Embed Size (px)
Citation preview

52 W . Walter und G . Randau Bd. 122 Liebigs Ann. Chem. 722, 52-79 (1969)
Uber die Oxydat ionsprodukte von Thiocarbondureamiden , XIX 1)
Thioharnstoff-S-monoxide von Wolfgang Waiter und Gert Randau
Aus dem Chemischen Staatsinstitut, Institut fur Organische Chemie der Universitat, 2 Hamburg
Eingegangen am 17. Juli 1968
Bei der Oxydation des Thioharnstoffs und seiner N-Monosubstitutionsprodukte sind Thio- harnstoff-S-monoxide in der Losung nachzuweisen. Die S-Monoxide der N.N-disubstituierten Thioharnstoffe sind chrornatographisch nachweisbar, lassen sich jedoch nicht isolieren. Dies gelingt bei zahlreichen S-Monoxiden 10 von N.N'-disubstituierten Thioharnstoffen, die sperrige Reste enthalten (vgl. Tab. 2). Bei den Thioharnstoff-S-Oxiden ist die Rotation um eine C-N- Bindung starker behindert als bei den Thioharnstoffen, was aus der Struktur der Verbindungen verstandlich wird. N-Acyl-Thioharnstoffe unterscheiden sich in ihren Oxydationsprodukten von den N-Alkyl- und N-Aryl-Derivaten. Oxidation Producrs of Thioamides, XIX 1) . Thiourea-S-monoxides
Thiourea-S-monoxides can be detected in solution on oxidation of thiourea and N-mono- substituted thioureas. The S-monoxides of N,N-disubstituted thioureas are sufficiently stable for chromatographic detection but cannot be isolated. Isolation is, however, possible with a number of S-monoxides 10 of N,N'-disubstituted thioureas containing bulky groups (compare table 2). Thiourea-S-monoxides exhibit a greater hindered rotation around a C-N-bond than do the thioureas. The oxidation products of N-acylated thioureas differ from those of alkyl and aryl derivatives.
Bei unseren Untersuchungen iiber Thioamid-S-oxide wurden die Substituenten an der Thioamid-Gruppierung X -CS -N( variiert und bisher u. a. neben aliphatischen, aromatischen und cyclischen Thioamiden auch Thiourethane (X = OR, SR) 2.3)
untersucht. Bei der Oxydation dieser Verbindungen mit Wasserstoffperoxid wurde die Bildung von S-Oxiden beobachtet. Die Oxydation von Thioharnstoff in neutraler Losung mit diesem Oxydationsmittel fiihrt dagegen zur ,,Formamidin-sulfinsaure"4), dern S-Dioxids-7) des Thioharnstoffs.
~~ ~
1) XVIII. Mitteilung: W . Walter und M . Sreflen, Liebigs Ann. Chem. 712, 53 (1968). 2 ) W. Walter und K.-D. Bode, Liebigs Ann. Chem. 681, 64 (1965). 3) W , Walrer und K.-D. Bode, Liebigs Ann. Chem. 698, 131 (1966). 4) E. de Barry Barnerr, J. chern. SOC. [London] 97, 63 (1910). 5 ) 5a) R. Kiramura, J. pharmac. SOC. Japan [Yakugakuzasshi] 55, 75 (1935) [C. 1935 11,
13461. - 5b) R. Kiramura, J. pharmac. SOC. Japan [Yakugakuzasshi] 59, 33 (1939) [C. 1939 I, 46071.
6 ) J. Eoeseken, Kon. Akad. Wetensch. Amsterdam, Proc. 39, 717 (1936) [C. 1936 II , 17081; 41, 70 (1938) [C. 1938 11, 12191.
7) R. A. Sullivan und A . Hargreaves, Acta crystallogr. [Copenhagen] 15, 675 (1962).

1969 Oxydationsprodukte von Thiocarbonsaureamiden, XIX 53
Substituierte Thioharnstoffe lassen sich ebenfalls mit Wasserstoffperoxid zu den S-Dioxiden oxydierensb.8). Der Befund, daB bei dieser Reaktion offenbar die Stufe der S-Monoxide durchlaufen wird 9). wie aufgrund der fur Thioamid-S-monoxide chrakteristischen Farbung
mit Eisen(Il1)-chlorid erkannt werden kann, wirft die Frage nach der Existenzfahigkeit von S-Monoxiden substituierter Thioharnstoffe auf. Von den ebenfalls die Thioharnstoff- Gruppierung enthaltenden I-Aryl-thiosemicarbaziden ist bereits bekannt, daB sie rnit Wasser- stoffperoxid unter gleichzeitiger Oxydation der NH -NH-Gruppe stabile S-Monoxide bildenlo.11):
so t f P 2 II
Ar-NH-NH-CS-NH2 - Ar-N=N-C-NH2
N-Monosubstituierte Thioharnstoffe Bei der Einwirkung von Wasserstoffperoxid auf N-rnonosubstituierte Thioharn-
stoffe, jedoch nur in neutraler alkoholischer Losung bei O", laRt sich sofort nach Zu- gabe des Oxydationsrnittels mit Eisen(II1)-chlorid eine blaue bis blaurote Farbung erzielen, die auf das Vorhandensein von S-Monoxiden schl iekn laat. Die gebildeten S-Monoxide von N-Alkyl-, N-Aryl- und N-Acyl-thioharnstoffen reagieren sehr schnell weiter. Relativ lange lassen sich die S-Monoxide von N-[2.6-Dialkyl-phenyI]-thioharn- stoffen rnit dieser Farbreaktion nachweisen, jedoch nicht chrornatographisch. Als Endprodukt der Oxydation wurden bei diesen wie bei N-Alkyl-thioharnstoffen nur die entsprechenden Harnstoffe isoliert und Sulfat-Ionen nachgewiesen.
R ' = CH3, C,H,, CH(CH,),
Disubstituierte Thioharnstoffe
Die Oxydation von N.N-disubstituierten Thioharnstoffen in verdunnter alkoholi- scher Losung bei 0-25" fuhrt zu instabilen S-Monoxiden, die nicht isolierbar sind, sich aber dunnschichtchromatographisch durch ihre Eisen(lI1)-chlorid-Reaktion
8 ) W. Wulter und G . Randau, Liebigs Ann. Chern. 722, 80 (1969), nachstehend. 9) W. Wulrer, Habilitationsschrift Univ. Hamburg 1958.
10) E. Bulka, M . Zimmermann und H . Beyer, Z. Chem. 6 , 416 (1966). 11) E. W. Pluugers und J. Berg, Tetrahedron Letters [London] 1966, 3695.

54 W. Walter und G . Randau Bd. 122
(blauviolette Farbung) nachweisen lassen (Tab. 1). Neben den S-Monoxiden lassen sich jeweils noch die entsprechenden S-Dioxide 8) nachweisen.
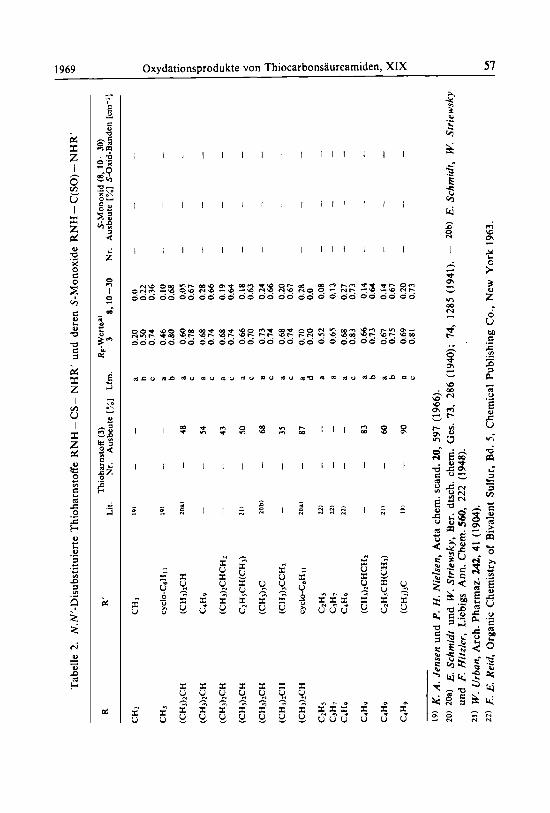
Tabelle 1. RpWerte der Oxydationsprodukte von N.N-disubstituierten Thioharnstoffen R2N - CS - NH2
- _ _ ~ ~ -
R Lit. Laufrnittela) RpWerte Thioharnstoff S-Monoxid
CH3 12) a 0.48 0.20
CH3CHzCHz 12) b 0.75 0.60 CH3CHzCHzCH2 13) a 0.72 0.55
b 0.78 0.62 (CHshCHCH2 b) b 0.78 0.62
8 ) Diinnwhichtchromatographie an Kieselgel GF; Laufmittel a = Chlorolorm/Mcihanol ( 5 : 1). b = Chloroform/ Methanol ( I : 4). b) Nach Lit.12) dargestellt, vgl. Lit.8).
N.N'-Disubshtuierte Thioharnstoffe
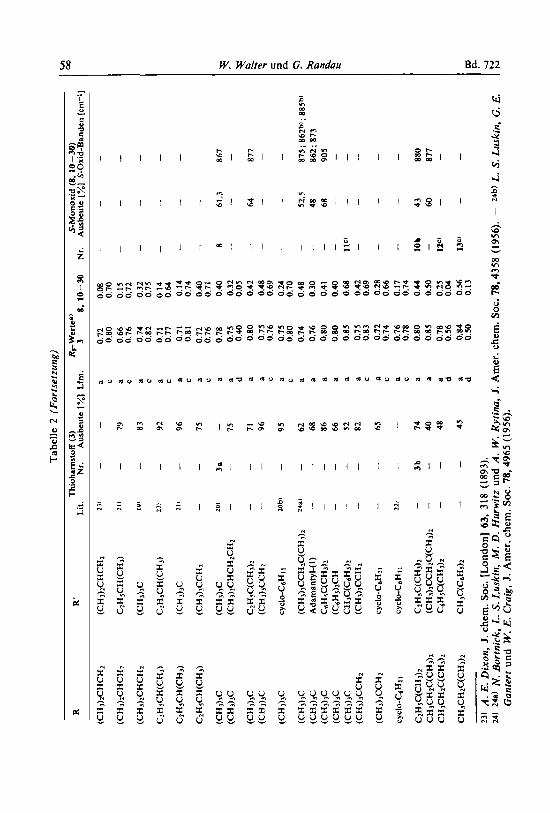
Die N.N'-disubstituierten Thioharnstoffe 3 wurden durch Umsetzung (1) von Iso- thiocyanaten 1 rnit den prirnaren Arninen 2 hergestellt (vgl. Tab. 2, S . 57).
a: R = R ' = (CH,),C R - N = C = S + R'NH2 RNHCSNHR' b: R = R ' = C2HSC(CH,), 1 2 3 C : R = R ' = CcHSC(CH3)z
Id + Id (l) \ J f
d: R = R ' CH3C(C6H5)2
c: R = R ' = (C,H,),C
f : R = R' = (CH3)3C-CH2-C(CI13)2 l e + za ' x l e + 2e
la-c + 2a-c
, la-b + 2d - 3 Niedrigsiedende, wasserlosliche Amine wurden in waRr. Losung, evtl. im Glasauto-
klaven, hohersiedende Amine in Aceton oder Chloroform umgesetzt. - Fur die Untersuchung des sterischen Einjlusses der Substituenten R bzw. R' in 3 auf die Oxydation wurden auch besonders sperrige Gruppen wie tert.-Butyl- (a), Neopentyl- (b) und 2.6-Dialkyl-phenyl- als R und R' in 3 eingefuhrt. Zur Darstellung von N.N'- Di-tert.-alkyl-thioharnstoffen fiihrt man die Reaktion am besten in Petrolather bei Raumtemperatur durch, wobei die substituierten Thioharnstoffe analysenrein aus- kristallisieren.
12) 0. Wulluch, Ber. dtsch. chern. Ges. 32, 1872 (I 899). 13) H . Beyer und R . Giebelrnann, J. prakt. Chern. [4] 20, 263 (1963).

