Embed Size (px)
Citation preview

Z. anorg. allg. Chem. 584 (1990) 12-20 J. A. Biirtli, Leipzig
Zur Synthese der Heptaphospha-nortricyclane R3P, R = Et, ii-Pr, n-Bu, i-Bu, SiH,Me, SIH, ,Et,P-SiMe,
G. FRITZ" und H.-W. SCHNEIDER
K a r l s r u h e , Institut fur Anorganischc Chemie der Universitiit
I n h a l t s u b e r s i c h t . Derpriiparative Zugang zu dcnVerbindungenEt,P, 1, i-Pr,P, 2, n-Bu3P,3, i-Bu,P, 4, (H,Si),P, 5 , (MeH,Si),P, 6, (Et,P-SiMe,),P, 7 durch Umsetzung von Li,P, . 3 DME mit EtBr bzw. i-PrBr, n-BuBr, i-BuBr, H,SiI, MeSiH,Br, Et,P-SiMe,Cl wird beschrieben. Dic Verbin- dungcn 1 bis 4 sind bei 20°C gelb-griine, viskosc Fliissigkeiten (Viskositiit mit der GriiDe von R steigend), gut loslich in Etliern und unpolaren Losungsmitteln. (H,Si),P, 5 bildet farblose Kristalle, die sic11 (wie ihre Losungen) bei LichteinfluU zersetzen, ahnlich wie die von 6. 6 und 7 werden auch quantitativ gebildct, sind aber nicht unzersetzt isolierbar. Wahrend die Bildung von 1 iiber die rote Zwischenstufe Li,EtP, quantitativ erfolgt, ist bei der Umsetzung zum i-Pr,P, das Li(i-Pr)2P7 faDbar, auf das die Bildung P-reicherer Phosphane wie i-Pr,P, zuruckgefuhrt wird. Li,P, laat sich nicht mit (Me,C),SiBr zum [(Me,C),Si],P, umsetzcn. Das Verhaltnis R,P,(sym.) : (asym.) - beim Et,P, I: 3 (wie beim Me,P,) - verschiebt sich rnit groDerem R zugunsten des sym.-Isomers. Im (H,Si),P, sind - wic im (Me,Si),P, - keine Anzeichen fur die Bildung des asym-Isomers vorhanden.
Concerning tho Synthesis of the Heptaphospha-nortricyclanos R3P7 R = Et, i-Pr, n-Bu, i-Bu, SiHgMe, SiHg, EtgP -SiMoB
Abst rac t . The preparative access to the compounds Et,P, 1, i-Pr,P, 2, n-Bu,P, 3, i-Bu,P, 4, (H,Si),P, 5, (MeH,Si),P, 6, and (Et,P-SiMe,),P, 7 through the reaction of Li,P, . 3 DME with either EtBr, i-PrBr, n-BuBr, H,SiI, MeSiH,Br or Et,P-SiMe,Cl, respectively, is described. At 20°C the compounds 1 to 4 are yellow-greenish, viscid liquids (viscosity increases with the size of R), which are well soluble in ethers and non-polar solvents. 5 forms colorless crystals, which (similar to those of 6) decompose, when exposed to sunlight. 6 and 7 are generated quantitatively, these com- pounds, however, cannot be isolated undecomposed. While the formation of 1 occurs quantitatively via the red intermediate Li,EtP,, it is possible to isolate Li(i-Pr),P, from the residue of the reaction leading to i-Pr3P7. This Li-phosphide is said to cause the formation of higher, P-rich phosphanes like i-Pr3P9. Treatment of Li,P, with (Me,C),SiBr does not yield [(Me,C),Si],P,. The ratio R,P, (sym.): R,P,(asym.) - being 1: 3 in Et,P, or Me,P,-shifts with increasing size of R, favouring the symmetrical isomer. There are no hints for the formation of an asymmetrical isomer in (H3Si)3P7 - as already known from (Me,Si),P,, where an asymmetric isomer does not exist either.
Einleitung Aus weil3em Phosphor bildet sich durch Umsetzung mit Na/K und anschlie-
Bender Reaktion mit Me,SiCl Verbindung a [I], mit Me,SiCl, Verbindung b [a]. Die am der Chemie der Silylphosphane bekannten Reaktionen der Si-P-Bindung [ 3 ] lassen sich nur bedingt zur Bildung von Derivaten von a einsetzen, weil die

G. FRITZ u. H.-W. SCHNEIDER, Synthese der Heptaphospha-nortricyclane 13
Spaltung der Si-P-Bindung in a leicht unter Veranderung des P,-Gerustes ver- lauft. Die Bildung von Li,P, [4] und ihr leichter Zugang durch Umsetzung von weirjem Phosphor mit LiMe [5] bietet Moglichkeiten zur Derivatisierung des P,-Gerustes. Zwar 1aBt sich Li,P, rnit einem UberschuB an Me,SiCl problemlos in (Me,Si),P, uberfuhren [4], jedoch wird bei der Umsetzung mit einer geringercn Menge des Halogensilans (entsprechend der Stochiometrie) die quantitative Ab- scheidung von Li,P1, aus der Reaktionslosung beobachtet. Es interessiert deshalb neben dem komplexchemischen Verhalten der Heptaphospha-nortricyclane [6] der EinfluB der Substituenten auf das P,-Gerust.
