Embed Size (px)
Citation preview

Han
dbuc
h Bi
osim
ilars
ZUSAMMENFASSUNG DER VON DER EMA ZUGELASSENEN UND IN DEUTSCHLAND IM VERKEHR BEFINDLICHEN BIOSIMILARS
Stand: Januar 2017Alle Angaben ohne Gewähr
HerausgeberPro Generika e. V. | Arbeitsgemeinschaft Pro Biosimilars
Unter den Linden 32–34 | 10117 Berlin
Tel. +49 (0)30 - 81 61 60 9-0
[email protected] | www.probiosimilars.de
Konzept und Gestaltungwww.tack-design.de
DIE ARBEITSGEMEINSCHAFT PRO BIOSIMILARS
Die AG Pro Biosimilars ist die Interessenvertretung der Biosimilarunter-
nehmen in Deutschland. Sie steht allen Unternehmen offen, die Biosimilars
entwickeln, herstellen und für die Versorgung bereitstellen. Die Arbeits-
gemeinschaft unter dem Dach des Pro Generika e. V. engagiert sich für einen
bedarfsgerechten Zugang der Patientinnen und Patienten zu modernen bio-
pharmazeutischen Arzneimitteltherapien, für eine bezahlbare Versorgung
und für faire und nachhaltige Wettbewerbsbedingungen.
Prof. Dr. Theodor DingermannSeniorprofessor am Institut für
Pharmazeutische Biologie,
Goethe-Universität Frankfurt a. M.
Michael DilgerPartner des Beratungsunternehmens
Simon-Kucher & Partners
Detlef BöhlerLeiter Arzneimittel der BARMER GEK
Johann FischaleckTeamleiter Arzneimittel
im Referat Vertrag und Arzneimittel,
Kassenärztliche Vereinigung Bayerns
Dr. Ilse ZündorfAkademische Oberrätin am Institut
für Pharmazeutische Biologie,
Goethe-Universität Frankfurt a. M.
2017 2017
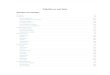
Handelsname /Hersteller
INN Referenzprodukt Datum der Zulassung
Omnitrope® / Sandoz Somatropin Genotropin® 12.04.2006
Binocrit® / Sandoz
Epoetin alfa Hexal® / Hexal
Abseamed® / Medice
Epoetin alfa Eprex® 28.08.2007
Retacrit® / Hospira
Silapo® / STADA
Epoetin zeta Eprex® 18.12.2007
Biograstim® / AbZ
Filgrastim Neupogen® 15.09.2008
Filgrastim Hexal® / Hexal
Filgrastim Neupogen® 06.02.2009
Nivestim® / Hospira Filgrastim Neupogen® 08.06.2010
Grastofil® / STADA / cell pharm
Filgrastim Neupogen® 18.10.2013
Accofil® / Accord Healthcare
Filgrastim Neupogen® 18.09.2014
Inflectra® / Hospira
Remsima® / Mundipharma
Infliximab Remicade® 10.09.2013
Flixabi® / Biogen Infliximab Remicade® 30.05.2016
Ovaleap® / Teva Follitropin alfa GONAL-f® 27.09.2013
Bemfola® / Finox Follitropin alfa GONAL-f® 27.03.2014
Abasaglar® / Lilly/Boehringer Ingelheim
Insulin Glargin Lantus® 09.09.2014
Benepali® / Biogen Etanercept Enbrel® 14.01.2016
HandbuchBiosimilars
ProBio-Biosimilars-Handbuch-Umschl-RZ.indd 1 22.12.16 14:45
Han
dbuc
h Bi
osim
ilars
ZUSAMMENFASSUNG DER VON DER EMA ZUGELASSENEN UND IN DEUTSCHLAND IM VERKEHR BEFINDLICHEN BIOSIMILARS
Stand: Januar 2017Alle Angaben ohne Gewähr
HerausgeberPro Generika e. V. | Arbeitsgemeinschaft Pro Biosimilars
Unter den Linden 32–34 | 10117 Berlin
Tel. +49 (0)30 - 81 61 60 9-0
[email protected] | www.probiosimilars.de
Konzept und Gestaltungwww.tack-design.de
DIE ARBEITSGEMEINSCHAFT PRO BIOSIMILARS
Die AG Pro Biosimilars ist die Interessenvertretung der Biosimilarunter-
nehmen in Deutschland. Sie steht allen Unternehmen offen, die Biosimilars
entwickeln, herstellen und für die Versorgung bereitstellen. Die Arbeits-
gemeinschaft unter dem Dach des Pro Generika e. V. engagiert sich für einen
bedarfsgerechten Zugang der Patientinnen und Patienten zu modernen bio-
pharmazeutischen Arzneimitteltherapien, für eine bezahlbare Versorgung
und für faire und nachhaltige Wettbewerbsbedingungen.
Prof. Dr. Theodor DingermannSeniorprofessor am Institut für
Pharmazeutische Biologie,
Goethe-Universität Frankfurt a. M.
Michael DilgerPartner des Beratungsunternehmens
Simon-Kucher & Partners
Detlef BöhlerLeiter Arzneimittel der BARMER GEK
Johann FischaleckTeamleiter Arzneimittel
im Referat Vertrag und Arzneimittel,
Kassenärztliche Vereinigung Bayerns
Dr. Ilse ZündorfAkademische Oberrätin am Institut
für Pharmazeutische Biologie,
Goethe-Universität Frankfurt a. M.
2017 2017
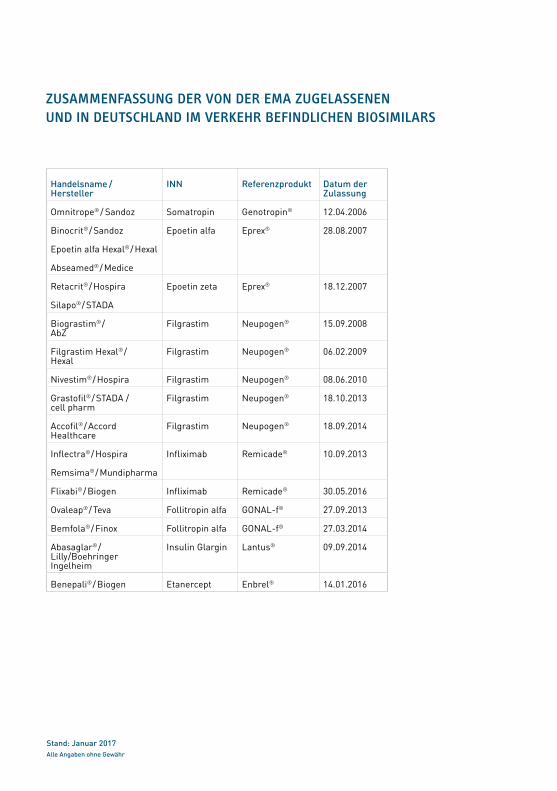
Handelsname /Hersteller
INN Referenzprodukt Datum der Zulassung
Omnitrope® / Sandoz Somatropin Genotropin® 12.04.2006
Binocrit® / Sandoz
Epoetin alfa Hexal® / Hexal
Abseamed® / Medice
Epoetin alfa Eprex® 28.08.2007
Retacrit® / Hospira
Silapo® / STADA
Epoetin zeta Eprex® 18.12.2007
Biograstim® / AbZ
Filgrastim Neupogen® 15.09.2008
Filgrastim Hexal® / Hexal
Filgrastim Neupogen® 06.02.2009
Nivestim® / Hospira Filgrastim Neupogen® 08.06.2010
Grastofil® / STADA / cell pharm
Filgrastim Neupogen® 18.10.2013
Accofil® / Accord Healthcare
Filgrastim Neupogen® 18.09.2014
Inflectra® / Hospira
Remsima® / Mundipharma
Infliximab Remicade® 10.09.2013
Flixabi® / Biogen Infliximab Remicade® 30.05.2016
Ovaleap® / Teva Follitropin alfa GONAL-f® 27.09.2013
Bemfola® / Finox Follitropin alfa GONAL-f® 27.03.2014
Abasaglar® / Lilly/Boehringer Ingelheim
Insulin Glargin Lantus® 09.09.2014
Benepali® / Biogen Etanercept Enbrel® 14.01.2016
HandbuchBiosimilars
ProBio-Biosimilars-Handbuch-Umschl-RZ.indd 1 22.12.16 14:45


HANDBUCH
Biosimilars

3Inhalt
INHALT
Kapitel 1
WAS SIND BIOSIMILARS? 04
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
Kapitel 2
ZULASSUNG, WIRKSAMKEIT UND QUALITÄT VON BIOSIMILARS 30
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
Kapitel 3
BIOSIMILARS IN DER VERSORGUNG 56
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
Kapitel 4
BIOSIMILARS 2.0: MONOKLONALE ANTIKÖRPER 68
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
Kapitel 5
10 JAHRE BIOSIMILARS – LESSONS LEARNED 82
Michael Dilger
Kapitel 6
DIE BEDEUTUNG VON BIOSIMILARS IN DER 98
VERTRAGSÄRZTLICHEN VERSORGUNG
Johann Fischaleck
Kapitel 7
DIE INITIATIVE „BIOLIKE“ DER BARMER GEK 106
Detlef Böhler
Anhang
GLOSSAR 115
IMPRESSUM / ABBILDUNGSNACHWEIS 120
HANDBUCH
Biosimilars

4 Biosimilars – Ein Handbuch
WAS SIND BIOSIMILARS?
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
Kapitel
1

5Was sind Biosimilars?
Einleitung
Mit der Entdeckung des Kodierungsprinzips biologischer In-
formation in Form der DNA-Doppelhelix (Abb. 1) durch Watson
und Crick im Jahre 1953 und mit der Entschlüsselung des ge-
netischen Codes (Abb. 2) acht Jahre später wurden zwei uni-
verselle Prinzipien beschrieben. Danach wird eine beliebige
genetische Informationseinheit in jedem biologischen Orga-
nismus eindeutig verstanden und kann somit prinzipiell auch
in jedem biologischen Organismus exakt in das entsprechende
Protein übersetzt werden.
Dies bildet die Basis für die ab 1975 sich entwickelnde Über-
legung, eine neue Klasse von Arzneimitteln zu konzipieren, die
als Biologicals, Biopharmazeutika oder gentechnisch herge-
stellte Arzneimittel beschrieben werden. Hierzu wird die ge-
netische Informationseinheit (das Gen) für ein therapeutisch
relevantes Protein in aller Regel aus dem menschlichen Ge-
nom isoliert und mithilfe gentechnischer Methoden so modifi-
ziert, dass von dieser Informationseinheit dann in einem an-
deren Organismus das menschliche Protein synthetisiert
werden kann.
Dieses Prinzip wurde 1982 erstmals mit der Produktion von
Humaninsulin in dem Darmbakterium Escherichia coli reali-
siert. Heute ist die Zahl gentechnisch hergestellter Wirkstoffe
auf über 120 angestiegen, die in mindestens 160 Arzneimitteln
enthalten sind. Fast alle diese Wirkstoffe sind von natürlichen,
humanen Biomolekülen abgeleitet, die allerdings natürlicher-
weise nur in so geringer Konzentration vorkommen, dass sie
konventionell aus den klassischen Quellen (Blut, Urin, Gewe-
be) nicht sicher isoliert werden konnten, obwohl sie therapeu-
tisch dringend gebraucht werden.
Abb. 2: Die Code-„Sonne“ als Illustration des uni- versellen genetischen Codes
Abb. 1: Die DNA-Doppel-helix
WAS SIND BIOSIMILARS?
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf

6 Biosimilars – ein Handbuch
Vom Biopharmazeutikum zum Biosimilar
Nach Einführung der ersten Biopharmazeutika in den frühen
achtziger Jahren ist deren Bedeutung immer weiter gestie-
gen. Im Jahre 2015 stieg der Umsatz von Biopharmazeutika
in Deutschland auf rund 8,2 Milliarden Euro (zu Hersteller-
abgabepreisen), was einem Wachstum gegenüber 2014 von
9,7 % entspricht. Damit erreichen Biopharmazeutika deutlich
mehr als ein Fünftel (22,9 %) des Marktanteils aller Arznei-
mittel in Deutschland.1
Seit 2001 begannen nach und nach die Patente einiger wich-
tiger, umsatzstarker Biopharmazeutika auszulaufen. Ab 2006
betraten Nachahmerprodukte, sogenannte Biosimilars, die
Bühne. Dies war konsequent und notwendig zur Entlastung
des Gesundheitssystems, denn Biopharmazeutika gehören
fast ausnahmslos zu den sehr hochpreisigen Arzneimitteln.
Der Prozess der Patentabläufe wichtiger Biopharmazeutika
beschleunigt sich zurzeit rasant (siehe Tab. 1).
Handelsname INN Jahr Patentablauf
Humulin® Human Insulin 2001
Cerezyme® Imiglucerase 2001
Intron A® Interferon alfa-2b 2002
Nutropin® / Nutropin AQ Somatropin 2003
Avonex® Interferon beta-1a 2003
Humatrope® Somatropin 2003
Epogen® / Procrit® Epoetin alfa 2004
Synagis® Palivizumab 2005
Tab. 1: Übersicht über Biopharmazeutika
mit Blick auf die Patentlaufzeiten
Nachahmer- produkte von
Biopharma zeu tika nennt man
Biosimilars.
Seit mehr als 30 Jahren gibt es gentechnisch her-
gestellte Arznei-mittel.
1 Medizinische Biotechnologie in Deutschland: BCG Biotech-Report 2016
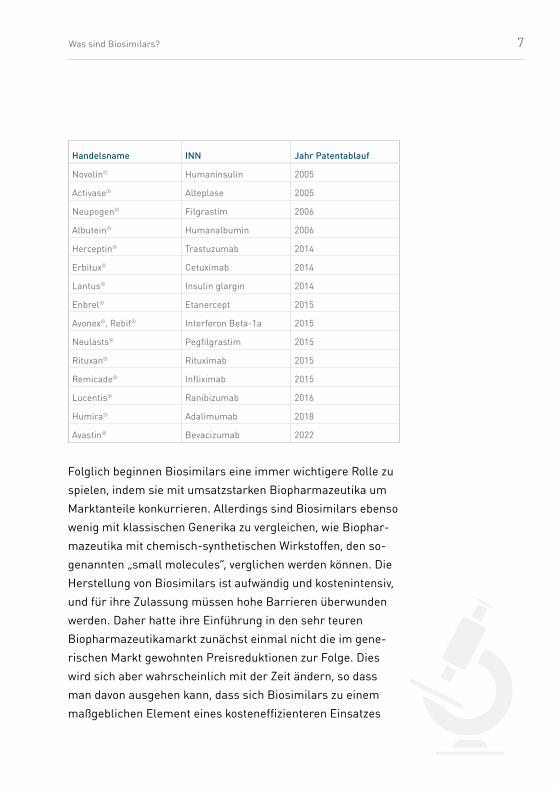
7Was sind Biosimilars?
Handelsname INN Jahr Patentablauf
Novolin® Humaninsulin 2005
Activase® Alteplase 2005
Neupogen® Filgrastim 2006
Albutein® Humanalbumin 2006
Herceptin® Trastuzumab 2014
Erbitux® Cetuximab 2014
Lantus® Insulin glargin 2014
Enbrel® Etanercept 2015
Avonex®, Rebif® Interferon Beta-1a 2015
Neulasts® Pegfilgrastim 2015
Rituxan® Rituximab 2015
Remicade® Infliximab 2015
Lucentis® Ranibizumab 2016
Humira® Adalimumab 2018
Avastin® Bevacizumab 2022
Folglich beginnen Biosimilars eine immer wichtigere Rolle zu
spielen, indem sie mit umsatzstarken Biopharmazeutika um
Marktanteile konkurrieren. Allerdings sind Biosimilars ebenso
wenig mit klassischen Generika zu vergleichen, wie Biophar-
mazeutika mit chemisch-synthetischen Wirkstoffen, den so-
genannten „small molecules“, verglichen werden können. Die
Herstellung von Biosimilars ist aufwändig und kostenintensiv,
und für ihre Zulassung müssen hohe Barrieren überwunden
werden. Daher hatte ihre Einführung in den sehr teuren
Biopharmazeutikamarkt zunächst einmal nicht die im gene-
rischen Markt gewohnten Preisreduktionen zur Folge. Dies
wird sich aber wahrscheinlich mit der Zeit ändern, so dass
man davon ausgehen kann, dass sich Biosimilars zu einem
maßgeblichen Element eines kosteneffizienteren Einsatzes

8 Biosimilars – ein Handbuch
von Biopharmazeutika entwickeln werden, wovon Patienten
ebenso profitieren werden wie das Gesundheitssystem.
Um sich einen Überblick über Biosimilars verschaffen zu kön-
nen, ist es sinnvoll, sich vorab mit ein paar grundlegenden
Fragen zu beschäftigen:
1. Was unterscheidet Biopharmazeutika von „small
molecules“?
2. Nach welchen Prinzipien werden Biopharmazeutika
hergestellt?
3. Was sind Biosimilars?
4. Warum sind Biosimilars keine Generika?
5. Welche Biosimilars gibt es derzeit in der EU / Deutschland
und seit wann gibt es sie?

9Was sind Biosimilars?
Was unterscheidet Biopharmazeutika von „small molecules“?
Offensichtlich liegt ein Unterschied zwischen Biopharmazeu-
tika und den sogenannten „small molecules“ zunächst einmal
in der Größe der Moleküle. So unterscheiden sich beispiels-
weise die Molekulargewichte der Acetylsalicylsäure (0,18 kDa)
und des Biopharmazeutikums Interferon alfa (19 kDa), die
jeweils als kleine Vertreter in den beiden Klassen gelten, um
den Faktor 100 (Abb. 3). Nochmals 15-mal größer als Interfe-
ron alfa, das bei bestimmten Tumorformen angewendet wird,
ist der Gerinnungsfaktor VIII mit einem Molekulargewicht von
ca. 330 kDa, der für Patienten mit einer Gerinnungsstörung
(Hämophilie A) lebenswichtig ist.
ChemischerWirkstoff
(Acetylsalicylsäure0,18 kDa)
„Kleiner“Proteinwirkstoff(Interferon alfa
19 kDa)
„Großer“Proteinwirkstoff
(Gerinnungsfaktor VIIIca. 330 kDa)
Die Unterschiede in Größe und Komplexität sind so gewaltig,
dass sich nur die „small molecules“ mit den Methoden der
klassischen organischen Chemie rentabel synthetisieren
lassen. Biopharmazeutika werden hingegen immer biologisch,
d. h. vom Biosynthese-Apparat lebender Zellen, synthetisiert
und dann aus diesen Zellen isoliert.
Abb. 3: Unterschiede der Molekulargewichte verschiedener Wirkstoffe aus der Gruppe der che-misch hergestellten bzw. der biotechnologisch hergestellten Moleküle

10 Biosimilars – ein Handbuch
Weitere Unterschiede zwischen beiden Wirkstoffklassen be-
stehen darin, dass „small molecules“ in den meisten Fällen
keine Kopien von Biomolekülen sind, sondern der kreativen
Fantasie von Chemikern entstammen. Hingegen lehnen sich
Biopharmazeutika immer an biologische Vorbilder an, die
zudem in den allermeisten Fällen im Menschen vorkommen.
Die enorme Größe von Biopharmazeutika und die chemisch-
physikalischen Eigenschaften dieser Molekülklasse bewirken
einige Besonderheiten, die „small molecules“ gar nicht oder
nicht in einem solchen Ausmaß aufweisen (Tab. 2).
Parameter Kleine Moleküle Biologika
Herstellung Meist chemische Synthese
Biochemische Synthese in lebenden Zellen
Chemisch-physikali-sche Eigenschaften
Einfach / Stabil / Gut definiert
Komplex / Labil / Heterogen
Analytik (Struktur) Einfache Bestimmung der chemischen Struk-tur
Schwierige Bestimmung der komplexen, hetero-genen Struktur
Analytik (Reinheit) Einfache Bestimmung der Reinheit
Schwierige Bestimmung der Reinheit
Immunogenität (Auslösen einer uner-wünschten immunolo-gischen Reaktion)
Selten Prinzipiell immunogen
Biologische Verunreinigungen
Keine oder selten Aufwändige Maßnah-men müssen getroffen werden, um virale / bakterielle / fungale Verunreinigungen auszuschließen
Applikationsform Überwiegend (bevor-zugt) orale Applikation
Überwiegend parente-rale, selten lokale Appli-kation
• Die Wirkung der Biopharmazeutika kommt immer durch ei-
ne sehr einzigartige Faltung der großen, linearkettigen Ami-
nosäurepolymeren – einer Art „Superstruktur“ – zustande.
Tab. 2: Unterschiede zwischen kleinen
Molekülen und Biopharmazeutika

11Was sind Biosimilars?
Dieses als Tertiärstruktur bezeichnete Molekülknäuel wird
durch schwache Wechselwirkungen zusammengehalten
und kann bereits durch geringe Energieeinträge wie
Erwärmung oder Agitation gestört werden. Dann geht die
aktive, „native“ Struktur in eine inaktive, „denaturierte“
Struktur über, ohne dass auch nur eine kovalente che-
mische Bindung gelöst wurde. Dies ist der Grund dafür,
dass Biopharmazeutika in der Regel kühl zu lagern und zu
transportieren sind und nicht geschüttelt werden dürfen.
• Die relative Labilität der Peptidbindungen, durch die die
Aminosäuren der Proteine miteinander verknüpft sind und
die Anfälligkeit einzelner Aminosäuren für chemische Reak-
tionen bedingen, dass Proteine generell nicht in Form einer
einzelnen Molekülspezies, sondern in Form von Molekül-
gruppen mit einer charakteristischen Heterogenität vorlie-
gen. Diese Tatsache stellt ganz erhebliche Anforderungen
an die molekulare Charakterisierung von Biopharmazeutika.
• Ferner neigen die großen Proteine zur Aggregatbildung,
was durch stabilisierende Formulierungen so weit wie mög-
lich zu unterbinden ist.
• Da Biopharmazeutika keine extremen pH-Werte tolerieren,
sind diese Wirkstoffe immer parenteral zu applizieren. Dies
stellt hohe Anforderungen an eine aseptische Herstellung.
• Schließlich verlangen die großen, strukturell heterogenen
Molekülgruppen immer ein wachsames Beobachten einer
eventuellen immunologischen Reaktivität. Diese potenzielle,
unerwünschte Eigenschaft lässt sich nur durch eine hohe
Prozessstabilität und der daraus folgenden Produktkon-
Biopharmazeutika und Biosimilars sind wesentlich größer, schwieriger herzustellen und deutlich labiler als chemisch-synthe-tische Wirkstoffe.

12 Biosimilars – ein Handbuch
stanz kontrollieren. Unter Beachtung dieser Anforderungen
und durch den zunehmenden Erkenntnisgewinn durch
Studien und Überwachung wird das immunologische Poten-
zial von Biopharmazeutika heute deutlich weniger kritisch
gesehen, als dies noch vor Jahren der Fall war.
Biopharmazeutika sind also hochkomplexe Wirkstoffe, die heu-
te in Prozessen hergestellt werden, die sehr detailliert spezi-
fiziert sind. Dazu gehören unter anderem die Auswahl der für
die Biosynthese erforderlichen Zelllinie, die technische Aus-
gestaltung der Produktionsanlage, die komplexe Zusammen-
setzung und die Charakterisierung der Nährsubstanzen, die
Temperaturverhältnisse während der Fermentation und
schließlich der anspruchsvolle Aufreinigungsprozess aus einer
sehr komplexen Matrix, um nur einige Spezifikationen zu nen-
nen. Die Einhaltung der im Rahmen der Prozessentwicklung
definierten Spezifikationsgrenzen, gewissermaßen das Spezi-
fikationsfenster (Abb. 4), bildet die Basis für die große Produkt-
reproduzierbarkeit der komplexen Moleküle.
Dies ist von so immenser Bedeutung, dass Biopharmazeutika
in den entsprechenden Wirkstoff-Monografien, beispiels weise
im Europäischen Arzneibuch (Ph. Eur.), nicht nur über Mole-
kül-Charakteristika, sondern auch über Prozess-Charakteri-
stika definiert sind. So lautet das neue Paradigma „the pro-
duct is the process“. Dies ist eine signifikante Erweiterung des
alten, für kleine, chemisch synthetisierte Moleküle geltenden
Paradigmas „the product is the molecule“.
13Was sind Biosimilars?
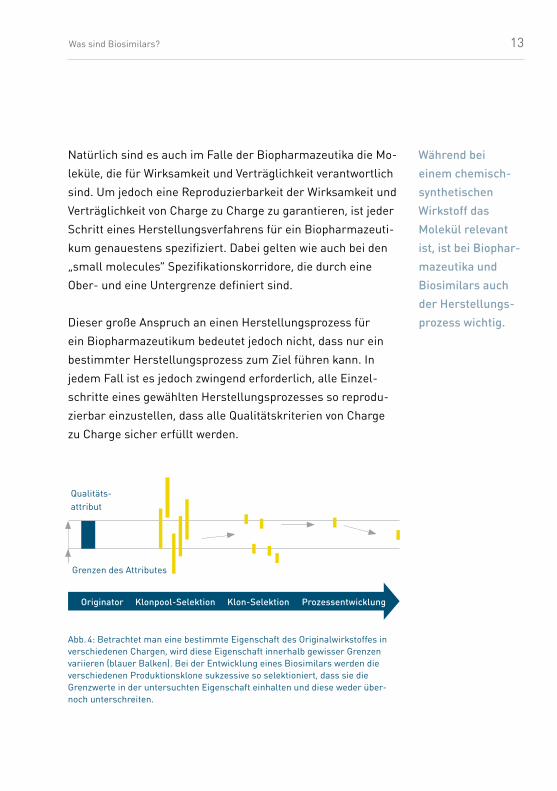
Natürlich sind es auch im Falle der Biopharmazeutika die Mo-
leküle, die für Wirksamkeit und Verträglichkeit verantwortlich
sind. Um jedoch eine Reproduzierbarkeit der Wirksamkeit und
Verträglichkeit von Charge zu Charge zu garantieren, ist jeder
Schritt eines Herstellungsverfahrens für ein Biopharmazeuti-
kum genauestens spezifiziert. Dabei gelten wie auch bei den
„small molecules“ Spezifikationskorridore, die durch eine
Ober- und eine Untergrenze definiert sind.
Dieser große Anspruch an einen Herstellungsprozess für
ein Biopharmazeutikum bedeutet jedoch nicht, dass nur ein
bestimmter Herstellungsprozess zum Ziel führen kann. In
jedem Fall ist es jedoch zwingend erforderlich, alle Einzel-
schritte eines gewählten Herstellungsprozesses so reprodu-
zierbar einzustellen, dass alle Qualitätskriterien von Charge
zu Charge sicher erfüllt werden.
Während bei einem chemisch-synthe tischen Wirkstoff das Molekül relevant ist, ist bei Biophar-mazeutika und Biosimilars auch der Herstellungs-prozess wichtig.
Qualitäts-attribut
Grenzen des Attributes
Originator Klonpool-Selektion Klon-Selektion Prozessentwicklung
Abb. 4: Betrachtet man eine bestimmte Eigenschaft des Originalwirkstoffes in verschiedenen Chargen, wird diese Eigenschaft innerhalb gewisser Grenzen variieren (blauer Balken). Bei der Entwicklung eines Biosimilars werden die verschiedenen Produktionsklone sukzessive so selektioniert, dass sie die Grenzwerte in der untersuchten Eigenschaft einhalten und diese weder über- noch unterschreiten.

