Embed Size (px)
Citation preview

-___--- Z. anorg. allg. Chem. 619 (1997) 437-441
Zeitschrift fur anorganische und allgerneine Chemie
@) Johann Arnhrosius Barth 1993 ------
Die Elektronen-Lokalisierungs-~unktion in closo-Bor-Clustern
Armin Burkhardt, Ulrich Wedig und Hans Georg von Schncring*
Stuttgart, Max-Planck-Institut fur Festkorperforschung
Andreas Savin
Sluttgart, Institut fur Theoretische Chemic der Universitat
I3ci der Redaktiori eingegangeri am 5. Oktober 1992.
Professor Rudolf Hoppe ziim 70. Gehurtstage gewidmet
Inhaltsiihersichl. Die Struktur und die Elektronendichte der closo-Bor-Cluster B,X, (X = H, Cl, Br, I), f3,X:- (X = H, C1, B1; I) und R,,H,?- wurden rnittels Pseudo- potential-Hartree-Fock-Rechnungen bestimmt. Die Tn- terpretation der Bindungsverhaltnisse erfolgte durch Aus- wertung der Elektronen-Lokalisierungs-Funktion (ELF). Die Bcreichc hohen ELF-Wertes besitzen in allen Fallen
die Form dcs zum Bor-Geriist dualen Polyeders. Sie las- scn die 3-Zentren-2-Elektroneri-Rindunge1i bcsonders gut crkennen. Der Vergleich Lwischen tlartree-Fock- und Ex- tended-Hiickel-Kechllungen zeigl, dali auch semienipiri- schc Verfahren eine gute Gruridlapc fur cine ELF-Inler- pretation sein konnen.
The Electron Localization Function in clmo Boron Clusters
Abstracl. The structure and the electron density in the closo boron clusters B,X, (X = H, CI, Br, I), B,X,’- (X = H, Cl, Br, 1) and B,,H,2L- were determined by pseudopotential Hartree-Fock calculations. The Electron Localization Function (ELF) was used to interprct the bonding characteristics. The regions of high ELF values in all cases have the form of the dual polyhedron of thc boron cagc. They show perfectly the 3 center 2 electron
bonds. The comparison betwcen Hartree-Fock and Ex- tendcd I-Tuckel calculalions point nut that semiempirical calculations can also be a good ha45 for ELF interpreta- tions.
Keywords: cfoso boron clusters; struclure; Hartrec-Fock; pscudopotential; Electron Localization Function
1 Einleitung
Die von Becke und Edgecornbe [2] eingefuhrte Elektro- nen-I~kalisierungs-Funklion (ELF) steht in Verbindung mit der Pauli-AbstoRung und quantifiziert das physi kali- sche BiId, das den Gillespie-Nyfiolm-Regeln [ 1 ] zugrunde liegt. Diese Funktion wurde erstmals von Savin et al. sy- slematisch auf Molckule angewendet und erwies sich als eine aufierordentlich wertvolle Hilfe bei der Tnterpreta- lion der vorliegenden Bindiingsverh~ltnisse. In den ersten Arbciten wurde diese Methode zur Untersuchung von kleinen Molekulen 131, von speLiellen Carbosilanen f4j
* jeweils Korrespondenzaulor
und auch von den Kristallstrukturen dcr Eleineritc der 4. Hauptgruppc eingcsetzt [ 5 ] . Nachdem in [3] mit B,H, bcreits ein Vertreter der ElekIronenniangel-Verbindungen behandell wurde, wcrden in der vorlicgendea Arbeit die Ror-Cluster B4X4 (X = 14, f l , Br, I), B,X:- {X = H, C1, Br, 1) und B,,H,,’ untcrsucht.