1969 Oxydationsprodukte von Thiocarbonslureamiden, XIX 55
Die Darstellung von beidseitig sperrig substituiertem 3 ist schwierig. a.a-Diphenyl-athylamin (Zd) reagiert zwar noch mit 1 a und 1 b (vgl. Tab. 2). aber nicht mehr mit a.a-Diphenyl-athyl- senfol ( ld ) . Triphenylmethylisocyanat (le) Iaint sich nach Bacon und Kb'chling 14) mit einem UberschuD primaren aliphatischen Amins ohne Losungsmittel bei Raumtemperatur zur Re- aktion bringen; aliphatische tert.-Alkylamine reagieren ebenfalls, nicht aber festes Trityl- amin (2e), auch nicht bei Zusatz von Losungsrnitteln. Auch rnit Thiophosgen lassen sich 2d und e nicht zu 3d bzw. e umsetzen. Andere vergebliche Versuche zur Darstellung von 3e haben Bredereck und ReiJl5) durchgefiihrt.
N-Aryl-N'-alkyl-substituierte Thioharnstoffe lassen sich aus aromatischen Iso- thiocyanaten rnit aliphatischen Arninen in guten Ausbeuten darstellen (Tab. 2). Aliphatische Senfole wie 1 a reagieren dagegen nur langsam mit 2.6-Dialkyl-anilinen. So liefert z. B. l a rnit 4a nach 4 Stunden bei 120" irn Glasautoklaven nu r 2% 5. Bei 160" entsteht 6a (56% d . Th.).
6a
Das gleiche Produkt tritt auch bei Reaktion von 4a mit anderen aliphatischen Senf- olen auf.
N.N'-Diaryl-thioharnstoffe erhalt man noch glatt aus aromatischen Isothiocyanaten und aromatischen Aminen. Selbst N.N'-Bis-[2.6-dialkyI-phenyl]-thioharnstoffe (6a - c) sind so praparativ zuganglich (Tab. 2).
7a-c 4a-c 6a-c a: it = C H ~ ; b: I< = c21i,; c: ti = (cI13)2c€r
Die benotigten lsothiocyanate 7a- c sind aus den entsprechenden Aminen nach der durch Hodgkins und Mitarbeitern 16) modifizierten Methode von Kaluza17) zuganglich.
14) R . C . R . Bacon und J. Kochling, J. chern. SOC. [London] 1965, 5366. 15) H . Bredereck und E. Rei' Chem. Ber. 81, 426 (1948). 16) 1. E. Hodgkins und M . G. Errlinger, J. org. Chemistry 21, 404 (1956); J . E. Hodgkins,
W. P . Reeves und Y-T. Liu, J. Amer. chem. SOC. 83, 2532 (1961); J . E. Hodgkins und W. P. Reeves, J. org. Chemistry 29, 3098 (1964).
17) L. Kaluza, Mh. Chern. 33, 363 (1912).

56 W . Walter und G . Randau Bd. 722
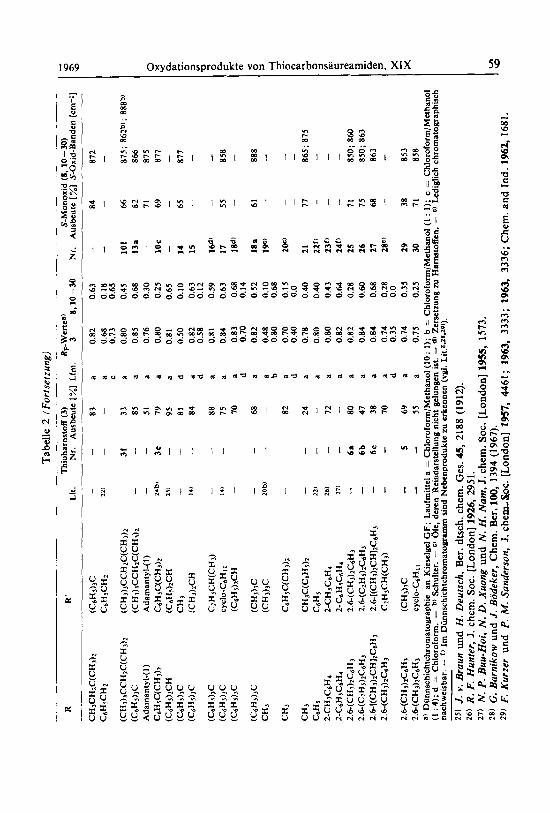
S-Monoxide Oxydiert man N.N'-disubstituierte Thioharnstoffe mit Wasserstoffperoxid, so
farben sich die Produkte mit Eisen(l1I)-chlorid tiefblauviolett 9) . Die S-Monoxide dieser Reihe sind erheblich stabiler als N-mono- und N.N-disubstituierte Thioharn- stoff-S-monoxide, wie sich papier- oder dunnschichtchromatographisch zeigen 1aBt. Bei der Oxydation von N.N'-Di-tert.-butyl-thioharnstoff (3a) mit Wasserstoffperoxid in Pyridin laBt sich in geringer Ausbeute N.N'-Di-tert.-butyl-thioharnstoff-S-monoxid9) (8) isolieren.
s 1I:O: I I - ( C €iJ 3C - N H- C -NH C ( C H 3 ) 3
S II
( C H3) ,C - N I I - C - N I I - C ( C I I,)
3a 8: x = s o 9: x = <)
Verbindung 8 zerfallt selbst bei -20" in einigen Wochen unter Schwefel-Ausscheidung und Bildung des entsprechenden Harnstoffs 9. Charakteristisch fur 8 ist die intensive Absorptionsbande im IR-Spektrum bei 867 cm-1 (S-Oxid-Bande) 18).
Fur die Isolierung von 8 wurden optimale Bedingungen (Oxydation mit Uber- schuB an Wasserstoffperoxid bei 0-20" in verdunnter alkoholischer Losung) erarbeitet und auf viele N.N'-disubstituierte Thioharnstoffe angewendet. Die Stabilitat der N.N'-disubstituierten Thioharnstoff-S-monoxide 10 zeigt eine deutliche Abhangigkeit von den Substituenten R und R'. In Chloroform und Methylenchlorid sind sie bei
S SO I1 11.0, II
3 11-NH-C-NII-R' - K - N I I - C - N H - R ' 10
niedriger Temperatur einige Zeit chromatographisch nachweisbar (Tab. 2). Sind R und R' in 10 primare oder sekundare Alkyl-Gruppen, so zersetzen sie sich beim Ein- engen auch bei niedriger Temperatur oder bei Zugabe von Petrolather (30-50") unter Schwefelausscheidung und Bildung der entsprechenden Harnstoffe (vgl. S . 73). N.N'-Di-tert.-alkyl-thioharnstoff-S-monoxide lassen sich dagegen isolieren (Tab. 2). Offenbar stabilisieren sperrige Gruppen die Thioharnstoff-S-monoxide.
Acylierte Thioharnstoffe
Bei offenkettigen und cyclischen Thioamiden hatte sich gezeigt, daB sich Acyl- Derivate hinsichtlich der Neigung zur Bildung von S-Monoxiden charakteristisch
18) W. Walrer und H.-P. Kubersky, Liebigs Ann. Chern. 694, 70 (1966). - Die IR-Spektren von N.N'-Di-tert.-butyl-thioharnstoff (7) und seinem S-Monoxid (8) werden voraussicht- lich in der Dokumentation der Molekiil-Spektroskopie, Verlag Chemie GrnbH, Weinheirn, veroffentlicht.
Tab
elle
2.
N.N
’-D
isub
stit
uier
te T
hioh
arns
toff
e R
NH
- C
S- N
HR
’ und
der
en S
-Mon
oxid
e R
NH
- C
(S0)
- N
HR
’
-
a b a b 48
a
54
a
43
a
50
a
68
a
35
a
87
a d a a a
83
a b 60
a b
90
a C
-
C
C
C C
C
C
-
-
-
C
C ..
R=-
Wer
tea’
8.10
-30
‘3
- .-
0.20
0.
50
0.74
0.
46
0.80
0.
60
0.78
0.
68
0.74
0.
68
0.74
0.
66
0.70
0.
73
0.74
0.
68
0.74
0.
70
0.20
0.
52
0.65
0.
68
0.83
0.
66
0.73
0.
67
0.75
0.
69
0.8
I
._
~~ 0.
0 0.
22
0.36
0.
10
0.68
0.
05
0.67
0.
28
0.66
0.
19
0.64
0.
18
0.63
0.
24
0.66
0.
20
0.67
0.
28
0.0
0.08
0.
13
0.27
0.
73
0.14
0.
64
0.14
0.
67
0.20
0.
73
S-M
onox
id (
8, 1
0-30
) N
r.
Au
sbeu
tc [%] S
-Oxi
d-B
ande
n [c
m-’
1 -
- -
-
19)
K. A
. Jen
sen
und
P. H
. Nie
lsen
, A
cta
chem
. sca
nd. 2
0, 591 (1966).
20)
2W
E. S
chm
idr
und
W. S
trie
wsk
y, B
er. d
tsch
. che
m. G
es. 7
3, 286 (1940); 74, 1285 (1941). - 2
Ob)
E
. Sch
mid
t, W
. Srr
iew
sky
21)
W. U
rban
, Arc
h. P
harm
az. 2
42, 41 (1904).
22)
E. E
. Rei
d, O
rgan
ic C
hem
istr
y of
B
ival
ent
Sulfu
r, B
d. 5
, C
hem
ical
Pub
lishi
ng C
o., N
ew Y
ork 1963.
und
F. H
irzl
er,
Lie
bigs
Ann
. Che
m. 5
60, 222 (1948).
Tab
elle
2 (
For
rser
zung
) ~
__
- -
-.
- ._
_-
--
-
__
T
hioh
arns
toff
(3)
RrW
ertc
a’
S-M
onox
id (8
. 10
-30)
N
r.
Aus
beut
e [%
I L
fm.
3 8.
10-3
0 N
r.
Aus
beut
e [%
I S-
Oxi
d-B
ande
n [c
m-1
1 .. _.
-
a
79
a
83
a
92
a
96
a
75
a a 75
a d
71
a 96
a
95
a
62
a 68
a
86
a 66
a
52
a 82
a
65
a a
74
a 40
a
48
a d 45
a d C C C
C C C
-
C C C C
-
C
0.72
0.
80
0.66
0.
76
0.74
0.
82
0.71
0.
77
0.71
0.
81
0.72
0.
76
0.78
0.
75
0.40
0.
80
0.75
0.
76
0.75
0.
80
0.74
0.
76
0.80
0.
80
0.85
0.
75
0.83
0.
72
0.74
0.
76
0.78
0.
80
0.85
0.
78
0.56
0.
84
0.50
0.08
0.
70
0.15
0.
72
0.32
0.
75
0.14
0.
64
0.14
0.
74
0.40
0.
71
0.40
0.
32
0.05
0.
42
0.48
0.
69
0.24
0.
70
0.48
0.
30
0.41
0.
40
0.68
0.
42
0.69
0.
28
0.66
0.
17
0.74
0.
44
0.50
0.
25
0.W
0.
56
0.13
23)
A. E
. Dix
on, J
. che
m. S
OC
. [Lo
ndon
] 63, 3
18 (
1893
). 24
) N. Bo
rtni
ck, L
. S. L
uski
n, M
. D. H
urw
irr un
d A
. W
. Ryr
ina,
J. A
mer
. che
m. S
OC.
78,
4358
(19
56).
-
Gan
rerr
und
W. E
. Cra
ig, J
. Am
er. c
hem
. SO
C. 78
,496
5 (1
956)
. 24b) L
. S. L
uski
n, G
. E.
Tab
elle
2 (
Fo
rtse
rzu
ng
)
R R’
T
hioh
arns
toff
(3)
R
F-W
erte
a)
S-M
onox
id (
8. 1
0-30
) Li
t. N
r. A
usbc
utc
[%I
Lfm
. 3
8.10
-30
Nr.
A
usbe
ute
[%I
S-O
xid-
Ban
dcn
[ern
-:] -
-
83 - 33
85
51
79
95
81
84
88
75
70
68
-
82
24
72
80
47
38
70
69
55
- -
a a a a a a a d a d a a a d a a b a d a a a a a a a a d a a C
0.82
0.
68
0.73
0.
80
0.85
0.
76
0.80
0.
81
0.50
0.
82
0.58
0.
8 I
0.84
0.
83
0.70
0.
82
0.48
0.
80
0.70
0.
40
0.78
0.
80
0.80
0.
82
0.82
0.
84
0.84
0.
74
0.35
0.
74
0.75
0.63
0.
18
0.65
0.
45
0.68
0.30
0.25
0.
65
0.10
0.
63
0.12
0.
59
0.63
0.
68
0.14
0.
52
0.10
0.
68
0.15
0.
0 0.
40
0.40
0.
43
0.64
0.
28
0.60
0.
68
0.28
0.
0 0.
35
0.25
84
-
66
82
71
69
65
- - -
55
-
61
-
- - 77
- -
-
71
75
68
-
38
71
872
-
875;
862
b); 8
88b)
86
6 87
5 87
7
877 - - - 858
-
888 - - - 865;
875
-
- - 850;
860
85
0; 8
63
863 - 853
858
a) D
unnr
hich
tchr
omat
ogra
phic
an
Kie
wlg
cl G
F;
Lau
fmitt
el a
= C
hlor
ofor
m/M
ctha
nol (
10:
I);
b =
Chl
orof
orm
/Met
hano
l (I :
I);
c =
Chl
orof
orm
/Met
hano
l (I
: 4)
; d
= C
hlor
ofor
m. -
b) S
chul
tcr.
- C)
ole
. dcr
cn R
eind
arst
cllu
ng n
icht
gel
ungc
n is
t. -
d) Z
crw
tzun
g N
Ham
stof
fen.
- 5
) Lc
digl
ich
chro
mat
ogra
phis
ch
nach
wci
sbar
. -
f) I
rn D
tinns
chic
htch
rom
atog
runr
n si
nd N
cben
prod
ukte
N c
rkcn
nen
(vgl
. Li
t.aJa
s91)
.
25) J.
v. B
rau
n u
nd H
. Deu
rsch
, Ber
. dts
ch. c
hern
. Ges
. 45,
218
8 (1
912)
. 26)
R. F
. Hun
ter,
J. c
hern
. SO
C. [L
ondo
n] 1
926,
295
1.
27) N. P
. Bu
u-H
oi,
N. D
. Xuo
ng u
nd N
. H. N
om, J
. che
rn. S
OC
. [Lon
don]
195
5, 1
573.
28
) G
. Bar
nik
ow
und
J. B
odek
er,
Che
m. B
er. 1
00.
1394
(196
7).
291
F. K
urz
er u
nd P. M
. San
ders
on,
J. c
hem
,!&oc
. [L
ondo
n] 1
957,
446
1; 1
963,
333
3; 1
963,
333
6; C
hern
. and
Ind
. 196
2,16
81.

