Embed Size (px)
Citation preview

VIERTELJAHRSSCHRIFTDER NATURFORSCHENDEN
GESELLSCHAFT IN ZÜRICHunter Mitwirkung von
A.U. DANIKER, P. FINSLER, H. FISCHER, A. FREY-WYSSLING
H. GUTERSOI-IN, P. KARRER, B. MILT, P. NIGGLI, P. SCHERRER
H. R. SCHINZ, FR. STUSSI und M. WALDMEIER
herausgegeben von
HANS STEINER
Beiheft Nr. 1
Das Polyhydrat des Kalzium-OxalatesVon
ROLAND HONEGGER
Preis Fr. 4.®
DRUCK UND VERLAG GEBR. FRETZ AG. ZÜRICH


Vierteljahrsschrift der NaturforschendenGesellschaft in Zürich
unter Mitwirkung von
A.U. DÄNIKER, P. FINSLER, H. FISCHER, A. FREY-WYSSLING, H. GÜTERSOHN, P. KARRER, B. MILT,P. NIGGLI, P. SCHERRER, H. R. SCHINZ, FR. STÜSSI und M. WALDMEIGR
herausgegeben von
HANS STEINER, ZÜRICH 7Druck und Verlag: Gebr. Frett AG., Zürich
Nachdruck auch auszugsweise nur mit Quellenangabe gestattet
Jahrgang 97 Beiheft Nr. 1
30. Juni 1952
Das Polyhydrat des Kalzium-OxalatesVon
ROLAND HONEGGER t (Zollikon-Zch.)(Mit 15 Abbildungen im Text)
Inhaltsverzeichnis
I. Tell:Problemstellung
II. Teil:Synthese und röntgenographische UntersuchungenA. Einleitung B. Herstellung synthetischer Kristalle 6
a) Herstellung des Kalzium-Oxalat-Monohydrates 7b) Herstellung des Polyhydrates 7
aa) Herstellung eines Kristallpulvers für die Pulveraufnahmen . 7bb) Herstellung von grösseren Einkristallen des Polyhydrates . . 9
C. Röntgenuntersuchungen 101. Pulveraufnahmen 10
a) Die Pulveraufnahmen des Monohydrates 10b) Pulveraufnahmen am Anhydrat 12c) Pulveraufnahmen des Polyhydrates 12
2. Ermittlung der Präzisionsgitterkonstanten und Indizierung der Polyhydrat-reflexe 17
3. Drehkristallaufnahmen 18a) Bestimmung der Gitterkonstanten 18
aa) Bei Drehung um [001] 18bb) Drehung um [100] 20cc) Drehung um [110] 20dd) Drehung um [111] 20
3
5
1

b) Indizierung der Drehkristallaufnahmen 21aa) Indizierung des Äquators bel Drehung um [001] 21bb) Indizierung der Schichtlinien 21
4. Goniometeraufnahmen 23a) Drehung um [001] 24b) Drehung um [100] 26c) Drehung um [110] 26
5 Laueaufnahmen 26a) Durchstrahlung parallel der c-Achse 27b) Durchstrahlung parallel der a-Achse 28
6. Ermittlung der Raumgruppe 287. Ermittlung der Anzahl Moleküle in der Elementarzelle . 28
a) Bestimmung der Dichte 28b) Berechnung der Anzahl Moleküle N in der Elementarzelle . 29c) Verteilung der Ca-Atome und C 2O4-Radikale auf die Punktlagen 30
Zusammenfassung des II. Teils 30
III. Teil:Chemisch-physikalische UntersuchungenA. Entwässerungskurven 31
1. Allgemeines 312. Beschreibung der Apparaturen 32
a) Bestimmung des Gewichtsverlustes mit einer analytischen Waage . 32b) Qualitative Bestimmung der wasserärmeren Hydrate des Kalzium-Oxa-
lates auf manometrischem Wege 323. Besprechung der Resultate 33
a) Quantitative Entwässerung mit einer analytischen Waage 33b) Ergebnisse der manometrischen Messungen 37
B. Analytische Wasserbestimmungen 371. Direkte Wasserbestimmungen mit Mikroanalysen 372. Indirekte Wasserbestimmungen 38
Zusammenfassung des III. Teils 39
IV. Teil:Untersuchung pflanzlicher KristalleA. Einleitung 40B. Debye-Scherrer-Aufnahmen von Raphiden und Kristallsand 40
1. Untersuchung von Raphiden 402. Untersuchung des Kristallsandes 40
Zusammenfassung des IV. Teils 42Schlussfolgerungen 42Literaturverzeichnis 43
2

I. Teil
Problemstellung
Im Jahre 1687 entdeckte MALPIGHI in der Pflanze kristallisierte Ausschei-dungen, die später als Kalzium-Oxalat identifiziert wurden. Lange Zeit be-fasste man sich überhaupt nicht oder dann ohne Erfolg mit der chemischenZusammensetzung, bis E. E. SCHMID (1856) auf Grund seiner Untersuchungenan isolierten Kristallen aus M ami l l a r i a fand, dass es sich um das Tri-hydrat CaC 204 . 3H20 handle, während synthetisch hergestellte Kristalle sichals Monohydrat CaC204 . 1 H2O erwiesen. Die beiden Salze unterscheiden sichmikroskopisch durch ihre verschiedene Kristallform, indem die höhere Hydrat-stufe dem tetragonalen System angehört, während das Salz mit niedererHydratstufe monoklin kristallisiert. Polarisationsoptisch unterscheiden sichdie beiden Hydrate durch eine auffallend verschiedene Stärke der Doppel-brechung.
Besonders ausführlich untersuchte A. FREY (1925) die beiden Salze, wobeisich das monoklin kristallisierende Kalzium-Oxalat wiederum als Monohydratund die höhere Hydratstufe als ein Trihydrat erwies. FREY führte seine Unter-suchungen an synthetischem Material durch und erkannte dabei die Schwie-rigkeit, das tetragonal kristallisierende Salz rein herzustellen. Er gewann dieKristalle nach dem Verfahren von A. SOVCHAY und E. LENSSEN (1856), dieerstmals die Synthese des Trihydrates mit Erfolg durchführten. Die bis 0,5 mmgrossen Kriställchen trennte er von dem verunreinigenden Monohydrat abund analysierte das so separierte Material.
Im Jahre 1930 veröffentlichten V. KOHLSCHÜTTER und J. MARTI (1930) eineArbeit, die sich mit den «Bildungsformen des Kalzium-Oxalates» beschäftigte.Sie bestätigten in ihrer Arbeit die Existenz des Mono- und des Trihydrates,fanden aber noch ein Salz, dessen Wassergehalt mit 2,5 Mol angegeben wurde.Allerdings konnten die beiden Forscher nicht mit Sicherheit feststellen, obes sich bei dem genannten Salz, das sie dem triklinen Kristallsystem zuordne-ten, um ein Kalzium-Oxalat handle.
In neuerer Zeit begannen auch die Mediziner sich für das Kalzium-Oxalat,vor allem für die Entstehungsbedingungen dieses Salzes zu interessieren, daes einen integrierenden Bestandteil der Nieren- und Blasensteine bildet. Eingrundlegendes Werk darüber erschien im Jahre 1937 von G. HAMMARSTEN(1937) «Kalzium-Oxalat als Steinbildner in den Harnwegen». HAMMARSTENbeschreibt in ihrer Arbeit wiederum drei Formen des Kalzium-Oxalates, wo-bei das monoklin kristallisierende Salz als Monohydrat bestätigt wird, wäh-rend sie dem tetragonal kristallisierenden Salz nur 2 Mol Wasser zuschreibt.Als Trihydrat bezeichnet sie die nämliche Kristallform, die KOHLSCHÜTTERals ein 2,5-Hydrat betrachtet hat. Nur ordnet HAMMARSTEN ihr Trihydratdem monoklinen System zu, während KOHLSCHÜTTER, wie oben erwähnt, dieseBildungsform dem triklinen System zuweist. Soweit mir bekannt, bezeichnet
3

HAMMARSTEN zum ersten Male die tetragonalen Kristalle als ein Dihydrat.Sie schreibt zwar in ihrer Arbeit:
«Gleichzeitig, jedoch unabhängig voneinander, gelang es JAKOR und LUECZAK und demVerfasser im Jahre 1928 das wirkliche Trihydrat CaC2 O 4 . 3H20 darzustellen, es kristalli-siert in grossen monoklinen Tafeln. Die Oktaederform ist nach JAKOB und LUECZAK's Ana-lysen CaC 2O4 21/4H20, nach Analysen des Verfassers CaC 2O4 2H20. Weil indessen keinervon uns für die Darstellung dieses Hydrates ein Verfahren gefunden hat, das mit SicherheitVerunreinigungen mit Formen der übrigen Kalzium-Oxalate ausschliesst, scheint mir einDisput über die Richtigkeit des Kristallwassergehaltes wenig am Platze. Der Einfachheithalber wird letztere Kristallform hier Dihydrat genannt.»
Das monokline Salz, welches HAMMARSTEN als Trihydrat bezeichnete, be-obachteten wir bei unseren Untersuchungen auch. Da es in der Natur bis anhinnicht festgestellt wurde, befassten wir uns mit dieser Hydratstufe nicht näher.
Auf Grund dieser Äusserungen scheint ein Dihydrat auch nach der An-sicht von HAMMARSTEN nicht mit absoluter Sicherheit vorhanden zu sein. Inden Jahren 1940 und 1941 erfolgten Veröffentlichungen von A. T. JENSEN (1940,1941), der sich intensiv mit der chemischen Zusammensetzung der Harnsteinebefasste. In seinen Darlegungen tritt er für zwei Hydratstufen des wasser-führenden Salzes ein, nämlich für das monokline Monohydrat und für eintetragonal kristallisierendes Dihydrat, wie es schon HAMMARSTEN angenom-men hat. Die Kristallarten, welche in Blasensteinen vorkommen, identifizierteJENSEN mit Hilfe von Debye-Scherrer-Röntgendiagrammen. In seinen Publi-kationen tritt er überzeugt für ein Dihydrat ein. Nach den Angaben der«X-Ray Diffraction Cards» der American Society for Testing Materials han-delt es sich bei den tetragonalen Kristallen jedoch um ein Trihydrat.
Die neuesten Handbücher der Chemie und der Physik bezeichnen die zu-letzt genannte Kristallart ebenfalls als ein Trihydrat. Für den Mineralogenist das Kalzium-Oxalat mit seinen beiden Hydraten insofern interessant, alses in der Natur als Mineral auftritt. Das monokline Monohydrat wird alsWhewellit bezeichnet, während die tetragonale Hydratstufe, die nachF. A. BANNISTER, M. H. HEY, K. P. OAKLEY (1947) im Weddell Sea vorkommt,W e d d e 1 i t genannt wird und nach den erwähnten Verfassern einKalzium-Oxalat-Dihydrat ist. Der Whewellit ist schon seit längerer Zeit be-kannt. Seine Flächenformen sind schon im Jahre 1924 von A. FREY (1924) mitdenjenigen der pflanzlichen Kristalle verglichen worden. Beim Weddelit hin-gegen handelt es sich um eine neuere Bezeichnung für ein tetragonales Ca(C00) 2 •2 H2O.
Die bisherigen Ausführungen zeigen, dass nur hinsichtlich der höherenHydratstufe des Kalzium-Oxalates, die wir im folgenden als Polyhydrat be-zeichnen, Meinungsverschiedenheiten bestehen. Der eine Teil der Forscherentscheidet sich für ein Dihydrat, während ein anderer Teil sich zu einemTrihydrat bekennt. Wiederum andere behaupten die Existenz nicht nur vonzwei, sondern sogar von drei Hydratstufen des Kalzium-Oxalates. Die Auf-gabe der vorliegenden Arbeit soll nun darin bestehen, einen Beitrag zur Klä-
4

rung dieser Streitfragen zu liefern, d. h. zu bestimmen, um welche Hydratstufees sich beim tetragonalen Polyhydrat handelt.
Bei meinen Untersuchungen bin ich von folgenden Überlegungen ausgegan-gen: Den Hydratstufen des Kalzium-Oxalates kommt in verschiedenen Ge-bieten der Naturwissenschaften Bedeutung zu:
1. In der Biologie spielen diese Salze mit ihrem Formenreichtum als Rekreteder Pflanzen eine grosse Rolle.
2. In der Medizin erhalten sie Bedeutung als wichtige Bestandteile derHarnsteine.
3. Da sie in der Natur als Minerale auftreten, werden sie auch Forschungs-objekt der Mineralogie.
Es ist daher notwendig, in einem zweiten Teil diese Hydrate des Kalzium-Oxalates feinstrukturell eindeutig zu charakterisieren, und zwar vorerst durchihre Debye-Scherrer-Röntgendiagramme. An Hand anderer Röntgenaufnah-men soll die Raumgruppe der umstrittenen Hydratstufe bestimmt werden, ummit ihrer Hilfe Aussagen über den möglichen Wassergehalt machen zu können.In einem dritten chemisch-physikalischen Teil soll mit verschiedenen Metho-den der Wassergehalt der tetragonalen Hydratstufe direkt und indirekt be-stimmt werden. Der Wassergehalt wird an Hand von Titrationen und Entwäs-serungskurven ermittelt werden. In einem vierten Teil werden die Resultatedes zweiten Teiles dazu benützt, um bestimmte pflanzliche Exkrete zu charak-terisieren.
II. Teil
Synthese und röntgenographische Untersuchungen
A. Einleitung
Die Tatsache, dass der kristalline Zustand bei biologischen Objekten vielhäufiger auftritt, als man früher angenommen hatte, führte in neuerer Zeitdazu, diesen Zustand mit mineralogischen Methoden zu charakterisieren. Oftist der kristalline Anteil bei bestimmten Stoffen weder makroskopisch nochmikroskopisch zu erkennen, und es ist daher notwendig, Art, Grösse und Güteder Kristalle mit Röntgenstrahlen zu bestimmen, mit denen noch Kristallitebis hinunter zu Abmessungen von 10-' cm identifiziert werden können. In-folgedessen lassen sich mit Hilfe der Röntgenstrahlen Aussagen über den sub-mikroskopischen Bau von kristallinen Stoffen machen. Die Anwendung derRöntgenographie auf dem Gebiete der Biologie spielt eine dreifache Rolle:Sie dient zur Feststellung von kristallinen Bereichen, zur Identifizierung vonStoffen und zur Strukturbestimmung von Kristallen; letztere ermöglicht esunter Umständen, die chemische Zusammensetzung abzuklären, wenn mannicht auf rein chemischem Wege zum Ziele kommt. Der Vorteil der röntgeno-graphischen Methode gegenüber der chemisch-analytischen besteht vor allemdarin, dass sich Pulveraufnahmen bereits an sehr geringen Mengen (1 mm8)durchführen lassen.
5

