Embed Size (px)
Citation preview

Zur Kenntnis der lyophilen Kolloide. I. M i t t e i l u n g .
Allgemeine Einleitung: Das Agarsol. Von H. R. K r u y t und H. G. B u n g e n b e r g de J o n g .
(van 't Hoff Laboratoriam, Utrecht.)
(Mit 10 Figuren.) (Eingegaugcu am 4. August 1928.)
E i n l e i t u n g .
Die in dieser Serie erscheinenden Mitteilungen enthalten die Er-
gebnisse yon Untersuchungen und Llberlegungen der letzten sieben Jahre aus dem hiesigen Laborator ium. Sie sind aus 5~ugeren Grtinden bisher nut erst in vorl~iufiger Form verSffentlieht worden 1) und bilden daher zum ersten Male eine mehr definitive Darstellung. Seit die bier
zu bespreehenden Untersuchungen abgesehlossen wurden (1921), sind so vie1 neue Erfahrungen gesammelt, dab wir jetzt mit besserem Ge- wissen die yon den fiblichen Ansichten abweiehenden Auffassungen vor-
zutragen wagen. Der Grundgedanke dieser ~Untersuchung war folgender: Wir besitzen zurzeit ein ziemlieh Hares Bild fiber die Bau- und
Stabilit~itsverh~iltnisse suspensoider und lyophober Kolloide: diese
1) H. R. Kruy t u. H. G. de Jong , Zeitschr. f. physik. Chem. 100, 250 (1922); H.R. Kruyt , Koll.-Zeitschr. 81, 338 (1922); die Utrechter Doktorarbeiten von H. G. de Jong, Het Agarsol (1921), H. Lier , Het Caseinesol (1924), H. J. c. T e n d e l o o , Hydratatie en lading (1926), und in geringerem Maf~e J. E. M. van der Made, Het Ceriumhydroxidesol [Rec. Tray. Chim. 42, 277 (1923)], undJ. P o s t m a (Leiden), Het Kielzuursol [Rec. Tray. Chim. 14; 765 (1925)]. Ferner H. G. B u n g e n b e r g de Jong, Rec. Tray. Chim. 42, 437 (1923), 43, 35 u. 189 (1924); H. R. K r u y t u. W. A. N. E g g i n k , Proc. Roy. AcacL Amsterdam 26, 43 (1922); H. R. Kruy t u. H. J. C. T e n d e l o o , Proc. Roy. Acad. Amsterdam 27, 377 (1924), Journ. of Physical Chem. 29, 1303 (1925); H. R, Kruyt , Nature (1923), 827; Natuurwetenschappelijk Tijdschrift 1924 (MS.rz-April.Heft); Chem. Weekblad 22, 473 (1925); Einfiihrung in die physikalische Chemie und Kolloid- chemie, Kapitel VII (holl/indisch 1924, 1925 und !926, deutsch 1926). Vgl. auch H. R. Kruyt , Colloids, a Textbook (New York 1927), Kapitel XII und folgertde.

,) KOLLO1DCHEMISCHE B E I H E F T E BAND X X V I I I , H E F T 1 - - 2
bestehen aus polymolekularen Teilchen, wclchc durch ihre elektrische Ladung daran behindert wcrden, bei den dutch die Brownsche Be- wegung viclfach verursachtcn Zusammenst613en zusammenzuklebcn. Nur ,,unl6sliche K6rper" sind imstande, dcrartigc L6sungen zu bilden, d.h. dicse polymolekularen Partikelchen kOnnen nur dann cxisticren, wenn eine molekulardispcrse Verteilung nicht einc Hcrabsetzung tier freien Energie des Systems herbciftihrt. Es liegt also cntwedcr vOllige Unl6slichkeit des dispcrgierten K6rpcrs im I)ispcrsionsmittel vor odor letzteres ist eine gcs/ittigte LSsung dcr dispcrgiertcn Substanz. In beiden F~illcn k6nnen wit for ein richtiges VersUindnis des kolloiden Systems von dicscr wahren L6slichkeit absehen und die kolloiden Teilchen als dispergiert in cinem Medium, in dcm sic nicht (bzw. nicht mehr welter) 16slich sind, bctrachten.
Die elektrische Ladung dicscr Tcilchen, so wie wir sie vor allcm aus tier Kataphorese kcnncn, zeigt den Charakter ciner Doppelschicht mit allen Eigentiimlichkciten tier kapillar-clcktrischen oder elektro- kinetischen Prozesse wic Elcktroendosmose und Str6mungspotentialc. Dcr Parallelismus zwischcn dcm Einflul3 kleincr Elektrolytkonzen- trationcn auf dicse Prozcssc und dcr Wirkung auf die Suspcnsoidc hat die Identit/tt dcr elektrischen Verh~iltnisse deutlich gezcigt. Die Art und Wcise, wie dic Elektrolyten die Vcrh~iltnissc bei den elektro- endosmotischen Untersuehungen E l i s s a f o f f s l ) , bei denjenigen fiber StrSmungspotentiale yon K r u y t 2) und bei den Versuchen yon P o w i s 3) iibcr Kataphorese beherrschen, ist dureh den tibcrragenden Einflutl des dem Teilchen entgegengesetzt geladenen Ions bzw. durch desscn Adsorbierbarkeit gckennzeichnet; dasselbe zeigtcn die Untersuchungen yon S c h u l z e 4) und L i n d e r und P i c t o n 5) und die mannigfaehen Arbeiten yon F r e u n d l i c h und seinen Sehfilern. 6) Es kann also kaum einem Zweifcl unterliegen, dab die elcktrisehe Ladung der Suspensoide und die der W~nde, welche in den genannten Prozesscn cine Rolle spielen, den gleichen Charakter haben. In beiden F~llen handelt es sich um Vorg~.nge, welche in Hinsicht auf die totalen Elektrolytmengen die gleiche Entladung bewirken und dabei keine stSehiometrischell Vet-
1) G. v. El issafoff , Zeitschr. f. physik. Chem. 79, 385 (1912). z) H. R. Kruyt , Proc. Roy. Acad. Amsterdam 17, 615 (1914); 19, 1023
(1916); Kolh-Zeitschr. $$, 81 (1918). 3) F. Powis, Zeitschr. f. physik. Chem. 89, 91 (1915'. *) H. Schulze, Journ. f. prakt. Chem. (2) 25, 431 (1882); 27, 320 (1883). ~) S. E. Linder u. H. Picton, Journ. Chem. Soc. 67, 62 (1895); 87. 1906
(1905). 6) Vgl. H. Freundl ich , Kapillarchemie (2. u. 3. Aufl.), 569fl.

KRUYT u. BUNOENBERO DE JONO, ZUR KENNTNIS D. LYOPHILEN KOLLOIDE 3
h~iltnisse zeigen: der EinfluB von z. B. K.-, Ba- und Al-Ion, wenn dieselben e i n e m negativen Kolloid in ~tquimolekularen Mengen hinzugesetzt werden, verh~ilt sich nicht, wie man erwarten k6nnte, wie 1 : 2 : 3 1 sondern etwa wie 1 : 7 : 500 im Falle eines As2Ss-Sols oder wie 1 : 30 :~50 im Falle eines Au-Sols. Dennoch haben die Untersuchungen F r e u n d l f c h s , vor allem an dem As2Sa-Sol 1) gezeigt, dab die tatsS~eh- lich yon den Teilchen bis zur bestimmten Herabsetzung der Ladung gebundene Menge Kation immer st6chiometrisch best immt ist. Bei un- 16slichen Verbindungen zwisehen dem fS.11enden Ion und dem, welches in der Doppelschicht die innere Belegung (vom Teilchen aus gerech- net) anbietet, kann dieses zu einer Vereinfachung, wie es die Schtiler Z s i g m o n d y s i) beim SnO2-Sol gefunden haben, fiihren,
Dieses ist also im allgemeinen das Bild, welches wir yon den Sus- pensoiden, ihrer Ladung und deren Bedeutung bekommen. In der kolloidchemischen Welt denkt man sich j edoch die Verhi~ltnisse ganz anders, sobald nicht mehr lyophobe, sondern lyophile Kolloide in Betracht kommen. Es wird bier das System als molekulardispers betrach- tet; man nimmt an, dab die Ladung die gleiche Natur wie bei einer
Elektrolytl6sung habe und dab diese Ladung gar nicht die Stabilit~it wie bei den Suspensoiden bedingt. Es liegt also eigentlich keine Kon- tinuititt zwischen diesen Gebieten der Kolloidchemie vor, und man fragt sich, ob es eigentlich Zweck hat, ein Goldsol und ein Albuminsol beide als Kolloidl6sungen zu betraehten, da bei so weir gehenden Unter- schieden das Wort Kolloid jede Bedeutung verliert.
DaB so weit gehende Gegens~tze, wie in der Entwicklung der Kol- loidchemie allm~hlich zutage traten, in Wirklichkeit bestehen, schien uns sehr zweifelhaft, und der eine yon uns hat deshalb auf diesen Zweifel in seinen Vortriigen in den letzten 15 Jahren immer hingewiesen. Die DualitS.t der ErklSxungen schien vielmehr darauf zuriickgeftihrt werden zu miissen, dab die Kolloidchemie der Suspensoide vor allem yon Phy- sikochemikern, die Kolloidchemie der Emulsoide aber von Physiologen betrieben worden war. Das Bedtirfnis, die Resultate schnell m6gliehst auf biologische Systeme anwenden zu k6nnen und mehr noch die leicht verst~ndliche Vorliebe far die EiweiB16sungen hat deshalb sein Ge- pr~ige auf die Entwicklung des letzten Teiles der Kolloidchemie ge- drfiekt.
1) H. F r e u n d I i c h, Zeitschr. f. physik. Chem. 78, 385 (1910) ; W. R. W h i t n e y u. J. E. O b e r , Zeitschr. f. physik. Chem. 89, 630 (1902).
2) Vgl. fiir die Arbeiten yon E. H e i n z , R. F r a n z , G, V a r g a usw. R. Z s i g m o n d y , Kolloidchemie (5. Aufl.), lI, 98tt.
1"

4 KOLLOIDCHEMISCHE BEIHEFTE BAND XXVlll, HEFT 1--2
Hierzu kommt noch folgendes. Die Bestimmung der elektrischen Ladung ist bei Suspensoiden gar nicht einfachl), bei lyophilen Systemen ist sie aber noch vie1 schwieriger (in der dritten Mitteilung dieser Serie werden wir ausftihrlich hierauf zuriickkommen). Direkte Bestimmungen der Ladung bei Zusatz n e u t r a l e r Elektrolyten lagen kaum vor, als wir mit diesen Untersuehungen begannen. Eine (vom physiologischen Standpunkt wieder sofort verst~ndliehe) Vorliebe ffir das H- und OH- Ion beherrschte die Untersuchungen, und die immer bestehende M6g- lichkeit einer Betrachtung der Eiweifisysteme als L6sungen yon Amino- sfiuren beherrschte die Theorie. Und was die Untersueher auf diesem Gebiete wohl am meisten davon abhielt an die sich allm~thlich aus- bildende Theorie der Suspensoide anzukntipten, war die Tatsache, dab die Proteine bei ElektroneutralitS.t der Teilchen vielfach nicht aus- fielen, was in ganz schroffem Gegensatz zum Verhalten aller Sus- pensoide steht. Es schien also, als ob vielleicht infolge der unverkenn- bar vorliegenden Lyophilie der Systeme, d.h. infolge der Hydratisie- rung yon Ionen und Molektilen der gel6sten AminosS.uren hier ganz andere VerhSAtnisse das System beherrsehten.
Es wird verstiindlich sein, warum wir bei dem erwlihnten Zweifel unsere Untersuchungen mit einem lyophilen System, welches keine Aminosiiuren entMilt, bei dem also yon vornherein eine Ionisations- theorie ausgeschlossen war, anfingen; dieses System sollte speziell bei H i n z u f t i g e n n e u t r a l e r E l e k t r o l y t e n s t u d i e r t w e r d e n .
Wir w~thlten hierzu Agar, welcher ein Kohlehydrat und zwar wahrscheinlich eine Polygalaktose ist.
Die unter dem Einflul3 yon S~uren und Basen, weniger von Salzen, eintretenden 2~nderungen in der Viskosit~it der Eiweifll6sungen haben eine besondere Bedeutung ftir die Entwicklung der obenerwS.hnten Theorie der lyophilen Kolloide gebabt. Wir wollten darum ebenfalls mit ViskositS.tsmessungen anfangen, legten aber grol3en Wert auf eine einwandfreie Methodik und auf die Gewiflheit, dab wirklich Viskosit~ts- messungen vorlagen, damit wir auch berechtigt wSoren, unsere Resultate vom Standpunkt der Viskositiitstheorie aus zu interpretieren. Uber die angewandte Methodik ist an anderer Stelle schon berichtet worden. 2) Es sei hier nur erwS~hnt, daft wir immer mit O s t w a l d s c h e n Viskosi- metern gearbeite t haben, welche den Anforderungen G r u n e i s e n s bis
auf 1 Promille Gentige leisteten.
1) Vgl. H. R. Kruyt , Koll.-Zeitschr. 37, 358 (1995) und (mit P. C. van der Willigen) 44, 22 (1928).
2) H. G. Bungenberg de Jong, Rec. Trav. Chim. 42, 1 (1923).

KRUYT u. BUNOENBERG DE JONG, ZUR KENNTNIS D. LYOPHILI~N KQLLOIDE 5
I. R e i n i g u n g d e s Agars .
Es lagen verschiedene Untersuchungen tiber die chemische Zu- sammensetzung bzw. den Molekularbau des aus japanischen Rotalgen gewonnenen Agars rot . l) ~) Viele Autoren batten auch schon Versuche
zu einer Reindarstellung angestellt. C o o p e r , C a n t a b und Nut ta l lS) , welche zum ersten Male eine
eingehendere und mehr systematische Obersicht tiber die Eigenschaften des Agars gegeben haben, beobachteten, dab mit destilliertem Wasser 18 Proz. fester Stoffe ausgewaschen wurden. Ir~ einem Patent yon M e r c k a) wird ebenfalls yon einem in kal tem Wasser und einem erst in kochendem Wasser l~slichen Tell, welch letzterer der Tr~ger der emul- soiden Eigenschaften sei, gesprochen.
Ftir kolloidchemische Zwecke ist es yon Wichtigkeit, dab die d i f f u n d i e r e n d e n Elektrolyten so gut wie msglich entfernt werden. F~llung einer AgarlSsung mit Alkohol fiihrt iedoeh nicht zu einer Ab- nahme des Asehegehaltes6), was tibrigens aus den nachstehenden Kapiteln hervorgehen wird. K e d i n g e) extrahierte hintereinander mit verdtinnter Salzs~ure, destilliertem Wasser, lprozentigem K 0 H und da- nach wieder mit destilliertem Wasser. Der Aschegehalt sank yon 3,92 Proz. bis auf 1,84 Proz. Der getrocknete Agar ist aber braun und sprSde und daher wahrscheinlieh doeh stark angegriffen. L e v i t e s ~) behandelt Agar mit schwacher Essigs~ture, wlischt bis zur neutralen Reaktion des Wasehwassers aus und behandelt danaeh mit 1 Proz. Arnmoniak. Darauf wird der Agar in koehendem Wasser aufgelSst und in Alkohol filtriert. Der Aschegehalt wurde nieht angegeben.
Aus unseren eigenen Untersuehungen geht hervor, dab man den Asehegehalt dureh Behandeln mit verdtinntem tlC1 zwar sehr schnell bis auf 0,4 Proz. herabsetzen kann, dab dabei aber das HC1 sehr lest gebunden bleibt und praktisch kaum auszuwaschen ist. Unserer Mei-
t) C. R. Po rumbaru , 90, 1081 (1880); C. R. Morin, 90, 924 (1880); Bauer , Journ. f. prakt. Chem. (2) 30, a75 (1884); K6nig u. Be t te l s , Zeitschr. f. Unters. d. Nahr.- u. Gen. 10, 457 (1905); Samec u. Isa javic , Kolloidchem. Beih. 16, 285 (1922).
a) Tschi rch , Handbuch der Pharmakognosie, Bd. II, 306 (Leipzig 1919.). 3) Cooper, Cantab u. Nuttal l , The Photograph. Journ. 48, 11 (1908). 4) Zeitschr. f. angew. Chem. 27, II, 117 (1914). 5) Cooper , Cantab u. Nut ta l l , loc. cit. Sic geben fiir den urspriing-
lichen Aschegehalt 0,487 Proz. an. Dieses mug ein Irrttim sein und wird wahr- seheinlich 4,87 heiI~en miissen, da nach Priizipitieren mit Alkohol 4,06 Proz. Asche gefunden wurde.
e) Keding, Wiss. Meeresuntersuch., Abt. Kiel, N. F. 9 (1906), zit. naeh Kiister, Kultur der Mikro-Organismen (Leipzig u. Berlin 1913).
~) Levi tes , Koll.-Zeitschr. 2, 161 (1907).

6 KOLLOIDCIqEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
nung nach ist der Gebrauch von S/turen und Basen bei dem Reinigungs- prozeg ohne Vorteil. 1)
Es erscheint uns aut3erdem nicht wtinschenswert, die Agarl6sung lange Zeit h6heren Temperaturen 2) auszusetzen [wie das Mac D o u ga 13)
bei seiner zehn Tage andauernden Dialyse tat]. Nach diesem Unter- sueher enth~tlt gereinigter Agar nur Spuren von Asche, die Tatsache
aber, daft die Agarl6sung eine hellbraune Farbe zeigt und ein 8/4pro- zentiges Gel sehr sehwaeh ist, scheint doch auf eine VerAnderung der ursprtinglichen Eigensehaften infolge der Behandlung zu schlieften.
In einem Patent von M e r c k 4) ist yon ciner Reinigung des Agars
durch wiederholtes Ausfrieren nach AuflSsen in heitlem Wasser und Gelatinieren die Rede. Solange aber die Natur derartiger Zustands- ~inderungen nicht aufgeklltrt ist, scheint diese Reinigungsmethode nicht die geeignete zu sein.
Wir w~hlten schliet31ich eine Reinigungsmethode, welehe der von H a r d y 5) sehr iihnlich ist, mit dem Untersehied, datl wir die ur-
sprtinglichen Agarstreifen 15.ngere Zeit mit frischem destillierten Wasser auswusehen, e) Dabei wurdea auf 35 g Agarstreifen immer 2 Liter
destilliertes Wasser genommen. Dieses wurde w~hrend der ersten zwei Tage f0nfmal am Tage erneuert und immer 10 ecru Ather hinzugeftigt. W~ihrend der letzten zwei Tage war der Asehegehalt konstant geblieben
1) Die ausgewaschenen Agarstreifen miissen, bevor sie in den Thermostat gebracht werden, einige Minuten lang N Ha-Dampf ausgesetzt werden, da sonst das adsorbierte H C1 den Agar bei der h/3heren Temperatur sehr stark angreift (die Stiickchen werden griinschwarz und sehr spr6de). Nun findet man z. B. einen Aschegehalt yon 0,4 Proz.; dieser geringe Wert ist jedoch kein sicheres Zeichen fiir einen niedrigen Elektrolytgehalt, da das g e b u n d e n e HCI jetzt als NH4CI verdunstet.
Bei der Abnahme des Asehegehaltes spielt wahrscheinlich eine Umtauseh. adsorption mit, wobei fliichtige Elektrolytbestandteile an Stelle der nicht. fliichtigen treten. Diese Meinung wird noch dutch die Beobachtung verst~rkt, dal3 man alles an den Agarstreifen adsorbierte HC1 zwar durch Behandeln mit 10prozentiger NaCI-L6sung austreiben kann, dab jedoch der Aschegehalt nach langem Auswasehen nieht tieier sinkt als bis 9~ Proz. Das fliichtige HCI ist bier wieder durch nichtfliichtiges NaC1 ersetzt worden. Die eingeschaltete Behandlung mit S~iuren erzielt also kein besseres Ergebnis als die im folgen- den beschriebene Behandlung ausschliel31ich mit destilliertem Wasser.
2) Levi tes , Koll.-Zeitschr. g, 161, 237 (1907/08): Andauerndes Kochen einer Agarl6sung fiihrt zu einer tlydrolyse (Reduktion des Fehling-Reagens) mid zu einer Abnahme der Gelatinierungsfiihigkeit.
8) Mac Douga l , The Botan. Gazette 70, 126 (1920). 4) Zeitschr. f. angew. Chem. 27, 11, 117 (1914). n) H a r d y , Zeitschr. f. phys. Chem. 38, 326 (1900). e) Spiiter entwarf der eine yon uns einen fortlaufend betriebenen Waseh-
apparat, mit welchem eine sehnelle Reinigung m6glieh wurde: H. G. Bungen- be rg de Jong , Rec. Tray. Chim. 42, 1074 (1983).