14 Biosimilars – ein Handbuch
Prinzipien der Herstellung von Biopharmazeutika
Biopharmazeutika sind per definitionem Proteine oder Pepti-
de, die in lebenden Zellen produziert werden. Jedoch werden
sie nicht aus den Organismen oder Zellen isoliert, in denen
sie natürlicherweise vorkommen. Vielmehr handelt es sich
bei diesen Molekülen um „ektopische“, d. h. in einem fremden
Organismus oder in einer fremden Zelle hergestellte Proteine
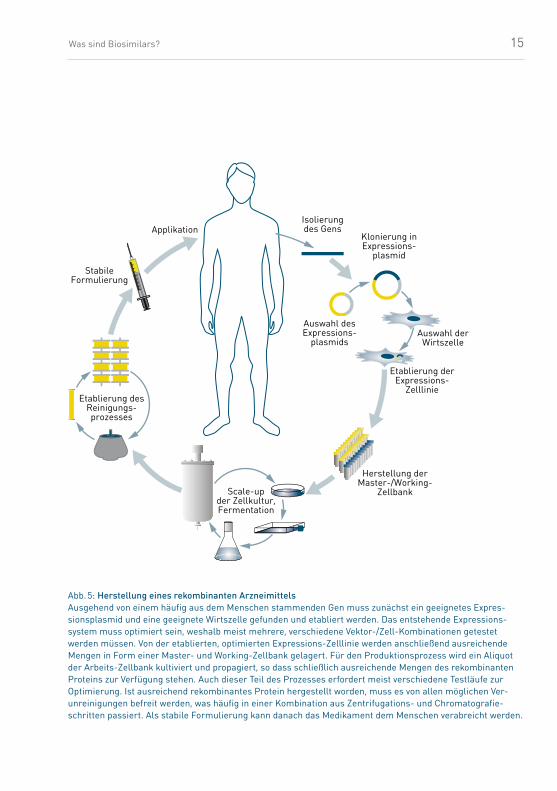
(Abb. 5).
15Was sind Biosimilars?
Klonierung inExpressions-
plasmid
Herstellung derMaster-/Working-
Zellbank
Isolierungdes Gens
Etablierung derExpressions-
Zelllinie
Auswahl desExpressions-
plasmidsAuswahl der
Wirtszelle
Scale-upder Zellkultur,Fermentation
Etablierung desReinigungs-prozesses
StabileFormulierung
Applikation
Abb. 5: Herstellung eines rekombinanten ArzneimittelsAusgehend von einem häufig aus dem Menschen stammenden Gen muss zunächst ein geeignetes Expres-sionsplasmid und eine geeignete Wirtszelle gefunden und etabliert werden. Das entstehende Expressions-system muss optimiert sein, weshalb meist mehrere, verschiedene Vektor-/Zell-Kombinationen getestet werden müssen. Von der etablierten, optimierten Expressions-Zelllinie werden anschließend ausreichende Mengen in Form einer Master- und Working-Zellbank gelagert. Für den Produktionsprozess wird ein Aliquot der Arbeits-Zellbank kultiviert und propagiert, so dass schließlich ausreichende Mengen des rekombinanten Proteins zur Verfügung stehen. Auch dieser Teil des Prozesses erfordert meist verschiedene Testläufe zur Optimierung. Ist ausreichend rekombinantes Protein hergestellt worden, muss es von allen möglichen Ver- unreinigungen befreit werden, was häufig in einer Kombination aus Zentrifugations- und Chromatografie-schritten passiert. Als stabile Formulierung kann danach das Medikament dem Menschen verabreicht werden.

16 Biosimilars – ein Handbuch
Dies bringt gewaltige Vorteile mit sich. Denn unter Sicher-
heitsaspekten ist es alles andere als trivial, ein aus dem Men-
schen isoliertes Protein beim Menschen therapeutisch einzu-
setzen, speziell im Hinblick auf eine Kontamination mit
menschlichen Krankheitserregern. Viel sicherer ist es da, ein
Protein als Therapeutikum für den Menschen zu entwickeln,
das zwar natürlicherweise im Menschen vorkommt, das
jedoch aus einem Bakterium, einer Hefezelle oder einer
Säuger-Zelllinie isoliert wurde. Hier bildet das ausgeklügelte
Methodenspektrum der Gentechnologie die Basis für die
Herstellung dieser Therapeutika.
Während die codierende Region eines Gens für ein bestimm-
tes Protein wegen der Universalität des genetischen Codes in
der gesamten belebten Natur eindeutig verständlich ist, ist die
Kontrolle des Abrufens dieser Information extrem spezifisch.
Daher müssen die ursprünglichen Kontrollregionen durch
spezifische und effiziente Kontrollregionen für den Wirtsorga-
nismus ersetzt werden.
Die Wahl des Wirtssystems ist eine kritische Entscheidung,
die zunächst einmal bestimmt wird durch die strukturellen
Anforderungen, die das zu isolierende Protein vorgibt. Darü-
ber hinaus spielt bei solchen Entscheidungen aber immer
auch die alles beherrschende Frage nach dem Sicherheitspro-
fil des Therapeutikums eine Rolle. Aus diesem Grund ist das
Wirtsspektrum, aus dem heute zugelassene Biopharmazeuti-
ka isoliert werden, sehr übersichtlich und deutlich kleiner als
das, was biologisch möglich wäre und was sich für die Lösung
wissenschaftlicher Problemstellungen technisch bewährt hat.

17Was sind Biosimilars?
Im Wesentlichen werden verwendet:
• das Bakterium Escherichia coli (ca. 35 Wirkstoffe)
• die Hefe Saccharomyces cerevisiae (ca. 15 Wirkstoffe)
• eine Insekten-Zelllinie (1 Wirkstoff)
• eine kleine Gruppe verschiedener Säuger-Zelllinien
(ca. 80 Wirkstoffe)
• eine humane Fibroblasten-Zelllinie (3 Wirkstoffe)
• eine Herde transgener, geklonter Ziegen (1 Wirkstoff) und
eine Herde transgener, geklonter Kaninchen (1 Wirkstoff)2,3
Alle diese biologischen Systeme haben ihre Vorteile, aber auch
ihre Limitationen. So ist die Produktion eines Proteins in E. coli
oder auch in der Hefe Saccharomyces cerevisiae relativ günstig
und biologisch sehr sicher, weil die Bakterien und Hefepilze
phylogenetisch so weit vom Menschen entfernt sind, dass bei-
spielsweise virale Kontaminationen hier keine Rolle spielen.
Allerdings ist die Aufreinigung zuweilen kompliziert, und es
lassen sich keine Proteine herstellen, die ein bestimmtes Mo-
difikationsmuster benötigen, um hinreichend wirksam zu sein.
Modifizierte Proteine lassen sich am besten in Säugerzellen
produzieren. Diese sind aber hinsichtlich der Fermentations-
bedingungen sehr anspruchsvoll, was deutlich höhere Kosten
verursacht. Und je näher man sich mit dem Wirtssystem phylo-
genetisch dem Menschen annähert, umso größer werden die
Anforderungen, eine Kontamination mit Humanpathogenen
sicher auszuschließen. Dieses Problem ist natürlich dann am
relevantesten, wenn man tatsächlich menschliche Zellen (z. B.
Die Wahl eines bestimmten Wirts-systems wird beeinflusst von Moleküleigen-schaften, von Produktionskosten und von Sicher-heitsaspekten.
Für die gentech-nische Herstellung von Wirkstoffen werden bisher nur wenige Zelllinien und Organismen verwendet.
2 Dingermann, Zündorf (2013). Vom Kopieren zum Kreieren (Teil 1). PharmInd 75, 835–8413 Dingermann, Zündorf (2013) Vom Kopieren zum Kreieren (Teil 2). PharmInd 75, 1034–1041

18 Biosimilars – ein Handbuch
eine humane Fibroblasten-Zelllinie) als Wirtszelle für die Her-
stellung eines therapeutischen Proteins verwendet. Schließlich
sind zurzeit zwei Wirkstoffe zugelassen, die in der laktierenden
Milchdrüse transgener Ziegen bzw. transgener Kaninchen pro-
duziert werden.
Anspruchsvoller als die Etablierung eines heterologen Produk-
tionssystems sind die eigentliche Fermentation und vor allem
die Aufreinigung der Wirkstoffe. Jeder Schritt in diesem kom-
plizierten Prozess ist hier durch Spezifikationsgrenzen defi-
niert und im Sinne eines antizipierenden Sicherheitssystems
muss ein Wirkstoff dann verworfen werden, wenn Spezifikati-
onsgrenzen in die eine oder andere Richtung überschritten
werden. Hier geht man von der Überlegung aus, dass eine
Reproduzierbarkeit zur Herstellung eines in sich zwangsläufig
inhomogenen Produktes dann am besten gewährleistet ist,
wenn jeder einzelne Produktionsschritt mit höchstmöglicher
Präzision reproduziert wird. Dies ist die Basis für das Paradig-
ma „the product is the process“.
Überraschen mag, dass die strukturelle Übereinstimmung
mit dem molekularen Vorbild aus dem Menschen heute viel-
fach von untergeordneter Bedeutung ist. Tatsächlich hat man
sich von Authentizität größtenteils sogar verabschiedet.
Mehr als die Hälfte aller derzeit zugelassenen rekombinan-
ten Wirkstoffe muss heute als nicht naturidentisch eingestuft
werden.
Kaum vorhersehbar war das erstaunliche Ausmaß an Modi-
fikationen, das offensichtlich noch vom Menschen vertragen
wird. So sind heute Biologicals zugelassen, für die es keine
Gentechnisch her-gestellte Arznei-
mittel sind häufig nicht naturiden-
tisch und werden trotzdem erfolg-
reich angewendet.

19Was sind Biosimilars?
Vorbilder in der Natur gibt. Teilweise werden auch Proteine
therapeutisch eingesetzt, die beim Menschen in dieser Form
gar nicht vorkommen, dennoch aber hinreichend vertragen
werden und bei den angezeigten Indikationen von großem the-
rapeutischen Nutzen sind. Beispielsweise besteht der Wirk-
stoff Etanercept aus Teilen des humanen TNF--Rezeptors
und Teilen eines humanen Antikörpermoleküls. Diese Kombi-
nation verleiht dem Wirkstoff eine für die Anwendung am
Menschen ausreichende Stabilität, so dass sich dieses artifi-
zielle Protein als wichtige Option für die Therapie der rheuma-
toiden Arthritis etablieren konnte. Diese Art Moleküldesign
hat sich zwischenzeitlich auch für Wirkstoffe bei anderen
Indikationen durchgesetzt.
Was sind Biosimilars?
Die EMA (European Medicines Agency – Zentrale europäische
Zulassungsbehörde) definiert Biosimilars wie folgt:
„Ein Biosimilar-Arzneimittel ist ein biologisches Arznei-
mittel, das derart entwickelt wurde, dass es einem bereits
existierenden Arzneimittel („dem Referenzarzneimittel“)
ähnelt. Biosimilar-Arzneimittel unterscheiden sich von
Generika, da Letztere einfachere chemische Strukturen
aufweisen und als identisch mit ihren Referenzarzneimit-
teln gelten.“
In erster Näherung sind Biosimilars also „Kopien“ eines seit
Jahren bereits zugelassenen Biopharmazeutikums („Referenz-
arzneimittel“). Prinzipiell sind das Biosimilar-Arzneimittel und

20 Biosimilars – ein Handbuch
das entsprechende Referenzarzneimittel strukturell vergleich-
bar. Jedoch können wegen der komplexen Natur von Bio phar- mazeutika und der aufwändigen Herstellungsverfahren dieser
Arzneimittel geringfügige Abweichungen möglich sein. Denn
sowohl das Referenzarzneimittel als auch das Biosimilar wei-
sen natürlicherweise eine gewisse molekulare Variabilität auf.
Eine solche Variabilität, die im Übrigen auch zwischen unter-
schiedlichen Chargen des Referenzarzneimittels nicht ver-
meidbar ist, darf jedoch keine Auswirkungen auf die Sicherheit
oder Wirksamkeit des Arzneimittels haben. Dies muss durch
Daten im Rahmen des Zulassungsverfahrens nachgewiesen
werden und wird dann zentral durch die europäische Zulas-
sung offiziell bestätigt.
Ein zugelassenes Biosimilar ist demnach genauso wirksam
und sicher wie das Referenzarzneimittel. Es wird gewöhnlich
in derselben Dosis zur Behandlung derselben Krankheiten
verwendet wie das Referenzarzneimittel. Und Warnhinweise,
die bei der Verabreichung des Referenzarzneimittels zu be-
achten sind, müssen generell auch beim Einsatz des Biosimi-
lars beachtet werden.
Der Unterschied zwischen innovativen Biologicals und Bio-similars aus unternehmerischer SichtEin relevantes Problem, mit dem sich neue Biologicals kon-
frontiert sehen, ist die Frage, inwieweit der Wirkstoff tatsäch-
lich ein pathologisches Geschehen zu korrigieren vermag und
ob und in welchem Ausmaß die Intervention ernsthafte uner-
wünschte Wirkungen nach sich zieht. „Drugability“ und „gene-
ral safety“ stellen, wie bei jeder pharmazeutischen Neuent-
wicklung, die ganz großen Risiken bei der Entwicklung neuer
Ein zugelassenes Biosimilar ist
genauso wirksam und sicher wie das
Referenzarznei- mittel.

21Was sind Biosimilars?
Biopharmazeutika dar, die im Vorfeld einer klinischen Ent-
wicklung kaum sicher auszuräumen sind.
Dieses Problem hat sich allerdings nach einer Marktpräsenz
von mehreren Jahren erledigt, so dass „efficacy“ und „general
safety“ eines bestimmten Moleküls hinreichend bekannt sind.
Dies ist zudem prozessunabhängig, wenn sichergestellt ist,
dass die Moleküle prinzipiell identisch sind. Dieser Nachweis
kann mit heutigen Methoden sicher geführt werden, so dass
in dieser Hinsicht der Zulassung einer „generischen“ Version
eines gut eingeführten Biologicals nichts im Wege steht.
Auf den Punkt gebrachtBiosimilars lassen sich wie folgt charakterisieren:4
• Ein Biosimilar ist ein Arzneimittel, das vergleichbar ist mit
einem bereits zugelassenen rekombinanten Arzneimittel
(Referenzarznei). Entsprechend sind die Wirkstoffe in bei-
den Arznei mitteln ähnlich.
• Biosimilar und Referenzarznei werden in gleicher Dosie-
rung bei gleichen Indikationen eingesetzt.
• Wie für alle anderen Arzneimittel muss auch für Biosimi-
lars eine Zulassung beantragt und erteilt werden, bevor sie
verkehrsfähig werden. Die Zulassung wird in Europa durch
die Europäische Kommission erteilt, nachdem die EMA als
Zulassungsbehörde eine wissenschaftliche Überprüfung
der Wirksamkeit, Sicherheit und Qualität des Arzneimittels
vorgenommen hat.
• Jeder Arzneimittelhersteller kann einen Antrag auf Zulas-
sung eines Biosimilars stellen, um nach Ablauf des Patents
eines „Originals“ und nach eingehender Prüfung einen
Marktzugang zugesprochen zu bekommen.
Hersteller von Bio-similars haben nicht das Risiko, dass das Protein im Patienten keine Wirksamkeit zeigt.
4 http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/q_and_a/q_ and_a_detail_000125.jsp&mid=WC0b01ac0580533e0c

22 Biosimilars – ein Handbuch
• Da der Wirkstoff seit Jahren bekannt ist, müssen nicht alle
Informationen eingeholt werden, die bei der Zulassung
eines komplett neuen Wirkstoffs erforderlich sind. Verbind-
liche Richtlinien legen fest, welche Studien durchzuführen
sind, die belegen, dass das Biosimilar ähnlich und ebenso
sicher und wirksam ist wie das Referenzarzneimittel.
• Wegen des komplexen Herstellungsprozesses eines Biologi-
cals können sich das Referenzarzneimittel und das Biosimi-
lar geringfügig unterscheiden, wie sich im Übrigen auch
einzelne Chargen des Referenzarzneimittels leicht unter-
scheiden. Aus diesem Grund sind Vergleichsstudien durch-
zuführen. Hierbei handelt es sich um einen „Schritt-für-
Schritt-Prozess“, der mit dem Vergleich der Qualität und der
Stabilität des Wirkstoffs und des Herstellungsprozesses be-
ginnt. Hierdurch wird belegt, dass keine relevanten Unter-
schiede hinsichtlich Sicherheit und Wirksamkeit zwischen
dem Biosimilar und dem Referenzarzneimittel bestehen.
• Generell folgen die Herstellungsverfahren von Biosimilars
denselben Qualitätsstandards, wie sie auch für alle anderen
neuen Wirkstoffe gelten. Dazu zählt auch, dass die Zulas-
sungsbehörden in bestimmten Abständen die Produktions-
stätten inspizieren.
• Da Biosimilar und Referenzarznei ähnlich, aber nicht iden-
tisch sind, kann es zumindest bei chronisch kranken Pati-
enten Vorbehalte hinsichtlich der generellen Austauschbar-
keit der beiden Arzneimittel geben. Die Entscheidung, einen
Patienten mit der Referenzarznei oder mit dem Biosimilar
zu behandeln, muss von dem behandelnden Arzt getroffen
werden.
• Alle Arzneimittel – also auch Biosimilars – werden nach ihrer
Zulassung hinsichtlich ihrer Sicherheitsprofile beobachtet.
Qualität und Sicherheit müssen
sowohl beim Original präparat
als auch beim Bio-similar gewährleis-
tet sein.
Die Entscheidung, einen Patienten mit der Referenzarznei
oder mit dem Biosimilar zu
behandeln, muss von dem behan-
delnden Arzt getroffen werden.

23Was sind Biosimilars?
• Hierzu muss jeder pharmazeutische Hersteller ein Pharma-
kovigilanz-System etablieren, das jede Art von Auffälligkeiten
– besonders auch immunologische Auffälligkeit – registriert.
Dieses System wird ebenfalls von der Zulassungsbehörde
überprüft. Sollte es Anlass zu besonderer Vorsicht geben, so
muss das Biosimilar die gleichen Auflagen erfüllen wie das
Referenzarzneimittel. Hierzu gehört beispielsweise die Er-
stellung eines besonderen Riskmanagement-Plans.
Warum sind Biosimilars keine Generika?
Während in der Gruppe der „small molecules“ der innovative
Wirkstoff und das entsprechende Generikum molekular abso-
lut identisch sind, ist dies aufgrund der komplexen Natur der
Biopharmazeutika nicht zwingend der Fall (Tab. 3).
Tab. 3: Unterschiede zwischen Referenzarznei, Biosimilar und Generikum5
5 Modifiziert nach http://www.vfa-bio.de/vb-de/aktuelle-themen/branche/ biosimilars-nachbildungen-patentfrei-gewordener-biopharmazeutika
Referenzarznei Biosimilar Generikum
Der Wirkstoff ist … … von Charge zu Charge innerhalb definierter Grenzen identisch
… ähnlich, aber nicht identisch mit der Refe-renzarznei. Von Charge zu Charge ist der Wirk-stoff innerhalb definier- ter Grenzen identisch
Verglichen mit dem Ori-ginal innerhalb defi-nierter Varianzen iden-tisch
Unter Patentschutz Ja: für eine begrenzte Zeit
Nein: Vermarktung erst nach Patentablauf bei der Referenzarznei möglich
Nein: Vermarktung erst nach Patentablauf beim Original möglich
Entwicklungszeit Sehr lang und unsicher, um Wirksamkeit und Si-cherheit in 4 klinischen Studienphasen zu zeigen
Lang, um Vergleichbar-keit mit Referenzarznei zu zeigen
Relativ kurz

24 Biosimilars – ein Handbuch
Mikroheterogenitäten und auch potenzielle Verunreinigungen
des Wirkstoffs, die aus dem Produktionsprozess stammen,
stellen Anforderungen an die Qualität dieser Wirkstoffe, die in
dem Maße beim Kopieren kleiner, chemisch definierter Mole-
küle unbekannt sind.
Bei der Herstellung von Biopharmazeutika – und damit auch
bei der Herstellung von Biosimilars – erlangt jeder einzelne
Schritt des gesamten Herstellungsprozesses höchste Rele-
vanz. Und in der Tat ist es für einen Hersteller, der ein zuge-
lassenes Biopharmazeutikum „kopieren“ will, unmöglich,
den Originalherstellungsprozess in jedem Detail exakt nach-
zu ahmen. Im Gegenteil: Da Verfahrensdetails in aller Regel
als strenges Geheimnis gehütet werden und patentrechtlich
geschützt sind, ist gezieltes Kopieren in diesem Bereich prak-
tisch ausgeschlossen. Folglich lassen sich auch Unterschiede
in der Mikroheterogenität zwischen Referenzarzneimittel und
Nachahmerprodukt nicht vermeiden. Andererseits sind Mikro-
heterogenitätsunterschiede bei unterschiedlichen Chargen
ein und desselben Originalproduktes ebenfalls nicht vermeid-
bar, weshalb alle Prozess- und Produkt-Parameter in Form
von Spezifika tionen mit Ober- und Untergrenzen festgelegt
werden.
Somit liegt ein Hauptproblem bei der Herstellung von Bio-
pharmazeutika in der Sicherstellung der „Produktsicherheit“,
die nach allgemeinem Verständnis dann gewährleistet ist,
wenn diese auf Basis eines hoch standardisierten und im
Detail spezifizierten Prozesses analytisch und klinisch belegt
wurde. Dieser Beleg muss konsequenterweise vom Referenz-
arzneimittel wie von Nachahmerprodukten eingefordert
Biosimilars können infolge eines ande-
ren Herstellungs-prozesses nie iden-
tisch zum Referenz- arzneimittel sein.

25Was sind Biosimilars?
werden, wenn sie eine Marktzulassung
erhalten wollen.
Hier müssen sich also offensichtlich eta-
blierte Generikakonzepte von neuen Kon-
zepten zur Zulassung von „generischen“
Biopharmazeutika unterscheiden, und
diese Unterschiede sollten auch seman-
tisch erkennbar sein. In Europa hat man
sich dazu entschieden, „generische Biolo-
gicals“ als „similar biological medicinal
products“ oder kurz als „Biosimilars“
zu bezeichnen.
In jedem Fall muss gefordert und akzep-
tiert werden, dass für eine Zulassung
eines Biosimilars ein klinisches Pro-
gramm zu durchlaufen ist, das vor allem die Produktsicherheit
dokumentiert. Die Wirksamkeit und generelle Verträglichkeit
wurde bereits für das Referenzprodukt gezeigt.
Europa hat diese konzeptionelle Herausforderung angenom-
men und gemeistert und ein Regelwerk etabliert, das den
Marktzugang qualitativ hochwertiger und sicherer Biosimilars
seit 2003 regelt.6
6 Wiecek A, Mikhail A. (2006). European regulatory guidelines for biosimilars. Nephrol. Dial. Transplant. 21, Suppl. 5, v17–20

26 Biosimilars – ein Handbuch
Welche Biosimilars gibt es in der EU /Deutschland und seit wann?
Derzeit gibt es in der EU und in Deutschland 23 Biosimilars
in acht verschiedenen Wirkstoffgruppen: Enoxaparin Natrium,
Epoetin, Etanercept, Filgrastim, Follitropin, Infliximab, Insulin
glargin und Somatropin.
Bisher sind die in Tabelle 4 dargestellten Biosimilars von der
EMA zuge lassen worden.
Handelsname(n) Wirkstoff Referenzarznei Zulassung
Omnitrope® Somatropin Genotropin® 2006
Abseamed®
Epoetin alfa Hexal®
Binocrit®
Epoetin alfa Erypo® 2007
Retacrit®
Silapo®Epoetin zeta Erypo® 2007
Biograstim®
Ratiograstim®
Tevagrastim®
Filgrastim Neupogen® 2008
Filgrastim Hexal®
Zarzio®Filgrastim Neupogen® 2009
Nivestim® Filgrastim Neupogen® 2010
Grastofil® Filgrastim Neupogen® 2013
Accofil® Filgrastim Neupogen® 2014
Inflectra® Remsima®
Infliximab Remicade® 2013
Flixabi® Infliximab Remicade® 2016
Ovaleap® Follitropin alfa GONAL-f® 2013
Bemfola® Follitropin alfa GONAL-f® 2014
Abasaglar® Insulin glargin Lantus® 2014
Tab. 4: In Europa /Deutschland
zugelassene Biosimilars

27Was sind Biosimilars?
Handelsname(n) Wirkstoff Referenzarznei Zulassung
Benepali® Etanercept Enbrel® 2016
Inhixa® EnoxaparinNatrium
Clexane® 2016
Thorinane® EnoxaparinNatrium
Clexane® 2016
Diese Arzneimittel wurden von der Europäischen Zulassungs-
behörde nach strengsten Kriterien geprüft, so dass man kon-
statieren kann, dass ein Biosimilar höchsten Ansprüchen
genügt, und dass man sich bei zugelassenen Biosimilars auf
die zur Referenzarznei vergleichbare Qualität, Wirksamkeit
und Verträglichkeit verlassen kann.
Zudem wird die Sicherheit von Biosimilararzneimitteln, wie
bei allen Arzneimitteln, nach Erteilung der Genehmigung fort-
laufend überwacht. Jedes Unternehmen muss ein System zur
Überwachung von Nebenwirkungen einrichten, die im Zusam-
menhang mit seinen Arzneimitteln berichtet werden. Pati-
enten können verdächtige Nebenwirkungen auch selbst
berichten.
Die Genehmigungsbehörden prüfen sowohl die erfassten
Sicherheitsdaten als auch das Sicherheitsüberwachungs-
system des Unternehmens. Bei Anzeichen für Sicherheits-
bedenken werden von den Genehmigungsbehörden Unter-
suchungen durchgeführt und geeignete Maßnahmen
ergriffen.
Nicht von allen bereits patent-freien Wirkstoffen gibt es Biosimilars.