Sodann können Verbindungen voneinander unterschieden werden, welchegleiche chemische Zusammensetzung aufweisen, aber in verschiedenen Er-scheinungsformen (Modifikationen) kristallisieren. E. BRANDENBERGER undH. R. SCHINZ (1948).
Für die biologischen Probleme ist es von Bedeutung, diese Modifikationenbestimmen zu können, da deren Entstehungsbedingungen voneinander abwei-chen, so dass Rückschlüsse auf die physikalisch-chemischen Verhältnisse inden Geweben des betreffenden Organismus gezogen werden können.
Auch zur Identifizierung der verschiedenen Kalzium-Oxalathydrate eignensich die Röntgenmethoden vorzüglich, da sich Debye-Scherrer-Aufnahmen vonreinen synthetischen und von aus Pflanzen isolierten Kristallen miteinandervergleichen lassen. Die kleinen Mengen verunreinigender Substanzen spielendabei keine Rolle. Demgegenüber würde eine chemische Analyse von pflanz-lichen Stoffen (vgl. IV. Teil) infolge der kleinen verfügbaren Mengen und dergrossen Unreinheit des Ausgangsmaterials, die der Vermischung verschiede-ner Pflanzensäfte bei der Isolation zuzuschreiben ist, ihren Zweck vollkom-men verfehlen. Entsprechend wurde gefunden, dass rein chemische Unter-suchungen von Blasensteinen nicht zum Ziele führen, da die Steine öfters ausmehreren Kristallarten bestehen. W. EPPRECHT und H. R. SCHINZ (1950). Sosind z. B. die Oxalatsteine oft ein Gemisch von Mono- und Polyhydrat. Ge-mische von kristallinen Stoffen können nun aber besonders gut an Hand derPulveraufnahmen unterschieden werden. Im folgenden Abschnitt sollen diePulveraufnahmen des Mono- und Polyhydrates erläutert werden.
B. Herstellung synthetischer Kristalle
Um Testaufnahmen anfertigen zu können, ist es notwendig, dass ein che-misch reines, homogenes Material vorliegt. Aus diesem Grunde mussten diebeiden Hydrate des Kalzium-Oxalates synthetisch hergestellt werden. Dabeiwar es notwendig, zunächst die Bildungsbedingungen des Mono- und desPolyhydrates einzuhalten, die für die beiden Hydratstufen verschieden sind.
Nach A. FREY (1925) bildet sich das Monohydrat bei Temperaturen von0-100° C in Lösungen von beliebigem Dampfdruck, und es ist im erwähntenBildungsbereich absolut stabil. Das Polyhydrat hingegen entsteht nur ineinem Temperaturbereich von 0 bis ca. 30° C in verdünnten Lösungen undbei Übersättigung an Kalzium-Oxalat. Im Gegensatz zum Monohydrat ist dasPolyhydrat in dem erwähnten Bildungsbereich nur metastabil; es hat dieTendenz, sich in das Monohydrat umzuwandeln. Von den beiden heteromor-phen Kristallarten der Hydrate des Kalzium-Oxalates ist also nur eine Formbeständig, da sich das Polyhydrat in einem irreversiblen, monotropen, d. h.nur in einer Richtung ablaufenden Vorgang, in das Monohydrat umwandelt.Wenn dieser Prozess bei einem bestimmten Umwandlungspunkt monotropabläuft, so ist dies nur möglich, wenn die höhere Hydratstufe die energie-reichere, d. h. entropieärmere und unbeständigere Form darstellt. Diese un-beständige Form hat die Tendenz, sich in die beständige Form mit dem Maxi-
6

mum an Entropie und einem Minimum an Energie umzuwandeln. Da dasPolyhydrat einen höheren Dampfdruck aufweist als das Monohydrat, sind dieEntstehungs- und Haltbarkeitsbedingungen für die höhere Hydratstufe ge-geben. Im übrigen ist es eine vielfach bestätigte Tatsache, dass sich Salze mithöheren Hydratstufen allgemein aus stark verdünnten Lösungen und bei nie-deren Temperaturen bilden.
a) Herstellung des Kalzium-Oxalat-Monohydrates
Die Kalzium-Oxalat-Monohydratkristalle gehören, wie schon betont, demmonoklinen System an. Sie sind optisch positiv und lassen sich infolge ihrerschiefen Auslöschung leicht identifizieren.
Zur Herstellung von Kristallen mit einheitlichem Habitus bediente ich michdes folgenden Verfahrens, welches H. A. KLASENS, W. G. PERDOK, P. TERPSTRA(1937) zur Gewinnung von Strontiumoxalat-2,5-Hydrat verwendeten:
Ein Becherglas, das zum Teil mit 200 cm 3 Wasser gefüllt ist, und in welchem 10 g Oxal-säure gelöst sind, wird in ein grösseres Becherglas gestellt, das eine Lösung von 15 gKalziumnitrat und 20 cm3 Salpetersäure im Verhältnis 1:5 in 200 cm3 Wasser enthält.Diese beiden Lösungen werden durch Diffusion miteinander in Kontakt gebracht, indemlangsam mit Hilfe einer Kapillare Wasser in das grössere Becherglas zugeführt wlrd. So-bald das grössere Becherglas soweit mit Wasser gefüllt ist, dass die beiden Wasserspiegeldas gleiche Niveau erreichen, setzt die Bildung von Kalzium-Oxalat ein. Dieses Verfahrenwird als diachrone Fällung bezeichnet. Nach ein paar Wochen ist die Oberfläche mit einerSchicht bedeckt, die aus lauter monoklinen, isodiametrischen Kristallen besteht, die eineGrösse von ca. 0,01 mm besitzen.
Diese Kristalle haben den gleichen Habitus wie diejenigen im Assimila-tionsgewebe der Pflanze H e d y c h i u m gar d e n a r i u m. Zur Reinigungwerden die in Wasser praktisch unlöslichen Kristalle etwa eine Stunde langgründlich unter einem Rührwerk gewaschen. Auf diese Weise werden dieKristalle von noch anhaftenden Fremdionen getrennt und können nun alsreines Monohydrat der Röntgenanalyse unterworfen werden.
Es handelt sich bei diesem Verfahren um eine diachrone Fällung (vgl.V. KOHLSCHÜTTER und J. MARTI [1930]), weil die beiden Komponenten nurnach und nach in das System eingeführt werden und miteinander reagieren.
b) Herstellung des PolyhydratesDas Polyhydrat wurde auf zwei verschiedene Arten hergestellt. Durch das
eine Verfahren wurde Substanz gewonnen, die zur Anfertigung von Pulver-aufnahmen genügte, aber für die Herstellung von Drehkristall- und Röntgen-goniometeraufnahmen infolge der Kleinheit der Kristalle nicht taugte. Ausdiesem Grunde musste ein weiteres Verfahren berücksichtigt werden, das dieSynthese von mindestens 0,1 mm grossen Kristallen ermöglichte.
aa) Herstellung eines Kristallpulvers für die Pulveraufnahmen
Anfänglich wurden verschiedene Methoden ausprobiert, die aber infolgeder Unreinheit des Produktes (Gemisch von Mono- und Polyhydrat) nicht
7

Kältelösung
Luft
Harn
Abb.1 Darstellung der Apparatur zur Herstellung von Polyhydrat.
brauchbar waren. Schliesslich wurde ein Apparat gebaut, mit welchem durcheine diachrone Fällung reines Polyhydrat erhalten werden konnte. Hiezuwurde eine Druckflasche mit drei Öffnungen benützt, von denen zwei mit jeeinem Scheidetrichter versehen wurden. Der eine dieser Trichter enthielt100 cm 3 einer 1/100 normalen Lösung von Kalziumhydrat in einer 1%igenLösung von Kalziumpektinat vom Veresterungsgrad 78, der andere 50 cm3einer 1/100 normalen Oxalsäurelösung. In der auf —4° C gekühlten Druckflaschewaren 200 cm3 Harn vorhanden. Das ganze System wurde unter eine Atmo-sphäre Überdruck gesetzt, indem mit Wasserdampf gesättigte Luft in dasSystem eingepresst wurde. Durch die Zugabe der Lösungsgenossen Harnstoffund Kalziumpektinat wurden Bedingungen geschaffen, die sich für eine Ent-stehung des Polyhydrates günstig auswirkten (s. Abb. 1).
Durch die Scheidetrichter tropften nun die beiden Reaktionskomponentenlangsam in das System, um dort miteinander in Kontakt zu treten. Der fürdie Bildung des Polyhydrates nötige Überschuss an Ca-Ionen wurde durchdas Kalziumpektinat und das Kalziumnitrat gewährleistet. Nach Beendigungder Reaktion wurde. das Produkt in einer Zentrifuge von der Harnflüssigkeitgetrennt und durch mehrmaliges Waschen mit destilliertem Wasser und durchZentrifugieren gereinigt. Die erhaltenen Kriställchen hatten eine Grösse vonca. 10 1u (s. Abb. 2).
Sie zeigten bei Zimmertemperatur schon nach 20 Stunden eine begin-nende Umwandlung ins Monohydrat; im Kühlschrank hingegen liessen sie sichein paar Monate halten, ohne dass Korrosionen wahrgenommen werden konn-ten. Pulveraufnahmen von solchen Präparaten bestätigen, dass bei tiefen Tem-peraturen solche metastabile Verbindungen längere Zeit ohne Veränderungenaufbewahrt werden können. (Siehe Diss. A. FREY, 1925).
8

Abb. 2 Ca. 10 μ grosse Polyhydratkriställchen mit Kristallkeimen. Vergr. 500 mal.
bb) Herstellung von grösseren Einkristallen des Polyhydrates
Schon SOUCHAY und LENSSEN (1856) betonten, dass sich Polyhydrat bilde,wenn man Kalzium-Oxalat nahe bis zur Sättigung in heisse, konzentrierteSalzsäure vom spezifischen Gewichte 1,1 eintrage. Ein besonders günstigesVerfahren beschrieben H. A. KLASENS, W. G. PERDOK, P. TERPSTRA (1937).Danach werden 5 g reines Kalzium-Oxalat-Monohydrat in 100 cm 3 20%iger,siedender Salzsäure gelöst. Beim Abkühlen scheiden sich schöne tetragonaleBipyramiden mit Prismen aus. Die Reinheit des Produktes ist sehr von derForm der Kristallisierschale abhängig. Als günstig erwies sich eine rundeSchale von 14 cm Durchmesser. Die so gewonnenen Kristalle sind von Ver-unreinigungen mit Oxalsäure frei. Die grössten Kristalle erreichten eine Längevon 3 mm. Sie wurden in destilliertem Wasser etwa eine halbe Stunde langgewaschen, bis das Wasser mit AgNO 3 keine Chlorreaktion mehr ergab.
Als Ausgangsstoff wurde reinstes Kalzium-Oxalat-Monohydrat, geliefertvon J. T. Baker Chemical Co., Phillipsburg, New Jersey, verwendet. Diesehr geringen Verunreinigungen bestanden in
Cl 0,001 % Fe 0,002 %SO4 0,02 % Ba 0,01N 0,001 % Mg und Alk 0,2 %
Bei diesem Verfahren handelt es sich um eine Umkristallisation, bei dernach Auflösung des monoklinen Monohydrates in Salzsäure das tetragonalePolyhydrat entsteht. KOHL (1889) beschreibt zahlreiche Verfahren zur Her-stellung von reinem Polyhydrat, die sich aber bei meinen Versuchen nichtmit Erfolg anwenden liessen, da sich immer Verunreinigungen mit Monohydratergaben. Für die Anfertigung von Test-Pulveraufnahmen würden solche Prä-parate, d. h. also Gemische von Mono- und Polyhydrat, eventuell noch taugen,wenn das Monohydrat in Gehalten unter seiner röntgenometrischen Nachweis-
9

barkeit vorläge. Für die späteren, im III. Teil zu behandelnden chemischenUntersuchungen könnten solche Mischpräparate nicht verwendet werden, weildie Verunreinigungen die chemischen Analysen wesentlich beeinflussten.
C. Röntgenuntersuchungen
1. Pulveraufnahmen
Die Pulveraufnahmen wurden mit einer Siemens-Feinstrukturröhre mitChromanode angefertigt (mittlere Wellenlänge der K«-Strahlung = 2,2869 Å).Der Durchmesser der Kamera betrug 114,4 mm. Die Belichtungszeiten warenauf zwei Stunden angesetzt, und die Röhre wurde mit 37 KV Spannung und9 mA Stromstärke betrieben. Die Chromstrahlung hat gegenüber der Kupfer-strahlung den Vorteil, dass infolge der grösseren Wellenlänge die Interferenzenweiter auseinander gezogen werden, so dass nahe beieinanderliegende Reflexebesser gemessen werden können.
a) Die Pulveraufnahmen des Monohydrates (Whewellit)
Von den synthetischen Monohydratkristallen wurden nur Pulveraufnahmenangefertigt, da zur Herstellung von Dreh- und Röntgen-Goniometerdiagram-men keine grösseren Kristalle zur Verfügung standen. Eine Raumgruppen-bestimmung der niedrigen Hydratstufe ist zur Abklärung des vorliegendenProblems auch nicht notwendig, weil die monokline Form als Monohydratsichergestellt ist. Die Netzebenenabstände, die sich aus der BRAGGSChen Glei-chung n • λ = 2 • R oaco . sin 19 und an Hand der Internationalen Tabellen(II. Band 1935b) ermitteln lassen, wurden mit denjenigen verglichen, die vonanderen Verfassern gewonnen worden sind. Es zeigte sich, dass die von ihnengefundenen Werte nur teilweise mit meinen Ergebnissen übereinstimmten.Die Aufnahme eines Harnsteines, der aus Whewellit bestand, ergab eine voll-kommene Identität mit dem Interferenzbild der synthetischen Kristalle.
In Tabelle 1 sind die d-Werte des Monohydrates verschiedener Verfassereinander gegenübergestellt. Da jeder Röntgenreflex nicht nur durch seineLage, sondern auch durch die Intensität charakterisiert ist, wurde die Intensi-tät der Reflexe durch Buchstaben gekennzeichnet, wobei stst = sehr stark,st = stark, mst = mittelstark, m = mittel, s = schwach usw. bedeuten. (E.BRANDENBERGER, FR. DE QUERVAIN, H. R. SCHINZ [1947]) . Diese Intensitätszah-len geben aber keine absoluten Werte an, da die Reflexe nicht photometriertwurden. Diese Art der Intensitätsschätzung ist natürlich sehr grob und eigent-lich nur für die stärksten Interferenzen brauchbar.
Zur Bestimmung der genauen Lage der Interferenzen werden normaler-weise dem Pulver Eichstoffe wie Au oder NaC1 beigegeben. Bei den Mono-hydrataufnahmen wurden diese weggelassen, da zur Charakterisierung derSubstanz die unkorrigierten d-Werte für unsere Zwecke vollauf genügten. Da
10
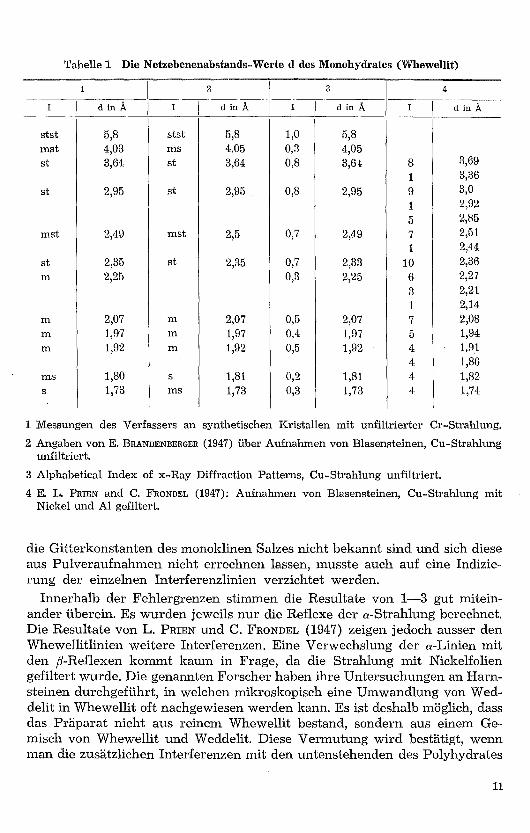
Tabelle 1 Die Netzebenenabstands -Werte d des Monohydrates (Whewellit)
1 2 3 4
I d in A I d in A I d in A I d in A
stst 5,8 stst 5,8 1,0 5,8mst 4,03 ms 4.05 0,3 4,05st 3,64 st 3,64 0,8 3,61 8 3,69
1 3,36
st 2,95 st 2,95 0,8 2,95 9 3,01 2,925 2,85
mst 2,49 mst 2,5 0,7 2,49 7 2,511 2,44
st 2,35 st 2,35 0,7 2,33 10 2,36
m 2,25 0,3 2,25 6 2,273 2,21
1 2,14m 2,07 m 2,07 0,5 2,07 7 2,08m l,97 m 1,97 0,4 1,97 5 1,94m 1,92 m 1,92 0,5 1,92 4 1,91
4 1,86ms 1,80 s 1,81 0,2 1,81 4 1,82s 1,73 ms 1,73 0,3 1,73 4 1,74
1 Messungen des Verfassers an synthetischen Kristallen mit unfiltrierter Cr-Strahlung.
2 Angaben von E. BRANDENBERGER (1947) über Aufnahmen von Blasensteinen, Cu-Strahlungunfiltriert.
3 Alphabetical Index of x-Ray Diffraction Patterns, Cu-Strahlung unfiltriert.
4 E. L. PRIEN and C. FRONDEL (1947): Aufnahmen von Blasensteinen, Cu-Strahlung mitNickel und Al gefiltert.
die Gitterkonstanten des monoklinen Salzes nicht bekannt sind und sich dieseaus Pulveraufnahmen nicht errechnen lassen, musste auch auf eine Indizie-rung der einzelnen Interferenzlinien verzichtet werden.
Innerhalb der Fehlergrenzen stimmen die Resultate von 1-3 gut mitein-ander überein. Es wurden jeweils nur die Reflexe der a-Strahlung berechnet.Die Resultate von L. PRIEN und C. FRONDEL (1947) zeigen jedoch ausser denWhewellitlinien weitere Interferenzen. Eine Verwechslung der a-Linien mitden β-Reflexen kommt kaum in Frage, da die Strahlung mit Nickelfoliengefiltert wurde. Die genannten Forscher haben ihre Untersuchungen an Harn-steinen durchgeführt, in welchen mikroskopisch eine Umwandlung von Wed-delit in Whewellit oft nachgewiesen werden kann. Es ist deshalb möglich, dassdas Präparat nicht aus reinem Whewellit bestand, sondern aus einem Ge-misch von Whewellit und Weddelit. Diese Vermutung wird bestätigt, wennman die zusätzlichen Interferenzen mit den untenstehenden des Polyhydrates
11

