Embed Size (px)
Citation preview

264
Bestimmung der Kohlenstoffdreifachbindung dutch quantitative selektive Hydrierung* WOLrGAI~G Mm~z und KARL MOLL~
Untersuchungslaboratorium der Badischen Anilin- u. Soda-Fabrik AG Ludwigshafen (Rhein)
Eingegangen am 12. Januar 1968
Sum~zary. C~C triple bonds are determined by selective hydrogenation; 1 triple bond adds I mol hydrogen. The hydrogenation is most successfully carried out in the micro scale in water as solvent. A Pd/CaC03 catalyst, inactivated by zinc, is used. The rate of hydrogenation is significantly increased by addition of traces of Mcaline substances. Sufficiently activated olefinic double bonds add also hydrogen ~nd simulate therefore C-~C triple bonds. -- By use of acetic acid as solvent acetylenic alcohols add hydrogen in presence of the same catalyst forming the corresponding saturated alcohols without attacking the OH-group.
Acetylenverbindungen, insbesondere Acetylenalkohole, sind dureh die Arbeiten yon R~PP]~ [11] zu technisch leieht zug/~nglichen Verbindungen geworden. Als groBtechnisehe Produkte seien hier Butin-2-diol-l,4 und Propargylalkohol erw~hnt, lqeuerdings gewinnen auch kompliziertere Aeetylenalkohole (z. B. Dehydronerolidol, Dehydroisophytol, ~thinyl- jonol) als Vor- und Zwischenprodukte f/Jr die Synthesen der Vitamine A tmd E sowie yon Rieehstoffen an Bedeutung. UV- und IR-spektroskopische Methoden kSnnen zum Nachweis und zur Konstitutionsaufkl~rung von Acetylenverbindungen herangezogen werden. Die UV-Spektroskopie hat te beispielsweise einen wichtigen An- teil an der Konsti tut ionsermitt lung natfirlich vorkommender Polyine [2]. Die II~-Spektren monosubstituierter Acetylene zeigen eine =CH-VMenz- schwingung bei 3300 em -1 und eine C~C-VMenzsehwingung bei 2100 bis 2140 cm-1; disubstituierte Acetylene haben eine (variable) C~C- Valenzschwingung bei 2190--2260 em -1. Die ehemisehen Methoden zur Best immung yon monosubstituierten Aeetylenverbindungen des Typs R �9 C ~ Ctt beruhen vornehm]ich auf der Acetylidbfldung mit Schwermetallsalzen, z. B. die Silberbenzoatmethode yon M~I~SZAK u. KOULKES [9], die einfaehe und vielseitig anwendbare Sflbernitratmethode yon B A ~ s u. M o L I ~ I [1], die wir friiher ein- gehend iiberprtiften [10] sowie die Kaliumquecksilberjodidmethode von H ~ N A u. SIGGIA [4]. Disubstituierte Aeetylenverbindungen werden durch katalytische Anlagerung yon Wasser zum entsprechenden Keton [14,15] oder katalytische Anlagerung yon Alkoholen zu den ent- sprechenden Ketalen [7,18] bes t immt oder nachgewiesen. Mono- und
�9 Herrn Prof. Dr. A. ST~r~OF~R zum 60. Geburtstag gewidmet.

W. MERZ u. K . MI)LLEI~: Kohlenstoffdreifachbindung durch Hydrierung 265
disubstituierte Acetylene k6nnen auch nach SIGGIA u. STAtrL [i6] dnrch UV-Spektroskopie der sich mit Hg(II)-Acetat bildenden Komplexe be- stimmt werden. Zur Vervollstgndigung der analytischen M6glichkeiten wurde nun ver- sucht, Hydrierungsmethoden, die sich als weitgehend universelle Metho- den zur Bestimmung olefinischer Doppclbindungen erwiesen haben [10,12], auch zur Bestimmung yon Kohlenstoffdreifachbindungen heranzuziehen. Unser Ziel war es, mit Hilfe eines geeigneten Katalysators 1. die Kohlenstoffdreifachbindung selektiv so zu hydrieren, daft 1 Mol aufgenommener Wasserstoff einer Dreifachbindung entspricht:
--C--C-- + H 2 -~ --CH=CH--
2. Acetylenalkohole zu" gesgtt. Alkoholen zu hydrieren, ohne daft dabei die OI{-Gruppe partiell angegriffen wird.
1. Experimentelles Arbeiten fiber die quantitative selektive Hydrierung yon Dreifaeh- bindungen im analytischen Magstab sind uns nieht bekannt. Es sind jedoch Katalysatoren beschrieben, welche die partielle Hydrierung von Acetylenderivaten im technisehen bzw. pr/~parativen Maftstab erm6g- lichen. Genannt seien die Katalysatoren yon LINDLAR [8] (Pd auf Cal- eiumearbonat, inaktiviert mit Bleiaeetat und Chinolin), H~xNIo~ [5] (sehr geringe Mengen Pd auf Barinmcarbonat) und TEDESOHI U. CLARK [17], die tertigre )~thinylcarbinole mit Hilfe yon Pd-Katalysatoren auf ge- eignetem Trggermaterial in Gegenwart geringer Mengen starker Basen unter schwaehem Druck und etwas erh6hter Temperatur loartiell hydrie- ren. F fir nnsere Zweeke erschien besonders der in einem Patent be- schriebene, durch Metalle der II. und/oder III . Nebengruppe inaktivierte Pd/CaCOa-Kata]ysator yon PaS]~DACH u. S]~F]~LDEl~ [3] geeignet, der auch zur partiellen Hydrierung yon 3-Methylbutin-l-ol-3 oder Bntin-2- diol-l,4 in teehnischem Maftstab eingesetzt wird. I)ieser Katalysator erffillt Bedingungen, die uns ffir seine analytische Handhabung wiehtig erschienen; er ist bereits bei Raumtemperatur und Normaldruck wirk- sam. Als L6sungsmittel dicnt Wasser. Naeh Patentangabe ist es zweck- m/tgig, dem wggrigen Medium geringe Mengen Ammoniak oder Amin als Besehleuniger znzusetzen; wir ffihrten deshalb auch Versuehe in Gegenwart yon Spuren Ammoniak oder ]4thanolamin aus. AnBerdem fiberprfiften wir in Anlehnung an T~DESCHI U. CLARK [17] den Einflug yon Spuren Alkali bzw. von Bariumhydroxid. Als weiteres L6sungs- mittel verwendeten wir Eisessig, obwohl bekannt ist, daft selektive Hydriernngen in saurem Medium nicht erfolgreieh ausffihrbar sind. ~Vir erwarteten, dab der Katalysator in Eisessig entgiftet wird und fanden, dab dabei ca. 25~ des aufgetragenen Zinks in L6sung gehen. Damit

266 W. M~gz u. K. Mi~LLEg:
war die Hoffnung gegeben, dab die bei katalytisehen Hydrierungen beobaehteten Teilhydrierungen der OH-Gruppe [10] vermieden werden. Neben der Hydrierung ausgew/~hlter Aeetylenderivate wurden aueh I-Iydrierversuehe mit einigen olefiniseh ungesgtt. Verbindungen und Nitrilen (C=N-Dreifaehbindung) durehgefiihrt, um die Selektivitat des Katalysators zu fiberpriifen und um eventuelle St6rungen erkennen zu k6nnen. Die t tydrierversuehe erfolgten vornehmlieh im Mikromal]stab mit Einwaagen yon ca. 5 rag, in einigen Beispielen aueh im MakromaB- stab mit Einwaagen yon 50--100 mg. Im einzelnen wurden die Versuehe folgendermaBen durehgefiihrt :
1.1. Reagentien 1.1.1. WasserstoH. Es wurde Elektrolytwasserstoff verwendet, der vor der Hydrie- rung dutch alkalisehe Plumbitl6sung geleitet wurde.
