Embed Size (px)
Citation preview

438 Berieht: Analyse organischer Stoffe
Weitere Untersuehungen fiber die Papierchromatographie der Fettsiiuren haben H. P. KArTF~A~ und E. Mom~ 1 angestellt. Verff. besch~ftigen sich mit der Fort- entwicklung und Verbesserung der papierchromatographischen Fettan~lyse sowie mit Fragen der theoretischen Deutung der Verteilungsvorg~nge. Die beschriebenen Versuche erweitern die Kenntnis der Umkehrphasensysteme und zeigen speziell bei der Trennung yon Fetts~uren die I~I6gliehkeit der Stand~rdisierung mid Ver- besserung der yon II. P. KAVF~A~ und W. NITSCH 2 angegebenen Methode auf. Diese Methode benutzt als st~tioni~re Phase und hydrophobierendes Nittel eine dem Undecan entspreehende Kohlenwasserstofffraktion, als mobile Phase eine mit dem gleiehen Pr/~parat ges/~ttigte Essigs/~ure versehiedener Konzentration. Zum Naehweis der getrennten Fetts/~uren eignet sieh die Uberfiihrung in farbige Verbindungen, insbesondere Kupferseifen, die dureh I~eakgion mit Kaliumhex~eyanoferr~t (II) [Umwandlung des Kupfers in Kupferhexaeyanoferrat (II)] versti~rkt angef/irbt werden k6rmen. Die Entwicklung der Chromatogramme wird naeh der aufsteigenden, horizontalen oder absteigenden Arbeitsweise durehgefiihrt. Es hat sich gezeigt, dab die Impr/ignierung yon wesentlichem EinfluB auf die Giite der Trennungen ist und dab dureh eine geeignete Impr~gnierung sowohl die Entwicklungsdauer als aueh die l%t-Werte der Fettsiiuren erheblich beeinfluBt werden k6nnen. Verff. halten es daher ffir zweekm/~Big, zur Standardisierung der Methode den Impr/~gnierungsgrad anzugeben, worunter die Menge des unpolaren Impr~gnierungsmittels (in Grammen), bezogen &uf 1 g Papier, zu verstehen ist. Der Anwendungsbereieh des grund- legenden Kohlenwasserstoff-S~ure-Wasser-Systems kann dureh Zus~tze polarer Stoffe zur Steigfltissigkeit erweitert werden. Von den untersuehten LSsungsmitteln eignen sieh am besten Zus&tze yon Aeeton und Aeetonitril. Beide verringern die Entwicklungsdauer und ermSglichen die Chrom~tographie bei hSheren Temperatu- ren. Dariiber hinaus erhSht ein Aeetonzusatz die Rf-Werte und die R~-Differenz der getrennten Fettsauren. Zur Chromatographie bei tiefen Temperaturen bietet das System Undecan-Aeeton-Propions/~ure-Wasser die giinstigsten Bedingungen, so z.B. zur Auftrennung der wichtigsten kritisehen Fettsaurepaare. Je naeh der Art der zu trennenden Fetts&uren, der zur Verfiigung stehenden Zeit und der Besehaffen- heir der benStigten LSsungsmittel mul~ also die Steigfltissigkeit variiert werden. Die versehiedenen Ausfiihrungsformen der papierehromatographisehen Analyse von Fettsauregemisehen werden eingehend in der Originalarbeit besehrieben.
Fette u. Seifen 60, 165--177 (1958). Univ. M/inster und Dtsch. Inst. Fett- forsehung, Mfinster. -- e Fette u. Seifen 56, 154 (1954); ~7, 473 (1955); 58, 234 (1956); vgl. diese Z. 146, 115 (1955); 150, 319 (1956); 157, 450 (1957).
K. M.~oI~EI~
Zur Best immung yon Urons~uren wird yon S. A. BARKER, A. B. FOSTER, I. R. SIDDIQUI und ~{. STACEY 1 die yon R. M. MCCREADY, H. A. SWENSEN und W. D. MACLAY 2 beschriebene Decarboxytierungsmethode so modifiziert, da8 die Proben- einwaagen yon 250 mg auf 20 mg reduziert werden kSnnen. -- Arbeitsweise. Die erforderliche Apparatur (Abb. 1) wird vor Gebrauch mit warmer Chromschwefel- saure gereinigt und getrocknet. Samtliehe Schliffverbindungen werden mit Silicon- fett ubgedichtet. Die ganze Apparatur wird durch AsearitrShrchen yon atmo- spharischer Kohlensaure freigehMten. 20 mg Probe werden in ein 1 cm langes enges I~Shrchen eingewogen und in das ReaktionskSlbehen iibergefiihrt. ]:)ann gibt man mittels Pipette 3 ml 19% ige Salzsaure zu und spfilt die Apparatur mit Stiekstoff C02-frei, wobei das AbsorptionsrOhrchen nicht angeschlossen ist. Letzteres wird ebenfalls mit Stiekstoff ausgesp~It, mit 5 ml 0,25 n Natrenlauge beschiekt und mit der Apparatur verbunden. M~n reguliert dell Stickstoffstrom auf t Blase/2--3 sec ein und taucht den Reaktionskolben so in ein 145~ heiges 01bad ein, dab sieh der