2 Rcch enver f ahren
Die der ELF zugrunde licgeridc blektronendichle wurdc auC 7weierlci Weke bercchnet (HP bzw. EH?’). Die ub initio-Dichte wuIcle aus Hartr~e-Fock-Rechnungen (H F) unter Verweiidung cines DZP-Basissatzes [6] rnit dern Programmpaket TURBO- MOLE 17) erhallcn. Ilabei wurde dcr 1 s-Rumpf des Bors durch eiti Pseudopotential [8] dargestellt. Die Clustcr-Slrukturen WUJ-


43 9 A. Burkhardt u. a., Elektronen-Lokalisierungs-b’unktion in closo-Bor-Clustcrn -
den untcr Einhaltung der h$hstmoglichen Punktsymmetrie (B,H4: 43m-T,; B6Hs2-: m3m-0,; R,2H12 : 235-1) opti- miert. Urn zu ubcrprdfcn, ob die mit Hilfe von Hartree- Fock-Rechnungcn erhaltenen Resultate auch durch weniger auf- wendige scmiempirische Verfahren zuganglich sind, wurden zii- satzlich fur die optimierten Hartree-FocL-Struk turcn Extended- Hiickel-Rechnungen (EHT) [9] durchgcfilhrt. Die Darstellung der ELF erfolgt zum einen durch zweidimensionale Schnitte, in denen die als Punktwolke wicdergegebene Elektronendichte entsprechend dcs zugchorigen ELF-Wertes (0 I ELF 5 1) ein- gefarbt wird [lo]. Bereiche hoher Lokalisierung sind weil3 dar- gestellt, der Ubergang zu kleinen ELF-Werten erfolgt uber rot, braun, gelb und griin nach blau. Cine andere hiet verwendete Darstellung sind dreidimensionale iso-ELF-Flachen [ 1 1 , 121.
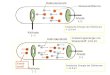
4 Abb. 1 Bild a) B4H4 (HF): Die Elektronendichte wird als Punktwolke dargestellt. Bereiche hoher Dichte crscheinen dabei we$, Bcreiche niederer Dichte schwarz. Dicser Punktwolke ist dic ELF als Rrbmuster uberlagert (klcine Elf-Werte sind blau, grolje weiB dargestcllt, die ubergange erfolgen von blau ubcr grun, braun, nach weil3; der Hintergrund ist schwarz). Die Schnittebene enthalt zwei B-Alome (tiefblaue Bereiche halb- rechts), zwei B-H-Bindungen (weiBe Bereiche in der rechtcn oberen und unteren Ecke) und zwei Dreizcnlren-Bindungen oberhalb der Tetraederflacfien (weiflc Bereiche in dcr linkcn Bildhalfte). Bild b) B4H4 (HF): Dreidirnensionale Darstellung der EL3 an- hand der Isoflache ELF = 0,8. Deutlich sind die den vier 3-Zentren-Bindungen zuzuordnenden 13ereiche hohcr ELF iiber den Flachen des Tetraeders zu erkennen. Hild c) H,H, (EHT): Isoflache ELF = 0,8 a u ~ eincr Extended Hiickel-Rechnung. Der einzige Unterschied zu Bild b) licgl dar- in, daR die ELF-Maxima etwas weiter von det Flache entfernt sind. Bild d) B6H>- (HF Isoflache ELF = 0,X): Die Bereiche hoher ELF iiber den FlBchen des Oktaeders bilden einen Wiirfel (dua- les Polyeder eines Oktaeders). Urn den Rlick auf den Cluster freizuhalten, wurden wie schon in b) und c) dic den H-Atomen 7uzuordnenden iso-ELF-Flachen angeschnitten, wodurch sie an ,, Parabol-Antennen“ erinnern. Bild e) B6H2- (HF): Darstcllung analog zu a). Die Schnitt- ebenc enthalt vier B-Atome (tiefblau), vier B-H-Bindungen (weiBe Bereiche an den Kantenmitten), sowie vicr lokalc ELF- Maxima iiber den Oktaeder-Kanten, die jedoch nur einen klei- nen Teil der Cluster-Bindung ausmacheri (vgl. Bild d). Bild f ) B6H& (HF): Darstellung der Orbilaldichte eincs der LMO’s (Isoflache p = 0,05). Die deutliche Verzerrung wird durch das Fehlen eines LMO’s fur die benachbarte Oktaederfla- che verursacht (vgl. lkxtj. Bild g) BIZHlz2- (HF Isoflache ELF = 0,8): Die Bereiche hohcr ELF bilden einen Pentagon-Dodekaeder (Dudes Polyeder eincs Ikosaedersj, der sich von innen der Clusterperipherie an- schmiegt. Bild h) BjzH,,2- (EHT Isoflache ELF = 0,8): Im Gcgensatz zur HF-Rechnung liegt der ELF-Pentagon-Dodekaeder bei der EHT-Rechnung etwas auRerhalb der Clust erperipherie. Bild i) B4C14 (HF Isoflache ELF = 0’8): Neben der eigentli- chen Cluster-Bindung (vgl. Bild b) sind die freien Elektronen- paare, sowic die polare B-Cl-Bindung deutlich zu erkennen.
