ermöglicht eine ersteEinschätzung. Für die Erstellung desMeilensteinterminplanes ist dernächstfolgende Schritt die Adaptionder notwendigen Produktionsunter-brechungen an den zuvor in der Jah-resplanung festgelegten Stillständen.
Tipps und Tricks bei derPlanung
Vorab wäre die Planung der Realisie-rungssequenzen genauer zu betrach-ten:
Der erste Schritt ist die Prüfungder vorhandenen Bestandsunterla-gen, die Identifizierung der Abwei-chungen und fehlenden Informatio-nen und deren Abgleich mit dem rea-len Zustand. Diese Erfassung des Ist-Zustandes kann sich, je nach Quali-tät der Dokumente, sehr aufwändiggestalten. Erst nach der Bestandsauf-nahme beginnt die eigentliche Pla-nung.
Bei den Montage- oder Installati-onstätigkeiten kann der Montagebe-reich in vielen Fällen interimsweisedurch sinnvoll gestellte Staubwändevom Produktionsbereich abgetrenntwerden. Eine Planung der einzelnenSequenzen anhand eines Pikto-gramm-Layouts erleichtert denÜberblick.
In jeder Bausequenz ist das ge-plante Montage-Layout auf essenzielleinzuhaltende Grundvoraussetzun-gen hin zu prüfen und zu optimieren:. Der Personalfluss zur Produktionmuss weiterhin gewährleistet sein.Aber auch der Personalzugangzum neuen Montagebereich mussgeregelt sein. Ist der Zugang nichtvon außen oder einem schwarzenBereich zu erschließen, sind tem-
poräre (mobile)Personalschleu-sen unabdingbar.
. Die Einbringungdes neuen sowiedie Entsorgungdes alten Equip-ments müssensichergestelltsein. Gegebe-nenfalls sindhierzu zusätz-liche vorberei-tende Tätigkei-ten notwendig,zum Beispiel zu-sätzliche Last-haken oder Ein-bringöffnungen,die späterschnell und sau-ber geöffnet undwieder ver-schlossen wer-den können.
Diese Maßnahmen können auchfür zukünftige Projekte Synergiendarstellen.
. Während der Montage muss eineinfacher Transport von schweremBaumaterial in den Montagebe-reich möglich sein. Ist dies nichtohne weiteres machbar, so ist zuprüfen, ob das Baumaterial in einerzügig durchgeführten Aktion zwi-schen zwei Produktionszyklenentsprechend verpackt und gerei-nigt direkt durch den Produkti-onsbereich geschleust werdenkann.
. Bei jedem Abschnitt müssen derBereiche den notwendigen GMP-Anforderungen entsprechen. DasZonenkonzept muss bei jederBausequenz in vollem Umfang er-halten bleiben. Sicherstellung einergerichteten Luftströmung vomProduktions- zum abgetrenntenBaubereich, Abschottung des Lüf-tungssystems, bei Bedarf zusätz-liche Installation von provisori-schen Lüftungs- bzw. Abluftsyste-men zur Vermeidung von be-reichsübergreifende Mischluftsys-temen sind Beispiele entsprechen-der GMP-Maßnahmen.
TechnoPharm 4, Nr. 4, 186–190 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 187Enderlein – Umbau während laufender Produktion . na und!
Abb. 1: Planung der Staubwand zur Trennung des Montagebereiches (grün schraffiert) zum Produktionsbereich(Quelle alle: Chemgineering GmbH).

. Der Produktionsfluss ist währendder gesamten Umbauphase stö-rungsfrei zu gewährleisten. Im Be-darfsfall müssen temporäre Lager-stätten für Material und Produktneu gefunden und eingerichtetwerden.
. Die gute Zugänglichkeit der Pro-zessanlagen für Inspektion, War-tung, Instandhaltung sowie für diemanuelle oder automatische Be-dienung muss zu jeder Zeit gege-ben sein.
Die notwendige Detaillierungtiefe derKonzeptplanung zeigt folgendes Bei-spiel: Die Abbildung 1 stellt die Pla-nung der Staubwand zur Trennungdes Montagebereiches (grün schraf-fiert) zum Produktionsbereich dar.Zu sehen ist der Personal- und Mate-rialfluss für die ge-sicherte Produk-tion (linke Flur-hälfte) und die Zu-gänglichkeit fürden Montagebe-reich mit Schleusegemäß den GMP-Anforderungen.Das Einbringenvon Material mussentweder über dasschwarze Trep-penhaus oder amWochenende überden weißen Fahr-stuhl (hier nichtsichtbar) durch den Produktions-bereich erfolgen, dann allerdings ent-sprechend verpackt und gereinigt.
Um die Prozessbehälter im abge-trennten Produktionsbereich (linkeFlurhälfte) automatisch mit dem fah-rerlosen Transportsystem (FTS) an-fahren und bedienen zu können, be-durfte es einer genauen Prüfung derMachbarkeit. In diesem Fall war dieAusbildung einer Ecke unumgäng-lich. Nur so war die notwendige Fahr-fläche für die Hüllkurve des FTS si-cherzustellen. Entsprechend wurdedies bei der Realisierung ausgeführt.(Abbildung 2). Die automatische Be-dienung der Behälter auf der Produk-tionsseite (weiße Seite) war somitdurchgehend gesichert.
Von allen Projektphasen ist dieKonzeptphase zwar die kürzeste,aber die ausschlaggebende Phase: Al-len Projektbeteiligten muss bewusstsein, dass in dieser Projektphase diesubstanziellen Grundlagen ermitteltund die wichtigsten Entscheidungengetroffen werden müssen. Oft liegendie endgültigen Daten und Fakten zudiesem Zeitpunkt aber noch nichtvor. Die Entscheidungen sind dannanhand von Annahmen zu treffen.Sind die Weichen erst einmal gestellt,so verursacht jede Änderung derBasis Terminverzögerungen undzwangsläufig Zusatzkosten.
Ein kompetentes Projektteam unddas frühzeitigen Einbindung der Pro-zesseigner gewährleistet ein auf dienotwendigen Bedürfnisse maßge-
schneidertesProjekt.
Ein fun-diert geplan-tes Konzeptbringt Klar-heit darüber,welche mögli-chen Varian-ten des Pro-jektes umge-setzt werdenkönnen, wiehoch die In-vestitionskos-ten dafür an-gesetzt wer-
den müssen, aber auch welche be-sonderen Schwierigkeiten zu erwar-ten sind. Ein ganz entscheidendes Er-gebnis dieser Phase ist der Termin-plan, der den weiteren Verlauf desProjektes bestimmt.
Der Terminplan – dasRückgrat des Projektes
Ist ein Projekt während laufenderProduktion zu realisieren, so sinddie fixen Bestandsgrößen des Pro-jektterminplanes die definierten Pro-duktionsstillstände wie Feiertage,Brücken- undWartungstage. Die pro-duktionsfreien Zeiten werden unter-nehmerisch meist frühzeitig fest-gelegt und sind in der Jahresproduk-
tion bereits einkalkuliert. Diese Still-standzeiten sind die Eckpunkte desProjektterminplans, die den Takt unddie mögliche Geschwindigkeit desProjektes maßgeblich vorgeben. Da-bei spielt auch jeder kleinste Still-stand eines Teilgewerkes eine Rollewie zum Beispiel einige StundenSAP-Wartungsfenster, Reinigungs-und Rüstzeiten der Maschinen, Aus-tausch von Filtern oder nicht kom-plett ausgenutzte Schichtmodelle.
Heutzutage müssen Investitions-projekte in immer kürzerer Zeit ab-gewickelt werden: Die Investitions-entscheidungen fallen später, derZeitraum bis zur Fertigstellung wirdkürzer.
Trotzdem dürfen die Termine desProjektterminplanes rund um dieseMeilensteine nicht „politisch ideali-siert“ sein, sondern müssen realis-tisch und belastbar geplant werden.
In einem vollständigen Termin-plan sind die einzelnen Projektse-quenzen, die integrierte Qualifizie-rung sowie die jeweiligen Halte-punkte enthalten.
Einige Maßnahmen, die helfenkönnen, einen Terminplan realistischzu erstellen und optimal zu verfol-gen, sind:1. Identifizierung der Langläufer
Equipments mit langen Lieferzei-ten müssen identifiziert werden,um diese mit höchster Prioritätvoranzutreiben. Um böse Überra-schungen zu vermeiden, solltehierbei nicht mit Lieferzeiten-benchmarks aus vergangenenProjekten gearbeitet werden. DieLieferzeiten mancher Equipmentsverlängerten sich innerhalb vonfünf Jahren um über 50 Prozent.Ein konkretes Anfragen der po-tentiellen Lieferanten ist unbe-dingt erforderlich und ermöglichtgesicherte und aktuelle Informa-tionen.
2. Beachtung der Rahmenbedin-gungenSo manches Projekt ist geschei-tert, weil bei enthusiastischerPlanung davon ausgegangen wird,dass das Jahr tatsächlich 365Arbeitstage hat. Auch wenn die
Maschinen- und Anlagenbau
TechnoPharm 4, Nr. 4, 186–190 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)188 Enderlein – Umbau während laufender Produktion . na und!
Abb. 2: Realisierter Umbau

produktionsfreien Zeiten – undsomit viele möglichen Projekt-aktivitäten – meist auf Feiertagenliegen, so ist doch nach Abzug vonUrlaub, krankheitsbedingtenFehltagen und Feiertagen realis-tisch „nur“ mit 250 Tagen zurechnen.
3. Berücksichtigung von Pla-nungsungenauigkeitenDie moderne Projektmana-gementsoftware ermöglicht esheute, auf den Tag genau zu pla-nen. In der Realität lassen sichviele Projektteams von dieservermeintlichen Exaktheit täu-schen. Es wird übersehen, dassjedes Projekt Risiken und Pla-nungsunsicherheiten beinhaltet.Vor allem in alten Gebäuden tre-ten bei Umbauarbeiten Überra-schungen auf, die zusätzlich Zeitkosten. Deshalb ist es von Vorteil,den Zeitplan zu diesem frühenZeitpunkt noch nicht zu eng zuschnüren.
4. Klare Analyse des kritischenPfades (critical path method)Der kritische Pfad beinhaltet Ar-beitspakete wie zum Beispiel dieAusschreibung und Bestellungvon entscheidenden Equipments,die Durchführung der Passivie-rung einer Gesamtanlage oderQualifizierungstätigkeiten. Jegli-che zeitliche Verzögerung der Ar-beitspakete, die sich auf dem kri-tischen Pfad befinden, beeinflusstletztendlich den Gesamttermin-plan. Aus diesem Grund ist eswichtig, diese Aktivitäten beson-ders sorgfältig zu kontrollieren, zusteuern und gegebenenfallsrechtzeitig Gegenmaßnahmeneinzuleiten. Nur so ist zu vermei-den, dass sich frühe Versäumnissebis zum Projektende durch-schleppen und sich ihre Auswir-kungen am Ende dramatisch po-tenzieren können.
5. Rechtzeitige Vor-Reservierungder FertigungskapazitätenStrategisch geht der voraus-schauende Projektplaner immermehr dazu über, sich bei kriti-schen Anlagen und notwendigen
Aktivitäten früh auf eine einge-grenzte, gegebenenfalls schon be-kannte und auditierte Lieferan-tenvorauswahl zu konzentrieren.Eine frühzeitige Kontaktauf-nahme kann schon vorab Fer-tigungskapazitäten sichern. Spe-ziell für die Zeiträume an undzwischen Feiertagen sollten Res-sourcen und Kapazitäten min-destens ein halbes Jahr vorhergeplant und reserviert werden.
6. Beachtung von limitierten Res-sourcenAuch wenn es wünschenswert undsinnvoll ist, bestimmte Tätigkeitenparallel ablaufen zu lassen, somuss schon bei der Erstellung desTerminplanes geprüft werden, obdie notwendigen Ressourcenhierfür auch vorhanden sind. Eintypischer restriktiver Faktor ist dieGröße des Montagebereiches: Istder Montagebereich sehr beengt,so hat es wenig Sinn, dort mehrereGewerke gleichzeitig einzusetzen.Weitere Limitierungen sind dieAnzahl der Teststände, die Aus-lastung von Laborkapazitätenoder das zur Verfügung gestellteTestprodukt, um nur einige Bei-spiele zu nennen.Liegen diese Informationen beider Erstellung des Terminplansnoch nicht vor, so sollte hier eherkonservativ gerechnet werden.
7. Sinnvolle Aufteilung der Aus-schreibungspaketeJe nach Auslastung der Lieferan-ten bzw. je nach Zwang des Ter-minplanes müssen die Ausschrei-bungspakete gegebenenfalls auf-geteilt und getrennt ausgeschrie-ben werden. Oft können be-stimmte Aktivitäten wie die Ein-bringung des Equipments oder dieAnbindung einer WFI-Leitung mitanschließender Requalifizierungnur innerhalb der produktions-freien Zeit durchgeführt werden.Diese Aktivitäten müssen strate-gisch möglichst unabhängig vomGesamtgewerk geplant unddurchgeführt werden, um dieproduktionsfreie Zeit optimalnutzen zu können.
8. Geeignete Vertragsform mitden LieferantenOftmals ist ein Lieferant aus Ku-lanzgründen ohne vorherige Prü-fung der Realisierbarkeit zuschnellen terminlichen Zusagenbereit. Es ist extrem wichtig, dieLieferanten frühzeitig in die Ter-minplanung einzubeziehen unddurch die Wahl einer geeignetenVertragsform möglichst eng anden gemeinsam besprochenenTerminplan zu binden. Musste dieRealisierung in einzelne von-einander unabhängige Projekt-phasen aufgeteilt werden, so solltedie Gültigkeit der Lieferanten-angebote entsprechend angepasstwerden. Es lohnt sich, in dieschriftliche Vereinbarung mehrZeit zu investieren, um ein ein-deutig abgesprochenes Projekt-verständnis zu erreichen.
9. Durchführung eines intensivenField-ExpeditingUm den FAT (Factory AcceptanceTest) gut vorzubereiten, ist es un-bedingt notwendig, nach der Lie-ferantenbeauftragung regelmä-ßige gemeinsame Meetings mitdem Lieferanten und auch mitden entsprechend angrenzendenGewerken durchzuführen. Vor-Ort-Besuche und Vor-FAT-Prü-fungen beim Lieferanten dienender Qualitäts- und Terminkon-trolle.
10.Einhaltung der GMP-Spiel-regelnPharmaprojekte sind gekenn-zeichnet durch intensive Qualifi-zierungsarbeiten, die sehr häufigzeitlich unterschätzt werden. Fürdie Erstellung des Terminplanssind die Qualifizierungszeiten zuverifizieren. Dazu muss vorab diedetaillierte Vorgehensweise derintegrierten Qualifizierung unddes Change-Managements klarformuliert sein. Während desProjekts sind effiziente Zeitein-sparungen möglich, wenn dasEngineering und die Qualifizie-rung Hand in Hand arbeiten, zumBeispiel bei der gemeinsamenNutzung der FAT-Prüfprotokolle.
TechnoPharm 4, Nr. 4, 186–190 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 189Enderlein – Umbau während laufender Produktion . na und!

11.Effektives Construction Ma-nagementTrotz guter Planung kennzeich-nen parallel ablaufende Baupha-sen zunehmend den Realisie-rungsprozess von Projekten.Oberstes Gebot ist es, die Rah-menbedingungen für einen rei-bungslosen und sicheren Ablaufauf der Baustelle zu schaffen. DieSchutzmaßnahmen zur Einhal-tung der GMP-Anforderungenmüssen rechtzeitig festgelegt undkolportiert werden, wie zum Bei-spiel die Durchführung staubrei-cher Bautätigkeiten außerhalb derMontageinnenbereiche und dieIntensivierung des Partikelmoni-torings innerhalb der Produkti-onsbereiche. Eine stetige Wach-samkeit, viel Erfahrung und eineausgeprägte Problemlösungskom-petenz sind erforderlich, umMontagetätigkeiten zu koordinie-ren und auftauchende Schwierig-keiten zeitnah zu beseitigen.
Der Eingriff in dieProduktion – eineOperation am offenenHerzen
Zur Vorbereitung der produktions-freien Zeiten muss jeweils ein Ab-laufplan erstellt werden. Empfeh-lenswert ist hierbei die Form einesMaßnahmenkatalogs, individuellaufgeteilt in einzelne Bereiche miteindeutig zugeordneten Verantwort-lichkeiten.
Bei der Ermittlung der Tätigkeits-liste sind alle beteiligten Gewerkewie Bau, Produktion, Raumlufttech-nik, Medien, Qualifizierung inklusivedie Peripherie der Automation undProzessleittechnik zu berücksichti-gen, nicht zu vergessen die angren-zenden Bereiche wie Labor, Logistik,Validierung und die eigentlichenWartungsarbeiten. Alle notwendigenAktivitäten müssen identifiziert undzeitlich definiert werden. Auswirkun-gen auf andere Bereiche sind zu son-dieren und möglichst zu minimieren.Aufgrund der hohen spezifischen Ab-hängigkeiten der einzelnen Gewerke
ist eine enge Abstimmung unerläss-lich.
Die regulatorischen Vorschriftenund Richtlinien unterscheiden nichtzwischen Neu- oder Umbau einerpharmazeutischen Produktionsstät-te. Mit Hilfe von frühzeitig durch-geführten Risikobetrachtungen undkonsolidierten Risikobewertungender kritischen Systeme sind alle Maß-nahmen GMP-konform festgelegtund terminlich fixiert. Das Ergebnisist ein auf die Stunde präzisierterTerminplan sowie ein gemeinsamentwickeltes Not-Szenario.
Diese lückenlose Vorbereitung istimmens wichtig, um den eigentli-chen Ablauf während der produkti-onsfreien Zeiten störungsfrei durch-führen zu können.
Nichts darf dem Zufall überlassenwerden. Alle Voraussetzungen fürdie Ausführung der Tätigkeiten müs-sen vorab geschaffen werden. Diesreicht von völlig banalen Punktenwie beispielsweise Parkmöglichkei-ten für die Lieferanten, Anmeldun-gen, Kennzeichnung und eindeutigeAbgrenzung der Baubereiche, gere-gelte Verkehrswege, festgelegte Zu-trittsberechtigungen, Zutrittskarten,Schulung und Baustelleneinweisung,Arbeitsgenehmigungen für Wochen-end- und Nachtarbeiten, Verfügbar-keit der notwendigen Medien, Si-cherheits- und Schleusenkleidung,Hebehilfen und Sicherheitsequip-ment, Prüfung der Schweißerlaub-nis, fixierte Unterschriften- und Ver-tretungsregelungen bis zum Notfall-plan bei Evakuierung.
Mancher Terminplan wäre schonfast geplatzt, weil fehlende Voraus-setzungen für die Tätigkeiten erstan Ort und Stelle geschaffen werdenmussten und dadurch wertvolle Zeitverschenkt wurde.
Der Zeitrahmen der Produktions-stillstände ist unverrückbar. Der Pro-jektleiter muss schon bei Verdachtvon Terminverzug entsprechendagieren. Alle Aktivitäten müssenexakt in ihren zeitlichen Vorgabenbleiben und gut ineinander greifen.Das erfordert von allen Beteiligtenein sehr hohes Maß an Flexibilität,
Engagement sowie eine gute undschnelle Projektkommunikation. EinLaufplan für die Lieferanten erleich-tert die Übersicht, klar definiertekurze Kommunikationswege verbes-sern die Reaktionszeiten bei kurzfris-tigen Problemen.
Und doch ist – trotz aufwändigerIst-Zustandsaufnahme und genau-ster Planung – keine Baustelle vorÜberraschungen gefeit. Was dann?
Pufferzeiten imTerminplan – die Rettungdes Projektes?
Grundsätzlich empfiehlt sich für je-den Terminplan immer ein gewisserPuffer. Zeitpuffer im Projekt sindaber ein zweischneidiges Schwert.Auf der einen Seite sind sie wichtig,um nicht gleich bei der kleinsten Ter-minverschiebung das gesamte Pro-jekt zu gefährden. Andererseits ver-führen zu großzügig bemessene Puf-fer oder überhaupt bekannte Puffer-zeiten im Projektteam zu einer nach-lassenden Termindisziplin. Um die-sem Problem zu begegnen, bietet essich an, nicht jedes Arbeitspaket mitZeitpuffern auszustatten, sondernnur einige wenige Ketten von mehre-ren Arbeitspaketen ganz am Ende.Das hat zur Folge, dass Terminver-schiebungen am Anfang der Kettesofort ins Auge fallen und gerechtfer-tigt werden müssen.
Fazit und Ausblick
Mit einer detaillierten Planung auchschon in der Konzeptphase sowie ei-ner guten Vorbereitung und enga-giertem Einsatz lässt sich auch dieKönigsdisziplin, der Umbau bei lau-fender Produktion im Rahmen derGMP, sauber bewerkstelligen. In ei-nem Lessons Learned nach jeder kri-tischen Phase werden die zum Teiltypischen Probleme und Schwierig-keiten genauer untersucht. Das Teamwird dadurch für die nächsten ge-planten Arbeitssequenzen sensibili-siert; der Ablauf während der Pro-duktionsstillstände wird perfektio-niert.
Maschinen- und Anlagenbau
TechnoPharm 4, Nr. 4, 186–190 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)190 Enderlein – Umbau während laufender Produktion . na und!

Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2014-04\Anzeigensatz-keine-Druck-PDFs\west-TP-2014-04-216x303.indd Zuletzt gesichert: 16.07.14 (08:57:13 Uhr)
West and the diamond logo, FluroTec® and By your side for a healthier world™ are trademarks or registered trademarks of West Pharmaceutical Services, Inc.,
in the United States and other jurisdictions.FluroTec technology is licensed from Daikyo Seiko, Ltd.
Copyright © 2014 West Pharmaceutical Services, Inc. #8676
An Investment in Drug Protection
Pharmaceutical manufacturers around the world choose components with FluroTec barrier film
for their injectable drugs. FluroTec film can help reduce the risk of packaging-related
failures and may improve component performance in filling lines. With West,
you have a partner by your side from discovery to the patient.
Help improve your drug’s shelf life
Help reduce visible and subvisible particulates
Help improve drug/closure compatability
Drug Protection
Contact West today.
North America +1 800-345-9800 Europe +49 2403 7960 Asia Pacific +65 6860 5879 www.westpharma.com
Visit us at Pharmaceutical Freeze Drying Conference Brussels, September 16–17, 2014, Booth #8.
8676 FluroTec Ad .indd 1 7/16/2014 7:43:34 AM

Serie: Sauberkeit und Reinigungin der ProduktionTeil 2: Technische Verunreinigungen bestimmen
Prof. Dr. Juliane König-Birk . PAMAS GmbH, Rutesheim
Korrespondenz: Prof. Dr. Juliane König-Birk, PAMAS Partikelmess- und Analysesysteme GmbH, Dieselstr.10, 71277 Rutesheim,e-mail: [email protected]
ZusammenfassungUm bestimmen zu können, ab wann eine Probe, eine Verpackung oder eine Oberflächeals verschmutzt zu gelten hat, müssen bestimmte Festlegungen getroffen werden. ZumTeil kann man sich an Normen orientieren, die genaue Vorgaben machen, was als Ver-unreinigung zählt und wie diese zu analysieren sind. Für manche Bereiche existierenjedoch keine Normen oder man entscheidet sich bewusst dafür, strengere Richtlinien alsdie Norm anzuwenden. In anderen Fällen ist es notwendig und sinnvoll, branchenweitübliche oder firmeninterne Festlegungen zu vereinbaren.Um genau definieren zu können, was als Verunreinigung gilt, sind oft schon Vorkennt-nisse darüber notwendig, woher die Verschmutzung kommt und ob diese überhaupt ver-meidbar ist. Kann die Verunreinigung im Produktions- oder Verarbeitungsprozess nichtumgangen werden, so muss eine Methode gefunden werden, um sie z. B. nach Art oderGröße zu klassifizieren. Dabei ist darauf zu achten, dass ein geeignetes Verfahrengewählt wird. Das gewählte Analyseverfahren dient einerseits dazu, den Verschmut-zungsgrad verschiedener Proben miteinander vergleichen zu können. Andererseits sollendamit eventuell notwendige Reinigungsschritte auf ihre Effektivität hin überprüfbar sein.Die einzusetzenden Verfahren müssen genau auf die zu reinigende Probe abgestimmtwerden. Ansonsten können nach der Reinigungsprozedur unerwünschte Effekte auf-treten.Der nachfolgende Artikel wird insbesondere auf die Voraussetzungen und Fehlermöglich-keiten verschiedener Analyseverfahren eingehen. Weiter werden unterschiedliche Rei-nigungsverfahren und die dabei zu beachtenden Besonderheiten vorgestellt. Somit wirddas entsprechende Wissen vermittelt, um die für die jeweilige Anwendung geeigneteMöglichkeit auszuwählen.
Einleitung
Da der Begriff „Technische Verunrei-nigung“ nicht definiert ist, ist nichtfestgelegt, was genau er beinhaltet.Auch für Formulierungen wie „Tech-nische Verschmutzung“ oder – wennman nicht die Verschmutzung, son-dern die Sauberkeit betrachtenmöchte – „Technische Sauberkeit“
und „Technische Reinheit“ existierenkeine Definitionen. So kann die Kon-tamination einer Flüssigkeit beimAbfüllprozess eine technische Ver-unreinigung darstellen, wenn Par-tikel im Größenbereich von wenigenMikrometern von den Behälterwän-den in die Flüssigkeit gelangen.Ebenso kann die filmische Kontami-nation einer Oberfläche, die im Her-
stellungsprozess entsteht, darunterfallen.
An diesen beiden Beispielen zeigtsich, dass Verschmutzungen in ver-schiedene Arten eingeteilt werdenkönnen. Üblicherweise wird zwi-schen partikulären und filmischenKontaminationen unterschieden.
Um festlegen zu können, wasüberhaupt als Verschmutzung gilt,
Maschinen- und Anlagenbau
TechnoPharm 4, Nr. 4, 192–197 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)192 König-Birk . Technische Verunreinigungen bestimmen
Autor
Prof. Dr. Juliane König-Birk
Nach einem Stu-dium der Physik ander UniversitätKonstanz pro-movierte Frau Prof.Dr. König-Birküber „Nahfeldpho-tographie mit kur-zen Laserpulsen“.Nach der Pro-motion ging sie indie Industrie zurPamas GmbH(Partikelmess- und
Analysesysteme). Dort war sie in der Entwick-lungsabteilung für die Neuentwicklung von Sen-soren zuständig. Dies umfasste den gesamtenProzess von der Idee, über die Entwicklung bis zurSerienreife. Frau Prof. Dr. König-Birk war stellver-tretende Leiterin der Entwicklungsabteilung derPamas GmbH. Seit November 2011 ist Frau Prof.Dr. König-Birk im Vorstand des Cleaning Excel-lence Center - Kompetenznetzwerk für IndustrielleBauteil- und Oberflächenreinigung Leonberg e.V.tätig. Dort haben sich mittlerweile über 40 Firmenvor allem aus dem süddeutschen Raum zusam-mengeschlossen. Seit April 2012 ist Frau Prof. Dr.König-Birk als Professorin für mathematisch-na-turwissenschaftliche Grundlagen im StudiengangProduktion und Prozessmanagement an derHochschule Heilbronn tätig.