a b
Ergebnisse der Untersuchung 1. Alkylsubstituierte Heptaphospha-nortricyclane
(Me,C),P, (bisher nicht beschrieben) als C-analoges zum (Me,Si),P, und das (H,Si)3P, als Xi-analoges zum Me,P, von Interesse. Das Me,P, [4] erwies sich aufgrund seiner eingeschrankten Loslichkeit fur derartige Untersuchungen als weniger geeignet, so daB die Darstellung von Et,P, und i-Pr,P, u. a. wegen der zu erwarteuden besseren Loslichkeit angestrebt wurde.
Die Natur der Substituenten R bestimmt in nortricyclischen R,P,-Systemen nicht nur die chemischen Verschiebungen und das Aufspaltungsmuster, sondern auch die Zahl der Signalgruppen in den 31P-NMR-Spektren. Ursache dafur ist die mogliche Anordnung der Substituenten an den Brucken-P-Atomen (P,) in Bezug auf die dreizahlige Achse des Grundgerustes ; symmetrisches Isomer (alle drei Substituenten im Uhrzeigersinn) , asymmetrisches Isomer (nur zwei Substituenten in Richtung des Uhrzeigers, der Dritte in entgegengesetzter Richtung) . Bei den Substituenten Me,&-, Ph,Si- u. 8. wird ausschlierjlich das symmetrische Isomer gebildet, das im 31P-NMR-Spektrum wegen der magnetischen Aquivalenz der P,-Atome erwartungsgemaB mit drei komplexen Signalen in Erscheinung tritt. Fur Substituenten H und Me wird die Bildung eines Isomerengemisches beob- achtet ; in den 3lP-NMR-Spektren solcher Reaktionslosungen sind jeweils zehn teilweise iiberlagerte Signale zu sehen, wobei drei dem symmetrischen (s) und sieben dem asymmetrischen (as) Isomer entsprechen. Fur kleine Substituenten R (H, Me) tritt nach statistischen uberlegungen bei der Bildung von R,P, ein Iso- merenverhaltnis R,P, (s) : R3P7 (as) von 1 : 3 auf, wie von BAUDLER experimentell bestatigt wurde [7, 81. Dieses Verhaltnis verschiebt sich mit GroBe des Substi- tueriten R (i-Pr, n-Bu, i-Bu) zugunsten des sterisch gunstigeren (s) Isomers, wie die folgenden Ergebnisse zeigen.
Im Hinblick auf die Untersuchungen zur Komplexbildung [ 61 ist das
1 4 Z. anorg. allg. Chem. 584 (1990)
1.1. Die Umsetzungen von Li,P, m i t Alkylha logeniden
Die Umsetzungen sollten verlaufen nach Lisp, + 3 RX + R,P, + 3 LiX (X = C1, Br). (1)
Die temperaturabhangige Bindungsfluktuation des Li,P, wirkt sich nicht auf diese Umsetzungen aus (Reaktionen Fei - 7 O O C und 23OC fiihren zu ahnlichen Ergebnissen) .
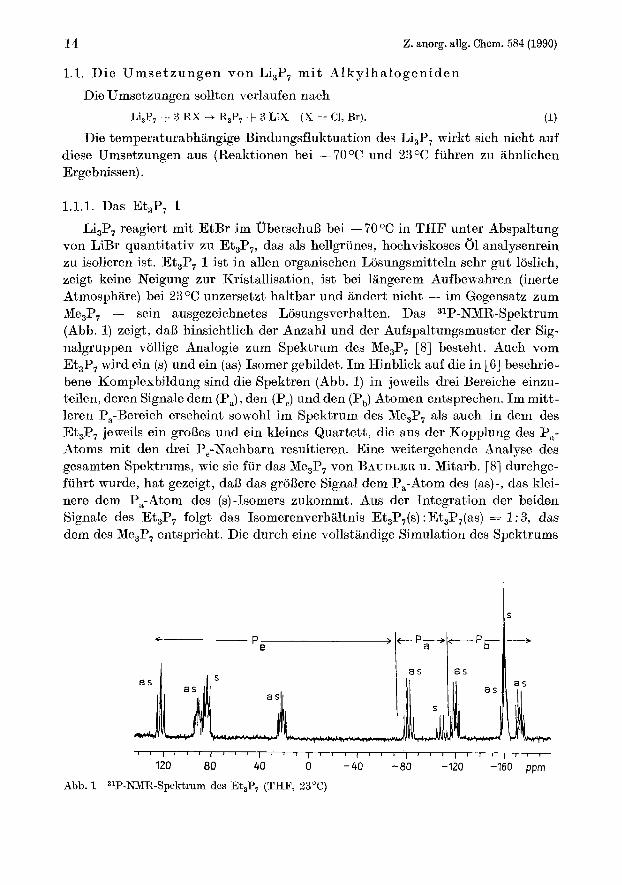
1.1.1. Das Et,P, 1 Li,P, reagiert mit EtBr im Uberschuli bei - 7 O O C in THF unter Abspaltung