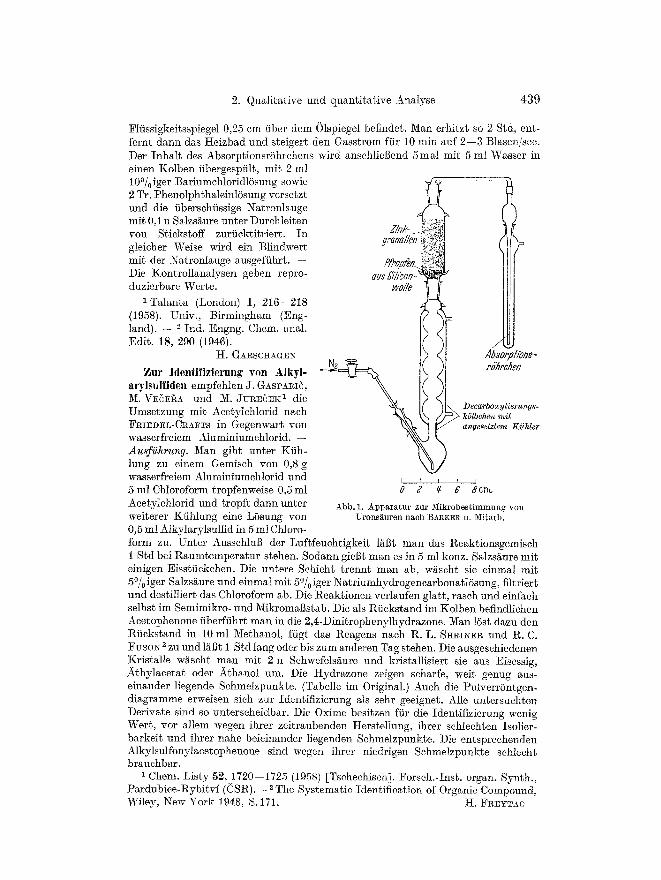
2. Qualitative und quantitative Analyse 439
F1/issigkeitsspiegel 0,25 em fiber dem 01spiegel befindet. Man erhitzt so 2 Std, ent- fernt dann das Heizbad und steigert den Gasstrom ffir 10 min auf 2--3 Btasen/see. Der Inhalt des AbsorptionsrShrehens wird anschliegend 5mal mit 5 ml Wasser in einen Kolben iibergespfilt, mit 2 ml 10~ BariumehloridlSsung sowie 2 Tr. PhenolphthaleinlSsung versetzt und die iiberschfissige Natronlauge mit 0,1 n Salzs/~ure unter Durehleiten yon Stickstoff zurfiektitriert. In gleieher Weise wird ein Blindwert mit der Natronlauge ausgeffihrt. -- Die Kontrollanalysen geben repro- duzierbare Werte.
1 Talanta (London) 1, 216--218 (1958). Univ., Birmingham (Eng- land). -- 2 Ind. Engng. Chem. anal. Edit. 18, 290 (1946).
I-I. G~A~SCKAOEX
Zur Identifizierung yon Alkyl- arylsulfiden empfehlen J. GASP~a~I5, IVl. V~SE~A und M. JV~E~E~: ~ die Umsetzung mit Acetylchlorid nach FRIEI)~L-C~FTS in Gegenwart yon wasserfreiem Alumin iumeh lo r id . - Ausfiihrung. Man gibt unter Kiih- lung zu einem Gemiseh yon 0,8 g wasserfreiem Aluminiumehlorid und 5 ml Chloroform tropfenweise 0,5 ml Aeetylehlorid und tropft dann unter weiterer Kfihlung eine LSsung yon 0,5 ml Alkylarylsulfid in 5 ml Chloro-
Z/nk- yp~nelien
xi~ ~i~'con -~ wolle
Absopp/ioz~- e~echen
Decarboxylieru~gs- k51behen mit angesetztem Kiihler
I r r r f
0 2 g r 8crr~
Abb. 1. Apparatur ztu. 2~Iikrobestimmung yon Uronsiiurea nach BA]gKEI~ U. h~[itarb.
form zu. Unter Aussehlug der Luftfeuehtigkeit 1/~Bt man das lC~eaktionsgemiseh 1 Std bei Raumtemperatur stehen. Sodann giegt man es in 5 ml konz. Salzs/~ure mit einigen Eisstfiekchen. Die nntere Sehicht trennt man ab, w/~seht sie einmal mit 50/0iger Salzs/~ure und einmal mit 50/0 iger NatriumhydrogenearbonatlSsung, filtriert und destilliert das Chloroform ab. Die Reaktionen verlaufen glatt, raseh -and einfach selbst im Semimikro- und Mikromagstab. Die als Riiekstand im Kolben befindliehen Aeetophenone fiberffihrt man in die 2,4-Dinitrophenylhydrazone. Man 15st dazu den Rfiekstand in l0 ml l~Iethanol, ffigt das Reagens naeh R. L. Sm~I~E~ nnd R. C. Fvso~ 2 zu nnd 1/iBt 1 Std lang oder bis zum anderen Tag stehen. Die ausgesehiedenen Kristalle w/iseht man mit 2 n Schwefels/~ure und kristallisiert sic aus Eisessig, Athylaeetat oder Athanol urn. Die ttydrazone zeigen seharfe, welt genug aus- einander liegende Sehmetzpunkte. (Tabelle im Original.) Aueh die PulverrSntgen- diagramme erweisen sieh zur Identifizierung als sehr geeignet. Alle untersuehten Derivate sind so unterseheidbax. Die Oxime besitzen fiir die Identifizierung wenig Wert, vor allem wegen ihrer zeitraubenden Herstellung, ihrer schleehten Isolier- barkeit und ihrer nahe beieinander liegenden Sehmelzpunkte. Die entspreehenden Alkylsulfonylaeetophenone sind wegen ihrer niedrigen Schmelzpunkte sehlecht brauehbar.
1 Chem. Listy 52, 1720--1725 (1958) [Tseheehiseh]. Forseh.-Inst. organ. Synth., Pardubiee-Rybi tv/ (~SR). - - 2 The Systematic Identification of Organic Compound, YViley, New York 1948, S. 171. H. FREYTAG





