wird häufig die Ausdehnung und Di-cke einer Schicht auf einer Oberflä-che oder die Ausdehnung und Be-schaffenheit von Partikeln auf undin Feststoffen und in Flüssigkeitenund Gasen als Parameter angegeben.Damit ist der erste Schritt, der aufdem Weg zur Bestimmung von tech-nischen Verunreinigungen gegangenwerden muss, festgelegt: Es musseine Grenze definiert werden, dieden Übergang von „sauber“ zu „kon-taminiert“ markiert. Diese Fest-legung kann auf Grund von Normengeschehen. Ebenso kann sie sich anprozessgesteuerten oder technischenGegebenheiten orientieren. Je nachKomplexität der Aufgabe sollte mansich im Vorfeld schon informieren,mit welcher Genauigkeit gewisse Pa-rameter unter welchem Aufwand ge-messen werden können. Unter Um-ständenmussman auf einen anderenMesswert ausweichen, über den sichdann der eigentlich gewünschteGrenzwert bestimmen lässt.
Filmische Verunreinigungen las-sen sich zum Beispiel mittels Test-tinten, Kontaktwinkelbestimmung,Fluoreszenzmessung, FTIR-Spektro-metern oder Ellipsometrie analysie-ren:. Testtinten erlauben eine Aussageüber die Oberflächenspannung derfilmischen Verunreinigung. JedeTesttinte liefert einen Messwert.Folglich ist das Verfahren mehr-fach durchzuführen, wenn einexakter Wert bestimmt werdensoll. Da sich die Oberflächenspan-nung der Verschmutzung unterUmständen mit der Zeit ändernkann, sollte der Messzeitpunktimmer im gleichen zeitlichen Ab-
stand zur Entstehung der fil-mischen Kontamination und unteransonsten gleichen Bedingungengewählt werden.Soll die Oberflächenspannungmittels der Testtinten-Methodeermittelt werden, so muss es sichzwangsweise um eine Oberflächehandeln, die unempfindlich genugist, um das Aufbringen der Tintemittels eines Pinsels ohne Beschä-digungen zu überstehen.
. Auch bei der Kontaktwinkel-bestimmung wird ausgenutzt, dassunterschiedliche Materialien einunterschiedliches Benetzungsver-halten zeigen. Die dahinter ste-hende Theorie geht davon aus,dass die Oberfläche eben undchemisch homogen ist. Das istnicht notwendigerweise der Fall(Abbildung 1). Inzwischen gibt esMessgeräte, die die Rauigkeit derOberfläche an der gleichen Pro-benstelle wie die Kontaktwinkel-messung durchführen. Damit kannder tatsächliche Winkel und somitdie tatsächliche Oberflächenspan-nung bzw. -energie ermittelt wer-den. Mit den exakten Werten istdann eine genaue Bestimmung derVerunreinigung möglich.Bei der Fluoreszenzmessung wirdUV-Licht eingesetzt. Die filmischeVerschmutzung wird zur Fluores-zenz angeregt und das dabei auf-tretende Signal detektiert. Je stär-ker die Verunreinigung, destostärker ist das Signal.Bevor die erste Messung gestartetwerden kann, muss eine absolutsaubere Oberfläche zur Kalibrie-rung zur Verfügung stehen. DieWahl dieses Referenzpunktes muss
somit sehrsorgsam erfol-gen. Weitermuss sicher-gestellt werden,dass keine an-deren Substan-zen wie z.B. derUntergrundzum Fluores-zenzsignal bei-tragen, da sonst
keine gesicherte Aussage über diezu messende Verschmutzung mehrmöglich ist.Mittels der Fluoreszenzmessungist eine Aussage über die Kon-tamination am Punkt, der mit demUV-Licht beleuchtet wird, möglich.Sollen größere Oberflächen unter-sucht werden, müssen Probe undMessgerät gegeneinander bewegtwerden.
. Wie der Name schon sagt, arbeitetdas FTIR-Spektrometer mit Wel-lenlängen im infraroten Wellen-längenbereich (IR). Die AbkürzungFT steht für Fourier-Transformati-on. Dies bezeichnet den mathe-matischen Weg der Umwandlungdes gemessenen Signals in dasausgegebene Spektrum. DieseMessmethode wird vor allem fürorganische Substanzen eingesetzt.Das Spektrum, das die Kontami-nation liefert, wird mit in einerDatenbank hinterlegten Spektrenverglichen. Folglich können nurSubstanzen erkannt werden, dieauch in der Datenbank vorhandensind.Befindet sich auf der zu unter-suchenden Probe Wasser, so kön-nen die Messwerte in nicht uner-heblichem Maße verfälscht wer-den. Dies liegt darin begründet,dass im Wassermolekül verschie-dene Schwingungsmoden angeregtwerden können. Durch Übergängein den Rotationszuständen, Streck-und Biegeschwingungen werdensowohl Frequenzen im fernen IRals auch im nahen und mittlerenFrequenzbereich absorbiert. Somitist sicherzustellen, dass die Probenahezu wasserfrei ist und auch
TechnoPharm 4, Nr. 4, 192–197 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 193König-Birk . Technische Verunreinigungen bestimmen
Abb. 1: Kontaktwinkelmessung auf nicht ebenen Oberflächen.
keine Anlagerung von Wasser z.B.aus der Luftfeuchtigkeit an der zumessenden Oberfläche möglich istoder dass dadurch entstehendeEffekte bei den Messwerten be-rücksichtigt werden.
. Das Messverfahren der Ellipsome-trie kann ganz unterschiedlicheFrequenzbereiche nutzen. Somitkönnen verschiedenste Materialienuntersucht werden. Die Ellipsome-trie wird vor allem dazu verwendet,Schichtdicken zu bestimmen. DerMessaufbau funktioniert so, dassLicht eines bestimmten Polarisati-onszustandes auf die Probe trifft. Jenach dort vorhandenem Materialund Schichtdicke wird die Polari-sationsrichtung des eintreffendenLichts verändert. Über die Detek-tion dieser Änderung können nundie Schichtdicke oder der Bre-chungsindex des Materials (unddamit das Material selber) be-stimmt werden. Hierfür ist aller-dings ein Modell zu erstellen, in dasdie Materialeigenschaften als Mo-dellparameter eingehen. Trotz derdamit einhergehenden Ungenau-igkeiten, liegt die Empfindlichkeitvon Ellipsometern bei unter 1 nm.
Zusammenfassend lässt sich für dieBestimmung von filmischen Verunrei-nigungen sagen,dass es je nachAnwendung ber-ührungslose undzerstörungsfreieMessmethodenwie z.B. die Ellip-sometrie oder dieFTIR-Spektrosko-pie gibt, die aller-dings eher teuersind. Außerdemstehen günstigeVerfahren wie dieTesttinten-Me-thode zur Ver-fügung, die immerdann eingesetztwerden können,wenn keine berüh-rungslose Mes-sung gefordertwird.
Zur Bestimmung von partikulärenVerunreinigungen werden meist dieMethoden Mikroskopie, Gravimetrie,optische Partikelzählung, Raster-Elektronen-Mikroskopie und Ra-man-Spektroskopie eingesetzt. ImGegensatz zu filmischen Verunrei-nigungen, die direkt auf der Oberflä-che, auf der sie sich befinden, detek-tiert werden, müssen partikuläre Ver-schmutzungen nicht auf der Oberflä-che, auf der sie sich zunächst befin-den untersucht werden. Ein Ablösender Partikel kann mittels einer ande-ren Oberfläche, an der die Partikelbesser haften, geschehen. Sinnvollist diese Vorgehensweise, wenn dieVerunreinigungen auf der nun zu un-tersuchenden Oberfläche z.B. aufGrund von optischen Eigenschaftenbesser detektiert werden könnenoder wenn die eigentliche Probe füreine direkte Nachweismethode zuempfindlich oder unhandlich ist. Al-lerdings ist darauf zu achten, dass dieProbenoberfläche durch die ge-wählte Methode zum Entfernen derPartikel nicht anderweitig verunrei-nigt oder beschädigt wird. Physika-lisch gesehen müssen zur Beschrei-bung des Anhaftungs- und Ablöse-prozesses die Adhäsions- und Kohä-sionskräfte betrachtet werden. Hier-
bei spielt es keine Rolle, ob die Par-tikel mittels eines anderen Feststof-fes, einer Flüssigkeit oder durch Ab-blasen entfernt werden.
Verfahren, die eine Flüssigkeitverwenden sind Abspülen, Absprit-zen, Fluten oder Ablösen im Ultra-schallbad. Dies bietet sich z.B. dannan, wenn die Oberflächenrauigkeitund die Partikel eine ähnliche Grö-ßenordnung aufweisen. Damit istein Ablöseprozess mittels einer an-deren Oberfläche praktisch unmög-lich und direkte Nachweismethodenliefern keine guten Ergebnisse mehr.Die Methode, mit der die Verunrei-nigung in eine Flüssigkeit gebrachtwird, ist sowohl auf die ver-schmutzte Oberfläche als auch aufdie Verunreinigung selbst abzustim-men: Die Partikel dürfen dabei nichtzerstört werden, da eine weitere Un-tersuchung ansonsten unmöglichist. Meist soll auch die verunreinigteOberfläche durch den Ablösepro-zess nicht zerstört werden. Anderer-seits müssen die Kräfte, die ein An-haften bewirken, überwunden wer-den. Im Ultraschallbad z.B. basiertdie Reinigung auf Kavitation. Überdie dabei wirkenden Kräfte könnenBeschichtungen mikroperforiertoder Oberflächen mit winzigsten
Maschinen- und Anlagenbau
TechnoPharm 4, Nr. 4, 192–197 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)194 König-Birk . Technische Verunreinigungen bestimmen
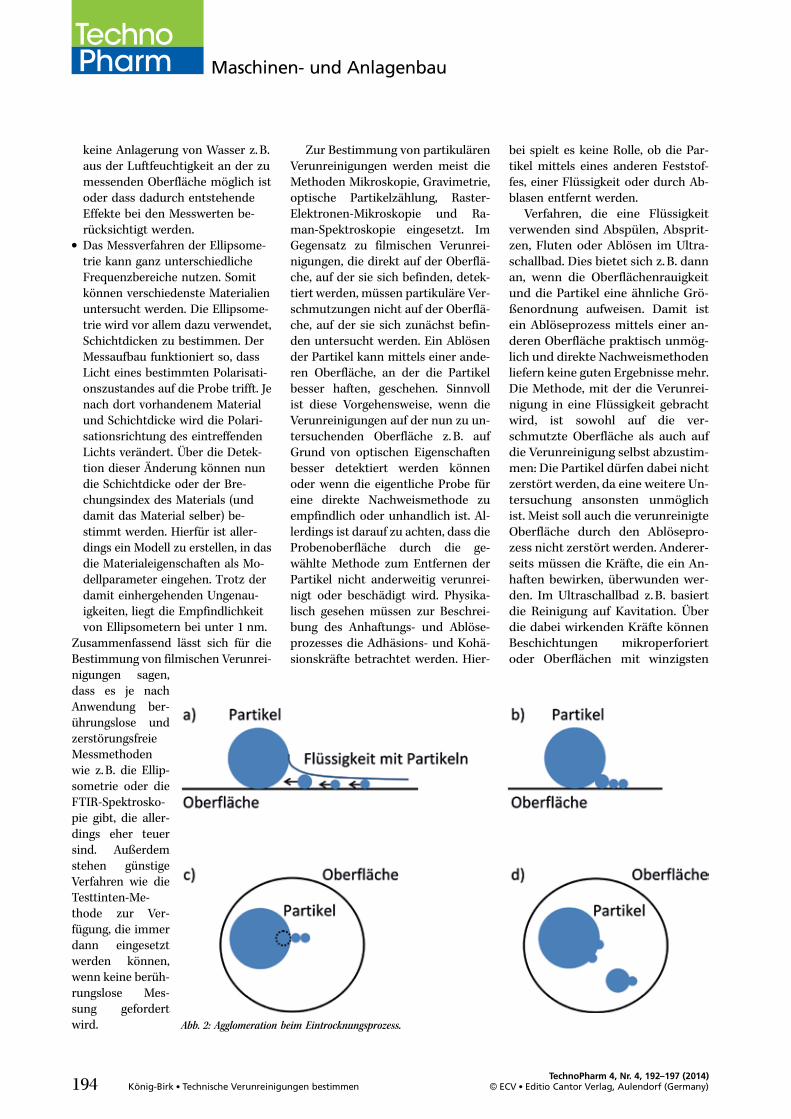
Abb. 2: Agglomeration beim Eintrocknungsprozess.

ecv
ECV · Editio Cantor Verlag
Ihr UnternehmenIhr Know-how
Ihr Buch
www.insights.ecv.de
Alles was Sie wissen müssen:Lara Lehmann Tel. +49 (0)8191-985 78 11eMail: [email protected]
Die Corporate Books-Reihe ecvINSIGHTS!
Ihr Buchdigital | gedruckt
NEU!

Löchern versehen werden. Folglichmuss vorher festgestellt werden,welche Behandlung die geeigneteist.
Nun ist entweder die Flüssigkeit,in der sich die Partikel jetzt befinden,zu analysieren oder wieder eineOberfläche, wenn die Teilchen ausder Flüssigkeit auf einen Filter auf-gebracht werden. Hierbei ist zu be-achten, dass der Eintrocknungspro-zess auf dem Filter so gesteuert wird,dass die nachfol-gende Ana-lysemethodeauch anwendbarist. Zur Bestim-mung der Grö-ßenverteilungmüssen die Par-tikel einzeln lie-gen. Betrachtetman die Kräftefür diesen Pro-zess, so zeigtsich, dass kleinePartikel durchden verdunsten-den Flüssigkeitsfilm zu größeren Par-tikeln hingezogen werden (Abbil-dung 2a+b). Im Mikroskop könnendie kleineren Partikel dann nicht de-tektiert werden, wenn sie sich zwi-schen einem großen Partikel undder Oberfläche befinden (Abbil-dung 2c). Sind die Partikel teilweiseverdeckt oder liegen zu nahe bei-einander, so wird anstatt mehreren,ein großes Teilchen erkannt (Abbil-dung 2d). Ist die nachfolgend ge-wählte Analysemethode die Gravi-metrie, so wird die Gesamtmasseder Verunreinigung bestimmt. Folg-lich ist es unerheblich, ob sich diePartikel einzeln oder als Agglomerateauf dem Filter befinden.
Auch Partikel, die von der Ober-fläche mittels eines Verfahrens ent-fernt werden, das diese in ein Gasbringt, werden danach oft auf einemFilter abgeschieden, um sie unter-suchen zu können. Ebenso wie fürdie Methoden, die eine Flüssigkeitverwenden, ist darauf zu achten, dassdie Oberfläche und die Partikel nichtverändert werden. Beispielhaft sei
hier das sogenannte Schneestrahlengenannt: Verwendet wird ein CO2-Schneestrahl. Dieser gilt als umwelt-freundlich, da das verwendete Koh-lenstoffdioxid oft als Nebenproduktin Prozessen anfällt und keine wei-teren Chemikalien eingesetzt wer-den. Ebenso wie oben für die Ultra-schallreinigung beschrieben, könnenaber auch hier durch die wirkendenKräfte kleinste Löcher in der zu rei-nigenden Oberfläche entstehen.
Oft bereiten partikuläre Ver-schmutzungen direkt in Flüssigkei-ten oder Gasen Probleme. Genanntseien in diesem Zusammenhang z.B.Infusionslösungen, Flow-Boxen oderReinräume.
Bei der Bestimmung des Partikel-gehalts in der Raumluft kommen Par-tikelfallen und Partikelzähler zumEinsatz. Partikelfallen müssen so auf-gestellt werden, dass sie an repräsen-tativen Orten stehen und nicht be-sonderen Strömungsverhältnissenausgesetzt sind, die für andere Pro-ben nicht gelten. Nur so kann sicher-gestellt werden, dass in den Partikel-fallen tatsächlich die Art und Anzahlder Partikel eingefangen wird, dieauch im übrigen Prozess anfallen.Die eingefangenen Teilchen müssenanschließend untersucht werden.
Partikelzähler hingegen gehörenzu den direkten Detektions- undAnalysegeräten.
Im Folgenden werden verschie-dene Detektions- und Analysemetho-den vorgestellt, je nachdem, welcheEigenschaft untersucht werden soll:
. Die Gravimetrie stellt ein einfachesAbwiegen der Partikelfracht aufeinem Filter dar. Um tatsächlichnur die Gesamtmasse der Ver-unreinigung zu bestimmen, mussvorher der Blindwert ermitteltwerden. Dieser setzt sich aus derMasse des Filters, eventuell einesHalters und der Masse der Partikelzusammen, die sich auf dem sau-beren Filter befinden. Die Gravi-metrie liefert damit keine Aus-
sagen über die Größenverteilungund die Beschaffenheit der par-tikulären Verunreinigung.
. Mittels eines Mikroskops könnenebenfalls Partikel auf einem Filteruntersucht werden (Abbildung 3a).Hierbei können die ParameterGröße, Form und Anzahl pro Flä-cheneinheit bestimmt werden. MitEinschränkungen ist auch eineAussage über das Material möglich(z.B. metallischer Glanz oderdurchsichtig im sichtbaren Wel-lenlängenbereich).
. Optische Partikelzähler (Abbil-dung 3b) analysieren Partikel inFlüssigkeiten oder Gasen. Hierbeiist zwischen Partikelzählern, diedas gesamte Volumen messen undsolchen, die lediglich einen Teil derFlüssigkeit bzw. des Gases unter-suchen, zu unterscheiden. Im Ge-gensatz zu Trübungsmessern, dieam Kollektiv messen und somitnur einen Trübungswert unab-hängig von der Anzahl und derGröße der Teilchen ausgeben,können mittels Partikelzählern
Maschinen- und Anlagenbau
TechnoPharm 4, Nr. 4, 192–197 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)196 König-Birk . Technische Verunreinigungen bestimmen
Abb. 3: Detektions- und Analysegeräte.

einzelne Partikel analysiert wer-den. Der ausgegebene ParameterGröße entspricht dem Durchmes-ser eines Kreises mit zum gemes-senen Partikel äquivalenter Fläche.Um eine Probe mit dem Raster-Elektronen-Mikroskop (REM) un-tersuchen zu können, muss sicher-gestellt sein, dass es nicht zu Auf-ladungseffekten kommt. Daher wirddie Probe entsprechend präpariert.Eine Möglichkeit stellt das Auf-dampfen eines dünnen Goldfilmesdar. Weiter muss die Probe so be-schaffen sein, dass sie ins Vakuumdes REM gebracht werden kann.Meist wird eine kombinierte REM-EDX-Analyse durchgeführt, bei derauch die entstehende Röntgen-strahlung detektiert wird. Damitkann sowohl die Größe, die Ober-flächenbeschaffenheit als auch die
Elementzusammensetzung derPartikel bestimmt werden.
. Die Raman-Spektroskopie nutztdie inelastische Streuung, um Ma-terialeigenschaften zu bestimmen.Dabei kann es zu einem Aufheizender Probe und somit zur ther-mischen Zerstörung kommen.Durch das thermische Signal oderein ebenfalls entstehendes Fluo-reszenzsignal kann das eigentlicheSignal überdeckt werden.
Da einige der aufgeführten Ana-lysemethoden speziell geschultesPersonal und teure Geräteinvestitio-nen erfordern, ist eine Abwägungsinnvoll, ob die Untersuchungen imeigenen Haus durchgeführt oder obexterne Dienstleister eingeschaltetwerden sollen.
Geht es um die Festlegung von zubestimmenden Parametern und
Grenzwerten, ist genau zu überlegen,welcher Nutzen aus der Kenntniswelches Parameters gezogen werdenkann, welche Grenzwerte noch tole-rierbar sind, mit welchen Methodensie zu bestimmen sind und welcheFehlertoleranzen damit einhergehen.Unabdingbar bei jedem eingesetztenAnalyseverfahren ist, dass es repro-duzierbare Ergebnisse liefert. Nur sokönnen Messungen an unterschiedli-chen Proben miteinander verglichenwerden.
In manchen Fällen kann es sinn-voll sein, über die eingesetzten Mate-rialien der Bauteile nachzudenken.Betrachtet man die oben erwähntenAdhäsions- und Kohäsionskräfte, solassen sich manche Verunreinigun-gen von Anfang an durch eine geeig-nete Wahl der Materialien vermei-den.
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2014-04\Anzeigensatz-keine-Druck-PDFs\belimed-TP-2014-04-216x149.indd Zuletzt gesichert: 21.07.14 (10:44:24 Uhr)
Belimed Deutschland: +49 8631 9896 0, Österreich: +43 3155 40699 0, Schweiz: 0848 55 88 11, www.belimed.com
Maßgeschneidert auf Ihre Bedürfnisse
Gesamtlösungen für Reinigung, Desinfektion und Sterilisation in Medizin, Pharma und Labor
GMP-konforme Reinigungs- und SterilisationsanlagenAngepasst an Ihre Anforderungen, für höchste Sicherheit Ihrer Produkte.

Reinraum-Qualifizierungs-messungen im Sterilbereich – Teil 2Regulatorische Ableitung und risikobasierte Festlegung
Stefan Erens . Testo industrial services GmbH, Kirchzarten
Korrespondenz: Stefan Erens, Testo industrial services GmbH, Gewerbestraße 3, 79199 Kirchzarten;e-mail: [email protected]
Risk based approach,Risikobasierter Ansatz zurDefinition von Qualifizie-rungsumfängen fürReinräume und reinluft-technische Anlagen
Eine risikobasierte Festlegung derQualifizierungsumfänge (Abb. 1) istsowohl in der DIN EN ISO 14644-2[3] als auch im PIC/S PI 032-2 [2]empfohlen. Mit Inkrafttreten desQuality Risk Management (ICH Q9),Teil III, EU GMP Leitfaden [5], wur-den richtungsweisende, strukturie-rende Elemente zur Steuerung vonRisiken definiert.
Diese setzen nicht erst mit derQualifizierung der Reinraumbereicheein, sondern schon in der Planungs-phase für lufttechnische Anlagen undReinräume. So können manche Risi-ken nicht über einen hohen Test-oder Monitoringaufwand innerhalbvertretbarer Grenzen minimiert wer-den, sondern sind unter Umständennur durch Eingriffe in Technik, Lay-out oder Prozessfluss unter Kontrollezu bringen. Wie bei allen GMP-Com-pliance induzierten Maßnahmen, istauch bei der Definition der reinluft-technischen Umgebungsbedingun-gen ein Life cycle Approach rich-tungsweisend für die Umsetzungder qualitätsdefinierenden Parame-ter.
Dabei sind die beiden Grundprin-zipien eines Qualitätsrisikomanage-
ments führend in der strukturiertenVorgehensweise:. Die Evaluierung von qualitätsin-duzierenden Risiken, basierend aufwissenschaftlichem Wissen undder bewusste und direkte Bezugzum Schutz des Patienten.
. Das Niveau des Aufwandes, derformalen Umsetzung und Doku-mentation des Qualitätsmanage-mentprozesses sollte angemessensein zum Niveau des Risikos.
Dies gilt natürlich im risikobasiertenRahmen sowohl für bestehende Rein-räume und raumlufttechnische Anla-gen, als auch für neuprojektierte Ge-werke.
Eine detailliertere Definition sollim Rahmen einer Risikobewertungfür die einzelnen Zonen, Räumeund reinlufttechnischen Anlagenfrühzeitig erfolgen.
Da die Umsetzung des Risikoma-nagementprozesses stark vom Pro-dukt und Einfluss der Umgebungauf die Arzneimittel- und Patienten-sicherheit abhängt und unterschied-liche pharmazeutische Hersteller dif-ferierende Systeme zum Risikomana-gement abbilden, soll im Folgendeneine strukturierte Vorgehensweisevorgestellt werden, die sich anhandder Basiselemente des ICH Q9 (vgl.Abb. 1) orientieren:. RisikobeurteilungMit den Teilelementen Risikoidenti-fizierung, Risikoanalyse und Risiko-bewertung
Reinraum
TechnoPharm 4, Nr. 4, 198–206 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)198 Erens – Reinraum-Qualifizierungsmessungen im Sterilbereich – Teil 2
Autor
Stefan Erens
Stefan Erens wurde 1970 in Kempen (Ndrh.) ge-boren und studierte Chemieingenieurwesen an derFachhochschule Niederrhein. Anschließend star-tete er bei Shimadzu im Vertriebs-Außendienst füranalytische Geräte ins Berufsleben.1996 wechselte er zur Astrid Twardy GmbH – ei-nem mittelständischen OTC-Arzneimittelhersteller– und übernahm dort die Leitung des analytischenLabors. Die ersten Erfahrungen im Engineering-Bereich sammelte er ab dem Jahr 2000 bei derTriplan AG, wo er Projekte im Qualifizierungs- undValidierungsbereich bei namhaften pharmazeuti-schen Unternehmen umsetzte.Drei Jahre später koordinierte er als Projektleiterbei der LSMW GmbH (heute M+W Process In-dustries GmbH) Projekte im Bereich Reinraum undraumlufttechnischen Anlagen. Seit 2005 ist er beider Testo Industrial Services tätig, zunächst fürBusiness Development und Projektleitung im Be-reich Reinräume und GMP-Compliance. Seit 2009leitet er den technischen Außendienst mit mitt-lerweile 80 Mitarbeitern und ab 2011 den Vertriebfür GMP-Dienstleistungen. 2012 wurde er zumProkuristen ernannt.

. RisikosteuerungMit den Teilelementen Risikoredu-zierung und Risikoakzeptanz. RisikoüberwachungMit dem Teilelement Über-wachungserkenntnisse.
Als rationale Entscheidungshilfekönnen dabei diverse Einflussgrö-ßen (Abb. 2) angesehen werden.Gruppiert man diese ergeben sichdie vier unten aufgeführten grund-sätzlichen Einflussgrößen:
Risikobeurteilung
Die strukturierte und am Life cycleApproach angelehnte Risikobeurtei-lung ist das zentrale Basiselementfür einen Qualitätsrisikomanage-mentprozess. Bereits hier werdenwährend der Design- und Planungs-phase von Anlagen oder Prozessendie Grundlagen zur späteren Beherr-schung qualitätsinduzierender Para-meter gelegt. Dabei wird der Prozessder Risikobeurteilung auch auf-grund der Festlegungen aus derICH Q9 Guideline nicht als stati-scher Vorgang, sondern als beglei-tende und im Life Cycle des Prozes-ses implementierte Maßnahme ge-sehen. Eine permanente Kommuni-kation unter den Prozess-, System-und Compliance Verantwortlichenist Grundlage für diesen gesteuertenRisikomanagementprozess („Risiko-kommunikation“). Eine Vernetzungder in Risikoanalysen festgelegten Ri-siken über den gesamten Life Cyclewird üblicherweise über Tracbility-dokumente hergestellt. Auf eine de-tailliertere Ausführung soll hier ver-zichtet werden.
Risikoidentifizierung, Risiko-analyse und RisikobewertungBeim ersten Basiselement geht es zu-nächst um die Konzentrierung vonWissen, Erfahrung und natürlichauch menschlichen Entscheidungs-und Bewertungsfaktoren. Die ver-ständliche und nachvollziehbare For-derung und Erwartung nach einem„0-Risiko“ ist aus der theoretischenAbleitung und praktischen Erfahrungniemals umsetzbar. Somit sind Risi-
komanagementprozesse „nur“ eineMöglichkeit Risiken beherrschbarzu machen und diese auf ein akzep-tables Niveau zu reduzieren.
Die strukturierte Vorgehensweiseeines Risikomanagementprozessesbietet mit verschiedenen Risikoana-
lysetools (z.B. FMEA, Failure ModeEffects Analysis) dann auch Möglich-keiten, einen für die Risiken adäqua-ten, organisatorischen und doku-mentatorischen Rahmen zu geben.
Angewendet auf die bereits for-mulierten Einflussgrößen lässt sich
TechnoPharm 4, Nr. 4, 198–206 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 199Erens – Reinraum-Qualifizierungsmessungen im Sterilbereich – Teil 2
Abb. 1: Darstellung Risikomanagementprozess nach „Quality Risk Management (ICH Q9)“,EU GMP Leitfaden Teil III, deutsche Übersetzung aus Bekanntmachung des BMG.
Abb. 2: Einflussgrößen für die Beurteilung von Risiken aus den Umgebungsbedingungen.

so eine breite Basis zur Beherrschungder Risiken formulieren:
Einflussgröße ProzessHat der Prozess oder Teile der Pro-zessführung bestimmte Anforderun-gen an die Umgebung zur Folge?
Hier können als Beispiel Räume mithohen Wärmelasten, z. B. freigesetztdurch Autoklaven, genannt werden,die mittels hohen Luftwechselraten eli-miniert werden müssen.
Einflussgröße ProduktWelche Einflussfaktoren der Umge-bung beeinträchtigen die Qualitätdes hergestellten Produktes negativ?
Beispielhaft kann hier die Sterilpro-duktion angeführt werden, in der unterallen Umständen partikuläre Ver-unreinigungen des Produktes vermie-den werden müssen.
Einflussgröße Person/UmweltGibt es für das Produkt oder dessenAusgangsstoffe/Zerfallsprodukte be-sondere Schutzmaßnahmen hin-sichtlich des Mitarbeiterschutzesoder des Schutzes der Umwelt (EHS,Environment/Health/Safety) zu be-achten?
Als Beispiel wäre die Produktioneines Zytostatikaproduktes oder dieArbeit mit anderen hochaktiven Subs-tanzen zu nennen.
Einflussgröße RegulatorischerHintergrundGibt es für die Prozessführung regu-lative Vorgaben, entweder aus phar-mazeutischen oder technischen Re-gelwerken?
Als Beispiel kann hier die Forde-rung nach definierter Strömung in A-Bereichen herangezogen werden oderdie strahlungsgeschirmte Prozessfüh-rung bei Radiopharmaka.
Risikosteuerung
Im zweiten Basiselement des Risiko-managementprozesses geht es in ers-ter Linie darum, auf der Grundlageder zuvor definierten Risiken dieseauf ein akzeptables Maß der Beein-flussung der Qualität zu bringen.
Salopp kann man definieren, dassdie Risikosteuerung durch Maßnah-mendefinition ein akzeptables „Rest-risiko“ für die Qualität des Produktesherbeiführen soll.
Die zu definierenden Maßnahmenhängen individuell zunächst vom Ri-sikopotential des Produktes und demzur Herstellung verwendeten Prozessab. Regulatorische Rahmenbedingun-gen und das Schutzbedürfnis vonMensch und Umwelt ergänzen dieseMaßnahmen. Nicht ganz unerwähntsollte aber auch der menschliche Ein-fluss, gerade im Hinblick auf die Risi-koakzeptanz bleiben. Unterschiedli-che Betreiber und vor allem unter-schiedlich beteiligte Personen habenindividuelle Sichtweisen auf Risiken,Maßnahmen und die Risikoakzep-tanz. Daher ist eine teamorientierteHerangehensweise bei der Realisie-rung des Risikomanagementprozes-ses zielführend und nutzbringend.
Betrachtet man die Maßnahmen,die grundsätzlich zur Risikoreduktiondefiniert werden können, kann mandiese in zwei Kategorien einteilen:. Maßnahmen, welche die Auftre-tenswahrscheinlichkeit herabsetzen
. Maßnahmen, welche die Ent-deckungswahrscheinlichkeit he-raufsetzenAnmerkung: In der Praxis lassensich keine risikoreduzierenden
Maßnahmen im Hinblick aufdie Bedeutung der Schwere ei-nes Fehlers ableiten.
Maßnahmen zur Senkung derWahrscheinlichkeit desAuftretensPrimär sollte immer versucht werdenein potentielles Risiko für die Quali-tät des Produktes erst gar nicht ent-stehen zu lassen. Damit sind Maß-nahmen, welche die Auftretenswahr-scheinlichkeit von Risiken minimie-ren stets allen anderen Maßnahmenvorzuziehen.
Beispiele für solche Maßnahmensind Barrieresysteme, gerichteteLuftströmungen, aber auch die Defi-nition von ausreichenden Raumluft-wechseln.
Die hier hauptsächlich definiertenMaßnahmen beziehen sich auf tech-nische und/oder organisatorische Ele-mente, die in der Designphase zu de-finieren sind. Durch Schutzsysteme(z.B. TAV Strömungen) wird das Auf-treten bestimmter Risiken soweit mi-nimiert, dass ein akzeptables Risiko-niveau verbleibt. Auch Schulungenund die Ausbildung der handelndenPersonen z.B. im Reinraum tragenmaßgeblich zur Risikoreduktion bei.Der Nachweis der Umsetzung undWirksamkeit organisatorischer Maß-nahmen wie Ausbildung und Schu-
Reinraum
TechnoPharm 4, Nr. 4, 198–206 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)200 Erens – Reinraum-Qualifizierungsmessungen im Sterilbereich – Teil 2
Abb. 3: Direkte Beeinflussung des Produktes („direct Impact“) durch ein Leck im TAV Filter(z. B. A-Bereiche nach Annex 1).