3 Ergebnisse und Diskussion
3.1 B4H4
Abbildung 1 a zeigt das Ergebnis der ah-inito-Rechnung fur B,Ii, anhand eines Schnittes durch den B,-Tetraeder. Die weioen Bereiche in den Ecken rechts oben und unten markieren die Bindungen zwischen zwei Wasserstoff-Ato- men und zwei Bor-Atomen in der Bildebene. Die in der Bildebene liegenden Bor-Atome sind in den tiefblauen Bereichen halbrechts zu finden. Die niedrigen ELF-Werte an den Kernpositionen sind durch das Fehlen dcs Is- Rumpfes in der Pseudoputentialrechnung bedingt. Die beiden anderen Bor-Atome dcs Tetraeders liegen in der Bildmitte oberhalb und unterhalb der Bildebene. Das hervorstechendste Merkmal in Abbildung 1 a sind jedoch die ausgedehnten Bereiche hoher Lokalisierung (weil3) auf der linken Bildhalfte. Die raumliche Darstcllung am Beispiel der Isoflache ELF = 0 3 (hbb. 1 b) zeigt, dal3 diese Bereiche hoher Lokalisierung oberhalb der Poly- edertlachen liegen und durch Briicken verbunden sind, welche die Kanten des B,-Tetraeders krcuzen. U beraus bernerkenswert ist die gute Ubereinstimmung zwischen dieser - auf einer ah-initio-Rechnung basierenden - ELF-Darstellung und dem Ergcbnis einer Extended Hiickcl-Rechnung (Abb. 1 c). Die Bilder legen nahe, die Clusterbindung in B4H4 mit Hilfe von vier 2-Elektronen-3-Zentren-Bindungen zu beschrciben, wie dies schon Swanton und Ahlrichs [ 131 vorgeschlagen ha- ben. Festzuhalten bleibt, daO sich die Bcreiche hoher Lo- kalisierung bei B,H, besonders weit nadh auOen er- strecken. Dies ist ein bemerkcnswertes Detail, denn der B,-Tetraedcr wird experimentell nur dann beobachtet, wenn er durch sterisch anspruchsvolle Substituenten ab- geschirmt ist [14].
3.2 &Hi
Auch bei B,,H;- liegen die Maxima der ELF oberhalb der Oktaeder-Flachen, wiederum durch Oktaederkanten kreuzende Briicken rniteinander verbundcn (Abb. 1 d). Die zu den Briicken gehorenden hohen ELF-Werle zwi- schen den Bor-Atomen, die ein Schnitt durch die Okta- eder-Basis (Abb. le) zeigt, stehen nur fur einen kleinen Teil der Cluster-Bindung. Es fallt schwer, diese Bindungs- verhaltnissc mit Wilfe von lokalisierten Orbitalen (LMO’s) zu verstehen, denn bci B,H?- steheri nur vier- Lehn gerustbindende Elektronen zur Verf‘iigung ( Wade- schc Regeln fur n = 6 + 1 = 7 geriistbindende Elektronen- paare). Somit gibt es nur sicben LMO’s fur acht Okta- edertlachen. In der Tat zeigt die nahcre Betrachtung der LMO’s [IS], da13 in dieser Beschreibung eine Oktaeder- flache leer bleibt und dal3 diese Liicke durch eine dcutli- che Verzerrung der benachbarten sechs LMO’s geschlos- sen wird, so dalj nur noch das der Liicke gegenuberliegen- de Orbital eine dreizithlige Symmetrie aufweist. Abb. i f reigt die Orbitaldichte eines der Liicke direkt benachbar- ten LMO’s.