60 W. Walter und G . Randau Bd. 122
von Alkyl- und Aryl-substituierten Thioamiden unterscheiden3.30.31). N-Acetyl-thio- harnstoff (31) in Pyridin ergibt rnit Wasserstoffperoxid 3.5-Diacetamino-1.2.4-thia- diazo1 (32)9.3*); fuhrt man die Reaktion in alkoholischer Losungaus, so lant sich sofort nach Zugabe des Oyxdationsmittels rnit Eisen(II1)-chlorid eine Blaufarbung beobach-
31 32
ten, aber kein S-Monoxid isolieren. Als Oxydationsprodukt wird 32 gefunden. Das gleiche gilt fur die Oxydation von N-Benzoyl-thioharnstoff (33) mit Wasserstoff-
NH-CO-CBH, A r 0 0 1 1 t- C6H,-CO-NH-CS-NII2 .% C6H5-CO-NH-CO-NH2
33 34 If J$
I c o - C 6 € I ,
35
peroxid (in geringer Ausbeute) zu N-Benzoyl-harnstoff (34). Die Oxydation von 33 rnit Peressigsaure fuhrt dagegen zu dem bereits bekannten33) 3.5-Dibenzamido-l.2.4- thiadiazol (35).
Eigenschaften der Thioharnstoff-S-monoxide Thioharnstoff-S-monoxide geben rnit Eisen(ll1)-chlorid eine tiefblauviolette Farbung. D a
mono- und disubstituierte Thioharnstoffe nicht mit Eisen(ll1)-chlorid reagieren, ist diese Farbreaktion wie die Reaktion mit Jod/Azid-LGsungM) zum Nachweis der S-Monoxide, speziell auf Diinnschichtplatten, gut geeignet. Silbernitrat wird in ammoniakalischer Losung durch Thioharnstoff-S-monoxide in der Warme zu Silber reduziert (schwarz). Thioharnstoffe ergeben unter diesen Bedingungen augenblicklich Silbersulfid (braunlich). Substituierte Thioharnstoffe Iassen sich von den S-Monoxiden auch aufgrund von RF-Werten unterscheiden (vgl. Tab. 2) .
In unpolaren Losungsmitteln wie Hexan, Cyclohexan und Tetrachlormethan sind S- Monoxide schlecht loslich, in Chloroform, Methanol und Athano1 hingegen gut. - Sperrige aliphatische Substituenten, wie die Adamantyl-(1)-Gruppe, bewirken eine geringere Polaritat, d a die lipophilen Substituenten die polare Thioharnstoff-S-monoxid-Gruppierung offenbar abschirmen.
Analog den Thioamid-S-oxiden und den I-Arylazo-thioformamid-S-oxidenlO.11) las- sen sich Thioharnstoff-S-monoxide mit Schwefelwasserstoff bei Raumtemperatur zu den entsprechenden Thioharnstoffen reduzieren, wobei Schwefel gebildet wird.
30) W . Walter und K.-D. Bode, Liebigs Ann. Chem. 660, 74 (1962). 31) W . Walter und G . Randau, Liebigs Ann. Chem. 681, 5 5 (1965). 32) W . Walter, Angew. Chem. 70, 371 (1958). 33) F. Kurzer, J . chem. SOC. [London] 1955, 2288. 34) W. Walter, Liebigs Ann. Chem. 633, 35 (1960).

1969 Oxydationsprodukte von Thiocarbonsaurearniden, XIX 61
Die Farblosen, kristallinen Thioharnstoff-S-monoxide (Tab. 2) sind thermisch labil. Die reinen Substanzen zersetzen sich zu Harnstoff und Schwefel, und zwar je nach Substituenten
so S It It
R-NH-C-NH-R ' + H2S --D R-NH-C-NH-R ' + S + H 2 0
verschieden schnell (vgl. S. 75); daneben bildet sich teilweise der Thioharnstoff zuriick. Ba- sen und Sauren sowie Erwiirrnung auf > 40" beschleunigen die Zersetzung.
0 S Z0 ti It R-NH-C-NH-R ' 4 R-NH-C-NH-R ' + S + RNH-C-NHR'
Der Nachweis der stabilen S-Monoxide gelingt nicht bei Oxydation in saurem Milieu. Die stabilen S-Monoxide lassen sich in der Regel nicht mit Peressigsaure zu den S-Trioxiden35) oxydieren. Eine Ausnahrne bildet 8, dessen S-Trioxid als Methanol-Addukt isolierbar ist35). Anstatt instabiler S-Monoxide entstehen dagegen rnit Peressigsaure glatt die S-Trioxide3s). Die Stabilitat der isolierbaren S-Monoxide durfte daher zurnindest z. T. darauf beruhen, daB der Angriff durch die sperrigen Gruppen an der SO-Gruppe erschwert ist.
IR- und NMR-Spektren der Thioharnstoff-S-monoxide Die IR-Spektren der Thioharnstoff-S-monoxide zeigen eine charakteristische, inten-
sive Bande im Bereich 850-905 cm-1, die hauptsachlich der SO-Valenzschwingung zugeordnet wird I*) (vgl. Tab. 2).
Bei einigen Verbindungen ist die Bande aufgespalten (vermutlich Kristalleffekte; vgl. Tab. 2). Die bathochrome Verschiebung der S-Oxid-Bande der Thioharnstoff-S-monoxide gegenuber den Thioamid-S-oxiden (900- 1000 cm-1) und den Sulfinen (> 1000 cm-I)M) kann als ein lndiz fur eine Erniedrigung der Bindungsordnung, d. h. eine starkere Polarisierung der SO- Gruppe angesehen werden. Die ,,Thioamid-B-Bande" 19) des Thioharnstoffs ist dagegen bei den entsprechenden S-Monoxiden hypsochrom verschoben, was durch eine Verstarkung des Doppelbindungscharakters der CN-Bindung erklart werden kann 19).
0 , B
II I s-00 s - 0 s-0" I - - c
>N N< >N N< :N/'\N: >N/e\ N: @4'\
"?=" y, - A B C D
Diese Befunde sprechen fur eine Beteiligung der polaren Grenzforrneln B und C an der Mesomerie der Thioharnstoff-S-monoxide. Irn Einklang hiermit ist die Steigerung der Stabili- tat der N.N'-Dialkyl-thioharnstoff-S-monoxide mit steigendern (+)-I-Effekt der Substituenten (primare < sekundare < tertiare Alkyl-Gruppen), jedoch 1st anzunehrnen, daR hierbei zu- satzlich sterische Effekte eine Rolle spielen. Ein weiterer Hinweis fur die Beteiligung der Grenz- forrneln B-D an der Mesomerie der S-Monoxide ist der Befund, daR bei 8 die Weiteroxy- dation ohne Eliminierung des Schwefels rn6glich ist'*), wahrend die Sulfine, bei denen die
35) W. Wulrer und C. Randuu, Liebigs Ann. Chern. 722, 98 (1969). nachstehend. 36) C. Opitz, Angew. Chern. 79, 161 (1967); Angew. Chem., internat. Edit. 6, 107 (1967).
62 W. Walter und G . Randau Bd. 122
Ylid-Grenzformel (analog A) zur Beschreibung der Mesomerie herangezogen wird37), sich nicht zu den Sulfenen oxydieren lassen 3 6 ~ 3 8 ) .
Der Angriff des elektrophilen Oxydationsmittels erfolgt bei den Sulfinen offenbar am Kohlenstoff-Atom und fuhrt zur Schwefel-Eliminierung. Bei den Thioharnstoff-S-monoxiden sind die Grenzformeln B und D durch den Elektronendruck der N-Atome gegenuber A so stark begunstigt, daR der Angriff des elektrophilen Oxydationsmittels am Schwefel, d. h. die Bildung von S-Dioxiden 8) und S-Trioxiden35), maglich wird.
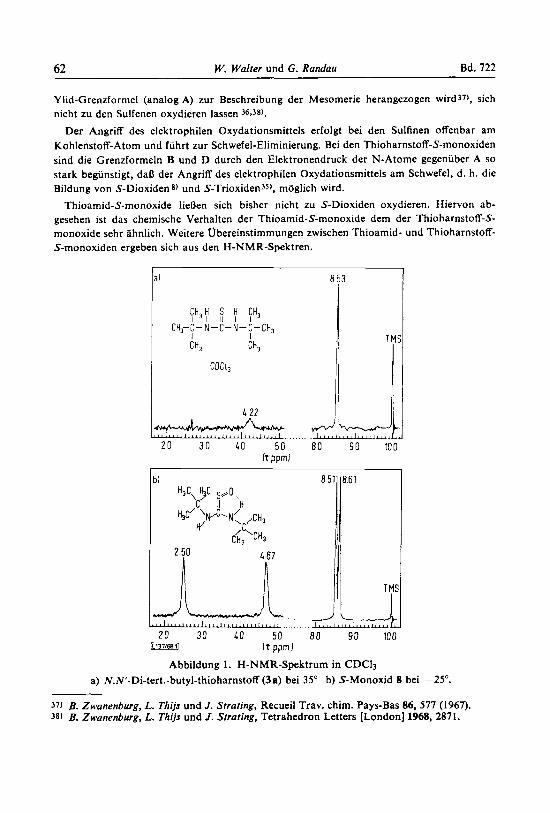
Thioamid-S-monoxide IieDen sich bisher nicht zu S-Dioxiden oxydieren. Hiervon ab- gesehen ist das chemische Verhalten der Thioamid-S-monoxide dem der Thioharnstoff-S- monoxide sehr ahnlich. Weitere Ubereinstimmungen zwischen Thioamid- und Thioharnstoff- S-monoxiden ergeben sich aus den H-NMR-Spektren.
CH,H S H CH, I I I I I I
CH2-C- N-C-N-C-CH, I I CH, 4 TM
I CDCI,
2 0 30 40 50 8 0 90 100 hpprnl
bl 85111861
2 ?I 4 67 1/11
I f pprn I Abbildung 1. H-NMR-Spektrum in CDCI3
a) NN-Di-tert.-butyl-thioharnstoff (3a) bei 35" b) S-Monoxid 8 bei -25".
37) B. Zwanenburg, L. Thus und J. Strating, Recueil Trav. chim. Pays-Bas 86, 577 (1967). 38) B. Zwanenburg, L. Thijs und J. Strating, Tetrahedron Letters [London] 1968, 2871.
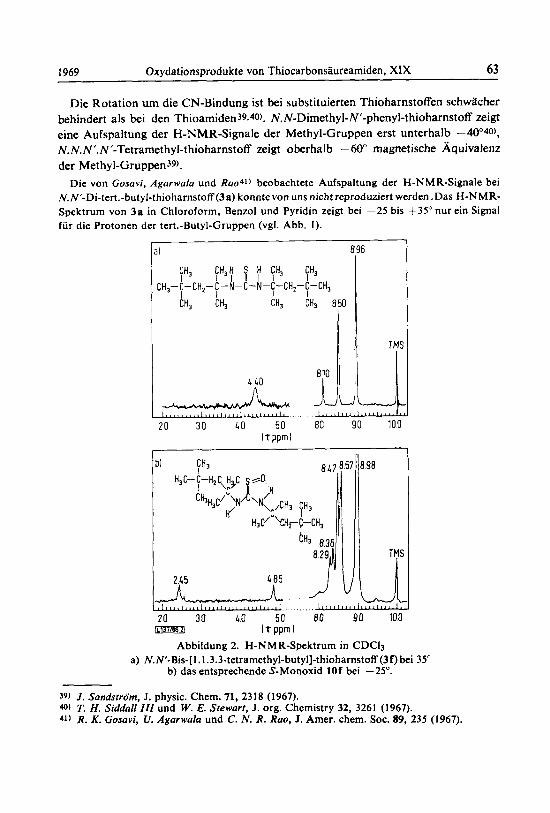
I969 Oxydationsprodukte von Thiocarbonsaureamiden, XlX 63
Die Rotation urn die CN-Bindung ist bei substituierten Thioharnstoffen schwacher behindert als bei den Thioamiden39.40). N.N-Dimethyl-N'-phenyl-thioharnstoff zeigt eine Aufspaltung der H-NMR-Signale der Methyl-Gruppen erst unterhalb -4O040), N.N.N'.N'-Tetramethyl-thioharnstoff zeigt oberhalb -60" magnetische Aquivalenz der Methyl-Gruppen39).
Die von Gosavi, Agurwala und Rao41) beobachtete Aufspaltung der H-NMR-Signale bei N.N'-Di-tert.-butyl-thioharnstoff (3a) konntevon uns nicht reproduziert werden .Das H-NMR- Spektrum von 3a in Chloroform, Benzol und Pyridin zeigt bei -25 bis +35" nur ein Signal fur die Protonen der tert.-Butyl-Gruppen (vgl. Abb. I ) .
8 96 1 1
20 30 LO 50 80 90 100 ITppml
20 30 4 0 50 80 90 100 m I T ppm I
Abbildung 2. H-NMR-Spektrum in CDC13 a) N.N'-Bis-[1.1.3.3-tetramethyl-butyl]-thioharnstoff (3f)bei 35"
b) das entsprechende S-Monoxid 10f bei -25".
39) J . Sandstriim, J. physic. Chem. 71, 2318 (1967). 40) T. H. Siddull 111 und W. E. Stewart, J. org. Chemistry 32, 3261 (1967). 41) R. K. Gosuvi, U. Agorwula und C. N. R. Roo, J. Amer. chem. SOC. 89, 235 (1967).