28 Biosimilars – ein Handbuch
Mythen zu Biosimilars und deren Entmystifizierung
Bei Ärzten und Apothekern herrscht nach wie vor ein großer
Informationsbedarf zu Biosimilars. Nicht selten begegnen
diese Berufsgruppen den Biosimilars mit einem Zerrbild, was
sicherlich auch daran liegt, dass immer noch Mythen über
Biosimilars verbreitet werden, von denen einige hier genannt
und enttarnt werden sollen (Tab. 5).

29Was sind Biosimilars?
Mythen und Fakten zu Biosimilars
Mythos Fakt
Ein Biosimilar unterscheidet sich
grundlegend von der Referenz-
arznei.
Biosimilars haben mit Generika
nichts gemein.
Die Referenzarznei bildet den
Goldstandard.
Die Referenzarznei enthält eine
definierte Molekülspezies. Das
Biosimilar enthält eine ähnliche,
aber nicht die identische Mole-
külspezies.
Biosimilars sind Biopharma
zeutika 2. Klasse.
In erster Näherung sind Biosimilars tatsächlich Kopien der
Referenzarznei.
Tatsächlich sind Biosimilars im Prinzip Generika von
Biopharmazeutika. Allerdings ist deren Herstellung,
Prüfung und Zulassung deutlich anspruchsvoller, als dies
für niedermolekulare-Generika der Fall ist, was letztlich
auch durch den „neuen“ Begriff „Biosimilar“ zum Ausdruck
gebracht wird.
Es gibt keinen Goldstandard. Alle Biopharmazeutika variie-
ren in einem gewissen Rahmen von Charge zu Charge bzw.
von Produktionsstandort zu Produktionsstandort. Das gilt
auch für unterschiedliche Chargen einer Referenzarznei.
Zwischen Referenzarznei und Biosimilar bestehen keine
prinzipiellen Unterschiede, weshalb es auch keine Biophar-
mazeutika 1. oder 2. Klasse geben kann und geben darf.
Biopharmazeutika enthalten immer strukturell variierende
Vertreter einer Molekülklasse. Diese Heterogenität ist je-
doch nicht beliebig, sondern streng spezifiziert. Biosimilars
bewegen sich in dem gleichen Variationskorridor wie die
Referenzarznei.
Tab. 5: Mythen und Fakten zu Biosimilars

30 Biosimilars – Ein Handbuch
ZULASSUNG, WIRKSAMKEIT UND
QUA LITÄT VON BIOSIMILARS
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
Kapitel
2

31
Zulassung eines Biosimilars: zwingend zentralisiert bei der Euro päischen Arzneimittel-Agentur (EMA)
Alle gentechnisch hergestellten Arzneimittel müssen in einem
zentralisierten Verfahren über die in London ansässige Europä-
ische Arzneimittel-Agentur (European Medicines Agency – EMA)
zugelassen werden. Daraus folgt, dass auch Biosimilars bei die-
ser Behörde ein Zulassungsverfahren durchlaufen müssen, in
dem die Qualität, die Wirksamkeit, die Sicherheit und die Ver-
träglichkeit der Präparate nach festen Regeln überprüft werden.
Allerdings galt es, für Biosimilars ein eigenes gesetzliches
Regelwerk zu entwickeln. Denn von Beginn an war klar, dass
es sich bei Biosimilars nicht um Generika im klassischen
Sinne handeln kann. Daher verbietet es sich auch aus wis-
senschaftlichen Gründen, das lang bewährte Zulassungs-
verfahren für Generika auf die Biosimilars zu übertragen.
Seit 2003 wurde bei der EMA an diesem Zulassungsprozess
gearbeitet. Die Basis bildete die Directive 2001/83/EC, die um-
fassend überarbeitet wurde. Zusätzlich wurde eine ganze Rei-
he von regulatorischen Leitlinien erstellt, in denen die Details
für das Zulassungsverfahren beschrieben werden. Neben die-
sen allgemeinen Leitlinien für Biosimilars hat die EMA auch
noch Leitlinien bezüglich Qualität, präklinischen und kli-
nischen Anforderungen sowie Produktspezifikationen für
bereits zugelassene Biologicals publiziert (Abb. 6).
Diese gesetzlichen Bestimmungen wurden 2005 in der EU für
die Zulassung von Biosimilars implementiert. Damit hat die
EMA auf dem Gebiet der Zulassung von Biosimilars weltweit
die Vorreiterrolle übernommen.
Biosimilars sind keine Generika und haben ein ei genes Regelwerk für das Zulas-sungsverfahren.
Zulassung, Wirksamkeit und Qualität von Biosimilars
ZULASSUNG, WIRKSAMKEIT UND
QUA LITÄT VON BIOSIMILARS
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf

32 Biosimilars – ein Handbuch
Als Resultat dieser regulatorischen Pionierarbeit stehen heu-
te qualitativ hochwertige Nachfolgepräparate von ehemals
innovativen Biopharmazeutika zur Verfügung, die nach Beste-
hen dieses Zulassungsverfahrens als sogenannte Biosimilars
vermarktbar sind.
Durch dieses Zulassungsverfahren unterscheiden sich Biosi-
milars auch grundlegend von anderen, im globalen Markt ver-
fügbaren Nachahmerprodukten eines Biopharmazeutikums.
Diese sind in Europa nicht verkehrsfähig und zeigen zum Teil
gefährliche Qualitätsmängel.
RICHTLINIEN MIT RELEVANZ FÜR BIOSIMILARS
7 http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/ general_content_000408.jsp
Abb. 6: Richtlinien mit Relevanz für die Zulassung von Biosimilars durch die EMA7 EPO: Erythropoetin, FSH: Follikel-stimulierendes Hormon, G-CSF: Granulozyten- Koloniestimulierender Faktor, IFN: Interferon, LMWH: niedermolekulares Hepa-rin, MAB: Monoklonale Antikörper
Übergreifende Richtlinien für Biosimilars
Allgemeine Fragen
Qualität
Präklinische und klinische Aspekte
Die EMA hat bei der Zulassung von Bio-
similars weltweit die Vorreiterrolle.
Andere Richtlinien mit Relevanz für Biosimilars
Vergleichbarkeit – Qualität
Vergleichbarkeit – Präklinische
und klinische Fragen
Immunogenität
Produktspezifische Richtlinien
Insulin Somatropin
G-CSF EPO LMWH IFN-alpha
FSH IFN-beta MAB

33
Auch passiert nicht jeder zur Zulassung bei der EMA einge-
reichte Biosimilarkandidat die anspruchsvollen Anforderun-
gen des zentralisierten Verfahrens zum Nachweis einer
Gleichwertigkeit mit der Referenzarznei. Dies belegt die hohen
Ansprüche, die an diese Arzneimittel gestellt werden, und es
unterstreicht die Gleichwertigkeit von Biosimilars mit den ent-
sprechenden Referenzprodukten nicht nur hinsichtlich
ihrer Wirksamkeit, sondern auch hinsichtlich ihrer Qualität
und Unbedenklichkeit. Gleichzeitig zeigt die Tatsache, dass
nicht jeder Biosimilarkandidat die anspruchsvollen Anforde-
rungen des zentralisierten Verfahrens erfüllt, wie wichtig es
ist, dass nur in der EU zugelassene Biosimilars therapeutisch
eingesetzt werden dürfen.
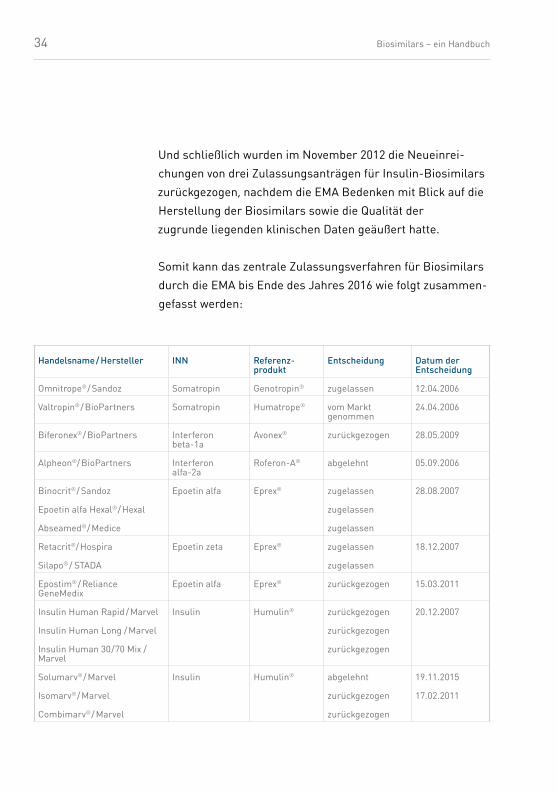
Die Tabelle (Tab. 6) fasst die bisherigen zentralen Zulassungs-
verfahren für Biosimilars bei der EMA zusammen. Zum Bei-
spiel listet die EMA-Homepage8 einen Interferon-alfa-2a-
Wirkstoff, dem die Zulassung 2006 versagt wurde. Ende 2007
wurden die Zulassungsanträge für drei Insulin-Biosimilars
zurückgezogen, da das Unternehmen die ihm vom Ausschuss
für Humanarzneimittel der EMA (Committee for Medicinal
Products for Human Use – CHMP) gestellten Fragen zur
Herstellung und zur Wirksamkeit nicht innerhalb der vorge-
gebenen Frist beantworten konnte.
Im März 2011 wurde der Zulassungsantrag für ein Epoetin-
Biosimilar zurückgezogen, da das Unternehmen auch die
Fragen des CHMP nicht fristgerecht beantworten konnte.
Nicht durch die EMA zugelassene Nachahmerpro-dukte sind in Europa nicht verkehrsfähig.
Das zentralisierte Verfahren bei der EMA gewährleistet einen hohen Quali-täts- und Sicher- heitsstandard der zugelassenen Biosimilars.
8 http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/ epar_search.jsp&mid=WC0b01ac058001d125
Zulassung, Wirksamkeit und Qualität von Biosimilars
34 Biosimilars – ein Handbuch
Und schließlich wurden im November 2012 die Neueinrei-
chungen von drei Zulassungsanträgen für Insulin-Biosimilars
zurückgezogen, nachdem die EMA Bedenken mit Blick auf die
Herstellung der Biosimilars sowie die Qualität der
zugrunde liegenden klinischen Daten geäußert hatte.
Somit kann das zentrale Zulassungsverfahren für Biosimilars
durch die EMA bis Ende des Jahres 2016 wie folgt zusammen-
gefasst werden:
Handelsname / Hersteller INN Referenz-produkt
Entscheidung Datum der Entscheidung
Omnitrope® / Sandoz Somatropin Genotropin® zugelassen 12.04.2006
Valtropin® / BioPartners Somatropin Humatrope® vom Markt genommen
24.04.2006
Biferonex® / BioPartners Interferon beta-1a
Avonex® zurück gezogen 28.05.2009
Alpheon®/ BioPartners Interferon alfa-2a
Roferon-A® abgelehnt 05.09.2006
Binocrit® / Sandoz
Epoetin alfa Hexal® / Hexal
Abseamed® / Medice
Epoetin alfa Eprex® zugelassen
zugelassen
zugelassen
28.08.2007
Retacrit®/ Hospira
Silapo® / STADA
Epoetin zeta Eprex® zugelassen
zugelassen
18.12.2007
Epostim® / Reliance GeneMedix
Epoetin alfa Eprex® zurück gezogen 15.03.2011
Insulin Human Rapid / Marvel
Insulin Human Long / Marvel
Insulin Human 30/70 Mix / Marvel
Insulin Humulin® zurück gezogen
zurück gezogen
zurück gezogen
20.12.2007
Solumarv® / Marvel
Isomarv® / Marvel
Combimarv® / Marvel
Insulin Humulin® abgelehnt
zurück gezogen
zurück gezogen
19.11.2015
17.02.2011
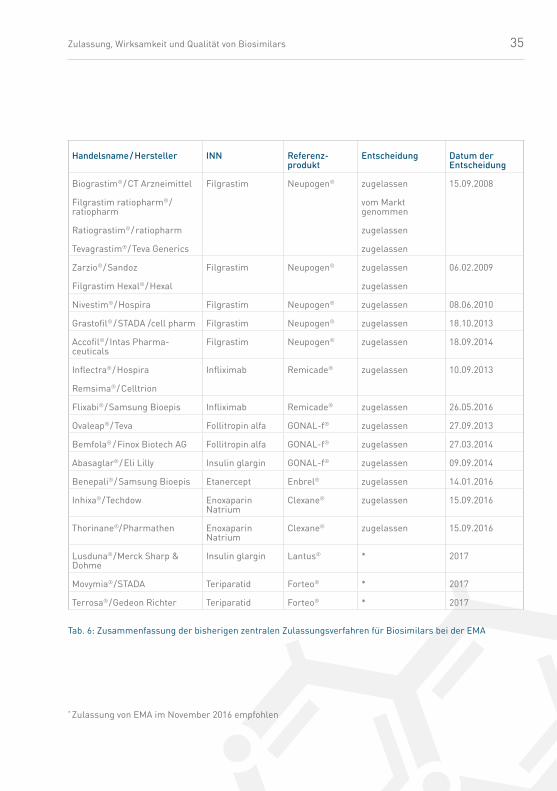
35Zulassung, Wirksamkeit und Qualität von Biosimilars
Handelsname / Hersteller INN Referenz-produkt
Entscheidung Datum der Entscheidung
Biograstim® / CT Arznei mittel
Filgrastim ratiopharm® / ratiopharm
Ratio grastim® / ratiopharm
Tevagrastim® / Teva Generics
Filgrastim Neupogen® zugelassen
vom Markt genommen
zugelassen
zugelassen
15.09.2008
Zarzio® / Sandoz
Filgrastim Hexal® / Hexal
Filgrastim Neupogen® zugelassen
zugelassen
06.02.2009
Nivestim® / Hospira Filgrastim Neupogen® zugelassen 08.06.2010
Grastofil® / STADA /cell pharm Filgrastim Neupogen® zugelassen 18.10.2013
Accofil® / Intas Pharma-ceuticals
Filgrastim Neupogen® zugelassen 18.09.2014
Inflectra® / Hospira
Remsima® / Celltrion
Infliximab Remicade® zugelassen 10.09.2013
Flixabi® / Samsung Bioepis Infliximab Remicade® zugelassen 26.05.2016
Ovaleap® / Teva Follitropin alfa GONAL-f® zugelassen 27.09.2013
Bemfola® / Finox Biotech AG Follitropin alfa GONAL-f® zugelassen 27.03.2014
Abasaglar® / Eli Lilly Insulin glargin GONAL-f® zugelassen 09.09.2014
Benepali® / Samsung Bioepis Etanercept Enbrel® zugelassen 14.01.2016
Inhixa® / Techdow EnoxaparinNatrium
Clexane® zugelassen 15.09.2016
Thorinane®/ Pharmathen EnoxaparinNatrium
Clexane® zugelassen 15.09.2016
Lusduna® / Merck Sharp & Dohme
Insulin glargin Lantus® * 2017
Movymia® / STADA Teriparatid Forteo® * 2017
Terrosa® / Gedeon Richter Teriparatid Forteo® * 2017
Tab. 6: Zusammen fassung der bisherigen zentralen Zulassungsverfahren für Biosimilars bei der EMA
* Zulassung von EMA im November 2016 empfohlen

36 Biosimilars – ein Handbuch
Die EMA als zentrales Kompetenzzentrum für Biopharmazeutika
Durch das zentralisierte Zulassungsverfahren aller in der EU
zugelassenen Biopharmazeutika bei der EMA liegen sämtliche
Daten zu allen Wirkstoffen bei dieser Behörde. Hierbei handelt
es sich zum einen um die primären Daten, die zur ursprüng-
lichen Zulassung der Wirkstoffe geführt haben. Hinzu kommen
jedoch auch Daten, mit denen Prozess- oder Produktände-
rungen angezeigt wurden. Dies kommt deutlich häufiger vor,
als hinlänglich bekannt ist.
Biopharmazeutikum Lizensierung Prozessänderungen
MabThera® 1998 6
Remicade® 1999 36
Enbrel® 2000 21
Humira® 2003 19
Derartige Änderungen werden von der Fachöffentlichkeit oft
gar nicht wahrgenommen, obwohl sie anzeige- und genehmi-
gungspflichtig und zum Teil erstaunlich signifikant sind.
Die zulassungs- relevanten Daten aller Biopharma-
zeutika sind bei der EMA hinterlegt.
Tab. 7: Prozessände-rungen nach der Zulas-
sung für verschiedene Biopharmazeutika9.
Orencia®
Enbrel®
Humira®
Remicade®
MabThera®
RoActemra®
Simponi®
Cimzia®
Rilonacept Regeneron®
Ilaris®
Benlysta®
9 Schneider CK: Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 72(3):315-8 (Data source: EPARs on EMA website)
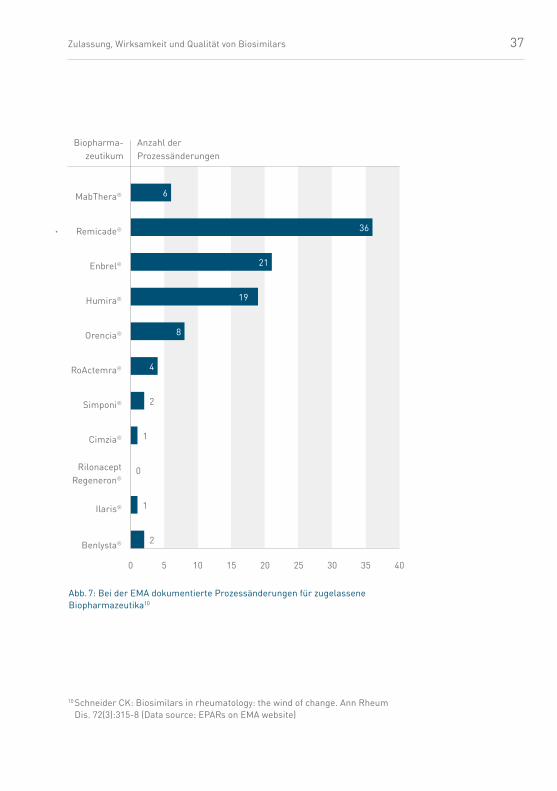
37
Abb. 7: Bei der EMA dokumentierte Prozessänderungen für zugelassene Biopharmazeutika10
Anzahl der Prozessänderungen
Bio pharma- zeutikum
Orencia®
Enbrel®
Humira®
Remicade®
MabThera®
RoActemra®
Simponi®
Cimzia®
Rilonacept Regeneron®
Ilaris®
Benlysta®
0 10 15 20 25 30 35 40 5
36
21
19
8
4
2
1
1
0
2
6
10 Schneider CK: Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 72(3):315-8 (Data source: EPARs on EMA website)
Zulassung, Wirksamkeit und Qualität von Biosimilars
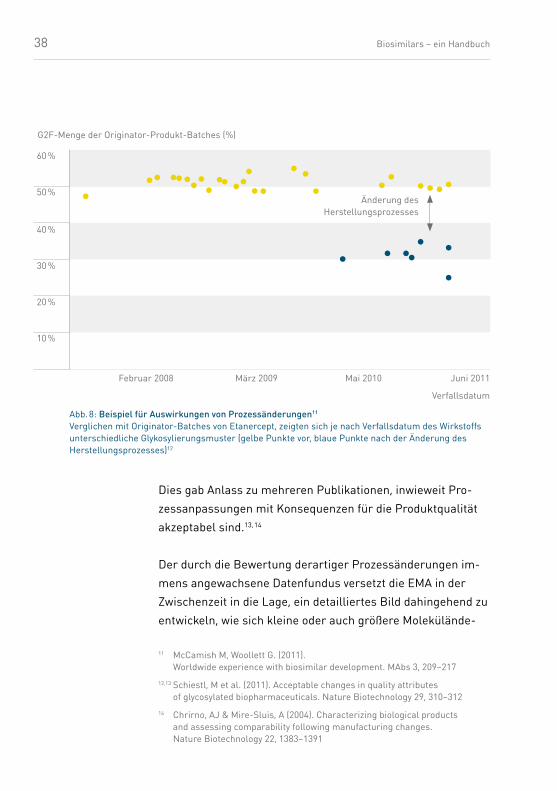
38 Biosimilars – ein Handbuch
Dies gab Anlass zu mehreren Publikationen, inwieweit Pro-
zessanpassungen mit Konsequenzen für die Produktqualität
akzeptabel sind.13, 14
Der durch die Bewertung derartiger Prozessänderungen im-
mens angewachsene Datenfundus versetzt die EMA in der
Zwischenzeit in die Lage, ein detailliertes Bild dahingehend zu
entwickeln, wie sich kleine oder auch größere Molekülände-
11 McCamish M, Woollett G. (2011). Worldwide experience with biosimilar development. MAbs 3, 209–217
12,13 Schiestl, M et al. (2011). Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nature Biotechnology 29, 310–312
14 Chrirno, AJ & Mire-Sluis, A (2004). Characterizing biological products and assessing comparability following manufacturing changes. Nature Biotechnology 22, 1383–1391
60 %
50 %
40 %
30 %
20 %
10 %
Februar 2008 März 2009 Mai 2010 Juni 2011
Änderung des Herstellungsprozesses
Verfallsdatum
G2F-Menge der Originator-Produkt-Batches (%)
Abb. 8: Beispiel für Auswirkungen von Prozessänderungen11
Verglichen mit Originator-Batches von Etanercept, zeigten sich je nach Verfallsdatum des Wirkstoffs unterschiedliche Glykosylierungsmuster (gelbe Punkte vor, blaue Punkte nach der Änderung des Herstellungsprozesses)12

39
rungen auf die klinische Wirksamkeit, die Sicherheit und die
Verträglichkeit der Wirkstoffe auswirken.
Zusätzlich kennt die Zulassungsbehörde natürlich auch sämt-
liche Spezifikationsfenster aller Biopharmazeutika. Dieses
Wissen gestattet es der Behörde, die Spezifikationsgrenzen
eines zur Zulassung eingereichten Biosimilars auf Plausibi-
lität einerseits und auf biologische Toleranz andererseits zu
überprüfen und zu bewerten. Zusammen mit den ebenfalls
eingereichten empirisch erhobenen Daten für das zuzulas-
sende Biosimilar kann so eine souveräne und sichere Bewer-
tung des neuen Arzneimittels erfolgen.
Dies ist ein völlig neues Vorgehen einer Zulassungsbehörde,
das getragen wird durch äußerst kompetente und souverän
agierende Experten und durch die Vorteile eines einheitlichen,
zentralen Zulassungsprozesses.
Was muss ein Biosimilar nachweisen, bevor es von der EMA zugelassen wird?
Um die Zulassung für ein Biosimilar zu erhalten, muss der
Hersteller die Qualität, Sicherheit und die Wirksamkeit im
Vergleich zur Referenzarznei belegen. Dieses Verfahren ist
wesentlich aufwändiger und kostspieliger als das Zulassungs-
verfahren für klassische Generika.
Es müssen Daten zur pharmazeutischen Qualität und Daten
aus präklinischen Untersuchungen ebenso erhoben werden
wie Daten aus breit angelegten klinischen Studienprogrammen
Zulassung, Wirksamkeit und Qualität von Biosimilars
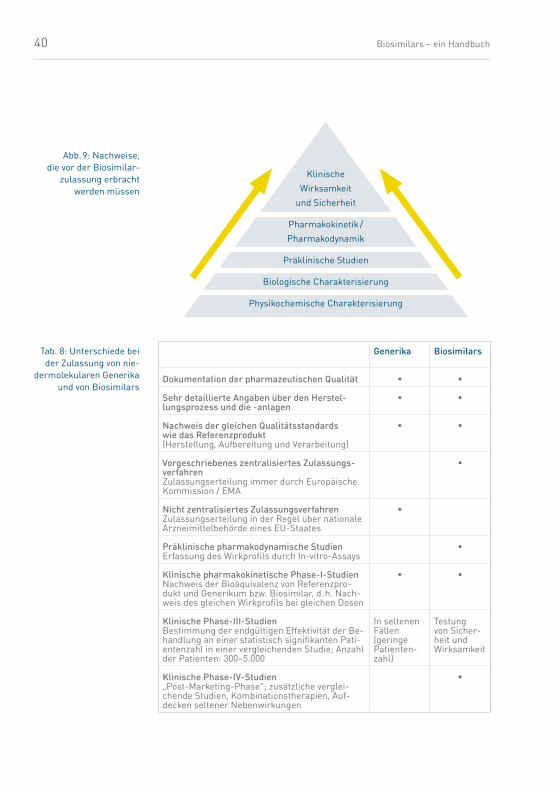
40 Biosimilars – ein Handbuch
Abb. 9: Nachweise, die vor der Biosimilar-
zulassung erbracht werden müssen
Tab. 8: Unterschiede bei der Zulassung von nie-
dermolekularen Generika und von Biosimilars
Generika Biosimilars
Dokumentation der pharmazeutischen Qualität • •
Sehr detaillierte Angaben über den Herstel-lungsprozess und die -anlagen
• •
Nachweis der gleichen Qualitätsstandards wie das Referenzprodukt (Herstellung, Aufbereitung und Verarbeitung)
• •
Vorgeschriebenes zentralisiertes Zulassungs-verfahrenZulassungserteilung immer durch Europäische Kommission / EMA
•
Nicht zentralisiertes ZulassungsverfahrenZulassungserteilung in der Regel über nationale Arzneimittelbehörde eines EU-Staates
•
Präklinische pharmakodynamische StudienErfassung des Wirkprofils durch In-vitro-Assays
•
Klinische pharmakokinetische Phase-I-StudienNachweis der Bioäquivalenz von Referenzpro-dukt und Generikum bzw. Biosimilar, d. h. Nach-weis des gleichen Wirkprofils bei gleichen Dosen
• •
Klinische Phase-III-StudienBestimmung der endgültigen Effektivität der Be-handlung an einer statistisch signifikanten Pati-entenzahl in einer vergleichenden Studie; Anzahl der Patienten: 300–5.000
In seltenen Fällen (geringe Patienten-zahl)
Testung von Sicher-heit undWirksamkeit
Klinische Phase-IV-Studien„Post-Marketing-Phase“; zusätzliche verglei-chende Studien, Kombinationstherapien, Auf-decken seltener Nebenwirkungen
•
Klinische
Wirksamkeit
und Sicherheit
Pharmakokinetik /
Pharmakodynamik
Präklinische Studien
Biologische Charakterisierung
Physikochemische Charakterisierung

41
an Patienten. Alle zu erhebenden Daten zielen darauf ab,
Vergleichbarkeit mit der Referenzarznei zu belegen.
Das Verfahren ist sequenziell ausgelegt und wird als
„comparability exercise“ bezeichnet.
• Im ersten Schritt werden die Vergleichbarkeit der Qualität
bzw. die physikalisch-chemische und die biologische Ver-
gleichbarkeit demonstriert. Hier wird die strukturelle
Übereinstimmung von Biosimilar und Referenzarznei in
allen relevanten Details mit einem riesigen Spektrum an
analytischen Methoden belegt. Mögliche Abweichungen
von den Daten der Referenzarznei müssen plausibel erklärt
werden. Ferner wird die Reinheit des Biosimilars überprüft.
Das Produkt wird nur freigegeben, wenn im Vorfeld
definierte Spezifikationskriterien erfüllt werden.
• Im zweiten Schritt werden Biosimilar und Referenzarznei
im präklinischen Setting miteinander verglichen. In aller
Regel kann hier auf ein verkürztes Verfahren in Form von
In-vitro-Untersuchungen zugegriffen werden. Diese Unter-
suchungen sind in produktspezifischen Richtlinien durch
die EMA vorgegeben. Die Pharmakokinetik- und die Phar-
makodynamik-Parameter und deren vordefinierter Grad
der Ähnlichkeit mit der Referenzarznei müssen begründet
und getroffen werden.
• Im dritten Schritt wird dann die klinische Vergleichbarkeit
belegt. Diese Studien haben mehr den Charakter von Si-
cherheitsstudien als von Wirksamkeitsstudien. Denn wenn
im ersten Schritt der „comparability exercise“ belegt ist,
Biosimilars müssen für ihre Zulassung deutlich umfang reichere Nachweise erbrin-gen als Generika.
Zulassung, Wirksamkeit und Qualität von Biosimilars

42 Biosimilars – ein Handbuch
dass Biosimilar und Referenzarznei ausreichend ähnlich
sind, dann ist auch damit zu rechnen, dass sie klinisch
äquivalent wirken. Zwingend sind diese klinischen Studien
jedoch gefordert, um die Verträglichkeit der Biosimilars zu
belegen. Denn die Herstellungsprozesse von Biosimilar und
Referenzarznei sind zwangsläufig unterschiedlich, so dass
nicht ausgeschlossen werden kann, dass analytisch nicht
oder nur schwer fassbare Komponenten eine klinische
Auffälligkeit provozieren. In klinischen Phase-I-Studien liegt
der Fokus zunächst auf der Toxikologie, der Pharmakokine-
tik und der Pharmakodynamik. Das heißt, der Wirkstoff wird
auf seine Reinheit und Unbedenklichkeit hin geprüft, seine
Verarbeitung im Körper nachvollzogen (Aufnahme, Vertei-
lung im Körper, biochemischer Auf- und Umbau sowie
Ausscheidung) und ein Wirkprofil erstellt. Daran schließen
sich Studien zur Wirksamkeit und Sicherheit im Sinne von
Schwere und Häufigkeit verschiedener Nebenwirkungen bei
einer oder mehreren repräsentativen Indikationen an, um
ein vergleichbares Wirksamkeits- und Sicherheitsprofil zu
demonstrieren. Hierzu zählt auch ein vergleichbares
Im munogenitätsprofil von Biosimilar und Referenzarznei.
Schwerpunkte und Anforderungen an diese Phase-III-Studi-
en sind je nach Biosimilarklasse verschieden. Entsprechend
der Unterschiedlichkeit und Komplexität von biologischen
Pharmazeutika legt die EMA die Anforderungen jedoch
individuell und an den Leitlinien orientiert mit den Herstel-
lern fest.
Relevant für die Zulassung eines
Biosimilars ist vor allem der umfang-
reiche Nachweis der Vergleichbar-
keit zum Referenz-arzneimittel.