Abb. 1. Aploaratur zur Mikrohydrierung
1.1.2. LSsungsmittel. Wasser, Wasser -k NIt8 (5 ixg NH~/ml), Wasser @ J~th~nol- amin (20 ~zg NI-I2CH~CI-I2OH/ml), Wasser ,-k KOII (16,5 ~zg KOH/ml), Wasser -k NaOH (12,5 ~g NaOH/ml), Wasser -i- Bariumhydroxid (50 ~zg Ba(OH)Jm]), Eis- essig (p. a.).

Kohlenstoffdreifachbindung dutch quantitative selektive Hydrierung 267
Ffir die iikrohydrierung wurden 2 ml, ffir die Makrohydrierung 15 ml L6sungs- mittel angewandt. 1.1.3. Katalysator. Es wurde ein Zn-inaktivierter Pd/CaCOa-Katalysator verwendet, der 0,6--0,7~ Pd und ca. 90/0 Zn enthiilt. Er wurde nach den Angaben des DBP 1115238 hergestellt, indem ein Pd/CaCO3-Katalysator mit ZinkaeetatlSsung in Wasser verriihrt und 1--2 h zum Sieden erhitzt wurde. Der abgesaugte Katalysator wurde mit Wasser ausgekoeht, gewaschen und ansehlieBend getrocknet. Ffir die Mikrohydrierung werden 20--25 mg, fiir die Makrohydrierung 200 mg Katalysator eingesetzt.
1.2. A pparatur Es wurden bew~hrte Apparaturen verwendet, die auf dem Prinzip des volumetrisch gemessenen Wasserstoffverbrauchs beruhen, und zwar ffir die Mikrohydrierung die Apparatur yon SC~5~mER bzw. SOLTYS [13] (Abb. 1).
Abb. 2. I-Iydriergefgg
Durch Verwendung eines besonderen HydriergefgA3es (Abb. 2) kormte die fiir Mikro- hydrierungen notwendige Vorhydrierung yon LSsungsmittel und Kontakt auf sehr einfache Weise durchgefiihrt werden. Es sei noch bemerkt, dal] zur Vermeidung eines Wasserstoffverlustes ,,Glas an Glas" zu arbeiten ist. Unges~ttigte Bestandteile
268 W. MEI~Z u. •. Mi)LLER:
des Hahnfettes fiihren zu erhShten Werten; desh~lb wurde als Diehtungsmittel ein Gemiseh aus festem Paraffin und Vaseline (1:4) verwendet. Ffir das Arbeiten im Makrobereieh wurde die yon WURZSO~MITT U. KERCXOW [10] modifizierte Itydrierapparatur yon KAUFMA~X U. BAriTES [6] verwendet.
2. Ergebnisse
2.1. Die Versuchsdaten, ausgedriickt in Mol aufgenommenem Wasser- stoff pro Mol Acetylenverbindung, die wir unter Verwendung des mit Zn inakt ivier ten Pd/CaCO3-Katalysators in den LSsungsmit teln Wasser (auch in Gegenwart yon Spuren hTHs, Xthanolamin, K O H ) und Eisessig erzielten, sind in Tab. 1 zusammengefaBt. Die angefiihrten, durch Mikro-
Tabelle 1. Mikrohydrierung yon Acetylenderivaten
Substanz Wasser LSsungsmittel Eisessig
rein -~ NH3 + NH~CH2CH2OH + KOH
Phenylacetylen -- -- -- 1,0 2,0 Propargyla]kohol 1,1 1,0 1,2 1,2 1,0 2,0 3-Methylbutin-l-ol-3 1,0 1,0 1,0 1,0 1,0 2,0 2,1
0,9 1,9 3-Methylpentin-l-ol-3 1,0 0,9 1,1 1,0 1,0 2,0
0,9 4-Athyloctin-l-ol-3 1,1 1,0 1,0 1,0 2,0 1-_~thinylcyclohexanol 1,0 1,0 1,0 0,9 1,0 2,0 2,0 2-Methy1-1-~ithinyl- 1,0 1,0 1,0 1,0 1,0 2,0 2,0
cyclohexanol 0,9 1,9 Butin-2-diol-l,4 1,9 1,0 1,0 1,0 1,0 1,0 2,0 2,0
1,0 1,0 2-Methylpentin-3- 1,0 1,0 1,0 1,0 2,0
diol-2,5 Dehydronerolidol 1,0 1,0 1,2 1,1 1,0 2,0
1,0
~thinyljonol 1,1 1,0 0,9 1,0 1,0 1,1 2,0 2,0 1,9
Dehydroisophytol 1,1 1,0 1,9 0,9 1,0 2,2 2,0 2,0
\
Kohlenstoffdreifaehbindung dureh qua.ntitative selektive Itydrierung 269
h y d r i e r u n g e r h a l t e n e n W e r t e d e c k e n sich in d e n m e i s t e n Fa l l en m i t den
d u t c h H y d r i e r u n g im Makroma l3s t ab g e w o n n e n e n E rgebn i s s en .