lung stellt jedoch eine große Heraus-forderung dar und ist nur über eineffizientes und strukturiertes Schu-lungsmanagement möglich.
Häufig kann es nötig sein be-stimmte technische oder organisato-rische Maßnahmen um Maßnahmenzur Erhöhung der Entdeckungswahr-scheinlichkeit zu ergänzen.
Maßnahmen, welche dieEntdeckungswahrschein-lichkeit heraufsetzenWird durch technische und/oder or-ganisatorische Maßnahmen keineausreichend akzeptierte Risikoreduk-tion erreicht, sind zusätzliche, ergän-zende Maßnahmen zur Risikoreduk-tion erforderlich.
Als Beispiel für diese Maßnahmenkönnen alle Monitoring-, Qualifizie-rungs- und Inprozesskontrollaktivi-täten genannt werden.
Die Maßnahmen zur Heraufset-zung der Entdeckungswahrschein-lichkeit sind als zusätzliche Maßnah-men zu verstehen, da ihre Wirksam-keit erst eintritt, wenn ein potentiel-les unerwünschtes Ereignis (= Risiko)bereits aufgetreten ist. Aus dergrundsätzlichen Auslegung vonGMP ist es daher ausgeschlosseneine Risikoreduktion- und Akzeptanzdurch „Hereinprüfungsmaßnahmen“zu erreichen.
Die genannten Maßnahmen sinddeshalb aber nicht als 2. Wahl anzu-sehen. Vielmehr erhöhen diese Maß-nahmen zur Heraufsetzung derEntdeckungswahrscheinlichkeit dieSicherheit des Prozesses zusätzlichund liefern vor allem wichtige Infor-mationen über die Langzeitstabilitätvon Umgebungsbedingungen. Dieparametrische Überwachung, sei esdurch Monitoring oder Qualifizie-rungsmaßnahmen, stellt somit einenwichtigen Baustein in der Beurtei-lung der Prozessfähigkeit von umge-bungskontrollierten Bereichen (z.B.Sterilabfüllungen) dar.
Risikoüberwachung
Ein modernes Risikomanagementbildet den Life Cycle Approach ab,
TechnoPharm 4, Nr. 4, 198–206 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 201Erens – Reinraum-Qualifizierungsmessungen im Sterilbereich – Teil 2
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2014-04\Anzeigensatz-keine-Druck-PDFs\cas-TP-2014-04_105x303.indd Zuletzt gesichert: 23.06.14 (03:14:41 Uhr)
CAS Clean-Air-Service AGCH–9630 WattwilT +41 (0)71 987 01 01
D-52134 HerzogenrathT +49 (0)2407 5656 - 0
A-1120 WienT +43 (0)1 71728 285
www.cas.ch
Reine Luft ist unser Business.
R
Akkreditierte Prüfstelle STS 566für die Qualifizierung von Reinraum-systemen und thermischen Prozessen.
Akkreditierte Prüfstelle SCS 118 für die Kalibration von Luftgeschwindigkeits- sensoren, CLiMET-Partikelzählern und Volumenstrom-Messhauben.
Handel von CLiMET-Partikelzählern, Dwyer-Produkte und Kanomax-Luft-geschwindigkeitssensoren.
Des weiteren bieten wir Strömungs-visualisierung, Qualitätssicherungsmass-nahmen wie auch Kundenseminare und Workshops an.
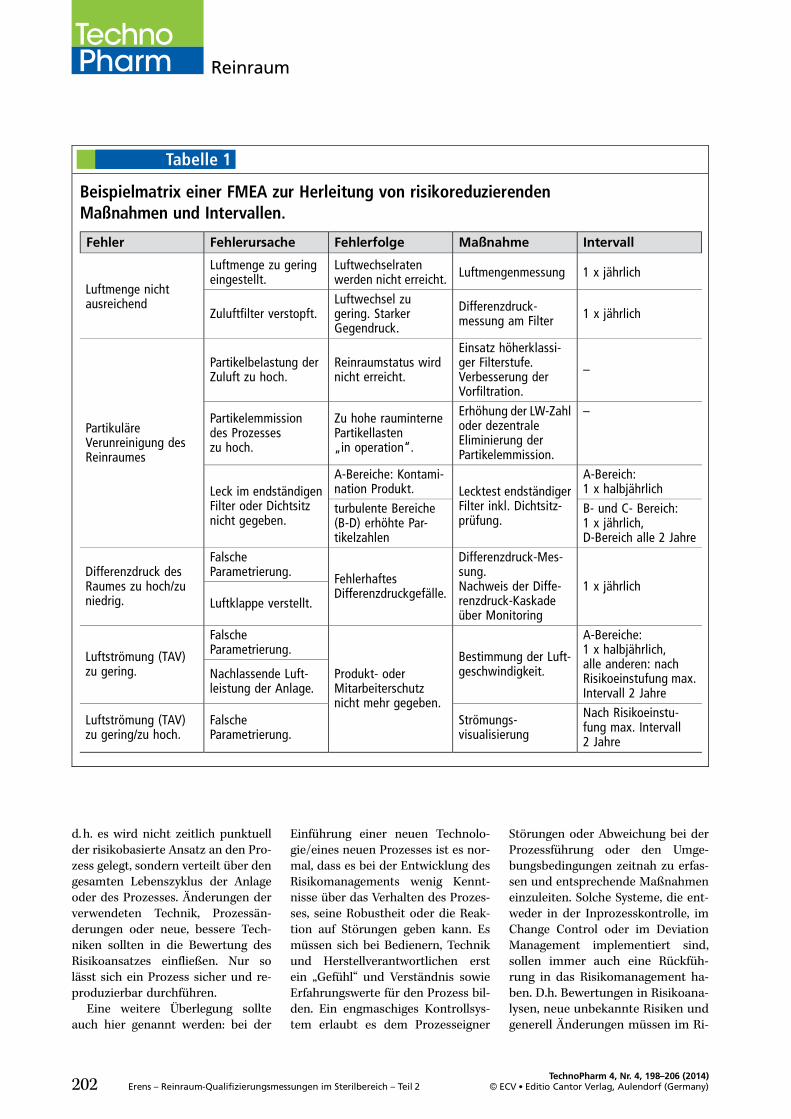
d.h. es wird nicht zeitlich punktuellder risikobasierte Ansatz an den Pro-zess gelegt, sondern verteilt über dengesamten Lebenszyklus der Anlageoder des Prozesses. Änderungen derverwendeten Technik, Prozessän-derungen oder neue, bessere Tech-niken sollten in die Bewertung desRisikoansatzes einfließen. Nur solässt sich ein Prozess sicher und re-produzierbar durchführen.
Eine weitere Überlegung sollteauch hier genannt werden: bei der
Einführung einer neuen Technolo-gie/eines neuen Prozesses ist es nor-mal, dass es bei der Entwicklung desRisikomanagements wenig Kennt-nisse über das Verhalten des Prozes-ses, seine Robustheit oder die Reak-tion auf Störungen geben kann. Esmüssen sich bei Bedienern, Technikund Herstellverantwortlichen erstein „Gefühl“ und Verständnis sowieErfahrungswerte für den Prozess bil-den. Ein engmaschiges Kontrollsys-tem erlaubt es dem Prozesseigner
Störungen oder Abweichung bei derProzessführung oder den Umge-bungsbedingungen zeitnah zu erfas-sen und entsprechende Maßnahmeneinzuleiten. Solche Systeme, die ent-weder in der Inprozesskontrolle, imChange Control oder im DeviationManagement implementiert sind,sollen immer auch eine Rückfüh-rung in das Risikomanagement ha-ben. D.h. Bewertungen in Risikoana-lysen, neue unbekannte Risiken undgenerell Änderungen müssen im Ri-
Reinraum
TechnoPharm 4, Nr. 4, 198–206 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)202 Erens – Reinraum-Qualifizierungsmessungen im Sterilbereich – Teil 2
Tabelle 1
Beispielmatrix einer FMEA zur Herleitung von risikoreduzierendenMaßnahmen und Intervallen.
Fehler Fehlerursache Fehlerfolge Maßnahme Intervall
Luftmenge nichtausreichend
Luftmenge zu geringeingestellt.
Luftwechselratenwerden nicht erreicht. Luftmengenmessung 1 x jährlich
Zuluftfilter verstopft.Luftwechsel zugering. StarkerGegendruck.
Differenzdruck-messung am Filter 1 x jährlich
PartikuläreVerunreinigung desReinraumes
Partikelbelastung derZuluft zu hoch.
Reinraumstatus wirdnicht erreicht.
Einsatz höherklassi-ger Filterstufe.Verbesserung derVorfiltration.
–
Partikelemmissiondes Prozesseszu hoch.
Zu hohe rauminternePartikellasten„in operation“.
Erhöhung der LW-Zahloder dezentraleEliminierung derPartikelemmission.
–
Leck im endständigenFilter oder Dichtsitznicht gegeben.
A-Bereiche: Kontami-nation Produkt. Lecktest endständiger
Filter inkl. Dichtsitz-prüfung.
A-Bereich:1 x halbjährlich
turbulente Bereiche(B-D) erhöhte Par-tikelzahlen
B- und C- Bereich:1 x jährlich,D-Bereich alle 2 Jahre
Differenzdruck desRaumes zu hoch/zuniedrig.
FalscheParametrierung. Fehlerhaftes
Differenzdruckgefälle.
Differenzdruck-Mes-sung.Nachweis der Diffe-renzdruck-Kaskadeüber Monitoring
1 x jährlichLuftklappe verstellt.
Luftströmung (TAV)zu gering.
FalscheParametrierung.
Produkt- oderMitarbeiterschutznicht mehr gegeben.
Bestimmung der Luft-geschwindigkeit.
A-Bereiche:1 x halbjährlich,alle anderen: nachRisikoeinstufung max.Intervall 2 Jahre
Nachlassende Luft-leistung der Anlage.
Luftströmung (TAV)zu gering/zu hoch.
FalscheParametrierung.
Strömungs-visualisierung
Nach Risikoeinstu-fung max. Intervall2 Jahre
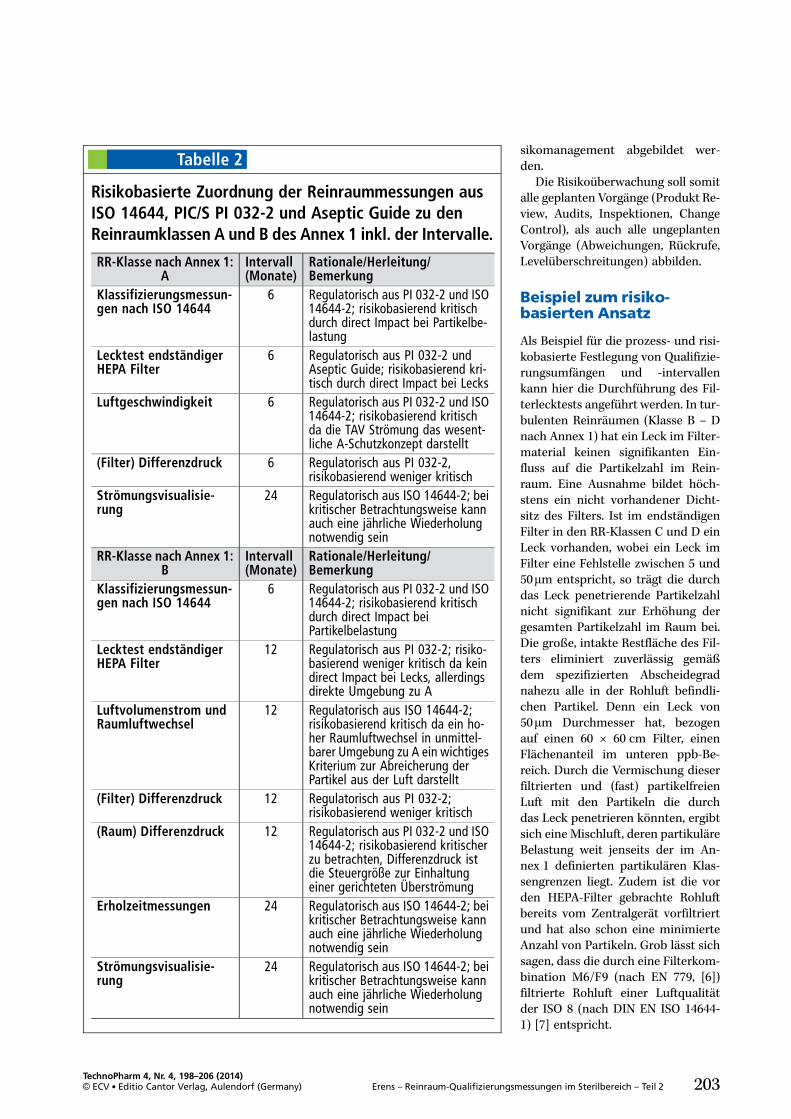
sikomanagement abgebildet wer-den.
Die Risikoüberwachung soll somitalle geplanten Vorgänge (Produkt Re-view, Audits, Inspektionen, ChangeControl), als auch alle ungeplantenVorgänge (Abweichungen, Rückrufe,Levelüberschreitungen) abbilden.
Beispiel zum risiko-basierten Ansatz
Als Beispiel für die prozess- und risi-kobasierte Festlegung von Qualifizie-rungsumfängen und -intervallenkann hier die Durchführung des Fil-terlecktests angeführt werden. In tur-bulenten Reinräumen (Klasse B – Dnach Annex 1) hat ein Leck im Filter-material keinen signifikanten Ein-fluss auf die Partikelzahl im Rein-raum. Eine Ausnahme bildet höch-stens ein nicht vorhandener Dicht-sitz des Filters. Ist im endständigenFilter in den RR-Klassen C und D einLeck vorhanden, wobei ein Leck imFilter eine Fehlstelle zwischen 5 und50μm entspricht, so trägt die durchdas Leck penetrierende Partikelzahlnicht signifikant zur Erhöhung dergesamten Partikelzahl im Raum bei.Die große, intakte Restfläche des Fil-ters eliminiert zuverlässig gemäßdem spezifizierten Abscheidegradnahezu alle in der Rohluft befindli-chen Partikel. Denn ein Leck von50μm Durchmesser hat, bezogenauf einen 60 × 60 cm Filter, einenFlächenanteil im unteren ppb-Be-reich. Durch die Vermischung dieserfiltrierten und (fast) partikelfreienLuft mit den Partikeln die durchdas Leck penetrieren könnten, ergibtsich eine Mischluft, deren partikuläreBelastung weit jenseits der im An-nex 1 definierten partikulären Klas-sengrenzen liegt. Zudem ist die vorden HEPA-Filter gebrachte Rohluftbereits vom Zentralgerät vorfiltriertund hat also schon eine minimierteAnzahl von Partikeln. Grob lässt sichsagen, dass die durch eine Filterkom-bination M6/F9 (nach EN 779, [6])filtrierte Rohluft einer Luftqualitätder ISO 8 (nach DIN EN ISO 14644-1) [7] entspricht.
TechnoPharm 4, Nr. 4, 198–206 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 203Erens – Reinraum-Qualifizierungsmessungen im Sterilbereich – Teil 2
Tabelle 2
Risikobasierte Zuordnung der Reinraummessungen ausISO 14644, PIC/S PI 032-2 und Aseptic Guide zu denReinraumklassen A und B des Annex 1 inkl. der Intervalle.
RR-Klasse nach Annex 1:A
Intervall(Monate)
Rationale/Herleitung/Bemerkung
Klassifizierungsmessun-gen nach ISO 14644
6 Regulatorisch aus PI 032-2 und ISO14644-2; risikobasierend kritischdurch direct Impact bei Partikelbe-lastung
Lecktest endständigerHEPA Filter
6 Regulatorisch aus PI 032-2 undAseptic Guide; risikobasierend kri-tisch durch direct Impact bei Lecks
Luftgeschwindigkeit 6 Regulatorisch aus PI 032-2 und ISO14644-2; risikobasierend kritischda die TAV Strömung das wesent-liche A-Schutzkonzept darstellt
(Filter) Differenzdruck 6 Regulatorisch aus PI 032-2,risikobasierend weniger kritisch
Strömungsvisualisie-rung
24 Regulatorisch aus ISO 14644-2; beikritischer Betrachtungsweise kannauch eine jährliche Wiederholungnotwendig sein
RR-Klasse nach Annex 1:B
Intervall(Monate)
Rationale/Herleitung/Bemerkung
Klassifizierungsmessun-gen nach ISO 14644
6 Regulatorisch aus PI 032-2 und ISO14644-2; risikobasierend kritischdurch direct Impact beiPartikelbelastung
Lecktest endständigerHEPA Filter
12 Regulatorisch aus PI 032-2; risiko-basierend weniger kritisch da keindirect Impact bei Lecks, allerdingsdirekte Umgebung zu A
Luftvolumenstrom undRaumluftwechsel
12 Regulatorisch aus ISO 14644-2;risikobasierend kritisch da ein ho-her Raumluftwechsel in unmittel-barer Umgebung zu A ein wichtigesKriterium zur Abreicherung derPartikel aus der Luft darstellt
(Filter) Differenzdruck 12 Regulatorisch aus PI 032-2;risikobasierend weniger kritisch
(Raum) Differenzdruck 12 Regulatorisch aus PI 032-2 und ISO14644-2; risikobasierend kritischerzu betrachten, Differenzdruck istdie Steuergröße zur Einhaltungeiner gerichteten Überströmung
Erholzeitmessungen 24 Regulatorisch aus ISO 14644-2; beikritischer Betrachtungsweise kannauch eine jährliche Wiederholungnotwendig sein
Strömungsvisualisie-rung
24 Regulatorisch aus ISO 14644-2; beikritischer Betrachtungsweise kannauch eine jährliche Wiederholungnotwendig sein
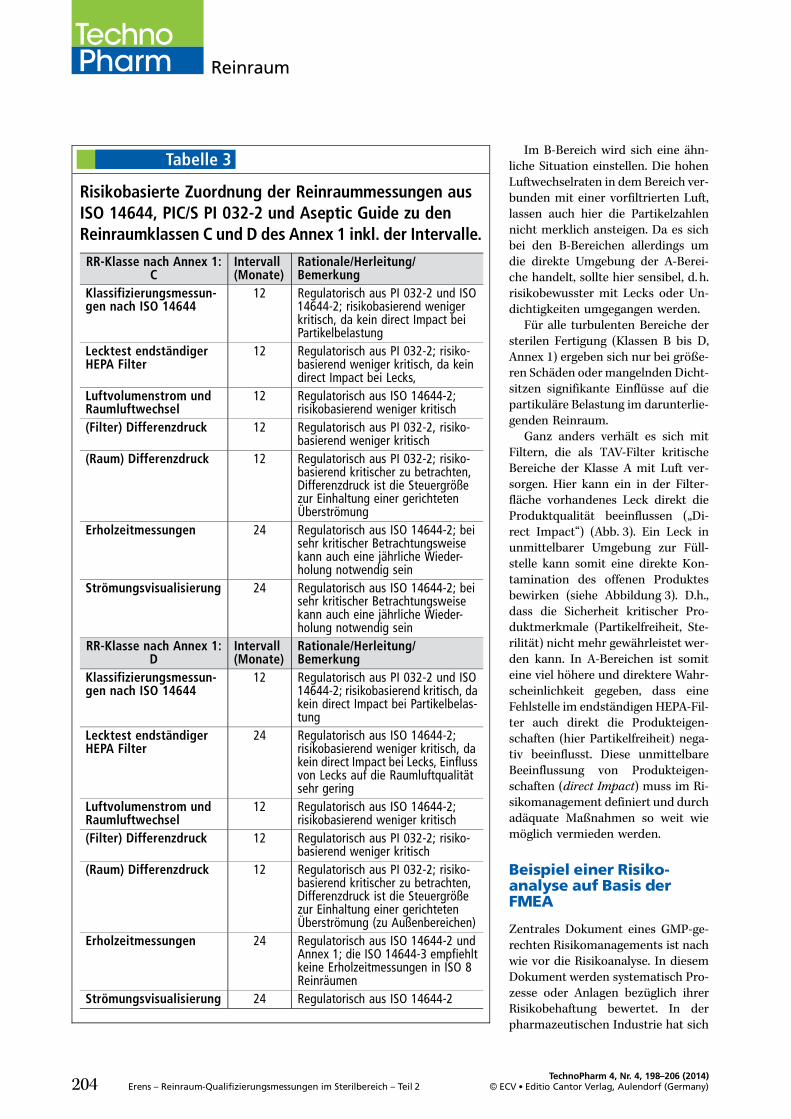
Im B-Bereich wird sich eine ähn-liche Situation einstellen. Die hohenLuftwechselraten in dem Bereich ver-bunden mit einer vorfiltrierten Luft,lassen auch hier die Partikelzahlennicht merklich ansteigen. Da es sichbei den B-Bereichen allerdings umdie direkte Umgebung der A-Berei-che handelt, sollte hier sensibel, d. h.risikobewusster mit Lecks oder Un-dichtigkeiten umgegangen werden.
Für alle turbulenten Bereiche dersterilen Fertigung (Klassen B bis D,Annex 1) ergeben sich nur bei größe-ren Schäden oder mangelnden Dicht-sitzen signifikante Einflüsse auf diepartikuläre Belastung im darunterlie-genden Reinraum.
Ganz anders verhält es sich mitFiltern, die als TAV-Filter kritischeBereiche der Klasse A mit Luft ver-sorgen. Hier kann ein in der Filter-fläche vorhandenes Leck direkt dieProduktqualität beeinflussen („Di-rect Impact“) (Abb. 3). Ein Leck inunmittelbarer Umgebung zur Füll-stelle kann somit eine direkte Kon-tamination des offenen Produktesbewirken (siehe Abbildung 3). D.h.,dass die Sicherheit kritischer Pro-duktmerkmale (Partikelfreiheit, Ste-rilität) nicht mehr gewährleistet wer-den kann. In A-Bereichen ist somiteine viel höhere und direktere Wahr-scheinlichkeit gegeben, dass eineFehlstelle im endständigen HEPA-Fil-ter auch direkt die Produkteigen-schaften (hier Partikelfreiheit) nega-tiv beeinflusst. Diese unmittelbareBeeinflussung von Produkteigen-schaften (direct Impact) muss im Ri-sikomanagement definiert und durchadäquate Maßnahmen so weit wiemöglich vermieden werden.
Beispiel einer Risiko-analyse auf Basis derFMEA
Zentrales Dokument eines GMP-ge-rechten Risikomanagements ist nachwie vor die Risikoanalyse. In diesemDokument werden systematisch Pro-zesse oder Anlagen bezüglich ihrerRisikobehaftung bewertet. In derpharmazeutischen Industrie hat sich
Reinraum
TechnoPharm 4, Nr. 4, 198–206 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)204 Erens – Reinraum-Qualifizierungsmessungen im Sterilbereich – Teil 2
Tabelle 3
Risikobasierte Zuordnung der Reinraummessungen ausISO 14644, PIC/S PI 032-2 und Aseptic Guide zu denReinraumklassen C und D des Annex 1 inkl. der Intervalle.
RR-Klasse nach Annex 1:C
Intervall(Monate)
Rationale/Herleitung/Bemerkung
Klassifizierungsmessun-gen nach ISO 14644
12 Regulatorisch aus PI 032-2 und ISO14644-2; risikobasierend wenigerkritisch, da kein direct Impact beiPartikelbelastung
Lecktest endständigerHEPA Filter
12 Regulatorisch aus PI 032-2; risiko-basierend weniger kritisch, da keindirect Impact bei Lecks,
Luftvolumenstrom undRaumluftwechsel
12 Regulatorisch aus ISO 14644-2;risikobasierend weniger kritisch
(Filter) Differenzdruck 12 Regulatorisch aus PI 032-2, risiko-basierend weniger kritisch
(Raum) Differenzdruck 12 Regulatorisch aus PI 032-2; risiko-basierend kritischer zu betrachten,Differenzdruck ist die Steuergrößezur Einhaltung einer gerichtetenÜberströmung
Erholzeitmessungen 24 Regulatorisch aus ISO 14644-2; beisehr kritischer Betrachtungsweisekann auch eine jährliche Wieder-holung notwendig sein
Strömungsvisualisierung 24 Regulatorisch aus ISO 14644-2; beisehr kritischer Betrachtungsweisekann auch eine jährliche Wieder-holung notwendig sein
RR-Klasse nach Annex 1:D
Intervall(Monate)
Rationale/Herleitung/Bemerkung
Klassifizierungsmessun-gen nach ISO 14644
12 Regulatorisch aus PI 032-2 und ISO14644-2; risikobasierend kritisch, dakein direct Impact bei Partikelbelas-tung
Lecktest endständigerHEPA Filter
24 Regulatorisch aus ISO 14644-2;risikobasierend weniger kritisch, dakein direct Impact bei Lecks, Einflussvon Lecks auf die Raumluftqualitätsehr gering
Luftvolumenstrom undRaumluftwechsel
12 Regulatorisch aus ISO 14644-2;risikobasierend weniger kritisch
(Filter) Differenzdruck 12 Regulatorisch aus PI 032-2; risiko-basierend weniger kritisch
(Raum) Differenzdruck 12 Regulatorisch aus PI 032-2; risiko-basierend kritischer zu betrachten,Differenzdruck ist die Steuergrößezur Einhaltung einer gerichtetenÜberströmung (zu Außenbereichen)
Erholzeitmessungen 24 Regulatorisch aus ISO 14644-2 undAnnex 1; die ISO 14644-3 empfiehltkeine Erholzeitmessungen in ISO 8Reinräumen
Strömungsvisualisierung 24 Regulatorisch aus ISO 14644-2

ecv
ECV · Editio Cantor Verlag
Bestellung:Tel. +49 (0)8191-97000 358, Fax +49 (0)8191-97000 293eMail: [email protected], Leseproben und Inhaltsverzeichnisse – www.ecv.de
Der Schutz geistigen Eigentums hat für forschende Unternehmen in-zwischen zentrale Bedeutung. Der Pharma-Markt teilt sich in zweiBereiche: den generischen und den der Originatoren. Der generischeMarkt ist hart umkämpft. Der Wettbewerb in diesem Markt setzt ein,sobald die Schutzrechte des Originators erloschen sind. Daher ist dieFrage des Schutzes geistigen Eigentums von außerordentlich großerBedeutung.
Die Autoren sind ausgewiesene Experten auf dem Gebiet des Schut-zes geistigen Eigentums in der Zulassung von Arzneimitteln und dessen Bedeutung für die Preisbildung nach dem Arzneimittelneu-ordnungsgesetz – AMNOG. Sie behandeln Patente und ergänzendeSchutzzertifikate in Abgrenzung zur Marktexklusivität für OrphanArzneimittel und den Unterlagenschutz im Rahmen von Zulassungs-verfahren.
Das Buch wendet sich an Mitarbeiter in pharmazeutischen Unter-nehmen, insbesondere an Regulatory Affairs Manager sowie Mitar-beiter in den Patent- und Rechtsabteilungen sowie in Market Access,die für die frühe Nutzenbewertung von Arzneimitteln zuständig sind.Bedeutung haben diese Rahmenbedingungen auch für die Konzepteder Forschung in den R&D-Abteilungen.
Von Interesse ist das Buch auch für Mitarbeiter in Behörden der Mit-gliedstaaten der Europäischen Union. Die Frage der Nutzenbewer-tung ist sicher eine nationale Orientierung, sie hat jedoch aufgrundder Bedeutung der deutschen Preise als Reference Price indirekt auchAuswirkungen auf andere Mitgliedstaaten der Europäischen Union,jedenfalls für die Preisbildung in diesen Staaten.
Schutz geistigen Eigentums an Arzneimitteln• in der Zulassung für Generika nach AMG• bei pädiatrischen und Orphan-Arzneimitteln• in der frühen Nutzenbewertung und Preisbildung nach SGB V• für Strategien in der Produktentwicklung und für
den Market Access in Europa
Burkhard Sträter, Claus Burgardt, Marion Bickmann
ISBN 978-3-87193-426-1• € 239,00• 1. Auflage 2014• 17 x 24 cm, 151 Seiten, gebunden
Neu!
EW 1-2 Seite hoch Buch Schutz geistigen Eigentums 07_14_Layout 1 07.07.14
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2014-04\Anzeigensatz-keine-Druck-PDFs\testo-saveris-TP-2014-04_105x303.indd Zuletzt gesichert: 21.07.14 (10:33:45 Uhr)
Ein kleines Stück
Verantwortung können
wir Ihnen abnehmen.
testo Saveris. Datenmonitoring-System mit umfassendem Alarm-Management.• Automatisierte und lückenlose
Messdaten-Erfassung • Validierfähige 21 CFR Part 11 Software • Hohe Sicherheit durch redundante Datenarchivierung
www.testo.de/saveris
Saveris_105x303_Technopharm.indd 1 17.07.2014 11:10:13