64 W. Walter und G . Randau Bd. 122
Bei den Thioharnstoff-S-monoxiden ist aufgrund der starkeren Beteiligung polarer Grenzformeln an der Mesomerie sowie infolge der Ausbildung intramolekularer Wasserstoff-Briicken, wie sie bei S-Oxiden von Thioamiden, Thio- und Dithiocarbon- saureestern nachgewiesen wurden I), mit einer Erhohung der Aktivierungsenergien der Rotation urn die CN-Bindung zu rechnen. Die H-NMR-Spektren der Thioharn- stoff-S-monoxide bei - 25 bis 0" zeigen deutlich die magnetische Nichtaquivalenz der
' ( 2 ) ' 1 (E) B I I H(2) (E)R
Substituenten an N-l und N-3 (Abb. 1-2). die sich mit einer Fixierung der SO- Bindung in der Thioharnstoff-Ebene in Richtung auf ein N-Atom erklaren lafit. Die magnetische Anisotropie der S-Oxid-Gruppe wirkt sich am starksten auf die in a- Stellung zum N befindlichen CH3- oder CHz-Gruppen aus, wahrend die weiter ent- fernt stehenden Gruppen (z. B. y-CH3 in lOf, Abb. 2b) magnetisch aquivalent sind.
Fur eine intramolekulare Wasserstoff-Briicke, die eine zusatzliche Begunstigung der fixierten Konfiguration bewirken wiirde, sprechen je zwei breite Signale bei stark unterschiedlicher chemischer Verschiebung fur die NH-Protonen. 1st fur die partielle Doppelbindung N( 1) -C(2), der die SO-Bindung benachbart ist, unter Annahme einer Wasserstoff briicke die E-Konfiguration'Q) vorauszusetzen, so ist fur die Bindung C(2)-"(3), die der SO-Gruppe abgewandt ist, aus sterischen Griinden die Z-Kon- figuration am giinstigsten. Die E.E-Konfiguration bzw. ein sich schnell einstellendes Gleichgewicht zwischen Z.E- und EX-Konfiguration ist jedoch nicht auszuschliekn.
Wir danken dem fonds der Chemischen Industrie fur die Farderung dieser Arbeit und den Furbenfubriken Buyer fur die uberlassung von Chemikalien.
Beschreibung der Versuche Die Schmelzpunkte wurden mit dem Leitz-Heizmikroskop bestimmt. - Die H-NMR-Spek-
tren wurden mit dem Spektrometer Varian A 60 aufgenommen. - Die Dlinnschichtchromatoo- grumme wurden auf Kieselgel GFzsr (Merck) hergestellt. - Zur Messung der IR-Spektren wurden die Gerate 137, 21 und 421 der Firma Perkin-Elmer verwendet.
Isothiocyanate Allgemeine Durstellung nach Hodgkins16): 0.2 Mol primores Amin wurden mit 28 ccm Tri-
athylamin in 10 ccm Benzol gemischt. Unter Riihren und Kuhlen mit Eis/Kochsalz wurden 13.2 ccm Schwefelkohlenstoff langsam zugetropft. Danach lie13 man in 30 Min. auf Raum- temperatur kommen. Nach 1-3 Tagen bei 5" bis -20" wurde das ausgefallene Carbamat abfiltriert und mit Ather fast farblos gewaschen. Das trockene Carbamat wurde in 150 ccm
42) Zur Nomenklatur s. J. E. Blackwood, C. L. Gladys, K . L. Loening, A. E. Petrarca und J. E. Rush, J. Amer. chem. SOC. 90, 509 (1968).

1969 Oxydationsprodukte von Thiocarbonsaureamiden, XIX 65
Chloroform gelost, dann wurden 28 ccm Triathylamin und unter Riihren bei 0" 20.4 ccm Chlorameisensuureuihylesier zugefiigt. Die Reaktionslosung, aus der sich meistens etwas feste Substanz abschied, wurde 1 Stde. bei Raumtemperatur geriihrt, rnit 60 ccm 2 n HCI versetzt und weitere 30 Min. geriihrt, bis sich zwei klare Phasen gebildet hatten. Die Chloroformphase wurde 2mal rnit je 50 ccrn Wasser gewaschen, rnit Natriumsulfat getrocknet, eingedampft und das lsothiocyanat i. Vak. fraktioniert.
Tabelle 3. Isothiocyanate R-N=C=S c____- - - _- I___
R Sdp./Torr (Schmp.) Nr. Ausbeute Analysen
N S
)43)
64 % 60 -65"/ 1 5 Ber. 10.84 24.79 Gef. 10.38 24.40
82 (173-174") Ber. 7.25 16.58 Gef. 7.43 16.41
- - C6H&(CH,)z Mb) l c 80 125 - 127"jI 4 Lit.: 83"/0.7
2.6-(CH~)zCsH3 la 94 127 - 1 3O0/I4 Ber 8.58 19.63 (1 5 - 16') Gef. 8.15 19.23
~ . ~ - ( C Z H S ) & H ~ 7b 73 130- 134"/11 Ber. 7.32 16.75 Gef. 8.06 16.97
2.6-[(CH3)2CH]2C6H34' 7 C 45 143--146O/14 Ber. 6.39 14.60 Gef. 6.18 14.72
CH3C(C6H5)za) I d 32 123 -127"/0.2 - -
a) Dithiocarbarnat nicht kristallin, ohne Isolierung umgesetzt.
N.N'-Disubstituierte Thioharnstoffe Allgemeine Vorschrifi (vgl. Tab. 2 ) . - 0.05 --0.4 Mol Isoihiocyanai wurden rnit der aqui-
valenten Menge primarem Ainin in 10 ccm Losungsmittel (Petrolather, Chloroform, Aceton oder Wasser) pro 0.1 MolO.5-2 Stdn. bei Raumtemperatur oder unter Erhitzen zum Sieden umgesetzt. Nach dem Abkiihlen wurde i. Vak. bis zur beginnenden Kristallisation eingeengt. Die bei -20" bis 5' auskristallisierten Thioharnstoffe wurden umkristallisiert, evtl. rnit Aktiv- kohle. N'-teri.-Alkyl-subsiituierie N-Methyl-ihioharnsro~e N-Methyl-N'-teri.-butyl-thioharnstoff. - Aus ieri.-Buiylsenfol ( 1 a) und 33 proz. waBr.
Methylurnin-LCisung. Ausbeute 77 % farblose Kristalle vom Schmp. I 13" (Petrollther [30 -SO"]/ Essigester = 5 : 2; Lit, ?Ob) 1 1 lo). N-Meihy~-N'-~a.a-dimeihylbenzyll-ihioharnsioff. - Aus Meihylsenfol und a.a-Dimethyl-
benzylamin (212) (dargestellt nach Lit.45)) in Chloroform durch 7stdg. Erhitzen. Farblose Kri- stalle (82 %) vom Schmp. 128 - 130" (Petrolather [30-5O0]/Essigester = 2 : 1). - IR-Banden (Bezeichnung nach Lit.19)): B 1543, 1490, C 1375,1355, D 1267 cm-1.
c l lH16N~S (208.3) Ber. C 63.42 H 7.74 N 13.45 S 15.38 Gef. 63.11 7.49 13.40 15.24
43) E. I. Du Pont de Nernours u. Co. (Erf. M. Paulshock und J. C. Watrs), Amer. Pat. 3 203 970 v. 31.8. 1965 [C. A. 64, 615 g (1966)J
44) W . Walter und K . Wohlers, unveroffentlicht. 45) J. W. Baker und C. K . Inzcld, J. chem. SOC. [London] 1927. 261. - . . . . - ..
5 Liebigs AM. Chcrn. Bd. 722

66 W. Walter und G. Randau Bd. 722
N-Methyl-N'-[a.a-diphenylathylJ-thioharnstofl - Aus MethylsenfoI und a.a-Diphenylathyl- arnin (2d) (dargestellt nach Lit.46) aus a.a-Diphenylathylchlorid und fliiss. Ammoniak bei Raumtemperatur unter Druck, Sdp.o.2 102- 103.5"). Hellgelbe Nadeln (24%) vom Schmp. 124-127" (Petrolather[30-50°]/Essigester = 10: I ) . - IR-Banden: B 1553,1495, C 1265, D 1070 (?), 1020 cm-1.
C16HlaNzS (270.4) Ber. c 71.07 H 6.71 N 10.36 s 11.86 Gef. 70.95 6.65 10.31 11.71
sek.-AIkyl-substituierte Thioharnstoffe N-Isopropyl-N'-n-butyl-thioharnstoff. - Aus Isopropylsenfol und n-Butylarnin in Chloroform.
Farblose Kristalle (54%) vom Schmp. 61 -65" (Benzol/PetrolBther). - la-Banden: B 1570, C 1242, 1210, D 1060 cm-1.
CsHlsN2S (174.3) Ber. C 55.12 H 10.41 N 16.07 s 18.40 Gef. 55.07 10.42 16.04 18.19
N-Isopropyl-N'-isobutyl-thioharnstoff. - Aus Isopropylsenfol und lsobutylarnin in Chloro- form. Farblose Kristalle (43 %) vom Schmp. 81 -83" (Cyclohexan/Petrolather). - IR- Banden: B 1585, C 1235, D 1072 cm-1.
CaHlgNzS (174.3) Ber. C 55.12 H 10.41 S 18.40 Gef. C 54.69 H 10.16 S 18.24
N-lsopropyl-N'-neopentyl-thioharnstoff. - Aus Isopropylsenfol und Neopentylarnin47') in Chloroform. Hellgelbe Nadeln (35%) vom Schmp. 11 I" (Petrolather).
C9H20NzS (188.3) Ber. C 57.40 H 10.70 S 17.03 Gef. C 57.84 H 10.59 S 16.66
N-n-Buty.1-N'-isobutyl-thioharnstoff. - Aus n-Butylsenfol und Isobutylamin in Petrolather (30-50") bei -5'. Farblose Kristalle (83%) vom Schmp. 41 -44".
C ~ H ~ O N Z S (188.3) Ber. C 57.40 H 10.70 N 14.88 S 17.03 Gef. 56.98 10.52 14.89 16.92
tert.-Alkyl-substituierte Thioharnstoffe
N-lsobutyl- N'-tert.-butyl-thioharnstoff. Wasser. Farblose Kristalle (83 %) vom B 1565, 1548, C 1355, D ll00cm-1.
C9H20NzS (188.3) Ber. Gef.
- Aus tert.-Butylsenfol ( l a ) und Isobutylarnin in Schmp. 101 - 102.5" (Petrolather). - IR-Banden:
C 57.40 H 10.70 N 14.88 S 17.03 57.68 10.74 15.19 17.18
N-sek.-Butyl-N'-tert.-butyl-thioharnstoff. - Aus 1 a und sek.-Buiylamin in Wasser. Farblose Kristalle(96%)vomSchmp. 139" (Methanol/Wasser; Lit.21) 132" fur (+) N-sek.-Butyl-N'-tert.- butyl-). - IR-Bunden: B 1545, C 1340 crn-1.
C9H20NzS (188.3) Ber. C 57.40 H 10.70 S 17.03 Gef. C 57.04 H 10.60 S 16.80
N-sek.-Butyl-N'-neopentyl-thioharnstoff. - Aus sek.-Butylsenfol und Neopentylaniin i n Wasser. Farblose Kristalle (75%) vom Schmp. 118-122" (Athanol). - IR-Bunden: B 1550, C 1360, D 1105 cm-1.
Cl0H22N2S (202.4) Ber. C 59.35 H 10.96 S 15.85 Gef. C 58.85 H 10.85 S 15.90
46) M. Brander, Recueil Trav. chim. Pays-Bas 37, 67 (1918). 47) G. Vexlearschi, C. R. hebd. Seances Acad. Sci. 228, 1655 (1949).