43
Gelten dieselben Anforderungen an Biosimilars wie an Referenzpräparate?
Prinzipiell gelten für die Zulassung von Biosimilars die
gleichen Anforderungen wie für die Zulassung von Innova-
torpräparaten. Für die Qualitätsaspekte gilt dies ohne
Einschränkungen. Lediglich die klinischen Untersuchungs-
programme können in einem verkürzten Setting absolviert
werden. So können Phase-II-Studien der Dosisfindung in der
Regel entfallen, da die pharmazeutische Formulierung, die
Wirkstärke und der Darreichungsweg des Biosimilars mit
dem Referenzprodukt übereinstimmen müssen, so dass hier
keine Abweichungen zu erwarten sind und die Vorgaben der
Referenzarznei auch für das Biosimilar gelten. Der Schwer-
punkt der Phase-III-Studien liegt eher auf dem Nachweis
einer guten Verträglichkeit und weniger auf dem Nachweis
der Wirksamkeit, die ja durch den Wirkstoff vorgegeben ist,
dessen Vergleichbarkeit mit der Referenzarznei bereits
belegt wurde.
Auf Basis der Studiendaten, der umfangreichen
Dokumentation des Herstellungs- und des Ana-
lyseprozesses sowie durch eine Inspektion vor
Ort bewertet die EMA in einem zentralen Verfah-
ren abschließend das Biosimilar.
Eine Zulassung erfolgt erst dann, wenn die EMA
anhand der eingereichten Dokumentation die
prinzipielle Ähnlichkeit zum Referenzpräparat und
damit die Qualität, Wirksamkeit und Verträglich-
keit bescheinigt hat.
Biosimilars müs-sen die gleichen Zulassungsanfor-derungen erfüllen wie das Referenz-präparat.
Zulassung, Wirksamkeit und Qualität von Biosimilars

44 Biosimilars – ein Handbuch
Marktzugang erhält das Biosimilarpräparat dann, wenn das
Referenzpräparat seinen Patentschutz verloren hat. Unter-
nehmen, die Biosimilars entwickeln, können jedoch bereits
vor Ablauf des Patents Studien, Versuche und weitere erfor-
derliche Schritte unternehmen, um zeitnah nach dem Stichtag
des Patentschutzverlustes mit ihrem Produkt auf den Markt
gehen zu können.
INN Anzahl der Anträge
Adalimumab 4
Etanercept 2
Insulin glargin 1
Pegfilgrastim 3
Rituximab 2
„Ähnlich“ statt „identisch“:Wie „gleich“ sind Biosimilars?
Die enorme strukturelle Komplexität von Proteinen lässt es
praktisch nicht zu, dass eine Präparation eines bestimmten
Proteins in absolut reiner Form vorliegt. Einen derartigen
Reinheitsgrad kann man nur in einem Kristall eines Proteins
erwarten. Eine Proteinlösung enthält dagegen immer auch
strukturell leicht unterschiedliche Molekülformen, die dadurch
entstehen, dass einzelne Aminosäuren durch chemische Reak-
tionen (Deamidierung oder Oxidation) verändert wurden, dass
an den Enden Aminosäuren abgeschnitten wurden, dass sich
Proteinlösungen enthalten immer und natürlicher-
weise eine hetero-gene Population
eines bestimmten Moleküls.
15 http://www.ema.europa.eu/docs/en_GB/document_library/Report/2016/12/ WC500217738.pdf
Tab. 9: Biosimilars, die derzeit von der
EMA evaluiert werden (Stand Dezember 2016)15

45
die Tertiärstruktur in Teilen verändert hat oder dass sich
Aggregate gebildet haben.
Alle diese Modifikationen, die letztlich nur einen sehr kleinen
Teil der Proteinpräparation ausmachen, lassen sich heute
analytisch nachweisen. Da sie de facto unvermeidlich sind,
müssen und können sie auch akzeptiert werden, vorausge-
setzt, sie verursachen keine auffälligen Reaktionen, wenn die
Proteinpräparation beim Menschen zu Therapiezwecken
eingesetzt wird.
Um hier Sicherheit zu garantieren, werden die Bedingungen
der Herstellung und Lagerung von Biopharmazeutika extrem
konstant gehalten. Dies wiederum garantiert auch eine ge-
wisse Konstanz der Heterogenität der Präparationen, die
durch Spezifikationsgrenzen nach oben und nach unten
definiert sind.
Untersucht man einzelne Herstellungschargen eines Bio-
pharmazeutikums hinsichtlich einer bestimmten Spezifika-
tion, so erkennt man sehr klar die unvermeidbare Variation,
bei recht großer Ähnlichkeit.
Innerhalb dieser Spezifikationsgrenzen haben sich heute auch
Biosimilars zu bewegen. Man kann daher in erster Näherung
konstatieren, dass Biosimilars in dem Maße der Referenz-
arznei ähneln, wie sich einzelne Chargen der Referenzarznei
untereinander ähneln.
Biosimilars können der Referenzarznei so gleichen, wie sich unter-schiedliche Chargen der Referenzarznei unterein ander gleichen.
Zulassung, Wirksamkeit und Qualität von Biosimilars
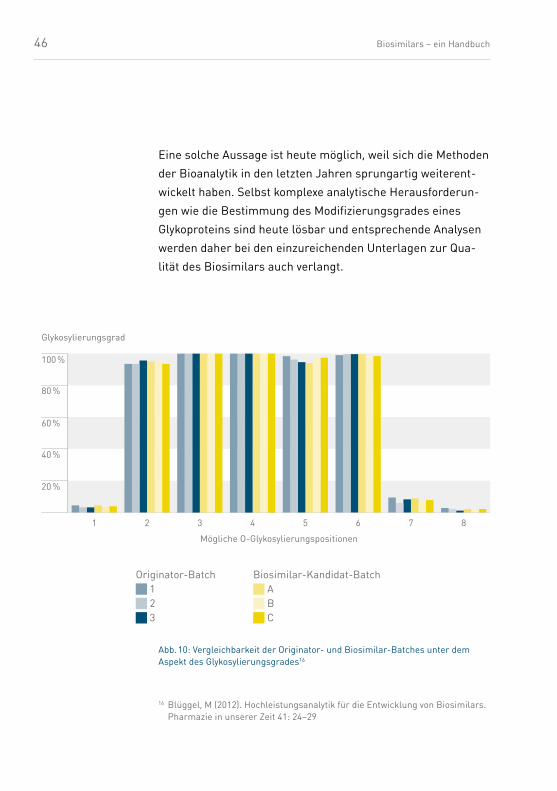
46 Biosimilars – ein Handbuch
Eine solche Aussage ist heute möglich, weil sich die Methoden
der Bioanalytik in den letzten Jahren sprungartig weiterent-
wickelt haben. Selbst komplexe analytische Herausforderun-
gen wie die Bestimmung des Modifizierungsgrades eines
Glykoproteins sind heute lösbar und entsprechende Analysen
werden daher bei den einzureichenden Unterlagen zur Qua-
lität des Biosimilars auch verlangt.
Abb. 10: Vergleichbarkeit der Originator- und Biosimilar-Batches unter dem Aspekt des Glykosylierungsgrades16
Originator-Batch 1 2 3
Biosimilar-Kandidat-Batch A B C
2 3 4 5 6 7 81
Glykosylierungsgrad
20 %
40 %
60 %
80 %
100 %
Mögliche O-Glykosylierungspositionen
16 Blüggel, M (2012). Hochleistungsanalytik für die Entwicklung von Biosimilars. Pharmazie in unserer Zeit 41: 24–29

47
Wie wird die Qualität von Biosimilars sichergestellt? Besondere Rolle der EMA
Biosimilars werden in Europa ausnahmslos von der Europä-
ischen Kommission unter Mitwirkung der Europäischen Arz-
neimittel-Agentur (EMA) und des dort angesiedelten Aus-
schusses für Humanarzneimittel (CHMP) zugelassen.17 Für
die EMA arbeiten insgesamt rund 3.500 Sachverständige. Erst
wenn der Ausschuss für Humanarzneimittel (CHMP) feststellt,
dass die Qualität, Sicherheit und Wirksamkeit des zu beurtei-
lenden Arzneimittels ausreichend belegt wurden, gibt er
„grünes Licht“ für die Zulassungserteilung.
Dem CHMP arbeitet unter anderem die Arbeitsgruppe für
Biosimilars (Similar Biological (Biosimilar) Medicinal Products
Working Party; BMWP) zu. Diese erstellt die Leitlinien zur
Qualität, Wirksamkeit und Sicherheit von Biosimilars. Die
BMWP-Arbeitsgruppe gibt dem CHMP wissenschaftlichen Rat
zu Fragen der generellen Sicherheit von Biosimilars und zu
deren Vergleichbarkeit zum Original. Außerdem unterhält sie
Kooperationen mit Nicht-EU-Arzneimittelbehörden, mit der
Weltgesundheitsorganisation, mit Patientenorganisationen,
mit Arzneimittelentwicklern und mit Fachkreisen.
Durch das bei der EMA angesiedelte zentrale Zulassungsver-
fahren wird sichergestellt, dass alle in Europa zugelassenen
Biosimilars einem einheitlichen, extrem hohen Qualitätsstan-
dard genügen. Dieser wiederum ist die Basis dafür, dass über
Alle Biosimilars werden in Europa durch die Euro-päische Kommission zuge lassen.
Die von der EMA zur Zulassung empfoh-lenen Wirkstoffe erfüllen einen hohen Sicherheitsstandard.
17 Die Richtlinie 93/41/EWG sah die Einführung eines zentralisierten Zulas- sungsverfahrens auf der Grundlage der Verordnung (EWG) Nr. 2309/93 vor. Das zentralisierte Verfahren trat 1995 in Kraft, im selben Jahr nahm die EMA ihre Tätigkeit auf. An Stelle der Verordnung Nr. 2309/93 trat später die Verord- nung (EG Nr. 726/2004 )
Zulassung, Wirksamkeit und Qualität von Biosimilars

48 Biosimilars – ein Handbuch
mehr als zehn Jahre weder neue unerwünschte Arzneimittel-
wirkungen durch Biosimilars gemeldet wurden noch dass
einem Biosimilar aus Sicherheitsgründen der Marktzugang
entzogen werden musste.
Wie weisen Biosimilars ihre gleiche Wirksamkeit nach?
Da Biosimilars per definitionem einer Referenzarznei ähnlich
sein müssen und dies auch mit einem umfangreichen Daten-
satz zu anspruchsvollen Analysen dokumentiert haben, kann
bereits theoretisch gefolgert werden, dass die Wirksamkeit
des Biosimilarwirkstoffs dem der Referenzarznei entsprechen
muss. Dies wird jedoch in einem präklinischen und
klinischen Untersuchungsprogramm auch empirisch belegt,
wobei das zu absolvierende Programm von der EMA in Form
produktspezifischer Richtlinien bindend vorgegeben ist.
Das Ergebnis aller Prüfungen muss die Äquivalenz der Wirk-
samkeit von Biosimilar und Referenzarznei sein. Das Biosimi-
lar darf weder schlechter noch besser abschneiden. Sollte
dies der Fall sein, kann eine Zulassung nach den Regeln
dieses Verfahrens nicht erteilt werden. Somit können Ärzte,
Apotheker und Patienten davon ausgehen, dass ein als Bio-
similar ausgewiesenes Biopharmazeutikum mit einer Zulas-
sung durch die Europäische Kommission in seiner Wirksam-
keit tatsächlich der Referenzarznei analog ist.
Biosimilars müs-sen die gleiche
Wirksamkeit zeigen wie die Referenz-
arznei.

49
Extrapolation der Indikationen
Ist das Referenzprodukt für mehr als eine Indikation zugelas-
sen, müssen im strengen Sinne die Wirksamkeit und Sicher-
heit des Biosimilars für jede einzelne Indikation über entspre-
chende Tests und Studien nachgewiesen werden.
Unter bestimmten Bedingungen, basierend auf einer Fall-zu-
Fall-Bewertung, erlaubt die EMA jedoch eine Extrapolation ei-
ner Indikation. Dies ist z. B. dann möglich, wenn ein Biosimilar
bereits eine zur Referenzarznei vergleichbare Sicherheit und
Wirksamkeit in einer sehr sensitiven Indikation gezeigt hat.
Voraussetzung ist allerdings, dass den unterschiedlichen
Indikationen der gleiche Wirkmechanismus des Wirkstoffs
zugrunde liegt und es keine wissenschaftlichen Einwände gibt.
Somit muss der Hersteller seinen Antrag auf Indikations-
erweiterung ohne zusätzliche klinische Studien detailliert
wissenschaftlich begründen. Auf Basis dieser Begründung
entscheidet dann die EMA und erteilt oder versagt die bean-
tragte Indikationserweiterung.
Zulassung, Wirksamkeit und Qualität von Biosimilars

50 Biosimilars – ein Handbuch
Welche Vorgaben der Arzneimittel überwachung gibt es für Biosimilars?
Wie bei den meisten neu zugelassenen Arzneimitteln ist der
Hersteller eines Biosimilars nach der Zulassung durch die
Europäische Kommission verpflichtet, einen Riskmanage-
ment-Plan vorzulegen. Dieser umfasst ein Pharmakovigilanz-
System, ein Meldesystem für unerwünschte Arzneimittel-
wirkungen (adverse event reporting), Sicherheitsstudien, die
auch von der Referenzarznei durchzuführen sind, Folgeunter-
suchungen bei Studienpatienten sowie spezielle pharma-
koepidemiologische Studien (z. B. Auswertung von Patienten-
Datenbanken). So werden über 2.000 Patienten beobachtet,
die mit rekombinantem Wachstumshormon behandelt werden,
ob sie beispielsweise einen Diabetes entwickeln. Es werden
über 4.000 Patienten beobachtet, die mit Epoetin-Wirkstoffen
behandelt werden, ob sie unter Thrombosen leiden, eine PRCA
(pure red cell aplasia) entwickeln, oder ob eventuell ein
Tumorwachstum stimuliert wird. Und es werden Patienten
beobachtet, die mit Filgrastim-Wirkstoffen behandelt werden,
wobei die Stimulierung bei chronischer Neutropenie oder die
Mobilisierung von Stammzellen gemeldet werden.
Solche Maßnahmen werden verlangt, obwohl durch den jah-
relangen Einsatz der Referenzarznei bereits sehr viel über
potenzielle unerwünschte Arzneimittelwirkungen bekannt
ist. Und grundsätzlich ist bei einem zugelassenen Biosimi-
lar auch von keinem höheren Sicherheitsrisiko auszugehen,
als dies dem Referenzprodukt zu eigen ist.
Von zugelassenen Biosimilars geht
kein höheres Sicherheitsrisiko aus als vom Refe-renzarzneimittel.
Teilnehmer an kli-nischen Studien werden auch bei Biosimilars über
Jahre beobachtet und untersucht.

51
Bei Biosimilars muss wie bei an- deren Biopharma- zeu tika auf die Immunogenität der Moleküle geachtet werden.
Zulassung, Wirksamkeit und Qualität von Biosimilars
Dennoch gehört die Überwachung eines Arzneimittels
nach seiner Einführung mit Blick auf Sicherheit und Neben-
wirkungen zu einem wichtigen Standbein der generellen
Arzneimittelsicherheit, dem sich natürlich auch Biosimilars
stellen.
Besondere Aufmerksamkeit verdienen dabei unerwünschte
immunologische Reaktionen. Hier liegt keine besondere
Gefahr bei den Biosimilars. Vielmehr muss bei Biopharma-
zeutika generell ein besonderes Augenmerk auf immunolo-
gische Unverträglichkeiten gelegt werden. Dies liegt zum
einen in der makromolekularen Struktur begründet. Zum
anderen können minimale Verunreinigungen aus der Wirts-
zelle oder dem Fermentationsmedium eine immunologische
Reaktion provozieren.
Die Erfahrung hat aber gezeigt, dass dieses Problem in der
Praxis viel kleiner ist, als es ursprünglich antizipiert wurde.
Biosimilars, so das Resümee aus millionenfachem Einsatz,
sind ebenso sicher einzusetzen wie die entsprechende Refe-
renzarznei.

52 Biosimilars – ein Handbuch
Situation außerhalb der EU
Nicht nur in Europa gibt es ein Regelwerk für die Zulassung
von Nachfolgeprodukten von patentfreien Biopharmazeutika.
Allerdings sind die Standards bisher keinesfalls harmonisiert,
weshalb eine gegenseitige Anerkennung auch nicht möglich ist.
Die EMA kooperiert mit folgenden Partnern:
• Health Canada (eine finalisierte Richtlinie „Guidance on
Subsequent Entry Biologics“ wurde im März 2010 publiziert)
• Japan (eine Richtlinie „Guideline on quality, safety and effi-
cacy of follow-on biologics“ wurde im März 2009 publiziert)
• WHO (eine Richtlinie „Guidelines on Evaluation of Similar
Biotherapeutic Products“ wurde im Oktober 2009 verab-
schiedet)
• FDA (Die Richtlinie „Scientific Considerations in Demons-
trating Biosimilarity to a Reference Product“18 und die
Richtlinie „Biosimilars: Questions and Answers Regarding
Implementation of the Biologics Price Competition and
Innovation Act of 2009“19 wurde am 28.4.2015 publiziert.
Andere Richtlinien liegen bisher noch als Diskussions-
entwurf vor20).
18 http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatory Information/Guidances/UCM291128.pdf19 http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatory Information/Guidances/UCM444661.pdf20 http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/ Guidances/ucm290967.htm

53
Ferner wurden die CHMP-Guidelines der EMA von den Zulas-
sungsbehörden in Australien und Malaysia übernommen.
Letztlich wird aber zu Recht gefordert, dass in Europa ver-
kehrsfähige Nachfolgeprodukte von Biopharmazeutika das
europäische Zulassungsverfahren durchlaufen müssen, um
hier den Status eines Biosimilars zu erlangen. Das ist für viele
im Weltmarkt befindliche Produkte nicht der Fall. Vor allem
aus Indien kommen eine ganze Reihe solcher Produkte, von
denen es zum Teil in Europa noch keine Vertreter gibt.
So ist z. B. in Indien bereits ein monoklonaler Antikörper
Reditux®, ein Nachahmerprodukt für Rituximab, seit 2007
zugelassen. Mit der Zulassung eines Rituximab-Biosimilars
in Europa wird für 2017 gerechnet. Auch ist seit 2007 ein
Nachahmerprodukt des Interferons alfa-2b in Indien auf dem
Markt. Seit 2003 sind auch Insulin-Nachahmerprodukte in
Indien verfügbar. Diese Präparationen haben die hohen Hür-
den der EMA nicht nehmen können und wurden entweder von
der Behörde abgelehnt, oder der pharmazeutische Hersteller
hat den Zulassungsantrag zurückgezogen. Ähnliches gilt für
eine rekombinante Hepatitis-B-Vakzine.
Biosimilars werden weltweit recht unterschiedlich reguliert.
Zulassung, Wirksamkeit und Qualität von Biosimilars

54 Biosimilars – ein Handbuch
Die Zulassung durch die EMA ist ein „Gütesiegel“,
auf das sich Ärzte und Patienten ver-
lassen können.
Diese Punkte machen einiges deutlich:
1. Die Zulassung eines Biosimilars durch die EMA ist ein an-
spruchsvoller und ein strikt auf Qualität und Sicherheit aus-
gelegter Prozess, der alles andere als ein „Selbstläufer“ ist.
2. Nicht jedes Nachahmermolekül aus der Klasse der Bio-
pharmazeutika ist ein Biosimilar nach europäischem Stan-
dard. Nicht in Europa zugelassene Nachahmerpräparate
besitzen hier auch keine Verkehrsfähigkeit und dürfen am
Patienten nicht eingesetzt werden.
3. Das zeigt: Die Zulassung durch die EMA ist ein „Gütesiegel“,
auf das sich Ärzte und Patienten verlassen können. Sie kön-
nen in aller Regel in den gleichen Indikationen eingesetzt
werden, für die die Referenzarznei eine Zulassung besitzt.
Bei nicht vortherapierten Patienten macht es faktisch kei-
nen Unterschied, ob die Referenzarznei oder ein Biosimilar
eingesetzt wird. Dies gilt für die Wirksamkeit ebenso wie
für die Verträglichkeit.