IKRUYT u. BUNGENBERG DE JONG, ZUR KENNTNIS D. LYOPHILEN KOLLOIDE 7
und also praktisch alle Irei diffundierenden Elektrolyten entfernt worden. Der ausgewaschene Agar wurde auf Glasplatten bei Zimmer- temperatur an der Luft getrocknet, wobei dafiir gesorgt war, dab er nicht mit Staub in Bertihrung kommen konnte. Um wS.hrend der langen Trockenperiode etwaige Verschimmelung zu verhfiten, wurden dem letzten Waschwasser 300 ccm mit Hg J2 bei Zimmertemperatur ges/~ttig- tes Wasser 1) beigegeben.
In dem Kapitel: ,,Der Emflug yon Elektrolyten auf die Viskosi- tgtt des Agarsols" ist der eventueIle Einflug dieser geringen Menge Hg J2 experimentell .untersucht worden (vgl. S. 2,6).
Der getrocknete Agar war glitzernd hell, enthielt 24,1 Proz. Wasser (bestimmt naeh 21/2 Stunden Erhitzen auf 1350 ) und einen Gehalt an Asche yon 1,86 Proz., d.h. 2,05 Proz. im Verhgltnis zu wasserfreiem Agar.
lI. Methodik und V o r u n t e r s u c h u n g .
Wenn man die in der Literatur verSffentliehten Viskosit~ts- messungen an lyophilen Solen in Betracht zieht, so wird es bald deut- lich, dab ftir eine groBe Anzahl dieser Untersuchungen eine Viskosi- tgtsmethode angewendet wurde, welche sehon ffir homogene Fltissig- keiten systematische Fehler aufweist. Unsere erste Sorge war nun, cIiese systematisehen Fehler zu vermeiden. Die yon uns verwendete Methode ist sehon frtiher an anderer Stelle erOrtert worden. 2)
Abgesehen yon diesen Fehlerquellen, welche appara t iver Art sind, begegnet man oft Komplikationen, welehe jedes Konkludieren aus den fibrigens so einfaehen ViskositS.tsmessungen sehr erschweren und deren Ursprung in dem lyophilen System selbst zu suchen ist. Vqir denken hierbei an die Rolle, welehe bekanntlich die thermisehe u die Hysteresis-Erscheinungen, das Sehfitteln, dic Ab- hgngigkeit der relativen DurchstrSmungszeit yon den StrSmungskon- ditionen usw. bei derartigen Versucben spielen, welehe ebensogut bei Agarsolen stSrend auftreten k~nnen, wenn man nicht daffir sorgt, dab alas System w'~ihrend der ganzen Versuchszeit fiber 40 ~ C b l e i b t .
Unter dieser Grenztemperatur a) zeigen sieh nl. ZustandsS.nderun- gen in dem 1/Tproz. Sol (die stets verwendete Konzentration), welche
x) Naeh Morse [Zeitschr. f physik. Chem. 43., 708 (1902)] betr/igt die LSslichkeit des HgJ2 in Wasser bei 250 C 0,0591 g pro Liter.
~) Ausfiihrlich beschrieben in H. G. Bungenbe rg de Jong, Rec. Tray. Chim, 42, ! (1923).
s) Die sich auf die Gelatinierung der Ag~rsole beziehenden Untersuchungen sind vor kurzer Zeit an anderer Stelle verSt~entlieht: H. G. Bungenbe rg .de Jong Rec. Tray. Chim. 47, 797 (1928).

8 KOLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
sich in konzentrierten Systemen als Gelatinierung /iuBern und ihrer Natur nach, w i e sich bei den ultramikroskopischen Untersuchungen B a c h m a n n s 1) zeigte, Aggregationserscheinungen sind.
Es ist sehr wahrscheinlich, dab die meisten der genannten JKom- plikationen auch bei anderen lyophilen Systemen (z. B. bei Gelatine- solen) durch derartige Gelatinierungs-Aggregrationen hervorgerufen werden, und dab sie verschwinden, wenn man die Messungen fiber der Gelatinierungstemperatur anstellt. 2)
Diese Voruntersuchungen sind schon an anderer Stelle ausffihrlich besprochen worden, ~) und wir wollen daher hier nur die Resultate er- wS~hnen.
a) Wenn man eine 2/vprozentige Agarl6sung bei 500 C mit einem ebenso groBen Volum an Wasser oder einer neutralen Kochsalz- 16sung verdtinnt, so wird sofort naeh der Vermisehung ein Endwert der Viskosit~it erreicht .
b) Sehtitteln hat keinerlei EinfluB auf die Viskosit/it. c) Ein 1/Tprozentiges Agarsol gehorcht bei 500 C dem Gesetz von
P o i s e u i l l e . 4)
d) Bei Temperaturwechsel (nicht unter 40 ~ wird bei jeder einzelnen Temperatur momentan ein gewisser Viskosit~itswert gefunden. Hysteresiserscheinungen fehlen v611ig.
Verdtinnte Agarsole verhalten sich also bei Temperaturen tiber 400 wie einfache Systeme. Wohl beobachtet man eine mit der Zeit gleichm/~Bige Abnahme der Viskosit~t, diese tr~gt jedoeh einen v611ig irreversiblen Charakter und mut3 einem fortschreitenden Abbau der Solteilchen zugeschrieben werden. 5) Diese Verminderung mit der Zeit verl/~uft bei 100 ~ so schnell, dab Agarsole derselben Konzentration und (durch Kochen) so viel wie m6glieh auf gleiche Weise hergestellt nie v611ig die gleiche Viskosit/it zeigen.
1) Bachmann , Zeitschr. f. anorg. Chem. 78, 125 (1912). -2) Davis u. Oakes [Journ. Amer. Chem. Soc. 44, 464 (1922)] tanden, dag
fiir Gelatine die Gelatinierung fiber 38,10 nicht mehr stattfindet. 8) H. G. Bungenbe rg de Jong, Rec. Tray. Chim. 4~, 1 (1923), ins-
besondere S. 17--23. Vgl. H. R. Kruyt , Koll.-Zeitschr. (Zsigmondy-Band) 36, 218 (19241.
4) Roth l in [Biochem. Zeitschr. 98, 34 (1919)] land starke Abweichungen des Gesetzes yon Poiseuil le. Da dieser Untersueher jedoch mit 0,2 Proz. Agarsolen bei 270 arbeitete, hat er nicht die Eigenschaften der Agarsole im strengsten Sinne, sondern die yon fliissigen, halb gelatinierten Systemen gemessen.
6) Levi tes , Koll.-Zeitschr. $, 161, 208, 237 (1907/08). v. Schr6der Iand bei Gelatinel6sungen eine ~ihnliche irreversible Reaktion, Zeitschr. f. phys. Chem. 45, 75 (1903). Er spricht yon einem Verseifungsprozelt.

KRUYT u. BUNGENBERG DE JONG, ZUR KENNTNIS D.LYOPHILEN KOLLOIDE 9
Hieraus geht hervor, (tag nur (lie an einem und demselben Sol an-
gestellten Messungen streng vergleichbar sin& Es ist jedoch m6glich, die an versehiedenen Solon gemessenen Serien mit Hilfe der Blanko- werte aufeinander umzureehnen.
Bei 500 C betr~igt die genannte Abnahrne mit der Zeit beJ 1/Tpro-
zentigen Solen ungef~ihr sechs Einhciten der dritten Dezimale in zw61f Stunden. W~thrend diese Abnahme bci jeder individuellen ViskositS.ts- messung inncrhalb des Versuchsfehlers fiillt, muB jede Serie hieraufhin korrigiert werden.
Die I)iehte yon Agarsolen ist linear yon der Agarkonzentration abh~.ngig. Ftir ein 2/Tprozentiges Sol fanden wir bei 500 C = 1,00103 und ffir ein 1/Tprozentiges Sol = 1,00048 (Wasser bei 500 = 1).
Bei der Berechnung der relativen ViskositS.t yon 1/Tprozentigen Agarsolen kann man diese geringe Dichtezunahme also vernachlS.ssigen, und die Forrnel wird dann numerisch dem Quotient der Durehstr6mungs-
zeit des Sols und der des Wassers gleich. Zum Schlut3 sei hier noch die I-lerstellungsweise der Agarsole er6rtert.
Die abgewogene Menge Agar wurde in einem Jena-Kolben yon bekanntem Gewicht mit der n~Stigen Menge destiiliertem Wasser eine Stunde lang in einem Thermostat bei 50 o C erw~rmt. Der Kolben wurde dann erhitzt und sechs Minuten lang gekocht. Darauf kfihlten wir den Agar wieder
auf 50 o ab und wogen das verdunstete Wasser zu. Auf diese Weise erh~lt man eine yon Fasern und Flocken getr/ibte Fliissigkeit. Wird nun direkt
Iiltriert, so zeigt sich doch im Filtrat innerhalb zehn Stunden wieder eine zusammenh~ingende floekige F~illung. Man rut also gut, den Kolben erst wS~hrend zehn bis zw61f Stunden in einemThermostat yon 500 zu behaltefi und erst nachher zu filtrieren. Fiir die Filtration verwendeten wir Filtrier-
papier Nr. 250A yon S c h l e i e h e r und Schfi l l , welches erst auf dem Trichter abwechselnd mit destilliertem Wasser ausgewaschen und aus- ged~mpft werden muB. Bei dem Filtrieren soll darauf geachtet werden,
dab die Temperatur der durcbstr6menden Flfissigkeit hie unter z~0~ sinkt.
IIl. Die Viskosit l i t von Agarso len als Funkt ion der Temperatur und der Agarkonzentrat ion.
E i n s t e i n 1) hat eine theoretischc Formel ftir die ViskositS.t eines fltissigen Systems, in welchem inkompressible Teilchen dispergiert sind, entwickelt : '~, = r~0 (1 + 5/., ~).
1) Einstein, Ann. d. Phys. 19, 289 11906); 34, 591 (1911); diese Formel wurde yon Bancelin gepriift, welcher als Konstante jedoch 2,9 land. Koll.- Zeitschr. 9, 154 (1911); Compt. rend. 152, 1382 (1911).

10 KOLLOIDCHEMISCHE BE1HEFTE BAND XXVIII, HEFT 1--2
Hierbei stellt ~]s die Viskosit~t des ganzen Systems und ~]o die der F]fissigkeit dar, w~hrend ~ das Volumen der Teilehen a]s Frakt ion des Totalvolumens ist. V o n S m o l u e h o w s k i 1) hat diese Formel ftir Teilchen einer willktirlichen Gestalt erweitert zu:
~/s = ~o (1 + K " ~0),
wobei K eine yon der Gestalt der Teilehen abh~ingende Konstante ist und ein Minimum ( = 5/2 ) ftir die Kugelform bildet. 2)
Diese Formel gilt nur, wenn der Radius der Teilchen im Verh~ilt- his zu ihrer gegenseitigen Entfernung klein ist, wenn also 99 nur einige Volumprozente betr/tgt.
Bei gr6t3eren Konzentra t ionen mug die Entwicklungsreihe naeh q0 weiter geftihrt werden:
rls == ~0 (1 + K(p + K I ~ ~/ . . . . . ).
Diese Formel deutet also an, dab die Viskosit/~tskonzentrati0ns- k u r v e bei kleinen Konzentra t ionen linear verl~uft und dab die Vis- kosit/tt bei gr6Beren Konzentrat ionen stets schneller wie linear steigen wird.
Die oben angeftihrten Formeln sind an erster Stelle auf Sus- pensoide anzuwenden. Bei den Emulsoiden ist es nicht sicher mehr, ob man den PrS~missen, auf die sie aufgebaut wurden, auch v611ig gerecht
wurde (z. B. in Hinsicht auf die Inkompressibilit~it der Teilehen). Ofters zeigen die Emulsoide schon bei sehr kleinen Konzentrat ionen eine viel h6here ViskositS& als man mit Hilfe der E i n s t e i n s c h e n Formel
berechnen kann. So fanden wir experimentell in der nachstehenden Versuchsreihe ftir ein 0,279prozentiges Agarsol bei 50 o C eine relative
Viskositfit von 2,392, w~hrend sieh, davon ausgehendl daft die Dichte
yon Agar 1 sei (ein etwas zu niedriger Wert), 1,007 berechnet. Die Vis-
kosit~itserh6hung ist also 1392 7 - - • 200mal zu grog.
Eine ~ihnliche Disproportion zwischen experimentell gefundener und bereehneter Viskosit~t finden wir bei allen fibrigen Konzentrat ionen,
so z . B . ftir eine experimentell gefundene Visk0s!t~t yon 1,048 bei der Konzentra t ion 0,03111,0,279 - - 0,00868 Proz., for welehe man theoretiseh eine relative ViskositS~t yon 1,000217 erwarten k6nnte.
Die Viskosit~ttserhOhung ist hier also 4800 217 - - i 220real zu grog.
Wenn man diese Resultate zu erkli~ren sucht, so erweisen sich
1) v. S m o l u e h o w s k i , K011..Zeitsehr. 18, 190 (1916). 2) In Flfissigkeiten mit grogem Widerstand ist k ebenfaUs yon der Ladung
der Teilehen abh~ingig. Siehe n~ichstes KapiteL

t(IRUYT u. B U N G E N B E R G DE J O N G , Z U R KENNTNIS D . L Y O P H I L E N K O L L O I D E 11
zweierlei M6glichkeiten, welche jede f~r sich zu der abnormen Steige- rung beitragen kSntlten.
1. Die Hydrata t ion der Teilchen. 2. Der schwammartige Bau der Teilchen. 1)
Im ersteren Fall handelt es sich um Wasser, welches in seiner freien Beweglichkeit behindert ist und dessen Volum zu dem des trok- kenen Agars gez~hlt werden mug.
Im letzteren Fall kann das Wasser zwar frei beweglich sein, es ist jedoch in kapillaren R~iumen eingeschlossen und wirkt daher in ge- wissem Sinne stagnierend. Das Volum dieses Wassers muff daher eben- falls bei 9 in Rechnung gebracht werden.
Es ist nicht immer einfach zu entscheiden, ob in einem gewissen Fall beide Faktoren zu der abnormen ViskositiitserhShung beitragen. Beim Agar spielt jedenfalls der erste Faktor eine grot3e Rolle, was schon daraus hervorgeht, daft die abnorme Viskosit~itssteigerung bei Entw~isse- rung mit Alkohol (siehe Kapitel V) sehr stark abnimmt. Die Abh~ingig- keit der relativen Viskosit~it yon der Temperatur spricht ebenfalls hierfiir.
Wir verwendeten ftir diese Versuchsreihe drei Thermostate yon 50 ~ 450 und 41 ~ welche um das Fixierstativ des Viskosimeters aufge- stellt waren. So konnte das Uberbringen eines Viskosimeters yon einem Thermostaten in den anderen innerhalb zehn Sekunden vor sich gehen.~) Schon drei Minuten nach dem Wechseln der Thermostaten erhiilt man so bei der neuen Temperatur eine konstante Durchstr6mungszeit.
Mit dreierlei Agarsolen (2/7 , 1/7 und 1/t 4 Proz.) wurde nun die relative Viskosit~t an einer Ftillung des Viskosimeters hinterein- ander bei 500 , 45 ~ 40 ~ 45 ~ und 50 ~ gemessen. In Tabelle I sind die Resultate dieser Messungen verzeichnet.
T a b e l l e I.
Temperatur 2/Tproz. Sol 1/TPrOz. Sol 1/14proz. Sol
50 45 40 45 50
2,364 2,393 2,419 2,393 2,362
1,652 1,662 1,674 1,663 1,652
1,322 1,325 1,330 1,326 1,321
Die Viskosit~Lten der Tabelle sind relative Werte, welche im Ver- h~iltnis zu Wasser der jeweiligen Temperatur (ira Sinne d e r - E i n s t e i n - se,hen Formel) berechnet wurden.
1) v. Smoluchowski , loc. tit. 2) Rec. Tray. Chim. 42, 1 (19t3).

1 ~ KOLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
Wir sehen also, dab die relative ViskositgtserhGhung ~ - -~o durch"
die gleiche Agarkonzentration bei jeder bestimmten Temperatur einen gewlssen Wert hat und dab sie bei einer Temperatursteigerung abnimmt. Zieht man nun in Betracht, dab die Quellungswgrme posi t iv is t , d.h.. dab die Hydrata t ion bei TemperaturerhGhung abnimmt, dann zeigt diese Temperaturabhgngigkeit yon der VJskosit~tserhShung also tat-- s~chlich, dab diese abnorme Viskosit~tserhShung r~ach unserer Ansicht yon einer Hydrata t ion der Agarteilchen verursacht wird.
Wir wo]len uns nun welter mit der Frage besch~ftigen, inwieferrr die Viskosit~t eines Emulsoides yon der Konzentration der dJspersen Phase abhiingt.
Eine streng mathematische L6sung dieses Problems besteht noch, nicht*), wir vermuten jedoch, dab bier in groBen Ztigen eine im Weserl gleiehe funktionelle Beziehung zwischen Viskosit~t und Konzentration wie ftir die Suspensoide besteht. Eine Anzahl Untersuchungen scheinen denn aueh darauf zu deuten, dab die ViskositgtserhShung in kleinen
Konzentra t ionen linear und bei gr6i~eren stiirker als linear zunimmt. Ein Punkt wirkt hier jedoeh befremdend. Die lineare Formel darf
nm strenggenommen bis zu qJ --= 2 Proz., mit anderen Worten bis zu einer relativen Viskosit/~t yon ungefghr 1,05 bestehen, wghrend der lineare Verlauf bei Emulsoiden sich auf den ersten Blick zu einer viel' grSfleren Viskosit~it auszustrecken scheint.
Dieser scheinbare Widerspruch veranlaBte uns zu einer m6glichst genauen Messung dieser Konzentrationsfunktion. Die Messungen x~urden bei 500 angestellt. Bei der ersten Serie gingen wir v0n einem Agarsol aus, welches pro 310 g Sol 1,14 g lufttrockenen Agar enthielt. Wir maehten die verdtinnungen mit Hilfe einer in einen Glasmantel~ gestellten Staspipette yon t5 ccm. I)urch diesen Glasmantel wurde Thermostatwasser yon 500 gepumpt. Ftir die erste Verdtinnung wurde die Mischung: ei n e Pipette Agarsol und ei n e Pipette destilliertes Wasser "yon 50 ~ in einem ausgedgmpften trockenen Jena-Kolben gebracht. Bei folgenden Verdtinnungen wurde immer ei n e Pipette der vorigen Verdiin- nung auf gleiche Weise mit e in e r Pipette destilliertem Wasser vermiseht.
Die Pipette wurde ftir destilliertes Wasser, ftir Agarsol und ftir die erste Verdtinnung geeicht. Hieraus ergab s]ch ein lineares Ver- hiiltnis ftir das Gewicht des ausgestrSmten Agarsols, welches gleich-
J) H a t s c h e k [Koll.-Zeitschr. 8, 84 (1911); 11, 284 (1912); lg, 238 (1913)] machte einen Versuch in dieser Richtung, man beachte jedoch die Kritik v. Smoluchowski [Koll.-Zeitschr. 18, 190 (1916)] und die v. Rossem [Chem. Weekblad 11, 579 (1914)].