1969 Oxydationsprodukte von Thiocarbonsaureamiden, XIX 67
N-sek.-Butyl-N'-[2.6-di~nethyl-phenylJ-thioharnstoff - Aus 2.6-Dimethyl-phenylsenfol (7a) und sek.-Butylamin in Aceton. Farblose Kristalle (70%) vom Schmp. 124.5- 126" (Methanol/ Wasser). - IR-Banden: B 1530, C 1270, 1240, D 1165 cm-I.
C13H2oNzS (236.4) Ber. C 66.05 H 8.53 N 1 I .85 S 13.56 Gef. 66.20 8.50 12.01 13.68
N-rert.-Butyl-N'-isoamyl-thioharnstoff. - Aus 1 a und Isoamylamin in Wasser. Farblose Kristalle (75%) vorn Schmp. 75.5-76.5" (Alkohol/Wasser). - IR-Banden: I3 1540, C 1340, D I208 cm-1.
CloH22N2S (202.4) Ber. C 59.35 H 10.96 S 15.85 Gef. C 58.89 H 10.87 S 16.00
N-tert.-Butyi-N'-neopentyl-thiohornstoff. - Aus 1 a und Neopentyiamin in Wasser. Farblose Nadeln (96%) vom Schmp. 167" (Athanol/Wasser).-IR-Banden: B 1540,C 1340, D 1130 cm-1.
CloH22N2S (202.4) Ber. C 59.35 H 10.96 N 13.84 S 15.85 Cef. 59.15 10.81 14.06 15.77
Di-tert.-alkyl-subsrituierte Thioharnstoffe N-tert.-Butyl-N'-tert.dmy1-thioharnstoff- - Aus tert.-Burylsenfof (1 a) und tert.-Amylamin
(2b) in Petrolather bei Raumtemperatur. Farblose Nadeln (71 %) vorn Schmp. 120" (Subl.). - IR-Bunden: B 1545, 1535, C 1320, D l200cm-1.
CloH22NzS (202.4) Ber. N 13.84 S 15.85 Gef. N 13.86 S 15.78
N-tert.-Butyl-N'-(l.I.3.3-tetramethyl-bu~yl]-~hioharnst0ff~~~~. - Aus I.l.3.3-Tetramethyf- butylsenfol(1 f ) und tert.-Butylamin (Za) in Petrolather (30-50") bei Raumtemperatur. Farb- lose Nadeln (62%) vom Schmp. 103- 106"(Petrolather [30-550']; Lit.24a) loo0). - fR-Banden: B 1530, C 1320, D 1195, 1 I10 cm-1. - H-NMR-Spektrurn (CDCI,): T = 8.95 Is, y'(CH3)3], 8.50 Is. a(CH3)3 + a'(CH3)2J, 8.13 (s, $-CH2), 4.23 (breit, NH) im Verhaltnis 9 : 15 : 2 : 2. Thioharnstoffe init sperrigen Substituenten
N-tert.-Bvtyl-N'-benzhydryl-thioharnstoff. - Aus 1 a und Benzhydrylamin in Aceton. Farb- lose Kristalle (66%) vom Schmp. 169- 171" (AthanoljWasser). - IR-Banden: B 1535, C 1300, D 1195, 1070cm-1.
ClsHzzN2S (298.4) Ber. C 72.44 H 7.43 N 9.39 S 10.74 Gef. 72.62 7.51 9.41 10.83
N-tert.-Butyl-N'-[a.a-dimethylbenzyll-thioharnstoff. - Aus 1 a und ma-Dimethylbenzylamin (2c) in Petrolather (50-70") bei Raumtemperatur. Farblose Kristalle (86%) vorn Schmp. 145-147.5'. - fR-Bander B 1550cm-1. - H - N M R - S p e k t r u m (CDCI,): T = 8.82 (s), 8.36 (s), 4.90 (breit, NH), 3.67 (breit, NH), 2.54 ppm (s) im Verhaltnis 9 : 6 : 1 : 1 : 5 .
C I ~ H ~ ~ N Z S (250.4) Ber. C 67.15 H 8.86 N 11.19 S 12.80 Gef. 67.57 8.86 10.99 13.10
N-rerr.-Butyl-N'-ia.a-diphenylathyll-thioharnstoff. - Aus l a und a.a-Diphenylathylamin (2d) in Tetrachlormethan oder Aceton nach 8stdg. Erhitzen. Farblose Kristalle (52%) vom Schmp. 130- 133.5" (Essigester/Petroilther [30-50°]). - If?-Bunde: B 1535 cm-1.
C19H24N2S (312.5) Ber. C 73.03 H 7.74 N 8.97 S 10.26 Gef. 72.23 7.78 9.65 10.39
Y

68 W . Walter und G. Randau Bd. 722
N-rert.-Butyl-N'-[2.6-dimethyl-phenyl]-thioharnstoff (5). - Aus 2.6-Dimethyl-phenylsenfol (7s) und tert.-Butylamin (2a) in Aceton. Farblose Kristalle (69%) vom Schmp. 135- 137" (Methanol). - IR-Banden: B 15 15, C 1340 crn--l.
C13H20NZS (236.4) Ber. C 66.05 H 8.53 N 11.85 S 13.56 Gef. 65.57 8.54 11.76 13.61
N.N'-Dineopentyl-thioharnstofl - Aus Neopentylsenful und Neopentylamin in Chloroform. Farblose Blattchen (82%) vom Schmp. 192.5 - 194.5" (Zers.; Methanol). - IR-Bunden: B 1590, C 1365, D 1200. 1140cm-1.
ClIH24N2S (216.4) Ber. C61.05 H 11.18 N 12.95 S 14.82 Gef. 60.93 10.94 13.23 14.79
N-Neopentyl-N'-cyclohexyl-thioharnstoff. - Aus Neopentylsenfol und Cyclohexylamin in Chloroform. Farblose Kristalle (65 %) vorn Schmp. 157 - 159" (bithanol). - IR-Banden: B 1590, 1580, C 1285, D 1115, F (?) 757, 746cm-1.
C12H24N2S (228.4) Ber. C 63.10 H 10.59 N 12.27 S 14.04 Gef. 62.65 10.58 12.25 14.11
N.N'-Di-tert.-amyl-thioharnstofl (3 b). - Aus tert.-Amylsenfol (1 b) und tert.-Amylamin (2b) in Wasser. Farblose Blattchen (74%) vom Schmp. 102-104" (verd. Athanol). - IR- Banden: B 1525, C 1311, 1175, D 1063, 1008, F (?) 788, 770, G (?) 672cm-1.
C I I H ~ ~ N $ ~ (216.4) Ber. C 61.05 H 11.18 N 12.95 S 14.82 Gef. 61.14 11.37 12.88 14.86
N-tert.-Amyl-N'-[I.I.3.3-tetramethyl-butyl]-thioharnstoff. - Aus 1 f und 2 b in Petrolather (30-50") bei Raumternperatur. Farblose Nadeln (40%) vom Schmp. 86-89".
C I ~ H ~ ~ N ~ S (258.5) Ber. C 65.05 H 11.70 N 10.84 S 12.40 Gef. 64.65 11.43 11.02 12.20
N-tert.-Amyl-N'-(a.a-dimethylbenzyll-tltioharnstoff. - Aus 1 b und a.a-Dimethylbenzylamin (2c) in Chloroform nach 8 stdg. Erhitzen. Farblose Blattchen (47%) vorn Schmp. 136- 139" (Methanol/Wasser).
CjsH24NzS (264.4) Ber. N 10.60 S 12.12 Gef. N 10.61 S 12.10
N-tert.-Amyl-N'-~a.a-diphenylathyl~-thioharnsro~. - Aus tert.-Amylsenfol (1 b) und a.a- Diphenylathylamin (2d) in Tetrachlormethan ; kristallisiert erst nach 3 Wochen bei Raum- temperatur. Farblose Kristalle (49 %) vom Schmp. 122- 124" (Petrolather). - IR-Banden: B 1545, C 1385, 1370, D 1255 cm-1.
C ~ O H ~ ~ N ~ S (326.5) Ber. C 73.57 H 8.03 N 8.58 S 9.82 Gef. 73.66 8.06 8.89 10.04
N-Cyclohexyl-N-[2.6-ditnethyl-phenyll-thioharnsto~ - Aus Cyclohexylsenfol und 2.6- Dimethyl-anilin (4a) ohne Losungsmittel bei 170 - 180" irn Glasautoklaven (23 %) oder aus Cyclohexylamin und 2.6-Dimethyl-phenylsenfol in Aceton ( 5 5 %). Hellgelbe Kristalle vorn Schmp. 129--130° (Methanol/Wasser). - IR-Banden: B 1545, C 1350, 1330, D (?) 1085, 980 cm-1.
C I S H ~ ~ N ~ S (262.4) Ber. C 68.65 H 8.45 N 10.68 S 12.22 Gel. 68.62 8.59 10.62 12.35

1969 Oxydationsprodukte von Thiocarbonsaureamiden, XIX 69
N.N'-Bis-[I . 1.3.3-tetrarnethyl-butyl]-thioharnstoff (3f). - Aus I f und 2f in Chloroform. Farblose Prismen (33 %) vom Schmp. 107- 109"(Petrolather [30-50°]/Essigester = 40: 1). - IR-Banden: B 1522, D 1212 cm-1.
C ~ ~ H ~ ~ N Z S (300.5) Ber. C 67.93 H 12.08 N 9.32 S 10.67 Gef. 67.78 12.48 9.86 10.79
N. N'-Di-odamantyl-(I j-fhioharnstoff. - Aus I-Amino-adamantan und I-lsoihiocyonaro- adomontan in Aceton nach 7stdg. Erhitzen. Farblose Prismen (51 %) vom Schmp. 166-171" (Zers.; Aceton). - IR-Banden: B 1545, C 1390, 1350, D 1218 (?), 1090cm-1.
C ~ I H ~ ~ N ~ S (344.6) Ber. C 73.20 H 9.36 N 8.13 S 9.31 Gef. 73.14 9.33 8.64 9.35
N.N'-Dibenzhydryl-thioharnstoff*5). - Aus Benzhydrylsenfol und Benzhydrylamin in Aceton. Farblose Kristalle (95%) vom Schmp. 221" (Aceton/Wasser; Lit.25) 210-211°).
C Z ~ H Z ~ N ~ S (408.6) Ber. C 79.38 H 5.92 N 6.86 S 7.84 Gef. 79.05 6.04 6.79 7.94
N . N'-Bis-[a.a-dime~hylbenzylJ-thioharnstoff~~~~ (3c). - a) Aus a.a-Dimethylbenzylsenfol (lc) und a.a-Dimethylbenzylamin (2c) in Ather. Farblose Kristalle (79 %) vom Schmp. 152-155" (Methanol/Wasser; Lit.*4b) 150- 152'). - H-NMR-Spektrum (CDCI,): T =
8.45 (s), 4.10 (breit, NH), 2.82 ppm (s) im Verhaltnis 6 : 1 : 5. b) Je 10 mMol 2c und Triathylamin wurden in 20ccm Ather gelost und eine Losungvon
5 mMol Thiophosgen in 10 ccm Ather zugetropft. Nach 15min. Erhitzen unter Ruckflu6 wurde eingedampft, der olige braune Riickstand in Chloroform aufgenommen, mit Wasser ge- waschen, mit Natriumsulfat getrocknet und i.Vak. eingeengt. Dieser Ruckstand wurde in heiBem Methanol mit Aktivkohle behandelt ; bei Zugabe von Wasser kristallisiert das Pro- dukt nach langerem Stehenlassen bei -20" aus. Nochmaliges Umkristallisieren aus Methanol/ Wasser ergab 380 mg (24%) hellgelbes Produkt vom Schmp. 150-152". N.N'-Bis-[2.6-dimethyl-phenyll-thiohornsto~48) (6a). - Aus 2.6-Dimethyl-anilin (48) und
2.6-Dimethyl-phenylsenfol (7a) nach 10stdg. Erhitzen in Aceton. Farblose Kristalle (80%) vom Schmp. 235" (Zers.; Aceton; Lit.48) 208"). - IR-Banden: B 1520, 1480, C 1235, D 1030 cm-1.
C I ~ H ~ O N ~ S (284.4) Ber. C 71.79 H 7.09 N 9.85 S 11.27 Gef. 71.41 7.11 9.81 11.40
N. N'-Bis-I2.6-diathyI-phenyli-thioharnstoff (6 b). - Aus 2.6-DiathyI-phenyIsenfol (7 b) und 2.6-Diathyl-anilin (4b) ohne Losungsmittel nach 5stdg. Erhitzen. Farblose Kristalle (47 %). subl. > 160",Schmp. 194"(Athanol/Wasser). - IR-Banden: B 1525,C 1265,1230,D 1058 cm-1.
C Z I H ~ R N Z S (340.5) Ber. C 74.07 H 8.29 N 8.23 S 9.42 Gef. 13.79 8.30 8.23 9.42
N. N'-Bis-~2.6-diisoprvpyI-phenyl!-rhioharnstoff (612). - Aus 2.6-Diisopropyl-phenylsenfol(7c) und 2.6-Diisopropyl-anilin (4c) in Aceton nach 7stdg. Erhitzen. Farblose Kristalle (38 %), subl. >19O0.Schmp.242"(Athanol/Wasser). - IR-Banden: B 1520,C 1253,1225.D 1050cm-1.
C ~ S H ~ ~ N ~ S (396.6) Ber. C 75.70 H 9.15 N 7.06 S 8.09 Gef. 74.85 8.97 7.1 1 8.38
48) G. M. Dyson, H . J. George und R. F. Hunter, J. chem. SOC. [London] 1927,436.