55Zulassung, Wirksamkeit und Qualität von Biosimilars
Mythen und Fakten zu Biosimilars
Biosimilars können ebenso wie
Generika zu einem sehr geringen
Preis angeboten werden.
Biosimilars werden irgendwo
auf dieser Welt produziert und
zugelassen und können dann in
Deutschland eingesetzt werden.
Biosimilars sind von schlech-
terer Qualität als das Referenz-
arzneimittel.
Bei Biosimilars treten häufiger
Nebenwirkungen auf.
Die Einführung von Biosimilars
birgt die Gefahr, dass neue uner-
wünschte Arzneimittelwirkungen
für einen lange eingeführten
Wirkstoff auftreten.
Biosimilars sind wesentlich
immunogener.
Auch Biosimilars müssen in einem aufwändigen Verfahren
hergestellt und extrem umfangreich getestet werden. Des-
halb können Biosimilars zwar etwas preiswerter als die Refe-
renzarznei sein, aber nicht beliebig billig angeboten werden.
Nur Nachahmerprodukte, die ein Zulassungsverfahren bei
der EMA erfolgreich durchlaufen haben, sind in Deutsch-
land verkehrsfähig und dürfen als Biosimilars bezeichnet
werden.
Biosimilars müssen in den Zulassungsstudien zeigen, dass
sowohl das Wirkungs- als auch das Nebenwirkungsspek-
trum vergleichbar ist mit dem des Referenzpräparats.
Hersteller von Biosimilars müssen für die Zulassung, die
Qualität, Sicherheit und Wirksamkeit nach den gleichen
Standards nachweisen, die auch für die Zulassung der
Referenzarznei gegolten haben.
Biopharmazeutika müssen generell hinsichtlich ihrer
Immunogenität bewertet werden. Dies gilt auch für Biosimi-
lars. Und auch hier gilt das Ähnlichkeitsprinzip zwischen
Referenzarznei und Biosimilar.
Seit Zulassung der ersten Biosimilars wurden weder
neue unerwünschte Arzneimittelwirkungen durch diese
Wirkstoffe gemeldet, noch musste einem Biosimilar aus
Sicherheitsgründen die Marktzulassung entzogen werden.
Mythos Fakt
Tab. 10: Mythen und Fakten zu Biosimilars

56 Biosimilars – Ein Handbuch
BIOSIMILARS IN DER VERSORGUNG
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
Kapitel
3

57
BIOSIMILARS IN DER VERSORGUNG
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
In welchen Indikationen kommen Biosimilars aktuell zum Einsatz?
Derzeit gibt es in Deutschland Biosimilars in sieben verschie-
denen Wirkstoffgruppen: Epoetin, Etanercept, Filgrastim,
Follitropin, Infliximab, Insulin glargin und Somatropin.
Zudem befinden sich aktuell (Stand Dezember 2016) zwölf
Wirkstoffe (Adalimumab (4), Etanercept (2), Insulin glargin,
Pegfilgrastim (3) und Rituximab (2)) im Zulassungsprozess.21
In einer fortgeschrittenen Entwicklung befinden sich außer-
dem Biosimilars zu Bevacizumab, Cetuximab, Darbepoetin
alfa, Insulin aspart, Peginterferon alfa-2a, Ranibizumab und
Trastuzumab.
Insgesamt wurden bisher 32 Anträge durch die EMA abschlie-
ßend begutachtet. In 23 Fällen wurde die Zulassung erteilt,
sieben Anträge wurden von den Herstellern zurückgezogen,
und in zwei Fällen wurde eine Zulassung versagt.
Diese Zusammenfassung lässt wichtige Schlüsse zu:
1. Der Zulassungsprozess ist eine substanzielle Hürde, bei
der alle Aspekte der Konzipierung und der Realisierung der
Herstellung eines Biosimilars auf einen kompromisslosen
Prüfstand gestellt werden. Eine Zulassung wird nur dann
erteilt, wenn Qualität, Wirksamkeit und Verträglichkeit auf
Basis vordefinierter Kriterien belegt sind. Die Tatsache,
Bisher haben Biosimilars von Enoxaparin, Epoe-tin, Etanercept, Fil-grastim, Infliximab, Insulin glargin und Somatropin das strenge Zulassungsver-fahren der EMA erfolgreich durch-laufen.
21 http://www.ema.europa.eu/docs/en_GB/document_library/Report/2016/12/ WC500217738.pdf
Biosimilars in der Versorgung

58 Biosimilars – ein Handbuch
dass Anträge auch negativ beschieden wurden (siehe Kap. 2,
Tab. 6), zeigt, dass Zulassungsanträge für Biosimilars von
der EMA nicht einfach „durchgewinkt“ werden, und dass
man sich auf das Urteil der EMA verlassen kann.
2. Das noch relative kleine Indikationsspektrum, das derzeit
durch Biosimilars bedient werden kann, wird sich in abseh-
barer Zeit deutlich ausweiten. Das ist eine gute Nachricht
für viele chronisch Kranke, die künftig noch besser mit
kostengünstigen Medikamenten versorgt werden können.
3. Auch werden etliche Biosimilars aus der Klasse der Tumor-
therapeutika die Therapieoptionen signifikant erweitern.
Hierbei handelt es sich in erster Linie um rekombinante,
monoklonale Antikörper.
4. Es ist zudem damit zu rechnen, dass durch die Verfügbar-
keit von Biosimilars im Bereich dieses hochpreisigen Arz-
neimittelsegments Interventionsstrategien, die derzeit noch
wegen limitierter klinischer Erfahrung als Second- oder
Third-Line-Option eingestuft werden, künftig als erste Inter-
ventionsoption zugelassen werden.
5. Vor allem wird das Gesundheitssystem von der Ausweitung
des Wirkstoffspektrums an Biosimilars profitieren. Die
Kostenträger rechnen mit einem Einsparungspotenzial in
Milliardenhöhe.
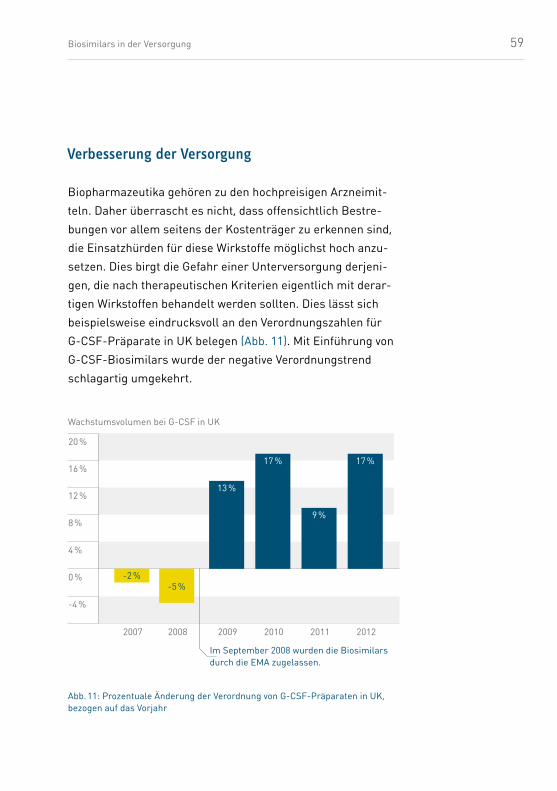
59
Verbesserung der Versorgung
Biopharmazeutika gehören zu den hochpreisigen Arzneimit-
teln. Daher überrascht es nicht, dass offensichtlich Bestre-
bungen vor allem seitens der Kostenträger zu erkennen sind,
die Einsatzhürden für diese Wirkstoffe möglichst hoch anzu-
setzen. Dies birgt die Gefahr einer Unterversorgung derjeni-
gen, die nach therapeutischen Kriterien eigentlich mit derar-
tigen Wirkstoffen behandelt werden sollten. Dies lässt sich
beispielsweise eindrucksvoll an den Verordnungszahlen für
G-CSF-Präparate in UK belegen (Abb. 11). Mit Einführung von
G-CSF-Biosimilars wurde der negative Verordnungstrend
schlagartig umgekehrt.
Abb. 11: Prozentuale Änderung der Verordnung von G-CSF-Präparaten in UK, bezogen auf das Vorjahr
Wachstumsvolumen bei G-CSF in UK
-4 %
4 %
12 %
0 %
8 %
16 %
20 %
9 %
17 %
-2 %-5 %
17 %
13 %
2007 2008 2009 2010 2011 2012
Im September 2008 wurden die Biosimilars durch die EMA zugelassen.
Biosimilars in der Versorgung

60 Biosimilars – ein Handbuch
Ähnliche Entwicklungen sind auch für andere Indikationen
zu erwarten, sobald günstigere Biosimilarpräparate verfügbar
werden. Hiervon werden im besonderen Maße Tumorpatien ten
profitieren. Ferner können auch Rheuma- und Psoriasis-
Patienten mit einer besseren medikamentösen Versorgung
rechnen.
Erweiterung des Indikationsspektrums
Während der ersten zehn Jahre seit der Verfügbarkeit von Bio-
similars wurden in erster Linie die Indikationsgebiete Wachs-
tumsstörungen (Somatropin), Onkologie (Epoetine, Granulo-
zyten-Koloniestimulierender Faktor) und Nephrologie (Epoe-
tine) durch Biosimilars bedient. Das beginnt sich nun deutlich
zu ändern. Hinzugekommen sind bereits die Indikationsgebiete
Rheumatologie, Gastroenterologie und Dermatologie (Inflixi-
Abb. 12: Indikationsspektrum, das durch zugelassene Biosimilars und durch Biosimilars in der fortgeschrittenen Entwicklung abgedeckt wird
BIO-SIMILARS
Nephro logie
Diabetes
Onkologie
Wachstums-störungen
Fertilitäts-störun gen
Alters- bedingte Makula-
Dege nera- tion
Chro nisch entzündliche
Erkrankungen
Hepatitis-C-Infektion

61
mab, Etanercept), Diabetes (Insulin glargin) und Fertilitätsstö-
rungen (Follitropin). Weitere Indikationen werden durch Biosi-
milars in fortgeschrittener Entwicklung zeitnah hinzukommen.
Mit einem Insulin-aspart-Biosimilar wird in absehbarer Zeit
neben dem bereits zugelassenen Insulin-glargin-Biosimilar
ein weiteres modernes Insulin-Biosimilar für den Diabetes-
Markt verfügbar. Zwar zählen Insulin-Präparate zu den eher
preisgünstigeren Biopharmazeutika. Wegen der enormen Grö-
ße des Diabetes-Marktes und der weiter steigenden Prävalenz
des Typ-2-Diabetes wird mit Einführung der Insulin-Biosimi-
lars dennoch mit einer deutlichen Entlastung bei den Arznei-
mittelkosten gerechnet. Zudem werden zunehmend mehr Pa-
tientinnen und Patienten, die heute noch aus Kostengründen
mit unmodifiziertem Humaninsulin behandelt werden, von den
modernen Analog-Insulinen profitieren können.
Weiter wird die Onkologie von den in der Entwicklung befind-
lichen Biosimilars in erheblichem Maße profitieren. Hierbei
handelt es sich fast durchweg um sehr hochpreisige Wirk-
stoffe. Durch die Zulassung weiterer Biosimilars in diesem
Bereich ist nicht nur mit erheblichen Einsparungen zu rech-
nen. Man kann auch davon ausgehen, dass bestimmte Wirk-
stoffe aus der Second- oder Third-Line-Therapie den Sprung
in die First-Line-Therapie schaffen werden, wodurch das Ver-
ordnungsvolumen deutlich gesteigert wird. Auch dies wird
letztlich wieder den schwer kranken Patienten zugutekommen.
Unter den neuen Krebstherapeutika befinden sich mit Beva-
cizumab, Cetuximab, Rituximab und Trastuzumab vier Anti-
körper. Lange Zeit wurde bezweifelt, dass sich rekombinante,
Mit Insulin-Bio-similars werden bei dem steigenden Bedarf erhebliche Therapiekosten eingespart.
Biosimilars in der Versorgung

62 Biosimilars – ein Handbuch
monoklonale Antikörper als Biosimilars herstellen lassen. Die
aktuellen Entwicklungen widerlegen diese pessimistische
Prognose, denn mit Infliximab hat tatsächlich das erste mono-
klonale Antikörper-Biosimilar die Hürde der europäischen
Zulassung genommen. Ferner wurde kürzlich mit einem
Etanercept-Biosimilar ein Fusionsprotein als Biosimilar zuge-
lassen. Zudem wird mit PEG-Filgrastim auch ein Biosimilar
für die supportive Therapie von Tumorerkrankungen verfügbar.
Mit Ranibizumab als Biosimilar wird ein Antikörperfragment
eingeführt, das große Bedeutung bei der Behandlung der
Altersbedingten Makula-Degeneration (AMD) erlangt hat.
Diese Therapie noch weiter verfügbar zu machen, kann als
echter Fortschritt bei der Bedienung wichtiger „medical
needs“ bezeichnet werden, denn nach wie vor bildet die
AMD die primäre Ursache für Altersblindheit, obwohl sie
mit einer lokalen Antiangiogenese-Strategie sehr gut thera-
pierbar ist.
Ein Adalimumab-Biosimilars wird neben den bereits verfüg-
baren Biosimilars für Infliximab und Etanercept das Gesund-
heitssystem im Bereich vieler chronisch entzündlicher Krank-
heiten deutlich entlasten. Dies trifft auch für Antikörper zu, die
gegen B-Zellen gerichtet sind und die heute noch meist aus-
schließlich bei B-Zell-Tumoren eingesetzt werden. So hat kürz-
lich der Anti-CD20-Antikörper Rituximab auch eine Zulassung
für den Einsatz bei der rheumatoiden Arthritis bekommen.
Und mit Darbepoetin alfa und Peginterferon alfa-2a werden
Biosimilars die Behandlungsoptionen für Dialysepatienten
und von Hepatitis-C-Infektionen erweitern.
In der Tumor - therapie stehen
bald Biosimilars von monoklonalen
Antikörpern zur Verfügung.

63
Wann ist der Einsatz von Biosimilars sinnvoll?
Biosimilars sind nach strengen Kriterien auf Qualität, Wirk-
samkeit und Sicherheit geprüfte Arzneimittel, die zudem
durch die Bezeichnung „Biosimilar“ von der EMA, der zentra-
len Zulassungsbehörde in der EU, als „ähnlich“ zu einem seit
vielen Jahren im Markt befindlichen Referenzarzneimittel
klassifiziert wurden. Ihr Einsatz ist immer dann sinnvoll, wenn
bei einem Patienten oder einer Patientin eine Indikation für
das Arzneimittel angezeigt ist.
In aller Regel entsprechen die Indikationen, für die ein Biosi-
milar zugelassen ist, auch den Indikationen, die für die Refe-
renzarznei zum Zeitpunkt der Zulassung des Biosimilars aus-
gewiesen sind. Somit kann man schlussfolgern, dass die
Referenzarznei und ein Biosimilar therapeutisch äquivalent
sind. Daraus ergibt sich, dass für therapienaive Patientinnen
und Patienten sowohl die Referenzarznei als auch das Biosi-
milar eingesetzt werden können. Aus ökonomischen Überle-
gungen sollte allerdings die Wahl auf das Biosimilar fallen.
Der Beleg der prinzipiellen pharmazeutischen Äquivalenz von
Referenzarznei und dem entsprechenden Biosimilar durch die
EMA legt nahe, dass sich beide Präparate auch austauschen
lassen. Dies gilt in besonderem Maße dann, wenn es sich um
einen therapienaiven Patienten handelt. Hier macht es
keinen Unterschied, ob die Referenzarznei oder das Biosimilar
eingesetzt wird, so dass sich der Therapeut künftig in der
Biosimilars sind dann angezeigt, wenn die entspre-chende Indikation vorliegt.
Wo möglich, sollte aus Kostengründen das Biosimilar eingesetzt werden.
Biosimilars in der Versorgung
64 Biosimilars – ein Handbuch
Regel sicher für das kostengünstigere Präparat entscheiden
wird. Die Entscheidung, welches Arzneimittel eingesetzt wird,
sollte dabei ausschließlich vom behandelnden Arzt getroffen
werden.
Ist ein Patient hingegen auf ein bestimmtes Arzneimittel ein-
gestellt, sollte ein Austausch – obwohl prinzipiell möglich,
wie immer mehr empirische Daten auch tatsächlich zeigen –
kritisch hinterfragt werden. Ein solcher Austausch kann Adhä-
renzprobleme nach sich ziehen, denn nicht selten kennen
die chronisch kranken Patientinnen und Patienten ihre Arznei-
mittel sehr genau.
Warum ist der Einsatz von Biosimilars sinnvoll?
Die Kosten im Gesundheitswesen steigen rasant. Zwar ist
der Anteil der Arzneimittelkosten an den Gesamtkosten mit
ca. 16 % relativ gering. Allerdings sind die Kostensteige-
rungen in diesem Segment überproportional. Das liegt
daran, dass Innovationen, und im besonderen Maße innova-
tive Biopharmazeutika, ausgesprochen kostspielig sind.
Dies ist keineswegs unberechtigt, denn die Entwicklungs-
kosten sind immens. Zudem ist die Entwicklung eines Bio-
pharmazeutikums mit einem erheblichen unternehme-
rischen Risiko verbunden. „Drugability“, d. h. die Eignung
des zu adressierenden Targets, nach Modulation durch
einen Wirkstoff auch das Pathogenitätsgeschehen zu beein-
flussen, einerseits, als auch die Verträglichkeit des Wirk-
Biopharmazeutika sind nicht zuletzt
wegen der sehr ho-hen Entwicklungs-kosten sehr teuer.

65
stoffs andererseits, sind im Vorfeld der klinischen Testung
eines solchen Wirkstoffs – also bis kurz vor Abschluss der
Entwicklung – kaum zuverlässig vorhersehbar.
Besteht allerdings die Möglichkeit, in diesem innovativen
Segment mehr Wettbewerb zu generieren, muss die-
se Möglichkeit genutzt werden. Das ist der Fall, wenn der
Patentschutz für den Wirkstoff abgelaufen ist und der Wirk-
stoff somit formal den Status der Innovation verlassen hat.
Wie hoch beläuft sich bei konsequentem Einsatz von Biosimilars das Einsparpotenzial?
Biosimilars werden selbstverständlich preiswerter sein als
die entsprechenden Referenzarzneimittel. Und das muss
auch so sein. Zwar ist der Aufwand zur Herstellung beider
Arzneimittelgruppen ganz ähnlich und würde daher einen
Preisabschlag nicht zwingend rechtfertigen. Allerdings ent-
fällt bei der Herstellung eines Biosimilars ein guter Teil des
unternehmerischen Risikos, das eingegangen werden muss,
wenn man sich entschließt, ein neues Biopharmazeutikum
zu entwickeln. Denn die Fragen nach „drugability“ einerseits
und „general safety“ andererseits wurden durch den jahre-
langen klinischen Einsatz der Referenzarznei eindeutig po-
sitiv beantwortet. Hier liegt eines der ganz großen Risiken
für den Hersteller eines neuen Biopharmazeutikums, und
diese Risiken rechtfertigen einen großen Anteil der teils
extremen Kosten vieler dieser Wirkstoffe.
Biosimilars sind eine Möglichkeit, Wettbewerb bei den teuren Bio-pharmazeutika zu generieren.
Biosimilars in der Versorgung

66 Biosimilars – ein Handbuch
Biosimilars könnten
ca. 25 % günstiger sein als die
Referenz - arzneimittel.
Erste Einschätzungen für zu realisierende Einsparpotenzi-
ale durch den verstärkten Einsatz von Biosimilars gibt es
bereits:
Laut BARMER GEK Arzneimittelreport 2016 lässt sich in den
nächsten fünf Jahren durch eine konsequente Verschrei-
bung von Biosimilars eine halbe Milliarde Euro allein bei der
BARMER GEK an Arzneimittelausgaben einsparen.
Grund dafür ist, dass einige der umsatzstärksten biotechno-
logisch hergestellten Medikamente kürzlich ihren Patent-
schutz verloren haben oder diesen in Kürze verlieren wer-
den. Damit werden mehr Biosimilars auf den Markt
drängen.
Ein Biosimilar ist dabei im Schnitt etwa 25 % günstiger als
das entsprechende Referenzarzneimittel.
Die Biosimilarquoten differieren nach den Auswertungen
der BARMER GEK dabei je nach Kassenärztlicher Vereini-
gung um fast 100 %. Während die Ärztinnen und Ärzte in
Bremen in 54,2 % der Fälle Biosimilars verordnen, sind es
im Saarland nur 27,4 %. Wenn man die einzelnen Präparate
betrachtet, unterscheiden sich die Verschreibungsquoten
sogar um das bis zu 19-Fache. Mecklenburg-Vorpommern
weist gar eine „Null-Quote“ für den biosimilaren Wirkstoff
Somatropin aus. Die BARMER GEK weist darauf hin, dass
sich diese enormen regionalen Differenzen bei den Verord-
nungsquoten nicht medizinisch erklären lassen.

67
Mythen und Fakten zu Biosimilars
Mit den derzeit verfügbaren
Biosimilars ist das Behandlungs-
spektrum bereits ausgereizt.
Von Biosimilars werden nur die
Kostenträger profitieren.
Biosimilars werden das Gesund-
heitssystem ökonomisch kaum
entlasten, da sie kaum günstiger
angeboten werden als die Refe-
renzpräparate.
Das Spektrum der Biosimilars wird sich deutlich erweitern.
Im Zulassungsverfahren befinden sich derzeit u. a. mit Adali-
mumab, Trastuzumab und Rituximab drei neue Wirkstoffe.
Etliche weitere Biosimilars befinden sich in einer fortge-
schrittenen Entwicklungsphase.
Vielfach muss man bei den hochpreisigen Biopharmazeu-
tika eine therapeutische Unterversorgung feststellen, die
vor allem durch die hohen Kosten begründet ist. Durch
die immer bessere Verfügbarkeit von Biosimilars in vielen
weiteren Indikationen wird sich auch die Versorgung der
Patienten verbessern, die somit als Profiteure dieser Ent-
wicklung zu bezeichnen sind.
Wegen des hohen Preisniveaus im Bereich der Biopharma-
zeutika führen bereits prozentual eher geringe Preisnach-
lässe zu absolut hoch signifikanten Einsparungen. Zwar
können Biosimilars nicht mit den Wirtschaftlichkeitspoten-
zialen der niedermolekularen Generika angeboten werden,
aber die Einsparungen werden erheblich sein.
Mythos Fakt
Tab. 11: Mythen und Fakten zu Biosimilars
Biosimilars in der Versorgung

BIOSIMILARS 2.0: MONOKLONALE ANTIKÖRPER
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
Kapitel
4

69Biosimilars 2.0: Monoklonale Antikörper
Was unterscheidet die kommenden Biosimilars von den bislang zugelassenen?
Bis Ende 2016 waren acht Wirkstoffklassen als Biosimilars
verfügbar: Enoxaparin-, Epoetin-, Etanercept-, Filgrastim-,
Follitropin-, Infliximab-, Insulin glargin- und Somatropin-
Biosimilars. Vertreten sind demnach unmodifizierte Proteine
(Insulin glargin- und Somatropin-Biosimilars) ebenso wie
Glykoproteine (Epoetin-, Etanercept-, Follitropin- und Inflixi-
mab-Biosimilars). Dieses Spektrum wird in absehbarer Zeit
signifikant erweitert.
Die Weichen hat die EMA bereits gestellt, die Richtlinien für
die Prüfung und Zulassung von Insulin-, Interferon-alfa- und
Interferon-beta-Biosimilars bereits publiziert hat.
Es sind aber vor allem die Biosimilars von monoklonalen
Antikörpern, die die Einsatzmöglichkeiten von Biosimilars
massiv erweitern. War die Herstellung von Somatropin- und
Filgrastim-Biosimilars bis vor einigen Jahren nur schwer
vorstellbar, kam wegen der erforderlichen Glykosylierung die
Herstellung von Epoetin- und Follitropin-Biosimilars fast einer
Provokation gleich. So wurden mit der Herstellung des ersten
Biosimilars eines monoklonalen Antikörpers (Infliximab) und
des ersten Biosimilars eines Rezeptor-Antikörper-Fusions-
proteins (Etanercept) die letzten Zweifel ausgeräumt, dass
praktisch jedes Biopharmazeutikum so kopiert werden kann,
dass es eine analoge klinische Wirksamkeit und Verträglich-
keit aufweist, wie die entsprechende Referenzarznei.
BIOSIMILARS 2.0: MONOKLONALE ANTIKÖRPER
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf
Für neue Biosimi-lars sind bereits Prüfrichtlinien vorbereitet.

70 Biosimilars – ein Handbuch
Monoklonale Antikörper als Paradebeispiel für Biosimilarität
Monoklonale Antikörper sind noch einmal um vieles größer als
die „klassischen“ Biopharmazeutika. So besteht beispielswei-
se gentechnisch hergestelltes Insulin aus knapp 800 Atomen.
Somatropin oder Epoetin sind im Vergleich dazu ungefähr
viermal größer. Aber monoklonale Antikörper weisen mehr als
20.000 Atome auf. Sie sind zudem komplex glykosyliert, wobei
die Zuckerketten im Falle der Antikörper nicht nur die biolo-
gische Halbwertszeit beeinflussen, sondern auch spezielle
Reaktionen von Komponenten des Immunsystems auf den
Vor allem monoklonale
Antikörper sind als Biosimilars
interessant.
Abb. 13: Physikochemische und biologische Charakteristika von Antikörpern
Variable Region• Deamidierung• Oxidation• N-terminales Pyro-Glu • Glykosylierung • Glykierung • …
Bindung• Affinität• Avidität• Immunreaktivität / Kreuzreaktivität• Unerwünschte Reaktivität• …
Effektorfunktion• Komplement-Interaktion• FcRn, FcR-Interaktion• Interaktion mit dem Mannan- bindenden Liganden• Interaktion mit Mannose- Rezeptor• …
Andere biologische Eigenschaften• PK-Eigenschaften• Epitop / Immunogenität• Modulierende Region (Tregitope …)• …
Konstante Region• Deamidierung • Oxidation• Acetylierung• Glykierung• Glykosylierung (Fucosylierung, Sialylierung, Galactosylierung, Mannosylierung)• C-terminales Lys• Verschiebung / Auflösung der Disulfidbrücken• Fragmentierung / Clipping • …
Physikochemische Charakteristika Biologische Charakteristika

71
Immunkomplex zwischen Antikörper und beispielsweise atta-
ckierter Tumorzelle steuern. Hier entscheidet sich maßgeb-
lich die Reaktion mit dem Komplement-System, die Bindung
an sogenannte Fc-Rezeptoren, durch die Immunreaktionen in-
duziert, aber auch gebremst werden und die Interaktion mit
einer Reihe anderer Rezeptoren.
Eine kleine Auswahl kritischer Positionen an einem Modell-
Antikörpermolekül soll hier erwähnt werden, die zeigen, wel-
che Herausforderungen bei der Entwicklung eines Antikörper-
Biosimilars zu meistern sind, um das Molekül der
Referenzarznei zu kopieren:
Biosimilars 2.0: Monoklonale Antikörper
• Pyro-E (2)• Deamidierung (D; 3 x 2)• Methionin-Oxidation (O; 2 x 2)• Glykierung (G; 2 x 2)
• Mannose-reich, G0, G1, G1, G2 (5)• Sialylierung• C-terminales Lysin (2)• 2 x 4 x 4 x 5 x 5 x 2 = 9600
(9600)2 ≈ 108
potenzielle Varianten
Pyro-EPyro-E
K K
GG
Abb. 14: Variable Posi-tionen an einem Modell-antikörper22
22 Kozlowski S, Swann (2006). Current and future issues in the manufacturing and development of monoclonal antibodies. Advanced Drug Delivery Reviews 58, 707–722
GG D D
DD
OO
OO
DD

72 Biosimilars – ein Handbuch
• Ganz oben, am N-Terminus des Antikörpers, kann die Ami-
nosäure Glutaminsäure zu Pyroglutaminsäure zyklisieren
(Pyro-E). Diese Position ist zweimal pro Antikörper vorhan-
den.
• Asparagin- und Glutamin-Reste können relativ leicht dea-
midiert werden, was deutliche Strukturänderungen nach
sich zieht. Sechs Positionen sind in dem Modell-Antikörper
gekennzeichnet.
• Methionin-Reste lassen sich oxidieren. Hier wurden zwei
Positionen markiert.
• Zuckerketten findet man in einem IgG-Mokekül an vier Po-
sitionen.
• Glykosylierungsheterogenität ergibt sich aus teils partiell
aufgebauten Zuckerbäumen.
• Eine endständige Sialinsäure trägt nicht jeder Zuckerbaum.
Dies führt wegen der negativen Ladung der Sialinsäure zu
Ladungsvariabilitäten. Zudem sind die endständigen Sialin-
säurereste an der Auswahl beteiligt, an welchem Fc-
Rezeptor der Antikörper bindet.
• Schließlich kann auch über die C-terminale Aminosäure
(K = Lysin) Strukturheterogenität in das Antikörpermolekül
eingeführt werden.
Für den Modell-Antikörper ergeben sich so nahezu 108 mög-
liche Strukturvariationen (Abb. 14). Da man jedoch so genau
über die Variationen Bescheid weiß und die einzelnen Positi-
onen mit modernen analytischen Verfahren exakt bestimmen
kann, dienen sie als Gradmesser für die Zuerkennung des
Prädikats „Biosimilarität“, was noch einmal unterstreicht,
wie engmaschig hier entschieden wird.
An etlichen Positionen eines
Antikörper - moleküls
können chemische Modifikationen
auftreten.