weitestgehend die FMEA (FailureMode Effects Analysis) als Risikoana-lysemethode etabliert.
Diese bietet den großen Vorteil,dass auch komplexe Anlagen oderProzesse in kleine Einheiten undTeilprozesse gegliedert werden kön-nen, die dann strukturiert und syste-matisch auf Risiken untersucht wer-den. Ein weiterer Vorteil ist, dass dieFMEA ideal die Risikoanalysetech-niken der Fault Tree Analysis (FTA)und der Event Tree Analysis (ETA)verbindet. D.h. in der Praxis, dassein und der selbe Fehler verschie-dene Ursachen und/oder verschie-dene Auswirkungen haben kann. Allediese verschiedenen Möglichkeitenkönnen mit der FMEA strukturiertbetrachtet werden, um daraufhindie Risikoeinstufung vorzunehmenund Maßnahmen abzuleiten.
Die Maßnahmen können, wie zu-vor beschrieben, entweder die Auf-tretenswahrscheinlichkeit verrin-gern, oder die Entdeckungswahr-scheinlichkeit erhöhen. Bei Maß-nahmen zur Erhöhung der Ent-deckungswahrscheinlichkeit ist alswichtiger Faktor noch das Intervalloder die Wiederholfrequenz zu be-achten. So können systemimma-nente Risiken nur durch per-manente Monitoringmaßnahmenüberwacht werden. Je weniger dasRisiko direkte Eigenschaften desProduktes oder des Prozesses beein-flusst, umso weniger häufig und res-triktiv sind Maßnahmen zu definie-ren.
Beispielhaft ist in Tabelle 1 eineFMEA Tabelle (auszugsweise) dar-gestellt.
Ableitung der Qualifizie-rungsumfänge fürReinräume oder reinluft-technische Anlagenkombiniert aus regulato-rischem und risikoba-siertem Ansatz
Fasst man die Erkenntnisse aus denregulatorischen Kapiteln mit denendes risikobasierenden Ansatzes zu-sammen ergeben sich für die Berei-
che A-D nach Annex 1 die in denTabellen 2 und 3 dargestellten rein-raumspezifischen Qualifizierungs-messungen.
Natürlich sind diese als „abgelei-tete Empfehlung“ zu sehen und kön-nen in verschiedenen Produktions-umgebungen sicher divergieren.Letztendlich ist auch die risikobasie-rende Einstufung individuell von Er-fahrungen und Einschätzungen un-ter der am Risikomanagement betei-ligten Personen abhängig.
Fazit undSchlussbemerkungen
Die Anforderungen an qualifizierteUmgebungsbedingungen in derpharmazeutischen und (GMP)Com-pliance regulierten Industrie sindbreit gefächert und haben den best-möglichen Schutz des Produktesoder des Prozesses im Fokus. Eineeinheitliche Herleitung oder ein regu-latives Dokument, welches alle An-forderungen definiert ist derzeitnicht vorhanden.
Die regulatorischen Rahmenbe-dingungen werden gerade für die ste-rile Herstellung von Arzneimittelndurch die Guidelines/Normen. ANNEX 1 (EU GMP Leitfaden) [1]. ASEPTIC GUIDE [4]. EN ISO 14644-2 [3]. PIC Dokument, PI 032-2 [2]gegeben. Aber auch diese Doku-mente liefern oft nur für Teilbereicheder RR-Qualifizierung eine konkreteAussage. Eine detaillierte Auflistung,gestaffelt nach prozessualem Risikoder Beeinflussung findet sich in kei-nem der Dokumente.
Der ideale Umfang der Reinraum-qualifizierung, mit risikominimieren-den aber noch bezahlbaren Maßnah-men kann über einen „Risk Based Ap-proach“ abgeleitet werden. Hier bie-tet ein Risikomanagement nach EUGMP Leitfaden Teil III (Quality RiskManagement, (ICH Q9)) [5] das rich-tige Tool, um die Umgebungsbedin-gungen anhand der Prozessanforde-rung zu bestimmen und die Maßnah-men zur Aufrechterhaltung des defi-nierten Zustandes zu benennen.
Grundsätzlich gelangt man zu denvier Einflussgrößen. Prozess. Produkt. Person/Umwelt. Regulatorischer HintergrundDie negative Beeinflussung dieservier Einflussgrößen durch die Umge-bungsbedingungen muss in einemgesteuerten Risikoanalyseprozesssystematisch untersucht und ent-sprechend ihrer Kritikalität Maßnah-men definiert werden. Hier bietet dieRisikoanalysemethode der FMEA dienotwendige Struktur zur risiko-gestützten Definition und Steuerungvon Maßnahmen zur Minimierungder Einflüsse aus der Umgebung.
Über einen risikobasierten LifeCycle Approach können die aus denUmgebungsbedingungen resultieren-den Risiken sicher über den gesam-ten Lebenszyklus des Produktes oderdes Prozesses beherrscht werden.
Fachliteratur[1] EudraLex The Rules Governing Medicinal
Products in the European Union, Volume4, EU Guidelines to Good ManufacturingPractise, Medicinal Product for Humanand Veterinary Use: Annex 1: Manufactureof Sterile Medicinal Products,25. November 2008
[2] PI 032-2, GMP Annex 1 Revision 2008,Interpretation of the most importantchanges for the manufacture of medicinalproducts Pharmaceutical Inspection Co-Operation Scheme, Januar 2010,
[3] DIN EN ISO 14644-2, Reinräume und zu-gehörige Reinraumbereiche, Teil 2: Fest-legungen zur Prüfung und Überwachungzum Nachweis der fortlaufenden Übe-reinstimmung mit ISO 14644-1 (ISO14644-2:2000); Deutsche Fassung EN ISO14644-2:2000
[4] Guidance for Industry, Sterile Drug Pro-ducts Produced by Aseptic Processing –Current Good Manufacturing Practice(Aseptic Guide), U.S. Department of He-alth and Human Services, Food and DrugAdministration, September 2004, Phar-maceutical CGMPs,
[5] European medicinal Agency Quality RiskManagement (ICH Q9), Teil III, EU-GMPLeitfaden, Januar 2011,
[6] DIN EN 779 Partikel-Luftfilter für die all-gemeine Raumlufttechnik –Bestimmungder Filterleistung Mai 2009
[7] DIN EN ISO 14644-1, Reinräume und zu-gehörige Reinraumbereiche Teil 1: Klas-sifizierung der Luftreinheit(ISO 14644-1:1999); Deutsche FassungEN ISO 14644-1:1999
Reinraum
TechnoPharm 4, Nr. 4, 198–206 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)206 Erens – Reinraum-Qualifizierungsmessungen im Sterilbereich – Teil 2

Media FillsRita Welser . Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Deutschland
Korrespondenz: Rita Welser, Boehringer Ingelheim Pharma GmbH & Co. KG, Birkendorferstraße 65, 88397 Biberach an der Riß,Deutschland; e-mail: [email protected]
ZusammenfassungEin Hauptbestandteil der Validierung in der Sterilfertigung ist die Durchführung vonMedia Fills. Diese dienen dazu den Nachweis zu erbringen, dass sterile Arzneimittel sterilhergestellt werden können. In jeder Behördeninspektion oder Kundenaudit wird hieraufein besonderes Augenmerk gelegt.
Einleitung
Durchführung von Media Fills sindein muss in der Herstellung sterilerArzneiformen. Unter Media Fills ver-steht man eine Abfüllung von Nähr-medium (üblicherweise eine Lösungaus Casein Sojamehl Pepton) bzw.Tryphic Soy Broth (TSB) unter Pro-duktionsbedingungen mit Simula-tion des „worst case“. Media Fills sindein Hauptbestandteil der Validierungin der Sterilfertigung und werden fürneue Anlagen als Erstvalidierung mitdrei Abfüllungen und für die Revali-dierung alle sechs Monate mit einerAbfüllung durchgeführt. Für MediaFills sind die strengsten und am bes-ten festgelegtesten Vorgaben in derLiteratur [1] zu finden. Eine sterileAbfüllung kann z.B.: auf einer Abfüll-anlage im konventionellen Serilraum(A/B Bereich), einer RABS (Res-tricted Access Barrier System) Abfüll-anlage oder auf einer Abfüllanlage ineinem Isolator durchgeführt werden.Im konventionellen Sterilraum istdas Personal direkt im Raum undgreift bei z.B.: Störungen direkt inden Prozess ein. Bei einer RABS Ab-füllanlage und Isolator werden not-wendige Eingriffe über spezielleHandschuhe durchgeführt.
Im Folgenden wird hauptsächlichauf die Abfüllung von Media Fills aufeiner Abfüllanlage in einem konven-tionellen Sterilraum eingegangen, dadies durch den direkten Eingriff von
Personal am kritischsten zu sehenist. Das Vorgehen kann jederzeit aufeine RABS oder Isolator Anlage üb-ertragen werden.
„Worst CaseBedingungen“
Media Fills sollen inkl. aller ungünst-igen Bedingungen unter „worst case“Bedingungen durchgeführt werden.
Zu den „worst case“ Bedingungenzählen z.B.:. Teilnahme maximale Anzahl Per-sonen im Sterilraum, die für dieRoutine Produktion definiert sind.Jede Person muss eine ihrer ent-sprechenden Position zugewiese-nen Tätigkeit durchführen. Tech-nisches Personal (z.B.: Einricht-tätigkeit), Bediener (z.B.: Wechseleiner Abfüllnadel)
. Reduzierung der Abfüllgeschwin-digkeit in Bereichen mit hohemAutomatisierungsgrad. Somit er-höht sich die Expositionszeit dernoch nicht verschlossenen abge-füllten Einheiten.
. Bei Abfüllanlagen mit niedrigemAutomatisierungsgrad und da-durch mehr manuellem handlingsollte die Abfüllgeschwindigkeitmind. die der Produktions-geschwindigkeit entsprechen.
. Produktionsunterbrechungen(Pausen), sollen durchgeführtwerden, um mögliche Störungenund dadurch Stillstände an der
Anlage zu simulieren. Währenddieser Zeit sollen sämtliche Ein-heiten (leer wie auch abgefüllt) aufder Abfüllanlage verbleiben.
. Längste Prozesszeiten inkl. Unter-brechungen sollen im Media Fillabgedeckt werden, um die Prozess-zeiten aller Produkte, die auf einerLinie abgefüllt werden abzudecken.
. Wechsel Personal (Schichtwechsel)soll durchführt werden, um mög-lichst viel Personal abzudeckenund auch einen Schichtwechsel zusimulieren.
. Durchführung von Routine – In-terventionen (kommen in jederAbfüllung vor – z.B.: Setup Tätig-keiten) sowie auch Nicht Routine –Interventionen (kommen spora-disch vor – z.B.: Ausbau Abfüll-nadel).
Einstufung undBewertung von Eingriffen
Während einer Abfüllung sind Ein-griffe unumgänglich. Sämtliche mög-liche Eingriffe, die im Rahmen einerRoutine Produktion auftreten oderauch potentiell auftreten könnenmüssen im Media Fill durchgeführtoder Simuliert werden. Die Eingriffemüssen aufgenommen und beschrie-ben werden damit die Kritikalität be-wertet werden kann. Eingriffe, diedazu führen, dass Einheiten verwor-fen werden müssen, sollen simuliertwerden.
Prozess- und Verfahrenstechnik
TechnoPharm 4, Nr. 4, 207–209 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 207Welser . Media Fills

Aufnahme der Eingriffe (Interven-tionen) können über folgende Punkteerfolgen z.B.:. Bezeichnung des Eingriffes – Je-dem Eingriff wird eine Bezeich-nung zugeordnet
. Beschreibung des Eingriffes – DerEingriff wird im Detail beschriebenwie er durchzuführen bzw. zu si-mulieren ist.
. Häufigkeit des Eingriffes – Be-schreibung wie oft ein Eingriff inder Routine Produktion auftritt.
. Eingriff mit Hand, Pinzette odersonstigem Hilfsmittel – Beschrei-bung mit welchem Hilfsmittel derjeweilige Eingriff durchzuführenist.
. Eingriff über offene Einheiten –Hier wird beschrieben, ob der Ein-griff über offene Einheiten erfolgt.
. Eingriff über oder in der Nähe vonproduktberührendem Equipment– Hier wird beschrieben, ob derEingriff in der Nähe von produkt-berührendem Equipment erfolgt.
Beispiele zur Bewertungder Eingriffe
. Routine Eingriff – kritisch / nichtkritisch?
. kein Routine Eingriff – kritisch /nicht kritisch?
. Bewertung der Einstufung mitEinplanung im Media Fill
. Monitoring (Händeabklatsch not-wendig – ja / nein? (mit Begrün-dung bei nein)
Die Bewertung der Eingriffe erfolgtanhand der Kritikalität ob z.B.: einEingriff mit Hand, über offene Ein-heiten oder sehr häufig in der Rou-tine Produktion vorkommt. Je öfterein Eingriff oder der Eingriff über of-fenen Einheiten oder mit der Handdurchgeführt werden muss, destokritischer ist dieser zu bewerten. Je-der Eingriff sollte zu einem Monito-ring der Hände führen. Ausnahmensollen begründet werden.
QA Oversight
Während den Media Fills wird vonden Behörden ein QA Oversight er-
wartet. Unter QA Oversight verstehtman, dass erfahrenes Personal derQuality Assurance (QA) also der Qua-litätssicherung vor Ort im Betrieb an-wesend ist. Das QA Oversight soll si-cherstellen, dass alle vorgegebenenTätigkeiten korrekt durchgeführtwerden sowie alle abgefüllten Einhei-ten bebrütet (inkubiert) werden. EinQA Oversight kann wie folgt durch-geführt werden:
Durchführung „QA Oversight“ an-hand folgenden Punkten:. Schulung der Herstellvorschriften(HV) und Media Fill Protocol –Hiermit soll sichergestellt werden,dass sämtliches Personal, das imMedia Fill involviert ist in der HVsowie im Media Fill Protocol ge-schult ist und die Vorgaben kennt.
. Während der gesamten Abfüllungsollen sämtliche Behältnisse, diebei folgenden Schritten aussortiertwurden überprüft werden, ob diesetatsächlich nicht bebrütet werdenkönnen.– während der Abfüllung– während dem Transport undBeladen sowie Entladen des Ge-friertrockners (sofern es sich umeine Abfüllung mit Gefriertrock-nung handelt)
– während dem Verschließen derEinheiten z.B.: dem Bördeln
– während der Palettierung (Vor-bereitung zum Transport zurEinlagerung)
– Bebrütung (Inkubieren) in Wär-mezelle (20-25 °C und 30-35 °C)
– während der visuellen Prüfung(nach Bebrütung 20-25 °C und30-35 °C)
Während den oben genannten Min-destschritten soll eine Person derQuality Assurance (QA) anwesendsein und soll die Schritte dokumen-tieren. Durch den QA Oversight sollsichergestellt werden, dass keine Ein-heiten aussortiert werden, die integersind und dass alle Vorgaben gemäßHV und Protocol eingehalten werden.
Bilanzierung
Es muss eine Bilanzierung der abge-füllten Behältnisse durchgeführt wer-
den. Dabei muss klar ersichtlich sein,wie viele Behältnisse insgesamt abge-füllt und wie viele davon insgesamtbebrütet worden sind. Zur Bilanzie-rung werden alle abgefüllten Behält-nisse herangezogen. Falls Behältnissenicht bebrütet wurden, muss derenVerbleib dokumentiert werden. Be-hältnisse, bei denen die Integritätder Einheit nicht gewährleistet wer-den können z.B.: Behältnis ohneGummistopfen oder zerbrochenesBehältnis werden nicht bebrütet.Jede Einheit, die verworfen wurdemuss begründet werden!
Vorgehen bei kontami-nierten Einheiten
Wenn kontaminierte Einheiten vor-kommen muss sofort das Abwei-chungsprozedere eingesetzt werden.Im Rahmen des Abweichungspro-zedere wird die Abweichung schrift-lich festgehalten unter Einbindungvon allen Bereichen wie Produktion,QA, Validierung und Mikrobiologie,die im Rahmen des Media Fills invol-viert sind.
Es muss eine Bewertung von bereitsgefertigter Chargen bis zum letztenerfolgreichen Media Fill, sowie Bewer-tung des Mikrobiologisches und Phy-sikalisches Monitorings und Auswer-tung von Störungen, Eingriffe erfolgen.
Die Ursache der Kontaminationmuss begründet werden und zur Ver-meidung des Fehlers müssen Vorbeu-gende Maßnahmen (Corrective Pre-ventive Actions – CAPAs) ergriffenwerden.
Je nach dem wie viele Kontami-nierte Einheiten aufgetreten sindund was als Ursache ermittelt wurde,muss der Media Fill mit einer oderdrei Abfüllungen wiederholt werden.. Ursachenforschung muss lücken-los dokumentiert und begründetwerden.
Erfahrungen aus Inspek-tionen (speziell FDA)
– In der Vorschrift zu Media Fillswurde der Begriff „worst case“nicht erwähnt.
Prozess- und Verfahrenstechnik
TechnoPharm 4, Nr. 4, 207–209 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)208 Welser . Media Fills

. Hier ist zu erwähnen, dass „worstcase“ keine deutsche Bezeichnungist.
. Es wurden leere Einheiten ver-worfen.– Hier muss man sich fragen wasan leeren Einheiten kritisch ist.
– Diskussion über ReduzierungAbfüllgeschwindigkeit.
– Dazu sollte eine entsprechendeRationale zur Abfüllgeschwin-digkeit vorliegen.
– QA Oversight wird erwartet.– QA Oversight sollte in sinnvollerweise durchgeführt werden.
– Diskussion über Video Aufnah-men während Media fill.
– Dazu muss zumindest inDeutschland auf Grund per-sonenbezogenen Daten der Be-triebsrat eingebunden werden.
– Kritische Interventionen sollenMitarbeiter bezogen durch-geführt werden.
– Dazu sollte eine separate Pla-nung vorliegen.
Fazit
Media fills stehen in einer Inspektionzur aseptischen Produktion als einmuss auf der Inspektionsagenda. Siesind ein wichtiger Bestandteil in derSterilfertigung, um eine sterile Abfül-lung nachweisen zu können. Um denNachweis zu erbringen, dass alle in-tegeren Behältnisse ausgewertetwurden, ist eine umfangreiche undlückenlose Bilanzierung der abgefüll-ten Behältnisse absolut notwendig.
Fachliteratur[1] Guideline for Industry „Sterile Drug Pro-
ducts Produced by Aseptic Processing –Current Good Manufacturing Practice“(Leitlinien für die Industrie „Im asepti-schen Verfahren hergestellte sterile Arz-neimittel – Gegenwärtige gute Herstel-lungspraxis), FDA , September 2004 ECGuide for Good Manufacturing Practice,Annex 1: Manufacture of sterile medicinalproduct (EU Richtlinie zur guten Herstel-lungspraxis, Anlage 1: Herstellung sterilermedizinischer Produkte), März 2009
HOSOKAWA ALPINE AktiengesellschaftPostfach 10 11 51 86001 Augsburg Tel: + 49 821 5906-0 Fax: + 49 821 5906-620 E-Mail: [email protected] www.hosokawa-alpine.de
LUPENREIN SAUBER.GARANTIERT.
› Intensive Abreinigung des geschlossenen Prozessraums › Kontrolle des Reinigungsergebnisses durch einfache Zerlegbarkeit aller produktberührten Komponenten › Vollaseptische Ausführung von Wellendurchführungen und Clampverbindungen › Glatter und totraumfreier Gesamtaufbau, um perfekte Drainage und Sterilisierbarkeit zu gewährleisten › Entlüftung von Räumen mit geringem Dampfaustausch › Armaturen und Instrumente in steriler Ausführung
160 UPZ STERILE MAHLANLAGE UND
DOSIERUNG PDD 35 IN VOLL CIP/SIP-FÄHIGER AUSFÜHRUNG

ProzessorientiertenWiegesystemen gehört die ZukunftPraktische Hilfestellungen zur systematischen Lösungskonzeption
Konrad Steinmetz . FELTEN Group GmbH, Serrig
Korrespondenz: Konrad Steinmetz, FELTEN Group GmbH, In den Dörrwiesen 31, 54455 Serrig;e-mail: [email protected]
ZusammenfassungObwohl Wiege- und Dosiervorgänge in der Fertigung der Prozessindustrie eine wesent-liche Bedeutung haben, sind diese derzeit vielfach noch sehr manuell geprägt.Notwendig ist stattdessen eine hohe Integration und Automatisation dieser Abläufe, umein Höchstmaß an Produktivität und Qualität zu erlangen. Auch den Dokumentations-pflichten für die Rückverfolgbarkeit lässt sich nur durch automatisierte Prozesseentsprechen.In dem Beitrag wird deshalb anhand eines Modellunternehmens ein produktneutralesKonzept zur Einführung eines prozessorientierten Wiege- und Dosiermanagements unterbeispielhaft angenommenen Implementierungsbedingungen skizziert. Es schließt dietechnische Integration in die Infrastruktur und das ERP-System bis hin zur Schnittstellen-thematik mit ein. Das Konzept beschränkt sich zudem nicht nur auf das Wiege- undDosiersystem (WDS), sondern nimmt gleichzeitig eine Integration mit einem OEE-Systemvor, das der Leistungssteuerung und -verbesserung der Prozesse im Produktionsalltagdient. Seine Betrachtung reicht bis zur Frage der notwendigen Kennzahlen und Reports.Aufgrund des generellen Charakters des Modellbeispiels lässt sich eine systematischePlanung der gesamten Projektierung zur WDS-Einführung und -Weiterentwicklung vor-nehmen. Der Beitrag bietet dadurch einen hohen Nutzwert für Verantwortliche imProduktionsmanagement.
Einleitung
Es gehört zu den unbestrittenen Er-kenntnissen, dass sich durch einehohe Integration und Automationder Prozesse sowohl Produktivitätals auch Qualität deutlich steigernlassen. Trotzdem erfolgen die Wiege-und Dosiervorgänge in der Industrieheutzutage noch in hohem Maßmanuell. Notwendig sind deshalbKonzepte zur softwaregestütztenProzessorientierung, die in einerKombination mit OEE-Systemen einetransparente Leistungssteuerungermöglichen.
Lösungen für ein prozessorientier-tes Wiegen und Dosieren von Roh-stoffen müssen dialoggeführt erfol-gen und eine automatische wie lü-ckenlose Dokumentation gewährleis-ten. Zu den wichtigsten Funktionen
zählen die Möglichkeit einer auf-trags- oder rohstoffbezogenen Ver-wiegung in einem vollautomatischenProzess samt einer Tarierung oderTeilverwiegung im Falle von Behäl-ter- oder bei LOT-Nummer-Wechsel.Auch Plausibilitätsprüfungen gehö-ren zu den Basisfunktionen von Wie-ge- und Dosiersystemen (WDS), umdie Bediener zu unterstützen und dieQualität der Bearbeitung zu sichern.Zur Vereinfachung der Bedienopera-tionen und Minimierung der manu-ellen Eingaben sollten außerdem alleAktionen mittels Barcode unter-stützt werden.
Das OEE-System für OverallEquipment Effectiveness wiederumdient der Erfassung, Auswertungund Archivierung von Betriebs- undMaschinendaten. Es soll die Wirt-schaftlichkeit der Produktion ermit-
teln und es möglich machen, Res-sourcen gezielt auszuschöpfen unddie Nutzung von Maschinen bzw. An-lagen zu optimieren. Dazu gehörtauch die frühzeitige Ermittlung undBeseitigung von Produktionsstörun-gen zur Steigerung der Gesamtpro-duktivität. Durch die Integrationvon WDS- und OEE-System werdennicht nur notwendige Funktionalitä-ten für den Wiegeprozess bereit ge-stellt, sondern es werden zusätzlichdie Voraussetzungen für eine Pro-zesssteuerung geschaffen.
Fallbeispiel mitAusgangssituation undZielsetzungen
Um die produktneutrale Konzeptionund Einführung eines prozessorien-tierten Wiegemanagements mit um-fassender Integration und Automati-sierung darzustellen, wird beispiel-haft ein Produktionsbetrieb der Le-bensmittelindustrie herangezogen.Damit eine hohe Sicherheit in denbisher manuellenWiege- und Dosier-prozessen sowie Transparenz in denProduktionsprozessen erlangt wird,ist die Einführung einer WDS-Lösungin Verbindung mit einem OEE-System notwendig. Das WDS soll dia-loggeführt die Verwiegung von Roh-stoffen für Herstellprozesse inner-halb der Wertschöpfungskette ge-währleisten, ebenso ist eine tech-nische Kommunikation mit gängigenWaagen und Terminals vorgesehen.Außerdem wird eine Kommunika-tion mit der ERP-Anwendung, ausder sich die Produktionsmanage-
Prozess- und Verfahrenstechnik
TechnoPharm 4, Nr. 4, 210–213 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)210 Steinmetz . Prozessorientierten Wiegesystemen gehört die Zukunft

mentsysteme mit den Auftrags- undweiteren relevanten Informationenspeisen, als notwendig erachtet.
Aus Gründen einer sicheren Rück-verfolgung sind weiterhin alle Ab-läufe und die damit verbundenen In-formationen präzise zu dokumentie-ren. Denn gesetzliche Vorgaben undandere Regularien verlangen inzwi-schen valide Nachweise der Prozesse.Diese Anforderung kann nur über au-tomatisch generierte Dokumente si-chergestellt werden, da ein Handver-wiegen mit manueller Aufschreibungmanipulierbar ist. Die erfassten Pro-duktionsdaten sollen letztendlich ineinem Herstellprotokoll dargestelltwerden, das nicht manipulierbar ist,weil die Dokumentation automati-siert erfolgt.
In dem Beispielmodell wird hin-sichtlich der Infrastruktur angenom-men, dass das Unternehmen auf einehohe technische Integration abzieltund die Entstehung einer heteroge-nen IT-Landschaft vermeiden will.Gleichzeitig legt es Wert auf die Ein-führung eines Web-basierten Sys-tems, das auch werksübergreifendan weiteren Standorten genutzt wer-den kann. Dies führt zu verschiede-nen Anforderungen an die tech-nische Landschaft mit einemSchnittstellenkonzept.
Da die Einführung eines Wiegesys-tems im Regelfall auf bestimmte tech-nische Ausgangsbedingungen trifft,wird hier eine bestimmte und in derPraxis häufig anzutreffende Infrastruk-tur angenommen. Zu ihren Merkma-len gehört, dass einzelne Maschineneiner Linie nicht miteinander gekop-pelt sind sowie über keine Steuerung(SPS) und TCP/IP Kommunikationverfügen. Auch ein übergeordnetesSCADA-System zur Steuerung der ein-zelnen Linien und eine ERP-Schnitt-stelle existieren nicht.
Anforderungen an denWiegeprozess
Der Wiegeprozess unterteilt sich ty-pischerweise in verschiedene syste-matische Schritte. Vorausgesetztwird dabei, dass sich das System
aus dem Auftrag die wiegepflichtigenPositionen selektieren und mögli-cherweise auch die Nutzung einerbestimmten Waage vorschreibenkann.
1. Wiegeaufträge:Das Wiegesystem soll unmittelbardie vom ERP-System übertragenenProduktionsaufträge in Wiegeauf-träge umsetzen und dabei auchgleichzeitig die Rohstoff- und Char-gendaten mit übernehmen. Es erfolgteine Status-Kennzeichnung der Auf-träge, die automatisch aktualisiertoder im Leitstand manuell bearbeitetwerden kann. Die Status-Informatio-nen beschreiben den jeweiligen Bear-beitungsstand und geben Auskunftdarüber, ob ein Auftrag beispiels-weise in Planung, zur Produktion be-reit, übertolerant, teilverwogen oderbeendet ist. Die Aufträge lassen sichaußerdem einem bestimmtenWiege-terminal zuordnen. Mit der Anwahleines Wiegeauftrags schlägt das Sys-tem auf Basis der zu verwiegendenSollmenge eine adäquate Waage mitentsprechendem Wiegebereich vor.Aus Flexibilitätsgründen soll das Sys-tem zwischen auftragsbezogener undrohstoffbezogener Verwiegung un-terscheiden können. Ebenso ist eswichtig, einzelne Produktionsauf-träge in weiteren Auftragslisten zu-sammenzustellen, um den Anforde-rungen von Wiegekampangen ge-recht zu werden.
2. Komponentenprüfung:In den Wiegeaufträgen wird definiert,welche Rohstoff-Chargen für die Ver-wiegungen verwendet werden dürfen.Vor Beginn jeder Verwiegung unter-stützen Plausibilitätsprüfungen beider Auswahl der für diesen Vorgangzugewiesenen Rohstoff-Chargen undBehälter. Hierzu gehört das Scannender entsprechend gekennzeichnetenGebinde. Außerdem werden die Ein-gaben überwacht und bewirken beiFalscheingaben eine automatischeSperrung der weiteren Bearbeitungdurch das System. In der Komponen-tenprüfung werden darüber hinaus zuden produktions- und ablaufrelevan-
ten Daten Gefahrensymbole und Si-cherheitshinweise angezeigt.
3. Wiegevorgang:Im Anschluss an die erfolgreich abge-schlossene Komponentenprüfungwird der eigentliche Wiegedialog(Wiegevorgang) automatisch frei ge-schaltet. Dieser Vorgang charakteri-siert sich durch folgende systemati-sche Schritte: Anhand der zu verwie-genden Komponente und des zuge-hörigen Sollwertes schlägt das Sys-tem die geeignete oder durch die Re-zeptur zwingend vorgeschriebeneWaage vor und tariert entweder au-tomatisch oder manuell. Anschlie-ßend erfolgt eine Korrekturberech-nung auf Basis der Wirkstoffkonzen-tration eines Rohstoffes. Im anschlie-ßenden Wiegedialog werden nebenden für den aktuellen Vorgang benö-tigten Auftrags- und Produktions-daten derWaagen der Istwert (nume-risch und grafisch) sowie die Tole-ranzgrenzen angezeigt. Vorteilhafter-weise sollte eine farbliche Darstel-lung der Istwerte erfolgen. Währenddes folgenden Wiegevorganges wirdeine automatische Überwachung derToleranzgrenzen vorgenommen, einÜberschreiten der oberen Toleranz-grenze muss zum Sperren des Wie-geauftrags führen. Nach Abschlusseines Wiegevorgangs werden die Da-ten zu den real verwogenen Mengenan das ERP-System übertragen, da-mit dort die Bestandsbuchung vor-genommen werden kann.
4. Wiegemethoden:Im Beispielfall soll das WDS drei un-terschiedliche Wiegemethoden ab-bilden können, die je nach Auftrags-bzw. Rezepturvorgaben oder durchtechnische Gegebenheiten zum Ein-satz kommen. Dazu gehört zunächstdie Einwaage, bei der die Waage-oder Mischbehälter auf der Waagestehen und bis zu Erreichung desSollwerts mit Rohstoff gefüllt wer-den. Zum anderen betrifft es die Ent-nahmewiegung. Hier wird einem Be-hälter Rohstoff entnommen und inden Einwaage- oder Mischbehältereingefüllt. Die dritte Methode betrifft
TechnoPharm 4, Nr. 4, 210–213 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 211Steinmetz . Prozessorientierten Wiegesystemen gehört die Zukunft