70 W. Walter und G. Randau Bd. 122
N'-Substituierte N-Trityl-thioharnstoffe
N-Methyl-N'-trityl-thioharnstoff. - 20 mMol Tritylisothiocyanat (1 e) wurden fein ge- pulvert und im Glasautoklaven bei Raumtemperatur in ca. 20 ccm wasserfreiem Methylamin geriihrt. Es bildete sich zuerst eine klare Losung, die nach einiger Zeit Kristalle abschied. Nach 3 Stdn. wurde iiberschiissiges Methylamin abgedampft. Der Ruckstand wurde aus 80ccm Athanol/Aceton(l : 1) umkristallisiert. Fast farblose, grofle, sechseckige,flacheKristalle (81 %) vom Schmp. 192-195". - IR-Banden; B 1538, C 1348 (?), 1242, D 1065. 1030cm-1.
CzlHzoN2S (332.5) Ber. C 75.86 H 6.06 N 8.43 S 9.65 Gef. 75.32 5.89 8.33 9.65
N-lsopropyl-N'-trityI-thioharnstoff~4). - 3 mMol feingepulvertes l e wurden 10 Min. bei 10" in 5 ccm Isopropylamin geriihrt, nach Stehenlassen iiber Nacht wurde abfiltriert und aus Benzol/Petrolather umkristallisiert. Farblose Kristalle (84%) vom Schmp. 187- 190.5" (Lit.14) 188-190"). - IR-Bnnden: B 1520, 1475, C 1240cm-I.
Analog wurden dargestellt :
N-sek.-Buryl-N'-triryl-rhioharnstoff - Hellgelbe Prismen (88 %) vom Schmp. 168- 170" (Methanol/Wasser). - IR-Banden; B 1515 , C 1360, D 1215 cm-1.
C Z ~ H ~ ~ N Z S (374.6) Ber. C 76.96 H 7.00 N 7.48 S 8.56 Gef. 76.71 7.03 7.73 8.71
N-rerr.-Butyl-N'-trityl-rhioharnstoff. - Farblose Kristalle (68 %) vom Schmp. 161 - 166" (Zers. : Methanol). - IR-Bande: B I525 cm-1.
c24H26N~S (374.6) Ber. C 76.96 H 7.00 N 7.48 S 8.56 Gef. 76.23 6.98 7.37 8.54
N-tert.-Amyl-N'-trityl-thioharnstoff. - Grobe, farblose Prismen (83 %) vom Schmp. 183 bis 185" (Methanol/Aceton = 5 : I ) . - IR-Banden: B 1520, C 1370 cm-1.
C25HzsNzS (388.6) Ber. N 7.21 S 8.25 Gef. N 7.25 S 8.28
N-Cyclohexyl-N'-rrityl-thioharnstoff~~~. - Farblose Kristalle (75 %) vom Schmp. 194- 19s" (Athanol; Lit.14) 191-192'). - IR-Banden: B 1527, C 1380, 1350, D l2l5cm-1.
N-[I.I.3.3-Tetramethyl-butylJ-N'-rrityl-thioharnstoff. - Farblose Kristalle (85%) vom Schmp. 144-147' (Methanol/Aceton = 1 : 1). - IR-Banden; B 1523, 1488, C 1375, 1360, D 1245 cm-1.
CzsH34N2.S (430.7) Ber. C 78.09 H 7.96 N 6.51 S 7.44 Gef. 77.92 7.88 6.87 7.74
N-Benzhydryl-N'-trityyl-thioharnstoff. - Grobe, farblose Kristalle (70%) vom Schmp. 194-196" (Aceton/Benzol = I : I). - IR-Banden: B 1511, C 1360, D 1238, ItOOcm-*.
C33HzsNzS (484.7) Ber. C 81.78 H 5.82 N 5.78 S 6.62 Gef. 81.00 5.86 5.82 6.65
N-tert.-Butyl-N'-adatnanryl- I I) -thioharnstoff. - I0 m Mol Adamanryl- ( I ) -isothiocyanat wurden fein gepulvert und in 17 ccm terl.-Burylamin 25 Min. bei Raumtemperatur geriihrt. Aufarbeitung analog der Isopopyl-Verbindung. Farblose Kristalle (68 %) vom Schmp. 178.5"

I969 Oxydationsprodukte von Thiocarbonsaureamiden, XIX 71
(Kristallumwandlung unter Verlust der Doppelbrechung bei 165"; Methanol/Aceton/ Wasser = 10: 10: I) . - IR-Banden: B 1530, C 1290, D 1130, 1092cm-I.
C15H26N2S (266.5) Ber. C67.61 H9.84 N 10.52 S 12.03 Gef. 67.70 9.91 10.80 11.92
Umserzungen sperrig subsrituierrer Amine (2) Umsetzung von Tritylamin (2e ) rnir Thiophosgen. - a) 10 mMol2e wurden in 50 ccm Chloro-
form gelost und 20 mMol Triathylamin zugefiigt. Bei 20" wurde Thiophosgen (5 mMol) in 10 ccm Chloroform langsam zugetropft und bei Raumtemperatur I2 Stdn. stehengelassen. Danach wurde 3 Stdn. unter RiickfluB erhitzt. Nach dem Eindampfen i.Vak. wurde der braune Riickstand in Methanol mit Aktivkohle entfarbt. Der nach Einengen verbliebene gelbe Riickstand wurde aus AthanollEssigesterlPetrolather (30 -50") umkristallisiert. Es wurden 0.98 g (33 %) fast farbloses kristallines Tritylamin-hydrochloridvom Schmp. 235-242" erhalten. - IR-Bunden: 3020, 2860 v(NH3Q). I595 F(NH3@)? cm-1.
C19Hl&IN (295.8) Ber. C 77.15 H 6.13 CI 11.99 N 4.74 Gef. 76.92 6.18 11 .55 4.71
b) 10 mMol feingepulvertes 2e wurden in 20 ccm Pyridin gelost und bei Raumtemperatur mit 5 mMol Thiophosgen versetzt. Es schied sich eine schwarze, amorphe Masse ab. Nach 15 Min. wurde mit 200 ccm Wasser verdiinnt, rnit verd. Salzsaure neutralisiert und die violette Losung rnit Chloroform extrahiert; die Chloroformlosung wurde mit Wasser gewaschen, ge- trocknet und i. Vak. eingeengt. Der dunkelbraune Riickstand ergab beim Umkristallisieren aus Methanol 650 mg (22 %) briunliches Tritylumin-hydrochlorid (IR-Spektrum und Misch- Schmp. 231 -235").
C19H18CIN (295.8) Ber. N 4.74 Gel. N 4.74
Umserzung von 2e rnir Trirylisorhiocyanor (le). - 1.5 g 2e und 1.73 g l e wurden in 100 ccm Dimethylformamid unter RiickfluR zum Sieden erhitzt. Nach 2.5 Stdn. wurde abgekiihlt und i n 200 ccm Wasser gegossen. Es fielen 2.8 g Triphenylcarbinol aus, Schmp. 160- 164", Misch-Schmp. 160- 162".
Umsetzung von Alkylsenfolen mir 2.6-Dimethyl-anilin (48). - Je 0. I Mol terr.-Burylsenjol(1 a) und 4a in IOccm Aceton wurden im Glasautoklaven 4Stdn. auf 120" erwarmt. Nach dem Ab- kiihlen wurde vom ausgeschiedenen 5 (0.47 g, 2 %) abgesaugt und i. Vak. eingedampft. Derolige, nicht kristallisierende Riickstand schied innerhalb 3 Stdn. im Glasautoklaven bei 160" all- mahlich Kristalle ab. Nach dem Abkiihlen wurde aus Aceton umkristallisiert und 8.0 g (56 %) N. N'-Bisl2.6-dimefhyl-phenyli-thioharnstoff (68) erhalten. Schmp. > 240" (Zers.). 1R- Spektrum identisch mit dern von authent. Produkt (S. 69).
C I ~ H ~ O N ~ S (284.4) Ber. C71.79 H 7.09 N9.85 S 11.27 Gef. 71.04 7.09 9.85 11.46
Analog wurde bei der Umsetzung von Merhylsenfol (5 Stdn. bei 160"), k'rhylsenfdl (4 Stdn. bei 150") und Cyclohexylsenfol(4 Stdn. bei 150") mit 4a jeweils nur 6 a in 30-, 77- bzw. 23 proz. Ausbeute isoliert. N-Phenyl-N'-12.6-dintethyl-~lienylf -rhiohornsioS: - Die Mischung von je 0.1 Mol Phenyl-
senfol und 4a reagierte nach kurzer Zeit heftig; der dabei entstehende feste Kristallkuchen wurde mit 50 ccm Aceton im siedenden Wasserbad kurz erhitzt, abgekiihlt und filtriert. Die

72 W . Walter und G. Randau Bd. 122
Umkristallisation aus 50 ccm Dimethylformamid /5 ccm Wasser ergab 20.3 g (79%) farblose Kristalle vom Schmp. 226-227", Kristallurnwandlung bei 200". - IR-Banden: B 1525, 1490, C 1240, D 1025 cm-1.
C ~ ~ H ~ ~ N Z S (256.4) Ber. C 70.27 H 6.29 N 10.93 S 12.51 Gef. 69.89 6.31 10.87 12.53
Umsetzung von a.a-Diphenylathylamin (2d) mil a.a-Diphenylathylisothiocyanat (1 d): a) 20 mMol 2d wurden in 20 ccm Benzol gelost und je 20 mMol Triathylarnin und Schwefel- kohlenstof zugefiigt. Nach Zugabe von 50 ccm Petrolither (30-50") schied sich bei -20' orangerotes Dithiocarbamat ab. Das iiberstehende Losungsmittel wurde abgegossen, der Riickstand in 20 ccm Chloroform gelost und nach Zugabe von 20 mMol Triathylamin unter Eiskiihlung 20 mMol Chlorameisensuureuthylester + 10 ccm Chloroform zugetropft. Es wurde langsam auf Raurntemperatur erwarmt, nach 1 Stde. Riihren 2maI rnit 10 ccm 2 n HCI und 3rnal rnit 10 ccm Wasser gewaschen, getrocknet und i. Vak. eingeengt. Die Fraktionierung des Riickstands ergab neben reichlich Vorlauf (Sdp.3.0 <80") 1.5 g Id, Sdp.o.2 123-127". Ausbeute 32% d. Th. Breite, intensive IR-Absorptionsbande bei 2140 cm-1 (-NCS).
b) 1.5 g I d wurden mit 1.3 g 2d in 15 ccm Chloroform unter RiickfluB 10 Stdn. zum Sieden erhitzt. Nach dem Abkiihlen wurde eingedampft; der Riickstand kristallisierte nach Zugabe von etwas Ather bei 5". Die Umkristallisation aus Methanol ergab 600 mg farblose Prismen vom Schmp. 187". Die Substanz, die nicht identifiziert werden konnte, enthielt keinen Schwefel.
Umsetzung von Tritylisothiocyanat (1 e) rnit 2.6-Dimethyl-anilin (4a). - 3.0 g l e wurden fein- gepulvert und bei Raumtemperatur in 3 g 4a geriihrt. Nach 2 Stdn. wurde das breiige Re- aktionsgemisch filtriert und getrocknet. Der Riickstand erwies sich als unverandertes 1 e (IR-Spektrum und Misch-Schmp. 137- 138").
Oxydation von substituierten Thioharnstoffen zu Harnstoffen
N-Monosubstituierte Harnstoffe
N-tert.-Butyl-harnstos - 0.1 g N-tert.-Butyl-thioharnstof wurde in 5 ccm Athanol bei 0" rnit 0.2 ccm Perhydrol unter diinnschichtchromatographischer Kontrolle oxydiert. Nach 2 Stdn. bei 0" trat eine Triibung auf. Es wurde in 20 ccm Wasser gegossen, 3mal rnit 10 ccm Chloroform extrahiert, die Chloroformphasen getrocknet und i. Vak. eingeengt. Der Riick- stand wurde aus verd. khan01 umkristallisiert. GroBe, farblose Plattchen vom Schmp. 182" (Zers. ; Lit.49) 182"). Im IR-Spektrum identisch mit authent. Praparat.
Analog wurden folgende substituierte Harnstoffe als Oxydationsprodukte isoliert und durch Schmp., IR-Spektrum und evtl. Elementar-Analyse charakterisiert :
N-sek.-Butyl-hornstoff. - Schmp. 165-170" (Lit.50) 169- 170").
N-Neopentyl-harnstos - Schmp. 153-155" (verd. Athanol; Lit.51) 157").
N-Benzyl-harnstof. - Schmp. 140- 142" (Lit.52) 146.6").
49) L. I. Smith und 0. H. Emerson, Org. Syntheses, Collect. Vol. 111, S. 151 (1955). 50) A . E. Dixon, J. chem. SOC. [London] 67, 556 (1895). 51) J. S. Buck und A . M. Hjort, J. Amer. chem. SOC. 59, 2567 (1937). 52) T. L. Davidson und D. A . Peak, J. chem. SOC. [London] 1963, 3327.