440 Z. anorg. allg. Chem. 619 (1993)
Die Zulassigkeit von ah-initio-Rechnungen fur isolierte anionische Systeme wird zur Zeit kontrovers diskutiert [16, 171. Um unsere Resultate diesbeziiglich abzusichern, wurde zusatzlich zur Rcchnung fur das isolicrte Dianion B,Hh2 eine Rechnung durchgefiihrt, bei der positive La- dungen (q = 0,25) oberhalb der Oktaederfliichen sitzen. Die Lage dieser Ladungen wurdc aus der Kristallstruktur- bestimmung von K2B,H, [ 181 iibernomrnen; ihr Betrag stiinmt weitgehend mit dem durch cine Madelung-kech- nung bestimmten Potential an den Kalium-Positionen (t 0,23) iiberein. Die aus dieser Rechnung erhaltene ELF- Verteilung ist mit bloljen Auge nicht von der des isolier- ten Clusters (Rbb. 1 d) 7u unterscheidcn.
In B6H7-, durch Protonierung von B,H;- erhalten, wandert das zusatzliche H-Atom uber die Clusteroberfla- che 1191. Ab-initio Rechnungen von Kuznetsov et al. [20J ergeben, daO dieses Proton in der energetisch giinstigsten Konfiguration oberhalb einer Fliiche des B,-Oktaeders liegt. Die Energiebarriere fur den Platzwechsel auf eine andere Oktaedcrflache ist am niedrigsten (ca. 10 kcal mol I ) , wenn der Wcg uber eine Oktaeder-Kante fuhrt. Vergleicht man dieses Ergebnis rnit der ELF von B6H:-, so zeigt sich, daO sich das zusiitzliche Proton bevorLugt entlang der Bcreiche hoher ELF-Werte bewegt.
3.3 B,&-
Fur B,2H,22 bilden die hohen ELF-Werte einen Penta- gon-Dodekaeder, der je nach der verwendeten Berech- nungsmethode nur noch entweder knapp innerhalb (HF) oder knapp aunerhalb des Clusters (EHT) liegt (Abb. 1 g, 1 h).
3.4 Hulogenoborune
Neben fruheren thcoretischen Arbeiten [13, 21, 221 sind in der Xwischenzeit auch neuere Strukturbcstimmungen fur einige Vertreter der Tetrahalogeno-doso-tetraborane B,X, 1231 und der Hexahalogeno-doso-hexaborate B,X:- (X = C1, Br, I) vcrfugbar [24], welche auch Rechnungen fur diese Substanzklassen sinnvoll machen. Da es fur die Existenz der entsprechenden Fluoro-Verbin- dungen bisher keinerlei cxperimentelle Hinweise gibt und fruhere theoretische Arbciten bereits deren geringe Stabi- litat bestgtigten, wurde auf ihre Behandlung verzichtet. Die Strukturen der ubrigen Halogeno-doso-Cluster wur- den unter Beibchaltung der hochstrnoglichen Punktsym- mctrie auf HF-Niveau optimierl. Die Ergebnisse sind in Tabelle 1 zusmimengefaBt. Bei den Tetrahalogcnotetra- boranen B,X4 schwanken die berechneten Bor - Bor-Ab- stande nur uin 0,5 pm; auch die gemeinsamen Elektro- nenzahlen [25] fur EX und EBB sind weitgehend unab- hangig vom Liganden. Die berechneten B-B-Abstande in 13,Cl4 sind allerdings urn 4 prn Ianger als die von Thie- ry riintgenographisch bestimrnten. Dabei ist jedoch zu beriicksichtigen, da8 B,Cl, im Kristall nuf noch die Symmetrie 42m besitzt und die mittleren Bindungsab-
stiinde (1 64( 1) pm) eine relativ grone Standardabwei- chung aufweisen. Abb. l i zeigl die Darstellung der ELF fur B,C1, anhand der Isolliiche ELF = 0,8. Neben der bereits oben diskutierten Cluster-Bindung sind dic freien Elektronenpaare an C1, sowie die polarc B-C1-Bindung deutlich zu erkennen. Da die ELF der Hexahalogenohe- xaborate(2 - ) nur bereits diskutierte Charakteristika auf- weist, wurde auf ihre gesonderte Darstellung vcrzichtet.