den Zählmodus. Sie findet dann An-wendung, wenn ein komplettes Ge-binde, dessen Gewichtswert exaktbekannt ist, in den Einwaage- oderMischbehälter eingefüllt wird. DerGewichtswert wird über die einge-gebene Stückzahl ohne echten Wie-gevorgang im Auftrag protokolliertund verbucht
5. Teilverwiegung /Chargenwechsel:Sofern das Sollgewicht bei einem Wie-gevorgang nicht erreicht wird, stelltdas WDS automatisch eine erforderli-che Teilverwiegung fest und kalkuliertdie Restmenge unter Berücksichti-gung der jeweiligen Toleranzgrenzen.Teilverwiegungen können beispiels-weise erforderlich werden bei Behäl-ter-, Lot-Nummer- oder Chargen-wechsel. Es ist jedoch sicherzustellen,dass jede Auftragsposition gemäß derTeilcharge dokumentiert wird.
6. Dokumentation:Zur Gewährleistung der Rückverfolg-barkeit sollen sämtliche Bediener-aktionen des am Wiegeterminal ange-meldeten Bedieners sowie die produk-tionsrelevanten Buchungen im Systemgeloggt werden. Voraussetzung hierfürist ein detailliertes User- und Rechte-system. Die Dokumentation beginntdamit, dass alle Daten der verwoge-nen Komponenten sowie die jeweili-gen Verbräuche an das ERP Systemübertragen und dort verbucht werdenkönnen. Über diese Daten und die au-tomatische Dokumentation aller Ar-beitsschritte erstellt das Wiegesystemautomatisch ein Wiegeprotokoll undermöglicht somit die Daten für die Be-reitstellung des Chargennachweises.Die Kennzeichnung aller verwogenenKomponenten, Subchargen und Intra-materialien (INTR) mittels Barcode-Label soll nach Ende eines jeden Wie-geschritts möglich sein. Schließlichwerden die im Labor verwogenenKleinmengen über manuelle Eingabenim Wiegeprotokoll dokumentiert.
7. IPC, CCP und CP:Das WDS muss die Fähigkeit haben,die Anforderungen an die IPC (In
Process Controll) sowie CCP (CriticalControl Point) und CP (ControlPoint) hinsichtlich der Abfragenund Dokumentation aus dem Systemheraus zu automatisieren. Diese Ab-fragen müssen anschließend durchden User bzw. durch Vier-Augen-Prinzip bestätigt und im Herstellpro-tokoll dokumentiert werden.
Darüber hinaus können unterneh-mensindividuell weitere Anforderun-gen entstehen, die im Wiegeprozessabgebildet werden müssen. Dies kannetwa die modulare Konzeption desSystems betreffen, um zukünftig wei-tere Waagen oder neue Funktionalitä-ten aufwandsarm integrieren zu kön-nen. Im Falle der Nachweise zur Ar-beitssicherheit können zudem zusätz-liche Dokumentationen erforderlichsein, wenn Gefahrstoffe zum Einsatzkommen. Aber auch die Entscheidung,ob mittels EBR (Eletronic BatchRecord) auf Papier in den gesamtenDokumentationen verzichtet werdenkann, ist ergänzend zu treffen.
Technische Infrastrukturund Schnittstellen
Ein Schwerpunkt stellt die Vernet-zung und Kopplung der einzelnenMaschinensteuerungen an das Systemsowie die Integration der Wiegepro-zesse dar. Damit ein zukunftssicheresSystem aufgebaut werden kann, isteine Vernetzung der Maschinen überein Netzwerk (TCP/IP) sowie eineKommunikation beispielsweise überOPC Server-Client Technologie sinn-voll. Zu den nachfolgend beschriebe-nen Systemen sind entsprechendeSchnittstellen zu realisieren.. ERP-Schnittstelle: Das WDS mussdie bidirektionale Kommunikationmit dem ERP-System des Unter-nehmens gewährleisten. Hierbeisind Standard Prozeduren der je-weiligen Hersteller zu bevorzugen.
. E-Mail-Schnittstelle: Zur auto-matischen Verteilung von Infor-mationen und Berichten via E-Mailist eine Schnittstelle zu beispiels-weise Microsoft Outlook oder Lo-tus Notes zu realisieren, die Kom-munikation erfolgt über das
SMTP-Protokoll (Simple MailTransfer Protocol).
. Schnittstelle zum QS-System: UmQualitätsdaten zu erfassen und zuarchivieren, soll im WDS/OEE-System eine Schnittstelle zum be-stehenden QS-System des Unter-nehmens integriert werden. DieTechnologie sollte sich dabei nachden Möglichkeiten der QS-Systemerichten. Beispielsweise XML-Tech-nologie.
. Waagen-Schnittstellen: Für dieKopplung an das Wiegesystemmüssen alle Waagen mit einerSchnittstelle ausgerüstet sein. Eswerden Waagen mit unterschied-lichen Wiegebereichen an einWiegeterminal angeschlossen. DasSystem ist so aufzubauen, dassimmer nur eine Waage eines Ter-minals betrieben werden kann. Siewerden von einem Wiegeterminalaus bedient. Dadurch könnenmehrere Mitarbeiter an unter-schiedlichen Aufträgen arbeitenbzw. unterschiedliche Rohstoffeeinwiegen.
. Labor: Für die Verwiegung vonKleinstmengen wird im Labor eineentsprechende Waage zur Ver-fügung gestellt, optional kann sie andas WDS angeschlossen werden.
. Maschinenschnittstellen: Die Ma-schinenschnittstellen sind in ersterLinie für die automatische Daten-erfassung des OEE-Systems erfor-derlich. Da die vorhandenen Ma-schinensteuerungen an älterenSystemen größtenteils ohne TCP/IP Schnittstellen und die Maschi-nen teilweise auch nicht mitSteuerungssystemen ausgerüstetsind, kann die Datenerfassung hierüber verdrahtete Signale auf IO-Klemmen erfolgen. Es empfiehltsich eine Realisierung der einzel-nen Steuerungsschnittstellen übernetzwerkfähige Kopf-SPSen.
OEE-Integration zurLeistungssteuerung
Das OEE-System dient dazu, durchdie Ermittlung von Prozess-, Be-triebs- und Maschinendaten Trans-
Prozess- und Verfahrenstechnik
TechnoPharm 4, Nr. 4, 210–213 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)212 Steinmetz . Prozessorientierten Wiegesystemen gehört die Zukunft

parenz in den Fertigungsprozesseninsgesamt und damit auch in denWiegeprozessen zu schaffen. Die er-mittelten Daten erzeugen eine be-lastbare Basis, um bei Bedarf Initia-tiven zur gezielten Verbesserung derEffizienz einleiten zu können.
Grundlage eines optimalen Be-triebs des OEE-Systems ist die Daten-erfassung über Maschinenschnittstel-len. Hierfür sollte jede Produktions-einheit (Linie, Wiegestation, etc.) in-dividuell im OEE-System konfiguriertwerden. Soweit die Daten nicht überSchnittstellen automatisch erfasstwerden, müssen sie durch manuelleEingaben an feststehenden bzw. mo-bilen Terminals komplettiert werdenkönnen. Zu den zu erfassenden Datengehören typischerweise allgemeineAuftragsdaten wie Auftrags-Nr., Linie,Chargen-Nr., Solltakt etc., Betriebs-arten wie Reinigung / Desinfektionund Störungsursachen an den Ma-schinen. Aber auch organisatorischeStillstände wie etwa Pausen, Rüstzei-ten, Massemangel oder fehlendes Per-sonal und geplante Wartungszeitenwerden ebenso erfasst wie personal-bezogene Informationen zu Schicht-wechsel oder Ausschuss. Im Ereignis-fall werden diese Signale automatischvom OEE erkannt, mit einem Zeit-stempel versehen und der entspre-chenden Kategorie zugeordnet:1. Kennzahlen:Das OEE-System sollte standard-mäßig etablierte Kennzahlen zurVerfügung stellen, die sich in dieGruppen Produktionszeit, Verhältnisvon Schicht- und Produktionszeit,Verhältnis maximal möglicher undeffektiver Produktionszeit sowie An-zahl der Mitarbeiter pro Schicht bzw.Auftrag in Stunden teilen. DieseKennzahlen sollten mittels Filternnach Auftragsnummern, Schichten,Zeiten etc. zusammengestellt und ta-bellarisch in Webmasken oder in Re-porten darstellbar sein. Außerdembedarf es der Möglichkeit, die Kenn-zahlenformeln eigenständig zumodi-fizieren bzw. neu zu erstellen. Eineklassische Kennzahl stellt dabei dieOEE (Overall Equipment Effective-ness) dar.
2. Maschinenlaufdiagramme:Hierüber lassen sich die Stillständeeiner Produktionseinheit grafisch ineinem Gantt-Diagramm darstellen.Durch frei definierbare Zeitbereiche,Kostenstellen oder auch Auftrag bzw.Schicht etc. soll eine detaillierte Dar-stellung und Auswertung ermöglichtwerden.3. Störstatistiken:Mit ihrer Hilfe werden Stillstandgrün-de, deren Häufigkeit und die Still-standdauer angezeigt. Störstatistikensollen für bestimmte Zeitkonten, Zeit-bereiche und Aufträge erstellt werden.Zur weiteren Detaillierung sollen Pro-duktionslinie, Maschine sowie Sortie-rungen auswählbar sein.4. Trenddarstellung:Diese Funktion dient dazu, dass zuallen definierten Daten mehrere
Trends mit unterschiedlichen Ach-sen angezeigt werden können.
Darüber hinaus werden die vomSystem generierten Daten in Re-ports in Form von Grafiken, Tabel-len oder Charts dargestellt. Dazu ge-hören Wiege- und Herstellpro-tokolle ebenso wie Kennzahlen-,Schicht-, Tages- und Störungs-reports, aber auch Barcode Labelund Reports zur Mitarbeiterauslas-tung und den Betriebsstunden sindrelevant. Deren Umfang und Aus-führungszeitpunkte lassen sich indi-viduell festlegen und sind wesent-lich von den organisatorischen Be-dingungen abhängig, in denenReports als Instrument zur Leis-tungsoptimierung und für Manage-mententscheidungen eingesetztwerden.
Cleanroom SolutionsSicherheit & Compliance für Ihre Reinräume
www.testotis.de/reinraumTesto Industrial Services GmbH · [email protected] · 07661 90901-8000
Qualifi zierung von Reinräumen und Lüftungsanlagen
Qualifi zierungsmessungen von Druck- und Prozessgasen
Mikrobiologisches Monitoring - Luft, Oberfl ächen und Gase
Durchführung von Schulungen & Workshops in unserem Reinraum- Trainingscenter

Prozessdaten: Komplexität wirdtransparent und beherrschbarDr.-Ing. Ulrich Lettau und Dr.-Ing. Andreas Quick . iba AG, Fürth
Korrespondenz: Dr.-Ing. Andreas Quick, iba AG, Koenigswarterstr. 44, 90762 Fürth;e-mail: [email protected]
ZusammenfassungKomplexität ist keine absolute Größe. Vielmehr lässt sich die Wahrnehmung von Komple-xität durch technische Verfahren reduzieren und durch ordnende Verfahren der Prozess-datenaufzeichnung und Analyse beherrschbar machen. Dies ist besonders für Prozesseder pharmazeutischen Industrie wichtig, da hier zum einen die regulatorischen undgesetzlichen Anforderungen bezüglich des Produktions- und Verpackungsprozesses ein-gehalten werden müssen und zum anderen Sicherheitsaspekte eine entscheidende Rollespielen. So müssen kritische Zustände der Prozessanlage, die zu akuten und längerfris-tigen Gefahren für Mensch und Umwelt führen könnten, nach Möglichkeit im Vorauserkannt und kritische Zustände unter allen Umständen vermieden werden. Hierbei kommtin der chemisch-pharmazeutischen Industrie der Auswahl geeigneter Sensoren zurErfassung des Prozesses eine besondere Rolle zu [3], [13].Voraussetzung für die Reduktion und Beherrschbarkeit der Komplexität ist ein drei-pha-siges Vorgehensmodell: Erfassung des dynamischen Prozessverhaltens durch geeigneteMessverfahren und Sensorik, Analyse der Daten durch multimediale Analyseinstrumentesowie Extraktion von Information und Wissen aus den Mess- und Prozessdaten sowieden Analyseergebnissen. Das Zusammenspiel dieser drei Phasen sowie deren Ausführungin einer geschlossenen Tool-Umgebung ermöglicht es, die semantische Ordnung einesProzesses zusammen mit dem dynamischen Zeitverhalten transparent und damitbeherrschbar zu machen. Aus dieser wahrgenommenen Reduktion der Komplexitätkönnen dann Störungen und deren Ursachen erkannt sowie Prozess- und Produktverbes-serungen unmittelbar abgeleitet werden.
Die menschlicheWahrnehmung vonKomplexität
Ob und zu welchem Grad Menscheneinen Sachverhalt und einen zeitli-chen Ablauf eines verfahrenstech-nischen Prozesses wie z.B. in derpharmazeutischen Industrie fürkomplex halten, hängt von einerFülle an Faktoren ab und ist indivi-duell stark unterschiedlich. Von zen-traler Bedeutung für die Komplexi-tätswahrnehmung sind Beobach-tungsmittel und Ordnungskriterien.Ordnende und visualisierende Ver-fahren ermöglichen eine erheblicheReduktion der wahrgenommenenKomplexität und erlauben Entschei-dungsträgern den Überblick über
eine große Zahl interdependenterEinzelgrößen und deren kausalenWechselwirkungen zu bewahren. Be-sonders deutlich wird diese Abhän-gigkeit von Komplexitätswahrneh-mung und medialer Aufbereitung inder Automatisierungstechnik. In denBereichen Produktion, Störungs-suche und Prozessoptimierung lässtsich durch geeignete Verfahren zurErfassung des zeitlichen dyna-mischen Verhaltens eine erheblicheKomplexitätsreduktion erzielen. Be-dingt durch die Vielfalt der verwen-deten Automatisierungssysteme(siehe [7]), Komponenten (Funk-tionsplan, Schaltschrank, Maschi-ne,…), Betriebssysteme, deren jewei-lige Versionen, verschiedene Mess-und Sensorsysteme und anderen
Variablen entsteht eine Heterogeni-tät, die einen hohen Anspruch andie Konnektivität der verwendetenMessdatenerfassungsgeräte stellt.Die Zustände und Daten der unter-schiedlichen Systeme müssen ohneAuswirkungen auf das Gesamtsys-tem (rückwirkungsfrei) und zeitlichsynchronisiert (isochron) aufge-zeichnet werden. Nur wenn es ge-lingt, interdependente Signale undSensorwerte mit einer hohen zeitli-chen Auflösung und ohne eine Verän-derung der Signale durch die Mes-sung zu erfassen, kann deren Wech-selwirkung aufgedeckt und analysiertwerden.
Aus Gründen der Rückverfolgbar-keit dürfen aufgezeichnete Datenund Signale nach der Aufzeichnung
Prozess- und Verfahrenstechnik
TechnoPharm 4, Nr. 4, 214–221 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)214 Lettau und Quick . PD: Komplexität wird transparent und beherrschbar

nicht mehr verändert und manipu-liert werden. Hierzu sind die binäreVerschlüsselung der Messdaten undeine Signierung der Messdateien not-wendig, um zu verhindern, dass auf-gezeichnete Daten im Nachhineinmit einem Editor verändert werden.Nur dann können die Messdatendazu verwendet werden, die Pro-zess-Compliance gemäß GMP (GoodManufacturing Practice) und 21 CFRPart 11 nachzuweisen. Der professio-nelle Umgang mit dem erhobenenDatenmaterial steigert die Produkti-vität und hat mithin erhebliche Aus-wirkungen auf den wirtschaftlichenErfolg der pharmazeutischen Unter-nehmen, die mit automatisiertenProzessen arbeiten.
Die Auswirkungen nichtbeherrschter Komplexität
Nicht beherrschte Komplexität beiAutomatisierungslösungen birgt dieGefahr potenzieller Instabilitäten inallen Phasen des Lebenszyklus’ einerAnlage, die besonders herausfor-dernd sind bei der Inbetriebsetzungund gefährlich während der Produk-tion. Im Produktionsalltag zeigt sichnicht beherrschte Komplexität einesSystems insbesondere dann, wennStörungen auftreten, deren Ursachennicht sofort lokalisierbar sind, unddie damit erhebliche Auswirkungenauf die Sicherheit, die Produktquali-tät [12] oder die Anlagenverfügbar-keit haben. Störungen sind Auswir-kungen systemimmanenter Instabili-
täten. Beispiele für Störungstypensind sogenannte sporadische Fehler,deterministische Fehler im Zeitver-halten, die langsame Verschlechte-rung der Qualität der Produkte bzw.der Anlagen sowie Störmeldungenohne erkennbare Ursache. Fast allesporadischen Fehler (Brüche, Ver-stopfungen, plötzliche Schwankun-gen, Kurzschlüsse, Kommunikations-störungen, etc.) sind in Wahrheit sys-tematische Fehler, die aber aufgrundunvollständiger Systemdurchdrin-gung nicht ausreichend durchschautwerden konnten. Nehmen determi-nistische Fehler zu oder Qualitätund Maßhaltigkeit ab (Ermüdungen,Abriebe, Verschmutzungen, Korrosi-on, Alterung, etc.), so sind die Gründehierfür ebenfalls systematischer Na-tur. Es handelt sich folglich um spo-radische Störungen, die aufgrund derSystemkomplexität hinsichtlich ihrerKausalität als nicht durchdringbarund damit nicht vorhersehbar odervermeidbar erscheinen.
Ein wichtiger Aspekt in der phar-mazeutischen Industrie ist die Ver-meidung kritischer Prozesssituatio-nen, die Auswirkungen auf die Anla-gensicherheit, die Arbeitssicherheitund den Umweltschutz haben kön-nen. Hier muss es mit Hilfe der Pro-zessanalyse nicht nur möglich sein,Störungen frühzeitig zu detektierenund zu melden, sondern Störungendurch eine kontinuierliche Über-wachung des Anlagenzustands ganzzu vermeiden und die Ursachen, diezu einer Störung führen können, bei
deren Auftreten sofort aufzuzeigen(root cause analysis [1]).
Störungen, die im Allgemeinensukzessiv und schleichend eintreten,sind schwer zu erkennen. Dies kanndazu führen, dass diese Störungenmitunter erst beim Abnehmer desProdukts (Endkunden) aufgedecktwerden. Fehler bei der Kennzeich-nung von Verpackungen könnenmassive finanzielle und wirtschaftli-che Auswirkungen bzgl. der Produkt-haftung für das Pharmaunterneh-men haben. Gemäß der Richtliniender FDA bzw. der EU müssen alleProdukte während der Produktioneindeutig gekennzeichnet sein (z.B.Barcode, Chargennummer und Halt-barkeitsdatum), sodass eine Rück-verfolgbarkeit eindeutig und leichtmöglich ist [9]. Kommt es zu Fehlernbei der Kennzeichnung (z.B. falscheChargennummer oder Nicht-Lesbar-keit des Barcodes), so hat dies eineenorme wirtschaftliche Tragweite.Dies führt vor Augen, wie wichtigdie reale oder gefühlte Reduktionvon Komplexität innerhalb auto-matisierter Vorgänge ist. Qualitäts-störungen sind daher durch die On-line-Extraktion von Qualitätsdatenaus den Prozessdaten bereits wäh-rend des Produktionsprozesses früh-zeitig zu erkennen und zu melden.
Wege zur Reduktion vonKomplexität
Um ein System mit vielen aneinandergebundenen prozessrelevanten Ein-zeldaten überschauen zu können,und damit die wahrgenommeneKomplexität des Systems zu reduzie-ren, ist eine Vorverarbeitung und Ag-gregierung der Informationen erfor-derlich. Um zu einer echten Erleich-terung in der Auswertung des Daten-materials zu führen, muss diese Bün-delung entlang überindividuell nach-vollziehbarer Ordnungskriterien er-folgen. Es ist folglich unumgänglichdie semantische Ordnung des Sys-tems auch bei der Datenerfassungund damit auch im Datenmaterialzu etablieren. Hierfür ist ein drei-pha-siges Vorgehensmodell notwendig
TechnoPharm 4, Nr. 4, 214–221 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 215Lettau und Quick . PD: Komplexität wird transparent und beherrschbar
Abb. 1: Drei-phasiges Vorgehensmodell (Quelle: alle iba AG).

(siehe Abb. 1): Erfassung des dyna-mischen Prozessverhaltens durch ge-eignete Messverfahren, Analyse derDaten durch multimediale Analy-seinstrumente sowie Extraktion vonInformation undWissen aus den Pro-zessdaten und Analyseergebnissen.
Die Art und Weise wie sich diesesemantische Ordnung auf die Einzel-daten bezieht, erlaubt Vergleiche mitdem Verhältnis zwischen Sprechak-ten und der zu Grunde liegendenGrammatik der Sprache, in der siegesprochen werden. Beim Erlerneneiner Fremdsprache werden die ein-zelnen Aussagen nur dann trans-parent, wenn die zugrunde liegendeRegelhaftigkeit erkannt wurde.
Analog ist es dem Prozessanalyti-ker im Bereich der Automation nurdann möglich aus einzelnen Mess-daten die richtigen Schlüsse zu zie-hen, wenn sich die zugrunde liegendetechnische Struktur des beobachte-ten Gesamtzusammenhangs in denerfassten Messdaten wiederspiegelt.Finden sich die funktionalen Zusam-menhänge des Systems in den Mess-daten wieder, so ist dies der ersteSchritt, die wahrgenommene Kom-plexität zu reduzieren und bietetdie Voraussetzung, das dynamischeZeitverhalten des Systems zu ana-lysieren, um die temporale Ordnungund das kausale Zeitverhalten zwi-schen Ereignisfolgen zu überprüfenund zu analysieren.
Darüber hinaus könnenMethodender mathematischen Statistik [2], desData Mining [4], [11] und der Zeitrei-henanalyse [5] implementiert undangewendet werden, die es erlauben,Qualitätsdaten sowie Informationenund Wissen aus den aufgenom-menen Prozessdaten zu extrahieren,was die wahr-genommeneKomplexität desGesamtsystemsweiter reduziert.Geeignet auf-bereitet und vi-sualisiert dienendiese Daten da-zu, die Produkt-qualität zu be-
werten bzw. den Produktionsprozesszu optimieren.
Standardisierung:Versuch eines komplexi-tätsreduzierendenVerfahrens
Eine der bedeutendsten Variantender Komplexitätsreduktion ist dieStandardisierung. Experten aus denverschiedensten Bereichen versuchenweltweit einheitliche Standards zuetablieren, um in ihren jeweiligen Be-reichen die Komplexität von Konfigu-rationen und Kommunikation zu re-duzieren. In der Vergangenheit hatsich jedoch immer wieder gezeigt,dass die Standardisierung nur be-dingt zur Reduktion der Gesamtkom-plexität von Systemen beiträgt. Mitder Standardisierung von Schnittstel-len, Kommunikation, Programmier-sprachen und Prozessketten sinktzwar die Komplexität auf den zuge-hörigen Ebenen zunächst ab, auf dendarüber liegenden Ebenen steigt siejedoch wieder an, weil die Anzahlder miteinander kombinierbaren Ob-jekte und Komponenten zunimmt,und sich der Integrationsgrad vonAutomatisierungslösungen tenden-ziell weiter erhöht.
Durch die Verwendung von Stan-dardkomponenten kann sich dieKomplexität also erhöhen, da die An-zahl der Komponenten und die An-zahl der Schnittstellen im Allgemei-nen zunehmen. In Abb. 2 ist exem-plarisch dargestellt, wie die Anzahlder Komponenten und Schnittstellenzunimmt, wenn statt einer projekt-spezifischen Lösung (Ansatz A) einestandardisierte Lösung realisiertwird (Ansatz B).
In der projektspezifischen Lösungwerden eine projektspezifische Kom-ponente P und zwei Standardkom-ponenten S1 und S2 eingesetzt. Sol-len nun ausschließlich Standard-komponenten verwendet werden(Ansatz B), so entstehen dadurch so-wohl neue Schnittstellen, die bisherim projektspezifischen Modul P ge-kapselt waren (rote Pfeile), als auchweitere Schnittstellen zu den schoneingesetzten StandardkomponentenS1 und S2 (blaue Pfeile). Da eine voll-ständige Realisierung mit Standard-komponenten im Allgemeinen nichtmöglich ist, lässt sich ein projektspe-zifischer Anteil P‘ nicht ganz vermei-den. So stehen den drei Komponen-ten und zwei Schnittstellen in AnsatzA nun sechs Komponenten und neunSchnittstellen in Ansatz B gegenüber.
Durch eine Lösung mit Standard-komponenten wird der projektspezi-fische Anteil zwar reduziert, dieKomplexität erhöht sich jedoch vorallem durch die Zunahme der Kom-munikation zwischen den einzelnenKomponenten. Wird die Komplexitätjedoch durch die hier beschriebeneTechnik beherrschbar gemacht, kannAnsatz B durchaus ein kostengünsti-ger und effizienterer Ansatz sein.
Datenerfassung in Auto-matisierungssystemen
Moderne Automatisierungstechnikbasiert auf digitalen Verarbeitungs-geräten, Kommunikation zu Feldgerä-ten über digitale Bussysteme sowieKommunikationstelegrammen vonLeitrechnern zu Ausgabegeräten wiebeispielsweise Etikettierer, etc. Daherliegen die meisten interessierendenSignale bereits in digitaler Form vor.
Trotzdem sindfür signifikanteSignale, bei denenman Verfälschun-gen durch dieVerarbeitung inder Automatisie-rung ausschlie-ßen will, hoch-wertige analogeEinkopplungen
Prozess- und Verfahrenstechnik
TechnoPharm 4, Nr. 4, 214–221 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)216 Lettau und Quick . PD: Komplexität wird transparent und beherrschbar
Abb. 2: Komplexitätszunahme durch Standardisierung.

sinnvoll. Dies betrifft vor allem Vibra-tions- und Schwingungssignale zurMaschinendiagnose (Condition Moni-toring) sowie elektrische Größen, beidenen Qualitätskennwerte im Zeit-und Frequenzbereich beobachtet wer-den sollen. Die Erfassung dieser Datenerfolgt hier nach dem Abtastprinzip[14]. Dabei wird das zu messende ana-loge Signal zeitdiskret abgetastet undder Wert zum Zeitpunkt der Abtas-tung quantisiert, das heißt in ein digi-tales Signal umgewandelt.
Die beiden wesentlichen Größenbei der Digitalisierung von Signalensind die Abtastfrequenz und die digi-tale Auflösung: Nach dem Nyquist-Theorem [10] muss die Abtastfre-quenz mindestens doppelt so hochsein wie die maximal im analogenSignal vorkommende Frequenz, umdas analoge Signal eindeutig be-schreiben und rekonstruieren zukönnen. In der Praxis wählt manmeistens mindesten die 2,5- bis 2,7-
fache Frequenz. Wird das Nyquist-Theorem verletzt, so treten im abge-tasteten Signal Artefakte auf. Es han-delt sich hierbei um das Phänomen,das als Aliasing bekannt ist. In derindustriellen Automatisierung habensich Abtastfrequenzen von 1 kHz alssinnvoll erwiesen, um auch schnelleRegelvorgänge messen zu können.Für Vibrationsanalysen an Maschi-nen werden entsprechend den auf-tretenden Maschinenschwingungenmeist 10 kHz bis 20 kHz benötigt.Für Schallmessungen sogar bis 100kHz. Auch bei Transientenrecordernin der elektrischen Energiemesstech-nik werden hohe Abtastfrequenzenbenötigt. Die Auflösung der Digitali-sierung wird in bit definiert. Wird dasSignal mit 14 bit quantisiert, so ent-spricht das 214 = 16384 möglichenStufen, mit dem der Signalbereichaufgelöst wird, das entspricht einerGenauigkeit von 0,06‰ Heutige Sys-teme sind mit 16 bit Auflösung schon
so genau, dass die Quantisierungs-unschärfe deutlich kleiner ist alsdas Signalrauschen des zu messen-den Signals. Was darüber liegt, istpraktisch nicht mehr relevant. Diezu messenden Signale werden in ei-nem digitalen Messsystem äquidis-tant abgetastet, quantisiert und alsZeitreihen, die aus einer Zahlenfolgedieser zeitlich äquidistanten Abtast-werte bestehen, abgespeichert.
In der pharmazeutischen Indus-trie ist darüber hinaus die Erfassungeines weiteren Datentyps, nämlichdie Erfassung der Kommunikations-telegramme notwendig. Da es zwin-gend vorgeschrieben ist, dass alleDruckdaten von einem übergeord-neten System kommen und nichtam Drucker direkt eingegeben wer-den dürfen [9], müssen alle Druckerund Etikettierer, etc. in die vorhan-dene IT-Landschaft integriert wer-den. Für die Prozessanalyse bedeutetdies, dass Messwerte und Prozesssig-
TechnoPharm 4, Nr. 4, 214–221 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 217Lettau und Quick . PD: Komplexität wird transparent und beherrschbar
Abb. 3: Beispiele für Konnektivität in heterogenen Automatisierungssystemen.