vergleicht, welche ziemlich genau mit den Reflexen des Polyhydrates über-einstimmen:
PRIEN und FRONDEL:zusätzliche Linien (a-Werte)
Polyhydrat (Weddelit):
3,69 A 3,68 A3,36 A 3,38 A2,44 A 2,41 A2,21 A 2,20 A2,14 A 2,12 A1,91 A 1,90 A
Das Präparat von PRIEN und FRONDEL kann daher als ein Weddelit aufge-fasst werden, der sich grösstenteils schon in das Monohydrat umgewandelt hat.Die Umwandlungserscheinungen des Polyhydrates werden weiter unten nochgenauer untersucht.
Bei der Entstehung der Harnsteine hat man anzunehmen, dass zuerst diehöhere Hydratstufe gebildet wird und die niedrige durch Umwandlung ausder höheren zustande kommt Der umgekehrte Fall ist unmöglich, weil dieUmwandlung Polyhydrat-Monohydrat monotrop ist, also nur in einer Rich-tung vor sich gehen kann.
b) Pulveraufnahmen am Anhydrat
Eine interessante Erscheinung ergab sich beim Entwässern des Monohydra-tes, das in das wasserfreie Kalzium-Oxalat übergeht, welches als Anhydratbezeichnet werden soll. Eine Pulveraufnahme dieses Anhydrates ergab dasgleiche Interferenzbild wie das Monohydrat. Sie ist noch interessanter, wennman in Betracht zieht, dass die Wassermoleküle im Kristallgitter sehr wahr-scheinlich genau definierte Stellen einnehmen, da das Monohydrat als einestabile Hydratstufe aufgefasst werden muss. Umwandlungserscheinungen anden Monohydrat-Kristallen konnten nirgends beobachtet werden, auch nichtAnzeichen, die auf einen zeolithischen Charakter mit vagabundierendem Was-ser hinzuweisen vermöchten (vgl. Abb. 3) .
Das Anhydrat wurde bis zur Röntgenaufnahme in zugeschmolzenen Glas-kapillaren aufbewahrt. Die Intensitäten der Monohydrat-Interferenzen undder Anhydratlinien wurden mit dem MoLLschen Photometer gemessen, wobeies sich herausstellte, dass die Intensitäten des Anhydrates gegenüber denendes Monohydrates nach höheren Beugungswinkeln hin wesentlich schneller ab-fallen, was darauf schliessen lässt, dass das Anhydrat aus kleineren Kristallenbesteht, die zudem gestörten Gitterbau aufweisen dürften.
c) Pulveraufnahmen des Polyhydrates (Weddelit)
Wie früher beschrieben, wurde das Polyhydrat auf zwei verschiedene Artenhergestellt: durch eine diachrone Fällung und durch Umkristallisation. Esfragte sich zunächst, ob die nach den verschiedenen Methoden hergestellten
12

Abb. 3 Vergleich zwischen Anhydrat und Monohydrat.a) Pulverdiagramm des Monohydrates. Cr-K-Strahlung, Kameraradius 57,2 mm.
b) Pulverdiagramm des Anhydrates.
Kristalle das nämliche Interferenzsystem ergeben, oder ob sich Unterschiedezeigen, aus denen auf eine Störung des Gitters bei der einen Herstellungsartzu schliessen wäre. Eine Umwandlung des Polyhydrates in das Monohydratkonnte beim Pulverisieren der Kristalle nicht festgestellt werden.
Demgegenüber schreiben KOHLSCHÜTTER und J. MARTI (1930) in ihrer Ar-beit, dass sich das tetragonale Trihydrat (Polyhydrat) beim Pulverisieren indas Monohydrat umwandle. Dies konnte bei meinen Aufnahmen nicht be-stätigt werden. Die Instabilität dieser Polyhydratkristalle spielt unseres Er-achtens beim Pulverisieren keine Rolle. Die Kristalle, von denen KOHLSCHüT-TER Diagramme hergestellt hat, haben sich vermutlich infolge ihrer mikro-skopischen Grösse schon vor der Röntgenanalyse umgewandelt.
Da die vorliegende Arbeit ihren Zweck nur dann erfüllt, wenn die an syn-thetischem Material gewonnenen Resultate auch für die in der Natur auf-tretenden Kristalle Geltung haben, war es nötig, die Diagramme der synthe-tischen und der natürlichen Polyhydratkristalle zu vergleichen. Aus diesemGrunde isolierte ich aus der Schale von A 11 i u m c e p a Kristalle, die etwaeine Länge von 66 μ und eine Breite von 8 μ aufwiesen. Das Isolationsver-fahren war sehr langwierig, doch nach mehreren Versuchen gelang es, einefür die Pulveraufnahme notwendige Menge anzureichern. Die trockenen Zwie-belschalen wurden in einem Blendar fein pulverisiert und hierauf in Chloro-form geschüttelt. In dieser Flüssigkeit sanken die Polyhydratkristalle, die eingrösseres spezifisches Gewicht als die Flüssigkeit aufweisen, zu Boden, wäh-rend die spezifisch leichteren Zellanteile an die Oberfläche stiegen. In einemScheidetrichter setzten sich die Kristalle langsam und konnten abgelassen undin einem Gefäss aufgefangen werden. Da der mikroskopische Befund immernoch Verunreinigungen mit Gewebeteilen zeigte, wurden die Kristalle durchWiederholung der Trennung und unter Benützung von spezifisch noch leich-teren und schwereren Flüssigkeiten wie Bromoform und Tetrachlorkohlen-stoff von fremden Bestandteilen getrennt. Infolge der zahlreichen Wiederho-
b
13
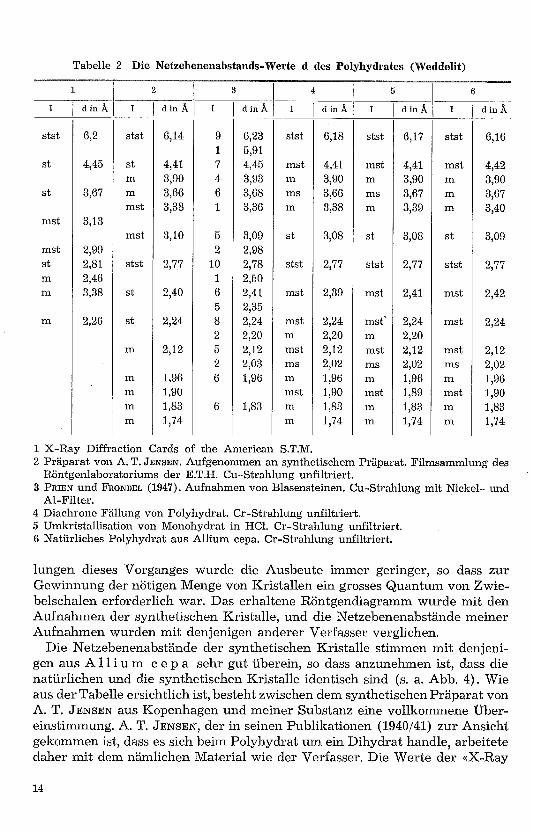
Tabelle 2 Die Netzebenenabstands -Werte d des Polyhydrates (Weddelit)
1 2 3 4 5 6
I dinA I dinA I dinA I dinA I dinA I dinA
stst 6,2 stst 6,14 9 6,23 stst 6,18 stst 6,17 stst 6,161 5,91
st 4,45 st 4,41 7 4,45 mst 4,41 mst 4,41 mst 4,42m 3,90 4 3,93 m 3,90 m 3,90 m 3,90
st 3,67 m 3,66 6 3,68 ms 3,66 ms 3,67 m 3,67mst 3,38 1 3,36 m 3,38 m 3,39 m 3,40
mst 3,13mst 3,10 5 3,09 st 3,08 st 3,08 st 3,09
mst 2,99 2 2,98st 2,81 stst 2,77 10 2,78 stst 2,77 stst 2,77 stst 2,77m 2,46 1 2,50m 3,38 st 2,40 6 2,41 mst 2,39 mst 2,41 mst 2,42
5 2,35m 2,26 st 2,24 8 2,24 mst 2,24 rose 2,24 mst 2,24
2 2,20 m 2,20 m 2,20m 2,12 5 2,12 mst 2,12 mst 2,12 mst 2,12
2 2,03 ms 2,02 ms 2,02 ms 2,02m 1,96 6 1,96 m 1,96 m 1,96 m 1,96m 1,90 mst l,90 rust 1,89 mst 1,90m 1,83 6 1,83 m 1,83 m 1,83 m 1,83m 1,74 m 1,74 m 1,74 m l,74
1 X-Ray Diffraction Cards of the American S.T.M.2 Präparat von A. T. JENSEN. Aufgenommen an synthetischem Präparat. Filmsammlung des
Röntgenlaboratoriums der E.T.H. Cu-Strahlung unfiltriert.3 PRIEN und FRONDEL (1947). Aufnahmen von Blasensteinen. Cu-Strahlung mit Nickel- und
Al-Filter.4 Diachrone Fällung von Polyhydrat. Cr-Strahlung unfiltriert.5 Umkristallisation von Monohydrat in HCl. Cr-Strahlung unfiltriert.6 Natürliches Polyhydrat aus Allium cepa. Cr-Strahlung unfiltriert.
lungen dieses Vorganges wurde die Ausbeute immer geringer, so dass zurGewinnung der nötigen Menge von Kristallen ein grosses Quantum von Zwie-belschalen erforderlich war. Das erhaltene Röntgendiagramm wurde mit denAufnahmen der synthetischen Kristalle, und die Netzebenenabstände meinerAufnahmen wurden mit denjenigen anderer Verfasser verglichen.
Die Netzebenenabstände der synthetischen Kristalle stimmen mit denjeni-gen aus Allium c e p a sehr gut überein, so dass anzunehmen ist, dass dienatürlichen und die synthetischen Kristalle identisch sind (s. a. Abb. 4). Wieaus der Tabelle ersichtlich ist, besteht zwischen dem synthetischen Präparat vonA. T. JENSEN aus Kopenhagen und meiner Substanz eine vollkommene Über-einstimmung. A. T. JENSEN, der in seinen Publikationen (1940/41) zur Ansichtgekommen ist, dass es sich beim Polyhydrat um ein Dihydrat handle, arbeitetedaher mit dem nämlichen Material wie der Verfasser. Die Werte der «X-Ray
14

Abb. 4 Vergleich zwischen synthetischem und natürlichem Polyhydrat.a) Synthetisches Polyhydrat. Cr-K-Strahlung, Kameraradius 57,2 mm.
b) Polyhydrat aus Allium cepa.
Abb. 5 Vergleich der beiden auf verschiedenen Wegen synthetisierten Polyhydratkristalle.a) Polyhydrat durch Umkristallisation in HCl hergestellt. Cr-K-Strahlung. b) Polyhydratdurch diachrone Fällung gewonnen. Aufnahme an 4 Monate alten Kristallen (bei niedriger
Temperatur aufbewahrt).
Diffraction Cards» der American Society for Testing Materials stimmen mit denübrigen Werten nur teilweise überein, so dass anzunehmen ist, dass die Un-tersuchungen nicht an frischem Material vorgenommen worden sind, weshalbauch Netzebenenabstände auftreten, welche dem Monohydrat zuzuordnen sind.Die Netzebenenabstände der beiden synthetischen Präparate (diachrone Fäl-lung, Umkristallisation) stimmen miteinander überein. Es ist daher nachge-wiesen, dass die beiden synthetischen Präparate einander gleich sind. (SieheAbbildung 5.)
Die Umwandlungserscheinungen des Polyhydrates wurden röntgenogra-phisch festgehalten:
Frisch gefällte, nicht pulverisierte Polyhydratkristalle wurden währendeiner Stunde bei 100° C im Trockenschrank getrocknet, worauf eine Pulver-aufnahme angefertigt wurde. Nach zwei, drei und vier Stunden wurden wei-
15