von LiBr quantitativ zu Et,P,, das als hellgrunes, hochviskoses 01 analysenrein zu isolieren ist. Et,P, 1 ist in allen organischen Liisungsmitteln sehr gut loslich, zeigt keine Neigung zur Kristallisation, ist bei langerem Aufbewahren (inerte Atmosphiire) bei 23 O C unzersetzt haltbar und andert nicht - im Gegensatz zum Me,P, - sein ausgezeichnetes Losungsverhalten. Das 31P-NMR-Spektrum (Abb. 1) zeigt, daB hinsichtlich der Anzahl und der Aufspaltungsmuster der Sig- nalgruppen vollige Analogie zum Spektrum des Me3P, [8] besteht. Auch vom Et,P, wird ein (s) und ein (as) Isomer gebildet. Im Hinblick auf die in [6] beschrie- bene Komplexbildung sind die Spektren (Abb. 1) in jeweils drei Bereiche einzu- teilen, deren Signale dem (Pa), den (P,) und den (Pb) Atomen entsprechen. Im mitt- leren Pa-Bereich erscheint sowohl im Spektrum des Me,P, als auch in dem des Et,P, jeweils ein grolies und ein kleines Quartett, die aus der Kopplung des Pa- Atoms mit den drei P,-Nachbarn resultieren. Eine weitergehende Analyse des gesamten Spektrums, wie sie fur das Me3P, von BAUDLER u. Mitarb. [S] durchge- fuhrt wurde, hat gezeigt, dali das groliere Signal dem Pa-Atom des (as)-, das klei- nere dem Pa-Atom des (s)-Isomers zukommt. Aus der Integration der beiden Signale des Et,P, folgt das Isomerenverhaltnis Et,P,(s) : Et3P7(as) = 1 : 3, das dem des Me,P, entspricht. Die durch eine vollstandige Simulation des Spektrums
Abb. 1 31P-NMR-Spektrum des Et,P, (THF, 23°C)
G. FRITZ u. H.-W. SCHNEIDER, Synthese der Heptaphospha-nortricyclane 15
von Me,P, ermittelten Zuordnungen der P,- und P,-Signale [8], die aufgrund ihrer Komplexitat und teilweisen Uberlagerung einer naheren Interpretation nicht direkt zuganglich waren, sind sinngemaB auf das Spektrum des Et,P, uber- tragen worden. Aus einem Vergleich der chemischen Verschiebungen beider Ver- bindungen geht hervor, daB die Pa- und P,-Signale des Et,P, nach hoherem Feld, die P,-Signale nach tieferem Feld verschoben sind.
1.1.2. Die Umsetzung von Li,P, mit i-PrBr
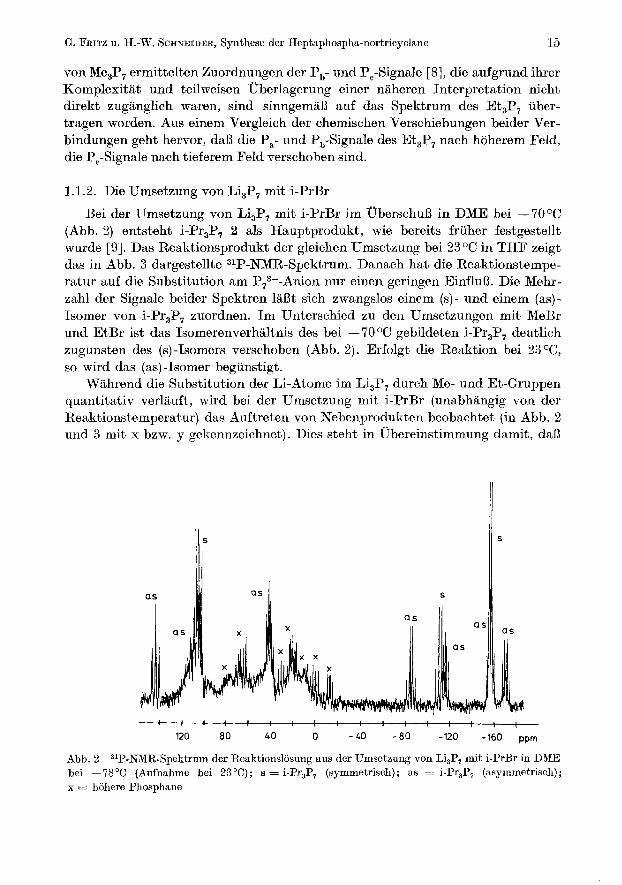
Bei der Umsetzung von Li,P, mit i-PrBr im UberschuB in DME bei - 70 OC (Ahb. 2) entsteht i-Pr,P, 2 als Hauptprodukt, wie bereits friiher festgestellt wurde [9]. Das Reaktionsprodukt der gleichen Umsetzung bei 23OC in THF zeigt das in Abb. 3 dargestellte 31P-NMR-Spektrum. Danach hat die Reaktionstempe- ratur auf die Substitution am P,,--Anion nur einen geringen EinfluB. Die Mehr- zahl der Signale beider Spektren laBt sich zwangslos einem (s)- und einem (as)- Isomer von i-Pr,P, zuordnen. Im Unterschied zu den Umsetzungen mit MeBr und EtBr ist das Isomerenverhiiltnis des bei - 70 O C gebildeten i-Pr,P, deutlich zugunsten des (s)-Isomcrs verschoben (Abb. 2). Erfolgt die Reuktion bei 23 O C ,