/ , (RUYT u. B U N O E N B E R G DE J O N G , Z U R KENNTNIS D. L Y O P H I L E N I~OLLOIDE 13
falls bei den fibrigen Verdfinndngen angewendet wurde. Ftir die Dichte des Sols wurde ebenfalls ein lineares Verhiiltnis ds -- dw (1 + 0,00103 C) aufgestellt, wobei die Konzentration des ursprtinglichen Sols gleich 1,000 ge- nommen wurde; die Konzentrationen sind auf die Volumeneinheit bezogen.
Wird also a Gramm Sol der Konzentration C rnit b Gramm Wasser Verdfinnt, so ist die Konzentration der Verdfinnung:
a dl �9 C, Cl~---a+ b "
wobei d die Dichte des zu verdfinnenden Sols und d 1 die Dichte der Verdiinnungen darstellea. Auf diese Weise sind die Konzentrationen in Tabelle I I berechnet.
Die relative Durchstr6mungszeit des ursprfinglichen Sols und jeder .einzelnen Verdfinnung wurde mit zwei verschiedenen Viskosimetern bestimmt, die relative Dichte wurde ebenfalls mit eingerechnet, so dab das Mittel der beiden Viskosit~itszahlen deshalb als bis auf 0,1 Proz genau betrachtet werden daft.
Am Schlufl dieser Versuchsreihe wurde die Viskosit~it des ursprting- lichen Sols von neuem bestimmt. Diese hatte in diesem Fall nicht abgenommen. Die Zahlen brauchten also nicht ftir die irreversible Reaktion korrigiert zu werden. Die Ergebnisse sind in nachstehender Tabelle wiedergegeben.
T a b e l l e II.
Konzentration Relative Viskositi it
1,0000 0,4988 0,2491 0,1245 0,06222 0,03111
2,392 2,392 1,653 1,655 1,338 1,337 1,180 1,179 1,095 1,095 1,049 1,047
Bei Beendigung der Versuchsreihe war noch eine gentigende Menge des Sols fibrig, um eine zweite Versuchsserie hiermit vornehmen zu kSnnen. Diese wtirde 71 Stunden nach der ersten angestellt. Bei den ersten zwei Verdfinnungen wurde ftir das destillierte Wasser keine Pipette verwendet; die nStigen Mengen Wasser wurden in Jena-K61bchen abgewogen. Die iibrigenVer dfinnungen wurden auf gleiche Weise wie bei der ersten Versuchs- reihe hergestellt. W~ihrend der Dauer dieser Serie (8 Stunden) betrug die irreversible Reaktion des urspriinglichen Sols vier Einheiten der dritten Dezimale, wodurch die Messungen einer kleinen Korrektion bedurften.
Das Ergebnis der zweiten Versuchsreihe ist in Tabelle I I I ver- zeichnet.
14 I~OLLOIDCHEMISCHE BEIHEFTE BAND X X V I I I , HEFT 1--2
Tabel le III.
Konzentration Relative Viskosit/it
1,OOI)O 0,7582 0,4952 0,2473 0,1236 0,06177 0,03088
2,351 2,350 1,982 1,985 1,629 1,627 1,324 1,324 1,171 1,169 1,090 1,092 I 1,047 i 1,048
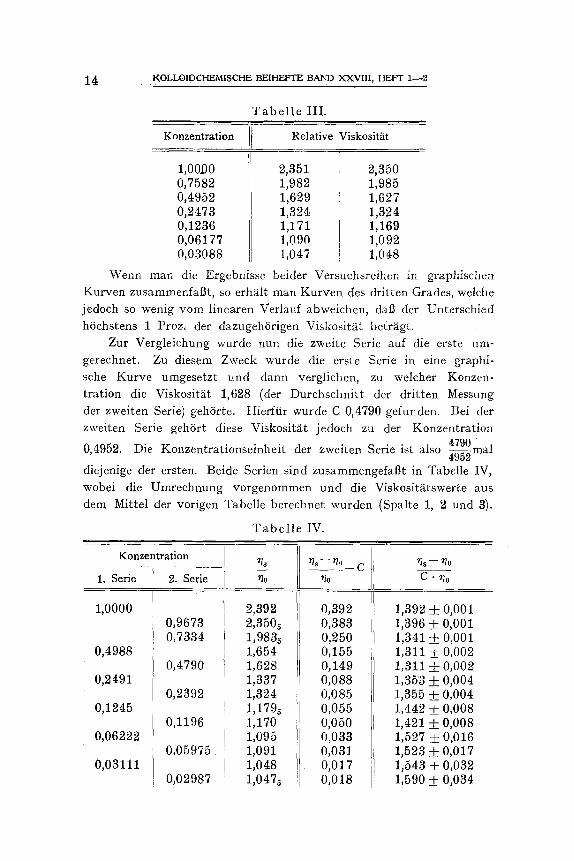
Wenn man die Ergebnisse beider Versuchsreihen in graphischerL Kurven zusammenfal3t, so erh~ilt man Kurven des dritten Grades, welche jedoch so wenig yore linearen Verlauf abweichen, dab der Unterschied hSchstens 1 Proz, der dazugeh6rigen Viskosit~t betrS.gt.
Zur Vergleichung wurde nun die zweite Serie auf die erste um- gerechnet. Zu diesem Zweck wurde die erste Serie in eine graphi- sche Kurve umgesetzt und dann verglichen, zu welcher Konzen- tration die Viskosit~t 1,628 (der Durchschnitt der dritten Messung der zweiten Serie) gehOrte. Hierfiir wurde C 0,4790 gefunden. Bei der zweiten Serie geh6rt diese Viskosit~t jedoch zu der Konzentration
4790 . 0,4952. Die Konzentrationseinheit der zweiten Serie ist also ~ m a l
diejenige der ersten. Beide Serien sind zusammengefaflt in Tabelle IV, wobei die Umrechnung vorgenommen und die Viskosit~tswerte aus dem Mittel der vorigen Tabelle berechnet wurden (Spalte 1, 2 und 3).
Tabel le IV.
Konzentration
1. Serie
1,0000
0,4988
0,2491
0,1245
0,06222
0,03111
2. Serie
0,9673 0,7334
0,4790
0,2392
0,1196
0,05975
0,02987
2,392 2,3505 1,9835 1,654 1,628 1,337 1,324 1,1795 1,170 1,095 1,091 1,048 1,0475
0,392 0,383 0,250 0,155 0,149 0,088 0,085 0,055 O,05O 0,033 0,031 0,017 0,018
C �9 *1o
1,392 _--4- 0,001 1,396 -t- 0,001 1,341 --4- 0,001 1,311 _+ 0,002 1,311 _ 0,002 1,353 i- 0,004 1,355 _4- 0,004 1,442 _+ 0,008 1,421 -4- 0,008 1,527 • 0,016 1,523 -t- 0,017 1,543 _+ 0,032 1,590 _+ 0,034
K R U Y T u. B U N G E N B E R O DE J O N O , Z U R KENNTN1S D. LYOPI-IILF.,N E O L L O I D E 1 5
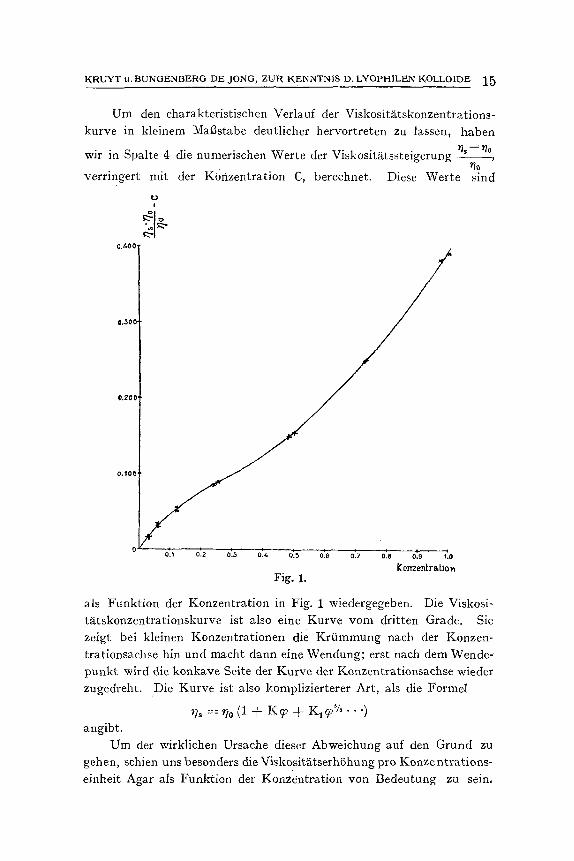
Um den charakteristischen Verlauf der Viskosit~itskonzentrations- kurve in kleinem Mat3stabe deutlicher bervortreten zu lassen, haben
wir in Spalte 4 die numerischen Werte der Viskosit~itssteigerung ~- -~~ *lo
verringert mit der Konzentration C, berechnet. Diese Werte sind
t~ J
0.t-~0 0.
0.30ff
O.20D
o.100
o o:1 o:~ o~,~ D:/~ " " ~ : ~ o.o o:,7 o:.8 o'.~ I'.0 Ko~zenLraLio~
Fig. 1.
als Funktion der Konzentration in Fig. 1 wJedergegeben. Die Viskosi- t~Ltskonzentrationskurve ist also eine Kurve yore dritten Grade. Sie zeigt bei kleinen Konzentrationen die Kriimmung nach der Konzen- trationsachse hin und macht dann eine Wendung; erst naeh dem Wende- punkt wird die konkave Seite der Kurve der Konzentrationsachse wieder zugedreht. Die Kurve ist also komplizierterer Art, als die Formel
~/~ =~/0 (1 + K ~ + K1~ ~/ . . . . ) angibt.
Um der wirkliehen Ursaehe dieser Abweichung auf den Grund zu gehen, sehien uns besonders die Viskosit~.tserhShung pro Konzentrutions- einheit Agar als Funktion der Konzeatration yon Bedeutung zu sein,
16 KOLLOIDCHEMISCHE BEIHEFTE BAND XXVlII, HEFT 1--2
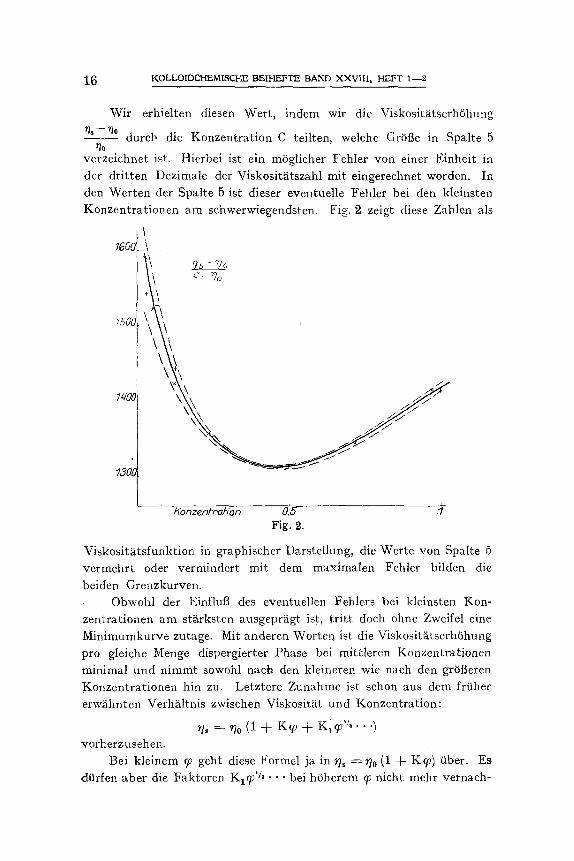
Wir erhielten diesen Weft, indem wir die Viskosit~itserh6hung
~ s - - ~ dutch die Konzentration C teilten, welche GrSfie in Spalte 5 ~o
verzeichnet ist. Hierbei ist ein m6glicher Fehler yon einer Einheit in der dritten Dezimale der Viskosit~tszahl mit eingerechnet worden. In den Werten der Spalte 5 ist dieser eventuelle Fehler bei den kleinsten Konzentrationen am sehwerwiegendsten. FiE. 9, zeigt diese Zahlen als
~o \
~\ g5 - Vo
k \ c . "%
(% \ \
\ \ \ 73~
Konzenbna/'/on 0,,.~ l - Fig. 2.
Viskosit~tsfunktion in graphischer Darstellung, die Werte von Spalte 5 vermehr t oder vermindert mi t dem maximalen Fehler bilden die
beiden Grenzkurven. Obwohl der Einflutt des eventuellen Fehlers bei kleinsten Kon-
zentrationen am stXrksten ausgepr~gt ist, tri t t doch ohne Zweifel eine Minimumkurve zutage. Mit anderen Worten ist die Viskosit~tserh6hung pro gleiche Menge dispergierter Phase bei mittleren Konzentrationen minimal und nimmt sowohl nach den kleineren wie nach den gr6t3eren Konzentrationen hin zu. Letztere Zunahme ist schon aus dem frfiher erw~ihnten Verh~iltnis zwisehen Viskosit~it und Konzentration:
~ = ~o (1 + K~o + Kt,T v . . . . ) vorherzusehen.
Bei kleinem 9~ geht diese Formel ja in ~s ---~ ~o (i -}- KgJ ) fiber. Es
dfirfen aber die Faktoren K19 ~5/3 �9 �9 �9 bei h6herem 9~ nicht mehr vernaeh-

KRUYT u. BUNGENBERG DE JONG, ZUR KENNTNIS D. LYOPHILEN K.OLLOIDE ] .~
li~ssigt werden, sie sind bei grSgeren Konzentrationen der Anlat3 einer stets zunehmenden Steigerung der Viskosit~ttserh6hung pro gleiche Agarmenge (welche bei kleineren Konzentrationen konstant sein mtiflte).
Die Zunahme nach den kleineren Konzentrationen ist so jedoch noch nicht erklgrt. Es wird wohl am wahrscheinlichsten sein, dab ~0 selber bei Konzentrationsabnahme zunimmt. Wenn wir annehmen, dab die abnorm hohe Viskositgt der Agarsole einer sehr kr~ftigen Hy- dratation zuzuschreiben ist, dann w~rde man aus dem Vorhergehenden schliegen kSnnen, dab die H y d r a t a t i o n bei Verdt in1~ung pro T e i l c h e n z u n i m m t .
Wahrscheinlich ist die Hydratationshtille nicht homogen und wird die Wasserbindung nach augen stets lockerer. Infolgedessen ist der Einfluf3 jedes einzelnen Teilchens in dem ihn umgebenden Wasser auf gr6i3ere Entfernung ftihlbar und tritt daher die gegenseitige StSrung in dem Hydratationszustand bei KonzentrationserhShung schon bei kleinen Konzentrationen ein.
IV. Einflul3 dcr Elektrolyte auf die Viskositl it des Agarsols .
L e v i t e s 1) untersuchte den EinfluB einzelner Elektrolyte in 1/2- und 1-n-Konzentrationen auf die Viskosit~t yon 0,2prozentigem Agar- sol. Dabei fand er als allgemein gtiltige Regel, dab die Viskosit~t durch diejenigen Elektrolyte, welche auch die Viskosit~t von Wasser steigern, erhSht wird. Weitere Regelm~Bigkeiten gehen aus diesen Versuchen nicht hervor. Dieser Autor hat also die Viskosit~t nicht relativ im Verh~,ltnis zu dem dazugehSrigei~ Dispersionsmittel (d. h. im Vergleich mit der t /_ und 1-n-Elektrolytl6sung) gemessen. Erst wenn die Mes- sungen in dieser Weise angestellt werden, darf die Viskositatssteigerung
~s-- ~0 bei konstanter Agarkonzentration als ein MaBstab ftir den inneren ~o
Zustand des Sols (als o z. B. fiir den Hydratationszustand der Teilchen) angesehen werden. Auf Grund dieser Oberlegung wollten wir den Ein- Ilufl yon Elektrolyten auf die Hydratation der Teilchen messen. Wir stieflen dabei jedoch bald auf Erscheinungen, welche nicht yon Hy- dratationseinfltissen herrtihren konnten. Diese Erscheinungen sind in diesem Kapitel beschrieben. Bevor wir jedoch zu den eigentlichen Versuchen tibergehen, s011 erst die verwendete Methodik erOrtert werden.
~) Levi tes , Koll.-Ze[tschr. 2, 161, 208, 237 (1907~08.

1~ KOLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
M e t h o d i k . Die Versuche sind alle bei 500 C angestellt worden. Die Agar-
sole, welche mit Elektrolytl6sungen verdfinnt wurden, waren wie auf S. 9 beschrieben hergestellt und enthielten 1,14 g lufttrockenen Agar auf 310 g Sol.
Ffir die Bereitung der Agar-Elektrolytmischungen wurde ein Jena-Mat~kolben von 50 ccm (dureh W~gen mit Wasser bei bO ~ geeicht) verwendet. Die K61bchen hatten oben einen engen Hals (0,79, cm Durchmesser), so dab wir innerhalb eines Tropfens ( + 30 mg Wasser) aus einer Ballonspritzflasche auf den Teilstrieh einstellen konnten. Das Reproduzierverm6gen des gleichen Endvolumens betrug also ungef~hr
1I~ o Proz. Zu der Herstellung der Mischungen wurde die benStigte Elektrolyt-
16sungsmenge aus einer Bfirette in den Maflkolben gebracht, welcher auf 50 o erwfirmt wurde. Nun ftillten wit mit ausgekochtem destillierten Wasser yon 50 o so lange auf, bis das KSlbehen ungefXhr 23 ccm ent- hielt. Darauf liet3en wir aus der auf S. 12 beschriebenen Pipette immer das gleiche Volum Agarsol 7ufliet3en. Eine erste grobe Einstellung wurde durch Hinzuffigen v0n ausgekochtem destillierten Wasser yon 50 o erreicht, dann der Inhalt gut vermischt, und nachdem der Kolben -+- 20 Minuten in einem Thermostat yon 50 o gestanden hatte, stellten wit mit einigen Tropfen destilliertem Wasser aus einer Spritzflasche genau ein. Nun wurden die Mischungen in ausged~mpfte Jena-Kolben yon 100 cem fibertragen und bis zu der 1/2 Stunde nach der Mischung vorgenommenen Messung bei 50 o C aufbewahrt. Wir fiberzeugten uns, dab schon gleich nach der Mischung die relative Durehlaufszeit ihren Endwert erreicht hatte.
Die Dichtemessungen der fibereinstimmenden Elektrolytwasser- mischungen wurden bei 500 mit einem Flaschenpyknometer von un- gef~hr 25 ccm vorgenommen. Die durch den Agar verursachte Dichte- zunahme des Wassers bzw. des elektrolythaltigen Dispersionsmittels konnte vernachl~ssigt werden. Wir berechneten die Dichte im Ver- h~ltnis zu ausgekochtem destiltierten Wasser yon 50 o C.
Um eine gr6f3ere irreversible Reaktion w~hrend der Messungen vermeiden zu k6nnen, wurde jede Versuchsreihe an einem und dem- selben "I age beendet. Daher konnten die Viskosit~tsbestimmungen aus Zeitmangel nur an je einer Ffillung des Viskosimeters ausgeffihrt werden. Wir fiberzeugten uns davon, dab die Versuche sowohl in bezug auf die Herstellung der Mischungen als auf die ViskositXtsmessungen leicht zu wiederholen sind.

K R U Y T u, B U N O E N B E R G DE J O N O , Z U R KENNTNIS D. L Y O P H I L E N I~OLLOIDE 19
Jede Messungsserie wurde mit der Elektrolytkonzentration 0 begonnen und beendigt. Die tibrigen Messungen mugten ftir die irre- versible Reaktion korrigiert werden. Ein Beispiel soll dies verdeut- lichen.
Nehmen wir an, das Ergebnis des ersten Nullversuches w~re */_A
---:-1,700 und acbt Stunden spS.ter das des zweiten Nullversuches
*/8 _ 1,692. Finden wir nun vier Stunden nach Beginn der Versuchsreihe
ftir eine Agarmischung den Wert 1,500, so ist dieser Wert nur mit einem Nullversuch 1,696 vergleichbar. Um diesen Wert auf den Anfang der Versuchsreihe zurtickftihren zu k6nnen, mtissen wir eine Korrektion yon
4 . 500 = 3 Einheiten der dritten Dezimale anbringen. Der korri- 696 gierte Weft ist also 1,503.
Alle Elektrolyte, welche in wS.sseriger L6sung weniger oder mehr hydrolytisch gespalten sind, sollten vermieden werden, da die irre- versible Reaktion durch t t -Ionen sehr beschleunigt wird.
Daher verwendeten wir ansta t t A1Cl a und Th C14 Luteokobalt chlorid [Co(N H3)6]C1 a 1) undTri~thylendiamin-Platinchlorid [Pt(Aein)a ](N Oa) 42), welche starke Elektrolyte sind. Auger dem La(NOa) a waren alle tibrigen Elektrolyte umkristallisierte Pr~parate yon M e r c k und K a h l b a u m . Das K4Fe (CN)6 war ein dreimal umkristallisiertes Handelspr~parat.
O r i e n t i e r u n g s v e r s u c h e t iber ein a u s g e d e h n t e s K o n z e n t r a t i o n s g e b i e t .
Der Einflufl einiger Elektrolyte (KC1, KNOa, KCNS, K2SO4, BaC12) wurde tiber ein ausgedehntes Konzentrationsgebiet untersucht. Als Beispiel des Allgemeinverbaltens soll hier die Serie mit Ba CI 2 folgen (siehe Tabelle V).
In Spalte 1 linden wir die Konzentrationen in ~-quivalenzen, in Spalte 2 und 3 die Viskosit~t der entsprechenden Elektrolyt-Wasser- und Elektrolyt-Solmischungen (relativ im Verh~ltnis zu der Viskosit~tt yon Wasser bei 500 C ausgedrtickt: ). Letztere Werte sind schon ftir die irreversible Reaktion k0rrigiert. Die Indizes e, s + e und w sollen
1) Umkristallisiertes Laboratoriumspriiparat. Das Luteokobaltchlorid hydroly- siert nicht und ist ira Wasser in vier lonen gespalten; siehe Hark ins , Hall u. Rober ts , Journ. Amer. Chem. Soc. 38, 2648 (1916).
2) Prof. Dr. F. M. Jaeger in Groningen steUte uns freundlicherweise ein Priiparat zur Verfiigung.
2"