tere Aufnahmen gemacht. Es konnte dabei an Pulvern mit vier Stunden Trock-nungszeit die vollständige Umwandlung des Polyhydrates in das Monohydratnachgewiesen werden. Die Aufnahmen nach ein-, zwei- und dreistündigerEntwässerung ergaben bereits Zwischenstadien, wie sie oft bei Gemischenvon Weddelit und Whewellit in Harnsteinen beobachtet wurden. Die inten-siveren Interferenzen (z. B. Reflex (411) mit einem Netzebenenabstand von2,77 Å) bleiben während 3 Stunden noch sichtbar und zeigen höchstens kleineLageverschiebungen. Die schwachen Interferenzlinien verschwinden nachkurzer Zeit. Nach einer Stunde Trocknungszeit ergibt sich aus der Verschie-bung des Reflexes (411) nach einem grösseren Glanzwinkel eine Gitter-schrumpfung.
Nach zwei Stunden ist aus der Lage von (411) eine Gitteraufweitung nach-zuweisen. Der Netzebenenabstand ändert sich von 2,76 Å bis 2,82 Å. Nachdrei Stunden ist die Mehrzahl der Polyhydratinterferenzen verschwunden; da-gegen sind die Reflexe des' Monohydrates neu aufgetreten. Es handelt sichdaher um eine zweiphasige Umwandlung.
Interessante Resultate zeigten auch die Diagramme von natürlich gealter-ten Kristallen. Von 6 Monate alten Kristallen wurden Pulveraufnahmen an-gefertigt unter Zugabe von NaCl als Eichsubstanz. Die mit Hilfe der Stein-salzlinien korrigierten Polyhydrat-Interferenzen frischer Kristalle ergabenfolgende Gitterkonstanten:
aa = 12,302 ± 0,007 Å co = 7,381 ± 0,003 Å.
Die Gitterkonstanten der 6 Monate alten Kristalle wurden zu
ao = 12,34 ± 0,01 Å co = 7,45 ± 0,04 Å ermittelt.
Werden die Kristalle während längerer Zeit an der Luft bei Zimmertem-peratur liegen gelassen, so tritt eine Gitteraufweitung in Richtung der c-Achseein. Bei metastabilen Kristallen kommt somit eine längere Ruhezeit einerTemperaturerhöhung gleich. Das Pulverdiagramm dieser 6 Monate altenKristalle weist gegenüber Aufnahmen frischer Kristalle im übrigen keinemerklichen Differenzen auf.
Werden grössere Einkristalle bei etwa 110° C längere Zeit getrocknet, sobleibt ihre äussere Form erhalten. Die Untersuchung mit dem Polarisa-tionsmikroskop zeigt aber sehr viele kleine, stark doppelbrechende Kriställ-chen an der Oberfläche des nun nicht mehr durchsichtigen Kristalles. Es kanndaraus gefolgert werden, dass der Polyhydratkristall bei der Umwandlung inzahlreiche Monohydratkristalle zerfällt.
Alle Pulverdiagramme weisen bei grossen Beugungswinkeln das charakteri-stische Ka-Doublett auf, was auf eine hohe Güte der Kristalle und eine nor-male Gitterordnung hindeutet.
16
2. Ermittlung der Präzisionsgitterkonstanten undIndizierung der Polyhydratreflexe
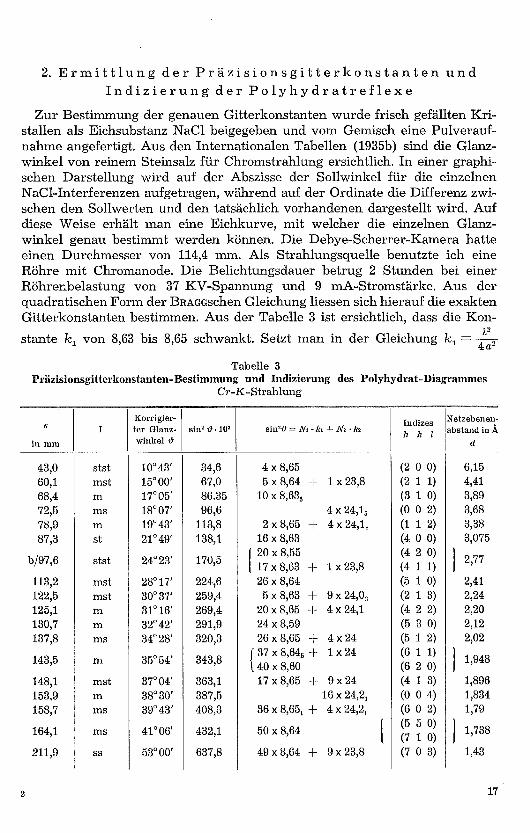
Zur Bestimmung der genauen Gitterkonstanten wurde frisch gefällten Kri-stallen als Eichsubstanz NaC1 beigegeben und vom Gemisch eine Pulverauf-nahme angefertigt. Aus den Internationalen Tabellen (1935b) sind die Glanz..winkel von reinem Steinsalz für Chromstrahlung ersichtlich. In einer graphi-schen Darstellung wird auf der Abszisse der Sollwinkel für die einzelnenNaCl-Interferenzen aufgetragen, während auf der Ordinate die Differenz zwi-schen den Sollwerten und den tatsächlich vorhandenen dargestellt wird. Aufdiese Weise erhält man eine Eichkurve, mit welcher die einzelnen Glanz-winkel genau bestimmt werden können. Die Debye-Scherrer-Kamera hatteeinen Durchmesser von 114,4 mm. Als Strahlungsquelle benutzte ich eineRöhre mit Chromanode. Die Belichtungsdauer betrug 2 Stunden bei einerRöhrenbelastung von 37 KV-Spannung und 9 mA-Stromstärke. Aus derquadratischen Form der BRAGGschen Gleichung liessen sich hierauf die exaktenGitterkonstanten bestimmen. Aus der Tabelle 3 ist ersichtlich, dass die Kon-stante k
Zal von 8,63 bis 8,65 schwankt. Setzt man in der Gleichung 1c, = 12
Tabelle 3Präzisionsgitterkonstanten-Bestinnung und Indizierung des Polyhydrat-Diagrammes
Cr-K-Strahlung
s
in mm
IKorrigierter Glanz-winkel /9
sin' I. 10' sin' I = N - k, + Nz • laIndizesh h 1
Netzebenen-abstand in A
d
43,0 stst 10'43' 34,6 4 x 8,65 (2 0 0) 6,1560,1 mst 15° 00' 67,0 5 x 8,64 + 1 x 23,8 (2 1 1) 4,4168,4 m 17°05' 86,35 10 x 8,635 (3 1 0) 3,8972,5 ms 18° 07' 96,6 4 x 24,1 5 (0 0 2) 3,6878,9 m 19° 43' 113,8 2 x 8,65 + 4 x 24,1 2 (1 1 2) 3,3887,3 st 21°49' 138,1 16 x 8,63 (4 0 0) 3,075
b/97,6 stst 24°23' 170,5 17 x 8,63 + 1 x 23,8 (4 1 1) 2,77
113,2 mst 28°17' 224,6 26 x 8,64 (5 1 0) 2,41122,5 mst 30° 37' 259,4 5 x 8,63 + 9 x 24,03 (2 1 3) 2,24125,1 m 31° 16' 269,4 20 x 8,65 + 4 x 24,1 (4 2 2) 2,20130,7 m 32° 42' 291,9 24 x 8,59 (5 3 0) 2,12137,8 ms 34° 28' 320,3 26 x 8,65 + 4 x 24 (5 1 2) 2,02
143,5 m 35° 54' 343,8 { x 8,60 540+ 1 x 24 (6 1
)1,948
148,1 mst 37° 04' 363,1 17 x 8,65 + 9 x 24 (4 1 3) 1,896153,9158,7
164,1
mms
ms
38° 30'39° 43'
41° 06'
387,5408,3
432,1
36 x 8,65,
50 x 8,64
16 x 24,2,+ 4 x 24,2,
(
(0 0(6 0(5 5
4)2)0)
1,8341,79
11,738l (7 1 0) J
211,9 ss 53° 00' 637,8 49 x 8,64 + 9 x 23,8 (7 0 3) 1,43
2 17

für k1 den Wert 8,63, so ergibt sich eine Gitterkonstante für ao von 12,295 Å.Wird die Rechnung mit dem k1-Wert von 8,65 durchgeführt, so wird ao =12,309 Å. Die Gitterkonstante ao errechnet sich daher zu 12,302 ± 0,007 A.Analog finden wir für co = 7,381 ± 0,003 Å. Dies ergibt ein Achsenverhältnisvon a : c = 1 : 0,6000, welches mit den von FREY angegebenen Werten unterBerücksichtigung der Winkel-Messgenauigkeit an Kleinkristallen im Mikro-skop übereinstimmt.
3. Drehkristallaufnahmen
Die Aufstellung des Kristalles und die Achsenwahl ist aus Abb. 6 ersicht-lich. Im Gegensatz zu FREY (1925) haben wir eine Bipyramide-Il.- und einePrisma-II.-Stellung angenommen. Der Fläche a wird daher der Indizes (100)und der Fläche e der Indizes (101) zugeschrieben. Der Winkel zwischen (100)und (101) berechnet sich aus den Gitterkonstanten zu 59° 02'.
Für das Drehkristallverfahren sind gut ausgebildete Einkristalle von min-destens 0,1 mm Grösse notwendig. Die mikroskopisch als geeignet befundenenKristalle wurden auf einen Glasfaden gekittet. Über die theoretischen Grund-lagen und über die Technik des Drehkristallverfahrens siehe E. BRANDEN-
BERGER (1945) und P. GLOCKER (1949).
a) Bestimmung der Gitterkonstanten
aa) Bei Drehung um [001]
Die Aufnahme wurde in einer Kamera mit dem Radius 28,6 mm und unterVerwendung von Co-Strahlung mit einer mittleren Ka-Wellenlänge von 1,787 Ådurchgeführt (Abb. 7).
Abb. 6 Achsenwahl und Aufstellungdes Kristalles. a = (100), e = (101).
18

Abb. 7 Drehkristallaufnahme um [001].
Der Identitätsabstand c o wurde aus der ersten, zweiten und dritten Schicht- 'linie (Schichtlinienabstand = en ) ermittelt. Die Gitterkonstante wurde alsMittel aus den drei Werten angenommen. Zu ihrer Bestimmung wurden dieZen Werte je an vier Stellen auf der linken und rechten Diagrammseite ge-messen, und daraus wurde das arithmetische Mittel berechnet.
Beispiel für die Berechnung der Gitterkonstante co zwischen den ersten Schichtlinien:
1. Schichtlinie
links (2e1) rechts (2e1)mm mm14,2 14,2.14,3. 14,214,4 14,414,5 14,6
Summe: 57,4 57,4Mittel: 14,35 14,35
2e1 = 14,35 mm e1 = 7,175 mm
Aus der allgemeinen Beziehung co —n •?
e lässt sich co berechnen.sin
1'
log e, = 0,85582log r = 1,45332
log tg l' = 9,39945
19

= = 14°05'
log sin µ = 9,38620
log l,co—u = 0,25212
log sln k = 0,86592
co = 7,344 XAus den nächsten Schichtlinien lassen sich folgende Gitterkonstanten berechnen:
2. Schichtlinie = 7,42 3 A3. Schichtlinie = 7,49, X
Die ermittelte Translationsperiode in c-Richtung streut somit stark, je nachder Schichtlinie, aus der sie bestimmt wurde. co beträgt im Mittel 7,42 ± 0,08 Å.(Genauer Wert siehe Seite 18.)
bb) Drehung um [100]Es wurde eine Kamera vom Radius 57,2 mm verwendet. Es ergab sich für:
ao = 12,29 Å.cc) Drehung um [110]
Um zu entscheiden, ob die gewählten Achsen gemäss Abbildung 6 Seite 18keine Flächenzentrierung ergeben, wurde zunächst abgeklärt, ob das obenbestimmte a° den kleinstmöglichen Wert darstellt. Der Kristall wurde hiezuso justiert, dass die von der a-Richtung um 45° abweichende [110]-RichtungDrehachse wurde (s. Abb. 8) .
Der Radius der Kamera und die Versuchsanordnung waren gleich wiebei bb). Für die Identitätsperiode dieser Flächendiagonale wurde der Wert
= 17,33 Å gefunden. Bei einfach-primitiver Zelle ist 2a 0 2 =τ2. In unseremFalle ergibt sich: 2 X 12,29 2 = 17,382, was beweist, dass das Gitter der Ele-mentarzelle nicht flächenzentriert ist, da sonst der Abstand τ nur halb sogross sein dürfte. Somit führt die gewählte Aufstellung zu einer kleinstmög-lichen Masche der Netzebene (001), was zeigt, dass unsere Aufstellung der-jenigen von FREY vorzuziehen ist.
dd) Drehung um [111]
Schliesslich wurde noch eine Drehkristallaufnahme mit Drehung um [111]hergestellt, um zu entscheiden, ob eine Innenzentrierung vorliege. Es ergabsich hiebei eine Identitätsperiode von 9,4 Å. Im einfach-primitiven Gitterwäre dieselbe 18,9 il (τ 2 = 17,332 + 7,42 2); es liegt somit Innenzentrierung vor.
Abb. 8 Drehung um [110].
20

Abb. 9 Drehung um [001].Vergr. 100 mal.
Abb. 10 Drehung um [100].Vergr. 100 mal.
Man unterscheidet beim tetragonalen System allgemein ein einfach-primi-tives und ein innenzentriertes Elementarparallelepiped. P. NIGGLY (1924).Die basiszentrierte Elementarzelle lässt sich stets in kleinere, einfach-primitiveZellen und das allseitig flächenzentrierte Elementarparallelepiped in ein innen-zentriertes Gitter überführen.
Wie an Hand obiger Ausführungen gezeigt wurde, haben wir eine doppelt-primitive, innenzentrierte Zelle vor uns.
b) Indizierung der Drehkristallaufnahmen
aa) Indizierung des Äquators bei Drehung um [001]
Die quadratische Form der BRAGGschen Gleichung für das tetragonale Systemlautet:
sinn =(h2 a2 (h2 + k 2 ) + 4 ca 12.
Da I für den Äquator = 0 ist, fällt das letzte Glied weg. λ, ao und co sindbekannt. Durch Ausmessung des Filmes für jeden Reflex lässt sich z9 , be-stimmen; die Indizes können somit ermittelt werden, wie dies aus Tabelle 4hervorgeht. Die rechnerisch ermittelten Indizes wurden graphisch mit einerBernal Chart nachgeprüft, J. D. BERNAL (1926). Dabei wurde auch aa gra-phisch ermittelt. Aus dem Wert sin g 79, = 0,0505 ergibt sich eine Gitterkon-stante von ao = 12,6 Å gegenüber der aus der Pulveraufnahme mit Steinsalz-eichung ermittelten von ao = 12,302 Å. Aus der Indizierung des Äquators gehtdas folgende zonale Auslöschungsgesetz hervor:
(hk0) ist nur vorhanden, wenn h + k = 2n ist.
bb) Indizierung der Schichtlinien
Der Glanzwinkel für die Interferenzen auf den Schichtlinien ergibt sich ausfolgender Formel der sphärischen Trigonometrie: cos 2z9 = cos μ • cos a, wobeiμ den Schichtwinkel bedeutet und 2 a den zwischen zwei entsprechendenInterferenzen, z. B. (101) und (101), liegenden Winkel.
21