so wird das (as)-Isomer begunstigt. Wahrend die Substitution der Li-Atorne im Ei,P, durch Me- und Et-Gruppen
quantitativ verlauft, wird bei der Umsetzung mit i-PrBr (unabhangig von der Reaktionstemperatur) das Auftreten von Nebenprodukten beobachtet (in Abb. 2 und 3 mit x bzw. y gekennzeichnet). Dies steht in Ubereinstimmung damit, daB
120 80 40 0 -40 -80 -120 -160 ppm
Abb. 2 31P-NMR-Spektrum der Reaktionslosung aus dcr Umsctzung von Li,P, mit i-PrBr in DME bei -78°C (Aufnahme bei 23°C); s = i-Pr3P, (symmetrisch); as = i-Pr3P, (asymmetrisch); x = hohere Phosphane
16 Z. anorg. allg. Chem. 584 (1990)
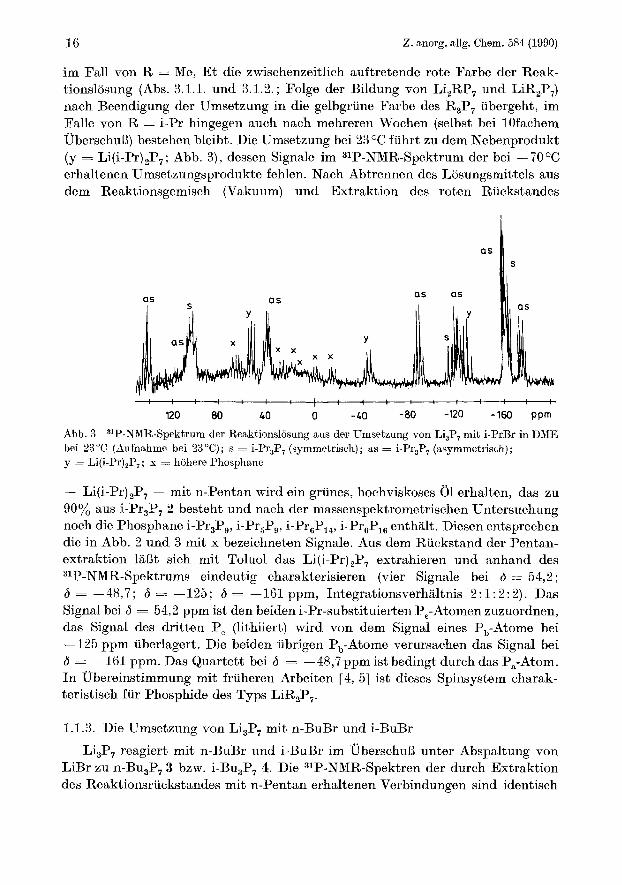
im Fall von R = Me, Et die zwischenzeitlich auftretende rote Farbe der Reak- tionslosung (Abs. 3.1.1. und 3.1.2.; Folge der Bildung von Li,RP, und LiR,P,) nach Beendigung der Umsetzung in die gelbgriine Farbe des R3P, ubergeht, im Falle von R = i-Pr hingegen auch nach mehreren Wochen (selbst bei lOfachem UberschuB) bestehen bleibt. Die Umsetzung bei 23 O C fiihrt zu dem Nebenprodukt (y = Li(i-Pr) 2P7 ; Abb. 3), dessen Signale im 31P-NMR-Spektrum der bei - 70 O C
erhaltenen Umsetzungsprodukte fehlen. Nach Abtrennen des Liisungsmittels aus dem Reaktionsgemisch (Vakuum) und Extraktion des roten Ruckstandes
l , . * . , . n c ~ ~ I ~ - . . ' ' ' ' ' ~
120 80 40 0 -LO -80 -120 -160 ppm
Abb. 3 bei 23°C (Aufnahmo bei 23°C); s = i-Pr,P, (symmetrisch); as = i-Pr,P, (asymmetrisch); y = Li(i-Pr)2P,; x = hohere Phosphane
31P-NMR-Spektrum der Reaktionslosung aus der Umsetzung von Li,P, mit i-PrBr in DME
- Li(i-Pr)2P7 - mit n-Pentan wird ein griines, hochviskoses 01 erhalten, das zu 90% aus i-Pr,P, 2 besteht und nach der massenspektrometrischen Untersuchung noch die Phosphane i-Pr3Pg, i-Pr5Pg, i-Pr6P14, i-Pr6P16 enthalt. Diesen entsprechen die in Abb. 2 und 3 mit x bezeichneten Signale. Aus dem Riickstand der Pentan- extraktion la& sich mit Toluol das Li(i-Pr)2P, extrahieren und anhand des 31P-NMR-Spektrums eindeutig charakterisieren (vier Signale bei 6 = 54,2 ; 6 = -48,7; 6 = -125; 6 = -161 ppm, Integrationsverhaltnis 2:1:2:2). Das Signal bei 6 = 54,2 pprn ist den beiden i-Pr-substituierten P,-Atomen zuzuordnen, das Signal des dritten P, (lithiiert) wird von dem Signal eines P,-Atome bei -125 ppm uberlagert. Die beiden iibrigen P,-Atome verursachen das Signal bei 6 = -161 ppm. Das Quartett bei 6 = -48,7 ppm ist bedingt durch das Pa-Atom. In Ubereinstimmung mit friiheren Arbeiten [4, 51 ist dieses Spinsystem charak- teristisch fur Phosphide des Typs LiR2P,.