1969 Oxydationsprodukte von Thiocarbonsaureamiden, XIX 73
N-[2.6-Diathyl-phenyl]-harns/off: Die Oxydation von 0.68 g N-[2.6-Diathyl-phenyll-thio- harnstoff in 40 ccm Methanol bei - 10" rnit 0.86 ccm 30proz. Wasserstofperaxid ergab in 2 Stdn. nach Einengen i. Vak. 320 mg (51 %) farblose Nadeln vom Schmp. 207' (Aceton).
C I ~ H ~ ~ N ~ O (192.3) Ber. C 68.72 H 8.39 N 14.57 Gef. C 68.45 H 8.49 N 14.25
N . N'-Disubslituierte Harns/ofe Allgemeine Vorschrift: 0.25 g Thioharnstoff in 20 ccrn Methanol wurden bei 0" unter
chrornatographischer Kontrolle (Rp-Werte vgl. Tab. 2) rnit I ccm 30prOZ. Wasserstoffperoxid versetzt. Nach Erreichung einer moglichst hohen Konzentration an S-Monoxid wurde in 50 ccrn Eiswasser gegossen und rnit rnindestens je 20 ccm kaltem Tetrachlormethan, Chloro- form und Dichlormethan ausgeschiittelt. Die organischen Phasen wurden rnit Natriumsulfat getrocknet und i. Vak. bei 20" (Bad) eingeengt. Dabei schied sich aus der vorher klaren und fast farblosen Losung Schwefel ab. Nach Behandlung rnit kaltem Methanol wurde das Filtrat eingeengt, der Ruckstand evtl. in heiBem Methanol mit Aktivkohle geklart und nach Ein- engen umkristallisiert. Die erhaltenen Harnstoffe wurden durch Schmp., IR-Spektrum und evtl. Elernentar-Analyse identifiziert.
N-Methyl-N'-ter/.-butyl-harnstoff - Farblose Kristalle vom Schmp. 146" (Methanol/ Wasser), Ausbeute 38 % d. Th.
C ~ H ~ ~ N Z O (130.2) Ber. C 55.35 H 10.84 N 21.52 Gef. C 54.58 H 10.86 N 21.43
N.N'-Di-n-propyl-hornstoff. - Farblose Nadeln vom Schmp. 105- 110" (Methanol; Lit.53)
N.N'-Diisopropyl-harnstoff. - Farblose Kristalle (50%) vom Schmp. 190- 191" (Methanol;
N-Isopropyl-N'-n-butyl-harns/ofl - Farblose Nadeln vom Schrnp. 75 -76' (Athanol/
107.5 - 112.5"). Im IR-Spektrum identisch rnit authent. Produkt.
Lit.54) 192"). Die Oxydation in IOproz. Natronlauge ergab 64% d. Th.
Wasser).
CaHlsNzO (158.3) Ber. C60.72 H 11.47 N 17.70 Gef. C 59.71 H 11.34 N 17.85
N-Isopropyl-N'-ter/.-butyl-harnstoff. - Farblose Nadeln vorn Schrnp. 185" (Methanol).
CsHlsNzO (158.3) Ber. C 60.72 H 11.47 Gef. C61.23 H 11.38
N-Isopropyl-N'-cyrlohexyl-harnstofl - Farblose Kristalle vorn Schmp. 144 - 148" (Athanol/Wasser; Lit.2oa) 148.5- 149.5").
(Methanol/ Wasser). N-n-Butyl-N'-/er/.-butyl-hornstoff. -- Farblose Kristalle (55 %) vom
C9H2oN20 (172.3) Ber. C 62.75 H 11.70 Gef. C 62.07 H I
N-Isobutyl-N'-tert.-butyl-harnstoff. - Farblose Kristalle vom Schmp. Wasser; Lit.55) 163").
Schmp. 77-82"
.27
77" (Methanol/
C9HzoNzO (172.3) Ber. C62.75 H 11.70 N 16.26 Gef. C 62.76 H 11.71 N 17.08
53) 0. Hech/, Ber. dtsch. chem. Ges. 23, 281 (1890). 54) A. W. Hofmanrr, Ber. dtsch. chem. Ges. 15, 756 (1882). 5 5 ) B. Brauner, Ber. dtsch. chem. Ges. 12, 1874 (1879).

14 W . Walter und C. Randau Bd. 122 ~ ~~~
N-sek.-Butyl-N'-tert.-butyl-harnstoff. - Farblose Kristalle (76 %) vom Schmp. 203" (Athanol/Wasser).
C9HzoNzO (172.3) Ber. C 62.75 H 11.70 Gef. C 62.98 H 11.70
N-sek.-Butyl-N'-neopenfyl-harnstoff. - Farblose Kristalle vom Schmp. 143 - 145" (durch Subl. i. Vak. gereinigt).
CloHz2NzO (186.3) Ber. C 64.47 H 11.90 N 15.04 Gef. C 64.89 H 12.30 N 14.52
N-sek.-Bufyl-N'-[Z.6-dimethyl-phenyl]-harnstoff. - Farblose Kristalle (27 %) vom Schmp. 210", sintern bei 178- 180" (Aceton/Wasser).
C13H2oN20 (220.3) Ber. C 70.87 H 9.15 N 12.72 Gef. C 70.27 H 9.21 N 12.88
N-terf.-Bufyl-N'-isoamyl-harnstoff. - Farblose, diinne Blattchen (27 %) vom Schmp. 101 - 103" (Methanol/Wasser).
CloH22NzO (186.3)
(Zers. ; Methanol/Wasser). CloHzzNzO (186.3)
Ber. C64.47 H 11.90 N 15.04 Gef. C64.39 H 11.76 N 15.71
N-tert.-Butyl-N'-neopentyl-harnstoff. - Farblose Kristalle (56 %) vom Schmp. 220-225"
Ber. C 64.47 H 11.90 N 15.04 Gef. C 64.30 H 11.65 N 14.96
N.N'-Dineopenfyl-harnstofl - Farblose Nadeln (75 %) vom Schmp. 212" (Athanol/Wasser).
CllH24NzO (200.3) Ber. C 65.95 H 12.08 N 13.98 Gef. C 66.00 H 11.99 N 14.04
N-!err.-Butyl-N'-benzhydryl-harnsfoff. -- Farblose Nadeln vom Schmp. 223.5 -225" (Methanol/ Wasser).
Cl~H2zN20 (282.4) Ber. C 76.56 H 7.85 N 9.92 Gef. C 76.25 H 7.95 N 9.65
N-tert.-Butyl-N'-[a.a-diphenylathylj-harnstoff. - Farblose Nadeln vom Schmp. I83 - 186" (Subl. ; Essigester/Petrolather).
C19H24N20 (296.4) Ber. C 76.99 H 8.16 N 9.45 Gef. C 76.08 H 8.26 N 8.70
N.N'- Disubstituierte Harnstoffe aus den entsprechenden Thioharnstoff-S-monoxiden (10): Die S-Monoxide 10 zerfielen bei langerem Aufbewahren (auch bei niedriger Temperatur) in die entsprechenden Harnstoffe, wurden in kaltem Methanol aufgenommen, vom ungelasten Schwefel abgesaugt und gekocht. Wenn hierbei Triibung oder Gelbfarbung eintrat, wurde mit Aktivkohle geklart und i. Vak. eingeengt. Der verbleibende Riickstand wurde umkristal- lisiert. Die Identifizierung erfolgte durch IR-Spektren und Elementar-Analysen.
199-201", Subl. bei 150" (Methanol/Wasser). N-tert.-Butyl-N'-[a.a-dimethylbenzyl]-harnstoff. - Farblose, feine Nadeln vom Schmp.
C I ~ H ~ ~ N ~ O (234.3) Ber. C 71.76 H 9.46 N 11.95 Gef. C 70.76 H 9.25 N 11.94
N. N'-Di-terf.-amyl-harnstoff. - Farblose SpieBe vom Schmp. 228 -230" (Subl. ; Methanol/ Wasser).
CllH24NzO (200.3) Ber. C 65.95 H 12.08 N 13.98 Gef. C66.00 H 11.93 N 13.67
N-terf.-Amyl-N'-[a.a-dimethylbenzyll-harnstoff. - Farblose Kristalle (64%) vom Schmp. 183 - 184" (Methanol/Wasser).
C15H24N20 (248.4) Ber. C 72.54 H 9.74 N 11.28 Gef. C 72.32 H 9.77 N 11.60

1969 Oxydationsprodukte von Thiocarbonsaureamiden, XIX 75
N-tert.-Amyl-N'-ia.a-diphenylathyll-harnstoff. - Farblose Nadeln und Prismen (87 %) vom Schmp. 172- 176" (Methanol/Wasser).
C20H26N20 (310.4) Ber. C 77.38 H 8.44 N 9.02 Gef. C 77.85 H 8.25 N 9.13
N.N'-Bis-[a.a-dimethylbenzyl]-harnsroff, - Farblose Nadeln vom Schmp. 225 -226" (Methanol/Wasser). Ausbeute 22 % d. Th. neben 69% S-Monoxid (S. 77).
C19H24N20 (296.4) Ber. C 76.99 H 8.16 N 9.45 Gef. C 75.38 H 7.79 N 9.46
N.N'-Bis-[Z.6-dimethyl-phenyl/-harnstoff. - Farblose Prismen, Subl. bei 270-3325",
C17HzoN20 (268.4) Ber. C 76.09 H 7.51 N 10.44 Gef. C 75.14 H 7.60 N 10.47
N-Benzhydryl-N'-trityl-hornstoff - Farblose Kristalle (43 %) vom Schmp. 202 -206" (khanol ) .
C33H28N20 (468.7) Ber. N 5.98 Gef. N 5.86
N-tert.-Butyl-N'adamantyl-(1)-harnstoff. - Farblose Kristalle, Subl. bei 240 -260" (Tetrachlormethan/Petrolat her).
C I S H ~ ~ N ~ O (250.4) Ber. C 71.96 H 10.47 N 11.19 Gef. C69.80 H 10.17 N 10.97
N.N'-Disubstituierte Thioharnstoff-S-monoxide
Allgemeine Darstellung: I g Thioharnstoff in 50- 100 ccm .&than01 wurde bei 0-40" mit 3 ccm 30proz. H202 unter diinnschichtchromatographischer Kontrolle (RF-Werte vgl. Tab. 2) oxydiert. Nach Erreichen einer maximalen S-Monoxid-Konzentration (0.5 -2 Stdn.) wurde in 150-250 ccrn Eiswasser gegossen und unter diinnschichtchrornatographischer Kontrolle mit rnehreren 50-ccm-Portionen kaltem Tetrachlormethan, Chloroform und Dichlormethan extrahiert. Die Extrakte mit den hochsten S-Monoxid-Gehalten wurden vereinigt, mit Eis- wasser gewaschen, mit Natriumsulfat getrocknet und bei 10" (Bad) i. Vak. bis zur beginnenden Kristallisation oder auf ca. 5 ccm eingeengt. Durch Zugabe von Essigester, Cyclohexan oder Petrolather wurde bei -20" das S-Monoxid analysenrein zur Kristallisation gebracht, nach einigen Stdn. bei -20" schnell abfiltriert und mit kaltem Essigester/Petrolather (1 : I ) ge- waschen.
N.N'-Di-tert.-butyl-rhioharnstoff-S-monoxid9) (8). - 665 mg (61 %) farblose, wiirfelige Kristalle vom Zers.-P. 105".
C9HzoN20.S (204.3) Ber. C 52.90 H 9.87 N 13.72 S 15.69 Gef. 52.94 9.88 13.67 15.47
Beim Aufbewahren von 8 bei -20" erfolgte Zersetzung; nach I Woche lieBen sich diinnschicht- chrornatographisch neben 8 Schwefel, N.N'-Di-tert.-butyl-thioharnstoff (38) und N. N'-Di- tert.-butyl-hornstoff (9) nachweisen. Schmp. im geschlossenen Rohr 240" (Lit.55) 242").
N-tert.-Butyl-N'-tert.-amyl-thioharnst~ff-S-monoxid. - Farblose, wiirfelige Kristalle (64 %) vom Schmp. 94-95" (Zers.), aus der Schmelze Nadeln vom Schmp. 210-220" (vgl. entspr. Harnstoff, Subl. bei 21 5 ' ) .
CtoH22N20S (218.4) Ber. C 55.00 H 10.16 N 12.83 S 14.68 Gef. 55.25 9.79 13.10 15.06

76 W. Walter und C. Randau Bd. 122
N-~ert.-Bu~yl-N'-[I.I.3.3-lelrame~hyl-bufyl]-~hioharnsfo~-S-monoxid. - Farblose Krktalle (78 %) vom Schrnp. 90" (Zers.), in Tetrachlorrnethan loslich. - H-NMR-Spekrrum (CDC13, -20"): 7 = 8.95 (s), 8.53-8.50 (rn), 8.30 (s), 4.80 (breit, NH), 2.30 pprn (breit, N H . . .O) i rnVerha l tn is9 :15:2 :1 : I .
C I ~ H ~ ~ N ~ O S (260.4) Ber. C 59.95 H 10.84 N 10.76 S 12.31 Gef. 59.72 10.91 11.42 12.51
N-terr.-Bu~yl-N'-[u.u-dime/hylbenzyll-rhioharnstoff-S-monoxid. - Farblose, verfilzte Na- deln (68%) vorn Schrnp. 80-82" (Zefs.), aus der Schrnelze Kristalle vorn Schmp. 150-190" (vgl. Harnstoff, Schrnp. 199-201"). - H-NMR-Speklrum (CDCI3, 0'): T = 9.02 (s), 8.22 (s), 5.53 (breit, NH), 2.50 (s), 2.05 ppm (breit, N H . . .O) irn Verhaltnis 9 : 6 : I : 5 : 1.
C14H22N20S (266.4) Ber. C 63.12 H 8.33 N 10.52 S 12.04 Gef. 63.39 8.83 10.55 11.97
N-rerf.-Butyl-N'-trityl-thiohornstoff-S-munoxid (Ha). - Farblose Plattchen (61 %) vorn Schrnp. 119 - 122" (Zers.), aus der Schmelze Kristalle.
C24H26NZOS (390.6) Ber. C 73.80 H 6.71 N 7.17 S 8.22 Gef. 73.03 6.67 7.37 8.43
N. N'-Di-terr.-omyl-thioharnstoff-S-monoxid (10 b). - Farblose Kristalle (43 %) vom Schrnp. 86-88" (Zers.), aus der Schrnelze Kristalle vorn Schmp. 220" (vgl. entspr. Harnstoff, Schmp.
C,lH24N20S (232.4) Ber. C56.85 H 10.41 N 12.06 S 13.80 Gef. 57.03 10.31 12.10 13.73
228 -230", Subl.).
N-/err. -Amyl- N'-[ I. I .3.3-1etramethyl-butyl] -/hiohorns/ofl-S-monoxid. - Hellgelbe Kristalle (60%) vorn Schmp. 71 -74" (Zers.).
C ~ ~ H ~ ~ N Z O S (274.5) Ber. N 10.21 S 11.68; Gef. N 10.03 S 11.21
N-fert.-Amyl-N'-trity/-rhiohorns/o~-S-monoxid. - Farblose, feine Kristalle (84%) vorn Schrnp. 134"; in Tetrachlormethan gut Ibslich.
C2sH28NzOS (404.6) Ber. C 74.22 H 6.98 N 6.93 S 7.92 Gef. 72.69 7.00 7.15 8.05
N-Cyclohexyl-N'-triryl-rhioharnstoff-S-monoxid (17). - Farblose, rechteckige Blattchen (56%) vorn Urnwandlungspunkt 112". Schmp. 180".
C26H28N20S (416.6) Ber. C 74.96 H 6.78 N 6.73 S 7.70 Gef. 73.36 6.83 6.48 7.43
N.N'-Bis-[I.I.3.3-/etrame/hyl-butylJ-?hioharns/off-S-monoxid (10f). - Farblose Prismen (66%) vorn Schrnp. 99- 101" (Zers.), in Tetrachlorrnethan ldslich.
C I ~ H ~ ~ N ~ O S (316.5) Ber. C64.50 H 11.47 N 8.85 S 10.12 Gef. 64.75 11.37 8.83 10.35
N-[ I . I.3.3-~e1rame1hyl-butyl]-N'-rri/~l~1hiohorns~o~-S-monoxid (13 a). - Farblose Kristalle (82%) vom Schrnp. 116-119" (Zers.).
C2eH34N20S (446.7) Ber. C 75.30 H 7.67 N 6.27 S 7.18 Gef. 74.33 7.88 6.36 6.92