Tabelle 4 Indizierung des Äquators bei Drehung um [001]
2 s I V sin' 09 . 10'a
• N4aaIndizes
16,3 st 8°09' 20,1 5,03 x 4 (2 0 0)26,0 mst 13° 00' 50,6 5,06 x 10 (3 1 0)33,0 st 16°30' 80,7 5,04x 16 (4 0 0)37,1 ss 18°33' 101,2 5,06x 20 (4 2 0)42,5 mst 21°15' 131,4 5,06 x 26 (5 1 0)48,8 m 24° 24' 170,7 5,02 x 34 (5 3 0)53,4 ss 26°42' 201,9 5,05 x 40 (6 2 0)
60,2 mst 30°06' 251,5 5,02 x 50{ (
7 1 0)t (550)
65,2 m 32°36' 290,3 5,01 x 58 (7 3 0)71,4 mst 35°42' 346,5 5,01 x 68 (8 2 0).75,2 m 37° 36' 372,3 5,03 x 74 (7 5 0)79,0 ss 39°30' 404,6 5,06 x 80 (8 4 0)
107,0 m 53° 30' 646,2 5,05 x 128 (8 8 0)112,0 m 56°00' 687,3 5,05 x 136 (10 6 0)117,0 s 58° 30' 727,0 5,05 x 144 (12 0 0)128,0 s 64°00' 807,8 5,05 x 160 (12 4 0)
Für die erste Schichtlinie ist 1 in der quadratischen Form der BRAGGSChenGleichung nicht mehr 0 sondern 1, für die zweite 2 usw.
2Die Gleichung wird nun nach dem Ausdruck 4 ä2 (h 2 + k') aufgelöst und er-
gibt 4^a2 (h 2 + k2) = sin g 9 - 42c 2
Da nach Seite 20 c„ = 7,42 A ist, ergibt sich für die erste Schichtlinie folgende
Gleichung: 122 (h 2 + k2) = sin' - X212 , wobei 4212 = 14,5 • 10-3 ist.4a 4c 4c
Der Winkel μ ist aus der Berechnung der Gitterkonstanten für jede Schicht-linie bekannt. Die Indizes wurden wiederum rechnerisch und graphisch be-stimmt. Da nun 1 nicht mehr 0 ist, so gibt uns die Indizierung der höherenSchichtlinien über die integralen Auslöschungen (hkl) Auskunft.
Die zweite Schichtlinie wurde ebenfalls indiziert, wobei für 1 der Wert 2gesetzt wurde. Der Ausdruck 4 c2 errechnet sich zu 58,0 X 10-'. Aus den dabeierhaltenen Indizes ist ein weiteres Auslöschungsgesetz zu erkennen, das durchdie Goniometeraufnahmen bestätigt wird: hol oder Oki, (was das nämlicheist, da im tetragonalen System infolge der Gleichheit von a 1 und a, h und kvertauschbar sind) kommen nur vor, wenn h + 1 respektiv k + 1 = 2n ist.
hhl kommt nur vor, wenn 1 = 2n ist. Ferner zeigen die Drehkristallaufnah-men, dass das integrale Auslöschungsgesetz h + k + 1 = 2n gilt, was auf Innen-zentrierung des Gitters hinweist (Tabellen 5 und 6) .
22
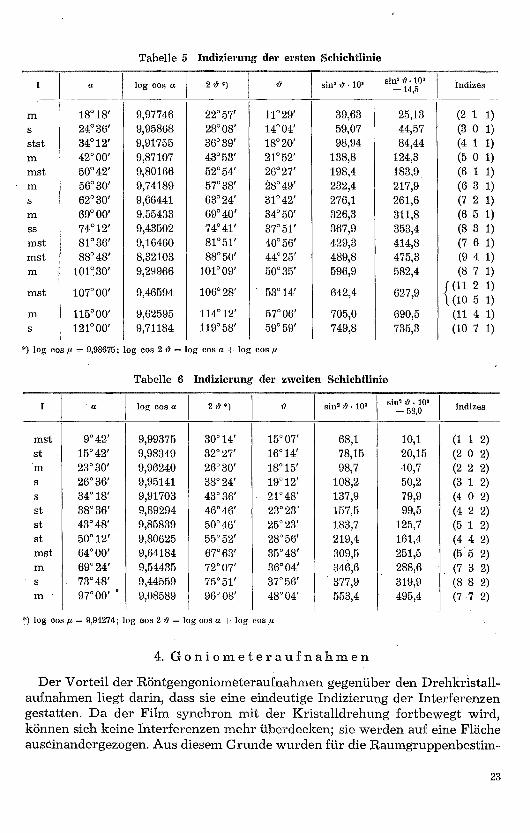
Tabelle 5 Indizierung der ersten Schichtlinie
I a log cos a 2 V *) V sin V • 10' sin' V • 10'-14,5 Indizes
m 18°18' 9,97746 22°57' 11°29' 39,63 25,13 (2 1 1)s 24°36' 9,95868 28° 08' 14°04' 59,07 44,57 (3 0 1)stst 34°12' 9,91755 36°39' 18°20' 98,94 84,44 (4 1 1)m 42°00' 9,87107 43°53' 21 0 52' 138,8 124,3 (5 0 1)mst 50° 42' 9,80166 52° 54' 26°27' 198,4 183,9 (6 1 1)m 56°30' 9,74189 57°38' 28°49' 232,4 217,9 (6 3 1)s 62°30' 9,66441 63°24' 31°42' 276,1 261,6 (7 2 1)m 69°00' 9,55433 69°40' 34°50' 326,3 311,8 (6 5 1)ss 74°12' 9,43502 74°41' 37°51' 367,9 353,4 (8 3 1)mst 81°36' 9,16460 81°51' 40°56' 429,3 414,8 (7 6 1)mst 88°48' 8,32103 88°50' 44°25' 489,8 475,3 (9 4 1)m 101°30' 9,29966 101°09' 50°35' 596,9 582,4 (8 7 1)
mst 107° 00' 9,46594 106° 28' 53° 14' 642,4 627,9 j (11 2 1)l (10 5 1)
m 115°00' 9,62595 114°12' 57°06' 705,0 690,5 (11 4 1)s 121° 00' 9,71184 119° 58' 59° 59' 749,8 735,3 (10 7 1)
*) log cos µ = 9,98675; log cos 2 U = log cos a + log cos µ
Tabelle 6 Indizierung der zweiten Schichtlinie
I a log cos a 2 6 5 ) d sin' 6U . 10' sin' • 10'- 58,0 Indizes
mst 9° 42' 9,99375 30° 14' 15° 07' 68,1 10,1 (1 1 2)st 15° 42' 9,98349 32°27' 16° 14' 78,15 20,15 (2 0 2)m 23° 30' 9,96240 26° 30' 18°15' 98,7 40,7 (2 2 2)s 26° 36' 9,95141 38° 24' 19° 12' 108,2 50,2 (3 1 2)s 34° 18' 9,91703 43° 36' 21° 48' 137,9 79,9 (4 0 2)st 38°36' 9,89294 46°46' 23°23' 157,5 99,5 (4 2 2)st 43°48' 9,85839 50°46' 25°23' 183,7 125,7 (5 1 2)st 50°12' 9,80625 55°52' 28°56' 219,4 161,4 (4 4 2)mst 64°00' 9,64184 67°63' 35°48' 309,5 251,5 (5 5 2)m 69°24' 9,54435 72°07' 36°04' 346,6 288,6 (7 3 2)s 73°48' 9,44559 75°51' 37°56' 377,9 319,9 (8 8 2)m 97°00' 9,08589 96° 08' 48°04' 553,4 495,4 (7 7 2)
*) log oos µ = 9,94274; log cos 2 U = log cos a + log cos µ
4. Goniometeraufnahmen
Der Vorteil der Röntgengoniometeraufnahmen gegenüber den Drehkristall-aufnahmen liegt darin, dass sie eine eindeutige Indizierung der Interferenzengestatten. Da der Film synchron mit der Kristalldrehung fortbewegt wird,können sich keine Interferenzen mehr überdecken; sie werden auf eine Flächeauseinandergezogen. Aus diesem Grunde wurden für die R,aumgruppenbestim-
23
Abb. 11 Goniometeraufnahme bei Drehung um [001].
mung, welche eine eindeutige Indizierung voraussetzt, noch einige Gonio-meteraufnahmen hergestellt.
Zur Verwendung gelangte ein Goniometer nach WEISSENBERG-BOEHM. Bei diesem Gonio-meter entspricht 1 Grad Kristalldrehung 1 mm Filmbewegung. Sämtliche Re flexe, welcheauf einer Geraden («Schräglinie.) liegen, stellen die verschiedenen Ordnungen der Inter-'ferenzen von ein und derselben Netzebene dar. Es wurden nur Aufnahmen des Aquators mitausgeblendeten Schichtlinien angefertigt. Der Innendurchmesser der Kamera betrug68,2 mm. Zur Verwendung gelangte eine Röhre mit einer Cobalt-Antikathode. Der Kristallwurde wiederum um die Achsen [100], [001] und [110] gedreht.
a) Drehung um [001]
Die Belichtungszeit dieser Aufnahme betrug 10 Stunden. Da die Schicht-linien ausgeblendet wurden, kommen nur Reflexe mit l = 0 vor. Für die Indi-
24

zierung galt es, zuerst die Koordinate e des (100)-Reflexes zu suchen unddamit die h00-Gerade zu finden. Ist der Kristall tetragonal, so muss die Ok0-Schräglinie in einem Abstand von 90 mm = 90° von der h00-Geraden gefun-den werden. Aus der BRAGGschen Gleichung sin 29' = 2 • a lässt sich sin 6 vonh00 berechnen; da wir den Reflex erster Ordnung ermitteln wollen, ist n = 1.
Setzen wir für den Wert a o = 12,3 A ein, so ergibt sich für sin 4 = 4° 10'.Die Koordinate berechnet sich nach folgender Formel: - 4 •
60 r , wobei r
den Radius der Kamera bedeutet. Setzt man die entsprechenden Werte indie Gleichung ein, so ergibt sich für = 5 mm. Auf dem Film findet sichaber erst in einem senkrechten Abstand von 10 mm von der Mittellinie einInterferenzpunkt. Der Reflex (100) kommt also nicht vor, sondern erst derReflex (200). Die rechnerische Indizierung der Interferenzen auf dieserSchräglinie ist in Tabelle 7 durchgeführt (vgl. Abb. 11) .
Tabelle 7 Indizierung der h00-Geraden
I 2 sa log s - 0,1884 2 v2 12 sin° 12 • 10' Indizes
stst 26,8 1,2397 17,37 8° 41' 22,79 (2 0 0)stst 53,4 1,5391 34,6 17° 18' 88,4 (4 0 0)m 81,1 1,7206 52,56 26° 17' 196,1 (6 0 0)m 111,2 1,8577 72,06 36° 02' 346,3 (8 0 0)s 146,0 1,9760 94,62 47° 31' 540,4 (10 0 0)ss 188,6 2,0869 122,2 61°06' 766,4 (12 0 0)
Der Faktor 0,1884 ist eine Apparatenkonstante. Durch Vertauschen derh-Werte mit den k-Werten kann man die Indizierung der OkO-Reflexe durch-führen, die genau denjenigen der h0O-Schräglinie entsprechen. Im Abstand45° von h00 findet sich die hh0-Schräglinie; ihre Indizierung geht aus Tabelle 8hervor.
Tabelle 8 Indizierung der hhO-Schräglinie
I 2 sa log s - 0,1884 2 0 'I sin' d • 10' Indizes
ss 19,5 1,1016 12,64 6° 19' 12,1 (1 1 0)sss 38,0 1,3914 24,63 12° 19' 46,58 (2 2 0)m 57,0 1,5675 36,9 18° 27' 101,1 (3 3 0)m 97,1 1,7988 62,9 31°27' 277,5 (5 5 0)s 144,0 1,9700 93,3 46° 39' 528,8 (7 7 0)mst 171,9 2,0469 111,4 55°42' 682,4 (8 8 0)
25

Zusammenstellung der beobachteten Reflexe desÄquators bei Drehung um [001]
h00: 200, 400, 600, 800, 10 00, 12 00.OkO: 020, 040, 060, 080, 010 0,012 0.hh0: . 110, 220, 330, 550, 770 880.hk0: 130, 150, 170, 240, 260, 280, 310, 350, 370, 420, 460, 510, 530, 570, 710, 730, 750, 820, 840, 860.
Daraus gehen folgende Auslöschungsgesetze hervor:h00: nur vorhanden, wenn h = 2nhk0: nur vorhanden, wenn h k = 2n.
b) Drehung um [100]
Die Belichtungszeit dieser Aufnahme betrug 12 Stunden. Als leitende Ge-raden sind 001 und Ok0 zu finden. Die Wiederholung des Interferenzbildes istnach 180° Drehung sichtbar, infolge der Drehung um eine Digyre. Die Indi-zierung der leitenden Schräglinien wurde rechnerisch durchgeführt, währenddie übrigen Reflexe graphisch indiziert wurden.
Zusammenstellung der gefundenen Reflexe
001: 002, 004, 006.h00: 200. 400, 600, 800, 10 00, 12 00.h01: 101, 301, 501, 901, 202, 402, 602, 802, 10 02, 12 02, 103, 303, 503, 703, 903, 204, 404, 604,
804, 705, 206, 406, 606.
Es gehen daraus folgende weitere Auslöschungsgesetze hervor:001: nur vorhanden, wenn l = 2nh01: nur vorhanden, wenn h 1 = 2n.
c) Drehung um [110]
Belichtungszeit: 12 Stunden.Als leitende Schräglinien finden sich 001 und hh0.
Zusammenstellung der gefundenen Reflexe:
hhl: 112, 222, 332, 552, 772, 882, 224, 334, 554, 336, 776.
Als Auslöschungsgesetz wurde gefunden:hhl: nur vorhanden, wenn 1= 2n.
5. Laueaufnahmen
Für die Raumgruppenbestimmung ist es notwendig, dass die Laueklasse,d. h. die speziellen Symmetrieverhältnisse unseres Kristalles, bekannt sind.Im Gegensatz zum Dreh- und Röntgengoniometerverfahren arbeitet man beiLaueaufnahmen mit weissem Röntgenlicht, d. h. mit einem Röntgenstrahl, wel-
26

Abb. 12 Laueaufnahme bei Einstrahlung parallel [001].
cher verschiedene Wellenlängen enthält. Der ruhende Kristall wird parallelzu einer seiner Achsen durchstrahlt, wobei infolge der Reflexion der verschie-denen Wellenlängen an verschiedenen Netzebenen das Lauebild zustandekommt. Es wurde Mo-Strahlung bei einer Belichtungszeit von 30 Minutenverwendet.
a) Durchstrahlung parallel der c-Achse
Die Abbildung 12 zeigt die Symmetrieverhältnisse der erhaltenen Laue-aufnahme. Die Tetragyre ist sehr gut erkennbar. Es ergibt sich somit dieSymmetrie C4. H. A. KLASENS, W. G. PERDOK, P. TERPSTRA (1937) haben beider Durchstrahlung des Kalzium-Oxalates (Polyhydrat) parallel zur Tetra-gyre ebenfalls die Symmetrie C 4 gefunden.
27

b) Durchstrahlung parallel der a-Achse
Alle Laueaufnahmen mit Einstrahlung senkrecht zur c-Achse, somit auchjene mit Einstrahlung parallel der a-Achse, liefern ein in bezug auf eine hori-zontale Symmetrieebene symmetrisches Bild. Wir haben daher die C4a-Laue-klasse vor uns.
6. Ermittlung der Raumgruppe
An Hand der Internationalen Tabellen I. Band (1935a) kann bei bekannterLauesymmetrie mit Hilfe der Auslöschungsgesetze die Raumgruppe gefundenwerden. Die oben erhaltenen Auslöschungsgesetze führen nach dem Bestim-mungsschlüssel der Internationalen Tabellen I. Band (1935a), Seite 394, zufolgendem:
C. (hkl) nur vorhanden, wenn h + k + l = 2n, II. (hk0) nur vorhanden, wenn h + k = 2n, I =ml. (001) nur vorhanden, wenn l = 2n, I 4/m—C; h , C4 i S
Die wahrscheinlichen Raumgruppen ermitteln sich daher als CL, C4 und S.Morphologische Betrachtungen sprechen dafür, dass die Raumgruppe CL diewahrscheinlichste ist. Als mögliche Raumgruppen kämen noch in Betracht:C4li (1-4), C4 (1,3), S4 sowie S. H. A. KLASENS, W. G. PERDOK, P. TERPSTRA(1937) bestimmten für das Sr(COO) 2 •2,5 H2O als wahrscheinlichste Raum-gruppe C AL = I 4/m, wobei ebenfalls 8 Formeleinheiten Sr(COO) 2 • 2,5 H2Oin der Elementarzelle liegen. Die beiden Oxalate haben somit offenbar die-selbe Raumgruppe.
7. Ermittlung der Anzahl Moleküle in der Elementarzelle
Die Anzahl der Moleküle errechnet sich nach folgender Formel:N_ a2 • c • • 10-24
N M. 1,66 . 10-24
N Anzahl der Moleküle pro Elementarzellea, c GitterkonstantenQ DichteM Molekulargewlcht1,66 •10-24 g absolute Masseneinheit des Atomgewichtes
a) Bestimmung der Dichte
Wie aus obenstehender Gleichung hervorgeht, wird zur Bestimmung derElementarmasse der Dichtewert für die betreffende Kristallart benötigt. Ver-suche, die Dichte pyknometrisch durch die Volumenverdrängung der Flüssig-keit zu ermitteln, ergaben sehr niedrige und stark schwankende Werte. Eswurde daher die Suspensionsmethode angewandt, deren Prinzip darauf be-ruht, dass ein einzelner, sehr schön ausgebildeter und mikroskopisch störungs-freier Kristall in eine spezifisch schwerere Flüssigkeit getaucht wird. Nun
28