2.2. I n T a b . 2 s ind e in ige E r g e b n i s s e w i e d e r g e g e b e n , die bei de r Mikro-
h y d r i e r u n g y o n D o p p e l b i n d u n g e n e n t h a l ~ e n d e n V e r b i n d u n g e n sowie y o n
N i t r i l e n u n d e i n e m K e t o n e r h a l t e n w u r d e n .
Tabelle 2. Milcrohydrierung vor~ Verbindungen ~nit C= C-, C O- und C =_N-Gruppen
Substa.nz LSsungsmittel Wasser Eis-
rein + NH a + NH2CtI2CI-I2Ott q- K 0 H essig
Styrol -- -- -- 0 1,0 Cyelohexadien- 1,3 0 -- -- 0 0,6 Buten-2-diol-l,4 0 0 0 0 1,0 1,0 Zimts~ure 0 -- -- -- 1,0 1,0 Acrylsiiuremethylester -- -- -- 1,0 -- Aeryls~ureamid -- 1,0 -- Cyclohexanon 0 0 -- -- 0 Aerylonitril - - 1,0 1,0 1,0 1,0 Aeetonitril . . . . 0 Adipodinitril . . . . 0
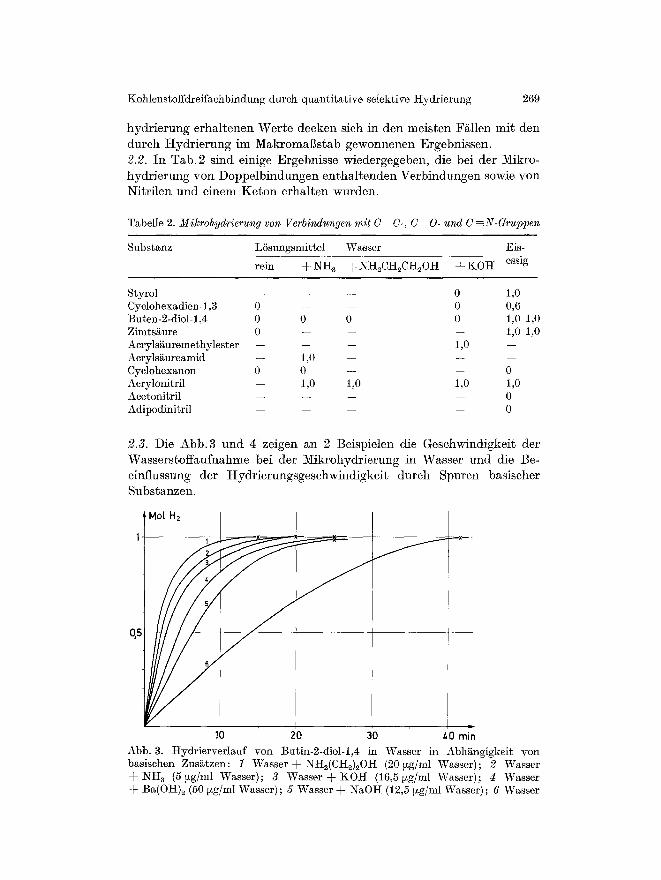
2.3. Die A b b . 3 u n d 4 ze igen an 2 Be i sp ie l en die G e s c h w i n d i g k e i t de r
W a s s e r s t o f f a n f n a h m e bei de r M i k r o h y d r i e r u n g in W a s s e r u n d die ]3e-
e inf lussung der H y d r i e r u n g s g e s e h w i n d i g k e i t d u r c h S p u r e n bas i sche r
S u b s t a n z e n .