~0 KOLLO1DCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
angeben, ob der ~-Wert sich auf die Elektrolytl6sung, auf das System Sol q- Elektrolyt oder auf reines Wasser bezieht.
Tabel le V.
Konzentration in.~quivaIenzen
0,000 0,002 0,020 0,040 0,096 0,199 0,513 0,954
He
1,000 1,000 1,003 1,006 1,016 1,034 1,080 1,152
~s -t- e
1,686 1,512 1,460 1,460 1,470 1,493 1,563 1,679
~s ~-- e
7/e
1,686 1,512 1,456 1,451 1,447 1,444 1,447 1,457
Im Hinblick aaf die E ins te in -Smoluchowskische Formel sind
jedoch nicht die Werte ~/*+_~2, sondern gerade die Werte ~s+~ ftir uns Uw ~e
yon Bedeutung. Diese Werte, vermindert um die Einheit, sind gleich f(r und
also ein Mat3 ftir die Hydratation der Teilchen. Hierzu kommt jedoch folgende Schwierigkeit.
Wir mtissen uns zuerst die Frage vorlegen, ob die Elektrolyt- konzentration im Dispersionsmittel tiberhaupt der normalen Elektro- lytkonzentration gleiehzustellen ist. Dieses wird nu r dann der Fall sein, wenn die Elektrolytkonzentration in der Hydratationshtille die gleiche ist wie die des freien Dispersionsmittels. Letzteres wurde bei
~/s+e der Berechnung yon vorausgesetzt.
Die in Spalte 4 der Tabelle V verzeiehneten Werte sind das Ergebnis der Teilung der Zahlen aus Spalte 3 durch die aus Spalte 2. Fig. 3 gibt diese Resultate graphisch wieder.
Die Ordinate der Kurve zeigt die Werte ~+~ 1 - ~ + ~ - - ~ ~e ~e
in graphischer Darstellung. Diese Werte sind also gleich f (q)). Nach unserer ersten Auffassung mfit3te diese Kurve folgender-
mat3en erkl~rt werden: Bei Hinzuttigen geringer Elektrolytmengen bis ungefS.hr 0,1 .Aqui-
valent nimmt die Hydratation stark ab, bei gr6Beren Konzentrationen ~ndert sie sich kaum mehr. Einen gleiehen Verlauf zeigeri nun aueh die Kurven naeh KCNS, KNO3, KC1 und K2S04, n. 1. bei kleinen
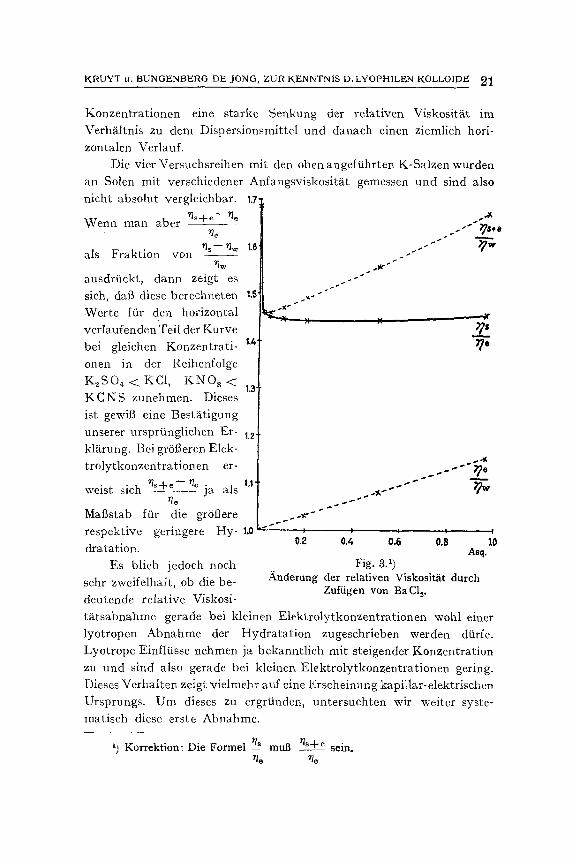
K R U Y T u. B U N O E N B E R G DE 3 0 N G , Z U R K E N N T N I S D .LYOPH1LEN KOLLOIDE ~
Konzentrationen eine starke Senkung der relativen Viskositgt im Verh~iltnis zu dem Dispersionsmittel und danach einen ziemlich hori- zontalen Verlauf.
Die vier Versuchsreihen mit den oben angefiihrten K-Salzen wurden an Solen mit verschiedener Anfangsviskosit~tt gemessen und sind also nicht absolut vergleichbar.
Wenn man aber r~s+~ ~ e
als Fraktion yon
ausdrtickt, dann zeigt es sich, dab diese berechneten Werte ftir den horizontal verlaufenden Teil der Kurve bei gleichen Konzentrati- onen in der Reihenfolge K ~ S O ~ < K C 1 , K N O a < K C N S zunehmen. Dieses ist gewiB eine Best~itigung unserer urspriingliehen Er- kl~rung. Bei grOi3eren Elek- trolytkonzentrationen er-
�9 . . r / s + e - - 7/e . , wezst smh . . . . . ja als ~ e
Mafistab fiir die gr6fiere respektive geringere Hy- dratation.
Es blieb jedoch noch sehr zweifelhaft, ob die be- deutende relative Viskosi-
1.7
1,6
1.5 �84
1,4"
1.3"
i
! ~
1.2 ! / t.1
1.0 L - " " - 0.2
S ~
. . . . - ~'--~
0.4 0.6 0.$ w A e q .
Fig. 3.1 ) A.nderung der relativen Viskositiit dureh
Zuftigen yon BaG1 v
t~tsabnahme gerade bei kleinen Elektrolytkonzentrationen wohl einer lyotropen Abnahme der Hydratat ion zugeschrieben werden dtirfe. Lyotrope Einfltisse nehmen ja bekanntlich mit steigender Konzentration zu und sind also gerade bei kleinen Elektrolytkonzentrationen gering. Dieses Verhalten zeigt vielmehr auf eine Erscheinung kapillar-elektrischen Ursprungs. Um dieses zu ergrtinden, untersuchten wir welter syste- matisch diese erste Abnahme.
1) Korrektion: Die Formel rt~ mug %+e sein. ~ e ~ e

9,~ K O L L O I D C I - ~ M i ~ ,]~__$~[~TE BAND XXVIII, HEFT 1--2
Einflut3 k l e i n e r E l e k t r o l y t k o n z e n t r a t i o n e n a u f die Viskosi t~i t von Agarsol .
Bei kleinen Elektrolytkonzentrationen gestalten sich die Messungen viel einfacher. Man kann die relative ViskositRt der Elektrolytl6sungen bis zu vier Milli~iquivalenten mit 1,000 gleichstellen, so dab die Formel ~ s + e ~ s + e - - zu vereinfaeht wird.
~e ~w Ebenso kann man die Dichten denen yon Wasser gleichstellen,
wodurch nur die Messung der relativen Durchlaufszeit im Verh~ltnis zu Wasser tibrigbleibt.
Oberdies verschwindet die auf S. 20 angeftihrte Ungewiilheit in der Berechnung der wiehtigen Formel
~ s - - ~ o ~ s + o - ~ e , da es hier _~s+e- -~w
In Tabelle VI - -XI I sind die Ergebnisse einiger Versuchsreihen
verzeichnet. Die Werte ~-~ sind sehon ffir die irreversible Reaktion korrigiert. ~/w
Wir wollen diese Serien einzeln n~her betrachten.
T a b e l l e VI.
Elektrolyt
Nullversuch
KCI
BaC12
Co (N H3) Cla
Konzentration in ~-Mol
Konzentration ill
#-~quivalenten
0
~ s + e
~w
1,704
50 100
1000
50 100
1000
~s - - 7/o - - 1 ) *]o
100,0
1,698 1,689 1,600
50 100 500
99,2 97,9 85,2
100 200
1000
1,669 1,638 1,556
15 50
100 333
95,0 90,6 79,0
45 i 1,673 150 I 1,620 300 I 1,571
1000 L 1,498
1) AbkiJrzung fiJr: 100. (~s--~w~. ( ~w+o
,~w+e=~wstellen diirfen: fiir 100.(~s--~w') ( ~w ) k 71w / ~s+~---~0 "
od er
95,6 88,1 81,1 70,7
falls wir

KRUYT u. BUNOENBERG DE JONG, ZUR KENNTNIS D. LYOPHILEN KOLLOIDE ~3
Bei gleichen Konzentrationen (Spalte 2, Tabelle VI), ausgedrtickt in #-Molen pro Liter (1/,-Mol-= 1,10-6Mol), sind also die relativen Viskositgtsabnahmen nicht gleich, sondern nehmea in tier Reihenfolge KCI - - -B aC12-Co(NH~)CI a zu.
Diese Elektrolyte haben das Cl-Ion gemein. Die Viskosit~tts- abnahmen stimmen bei gleichen Elektrolytkonzentrationen ebenso- wenig fiberein, wenn die Konzentrationen in Aquivalenten ausge- drtickt werden, wenn also die Konzentration der Cl-Ionen gleich ist (Tabelle VI). Der groBe Unterschied wird durch die Kationen ver- u rsaeht.
Tabe l l e VII.
Elektrolyt Konzentration Konzentration
in m-Mol m.&quilvnalenten
Nullversuch
1,00 1,00 1,617 85,8 K CI 4,04 4,04 1,552 76,8
10,102 ) 10,10 1,515 71,6
BaCI~
Co (NHa)~C1 a
1,00 2,06 4,04
10,10
0,50 1,00 2,00 4,04
10,10
2,00 4,12 8,08
20,20
1,50 3,00 6,00
12,12 30,30
1,719 100,0
1,537 1,511 1,492 1,487
1,499 1,483 1,471 1,458 1,453
74,7 71,1 68,4 67,7
69,4 67,2 65,5 63,7 63,0
Bei h6heren Konzentrationen (Tabelle vii) bleibt der Unterschied
bestehen, obwohl die Werte n~--n0 dort bei gleicher Km~zentration ~0
weniger voneinander abweichen, als gerade in dem ersten Konzen- trationsgebiet.
x) Bei 10 Millimol betriigt der Unterschied zwisehen der Viskosit~it der Elektrolytl6sungen und der des Wassers einige Zehntelprozente. Bei den Zahlen in Tabelle IX ist diese Korrektion jedoch nicht angebracht worden.
24 KOLI .OIDCHEMISCHE B E I H E F T E BAND X X V I I I , HE.FT 1 ~ 2
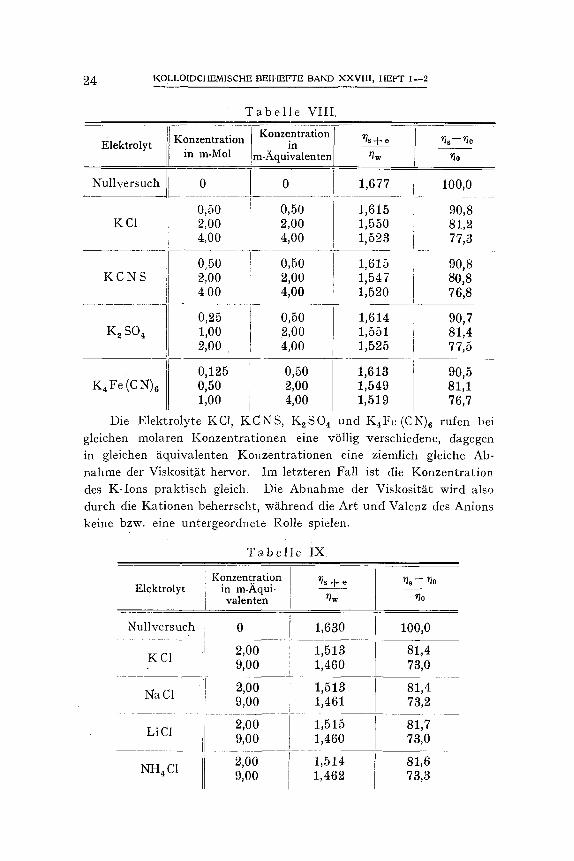
T a b e l l e VIII.
Elektrolyt
Nullversuch
KCI
KCNS
K s SOa
K4 Fe (C N)~
Konzentration I K~176 / n~-be in m-Mol m.,~.quivalenten / ~w
0,50 2,00 4,00
0~50 2,00 400
0,25 1,00 2,00 .
0,125 0,50 1,00
0,50 2,00 4,00
0,50 2,00 4,00
0,50 2,00 4,00
0,50 2,00 4,00
1,677
1,615 1,550 1,523
1,615 1,547 1,520
1,614 1,551 1,525
1,613 1,549 1,519
~o
100,0
90,8 81,2 77,3
90,8 80,8 76,8
90,7 81,4 77,5
90,5 81,1 76,7
Die Elektrolyte KC1, KCNS, K2SO 4 und K4Fe (CN)6 rufen bei gleichen molaren Konzentrationen eine vOllig verschiedene, dagegen in gleichen ~tquivalenten Konzentrationen eine ziemlich gleiche Ab- nahme der Viskosit.~it hervor. Im letzteren Fall ist die Konzentration des K-Ions praktisch gleich. Die Abnahme der Viskosit~it wird also durch die Kationen beherrscht, w~hrend die Art und Valenz des Anions keine bzw. eine untergeordnete Rolle spielen.
T a b e l l e IX
Elektrolyt Konzentration
in m-,~qui- valenten
"~s -+- e
r/w r / s - r/o
r/o
Nullversuch 0 1,630 100,0
K C1 2,00 1,513 81,4 9,00 1,460 73,0
2,00 9,00 Na C1
LiC1
1,513 1,461
1,515 1,460
2,00 9,00
81,4 73,2
81,7 73,0
81,6 73,3
2,00 1,514 NH4Cl 9,00 1,462

K R U Y T u . B U N O E N B E R O D E J O N O , Z U R K E N N T N I S D. L Y O P H I L E N K O L L O I D E "25
W~ihrend sich die Chloride KC1, BaG1 z und Co (NtIa)6Cl a sehr verschieden verhalten, auch wenn die Konzentration in Milli~iquivalenten ausgedrfickt wird, tiben KC1, NaC1, LiC1 und NH4C1 einen gleiehen EinfluIl aus. Im letzteren Fall wechselt zwar die Art, aber nieht die Valenz des Kations.
Aus untenstehender Versuchsserie (Tabelle X) geht hervor, daft BaC12, SrC12, MgSO 4 und CdSO~ eine ziemlich gleiche Viskosi- t~tsabnahme verursachen. Die Art des Kations, die Valenz und Natur des Anions wechseln, abet die Valenz des Kations zeigt sich wiederum als ausschlaggebend.
T a b e l l e X.
Elektrolyt Konzentration
in m Aqui- valenzen
i
Nullversuch [[ 0
BaCl~
SrCI 2
Mg S O 4
Cd S 04
1,00 4,00
r/s + e r / S - 7/o
qw r/o
1,574 100,0
1,449 78,2 1,405 70,6
1,00 4,00
1,00 4,00
1,00 4,00
1,447 77,9 1,407 70,9
1,451 78,6 1,411 71,6
1,450 78,4 1,411 71,6
Bei dem Reinigungsprozet3 des Agars verwendeten wit, um Ver- schimmelung zu verhtiten, eine gewisse Menge HgJ2 , welche in allen in diesem Kapitel angefiihrten Elektrolytagarmischungen bis zu Kon- zentrationen yon h6chstens 2/~-Mol vorhanden sind. 1)
In Kontrollversuchen haben wir uns davon tiberzeugt, dab diese Spur yon HgJ2 keinen Einflufi auf unsere Ergebnisse haben konnte.
T a b e l l e XI.
Elektrolyt Konzentration
in /z-Mol
Nullversuch 0
H g J 2 50
"r/s .t_ e
~w
1,659
1,640
1) S iehe S. 7,

3 6 I~OLLOIDCHEMISCHE BEIHEFTE BAND XXVlll, HEFT 1--2
Bei einer Konzentration von 2/~-Mol wtirde die Viskosit~it also noch um keine Einheit der dritten Dezimale gesunken sein.
In einer anderen Serie untersuchten wir die Wirkung yon Co (NHs)6Cla, La (NO8)a und Pt (Atqn)a (NO3) 4. Das Ergebnis entsprach unseren Erwartungen v611ig.
T a b e l l e XII.
Elektroly t Konzentration
in m Aqui- valenzen
1,667 100,0 Nullversuch 0
0,50 1,512 76,8 Co (NH~) 6 C18 2,00 1,457 68,5
4,00 1,445 66,7
0,50 1,511 76,6 La (NOa) 3 2,00 1,454 68,1
4,00 1,448 l 67,2
0,125 0,25 0,50 1,00 2,00 4,00 8,00
Pt (Atqn)a(NOa) 4 1)
1,580 87,0 1,528 79,2 1,458 , 68,7 1,427 I 64,0 1,404 i 60,6 1,400 ~ 60,0 1,398 i 59,7
Die Viskosit~tt nimmt durch die beiden Elektrolyte mit einer trivalenten Kation ungefiihr gleich viel ab, wiihrend das komplexe Pt-Salz mit einer tetravalenten Kation wieder eine st~irkere Wirkung ausiibt.
Um alle Versuchsreihen miteinander vergleichen zu k6nnen, haben
wir als letzte Spalte in jeder Tabelle die Formel ~s--~0 prozentual im */o
Verhiiltnis zu dem gleichen Weft des Kontrollversuches berechnet. Diese Zahlenwerte diirfen n~imlich untereinander verglichen werden. Sie sind graphiseh in Fig. 4 (S. 28) zusammengestellt. Innerhalb der schwarzen Streifen fallen alle Elektrolytkurven zusammen.
1) Die Agarmischungen mit diesem Elektrolyt opaleszierten. Bei geniigen- tier Verdiinnung zeigte sich im Ultramikroskop ein entstandenes Suspensoid. Die Triibung ist jedoch ganz anderer Art als die, welche in nachstehenden Kapiteln er6rtert werden soil. Sie wird vielleicht dutch das Entstehen einer unl6slichen Verbindung durch Wechselwirkung des komplexen Pt-Salzes und der Asehebestandteite verursacht sein, da sie nach Hinzuftigen eines Tropfens HC1 verschwindet.