1.1.3. Die Umsetzung von Li,P, mit n-BuBr und i-BuBr
Li,P, reagiert mit n-BuBr und i-BuBr im UberschuB unter Abspaltung von LiBr zu n-Bu,P, 3 bzw. i-Bu,P, 4. Die 3lP-NMR-Spektren der durch Extraktion des Reaktionsriickstandes mit n-Pentan erhaltenen Verbindungen sind identisch

G. FRITZ u. H.-W. SCHNEIDER, Syntheso der Heptaphospha-nortricyclane 17
mit den Spektren der Reaktionslosungen. Die im Verlauf der Umsetzung auf- tretende rote Farbe der Losung (Zwischenstufen Li,RP, bzw. LiR,P,) , geht nach Beendigung der Umsetzung in die gelbgrune Farbe des n-Bu,P, bzw. i-Bu,P, uber. Dariach ist die Reaktivitat der Phosphide Li,R,,P, (n = 1, 2 ) fur R = n-Bu, i-Bu groBer als die fur R = i-Pr. Die in beiden Umsetzungen auftretenden Verun- reinigungen (sichtbar in den ,lP-NMR-Spektren) sind voraussichtlich Umlage- rungsprodukte der teillithierten Phosphide. Ein Vergleich der ,lP-NMR-Spektren des n-Bu,P, bzw. i-Bu,P, mit dem des Et,P, zeigt, daB bei der Substitution des P,3--Anions durch sterisch anspruchsvollere Butylreste die sterisch gunstigere symmetrische Anordnung an den P,-Atomen zunehmend bevorzugt wird. Bei der Bilclung des i-Bu3P, steigt der Anteil des (s)-Isomers auf 50%.
2. IJmsetzung yon LiZP7 mit Halogensilanen
In der Reihe (H,Si),P,, (Me,Si),P,, (Ph3Si),P, [lo], [(Me,C),Si],P, nimmt der Raumanspruch der Substituenten am Silicium zu. Der gleichzeitig steigende +I- Effekt sollte eine nbertragung negativer Ladung vom Alkylsubstituenten uber das Si-Atom auf das benachbarte Brucken-Phosphoratom bewirken und dessen Basizitat erhohen. WSihrend die elektronischen Verhaltnisse eine Komplexbildung begunstigen sollten, stehen dem die sterischen entgegen. Die Abwagung der steri- schen und elektronischen Einflusse in der Reihe (R3Si),P7 bedingt das Interesse an ihrer Synthese.
2.1. Das (H3Si),P7 5 Die Umsetzung von Li,P, mit H,SiI bei - 7 O O C in Toluol fuhrt zum
(H,Si),P,, wie aus der 31P-NMR-Untersuchung zu erkennen ist [9]. Es gelang nun erstmals, (H,Si),P7 in Form weilJer Kristalle und in guter Ausbeute zu isolieren. Das 31P-NMR-Spektrum des isolierten (H,Si) ,P7 ist identisch rnit dem Spektrum der Reaktionslosung. Hinsichtlich der chemischen Verschiebung und des Auf- spaltungsmusters der drei Signalgruppen entspricht es den Spektren der bereits bekannten Derivate (R,Si) ,P7. Der im Vergleich zur Me,Si-Gruppe sterisch weit weniger anspruchsvolle H,Si-Rest ordnet sich in Bezug auf die dreizahlige Achse des P,-Gerustes ausschlieBlich symmetrisch an. Im 31P-NMR-Spektrum der Reak- tionslosung sind keine Anzeichen fur die Bildung eines (as) -Isomers. Das (1H)Wi- NMR-Spektrum zeigt bei 6 = -39,2 ppm (Standard OMCT) fur die drei magne- tisch aquivalenten Si-Atome ein Dublett mit der Kopplungskonstante Jsi,p =
42 Hz. Das im Vergleich zum (Me,&) ,P7 erheblich reaktionsfahigere (H,Si),P, ist
in Toluol gut loslich, wird aber in THF spontan unter Abbau des P,-Gerustes zersetzt. Aus der toluolischen Losung scheidet sich bei 23 O C unter LichteinfluB ein gelber Feststoff ab. Die unter LichtausschluB dargestellten farblosen Kristalle des (H,Si),P werden unter der Einwirkung von Sonnenlicht an der Oberflache gelb, womit sich die Loslichkeit zusehends verschlechtert.