1969 Oxydationsprodukte von Thiocarbonsaureamiden, XIX 77
N. N'-Bis-[a.a-dimerhylbenzylJ-thioharnsroff-S-monoxid (toe). - Farblose Nadeln (69 %, neben 22 % N. N'-Bis-[a.a-dimerhyl-benzyll-harnsfoff, S . 75) vom Schmp. 84 -86" (aus Ather umkristallisierbar). - H-NMR-Spekrrutn (CDC13, -25"): 7 = 8.72 (s), 8.17 (s), 5.15 (breit, NH), 2.83 (m, aromat. H), 2.43 (s, aromat. H) 2.15 ppm (breit, N H . . .O) im Verhaltnis 6 : 6 : I : 5 : 5 : 1.
ClyH24NzOS (328.5) Ber. C 69.47 H 7.37 N 8.53 S 9.76 Gef. 69.47 7.41 8.45 10.07
N-Me~hyl-N'-[a.a-diphenyla~hylJ-thioharnsfoff-S-monoxid (21). - Farblose, verfilzte Na- deln (77 %) vom Schmp. 108 - 1 12" (Zers.).
C ~ ~ H I ~ N ~ O S (286.4) Ber. C 67.10 H 6.34 N 9.78 S 11.19 Gef. 66.24 6.21 9.72 10.87
N- Methyl-N'-frityl-rhioharnstoff-S-monoxid (14). - Farblose Kristalle (65 %) vom Schmp. 170- 172" (Zers.).
C ~ ~ H Z O N Z O S (348.5) Ber. C 72.39 H 5.79 N 8.04 S 9.20 Gef. 71.72 5.79 7.73 8.89
N-ferf.-Bufyl-N'-adamanfyl-(l)-fhioharnsfo~-S-monoxid. - 0.5 g des entsprechenden Thioharnsfoffs wurden in 10 ccm Chloroform + 50 ccm Athanol rnit 4 ccm 30proz. Wasser- sfoffperoxid bei Raumtemperatur oxydiert. Nach 30 Min. wurde in 150 ccm Eiswasser ge- gossen, noch 10 ccm Chloroform zugefiigt, die Chloroform-Phase getrocknet und auf ca. 2 ccm eingeengt. Nach Zugabe von Essigester und Petrolather in kleinen Portionen bei -20" kristallisierte das Produkt langsam aus. Farblose Kristalle (48 %) vom Schmp. 84-86" (Zers.).
ClsH26NzOS (282.5) Ber. C 63.78 H 9.28 N 9.92 S 11.35 Gef. 62.68 9.05 9.97 10.69
N.N-Di-adamantyl-(I)-rhioharnstoff-S-monoxid. - Analog voranstehender Verbindung. Farblose Kristalle (71 %) vom Umwandlungspunkt > 140; bis 200' ist alles geschmolzen, dann bilden sich erneut Kristalle aus der Schmelze.
C ~ ~ H ~ Z N ~ O S (360.6) Ber. C 69.95 H 8.95 N 7.77 S 8.89 Gef. 70.23 8.99 7.62 8.50
N-terf.-Bufyl-N'-[2.6-dimethyl-phenylJ-fhioharnstoff-S-monoxid (29). - I mMol des Thio- harnstoffs 5 in 20 ccm Methanol wurde bei - 10" mit 2 mMol WasserstoJperoxid in 5 ccm Methanol versetzt. Nach 1 Stde. wurde i. Vak. auf ca. 1 ccm eingeengt, mit 2 ccm Aceton versetzt und bis zur beginnenden Kristallisation eingeengt. Die nach 40 Min. bei -20" aus- kristallisierte Substanz wurde abfiltriert und mit kaltem Aceton, Essigester/Petrolather und Petrolather gewaschen. Farblose, sehr feine Krktalle (37%) vom Schmp. 110-1 12" (Zers.); Kristalle aus der Schmelze mit Schmp. 160".
C I ~ H ~ ~ N Z O S (252.4) Ber. C 61.86 H 7.99 N 11.10 S 12.70 Gef. 61.83 8.16 10.94 12.47
N-Cyclohexyl-N'-[2.6-dimerhyl-phenylj-~hioharns~off-S-monoxid (30). - Analog 29. Als Hydrar isoliert; farblose Saulen (71 %) vom Zers.-P. 110- 115", Subl. bei 150", Schmp. 227 -230". Trocknen bei 40" i. Vak. iiber Phosphorpentoxid gab kein wasserfreies S-Monoxid.
C I ~ H ~ ~ N Z O ~ S (296.4) Ber. C 60.78 H 8.16 N 9.45 S 10.81 Gef. 60.85 7.84 9.48 10.84

78 W. Walter und G . Randau Bd. 122
N.N'-Bis-i2.6-dimethyl-phenyl/-rhioharnstoff-S-monoxid (25). - Analog 29 wurden 2 rnMol 6a in 60 ccm Methanol und 10 ccm Aceton gelost und bei -15" rnit 2 rnMol Wasserstof- peroxid in 2 Stdn. oxydiert. Dann wurde i. Vak. auf ca. 5 ccm eingeengt, 5 ccm Aceton zu- gefiigt und auf 2 ccrn eingeengt. Das bei -20" ausgefallene S-Monoxid-hydroi wurde mit Essigester/Petrolather ( 1 : I ) und Petrolather gewaschen. Farblose Prismen (71 %) vorn Urn- wandlungspunkt > 120°, vollstandiges Schrnelzen unter Rotfarbung (225". Das Hydrat konnte nicht wasserfrei erhalten werden.
Cl7H22N202S (318.4) Ber. C 64.1 I H 6.97 N 8.80 S 10.07 Gef. 64.71 6.70 8.75 10.28
N.N'-Bis-j2.6-diarhyl-phenylj-rhioharns~off-S-monoxid (26). - Analog 29 als S-Monoxid- hydrar isoliert. Farblose Nadeln (75 %) vorn Urnwandlungspunkt > 140". vollstandiges Schmelzen (240".
Cz~H3oN202S (374.5) Ber. C 67.34 H 8.07 N 7.48 S 8.56 Gef. 67.08 7.91 7.31 8.66
N.N'-Bis-[2.6-diisopropyl-phenyll-thioharnstoff-SS-monoxid (27). - Analog 29, jedoch wasserfreie, hellrote Blattchen (68 %) vorn Urnwandlungspunkt 185", vollstandiges Schrnelzen bei 210".
C ~ S H ~ ~ Y ~ O S (412.6) Ber. C 72.76 H 8.80 N 6.79 S 7.78 Gef. 72.41 8.93 6.75 7.86
Oxydation von N-Acyl-thioharnstoffen
Oxydation von N-Aceryl-thioharnstoff (31). - Z u 0.5 g 31 in 50 ccrn Athanol wurde bei 0" 1 ccrn 30proz. Wasserstofperoxid gegeben. Nach 1 Stde. wurde auf Raurnternperatur er- wiirrnt, noch I ccrn 30proz. HzO2 zugefiigt und I Stde. stehengelassen. Der ausgefallene Niederschlag von 3.5-Diocetamino-I.Z.4-thiadiazol (32) wurde abfiltriert und mit Wasser ge- waschen. Farblose Nadeln, 90 mg (23 %), vorn Schrnp. 336 -338", Subl. bei 320" (Lit.33) 314 bis 315", Subl. bei 320"). Irn IR-Spektrurn identisch rnit authent. 3232).
Die Oxydation von 2 rnMol 31 in 25 ccm Methanol bei -12" rnit 6 mMol Peressigsuure ergab 32 zu 12% d.Th.
Oxydation von N-Benzoyl-thioharnstoff (33). - a) Zu 0.5 g 33, gelost in 50 ccm Athanol, wurde bei 0" 1 ccm 30prOZ. Wassersroffperoxid gegeben und langsarn auf Raurnternperatur erwarrnt. Nach Stehenlassen iiber Nacht wurden 5 ccm 2n HCI zugefiigt und i. Vak. auf 5 ccrn eingeengt. Bei 5" kristallisierten 85 mg (18%) N-Benzoyl-hornstoff(34) aus, Schmp. 210-212" (Lit.56) 214-215"). Im IR-Spektrum identisch rnit authent. Praparat.
b) 2 mMol 33 in 30 ccrn Methanol wurden bei - 10" rnit 6 mMol Peressigsuure versetzt. Nach 1 Stde. waren farblose Flocken ausgefallen. Es wurde i. Vak. auf die Halfte des Vo- lumens eingeengt und die nach 24 Stdn. bei -20" ausgefallenen Kristalle (100 rng, 31 %) von 3.5-Dibenzarnino-1.2.4-thiudiazol(35) abfiltriert und getrocknet. Hellgelbe Nadeln vom Schrnp. 267-2269' (Lit.33) 263-265").
C ~ ~ H I ~ N ~ O ~ S (324.3) Ber. N 17.28 S 9.88 Gef. N 17.81 S 10.48
56) H. L. Wheeler und T. B. Johnson, Amer. chem. J. 24, 189 (1900).

1969 Oxydationsprodukte von Thiocarbonsaureamiden, XIX 79
Reduktion von substituierten Thioharnstoff-S-oxiden
Reduktion von Phenylazothioformamid-S-oxid57): 100 mg S-Oxid wurden in 10 ccm Chloro- form suspendiert und 10 Min. mit Schwefelwasserstoff gesattigt. Nach 1 Stde. bei Raum- temperatur war bereits I-Phenyl-thiosemicarbazid dunnschichtchromatographisch ( R P =
0.48) nachweisbar; nach 12 Stdn. (Entfarbung) hatte sich ein flockiger, farbloser Niederschlag gebildet. In der Losung war neben Schwefel (RF = 0.90) kein Ausgangsmaterial (RF =
0.63, jeweils mit Chloroform/Methanol = 10 : 1) mehr nachweisbar. Der Niederschlag wurde in Methanol mit Aktivkohle behandelt und nach Einengen aus 1.5 ccm bithanol umkristalli- siert. Farblose Kristalle vom Schmp. 204" (Zers.), Misch-Schmp. mit authent. Materialss) 204-205" (Zers.), Ausbeute 65 mg (67%).
Reduktion von N.N'-Di-tert.-butyl-thioharnstoff-S-monoxid (8). - 100 mg 8 wurden in 5 ccm Chloroform gelost, bei 0" wurde 10 Min. Schwefelwasserstoff eingeleitet. Nach 2 Stdn. waren diinnschichtchromatographisch neben 8 Schwefel und 3a nachweisbar. Es wurde i. Vak. ein- gedampft, der Ruckstand in Methanol/Aceton aufgenommen, ungeloster Schwefel abfiltriert und nach Einengen aus Methanol umkristallisiert. Farblose KristalIe von N.N'-Di-tert.- butyl-thioharnstoff (3a), Schmp. 146- 150" (Zers.; Lit. 20b) 150", Zers.). Im IR-Spektrum iden- tisch rnit authent. 3a.
Reduktion von N-Cyclohexyl-N'-trityl-thioharnstoff-S-monoxid (17). - Analog 8 + 3a ergaben 100 mg 17 50 mg (52%) N-Cyclohexyl-N'-trityl-thioharnstoff vom Schmp. 187 - 192", im IR-Spektrum identisch mit authent. Produkt.
57) Herrn Prof. H. Beyer, Greifswald, sei fur die Uberlassung einer Probe 37 gedankt. 5 8 ) E. Fischer und E. Besfhorn, Liebigs Ann. Chem. 212, 316 (1882).
[ I 37/68] _- .




























![Kurzpräsentation über S-Monitoring Konzept und Produkte [DE]](https://img.pdfslide.org/doc/110x75/559ea42d1a28abff618b4789/kurzpraesentation-ueber-s-monitoring-konzept-und-produkte-de.jpg)