KRUYT u. BUNOENBIERO DE JONO, ZUR KIENNTNIS D. LYOPHILIEN KOLLOIDE ~7
D i s k u s s i o n der V e r s u c h s e r g e b n i s s e .
Die durch ~iquivalente Elektrolytmengen hervorgerafene Viskosi- t~ t sabnahme n immt also nm so starker zu, :je,naehdem alas Nation ein-, zwei-, drei- oder viermal geladen ist. Ersetzt man das Wort ,,Viskosit~it" aus dem vorigen Satz durch ,,Potentialdifferenz" in der Doppelschicht, so gibt dieser Satz das Allgemeinresultat, welches das Studium der kapillarelektrischen Erscheinungen ftir den Einflut3 von Elektrolyten auf negativ geladene Grenzfl~ichen ergeben hat, wieder.
Nun hat v. S m o l u c h o w s k i 1) theoretisch abgeleitet, dab die Viskosit~tt eines geladenen Suspensoids griSger als die eines entladenen
sein mut3. Nehmen wir an, daft ftir letzteres die E i n s t e i n s e h e Formel
ns - ~_0 = 5/2 %
gilt, dann muB man far ein geladenes Suspensoid die Formel
~ s - ~ o = s / , q 0 1 1 + 1 (D~]e] ~o ~ \2 ~I J
annehmen, wobei = I,eitfS~bigkeit
r = Radius der Teilchen --Potent ialdifferenz der Doppelschicht
D = Dielektrizit~ttskonstante. Diese Formel k6nnen wir nun zwar nicht quanti tat iv auf unseren
Fall anwenden, da wir hier kein Suspensoid haben und die Volumkon- zentration ~ der Teilchen sieberlich so groB ist, dab die Serienentwick- lung nach ~0 um einige Glieder weitergeftihrt werden mtiBte (siehe S. 10). Sic wird uns aber qualitativ bei der Beschreibung der Ergebnisse nach Hinzuftigen yon Elektrolyten yon Nutzen sein. Der ,,quasi-visk6se Effekt" (v. S m o l u c h o w s k i ) , besser gesagt: der e l e k t r o - v i s k 6 s e
Effekt
kann nur dann zur Geltung kommen, wenn n nicht zu grog ist. Bei gr6Beren Elektrolytkonzentrationen muff der elektro-visk6se Effekt also, abgesehen yon einer positiVen oder negativen Ladung der Teilchen, versehwinden. Die Bedeutung der ~-nderung der I.eitf~higkeit wird in der dritten Abhandlung dieser Serie ( K r u y t und T e n d e l o o ) aus- ftihrlieh diskutiert werden.
Tats~ichlich nimmt die Viskosit~it eines Agarsols nach Hinzuftigen aller Elektrolyte ab, w~ihrend sich die relative Viskosit~it, durch Nieht-
1) v. Smoluchowski , Koll.-Zeitschr. 18, 190 (1916).
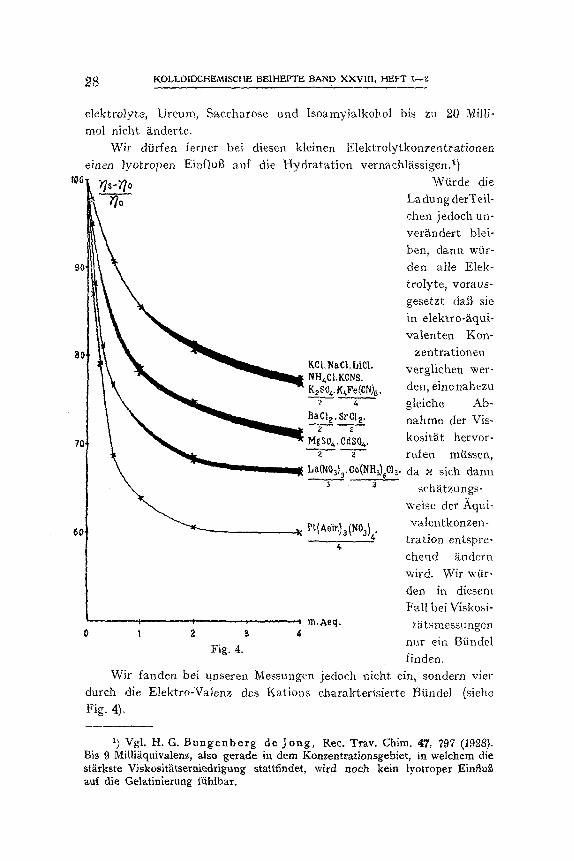
~ KOLLOIDCHEMISCHE BE1HEFTE BAND XXVII1, HEFT 1--~
elektrolyte, Ureum, Saccharose and Isoam'ytalkohol bis zu 20 Milli- tool nicht anderte.
Wir dfirfen fet ter bei diesen kleinen Elektrolytkonzentrat-ionen eiacn )yotropen Ei~/lug auf die Hydratat ion vernachl~tssigen. 1)
tO0 1~ ~7s#/___2
9O
8 0 ' K e l . N a e i . b i C l .
NHhCI.KCNS.
2 70 M~S0~. CdS%.
2
ha(R0a)a. Co(Rlta)6Clv 3 3
~, NAo%(N%). 4
6 0
; -; - : - - . i m . A ~ q . o 1 2 ~ 4
Fig. 4.
Wtirde die Ladung derTeii- chen jedoch un- ver~.ndert blei- ben, dann war- den alle Elek- trolyte, voraus- gesetzt dab sie in elektro-{iqui- valenten Ken-
zentrationen verglichen wer- den, einenahezu gleiche Ab- nahme der Vis- kositg, t hervor- ruien miissen, da x sich dann
sch~tzungs- weise der Aqui- valentkonzen-
tration entspre- chend ~ndern wird. Wir w(ir- den in diesem Fall bei ViskOsi-
t~tsmessungen nur ein BfindeI linden.
Wit fandert bei unseren Messungen jedoch nicht ein, sondern vier durch die Elektro-Valenz des Kations charakterisierte BOndel (siehe Fig. 4),
1) VgL H. G. Bungenbe rg de Jong, Rec. Tray. Chim. 47, 797 (1928). Bis 9 Milli~iquivalenz, also gerade in dem Konzentrafionsgebiet, in welchem die st~irkste Viskositiitserniedrigung stattfindet, wird noch kein lyotroper Einflug auf die Gelatinierung fiihlbar,

KRUYT u. BUNGIgNBERG DE JONG, ZUR KENNTNIS D.LYOPHILEN KOLLOIDE 29
Dann nmt3 die Ladung also bei ~quivalenten Elektrolytkonzen- trationen sprungweise kleiner werden, je nachdem das Kation ein-, zwei-, drei- oder viermal geladen ist.
Die Ladung ist also fgr ttquivalente Mengen yon
K+ > Ba + + Co (N Ha~+ + + Pt (Aein).~ + + + + > 3 > 4
Diese Reihenfolge zeigt scbon an, dab das Agarsol negativ ge- laden ist, welches naeb eingehender Untersuchung tats~chlich be- stS~tigt wurde.
Wir bestimmten die Ladung des Agarsols bei 500 C in einer Kata- phoresertihre naeh B u r t o n , welche in einem Thermostat in dunklem Zimmer aufgestellt war und verwendeten dazu einen Gleichstrom yon 220 Volt. Ein kleiner Spiegel warf von oben ein Bogeniichtbtinde.l in einen der R6hrenschenkel. Die Gren~zflS~che wird auf diese Weise durch das ~1 yndall-Licht, welches das Sol seitw~irts ausstrahlt, gut sichtbar. I)
Das Sol war negativ geladen. Wir fanden dann auch, dag die Ladung tffts/~chlich durch Hinzu-
ftigen yon Elektrolyten abnimmt. Hierbei waren einige Sehwierigkeiten zu iiberwinden, welche kurz
er6rtert werden sollen. Erstens muB daftir gesorgt werden, dab der Potentialabfall durch die ganze R6hre konstant ist. Als FRissigkeit, welehe die Elektroden umsptilt, muff man also eine L6sung mit dem Elektrolytgehalt des Sols nehmen. Dann wird die Leitungsf/~higkeit jedoch so grot3, dab die Grenzfl/~che infolge yon Konvektionsstr6men, welche dureh die Str6mungsw~trme und die gasf6rmigen Polarisations- produkte an den Pt-Elektroden hervorgerufen sindl schnell verschwindet. Wir verwendeten darum umkehrbare Elektroden (Cu--Cu SO4) , und um die Str6mungsw/~rme so gering als miiglich zu erhalten, wurde eine geringe Potentialdifferenz an den Elektroden (14 Volt) eingeschaltet.
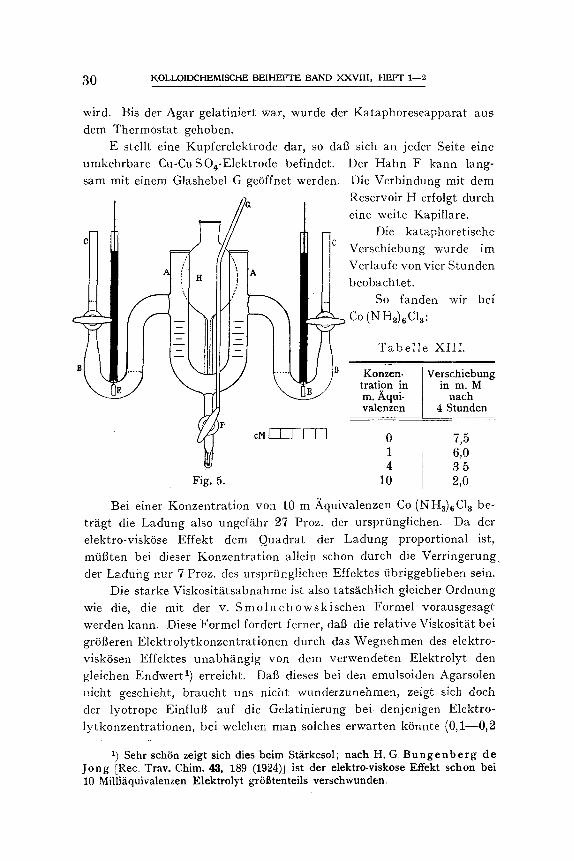
Aut]erdem brachten wir folgende kleine ~-nderungen an dem ur- spriinglichen Apparat yon B u r t o n an (siehe Fig. 5).
Bei jedem der beiden Schenkel A isi: eine gleich weite U-f6rmig gebogene Glasr~Shre angeschmolzen, welche sieh nacb oben hin lang- sam verengt. Der unterste Teil jedes Seitenrohres wird mit 2 Proz. Agarsol, welches ungef~hr 1 )kquivalent CuSO4 enth/~lt, vollgegossen. Danach wird die R6hre mit einem Gummisehlauch verbunden und die AgarlOsung bis tiber den Hahn D aufgesogen, wonach dieser geschlossen
1) Die Methodik wurde sp~iter verbessert. Vgl. die direkte Abhandlung dieser Serie und H. R. Kruyt, KoU.-Zeitschr. 37, 358" (1925).
3 0 KOLLOIDCHEMISCHE BEIHEFTE BAND XXVlll, HEFT 1--2
i B
wird. Bis der Agar gelatiniert war, wurde der Kataphoreseapparat aus dem Thermostat gehoben.
E stellt eine Kupferelektrode dar, so dab sich an jeder Seite eine umkehrbare Cu-CuSO,-Elektrode befindet. Der Hahn F kann lang- sam mit einem Glashebel G ge6ffnet werden. Die Verbindung mit dem
c. Reservoir H erfolgt durch
t ,J eine welte Kapillare. Die kataphoretische
m Verschiebung wurde im Verlaufe von vier Stunden
C
B
cM UVT-V-I-I
beobachtet. So fanden wir
Co (N I-la)6 (~13:
J
bei
T a b e l l e XIII.
Konzen. Verschiebung tration in in m. M m. ~qui- nach valenzen 4 Stunden
0 7,5 1 6,0 4 35
Fig. 5. 10 2,0
Bei einer Konzentration von 10 m Aquivalenzen Co (NH8)6CI abe- tr~igt die Ladung also ungef~thr 27 Proz. der ursprtingliehen. Da der elektro-viskSse Effekt dem Quadrat der Ladung proportional ist, mtit3ten bei dieser Konzentration allein sehon durch die Verringerung der Ladung nur 7 Proz. des ursprtinglichen Effektes tibriggeblieben sein.
Die starke ViskositS.tsabnahme ist also tats~tehlich gleieher Ordnung wie die, die mit der v. S m o l u c h o w s k i s c h e n Formel vorausgesagt werden kann. Diese Formel fordert ferner, daft die relative Viskosit~it bei gr6t3eren Elektrolytkonzentrationen durch das Wegnehmen des elektro- viskSsen Effektes unabhSmgig yon dem verwendeten Elektrolyt den gleichen Endwert 1) erreicht. DaB dieses bei den emulsoiden Agarsolen nicht geschieht, braucht uns nicht wunderzunehmen, zeigt sich doeh der lyotrope Einflut3 auf die Gelatinierung bei denjenigen Elektro- lytkonzentrationen, bei welehen man solches erwarten kSnnte (0,1--0,2
x) Sehr schSn zeigt sich dies beim St~irkesol; nach H. G Bungenberg de Jong ~Rec Trav. Chim. 48, 189 (1924)] ist der elektro-viskose Effekt schon bei 10 Milli~iquivalenzen Elektrolyt grSgtenteils verschwunden.

KRuYT u. BUNGISNBERO DE .JONG, ZUR I4[ENNTNIS D, LYOPHILEN KOLLOIDE 31
Aquivalenzen) schon sehr bedeutendl), mit anderen Wor ten ist also die Hydratation der Teilchen und damit das scheinbare Volumen bei glejchen Agarkonzentrationen nicht mehr gleieh groB. {3berdies besteht bei den Elektrolytkonzentrationen wieder die Schwierigkeit, zu ent- scheiden, wie grot3 die wirkliche Konzentration in dem freien Disper- sionsmittel ist.") Je kleiner die Elektrolytkonzentrationen werden, um so mehr nS~hern wir uns in jeder Hinsicht einem Zustand, auf welchen die S m o l u c h o w s k i s e h e Formel, wenn nicht quantitativ, so doch halb- quanti tat iv angewendet werden kann.
Aus dem Vorhergehenden k6nnen wir also ohne weiteres sehlieflen, dab die e l e k t r i s e h e n E i g e n s c h a f t e n des t y p i s c h emulso ide~ l A g a r s o l s d e n j e n i g e n der t y p i s c h e n S u s p e n s o i d e g l e i eh s ind .
F r e u n d l i c h gibt in seiner Adsorptionstheorie eine Erklgrung ffir das Phiinomen, bei welchem, je nachdem das entgegengesetzt geladene ]on eine gr6Bere Ladung tragt, eine zunehmende Entladung durch gtquivalente Konzentrationen erfolgt. Er geht dabei yon der Tatsache aus, dat3 alle anorganischen Elektrolyte gleich stark adsorbiert werden.
Das H-Ion, OH-Ion, die lonen der schweren Metalle und die orga- nischen Ionen werden jedoch stgrker adsorbiert. In den nachstehenden zwei Versuchsreihen wurde nun untersucht, ob H C1, K O Ha), Strychnin- nitrat und Fuchsin die Viskosit/~t des Agarsols starker herabsetzen ats KCl.
Die Agarmischungen mit K O H erreichten sofort ihren Endwert, bei HC1 nahm die Viskositgt linear mit der Zeit ab. Die Anfangswerte der Misehungszeitpunkte wurden aus drei Messungen jeder Mischung extrapoliert.
T a b e l l e XIV.
Elektrolyt Konzentration
in o,-Aquivalenzen
Nullversuch 0 1,699
53 1,691 98,9 HC1 106 1,665 95,1
318 1,610 87,3
qs -b e qs -- ~o
~w ~o
I00
54 1,690 98,7 K O H 107 1,676 96,7
322 1,638 91,3
1) H. G. Bungenberg de Jong, Rec. Tray. Chim. 47, 797 (.1928). �9 2) Siehe S. 20. 3) KOH, alc. dep. (de Haen), Karbonatgehalt 1--2 Proz.

3 2 I4,OLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
Vergleicht man diese Tabelle mit Tabelle VI, so reihen sich die Kurven ftir HCI und K O H zwischen die for KCI und BaC12.
Die Tatsache, dab diese zwei Elektrolyte nicht in das erste Biindel fallen, braucht noch nicht auf eine stfirkere Entladung hinzuweisen. Denn schon durch die grotle Beweglichkeit des H-und OH-Ions mtissen diese Elektrolyte infolge ihrer besonderen Wirkung auf z den elektro- viskSsen Effekt stttrker herabsetzen als KC1.
Die HC1-Kurve scheint in der Richtung der Konzentrationsachse um ungef~thr 40 be fi~quivalenzen in positivem Sinne verschoben zu sein. Es ist wahrseheinlich, dab diese ersten HC1-Mengen in Wechselwirkung mit den Aschebestandteilen des Agarsols treten, so dab man dieser Verschiebung nicht zu viel Bedeutung beimessen darf.
In einer anderen Serie wurde der Einflufl des K C1 auf die Vis- kosit~t mit dem des Stryelaninnitrats 1) und dem des Fuchsins2) 3) ver- glichen.
T a b e l l e XV.
Elektrolyt
Nullversuch
KCI
S trychnin- rfitrat
Fuchsin
Konzentration ~s + ~ ~s -- ~o
m..A.q uilvnlenzen ~w ~o
0 1,649 100,0
1 4
0,5 1 4
0,5 1 2 3 4 7
1,557 85,8 1,504 77,7
1,589 90,8 1,559 86,1 1,508 78,3
1,550 s4,7 1,489 75,3 1,408 62,9 1,380 58,6 1,368 56,7 1,361 55,6
x) Strychninnitrat, Handelspraparat, dessen Gehalt durch azidimetrische Titration kontrolliert war.
3) Magentarot yon Dr. G. Griibler & Co., Leipzig. 3) Die geringe L6slichkeit des Fuchsins bei Zimmertemperatur liefl die
auf S. 18 beschriebene Behandlung der Mischungen fftr gr6flere Konzentrationen als 2 Milligramm~iquivalenz nicht zu. Die iibrigen Mischungen wurden hergestellt, indem zu den mit der Konzentration 2 m-Aquivalenz gefiillten Viskosimetern abgewogene Mengen Fuehsin gebracht wurden. Die Konzentrationen 3, 4 und 7 m-,~quivalenz sind also nicht vSllig genau.

:V,,RUYT u.BUNGENBERG DE JONG, ZUR KENNTNIS D. LYOPHILEN KOLLOIDE ~
Das einwertige Farbstoffkation verringert die Ladung viel st~trker als das K-Ion. Die Kurve 15st sich yon dem ersten Biindel und durch- quert die tibligen drei.
Gegen jede kolloidci~emische Erfahrung auf anderen Gebieten schlot3 das Strychninion sich bei den einwertigen anorganischen Kat- ionen an.
Z u s a m m e n f a s s u n g .
Alle Elektrolyte verringern in kleinen Konzentrationen die Vis- kositgt vor~ Agarsol stark.
KC1, Nat1, LiCl, NH4C1, KCNS, K2SO 4 und K4Fe(CN)6 er- geben in tiquivalenten Konzentrationen eine praktisch gleiche Viskosi- t~tsabnahme.
Eine starkere, aber gegenseitig wieder fast gleiche Senkung wird durch BaCle, SrC12, MgSO 4 und CdSO 4 hervorgerufen.
Eine dritte Gruppe bilden Co (NHa)6C1 a und La (NOn)s, wS.hrend die gr/Sfite Senkung, immer bei gquivalenten Konzentrationen berechnet, durch Pt (Aein)a (NO3) 4 verursacht wird.
Die vier Gruppen sind durch den Besitz eines ein-, zwei-, drei- und viermal geladenen Kations gekennzeichnet. Aus Kataphoreseversuchen erwies sich, dab Agarsol negativ geladen ist und dab die Ladung durch Hinzuftigen von Co (NH3)6C1 ~ abnimmt.
v. Smoluchowsk i hat nun theoretisch abgeleitet, dab die Vis- kositS.t eines geladenen Suspensoids grOfier sein mull als die eines ent- ladenen. Seine Formel ist bier zwar nicht quantitativ anzuwenden, kann aber doch qualitativ die Ph~inomene erkl~iren. Aufierdem ergibt sich hieraus, dab die elektrischen Eigensehafter~ des typisch emul- soiden Agarsols die gleichen sind wie die der Suspensoide, d. h. die elek- trische Ladung ist kapillar-elektrischer Natur.
Nach dem Entfernen der Ladung flockt das Agarsol nicht aus. Es dankt also seine Stabilit~it noch einem anderen Faktor.
Die ViskositS~tsabnahme durch HC1 und KO H ist starker als durch BaCI~. Diese ist ohne Anwendung neuer Hypothesen schon aus der v. Smoluchowskischen Formel deutlich.
Durch Fuchsin wird eine viel stS~rkere ViskositS.tsabnahme erzielt als durch KC1, welches in {)bereinstimmung mit der Tatsache, dab im allgemeinen organische Ionen stSxker als ~norganische adsorbiert werden, steht.
Wider alle Erwartung verh~ilt sieh jedoch Strychninnitrat wie KCI. Bei gr/513eren Elektrolytkonzentrationen, bei welehen der elektro-
visk~sse Effekt nicht mehr anwesend ist, bieibt die Formel *1~--*~" als % 3

3 4 KOLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
Ma/3stab far die Hydratat ion bestehen. Diese nimmt zu in der lyotropen
Reihenfolge: K2SO 4 ~< KC1 ~< K N O a.~ KCNS.
V. H y d r a t a t i o n u n d S t a b i l i t g t .
E i n l e i t u n g .
In den vorigen Kapiteln haben wir gesehen, dab die elektriscben Eigenschaftel~ des Agarsols denjenigen der im allgerneinen bei Sus- pensoiden beobachteten gleich sind. Bei letzteren Systemen ist die elektrische Ladung der einzige Stabilitgtsfaktor. Wird clie Ladung bis zu einem gewissen kritischen Wert herabgesetzt, dann flocken ctie Suspeasoide sehnell aus. Bei Agar end ebenfalls bei vielea anderen Emulsoiden geschieht dieses naeh Entladung nicht.
Man muB also annehmen, dab Emulsoide aut3er der elektrischen Ladung noch einen anderen Stabilit~ttsfaktor besitzen, welcher oft einflut3- reich genug ist, um die Stabilit~tt der entladenen Teilehen aufrecht- zuerhalten. Aus den folgenden Zeilen wird sich zeigen, dab die Hydra- ration der Teilehen dieser Stabilit~ttsfaktor ist.
O r i e n t i e r u n g s v e r s u c h e .
Wenn man zu einem 1/Tprozentigen Agarsol, welches schon neun Milli~tquivalent KC1, BaC12 oder Co(NHa)sC1 a enthglt, eine ffinffache Menge Alkohol 1) hinzuftigt, so flockt der Agar sehr schnell voll- st~tndig aus.
Enthglt das Agarsol jedoch keine Elektrolyten, dann erhS~lt man eine hellblaue opaleszierende Fliissigkeit, welche sich, wie sp~tter gezeigt wird, vOllig wie ein typisehes Suspensoid verhglt.
Nimmt man Azeton anstatt Alkohol, so ist das Versuchsergebnis alas gleiehe.
Eigenschaften der blau opaleszierenden S y s t e m e : A g a r - W a s s e r - A l k o h o l und Agar-Wasser-Azeto'n. 2)
U l t r a m i k r o s k o p i e .
Verdtinnt man das System Agarsol-Alkohol (1 : 5) oder Agarsol- Azeton (1 : 5) bei 50 o C 60mal mit einer Alkohol- oder Azeton-Wasser- mischung derselben Konzentration, so zeigen sich nach Abkfihlung unter dem Ultramikroskop (Zeit3-Mikroskop mit Paraboloidkondensor, Licht- quelle: Weule-Lampe von Zei/3) eine grofle Anzahl stark leuchtender Partikelcherl mit lebhafter B r o w n s c h e r Bewegung.
1) Der verwendete Alkohol war dutch Destillieren gereinigt worden. 2) Handelspr~iparat, iiberdestillierte Fraktion 56,2--56,8 o C.