73
Neben der terminalen Sialinisierung der Zuckerketten-Variati-
on steht dabei eine weitere Variante unter ganz besonderer
Beobachtung. Dies ist der Fucose-Rest relativ nah in Nachbar-
schaft zur Asparaginsäure, die den Zuckerbaum fixiert. Von
diesem Rest weiß man, dass er ganz direkt das biologische
Verhalten des Antikörpers beeinflussen kann. Je weniger von
diesem Fucose-Rest in einer Antikörperpopulation vorhanden
ist, umso fulminanter fällt die sogenannte antikörperabhän-
gige zelluläre Zytotoxizität (ADCC) aus. Darunter versteht man
die Zerstörung einer Zelle (beispielsweise einer Tumorzelle)
durch natürliche Killerzellen, die über einen an die Zelle ange-
dockten Antikörper angelockt werden. Von einem in der Tu-
mortherapie eingesetzten biosimilaren Antikörper erwartet
man, dass er genau die ADCC-Aktivität entfaltet, wie der
Referenz antikörper. Auch dies bedeutet, dass die Strukturvor-
gaben sehr genau bekannt sein müssen und zu treffen sind.
Für die Entwicklung solcher Biosimilars bedeutet das riesige
Herausforderungen. So müssen die Moleküle möglichst iden-
tische Strukturen aufweisen und dies gilt auch für Mikrohe-
terogenitäten. Es muss gezeigt werden, dass mögliche, kleine
Unterschiede zwischen dem biosimilaren Antikörper und dem
Referenzprodukt keine signifikanten Auswirkungen auf die
klinische Wirksamkeit und / oder die klinische Sicherheit
besitzen. Dazu müssen die Daten aus physikochemischen
Analysen und Strukturanalysen mit Daten aus Funktions-
assays kombiniert werden. Und letztlich müssen die Wirk-
samkeit und vor allem die Verträglichkeit durch ein sorgfältig
geplantes klinisches Studienprogramm belegt werden.
Biosimilars 2.0: Monoklonale Antikörper
Für die Funktio- nalität des Anti- körpers ist die Zusammensetzung der Zuckerketten extrem wichtig.
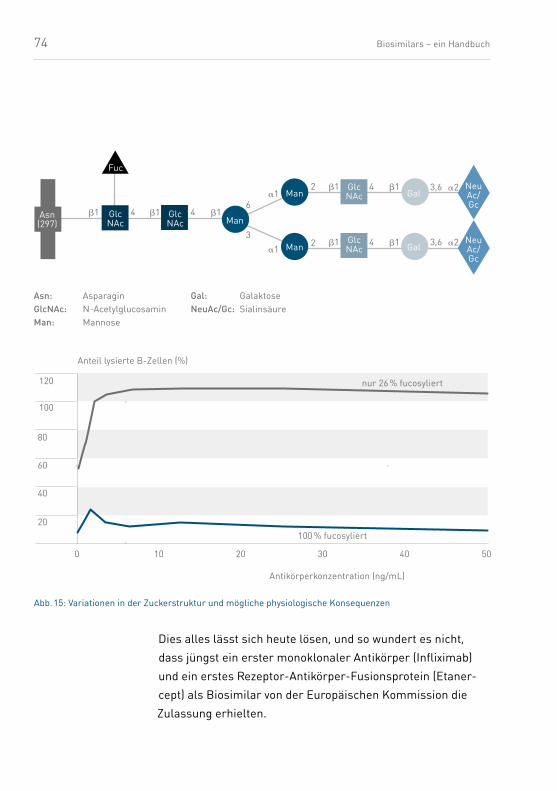
74 Biosimilars – ein Handbuch
Dies alles lässt sich heute lösen, und so wundert es nicht,
dass jüngst ein erster monoklonaler Antikörper (Infliximab)
und ein erstes Rezeptor-Antikörper-Fusionsprotein (Etaner-
cept) als Biosimilar von der Europäischen Kommission die
Zulassung erhielten.
Abb. 15: Variationen in der Zuckerstruktur und mögliche physiologische Konsequenzen
Asn(297)
GlcNAc
Fuc
GlcNAc Man
Man
Man Gal
GalNeuAc/Gc
NeuAc/Gc
GlcNAc
GlcNAc
Asn: AsparaginGlcNAc: N-AcetylglucosaminMan: Mannose
Gal: GalaktoseNeuAc/Gc: Sialinsäure
Anteil lysierte B-Zellen (%)
Antikörperkonzentration (ng/mL)
nur 26 % fucosyliert
100 % fucosyliert
100
20
40
60
80
100
120
20 30 40 50
1 1 1
11 1
1 2
2
11
4 46
2 4
4 3,6
3,6
23

75
Wann werden die nächsten Biosimilars den Markt erreichen?
Der erste biosimilare monoklonale Antikörper (Inflectra® /
Remsima®) wurde im September 2013 von der Europäischen
Kommission zugelassen. Als Referenzarznei für die Entwick-
lung dieses Infliximab-Biosimilars diente Remicade® der
Firma MSD. Inflectra® / Remsima® und das 2016 zugelassene
Flixabi® sind für folgende Indikationen zugelassen: Rheuma-
toide Arthritis (RA), Ankylosierende Spondylitis, Morbus Crohn
(CD), Colitis ulcerosa (UC), Psoriasis-Arthritis (PsA) und Pso-
riasis.
Kurz vor der Zulassung dieses ersten biosimilaren Antikörpers
konnte die Firma Janssen Biotech Inc. eine Verlängerung
ihres Patents für Remicade® bis Anfang 2015 erwirken, so
dass sich die Einführung von Inflectra® / Remsima® in Zentral-
europa noch bis Anfang 2015 verzögerte.
Anfang 2016 wurde zudem ein Etanercept-Biosimilar (Bene-
pali®) zugelassen. Bei Etanercept handelt es sich um ein hu-
manes Tumornekrosefaktor-Rezeptor-p75-Fc-Fusionsprotein.
Etanercept ist ein Dimer eines chimären Proteins, das durch
Fusion der extrazellulären Ligandenbindungsdomäne des hu-
manen Tumornekrosefaktor-Rezeptor-2 (TNFR2/p75) mit der
Fc-Domäne eines humanen IgG1-Antikörpers gentechnisch
hergestellt wird. Die Fc-Komponente enthält die Scharnier-,
CH2- und CH3-Regionen, nicht aber die CH1-Region des IgG1-
Antikörpers.
Biosimilars 2.0: Monoklonale Antikörper
Ein monoklonaler biosimilarer Anti-körper wurde bereits von der EMA zugelassen.

76 Biosimilars – ein Handbuch
Damit ist ein starker Anfang gemacht. Das lange beschworene
„Dogma“, rekombinante, monoklonale Antikörper ließen sich
nicht kopieren, ist seit der Erteilung der Zulassung dieser bei-
den Biosimilars nicht mehr zu halten.
So wundert es nicht, dass weitere monoklonale Antikörper als
Biosimilars in der fortgeschrittenen Entwicklung sind. Mit
deren Markteinführung muss also gerechnet werden, wenn
die Patente der Referenzprodukte auslaufen und die komplexe
Entwicklung abgeschlossen ist.
Abb.16: Schematischer Aufbau von Etanercept
HOOC
Bindungsstelle des
TNF-alfa-Rezeptors
Etanercept
COOH
SS S
S
SS S
S
CH3
CH2 CH2
H H
CH3
SS
SS

77
In welchen Indikationen werden die kommenden Biosimilars eine Rolle spielen?
Neben den Indikationen, für die Inflectra®, Remsima®, Flixabi®
und Benepali® ihre Zulassung erhalten haben, werden vor
allem Indikationen im Bereich unterschiedlicher Tumorer-
krankungen durch biosimilare Antikörper bedient werden.
Dazu zählen sowohl lymphatische als auch solide Tumore.
Konkret kann man mit biosimilaren Antikörpern zu den Refe-
renzmolekülen Adalimumab (Rheumatoide Arthritis, Psoriasis,
Morbus Crohn), Rituximab (niedrig maligne und follikuläre
Non-Hodgkin-Lymphome, Rheumatoide Arthritis), Trastu-
zumab (Mamma-Karzinom), Bevacizumab / Ranibizumab (Anti-
angiogenese) und Cetuximab (Kolon-Karzinom) rechnen.
Ferner sind Biosimilarvarianten von Pegfilgrastim, Darbe-
poetin alfa, Peginterferon alfa und modifizierten Insulinen in
der fortgeschrittenen Entwicklung (siehe auch Kap. 3).
Wie verbessern Biosimilars die Versorgung und den Zugang der Patienten zu hochwirksamen Therapien?
Biosimilars werden zu einer besseren Versorgung von Patien-
tinnen und Patienten mit biotechnologisch hergestellten
Arzneimitteln beitragen. Aus Kostengründen erhalten nach
wie vor noch längst nicht alle Patienten diese Wirkstoffe,
obwohl sie bei ihnen indiziert wären. Diese Situation wird
sich durch die günstiger angebotenen Biosimilars mit Sicher-
heit verbessern.
Biosimilare Anti-körper werden vor allem in der Tumortherapie eingesetzt.
Biosimilars 2.0: Monoklonale Antikörper

78 Biosimilars – ein Handbuch
Biosimilars sind Hoffnungsträger der modernen Medizin.
Denn durch die Verfügbarkeit von Biosimilars wird sich auch
in dem Segment der rekombinanten Wirkstoffe und damit in
einem wichtigen und zudem extrem hochpreisigen Segment
der Arzneimittelversorgung ein Preiswettbewerb etablieren.
Das Potenzial erkennt man an den kürzlich zugelassen Inflixi-
mab-Biosimilars. Die Referenzarznei Remicade® gehört
zweifelsfrei zu den Blockbuster-Wirkstoffen. Dieser Wirkstoff
erwirtschaftete im Jahr 2012 einen Umsatz von ca. 2 Milliar-
den USD. Daraus leitet sich ab, dass sich bereits durch eine
relativ geringe prozentuale Preisreduktion von beispielsweise
25 % ein signifikanter absoluter Geldbetrag einsparen lässt.
Welche Kostenersparnisse könnten generiert werden?
Wie groß das Einsparpotenzial durch Biosimilars sein könnte,
haben Wissenschaftler des IGES Instituts anhand von Modell-
rechnungen ermittelt. Untersucht wurden die Einsparpoten-
ziale für die Wirkstoffgruppen Erythropoetin, Granulozyten-
Koloniestimulierende Faktoren und monoklonale Antikörper
in acht europäischen Ländern (Deutschland, Frankreich,
Italien, Polen, Großbritannien, Rumänien, Schweden, Spanien).
Betrachtet wurde dabei der Zeitraum 2007 bis 2020.
Auch wenn mit Biosimilars nur
25 % der Kosten eingespart werden
können, resultieren daraus signifikante
Beträge.

79
Die Studie23 ergab, dass sich die aus der Modellrechnung
abgeleiteten Einsparungen zwischen 11,8 Milliarden Euro
und 33,4 Milliarden Euro bewegen – in Abhängigkeit von der
Marktpenetrationsrate, der Markteintrittsgeschwindigkeit
sowie dem Preisniveau der Biosimilars. Das Einsparpotenzial
in Deutschland beträgt laut dieser Studie bis zu 11,7 Milliar-
den Euro – falls Ärzte Biosimilars immer dann verordnen,
wenn dies medizinisch möglich ist und keine Marktbarrieren
in Deutschland bestehen.
Das sind riesige Summen, die es zukünftig zu realisieren gilt,
um die Versorgung der Patientinnen und Patienten weiter zu
verbessern und anstehende Herausforderungen im Gesund-
heitssystem durch eine immer älter werdende Gesellschaft zu
meistern.
Was muss dafür geschehen?
Um diese Effizienzreserven zu heben und dabei gleichzeitig
den Patientinnen und Patienten eine noch bessere Versorgung
mit hochwirksamen Arzneimitteln zukommen zu lassen, muss
vor allen Dingen das Vertrauen in die absolute Gleichwertig-
keit der Biosimilars im Vergleich zu den Referenzprodukten
bei den Heilberuflern gestärkt werden. Nach wie vor wird den
Biosimilars mit einer Skepsis begegnet, die auch nicht nähe-
rungsweise gerechtfertigt ist.
23 Haustein R, de Millas C, Höer A, Häussler B (2012. Saving Money in the European Health Care System with Biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 1(3-4):120–126. doi: 10.5639/gabij.2012
Biosimilars 2.0: Monoklonale Antikörper

80 Biosimilars – ein Handbuch
Durch den zentralen und kompromisslosen Zulassungspro-
zess durch die Europäische Arzneimittel-Agentur (EMA) ist si-
chergestellt, dass sich das Biosimilar und die Referenzarznei
auf molekularer Ebene so sehr ähneln, dass Vergleichbarkeit
bescheinigt werden kann. Zudem garantieren die Ergebnisse
klinischer Studien auch eine vergleichbare Wirksamkeit und
Verträglichkeit. Keine sachlichen Gründe sprechen daher ge-
gen die Äquivalenz der beiden Wirkstoffe. Zudem muss gerade
in der Zeit, wo Biosimilars versuchen, im Markt Fuß zu fassen,
ein fairer Wettbewerb garantiert sein.
Noch immer werden Biosimilars
sehr kritisch betrachtet – zu Unrecht.

81
Mythen und Fakten zu Biosimilars
Biosimilars von monoklonalen
Antikörpern lassen sich nicht
herstellen.
Biosimilare Antikörper verhalten
sich in der Klinik unzuverlässig,
da diese Moleküle strukturell
zu aufwändig sind, um sie
angemessen exakt kopieren zu
können.
Mit Infliximab-Biosimilars (Inflectra®, Remsima®, Flixabi®)
sind bereits erste Biosimilars eines monoklonalen Anti-
körpers zugelassen. Diese erfüllen, wie alle anderen
Biosimilar-Präparate auch, alle regulatorischen Anforde-
rungen, die an diese neue Klasse von Biopharmazeutika
gestellt werden.
Durch die Zulassung durch die EMA wird garantiert, dass
sich Biosimilars von monoklonalen Antikörpern in der Klinik
ganz analog zur Referenzarznei verhalten. Darauf wurde in
speziell hierfür ausgelegten klinischen Studien kompro-
misslos getestet.
Mythos Fakt
Tab. 12: Mythen und Fakten zu Biosimilars
Biosimilars 2.0: Monoklonale Antikörper

Kapitel
5
10 JAHRE BIOSIMILARS –
LESSONS LEARNED Michael Dilger

8310 Jahre Biosimilars – Lessons learned
10 Jahre Biosimilars – Lessons learned
Mit Omnitrope wurde 2006 das erste „Nachahmerpräparat“ zu
einem Biologikum in Deutschland zugelassen. Es folgten Bio-
similars zu Epoetin und Filgrastim. Der Versorgungsanteil mit
Biosimilars entwickelte sich sehr unterschiedlich. Im 2. Quar-
tal 2016 liegt der bundesweite Durchdringungsgrad von Epo-
etin bei 40 %, Filgrastim 74 % und Somatropin 15 %.24 Dabei
sind regionale Unterschiede erkennbar.
Aus Sicht vieler Experten wurde das durch diese günstigeren
Biosimilars mögliche Sparpotenzial allerdings nicht voll aus-
geschöpft. Das lag zum einen an spezifischen Marktbedin-
gungen (z. B. Struktur der Versorgungseinrichtungen in den
entsprechenden Indikationen), der untergeordneten Bedeu-
tung der Biosimilars in einzelnen Produktgruppen, aber auch
an Unsicherheiten der Ärzte bezüglich der Verordnung von
Biosimilars aufgrund mangelnder Erfahrung und dem nicht
immer konsequenten Steuerungs- und auch Informations-
management seitens der Kostenträger.
„Biosimilars 2.0“
Seit Anfang 2015 sind für Infliximab und seit März 2016 für
Etanercept die ersten biosimilaren monoklonalen Antikörper
in Deutschland erhältlich. Seit Einführung der Infliximab-Bio-
similars wurden bis Juni 2016 bereits 13 Millionen Euro25 nur
24, 25 AG Pro Biosimilars, Insight Health, Simon-Kucher & Partners: eigene Berechnung, basierend auf GKV-Verordnungen
Aus Sicht vieler Experten wurde das durch die günstigeren Biosimilars mög-liche Sparpotenzial nicht voll ausge-schöpft.
10 JAHRE BIOSIMILARS –
LESSONS LEARNED Michael Dilger

84 Biosimilars – ein Handbuch
durch den preislichen Vorteil für das GKV-System eingespart.
Doch es ist noch viel Luft nach oben, regional schwankt der
Verordnungsanteil deutlich.
In den nächsten Jahren werden weitere umsatzstarke Biologi-
ka ihren Patentschutz verlieren. Das GKV-Umsatzvolumen von
Humira®, Lucentis® und Avastin® belief sich 2015 auf 1,6 Milli-
arden Euro.26 Dies lässt erahnen, welches weitere Einsparpo-
tenzial durch die Einführung von Biosimilars erwartet werden
kann, da die neuen Biosimilars in großen, kostenintensiven In-
dikationen wie Rheuma und Krebs eingesetzt werden. Die dort
zu erwartenden signifikanten Einsparungen haben die Auf-
merksamkeit der Kostenträger „neu geweckt“ und es ist davon
auszugehen, dass sich die Kostenträgerseite intensiver als in
der Vergangenheit dem Thema Biosimilars 2.0 widmen wird.
Steuerung soll nachhaltigen Wettbewerb ermög lichen
Um die erwarteten Kosteneinsparungen zu realisieren, wol-
len Krankenkassen und Kassenärztliche Vereinigungen die
Verordnung von Biosimilars durch verbesserte Informations-
maßnahmen und optimierte Steuerung des Biologikamarktes
fördern. Aktivitäten seitens der Kostenträger sollten hierbei
berücksichtigen, dass sich Biosimilarhersteller nicht nur als
„Nachahmer“ sehen, sondern in Infrastruktur wie beispiels-
weise die Finanzierung von Registerstudien investieren und
damit ihr Engagement zu einer langfristigen Versorgung beto-
nen. Die Steuerung von Biologika sollte daher einen nachhal-
26 Simon-Kucher & Partners, Bundesbericht GAmSi 12/2015; IMS Patent focus 2014
Es ist noch viel Luft nach oben, regional
schwankt derVerordnungsanteil
deutlich.

85
tigen Wettbewerb und eine langfristige Perspektive für Bio-
similars ermöglichen. Dies ist auch ein Ergebnis des
Pharmadialogs 2016:
„Biosimilars werden in den nächsten Jahren eine immer
wichtigere Rolle bei der Behandlung von schweren Erkran-
kungen wie Rheuma, Multiple Sklerose oder Krebs und für
die Finanzierbarkeit des Gesundheitssystems einnehmen.
Daher sind Rahmenbedingungen für eine nachhaltige und
sichere Versorgung mit generischen Arzneimitteln und
Bio similars fortzuentwickeln.“27
ERHALTUNG DER MARKTATTRAKTIVITÄTfür den Eintritt von Biosimilars für die Hersteller
Ermöglichung eines fairen Preis-/ Volumenverhältnisses für Biosimilars
WETTBEWERBS - DYNAMIKhinsichtlich Preis und Leistung für Biosimilars
ERHÖHTE AKZEPTANZgegenüber Biosimilars, z. B. bei Ärzten und Patienten
NACHHALTIGE RAHMENBEDIN-GUNGEN zur Förderung von Biosimilars
Einsparpotenzial für Krankenkassen bei gleichbleibend hoher Versorgungs - qualität und somit mehr freie Ressourcen zur Patientenversorgung für das Gesundheitssystem
BIOSIMILAR- HERSTELLER ALS MARKTPARTNERfür ein zusätzliches Pro-duktangebot, Stu dien und Arztinformation
10 Jahre Biosimilars – Lessons learned
Die Steuerung von Biologika sollte einen nachhaltigenWettbewerb und eine langfristige Perspektive für Biosimilarsermöglichen.
Abb. 17: Ein Steuerungsrahmen für Biologika sollte einen nachhaltigen Wett-bewerb und eine langfristige Perspektive für Biosimilars ermöglichenSimon-Kucher & Partners: „10 Jahre Biosimilars – Lessons learned“, September 2016
27 Bericht zu den Ergebnissen des Pharmadialogs (Bundesministerium für Gesundheit, April 2016)

86 Biosimilars – ein Handbuch
Zielvereinbarungen für Biosimilars: bei umfassender Information und richtiger Umsetzung generell ein nachhaltiges Steuerungs-instrument zur Förderung von Biosimilars
Viele KV-Regionen haben seit 2016 regionale Zielverordnungs-
anteile für Infliximab-Biosimilars, die realisierten Verord-
nungsanteile unterscheiden sich aber deutlich. Die vereinbar-
ten Ziele sind in vielen Regionen bereits übertroffen – eine
klare Korrelation zwischen Höhe des Zielverordnungsanteils
und dem realisierten Biosimilarverordnungsanteil besteht
nicht. Dies könnte auch daran liegen, dass die Höhe des Ziel-
verordnungsanteils eher konservativ definiert wurde. Optima-
lerweise erfolgt die Definition datenbasiert und berücksichtigt
regions-, indikations- und produktspezifische Besonderheiten.
Abb. 18: Konzeptioneller Aufbau einer Zielvereinbarung für BiosimilarsSimon-Kucher & Partners: „10 Jahre Biosimilars – Lessons learned“, September 2016
Zielvereinbarungfür Biosimilars
Maßnahmen zur konsequenten Umsetzung
InformationSicherheit / Wirksamkeit von Biosimilars
Regionale Vereinbarungen zwischen Krankenkassen und Kassenärztlichen Vereinigungen
BeratungWirtschaftlichkeit von Biosimilars
DefinitionVereinbarung eines Zielverordnungsanteils
Reporting Definitionvon Folgen
Überprüfung Zielerreichung

87
Zielvereinbarungen für Biosimilars umfassen Information, Beratung und die Definition eines Zielverordnungsanteils
Experten befürworten Zielvereinbarungen als Steuerungsin-
strument zur Förderung von Biosimilars. Allerdings nur, wenn
diese zusätzlich zu einem definierten Zielverordnungsanteil
auch Informationen und Beratungsmaßnahmen umfasst und
des Weiteren durch konsequente Umsetzungsmaßnahmen be-
gleitet werden.
Überdurchschnittliche Infliximab-Biosimilar-Verordnungsan-
teile sind demnach vor allem auf eine gründliche Information
und Umsetzung der Zielvereinbarung zurückzuführen.
In allen KV-Regionen sind Maßnahmen wie Reporting und
Informationspflicht definiert, allerdings gibt es Unterschiede
in Detailgrad und Nachdrücklichkeit. Für eine konsequente
Umsetzung der Zielvereinbarungen sind auch der Aufbau und
die Pflege von Datenbanken und Analysesoftware sowie regel-
mäßige Auswertung von Verordnungsdaten zur Ableitung von
Handlungsempfehlungen notwendig.
Das erfordert ausreichende personelle und technische Res-
sourcen in den KVen; diese sind nach Meinung von Experten
jedoch regional sehr heterogen.
Dennoch gibt es Regionen ohne definierte Zielverordnungs-
anteile, die trotzdem hohe Biosimilaranteile erzielen. Folglich
muss es weitere Einflussfaktoren geben, die die Verordnung
von Biosimilars positiv beeinflussen können.
10 Jahre Biosimilars - Lessons learned
Experten befürworten Zielvereinbarungen als Steuerungs- instrument zur Förderung von Biosimilars.
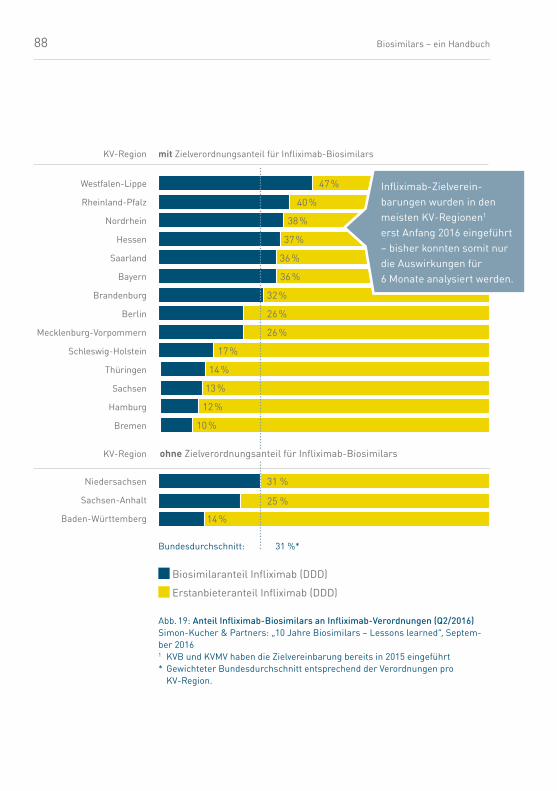
88 Biosimilars – ein Handbuch
Biosimilaranteil Infliximab (DDD)
Erstanbieteranteil Infliximab (DDD)
Westfalen-Lippe
Rheinland-Pfalz
Nordrhein
Hessen
Saarland
Bayern
Brandenburg
Berlin
Mecklenburg-Vorpommern
Schleswig-Holstein
Thüringen
Sachsen
Hamburg
Bremen
Niedersachsen
Sachsen-Anhalt
Baden-Württemberg
KV-Region mit Zielverordnungsanteil für Infliximab-Biosimilars
KV-Region
47 %
40 %
38 %
37 %
36 %
36 %
32 %
26 %
26 %
17 %
14 %
13 %
12 %
10 %
31 %
25 %
14 %
Abb. 19: Anteil Infliximab-Biosimilars an Infliximab-Verordnungen (Q2/2016)Simon-Kucher & Partners: „10 Jahre Biosimilars – Lessons learned“, Septem-ber 20161 KVB und KVMV haben die Zielvereinbarung bereits in 2015 eingeführt* Gewichteter Bundesdurchschnitt entsprechend der Verordnungen pro KV-Region.
Infliximab-Zielverein- barungen wurden in den meisten KV-Regionen1
erst Anfang 2016 eingeführt – bisher konnten somit nur die Auswirkungen für 6 Monate analysiert werden.
ohne Zielverordnungsanteil für Infliximab-Biosimilars
Bundesdurchschnitt: 31 %*