, Mot H2
0,E
3
10
Y
f _ _ ^ m
f
30 Abb. 3. Hydrierverlauf yon in Wasser basischen Zus~tzen: 1 Wasser d-NH2(CIt2)20I-I (20 ~zg/ml V~rasser); 2 Wasser d- NI-I~ (5 ~xg/ml Wasser); 3 Wasser -4- ] tOH (16,5 ~xg/ml Wasser); ~ Wasser d- Ba(OH)2 (50 ~xg/ml Wasser); 5 Wasser d- N~O]-I (12,5 ~zg/ml Wasser); 6 Wasser
20 40 rain
Butin-2-diol-l,4 in Abh~ngigkeit yon
270 W. MERE U. K. Mi~LL~R:
0,~
, Mot H2
/
|0 20 30 40 rain
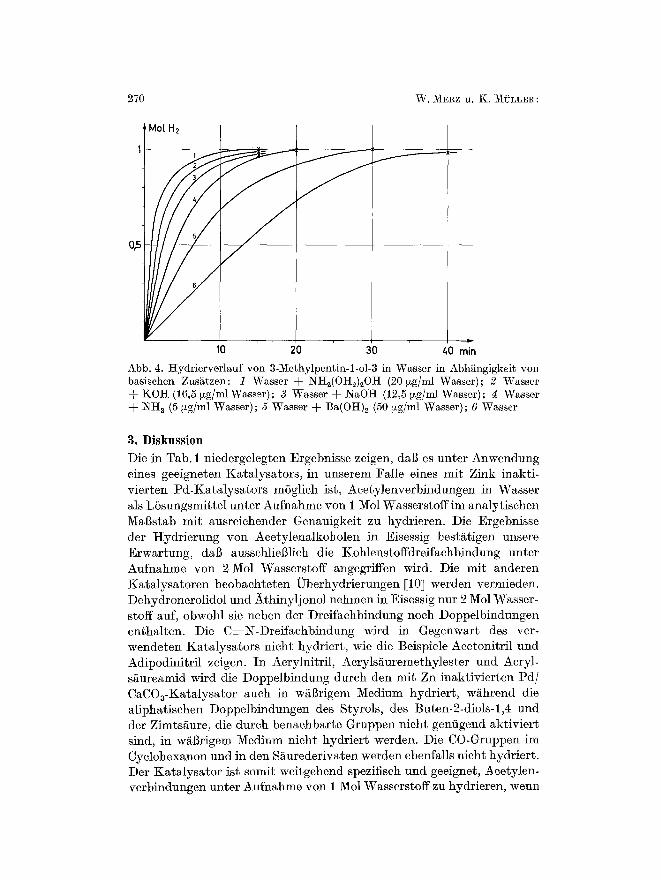
Abb. 4. Hydrierverl~uf yon 3-Me~hylpentin-l-o]-3 in Wasser in Abh~ngigkeit yon basischen Zusgtzen: 1 Wasser + NH2(OH2)zOH (20 ~g/m] Wasser); 2 Wasser + KOH (16,5 ~zg/ml Wasser); 3 Wasser + NaOH (12,5 ~g/ml Wasser); 4 Wasser -~- NH a (5 ~g/ml Wasser); 5 Wasser -~ Ba(OH)2 (50 ~g/ml Wasser); 6 Wasser
3. Diskuss ion
Die in Tab. 1 niedergelegten Ergebnisse zeigen, dal] es unter Anwendung eines geeigneten Katalysators, in unserem Falle eines mit Zink inakti- vierten Pd-Katalysators m6glich ist, Acetylenverbindungen in Wasser als L6sungsmittel unter Aufnahme yon i Mol Wasserstoffim analytischen Magstab mit ausreichender Genauigkeit zu hydrieren. Die Ergebnisse der Hydrierung yon Acetylenalkoholen in Eisessig best~tigen unsere Erwartung, dab ausschlieBlich die Kohlenstoffdreffachbindung unter Aufnahme von 2 Mol Wasserstoff angegriffen wird. Die mit anderen Katalysatoren beobachteten 1Jberhydrierungen [10] werden vermieden. Dehydronerolido] und Xthinyljono] nehmen in Eisessig nur 2 Mol Wasser- stoff auf, obwohl sie neben deI ~ Dreifachbindung noch Doppelbindungen enthalten. Die C~N-Dreffachbindung wird in Gegenwart des ver- wendeten Katalysators nicht hydriert, wie die Beispiele Acetonitril und Adipodinitril zeigen. In Acrylnitril, Acrylsguremethylester und Acryl- sgureamid wird die Doppelbindung durch den mit Zn inaktivierten Pd/ CaCOa-Katalysator auch in wgBrigem Medium hydriert, w~hrend die aliphatischen Doppelbindungen des