KRUYT U. BUNGENBERG DE JONG, ZUR KENNTNIS D. LYOPHILEN I~OLLOIDE 3 ~
Das ursprtingliche, 60mal mlt destilliertem Wasser verdtinnte Agarsol l~iflt uns keine Differenzierung des Ultrabildes erkennen, sogar nicht unter dem ZeiBschen Spalt-Ultramikroskop, mit welchem tab- gesehen von einigen deutlichen qptischen Verunreinigungen) nur ein Tyndallkegel beobachtet wurde.
E l e k t r i s c h e L a d u n g .
Die elektrische Ladung wurde in einer Kataphoreser6hre nach B u r t o n bestimmt, wobei wieder einer der Schenkel yon oben mit Bogenlicht beleuchtet war. Ftir die Agarsol-Alkohol-Mischung betrug die Versehiebung der Grenzfl~iehe naeh der Anode hin bei einer Po- tentialdifferenz yon 10 Volt pro Zentimeter durchschnittlieh 6 mrn pro 20 Minuten.
Die Agarsol-Azeton-Mischung enth~lt ebenfalls negativ geladene Teilchen.
G r e n z w e r t e .
Ein Vorrat einer 2/Tprozentigen Agarsol-Alkohol-Mischung (i : 5) wurde naeh Herstellung bei 500 C in dem Thermostat aufbewahrt.
In eine Anzahl mit Dampf gereinigte und mit geschliffenen St6pseln versehene Glasr6hrchen (10 cem Inhalt) wurden mit einer Pipette 3 ccm einer Alkohol-Elektrolyt-Wasser-Mischung gebracht. Das VerhS~ltnis Alkohol :E!ektrolyt l6sung war immer 1 : 5 , w~hrend die Konzentration der Elektrolytl~Ssung wechselte. Diese R~Shrehen wurden danach in einen Thermostaten yon 50 ~ C gehS~ngt, zu jedem 3 cem der obengenannten Agarsol-Alkohol-Mischung hinzugefiigt und etwas ge- schiittelt. Die Zusammensetzung j edes R6hrchens war alsoann:
i1 ecm Agars~ + 5 ccm A l k ~ 1 7 6 50 o C -~ I 1 ecm Elektr~ + 5 ecm A l k ~ 1 7 6 Zimmertemperatur J
Nach sechs Stunden trat in der Flockung praktisch keine Ver- 5mderung mehr ein. In Tabelle XVI findet man bei jedem Elektrolyt zwei Zahlen. Die h6here bezieht sich auf die Konzentration, in welcher sich gerade noch v611ige Flockung des Agars zeigte und die tiberstehende Fltissigkeit vollkommen Mar war, die niedrige deutet die Konzentration in dem n~ichsten R6hrchen an, in welehem die tiberstehende Fltissigkeit noch etwas trtibe aussah.
Die Zahlen geben die Konzentration der ElektrolytlSsung, yon welcher 1 e e m verwendet worden war, in Millimolen an. Es sind also relative Zahlen der wirklichen 12real kleineren Konzentrationen.
In Tabelle XVI sind tiberdies~die Ergebnisse fiir die ~/Tprozentige Agarsol-Azeton-Miscbung (1: 5), sowohl bei 50 ~ C als bei Zimmer- temperatur (~- 18 o C) hergestellt, verzeichnet.
3"
36 KOLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1 - -2
Der Ausflockungsmechanismus verlief bei Zimmertemperatur langsamer, so dab die Ergebnisse erst nach 12 Stunden beurteilt werden konnten.
T a b e l t e XV1.
System
Agarsol-Alkohol (1: 5) 500 C
Agarsol-Azeton (1: 5) 500 C
Agarsol-Azeton
Konzentration
K CI Ba C12 Co (NHs) 6 CI~
14 - -16 0,53--0,61 0,33--0,37
8 - -10 0,26--0,28 0,17--0,19
(1: 5) 3,5-- 4 0,20--0,22 0,11--0,12 180 C I
Die Grenzwerte (Durchschnitt jedes Zahlenpaares) liegen tiefer, je nachdem das Kation ein-, zwei- oder dreimal geladen ist. Die elektrische Ladung hat also nach dem Hinzuftigen von AIkohol oder Azeton das gleiche Zeichen behalten und ist ebenso wie bei dem emul- soiden Agarsol kapillar-elektrischer Natur.
Sowohl die Agarsol-Alkohol- wie die Agarsol-Azeton-Mischung behS~It beim Abkiihlen auf Zimmertemperatur ihre Stabilit~it, w~ihrend das emulsoide Agarsol bei Abktihlung gelati~iert.
Vier Tage nach der Abktihlung der in Tabelle XVI angegebenen Agarsol-Azeton-Mischung auf Zimmertemperatur wurden die Grenzwerte von neuem bestimmt, und zwar fiir KC1 und ftir KCNS. Wir fanden ftir beide Elektrolyte wieder die gleichen Grenzen (3,5--4) wie frfiher.
I r r e v e r s i b i l i t ~ i t der F l o c k u n g .
Wenn man nach der Flockung die tiberstehende Alkohol-Wasser- oder Azeton-Wasser-Mischung abgiel3t und die klaren weiBen Flocken mit destilliertem Wasser tibergiei3t, so werden diese plbtzlich unsichtbar. Es hardelt sich bier jedoch nicht um eine Peptisation, da die Flocken nach Schtitteln der R6hrchen deutlich zu erkennen sind. Sie gleiten als durchsichtige Klumpen an der Glaswand herunter. Nicht die Flockung, sondern das Verhalten der optischen Eigenschaften ist also reversibel. Man kann diese optischen Eigenschaften mit Hydratations- htillen in Verbindung bringen. Der optische Kontrast der Teilchen

KRUYT u. BUNGENBERG DE JONO, ZUR KENNTNIS D. LYOPHILEN KOLLO1DE 3~
mit dem Dispersionsmittel wird hierdurch bei dem Agarsol stark ver- ringert (schwaches Tyndallph~nomen, amikroskopisches Sol).
Nimmt man nun an, dab die Teilehen in dem Alkohol-Wasser- und dem Azeton-Wasser-Milieu ihre Hydratationshtille zum gr613ten Tell verlieren, dann wird es verstS~ndlich, warum sie jetzt einen viel griSgeren optisehen Kontrast rnit dem Dispersionsmittel zeigen (starkes Tyndall- phgnomen, Ultramikronen). Unsere Voraussetzung wird durch die Tat- sache untersttitzt, dab die bier oben besprochenen Sole nach Entladung ihre Stabilit~tt verlieren.
R e v e r s i b i l i t g t der D e h y d r a t a t i o n .
Aus dem System Agarsol-Azeton (1 : 5) kann man bei 500 C unter niedrigem Druck das Azeton auskochen. Man behgtt dann das Agar- Wasser fibrig, welches durch Abkiihlung zu dem typisch kriimeligen Agargel gelatiniert und dadurch anzeigt, dab es seine emulsoiden Eigen- schaften v611ig zur/ickerhalten hat.
Auch hieraus geht hervor, d a b die Entw~sserung ein re~,~ersibler Prozet3 ist (siehe Ierner S. 41).
Setzt man bei 500 C einem 2/@rozentigen Agarsol Alkohol oder Azeton zu, so zeigt es sich, dab bei einem Gehalt yon + 50 Gewichts- prozenten Alkohol die Opaleszenz pl6tzlich auftritt. Wird dann ein wenig destilliertes Wasser hinzugeftigt, so verschwindet die Opaleszenz, um nach weiterem Hinzuftigen yon AIkohol pl6tzlich wieder auf- zutreten, usw.
Unserer Meinung nach nimmt also die Entwasserung der Teilehen durch Alkohol nicht allmS~hlich, sondern ziemlich genau bei dem Mischungsverhaltnis 50 Proz. sehr sehnell zu. Wir werden sehen, dab die Viskosit~Ltsmessungen diese Vermutung bestgtigen.
V i s k o s i t ~ t s m e s s u n g e n .
Das emulsoide Agarsol ver/~ndert sich durch Hinzuftigen yon AIkohol oder Azeton in ein typisehes Suspensoid. Wir haben schon unsere Ver- mutung ausgesproehen, dab die Ursache in einer Dehydratation der Teilchen gelegen sei, was sich dann in einer bedeutenden Abnahme der relativen ViskositS~t imVerh~ltnis zu dem Dispersionsmittel ~uBern mtiBte.
Die Viskositgtsmessungen wurden bei 45 o C an versehiedenen Agar- Alkohol-Wasser-Misehungen mit konstanter Agar- aber weehselnder Alkoholkonzentration vorgenommen.
Nebenher wurde die Viskositgt der entsprechenden Alkohol-Wasser- Mischungen gemessen.

3 8 KOLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
Um die Alkoholkonzentration m6glichst reproduzierbar machen zu kSnnen, weil die ViskositS~t yon dem bin~iren System Alkohol--Wasser dureh ein ziemlich hohes Maximum 1) geht, wurden die Mischungen nicht mehr in einem Mat3kolben yon 50 ecm, sondern yon t00 ccm hergestellt. 80, 100 und 120 g Alkobol wurden zuerst zugewogen und danach so viel ausgekochtes destilliertes Wasser beigeftigt, dab noch gentigend Raum far den Inhalt einer Pipette Agarsol tibrigblieb. Bei Eiehung der Pipette, welche iibrigens ganz, wie auf S. 12 beschrieben, eingerichtet war, betrug das Gewicht des ausfliet3enden Wassers 39,021 g. Die Konzentration des ursprtingliehen Sols war 1,71 g lufttrockener Agar pro 310 g Sol. Dieses Sol wurde bei 500 C in dem Thermostat auf- bewahrt. Zu der auf 450 C gebrachten Alkohol-Wasser-Mischung wurde nun mit dieser Pipette jedesmal eine reproduzierbare Menge Agarsol beigeftigt, der MaBkolben bei 450 C l~ingere Zeit in den Thermostat gestellt und darauf die feinere Einstellung auf den Teilstrieh durch Hinzuftigen yon Wasser erreicht.
Die entsprechenden Alkohol-Wassermischungen sind ebenfalls bei 450 C hergestellt. Zur Feststellung der Alkoholkonzentration bestimm- ten wir mit HiKe eines Flaschenpyknometers yon @ 25 ecru bei 150 C die relative Dichte dieser letzten Misehungen im VerhMtnis zu Wasser der gleichen Temperatur (siehe Spalte 1, Tabelle XVII).
Die mit HiKe der Alkoholtabellen aus dem ,,Chemisch Jaarboekje ''~-) berechnete Konzentration in Gewichtsprozenten ist in Spalte i ver- zeiehnet.
Die relative Diehte und die relative Viskosit/it (Doppelbestim- mung) bei 450 C im Verh~tltnis zu Wasser der gleichen Temperatur finden sieh in Spalte 3 und 4.
T a b e l l e XVII.
(%% Proz. ( d d w /15 Alkohol \ -dw - J 4 5 \ ~/w /45
1,0000 0,9384 0,9138 0,8842 0,8283
0 40,6 52,1 65,0 87,9
1,0000 0,9235 0,8967 0,8659 0,8088
1,000 2,105 2,130 2,019 1,567
Bei der Berechnung der Viskosit~it derAgar-Alkohol-Wasser-Mischun- gen haben wir die Dichte dieser Systeme denen der entsprechenden
1) Dunstan, Zeitschr. f. phys. Chem. 49, 590 (1904). 2) Chem. Jaarboekje II (Amsterdam 1920).
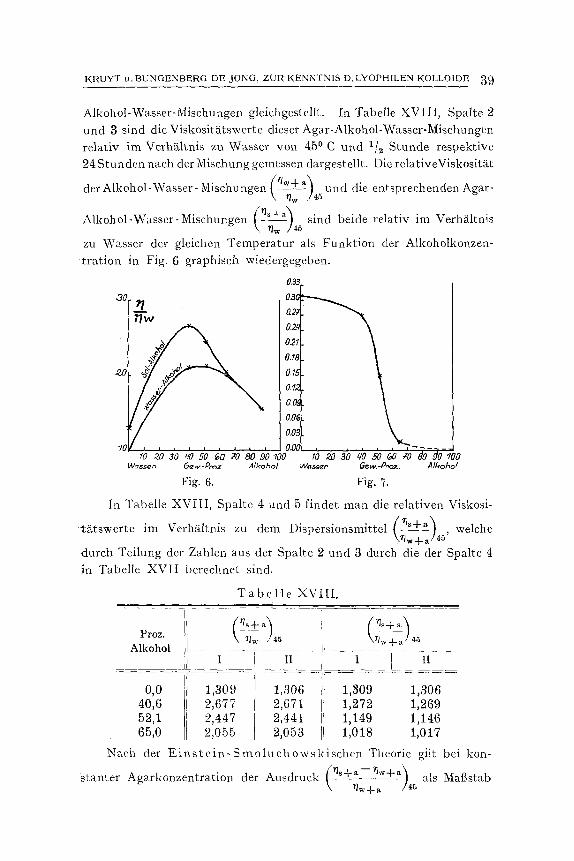
KRUYT u. BUNGENBERG DE JONG, ZUR KENNTNIS D.LYOPHILEN I~OLLOIDE ~9
Alkohol-Wasser-Mischungen gleichgestellt. In Tabelle XViII, Spalte 2 und 3 sind die ViskositS, tswerte dieser Agar-Alkohol-Wasser-Mischungen relativ im VerhSAtnis zu Wasser yon 450 C und 1/2 Stunde respektive
24 Stunden nach der Mischung gemessen dargestellt. Die relativeViskositXt �9 ~ w + a der Alkohol-Wasser-Mlschungen ( -~-- )45und die entsprechenden Agar-
Alkohol-Wasser-Mischungen (~-' +~] sind beide relativ im Verh~Itnis / %
\ ~w /45
zu Wasser der gleichen Temperatur als Funktion der Atkoholkonzen- tration in Fig. 6 graphisch wiedergegeben.
033_ ao ~/ o s ~ rl--~ a2
x~*~176 ~ 0.~ 0.2 O'8 t
o off \ 0.031 ~ ,~
10 . . . . . . . ~ , 0 . 0 ~ " ~0 ~ ~0 ~0 50 ~0 70 ~ ~0 100 10 ~ @0 l~o 20 ~ ;0--~I~0}00 Wa.~r Gr ~ - P,'oZ Alkohol W~, ,~Cr, Gew.-P,~z. Alltohol
Fig. 6. Fig. 7.
In Tabelle XVIII, Spalte 4 und 5 finder man die relativen Viskosi-
tgtswerte im Verh~iltnis zu dem Dispersionsmittel (%+-~) , welche \ ~ w "-b a/45
durch Teilung der Zahlen aus der Spalte 2 und 3 durch die der Spalte 4 in Tabelle XVII berechnet sind.
T a b e l l e XVIII.
Proz . k ~w /45 \~w--a 45 Alkohol
I I[
0,0 40,6 52,1 65,0
I II
1,309 1,306 2,677 2,671 2,447 2,441 2,055 2,053
1,809 1,272 1,149 1,018
1,306 1,269 1,146 1,017
Nach der E i n s t e i n - S m o l u c h o w s k i s c h e n Theorie gilt bei kon-
stanter Agarkonzentration der Ausdruck (~s+a--~,v+.~] als Mafistab \ ~w+a /45

4 0 KOLLOIDCHEMIECHE BEIHEFTE BAND XXVIII, HEFT 1--2
ftir die Hydrata t ion der Teilchen (eine mSgliche VerSmderung des elektroviskosen Effektes kann dabei in unserem Falle vernachltissigt werden).
Diese durch Vermindern der Zahlen aus Tabelle XVII I , Spalte 4 und 5 mit der Einheit erhaltenen Werte finder man al~ Funktion der Alkoholkonzentration in Fig. 7 (S. 39) graphisch daigestellt.
Wir haben die relative Viskositat bei der Alkoholkonzentration 100 Proz. ftir diese Figur auf die nach der E i n s t e i n s c h e n Formel ftir ganz entw~tsserte Teilchen berechnete Zahl 1,002 gestellt.
Es ergibt sich also tats~ichlich, daft die Hydrata t ion bei grSt3eren Alkoholkonzentrationen abnimmt, und dab diese Entw~tsserung gerade in dem mittleren Konzentrationsgebiet, in welchem ftir das binare System Alkohol-Wasser das ViskositS~tsmaximum liegt, sehr ausge- sprochen ist.
Wir kontrollierten dann noch auf anderem Wege die relative Vis- kositS~t im Verh~iltnis zu dem Dispersionsmittel ftir die 65,0prozentige Alkoholsolmischung. Zu diesem Zwecke wurde aus einer L6sung 25 r Mol Co (NH3)eC13 verdunstet, welches sieh an der Wand des Jena- KSlbchens absetzte. Darauf wurden 50 ccm der 65,0prozentigen Alkohol- solmischung bei 450 C hinzugeftigt; der Agar flockte sofort aus und wir konnten die Viskosit~t des sich auf diese Weise absetzenden tiberstehen- den Dispersionsmittel bestimmen.
Hieraus wurde ftir die Alkoholsolmischung eine relative Viskosi- t~t 1,015 berechnet, welche Zaht gut mit der in Tabelle X V I I I vcr- zeichneten tibereinstimmt.
R e v e r s i b i l i t ~ t de r D e h y d r a t a t i o n .
Um uns fiber dieses Problem weitere Klarheit zu verschaffen, stell- ten wir noch folgenden Versuch an. Wir gingen von einem Agarsol~ welches 2,5 g an der Luft getrockneten Agar pro 200 g Sol enthielt, aus. Bei der Herstellung der Mischungen bedienten wir uns zweier mit einem Glasmantel umgebenen Pipetten A und B (siehe S. 12). Bei Eichung der Pipetten mitWasser yon 50 ~ ergab sich, dat3 A = 24,832 g und B = 32,021 g wog. In drei ausgedS.mpften Jena-Kolben yon 200 ccm wurden genau 60 g Alkohol abgewogen und danach die gut verschlossenen Kolben in einem Thermostat von 50 ~ C auf Temperatur gebracht.
Nun stellten wir bei 500 C die folgenden drei Mischungen, deren Durchlaufszeit gemessen wurde, her. Die Mischungen I und III , welche die gleiche Zusammenstellung besitzen, sind jedoch in einer anderen Reib enfolge bereitet.

K R U Y T u. BUNGENBERG DE JON(3, ZUR KENNTNIS D.LYOPHILEN KOLLOtDE 41
I 60 g Alkohol 4-. A Sol + B Wasser ~:- A Wasser, II 60 g Alkoholq- A Wasser + B W a s s e r " A Sol,
III 60 g Alkohol + A Wasser @ ]3 Wasser -}- A Wasser.
Wenn wir die durch den Agar verursachte Dichtezunahme ver- naehl~issigen und immer mit dem gleichen Viskosimeter messen, so i'st die relative ViskositXt der Mischungen 1 und II im Verh~iltnis zu denl Dispersionsmittel III dem jeweiligen Quotienten der Durchlaufszeit I / I I I und I I / I I l gleich.
W~ihrend die Misehung I bei der Herstellung zeitweise den sus- pensoiden Zustand passiert und danach durch Hinzuftigen der 56,853 g Wasser wieder den emulsoiden Zustand annimmt, befindet die Mischung If sich w~hrend der ganzen Herstellung in letzterem Zustand.
Wir fanden als relative Viskosit~it im Verti~iltnis zu dem Disper- sionsmittel ftir die Mischung I den Wert 1,380 und ftir die Misehung II 1,382. Die Differenz dieser Werte ist gteieher Ordnung als die der Versuehsfebler. Wit wiederholten diese Versuehe sechs Stunden sp~iter und fanden als Mittel ftir I ~ 1,376 und fiir II = 1,377.
I n d i v i d u a l i t ~ t t der A g a r t e i l c h e n .
W~thrend wir erst meinten, aus obigen Versuchen aufierdem schlie- fien zu k6nnen, dab die Individualit~tt der Agarteilchen bei der Ent- w~isserung bestehen bleibt, und also die ursprtinglichen Agarteilcherf ultramikroskopisch dadureh sichtbar werden, haben spS~tere Versuche gezeigt, dab unter gewissen UmstS~nden gleichzeitig Aggregation statt- finden kann. Dieses behindert uns jedoch nicht, an der Meinung fest- zuhalten, dab die Ursache der auf viskosimetrischem Wege gefundenen Viskosit~itsabnahme in einer Entw~isserung der Teilehen zu suchen ist. Aggregate verursachen an sich bekanntlich schon eine 9)-Zunahme (infolge eingeschlossener H6hlungen), so dab die aut3ergewOhnliche
Senkung in ~ - - afor t ior i auf eine gleichzeitige starke Abnahme von 99
der ursprtinglichen Solteilchen schliegen l~fit.
U r s a c h e der D e b y d r a t a t i o n .
Vergleicht man Fig. 6 und Fig. 7 (S. 39), so zeigt sich, dab die EntwS~sserung der Agarteilchen gerade in dem Gebiet, wo das Viskosi- t~ttsmaximum des bin~ren Systems Alkohol--Wasser gelegen ist, schnell zunimmt.
Auch das bin~ire System Azeton--Wasser 1) zeigt ein Viskosit~its-
~) Jones u. Mahin, Zeitschr. f. physik. Chem. 89, 389 (1909).