Tabetlcl B4X4, B6X,Z- (X = H, C1, Br, I ) und BfLHlz' : Bc- rechnetc Abstande r in pm, sowie Oemcinsamc Elektronen-Zah- leri (SEN) nacli Erhurdf urid Ahlrichs [25].
B J b 167,9 I18,7 1,28 037
B,Br4 1 G8,O 187,7 1,35 0,50 BX14 168,3 i 72,7 I ,38 0,41
167,8 209,6 1,35 0,50
B J L L 172,9 1223 1,32 0,6t
B A - 170,l 218,8 0,98 0,66
B,,C'162- 170,l 183,6 0,58 0,7 1 l3,,Br2- 170,l 1973 0,55 O,60
Bt,Hj,Z 178,O 120,9 0,73 0,53
Sowohl bei den Hexaboraten(2 -) als auch bei den Tetta- boranen erfolgt beim Ubergang vom Wasserstofl' zu den hoheren Halogenen cine leichte Kontraktion des Clusters mit zunehmender CrroOe der Substituenten. Dieser Be- fund stcht im Einklang mit riintgenographischci Ergeb- nisscn, wenngleich die dort gefundene Kontraktion gro- Der ist. Bei allen untersuchten Clustrrn ist die gemein- same Elektronenzahl der Drei-Zentren-Bindung BBB weitgehend unabh5ngig von der Art des Liganden.
4 Zusammenfassung
Allen betrachteten Clustern gcmeinsam ist einc hohe Elektronen-Lokalisierung oberhalb der Flachen der Bor- Polyeder. Wegen ihrer starken Strukturierung la& die ELF die 3-Zentren-2-Elektronenbindungen besonders gut erkenncn. In der reinen Dichtedarstellung sind diese kaum erkennbar. Wie das Beispiel der Isoflache ELF = 0,8 zeigt, besitzen die Bereiche hoher Lokalisie- rung in allen Fallen die Form des zum jeweiligen Bor-Ge- rust dualen Polyeders. Mit zunehmender Cluster-GroBe nahern sich die Bereiche groOer ELF-Werte immer mehr der Oberflache des Clusters. Gleichzeitig sleigt der Grad dcr Lokalisierung xwischen den Atomen an. Bei B,2H,,2 ist er rnit den Werten, die im Bereich der Drei-Zen- tren-Bindung gefundcn werden, vergleichbar. Dics kann als Hinweis auf den dreidimensional aromatischen Cha- rakter des Systems angesehen werden. Im Hinblick auf die Untersuchung groOer Systeme ist
der Vergleich zwischen scmiempirischen und ab-initio-Er- gebnissen wichtig. Hier zeigt sich uberraschenderweise,

A. Rurkhardt u. a., Elektroncn-Lokalisicrungs-Funktion in cl~.sn-Bor-Clusterii 44 1 _- -- ____-------------
da13 die Topologie der ELF kaum verdndert wird. Fur die betrachleten Cluster reichcn ofienbar einfache Extended Hiickcl-Rechnungen aus.
Die Elcktronen-Lokalisierungs-Funktion ELF eignet sich hervorragend zur Interpretation der Bindungsver- hailtnisse in Bor-Clustern, Diese und friihere Arbeiten zei- gcn, da13 die Beschreibung unterschiedlichster Bindungs- typcn durch die ELF mijglich ist.
Ilicse Arbcil wurde vom Foiids dcr Chcmischen Iiidustrie geffir- dert. Xu bcsondcreni Dank sind wir J. Flcrd fiir die Ubcrlassung des Programmes GRAPA vcrpflichlet.