Styrols, des Buten-2-diols-l,4 und der Zimtsgure, die durch benachbarte Gruppen nicht geniigend aktiviert sind, in wgBrigem Medium nicht hydriert werden. Die CO-Gruppen im Cyclohexanon und in den S~urederivaten werden ebenfalls nicht hydriert. Der Katalysator ist somit weitgehend spezifisch und geeignet, Acetylen- verbindungen unter Aufnahme yon 1 Mol Wasserstoff zu hydrieren, wenn

Kohlenstoffdreifachbindang durch quantitative selektive Hydrierung 271
auch E insch rgnkungen bei gewissen ak t i v i e r t en Doppe lb indungen zu machen sind.
Die H y d r i e r k u r v e n zeigen die auch an den anderen un t e r such ten Ace- t y l e n d e r i v a t e n beobach t e t e Verkf i rzung der H y d r i e r d a u e r , wenn Spuren Nt ta , A thano lamin , Alka l i oder B a r i u m h y d r o x i d , dem L6sungsmi t t e l Wasse r zugese tz t werden.
Zusammenfassung C- -C-Dre i f achb indungen k6nnen durch sclekt ive H y d r i e r u n g b e s t i m m t werden, wobei pro Dre i fachb indung 1 Mol W~ssers toff au fgenommen wird. Die H y d r i e r u n g erfolgt a m bes ten im MikromM~stab. Sie wird in Wasse r in Gegenwar t eines mi t Zn i na k t i v i e r t e n P d / C a C 0 s - K a t a l y s a t o r s durchgeff ihr t . Die Hydr i e rungsgeschwind igke i t kanrL durch Zusa tz yon Spuren basischer Stoffe wesent l ich e rh6ht werden. Genfigend s t a rk ak t iv i e r t e Doppe lb indungen werden u n t e r den Versuchsbcd ingungen ebenfal ls h y d r i e r t und t~uschen d a n n Kohlens to f f -Dre i fachb indungen vor. - - Der gleiche K a t a l y s a t o r h y d r i e r t Ace ty lena]kohole in Eisessig als L6sungsmi t t e l ohne Angri f f der Ot t -Grulopen zu en t sprechenden
ges~tt . Alkoholen.
Wir danken I-Ierrn Dr. PAS~D.~C~ und I-Ierrn Dr. PFAB ffir wertvol]e Anregungen. Ffir die sorgf~iltige Durchffihrung der Versuche danken wir Herrn G. NICKLAS, A. SCHLI~DWEI~ and H. TRARBACH.