4 2 I(OLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT /..-.-2
maximum, und es liegt auf der Hand, die Ursache for die Entw/~sserung der Teilehen hierauf zurtiekzufiJhren.
Jede einfache Erkl~trung l) tier ViskositS~tsmaxima wird jedoch durch die Tatsache erschwert, dab sowohl Wasser wie Alkohol au den stark assoziierten Fltissigkeiten geh6ren.
Vermuttich zeigen diese Maxima auf ein Vorhandensein stark kon- stituierter Hydrate hin, wodurch die Konzentration des ftir die Hydra- ration der Agarteilchen nStigen Wassers gerade bei der Konzentration dieser Maxima sehr gering wird.
A g a r s o l q - E s s i g s ~ u r e .
Das bin~tre System Essigsgure--Wasser zeigt ebenso wie Alkohol - - Wasser und Azeton--Wasser ein Maximum in der Viskosit~ttskurve. 2)
Ftigt man zu einem Agarsol der Konzentration 2/7 Proz. bei 50 o C eine ftinffache Menge Essigsgure, so flockt der Agar ziemlich bald aus. Das Entw/~sserungsmittel wirkt hier also ebenfalls als Elektrolyt, so dab nach der Dehydratat ion keine schtitzende Ladung mehr tibrig- bleibt.
Der AussalzungsprozeB.
Eine Anzahl Elektrolyte (NH 4F, K 2 S 04, Na 2 S 04, (NH4) 2 SOa, CdSO4, MgSO4, Na-Zitrat) besitzen die F~higkeit, ein Agarsol aus- zusalzen, w/~hrend dieses mit anderen Elektrolyten wie N H4C1 , K C N S, K J, MgCI~ sogar bei S~tttigung nicht gelingt.
Im Zusammenhang mit den oben erw~ihnten Erscheinungen nahmen wir an, dab der Aussalzungsprozel3 auf einer Dehydratat ion der schon bei viel kleineren Elektrolytkonzentrationen entladenen Teilehen beruhe. Unsere Vermutung wurde tatsgchlich durch folgende Viskosit~tsmes- sungen bestgtigt.
Als Elektrolyte wurden wegen ihrer neutralen Reaktion und gr6fieren L6slichkeit MgC12 und MgSO 4 gewS~hlt. Beide Pr~tparate waren yon K a h l b a u m , das MgSOa.wurde nochmals umkristallisiert.
Wir stellten die Untersuchungen bei 500 C an; die Mischungen wurden in einem Jena-MaBkolben yon 50 ccm auf die auf S. 18 be- schriebene Weise bereitet.
Die Konzentration der ursprtinglichen MgC12-L6sung wurde durch Titration des Cl-Ions, nach V o l h a r d , die der MgSQ-L6sung gravi- metrisch als BaSO 4 bestimmt.
1) Kremann , Die Eigerischaften der bingren Fliissigkeitsgemisehe (Stutt- gart 1916).
2) Dunstan , Zeitschr. f. phys. Chem. 49, 590 (1904).
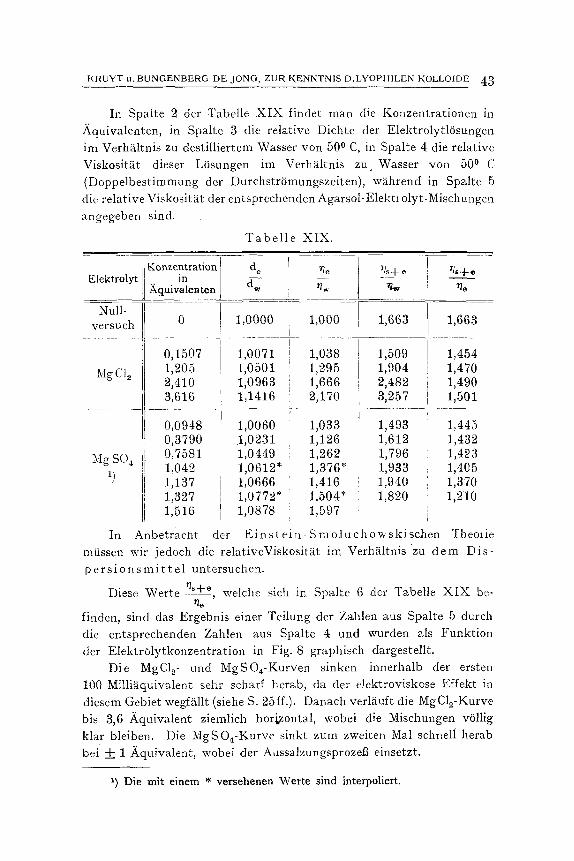
IKRUYT u. B U N G E N B E R G DE J O N G , ZUR KENNTNIS D . L Y O P H I L E N KOLLOIDE 43
111 Spalte 2 der Tabelle XIX findet man die Konzentrationen in Aquivalenten, in Spalte 3 die relative Dichte der ElektrolytlSsungen im Verhaltnis zu destilliertem Wasser von 500 C, in Spalte 4 die relative Viskosit~tt dieser LSsungen im Verhgltnis z u Wasser von 500 C (Doppelbestimmung der DurchstrSmungszeiten), w~thrend in Spalte 5 die relative ViskositSot der entsprechenden Agarsol-Elekt~ olyt-Mischungen angegeben sind.
Tabel le XIX.
Elektrolyt
Null-
K o n z e n t r a t i o n in
Aquivalenten
versuch 0
0,1507 MgC12 1,205
2,410 3,616
big SO~
dw ~ ~ "~
1,0000 1,000 1,663 1,663
0,0948 0,3790 0,7581 1,042 1,137 1,327 1,516
1,0071 1,0501 1,0963 1,1416
1,0060 1,0231 1,0449 1,0612" 1,0666 1,0772" 1,0878
1,038 1,295 1,666 2,170
1,033 1,126 1,262 1,376" 1,416 1.504" 1,597
1,509 1,454 1,904 1,470 2,482 1,490 3,257 1,501
1,493 1,445 1,612 1,432 1,796 1,423 1,933 1,405 1,940 1,370 1,820 1,210
In Anbetracnt der E ins t e ln -Smoluchowsk i schen Theotie mtissen wir jedoeh die relativeViskosit~tt ira Verh~tltnis zu dem Dis- p e r s i o n s m i t t e l untersuehen.
Diese Werte rls+e, welche sich il~ Spalte 6 dcr Tabetle XIX be- r/e
finden, sind das Ergebnis einer Teilung der Zahlen aus Spalte 5 durch die entsprechenden Zahlen aus Spalte 4 und wurden als Funktion der Elektrolytkonzentration in Fig. 8 graphisch dargestellt.
Die MgC12- und MgSO4-Kurven sinken innerhalb der ersten 100 Milli{iquivalent sehr scbarf herab, da der eJektroviskose Effekt in diesem Gebiet wegf~illt (siehe S. 25ff.). Danach verl~uft die MgCl2-Kurve bis 3,6 Jkquivalent ziemlich horizontal, wobei die Mischungen vSllig klar bleiben. Die MgSOa-Kurve sinkt zum zweiten Mal schneli herab bei 4- 1 Aquivalent, wobei der AassalzungsprozeB einsetzt.
x) Die mit einem * versehenen Werte sind interpoliert.
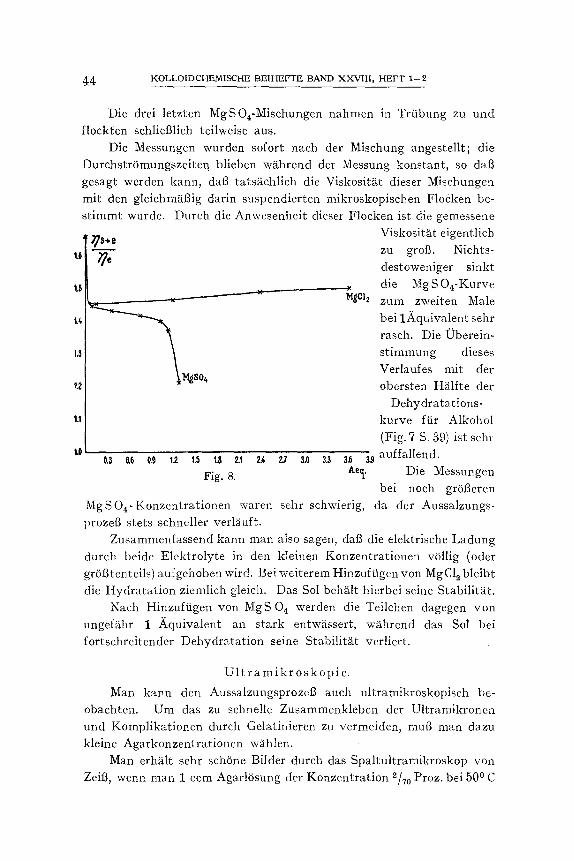
~ 4 KOLLOIDCHEMISCHE BEIHEFTE BAND XXVlII , HEFT 1--2
1.6
1.5
1.4
1.3
1.2
1.1
tO
Die drei letzten MgSO4-Mischungen nahmen in Triibung zu und flockten schlieglich teilweise aus.
Die Messungen wurden sofort nach der Mischung angestetlt; die Durchstr6mungszeiten. blieben wghrend der Messung konstant, so dab gesagt werden kann, dab tatsgchlich die Viskositgt dieser Mischungen mit den gleichm~igig darin suspendierten mikroskopiscben Flocken be- s t immt wurde. Durch die Anwesenheit dieser Flocken ist die gemessene
: ~'/$+ e
MFI2
0.3 0.6 0.9 t2 t.5 l a 2.t 2.4 z.7 ~0 33 a.6 a~ Fig. 8. Ae~[.
Viskosit~tt eigentlich zu grog. Nichts- destoweniger sinkt die MgSO4-Kurve zum zweiten Male bei 1Aquivalent sehr rasch. Die LTberein- st immung dieses Verlaufes mit der obersten Halfte der
Dehydratations- kurve fiir Alkohol (Fig. 7 S, 39) ist sehl auffallend.
Die Messungen bei noch gr6fieren
MgSO 4-Konzentrationen waren sehr schwierig, da der Aussalzungs- prozet3 stets schneller verlguft.
Zusammenfassend kann man also sagen, dab die elektrische Ladung durch beide Elektrolyte in den kleinen Konzentrationen v611ig (oder gr6Btenteils) aufgehoben wird. Bei weiterem Hinzuffigen yon MgC12 bleibt die Hydrata t ion ziemlich gleich. Das Sol behglt hierbei seine Stabilit~it.
Nach Hinzuftigen yon MgS 0 4 werden die Teilchen dagegen yon ungefghr 1 Aquivalent an stark entwgssert, wghrend das Sol bei fortschreitender Dehydratat ion seine StabilitS.t verliert.
U l t r a m i k r o s k o p i e .
Man kann den Aussalzungsprozet3 auch ultramikroskopisch be- obachten. Um das zu schnelle Zusammenkleben der Ultramikronen und Komplikationen durch Gelatinieren zu vermeiden, mug man dazu kleine Agarkonzentrationen wS~hlen.
Man erhglt sehr sch6ne Bilder durch das Spaltultramikroskop von Zeit3, wenn man 1 ccm Agarl6sung der Konzentration 2/7 0 Proz. bei 50 0 C

KRUYT u. BUNGENBERG DE JONG, ZUR t~ENNTNIS D.LYOPHILEN KOLLOIDE 4~
mit 30 ccm einer MgSO4-L6sung der Konzentration ~ )kquivalent pro Liter mischt, so dab die Agarkonzentration in der Mischung ungef~ihr 0,001 Proz. betr~igt. Nach Abktihlen auf Zimmertemperatur sieht man hell leuchtende Partikelchen mit starker B ro w n sch e r Bewegung, welche sich auf die Dauer zu mehrfachen Teilchen vereinigen.
Kontrollmischungen aus destilliertem Wasser oder konzentrierter MgCl~-LSsnng mit einer ebenfalls 0,001prozentigen Agarkonzentration zeigten unter dem Ultramikroskop nur einen diffusen Tyndallkegel.
S t a b i l i t ~ t t .
In nachstehender Tabelle ist eine Ubersicht tiber die Eigenschaften des emulsoiden Agarsols und der durch Hinzuftigen yon Alkohol oder Azeton davon abgeleiteten suspensoiden Sole gegeben.
T a b e l l e XX
glektrische
Eigenschaften
Eigenschaften, welche mit der Hydrata t ion zusammenhgngen.
Agar dispergiert in:
W a s s e r
Ladung negativ.
gntladung durch kleine Elektrolytkonzen-
trationen.
Valenz des Kations ent- scheidend.
Alkohol--Wasser (5 : 1) Azeton - Wasser ~5 : 1)
Ladung negativ.
Entladung durch kleine EIektrolyt- konaentrationen.
Valenz des Kations entscheidend.
Nach Entladung bleibt das Sol stabil.
Einige Elektrolyte sal- zerl das Sol bei h6heren
Konzentrationen aus.
Gelatiniert bei Abktih- lung, was Elektrolyte
lyotrop beeinflussen k6nnen.
Schwaches Tyndall, Ph~nomen.
rs-- 70 grofi.
Nach gnt ladung flockt das Sol aus.
Gelatiniert bei Ab- ktthlung nieht.
Starkes Tyndall- Ph~nomer!.
~ - - ~2 klein %
Befindet sich in dem emulsoiden Zustande.
Befir!det sich in dem suspensoiden
Zustande.

46 IKOLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
Die Hydratat ion wird haupts~ichlich durch die yon den disper- gierten Teilehen ausgehenclen Kr~iften bestimmt. Elektrolyte tiben indirekt einen lyotropen EinfluB auf die Hydratat ion aus, indem sie wahrscheinlich den inneren Zustand des Wassers ~indern.
Bei Fortfall der Hydratat ion gehen gleichzeitig alle emulsoiden Eigenschaften verloren, und man erh~tlt ein typisches Suspensoid zurtick, dessen Stabilitgt nur noch dutch die Ladung der Teilehen aufrecht- erhalten wird. Die elektrischen Eigenschaften sind vm und nach der Entwgsserung gleich, die Ladung ist kapillarelcktrischer Natur. Das emu!soide Agarsol ist also als ein Suspensoid mit hydratisierten Teilchen zu betrachten.
Die suspensoiden Sole werden ebenso wie das enmlsoide Sol durch kleine Elektrolytkonzentrationen entladen, erstere verlieren dabei ihre Stabilitgt, letztere nicht.
Das e m u l s o i d e A g a r s o l b e s i t z t a lso zwei S tab i l i tS~ts - f a k t o r e n : die e l e k t r i s c h e L a d u n g und die H y d r a t a t i o n der T e i l c h e n .
Jeder dieser beiden Faktoren ist an sich bedeutend genug, um die Stabilitgt allein aufrechterhalten zu kSnnen.
Ein effektiver Zusammenstot3 infolge der Brownschen Bewegung kann im ersteren Falle durch elektrische Ursache, im letzteren durch die Eigenschaften der Hydratationshtille verhindert werden.
Das emulsoide Agarsol kann also erst, wenn beide Faktoren entfernt sind, ausflocken.
Man kann diese Flockung, je nachdem die Stabilitatsfaktoren in verschiedener Reihenfolge entfernt werden, auf zweierlei Wegen er- zielen :
1. Durch Entladung mit kleinen Mengen Elektrolyt und darauf- folgender Entw~tsserung mit Alkohol, Azeton oder gr6Beren Mengen MgS04, K 2SO4, NaSO 4 usw.
9.. Durch Dehydratation mit Alkohol oder Azeton und darauffolgender Entladung mit kleinen Men gen Elektrolyt.
}rig. 9 zeigt eine schematische {)bersicht der einzelnen Zustands- gnderungen. In vertikaler Richtung sind Abgnderungen kapillar- elektrischer Natur, in horrizontaler Richtung die Anderungen in dem Hydratationszustand verzeichnet.
Die in diesem Kapitel er/3rterten Dehydratationserscheinungen sind nicht spezifisch ftir Agar. O. S c a r p a 1) land, dab Gelatine-und Gummi-
1) Scarpa, Koll.-Zeitschr. 15, 8 (1914).

t(RUYT u. BUNGENBERG DE JONG, ZUR KENNTNIS D. LYOPHILEN KOLLOIDE 47
arabicum-L6sungen in den reversibel suspensoiden Zustand tibergehen
kiJnnem Derselbe Autor untersuchte zasammen mit D ' A q u i n o z) den Ein-
flui3 yon Kolloiden auf die Viskositgt des bin{iren Systems Alkoho l - - Wasser. Sie fanden, dab die Viskositgt bei gr6Beren Alkoholkonzen-
t rat ionen nur wenig h/Sher ist als die der entsprechenden Alkohol-Wasser-
Mischung. Sie ftigten danr~ zu 2 Proz. Gummi arabicum hinter-
einander 10, 20, 30- -140 ccm absoluten Alkohol und mal3en die Viskosit~tt. Die Kolloidkon- ,.qeladene,~
gmulaoid zentrat ion selber ~tndert sich also, / und die gemessene Viskosit/it / 2~
Ng bezieht sich bei jeder Misehung ~ ] ,
sowohl auf eine andere Alkohol- ..~1-~ "~.l.e als auI eine andere Koiloid- v . ~
konzentrat ion. Die Berechnung einer De-
hydra ta t i anskurve im Sinne der Fig. 9 ist deshalb aus diesen Ver- ungeladenea suchen 1~icht m6glich.
100 ccm 1,5 Proz. Gelatine oder
+(~Deh~,d~la'h'on duc.ch.41kohol" ~ x ~ R e h ~ r e f a f / ~ n durch Wo~oer . + =
~uapens elgLdenea old
@ Oel~ldrm~ah'on dU.~kohoI 0
aus.r ElwulaoM TeJlchen " Fig. 9.
VI. A l lgeme ine Stabilit~ttstheorie der H y d r o s o l e .
Die in den Kapiteln tiber Agarsole entwickelte Stabilit~ttstheorie kann nun far alle Hydrosole erweitert werden, wenn wir voraussetzen, dab die disperse Phase stets von einer werm auch in ihren Dimensionen
wechselnden HydratationshOlle umgeben ist. In diesem Falle wird es dann m6glich, die Stabilit~tt der Suspensoide, Emulsoide und der {)bergangsstadien zwischen beiden yon einem GesichtspunkL aus zu
betrachten. Die t y p i s c h e n S u s p e n s o i d e wie Gold-2), Pt -a) und As2Sa-Sole 4)
besitzen gar keine oder nur sehr wenig hydratisierte Teilchen. Wird die Ladung durch' Hinzuftigen yon Elektrolyten zu einer kritischen
herabgesetzt, so flocken sie v611ig aus. Die Valenz des entgegengesetzt geladenen Ions allein best immt den Grenzwert der Elektrolyten. Eine
lyotrope Reihenfolge der Grenzwerte ist nicht oder kaum vorhanden.
1) S c a r p a u. D 'Aquino, Rendiconti Soc. Chim. ltaliana (II) 5, 370 (1913). a) Morawitz , Kolloidchem. Beih. 1, 301 (1910); v. Ga leck i , Zeitschr. f.
Elektrocnem. 14, 767 (1908). a) F r e u n d l i c h , Zeitschr. f. physik. Chem. 44, 129 (1903) ~) F r e u n d l i c h , loc. cit.