89
Selektivverträge können bei entsprechender Ausge-staltung die Verordnung von Biosimilars fördern und damit Kosten einsparen
Ein Beispiel ist der Selektivvertrag der BARMER GEK in Koo-
peration mit der KV Westfalen-Lippe und dem Bundesverband
der niedergelassenen Gastroenterologen. Der Vertrag soll
einerseits eine intensivere Betreuung von CED-Patienten
ermöglichen und zielt andererseits auf Kosteneinsparungen
durch die vermehrte Verordnung von Biosimilars ab. Ärzte
werden dabei vertraglich angehalten, neue Patienten mög-
lichst auf wirtschaftliche Arzneimittel, z. B. Biosimilars, ein-
zustellen. Zusätzlich sollen die Ärzte prüfen, ob Patienten,
die auf das Referenzprodukt eingestellt sind, auf ein Biosimi-
lar umgestellt werden können.
Auch die Ärzte profitieren von diesem Programm: Sie erhalten
eine Pauschale für ihren erhöhten Betreuungsaufwand und
werden durch Rückzahlungen an den durch Biosimilareinsatz
realisierten Einsparungen beteiligt. Zusätzlich ermöglicht ein
umfangreiches Informationsprogramm sowie eine
intensivere Patientenbetreuung eine insgesamt qualitativ
bessere Versorgung.
Die BARMER GEK verknüpft den Vertrag mit einem Rabatt-
vertrag (Open-House-Vertrag), d. h., sie empfiehlt den Ärzten,
vorrangig die rabattierten Therapien einzusetzen. Zusätzlich
informiert die BARMER GEK die Ärzte über die tatsächliche
Wirtschaftlichkeit der Arzneimittel – unter Berücksichtigung
von Rabatt und Listenpreis. Dies soll Ärzte in ihrer preisbe-
wussten Verordnungsweise unterstützen.
10 Jahre Biosimilars - Lessons learned

90 Biosimilars – ein Handbuch
Abb. 20: Beispiel BARMER-GEK CED-VertragSimon-Kucher & Partners: „10 Jahre Biosimilars – Lessons learned“, September 20161 Berufsverband für niedergelassene Gastroenterologen2 Kassenärztliche Vereinigung Westfalen-Lippe
Inzwischen hat die BARMER GEK ein ähnliches Programm für
Rheuma aufgelegt, weitere Module und eine regionale Aus-
weitung sind in Planung. Dabei sollen die jeweils indikations-
und regionsspezifischen Bedürfnisse berücksichtigt werden.
Um Biosimilars zu fördern, sollte ein Selektivvertrag eine ent-
sprechende Zielvereinbarung enthalten, Neueinstellungen auf
Biosimilars fordern und Umstellungen auf Biosimilars fördern.
BNG1
Hersteller von Biologika
KVWL2
CED-Versorgungs-
vertrag
Open-House-Vertrag
BARMER GEK
BARMER GEK

91
Insgesamt gilt: Steuerungsinstrumente der Kosten-träger müssen transparent und diskriminierungsfrei gestaltet sein, um den Wettbewerb nicht zu behin-dern
Voraussetzung für die Transparenz von Steuerungsinstru-
menten ist, dass die Inhalte an alle Marktteilnehmer kommu-
niziert werden und allgemein zugänglich sind. In der Praxis
heißt dies z. B., dass Rabattverträge transparent bekannt
gemacht werden und Ärzte über die tatsächliche Wirtschaft-
lichkeit (unter der Berücksichtigung von Listenpreis und
Rabatten) informiert werden müssen. Nur so können Ärzte
auch wirtschaftlich verordnen und die erhofften Einsparungen
generieren.
Open-House-Verträge fördern den Einsatz der preis-günstigeren Biosimilars in der Regel nicht
Im Rahmen eines Open-House-Vertrags bietet die Kranken-
kasse allen Anbietern einen Vertrag mit festgelegtem Rabatt-
satz an. Jeder Anbieter (Original- und Biosimilarhersteller)
kann sich am Open-House-Vertrag beteiligen, wenn er diesen
Rabatt auf den jeweiligen Listenpreis gewährt. In der Folge
gelten alle Arzneimittel, die dem Open-House-Vertrag beige-
treten sind, trotz Preisunterschiede als (gleich) wirtschaftlich.
Der Wettbewerbsvorteil der Biosimilars wird dadurch
gemindert.
10 Jahre Biosimilars – Lessons learned

92 Biosimilars – ein Handbuch
Aus Sicht der Krankenkassen dienen Open-House-Verträge
dazu, kurzfristig Einsparungen zu generieren. Sie sind einfach
in der Umsetzung und diskriminierungsfrei. Als langfristiges
Vertragsmodell werden sie allerdings nicht betrachtet, da
sie nach Meinung der Experten den Preiswettbewerb nicht
fördern.
Biosimilarhersteller sehen sich oft in einer „Zwickmühle“:
Wenn sie sich am Vertrag beteiligen, ist der Preisvorteil des
Biosimilars in der Praxissoftware meist nicht erkennbar,
obwohl es tatsächlich günstiger ist als das Referenzprodukt
(gleicher Rabattsatz bei niedrigerem Listenpreis). Beteiligt
sich der Biosimilarhersteller nicht am Vertrag, gilt das Bio-
similar als nicht wirtschaftlich, auch wenn das Referenz-
produkt trotz Rabatt teurer ist.
Diese Fehlanreize können vermieden werden, wenn Ärzte über
die tatsächliche Wirtschaftlichkeit der Arzneimittel im Vertrag
informiert werden. Dann können auch innerhalb eines Open-
House-Vertrags Anreize für Biosimilarverordnungen gesetzt
werden. Einige Krankenkassen handhaben dies auch heute
schon so und senden beispielsweise entsprechende Informa-
tionsschreiben an die Vertragsärzte.
Fehlanreize können vermieden
werden, wenn Ärzte über
die tatsächliche Wirtschaftlichkeit
der Arzneimittel im Vertrag informiert
werden.

93
Das Vertrauen der Ärzte in Biosimilars ist im Laufe der Zeit gestiegen – der Informationsbedarf ist jedoch immer noch hoch
Informationsmaßnahmen werden von allen Experten als
genauso wichtig angesehen wie die Steuerungsinstrumente
selbst.
Die Unsicherheiten bezüglich Wirksamkeit und Sicherheit von
Biosimilars haben bei Ärzten abgenommen. Dennoch sind sie
als Entscheider bei der Therapieauswahl wichtigstes Ziel von
Informationsmaßnahmen. Informationsbedarf wird vor allem
bei Arztgruppen und Indikationen gesehen, die bisher keinen
Berührungspunkt mit Biosimilars hatten.
Die Information des Patienten ist vor allem Aufgabe des Arztes
Bei Patienten wird der Informationsbedarf dann als hoch ein-
geschätzt, wenn es sich um chronische oder sensible Krank-
heiten oder um Medikamente handelt, die sich der Patient
selbst verabreicht. Der Arzt hat den direkten Kontakt zum
Patienten und ist damit seine wichtigste Informationsquelle.
Es wird daher als wichtige Aufgabe der Krankenkassen, KVen
und Biosimilarhersteller gesehen, den Arzt durch das Bereit-
stellen von auf den Patienten zugeschnittenem Informations-
material zu unterstützen.
10 Jahre Biosimilars – Lessons learned
Informationsbedarf wird vor allembei Arztgruppen und Indikationen gesehen, die bisher keinen Berüh-rungspunkt mit Biosimilars hatten.

94 Biosimilars – ein Handbuch
Informationen zu Biosimilars: einfach verständlich, klar strukturiert und objektiv
Neben dem Konzept der Biosimilarität sind Themen wie Zu-
lassungsverfahren sowie Regularien zur Sicherstellung der
klinischen Sicherheit relevant, um das Vertrauen in Biosimi-
lars zu stärken. Für Ärzte sind zusätzliche Informationen zur
Wirtschaftlichkeit von Biosimilars, vor allem im Zusammen-
hang mit Verträgen, wichtig.
Beispiele erfolgreicher Kommunikation über Biosimilars
Kommunikationsmaßnahmen werden in verschiedenen KVen
schon gut umgesetzt. Das Ziel ist immer, das Vertrauen der
Ärzte in Biosimilars zu stärken und somit den Anteil der Bio-
similarverordnungen zu fördern. Beispielsweise führt die KV
Nordrhein Pharmakotherapie-Workshops speziell zum Thema
Biosimilars durch.
Auch einige Krankenkassen übernehmen intensiv Informa-
tions- und Kommunikationsaufgaben. Die AOK Niedersachsen
platziert beispielsweise über den Außendienst die Biosimilar-
thematik in persönlichen Gesprächen mit Ärzten. Andere
Krankenkassen veröffentlichen Berichte (z. B. BARMER GEK:
Arzneimittelreport – Spezialthema Biosimilars, Techniker
Krankenkasse: Faktenbuch zur Rheumatherapie) zur ein-
fachen Orientierung in der Biosimilarthematik.

95
Ein Beispiel für eine gelungene Zusammenarbeit zwischen
Krankenkasse und KV ist die „Biolike“-Initiative der BARMER
GEK und der KV Westfalen-Lippe. Die Maßnahme ist an die
oben angesprochenen Selektivverträge gekoppelt und umfasst
speziell auf Indikationen abgestimmte Pakete mit medizi-
nischen und wirtschaftlichen Informationen, z. B. für Gastro-
enterologie und Rheuma.
Auch Zulassungsbehörden können dazu beitragen, Unsicher-
heiten bezüglich Biosimilars abzubauen und Fragen zu beant-
worten. Sie sind wichtig, um Vertrauen in die klinische Wirk-
samkeit und Sicherheit von Biosimilars zu bilden, da sie als
unabhängige Experten und objektive Informationsquelle als
besonders glaubwürdig angesehen werden. Für Krankenkas-
sen, KVen und Biosimilarhersteller sind sie wichtige Dialog-
partner. Veröffentlichungen und Dialogveranstaltungen zeigen,
dass sich BfArM und Paul-Ehrlich-Institut verstärkt in die
Diskus sion um Biosimilars einbringen.
Mit der Veranstaltung „BfArM im Dialog: Biosimilars“ im Juni
2016 hat das Institut das Thema aufgegriffen und wichtige
Fragestellungen mit Bezug auf Biosimilars adressiert. Diese
Art von Veranstaltungen fördert den Austausch von Perspekti-
ven bezüglich Biosimilarthemen zwischen Herstellern, Kran-
kenkassen und Zulassungsbehörden.
10 Jahre Biosimilars – Lessons learned
Auch Zulassungs-behörden können dazu beitragen, Unsicherheiten be-züglich Biosimilars abzubauen und Fragen zu beant-worten.
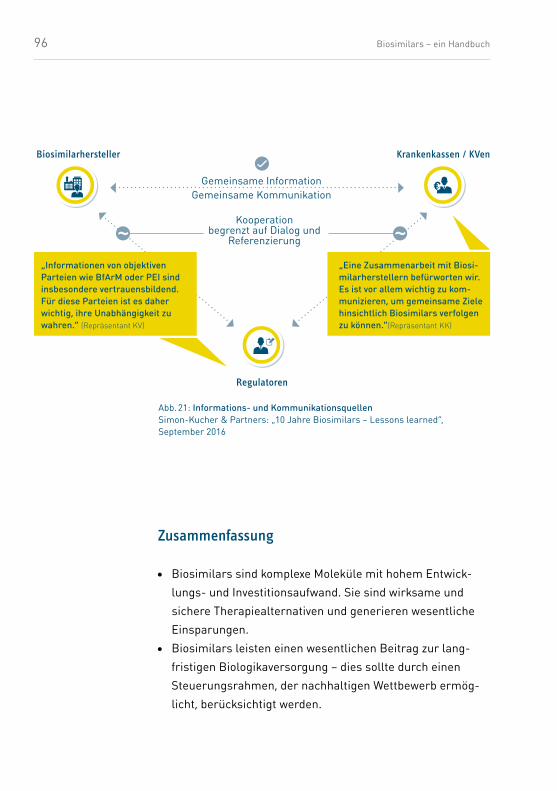
96 Biosimilars – ein Handbuch
Zusammenfassung
• Biosimilars sind komplexe Moleküle mit hohem Entwick-
lungs- und Investitionsaufwand. Sie sind wirksame und
sichere Therapiealternativen und generieren wesentliche
Einsparungen.
• Biosimilars leisten einen wesentlichen Beitrag zur lang-
fristigen Biologikaversorgung – dies sollte durch einen
Steuerungsrahmen, der nachhaltigen Wettbewerb ermög-
licht, berücksichtigt werden.
Krankenkassen / KVenBiosimilarhersteller
Gemeinsame InformationGemeinsame Kommunikation
Kooperation begrenzt auf Dialog und
Referenzierung
Regulatoren
„Informationen von objektiven Parteien wie BfArM oder PEI sind insbesondere vertrauensbildend. Für diese Parteien ist es daher wichtig, ihre Unabhängigkeit zu wahren.“ (Repräsentant KV)
„Eine Zusammenarbeit mit Biosi-milarherstellern befürworten wir. Es ist vor allem wichtig zu kom-munizieren, um gemeinsame Ziele hinsichtlich Biosimilars verfolgen zu können.“(Repräsentant KK)
Abb. 21: Informations- und KommunikationsquellenSimon-Kucher & Partners: „10 Jahre Biosimilars – Lessons learned“, September 2016

97
• Zielvereinbarungen, die Informations- und Beratungsmaß-
nahmen sowie einen definierten Zielverordnungsanteil für
Biosimilars umfassen, sind bei konsequenter Umsetzung
ein nachhaltiges Steuerungsinstrument zur Förderung von
Biosimilars, da Ärzten eine klare Zielvorgabe gesetzt wird.
• Selektivverträge sind flexibel ausgestaltbar und eignen sich
bei entsprechender Ausgestaltung zur Förderung von Bio-
similars, indem sie dem Arzt positive Anreize zur Verord-
nung von Biosimilars setzen.
• Open-House-Verträge sowie Rabattverträge mit dem Refe-
renzprodukt sind nicht geeignet, Biosimilars zu fördern,
können jedoch durch Information zur Wirtschaftlichkeit von
Biosimilars verbessert werden (z. B. Informationsschreiben
der Krankenkassen).
• Begleitende Informations-/Beratungsmaßnahmen von
Krankenkassen, KVen und Herstellern sind unerlässlich, um
die Förderung der Biosimilarverordnungen zu unterstützen.
• Krankenkassen und KVen stehen der gemeinsamen sowie
gegenseitigen Information mit Biosimilarherstellern positiv
gegenüber. Regulatoren sind wertvolle Dialogpartner und
als unabhängige und objektive Informationsquelle wichtig,
um das Vertrauen in Biosimilars weiter zu fördern.
• Das Vertrauen in Biosimilars ist im Laufe der Zeit gestiegen.
Der Informationsbedarf wird aber immer noch als hoch ein-
geschätzt.
10 Jahre Biosimilars – Lessons learned

98 Biosimilars – Ein Handbuch
DIE BEDEUTUNG VON BIOSIMILARS IN DER
VERTRAGS ÄRZTLICHEN VERSORGUNG
Johann Fischaleck
Kapitel
6

99
DIE BEDEUTUNG VON BIOSIMILARS IN DER
VERTRAGS ÄRZTLICHEN VERSORGUNG
Johann Fischaleck
Was sind Biosimilars?
Seit 2001 begannen nach und nach die Patente einiger wich-
tiger, umsatzstarker Biopharmazeutika auszulaufen. Da sich
der Prozess der Patentabläufe wichtiger Biopharmazeutika
rasant beschleunigt, beginnen Biosimilars eine immer ent-
scheidendere Rolle zu spielen, indem sie mit umsatzstarken
Biopharmazeutika um Marktanteile konkurrieren.
Zehn Jahre sind nun seit der Zulassung des ersten Biosimilars
in Deutschland vergangen. Wie sieht der Markt heute aus?
Welche biosimilaren Wirkstoffe stehen zur Verfügung und
welche Vorteile bietet deren Verordnung?
Wie erfolgt der Austausch eines Originals in ein Biosimilar?
Originale und Biosimilars sind nicht im Rahmen der Aut-
Idem-Regelung austauschfähig. Daher wird in der Apotheke
grundsätzlich das verordnete Medikament an den Patienten
abgegeben. Auch das Bestehen von Rabattverträgen bewirkt
keinen Austausch des Präparates. Dabei spielt es keine Rolle,
dass die Biosimilars denselben International Non-Proprietary
Name (INN) tragen wie das Original. Durch die Dokumentation
des Arztes ist die Therapie eindeutig rückverfolgbar. Ein ande-
rer Name als derselbe INN-Name wie das Original würde eher
zur Verwirrung führen und den Marktzutritt für Biosimilars
vielleicht behindern. Auch die Akzeptanz durch den Patienten
wird durch den gleichen INN-Namen erhöht.
Die Bedeutung von Biosimilars in der vertragsärztlichen Versorgung
Durch die Dokumentationdes Arztes ist die Therapie eindeutig rückverfolgbar.

100 Biosimilars – ein Handbuch
Lediglich identische biosimilare Präparate aus der gleichen
Fabrikation, wie bspw. Biograstim®, Ratiograstim® und Teva-
grastim®, bei denen es sich allesamt um identische Arznei-
mittel handelt, die aus einer Produktion kommen, kann ein
Austausch auch in der Apotheke vorgenommen werden. Aus
gleichem Grund können auch Filgrastim Hexal® und Zarzio®
in den Apotheken gegeneinander ausgetauscht werden oder
Inflectra® und Remsima®, die Biosimilars zu Remicade® sind,
eingesetzt werden – v. a. in der Rheumatologie und der
Gastroentero logie.
Die Einstellung auf ein Biosimilar erfolgt ausschließlich durch
den verordnenden Arzt. Besonders bei der Neueinstellung von
Patienten kann ohne Mehraufwand von den ökonomischen
Vorteilen der Verordnung eines Biosimilars profitiert werden.
Je nach Wirkstoff sind die Biosimilars ca. 20 % bis 30 % preis-
günstiger als die dazugehörigen Originalpräparate.
Umstellung bei laufender Therapie
Die FDA verlangt im Rahmen der Markteinführung von Bio-
similars auch sogenannte Interchangeability-Studien, die den
Nachweis erbringen sollen, dass Originale und Biosimilars
austauschbar sind.
Der Ausschuss für Humanarzneimittel CHMP bei der Euro-
päischen Arzneimittel-Agentur („European Medicines Agency“
– EMA) bewertet als Teil des Zulassungsverfahrens, bei dem
Nutzen und Risiko eines Arzneimittels gegenübergestellt wer-
den, primär die pharmazeutische Qualität, Wirksamkeit und
Die Einstellung auf ein Biosimilar
erfolgt ausschließ-lich durch den
verordnenden Arzt.

101
auch Sicherheit von Biosimilarkandidat und Originatormolekül
im direkten Vergleich, und nicht die Austauschbarkeit.
Nach derzeitigem Diskussionsstand im CHMP und seinen
Arbeitsgruppen können Biosimilars grundsätzlich nach er-
wiesener Äquivalenz und erfolgter Zulassung so eingesetzt
werden, wie Originatorprodukte auch. Dies beinhaltet implizit
daher sowohl Patienten, die vorher noch keine Therapie mit
Biologika erhalten, als auch solche Patienten, die vorher das
Originatormolekül bekommen haben.
Dieser Austausch wird nach unseren Analysen im Bereich der
Erythropoetine von den Vertragsärzten bereits seit einigen
Jahren vorgenommen und kann sicherlich auch bei anderen
Produkten in Erwägung gezogen werden. Für die Biosimilars
von Infliximab und neuerdings auch für das biosimilare
Etanercept trifft dies in Bayern in zunehmendem Maße zu.
Erythropoetin wird bei Patienten mit Nierenversagen, soge-
nannter terminaler Niereninsuffizienz, im Rahmen einer Blut-
wäsche sowie zur Behandlung der Blutarmut eingesetzt. Sei-
ne Wirkung erkennt man am Anstieg des Hämoglobinwertes,
so dass die zum Erreichen des Zielwertes notwendige Dosis
des Erythropoetins als Surrogatparameter für die Wirksamkeit
des Erythropoetins dienen kann. Immer wieder kam insbeson-
dere bei Erythropoetin-Biosimilars die Behauptung auf, dass
Biosimilars zu einem Mehrverbrauch an Einheiten im Ver-
gleich zu den Erstanbieterprodukten führen.
Die KV Bayerns hat dazu eine retrospektive Versorgungsfor-
schungsstudie aufgelegt, um der Fragestellung nachzugehen,
Die Bedeutung von Biosimilars in der vertragsärztlichen Versorgung

102 Biosimilars – ein Handbuch
ob es einen Unterschied im Verbrauch zwischen den verschie-
denen Erythropoetinen-H gibt.28
Vergleichende Untersuchung von Erythropoetin- Biologika und Biosimilars in Bayern
Aus der Grundgesamtheit der 10,4 Millionen gesetzlich Ver-
sicherten in Bayern wurden 16.895 Patienten mit terminaler
Niereninsuffizienz und chronisch intermittierender Hämodia-
lyse-Behandlung selektiert. Von diesen wurden 6.177 Patien-
ten über mindestens 1,5 Jahre während des Untersuchungs-
zeitraumes mit Erythropoetin behandelt. Diese Patienten
wurden analysiert. 64,4 % dieser Patienten wurden mit
Erythro poetin-Originatorarzneimitteln, 21,1 % mit Erythropo-
etin-Biosimilars und 14,6 % mit Erythropoetin-Originator und
Bio similar nacheinander behandelt. Insgesamt 35,7 % aller
Patienten wurden dabei mit einem Biosimilar behandelt. Für
507 Patienten liegen Daten zum „Switch“ vor, für 450 zum
Wechsel vom Originator zum Biosimilar, für 57 vom Biosimilar
zum Originator. Es zeigten sich folgende Ergebnisse: Die Do-
sierung der Erythopoetin-Biosimilars entsprach der Dosierung
der Originatoren. Ein Wechsel von einem Originator zu einem
Erythropoetin-Biosimilar führte nicht zu einer Erhöhung der
Dosierung. Insbesondere die Dosiskonstanz bei einer Beo-
bachtungszeit der Patienten über 1,5 Jahre unterstreicht die
Schlussfolgerung der gleichen Wirksamkeit von Eryhtropoetin-
28 Hörbrand F. Bramlage P. Fischaleck J. Hasford J. Brunkhorst R. A. Population-based study comparing biosimilar versus originator erythropoiesis-stimulating agent consumption in 6,6177 patients with renal anaemia. Eur J Clin Pharmacol, March 2013

103
Originator und Biosimilar. Die Patienten der mit Biosimilars
behandelten Gruppe waren etwas älter und hatten etwas mehr
Begleit erkrankungen, einen Einfluss auf die Dosierung hatte
dies jedoch nicht.
Dies ist die erste populationsbasierte Studie zum Vergleich
der Wirksamkeit von Erythropoetin-Originatoren und ihren
korrespondierenden Biosimilars. Sie belegt die therapeutische
Gleichwertigkeit und unterstützt die Empfehlung der KV
Bayerns zur Umstellung von Patienten vom Originator auf das
Biosimilar.
Wie gehen wir heute mit Biosimilars um?
Es ist ein wichtiges Ziel von Ärzteschaft und Krankenkassen,
hochwertige Medizin bezahlbar zu halten. Bei den meisten
Kassenärztlichen Vereinigungen sind mittlerweile Biosimilar-
quoten in den Arzneimittelvereinbarungen eine Selbstver-
ständlichkeit. Der Verband der forschenden Arzneimittelher-
steller (vfa) hingegen mahnt zum „vorsichtigen Umgang“ mit
Biosimilars. Dies scheint eher ökonomisch motiviert als evi-
denzbasiert zu sein, denn bei genauer Betrachtung gibt es das
Original ja gar nicht. Jede neue Charge eines biologischen
Originalproduktes unterscheidet sich mehr oder weniger von
seinem Vorgängerprodukt. Viele Biologika haben mittlerweile
zahlreiche Änderungen im Herstellungsverfahren durchlaufen,
Die Bedeutung von Biosimilars in der vertragsärztlichen Versorgung
Die Studie belegt die therapeutischeGleichwertigkeit und unterstützt die Empfehlung der KVBayerns zur Um-stellung von Pati-enten vom Origina-tor auf das Biosimilar.