Literatur 1. BARNES jr., L., and L. J. ~r Anal. Chem. 27, 1025 (1955); vgl. diese
Z. 152, 301 (1956). 2. BO~L_~A~N, F.: Angew. Ch~m. 67, 389 (1955). 3. Deutsches Bundespatent 1115238 (BASF; Erfinder: H. PAS~DAC~ u. M. SEE-
FELDER). 4. HANNA, J. G., and S. SIGGI~: Anal. Chem. 21, 1469 (1949); vgl. dies~ Z. 131,
381 (1950). 5. H ~ i o ~ , G. F., u. Mitarb. : J. Org. Chem. 21, 1142 (1956). 6. KAVFMA~, H. P., u. J. BALTES: Ber. Deut. Chem. Ges. 70, 2537 (1938). 7. Kui2oP.n, R., u. T. G6ssL: Angew. Chem. 65, 557 (1953); vgl. diese Z. 143,
66 (1954). 8. LINDLAn, H.: ttelv. Chim. Acta 35, 446 (1952). 9. MARSZAK, J., et M. KOULKES: Bull. Soc. Chim. France (5) 17, 364 (1950);
vgl. diese Z. 132, 189 (1951). 10. ~i3LL~a, K.: dies~ Z. 181, 126 (1961). 11. R~eeE, W., u. Mitarb.: Ann. Chem. 596, 1 (1955). 12. I~EVTEI~, A.: diese Z. 231, 356 (1967). 13. SCH6~IOER, W.: Mikrochem. Verein. Mikrochim. Acb~ 38, 132 (1951). 14. S~AREF~:I~, J. G., and E. W. BOG~OSlA~: Anal. Chem. 33, 640 (1961); vgl.
diese Z. 189, 214 (1962). 15. SIOGIA, S.: Aria.1. Chem. 28, 1481 (1956); vgl. diese Z. 157, 39 (1957).

272 W . M ~ z :
16. - - , and C. 1%. STAEL: Anal. Chem. 8g, 1740 (1963); vgl. diese Z. 214, 364 (1965).
17. TED~.SCtII, P~. J., and G. CLARK jr.: J. Org. Chem. 27, 4323 (1962). 18. WAGNER, C. ])., T. GOLDSTEIN, and E. D. PETERS: Anal. Chem. 19, 103 (1947).
Dr. W. MERZ und Dr. K. Mi3LLER Badische Anilin- u. Soda~ AG, Untersuchungslaboratorium 6700 Ludwigshafen (Rhein)
Automatische Schnellmethode zur Stickstoffbestimmung*
WOLFGANG 7~/~ E R Z
Untersuchungslaboratorium der Badischen Anilin- u. Soda-Fabrik AG Ludwigshafen (l%hein)
Eingegangen am 4. Januar 1968
Summary. A very rapid fully automatic method for the micro-determination of nitrogen in organic compounds is described. The complete analysis time is only two and a half minutes, and the results are at least as precise as those obtained with classical procedures, which this method replaces. The sample is dropped into a vertical quartz tube, where it is burned in a stream of pure oxygen. On completion of combustion the oxygen stream is replaced by carbon dioxide, and the gaseous products are driven through copper oxide and copper fillings, and into an auto- matic azotometer, where the nitrogen volume is read from the digital counter or printed out. The complete cycle is preset and controlled by an electronic pro- grammer, which actuates the magnetically operated three-way valves through which oxygen or carbon dioxide is introduced, and that through which the nitrogen is led into the azotometer.
Die beiden grundlegenden Fehler der klassischen StickstoffanMyse nach DtrMAs sind a) zu niedere Wer te durch unvol ls t / indigen Aufsehlug ther- miseh stabiler Subs tanzen u n d b) zu hohe Werte, durch Bi ldung leieht fliiehtiger Craekgase, die n ich t vollst/~ndig oxydier t werden. U~TE~- ZAVCln~I~ [6] e rkann te als erster, dab ein Zusatz von Sauerstoff in beiden F/tllen zu r icht igen Wer t en fiihrt. Diese Methode, die mi t feuehtem, aus Wasserstoffperoxid gebildetem Sauerstoff arbeitete, wurde zwischen- zeitlich mehrfach modifiziert [2, 5] u n d zuletzt yon EI{R~SIBE~G~I~ [1] in einer zweekm/~gigen Ausffihrung besehrieben. Aber auch hier war die Ana lysendauer noah dureh den Vorsehub des beweglichen Brenners und vor allem der re la t iv langen Zeit der 0 x y d a t i o n der Substanz gegeben. U m diese Zeit zu verkiirzen, suehten wJr naeh einer Methode, die bei
* I-Ierrn Prof. Dr. A. S~]~I~r zum 60. Geburtstag gewidmet.