~ 8 KOLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
Unter den k o l l o i d e n H y d r o x y d e n bilden das Vanadium- und das Molybd~inpentoxydsoD) einen Obergang zu den Emulsoiden. Hier fiberwiegt meistens noch die elektrische Ladung als Stabilit~tsfaktor, so dab die Valenz des entgegengesetzt geladenen Ions deutlich in den Grenzwerten zu erkennen ist. Neben der elektrischen Ladung ist jedoch eine mehr oder weniger starke Hydrata t ion vorhanden, welche meistens nicht gentigt, um die Stabilit~it nach der Entladung aufrechtzuerhalten. Zugeftigte Elektrolyte bewirken also bei der Entladung ebenfalls eine fi~nderung in der Hydratat ion. Die Stabilit~tt wird gewahrt, solange eine gewisse Ladungs- und Hydratat ionsfunktion tiber einem bestimmten kritischen Wert bleibt. Die Grenzwerte haben also hier nicht mehr die gleiche Bedeutung wie bei den typischen Suspensoiden und bilden deut- lich lyotrope Reihen.
Im allgemeinen haben die t y p i s c h e n E m u l s o i d e stark hydra- tisierte Teilchen. Nach Entladung verlieren diese Sole 6fters ihre Stabilitlit noch nicht. Bei grSBeren Konzentrationen tr i t t Flockung erst dann ein, wenn die Hydrata t ion bis zu dem kritischen Wert herab- gesetzt wird.
Die Konzentrationen, bei welchen die einzelnen Elektrolyte aus- flocken (aussalzen), liegen also in lyotroper Reihenfolge, wS~hrend die Valenz des entgegengesetzt geladenen Ions in den Hintergrund ge- schoben wird.
VII . D a s k o l l o i d c h e m i s c h e V e r h a l t e n d e r E i w e i l 3 1 t l s u n g e n .
Nachdem unser Standpunkt tiber das Verhalten der Eiweit315sungen in der allgemeinen Einleitung schon kurz er6rtert wurde und danach unsere Versuche mit Agarsol besprochen sind, wollen wir nun in diesem Kapitel wieder auf die Eiwei1316sungen zurtickkommen und unsere Ansicht n~iher umschreiben.
Als Beispiel soll das Gelatinesol gew~hlt werden. Obwohl eine vollst~tndig systematische Untersuchung unter den Temperaturverh~lt- nissen, welche den wirklich reinen Solzustand e) bedingen, noch nicht vorliegt, darf man doch aus den zahlreichen in der Literatur verbreiteten
1) F reund l i ch u. Leonha rd t , Kolloidchem. Beih. 7, 172 (1915). 3) Davis u. Oakes ~Journ. Amer. Chem. Soc. 44, 464 (1922)] fanden, dab
die Viskositiitszunahme mit der Zeit, welche infolge des Gelatinierungsprozesses eintritt, nur unter 38,10 stattfindet. Ober dieser Temperatur ist also nur der reine Sohustand vorhanden. Tatsiichlich beobachtete einer von uns, dab asche- freie Gelatinesole sowohl bei dem isoelektrischen Punkt wie bei dem Viskositiits- maximum mit HC1 dem Gesetz yon Poiseui l le bei 40 o gehorchen; siehe H. G. B u n g e n b e r g de Jong, Rec. Trav. Chim. 48, 38 (1924).

I.~RUYT u. BUNGENBERG DE JONG,. ZUR KENNTNIS D. LYOPHILEN t'(OLLOIDE 4 9
Angaben schliegen, dab ebenso wie bei dem Agarsol auch hier zwei Stabilit~tsfaktoren, die kapillarelektrische Ladung und die Hydra- ration eine Rolle spielen.
Nur in einer Hinsicht ist das Verhalten der EiweiBsole kompli- zierter. W~hrend n. 1. die Konzentration der H-Ionen bei dem Agarsol keinen auffallenden EinfluB auf den kolloidchemischen Zustand aus- fibt, ist das variable PH ffir den Zustand der EiweiBsole yon der aller- grSt3ten Bedeutung.
Ftir dasAgarsol gentigt ein Schema vonvier Symbolzeichen (Fig. 9), um die verschieden extremen kolloidchemischen Zu- stgndezuumschreiben;umimallgemeinendasVer- ': I . ,6 O halten der EiweiBsole fibersehen zu kSnnen, muff . .... " dieses Schema auf zehn Zeichen erweitert werden. 1 1
Fig. 10 zeigt dieses Stabilit~itssehema, wel- - . ' - : ' - - ' . ~ " 2 , 7 . ' ~ .
ches nun an Hand der Literaturangaben betreffs -'., ~J~* ,,- ' . '~'.. des Gelatinesols n~ther beleuchtet werden soll. - " - - " -
Bei dem isoelektrischen Punkt (PI-I-~ 4,7) ] [ ist das Gelatinesol ungeladen. 1) Nichtsdesto- ,"g,: 8 @ weniger bleibt es dabei stabil (namentlich bei "-
L
Bedingungen, bei welchen man im strengsten 1 [ Sinne des Wortes yon dem reinen Solzustand _ -
, / ~O_ sprechen darf, d.h. oberhalb 380). -_~_-'~'F ' ,, 4 �9 ,9, '_ : .
Das Teilchen wird noch durch seine Wechsel- : . . . . - : ' : ' wirkung mit dem Dispersionsmittel, d. h. durch l l seine Hydrata t ion vor Ausflockung geschfitzt. - . . . . Dieser Zustand wird in unserem Schema durch Symbol 3 dargestellt.
Bei zunehmender Wasserstoffionenkonzen-
[ ~ }s ,____.1o
Fig. 10.
tration nimmt das Teilchen eine positive Ladung an (Symbol 2), welche nach Erreichung ihres maximalen Wertes bei weiterer Zunahme der Wasserstoffionenkonzentration wiederum abnimmt, um sich nach und nach dem durch Symbol 1 angedeuteten Zustand zu ng.hern.
Eine Entladung durch verhS~ltnismSs kleine Salzkonzentrationen kann nicht nur bei oben genanntem Maximum, sondern auch schon vor diesem erfolgen. Das entladene Gelatinesol ist aber noch immer stabil und mug also noch einen anderen Stabiliti~tsfaktor besitzem Dieser ist die Hydratat ion, was durch Symbol 3 dargestellt ist.
1) u. a. Pauli , Kolloidchemie der Eiweil~k6rper I (Dresden und Leipzig 1920); Loeb, Proteins and the theory of colloidal behavior. Mc Graw Hill. Publishing Co. London (199.2).

O0 KOLLOIDCHEMISCHE BEIHBFTE BAND XXVll l , HEFT 1--2
Das oben ftir die pn-Abnahme Gesagte gilt gleichfalls ftir die PH Zunahme, mit dem Unterschied, dab die kapillarelektrische Ladung jetzt das negative Zeichen tr~Lgt (Symbol 4 und 5):
In unserem Schema stellt also die linke Symbolreihe (1--5) die ftinf extremen kolloidchemischen ZustSmde, in welchen wfifirige Gelatine- sole bestehen k6nnen, dar (bei wechselndem PFI und bei wechselnden, aber relativ kleinen Konzentrationen neutraler ElektrolytlGsungen). Das Gelatinesol ist dabei stets stabil, weil mindestens ein Stabilit/~ts- faktor (die Hydra tation) erhalten bleibt.
Heben wir aber die Wechselwirkung zwischen Teilchen und Di- spersionsmittel z. B. durch Hinzuftigen einer reichlichen Alkoholmenge zum gr6flten Teile auf, so erhalten wir ZustSmde der dispersen Phase, ,~.elche dutch die reehte Symbolreihe (6--10) dargestellt sind.
Nur die Symbole 7m9 stellen, da sic noch einen Stabilit/~tsfaktor (die kapillarelektrische Ladung) ~ besitzen, stabile Solzust/inde dar. Isoelektrische Gelatine (Symbol 3) oder gentigend entladene Sole (Symbol 1 und 5) flocken nach der Hydratation aus, weil der Zustand der: TeiIchen nun der der Symbole 6, 8 und 10, bei weIchen der Stabili- t~tsfaktor fehlt, geworden ist.
Auch der Aussalzungsprozef3 patBt gut in dieses Schema. Das hin- zugeftigte Salz wirkt bei den ersten gegebenen Mengen in der Hauptsache entladend (z. B. 2 > 1) und bei gr6t3eren Konzentrationen in der Haupt- saehe 'dehydratisierend (z. B. 1 ~ 6), bei welch letzterer Zustands- /~nderung der letzte Stabilit/~tsfaktor verschwindet.
Beide Stabilit~tsfaktoren 5mBern sich ebenfalls in der Viskosit~t des Gelatinesols. Die Sole, deren Konditionen durch Symbol 1--5 charakterisiert werden, zeigen ebenso wie das w/~t3rige Agarsol einen
abnorm groBen Wert far die Formel % - ~o, was beweist, dab das Volum c ~/o
der dispersen Phase viel gr6ger ist als dasjenige des darin vorhandenen trockenen Gelatins, dab mit anderen Worten die Partikelchen die freie Beweglichkeit eines Teiis des Dispersionsmittels mit Beschlag belegen. Dieses kann teilweise stagnierendes, in den Kapillarh6hlungen des Partikelchens eingeschlossenes Wasser, zum~I'eil sicherlich Hydra- tationswasser sein.
Das geht schon aus der starken Abnahme genannter Formeln hervor, wenn durch Hinzuftigen eines Oberschusses an Alkohol die Hydratat ion zum gr6tBten Teil aufgehoben wird.
Durch eine derartige Dehydratation n~hert sieh die relative Vis- kositiit (im V6rh/~ltnis zum dazugeh6rigen Dispersionsmittel berechnet)

K R U Y T u. B U N G E N B E R G D E J O N G , ZUR K E N N T N I S D . L Y O P H I L E N KOLLO1DE 5 1
einem Weft, welchen wir bereits aus der E i n s t e i n s c h e n Formel er- warren konnten. 1)
Aus der E i n s t e i n - S m o l u c h o w s k i s c h e n Gleichung l~tfit sich schon feststellen, dab Sole mit den durch die Symbole 2 und 4 repra- sentierten Teilchen (respektive + u n d - - geladen) eiae hShere Viskosit~tt besitzen miissen wie 1, 3 und 5 (ungeladene Teilchen).
Infolge der Aufladung mit HC1 wird die Viskositgt also bis zu einem Maximum zunehmen. Der HC1 wird dann chemisch an die aus der TeilehenoberflS~che herausragenden N H 2-Gruppen des EiweiBteilchens gebunden und die dadurch entstandenen N HaC1-Gruppen bilden dann die Zentren der positiven kapillarelektrisehen Ladung. Wenn alle N H 2- Gruppen auf diese Weise umgesetzt sind, so wirkt neu hinzugesetzter
HCI in gleicher Weise entladend wie NaC1, b_;c~ USW.
Sehr wahrscheinlich superponieren sich die beiden Erscheinungen, so dab z. B. die HC1-Bindung bei dem Viskosit/~tsmaximum noch nicht vOilig abgesehlossen ist, wenn die kapillarelektrische Ladung schon an- gefangen hat. Das Viskositatsmaximum ist dann jener Zustand der dispersen Phase, bei welchem der viskosit/itserh61~ende EinfluB weiterer H C1-Bindung infolge Bildung neuer Zentren der kapillarelektrisehen Ladung gerade ebenso grog und dem viskositgtsherabsetzenden Einflug der extra hinzugeftigten Fraktion des HC1, welche nicht gebunden wird und daher enttadend auf die vorhandene kapillarelektrische Ladung wirkt, entgegengesetzt ist.
Auf diese Weise ist weiter zu sehen, dab H 2SO 4 mit ihrem viel stgrker entladenden zweiwertigen Anion ein Maximum gibt, welches viel tiefer liegt. ~) Das gleiche gilt far die negativ geladenen Sole von Ca (OH)2 und Ba (OH)2 im Verh~ltnis zu NaOH. a)
Der starke Einflug, welchen neutrale Salze auf die ViskositS.t haben, beruht dann an erster Stelle auf der Fortnahme des elektroviskosen Effektes. Da das Sol mit Symbol i positiv geladen ist, ist die Valenz des Anions an jener Seite des isoelektrischen Punktes der ausschlag- gebende Faktor. Bei dem Sol mit Symbol 4 ist dagegen die Valenz des Kations entscheidend.
1) Vgl. Messungen von Loeb, loc. cit. S. 280. Diese Messungen sind jedoch unter Gelatinierungstemperatur angestellt. Vgl. auch Die Messung von Casein von Bra i l s ford Rober tson , Journ. of Physical Chem. 15, 179 {1911), welcher jedoch nicht die Viskosit~t relativ im Verh~ltnis zu dem dazugehSrigen Dispersionsmittel ausdriickte, sondern eine Differenzkurve konstruiert hat.
2) Loeb , loc. cit. 3) L o e b, loc. cit.

5 2 KOLLOIDCHEMSCHE BEIHEFTE BAND XXVIII, HEFT 1--2
So land L o e b 1), dab NaC1, CaCI 2 und LaC1 abe i einem Gelatinesol von PH = 3 die gleiche Viskosit~Ltsabnahme verursachen, wenn man gleiche J~quivalentkonzentrationen miteinander vergleicht. Dagegen ist diese Viskosit~tsabnahme bei den Salzen K Cl, K 2 S 0 4 und K 4 Fe (CN)6 gr6Ber als mit der J~quivalentrechnung tibereinstimmt.
Diese ftir elektrokinetische Erscheinungen kennzeichnenden Regel- m~tt3igkeiten best~irken uns in unserer Uberzeugung, daft man bei allen Eiweigsolen und bei dem Gelatinesol im besonderen nicht mit ion- dispersen Systemen, sondern im Gegenteil mit kolloiddispersen Systemen zu tun hat, welche eine gentigende GrenzflS~ehenentwicklung gegentiber dem Dispersionsmittel entfalten, um die typischen kapillarelektrisehen Erseheinungen auftreten zu lassen.
Sogar L o e b, welcher sonst ein ausgesprochener Vork~impfer der erst- genannten Theorie ist, kann den Einflut3 neutraler Salze auf die ViskositS~t des Gelatinesols nicht erkl~tren ; er nimmt daher in dieser L6sung Struktur- elemente, d. h. kolloiddispergierte Materie an, zwischen deren Inhalt und der freien Fltissigkeit sich ein Donnan-Gle ichgewich t einstellen soll.
Diese Erklgrung ist jedoeh unn6tig kompliziert, da ja eine Be- ziehung zwischen Viskosittit und kapillarelektrischer Ladung direkt durch die S m o l u c h o w s k i - E i n s t e i n s c h e n Formel gegeben wird.
L o e b unterscheidet ferner zwischen Eiweifll0sungen des Gelatine- typus und des Albumintypus. Bei letzterem soil man mit einem rein iondispersen S3istem zu tun haben, wghrend ersterer nur das ,,colloidal behavior" dutch die Anwesenheit der Strukturelemente zeige. Als Argument ftihrt L o e b unter anderem an, dab die Viskositat yon Gela- tinel6sungen sehr viel h6here Werte zeigt, als aus der E i n s t e i n s c h e n Formel berechnet werden kann, dab dieser Unterschied bei Albumin jedoch nicht so grog ist.
Unserer Meinung nach bertihrt diese Beweisftihrung jedoch nicht den wichtigsten Punkt. Zum ersten ergaben verschiedene Diffusions- messungen, dab der Diameter der EiweiBteilchen, auch der des Albumin- typus stets einige/,b, betragt, d. h. dieses Teilchen ist (ganz davon ab- gesehen, ob dieses, chemisch gesprochen, nur ein Molektil ist, oder dagegen aus einem Aggregat mehrerer Eiweifimolekiile oder einem Assoziat verschiedenster niedriger molekularer Bausteine besteht) beztiglich seiner Dimensionen in das Gebiet der kolloiddispersen Systeme ein- zureihen, so dab also alle Eigenttimlichkeiten det GrenzflS~chenerschei- nungen, wie Adsorption, Hydrata t ion und kapillarelektrische Er- scheinungen zu erwarten sind.
1) Loeb, loc. cit.

KRUYT u. BUNGENBERO DE JONO, ZUR KENNTNI$ D. LYOPHILEN KOLLOIDE ~
Wenn wir im Auge behalten, dab sogar die EiweiBsole des Albumin- typus kolloide Systeme sind, welche alle Eigentfimlichkeiten des Agars und Gelatinesols aufweisen mtissen, dann zeigt es sich, dab die L o e b - sche Differenzierung der EiweiBsole in genannte zwei Klassen sich nicht auf eine Unterscheidung der iondispersen Systeme bezieht, in welehen wohl oder nicht tiberdies noch Strukturelemente vorhanden sein mtissen, sondern auf die Volumin/Ssit~tt der in den L/Ssungen an- wesenden dispersen Phase.
Die in der E i n s t e i n s c h e n Formel vorkommende Gr6Be go kann n~imlieh formell als aus drei einzelnen Volumina bestehend aufgefaBt werden:
I. das Volum der wasserfreien dispergierten Substanz selber; II. das Volum des Hydratationswassers;
I l l . das Volum des stagnierenden in eventuellen H6hlungen ein- gesehlossenen kapillaren Wassers.
Unserer Meinung nach ist die abnorme Viskositgtserh6hung bei Agar und dem Gelatinesol sicher r ~ t e n t e i l s dem unter I I I genannten Volum zuzuschreiben; d.h. die Teilchen in den Solen sind nicht kom- pakt, sondern schwammig gebaut und bestehen eventuell aus einzeln gebauten sekundaren Teilchen. Die giweiBsole, deren Viskositgt un- gefghr mit dem aus der Ei n s t e i n schen Formel berechneten Wert fiber- einstimmt, haben dagegen Teilchen, welche wohl kompakt gebaut sind, wobei also das stagnierend eingeschlossene Wasser mehr oder weniger (ehlt.
Wir sehen dann in der yon L o eb gemachten Unterseheidung nicht eine absolute, sondern nur eine relative Differenz, welche in dem Bau der Teilchen zu suchen ist.
Das Stabilitatsverhalten wird, wie immer der innere Bau des Teil- chens sein m/Sge, nur yon dem Zustand seiner Oberflgche, d. h. durch seine Hydratat ion und kapillarelektrische Ladung beherrscht sein.
Wir haben bei den bier oben beschriebenen Stabilitatsbetraeh- tungen vorausgesetzt, dab die Hydrata t ion unabhgngig yon der Ladung vor sich geht. Es sind jedoch Anweisungen dafOr vorhanden, dab dieses bei Gelatine nicht der Fall ist und dab die Hydra ta t ion yon dem iso- elektrischen Punkt sowohl bei der positiven Aufladung mit S~iuren wie bei der negativen mit Basen zu einem Maximum steigt, um danach wieder mit der Ladungsabnahme zu sinken.
Zieht man das oben fiber den Bau der Gelatineteilchen Gesagte in Betracht, so ist es noch sehr gut mtSglich, dab auch eine Vergr6fierung des stagnierend eingeschlossenen Wassers zu dem h~Sher gelegenen Viskosit~itsmaximum beitragen kann.

54= KOLLOIDCHEMISCHE BEIHEFTE BAND XXVIII, HEFT 1--2
Gleichfalls wS~re es nicht ausgeschlossen, dab die enorme Herab- setzung der relativen ViskositSJc des Agarsols durch Dehydratation mit Alkohol zum Teil auf einer Abnahm~,des eingeschlossenen Dispersions- mittels beruht. Wie dem auch sei, so sind unserer Meinung nach die Vergnderungen d e r Hydratat ion und der kapillarelektrischen Ladung stets primer; die eventuelle AbhSmgigkeit der t-Iydratation von der Ladung spielt keine Rolle bei den Stabilit~tsbetrachtungen des Gelatine- sols, da die tibrigbleibende Hydratat ion in dem ungeladenen Zustand an sich schon groB genug ist, um die Stabilitgt als unabhgngiger Faktor aufrechtzuerhalten.
Wir haben darum in unserem Schema die eventuellen Intensitgts- ver~inderungen der Hydratation in Abh~tngigkeit der Ladung nicht in Betracht gezogen. Ebensowenig wurde der eventuelle schwammige Bau der Gelatineteilchen zum Ausdruck gebracht.
Das Schema ist unserer Meinung nach also auch auf Eiweit3sole des Albumintypus, in welchem die Teilchen vermutlich kompakt gebaut sind, anzuwenden.
Wo. P a u l g ) unterscheidet die EiweiBe in isostabile (z. B. Gelatine, Albumin) und isolabile (z. B. Kasein). Wit kSnnen diese Unterscheidung v511ig best ~ttigen.
Bei den isostabilen gentigt die Hydratation bei Abwesenheit der Ladung noeh, um als unabhgngiger Stabilit~ttsfaktor auftreten zu kSnnen, bei den isolabilen ist sie dazu nicht imstande.
Erst bei einer gewissen positiven oder negativen Aufladung gentigt die Kombination yon Ladung und Hydratat ion gerade, um die Stabilitgt zu erhalten. ~)
Wir verweisen jedoeh ftir obengenannte Ph~tnomene, welche iibrigens vSllig mit der hier umschriebenen Theorie der Eiweil3sole tiberein- stimmen, auf eine weitere Abhandlung yon H. R. K r u y t und H. Lier~ welche sich speziell mit dem Kaseinsol besch~tftigen wird.
1) Pauli, loc. cit. 2) H. R. Kruyt, Koll.-Zeitschr. 31, 338 ~,1922).