nale mit den Druck-daten synchronisiertwerden müssen, d. h.der Dateninhalt derKommunikationstele-gramme vom Leitrech-ner zu den Ausgabe-geräten muss ebensozeitsynchron auf-gezeichnet werden wiez.B. Sensordaten undAntriebs- und Produkti-onsdaten.
PraktischeProbleme bei der Mess-datenerfassung inverteilten heterogenenSystemen
Bei größeren Anlagen liegt meist einesehr heterogene Landschaft aus Au-tomatisierungs- und Bussystemenverschiedener Hersteller und Gene-rationen vor. Besonders in der che-misch-pharmazeutischen Industriewird eine Vielzahl verschiedener Sen-soren eingesetzt [15], um unter-schiedlichste Messgrößen zu erfas-sen. Um Prozesse weiter zu optimie-ren, dürfen die Sensoren nicht iso-liert eingesetzt, sondern in die Auto-matisierungslandschaft als inline-Messung integriert werden (Zielstel-lung XIII in [15]). Auch wenn die Sen-soren immer robuster und langzeit-stabiler werden sollen, sodass derWartungsaufwand in Zukunft immergeringer wird (Zielstellung I in [15],so haben sie im Allgemeinen herstel-lerbedingt und in Abhängigkeit vonder Messaufgabe unterschiedlicheSchnittstellen, um die Messergeb-nisse online in die Automatisierungeinzukoppeln.
Um eine einheitliche Diagnoseund Beschreibung der Vorgänge inder Anlage vornehmen zu können,ist eine Konnektivität zu all diesenSystemen erforderlich. Einzelne Her-steller widmen sich bereits seit län-gerem diesem Thema und könnenheute Zugangsmöglichkeiten für allegängigen Automatisierungssystemeanbieten. In Abb. 3 ist beispielhaftdargestellt, welche Konnektivität für
heterogene Automatisierungssys-teme notwendig ist.
Eine exakte Analyse von Vorgän-gen in einer Großanlage mit mehre-ren Steuerungen und Prozessorenkann nur dann erfolgen, wenn alleSignale isochron abgetastet werden.Ansonsten muss mit nichtkausalenZusammenhängen und Artefaktendurch Schwebungen zwischen ver-schiedenen Abfragetakten gerechnetwerden. Messtechnische Unterneh-men verwenden daher häufig zurÜbertragung der Messwerte starreKommunikationsmechanismen überLichtwellenleiter-Strecken. Der Kom-munikationstakt wird bei diesem Ver-fahren für alle angeschlossenen Sys-teme von den PC-Anschaltbaugrup-pen synchronisiert. Auf dem PC selbstwird dann die Software zur Mess-datenerfassung, Visualisierung undAufzeichnung der Messdaten aus-geführt.
Praktische Herausforde-rungen beim Aufzeichnengroßer Datenmengen
Die Aufzeichnungsmethodik ist vongroßer Relevanz für die Reduktionder Komplexitätswahrnehmung.Denn bei der anfallenden Daten-menge ist ein hoch performanterSpeicheralgorithmus mit einer ver-lustfreien Komprimierung der Datenvon größter Wichtigkeit. Die Pro-zessdatenaufzeichnung muss es er-möglichen, mehrere Datenaufzeich-nungen parallel ablaufen zu lassen[8]. Dies wird in vielen Anwendungengenutzt, um verschiedene Sichten auf
die Messwerte zu erlauben. Beispiels-weise können parallel kontinuierlichDaten als „Flugschreiberfunktion“abgespeichert werden und gleichzei-tig mittels getriggerter Aufzeichnung„Produktdateien“ erzeugt werden,welche die auf eine zeitlich begrenzteCharge bezogenen Prozessdaten ent-halten. Die dateibasierte Aufzeich-nung der Messdaten bietet bei einersinnvollen Strukturierung der Datei-ablage, einer angemessenen Dimen-sionierung der Aufzeichnungsdauerpro Datei und einer verständlichenDateibenennung, die aus den Kom-munikationsdaten des übergeord-neten Leitrechners extrahiert werdenkönnen (z.B. Chargennummer), einepraktikable Lösung für kurz- undmittelfristige Analysen, wie sie oftin der Instandhaltung benötigt wer-den. Sie eignet sich besonders füreine umfassende interaktive Offline-Analyse. Die Daten können leicht ar-chiviert und kopiert werden und sindfür eine produkt- oder ereignis-abhängige Nachbearbeitung beson-ders geeignet.
Allerdings kann es bei der char-gen-orientierten Sichtweise schwie-rig sein, den Überblick über langeZeiträume und über Messdateigren-zen hinweg zu behalten. Bei kontinu-ierlichen Prozessen, wie sie etwa inverfahrenstechnischen Anlagen derchemisch-pharmazeutischen Indus-trie, Prüfständen für Langzeittests,Energieversorgungsanlagen oderauch Papiermaschinen vorliegen,wünschen sich Instandhalter, Pro-zess-Ingenieure wie auch Produkti-onspersonal eine aus Anwendersicht
Prozess- und Verfahrenstechnik
TechnoPharm 4, Nr. 4, 214–221 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)218 Lettau und Quick . PD: Komplexität wird transparent und beherrschbar
Abb. 4: Typische Auswerteumgebung zur interaktiven Messdatenanalyse.

lückenlose und kontinuierliche Auf-zeichnung der Daten. Hierzu gibt esneuerdings Lösungen, die Messdatenüber lange Zeiträume zeitlich hochaufgelöst aufzeichnen (high defini-tion historical data). Die Daten wer-den bei dieser Aufzeichnungsartnicht in einzelne Messdateien zer-legt, sondern mit einem speziellenKompressionsalgorithmusauf dem sog. HD-Server(Historical Data Server)gespeichert. In Analyse-Clients können sowohldie historischen als auchdie aktuell online erfasstenSignalwerte in einerTrenddarstellung ange-zeigt werden. Mit wenigenMausklicks gelangt manvon der Jahres-, Monats-oder Wochenübersichtbis hin zu Details im Milli-sekundenbereich. Die Na-vigation wird über Zeit-marker, Blätterfunktionsowie über die Eingabe ei-nes bestimmten Zeit-punkts bewerkstelligt. Be-sonders hilfreich ist eineFunktion, die es erlaubtvorher definierte Ereig-nisse durch Mausklick ineiner Ereignistabelle anzu-springen.
Messdaten-analyse
Um die erfassten Mess-daten interaktiv analysie-ren zu können, müssendiese geeignet medial vi-sualisiert werden. Dabeimuss es sowohl möglichsein, Signale miteinanderzu verknüpfen, um sowohlWechselwirkungen zwi-schen Signalen aufzuzei-gen als auch signalinhä-rente Information zu ex-trahieren und darzustel-len. Diese Verknüpfungenkönnen von einfachenarithmetischen Operatio-nen bis hin zur Bildung
von komplexen Kennwerten für An-wendungen in der elektrischen Ener-gieübertragung (Power Quality Moni-toring) oder der zustandsbasiertenInstandhaltung (Condition Monito-ring) reichen. Condition Monitoringist geeignet, beispielsweise Antriebs-und Lagerschäden frühzeitig zu er-kennen und basierend auf den Ergeb-
nissen der Messdatenanalyse die not-wendigen Maßnahmen einzuleiten.Nur so können Störungen mit Aus-wirkungen auf die Sicherheit vermie-den und gleichzeitig die Anlagenver-fügbarkeit erhöht werden.
Mit Hilfe dieser virtuellen Signalelassen sich komplexe Zusammen-hänge analysieren, darstellen und
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2014-04\Anzeigensatz-keine-Druck-PDFs\optima-TP-2014-04_146x213.indd Zuletzt gesichert: 13.06.14 (02:25:08 Uhr)
Daniel DrosselMaschinenbautechniker(Konstruktion)
„Lösungen von der Stange? Nein, danke. Schließlich sind unsere Kunden ja auch etwas ganz Besonderes.“
EXCELLENCE IN PHARMA
OPTIMAJeder Kunde hat besondere Anforderungen. Darum bauen wir bei Optima Abfüllanlagen, die für jeden Bedarf maßgeschneidert sind und als Komplett-linien doch alles bieten: die vollständige Maschinenbandbreite, die einheitliche Dokumentation und die ideale Software-Lösung gleich mit dazu. Und all das bei einem Ansprechpartner, der sich um jedes Detail kümmert. Besonders bleibt eben besonders.
OPTIMA pharma GmbH | Otto-Hahn-Straße 1 | 74523 Schwäbisch Hall E-Mail [email protected] | www.optima-pharma.com
Member of

verstehen. Die zentrale Herausforde-rung der Messdatenanalyse bei einerStörungssuche liegt darin, die zeitli-chen Verläufe der funktional zusam-mengehörigen Signale zu unter-suchen und mit dem erwarteten Ver-halten zu vergleichen. Diese Musterzu identifizieren erfordert jedochausreichend Zeit und Erfahrungund muss durch Werkzeuge unter-stützt werden (siehe Abb. 4).
Aufgrund grober Vermutungenwird die erste Messaufgabe gestellt.Abhängig von den gefundenen Indi-zien muss die Messaufgabe für dieFortsetzung der Suche immer wiederinteraktiv modifiziert und überprüftwerden. Zur effizienten Analyse müs-sen verschiedene Filter zum Glättenvon Signalen sowie Transformations-werkzeuge wie beispielsweise dieFourier-Transformation zur Erken-nung frequenzabhängiger Effekteverfügbar sein. Die mediale Visuali-sierung der Messdaten, der verknüpf-ten Messdaten (virtuelle Signale) so-wie der extrahierten Größen, wie sta-tistische Kenngrößen, ist unabding-bar für die Aufbereitung und Analyseder Messdaten [6].
SynchronisierteErfassung von Messsig-nalen und Videobildern
Beim Betrieb einer komplexen Anlagetreten immer wieder Situationen auf,die mithilfe der digitalen und analo-gen Anlagensignalenicht oder nurschwer zu interpre-tieren sind. Dies istnicht nur bei der Stö-rungssuche der Fall,sondern auch beimAuftreten von tech-nologischen Proble-men oder Qualitäts-beanstandungen derKunden. Hierfür hatsich die zeitsyn-chrone Aufzeichnungder Anlagendatenmit Videosignalenals effizientes Hilfs-mittel erwiesen.
Die gleichzeitige Aufzeichnungvon Messsignalen und Videobildernreduziert die wahrgenommene Kom-plexität eines Systems erheblich undvisualisiert Ursache und Wirkung,wenn beide Signalarten zeitsynchro-nisiert gemeinsam wiedergegebenwerden (siehe Abb. 5). So weiß mangenau, was zum Zeitpunkt einer Sig-naländerung in den aufgezeichnetenMesssignalen in der Anlage tatsäch-lich geschehen ist. Um sehr schnelleAbläufe ebenfalls getriggert aufzeich-nen zu können, müssen neben klas-sischen analogen Kameras und IP-Cams ebenfalls Lösungen mit Hoch-geschwindigkeitskameras (300 fps)realisiert werden. Hinsichtlich derBedien- und Visualisierungsoberflä-che sowie der anschließenden Signal-analyse mit Analysetools müssenderartige „Kamerasignale“ für denAnwender genauso einfach wie nor-male Signale behandelt werden undbei der Auswertung in den verschie-densten Modi dargestellt werden.Pharmaunternehmen setzen bereitsseit einiger Zeit Kamerasignale zurValidierung des Packungsaufdrucks
ein. Werden diese Videosignale mitden Prozesssignalen und den Kom-munikationsdaten synchronisiert, sokann nicht nur die Lesbarkeit desAufdrucks validiert, sondern im Feh-lerfall die Ursache leicht bestimmtwerden. Die synchronisierte Auf-zeichnung der drei Datentypen Vi-deobilder, Kommunikationsdatenvom übergeordneten Leitrechner so-wie Prozessdaten bildet die Basis füreine erfolgreiche Prozessanalyse.
Online-Visualisierung
Neben der interaktiven Offline-Ana-lyse spielt auch die direkte Betrach-tung der medial aufbereiteten Mess-daten sowie der daraus online extra-hierten Qualitätsdaten eine wichtigeRolle. Nur so kann die Qualität desProdukts direkt während der Entste-hung beobachtet und im Falle vonQualitätsabweichungen entspre-chend signalisiert werden. Hierzusollte ein Werkzeug zur grafischenOnline-Darstellung von Messgerätenwie Multimeter und Oszilloskop,aber auch von Trends und Signalen
Prozess- und Verfahrenstechnik
TechnoPharm 4, Nr. 4, 214–221 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)220 Lettau und Quick . PD: Komplexität wird transparent und beherrschbar
Abb. 6: Beispiele grafischer online-Prozessvisualisierungen.
Abb. 5: Synchronisierte Darstellung von Messsignalen und Videobildern.

im Frequenzbereich zur Verfügungstehen (siehe Abb. 6).
AutomatischeInformationsgenerierung
Neben der interaktiven, manuellenAuswertung der Messdaten könnenMessdaten auch nach vorgegebenenRegeln automatisch ausgewertet wer-den, um beispielsweise Aussagen überdie Produktqualität zu generierenoder auch den Anlagenzustand zu be-werten. Der Unterschied zwischenProzess- und Qualitätsdaten lässt sichwie folgt erklären: Während Prozess-daten schnelle Veränderungen inner-halb des Prozesses darstellen könnenund daher zeitlich möglichst hochauf-lösend erfasst werden, handelt es sichbei Qualitätsdaten um Kennwerte(z.B. Mittelwerte, Standardabwei-chungen und andere statistische Grö-ßen), die aus den Prozessdaten überAnalysevorschriften berechnet wer-den. Sie erlauben allgemeine Aus-sagen über die Produkt- und Prozess-qualität, insbesondere im Hinblick aufProduktfreigaben und eine langfris-tige Entwicklung der Qualität. DieProzessdaten hingegen lassen exakteRückschlüsse auf die Ursachen even-tueller Qualitätsdefekte zu. So kannder aktuelle Anlagenzustand währendder Produktion mit bereits früher ge-lernten Daten korreliert werden, umFehlerzustände im Prozess und in derAnlage automatisch zu diagnostizie-ren. Auf diese Weise können dem In-standhalter wertvolle Hinweise zurWartung der Anlage gegeben unddem schleichenden Qualitätsverlustentgegen gewirkt werden.
Als besonders hilfreich erweist essich bei dieser Form der diagnosti-schen Analyse, wenn aus den aggre-gierten Qualitätsdaten und Wartungs-hinweisen automatisch in einer in sichgeschlossenen Tool-Umgebung auf die„Rohdaten“, d. h. auf die aufgezeichne-ten Messdaten zugegriffen werdenkann. Nur so ist es möglich, den Ur-sachen für das gezeigte Verhalten aufden Grund zu gehen („drill down“).
Bestimmte Anbieter stellen Ana-lysefunktionen bereit, die die gemes-
senen Daten erst konditionieren, an-schließend zu Kennwerten umrech-nen und schließlich die so gewonne-nen Kennwerte in eine definierte, of-fene Datenbankstruktur ablegen. Da-mit können andere IT-Systeme aufsauber vorbehandelte Kennwerte zu-greifen, deren Herkunft genau be-kannt ist und die vom Prozessexper-ten mittels einfacher grafischer Ana-lysen definiert werden können.
Fazit
Heutige Produktionsprozesse derpharmazeutischen Industrie enthal-ten eine Vielzahl gleichzeitig einströ-mender Messsignale, die alle auf-gezeichnet und analysiert werdenmüssen, um den Verantwortlicheneine verlässliche Entscheidungsbasiszu liefern. Je größer die Anzahl dererhobenen Messwerte ist, und je he-terogener die Signale sind, die ver-arbeitet werden müssen, desto größerist die Herausforderung, durch struk-turierende Maßnahmen und multi-mediale Visualisierungshilfen die Pro-zesskomplexität beherrschbar zu ma-chen, besonders dann, wenn Anlagen-sicherheit und Rückverfolgbarkeiteine wesentliche Rolle spielen. Eszeigt sich im Produktionsalltag füh-render Unternehmen immer wieder,dass Komplexität keine absolute Grö-ße, sondern eine menschliche Wahr-nehmung ist. Insbesondere durch Op-timierung der Aufzeichnungsverfah-ren, durch ständiges Hinterfragender Prozess-Semantik und durch kon-sequente Störungsanalyse kann unterZuhilfenahme multimedialer Analyse-tools eine Reduktion der wahrgenom-menen Komplexität erfolgen. Die re-sultierende Durchdringung der Pro-zesslogik steigert insbesondere danndie Produktions- und Anlagensicher-heit sowie die Rentabilität der Pro-duktion in erheblichem Maße, wenndie gewonnenen Erkenntnisse kon-sequent und zeitnah visualisiert undfür die automatische Prozess- undProduktdiagnose herangezogen wer-den. Die Verwendung einer geschlos-senen Tool-Umgebung für die Mess-datenerfassung, die Analyse der Mess-
daten sowie die Extraktion von Qua-litätsdaten hat sich als hilfreich erwie-sen [6], um die Komplexität eines Pro-zesssystems transparent und be-herrschbar zu machen.
Fachliteratur[1] Andersen, Bjørn; Fagerhaug, Tom. Root
Cause Analysis: Simplified Tools andTechniques. ASQ Quality Press, 2nd editi-on, 2006.
[2] Dietrich, Edgar; Schulze, Alfred. Statisti-sche Verfahren zur Maschinen- und Pro-zessqualifikation. Carl Hanser VerlagGmbH & Co. KG, 6. Auflage, 2009.
[3] Gerlach, Martin u.a. Trends in der Online-Analytik – Bessere Daten für effizientereProzesse, 65. NAMUR-Hauptsitzung, 2002.
[4] Kaiser, Volker; Otte, Viktor. Data Miningfür die industrielle Praxis. Carl HanserVerlag GmbH & Co. KG, 1. Auflage, 2004.
[5] Kreiß, Jens-Peter. Einführung in die Zeit-reihenanalyse (Statistik und ihre Anwen-dungen). Springer, 1. Auflage, 2006.
[6] Krumbach, Robert; Lettau, Ulrich. Trans-parente Erfassung und Aufbereitung vonMessdaten. In: Stahl und Eisen 132, Nr. 7,2012.
[7] Langmann, Reinhard. Taschenbuch derAutomatisierung. Carl Hanser VerlagGmbH & Co. KG, 2. neu bearbeitete Auf-lage, 2010.
[8] Lettau, Ulrich. Die Evolution der Mess-datenerfassung, In: Automatisierung undMesstechnik, 2009.
[9] Mühlenkamp, Sabine u. a. Neue gesetzli-che Anforderungen stellen neue Heraus-forderungen an Kennzeichnungssystemeder Pharma-Industrie. Process, Portal fürChemie und Pharmatechnik, Februar2008.
[10] Nyquist, Harry. Certain Topics in Tele-graph Transmission Theory. In: Trans-actions of the American Institute ofElectrical Enigneers. Vol. 47, April 1928.Wiederabdruck in: Proceedings of theIEEE, Vol. 90, No. 2, 2002.
[11] Peters, Harald et al. Industrielles DataMining in der Stahlindustrie, In: Stahl undEisen 132, Nr. 2, 2012.
[12] Pfeifer, Tilo; Schmitt, Robert. Qualitäts-management: Strategien, Methoden,Techniken, Carl Hanser Verlag GmbH &CO. KG; 4. vollständig überarbeitete Auf-lage, 2010.
[13] Sanden, Josef u. a. Anforderungen anSensorsysteme zur Prozessführung. 66.NAMUR-Hauptsitzung, 2003.
[14] Schüßler, Hans W. Digitale Signalver-arbeitung 1: Analyse diskreter Signale undSysteme. Springer, 5. Auflage 2008.
[15] VDI/VDE-Gesellschaft Mess- und Auto-matisierungstechnik (GMA), NAMUR –Interessengemeinschaft Automatisie-rungstechnik in der Prozessindustrie.Sensoren 2015+. Technologie-Roadmapfür Prozess-Sensoren in der chemisch-pharmazeutischen Industrie. November2009.
TechnoPharm 4, Nr. 4, 214–221 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 221Lettau und Quick . PD: Komplexität wird transparent und beherrschbar

50 Jahre rommelag®
und bottelpack®
Geschichte
Vor fünfzig Jahren brachte die rom-melag die Kunststoff-Verpackungs-technologie einen Schritt voran. Ver-antwortlich für diese Entwicklungwar Gerhard Hansen (geb. 1921) ausder Oberlausitz. Angefangen hat ermit einer Lehre zum Elektriker, dannmit einem Ingenieursstudium zumFlugzeugkonstrukteur. Wie bei vielenseiner Generation hat der ZweiteWeltkrieg seine berufliche Laufbahnabrupt und schmerzhaft unterbro-chen. Wie wenigen dagegen gelangihm gleich zweimal die Flucht ausrussischer Gefangenschaft. Nach sei-ner Rückkehr nach Sachsen ließ seinFreiheitsdrang auch in der Sowjeti-schen Besatzungszone nicht nach.1950, kurz nach Gründung der DDR,floh er nach Westdeutschland, ent-wickelte Maschinen für die Extrusionund das Bedrucken von Kunststoff-folien und gründete 1952 seine ersteFirma, die thermo-pack. 1963 erfandund baute er seine erste Blow-Fill-Seal-Anlage. Dieser Erfolg ließ ihnnach neuen Vertriebswegen suchen.Von der Schweiz aus sollte der inter-nationale Verkauf seiner Maschinenaufgebaut werden – mit der am1. Mai 1964 gegründeten Handels-gesellschaft rommelag in Aarau. In-zwischen umfasst rommelag als Teilder Hansen-Gruppe mit BerndHansen als CEO vier Vertriebsfirmenmit Sitz in der Schweiz, in Deutsch-land, den USA und in China. Weltweitist das Unternehmen der führendeLieferant von Blow-Fill-Seal-Anlagen,die unter dem Markennamen bottel-pack vertrieben werden.
Technologie
Aus drei mach eins. Alle bottelpackAnlagen arbeiten nach dem soge-
nannten Blow-Fill-Seal-Prinzip: Siestellen in einem einzigen automati-schen Prozess die zu befüllenden Be-hältnisse aus einem thermoplas-tischen Kunststoffgranulat her(Blow), füllen diese (Fill) und ver-schließen sie anschließend (Seal).Die Vorteile dieser BFS-Technologieim Vergleich zu konventionellen Me-thoden sind z.B. konservierungsmit-tel- und kontaminationsfreie Produk-te, Zeit-, Platz- und Personalerspar-
nis, Vollautomatisierung, geschlosse-ne, bruchsichere Behälter sowie einegleichbleibend Produktsicherheitz.B. durch Keimfreiheit und Fäl-schungssicherheit.
Ruhelos ist die Idee, weil sie auchnach fünf Jahrzehnten immer nochneue Innovationen hervorbringt. AlsBeispiel seien hier die aseptischenBFS-bottelpack CoEx-Verpackungs-anlagen genannt. Wurden zu Anfangdie BFS-Anlagen überwiegend fürMilch, Speiseöl, Softgetränke, Körper-
pflegemittel, Flüssigreiniger, Che-mikalien, Pflanzenschutzmittel, tech-nische Flüssigprodukte u. a. einge-setzt, liegt heutzutage der Einsatz-schwerpunkt weltweit überwiegendim Pharmabereich zur aseptischenVerpackung steriler Flüssigprodukte,Cremes und Salben. Die CoEx-Ma-schinen ermöglichen die sterile, pyro-genfreie Behälterherstellung, dieaseptische Abfüllung steriler Pro-dukte und das hermetische Verschlie-
ßen in einem Arbeitszyklus in einemanlageneigenen Reinraum der US-Klasse 100 (ISO 3) im Abfüllbereich.Darüber hinaus stellen sie Mehr-schichtbehälter mit speziellen Sperr-schichten zur Verpackung besondersempfindlicher Produkte her. In derCo-Extrusion werden verschiedeneKunststoffe als mehrschichtigeSchläuche extrudiert, um deren un-terschiedliche Materialeigenschaften,wie z.B. Gasdiffusion, Elastizität oderSteifheit zu kombinieren.
Spektrum
TechnoPharm 4, Nr. 4, 222–223 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)222 Spektrum
Produkte aus 50 Jahren rommelag® und bottelpack® (Quelle: rommelag).

Weiterhin ist die Idee auch inte-ressant, weil sie in einem permanen-ten Entwicklungs- und Optimie-rungsprozess immer neue Behält-nisse und Verschlüsse hervorbringt,um sowohl kundenspezifische Be-dürfnisse als auch die steigenden An-forderungen des modernen Gesund-heitswesens und der zunehmendenMobilität der Menschen zu erfüllen.Zu nennen sind hier z.B. Ready-to-use-Produkte wie Injektionsampul-len mit integrierter Injektionsnadel.Oder Ampullen mit Luer-Anschluss,bei denen der Gebrauch von Entnah-menadeln entfällt. Oder Unitdose-Ampullen für Augen-, Nasen- undOhrentropfen mit integrierter Do-sierkammer, so dass auf Konservie-rungsstoffe verzichtet werden kann.Der Entwicklung neuer Funktionen,meist in enger Zusammenarbeit mitdem Kunden, scheinen dabei keineGrenzen gesetzt.
Prozess- undProduktsicherheit
Denn das experimentum crucis fürjede Neuentwicklung bleibt die Si-cherheit – die Prozess- wie Produkt-sicherheit, also die Gewährleistungabsoluter Sterilität sowohl währenddes aseptischen Abfüllprozesses alsauch dank absolut hermetischer Ver-schlüsse während der Aufbewah-rungszeit. Letztendlich geht es umdie Sicherheit des Menschen. Dieserexistenziellen Verantwortung sindsich alle rommelag Mitarbeiter be-wusst.
Und dafür wird einiges getan.Zwar entfallen bei der BFS-Technolo-gie die bei vorgefertigten Behälternunumgänglichen Reinigungs- undEntkeimungsprozesse. Um aber eine100 %-Sicherheit der aseptischenProduktion zu gewährleisten, lassensich die bottelpack Anlagen mit Si-
cherheits-, Qualitäts- und Kontroll-modulen bestücken, um die vonstaatlicher Seite, wie z.B. der US-amerikanischen Food and DrugAdministration (FDA), gefordertenStandards zu erfüllen:. Aseptikanlagen zur automatischenCIP/SIP-Reinigung aller produkt-berührten Leitungen, die mitDruckdampf sterilisiert und mitSterilluft getrocknet werden
. Behälterdichtigkeitsprüf- und Par-tikelinspektionsanlagen mit auto-matischem Ausstoß fehlerhafterBehälter
. Ampullen-Prüfanlagen fürIn-Prozesskontrollen
. Reinraumluftmonitoring usw.Dass die BFS-Anlagen trotz all dieserSicherheitsmechanismen bis über34000 Behälter/h mit Behältergrö-ßen von 0,1 ml bis zu über 1000 mlproduzieren, zeugt von der technolo-gischen Relevanz des bottelpack Sys-tems. Um diese Effektivität zu errei-chen, werden Planung, Inbetrieb-nahme und Wartung der Anlagen in-dividuell auf die Kundenbedürfnisseausgerichtet. Diese enge Kundenbe-treuung gehört mit zu den wichtigs-ten Erfolgsfaktoren der rommelag.Dazu gehören Remote-Diagnostics-Systeme für die Fernwartung ge-nauso wie Kundenschulungen im ei-genen bottelpack Training Centeroder weltweit beim Kunden vor Ortdurch eigens qualifiziertes Service-personal. Nicht zuletzt dank ihrerSchwesterfirmen für Lohnherstel-lung – der Holopack in Deutschlandund der Maropack in der Schweiz –kann rommelag ein ausdifferenzier-tes Produkt- und Serviceportfolio beiFlüssigverpackungen einschließlichImpfstoffverpackungen anbieten. Sofinden Kunden Unterstützung beiVersuchs- und Lohnabfüllungen überdie Prozessentwicklung, Qualifizie-rung und Validierung bis hin zur Aus-
lagerung ihrer bottelpack Anlagenzur Produktion bei Holopack.
Eigenschaften vonKunststoffen
Kunststoffmaterialien bringen fürHersteller und Anwender von Infusi-onsbehältnissen und Ampullen ei-nige Vorteile mit sich. Heutige Kunst-stoffe erlauben dünne Wandstärkenbei sowohl hoher Bruchsicherheit alsauch guter Kollabierbarkeit, so dasseine Belüftung überflüssig wird. Siesind chemisch inert, enthalten keineAdditive, weisen eine geringe Was-serdampfdurchlässigkeit auf undsind nicht nur in Krankenhäusern,sondern auch bei mobilen Einsätzenleicht und sicher zu handhaben. Da-her kommen gerade in Ländern, indenen sich ein flächendeckendes Ge-sundheitswesen erst entwickelt, umdie Bevölkerung mit qualitativ hoch-wertigen Produkten zu versorgen,bottelpack Anlagen zunehmendzum Einsatz. Nicht ohne Grund sindChina und Indien zu wichtigen Ab-satzmärkten der rommelag gewor-den. Dabei sind die Kunststoff-Ver-packungen umweltfreundlich undEntsorgung oder Recycling erfolgenrückstandsfrei. Die bottelpack Tech-nologie ermöglicht es, solche reinenMaterialien weiterzuverarbeiten,ohne dass zusätzlich aktive Additivebenötigt würden.
Weitere Informationen:rommelag Kunststoff-MaschinenVertriebsgesellschaft mbHRudolf GeissMayenner Str. 18-2071332 Waiblingen (Germany)Tel: +49 (0)172 26 62 927e-mail: [email protected]
TechnoPharm 4, Nr. 4, 222–223 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 223Spektrum

„Bio on Demand“Vom Konzept bis zur ersten Charge: ein schneller, offener und kosteneffizienter Weg zurOptimierung der zukünftigen Marktposition
Da die globale Nachfrage nach be-zahlbaren Medikamenten steigt,wächst der Druck auf Pharma- undBiotechunternehmen, ihre Produk-tion zu überdenken. NNE Pharma-plan sieht hier einen Bedarf an klei-neren, flexibleren Biotech-Anlagenund hat als Antwort auf diese Anfor-derungen das „Bio on Demand“-Kon-zept entwickelt. Ein flexibles Kon-zept, dass ein weites Portfolio vonAnwendungen abdeckt und mit ei-nem vorhersehbaren Zeitplan undBudget, von den ersten Konzeptions-und Planungsschritten bis zur Pro-duktion begleitet.
NNE Pharmaplans „Bio on de-mand“-Anlagen basieren auf einerReihe von einfach einsetzbaren Modu-len, die dort gebaut werden können,wo auch immer die Herstellungsketteeiner biopharmazeutischen Firmadies fordert. Standardisierte Prozesseund Versorgungsmodule werden in
verschiedenen Varianten kombiniert,um den unterschiedlichen Funktioneneiner modernen Biotechproduktionzu entsprechen. Das Design der stan-dardisierten, funktionalen Module er-möglicht ein hohes Maß an Flexibili-tät. Somit können die Anlagen strate-gisch an die jeweilige Prozess- undAnlagensituation angepasst werdenund die spezifischen technischen, pro-duktionsbezogenen und regulatori-schen Anforderungen erfüllen. Eineflexible Anpassung an geänderteMarktanforderungen ist jederzeitmöglich.
„Bio on Demand“ beinhaltet das En-gineering, die Lieferung der Anlagesowie die entsprechenden Qualitäts-systeme, SOPs (Standard OperatingProcedures) und die Organisation dererforderlichen Qualitätsprüfungen.
Vorteile von „Bio on Demand“sind u. a. ein hohes Flexibilitätslevelfür Produktionsumstellungen und
die schnelle Einführung von neuenProdukten, Multiproduktionsanla-gen, eine offene Prozessarchitektur,optimale Ergonomie und Abläufe,Verkürzung der Bauzeit, schnelleEntscheidungen sowie die Reduzie-rung des Investitionsrisikos. Das fle-xible Konzept ermöglicht es, mit Pro-duktionserweiterungen zu warten,bis sich das Produkt auf dem Marktetabliert hat.
„Bio on demand“ wurde bereitserfolgreich bei der Planung von Bio-techproduktionsanlagen in verschie-denen Ländern weltweit umgesetzt.
NNE Pharmaplan GmbHSiemensstr. 2161352 Bad HomburgTel: +49 6172 8502-100Fax: +49 6172 8502-501E-Mail:[email protected]
Spektrum
Beispiel eines Grundrisses einer „Bio on Demand“-Anlage.