gibt man eine spezifisch leichtere Flüssigkeit zu, bis das Gemisch das gleichespezifische Gewicht aufweist wie der Kristall, d. h. bis dieser darin schwebt.Darauf wird das spezifische Gewicht des Gemisches bestimmt.
Es wurde dazu ein Präzisionspyknometer mit kalibriertem Stiel nachFURTER aus Jenaglas 16 III verwendet.
Aus 40 Dichtebestimmungen an 20 Kristallen wurde das Mittel genommen,welches den Wert 2,077 ± 0,012 ergab. Als Flüssigkeiten wurden Bromoform(spezifisches Gewicht 2,8) und Chloroform (spezifisches Gewicht 1,5) ver-wendet.
b) Berechnung der Anzahl Moleküle N in der Elementarzelle
Nimmt man zunächst an, das Polyhydrat sei ein Kalzium-Oxalat-Dihydrat,so beträgt das Molekulargewicht 164,13. Somit ergibt sich nach der Formel aufSeite 28 8,51 Ca (COO) 2 • 2 H 2O in der Elementarzelle.
N _ (12, 302) 2 . 7,381 • 2,077.10-24 = 8,51 Ca'COO' 2 • 2 H2O164,13 . 1,66 . 10-24
Berechnet man andererseits die Anzahl Moleküle in der Elementarzelle fürein Trihydrat, so ergibt sich:
N = (12 ,302) 2 .7,381.2,077.10-24 = 7,67 Moleküle Ca(C00) 2 . 3 H2O182,15.1,66.10-24
Die Anzahl der Moleküle in der Elementarzelle kann aber nur ganzzahligsein. Für das Dihydrat müssten daher 8 oder 9 Moleküle in der Elementarzelleangenommen werden. Da sowohl die Bestimmung der Gitterkonstanten alsauch der Dichte an frischen Kristallen sorgfältig ausgeführt wurde, ist dieAbweichung von N = 8 respektiv = 9 zu gross, als dass die Annahme einesDihydrates gerechtfertigt wäre. Ein D i h y d r a t kann daher nachden röntgenographischen Befunden nicht vorliegen.
Das Trihydrat hätte beim Aufrunden von 7,67 auf 8 eine Molekülzahl, diemit der Zähligkeit der CL Raumgruppe vereinbar wäre, doch ist auch hierdie Abweichung von 8 allzugross. Rechnet man andererseits aus der allge-meinen Formel Seite 28 mit einer Molekülzahl von 8 pro Elementarzelle dasMolekulargewicht aus, so ergibt sich der Wert 174,7, welcher nun aber ehereinem Kalzium-Oxalat-2,5-Hydrat entspricht. Benützt man zur Berech-nung von N ein Molekulargewicht von 173,14, welches einem 2,5-Hydrat ent-spricht, so erhält man 8,07 Moleküle pro Elementarzelle. Dieser Wert liegtsehr viel näher bei 8 als 7,67 oder 8,51.
Es ist daher für das Polyhydrat (durch Umkristalli-sation aus HC1 gewonnen) nach den röntgenometri-schen Messungen ein Ca(COO) 2 . 2,5H20 als wahrscheinlichanzunehmen.
Der Fehler bei der Molekulargewichtsbestimmung beträgt in diesem Falle0,9 %.
29

c) Verteilung der Ca-Atome und C2O4-Radikale auf die Punktlagen
Wenn wir 8 Kalzium-Oxalat-2,5-Hydrat-Moleküle in der Elementarzelle an-nehmen, so müssen in der Zelle 8 Ca-Atome, 8 Oxalat-Radikale und 20 H 20-Moleküle vorhanden sein. Betrachten wir nun die Zähligkeit der Punktlagenin der in Frage kommenden wahrscheinlichsten Raumgruppe C4 11 , so ist er-sichtlich, dass sich die verschiedenen Atome und Radikale folgendermassenauf die Punktlagen verteilen lassen:8 Ca-Atome a) auf eine achtzählige Punktlageoder b) auf zwei vierzählige Punktlagenoder c) auf eine vierzählige und zwei zweizählige Punktlagen
Theoretisch können sich die 8 C,0, -Radikale wie die Ca-Atome verteilen.Aus den Arbeiten von W. H. ZACHARIASEN (1934), ST. B. HENDRICKS (1935)und ST. B. HENDRICKS und W. E. DEMING (1935) geht hervor, dass das C2O4Radikal eine Punktsymmetrie von C 2 besitzt, wenn angenommen wird, dassdie Abstände von C-0 1 und C-02 die nämlichen sind. Falls die C-01 undC-02-Abstände verschieden sind, kommt für das C,0 4-Radikal eine Punkt-symmetrie von C i in Frage. Auf Seite 161 der Internationalen Tabellen I. Band(1935a) ist bei der C411 Raumgruppe ersichtlich, dass die Punktsymmetrie C;nur für die 8-zählige Punktlage f erfüllt ist; die Punktsymmetrie C 2 nur fürdie 8-zählige Punktlage g.
Die 20 H2O-Moleküle können z. B. aufa) 1 X 16-zählige Punktlage und eine 4zählige Lage oderb) 2 X 8-zählige Punktlagen und eine 4-zählige Lage oderc) 2 x 8-zählige Punktlagen und 2 X 2-zählige Lagen usw.
verteilt werden.
Zusammenfassung des II. Teils
1.Es wurden an Hand von reinen synthetischen Substanzen Testaufnahmenvom Monohydrat und vom tetragonalen Polyhydrat des Kalzium-Oxalatesnach dem Verfahren von DEBYE und SCHERRER angefertigt. Dabei konnte ge-zeigt werden, dass sich die erhaltenen Diagramme nicht in allen Fällen mitdenjenigen anderer Verfasser decken.
2. Die durch Erhitzung oder Alterung stattfindende Umwandlung des Poly-hydrates in das Monohydrat wurde an Hand von Pulveraufnahmen verfolgt.Es zeigte sich dabei, dass dieser Vorgang zweiphasig abläuft. Das Auftretenvon Mono- und Polyhydrat-Interferenzen bei nicht frischen Polyhydratprobenwird als Grund dafür angenommen, dass in der Literatur oft Netzebenenab-stände angegeben werden, die von zum Teil umgewandeltem Polyhydrat her-rühren.
3. Wird das Monohydrat durch Erhitzen entwässert, so geht es in dasAnhydrat über. Pulveraufnahmen dieses Anhydrates zeigten das nämlicheInterferenzsystem wie das Monohydrat.
30

4. Die röntgenographisch richtige Aufstellung (innenzentrierte Elementar-zelle) ist diejenige, welche die einzig vorhandenen Prismen- und Pyramiden-flächen als solche II. Stellung annimmt. Das Achsenverhältnis a : c errechnetsich in diesem Falle zu 0,6000, womit der Winkel zwischen (100) und (101)59° 02' beträgt.
5. An Hand von Drehkristallaufnahmen am tetragonalen Polyhydrat umdie [001], [100] und [110]-Achsen konnte eine erste Gitterkonstantenbe-stimmung durchgeführt werden. Pulveraufnahmen ergaben die Werte a o =12,302 ± 0,007 Å , co = 7,381 ± 0,003 A.
6.Durch die Indizierung der Drehkristallaufnahmen konnte gefunden wer-den, dass das integrale Auslöschungsgesetz h + k + l = 2n besteht.
7.Aus den Goniometeräquatoraufnahmen beim Drehen um [001], [100] und[110] gehen folgende Auslöschungsgesetze hervor:
h00: nur vorhanden, wenn h = 2nhk0: nur vorhanden, wenn h + k = 2n001: nur vorhanden, wenn 1 = 2nh01: nur vorhanden, wenn h + l = 2nhhl: nur vorhanden, wenn l = 2n
9. Aus der Laueklasse, den Auslöschungsgesetzen und der Morphologieergab sich als wahrscheinlichste Raumgruppe C2h-I 4/m; weitere wahrschein-liche Raumgruppen sind C2 und S.
10. An Hand des röntgenographisch ermittelten Molekulargewichtes konntegezeigt werden, dass das tetragonale Polyhydrat (Weddelit) offenbar ein Kal-zium-Oxalat-2,5-Hydrat ist, während ein Dihydrat und ein Trihydrat für dieseKristallart ausgeschieden werden müssen.
11. An Hand der Zähligkeiten der verschiedenen Punktlagen wurde ge-zeigt, dass ein Ca (COO) 2 • 2,5 H2O mit der Raumgruppe C2 h verträglich ist.
III. Teil
Chemisch-physikalische Untersuchungen
A. Entwässerungskurven
1. Allgemeines
Entwässerungskurven zeigen, welche wasserärmeren Hydrate ein Salz bil-den kann. Sie geben ferner Aufschluss über die Stabilität einzelner Hydratebei verschiedenen Temperaturen und lassen erkennen, ob das Wasser imKristall aktiv am Gitteraufbau teilnimmt, oder ob es sich um vagabundieren-des Kristallwasser handelt, wie es in den Zeolithen vorkommt. Durch Erwär-mung der Kristalle wird die Schwingung der Atome und Moleküle verstärkt,so dass schliesslich der Austritt von Wasser bewirkt wird. Handelt es sich
31

dabei um echtes Kristallwasser, so tritt dieses bei Erreichung einer bestimmtenTemperatur plötzlich aus dem Gitter aus, was sich in einem steilen Anstiegder Kurve bekundet. Es entsteht dabei eine niedrigere Hydratstufe oder dasAnhydrat. Ist innert eines bestimmten Temperaturintervalls eine gewisse Sta-bilität vorhanden, so wird der Steigungswinkel der Kurve kleiner. Ist hin-gegen das Wasser nicht am Gitteraufbau beteiligt, so wird die Entwässerungs-kurve mehr oder weniger gleichmässig ansteigen, was auf den stetigen Wasser-verlust zurückzuführen ist. Die typischen Sprünge, wie sie sich bei der Ent-wässerung von Substanzen mit echtem Kristallwasser ergeben, fehlen hier.
Die Entwässerungskurven können auf zwei verschiedenen Wegen ermitteltwerden:
1. durch kontinuierliche Erhitzung und Gewichtsverlustbestimmung,2. auf manometrischem Wege, wobei der aus der erhitzten Substanz ent-
weichende H 2O-Dampf auf ein Manometer drückt. Wird bei einer bestimm-ten Temperatur der Druck und damit der Steigungswinkel der Temperatur-Druckkurve grösser, so ist dies die Folge einer verstärkten H 2O-Abgabe durchdas Kristallpulver.
Die erste Methode bietet die Möglichkeit, den Gewichtsverlust und damitden Wassergehalt quantitativ zu bestimmen. Das zweite Verfahren lässt nurqualitative Aussagen über die Anzahl Mole der wasserärmeren Hydrate zu.
2. Beschreibung der Apparaturen
a) Bestimmung des Gewichtsverlustes mit einer analytischen Waage
Bei der ersten der oben erwähnten Methoden wird zur Bestimmung des Gewichtsver-lustes eine analytische Waage benutzt. An der einen Waagschale wird ein Platlndraht be-festigt, der durch ein Loch in der Tischplatte zu einem darunter stehenden Ofen führt. AmEnde dieses Drahtes wird ein Platintiegel befestigt, der die eingewogene Substanz enthält.Die im Ofen erzeugte Temperatur wird mit einem Thermoelement gemessen. Ein Wider-stand ermöglicht, jede beliebige Temperatur bis 1000° C einzustellen. Zu Beginn des Ver-suches wird auf der zweiten Waagschale so viel Gewicht aufgelegt, bis die Waage im Gleich-gewicht ist.
Bei der Erhitzung wird der Gewichtsverlust der Probe sukzessive austariert. Durch dieErwärmung wird der Tiegel infolge des Auftriebes der warmen Luft etwas leichter, so dassdie Messungen mit einem Fehler von etwa einem Prozent des Gewichtsverlustes behaftetsind. Trotzdem können die Resultate noch sehr gut quantitativ verwertet werden. Die Sub-stanz wurde in unserem Falle bis zu einer Temperatur von 950° C ausgeglüht.
b) Qualitative Bestimmung der wasserärmeren Hydrate des Kalzium-Oxalatesauf manometrischem Wege
Zu diesem Zwecke wurde ein Apparat zur Messung des Dampfdruckes gebaut (Abb. 13).In einem Rundkolben, der sich in einem heizbaren Glyzerinbad befindet und mit einemThermostaten verbunden ist, wird die pulverisierte Substanz erwärmt. Der Kolben ist miteinem Quecksilbermanometer durch einen Kugelschliff verbunden. Die Schenkel des Mano-meters haben eine Höhe von 80 cm und besitzen auf der einen Seite eine Öffnung, die miteiner Vakuumpumpe verbunden ist. Das Manometer ist oben luftdicht abgeschlossen, sodass das ganze System vom Luftdruck unabhängig ist. Wird nun die Substanz mit Hilfe
32