18 Z. anorg. allg. Chem. 584 (1990)
2.2. (MeH,Si),P, 6 Li,P, reagiert rnit MeH,SiBr im UberschuB bei - 70 OC in Toluol quantitativ
zu (MeH,Si) ,P7, das anhand des 31P-NMR-Spektrums identifiziert wurde. Der EinfluS der Methylgruppe auf die chemische Verschiebung macht sich in einer - bezogen auf die Signale des (H,Si),P, - geringfiigigen Verlagerung des Pe- Signals nach Tieffeld und der Pa- und P,-Signale nach Hochfeld bemerkbar. Die Losungen von (MeH,Si) ,P7 in Toluol zersetzen sich unter LichteinfluB. LiBr- freie, frisch bereitete Toluollijsungen von (MeH,Si),P, lassen sich als Stamm- losungen fur weitere Umsetzungen einsetzen.
2.3. Die Umsetzung von Li,P, rnit (Me,C),XiBr
Wahrend Ph,SiBr mit Li,P, unter Bildung von (Ph,Si) ,P7 reagiert [ 101, erfolgt die entsprechende Umsetzung rnit (Me,C) ,SiBr weder bei - 70 O C noch bei 23 OC. Erst beim Kochen der Reaktionslosung am RuckfluB uber mehrere Stunden zeigt ihr NMR-Spektrum Vertinderungen, die auf die Bildung von Li2[ (Me,C) Si]P, hin- weisen, dessen quantitative Bildung aber aufgrund der Zersetzung des Li,P, unter den gegebenen Bedingungen nicht erreicht wird.
2.4. Das (Et,PSiMe,),P, 7 Li,P, reagiert mit Et,PSiMe,Cl im UberschuIj bei - 70 O C in Toluol quantitativ
zu (Et,PSiMe,),P,. Die Integration der im 31P-NMR-Spektrum auftretenden Signale bei 6 = - 1, - 75, -96 und 156 ppm ergibt das Verhaltnis 3 : 3 : 1 : 3. Die Multipletts bei 6 = -1, -96 und -156 ppm sind die fur das (s)-Isomer einer (R,Si) ,P,-Verbindung charakteristischen Pe-, Pa- bzw. P,-Signale, das Singulett bei 6 = -75 ppm wird von den drei exocyclischen P-Atomen verursacht. Das schrittweise Einengen und Abkuhlen der Reaktionslosung auf - 30 OC fuhrte nicht zur Abscheidung von Kristallen der Verbindung 7. Beim Versuch der Ab- trennung von 7 aus dem Reaktionsriickstand durch Sublimation (10-6 Ton/ 40 "C) tritt Zersetzung ein.
3. Experimentelle Einzelheiten
3.1. Die Umsetzung von Li,P, - 3 DME rnit Alkylhalogeniden
3.1.1. Darstellung von Et,P, 1, n-Bu,P, 3, i-Bu,P, 4. IJ,P, wird in THF gelost, die klare, gelbe Losung auf -70°C gekuhlt (wobei der grol3te Teil des Lisp, ausfallt; rein woil3) und zu dieser Sus- pension tropfenweise ein ifberschul3 des unverdunnten Alkylhalogenids innerhalb 1 h zugesetzt. Beim Molverhaltnis 1: 1 ist die nun homogene Reaktionslosung intensiv rot und wird beim Erreichen des Molverhaltnisses 1 : 3 gelb-grun (Farbe des Produktes R,P,). Zur Vervollstandigung der Reoktion wird noch 4 h bei -70 bis -60°C geruhrt. Dabei scheidet sioh ein Teil des gebildeten LiBr kristallin ab. h'rch dem Erwiirrnen des Reaktionsgemisches auf 20 "C wird das Losungsmittel bei Unterdruck abdestilliert, der gelbgrune, hochviskose Reilktiorisruckstand viermal rnit je 60 ml n-Pentan extra- Iiiert und das verbleibende LiBr bei der letzten Extraktion uber cine G4-Fritte abfiltriert. Die Ex-

G. FRITZ u. H.-W. SCHNEIDER, Synthese der Hcptaphospha-nortricyclane 19
trakte werden vereinigt und das Pentan bei Unterdruck vollstandig abdestilliert. Es verbleibt ein gelb-grunes, hochviskoses 01. Die so erhaltenen Verbindungen 1, 3, 4 sind analysenrein (Kontrolle durch 31P-NMR-Spektren bzw. massenspektrometrische Untersuchung) und losen sich gut in THF, Toluol und uupolaren Kohlenwasserstoffen. Aus keiner dieser Losungen wird die Abscheidung der kristallinen Verbindungcn erreicht. Typischer Ansatz: 10,l g (19,9 mmol) Li,P, . 3 DNE; 5 ml = 7,3 g (67 mmol) EtBr in 100 ml THF. Ausbeute 5,7 g (18,9 mmol) Et,P, (95%).