Schweizer Fachmesse für integrierte Logistiklösungen und Verpackungstechnik | Messe Basel | www.packmove.ch
IHRE KONKURRENZ KOMMT. SIE AUCH?PACK&MOVE IN BASEL9. BIS 12. SEPTEMBER 2014
Kompetenzpartner Fachpartner
GRATISTICKET
packmove.ch/free
PM14_Inserat_210x146_d_sc.indd 1 27.05.14 20:37
ecvDie Tablette – Handbuch der Entwicklung, Herstellung und QualitätssicherungAnnette Bauer-Brandl, Wolfgang A. Ritschl†
ISBN 978-3-87193-407-0• € 186,00• 3. vollständig überarbeitete und erweiterte Auflage 2012• 17 x 24 cm, 736 Seiten, gebunden
Berücksichtigt wurden neben den grund legen-den, zum Teil schwer zugänglichen älteren Stan-dardwerken die neuesten wissenschaftlichenArbeiten und die Änderungen der Arzneibücher.Altbewährte Techniken des Tabletteurs werdenebenso behandelt wie etwa Verbesserungen beiden Tablettenmaschinen und deren Instrumen-tierung, bei den Herstellprozessen und Prüfme-thoden oder Entwicklungen bei den Hilfsstoffenund Tablettenarten. Zahlreiche neue Abbildungen und überabeiteteTabellen unterstreichen die Aktualität des mo-numentalen Werkes.
Der Standard seit über 4 Jahrzehnten fürEntwicklung, Herstellung und Qualitätssi-cherungDie 3. Auflage der Monographie „Die Tablette“führt einerseits die Tradition der wegweisendenErstauflage fort, wurde andererseits aber akri-bisch überarbeitet und auf den aktuellen Standder Technik gebracht. So informiert sie denLeser weiterhin umfassend über alle Aspekte derTablettenherstellung, aber auch über wichtigeInnovationen des letzten Jahrzehnts.
Bestellung:Tel. +49 (0)8191-97000 358, Fax +49 (0)8191-97000 293, eMail: [email protected], Leseproben und Inhaltsverzeichnisse – www.ecv.de
ECV · Editio Cantor Verlag
Zielgruppen• Pharmaunternehmen• Auftragshersteller• Forschungs- / Prüflabore• Maschinen- und Anlagen-
bauer• Universitäten und
Fachhochschulen• Behörden

Lounges, Vision Pharma,Innovation FoodEine Retrospektive
Hans-Hermann Letzner . Letzner Pharmawasseraufbereitung GmbH, Hückeswagen
Korrespondenz: Hans-Hermann Letzner, Letzner Pharmawasseraufbereitung GmbH Robert-Koch-Str. 1, 42499 Hückeswagen;e-mail: [email protected]
Aussteller und Besucher
Vom 3. bis 5. Juni 2014 trafen in Stutt-gart 270 ausstellende Firmen undInstitutionen auf 8200 Fachbesucheraus 28 Ländern bei 250 Fachvorträ-gen und 56 Aktionsbühnen. Zum Ver-gleich: 2013 waren es in Karlsruhe260 Firmen und 8000 Fachbesucher.Im Mittelpunkt stand die Darstellungund Vermittlung von Fachwissen,Anwendung, Erfahrung, Kundenori-entierung und Kompetenz der aus-stellenden Firmen. Diese hatten dieMöglichkeiten der mehrschichtigenDarstellung mit Vortrag, Vorführungund Gespräch bei dieser Kommuni-kationsplattform genutzt und sichdamit Kunden und Besuchern inte-ressanter als „messeüblich“ präsen-tiert.
Mit dem Reinheitstechnikpreis„CLEAN!“ des Fraunhofer Institutfür Produktionstechnik und Auto-matisierung IPA, wurden drei Firmenfür herausragende Innovationen beider Erreichung der Sauberkeitsspezi-fikationen ausgezeichnet.
Die Preisträger und deren Beiträ-ge: PartikelXpert „Gebrauchsnor-mal für eine Qualifizierung von Sau-berkeitsuntersuchungen“, Pharma-serv „Wand- und Deckendurchfüh-rung clean-shut – GMP-gerechte Ab-dichtung von Rohren und Strangma-terialien durch Reinraum-Wändeund -Decken“ und Berner Interna-tional „Claire-die neue Generationvon Sicherheitswerkbänken“.
In der „Recruiting Lounge“ wur-den hunderte von Jobs sowie Arbeit-geberpräsentationen aus den Life
Sciences Bereichen angeboten. Vom19. bis 21. Mai 2015 finden deshalbdie drei Veranstaltungen LOUNGES,VISION PHARMA und INNOVATIONFOOD erneut in der Halle 1 derMesse Stuttgart statt.
Partner bei Drees & Sommer undVize Präsident der Interessen-gemeinschaft Pharmabau e.V. (VIP3000) Rino Woyczyk: „Das bewährteKonzept der Lounges fand in diesemJahr wieder einen bemerkenswertenpositiven Anklang. Dass die Messe-halle am neuen Standort in Stuttgarthierfür eine bessere Basis bot als inKarlsruhe, war schnell erkennbar.Wir konnten uns als Drees & Som-mer gut in das Messekonzept einbin-den und waren sehr glücklich, dassmit dem VIP 3000-Areal, in welchemwir unseren Stand hatten, auch eineSonderfläche geschaffen wurde, diehohe Aufmerksamkeit und positivesFeedback bei unseren Kunden undBesuchern erfuhr. Auch bietet dieHalle in Stuttgart enormes Potenzialfür weitere Aussteller und ist somitzukunftsträchtig.“
„Messeunüblich“ wurde auch desÖfteren die Standardfrage des Autors„Was gibt es Neues“ beantwortet mit:„Leider nicht Neues“.
Wasseraufbereitung und„hygienic Design“
Das Risiko einer „Kontamination“mit Öl, welches als Druckmittler-Flüssigkeit für die totraumfreie Üb-ertragung des anstehenden Druckesauf das Messsystem üblicherweisebenötigt wird, vermeidet ARMATU-
RENBAU mit einem neuen komplettverschweißten Plattenfeder-Mano-meter aus Edelstahl. Ölfrei wird übereine mechanische Verbindung dieKraft übertragen, das Messsystemist komplett trocken. Der Membrand-ruckmittler ist aus PTFE. Mit einemDurchmesser von nur 40 mm ist esbesonders kompakt gebaut. Das Sig-nal kann einen elektrischem Ausgangverarbeitet werden. Auch die mo-derne Kupplungsart gemäß „hygienicdesign“ nach der DIN 11864, wirdstandmäßig angeboten, alternativdie herkömmliche Tri Clamp Verbin-dung. Der Zusatzfehler im Vergleichzu einem Druckmittler-manometerbeträgt 0,8 % / 10K.
Bürkert kombiniert jetzt auch dieRobolux-Ventile mit den Steuerköp-fen der Serien Elemente. Damit ent-steht eine umfassende Basis für diedezentrale Automatisierung. Allernötigen Automatisierungsfunktionenbefinden sich im eigentlichen Steuer-kopf, dieser übernimmt alle pneuma-tischen Stell-, Feedback- und Diagno-sefunktionen. Empfohlen wird dieVerwendung eines AS-Interface alsFeldbus-Schnittstelle. Für die Strom-versorgung, Signalrückmeldung undKommunikation wird nur eine Dop-pelader benötigt, welche die SPS mitbis zu 62 Ventilen verbindet. Basie-rend auf der Membranventiltechnikverbinden die Bauteile unabhängigeUmschaltfunktionen für zwei Pro-zesse in einem Gehäuse mit nur einerMembran und einem Stellantrieb.Das kompakte Multiportventil solletwa 40 % weniger Platz brauchenals herkömmliche Ventilverteiler.
Lounges
TechnoPharm 4, Nr. 4, 226–230 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)226 Letzner . Retrospektive Lounges 2014

Neu ist das GEMÜMembranventilSUMONDO, ein regelbares „Single-Use“ Membranventil. Der Single-UseMembranventilkörper verfügt übereine aufgeschweißte Membrane. ZurMontage wird der Ventilkörper ampneumatischen Edelstahlantrieb be-festigt und mittels Rastbolzen verrie-gelt. Nach der Anwendung kann derVentilkörper mitsamt der Membranevom pneumatischem Edelstahl-antrieb getrennt und entsorgt wer-den. Der pneumatischer Edelstahl-antrieb, basierend auf dem Typ 650,verbleibt in der Anlage. Der Ventil-körper ist aus dem Werkstoff PP-R,die Membran aus dem WerkstoffTPE.
Gedacht ist das Ventil auch als Re-gelventil zur Überwachung des TMP(Transmembrandruck) bei Ultrafil-trationsanlagen.
Hager + Elsässer haben einen In-vestor gefunden: die Aquarion AG.Technologisch sieht man insbeson-dere, als sinnvolle Neuerung, denEinsatz einer nachgeschalteten Kon-zentratstufe, um eine Umkehr-osmose-Ausbeute von 85 bis 90 %,unterhalb des Scaling-bereiches zu
erzielen, weil bei den Be-triebskosten der Trink-wassser- und Abwasser-preis der bei weitemgrößte Faktor darstellt.Desweitern wird heraus-gestellt das vom Marktweniger Anlagen „ausder Schublade“ gefragtsind, sondern kunden-spezifische Ausführun-gen.
Letzner Pharmawas-seraufbereitung stelltfür die Vorbehandlungvor Umkehrosmose-Elektrodeionisationsan-lagen eine Enthärtungs-anlage komplett im „hy-gienic design“ vor. Tot-raumfreiheit unter ande-rem auch durch den Ein-satz sehr hochwertigerGemü-Blockventile, Ver-bindungstechnik mit Ste-rilclamps (DIN 11864),
Entleerbarkeit, Gefälle bei der Ver-rohrung, abflammbare Probenahme-stellen zusammengefasst in Augen-höhe und die vollautomatische Auf-heizung auf 90 °C sieht man als ein-
zige Antwort auf dieses – aufgrundder extrem großen Oberflächen –mi-krobiologisch sehr sensiblen Verfah-rens. Auch aufgrund der häufigerenMonitorings auf Pseudomonas Aeru-ginosa wird hiermit eine Lösung an-geboten.
Als Hersteller von CIP-Anlagenund von TOC-Online Geräten ist inder logischen Weiterentwicklung dasLetzAnalyse-CIP entstanden.
Im neuen Draft (Feb. 2014) zumAnnex 15 EU GMP Leitfaden wirddas ADE/PDE Kriterium (acceptabledaily exposure / permitted daily ex-posure) beim Schwerpunkt Rei-nigungsvalidierung explizit genannt.Der PDE ist die Dosis eines Stoffes,bei der bei täglicher Aufnahme überden gesamten Lebenszeitraum keinnegativer Effekt beobachtet wird.Für diese neuen Herausforderungenwerden in Zukunft für die Berech-nung von Rückständen toxikologi-sche Daten benötigt. Zur Beschleuni-gung der Qualifizierung und zur kon-tinuierlichen Überwachung des kom-pletten CIP-Prozesses, entsprechendder PAT (Process Analytical Tech-nologies) Initiative der FDA, wurdedie Neuentwicklung LetzAnalyse-
CIP vorgestellt. Diese be-steht aus einer reinraum-tauglichen fahrbaren Ein-heit, komplett im hygienicDesign, ist dampfsterili-sierbar etc. Zurzeit kön-nen parallel zwei Pro-zesse angeschlossen wer-den um das „Final Rinse“Wasser zu überwachen.Wenn Wasser das Lö-sungsmittel für den FinalRinse ist, kann nach demErreichen einer definier-ten Leitfähigkeit die TOCMessung mit demLetzTOC Gerät erfolgen.Dabei wird der TOC-Wertdirekt durch die Messungdes durch die UV – Oxi-dation entstandenenKohlendioxids ermittelt.Die Messung erfolgt miteinem Laser (NDIR De-tektion).
TechnoPharm 4, Nr. 4, 226–230 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 227Letzner . Retrospektive Lounges 2014
Abb. 1: (Quelle: Bürkert.).
Abb. 2: (Quelle: Gemü.).

Das Prinzip gestattet neben ande-ren Merkmalen auch hohe TOCWerte bis 8.000 ppb zu messen. Istein definierter TOC beim Final Rinseerreicht, erfolgt die vollautomatischeFlaschenabfüllung im geschlossenenSystem. Diese Flaschen können dann,ohne Kontaminationsrisiko, im La-bor per HPLC analysiert werden.
Die Qualifizierung von neuen CIP-Prozessen wird stark beschleunigt,weil die online TOC-Analyse die La-boranalysezeiten für TOC eliminiert.Die Reduzierung des Reinigungsmit-teleinsatz und ein größeres Einspa-rungspotential an Pharmawassersind gegeben.
Die Steuerung erfolgt mit einerSiemens SPS, die Visualisierung undDatendokumentation mit einem In-dustrie-PC. Interessanter Neben-effekt, die TOC Werte vom FinalRinse Wasser können direkt mitübernommen werden. Da beimLetzTOC Gerät insgesamt 7 Ein-gangskanäle zur Verfügung stehen,können auch Proben separat gemes-sen werden, bzw. kann hier der SST(System Suitability Test) und die Ka-
librierung erfolgen,ohne das geschlos-sene System zu öff-nen.
Mettler-Toledo:Vorgestellt wirddas tragbare TOCGerät 450TOC wel-ches eine beson-ders kurze An-sprechzeit hat. DasMessergebnis kannsofort ausgedrucktoder auf einemUSB Stick gespei-chert werden. Da-mit kann in einerPharmawasser-Ringleitung an je-der Entnahmestellegeprüft werden,ohne Verfälschun-gen durch die ma-nuelle Probenahmezu riskieren.
Für den von den Arzneibücherngeforderten Systemeignungstest istein SST Modul als Option erhältlich.Die Leitfähigkeit des zu prüfendenWassers muss kleiner 2μS/cm sein.
PMT stellt die Echtzeit Detektionvon Mikroorganismen zum Beispielin Pharmawasser mittels laserindu-zierter Fluoreszenz vor. Das Einsatz-spektrum reicht von der Vermessungeinzelner Wasserproben, bis hin zurkontinuierlichen Analyse an Pharma-wasseranlagen. Das Monitoring inheißem WFI erfolgt durch die spe-zielle ON 90 Gerätekonfiguration.Die Detektionsgrenze für die Ge-samtkeimzahl beträgt 1 Zelle/100 ml.Als „Nebeneffekt“ zeigen die RMSAnalysatoren auch die nichtbiologi-sche Partikelzählrate an.
Ruland hat eine neue Prüf-methode zur Dichtheitsprüfungzum Beispiel von Anlagenkom-ponenten wie Wärmetauscher vor-gestellt. Dabei handelt es sich umeine Wasserstoff-Dichtheitsprüfung,
da Wasserstoff bes-ser verfügbar ist alsHelium (bisherigesPrüfverfahren). Kon-zentration von 5 %Wasserstoff sindausreichend, > 10ppm können in at-mosphärischer Luftgemessen werden.Anwendungsbei-spiel: Wärmetau-scher, das Gas-gemisch zirkuliertim Kreislauf, auf deranderen Seite befin-det sich die Sonde.Mikrorisse in derSchweißnaht wer-den mit dieser ein-fachen, preisgüns-tigen Methode er-kannt. Innerhalbvon einer Stundekönnen Fehler auf-gedeckt werden. DasVerfahren wird auchzur Leckortung beiRohrleitungen einge-setzt.
Lounges
TechnoPharm 4, Nr. 4, 226–230 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)228 Letzner . Retrospektive Lounges 2014
Abb. 3: (Quelle: Letzner).
Abb. 4: (Quelle: Letzner).

Stilmas hebt hervor, dass mit derÜbernahme von Olsa, ein großesLeistungsspektrum angeboten wird.Vom Vakuummixer ab 2,7 kg bis10000 kg, Lösungsansatz-Prozess-anlagen über Fermenter bis hin zuTrocknungsanlagen für Granulate.Bei der Her-stellung vonWFI ist dieThermokom-pression, inAbhängigkeitvon den ört-lichenStromkosten,wirtschaftli-cher als eine6 KolonnenDestillati-onsanlage.Insbeson-dere dann,wenn dieMöglichkeitbesteht,diese nur miteiner Ionentauscher- Enthärtung zubetrieben und auf die Umkehrosmo-setechnik zu verzichten. Thermo-kompressionsanlagen sind seit Jahr-zehnten insbesondere in den USA be-währt. Der Anteil der Mehrstufen-Druckkolonnendestillationsanlagenwird weltweit auf 70-80 % geschätzt.
WIKA stellt eine trockene Mess-zelle vor (zur Vermeidung des Risikoeiner „Kontamination“ mit Öl) bei
der mit einer Plattenfeder die Kraftübertragen wird. Die Überwachungeines Membranbruches erfolgt durchein angelegtes Vakuum, wenn diesesdurch einen Membranbruch auf-gehoben wird erfolgt eine Signalisie-rung.
Monitoring
CAS stellt für die kontinuierlicheMessung im Isolator das neue mikro-biologische Monitoringsystem CLi-MET CL-99 vor. Eine Durchflussratevon 100 l/min wird kontinuierlichdurch die Probe gezogen und derLuftstrom über eine Agarplatte, zurMessung der Gesamtkeimzahl, gelei-
tet. Der Messkopf befindet sich imIsolator, die weitere Technik außer-halb des Isolators. Visualisiert wer-den können der Status der Messung,der Durchfluss, die Verzögerung so-wie Alarmwerte.
DEHA bietet Reinraum-Monito-ring an. Es werdenPartikelzahlenmittels LaserStreuung gemes-sen. Des Weiterenwerden Klima-parameter, Luft-temperatur, rel.Luftfeuchte unddie Druckdifferenzzwischen denReinraum-Berei-chen erfasst. Neuist der Partikelsen-sor AeroTrak Mo-dell Reihe 6510-VHP mit integrier-ter Vakuumpum-pe. Die Empfind-lichkeit beträgt
0,3μm, wobei bis zu vier Partikelgrö-ßen gleichzeitig erfasst werden kön-nen.
ELPRO: Für GMPApotheken wer-den alle relevanten Klimadaten: Dif-ferenzdruck, Temperatur, relativeLuftfeuchtigkeit, Strömungs-geschwindigkeit oder Partikelkon-zentration kontinuierlich aufgezeich-net und abgespeichert. KomfortableFunktionen zur Datenvisualisierung
Abb. 5: (Quelle: CAS).
IT-ValidierungOhne Umwege zum Ziel.
pragmatisch · verständlich · transparent

sorgen dafür, dass zu jedem Zeit-punkt die aktuellen Bedingungen ge-zeigt werden mittels Visualisierungs-panels, dieser ist für die Wandmon-tage in reinraumgerechtem Designausgeführt. Im Falle einer Abwei-chung kann man vollautomatisch in-formiert werden.
Testo: Für die Qualitätsdaten-dokumentation beim Transport(GDP) ist der neue testo184 Daten-logger. Er erfasst beim Transport dieMessgrößen Temperatur, Feuchteund Schock.
Reinraum und Co
AZO zeigt eine neue „Glovebox“ fürhochsensible Wirkstoffe, bei denender Bediener mit Hilfe von Hand-schuhen in den geschlossenen Innen-raum des Einfülltrichters greifenkann. Über ein Sichtfenster kannman ins Innere der Station schauenund so die Gebinde völlig kontami-nationsfrei entleeren. Ein integriertesSieb verhindert, dass Packmittelresteoder Verklumpungen ins Produkt ge-langen. Diese Speziallösung ermög-lichte die Einschleusung von bis zu300 kg Substanz. Vorgehoben wirdder Bedienerschutz.
Comprei ist spezialisiert auf dieSchulung von Reinraumpersonal, ob„inhouse“ in der tatsächlichen Pro-
duktionsumgebung, in eigenen Trai-ningsräumen oder in mobilen Rein-raumkabinen. Alle Fragestellungenzur Praxis, Sterilabfüllung, Formate,Desinfektion etc. werden behandelt.
IAB zeigt einen komplett funk-tionsfähigen Reinraum mit Schleu-sentür und Möbel aus PP. Neu istdie Tücher-Serie VIPERS.
Nora stellt Bodenbeläge mit elek-trostatischer Ableitfähigkeit (ESD-Schutz) vor. Eine hohe Abriebfestig-keit, glatte, dichte Oberflächen ohneRisse und Fugen sowie gute Rei-nigungs- und Desinfektionseigen-schaften sind die Merkmale.
Sika stellt Wandbeschichtung mitEpoxidharz alternativ zu Wandpa-neelen aus Edelstahl vor.
Zusammenarbeit
Becker, Dorfner, Elpro,Klima Becker, Mach4, Pure11,ReinRaumTechnik, SLKB, STO,TÜV-SÜD, WZBhaben einen Leitfaden für sämtlicherSchleusenparameter mit der Ziel-gruppe „GMP Apotheken“ erstellt. Da-mit soll eine Umsetzung der Apothe-kenbetriebsordnung ApoBetrO von2012 sichergestellt werden.
Beschrieben werden baulichenVarianten, das Bekleidungskonzept,der Schleusenablauf, die Möblierung,das Monitoringsystem über Visuali-sierungspanels und die Reinraumrei-nigung.
Diverses
Netzsch: Mit der neuenRührwerkskugelmühleDelta VITA können Vo-lumina zwischen 15 mlbis 300 ml gemahlenund dispergiert werden.Die Herstellung vonWirkstoffen im Nano-meter-Bereich bis150 nm mit enger Par-tikelgrößenverteilungist möglich.
Watson – Marlowhat den Exklusivvertriebvon Flexicon Anlagen.Vollautomatische Sys-teme werden maß-geschneidert für Vials,Kunststoffflaschen, etc.angeboten. Flexicon Ac-cusil dient dem Trans-port des Mediums imEinwegsystem.
Lounges
TechnoPharm 4, Nr. 4, 226–230 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)230 Letzner . Retrospektive Lounges 2014
Abb. 7: (Quelle: AZO).
Abb. 6: (Quelle: Elpro).

Produkte
TechnoPharm 4, Nr. 4, 231 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 231pharmind . Produkte
TiefziehverpackungsmaschinenMit der R 900 bringt Multivac* einneues Modell seiner Tiefziehver-packungsmaschinen auf den Markt,welches sich besonders für großeProdukte eignet, die eine hohe Pa-ckungskavität benötigen. Die Ma-schine wurde entwickelt, um Mehr-komponentensysteme (OP-Sets)fürden OP-Saal, bestehend aus ge-brauchsfertigen Elementen wie Sprit-zen, Skalpellen, Klemmen, Naht-material etc., zu verpacken. DieseMehrkomponentensysteme werdenzunächst in einer Plastikbox bzw. ei-ner flexiblen Packung verpackt undanschließend in sterile OP-Tücher
eingewickelt. Da-mit das kompletteSet, das bis zu15 cm hoch istund häufig aus 2oder 3 Lagen be-steht, automati-siert auf einerHochleistungs-Tiefziehverpackungs-maschine verpackt werden kann,wurde ein neues Maschinenkonzeptentwickelt, welches einen Tiefzugvon 450 mm ermöglicht. Die Maschi-nenrahmenhöhe liegt deshalb bei1300 mm. Mit der neuen Tiefzieh-technologie kann die Zahl der Fehl-packungen reduziert werden. Auch
das Einlegen der Produkte in die Ver-packung lässt sich mittels Roboternautomatisieren.
Scan App für Pharma-CodesREA Elektronik* stellt ein effizientesund kostenloses Tool zum Entschlüs-seln von Codes zur Verfügung. Mitder PharmaScan App werdenStrich-, Matrix- und QR-Codes aufMedikamentenverpackungen perSmartphone decodiert.
Die App erkennt PPN Codes, dievon den Organisationen IFA und EF-PIA für die Serialisierung vergebenworden sind. Ebenfalls erfasst werdenQR- und Data Matrix Codes, die eineGTIN oder eine NTIN nach GS1 Normenthalten. Pharmafälschungen kön-nen durch die Identifizierung nichtvalidierter Codes erkannt werden.
Die App überprüft die Zeichen ge-mäß den Vorgaben von EFPIA undSecurpharm, übersetzt den Inhalt inKlartext und lokalisiert darüber hi-naus etwaige Fehler in der Daten-struktur. Dadurch ist nicht nur dieKontrolle firmenfremder Codes, son-dern auch die Überprüfung selbst-produzierter Symbole auf korrekteVerschlüsselung möglich. Alle Scan-
Ergebnisse werden in einer Historiechronologisch gespeichert. Die Code-arten werden namentlich benannt,Rohdaten ohne Steuerzeichen wer-den unter „Codeinhalt“ dargestellt.
Die App ist ein nützliches Werk-zeug für den pharmazeutischen Ver-packungs- und Vertriebsprozess: Sieeignet sich z.B. zur Erstellung derLayouts für Primär- und Sekundärver-packungen, zur Erreichung eines Pro-duktions-Auftrags in der Arbeitsvor-bereitung, für Stichproben in der Qua-
litätssicherung oder zur Kontrolle vonProofs in Druckereien. Sie identifiziertArtikel- und Seriennummer, Chargen-bezeichnung, Verfallsdatum und – so-fern vorhanden – auch das Produkti-onsdatum. Die App ist als AndroidVersion im Playstore und als iOS Ver-sion im App Store erhältlich.
Die Tiefziehverpackungsmaschine R 900 ermöglicht die wirtschaftlicheVerpackung von Mehrkomponentensystemen für den OP-Saal.
* Multivac Sepp HaggenmüllerGmbH & Co. KGBahnhofstr. 487787 Wolfertschwendenwww.multivac.de
Die PharmaScan App von REA.
* REA Elektronik GmbHTeichwiesenstr. 164367 Mühltalwww.rea.de