des Glyzerinbades erhitzt, so entweicht bei einer bestimmten Temperatur das Wasser inDampfform. Mit der Pumpe wird das System so lange evakuiert, bis die beiden Quecksilber-säulen des Manometers das gleiche Niveau haben.
Damit der Wasserdampf nicht vorzeitig kondensiert, werden die Schenkel des Mano-meters mit Hllfe einer Glühspirale erhitzt. Diese steht mit einem Thermostaten in Verbin-dung, welcher dafür sorgt, dass die Wasserdampfmoleküle keine Beschleunigung oder Ver-zögerung erfahren, was sich in einer Veränderung des Druckes äussern müsste. Auf dieseWeise wird im oberen Teil der Apparatur, in welcher sich H 2O-Dampf ansammelt, einekonstante Temperatur von 80° C erreicht. Zur Bestimmung des Druckes wird die Differenzzwischen den beiden Quecksilberniveaus gemessen und bei jeder Ablesung die zugehörigeTemperatur notiert.
Aus der Anzahl der Sprünge, die sich ergeben, kann auf die Zahl der möglichen, stabilenHydratstufen geschlossen werden.
3. Besprechung der Resultate
a) Quantitative Entwässerung mit einer analytischen Waage
In Tabelle 9 sind die Ergebnisse der Entwässerung zusammengestellt.
Tabelle 9 Entwässerung des Polyhydrates
Temperatur°C
Totaler Ge-wichtsverlust
in mg
Gewichtsverlustin mg bei der
betr. Temperatur
Totaler Ge-wiehtsverlust
60° 0 0 070° 6 6 0,54
110° 117 111 10,6115° 126 9 11,4120° 166 40 15,1135° 181 15 16,4140° 191 10 17,3145° 201 10 18,3165° 221 20 20,0170° 241 20 21,9180° 251 10 22,8190° 271 20 24,6230° 301 30 27,3240° 301 0 27,3340° 301 0 27,3380° 461 150 43,7420° 491 30 44,6440° 491 0 44,6560° 491 0 44,6610° 501 10 45,5720° 541 40 49,1790° 711 170 64,2830° 751 40 68,2
3 33
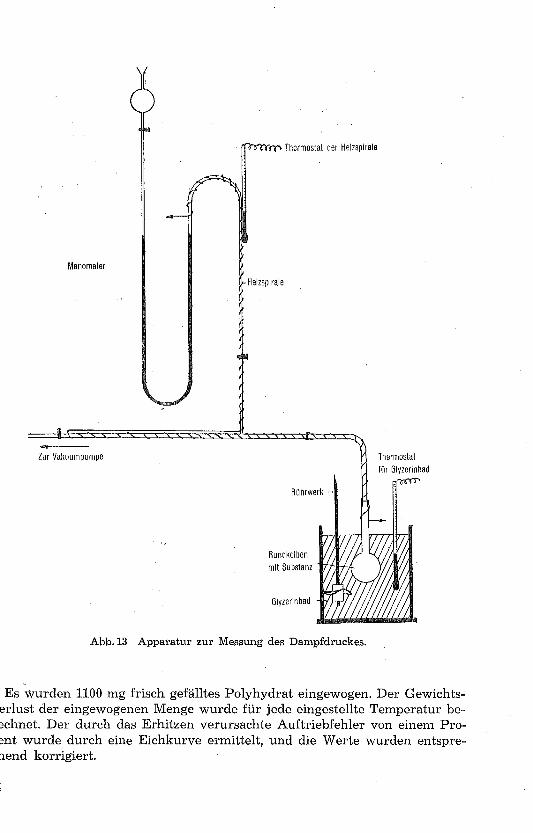
Abb. 13 Apparatur zur Messung des Dampfdruckes.
Es wurden 1100 mg frisch gefälltes Polyhydrat eingewogen. Der Gewichts-erlust der eingewogenen Menge wurde für jede eingestellte Temperatur be-=_chnet. Der durch das Erhitzen verursachte Auftriebfehler von einem Pro->nt wurde durch eine Eichkurve ermittelt, und die Werte wurden entspre-zend korrigiert.
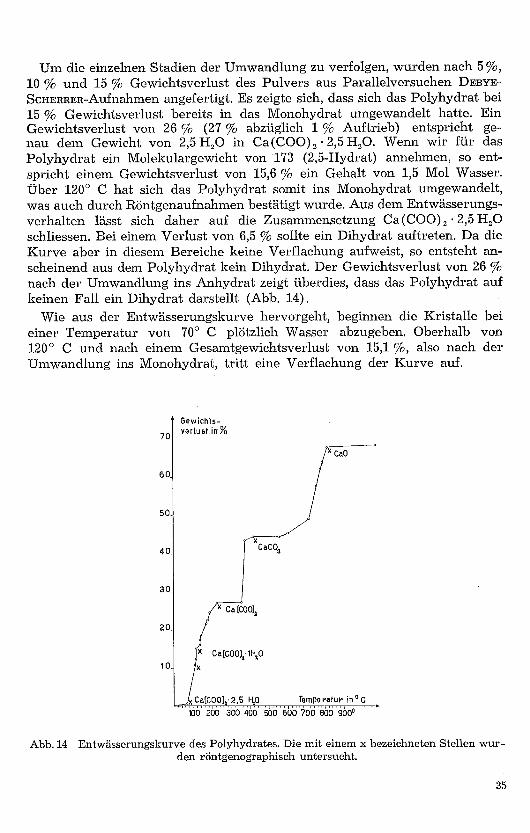
Um die einzelnen Stadien der Umwandlung zu verfolgen, wurden nach 5 %,10 % und 15 % Gewichtsverlust des Pulvers aus Parallelversuchen DEBYE-SCHERRER-Aufnahmen angefertigt. Es zeigte sich, dass sich das Polyhydrat bei15 % Gewichtsverlust bereits in das Monohydrat umgewandelt hatte. EinGewichtsverlust von 26 % (27 % abzüglich 1 % Auftrieb) entspricht ge-nau dem Gewicht von 2,5 H2O in Ca(COO) 2 • 2,5 H2O. Wenn wir für dasPolyhydrat ein Molekulargewicht von 173 (2,5-Hydrat) annehmen, so ent-spricht einem Gewichtsverlust von 15,6 % ein Gehalt von 1,5 Mol Wasser.Über 120° C hat sich das Polyhydrat somit ins Monohydrat umgewandelt,was auch durch Röntgenaufnahmen bestätigt wurde. Aus dem Entwässerungs-verhalten lässt sich daher auf die Zusammensetzung Ca (COO) 2 • 2,5 H2Oschliessen. Bei einem Verlust von 6,5 % sollte ein Dihydrat auftreten. Da dieKurve aber in diesem Bereiche keine Verflachung aufweist, so entsteht an-scheinend aus dem Polyhydrat kein Dihydrat. Der Gewichtsverlust von 26 %nach der Umwandlung ins Anhydrat zeigt überdies, dass das Polyhydrat aufkeinen Fall ein Dihydrat darstellt (Abb. 14) .
Wie aus der Entwässerungskurve hervorgeht, beginnen die Kristalle beieiner Temperatur von 70° C plötzlich Wasser abzugeben. Oberhalb von120° C und nach einem Gesamtgewichtsverlust von 15,1 %, also nach derUmwandlung ins Monohydrat, tritt eine Verflachung der Kurve auf.
Gewichts-Verlust in
x Ca [C00),
x Ga(C00)21H,,0x
Ca[C00), 2,5 H,0 Tempe ratio in ° G160 260 300 400 500 660 760 e60 900°
Abb. 14 Entwässerungskurve des Polyhydrates. Die mit einem x bezeichneten Stellen wur-den röntgenographisch untersucht.
70_
60_
50
40
30
20
10
35

Zwischen 130° C und 135° C setzt hierauf die Abgabe von Wasser aus demMonohydrat ein, die bei 230° C beendet ist. Um diesen Entwässerungsvorgangvom Monohydrat ins Anhydrat zu prüfen, wurden Entwässerungen mit reinem,gekauftem Monohydrat durchgeführt, welche die erwähnten Angaben bestä-tigten. Von 230° C bis zu 360° C tritt praktisch kein Gewichtsverlust mehrein. Bei weiterem Erhitzen entsteht ein grosser Sprung, der bis 420° C reicht.Das Anhydrat wandelt sich hier in Kalziumkarbonat um. Eine Pulverauf-nahme zeigte, dass es sich dabei um die Modifikation Kalzit handelt. Bei einerweiteren Erwärmung auf 560-610°C beginnt ein neuer Anstieg der Kurve,der bis gegen 830° C anhält. Eine röntgenographische Prüfung ergab, dass sichin diesem Bereiche Kalziumoxyd (CaO) bildet. Bei einer weiteren Tempera-turerhöhung ergibt sich kein Gewichtsverlust mehr.
Beim Erhitzen wandelt sich das Ca (COO) , • 2,5 11,0 somit über das Mono-hydrat und Anhydrat in Kalzit und schlussendlich in das Kalziumoxyd um.
JEAN LECOMPTE, THERESE PROBEGUIN und JEAN WYART (1943) beschreibenin ihrer Arbeit drei verschiedene Polyhydrate des Kalzium-Oxalates. Beieinem davon handelt es sich um das eingangs erwähnte Trihydrat von HAM-MARSTEN, das von uns nicht näher untersucht wurde. Beim zweiten der dortbeschriebenen Hydrate handelt es sich um Kristalle, welche durch eine Um-kristallisation aus HC1 gewonnen wurden; die dritte Art des Polyhydrateswurde durch eine diachrone Fällung gewonnen. Die Verfasser erwähnen,dass dieses Polyhydrat einen etwas kleineren Wassergehalt aufweist, als dasdurch Umkristallisation gewonnene.
Sie stellen immerhin fest, dass es ihnen nie ganz geglückt sei, die kleinenKristalle ganz rein herzustellen. Bei einer Wasserbestimmung von verun-reinigten Substanzen ergibt sich ein zu grosser Einwaagewert, woraus ein zukleiner Wassergehalt resultiert. Dass diese kleinen Kriställchen ihr Wasserschneller abgeben als die grösseren, durch Umkristallisation hergestellten,ist verständlich, wenn man die grosse Instabilität der kleinen Kristalle in Be-tracht zieht. Untersuchungen über den Wassergehalt der mikroskopisch klei-nen «envelope»-Kristalle wurden daher von uns nicht vorgenommen. DiePulveraufnahmen von Polyhydraten, welche durch Fällung und Umkristalli-sation gewonnen werden, sind jedoch identisch. Dies lässt vermuten, dass derWassergehalt in beiden Fällen gleich sei, so dass die von den erwähnten Ver-fassern getroffene Unterscheidung der synthetisch hergestellten Kristalle hin-sichtlich des Wassergehaltes sehr problematisch ist. Jedenfalls handelt es sichbei den diachron gefällten und den durch Umkristallisation gewonnenen Poly-hydratkristallen um dieselbe Kristallart.
Es wurde schon im röntgenographischen Teil betont, dass das Trihydratnach HAMMARSTEN von uns nicht näher untersucht wurde, da es in der Naturbis anhin nirgends beobachtet werden konnte. Auffallend ist die grosse Lös-lichkeit dieses Salzes gegenüber dem Ca(COO)., • 2,5 H 2O. Nach Untersuchun-gen von LECOMPTE (1943) unterscheidet sich jenes monokline Trihydratröntgenographisch stark von den anderen • Polyhydratkristallen. Ob es sicheffektiv um ein reines Kalzium-Oxalat handelt, scheint noch nicht sicher
30

festzustehen. F. T. JONES und M. WHITE (1946) beschreiben in ihrer Arbeitein Doppelsalz des Kalzium-Oxalates mit Kalziumchlorid mit der FormelCa (COO), Ca C12 • 7 H2O, welches seiner Kristallform nach mit dem Kalzium-Oxalat-Trihydrat von HAMMARSTEN übereinstimmt. Beim Auflösen von Mono-hydrat in zu konzentrierter Salzsäure fand ich jenes Salz ebenfalls. Ein Pulver-diagramm ergab sowohl die Interferenzen unseres Polyhydrates als auch diedes genannten Doppelsalzes.
b) Ergebnisse der manometrischen Messungen
Die Substanz wurde im Rundkolben von 20° C an in Stufen von 10° C suk-zessive erhitzt. Dabei wurde zu jeder Temperaturstufe der zugehörige Druckabgelesen. Es zeigte sich, dass die Kurve bis zu etwa 65° C langsam ansteigt,worauf ein starker Anstieg bis zu 80° C erfolgt. Dann verflacht sich die Kurvewieder bis 100° C. Bei 110° C wird ein neuer Anstieg registriert. Das allmäh-liche Ansteigen der Kurve ist, wie die Entwässerung zeigt, der Erhöhung desDampfdruckes der Substanz bei Erhöhung der Temperatur zuzuschreiben.Obwohl keine quantitativen Resultate aus der Kurve hervorgehen, sind wie-derum die beiden Sprünge typisch. Der erste zeigt den Übergang des Poly-hydrates in das Monohydrat an, während der zweite auf die Umwandlung desMonohydrates in das Anhydrat zurückzuführen ist. Röntgenographische Auf-nahmen bestätigen diese Feststellung.
B. Analytische Wasserbestimmungen
1. Direkte Wasserbestimmungen mit Mikroanalysen
Analysenresultate:
1. Analyse
Von den pulverisierten, bei. 30° C getrockneten Polyhydratkristallen wurde eine Mengevon 5,428 mg bei 150° C während 22 Stunden im Hochvakuum entwässert. Es ergab sichein Gewichtsverlust von 1,378 mg, was 25,39 % der Einwaage ausmacht. Unter der Vor-aussetzung, dass die Kristalle keine Verunreinigungen aufwiesen, entspricht dies 25,39 %H2O und 74,61 % Ca(COO) 2 . Daraus lässt sich ein Wassergehalt von 2,42 Mol berechnen.
2. Analyse
Das trockene Kristallpulver wurde während 12 Stunden im Hochvakuum bei 150° Centwässert.
Einwaage 4,890 mg PolyhydratGewicht des Rückstandes 3,640 mg
Wasserverlust
1,250 mg
Der Gehalt an H2O beträgt daher 25,56 % der Einwaage, was 2,44 Mol H 2O entspricht.
37

3. Analyse
Quantitative Bestimmung von Ca, H, 0 und C.
Einwaage 4,124 mg Ca(COO) 2 x H2OCO2 -Gehalt 2,060 mgH20-Gehalt 1,110 mgCa-Gehalt 0,954 mg
Aus diesen Angaben wurden die Gewichtsprozente für Ca, C, H und 0 berechnet undmit den theoretischen Werten von Kalzium-Oxalat-Dihydrat, Kalzium-Oxalat-2,5-Hydratund Kalzium-Oxalat-Trihydrat verglichen (siehe Tabelle 10). Die gefundenen Werte stim-men weder mit einem Di- noch mit einem Trihydrat überein, decken sich aber mit denerrechneten Werten des 2,5-Hydrates. Der Wassergehalt dieses Kristallpulvers ergab 2,6 Mol.Dieser Wert liegt aber immer noch viel näher bei einem 2,5-Hydrat als bei einem Tri-hydrat.
Aus der prozentualen Zusammensetzung wurde die chemische Molekularformel be-rechnet und mit der theoretisch ermittelten verglichen.
Tabelle 10 Vergleich des C-, H-, 0- und Ca-Gehaltes (Gewicht %) verschiedenertheoretischer Kalzium-Oxalat-Hydrate mit den Analysenresultaten
Ca (C00)2 .2H20Ca (C00)2 .2,5 H2 0 Ca (C00)2 .3 H 20 «Polyhydrat»theoretisch theoretisch theoretlsch Analysenresultate
Ca = 24,39 % Ca = 23,12% Ca = 21,97% Ca = 23,13 %C = 14,63 % C = 13,87 % C = 13,19 % C = 13,630/0H = 2,44 % H = 2,89 % H = 3,32 % H = 2,99 %0 = 58,53 % 0 = 60,11 % 0 = 61,51 % 0 = 60,24 %
99,99 % 99,99 °/099,99 % 99,99 %
Ca,,.: Cx : H Oz = 23,13 13,63 2,99 60,24 = 0,57 :1 14:2,99 :3,76 .Y ' 40 ' 12 ' 1 • 16
Aus der Analyse ergibt sich folgende chemische Formel: CaC2H5.200.0• Kalzium-Oxalat-2,5-Hydrat hat theoretisch die Zusammensetzung CaC2H°00.6.
Aus den oben genannten Wassergehaltsbestimmungen ist wiederum ersicht-lich, dass das Polyhydrat einem Kalzium-Oxalat-2,5-Hydrat sehr nahe kommt.Eine Prüfung der Kristalle in bezug auf Verunreinigungen ergab, dass manch-mal Chlorionen nachgewiesen werden konnten. Diese waren aber nicht beiallen Umkristallisationen vorhanden. In welcher Form das Chlor in den Kri-stallen enthalten ist, wurde nicht weiter untersucht, da die Berücksichtigungder kleinen Chlormenge keine wesentliche Änderung der Resultate bewirkt.Möglicherweise enthält das durch Umkristallisation aus HC1 gewonnene Poly-hydrat Verunreinigungen von Kalziumchlorid.
2. Indirekte Wasserbestimmungen
Durch Titration mit KMnO4
Die Herstellung des Titers der Permanganatlösung erfolgte mit handelsüblichem Natri-um-Oxalat, welches nach SöRENSEN gereinigt wurde. Das Natrium-Oxalat hat gegenüberder Oxalsäure den Vorteil, dass es wasserfrei kristallisiert. Siehe F. P. TREADWELL (1919).Es wurde ein Titer einer 1/10 normalen Kaliumpermanganatlösung von 1,069 ermittelt.
38