3.1.2. Darstellung von i-Pr,P, 2 Zu einer Losung von Li,P, in THF wird bei 20°C unter Ruhren unverdiinntes 2-Brom-propan im UberschuR zugetropft. Da die Reaktion exotherm verlauft, sol1 die Zugebe langsam erfolgen, um ein Aufheizen des Reaktionsgemisches zu vermeiden. Die Reaktions- losung nimmt eine intensiv rote Farbe an, die aach bei einem tfberschuB an 2-Brom-propan bestehen bleibt. Nach beendeter Zugabe wird das THF im Vakuum vollstandig abkondensiert, der rote Ruckstand viermal mit 50 ml n-Pentan extrahiert und bei der letzten Extraktion uber eine G4-Fritte filtriert. Aus den vereinigten Extrakten wird das Losungsmittel vollstandig abkondensiert. Es ver- bleibt i-Pr,P, als gelb-griines, zahes 01, das nur geringfugig durch hohere Phosphane verunreinigt ist. Das Reaktionsprodukt ist in allen organischen Losungsmitteln gut Ioslich, laBt sich aber nicht kristallin erhalten. Typischer Ansatz: 10 g (19,7 mmol) Li,P, . 3 DME; 6 ml = 7,9 g (64,l mmol) i-PrBr; 100 ml THF. Ausbeute 4,2 g (12,2 mmol) i-Pr,P, (62%).
Durch erneute Extraktion des Reaktionsriickstandes rnit Toluol ist die rote Komponente [das reine Li(i-Pr)zP7] quantitativ vom LiBr zu trennen. Da von diesem Phosphid die Bildung hoherer Phosphorgeriiste ausgeht, hangen Ausbeute und Reinheit des i-Pr,P, davon ab, wie schnell das i-Pr,P, von dem Li(i-Pr),P, abgetrennt wird. Wird die Reaktion von Li,P, mit 2-Brom-propan bei -70°C durchgefuhrt, so ist ein hoherer Anteil an Verunreinigungen zu beobachten, wahrend die Signale des Li(i-Pr)zP7 mit deutlich geringerer Intensitit auftreten. Unabhangig von der Reaktions- temperatur kann auch bei zehnfachem UberschuB an i-PrBr ein quantitatives Abreagieren zu i-Pr8P, nicht erreicht werden. Bei relativ kurzen Reaktionszeiten ist der Anteil an Li(i-P&P, groBer, der Anteil der hoheren i-Pr-Phosphane vernachlaaigbar klein; bei langeren Reaktionszeiten sind die Mengenverhaltnisse umgekehrt.
Massenspektrum yon 2: M+, i-Pr3P7, 346: M+-i-Pr, 303; i-Pr3Pg, 408; i-Pr5Pg, 494, i-Pr6P14, 692; i-Pr6P16, 754.
3.2. Die Umsetzung von Lisp, * 3 DME mit Hslogensilanen
3.2.1. Derstellung von (H,Si),P, 6 und (MeHzSi),P, 6. Auf eine eingefrorene Suspension von Li,P, . 3 DME in Toluol (fl. Stickstoff) wird ein UberschuU an H,SiI [ll] aufkondensiert, das Reak- tionsgemisch auf -70°C aufgetaut und bei dieser Temperatur 1 2 h geriihrt. Danach wird auf 20°C erwarmt, vom ausgefallenen LiI * DME (quantitative Abscheidung bei dem angegebenen Ansatz) abfiltriert und das Filtrat ohne vorheriges Einengen auf -30°C gekuhlt. Innerhalb von 10 h scheiden sich weiBe, nadelformige Kristalle des (H,Si),P, ab, die im Vakuum getrocknet werden. (I€,Si),P, lost sich gut in Toluol (beim Zusatz von THF erfolgt spontane Zersetzung unter Braunfarbung und Gasentwicklung). Die frisch dargestellte Verbindung ist ruckstandsfrei verdampfbar. Nach dem 31P-NMR-Spektrum und der massenspektrometrischen Untersuchung ist die Verbindung 5 rein. Ublicher Ansatz: 5,2 g (l0,2 mmol) Li,P, . 3 DME; 5,8 g (36,7 mmol) H,SiI; 120 ml Toluol. Ausbeute 1,7 g (5,6 mmol) (H,Si),P, (54%). Massenspektrum von (H,Si),P,: M+, P,28Si29SizH,, 311,8161 (Diff. zum ber. Wert --0,6) (Int. 1,02%); Mf-SiH,, P,28Si,H,, 278,8174 (0,3) (19,7%); P,Si,H,,
Li,P, . 3 DME reagiert unter den gleichen Bedingungen rnit &!IeH,SiBr zum (MeH,Si),P, 6. 6 konnte nicht durch Kristallisation abgetrennt werden und zersetzt sich bei der Destillation. Nach Abtrennung des LiBr kann ihre Toluol-Losung als Stammlosung fur weitere Reaktionen Verwendung finden, da Verbindung 6 quantitativ entsteht und sich somit die Molaritaten der Losungen errechnen lessen.