104 Biosimilars – ein Handbuch
die jeweils ein ähnliches, aber kein gleiches Produkt zur Folge
haben. Ärzte, die das „Original“ über Jahre hinweg verordnen
und selbstverständlich die gleiche Wirksamkeit erwarten,
haben in der Realität immer quasi Biosimilars zur ersten
Charge des Biologikums verordnet. Es verwundert, wenn dann
beim Einsatz von „echten“ Biosimilars der Arzt Zweifel an der
Qualität anmeldet, obwohl die Produktion innerhalb der glei-
chen Spezifikationsgrenzen gehalten wird, die auch für die
Originalprodukte gelten und die eine gleiche Wirksamkeit
gewährleisten.
Biosimilarquoten auf DDD-Basis sind in den Arzneimittelver-
einbarungen auf KV-Ebene ein probates Mittel, sollten jedoch
von KV und Krankenkassenseite immer positiv begleitet wer-
den und bei Erreichen der Quote mit entsprechenden Entlas-
tungen in der Wirtschaftlichkeitsprüfung belohnt werden.
Der Arzt hat damit den Hebel in der Hand, über das Erreichen
einer Zielquote bei Generika und Biosimilars die Wirtschaft-
lichkeitsziele zu erreichen. So werden auch in Bayern zuneh-
mend Ziele formuliert, die eine Biosimilarquote ausweisen
und die Steuerung des Marktes ermöglichen. Demnächst wird
die bisherige Leitsubstanzregelung bei den TNF--Blockern
durch eine reine Biosimilarquote ersetzt werden. Das Prüfen
nach Kosten gehört somit der Vergangenheit an. Wichtig ist,
dass die Ärzte jederzeit aktuell über die entsprechenden
Märkte und deren Neuerungen informiert und immer wieder
auf diese Thematik angesprochen werden. Das erledigen im
Gebiet der KV Bayerns 15 Pharmakotherapie-Berater/-innen,
die ständig entsprechende Fragen von Ärzten beantworten,
aber auch in persönlichen Gesprächen oder bei Qualitäts-
zirkeln zu Biosimilars informieren. Als Naturwissenschaftler

105
sind die Apothekerinnen und Apotheker bestens geeignet, über
die Herstellung, Strukturaufklärung und Spezifikationen von
Biologikas ganz allgemein zu informieren. So werden evtl.
bestehende Vorbehalte abgebaut und die emotionale Ebene
durch die Sachebene ersetzt.
Auch der Gesetzgeber kann Hürden abbauen helfen, die
derzeit einen Marktzutritt von Biosimilars behindern. So ist
zumindest eine der Forderungen aus dem eben abgeschlos-
senen Pharmagipfel, dass Originalhersteller keine Rabatt-
verträge mit Krankenkassen über den Ablauf des Patentschut-
zes hinaus schließen dürfen, um den Markteintritt der Bio-
similars auf diese Weise nicht zu behindern. Im Abschluss-
bericht zum Pharmadialog aus dem April 2016 wurden
Zielvereinbarungen für Biosimilars von den beteiligten Minis-
terien und Herstellerverbänden als probates Mittel gesehen,
um Biosimilars schneller in die Versorgung zu bringen. Daher
erfolgte zumindest der Appell an die Vertragspartner, dieses
Instrument stärker zu nutzen.
Die Bedeutung von Biosimilars in der vertragsärztlichen Versorgung

Kapitel
7
DIE INITIATIVE „BIOLIKE“
DER BARMER GEK Detlef Böhler

107Die Initiative „Biolike“ der BARMER GEK
Die Initiative „Biolike“ der BARMER GEK
Ziel der Initiative „Biolike“ ist die Förderung einer aus medizi-
nischer und wirtschaftlicher Sicht angemessenen Verordnung
biologisch hergestellter Arzneimittel. Dazu gehört ein struktu-
riertes Arzneimittel-Management zu Biologika und Biosimi-
lars auf Basis gemeinsamer Vereinbarungen mit Kassenärzt-
lichen Vereinigungen. Die erste Vereinbarung wird bereits seit
dem Oktober 2014 in der Pilotregion der Kassenärztlichen
Vereinigung Westfalen-Lippe umgesetzt. Nach erfolgreichem
Abschluss der Pilotphase, wird die Initiative derzeit auf wei-
tere interessierte KV-Regionen ausgerollt.
Gegenstand des „Biolike-Vertrages“ ist ein mehrstufiges Kon-
zept zur Förderung der Verordnung von Biosimilars und zum
rationalen Einsatz von Biologika. Biotechnologisch hergestell-
te Arzneimittel (Biologika) haben die Therapiemöglichkeiten
erweitert. Diese medizinischen Innovationen, kombiniert mit
den meist hohen Kosten, führen nicht selten zu Unsicher-
heiten im Verordnungsverhalten niedergelassener Ärzte. Bio-
similars (also die Nachahmerpräparate von Biologika) können
dazu beitragen, die Kosten einer biologischen Therapie zu
senken. Durch den spezifischen Zulassungsprozess und den
anschließenden Überwachungsmaßnahmen werden Wirksam-
keit und Unbedenklichkeit nachgewiesen.
Im ersten Schritt fokussieren sich die Aktivitäten auf den
Bereich der sogenannten TNF-alpha-Inhibitoren, die im Be-
reich der Rheumatoiden Arthritis, Psoriasis und Morbus Crohn
eingesetzt werden. In den nächsten Jahren werden weitere
Patentabläufe von umsatzstarken Präparaten erwartet und so-
mit ist mit dem Markteintritt von Biosimilars zu rechnen.
Biosimilars können dazu beitragen, die Kosten einer biologischen Therapie zu senken.
DIE INITIATIVE „BIOLIKE“
DER BARMER GEK Detlef Böhler

108 Biosimilars – ein Handbuch
In einem ersten Schritt werden die Vertragsärzte zunächst
umfassend und objektiv informiert. Dazu gibt es ein gemein-
sames Informations- und Beratungskonzept zu Biosimilars für
die Vertragsärzte – initial für TNF-alpha-Inhibitoren verord-
nende Ärzte und Arztgruppen – welches die Verordnung von
Biosimilars innerhalb des jeweiligen Wirkstoffes fördern soll.
Die KVen informieren ihre Vertragsärzte und bieten persön-
liche, individuelle Beratungsgespräche zu diesem Thema an.
Ärzte können nach der Beratung den Rabattverträgen der
Kasse zu den besonders wirtschaftlichen Arzneimitteln bei-
treten. Der Beitritt zum Rabattvertrag wirkt sich für den Arzt
positiv in der Wirtschaftlichkeitsprüfung aus.
In einem zweiten Schritt werden dann konkrete Steuerungs-
maßnahmen unter Einbeziehung der jeweiligen Facharzt-
gruppe vereinbart.
Seit dem 01.07.2015 gibt es im Bereich der KV Westfalen-
Lippe einen ersten Vertrag zu den chronisch entzündlichen
Darmerkrankungen (CED).
Colitis ulcerosa (CU) und Morbus Crohn (MC) werden als chro-
nisch entzündliche Darmerkrankungen (CED) zusammenge-
fasst. In Deutschland sind etwa 400.000 Patienten an einer
CED erkrankt. Ungefähr 50–70 % der Patienten haben einen
eher schweren, komplexen Verlauf, die intensive Therapie-
maßnahmen, die teilweise auch nebenwirkungsbehaftet sein
können, benötigen.
Die KVen informie-ren ihre Vertrags-
ärzte und bieten persönliche, indivi-duelle Beratungs-
gespräche an.

109
Durch die Einführung von Infliximab(IFX)-Biosimilars wurde
seit Anfang 2015 die Diskussion bezüglich der Indikationsbrei-
te und der Sinnhaftigkeit des Einsatzes von IFX-Biosimilars
innerhalb des gesamten Therapiekonzeptes in die Informa-
tionsarbeit vermehrt einbezogen.
Gemeinsam wollten der Berufsverband Niedergelassener
Gastroenterologen Deutschlands e. V. (bng) mit seiner Regio-
nalgruppe in Westfalen, die KVWL und die BARMER GEK mit
diesem Vertrag die fachlichen und organisatorischen Voraus-
setzungen für eine am individuellen Krankheitsverlauf abge-
stimmte, qualitätsgesicherte und passgenaue Behandlung
nach den allgemein anerkannten Standards der medizini-
schen Erkenntnisse durch in der CED-Therapie erfahrenen
Ärzte etablieren. Dabei soll auch der medizinische und phar-
makologische Fortschritt berücksichtigt und die therapeu-
tische Vorgehensweise insbesondere auch in Qualitätszirkeln
diskutiert werden.
Teilnahmeberechtigt sind im Bereich der KV zugelassene, an-
gestellte sowie ermächtigte Fachärzte für Innere Medizin mit
dem Schwerpunkt Gastroenterologie oder mit einer Geneh-
migung zur Führung der Facharztbezeichnung für Innere
Medizin mit fachärztlicher Niederlassung und der Genehmi-
gung zur Durchführung der Vorsorge-Koloskopie, soweit sie
festgelegte persönliche / sachliche Voraussetzungen erfüllen.
Versicherte mit gesicherter Diagnose einer CED gemäß ICD 10
(K50.-, K51.-) können unabhängig von ihrer Medikation an die-
sem Vertrag teilnehmen.
Die Initiative „Biolike“ der BARMER GEK

110 Biosimilars – ein Handbuch
Die am Vertrag teilnehmenden Ärzte werden die gegebenen-
falls erforderlichen Arzneimittel zur Therapie der chronisch
entzündlichen Darmerkrankungen wirtschaftlich, entspre-
chend der aktuellen medizinischen Leitlinien der Fachgesell-
schaft und der im Vertrag festgelegten Regelungen verordnen.
Dabei steht bei dem medizinisch notwendigen Einsatz von bio-
logischen Arzneimitteln die Verordnung von Biosimilars bzw.
der Rabattvertrags-Biologika im Vordergrund. Um die Wirt-
schaftlichkeit der Biosimilars zu unterstreichen, werden diese
Verordnungen von teilnehmenden Ärzten komplett aus der
Wirtschaftlichkeitsprüfung herausgenommen.
Ab dem 01.07.2016 gibt es im Bereich der KV Westfalen-Lippe
zur Initiative „Biolike“ einen weiteren Vertrag zu den entzünd-
lichen rheumatischen Erkrankungen.
Die heute verfügbaren Therapiestrategien ermöglichen zu-
nehmend eine effektive Kontrolle der Krankheitsaktivität und
somit die Verhinderung der Zerstörung der Gelenke sowie der
damit verbundenen Folgen. Entscheidend für einen guten
Therapieerfolg sind eine möglichst frühzeitige Diagnosestel-
lung, eine frühzeitige Einleitung einer antirheumatischen,
krankheitsmodifizierenden Therapie und eine engmaschige,
konsequente Therapieüberwachung. Hierbei spielt eine quali-
tätsgesicherte Arzneimitteltherapie, in der auch hochpreisige
biotechnologisch hergestellte Antirheumatika (Biologicals und
Biosimilars) einen in Leitlinien festgelegten Platz haben, eine
wichtige Rolle.

111
Durch die Einführung von Infliximab(IFX)- und Etanercept(ETA)-
Biosimilars erweitert sich das Spektrum der zur Verfügung
stehenden biotechnologisch hergestellten Antirheumatika.
Erstmalig wird eine Konkurrenzsituation zwischen weitgehend
wirkgleichen Präparaten entstehen, die eine Senkung der z. T.
sehr hohen Therapiekosten erwarten lässt. Daher ergeben
sich Optimierungspotenziale in der Behandlung mit biotech-
nologisch hergestellten Antirheumatika bei Patienten mit ent-
zündlichen rheumatischen Erkrankungen.
Teilnahmeberechtigt sind im Bereich der KVWL zugelassene,
angestellte sowie ermächtigte Fachärzte für Innere Medizin
und Rheumatologie (Facharztgruppe 31), soweit sie festge-
legte persönliche / sachliche Voraussetzungen erfüllen.
Die Teilnahme der Versicherten ist an das Vorliegen einer füh-
renden gesicherten endstelligen Diagnose für eine entzünd-
liche Rheumaerkrankung entsprechend vereinbarter ICD-ID,
sowie an eine hohe Krankheitsaktivität definiert, durch As-
sessment-Scores für Aktivität und Funktion (DAS28, BASDAI)
und eine Notwendigkeit der Optimierung der Basistherapie
geknüpft. Auch eine geplante Umstellung auf ein wirtschaft-
liches Arzneimittel gemäß der vereinbarten Aufstellung bzw.
eine geplante Deeskalation der Dosis eines Biologicals oder
eines Biosimilars berechtigen zur Teilnahme.
Die am Vertrag teilnehmenden Ärzte werden die gegebenen-
falls erforderlichen Arzneimittel zur Therapie der entzündlich
Die Initiative „Biolike“ der BARMER GEK

112 Biosimilars – ein Handbuch
rheumatischen Erkrankungen wirtschaftlich, entsprechend
den aktuellen Leitlinien und der im Vertrag festgelegten
Regelungen verordnen.
Bei der Verordnung von generikafähigen Arzneimitteln soll
kein Aut-idem-Kreuz gesetzt oder nur der Wirkstoff verordnet
werden. Natürlich steht auch bei dem medizinisch notwendi-
gen Einsatz von biologischen Arzneimitteln die Verordnung
von Bio similars bzw. der Rabattvertrags-Biologika im Vorder-
grund. Um die Wirtschaftlichkeit der Biosimilars zu unter-
streichen, werden diese Verordnungen von teilnehmenden
Ärzten komplett aus der Wirtschaftlichkeitsprüfung herausge-
nommen.
Unter Berücksichtigung der vorstehenden Erläuterungen ge-
währleistet die Initiative „Biolike“ die Verbesserung der Ver-
sorgungsqualität und das Erschließen der Wirtschaftlichkeits-
potenziale im Bereich der biologischen Arzneimittel. Die
Erfahrungen in der Umsetzung des ersten Vertrages zeigen
eine hohe Akzeptanz auf ärztlicher Seite und Erfolge bei der
Erreichung der Vertragsziele für alle Parteien.
Die Initiative „Biolike“ gewähr-leistet die Verbes-
serung der Ver- sorgungsqualitätund das Erschlie-
ßen der Wirtschaft-lichkeitspotenziale
im Bereich der biologischen Arzneimittel.

113Die Initiative „Biolike“ der BARMER GEK

114 Biosimilars – ein Handbuch
ANHANG
115
ANHANG
Anhang
Glossar
Adhärenz Einverständnis der Patienten, die vom Arzt empfohlene Therapie einzuhalten
Agitation Heftige Bewegung
Aminosäurepolymere Synthetische Proteine, Eiweiße
Angiogenese bezeichnet das Wachstum von Blutgefäßen, durch Sprossungs- oder Spaltungsvorgänge aus bereits vorgebildeten Blutgefäßen.
AVP Apothekenverkaufspreis
AVP real Apothekenverkaufspreis unter Berücksichtigung aller Zwangsrabatte für Hersteller und Apotheker, inkl. Berücksichtigung der Zusatzabschläge infolge des Preismoratoriums
BatchEin Produktionsansatz zur Herstellung eines Arzneimittels, auch „Charge“ genannt
BioäquivalenzBioäquivalenz bedeutet, dass der Arzneimittelwirkstoff des Generikums identisch mit dem des vergleichbaren Originalpräparates ist. Beide sind somit miteinander austauschbar.
Biological Proteinwirkstoff, der gentechnisch in einer Zelle hergestellt wird
Biopharmazeutikum Arzneimittel, das mithilfe der Biotechnologie und in gentechnisch verän-derten Organismen hergestellt wird
Biosimilar Arzneimittel, das von den Zulassungsbehörden wegen seiner Ähnlichkeit in Bezug auf Qualität, Sicherheit und Wirksamkeit mit einem biologischen Referenzarzneimittel, mit dem es verglichen worden ist, zugelassen wird

116 Biosimilars – ein Handbuch
BMWP Similar Biological (Biosimilar) Medicinal Products Working Party; Arbeitsgruppe für Biosimilars an der Europäischen Arzneimittelagentur
CHMP Committee for Medicinal Products for Human Use; Ausschuss für Humanarzneimittel an der Europäischen Arzneimittelagentur
Code, genetischer Allgemein in der belebten Natur gültige Regelung, wonach jeweils drei Basen in der DNA für eine bestimmte Aminosäure stehen
DDDDefined daily dose, definierte Tagestherapiedosis
DNA Desoxyribonucleic acid, das Erbmaterial von Zellen
Drugability Zielstruktur, die sich für die therapeutische Anwendung eines Wirkstoffes eignet
Efficacy Wirksamkeit einer Substanz
ektopisch Befindet sich nicht am physiologischen Ort
EMA European Medicines Agency; Europäische Arzneimittelagentur
Europäisches Arzneibuch (PhEur) In Europa gültige Sammlung von Beschreibungen einzelner Arzneistoffe hinsichtlich Qualität, Prüfung, Lagerung und Bezeichnung sowie der dazu nötigen Materialien und Methoden
FestbetragHöchstbetrag, den die gesetzlichen Krankenkassen für ein Arzneimittel übernehmen, und zwar unabhängig vom tatsächlichen Preis des Arznei-mittels. Das heißt: Ist der Preis eines Arzneimittels höher als der von den Krankenkassen dafür erstattete Betrag, müssen Patienten in der Apotheke eine sogenannte Aufzahlung leisten. Senkt der Hersteller dagegen den Preis für sein Arzneimittel um 30 % unter den Festbetrag, entfällt für den Pa tienten die Arzneimittelzuzahlung in der Apotheke.

117Anhang
Gentechnik Isolation und Neukombination von DNA, die in einem Wirtsorganismus in ein Protein umgeschrieben wird
General Safety Allgemeine Sicherheitsaspekte
Generikum Arzneimittel, das hinsichtlich des Arzneimittelwirkstoffs identisch mit dem Erstanbieterpräparat ist
GKV-MarktMarkt, der die Verordnungen zulasten der Gesetzlichen Krankenversicherung (GKV) abdeckt
GKV-SpitzenverbandDach- und Lobbyorganisation der gesetzlichen Krankenkassen in Deutschland
Hämophilie Erbkrankheit, die auch „Bluterkrankheit“ genannt wird, bei der die Blut gerinnung gestört ist
HAPHerstellerabgabepreis
HAP realHerstellerabgabepreis unter Berücksichtigung des Herstellerzwangs rabattes, inkl. Zusatzabschläge infolge des Preismoratoriums
INN (International Non-proprietary Name)Wissenschaftliche oder generische Bezeichnung eines Wirkstoffes; INN für neue Wirkstoffe werden von der WHO in Genf vergeben. Der INN ist ein einmaliger (eindeutiger) und allgemein verfügbarer Name.
Immunogenität Fähigkeit, eine Abwehrreaktion des menschlichen Immunsystems anzu regen
klonen Herstellen von mehreren genetisch identischen Organismen

118 Biosimilars – ein Handbuch
Kontrollregion, genetische kurze Bereiche auf der DNA, die das Abschreiben des Gens in mRNA regeln; Promotor am Anfang und Terminator am Ende des Gens
medical need Bedarf an einer Therapieoption
Mikroheterogenität Kleinste Unterschiede zwischen zwei gleichen Molekülen
MolekülVerbindung, die aus Atomen besteht, die durch starke chemische Bindungen in einer festen und bestimmten Anordnung zusammengehalten werden
Monoklonaler AntikörperVon einer Zelllinie („Zellklon“) produziert, die auf einen einzigen B-Lymphozyten zurückgeht; sie richten sich gegen ein bestimmtes, einzelnes Epitop; d. h. eines Molekülabschnittes, der eine spezifische Immunantwort auslösen kann.
Pharmakodynamik Biochemische und physiologische Effekte eines Arzneistoffes in einem Organismus
Pharmakoepidemiologie Untersuchungen des Arzneimittelgebrauchs und der (unerwünschten) Arzneimittelwirkungen in der Bevölkerung im Hinblick auf die Effizienz und Sicherheit der Arzneimitteltherapie
Pharmakokinetik Gesamtheit aller Prozesse, die in einem Organismus auf einen Arzneistoff wirken, z. B. Aufnahme, Verteilung, Umbau und Ausscheidung des Arzneistoffes
Pharmakovigilanz Laufende Überwachung der Sicherheit eines Arzneimittels in der Therapie
phylogenetisch stammesgeschichtlich verwandt
119Anhang
Praxisbesonderheit liegt vor, wenn in einer Arztpraxis z. B. überdurchschnittlich viele Patien ten mit einer besonders kostenintensiven Behandlung betreut werden; so kann ein Arzt diese Praxisbesonderheiten geltend machen, um einen finanziellen Regress der Krankenkassen abzuwehren, wenn er die arztgruppenspezifischen Richtgrößen überschreitet. Diese werden zwischen den Krankenkassen und den Kassenärztlichen Vereinigungen vereinbart, um die vertragsärztliche und damit auch wirtschaftliche Versorgung der Patienten sicherzustellen.
ProteinGroßes Molekül, das aus zu einer Kette angeordneten Aminosäuren besteht
Referenzarzneimittel Ausgangsprodukt, auf das sich Hersteller eines Nachahmerprodukts beziehen
rekombinant Mit einer Neukombination von DNA-Stücken ausgestattet
small molecules Kleine, chemisch-synthetische und meist oral einzunehmende Wirkstoffe
transgen Organismus, in dessen Genom ein fremdes Gen integriert wurde
WirkstoffArzneilich wirksamer Inhaltsstoff oder Molekül in einem Arzneimittel, der / das diesem Arzneimittel Eigenschaften zur Behandlung oder Vergütung einer oder mehrerer Erkrankungen verleiht
Wirtssystem Organismus, der mithilfe einer rekombinanten DNA ein neues Protein herstellt
Zwangsrabatt Gesetzlich vorgeschriebener Rabatt, den pharmazeutische Unternehmen den Krankenkassen einräumen müssen

120 Biosimilars – ein Handbuch
Impressum
Herausgeber: Pro Generika e.V. | Arbeitsgemeinschaft Pro Biosimilars Unter den Linden 32–34 | 10117 BerlinTel. +49 (0)30 - 81 61 60 9-0 [email protected] | www.probiosimilars.de
V. i. S. d. P.: Bork Bretthauer, Geschäftsführer Pro Generika e. V.
Konzept und Gestaltungwww.tack-design.de
BildnachweisUmschlag und KapiteltrennerTack Design GmbH, Berlin
Abb. 1 bis 16 Prof. Dr. Theo Dingermann / Dr. Ilse Zündorf
Seite 14 Fabrik, Seite 25 ProduktionSandoz International GmbH, Holzkirchen
Bild Seite 43Illustrationen Seite 7, 8, 16, 27, 28–29, 35, 36, 45, 49, 51, 53, 54–55, 65, 66–67, 76, 78, 80–81, 92, 97, 101, 105, 111, 112–113Shutterstock Inc.
Stand Januar 2017

Han
dbuc
h Bi
osim
ilars
ZUSAMMENFASSUNG DER VON DER EMA ZUGELASSENEN UND IN DEUTSCHLAND IM VERKEHR BEFINDLICHEN BIOSIMILARS
Stand: Januar 2017Alle Angaben ohne Gewähr
HerausgeberPro Generika e. V. | Arbeitsgemeinschaft Pro Biosimilars
Unter den Linden 32–34 | 10117 Berlin
Tel. +49 (0)30 - 81 61 60 9-0
[email protected] | www.probiosimilars.de
Konzept und Gestaltungwww.tack-design.de
DIE ARBEITSGEMEINSCHAFT PRO BIOSIMILARS
Die AG Pro Biosimilars ist die Interessenvertretung der Biosimilarunter-
nehmen in Deutschland. Sie steht allen Unternehmen offen, die Biosimilars
entwickeln, herstellen und für die Versorgung bereitstellen. Die Arbeits-
gemeinschaft unter dem Dach des Pro Generika e. V. engagiert sich für einen
bedarfsgerechten Zugang der Patientinnen und Patienten zu modernen bio-
pharmazeutischen Arzneimitteltherapien, für eine bezahlbare Versorgung
und für faire und nachhaltige Wettbewerbsbedingungen.
Prof. Dr. Theodor DingermannSeniorprofessor am Institut für
Pharmazeutische Biologie,
Goethe-Universität Frankfurt a. M.
Michael DilgerPartner des Beratungsunternehmens
Simon-Kucher & Partners
Detlef BöhlerLeiter Arzneimittel der BARMER GEK
Johann FischaleckTeamleiter Arzneimittel
im Referat Vertrag und Arzneimittel,
Kassenärztliche Vereinigung Bayerns
Dr. Ilse ZündorfAkademische Oberrätin am Institut
für Pharmazeutische Biologie,
Goethe-Universität Frankfurt a. M.
2017 2017
Handelsname /Hersteller
INN Referenzprodukt Datum der Zulassung
Omnitrope® / Sandoz Somatropin Genotropin® 12.04.2006
Binocrit® / Sandoz
Epoetin alfa Hexal® / Hexal
Abseamed® / Medice
Epoetin alfa Eprex® 28.08.2007
Retacrit® / Hospira
Silapo® / STADA
Epoetin zeta Eprex® 18.12.2007
Biograstim® / AbZ
Filgrastim Neupogen® 15.09.2008
Filgrastim Hexal® / Hexal
Filgrastim Neupogen® 06.02.2009
Nivestim® / Hospira Filgrastim Neupogen® 08.06.2010
Grastofil® / STADA / cell pharm
Filgrastim Neupogen® 18.10.2013
Accofil® / Accord Healthcare
Filgrastim Neupogen® 18.09.2014
Inflectra® / Hospira
Remsima® / Mundipharma
Infliximab Remicade® 10.09.2013
Flixabi® / Biogen Infliximab Remicade® 30.05.2016
Ovaleap® / Teva Follitropin alfa GONAL-f® 27.09.2013
Bemfola® / Finox Follitropin alfa GONAL-f® 27.03.2014
Abasaglar® / Lilly/Boehringer Ingelheim
Insulin Glargin Lantus® 09.09.2014
Benepali® / Biogen Etanercept Enbrel® 14.01.2016
HandbuchBiosimilars
ProBio-Biosimilars-Handbuch-Umschl-RZ.indd 1 22.12.16 14:45

Han
dbuc
h Bi
osim
ilars
ZUSAMMENFASSUNG DER VON DER EMA ZUGELASSENEN UND IN DEUTSCHLAND IM VERKEHR BEFINDLICHEN BIOSIMILARS
Stand: Januar 2017Alle Angaben ohne Gewähr
HerausgeberPro Generika e. V. | Arbeitsgemeinschaft Pro Biosimilars
Unter den Linden 32–34 | 10117 Berlin
Tel. +49 (0)30 - 81 61 60 9-0
[email protected] | www.probiosimilars.de
Konzept und Gestaltungwww.tack-design.de
DIE ARBEITSGEMEINSCHAFT PRO BIOSIMILARS
Die AG Pro Biosimilars ist die Interessenvertretung der Biosimilarunter-
nehmen in Deutschland. Sie steht allen Unternehmen offen, die Biosimilars
entwickeln, herstellen und für die Versorgung bereitstellen. Die Arbeits-
gemeinschaft unter dem Dach des Pro Generika e. V. engagiert sich für einen
bedarfsgerechten Zugang der Patientinnen und Patienten zu modernen bio-
pharmazeutischen Arzneimitteltherapien, für eine bezahlbare Versorgung
und für faire und nachhaltige Wettbewerbsbedingungen.
Prof. Dr. Theodor DingermannSeniorprofessor am Institut für
Pharmazeutische Biologie,
Goethe-Universität Frankfurt a. M.
Michael DilgerPartner des Beratungsunternehmens
Simon-Kucher & Partners
Detlef BöhlerLeiter Arzneimittel der BARMER GEK
Johann FischaleckTeamleiter Arzneimittel
im Referat Vertrag und Arzneimittel,
Kassenärztliche Vereinigung Bayerns
Dr. Ilse ZündorfAkademische Oberrätin am Institut
für Pharmazeutische Biologie,
Goethe-Universität Frankfurt a. M.
2017 2017
Handelsname /Hersteller
INN Referenzprodukt Datum der Zulassung
Omnitrope® / Sandoz Somatropin Genotropin® 12.04.2006
Binocrit® / Sandoz
Epoetin alfa Hexal® / Hexal
Abseamed® / Medice
Epoetin alfa Eprex® 28.08.2007
Retacrit® / Hospira
Silapo® / STADA
Epoetin zeta Eprex® 18.12.2007
Biograstim® / AbZ
Filgrastim Neupogen® 15.09.2008
Filgrastim Hexal® / Hexal
Filgrastim Neupogen® 06.02.2009
Nivestim® / Hospira Filgrastim Neupogen® 08.06.2010
Grastofil® / STADA / cell pharm
Filgrastim Neupogen® 18.10.2013
Accofil® / Accord Healthcare
Filgrastim Neupogen® 18.09.2014
Inflectra® / Hospira
Remsima® / Mundipharma
Infliximab Remicade® 10.09.2013
Flixabi® / Biogen Infliximab Remicade® 30.05.2016
Ovaleap® / Teva Follitropin alfa GONAL-f® 27.09.2013
Bemfola® / Finox Follitropin alfa GONAL-f® 27.03.2014
Abasaglar® / Lilly/Boehringer Ingelheim
Insulin Glargin Lantus® 09.09.2014
Benepali® / Biogen Etanercept Enbrel® 14.01.2016
HandbuchBiosimilars
ProBio-Biosimilars-Handbuch-Umschl-RZ.indd 1 22.12.16 14:45