Verlag / PublisherECV · Editio Cantor Verlag für Medizinund Naturwissenschaften GmbHBaendelstockweg 2088326 Aulendorf (Germany)GF/MD: Claudius Arndt,Andreas GerthEingetragen/Registered:Amtsgericht Ulm, HRB 600174Tel. +49 (0) 7525-9400Fax +49 (0) 7525-940 180www.ecv.de
Redaktion / Editorial officeChefredakteur / Editor-in-Chief:Claudius ArndtTel. +49 (0) 7525-940 159Leitende Redakteurin/Managing Editor:Kerstin Jarosch (V. i. S. d. P.)Tel. +49 (0) 8191-98578 12Fax +49 (0) 8191-98578 19e-mail: [email protected]
Anzeigen / AdvertisementsVerantwortlich/Responsible:Lara LehmannTel. +49 (0) 8191-98578 11Fax +49 (0) 8191-98578 19e-mail: [email protected]
Herstellung / ProductionRombach Druck- und VerlagshausGmbH & Co. KGUnterwerkstr. 5, 79115 FreiburgTel. +49 (0) 761-4500-0Fax +49 (0) 761-4500-2122e-mail: [email protected]
Druck / PrintHOLZMANN DRUCK GmbH & Co. KGGewerbestraße 2, 86825 Bad WörishofenTel.: +49-(0) 8247-993 0Fax:: +49-(0) 8247-993 208e-mail: [email protected]
Bezugsbedingungen (gültig ab Januar 2011)Die Zeitschrift erscheint sechsmal pro Jahr und kannvom Verlag, von der Arbeitsgemeinschaft für Pharma-zeutische Verfahrenstechnik e. V. (APV) oder durch eineBuchhandlung (ISSN 2191-8341) bezogen werden.Preise für das Jahresabonnement als Printausgabe ein-schließlich Online-Zugang (inkl. MwSt., zzgl. Versand):Inland: 72,00 €, APV-Mitglieder und Studenten 54,00 €,zzgl. 14,00 € Versand; Ausland (Europa mit VAT Ident.Nr.): 67,29 €, APV-Mitglieder und Studenten 50,47 €,Versandkosten 16,82 €Ausland (Europa ohne VAT Ident. Nr. und weiteresAusland): 72,00 €, APV-Mitglieder und Studenten 54,00 €,Versandkosten 18,00 €Preis für das Einzelheft: 19,00 € (inkl. MwSt., zzgl.Versand). Das Abonnement ist weiter rechtsverbindlich,wenn es nicht mindestens 3 Monate vor Ende desBerechnungszeitraums gekündigt wird.Konten des Verlages: Commerzbank Friedrichshafen(BLZ 651 400 72) 17 080 80;IBAN: DE21 6514 0072 0170 8080 00;SWIFT-BIC: COBADEFF651.Landesbank Baden-Württemberg(BLZ 600 501 01) 4 508 560;IBAN: DE57 6005 0101 0004 5085 60;SWIFT-BIC: SOLADEST.Deutsche Postbank AG(BLZ 600 100 70) 29 487 703;IBAN: DE08 6001 0070 0029 4877 03;SWIFT-BIC: PBNKDEFF600.
ManuskripteManuskripte sind an die Redaktion zu senden. Es werdennur Beiträge zur Erstveröffentlichung angenommen.Die Autoren versichern, dass sie allein berechtigt sind,über die urheberrechtlichen Nutzungsrechte an ihrenArbeiten einschließlich etwaiger Bilder, Tabellen etc. zuverfügen und dass keine Rechte Dritter verletzt werden.Sie versichern außerdem, dass keine Doppelpublikation(Veröffentlichung auch in anderen Zeitschriften)beabsichtigt ist bzw. erfolgt. Mit der Annahme desManuskriptes zur Veröffentlichung räumt der Autor demVerlag das ausschließliche, zeitlich, räumlich und in-haltlich unbeschränkte Recht zur Veröffentlichung, Ver-vielfältigung, Verbreitung und öffentlichen Wiedergabe inallen Sprachen und Ländern ein, einschließlich desRechts zur Speicherung in und Nutzung durch Daten-banken jeder Art (Online, auch Internet, und Offline)sowie der weiteren Vervielfältigung zu gewerblichenZwecken im Wege des fotomechanischen oder einesanderen Verfahrens.
Urheber- und VerlagsrechteSämtliche in dieser Zeitschrift veröffentlichten Beiträgegenießen urheberrechtlichen Schutz. Kein Teil der Zeit-schrift darf außerhalb der engen Grenzen des Urheber-rechtsgesetzes ohne schriftliche Einwilligung des Ver-lages in irgendeiner Form vervielfältigt, verbreitet odersonst verwertet werden oder in eine von Maschinenverwendbare Sprache übertragen oder übersetzt werden.Insbesondere ist jede Digitalisierung, Speicherung undNutzung in und durch elektronische Datenbanken jederArt untersagt.
HaftungDer Inhalt dieses Heftes wurde sorgfältig erarbeitet.Dennoch übernehmen Autoren, Beirat, Redaktion undVerlag für die Richtigkeit von Angaben sowie für even-tuelle Satz- oder Druckfehler keine Haftung.
WarenzeichenDas Fehlen des Symbols ® nach Namen bedeutet nicht,dass der Name nicht durch Warenzeichen geschützt ist.
SonderdruckeVon Veröffentlichungen erhält der Korrespondenzautor20 Sonderdrucke in Form guter Fotokopierqualität ohneBerechnung. Weitere Sonderdrucke oder Ausführungenin höherwertiger Form oder Aufmachung können gegenÜbernahme der Kosten beim Verlag bezogen werden.
TechnoPharm® ist eine eingetragene Marke
Terms of subscription (valid from January 2011)The journal is published bimonthly beginning in 2012and can be obtained from the Publisher, from the Ar-beitsgemeinschaft Pharmazeutische Verfahrenstechnike. V. (APV) or via bookstores (ISSN 2191-8341).Rates per annum for the print + online subscription(including VAT, plus postage):Germany: 72.00 €, APV members and students 54.00 €,plus postage 14.00 €;Abroad (Europe with VAT ID number): 67.29 €, APV mem-bers and students 50.47 €, plus postage 16,82 € (surface mail);Abroad (Europe without VAT ID number and othercountries): 72.00 €, APV members and students 54.00 €,plus postage 18.00 € (surface mail);Price for single copy: 19.00 € (including VAT, plus postage).A subscription will continue unless cancelled with threemonths’ notice prior to the end of the invoiced period.Publisher’s bank accounts:Commerzbank Friedrichshafen(Bank Code 651 400 72) Account no. 17 080 80;IBAN: DE21 6514 0072 0170 8080 00;SWIFT-BIC: COBADEFF651.Landesbank Baden-Württemberg(Bank Code 600 501 01) Account no. 4 508 560;IBAN: DE57 6005 0101 0004 5085 60;SWIFT-BIC: SOLADEST.Deutsche Postbank AG(Bank Code 600 100 70) Account no. 29 487 703;IBAN: DE08 6001 0070 0029 4877 03;SWIFT-BIC: PBNKDEFF600.
ManuscriptsManuscripts have to be sent to the editorial office. Onlypreviously unpublished papers are eligible for publicati-on. Authors affirm that they are the sole owners of thecopyright to their manuscripts including any figures,tables, etc., if applicable, and that they do not infringe anyrights of third parties. They further affirm that no doublepublication (additional publication in other journals) isintended or will occur. The acceptance of a manuscriptfor publication implies that the author assigns to thePublisher the copyright to the paper whereby the Pu-blisher acquires the exclusive right – unrestricted withregard to time, territory and contents – of publication,reproduction, distribution and public communication inany languages and countries, including the right of sto-rage and use in databases of any kind (online includinginternet and offline) and the right of reproduction forcommercial purposes by photomechanical or other me-ans.
Copyright and the publisher’s rightAll articles published in this journal are protected bycopyright. No part of this journal may be reproduced,distributed or otherwise exploited in any form or trans-ferred into a machine-readable language or translatedbeyond the narrow scope of the Copyright Act withoutthe Publisher's consent in writing. In particular, any kindof digitizing, storage or utilization in and by means ofdatabases is prohibited.
LiabilityThe contents of this issue have been carefully compiled. Ne-vertheless, the authors, editorial advisory board, editorial officeand Publisher do not assume any liability for the correctness ofinformation or for typographical errors or misprints.
TrademarksThe absence of the symbol ® after any name does notimply that this name is not under trademark protection.
ReprintsThe corresponding author receives 20 reprints of a pu-blished article in good photocopy quality free of charge.Additional reprints or reprints in higher quality form ordesign can be obtained from the Publisher against pay-ment.
TechnoPharm® is a registered trademark
TechnoPharm 4, Nr. 4, 232 (2014)© ECV . Editio Cantor Verlag, Aulendorf (Germany)232 Impressum / Masthead
Impressum / Masthead

APVNEWS 04 • 2014
Nachrichten und Mitteilungen
APV – Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e. V.
MAKING SCIENCE WORK
Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e.V.Gemeinnütziger wissenschaftlicher VereinInternational Association for Pharmaceutical Technology
APV NEWS 04_2014_APVnews TP 26.06.2014 15:41 Seite 1

APV NEWS 4-2014
APV NEWS – Aus der Geschäftsstelle
Vom 30. September bis 2. Oktober 2014 dreht sich in Nürnbergauf der TechnoPharm wieder alles um „Pharma. Manufacturing.Excellence“. Knapp drei Monate vor der Veranstaltung werfenwir in den APV news einen ersten exklusiven Blick ins Fachpro-gramm.
Fachforum TechnoPharmHier tauschen sich Pharma-Produktionsspezialisten über dieneuesten Entwicklungen aus. Highlights sind das Forum zur Se-rialisierung für Pharmahersteller, eine Vortragsreihe des Deut-schen Verpackungsinstituts und mehrere Vortragsreihen zuorodispersiblen und festen Arzneiformen sowie zu GMP, GDPund innovativen Verpackungslösungen.
Fachforum „Focus Cleanroom“Auch in diesem Jahr gibt es wieder ein Expertenforum mit spe-ziellen Vorträgen rund um die Planung, den Bau und den Betriebreiner und reinster Räume. Highlights sind ein Vortrag von Bo-ehringer Ingelheim zum Vergleich von RABS und Isolatoren sowieVorträge zu den aktuellen behördlichen Anforderungen an Rein-räume.
Sonderforum zum ExplosionsschutzEinen Nachmittag lang dreht sich alles um effizienten Explosions-schutz. Es gibt Fachvorträge zum Thema Brand- und Explosions-schutz mit anschließender Podiumsdiskussion und einerexklusiven Guided Tour präsentiert durch die Fachzeitschrift Ver-fahrenstechnik. Ebenfalls sehenswert: die Liveexplosionen imMessepark.
Innovations AwardBereits zum siebten Mal verleiht der Vogel Business Verlag den Innovation Award. Prämiert werden am Abend des ersten Mes-setags die innovativsten Apparate und Verfahren, u. a. in den Kategorien Abfüll-/Verpackungstechnik und Pharmatechnik/Rein-raumtechnik. Am zweiten Messetag gibt es erstmals 90-minütige Rundgänge, in denen die Gewinner sowie dieShortlist-Platzierten ihre Produkte vorstellen.
Fachforum „Serialisierung für Pharmahersteller“Die Uhr tickt: Ab dem Jahr 2017 stehen alle Pharmahersteller in der Pflicht, Verpackungen verschreibungspflichtiger Medika-mente sowohl mit einer eindeutigen Serialisierung als auch mit einem Erstöffnungsschutz zu versehen. Die Vortragsreihe wirddurch den Hüthig Verlag organisiert und gibt am ersten Messetag den Betreibern die nötigen Informationen, die Richtlinie ef-fizient umzusetzen.
Fachforum „Feststoff Förderung“Nicht selten kommen in der Pharmaindustrie empfindliche Schüttgüter zum Einsatz, die einen sensiblen Umgang erfordernund schonend und entmischungsfrei von A nach B transportiert werden müssen. Das Fachforum des Konradin Verlags gibt amersten Messetag einen umfassenden Überblick über die derzeit angebotenen Technologien.
Mehr Informationen zum Fachprogramm unter: www.technopharm.de/fachprogramm
Ansprechpartner für Aussteller Ansprechpartner für Referenten im FachforumPhillip Blass Dr. Martin BornhöftTel +49 (0) 9 11. 86 06-82 31 Tel +49 (0) 61 31. 97 69 [email protected] [email protected]
Blick ins Fachprogramm der TechnoPharm 2014
Termin jetzt schon vormerken: Die TechnoPharm 2014 glänzt miteinem reichhaltigen Fachprogramm.
APV NEWS 04_2014_APVnews TP 26.06.2014 15:41 Seite 2

APV NEWS – Aus dem Vereinsleben
APV NEWS 6-2013
Lokale Gruppen
Mittwoch, 23. Juli 2014
Lokale APV-Gruppe Rhein-Main ab 19:30 Uhr. Ort wird noch bekanntgegeben.
Weitere Informationen erhalten Sie bei Cathrin Pauly.
Donnerstag, 24. Juli 2014
Lokale APV-Gruppe Oberbayern ab 19:30 Uhr im Restaurant Pardi, Volkartstr. 24, 80634 München, Telefon 089-131850mit dem Themenschwerpunkt Südosteuropa.
Anmeldung erforderlich bis zum 15. Juli 2014 bei Dr. (USA) Julia Schulze-Nahrup.
Montag, 25. August 2014
Lokale APV-Gruppe Bonn/Köln/Aachen ab 18:30 Uhr im Restaurant Rietbrocks Weinhaus (Königstrasse 84, 53115 Bonn)
Anmeldung erforderlich bis zum 15. August 2014 bei Dr. Michael Horstmann.
Mittwoch, 27. August 2014
Lokale APV-Gruppe Nord ab 18:30 Uhr im Hofbräuhaus (Esplanade 6, 20354 Hamburg)
Anmeldung erforderlich bis zum 15. August 2014 bei Birgit Mootz.
Montag, 01. September 2014
Lokale APV-Gruppe Westfalen ab 19:30 Uhr in der Hövels-Hausbrauerei (Hoher Wall 5, 44137 Dortmund).
Anmeldung erforderlich bis zum 25. August 2014 bei Dr. Kathrin Bartscher.
Dienstag, 02. September 2014
Lokale APV-Gruppe Berlin um 19:00 Uhr, Treffpunkt wird noch bekanntgegeben.
Weitere Informationen erhalten Sie bei Dr. Andreas Sachse.
Prof. Gerhard Ross verstorben
Die APV nimmt Abschied von ihrem langjährigen Mitglied, ehemaligem Vorstandsmitglied und Träger der APV Medaille HerrnProf. Gerhard Ross, der am 15. Mai 2014 im Alter von 89 Jahren verstorben ist.
Die APV wird ihn und sein Wirken in dankbarer, dauernder Erinnerung behalten.
APV NEWS 04_2014_APVnews TP 26.06.2014 15:41 Seite 3

APV NEWS 4-2014
APVnews – Infos aus der Hochschule
European Journal of Pharmaceutics and Biopharmaceutics87 (2014) 1–18Nanotoxicology applied to solid lipid nanoparti-cles and nanostructured lipid carriers – A syste-matic review of in vitro dataSlavomira Doktorovova, Eliana B. Souto, Amélia M. Silva
abstractSolid lipid nanoparticles (SLN) and nanostructured lipid car-riers (NLC) were developed as alternative to other colloidalcarriers. They were designed to overcome lipid nanoemul-sions and liposomes in stability and ability to control therelease of an encapsulated substance, and at the sametime to be better tolerated than polymeric nanoparticles.Since the patenting of SLN discovery, large amount of databecame available on the behaviour of these systems invitro. SLN/NLC have many prerequisites to be a well tolera-ted carrier – the currently available data seem to confirm it,but there are also some contradictory results. In thisreview, we collected the available data from cytotoxicity,oxidative stress and hemocompatibility studies in vitro andanalysed their outcomes. We also provide a summary ofthe available data in a form of reference table.
European Journal of Pharmaceutics and Biopharmaceutics87 (2014) 19–29Surfactants, not size or zeta-potential influenceblood–brain barrier passage of polymericnanoparticlesNadine Voigt, Petra Henrich-Noack, Sarah Kockentiedt,Werner Hintz, Jürgen Tomas, Bernhard A. Sabel
abstractNanoparticles (NP) can deliver drugs across the blood–brain barrier (BBB), but little is known which of the factorssurfactant, size and zeta-potential are essential for allo-wing BBB passage. To this end we designed purpose-builtfluorescent polybutylcyanoacrylate (PBCA) NP and imagedthe NP’s passage over the blood-retina barrier – which is amodel of the BBB – in live animals. Rats received intrave-nous injections of fluorescent PBCA-NP fabricated by mini-emulsion polymerisation to obtain various NP’s compositi-ons that varied in surfactants (non-ionic, anionic, cationic),size (67–464 nm) and zeta-potential. Real-time imaging ofretinal blood vessels and retinal tissue was carried out within vivo confocal neuroimaging (ICON) before, during and
after NP’s injection. Successful BBB passage with subse-quent cellular labelling was achieved if NP were fabricatedwith non-ionic surfactants or cationic stabilizers but notwhen anionic compounds were added. NP’s size and char-ge had no influence on BBB passage and cell labelling. Thistransport was not caused by an unspecific opening of theBBB because control experiments with injections of unla-belled NP and fluorescent dye (to test a ‘‘door-opener’’effect) did not lead to parenchymal labelling. Thus, neitherNP’s size nor chemo-electric charge, but particle surface isthe key factor determining BBB passage. This result hasimportant implications for NP engineering in medicine:depending on the surfactant, NP can serve one of twoopposite functions: while non-ionic tensides enhancebrain up-take, addition of anionic tensides prevents it. NPcan now be designed to specifically enhance drug deliveryto the brain or, alternatively, to prevent brain penetrationso to reduce unwanted psychoactive effects of drugs orprevent environmental nanoparticles from entering tissueof the central nervous system.
European Journal of Pharmaceutics and Biopharmaceutics87 (2014) 279–289Mixing and transport during pharmaceuticaltwin-screw wet granulation: Experimental ana-lysis via chemical imagingAshish Kumar, Jurgen Vercruysse, Maunu Toiviainen,Pierre-Emmanuel Panouillot, Mikko Juuti,Valérie Vanhoorne, Chris Vervaet, Jean Paul Remon, KristV. Gernaey, Thomas De Beer, Ingmar Nopens
abstractTwin-screw granulation is a promising continuous alterna-tive for traditional batch high shear wet granulation(HSWG). The extent of HSWG in a twin screw granulator(TSG) is greatly governed by the residence time of the gra-nulation materials in the TSG and degree of mixing. Inorder to determine the residence time distribution (RTD)and mixing in TSG, mostly visual observation and particletracking methods are used, which are either inaccurateand difficult for short RTD, or provide an RTD only for afinite number of preferential tracer paths. In this study,near infrared chemical imaging, which is more accurateand provides a complete RTD, was used. The impact ofchanges in material throughput (10–17 kg/h), screw speed(500–900 rpm), number of kneading discs (2–12) and
What’s hot in European Journal ofPharmaceutics and Biopharmaceutics?Stefanie Funke, Ludwig-Maximilians-Universität, D-München
APV NEWS 04_2014_APVnews TP 26.06.2014 15:41 Seite 4

APVnews – Infos aus der Hochschule
APV NEWS 2-2012
Impressum:
Redaktion
Prof. Dr. Jörg Breitkreutz (Präsident)Dr. Martin Bornhöft (Leiter Geschäftsstelle)
Vorstand der APV
Dr. Rainer Alex · Dr. Hermann Allgaier ·Prof. Dr. Jörg Breitkreutz · Dr. HubertusFolttmann · Prof. Dr. Achim Göpferich · Prof. Dr. Heribert Häusler · Dr. Hermann P.Osterwald · Dr. Andreas Rummelt
Arbeitsgemeinschaft für Pharmazeu tischeVerfahrenstechnik e. V. (APV) Kurfürstenstraße 5955118 Mainz (Germany)Telefon +49 6131 9769-0Telefax +49 6131 9769-69e-mail: [email protected]://www.apv-mainz.de
Verlag
ECV · Editio Cantor Verlag für Medizinund Naturwissenschaften GmbHBaendelstockweg 2088326 Aulendorf, Germany
Telefon +49 7525 940-0Telefax +49 7525 940-180
e-mail: [email protected]://www.ecv.de
Alle Rechte bei APV e. V.All rights reservedPrinted in GermanyJede Form des Nachdrucks verboten
Druck
Holzmann Druck GmbH & Co. KGGewerbestr. 286825 Bad Wörishofen, Germany
Satz
Arbeitsgemeinschaft für Pharmazeu tischeVerfahrenstechnik e. V. (APV) Kurfürstenstraße 5955118 Mainz (Germany)
stagger angle (30–90) on the RTD and axial mixing of thematerial was characterised. The experimental RTD curveswere used to calculate the mean residence time, meancentred variance and the Péclet number to determine theaxial mixing and predominance of convective over disper-sive transport. The results showed that screw speed is themost influential parameter in terms of RTD and axialmixing in the TSG and established a significant interactionbetween screw design parameters (number and staggerangle of kneading discs) and the process parameters(material throughput and number of kneading discs). Theresults of the study will allow the development and valida-tion of a transport model capable of predicting the RTDand macro-mixing in the TSG. These can later be coupledwith a population balance model in order to predict granu-lation yields in a TSG more accurately.
European Journal of Pharmaceutics and Biopharmaceutics87 (2014) 357–365That’s cool! – Nebulization of thermolabile pro-teins with a cooled vibrating mesh nebulizerSebastian Hertel, Thomas Pohl, Wolfgang Friess, GerhardWinter
abstractDespite contrary reports, heating inside the medicationreservoir was observed for several vibrating mesh nebuli-zers, which may be detrimental when nebulizing biophar-maceuticals. In this study we evaluated different strategiesto reduce reservoir heating during nebulization with a PARIeFlow regarding cooling efficiency, impact on nebulizerperformance and on protein stability after nebulization.Passive cooling was achieved by solution pre-cooling, over-charging of the reservoir with 1 mL additional solution orintermittent nebulization. Active cooling was realized witha micro Peltier element attached to the nebulizer reservoir.Passive cooling was most effective when the reservoir wasovercharged with pre-cooled solution reducing the avera-ge reservoir temperature (T RES AVG) by 8.4 °C. Activecooling enabled nebulization at a constant reservoir tem-perature (TRES) as low as 15 °C. T RES manipulation had alinear impact on nebulizer performance. While the outputrate decreased with decreasing T RES, the inhalable fracti-on increased resulting in an inhalable aerosol rate constantover a large T RES range. The effect on protein stabilitydepended on the susceptibility to thermal stress and waspredicted by Tm values. For lactic dehydrogenase andSM101, both exhibiting a Tm below 60 °C, cooling wasprotective in increasing the residual activity and reducingprotein aggregation. A more thermostable IgG1 did notbenefit from cooled nebulization. Nebulizer cooling is aprerequisite to retain the activity and stability of thermola-bile proteins during vibrating mesh nebulization. It is bestachieved by micro Peltier based active cooling or by simplepassive cooling strategies.
APV NEWS 04_2014_APVnews TP 26.06.2014 15:41 Seite 5

Hersteller/Typ Listenpreis mtl. Rate
Audi A3 Cabrio 103kW/140PS „Vfw“ inkl. Connectivity-Paket, Klimaautomatik, Xenon, Einparkhilfe, Sitzheizung, 17" LM-Felgen etc. 34.265,00 € 399,00 €
Audi A5 Sportback 2.0 TDI 130kW/177PS multitronic inkl. Businesspaket, S line Sportpaket, S line Exterieurpaket, Navi plus etc. 44.168,00 € 449,00 €
Audi A6 Avant 2.0 TDI 130kW/177PS multitronic inkl. Alcantara/Leder, Metallic, Businesspaket, S line Sportpaket, S line Selection, Navi etc. 47.517,00 € 479,00 €
BMW 320d Touring „DW“ EZ: 07-2013 inkl. Metallic, Leder, Automatik, Navi, LM-Felgen, PDC, Xenon, Klimaautomatik, Lichtpaket etc. 28.546,00 € 415,00 €
BMW 520d Touring „DW“ EZ: 07-2013 inkl. Automatik, Leder, Navi, Xenon-Licht, PDC, Panorama-Glasdach, Klimaautomatik, Sitzheizung etc. 30.168,00 € 391,00 €
BMW X3 sDrive 18d 110kW/150PS inkl. Klimaautomatik, PDC, 17" LM-Räder, Lederlenkrad mit Multifunktion, Lichtpaket, Bluetooth etc. 33.529,00 € 425,00 €
Ford Kuga „Titanium“ 1,6 EcoBoost 2x4 110kW/150PS inkl. Metallic, Navigationssystem, Klimaautomatik, Überführungskosten etc. 24.164,00 € 199,00 €
Jaguar XF Sportbrake „Vfw“ 2.2 L Diesel 147kW/200PS inkl. Automatik, Leder, Navigation, Einparkhilfe mit Rückfahrkamera, Sitzheizung etc. 47.790,00 € 349,00 €
Land Rover Range Rover Evoque „Vfw“ TD4 Pure 110kW/150PS inkl. Automatik, Navi, Leder, Winterpaket, Technikpaket, PDC vorn und hinten etc. 39.706,00 € 489,00 €
MINI One Cabrio 72kW/98PS inkl. Radio/CD, Park Distance Control (PDC) hinten, Frontscheibenwaschdüsen und Aussenspiegel beheizt etc. 17.773,00 € 225,00 €
Seat Leon ST FR 2.0 TDI 110kW/150PS DSG inkl. Navigationssystem, Voll-LED-Scheinwerfer, Alcantara/Leder, PDC, Winter-Paket etc. 28.143,00 € 269,00 €
Skoda Citigo 3-Türer 1.0 MPI „Active“ 44kW/60PS inkl. Klimaanlage, Musiksystem, Easy Entry System, Servolenkung, ABS, ESC, ASR etc. 8.890,00 € 89,00 €
Skoda Roomster 1.2 „Active“ 51kW/70PS inkl. Musiksystem Blues, Klimaanlage, Sitzsystem VARIOFLEX, Reifendrucküberwachung etc. 12.466,00 € 135,00 €
Toyota Yaris Hybrid 5-Türer „Club“ 74kW/100PS inkl. Automatik, Multimedia Navigationssystem Toyota Touch, Klimaautomatik, LM-Felgen etc. 16.924,00 € 169,00 €
VW Golf Trendline 3-Türer BMT 1,2l TSI 63kW/85PS 5-Gang inkl. Klimaanlage, Radio/CD, Aussenspiegel elektr. einstell- u.beheizbar etc. 15.075,00 € 159,00 €
VW Tiguan Trend & Fun BMT 2,0l TDI 81kW/110PS 6-Gang inkl. Radio/CD 310, Klimaanlage, ParkPilot hinten, LM-Räder „Portland“ etc. 22.903,00 € 219,00 €
Vfw = Vorführwagen zu Sonderkonditionen, DW = Dienst-/Werkswagen (genannter Listenpreis=Kaufpreis)
Kfz-Leasing: Vorteile für APV-MitgliederDie APV hat für ihre Mitglieder einen Rahmenvertrag mit einem bekannten Leasing-Unternehmen geschlossen. Als Koopera-tionspartner der APV bietet das Unternehmen Leasing von Neu- und Gebrauchtfahrzeugen zu Sonderkonditionen. Alle Markenund Modelle sind lieferbar. Die nachfolgende Tabelle gibt nur wenige aktuelle Beispiele möglicher Modelle und Marken wieder.NEU: Vorführwagen (VFW) aus dem Leasing-Pool und Dienst-/Werkswagen (DW) zu attraktiven Konditionen erhältlich.
Alle Preise in Euro zuzüglich gesetzlicher Mehrwertsteuer. Beschaffung durch die Leasing-Gesellschaft. 36 Monate Laufzeit,15.000 km pro Jahr, Angebote freibleibend. Der Nachlass auf den Listenpreis ist in die ermäßigte Rate einkalkuliert.
Anfragen bitte an [email protected], das Leasing-Unternehmen wird sich dann mit Ihnen in Verbindung setzen.
Leasing und Finanzierung zu günstigen Konditionen sind auch für Investitionsgüter wie Walzenpressen,Verpackungsmaschinen, Laboreinrichtungen etc. über die APV möglich. Sprechen Sie uns an.
JETZT NEU: Leasing auch für andere Investitionsgüter
APV NEWS – Leasing-Highlights zu Sonderkonditionen
APV NEWS 04_2014_APVnews TP 26.06.2014 15:41 Seite 6

Abgelegt auf: Vorstufe:GK:ECV:Satz:Pharmind:PI_2014-07:Anzeigensatz-keine-Druck-PDFs:APV-pi-2014-07-216x303.indd Zuletzt gesichert: 08.07.14 (10:26:17 Uhr)
Skin Forum 14th Annual MeetingPercutaneous penetration -
measurement, modulation and modelling4 to 5 September 2014 · IMG Conference Centre, Prague, Czech Republic · Course No. 6557
A conference organised byThe International Association forPharmaceutical Technology in partnership with Skin Forum
Keynote Speakers:• Gopinathan K. Menon, Ashland Specialty Ingredients, United States• Michael Horstmann, Acino-Pharma AG, Switzerland• Majella Lane, UCL School of Pharmacy, United Kingdom• Georgios Stamatas, Johnson & Johnson, France• Jacob Thyssen, University of Copenhagen, Denmark
Further information at www.apv-mainz.de
Further information at www.apv-mainz.de
Formulating better medicines for childrenMeeting the needs of the children
6th conference of the European Paediatric Formulation InitiativeA conference organised by the International Association for Pharmaceutical Technology in partnership with the European Paediatric Formulation Initiative
17 to 18 September 2014
Athens, Greece · Course No. 6558
Keynote Speakers• Natelle Rakhmanina, Children's National Medical Center• Sandra Klein, University of Greifswald• Sonja Skopp, Merck KGaA• Terry Ernest, GSK• Herbert Wachtel, Boehringer Ingelheim
Preconference workshops• Introduction to developmental pharmacology• Introduction to regulatory framework/quality section of PIPs
Soap box sessions – Poster session – Exhibition
Preliminary conference programme
International Association for Pharmaceutical Technology
IPEC_APV-goes-green_AnzDinA4_Layout 1 06.06.2014 15:49 Seite 2

Abgelegt auf: Vorstufe:GK:ECV:Satz:TechnoPharm:TP_2014-04:Anzeigensatz-keine-Druck-PDFs:Bohle-TP-2014-04-216x303.indd Zuletzt gesichert: 10.07.14 (02:20:49 Uhr)
DIE TECHNOLOGISCHE ZUKUNFT HAT BEGONNEN:
Bohle Uni Cone BUC® produktiver, praktischer, präziser
Ihre Vorteile:
• Hohe Ausbeute und Vermeidung von Zwillingsbildung beim Coaten
• Trocknen, Granulieren und Coaten ohne Umbau im selben Gerät
• Bequeme Bedienung durch seitlich angebrachte Düsen
• Optimierte Prozessführung mittels innovativer PAT-Methoden (Partikelgröße und Feuchte)www.lbbohle.de
Die Alternative
zum herkömmlichen
Wurster-Verfahren
Herkömmliches Wurster-Verfahren
Wegweisende Bohle Uni Cone BUC®-Technologie
Bohle-BUC_D_A4_1.indd 1 23.04.14 17:31





