246,5339 (-1:7) (10,67‘)”).

20 Z. anorg. allg. Chem. 584 (1990)
3.2.2. Die Urnsetzung von Li,P, * 3 DME rnit (Me,C),SiBr. Li,P7 * 3 DME wird in Tolnol gelost und die auf -GO "C gekuhlte Suspension tropfenweise mit einem UberschuW von (Me,C),SiBr in Toluol versetzt. Nach 16 h bei -GO"C unter Ruhren wird das Rcaktionsgemisch auf 23°C erwarmt. I m 31P-NMR-Spektrum der Losung sind nur die Signale des unumgesetzten Li3P, zu erkennen. Nach mehrstundigem Kochen am RuckfluS ergeben sich aus dem 31P-NMR-Spektrum des Reaktions- gemisches Hinweise auf das in geringem Umgang gebildete Li,[(Me,C)Si]P,. Eine vollstandige Sub- stitution konnte nicht erreicht wcrden, zumal sich Li,P, unter diesen Bedingungen schon langsam zersetzt. Ansatz: 4,2 g (8,3 mmol) Li,P, . 3 DME; 8,4 g (30 mmol) (Me,C),SiBr; 60 ml Toluol.
3.8.3. Die Umsetzung von Li:$P7 * 3 DME rnit Et,P- SiMe,Cl. Li,P, . 3 DME wird in Toluol vorgelegt und die auf - 60 "C gekuhlte Suspension tropfenweise mit uberschussigem Et,PSiMe,CI in Toluol versetzt. Kach beendeter Zugabe wird das Reaktionsgemisch noch 4 h bei -60°C geruhrt, dann auf 23°C erwarmt und das ausgcfallene LiCl abfiltriert. Das Filtrat enthilt nach dem ,lP-NMR- Spektrums das quantitativ gebildete (Et,PSiMe,),P, 7. 7 konnte nicht zur Kristallisation gcbracht werden und ist auch nicht durch Sublimation in rciner Form zu erhalten, da es sich bereits bei 30-440°C/10-6 Torr unter Abbau des P,-Gerustes zersetzt. Ansatz: 3,G g (7,l mmol) Li,P, . 3 DNE in 50 ml Toluol; 4,O g (22,O mmol) Et,PSiMe,CI in 100 ml Toluol. Massenspektram von 7 : M+ (nicht beobachtet); Mf-PEt,, 569; M+-SiMc,-PEt,, 511; M+-2 PEt,, 480; M+-SiMe,PEt,-PEt,, 422; M+ -3 PEt,, 391; M+-2 SiMe,PEt,, 364.
Dem Fonds der Chemie und dcr Dcutschen Forschungsgemeinschaft danken wir fur die Forde- rung. Die Bayer AG, Leverkusen, und die Hans-Heinrich-Hutte, Langelsheim/Harz, unterstutzten uns mit Chemikalien. Herrn DOMNICK danken wir fur die Aufnahmen der NMR-Spektren, Herrn Dr. SCHEER fur die massenspektrometrischc Untersuchung und Herrn Dr. MATERN fur die Spektren- simulation.
Literatur [l] FRITZ, G.; H~LDERICH, W.: Naturwissenschaften 62 (1975) 573. [a] FRITZ, G.; UHLMANN, R.: Z. anorg. allg. Chem. 440 (1978) 168. [3] FRITZ, G.: Comments Inorg. Chem. 1 (1982) 329. [4] BAUDLER, M.: Angew. Chem. '34 (1982) 520; Angew. Chem. Int. Ed. Engl. 21 (1982) 492. [5] FRITZ, G.; HARER, J.: Z. anorg. allg. Chem. 504 (1983) 23. [6] FRITZ, G.; SCHNEIDER, H.-W.; HONLE, W.; v. SCHNERINC-, H. G.: Z. anorg. allg. Chem. 584
[7] BAUDLER, M.; FADER, W.; HAHN, H.: Z. anorg. allg. Chem. 469 (1980) 15. [8] BAUDLER, M.; PONTZEN, TH.: Z. Naturforsch. 38b (1983) 955. [9] HOPPE, H.-D. : Dissertation, Univ. Karlsruhe 1988.
(1990) 21.
[lo] FRITZ, G.; HOPPE, K.-D.; HONLE, W.; MUJICA, C.; NIANRIQUEZ, V.; v. SCHNERING, H. G.:
[ll] FRITZ, 0.; KUMMER, D.: Z. anorg. allg. Chem. 304 (1960) 322. J. Oganomet. Chem. 24'3 (1983) 63.
Bei der Redaktion eingegangen am 18. Juli 1989.
Anschr. d. Verf.: Prof. Dr. Dr. h. c. G. FRITZ, Dr. H.-W. SCHNEIDER, Inst. f. Anorg. Chemie d. Univ., EngesserstraSe, Geb.-Nr. 30.45, D-7500 Karlsruhe





