1. Analyse
Die Einwaage betrug 0,1927 g Polyhydratpulver. Dieses wurde in 10 cm 3 konzentrierterSalzsäure gelöst und darauf mit 200 cm 3 destilliertem Wasser verdünnt. Bis zum Um-schlagspunkt in Rot wurden 22,18 cm 3 exakt 1/im normale KMnO 4-Lösung verbraucht. EinGramm Kalzium-Oxalat-Polyhydrat verbraucht infolgedessen 115,1 cm 3 1/io normaleKMnO4-Lösung. Es folgt daraus, dass ein Gramm Polyhydrat 11,51 cm 3 einer normalenKMnO4-Lösung zur Oxydation benötigt. Da ein Äquivalent Kalzium-Oxalat 64 g wiegt,ergibt sich ein Milliäquivalent von 0,064. Durch Multiplikation von 0,064 mit 11,51 erhältman eine Menge von 0,7366 g Kalzium-Oxalat und 0,2634 g H 2O. Daraus resultiert einWassergehalt von 2,54 Mol. Dieser Wert stimmt mit einem Fehler von 1,6 % mit einemKalzium-Oxalat-2,5-Hydrat überein.
2. Analyse
Nach dem oben beschriebenen Verfahren wurden 0,1968 g Polyhydrat in HCl auf-gelöst und gelangten zur Titration. Es ergab sich ein Wassergehalt des Kalzium-Oxalatesvon 2,55 Mol. Dieser Wert stimmt mit einem Fehler von 2 % mit einem Kalzium-Oxalat-2,5-Hydrat überein.
Aus diesen Resultaten geht hervor, dass das tetragonale Polyhydrat als ein2,5-Hydrat angesprochen werden muss.
Zusammenfassung des III. Teils
1. Mit Hilfe von Entwässerungskurven konnte gezeigt werden, dass dasPolyhydrat über das Monohydrat ins Anhydrat übergeht. Ein Dihydrat trittscheinbar nicht auf, da sich das Polyhydrat in einem direkten Sprung insMonohydrat umwandelt. Das tetragonale Polyhydrat selber stellt kein Di-hydrat dar, wie der Gewichtsverlust von 26 % bei der Umwandlung ins An-hydrat zeigt.
2. Mit Mikronanalysen wurde nachgewiesen, dass es sich beim Polyhydratum ein Ca (COO) 2 • 2,5 H 2O handelt. Die Analysenresultate zeigen Werte von2;44 und 2,42 Mol Wasser, stimmen also mit einem Fehler von 2,4 % bzw. 3,2%mit einem Kalzium-Oxalat-2,5-Hydrat überein.
3. Die chemische Zusammensetzung des tetragonalen Polyhydratpulverswurde durch eine analytische Bestimmung von C, H, 0 und Ca ermittelt.Die Untersuchung ergab, dass die erhaltenen Werte einem Ca (COO) 2 . 2,5 H2Osehr nahe kommen.
4. KMnO4 Titrationen ergaben Wassergehalte von 2,54 und 2,55 Mol, stimmenalso mit einem Fehler von 1,6 % bzw. 2 % mit dem Ca (COO) 2 •2,5 H2Oüberein.
5. H. A. KLASENS, W. G. PERDOK, P. TERPSTRA (1937) fanden für das tetra-gonale Strontium-Oxalat einen Wassergehalt von 2,5 H 2O. Da dieses Salzwahrscheinlich mit dem tetragonalen Polyhydrat des Kalzium-Oxalates iso-morph ist, können dadurch unsere Befunde, die ein Ca(COO) 2 • 2,5 H2O er-geben, gestützt werden.
.39

IV. Teil
Untersuchung pflanzlicher Kristalle
A. Einleitung
Da die DEBYE-SCHERRER-Testaufnahmen von reinem, synthetischem Ca(COO) 2 •2,5 H2O und von Ca (COO) 2 • H2O angefertigt wurden, können siezur Identifizierung von Kalziumrekreten in der Pflanze verwendet werden.Wie schon im I. Teil der Arbeit erwähnt wurde, sind chemische Analysen zurKennzeichnung pflanzlicher Rekrete viel zu ungenau, als dass mit diesen Me-thoden zuverlässige Resultate gewonnen werden könnten.
Tetragonale Kristalle aus der Zwiebelschale von Allium cepa gelangtenzur Untersuchung, um zu erkennen, ob die synthetischen Präparate mit dennatürlichen übereinstimmen. Das Isolationsverfahren und die Resultate wur-den im II. Teil beschrieben. Da die tetragonalen Kristalle infolge ihrer Formund ihrer Doppelbrechung im Polarisationsmikroskop relativ leicht erkanntwerden können, wurde darauf verzichtet, noch weitere Kristalle des Poly-hydrates aus anderen Pflanzen zu isolieren. Die vorliegende Untersuchungbeschränkt sich auf die röntgenographische Analyse der Raphiden sowie aufdie Bestimmung des H 2O-Gehaltes von Kristallsand.
B. Debye-Scherrer-Aufnahmen von Raphiden und Kristallsand
1. Untersuchung von Raphiden
Diese Kristalle, welche für die Monokotyledonen charakteristisch sind, be-finden sich in grossen Mengen in den sukkulenten Blättern der Aloe. DurchAuspressen des Saftes in einen Sedimentierapparat konnten die Kristalle iso-liert und der röntgenographischen Analyse unterzogen werden. Die nadel-förmigen Kristalle sind optisch positiv. Auf Grund optischer Untersuchungenbewies schon A. FREY (1929), dass es sich bei den Raphiden um das Mono-hydrat des Kalzium-Oxalates handelt. Das Röntgendiagramm ergab eindeutigeine Bestätigung dieses Befundes, denn die Interferenzen stimmen mit jenendes Monohydrates überein. Neben den Monohydratlinien sind Interferenzenanderer, wahrscheinlich organischer Stoffe zu erkennen. Die zusätzlichenLinien sind auf Verunreinigungen des Ausgangsmaterials durch Zellbestand-teile zurückzuführen.
2. Untersuchung des KristallsandesDer Kristallsand tritt bei Solanum tuberosum, Aucuba japonica, Allium
globosum sowie bei Sambucus nigra auf. Da die Form der Kriställchen imKristallsand bis anhin nie genau ermittelt werden konnte, kann auch mitoptischen Methoden über ihre chemische Zusammensetzung nichts Bestimm-tes ausgesagt werden. Die Stärke der Doppelbrechung spricht für ein Kalzium-Oxalat-Monohydrat. Über die Flächenformen des Whewellit und über Beob-
40

achtungen an Kristallsand siehe KOLLBECK, GOLDSCHMIDT Und SCHRODER (1918),A. FREY-WYSSLING und F. BLANK (1938), G. ARCANGELI (1891), J. VESQUES(1874).
Die Kriställchen wurden aus dem Mark von Aucuba japonica isoliert. Dasfrische Mark wurde zur Sprengung der Zellwände gefroren und nach demAuftauen durch Gaze auf einen Objektträger gepresst. Auffallend ist diestarke Doppelbrechung der kleinen, ca. 3 i.4 grossen Kriställchen, die, wieerwähnt, auf Monohydrat hindeutet. KOHL (1889) behauptet dagegen, dasses sich bei diesen Kristallen nicht um das Monohydrat, sondern um die tetra-gonale Hydratstufe des Kalzium-Oxalates handle. Da die Kriställchen zumTeil «Tetraeder»form aufweisen, ordnet er sie der tetragonalen Hemiedrie zu.P. JACCARD und A. FREY (1928) fanden bei Allium globosum Kristallsand-zellen in der äusseren Zwiebelschale. Sie entdeckten indessen in diesem Kri-stallsand neben tetraedrischen Kriställchen eine Anzahl gut ausgebildeter,monokliner Kristalle. Sie ordneten daher den Kristallsand vorläufig dem mono-klinen Kristallsystem zu.
Die röntgenographische Analyse ergab eindeutig ein Monohydrat. Damitist die Streitfrage um die Hydratstufe des Kristallsandes entsChieden. Um dieKristallform zu deuten, wurde der Versuch unternommen, mit Hilfe der stereo-graphischen Projektion die Flächen des sogenannten «Sable tetraédrique» zubestimmen.
Abbildung 15 zeigt eine Mikroaufnahme der Kriställchen. Es ist daraus zu
Abb. 15 Kristallsand im polarisierten Licht photographiert. Vergr. 1500 mal.
41

ersehen, dass sie, auf flache Unterlage gelegt, zwei verschiedene Lagen bevor-zugen, die mit A und B bezeichnet werden sollen. Lage A weist einen spitzenWinkel, Lage B einen stumpfen Winkel auf. Der Winkel bei A misst ca. 65°+ 8°, der Winkel bei B ca. 93° ± 4°. Da eine Winkelmessung an solch kleinenKriställchen sehr ungenau ist, sind die erwähnten Werte nicht sehr zuver-lässig. Die Auslöschung der sichtbaren Fläche bei Lage A ist schief, währendLage B eine gerade Auslöschung zeigt. A. FREY (1924) hat die Flächenformender monoklinen Pflanzenkristalle nach ihrer Häufigkeit zusammengestellt.
Herr Prof. Dr. R. L. PARKER war so freundlich, zur Bestimmung der Winkelbei den verschiedenen Kombinationen der häufigsten Flächen die stereogra-phischen Projektionen durchzuführen. Als eine wahrscheinliche Form resul-tierte daraus eine Kombination einer e-Fläche (101) mit einer c-Fläche (001)und einer m-Fläche (110). Bei dieser Kombination setzt sich der Kristall ausacht dreieckigen Flächen zusammen.
Der stumpfe Winkel der c-Fläche beträgt 96° und stimmt daher gut mitdem stumpfen Winkel bei Lage B überein. Es ist anzunehmen, dass der Kri-stall in diesem Falle auf der Gegenfläche von c = c' läge. Für die Lage Akommt als sichtbare Fläche e kaum in Frage, da die m-Flächen im Polarisa-tionsmikroskop zu erkennen wären. Es handelt sich daher möglicherweise umeine m-Fläche. In diesem Falle aber müsste der spitze Winkel 78° betragen;die Messung ergibt aber einen Wert von 65°. Es ist daher fraglich, ob die obengenannte Kombination der Flächen der Form des Kristallsandes entspricht.
Zusammenfassung des IV. Teils
1. Es konnte mit DEBYE-SCHERRER-Aufnahmen nachgewiesen werden, dassdie R a p h i d en sowie der Kristallsand aus dem Monohydrat des Kalzium-Oxalates bestehen. Die Kristalle aus der Zwiebelschale von Allium cepa er-wiesen sich als identisch mit dem Ca (COO) 2 • 2,5 H2O (Polyhydrat) .
2. Es wurde der Versuch unternommen, die Form des Kristallsandes zubestimmen. Dabei gingen wir von der Annahme aus, dass der Kristall nurvon dreieckigen Flächen begrenzt sei. In diesem Falle ist eine Kombinationvon c-, e- und m-Flächen wahrscheinlich.
Schlussfolgerungen
1. Die röntgenographische Molekulargewichtsbestimmung lässt daraufschliessen, dass das tetragonale Kalzium-Oxalat-Hydrat (Weddelit) 2,5 MoleH2O enthält [Ca (COO) 2 . 2,5 H2O].
2. Chemische Untersuchungen ergaben für das tetragonale Kalzium-Oxalatebenfalls einen Wassergehalt von 2,5 H2O.
3. Es konnte nachgewiesen werden, dass die Raphiden und der Kristall-sand dem Monohydrat des Kalzium-Oxalates zuzuordnen sind; demgegen-über sind die in Allium cepa vorhandenen Kristalle als Kalzium-Oxalat-2,5-Hydrat bestimmt worden.
42

Literaturverzeichnis
NGELI, G., 1891. Bot. Ital. 23, 489.ISTER, F. A., 1947. Identification of a Calculus from a Hippopotamus. Nature 160, 470.AL, J. D., 1926. Proc. Roy. Soc. A 113, 117.DENBERGER, E., 1945. Röntgenographisch -analytische Chemie.DE QERVAIN, SCHINZ, H. R., 1947. Röntgenographische und mikroskopisch-kristall-optische Untersuchung an Harnsteinen. Helv. Med. Acta, 14, 195.SCHINZ, H. R., 1948. Zielsetzung und Ergebnisse systematischer Feinstrukturunter-suchungen mittels der Röntgeninterferenzen beim Menschen. Bull. Schweiz. Akad.med. Wiss. 3, 262.
CHT, W., SCHINZ, H. R., 1950. Ergebnisse der Feinstrukturuntersuchung von Blasen-steinen aus dem vorderen Orient. Schweiz. med. Wschr. 80, 792.A., 1924. Vergleich des Whewellit mit Mikrokristallen von Kalzium-Oxalat-Mono-hydrat. Schweiz. Mineralog. u. petrogr. Mitt. 4, 16.1925. Kalzium-Oxalat-Monohydrat und -Trihydrat in der Pflanze. Diss. Zürich. Vier-teljahrsschr. Naturf. Ges. Zürich, 70, 1-65.1929. Kalzium-Oxalat-Monohydrat und -Trihydrat. LINSBAUER'S Hdb. d. Pflanzen-anatomie, Bd. 3/la, 90.
-WYSSLING, A., BLANK, F., 1938. Über knieförmige Oxalatkristalle von Caryocar nuci-ferum L. Protoplasma 31, 194.
HER, R., 1949, Materialprüfung mit Röntgenstrahlen. Berlin 1949.YIARSTEN, G., 1937. Kalzium-Oxalat als Steinbildner in den Harnwegen. Lunds Univ.
Arsskr. N. F. Avd. 2. Bd. 32, Nr. 12.RICKS, ST. B., 1935. The Orientation of the Oxalate-Group in oxalic Acid and some ofits Salts. Z. Kristallogr. 91, 48.DEMING, W. E., 1935. On the Optical Anisotropy of Molecular Crystals as Illustratedby some Oxalates. Z. Kristallogr. 91, 290.
ARD, P., FREY, A., 1928. Kristallhabitus und Ausbildungsformen des Kalzium als Art-merkmal, Schinz-Festschr. d. Naturf. Ges. Zürich. 53, 142.
nationale Tabellen zur Bestimmung von Kristallstrukturen. 1935. Band I (1935a) undIT (1935b). Berlin.
EN, A. T., 1940. On Concrements from the Urinary Tract. II. Acta chir. scand. 84, 207.1941. On Concrements from the Urinary Tract. III. Acta chir. scand. 85, 473.
s, F. T., WHITE, L. M., 1946. The Composition, Optical and Crystallographic Propertiesof two Calcium-Oxalate-Chloride Double Salts. J. Am. Chem. Soc. 68, 1339.
ENS, H. A., PERDOK, W. G., TERPSTRA, P., 1937. Crystallography of Strontium Oxalate.Z. Kristallogr. 96, 227.FR. G., 1889. Anat.-physiol. Unters. d. Kalksalze u. Kieselsäure i, d, Pflanze, Mar-burg.
t.SCHÜTTER, V., MARTI, J., 1930. Über Bildungsformen des Kalzium-Oxalats. Helv. Chim.Acta, 13, 929.
BECK, GOLDSCHMIDT, SCHRÖDER, 1918. Über Whewellit. Beitr. Z. Krist. U. Mineral. 1, 199.■MPTE, J., PROBEGUIN, T., WYART, J., 1943. Sur les sell de calcium de l'acide oxalique.
C. R. Acad. Sc. Paris. 216, 808.LI, P., 1924. Lehrbuch der Mineralogie. Berlin 1924.sr, L., FRONDEL, C., 1947. Studies in Urolithiasis I. The Composition of urinary calculi.
J. Ur. 57, 961.
43

SCHINZ, H. R., 1943. Röntgenographische Feinstrukturuntersuchungen der Verkalkungen immenschlichen Körper. Radiologia Clinica. 12, Nr. 3, 129.
SCHMID, E. E., 1856. Über die oxalsaure Kalkerde. Ann. d. Chemie u. Pharm., 97, 225.SoUcHAY, A., LENSSEN, E., 1856. Über die Oxalate der Alkalien und alkalischen Erden. Ann.
d. Chemie u. Pharm., 100, 308.TREADWELL, F. P., 1919. Lehrbuch der analytischen Chemie II. Leipzig u. Wien 1919.VESQUES, J., 1874. Ann. sc. nat. 19, 300.ZACHARIASEN, W. H., 1934. The Crystal Lattice of oxalic Acid Dihydrate H 2C2O4 •2 H2O and
the Structure of the Oxalate Radical. Z. Kristallogr. 89, 442.
44






















![Physikalische Chemie - Grundmodul...Joh Ins 0 I / [a.u.] Abbild @647n scheide 1.2 Z Trotz z stark s ein Üb Versch wieder oder T detektie Abbild In eine Intensit annes Gute titut für](https://img.pdfslide.org/doc/110x75/5e5994677a63f40a521d2029/physikalische-chemie-grundmodul-joh-ins-0-i-au-abbild-647n-scheide.jpg)

![L = TMOGA · Hb — a.u. [NpO2]+ GAS SMD ∠O-Np-O 179.2 180.0 d d1/d2 MBO DI ρb 2ρ b Hb ε d d1/d2 MBO DI ρb 2ρb Hb ε Np-Oyl-1 1.736 0.965/0.771 2.25 2.97 0.336 0.117 -0.35](https://img.pdfslide.org/doc/110x75/5f302344bdbfe00f40656003/l-hb-a-au-npo2-gas-smd-ao-np-o-1792-1800-d-d1d2-mbo-di-b-2-b-hb.jpg)