Litera tur
[ 11 K. .Z G i k p i e , R. S . Nvholrrt, Q. Ilev. Chcm. Soc. 11 ( 1 957) 329
[2] A. D. Becke, K. E. Edgecumbe, J, Chcrri. Phya. 92 (1990) 5397
131 A . Savin, A . U. Becke. J. Flalad, R. Nesper, H . Preup, ZJ. G. von Schnermg, Angew. Chern. 103 (1991) 421; An- gcw. Chem. Int. Ed. Engl. 30 (1991) 409
141 A. Suvin, H. J. Had, J . Had, H . PreuJ, II. G. von Schne- ring, Angew. Chem. 104 (1992) 185; Angew. Chem. hit. Ed. Engl. 31 (1992) 185
[ 5 ) A . Suvin, 0. Jepsen, .I F h d , 0. U. Andersen, ii. PreuJ, H. G. vonSchnrring, Angcw. Chcrri. 104 (1992) 186; An- gew. Chem. Int. Ed. Engl. 31 (1992) 187
[61 G. Igel-Munn, H . J . Poppr, personlichc Mitteilung [7] 12. Ahlrrcbs, M. Bur, M. Ifuser, H. / lorn, C. Kolrrrel,
Chetn. Phys. Lett. 162 (1989) 165 [81 G. fgel-Mcrnn, H. Sfofl, If. Preiij’, Mol. Pfiys. 65 ( 7 988)
f 321 [9] I<. Hoffrriann, Progranim ICON QCPL 344, lndlaria Uni-
versity, 13loomington [lo] J. Flud, EX. Praschio, f3. Mieblic-b, Prograrrim GRAPA,
lristitul fur Theoretisclic C’hcmie dcr Univcrsitat Stuttgarl 1989
[ I 11 A. Burkhardf, F J++/,qwr, Prograrnm KOBY ELF, Max- Planck-Imtitut fur ~esthorperfor ichui i~ i 991
[I 21 U. Wtdig, R. Nesper, H. G. von Sclinering, Prograrnmpa- kct VISl-’, Max-Planck-Itis1 ilut fiir ~estk8rpcrrorschung 1990
[13] D. J. Swanfon, R. Ab/ric,bs, Thcor. C:him. Acla 75 (1 989) 163
[I41 I: Menaekes, I? Pae/mld, R. UOEW, D. Bla,x?r, Angcw. Chcni. 103 (1991) 199
[ 151 MOIPRO ist ein Ab-initio-Pro~raIIIm-Pak~t von H.J. Werni,r und P J, Knowler mit Bcitragcn von .I AlrnIOf, R. Amos, S. Elbert, K. Iiampel, W. Meycr, K. I’elcrson, R. Pikyr iind A . Stone (0ibilal-L.cikalisier~iig vori W. Mqyer, installierl auf der (‘ray-2 diirch H. Sloll)
[I61 P Py.ykkii, Y . Zuo, J. Plys. Chern. 94 (1990) 7753 [I71 I?. Jonoscbek, Nachr. Cheiii. Tech. Lab. 40 (1992) 421 I 181 I . l! KuZnPlsov, 0. hl. Vinitskii. K . A . Solntsrv,
N. 7: Kuznelsov, L. A. Bulriian, Kuss. J. Iriorg. Chcrn. 32 (1987) 1803
[I91 V, I. Privalov, K P 7hasov. M. A. Mdadzize, D. h% V’inits- kii, K. A. Solnlsev, Y. A. BiisIoev, N. T Uuznetsov, Russ. J. Inorg. Chem. 31 (1986) 633
[201 A. M. Mefwl , 0. l? Cburkiri. h’. A. Solnlwv, N T. Kuznet- sov, Russ. 5. Inorg. Chcrn. 33 (1988) 1292
[21] 0. A. Kkier, J . Bicrruno, I+’. N. Lipscomb, Inorg. Chem. 19 (1980) 216
[22] J. I!. Hall, Jr., W. ff. I,ipscomh, Inorg. Chem. 13 (1974) 710
[23] U. 7biwy, Dissertation, Llnivcrsitiit Stuttgart (1 992) 1241 J. Tlzi:sirrg, U.: Prep&, D. 7hkrJJ. H . G. von Sohnering,
1251 ( 2 Erhard/, R. Abli-ichs, Theo. Chim. Acta 68 (1985) 231 Z. Naturrorsch. 4Sb (1990) I84
Anschr. d . Verf.: Dr. A. Ikirkhardt, Dr. U. Wedig, Prof. Dr. H. G. von Schnering Max-l’lanck-Inslitut Cur ~~stkorirerfotschullg Heisenbergsir. 1 W-7000 StutIgart 80
Priv. Doz. Dr. A. Savin lrirtitut fui Theoreliwhc Chcmie der Uiiiversital P faffciiwaldririg 5 5 W-7000 Siuttgart 80